Attached files
| file | filename |
|---|---|
| EX-10.36 - EX-10.36 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex103676696.htm |
| EX-10.8 - EX-10.8 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex108496d9b.htm |
| EX-21.1 - EX-21.1 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex2115fefc7.htm |
| EX-31.1 - EX-31.1 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex3114f7fef.htm |
| EX-10.9 - EX-10.9 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex10939864a.htm |
| EX-10.16 - EX-10.16 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex1016101f8.htm |
| EX-10.25 - EX-10.25 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex1025d29f7.htm |
| EX-32.1 - EX-32.1 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex321f2a4aa.htm |
| EX-31.2 - EX-31.2 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex3120ca05e.htm |
| EX-23.1 - EX-23.1 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex231daab4b.htm |
| EX-10.10 - EX-10.10 - EPIRUS Biopharmaceuticals, Inc. | eprs-20151231ex10109e63d.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
☒ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2015
◻TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File No. 000-51171
EPIRUS BIOPHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
04-3514457 |
|
(State or other jurisdiction of Incorporation or organization) |
|
(IRS Employer Identification Number) |
|
|
|
|
|
699 Boylston Street Eighth Floor Boston, MA 02116 |
|
02116 |
|
(Address of Principal Executive Offices) |
|
(Zip Code) |
(617) 600-3497
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Exchange Act:
|
Common Stock, $0.001 par value per share |
|
The NASDAQ Stock Market LLC |
|
(Title of each class) |
|
(Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ◻ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ◻ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ◻
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ◻
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
◻ |
|
|
Accelerated filer |
☒ |
|
|
|
|
|
|
|
|
Non-accelerated filer |
◻ |
(Do not check if a smaller reporting company) |
|
Smaller reporting company |
◻ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes ◻ No ☒
As of June 30, 2015, the last business day of the registrant’s last completed second fiscal quarter, the aggregate market value of the Common Stock held by non-affiliates of the registrant was approximately $97,043,791 based on the closing price of the registrant’s Common Stock, as reported by the NASDAQ Capital Market, on such date. Shares of Common Stock held by each executive officer and director and certain affiliate stockholders known by the registrant to own 5% or more of the outstanding stock based on public filings and other information known to the registrant have been excluded since such persons may be deemed affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares of the registrant’s Common Stock, par value $0.001 per share, outstanding as of March 25, 2016 was 26,184,349.
The exhibit index as required by Item 601(a) of Regulation S-K is included in Item 15 of Part IV of this report.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s proxy statement with respect to the registrant’s 2016 Annual Meeting of Stockholders, which is to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2015, are incorporated by reference into Part III of this Form 10-K.
EPIRUS Biopharmaceuticals, Inc.
Form 10‑K
Explanatory Note
The registrant qualified as a smaller reporting company for the fiscal year ended December 31, 2015. Because the registrant's public float exceeded $75.0 million on the last business day of the registrant’s last completed second fiscal quarter, the registrant will qualify and report as an accelerated filer in 2016, starting with its quarterly report on Form 10-Q for the quarter ending March 31, 2016. Pursuant to the rules of the Securities and Exchange Commission, the registrant is relying upon the smaller reporting company scaled disclosure rules for portions of this annual report on Form 10-K.
1
CAUTIONARY NOTE REGARDING FORWARD‑LOOKING STATEMENTS
Various statements throughout this report are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements may appear throughout this report. Words such as “believe,” “anticipate,” “expect,” “estimate,” “intend,” “plan,” “project,” “will be,” “will continue,” “will result,” “seek,” “could,” “may,” “might,” or the negative of these terms and similar expressions or words, identify forward-looking statements. Forward-looking statements are based upon current expectations that involve risks, changes in circumstances, assumptions and uncertainties. Important factors that could cause actual results to differ materially from those reflected in our forward-looking statements include, among others:
|
· |
our inability to obtain regulatory approval for, or successfully commercialize, our leading product candidate, BOW015, or our other product candidates, BOW050, BOW070, BOW080, BOW090 and BOW100; |
our inability to access sufficient capital resources to fund our operations as a going concern beyond the second quarter of 2016;
our inability to secure and maintain relationships with collaborators and single-source contract manufacturers;
our history of operating losses and inability to ever become profitable;
our limited history of complying with public company reporting requirements;
our limited sales and marketing infrastructure;
uncertainty and volatility in the price of our common stock;
our inability to develop, implement and maintain appropriate internal controls;
our inability to remediate the material weaknesses in our internal control over financial reporting;
uncertainty as to our employees’, independent contractors’ and other collaborators’ compliance with regulatory standards and requirements and insider trading rules;
uncertainty as to the relationship between the benefits of our product candidates and the risks of their side‑effect profiles;
our inability to effectively compete with the reference biologic products for our biosimilar product candidates and from other pharmaceuticals approved for the same indication as the reference biologic products;
our inability to complete our in-house cell line and process development activities;
dependence on the efforts of third-parties to conduct and oversee our clinical trials for our product candidates, to manufacture nonclinical and clinical supplies of our product candidates, to store critical components of our product candidates, and to commercialize our product candidates;
uncertainty relating to U.S. regulatory and legal landscapes and a fragmented payer market;
2
the extent of government regulations;
uncertainty of clinical trial results;
a loss of any of our key management personnel;
our inability to develop or commercialize our product candidates due to intellectual property rights held by third parties and our inability to protect the confidentiality of our trade secrets; and
the cost and effects of potential litigation.
All written and verbal forward‑looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We caution investors not to rely too heavily on the forward‑looking statements we make or that are made on our behalf. We undertake no obligation, and specifically decline any obligation, to update or revise publicly any forward‑looking statements, whether as a result of new information, future events or otherwise.
We encourage you to read Management’s Discussion and Analysis of our Financial Condition and Results of Operations and our consolidated financial statements contained in this annual report on Form 10‑K. We also encourage you to read Item 1A of Part I of this annual report on Form 10‑K, entitled “Risk Factors,” which contains a more complete discussion of the risks and uncertainties associated with our business. In addition to the risks described above and in Item 1A of this report, other unknown or unpredictable factors also could affect our results. Therefore, the information in this report should be read together with other reports and documents that we file with the Securities and Exchange Commission (“SEC”) from time to time, including on Form 10‑Q and Form 8‑K, which may supplement, modify, supersede or update those risk factors. As a result of these factors, we cannot assure you that the forward‑looking statements in this report will prove to be accurate. Furthermore, if our forward‑looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward‑looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all.
Explanatory Note
On July 15, 2014, EPIRUS Biopharmaceuticals, Inc., a Delaware corporation and private company, or Private Epirus, completed its merger with EB Sub, Inc., a wholly-owned subsidiary of Zalicus Inc., a Delaware corporation, or Zalicus, or the Merger. As part of the Merger, Zalicus was renamed EPIRUS Biopharmaceuticals, Inc., or Public Epirus, and Private Epirus was renamed EB Sub, Inc., or EB Sub.
In this annual report, unless the context specifically indicates otherwise, “the Company,” “we,” “us,” “our,” and “Epirus,” refer to Public Epirus and its subsidiaries following the Merger, effective on July 15, 2014, and to Private Epirus and its subsidiaries prior to the Merger.
“EPIRUS Biopharmaceuticals”, “In Market, For Market”, “SCALE” and “Infimab” are registered trademarks of EPIRUS Biopharmaceuticals, Inc. Our logos and trademarks are the property of EPIRUS Biopharmaceuticals, Inc. All other brand names or trademarks appearing in this annual report are the property of their respective holders. Use or display by us of other parties' trademarks, trade dress, or products in this prospectus supplement is not intended to, and does not, imply a relationship with, or endorsements or sponsorship of, us by the trademark or trade dress owners.
3
Overview
We are a global biopharmaceutical company focused on building a pure-play, sustainable and profitable biosimilar business by improving patient access through cost-effective medicines. Our ability to deliver on this vision is anchored in our strong technical platform and pragmatic approach to development and commercialization.
Our business focuses on biosimilars, which are biologic drugs that are demonstrated to be “highly similar” to a previously approved biologic drug, known as a “reference product”. A biosimilar has to undergo extensive analytical characterization and prove similarity in efficacy and safety to the reference product in order to be approved by regulators.
Global healthcare systems continue to be challenged with expensive therapies. According to Express Scripts®, in the United States, the cost of a biologic drug accounted for approximately 40% of total drug spending in 2014. These cost pressures provide a large opportunity for biosimilars. Over the next decade, biologic drugs with approximately $90 billion worth of annual sales will lose patent protection, based on projected global sales estimates from EvaluatePharma®. Approximately half of the biosimilar market opportunity is outside the United States. We believe that biosimilars are likely to play a crucial role in reducing healthcare costs and improving patient access.
We are headquartered in Boston, Massachusetts, with laboratories and technical capabilities, including our proprietary CHOBC® cell line platform, in Utrecht, the Netherlands, and business operations, clinical and regulatory teams based in Zug, Switzerland.
As a biopharmaceutical company charting a course for long-term success, we are deeply committed to building a focused, sustainable and profitable pure-play biosimilar business. Specifically, we believe the path to achieving our goals has a foundation in the following five key strategies:
|
· |
build and utilize technical capabilities to optimize product characteristics and manage costs; |
|
· |
plan our pipeline to gain cost synergies from product development, through clinical development and to eventual commercialization; |
|
· |
focus on global markets where the regulatory, legal and commercial landscapes are more evolved while evaluating and preparing in the long term for potential United States market entry; |
|
· |
enter into partnership arrangements where we expect to retain substantial economics over time; and |
|
· |
implement an efficient global tax structure. |
Since our inception, we have maintained in-house technical capabilities supported by external vendor laboratories. In September 2015, we acquired our primary vendor laboratory, Bioceros Holding B.V., a Netherlands company, or Bioceros, to expand our biosimilar pipeline and to vertically integrate our product development capabilities. As a result of the acquisition, Bioceros has become our wholly-owned subsidiary, renamed Epirus Biopharmaceuticals (Netherlands) B.V.
Bioceros was focused on the development of monoclonal antibodies (mAbs). We acquired Bioceros’ proprietary CHOBC cell line platform, all related intellectual property rights, a fully equipped laboratory and bioreactor capabilities designed for the development of mAbs and protein therapeutics, with a focus on biosimilars, including capabilities for the production of three new biosimilar product candidates to add to our pipeline of biosimilar product candidates, as further described below. We believe that the addition of Bioceros staff and capabilities, combined with existing in-house expertise, provides us with a strong technical foundation to accelerate product development and optimize product quality. Specifically, we believe that our ability to rapidly and efficiently conduct and repeat experiments to optimize product quality and cell line titers and process yields will help us to reduce regulatory risk and manage long term cost of goods.
4
We currently have a robust and cohesive pipeline of five biosimilar product candidates in the autoimmune and inflammation areas, including BOW015, a biosimilar version of Remicade® (infliximab), BOW050, a biosimilar version of Humira® (adalimumab), BOW070, a biosimilar version of Actemra® (tocilizumab), BOW090, a biosimilar version of STELARA® (ustekinumab) and BOW100, a biosimilar version of SIMPONI® (golimumab).
We expect the standardization of our technology platform and the clustering of the therapeutic area will allow us to have a greater degree of focus, synergy and cost management across product development, manufacturing, clinical and commercialization. Additionally, we have expanded our therapeutic focus with a rare disease product candidate, BOW080, a biosimilar version of Soliris® (eculizumab). We believe there will be a significant unmet need for cost-effective therapies for rare diseases. Cumulatively, these six reference products generated approximately $29.2 billion in global innovator sales in 2014, according to EvaluatePharma. We estimate this to be a biosimilar market opportunity of more than $9 billion. Furthermore, there are over 20 other mAbs with near-term patent expiries clustered in five therapeutic areas, which we believe will provide mid- to long-term pipeline growth options for us, including expanding our rare disease business.
For our lead product candidate, BOW015, we have reported bioequivalence, efficacy and safety data from a previous Phase 1 study in healthy volunteers and a Phase 3 study in active rheumatoid arthritis patients, both of which demonstrated the equivalence of BOW015 to Remicade. Our Phase 3 study included an open label phase, where we demonstrated that patients can be safely initiated and effectively maintained on BOW015 for 58 weeks, and that patients can be safely switched from Remicade to BOW015 and effectively maintained out to 58 weeks. Using this regulatory package, in 2014, in collaboration with our commercialization partner Sun Pharmaceutical Industries Ltd., or Sun, we launched BOW015 as the first infliximab biosimilar in India, sold under the brand name InfimabTM. We have also filed for approval in additional Southeast Asian and Latin American countries.
To date, nearly 1,000 patients have been treated with BOW015. We expect this early experience to provide valuable in-market experience while we continue making BOW015 available to patients globally, to support filings in developed markets, and to build important commercial experience.
In November 2015, we completed manufacturing readiness for BOW015 to support the initiation of a global clinical program for BOW015. In February 2016, we initiated this global clinical program, which we expect to support regulatory filings in developed markets such as Europe and North America. The UNIFORM (Understanding BOW015 (infliximab-EPIRUS) and reference infliximab (Remicade®) in patients with active rheumatoid arthritis on stable doses of methotrexate) study is a 58-week, double-blind, one-to-one randomized, comparator-controlled multi-center global study to compare safety, efficacy and immunogenicity and demonstrate clinical equivalence of BOW015 with Remicade. We plan to enroll over 500 patients with active rheumatoid arthritis in the UNIFORM study, which will be conducted at sites in Europe, North America and Latin America. The primary endpoint at week 16 for the study will assess the proportion of patients that meet ACR20, which is a 20 percent or greater improvement in American College of Rheumatology assessment. We have been designing this clinical program in consultation with European regulatory bodies in order to obtain the data that may be necessary to support potential approval of BOW015 in developed markets.
Leveraging early commercial experience and our robust global clinical program, we expect to file for approval of BOW015 in developed markets, such as Europe and North America, in 2017. For the remainder of our pipeline, we expect that our filings will be global and harmonized.
We believe that the biosimilar market in the United States will likely be attractive over the long term. Currently there are near term challenges in the United States, including an evolving but uncertain regulatory framework and legal situation, a complex commercial environment, a fragmented payor market and a market which focuses on switching patients off existing reference drugs.
We believe that the most attractive market opportunity for biosimilars in the near term is outside of the United States. Many global markets outside the United States have more defined regulatory environments, limited legal encumbrance, commercial precedence and payors seeking lower cost biologics. For each of the markets outside of the United States, which collectively represent approximately 50% of the global opportunity for biosimilars, we intend to
5
take specific approaches to build profitable partnerships and, to date, we have grown the global distribution coverage of BOW015 in over 70 countries.
For European markets and select additional territories, in July 2015, we entered into an agreement with Swiss Pharma International AG, an affiliate of Pharmaceutical Works Polpharma S.A., or, together with Swiss Pharma International AG, Polpharma, for the development and commercialization of BOW015, BOW050 and BOW070 in certain designated territories. The designated territories include the European Union (with the exception of Austria, Belgium, Denmark, Finland, Luxembourg, the Netherlands and Sweden, which are reserved for us, along with Switzerland and Norway), together with certain countries in the Middle East, Turkey, Russia, and other countries comprising the Commonwealth of Independent States, or CIS.
In local production markets, such as China, where local authorities mandate or strongly encourage local production as a condition for regulatory and/or commercial acceptance, we intend to collaborate with local partners to enable in-country production of our products. We have entered into agreements with Livzon Mabpharm Inc., or Livzon, for the global development and commercialization of certain antibodies or related biological compounds, including BOW015 and BOW070, for China and associated markets.
In India, the current market and near-term markets, such as Latin America, in which our current BOW015 data package is expected to be sufficient for approval, we intend to pursue near-term product launches. This would enable us to gain valuable in-market patient exposure and commercial experience while we continue to work on making BOW015 available globally. This early commercial experience will strengthen our regulatory filings for Europe, the United States and additional global markets. In collaboration with our commercialization partner Sun, we launched BOW015 in India in November 2014, under the brand name Infimab. We expect to utilize our existing regulatory data package to gain regulatory approval for BOW015 in additional countries. In May 2015, we also entered into a development and future distribution agreement with mAbxience S.A., or Mabxience, a company organized and existing under the laws of Uruguay and a wholly owned subsidiary of CHEMO Group, for BOW015 in Latin American markets, including Argentina, Chile, Ecuador, Paraguay, Uruguay and Venezuela.
Finally, in the United States, we intend to retain rights and evaluate market conditions prior to determining a commercial plan commensurate with our objectives to create value. We are currently evaluating development and commercial options including partnerships, hybrid profit sharing approaches and potentially building or acquiring commercial infrastructure.
The combination of a global sales approach and a focus on deals which would allow us to retain substantial economic value is supported by our Swiss-based tax structure, which we believe will allow us to efficiently manage future cash for operations outside the United States.
Our long-term ability to deliver these important medicines to patients worldwide can only be achieved through a disciplined approach. Our experienced management team plans to address the diverse regulatory, legal and commercial landscape by gaining near-term market access, creating a strong technical platform with opportunities for sustainable pipeline growth, and finding tax-optimized partnership deals that maximize future value to provide us with a path forward to build a sustainable and profitable biosimilar business.
Going Concern
We believe that our existing cash and cash equivalents will be sufficient to fund our operations into the second quarter of 2016. As of December 31, 2015, our total unrestricted cash and cash equivalents was $31.5 million. We will need to raise additional capital in the near term to fund our operating requirements and continue as a going concern. If we cannot raise the money that we need in order to continue to operate our business, we will be forced to delay, scale back or eliminate some or all of our proposed operations. If any of these were to occur, there is a substantial risk that our business would fail.
This need to raise additional capital in the near term coupled with our recurring losses from operations and our stockholders’ deficit raise substantial doubt about our ability to continue as a going concern. As a result, our independent
6
registered public accounting firm included an explanatory paragraph in its report on our consolidated financial statements for the year ended December 31, 2015, which appear elsewhere in this annual report, with respect to this uncertainty. This uncertainty may adversely affect our ability to raise additional capital.
Products
Our product pipeline contains six products at different stages of development. The most advanced of these is BOW015 (infliximab), which has received marketing and manufacturing approval in India, and for which we have reported favorable Phase 1 and Phase 3 clinical data. Our other pipeline products, BOW050 (adalimumab), a proposed biosimilar to Humira, BOW070 (tocilizumab), a proposed biosimilar to Actemra, BOW080 (eculizumab), a proposed biosimilar to Soliris, BOW090 (ustekinumab), a proposed biosimilar to STELARA and BOW100 (golimumab), a proposed biosimilar to SIMPONI, are in preclinical development.
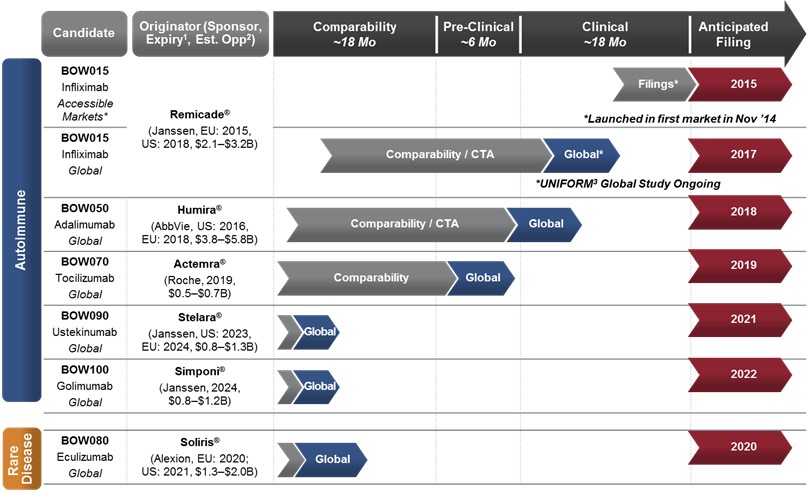
1. Ark Patent Intelligence and other public databases 2. Assumes 40% price discount, 40-60% biosimilar. 3. UNIFORM Study – Understanding BOW015 ( infliximab-EPIRUS) and reference infliximab (Remicade®) in patients with active rheumatoid arthritis on stable doses of methotrexate.
*BOW015 launched in its first market in collaboration with Sun under the trade name Infimab™ in India; additional near-term filings targeted for select accessible markets; Remicade is a registered trademark of Johnson and Johnson; Humira is a registered trademark of AbbVie; Actemra is a registered trademark of Chugai Seiyaku Kabushiki Kaisha Corp., a member of the Roche Group; Stelara is owned and marketed by Centocor Ortho Biotech Inc, a wholly owned subsidiary of Johnson & Johnson; Simponi is marketed by Janssen Biotech Inc; Soliris is a registered trademark of Alexion Pharmaceuticals, Inc.
BOW015 (Infliximab)
Our lead product candidate is BOW015 (infliximab, reference biologic Remicade), a monoclonal antibody against tumor necrosis factor alpha (TNF-α). It is intended for the treatment of inflammatory diseases including rheumatoid arthritis, Crohn's Disease, ankylosing spondylitis, psoriatic arthritis and psoriasis. According to EvaluatePharma, Remicade, a prescription product marketed globally by Johnson & Johnson, Merck Schering and Mitsubishi Tanabe, generated approximately $8.8 billion in global innovator sales in 2014. We have conducted extensive bioanalytical and physicochemical comparisons of BOW015 to Remicade and have data from a Phase 1 study in the
7
United Kingdom and a Phase 3 double blind comparator study in India demonstrating bioequivalence, safety, quality and efficacy of BOW015.
On September 23, 2014, we announced positive long-term data (58 week) from an efficacy and safety trial comparing BOW015 to Remicade. The study consisted of a 16 week, double blinded, head to head comparison with Remicade for safety and efficacy followed by an open label phase where Remicade responders were switched to BOW015 and all patients were followed for the duration of the study. The study met its primary endpoint of ACR20 response, the American College of Rheumatology criteria for clinical improvement in patients with rheumatoid arthritis, indicating a 20% improvement across a series of diagnostic parameters. These patients were then followed out to week 58 in an open label phase of the trial, with BOW015 patients remaining on BOW015 and Remicade responders being switched to BOW015 for the remainder of the 58 weeks. Furthermore, these data suggest that patients can safely be started and durably maintained on BOW015 and that patients can safely be switched from Remicade to BOW015.
Using this regulatory package, we filed for and received approval in India in September 2014, and have also filed for approval in additional Southeast Asian and Latin American countries. In collaboration with our commercialization partner Sun, we launched BOW015 in India in November 2014. BOW015 is sold under the brand name Infimab and is the first infliximab biosimilar approved in India. To date, nearly 1,000 patients have been treated with BOW015.
The early commercial experience in India and near-term anticipated experience in select Southeast Asian and Latin American countries, will provide valuable in-market experience while we continue making BOW015 available to patients globally. This experience will support filings in developed markets, such as the United States and Europe, and will build important commercial experience.
In November 2015, we completed manufacturing readiness for BOW015 to support the initiation of a global clinical program for BOW015. In February 2016, we initiated this global clinical program, which will support regulatory filings in established markets such as Europe and North America. The UNIFORM Study is a 58-week, double-blind, one-to-one randomized, comparator-controlled multi-center global study to compare safety, efficacy and immunogenicity and demonstrate clinical equivalence of BOW015 with Remicade. We plan to enroll over 500 patients with active rheumatoid arthritis in the UNIFORM study, which will be conducted at sites in Europe, North America and Latin America. The primary endpoint at week 16 for the study will assess the proportion of patients that meet ACR20. We have been designing this clinical program in consultation with European regulatory bodies in order to obtain the data that may be necessary to support potential approval of BOW015 in developed markets. We expect to file for approval of BOW015 in developed markets, including Europe and North America, in 2017.
We have agreements with Polpharma for the development and commercialization of BOW015 in certain developed markets, with Livzon for the development and commercialization of BOW015 in certain local production markets, and with Sun and Mabxience for the development and commercialization of BOW015 in certain accessible markets. In total, over 70 countries have global distribution coverage for BOW015.
BOW050 (Adalimumab)
We are currently developing and evaluating commercialization opportunities for BOW050 as a proposed biosimilar to Humira in a range of target markets, including our collaboration with Polpharma for the development and commercialization of BOW050, among our other product candidates, in the territories designated under the Polpharma agreement. Humira (marketed by AbbVie) is an inhibitor of TNF-α used to treat inflammatory diseases, including rheumatoid arthritis and certain other forms of adult and pediatric arthritis, ankylosing spondylitis, inflammatory bowel disease, and chronic psoriasis and psoriasis. According to EvaluatePharma, Humira generated approximately $12.9 billion in global innovator sales in 2014. Physiochemical characterization of BOW050 is ongoing and the product may enter clinical trials in late 2016, providing a path to an anticipated global filing in 2018.
8
BOW070 (Tocilizumab)
We are in the comparability phase of development of BOW070, a proposed biosimilar version of Actemra (marketed by Genentech/Roche). Actemra is an immunosuppressive drug for the treatment of rheumatoid arthritis, polyarticular arthritis and systemic juvenile idiopathic arthritis. According to EvaluatePharma, Actemra generated approximately $1.3 billion in global innovator sales in 2014. We are targeting a global filing for BOW070 in 2019.
We have agreements with Polpharma for the development and commercialization of BOW070 in certain developed markets and with Livzon for the development and commercialization of BOW070 in certain local production markets.
BOW080 (Eculizumab)
We are in the comparability phase of development of BOW080, a proposed biosimilar version of Soliris (marketed by Alexion Pharmaceuticals, Inc.). Soliris is currently indicated to treat ultra-rare blood disorders, including paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS). According to EvaluatePharma, Soliris generated approximately $2.2 billion in global innovator sales in 2014. We are targeting a global filing for BOW080 in 2020.
BOW090 (Ustekinumab)
We are in the comparability phase of development of BOW090, a proposed biosimilar version of STELARA (marketed by Janssen Pharmaceuticals, Inc.). STELARA is an immunosuppressant drug for the treatment of plaque psoriasis and psoriatic arthritis. According to EvaluatePharma, STELARA generated approximately $2.1 billion in global innovator sales in 2014. We are targeting a global filing for BOW090 in 2021.
BOW100 (Golimumab)
We are in the comparability phase of development of BOW100, a proposed biosimilar version of SIMPONI (marketed by Janssen Pharmaceuticals, Inc.). SIMPONI is an inhibitor of TNF-α used to treat inflammatory diseases, including rheumatoid arthritis, ankylosing spondylitis, ulcerative colitis, and psoriatic arthritis. According to EvaluatePharma, SIMPONI generated approximately $1.9 billion in global innovator sales in 2014. We are targeting a global filing for BOW100 in 2022.
Management
Our senior management team and our board of directors have worked for prominent biotechnology and pharmaceutical companies including Amgen, Biogen Idec, Pfizer, Quintiles, Wyeth (acquired by Pfizer), Genzyme (acquired by Sanofi), Shire, Cephalon, Millennium, Takeda, BioAssets (acquired by Cephalon), Cubist Pharmaceuticals (acquired by Merck), Invida (acquired by Menarini), Therion Biologics and ToleRx. Our president and chief executive officer, Amit Munshi, was a co‑founder, and the chief business officer at Kythera Biopharmaceuticals (acquired by Allergan Plc), which underwent a successful initial public offering on NASDAQ in 2013. Mr. Munshi has more than 25 years of pharmaceutical and biotechnology experience in both the United States and internationally, including general management, product development, licensing and business development.
Strategy
We believe that successfully building a pure-play biosimilar business requires a clear path toward sustainability and profitability. We have cultivated a therapeutically focused product portfolio and are diligently aware of profitability and sustainability. In order to reach our objectives, we have chosen to focus on markets that meet three important criteria: (1) clear, precedent‑driven regulatory pathway; (2) minimal exposure to potential patent encumbrances; and (3) a commercially viable path. We believe that the most attractive market opportunity for biosimilars in the near term is outside of the United States. Europe and the markets in the Middle East, Latin America and Asia have defined regulatory environments, limited legal encumbrance, commercial precedence and payors seeking lower cost biologics. For each of
9
the markets outside of the United States, which collectively represent approximately 50% of the global opportunity for biosimilars, we intend to take specific approaches to build profitable partnerships.
We believe the market in the United States is large and attractive over the long term, however, in the near term, challenges include an evolving but uncertain regulatory framework and legal situation, a complex commercial environment with a need to focus on switching patients off existing reference drugs, and a fragmented payor market.
Our key priorities in executing on our strategy include:
|
· |
In Europe, we intend to commercialize our products using a licensing or distribution model in conjunction with direct sales. These markets are expected to be the financial anchor of our business in the near- to mid-term. Europe has an existing regulatory approval pathway and a patent environment we believe offers a clear path forward for our pipeline. From a commercial perspective, the European market for biosimilars is strengthened by the desire of governments to reduce overall healthcare expenditure. Through a combination of substitution rules, regional tenders and political pressure to introduce biosimilars, Europe represents a commercially tractable market. For European markets, in July 2015, we entered into an agreement with Swiss Pharma International AG, an affiliate of Pharmaceutical Works Polpharma S.A., for the development and commercialization of BOW015, BOW050 and BOW070 in certain designated territories. The designated territories include the European Union (with the exception of Austria, Belgium, Denmark, Finland, Luxembourg, the Netherlands and Sweden, which are reserved for us, along with Switzerland and Norway), together with certain countries in the Middle East, Turkey, Russia, and other countries comprising the Commonwealth of Independent States, or CIS. |
|
· |
In local production markets, such as China, we intend to collaborate with local partners to enable in‑country production of our products. In local production markets, local authorities mandate or strongly encourage local production, and additional clinical work above and beyond that submitted for Indian regulatory approval will likely be necessary to secure approval for BOW015. In these markets, several biologics have already seen their key patents expire. Furthermore, we expect to establish local commercialization partnerships in these, often tender driven, markets. Any necessary additional local studies would likely be supported by our current or future local partners. For instance, in China, we have entered into an Exclusive License and Collaboration Agreement and a New Collaboration Compound Supplement to the Collaboration Agreement with Livzon for the global development and commercialization of certain antibodies or related biological compounds, including BOW015 and BOW070. Finally, where appropriate, we will leverage our proprietary SCALE manufacturing technology (as described further below) to generate In Market, For MarketTM manufacturing solutions in these markets. |
|
· |
In near-term markets, such as India and Latin America, in which our current regulatory data are expected to be sufficient for approval, we intend to pursue product launches. These markets are likely to reference and accept as the basis for approval the Indian BOW015 regulatory package and the current data set and also allow importation of BOW015 manufactured outside of such markets. These markets often present no innovator patent protection. We expect that this will enable us to gain in-market patient exposure needed to support eventual filings in developed markets and allow us to build important commercial and commercial support services with and for our partners. Under the terms of our existing license agreement, Sun is responsible for the commercialization of BOW015 in India and selected Southeast Asian and North African countries. In collaboration with Sun, we launched BOW015 in India in November 2014. BOW015 is sold under the brand name Infimab and is the first infliximab biosimilar approved in India. We have also filed for approval in additional Southeast Asian and Latin American countries. In May 2015, we also entered into a development and future distribution agreement with mAbxience S.A., a company organized and existing under the laws of Uruguay and a wholly owned subsidiary of CHEMO Group, for BOW015 in Latin American markets, including Argentina, Chile, Ecuador, Paraguay, Uruguay and Venezuela. |
|
· |
Finally, in the United States, we intend to evaluate our commercial plan commensurate with our objectives to create value. As the regulatory and legal environments in the United States become clearer, |
10
we anticipate seeking a commercial partner or alternative commercial model for BOW015, which may include contracting directly with payors or other third‑party entities. We are currently evaluating development and commercial options including partnerships, hybrid profit sharing approaches and potentially building or acquiring commercial infrastructure. |
Our strategy for commercial success relies on tailored approaches to address the diversity of our target global markets. Additionally, we intend to leverage our development and commercial experience with BOW015 to both advance our pipeline and our overall direct sales infrastructure.
Europe
Europe has an established regulatory framework for biosimilars, and the European Medicines Agency (EMA) has approved Celltrion's infliximab program under the trade names Inflectra/Remsima®. The initial launch of Inflectra/Remsima and the approval of over 20 biosimilars over the last decade suggest that the legal landscape in Europe is conducive to the introduction of biosimilars. In general, the European patent landscape provides for far fewer patent extensions for manufacturing, method of use, or processes, as compared to the patent landscape in the United States. Finally, the European market for biosimilars is strengthened by the desire of governments to reduce overall healthcare expenditure. Through a combination of substitution rules, regional tenders and political pressure to introduce biosimilars, Europe represents a commercially attractive market.
Our operating strategy across Europe and other select territories is to enter into profit-sharing collaborations in order to retain maximum value. In July 2015, we entered into a Collaboration Agreement with Polpharma, for the development and commercialization in certain designated territories of our product candidates, BOW015, BOW050 and BOW070. The designated territories include the European Union (with the exception of Austria, Belgium, Denmark, Finland, Luxembourg, the Netherlands and Sweden, which are reserved for us), together with certain countries in the Middle East, Turkey, Russia, and other countries comprising the Commonwealth of Independent States, or CIS. We will also retain exclusive rights for the three products in North America and other markets outside of the designated territories, including Switzerland and Norway. Polpharma is a leading generics player based in Poland, operating across Europe, the Caucasus and Central Asia, with manufacturing subsidiaries in Russia and Kazakhstan. Polpharma is among the top 20 generic drug manufacturers in the world with annual sales of approximately $1 billion. Polpharma’s portfolio includes about 600 products with another 200 in pipeline.
Local production markets, including China
In local production markets, such as China, governments either mandate or have a strong preference for local manufacture and supply of pharmaceutical products and have implemented frameworks and/or established various incentives for such local production. These incentives may include facilitating access to funding, acceleration of the regulatory review process, improved or preferential access to government tenders and direct or indirect trade barriers on imported products. In these markets, there is a clear regulatory and patent landscape. Also, the commercial opportunity is substantial, tractable and protectable. In most of these markets, the transference of product manufacturing is rewarded by government incentives, access to tenders, and ability to restrict competition from imported products.
In China, the central and provincial governments encourage local production of biopharmaceuticals. In September 2014, we entered into an Exclusive License and Collaboration Agreement (the “Livzon Collaboration Agreement”) with Livzon for the global development and commercialization of certain antibodies or related biological compounds, including BOW015. In September 2015, we entered into a New Collaboration Compound Supplement with Livzon to add BOW070 as a compound to be governed by the Livzon Collaboration Agreement. Livzon is a fully integrated pharmaceutical company based in Guangdong Province, China, with over 5,000 employees, multiple production facilities across China, and approximately $730 million in revenue from over 200 marketed products across a range of therapeutic areas. Livzon is focused on monoclonal antibody development and production, leveraging single use disposable systems that we expect will be compatible with the optimized processes we are currently developing with our partners for BOW015 and BOW070.
11
Near-term markets, including India and Latin America
In India, our current market and near-term markets, including Southeast Asia and Latin America, regulatory frameworks are clear and patent environments allow for freedom to operate. From a commercial perspective, innovator drugs have had limited market penetration in these markets due, in part, to the relatively high cost of these branded products. Further, as evidenced by several products already launched, biosimilars may actually be able to significantly expand the accessible patient populations in these markets. The commercial focus in these markets is market development and expansion. As further discussed below, in March 2014, our manufacturing partner, Reliance Life Sciences Pvt Ltd, or RLS, obtained manufacturing and marketing approval on our behalf in India for BOW015 as a treatment for rheumatoid arthritis. In September 2014, again with RLS, we received final manufacturing clearance from the Drug Controller General of India, or DCGI.
In January 2014, we entered into an agreement with our commercialization partner, Sun, to commercialize BOW015 in India and other selected Southeast Asian and North African markets, pending marketing authorization in those jurisdictions. These markets, including India, do not require that BOW015 be manufactured within the applicable country. In collaboration with Sun, we launched BOW015 in India in November 2014. BOW015 is sold under the brand name Infimab and is the first infliximab biosimilar approved in India. To date, nearly 1,000 patients have been treated with BOW015. We will be responsible for any additional development activities required by Indian regulatory authorities. Sun is responsible for all marketing and commercialization activities with respect to BOW015 in India, as well as any costs associated with development, regulatory filings and marketing and commercialization in the additional countries covered by the agreement. Under the terms of the agreement, we will supply Sun with commercial products, and Sun will be required to make payments to us upon achievement of certain development and sales milestones for BOW015, as well as to pay us a royalty on net sales of BOW015 in all territories covered by the agreement.
In addition to India and the other countries covered by our agreement with Sun, we believe that our existing regulatory dossier for BOW015 will be sufficient to achieve regulatory approval in a range of South and Central American countries. In late 2015, we also filed for approval in additional Southeast Asian and Latin American countries. We expect to grant rights to commercialize BOW015 in these countries, pending receipt of marketing authorization, through a licensing structure similar to the approach taken in India.
In May 2015, we entered into an Exclusive License Agreement (the “Mabxience License Agreement”) with Mabxience for the development, manufacturing and commercialization of BOW015. Under the Mabxience License Agreement, we granted to Mabxience an exclusive, royalty-bearing, non-transferable, sublicenseable license to develop, manufacture, commercialize and distribute BOW015 in the Mabxience territory. The Mabxience territory consists of Argentina, Chile, Ecuador, Paraguay, Uruguay and Venezuela. Pursuant to the Mabxience License Agreement, Mabxience will be solely responsible, at its expense, for all aspects of commercialization of licensed product in the Mabxience territory. Mabxience is a global fully integrated biopharmaceutical company specialized in research, development and manufacturing of biosimilars for the treatment and/or prevention of several diseases in diverse therapeutic areas. Mabxience is acting worldwide, creating a broad and balanced presence in all major pharmaceutical markets to address global opportunities and customers’ needs, with full-line research and manufacturing capabilities and highly qualified professionals worldwide. Mabxience is a wholly-owned subsidiary of CHEMO Group, a well-established Spanish-based global healthcare corporation.
United States
Given the still nascent and uncertain biosimilar market in the United States, our strategy remains focused on evaluating partnerships or an alternative commercial model, which may include contracting directly with payers or other third-party entities. While the recently completed FDA advisory committee hearing to weigh the approvability of Celltrion’s Remsima for commercial sale in the United States resulted in very encouraging regulatory signals in the United States, the need for greater legal and commercial clarity remain as important considerations in our partnership discussions and decisions.
12
Industry Overview
Biosimilars Definition
Biosimilars are highly similar versions of approved biological drug products, referred to as reference or innovator products. Because a biosimilar product may reference existing information regarding the structure, safety, and efficacy of a previously approved reference product, a biosimilar product application emphasizes analytical characterization to demonstrate similarity between the proposed biosimilar and the reference product. In addition, preclinical and clinical studies may be required to support an application for approval. Biosimilars are often characterized as fitting within one of two categories: first generation, less complex biologics; and second generation, more complex biologics, including fusion proteins and monoclonal antibodies, or mAbs.
Both first and second generation biosimilars are significantly more complex and difficult to characterize, manufacture and develop than small molecule generics. For example, the first generation biological drug, Epogen (epoetin alfa) has a molecular weight that is 25x greater than that of small molecule drug Lipitor (atorvastatin calcium). The second generation mAb biologics—e.g., Remicade (infliximab) and Humira (adalimumab)—are, in turn, nearly 5x larger than the first generation biologicals. MAb biosimilars are complex to manufacture in part because they require the use of living organisms to produce them, and this introduces challenges in manufacturing and production on a commercial scale. Glycosylation (complex carbohydrate branches that are added by the cellular machinery) and other forms of molecular modification are hallmarks of proteins produced in living cells, in particular mammalian cells. Compared to first generation biologics, mAbs are not only larger but also have greater structural complexity, including complex glycosylation patterns which may be critical for the function and activity of the molecule. MAb biosimilars therefore must be rigorously and accurately characterized to establish their biosimilarity to reference biologics in terms of glycosylation patterns, or glycoforms, and other important molecular modifications. Biosimilars—both first and second generation—also require significantly more clinical testing and regulatory review than small molecule generics, as described in more detail below.
The manufacturing, clinical and regulatory complexity and challenges of developing “second generation” biosimilars create barriers to market entry. As such, these “second generation” biosimilars can usually be sold at relatively higher prices, and with better margins, than small molecule generics and first generation biosimilars.
Regulatory Aspects of Biosimilars
Similar to other follow‑on product opportunities, product development proceeds differently with biosimilars than with innovative biologic candidates. This is a result of abbreviated development requirements for the approval of biosimilars as compared with innovative biologic products. Because the structure/function and target characteristics of the reference biologic are already known, a biosimilar product application emphasizes analytical characterization to demonstrate similarity between the biosimilar and the reference biological product, which regulators have already determined to be safe and effective.
Early Reduction of Risk in Drug Development
In the development of novel pharmaceuticals and biologic products, the question of whether or not a product will ever reach the point of being commercially viable remains largely unanswered. As such, the innovator company assumes significant risk through the completion of Phase 3 trials, in which the drug or biologic therapeutic is administered to a sufficient number of subjects to make a definitive determination of a drug’s safety and efficacy. Comprehensive Phase 3 trials are conducted for novel biologics at significant cost and exposure for the innovator company. In contrast, a large proportion of de‑ risking occurs much earlier in the development process of biosimilars through analytic testing. As the antibody, its target, and its mechanism of action have already been validated clinically by the reference product sponsor, and the dose, regimen, and indications are already known, early testing provides greater insight into the future regulatory success for biosimilars. Preclinical and early clinical studies required to demonstrate
13
comparability to the originator drug’s pharmacokinetic/pharmacodynamic (PK/PD), safety, and potency profile may provide early indications of a product’s eventual regulatory outcome.
The regulatory standards applicable to establish such biosimilarity vary by jurisdiction. Over the last 10 years, many jurisdictions globally have established formal regulatory regimes for review and approval of biosimilar products, but these regimes are at differing stages of development, with limited harmonization among jurisdictions.
Technical Complexity of Biosimilars
Historically, biologics have not yielded readily to the development of generic versions. Because biologics are structurally far larger and more complex than small molecule drugs, their manufacturing processes and requirements are correspondingly more complex as well. Biologics are produced through a technologically challenging five‑step process:
|
· |
Scientists isolate and identify the genetic code of the protein they want to produce. |
|
· |
This genetic code is inserted into living cells (bacteria, yeast, or cultured mammalian cells). Once inside the cell, the genetic code instructs the cell to produce the protein, or biotech medicine, which will later be used to treat a specific disease. Mammalian cells are the most complex of the various cell lines and generate complexities in protein structure through various biochemical modifications. |
|
· |
These genetically modified cells (known as cell lines) are carefully selected and cultured over time in bioreactor tanks, surrounded by nutrients designed to encourage protein production. The specific conditions for production determine important physical attributes of the protein—such as glycosylation (complex carbohydrate branches that are added by the cellular machinery)—for its function. The structure and function of the final product is therefore highly sensitive to the specific conditions under which it is generated. |
|
· |
The protein is then isolated from the cells and the nutrients through a sequence of purification processes. |
|
· |
Finally, the isolated protein is packaged, typically after a number of steps to ensure sterility and stability, into sterile vials or syringes for use by doctors and patients. |
Because the structure and function of a biologic, such as a monoclonal antibody, are directly linked to its production process in living systems, companies that intend to develop a biosimilar must go through all five steps above, from isolation and identification of the target protein and creation of a proprietary cell line, to final packaging of the drug product. Furthermore, companies must go to great lengths to characterize their molecules relative to the reference biologic. They must also prove to regulators that the proposed biosimilar is highly similar to the original in terms of safety and efficacy through a combination of laboratory and clinical studies. In summary, biosimilars must be shown to be comparable to their reference biologics in terms of structure, purity, safety and efficacy.
Importantly, just as there is variation among individual human beings at the biochemical level, there is also natural variation among biologic molecules created by cell systems, because they are derived directly from living systems. Even originator biologics are characterized by inherent structural and functional variability. This leads to a range of profiles and performance for the innovator molecules, themselves, even among batches produced at the same facility. As a result, “identical” copies of biologics such as antibodies are not the objective for biosimilars. Instead, biosimilars must fall within a range of values across important structural and functional parameters compared to those of the reference drug.
Barriers to Entry
The high technical and performance standards that every biosimilar must achieve to gain regulatory approval provide two advantages to biosimilar developers and manufacturers, such as our company. First, such standards create a significant barrier to entry in the form of both regulatory and clinical hurdles that a company must overcome to bring biosimilar products to market, thus increasing the complexity and related costs for potential competitors. Second, these
14
high standards are expensive to achieve, thus the limited number of producers who can meet these standards can then command pricing for biosimilar products that, while lower than the pricing for the reference product, is still high enough to generate meaningful revenues and profits.
Competition
Based on our market analysis, we may be subject to competition for BOW015 in various jurisdictions from the following groups:
(1)Johnson & Johnson developed the reference product for BOW015, Remicade, along with its partners Merck Schering and Mitsubishi Tanabe, which are responsible for sales outside the United States. Remicade is one of the longest established biologics, supported by extensive safety and efficacy data and widespread use in multiple indications. We expect that Remicade’s market share will gradually erode over time with the advent of biosimilars; however, Johnson & Johnson will seek to defend its market share against biosimilar entry, which may include reduction in prices and other incentives.
(2)Celltrion (Korea) and Hospira, Inc. (acquired by Pfizer) co-commercializes a biosimilar infliximab product in European and other global markets. Celltrion markets its product under the name Remsima® and Hospira under the name Inflectra®. Remsima and Inflectra were approved by the EMA in 2013 with all approved indications as Remicade and represent the first biosimilar infliximab to be launched in major markets. Celltrion’s Biologics License Application in the United States is currently under FDA review with a decision expected in the first half of 2016. It is likely that Celltrion, if approved, will launch Remsima in the United States upon expiration of the patent protection on Remicade in 2018.
(3)We are aware that other companies, including Samsung Bioepis, Pfizer and Amgen, are in pre-clinical and clinical stages of development and may become competitors for BOW015 in various markets over time. In March 2015, Samsung Bioepis submitted a Marketing Authorization Application to the EMA for its biosimilar infliximab. A decision from the EMA has yet to be announced. Samsung Bioepis and Biogen Idec also recently entered into a joint venture to develop, manufacture and market biosimilars. For the other companies, there is limited data available and we cannot currently predict if and when the potential competitors will launch in our target markets.
Based on our market analysis, we may be subject to competition for BOW050 in various jurisdictions from the following groups:
(1)AbbVie developed the reference product for BOW050, Humira. Humira is supported by extensive safety and efficacy data and widespread use in multiple indications. We expect that Humira’s market share will gradually erode over time with the advent of biosimilars; however, AbbVie will seek to defend its market share against biosimilar entry, which may include reduction in prices and other incentives.
(2)Currently Cadila Healthcare Ltd., is the only company that markets a biosimilar adalimumab under the brand name Exemptia™. Exemptia is only approved and available in India with the full label as Humira.
(3)We are aware that other companies, including Amgen, Samsung Bioepis, Pfizer, Boehringer Ingelheim, Sandoz, Momenta/Baxter, Coherus, Biocon/Mylan, and Therapeutics Proteins International are in pre-clinical and clinical stages of development and may become competitors for BOW050 in various markets over time. In November 2015, Amgen submitted a Biologics License Application to the FDA for its biosimilar adalimumab and subsequently submitted a Marketing Authorization Application to the EMA in December. Both applications are currently
15
pending regulatory review. For the other companies, there is limited data available and we cannot currently predict if and when the potential competitors will launch in our target markets.
Based on our market analysis, we may be subject to competition for our other biosimilar product candidates in the early stages of development, including BOW070, BOW080, BOW090 and BOW100. There is limited publicly available data on potential competitive molecules for these product candidates at this time.
Clinical Development of BOW015 (Infliximab)
We have conducted extensive bioanalytical and physicochemical comparisons of BOW015 to Remicade and have data from a Phase 1 study in the United Kingdom and a 189-patient Phase 3 double blind comparator study in India demonstrating bioequivalence, safety and efficacy of BOW015. The Phase 3 study in India met the primary endpoint of efficacy at an interim analysis conducted at 16 weeks and finished its open label phase with a 54-week end-point in the third quarter of 2014. In March 2014, BOW015 was granted manufacturing and marketing approval in India through our manufacturing partner RLS as a treatment for rheumatoid arthritis. Final manufacturing site authorization was received in July 2014. The approval was based on the 16-week, double blind safety and efficacy data comparing BOW015 against Remicade as the active comparator and reference product, in accordance with the required regulatory process in India. In November 2014, we launched BOW015, under the brand name Infimab, in India with Sun and intend to launch in additional territories thereafter by leveraging our existing regulatory data package.
In November 2015, we completed manufacturing readiness for BOW015 to support the initiation of a global clinical program for BOW015. In February 2016, we initiated this global clinical program, which will support regulatory filings in established markets such as Europe and the United States. The UNIFORM Study is a 58-week, double-blind, one-to-one randomized, comparator-controlled multi-center global study to compare safety, efficacy and immunogenicity and demonstrate clinical equivalence of BOW015 with Remicade. We plan to enroll over 500 patients with active rheumatoid arthritis in the UNIFORM study, which will be conducted at sites in Europe, North America and Latin America. The primary endpoint at week 16 for the study will assess the proportion of patients that meet ACR20. We have been designing this clinical program in consultation with European regulatory bodies in order to obtain the data that may be necessary to support potential approval of BOW015 in developed markets. We expect to file for approval of BOW015 in developed markets, including Europe and North America, in 2017.
Since BOW015 is a biosimilar molecule, its characterization requires comparative analysis to the reference product, Remicade. The BOW015 drug substance and drug product manufacturing processes have been designed in conjunction with our manufacturing partners to make the final BOW015 drug product comparable to that of Remicade, and with comparable safety and efficacy in accordance with regulatory requirements.
We have produced a data package to demonstrate biosimilarity of BOW015 to Remicade. Its comparability data set, developed under a comparability and characterization protocol, addresses the physicochemical, biochemical and biological properties of infliximab and has been designed to assess biosimilarity between the reference product and BOW015. Critical Quality Attributes, or CQAs, are physical, chemical, biological or microbiological properties or characteristics that should be within appropriate limits to ensure the desired product quality. The CQAs of infliximab have been identified based on the mechanism of action, clinical experience, impact/risk assessment of production processes and the assessed ranges of specific attribute data generated by analysis of multiple lots of Remicade. The CQAs are supported by Annex I of the Summary of Product Characteristics of the Remicade European Public Assessment Report. Full side-by-side characterization of BOW015 and Remicade, including all CQAs for infliximab, has been completed. The data set includes all known attributes that have the potential to impact safety, potency and efficacy.
The types of assays used to assess biosimilarity include the following:
|
· |
Physicochemical. These are assays that measure the physical and chemical structure of the molecule. |
|
· |
In vitro biochemical. These are assays that measure the interaction of the molecule with other molecules (e.g. target binding). |
16
|
· |
In vitro biological. These are assays that measure the interaction of the molecule with biological media (e.g. cellular or animal test systems). |
These three levels of characterization are complementary and correlations are drawn from the individual and aggregate findings. For example, glycosylation heterogeneity (a physicochemical attribute) has direct impact on FcᵞRIIIa binding (a biochemical attribute), which in turn drives ADCC (antigen-dependent cellular cytotoxicity) activity (a biological attribute). In this way, multiple data sets support and confirm each other and the biosimilarity of BOW015 to Remicade.
The assessment of CQAs demonstrated similarity between BOW015 and Remicade. Both BOW015 and Remicade are produced using similar manufacturing processes. Minor differences between BOW015 and Remicade in the non-critical quality attributes may be consequences of differing manufacturing technologies and have not demonstrated adverse impact on the biology and efficacy of BOW015 in either in vitro or clinical studies.
We have also conducted multiple preclinical studies on BOW015. These studies include:
|
· |
Single dose toxicity of BOW015 in Swiss albino mice |
|
· |
Single dose toxicity of BOW015 in Wistar rats |
|
· |
Repeat dose (4-week) study in Wistar rats |
|
· |
Repeat dose (4-week) study in New Zealand White Rabbits |
|
· |
Skin sensitization study of BOW015 in Dunkin Hartley Guinea Pigs (Maximization Test) |
We believe that BOW015 has yielded satisfactory results in each of the preclinical studies to support regulatory filings in each of the additional markets we are seeking to target. Infliximab binds selectively to human and chimpanzee tumor necrosis factor-alpha (TNF-α), and thus additional preclinical studies in non-relevant species were not required by Medicines and Healthcare Products Regulatory Agency (UK) prior to initiating Phase 1.
BOW015 Phase 1 Study
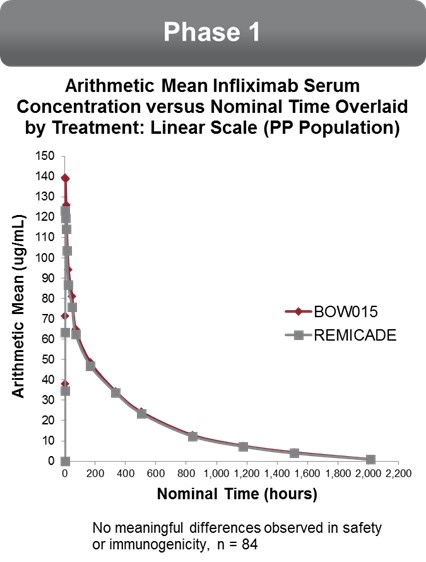
Our Phase 1 bioequivalence study was conducted in the United Kingdom in 2012 under the authority of the Medicines and Healthcare Products Regulatory Agency. The primary objective of the study was to compare the pharmacokinetics of infliximab administered by intravenous infusion. The secondary objectives of the study were to assess (i) the safety and tolerability and (ii) immunogenicity of BOW015 compared to Remicade. The study was conducted at a single clinical site and compared the safety and pharmacokinetic profile of BOW015 to Remicade after a single intravenous dose. The two drugs were considered to be similar if at various timepoints the concentrations of the drugs were comparable and were within the specific statistical parameters of 80%‑125%. The study design and criteria for success were based on standard bioequivalence requirements.
Eighty‑four healthy volunteers were randomized one‑to‑one and given either BOW015 or Remicade via intravenous infusion at a dose level of 5mg/kg with a 12‑week follow‑up period. The study was to detect bioequivalence at 90% confidence interval of BOW015 to Remicade. Out of the 84 subjects, 43 evaluable subjects received the test product BOW015 and 41 subjects received the reference product Remicade.
The profile of BOW015 and Remicade is shown in the graph below. The pre‑ defined pharmacokinetic values for the maximum height of the drug concentration as well as the pattern of elimination are similar. Thus, the study demonstrated similarity in PK profiles between BOW015 and the reference product Remicade. A single severe adverse
17
event was reported in one of the patients receiving Remicade. This was considered by the investigator as unlikely to be related to the experimental protocol.
No relevant differences in immunogenicity test results between the two treatment groups were observed, nor were any differences observed between the two groups in safety or tolerability.
BOW015 Phase 3 Study
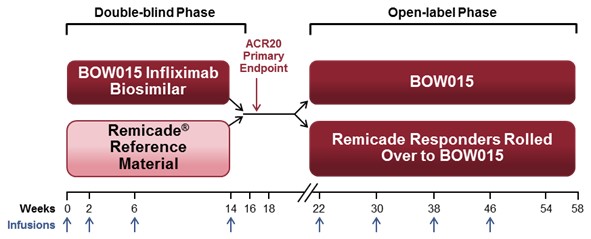
We conducted a randomized, double-blind, active comparator Phase 3 study in India of the efficacy and safety of BOW015 in patients with severe, active rheumatoid arthritis on stable doses of methotrexate. The study randomized subjects to the two treatment arms in a 2:1 allocation. Out of 189 total subjects, 127 were given BOW015 and 62 were given Remicade during the first 16 weeks of the study. The primary endpoint of the study was equivalence of both arms on the standardized American College of Rheumatology 20% improvement (ACR20) scoring system—a composite scoring system that includes objective laboratory measures as well as physician and patient assessments of well-being. Secondary endpoints included the ACR50 and ACR70 (50% and 70% improvement respectively) and the various components of the ACR20 scoring system. From week 22, BOW015 responders were administered BOW015 in an open-label phase for the study duration of 54 weeks, while Remicade responders were crossed over into the open label phase and switched to BOW015 for the study duration of 54 weeks. Non-responders immediately entered a three-month follow-up phase.
Both BOW015 and Remicade were administered at a dose of 3mg/kg given as an intravenous infusion at week 0, followed with similar doses at weeks 2, 6 and 14. Subjects were assessed at week 16 and responders were able to enter an open‑label phase. In the open‑label phase, subjects received BOW015 at a dose of 3mg/kg given as an intravenous
18
infusion at weeks 22, 30, 38 and 46 and were followed up at Weeks 54 and 58. Subjects who were non‑responders at week 16 entered a follow‑up phase for immunogenicity, PK and safety for an additional 3 months.
The 16‑week data showed that patients responded to BOW015 at a rate of 89.8% ACR20 compared to an 86.4% ACR20 response rate to Remicade. This outcome met its statistical endpoint within the pre-specified equivalence margin at a 95% confidence interval. The results met the 23% equivalence margin authorities required for approval by the Indian
19
regulatory authorities. There was no difference reported in safety or immunogenicity between the treatment groups. There was also no reported difference between the groups on the secondary endpoints.
We measured the patients’ responses on an ACR20 scoring system to BOW015 and Remicade at multiple time points. The data suggest that BOW015 and Remicade patients responded similarly at all time‑points up to the final 16 week efficacy endpoint.
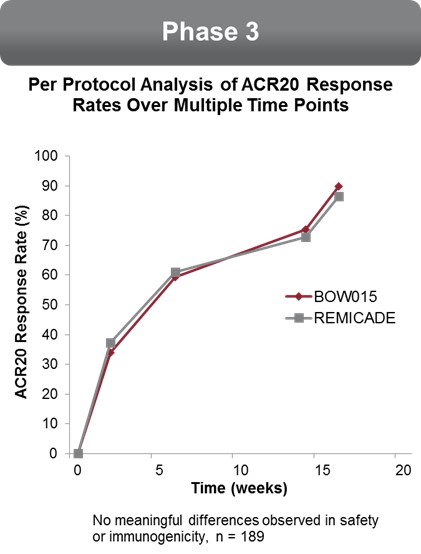
Comparison of BOW015 and Remicade at multiple time points
In September 2014 we announced 58 week data, which demonstrated therapeutic equivalence to Remicade and confirmed the safety of switching from Remicade to BOW015. In the open‑label phase, patients who continued on BOW015 were compared to patients who received four doses of Remicade, followed by a switch to four doses of BOW015. Immune responses as well as overall safety and tolerability for BOW015 were comparable to the arm
20
switched from Remicade to BOW015 and were consistent with the expected profile of Remicade. Further, ACR20 responses were durably maintained to 54 weeks from the week 16 primary endpoint previously reported.
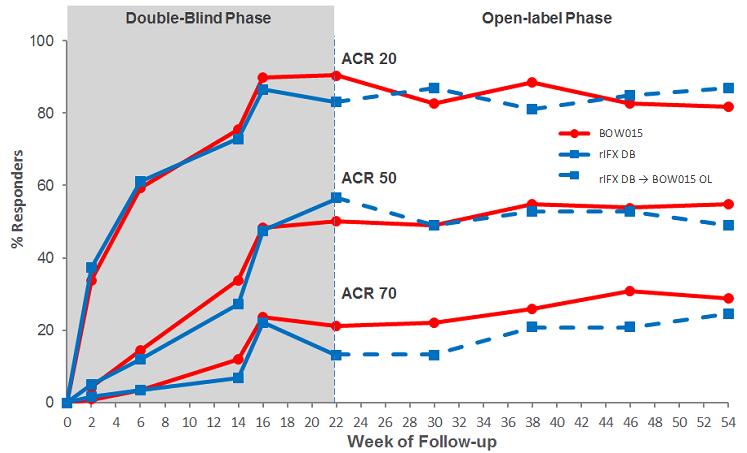
Equivalence in ACR20 Response Rates—Double Blind (DB) and Open Label (OL) Phase
License and Acquisition Agreements
Acquisition of Bioceros
On September 9, 2015, we entered into and closed a definitive Stock Purchase Agreement (the “Stock Purchase Agreement”) with Bioceros Holding B.V., a Netherlands company and our primary vendor for preclinical development work (“Bioceros”), the sellers identified therein (the “Sellers”) and Oscar Schoots and Carine van den Brink as the representatives of the Sellers. Under the Stock Purchase Agreement, we purchased all of the outstanding shares of the share capital of Bioceros (the “Share Transfer”), with the result that Bioceros has become our wholly-owned subsidiary following the Share Transfer. Bioceros is a leading provider of services related to the preclinical development of monoclonal antibodies and generation of good manufacturing practices (GMP)-ready protein producing cell lines. The assets acquired include Bioceros’ proprietary CHOBC® cell line platform, all related intellectual property rights, and a fully equipped laboratory together with Bioceros’ staff of scientists and bioreactor capabilities designed for the development of monoclonal antibodies and protein therapeutics, with a focus on biosimilars, including capabilities for the production of three new biosimilar product candidates to add to our pipeline of biosimilar product candidates, as further described above.
As a result of the Share Transfer, the Sellers will receive a total of $14.1 million in consideration, consisting of: (i) an initial cash payment of $3.4 million, (ii) a second cash payment of $1.7 million, to be paid on the first anniversary of the closing date, (iii) an initial issuance of shares of our common stock, $0.001 par value per share (the “Common Stock”) with a value of $4.0 million, calculated as set forth in the Stock Purchase Agreement and (iv) a second issuance of shares of Common Stock with a value of $5.0 million, calculated as set forth in the Stock Purchase Agreement, to be issued on the date that is six months after the closing date, subject to our setoff rights with regard to the second cash payment and the second installment of shares of Common Stock. In addition, the Sellers will receive, if any such
21
payment is due, a pro rata share of a net cash payout equal to the aggregate amount of Bioceros’ net cash at closing in excess of $1.2 million, calculated as set forth in the Stock Purchase Agreement.
In connection with the Stock Purchase Agreement, Bioceros granted to a newly-formed Netherlands company (“NewCo”), which is owned by the Sellers, a fully paid-up, non-exclusive, transferable (under limited, specified circumstances) license to Bioceros’ CHOBC® platform and future improvements thereto, for NewCo’s use in the development of monoclonal antibodies and proteins other than biosimilar monoclonal antibodies and Ig fusion proteins.
Prior to the Share Transfer, we were party to several license and master services agreements with Bioceros, for non-exclusive licenses under its rights in the cell line and associated intellectual property relating to molecules such as adalimumab and tocilizumab for certain designated territories.
Sun Pharmaceutical Industries Ltd.
In January 2014, we entered into a license agreement with Sun Pharmaceutical Industries Ltd., formerly known as Ranbaxy Laboratories Limited (“Sun”), pursuant to which we granted to Sun exclusive rights under our intellectual property and regulatory materials relating to BOW015, to develop and commercialize BOW015 in India and certain other countries in Southeast Asia and North Africa. Pursuant to the agreement, Sun has agreed to distribute and sell BOW015 in India. Under our agreement with Sun, we will be responsible, through RLS, for supplying BOW015 to Sun for sale in the licensed territory.
Under our agreement with Sun, Sun paid us an up-front payment of $0.5 million, and will be required to make payments to us upon the achievement of certain regulatory and commercialization milestones of up to $1 million in the aggregate. Sun is also required to make payments to us upon the achievement of specified levels of aggregate gross sales of BOW015 in the licensed territory totaling up to $10 million in the aggregate, and to pay to us a royalty on net sales of BOW015 at a percentage in the mid to high teens, subject to reductions in certain circumstances. Sun's obligation to pay us royalties will expire 20 years following the first commercial sale of BOW015 in the licensed territory. On September 9, 2014, we amended the license agreement to revise the royalty tiers for net sales based on final product sale forecasts and supply costs in the territory. Under the amendment, we also revised Sun's obligations to achieve specified minimum annual sales targets in any given country such that these obligations will now apply following the final grant of approval by the applicable regulatory authority for BOW015 for all indications for which the innovator product has received approvals in such country.
Our agreement with Sun will remain in force, absent earlier termination, for 20 years following November 2014, the date of the first commercial sale of BOW015 in the licensed territory. Either party may terminate the agreement on 60 days' notice for the other party's uncured material breach or insolvency, or upon 30 days' notice in the event that our rights in the BOW015 cell line under a separate agreement are terminated, and we may terminate the agreement upon 45 days' notice in the event of a patent challenge brought by Sun in relation to any patents licensed under the agreement. Sun may also terminate the agreement on 60 days' notice to us in the event of certain failures relating to the qualification of the manufacturing facility for production of BOW015, unless we provide an appropriate plan to remedy such issues, and use commercially reasonable efforts to execute such plan, in which case the cure period for such manufacturing related breaches is extended for two years following the date of the remedial plan. If we fail to cure the breaches within the two-year period, and Sun elects to terminate the agreement, we may, under certain circumstances, be required to pay specified amounts in damages to Sun as Sun's sole remedy for the breach.
Livzon Mabpharm Inc.
In September 2014, we entered into an Exclusive License and Collaboration Agreement (the “Livzon Collaboration Agreement”) with Livzon for the global development and commercialization of certain antibodies or related biological compounds, including BOW015.
Under the Livzon Collaboration Agreement, we and Livzon agreed to grant each other, in the other party's territory, exclusive, royalty-bearing licenses under certain patent rights and know-how to develop, manufacture and commercialize BOW015 and up to four additional compounds chosen by mutual agreement of the parties, which we
22
refer to as the Collaboration Compounds. Livzon's territory consists of China, Hong Kong, Macau and Taiwan, and our territory contains the rest of the world. We share pre-clinical development expenses with Livzon for each Collaboration Compound based on certain factors specific to each such compound. Each party bears the responsibility and expenses for clinical development and commercialization of Collaboration Compounds in its territory. Livzon will be the preferred supplier of each Collaboration Compound for pre-clinical, clinical, and commercialization purposes, subject to Livzon's satisfaction of certain performance criteria. Our agreement with Livzon will remain in force, absent earlier termination, for 20 years. Either party may terminate the agreement in the event of a challenge brought by the other party against any patents licensed under the agreement, in the event of the other party's insolvency, or upon 60 days' notice in the event of material breach by the other party.
In consideration for the license granted to Livzon to develop and commercialize BOW015, we are eligible to receive from Livzon a milestone payment of $2.5 million upon the achievement of a specified regulatory milestone. We are also eligible to receive from Livzon tiered royalties at a percentage in the low to high single digits based on net sales of BOW015 products in the Livzon Territory. Any future Collaboration Compounds have cross-milestone and royalty obligations in amounts to be mutually agreed upon at a later date.
In September 2015, we entered into a New Collaboration Compound Supplement (the “Supplement Agreement”) to the Livzon Collaboration Agreement to add BOW070 to the list of collaboration compounds included under the terms of the Livzon Collaboration Agreement. Livzon will be responsible for certain pre-clinical development activities, including innovator analysis, drug substance process development, characterization work and formulation development, and each party will be responsible at its sole expense for clinical development, regulatory filings and approvals, and commercialization of BOW070 in each such party’s territory.
We will pay a total of $4.5 million to Livzon to complete the pre-clinical development activities, consisting of: (i) an initial cash payment of $1.5 million paid within ten days of signing the Supplement Agreement and (ii) three additional cash payments of $1 million each within ten days upon the achievement of certain pre-clinical development milestones. Each of the parties is eligible to receive from the other party tiered royalty payments, based on a percentage of sales of BOW070 in the other party’s territory, ranging from the low- to mid-single digit percentages.
Polpharma
In July 2015, we entered into a Collaboration Agreement (the “Polpharma Collaboration Agreement”) with Polpharma for the development and commercialization in certain designated territories of our product candidates, BOW015, BOW050 and BOW070. The designated territories include the European Union (with the exception of Austria, Belgium, Denmark, Finland, Luxembourg, the Netherlands and Sweden, which are reserved for us), together with certain countries in the Middle East, Turkey, Russia, and other countries comprising the Commonwealth of Independent States, or CIS. We will also retain exclusive rights for the three products in North America and other markets outside of the designated territories, including Switzerland and Norway. Within the designated territories, the Polpharma Collaboration Agreement contains exclusivity provisions such that we and Polpharma agree not to exploit any other biosimilar product based on reference product other than the three products licensed under the Polpharma Collaboration Agreement, subject to limited exceptions.
Under the Polpharma Collaboration Agreement, we and Polpharma have cross-licensed all intellectual property rights and technology relating to our applicable biosimilar development programs to enable the parties to develop and commercialize the three above-mentioned biosimilar products in the designated territories and for us to develop and commercialize these biosimilar products outside of the designated territories. The three product programs will consist of process development, clinical trials, regulatory, manufacturing and commercialization activities in the designated territories. For such programs, we will lead the global product development and clinical programs and will also be responsible for process development, scale-up and manufacturing in the designated territories, both parties will collaborate on regulatory filings in the designated territories, and Polpharma will be responsible for the commercialization of the products in the designated territories. Polpharma has the right to be a second source
23
manufacturer for any of the products, including a right to perform fill and finish production for all three products, subject to certain conditions. A joint management committee will oversee the collaboration on behalf of both parties.
Polpharma will bear 51% and we will bear 49% of the costs associated with the development programs for the three products in the designated territories. Following commercial launch, profits and losses for the three products in the designated territories will be split 51% for Polpharma and 49% for us.
Manufacturing
We currently manufacture BOW015 through a manufacturing and supply agreement with Reliance Life Sciences Pvt Ltd, or RLS, our BOW015 contract manufacturing partner for India, which we refer to as the RLS Agreement. In order to migrate from traditional stainless steel manufacturing to single use disposable systems for our SCALE process, we also have an agreement with Fujifilm Diosynth Biotechnologies U.S.A., or Fujifilm, for BOW015 process development with a view toward establishing Fujifilm as a source of future clinical and commercial supply of BOW015. We believe that our relationship with Fujifilm will allow us to expand future capacity and provide a back-up secondary manufacturing site. We are also currently working with our manufacturing partners to establish appropriate arrangements for the necessary scale-up of manufacturing operations for longer term commercial supply in markets where we or our licensees are developing and commercializing products. For markets requiring in-country manufacturing, we expect to work with local partners to deploy our SCALE manufacturing platform in whole or in part to enable our In Market, For Market solution. We expect that this strategy will provide us with multiple sourcing options to enable uninterrupted product supply to our partners and therefore patients, and to meet the needs of countries requiring locally-based manufacturing. In December 2014, RLS exercised its three-year termination for-convenience right with respect to the RLS Agreement, which would have caused the RLS Agreement to terminate in December 2017. In addition, in March 2015, RLS initiated an arbitration proceeding under the RLS Agreement alleging that the RLS Agreement grants RLS exclusive global supply rights for BOW015, that the terms of our collaboration agreements with Fujifilm Diosynth Biotechnologies U.S.A., Inc. and Livzon MabPharm Inc. violate the RLS Agreement, and sought to terminate the RLS Agreement. The proceeding initiated by RLS sought a declaratory judgment, but did not seek any monetary damages.
On April 22, 2015, we entered into a Settlement Agreement with RLS (the “Settlement Agreement”). Pursuant to the Settlement Agreement, the parties finalized the remaining BOW015 batch production schedule with RLS agreeing to manufacture 15 final batches of BOW015 on or before June 30, 2016 with our option to have RLS manufacture an additional 5 batches on or before December 31, 2016. In January 2016, we amended the Settlement Agreement to extend the time for RLS to manufacture the 15 final batches to June 30, 2017. RLS has also agreed to use reasonable commercial efforts to transfer the existing Indian marketing authorization for BOW015 granted by the Drug Controller General of India (“DCGI”) to us or our designee, with all costs of the transfer to be borne by us and subject to the DCGI’s approval of such transfer. We have agreed to pay to RLS a fee of approximately $2.3 million payable in four installments over the course of the final batch manufacture, with the final installment to be paid no later than ten days after June 30, 2016. Pursuant to the Settlement Agreement, on May 4, 2015, RLS withdrew the arbitration proceeding it initiated, and the parties have agreed to release each other from all causes of actions or claims, present and future, arising under the RLS Agreement. Following the satisfaction by both parties of all of the terms of the Settlement Agreement, the RLS Agreement and related agreements between the parties shall be terminated and neither party shall have any further rights or obligations thereunder.
SCALE Manufacturing Platform and In Market, For Market Solution
Manufacturing of biologics is currently shifting away from traditional methods involving steel bioreactors to small, single‑use bioreactors. Until recently, production facilities relied on the use of relatively inflexible, hard‑piped equipment including large, stainless steel bioreactors and tanks to manufacture product intermediates and buffers. However, there is an increasing trend towards the adoption of single‑use technologies across the manufacturing process.
There are several key advantages to single‑use technologies. These include:
|
· |
Reduced capital costs for plant construction; |
24
|
· |
Reduced risk for product cross‑contamination in a multiproduct facility; |
|
· |
Increased flexibility to change rapidly from one product to another; |
|
· |
Lower utility costs due to reduced need for steaming‑in‑place sterilization; and |
|
· |
Reduced time for a new facility to become operational. |
Manufacturing using single‑use bioreactors is at the heart of SCALE, our solution for manufacturing biosimilars in emerging markets. The modular nature of the SCALE manufacturing facilities enables our business plan of building manufacturing facilities to suit the local requirements of our partners. Investment into a SCALE facility can range from $20 million to $40 million for a facility that, once up and running, could produce up to 150 kilograms per year of biologic material. Multiple biologic products can easily be manufactured in a single SCALE facility. Single‑use bioreactors enable smaller production runs and facilitate operation of a multi‑product facility that is well suited to the evolving market for biologics. Once the market demand exceeds the production output of a single bioreactor, additional equipment can easily be ordered and installed.
Intellectual Property
“EPIRUS Biopharmaceuticals”, “In Market, For Market”, “SCALE” and “Infimab” are our registered trademarks. As a company focused on biosimilars, we do not own any product related patents.
In January 2009, our predecessor, Moksha8, acquired rights in and title to the GPEx Cell Line from Catalent Pharma Solutions, LLC, or Catalent, for the gene expression product M80015 (renamed BOW015), subject to our obligation to pay certain milestones and royalties on net sales to Catalent with respect to the development and commercialization of BOW015. Moksha8 assigned their agreement with Catalent to us on May 14, 2009. Following the Share Transfer with Bioceros in September 2015, we are the owners of certain proprietary cell lines applicable to our pipeline of biosimilar product candidates, including the cell lines for BOW050 (adalimumab), BOW070 (tocilizumab), BOW080 (eculizumab), BOW090 (ustekinumab) and BOW100 (golimumab). Please see “Business—License and Acquisition Agreements” for a more detailed description of our rights and obligations with respect to our product candidates.
Government Regulation
We and our partners are subject to a variety of laws and regulations governing the development, manufacture, marketing, and distribution of biosimilars. Regulatory authorities around the world regulate, among other things, the research and development, testing, manufacture, quality control, safety, purity, potency, labeling, storage, record keeping, approval, advertising and promotion, distribution, post‑approval monitoring and reporting, sampling, and import and export of our products and product candidates. The regulatory requirements and approval processes vary from country to country, and the various regulatory regimes are at various stages of maturity depending on the jurisdiction.
United States
FDA
The Biologics Price Competition and Innovation Act, or BPCIA, enacted in 2010, established an abbreviated approval pathway for biosimilars in the United States. The law defines biosimilars as products that are highly similar to biologics already licensed by the FDA pursuant to Biologic License Applications, or BLAs, notwithstanding minor differences in clinically inactive components, and that have no clinically meaningful differences from the reference product in terms of safety, purity and potency. A biosimilar application submitted pursuant to the BPCIA must contain information demonstrating biosimilarity based upon the following, unless the FDA determines otherwise:
25
|
· |
data derived from analytical studies, demonstrating that the proposed biosimilar product is highly similar to the approved product notwithstanding minor differences in clinically inactive components; |
|
· |
animal studies (including an assessment of toxicity); and |
|
· |
a clinical study or studies (including an assessment of immunogenicity and pharmacokinetics or pharmacodynamics), sufficient to demonstrate safety, purity and potency in one or more conditions for which the reference product is licensed and intended to be used. |
In addition, a biosimilar application must include information demonstrating (1) sameness of strength, dosage form, route of administration and mechanism(s) of action with the reference product (where known), (2) approval of the reference product for the condition(s) of use prescribed, recommended or suggested in the labeling proposed for the biosimilar product, and (3) appropriate manufacturing, processing, packing, and holding facilities that meet the standards designed to ensure a safe, pure and potent medicine. The FDA will approve a biosimilar application based on a finding of biosimilarity with the reference product.
A pending biosimilar application or a supplement to an approved biosimilar application may seek an FDA determination that the proposed or approved product is “interchangeable” with the reference product. FDA may determine that the product is interchangeable with the reference product if the application includes sufficient information to show that the product is biosimilar to the reference product, and that it can be expected to produce the same clinical result as the reference product in any given patient. Further, if the product may be administered more than once to a patient, the applicant must demonstrate that the risk in terms of safety or diminished efficacy of alternating or switching between the biosimilar and reference product is not greater than the risk of using the reference product without such alternation or switch. The determination of interchangeability means that the biosimilar product may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
The timing of final FDA approval of a biosimilar for commercial distribution depends on a variety of factors, including whether the reference product is entitled to one or more of a number of marketing exclusivity periods available for reference products under the BPCIA and the Food, Drug and Cosmetic Act that may delay submission and approval of biosimilar applications. The BPCIA prohibits the submission of a biosimilar application until four years after the date on which the reference product was first licensed, and delays the approval of a biosimilar application from becoming effective until twelve years after the date on which the reference product was first licensed. The first‑licensure exclusivity provisions are not triggered by a supplement to the approved original application for the reference product, or by the submission of an entirely new BLA submitted by the same sponsor or manufacturer of the reference product that proposes certain changes to the reference product. Such changes include (1) a non‑structural change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength, and (2) a structural change that does not result in a change in safety, purity or potency. In addition, as in the case of applications for approval of small molecule drugs in the United States, BLAs may be entitled to other periods of exclusivity. For example, a reference product that receives an orphan drug designation will typically be entitled to seven years of market exclusivity, in which case no product that is biosimilar to the reference product may be approved until either the end of the twelve year period provided under the BPCIA or the end of the seven year orphan exclusivity period, whichever occurs later. In certain circumstances, a regulatory exclusivity period can extend beyond the life of a patent, and thus block biosimilar applications from being approved on or after the patent expiration date. In addition, the FDA may under certain circumstances extend the exclusivity period for the reference product by an additional six months if the FDA requests, and the manufacturer undertakes, studies on the effect of its product in children, referred to as a pediatric extension.
The first biosimilar determined to be interchangeable with a particular reference product for any condition of use is also protected by a period of exclusivity that delays an FDA determination that a second or subsequent biosimilar product is interchangeable with that reference product for a period of time generally ranging from 12 to 42 months from approval or as determined by a number of patent litigation triggers. Specifically, this exclusivity period extends until the earlier of: (1) one year after the first commercial marketing of the first interchangeable product; (2) 18 months after resolution of a patent infringement suit instituted under 42 U.S.C. § 262(l)(6) against the applicant that submitted the
26
application for the first interchangeable product, based on a final court decision regarding all of the patents in the litigation or dismissal of the litigation with or without prejudice; (3) 42 months after approval of the first interchangeable product, if a patent infringement suit instituted under 42 U.S.C. § 262(l)(6) against the applicant that submitted the application for the first interchangeable product is still ongoing; or (4) 18 months after approval of the first interchangeable product if the applicant that submitted the application for the first interchangeable product has not been sued under 42 U.S.C. § 262(l)(6).
After obtaining regulatory approval of a biosimilar product, sponsors will be required to comply with many of the post‑approval requirements that apply to approved small molecule drugs. For example, the holder of an approved BLA or biosimilar application is required to report certain adverse reactions and production problems involving its product to the FDA, provide updated safety and efficacy information and comply with requirements concerning advertising and promotional labeling for the product. It is also required to comply with FDA’s cGMPs after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMPs. In addition, in some cases, as a condition of approval of a BLA or a biosimilar application, the FDA may require post-marketing testing and surveillance to monitor the product’s safety or efficacy.
The BPCIA also establishes a detailed framework for addressing potential patent disputes between biosimilar product sponsors and reference product sponsors. Once the biosimilar applicant has received notification that the FDA has accepted its application for review, the BPCIA establishes a 20‑day time frame within which time the biosimilar applicant must provide a copy of the application, and may provide any other information that describes the manufacturing processes for the biosimilar product, to the reference product sponsor’s in‑house counsel, the reference product sponsor’s outside counsel, and/or a representative of the owner of a patent exclusively licensed to the reference product sponsor with respect to the reference product who has retained a right to assert the patent or participate in litigation. The copy of the application and any other information provided are considered “confidential information,” and recipients are generally prohibited from disclosing anything contained therein and from using the information for any purposes other than to determine whether a patent infringement claim may reasonably be asserted. The reference product sponsor then has sixty days within which to provide the biosimilar applicant with a list of patents for which it believes a patent infringement claim could reasonably be brought. The reference product sponsor may also choose to designate patents that it would be willing to license to the biosimilar applicant.
Subsequently, the parties must engage in a back and forth negotiation regarding which patents will be part of the anticipated litigation, during which time the FDA continues to review the biosimilar application. Unlike in the context of applications for small molecule generics submitted under the Hatch‑Waxman Act, the BPCIA does not require the FDA to stay approval of a follow‑on biologic application for 30 months once patent litigation has been initiated.
In September 2014, the FDA published the “Purple Book,” which lists biological products licensed by the FDA pursuant to approved BLAs and identifies the date of licensure and whether the FDA has evaluated the biological product for reference product exclusivity. The Purple Book will also enable users to identify whether a biological product has been determined by the FDA to be biosimilar to or interchangeable with a reference biological product. Biosimilar and interchangeable biological products will be listed under the reference product to which biosimilarity or interchangeability was demonstrated.
To date, the FDA has published several final and draft guidance documents and a proposed rule regarding implementation of the BPCIA and other issues that will affect biosimilars. These guidance documents and proposed rule focus on scientific considerations, quality considerations and clinical pharmacology data related to demonstrating biosimilarity; meetings between the FDA and biosimilar product sponsors; requests for reference product exclusivity; common questions and answers regarding implementation of the BPCIA; the standards for interchangeability; and naming of biological products.
On March 6, 2015, the FDA approved Zarxio®, Sandoz’s biosimilar version of Amgen’s Neupogen (filgrastim) for all of the reference product’s approved indications.
27
Coverage and Reimbursement
The commercial success of our biosimilar product candidates and our ability to commercialize those products successfully if approved will depend in part on the extent to which governmental payor programs at the federal and state levels, including Medicare and Medicaid, private health insurers and other third‑party payors provide adequate coverage and reimbursement. These third‑party payors generally develop their own policies as to which drugs they will pay for and the reimbursement levels for the drugs. For example, governmental programs in the United States often require manufacturers to pay certain rebates or otherwise provide discounts to secure coverage of drug products. To control healthcare expenditures generally, in the United States, the EU and other potentially significant markets for our product candidates, government authorities and third party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies. The measures taken often have resulted in lower average selling prices. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the EU places additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, as well as drug coverage and reimbursement policies and pricing in general.
Some of the additional requirements and restrictions on coverage and reimbursement levels imposed by third‑party payors influence the purchase of healthcare services and products. For example, there may be limited coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA‑approved drug products for a particular indication. There may also be formulary placements that result in lower reimbursement levels and higher cost‑sharing borne by patients. Further, third‑party payors are increasingly examining the medical necessity and cost‑effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost‑effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our products may not be considered medically necessary or cost‑effective. Even if a third‑party payor determines to provide coverage for a drug product, adequate reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in drug development.
Healthcare legislative proposals to reform healthcare or reduce costs under government insurance programs may also result in lower reimbursement for our drugs and drug candidates or exclusion of our drugs and drug candidates from coverage altogether. The cost containment measures that healthcare payors and providers are instituting and any healthcare reform could significantly reduce our revenues from the sale of any of our drug candidates, if approved. We cannot provide any assurances that we will be able to obtain and maintain third party coverage or adequate reimbursement for any of our approved drug candidates in whole or in part.
Healthcare Reform
With respect to legislative reform, in the United States, there have been and continue to be a number of significant legislative initiatives to contain healthcare costs. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, was passed, which substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things, subjects biologic products to potential competition by lower‑cost biosimilars, and introduced a new methodology for determining the payment amount for a biosimilar product under Medicare Part B. In October 2015, the Center for Medicare & Medicaid Services, or CMS, finalized the rule for reimbursement of biosimilars. Under the rule, all biosimilars relying on a common reference product will be grouped into the same payment calculation for determining the single average sales price, or ASP, payment limit. Until such time as there are additional sales data for subsequent biosimilars to determine an ASP, a newly approved biosimilar price will be determined by local contractors using available pricing information. As with newly approved drugs and biologicals, Medicare Part B payment will generally be available once the product is approved by the FDA provided the claim is reasonable and necessary and meets applicable coverage and claims submission criteria. CMS has not yet issued guidance for interchangeable biologics since there are currently no interchangeable biologics being marketed.
28
In addition, other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013, and will remain in effect through 2024 unless additional Congressional action is taken.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Other Healthcare Laws
We may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we conduct our business. These laws include, without limitation, state and federal anti‑kickback, fraud and abuse, false claims, privacy and security and physician sunshine laws and regulations.
The federal Anti‑Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The Anti‑Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti‑Kickback Statute to reach large settlements with healthcare companies based on alleged sham consulting and other financial arrangements with physicians. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti‑Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. The majority of states also have anti‑kickback laws which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third‑party payor, including commercial insurers.
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government. Under the False Claims Act, a person acts knowingly if he has actual knowledge of the information or acts in deliberate ignorance or in reckless disregard of the truth or falsity of the information. Specific intent to defraud is not required. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the U.S., for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi‑million and multi‑billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created new federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third‑party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti‑Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
29
The Physician Payments Sunshine Act, or the Sunshine Act, which was enacted as part of the Affordable Care Act, requires applicable manufacturers of drugs, devices, biologicals, or medical supplies covered under Medicare and other programs to report annually payments or other transfers of value made by that entity, or by a third party as directed by that entity, to physicians and teaching hospitals, or to third parties on behalf of physicians or teaching hospitals, during the course of the preceding calendar year. Failure to comply with the reporting requirements can result in significant civil monetary penalties. We are required to collect data on these payments and report such payments.
HIPAA’s privacy and security provisions, and regulations implementing these provisions, apply to “covered entities” or health care providers engaging in certain standard electronic transactions (such as electronic billing), health plans and health care clearinghouses and limit the permissible uses and disclosures of individually identifiable health information, also called protected health information or PHI. Amendments to HIPAA under the Health Information Technology for Economic and Clinical Health Act, or HITECH, require notification to the government and affected individuals of certain breaches of PHI that has not been secured or encrypted in accordance with federal standards. HIPAA does not directly regulate us, but it does limit the types of communications and data sharing that health care providers and plans may engage in with us. HITECH created liability for third parties in possession of PHI in violation of HIPAA, so we must ensure that our own uses or disclosures of, and requests for, PHI comply with these provisions. Penalties for HIPAA violations include fines, civil monetary penalties, criminal prosecution and imprisonment as well as significant reputational damage for reported breaches and other violations.
The Foreign Corrupt Practices Act
We are subject to the U.S. Foreign Corrupt Practices Act, or FCPA, which generally prohibits certain individuals and entities subject to its jurisdiction from paying, offering, or authorizing payment of anything of value, directly or indirectly to a “foreign official” with the corrupt intent of improperly influencing any act or decision of the official in order to assist the individual or business in obtaining or retaining business or any improper business advantage. The activities of our business partners and intermediaries could also subject us to potential liability under the FCPA, as we could be held responsible for their improper conduct while acting on our behalf. In addition, the FCPA requires companies whose securities are publicly listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect transactions of the corporation and to devise and maintain an adequate system of internal accounting controls for its operations. Activities that violate the FCPA, even if they occur outside of the United States, can result in criminal and civil fines, imprisonment, disgorgement, mandatory compliance monitor oversight, debarment from government contracts, and reputational damage.
We are also subject to similar anti‑bribery laws in other jurisdictions in which we conduct or expect to conduct business, including the United Kingdom’s Bribery Act of 2010, which also prohibits commercial bribery. These laws are complex and far‑reaching in nature, and may require us in the future to alter one or more of our practices to be in compliance with these laws or any changes in these laws or the interpretation thereof. Any violations of the FCPA or other anti‑corruption laws, or allegations of such violations, could disrupt our operations, involve significant management distraction, involve significant costs and expenses, including legal fees, and could result in a material adverse effect on our business, prospects, financial condition, or results of operations.
Europe
The European Medicines Agency, or EMA, is responsible for the evaluation and approval of marketing authorization applications for human and veterinary medicines in the European Union, or EU, under the “centralized procedure” set forth in Regulation (EC) No. 726/2004. The centralized procedure consists of “a single application, a single evaluation and a single authorisation” that allows applicants to obtain a marketing authorization that is valid throughout the entire European Economic Area, which is comprised of the 28 Member States of the EU plus Iceland, Norway, and Liechtenstein. In addition, each EU Member State also has its own procedures for authorizing, within its territory, medicines that fall outside of the scope of the centralized procedure.
Use of the centralized procedure is mandatory for biosimilars that are developed using one of the biotechnological processes enumerated in the Annex of Regulation (EC) No 726/2004.284. These include recombinant
30
DNA technology, hybridoma and monoclonal antibody methods, and “controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes including transformed mammalian cells.” Use of the centralized procedure is permitted, but not mandatory, where the reference medicinal product was authorized via the centralized procedure. It may also be used in cases where the reference medicinal product was not authorized via the centralized procedure, if the biosimilar product applicant demonstrates that either: (1) its product constitutes a significant therapeutic, scientific, or technical innovation; or (2) granting a single European Community marketing authorization is in the interest of patients in the European Community as a whole. Other biosimilar products may be authorized by individual member countries on a national level.
The legal basis for authorization of biosimilars in the EU is set forth in Article 10(4) of Directive 2001/83/EC, as amended by Directive 2004/27/EC,292 and Article 6 of Regulation (EC) No. 726/2004. The primary objective of the evaluation of an application pursuant to Article 10(4) is to determine the similarity of a proposed biosimilar to a reference product. The regulation does not, however, establish an explicit standard for determining biosimilarity. Rather, “[w]hether a medicinal product would be acceptable using the ‘similar biological medicinal product’ approach depends on the state of the art of analytical procedures, the manufacturing processes employed, as well as clinical and regulatory experiences.” In addition, comparability studies are needed to generate evidence substantiating the similar nature, in terms of quality, safety and efficacy, of the new similar biological medicinal product and the chosen reference medicinal product authorized in the Community.
Article 10(4) of Directive 2001/83/EC establishes the pre‑clinical and clinical testing requirements to support applications for biosimilars that do not meet the definition of generic medicinal products due to differences in raw materials or manufacturing processes. Annex I of the Directive sets forth the type and quantity of supplementary data that must be provided. To demonstrate biosimilarity between a proposed biosimilar medicinal product and a reference medicinal product, the EMA expects a robust head‑to‑head comparison that assesses quality, safety and efficacy. To demonstrate similar quality, the EMA requires studies comparing the structure and biological activity of the active ingredients in the proposed biosimilar and the reference medicinal product. Comparisons of safety and effectiveness must demonstrate the absence of significant differences between the proposed and reference products in terms of benefits and risks, including the risks of adverse immune reactions. Similar biological medicinal products are also subject to post‑marketing monitoring requirements consistent with those applicable to the reference product. The EMA requires clinical safety of biosimilars to be monitored closely on an ongoing basis during the post‑approval phase including continued benefit‑risk assessment.
The EMA has issued a number of general and product‑specific guidance documents clarifying its regulation of biosimilars. These guidance documents include guidance for industry on the non‑clinical and clinical aspects of the development of biosimilars, as well as product‑class‑specific guidelines for the development of biosimilar epoetins, filgrastims, insulins, growth hormones, alfa interferons, monoclonal antibodies, beta interferons, follitropins, and low‑molecular‑weight heparins. Although the EMA recognizes that the diversity of biological medicinal products requires a case‑by‑case determination of the amount of data required for a particular application, the guidelines issued to date indicate that the demonstration of comparability requires, among other things, an assessment of purity and impurity profiles of the active substance and medicinal product, characterization of the physicochemical properties including composition and primary and higher order structures, and an assessment of biological activity with “biological assays using different approaches to measure the biological activity,” as appropriate.
The EU regulatory framework does not provide authority for the EMA to determine whether the biosimilar may be used interchangeably with the reference product. Instead, the EMA advises patients to speak with their doctors and pharmacists about the possibility of switching between a reference product and a biosimilar product. In addition, individual Member States have the authority to determine whether or not a biosimilar is interchangeable with, or should automatically be substituted for, the reference product. Throughout the EU, all similar biological medicinal products must include the following statement in their Summary of Product Characteristics, or SmPC: “[Invented Name] is a Biosimilar medicinal product. Detailed information is available on the website of the European Medicines Agency http://www.ema.europa.eu.”
A reference biological product receives a period of eight years of data exclusivity during which time applications for biosimilars may not be filed, starting from the date of the reference product’s initial authorization. In
31
addition, the EMA may not issue a marketing authorization for a similar biological medicinal product application for a total of 10 years after the reference product’s approval, providing the reference product with an additional two years of market exclusivity after the initial eight years of data exclusivity. Market exclusivity may be extended for an additional year if the reference product sponsor obtains approval for a second significant new indication during the data exclusivity period. This framework is known as the “8 + 2 (+ 1)” exclusivity period. In addition, the patent protections available for reference products may further restrict or delay the development and approval of biosimilars. However, Article 11 of Directive 2001/83/EC and Article 3.3(b) of Regulation No. 726/2004 allow applicants and marketing authorization holders to exclude from their proposed product information those parts of the reference product’s SmPC that refer to indications or dosage forms that are covered by unexpired patents. Where patent protection of the reference product differs across Member States, the EMA allows the submission of duplicate applications for the biosimilar product to the extent that the reference product is protected by patents for certain therapeutic indications or pharmaceutical forms. Therefore, the duplicate application may contain more or fewer indications or pharmaceutical forms than the original application, as necessary to market the product in Member States where specific indications or pharmaceutical forms are protected by patents. An applicant that submits such applications must commit to extend the indications or pharmaceutical forms of the duplicate marketing authorization, or to withdraw the duplicate marketing authorization, as soon as the patents expire to ensure harmonization of the SmPCs across the EU.
China
China’s State Food and Drug Administration, or SFDA, has established a number of regulatory requirements for the registration, manufacture, quality standards, marketing, distribution, importation, pricing control, advertising and labeling of pharmaceutical products, including biosimilars, in China.
Pursuant to the Administrative Measures Governing Registration of Medicines issued by SFDA on July 10, 2007 and effective on October 1, 2007, a pharmaceutical product, whether manufactured domestically or imported from outside of China, must be registered and approved by SFDA or its local counterparts before it may be manufactured in or imported into China. Different registration requirements and formalities are applicable to the registration of new pharmaceutical products, generic pharmaceutical products and imported pharmaceutical products respectively. Biosimilar products manufactured within China and imported into China are treated as new pharmaceutical products and imported pharmaceutical products, respectively, in respect of registration requirements and formalities. A new pharmaceutical product must be registered and approved by SFDA before it can be manufactured. Once on the market, any change in dosage form or route of administration of the approved new pharmaceutical product, or any claim of a new indication for such product, shall be subject to the same scrutiny and procedure as if such change or new indication were a registration of a new pharmaceutical product. As a part of the registration process, the manufacturer of a new pharmaceutical product is also required to conduct pre‑clinical trials, apply to SFDA for permission to conduct clinical trials, and, after clinical trials are completed, file clinical data with the SFDA for approval. Once a pharmaceutical product is approved by SFDA as a new pharmaceutical product and its manufacturer has met the requisite manufacturing requirements and is in possession of a manufacturing permit, SFDA will issue a New Medicine Certificate together with an approval number to the manufacturer and may, at its discretion, impose a monitoring period of not more than five years. During the monitoring period, SFDA will monitor the safety of the new pharmaceutical product, and will refrain from both (i) registering an identical pharmaceutical product manufactured or imported by another pharmaceutical company, and (ii) approving applications made by another pharmaceutical company for changes to the ingredients of its registered product which will render it identical to the new product. On January 7, 2009, SFDA promulgated the Administrative Measures Governing the Special Examination and Approval of New Medicine Registration, under which manufacturers of certain types of new pharmaceutical products may apply to follow the special examination and approval procedure with respect to clinical trial applications or production applications as the case may be. Under such special examination and approval procedure, SFDA will strengthen communications with the applicant and the application will be reviewed on an accelerated basis. On October 29, 2014, the Center for Drug Evaluation of the SFDA issued a draft for public comment called “Technical Guidelines for Biosimilar R&D and Evaluation.” If and when the draft is finalized and implemented into law, whether in the currently published form or otherwise, the requirements for registration of biosimilars in China may change in ways that we cannot predict.
A manufacturer is required to obtain a manufacturing permit from the relevant provincial counterpart of SFDA before it may carry out manufacturing operations. The issuance of such permit is conditional upon satisfactory inspection
32
of the applicant’s manufacturing facilities and the applicant’s satisfaction of certain personnel qualifications, sanitary conditions, quality assurance systems, management structure and equipment standards requirements. Pursuant to the Regulations on the Implementation of the Law of the People’s Republic of China on the Administration of Medicines, which took effect on September 15, 2002, and the Measures on the Supervision and Administration of the Manufacture of Medicines, which took effect on August 5, 2004, a manufacturing permit is valid for five years and application for its renewal should be made to the relevant provincial counterpart of SFDA at least six months prior to its expiry. Manufacturers must also obtain GMP certification for production of pharmaceutical products in China. The Good Manufacturing Practice for Medicines (2010 revised edition) promulgated by the Ministry of Health and effective on March 1, 2011 is made up of a set of detailed guidelines on practices governing different aspects of the production of pharmaceutical products, including institutional qualifications, staff qualifications, production premises and facilities, equipment, hygiene, production management, quality control and assurance, product distribution and recall, documentation and raw material management. A GMP certificate is valid for a term of 5 years and application for its renewal should be made to the provincial counterpart of SFDA at least six months prior to its expiry.
India
In India, biosimilars (which are more commonly known as “similar biologics”) are regulated pursuant to (i) the Drugs and Cosmetics Act, 1940; (ii) the Drugs and Cosmetics Rules, 1945 as amended from time to time; (iii) Rules for the Manufacture, Use, Import, Export and Storage of Hazardous Micro Organisms, Genetically Engineered Organisms or Cells, 1986 notified under the Environment Protection Act, 1986; (iv) Recombinant DNA Safety Guidelines & Regulations, 1990; (v) Guidelines for generating Pre‑Clinical & Clinical Data for R‑DNA vaccines, diagnostics & other Biologicals, 1999; (vi) Good Environmental Practice guidelines issued by the Ministry of Environment and Forests, including Good Practices in Environmental Regulation and Good Practices in Animal Experimentation; (vii) CDSCO Guidance for Industry, 2008 (read with clarificatory Circular dated 5th August, 2010), which covers: (a) Submission of Clinical Trial Application for Evaluating Safety and Efficacy; (b) Requirements for permission of New Drugs Approval; (c) Post approval changes in biological products: Quality, Safety and Efficacy documents; and (d) Preparation of the Quality Information for Drug Submission for New Drug Approval: Biotechnological/Biological Products. issued by the CDSCO with respect to the requirements for post‑approval changes; (viii) Guidelines and Handbook for Institutional Biosafety Committees, 2011; and (ix) Guidelines on Recall & Rapid Alert System for Drugs (Including Biologicals & Vaccines) (effective from 23rd November 2012). In addition, the Central Drugs Standard Control Organization, or CDSCO, established by the Ministry of Health and Welfare, Government of India, which is the overarching regulatory body in the Ministry for the regulation of drugs, issues additional requirements for approval of drugs or clarification on the requirements from time to time.
In June 2012, the CDSCO and the Department of Biotechnology issued a guidance document titled “Guidelines on Similar Biologics.” The Guidelines lay down the regulatory pathway for a biologic claiming to be similar to an already authorized reference biologic. Pursuant to the Guidelines, to obtain approval for a biosimilar, the sponsor must establish that the proposed biosimilar is similar to the reference biologic, which is authorized for marketing in India. The similarity of the proposed biosimilar to the reference product is demonstrated through quality characterization studies that compare the molecular and quality attributes of the proposed biosimilar with those of the reference biologic. The testing of the proposed biosimilar should also be sufficient to ensure that the proposed biosimilar meets acceptable levels of safety, quality and efficacy. Biosimilars are subject to a number of premarket regulatory requirements, including comparability exercises for quality, pre‑clinical and clinical studies, along with post‑market regulatory obligations, including post‑approval testing. The Guidelines are applicable both to similar biologics developed in India and similar biologics imported into India.
The authorities involved in the approval process of biosimilars include the Review Committee on Genetic Manipulation, or RCGM, set up in the Department of Biotechnology Ministry of Science and Technology, Government of India; the Genetic Engineering Appraisal Committee, or GEAC, in the Ministry of Environment and Forests; and CDSCO, in the Ministry of Health and Welfare. The RCGM is responsible for authorizing import and export for research and development and review of data up to preclinical evaluation. The GEAC is a statutory body established for review and approval of activities involving large‑scale use of genetically engineered organisms (also referred as living modified organisms) and products thereof in research and development, industrial production, environmental release and field applications. The CDSCO is the apex regulatory body for the regulation of drugs, including biosimilars. It is responsible
33
for the grant of import and export licenses, clinical trial approvals and authorizations for marketing and manufacturing. The State Food & Drug Administration works with CDSCO in each state within India and is responsible for the issuance of licenses to manufacture similar biologics in India. Other bodies involved in the approval process for biosimilars are the Recombinant DNA Advisory Committee, the Institutional Biosafety Committees, the State Biosafety Co‑ordination Committees, the District Level Committees, and the State Licensing Authority.
Employees
As of March 2016, we had 73 employees, of which 45 were engaged in research and development activities and 28 were engaged in finance, business development, legal, human resources and other general administrative activities. None of our employees are represented by a labor union and we consider our employee relations to be good.
Research and Development
We have built a research and development organization that includes extensive expertise in the scientific disciplines of biosimilars. We operate cross‑ functionally and are led by an experienced research and development management team. We use rigorous project management techniques to assist us in making disciplined strategic research and development program decisions and to help limit the risk profile of our product pipeline. We also access relevant market information and key opinion leaders in creating target product profiles and, when appropriate, as we advance our programs towards commercialization. We engage third parties to conduct portions of our preclinical research. In addition, we utilize multiple clinical sites to conduct our clinical trials; however we are not substantially dependent upon any one of these sites for our clinical trials nor do any of them conduct a major portion of our clinical trials.
We incurred $30.9 million and $16.3 million in research and development expenses in the years ended December 31, 2015 and 2014, respectively.
Corporate Information
We were incorporated in Delaware in 2000. Our principal executive offices are located at 699 Boylston Street, 8th Floor, Boston, MA 02116, and our telephone number is (617) 600-3497. We have the following subsidiaries: EB Sub, Inc., a Delaware corporation, Epirus Biopharmaceuticals Ltd., a United Kingdom corporation, Epirus Biopharmaceuticals (Switzerland) GmbH, a Swiss corporation, Epirus Brasil Tecnologia Ltda, a Brazilian corporation and Epirus Biopharmaceuticals (Netherlands) B.V., a Netherlands corporation. We maintain an internet website at www.epirusbiopharma.com and the information contained in, or that can be accessed through, our website is not part of this annual report and should not be considered part of this annual report.
Private Epirus was formerly known as Fourteen22, Inc. and changed its name to EPIRUS Biopharmaceuticals, Inc. in January 2013. Fourteen22, Inc. was originally incorporated in November 2008 in the Cayman Islands. In January 2011, Fourteen22, Inc. became a Delaware corporation. On July 15, 2014, in connection with the merger with Zalicus Inc., Zalicus was renamed EPIRUS Biopharmaceuticals, Inc. and we began operating under our current corporate structure.
Available Information
We file annual, quarterly, and current reports, proxy statements, and other documents with the Securities and Exchange Commission (SEC) under the Securities Exchange Act of 1934 (the Exchange Act). The public may read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1‑ 800‑SEC‑0330. Also, the SEC maintains an internet website at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers, including us, that file electronically with the SEC.
We also make available free of charge on our Internet website at www.epirusbiopharma.com our annual reports on Form 10‑K, quarterly reports on Form 10‑Q, current reports on Form 8‑K, and, if applicable, amendments to those
34
reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
Our code of business conduct and ethics, and the charters of our Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee are available through our Internet website at www.epirusbiopharma.com.
Investing in our common stock involves a high degree of risk. The risks described below and elsewhere in this annual report on Form 10-K are some of the important factors that, individually or in the aggregate, we believe could make our actual results differ materially from those described in any forward-looking statements. The risks described below are not the only ones we face. Our business operations could also be affected by additional factors that apply to all companies operating in the U.S. and globally, as well as other risks that are not presently known to us or that we currently consider to be immaterial to our operations.
In the following discussion of risk factors, references to "Zalicus" refer to the business of Zalicus Inc. as it existed prior to the merger on July 15, 2014, and references to "Private Epirus" refer to the business of EPIRUS Biopharmaceuticals, Inc. prior to the merger on July 15, 2014. References to "we," "us," "our," and similar terms refer to the combined business of EPIRUS Biopharmaceuticals, Inc. after the merger on July 15, 2014.
Risks Related to Financial Results, Need for Additional Financing and Debt Arrangements
We have a history of operating losses. We expect to incur significant operating losses and may never be profitable. Our common stock is a highly speculative investment.
Prior to the July 15, 2014 merger with Zalicus Inc. (the “Merger”), we incurred operating losses since its inception, and was never cash-positive. Similarly, following the Merger, we have continued to incur net losses and has not been cash-positive. As of December 31, 2015, we had an accumulated deficit of $138.8 million. We have spent, and expect to continue to spend, significant resources to fund research and development of our product candidates and to enhance our drug development technologies. We expect to incur substantial operating losses over the next several years due to our ongoing research, development, pre-clinical testing, and clinical trial activities. As a result, our accumulated deficit will continue to increase.
Except for BOW015, our product candidates are in the early stages of development and may never result in any revenue. We will not be able to generate product revenue unless we or our partners successfully commercialize BOW015 or our early stage product candidates. We may seek to obtain revenue from collaboration or licensing agreements with third parties. Our current collaboration and license agreements may not provide us with material, sustainable ongoing future revenue, and we may not be able to enter into additional collaboration agreements. Even if we eventually generate product revenues, we may never be profitable, and if we ever achieve profitability, we may not be able to sustain it.
We will require substantial additional funds in the near term to continue as a going concern, and to obtain regulatory approval for and commercialize our current and any future biosimilar product candidates and, if additional capital is not available, we may need to limit, scale back or cease our operations.
Since our inception, we have devoted substantial resources to the nonclinical and clinical development of our most advanced biosimilar product candidate, BOW015, a proposed biosimilar of Remicade® (infliximab). We will incur substantial costs for our global clinical program for BOW015, which we initiated in February 2016. We believe that we will continue to expend substantial resources for the foreseeable future for the clinical development of our current pipeline of biosimilar product candidates, including for development of any other indications and product candidates we may choose to pursue. These expenditures will include costs associated with research and development, conducting nonclinical studies and clinical trials, and manufacturing and supply as well as marketing and selling any products approved for sale. In addition, other unanticipated costs may arise. Because the outcome of any clinical trial is highly
35
uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of BOW015 in all of the markets in which we plan to commercialize the product and our pipeline of other product candidates.
We believe that our existing cash and cash equivalents will be sufficient to fund our operations into the second quarter of 2016. As of December 31, 2015, our total unrestricted cash and cash equivalents was $31.5 million. We will need to raise additional capital in the near term to fund our operating requirements and continue as a going concern. We may also seek to raise additional funds if our research and development expenses exceed current expectations, our collaboration funding is less than current assumptions or expectations, or we encounter obstacles to our development and commercialization of our product candidates that were not anticipated. This could occur for many reasons, including:
our product candidates require more extensive clinical or pre-clinical testing, our research and development programs for our product candidates do not proceed as expected, our clinical trials take longer to complete than we currently expect or our clinical trials are not successful;
we advance more of our product candidates than expected into costly later stage clinical trials;
we advance more of our pre-clinical product candidates than expected into early stage clinical trials;
our revenue-generating collaboration agreements are terminated;
we are unable to reduce our cost structure while simultaneously pursuing new business opportunities and enhancing other aspects of our business;
the time and costs involved in obtaining regulatory approvals are higher than anticipated in one or more jurisdictions where we are seeking to manufacture and/or market our products;
some or all of our product candidates fail in clinical or pre-clinical studies or prove to be less commercially promising than we expect or we are forced to seek additional product candidates;
we are required, or consider it advisable, to acquire or license rights from one or more third parties;
we determine to enter into additional business combinations or acquire or license rights to additional product candidates or new technologies;
the cost of regulatory, manufacturing and commercialization activities, if any, relating to our product candidates, are higher, or take longer to establish than anticipated; or
we are subject to litigation in relation to our activities with respect to our product candidates, including potential patent litigation with innovator companies or others who may hold patents.
These factors raise substantial doubt about our ability to continue as a going concern.
Our consolidated financial statements for the year ended December 31, 2015 have been prepared assuming that we will continue as a going concern. The factors describe above coupled with our recurring losses from operations and our stockholders’ deficit raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our consolidated financial statements for the year ended December 31, 2015 with respect to this uncertainty. The perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations and may adversely affect our ability to raise additional capital.
36
While we expect to seek additional funding through public or private financings, we may not be able to obtain financing on acceptable terms, or at all. In addition, the terms of any financings may be dilutive to, or otherwise adversely affect, holders of our common stock. We may also seek additional funds through arrangements with collaborators or others. These arrangements would generally require us to relinquish rights to some of our technologies, product candidates or products, and we may not be able to enter into such agreements on acceptable terms, if at all. The arrangements also may include issuances of equity, which may also be dilutive to, or otherwise adversely affect, holders of our common stock. There can be no assurance that we will be able to access equity or credit markets in order to finance our operations or expand development programs for any of our product candidates, or that there will not be a deterioration in financial markets and confidence in economies that could make it difficult or impossible to access these markets. We may also have to scale back or further restructure our operations. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all of our research or development programs.
With the filing of this annual report on Form 10-K, unless and until the value of our public float (which refers to the shares held by shareholders who are not affiliates of ours) increases to an amount equal to or greater than $75 million, we will be subject to limitations imposed by SEC rules on the value of shares we may issue pursuant to our Form S-3 shelf registration statement in a primary offering. Specifically, SEC rules provide that a company with less than $75 million in public float may issue securities with a value of no more than one-third of the company’s public float value in any twelve-month period. This may further affect the amount of capital we can raise to pursue the development of our programs.
Our term loans include certain covenants and other events of default. Should we not comply with these covenants or incur an event of default, we may be required to repay our obligation in cash, which could have an adverse effect on our liquidity.
Our term loans from Hercules Technology Growth Capital, Inc. include certain customary covenants, including limitations on other indebtedness, liens, acquisitions, investments and dividends.
If we fail to stay in compliance with our covenants or suffer some other event of default under the term loans, we may be required to repay the outstanding principal. Should this occur, our liquidity would be adversely impacted.
The requirements of being a public company have required and will continue to require significant resources, increase our costs and occupy our management’s time and efforts, and we may be unable to continue to comply with these requirements in a timely or cost-effective manner.
Our merger with Zalicus was determined to constitute a reverse acquisition and Private Epirus, a privately held company, was determined to be the acquirer for accounting purposes. Although Zalicus was an operating company, its operations were deemphasized following the consummation of the merger with the post-merger entity, which focuses on Private Epirus’ biosimilar business. Because the financial statements and information relating to Private Epirus now constitute our financial statements and information, we are in a position similar to a newly public company.
As a company with public reporting responsibilities, we have incurred and will continue to incur significant legal, accounting, and other expenses that Private Epirus did not incur as a private company. Complying with rules, regulations and requirements applicable to public companies has required substantial effort and has increased and may continue to increase our costs and expenses. We have been required to:
institute a more formalized function of internal control over financial reporting;
prepare, file and distribute periodic and current reports under the Securities Exchange Act of 1934, or the Exchange Act, and comply with other Exchange Act requirements applicable to public companies;
formalize old and establish new internal policies, such as those relating to insider trading and disclosure controls and procedures;
37
involve and retain to a greater degree outside counsel and accountants in the above activities; and
establish and maintain an investor relations function, including the provision of certain information on our website.
Compliance with these rules and regulations has increased, and will continue to increase, our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, these rules and regulations have made it more expensive for us to obtain director and officer liability insurance and we have been required to incur substantial costs to maintain the same or similar coverage.
In connection with our merger, the Securities and Exchange Commission, or the SEC, granted us a waiver of the requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, for a period of one year, and therefore we were not required to make a formal assessment of the effectiveness of our internal controls over financial reporting for that purpose until the filing of this annual report on Form 10-K for the fiscal year ending December 31, 2015. In anticipation of this formal assessment, we have been required to comply with Section 404 since January 1, 2015. In connection with the audit of our 2015 financial statements and in connection with the preparation of certain of our 2014 interim financial statements, our management team and independent registered public accounting firm identified certain weaknesses in our internal controls that were considered to be material weaknesses. For a discussion of our internal control over financial reporting and a description of the identified material weaknesses, see "Management's Report on Internal Control over Financial Reporting" under Item 9A, "Controls and Procedures."
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2015, excluding the internal controls of Bioceros, B.V., which the Company acquired on September 9, 2015. This assessment identified material weaknesses in our internal control over our financial statement close process and the financial reporting processes related to business combinations, income taxes and accrued development costs. Because of these material weaknesses, management believes that, as of December 31, 2015, our internal control over financial reporting was not effective based on those criteria.
We may subsequently identify additional errors or deficiencies in our internal control over financial reporting, including any such errors or deficiencies in connection with the Bioceros financial statements that are deemed to be material weaknesses, such as the material weakness discussed in the other risk factors described in this section. Additionally, in complying with Section 404, we have incurred, and expect to continue to incur, significant expenses and devote substantial management effort toward ensuring compliance with these requirements. If we are not able to comply with these requirements in a timely manner, or if we identify deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our common stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would entail expenditure of additional financial and management resources. See “Risks Related to an Investment in Our Common Stock—Our management and independent auditors have identified material weaknesses in our internal controls, and we may be unable to develop, implement and maintain appropriate internal and disclosure controls in future periods, which may lead to errors or omissions in our financial statements.”
Our international operations subject us to potentially adverse tax consequences.
We generally conduct our international operations through wholly-owned subsidiaries and report our taxable income in various jurisdictions worldwide based upon our business operations in those jurisdictions. Our intercompany relationships are subject to complex transfer pricing regulations administered by taxing authorities in various jurisdictions. The relevant taxing authorities may disagree with our determinations as to the income and expenses attributable to specific jurisdictions. If such a disagreement were to occur, and our position were not sustained, we could be required to pay additional taxes, interest and penalties, which could result in one-time tax charges, higher effective tax rates, reduced cash flows and lower overall profitability of our operations.
38
Risks Relating to the Development, Manufacturing and Commercialization of Our Products
We are largely dependent on the success of BOW015. All of our other product candidates are still in pre-clinical development. If we are unable to obtain regulatory approval in additional jurisdictions for, or successfully commercialize, BOW015, our business will be materially harmed.
Our business prospects and potential product revenues are largely dependent upon our ability to obtain regulatory approval of, and successfully commercialize, BOW015. We have reported positive bioequivalence and efficacy data in the clinical development program for BOW015, including a Phase 1 clinical trial in the United Kingdom and a Phase 3 clinical trial in India, in each case showing equivalence with Remicade as the reference product. We also announced positive 58 week follow up data from the Phase 3 trial. In this open label phase, we demonstrated that patients can be initiated and maintained on BOW015 for 58 weeks, and that patients can be switched from Remicade to BOW015 and maintained out to 58 weeks, with a favorable safety and efficacy profile. In March 2014, our manufacturing partner, Reliance Life Sciences Pvt Ltd, or RLS, obtained marketing approval on our behalf in India for BOW015 for rheumatoid arthritis on the basis of our Phase 3 trial, and in September 2014, RLS received final manufacturing and market approval on our behalf from the Drug Controller General of India, or the DCGI. We currently have an agreement to commercialize BOW015 in India with our partner Sun Pharmaceutical Industries Ltd., or Sun, and in November 2014, we launched BOW015 in India with Sun. As discussed in the other risk factors described in this section, we settled a dispute with RLS that may adversely affect our ability to rely on the approvals that have been obtained by RLS for our benefit and that we currently rely upon for selling BOW015 in India. In the event that the DCGI does not approve the transfer of the marketing approval to us or our designee, pursuant to the terms of our Settlement Agreement with RLS (as described below) and it becomes necessary to obtain new manufacturing and marketing approvals from the DCGI, no assurance can be given as to whether we or our partners will be able to obtain the required approvals or that any such approvals will be obtained in a timely manner. If Sun encounters delays or difficulties in commercializing BOW015 pursuant to our agreement, it may adversely affect our ability to successfully commercialize BOW015 in India. Additionally, in conjunction with other potential partners with whom we may form alliances, we intend to file for regulatory approval in additional markets where our current data are expected to be sufficient for approval. In February 2016, we initiated this global clinical program, which we refer to as the UNIFORM Study. The UNIFORM Study is a 58-week, double-blind, one-to-one randomized, comparator-controlled multi-center global study to compare safety, efficacy and immunogenicity and demonstrate clinical equivalence of BOW015 with Remicade. As discussed in the other risk factors described in this section, obtaining regulatory approval is costly and uncertain. Even if we obtain regulatory approval for BOW015 in a jurisdiction, we may not be successful in commercializing it in the jurisdiction. If we fail to successfully commercialize BOW015, or encounter significant delays in doing so, we may be required to curtail or terminate some or all of our research or development programs and our business prospects, financial condition and results of operations would be materially harmed. To date, we have not yet commercialized BOW015 in any of our target markets other than India.
Our other product candidates, BOW050, BOW070, BOW080, BOW090 and BOW100 are in pre-clinical development. Before we can commercialize these product candidates we need to:
conduct substantial research and development;
undertake nonclinical and clinical testing, for the purpose of demonstrating bioequivalence with the applicable reference products, and engage in sampling activity and other costly and time consuming measures;
scale-up manufacturing processes; and
pursue and obtain marketing and manufacturing approvals and, in some jurisdictions, pricing and reimbursement approvals.
This process involves a high degree of risk and takes many years, and success is never guaranteed. Our product development efforts with respect to these product candidates may fail for many reasons, including:
39
failure of the product candidate in nonclinical studies, including those required to demonstrate bioequivalence with the reference product;
inability to obtain on a timely basis supplies of the applicable reference products to which our product candidates must be compared;
delays or difficulty enrolling patients in clinical trials, particularly for disease indications with small patient populations;
patients exhibiting adverse reactions to the product candidate or indications of other safety concerns;
insufficient clinical trial data to support the bioequivalence of one or more of our product candidates with the applicable reference product;
inability to manufacture sufficient quantities of the product candidate for development or commercialization activities in a timely and cost-efficient manner, if at all;
potential patent litigation with innovator companies or others who may hold patents;
failure to obtain, or delays in obtaining, the required regulatory approvals for the product candidate, the facilities or the processes used to manufacture the product candidate; or
changes in the regulatory environment, including pricing and reimbursement that make development of our biosimilar product candidates or development of such product candidates for a new indication no longer desirable.
If our product development efforts fail for any of these or other reasons, or we decide to abandon development of a product candidate at any time, we would never realize revenue from those programs and our business could be materially harmed.
If an improved version of a reference product, such as Remicade, is developed, or if the market for a reference product significantly declines, sales or potential sales of our biosimilar product candidates may suffer.
We are developing and commercializing follow-on versions of approved, reference biological products. Such follow-on products are known as biosimilars. Innovator companies may develop improved versions of a reference product as part of a life cycle extension strategy, and may obtain regulatory approval of the improved version under a new or supplemental application filed with the applicable regulatory authority. Should the innovator company succeed in obtaining an approval of an improved biologic product, it may capture a significant share of the collective reference product market in the applicable jurisdiction and significantly reduce the market for the reference product and thereby the potential size of the market for our product candidates. In addition, the improved product may be protected by additional patent rights and thereby subject our follow-on biosimilar to claims of infringement.
Additionally, competition in the biotechnology industry is intense. Reference biologic products face competition on numerous fronts as technological advances are made that may offer patients a more convenient form of administration or increased efficacy, or as new products are introduced. As new products are approved that compete with the reference products to our biosimilar product candidates, such as Remicade, or our pipeline products, such as Humira and others, sales of the reference biologic products may be significantly and adversely impacted and may render the reference products obsolete. If the market for the reference product is impacted, we in turn may lose significant market share or market potential for our biosimilar products and product candidates, and the value for our product pipeline could be negatively impacted. As a result of the above factors, our business, prospects and financial condition could suffer.
40
Our biosimilar product candidates, if approved, will face significant competition from the reference products and from other biosimilars approved for the same indication as the reference biologic products. Our failure to effectively compete may prevent us from achieving significant market penetration and expansion.
Our business involves highly competitive pharmaceutical and biotechnology markets. Successful competitors in the pharmaceutical market have the ability to effectively discover, obtain patents, develop, test and obtain regulatory approvals for products, as well as the ability to effectively commercialize, market and promote approved products, including communicating the effectiveness, safety and value of products to actual and prospective customers and medical staff. Numerous companies, universities, and other research institutions are engaged in developing, patenting, manufacturing and marketing of products competitive with those that we are developing. Many of these potential competitors, such as Celltrion, Inc. and Hospira, Inc. (acquired by Pfizer, Inc.), are large, experienced companies that enjoy significant competitive advantages, such as substantially greater financial, research and development, manufacturing, personnel and marketing resources, greater brand recognition and more experience and expertise in undertaking nonclinical testing and clinical trials of product candidates, and obtaining regulatory approvals of products. If we do not effectively compete with these potential competitors, the value of our product pipeline could be negatively impacted and our business, prospects and financial condition could suffer.
Use of our product candidates could be associated with side effects or adverse events.
As with most pharmaceutical and biological drug products, use of our product candidates could be associated with side effects or adverse events, which can vary in severity (from minor reactions to death) and frequency. Side effects or adverse events associated with the use of our product candidates or those of our collaborators may be observed at any time, including in clinical trials or later when a product is commercialized, and any such side effects or adverse events may negatively affect our ability to obtain regulatory approvals or market our product candidates. Side effects such as toxicity or other safety issues associated with the use of our product candidates could require us or our collaborators to halt development or sale of these product candidates or expose us to product liability lawsuits that would harm our business. We may also be required by regulatory agencies to conduct additional nonclinical studies or clinical trials that we have not planned or anticipated. There can be no assurance that we will resolve any issues related to any product-related adverse events to the satisfaction of any regulatory agency in a jurisdiction where we are seeking to commercialize our products or obtain approval in a timely manner or ever, which could harm our business, prospects and financial condition.
In addition, if we are successful in commercializing the most advanced biosimilar in our pipeline, BOW015, or any other of our pipeline product candidates, then laws and regulations may require that we report certain information about adverse medical events according to timelines that may vary from jurisdiction to jurisdiction. The timing of our obligation to report may be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, regulatory agencies could take action including, without limitation, criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or delay in approval of future products. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
We may have significant product liability exposure that may harm our business and our reputation.
Although our product candidates are biosimilar products, and are designed to be equivalent against reference products that have an established safety record in the indications in which they are marketed and used, we face a risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk as we commercialize our products. For example, we may incur liability if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts or comparable statutes abroad. If we cannot successfully defend ourselves against product liability claims,
41
we may incur substantial liabilities or be required to limit commercialization of our products. Regardless of the merits or eventual outcome, liability claims may result in:
decreased demand for BOW015, or any future product candidates that may obtain regulatory approval;
injury to our reputation and significant negative media attention;
withdrawal of clinical trial participants or cancellation of clinical trials;
costs to defend the related litigation;
a diversion of management’s time and our resources;
substantial monetary awards to trial participants or patients;
regulatory investigations, product recalls, withdrawals or labeling, marketing or promotional restrictions;
loss of revenue; and
the inability to commercialize any products we develop.
Our inability to obtain and maintain sufficient product liability insurance at an acceptable cost and scope of coverage to protect against potential product liability claims could impact the commercialization of BOW015, and any of our other product candidates. We currently carry product liability insurance covering our clinical trials in the amount of $5 million in the aggregate. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions and deductibles, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses in all the jurisdictions in which we seek to market and sell our products. As we obtain approvals for marketing BOW015 and any other product candidates in more jurisdictions, we intend to expand our insurance coverage to include the sale of such products; however, we may be unable to obtain this liability insurance on commercially reasonable terms.
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop our pipeline product candidates, conduct our clinical trials and commercialize BOW015, or any future pipeline product candidates we develop.
Our success depends in large part on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. We believe that our future success is highly dependent upon the contributions of our senior management, particularly our chief executive officer, chief financial officer, chief technical officer, vice president of program management, and vice president of manufacturing and quality, as well as our senior scientists and other members of our senior management team. The loss of services of any of these individuals could delay or prevent the successful development of our product pipeline, completion of our planned clinical trials or the commercialization of BOW015, or any future products we develop.
Although we have not historically experienced significant difficulties attracting and retaining qualified employees, we could experience such problems in the future. For example, competition for qualified personnel in the biotechnology and pharmaceuticals field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel as we expand our process and clinical development and commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all, which may cause our business and operating results to suffer.
42
We expect to enter into alliances with other companies that can provide capabilities and funds for the development and commercialization of our product candidates, as well as local market access in jurisdictions where we intend to pursue our In Market, For Market commercialization strategy. If we are unsuccessful in forming or maintaining these alliances on favorable terms, or if such alliances do not prove to be as successful as we anticipate, our business could be adversely affected.
Our In Market, For Market strategy for development and commercialization of our biosimilar product candidates in key emerging markets is based on the assumption that we will enter into collaborative relationships with local entities to facilitate our access to and penetration into such markets. We also have limited or no capabilities for independent manufacturing, sales, marketing and distribution. In September 2014, we entered into a collaboration agreement with Livzon MabPharm Inc., or Livzon, for the development and commercialization of BOW015 and other pipeline biosimilar products to be agreed with Livzon in China, using our In Market, For Market strategy, and in September 2015, added BOW070 as a product candidate to be developed pursuant to the terms of this collaboration agreement. We retain global rights to commercialize BOW015, BOW070 and the other pipeline biosimilar products outside of China, subject to our other agreements with third parties.
In the future, we expect to form additional alliances with other companies that are based in our target markets and that have expertise in development, manufacture and commercialization of biologics and biosimilar products in our target markets to enable us to expand the commercialization opportunities for our product candidates. In such alliances, we would expect our partners to provide substantial capabilities in clinical development, manufacturing, regulatory affairs, sales and marketing. We may not be successful in entering into any such alliances. Even if we do succeed in securing such alliances, we may not be able to maintain them if, for example, development or approval of a product candidate is delayed or sales of an approved product are disappointing. If we are unable to secure or maintain such alliances we may not have the capabilities necessary to continue or complete development of our product candidates and bring them to market, which may have an adverse effect on our business.
In addition to product development and commercialization capabilities, we may depend on our alliances with other companies, such as our current alliances with Livzon, Polpharma, and Mabxience, among others, to provide substantial additional funding for development and potential commercialization of our product candidates. We may not be able to obtain funding on favorable terms from these alliances, and if we are not successful in doing so, we may not have sufficient funds to develop a particular product candidate internally, or to bring product candidates to market. Failure to bring our product candidates to market would prevent us from generating sales revenue, and this would substantially harm our business. Furthermore, any delay in entering into these alliances could delay the development and commercialization of our product candidates and reduce their competitiveness even if they reach the market. As a result, our business may be adversely affected.
Failure to obtain or maintain adequate coverage and reimbursement for our product candidates could limit our ability to market those products and decrease our ability to generate revenue.
Sales of our product candidates will depend substantially, both domestically and abroad, on the extent to which our product candidates will be covered by third party payors such as private health insurers, managed care organizations, and government healthcare programs such as Medicare and Medicaid and the adequacy of the payments for those products. If coverage and reimbursement are not available, or are available only to limited levels, we may not be able to successfully commercialize our product candidates. Because there are not many approved biosimilars available, many countries and payors have not yet established policies for the coverage and payment for those types of drugs, even if the reference biologic is covered. Moreover, even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a return on our investment.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, third-party payors, including private and governmental payors, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs and biologics will be covered and reimbursed. The Medicare program covers most individuals aged 65 or older, individuals suffering from end-stage renal disease and certain disabled individuals. The Medicaid program, which varies from state-to-state, covers individuals and families who have limited financial means or meet other eligibility requirements. Medicare is often used
43
as a model for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada, and other countries has and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. In general, the prices of medicines under such systems are substantially lower than in the United States. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenues and profits.
Moreover, increasing efforts by governmental and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Healthcare legislative reform measures may have a material adverse effect on our business and results of operations.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the Affordable Care Act, was passed, which substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things, subjects biologic products to potential competition by lower-cost biosimilars, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations, establishes annual fees and taxes on manufacturers of certain branded prescription drugs, and a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
If we obtain approval from the U.S. Food and Drug Administration, or FDA, for any of our product candidates and begin commercializing those products in the United States, our operations may be directly, or indirectly through our customers, subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal False Claims Act, and physician sunshine laws and regulations. These laws may impact, among other things, our proposed sales, marketing, and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
44
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs;
federal, civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent;
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters;
HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and its implementing regulations, which imposes certain requirements relating to the privacy, security, and transmission of individually identifiable health information;
the federal physician sunshine requirements under the Affordable Care Act, which require manufacturers of drugs, devices, biologics, and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations; and
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers, state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our current or future business activities could be subject to challenge under one or more of such laws. In addition, recent health care reform legislation has strengthened these laws. For example, the Affordable Care Act, among other things, amends the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. Moreover, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from participation in government health care programs, such as Medicare and Medicaid, imprisonment of our employees, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Failure to comply with the Foreign Corrupt Practices Act, or FCPA, and other similar anti-corruption laws and anti-money laundering laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations could subject us to significant penalties and damage our reputation.
45
We are subject to the FCPA, which generally prohibits U.S. companies and intermediaries acting on their behalf from offering or making corrupt payments to “foreign officials” for the purpose of obtaining or retaining business or securing an improper business advantage. The FCPA also requires companies whose securities are publicly listed in the United States to maintain accurate books and records and to maintain adequate internal accounting controls. We are also subject to other similar anti-corruption laws and anti-money laundering laws, as well as export control laws, customs laws, sanctions laws and other laws that apply to our activities in the countries where we operate. Certain of the jurisdictions in which we conduct or expect to conduct business have heightened risks for public corruption, extortion, bribery, pay-offs, theft and other fraudulent practices. In addition, our international business is heavily regulated and therefore involves significant interactions with “foreign officials.” We are also subject to other federal statutes and regulations, including those established by the Office of Foreign Assets Control, or OFAC. In many countries, health care professionals who serve as investigators in our clinical studies, or prescribe or purchase our commercialized products, are employed by a government or an entity owned or controlled by a government. Dealings with these investigators, prescribers and purchasers are subject to regulation under the FCPA. Under these laws and regulations, as well as other anti-corruption laws, anti-money-laundering laws, export control laws, customs laws, sanctions laws and other laws governing our operations, various government agencies may require export licenses, may seek to impose modifications to business practices, including cessation of business activities in sanctioned countries or with sanctioned persons or entities and modifications to compliance programs, which may increase compliance costs, and may subject us to fines, penalties and other sanctions.
Recent events in Ukraine and Crimea have resulted in the European Union and the United States imposing and escalating sanctions on Russia and certain businesses, sectors and individuals in Russia. The European Union and the United States have also suspended the granting of certain types of export licenses to Russia. Russia has imposed its own sanctions on certain individuals in the United States and may impose other sanctions on the United States and the European Union and/or certain businesses or individuals from these regions. Although our products are not currently subject to such sanctions, we cannot assure you that the current sanctions or any further sanctions imposed by the European Union, the United States or other international interests will not materially adversely affect our operations. We rely on local and strategic business partners and collaborators to produce and commercialize our products in certain markets outside of the United States, and we rely on distributors and other intermediaries to sell and distribute our products internationally.
We maintain policies, procedures and internal controls designed to promote compliance with the FCPA, OFAC restrictions and other anti-corruption laws and anti-money laundering laws, as well as export control laws, customs laws, sanctions laws and other laws. However, if we, our business partners, collaborators or intermediaries fail to comply with the requirements of the FCPA, OFAC restrictions or similar laws of other countries, or our controls and procedures fail to detect such noncompliance, governmental authorities in the United States or elsewhere, as applicable, could seek to impose civil penalties, criminal fines, and other collateral consequences. As we continue to expand internationally, there continues to be risk that the policies, procedures and internal controls on which we currently rely to promote compliance with these laws, regulations and restrictions will fail to be sufficiently effective and we will need to continue to develop those policies. A violation of these laws or regulations could damage our reputation and have a material adverse effect on our business, financial condition, and results of operations.
Our employees, independent contractors, principal investigators, contract research organizations, or CROs, consultants and collaborators may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk that our employees, independent contractors, principal investigators, CROs, consultants and collaborators may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or unauthorized activities that violate: (1) regulations of regulatory authorities in jurisdictions where we are performing activities in relation to our product candidates, including those laws requiring the reporting of true, complete and accurate information to such authorities; (2) manufacturing regulations and standards; (3) applicable regulatory laws prohibiting the promotion of a medical product for a use that has not been cleared or approved by the applicable regulator; (4) fraud and abuse, anti-corruption laws and anti-money laundering laws, as well as similar laws and regulations and other laws; or (5) laws that require the reporting of true and accurate financial information and data. In particular, sales, marketing and business arrangements in the healthcare
46
industry are subject to extensive laws and regulations, as well as various corruption investigations, intended to prevent fraud, bias, misconduct, kickbacks, self-dealing and other abusive practices, and these laws may differ substantially from country to country. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct by these parties could also include the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. While we have in place a Code of Business Conduct and Ethics, it is not always possible to identify and deter misconduct by employees and other third parties. Moreover, the precautions we take to detect and prevent this activity may not be effective in controlling misconduct or preventing governmental investigations or proceedings or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in subsidized healthcare programs in a given country, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Risks Related to Regulatory Approvals
The regulatory approval process is costly and lengthy and, for biosimilar products, still evolving. We may not be able to successfully obtain all required regulatory approvals or may be delayed in commercializing our products even after regulatory approvals are obtained.
The pre-clinical development, clinical trials, manufacturing, marketing, testing, approval, and labeling of pharmaceutical and biological products, including biosimilars, are all subject to extensive regulation by numerous regulatory authorities throughout the world. We or our collaborators must obtain regulatory approval for product candidates before marketing or selling any of them. The approval process is typically lengthy and expensive, and approval is never certain. Moreover, only one biosimilar has been approved in the United States to date, and few biosimilars have been approved in other countries. It is not possible to predict with any specificity how long the approval processes of the applicable regulatory authorities for any of our products will take or whether any such approvals ultimately will be granted on a timely basis or at all. Moreover, we expect to seek regulatory approvals in jurisdictions in which we have limited experience navigating the regulatory frameworks, or where the regulatory frameworks for the approval of biosimilars are not well-developed and are constantly evolving. In particular, the regulatory standards applicable to establish biosimilarity vary by jurisdiction. The last ten years have seen the establishment in many jurisdictions of a formal regulatory process for review and approval of biosimilar products, but these procedures are at differing stages of development, with limited harmonization among major jurisdictions. We plan to continue to analyze and incorporate into our biosimilar development plans any new laws, regulations, or policies promulgated or established by relevant authorities. The costs of development and approval, along with the probability of success for our biosimilar product candidates, will be dependent upon application of any laws and regulations issued by the relevant regulatory authorities.
Regulatory agencies generally have substantial discretion in the biologic and biosimilar approval processes, and positive results in pre-clinical testing or early phases of clinical trials offer no assurance of success in later phases of such tests or trials. Generally, pre-clinical and clinical testing of products can take many years and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Any delay in obtaining, or failure to obtain, approvals could prevent or adversely affect the marketing of our products or our collaborator’s products and our ability to generate product revenue. The risks associated with the approval process include delays or rejections in the regulatory approval process based on the failure of clinical or other data to meet expectations, or the failure of the product or candidate to meet a regulatory agency’s requirements for safety, efficacy and quality.
Clinical trials can be delayed for a variety of reasons, including delays related to:
ongoing discussions with the regulatory authorities regarding the scope or design of clinical trials;
47
delays or the inability to obtain required approvals from institutional review committees or ethics committees at clinical sites selected for participation in our clinical trials;
delays in enrolling patients and volunteers into clinical trials;
lower than anticipated retention rates of patients and volunteers in clinical trials;
the need to repeat clinical trials as a result of inconclusive or negative results or poorly executed testing;
lack of sufficient funds for further clinical development;
insufficient supply or deficient quality of product candidate materials or other materials necessary to conduct clinical trials;
unsatisfactory regulatory inspection of a manufacturing, testing, labeling or packaging facility;
unsatisfactory regulatory inspection of a clinical trial site or the records generated or maintained by such site;
serious and unexpected drug-related side effects or serious adverse safety events experienced by participants in clinical trials or by patients following commercialization; or
the placement of a clinical hold on a product candidate in a proposed or an ongoing clinical trial.
We may not be able to conduct clinical trials at preferred sites, enlist clinical investigators, enroll sufficient numbers of patients, or demonstrate equivalence of our product candidates with the applicable reference products, or begin or successfully complete clinical trials in a timely fashion, if at all. Any failure to perform may delay or terminate the trials. Our current clinical trials may be insufficient to demonstrate that our potential products are equivalent to the applicable reference products, or sufficiently active, safe, or effective and as a result we may decide to abandon further development of such product candidates. Additional clinical trials may be required if clinical trial results are negative or inconclusive, or insufficient to obtain regulatory approval in one or more countries under applicable laws regulating biosimilar products, which will require us to incur additional costs and significant delays. If we do not receive the necessary regulatory approvals, we will not be able to generate product revenues and will not become profitable. We may encounter significant delays in the regulatory process that could result in excessive costs that may prevent us from continuing to develop our product candidates. In addition, the failure to comply with applicable regulatory requirements may result in criminal prosecution, civil penalties, product recalls, withdrawal of product approval, mandatory restrictions or other actions that could impair our ability to conduct our business.
The development, manufacture and commercialization of biosimilar products in the United States poses unique risks, and our failure to successfully introduce biosimilar products in the United States could have a negative impact on our business and future operating results.
We intend to pursue market authorization for our biosimilar product candidates in numerous jurisdictions, including the United States. In the United States, an abbreviated pathway for approval of biosimilar products was established by the Biologics Price Competition and Innovation Act of 2009, or BPCIA, enacted on March 23, 2010, as part of the Patient Protection and Affordable Care Act. Subsequent to the enactment of the BPCIA, the FDA has released several final and draft guidance documents regarding implementation of the BPCIA regulatory pathway.
These guidance documents focus on scientific considerations, quality considerations and clinical pharmacology data related to demonstrating biosimilarity; meetings between the FDA and biosimilar product sponsors; requests for reference product exclusivity; naming conventions; common questions and answers regarding implementation of the BPCIA; and the standards for interchangeability.
48
Although the FDA is still in the process of implementing the BPCIA, it approved the first biosimilar drug, Zarxio®, Sandoz’s biosimilar version of Amgen’s Neupogen® (filgrastim) for all of the reference product’s approved indications on March 6, 2015.
Moreover, market acceptance of biosimilar products in the United States is unclear. Numerous states are considering or have already enacted laws that regulate or restrict the substitution by state pharmacies of biosimilars for biological products already licensed by the FDA pursuant to biologic license applications, or “reference products.” Market success of biosimilar products will depend on demonstrating to patients, physicians, payors, and relevant authorities that such products are safe and efficacious compared to other existing products.
We plan to continue to analyze and incorporate into our biosimilar development plans any final regulations issued by the FDA, pharmacy substitution policies enacted by state governments, and other applicable requirements established by relevant authorities. The costs of development and approval, along with the probability of success for our biosimilar product candidates, will be dependent upon application of any laws and regulations issued by the relevant regulatory authorities.
Biosimilar products may also be subject to extensive patent clearances and patent infringement litigation, which will likely delay and could prevent the commercial launch of a product. Moreover, the BPCIA prohibits the FDA from accepting an application for a biosimilar candidate to a reference product within four years of the reference product’s licensure by the FDA. In addition, the BPCIA provides innovative biologics with twelve years of exclusivity from the date of their licensure, during which time the FDA cannot approve any application for a biosimilar candidate to the reference product. However, in his proposed budget for fiscal year 2015, President Obama proposed to cut this twelve-year period of exclusivity down to seven years. He also proposed to prohibit additional periods of exclusivity due to minor changes in product formulations, a practice often referred to as “evergreening.” It is possible that Congress may take these or other measures to reduce or eliminate periods of exclusivity.
In July 2015, the U.S. Court of Appeals for the Federal Circuit interpreted the BPCIA as requiring biosimilar manufacturers to send a required pre-launch notice to the manufacturer of the reference biologic only after the FDA has approved the biosimilar for licensure. This ruling potentially provides the reference product manufacturer with an additional 180 days of marketing exclusivity for the innovator biologic, depending on whether the product’s 12-year exclusivity has expired at the time that the pre-launch notice is received from the biosimilar manufacturer.
The BPCIA is complex and only beginning to be interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning is subject to significant uncertainty. Future implementation decisions by the FDA could result in delays in the development or commercialization of our product candidates or increased costs to assure regulatory compliance, and could adversely affect our operating results by restricting or significantly delaying our ability to market new biosimilar products in the United States.
Even if we receive regulatory approvals for our product candidates, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. If we fail to comply with continuing regulatory requirements, we could lose regulatory approvals, and our business would be adversely affected.
Even if we obtain regulatory approvals for our product candidates, we will be subject to extensive and continued regulatory review regarding manufacturing, labeling, packaging, distribution, adverse event reporting, storage, import, export, advertising, promotion and recordkeeping, among other activities. These requirements include submissions of safety and other post-marketing information and reports, manufacturing facility registration and inspection, as well as continued compliance with current Good Manufacturing Practices, or cGMPs and Good Clinical Practice, or GCPs for any clinical trials that we conduct post-approval. Any regulatory approvals that we receive for our product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing and surveillance to monitor the safety and efficacy of the product candidate. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
49
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls;
fines, warning letters or holds on clinical trials;
refusal by regulatory authorities to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product approvals;
product seizure or detention, recalls, or refusal to permit the import or export of products; and
injunctions or the imposition of civil or criminal penalties.
The laws, regulations, and policies of the regulatory authorities may change and additional laws, regulations, and policies may be enacted or established that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability. See “Risks Relating to our Reliance on Third Parties—We rely on third parties to conduct and oversee our clinical trials for our biosimilar product candidates, and expect to continue to do so as we progress the development of our pipeline product candidates. If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or otherwise conduct the trials as required or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates when expected or at all, and our business could be substantially harmed.”
If we are not able to demonstrate biosimilarity of our biosimilar product candidates to the satisfaction of regulatory authorities, we will not obtain regulatory approval for commercial sale of our biosimilar product candidates and our future results of operations would be adversely affected.
Our future results of operations depend, to a significant degree, on our ability to obtain regulatory approval for and to commercialize our proposed biosimilar products. To obtain regulatory approval for the commercial sale of these product candidates, we will be required to demonstrate to the satisfaction of regulatory authorities, among other things, that our proposed biosimilar products are highly similar to biological reference products already licensed by the regulatory authority pursuant to marketing applications, notwithstanding minor differences in clinically inactive components, and that they have no clinically meaningful differences as compared to the marketed biological products in terms of the safety, purity and potency of the products. Each individual jurisdiction may apply different criteria to assess biosimilarity, based on a preponderance of the data that can be interpreted subjectively in some cases. In the European Union, the similar nature of a biosimilar and a reference product is demonstrated by comprehensive comparability studies covering quality, biological activity, safety and efficacy.
It is uncertain if regulatory authorities will grant the full originator label to biosimilar product candidates when they are approved. For example, Hospira’s INFLECTRA™, an infliximab biosimilar molecule, was approved in Europe for the full originator label but did not receive the full originator label when approved in Canada. A similar outcome could occur with respect to one or more of our product candidates.
In the event that regulatory authorities require us to conduct additional clinical trials or other lengthy processes, the commercialization of our proposed biosimilar products could be delayed or prevented. Delays in the commercialization of or the inability to obtain regulatory approval for these products could adversely affect our operating results by restricting or significantly delaying our introduction of new biosimilars.
The structure of complex proteins used in protein-based therapeutics is inherently variable and highly dependent on the processes and conditions used to manufacture them. If we are unable to develop manufacturing processes that achieve a requisite degree of biosimilarity to the originator drug, and within a range of variability considered acceptable by regulatory authorities, we may not be able to obtain regulatory approval for our products.
50
Protein-based therapeutics are inherently heterogeneous and their structures are highly dependent on the production process and conditions. Products from one production facility can differ within an acceptable range from those produced in another facility. Similarly, physicochemical differences can also exist among different lots produced within a single facility. The physicochemical complexity and size of biologic therapeutics create significant technical and scientific challenges in the context of their replication as biosimilar products.
The inherent variability in protein structure from one production lot to another is a fundamental consideration with respect to establishing biosimilarity to an originator product to support regulatory approval requirements. For example, the glycosylation of the protein, meaning the manner in which sugar molecules are attached to the protein backbone of a therapeutic protein when it is produced in a living cell, is critical to half-life (how long the drug stays in the body), efficacy and even safety of the therapeutic and is therefore a key consideration for biosimilarity. Defining and understanding the variability of an originator molecule in order to match its glycosylation profile requires significant skill in cell biology, protein purification and analytical protein chemistry. Furthermore, manufacturing proteins with reliable and consistent glycosylation profiles at scale is challenging and highly dependent on the skill of the cell biologist and process scientist.
There are extraordinary technical challenges in developing complex protein-based therapeutics that not only must achieve an acceptable degree of similarity to the originator molecule in terms of characteristics such as the unique glycosylation pattern (attachment of sugars to the protein) critical to therapeutic efficacy, but also the ability to develop manufacturing processes that can replicate the necessary structural characteristics within an acceptable range of variability sufficient to satisfy regulatory authorities.
Given the challenges caused by the inherent variability in protein production, we may not be successful in developing our products if regulators conclude that we have not achieved a sufficient level of biosimilarity to the originator product, or that the processes we use to manufacture our products are unable to produce our products within an acceptable range of variability.
Risks Relating to our Reliance on Third Parties
We rely on third parties to conduct and oversee our clinical trials for our biosimilar product candidates, and expect to continue to do so as we progress the development of our pipeline product candidates. If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or otherwise conduct the trials as required or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates when expected or at all, and our business could be substantially harmed.
We rely upon third-party CROs to monitor and manage the clinical sites and investigators for our ongoing clinical programs, and to audit and verify the data produced by these parties. We also rely upon medical institutions, clinical investigators and contract laboratories to conduct our trials in accordance with our clinical protocols and in accordance with applicable legal and regulatory requirements. These third parties play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials. These third parties are not our employees, and except for remedies available to us under our agreements with such third parties, there is no guarantee that any such third party will devote adequate time and resources to our clinical trial. If any CROs engaged by us or any other third parties upon which we rely for administration and conduct of our clinical trials do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements, or for other reasons, or if they otherwise perform in a substandard manner, our clinical trials may be extended, delayed, suspended or terminated, and we may not be able to complete development of, obtain regulatory approval for, or successfully commercialize our product candidates. We plan to rely heavily on these third parties for the execution of our planned global clinical program for BOW015 and for other clinical trials for products we are developing or may develop in the future, and will control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on our CROs does not relieve us of our regulatory responsibilities.
51
We and any CROs that we engage are required to comply with GCP requirements, which are regulations and guidelines enforced by regulatory authorities around the world for products in clinical development. Regulatory authorities enforce these GCP regulations through periodic inspections of clinical trial sponsors, principal investigators and clinical trial sites. If we or any of our CROs fail to comply with applicable GCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and our submission of marketing applications may be delayed or the regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot provide any assurance that, upon inspection, a regulatory authority will determine that any of our clinical trials comply or complied with applicable GCP regulations. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
Clinical trials require a substantial number of patients that can allow statistically significant results. Delays in site initiation or unexpectedly low patient recruitment rates may delay the results of the clinical trial. CROs may also generate higher costs than anticipated. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase, and our ability to generate revenue could be delayed. Further, if our relationship with any CRO that we engage to perform services on our behalf is terminated, we may be unable to enter into arrangements with an alternative CRO on commercially reasonable terms, or at all. Switching or adding CROs can involve substantial cost and require extensive management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays may occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, prospects, financial condition or results of operations.
We rely on third parties, and in some cases a single third party, to manufacture nonclinical and clinical supplies of our product candidates, to store critical components of our product candidates for us, and also to commercialize our product candidates. Our business could be harmed if those third parties fail to provide us with sufficient quantities of product candidates at acceptable quality levels and on commercially reasonable terms, or fail to commercialize our products in accordance with the terms of our agreements with them.
We do not have the infrastructure or capability internally to manufacture supplies of our biosimilar product candidates for use in the conduct of our nonclinical and clinical studies or on a commercial scale. We rely on third-party manufacturers, including our India-based contract manufacturer RLS, to manufacture and supply us with BOW015 in India. As discussed in the other risk factors described in this section, we may need to seek additional third-party manufacturers following RLS’ completion of its manufacture obligations pursuant to the Settlement Agreement (as described below). In May 2015, we entered into a development and future distribution agreement with mAbxience, S.A., or Mabxience, a wholly owned subsidiary of CHEMO Group, for BOW015 in Latin American markets, including Argentina, Chile, Ecuador, Paraguay, Uruguay and Venezuela. We have an established partnership with Livzon for the production of biosimilars for China and associated markets. In July 2015, we also entered into an agreement with Swiss Pharma International AG, an affiliate of Pharmaceutical Works Polpharma S.A., or, together with Swiss Pharma International AG, Polpharma, for the development and commercialization of BOW015, BOW050 and BOW070 in certain designated territories, including most of the European Union, certain countries in the Middle East, Turkey, Russia, and other countries comprising the CIS. Further, we are currently party to an agreement with Sun, pursuant to which we granted to Sun exclusive rights under our intellectual property and regulatory materials relating to BOW015 to develop and commercialize BOW015 in India and certain other countries in Asia and North Africa. Pursuant to this agreement, Sun has agreed to distribute and sell BOW015 in India under the marketing authorization granted to RLS for BOW015. We also have an agreement with Fujifilm Diosynth Biotechnologies U.S.A., Inc. (“Fujifilm”) for BOW015 process development with a view toward establishing Fujifilm as a source of future clinical and commercial supply of BOW015. Developing successful scale up techniques for manufacture of our biosimilar product candidates for commercial quantities will be time consuming and may not yield the quantities of product that we anticipate, which may have a material impact on our ability to successfully commercialize our product candidates. Moreover, the market for contract manufacturing services for protein therapeutics is highly cyclical, with periods of relatively abundant capacity alternating with periods in which there is little available capacity. If any need we have for contract manufacturing services increases during a period of industry-wide production capacity constraints, we may not be able to produce our product candidates on a timely basis or on commercially viable terms. Although we generally do not begin a clinical
52
study unless we believe we have a sufficient supply of a product candidate to complete such study, any significant delay or discontinuity in the supply of a product candidate for an ongoing clinical study due to the need to replace a third-party manufacturer could considerably delay completion of our clinical studies, product testing, and potential regulatory approval of our product candidates, which could harm our business and results of operations.
Any failure or refusal to supply the components for product candidates that we may develop could delay, prevent or impair our clinical development or commercialization efforts. Although we endeavor, and will continue to do so in the future, to establish alternative sources of supply for BOW015 and our other product candidates in order to protect against any disruption in product supply, we cannot assure you that we will be able to find a suitable alternative manufacturer who is permitted to supply product under applicable regulatory laws, or to enter into appropriate agreements with such third parties on commercially reasonable terms, or at all. If our contract manufacturers were to breach or terminate their manufacturing arrangements with us, the development or commercialization of the affected products or product candidates could be delayed, which could have an adverse effect on our business. Any change in our manufacturers could be costly because the commercial terms of any new arrangement could be less favorable and the expenses relating to the transfer of necessary technology and processes could be significant.
If any of our product candidates are approved for any given jurisdiction, in order to produce the quantities necessary to meet anticipated market demand, any contract manufacturer that we engage may need to increase manufacturing capacity. If we are unable to produce our product candidates in sufficient quantities to meet the requirements for the launch of these products or to meet future demand, our revenue and gross margins could be adversely affected. Although we believe that we will not have any material supply issues, we cannot be certain that we will be able to obtain long-term supply arrangements for our product candidates or materials used to produce them on acceptable terms, if at all. If we are unable to arrange for third-party manufacturing, or to do so on commercially reasonable terms, we may not be able to complete development of our products or market them.
We also rely on third parties to store the BOW015 master and working cell bank, and those of our pipeline product candidates. We have one master cell bank and one working cell bank and believe we would have adequate backup should any cell bank be lost in a catastrophic event. However, it is possible that we could lose multiple cell banks and have our manufacturing severely impacted by the need to replace the cell banks, which could materially and adversely affect our business, financial condition and results of operations.
If we encounter delays or difficulties in executing our agreements with Mabxience, Polpharma or Sun to commercialize our product candidates in the various territories covered by our agreements, we may be required to seek alternative commercialization partners and/or develop a plan to commercialize our product candidates through direct sales into the various markets, either of which may adversely affect our ability to successfully commercialize our product candidates. We have no direct sales experience in these territories. Therefore, if we decide to commercialize our product candidates through direct sales, we cannot assure you that we would be successful.
Disputes under key agreements with third parties could adversely affect, delay or prevent development or commercialization of our product candidates.
Any agreements we have or may enter into with third parties, such as collaboration, license, formulation supplier, manufacturing, testing, clinical research organization or clinical trial agreements, may give rise to disputes regarding the rights and obligations of the parties to such agreements. Disagreements may occur relating to the parties’ rights and obligations under these agreements, such as the scope of development or commercialization rights, ownership or use of intellectual property, the approach to obtaining regulatory approvals or commercialization strategy. Any disputes or commercial conflicts could lead to the termination of these agreements, delay progress of our product development programs, compromise our ability to renew agreements or obtain future agreements, lead to the loss of suppliers of our products, harm our intellectual property rights or result in costly litigation.
In December 2014, RLS exercised its three-year termination for-convenience right with respect to our manufacturing and supply agreement with RLS, which we refer to as the RLS Agreement, which would have caused the RLS Agreement to terminate in December 2017. In addition, in March 2015, RLS initiated an arbitration proceeding under the RLS Agreement alleging that the RLS Agreement grants RLS exclusive global supply rights to BOW015, that
53
the terms of our collaboration agreements with Fujifilm and Livzon (described above) violate the RLS Agreement, and sought to terminate the RLS Agreement. On April 22, 2015, we entered into a Settlement Agreement with RLS. Pursuant to the Settlement Agreement, on May 4, 2015, RLS withdrew the arbitration proceeding it initiated, and the parties have agreed to release each other from all causes of actions or claims, present and future, arising under the RLS Agreement. Pursuant to the Settlement Agreement, the parties finalized the remaining BOW015 batch production schedule with RLS agreeing to manufacture 15 final batches of BOW015 on or before June 30, 2016 with our option to have RLS manufacture an additional 5 batches on or before December 31, 2016. In January 2016, we amended the Settlement Agreement to extend the time for RLS to manufacture the 15 final batches to June 30, 2017. RLS has also agreed to use reasonable commercial efforts to transfer the existing Indian marketing authorization for BOW015 granted by the DCGI to us or our designee, with all costs of the transfer to be borne by us and subject to the DCGI’s approval of such transfer. We have agreed to pay to RLS a fee of $2.3 million, payable in four installments over the course of the final batch manufacture, with the final installment to be paid no later than ten days after June 30, 2016. Following the satisfaction by both parties of all of the terms of the Settlement Agreement, the RLS Agreement and related agreements between the parties shall be terminated and neither party shall have any further rights or obligations thereunder.
While we believe that, based on forecasts from our Indian commercial partner, Sun, the current inventory, and future inventory as agreed to under the Settlement Agreement, of BOW015 is sufficient to fulfill anticipated demand there through the middle of 2018, it is possible that demand will increase in excess of our current inventory and that we will not be able to meet these demands if the agreed-upon batches to be manufactured by RLS pursuant to the Settlement Agreement are not sufficient to meet such increased needs. We intend to continue to build up additional BOW015 inventory via the agreed upon production runs by RLS, as necessary to establish sufficient product for the Indian territory, but we cannot be certain that this additional supply will be adequate. It is possible that RLS could stop providing us with BOW015 entirely, despite the terms of the Settlement Agreement and regardless of whether this causes RLS to breach the RLS Agreement or the Settlement Agreement. We and Sun intend to transfer BOW015 manufacturing for Indian territory to an alternate contract manufacturer, but there can be no assurances that we will be successful in making such a transfer on a timely basis, or at all. This, in turn, could materially and adversely affect our ability to manufacture or supply BOW015 for use in commercial sales in India and other markets currently contemplated to be served by Sun.
Where we apply our In Market, For Market strategy for commercialization of our product candidates in emerging markets, where approved, we expect to transfer our cell lines and certain proprietary manufacturing technology to our third-party partners in order to permit locally-based manufacture of our biosimilar products by our partners. If our alliances with these partners terminate, our business could be harmed if such former partners continue to use our technology in such countries following termination, in breach of our agreements with them.
Under our collaboration with Livzon, and under future In Market, For Market collaborations, we expect to transfer the manufacture and certain development and commercialization responsibilities for BOW015 and certain of our pipeline product candidates to Livzon and other future collaborators, along with certain of our SCALE manufacturing technology, and to provide technical assistance in such territories to assist our partners in establishing manufacturing capacity or modifying their existing manufacturing capacity for the production of our biosimilar product candidates. If our alliances with Livzon and any future partners are not successful, or our agreements with such partners are terminated, our agreements will include terms that require the partners to immediately cease any use of our technology, and to return to us any tangible embodiments of such technology, including our cell lines. However, our former partners may fail to comply with these terms, and may continue to use our technology to manufacture and sell biosimilar products in such countries, in breach of our agreements with them. In such case, we may be forced to enforce our contractual and intellectual property rights in the applicable countries, which would be time-consuming and costly, and would divert substantial management time and resources from other aspects of our business. Further, although we endeavor to include dispute resolution provisions in our agreements that we believe are fully enforceable irrespective of the jurisdiction, including by the use of arbitration, countries outside the United States where we have In Market, For Market collaborations may not provide the same level of protection for our contractual and intellectual property rights, and there is no guarantee that we would be successful in preventing a party to whom we have transferred our technology from continuing to use our cell lines and technology to manufacture biosimilar products, in which case our business in those countries could be adversely affected.
54
In certain countries where we expect to commercialize our product candidates, under applicable laws, it is necessary for us to rely on locally established third parties to act as the holder of marketing approvals or authorizations for our product candidates in such countries. Our business could be harmed if those third parties fail to comply with the terms of such marketing authorizations or approvals, or fail to act in accordance with our instructions in relation to the marketing and sale of our product candidates covered by such authorizations or approvals.
In some circumstances, it may be necessary as a matter of applicable local law, for the marketing and manufacturing approvals for our biosimilar products to be held by a third party that is not one of our affiliates. For example, our marketing approval in India for BOW015 is currently held in the name of our manufacturing partner, RLS, which also holds the manufacturing approval for BOW015. Pursuant to the Settlement Agreement described above, RLS has agreed to use reasonable commercial efforts to transfer such authorization to us or our designee, subject to approval by the DCGI. The marketing approval held by RLS for manufacture and sale of infliximab in India is based on and is specific to the data arising from our clinical trials conducted with respect to BOW015, which data is owned solely by us, and RLS therefore has no right to commercialize BOW015 in India or in any other country independently of us on the basis of such marketing authorization. We have a manufacturing and supply agreement in place with RLS that includes provisions that permit us to have oversight over RLS’s activities in order to protect against breach by RLS of the terms of such marketing approval, and we will seek to include equivalent contractual protections in future agreements with third parties that hold regulatory approvals on our behalf. However, there is no guarantee that RLS or any other such third party will comply with the terms of the marketing authorization for the applicable biosimilars or with applicable laws. Where permissible under applicable laws, we also seek to provide for transfer of rights in any regulatory approval to an alternative holder on termination of our agreements with such third parties, and we also endeavor, and will continue to do so in the future, to establish alternative sources of supply for BOW015 and our future product candidates in order to protect against any disruption in product supply. However, we cannot assure you that we will be able to find an alternative holder for any such regulatory approvals in a timely fashion, or to enter into appropriate agreements with such third parties on commercially reasonable terms, or at all. Any non-compliance with applicable laws or the terms of regulatory approvals by such third parties could result in the permission to market and sell the applicable product candidate being revoked in one or more countries, which would materially jeopardize our right to commercialize BOW015 in India and such other countries.
We expect to partner with third parties in connection with the development of certain of our product candidates. Even if we enter into such collaborations, and we believe that the development of our technology and product candidates is promising, our partners may choose not to proceed with such development.
Our agreements with potential partners may include the right for our partner to terminate the agreement on short notice upon the occurrence of certain circumstances. Accordingly, even if we enter into agreements with third-party partners in all of the territories where we intend to pursue commercialization of our product candidates, and we believe that the development of such product candidates are worth pursuing, our partners may choose not to continue with such development. If any of our partnerships are terminated, we may be required to devote additional resources to the development of our product candidates or seek a new partner on short notice, and the terms of any additional partnerships or other arrangements that we establish may not be favorable to us, if an alternative partner is even available.
We are also at risk that our partnerships or other arrangements may not be successful. Factors that may affect the success of our partnerships include the following:
our partners may incur financial and cash-flow difficulties that force them to limit or reduce their participation in our joint projects;
our partners may be pursuing alternative technologies or developing alternative products that are competitive to our technology and products, either on their own or in partnership with others;
our partners may terminate their partnerships with us, which could make it difficult for us to attract new partners or adversely affect perception of us in the business and financial communities; and
55
our partners may pursue higher priority programs or change the focus of their development programs, which could affect their commitment to us.
We have entered into agreements with Mabxience, for the development and future distribution of BOW015 in certain Latin American countries, Polpharma, for the development and commercialization of BOW015, BOW050 and BOW070 in certain European and Middle Eastern countries, and Livzon, for development and commercialization of BOW015, BOW070 and up to three other biosimilar pipeline products in China and related territories. If our collaborations are not successful or if we or our partners terminate our agreements, we may not be able to progress our plans to develop and commercialize our product candidates in the related territories. Although we believe that there are appropriate alternative partners in each territory, there is no guarantee that we could identify and negotiate satisfactory terms with any such alternative partner, or that we could do so in a timeframe that would not compromise our ability to commercialize our product candidates in a timely manner or at all.
If we cannot maintain successful partnerships, our business, financial condition and operating results may be adversely affected.
International expansion of our business exposes us to business, regulatory, political, operational, financial, and economic risks associated with doing business outside of the United States.
Our business strategy incorporates significant international expansion, with a primary focus on developed markets, such as Europe, as well as local production markets, such as China and Brazil and accessible markets such as India and Latin America. We plan to engage in manufacturing, maintain sales representatives and conduct clinical trials outside of the United States. Doing business internationally involves a number of risks, including but not limited to:
multiple, conflicting, and changing laws and regulations such as privacy regulations, tax laws, export and import restrictions, employment laws, intellectual property laws, regulatory requirements, and other governmental approvals, permits, and licenses;
foreign laws favoring businesses that are owned by nationals of those countries as opposed to foreign-owned businesses operating locally;
failure by us to obtain and maintain regulatory approvals for the use of our products in various countries;
additional potentially relevant third-party patent or other rights;
complexities and difficulties in obtaining protection and enforcing our intellectual property;
difficulties in staffing and managing foreign operations;
limits in our ability to penetrate and compete in international markets;
financial risks, such as longer payment cycles, difficulty collecting accounts receivable, difficulty obtaining insurance coverage, the impact of local and regional financial crises on demand and payment for our products, and exposure to foreign currency exchange rate fluctuations and controls;
natural disasters, political and economic instability, including wars, terrorism, and political unrest, outbreak of disease, boycotts, curtailment of trade, and other business restrictions;
certain expenses including, among others, expenses for travel, translation, and insurance; and
regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the FCPA, its books and records provisions, or its anti-bribery provisions, OFAC restrictions, anti-money laundering laws, export control laws, customs laws, sanctions laws and other laws governing our operations.
56
Any of these factors could significantly harm any future international expansion and operations and, consequently, could adversely affect our business prospects.
Our business could be harmed as a result of the risks associated with our acquisitions.
As part of our business strategy, we may from time to time seek to acquire businesses or assets that provide us with additional intellectual property, product candidates, customer relationships or geographic coverage. We can provide no assurances that we will be able to find and identify desirable acquisition targets or that we will be successful in entering into a definitive agreement with any one target. In addition, even if we reach a definitive agreement with a target, there is no assurance that we will complete any future acquisition.
Any acquisitions we undertake or have recently completed, including the acquisition of Bioceros Holding B.V. (“Bioceros”), will likely be accompanied by business risks which may include, among other things:
|
· |
the effect of the acquisition on our financial and strategic position and reputation; |
|
· |
the failure of an acquisition to result in expected benefits, which may include benefits relating to enhanced revenues, technology, human resources, costs savings, operating efficiencies, goodwill and other synergies; |
|
· |
the difficulty, cost and management effort required to integrate the acquired businesses, including costs and delays in implementing common systems and procedures and costs and delays caused by communication difficulties; |
|
· |
the assumption of certain known or unknown liabilities of the acquired business, including litigation-related liabilities; |
|
· |
the reduction of our cash available for operations and other uses, the increase in amortization expense related to identifiable assets acquired, potentially dilutive issuances of equity securities or the incurrence of debt; |
|
· |
a lack of experience in new markets, new business culture, products or technologies or an initial dependence on unfamiliar distribution partners; |
|
· |
the possibility that we will pay more than the value we derive from the acquisition; |
|
· |
the impairment of relationships with our customers, partners or suppliers or those of the acquired business; and |
|
· |
the potential loss of key employees of the acquired business. |
These factors could harm our business, results of operations or financial condition.
In addition to the risks commonly encountered in the acquisition of a business or assets as described above, we may also experience risks relating to the challenges and costs of closing a transaction. The risks described above may be exacerbated as a result of managing multiple acquisitions at once.
Industry Related Risks
If efforts by manufacturers of reference products to delay or limit the use of biosimilars are successful, our sales of biosimilar products may suffer.
57
Many manufacturers of reference products have increasingly used legislative, regulatory and other means in attempts to delay regulatory approval of and competition from biosimilars. These efforts have included sponsoring legislation to prevent pharmacists from substituting biosimilars for prescribed reference products or to make such substitutions more difficult by establishing notification, recordkeeping, and/or other requirements, as well as seeking to prevent manufacturers of biosimilars from referencing the brands of the innovator products in biosimilar product labels and marketing materials. If these or other efforts to delay or block competition are successful, we may be unable to sell our biosimilar product candidates, which could have a material adverse effect on our sales and profitability.
Foreign governments tend to impose strict price controls on pharmaceutical products, which may adversely affect our revenue, if any.
In some foreign countries, the pricing of prescription pharmaceuticals is subject to governmental control. This is likely also to apply to pricing of biosimilar products. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
We use and generate materials that may expose us to expensive and time-consuming legal claims.
Our development programs involve the use of hazardous materials, chemicals, and biological materials. We are subject to foreign, federal, state and local laws and regulations governing the use, manufacture, storage, and disposal of materials and waste products. We believe that our safety procedures for handling these materials comply with the standards prescribed by laws and regulations. However, we may incur significant costs to comply with current or future environmental laws and regulations. In addition, we cannot completely eliminate the risk of contamination or injury from hazardous materials. In the event of an accident, an injured party may seek to hold us liable for any damages that result. Any liability could exceed the limits or fall outside the coverage of our insurance, and we may not be able to maintain insurance on acceptable terms, if at all.
Risks Related to Intellectual Property
We may not be able to develop or commercialize our product candidates due to intellectual property rights held by third parties.
Although we aim to launch our biosimilar product candidates in markets where there is no valid third party patent protection covering the reference biologic product, or in some cases where the fundamental patent protection for the reference product has expired, if a third party holds a patent to a formulation technology related to our planned formulation of a product candidate, we may not be able to develop or commercialize such product candidates without first obtaining a license to such patent, or waiting for such patent to expire. In addition, in the United States, under certain limited circumstances, patents known as “submarine” patents may issue without the corresponding application being previously published. If such a submarine patent were to issue and cover aspects of any one or more of our product candidates, it is possible that such patents could affect our ability to commercialize the affected product candidate(s). Our business will be harmed if we are unable to use the optimal formulation of our product candidates, which may occur because the formulations or methods of use are covered by one or more third-party patents, and a license to such patents is unavailable or is only available on terms that are unacceptable to us. In the case of our biosimilar product candidates, this may prevent us from establishing the equivalence to the reference biologic product required to gain regulatory approval for sale of such biosimilar product within a time frame that would make the commercialization of the affected biosimilar product candidate commercially viable.
We may not have identified all the relevant patents covering our product candidates, or the scope or term of identified patents may differ from our assessment, which may affect our ability to commercialize our product candidates.
Although we have conducted searches in commercially available patent databases directed to patents that would cover reference products in our pipeline, we may not have identified all of the relevant patents. Patent filings can be
58
complicated and involve many progeny cases in different jurisdictions around the world. Patents and patent applications are published weekly and if the databases that we use are not up to date when the search is conducted, then we may not identify relevant patents that may cover our product patents. If we fail to identify relevant patent filings in the jurisdictions in which we plan to commercialize, or if we incorrectly interpret the claims or scope of coverage afforded by any patent identified in our searches in any jurisdiction, such that our product candidates in fact infringe such patents, then our ability to commercialize our products in the applicable countries could be prevented or delayed. Further, the patents covering the reference products may be subject to adjustments or extensions under applicable law that may prolong the life of such patents. While we strive to avoid litigation by launching our product candidates in markets where the reference product is no longer subject to patent protection, if we incorrectly assess the applicable term of the patent, or if such term is extended in any jurisdiction via an appeal or otherwise, our ability to commercialize in that such jurisdiction could be adversely affected.
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be materially adversely affected and our business would be harmed.
We do not currently own any patents or have any patent filings with any patent offices in the world. We rely solely on trade secret protection and confidentiality agreements to protect proprietary know-how that may not be patentable, processes for which patents may be difficult to obtain or enforce and any other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. This includes our rights in our proprietary SCALE technology, which is critical to the success of our In Market, For Market strategy for commercialization of our product candidates in emerging markets. Trade secret protection and confidentiality agreements may afford only limited protection and may not: prevent our competitors from duplicating our products; prevent our competitors from gaining access to our proprietary information and technology; or permit us to gain or maintain a competitive advantage.
As part of our efforts to protect our trade secrets and other confidential information, we require our employees, consultants, collaborators and advisors to execute confidentiality agreements upon the commencement of their relationships with us, and we include equivalent provisions in all of our material collaboration and license agreements. These agreements require that all confidential information developed by the individual or made known to the individual by us during the course of the individual’s relationship with us be kept confidential and not be disclosed to third parties. These agreements, however, may not provide us with adequate protection against improper use or disclosure of confidential information, and/or these agreements may be breached. Adequate remedies may not exist in the event of unauthorized use or disclosure of our confidential information. A breach of confidentiality could significantly affect our competitive position. Also, third parties, including our competitors, may independently develop substantially equivalent proprietary information and technologies or otherwise lawfully gain access to our trade secrets and other confidential information. In such a case, we would have no right to prevent such third parties from using such proprietary information or technologies to compete with us, which could harm our competitive position.
In addition, where we enter into In Market, For Market collaborations to develop and commercialize our product candidates in emerging markets, these collaborations are expected to involve the establishment in such countries of independent manufacturing capacity for the applicable product candidates. Establishing such manufacturing capacity is expected to involve certain technology transfer by us to our strategic partners, and the provision by us of technical assistance in connection with such transfer. If our agreements with our collaborators are terminated, although these agreements provide that our former partners have no further rights in the transferred technology, such third parties could use the technology and know-how they received from us to continue to develop and commercialize the applicable products, in breach of our agreements with them, and which would materially impact our revenues from commercialization of our product candidates. In such case, it may be necessary for us to become involved in litigation or arbitration to retain and defend our rights in such technology, and to prevent such commercialization. Any such proceedings, which would be likely to be conducted in a country outside the United States, would be costly and time consuming, and would divert significant management time and resources, with no guarantee of a successful outcome.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
59
We rely on trademark protection to protect our business and our products. Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build brand name recognition by potential customers or partners in our markets of interest. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of our employees’ former employers.
Some of our employees, consultants, collaborators, and advisors were previously employed at other biotechnology companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these individuals or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money damages, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize certain product candidates, which would adversely affect our commercial development efforts.
We are dependent on proprietary technology licensed from others. If we lose our licenses, we may not be able to continue developing our products.
We have obtained licenses that give us rights to third party intellectual property that is necessary or useful to our business. These license agreements covering our product candidates impose various royalty and other obligations on us. One or more of our licensors may allege that we have breached our license agreement with them, and accordingly seek to terminate our license. If we materially breach the obligations in our license agreements, the licensor typically has the right to terminate the license and we may not be able to market products that were covered by the license, which could adversely affect our competitive business position and harm our business prospects. In addition, any claims brought against us by our licensors could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations.
We may not be able to protect against third-party copying of our product candidates.
We may need to resort to litigation to enforce or defend our intellectual property rights, including any misappropriation of our trade secrets or know how. As noted above, we currently rely upon trade secrets to protect our product candidates. Our product candidates utilize the same formulations and ingredients as others used in the marketplace and, as such, may not be eligible for any patent protection. In addition, there may be third-party patents and patent applications directed to formulations, processes of manufacture or other processes that could cover our product candidates. If these third-party patent applications covering our product candidates are approved, our business, prospects and financial condition would be materially adversely affected.
Litigation or third-party claims of intellectual property infringement could require substantial time and money to resolve. Unfavorable outcomes in these proceedings could limit our activities.
Our commercial success will depend in part on not infringing or violating the intellectual property rights of others. The manufacture, use and sale of new biosimilars that are the subject of conflicting patent rights have been the subject of substantial litigation in the biosimilars industry. These lawsuits relate to the validity and infringement of patents or proprietary rights of third parties. We may have to defend against charges that we violated patents or proprietary rights of third parties. This is especially true in the case of generic products on which the patent covering the reference product is expiring, an area where infringement litigation is prevalent, and in the case of new reference products where a competitor has obtained patents for similar products. We cannot guarantee that our product candidates will be free from claims by third parties alleging that we have infringed their intellectual property rights. Our competitors, some of which have substantially greater resources than we do and have made substantial intellectual
60
property investments in competing technologies, may have applied for or obtained, or may in the future apply for and obtain, patent rights and other intellectual property that will prevent, limit or otherwise interfere with our ability to make, use and sell our products. Third parties may assert that we are employing their proprietary technologies without authorization and they may resort to litigation to attempt to enforce their rights. We may not be aware of whether our products do or will infringe existing or future patents or the intellectual property rights of others. Third parties may have or obtain patents in the future and claim that the use of our technology or any of our product candidates infringes their patents. Any lawsuits resulting from allegations that the operation of our business infringes the intellectual property rights of third parties could subject us to significant liability for damages and invalidate our proprietary rights, even if the lawsuits lack merit. We may not be able to develop or commercialize product candidates because of patent protection others have. Our business will be harmed if we cannot obtain a necessary or desirable license, can obtain such a license only on terms we consider to be unattractive or unacceptable, or if we are unable to redesign our product candidates or processes to avoid actual or potential patent or other intellectual property infringement.
Our efforts to register, protect and defend our intellectual property rights, whether we are successful or not, may require us to incur substantial costs, including the diversion of management and technical personnel. An unfavorable ruling in intellectual property litigation could subject us to significant liabilities to third parties, require us to cease developing, manufacturing or selling the affected products or using the affected processes, require us to license the disputed rights from third parties, or result in awards of substantial damages against us. In addition, intellectual property litigation, whether we are successful or not, can be very expensive and may require us to incur substantial costs, including the diversion of management and technical personnel. During the course of any litigation, there may be public announcements of the results of hearings, motions, and other interim proceedings or developments in the litigation. If securities analysts or investors regard these announcements as negative, the market price of our common stock may decline. Any potential intellectual property litigation also could force us to do one or more of the following:
|
· |
stop making, selling or using products or technologies that allegedly infringe the asserted intellectual property; |
|
· |
lose the opportunity to license our technology to others or to collect royalty payments based upon successful protection and assertion of our intellectual property rights against others; |
|
· |
pay substantial damages or royalties to the party whose intellectual property rights we may be found to be infringing; |
|
· |
pay the attorney fees and costs of litigation to the party whose intellectual property rights we may be found to be infringing; |
|
· |
redesign or rename, in the case of trademark claims, those products that contain the allegedly infringing intellectual property, which could be costly, disruptive and/or infeasible; or |
|
· |
attempt to obtain a license to the relevant intellectual property from third parties, which may not be available on reasonable terms or at all. |
If we fail to obtain any required licenses or make any necessary changes to our products or technologies, we may have to withdraw existing products from the market or may be unable to commercialize one or more of our products, all of which could have a material adverse effect on our business, results of operations and financial condition. There can be no assurance that we would prevail in any intellectual property infringement action, will be able to obtain a license to any third-party intellectual property on commercially reasonable terms, successfully develop non-infringing alternatives on a timely basis, or license non-infringing alternatives, if any exist, on commercially reasonable terms. Any significant intellectual property impediment to our ability to develop and commercialize our products could seriously harm our business and prospects.
Risks Related to an Investment in Our Common Stock
Our management and independent auditors have identified material weaknesses in our internal controls, and we may be unable to develop, implement and maintain appropriate internal and disclosure controls in future periods, which may lead to errors or omissions in our financial statements.
61
As of December 31, 2015, there were material weaknesses in our controls over our financial statement close process and the financial reporting processes related to business combinations, income taxes and accrued development costs.
These material weaknesses have not been fully remediated. Prior to the complete remediation of these material weaknesses, there remains risk that the transitional controls on which we currently rely will fail to be sufficiently effective, which could result in a material misstatement of our financial position or results of operations and require a restatement of our financial statements.
As of December 31, 2015, our management, including our Chief Executive Officer and Chief Financial Officer, evaluated our internal controls and procedures, including the existence of the material weaknesses in our internal control over financial reporting, and concluded that our disclosure controls and procedures were not effective to provide reasonable assurance that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, or that such information is accumulated and communicated to our management, including the CEO and CFO, as appropriate to allow timely decisions regarding required disclosure. We are currently designing and implementing new procedures and controls intended to address the material weaknesses described above. While this design and implementation phase is underway, we are relying significantly on outside accounting professionals until permanent employees can be hired to fill these roles and on manual procedures to assist us with meeting the objectives otherwise fulfilled by an effective control environment. The implementation of new procedures and controls could be costly and distract management from other activities.
We note that a system of procedures and controls, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all systems of procedures and controls, no evaluation can provide absolute assurance that all control issues, including instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and breakdowns can occur because of simple error or mistake. Additionally, procedures and controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override. The design of any system of procedures and controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, our systems of procedures and controls, as we further develop and enhance them, may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective system of procedures and controls, misstatements due to error or fraud may occur and not be detected and could be material and require a restatement of our financial statements.
Following the expiration of the SEC waiver granted to us in connection with the Merger, we are now required to make a formal assessment of the effectiveness of our internal controls over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act, beginning with this annual report on Form 10-K for the fiscal year ending December 31, 2015. If we are unable to establish appropriate internal controls, we may not have adequate, accurate or timely financial information, and we may be unable to meet our reporting obligations or comply with the requirements of the SEC or the Sarbanes-Oxley Act, which could result in the imposition of sanctions, including the inability of registered broker dealers to make a market in our common shares, or investigation by regulatory authorities. Any such action or other negative results caused by our inability to meet our reporting requirements or comply with legal and regulatory requirements or by disclosure of an accounting, reporting or control issue could adversely affect the trading price of our securities. Further and continued determinations that there are significant deficiencies or material weaknesses in the effectiveness of our internal controls could also reduce our ability to obtain financing or could increase the cost of any financing we obtain and require additional expenditures to comply with applicable requirements.
Our failure to meet the continued listing requirements of The NASDAQ Capital Market could result in a delisting of our common stock.
In connection with the Merger, we effected a reverse split of our common stock to enable us to comply with The NASDAQ Capital Market’s minimum bid price listing requirement. Notwithstanding the reverse stock split and
62
compliance with The NASDAQ Capital Market’s minimum bid price listing requirement, we may not be able to maintain a price per share of our common stock in excess of $1.00 per share or the additional criteria for continued listing of our common stock set forth by The NASDAQ Capital Market. The occurrence of any future non-compliance with The NASDAQ Capital Market’s minimum bid price or other listing requirements may have a material adverse effect on our stock price, our business and our prospects.
Our stock price is expected to continue to be volatile and you may not be able to resell your shares.
We cannot assure you that an active trading market for shares of our common stock will develop or be sustained. If an active market for our common stock does not develop, or is not sustained, it may be difficult to sell shares without depressing the market price for the shares or to sell your shares at all.
The market price for our common stock has been volatile, and market prices for securities of companies comparable to us have been highly volatile. In addition, the stock market as a whole and biotechnology and other life science stocks in particular have experienced significant price volatility. Like our common stock, these stocks have experienced significant price and volume fluctuations for reasons unrelated to the operating performance of the individual companies. Factors giving rise to this volatility may include:
regulatory and other developments in both the United States and abroad;
developments concerning proprietary rights, including patents and litigation matters;
disclosure of new collaborations or other strategic transactions;
public concern about the safety or efficacy of our product candidates or technology, their components, or related technology or new technologies generally;
public announcements by our competitors or others regarding new products or new product candidates;
general market conditions and comments by securities analysts and investors; and
developments relating to our key partners.
In the past, securities class action litigation has often been brought against companies that experience volatility in the market price of their securities. Whether or not meritorious, litigation brought against us could result in substantial costs and a diversion of management’s attention and resources, which could adversely affect our business, operating results and financial condition.
Fluctuations in our operating losses could adversely affect the price of our common stock.
Our operating losses may fluctuate significantly on a quarterly basis. Some of the factors that may cause our operating losses to fluctuate on a period-to-period basis include the status of our clinical and pre-clinical development programs, level of expenses incurred in connection with our clinical and pre-clinical development programs, restructuring costs, implementation or termination of collaboration, licensing, manufacturing or other material agreements with third parties, non-recurring revenue or expenses under any such agreement, and compliance with regulatory requirements. Period-to-period comparisons of our historical and future financial results may not be meaningful, and investors should not rely on them as an indication of future performance. Our fluctuating losses may fail to meet the expectations of securities analysts or investors. Our failure to meet these expectations may cause the price of our common stock to decline.
Anti-takeover provisions in our charter documents and provisions of Delaware law may make an acquisition more difficult and could result in the entrenchment of management.
63
We are incorporated in Delaware. Anti-takeover provisions of Delaware law and our charter documents may make a change in control or efforts to remove management more difficult. Also, under Delaware law, our board of directors may adopt additional anti-takeover measures. The existence of the following provisions of Delaware law and our sixth amended and restated charter, as amended, or our amended and restated bylaws could limit the price that investors might be willing to pay in the future for shares of our common stock.
Our charter authorizes our board of directors to issue up to 5,000,000 shares of preferred stock and to determine the terms of those shares of stock without any further action by our stockholders. If the board of directors exercises this power to issue preferred stock, it could be more difficult for a third party to acquire a majority of our outstanding voting stock and vote the stock they acquire to remove management or directors.
Our charter also provides staggered terms for the members of our board of directors. Under Section 141 of the General Corporation Law of the State of Delaware, or the DGCL, and our charter, our directors may be removed by stockholders only for cause and only by vote of the holders of 75% of voting shares then outstanding. These provisions may prevent stockholders from replacing the entire board in a single proxy contest, making it more difficult for a third party to acquire control without the consent of our board of directors. These provisions could also delay the removal of management by the board of directors with or without cause. In addition, our amended and restated bylaws limit the ability our stockholders to call special meetings of stockholders.
Our equity incentive plans generally permit our board of directors to provide for acceleration of vesting of options granted under these plans in the event of certain transactions that result in a change of control. If our board of directors uses its authority to accelerate vesting of options, this action could make an acquisition more costly, and it could prevent an acquisition from going forward.
Under Section 203 of the DGCL, a corporation may not engage in a business combination with any holder of 15% or more of its capital stock until the holder has held the stock for three years unless, among other possibilities, the board of directors approves the transaction in advance. These provisions may have the effect of entrenching our management team and may deprive you of the opportunity to sell your shares to potential acquirers at a premium over prevailing prices. The potential inability to obtain a control premium could reduce the price of our common stock.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
Our headquarters is located in Boston, MA. The term of our lease was originally set to expire in 2016, and, following the amendment we entered into in May 2014, the term of our lease was extended until 2021, and, following the amendment we entered into in May 2015, we expanded our lease to include an additional approximately 3,000 square feet of space, for a total of approximately 11,000 square feet. In addition, we lease space in Zug, Switzerland and Utrecht, The Netherlands. The premises are anticipated to be sufficient for current and future operations and we expect to be able to renew the leases for these premises upon expiration.
Reliance Life Sciences Dispute
In December 2014, Reliance Life Sciences Pvt Ltd (“RLS”), our BOW015 contract manufacturing partner for India, exercised its three-year termination for-convenience right with respect to the parties’ Manufacturing and Supply Agreement, dated as of May 14, 2014 and as amended from time to time (the “RLS Agreement”), which would have caused the RLS Agreement to terminate in December 2017. In addition, in March 2015, RLS initiated an arbitration proceeding under the RLS Agreement alleging that the RLS Agreement grants RLS exclusive global supply rights for BOW015, that the terms of our collaboration agreements with Fujifilm Diosynth Biotechnologies U.S.A., Inc. and
64
Livzon MabPharm Inc. violate the RLS Agreement, and sought to terminate the RLS Agreement. The proceeding initiated by RLS sought a declaratory judgment, but did not seek any monetary damages.
On April 22, 2015, we entered into a Settlement Agreement with RLS (the “Settlement Agreement”). Pursuant to the Settlement Agreement, the parties agreed to have RLS manufacture 15 final batches of BOW015 on or before June 30, 2016 with the Company’s option to have RLS manufacture an additional 5 batches on or before December 31, 2016. In January 2016, we amended the Settlement Agreement to extend the time for RLS to manufacture the 15 final batches to June 30, 2017. RLS has also agreed to use reasonable commercial efforts to transfer the existing Indian marketing authorization for BOW015 granted by the Drug Controller General of India (“DCGI”) to us or our designee, with all costs of the transfer to be borne by us and subject to the DCGI’s approval of such transfer. We have agreed to pay to RLS a fee of approximately $2.3 million payable in four installments over the course of the final batch manufacture, with the final installment to be paid no later than ten days after June 30, 2016. Pursuant to the Settlement Agreement, on May 4, 2015, RLS withdrew the arbitration proceeding it initiated, and the parties have agreed to release each other from all causes of actions or claims, present and future, arising under the RLS Agreement. Following the satisfaction by both parties of all of the terms of the Settlement Agreement, the RLS Agreement and related agreements between the parties shall be terminated and neither party shall have any further rights or obligations thereunder.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
65
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock has been publicly traded on The NASDAQ Capital Market under the symbol “EPRS” since July 16, 2014, prior to which it was traded under the symbol “ZLCS”. The following table sets forth, for the periods indicated, the high and low intraday sale prices of our common stock as reported by The NASDAQ Capital Market. The per share prices below reflect the adjustments made for a 1‑for‑10 reverse stock split effected at 12:01 a.m. ET on July 16, 2014:
|
Year Ended December 31, 2015 |
|
High |
|
Low |
|
||
|
First quarter |
|
$ |
11.75 |
|
$ |
4.72 |
|
|
Second quarter |
|
|
9.60 |
|
|
5.26 |
|
|
Third quarter |
|
|
8.75 |
|
|
4.02 |
|
|
Fourth quarter |
|
|
5.95 |
|
|
2.62 |
|
|
Year Ended December 31, 2014 |
|
High |
|
Low |
|
||
|
First quarter |
|
$ |
22.50 |
|
$ |
11.00 |
|
|
Second quarter |
|
|
17.40 |
|
|
8.60 |
|
|
Third quarter |
|
|
12.90 |
|
|
6.75 |
|
|
Fourth quarter |
|
|
7.14 |
|
|
3.78 |
|
As of March 25, 2016, there were 53 holders of record of our common stock.
Dividends
We have not paid dividends to our stockholders since our inception and do not plan to pay dividends in the foreseeable future. We currently intend to retain earnings, if any, to finance our growth.
Securities Authorized For Issuance Under Equity Compensation Plans
|
|
|
|
|
|
|
|
Number of Securities |
|
|
|
|
|
|
|
|
|
Remaining Available |
|
|
|
|
Number of Securities |
|
|
|
|
for Future Issuance |
|
|
|
|
to be Issued upon |
|
Weighted Average |
|
Under Equity |
|
|
|
|
|
Exercise of |
|
Exercise Price of |
|
Compensation |
|
|
|
|
|
Outstanding |
|
Outstanding |
|
Plan (Excluding |
|
|
|
|
|
Options, Warrants |
|
Options, Warrants |
|
Securities Reflected |
|
|
|
|
|
or Rights(1) |
|
or Rights(2) |
|
in Column(a))(1)(3) |
|
|
|
Plan Category |
|
(a) |
|
(b) |
|
(c) |
|
|
|
Equity compensation plans approved by security holders |
|
4,078,854 |
|
$ |
8.59 |
|
394,402 |
|
|
Equity compensation plans not approved by security holders |
|
— |
|
|
— |
|
— |
|
|
Total |
|
4,078,854 |
|
$ |
8.59 |
|
394,402 |
|
|
(1) |
As of December 31, 2015. |
|
(2) |
For outstanding restricted stock units, the exercise price was deemed to be $0. |
|
(3) |
Our 2015 Equity Incentive Plan (the “2015 Plan”) contains an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2015 Plan, which annual increase is and will be added on the first day of each fiscal year from 2016 through 2025, inclusive, and will be equal to the |
66
least of: (i) 1,000,000 shares of common stock, (ii) 4% of the outstanding shares on that date, or (iii) such lesser amount determined by the compensation committee of the board of directors. |
ITEM 6. SELECTED CONSOLIDATED FINANCIAL DATA
Not applicable.
67
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes appearing in this annual report on Form 10‑K. Some of the information contained in this discussion and analysis or set forth elsewhere in this annual report on Form 10‑K include historical information and other information with respect to our plans and strategy for our business and contain forward‑looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward‑looking statements as a result of certain factors, including but not limited to those set forth under the “Risk Factors” section of this report and elsewhere in this annual report on Form 10‑K.
In the following discussion the terms “Epirus,” “we,” “our,” “us” or the “Company” refer to EPIRUS Biopharmaceuticals, Inc., a Delaware corporation and private company, prior to the July 15, 2014 merger described below, and EPIRUS Biopharmaceuticals, Inc., formerly known as Zalicus Inc., a Delaware corporation and public company, subsequent to the July 15, 2014 merger described below.
Overview
Recent Developments
Merger with Zalicus
On July 15, 2014, EPIRUS Biopharmaceuticals, Inc., a Delaware corporation and private company (“Private Epirus”), completed its merger with EB Sub, Inc., a wholly‑owned subsidiary of Zalicus Inc., a Delaware corporation (“Zalicus”), pursuant to the terms of that certain Agreement and Plan of Merger and Reorganization (as amended, the “Merger Agreement”), dated as of April 15, 2014, by and among Private Epirus, Zalicus and EB Sub, Inc., formerly known as BRunning, Inc. (the “Merger”). As part of the Merger, Zalicus was renamed EPIRUS Biopharmaceuticals, Inc. (“Public Epirus”) and Private Epirus was renamed EB Sub, Inc. (“EB Sub”). The boards of directors of Private Epirus and Zalicus approved the Merger on April 15, 2014, and the stockholders of Private Epirus and Zalicus approved the Merger and related matters on July 15, 2014. Following completion of the Merger, Private Epirus, now known as EB Sub, Inc., is the surviving corporation of the Merger and a wholly‑owned subsidiary of Public Epirus. The Merger has been accounted for as a “reverse merger” under the acquisition method of accounting for business combinations with Private Epirus treated as the accounting acquirer of Zalicus. The historical financial statements of Private Epirus have become the historical financial statements of Public Epirus, or the combined company, and are included in this filing labeled EPIRUS Biopharmaceuticals, Inc. As a result of the Merger, historical common stock, stock options and additional paid‑in capital, including share and per share amounts, have been retroactively adjusted to reflect the equity structure of Public Epirus, including the effect of the Merger exchange ratio and the Public Epirus common stock par value of $0.001 per share. See “Merger and Exchange Ratio” within Note 2 of the notes to our audited consolidated financial statements for the year ended December 31, 2014 included in this annual report on Form 10‑K for additional discussion of the Merger and the exchange ratio.
Pursuant to the terms of the Merger Agreement and after giving effect to a reverse stock split, at the effective time of the Merger (the “Effective Time”), each outstanding share of Private Epirus capital stock was converted into the right to receive approximately 0.13259 shares of Zalicus’ common stock (the “Exchange Ratio”). In addition, at the Effective Time: (i) Zalicus assumed all outstanding options to purchase shares of Private Epirus common stock, which were exchanged for options to purchase shares of Zalicus’ common stock, in each case appropriately adjusted based on the Exchange Ratio; and (ii) Zalicus assumed all outstanding warrants to purchase shares of Private Epirus capital stock, which were exchanged for warrants to purchase shares of Zalicus, or Public Epirus, common stock, in each case appropriately adjusted based on the Exchange Ratio. As of the Effective Time, Private Epirus former stockholders held approximately 81% of the combined company, calculated on a fully‑diluted basis, and former stockholders of Zalicus held approximately 19% of the combined company, calculated on a fully‑diluted basis. No fractional shares of common stock were issued in connection with the Merger. Instead, Private Epirus stockholders received cash in lieu of any
68
fractional shares of common stock that they would otherwise have been entitled to receive in connection with the Merger.
Presentation for Reverse Stock Split
On July 16, 2014, Public Epirus effected a 1‑for‑10 reverse stock split of its outstanding common stock, par value $0.001 per share (“Public Epirus Common Stock”) (the “Reverse Stock Split”). As a result of the Reverse Stock Split, each ten shares of Public Epirus Common Stock issued and outstanding immediately prior to the Reverse Stock Split were automatically combined into and became one share of common stock. No fractional shares were issued as a result of the Reverse Stock Split and any stockholder who otherwise would have been entitled to receive fractional shares was entitled to receive cash in an amount, without interest, determined by multiplying such fraction of a share by $11.80, the closing price of a share of Public Epirus Common Stock on the Nasdaq Stock Market on July 15, 2014, after giving effect to the Reverse Stock Split. Also, as a result of the Reverse Stock Split, the per share exercise price of, and the number of shares of common stock underlying, our stock options, warrants and other derivative securities outstanding immediately prior to the Reverse Stock Split were automatically proportionally adjusted based on the 1‑for‑10 split ratio in accordance with the terms of such options, warrants or other derivative securities, as the case may be. Share and per‑share amounts of Public Epirus Common Stock, options and warrants included herein have been adjusted to give effect to the Reverse Stock Split. The Reverse Stock Split did not alter the par value of Public Epirus Common Stock, $0.001 per share, or modify any voting rights or other terms of the common stock. The accompanying consolidated financial statements and notes to the consolidated financial statements, including the Exchange Ratio applied to historical Private Epirus common stock and stock options, give retroactive effect to the Reverse Stock Split for all periods presented.
After giving effect to the Reverse Stock Split and the Merger, Public Epirus had approximately 12.9 million shares of common stock outstanding.
Business
We are a global biopharmaceutical company focused on building a pure-play, sustainable and profitable biosimilar business by improving patient access through cost-effective medicines. Our ability to deliver on this vision is anchored in our strong technical platform and pragmatic approach to development and commercialization.
Our business focuses on biosimilars, which are biologic drugs that are demonstrated to be “highly similar” to a previously approved biologic drug, known as a “reference product”. A biosimilar has to undergo extensive analytical characterization and prove similarity in efficacy and safety to the reference product in order to be approved by regulators.
Global healthcare systems continue to be challenged with expensive therapies. According to Express Scripts®, in the United States, the cost of a biologic drug accounted for approximately 40% of total drug spending in 2014. These cost pressures provide a large opportunity for biosimilars. Over the next decade, biologic drugs with approximately $90 billion worth of annual sales will lose patent protection, based on projected global sales estimates from EvaluatePharma®. Approximately half of the biosimilar market opportunity is outside the United States. We believe that biosimilars are likely to play a crucial role in reducing healthcare costs and improving patient access.
We are headquartered in Boston, Massachusetts, with laboratories and technical capabilities, including our proprietary CHOBC® cell line platform, in Utrecht, the Netherlands, and business operations, clinical and regulatory teams based in Zug, Switzerland.
As a biopharmaceutical company charting a course for long-term success, we are deeply committed to building a focused, sustainable and profitable pure-play biosimilar business. Specifically, we believe the path to achieving our goals has a foundation in the following five key strategies:
|
· |
build and utilize technical capabilities to optimize product characteristics and manage costs; |
69
|
· |
plan our pipeline to gain cost synergies from product development, through clinical development and to eventual commercialization; |
|
· |
focus on global markets where the regulatory, legal and commercial landscapes are more evolved while evaluating and preparing in the long term for potential United States market entry; |
|
· |
enter into partnership arrangements where we expect to retain substantial economics over time; and |
|
· |
implement an efficient global tax structure. |
Since our inception, we have maintained in-house technical capabilities supported by external vendor laboratories. In September 2015, we acquired our primary vendor laboratory, Bioceros Holding B.V., a Netherlands company, or Bioceros, to expand our biosimilar pipeline and to vertically integrate our product development capabilities. As a result of the acquisition, Bioceros has become our wholly-owned subsidiary, renamed Epirus Biopharmaceuticals (Netherlands) B.V.
Bioceros was focused on the development of monoclonal antibodies (mAbs). We acquired Bioceros’ proprietary CHOBC cell line platform, all related intellectual property rights, a fully equipped laboratory and bioreactor capabilities designed for the development of mAbs and protein therapeutics, with a focus on biosimilars, including capabilities for the production of three new biosimilar product candidates to add to our pipeline of biosimilar product candidates, as further described below. We believe that the addition of Bioceros staff and capabilities, combined with existing in-house expertise, provides us with a strong technical foundation to accelerate product development and optimize product quality. Specifically, we believe that our ability to rapidly and efficiently conduct and repeat experiments to optimize product quality and cell line titers and process yields will help us to reduce regulatory risk and manage long term cost of goods.
We currently have a robust and cohesive pipeline of five biosimilar product candidates in the autoimmune and inflammation areas, including BOW015, a biosimilar version of Remicade® (infliximab), BOW050, a biosimilar version of Humira® (adalimumab), BOW070, a biosimilar version of Actemra® (tocilizumab), BOW090, a biosimilar version of STELARA® (ustekinumab) and BOW100, a biosimilar version of SIMPONI® (golimumab).
We expect the standardization of our technology platform and the clustering of the therapeutic area will allow us to have a greater degree of focus, synergy and cost management across product development, manufacturing, clinical and commercialization. Additionally, we have expanded our therapeutic focus with a rare disease product candidate, BOW080, a biosimilar version of Soliris® (eculizumab). We believe there will be a significant unmet need for cost-effective therapies for rare diseases. Cumulatively, these six reference products generated approximately $29.2 billion in global innovator sales in 2014, according to EvaluatePharma. We estimate this to be a biosimilar market opportunity of more than $9 billion. Furthermore, there are over 20 other mAbs with near-term patent expiries clustered in five therapeutic areas, which we believe will provide mid- to long-term pipeline growth options for us, including expanding our rare disease business.
For our lead product candidate, BOW015, we have reported bioequivalence, efficacy and safety data from a previous Phase 1 study in healthy volunteers and a Phase 3 study in active rheumatoid arthritis patients, both of which demonstrated the equivalence of BOW015 to Remicade. Our Phase 3 study included an open label phase, where we demonstrated that patients can be safely initiated and effectively maintained on BOW015 for 58 weeks, and that patients can be safely switched from Remicade to BOW015 and effectively maintained out to 58 weeks. Using this regulatory package, in 2014, in collaboration with our commercialization partner Sun Pharmaceutical Industries Ltd., or Sun, we
70
launched BOW015 as the first infliximab biosimilar in India, sold under the brand name InfimabTM. We have also filed for approval in additional Southeast Asian and Latin American countries.
To date, nearly 1,000 patients have been treated with BOW015. We expect this early experience to provide valuable in-market experience while we continue making BOW015 available to patients globally, to support filings in developed markets, such as Europe and North America, and to build important commercial experience.
In November 2015, we completed manufacturing readiness for BOW015 to support the initiation of a global clinical program for BOW015 in early 2016. In February 2016, we initiated this global clinical program, which we expect to support regulatory filings in established markets such as Europe and the United States. The UNIFORM (Understanding BOW015 (infliximab-EPIRUS) and reference infliximab (Remicade®) in patients with active rheumatoid arthritis on stable doses of methotrexate) study is a 58-week, double-blind, one-to-one randomized, comparator-controlled multi-center global study to compare safety, efficacy and immunogenicity and demonstrate clinical equivalence of BOW015 with Remicade. We plan to enroll over 500 patients with active rheumatoid arthritis in the UNIFORM study, which will be conducted at sites in Europe, North America and Latin America. The primary endpoint at week 16 for the study will assess the proportion of patients that meet ACR20, which is a 20 percent or greater improvement in American College of Rheumatology assessment. We have been designing this clinical program in consultation with European regulatory bodies in order to obtain the data that may be necessary to support potential approval of BOW015 in developed markets.
Leveraging early commercial experience and our robust global clinical program, we expect to file for approval of BOW015 in developed markets, such as Europe and North America, in 2017. For the remainder of our pipeline, we expect that our filings will be global and harmonized.
We believe that the biosimilar market in the United States will likely be attractive over the long term. Currently there are near term challenges in the United States, including an evolving but uncertain regulatory framework and legal situation, a complex commercial environment, a fragmented payor market and a market which focuses on switching patients off existing reference drugs.
We believe that the most attractive market opportunity for biosimilars in the near term is outside of the United States. Many global markets outside the United States have more defined regulatory environments, limited legal encumbrance, commercial precedence and payors seeking lower cost biologics. For each of the markets outside of the United States, which collectively represent approximately 50% of the global opportunity for biosimilars, we intend to take specific approaches to build profitable partnerships and, to date, we have grown the global distribution coverage of BOW015 in over 70 countries.
For European markets and select additional territories, in July 2015, we entered into an agreement with Swiss Pharma International AG, an affiliate of Pharmaceutical Works Polpharma S.A., or, together with Swiss Pharma International AG, Polpharma, for the development and commercialization of BOW015, BOW050 and BOW070 in certain designated territories. The designated territories include the European Union (with the exception of Austria, Belgium, Denmark, Finland, Luxembourg, the Netherlands and Sweden, which are reserved for us, along with Switzerland and Norway), together with certain countries in the Middle East, Turkey, Russia, and other countries comprising the Commonwealth of Independent States, or CIS.
In local production markets, such as China, where local authorities mandate or strongly encourage local production as a condition for regulatory and/or commercial acceptance, we intend to collaborate with local partners to enable in-country production of our products. We have entered into agreements with Livzon Mabpharm Inc., or Livzon, for the global development and commercialization of certain antibodies or related biological compounds, including BOW015 and BOW070, for China and associated markets.
In near-term markets, such as India and Latin America, in which our current BOW015 data package is expected to be sufficient for approval, we intend to pursue near-term product launches. This would enable us to gain valuable in-market patient exposure and commercial experience while we continue to work on making BOW015 available globally. This early commercial experience will strengthen our regulatory filings for Europe, the United States and additional
71
global markets. In collaboration with our commercialization partner Sun, we launched BOW015 in India in November 2014, under the brand name Infimab. We expect to utilize our existing regulatory data package to gain regulatory approval for BOW015 in additional countries. In May 2015, we also entered into a development and future distribution agreement with mAbxience S.A., or Mabxience, a company organized and existing under the laws of Uruguay and a wholly owned subsidiary of CHEMO Group, for BOW015 in Latin American markets, including Argentina, Chile, Ecuador, Paraguay, Uruguay and Venezuela.
Finally, in the United States, we intend to retain rights and evaluate market conditions prior to determining a commercial plan commensurate with our objectives to create value. We are currently evaluating development and commercial options including partnerships, hybrid profit sharing approaches and potentially building or acquiring commercial infrastructure.
The combination of a global sales approach and a focus on deals which would allow us to retain substantial economic value is supported by our Swiss-based tax structure, which we believe will allow us to efficiently manage future cash for operations outside the United States.
Our long-term ability to deliver these important medicines to patients worldwide can only be achieved through a disciplined approach. Our experienced management team plans to address the diverse regulatory, legal and commercial landscape by gaining near-term market access, creating a strong technical platform with opportunities for sustainable pipeline growth, and finding tax-optimized partnership deals that maximize future value to provide us with a path forward to build a sustainable and profitable biosimilar business.
Critical Accounting Policies and Estimates
Our financial statements are prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, costs and expenses and related disclosures. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. In many instances, we could have reasonably used different accounting estimates, and in other instances changes in the accounting estimates are reasonably likely to occur from period to period. Accordingly, actual results could differ significantly from the estimates made by our management. To the extent that there are material differences between these estimates and actual results, our future financial statement presentation, financial condition, results of operations and cash flows could be affected. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Our significant accounting policies are described in more detail in the notes to our financial statements contained elsewhere in this annual report on Form 10‑K. However, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our financial condition and results of operations.
We believe that several accounting policies are important to understanding our historical and future performance. We refer to these policies as “critical” because these specific areas generally require us to make judgments and estimates about matters that are uncertain at the time we make the estimate, and different estimates—which also would have been reasonable—could have been used, which would have resulted in different financial results.
Fair Value of Financial Instruments
We are required to disclose information on all assets and liabilities reported at fair value that enables an assessment of the inputs used in determining the reported fair values. We determine the fair market values of our financial instruments based on the fair value hierarchy, which is a hierarchy of inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the financial instrument based on market data obtained from sources independent of us. Unobservable inputs are inputs that reflect our assumptions about the inputs that market participants would use in pricing the financial instrument and are developed
72
based on the information available in the circumstances. The fair value hierarchy applies only to the valuation inputs used in determining the reported or disclosed fair value of the financial instruments and is not a measure of the investment credit quality. The three levels of the fair value hierarchy, and its applicability to our financial assets, are described below:
Level 1 Inputs: Unadjusted quoted prices in active markets that are accessible at the measurement date of identical, unrestricted assets and liabilities.
Level 2 Inputs: Quoted prices for similar assets and liabilities, or inputs that are observable, either directly or indirectly, for substantially the full term through corroboration with observable market data. Level 2 includes investments valued at quoted prices adjusted for legal or contractual restrictions specific to the security.
Level 3 Inputs: Pricing inputs are unobservable for the assets and liabilities, that is, inputs that reflect the reporting entity’s own assumptions about the assumptions market participants would use in pricing the assets. Level 3 includes private investments that are supported by little or no market activity.
The fair value hierarchy level is determined by asset class based on the lowest level of significant input. In periods of market inactivity, the observability of prices and inputs may be reduced for certain instruments. This condition could cause an instrument to be reclassified between levels. During the years ended December 31, 2015 and 2014, there were no transfers between levels.
We measure cash equivalents at fair value on a recurring basis. The fair value of cash equivalents is determined based on “Level 1” inputs, which consist of quoted prices in active markets for identical assets.
The fair value of our note is determined using current applicable rates for similar instruments with similar settlement features as of the balance sheet date. The carrying value of our short-term debt approximates its fair value as our interest rate is near current market rates for instruments with similar settlement features. The fair value of our note and contingent consideration were determined using Level 3 inputs.
Revenue Recognition
Multiple‑Element Arrangements
Under the authoritative guidance for revenue recognition related to multiple‑element revenue arrangements, each deliverable within a multiple‑element revenue arrangement is accounted for as a separate unit of accounting if both of the following criteria are met: (1) the delivered item or items have value to the customer on a standalone basis and (2) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in the Company’s control. We consider a deliverable to have standalone value if we sell this item separately or if the item is sold by another vendor or could be resold by the customer. Deliverables not meeting the criteria for being a separate unit of accounting are accounted for on a combined basis.
In the event we enter into a contract in which the deliverables are required to be separated, we will allocate arrangement consideration to each deliverable in an arrangement based on its relative selling price. We determine selling price using vendor‑specific objective evidence (“VSOE”), if it exists; otherwise, we use third‑party evidence (“TPE”). If neither VSOE nor TPE of selling price exists for a deliverable, we use estimated selling price (“ESP”) to allocate the arrangement consideration. We apply appropriate revenue recognition guidance to each unit of accounting.
Milestones
Contingent consideration from research and development activities that is earned upon the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved. At the inception of each arrangement that includes milestone payments, we evaluate whether each milestone is substantive. This evaluation includes an assessment of whether: (a) the consideration is commensurate with either (1) the entity’s performance to
73
achieve the milestone, or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, (b) the consideration relates solely to past performance and (c) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. We evaluate factors such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone, the level of effort and investment required and whether the milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement in making this assessment.
Royalty Revenue
Under certain license agreements to which we are a party, we receive royalty payments based upon our licensees’ net sales of products. Generally, under these agreements, we receive royalty payments from licensees approximately one quarter in arrears after the licensee has sold the income generating product or products. We recognize royalty revenues when we can reliably estimate such amounts and collectability is reasonably assured. As such, we generally recognize royalty revenues in the quarter reported to us by its licensees. Therefore, royalty revenues are recognized one quarter in arrears from the quarter in which sales by our licensees occurred. Under this accounting policy, the royalty revenues we report are not based upon estimates and are typically reported in the same period in which we receive payment from its licensees.
In‑process Research & Development
In‑process research & development (“IPR&D”) represents the fair value assigned to research and development assets that were not fully developed at the date of acquisition. IPR&D acquired in a business combination or recognized from the application of push‑down accounting is capitalized on our consolidated balance sheet at its acquisition‑date fair value. Until the project is completed, the assets are accounted for as indefinite‑lived intangible assets and subject to impairment testing. Upon completion of a project, the carrying value of the related IPR&D is reclassified to intangible assets and is amortized over the estimated useful life of the asset.
When performing the impairment assessment, we first assess qualitative factors to determine whether it is necessary to recalculate the fair value of our acquired IPR&D. If we believe, as a result of the qualitative assessment, that it is more likely than not that the fair value of acquired IPR&D is less than its carrying amount, we calculate the asset’s fair value. If the carrying value of our acquired IPR&D exceeds its fair value, then the intangible asset is written down to its fair value. For the year ended December 31, 2015, we determined that there was an impairment related to IPR&D acquired in the Merger. As a result, we recorded a loss on impairment of IPR&D in the amount of $1,671 for the year ended December 31, 2015. This impairment charge was recorded within research and development expense. For the year ended December 31, 2014, we determined that there was no impairment of IPR&D.
Intangible Assets
We amortize our intangible assets using the straight‑line method over its estimated economic life, which range from six to 16.5 years. We evaluate the potential impairment of our other intangible assets if events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful life of the assets is no longer appropriate. The impairment test is first based on a comparison of the undiscounted cash flows expected to be generated from the use of the asset group and its eventual disposition to the carrying value of the asset group. If impairment is indicated, the assets are written down by the amount by which the carrying value of the assets exceeds the related fair value of the assets. Any write‑downs are treated as permanent reductions in the carrying value of the assets. For the years ended December 31, 2015 and 2014, we determined that there was no impairment of our intangible assets.
Goodwill
Goodwill represents the difference between the consideration transferred and the fair value of the net assets acquired under the acquisition method of accounting for push‑down accounting. Goodwill is not amortized but is evaluated for impairment within our single reporting unit on an annual basis, during the fourth quarter, or more frequently if an event occurs or circumstances change that would more likely than not reduce the fair value of our
74
reporting unit below its carrying amount. When performing the impairment assessment, the accounting standard for testing goodwill for impairment permits a company to first assess the qualitative factors to determine whether the existence of events and circumstances indicates that it is more likely than not that the goodwill is impaired. If we believe, as a result of the qualitative assessment, that it is more likely than not that the fair value of goodwill is impaired, we must perform the first step of the goodwill impairment test. We have determined that goodwill was not impaired as of December 31, 2015. To date, we have not recognized any impairment charges related to goodwill.
Research and Development Costs
Research and development costs are charged to expense as incurred in performing research and development activities. The costs include employee compensation, facilities and overhead, clinical study and related clinical manufacturing costs, regulatory and other related costs. Nonrefundable advanced payments for goods or services to be received in the future for use in research and development activities are deferred and capitalized. The capitalized amounts are expensed as the related goods are delivered or the services are performed.
Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided to the Company by its vendors with respect to their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid or accrued research and development expense, as the case may be.
Stock‑Based Compensation
We account for grants of stock options and restricted stock based on their grant date fair value and recognize compensation expense over the vesting period. We estimate the fair value of stock options as of the date of grant using the Black‑Scholes option‑pricing model and restricted stock based on the fair value of the underlying common stock, which, subsequent to the Merger, is determined as the closing trading price of the our common stock and, prior to the merger due to the absence of an active market for our common stock, the fair value of our common stock was determined in good faith by our board of directors, with the assistance and upon the recommendations of management, based on objective and subjective factors consistent with the methodologies outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. See Note 15 of the notes to our audited consolidated financial statements for the year ended December 31, 2015 included in this annual report on Form 10‑K for additional information.
Stock‑based compensation expense represents the cost of the grant date fair value of employee stock option grants recognized over the requisite service period of the awards (usually the vesting period) on a straight‑line basis, net of estimated forfeitures. The expense is adjusted for actual forfeitures at the end of each reporting period. Stock‑based compensation expense recognized in the consolidated financial statements is based on awards that are ultimately expected to vest.
Stock‑based awards issued to non‑employees are accounted for based on the fair value of such services received or of the equity instruments issued, whichever is more reliably measured. Awards are revalued at each reporting date and upon vesting and are expensed on a straight‑line basis over the vesting period.
We recognized stock‑based compensation expense of $3.5 million and $1.7 million for the years ended December 31, 2015 and 2014, respectively. As of December 31, 2015, there was approximately $12.3 million of total stock‑based compensation expense not yet recognized relating to non‑vested awards granted under our equity incentive plan. This expense is net of estimated forfeitures and is expected to be recognized over a weighted‑average period of approximately 3.10 years. The total unrecognized share‑based compensation cost will be adjusted for future changes in estimated forfeitures. The amount of stock‑based compensation expense to be recorded in any future period cannot be accurately predicted due to the uncertainty of future grant levels and actual forfeitures to be recorded. Additionally, changes to the assumptions used in the Black‑Scholes model could cause a material change in the amount of stock‑based compensation expense to be recorded in future reporting periods.
75
Income Taxes
We account for income taxes under the liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, deferred tax assets and liabilities are determined on the basis of the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in operations in the period that includes the enactment date.
We recognize net deferred tax assets through the recording of a valuation allowance to the extent that we believe these assets are more likely than not to be realized. In making such a determination, we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax‑planning strategies, and results of recent operations. If we determine that we would be able to realize our deferred tax assets in the future, in excess of its net recorded amount, we would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes.
We record uncertain tax positions on the basis of a two‑step process whereby (1) we determine whether it is more likely than not that the tax positions will be sustained on the basis of the technical merits of the position and (2) for those tax positions that meet the more‑likely‑than‑not recognition threshold, we recognize the largest amount of tax benefit that is more than 50% likely to be realized upon ultimate settlement with the related tax authority. We recognize interest and penalties related to unrecognized tax benefits within income tax expense. Any accrued interest and penalties are included within the related tax liability.
Recent Accounting Pronouncements
Going Concern
In August 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standard Update (“ASU”) No. 2014-15, Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to continue as a Going Concern. This update is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern within one year of the date of issuance of the entity’s financial statements and to provide related footnote disclosures. This guidance is effective for fiscal years beginning after December 15, 2016, with early application permitted. We are currently evaluating what effect, if any, the adoption of this guidance will have on the disclosures included in our consolidated financial statements.
Debt Issuance Costs
In April 2015, the FASB issued ASU No. 2015-03, Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs, which requires that debt issuance costs be presented in the balance sheet as a deduction from the carrying amount of the related liability, rather than as a deferred charge. The updated guidance is effective retroactively for financial statements covering fiscal years beginning after December 15, 2015, and interim periods within those fiscal years. We early adopted ASU No. 2015-03 effective December 31, 2015. The adoption of this standard did not have a material impact on our consolidated financial statements and footnote disclosures.
Technical Corrections and Improvements
In June 2015, the FASB issued ASU No. 2015-10, Technical Corrections and Improvements. This update covers a wide range of Topics in the ASC. The amendments in this update represent changes to clarify the ASC, correct unintended application of guidance, or make minor improvements to the ASC that are not expected to have a significant effect on current accounting practice or create a significant administrative cost to most entities. Additionally, some of the amendments will make the ASC easier to understand and easier to apply by eliminating inconsistencies, providing needed clarifications, and improving the presentation of guidance in the ASC. The amendments in this update that
76
require transition guidance are effective for all entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2015. All other amendments will be effective upon the issuance of this update. We do not anticipate that the adoption of this standard will have a material impact on our consolidated financial statements and footnote disclosures.
Revenue Recognition
In July 2015, the FASB deferred the effective date for ASU No. 2014-09, Revenue from Contracts with Customers, by one year. This update will supersede the revenue recognition requirements in Revenue Recognition (Topic 605) and requires entities to recognize revenue in a way that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled to in exchange for those goods or services. This update is effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period, which for us is January 1, 2018. Early application is permitted for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period. The update can be applied retrospectively to each prior reporting period presented or retrospectively with the cumulative effect of the change recognized at the date of the initial application in retained earnings. We are currently evaluating the potential impact of the adoption of this update will have on our consolidated financial statements and have not yet selected a transition method.
Business Combinations
In September 2015, the FASB issued ASU No. 2015-16, Business Combinations (Topic 805): Simplifying the Accounting for Measurement - Period Adjustments. This update eliminates the requirement to restate prior period financial statements for measurement period adjustments following a business combination. The update requires that the cumulative impact of a measurement period adjustment (including the impact on prior periods) be recognized in the reporting period in which the adjustment is identified. The prior period impact of the adjustment should be either presented separately on the face of the income statement or disclosed in the notes. The update is effective for fiscal years beginning after December 15, 2015. The adoption of this update is not expected to have a material impact on our financial position or results of operations.
Income Taxes
During November 2015, the FASB issued ASU 2015-17, “Balance Sheet Classification of Deferred Taxes”, which simplifies the presentation of deferred income taxes. This ASU requires that deferred tax assets and liabilities be classified as non-current in a statement of financial position. The standard is effective for public companies for fiscal years beginning after December 15, 2016, including interim periods within that reporting period. Early adoption is permitted for any interim and annual financial statements that have not yet been issued. We early adopted ASU 2015-17 effective December 31, 2015 on a prospective basis. Adoption of this ASU resulted in a reclassification of the net current deferred tax asset of $109 and the net current deferred tax liability of $80 to the net non-current deferred tax liability in our consolidated balance sheet as of December 31, 2015. No prior periods were retrospectively adjusted.
Leases
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842). This update improves financial reporting around leasing transactions and more closely aligns accounting for leases with the recently issued International Financial Reporting Standard. The update requires a lessee to recognize assets and liabilities on the balance sheet for operating leases and changes many key definitions, including the definition of a lease. The update includes a short-term lease exception for leases with a term of 12 months or less, in which a lessee can make an accounting policy election not to recognize lease assets and lease liabilities. Lessees will continue to differentiate between finance leases (previously referred to as capital leases) and operating leases, using classification criteria that are substantially similar to the previous guidance. This update is effective for fiscal years beginning after December 15, 2018 and interim periods within those fiscal years, with earlier application permitted. We are currently evaluating the effect of this update on our consolidated financial statements.
77
Financial Overview
Research and Development Expenses
Research and development expenses consist primarily of costs incurred for the research and development of our preclinical and clinical candidates, and include:
|
· |
employee‑related expenses, including salaries, benefits and stock‑based compensation expense; |
|
· |
expenses incurred under agreements with contract research organizations, or CROs, contract manufacturing organizations, or CMOs, and consultants that conduct our clinical trials and preclinical activities; |
|
· |
payments made under our licensing agreements; |
|
· |
the cost of acquiring, developing and manufacturing clinical trial materials and lab supplies; |
|
· |
facility, depreciation and other expenses, which include direct and allocated expenses for rent, maintenance of our facility, insurance and other supplies; and |
|
· |
costs associated with preclinical activities and regulatory operations. |
We expense research and development costs to operations as incurred. We recognize costs for certain development activities, such as clinical trials, based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations or information provided to us by our vendors.
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later‑stage clinical trials.
For the period January 25, 2011 through December 31, 2015, we have incurred an aggregate of $73.9 million in research and development costs of which $49.1 million related to BOW015, $6.4 million related to BOW050, $0.8 million related to BOW030 (our bevacizumab program), $1.5 million related to BOW070, $9.5 million related to headcount and related expenses and $6.6 million related to overhead and other expenses. We expect that our research and development expenses will increase substantially as we continue development of BOW015 and our pipeline products.
Because of the numerous risks and uncertainties associated with product development, however, we cannot determine with certainty the duration and completion costs of these or other current or future clinical trials of BOW015 or our other product candidates. The duration, costs and timing of clinical trials and development of our product candidates will depend on a variety of factors, including the uncertainties of future clinical and preclinical studies, uncertainties in clinical trial enrollment rate and significant and changing government regulation. In addition, the probability of success for each product candidate will depend on numerous factors, including competition, manufacturing capability and commercial viability.
General and Administrative Expenses
General and administrative expenses, including costs associated with mergers and acquisition and legal settlement costs, consist principally of salaries and related costs such as stock‑based compensation for personnel in executive, finance, business development, corporate communications and human resource functions, facility costs not otherwise included in research and development expenses, patent filing fees and professional legal fees. Other general and administrative expenses include travel expenses and professional fees for consulting, auditing and tax services. We anticipate that our general and administrative costs will increase in future periods as compared to 2015 due to resources required to support our development and clinical programs.
78
Interest Expense
Interest expense primarily reflects the amortization of debt discounts and interest expense in connection with a term loan, which was funded in October 2014 and May 2015.
Gain on Sale of Subsidiary
The gain on sale of subsidiary related to the sale of Zalicus Ltd.
Results of Operations
The following discussion summarizes the key factors our management team believes are necessary for an understanding of our financial statements. The results for the years ended December 31, 2013 are the results of Private Epirus. The results for the year ended December 31, 2014 include our pre and post‑Merger results. The results for the year ended December 31, 2015 include our post-Merger results.
Comparison of the Fiscal Years Ended December 31, 2015 and December 31, 2014:
The following table sets forth our results of operations for each of the periods set forth below (in thousands):
|
|
|
Year Ended |
|
|
|
|
|
||||
|
|
|
December 31, |
|
Change |
|||||||
|
|
|
2015 |
|
2014 |
|
$ |
|
% |
|||
|
Revenue, net of costs |
|
$ |
576 |
|
$ |
4 |
|
$ |
572 |
|
14300% |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
$ |
30,925 |
|
$ |
16,286 |
|
$ |
14,639 |
|
90% |
|
General and administrative |
|
|
21,951 |
|
|
22,973 |
|
|
(1,022) |
|
-4% |
|
Total operating expenses |
|
|
52,876 |
|
|
39,259 |
|
|
13,617 |
|
35% |
|
Loss from operations |
|
|
(52,300) |
|
|
(39,255) |
|
|
(13,045) |
|
33% |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense, net |
|
|
(1,332) |
|
|
(2,672) |
|
|
1,340 |
|
-50% |
|
Gain on sale of subsidiary |
|
|
1,016 |
|
|
— |
|
|
1,016 |
|
100% |
|
Change in fair value of warrant liability |
|
|
— |
|
|
(222) |
|
|
222 |
|
-100% |
|
Other income (expense): |
|
|
(116) |
|
|
262 |
|
|
(378) |
|
-144% |
|
Total other expense, net |
|
|
(432) |
|
|
(2,632) |
|
|
2,200 |
|
-84% |
|
Loss before income taxes |
|
$ |
(52,732) |
|
$ |
(41,887) |
|
$ |
(10,845) |
|
26% |
|
Benefit from income taxes |
|
|
544 |
|
|
43 |
|
|
501 |
|
1165% |
|
Net loss |
|
$ |
(52,188) |
|
$ |
(41,844) |
|
$ |
(10,344) |
|
25% |
Revenue — For the year ended December 31, 2015, we recorded net revenue of $576,000 consisting of revenues under our agreement with Polpharma of $236,000, development services performed by our newly acquired subsidiary, Epirus Netherlands, of $186,000, royalties of $119,000 and amortization of deferred revenue of $35,000 under our agreement with Sun.
79
Research and development expenses—For the year ended December 31, 2015, research and development expense was $30.9 million compared to $16.3 million for the year ended December 31, 2014, an increase of $14.6 million, or 90%. This increase was primarily the result of increased development costs of $3.9 million, $3.8 million and $1.0 million related to BOW015, BOW050 and BOW070, respectively, increased headcount related costs of $2.4 million, increased non‑cash expense including stock‑based compensation of $3.8 million offset in part by a decrease in overhead costs of $0.3 million. We expect research and development expense to increase in future periods as compared due to increased development costs related to BOW015 and our pipeline products.
|
|
|
Year Ended |
|
|
|
|
|
|
||||
|
|
|
December 31, |
|
Change |
|
|||||||
|
Project |
|
2015 |
|
2014 |
|
$ |
|
% |
|
|||
|
BOW015 |
|
$ |
14,897 |
|
$ |
11,042 |
|
$ |
3,855 |
|
35% |
|
|
BOW050 |
|
|
5,019 |
|
|
1,243 |
|
|
3,776 |
|
304% |
|
|
BOW030 |
|
|
50 |
|
|
386 |
|
|
(336) |
|
-87% |
|
|
BOW070 |
|
|
1,234 |
|
|
277 |
|
|
957 |
|
345% |
|
|
Overhead/Other |
|
|
9,725 |
|
|
3,338 |
|
|
6,387 |
|
191% |
|
|
Total |
|
$ |
30,925 |
|
$ |
16,286 |
|
$ |
14,639 |
|
90% |
|
General and administrative expenses—For the year ended December 31, 2015, general and administrative expense was $22.0 million compared to $23.0 million for the year ended December 31, 2014, a decrease of $1.0 million, or 4%. This decrease was primarily the result of $5.8 million in professional fees related to the Merger, severance paid in connection with the Merger of $1.8 million in 2014 and a gain related to the change in contingent consideration of $0.4 million. Offsetting these decreases were a one-time settlement fee of $2.3 million related to the Settlement Agreement (Note 21) with Reliance which occurred in the second quarter of 2015, increased headcount and related expenses of $1.6 million, increased non‑cash expenses of $1.0 million, increased professional fees of $1.0 million, costs related to our recently acquired subsidiary in the Netherlands of $0.4 million, and overhead and other expenses of $0.7 million. We anticipate that general and administrative costs will increase in future periods as compared to 2015 due to resources required to support our development and clinical programs.
Interest expense—For the year ended December 31, 2015, interest expense was $1.3 million compared to $2.7 million for the year ended December 31, 2014, a decrease of $1.4 million, or 50%. Interest expense in 2015 consists entirely of interest expense and the amortization of debt discounts related to the term loan funded in October 2014 and May 2015. Interest expense in 2014 primarily reflects interest expense and the amortization of debt discounts related to convertible notes issued during 2013, and in 2014, to a lesser extent, interest expense and the amortization of debt discounts related to the term loan funded in October 2014. All of our convertible notes converted to equity in March and April of 2014.
Gain on sale of subsidiary—For the year ended December 31, 2015, the gain on the sale of subsidiary is a result of the sale of Zalicus Ltd.
Change in fair value of warrant liability—For the year ended December 31, 2015, change in fair value of warrant liability was $0. For the year ended December 31, 2014, change in fair value of warrant liability was $0.2 million. We do not anticipate that we will recognize further amounts with respect to fair value adjustments of warrants as a result of the conversion of all outstanding warrants to purchase our preferred stock into warrants to purchase our common stock in connection with the completion of the Merger.
Other income (expense), net—For the year ended December 31, 2015, other expense, net was $(0.1) million.
Benefit from income taxes—For the year ended December 31, 2015, the benefit from income taxes of $0.5 million reflected an adjustment of deferred taxes of $0.4 million resulting from the impairment of intangible asset and
80
amortization of a deferred tax benefit of $0.2 million offset in part by tax expense generated in foreign territories and minimum state taxes due in the United States.
Comparison of the fiscal years ended December 31, 2014 and December 31, 2013:
The following table sets forth our results of operations for each of the periods set forth below (in thousands):
|
|
|
Year Ended |
|
|
|
|
|
||||
|
|
|
December 31, |
|
Change |
|||||||
|
|
|
2014 |
|
2013 |
|
$ |
|
% |
|||
|
Revenue, net of costs |
|
$ |
4 |
|
$ |
— |
|
$ |
4 |
|
100% |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
$ |
16,286 |
|
$ |
9,659 |
|
$ |
6,627 |
|
69% |
|
General and administrative |
|
|
22,973 |
|
|
4,809 |
|
|
18,164 |
|
378% |
|
Total operating expenses |
|
|
39,259 |
|
|
14,468 |
|
|
24,791 |
|
171% |
|
Loss from operations |
|
|
(39,255) |
|
|
(14,468) |
|
|
(24,787) |
|
171% |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense, net |
|
|
(2,672) |
|
|
(5,543) |
|
|
2,871 |
|
-52% |
|
Change in fair value of warrant liability |
|
|
(222) |
|
|
(789) |
|
|
567 |
|
-72% |
|
Other income (expense): |
|
|
262 |
|
|
5 |
|
|
257 |
|
5140% |
|
Total other expense, net |
|
|
(2,632) |
|
|
(6,327) |
|
|
3,695 |
|
-58% |
|
Loss before income taxes |
|
$ |
(41,887) |
|
$ |
(20,795) |
|
$ |
(21,092) |
|
101% |
|
Benefit from income taxes |
|
|
43 |
|
|
— |
|
|
43 |
|
100% |
|
Net loss |
|
$ |
(41,844) |
|
$ |
(20,795) |
|
$ |
(21,049) |
|
101% |
Research and development expenses—For the year ended December 31, 2014, research and development expense was $16.3 million compared to $9.7 million for the year ended December 31, 2013, an increase of $6.6 million, or 69%. This increase was primarily the result of increased development costs of $3.5 million, $1.1 million and $0.3 million related to BOW015, BOW050 and BOW070, respectively, increased headcount related costs of $1.2 million, increased non‑cash stock‑based compensation of $0.3 million and overhead costs of $0.2 million. We expect research and development expense to increase in future periods as compared due to increased development costs related to BOW015 and our pipeline products.
|
|
|
Year Ended |
|
|
|
|
|
|
||||
|
|
|
December 31, |
|
Change |
|
|||||||
|
Project |
|
2014 |
|
2013 |
|
$ |
|
% |
|
|||
|
BOW015 |
|
$ |
11,042 |
|
$ |
7,574 |
|
$ |
3,468 |
|
46% |
|
|
BOW050 |
|
|
1,243 |
|
|
129 |
|
|
1,114 |
|
864% |
|
|
BOW030 |
|
|
386 |
|
|
332 |
|
|
54 |
|
16% |
|
|
BOW070 |
|
|
277 |
|
|
— |
|
|
277 |
|
100% |
|
|
Overhead/Other |
|
|
3,338 |
|
|
1,624 |
|
|
1,714 |
|
106% |
|
|
Total |
|
$ |
16,286 |
|
$ |
9,659 |
|
$ |
6,627 |
|
69% |
|
General and administrative expenses—For the year ended December 31, 2014, general and administrative expense was $23.0 million compared to $4.8 million for the year ended December 31, 2013, an increase of $18.2 million, or 378%. This increase was primarily the result of $5.8 million in costs related to the Merger and increased professional fees of $5.0 million related to professional services related primarily to increased consulting costs related to public reporting activities and corporate tax structure, increased insurance costs as a result of becoming a public company and increased legal costs related to business development activities. Additional increases included $1.2 million related to
81
commercialization costs for BOW015, severance paid in connection with the Merger of $1.8 million, $2.4 million related to increased headcount, increased non‑cash stock‑based compensation of $1.4 million and overhead expenses of $0.8 million. We expect general and administrative expense to decrease in future periods as costs incurred related to the Merger in 2014 will not recur.
Interest expense—For the year ended December 31, 2014, interest expense was $2.7 million compared to $5.5 million for the year ended December 31, 2013, a decrease of $2.9 million, or 52%. Interest expense in 2014 and 2013 primarily reflects interest expense and the amortization of debt discounts related to convertible notes issued during 2013, and in 2014, to a lesser extent, interest expense and the amortization of debt discounts related to the term loan funded in October 2014. All of our convertible notes converted to equity in March and April of 2014.
Change in fair value of warrant liability—For the year ended December 31, 2014, change in fair value of warrant liability was $0.2 million compared to $0.8 million for the year ended December 31, 2013, a decrease of $0.6 million, or 72%, due to the change in fair value of our preferred stock offset in part by the exercise of certain warrants in April 2014. We do not anticipate that we will recognize further amounts with respect to fair value adjustments of warrants as a result of the conversion of all outstanding warrants to purchase our preferred stock into warrants to purchase our common stock in connection with the completion of the Merger.
Other income, net—For the year ended December 31, 2014, other income, net was $0.3 million. Other income, net primarily reflects a payment received from a former partner recognized as income upon termination of our agreement.
Benefit from income taxes—For the year ended December 31, 2014, the benefit from income taxes was less than $0.1 million. The benefit from income taxes primarily reflects the realization of a deferred tax benefit offset in part by tax expense for cash flows generated in foreign territories.
Liquidity and Capital Resources
From January 25, 2011 to December 31, 2015, we have incurred an accumulated deficit of $138.8 million, primarily as a result of expenses incurred through a combination of research and development activities related to our lead product candidate, BOW015 and other product candidates, and expenses supporting those activities. We have financed our operations since inception primarily through the sale of our equity securities, cash received in connection with the Merger and the issuance of debt securities. In February 2015, we closed on an underwritten public offering of approximately 10.6 million shares of our common stock, offered at a price of $5.00 per share. Net proceeds to us from this offering were approximately $48.0 million after deducting underwriting discounts and commissions and offering expenses paid by us.
Our total unrestricted cash and cash equivalents balance as of December 31, 2015, was $31.5 million.
We believe that our existing cash and cash equivalents will be sufficient to fund our current operating plan and capital expenditure requirements into the second quarter of 2016. We have based this estimate on assumptions that may prove to be wrong, and we could use up our available capital resources sooner than we currently expect. Based on our planned use of our net cash and cash equivalents and our existing cash resources, we believe that our available funds will be sufficient to enable us to continue our global clinical program for BOW015 in early 2016 and continue our pre-clinical development of BOW050 and BOW070. We expect that these funds will not be sufficient to enable us to complete this global clinical program or seek additional marketing approvals.
We have based our projections of operating capital requirements on assumptions that may prove to be incorrect and we may use all of our available capital resources sooner than we expect. Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amount of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
|
· |
the timing and costs of our planned global clinical program for BOW015; |
82
|
· |
the progress, timing and costs of manufacturing BOW015, BOW050, BOW070, BOW080, BOW090 and BOW100 for current and planned clinical trials; |
|
· |
the initiation, progress, timing, costs and results of pre-clinical studies and clinical trials for our other product candidates and potential product candidates; |
|
· |
the outcome, timing and costs of seeking regulatory approvals; |
|
· |
the costs of commercialization activities for BOW015 and other product candidates if we receive marketing approval, including the costs and timing of establishing product sales, marketing, distribution and manufacturing capabilities; |
|
· |
subject to receipt of marketing approval, revenue received from commercial sales of our product candidates by us or our partners; |
|
· |
the terms and timing of any future collaborations, licensing, consulting or other arrangements that we may establish; |
|
· |
the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights, including milestone and royalty payments and patent prosecution fees; |
|
· |
the costs of hiring additional employees; |
|
· |
the costs of maintaining and protecting our intellectual property rights and defending against intellectual property related claims; and |
|
· |
the extent to which we in-license or acquire other products and technologies. |
We expect that we will need to obtain substantial additional funding in order to commercialize BOW015, BOW050, BOW070, BOW080, BOW090, BOW100 and other future product candidates. To the extent that we raise additional capital through the sale of common stock, convertible securities or other equity securities, the ownership interests of our existing stockholders may be materially diluted and the terms of these securities could include liquidation or other preferences that could adversely affect the rights of our existing stockholders. In addition, debt financing, if available, would result in increased fixed payment obligations and may involve agreements that include restrictive covenants that limit our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends, that could adversely affect our ability to conduct our business. If we are unable to raise capital when needed or on acceptable terms, we could be forced to significantly delay, scale back or discontinue the development or commercialization of BOW015, BOW050, BOW070, BOW080, BOW090, BOW100 and other future product candidates, seek collaborators at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available, and relinquish or license, potentially on unfavorable terms, our rights to BOW015, BOW050, BOW070, BOW080, BOW090, BOW100 and other future product candidates that we otherwise would seek to develop or commercialize ourselves.
Without additional sources of cash and/or the deferral, reduction, or elimination of significant planned expenditures, we will not have the cash resources to remain as a going concern thereafter.
The factors that can impact our ability to continue to fund our operating needs through fiscal 2016 include, but are not limited to:
|
· |
our ability to raise additional capital through equity or debt financings; |
|
· |
our ability to enter into collaboration agreements which include immediate revenue payments; and; |
83
|
· |
our continued need to reduce our cost structure while simultaneously expanding the breadth of our business, enhancing our technical capabilities, and pursuing new business opportunities. |
If we cannot effectively manage these factors, we will need to raise additional capital to support our business. We have no commitments for any such funding, and there are no assurances that such additional sources of liquidity can be obtained on terms acceptable to us, or at all. If we are unable to obtain adequate financing or financing on terms satisfactory to us, we will not have the cash resources to continue as a going concern.
We expect that our operating expenses will increase over the next twelve months to continue our development activities. As of the date of this filing, we had cash and cash equivalents of approximately $21.8 million. If we cannot raise the money that we need in order to continue to operate our business, we will be forced to delay, scale back or eliminate some or all of our proposed operations. If any of these were to occur, there is a substantial risk that our business would fail. If we are unsuccessful in raising additional financing, we may need to curtail, discontinue or cease operations.
While we will actively seek to identify sources of liquidity, there are no assurances that such additional sources of liquidity can be obtained on terms acceptable to us on a commercially reasonable basis, or at all. These factors raise substantial doubt about our ability to continue as a going concern as of December 31, 2015. Furthermore, the 2015 audit opinion from our independent registered public accounting firm contains a going concern explanatory paragraph which may make it more difficult for us to raise funds.
We will require additional funds to implement our growth strategy for our business. Therefore, we will need to raise additional capital to fund our operations. These funds may be raised through equity financing, debt financing, or other sources, which may result in further dilution in the equity ownership of our shares of common stock. There can be no assurance that additional financing will be available to us when needed or, if available, that it can be obtained on commercially reasonable terms. If we are not able to obtain the additional financing on a timely basis should it be required, or generate significant material revenues from operations, we will not be able to meet our other obligations as they become due and will be forced to scale down or perhaps even cease our operations.
Cash Flows
The following table sets forth the major sources and uses of cash for each of the periods set forth below (in thousands):
|
|
|
|
|
||||
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
Net cash used in operating activities |
|
$ |
(46,582) |
|
$ |
(35,470) |
|
|
Net cash provided by investing activities |
|
|
841 |
|
|
12,293 |
|
|
Net cash provided by financing activities |
|
|
55,817 |
|
|
42,841 |
|
|
Effect of exchange rate on cash |
|
|
(23) |
|
|
— |
|
|
Net increase in cash and cash equivalents |
|
$ |
10,053 |
|
$ |
19,664 |
|
Operating Activities. Our operating activities used $46.6 million and $35.5 million for the years ended December 31, 2015 and 2014, respectively. The increase in net cash used in operating activities for the year ended December 31, 2015 is primarily attributable to the $10.3 million increase in net loss as described above.
Investing Activities. Our investing activities provided cash of $0.8 million and $12.3 million for the years ended December 31, 2015 and 2014, respectively. The cash provided by investing activities for the year ended December 31, 2015 is primarily attributable to cash received as a result of the sale of our subsidiary, Zalicus, Ltd. of $3.8 million and $0.9 million of cash acquired in the purchase of Bioceros in September 2015 offset in part by the payment to the shareholders of Bioceros for the acquisition of $3.4 million and the purchase of laboratory and office related equipment
84
of $0.5 million. The cash provided by investing activities for the year ended December 31, 2014 is primarily attributable to the $13.8 million of cash acquired in the Merger offset by approximately $0.2 million of purchases of office equipment and an addition to intangible assets of $1.3 million, resulting from a payment made to terminate our revenue sharing payment obligations under an agreement with Moksha8 Pharmaceuticals.
Financing Activities. Our financing activities provided cash of $55.8 million and $42.8 million for the years ended December 31, 2015 and 2014, respectively. Net cash provided by financing activities for the year ended December 31, 2015 is primarily related to $48.0 million of net proceeds from the issuance of common stock in February 2015, $7.5 million from the issuance of debt and $0.3 million of proceeds from the exercise of stock options. Net cash provided by financing activities for the year ended December 31, 2014 is primarily related to $30.5 million of net proceeds from the issuance of Series B preferred stock in April 2014, $7.3 million of net proceeds from the issuance of debt, $5.0 million of proceeds from the issuance of convertible notes in March 2014 and $0.4 million related to proceeds from the exercise of warrants and stock options offset in part by the repurchase of common stock of $0.3 million during the year ended December 31, 2014.
Net Operating Loss Carryforwards
As of December 31, 2015 and 2014, we had U.S. federal net operating loss carryforwards of approximately $38.8 million and $31.0 million, respectively and state net operating loss carryforwards of approximately $27.6 million and $21.3 million, respectively, which may be available to offset future income tax liabilities and expire at various dates through 2035. In addition, as of December 31, 2015 and 2014, we had foreign net operating loss carryforwards of approximately $82 million and $145 million, respectively, which may be available to offset future income tax liabilities. Net operating loss carryforward periods are not limited in the United Kingdom and Brazil, and Switzerland allows for a seven year carryforward period.
As of December 31, 2015 and 2014, we had federal research and development tax credit carryforwards of approximately $1.2 million and $0.5 million, respectively, available to reduce future tax liabilities which expire at various dates through 2035. As of December 31, 2015 and 2014, we had state research and development tax credit carryforwards of approximately $0.2 million and $0.2 million, respectively, available to reduce future tax liabilities, which expire at various dates through 2030.
Under the provision of the Internal Revenue Code, the net operating loss and tax credit carryforwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. Net operating loss and tax credit carryforwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three‑year period in excess of 50%, as defined under Sections 382 and 383 of the Internal Revenue Code, respectively, as well as similar state provisions. This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. The amount of the annual limitation is determined based on the value of the company immediately prior to the ownership change. Subsequent ownership changes may further affect the limitation in future years. The federal and state net operating loss carryforward and tax credit carryforward are net of the limitation due to Sections 382 and 383. A full valuation allowance has been provided against the net operating loss and tax credit carryforwards as of December 31, 2015.
Contractual Obligations and Commitments
Our contractual obligations consist primarily of debt principal payments totaling $15 million, operating lease payments totaling $4.8 million and payments due under license agreements (see Note 17 of the notes to our audited consolidated financial statements for the year ended December 31, 2015 included in this annual report on Form 10‑K for additional information).
Off‑Balance Sheet Arrangements
We do not have any off‑balance sheet arrangements or any relationships with unconsolidated entities of financial partnerships, such as entities frequently referred to as structured finance or special purpose entities.
85
ITEM 7A. QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISK
We do not hold or issue derivatives, derivative commodity instruments or other financial instruments for speculative trading purposes. Additionally, we do not believe our cash equivalents have any significant risk of default or illiquidity.
Our results of operations are subject to foreign currency exchange rate fluctuations due to the international nature of our operations. We have operations or maintain relationships with entities in the United States, United Kingdom, India, Switzerland, the Netherlands, and elsewhere in Europe. As a result, our financial position, results of operations and cash flows can be affected by market fluctuations in foreign exchange rates, primarily with respect to the British pound, Euro, and the Swiss franc. The current exposures arise primarily from cash, intercompany receivables, and payables. The financial results of our international activities are reported in United States dollars, and the functional currency for most of our foreign subsidiaries is United States dollars with the exception of Epirus Biopharmaceuticals (Netherlands) B.V. whose functional currency is the Euro. Fluctuations in the foreign currency exchange rates of the countries in which we do business will affect our operating results, often in ways that are difficult to predict.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The consolidated financial statements and related financial statement schedules required to be filed are listed in the Index to Consolidated Financial Statements and are incorporated herein.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES (in 000’s)
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining an adequate system of internal control over financial reporting, as defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting is a process designed under the supervision of our principal executive officer and principal financial officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external purposes in accordance with accounting principles generally accepted in the U.S. Management evaluated the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework (the 2013 Framework). All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Our management, under the supervision and with the participation of the principal executive officer and principal financial officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2015, excluding the internal controls of Bioceros B.V., acquired on September 9, 2015, which is included in the 2015 consolidated financial statements of EPIRUS Biopharmaceuticals, Inc. and constituted $16,251 and $13,522 of total and net assets, respectively, as of December 31, 2015 and $186 and $96 of net revenues and net loss, respectively for the year then ended. This assessment identified material weaknesses in our internal control over our financial statement close process and the financial reporting processes related to business combinations, income taxes and accrued development costs. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. Because of these material weaknesses, management believes that, as of December 31, 2015, our internal control over financial reporting was not effective at a reasonable assurance level.
We are committed to remediating the control deficiencies that constituted the above material weaknesses by implementing changes to our internal control over financial reporting. Management is responsible for implementing
86
changes and improvements in the internal control over financial reporting and for remediating the control deficiencies that gave rise to the material weaknesses. To remediate the material weaknesses described above, we are currently evaluating the controls and procedures we will design and put in place to address these material weaknesses and plan to implement appropriate measures as part of this effort. These controls and procedures may include engagement of independent consultants to aid us in our financial statement close process and in our processes related to business combinations, income taxes and accrued development costs.
Any actions we take or may take to remediate these material weaknesses are subject to continued management review supported by testing, as well as oversight by the Audit Committee of our Board of Directors. We cannot assure you, in any way, even if we involve an independent consultant, that material weaknesses or significant deficiencies will not occur in the future and that we will be able to remediate such weaknesses or deficiencies in a timely manner, which could impair our ability to accurately and timely report our financial position, results of operations or cash flows.
The effectiveness of our internal control over financial reporting as of December 31, 2015 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report included in this annual report on Form 10-K.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rule 13a‑15(e)), as of the end of the period covered by this report. Management recognizes that any disclosure controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost‑benefit relationship of possible controls and procedures.
Based on their evaluation, including the existence of the material weaknesses in our internal control over financial reporting referenced above, our management, including our Chief Executive Officer and Chief Financial Officer, concluded that our disclosure controls and procedures were not effective as of December 31, 2015.
Changes in Internal Control over Financial Reporting
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated any change in our internal control over financial reporting that occurred during the period covered by this annual report on Form 10‑K that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. Based on that evaluation, except as described in this Item 9A, there was no change in our internal control over financial reporting during the fiscal year ended December 31, 2015 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of EPIRUS Biopharmaceuticals, Inc.
We have audited EPIRUS Biopharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). EPIRUS Biopharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and
87
evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
As indicated in the accompanying Management’s Report on Internal Control Over Financial Reporting, management’s assessment of and conclusion on the effectiveness of internal control over financial reporting did not include the internal controls of Bioceros Holding B.V. acquired on September 9, 2015, which is included in the 2015 consolidated financial statements of EPIRUS Biopharmaceuticals, Inc. and constituted $16,251 and $13,522 of total and net assets, respectively, as of December 31, 2015 and $186 and $96 of net revenues and net loss, respectively for the year then ended. Our audit of internal control over financial reporting of EPIRUS Biopharmaceuticals, Inc. also did not include an evaluation of the internal control over financial reporting of Bioceros Holding B.V.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weaknesses have been identified and included in management’s assessment. Management has identified material weaknesses in controls related to its financial statement close process and the accounting for business combinations, income taxes and accrued development costs. We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of EPIRUS Biopharmaceuticals, Inc. as of December 31, 2015 and 2014, and the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2015. These material weaknesses were considered in determining the nature, timing and extent of audit tests applied in our audit of the 2015 financial statements, and this report does not affect our report dated March 28, 2016, which expressed an unqualified opinion on those financial statements and included an explanatory paragraph regarding EPIRUS Biopharmaceuticals, Inc.’s ability to continue as a going concern.
In our opinion, because of the effects of the material weaknesses described above on the achievement of the objectives of the control criteria, EPIRUS Biopharmaceuticals, Inc. has not maintained effective internal control over financial reporting as of December 31, 2015, based on the COSO criteria.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 28, 2016
88
89
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Information required under this item will be contained in our Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2015, under the captions “Election of Directors,” “Executive Officers,” “Corporate Governance”, and “Section 16(a) Beneficial Ownership Reporting Compliance” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10‑K.
ITEM 11. EXECUTIVE COMPENSATION
Information required under this item will be contained in our Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2015, under the captions “Corporate Governance” and “Executive Compensation,” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10‑K.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Information required under this item will be contained in our Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2015, under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10‑K.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
Information required under this item will be contained in our Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2015, under the captions “Corporate Governance” and “Certain Relationships and Related Persons Transactions” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10‑K.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Information required under this item will be contained in our Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2015, under the caption “Ratification of Selection of Independent Registered Public Accounting Firm” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10‑K.
90
ITEM 15. EXHIBITS AND FINANCIAL STATEMENTS SCHEDULES
The consolidated financial statements filed as part of this annual report on Form 10‑K are listed in the Index to Consolidated Financial Statements. Certain schedules are omitted because they are not applicable, or not required, or because the required information is included in the consolidated financial statements or notes thereto. The Exhibits are listed in the Exhibit Index and are filed as part of this annual report on Form 10‑K.
91
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this annual report on Form 10‑K to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
EPIRUS Biopharmaceuticals, Inc. |
|
|
|
|
|
|
March 28, 2016 |
By: |
/s/ Amit Munshi |
|
|
|
Amit Munshi |
|
|
|
President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this annual report on Form 10‑K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Name |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Amit Munshi |
|
President, Chief Executive Officer and Director |
|
March 28, 2016 |
|
Amit Munshi |
|
(principal executive officer) |
|
|
|
|
|
|
|
|
|
/s/ Thomas Shea |
|
Senior Vice President, Chief Financial Officer |
|
March 28, 2016 |
|
Thomas Shea |
|
and Treasurer (principal financial officer and |
|
|
|
|
|
principal accounting officer) |
|
|
|
|
|
|
|
|
|
/s/ Mark H.N. Corrigan, M.D. |
|
Chairman of the Board and Director |
|
March 28, 2016 |
|
Mark H.N. Corrigan, M.D. |
|
|
|
|
|
|
|
|
|
|
|
/s/ J. Kevin Buchi |
|
Director |
|
March 28, 2016 |
|
J. Kevin Buchi |
|
|
|
|
|
|
|
|
|
|
|
/s/ Geoffrey Duyk, M.D., Ph.D. |
|
Director |
|
March 28, 2016 |
|
Geoffrey Duyk, M.D., Ph.D. |
|
|
|
|
|
|
|
|
|
|
|
/s/ Daotian Fu, Ph.D. |
|
Director |
|
March 28, 2016 |
|
Daotian Fu, Ph.D. |
|
|
|
|
|
|
|
|
|
|
|
/s/ William Hunter, M.D. |
|
Director |
|
March 28, 2016 |
|
William Hunter, M.D. |
|
|
|
|
|
|
|
|
|
|
|
/s/ Julie McHugh |
|
Director |
|
March 28, 2016 |
|
Julie McHugh |
|
|
|
|
|
|
|
|
|
|
|
/s/ Scott Rocklage, Ph.D. |
|
Director |
|
March 28, 2016 |
|
Scott Rocklage, Ph.D. |
|
|
|
|
92
EPIRUS Biopharmaceuticals, Inc.
Index to Consolidated Financial Statements
|
|
|
Page |
|
|
|
94 |
|
|
|
Consolidated Financial Statements as of December 31, 2015 and 2014 and for the years ended December 31, 2015 and 2014: |
|
|
|
|
|
95 |
|
|
|
Consolidated Statements of Operations and Comprehensive Loss |
|
96 |
|
|
Consolidated Statements of Changes in Stockholders’ Equity (Deficit) |
|
97 |
|
|
|
98 |
|
|
|
|
99 |
|
93
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of EPIRUS Biopharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of EPIRUS Biopharmaceuticals, Inc. (the “Company”) as of December 31, 2015 and 2014, and the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of EPIRUS Biopharmaceuticals, Inc. at December 31, 2015 and 2014, and the consolidated results of its operations and its cash flows for each of the two years in the period ended December 31, 2015, in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has recurring losses from operations and insufficient cash resources that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), EPIRUS Biopharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 28, 2016 expressed an adverse opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 28, 2016
94
EPIRUS Biopharmaceuticals, Inc.
(in thousands, except share and per share amounts)
|
|
|
December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
31,515 |
|
$ |
21,462 |
|
|
Prepaids and other current assets |
|
|
3,732 |
|
|
1,556 |
|
|
Total current assets |
|
|
35,247 |
|
|
23,018 |
|
|
Property and equipment, net |
|
|
2,073 |
|
|
704 |
|
|
In-process research and development |
|
|
— |
|
|
5,500 |
|
|
Intangible assets, net |
|
|
6,410 |
|
|
3,605 |
|
|
Goodwill |
|
|
26,361 |
|
|
16,363 |
|
|
Restricted cash |
|
|
1,924 |
|
|
1,825 |
|
|
Other assets |
|
|
249 |
|
|
361 |
|
|
Total assets |
|
$ |
72,264 |
|
$ |
51,376 |
|
|
Liabilities, Convertible Preferred Stock and Stockholders' Equity |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
2,051 |
|
$ |
2,483 |
|
|
Accrued development costs |
|
|
4,438 |
|
|
876 |
|
|
Accrued expenses |
|
|
4,003 |
|
|
3,225 |
|
|
Short term debt, net of debt discount |
|
|
14,833 |
|
|
— |
|
|
Accrued purchase price liability |
|
|
5,493 |
|
|
— |
|
|
Deferred revenue |
|
|
957 |
|
|
50 |
|
|
Current portion of settlement of obligation |
|
|
978 |
|
|
— |
|
|
Other current liabilities |
|
|
343 |
|
|
557 |
|
|
Deferred tax benefit |
|
|
185 |
|
|
185 |
|
|
Total current liabilities |
|
|
33,281 |
|
|
7,376 |
|
|
Long term debt, net of debt discount |
|
|
— |
|
|
7,269 |
|
|
Deferred revenue, net of current portion |
|
|
1,173 |
|
|
946 |
|
|
Deferred tax liability |
|
|
648 |
|
|
2,166 |
|
|
Deferred tax benefit, net of current portion |
|
|
369 |
|
|
553 |
|
|
Other non-current liabilities |
|
|
425 |
|
|
665 |
|
|
Total liabilities |
|
|
35,896 |
|
|
18,975 |
|
|
Commitments and contingencies (Note 17) |
|
|
|
|
|
|
|
|
Stockholders' equity: |
|
|
|
|
|
|
|
|
Preferred stock, $0.001 par value; Authorized - 5,000,000 shares, Issued and Outstanding - 0 shares at December 31, 2015 and December 31, 2014 |
|
|
— |
|
|
— |
|
|
Common stock, $0.001 par value; Authorized - 70,000,000 and 300,000,000 shares at December 31, 2015 and December 31, 2014, respectively; Issued - 24,377,573 and 12,934,102 shares at December 31, 2015 and December 31, 2014, respectively; Outstanding - 24,377,573 and 12,920,843 shares at December 31, 2015 and December 31, 2014, respectively |
|
|
24 |
|
|
13 |
|
|
Additional paid-in capital |
|
|
175,126 |
|
|
118,959 |
|
|
Accumulated other comprehensive loss |
|
|
(23) |
|
|
— |
|
|
Accumulated deficit |
|
|
(138,759) |
|
|
(86,571) |
|
|
Total stockholders' equity |
|
|
36,368 |
|
|
32,401 |
|
|
Total liabilities, convertible preferred stock and stockholders' equity |
|
$ |
72,264 |
|
$ |
51,376 |
|
The accompanying notes are an integral part of these consolidated financial statements.
95
EPIRUS Biopharmaceuticals, Inc.
Consolidated Statements of Operations & Comprehensive Loss
(in thousands, except share and per share amounts)
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
Revenue |
|
$ |
701 |
|
$ |
4 |
|
|
|
|
|
|
|
|
|
|
|
Cost of revenues |
|
|
(125) |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
Research and development |
|
|
30,925 |
|
|
16,286 |
|
|
General and administrative |
|
|
21,951 |
|
|
22,973 |
|
|
Total operating expenses |
|
|
52,876 |
|
|
39,259 |
|
|
Loss from operations |
|
|
(52,300) |
|
|
(39,255) |
|
|
Other income (expense): |
|
|
|
|
|
|
|
|
Interest income (expense), net |
|
|
(1,332) |
|
|
(2,672) |
|
|
Gain on sale of subsidiary |
|
|
1,016 |
|
|
— |
|
|
Change in fair value of warrant liability |
|
|
— |
|
|
(222) |
|
|
Other income (expense), net |
|
|
(116) |
|
|
262 |
|
|
Total other income (expense), net |
|
|
(432) |
|
|
(2,632) |
|
|
Loss before income taxes |
|
|
(52,732) |
|
|
(41,887) |
|
|
Benefit from income taxes |
|
|
544 |
|
|
43 |
|
|
Net loss |
|
$ |
(52,188) |
|
$ |
(41,844) |
|
|
Net loss per share--basic and diluted |
|
$ |
(2.29) |
|
$ |
(6.81) |
|
|
Weighted-average number of common shares used in net loss per share calculation--basic and diluted |
|
|
22,747,826 |
|
|
6,141,605 |
|
|
Foreign currency translation adjustment |
|
|
(23) |
|
|
— |
|
|
Comprehensive loss |
|
$ |
(52,211) |
|
$ |
(41,844) |
|
The accompanying notes are an integral part of these consolidated financial statements.
96
EPIRUS Biopharmaceuticals, Inc.
Statements of Changes in Stockholders’ Equity (Deficit)
(in thousands, except share amounts)
|
|
|
Series A |
|
Series B |
|
|
|
|
|
|
|
Additional |
|
Accumulated Other |
|
|
|
|
Total |
|
|||||||||
|
|
|
Convertible Preferred Stock |
|
Convertible Preferred Stock |
|
|
Common Stock |
|
Paid-In |
|
Comprehensive |
|
Accumulated |
|
Stockholders’ |
|
|||||||||||||
|
|
|
Shares |
|
Amount |
|
Shares |
|
Amount |
|
|
Shares |
|
Amount |
|
Capital |
|
Income |
|
Deficit |
|
Equity (Deficit) |
|
|||||||
|
Balance at December 31, 2013 |
|
27,248,746 |
|
$ |
23,348 |
|
— |
|
$ |
— |
|
|
237,026 |
|
$ |
— |
|
$ |
5,348 |
|
$ |
— |
|
$ |
(44,727) |
|
$ |
(39,379) |
|
|
Exercise of preferred stock warrant |
|
5,101,816 |
|
|
6,436 |
|
787,402 |
|
|
993 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Issuance of preferred stock upon conversion of convertible debt |
|
13,184,383 |
|
|
13,184 |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Issuance of preferred stock |
|
— |
|
|
— |
|
24,409,444 |
|
|
30,478 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Issuance of convertible notes |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
2,071 |
|
|
— |
|
|
— |
|
|
2,071 |
|
|
Extinguishment of convertible notes |
|
— |
|
|
— |
|
3,947,363 |
|
|
5,013 |
|
|
— |
|
|
— |
|
|
(1,150) |
|
|
— |
|
|
— |
|
|
(1,150) |
|
|
Exchange of preferred stock for common stock in connection with the Merger |
|
(45,534,945) |
|
|
(42,968) |
|
(29,144,209) |
|
|
(36,484) |
|
|
9,901,700 |
|
|
10 |
|
|
79,442 |
|
|
— |
|
|
— |
|
|
79,452 |
|
|
Issuance of common stock in connection with the Merger |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
2,610,871 |
|
|
3 |
|
|
31,321 |
|
|
— |
|
|
— |
|
|
31,324 |
|
|
Exchange of warrants in connection with the Merger |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
167 |
|
|
— |
|
|
— |
|
|
167 |
|
|
Exercise of stock options |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
98,405 |
|
|
— |
|
|
90 |
|
|
— |
|
|
— |
|
|
90 |
|
|
Issuance of warrants in connection with Hercules notes |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
248 |
|
|
— |
|
|
— |
|
|
248 |
|
|
Issuance of common stock upon vesting of restricted stock units |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
27,540 |
|
|
— |
|
|
(297) |
|
|
— |
|
|
— |
|
|
(297) |
|
|
Stock-based compensation expense and vesting of restricted stock |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
45,301 |
|
|
— |
|
|
1,719 |
|
|
— |
|
|
— |
|
|
1,719 |
|
|
Net loss |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(41,844) |
|
|
(41,844) |
|
|
Balance at December 31, 2014 |
|
— |
|
$ |
— |
|
— |
|
$ |
— |
|
|
12,920,843 |
|
$ |
13 |
|
$ |
118,959 |
|
$ |
— |
|
$ |
(86,571) |
|
$ |
32,401 |
|
|
Issuance of common stock |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
10,558,208 |
|
|
11 |
|
|
47,951 |
|
|
— |
|
|
— |
|
|
47,962 |
|
|
Stock issued in connection with purchase of Bioceros |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
788,960 |
|
|
— |
|
|
4,568 |
|
|
— |
|
|
— |
|
|
4,568 |
|
|
Exercise of stock options |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
130,920 |
|
|
— |
|
|
355 |
|
|
— |
|
|
— |
|
|
355 |
|
|
Retirement of common stock |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
(34,617) |
|
|
— |
|
|
(177) |
|
|
— |
|
|
— |
|
|
(177) |
|
|
Stock-based compensation expense and vesting of restricted stock |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
13,259 |
|
|
— |
|
|
3,470 |
|
|
— |
|
|
— |
|
|
3,470 |
|
|
Foreign currency translation adjustment |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(23) |
|
|
— |
|
|
(23) |
|
|
Net loss |
|
— |
|
|
— |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(52,188) |
|
|
(52,188) |
|
|
Balance at December 31, 2015 |
|
— |
|
$ |
— |
|
— |
|
$ |
— |
|
|
24,377,573 |
|
$ |
24 |
|
$ |
175,126 |
|
$ |
(23) |
|
$ |
(138,759) |
|
$ |
36,368 |
|
The accompanying notes are an integral part of these consolidated financial statements.
97
EPIRUS Biopharmaceuticals, Inc.
Consolidated Statements of Cash Flows (in thousands)
|
|
|
Year Ended |
|
||||
|
|
|
December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(52,188) |
|
$ |
(41,844) |
|
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
Gain on sale of subsidiary |
|
|
(1,016) |
|
|
— |
|
|
Gain on the change in contingent consideration |
|
|
(409) |
|
|
— |
|
|
Depreciation and amortization |
|
|
597 |
|
|
165 |
|
|
Non-cash interest expense |
|
|
373 |
|
|
2,475 |
|
|
Non-cash accretion expense |
|
|
170 |
|
|
— |
|
|
Deferred taxes |
|
|
(608) |
|
|
(185) |
|
|
Stock-based compensation and vesting of restricted stock |
|
|
3,470 |
|
|
1,719 |
|
|
Other receivable recovery |
|
|
(177) |
|
|
— |
|
|
Deferred rent and lease incentive activity |
|
|
(43) |
|
|
95 |
|
|
Loss on disposal of equipment |
|
|
1 |
|
|
— |
|
|
Loss on impairment of asset held for sale |
|
|
1,671 |
|
|
— |
|
|
Extinguishment of convertible notes |
|
|
— |
|
|
(22) |
|
|
Change in fair value of warrants |
|
|
— |
|
|
222 |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
Prepaids and other current assets |
|
|
(1,982) |
|
|
(276) |
|
|
Restricted cash |
|
|
(99) |
|
|
50 |
|
|
Other non-current assets |
|
|
111 |
|
|
(304) |
|
|
Accounts payable |
|
|
(1,235) |
|
|
(174) |
|
|
Accrued expenses and other current liabilities |
|
|
3,302 |
|
|
1,823 |
|
|
Other non-current liabilities |
|
|
(196) |
|
|
(210) |
|
|
Deferred revenue |
|
|
698 |
|
|
996 |
|
|
Settlement obligation |
|
|
978 |
|
|
— |
|
|
Net cash used in operating activities |
|
|
(46,582) |
|
|
(35,470) |
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
Cash acquired in acquisition |
|
|
889 |
|
|
13,756 |
|
|
Payment to stockholders of Bioceros |
|
|
(3,400) |
|
|
— |
|
|
Proceeds from the sale of in-process research and development |
|
|
3,829 |
|
|
— |
|
|
Addition to intangible assets |
|
|
— |
|
|
(1,250) |
|
|
Proceeds from the sale of property & equipment |
|
|
— |
|
|
4 |
|
|
Purchases of property and equipment |
|
|
(477) |
|
|
(217) |
|
|
Net cash provided by investing activities |
|
|
841 |
|
|
12,293 |
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
Proceeds from the issuance of common stock, net of costs |
|
|
47,962 |
|
|
— |
|
|
Proceeds from the issuance of preferred stock and warrants |
|
|
— |
|
|
30,478 |
|
|
Proceeds from the issuance of convertible notes |
|
|
— |
|
|
5,000 |
|
|
Proceeds from the issuance of debt, net of fees paid to lender |
|
|
7,500 |
|
|
7,285 |
|
|
Payment of debt issuance costs |
|
|
— |
|
|
(49) |
|
|
Proceeds from the exercise of preferred stock warrants |
|
|
— |
|
|
334 |
|
|
Repurchases of common stock |
|
|
— |
|
|
(297) |
|
|
Proceeds from the exercise of common stock options |
|
|
355 |
|
|
90 |
|
|
Net cash provided by financing activities |
|
|
55,817 |
|
|
42,841 |
|
|
Effect of exchange rate on cash |
|
|
(23) |
|
|
— |
|
|
Net increase (decrease) in cash and cash equivalents |
|
|
10,053 |
|
|
19,664 |
|
|
Cash and cash equivalents—Beginning of period |
|
|
21,462 |
|
|
1,798 |
|
|
Cash and cash equivalents—End of period |
|
$ |
31,515 |
|
$ |
21,462 |
|
|
Supplemental disclosure of cash flow information |
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
913 |
|
$ |
101 |
|
|
Cash paid for income taxes |
|
$ |
40 |
|
$ |
177 |
|
|
Supplemental schedule of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
Amounts in accounts payable for capital expenditures |
|
$ |
657 |
|
$ |
5 |
|
|
Conversion of convertible debt and accrued interest into preferred stock and warrants |
|
$ |
— |
|
$ |
13,184 |
|
|
Extinguishment of convertible debt and accrued interest into preferred stock |
|
$ |
— |
|
$ |
5,013 |
|
|
Exercise of warrants into preferred stock |
|
$ |
— |
|
$ |
7,095 |
|
|
Conversion of preferred stock into common stock |
|
$ |
— |
|
$ |
79,452 |
|
|
Conversion of preferred stock warrants to equity |
|
$ |
— |
|
$ |
167 |
|
|
Issuance of warrants in connection with Hercules debt |
|
$ |
— |
|
$ |
248 |
|
|
Leasehold improvements incurred by landlord |
|
$ |
— |
|
$ |
477 |
|
|
Fair value of assets and liabilities acquired in the Acquisition (Note 5): |
|
|
|
|
|
|
|
|
Fair value of assets acquired in the Acquisition |
|
$ |
15,697 |
|
$ |
— |
|
|
Fair value of liabilities assumed in the Acquisition |
|
$ |
(2,084) |
|
$ |
— |
|
|
Fair value of net assets acquired in the Acquisition |
|
$ |
13,613 |
|
$ |
— |
|
|
Fair value of assets and liabilities acquired in the Merger (Note 4): |
|
|
|
|
|
|
|
|
Fair value of assets acquired in the Merger |
|
$ |
— |
|
$ |
36,223 |
|
|
Fair value of liabilities assumed in the Merger |
|
$ |
— |
|
$ |
(4,899) |
|
|
Fair value of net assets acquired in the Merger |
|
$ |
— |
|
$ |
31,324 |
|
The accompanying notes are an integral part of these consolidated financial statements.
98
EPIRUS Biopharmaceuticals, Inc.
Notes to the Consolidated Financial Statements
(In thousands, except share and per share amounts)
1. Organization
EPIRUS Biopharmaceuticals, Inc. (the “Company”) is a global biopharmaceutical company focused on building a pure-play, sustainable and profitable biosimilar business by improving patient access through cost-effective medicines. The Company’s ability to deliver on this vision is anchored in its strong technical platform and pragmatic approach to development and commercialization.
The Company is headquartered in Boston, Massachusetts, with laboratories and technical capabilities, including the Company’s proprietary CHOBC® cell line platform, in Utrecht, the Netherlands, and business operations, clinical and regulatory teams based in Zug, Switzerland.
Since its inception, the Company has maintained in-house technical capabilities supported by external vendor laboratories. In September 2015, the Company acquired its primary vendor laboratory, Bioceros Holding B.V., a Netherlands company (“Bioceros”), to expand its biosimilar pipeline and to vertically integrate its product development capabilities. As a result of the acquisition, Bioceros has become the Company’s wholly-owned subsidiary, renamed Epirus Biopharmaceuticals (Netherlands) B.V. See Note 5, “Acquisition,” for additional discussion of the acquisition of Bioceros.
Bioceros was focused on the development of monoclonal antibodies (mAbs). The Company acquired Bioceros’ proprietary CHOBC cell line platform, all related intellectual property rights, a fully equipped laboratory and bioreactor capabilities designed for the development of mAbs and protein therapeutics, with a focus on biosimilars, including capabilities for the production of three new biosimilar product candidates to add to the Company’s pipeline of biosimilar product candidates, as further described below. The Company believes that the addition of Bioceros staff and capabilities, combined with existing in-house expertise, provides the Company with a strong technical foundation to accelerate product development and optimize product quality. Specifically, the Company believes that its ability to rapidly and efficiently conduct and repeat experiments to optimize product quality and cell line titers and process yields will allow it to reduce regulatory risk and manage long term cost of goods.
On July 15, 2014, EPIRUS Biopharmaceuticals, Inc., a Delaware corporation and private company ("Private Epirus"), completed its merger with EB Sub, Inc., a wholly-owned subsidiary of Zalicus Inc., a Delaware corporation ("Zalicus") (the "Merger"). As part of the Merger, Zalicus was renamed EPIRUS Biopharmaceuticals, Inc. ("Public Epirus") and Private Epirus was renamed EB Sub, Inc. ("EB Sub"). The boards of directors of Private Epirus and Zalicus approved the Merger on April 15, 2014, and the stockholders of Private Epirus and Zalicus approved the Merger and related matters on July 15, 2014. Following completion of the Merger, EB Sub, Inc. (formerly Private Epirus), is the surviving corporation of the Merger and a wholly-owned subsidiary of Public Epirus. The historical financial statements of Private Epirus have become the historical financial statements of Public Epirus, or the combined company.
The terms “Company” and “Epirus” as used in these notes to consolidated financial statements refer to Private Epirus and its subsidiaries prior to the completion of the Merger and to Public Epirus and its subsidiaries subsequent to the completion of the Merger.
To date, the Company has devoted substantially all of its efforts to product research and development market research, and raising capital. The Company is subject to a number of risks similar to those of other development stage life science companies, including dependence on key individuals, competition from other companies, the need for development of commercially viable products, and the need to obtain adequate additional financing to fund the development of its product candidates. The Company is also subject to a number of risks similar to other companies in the industry, including rapid technological change, regulatory approval of products, uncertainty of market acceptance of products, competition from substitute products and larger companies, the need to obtain additional financing, compliance with government regulations, protection of proprietary technology, dependence on third parties, product liability, and dependence on key individuals. If the Company does not successfully commercialize any of its product candidates within
99
India or in additional countries, it will be unable to generate recurring product revenue or achieve profitability. As presented in the financial statements, at December 31, 2015, the Company had cash and cash equivalents of $31,515 and an accumulated deficit of $138,759. During the year ended December 31, 2015, the Company incurred a net loss of $52,188. The Company believes that its existing cash and cash equivalents will be sufficient to fund its current operating plan and capital expenditure requirements into the second quarter of 2016.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. The Company has net losses for the period from inception (January 2011) through December 31, 2015 of $138,759, as well as negative cash flows from operating activities. Presently, the Company does not have sufficient cash resources to meet its cash requirements for the twelve months following December 31, 2015. These factors raise substantial doubt about the Company's ability to continue as a going concern. Management is in the process of evaluating various financing alternatives for operations, as the Company will need to finance future research and development activities and general and administrative expenses through fund raising in the public or private equity markets.
The consolidated financial statements do not include any adjustments that may be necessary should the Company be unable to continue as a going concern. The Company’s continuation as a going concern is dependent on its ability to obtain additional financing as may be required and ultimately to attain profitability. If the Company raises additional funds through the issuance of equity, the percentage ownership of current shareholders would be reduced, and such securities might have rights, preferences or privileges senior to the common stock. Additional financing may not be available upon acceptable terms, or at all. If adequate funds are not available or are not available on acceptable terms, the Company may not be able to take advantage of prospective business endeavors or opportunities, which could significantly and materially restrict its future plans for developing its business and achieving commercial revenues. If the Company is unable to obtain the necessary capital, the Company may have to cease operations.
2. Summary of Significant Accounting Policies
Basis of Presentation and Use of Estimates
The preparation of the Company’s consolidated financial statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”) requires it to make estimates and assumptions that impact the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in the Company’s consolidated financial statements and accompanying notes. The most significant estimates in the Company’s consolidated financial statements relate to the valuation of equity awards, purchase price allocations including fair value estimates of intangible assets, estimated useful lives of fixed assets and intangible assets and accruals relating to development contracts, clinical trials and settlement obligations. The Company bases its estimates and assumptions on historical experience when available and on various factors that it believes to be reasonable under the circumstances. The Company evaluates its estimates and assumptions on an ongoing basis. The Company’s actual results may differ from these estimates under different assumptions or conditions.
Certain prior year amounts have been conformed to the current year presentation. These reclassifications had no impact on net loss or total equity.
Principles of Consolidation
The consolidated financial statements include the accounts of Epirus and its wholly owned subsidiaries: EB Sub, Inc., a Delaware Corporation, Epirus Biopharmaceuticals Ltd., a United Kingdom corporation, Epirus Biopharmaceuticals (Switzerland) GmbH, a Swiss corporation, Epirus Brasil Tecnologia Ltda, a Brazilian corporation, Epirus Biopharmaceuticals (Netherlands) B.V., a Netherlands corporation, and Zalicus Pharmaceuticals Ltd., a Canadian corporation. All significant intercompany balances and transactions have been eliminated in consolidation.
100
Merger and Exchange Ratio
The Merger has been accounted for as a “reverse merger” under the acquisition method of accounting for business combinations with Private Epirus treated as the accounting acquirer of Zalicus. The historical financial statements of Private Epirus have become the historical financial statements of Public Epirus, or the combined company, and are included in this filing labeled Epirus Biopharmaceuticals, Inc. As a result of the Merger, historical common stock, stock options and additional paid-in capital, including share and per share amounts, have been retroactively adjusted to reflect the equity structure of Public Epirus, including the effect of the Merger exchange ratio and the Public Epirus common stock par value of $0.001 per share. See Note 4, “Merger,” for additional discussion of the Merger and the exchange ratio.
Reverse Stock Split
On July 16, 2014, Public Epirus effected a 1-for-10 reverse stock split of its outstanding common stock, par value $0.001 per share (“Public Epirus Common Stock”) (the “Reverse Stock Split”). The accompanying consolidated financial statements and these notes to consolidated financial statements, including the Merger exchange ratio (Note 4) applied to historical Private Epirus common stock and stock options, give retroactive effect to the Reverse Stock Split for all periods presented. The shares of Public Epirus Common Stock retained a par value of $0.001 per share.
Segment and Geographic Information
Operating segments are defined as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker (“CODM”) or decision-making group in making decisions regarding resource allocation and assessing performance. The Company and the Company’s CODM view the Company’s operations and manage its business in one operating segment: the development and commercialization of biosimilar monoclonal antibodies for emerging markets. The Company's entire business is managed by a single management team, which reports to the Chief Executive Officer. As of December 31, 2015 and 2014, all of the Company's long-lived assets were held within the United States, Switzerland and The Netherlands.
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities of 90 days or less from the date of purchase to be cash equivalents. As of December 31, 2015 and 2014, all cash and cash equivalents are held in depository accounts at six commercial banks. Cash equivalents are reported at fair value.
Concentrations of Credit Risk and Off-Balance Sheet Risk
Financial instruments that potentially subject the Company to concentrations of credit risk are primarily cash, cash equivalents and restricted cash. The Company maintains its cash and cash equivalent balances in the form of checking and savings accounts with financial institutions that management believes are creditworthy. The Company’s investment policy includes guidelines on the quality of the institutions and financial instruments and defines allowable investments that the Company believes minimizes the exposure to concentration of credit risk. The Company has no financial instruments with off-balance-sheet risk of loss.
Fair Value of Financial Instruments
The Company is required to disclose information on all assets and liabilities reported at fair value that enables an assessment of the inputs used in determining the reported fair values. The Company determines the fair market values of its financial instruments based on the fair value hierarchy, which is a hierarchy of inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the financial instrument based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the inputs that market participants would use in pricing the financial instrument and are developed based on the information available in the circumstances. The fair value hierarchy applies
101
only to the valuation inputs used in determining the reported or disclosed fair value of the financial instruments and is not a measure of the investment credit quality. The three levels of the fair value hierarchy, and its applicability to the Company’s financial assets and liabilities, are described below:
Level 1 Inputs: Unadjusted quoted prices in active markets that are accessible at the measurement date of identical, unrestricted assets and liabilities.
Level 2 Inputs: Quoted prices for similar assets and liabilities, or inputs that are observable, either directly or indirectly, for substantially the full term through corroboration with observable market data. Level 2 includes investments valued at quoted prices adjusted for legal or contractual restrictions specific to the security.
Level 3 Inputs: Pricing inputs are unobservable for the assets and liabilities, that is, inputs that reflect the reporting entity’s own assumptions about the assumptions market participants would use in pricing the assets. Level 3 includes private investments that are supported by little or no market activity.
The fair value hierarchy level is determined by asset class based on the lowest level of significant input. In periods of market inactivity, the observability of prices and inputs may be reduced for certain instruments. This condition could cause an instrument to be reclassified between levels. During the years ended December 31, 2015 and 2014, there were no transfers between levels.
The Company measures cash equivalents at fair value on a recurring basis. The fair value of cash equivalents is determined based on “Level 1” inputs, which consist of quoted prices in active markets for identical assets.
The fair value of the Company's note is determined using current applicable rates for similar instruments with similar settlement features as of the balance sheet date. The carrying value of the Company's short term debt approximates its fair value as the Company's interest rate is near current market rates for instruments with similar settlement features. The fair value of the Company's note and contingent consideration was determined using Level 3 inputs.
Property and Equipment
Property and equipment consists of office furniture, office and computer equipment and leasehold improvements. Expenditures for repairs and maintenance are recorded to expense as incurred, whereas major betterments are capitalized as additions to property and equipment. Property and equipment are recorded at cost and are depreciated when placed into service using the straight-line method of depreciation, based on their estimated useful lives as follows:
|
Asset Description |
|
Estimated Useful Lives |
|
|
Furniture and fixtures |
|
5 |
years |
|
Office and computer equipment |
|
3 |
years |
|
Leasehold improvements |
|
Shorter of the useful life or remaining lease term |
|
Upon retirement or sale, the cost of assets disposed and the related accumulated depreciation is removed from the accounts and any resulting gain or loss is recorded in the consolidated statements of operations.
The Company evaluates the potential of its long-lived assets for impairment if events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful life of the assets is no longer appropriate. The impairment test is based on a comparison of the undiscounted cash flows expected to be generated from the use of the asset group and its eventual disposition to the carrying value of the asset group. If impairment is indicated, the assets are written down by the amount by which the carrying value of the assets exceeds the related fair value of the assets. Any write-downs are treated as permanent reductions in the carrying value of the assets. No such impairment losses have been recorded through December 31, 2015.
102
Restricted Cash
Restricted cash represents cash held in depository accounts at financial institutions to collateralize conditional stand-by letter of credits related to the Company’s Boston, Massachusetts and Zug, Switzerland facility lease agreements and the Company’s subleased Cambridge, Massachusetts facility. Restricted cash is reported as non-current unless the restrictions are expected to be released in the next 12 months.
Acquisitions
Business combinations are accounted for using the acquisition method of accounting. Under the acquisition method of accounting, the tangible and intangible assets acquired and the liabilities assumed are recorded as of the acquisition date at their respective fair values. The Company evaluates a business as an integrated set of activities and assets that is capable of being managed for the purpose of providing a return in the form of dividends, lower costs or other economic benefits and consists of inputs and processes that provide or have the ability to provide outputs. In an acquisition of a business the excess of the fair value of the consideration transferred over the fair value of the net assets acquired is recorded as goodwill. In an acquisition of net assets that does not constitute a business, no goodwill is recognized.
The Company’s consolidated financial statements include the results of operations of an acquired business after the completion of the acquisition.
In-process Research & Development
In-process research & development (“IPR&D”) represents the fair value assigned to research and development assets that were not fully developed at the date of acquisition. IPR&D acquired in a business combination or recognized from the application of push-down accounting is capitalized on the Company’s consolidated balance sheet at its acquisition-date fair value. Until the project is completed, the assets are accounted for as indefinite-lived intangible assets and subject to impairment testing. Upon completion of a project, the carrying value of the related IPR&D is reclassified to intangible assets and is amortized over the estimated useful life of the asset.
When performing the impairment assessment, the Company first assesses qualitative factors to determine whether it is necessary to recalculate the fair value of its acquired IPR&D. If the Company determines, as a result of the qualitative assessment, that it is more likely than not that the fair value of acquired IPR&D is less than its carrying amount, it calculates the asset’s fair value. If the carrying value of the Company’s acquired IPR&D exceeds its fair value, then the intangible asset is written down to its fair value. For the year ended December 31, 2015, the Company determined that there was an impairment related to IPR&D acquired in the Merger. As a result, the Company recorded a loss on impairment of IPR&D in the amount of $1,671 for the year ended December 31, 2015. This impairment charge was recorded within research and development expense. For the year ended December 31, 2014, the Company determined that there was no impairment of IPR&D.
Intangible Assets
The Company amortizes its intangible assets using the straight-line method over its estimated economic life, which ranges from six to 16.5 years. The Company evaluates the potential impairment of its intangible assets if events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful life of the assets is no longer appropriate. The impairment test is first based on a comparison of the undiscounted cash flows expected to be generated from the use of the asset group and its eventual disposition to the carrying value of the asset group. If impairment is indicated, the assets are written down by the amount by which the carrying value of the assets exceeds the related fair value of the assets. Any write-downs are treated as permanent reductions in the carrying value of the assets. For the years ended December 31, 2015 and 2014, the Company determined that there was no impairment of its intangible assets.
103
Goodwill
Goodwill represents the difference between the consideration transferred and the fair value of the net assets acquired under the acquisition method of accounting for push-down accounting. Goodwill is not amortized but is evaluated for impairment within the Company’s single reporting unit on an annual basis, during the fourth quarter, or more frequently if an event occurs or circumstances change that would more likely than not reduce the fair value of the Company’s reporting unit below its carrying amount. When performing the impairment assessment, the accounting standard for testing goodwill for impairment permits a company to first assess the qualitative factors to determine whether the existence of events and circumstances indicates that it is more likely than not that the goodwill is impaired. If the Company believes, as a result of the qualitative assessment, that it is more likely than not that the fair value of goodwill is impaired, the Company must perform the first step of the goodwill impairment test. The Company has determined that goodwill was not impaired as of December 31, 2015 and 2014. To date, the Company has not recognized any impairment charges related to goodwill.
Revenue Recognition
Multiple-Element Arrangements
Under the authoritative guidance for revenue recognition related to multiple-element revenue arrangements, each deliverable within a multiple-element revenue arrangement is accounted for as a separate unit of accounting if both of the following criteria are met: (1) the delivered item or items have value to the customer on a standalone basis and (2) for an arrangement that includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in the Company’s control. The Company considers a deliverable to have standalone value if the Company sells this item separately or if the item is sold by another vendor or could be resold by the customer. Deliverables not meeting the criteria for being a separate unit of accounting are accounted for on a combined basis.
In the event the Company enters into a contract in which the deliverables are required to be separated, the Company will allocate arrangement consideration to each deliverable in an arrangement based on its relative selling price. The Company determines selling price using vendor-specific objective evidence (“VSOE”), if it exists; otherwise, the Company uses third-party evidence (“TPE”). If neither VSOE nor TPE of selling price exists for a deliverable, the Company uses estimated selling price (“ESP”) to allocate the arrangement consideration. The Company applies appropriate revenue recognition guidance to each unit of accounting.
Milestones
Contingent consideration from research and development activities that is earned upon the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved. At the inception of each arrangement that includes milestone payments, the Company evaluates whether each milestone is substantive. This evaluation includes an assessment of whether: (a) the consideration is commensurate with either (1) the entity’s performance to achieve the milestone, or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, (b) the consideration relates solely to past performance and (c) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. The Company evaluates factors such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone, the level of effort and investment required and whether the milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement in making this assessment.
Royalty Revenue
Under certain license agreements to which the Company is a party, the Company receives royalty payments based upon its licensees’ net sales of products. Generally, under these agreements, the Company receives royalty payments from licensees approximately one quarter in arrears after the licensee has sold the income generating product or products. The Company recognizes royalty revenues when it can reliably estimate such amounts and collectability is
104
reasonably assured. As such, the Company generally recognizes royalty revenues in the quarter reported to it by its licensees. Therefore, royalty revenues are recognized one quarter in arrears from the quarter in which sales by its licensees occurred. Under this accounting policy, the royalty revenues the Company recognizes are not based upon estimates and are typically reported in the same period in which the Company receives payment from its licensees.
Deferred Revenue
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying consolidated balance sheets. Amounts not expected to be recognized within 12 months from the balance sheet date are classified as long-term deferred revenue.
Deferred Rent
Deferred rent, included within other non-current liabilities, net of the current portion recorded in accrued expenses in the consolidated balance sheet, consists of the difference between cash payments and the recognition of rent expense on a straight-line basis for the facilities the Company occupies. The Company’s lease for its Boston, Massachusetts, facility provides for a rent-free period as well as fixed increases in minimum annual rental payments. The total amount of rental payments due over the lease term is being charged to rent expense ratably over the life of the lease.
Other Liabilities
In conjunction with the Merger, as discussed in Note 4, the Company assumed a sublease liability for Zalicus’ Cambridge, Massachusetts facility, which is included within other non-current liabilities, net of the current portion recorded in other current liabilities on the consolidated balance sheet. The liability represents the fair value of the difference between the total sublease payments to be received by the Company and the total payment obligation for the Company per the lease agreement.
The Company has an operating lease for office space in Boston, Massachusetts. In connection with this lease, the Company and its third party lessor agreed that the lessor would pay for certain leasehold improvements on behalf of the Company. These leasehold improvements are accounted for as a lease incentive obligation, which is recorded in other non-current liabilities, net of the current portion recorded in other current liabilities. This liability is being amortized over the life of the lease and as a reduction to rent expense.
Organizational Costs
All organizational costs are expensed as incurred.
Research and Development Costs
Research and development costs are charged to expense as incurred in performing research and development activities. The costs include employee compensation, facilities and overhead, clinical study and related clinical manufacturing costs, regulatory and other related costs. Nonrefundable advanced payments for goods or services to be received in the future for use in research and development activities are deferred and capitalized. The capitalized amounts are expensed as the related goods are delivered or the services are performed.
Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided to the Company by its vendors with respect to their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid or accrued research and development expense, as the case may be.
105
Stock-Based Compensation
The Company accounts for grants of stock options and restricted stock based on their grant date fair value and recognizes compensation expense over the award’s vesting period. The Company estimates the fair value of stock options as of the date of grant using the Black-Scholes option-pricing model and restricted stock based on the fair value of the underlying common stock, which is determined as the closing trading price of the Company’s common stock subsequent to the Merger, in which the Company’s common stock became publicly traded, and was determined by management prior to the Merger (Note 15).
Stock-based compensation expense represents the cost of the grant date fair value of employee stock option grants recognized over the requisite service period of the awards (usually the vesting period) on a straight-line basis, net of estimated forfeitures. The expense is adjusted for actual forfeitures at the end of each reporting period. Stock-based compensation expense recognized in the consolidated financial statements is based on awards that are ultimately expected to vest.
Stock-based awards issued to nonemployees are accounted for based on the fair value of such services received or of the equity instruments issued, whichever is more reliably measured. Awards are revalued at each reporting date and upon vesting and are expensed on a straight-line basis over the vesting period.
Income Taxes
The Company accounts for income taxes under the liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, deferred tax assets and liabilities are determined on the basis of the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in operations in the period that includes the enactment date.
The Company recognizes net deferred tax assets through the recording of a valuation allowance to the extent that the Company believes these assets are more likely than not to be realized. In making such a determination, management considers all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax-planning strategies, and results of recent operations. If management determines that the Company would be able to realize its deferred tax assets in the future, in excess of its net recorded amount, management would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes.
The Company records uncertain tax positions on the basis of a two-step process whereby: (1) management determines whether it is more likely than not that the tax positions will be sustained on the basis of the technical merits of the position and (2) for those tax positions that meet the more-likely-than-not recognition threshold, management recognizes the largest amount of tax benefit that is more than 50% likely to be realized upon ultimate settlement with the related tax authority. The Company recognizes interest and penalties related to unrecognized tax benefits within income tax expense. Any accrued interest and penalties are included within the related tax liability.
Net Loss Per Share
Basic net loss per share is calculated by dividing net loss by the weighted average shares outstanding during the period, without consideration for common stock equivalents. During periods of income, the Company allocates participating securities a proportional share of income determined by dividing total weighted average participating securities by the sum of the total weighted average common shares and participating securities (the “two-class method”). The Company’s convertible preferred stock, which was exchanged for Public Epirus Common Stock in the Merger, participated in any dividends declared by the Company and were therefore considered to be participating securities. Participating securities have the effect of diluting both basic and diluted earnings per share during periods of income. During periods of loss, the Company allocates no loss to participating securities because they have no contractual obligation to share in the losses of the Company. Diluted net loss per share attributable to common stockholders is
106
calculated by adjusting weighted average shares outstanding for the dilutive effect of common stock equivalents outstanding for the period, determined using the treasury-stock and if-converted methods. For purposes of the diluted net loss per share calculation, common stock equivalents have been excluded from the calculation of diluted net loss per share, as their effect would be anti-dilutive for all periods presented. Therefore, basic and diluted net loss per share were the same for all periods presented.
Comprehensive Loss
Comprehensive loss is the change in equity of a company during a period from transactions and other events and circumstances, excluding transactions resulting from investments by owners and distributions to owners.
Recent Accounting Pronouncements
Going Concern
In August 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standard Update (“ASU”) No. 2014-15, Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to continue as a Going Concern. This update is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern within one year of the date of issuance of the entity’s financial statements and to provide related footnote disclosures. This guidance is effective for fiscal years beginning after December 15, 2016, with early application permitted. The Company is currently evaluating what effect, if any, the adoption of this guidance will have on the disclosures included in its consolidated financial statements.
Debt Issuance Costs
In April 2015, the FASB issued ASU No. 2015-03, Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issuance Costs, which requires that debt issuance costs be presented in the balance sheet as a deduction from the carrying amount of the related liability, rather than as a deferred charge. The updated guidance is effective retroactively for financial statements covering fiscal years beginning after December 15, 2015, and interim periods within those fiscal years. The Company early adopted ASU No. 2015-03 effective December 31, 2015. The adoption of this standard did not have a material impact on the Company’s consolidated financial statements and footnote disclosures.
Technical Corrections and Improvements
In June 2015, the FASB issued ASU No. 2015-10, Technical Corrections and Improvements. This update covers a wide range of Topics in the ASC. The amendments in this update represent changes to clarify the ASC, correct unintended application of guidance, or make minor improvements to the ASC that are not expected to have a significant effect on current accounting practice or create a significant administrative cost to most entities. Additionally, some of the amendments will make the ASC easier to understand and easier to apply by eliminating inconsistencies, providing needed clarifications, and improving the presentation of guidance in the ASC. The amendments in this update that require transition guidance are effective for all entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2015. All other amendments will be effective upon the issuance of this update. The Company does not anticipate that the adoption of this standard will have a material impact on its consolidated financial statements and footnote disclosures.
Revenue Recognition
In July 2015, the FASB deferred the effective date for ASU No. 2014-09, Revenue from Contracts with Customers, by one year. This update will supersede the revenue recognition requirements in Revenue Recognition (Topic 605) and requires entities to recognize revenue in a way that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled to in exchange for those goods or services. This update is effective for annual reporting periods beginning after December 15, 2017, including
107
interim periods within that reporting period, which for the Company is January 1, 2018. Early application is permitted for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period. The update can be applied retrospectively to each prior reporting period presented or retrospectively with the cumulative effect of the change recognized at the date of the initial application in retained earnings. The Company is currently evaluating the potential impact the adoption of this update will have on its consolidated financial statements and has not yet selected a transition method.
Business Combinations
In September 2015, the FASB issued ASU No. 2015-16, Business Combinations (Topic 805): Simplifying the Accounting for Measurement - Period Adjustments. This update eliminates the requirement to restate prior period financial statements for measurement period adjustments following a business combination. The update requires that the cumulative impact of a measurement period adjustment (including the impact on prior periods) be recognized in the reporting period in which the adjustment is identified. The prior period impact of the adjustment should be either presented separately on the face of the income statement or disclosed in the notes. The update is effective for fiscal years beginning after December 15, 2015. The adoption of this update is not expected to have a material impact on the Company’s financial position or results of operations.
Income Taxes
During November 2015, the FASB issued ASU 2015-17, “Balance Sheet Classification of Deferred Taxes”, which simplifies the presentation of deferred income taxes. This ASU requires that deferred tax assets and liabilities be classified as non-current in a statement of financial position. The standard is effective for public companies for fiscal years beginning after December 15, 2016, including interim periods within that reporting period. Early adoption is permitted for any interim and annual financial statements that have not yet been issued. The Company early adopted ASU 2015-17 effective December 31, 2015 on a prospective basis. Adoption of this ASU resulted in a reclassification of the net current deferred tax asset of $109 and the net current deferred tax liability of $80 to the net non-current deferred tax liability in the Company’s consolidated balance sheet as of December 31, 2015. No prior periods were retrospectively adjusted.
Leases
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842). This update improves financial reporting around leasing transactions and more closely aligns accounting for leases with the recently issued International Financial Reporting Standard. The update requires a lessee to recognize assets and liabilities on the balance sheet for operating leases and changes many key definitions, including the definition of a lease. The update includes a short-term lease exception for leases with a term of 12 months or less, in which a lessee can make an accounting policy election not to recognize lease assets and lease liabilities. Lessees will continue to differentiate between finance leases (previously referred to as capital leases) and operating leases, using classification criteria that are substantially similar to the previous guidance. This update is effective for fiscal years beginning after December 15, 2018 and interim periods within those fiscal years, with earlier application permitted. The Company is currently evaluating the effect of this update on its consolidated financial statements.
3. Business Agreements
Sun Pharmaceutical Industries (formerly known as Ranbaxy Laboratories)
In January 2014, the Company and Sun Pharmaceutical Industries Limited (formerly known as Ranbaxy Laboratories Limited) (“Sun”) executed a royalty-bearing and non-transferrable license agreement (“Sun License Agreement”) for BOW015 to Sun for a broad range of territories including India, selected Southeast Asian markets and North Africa. Under the terms of the Sun License Agreement, the Company and Sun will pursue the commercialization of BOW015 in India and Sun received the right to sell BOW015 in the territories specified in the agreement. Sun does not have the right to manufacture BOW015.
108
Sun made an upfront payment of $500 upon the execution of the Sun License Agreement and is obligated to pay the Company up to $1,000 if certain development and regulatory approval milestones are achieved and up to $10,000 if certain sales milestones are achieved. Additionally, the Company is entitled to receive royalties in the mid-teens on net sales in the territories, which royalties are subject to reduction or renegotiation in certain limited circumstances. Under certain circumstances of uncured breach by the Company and termination of the Sun License Agreement by Sun, monetary damages of $500, would be due to Sun.
Unless terminated earlier in accordance with the terms of the agreement, the Sun License Agreement expires 20 years following the first commercial sale in the territories.
Under the Sun License Agreement, Sun is responsible for all activities related to the commercialization of BOW015 in the territories. The Company is responsible for performing the development activities required for obtaining regulatory approval in India, joint steering committee participation and clinical and commercial supply of product. The Company also agreed to perform the development activities required to obtain regulatory approval in other parts of the territories. These activities and the related fees will be agreed upon by Sun and the Company in future written agreements.
The Company has determined that the deliverables under the Sun License Agreement are the license, development activities related to regulatory approval in India, joint steering committee services and the obligation to supply clinical and commercial product. The Company’s obligation to perform future development activities to obtain regulatory approval in other parts of the territories is not a deliverable at the inception of the agreement as the Company will be compensated separately for these activities, and it is uncertain at this time if the Company will ever be requested to perform these activities by Sun.
The Company determined that the license and the other activities and services do not have standalone value apart from the requirement for the Company to deliver commercial material. Sun has not been granted a license to manufacture BOW015, which prevents them from using the license and related services for their intended purpose without the involvement of the Company. Therefore, the deliverables are not separable and represent one single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, the Company bases its revenue recognition pattern on the last to be provided deliverable. As a result, no revenue was recognized until delivery of the commercial supply began. All amounts received from Sun were recorded as deferred revenue prior to the delivery of the commercial supply began.
The arrangement includes the delivery of commercial material and therefore does not qualify for the milestone method of revenue recognition as the agreement is not a research and development arrangement. As a result, upon achievement of a milestone, the amounts will be included in the total arrangement consideration. In January 2014, the Company invoiced Sun $125 related to the filing for approval in India. In March 2014, the Drug Controller General in India approved BOW015 for commercial sale to the Indian market, upon which a payment from Sun of $250 became due and payable and was invoiced in April 2014. In September 2014, the Company executed a supply agreement with Sun, upon which an additional milestone payment of $125 became due.
On September 9, 2014, the Company entered into an amendment (the “Sun License Amendment”) to the Sun License Agreement. The Sun License Amendment amended the Sun License Agreement to revise (i) the royalty tiers for net sales based on final product sale forecasts and supply costs in the territory, and (ii) Sun’s obligations with respect to minimum annual sales targets in any given country such that the obligations are subject to a final grant of approval for BOW015 by the applicable regulatory authority for all indications as have been granted for the innovator product in such country. Other than as set forth in the Sun License Amendment, the Sun License Agreement remains in full force and effect following the parties’ execution of the Sun License Amendment.
In September 2014, the Company and Sun entered into a supply agreement for BOW015 under which the Company, through its third party manufacturing supplier, will provide Sun with finished products for clinical use, regulatory use and commercialization in accordance with the Sun License Agreement. Under the terms of the supply agreement, the Company’s designated manufacturer will receive consideration from Sun for commercial finished
109
product. Unless terminated earlier in accordance with terms, the Company’s supply agreement with Sun expires 10 years from the effective date, subject to five two-year automatic renewal periods.
In November 2014, the Company launched InfimabTM (BOW015) and Sun completed the first commercial sale of Infimab in India, signifying commercial supply of product has commenced. Upon this event, an additional milestone of $250 became due and payable and the Company began to recognize revenue, over the remaining term of the Sun License Agreement, associated with the upfront and milestone payments which were initially recorded as deferred revenue, excluding $500 which will remain deferred until the previously discussed breach and termination right lapses. As subsequent milestones are achieved, these payments will be recognized as revenue over the remaining term of the Sun License Agreement, including the five two-year automatic renewal periods. Royalty revenues from sales of Infimab will be recognized one quarter in arrears in conjunction with the Company’s accounting policy (Note 2).
For the fiscal years ended December 31, 2015 and 2014, the Company recorded royalty revenue of $119 and $4, respectively, related to commercial sales under this agreement.
The Company has invoiced Sun for a total of $1,250 in milestone payments, including the upfront payment, and received payments totaling $1,250 through December 31, 2015.
Mabxience, S.A.
On May 13, 2015, the Company entered into an Exclusive License Agreement (the “MabX Agreement”) with mAbxience S.A., a company organized and existing under the laws of Uruguay and a wholly owned subsidiary of CHEMO Group (“Mabxience”), for the development, manufacturing and commercialization of BOW015.
Under the MabX Agreement, the Company licensed to Mabxience an exclusive, royalty-bearing, non-transferable, sublicenseable license to develop, manufacture, commercialize and distribute BOW015 in the Mabxience territory. The Mabxience territory consists of Argentina, Chile, Ecuador, Paraguay, Uruguay and Venezuela. Pursuant to the MabX Agreement, Mabxience will be solely responsible, at its expense, for all aspects of commercialization of licensed product in the Mabxience territory. As consideration for the license granted to Mabxience, the Company is eligible to receive certain non-refundable, non-creditable milestone payments up to $1,500 upon the achievement of certain commercial sales in the Mabxience territory. Specifically, the Company will earn $250 upon the first commercial sale for each first commercial sale in each of the territory countries. In addition, the Company is eligible to receive certain royalty payments in the high teen percentages and distribution payments from Mabxience.
These potential milestone and royalty payments have not been earned as of December 31, 2015.
Unless terminated earlier in accordance with the terms of the agreement or extended by mutual agreement of the parties, the MabX Agreement expires 15 years following the first commercial sale of BOW015 in the Mabxience territory.
Polpharma
On July 13, 2015, the Company entered into a Collaboration Agreement (the “Polpharma Collaboration Agreement”) with Swiss Pharma International AG, an affiliate of Pharmaceutical Works Polpharma S.A. (together with Swiss Pharma International AG, “Polpharma”), for the development and commercialization in certain designated territories of the Company’s product candidates, BOW015, BOW050 and BOW070. The designated territories include the European Union (with the exception of Austria, Belgium, Denmark, Finland, Luxembourg, the Netherlands and Sweden, which are reserved for the Company), together with certain countries in the Middle East, Turkey, Russia, and other countries comprising the Commonwealth of Independent States, or CIS. The Company will also retain exclusive rights for the three products in North America and other markets outside of the designated territories, including Switzerland and Norway. Within the designated territories, the Polpharma Collaboration Agreement contains exclusivity provisions such that each of the Company and Polpharma agrees not to exploit any other biosimilar product based on reference product other than the three products licensed under the Polpharma Collaboration Agreement, subject to limited exceptions.
110
Under the Polpharma Collaboration Agreement, the Company and Polpharma have cross-licensed all intellectual property rights and technology relating to their applicable biosimilar development programs to enable the parties to develop and commercialize the three above-mentioned biosimilar products in the designated territories and the Company to develop and commercialize these biosimilar products outside of the designated territories. The three product programs will consist of process development, clinical trials, regulatory, manufacturing and commercialization activities in the designated territories. For such programs, the Company will lead the global product development and clinical programs and will also be responsible for process development, scale-up and manufacturing in the designated territories, both parties will collaborate on regulatory filings in the designated territories, and Polpharma will be responsible for the commercialization of the products in the designated territories. Polpharma has the right to be a second source manufacturer for any of the products, including a right to perform fill and finish production for all three products, subject to certain conditions. A joint management committee will oversee the collaboration on behalf of both parties.
Polpharma will bear 51% and the Company will bear 49% of total costs associated with the development programs for the three products in the designated territories. For clinical trial costs incurred in connection with global development, the cost sharing ratio is changed from 100% to 38% of total costs. As a result, Polpharma will bear 19% and the Company will bear 81% of total clinical trial costs incurred. Following commercial launch, profits and losses for the three products in the designated territories will be split 51% for Polpharma and 49% for the Company.
For the fiscal year ended December 31, 2015, the Company recorded revenue of $235 related to this agreement. The Company invoiced and received a payment in the amount of $760 from Polpharma in November 2015. As of December 31, 2015, the Company had a deferred revenue balance of $525 related to the payment received for services that will be performed in the first quarter of 2016.
Livzon Mabpharm
In September 2014, the Company entered into an Exclusive License and Collaboration Agreement (the “Livzon Agreement”) with Livzon Mabpharm Inc. (“Livzon”) for the global development and commercialization of certain antibodies or related biological compounds, including the Company’s BOW015, a biosimilar version of infliximab.
Under the Livzon Agreement, the Company and Livzon each agreed to grant the other party, in the other party’s territory, exclusive, royalty-bearing licenses under certain patent rights and know-how to develop, manufacture and commercialize BOW015 and up to four additional compounds chosen by mutual agreement of the parties (collectively, the “Collaboration Compounds”). Livzon’s territory consists of China, Hong Kong, Macau and Taiwan (the “Livzon Territory”), and the Company’s territory consists of the rest of the world. The parties share pre-clinical development expenses for each Collaboration Compound based on certain factors specific to each compound. Each party bears the responsibility and expenses for clinical development and commercialization of each Collaboration Compound in its territory. Livzon will be the preferred supplier of each Collaboration Compound for pre-clinical, clinical, and commercialization purposes, subject to Livzon’s satisfaction of certain performance criteria and subject to the negotiation of a separate supply agreement.
As consideration for the license granted to Livzon to develop and commercialize BOW015, the Company is eligible to receive from Livzon a nonrefundable milestone payment of $2,500 upon the achievement of a specified regulatory milestone in the Livzon Territory. The Company is also eligible to receive from Livzon tiered royalties based on a percentage of net sales of BOW015 products in the Livzon Territory ranging from the low- to high-single digits. Any future Collaboration Compounds have cross-milestone and royalty obligations in amounts to be mutually agreed upon at a later date.
If not terminated earlier or extended by mutual agreement of the parties, the Livzon Agreement expires in its entirety 20 years from the effective date.
The Company determined that the substantive terms of the Livzon Agreement are those relating to the license granted to Livzon to develop and commercialize BOW015 and the related Technology Transfer, as defined in the Agreement, which is the know-how to develop and manufacture BOW015 as described above. Under the Livzon
111
Agreement, Livzon is responsible for all activities related to the commercialization of BOW015 in the Livzon Territory. The Company is responsible for the Technology Transfer and a small amount of advisory services to Livzon.
The Company has determined that the deliverables under the Livzon Agreement are the license, Technology Transfer and a small amount of advisory services. Additional obligations that may arise under the Livzon Agreement are not deliverables at the inception of the Livzon Agreement as the Company will be compensated separately for these activities, and it is uncertain at this time if the Company will ever be requested to perform these activities by Livzon.
The Company determined that the license, Technology Transfer and advisory services do not have standalone value. The Technology Transfer is required to enable Livzon to manufacture BOW015. Without such capability, Livzon would not be able to use the license and services for their intended purpose without the involvement of the Company. Therefore, the deliverables are not separable and represent one single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, the Company bases its revenue recognition pattern on the last to be provided deliverable.
The Company determined that the specified regulatory milestone is not substantive due to the lack of development activities required from the Company subsequent to the execution of the Livzon Agreement. As a result of the factors above, no revenue will be recognized until the milestone is achieved and the $2,500 will be recognized as revenue after the milestone is achieved, over the remaining period of the Technology Transfer. As of December 31, 2014, no amounts have been paid and no amounts have become due and payable under the Livzon Agreement.
On September 24, 2015, the Company entered into a New Collaboration Compound Supplement to the Collaboration Agreement (the “Livzon Supplement Agreement”) with Livzon. The Livzon Supplement Agreement amends the Livzon Agreement to add the Company’s product candidate, BOW070, a biosimilar version of Actemra® (tocilizumab), as one of the additional compounds to be developed and commercialized pursuant to the terms of the Livzon Agreement.
Under the Livzon Supplement Agreement, the Company and Livzon each granted the other party, in the other party’s territory, exclusive, royalty-bearing licenses under certain patent rights and know-how to develop, manufacture and commercialize BOW070. Livzon’s territory consists of China, Hong Kong, Macau and Taiwan, and the Company’s territory consists of the rest of the world. Livzon will be responsible for certain pre-clinical development activities, including analytical and process development, characterization work and formulation development, with the costs for such pre-clinical development activities to be borne by both parties, and each party will be responsible at its sole expense for clinical development, regulatory filings and approvals, and commercialization of BOW070 in each such party’s territory. Livzon will be a preferred supplier of BOW070 for clinical and commercialization purposes, subject to Livzon’s satisfaction of certain performance criteria. As part of such preferred supplier designation, the parties have agreed to certain minimum supply price and purchase commitments following commercial launch of BOW070.
The Company will pay a total of $4,500 to Livzon to complete the pre-clinical development activities, consisting of: (i) an initial cash payment of $1,500 within 10 days of entry into the Livzon Supplement Agreement, which amount was paid on October 10, 2015, and (ii) three additional cash payments of $1,000 each upon the achievement of certain pre-clinical development milestones. The Company is expensing costs under the Supplement Agreement, as incurred, over the period that the work is performed (currently estimated at 18 months). For the fiscal year ended December 31, 2015, the Company recorded expense of $500 under the Livzon Supplement Agreement.
Each of the parties is eligible to receive from the other party royalty payments, based on gross margin of BOW070 in the other party’s territory, ranging from the low- to mid-single digit percentages.
As of December 31, 2015, Livzon owned approximately 6.4% of the Company’s issued and outstanding common stock.
112
4. Merger
As described in Note 1, “Organization,” on July 15, 2014, the Company completed the Merger with Zalicus. Pursuant to the Merger Agreement, each share of capital stock of Private Epirus was exchanged for 0.13259 shares of Public Epirus Common Stock (the “Exchange Ratio”). All Private Epirus stock options granted under the Private Epirus stock option plans (whether or not then exercisable) that were outstanding prior to the effective time of the Merger, and all warrants to purchase Private Epirus capital stock that were outstanding prior to the effective time of the Merger were exchanged at the same ratio as described below. Upon the consummation of the Merger, equity holders of Private Epirus were issued an aggregate of 10,288,180 shares of Public Epirus Common Stock, plus 1,152,042 and 21,996 shares of Public Epirus Common Stock which could be issued in the future upon exercise of stock options and warrants, respectively.
After the Effective Time, all outstanding and unexercised Private Epirus stock options assumed by Zalicus may be exercised solely for shares of Public Epirus Common Stock and all outstanding and unexercised warrants to purchase Private Epirus capital stock may be exercised solely for shares of Public Epirus Common Stock. The number of shares of Public Epirus Common Stock subject to each Private Epirus stock option or warrant assumed by Zalicus was determined by multiplying (a) the number of shares of Private Epirus common stock that were subject to such Private Epirus stock option or warrant, as in effect immediately prior to the Effective Time, by (b) the exchange ratio, as defined in the merger agreement, and rounding the resulting number down to the nearest whole number of shares of Public Epirus Common Stock. The per share exercise price for the Public Epirus Common Stock issuable upon exercise of each Private Epirus stock option or warrant assumed by Zalicus was determined by dividing (a) the per share exercise price of Private Epirus common stock subject to such Private Epirus stock option or warrant, as in effect immediately prior to the Effective Time, by (b) the Exchange Ratio and rounding the resulting exercise price up to the nearest whole cent.
All Zalicus equity awards granted prior to the Merger remained legally outstanding.
The Merger has been accounted for as a reverse acquisition under the acquisition method of accounting with Private Epirus treated as the accounting acquirer and Zalicus treated as the acquired company for financial reporting purposes. Private Epirus was determined to be the accounting acquirer based upon the terms of the Merger and other factors, such as relative voting rights and the composition of the combined company’s board of directors and senior management. Accordingly, the Zalicus tangible and identifiable intangible assets acquired and liabilities assumed were recorded at fair value as of the date of acquisition, with the excess consideration transferred recorded as goodwill.
See Note 15, “Stock-Based Compensation,” for additional details regarding the accounting treatment for the equity awards of Private Epirus and Zalicus.
The acquisition-date fair value of the consideration transferred is as follows:
|
Number of shares of Public Epirus Common Stock owned by Zalicus stockholders (1) |
|
|
2,635,871 |
|
|
Multiplied by the price per share of Public Epirus Common Stock (2) |
|
$ |
11.80 |
|
|
Fair value of shares of Public Epirus Common Stock owned by Zalicus stockholders |
|
$ |
31,104 |
|
|
Acquisition-date fair value of assumed Zalicus equity awards attributable to precombination services (3) |
|
$ |
220 |
|
|
Total consideration transferred |
|
$ |
31,324 |
|
|
(1) |
The stock transferred in the table above is comprised of 2,610,871 common shares outstanding at the time of the Merger plus 25,000 restricted stock units (RSUs) that vested immediately upon the closing of the Merger. The fair value of shares of Public Epirus Common Stock owned by Zalicus shareholders of $31,104 includes approximately $1 related to fractional shares resulting from the Reverse Stock Split. |
|
(2) |
The shares outstanding are multiplied by the closing trading price of Public Epirus Common Stock as of the Merger date. |
113
|
(3) |
Consideration transferred includes approximately $220 of Zalicus equity awards assumed and deemed attributable to precombination services to Zalicus. The fair value of the equity awards at July 15, 2014 was determined using a Black-Scholes option-pricing model with the following ranges of assumptions: exercise price of $14.70 per share to $591.00 per share; underlying stock value of $11.80 per share; expected volatility of 66.02%; expected term of 0.1 years to 6.0 years; and risk-free interest rate of 1.91%. |
The following table summarizes the estimated fair values of the assets acquired and liabilities assumed at the date of acquisition:
|
|
|
July 15, 2014 |
|
|
|
Cash and cash equivalents |
|
$ |
13,756 |
|
|
In-process research and development |
|
|
5,500 |
|
|
Goodwill |
|
|
14,127 |
|
|
Restricted cash |
|
|
1,850 |
|
|
Other assets |
|
|
253 |
|
|
Total assets acquired |
|
|
35,486 |
|
|
Accounts payable |
|
|
(1,584) |
|
|
Accrued expenses |
|
|
(381) |
|
|
Other current liabilities |
|
|
(376) |
|
|
Other liabilities |
|
|
(391) |
|
|
Deferred tax liabilities |
|
|
(1,430) |
|
|
Total liabilities assumed |
|
|
(4,162) |
|
|
Total net assets acquired |
|
$ |
31,324 |
|
The purchase price allocation was prepared on a preliminary basis and was subject to change as additional information becomes available concerning the tax basis of the assets acquired and the liabilities assumed. All adjustments to the purchase price allocation were made within one year from July 15, 2014, the acquisition date.
For acquired working capital accounts such as accounts receivable, prepaid expenses and other current assets, accounts payable and certain accrued expenses, the Company determined that no fair value adjustments were required due to the short timeframe until settlement for these assets and liabilities.
The fair value of the IPR&D was determined using the multi-period excess earnings method, a variation of the income approach, including a discount rate of 13.1% applied to the projected cash flows. The Company believes the assumptions used are consistent and representative of those a market participant would use in estimating the fair value of the IPR&D. The Company will not begin amortizing the IPR&D asset until the research and development is complete and the asset is reclassified to a definite-lived amortizing asset.
Goodwill is calculated as the difference between the acquisition-date fair value of the consideration transferred and the fair values of the assets acquired and liabilities assumed. The goodwill is not expected to be deductible for income tax purposes. Goodwill was recorded as an indefinite-lived asset and is not currently being amortized but is tested for impairment on an annual basis or when indications of impairment exist.
Other liabilities of $391 and $376 of other current liabilities relates to a sublease liability assumed for Zalicus’ Cambridge, Massachusetts facility. The liability represents the fair value of the difference between the total sublease payments and the total payments for the facility per the lease agreement. The original lease and the sublease agreement terminate in January 2017.
The deferred tax liability represents the temporary difference associated with the $5,500 value of the IPR&D asset, which is not deductible for tax purposes. During the year ended December 31, 2015, the Company completed its analysis of the tax basis of the IPR&D asset and reduced the preliminary deferred tax liability of $2,166 to $1,430 resulting in an adjustment to goodwill of $736.
114
The operating results of Zalicus for the period from July 16, 2014 to December 31, 2014, including an operating loss of $25, have been included in the Company’s consolidated financial statements as of and for the year ended December 31, 2014. During the year ended December 31, 2015, the operating results for Zalicus were immaterial.
The Company incurred a total of $5,779 in transaction costs in connection with the Merger, excluding Zalicus transaction costs, which were included in general and administrative expense within the consolidated statements of income for the year ended December 31, 2014. The following supplemental unaudited pro forma information presents the Company’s financial results as if the acquisition of Zalicus had occurred on January 1, 2013:
|
|
|
Year Ended |
|
||||
|
|
|
December 31, |
|
||||
|
|
|
2014 |
|
2013 |
|
||
|
|
|
|
|
|
|
|
|
|
Total revenues, net |
|
$ |
2,739 |
|
$ |
14,731 |
|
|
Net loss |
|
$ |
(32,410) |
|
$ |
(69,630) |
|
The above unaudited pro forma information was determined based on the historical GAAP results of the Company and Zalicus. The unaudited pro forma consolidated results are not necessarily indicative of what the Company’s consolidated results of operations actually would have been if the acquisition was completed on January 1, 2013. The unaudited pro forma consolidated net loss includes pro forma adjustments primarily relating to the following non-recurring items directly attributable to the business combination:
|
(1) |
Elimination of $8,673 of transaction costs for both the Company and Zalicus from the year ended December 31, 2014, and inclusion of these transaction costs in the year ended December 31, 2013; |
|
(2) |
Elimination of $355 of stock-based compensation expense related to the acceleration of vesting of previously unvested Zalicus awards in connection with the Merger from the year ended December 31, 2014, and inclusion of this charge in the year ended December 31, 2013; |
|
(3) |
Elimination of $1,980 of expense related to severance agreements from the year ended December 31, 2014, and inclusion of this charge in the year ended December 31, 2013; and |
|
(4) |
Elimination of $222 and $789 of other expense related to the change in the fair values of liability-classified warrants from the years ended December 31, 2014 and 2013, respectively, as the Company’s outstanding warrants were reclassified to equity in connection with their exchange in the Merger. |
5. Acquisition
On September 9, 2015, the Company entered into a stock purchase agreement (the “Stock Purchase Agreement”) whereby it acquired 100% of the voting interests of Bioceros Holding B.V. (“Bioceros”), enabling the Company to expand its biosimilar pipeline and vertically integrate product development capabilities.
Bioceros was a privately-held, Netherlands-based biopharmaceutical R&D company focused on the development of mAbs and generation of GMP-ready cell lines. From the Bioceros platform, the Company will expand its pipeline with the addition of three preclinical product candidates: BOW080, a proposed biosimilar to eculizumab (reference biologic Soliris®); BOW090, a proposed biosimilar to ustekinumab (reference biologic STELARA®); and BOW100, a proposed biosimilar to golimumab (reference biologic SIMPONI®). Soliris, marketed by Alexion Pharmaceuticals, is currently indicated to treat ultra-rare blood disorders, including paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS). SIMPONI and STELARA are marketed by Janssen Pharmaceuticals and indicated in inflammatory and immune mediated disorders.
The Company will pay a total of $14,100 in consideration consisting of: (i) an initial cash payment of $3,400 at closing, (ii) a second cash payment of $1,700, to be paid on the first anniversary of the closing date , (iii) an initial issuance of 788,960 shares of the Company’s common stock, $0.001 par value per share (the “Common Stock”), with a value of $4,000 using a 10 day pre-closing date average price for issuance of $5.07 (value of $4,568 on acquisition date)
115
and (iv) a second issuance of shares of Common Stock with a value of $5,000, calculated based on the Company’s stock price as set forth in the Stock Purchase Agreement, to be issued on the date that is six months after the closing date (“Second Installment Shares”) , subject to the Company’s setoff rights with regard to the second cash payment and Second Installment Shares. Of the Second Installment Shares, shares with an undiscounted value of $1,015 are to be issued to Bioceros key employees and are subject to those employees’ continued employment. In addition, the shareholders of Bioceros would receive a pro rata share of a net cash payout equal to the aggregate amount of: (a) Bioceros’ net working capital as the acquisition date; plus (b) revenue agreements signed from select Bioceros customers subsequent to the acquisition date and through December 31, 2015; in excess of (c) $1,200, calculated as set forth in the Stock Purchase Agreement. As of September 9, 2015, the Company estimated this amount to be $409.
Second Installment Shares
As described above, shares with an undiscounted value of $1,015 from the Second Installment Shares are to be issued to Bioceros key employees and are subject to those employees’ continued employment. Pursuant to the Stock Purchase Agreement, 75% of the shares to be issued to Bioceros key employees on March 9, 2016 in connection with the Second Installment Shares will be held in an escrow account and released in three equal installments 12, 18 and 24 months after the closing date, unless the employee is terminated for cause or voluntarily resigns prior to each release date. The other 25% will be issued as of March 9, 2016, unless the employee is terminated for cause or voluntarily resigns prior to this date. As a result, the consideration related to such $1,015 of shares was determined to be attributable to post-combination service to Epirus and will be recognized as stock-based compensation expense by the combined company as the shares vest. The Company recognized $126 of stock-based compensation expense related to the Second Installment Shares for the key employees in the consolidated statement of operations and comprehensive loss for the year ended December 31, 2015.
The acquisition-date fair value of the consideration transferred is as follows:
|
Initial equity consideration paid at closing |
|
|
|
|
|
Initial shares issued to Bioceros shareholders |
|
|
788,960 |
|
|
Price per Epirus share at issuance |
|
$ |
5.79 |
|
|
|
|
$ |
4,568 |
|
|
Initial cash consideration paid at closing |
|
|
3,400 |
|
|
Total consideration paid at closing |
|
|
7,968 |
|
|
|
|
|
|
|
|
Settlement of preexisting Epirus accounts payable to Bioceros (1) |
|
|
(87) |
|
|
Second Installment Shares, excluding key employee stock compensation expense (2) |
|
|
3,788 |
|
|
Net cash payout (3) |
|
|
409 |
|
|
Final payment of cash consideration (4) |
|
|
1,535 |
|
|
Preliminary estimated purchase price |
|
$ |
13,613 |
|
|
(1) |
Represents the settlement of preexisting accounts payable to Bioceros as a result of activity prior to the transaction. |
|
|
|
|
(2) |
Represents the fair value of the undiscounted $3,985 of Second Installment Shares to be paid and issued on March 9, 2016, which excludes the undiscounted $1,015 of shares to be issued to Bioceros key employees contingent upon the delivery of postcombination service to the Company, as described above. This amount has been recorded as a liability on the Company’s consolidated balance sheet, because it represents an obligation to issue a variable number of shares based on a fixed dollar amount of $3,985, and recorded at fair value utilizing a discount rate of 10.7%. |
|
|
|
|
(3) |
Represents the fair value of the net cash payout to be made pursuant to the Stock Purchase Agreement. This amount has been recorded as a contingent consideration liability on the Company’s consolidated balance sheet as of September 9, 2015 and recorded at fair value utilizing a discount rate of 10.7%. The amount of the net cash payout is dependent on revenue agreements signed from select Bioceros customers subsequent to the acquisition date and through December 31, 2015. |
116
|
|
|
|
(4) |
Represents the fair value of the final cash payment of $1,700, which, pursuant to the Stock Purchase Agreement, the Company will pay on September 9, 2016. This amount has been recorded as a liability on the Company’s consolidated balance sheet as of September 9, 2016 and recorded at fair value utilizing a discount rate of 10.7%. |
The following table summarizes the preliminary estimated fair values of the assets acquired and liabilities assumed at the date of acquisition:
|
|
|
(in thousands) |
|
|
|
Cash and cash equivalents |
|
$ |
889 |
|
|
Deferred tax assets |
|
|
161 |
|
|
Accounts receivable |
|
|
49 |
|
|
Prepaid expenses and other current assets |
|
|
226 |
|
|
Property and equipment, net |
|
|
447 |
|
|
Intangible assets, net |
|
|
3,191 |
|
|
Goodwill |
|
|
10,734 |
|
|
Accounts payable |
|
|
(146) |
|
|
Accrued expenses and other current liabilities |
|
|
(704) |
|
|
Deferred revenue |
|
|
(436) |
|
|
Deferred tax liabilities |
|
|
(798) |
|
|
Net assets acquired |
|
$ |
13,613 |
|
During the measurement period, the Company made adjustments to the provisional amounts reported as the estimated fair values of assets acquired and liabilities assumed as part of the Acquisition. Compared to the provisional values reported as of September 30, 2015, the fair values presented in the table above reflect increases to cash and cash equivalents of $56, intangible assets of $1,199, accounts payable of $25 and decreases to prepaid expenses, other current assets and accounts receivable of $112, goodwill of $1,478, accrued expenses of $232, deferred tax assets of $168 and deferred tax liability of $300.
As of December 31, 2015, the Company evaluated the net cash payout liability of $409 and reduced the fair value to $0. This reduction was recorded as a credit to general and administrative expenses in the Company’s consolidated statement of operations for the fiscal year ended December 31, 2015.
As of December 31, 2015, the liability associated with the Second Installment shares and the final cash payment were $3,909 and $1,584, respectively, and are presented on the consolidated balance sheet as accrued purchase price liability.
In March 2016, there was an amendment to the Stock Purchase Agreement (Note 22).
The purchase price allocation has been prepared on a preliminary basis and is subject to change as additional information becomes available concerning the fair value and tax basis of the assets acquired and the liabilities assumed. The purchase price allocation will remain preliminary until Epirus completes a final valuation of the assets acquired and liabilities assumed, including deferred tax assets and liabilities, deferred revenue and the identification and valuation of all intangible assets, as of the transaction closing date. The excess of consideration transferred over the estimated fair value of net identifiable assets acquired will be allocated to goodwill, which represents the expected synergies between the companies and acquired workforce. Goodwill is not expected to be deductible for tax purposes. The final determination of the allocation of consideration transferred is expected to be completed as soon as practicable, but will in no event exceed one year from September 9, 2015, the closing date. The final amounts allocated to assets acquired and liabilities assumed could differ significantly from the amounts presented above.
For acquired working capital accounts such as accounts receivable, prepaid expenses and other current assets, including deferred tax assets, accounts payable and certain accrued expenses, Epirus determined that no preliminary fair value adjustments were required due to the short timeframe until settlement for these assets and liabilities. The Company
117
acquired gross contractual accounts receivable of $49, which it determined represents fair value and expects to collect in full.
Intangible asset, or acquired know how technology, currently represent the preliminary estimated fair value for the CHOBC® cell line platform and cell line technology platform which will be the platform for future research. Based on the estimated time to commercialization and other factors, the intangible asset is being amortized over a six year expected useful life, which is the estimated period of time the Company expects to receive benefit from the platform during its development processes. The estimated fair value of the intangible was determined based on a cost approach. The present value of the investment costs was then determined utilizing an estimated risk-adjusted discount rate. The estimated fair value may be adjusted based upon the final valuation and any such adjustments could be significant. The final valuation is expected to be completed within one year from the acquisition closing date. If the platform becomes impaired or is abandoned, the carrying value of the related intangible asset will be written down to its fair value and an impairment charge will be recorded in the period in which the impairment occurs.
Goodwill represents the excess of the preliminary estimated purchase price over the preliminary estimated fair value of the assets acquired and liabilities assumed and recognizes results from the expected synergies from combining operations of the Bioceros technical team’s expertise and the Company’s existing pipeline and management expertise. The Company believes that the addition of Bioceros staff and capabilities, combined with existing in-house expertise, provides the Company with a strong technical foundation to accelerate product development and optimize product quality. Goodwill will not be amortized, but will be evaluated for impairment on an annual basis or more frequently if an event occurs or circumstances change that would more-likely-than-not reduce the fair value below its carrying amount. In the event that Epirus determines that the value of goodwill has become impaired, an impairment charge will be recorded in the period in which the determination is made.
Deferred revenue balances were preliminarily adjusted to amounts that represent the estimated costs to fulfill the underlying obligations plus a normal profit margin, consistent with what the Company determined to be a market participant’s estimate of such costs and profit margin.
The deferred tax liability relates to the temporary difference associated with the preliminary estimated value of the intangible asset and has been estimated using a 25% effective tax rate.
The operating results of Bioceros for the period from September 10, 2015 to December 31, 2015, include net revenue of $186 and net loss of $96 which have been included in the Company’s consolidated financial statements as of and for the year ended December 31, 2015.
The Company incurred a total of approximately $754 in transaction costs in connection with the transaction, excluding Bioceros transaction costs, which were included in selling, general and administrative expense within the consolidated statement of operations and comprehensive loss for the year ended December 31, 2015. The following supplemental unaudited pro forma information represents the Company’s financial results as if the acquisition had occurred on January 1, 2014.
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31, |
||||
|
|
|
2015 |
|
2014 |
||
|
|
|
|
|
|
|
|
|
Total revenues, net |
|
$ |
1,728 |
|
$ |
2,474 |
|
Net loss |
|
$ |
(50,985) |
|
$ |
(41,720) |
The above unaudited pro forma information was determined based on the historical GAAP results of the Company and Bioceros. The unaudited pro forma consolidated results are not necessarily indicative of what the Company’s consolidated results of operations and comprehensive loss actually would have been if the acquisition was completed on January 1, 2014. The unaudited pro forma consolidated net loss includes pro forma adjustments primarily relating to the following non-recurring items directly attributable to the business combination.
118
|
(1) |
Elimination of $964 transaction costs for both the Company and Bioceros from the year ended December 31, 2015, and inclusion of these transactions costs in the year ended December 31, 2014; |
|
(2) |
The adjustment for stock compensation expense that would have been charged assuming the key employees of Bioceros, increased the expense by $374 and $500 for the years ended December 31, 2015 and 2014, respectively; |
|
(3) |
To eliminate intercompany net revenue from Bioceros to Epirus recorded in Bioceros’ historical financial statements and the corresponding amount recorded as research and development expense in the Epirus historical financial statements; |
|
(4) |
To eliminate the income recorded related to the change in the net cash payout contingent consideration liability from the year ended December 31, 2015, and include this gain in the year ended December 31, 2014. |
6. Disposition
On October 1, 2015, the Company entered into and closed a Share Purchase Agreement (the “Purchase Agreement”) with Taro Pharmaceuticals Inc. (“Taro”) for the sale by the Company to Taro of all of the shares of Zalicus Pharmaceuticals Ltd., the Company’s Canadian subsidiary that holds the Company’s product candidate Z944 and certain related assets (collectively, the “Z944 Assets”) which the Company acquired as a result of its merger with Zalicus Inc. in July 2014 (the “Zalicus Ltd. Sale”).
As a result of the Zalicus Ltd. Sale, the Company received from Taro payment comprised of Canadian Dollar (“CAD”) $5,000 in cash and a non-interest bearing, limited recourse promissory note in the amount of CAD $5,000 with a maturity date of July 1, 2017 (the “Promissory Note”). The Promissory Note is repayable either in cash or through Taro’s transfer back of the Z944 Assets, including any further developments made by Taro to such assets, to the Company. No repayment of the Promissory Note is required until such maturity date. If Taro elects to repay the Promissory Note in cash and continue development of the Z944 Assets, the Company will also be entitled to additional payments of up to USD $7,500 upon achievement of certain development milestones for the Z944 Assets, up to USD $30,000 upon achievement of certain sales milestones for the Z944 Assets, and mid-single digit percentage royalty payments upon global commercial sales of products developed with the Z944 Assets, with such percentage to be reduced in the event of commercial sales of competing generic products under certain circumstances. The Purchase Agreement further requires Taro to use customary commercially reasonable efforts to develop the Z944 Assets, subject to the Company’s reversion right to such assets for any material breach following standard notice and cure periods.
During the year ended December 31, 2015, the Company recognized a gain on the Zalicus Ltd. Sale of $1,016 representing the difference between the carrying value of the net assets of Zalicus Pharmaceuticals Ltd. and the initial cash payment received.
The Company will account for the Promissory Note, milestone payments and royalty payments from the Purchase Agreement as a contingent gain to be recorded when realizable.
7. Prepaid and Other Current Assets
Prepaid expenses and other current assets consisted of the following:
|
|
|
As of December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
Prepaid development expenses |
|
$ |
2,281 |
|
$ |
457 |
|
|
Prepaid other expenses |
|
|
668 |
|
|
638 |
|
|
Accounts receivable |
|
|
130 |
|
|
4 |
|
|
Other current assets |
|
|
653 |
|
|
457 |
|
|
Total prepaids and other current assets |
|
$ |
3,732 |
|
$ |
1,556 |
|
119
8. Property and Equipment, Net
Property and equipment consisted of the following:
|
|
|
As of December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
|
|
|
|
|
|
|
|
|
Laboratory equipment |
|
$ |
1,268 |
|
$ |
— |
|
|
Leasehold improvements |
|
|
518 |
|
|
504 |
|
|
Computer equipment |
|
|
303 |
|
|
173 |
|
|
Furniture and fixtures |
|
|
284 |
|
|
116 |
|
|
Total property and equipment |
|
|
2,373 |
|
|
793 |
|
|
Less: accumulated depreciation |
|
|
(300) |
|
|
(89) |
|
|
Property and equipment, net |
|
$ |
2,073 |
|
$ |
704 |
|
Depreciation expense related to property and equipment amounted to $211 and $83 for the years ended December 31, 2015 and 2014, respectively.
9. Goodwill, IPR&D and Intangible Assets, Net
Goodwill
The following table represents changes in goodwill for the fiscal year ended December 31, 2015:
|
Balance at December 31, 2014 |
|
$ |
16,363 |
|
Adjustment for Zalicus deferred tax liability |
|
|
(736) |
|
Goodwill related to Bioceros acquisition |
|
|
10,734 |
|
Balance at December 31, 2015 |
|
$ |
26,361 |
As of December 31, 2015, there were no accumulated impairment losses associated with goodwill. Goodwill has been assigned to the Company’s single reporting unit, which is the single operating segment by which the CODM manages the Company (Note 2).
IPR&D
In connection with the Merger in 2014, the Company recognized IPR&D related to the Company’s product candidate Z944. The carrying value of the IPR&D related to Z944 as of December 31, 2014 was $5,500. See Note 4 “Merger” for additional details. This asset was sold on October 1, 2015 as part of a Share Purchase Agreement with Taro Pharmaceuticals, Inc. As a result, in September 2015, the Company performed an impairment analysis of this asset and recognized an impairment charge of $1,671 which was recorded within research and development expense in the Company’s consolidated statement of operations and reduced the carrying value of the IPR&D asset to $3,829. The Company determined the fair value of the IPR&D utilizing the income approach.
Intangibles
In November 2014, the Company received the necessary approvals to manufacture and market BOW015 in India. Upon such approval for commercialization in India, the $2,437 of IPR&D associated with BOW015 was reclassified from an indefinite-lived asset to a finite lived asset.
During the year ended December 31, 2014 the Company began amortizing the intangible asset over its expected useful life of approximately 16.5 years. The Company determined the useful life of the asset based upon the period over which the Company expects to earn future economic benefit for BOW015. The Company recognizes amortization expense utilizing the straight-line method.
120
In September 2014 and October 2014, the Company entered into an agreement with Moksha8 to terminate the Company’s revenue sharing payment obligations with respect to products that are biosimilars to infliximab, in exchange for payments of $1,400. In connection with the payments under the Amendment and Second Amendment, in September 2014, the Company recorded an intangible asset of $1,250, which will be amortized over the expected useful life of the intangible asset associated with BOW015, described above.
In connection with the Bioceros acquisition in September 2015, the Company recognized an intangible asset related to Bioceros’ platform technology. During the year ended December 31, 2015, the Company began amortizing the intangible asset over its expected useful life of approximately six years. The carrying value of the intangible asset related to the platform technology as of December 31, 2015 was $3,191. See Note 5, “Acquisition,” for additional details.
Intangible assets, net of accumulated amortization is as follows:
|
|
|
As of December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
Intangible assets (excluding IPR&D) |
|
$ |
6,878 |
|
$ |
3,687 |
|
|
Less: accumulated amortization—intangible assets |
|
|
(468) |
|
|
(82) |
|
|
Intangible assets, net (excluding IPR&D) |
|
$ |
6,410 |
|
$ |
3,605 |
|
Amortization expense, which is included within research and development expense in the consolidated statements of operations and comprehensive loss, was $386 and $82 for the years ended December 31, 2015 and 2014, respectively.
The estimated aggregate amortization of intangible assets as of December 31, 2015, for each of the five succeeding years and thereafter is as follows:
|
|
|
December 31, 2015 |
|
|
|
|
|
|
|
|
|
2016 |
|
$ |
757 |
|
|
2017 |
|
|
757 |
|
|
2018 |
|
|
757 |
|
|
2019 |
|
|
757 |
|
|
2020 |
|
|
757 |
|
|
2021 and thereafter |
|
|
2,625 |
|
|
Total |
|
$ |
6,410 |
|
10. Accrued Expenses
Accrued expenses consisted of the following:
|
|
|
As of December 31, |
||||
|
|
|
|
2015 |
|
2014 |
|
|
Accrued compensation |
|
$ |
2,057 |
|
$ |
1,244 |
|
Accrued professional fees |
|
|
1,502 |
|
|
1,822 |
|
Accrued interest expense |
|
|
264 |
|
|
71 |
|
Other |
|
|
180 |
|
|
88 |
|
Total accrued expenses |
|
$ |
4,003 |
|
$ |
3,225 |
121
11. Debt
2013 Notes
On March 10, 2014, the $12,500 principal amount of convertible notes outstanding at December 31, 2013, plus accrued but unpaid interest of $684, were converted into 13,184,383 shares of Series A convertible preferred stock.
March 2014 Notes
On March 10, 2014, the Company issued and sold $5,000 of convertible notes to finance the operations of the Company. The notes bore interest at a rate of 8% per annum, had a maturity date of April 30, 2014 and provided the holders of the notes with 20% warrant coverage.
On April 15, 2014, the $5,000 principal amount of convertible notes issued in March 2014, plus accrued and unpaid interest of $13, were converted into 3,947,363 shares of Series B Preferred and the warrants became exercisable into 787,402 shares of Series B Preferred at an exercise price per share of $0.01.
Hercules Notes
On September 30, 2014, the Company entered into a Loan and Security Agreement (the “Loan Agreement”) with Hercules Technology Growth Capital, Inc. (“Hercules”). Under the Loan Agreement, Hercules will provide the Company with access to two term loans with an aggregate principal amount of up to $15,000 (collectively, the "Loan"). The first term loan ("Tranche 1"), with a principal amount of $7,500, was funded on October 1, 2014. The second term loan ("Tranche 2"), with a principal amount of up to $7,500, was funded on May 29, 2015. The Loan is repayable in monthly installments until April 1, 2018, including an initial interest-only period of 18 months after funding of Tranche 1. Any amounts remaining outstanding at April 1, 2018 will become due immediately. The interest rate is a floating interest rate equal to the greater of (i) 7.95%, or (ii) the sum of (A) 7.95%, plus (B) the Prime Rate minus 3.25% per annum. An end of term charge equal to 3.00% of the aggregate original principal balance of the Loan is payable at maturity, including in the event of any prepayment, and is being accrued as interest expense over the term of the loan using the effective interest method. Borrowings under the 2014 Loan Agreement are collateralized by substantially all assets of the Company.
If the Company repays all or a portion of the term loans prior to maturity, it will pay Hercules a prepayment fee as follows: (i) 2.00% if the prepayment occurs in the first 18 months following the funding date and (ii) no fee if the prepayment occurs after the first 18 months following the funding date.
Pursuant to the Loan Agreement, subject to certain terms, Hercules was granted the right to participate, in an amount of up to $1,000, in subsequent sales and issuances of the Company’s equity securities to one or more investors for cash for financing purposes in an offering that is broadly marketed to multiple investors and at the same terms as the other investors.
In connection with the Loan Agreement, on September 30, 2014, the Company issued to Hercules a warrant to purchase 64,194 shares of Public Epirus Common Stock at an exercise price of $7.01 per share. In addition, the Company has the ability to settle up to $2.0 million of the Note with its common stock at a fixed price that is 15% above the warrant exercise price or 248,093 shares at $8.06.
See Note 12, “Warrants,” for further description of the warrant.
As of December 31, 2015, the Company had outstanding borrowings under the Loan Agreement of $15,000. Due to the going concern opinion issued as of December 31, 2015 and the fact that the Note may be declared immediately due by Hercules, upon the occurrence of a material adverse change in the Company’s business, the Company has recorded all amounts due under the Note as a short-term debt obligation. As of December 31, 2015, the short-term debt obligation was $14,833, net of a debt discount of $167. Amortization of the debt discount, which was
122
recorded as interest expense in the statement of operations, was approximately $89 and $14 for the years ended December 31, 2015 and December 31, 2014, respectively.
Future principal payments, which exclude the 3% end of term charge that will be payable upon repayment of the notes in full, in connection with the Loan Agreement, as of December 31, 2015 are as follows:
|
|
|
Principal Payments |
|
|
|
2016 |
|
$ |
3,693 |
|
|
2017 |
|
|
5,940 |
|
|
2018 |
|
|
5,367 |
|
|
Total |
|
$ |
15,000 |
|
12. Warrants
Prior to the Merger, Zalicus issued warrants to purchase common stock to certain third parties. Zalicus issued warrants to purchase 148 shares of common stock to General Electric Capital Corporation with an exercise price of $405.00 per share that expired on June 28, 2015 and warrants to purchase 1,079, 1,887 and 3,737 shares of common stock to Oxford at exercise prices of $83.40, $135.00 and $68.40 per share, respectively. The Oxford warrants expire on December 22, 2017.
In March 2014, the Company issued warrants, in connection with a convertible notes issuance, to purchase 1,000,000 shares of Series A Preferred with an exercise price of $0.01 per share (Note 11). Upon the Series B Preferred financing described in Notes 11 and 14, these warrants became exercisable into 787,402 shares of Series B Preferred at an exercise price of $0.01 per share. The aggregate fair value of such warrants at their issuance date was $1,171. Each warrant was immediately exercisable and expires approximately seven years from the original date of issuance. Each warrant was exercisable on either a physical settlement or net share settlement basis.
In April 2014, warrants to purchase 5,101,816 shares Series A Preferred were exercised into 5,101,816 shares of Series A Preferred. In April 2014, the warrants to purchase Series B Preferred were exercised into 787,402 shares of Series B Preferred.
In connection with the Merger on July 15, 2014, all of the Company’s outstanding warrants exercisable for shares of preferred stock were exchanged for warrants exercisable for shares of Public Epirus Common Stock, which resulted in the reclassification of the liability-classified warrants to additional paid-in capital.
As described in Note 11, the Company entered into a Loan Agreement with Hercules under which the Company was given access to up to $15,000 of term loans. As consideration for the Loan Agreement, on September 30, 2014, the Company issued Hercules warrant to purchase 64,194 shares of its common stock at an exercise price of $7.01 per share, which expires five years from issuance. The Warrant’s fair value of $248 at September 30, 2014 was determined using a Black‑Scholes option‑pricing model with the following assumptions: expected volatility of 61.68%; contractual term of five years; and risk‑free interest rate of 1.78%.
As of December 31, 2015 and 2014, all of the Company’s outstanding warrants were classified in equity. The following warrants to purchase common stock were outstanding:
123
|
|
|
As of December 31, 2015 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
Number of Shares |
|
|
|
|
|
|
|
Underlying Warrants |
|
Exercise Price Per Share |
|
|
|
Common Stock |
|
1,379 |
|
$ |
0.08 |
|
|
Common Stock |
|
64,194 |
|
$ |
7.01 |
|
|
Common Stock |
|
20,616 |
|
$ |
7.55 |
|
|
Common Stock |
|
3,737 |
|
$ |
68.40 |
|
|
Common Stock |
|
1,079 |
|
$ |
83.40 |
|
|
Common Stock |
|
1,887 |
|
$ |
135.00 |
|
|
|
|
As of December 31, 2014 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
Number of Shares |
|
|
|
|
|
|
|
Underlying Warrants |
|
Exercise Price Per Share |
|
|
|
Common Stock |
|
1,379 |
|
$ |
0.08 |
|
|
Common Stock |
|
64,194 |
|
$ |
7.01 |
|
|
Common Stock |
|
20,616 |
|
$ |
7.55 |
|
|
Common Stock |
|
3,737 |
|
$ |
68.40 |
|
|
Common Stock |
|
1,079 |
|
$ |
83.40 |
|
|
Common Stock |
|
1,887 |
|
$ |
135.00 |
|
|
Common Stock |
|
148 |
|
$ |
405.00 |
|
13. Fair Value Measurements
|
|
|
As of December 31, 2015 |
|
||||||||||
|
|
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
31,515 |
|
$ |
— |
|
$ |
— |
|
$ |
31,515 |
|
|
|
|
$ |
31,515 |
|
$ |
— |
|
$ |
— |
|
$ |
31,515 |
|
|
|
|
As of December 31, 2014 |
|
||||||||||
|
|
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
21,462 |
|
$ |
— |
|
$ |
— |
|
$ |
21,462 |
|
|
|
|
$ |
21,462 |
|
$ |
— |
|
$ |
— |
|
$ |
21,462 |
|
The reconciliation of the contingent consideration measured at fair value on a recurring basis using unobservable inputs (Level 3) is as follows:
|
Balance at December 31, 2014 |
|
$ |
— |
|
Addition of contingent consideration related to Bioceros acquisition |
|
|
409 |
|
Remeasurement of Bioceros contingent consideration |
|
|
(409) |
|
Balance at December 31, 2015 |
|
$ |
— |
The contingent consideration obligation represents the fair value of the net cash payout to former Bioceros shareholders, which is equal to the aggregate amount of: (a) Bioceros’ net working capital as the acquisition date; plus (b) revenue generated from Bioceros customers subsequent to the acquisition date and through December 31, 2015; in excess of (c) $1,200, calculated as set forth in the Stock Purchase Agreement.
124
The amount of the contingent consideration obligation is dependent on the amount of revenue generated by the Company from Bioceros customers through December 31, 2015. The estimates of fair value may not be indicative of the amounts that could be realized in a third-party exchange. Accordingly, the use of different market assumptions and/or different valuation techniques could result in materially different fair value estimates. The significant unobservable inputs to the valuation were management’s expectation for the amounts of the revenues generated from customers and a discount rate of 10.7%.
The fair value of the contingent consideration obligation recognized on the acquisition date was estimated by applying a risk adjusted discount rate of 10.7% to the potential revenue generated resulting from probability weighted revenue projections. The Company revised its estimate related to the net cash payout based on actual revenue generated from Bioceros customers and net working capital as of the acquisition date and determined it would be less than $1,200. Accordingly, the Company adjusted the fair value of the contingent consideration obligation to reflect a zero balance as of December 31, 2015. The Company does not anticipate to make a payment in connection with the net cash payout.
As of December 31, 2015 and 2014, the Company did not have any liabilities measured at fair value on a recurring basis.
14. Capital Stock
As of December 31, 2015, the Company had authorized 70,000,000 shares of common stock, $0.001 par value per share, of which 24,377,573 shares were issued and outstanding. Additionally, as of December 31, 2015, the Company had authorized 5,000,000 shares of preferred stock, $0.001 par value per share, of which zero were issued and outstanding.
As of June 4, 2015, the Company amended its certificate of incorporation to decrease the authorized capital stock of the Company from 305,000,000 to 75,000,000 shares, which included 70,000,000 shares of common stock, $0.001 par value per share and 5,000,000 shares of preferred stock, $0.001 par value per share.
Reserved for Future Issuance
The Company has reserved for future issuances the following number of shares of common stock:
|
|
|
Year Ended December 31, |
|
||
|
|
|
2015 |
|
2014 |
|
|
|
|
|
|
|
|
|
Stock-based compensation awards |
|
4,132,271 |
|
4,403,266 |
|
|
Warrants to purchase common stock |
|
92,892 |
|
93,040 |
|
|
Settlement of short-term debt (Note 11) |
|
248,093 |
|
248,093 |
|
|
Total |
|
4,473,256 |
|
4,744,399 |
|
In addition, Second Installment Shares issuable to former Bioceros shareholders with a value of $5,000 to be issued in March 2016 are reserved for future issuance as of December 31, 2015 (Note 5).
Convertible Preferred Stock
In April 2014, Epirus closed an initial Series B Preferred financing with an issuance of 24,409,444 shares for aggregate proceeds of approximately $31,000. Upon the closing of such financing, the $5,000 principal amount of convertible notes issued in March 2014, plus accrued and unpaid interest of $13, were converted into 3,947,363 shares of Series B Preferred (Note 11).
Upon the closing of the Merger, all outstanding shares of preferred stock were exchanged for shares of Public Epirus Common Stock as part of the Merger at the Exchange Ratio. See Note 4, “Merger,” for additional details. As of December 31, 2015, the Company did not have any convertible preferred stock issued or outstanding.
125
15. Stock-Based Compensation
On April 2, 2015, the board of directors of the Company adopted the 2015 Equity Incentive Plan (“2015 Plan”) and on June 4, 2015, the stockholders of the Company approved the 2015 Plan. Pursuant to the 2015 Plan, 1,700,000 shares of common stock were initially authorized for issuance. The 2015 Plan provides for the grant of incentive stock options, non-statutory stock options, stock appreciation rights, restricted and unrestricted stock and stock units, performance awards, cash-based awards and awards that are convertible into or otherwise based on the Company’s common stock. As of December 31, 2015, the Company had 394,402 shares available for issuance under the 2015 Plan.
The 2015 Plan includes an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2015 Plan, which annual increase will be added on the first day of each fiscal year from 2016 through 2025, inclusive, and will be equal to the least of (i) 1,000,000 shares of common stock, (ii) 4% of the outstanding shares on that date or (iii) such lesser amount determined by the Compensation Committee.
Private Epirus adopted the 2011 Equity Incentive Plan, as amended (“2011 Plan”), pursuant to which 740,470 shares of common stock were initially authorized for issuance to employees, officers, directors, consultants and advisors of the Private Epirus. The 2011 Plan provides for the grant of incentive stock options, non-statutory stock options, rights to purchase restricted stock, stock appreciation rights, phantom stock awards and stock units. In connection with the issuance of restricted common stock, Private Epirus maintains a repurchase right and shares of restricted common stock are released from such repurchase right over a period of time of continued service by the recipient. Recipients of incentive stock options shall be eligible to purchase shares of the Private Epirus common stock at an exercise price equal to no less than the estimated fair value of such stock on the date of grant. In April 2014, the Private Epirus board of directors amended the terms of the 2011 Plan to increase the authorized number of shares of common stock available for issuance under the 2011 Plan from 740,470 to 1,504,752. As described in Note 4, through the Merger, all outstanding and unexercised Private Epirus stock options under the 2011 Plan were assumed by Public Epirus under the 2004 Plan.
In December 2004, the board of directors and stockholders of Zalicus adopted the 2004 Incentive Plan ("2004 Plan"), which was effective on November 9, 2005. The 2004 Plan provides for the grant of incentive stock options, non-statutory stock options, restricted stock and unrestricted stock awards, stock appreciation rights, cash awards, performance awards and restricted stock units. Awards under the 2004 Plan may be granted to employees, directors, consultants and advisors.
In August 2014, the Company submitted a Form S-8 Registration Statement (“S-8") to the SEC. As reflected in the S-8, the Company amended the 2004 Plan to increase the number of shares available for issuance under the 2004 Plan by 3,000,000 shares of common stock. As of December 31, 2015, the Company had no shares available for issuance.
Terms of stock award agreements are determined by the board of directors, subject to the provisions provided in the 2015 Plan, the 2011 Plan and the 2004 Plan. Stock options generally vest over a four -year period: 25% on the first anniversary of the original vesting date, with the balance vesting monthly over the remaining three years, unless they contain specific performance and/or market-based vesting provisions. Options are exercisable over a maximum term of 10 years from the grant date.
For the years ended December 31, 2015 and 2014, the Company recognized stock-based compensation expense of approximately $3,470 and $1,719, respectively, in connection with its stock-based payment awards. The Company recorded a reduction in stock-based compensation expense for the year ended December 31, 2015 of $100 resulting from the modification of an award for a former officer of the Company, Stock-based compensation expense for the year ended December 31, 2014 includes $355 of postcombination stock-based compensation expense resulting from the acceleration of vesting of previously unvested Zalicus stock options and RSUs, which occurred upon the termination of certain Zalicus employees following the Merger, and $303 related to restricted stock issued to a non-employee.
As described in Note 4, through the Merger, outstanding Zalicus equity awards remained legally outstanding without change to any agreement terms. Such Zalicus equity awards were assumed to have been exchanged for equity awards of the Company, the accounting acquirer. As a result, the portion of the acquisition-date fair value of the Zalicus
126
equity awards attributable to precombination service to Zalicus, $220, was treated as a component of consideration transferred. See Note 4, “Merger.”
The exchange of the Private Epirus stock options for stock options to purchase Public Epirus Common Stock was treated as a modification of the awards. The modification of the stock options did not result in any incremental compensation expense as the modification did not increase the fair value of the stock options.
Stock Options
A summary of stock option activity during the year ended December 31, 2015 is as follows:
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
|
|
|
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
|
|
|
Weighted- |
|
Remaining |
|
|
|
|
|
|
|
|
|
|
|
Average |
|
Contractual |
|
Aggregate |
|
||
|
|
|
Number |
|
Exercise |
|
Term |
|
Intrinsic |
|
|||
|
|
|
of Options |
|
Price |
|
(in years) |
|
Value |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2014 |
|
|
1,652,882 |
|
$ |
10.03 |
|
8.9 |
|
|
1,628 |
|
|
Granted |
|
|
2,269,247 |
|
|
7.50 |
|
|
|
|
|
|
|
Exercised |
|
|
(130,920) |
|
|
2.71 |
|
|
|
|
|
|
|
Cancelled |
|
|
(177,782) |
|
|
7.83 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2015 |
|
|
3,613,427 |
|
$ |
8.81 |
|
8.8 |
|
$ |
485 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested and expected to vest at: December 31, 2015 |
|
|
3,438,732 |
|
$ |
8.90 |
|
8.8 |
|
$ |
482 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at: December 31, 2015 |
|
|
699,459 |
|
$ |
14.73 |
|
7.7 |
|
$ |
316 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average grant-date fair value of options granted during the period |
|
$ |
4.86 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash received upon exercise of options |
|
$ |
355 |
|
|
|
|
|
|
|
|
|
The aggregate intrinsic value in the table above represents the value (the difference between the Company’s common stock fair value on December 31, 2015 and the exercise price of the options, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their options on December 31, 2015. The total intrinsic value of options exercised in the years ended December 31, 2015 and 2014 was $547 and $71, respectively. As of December 31, 2015, there was $11,284 of total unrecognized stock-based compensation expense related to stock options granted under the plans. The expense is expected to be recognized over a weighted-average period of 3.10 years.
In order to determine the fair value of each employee stock award on the grant date, the Company used the Black-Scholes option-pricing model. During the years ended December 31, 2015 and 2014, respectively, the range of assumptions used in the Black-Scholes pricing model for new grants and grants issued or accelerated in connection with the Merger were as follows:
|
|
|
Year Ended December 31, |
|
|
||
|
|
|
2015 |
|
2014 |
|
|
|
|
|
|
|
|
|
|
|
Risk-free interest rate |
|
1.48% - 1.96% |
|
1.80% - 2.09% |
|
|
|
Expected volatility |
|
61.40% - 89.05% |
|
63.96% - 66.07% |
|
|
|
Expected term (in years) |
|
6.00 |
|
6.00 |
|
|
|
Expected dividend yield |
|
0.0% |
|
0.0% |
|
|
127
Risk-Free Interest Rate. The risk-free interest rate assumption is based on observed interest rates appropriate for the expected term of the stock option grants.
Expected Volatility. Due to the Company’s limited operating history and lack of Company-specific historical or implied volatility, the expected volatility assumption is based on historical volatilities of a peer group of similar companies whose share prices are publicly available. The peer group was developed based on companies in the biotechnology and medical device industries.
Expected Term. The expected term represents the period of time that options are expected to be outstanding. The Company utilizes the expected terms from an analysis of peer group companies to support the expected term used in the Black-Scholes model, as it does not believe it has sufficient historical data to support this assumption.
Expected Dividend Yield. The expected dividend yield assumption is based on the fact that the Company has never paid cash dividends and has no intention to pay future cash dividends.
Restricted Stock Award
In April 2014, the Company granted 53,036 shares of restricted common stock to a non-employee for consulting services. The restricted stock award is subject to certain performance-based vesting conditions under which 25% of the award vested on April 15, 2014, 50% vested on September 25, 2014 and the remaining 25% of the award would vest following the successful achievement of certain performance milestones and the passage of time.
As of December 31, 2015, the Company had no outstanding shares of nonvested restricted common stock. As of December 31, 2014, the Company had outstanding 13,259 shares of nonvested restricted common stock at a weighted average grant date fair value of $5.08. During the years ended December 31, 2015 and 2014, the Company recorded $58 and $98, respectively, in stock compensation expense related to the nonvested restricted common stock award.
A summary of the restricted stock activity during the year ended December 31, 2015 is as follows:
|
|
|
|
|
Weighted- |
|
|
|
|
|
Restricted |
|
Average Grant |
|
|
|
|
|
Stock |
|
Date Fair Value |
|
|
|
|
|
|
|
|
|
|
|
Nonvested at December 31, 2014 |
|
13,259 |
|
$ |
5.08 |
|
|
Granted |
|
— |
|
|
|
|
|
Vested |
|
(13,259) |
|
|
5.08 |
|
|
Cancelled |
|
— |
|
|
— |
|
|
Nonvested at December 31, 2015 |
|
— |
|
$ |
— |
|
128
RSUs
A summary of the activity of nonvested restricted stock units (“RSUs”) for the year ended December 31, 2015 is as follows:
|
|
|
|
|
|
|
|
Weighted- |
|
|
|
|
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
Number |
|
|
Weighted- |
|
Remaining |
|
|
|
|
|
|
|
of |
|
|
Average |
|
Contractual |
|
|
Aggregate |
|
|
|
|
Restricted |
|
|
Grant Date |
|
Term |
|
|
Intrinsic |
|
|
|
|
Stock Units |
|
|
Fair Value |
|
(in years) |
|
|
Value |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nonvested at December 31, 2014 |
|
— |
|
$ |
— |
|
— |
|
$ |
— |
|
|
Granted |
|
126,621 |
|
|
10.53 |
|
|
|
|
— |
|
|
Vested |
|
— |
|
|
— |
|
— |
|
|
|
|
|
Cancelled |
|
(2,179) |
|
|
10.53 |
|
— |
|
|
|
|
|
Nonvested at December 31, 2015 |
|
124,442 |
|
$ |
10.53 |
|
2.9 |
|
$ |
385 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested and expected to vest at: |
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2015 |
|
116,964 |
|
$ |
10.53 |
|
2.9 |
|
$ |
361 |
|
|
Exercisable at: |
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2015 |
|
— |
|
$ |
— |
|
— |
|
$ |
— |
|
As of December 31, 2015, there was $972 of total unrecognized stock-based compensation expense related to nonvested RSUs. The expense is expected to be recognized over a weighted-average period of 2.93 years.
During February 2015, the Company issued 117,131 RSUs to certain employees, which vest with respect to one quarter (1/4) of the RSUs on the first anniversary of the grant date and an additional one quarter (1/4) on each anniversary thereafter until the fourth anniversary of the grant date. In addition, the Company issued 9,490 RSUs to certain employees as merit grants in lieu of annual raises. These RSUs vest in full on the first year anniversary of the grant date.
Employee Stock Purchase Plan
The Company has an Employee Stock Purchase Plan (“ESPP”) that permits eligible employees to enroll in a six-month offering period. Participants may purchase shares of the Company’s Common Stock, through payroll deductions, at a price equal to 85% of the fair market value of the Common Stock on the first day of the applicable six-month offering period, or the last day of the applicable six-month offering period, whichever is lower. Purchase dates under the ESPP occur on or about June 1 and December 1 of each year.
During the year ended December 31, 2015, the Company recorded stock-based compensation expense of $4. As of December 31, 2015, 352,722 shares of the Company’s Common Stock remain available for issuance under the ESPP. As of December 31, 2015, there was $21 of total unrecognized stock-based compensation expense related to the ESPP. The expense is expected to be recognized over a period of five months.
129
The fair value of the option component of the shares purchased under the ESPP was estimated using the Black-Scholes option-pricing model with the following weighted-average assumptions:
|
|
|
Year Ended December 31, |
|
|
|
2015 |
|
|
|
|
|
Risk-free interest rate |
|
0.42% |
|
Expected volatility |
|
76.01% |
|
Expected term (in years) |
|
0.50 |
|
Expected dividend yield |
0.0% |
16. Income Taxes
During 2013, the Company realized a deferred tax benefit as a result of the transfer of intellectual property to the Company’s subsidiary, Epirus Biopharmaceuticals (Switzerland) GmbH, a Swiss Corporation. However, because the transfer is an intercompany transaction and is eliminated in consolidation, the Company cannot recognize a tax benefit. As a result, the deferred tax benefit of $943 related to the sale of intellectual property was deferred and was presented on the balance sheet as a deferred tax benefit at December 31, 2013. It is being recognized in earnings as the intellectual property asset is amortized in Switzerland over a period of five years, which is the expected tax amortization period for the intellectual property.
For the year ended December 31, 2015, the Company has recorded an income tax benefit of approximately $544, which is primarily due to a tax benefit of approximately $185 from the amortization of the Company’s deferred tax benefit related to the 2013 transfer of intellectual property described above and approximately $434 of in-process research & development impairment in Canada and offset by approximately $37 of withholding tax in Switzerland and $32 in income tax in the Netherlands.
The component of U.S. and foreign loss from continuing operations before income taxes are as follows:
|
|
|
Year Ended December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
(6,145) |
|
$ |
(16,149) |
|
|
Foreign |
|
|
(46,587) |
|
|
(25,738) |
|
|
Loss before income taxes: |
|
$ |
(52,732) |
|
$ |
(41,887) |
|
130
Deferred taxes are recognized for temporary differences between the basis of assets and liabilities for financial statement and income tax purposes. The Company’s deferred tax assets and liabilities are comprised of the following:
|
|
|
As of December 31, |
|
||||
|
|
|
2015 |
|
2014 |
|
||
|
|
|
|
|
|
|
|
|
|
Deferred tax assets |
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Accrued rent |
|
$ |
- |
|
$ |
208 |
|
|
Accrued expenses |
|
|
- |
|
|
539 |
|
|
Gross current deferred tax assets |
|
|
- |
|
|
747 |
|
|
|
|
|
|
|
|
|
|
|
Non-current assets: |
|
|
|
|
|
|
|
|
Net operating loss - U.S. |
|
$ |
14,261 |
|
$ |
11,660 |
|
|
Research and development credits |
|
|
1,362 |
|
|
573 |
|
|
License/start-up expenditures |
|
|
205 |
|
|
222 |
|
|
Accrued rent |
|
|
125 |
|
|
111 |
|
|
Accrued expenses and other |
|
|
642 |
|
|
— |
|
|
Stock based compensation |
|
|
941 |
|
|
147 |
|
|
Capitalized research and development/patent intangibles |
|
|
4,645 |
|
|
5,611 |
|
|
Capitalized research and developmet RBIL Limitation |
|
|
714 |
|
|
— |
|
|
Foreign investment tax credit |
|
|
— |
|
|
4,370 |
|
|
Net operating loss - foreign |
|
|
16,458 |
|
|
35,546 |
|
|
Gross non-current deferred tax assets |
|
$ |
39,353 |
|
$ |
58,240 |
|
|
Gross Deferred tax assets |
|
$ |
39,353 |
|
$ |
58,987 |
|
|
|
|
|
|
|
|
|
|
|
Deferred tax liabilities |
|
|
|
|
|
|
|
|
Non-current: |
|
|
|
|
|
|
|
|
Depreciation |
|
|
(14) |
|
|
(14) |
|
|
Swiss IP amortization |
|
|
(153) |
|
|
(85) |
|
|
Bioceros acquired intangibles |
|
|
(757) |
|
|
0 |
|
|
Identifiable intangible assets - IPR&D |
|
|
- |
|
|
(2,166) |
|
|
Total non-current deferred tax liabilities |
|
$ |
(924) |
|
$ |
(2,265) |
|
|
Gross deferred taxes |
|
$ |
38,429 |
|
$ |
56,722 |
|
|
Valuation allowance |
|
|
(39,077) |
|
|
(58,888) |
|
|
Net deferred tax liability |
|
$ |
(648) |
|
$ |
(2,166) |
|
The Company has recorded a net deferred tax liability of $648 related to the acquired intangible assets, other accrual liabilities and deferred revenue in the Netherlands. The Company evaluated the positive and negative evidence bearing upon the realizability of the deferred tax assets. Based on the Company’s history of operating losses, the Company has concluded that it is more likely than not that the benefit of its deferred tax assets will not be realized in all jurisdictions except the Netherlands. The Netherlands is in a net deferred tax liability position as of December 31, 2015. The valuation allowance increase during the year ended December 31, 2015 is primarily due to the increase in the current year net operating losses offset by the disposition of Canadian deferred balances as a result of the Company’s sale of its Canadian entity.
131
A reconciliation of income tax expense computed at the statutory federal income tax rate to income taxes as reflected in the financial statements is as follows:
|
|
|
Year Ended December 31, |
||||
|
|
|
2015 |
|
2014 |
||
|
Income tax benefit using U.S. federal statutory rate |
|
$ |
(17,929) |
|
$ |
(14,241) |
|
State income taxes, net of federal benefit |
|
|
(270) |
|
|
(296) |
|
Other permanent differences |
|
|
(304) |
|
|
2,985 |
|
Foreign rate differential |
|
|
6,615 |
|
|
3,836 |
|
Tax credits |
|
|
(795) |
|
|
(223) |
|
Other items |
|
|
(811) |
|
|
503 |
|
Tax benefit related to the IPR&D transfer |
|
|
(185) |
|
|
(185) |
|
IPR&D impairment charge benefit |
|
|
(434) |
|
|
— |
|
Foreign withholding taxes |
|
|
37 |
|
|
101 |
|
Net change in valuation allowance |
|
|
13,532 |
|
|
7,477 |
|
Income tax benefit |
|
$ |
(544) |
|
$ |
(43) |
As of December 31, 2015 and 2014, the Company had U.S. federal net operating loss carryforwards of approximately $38.8 million and $31.0 million, respectively and state net operating loss carryforwards of approximately $27.6 million and $21.3 million, respectively, which may be available to offset future income tax liabilities and expire at various dates through 2035. In addition, as of December 31, 2015 and 2014, the Company had foreign net operating loss carryforwards of approximately $82 million and $145 million, respectively, which may be available to offset future income tax liabilities. The decrease in foreign net operating losses is primarily due to the loss of net operating losses as a result of the Company’s sale of its Canadian entity offset by the current year loss in Switzerland. Net operating loss carryforwards are unlimited in the United Kingdom and Brazil, Switzerland allows for a seven year carryforward.
The Company had generated NOL carryforwards from stock-based compensation deduction in excess of expense recognized for financial reporting purposes (“excess tax benefit”). Excess tax benefits are realized when they reduce the taxes payable, as determined by using a “with and without” method, and are credited to additional paid-in capital, rather than a reduction of the income tax provision. As of December 31, 2015 and 2014, the Company had approximately $21 and $77 excess tax benefits, respectively, which will be credited to additional paid-in capital when realized.
As of December 31, 2015 and 2014, the Company had federal research and development tax credit carryforwards of approximately $1.2 million and $0.5 million, respectively, available to reduce future tax liabilities which expire at various dates through 2035. As of December 31, 2015 and 2014, the Company had state research and development tax credit carryforwards of approximately $0.2 million and $0.2 million, respectively, available to reduce future tax liabilities, which expire at various dates through 2030.
Under the provision of the Internal Revenue Code, the net operating loss and tax credit carryforwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. Net operating loss and tax credit carryforwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three‑year period in excess of 50%, as defined under Sections 382 and 383 of the Internal Revenue Code, respectively, as well as similar state provisions. This could limit the amount of tax attributes that can be utilized annually to offset future taxable income or tax liabilities. The amount of the annual limitation is determined based on the value of the company immediately prior to the ownership change. Subsequent ownership changes may further affect the limitation in future years. The federal and state net operating loss carryforward and tax credit carryforward are net of the limitation due to Sections 382 and 383. A full valuation allowance has been provided against the net operating loss and tax credit carryforwards as of December 31, 2015.
For years through December 31, 2015, the Company generated research credits but has not conducted a study to document the qualified activities. This study may result in an adjustment to the Company’s research and development credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented
132
as an uncertain tax position. A full valuation allowance has been provided against the Company’s research and development credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the deferred tax asset established for the research and development credit carryforwards and the valuation allowance.
The Company will recognize interest and penalties related to uncertain tax positions in income tax expense. As of December 31, 2015 and 2014, the Company had no accrued interest or penalties related to uncertain tax positions and no amounts have been recognized in the Company’s statements of operations and comprehensive loss.
The Company files income tax returns in the U.S., various state jurisdictions and foreign jurisdictions for its foreign subsidiaries. The federal and state income tax returns are generally subject to tax examinations for the tax years ended 2012 through 2015. The foreign income tax returns are generally subject to examinations for the tax years ended 2011 through 2015. To the extent the Company has tax attribute carryforwards, the tax years in which the attribute was generated may still be adjusted upon examination by the Internal Revenue Service, state or foreign tax authorities to the extent utilized in a future period. There are no currently ongoing or pending examinations in any jurisdiction.
During November 2015, the FASB issued ASU 2015-17, “Balance Sheet Classification of Deferred Taxes”, which simplifies the presentation of deferred income taxes. This ASU requires that deferred tax assets and liabilities be classified as non-current in a statement of financial position. The standard is effective for public companies for fiscal years beginning after December 15, 2016, including interim periods within that reporting period. Early adoption is permitted for any interim and annual financial statements that have not yet been issued. The Company early adopted ASU 2015-17 effective December 31, 2015 on a prospective basis. Adoption of this ASU resulted in a reclassification of the net current deferred tax asset of $109 and the net current deferred tax liability of $80 to the net non-current deferred tax liability in the Company’s consolidated balance sheet as of December 31, 2015. No prior periods were retrospectively adjusted.
17. Commitments
On May 15, 2015, the Company amended its March 2013 operating lease for office space in Boston, Massachusetts (the “Second Amendment”). The Second Amendment provides the Company additional square footage of approximately 3,000 square feet. The Company has committed to lease this space for a period of six years. The Second Amendment contains rent escalation that is being accounted for as rent expense under the straight-line method. The Company will record rent expense of approximately $14 per month on a straight-line basis over the effective lease term.
On September 9, 2015, the Company acquired Bioceros, located in Utrecht, The Netherlands. As part of the acquisition, the Company acquired a lease for approximately 5,800 square feet consisting of lab and office space. The lease is for 7 years, commencing on January 1, 2011 and ending December 31, 2018. The Company will record approximately $25 per month on a straight-line basis over the effective lease term.
On October 15, 2015, the Company signed a lease for approximately 4,400 square feet of office space in Zug, Switzerland. The Company has committed to lease this space through March 31, 2017 and will record approximately $17 per month on a straight-line basis over the effective lease term.
As of December 31, 2015, future minimum lease payments under all of the Company’s operating leases are as follows:
133
|
|
|
December 31, 2015 |
|
|
|
2016 |
|
$ |
2,399 |
|
|
2017 |
|
|
1,092 |
|
|
2018 |
|
|
644 |
|
|
2019 |
|
|
655 |
|
|
2020 |
|
|
666 |
|
|
2021 and thereafter |
|
|
464 |
|
|
Total |
|
$ |
5,920 |
|
|
Less: Future sublease payments to be received |
|
|
(1,124) |
|
|
Net future lease payments |
|
$ |
4,796 |
|
Rent expense under operating lease agreements amounted to approximately $727 and $366 for the years ended December 31, 2015 and 2014, respectively. Rent expense is reported net of sublease income.
License Agreements
From time to time, the Company enters into various licensing agreements whereby the Company may use certain technologies in conjunction with its product research and development.
Catalent Pharma Solutions, LLC
In January 2009, Moksha8 entered into a cell line sale agreement (“the Cell Line Agreement”) with Catalent Pharma Solutions (“Catalent”) for the acquisition of a gene expression cell line for BOW015 (“the GPEx Cell Line,”) developed by Catalent for Moksha8 pursuant to a separate development and manufacturing agreement dated July 14, 2008. The Cell Line Agreement was assigned to the Company on May 14, 2009, and was amended July 31, 2009 to revise certain payment-related terms. Under the terms of the Cell Line Agreement, the Company exercised an option to acquire all rights in the GPEx Cell Line for development and commercialization of products using the GPEx Cell Line, namely BOW015, subject to certain obligations to make contingent payments to Catalent.
The Company paid Catalent approximately $100 on execution of the Cell Line Agreement, and paid a further approximately $100 upon exercise of its option to complete the purchase of the GPEx Cell Line. The Company is required to make additional payments to Catalent of up to approximately $700 in the aggregate upon the achievement of certain development and regulatory milestones. In March 2013, the Company paid approximately $200 to Catalent upon the occurrence of certain clinical trial events for BOW015. Pursuant to the agreement, the Company will be obligated to pay approximately $500 upon the achievement of certain development and regulatory milestones. In addition, the Company will be required to pay a contingent sale fee in the form of royalties on worldwide net sales of BOW015 and any other product manufactured using the GPEx Cell Line at a percentage in the very low single digits for a period of 20 years following the first commercial sale of such product, and thereafter at a rate of less than one percent.
Either the Company or Catalent may terminate the Cell Line Agreement on 60 days' notice for the other party's material breach of the agreement, or for the other party's insolvency, and in the event of Catalent's termination for the Company’s material breach of the Cell Line Agreement, the Company’s ownership rights in the GPEx Cell Line would revert to Catalent. If the Company terminates the Cell Line Agreement for Catalent's breach, it will retain ownership of the GPEX Cell Line, but its payment obligations to Catalent will terminate.
18. Net Loss Per Share
The Company computes basic and diluted loss per share using a methodology that gives effect to the impact of outstanding participating securities (the “two-class method”). As the years ended December 31, 2015 and 2014 resulted in net losses, there is no income allocation required under the two-class method or dilution attributed to weighted-average shares outstanding in the calculation of diluted loss per share.
134
The following potentially dilutive securities outstanding, prior to the use of the treasury stock method, have been excluded from the computation of diluted weighted-average shares outstanding for the years ended December 31, 2015 and 2014, as they would be anti-dilutive. In addition, Second Installment Shares issuable to former Bioceros shareholders with a value of $5,000 to be issued in March 2016 have been excluded from the computation of basic and diluted weighted-average shares outstanding for the year ended December 31, 2015 (Note 5).
|
|
|
As of December 31, |
|
||
|
|
|
2015 |
|
2014 |
|
|
Options to purchase common stock |
|
3,613,427 |
|
1,652,882 |
|
|
Settlement of loan agreement |
|
248,093 |
|
248,093 |
|
|
Nonvested restricted stock units |
|
124,442 |
|
— |
|
|
Warrants to purchase common stock |
|
92,892 |
|
93,040 |
|
|
Employee stock purchase plan |
|
2,871 |
|
— |
|
|
Nonvested restricted stock awards |
|
— |
|
13,259 |
|
19. Employee Benefits
Effective January 1, 2013, the Company adopted a defined contribution 401(k) plan for employees who are at least 21 years of age. Employees are eligible to participate in the plan beginning on the first day of the calendar quarter following their date of hire. Under the terms of the plan, employees may make voluntary contributions as a percentage of compensation. As of December 31, 2015, the Company has not made any matching contributions since the adoption of the 401(k) plan.
20. Related Parties
As discussed in Note 3, in September 2014, the Company entered into an Exclusive License and Collaboration Agreement (the “Livzon Collaboration Agreement”) with Livzon Mabpharm Inc. (“Livzon”) for the global development and commercialization of certain antibodies or related biological compounds, including the Company’s BOW015, a biosimilar version of infliximab. In September 2015, the Company entered into a New Collaboration Compound Supplement with Livzon to add BOW070, a biosimilar version of tocilizumab, as a compound to be governed by the Livzon Collaboration Agreement. As of December 31, 2015, Livzon owned approximately 6.4% of the Company’s issued and outstanding common stock.
As discussed in Note 3, in September 2014 and October 2014, the Company entered into a series of amendments (collectively, the “amendments”) to that certain Revenue and Negotiation Rights Agreement between the Company and Moksha8 Pharmaceuticals, Inc. (“Moksha8”), dated as of December 31, 2010 (the “Moksha8 Revenue Agreement”). Moksha8 is owned in part by Montreux Equity Partners and TPG Capital, which are also shareholders of the Company. As of December 31, 2015, Montreux Equity Partners owned approximately 1% of the Company’s issued and outstanding common stock and TPG Capital owned approximately 9% of the Company’s issued and outstanding common stock.
21. Legal Proceedings
From time to time, the Company is involved in legal proceedings and claims of various types and accounts for these legal proceedings in accordance with ASC Topic 450, “Contingencies.” The Company records a liability in its consolidated financial statements for these matters when a loss is considered probable and the amount of loss can be reasonably estimated. The Company reviews these estimates each accounting period as additional information is known and adjusts the loss provision when appropriate. If the loss is not probable or cannot be reasonably estimated, a liability is not recorded in its consolidated financial statements.
Reliance Life Sciences Dispute
In December 2014, Reliance Life Sciences Pvt Ltd, or RLS, the Company’s BOW015 contract manufacturer for India, exercised its three-year termination for-convenience right with respect to the parties’ Manufacturing and Supply
135
Agreement, dated as of May 14, 2014 and as amended from time to time (the “RLS Agreement”), which would have caused the RLS Agreement to terminate in December 2017. In addition, in March 2015, RLS initiated an arbitration proceeding under the RLS Agreement alleging that the RLS Agreement grants RLS exclusive global supply rights for BOW015, that the terms of the Company’s collaboration agreements with Fujifilm Diosynth Biotechnologies U.S.A., Inc. and Livzon MabPharm Inc. violate the RLS Agreement, and sought to terminate the RLS Agreement. The proceeding initiated by RLS sought a declaratory judgment, but did not seek any monetary damages.
On April 22, 2015, the Company entered into a Settlement Agreement with RLS (the “Settlement Agreement”). Pursuant to the Settlement Agreement, the parties finalized the remaining BOW015 batch production schedule with RLS agreeing to manufacture 15 final batches of BOW015 on or before June 30, 2016 with the Company’s option to have RLS manufacture an additional 5 batches on or before December 31, 2016. In January 2016, the Company amended the Settlement Agreement (Note 22). RLS has also agreed to use reasonable commercial efforts to transfer the existing Indian marketing authorization for BOW015 granted by the Drug Controller General of India (“DCGI”) to the Company or its designee, with all costs of the transfer to be borne by the Company and subject to the DCGI’s approval of such transfer. The Company has agreed to pay to RLS a fee of approximately $2,250, payable in four installments over the course of the final batch manufacture, with the final installment to be paid no later than ten days after June 30, 2016. Pursuant to the Settlement Agreement, on May 4, 2015, RLS withdrew the arbitration proceeding it initiated, and the parties have agreed to release each other from all causes of actions or claims, present and future, arising under the RLS Agreement. Following the satisfaction by both parties of all of the terms of the Settlement Agreement, the RLS Agreement and related agreements between the parties shall be terminated and neither party shall have any further rights or obligations thereunder. The Company made the first payment of $750 on April 27, 2015 and made an additional payment of $500 on September 10, 2015.
For the year ended December 31, 2015, the Company recorded a charge of $2,228 to general and administrative expenses, which represents the net present value of the installment payments due under the Settlement Agreement.
In addition, the Company recorded a one-time charge of $1,800 to research and development expense in the year ended December 31, 2015, which represents the Company’s portion of the purchase obligation of the 15 binding forecasted batches to be manufactured by RLS. As of December 31, 2015, the Company has paid $561 towards this obligation.
22. Subsequent Events
In January 2016, the Company entered into an amendment to the Settlement Agreement with RLS to extend the time for RLS to manufacture the 15 final batches from June 30, 2016 to June 30, 2017.
On February 29, 2016, the Company entered into an At-the-Market Sales Agreement (the “Sales Agreement”) with BTIG, LLC (“BTIG”), pursuant to which the Company may sell at its option from time to time up to an aggregate of $30 million in shares of the Company’s common stock, through BTIG, as sales agent (the “Offering”). Sales of the common stock made pursuant to the Sales Agreement, if any, will be made under the Company’s previously filed and currently effective Registration Statement on Form S-3 (File No. 333-205420) and a related prospectus supplement.
The Company will pay BTIG a commission equal to 3% of the gross proceeds from the sale of shares of
common stock under the Sales Agreement, if any. Pursuant to the terms of the Sales Agreement, the Company also provided BTIG with customary indemnification rights. The Company has also agreed to reimburse BTIG for its reasonable out-of-pocket expenses, including the fees and disbursements of counsel to BTIG, incurred in connection with the Offering. The Company intends to use the net proceeds from the Offering for general corporate purposes, which may include research and development expenditures, clinical trial expenditures, commercialization activities and working capital. As of March 25, 2016, proceeds under this Sales Agreement have not been material.
On March 8, 2016, the Company entered into the First Amendment to the Stock Purchase Agreement between the Company and seller representatives of Bioceros (the “First Amendment”). The First Amendment removes any future employment requirements of Bioceros key employees to receive the Second Installment Shares, removes the lock-up period for the Second Installment Shares and updates the maximum equity consideration to be issued pursuant to the
136
Stock Purchase Agreement from 10% to 19.99% of the aggregate number of the Company’s total shares outstanding on the date preceding the date of the Stock Purchase Agreement.
On March 9, 2016, the Company issued 1,772,107 shares of common stock with a 10 day trailing price of $2.82, in accordance with the Stock Purchase Agreement and the First Amendment.
137
EPIRUS BIOPHARMACEUTICALS, INC.
|
|
|
Incorporated by Reference to: |
|||
|
Exhibit |
Description |
Form or |
Exhibit No. |
Filing Date |
SEC File |
| 2.1 |
Stock Purchase Agreement Regarding the Sale and Transfer of All Outstanding Shares in Bioceros Holding B.V., dated as of September 9, 2015, by and among the registrant, the Sellers identified therein and Oscar Schoots and Carine van den Brink as the representatives of the Sellers |
8-K |
2.1 |
09/09/2015 |
000-51171 |
| 3.1 |
Sixth Amended and Restated Certificate of Incorporation of the registrant |
S-1/A |
3.2 |
11/04/2005 |
333-121173 |
| 3.2 |
Certificate of Amendment to the Sixth Amended and Restated Certificate of Incorporation of the registrant |
8-K |
3.1 |
12/21/2009 |
000-51171 |
| 3.3 |
Certificate of Amendment to the Sixth Amended and Restated Certificate of Incorporation of the registrant |
8-K |
3.1 |
09/09/2010 |
000-51171 |
| 3.4 |
Certificate of Amendment to the Sixth Amended and Restated Certificate of Incorporation of the registrant |
8-K |
3.1 |
10/02/2013 |
000-51171 |
| 3.5 |
Certificate of Amendment to the Sixth Amended and Restated Certificate of Incorporation of the registrant |
8-K |
3.1 |
07/15/2014 |
000-51171 |
| 3.6 |
Certificate of Amendment to the Sixth Amended and Restated Certificate of Incorporation of the registrant |
8-K |
3.2 |
07/15/2014 |
000-51171 |
| 3.7 |
Certificate of Amendment to the Sixth Amended and Restated Certificate of Incorporation of the registrant |
8-K |
3.1 |
06/09/2015 |
000-51171 |
| 3.8 |
Second Amended and Restated By-Laws of the registrant |
8-K |
3.1 |
07/18/2014 |
000-51171 |
| 4.1 |
Specimen certificate representing the common stock of the registrant |
10-Q |
4.1 |
08/11/2014 |
000-51171 |
| 4.2 |
Form of Warrant to purchase shares of the registrant's common stock, dated December 22, 2010, by and between the registrant and Oxford Finance Corporation |
8-K |
4.1 |
12/22/2010 |
000-51171 |
| 4.3 |
Form of Warrant to purchase shares of the registrant's common stock, dated June 27, 2011, by and between the registrant and Oxford Finance Corporation |
10-K |
4.4 |
03/31/2015 |
000-51171 |
| 4.4 |
Schedule of Warrants substantially identical to the Form of Warrant filed as Exhibit 4.3 |
10-K |
4.5 |
03/31/2015 |
000-51171 |
| 4.5 |
Warrant to Purchase Series A Preferred Stock, No. PSW-5, dated as of January 25, 2011, by and between the registrant and Dee Athwal |
10-Q |
4.2 |
08/11/2014 |
000-51171 |
138
| 4.6 |
Warrant to Purchase Series A Preferred Stock, No. PSW-9, dated as of April 3, 2012, by and between the registrant and 5AM Ventures III, L.P. |
10-Q |
4.3 |
08/11/2014 |
000-51171 |
| 4.7 |
Warrant to Purchase Series A Preferred Stock, No. PSW-11, dated as of April 3, 2012, by and between the registrant and 5AM Co-Investors III, L.P. |
10-Q |
4.4 |
08/11/2014 |
000-51171 |
| 4.8 |
Warrant to Purchase Series A Preferred Stock, No. PSW-16, dated as of April 3, 2012, by and between the registrant and Dee Athwal |
10-Q |
4.5 |
08/11/2014 |
000-51171 |
| 4.9 |
Warrant to Purchase Series A Preferred Stock, No. PSW-17, dated as of April 3, 2012, by and between the registrant and Dee Athwal |
10-Q |
4.6 |
08/11/2014 |
000-51171 |
| 4.10 |
Warrant to Purchase Series A Preferred Stock, No. PSW-22, dated as of August 21, 2012, by and between the registrant and Dee Athwal |
10-Q |
4.7 |
08/11/2014
|
000-51171 |
| 4.11 |
Warrant Agreement, dated as of September 30, 2014, by and between the registrant and Hercules Technology Growth Capital, Inc. |
8-K |
4.1 |
10/03/2014 |
000-51171 |
|
+10.1 |
2011 Equity Incentive Plan of the registrant |
S-4 |
10.5 |
05/08/2014 |
333-195818 |
|
+10.2 |
Form of Stock Option Agreement for awards granted under the 2011 Equity Incentive Plan of the registrant |
S-4 |
10.6 |
05/08/2014 |
333-195818 |
|
+10.3 |
Amended and Restated 2004 Incentive Plan of the registrant |
S-8 |
4.1 |
04/16/2010 |
333-166118 |
|
+10.4 |
Form of Incentive Stock Option Agreement under the 2004 Incentive Plan |
10-K |
10.4 |
03/31/2015 |
000-51171 |
|
+10.5 |
Form of Non-Qualified Option Agreement under the 2004 Incentive Plan |
10-K |
10.5 |
03/31/2015 |
000-51171 |
|
+10.6 |
Form of Restricted Stock Unit Agreement under the 2004 Incentive Plan |
10-K |
10.6 |
03/31/2015 |
000-51171 |
|
+10.7 |
2015 Equity Incentive Plan of the registrant |
S-8 |
99.1 |
07/01/2015 |
333-205418 |
|
+10.8 |
Form of Incentive Stock Option Agreement under the 2015 Equity Incentive Plan |
|
|
|
* |
|
+10.9 |
Form of Non-Qualified Option Agreement under the 2015 Equity Incentive Plan |
|
|
|
* |
|
+10.10 |
Form of Restricted Stock Unit Agreement under the 2015 Equity Incentive Plan |
|
|
|
* |
|
+10.11 |
2015 Employee Stock Purchase Plan of the registrant |
S-8 |
99.2 |
07/01/2015 |
333-205418 |
| 10.12 |
Form of Indemnification Agreement of the registrant |
8-K |
10.1 |
12/21/2009 |
000-51171 |
|
+10.13 |
Letter Agreement, by and between the registrant and Mark Corrigan dated as of May 27, 2014 |
8-K |
10.2 |
07/18/2014 |
000-51171 |
|
+10.14 |
Letter Agreement, by and between the registrant and William Hunter dated as of May 22, 2014 |
8-K |
10.3 |
07/18/2014 |
000-51171 |
139
|
+10.15 |
Letter Agreement, by and between the registrant and Julie McHugh dated as of July 1, 2014 |
8-K |
10.4 |
07/18/2014 |
000-51171 |
|
+10.16 |
Amended and Restated Non-Employee Director Compensation Policy of the registrant |
|
|
|
* |
|
+10.17 |
Offer Letter, dated as of May 17, 2012, by and between the registrant and Amit Munshi |
10-Q |
10.7 |
08/11/2014 |
000-51171 |
|
+10.18 |
Offer Letter, dated as of April 29, 2013, by and between the registrant and Thomas Shea |
10-Q |
10.9 |
08/11/2014 |
000-51171 |
|
+10.19 |
Offer Letter, dated as of April 28, 2014, by and between the registrant and Robert Ticktin |
10-Q |
10.10 |
08/11/2014 |
000-51171 |
|
+10.20 |
Offer Letter, dated as of June 15, 2012, by and between the registrant and Michael Wyand |
10-Q |
10.11 |
08/11/2014 |
000-51171 |
|
+10.21 |
Letter Agreement from the registrant to Amit Munshi, effective as of April 16, 2014 |
10-Q |
10.12 |
08/11/2014 |
000-51171 |
|
+10.22 |
Letter Agreement from the registrant to Thomas Shea, effective as of April 16, 2014 |
10-Q |
10.14 |
08/11/2014 |
000-51171 |
|
+10.23 |
Letter Agreement from the registrant to Michael Wyand, effective as of April 16, 2014 |
10-Q |
10.15 |
08/11/2014 |
000-51171 |
|
+10.24 |
Severance Plan of the registrant |
8-K |
10.1 |
01/26/2015 |
000-51171 |
|
+10.25 |
Employment Agreement, dated as of December 1, 2015, by and between the registrant and Vincent Aurentz |
|
|
|
* |
| 10.26 |
Lease Agreement, by and between the registrant and CPT One Exeter Plaza, LLC, dated as of March 8, 2013 |
10-Q |
10.16 |
08/11/2014 |
000-51171 |
| 10.27 |
First Amendment to Lease Agreement, by and between the registrant and CPT One Exeter Plaza, LLC, dated as of May 15, 2014 |
10-Q |
10.6 |
11/10/2014 |
000-51171 |
| 10.28 |
Second Amendment to Lease Agreement, by and between EB Sub, Inc. and CPT One Exeter Plaza, LLC, dated as of May 29, 2015 |
10-Q |
10.2 |
08/12/2015 |
000-51171 |
| 10.29 |
Loan and Security Agreement, dated as of September 30, 2014, by and between the registrant and Hercules Technology Growth Capital, Inc. |
8-K |
10.1 |
10/03/2014 |
000-51171 |
|
#10.30 |
License Agreement, dated as of January 3, 2014, by and between the registrant and Sun Pharmaceutical Industries Ltd. (formerly known as Ranbaxy Laboratories Limited) |
10-Q |
10.6 |
08/11/2014 |
000-51171 |
| 10.31 |
Amendment No. 1 to Sun-Epirus License Agreement, dated as of September 1, 2014, by and between the registrant and Sun Pharmaceutical Industries Ltd. |
10-Q |
10.1 |
11/10/2014 |
000-51171 |
| 10.32 |
Exclusive License and Collaboration Agreement, dated as of September 24, 2014, by and between the registrant and Livzon Mabpharm Inc. |
10-Q |
10.4 |
11/10/2014 |
000-51171 |
| 10.33 |
Settlement Agreement dated as of April 22, 2015 by and between the registrant and Reliance Life Sciences Pvt Ltd |
10-Q |
10.2 |
05/13/2015 |
000-51171 |
140
|
#10.34 |
Collaboration Agreement, dated as of July 13, 2015, by and between the registrant and Swiss Pharma International AG |
10-Q |
10.1 |
11/16/2015 |
000-51171 |
|
#10.35 |
New Collaboration Compound Supplement to the Collaboration Agreement, dated as of September 24, 2015, by and between the registrant and Livzon Mabpharm Inc. |
10-Q |
10.3 |
11/16/2015 |
000-51171 |
| 10.36 |
Share Purchase Agreement, dated as of October 1, 2015, by and between the registrant and Taro Pharmaceuticals Inc. regarding Zalicus Pharmaceuticals Ltd. |
|
|
|
* |
| 21.1 |
Subsidiaries of the registrant |
|
|
|
* |
| 23.1 |
Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm |
|
|
|
* |
| 31.1 |
Certification of the Chief Executive Officer, as required by Section 302 of the Sarbanes‑Oxley Act of 2002 |
|
|
|
* |
| 31.2 |
Certification of the Chief Financial Officer, as required by Section 302 of the Sarbanes‑Oxley Act of 2002 |
|
|
|
* |
| 32.1 |
Certifications of the Chief Executive Officer and Chief Financial Officer, as required by 18 U.S.C. 1350 |
|
|
|
** |
| 101 |
The following financial information from this annual report on Form 10-K for the fiscal year ended December 31, 2015, formatted in XBRL (eXtensible Business Reporting Language) and furnished electronically herewith: (i) Consolidated Balance Sheets as of December 31, 2015 and December 31, 2014; (ii) Consolidated Statements of Operations for the years ended December 31, 2015 and 2014; (iii) Consolidated Statements of Comprehensive Loss for the years ended December 31, 2015 and 2014; (iv) Consolidated Statements of Changes in Stockholders' Equity for the years ended December 31, 2015 and 2014; (v) Consolidated Statements of Cash Flows for the years ended December 31, 2015 and 2014; and (vi) Notes to the Consolidated Financial Statements |
|
|
|
* |
* Filed herewith.
**Furnished herewith.
# Confidential treatment has been granted with respect to certain provisions of this exhibit.
+ Indicates management contract or compensatory plan.
141