Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 CONSENT OF INDEPENDANT REGISTERED PUBLIC ACCOUNTING FIRM - GILEAD SCIENCES INC | gild2015form10-kexhibit231.htm |
| EX-21.1 - EXHIBIT 21.1 LISTING OF SUBSIDIARIES - GILEAD SCIENCES INC | gild2015form10-kexhibit211.htm |
| EX-31.2 - CFO CERTIFICATION - GILEAD SCIENCES INC | gild2015form10-kexhibit312.htm |
| EX-31.1 - CEO CERTIFICATION - GILEAD SCIENCES INC | gild2015form10-kexhibit311.htm |
| EX-10.53 - EXHIBIT 10.53 GILEAD SCIENCE CORPORATE BONUS PLAN - GILEAD SCIENCES INC | gild2015form10-kexhibit1053.htm |
| EX-32.1 - SECTION 906 CERTIFICATIONS - GILEAD SCIENCES INC | gild2015form10-kexhibit321.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2015
or
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 0-19731
GILEAD SCIENCES, INC.
(Exact name of registrant as specified in its charter)
Delaware | 94-3047598 |
(State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) |
333 Lakeside Drive, Foster City, California | 94404 |
(Address of principal executive offices) | (Zip Code) |
Registrant's telephone number, including area code: 650-574-3000
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
Title of each class | Name of each exchange on which registered |
Common Stock, $0.001 par value per share | The Nasdaq Global Select Market |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT: NONE
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer x | Accelerated filer ¨ | Non-Accelerated filer ¨ | Smaller reporting company ¨ |
(Do not check if a smaller reporting company) | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant based upon the closing price of its Common Stock on the Nasdaq Global Select Market on June 30, 2015 was $140,034,139,655.*
The number of shares outstanding of the registrant's Common Stock on February 12, 2016 was 1,366,845,691.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant's proxy statement, which will be filed with the Commission pursuant to Regulation 14A in connection with the registrant's 2016 Annual Meeting of Stockholders, to be held on May 11, 2016, are incorporated by reference into Part III of this Report.
* Based on a closing price of $117.08 per share on June 30, 2015. Excludes 276,651,262 shares of the registrant's Common Stock held by executive officers, directors and any stockholders whose ownership exceeds 5% of registrant's common stock outstanding at June 30, 2015. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
GILEAD SCIENCES, INC.
2015 Form 10-K Annual Report
Table of Contents
PART I | ||
Item 1 | ||
Item 1A | ||
Item 1B | ||
Item 2 | ||
Item 3 | ||
Item 4 | ||
PART II | ||
Item 5 | ||
Item 6 | ||
Item 7 | ||
Item 7A | ||
Item 8 | ||
Item 9 | ||
Item 9A | ||
Item 9B | ||
PART III | ||
Item 10 | ||
Item 11 | ||
Item 12 | ||
Item 13 | ||
Item 14 | ||
PART IV | ||
Item 15 | ||
We own or have rights to various trademarks, copyrights and trade names used in our business, including the following: GILEAD®, GILEAD SCIENCES®, AMBISOME®, CAYSTON®, COMPLERA®, EMTRIVA®, EVIPLERA®, GENVOYA®, HARVONI®, HEPSERA®, LETAIRIS®, RANEXA®, RAPISCAN®, SOVALDI®, STRIBILD®, TRUVADA®, TYBOST®, VIREAD®, VITEKTA®, VOLIBRIS®, and ZYDELIG®. ATRIPLA® is a registered trademark belonging to Bristol-Myers Squibb & Gilead Sciences, LLC. LEXISCAN® is a registered trademark belonging to Astellas U.S. LLC. MACUGEN® is a registered trademark belonging to Eyetech, Inc. SUSTIVA® is a registered trademark of Bristol-Myers Squibb Pharma Company. TAMIFLU® is a registered trademark belonging to Hoffmann-La Roche Inc. This report also includes other trademarks, service marks and trade names of other companies.
This Annual Report on Form 10-K, including the section entitled “Management's Discussion and Analysis of Financial Condition and Results of Operations,” contains forward-looking statements regarding future events and our future results that are subject to the safe harbors created under the Securities Act of 1933, as amended (the Securities Act), and the Securities Exchange Act of 1934, as amended (the Exchange Act). Words such as “expect,” “anticipate,” “target,” “goal,” “project,” “hope,” “intend,” “plan,” “believe,” “seek,” “estimate,” “continue,” “may,” “could,” “should,” “might,” variations of such words and similar expressions are intended to identify such forward-looking statements. In addition, any statements other than statements of historical fact are forward-looking statements, including statements regarding overall trends, operating cost and revenue trends, liquidity and capital needs and other statements of expectations, beliefs, future plans and strategies, anticipated events or trends and similar expressions. We have based these forward-looking statements on our current expectations about future events. These statements are not guarantees of future performance and involve risks, uncertainties and assumptions that are difficult to predict. Our actual results may differ materially from those suggested by these forward-looking statements for various reasons, including those identified in Part I, Item 1A of this Form 10-K under the heading “Risk Factors.” Given these risks and uncertainties, you are cautioned not to place undue reliance on forward-looking statements. The forward-looking statements included in this report are made only as of the date hereof. Except as required under federal securities laws and the rules and regulations of the Securities and Exchange Commission (SEC), we do not undertake, and specifically decline, any obligation to update any of these statements or to publicly announce the results of any revisions to any forward-looking statements after the distribution of this report, whether as a result of new information, future events, changes in assumptions or otherwise.
2
PART I
ITEM 1. | BUSINESS |
Overview
Gilead Sciences, Inc. (Gilead, we or us), incorporated in Delaware on June 22, 1987, is a research-based biopharmaceutical company that discovers, develops and commercializes innovative medicines in areas of unmet medical need. With each new discovery and investigational drug candidate, we strive to transform and simplify care for people with life-threatening illnesses around the world. Gilead's primary areas of focus include human immunodeficiency virus (HIV), liver diseases such as chronic hepatitis C virus (HCV) infection and chronic hepatitis B virus (HBV) infection, cardiovascular, hematology/oncology and inflammation/respiratory. We have operations in more than 30 countries worldwide, with headquarters in Foster City, California. We continue to add to our existing portfolio of products through our internal discovery and clinical development programs and through a product acquisition and in-licensing strategy.
2015 Highlights
Over the past year, we worked to bring best-in-class drugs to market that advance the standard of care by offering enhanced modes of delivery, more convenient treatment regimens, improved resistance profiles, reduced side effects and greater efficacy. In the HIV area, we received approval from U.S. Food and Drug Administration (FDA) and the European Commission of Genvoya® (elvitegravir 150 mg/cobicistat 150 mg/emtricitabine 200 mg/tenofovir alafenamide 10 mg or E/C/F/TAF), a once-daily single tablet regimen for the treatment of HIV-1 infection. Two other TAF-based regimens are currently under evaluation by FDA and the European Medicines Agency (EMA). The first is an investigational, fixed-dose combination of emtricitabine 200 mg and tenofovir alafenamide 25 or 10 mg (F/TAF) for use in combination with other antiretroviral agents. The second is an investigational, once-daily single tablet regimen that combines emtricitabine 200 mg, tenofovir alafenamide 25 mg and rilpivirine 25 mg (R/F/TAF). In the liver diseases area, we received approval from FDA to expand the use of Harvoni® in patients with genotype 4, 5 and 6 chronic HCV infection and in patients co-infected with HIV. In addition, Harvoni plus ribavirin (RBV) for 12 weeks was approved as an alternate therapy to 24 weeks of Harvoni for treatment-experienced, genotype 1 patients with cirrhosis. We also submitted marketing applications to FDA and the EMA for the approval of a once-daily fixed-dose combination of sofosbuvir (SOF), approved as Sovaldi® in December 2013, and velpatasvir (VEL), an investigational pan-genotypic NS5A inhibitor, for the treatment of chronic genotype 1-6 HCV. If approved, SOF/VEL would become the first pan-genotypic, all-oral single tablet regimen for the treatment of HCV and would complement our current HCV portfolio of Sovaldi and Harvoni, offering high cure rates and the potential to simplify treatment and eliminate the need for HCV genotype testing. In the hematology/oncology area, we submitted supplemental new drug applications to FDA and the EMA for approval of Zydelig® (idelalisib) in combination with ofatumumab in previously-treated patients with chronic lymphocytic leukemia (CLL). Zydelig was originally approved in combination with rituximab for the treatment of certain patients with CLL, small lymphocytic lymphoma and follicular lymphoma, the most common type of indolent non-Hodgkin's lymphoma (iNHL). We also advanced our research and development pipeline, with 180 active clinical studies at the end of 2015, of which 61 were Phase 3 clinical trials.
In addition to advancing treatment options across therapeutic areas, we also enabled access to our medications for people who need them around the world. During 2015, we expanded our generic licensing agreements with our India-based manufacturing partners to include SOF/VEL, once approved, for distribution in developing countries. A pan-genotypic therapeutic option for the treatment of HCV is particularly important for developing countries, where genotype testing is often unreliable or not readily available. We also expanded the geographic scope of our licensing agreements with our India-based manufacturing partners to include 101 developing countries. In 2015, we also updated our tiered pricing strategy to make our branded HCV medicines available at a significantly reduced public/government price in all of these 101 countries. By making our pricing in these countries clear and transparent, we hope to facilitate planning and encourage a meaningful public health response to HCV.
HIV Program
Our goal is to ensure that all HIV patients can choose a single tablet regimen that is right for them. Single tablet regimens allow patients to adhere to a fully suppressive course of therapy more easily and consistently, which is critical for the successful management of the disease. HIV patients are living longer, thus facing additional health challenges to those experienced by newly diagnosed patients. We are motivated to continue improving on existing treatment options. The need for efficacy together with improved long-term safety has driven our development programs and the design of the studies we have completed and those that are planned.
We look forward to introducing this new generation of TAF single tablet regimens that we have created to address the evolving needs of people living with HIV. TAF is a novel targeted prodrug of tenofovir that has demonstrated high antiviral efficacy similar to and at a dose less than one-tenth that of Viread® (tenofovir disoproxil fumarate, TDF), as well as
3
improvement in surrogate laboratory markers of renal and bone safety as compared in clinical trials to TDF in combination with other antiretroviral agents. With the launch of our first TAF-based regimen, Genvoya, we now have four single tablet regimens available for the treatment of HIV. Marketing approvals for two additional TAF-based product candidates, F/TAF and R/F/TAF, are pending in the United States and European Union. Our product candidate R/F/TAF has been assigned an approval date under the Prescription Drug User Fee Act (PDUFA) of March 1, 2016 and a European Commission decision is expected in the third quarter of 2016. F/TAF has been assigned a PDUFA date of April 7, 2016 and a European Commission decision is expected in the second quarter of 2016. Emtricitabine and TAF are from Gilead and rilpivirine is from Janssen Sciences Ireland UC (Janssen).
In addition, we are investigating two additional TAF-based single tablet regimens; TAF, emtricitabine and GS-9883, our proprietary integrase inhibitor currently in Phase 3 clinical studies; and TAF, emtricitabine, cobicistat and Janssen’s darunavir (D/C/F/TAF), which is being developed and commercialized by Janssen.
Liver Diseases
Our goal is to advance the treatment options and standard of care for the underserved HCV market. With the approval of Sovaldi, compared to the prior standard of care of up to 48 weeks, the duration of treatment has been shortened to as few as 12 weeks and the need for peg-interferon (peg-IFN) injections in certain viral genotype populations has been reduced or eliminated completely. In 2014, we received FDA and European Commission approval of Harvoni, the first once-daily single tablet regimen for the treatment of HCV genotype 1 infected patients, the most prevalent genotype in the United States. We received approval of Harvoni in Japan in 2015. Harvoni combines the NS5A inhibitor ledipasvir with sofosbuvir and is indicated for an eight, 12 or 24 week treatment duration depending on prior treatment history, cirrhosis status and baseline viral load and eliminates the need for peg-IFN and RBV, which can be challenging to take and tolerate. In 2015, FDA expanded the use of Harvoni to include patients with genotype 4, 5 and 6 chronic HCV infection and in patients co-infected with HIV. In addition, Harvoni plus ribavirin for 12 weeks was approved as an alternate therapy to 24 weeks of Harvoni for treatment-experienced, genotype 1 patients with cirrhosis.
Our long term goal is to develop an oral therapy for all HCV patients across genotypes. In the fourth quarter of 2015, we submitted marketing applications to FDA and the EMA for the approval of a once-daily fixed-dose combination of SOF/VEL for the treatment of chronic genotype 1-6 HCV. In the fourth quarter of 2015, we also initiated Phase 3 clinical trials evaluating the once-daily fixed-dose combination of SOF, VEL and GS-9857, an investigational NS3 protease inhibitor, for the treatment of chronic genotype 1-6 HCV.
We are evaluating TAF for the treatment of chronic HBV infection and based on data from two Phase 3 clinical trials, we filed marketing applications to FDA and the EMA in the first quarter of 2016. We are also conducting Phase 2 clinical trials of GS-9620, an oral TLR-7 agonist, and GS-4774, a Tarmogen T cell immunity stimulator, for the treatment of HBV.
We are evaluating simtuzumab, a monoclonal antibody, for the treatment of nonalcoholic steatohepatitis (NASH) and primary sclerosing cholangitis in Phase 2 clinical trials. We are also evaluating GS-4977, an ASK-1 inhibitor, for NASH in Phase 2 clinical trials. We are also evaluating GS-9674, a FXR Agonist, for NASH in Phase 1 clinical trials.
Cardiovascular
In 2015, we received FDA approval of the use of Letairis® (ambrisentan) in combination with tadalafil for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to reduce the risks of disease progression and hospitalization for worsening PAH, and to improve exercise ability. Letairis is an endothelin receptor antagonist that was first approved in 2007 in the United States as monotherapy for PAH to improve exercise ability and delay clinical worsening. Tadalafil is a PDE5 inhibitor that was initially approved for PAH in the United States in 2009 to improve exercise ability.
Eleclazine, formerly known as GS-6615, a late sodium channel inhibitor, is being evaluated in Phase 3 clinical trials for the treatment of Long QT-3 Syndrome. Eleclazine is also being evaluated in Phase 2 clinical trials for the treatment of hypertrophic cardiomyopathy and ventricular tachycardia/ventricular fibrillation. We are also evaluating GS-4977, an ASK-1 inhibitor, for pulmonary arterial hypertension in Phase 2 clinical trials.
Hematology/Oncology
In the oncology area, we are seeking to expand the use of Zydelig (idelalisib), a first-in-class PI3K delta inhibitor, for the treatment of patients with certain blood cancers. In 2015, we submitted supplemental new drug applications with FDA and the EMA for approval of Zydelig in combination with ofatumumab in previously-treated patients with CLL. Idelalisib is in Phase 3 clinical trials for the treatment of patients with frontline and relapsed refractory CLL and relapsed refractory
4
iNHL. We plan to submit supplemental regulatory filings with FDA and the EMA for approval of Zydelig in combination with bendamustine and rituximab for patients with previously treated CLL in the second quarter of 2016.
In the fourth quarter of 2015, we initiated Phase 3 clinical trials evaluating GS-5745, a MMP9 mAb inhibitor, for the treatment of gastric cancer. We are also conducting Phase 3 clinical trials evaluating momelotinib for the treatment of myleofibrosis and pancreatic cancer.
Inflammation/Respiratory
In the inflammation/respiratory area, we advanced several product candidates in clinical trials. Presatovir, formerly known as GS-5806, a fusion inhibitor, is currently in Phase 2 clinical trials for the treatment of respiratory syncytial virus. GS-5745, a MMP9 mAb inhibitor, is being evaluated in Phase 2 clinical trials for ulcerative colitis and Crohn’s disease. Filgotinib, a JAK1 inhibitor, is being evaluated in Phase 2 clinical trials for rheumatoid arthritis and Crohn's disease.
Our Products
HIV
• | Genvoya is an oral formulation dosed once a day for the treatment of HIV-1 infection in adults. Genvoya is our fourth complete single tablet regimen for the treatment of HIV and is a fixed-dose combination of our antiretroviral medicines, Vitekta® (elvitegravir 85 mg and 150 mg), Tybost® (cobicistat), Emtriva® (emtricitabine) and TAF 10 mg. Genvoya was approved by FDA and the European Commission in November 2015. |
• | Stribild® is an oral formulation dosed once a day for the treatment of HIV-1 infection in treatment-naive adults. Stribild is our third complete single tablet regimen for the treatment of HIV and is a fixed-dose combination of our antiretroviral medications, Vitekta, Tybost, Viread® and Emtriva. |
• | Complera®/Eviplera® is an oral formulation dosed once a day for the treatment of HIV-1 infection in adults. The product, marketed in the United States as Complera and in Europe as Eviplera, is our second complete single tablet regimen for the treatment of HIV and is a fixed-dose combination of our antiretroviral medications, Viread and Emtriva, and Janssen's non-nucleoside reverse transcriptase inhibitor, Edurant (rilpivirine). |
• | Atripla® is an oral formulation dosed once a day for the treatment of HIV infection in adults. Atripla is our first single tablet regimen for HIV intended as a stand-alone therapy or in combination with other antiretrovirals. It is a fixed-dose combination of our antiretroviral medications, Viread and Emtriva, and Bristol-Myers Squibb Company's (BMS's) non-nucleoside reverse transcriptase inhibitor, Sustiva (efavirenz). |
• | Truvada® (emtricitabine and tenofovir disoproxil fumarate) is an oral formulation dosed once a day as part of combination therapy to treat HIV infection in adults. It is a fixed-dose combination of our antiretroviral medications, Viread and Emtriva. FDA also approved Truvada, in combination with safer sex practices, to reduce the risk of sexually acquired HIV-1 infection in adults at high risk; a strategy called pre-exposure prophylaxis (PrEP). |
• | Viread is an oral formulation of a nucleotide analog reverse transcriptase inhibitor, dosed once a day as part of combination therapy to treat HIV infection in patients two years of age and older. The European Commission also approved the use of Viread in combination with other antiretroviral agents for the treatment of HIV-1 infected adolescent patients aged two to less than 18 years with nucleoside reverse transcriptase inhibitor resistance or toxicities precluding the use of first-line pediatric agents. Viread is also approved for the treatment of chronic HBV. |
• | Emtriva is an oral formulation of a nucleoside analog reverse transcriptase inhibitor, dosed once a day as part of combination therapy to treat HIV infection in adults. In the United States and Europe, Emtriva is also available as an oral solution approved as part of combination therapy to treat HIV infection in children. |
• | Tybost is a pharmacokinetic enhancer dosed once a day that boosts blood levels of certain HIV medicines. Tybost is indicated as a boosting agent for the HIV protease inhibitors atazanavir and darunavir as part of antiretroviral combination therapy in adults with HIV-1 infection. |
• | Vitekta is an oral formulation of an integrase inhibitor, dosed once a day as part of combination therapy to treat HIV infection in adults without known mutations associated with resistance to elvitegravir, the active ingredient of Vitekta. Vitekta is indicated for use as part of HIV treatment regimens that include a ritonavir-boosted protease inhibitor. |
5
Liver Diseases
• | Harvoni is an oral formulation of the NS5A inhibitor with a nucleotide analog polymerase inhibitor dosed once a day for the treatment of genotypes 1, 4, 5 and 6, HCV/HIV-1 co-infection, HCV genotype 1 and 4 liver transplant recipients, and genotype 1-infected patients with decompensated cirrhosis. In Europe, Harvoni is also indicated for certain patients with HCV genotype 4 infection, HCV genotype 3 infection with cirrhosis and/or prior treatment failure and those with HCV/HIV-1 co-infection. |
• | Sovaldi is an oral formulation of a nucleotide analog polymerase inhibitor dosed once a day for the treatment of HCV as a component of a combination antiviral treatment regimen. Sovaldi’s efficacy has been established in patients with HCV genotypes 1, 2, 3 or 4 infection (in United States and Europe) and genotypes 5 and 6 infection (in Europe), including those with hepatocellular carcinoma meeting Milan criteria (awaiting liver transplantation) and those with HCV/HIV-1 co-infection. |
• | Viread is an oral formulation of a nucleotide analog reverse transcriptase inhibitor, dosed once a day for the treatment of chronic HBV in adults with compensated and decompensated liver disease. We licensed to GlaxoSmithKline Inc. (GSK) the rights to commercialize Viread for the treatment of chronic HBV in China, Japan and Saudi Arabia. In 2012, the European Commission approved the use of Viread for the treatment of chronic HBV infection in adolescent patients aged 12 to less than 18 years with compensated liver disease and evidence of immune active disease. Viread is also approved for the treatment of HIV infection. |
• | Hepsera® (adefovir dipivoxil) is an oral formulation of a nucleotide analog polymerase inhibitor, dosed once a day to treat chronic HBV in patients 12 years of age and older. We licensed to GSK the rights to commercialize Hepsera for the treatment of chronic HBV in Asia Pacific, Latin America and certain other territories. |
Cardiovascular
• | Letairis (ambrisentan) is an oral formulation of an endothelin receptor antagonist (ERA) indicated for the treatment of pulmonary arterial hypertension (PAH) (World Health Organization (WHO) Group 1) in patients with WHO Class II or III symptoms to improve exercise capacity and delay clinical worsening. We sublicensed to GSK the rights to ambrisentan, marketed by GSK as Volibris (ambrisentan), for PAH in territories outside of the United States. |
• | Ranexa® (ranolazine) is an extended-release tablet for the treatment of chronic angina. We have licensed to Menarini International Operations Luxembourg SA the rights to Ranexa in territories outside of the United States. |
• | Lexiscan®/Rapiscan® (regadenoson) injection is indicated for use as a pharmacologic stress agent in radionuclide myocardial perfusion imaging (MPI), a test that detects and characterizes coronary artery disease, in patients unable to undergo adequate exercise stress. Astellas US LLC (Astellas) has exclusive rights to manufacture and sell regadenoson under the name Lexiscan in the United States. Rapidscan Pharma Solutions, Inc. (RPS) holds the exclusive right to manufacture and sell regadenoson under the name Rapiscan in Europe and certain territories outside the United States. We receive royalties from Astellas and RPS for sales in these territories. |
Hematology/Oncology
• | Zydelig is a first-in-class PI3K delta inhibitor for the treatment of certain blood cancers. In the United States, Zydelig is approved in combination with rituximab for patients with relapsed chronic lymphocytic leukemia (CLL) for whom rituximab alone would be considered appropriate therapy and as monotherapy for patients with relapsed follicular B-cell non-Hodgkin lymphoma (FL) and small lymphocytic lymphoma (SLL) who have received at least two prior systemic therapies. In the European Union, Zydelig is approved for the treatment of CLL and FL. |
Inflammation/Respiratory
• | Cayston® (aztreonam for inhalation solution) is an inhaled antibiotic for the treatment of respiratory systems in cystic fibrosis (CF) patients seven years of age and older with Pseudomonas aeruginosa (P. aeruginosa). |
• | Tamiflu® (oseltamivir phosphate) is an oral antiviral available in capsule form for the treatment and prevention of influenza A and B. Tamiflu is approved for the treatment of influenza in children and adults in more than 60 countries, including the United States, Japan and the European Union. Tamiflu is also approved for the prevention of influenza in children and adults in the United States, Japan and the European Union. We developed Tamiflu with F. Hoffmann-La Roche Ltd (together with Hoffmann-La Roche Inc., Roche). Roche has the |
6
exclusive right to manufacture and sell Tamiflu worldwide, subject to its obligation to pay us royalties based on a percentage of the net sales of Tamiflu.
Other
• | AmBisome® (amphotericin B liposome for injection) is a proprietary liposomal formulation of amphotericin B, an antifungal agent to treat serious invasive fungal infections caused by various fungal species in adults. Our corporate partner, Astellas Pharma US, Inc., promotes and sells AmBisome in the United States and Canada, and we promote and sell AmBisome in Europe, Australia and New Zealand. |
• | Macugen® (pegaptanib sodium injection) is an intravitreal injection of an anti-angiogenic oligonucleotide for the treatment of neovascular age-related macular degeneration. Macugen was developed by Eyetech Inc. (Eyetech) using technology licensed from us and is now promoted in the United States by Valeant Pharmaceuticals, Inc. (Valeant), which acquired Eyetech in 2012. Valeant holds the exclusive rights to manufacture and sell Macugen in the United States, and Pfizer Inc. (Pfizer) holds the exclusive right to manufacture and sell Macugen in the rest of the world. We receive royalties from Valeant and Pfizer based on worldwide sales of Macugen. |
Sales of our antiviral products, which include products in our HIV and liver diseases areas described above, were $30.2 billion in 2015, $22.8 billion in 2014 and $9.3 billion in 2013 and represented 93% of our total revenues in 2015, 92% of our total revenues in 2014 and 83% of our total revenues in 2013. Sales of our other products were $1.9 billion in 2015, $1.7 billion in 2014 and $1.5 billion in 2013 and represented 6% of our total revenues in 2015, 7% of our total revenues in 2014 and 13% of our total revenues in 2013. See Item 7, Management's Discussion and Analysis and Item 8, Note 15 Segment Information in our Consolidated Financial Statements included in this Annual Report on Form 10-K for further information related to sales by product.
Commercialization and Distribution
We have U.S. and international commercial sales operations, with marketing subsidiaries in over 30 countries. Our products are marketed through our commercial teams and/or in conjunction with third-party distributors and corporate partners. Our commercial teams promote our products through direct field contact with physicians, hospitals, clinics and other healthcare providers. We generally grant our third-party distributors the exclusive right to promote our product in a territory for a specified period of time. Most of our agreements with these distributors provide for collaborative efforts between the distributor and Gilead in obtaining and maintaining regulatory approval for the product in the specified territory.
We sell and distribute Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera, Viread, Genvoya, Emtriva, Tybost, Vitekta, Ranexa, AmBisome, Zydelig and Hepsera in the United States exclusively through the wholesale channel. Our product sales to three large wholesalers, Cardinal Health, Inc., McKesson Corporation and AmerisourceBergen Corporation, each accounted for more than 10% of total revenues for each of the years ended December 31, 2015, 2014 and 2013. On a combined basis, in 2015, these wholesalers accounted for approximately 89% of our product sales in the United States and approximately 58% of our total worldwide revenues. Letairis and Cayston are distributed exclusively by specialty pharmacies. These specialty pharmacies dispense medications for complex or chronic conditions that require a high level of patient education and ongoing counseling. We sell and distribute Harvoni, Sovaldi, Truvada, Atripla, Stribild, Eviplera, Viread, Emtriva, Tybost, Vitekta, Genvoya, Ranexa, AmBisome, Zydelig and Hepsera in Europe and countries outside the United States where the product is approved, either through our commercial teams, third-party distributors or corporate partners.
U.S. Patient Access
We make it a priority to increase access to our medicines for people who can benefit from them, regardless of their ability to pay. In the United States, our U.S. patient assistance programs help make our therapies accessible for uninsured individuals and those who need financial assistance. We also support programs for those unable to afford the co-payments associated with health insurance programs. Half of all patients taking our HIV medicines in the United States already receive them through federal and state programs at substantially discounted prices. We have a long history of working with state AIDS Drug Assistance Programs (ADAPs) to provide lower pricing for our HIV medicines. The price freeze we instituted for ADAPs in 2008 was extended in 2013 through the end of 2016, providing important support to these critical programs as they evolve in the changing U.S. healthcare environment.
7
Access in the Developing World
Through the Gilead Access Program, established in 2003, certain of our products for HIV/AIDS, viral hepatitis and visceral leishmaniasis are available at substantially reduced prices in the developing world. We work with a network of regional business partners, generic licensing partners, the Medicines Patent Pool and other stakeholders to expand treatment globally. We have also entered into a number of collaborations related to access to our products in the developing world, which include:
• | Licenses with Generic Manufacturers. We have entered into non-exclusive license agreements with Indian generic manufacturers, granting them rights to produce and distribute generic versions of TDF, emtricitabine, cobicistat, elvitegravir, including generic versions of combination product containing cobicistat, elvitegravir, TDF and emtricitabine for the treatment of HIV infection to low income countries around the world, which include India and many countries in our Gilead Access Program. We also included in these non-exclusive license agreements the ability to manufacture and distribute generic versions of TDF for the treatment of HBV in the same countries where they are authorized to sell generic versions of TDF for HIV. In 2014, we granted certain of our Indian partners direct licenses to produce and distribute generic TAF in the developing world, including single tablet regimens containing emtricitabine and fixed-dose combinations of TAF and emtricitabine co-formulated with our other HIV medicines. We also entered into collaborations with our Indian partners to produce and distribute generic versions in low-income countries and lower-middle income countries. In early 2015, we expanded our collaborations to allow our Indian partners to manufacture VEL and the single tablet regimen of SOF/VEL, once approved. |
• | Medicines Patent Pool (the MPP). In 2011, we entered into an agreement with the MPP, an organization that was established by the United Nations to increase global access to high-quality, low-cost antiretroviral therapy through the sharing of patents. We granted the MPP a non-exclusive license to identify generic pharmaceutical manufacturers in India who specialize in high-quality production of generic medicines and granted sublicenses to those Indian manufacturers to manufacture and distribute generic versions of our antiretrovirals in the developing world. Sublicensees through the MPP will be free to develop combination products and pediatric formulations of our HIV medicines. We also granted the MPP the right to grant sublicenses to generic versions of elvitegravir and cobicistat, the single tablet regimen consisting of elvitegravir, cobicistat, TDF and emtricitabine and TAF for HIV and HBV to developing countries, contingent on the medicine’s U.S. regulatory approval. |
• | Special Partnerships. We work with national governments and local organizations to increase access to our HIV and HCV medicines and strengthen healthcare systems. For example, we have established an agreement with the National AIDS Program of Myanmar to donate a generic version of our Atripla to 2,000 people living with HIV in the country, as well as provide HIV educational activities and financial support to strengthen the country’s health system. In Tanzania, we launched an HIV “test-and-treat” demonstration project with the Holy See’s Good Samaritan Foundation. The program's goal is to enable screening of 120,000 patients for HIV and provide HIV therapy to 20,000 HIV-positive individuals over five years. In Egypt, we have agreed to provide Sovaldi and Harvoni to the Egyptian Ministry of Health at a significantly reduced price. In addition, in partnership with the Ministry of Health, we invest in local HCV medical education and prevention efforts, as well as screening and patient awareness initiatives. In Georgia, we established an agreement with the Ministry of Labor, Health and Social Affairs of Georgia to help eliminate HCV in the country. The project aims to reduce the number of Georgians infected with HCV and lower the rate of new infections through universal screening, treatment, prevention and surveillance. |
Competition
Our marketed products target a number of areas, including HIV, liver diseases, hematology/oncology, cardiovascular, inflammation/respiratory and other diseases. There are many commercially available products for the treatment of these diseases. We face significant competition from large global pharmaceutical and biotechnology companies, specialized pharmaceutical firms and generic drug manufacturers. Our products compete with other available products based primarily on efficacy, safety, tolerability, acceptance by doctors, ease of patient compliance, ease of use, price, insurance and other reimbursement coverage, distribution and marketing.
8
Our HIV Products
The HIV landscape is becoming more competitive and complex as treatment trends continue to evolve. A growing number of HIV drugs are currently sold or are in advanced stages of clinical development. Competition from current and expected competitors may erode the revenues we receive from sales of our HIV products. Our HIV products compete primarily with products from ViiV Healthcare (ViiV), which markets fixed-dose combination products that compete with Genvoya, Stribild, Complera/Eviplera, Atripla and Truvada. For example, two products marketed by ViiV, Tivicay (dolutegravir), an integrase inhibitor, and Triumeq (dolutegravir/abacavir/lamivudine), a single tablet antiretroviral regimen, could adversely impact sales of our HIV products. In addition, ViiV's lamivudine competes with emtricitabine, the active pharmaceutical ingredient of Emtriva and a component of Genvoya, Stribild, Complera/Eviplera, Atripla and Truvada. For Tybost, we compete with ritonavir, marketed by AbbVie Inc. (AbbVie).
We also face competition from generic HIV products. Generic versions of lamivudine and Combivir (lamivudine and zidovudine) are available in the United States and certain other countries. Generic versions of Sustiva (efavirenz), a component of our Atripla, are now available in Canada and Europe and we anticipate competition from generic efavirenz in the United States in December 2017. We have observed some pricing pressure related to the Sustiva component of our Atripla sales.
Our Liver Diseases Products
Our HCV products, Harvoni and Sovaldi, compete with Viekira Pak (ombitasvir, paritaprevir and ritonavir tablets co-packaged with dasabuvir tablets) marketed by AbbVie, Zepatier (elbasvir and grazoprevir) marketed by Merck & Co. Inc. (Merck), Daklinza (daclastavir) marketed by Bristol-Myers Squibb Company (BMS) and Olysio (simeprevir) marketed by Janssen Therapeutics.
Our HBV products, Viread and Hepsera, face competition from existing and expected therapies for treating patients with HBV. Our HBV products face competition from Baraclude (entecavir), an oral nucleoside analog marketed by BMS, as well as generic entecavir. Our HBV products also compete with Tyzeka/Sebivo (telbivudine), an oral nucleoside analog marketed by Novartis Pharmaceuticals Corporation (Novartis).
Our Cardiovascular Products
Letairis competes with Tracleer (bosentan) and Opsumit (macitentan) marketed by Actelion Pharmaceuticals US, Inc. and also with Adcirca (tadalafil) marketed by United Therapeutics Corporation and Pfizer Inc. (Pfizer).
Ranexa competes predominantly with generic compounds from three distinct classes of drugs for the treatment of chronic angina in the United States, including generic and/or branded beta-blockers, calcium channel blockers and long-acting nitrates. In addition, surgical treatments and interventions such as coronary artery bypass grafting and percutaneous coronary intervention can be another option for angina patients, which may be perceived by healthcare practitioners as preferred methods to treat the cardiovascular disease that underlies and causes angina.
There are numerous marketed generic and/or branded pharmacologic stress agents that compete with Lexiscan/Rapiscan.
Our Hematology/Oncology Products
Zydelig competes with Imbruvica (ibrutinib) marketed by Pharmacyclics, Inc., Gazyva (obinutuzumab) marketed by Genentech (a member of the Roche Group) and Treanda (bendamustine hydrochloride) marketed by Cephalon, Inc.
Our Inflammation/Respiratory Products
Cayston competes primarily with Tobi (tobramycin inhalation solution), an inhaled medication marketed by Novartis for the treatment of cystic fibrosis patients whose lungs contain P. aeruginosa, a bacterial infection.
Tamiflu competes with Relenza (zanamivir), an influenza neuraminidase inhibitor marketed by GlaxoSmithKline, and products sold by generic competitors.
9
Our Other Products
AmBisome competes with Vfend (voriconazole) marketed by Pfizer and caspofungin, a product developed by Merck that is marketed as Cancidas in the United States and as Caspofungin elsewhere. AmBisome also competes with other lipid-based amphotericin B products, including Abelcet (amphotericin B lipid complex injection), sold by Enzon Pharmaceuticals, Inc. in the United States, Canada and Japan and by Zeneus Pharma Ltd. in Europe; Amphotec (amphotericin B cholesteryl sulfate complex for injection), sold by Three Rivers Pharmaceuticals, LLC worldwide; and Anfogen (amphotericin B liposomal), sold by Genpharma, S.A. in Argentina. BMS and numerous generic manufacturers sell conventional amphotericin B, which also competes with AmBisome. In addition, we are aware of at least three lipid formulations that claim similarity to AmBisome becoming available outside of the United States. These formulations may reduce market demand for AmBisome. Furthermore, the manufacture of lipid formulations of amphotericin B is very complex, and if any of these formulations are found to be unsafe, sales of AmBisome may be negatively impacted by association.
In addition, a number of companies are pursuing the development of technologies which are competitive with our existing products or research programs. These competing companies include specialized pharmaceutical firms and large pharmaceutical companies acting either independently or together with other pharmaceutical companies. Furthermore, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection and may establish collaborative arrangements for competitive products and programs. If any of these competitors gain market share on our products, it could adversely affect our results of operations and stock price.
Collaborative Relationships
As part of our business strategy, we establish collaborations with other companies, universities and medical research institutions to assist in the clinical development and/or commercialization of certain of our products and product candidates and to provide support for our research programs. We also evaluate opportunities for acquiring products or rights to products and technologies that are complementary to our business from other companies, universities and medical research institutions. For more information regarding certain of these relationships, including their ongoing financial and accounting impact on our business, see Item 8, Note 9 Collaborative Arrangements in our Consolidated Financial Statements included in this Annual Report on Form 10-K.
Commercial Collaborations
Although we currently have a number of collaborations with corporate partners for the manufacture, sale, distribution and/or marketing of our products in various territories worldwide, the following commercial collaborations are those that are most significant to us from a financial statement perspective and where significant ongoing collaboration activity exists.
• | BMS. In 2004, we entered into a collaboration arrangement with BMS to develop and commercialize a single tablet regimen containing our Truvada and BMS's Sustiva (efavirenz) in the United States. This combination was approved for use in the United States in 2006 and is sold under the brand name Atripla. We and BMS structured this collaboration as a joint venture that operates as a limited liability company named Bristol-Myers Squibb & Gilead Sciences, LLC, which we consolidate. We and BMS granted royalty-free sublicenses to the joint venture for the use of our respective company owned technologies and, in return, were granted a license by the joint venture to use any intellectual property that results from the collaboration. In 2006, we and BMS amended the joint venture's collaboration agreement to allow the joint venture to sell Atripla in Canada. The economic interests of the joint venture held by us and BMS (including share of revenues and out-of-pocket expenses) are based on the portion of the net selling price of Atripla attributable to efavirenz and Truvada. Since the net selling price for Truvada may change over time relative to the net selling price of efavirenz, both our and BMS's respective economic interests in the joint venture may vary annually. We and BMS shared marketing and sales efforts. Starting in 2011, except for a limited number of activities that are jointly managed, the parties no longer coordinate detailing and promotional activities in the United States, and the parties have reduced their joint promotional efforts since we launched Complera in August 2011 and Stribild in August 2012. Efavirenz purchased by the joint venture from BMS at BMS's estimated net selling price of efavirenz is included in inventories on our Consolidated Balance Sheets as of December 31, 2015 and 2014.The agreement will continue until terminated by the mutual agreement of the parties. In addition, either party may terminate the other party's participation in the collaboration within 30 days after the launch of at least one generic version of such other party's single agent products (or the double agent products). The terminating party then has the right to continue to sell Atripla and become the continuing party, but will be obligated to pay the terminated party certain royalties for a three-year period following the effective date of the termination. |
In 2007, Gilead Sciences Ireland Unlimited Company, our wholly-owned subsidiary, and BMS entered into a collaboration agreement under which we and BMS commercialize and distribute Atripla in the European Union, Iceland, Liechtenstein, Norway and Switzerland (collectively, the European Territory). The parties formed a
10
limited liability company which we consolidate, to manufacture Atripla for distribution in the European Territory using efavirenz that it purchases from BMS at BMS's estimated net selling price of efavirenz in the European Territory. Starting in 2012, except for a limited number of activities that are jointly managed, the parties no longer coordinate detailing and promotional activities in the region. Efavirenz purchased from BMS at BMS's estimated net selling price of efavirenz in the European Territory is included in inventories on our Consolidated Balance Sheets as of December 31, 2015 and 2014. The agreement will terminate upon the expiration of the last-to-expire patent which affords market exclusivity to Atripla or one of its components in the European Territory. In addition, either party may terminate the agreement for any reason and such termination will be effective two calendar quarters after notice of termination. The non-terminating party has the right to continue to sell Atripla and become the continuing party, but will be obligated to pay the terminating party certain royalties for a three-year period following the effective date of the termination. In the event the continuing party decides not to sell Atripla, the effective date of the termination will be the date Atripla is withdrawn in each country or the date on which a third party assumes distribution of Atripla, whichever is earlier.
• | Janssen. In 2009, we entered into a collaboration agreement with Janssen to develop and commercialize a fixed-dose combination of our Truvada and Janssen's rilpivirine. The agreement was amended in 2011, 2013 and 2014. The combination was approved in the United States and European Union in 2011 and is sold under the brand name Complera in the United States and Eviplera in the European Union. The 2014 amendment expanded the collaboration to include another single tablet regimen containing Janssen’s rilpivirine and our emtricitabine and tenofovir alafenamide (R/F/TAF). Under the agreement, Janssen granted us an exclusive license to Complera/Eviplera and R/F/TAF worldwide but has the right to distribute both combination products in 18 countries including Mexico, Russia and Japan. Neither party is restricted from combining its drugs with any other drug products except those which are similar to the components of Complera/Eviplera and R/F/TAF. |
We are responsible for manufacturing Complera/Eviplera and R/F/TAF and have the lead role in registration, distribution and commercialization of both products except in the countries where Janssen distributes. Janssen has exercised a right to co-detail the combination product in some of the countries where Gilead is the selling party. The selling party sets the price of the products and the parties share revenues based on the ratio of the net selling prices of the party’s component(s), subject to certain restrictions and adjustments. We retain a specified percentage of Janssen’s share of revenues, up to 30% in major markets.
Either party may terminate the collaboration agreement with respect to a product and a country if the product is withdrawn from the market in such country or with respect to a product in all countries if the other party materially breaches the agreement with respect to a product. The agreement and the parties’ obligation to share revenues will expire on a product-by-product and country-by-country basis as Janssen patents providing exclusivity for the product expire or, if later, on the tenth anniversary of the commercial launch for such product. We may terminate the agreement without cause with respect to the countries where we sell the products in which case Janssen has the right to become the selling party for such country if the product has launched but has been on the market for fewer than 10 years.
• | Japan Tobacco. In 2005, Japan Tobacco Inc. (Japan Tobacco) granted us exclusive rights to develop and commercialize elvitegravir, a novel HIV integrase inhibitor, in all countries of the world, excluding Japan, where Japan Tobacco retained such rights. Under the agreement, we are responsible for seeking regulatory approval in our territories and are required to use diligent efforts to commercialize elvitegravir for the treatment of HIV infection. We bear all costs and expenses associated with such commercialization efforts. |
We received approval of Stribild (an elvitegravir-containing product) from FDA in August 2012 and from the European Commission in May 2013. We received approval of Genvoya (an elvitegravir-containing product) from FDA and the European Commission in November 2015.
The agreement and our obligation to pay royalties to Japan Tobacco will terminate on a product-by-product basis as patents providing exclusivity for the product expire or, if later, on the tenth anniversary of commercial launch for such product. We may terminate the agreement for any reason in which case the license granted by Japan Tobacco to us would terminate. Either party may terminate the agreement in response to a material breach by the other party.
Research Collaborations
We have a number of collaborations with partners for the research and development (R&D) of certain compounds and drug candidates. None of our research collaborations are significant to us from a financial statement perspective.
11
Research and Development
Our R&D philosophy and strategy is to develop best-in-class drugs that improve safety or efficacy for unmet medical needs. We intend to continue committing significant resources to internal R&D opportunities and external business development activity.
Our product development efforts cover a wide range of medical conditions, including HIV/AIDS and liver diseases such as HBV and HCV, inflammation/oncology and serious cardiovascular and respiratory conditions. We have research scientists in Foster City, Fremont, San Dimas and Oceanside, California; Seattle, Washington; and Alberta, Canada engaged in the discovery and development of new molecules and technologies that we hope will lead to the approval of new medicines addressing unmet needs.
The development of our product candidates is subject to various risks and uncertainties. These risks and uncertainties include our ability to enroll patients in clinical trials, the possibility of unfavorable results of our clinical trials, the need to modify or delay our clinical trials or to perform additional trials and the risk of failing to obtain regulatory approvals. As a result, our product candidates may never be successfully commercialized. Drug development is inherently risky and many product candidates fail during the drug development process.
Below is a summary of our key product candidates and their corresponding current stages of development.
Product Candidates for the Treatment of HIV
Product Candidates | Description | |
Marketing Applications Pending | ||
Fixed-dose co-formulation of emtricitabine and TAF (F/TAF) | A fixed-dose co-formulation of emtricitabine and TAF is being evaluated for the treatment of HIV infection. | |
Single tablet regimen of emtricitabine, rilpivirine and TAF (R/F/TAF) | Under an agreement with Janssen, a single tablet regimen of emtricitabine, rilpivirine and TAF is being evaluated for the treatment of HIV infection. | |
Product in Phase 3 | ||
Single tablet regimen of GS-9883 (non-boosted integrase inhibitor) and F/TAF | A single tablet regimen of GS-9883 and F/TAF is being evaluated for the treatment of HIV infection. | |
Product in Phase 1 | ||
GS-9620 | GS-9620, a TLR-7 agonist, is being evaluated for the treatment of HIV infection. | |
Product Candidates for the Treatment of Liver Diseases
Product Candidates | Description | |
Market Applications Pending | ||
Single tablet regimen of sofosbuvir (SOF) and velpatasvir (VEL) | A single tablet regimen of sofosbuvir and velpatasvir, a nucleotide NS5B inhibitor/pan-genotypic NS5A inhibitor, is being evaluated for the treatment of HCV. | |
TAF | TAF is a nucleotide reverse transcriptase inhibitor being evaluated for the treatment of HBV. | |
Product in Phase 3 | ||
Single tablet regimen of GS-9857 and SOF/VEL | A single tablet regimen of GS-9857, a pan-genotypic NS3 protease inhibitor, and SOF/VEL is being evaluated for the treatment of HCV. | |
Products in Phase 2 | ||
GS-4774 | GS-4774, a Tarmogen T cell immunity stimulator, is being evaluated for the treatment of HBV. | |
GS-9620 | GS-9620 is being evaluated for the treatment of HBV. | |
Simtuzumab | Simtuzumab, a monoclonal antibody, is being evaluated for the treatment of NASH and primary sclerosing cholangitis. | |
GS-4997 | GS-4997, an ASK-1 inhibitor, is being evaluated for the treatment of diabetic nephropathy and NASH. | |
12
Product Candidates for the Treatment of Cardiovascular Diseases
Product Candidates | Description | |
Product in Phase 3 | ||
Eleclazine | Eleclazine, a late sodium current inhibitor, is being evaluated for the treatment of Long QT-3 Syndrome. | |
Products in Phase 2 | ||
Eleclazine | Eleclazine is being evaluated for the treatment of hypertrophic cardiomyopathy and ventricular tachycardia/ventricular fibrillation. | |
GS-4997 | GS-4997 is being evaluated for the treatment of pulmonary arterial hypertension. | |
Product Candidates for the Treatment of Hematology/Oncology
Product Candidates | Description | |
Products in Phase 3 | ||
Idelalisib | Idelalisib, a PI3K delta inhibitor, is being evaluated for the treatment of frontline and relapsed refractory CLL and relapsed refractory iNHL. | |
Momelotinib | Momelotinib, a JAK inhibitor, is being evaluated for the treatment of myelofibrosis and pancreatic cancer. | |
GS-5745 | GS-5745, a MMP9 maB inhibitor, is being evaluated for the treatment of gastric cancer. | |
Products in Phase 2 | ||
Entospletinib | Entospletinib, a spleen tyrosine kinase (Syk) inhibitor, is being evaluated for the treatment of hematological malignancies. | |
Idelalisib | Idelalisib is being evaluated for the treatment of frontline iNHL. | |
Products in Phase 1 | ||
GS-4059 | GS-4059, a Bruton’s tyrosine kinase inhibitor, is being evaluated for the treatment of B-cell malignancies. | |
GS-5745 | GS-5745 is being evaluated for the treatment of solid tumors. | |
GS-5829 | GS-5829, a bromodomain and extra-terminal (BET) inhibitor, is being evaluated for the treatment of solid tumors. | |
Product Candidates for the Treatment of Inflammation/Respiratory Diseases
Product Candidates | Description | |
Products in Phase 2 | ||
Filgotinib | Filgotinib, a JAK1-selective inhibitor, is being evaluated for the treatment of rheumatoid arthritis and Crohn's Disease. | |
GS-5745 | GS-5745 is being evaluated for the treatment of ulcerative colitis and Crohn’s Disease. | |
Presatovir | Presatovir, a fusion inhibitor, is being evaluated for the treatment of respiratory syncytial virus. | |
Products in Phase 1 | ||
GS-5745 | GS-5745 is being evaluated for the treatment of chronic obstructive pulmonary disease and rheumatoid arthritis. | |
GS-9876 | GS-9876, a Syk inhibitor, is being evaluated for the treatment of rheumatoid arthritis. | |
13
Other Product Candidates
Product Candidates | Description | |
Product in Phase 2 | ||
GS-4997 | GS-4997 is being evaluated for the treatment of Diabetic Nephropathy. | |
Product in Phase 1 | ||
GS-5734 | GS-5734, a nucleotide prodrug, is being evaluated for the treatment of Ebola. | |
In total, our R&D expenses were $3.0 billion for 2015, $2.9 billion for 2014 and $2.1 billion for 2013. In addition to our internal discovery and clinical development programs, we seek to add to our portfolio of products through product acquisitions, licenses and collaborations.
We entered into a license and collaboration agreement with Galapagos NV (Galapagos), a clinical-stage biotechnology company based in Belgium, for the development and commercialization of filgotinib, a JAK1-selective inhibitor being investigated for inflammatory disease indications. The agreement became effective on January 19, 2016. Phase 3 trials in rheumatoid arthritis and Crohn's Disease are expected to start in 2016.
Patents and Proprietary Rights
U.S. and European Patent Expiration
We have a number of U.S. and foreign patents, patent applications and rights to patents related to our compounds, products and technology, but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will result in issued patents.
The following table shows the estimated expiration dates (including Patent Term Extension, Supplementary Protection Certificates and/or Pediatric exclusivity where granted) in the United States and Europe for the primary (typically compound) patents for our Phase 3 product candidates. Patents do not cover the ranolazine compound, the active ingredient of Ranexa. Instead, when it was discovered that only a sustained-release formulation of ranolazine would achieve therapeutic plasma levels, patents were obtained on those formulations and the characteristic plasma levels they achieve. For our product candidates that are single tablet regimens, the estimated patent expiration date provided corresponds to the latest expiring compound patent for one of the active ingredients in the single tablet regimen.
Phase 3 Product Candidates | Patent Expiration | |||||
Product Candidates for the Treatment of HIV | U.S. | E.U. | ||||
Single tablet regimen of emtricitabine and TAF | 2022 | 2021 | ||||
Single tablet regimen of darunavir, cobicistat, emtricitabine and TAF | 2029 | 2027 | ||||
Single tablet regimen of emtricitabine, rilpivirine and TAF | 2022 | 2022 | ||||
Single tablet regimen of GS-9883 and F/TAF | 2033 | (2033) | ||||
Product Candidates for the Treatment of Liver Diseases | ||||||
Single tablet regimen of sofosbuvir and velpatasvir for the treatment of HCV | 2032 | 2032 | ||||
Single tablet regimen of sofosbuvir, velpatasvir and GS-9857 for the treatment of HCV | (2033) | (2033) | ||||
Single agent TAF for the treatment of HBV | 2022 | 2021 | ||||
Product Candidates for the Treatment of Oncology/Inflammation | ||||||
Idelalisib for the treatment of frontline and relapsed refractory CLL and relapsed refractory iNHL. | 2025 | (2025) | ||||
Momelotinib for the treatment of myelofibrosis and pancreatic cancer | 2030 | 2028 | ||||
GS-5745 for the treatment of gastric cancer | 2031 | (2031) | ||||
Product Candidates for the Treatment of Cardiovascular Diseases | ||||||
Eleclazine (formerly known as GS-6615) for the treatment of LQT-3 Syndrome | 2032 | (2032) | ||||
Dates in parentheses reflect the estimated expiration date of patents which may issue from currently pending applications. The estimated expiration dates do not include any potential additional exclusivity (e.g., patent term extension, supplementary protection certificates or pediatric exclusivity) that has not yet been granted. | ||||||
14
The following table shows the actual or estimated expiration dates (including Patent Term Extension, Supplementary Protection Certificates and/or Pediatric exclusivity where granted) in the United States and Europe for the primary (typically compound) patents for our marketed products. For our product that are fixed-dose combinations or single tablet regimens (e.g., Truvada, Atripla, Complera/Eviplera, Stribild and Genvoya), the estimated patent expiration dates provided correspond to the latest expiring compound patent for one of the active ingredients in the single tablet regimen.
Products | Patent Expiration | |||||
U.S. | E.U. | |||||
Hepsera | 2014 | 2016 | ||||
AmBisome | 2016 | 2008 | ||||
Macugen | 2017 | 2017 | ||||
Tamiflu | 2017 | 2016 | ||||
Letairis | 2018 | 2020 | ||||
Viread | 2018* | 2017 | ||||
Ranexa | 2019** | 2023 | ||||
Atripla | 2021 | 2017 | ||||
Cayston | 2021 | 2021 | ||||
Emtriva | 2021 | 2016 | ||||
Truvada | 2021 | 2017 | ||||
Lexiscan | 2022 | 2025 | ||||
Complera/Eviplera | 2022 | 2022 | ||||
Vitekta | 2023 | 2028 | ||||
Zydelig | 2025 | (2025) | ||||
Sovaldi | 2029 | 2028 | ||||
Stribild | 2029 | 2028 | ||||
Genvoya | 2029 | 2028 | ||||
Tybost | 2029 | 2027 | ||||
Harvoni | 2030 | 2030 | ||||
Dates in parentheses reflect the estimated expiration date of patents which may issue from currently pending applications. The estimated expiration dates do not include any potential additional exclusivity (e.g., patent term extension, supplementary protection certificates or pediatric exclusivity) that has not yet been granted. | ||||||
* | In 2013, Gilead and Teva Pharmaceuticals (Teva) reached an agreement in principle to settle the ongoing patent litigation concerning the four patents that protect tenofovir disoproxil fumarate in our Viread, Truvada and Atripla products. Under the agreement, Teva will be allowed to launch a generic version of Viread on December 15, 2017. |
** | In 2013, Gilead and Lupin Limited (Lupin) reached an agreement to settle the patent litigation prior to issuance of the court’s decision. Under the agreement, Lupin will be allowed to launch a generic version of Ranexa on February 27, 2019. |
Patent Protection and Certain Challenges
Patents and other proprietary rights are very important to our business. If we have a properly drafted and enforceable patent, it can be more difficult for our competitors to use our technology to create competitive products and more difficult for our competitors to obtain a patent that prevents us from using technology we create. As part of our business strategy, we actively seek patent protection both in the United States and internationally and file additional patent applications, when appropriate, to cover improvements in our compounds, products and technology.
Patents covering certain of the active pharmaceutical ingredients of Truvada, Atripla, Stribild, Complera/Eviplera, Genvoya, Vitekta, Emtriva, Letairis, and Hepsera are held by third parties. We acquired exclusive rights to these patents in the agreements we have with these parties. Patents do not cover the ranolazine compound, the active ingredient of Ranexa. Instead, when it was discovered that only a sustained-release formulation of ranolazine would achieve therapeutic plasma levels, patents were obtained on those formulations and the characteristic plasma levels they achieve. Patents do not cover the active ingredients in AmBisome.
15
We may obtain patents for certain products many years before marketing approval is obtained for those products. Because patents have a limited life, which may begin to run prior to the commercial sale of the related product, the commercial value of the patent may be limited. However, we may be able to apply for patent term extensions or supplementary protection certificates in some countries. For example, extensions for the patents or supplementary protection certificates on many of our products have been granted in the United States and in a number of European countries, compensating in part for delays in obtaining marketing approval. Similar patent term extensions may be available for other products that we are developing, but we cannot be certain we will obtain them in some countries.
It is also important that we do not infringe the valid patents of third parties. If we infringe the valid patents of third parties, we may be prevented from commercializing products or may be required to obtain licenses from these third parties. We may not be able to obtain alternative technologies or any required license on reasonable terms or at all. If we fail to obtain these licenses or alternative technologies, we may be unable to develop or commercialize some or all of our products. For example, we are aware of a body of patents that may relate to our operation of Letairis Education and Access Program (LEAP), our restricted distribution program designed to support Letairis and we are aware of patents and patent applications owned by other parties that may claim to cover the use of sofosbuvir and the use of the combination of sofosbuvir and ledipasvir.
Because patent applications are confidential for a period of time until a patent is issued, we may not know if our competitors have filed patent applications for technology covered by our pending applications or if we were the first to invent or first to file an application directed toward the technology that is the subject of our patent applications. Competitors may have filed patent applications or received patents and may obtain additional patents and proprietary rights that block or compete with our products. In addition, if competitors file patent applications covering our technology, we may have to participate in interference/derivation proceedings or litigation to determine the right to a patent. Litigation and interference/derivation proceedings are unpredictable and expensive, such that, even if we are ultimately successful, our results of operations may be adversely affected by such events.
Patents relating to pharmaceutical, biopharmaceutical and biotechnology products, compounds and processes such as those that cover our existing compounds, products and processes and those that we will likely file in the future, do not always provide complete or adequate protection. Future litigation or other proceedings regarding the enforcement or validity of our existing patents or any future patents could result in the invalidation of our patents or substantially reduce their protection. From time to time, certain individuals or entities may challenge our patents.
Our pending patent applications and the patent applications filed by our collaborative partners may not result in the issuance of any patents or may result in patents that do not provide adequate protection. As a result, we may not be able to prevent third parties from developing compounds or products that are closely related to those which we have developed or are developing. In addition, certain countries in South America, Africa and Asia, including Brazil and China, do not provide effective enforcement of our patents, and third-party manufacturers may be able to sell generic versions of our products in those countries.
Litigation Related to Sofosbuvir
In January 2012, we acquired Pharmasset, Inc. (Pharmasset). Through the acquisition, we acquired sofosbuvir, a nucleotide analog that acts to inhibit the replication of the hepatitis C virus (HCV). In December 2013, we received U.S. Food and Drug Administration (FDA) approval of sofosbuvir, now known commercially as Sovaldi. In October 2014, we also received approval of the fixed-dose combination of ledipasvir and sofosbuvir (LDV/SOF), now known commercially as Harvoni. We have received a number of contractual and intellectual property claims regarding sofosbuvir. While we have carefully considered these claims both prior to and following the acquisition and believe they are without merit, we cannot predict the ultimate outcome of such claims.
We own patents and patent applications that claim sofosbuvir (Sovaldi) as a chemical entity and its metabolites and the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni). Third parties may have, or may obtain rights to, patents that allegedly could be used to prevent or attempt to prevent us from commercializing Sovaldi or Harvoni. For example, we are aware of patents and patent applications owned by other parties that have been or may in the future be alleged by such parties to cover the use of Sovaldi and Harvoni. We cannot predict the ultimate outcome of intellectual property claims related to Sovaldi or Harvoni. We have spent, and will continue to spend, significant resources defending against these claims.
If third parties successfully obtain valid and enforceable patents, and successfully prove infringement of those patents by Sovaldi and/or Harvoni, we could be prevented from selling these products unless we were able to obtain a license under such patents. Such a license may not be available on commercially reasonable terms or at all.
16
Interference Proceedings and Litigation with Idenix Pharmaceuticals, Inc. (Idenix)
In February 2012, we received notice that the U.S. Patent and Trademark Office (USPTO) had declared Interference No. 105,871 (First Idenix Interference) between our U.S. Patent No. 7,429,572 (the ’572 patent) and Idenix's pending U.S. Patent Application No. 12/131,868. An interference is a proceeding before the USPTO designed to determine who was the first to invent the subject matter claimed by both parties. In January 2014, the USPTO Patent Trial and Appeal Board (PTAB) determined that Pharmasset and not Idenix was the first to invent the compounds in dispute and accordingly Gilead prevailed in the First Idenix Interference. Idenix has appealed the PTAB’s decisions to the U.S. District Court for the District of Delaware.
In December 2013, after receiving our request to do so, the USPTO declared Interference No. 105,981 (Second Idenix Interference) between our pending U.S. Patent Application No. 11/854,218 and Idenix’s U.S. Patent No. 7,608,600 (the ’600 patent). The ’600 patent is related to the Idenix patent application at issue in the First Idenix Interference and includes claims directed to methods of treating HCV with nucleoside compounds. The purpose of the Second Idenix Interference was to determine who was first to invent the claimed methods of treating HCV with compounds similar to those which were involved in the First Idenix Interference. In March 2015, the PTAB determined that Pharmasset and not Idenix was the first to invent the claimed methods of treating HCV. Idenix appealed this decision in both the U.S. District Court for the District of Delaware and the U.S. Court of Appeal for the Federal Circuit (CAFC). We have filed a motion to dismiss the appeal in Delaware and will respond to the appeal filed in the CAFC.
We believe that the Idenix claims involved in the First and Second Idenix Interferences, and similar U.S. and foreign patents claiming the same compounds, metabolites and uses thereof, are invalid. As a result, we filed an Impeachment Action in the Federal Court of Canada to invalidate Idenix Canadian Patent No. 2,490,191 (the ’191 patent), which is the Canadian patent that corresponds to the ’600 patent. Idenix asserted that the commercialization of Sovaldi in Canada will infringe its ’191 patent and that our Canadian Patent No. 2,527,657, corresponding to the ’572 patent involved in the First Idenix Interference, is invalid. A trial on these issues was held in January and February 2015, and in November 2015, the Federal Court of Canada rendered its public decision holding that Idenix's patent is invalid and that Gilead's patent is valid. In the same month, Idenix appealed the court's decision.
We filed a similar legal action in Norway in the Oslo District Court seeking to invalidate Idenix's Norwegian patent corresponding to the ’600 patent. In September 2013, Idenix filed an invalidation action in the Norwegian proceedings against our Norwegian Patent No. 333700 patent, which corresponds to Gilead's ’572 patent. In March 2014, the Norwegian court found all claims in the Idenix Norwegian patent to be invalid and upheld the validity of all claims in the challenged Gilead patent. In April 2014, Idenix appealed the March 2014 decision to the Norwegian Court of Appeal. The appeal hearing from the March 2014 decision took place in February 2016.
In January 2013, we filed a legal action in the Federal Court of Australia seeking to invalidate Idenix’s Australian patent corresponding to the ’600 patent. In April 2013, Idenix asserted that the commercialization of Sovaldi in Australia will infringe its Australian patent corresponding to the ’600 patent. A month-long trial was completed in October 2015 in Sydney. A decision is pending.
In March 2014, the European Patent Office (EPO) granted Idenix European Patent No. 1 523 489 (the ’489 patent), which corresponds to the ’600 patent. The same day that the ’489 patent was granted, we filed an opposition with the EPO seeking to revoke the ’489 patent. An opposition hearing was held in February 2016, and the EPO ruled in our favor and revoked the '489 patent. In March 2014, Idenix also initiated infringement proceedings against us in the United Kingdom (UK), Germany and France alleging that the commercialization of Sovaldi would infringe the UK, German and French counterparts of the ’489 patent. A trial was held in the UK in October 2014 to determine the issues of infringement and validity of the Idenix UK patent. In December 2014, the High Court of Justice of England and Wales (UK Court) invalidated all challenged claims of the ’489 patent on multiple grounds. The UK Court has granted Idenix permission to appeal the December 2014 judgment. The appeal of the UK Court's decision is scheduled for July 2016. In March 2015, the German court in Düsseldorf determined that the Idenix patent was highly likely to be invalid and stayed the infringement proceedings pending the outcome of the opposition hearing held by the EPO in February 2016. Idenix has not appealed this decision of the German court staying the proceedings. Upon Idenix's request, the French proceedings have been stayed.
Idenix has not been awarded patents corresponding to the ’600 patent in Japan or China. In the event such patents are issued, we expect to challenge them in proceedings similar to those we invoked in other countries.
In December 2013, Idenix, Universita Degli Studi di Cagliari (UDSG), Centre National de la Recherche Scientifique and L’Université Montpellier II sued us in U.S. District Court for the District of Delaware alleging that the commercialization of sofosbuvir will infringe the ’600 patent and that an interference exists between the ’600 patent and our U.S. Patent No. 8,415,322. Also in December 2013, Idenix and UDSG sued us in the U.S. District Court for the District of Massachusetts
17
alleging that the commercialization of sofosbuvir will infringe U.S. Patent Nos. 6,914,054 and 7,608,597. In June 2014, the court transferred the Massachusetts litigation to the U.S. District Court for the District of Delaware. The district court has set trial dates in October 2016 and December 2016 for resolution of these issues. A decision by the district court may be appealed by either party to the CAFC.
Idenix was acquired by Merck in August 2014. While the acquisition does not change our view of the lack of merit in the claims made by Idenix, Merck has greater resources than Idenix and may therefore choose to fund the litigation at higher levels than Idenix.
Litigation with Merck
In August 2013, Merck contacted us requesting that we pay royalties on the sales of sofosbuvir and obtain a license to U.S. Patent Nos. 7,105,499 and 8,481,712, which it co-owns with Isis Pharmaceuticals, Inc. In August 2013, we filed a lawsuit in the U.S. District Court for the Northern District of California seeking a declaratory judgment that the Merck patents are invalid and not infringed. Merck’s U.S. Patent Nos. 7,105,499 and 8,481,712 cover compounds which do not include, but may relate to, sofosbuvir. During patent prosecution, Merck amended its patent application in an attempt to cover compounds related to sofosbuvir. If the court determines that Merck’s patents are valid and that we have infringed those claims, we may be required to obtain a license from and pay royalties to Merck to commercialize sofosbuvir. The court has set a trial date of March 7, 2016 for this lawsuit. Either party may appeal a decision by the District Court to the CAFC.
Litigation with AbbVie, Inc. (AbbVie)
AbbVie has obtained U.S. Patent Nos. 8,466,159, 8,492,386, 8,680,106, 8,685,984, and 8,809,265 (AbbVie Patents) which purport to cover the use of a combination of LDV/SOF (or Harvoni) for the treatment of HCV. Gilead is aware that AbbVie has pending patent applications in the United States and granted and pending applications in other countries. We own published and pending patent applications directed to the use of combinations for the treatment of HCV, and, specifically, to the combination of LDV/SOF. Certain of our applications were filed before the AbbVie Patents. For this reason and others, we believe the AbbVie Patents are invalid.
Accordingly, in December 2013, we filed a lawsuit in the U.S. District Court for the District of Delaware seeking declaratory judgment that the AbbVie Patents are invalid and unenforceable, as well as other relief. We believe that Abbott Laboratories, Inc. and AbbVie conspired to eliminate competition in the HCV market by falsely representing to the USPTO that they, and not Gilead, invented methods of treating HCV using a combination of LDV/SOF. In February and March 2014, AbbVie responded to our lawsuit by also filing two lawsuits in the U.S. District Court for the District of Delaware alleging that our fixed-dose combination of LDV/SOF will infringe its patents. All of those lawsuits have been consolidated into a single action. In the United States, either party may appeal a decision by the District Court to the CAFC. The AbbVie Patents have not blocked or delayed the commercialization of our combination product in the United States, Canada, or Europe. We do not expect any other foreign patents to block or delay the commercialization around the world. The court has set a trial date of September 12, 2016 for this lawsuit.
Additionally, AbbVie has obtained U.S. Patent No. 9,034,832 which purports to cover a solid oral dosage form containing ledipasvir. Accordingly, in May 2015, we filed a lawsuit in the U.S. District Court for the District of Delaware seeking declaratory judgment that AbbVie’s patent is invalid, as well as other relief. We do not expect AbbVie’s patent to block the commercialization of our combination product. The court has set a trial date of July 31, 2017.
In August 2015, we brought an impeachment action seeking a declaration that AbbVie's Canadian Patent No. 2,811,250 ('250 Patent), which purports to cover the use of a combination of LDV/SOF for the treatment of HCV, is invalid. On the same day, AbbVie brought an infringement action which asserts that commercialization of Harvoni in Canada will infringe its '250 Patent. The impeachment action has been stayed and we have counterclaimed for invalidity in the infringement proceeding. A trial date has not been set.
In November 2015, AbbVie filed a lawsuit against us in the Regional Court Düsseldorf for infringement of two quasi-patents, known as “utility models.” Utility models are unexamined IP rights and are not the same as standard patents. One utility model, DE 20 2012 013 117, purports to cover the use of a combination of direct-acting antivirals which includes at least an HCV polymerase inhibitor and an HCV NS5A inhibitor in the treatment of HCV; the other utility model, DE 21 2012 000 197, purports to cover a solid dispersion that includes ledipasvir. A trial date has not been set.
If a court determines that the AbbVie Patents are valid and that we have infringed those claims, we may be required to obtain a license from and pay royalties to AbbVie to commercialize sofosbuvir combination products.
18
European Patent Claims
In February 2015, several parties filed oppositions in the European Patent Office requesting revocation of our granted European patent covering sofosbuvir that expires in 2028. While we are confident in the strength of our sofosbuvir patent, we cannot predict the ultimate outcome of these oppositions. If we are unsuccessful in defending these oppositions, some or all of our patent claims may be narrowed or revoked and the patent protection for sofosbuvir in Europe could be substantially shortened or eliminated entirely. If the sofosbuvir patent is revoked, and no other European patents are granted covering sofosbuvir, our exclusivity will be based entirely on regulatory exclusivity granted by the European Medicines Agency (EMA). Sovaldi has been granted regulatory exclusivity that will prevent generic sofosbuvir from entering the European Union for 10 years following approval of Sovaldi, or January 2024. If we lose exclusivity for Sovaldi prior to 2028, our expected revenues and results of operation could be negatively impacted for the years including and succeeding the year in which such exclusivity is lost, which may cause our stock price to decline.
Litigation with Generic Manufacturers
As part of the approval process for some of our products, FDA granted us a New Chemical Entity (NCE) exclusivity period during which other manufacturers' applications for approval of generic versions of our product will not be approved. Generic manufacturers may challenge the patents protecting products that have been granted NCE exclusivity one year prior to the end of the NCE exclusivity period. Generic manufacturers have sought and may continue to seek FDA approval for a similar or identical drug through an abbreviated new drug application (ANDA), the application form typically used by manufacturers seeking approval of a generic drug. The sale of generic versions of our products earlier than their patent expiration would have a significant negative effect on our revenues and results of operations.
HIV Products
In November 2011, December 2011 and August 2012, we received notices that Teva Pharmaceuticals (Teva) submitted an abbreviated new drug submission (ANDS) to the Canadian Minister of Health requesting permission to manufacture and market generic versions of Truvada, Atripla and Viread. In the notices, Teva alleges that the patents associated with Truvada, Atripla and Viread are invalid, unenforceable and/or will not be infringed by Teva's manufacture, use or sale of generic versions of those products. We filed lawsuits against Teva in the Federal Court of Canada seeking an order of prohibition against approval of these applications.
In December 2013, the court issued an order prohibiting the Canadian Minister of Health from approving Teva’s generic versions of our Viread, Truvada and Atripla products until expiry of our patents in July 2017. Teva has appealed that decision. That decision did not rule on the validity of the patents and accordingly the only issue on appeal is whether the Minister of Health should be prohibited from approving Teva’s products. The appeal will be heard by the Canadian Federal Court of Appeal after the trial in the Impeachment Action. The court will determine the validity of the patents in the pending Impeachment Action. A trial in the Impeachment Action is scheduled for November 2016. If Teva is successful in invalidating our patents, Teva may be able to launch generic versions of our Viread, Truvada and Atripla products in Canada prior to the expiry of our patents.
In April 2014 and July 2015, we received notices that Mylan Inc. (Mylan) submitted an ANDA to FDA requesting permission to manufacture and market generic versions of Truvada and Complera. In the notice, Mylan alleges that the patents associated with Truvada and Complera are invalid, unenforceable and/or will not be infringed by Mylan's manufacture, use or sale of a generic version of these products. We filed lawsuits against Mylan in U.S. District Court for the Northern District of West Virginia for infringement of our patents. In June 2014, we received notice that Mylan submitted petitions for Inter Partes Review (IPR) to the PTAB alleging that four patents associated with tenofovir disoproxil fumarate are invalid. We opposed Mylan’s petitions. In December 2014, the PTAB issued decisions denying each of Mylan’s petitions for IPR. In January 2015, Mylan requested a rehearing on the basis that it believes the PTAB decision is wrong. In August 2015 and November 2015, the PTAB denied Mylan's requests for a rehearing. In October 2015, we reached an agreement with Mylan to settle the proceedings. The terms of the settlement agreement are confidential.
In June 2014, we received notice that Apotex Inc. (Apotex) submitted an ANDS to the Canadian Minister of Health requesting permission to manufacture and market a generic version of Truvada and a separate ANDS requesting permission to manufacture and market a generic version of Viread. In the notice, Apotex alleges that three of the patents associated with Truvada and two of the patents associated with Viread are invalid, unenforceable and/or will not be infringed by Apotex's manufacture, use or sale of a generic version of Truvada or Viread. In August 2014, we filed a lawsuit against Apotex in the Federal Court of Canada seeking an order of prohibition against approval of this ANDS. A hearing in that case is scheduled for April 2016.
19
Letairis
In February 2015, we received notice that Watson Laboratories, Inc. (Watson) submitted an ANDA to FDA requesting permission to manufacture and market a generic version of Letairis. In the notice, Watson alleges that one of the patents associated with ambrisentan tablets is invalid, unenforceable and/or will not be infringed by Watson's manufacture, use or sale of a generic version of Letairis. In April 2015, we filed a lawsuit against Watson in the U.S. District Court for the District of New Jersey.
In June 2015, we received notice that SigmaPharm Laboratories, LLC (SigmaPharm) submitted an ANDA to FDA requesting permission to manufacture and market a generic version of Letairis. In the notice, SigmaPharm alleges that one of the patents associated with ambrisentan tablets is invalid, unenforceable and/or will not be infringed by SigmaPharm’s manufacture, use or sale of a generic version of Letairis. In June 2015, we filed a lawsuit against SigmaPharm in the U.S. District Court for the District of New Jersey.
We cannot predict the ultimate outcome of these actions, and we may spend significant resources enforcing and defending these patents. If we are unsuccessful in these lawsuits, some or all of our claims in the patents may be narrowed or invalidated and the patent protection for our products could be substantially shortened. Further, if all of the patents covering one or more products are invalidated, FDA or Health Canada could approve the requests to manufacture a generic version of such products in the United States or Canada, respectively, prior to the expiration date of those patents. The sale of generic versions of these products earlier than their patent expiration would have a significant negative effect on our revenues and results of operations.
TAF Litigation
In January 2016, AIDS Healthcare Foundation, Inc. (AHF) filed a complaint with the U.S. District Court for the Northern District of California against Gilead, Japan Tobacco, Inc., Japan Tobacco International, U.S.A. (together, Japan Tobacco), and Emory University (Emory). AHF claims that U.S. Patent Nos. 7,390,791; 7,800,788; 8,754,065; 8,148,374; and 8,633,219 are invalid under 35 U.S.C. §§ 101 et seq. In addition, AHF claims that Gilead, independently and together with Japan Tobacco and Emory, is violating federal antitrust laws in the market for sales of tenofovir alafenamide (TAF) by offering TAF as part of a fixed-dose combination product with elvitegravir, cobicistat, and emtricitabine. AHF seeks a declaratory judgment of invalidity against each of the patents as well as monetary damages.
Trade Secrets
We also rely on unpatented trade secrets and improvements, unpatented internal know-how and technological innovation. In particular, a great deal of our liposomal manufacturing expertise, which is a key component of our liposomal technology, is not covered by patents but is instead protected as a trade secret. We protect these rights mainly through confidentiality agreements with our corporate partners, employees, consultants and vendors. These agreements provide that all confidential information developed or made known to an individual during the course of their relationship with us will be kept confidential and will not be used or disclosed to third parties except in specified circumstances. In the case of employees, the agreements provide that all inventions made by an individual while employed by us will be our exclusive property. We cannot be certain that these parties will comply with these confidentiality agreements, that we have adequate remedies for any breach or that our trade secrets will not otherwise become known or be independently discovered by our competitors. Under some of our R&D agreements, inventions become jointly owned by us and our corporate partner and in other cases become the exclusive property of one party. In certain circumstances, it can be difficult to determine who owns a particular invention and disputes could arise regarding those inventions. If our trade secrets or confidential information become known or independent discovered by competitors or if we enter into disputes over ownership of inventions, our business and results of operations could be adversely affected.
Manufacturing and Raw Materials
Our manufacturing strategy is to contract with third parties to manufacture the majority of our active pharmaceutical ingredients (API) and solid dose products. We also rely on our corporate partners to manufacture certain of our products. Additionally, we own or lease manufacturing facilities in Foster City, San Dimas and Oceanside, California; Edmonton, Alberta, Canada and Cork, Ireland, where we manufacture certain products and API for clinical and commercial uses.
20
Manufacturing of our Products
We contract with third parties to manufacture certain products for clinical and commercial purposes, including Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya, Emtriva, Tybost, Vitekta, Ranexa, AmBisome, Zydelig and Cayston. We generally use multiple third-party contract manufacturers to manufacture the API in our products. We are the exclusive manufacturer of ambrisentan, the API of Letairis, although another supplier is qualified to make the API of Letairis.
We also rely on third-party contract manufacturers to manufacture our tablet or capsule products. For example, we use multiple third-party contract manufacturers to tablet Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya, Tybost, Vitekta, Letairis, Ranexa, Zydelig and Hepsera. Emtriva encapsulation is also completed by third-party contract manufacturers. In addition, we rely on third-party contract manufacturers to manufacture our aseptic products such as AmBisome and Cayston.
We also have manufacturing agreements with many of our corporate partners. Roche, by itself and through third parties, is responsible for manufacturing Tamiflu. Under our agreement with Roche, through a joint manufacturing committee composed of representatives from Roche and Gilead, we have the opportunity to review Roche's existing manufacturing capacity for Tamiflu and global plans for manufacturing Tamiflu. Astellas US LLC, our corporate partner for Lexiscan in the United States, is responsible for the commercial manufacture and supply of product in the United States and is dependent on a single supplier for the API of Lexiscan.
For our future products, we continue to develop additional manufacturing capabilities and establish additional third-party suppliers to manufacture sufficient quantities of our product candidates to undertake clinical trials and to manufacture sufficient quantities of any product that is approved for commercial sale.
Our Manufacturing Facilities
At our Foster City, California facility, we conduct process chemistry research and development activities, manufacture API for our clinical trials and oversee our third-party contract manufacturers.
At our San Dimas, California facility, we package and label solid dosage oral form products, including Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya, Emtriva, Ranexa and Zydelig, and label Hepsera at our facilities in San Dimas. We manufacture AmBisome and Cayston at our San Dimas facility. We depend on a single supplier for the high quality cholesterol and the API used in the manufacture of AmBisome. Because we are the exclusive supplier of key drug product intermediates of AmBisome, in the event of a disaster, including an earthquake, equipment failure or other difficulty, we may be unable to replace this manufacturing capacity in a timely manner and may be unable to manufacture AmBisome to meet market needs.
We utilize our Cork, Ireland facility primarily for solid dose tablet manufacturing of certain of our antiviral products, as well as product packaging activities. We package and label drug product for Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya, Tybost and Vitekta and label Hepsera and Emtriva at our facilities in Cork, Ireland. We also perform quality control testing, final labeling and secondary packaging of both AmBisome and Cayston and final release of many of our products for the European Union and elsewhere at this facility. We distribute our products to the European Union and other international markets from our Dublin, Ireland site.
At our Edmonton, Alberta facility in Canada, we carry out process research and scale-up of our clinical development candidates, manufacture API for both investigational and commercial products and conduct chemical development activities to improve existing commercial manufacturing processes. We also manufacture the API of Letairis and Hepsera at our Edmonton site.
Our Oceanside, California facility is designed and equipped to produce biologic compounds for toxicological, Phase 1 and Phase 2 clinical studies. We use the facility for the process development and manufacture of simtuzumab, an investigational monoclonal antibody candidate, GS-5745 bulk drug substance, an investigational MMP9 mAb inhibitor, and other biologics.
21
Third-party Manufacturers
Our third-party manufacturers and corporate partners are independent entities who are subject to their own unique operational and financial risks which are out of our control. If we or any of these third-party manufacturers or corporate partners fail to perform as required, this could impair our ability to deliver our products on a timely basis or receive royalties or cause delays in our clinical trials and applications for regulatory approval. Further, we may have to write-off the costs of manufacturing any batch that fails to pass quality inspection or meet regulatory approval. To the extent these risks materialize and affect their performance obligations to us, our financial results may be adversely affected. In addition, we, our third-party manufacturers and our corporate partners may only be able to produce some of our products at one or a limited number of facilities and, therefore, have limited manufacturing capacity for certain products.
We believe the technology we use to manufacture our products is proprietary. For products manufactured by our third-party contract manufacturers, we have disclosed all necessary aspects of this technology to enable them to manufacture the products for us. We have agreements with these third-party manufacturers that are intended to restrict these manufacturers from using or revealing this technology, but we cannot be certain that these third-party manufacturers will comply with these restrictions. In addition, these third-party manufacturers could develop their own technology related to the work they perform for us that we may need to manufacture our products. We could be required to enter into additional agreements with these third-party manufacturers if we want to use that technology ourselves or allow another manufacturer to use that technology. The third-party manufacturer could refuse to allow us to use their technology or could demand terms to use their technology that are not acceptable to us.
Regulation of Manufacturing Process
The manufacturing process for pharmaceutical products is highly regulated and regulators may shut down manufacturing facilities that they believe do not comply with regulations. We, our third-party manufacturers and our corporate partners are subject to current Good Manufacturing Practices, which are extensive regulations governing manufacturing processes, stability testing, record keeping and quality standards as defined by FDA and the European Medicines Agency (EMA). Similar regulations are in effect in other countries.
Our manufacturing operations are subject to routine inspections by regulatory agencies. For example, in 2014, we received a letter from FDA related to the extent of method revalidations being conducted, stability program oversight, audit trail review/data management and Quality Management System gaps. We completed and filed our responses to these observations with FDA. If we are unable to remedy the deficiencies cited by FDA or to the extent there are additional deficiencies cited by FDA in future inspections, our currently marketed products and the timing of regulatory approval of products in development could be adversely affected. Further, there is risk that regulatory agencies in other countries where marketing applications are pending will undertake similar additional reviews or apply a heightened standard of review, which could delay the regulatory approvals for products in those countries. If approval of any of our product candidates were delayed or if production of our marketed products was interrupted, our anticipated revenues and our stock price would be adversely affected.
Access to Supplies and Materials
We need access to certain supplies and products to manufacture our products. If we are unable to purchase sufficient quantities of these materials or find suitable alternate materials in a timely manner, our development efforts for our product candidates may be delayed or our ability to manufacture our products would be limited, which would limit our ability to generate revenues. For example, a significant portion of the raw materials and intermediates used to manufacture our antiviral products (Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya, Emtriva, Tybost and Vitekta) are supplied by China-based companies. As a result, an international trade dispute between China and the United States or any other actions by the Chinese government that would limit or prevent Chinese companies from supplying these materials would adversely affect our ability to manufacture and supply our HIV and HCV products to meet market needs and have a material and adverse effect on our operating results.
Seasonal Operations and Backlog
Our worldwide product sales do not reflect any significant degree of seasonality.
For the most part, we operate in markets characterized by short lead times and the absence of significant backlogs. We do not believe that backlog information is material to our business as a whole.
Government Regulation
Our operations and activities are subject to extensive regulation by numerous government authorities in the United States and other countries. In the United States, the European Union and other countries, drugs are subject to rigorous
22
regulation. Federal and state statutes and regulations govern the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. As a result of these regulations, product development and product approval processes are very expensive and time consuming.
A country's regulatory agency, such as FDA in the United States and European Medicines Agency for the European Union, must approve a drug before it can be sold in the respective country or countries. The general process for drug approval in the United States is summarized below. Many other countries, including countries in the European Union and Japan, have very similar regulatory structures.
Preclinical Testing
Before we can test a drug candidate in humans, we must study the drug in laboratory experiments and in animals to generate data to support the drug candidate's potential benefits and safety. We submit this data to FDA in an investigational new drug (IND) application seeking its approval to test the compound in humans.
Clinical Trials
If FDA accepts the IND, the drug candidate can then be studied in human clinical trials to determine if the drug candidate is safe and effective. These clinical trials involve three separate phases that often overlap, can take many years and are very expensive. These three phases, which are subject to considerable regulation, are as follows:
• | Phase 1. The drug candidate is given to a small number of healthy human control subjects or patients suffering from the indicated disease, to test for safety, dose tolerance, pharmacokinetics, metabolism, distribution and excretion. |
• | Phase 2. The drug candidate is given to a limited patient population to determine the effect of the drug candidate in treating the disease, the best dose of the drug candidate, and the possible side effects and safety risks of the drug candidate. It is not uncommon for a drug candidate that appears promising in Phase 1 clinical trials to fail in the more rigorous Phase 2 clinical trials. |
• | Phase 3. If a drug candidate appears to be effective and safe in Phase 2 clinical trials, Phase 3 clinical trials are commenced to confirm those results. Phase 3 clinical trials are conducted over a longer term, involve a significantly larger population, are conducted at numerous sites in different geographic regions and are carefully designed to provide reliable and conclusive data regarding the safety and benefits of a drug candidate. It is not uncommon for a drug candidate that appears promising in Phase 2 clinical trials to fail in the more rigorous and extensive Phase 3 clinical trials. |
FDA Approval Process
When we believe that the data from our clinical trials show an adequate level of safety and efficacy, we submit the appropriate filing, usually in the form of an NDA or supplemental NDA, with FDA seeking approval to sell the drug candidate for a particular use. FDA may hold a public hearing where an independent advisory committee of expert advisors asks additional questions and makes recommendations regarding the drug candidate. This committee makes a recommendation to FDA that is not binding but is generally followed by FDA. If FDA agrees that the compound has met the required level of safety and efficacy for a particular use, it will allow us to sell the drug candidate in the United States for that use. It is not unusual, however, for FDA to reject an application because it believes that the drug candidate is not safe enough or efficacious enough or because it does not believe that the data submitted is reliable or conclusive.
At any point in this process, the development of a drug candidate can be stopped for a number of reasons including safety concerns and lack of treatment benefit. We cannot be certain that any clinical trials that we are currently conducting or any that we conduct in the future will be completed successfully or within any specified time period. We may choose, or FDA may require us, to delay or suspend our clinical trials at any time if it appears that the patients are being exposed to an unacceptable health risk or if the drug candidate does not appear to have sufficient treatment benefit.
FDA may also require Phase 4 non-registrational studies to explore scientific questions to further characterize safety and efficacy during commercial use of our drug. FDA may also require us to provide additional data or information, improve our manufacturing processes, procedures or facilities or may require extensive surveillance to monitor the safety or benefits of our product candidates if it determines that our filing does not contain adequate evidence of the safety and benefits of the drug. In addition, even if FDA approves a drug, it could limit the uses of the drug. FDA can withdraw approvals if it does not believe that we are complying with regulatory standards or if problems are uncovered or occur after approval.
23
In addition to obtaining FDA approval for each drug, we obtain FDA approval of the manufacturing facilities for any drug we sell, including those of companies who manufacture our drugs for us. All of these facilities are subject to periodic inspections by FDA. FDA must also approve foreign establishments that manufacture products to be sold in the United States and these facilities are subject to periodic regulatory inspection. Our manufacturing facilities located in California, including our Oceanside and San Dimas facilities, also must be licensed by the State of California in compliance with local regulatory requirements. Our manufacturing facilities located in Canada, including our Edmonton, Alberta facility, and our facilities located near Dublin and in Cork, Ireland, also must obtain local licenses and permits in compliance with local regulatory requirements.
Drugs that treat serious or life threatening diseases and conditions that are not adequately addressed by existing drugs, and for which the development program is designed to address the unmet medical need, may be designated as fast track candidates by FDA and may be eligible for accelerated and priority review. Drugs for the treatment of HIV infection that are designated for use under the U.S. President's Emergency Plan for AIDS Relief may also qualify for an expedited or priority review. Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera, Viread, Genvoya and Zydelig received accelerated approval and priority reviews. Drugs receiving accelerated approval must be monitored in post-marketing clinical trials in order to confirm the safety and benefits of the drug.
Drugs are also subject to extensive regulation outside of the United States. In the European Union, there is a centralized approval procedure that authorizes marketing of a product in all countries of the European Union (which includes most major countries in Europe). If this centralized approval procedure is not used, approval in one country of the European Union can be used to obtain approval in another country of the European Union under one of two simplified application processes: the mutual recognition procedure or the decentralized procedure, both of which rely on the principle of mutual recognition. After receiving regulatory approval through any of the European registration procedures, separate pricing and reimbursement approvals are also required in most countries. The European Union also has requirements for approval of manufacturing facilities for all products that are approved for sale by the European regulatory authorities.
Pricing and Reimbursement
Successful commercialization of our products depends, in part, on the availability of governmental and third-party payer reimbursement for the cost of such products and related treatments in the markets where we sell our products. Government health authorities, private health insurers and other organizations generally provide reimbursement. In the United States, the European Union, Japan and other significant or potentially significant markets for our products and product candidates, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medical products and services. As a result, a significant portion of our sales of the majority of our products are subject to substantial discounts and rebate obligations.
In the United States, state AIDS Drug Assistance Programs (ADAPs), which purchase a significant portion of our HIV products, rely on federal, supplemental federal and state funding to help fund purchases of our products. If federal and state funds are not available in amounts sufficient to support the number of patients that rely on ADAPs, sales of our HIV products could be negatively impacted which would reduce our revenues. In prior quarters, because of the insufficiency of federal and state funds, and as many states reduced eligibility criteria, we saw an increase in the number of patients on state ADAP waitlists, and we may see similar increases in future periods as a result of any reduction in federal and state ADAP support. Until these patients are enrolled in an ADAP, they generally receive free product from industry-supported patient assistance programs or are unable to access treatment. The increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our product sales and profitability. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general.
We continue to experience global pricing pressure on our HCV products, which often results in increases in the amount of discounts required on our products or delayed reimbursement, which could negatively impact our future product sales and results of operations. Also, private and public payers can choose to exclude Harvoni or Sovaldi from their formulary coverage or limit the types of patients for whom coverage will be provided, which could negatively impact the demand for, and revenues of, Harvoni and Sovaldi. Any change in the formulary coverage, reimbursement levels or discounts or rebates offered on our HCV products to payers may impact our anticipated revenues. We expect pricing pressure in the HCV market to continue. See also our risk factor "A substantial portion of our revenues is derived from sales of products to treat HCV and HIV. If we are unable to maintain or continue increasing sales of these products, our results of operations may be adversely affected."
24
In July 2014, we received a letter from the U.S. Senate Committee on Finance (Senate Committee) requesting information and supporting documentation from us related to Sovaldi and the pricing of Sovaldi in the United States. The letter raised concerns about our approach to pricing Sovaldi, its affordability and its impact on federal government spending and public health. In December 2015, the Senate Committee released the results of the investigation, which alleged that we engaged in a revenue-driven pricing strategy in setting Sovaldi's price. Gilead disagrees with many of the conclusions in the report. In January 2016, we received a letter from the Massachusetts Attorney General that their office is considering whether our pricing of Sovaldi and Harvoni may constitute an unfair trade practice in violation of Massachusetts law. In February 2016, the Massachusetts Attorney General’s office served us with a Civil Investigative Demand requesting that we produce documents related to our HCV products. It is possible that the results of the Senate Committee investigation and any actions taken by the Massachusetts Attorney General or other state governments could result in negative publicity or other negative actions that could harm our reputation, reduce demand for Harvoni, Sovaldi or other sofosbuvir containing products and/or reduce coverage of Harvoni, Sovaldi or other sofosbuvir containing products, including by federal health care programs such as Medicare and Medicaid and state health care programs. If any or all of these events occur, our business and stock price could be materially and adversely affected.
In countries outside the United States, the success of our commercialized products, and any other product candidates we may develop, will depend largely on obtaining and maintaining government reimbursement, because in many countries patients are unlikely to use prescription drugs that are not reimbursed by their governments. Recently, many countries in the European Union have increased the level of discounting required on our products, and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union. Cost containment pressures in the European Union, especially in Southern Europe, could lead to delays in the treatment of patients and also delay pricing approval by 12 months or more, which could negatively impact the commercialization of new products. Reimbursement policies may adversely affect our ability to sell our products on a profitable basis. In many international markets, governments control the prices of prescription pharmaceuticals, including through the implementation of reference pricing, price cuts, rebates, revenue-related taxes, tenders and profit control, and they expect prices of prescription pharmaceuticals to decline over the life of the product or as volumes increase. For example, we anticipate the government of Japan will impose significant pricing discounts for Harvoni and Sovaldi that will start taking effect in the first half of 2016.
United States Healthcare Reform
Legislative and regulatory changes to government prescription drug procurement and reimbursement programs occur relatively frequently in the United States and foreign jurisdictions. In March 2010, healthcare reform legislation was adopted in the United States, requiring us to further rebate or discount products reimbursed or paid for by various public payers, including Medicaid and other entities eligible to purchase discounted products through the 340B Drug Pricing Program under the Public Health Service Act, such as ADAPs. In the United States, we, along with other pharmaceutical manufacturers of branded drug products, are required to pay a portion of an industry fee (also known as the Branded Prescription Drug (BPD) fee), calculated based on select government sales during the year as a percentage of total industry government sales. The amount of the annual BPD fee to be billed to the pharmaceutical industry as a whole is $3.0 billion in 2015 through 2016, which will increase to $4.0 billion in 2017, increase to a peak of $4.1 billion in 2018, and then decrease to $2.8 billion in 2019 and thereafter. The BPD fee is not tax deductible. In 2014, the Internal Revenue Service (IRS) issued final regulations which accelerated the expense recognition criteria for the fee obligation from the year in which the fee is paid, to the year in which the market share used to allocate the fee is determined. Our BPD fee expenses were $414 million in 2015, $590 million in 2014 and $110 million in 2013. We expect our portion of the BPD fee to increase as the total annual industry-wide fee increases through 2017 and drug patents expire on major drugs of other companies. In addition, even though not addressed in the healthcare reform legislation, discussions continue at the federal level on legislation that would either allow or require the federal government to directly negotiate price concessions from pharmaceutical manufacturers or set minimum requirements for Medicare Part D pricing. Further, certain states have proposed legislation that seek to regulate pharmaceutical drug pricing. If such proposed legislation is passed, we may experience additional pricing pressures on our products.
In addition, state Medicaid programs could request additional supplemental rebates on our products as a result of the increase in the federal base Medicaid rebate. Private insurers could also use the enactment of these increased rebates to exert pricing pressure on our products, and to the extent that private insurers or managed care programs follow Medicaid coverage and payment developments, the adverse effects may be magnified by private insurers adopting lower payment schedules.
Health Care Fraud and Abuse Laws and Anti-Bribery Laws
We are subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive or
25
pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the increasing attention being given to them by law enforcement authorities, it is possible that certain of our practices may be challenged under anti-kickback or similar laws. False claims laws generally prohibit anyone from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment by federal and certain state payers (including Medicare and Medicaid), or knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim. Our sales, marketing and medical activities may be subject to scrutiny under these laws. In addition, the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments for the purpose of obtaining or retaining business. Our policies mandate compliance with these anti-bribery laws. We operate in parts of the world that have experienced governmental corruption to some degree. In certain circumstances, strict compliance with anti-bribery laws may conflict with local customs and practices or may require us to interact with doctors and hospitals, some of which may be state controlled, in a manner that is different than local custom. Despite our training and compliance program, our internal control policies and procedures may not protect us from reckless or criminal acts committed by our employees or agents. Violations of fraud and abuse laws or anti-bribery laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, as well as the possibility of exclusion from federal health care programs (including Medicare and Medicaid). Violations can also lead to the imposition of a Corporate Integrity Agreement or similar government oversight program. If the government were to allege against or convict us of violating these laws, there could be a disruption on our business and material adverse effect on our results of operations.
Compulsory Licenses
In a number of developing countries, government officials and other interested groups have suggested that pharmaceutical companies should make drugs for HCV or HIV infection available at low cost. Alternatively, governments in those developing countries could require that we grant compulsory licenses to allow competitors to manufacture and sell their own versions of our products, thereby reducing our product sales. For example, there is growing attention on the availability of HCV therapies and some activists are advocating for the increased availability of HCV therapies through means including compulsory licenses. In the past, certain offices of the government of Brazil have expressed concern over the affordability of our HIV products and declared that they were considering issuing compulsory licenses to permit the manufacture of otherwise patented products for HIV infection, including Viread. In addition, concerns over the cost and availability of Tamiflu related to a potential avian flu pandemic and H1N1 influenza generated international discussions over compulsory licensing of our Tamiflu patents. For example, the Canadian government considered allowing Canadian manufacturers to manufacture and export the active ingredient in Tamiflu to eligible developing and least developed countries under Canada's Access to Medicines Regime. Furthermore, Roche issued voluntary licenses to permit third-party manufacturing of Tamiflu. For example, Roche granted a sublicense to Shanghai Pharmaceutical (Group) Co., Ltd. for China and a sublicense to India's Hetero Drugs Limited for India and certain developing countries. If compulsory licenses permit generic manufacturing to override our product patents for Harvoni, Sovaldi, our HIV products or Tamiflu, or if we are required to grant compulsory licenses for these products, it could reduce our earnings and cash flows and harm our business.
In addition, certain countries do not permit enforcement of our patents, and third-party manufacturers are able to sell generic versions of our products in those countries. For example, in July 2009, the Brazilian patent authority rejected our patent application for tenofovir disoproxil fumarate, the active pharmaceutical ingredient in Viread. This was the highest level of appeal available to us within the Brazilian patent authority. Because we do not currently have a patent in Brazil, the Brazilian government now purchases its supply of tenofovir disoproxil fumarate from generic manufacturers. Sales of generic versions of our products could significantly reduce our sales and adversely affect our results of operations, particularly if generic versions of our products are imported into territories where we have existing commercial sales.
Employees
As of January 31, 2016, we had approximately 8,000 employees. We believe we have good relations with our employees.
Environment, Health and Safety
We strive to incorporate sustainability in all phases of our drug development business, from the ethical sourcing of natural renewable materials to utilizing green chemistry practices. We continue to look for ways to minimize our impact on the environment. Some factors that contribute to our environmental impact include greenhouse gas emissions produced by employee commutes, the energy and water consumed by our facilities, and the use of hazardous materials such as chemicals, viruses and radioactive compounds in our R&D facilities. Please refer to our 2014 Sustainability Report found on our website at www.gilead.com under "Responsibility" for some of the measures we have taken to mitigate the environmental impact from our business.
26
We are subject to a number of laws and regulations that require compliance with federal, state, and local regulations regarding workplace safety and protection of the environment. We anticipate additional regulations in the near future. Laws and regulations are implemented and under consideration to mitigate the effects of climate change mainly caused by greenhouse gas emissions. Our business is not energy intensive. Therefore, we do not anticipate being subject to a cap and trade system or other mitigation measure that would materially impact our capital expenditures, operations, or competitive position. Based on current information, and subject to the finalization of proposed regulations, we believe that our primary risk related to climate change is increased energy costs.
Other Information
We are subject to the information requirements of the Exchange Act. Therefore, we file periodic reports, proxy statements and other information with the SEC. Such reports, proxy statements and other information may be obtained by visiting the Public Reference Room of the SEC at 100 F Street, NE, Washington, D.C. 20549 or by calling the SEC at 1-800-SEC-0330, by sending an electronic message to the SEC at publicinfo@sec.gov or by sending a fax to the SEC at 1-202-777-1027. In addition, the SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically.
The mailing address of our headquarters is 333 Lakeside Drive, Foster City, California 94404, and our telephone number at that location is 650-574-3000. Our website is www.gilead.com. Through a link on the “Investors” section of our website (under “SEC Filings” in the “Financial Information” section), we make available the following filings as soon as reasonably practicable after they are electronically filed with or furnished to the SEC: our Annual Reports on Form 10-K; Quarterly Reports on Form 10-Q; Current Reports on Form 8-K; and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act. All such filings are available free of charge upon request.
Transactions with Iran
We did not have any transactions with Iran during 2015 that would require disclosure in this Annual Report on Form 10-K.
ITEM 1A. | RISK FACTORS |
In evaluating our business, you should carefully consider the following risks in addition to the other information in this Annual Report on Form 10-K. A manifestation of any of the following risks could materially and adversely affect our business, results of operations and financial condition. We note these factors for investors as permitted by the Private Securities Litigation Reform Act of 1995. It is not possible to predict or identify all such factors and, therefore, you should not consider the following risks to be a complete statement of all the potential risks or uncertainties that we face.
A substantial portion of our revenues is derived from sales of products to treat HCV and HIV. If we are unable to maintain or continue increasing sales of these products, our results of operations may be adversely affected.
During the year ended December 31, 2015, sales of Harvoni and Sovaldi for the treatment of HCV, accounted for approximately 60% of our total product sales. We cannot be certain if prior year sales of our HCV products are indicative of future sales. The number of warehoused HCV patients has diminished since the first quarter of 2015, and we anticipate that the rate at which new patients started treatment during the second half of 2015 may be more indicative of the pace of new patient starts in 2016. With the approval and entry of competitors' HCV products in December 2014 and January 2016, we cannot predict whether, and to what extent, our HCV revenues may be adversely affected by competition from these products based on price and/or market share. As a result of the launch of competing regimens, we have experienced, and may continue to experience, increased pricing pressure. We have provided significant discounts or rebates to public and private payers in order to obtain formulary status and to expand access for patients to our HCV products.
In addition, future sales of Harvoni and Sovaldi are difficult to estimate because demand depends, in part, on the extent of reimbursement of our HCV products by private and government payers. In light of continued fiscal and debt crises experienced by several countries in the European Union (EU) and Japan, governments have announced or implemented measures to manage healthcare expenditures. We continue to experience global pricing pressure which often results in increases in the amount of discounts required on our products or delayed reimbursement, which could negatively impact our future product sales and results of operations. Also, private and public payers can choose to exclude Harvoni or Sovaldi from their formulary coverage lists or limit the types of patients for whom coverage will be provided, which would negatively impact the demand for, and revenues of, Harvoni and Sovaldi. Any change in the formulary coverage, reimbursement levels or discounts or rebates offered on our HCV products to payers may impact our anticipated revenues. We expect pricing pressure in the HCV market to continue. For example, we anticipate the government of Japan will impose significant pricing discounts for Harvoni and Sovaldi that will start taking effect in the first half of 2016. If we are unable to maintain our forecast for HCV
27
sales similar to prior years or obtain approval or reimbursement for additional HCV product candidates in the currently anticipated timelines, our results of operations and stock price could be negatively affected.
We receive a substantial portion of our revenue from sales of our products for the treatment of HIV infection, particularly our single tablet regimen products, Stribild, Complera/Eviplera and Atripla. During the year ended December 31, 2015, sales of our HIV products accounted for approximately 34% of our total product sales. Most of our HIV products contain tenofovir alafenamide (TAF), tenofovir disoproxil fumarate and/or emtricitabine, which belong to the nucleoside class of antiviral therapeutics. If the treatment paradigm for HIV changes, causing nucleoside-based therapeutics to fall out of favor, or if we are unable to maintain or continue increasing our HIV product sales, our results of operations would likely suffer and we would likely need to scale back our operations, including our spending on research and development (R&D) efforts.
We may not be able to sustain or increase the growth rate of sales of our HCV or HIV products for any number of reasons including, but not limited to, the following:
• | As our HCV and HIV products are used over a longer period of time in many patients and in combination with other products, and additional studies are conducted, new issues with respect to safety, resistance and interactions with other drugs may arise, which could cause us to provide additional warnings or contraindications on our labels, narrow our approved indications or halt sales of a product, each of which could reduce our revenues. |
• | As our products mature, private insurers and government payers often reduce the amount they will reimburse patients for these products, which increases pressure on us to reduce prices. |
• | If physicians do not see the benefit of our HCV or HIV products, the sales of our HCV or HIV products will be limited. |
• | As new HCV or new or generic HIV products are introduced into major markets, our ability to maintain pricing and market share may be affected. |
If we fail to commercialize new products or expand the indications for existing products, our prospects for future revenues may be adversely affected.
If we do not introduce new products or increase sales of our existing products, we will not be able to increase or maintain our total revenues nor continue to expand our R&D efforts. Drug development is inherently risky and many product candidates fail during the drug development process. For example, in January 2016 we announced that we terminated our Phase 2 study of simtuzumab for the treatment of idiopathic pulmonary fibrosis after data showed a lack of treatment benefit.
In the second quarter of 2015, we filed our NDA and MAA for the approval of two doses of a fixed-dose combination of emtricitabine and TAF for the treatment of HIV-1 infection in adults and pediatric patients age 12 years and older, in combination with other HIV antiretroviral agents, in the United States and European Union. In the third quarter of 2015, we filed our NDA and MAA for the approval of the single tablet regimen of rilpivirine, emtricitabine and TAF in the United States and European Union. In the fourth quarter of 2015, we filed our NDA and MAA for the approval of a single tablet regimen of sofosbuvir and velpatasvir for the treatment of HCV in the United States and European Union. In the first quarter of 2016, we filed our NDA and MAA for the approval of TAF for the treatment of chronic hepatitis B virus (HBV) infection in the United States and European Union. These marketing applications may not be approved by the regulatory authorities on a timely basis, or at all. Even if marketing approval is granted for these products, there may be significant limitations on their use. Further, we may be unable to file our marketing applications for new products.
Our inability to accurately predict demand for our products, the uptake of new products or the timing of fluctuations in the inventories maintained by customers makes it difficult for us to accurately forecast sales and may cause our revenues and earnings to fluctuate, which could adversely affect our financial results and our stock price.
We may be unable to accurately predict demand for our products, including the uptake of new products, as demand is dependent on a number of factors. For example, our HCV products, Harvoni and Sovaldi, represent a significant change in the treatment paradigm for HCV-infected patients due to the shortened duration of treatment and the reduction or elimination of the need for pegylated interferon injection and ribavirin in certain patient populations. Because these products represent a cure and are in a new therapeutic area for us, revenues from these products in 2016 and beyond are difficult for us and investors to estimate. Demand for Harvoni and Sovaldi will depend on the availability of HCV patients and the extent of reimbursement of our HCV products by private and public payers in the United States and other countries. Private and public payers can choose to exclude Harvoni or Sovaldi from their formulary coverage lists or limit the types of patients for whom coverage will be provided, which would negatively impact the demand for and revenues of Harvoni and Sovaldi. Also, because our HCV products represent a significant change in the treatment paradigm and in light of the launches of competitive products, sales levels or prescription growth rates may not be indicative of future sales. We have experienced, and may continue to experience, increased pricing pressure in the United States, European Union and other countries and in certain cases, have
28
provided significant discounts to private and public payers in order to obtain formulary status or to expand access for patients to our HCV products. Any change in the formulary coverage, reimbursement levels or discounts or rebates offered on our HCV products to payers may negatively impact our anticipated revenues. We expect pricing pressure to continue. Because HCV-related revenues are difficult to predict, investors may have widely varying expectations that may be materially higher or lower than our actual revenues. To the extent our HCV product revenues exceed or fall short of these expectations, our stock price may experience significant volatility.
In the year ended December 31, 2015, approximately 89% of our product sales in the United States were to three wholesalers, AmerisourceBergen Corp., McKesson Corp. and Cardinal Health, Inc. The U.S. wholesalers with whom we have entered into inventory management agreements make estimates to determine end user demand and may not be completely effective in matching their inventory levels to actual end user demand. As a result, changes in inventory levels held by those wholesalers can cause our operating results to fluctuate unexpectedly if our sales to these wholesalers do not match end user demand. In addition, inventory is held at retail pharmacies and other non-wholesaler locations with whom we have no inventory management agreements and no control over buying patterns. Adverse changes in economic conditions or other factors may cause retail pharmacies to reduce their inventories of our products, which would reduce their orders from wholesalers and, consequently, the wholesalers' orders from us, even if end user demand has not changed. For example, during the second half of 2014, strong wholesaler and sub-wholesaler purchases of our HIV products resulted in inventory draw-down by wholesalers and sub-wholesalers in the first quarter of 2015. As inventory in the distribution channel fluctuates from quarter to quarter, we may continue to see fluctuations in our earnings and a mismatch between prescription demand for our products and our revenues.
In addition, the non-retail sector in the United States, which includes government institutions, including state AIDS Drug Assistance Programs (ADAPs), Veterans Administration (VA), correctional facilities and large health maintenance organizations, tends to be even less consistent in terms of buying patterns and often causes quarter over quarter fluctuations that do not necessarily mirror patient demand for our products. Federal and state budget pressures, including sequestration, as well as the annual grant cycles for federal and state funds, may cause purchasing patterns to not reflect patient demand of our products. For example, in the first quarters of certain prior years, we observed large non-retail purchases of our HIV products by a number of state ADAPs that exceeded patient demand. We believe such purchases were driven by the grant cycle for federal ADAP funds. Additionally, during the second half of 2015, we experienced fluctuations in VA new patient starts and purchasing patterns due to VA funding. We expect to continue to experience fluctuations in the purchasing patterns of our non-retail customers which may result in fluctuations in our product sales, revenues and earnings in the future. In light of the global economic downturn and budget crises faced by many European countries, we have observed variations in purchasing patterns induced by cost containment measures in Europe. We believe these measures have caused some government agencies and other purchasers to reduce inventory of our products in the distribution channels, which has decreased our revenues and caused fluctuations in our product sales and earnings. We may continue to see this trend in the future.
Our results of operations may be adversely affected by current and potential future healthcare reforms.
Legislative and regulatory changes to government prescription drug procurement and reimbursement programs occur relatively frequently in the United States and foreign jurisdictions. In the United States, we, along with other pharmaceutical manufacturers of branded drug products, are required to pay a portion of an industry fee (also known as the Branded Prescription Drug (BPD) fee), calculated based on select government sales during the year as a percentage of total industry government sales. The amount of the annual BPD fee imposed on the pharmaceutical industry as a whole is $3.0 billion in 2015 through 2016, which will increase to $4.0 billion in 2017, increase to a peak of $4.1 billion in 2018, and then decrease to $2.8 billion in 2019 and thereafter. In 2014, the Internal Revenue Service (IRS) issued final regulations which accelerated the expense recognition criteria for the fee obligation from the year in which the fee is paid, to the year in which the market share used to allocate the fee is determined. Our BPD fee expenses were $414 million in 2015, $590 million in 2014 and $110 million in 2013. We expect our portion of the BPD fee to increase as the total annual industry-wide fee increases through 2017 and drug patents expire on major drugs of other companies. The BPD fee is not tax deductible. In addition, even though not addressed in the healthcare reform legislation, discussions continue at the federal level on legislation that would either allow or require the federal government to directly negotiate price concessions from pharmaceutical manufacturers or set minimum requirements for Medicare Part D pricing. Further, certain states have proposed legislation that seek to regulate pharmaceutical drug pricing. If such proposed legislation is passed, we may experience additional pricing pressures on our products.
In addition, state Medicaid programs could request additional supplemental rebates on our products as a result of the increase in the federal base Medicaid rebate. Private insurers could also use the enactment of these increased rebates to exert pricing pressure on our products, and to the extent that private insurers or managed care programs follow Medicaid coverage and payment developments, the adverse effects may be magnified by private insurers adopting lower payment schedules.
29
Our existing products are subject to reimbursement from government agencies and other third parties. Pharmaceutical pricing and reimbursement pressures may reduce profitability.
Successful commercialization of our products depends, in part, on the availability of governmental and third-party payer reimbursement for the cost of such products and related treatments in the markets where we sell our products. Government health authorities, private health insurers and other organizations generally provide reimbursement. In the United States, the European Union, Japan and other significant or potentially significant markets for our products and product candidates, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medical products and services. A significant portion of our sales of the majority of our products are subject to significant discounts from list price and rebate obligations. See also our risk factor "A substantial portion of our revenues is derived from sales of products to treat HCV and HIV. If we are unable to maintain or continue increasing sales of these products, our results of operations may be adversely affected."
In the United States, state AIDS Drug Assistance Programs (ADAPs), which purchase a significant portion of our HIV products, rely on federal, supplemental federal and state funding to help fund purchases of our products. If federal and state funds are not available in amounts sufficient to support the number of patients that rely on ADAPs, sales of our HIV products could be negatively impacted which would reduce our revenues. In prior quarters, because of the insufficiency of federal and state funds, and as many states reduced eligibility criteria, we saw an increase in the number of patients on state ADAP waitlists, and we may see similar increases in future periods as a result of any reduction in federal and state ADAP support. Until these patients are enrolled in an ADAP, they generally receive free product from industry-supported patient assistance programs or are unable to access treatment. The increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our product sales and profitability. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general.
We continue to experience global pricing pressure on our HCV products, which often results in increases in the amount of discounts required on our products or delayed reimbursement, which could negatively impact our future product sales and results of operations. Also, private and public payers can choose to exclude Harvoni or Sovaldi from their formulary coverage or limit the types of patients for whom coverage will be provided, which could negatively impact the demand for, and revenues of, Harvoni and Sovaldi. Any change in the formulary coverage, reimbursement levels or discounts or rebates offered on our HCV products to payers may impact our anticipated revenues. We expect pricing pressure in the HCV market to continue. See also our risk factor "A substantial portion of our revenues is derived from sales of products to treat HCV and HIV. If we are unable to maintain or continue increasing sales of these products, our results of operations may be adversely affected."
In July 2014, we received a letter from the U.S. Senate Committee on Finance (Senate Committee) requesting information and supporting documentation from us related to Sovaldi and the pricing of Sovaldi in the United States. The letter raised concerns about our approach to pricing Sovaldi, its affordability and its impact on federal government spending and public health. In December 2015, the Senate Committee released the results of the investigation, which alleged that we engaged in a revenue-driven pricing strategy in setting Sovaldi's price. Gilead disagrees with many of the conclusions in the report. In January 2016, we received a letter from the Massachusetts Attorney General that their office is considering whether our pricing of Sovaldi and Harvoni may constitute an unfair trade practice in violation of Massachusetts law. In February 2016, the Massachusetts Attorney General’s office served us with a Civil Investigative Demand requesting that we produce documents related to our HCV products. It is possible that the results of the Senate Committee investigation and any actions taken by the Massachusetts Attorney General or other state governments could result in negative publicity or other negative actions that could harm our reputation, reduce demand for Harvoni, Sovaldi or other sofosbuvir containing products and/or reduce coverage of Harvoni, Sovaldi or other sofosbuvir containing products, including by federal health care programs such as Medicare and Medicaid and state health care programs. If any or all of these events occur, our business and stock price could be materially and adversely affected.
In countries outside the United States, the success of our commercialized products, and any other product candidates we may develop, will depend largely on obtaining and maintaining government reimbursement, because in many countries patients are unlikely to use prescription drugs that are not reimbursed by their governments. Recently, many countries in the European Union have increased the level of discounting required on our products, and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union. Cost containment pressures in the European Union, especially in Southern Europe, could lead to delays in the treatment of patients and also delay pricing approval by 12 months or more, which could negatively impact the commercialization of new products. Reimbursement policies may adversely affect our ability to sell our products on a profitable basis. In many international markets, governments control the prices of prescription pharmaceuticals, including
30
through the implementation of reference pricing, price cuts, rebates, revenue-related taxes, tenders and profit control, and they expect prices of prescription pharmaceuticals to decline over the life of the product or as volumes increase. For example, we anticipate the government of Japan will impose significant pricing discounts for Harvoni and Sovaldi that will start taking effect in the first half of 2016.
Approximately 34% of our product sales occur outside the United States, and currency fluctuations and hedging expenses may cause our earnings to fluctuate, which could adversely affect our stock price.
Because a significant percentage of our product sales are denominated in foreign currencies, primarily the Euro and Yen, we face exposure to adverse movements in foreign currency exchange rates. When the U.S. dollar strengthens against these foreign currencies, the relative value of sales made in the respective foreign currency decreases. Conversely, when the U.S. dollar weakens against these currencies, the relative value of such sales increases. Overall, we are a net receiver of foreign currencies and, therefore, benefit from a weaker U.S. dollar and are adversely affected by a stronger U.S. dollar.
We use foreign currency exchange forward and option contracts to hedge a percentage of our forecasted international sales, primarily those denominated in the Euro and Yen. We also hedge certain monetary assets and liabilities denominated in foreign currencies, which reduces but does not eliminate our exposure to currency fluctuations between the date a transaction is recorded and the date that cash is collected or paid. Foreign currency exchange, net of hedges, had an unfavorable impact of $737 million on our 2015 revenues compared to 2014 and a favorable impact of $39 million on our 2014 revenues compared to 2013.
We cannot predict future fluctuations in the foreign currency exchange rates of the U.S. dollar. If the U.S. dollar appreciates significantly against certain currencies and our hedging program does not sufficiently offset the effects of such appreciation, our results of operations will be adversely affected and our stock price may decline.
Additionally, the expenses that we recognize in relation to our hedging activities can also cause our earnings to fluctuate. The level of hedging expenses that we recognize in a particular period is impacted by the changes in interest rate spreads between the foreign currencies that we hedge and the U.S. dollar.
We face significant competition.
We face significant competition from large global pharmaceutical and biotechnology companies, specialized pharmaceutical firms and generic drug manufacturers.
Our HCV products, Harvoni and Sovaldi, compete with Viekira Pak (ombitasvir, paritaprevir and ritonavir tablets co-packaged with dasabuvir tablets) marketed by AbbVie Inc. (AbbVie), Zepatier (elbasvir and grazoprevir) marketed by Merck & Co. Inc. (Merck), Daklinza (daclastavir) marketed by Bristol-Myers Squibb Company (BMS) and Olysio (simeprevir) marketed by Janssen Therapeutics.
Our HIV products compete primarily with products from ViiV Healthcare (ViiV), which markets fixed-dose combination products that compete with Genvoya, Stribild, Complera/Eviplera, Atripla and Truvada. For example, two products marketed by ViiV, Tivicay (dolutegravir), an integrase inhibitor, and Triumeq, a single-tablet triple-combination antiretroviral regimen, could adversely impact sales of our HIV products. In addition, lamivudine, marketed by this joint venture, competes with emtricitabine, the active pharmaceutical ingredient of Emtriva and a component of Genvoya, Stribild, Complera/Eviplera, Atripla and Truvada. For Tybost, we compete with ritonavir marketed by AbbVie.
We also face competition from generic HIV products. Generic versions of lamivudine and Combivir (lamivudine and zidovudine) are available in the United States and certain other countries. Generic versions of Sustiva (efavirenz), a component of our Atripla, are now available in Canada and Europe and we anticipate competition from generic efavirenz in the United States in December 2017. We have observed some pricing pressure related to the Sustiva component of our Atripla sales.
Our HBV products, Viread and Hepsera, face competition from Baraclude (entecavir) marketed by BMS as well as generic entecavir. Our HBV products also compete with Tyzeka/Sebivo (telbivudine) marketed by Novartis Pharmaceuticals Corporation (Novartis).
Zydelig competes with Imbruvica (ibrutinib) marketed by Pharmacyclics, Inc., Gazyva (obinutuzumab) marketed by Genentech (a member of the Roche Group) and Treanda (bendamustine hydrochloride) marketed by Cephalon, Inc.
Letairis competes with Tracleer (bosentan) and Opsumit (macitentan) marketed by Actelion Pharmaceuticals US, Inc. and also with Adcirca (tadalafil) marketed by United Therapeutics Corporation and Pfizer Inc. (Pfizer).
31
Ranexa competes predominantly with generic compounds from three distinct classes of drugs for the treatment of chronic angina in the United States, including generic and/or branded beta-blockers, calcium channel blockers and long-acting nitrates.
Cayston competes with Tobi (tobramycin inhalation solution) marketed by Novartis.
Tamiflu competes with Relenza (zanamivir) marketed by GlaxoSmithKline and products sold by generic competitors.
AmBisome competes with Vfend (voriconazole) marketed by Pfizer and caspofungin, a product developed by Merck that is marketed as Cancidas in the United States and as Caspofungin elsewhere. In addition, we are aware of at least three lipid formulations that claim similarity to AmBisome becoming available outside of the United States. These formulations may reduce market demand for AmBisome. Furthermore, the manufacture of lipid formulations of amphotericin B is very complex and if any of these formulations are found to be unsafe, sales of AmBisome may be negatively impacted by association.
In addition, a number of companies are pursuing the development of technologies which are competitive with our existing products or research programs. These competing companies include specialized pharmaceutical firms and large pharmaceutical companies acting either independently or together with other pharmaceutical companies. Furthermore, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection and may establish collaborative arrangements for competitive products or programs. If any of these competitors gain market share on our products, it could adversely affect our results of operations and stock price.
If significant safety issues arise for our marketed products or our product candidates, our future sales may be reduced, which would adversely affect our results of operations.
The data supporting the marketing approvals for our products and forming the basis for the safety warnings in our product labels were obtained in controlled clinical trials of limited duration and, in some cases, from post-approval use. As our products are used over longer periods of time by many patients with underlying health problems, taking numerous other medicines, we expect to continue to find new issues such as safety, resistance or drug interaction issues, which may require us to provide additional warnings or contraindications on our labels or narrow our approved indications, each of which could reduce the market acceptance of these products.
Regulatory authorities have been moving towards more active and transparent pharmacovigilance and are making greater amounts of stand-alone safety information and clinical trial data directly available to the public through websites and other means, e.g. periodic safety update report summaries, risk management plan summaries and various adverse event data. Safety information, without the appropriate context and expertise, may be misinterpreted and lead to misperception or legal action which may potentially cause our product sales or stock price to decline.
Further, if serious safety, resistance or drug interaction issues arise with our marketed products, sales of these products could be limited or halted by us or by regulatory authorities and our results of operations would be adversely affected.
Our operations depend on compliance with complex FDA and comparable international regulations. Failure to obtain broad approvals on a timely basis or to maintain compliance could delay or halt commercialization of our products.
The products we develop must be approved for marketing and sale by regulatory authorities and, once approved, are subject to extensive regulation by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA) and comparable regulatory agencies in other countries. We are continuing clinical trials for Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya, Emtriva, Tybost, Vitekta, Letairis, Ranexa, Cayston, Zydelig and Hepsera for currently approved and additional uses. We anticipate that we will file for marketing approval in additional countries and for additional indications and products over the next several years. These products may fail to receive such marketing approvals on a timely basis, or at all.
Further, how we manufacture and sell our products is subject to extensive regulation and review. Discovery of previously unknown problems with our marketed products or problems with our manufacturing, safety reporting or promotional activities may result in restrictions on our products, including withdrawal of the products from the market. If we fail to comply with applicable regulatory requirements, including those related to promotion and manufacturing, we could be subject to penalties including fines, suspensions of regulatory approvals, product recalls, seizure of products and criminal prosecution.
For example, under FDA rules, we are often required to conduct post-approval clinical studies to assess a known serious risk, signals of serious risk or to identify an unexpected serious risk and implement a Risk Evaluation and Mitigation Strategy for our products, which could include a medication guide, patient package insert, a communication plan to healthcare providers or other elements as FDA deems are necessary to assure safe use of the drug, which could include imposing certain
32
restrictions on the distribution or use of a product. Failure to comply with these or other requirements, if imposed on a sponsor by FDA, could result in significant civil monetary penalties and our operating results may be adversely affected.
The results and anticipated timelines of our clinical trials are uncertain and may not support continued development of a product candidate, which would adversely affect our prospects for future revenue growth.
We are required to demonstrate the safety and efficacy of products that we develop for each intended use through extensive preclinical studies and clinical trials. The results from preclinical and early clinical studies do not always accurately predict results in later, large-scale clinical trials. Even successfully completed large-scale clinical trials may not result in marketable products. For example, in January 2016, we announced that we terminated our Phase 2 trial of simtuzumab for the treatment of idiopathic pulmonary fibrosis after results showed a lack of treatment benefit. If any of our product candidates fails to achieve its primary endpoint in clinical trials, if safety issues arise or if the results from our clinical trials are otherwise inadequate to support regulatory approval of our product candidates, commercialization of that product candidate could be delayed or halted. In addition, we may also face challenges in clinical trial protocol design.
If the clinical trials for any of the product candidates in our pipeline are delayed or terminated, our prospects for future revenue growth would be adversely impacted. For example, we face numerous risks and uncertainties with our product candidates, including the single tablet regimen of GS-9883, emtricitabine and TAF for the treatment of HIV, the single tablet regimen of SOF, velpatasvir and GS-9857 for the treatment of chronic HCV, idelalisib for the treatment of relapsed refractory indolent non-Hodgkin lymphoma and frontline and relapsed refractory chronic lymphocytic leukemia; momelotinib for the treatment of myelofibrosis and pancreatic cancer; eleclazine (formerly GS-6615) for the treatment of long QT-3 syndrome; and GS-5745 for the treatment of ulcerative colitis, each currently in Phase 3 clinical trials, that could prevent completion of development of these product candidates. These risks include our ability to enroll patients in clinical trials, the possibility of unfavorable results of our clinical trials, the need to modify or delay our clinical trials or to perform additional trials and the risk of failing to obtain FDA and other regulatory body approvals. As a result, our product candidates may never be successfully commercialized. Further, we may make a strategic decision to discontinue development of our product candidates if, for example, we believe commercialization will be difficult relative to other opportunities in our pipeline. If these programs and others in our pipeline cannot be completed on a timely basis or at all, then our prospects for future revenue growth may be adversely impacted. In addition, clinical trials involving our commercial products could raise new safety issues for our existing products, which could in turn decrease our revenues and harm our business.
Due to our reliance on third-party contract research organizations to conduct our clinical trials, we are unable to directly control the timing, conduct, expense and quality of our clinical trials.
We extensively outsource our clinical trial activities and usually perform only a small portion of the start-up activities in-house. We rely on independent third-party contract research organizations (CROs) to perform most of our clinical studies, including document preparation, site identification, screening and preparation, pre-study visits, training, program management and bioanalytical analysis. Many important aspects of the services performed for us by the CROs are out of our direct control. If there is any dispute or disruption in our relationship with our CROs, our clinical trials may be delayed. Moreover, in our regulatory submissions, we rely on the quality and validity of the clinical work performed by third-party CROs. If any of our CROs' processes, methodologies or results were determined to be invalid or inadequate, our own clinical data and results and related regulatory approvals could be adversely affected.
We depend on relationships with other companies for sales and marketing performance, development and commercialization of product candidates and revenues. Failure to maintain these relationships, poor performance by these companies or disputes with these companies could negatively impact our business.
We rely on a number of significant collaborative relationships with major pharmaceutical companies for our sales and marketing performance in certain territories. These include collaborations with Janssen for Complera/Eviplera; BMS for Atripla in the United States, Europe and Canada; F. Hoffmann-La Roche Ltd. (together with Hoffmann-La Roche Inc., Roche) for Tamiflu worldwide; and GSK for ambrisentan in territories outside of the United States. In some countries, we rely on international distributors for sales of Truvada, Viread, Hepsera, Emtriva and AmBisome. Some of these relationships also involve the clinical development of these products by our partners. Reliance on collaborative relationships poses a number of risks, including the risk that:
• | we are unable to control the resources our corporate partners devote to our programs or products; |
• | disputes may arise with respect to the ownership of rights to technology developed with our corporate partners; |
• | disagreements with our corporate partners could cause delays in, or termination of, the research, development or commercialization of product candidates or result in litigation or arbitration; |
33
• | contracts with our corporate partners may fail to provide significant protection or may fail to be effectively enforced if one of these partners fails to perform; |
• | our corporate partners have considerable discretion in electing whether to pursue the development of any additional products and may pursue alternative technologies or products either on their own or in collaboration with our competitors; |
• | our corporate partners with marketing rights may choose to pursue competing technologies or to devote fewer resources to the marketing of our products than they do to products of their own development; and |
• | our distributors and our corporate partners may be unable to pay us, particularly in light of current economic conditions. |
Given these risks, there is a great deal of uncertainty regarding the success of our current and future collaborative efforts. If these efforts fail, our product development or commercialization of new products could be delayed or revenues from products could decline.
In addition, Letairis and Cayston are distributed through third-party specialty pharmacies, which are pharmacies specializing in the dispensing of medications for complex or chronic conditions that may require a high level of patient education and ongoing counseling. The use of specialty pharmacies requires significant coordination with our sales and marketing, medical affairs, regulatory affairs, legal and finance organizations and involves risks, including but not limited to risks that these specialty pharmacies will:
• | not provide us with accurate or timely information regarding their inventories, patient data or safety complaints; |
• | not effectively sell or support Letairis or Cayston; |
• | not devote the resources necessary to sell Letairis or Cayston in the volumes and within the time frames that we expect; |
• | not be able to satisfy their financial obligations to us or others; or |
• | cease operations. |
We also rely on a third party to administer our Letairis Education and Access Program (LEAP), the restricted distribution program designed to support Letairis. This third party provides information and education to prescribers and patients on the risks of Letairis, confirms insurance coverage and investigates alternative sources of reimbursement or assistance, ensures fulfillment of the risk management requirements mandated for Letairis by FDA and coordinates and controls dispensing to patients through the third-party specialty pharmacies. Failure of this third party or the specialty pharmacies that distribute Letairis to perform as expected may result in regulatory action from FDA or decreased Letairis sales, either of which would harm our business.
Further, Cayston may only be taken by patients using a specific inhalation device that delivers the drug to the lungs of patients. Our ongoing distribution of Cayston is entirely reliant upon the manufacturer of that device. This manufacturer could encounter other issues with regulatory agencies related to the device or be unable to supply sufficient quantities of this device. In addition, the manufacturer may not be able to provide adequate warranty support for the device after it has been distributed to patients. With respect to distribution of the drug and device to patients, we are reliant on the capabilities of specialty pharmacies. For example, the distribution channel for drug and device is complicated and requires coordination. The reimbursement approval processes associated with both drug and device are similarly complex. If the device manufacturer is unable to obtain reimbursement approval or receives approval at a lower-than-expected price, sales of Cayston may be adversely affected. Any of the previously described issues may limit the sales of Cayston, which would adversely affect our financial results.
Our success will depend to a significant degree on our ability to defend our patents and other intellectual property rights both domestically and internationally. We may not be able to obtain effective patents to protect our technologies from use by competitors and patents of other companies could require us to stop using or pay for the use of required technology.
Patents and other proprietary rights are very important to our business. Our success will depend to a significant degree on our ability to:
• | obtain patents and licenses to patent rights; |
• | preserve trade secrets; |
• | defend against infringement and efforts to invalidate our patents; and |
• | operate without infringing on the intellectual property of others. |
34
If we have a properly drafted and enforceable patent, it can be more difficult for our competitors to use our technology to create competitive products and more difficult for our competitors to obtain a patent that prevents us from using technology we create. As part of our business strategy, we actively seek patent protection both in the United States and internationally and file additional patent applications, when appropriate, to cover improvements in our compounds, products and technology.
We have a number of U.S. and foreign patents, patent applications and rights to patents related to our compounds, products and technology, but we cannot be certain that issued patents will be enforceable or provide adequate protection or that pending patent applications will result in issued patents. Patent applications are confidential for a period of time before a patent is issued. As a result, we may not know if our competitors filed patent applications for technology covered by our pending applications or if we were the first to invent or first to file an application directed toward the technology that is the subject of our patent applications. Competitors may have filed patent applications or received patents and may obtain additional patents and proprietary rights that block or compete with our products. In addition, if competitors file patent applications covering our technology, we may have to participate in litigation, interference or other proceedings to determine the right to a patent. Litigation, interference or other proceedings are unpredictable and expensive, such that, even if we are ultimately successful, our results of operations may be adversely affected by such events.
Patents do not cover the ranolazine compound, the active ingredient of Ranexa. Instead, when it was discovered that only a sustained-release formulation of ranolazine would achieve therapeutic plasma levels, patents were obtained on those formulations and the characteristic plasma levels they achieve. Patents do not cover the active ingredients in AmBisome.
We may obtain patents for certain products many years before marketing approval is obtained for those products. Because patents have a limited life, which may begin to run prior to the commercial sale of the related product, the commercial value of the patent may be limited. However, we may be able to apply for patent term extensions or supplementary protection certificates in some countries.
Generic manufacturers have sought, and may continue to seek, FDA approval to market generic versions of our products through an abbreviated new drug application (ANDA), the application form typically used by manufacturers seeking approval of a generic drug. See a description of our ANDA litigation in Note 11 Commitments and Contingencies - Legal Proceedings of the Notes to Consolidated Financial Statements included in Item 8 of this Annual Report on Form 10-K and risk factor entitled "Litigation with generic manufacturers has increased our expenses which may continue to reduce our earnings. If we are unsuccessful in all or some of these lawsuits, some or all of our claims in the patents may be narrowed or invalidated and generic versions of our products could be launched prior to our patent expiry." beginning on page 39.
Our success depends in large part on our ability to operate without infringing upon the patents or other proprietary rights of third parties.
If we infringe the valid patents of third parties, we may be prevented from commercializing products or may be required to obtain licenses from these third parties. We may not be able to obtain alternative technologies or any required license on reasonable terms or at all. If we fail to obtain these licenses or alternative technologies, we may be unable to develop or commercialize some or all of our products. For example, we are aware of patents that may relate to our operation of LEAP, our restricted distribution program designed to support Letairis and we are aware of patents and patent applications owned by other parties that may claim to cover the use of sofosbuvir. See a description of our litigation regarding sofosbuvir in Note 11 Commitments and Contingencies - Legal Proceedings of the Notes to Consolidated Financial Statements included in Item 8 of this Annual Report on Form 10-K and the risk factor entitled "If any party is successful in establishing exclusive rights to Harvoni and/or Sovaldi, our expected revenues and earnings from the sale of Harvoni and/or Sovaldi could be adversely affected" beginning on page 36.
Furthermore, we also rely on unpatented trade secrets and improvements, unpatented internal know-how and technological innovation. We protect these rights mainly through confidentiality agreements with our corporate partners, employees, consultants and vendors. These agreements provide that all confidential information developed or made known to an individual during the course of their relationship with us will be kept confidential and will not be used or disclosed to third parties except in specified circumstances. In the case of employees, the agreements provide that all inventions made by an individual while employed by us will be our exclusive property. We cannot be certain that these parties will comply with these confidentiality agreements, that we have adequate remedies for any breach or that our trade secrets will not otherwise become known or be independently discovered by our competitors. Under some of our R&D agreements, inventions become jointly owned by us and our corporate partner and in other cases become the exclusive property of one party. In certain circumstances, it can be difficult to determine who owns a particular invention and disputes could arise regarding those inventions. If our trade secrets or confidential information become known or independently discovered by competitors or if we enter into disputes over ownership of inventions, our business and results of operations could be adversely affected.
35
If any party is successful in establishing exclusive rights to Harvoni and/or Sovaldi, our expected revenues and earnings from the sale of Harvoni and/or Sovaldi could be adversely affected.
We own patents and patent applications that claim sofosbuvir (Sovaldi) as a chemical entity and its metabolites and the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni). Third parties may have, or may obtain rights to, patents that allegedly could be used to prevent or attempt to prevent us from commercializing Harvoni or Sovaldi. For example, we are aware of patents and patent applications owned by other parties that may be alleged by such parties to cover the use of Harvoni and Sovaldi. We cannot predict the ultimate outcome of intellectual property claims related to Harvoni or Sovaldi, and we have spent, and will continue to spend, significant resources defending against these claims. If these parties successfully obtain valid and enforceable patents, and successfully prove infringement of those patents by Harvoni and/or Sovaldi, we could be prevented from selling sofosbuvir unless we were able to obtain a license under such patents. Such a license may not be available on commercially reasonable terms or at all.
Interference Proceedings and Litigation with Idenix Pharmaceuticals, Inc. (Idenix)
In February 2012, we received notice that the U.S. Patent and Trademark Office (USPTO) had declared Interference No. 105,871 (First Idenix Interference) between our U.S. Patent No. 7,429,572 (the ’572 patent) and Idenix's pending U.S. Patent Application No. 12/131,868. An interference is a proceeding before the USPTO designed to determine who was the first to invent the subject matter claimed by both parties. In January 2014, the USPTO Patent Trial and Appeal Board (PTAB) determined that Pharmasset and not Idenix was the first to invent the compounds in dispute and accordingly Gilead prevailed in the First Idenix Interference. Idenix has appealed the PTAB’s decisions to the U.S. District Court for the District of Delaware.
In December 2013, after receiving our request to do so, the USPTO declared Interference No. 105,981 (Second Idenix Interference) between our pending U.S. Patent Application No. 11/854,218 and Idenix’s U.S. Patent No. 7,608,600 (the ’600 patent). The ’600 patent is related to the Idenix patent application at issue in the First Idenix Interference and includes claims directed to methods of treating HCV with nucleoside compounds. The purpose of the Second Idenix Interference was to determine who was first to invent the claimed methods of treating HCV with compounds similar to those which were involved in the First Idenix Interference. In March 2015, the PTAB determined that Pharmasset and not Idenix was the first to invent the claimed methods of treating HCV. Idenix appealed this decision in both the U.S. District Court for the District of Delaware and the U.S. Court of Appeal for the Federal Circuit (CAFC). We have filed a motion to dismiss the appeal in Delaware and will respond to the appeal filed in the CAFC.
We believe that the Idenix claims involved in the First and Second Idenix Interferences, and similar U.S. and foreign patents claiming the same compounds, metabolites and uses thereof, are invalid. As a result, we filed an Impeachment Action in the Federal Court of Canada to invalidate Idenix Canadian Patent No. 2,490,191 (the ’191 patent), which is the Canadian patent that corresponds to the ’600 patent. Idenix asserted that the commercialization of Sovaldi in Canada will infringe its ’191 patent and that our Canadian Patent No. 2,527,657, corresponding to the ’572 patent involved in the First Idenix Interference, is invalid. A trial on these issues was held in January and February 2015, and in November 2015, the Federal Court of Canada rendered its public decision holding that Idenix's patent is invalid and that Gilead's patent is valid. In the same month, Idenix appealed the court's decision.
We filed a similar legal action in Norway in the Oslo District Court seeking to invalidate Idenix's Norwegian patent corresponding to the ’600 patent. In September 2013, Idenix filed an invalidation action in the Norwegian proceedings against our Norwegian Patent No. 333700 patent, which corresponds to Gilead's ’572 patent. In March 2014, the Norwegian court found all claims in the Idenix Norwegian patent to be invalid and upheld the validity of all claims in the challenged Gilead patent. In April 2014, Idenix appealed the March 2014 decision to the Norwegian Court of Appeal. The appeal hearing from the March 2014 decision took place in February 2016.
In January 2013, we filed a legal action in the Federal Court of Australia seeking to invalidate Idenix’s Australian patent corresponding to the ’600 patent. In April 2013, Idenix asserted that the commercialization of Sovaldi in Australia will infringe its Australian patent corresponding to the ’600 patent. A month-long trial was completed in October 2015 in Sydney. A decision is pending.
In March 2014, the European Patent Office (EPO) granted Idenix European Patent No. 1 523 489 (the ’489 patent), which corresponds to the ’600 patent. The same day that the ’489 patent was granted, we filed an opposition with the EPO seeking to revoke the ’489 patent. An opposition hearing was held in February 2016, and the EPO ruled in our favor and revoked the '489 patent. In March 2014, Idenix also initiated infringement proceedings against us in the United Kingdom (UK), Germany and France alleging that the commercialization of Sovaldi would infringe the UK, German and French counterparts of the ’489 patent. A trial was held in the UK in October 2014 to determine the issues of infringement and validity of the Idenix UK patent. In December 2014, the High Court of Justice of England and Wales (UK Court) invalidated
36
all challenged claims of the ’489 patent on multiple grounds. The UK Court has granted Idenix permission to appeal the December 2014 judgment. The appeal of the UK Court's decision is scheduled for July 2016. In March 2015, the German court in Düsseldorf determined that the Idenix patent was highly likely to be invalid and stayed the infringement proceedings pending the outcome of the opposition hearing held by the EPO in February 2016. Idenix has not appealed this decision of the German court staying the proceedings. Upon Idenix's request, the French proceedings have been stayed.
Idenix has not been awarded patents corresponding to the ’600 patent in Japan or China. In the event such patents are issued, we expect to challenge them in proceedings similar to those we invoked in other countries.
In December 2013, Idenix, Universita Degli Studi di Cagliari (UDSG), Centre National de la Recherche Scientifique and L’Université Montpellier II sued us in U.S. District Court for the District of Delaware alleging that the commercialization of sofosbuvir will infringe the ’600 patent and that an interference exists between the ’600 patent and our U.S. Patent No. 8,415,322. Also in December 2013, Idenix and UDSG sued us in the U.S. District Court for the District of Massachusetts alleging that the commercialization of sofosbuvir will infringe U.S. Patent Nos. 6,914,054 and 7,608,597. In June 2014, the court transferred the Massachusetts litigation to the U.S. District Court for the District of Delaware. The district court has set trial dates in October 2016 and December 2016 for resolution of these issues. A decision by the district court may be appealed by either party to the CAFC.
Idenix was acquired by Merck in August 2014. While the acquisition does not change our view of the lack of merit in the claims made by Idenix, Merck has greater resources than Idenix and may therefore choose to fund the litigation at higher levels than Idenix.
Litigation with Merck
In August 2013, Merck contacted us requesting that we pay royalties on the sales of sofosbuvir and obtain a license to U.S. Patent Nos. 7,105,499 and 8,481,712, which it co-owns with Isis Pharmaceuticals, Inc. In August 2013, we filed a lawsuit in the U.S. District Court for the Northern District of California seeking a declaratory judgment that the Merck patents are invalid and not infringed. Merck’s U.S. Patent Nos. 7,105,499 and 8,481,712 cover compounds which do not include, but may relate to, sofosbuvir. During patent prosecution, Merck amended its patent application in an attempt to cover compounds related to sofosbuvir. If the court determines that Merck’s patents are valid and that we have infringed those claims, we may be required to obtain a license from and pay royalties to Merck to commercialize sofosbuvir. The court has set a trial date of March 7, 2016 for this lawsuit. Either party may appeal a decision by the District Court to the CAFC.
Litigation with AbbVie
AbbVie has obtained U.S. Patent Nos. 8,466,159, 8,492,386, 8,680,106, 8,685,984, and 8,809,265 (AbbVie Patents) which purport to cover the use of a combination of LDV/SOF (or Harvoni) for the treatment of HCV. Gilead is aware that AbbVie has pending patent applications in the United States and granted and pending applications in other countries. We own published and pending patent applications directed to the use of combinations for the treatment of HCV, and, specifically, to the combination of LDV/SOF. Certain of our applications were filed before the AbbVie Patents. For this reason and others, we believe the AbbVie Patents are invalid.
Accordingly, in December 2013, we filed a lawsuit in the U.S. District Court for the District of Delaware seeking declaratory judgment that the AbbVie Patents are invalid and unenforceable, as well as other relief. We believe that Abbott Laboratories, Inc. and AbbVie conspired to eliminate competition in the HCV market by falsely representing to the USPTO that they, and not Gilead, invented methods of treating HCV using a combination of LDV/SOF. In February and March 2014, AbbVie responded to our lawsuit by also filing two lawsuits in the U.S. District Court for the District of Delaware alleging that our fixed-dose combination of LDV/SOF will infringe its patents. All of those lawsuits have been consolidated into a single action. In the United States, either party may appeal a decision by the District Court to the CAFC. The AbbVie Patents have not blocked or delayed the commercialization of our combination product in the United States, Canada, or Europe. We do not expect any other foreign patents to block or delay the commercialization around the world. The court has set a trial date of September 12, 2016 for this lawsuit.
Additionally, AbbVie has obtained U.S. Patent No. 9,034,832 which purports to cover a solid oral dosage form containing ledipasvir. Accordingly, in May 2015, we filed a lawsuit in the U.S. District Court for the District of Delaware seeking declaratory judgment that AbbVie’s patent is invalid, as well as other relief. We do not expect AbbVie’s patent to block the commercialization of our combination product. The court has set a trial date of July 31, 2017.
In August 2015, we brought an impeachment action seeking a declaration that AbbVie's Canadian Patent No. 2,811,250 ('250 Patent), which purports to cover the use of a combination of LDV/SOF for the treatment of HCV, is invalid. On the same day, AbbVie brought an infringement action which asserts that commercialization of Harvoni in Canada will infringe its '250
37
Patent. The impeachment action has been stayed and we have counterclaimed for invalidity in the infringement proceeding. A trial date has not been set.
In November 2015, AbbVie filed a lawsuit against us in the Regional Court Düsseldorf for infringement of two quasi-patents, known as “utility models.” Utility models are unexamined IP rights and are not the same as standard patents. One utility model, DE 20 2012 013 117, purports to cover the use of a combination of direct-acting antivirals which includes at least an HCV polymerase inhibitor and an HCV NS5A inhibitor in the treatment of HCV; the other utility model, DE 21 2012 000 197, purports to cover a solid dispersion that includes ledipasvir. A trial date has not been set.
If a court determines that the AbbVie Patents are valid and that we have infringed those claims, we may be required to obtain a license from and pay royalties to AbbVie to commercialize sofosbuvir combination products.
European Patent Claims
In February 2015, several parties filed oppositions in the European Patent Office requesting revocation of our granted European patent covering sofosbuvir that expires in 2028. While we are confident in the strength of our sofosbuvir patent, we cannot predict the ultimate outcome of these actions. If we are unsuccessful in defending these oppositions, some or all of our patent claims may be narrowed or revoked and the patent protection for sofosbuvir in Europe could be substantially shortened or eliminated entirely. If the sofosbuvir patent is revoked, and no other European patents are granted covering sofosbuvir, our exclusivity will be based entirely on regulatory exclusivity granted by the EMA. Sovaldi has been granted regulatory exclusivity that will prevent generic sofosbuvir from entering the EU for 10 years following approval of Sovaldi, or January 2024. If we lose exclusivity for Sovaldi prior to 2028, our expected revenues and results of operation could be negatively impacted for the years including and succeeding the year in which such exclusivity is lost, which may cause our stock price to decline.
Manufacturing problems, including at our third-party manufacturers and corporate partners, could cause inventory shortages and delay product shipments and regulatory approvals, which may adversely affect our results of operations.
In order to generate revenue from our products, we must be able to produce sufficient quantities of our products to satisfy demand. Many of our products are the result of complex manufacturing processes. The manufacturing process for pharmaceutical products is also highly regulated and regulators may shut down manufacturing facilities that they believe do not comply with regulations.
Our products are either manufactured at our own facilities or by third-party manufacturers or corporate partners. We depend on third parties to perform manufacturing activities effectively and on a timely basis for the majority of our solid dose products. In addition, Roche, either by itself or through third parties, is responsible for manufacturing Tamiflu. We, our third-party manufacturers and our corporate partners are subject to Good Manufacturing Practices (GMP), which are extensive regulations governing manufacturing processes, stability testing, record keeping and quality standards as defined by FDA and the EMA. Similar regulations are in effect in other countries.
Our third-party manufacturers and corporate partners are independent entities who are subject to their own unique operational and financial risks which are out of our control. If we or any of these third-party manufacturers or corporate partners fail to perform as required, this could impair our ability to deliver our products on a timely basis or receive royalties or cause delays in our clinical trials and applications for regulatory approval. Further, we may have to write-off the costs of manufacturing any batch that fails to pass quality inspection or meet regulatory approval. In addition, we, our third-party manufacturers and our corporate partners may only be able to produce some of our products at one or a limited number of facilities and, therefore, have limited manufacturing capacity for certain products. To the extent these risks materialize and affect their performance obligations to us, our financial results may be adversely affected.
Our manufacturing operations are subject to routine inspections by regulatory agencies. For example, in 2014, we received a letter from FDA related to the extent of method revalidations being conducted, stability program oversight, audit trail review/data management and Quality Management System gaps. We completed and filed our responses to these observations with FDA. If we are unable to remedy the deficiencies cited by FDA or to the extent there are additional deficiencies cited by FDA in future inspections, our currently marketed products and the timing of regulatory approval of products in development could be adversely affected. Further, there is risk that regulatory agencies in other countries where marketing applications are pending will undertake similar additional reviews or apply a heightened standard of review, which could delay the regulatory approvals for products in those countries. If approval of any of our product candidates were delayed or if production of our marketed products was interrupted, our anticipated revenues and our stock price would be adversely affected.
38
We may not be able to obtain materials or supplies necessary to conduct clinical trials or to manufacture and sell our products, which would limit our ability to generate revenues.
We need access to certain supplies and products to conduct our clinical trials and to manufacture our products. If we are unable to purchase sufficient quantities of these materials or find suitable alternate materials in a timely manner, our development efforts for our product candidates may be delayed or our ability to manufacture our products would be limited, which would limit our ability to generate revenues.
Suppliers of key components and materials must be named in the NDA or MAA filed with FDA, EMA or other regulatory authority for any product candidate for which we are seeking marketing approval, and significant delays can occur if the qualification of a new supplier is required. Even after a manufacturer is qualified by the regulatory authority, the manufacturer must continue to expend time, money and effort in the area of production and quality control to ensure full compliance with GMP. Manufacturers are subject to regular, periodic inspections by the regulatory authorities following initial approval. If, as a result of these inspections, a regulatory authority determines that the equipment, facilities, laboratories or processes do not comply with applicable regulations and conditions of product approval, the regulatory authority may suspend the manufacturing operations. If the manufacturing operations of any of the single suppliers for our products are suspended, we may be unable to generate sufficient quantities of commercial or clinical supplies of product to meet market demand, which would in turn decrease our revenues and harm our business. In addition, if delivery of material from our suppliers were interrupted for any reason, we may be unable to ship certain of our products for commercial supply or to supply our products in development for clinical trials. In addition, some of our products and the materials that we utilize in our operations are made at only one facility. For example, we manufacture certain drug product intermediates utilized in AmBisome exclusively at our facilities in San Dimas, California. In the event of a disaster, including an earthquake, equipment failure or other difficulty, we may be unable to replace this manufacturing capacity in a timely manner and may be unable to manufacture AmBisome to meet market needs.
In addition, we depend on a single supplier for amphotericin B, the active pharmaceutical ingredient of AmBisome, and high-quality cholesterol in the manufacture of AmBisome. We also rely on a single source for the active pharmaceutical ingredients found in Letairis and Cayston. Astellas US LLC, which markets Lexiscan in the United States, is responsible for the commercial manufacture and supply of product in the United States and is dependent on a single supplier for the active pharmaceutical ingredient of Lexiscan. Problems with any of the single suppliers we depend on may negatively impact our development and commercialization efforts.
A significant portion of the raw materials and intermediates used to manufacture our antiviral products (including Harvoni, Sovaldi, Truvada, Atripla, Stribild, Complera/Eviplera, Viread, Genvoya and Emtriva) are supplied by China-based companies. As a result, an international trade dispute between China and the United States or any other actions by the Chinese government that would limit or prevent Chinese companies from supplying these materials would adversely affect our ability to manufacture and supply our antiviral products to meet market needs and have a material and adverse effect on our operating results.
Litigation with generic manufacturers has increased our expenses which may continue to reduce our earnings. If we are unsuccessful in all or some of these lawsuits, some or all of our claims in the patents may be narrowed or invalidated and generic versions of our products could be launched prior to our patent expiry.
As part of the approval process for some of our products, FDA granted us a New Chemical Entity (NCE) exclusivity period during which other manufacturers' applications for approval of generic versions of our product will not be approved. Generic manufacturers may challenge the patents protecting products that have been granted NCE exclusivity one year prior to the end of the NCE exclusivity period. Generic manufacturers have sought and may continue to seek FDA approval for a similar or identical drug through an ANDA, the application form typically used by manufacturers seeking approval of a generic drug. Current legal proceedings of significance with some of our generic manufacturers include:
39
Mylan
In April 2014 and July 2015, we received notices that Mylan Inc. (Mylan) submitted an ANDA to FDA requesting permission to manufacture and market generic versions of Truvada and Complera. In the notice, Mylan alleges that the patents associated with Truvada and Complera are invalid, unenforceable and/or will not be infringed by Mylan's manufacture, use or sale of a generic version of these products. We filed lawsuits against Mylan in U.S. District Court for the Northern District of West Virginia for infringement of our patents. In June 2014, we received notice that Mylan submitted petitions for Inter Partes Review (IPR) to the PTAB alleging that four patents associated with tenofovir disoproxil fumarate are invalid. We opposed Mylan’s petitions. In December 2014, the PTAB issued decisions denying each of Mylan’s petitions for IPR. In January 2015, Mylan requested a rehearing on the basis that it believes the PTAB decision is wrong. In August 2015 and November 2015, the PTAB denied Mylan's requests for a rehearing. In October 2015, we reached an agreement with Mylan to settle the proceedings. The terms of the settlement agreement are confidential.
Apotex
In June 2014, we received notice that Apotex Inc. (Apotex) submitted an abbreviated new drug submission (ANDS) to Health Canada requesting permission to manufacture and market a generic version of Truvada and a separate ANDS requesting permission to manufacture and market a generic version of Viread. In the notice, Apotex alleges that three of the patents associated with Truvada and two of the patents associated with Viread are invalid, unenforceable and/or will not be infringed by Apotex's manufacture, use or sale of a generic version of Truvada or Viread. In August 2014, we filed a lawsuit against Apotex in the Federal Court of Canada seeking an order of prohibition against approval of this ANDS. A hearing in that case is scheduled for April 2016.
Teva
In November 2011, December 2011 and August 2012, we received notices that Teva Pharmaceuticals (Teva) submitted an abbreviated new drug submission (ANDS) to the Canadian Minister of Health requesting permission to manufacture and market generic versions of Truvada, Atripla and Viread. In the notices, Teva alleges that the patents associated with Truvada, Atripla and Viread are invalid, unenforceable and/or will not be infringed by Teva's manufacture, use or sale of generic versions of those products. We filed lawsuits against Teva in the Federal Court of Canada seeking an order of prohibition against approval of these applications.
In December 2013, the court issued an order prohibiting the Canadian Minister of Health from approving Teva’s generic versions of our Viread, Truvada and Atripla products until expiry of our patent in July 2017. Teva has appealed that decision. That decision did not rule on the validity of the patents and accordingly the only issue on appeal is whether the Minister of Health should be prohibited from approving Teva’s products. The appeal will be heard by the Canadian Federal Court of Appeal after the trial in the Impeachment Action. The court will determine the validity of the patents in the pending Impeachment Action. A trial in the Impeachment Action is scheduled for November 2016. If Teva is successful in invalidating our patents, Teva may be able to launch generic versions of our Viread, Truvada and Atripla products in Canada prior to the expiry of our patents.
Watson
In February 2015, we received notice that Watson Laboratories, Inc. (Watson) submitted an ANDA to FDA requesting permission to manufacture and market a generic version of Letairis. In the notice, Watson alleges that one of the patents associated with ambrisentan tablets is invalid, unenforceable and/or will not be infringed by Watson's manufacture, use or sale of a generic version of Letairis. In April 2015, we filed a lawsuit against Watson in the U.S. District Court for the District of New Jersey.
SigmaPharm
In June 2015, we received notice that SigmaPharm Laboratories, LLC (SigmaPharm) submitted an ANDA to FDA requesting permission to manufacture and market a generic version of Letairis. In the notice, SigmaPharm alleges that one of the patents associated with ambrisentan tablets is invalid, unenforceable and/or will not be infringed by SigmaPharm’s manufacture, use or sale of a generic version of Letairis. In June 2015, we filed a lawsuit against SigmaPharm in the U.S. District Court for the District of New Jersey.
We cannot predict the ultimate outcome of the foregoing actions and other litigation with generic manufacturers, and we may spend significant resources enforcing and defending these patents. If we are unsuccessful in these lawsuits, some or all of our original claims in the patents may be narrowed or invalidated and the patent protection for Truvada, Viread and Letairis in the United States and Atripla, Truvada and Viread in Canada could be substantially shortened. Further, if all of the patents covering one or more products are invalidated, FDA or Health Canada could approve the requests to manufacture a generic version of such products in the United States or Canada, respectively, prior to the expiration date of those patents. The sale of
40
generic versions of these products earlier than their patent expiration would have a significant negative effect on our revenues and results of operations.
We face credit risks from our Emerging Market and Southern European customers that may adversely affect our results of operations.
We have exposure to customer credit risks in Emerging Markets and Southern Europe. Southern European product sales to government-owned or supported customers in Southern Europe, specifically Spain, Italy, Portugal and Greece have historically been subject to significant payment delays due to government funding and reimbursement practices. This has resulted and may continue to result in days sales outstanding being significantly higher in these countries due to the average length of time that accounts receivable remain outstanding. As of December 31, 2015, our accounts receivable in Southern Europe, specifically Greece, Italy, Portugal and Spain, totaled approximately $1.3 billion, of which $218 million were greater than 120 days past due, including $31 million greater than 365 days past due.
Historically, receivable balances with certain publicly-owned hospitals accumulate over a period of time and are then subsequently settled as large lump sum payments. This pattern is also experienced by other pharmaceutical companies that sell directly to hospitals. If significant changes were to occur in the reimbursement practices of these European governments or if government funding becomes unavailable, we may not be able to collect on amounts due to us from these customers and our results of operations would be adversely affected.
Our revenues and gross margin could be reduced by imports from countries where our products are available at lower prices.
Prices for our products are based on local market economics and competition and sometimes differ from country to country. Our sales in countries with relatively higher prices may be reduced if products can be imported into those or other countries from lower price markets. There have been cases in which other pharmaceutical products were sold at steeply discounted prices in the developing world and then re-exported to European countries where they could be re-sold at much higher prices. If this happens with our products, particularly Truvada and Viread, which we have agreed to make available at substantially reduced prices to more than 125 countries participating in our Gilead Access Program, or Atripla, which Merck distributes at substantially reduced prices to HIV infected patients in developing countries under our 2006 agreement, our revenues would be adversely affected. In addition, we have established partnerships with India-based generic manufacturers to distribute generic versions of tenofovir disoproxil fumarate and TAF, contingent on U.S. regulatory approval, to 112 developing world countries, including India. We expanded these agreements to include rights to Stribild, Tybost and Vitekta. We also entered into agreements with certain India-based generic manufacturers to produce and distribute generic emtricitabine in the developing world, including single tablet regimens containing emtricitabine and fixed-dose combinations of emtricitabine co-formulated with our other HIV medicines. Starting in September 2014, we entered into licensing agreements with India-based generic manufacturers to produce and distribute generic sofosbuvir and the fixed-dose combination of LDV/SOF to 101 developing countries. If generic versions of our HIV and HCV medications under these licenses are then re-exported to the United States, Europe or other markets outside of these developing world countries, our revenues would be adversely affected. As part of our commitment to make Sovaldi available in the developing world at discounted prices, we entered into an agreement to make Sovaldi available in Egypt, a country that has among the highest HCV prevalence in the world. If the discounted Sovaldi is re-exported from these developing countries into the United States or other higher price markets, our revenues could be adversely affected.
In addition, purchases of our products in countries where our selling prices are relatively low for resale in countries in which our selling prices are relatively high may adversely impact our revenues and gross margin and may cause our sales to fluctuate from quarter to quarter. For example, in the European Union, we are required to permit products purchased in one country to be sold in another country. Purchases of our products in countries where our selling prices are relatively low for resale in countries in which our selling prices are relatively high can affect the inventory level held by our wholesalers and can cause the relative sales levels in the various countries to fluctuate from quarter to quarter and not reflect the actual consumer demand in any given quarter. These quarterly fluctuations may impact our earnings, which could adversely affect our stock price and harm our business.
41
Expensive litigation and government investigations have increased our expenses which may continue to reduce our earnings.
We are involved in a number of litigation, investigation and other dispute-related matters that require us to expend substantial internal and financial resources. We expect these matters will continue to require a high level of internal and financial resources for the foreseeable future. These matters have reduced and will continue to reduce our earnings. Please see a description of our Litigation Related to Sofosbuvir and Litigation with Generic Manufacturers in Note 11 Commitments and Contingencies - Legal Proceedings of the Notes to Consolidated Financial Statements included in Item 8 of this Annual Report on Form 10-K. The outcome of such lawsuits or any other lawsuits that may be brought against us, the investigation or any other investigations that may be initiated, are inherently uncertain, and adverse developments or outcomes can result in significant expenses, monetary damages, penalties or injunctive relief against us that could significantly reduce our earnings and cash flows and harm our business.
In some countries, we may be required to grant compulsory licenses for our products or our patents may not be enforced.
In a number of developing countries, government officials and other interested groups have suggested that pharmaceutical companies should make drugs for HCV or HIV infection available at low cost. Alternatively, governments in those developing countries could require that we grant compulsory licenses to allow competitors to manufacture and sell their own versions of our products, thereby reducing our product sales. For example, there is growing attention on the availability of HCV therapies and some activists are advocating for the increased availability of HCV therapies through means including compulsory licenses. In the past, certain offices of the government of Brazil have expressed concern over the affordability of our HIV products and declared that they were considering issuing compulsory licenses to permit the manufacture of otherwise patented products for HIV infection, including Viread. In addition, concerns over the cost and availability of Tamiflu related to a potential avian flu pandemic and H1N1 influenza generated international discussions over compulsory licensing of our Tamiflu patents. For example, the Canadian government considered allowing Canadian manufacturers to manufacture and export the active ingredient in Tamiflu to eligible developing and least developed countries under Canada's Access to Medicines Regime. Furthermore, Roche issued voluntary licenses to permit third-party manufacturing of Tamiflu. For example, Roche granted a sublicense to Shanghai Pharmaceutical (Group) Co., Ltd. for China and a sublicense to India's Hetero Drugs Limited for India and certain developing countries. If compulsory licenses permit generic manufacturing to override our product patents for Harvoni, Sovaldi, our HIV products or Tamiflu, or if we are required to grant compulsory licenses for these products, it could reduce our earnings and cash flows and harm our business.
In addition, certain countries do not permit enforcement of our patents, and third-party manufacturers are able to sell generic versions of our products in those countries. For example, in July 2009, the Brazilian patent authority rejected our patent application for tenofovir disoproxil fumarate, the active pharmaceutical ingredient in Viread. This was the highest level of appeal available to us within the Brazilian patent authority. Because we do not currently have a patent in Brazil, the Brazilian government now purchases its supply of tenofovir disoproxil fumarate from generic manufacturers. Sales of generic versions of our products could significantly reduce our sales and adversely affect our results of operations, particularly if generic versions of our products are imported into territories where we have existing commercial sales.
We may face significant liability resulting from our products that may not be covered by insurance and successful claims could materially reduce our earnings.
The testing, manufacturing, marketing and use of our commercial products, as well as product candidates in development, involve substantial risk of product liability claims. These claims may be made directly by consumers, healthcare providers, pharmaceutical companies or others. We may be unable to maintain sufficient coverage for product liabilities that may arise. In addition, the cost to defend lawsuits or pay damages for product liability claims may exceed our coverage. If we are unable to maintain adequate coverage or if claims exceed our coverage, our financial condition and our ability to clinically test our product candidates and market our products will be adversely affected. In addition, negative publicity associated with any claims, regardless of their merit, may decrease the future demand for our products and impair our financial condition.
Business disruptions from natural or man-made disasters may harm our future revenues.
Our worldwide operations could be subject to business interruptions stemming from natural or man-made disasters for which we may be self-insured. Our corporate headquarters and Fremont locations, which together house a majority of our R&D activities, and our La Verne, San Dimas and Oceanside manufacturing facilities are located in California, a seismically active region. As we may not carry adequate earthquake insurance and significant recovery time could be required to resume operations, our financial condition and operating results could be materially adversely affected in the event of a major earthquake.
42
We are dependent on information technology systems, infrastructure and data.
We are dependent upon information technology systems, infrastructure and data. The multitude and complexity of our computer systems make them inherently vulnerable to service interruption or destruction, malicious intrusion and random attack. Likewise, data privacy or security breaches by employees or others may pose a risk that sensitive data, including our intellectual property, trade secrets or personal information of our employees, patients, customers or other business partners may be exposed to unauthorized persons or to the public. Cyberattacks are increasing in their frequency, sophistication and intensity. Cyberattacks could include the deployment of harmful malware, denial-of-service, social engineering and other means to affect service reliability and threaten data confidentiality, integrity and availability. Our business partners face similar risks and any security breach of their systems could adversely affect our security posture. While we have invested, and continue to invest, in the protection of our data and information technology infrastructure, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm to us. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyberattacks and other related breaches.
Changes in our effective income tax rate could reduce our earnings.
We are subject to income taxes in both the United States and various foreign jurisdictions including Ireland. Due to economic and political conditions various countries are actively considering changes to existing tax laws. We cannot predict the form or timing of potential legislative changes that could have a material adverse impact on our results of operations. In addition, significant judgment is required in determining our worldwide provision for income taxes. Various factors may have favorable or unfavorable effects on our income tax rate including, but not limited to, changes in forecasted demand for our HCV products, our portion of the non-tax deductible annual BPD fee, the accounting for stock options and other share-based awards, mergers and acquisitions, the ability to manufacture product in our Cork, Ireland facility, the amortization of certain acquisition related intangibles for which we receive no tax benefit, future levels of R&D spending, changes in the mix of earnings in the various tax jurisdictions in which we operate, changes in overall levels of pre-tax earnings and resolution of federal, state and foreign income tax audits. The impact on our income tax provision resulting from the above mentioned factors may be significant and could have a negative impact on our consolidated results of operations.
Our income tax returns are subject to audit by federal, state and foreign tax authorities. We are currently under examination by the Internal Revenue Service for the 2010, 2011 and 2012 tax years and by various state and foreign jurisdictions. There are differing interpretations of tax laws and regulations, and as a result, significant disputes may arise with these tax authorities involving issues of the timing and amount of deductions and allocations of income among various tax jurisdictions. Resolution of one or more of these exposures in any reporting period could have a material impact on the results of operations for that period.
If we fail to attract and retain highly qualified personnel, we may be unable to successfully develop new product candidates, conduct our clinical trials and commercialize our product candidates.
Our future success will depend in large part on our continued ability to attract and retain highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing, governmental regulation and commercialization. We face competition for personnel from other companies, universities, public and private research institutions, government entities and other organizations. Competition for qualified personnel in the biopharmaceutical field is intense, and there is a limited pool of qualified potential employees to recruit. We may not be able to attract and retain quality personnel on acceptable terms. If we are unsuccessful in our recruitment and retention efforts, our business may be harmed.
There can be no assurance that we will pay dividends or continue to repurchase stock.
In February 2016, we announced that our Board of Directors authorized an increase to our dividend program under which we intend to pay quarterly dividends of $0.47 per share, beginning in the second quarter of 2016 and subject to quarterly declarations by our Board of Directors, and that our Board of Directors also approved the repurchase of up to an additional $12.0 billion of our common stock to commence after completion of our existing $15.0 billion repurchase plan approved in January 2015. Any future declarations, amount and timing of any dividends and/or the amount and timing of such stock repurchases are subject to capital availability and determinations by our Board of Directors that cash dividends and/or stock repurchases are in the best interest of our stockholders and are in compliance with all respective laws and our agreements applicable to the declaration and payment of cash dividends and the repurchase of stock. Our ability to pay dividends and/or repurchase stock will depend upon, among other factors, our cash balances and potential future capital requirements for strategic transactions, including acquisitions, debt service requirements, results of operations, financial condition and other factors beyond our control that our Board of Directors may deem relevant. A reduction in or elimination of our dividend payments, our dividend program and/or stock repurchases could have a negative effect on our stock price.
43
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
Not applicable.
ITEM 2. | PROPERTIES |
Our corporate headquarters is located in Foster City, California, where we house our administrative, manufacturing and R&D activities. We also have R&D facilities in Oceanside, California; Fremont, California; Seattle, Washington; and Alberta, Canada and manufacturing facilities in San Dimas, California and Cork, Ireland. Our global operations include offices in Europe, North America, Asia, South America, Africa, Australia, India and the Middle East.
We believe that our existing properties, including both owned and leased sites, are in good condition and suitable for the conduct of our business. We believe our capital resources are sufficient to purchase, lease or construct any additional facilities required to meet our expected long-term growth needs.
ITEM 3. | LEGAL PROCEEDINGS |
For a description of our significant pending legal proceedings, please see Note 11 Commitments and Contingencies - Legal Proceedings of the Notes to Consolidated Financial Statements included in Item 8 of this Annual Report on Form 10-K, which is incorporated herein by reference.
ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
PART II
ITEM 5. | MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock is traded on The Nasdaq Global Select Market under the symbol “GILD”. The following table sets forth the high and low intra-day sale prices per share of our common stock on The Nasdaq Global Select Market for the periods indicated. These prices represent quotations among dealers without adjustments for retail mark-ups, markdowns or commissions and may not represent prices of actual transactions.
2015 | 2014 | |||||||
High | Low | High | Low | |||||
First Quarter | $107.77 | $93.18 | $84.88 | $67.63 | ||||
Second Quarter | $123.37 | $95.38 | $84.45 | $63.50 | ||||
Third Quarter | $120.37 | $86.00 | $110.64 | $83.32 | ||||
Fourth Quarter | $111.11 | $94.37 | $116.83 | $85.95 | ||||
As of February 12, 2016, we had 1,366,845,691 shares of common stock outstanding held by approximately 363 stockholders of record, which include shares held by a broker, bank or other nominee.
Dividends
We initiated a quarterly cash dividend of $0.43 per share that began in the second quarter of 2015. During 2015, we declared and paid aggregate cash dividends of $1.9 billion or $1.29 per common share. See Item 8, Note 12 Stockholders' Equity in our Consolidated Financial Statements included in this Annual Report on Form 10-K for additional information.
Performance Graph (1)
The following graph compares our cumulative total stockholder return for the past five years to two indices: the Standard & Poor's 500 Stock Index, labeled S&P 500 Index; and the Nasdaq Biotechnology Index, labeled NBI Index. The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
44
Comparison of Cumulative Total Return on Investment for the Past Five Years (2)
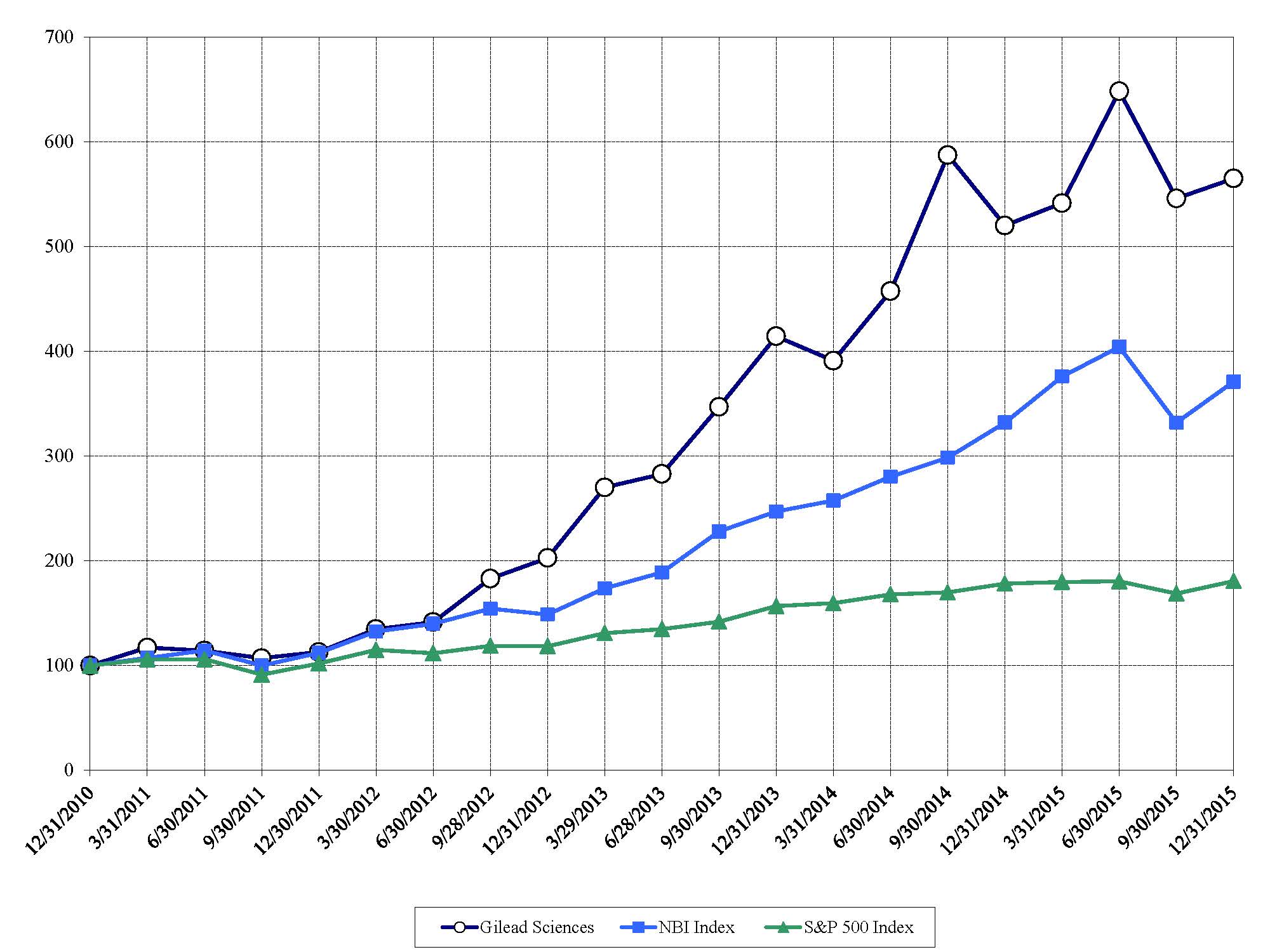
______________________________________________________
(1) | This section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any of our filings under the Securities Act or the Exchange Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
(2) | Shows the cumulative return on investment assuming an investment of $100 in our common stock, the NBI Index and the S&P 500 Index on December 31, 2010, and that all dividends were reinvested. |
Issuer Purchases of Equity Securities
In January 2015, our Board of Directors authorized a five-year, $15.0 billion stock repurchase program (2015 Program). Purchases under the 2015 Program may be made in the open market or in privately negotiated transactions. The 2015 Program commenced after the $5.0 billion stock repurchase program authorized by our Board of Directors in May 2014 was completed in the first quarter of 2015. In 2015, we spent $10.0 billion to repurchase 95 million shares of our common stock at an average purchase price of $104.91 per share. See Item 8, Note 12 Stockholders' Equity in our Consolidated Financial Statements included in this Annual Report on Form 10-K for more information regarding our stock repurchase programs. The table below summarizes our stock repurchase activity for the three months ended December 31, 2015:
Total Number of Shares Purchased (in thousands) | Average Price Paid per Share (in dollars) | Total Number of Shares Purchased as Part of Publicly Announced Program (in thousands) | (1) | Maximum Fair Value of Shares that May Yet Be Purchased Under the Program (in millions) | (1) | ||||||||||
October 1 - October 31, 2015 | 8,367 | $ | 102.25 | 8,313 | $ | 10,200 | |||||||||
November 1 - November 30, 2015 | 10,127 | $ | 106.88 | 9,792 | $ | 9,154 | |||||||||
December 1 - December 31, 2015 | 11,259 | $ | 102.69 | 11,239 | $ | 8,000 | |||||||||
Total | 29,753 | (2) | $ | 103.99 | 29,344 | (2) | |||||||||
(1) | Stock repurchases were made under the 2015 Program. |
(2) | The difference between the total number of shares purchased and the total number of shares purchased as part of publicly announced programs is due to shares of common stock withheld by us from employee restricted stock awards in order to satisfy applicable tax withholding obligations. |
In February 2016, we entered into an accelerated share repurchase program (“ASR”) to repurchase $5.0 billion of our common stock. We paid $5.0 billion and received 46 million shares of our common stock, which represents approximately 80% of the total shares expected to be delivered to us under the ASR. The total number of shares to be received under the ASR will be based on the average price of our common stock during the purchase period, which will end in April 2016.
In February 2016, our Board of Directors authorized a new $12.0 billion share repurchase program (2016 Program) which will commence upon the completion of our 2015 Program. Purchases under the 2016 Program may be made in the open market or in privately negotiated transactions.
45
ITEM 6. | SELECTED FINANCIAL DATA |
GILEAD SCIENCES, INC.
SELECTED CONSOLIDATED FINANCIAL DATA
(in millions, except per share data)
Year Ended December 31, | |||||||||||||||||||
2015 | 2014 | 2013 | 2012 | 2011 | |||||||||||||||
CONSOLIDATED STATEMENT OF INCOME DATA: | |||||||||||||||||||
Total revenues (1) | $ | 32,639 | $ | 24,890 | $ | 11,202 | $ | 9,702 | $ | 8,385 | |||||||||
Total costs and expenses (1) | $ | 10,446 | $ | 9,625 | $ | 6,678 | $ | 5,692 | $ | 4,596 | |||||||||
Income from operations | $ | 22,193 | $ | 15,265 | $ | 4,524 | $ | 4,010 | $ | 3,790 | |||||||||
Provision for income taxes | $ | 3,553 | $ | 2,797 | $ | 1,151 | $ | 1,038 | $ | 862 | |||||||||
Net income attributable to Gilead | $ | 18,108 | $ | 12,101 | $ | 3,075 | $ | 2,592 | $ | 2,804 | |||||||||
Net income per share attributable to Gilead common stockholders - basic | $ | 12.37 | $ | 7.95 | $ | 2.01 | $ | 1.71 | $ | 1.81 | |||||||||
Shares used in per share calculation-basic | 1,464 | 1,522 | 1,529 | 1,515 | 1,550 | ||||||||||||||
Net income per share attributable to Gilead common stockholders - diluted | $ | 11.91 | $ | 7.35 | $ | 1.81 | $ | 1.64 | $ | 1.77 | |||||||||
Shares used in per share calculation-diluted | 1,521 | 1,647 | 1,695 | 1,583 | 1,580 | ||||||||||||||
Cash dividends declared per share | $ | 1.29 | $ | — | $ | — | $ | — | $ | — | |||||||||
December 31, | |||||||||||||||||||
2015 | 2014 | 2013 | 2012 | 2011 | |||||||||||||||
CONSOLIDATED BALANCE SHEET DATA: | |||||||||||||||||||
Cash, cash equivalents and marketable securities (2) | $ | 26,208 | $ | 11,726 | $ | 2,571 | $ | 2,582 | $ | 9,964 | |||||||||
Working capital (2) | $ | 14,872 | $ | 11,953 | $ | 590 | $ | 1,918 | $ | 11,432 | |||||||||
Total assets (2) | $ | 51,839 | $ | 34,664 | $ | 22,579 | $ | 21,240 | $ | 17,303 | |||||||||
Other long-term obligations (3) | $ | 395 | $ | 586 | $ | 262 | $ | 281 | $ | 180 | |||||||||
Senior unsecured notes, convertible senior notes and credit facility (2) | $ | 22,178 | $ | 12,404 | $ | 6,636 | $ | 8,224 | $ | 7,606 | |||||||||
Retained earnings | $ | 18,001 | $ | 12,732 | $ | 6,106 | $ | 3,705 | $ | 1,777 | |||||||||
Total stockholders' equity | $ | 19,113 | $ | 15,819 | $ | 11,745 | $ | 9,544 | $ | 6,867 | |||||||||
(1) | See Item 7, Management's Discussion and Analysis for a description of our results of operations for 2015. | |
(2) | During 2015, we issued $10.0 billion principal amount of senior unsecured notes in a registered offering. We also repaid $213 million of principal balance of convertible senior notes due in 2016 and $784 million in cash related to the conversion spread of the notes. | |
During 2014, we issued $8.0 billion principal amount of senior unsecured notes in registered offerings. We also repaid $912 million of principal balance of convertible senior notes due in 2014, $2.5 billion in cash related to the conversion spread of the notes, $750 million for senior unsecured notes and $600 million under the five-year revolving credit facility agreement (the Five-Year Revolving Credit Agreement). | ||
During 2013, we repaid $1.5 billion of principal balance of convertible senior notes and repaid $150 million under our Five-Year Revolving Credit Agreement. | ||
During 2012, we completed the acquisition of Pharmasset, Inc. and we recognized consideration transferred of $11.1 billion which was primarily recorded in intangible assets. We financed the transaction with approximately $5.2 billion in cash on hand, $2.2 billion in bank debt issued in January 2012 and $3.7 billion in senior unsecured notes issued in December 2011. | ||
(3) | Prior year amounts have been reclassified to conform to current presentation. | |
46
ITEM 7. | MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following Management's Discussion and Analysis of Financial Condition and Results of Operations (MD&A) is intended to help the reader understand our results of operations and financial condition. MD&A is provided as a supplement to, and should be read in conjunction with, our audited Consolidated Financial Statements and the accompanying Notes to Consolidated Financial Statements and other disclosures included in this Annual Report on Form 10-K (including the disclosures under Item 1A, Risk Factors). Our Consolidated Financial Statements have been prepared in accordance with U.S. generally accepted accounting principles and are presented in U.S. dollars.
Management Overview
Gilead Sciences, Inc. (Gilead, we or us), incorporated in Delaware on June 22, 1987, is a research-based biopharmaceutical company that discovers, develops and commercializes innovative medicines in areas of unmet medical need. With each new discovery and investigational drug candidate, we strive to transform and simplify care for people with life-threatening illnesses around the world. Gilead's primary areas of focus include human immunodeficiency virus (HIV), liver diseases such as chronic hepatitis C virus (HCV) infection and chronic hepatitis B virus (HBV) infection, cardiovascular, hematology/oncology and inflammation/respiratory. We have operations in more than 30 countries worldwide, with headquarters in Foster City, California. We continue to add to our existing portfolio of products through our internal discovery and clinical development programs and through a product acquisition and in-licensing strategy.
Our portfolio of marketed products includes AmBisome®, Atripla®, Cayston®, Complera®/Eviplera®, Emtriva®, Genvoya®, Harvoni®, Hepsera®, Letairis®, Ranexa®, Sovaldi®, Stribild®, Tamiflu®, Truvada®, Tybost®, Viread®, Vitekta®, and Zydelig®. We have U.S. and international commercial sales operations, with marketing subsidiaries in North and South America, Europe and Asia-Pacific. We also sell and distribute certain products through our corporate partners under royalty-paying collaborative agreements.
2015 Business Highlights
During 2015, we continued to advance our product pipeline across our therapeutic areas with the goal of delivering best-in-class drugs that advance the current standard of care and/or address unmet medical needs. Highlights of our 2015 activities include:
Antiviral Program
• | U.S. Food and Drug Administration (FDA) and European Commission approved Genvoya for the treatment of HIV-1 infection. Genvoya is our first tenofovir alafenamide (TAF)-based regimen. |
• | We submitted marketing applications to FDA and European Medicines Agency (EMA) for an investigational, once-daily single tablet regimen that combines our emtricitabine 200 mg and TAF 25 mg with rilpivirine 25 mg (R/F/TAF) from Janssen Sciences Ireland UC, one of the Janssen Pharmaceutical Companies of Johnson & Johnson, for the treatment of HIV-1 infection in adult and pediatric patients 12 years of age and older. |
• | We submitted marketing applications to FDA and EMA for two doses of F/TAF (200/10 mg and 200/25 mg) for the treatment of HIV-1 infection in adults and pediatric patients age 12 years and older, in combination with other HIV antiretroviral agents. |
• | FDA approved Harvoni for expanded use in patients with genotype 4, 5 and 6 HCV infection and in patients co-infected with HIV. In addition, Harvoni plus ribavirin for 12 weeks was approved as an alternate therapy to 24 weeks of Harvoni for treatment-experienced, genotype 1 patients with cirrhosis. |
• | Japanese Ministry of Health, Labour and Welfare approved Sovaldi for the suppression of viremia in patients with genotype 2 chronic HCV infection with or without compensated cirrhosis and Harvoni, the first once-daily single-tablet regimen for the treatment of chronic HCV genotype 1 infection in adults with or without compensated cirrhosis, with a treatment duration of 12 weeks. |
• | We submitted marketing applications to FDA and EMA for an investigational, once-daily fixed-dose combination of the nucleotide analog polymerase inhibitor sofosbuvir (SOF) 400 mg and velpatasvir (VEL) 100 mg, an investigational pan-genotypic NS5A inhibitor, for the treatment of genotype 1-6 chronic HCV infection. |
• | We received reimbursement approval for Sovaldi and Harvoni in various countries in the European Union. |
47
Cardiovascular Program
• | FDA approved the use of Letairis (ambrisentan) in combination with tadalafil for the treatment of pulmonary arterial hypertension (PAH) to reduce the risks of disease progression and hospitalization for worsening PAH, and to improve exercise ability. |
Hematology/Oncology Program
• | We filed a supplemental new drug application for the use of Zydelig (idelalisib) in combination with ofatumumab in previously-treated patients with chronic lymphocytic leukemia. |
Inflammation/Respiratory Program
• | We entered into a collaboration and license agreement with Galapagos NV, which became effective January 2016, for the development and commercialization of the JAK1-selective inhibitor filgotinib for inflammatory disease indications. This collaboration represents an opportunity to add complementary clinical programs to our growing inflammation research and development efforts. |
2015 Financial Highlights
During 2015, total revenues increased to $32.6 billion and total product sales increased to $32.2 billion, compared to $24.9 billion and $24.5 billion respectively, in 2014, driven primarily by sales of Harvoni and increased sales of our HIV single tablet regimen products, Stribild, Complera/Eviplera and the recently launched Genvoya, partially offset by decreased sales of Sovaldi due to the uptake of Harvoni. Harvoni was approved in the United States in October 2014, in the European Union in November 2014 and in Japan in July 2015. For 2015, product sales in the U.S. were $21.2 billion compared to $18.1 billion in 2014. In Europe, product sales were $7.2 billion compared to $5.1 billion in 2014. Sales in other international locations were $3.8 billion in 2015 compared to $1.2 billion in 2014, primarily due to sales of Sovaldi and Harvoni in Japan.
R&D expenses increased 6% to $3.0 billion for 2015 compared to 2014 due to continued investment in the progression and expansion of our product pipeline. Selling, general and administrative (SG&A) expenses increased 15% to $3.4 billion for 2015 compared to 2014 due to increased costs to support our business expansion.
Net income attributable to Gilead for 2015 was $18.1 billion or $11.91 per diluted share, compared to $12.1 billion or $7.35 per diluted share in 2014, due primarily to the launch of Harvoni, partially offset by the declines in sales of Sovaldi and increases in operating expenses.
As of December 31, 2015, our cash, cash equivalents and marketable securities totaled $26.2 billion. During 2015, we generated $20.3 billion in operating cash flows, issued senior unsecured notes with a total aggregate principal amount of $10.0 billion (2015 Notes), and paid $3.9 billion to settle 46 million warrants related to our convertible senior notes due May 2016. We repurchased 95 million shares of our common stock in 2015 for an aggregate amount of $10.0 billion. We also initiated a quarterly cash dividend of $0.43 per share in the second quarter of 2015, and paid a total of $1.9 billion in dividends to our shareholders in 2015.
Outlook 2016
In 2016, we will continue to focus on our key operating objectives which include the progression of our product pipeline and continued uptake of our commercial products. From a research and development (R&D) perspective, we will continue to invest in conducting new and ongoing clinical studies, which support both our existing products and our product candidates. We expect to move forward on a number of late-stage clinical studies for new product candidates and plan to file marketing applications for product candidates in various therapeutic areas.
From a commercial perspective, we will continue to focus on supporting the uptake of Genvoya and prepare for additional anticipated launches of our new TAF-based regimens, F/TAF and R/F/TAF, and continue to promote the use of our existing commercial products. In HCV, we will continue to focus on advancing care of people with the disease regardless of genotype or disease severity. SOF/VEL, if approved, would become the first and only regimen offering high sustained viral response rates with 12 weeks of treatment for patients with all HCV genotypes. We also plan to further build-out and expand our commercial infrastructure globally.
Additionally, we will focus both on near term and longer term objectives to help many more patients around the world. Our progress is subject to a number of uncertainties, including, but not limited to, the continuation of an uncertain global macroeconomic environment; adoption of additional pricing measures to reduce healthcare spending particularly in HCV; volatility in foreign currency exchange rates; inaccuracies in our HCV patient start estimates; additional competitive launches in HCV; an increase in discounts, chargebacks and rebates due to ongoing private and public payer negotiations; and a larger than anticipated shift in payer mix to more highly discounted payer segments.
48
2015 Results of Operations
Total Revenues
The following table summarizes our product sales, and royalty, contract and other revenues:
(In millions, except percentages) | 2015 | Change | 2014 | Change | 2013 | |||||||||||||
Revenues: | ||||||||||||||||||
Product sales | $ | 32,151 | 31 | % | $ | 24,474 | 127 | % | $ | 10,804 | ||||||||
Royalty, contract and other revenues | 488 | 17 | % | 416 | 5 | % | 398 | |||||||||||
Total revenues | 32,639 | 31 | % | 24,890 | 122 | % | 11,202 | |||||||||||
Product Sales
Total product sales were $32.2 billion in 2015, compared to $24.5 billion in 2014 and $10.8 billion in 2013, driven primarily by an increase in antiviral product sales.
Antiviral product sales, which include products in our HIV and liver disease areas, were $30.2 billion in 2015, $22.8 billion in 2014 and $9.3 billion in 2013. The sequential increases in antiviral product sales in 2015 and 2014 were driven primarily by the launch of Sovaldi and Harvoni. The increases in 2015 sales from the launch of Harvoni across various geographies were partially offset by a year-over-year decline in Sovaldi sales, with patients being prescribed Harvoni instead of Sovaldi. HIV products also contributed to sales increases in 2015 and 2014 primarily due to increased sales of our newer HIV single-tablet regimens, Stribild, Complera/Eviplera and the recently launched Genvoya, partially offset by declines in Atripla sales volumes.
Other product sales, which include Letairis, Ranexa, AmBisome and Zydelig, were $1.9 billion in 2015, an increase of 16% compared to $1.7 billion in 2014, an increase of 15% over other product sales of $1.5 billion in 2013.
In 2015, approximately 34% of our product sales were generated outside the United States. We face exposure to adverse movements in foreign currency exchange rates, primarily in the Euro and Yen. We used foreign currency exchange contracts to hedge a percentage of our foreign currency exposure. Foreign currency exchange, net of hedges, had an unfavorable impact of $737 million on our 2015 revenues compared to 2014 and a favorable impact of $39 million on our 2014 revenues compared to 2013.
We record product sales net of estimated mandatory and supplemental discounts to government payers, in addition to discounts to private payers, including rebates, chargebacks, cash discounts for prompt payment, distributor fees and other related costs. These deductions are generally referred to as gross-to-net deductions and totaled $18.1 billion or 36% of gross product sales in 2015, $7.3 billion or 23% in 2014 and $3.9 billion or 26% in 2013. Of the $18.1 billion in 2015, $16.4 billion or 33% of gross product sales was related to government and other rebates and chargebacks, and $1.7 billion was related to cash discounts for prompt payment, distributor fees and other related costs. As anticipated, our 2015 gross-to-net deductions attributable to our HCV product sales exceeded our overall gross-to-net of 36% in order to obtain formulary status or expand access for patients. As a result of the launch of competing regimens, we have experienced, and may continue to experience, increased pricing pressure.
The decline in gross-to-net deductions as a percentage of gross product sales in 2014 compared to 2013 was primarily due to change in our payer mix reflecting a higher proportion of product sales to private payers compared to 2013 given the launch of Sovaldi in December 2013 and Harvoni in October 2014.
Product sales in the United States increased by 17% to $21.2 billion in 2015 compared to $18.1 billion in 2014, primarily due to sales of Harvoni and increases in sales of Stribild, Truvada and Complera, partially offset by declines in sales of Sovaldi. Product sales in the United States increased by 173% in 2014 compared to $6.6 billion in 2013, primarily due to sales of Sovaldi and Harvoni and increases in sales of Stribild and Complera.
Product sales in Europe increased by 39% to $7.2 billion in 2015 compared to $5.1 billion in 2014, primarily due to sales of Harvoni. Product sales in Europe increased by 54% to $5.1 billion in 2014 compared to $3.3 billion in 2013, primarily due to sales of Sovaldi and increases in sales of Eviplera and Stribild. Foreign currency exchange, net of hedges, had an unfavorable impact of $611 million on our European product sales in 2015 compared to 2014 and a favorable impact of $72 million on our European product sales for 2014 compared to 2013.
49
Product sales in other international locations increased to $3.8 billion in 2015 compared to $1.2 billion in 2014, primarily due to the launch in Japan of Sovaldi in March 2015 and Harvoni in July 2015. Product sales in other international locations increased by 47% in 2014 compared to $826 million in 2013, primarily due to the launch of Sovaldi in various geographies.
Since our HCV products, Harvoni and Sovaldi, were only recently launched, historical sales may not be indicative of future sales. In the United States, the number of HCV new patient starts has diminished since the first quarter of 2015, indicative of the rapid initiation of treatment for many warehoused patients, followed by a flattening of patients in the remaining quarters. We anticipate that the rate at which new patients start treatment in the second half of 2015 may be more indicative of the pace of new patient starts in 2016. In Europe, we expect early launch markets to stabilize and new markets to ramp up treatment. In Japan, patient numbers are difficult to predict because our HCV products were recently launched. Additionally, we anticipate the government of Japan will impose significant pricing discounts for Harvoni and Sovaldi that will start taking effect in the first half of 2016.
The following table summarizes the period over period changes in our product sales:
(In millions, except percentages) | 2015 | Change | 2014 | Change | 2013 | |||||||||||||
Antiviral products: | ||||||||||||||||||
Harvoni | $ | 13,864 | * | $ | 2,127 | * | — | |||||||||||
Sovaldi | 5,276 | (49 | )% | 10,283 | * | $ | 139 | |||||||||||
Truvada | 3,459 | 4 | % | 3,340 | 7 | % | 3,136 | |||||||||||
Atripla | 3,134 | (10 | )% | 3,470 | (5 | )% | 3,648 | |||||||||||
Stribild | 1,825 | 52 | % | 1,197 | 122 | % | 539 | |||||||||||
Complera/Eviplera | 1,427 | 16 | % | 1,228 | 52 | % | 810 | |||||||||||
Viread | 1,108 | 5 | % | 1,058 | 10 | % | 959 | |||||||||||
Genvoya | 45 | * | — | * | — | |||||||||||||
Other antiviral | 69 | (22 | )% | 88 | (21 | )% | 111 | |||||||||||
Total antiviral products | 30,207 | 33 | % | 22,791 | 144 | % | 9,342 | |||||||||||
Other products: | ||||||||||||||||||
Letairis | 700 | 18 | % | 595 | 14 | % | 520 | |||||||||||
Ranexa | 588 | 15 | % | 510 | 14 | % | 449 | |||||||||||
AmBisome | 350 | (10 | )% | 388 | 10 | % | 352 | |||||||||||
Zydelig | 132 | * | 23 | * | — | |||||||||||||
Other | 174 | 4 | % | 167 | 18 | % | 141 | |||||||||||
Total product sales | $ | 32,151 | 31 | % | $ | 24,474 | 127 | % | $ | 10,804 | ||||||||
* Percentage not meaningful
Antiviral Products
The following is additional discussion of our results by product:
• | Harvoni |
Harvoni sales accounted for 46% and 9% of our total antiviral product sales for 2015 and 2014, respectively. Harvoni was approved by FDA in October 2014, by the European Commission in November 2014 and by the Japanese Ministry of Health, Labour and Welfare (MHLW) in July 2015. Harvoni was approved as the first once-daily single tablet regimen for the treatment of chronic HCV genotype 1.
Net product sales of Harvoni in the United States were $10.1 billion in 2015 and $2.0 billion in 2014 driven by the product launch. Net product sales of Harvoni in Europe were $2.2 billion in 2015 and $103 million in 2014. We launched Harvoni in a number of countries including France, Germany, U.K., Italy and Spain. Net product sales of Harvoni in other international locations were $1.6 billion in 2015 and $23 million in 2014, primarily due to the product launch in Japan.
50
• | Sovaldi |
Sovaldi sales accounted for 17%, 45%, and 1% of our total antiviral product sales for 2015, 2014 and 2013, respectively. Sovaldi was approved by FDA in December 2013, by the European Commission in January 2014 and by Japan in March 2015.
Net product sales of Sovaldi decreased by 49% to $5.3 billion in 2015 compared to $10.3 billion in 2014 primarily due to volume declines in the United States with patients being prescribed Harvoni instead of Sovaldi, partially offset by volume increases in Japan and Europe as we continue to launch Sovaldi in various countries.
In 2015, net product sales of Sovaldi were $2.4 billion in the United States, $1.6 billion in Europe and $1.3 billion in other international locations, primarily Japan. In 2014, net products sales of Sovaldi were $8.5 billion in United States, $1.5 billion in Europe and $230 million in other international locations.
• | Truvada |
Truvada sales accounted for 11%, 15% and 34% of our total antiviral product sales for 2015, 2014 and 2013, respectively. Truvada sales increased by 4% to $3.5 billion in 2015 compared to $3.3 billion in 2014 and by 7% in 2014 compared to $3.1 billion in 2013, primarily due to sales volume growth and an increase in the average net selling price in the United States.
In 2015, net product sales of Truvada were $2.1 billion in the United States, $1.1 billion in Europe and $284 million in other international locations. In 2014, net products sales of Truvada were $1.8 billion in United States, $1.3 billion in Europe and $278 million in other international locations. In 2013, net products sales of Truvada were $1.6 billion in United States, $1.3 billion in Europe and $270 million in other international locations.
• | Atripla |
Atripla sales accounted for 10%, 15% and 39% of our total antiviral product sales for 2015, 2014 and 2013, respectively. Atripla sales decreased 10% to $3.1 billion in 2015 compared to $3.5 billion 2014 and 5% in 2014 compared to $3.6 billion in 2013, primarily due to declines in volume as doctors prescribed newer treatments such as Complera/Eviplera and Stribild. The efavirenz component of Atripla, which has a gross margin of zero, comprised $1.2 billion, $1.3 billion and $1.4 billion of our Atripla sales in 2015, 2014 and 2013, respectively.
In 2015, net product sales of Atripla were $2.2 billion in the United States, $694 million in Europe and $218 million in other international locations. In 2014, net products sales of Atripla were $2.4 billion in United States, $888 million in Europe and $225 million in other international locations. In 2013, net products sales of Atripla were $2.4 billion in United States, $1.1 billion in Europe and $231 million in other international locations.
A generic version of Bristol-Myers Squibb Company's Sustiva (efavirenz), a component of Atripla, was made available in Canada and Europe in 2013 and will be made available in the United States in 2017. While we have observed some pricing pressure related to the efavirenz component of our Atripla sales, we have not yet observed any meaningful splitting of the Atripla single tablet regimen.
• | Stribild |
Stribild sales accounted for 6%, 5% and 6% of our total antiviral product sales for 2015, 2014 and 2013, respectively. Stribild sales increased 52% to $1.8 billion in 2015 compared to $1.2 billion in 2014 and 122% compared to $539 million in 2013, primarily due to increased sales volume in the United States and Europe.
In 2015, net product sales of Stribild were $1.5 billion in the United States and $282 million in Europe. In 2014, net products sales of Stribild were $1.0 billion in United States and $145 million in Europe. In 2013, net products sales of Stribild were primarily attributable to sales in United States of $510 million.
• | Complera/Eviplera |
Complera/Eviplera sales accounted for 5%, 5% and 9% of our total antiviral product sales for 2015, 2014 and 2013, respectively. Complera/Eviplera sales increased by16% to $1.4 billion in 2015 compared to $1.2 billion 2014 and 52% compared to $810 million in 2013 driven primarily by sales volume growth in the United States and Europe.
In 2015, net product sales of Complera/Eviplera were $796 million in the United States and $576 million in Europe. In 2014, net products sales of Complera/Eviplera were $663 million in United States and $513 million in Europe. In 2013, net products sales of Complera/Eviplera were $503 million in United States and $268 million in Europe.
51
• | Viread |
Viread sales accounted for 4%, 5%, 10% of our total antiviral product sales for 2015, 2014 and 2013, respectively. Viread sales increased 5% in 2015 compared to 2014 and increased 10% in 2014 compared to 2013 driven primarily by sales volume in the United States and other international locations.
In 2015, net product sales of Viread were $541 million in the United States, $310 million in Europe and $257 million in other international locations. In 2014, net products sales of Viread were $484 million in United States, $336 million in Europe and $238 million in other international locations. In 2013, net products sales of Viread were $428 million in United States, $354 million in Europe and $177 million in other international locations.
Other Products
Other products which include Letairis, Ranexa, AmBisome and Zydelig, were $1.9 billion in 2015, $1.7 billion in 2014 and $1.5 billion in 2013. The year-over-year increases in other product sales were primarily due to increased sales volume of Letairis and Zydelig.
Royalty, Contract and Other Revenues
The following table summarizes the period over period changes in our royalty, contract and other revenues:
(In millions, except percentages) | 2015 | Change | 2014 | Change | 2013 | |||||||||||||
Royalty, contract and other revenues | $ | 488 | 17 | % | $ | 416 | 5 | % | $ | 398 | ||||||||
Royalty, contract and other revenues primarily includes royalty revenues from F. Hoffman-La Roche Ltd for sales of Tamiflu. The majority of our royalties are recognized in the quarter following the quarter in which the corresponding product sales occur.
Cost of Goods Sold and Product Gross Margin
The following table summarizes the period over period changes in our product sales, cost of goods sold and product gross margin:
(In millions, except percentages) | 2015 | Change | 2014 | Change | 2013 | |||||||||||||
Total product sales | $ | 32,151 | 31 | % | $ | 24,474 | 127 | % | $ | 10,804 | ||||||||
Cost of goods sold | $ | 4,006 | 6 | % | $ | 3,788 | 32 | % | $ | 2,859 | ||||||||
Product gross margin | 88 | % | 85 | % | 74 | % | ||||||||||||
Our product gross margin for 2015 increased compared to 2014 primarily due to changes in product mix, as Atripla sales, which include the efavirenz component at a gross margin of zero, declined and HCV sales increased as a percentage of product sales. Our product gross margin for 2014 increased compared to 2013 primarily due to changes in product mix, resulting from the launches of Sovaldi and Harvoni.
Research and Development Expenses
The following table summarizes the period over period changes in R&D expenses:
(In millions, except percentages) | 2015 | Change | 2014 | Change | 2013 | |||||||||||||
Research and development | $ | 3,014 | 6 | % | $ | 2,854 | 35 | % | $ | 2,120 | ||||||||
R&D expenses summarized above consist primarily of clinical studies performed by contract research organizations, materials and supplies, licenses and fees, milestone payments under collaboration arrangements, personnel costs, including salaries, benefits and stock-based compensation and overhead allocations consisting of various support and facilities-related costs.
We do not track total R&D expenses by product candidate, therapeutic area or development phase. However, we manage our R&D expenses by identifying the R&D activities we anticipate will be performed during a given period and then prioritizing efforts based on scientific data, probability of successful development, market potential, available human and capital resources and other considerations. We continually review our R&D pipeline and the status of development and, as necessary, reallocate resources among the R&D portfolio that we believe will best support the future growth of our business.
52
The following table provides a breakout of R&D expenses by major cost type:
(In millions, except percentages) | 2015 | 2014 | 2013 | |||||||||
Clinical studies and outside services | $ | 1,634 | $ | 1,688 | $ | 1,147 | ||||||
Personnel and infrastructure expenses | 1,041 | 900 | 714 | |||||||||
Facilities, IT and other costs | 339 | 266 | 259 | |||||||||
Total | $ | 3,014 | $ | 2,854 | $ | 2,120 | ||||||
In 2015, R&D expenses increased $160 million or 6% compared to 2014, primarily due to increases in personnel and infrastructure expenses of $141 million and facilities, IT and other costs of $73 million to support our ongoing clinical study activity and geographic expansion. As discussed below, 2014 clinical studies and outside services included one-time items of $350 million for collaboration and acquisition related expenses and the purchase of a FDA priority review voucher.
In 2014, R&D expenses increased $734 million or 35% compared to 2013, primarily due to an increase in clinical studies and outside services. The increase in clinical studies and outside services includes one-time items of $350 million for collaboration and acquisition related expenses and the purchase of a FDA priority review voucher and $191 million for expenses related to the progression of clinical study activity, primarily in the oncology and HIV areas. Personnel and infrastructure expenses increased $186 million to support our ongoing clinical study activity, geographic expansion and marketed product support.
In 2016, we expect R&D expenses to increase over 2015 to support the expansion of our clinical studies in various therapeutic areas including liver disease, HIV and inflammation.
Selling, General and Administrative Expenses
The following table summarizes the period over period changes in SG&A expenses:
(In millions, except percentages) | 2015 | Change | 2014 | Change | 2013 | |||||||||||||
Selling, general and administrative | $ | 3,426 | 15 | % | $ | 2,983 | 76 | % | $ | 1,699 | ||||||||
SG&A expenses relate to sales and marketing, finance, human resources, legal and other administrative activities. Expenses are primarily comprised of facilities and overhead costs, information technology infrastructure, outside marketing, advertising and legal expenses and other general and administrative costs. SG&A expenses also include the Branded Prescription Drug (BPD) fee, enacted with the Affordable Care Act in 2010.
In 2015, SG&A expenses increased $443 million or 15% compared to 2014 primarily due to an increase of $627 million in headcount-related, marketing and other expenses to support the growth and geographic expansion of our business, partially offset by a decrease in BPD fee expense of $100 million due to a change in estimate of our portion of the fee related to prior years.
In 2014, SG&A expenses increased $1.3 billion or 76% compared to 2013 primarily due to an increase in headcount-related and other expenses of $542 million to support the ongoing growth and expansion of our business, including commercial expansion related to the launches of Sovaldi and Harvoni and an increase in BPD fee expense of $480 million. During 2014, the Internal Revenue Service (IRS) issued final regulations which accelerated the expense recognition criteria for the fee obligation from the year in which the fee is paid, to the year in which the market share used to allocate the fee is determined. As a result, we recognized $460 million in our 2014 SG&A expenses that would have previously been accrued in 2015.
Our BPD fee expenses were $414 million in 2015, $590 million in 2014 and $110 million in 2013. The BPD fee is not tax deductible.
In 2016, we expect SG&A expenses to increase compared to 2015 to support our continued build-out and expansion of our commercial infrastructure globally to support our products and to increase by an estimated $200 million for the BPD fee.
Interest Expense
In 2015, interest expense increased to $688 million compared to $412 million in 2014. The increase was primarily due to the issuance of $10.0 billion aggregate principal amount of senior unsecured notes (the 2015 Notes) in 2015 and the issuance of $8.0 billion aggregate principal amount of senior unsecured notes (the 2014 Notes) in 2014.
53
In 2014, interest expense increased to $412 million compared to $307 million in 2013. The increase was primarily a result of the issuance of the 2014 Notes, offset by repayment of our senior unsecured notes issued in March and December 2011 (the 2011 Notes) and conversion and maturity of our convertible senior notes due in May 2014 (the May 2014 Notes) and partial conversion of our convertible senior notes due in May 2016 (the May 2016 Notes, and collectively with the May 2014 Notes, the May Notes).
Other Income (Expense), Net
Other income (expense), net increased to $154 million in 2015 compared to $3 million in 2014 primarily due to higher interest income as the result of our portfolio earning a higher yield and higher cash balances. Other income (expense), net was insignificant in 2014 and 2013.
Provision for Income Taxes
Our provision for income taxes was $3.6 billion, $2.8 billion and $1.2 billion in 2015, 2014 and 2013, respectively. The 2015 effective tax rate of 16.4% differed from the U.S. federal statutory rate of 35% primarily due to certain operating earnings from non-U.S. subsidiaries that are considered indefinitely reinvested and tax credits, partially offset by state taxes, our portion of the non-tax deductible BPD fee and amortization expense of the intangible asset related to sofosbuvir for which we receive no tax benefit. We do not provide for U.S. income taxes on undistributed earnings of our foreign operations that are intended to be indefinitely reinvested in our foreign subsidiaries.
The 2014 effective tax rate of 18.8% differed from the U.S. federal statutory rate of 35% primarily due to certain operating earnings from non-U.S. subsidiaries that are considered indefinitely reinvested and tax credits, partially offset by state taxes, our portion of the non-tax deductible BPD fee and amortization expense of the intangible asset related to sofosbuvir for which we receive no tax benefit.
The 2013 effective tax rate of 27.3% differed from the U.S. federal statutory rate of 35% primarily due to the retroactive extension of the 2012 federal research tax credit in January 2013, the 2013 federal research tax credit and certain operating earnings from non-U.S. subsidiaries that are considered indefinitely reinvested, partially offset by state taxes, our portion of the non-tax deductible BPD fee, amortization expense of the intangible asset related to sofosbuvir and contingent consideration expense related to certain acquisitions for which we receive no tax benefit.
Subsequent Event
Galapagos
We entered into a license and collaboration agreement with Galapagos NV (Galapagos), a clinical-stage biotechnology company based in Belgium, for the development and commercialization of filgotinib, a JAK1-selective inhibitor being investigated for inflammatory disease indications. Under the terms of the agreement, which became effective on January 19, 2016, we made an upfront license fee payment of $300 million and a $425 million equity investment in Galapagos. In addition, Galapagos is eligible to receive development and regulatory milestone-based payments of up to $755 million, sales-based milestone payments of up to $600 million, tiered royalties on global sales and a profit split in potential co-promotion territories.
Liquidity and Capital Resources
We believe that our existing capital resources, supplemented by our cash flows generated from operating activities will be adequate to satisfy our capital needs for the foreseeable future. The following table summarizes our cash, cash equivalents and marketable securities and working capital:
(in millions) | 2015 | 2014 | 2013 | ||||||||||
As of December 31: | |||||||||||||
Cash, cash equivalents and marketable securities | $ | 26,208 | $ | 11,726 | $ | 2,571 | |||||||
Working capital | $ | 14,872 | $ | 11,953 | $ | 590 | |||||||
Cash, Cash Equivalents and Marketable Securities
Cash, cash equivalents and marketable securities totaled $26.2 billion at December 31, 2015, an increase of $14.5 billion or 124% when compared to $11.7 billion at December 31, 2014. During 2015, we generated $20.3 billion in cash flows from operations, received $9.9 billion in net proceeds from the 2015 Notes and repurchased $10.0 billion of common stock. Additionally, we utilized $3.9 billion to settle 46 million warrants related to the May 2016 Notes (the 2016 Warrants) and paid cash dividends of $1.9 billion.
54
Cash, cash equivalents and marketable securities totaled $11.7 billion at December 31, 2014, an increase of $9.2 billion or 356% when compared to $2.6 billion at December 31, 2013. During 2014, we generated $12.8 billion in cash flows from operations, received $7.9 billion from the issuance of the 2014 Notes, repaid $2.3 billion in debt, net of convertible note hedges, repurchased $5.3 billion of common stock and paid approximately $4.1 billion to settle the warrants expiring in 2014 related to the May 2014 Notes (the 2014 Warrants).
Of the total cash, cash equivalents and marketable securities at December 31, 2015, approximately $15.7 billion was generated from operations in foreign jurisdictions and is intended for use in our foreign operations. We do not rely on unrepatriated earnings as a source of funds for our domestic business as we expect to have sufficient cash flow and borrowing capacity in the United States to fund our domestic operational and strategic needs.
Working Capital
Working capital was $14.9 billion at December 31, 2015. The increase of $2.9 billion from working capital as of December 31, 2014 was driven primarily by the increase in cash, cash equivalents and short-term marketable securities and an increase in accounts receivable, partially offset by increases in accrued government and other rebates.
Working capital was $12.0 billion at December 31, 2014. The increase of $11.4 billion from working capital as of December 31, 2013 was driven primarily by positive cash flows from operations and an increase in cash and cash equivalents due to the issuance of the 2014 Notes, partially offset by cash paid to settle convertible senior notes and the 2014 Warrants, repayment of our bank debt, and repurchases of common stock.
Cash Flows
The following table summarizes our cash flow activities:
(in millions) | 2015 | 2014 | 2013 | ||||||||||
Cash provided by (used in): | |||||||||||||
Operating activities | $ | 20,329 | $ | 12,818 | $ | 3,105 | |||||||
Investing activities | $ | (12,475 | ) | $ | (1,823 | ) | $ | (254 | ) | ||||
Financing activities | $ | (4,963 | ) | $ | (3,025 | ) | $ | (2,544 | ) | ||||
Cash Provided by Operating Activities
Cash provided by operating activities was $20.3 billion in 2015, consisting primarily of net income of $18.1 billion, adjusted for non-cash items such as $1.1 billion of depreciation and amortization expenses, $382 million for stock-based compensation expense and $1.2 billion of net cash inflow related to changes in operating assets and liabilities. Cash flows from operations may decrease in the future as we continue to make cash payments related to accrued government and other rebates.
Cash provided by operating activities was $12.8 billion in 2014, consisting primarily of net income of $12.1 billion, adjusted for non-cash items such as $1.1 billion of depreciation and amortization expenses and $360 million of stock-based compensation expenses. This was partially offset by $518 million of net cash outflow related to changes in operating assets and liabilities.
Cash provided by operating activities was $3.1 billion in 2013, consisting primarily of net income of $3.1 billion, adjusted for non-cash items such as $345 million of depreciation and amortization expenses and $252 million of stock-based compensation expenses. This was partially offset by $562 million of net cash outflow related to changes in operating assets and liabilities.
Cash Used in Investing Activities
Cash used in investing activities in 2015 was $12.5 billion, consisting primarily of $11.7 billion in net purchases of marketable securities and $747 million in capital expenditures related to the expansion of our business.
Cash used in investing activities in 2014 was $1.8 billion, consisting primarily of $1.2 billion in net purchases of marketable securities and $557 million in capital expenditures related to the expansion of our business.
Cash used in investing activities in 2013 was $254 million, consisting primarily of $379 million used in our acquisition of YM BioSciences, net of cash acquired and $190 million of capital expenditures primarily related to construction in progress associated with new facilities at our headquarters to support the ongoing growth of our business. This was partially offset by $315 million of net proceeds from sales of marketable securities.
55
Cash Used in Financing Activities
Cash used in financing activities in 2015 was $5.0 billion, consisting primarily of $10.0 billion used to repurchase common stock under our stock repurchase programs, $3.9 billion used to settle 46 million of the 2016 Warrants, and $1.9 billion used to pay dividends. These payments were primarily offset by $9.9 billion in net proceeds from the issuance of our 2015 Notes.
Cash used in financing activities in 2014 was $3.0 billion, consisting primarily of $2.3 billion used to repay debt, net of convertible notes hedges, $5.3 billion used to repurchase common stock under our stock repurchase programs and $4.1 billion to settle the 2014 Warrants. These payments were primarily offset by $7.9 billion in net proceeds from the issuance of our 2014 Notes.
Cash used in financing activities in 2013 was $2.5 billion, consisting primarily of $4.4 billion used to repay debt financing which includes the maturity of our convertible senior notes due in May 2013 (the May 2013 Notes) and conversions of our May Notes, $1.0 billion to settle the warrants related to our May 2013 Notes that settled in August 2013 and $582 million used to repurchase common stock under our stock repurchase program. This cash outflow was partially offset by proceeds of $2.8 billion related to our convertible note hedges.
Debt and Credit Facility
Long-Term Obligations
The summary of our borrowings under various financing arrangements is included in Item 8, Note 10 Debt and Credit Facility in our Consolidated Financial Statements included in this Annual Report on Form 10-K.
Debt Financing
In September 2015, we issued our 2015 Notes in the aggregate principal amount of $10.0 billion. In 2014, we issued our 2014 Notes in the aggregate principal amount of $8.0 billion. The 2015 Notes and 2014 Notes were issued for general corporate purposes, which may include the repayment of debt, working capital, payment of dividends and the repurchase of our outstanding common stock pursuant to our authorized share repurchase programs.
Convertible Senior Note Repayments and Warrant Settlements
During 2015, a portion of the May 2016 Notes were settled and we repaid $213 million of principal balance related to these notes. We also paid $784 million in cash related to the conversion spread of the May 2016 Notes, which represents the conversion value in excess of the principal amount, and received $784 million in cash from the convertible note hedges related to the May 2016 Notes. In 2015, we entered into modified agreements with our warrant counterparties which changed the timing of the expiration for 46 million of the 2016 Warrants. The agreements allowed us to settle the 46 million warrants at our option, in cash or shares. According to the terms of the agreements, these warrants expired during a 32 trading-day period which commenced on May 11, 2015 and ended on June 24, 2015. We exercised our option to settle in cash, and as a result, paid
$3.9 billion as the market value of our common stock at the time of the exercise of the warrants exceeded their strike prices.
During 2014, our May 2014 Notes matured and a portion of our May 2016 Notes was converted. During 2014, we repaid $912 million of principal balance relating to the May Notes. We also paid $2.5 billion in cash related to the conversion spread of the May Notes, which represents the conversion value in excess of the principal amount, and received $2.5 billion in cash from the convertible note hedges related to the May Notes. In 2014, we exercised our option to settle in cash the 2014 Warrants. As a result, we paid $4.1 billion to settle the warrants as the market value of our common stock at the time of the exercise of the warrants exceeded their strike price. There were 56 million shares of our common stock underlying the 2014 Warrants, which had a strike price of $28.38 per share and expired during the 40 trading-day period commencing August 1, 2014 and ending on September 26, 2014.
As of December 31, 2015 we had $283 million of outstanding convertible senior notes. The notes will mature in May 2016, unless earlier repurchased or converted. The remaining 9 million outstanding 2016 Warrants have a strike price of $28.76 per share, as adjusted for quarterly dividend distributions, and are due to expire during the 40 trading-day period commencing August 1, 2016. There were no other changes in terms for the remaining 9 million 2016 warrants.
Credit Facility
In January 2012, we entered into a five-year $1.3 billion revolving credit facility credit agreement (the Five-Year Revolving Credit Agreement) and borrowed $750 million thereunder. In 2013, we repaid $150 million under the Five-Year Revolving Credit Agreement. During 2014, we repaid the remaining balance of $600 million that was outstanding. The Five-Year Revolving Credit Agreement contains customary representations, warranties, affirmative, negative and financial maintenance covenants and events of default. The loan bears interest at either (i) the Eurodollar Rate plus the Applicable
56
Margin or (ii) the Base Rate plus the Applicable Margin, each as defined in the credit agreement. We may reduce the commitments and may prepay the loan in whole or in part at any time without premium or penalty. We are required to comply with certain covenants under the credit agreement and note indentures and as of December 31, 2015, we were not in violation of any covenants, and no amounts were outstanding under the credit facility.
Capital Return Program
Stock Repurchase Programs
In January 2015, our Board of Directors authorized a five-year, $15.0 billion stock repurchase program (2015 Program). The 2015 Program commenced after the $5.0 billion stock repurchase program authorized by our Board of Directors in May 2014 was completed in the first quarter of 2015. The $5.0 billion repurchase program authorized by our Board of Directors in January 2011 was completed in 2014. As of December 31, 2015, the remaining authorized repurchase amount under the 2015 Program was $8.0 billion.
The following table summarizes our stock repurchases under the above-described programs:
(in millions) | 2015 | 2014 | 2013 | ||||||||||
Shares repurchased and retired | 95 | 59 | 10 | ||||||||||
Amount | $ | 10,002 | $ | 5,349 | $ | 582 | |||||||
In February 2016, we entered into an accelerated share repurchase program (“ASR”) to repurchase $5.0 billion of our common stock. We paid $5.0 billion and received 46 million shares of our common stock, which represents approximately 80% of the total shares expected to be delivered to us under the ASR. The total number of shares to be received under the ASR will be based on the average price of our common stock during the purchase period, which will end in April 2016.
In February 2016, our Board of Directors authorized a new $12.0 billion share repurchase program (2016 Program) which will commence upon the completion of our 2015 Program. Purchases under the 2016 Program may be made in the open market or in privately negotiated transactions.
Dividends
In the second quarter of 2015, we began paying quarterly dividends on our common stock. During 2015, we paid cash dividends of $1.9 billion or $1.29 per share. On February 2, 2016, we announced that our Board of Directors declared a quarterly cash dividend of $0.43 per share of our common stock, with a payment date of March 30, 2016 to all stockholders of record as of the close of business on March 16, 2016.
Capital Resources
We believe our existing capital resources, supplemented by cash flows generated from our operations, will be adequate to satisfy our capital needs for the foreseeable future. Our future capital requirements will depend on many factors, including but not limited to the following:
• | the commercial performance of our current and future products; |
• | the progress and scope of our R&D efforts, including preclinical studies and clinical trials; |
• | the cost, timing and outcome of regulatory reviews; |
• | the expansion of our sales and marketing capabilities; |
• | administrative expenses; |
• | the possibility of acquiring additional manufacturing capabilities or office facilities; |
• | the possibility of acquiring other companies or new products; |
• | costs associated with the settlement and conversion of our convertible senior notes and related warrants; |
• | the establishment of additional collaborative relationships with other companies; and |
• | costs associated with the defense, settlement and adverse results of litigation and government investigations. |
We may in the future require additional funding, which could be in the form of proceeds from equity or debt financings. If such funding is required, we cannot guarantee that it will be available to us on favorable terms, if at all.
57
Critical Accounting Policies, Estimates and Judgments
The discussion and analysis of our financial condition and results of operations is based on our Consolidated Financial Statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosures. On an ongoing basis, we evaluate and base our estimates on historical experience and on various other market specific and other relevant assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates.
We believe the following critical accounting policies reflect the more significant judgments and estimates used in the preparation of our Consolidated Financial Statements.
Revenue Recognition
Product Sales
We recognize revenues from product sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured. We record product sales net of estimated mandatory and supplemental discounts to government payers, in addition to discounts to private payers, and other related charges. These are generally referred to as gross-to-net deductions and are recorded in the same period the related sales occur. Government and other rebates and chargebacks represent the majority of our gross-to-net deductions and require complex and significant judgment by management. Estimates are assessed each period and updated to reflect current information.
Government and Other Rebates and Chargebacks
Government and other rebates and chargebacks include amounts paid to payers and healthcare providers in the United States, including Medicaid rebates, ADAPs, Veterans Administration and Public Health Service discounts, and other rebates, as well as foreign government rebates. Rebates and chargebacks are based on contractual arrangements or statutory requirements which may vary by product, by payer and individual payer plans.
For qualified programs that can purchase our products through wholesalers or other distributors at a lower contractual price, the wholesalers or distributors charge back to us the difference between their acquisition cost and the lower contractual price. Our consolidated allowances for government and other chargebacks that are payable to our direct customers are classified as reductions of accounts receivable, and totaled $907 million as of December 31, 2015 and $220 million as of December 31, 2014.
Our consolidated allowance for government and other rebates that will be paid to parties other than our direct customers are recorded in accrued government and other rebates on our Consolidated Balance Sheets, and totaled $4.1 billion as of December 31, 2015 and $2.3 billion as of December 31, 2014.
Our allowances for government and other rebates and chargebacks are estimated based on products sold, historical utilization rates, pertinent third party industry information, estimated patient population, known market events or trends, channel inventory data and/or other market data. We also consider new information regarding changes in programs' regulations and guidelines that would impact the amount of the actual rebates and/or our expectations regarding future utilization rates for these programs. We believe that the methodology that we use to estimate our government and other rebates and chargebacks is reasonable and appropriate given the current facts and circumstances. However, actual results may differ significantly from our estimates. During the last three years, our actual government rebates and chargebacks claimed for prior periods have varied by less than 5% from our estimates.
58
The following table summarizes the consolidated activity in our government and other rebates and chargebacks accounts (in millions):
Accrued government and other rebates and chargebacks: | Balance at Beginning of Year | Decrease/(Increase) to Product Sales | Payments | Balance at End of Year | ||||||||||||
Year ended December 31, 2015: | ||||||||||||||||
Activity related to 2015 sales | $ | — | $ | 16,400 | $ | (11,597 | ) | $ | 4,803 | |||||||
Activity related to sales prior to 2015 | 2,536 | 7 | (2,321 | ) | 222 | |||||||||||
Total | $ | 2,536 | $ | 16,407 | $ | (13,918 | ) | $ | 5,025 | |||||||
Year ended December 31, 2014: | ||||||||||||||||
Activity related to 2014 sales | $ | — | $ | 6,113 | $ | (3,650 | ) | $ | 2,463 | |||||||
Activity related to sales prior to 2014 | 1,167 | (109 | ) | (985 | ) | 73 | ||||||||||
Total | $ | 1,167 | $ | 6,004 | $ | (4,635 | ) | $ | 2,536 | |||||||
The majority of the increase in accrued government and other rebates and chargebacks in 2015, compared to 2014, was driven by the increase in sales volume in 2015.
Allowance for Doubtful Accounts
We maintain an allowance for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments. This allowance is based on our analysis of several factors, including, but not limited to, contractual payment terms, historical payment patterns of our customers and individual customer circumstances, an analysis of days sales outstanding by geographic region and a review of the local economic environment and its potential impact on government funding and reimbursement practices. If the financial condition of our customers or the economic environment in which they operate were to deteriorate, resulting in an inability to make payments, additional allowances may be required. We believe that the allowance for doubtful accounts is adequate; however, significant deterioration in any of the above factors could materially change these expectations and may result in an increase to our allowance for doubtful accounts. As of December 31, 2015 and 2014, our accounts receivable, net were $5.9 billion and $4.6 billion and our allowances for doubtful accounts were $65 million and $31 million, respectively.
Valuation of Intangible Assets
In conjunction with our business combinations, we have recorded intangible assets primarily related to in-process research and development (IPR&D) projects. We had total intangible assets of $10.2 billion as of December 31, 2015 and $11.1 billion as of December 31, 2014.
The identifiable intangible assets are measured at their respective fair values as of the acquisition date. The models used in valuing these intangible assets require the use of significant estimates and assumptions including but not limited to:
• | estimates of revenues and operating profits related to the products or product candidates; |
• | the probability of success for unapproved product candidates considering their stages of development; |
• | the time and resources needed to complete the development and approval of product candidates; |
• | the life of the potential commercialized products and associated risks, including the inherent difficulties and uncertainties in developing a product candidate such as obtaining FDA and other regulatory approvals; and |
• | risks related to the viability of and potential alternative treatments in any future target markets. |
We believe the fair values used to record intangible assets acquired in connection with a business combination are based upon reasonable estimates and assumptions given the facts and circumstances as of the related valuation dates.
Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated R&D efforts. If and when development is complete, which generally occurs if and when regulatory approval to market a product is obtained, the associated assets would be deemed finite-lived and would then be amortized based on their respective estimated useful lives at that point in time. During the period the assets are considered indefinite-lived, they will not be amortized but will be tested for impairment on an annual basis as well as between annual tests if we become aware of any events or changes that would indicate that it is more likely than not that the fair value of the IPR&D projects below their respective carrying amounts. The fair value of our indefinite-lived intangible assets is dependent on assumptions such as the expected timing or probability of achieving the specified milestones, changes in projected revenues or changes in discount rates. Significant judgment is employed in determining these assumptions and changes to our assumptions could have a significant impact on our results of operations in any given period.
59
Intangible assets with finite useful lives are amortized over their estimated useful lives primarily on a straight-line basis. Intangible assets with finite useful lives are reviewed for impairment when facts or circumstances suggest that the carrying value of these assets may not be recoverable.
Tax Provision
We estimate our income tax provision, including deferred tax assets and liabilities, based on significant management judgment. We evaluate the realization of all or a portion of our deferred tax assets on a quarterly basis. We record a valuation allowance to reduce our deferred tax assets to the amounts that are more likely than not to be realized. We consider future taxable income, ongoing tax planning strategies and our historical financial performance in assessing the need for a valuation allowance.
If we expect to realize deferred tax assets for which we have previously recorded a valuation allowance, we will reduce the valuation allowance in the period in which such determination is first made.
We are subject to income taxes in both the United States and various foreign jurisdictions including Ireland. Due to economic and political conditions, various countries are actively considering changes to existing tax laws. We cannot predict the form or timing of potential legislative changes that could have a material adverse impact on our results of operations. In addition, significant judgment is required in determining our worldwide provision for income taxes.
We record liabilities related to uncertain tax positions in accordance with the guidance that clarifies the accounting for uncertainty in income taxes recognized in an enterprise's financial statements by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. We do not believe any such uncertain tax positions currently pending will have a material adverse effect on our Consolidated Financial Statements, although an adverse resolution of one or more of these uncertain tax positions in any period could have a material impact on the results of operations for that period.
At December 31, 2015 and 2014, we had total federal, state and foreign unrecognized tax benefits of $1.4 billion and $661 million, respectively. Of the total unrecognized tax benefits, $1.3 billion and $602 million at December 31, 2015 and 2014, respectively, if recognized, would reduce our effective tax rate in the period of recognition. As of December 31, 2015, we believe that it is reasonably possible that our unrecognized tax benefits will decrease by approximately $7 million in the next 12 months as we expect to have clarification from the IRS and other tax authorities regarding our uncertain tax positions. With respect to the remaining unrecognized tax benefits, we are currently unable to make a reasonable estimate as to the period of cash settlement, if any, with the respective tax authorities.
We file federal, state and foreign income tax returns in many jurisdictions in the United States and abroad. For federal income tax purposes, the statute of limitations is open for 2010 and onwards. For certain acquired entities, the statute of limitations is open for all years from inception due to our utilization of their net operating losses and credits carried over from prior years. For California income tax purposes, the statute of limitations is open for 2010 and onwards.
Our income tax returns are subject to audit by federal, state and foreign tax authorities. We are currently under examination by the IRS for the 2010, 2011 and 2012 tax years and by various state and foreign jurisdictions. There are differing interpretations of tax laws and regulations, and as a result, significant disputes may arise with these tax authorities involving issues of the timing and amount of deductions and allocations of income among various tax jurisdictions. We periodically evaluate our exposures associated with our tax filing positions.
Off Balance Sheet Arrangements
We do not have any off balance sheet arrangements as defined in Item 303(a)(4)(ii) of Regulation S-K.
60
Contractual Obligations
Our contractual obligations consist of debt obligations, operating leases, capital commitments, purchase obligations for active pharmaceutical ingredients and inventory-related items and clinical trials contracts. The following table summarizes our significant enforceable and legally binding obligations, future commitments and obligations related to all contracts that we are likely to continue regardless of the fact that certain of these obligations may be cancelable as of December 31, 2015 (in millions):
Payments due by Period | ||||||||||||||||||||
Contractual Obligations | Total | Less than one year | 1-3 years | 3-5 years | More than 5 years | |||||||||||||||
Debt (1) | $ | 36,003 | $ | 1,819 | $ | 2,645 | $ | 4,587 | $ | 26,952 | ||||||||||
Operating lease obligations | 317 | 66 | 114 | 74 | 63 | |||||||||||||||
Capital commitments (2) | 847 | 387 | 438 | 22 | — | |||||||||||||||
Purchase obligations (3)(4) | 3,084 | 1,759 | 1,034 | 208 | 83 | |||||||||||||||
Clinical trials (5) | 1,336 | 683 | 481 | 126 | 46 | |||||||||||||||
Total | $ | 41,587 | $ | 4,714 | $ | 4,712 | $ | 5,017 | $ | 27,144 | ||||||||||
(1) | Our debt obligations include senior unsecured notes and convertible senior notes. Interest payments are incurred and calculated based on terms of the related notes. For further information, see Item 8, Note 10 Debt and Credit Facility in our Consolidated Financial Statements included in this Annual Report on Form 10-K. |
(2) | At December 31, 2015, we had firm capital project commitments of approximately $847 million primarily relating to construction of new buildings. |
(3) | At December 31, 2015, we had firm purchase commitments related to active pharmaceutical ingredients and certain inventory-related items. These amounts include minimum purchase requirements. |
(4) | In addition to the above, we have committed to make potential future milestone payments to third parties as part of licensing, collaboration and development arrangements. Payments under these agreements generally become due and payable only upon achievement of certain developmental, regulatory and/or commercial milestones. Because the achievement of these milestones is neither probable nor reasonably estimable, such contingencies have not been recorded on our Consolidated Balance Sheets and have not been included in the table above. |
(5) | At December 31, 2015, we had several clinical studies in various clinical trial phases. Our most significant clinical trial expenditures are to contract research organizations (CROs). Although all of our material contracts with CROs are cancelable, we historically have not canceled such contracts. These amounts reflect commitments based on existing contracts and do not reflect any future modifications to, or terminations of, existing contracts or anticipated or potential new contracts. |
We had total gross unrecognized tax benefit liabilities including interest and penalties of $1.4 billion as of December 31, 2015. We believe that it is reasonably possible that our unrecognized tax benefits will decrease by approximately $7 million in the next 12 months as we expect to have clarification from the IRS and other tax authorities regarding our uncertain tax positions. With respect to the remaining unrecognized tax benefits, we are currently unable to make a reasonable estimate as to the period of cash settlement, if any, with the respective tax authorities. The unrecognized tax benefits were included in current and long-term income taxes payable and long-term deferred tax assets on our Consolidated Balance Sheets and have not been included in the table above.
Recent Accounting Pronouncements
The information required by this item is included in Item 8, Note 1 Organization and Summary of Significant Accounting Policies in our Consolidated Financial Statements included in this Annual Report on Form 10-K.
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are exposed to market risks that may result from changes in foreign currency exchange rates, interest rates and credit risks. To reduce certain of these risks, we enter into various types of foreign currency or interest rate derivative hedging transactions, follow investment guidelines and monitor outstanding receivables as part of our risk management program.
Foreign Currency Exchange Risk
Our operations include manufacturing and sales activities in the United States, Canada and Ireland as well as sales activities in countries outside the United States, including Europe and Asia Pacific. As a result, our financial results could be significantly affected by factors such as changes in foreign currency exchange rates or weak economic conditions in the foreign markets in which we distribute our products. Our operating results are exposed to changes in foreign currency exchange rates
61
between the U.S. dollar and various foreign currencies, the most significant of which are the Euro and Yen. When the U.S. dollar strengthens against these currencies, the relative value of sales made in the respective foreign currency decreases. Conversely, when the U.S. dollar weakens against these currencies, the relative amounts of such sales increase. Overall, we are a net receiver of foreign currencies and, therefore, benefit from a weaker U.S. dollar and are adversely affected by a stronger U.S. dollar relative to those foreign currencies in which we transact significant amounts of business.
Approximately 34% of our product sales were denominated in foreign currencies during 2015. To partially mitigate the impact of changes in currency exchange rates on net cash flows from our foreign currency denominated sales, we may enter into foreign currency exchange forward and option contracts. We also hedge certain monetary assets and liabilities denominated in foreign currencies, which reduces but does not eliminate our exposure to currency fluctuations between the date a transaction is recorded and the date that cash is collected or paid. In general, the market risks of these contracts are offset by corresponding gains and losses on the transactions being hedged.
As of December 31, 2015 and 2014, we had open foreign currency forward contracts with notional amounts of $9.1 billion and $6.4 billion, respectively. A hypothetical 10% adverse movement in foreign currency exchange rates compared with the U.S. dollar relative to exchange rates at December 31, 2015 would have resulted in a reduction in fair value of these contracts of approximately $893 million on this date and, if realized, would negatively affect earnings over the remaining life of the contracts. The same hypothetical movement in foreign currency exchange rates compared with the U.S. dollar relative to exchange rates at December 31, 2014, would have resulted in a reduction in fair value of these contracts of approximately $600 million on this date and, if realized, would negatively affect earnings over the remaining life of the contracts. The analysis does not consider the impact that hypothetical changes in foreign currency exchange rates would have on anticipated transactions that these foreign currency sensitive instruments were designed to offset.
Interest Rate Risk
Our portfolio of available-for-sale marketable securities and our fixed and variable rate liabilities create an exposure to interest rate risk. With respect to our investment portfolio, we adhere to an investment policy that requires us to limit amounts invested in securities based on credit rating, maturity, industry group and investment type and issuer, except for securities issued by the U.S. government. The goals of our investment policy, in order of priority, are as follows:
• | safety and preservation of principal and diversification of risk; |
• | liquidity of investments sufficient to meet cash flow requirements; and |
• | competitive after-tax rate of return. |
The following table summarizes the expected maturities and average interest rates of our interest-generating assets and fixed interest-bearing liabilities at December 31, 2015 (in millions, except percentages):
Expected Maturity | Total Fair Value | |||||||||||||||||||||||||||||||
2016 | 2017 | 2018 | 2019 | 2020 | Thereafter | Total | ||||||||||||||||||||||||||
Assets | ||||||||||||||||||||||||||||||||
Available-for-sale debt securities | $ | 1,759 | $ | 6,020 | $ | 4,466 | $ | 525 | $ | 384 | $ | 206 | $ | 13,360 | $ | 13,360 | ||||||||||||||||
Average interest rate | 0.75 | % | 1.21 | % | 1.58 | % | 1.36 | % | 1.48 | % | 1.71 | % | ||||||||||||||||||||
Liabilities | ||||||||||||||||||||||||||||||||
Debt (1) | $ | 985 | $ | — | $ | 1,000 | $ | 500 | $ | 2,500 | $ | 17,250 | $ | 22,235 | $ | 23,738 | ||||||||||||||||
Average interest rate | 2.64 | % | — | % | 1.85 | % | 2.05 | % | 2.51 | % | 4.24 | % | ||||||||||||||||||||
(1) | As of December 31, 2015 our debt consisted of senior unsecured notes and convertible senior notes with an aggregate carrying value of $22.2 billion. Since these instruments bear interest at fixed rates, changes in interest rates do not affect interest expense or cash flows. However, the fair value of these instruments fluctuates when interest rates change. See Note 10, Debt and Credit Facility in our Consolidated Financial Statements included in this Annual Report on Form 10-K for additional information. |
Credit Risk
We are subject to credit risk from our portfolio of cash equivalents and marketable securities. Under our investment policy, we limit amounts invested in such securities by credit rating, maturity, industry group, investment type and issuer, except for securities issued by the U.S. government. We are not exposed to any significant concentrations of credit risk from these financial instruments. The goals of our investment policy, in order of priority, are as follows: safety and preservation of principal and diversification of risk; liquidity of investments sufficient to meet cash flow requirements; and a competitive after-tax rate of return.
62
We are also subject to credit risk from our accounts receivable related to our product sales. The majority of our trade accounts receivable arises from product sales in the United States and Europe.
As of December 31, 2015, our accounts receivable in Southern Europe, specifically Greece, Italy, Portugal and Spain, totaled approximately $1.3 billion, of which $218 million were greater than 120 days past due, including $31 million greater than 365 days past due. As of December 31, 2014, our accounts receivable in Southern Europe, specifically Greece, Italy, Portugal and Spain, totaled approximately $504 million, of which $157 million were greater than 120 days past due, including $44 million greater than 365 days past due. To date, we have not experienced significant losses with respect to the collection of our accounts receivable.
63
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
GILEAD SCIENCES, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Years ended December 31, 2015, 2014 and 2013
CONTENTS
Report of Independent Registered Public Accounting Firm | ||
Audited Consolidated Financial Statements: | ||
Selected Quarterly Financial Information (Unaudited) | ||
64
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Gilead Sciences, Inc.
We have audited the accompanying consolidated balance sheets of Gilead Sciences, Inc. as of December 31, 2015 and 2014, and the related consolidated statements of income, comprehensive income, stockholders' equity, and cash flows for each of the three years in the period ended December 31, 2015. Our audits also included the financial statement schedule listed in the Index at Item 15(a). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Gilead Sciences, Inc. at December 31, 2015 and 2014, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2015, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Gilead Sciences, Inc.'s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated February 24, 2016 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Redwood City, California
February 24, 2016
65
GILEAD SCIENCES, INC.
Consolidated Balance Sheets
(in millions, except per share amounts)
December 31, | |||||||
2015 | 2014 | ||||||
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 12,851 | $ | 10,027 | |||
Short-term marketable securities | 1,756 | 101 | |||||
Accounts receivable, net of allowances of $1,032 at December 31, 2015 and $356 at December 31, 2014 | 5,854 | 4,635 | |||||
Inventories | 1,955 | 1,386 | |||||
Deferred tax assets | 828 | 508 | |||||
Prepaid and other current assets | 1,519 | 1,057 | |||||
Total current assets | 24,763 | 17,714 | |||||
Property, plant and equipment, net | 2,276 | 1,674 | |||||
Long-term portion of prepaid royalties | 400 | 466 | |||||
Long-term deferred tax assets | 324 | 236 | |||||
Long-term marketable securities | 11,601 | 1,598 | |||||
Intangible assets, net | 10,247 | 11,073 | |||||
Goodwill | 1,172 | 1,172 | |||||
Other long-term assets | 1,056 | 731 | |||||
Total assets | $ | 51,839 | $ | 34,664 | |||
Liabilities and Stockholders’ Equity | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 1,178 | $ | 955 | |||
Accrued government and other rebates | 4,118 | 2,316 | |||||
Other accrued liabilities | 3,172 | 1,873 | |||||
Deferred revenues | 440 | 134 | |||||
Current portion of long-term debt and other obligations, net | 983 | 483 | |||||
Total current liabilities | 9,891 | 5,761 | |||||
Long-term debt, net | 21,195 | 11,921 | |||||
Long-term income taxes payable | 1,243 | 562 | |||||
Other long-term obligations | 395 | 586 | |||||
Commitments and contingencies (Note 11) | |||||||
Equity component of currently redeemable convertible notes | 2 | 15 | |||||
Stockholders’ equity: | |||||||
Preferred stock, par value $0.001 per share; 5 shares authorized; none outstanding | — | — | |||||
Common stock, par value $0.001 per share; shares authorized of 5,600 at December 31, 2015 and December 31, 2014; shares issued and outstanding of 1,422 at December 31, 2015 and 1,499 at December 31, 2014 | 1 | 2 | |||||
Additional paid-in capital | 444 | 2,391 | |||||
Accumulated other comprehensive income | 88 | 301 | |||||
Retained earnings | 18,001 | 12,732 | |||||
Total Gilead stockholders’ equity | 18,534 | 15,426 | |||||
Noncontrolling interest | 579 | 393 | |||||
Total stockholders’ equity | 19,113 | 15,819 | |||||
Total liabilities and stockholders’ equity | $ | 51,839 | $ | 34,664 | |||
See accompanying notes.
66
GILEAD SCIENCES, INC.
Consolidated Statements of Income
(in millions, except per share amounts)
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Revenues: | ||||||||||||
Product sales | $ | 32,151 | $ | 24,474 | $ | 10,804 | ||||||
Royalty, contract and other revenues | 488 | 416 | 398 | |||||||||
Total revenues | 32,639 | 24,890 | 11,202 | |||||||||
Costs and expenses: | ||||||||||||
Cost of goods sold | 4,006 | 3,788 | 2,859 | |||||||||
Research and development expenses | 3,014 | 2,854 | 2,120 | |||||||||
Selling, general and administrative expenses | 3,426 | 2,983 | 1,699 | |||||||||
Total costs and expenses | 10,446 | 9,625 | 6,678 | |||||||||
Income from operations | 22,193 | 15,265 | 4,524 | |||||||||
Interest expense | (688 | ) | (412 | ) | (307 | ) | ||||||
Other income (expense), net | 154 | 3 | (9 | ) | ||||||||
Income before provision for income taxes | 21,659 | 14,856 | 4,208 | |||||||||
Provision for income taxes | 3,553 | 2,797 | 1,151 | |||||||||
Net income | 18,106 | 12,059 | 3,057 | |||||||||
Net loss attributable to noncontrolling interest | (2 | ) | (42 | ) | (18 | ) | ||||||
Net income attributable to Gilead | $ | 18,108 | $ | 12,101 | $ | 3,075 | ||||||
Net income per share attributable to Gilead common stockholders - basic | $ | 12.37 | $ | 7.95 | $ | 2.01 | ||||||
Shares used in per share calculation - basic | 1,464 | 1,522 | 1,529 | |||||||||
Net income per share attributable to Gilead common stockholders - diluted | $ | 11.91 | $ | 7.35 | $ | 1.81 | ||||||
Shares used in per share calculation - diluted | 1,521 | 1,647 | 1,695 | |||||||||
Cash dividends declared per share | $ | 1.29 | $ | — | $ | — | ||||||
See accompanying notes.
67
GILEAD SCIENCES, INC.
Consolidated Statements of Comprehensive Income
(in millions)
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Net income | $ | 18,106 | $ | 12,059 | $ | 3,057 | ||||||
Other comprehensive income (loss): | ||||||||||||
Net foreign currency translation gain (loss), net of tax | 9 | (9 | ) | (44 | ) | |||||||
Available-for-sale securities: | ||||||||||||
Net unrealized gain (loss), net of tax impact of $(17), $0 and $4 | (29 | ) | 1 | 5 | ||||||||
Reclassifications to net income, net of tax impact of $1, $0 and $0 | 1 | (1 | ) | — | ||||||||
Net change | (28 | ) | — | 5 | ||||||||
Cash flow hedges: | ||||||||||||
Net unrealized gain (loss), net of tax impact of $21, $16 and $4 | 389 | 430 | (60 | ) | ||||||||
Reclassification to net income, net of tax impact of $(19), $(4) and $(1) | (583 | ) | 4 | 21 | ||||||||
Net change | (194 | ) | 434 | (39 | ) | |||||||
Other comprehensive income (loss) | (213 | ) | 425 | (78 | ) | |||||||
Comprehensive income | 17,893 | 12,484 | 2,979 | |||||||||
Comprehensive loss attributable to noncontrolling interest | (2 | ) | (42 | ) | (18 | ) | ||||||
Comprehensive income attributable to Gilead | $ | 17,895 | $ | 12,526 | $ | 2,997 | ||||||
See accompanying notes.
68
GILEAD SCIENCES, INC.
Consolidated Statements of Stockholders' Equity
(in millions)
Gilead Stockholders' Equity | Noncontrolling Interest | Total Stockholders' Equity | |||||||||||||||||||||||||
Common Stock | Additional Paid-In Capital | Accumulated Other Comprehensive Income (Loss) | Retained Earnings | ||||||||||||||||||||||||
Shares | Amount | ||||||||||||||||||||||||||
Balance at December 31, 2012 | 1,519 | $ | 2 | $ | 5,642 | $ | (46 | ) | $ | 3,705 | $ | 241 | $ | 9,544 | |||||||||||||
Contributions from noncontrolling interest | — | — | — | — | — | 152 | 152 | ||||||||||||||||||||
Net income (loss) | — | — | — | — | 3,075 | (18 | ) | 3,057 | |||||||||||||||||||
Other comprehensive loss, net of tax | — | — | — | (78 | ) | — | — | (78 | ) | ||||||||||||||||||
Issuances under employee stock purchase plan | 3 | — | 55 | — | — | — | 55 | ||||||||||||||||||||
Issuances under equity incentive plans | 24 | — | 258 | — | — | — | 258 | ||||||||||||||||||||
Tax benefits from employee stock plans | — | — | 285 | — | — | — | 285 | ||||||||||||||||||||
Stock-based compensation | — | — | 254 | — | — | — | 254 | ||||||||||||||||||||
Repurchases of common stock | (12 | ) | — | (14 | ) | — | (674 | ) | — | (688 | ) | ||||||||||||||||
Warrants settlement | — | — | (1,040 | ) | — | — | — | (1,040 | ) | ||||||||||||||||||
Convertible notes settlement | — | — | (2,771 | ) | — | — | — | (2,771 | ) | ||||||||||||||||||
Convertible notes hedge settlement | — | — | 2,774 | — | — | — | 2,774 | ||||||||||||||||||||
Reclassification to equity component of currently redeemable convertible notes | — | — | (57 | ) | — | — | — | (57 | ) | ||||||||||||||||||
Balance at December 31, 2013 | 1,534 | 2 | 5,386 | (124 | ) | 6,106 | 375 | 11,745 | |||||||||||||||||||
Change in noncontrolling interest | — | — | — | — | — | 60 | 60 | ||||||||||||||||||||
Net income (loss) | — | — | — | — | 12,101 | (42 | ) | 12,059 | |||||||||||||||||||
Other comprehensive income, net of tax | — | — | — | 425 | — | — | 425 | ||||||||||||||||||||
Issuances under employee stock purchase plan | 3 | — | 72 | — | — | — | 72 | ||||||||||||||||||||
Issuances under equity incentive plans | 24 | — | 260 | — | — | — | 260 | ||||||||||||||||||||
Tax benefits from employee stock plans | — | — | 484 | — | — | — | 484 | ||||||||||||||||||||
Stock-based compensation | — | — | 362 | — | — | — | 362 | ||||||||||||||||||||
Repurchases of common stock | (62 | ) | — | (133 | ) | — | (5,475 | ) | — | (5,608 | ) | ||||||||||||||||
Warrants settlement | — | — | (4,093 | ) | — | — | — | (4,093 | ) | ||||||||||||||||||
Convertible notes settlement | — | — | (2,513 | ) | — | — | — | (2,513 | ) | ||||||||||||||||||
Convertible notes hedge settlement | — | — | 2,543 | — | — | — | 2,543 | ||||||||||||||||||||
Purchases of convertible note hedges | — | — | (26 | ) | — | — | — | (26 | ) | ||||||||||||||||||
Reclassification to equity component of currently redeemable convertible notes | — | — | 49 | — | — | — | 49 | ||||||||||||||||||||
Balance at December 31, 2014 | 1,499 | 2 | 2,391 | 301 | 12,732 | 393 | 15,819 | ||||||||||||||||||||
Change in noncontrolling interest | — | — | — | — | — | 188 | 188 | ||||||||||||||||||||
Net income (loss) | — | — | — | — | 18,108 | (2 | ) | 18,106 | |||||||||||||||||||
Other comprehensive loss, net of tax | — | — | — | (213 | ) | — | — | (213 | ) | ||||||||||||||||||
Issuances under employee stock purchase plan | 1 | — | 86 | — | — | — | 86 | ||||||||||||||||||||
Issuances under equity incentive plans | 21 | — | 235 | — | — | — | 235 | ||||||||||||||||||||
Tax benefits from employee stock plans | — | — | 586 | — | — | — | 586 | ||||||||||||||||||||
Stock-based compensation | — | — | 384 | — | — | — | 384 | ||||||||||||||||||||
Repurchases of common stock | (99 | ) | (1 | ) | (222 | ) | — | (10,115 | ) | — | (10,338 | ) | |||||||||||||||
Warrants settlement | — | — | (3,031 | ) | — | (834 | ) | — | (3,865 | ) | |||||||||||||||||
Convertible notes settlement | — | — | (782 | ) | — | — | — | (782 | ) | ||||||||||||||||||
Convertible notes hedge settlement | — | — | 784 | — | — | — | 784 | ||||||||||||||||||||
Dividends declared | — | — | — | — | (1,890 | ) | — | (1,890 | ) | ||||||||||||||||||
Reclassification to equity component of currently redeemable convertible notes | — | — | 13 | — | — | — | 13 | ||||||||||||||||||||
Balance at December 31, 2015 | 1,422 | $ | 1 | $ | 444 | $ | 88 | $ | 18,001 | $ | 579 | $ | 19,113 | ||||||||||||||
See accompanying notes.
69
GILEAD SCIENCES, INC.
Consolidated Statements of Cash Flows
(in millions)
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Operating Activities: | ||||||||||||
Net income | $ | 18,106 | $ | 12,059 | $ | 3,057 | ||||||
Adjustments to reconcile net income to net cash provided by operating activities: | ||||||||||||
Depreciation expense | 161 | 125 | 103 | |||||||||
Amortization expense | 937 | 925 | 242 | |||||||||
Stock-based compensation expense | 382 | 360 | 252 | |||||||||
Excess tax benefits from stock-based compensation | (585 | ) | (482 | ) | (279 | ) | ||||||
Tax benefits from exercise and vesting of stock-based awards | 586 | 484 | 285 | |||||||||
Deferred income taxes | (393 | ) | (236 | ) | (98 | ) | ||||||
Other | (24 | ) | 101 | 105 | ||||||||
Changes in operating assets and liabilities: | ||||||||||||
Accounts receivable, net | (1,397 | ) | (2,578 | ) | (315 | ) | ||||||
Inventories | (855 | ) | 143 | (343 | ) | |||||||
Prepaid expenses and other assets | (90 | ) | (371 | ) | (170 | ) | ||||||
Accounts payable | 226 | (289 | ) | (98 | ) | |||||||
Income taxes payable | 269 | 533 | 30 | |||||||||
Accrued liabilities | 2,632 | 2,013 | 312 | |||||||||
Deferred revenues | 374 | 31 | 22 | |||||||||
Net cash provided by operating activities | 20,329 | 12,818 | 3,105 | |||||||||
Investing Activities: | ||||||||||||
Purchases of marketable securities | (17,239 | ) | (2,107 | ) | (257 | ) | ||||||
Proceeds from sales of marketable securities | 4,792 | 807 | 494 | |||||||||
Proceeds from maturities of marketable securities | 719 | 52 | 78 | |||||||||
Other investments | — | (18 | ) | — | ||||||||
Acquisitions, net of cash acquired | — | — | (379 | ) | ||||||||
Capital expenditures | (747 | ) | (557 | ) | (190 | ) | ||||||
Net cash used in investing activities | (12,475 | ) | (1,823 | ) | (254 | ) | ||||||
Financing Activities: | ||||||||||||
Proceeds from debt financing, net of issuance costs | 9,902 | 7,932 | — | |||||||||
Proceeds from convertible note hedges | 784 | 2,543 | 2,774 | |||||||||
Purchases of convertible note hedges | — | (26 | ) | — | ||||||||
Proceeds from issuances of common stock | 319 | 331 | 313 | |||||||||
Repurchases of common stock | (10,002 | ) | (5,349 | ) | (582 | ) | ||||||
Repayments of debt and other obligations | (997 | ) | (4,779 | ) | (4,440 | ) | ||||||
Payments to settle warrants | (3,865 | ) | (4,093 | ) | (1,040 | ) | ||||||
Excess tax benefits from stock-based compensation | 585 | 482 | 279 | |||||||||
Payment of contingent consideration | (3 | ) | (101 | ) | — | |||||||
Payment of dividends | (1,874 | ) | — | — | ||||||||
Contributions from noncontrolling interest | 188 | 35 | 152 | |||||||||
Net cash used in financing activities | (4,963 | ) | (3,025 | ) | (2,544 | ) | ||||||
Effect of exchange rate changes on cash and cash equivalents | (67 | ) | (56 | ) | 2 | |||||||
Net change in cash and cash equivalents | 2,824 | 7,914 | 309 | |||||||||
Cash and cash equivalents at beginning of period | 10,027 | 2,113 | 1,804 | |||||||||
Cash and cash equivalents at end of period | $ | 12,851 | $ | 10,027 | $ | 2,113 | ||||||
Supplemental disclosure of cash flow information: | ||||||||||||
Interest paid, net of amounts capitalized | $ | 529 | $ | 330 | $ | 238 | ||||||
Income taxes paid | $ | 3,137 | $ | 2,060 | $ | 1,051 | ||||||
See accompanying notes.
70
GILEAD SCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. | ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Overview
Gilead Sciences, Inc. (Gilead, we or us), incorporated in Delaware on June 22, 1987, is a research-based biopharmaceutical company that discovers, develops and commercializes innovative medicines in areas of unmet medical need. With each new discovery and investigational drug candidate, we strive to transform and simplify care for people with life-threatening illnesses around the world. Gilead's primary areas of focus include human immunodeficiency virus (HIV), liver diseases such as chronic hepatitis C virus (HCV) infection and chronic hepatitis B virus (HBV) infection, cardiovascular, hematology/oncology and inflammation/respiratory. We have operations in more than 30 countries worldwide, with headquarters in Foster City, California. We continue to add to our existing portfolio of products through our internal discovery and clinical development programs and through a product acquisition and in-licensing strategy.
Our portfolio of marketed products includes AmBisome®, Atripla®, Cayston®, Complera®/Eviplera®, Emtriva®, Genvoya®, Harvoni®, Hepsera®, Letairis®, Ranexa®, Sovaldi®, Stribild®, Tamiflu®, Truvada®, Tybost®, Viread®, Vitekta®, and Zydelig®. We have U.S. and international commercial sales operations, with marketing subsidiaries in North and South America, Europe and Asia-Pacific. We also sell and distribute certain products through our corporate partners under royalty-paying collaborative agreements.
Basis of Presentation
The accompanying Consolidated Financial Statements include the accounts of Gilead, our wholly-owned subsidiaries and certain variable interest entities for which we are the primary beneficiary. All intercompany transactions have been eliminated. For consolidated entities where we own or are exposed to less than 100% of the economics, we record net income (loss) attributable to noncontrolling interests in our Consolidated Statements of Income equal to the percentage of the economic or ownership interest retained in such entities by the respective noncontrolling parties.
We assess whether we are the primary beneficiary of a variable interest entity (VIE) at the inception of the arrangement and at each reporting date. This assessment is based on our power to direct the activities of the VIE that most significantly impact the VIE's economic performance and our obligation to absorb losses or the right to receive benefits from the VIE that could potentially be significant to the VIE. As of December 31, 2015, the only material VIE was our joint venture with Bristol-Myers Squibb (BMS) which is described in Note 9, Collaborative Arrangements.
Significant Accounting Policies, Estimates and Judgments
The preparation of these Consolidated Financial Statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosures. On an ongoing basis, we evaluate our significant accounting policies and estimates. We base our estimates on historical experience and on various market specific and other relevant assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates.
Revenue Recognition
Product Sales
We recognize revenue from product sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured. Upon recognition of revenue from product sales, provisions are made for government and other rebates such as Medicaid reimbursements, customer incentives such as cash discounts for prompt payment, distributor fees and expected returns of expired products, as appropriate.
71
Items Deducted from Gross Product Sales
Rebates and Chargebacks
We estimate reductions to our revenues for amounts paid to payers and healthcare providers in the United States, including Medicaid rebates, ADAPs, Veterans Administration and Public Health Service discounts, and other rebates, as well as foreign government rebates. Rebates and chargebacks are based on contractual arrangements or statutory requirements which may vary by product, by payer and individual payer plans. Our estimates are based on products sold, historical utilization rates, and as available, pertinent third party industry information, estimated patient population, known market events or trends, and for our U.S. product sales, channel inventory data obtained from our major U.S. wholesalers in accordance with our inventory management agreements. We also take into consideration, as available, new information regarding changes in programs' regulations and guidelines that would impact the amount of the actual rebates and/or our expectations regarding future utilization rates for these programs. Government and other chargebacks that are payable to our direct customers are classified as reductions of accounts receivable on our Consolidated Balance Sheets. Government and other rebates that are invoiced directly to us are recorded in accrued government and other rebates on our Consolidated Balance Sheets.
Cash Discounts
We estimate cash discounts based on contractual terms, historical utilization rates, as available, and our expectations regarding future utilization rates.
Distributor Fees
Under our inventory management agreements with our significant U.S. wholesalers, we pay the wholesalers a fee primarily for the compliance of certain contractually determined covenants such as the maintenance of agreed upon inventory levels. These distributor fees are based on a contractually determined fixed percentage of sales.
Product Returns
We do not provide our customers with a general right of product return, but typically permit returns if the product is damaged or defective when received by the customer, or in the case of product sold in the United States and certain countries outside the United States, if the product has expired. We will accept returns for product that will expire within six months or that have expired up to one year after their expiration dates. Our estimates for expected returns of expired products are based primarily on an ongoing analysis of our historical return patterns, historical industry information reporting the return rates for similar products and contractual agreements intended to limit the amount of inventory maintained by our wholesalers.
Royalty, Contract and Other Revenues
Royalty revenue from sales of our other products is generally recognized when received, which is generally in the quarter following the quarter in which the corresponding sales occur or in the month following the month in which the corresponding sales occur.
Revenue from non-refundable up-front license fees and milestone payments, such as under a development collaboration or an obligation to supply product, is recognized as performance occurs and our obligations are completed. In accordance with the specific terms of our obligations under these arrangements, revenue is recognized as the obligation is fulfilled or ratably over the development or manufacturing period. Revenue associated with substantive at-risk milestones is recognized based upon the achievement of the milestones set forth in the respective agreements. Advance payments received in excess of amounts earned are classified as deferred revenue on our Consolidated Balance Sheets.
Research and Development Expenses
Research and development (R&D) expenses consist primarily of personnel costs, including salaries, benefits and stock-based compensation, clinical studies performed by contract research organizations (CROs), materials and supplies, licenses and fees, milestone payments under collaboration arrangements and overhead allocations consisting of various support and facility-related costs.
We charge R&D costs, including clinical study costs, to expense when incurred. Clinical study costs are a significant component of R&D expenses. Most of our clinical studies are performed by third-party CROs. We monitor levels of performance under each significant contract including the extent of patient enrollment and other activities through communications with our CROs. We accrue costs for clinical studies performed by CROs over the service periods specified in the contracts and adjust our estimates, if required, based upon our ongoing review of the level of effort and costs actually incurred by the CROs. All of our material CRO contracts are terminable by us upon written notice and we are generally only liable for actual services completed by the CRO and certain non-cancelable expenses incurred at any point of termination.
72
Advertising Expenses
We expense the costs of advertising, including promotional expenses, as incurred. Advertising expenses were $601 million in 2015, $393 million in 2014 and $216 million in 2013.
Cash and Cash Equivalents
We consider highly liquid investments with insignificant interest rate risk and an original maturity of three months or less on the purchase date to be cash equivalents. Eligible instruments under our investment policy that are included in cash equivalents primarily include commercial paper, money market funds, overnight repurchase agreements (repos) with major banks and authorized dealers and other bank obligations.
Marketable and Nonmarketable Securities
We determine the appropriate classification of our marketable securities, which consist primarily of debt securities, at the time of purchase and reevaluate such designation at each balance sheet date. All of our marketable securities are considered as available-for-sale and carried at estimated fair values and reported in cash equivalents, short-term marketable securities or long-term marketable securities. Unrealized gains and losses on available-for-sale securities are excluded from net income and reported in accumulated other comprehensive income (loss) as a separate component of stockholders' equity. Other income (expense), net, includes interest, dividends, amortization of purchase premiums and discounts, realized gains and losses on sales of securities and other-than-temporary declines in the fair value of securities, if any. The cost of securities sold is based on the specific identification method. We regularly review all of our investments for other-than-temporary declines in fair value. Our review includes the consideration of the cause of the impairment, including the creditworthiness of the security issuers, the number of securities in an unrealized loss position, the severity and duration of the unrealized losses, whether we have the intent to sell the securities and whether it is more likely than not that we will be required to sell the securities before the recovery of their amortized cost basis. When we determine that the decline in fair value of an investment is below our accounting basis and this decline is other-than-temporary, we reduce the carrying value of the security we hold and record a loss for the amount of such decline.
As a result of entering into collaborations, from time to time, we may hold investments in non-public companies. We record these nonmarketable securities at cost in other long-term assets, less any amounts for other-than-temporary impairment. We regularly review our securities for indicators of impairment. Investments in nonmarketable securities are not material for the periods presented.
Concentrations of Risk
We are subject to credit risk from our portfolio of cash equivalents and marketable securities. Under our investment policy, we limit amounts invested in such securities by credit rating, maturity, industry group, investment type and issuer, except for securities issued by the U.S. government. We are not exposed to any significant concentrations of credit risk from these financial instruments. The goals of our investment policy, in order of priority, are as follows: safety and preservation of principal and diversification of risk; liquidity of investments sufficient to meet cash flow requirements; and a competitive after-tax rate of return.
We are also subject to credit risk from our accounts receivable related to our product sales. The majority of our trade accounts receivable arises from product sales in the United States and Europe. As of December 31, 2015, our accounts receivable in Southern Europe, specifically Greece, Italy, Portugal and Spain, totaled approximately $1.3 billion, of which $218 million were greater than 120 days past due, including $31 million greater than 365 days past due. To date, we have not experienced significant losses with respect to the collection of our accounts receivable.
Certain of the raw materials and components that we utilize in our operations are obtained through single suppliers. Certain of the raw materials that we utilize in our operations are made at only one facility. Since the suppliers of key components and raw materials must be named in a new drug application (NDA) filed with U.S. Food and Drug Administration (FDA) for a product, significant delays can occur if the qualification of a new supplier is required. If delivery of material from our suppliers was interrupted for any reason, we may be unable to ship our commercial products or to supply our product candidates for clinical trials.
73
Accounts Receivable
Trade accounts receivable are recorded net of allowances for wholesaler chargebacks related to government and other programs, cash discounts for prompt payment and doubtful accounts. Estimates for wholesaler chargebacks for government and other programs and cash discounts are based on contractual terms, historical trends and our expectations regarding the utilization rates for these programs. Estimates of our allowance for doubtful accounts are determined based on existing contractual payment terms, historical payment patterns of our customers and individual customer circumstances, an analysis of days sales outstanding by geographic region and a review of the local economic environment and its potential impact on government funding and reimbursement practices. Historically, the amounts of uncollectible accounts receivable that have been written off have been insignificant and consistent with management's expectations.
Inventories
Inventories are recorded at the lower of cost or market, with cost determined on a first-in, first-out basis. We periodically review the composition of our inventories in order to identify obsolete, slow-moving or otherwise unsaleable items. If unsaleable items are observed and there are no alternate uses for the inventory, we will record a write-down to net realizable value in the period that the impairment is first recognized.
When future commercialization is considered probable and the future economic benefit is expected to be realized, based on management's judgment, we capitalize pre-launch inventory costs prior to regulatory approval. A number of factors are taken into consideration, including the current status in the regulatory approval process, potential impediments to the approval process such as safety or efficacy, anticipated research and development initiatives that could impact the indication in which the compound will be used, viability of commercialization and marketplace trends. As of December 31, 2015 and 2014, the amount of pre-launch inventory on our Consolidated Balance Sheets was not significant.
Property, Plant and Equipment
Property, plant and equipment is stated at cost less accumulated depreciation and amortization. Depreciation and amortization are recognized using the straight-line method. Repairs and maintenance costs are expensed as incurred. Estimated useful lives in years are generally as follows:
Description | Estimated Useful Life |
Buildings and improvements | 20-35 |
Laboratory and manufacturing equipment | 4-10 |
Office and computer equipment | 3-7 |
Leasehold improvements | Shorter of useful life or lease term |
Office and computer equipment includes capitalized software. We had unamortized capitalized software costs on our Consolidated Balance Sheets of $115 million as of December 31, 2015 and $80 million as of December 31, 2014. Capitalized interest on construction in-progress is included in property, plant and equipment. Interest capitalized in 2015, 2014 and 2013 was not significant.
Goodwill and Intangible Assets
Goodwill represents the excess of the consideration transferred over the estimated fair value of assets acquired and liabilities assumed in a business combination. Intangible assets with indefinite useful lives are related to purchased in-process research and development (IPR&D) projects and are measured at their respective fair values as of the acquisition date. We do not amortize goodwill and intangible assets with indefinite useful lives. Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated R&D efforts. If and when development is complete, which generally occurs if and when regulatory approval to market a product is obtained, the associated assets are deemed finite-lived and are amortized based on their respective estimated useful lives at that point in time. We test goodwill and other indefinite-lived intangible assets for impairment on an annual basis and in between annual tests if we become aware of any events or changes that would indicate the fair values of the assets are below their carrying amounts.
Intangible assets with finite useful lives are amortized over their estimated useful lives, primarily on a straight-line basis, and are reviewed for impairment when facts or circumstances suggest that the carrying value of these assets may not be recoverable.
74
Impairment of Long-Lived Assets
Long-lived assets, including property, plant and equipment and finite-lived intangible assets, are reviewed for impairment on a regular basis and whenever facts or circumstances either internally or externally may suggest that the carrying value of an asset or asset group may not be recoverable. Should there be an indication of impairment, we test for recoverability by comparing the estimated undiscounted future cash flows expected to result from the use of the asset or asset group and its eventual disposition to the carrying amount of the asset or asset group. Any excess of the carrying value of the asset or asset group over its estimated fair value is recognized as an impairment loss.
Foreign Currency Translation, Transaction Gains and Losses, and Hedging Contracts
Non-U.S. entity operations are recorded in the functional currency of each entity. Results of operations for non-U.S. dollar functional currency entities are translated into U.S. dollars using average currency rates. Assets and liabilities are translated using currency rates at period end. Foreign currency translation adjustments are recorded as a component of other comprehensive income (loss) within stockholders' equity. Foreign currency transaction gains and losses are recorded in other income (expense), net on our Consolidated Statements of Income. Net foreign currency transaction gains and losses were immaterial for the years ended December 31, 2015, 2014 and 2013.
We hedge a portion of our foreign currency exposures related to outstanding monetary assets and liabilities as well as forecasted product sales using foreign currency exchange forward and option contracts. In general, the market risk related to these contracts is offset by corresponding gains and losses on the hedged transactions. The credit risk associated with these contracts is driven by changes in interest and currency exchange rates and, as a result, varies over time. By working only with major banks and closely monitoring current market conditions, we seek to limit the risk that counterparties to these contracts may be unable to perform. We also seek to limit our risk of loss by entering into contracts that permit net settlement at maturity. Therefore, our overall risk of loss in the event of a counterparty default is limited to the amount of any unrecognized gains on outstanding contracts (i.e., those contracts that have a positive fair value) at the date of default. We do not enter into derivative contracts for trading purposes, nor do we hedge our net investment in any of our foreign subsidiaries.
Fair Value of Financial Instruments
We apply fair value accounting for all financial assets and liabilities and non-financial assets and liabilities that are recognized or disclosed at fair value in the financial statements on a recurring basis. We define fair value as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities which are required to be recorded at fair value, we consider the principal or most advantageous market in which we would transact and the market-based risk measurements or assumptions that market participants would use in pricing the asset or liability, such as risks inherent in valuation techniques, transfer restrictions and credit risks.
Derivative Financial Instruments
We recognize all derivative instruments as either assets or liabilities at fair value in our Consolidated Balance Sheets. Changes in the fair value of derivatives are recorded each period in current earnings or accumulated other comprehensive income (loss), depending on whether a derivative is designated as part of a hedge transaction and, if it is, the type of hedge transaction. We classify the cash flows from these instruments in the same category as the cash flows from the hedged items. We do not hold or issue derivative instruments for trading or speculative purposes.
We assess, both at inception and on an ongoing basis, whether the derivatives that are used in hedging transactions are highly effective in offsetting the changes in cash flows or fair values of the hedged items. We also assess hedge ineffectiveness on a quarterly basis and record the gain or loss related to the ineffective portion to current earnings to the extent significant. If we determine that a forecasted transaction is no longer probable of occurring, we discontinue hedge accounting for the affected portion of the hedge instrument, and any related unrealized gain or loss on the contract is recognized in current earnings.
Income Taxes
Our income tax provision is computed under the liability method. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Significant estimates are required in determining our provision for income taxes. Some of these estimates are based on interpretations of existing tax laws or regulations.
We record liabilities related to uncertain tax positions in accordance with the guidance that clarifies the accounting for uncertainty in income taxes recognized in an enterprise's financial statements by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. We do not believe any such uncertain tax positions currently pending will have a material adverse effect
75
on our Consolidated Financial Statements, although an adverse resolution of one or more of these uncertain tax positions in any period could have a material impact on the results of operations for that period.
Branded Prescription Drug (BPD) Fee
We, along with other pharmaceutical manufacturers of branded drug products, are required to pay a portion of the BPD fee, which is calculated based on select government sales during each calendar year as a percentage of total industry government sales. In 2014, the Internal Revenue Service (IRS) issued final regulations related to the BPD fee which accelerated the expense recognition criteria for the fee obligation from the year in which the fee is paid, to the year in which the related sales and market share used to allocate the fee is determined. Our BPD fee expenses were $414 million in 2015, $590 million in 2014 and $110 million in 2013 and are recorded as selling, general and administrative (SG&A) expense within our Consolidated Statements of Income. Our BPD fee accrual totaled $780 million as of December 31, 2015 and $500 million as of December 31, 2014 on our Consolidated Balance Sheets.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB), jointly with the International Accounting Standards Board, issued a comprehensive new standard on revenue recognition from contracts with customers. The standard's core principle is that a reporting entity will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. In August 2015, the FASB issued an accounting standard update which defers the effective date of the new standard by one year. The standard will become effective for us beginning in the first quarter of 2018. Early adoption is permitted in 2017. Entities have the option of using either a full retrospective or a modified retrospective approach to adopt this new guidance. We are evaluating the impact of the adoption of this standard on our Consolidated Financial Statements.
In April 2015, the FASB issued an accounting standard update which requires presentation of debt issuance costs as a direct deduction from the carrying amount of a recognized debt liability on the balance sheet. The update does not change the guidance on the recognition and measurement of debt issuance costs. This guidance will become effective for us beginning in the first quarter of 2016. At the time of adoption, we will reclassify debt issuance costs to a liability as a direct deduction from the carrying value of the debt, consistent with the presentation of a debt discount. We do not expect that the adoption of this update will have a material impact on our Consolidated Balance Sheets.
In November 2015, the FASB issued an accounting standard update which requires that deferred tax liabilities and assets be classified as noncurrent on the balance sheet. Previous guidance required deferred tax liabilities and assets to be separated into current and noncurrent amounts on the balance sheet. The guidance will become effective for us beginning in the first quarter of 2017 and may be applied either prospectively or retrospectively. Early adoption is permitted. At the time of adoption, we will reclassify current deferred tax amounts on our Consolidated Balance Sheets as noncurrent. We are evaluating the impact of the method of adoption of this standard on our Consolidated Financial Statements.
In January 2016, the FASB issued new guidance related to accounting for equity investments, financial liabilities under the fair value option, and the presentation and disclosure requirements for financial instruments. In addition, the FASB clarified guidance related to the valuation allowance assessment when recognizing deferred tax assets resulting from unrealized losses on available-for-sale debt securities. The guidance will become effective for us beginning in the first quarter of 2018. Early adoption is permitted. We are evaluating the impact of adopting this accounting guidance on our Consolidated Financial Statements.
2. | FAIR VALUE MEASUREMENTS |
We determine the fair value of financial and non-financial assets and liabilities using the fair value hierarchy, which establishes three levels of inputs that may be used to measure fair value, as follows:
• | Level 1 inputs which include quoted prices in active markets for identical assets or liabilities; |
• | Level 2 inputs which include observable inputs other than Level 1 inputs, such as quoted prices for similar assets or liabilities; quoted prices for identical or similar assets or liabilities in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the asset or liability. For our marketable securities, we review trading activity and pricing as of the measurement date. When sufficient quoted pricing for identical securities is not available, we use market pricing and other observable market inputs for similar securities obtained from various third-party data providers. These inputs either represent quoted prices for similar assets in active markets or have been derived from observable market data; and |
• | Level 3 inputs which include unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the underlying asset or liability. Our Level 3 assets and liabilities include those whose |
76
fair value measurements are determined using pricing models, discounted cash flow methodologies or similar valuation techniques and significant management judgment or estimation.
Our financial instruments consist principally of cash and cash equivalents, marketable securities, accounts receivable, foreign currency exchange contracts, accounts payable and short-term and long-term debt. Cash and cash equivalents, marketable securities and foreign currency exchange contracts that hedge accounts receivable and forecasted sales are reported at their respective fair values on our Consolidated Balance Sheets. Short-term and long-term debt are reported at their amortized cost on our Consolidated Balance Sheets. The remaining financial instruments are reported on our Consolidated Balance Sheets at amounts that approximate current fair values. There were no transfers among the fair value levels in the periods presented.
The following table summarizes the assets and liabilities measured at fair value on a recurring basis by level within the fair value hierarchy (in millions):
December 31, 2015 | December 31, 2014 | ||||||||||||||||||||||||||||||
Level 1 | Level 2 | Level 3 | Total | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||||||||||
Assets: | |||||||||||||||||||||||||||||||
Money market funds | $ | 10,161 | $ | — | $ | — | $ | 10,161 | $ | 7,926 | $ | — | $ | — | $ | 7,926 | |||||||||||||||
Corporate debt securities | — | 5,773 | — | 5,773 | — | 938 | — | 938 | |||||||||||||||||||||||
U.S. treasury securities | 4,389 | — | — | 4,389 | 363 | — | — | 363 | |||||||||||||||||||||||
Residential mortgage and asset-backed securities | — | 1,695 | — | 1,695 | — | 269 | — | 269 | |||||||||||||||||||||||
U.S. government agencies securities | — | 707 | — | 707 | — | 113 | — | 113 | |||||||||||||||||||||||
Certificates of deposit | — | 448 | — | 448 | — | — | — | — | |||||||||||||||||||||||
Non-U.S. government securities | — | 313 | — | 313 | — | — | — | — | |||||||||||||||||||||||
Municipal debt securities | — | 34 | — | 34 | — | 16 | — | 16 | |||||||||||||||||||||||
Foreign currency derivative contracts | — | 210 | — | 210 | — | 349 | — | 349 | |||||||||||||||||||||||
Deferred compensation plan | 66 | — | — | 66 | 54 | — | — | 54 | |||||||||||||||||||||||
$ | 14,616 | $ | 9,180 | $ | — | $ | 23,796 | $ | 8,343 | $ | 1,685 | $ | — | $ | 10,028 | ||||||||||||||||
Liabilities: | |||||||||||||||||||||||||||||||
Contingent consideration | $ | — | $ | — | $ | 59 | $ | 59 | $ | — | $ | — | $ | 133 | $ | 133 | |||||||||||||||
Deferred compensation plan | 66 | — | — | 66 | 54 | — | — | 54 | |||||||||||||||||||||||
Foreign currency derivative contracts | — | 41 | — | 41 | — | — | — | — | |||||||||||||||||||||||
$ | 66 | $ | 41 | $ | 59 | $ | 166 | $ | 54 | $ | — | $ | 133 | $ | 187 | ||||||||||||||||
Level 2 Inputs
We estimate the fair values of our investments in corporate debt securities, residential mortgage and asset-backed securities, government-related securities and certificates of deposit by taking into consideration valuations obtained from third-party pricing services. The pricing services utilize industry standard valuation models, including both income- and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. These inputs include reported trades of and broker/dealer quotes on the same or similar securities; issuer credit spreads; benchmark securities; prepayment/default projections based on historical data; and other observable inputs.
Substantially all of our foreign currency derivative contracts have maturities over an 18 month time horizon and all are with counterparties that have a minimum credit rating of A- or equivalent by Standard & Poor's, Moody's Investors Service, Inc. or Fitch, Inc. We estimate the fair values of these contracts by taking into consideration valuations obtained from a third-party valuation service that utilizes an income-based industry standard valuation model for which all significant inputs are observable, either directly or indirectly. These inputs include foreign currency rates, London Interbank Offered Rates (LIBOR) and swap rates. These inputs, where applicable, are at commonly quoted intervals.
The total estimated fair values of our convertible senior notes and senior unsecured notes, determined using Level 2 inputs based on their quoted market values, were approximately $23.7 billion at December 31, 2015 and $15.0 billion at December 31, 2014, and the carrying values were $22.2 billion at December 31, 2015 and $12.4 billion at December 31, 2014.
77
Level 3 Inputs
As of December 31, 2015 and 2014, the only assets or liabilities that were measured using Level 3 inputs were contingent consideration liabilities. Our policy is to recognize transfers into or out of Level 3 classification as of the actual date of the event or change in circumstances that caused the transfer. Our contingent consideration liabilities were immaterial as of December 31, 2015 and 2014.
3. AVAILABLE-FOR-SALE SECURITIES
Estimated fair values of available-for-sale securities are generally based on prices obtained from commercial pricing services. The following table is a summary of available-for-sale securities recorded in cash and cash equivalents or marketable securities in our Consolidated Balance Sheets (in millions):
December 31, 2015 | December 31, 2014 | |||||||||||||||||||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | |||||||||||||||||||||||||
Money market funds | $ | 10,161 | $ | — | $ | — | $ | 10,161 | $ | 7,926 | $ | — | $ | — | $ | 7,926 | ||||||||||||||||
Corporate debt securities | 5,795 | 1 | (23 | ) | 5,773 | 941 | — | (3 | ) | 938 | ||||||||||||||||||||||
U.S. treasury securities | 4,407 | — | (18 | ) | 4,389 | 363 | — | — | 363 | |||||||||||||||||||||||
Residential mortgage and asset-backed securities | 1,701 | — | (6 | ) | 1,695 | 269 | — | — | 269 | |||||||||||||||||||||||
U.S. government agencies securities | 709 | — | (2 | ) | 707 | 113 | — | — | 113 | |||||||||||||||||||||||
Certificates of deposit | 448 | — | — | 448 | — | — | — | — | ||||||||||||||||||||||||
Non-U.S. government securities | 315 | — | (2 | ) | 313 | — | — | — | — | |||||||||||||||||||||||
Municipal debt securities | 34 | — | — | 34 | 16 | — | — | 16 | ||||||||||||||||||||||||
Total | $ | 23,570 | $ | 1 | $ | (51 | ) | $ | 23,520 | $ | 9,628 | $ | — | $ | (3 | ) | $ | 9,625 | ||||||||||||||
The following table summarizes the classification of the available-for-sale securities on our Consolidated Balance Sheets (in millions):
December 31, 2015 | December 31, 2014 | ||||||
Cash and cash equivalents | $ | 10,163 | $ | 7,926 | |||
Short-term marketable securities | 1,756 | 101 | |||||
Long-term marketable securities | 11,601 | 1,598 | |||||
Total | $ | 23,520 | $ | 9,625 | |||
Cash and cash equivalents in the table above excludes cash of $2.7 billion as of December 31, 2015 and $2.1 billion as of December 31, 2014.
The following table summarizes our portfolio of available-for-sale securities by contractual maturity (in millions):
December 31, 2015 | |||||||
Amortized Cost | Fair Value | ||||||
Less than one year | $ | 11,921 | $ | 11,919 | |||
Greater than one year but less than five years | 11,442 | 11,395 | |||||
Greater than five years but less than ten years | 186 | 184 | |||||
Greater than ten years | 21 | 22 | |||||
Total | $ | 23,570 | $ | 23,520 | |||
78
The following table summarizes our available-for-sale debt securities that were in a continuous unrealized loss position, but were not deemed to be other-than-temporarily impaired (in millions):
Less Than 12 Months | 12 Months or Greater | Total | ||||||||||||||||||||||
Gross Unrealized Losses | Estimated Fair Value | Gross Unrealized Losses | Estimated Fair Value | Gross Unrealized Losses | Estimated Fair Value | |||||||||||||||||||
December 31, 2015 | ||||||||||||||||||||||||
Debt securities: | ||||||||||||||||||||||||
Corporate debt securities | $ | (23 | ) | $ | 4,891 | $ | — | $ | 43 | $ | (23 | ) | $ | 4,934 | ||||||||||
U.S. treasury securities | (18 | ) | 4,342 | — | — | (18 | ) | 4,342 | ||||||||||||||||
Residential mortgage and asset-backed securities | (6 | ) | 1,626 | — | 20 | (6 | ) | 1,646 | ||||||||||||||||
U.S. government agencies securities | (2 | ) | 707 | — | — | (2 | ) | 707 | ||||||||||||||||
Non-U.S. government securities | (2 | ) | 313 | — | — | (2 | ) | 313 | ||||||||||||||||
Municipal debt securities | — | 21 | — | — | — | 21 | ||||||||||||||||||
Total | $ | (51 | ) | $ | 11,900 | $ | — | $ | 63 | $ | (51 | ) | $ | 11,963 | ||||||||||
December 31, 2014 | ||||||||||||||||||||||||
Debt securities: | ||||||||||||||||||||||||
Corporate debt securities | $ | (3 | ) | $ | 802 | $ | — | $ | — | $ | (3 | ) | $ | 802 | ||||||||||
Residential mortgage and asset-backed securities | — | 227 | — | 1 | — | 228 | ||||||||||||||||||
U.S. treasury securities | — | 206 | — | — | — | 206 | ||||||||||||||||||
U.S. government agencies securities | — | 22 | — | — | — | 22 | ||||||||||||||||||
Municipal debt securities | — | 2 | — | — | — | 2 | ||||||||||||||||||
Total | $ | (3 | ) | $ | 1,259 | $ | — | $ | 1 | $ | (3 | ) | $ | 1,260 | ||||||||||
We held a total of 2,742 positions as of December 31, 2015 and 468 positions as of December 31, 2014 that were in an unrealized loss position. Based on our review of these securities, we believe we had no other-than-temporary impairments on these securities as of December 31, 2015 and 2014 because we do not intend to sell these securities and we believe it is not more likely than not that we will be required to sell these securities before the recovery of their amortized cost basis. Gross realized gains and gross realized losses were immaterial for the years ended December 31, 2015, 2014 and 2013.
4. | DERIVATIVE FINANCIAL INSTRUMENTS |
Our operations in foreign countries expose us to market risk associated with foreign currency exchange rate fluctuations between the U.S. dollar and various foreign currencies, the most significant of which are the Euro and Yen. In order to manage this risk, we may hedge a portion of our foreign currency exposures related to outstanding monetary assets and liabilities as well as forecasted product sales using foreign currency exchange forward or option contracts. In general, the market risk related to these contracts is offset by corresponding gains and losses on the hedged transactions. The credit risk associated with these contracts is driven by changes in interest and currency exchange rates and, as a result, varies over time. By working only with major banks and closely monitoring current market conditions, we seek to limit the risk that counterparties to these contracts may be unable to perform. We also seek to limit our risk of loss by entering into contracts that permit net settlement at maturity. Therefore, our overall risk of loss in the event of a counterparty default is limited to the amount of any unrecognized gains on outstanding contracts (i.e., those contracts that have a positive fair value) at the date of default. We do not enter into derivative contracts for trading purposes.
We hedge our exposure to foreign currency exchange rate fluctuations for certain monetary assets and liabilities of our entities that are denominated in a non-functional currency. The derivative instruments we use to hedge this exposure are not designated as hedges, and as a result, changes in their fair value are recorded in other income (expense), net on our Consolidated Statements of Income.
We hedge our exposure to foreign currency exchange rate fluctuations for forecasted product sales that are denominated in a non-functional currency. The derivative instruments we use to hedge this exposure are designated as cash flow hedges and have maturity dates of 18 months or less. Upon executing a hedging contract and quarterly thereafter, we assess prospective hedge effectiveness using a regression analysis which calculates the change in cash flow as a result of the hedge instrument. On
79
a quarterly basis, we assess retrospective hedge effectiveness using a dollar offset approach. We exclude time value from our effectiveness testing and recognize changes in the time value of the hedge in other income (expense), net. The effective component of our hedge is recorded as an unrealized gain or loss on the hedging instrument in accumulated other comprehensive income (OCI) within stockholders' equity. When the hedged forecasted transaction occurs, the hedge is de-designated and the unrealized gains or losses are reclassified into product sales. The majority of gains and losses related to the hedged forecasted transactions reported in accumulated OCI at December 31, 2015 are expected to be reclassified to product sales within 12 months.
The cash flow effects of our derivative contracts for the three years ended December 31, 2015, 2014 and 2013 are included within net cash provided by operating activities in the Consolidated Statements of Cash Flows.
We had notional amounts on foreign currency exchange contracts outstanding of $9.1 billion at December 31, 2015 and $6.4 billion at December 31, 2014.
While all of our derivative contracts allow us the right to offset assets or liabilities, we have presented amounts on a gross basis. Under the International Swap Dealers Association, Inc. master agreements with the respective counterparties of the foreign currency exchange contracts, subject to applicable requirements, we are allowed to net settle transactions of the same currency with a single net amount payable by one party to the other. The following table summarizes the classification and fair values of derivative instruments on our Consolidated Balance Sheets (in millions):
December 31, 2015 | ||||||||||||
Asset Derivatives | Liability Derivatives | |||||||||||
Classification | Fair Value | Classification | Fair Value | |||||||||
Derivatives designated as hedges: | ||||||||||||
Foreign currency exchange contracts | Other current assets | $ | 200 | Other accrued liabilities | $ | (32 | ) | |||||
Foreign currency exchange contracts | Other long-term assets | 9 | Other long-term obligations | (8 | ) | |||||||
Total derivatives designated as hedges | 209 | (40 | ) | |||||||||
Derivatives not designated as hedges: | ||||||||||||
Foreign currency exchange contracts | Other current assets | 1 | Other accrued liabilities | (1 | ) | |||||||
Total derivatives not designated as hedges | 1 | (1 | ) | |||||||||
Total derivatives | $ | 210 | $ | (41 | ) | |||||||
December 31, 2014 | ||||||||||||
Asset Derivatives | Liability Derivatives | |||||||||||
Classification | Fair Value | Classification | Fair Value | |||||||||
Derivatives designated as hedges: | ||||||||||||
Foreign currency exchange contracts | Other current assets | $ | 314 | Other accrued liabilities | $ | — | ||||||
Foreign currency exchange contracts | Other long-term assets | 35 | Other long-term obligations | — | ||||||||
Total derivatives | $ | 349 | $ | — | ||||||||
As of December 31, 2014, there were no material derivatives not designated as hedges.
80
The following table summarizes the effect of our foreign currency exchange contracts on our Consolidated Financial Statements (in millions):
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Derivatives designated as hedges: | ||||||||||||
Gains (losses) recognized in accumulated OCI (effective portion) | $ | 410 | $ | 446 | $ | (55 | ) | |||||
Gains (losses) reclassified from accumulated OCI into product sales (effective portion) | $ | 602 | $ | — | $ | (20 | ) | |||||
Gains (losses) recognized in other income (expense), net (ineffective portion and amounts excluded from effectiveness testing) | $ | 13 | $ | (7 | ) | $ | 2 | |||||
Derivatives not designated as hedges: | ||||||||||||
Gains (losses) recognized in other income (expense), net | $ | 117 | $ | 135 | $ | (17 | ) | |||||
From time to time, we may discontinue cash flow hedges and as a result, record related amounts in other income (expense), net on our Consolidated Statements of Income. There were no material amounts recorded in other income (expense), net for the years ended December 31, 2015, 2014 and 2013 as a result of the discontinuance of cash flow hedges.
As of December 31, 2015 and 2014, we held one type of financial instrument, derivative contracts related to foreign currency exchange contracts. The following table summarizes the potential effect of offsetting derivatives by type of financial instrument on our Consolidated Balance Sheets (in millions):
As of December 31, 2015 | ||||||||||||||||||||||||
Offsetting of Derivative Assets/Liabilities | ||||||||||||||||||||||||
Gross Amounts Not Offset in the Consolidated Balance Sheet | ||||||||||||||||||||||||
Description | Gross Amounts of Recognized Assets/Liabilities | Gross Amounts Offset in the Consolidated Balance Sheet | Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | Derivative Financial Instruments | Cash Collateral Received/Pledged | Net Amount (Legal Offset) | ||||||||||||||||||
Derivative assets | $ | 210 | $ | — | $ | 210 | $ | (38 | ) | $ | — | $ | 172 | |||||||||||
Derivative liabilities | (41 | ) | — | (41 | ) | 38 | — | (3 | ) | |||||||||||||||
As of December 31, 2014 | ||||||||||||||||||||||||
Offsetting of Derivative Assets/Liabilities | ||||||||||||||||||||||||
Gross Amounts Not Offset in the Consolidated Balance Sheet | ||||||||||||||||||||||||
Description | Gross Amounts of Recognized Assets/Liabilities | Gross Amounts Offset in the Consolidated Balance Sheet | Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | Derivative Financial Instruments | Cash Collateral Received/Pledged | Net Amount (Legal Offset) | ||||||||||||||||||
Derivative assets | $ | 349 | $ | — | $ | 349 | $ | — | $ | — | $ | 349 | ||||||||||||
Derivative liabilities | — | — | — | — | — | — | ||||||||||||||||||
5. | INVENTORIES |
Inventories are summarized as follows (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Raw materials | $ | 1,332 | $ | 909 | ||||
Work in process | 542 | 500 | ||||||
Finished goods | 852 | 466 | ||||||
Total | $ | 2,726 | $ | 1,875 | ||||
Reported as: | ||||||||
Inventories | $ | 1,955 | $ | 1,386 | ||||
Other long-term assets | 771 | 489 | ||||||
Total | $ | 2,726 | $ | 1,875 | ||||
81
Amounts reported as other long-term assets primarily consisted of raw materials as of December 31, 2015 and December 31, 2014.
The joint ventures formed by Gilead Sciences, LLC and BMS (See Note 9, Collaborative Arrangements), which are included in our Consolidated Financial Statements, held efavirenz active pharmaceutical ingredient in inventory. This efavirenz inventory was purchased from BMS at BMS's estimated net selling price of efavirenz and totaled $1.3 billion as of December 31, 2015 and $806 million as of December 31, 2014.
6. | PROPERTY, PLANT AND EQUIPMENT |
Property, plant and equipment is summarized as follows (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Buildings and improvements (including leasehold improvements) | $ | 1,320 | $ | 997 | ||||
Laboratory and manufacturing equipment | 377 | 327 | ||||||
Office and computer equipment | 395 | 305 | ||||||
Construction in progress | 554 | 411 | ||||||
Subtotal | 2,646 | 2,040 | ||||||
Less accumulated depreciation and amortization (including $0 for 2015 and $2 for 2014 related to capitalized leased equipment) | (763 | ) | (620 | ) | ||||
Subtotal | 1,883 | 1,420 | ||||||
Land | 393 | 254 | ||||||
Total | $ | 2,276 | $ | 1,674 | ||||
7. | INTANGIBLE ASSETS |
The following table summarizes the carrying amount of our intangible assets (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Finite-lived intangible assets | $ | 9,815 | $ | 10,641 | ||||
Indefinite-lived intangible assets | 432 | 432 | ||||||
Total intangible assets | $ | 10,247 | $ | 11,073 | ||||
Finite-Lived Intangible Assets
The following table summarizes our finite-lived intangible assets (in millions):
December 31, 2015 | December 31, 2014 | |||||||||||||||
Gross Carrying Amount | Accumulated Amortization | Gross Carrying Amount | Accumulated Amortization | |||||||||||||
Intangible asset - sofosbuvir | $ | 10,720 | $ | 1,456 | $ | 10,720 | $ | 757 | ||||||||
Intangible asset - Ranexa | 688 | 363 | 688 | 277 | ||||||||||||
Other | 455 | 229 | 455 | 188 | ||||||||||||
Total | $ | 11,863 | $ | 2,048 | $ | 11,863 | $ | 1,222 | ||||||||
Amortization expense related to finite-lived intangible assets, included primarily in cost of goods sold in our Consolidated Statements of Income, totaled $826 million in 2015, $818 million in 2014 and $143 million in 2013. As of December 31, 2015, the estimated future amortization expense associated with our finite-lived intangible assets for each of the five succeeding fiscal years is as follows (in millions):
82
Fiscal Year | Amount | ||
2016 | $ | 839 | |
2017 | 844 | ||
2018 | 849 | ||
2019 | 741 | ||
2020 | 713 | ||
Thereafter | 5,829 | ||
Total | $ | 9,815 | |
Indefinite-Lived Intangible Assets
In 2013, we completed our acquisition of YM BioSciences (YM). Of the total $488 million fair value of acquired assets and assumed liabilities for YM, we attributed $363 million to IPR&D related to momelotinib on our Consolidated Balance Sheets. The following table summarizes our indefinite-lived intangible assets as of December 31, 2015 and December 31, 2014 (in millions):
Amount | ||||
Indefinite-lived intangible asset - momelotinib | $ | 315 | ||
Indefinite-lived intangible assets - Other | 117 | |||
Total | $ | 432 | ||
8. | OTHER FINANCIAL INFORMATION |
Prepaid and other current assets
The components of prepaid and other current assets are summarized as follows (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Prepaid taxes | $ | 773 | $ | 391 | ||||
Prepaid expenses | 240 | 194 | ||||||
Other current assets | 506 | 472 | ||||||
Total prepaid and other current assets | $ | 1,519 | $ | 1,057 | ||||
Other accrued liabilities
The components of other accrued liabilities are summarized as follows (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Income taxes payable | $ | 65 | $ | 105 | ||||
Compensation and employee benefits | 380 | 316 | ||||||
Branded Prescription Drug Fee | 649 | 186 | ||||||
Accrued royalties | 237 | 355 | ||||||
Other accrued expenses | 1,841 | 911 | ||||||
Total other accrued liabilities | $ | 3,172 | $ | 1,873 | ||||
9. | COLLABORATIVE ARRANGEMENTS |
We enter into collaborative arrangements with third parties for the development and commercialization of certain products. Both parties are active participants in the operating activities of the collaboration and are exposed to significant risks and rewards depending on the commercial success of the activities. The following are our significant collaborative arrangements.
83
Bristol-Myers Squibb Company
North America
In 2004, we entered into a collaboration arrangement with BMS to develop and commercialize a single tablet regimen containing our Truvada and BMS's Sustiva (efavirenz) in the United States. This combination was approved for use in the United States in 2006 and is sold under the brand name Atripla. We and BMS structured this collaboration as a joint venture that operates as a limited liability company named Bristol-Myers Squibb & Gilead Sciences, LLC, which we consolidate. We and BMS granted royalty-free sublicenses to the joint venture for the use of our respective company owned technologies and, in return, were granted a license by the joint venture to use any intellectual property that results from the collaboration. In 2006, we and BMS amended the joint venture's collaboration agreement to allow the joint venture to sell Atripla in Canada. The economic interests of the joint venture held by us and BMS (including a share of revenues and out-of-pocket expenses) are based on the portion of the net selling price of Atripla attributable to efavirenz and Truvada. Since the net selling price for Truvada may change over time relative to the net selling price of efavirenz, both our and BMS's respective economic interests in the joint venture may vary annually.
We and BMS shared marketing and sales efforts. Starting in the second quarter of 2011, except for a limited number of activities that will be jointly managed, the parties no longer coordinate detailing and promotional activities in the United States, and the parties reduced their joint promotional efforts since we launched Complera in August 2011 and Stribild in August 2012. The parties will continue to collaborate on activities such as manufacturing, regulatory, compliance and pharmacovigilance. The daily operations of the joint venture are governed by four primary joint committees formed by both BMS and Gilead. We are responsible for accounting, financial reporting, tax reporting, manufacturing and product distribution for the joint venture. Both parties provide their respective bulk active pharmaceutical ingredients to the joint venture at their approximate market values. The agreement will continue until terminated by the mutual agreement of the parties. In addition, either party may terminate the other party's participation in the collaboration within 30 days after the launch of at least one generic version of such other party's single agent products (or the double agent products). The terminating party then has the right to continue to sell Atripla and become the continuing party, but will be obligated to pay the terminated party certain royalties for a three-year period following the effective date of the termination.
As of December 31, 2015 and 2014, the joint venture held efavirenz active pharmaceutical ingredient which it purchased from BMS at BMS's estimated net selling price of efavirenz in the U.S. market. These amounts were primarily included in inventories on our Consolidated Balance Sheets.
Selected financial information for the joint venture was as follows (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Total assets | $ | 2,464 | $ | 2,138 | ||||
Cash and cash equivalents | 166 | 250 | ||||||
Accounts receivable, net | 269 | 297 | ||||||
Inventories | 2,027 | 1,590 | ||||||
Total liabilities | 1,055 | 1,157 | ||||||
Accounts payable | 606 | 749 | ||||||
Other accrued liabilities | 449 | 408 | ||||||
These asset and liability amounts do not reflect the impact of intercompany eliminations that are included in our Consolidated Balance Sheets. Although we consolidate the joint venture, the legal structure of the joint venture limits the recourse that its creditors will have over our general credit or assets. Similarly, the assets held in the joint venture can be used only to settle obligations of the joint venture.
84
Europe
In 2007, Gilead Sciences Ireland UC, our wholly-owned subsidiary, and BMS entered into a collaboration agreement with BMS which sets forth the terms and conditions under which we and BMS commercialize and distribute Atripla in the European Union, Iceland, Liechtenstein, Norway and Switzerland (collectively, the European Territory). The parties formed a limited liability company which we consolidate, to manufacture Atripla for distribution in the European Territory using efavirenz that it purchases from BMS at BMS's estimated net selling price of efavirenz in the European Territory. We are responsible for manufacturing, product distribution, inventory management and warehousing. Through our local subsidiaries, we have primary responsibility for order fulfillment, collection of receivables, customer relations and handling of sales returns in all the territories where we and BMS promote Atripla. In general, the parties share revenues and out-of-pocket expenses in proportion to the net selling prices of the components of Atripla, Truvada and efavirenz.
Starting in 2012, except for a limited number of activities that will be jointly managed, the parties no longer coordinate detailing and promotional activities in the region. We are responsible for accounting, financial reporting and tax reporting for the collaboration. As of December 31, 2015 and 2014, efavirenz purchased from BMS at BMS's estimated net selling price of efavirenz in the European Territory is included in inventories on our Consolidated Balance Sheets.
The parties also formed a limited liability company to hold the marketing authorization for Atripla in Europe. We have primary responsibility for regulatory activities. In the major market countries, both parties have agreed to independently continue to use commercially reasonable efforts to promote Atripla.
The agreement will terminate upon the expiration of the last-to-expire patent which affords market exclusivity to Atripla or one of its components in the European Territory. In addition, since December 31, 2013, either party may terminate the agreement for any reason and such termination will be effective two calendar quarters after notice of termination. The non-terminating party has the right to continue to sell Atripla and become the continuing party, but will be obligated to pay the terminating party certain royalties for a three-year period following the effective date of the termination. In the event the continuing party decides not to sell Atripla, the effective date of the termination will be the date Atripla is withdrawn in each country or the date on which a third party assumes distribution of Atripla, whichever is earlier.
Japan Tobacco Inc.
In 2005, Japan Tobacco Inc. (Japan Tobacco) granted us exclusive rights to develop and commercialize elvitegravir, a novel HIV integrase inhibitor, in all countries of the world, excluding Japan, where Japan Tobacco retained such rights. Under the agreement, we are responsible for seeking regulatory approval in our territories and are required to use diligent efforts to commercialize elvitegravir for the treatment of HIV infection. We bear all costs and expenses associated with such commercialization efforts.
We received approval of Stribild (an elvitegravir-containing product) from FDA in August 2012 and from the European Commission in May 2013. We received approval of Genvoya (an elvitegravir-containing product) from FDA and the European Commission in November 2015.
The agreement and our obligation to pay royalties to Japan Tobacco will terminate on a product-by-product basis as patents providing exclusivity for the product expire or, if later, on the tenth anniversary of commercial launch for such product. We may terminate the agreement for any reason in which case the license granted by Japan Tobacco to us would terminate. Either party may terminate the agreement in response to a material breach by the other party.
Janssen
In 2009, we entered into a license and collaboration agreement with Janssen Sciences Ireland UC (Janssen), formerly Tibotec Pharmaceuticals, to develop and commercialize a fixed-dose combination of our Truvada and Janssen's non-nucleoside reverse transcriptase inhibitor rilpivirine. This combination was approved in the United States and European Union in 2011 and is sold under the brand name Complera in the United States and Eviplera in the European Union. Under this original agreement, Janssen granted us an exclusive license to Complera/Eviplera worldwide excluding certain middle income and developing world countries and Japan.
In 2011 and 2013, we amended the agreement to include distribution of Complera/Eviplera to the rest of the world. In 2014, we amended the agreement to expand the collaboration to include another product containing Janssen’s rilpivirine and our emtricitabine and tenofovir alafenamide (R/F/TAF). Under the amended agreement, Janssen granted us an exclusive license to Complera/Eviplera and R/F/TAF worldwide, but retained rights to distribute both combination products in 18 countries including Mexico, Russia and Japan. Neither party is restricted from combining its drugs with any other drug products except those which are similar to the components of Complera/Eviplera and R/F/TAF.
85
We are responsible for manufacturing Complera/Eviplera and R/F/TAF and have the lead role in registration, distribution and commercialization of both products except in the countries where Janssen distributes. Janssen has exercised a right to co-detail the combination product in some of the countries where Gilead is the selling party.
Under the initial agreement, the price of Complera/Eviplera was expected to be the sum of the price of Truvada and the price of rilpivirine purchased separately. The cost of rilpivirine purchased by us from Janssen for Complera/Eviplera was approximately the market price of rilpivirine, less a specified percentage of up to 30% in major markets. The 2014 amendment, effective in 2015, enables the selling party to set the price of the combined products and the parties share revenues based on the ratio of the net selling prices of the party’s component(s), subject to certain restrictions and adjustments. We will continue to retain a specified percentage of Janssen’s share of revenues, up to 30% in major markets.
Either party may terminate the collaboration agreement with respect to a product and a country if the product is withdrawn from the market in such country or with respect to a product in all countries if the other party materially breaches the agreement with respect to a product. The agreement and the parties’ obligation to share revenues will expire on a product-by-product and country-by-country basis as Janssen patents providing exclusivity for the product expire or, if later, on the tenth anniversary of commercial launch for such product. We may terminate the agreement without cause with respect to the countries where we sell the products in which case Janssen has the right to become the selling party for such country if the product has launched but has been on the market for fewer than 10 years.
10. | DEBT AND CREDIT FACILITY |
Financing Arrangements
The following table summarizes the carrying amount of our borrowings under various financing arrangements (in millions):
Stated | December 31, | |||||||||||||
Type of Borrowing | Issue Date | Due Date | Interest Rate | 2015 | 2014 | |||||||||
Convertible Senior | July 2010 | May 2016 | 1.625% | $ | 283 | $ | 483 | |||||||
Senior Unsecured | December 2011 | December 2016 | 3.05% | 700 | 700 | |||||||||
Senior Unsecured | September 2015 | September 2018 | 1.85% | 1,000 | — | |||||||||
Senior Unsecured | March 2014 | April 2019 | 2.05% | 499 | 499 | |||||||||
Senior Unsecured | November 2014 | February 2020 | 2.35% | 499 | 499 | |||||||||
Senior Unsecured | September 2015 | September 2020 | 2.55% | 1,997 | — | |||||||||
Senior Unsecured | March 2011 | April 2021 | 4.50% | 995 | 995 | |||||||||
Senior Unsecured | December 2011 | December 2021 | 4.40% | 1,248 | 1,248 | |||||||||
Senior Unsecured | September 2015 | September 2022 | 3.25% | 999 | — | |||||||||
Senior Unsecured | March 2014 | April 2024 | 3.70% | 1,748 | 1,747 | |||||||||
Senior Unsecured | November 2014 | February 2025 | 3.50% | 1,748 | 1,748 | |||||||||
Senior Unsecured | September 2015 | March 2026 | 3.65% | 2,739 | — | |||||||||
Senior Unsecured | September 2015 | September 2035 | 4.60% | 997 | — | |||||||||
Senior Unsecured | December 2011 | December 2041 | 5.65% | 998 | 998 | |||||||||
Senior Unsecured | March 2014 | April 2044 | 4.80% | 1,747 | 1,747 | |||||||||
Senior Unsecured | November 2014 | February 2045 | 4.50% | 1,740 | 1,740 | |||||||||
Senior Unsecured | September 2015 | March 2046 | 4.75% | 2,241 | — | |||||||||
Total debt, net | $ | 22,178 | $ | 12,404 | ||||||||||
Less current portion | 983 | 483 | ||||||||||||
Total long-term debt, net | $ | 21,195 | $ | 11,921 | ||||||||||
Senior Unsecured Notes
In 2015, we issued $10.0 billion aggregate principal amount of senior unsecured notes (the 2015 Notes) in a registered offering. In 2014, we issued $8.0 billion aggregate principal amount of senior unsecured notes (the 2014 Notes) in registered offerings in March and November 2014. The 2015 Notes and 2014 Notes were issued for general corporate purposes, which may include the repayment of debt, working capital, payment of dividends and the repurchase of our outstanding common stock pursuant to our authorized share repurchase programs.
86
We collectively refer to the 2015 Notes, 2014 Notes and our senior unsecured notes issued in March and December 2011 (the 2011 Notes) as our Senior Notes. Our Senior Notes may be redeemed at our option at a redemption price equal to the greater of (i) 100% of the principal amount of the notes to be redeemed and (ii) the sum, as determined by an independent investment banker, of the present value of the remaining scheduled payments of principal and interest on the notes to be redeemed (exclusive of interest accrued to the date of redemption) discounted to the redemption date on a semiannual basis at the Treasury Rate plus a make-whole premium as defined in the indenture. Our Senior Notes maturing after 2020 also have a call feature, exercisable at our option, to redeem the notes at par in whole or in part two to six months immediately preceding maturity. In each case, accrued and unpaid interest is also required to be redeemed to the date of redemption. In 2014, we repaid at maturity $750 million of principal balance related to the 2011 Notes.
In the event of the occurrence of a change in control and a downgrade in the rating of our Senior Notes below investment grade by Standard & Poor's Ratings Services and Moody's Investors Service, Inc., the holders may require us to purchase all or a portion of the Senior Notes at a price equal to 101% of the aggregate principal amount of the notes repurchased, plus accrued and unpaid interest to the date of repurchase.
We incurred debt issuance costs of $70 million in connection with the issuance of the 2015 Notes and $50 million for the 2014 Notes which are being amortized to interest expense over the contractual term of each of the respective notes. We recognized $605 million in 2015, $350 million in 2014 and $201 million in 2013 of interest expense on our Senior Notes related to the contractual coupon rates and amortization of the debt discount and issuance costs.
Convertible Senior Notes
In July 2010, we issued $1.3 billion of convertible senior notes due in May 2014 (the May 2014 Notes) and $1.3 billion of convertible senior notes due in May 2016 (the May 2016 Notes, and collectively with the May 2014 Notes, the May Notes) in a private placement pursuant to Rule 144A of the Securities Act of 1933, as amended.
The May Notes were issued at par. The May 2014 Notes bore an annual interest rate of 1.00% and the May 2016 Notes bear an annual interest rate of 1.625%. Debt issuance costs of $35 million were recorded in other long term assets and are being amortized to interest expense over the contractual terms of the May Notes. The initial conversion rate for the May 2014 Notes was 44.3690 shares per $1,000 principal amount (which represented an initial conversion price of approximately $22.54 per share), and the initial conversion rate for the May 2016 Notes was 44.0428 shares per $1,000 principal amount (which represents an initial conversion price of approximately $22.71 per share). The conversion rates are subject to customary anti-dilution adjustments, including quarterly dividend distributions. As of December 31, 2015, the conversion rate for the May 2016 Notes was 44.5680 (which represented a conversion price of approximately $22.44 per share).
The May 2016 Notes may be converted prior to April 1, 2016 only under the following circumstances: 1) during any calendar quarter commencing after September 30, 2010, if the closing price of the common stock for at least 20 trading days (whether or not consecutive) during the period of 30 consecutive trading days ending on the last trading day of the preceding calendar quarter is greater than 130% of the applicable conversion price on each applicable trading day, or 2) during the five business day period after any measurement period of ten consecutive trading days in which, for each trading day of such period, the trading price per $1,000 principal amount of notes was less than 98% of the product of the last reported sale price of our common stock and the applicable conversion rate on such trading day, or 3) upon the occurrence of specified corporate transactions, such as the distribution of certain stock rights, cash amounts, or other assets to all of our shareholders or the occurrence of a change in control. On and after April 1, 2016, in the case of the May 2016 Notes, holders may convert their notes at any time, regardless of the foregoing circumstances. Generally, upon conversion, a holder would receive an amount in cash equal to the lesser of (i) the principal amount of the note or (ii) the conversion value for such note, as measured under the indenture governing the relevant notes. If the conversion value exceeds the principal amount, we may also deliver, at our option, cash or common stock or a combination of cash and common stock for the conversion value in excess of the principal amount.
During 2015, a portion of the May 2016 Notes was converted. During 2014, the May 2014 Notes matured and a portion of the May 2016 Notes was converted. The following table summarizes information about the May Notes settlements (in millions):
Principal repayments | Conversion value paid in excess of principal | Net proceeds from convertible note hedges | ||||||||||||||||||||||
Year Ended December 31, | Year Ended December 31, | Year Ended December 31, | ||||||||||||||||||||||
2015 | 2014 | 2015 | 2014 | 2015 | 2014 | |||||||||||||||||||
May Notes | $ | 213 | $ | 912 | $ | 784 | $ | 2,517 | $ | 784 | $ | 2,517 | ||||||||||||
87
As of December 31, 2015, given their maturity date, the May 2016 Notes were classified as current. As of December 31, 2014, the May 2016 Notes were classified as current given that their conversion criteria were met. As a result, the related equity component equal to the unamortized discounts of $2 million and $15 million as of December 31, 2015 and 2014, respectively, was classified as an equity component of currently redeemable convertible notes on our Consolidated Balance Sheets.
If the May 2016 Notes are converted in connection with a change in control, we may be required to provide a make whole premium in the form of an increase in the conversion rate, subject to a stated maximum amount. In addition, in the event of a change in control, the holders may require us to purchase all or a portion of their notes at a purchase price equal to 100% of their principal amount, plus accrued and unpaid interest, if any. As of December 31, 2015, the if-converted value of the May 2016 Notes would exceed the principal amounts of the May 2016 Notes by $1.0 billion.
Concurrent with the issuance of the May Notes, we purchased convertible note hedges in private transactions at a cost of $363 million, which is tax deductible over the life of the notes. We also sold warrants in private transactions to acquire 111 million shares of our common stock and received net proceeds of $155 million from the sale of the warrants. The convertible note hedges and warrants are intended to reduce the potential economic dilution upon future conversions of the May Notes by effectively increasing our conversion price to $28.38 per share for the May 2014 Notes and $30.05 per share for the May 2016 Notes. The net cost of $207 million of the convertible note hedge and warrant transactions was recorded in stockholders' equity on our Consolidated Balance Sheets. In addition, because both of these contracts are classified in stockholders’ equity and are indexed to our common stock, they are not accounted for as derivatives.
The convertible note hedges covered, subject to customary anti-dilution adjustments, 111 million shares of our common stock at strike prices that initially correspond to the initial conversion prices of the May Notes and are subject to adjustments similar to those applicable to the conversion price of the related notes. If the market value per share of our common stock at the time of conversion of the May Notes is above the strike price of the applicable convertible note hedges, we will be entitled to receive from the counterparties in the transactions shares of our common stock or, to the extent we have made a corresponding election with respect to the related convertible notes, cash or a combination of cash and shares of our common stock, at our option, for the excess of the market value of the common stock over the strike price of the convertible note hedges. The convertible note hedges terminate upon the maturity of the May Notes or when none of the May Notes remain outstanding due to conversion or otherwise. There were 111 million shares of our common stock underlying the warrants, subject to customary anti-dilution adjustments. The warrants had a strike price of $28.38 per share for the warrants that expired in 2014 (the 2014 Warrants) and $30.05 per share for the warrants expiring in 2016 (the 2016 Warrants). Both the 2014 Warrants and the 2016 Warrants had terms whereby they were or will be exercisable only on their respective expiration dates. If the market value of our common stock at the time of the exercise of the applicable warrants exceeds their respective strike prices, we will be required to net settle in cash or shares of our common stock, at our option, with the respective counterparties for the value of the warrants in excess of the warrant strike prices.
In 2015, we entered into modified agreements with our warrant counterparties which changed the timing of the expiration for 46 million of our 2016 Warrants. The modified agreements allowed us to settle the 46 million warrants at our option, in cash or shares. According to the terms of the modified agreements, these warrants expired during a 32 trading-day period which commenced on May 11, 2015 and ended on June 24, 2015. We exercised our option to settle in cash, and as a result, paid $3.9 billion as the market value of our common stock at the time of the exercise of the warrants exceeded their strike prices. Because these warrants could have been settled at our option, in cash or shares of common stock, under both the original and the modified agreements and these contracts met all of the applicable criteria for equity classification, the settlement payments were recorded as a reduction to paid-in capital on our Consolidated Balance Sheets and the remainder allocated to retained earnings to the extent additional paid-in capital was reduced to zero. As of December 31, 2015, 9 million of the 2016 Warrants remained outstanding and have a strike price of $28.76 per share, as adjusted for quarterly dividend distributions, and are due to expire during the 40 trading-day period commencing August 1, 2016. There were no other changes in terms for the remaining 9 million 2016 warrants.
In 2014, we exercised our option to settle the 2014 Warrants in cash. As result, we paid $4.1 billion to settle the 2014 Warrants as the market value of our common stock at the time of the exercise of the 2014 Warrants exceeded their strike price. There were 56 million shares of our common stock underlying the 2014 Warrants, which had a strike price of $28.38 per share and expired during the 40 trading-day period commencing August 1, 2014 and ending on September 26, 2014. Because the 2014 Warrants could have been settled, at our option, in cash or shares of our common stock, and the related contracts met all of the applicable criteria for equity classification, the settlement was recorded as a reduction of additional paid-in capital in our Consolidated Balance Sheets.
88
Under current accounting guidance, we bifurcated the conversion option of the May Notes from the debt instrument, classified the conversion option in equity and are accreting the resulting debt discount as interest expense over the contractual terms of the May Notes. The following table summarizes information about the equity and liability components of the May 2016 Notes (in millions):
Carrying Value of Equity Component | Net Carrying Amount of Liability Component | Unamortized Discount of Liability Component | ||||||||||||||||||||||
December 31, | December 31, | December 31, | ||||||||||||||||||||||
2015 | 2014 | 2015 | 2014 | 2015 | 2014 | |||||||||||||||||||
May 2016 Notes | $ | 35 | $ | 61 | $ | 283 | $ | 483 | $ | (2 | ) | $ | (15 | ) | ||||||||||
We recognized interest expense of $16 million in 2015, $38 million in 2014 and $107 million in 2013 related to the contractual coupon rates and amortization of the debt discount and issuance costs for the May Notes. The effective interest rates on the liability components of the May 2014 Notes and May 2016 Notes were 3.50% and 4.00%, respectively.
Credit Facility
In January 2012, we entered into a five-year $1.3 billion revolving credit facility credit agreement (the Five-Year Revolving Credit Agreement), which expires in January 2017, and borrowed $750 million thereunder. In 2013, we repaid $150 million under the Five-Year Revolving Credit Agreement. During 2014, we repaid the remaining balance of $600 million that was outstanding.
The Five-Year Revolving Credit Agreement contains customary representations, warranties, affirmative, negative and financial maintenance covenants and events of default. The loan bears interest at either (i) the Eurodollar Rate plus the Applicable Margin or (ii) the Base Rate plus the Applicable Margin, each as defined in the credit agreement. We may reduce the commitments and may prepay the loan in whole or in part at any time without premium or penalty. The Five-Year Revolving Credit Agreement will terminate and all amounts owed under the agreement shall be due and payable in January 2017. There were no amounts outstanding under the credit agreement as of December 31, 2015 and December 31, 2014.
We are required to comply with certain covenants under the credit agreement and notes indentures and as of December 31, 2015, we were not in violation of any covenants.
Contractual Maturities of Financing Obligations
As of December 31, 2015, the aggregate future principal maturities of financing obligations for each of the next five years, based on contractual due dates, are as follows (in millions):
Year | 2016 | 2017 | 2018 | 2019 | 2020 | |||||||||||||||
Contractual Maturities | $ | 985 | $ | — | $ | 1,000 | $ | 500 | $ | 2,500 | ||||||||||
11. | COMMITMENTS AND CONTINGENCIES |
Lease Arrangements
We lease facilities and equipment related primarily to administrative, R&D, sales and marketing activities under various long-term non-cancelable operating leases in the United States and international markets. Our leases expire on various dates between 2016 and 2068, with many of our leases containing options to renew. Lease expense under our operating leases was approximately $78 million in 2015, $66 million in 2014 and $54 million in 2013.
Aggregate non-cancelable future minimum rental payments under operating leases are as follows (in millions):
2016 | $ | 66 | |
2017 | 63 | ||
2018 | 51 | ||
2019 | 43 | ||
2020 | 31 | ||
Thereafter | 63 | ||
Total | $ | 317 | |
89
Legal Proceedings
We are a party to various legal actions. The most significant of these are described below. It is not possible to determine the outcome of these matters, and we cannot reasonably estimate the maximum potential exposure or the range of possible loss.
Litigation Related to Sofosbuvir
In January 2012, we acquired Pharmasset, Inc. (Pharmasset). Through the acquisition, we acquired sofosbuvir, a nucleotide analog that acts to inhibit the replication of the hepatitis C virus (HCV). In December 2013, we received U.S. Food and Drug Administration (FDA) approval of sofosbuvir, now known commercially as Sovaldi. In October 2014, we also received approval of the fixed-dose combination of ledipasvir and sofosbuvir (LDV/SOF), now known commercially as Harvoni. We have received a number of contractual and intellectual property claims regarding sofosbuvir. While we have carefully considered these claims both prior to and following the acquisition and believe they are without merit, we cannot predict the ultimate outcome of such claims or range of loss.
We own patents and patent applications that claim sofosbuvir (Sovaldi) as a chemical entity and its metabolites and the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni). Third parties may have, or may obtain rights to, patents that allegedly could be used to prevent or attempt to prevent us from commercializing Sovaldi or Harvoni. For example, we are aware of patents and patent applications owned by other parties that have been or may in the future be alleged by such parties to cover the use of Sovaldi and Harvoni. We cannot predict the ultimate outcome of intellectual property claims related to Sovaldi or Harvoni. We have spent, and will continue to spend, significant resources defending against these claims.
If third parties successfully obtain valid and enforceable patents, and successfully prove infringement of those patents by Sovaldi and/or Harvoni, we could be prevented from selling these products unless we were able to obtain a license under such patents. Such a license may not be available on commercially reasonable terms or at all.
Interference Proceedings and Litigation with Idenix Pharmaceuticals, Inc. (Idenix)
In February 2012, we received notice that the U.S. Patent and Trademark Office (USPTO) had declared Interference No. 105,871 (First Idenix Interference) between our U.S. Patent No. 7,429,572 (the ’572 patent) and Idenix's pending U.S. Patent Application No. 12/131,868. An interference is a proceeding before the USPTO designed to determine who was the first to invent the subject matter claimed by both parties. In January 2014, the USPTO Patent Trial and Appeal Board (PTAB) determined that Pharmasset and not Idenix was the first to invent the compounds in dispute and accordingly Gilead prevailed in the First Idenix Interference. Idenix has appealed the PTAB’s decisions to the U.S. District Court for the District of Delaware.
In December 2013, after receiving our request to do so, the USPTO declared Interference No. 105,981 (Second Idenix Interference) between our pending U.S. Patent Application No. 11/854,218 and Idenix’s U.S. Patent No. 7,608,600 (the ’600 patent). The ’600 patent is related to the Idenix patent application at issue in the First Idenix Interference and includes claims directed to methods of treating HCV with nucleoside compounds. The purpose of the Second Idenix Interference was to determine who was first to invent the claimed methods of treating HCV with compounds similar to those which were involved in the First Idenix Interference. In March 2015, the PTAB determined that Pharmasset and not Idenix was the first to invent the claimed methods of treating HCV. Idenix appealed this decision in both the U.S. District Court for the District of Delaware and the U.S. Court of Appeal for the Federal Circuit (CAFC). We have filed a motion to dismiss the appeal in Delaware and will respond to the appeal filed in the CAFC.
We believe that the Idenix claims involved in the First and Second Idenix Interferences, and similar U.S. and foreign patents claiming the same compounds, metabolites and uses thereof, are invalid. As a result, we filed an Impeachment Action in the Federal Court of Canada to invalidate Idenix Canadian Patent No. 2,490,191 (the ’191 patent), which is the Canadian patent that corresponds to the ’600 patent. Idenix asserted that the commercialization of Sovaldi in Canada will infringe its ’191 patent and that our Canadian Patent No. 2,527,657, corresponding to the ’572 patent involved in the First Idenix Interference, is invalid. A trial on these issues was held in January and February 2015, and in November 2015, the Federal Court of Canada rendered its public decision holding that Idenix's patent is invalid and that Gilead's patent is valid. In the same month, Idenix appealed the court's decision.
We filed a similar legal action in Norway in the Oslo District Court seeking to invalidate Idenix's Norwegian patent corresponding to the ’600 patent. In September 2013, Idenix filed an invalidation action in the Norwegian proceedings against our Norwegian Patent No. 333700 patent, which corresponds to Gilead's ’572 patent. In March 2014, the Norwegian court found all claims in the Idenix Norwegian patent to be invalid and upheld the validity of all claims in the challenged Gilead patent. In April 2014, Idenix appealed the March 2014 decision to the Norwegian Court of Appeal. The appeal hearing from the March 2014 decision took place in February 2016.
90
In January 2013, we filed a legal action in the Federal Court of Australia seeking to invalidate Idenix’s Australian patent corresponding to the ’600 patent. In April 2013, Idenix asserted that the commercialization of Sovaldi in Australia will infringe its Australian patent corresponding to the ’600 patent. A month-long trial was completed in October 2015 in Sydney. A decision is pending.
In March 2014, the European Patent Office (EPO) granted Idenix European Patent No. 1 523 489 (the ’489 patent), which corresponds to the ’600 patent. The same day that the ’489 patent was granted, we filed an opposition with the EPO seeking to revoke the ’489 patent. An opposition hearing was held in February 2016, and the EPO ruled in our favor and revoked the '489 patent. In March 2014, Idenix also initiated infringement proceedings against us in the United Kingdom (UK), Germany and France alleging that the commercialization of Sovaldi would infringe the UK, German and French counterparts of the ’489 patent. A trial was held in the UK in October 2014 to determine the issues of infringement and validity of the Idenix UK patent. In December 2014, the High Court of Justice of England and Wales (UK Court) invalidated all challenged claims of the ’489 patent on multiple grounds. The UK Court has granted Idenix permission to appeal the December 2014 judgment. The appeal of the UK Court's decision is scheduled for July 2016. In March 2015, the German court in Düsseldorf determined that the Idenix patent was highly likely to be invalid and stayed the infringement proceedings pending the outcome of the opposition hearing held by the EPO in February 2016. Idenix has not appealed this decision of the German court staying the proceedings. Upon Idenix's request, the French proceedings have been stayed.
Idenix has not been awarded patents corresponding to the ’600 patent in Japan or China. In the event such patents are issued, we expect to challenge them in proceedings similar to those we invoked in other countries.
In December 2013, Idenix, Universita Degli Studi di Cagliari (UDSG), Centre National de la Recherche Scientifique and L’Université Montpellier II sued us in U.S. District Court for the District of Delaware alleging that the commercialization of sofosbuvir will infringe the ’600 patent and that an interference exists between the ’600 patent and our U.S. Patent No. 8,415,322. Also in December 2013, Idenix and UDSG sued us in the U.S. District Court for the District of Massachusetts alleging that the commercialization of sofosbuvir will infringe U.S. Patent Nos. 6,914,054 and 7,608,597. In June 2014, the court transferred the Massachusetts litigation to the U.S. District Court for the District of Delaware. The district court has set trial dates in October 2016 and December 2016 for resolution of these issues. A decision by the district court may be appealed by either party to the CAFC.
Idenix was acquired by Merck in August 2014. While the acquisition does not change our view of the lack of merit in the claims made by Idenix, Merck has greater resources than Idenix and may therefore choose to fund the litigation at higher levels than Idenix.
Litigation with Merck
In August 2013, Merck contacted us requesting that we pay royalties on the sales of sofosbuvir and obtain a license to U.S. Patent Nos. 7,105,499 and 8,481,712, which it co-owns with Isis Pharmaceuticals, Inc. In August 2013, we filed a lawsuit in the U.S. District Court for the Northern District of California seeking a declaratory judgment that the Merck patents are invalid and not infringed. Merck’s U.S. Patent Nos. 7,105,499 and 8,481,712 cover compounds which do not include, but may relate to, sofosbuvir. During patent prosecution, Merck amended its patent application in an attempt to cover compounds related to sofosbuvir. If the court determines that Merck’s patents are valid and that we have infringed those claims, we may be required to obtain a license from and pay royalties to Merck to commercialize sofosbuvir. The court has set a trial date of March 7, 2016 for this lawsuit. Either party may appeal a decision by the District Court to the CAFC.
Litigation with AbbVie, Inc. (AbbVie)
AbbVie has obtained U.S. Patent Nos. 8,466,159, 8,492,386, 8,680,106, 8,685,984, and 8,809,265 (AbbVie Patents) which purport to cover the use of a combination of LDV/SOF (or Harvoni) for the treatment of HCV. Gilead is aware that AbbVie has pending patent applications in the United States and granted and pending applications in other countries. We own published and pending patent applications directed to the use of combinations for the treatment of HCV, and, specifically, to the combination of LDV/SOF. Certain of our applications were filed before the AbbVie Patents. For this reason and others, we believe the AbbVie Patents are invalid.
Accordingly, in December 2013, we filed a lawsuit in the U.S. District Court for the District of Delaware seeking declaratory judgment that the AbbVie Patents are invalid and unenforceable, as well as other relief. We believe that Abbott Laboratories, Inc. and AbbVie conspired to eliminate competition in the HCV market by falsely representing to the USPTO that they, and not Gilead, invented methods of treating HCV using a combination of LDV/SOF. In February and March 2014, AbbVie responded to our lawsuit by also filing two lawsuits in the U.S. District Court for the District of Delaware alleging that our fixed-dose combination of LDV/SOF will infringe its patents. All of those lawsuits have been consolidated into a single action. In the United States, either party may appeal a decision by the District Court to the CAFC. The AbbVie Patents have not
91
blocked or delayed the commercialization of our combination product in the United States, Canada, or Europe. We do not expect any other foreign patents to block or delay the commercialization around the world. The court has set a trial date of September 12, 2016 for this lawsuit.
Additionally, AbbVie has obtained U.S. Patent No. 9,034,832 which purports to cover a solid oral dosage form containing ledipasvir. Accordingly, in May 2015, we filed a lawsuit in the U.S. District Court for the District of Delaware seeking declaratory judgment that AbbVie’s patent is invalid, as well as other relief. We do not expect AbbVie’s patent to block the commercialization of our combination product. The court has set a trial date of July 31, 2017.
In August 2015, we brought an impeachment action seeking a declaration that AbbVie's Canadian Patent No. 2,811,250 ('250 Patent), which purports to cover the use of a combination of LDV/SOF for the treatment of HCV, is invalid. On the same day, AbbVie brought an infringement action which asserts that commercialization of Harvoni in Canada will infringe its '250 Patent. The impeachment action has been stayed and we have counterclaimed for invalidity in the infringement proceeding. A trial date has not been set.
In November 2015, AbbVie filed a lawsuit against us in the Regional Court Düsseldorf for infringement of two quasi-patents, known as “utility models.” Utility models are unexamined IP rights and are not the same as standard patents. One utility model, DE 20 2012 013 117, purports to cover the use of a combination of direct-acting antivirals which includes at least an HCV polymerase inhibitor and an HCV NS5A inhibitor in the treatment of HCV; the other utility model, DE 21 2012 000 197, purports to cover a solid dispersion that includes ledipasvir. A trial date has not been set.
If a court determines that the AbbVie Patents are valid and that we have infringed those claims, we may be required to obtain a license from and pay royalties to AbbVie to commercialize sofosbuvir combination products.
European Patent Claims
In February 2015, several parties filed oppositions in the European Patent Office requesting revocation of our granted European patent covering sofosbuvir that expires in 2028. While we are confident in the strength of our sofosbuvir patent, we cannot predict the ultimate outcome of these oppositions. If we are unsuccessful in defending these oppositions, some or all of our patent claims may be narrowed or revoked and the patent protection for sofosbuvir in Europe could be substantially shortened or eliminated entirely. If the sofosbuvir patent is revoked, and no other European patents are granted covering sofosbuvir, our exclusivity will be based entirely on regulatory exclusivity granted by the European Medicines Agency (EMA). Sovaldi has been granted regulatory exclusivity that will prevent generic sofosbuvir from entering the European Union for 10 years following approval of Sovaldi, or January 2024. If we lose exclusivity for Sovaldi prior to 2028, our expected revenues and results of operation could be negatively impacted for the years including and succeeding the year in which such exclusivity is lost, which may cause our stock price to decline.
Litigation with Generic Manufacturers
As part of the approval process for some of our products, FDA granted us a New Chemical Entity (NCE) exclusivity period during which other manufacturers' applications for approval of generic versions of our product will not be approved. Generic manufacturers may challenge the patents protecting products that have been granted NCE exclusivity one year prior to the end of the NCE exclusivity period. Generic manufacturers have sought and may continue to seek FDA approval for a similar or identical drug through an abbreviated new drug application (ANDA), the application form typically used by manufacturers seeking approval of a generic drug. The sale of generic versions of our products earlier than their patent expiration would have a significant negative effect on our revenues and results of operations.
Current legal proceedings of significance with some of our generic manufacturers include:
HIV Products
In November 2011, December 2011 and August 2012, we received notices that Teva Pharmaceuticals (Teva) submitted an abbreviated new drug submission (ANDS) to the Canadian Minister of Health requesting permission to manufacture and market generic versions of Truvada, Atripla and Viread. In the notices, Teva alleges that the patents associated with Truvada, Atripla and Viread are invalid, unenforceable and/or will not be infringed by Teva's manufacture, use or sale of generic versions of those products. We filed lawsuits against Teva in the Federal Court of Canada seeking an order of prohibition against approval of these applications.
In December 2013, the court issued an order prohibiting the Canadian Minister of Health from approving Teva’s generic versions of our Viread, Truvada and Atripla products until expiry of our patents in July 2017. Teva has appealed that decision. That decision did not rule on the validity of the patents and accordingly the only issue on appeal is whether the Minister of Health should be prohibited from approving Teva’s products. The appeal will be heard by the Canadian Federal Court of
92
Appeal after the trial in the Impeachment Action. The court will determine the validity of the patents in the pending Impeachment Action. A trial in the Impeachment Action is scheduled for November 2016. If Teva is successful in invalidating our patents, Teva may be able to launch generic versions of our Viread, Truvada and Atripla products in Canada prior to the expiry of our patents.
In April 2014 and July 2015, we received notices that Mylan Inc. (Mylan) submitted an ANDA to FDA requesting permission to manufacture and market generic versions of Truvada and Complera. In the notice, Mylan alleges that the patents associated with Truvada and Complera are invalid, unenforceable and/or will not be infringed by Mylan's manufacture, use or sale of a generic version of these products. We filed lawsuits against Mylan in U.S. District Court for the Northern District of West Virginia for infringement of our patents. In June 2014, we received notice that Mylan submitted petitions for Inter Partes Review (IPR) to the PTAB alleging that four patents associated with tenofovir disoproxil fumarate are invalid. We opposed Mylan’s petitions. In December 2014, the PTAB issued decisions denying each of Mylan’s petitions for IPR. In January 2015, Mylan requested a rehearing on the basis that it believes the PTAB decision is wrong. In August 2015 and November 2015, the PTAB denied Mylan's requests for a rehearing. In October 2015, we reached an agreement with Mylan to settle the proceedings. The terms of the settlement agreement are confidential.
In June 2014, we received notice that Apotex Inc. (Apotex) submitted an ANDS to the Canadian Minister of Health requesting permission to manufacture and market a generic version of Truvada and a separate ANDS requesting permission to manufacture and market a generic version of Viread. In the notice, Apotex alleges that three of the patents associated with Truvada and two of the patents associated with Viread are invalid, unenforceable and/or will not be infringed by Apotex's manufacture, use or sale of a generic version of Truvada or Viread. In August 2014, we filed a lawsuit against Apotex in the Federal Court of Canada seeking an order of prohibition against approval of this ANDS. A hearing in that case is scheduled for April 2016.
Letairis
In February 2015, we received notice that Watson Laboratories, Inc. (Watson) submitted an ANDA to FDA requesting permission to manufacture and market a generic version of Letairis. In the notice, Watson alleges that one of the patents associated with ambrisentan tablets is invalid, unenforceable and/or will not be infringed by Watson's manufacture, use or sale of a generic version of Letairis. In April 2015, we filed a lawsuit against Watson in the U.S. District Court for the District of New Jersey.
In June 2015, we received notice that SigmaPharm Laboratories, LLC (SigmaPharm) submitted an ANDA to FDA requesting permission to manufacture and market a generic version of Letairis. In the notice, SigmaPharm alleges that one of the patents associated with ambrisentan tablets is invalid, unenforceable and/or will not be infringed by SigmaPharm’s manufacture, use or sale of a generic version of Letairis. In June 2015, we filed a lawsuit against SigmaPharm in the U.S. District Court for the District of New Jersey.
We cannot predict the ultimate outcome of these actions, and we may spend significant resources enforcing and defending these patents. If we are unsuccessful in these lawsuits, some or all of our claims in the patents may be narrowed or invalidated and the patent protection for our products could be substantially shortened. Further, if all of the patents covering one or more products are invalidated, FDA or Health Canada could approve the requests to manufacture a generic version of such products in the United States or Canada, respectively, prior to the expiration date of those patents. The sale of generic versions of these products earlier than their patent expiration would have a significant negative effect on our revenues and results of operations.
TAF Litigation
In January 2016, AIDS Healthcare Foundation, Inc. (AHF) filed a complaint with the U.S. District Court for the Northern District of California against Gilead, Japan Tobacco, Inc., Japan Tobacco International, U.S.A. (together, Japan Tobacco), and Emory University (Emory). AHF claims that U.S. Patent Nos. 7,390,791; 7,800,788; 8,754,065; 8,148,374; and 8,633,219 are invalid under 35 U.S.C. §§ 101 et seq. In addition, AHF claims that Gilead, independently and together with Japan Tobacco and Emory, is violating federal antitrust laws in the market for sales of tenofovir alafenamide (TAF) by offering TAF as part of a fixed-dose combination product with elvitegravir, cobicistat, and emtricitabine. AHF seeks a declaratory judgment of invalidity against each of the patents as well as monetary damages.
93
Department of Justice Investigation
In June 2011, we received a subpoena from the U.S. Attorney's Office for the Northern District of California requesting documents related to the manufacture, and related quality and distribution practices, of Complera, Atripla, Truvada, Viread, Emtriva, Hepsera and Letairis. We cooperated with the government’s inquiry. In April 2014, the United States Department of Justice informed us that, following an investigation, it declined to intervene in a False Claims Act lawsuit filed by two former employees. In April 2014, the former employees served a First Amended Complaint. In January 2015, the federal district court issued an order granting in its entirety, without prejudice, our motion to dismiss the First Amended Complaint. In February 2015, the plaintiffs filed a Second Amended Complaint and in June 2015, the federal district court issued an order granting our motion to dismiss the Second Amended Complaint. In July 2015, the plaintiffs filed a notice of appeal in the U.S. Court of Appeals for Ninth Circuit.
Other Matters
We are a party to various legal actions that arose in the ordinary course of our business. We do not believe that these other legal actions will have a material adverse impact on our consolidated business, financial position or results of operations.
Other Commitments
In the normal course of business, we enter into various firm purchase commitments primarily related to active pharmaceutical ingredients and certain inventory related items. As of December 31, 2015, these commitments for the next five years were approximately $1.5 billion in 2016, $553 million in 2017, $389 million in 2018, $125 million in 2019 and $83 million in 2020. The amounts related to active pharmaceutical ingredients represent minimum purchase commitments. Actual payments for the purchases related to active pharmaceutical ingredients were $2.2 billion in 2015, $1.8 billion in 2014 and $2.1 billion in 2013.
We also enter into letters of credit and bank guarantees to support our commercial activities. Our outstanding letters of credit and bank guarantees totaled $521 million as of December 31, 2015. A majority of these letters of credit and bank guarantees expire within the year and are not expected to be funded.
12. | STOCKHOLDERS' EQUITY |
Stock Repurchase Programs
In January 2015, our Board of Directors authorized a five-year, $15.0 billion stock repurchase program (2015 Program). Purchases under the 2015 Program may be made in the open market or in privately negotiated transactions. The 2015 Program commenced after the $5.0 billion stock repurchase program authorized by our Board of Directors in May 2014 (2014 Program) was completed in the first quarter of 2015. The $5.0 billion repurchase program authorized by our Board of Directors in January 2011 (2011 Program) was completed in 2014. As of December 31, 2015, the remaining authorized repurchase amount under the 2015 Program was $8.0 billion. The following table summarizes our stock repurchases under the above-described programs (in millions, except per share data):
Year ended December 31, | |||||||||||||
2015 (1) | 2014 (2) | 2013 (3) | |||||||||||
Shares repurchased and retired | 95 | 59 | 10 | ||||||||||
Amount | $ | 10,002 | $ | 5,349 | $ | 582 | |||||||
Average price per share | $ | 104.91 | $ | 90.29 | $ | 60.78 | |||||||
(1) | Includes 65 million shares repurchased for $7.0 billion under the 2015 Program and 30 million shares repurchased for $3.0 billion under the 2014 Program. |
(2) | Includes 19 million shares repurchased for $2.0 billion under the 2014 Program and 40 million shares repurchased for $3.3 billion under the 2011 Program. |
(3) | All shares repurchased under the 2011 Program. |
In February 2016, we entered into an accelerated share repurchase program (“ASR”) to repurchase $5.0 billion of our common stock. We paid $5.0 billion and received 46 million shares of our common stock, which represents approximately 80% of the total shares expected to be delivered to us under the ASR. The total number of shares to be received under the ASR will be based on the average price of our common stock during the purchase period, which will end in April 2016.
In February 2016, our Board of Directors authorized a new $12.0 billion share repurchase program (2016 Program) which will commence upon the completion of our 2015 Program. Purchases under the 2016 Program may be made in the open market or in privately negotiated transactions.
94
We use the par value method of accounting for our stock repurchases. Under the par value method, common stock is first charged with the par value of the shares involved. The excess of the cost of shares acquired over the par value is allocated to additional paid-in capital (APIC) based on an estimated average sales price per issued share with the excess amounts charged to retained earnings.
In addition to repurchases from our stock repurchase programs, we repurchased shares of common stock withheld by us from employee restricted stock awards to satisfy our applicable tax withholding obligations. The following table summarizes the reduction of common stock and APIC and the charge to retained earnings as a result of our stock repurchases (in millions):
Year ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Reduction of common stock and APIC | $ | 223 | $ | 133 | $ | 14 | ||||||
Charge to retained earnings | $ | 10,115 | $ | 5,475 | $ | 674 | ||||||
Dividends
In the second quarter of 2015, we began paying quarterly dividends on our common stock. The following table summarizes cash dividends declared on our common stock (in millions, except per share data):
Dividend Per Share | Amount | |||||||
2015: | ||||||||
Second quarter | $ | 0.43 | $ | 639 | ||||
Third quarter | 0.43 | 631 | ||||||
Fourth quarter | 0.43 | 620 | ||||||
Total | $ | 1.29 | $ | 1,890 | ||||
Our restricted stock and performance-based stock units have dividend equivalent rights entitling holders to dividend equivalents to be paid upon vesting for each share of the underlying units.
On February 2, 2016, we announced that our Board of Directors declared a quarterly cash dividend of $0.43 per share of our common stock, with a payment date of March 30, 2016 to all stockholders of record as of the close of business on March 16, 2016.
Preferred Stock
We have 5 million shares of authorized preferred stock issuable in series. Our Board is authorized to determine the designation, powers, preferences and rights of any such series. There was no preferred stock outstanding as of December 31, 2015 and 2014.
Accumulated Other Comprehensive Income
The following table summarizes the changes in accumulated OCI by component, net of tax (in millions):
Foreign Currency Items | Unrealized Gains and Losses on Available-for-Sale Securities | Unrealized Gains and Losses on Cash Flow Hedges | Total | |||||||||||||
Balance at December 31, 2013 | $ | (45 | ) | $ | 12 | $ | (91 | ) | $ | (124 | ) | |||||
Other comprehensive income (loss) before reclassifications | (9 | ) | — | 430 | 421 | |||||||||||
Amounts reclassified from accumulated other comprehensive income | — | — | 4 | 4 | ||||||||||||
Net current period other comprehensive income (loss) | (9 | ) | — | 434 | 425 | |||||||||||
Balance at December 31, 2014 | (54 | ) | 12 | 343 | 301 | |||||||||||
Other comprehensive income (loss) before reclassifications | 9 | (29 | ) | 389 | 369 | |||||||||||
Amounts reclassified from accumulated other comprehensive income | — | 1 | (583 | ) | (582 | ) | ||||||||||
Net current period other comprehensive income (loss) | 9 | (28 | ) | (194 | ) | (213 | ) | |||||||||
Balance at December 31, 2015 | $ | (45 | ) | $ | (16 | ) | $ | 149 | $ | 88 | ||||||
95
The amounts reclassified for gains (losses) on cash flow hedges were recorded as part of product sales on our Consolidated Statements of Income. Amounts reclassified for gains (losses) on available-for-sale securities were recorded as part of other income (expense), net on our Consolidated Statements of Income.
13. | EMPLOYEE BENEFITS |
We utilize share based compensation in the form of various types of equity-based awards, including restricted stock units (RSUs), performance-based restricted stock units (PSUs) and stock options. Compensation expense is recognized in the Consolidated Statements of Income based on the estimated fair value of the award on the grant date. The estimated fair value of RSUs is based on the closing price of our common stock. For PSUs, estimated fair value is based on either the Monte Carlo valuation methodology or the stock price on the date of grant. For stock option awards, estimated fair value is based on the Black-Scholes option valuation model.
2004 Equity Incentive Plan
In May 2004, our stockholders approved and we adopted the Gilead Sciences, Inc. 2004 Equity Incentive Plan (the 2004 Plan). The 2004 Plan is a broad based incentive plan that provides for the grant of equity-based awards, including stock options, restricted stock units, restricted stock awards and performance awards, to employees, directors and consultants. Under the 2004 Plan, we are authorized to issue a maximum of 50 million shares of full-value awards, such as restricted stock, restricted stock units, performance shares, performance units (to the extent settled in common stock) and phantom shares over the term of the plan. The 2004 Plan authorizes the issuance of a total of 243 million shares of common stock. As of December 31, 2015, a total of 67 million shares remain available for future grant under the 2004 Plan.
Stock Options
The 2004 Plan provides for option grants designated as either non-qualified or incentive stock options. Prior to January 1, 2006, we granted both non-qualified and incentive stock options, but all stock options granted after January 1, 2006 have been non-qualified stock options. Under the 2004 Plan, employee stock options granted prior to 2011 generally vest over five years and stock options granted starting in 2011 generally vest over four years. All options are exercisable over a period not to exceed the contractual term of ten years from the date the stock options are issued and are granted at prices not less than the fair market value of our common stock on the grant date. Stock option exercises are settled with common stock from the 2004 Plan's previously authorized and available pool of shares.
The following table summarizes activity and related information under our stock option plans. All option grants presented in the table had exercise prices not less than the fair value of the underlying common stock on the grant date:
Shares (in thousands) | Weighted- Average Exercise Price (in dollars) | Weighted-Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value (in millions) | ||||||||||
Outstanding at December 31, 2014 | 39,144 | $ | 22.63 | ||||||||||
Granted | 1,356 | $ | 102.97 | ||||||||||
Forfeited | (110 | ) | $ | 69.11 | |||||||||
Expired | (3 | ) | $ | 21.06 | |||||||||
Exercised | (12,974 | ) | $ | 18.11 | |||||||||
Outstanding at December 31, 2015 | 27,413 | $ | 28.56 | 3.6 | $ | 1,995 | |||||||
Exercisable at December 31, 2015 | 24,731 | $ | 23.11 | 3.1 | $ | 1,931 | |||||||
Expected to vest, net of estimated forfeitures at December 31, 2015 | 2,576 | $ | 78.10 | 8.2 | $ | 63 | |||||||
Aggregate intrinsic value represents the value of our closing stock price on the last trading day of the year in excess of the weighted-average exercise price multiplied by the number of options outstanding or exercisable. Total intrinsic value of options exercised was $1.1 billion for 2015, $1.2 billion for 2014 and $837 million for 2013.
The weighted-average grant date fair value of the stock options granted was $29.73 per share for 2015, $27.63 per share for 2014 and $12.41 per share for 2013.
As of December 31, 2015, there was $52 million of unrecognized compensation cost related to stock options, which is expected to be recognized over an estimated weighted-average period of 2.2 years.
96
Performance-Based Restricted Stock Units
Under the 2004 Plan, we grant PSUs which vest upon the achievement of specified market or performance goals, which could include achieving a total shareholder return compared to a pre-determined peer group or achieving revenue targets. The actual number of common shares ultimately issued is calculated by multiplying the number of PSUs by a payout percentage ranging from 0% to 200% and these awards generally vest only when a committee (or subcommittee) of our Board has determined that the specified market and performance goals have been achieved. The fair value of each PSU is estimated at the date of grant or when performance objectives are defined for the grants. Depending on the terms of the award, fair value on the date of grant is determined based on either the Monte Carlo valuation methodology or the closing stock price on the date of grant.
In addition, we have also granted other PSUs to certain of our employees under the 2004 Plan. The vesting of these awards is subject to the achievement of specified individual performance goals, typically within a one year period. The fair value of such an award is equal to the closing price of our common stock on the grant date.
The following table summarizes activity and related information for all of our PSUs:
Shares (1) (in thousands) | Weighted- Average Grant-Date Fair Value Per Share (1) (in dollars) | ||||||
Outstanding at December 31, 2014 | 827 | $ | 51.52 | ||||
Granted | 1,219 | $ | 61.71 | ||||
Vested | (1,554 | ) | $ | 48.60 | |||
Forfeited | (5 | ) | $ | 98.32 | |||
Outstanding at December 31, 2015 | 487 | $ | 85.83 | ||||
(1) Weighted-average grant-date fair value per share excludes shares related to grants that currently have no grant-date fair value as the performance objectives have not yet been defined.
The weighted-average grant date fair value of our PSUs granted was $61.71 per share for 2015, $56.38 per share for 2014 and $30.16 per share for 2013. The total grant date fair value of our vested PSUs was $76 million for 2015, $46 million for 2014 and $11 million for 2013, and total fair value as of the respective vesting dates was $160 million for 2015, $145 million for 2014 and $19 million for 2013.
We recognized stock-based compensation expenses of $40 million in 2015, $57 million in 2014 and $25 million in 2013 related to these PSUs. As of December 31, 2015, there was $12 million of unrecognized compensation costs related to these PSUs, which is expected to be recognized over an estimated weighted-average period of 1.0 years.
Restricted Stock Units
We grant time-based RSUs to certain employees as part of our annual employee equity compensation review program as well as to new hire employees and to non-employee members of our Board. RSUs are share awards that entitle the holder to receive freely tradable shares of our common stock upon vesting. For awards granted prior to 2011 to employees, RSUs vest ratably on an annual basis over five years from the date of grant. Starting January 1, 2011, RSUs vest over four years from the date of grant.
The fair value of an RSU is equal to the closing price of our common stock on the grant date. The following table summarizes our RSU activities and related information:
Shares (in thousands) | Weighted- Average Grant-Date Fair Value Per Share (in dollars) | ||||||
Outstanding at December 31, 2014 | 14,483 | $ | 49.37 | ||||
Granted | 4,065 | $ | 103.19 | ||||
Vested | (6,397 | ) | $ | 38.86 | |||
Forfeited | (1,123 | ) | $ | 62.96 | |||
Outstanding at December 31, 2015 | 11,028 | $ | 73.93 | ||||
97
The weighted-average grant date fair value of RSUs granted was $103.19 per share for 2015, $86.75 per share for 2014 and $48.61 per share for 2013.The total grant date fair value of our vested RSUs was $249 million for 2015, $182 million for 2014 and $118 million for 2013, and total fair value as of the respective vesting dates was $666 million for 2015, $535 million for 2014 and $253 million for 2013.
As of December 31, 2015, there was $558 million of unrecognized compensation cost related to unvested RSUs which is expected to be recognized over a weighted-average period of 2.1 years.
Employee Stock Purchase Plan
Under our Employee Stock Purchase Plan and the International Employee Stock Purchase Plan (together, as amended, the ESPP), employees can purchase shares of our common stock based on a percentage of their compensation subject to certain limits. The purchase price per share is equal to the lower of 85% of the fair market value of our common stock on the offering date or the purchase date. Prior to 2016, the ESPP offered a two-year look-back feature as well as an automatic reset feature that provides for an offering period to be reset to a new lower-priced offering if the offering price of the new offering period is less than that of the current offering period. Beginning in the first quarter of 2016, the look-back feature for future ESPP offering periods will be six-months. ESPP purchases are settled with common stock from the ESPP's previously authorized and available pool of shares. During 2015, 1 million shares were issued under the ESPP for $86 million. A total of 79 million shares of common stock have been reserved for issuance under the ESPP, and there were 14 million shares available for issuance under the ESPP as of December 31, 2015.
As of December 31, 2015, there was $19 million of unrecognized compensation cost related to the ESPP, which is expected to be recognized over an estimated weighted-average period of 0.5 years.
Stock-Based Compensation
The following table summarizes the stock-based compensation expenses included in our Consolidated Statements of Income (in millions):
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Cost of goods sold | $ | 11 | $ | 10 | $ | 7 | ||||||
Research and development expenses | 173 | 152 | 109 | |||||||||
Selling, general and administrative expenses | 198 | 198 | 136 | |||||||||
Stock-based compensation expense included in total costs and expenses | 382 | 360 | 252 | |||||||||
Income tax effect | (131 | ) | (64 | ) | (67 | ) | ||||||
Stock-based compensation expense, net of tax | $ | 251 | $ | 296 | $ | 185 | ||||||
We capitalized stock-based compensation costs to inventory totaling $13 million in 2015, $12 million in 2014 and $9 million in 2013. The capitalized stock-based compensation costs remaining in inventory were $8 million as of December 31, 2015, $6 million as of December 31, 2014 and $4 million as of December 31, 2013.
Stock-based compensation is recognized as expense over the requisite service periods in our Consolidated Statements of Income using the straight-line expense attribution approach for stock options, reduced for estimated forfeitures. We estimate forfeitures based on our historical experience. We recognize a tax benefit from stock-based compensation in APIC if an incremental tax benefit is realized after all other tax attributes currently available to us have been utilized. In addition, we have elected to account for the indirect benefits of stock-based compensation on the research tax credit and the extraterritorial income deduction through the Consolidated Statements of Income rather than through APIC.
98
Valuation Assumptions
Fair value of options granted under our 2004 Plan and purchases under our ESPP were estimated at grant or purchase dates using a Black-Scholes option valuation model. The Black-Scholes option valuation model was developed for use in estimating the fair value of traded options, which have no vesting restrictions and are fully transferable. In addition, option valuation models require the input of highly subjective assumptions, including expected stock price volatility and expected award life. We used the following assumptions to calculate the estimated fair value of the awards:
Year Ended December 31, | |||||||||
2015 | 2014 | 2013 | |||||||
Expected volatility: | |||||||||
Stock options | 35 | % | 34 | % | 29 | % | |||
ESPP | 32 | % | 32 | % | 31 | % | |||
Expected term in years: | |||||||||
Stock options | 5.7 | 5.5 | 5.7 | ||||||
ESPP | 1.2 | 1.2 | 1.2 | ||||||
Risk-free interest rate: | |||||||||
Stock options | 1.4 | % | 1.8 | % | 1.1 | % | |||
ESPP | 1.4 | % | 1.5 | % | 1.1 | % | |||
Expected dividend yield | 1.7 | % | — | % | — | % | |||
The fair value of stock options granted was calculated using the single option approach. We use a blend of historical volatility along with implied volatility for traded options on our common stock to determine our expected volatility. The expected term of stock-based awards represents the weighted-average period the awards are expected to remain outstanding. We estimate the weighted-average expected term based on historical cancellation and historical exercise data related to our stock options as well as the contractual term and vesting terms of the awards. The risk-free interest rate is based upon observed interest rates appropriate for the term of the stock-based awards. The dividend yield is based on our history and expectation of dividend payouts.
Deferred Compensation
We maintain a retirement saving plan under which eligible U.S. employees may defer compensation for income tax purposes under Section 401(k) of the Internal Revenue Code (the Gilead Sciences 401k Plan). In certain foreign subsidiaries, we maintain defined benefit plans as required by local regulatory requirements. Our total matching contribution expense under the Gilead Sciences 401k Plan and other defined benefit plans was $47 million during 2015, $40 million during 2014 and $32 million during 2013.
We maintain a deferred compensation plan under which our directors and key employees may defer compensation. Amounts deferred by participants are deposited into a rabbi trust. The total assets and liabilities associated with the deferred compensation plan were $66 million as of December 31, 2015 and $54 million as of December 31, 2014.
14. | NET INCOME PER SHARE ATTRIBUTABLE TO GILEAD COMMON STOCKHOLDERS |
Basic net income per share attributable to Gilead common stockholders is calculated based on the weighted-average number of shares of our common stock outstanding during the period. Diluted net income per share attributable to Gilead common stockholders is calculated based on the weighted-average number of shares of our common stock outstanding and other dilutive securities outstanding during the period. The potential dilutive shares of our common stock resulting from the assumed exercise of outstanding stock options, PSUs and the assumed exercise of warrants relating to our convertible senior notes, including the convertible senior notes that were due in May 2013 (May 2013 Notes), May 2014 Notes and the May 2016 Notes (collectively, the Convertible Notes) were determined under the treasury stock method.
Because the principal amount of the Convertible Notes has been or will be settled in cash, only the conversion spread relating to the outstanding Convertible Notes is included in our calculation of diluted net income per share attributable to Gilead common stockholders. Our common stock resulting from the assumed settlement of the conversion spread of the May 2013 Notes had a dilutive effect when the average market price of our common stock during the period exceeded the conversion price of $19.05. Our common stock resulting from the assumed settlement of the conversion spread of the May Notes had a dilutive effect when the average market price of our common stock during the period exceeded the conversion price for the May Notes. See Note 10 Debt and Credit Facility for additional information.
99
We included the dilutive impact of the outstanding warrants related to the Convertible Notes for the periods they had a dilutive effect on our net income per share calculations. The warrants related to our May 2013 Notes had a dilutive effect when the average market price of our common stock during the period exceeded the warrants' exercise price of $26.95. The 2014 Warrants and 2016 Warrants have a dilutive effect when the average market price of our common stock during the period exceeds the warrants' exercise price. See Note 10 Debt and Credit Facility for additional information.
We excluded stock options to purchase approximately 1 million weighted-average shares of our common stock that were outstanding during both 2015 and 2014 and less than 1 million weighted-average shares during 2013 in the computation of diluted net income per share attributable to Gilead common stockholders because their effect was antidilutive.
The following table is a reconciliation of the numerator and denominator used in the calculation of basic and diluted net income per share attributable to Gilead common stockholders (in millions):
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Net income attributable to Gilead | $ | 18,108 | $ | 12,101 | $ | 3,075 | ||||||
Shares used in per share calculation — basic | 1,464 | 1,522 | 1,529 | |||||||||
Effect of dilutive securities: | ||||||||||||
Stock options and equivalents | 23 | 33 | 40 | |||||||||
Conversion spread related to the Convertible Notes | 14 | 30 | 63 | |||||||||
Warrants related to the Convertible Notes | 20 | 62 | 63 | |||||||||
Shares used in per share calculation — diluted | 1,521 | 1,647 | 1,695 | |||||||||
Net income per share attributable to Gilead common stockholders — basic | $ | 12.37 | $ | 7.95 | $ | 2.01 | ||||||
Net income per share attributable to Gilead common stockholders — diluted | $ | 11.91 | $ | 7.35 | $ | 1.81 | ||||||
15. | SEGMENT INFORMATION |
We have one operating segment, which primarily focuses on the discovery, development and commercialization of innovative medicines in areas of unmet medical need. Therefore, our results of operations are reported on a consolidated basis consistent with internal management reporting reviewed by our chief operating decision maker, our chief executive officer. Enterprise-wide disclosures about product sales, revenues and long-lived assets by geographic area, and revenues from major customers are presented below.
100
Product Sales
Our product sales consist of the following (in millions):
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Antiviral products: | ||||||||||||
Harvoni | $ | 13,864 | $ | 2,127 | $ | — | ||||||
Sovaldi | 5,276 | 10,283 | 139 | |||||||||
Truvada | 3,459 | 3,340 | 3,136 | |||||||||
Atripla | 3,134 | 3,470 | 3,648 | |||||||||
Stribild | 1,825 | 1,197 | 539 | |||||||||
Complera/Eviplera | 1,427 | 1,228 | 810 | |||||||||
Viread | 1,108 | 1,058 | 959 | |||||||||
Genvoya | 45 | — | — | |||||||||
Other antiviral | 69 | 88 | 111 | |||||||||
Total antiviral products | 30,207 | 22,791 | 9,342 | |||||||||
Other products: | ||||||||||||
Letairis | 700 | 595 | 520 | |||||||||
Ranexa | 588 | 510 | 449 | |||||||||
AmBisome | 350 | 388 | 352 | |||||||||
Zydelig | 132 | 23 | — | |||||||||
Other | 174 | 167 | 141 | |||||||||
Total product sales | $ | 32,151 | $ | 24,474 | $ | 10,804 | ||||||
Revenues by Geographic Region
The following table summarizes total revenues from external customers and collaboration partners by geographic region (in millions). Product sales and product-related contract revenue are attributed to regions based on ship-to location. Royalty and non-product related contract revenue are attributed to regions based on the location of the collaboration partner.
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Revenues: | ||||||||||||
United States | $ | 21,234 | $ | 18,182 | $ | 6,695 | ||||||
Europe | 7,528 | 5,442 | 3,614 | |||||||||
Other countries | 3,877 | 1,266 | 893 | |||||||||
Total revenues | $ | 32,639 | $ | 24,890 | $ | 11,202 | ||||||
Long-lived Assets
The net book value of our property, plant and equipment (less office and computer equipment) in the United States was $1.8 billion as of December 31, 2015 and $1.3 billion as of December 31, 2014. The corresponding amount in international locations was $334 million as of December 31, 2015 and $275 million as of December 31, 2014. All individual international locations accounted for less than ten percent of the total balances.
Revenues from Major Customers
The following table summarizes revenues from each of our customers who individually accounted for 10% or more of our total revenues (as a percentage of total revenues):
Year Ended December 31, | |||||||||
2015 | 2014 | 2013 | |||||||
McKesson Corp. | 24 | % | 24 | % | 16 | % | |||
AmerisourceBergen Corp. | 19 | % | 25 | % | 13 | % | |||
Cardinal Health, Inc. | 15 | % | 14 | % | 17 | % | |||
101
16. INCOME TAXES
Income before provision for income taxes consists of the following (in millions):
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Domestic | $ | 7,953 | $ | 6,678 | $ | 3,470 | ||||||
Foreign | 13,706 | 8,178 | 738 | |||||||||
Total income before provision for income taxes | $ | 21,659 | $ | 14,856 | $ | 4,208 | ||||||
The provision for income taxes consists of the following (in millions):
Year Ended December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Federal: | ||||||||||||
Current | $ | 3,568 | $ | 2,810 | $ | 1,156 | ||||||
Deferred | (313 | ) | (190 | ) | (71 | ) | ||||||
3,255 | 2,620 | 1,085 | ||||||||||
State: | ||||||||||||
Current | 158 | 152 | 62 | |||||||||
Deferred | (21 | ) | (30 | ) | (22 | ) | ||||||
137 | 122 | 40 | ||||||||||
Foreign: | ||||||||||||
Current | 212 | 85 | 46 | |||||||||
Deferred | (51 | ) | (30 | ) | (20 | ) | ||||||
161 | 55 | 26 | ||||||||||
Provision for income taxes | $ | 3,553 | $ | 2,797 | $ | 1,151 | ||||||
The cumulative unremitted foreign earnings that are considered indefinitely reinvested in our foreign subsidiaries and for which no U.S. taxes have been provided, were approximately $28.5 billion as of December 31, 2015 and $15.6 billion as of December 31, 2014. The residual U.S. tax liability, if such amounts were remitted, would be approximately $9.7 billion as of December 31, 2015 and $5.5 billion as of December 31, 2014.
The reconciliation between the federal statutory tax rate applied to income before taxes and our effective tax rate is summarized as follows:
Year Ended December 31, | |||||||||
2015 | 2014 | 2013 | |||||||
Federal statutory rate | 35.0 | % | 35.0 | % | 35.0 | % | |||
State taxes, net of federal benefit | 0.5 | % | 0.6 | % | 0.5 | % | |||
Foreign earnings at different rates | (18.5 | )% | (16.9 | )% | (6.6 | )% | |||
Research and other credits | (0.7 | )% | (0.9 | )% | (3.0 | )% | |||
Net unbenefitted stock compensation | 0.1 | % | 0.2 | % | 0.6 | % | |||
Other | — | % | 0.8 | % | 0.8 | % | |||
Effective tax rate | 16.4 | % | 18.8 | % | 27.3 | % | |||
102
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets and liabilities are as follows (in millions):
December 31, | ||||||||
2015 | 2014 | |||||||
Deferred tax assets: | ||||||||
Net operating loss carryforwards | $ | 199 | $ | 215 | ||||
Stock-based compensation | 222 | 157 | ||||||
Reserves and accruals not currently deductible | 676 | 383 | ||||||
Deferred revenue | 55 | 46 | ||||||
Depreciation related | 63 | 55 | ||||||
Research and other credit carryforwards | 135 | 91 | ||||||
Other, net | 118 | 125 | ||||||
Total deferred tax assets before valuation allowance | 1,468 | 1,072 | ||||||
Valuation allowance | (6 | ) | (9 | ) | ||||
Total deferred tax assets | 1,462 | 1,063 | ||||||
Deferred tax liabilities: | ||||||||
Intangibles | (280 | ) | (328 | ) | ||||
Unremitted foreign earnings | — | (16 | ) | |||||
Other | (50 | ) | (34 | ) | ||||
Total deferred tax liabilities | (330 | ) | (378 | ) | ||||
Net deferred tax assets | $ | 1,132 | $ | 685 | ||||
The valuation allowance was $6 million as of December 31, 2015 and $9 million as of December 31, 2014 and December 31, 2013. It is more likely than not that we will not realize any benefit from the deferred tax assets related to certain state net operating loss and credit carryforwards.
At December 31, 2015, we had U.S. federal net operating loss carryforwards of approximately $354 million. The federal net operating loss carryforwards will start to expire in 2019, if not utilized. We also had federal tax credit carryforwards of approximately $8 million which will start to expire in 2017, if not utilized. In addition, we had state net operating loss and tax credit carryforwards of approximately $732 million and $234 million, respectively. The state net operating loss and tax credit carryforwards will start to expire in 2016 if not utilized.
Utilization of net operating losses and tax credits may be subject to an annual limitation due to ownership change limitations provided in the Internal Revenue Code of 1986, as amended, and similar state provisions. This annual limitation may result in the expiration of the net operating losses and credits before utilization.
We file federal, state and foreign income tax returns in many jurisdictions in the United States and abroad. For federal income tax purposes, the statute of limitations is open for 2010 and onwards. For certain acquired entities, the statute of limitations is open for all years from inception due to our utilization of their net operating losses and credits carried over from prior years. For California income tax purposes, the statute of limitations is open for 2010 and onwards.
Our income tax returns are subject to audit by federal, state and foreign tax authorities. We are currently under examination by the IRS for the 2010, 2011 and 2012 tax years and by various state and foreign jurisdictions. There are differing interpretations of tax laws and regulations, and as a result, significant disputes may arise with these tax authorities involving issues of the timing and amount of deductions and allocations of income among various tax jurisdictions. We periodically evaluate our exposures associated with our tax filing positions.
We have total federal, state and foreign unrecognized tax benefits of $1.4 billion as of December 31, 2015 and $661 million as of December 31, 2014. Of the total unrecognized tax benefits, $1.3 billion and $602 million at December 31, 2015 and 2014, respectively, if recognized, would reduce our effective tax rate in the period of recognition. We have continued to classify interest and penalties related to unrecognized tax benefits as part of our income tax provision in our Consolidated Statements of Income. We had accrued interest and penalties related to unrecognized tax benefits of $24 million as of December 31, 2015 and $18 million as of December 31, 2014.
103
As of December 31, 2015, we believe that it is reasonably possible that our unrecognized tax benefits will decrease by approximately $7 million in the next 12 months as we expect to have clarification from the IRS and other tax authorities regarding our uncertain tax positions. With respect to the remaining unrecognized tax benefits, we are currently unable to make a reasonable estimate as to the period of cash settlement, if any, with the respective tax authorities.
The following is a rollforward of our total gross unrecognized tax benefit liabilities (in millions):
December 31, | ||||||||||||
2015 | 2014 | 2013 | ||||||||||
Balance, beginning of period | $ | 661 | $ | 237 | $ | 157 | ||||||
Tax positions related to current year: | ||||||||||||
Additions | 675 | 430 | 112 | |||||||||
Reductions | — | — | — | |||||||||
Tax positions related to prior years: | ||||||||||||
Additions | 45 | 21 | 13 | |||||||||
Reductions | — | (20 | ) | — | ||||||||
Settlements | (24 | ) | (5 | ) | (39 | ) | ||||||
Lapse of statute of limitations | (7 | ) | (2 | ) | (6 | ) | ||||||
Balance, end of period | $ | 1,350 | $ | 661 | $ | 237 | ||||||
17. | SUBSEQUENT EVENT |
We entered into a license and collaboration agreement with Galapagos NV (Galapagos), a clinical-stage biotechnology company based in Belgium, for the development and commercialization of filgotinib, a JAK1-selective inhibitor being investigated for inflammatory disease indications. Under the terms of the agreement, which became effective on January 19, 2016, we made an upfront license fee payment of $300 million and a $425 million equity investment in Galapagos. In addition, Galapagos is eligible to receive development and regulatory milestone-based payments of up to $755 million, sales-based milestone payments of up to $600 million, tiered royalties on global sales and a profit split in potential co-promotion territories.
104
SELECTED QUARTERLY FINANCIAL INFORMATION (UNAUDITED)
The following amounts are in millions, except per share amounts:
1st Quarter | 2nd Quarter | 3rd Quarter | 4th Quarter | |||||||||||||
2015 | ||||||||||||||||
Total revenues | $ | 7,594 | $ | 8,244 | $ | 8,295 | $ | 8,506 | ||||||||
Gross profit on product sales | $ | 6,523 | $ | 7,128 | $ | 7,147 | $ | 7,347 | ||||||||
Net income | $ | 4,332 | $ | 4,497 | $ | 4,592 | $ | 4,685 | ||||||||
Net income attributable to Gilead | $ | 4,333 | $ | 4,492 | $ | 4,600 | $ | 4,683 | ||||||||
Net income per share attributable to Gilead common stockholders-basic | $ | 2.91 | $ | 3.05 | $ | 3.14 | $ | 3.26 | ||||||||
Net income per share attributable to Gilead common stockholders-diluted | $ | 2.76 | $ | 2.92 | $ | 3.06 | $ | 3.18 | ||||||||
2014 | ||||||||||||||||
Total revenues | $ | 4,999 | $ | 6,535 | $ | 6,042 | $ | 7,314 | ||||||||
Gross profit on product sales | $ | 4,058 | $ | 5,488 | $ | 4,981 | $ | 6,159 | ||||||||
Net income | $ | 2,223 | $ | 3,650 | $ | 2,724 | $ | 3,462 | ||||||||
Net income attributable to Gilead | $ | 2,227 | $ | 3,656 | $ | 2,731 | $ | 3,487 | ||||||||
Net income per share attributable to Gilead common stockholders-basic | $ | 1.45 | $ | 2.39 | $ | 1.80 | $ | 2.32 | ||||||||
Net income per share attributable to Gilead common stockholders-diluted | $ | 1.33 | $ | 2.20 | $ | 1.67 | $ | 2.18 | ||||||||
105
GILEAD SCIENCES, INC.
Schedule II: Valuation and Qualifying Accounts
(in millions)
Balance at Beginning of Period | Additions/Charged to Expense | Deductions | Balance at End of Period | |||||||||||||
Year ended December 31, 2015: | ||||||||||||||||
Accounts receivable allowances (1) | $ | 356 | $ | 6,934 | $ | 6,258 | $ | 1,032 | ||||||||
Sales return allowance | $ | 171 | $ | 219 | $ | 19 | $ | 371 | ||||||||
Valuation allowances for deferred tax assets (2) | $ | 9 | $ | — | $ | 3 | $ | 6 | ||||||||
Year ended December 31, 2014: | ||||||||||||||||
Accounts receivable allowances (1) | $ | 252 | $ | 2,867 | $ | 2,763 | $ | 356 | ||||||||
Sales return allowance | $ | 82 | $ | 104 | $ | 15 | $ | 171 | ||||||||
Valuation allowances for deferred tax assets (2) | $ | 9 | $ | — | $ | — | $ | 9 | ||||||||
Year ended December 31, 2013: | ||||||||||||||||
Accounts receivable allowances (1) | $ | 188 | $ | 1,870 | $ | 1,806 | $ | 252 | ||||||||
Sales return allowance | $ | 73 | $ | 21 | $ | 12 | $ | 82 | ||||||||
Valuation allowances for deferred tax assets (2) | $ | 9 | $ | — | $ | — | $ | 9 | ||||||||
(1) | Allowances are for doubtful accounts, cash discounts and chargebacks. |
(2) | Valuation allowance for deferred tax assets includes $4 million and $6 million as of December 31, 2015 and 2014, respectively, related to our acquisitions. |
106
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Not applicable.
ITEM 9A. | CONTROLS AND PROCEDURES |
(a) Evaluation of Disclosure Controls and Procedures
An evaluation as of December 31, 2015 was carried out under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our “disclosure controls and procedures,” which are defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, as amended (the Exchange Act), as controls and other procedures of a company that are designed to ensure that the information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission's rules and forms, and that such information is accumulated and communicated to the company's management, including its Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at December 31, 2015.
(b) Management's Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) of the Exchange Act. Our internal control system is designed to provide reasonable assurance regarding the preparation and fair presentation of financial statements for external purposes in accordance with generally accepted accounting principles. All internal control systems, no matter how well designed, have inherent limitations and can provide only reasonable assurance that the objectives of the internal control system are met.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting, based on criteria established by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in its 2013 Internal Control-Integrated Framework. Based on our evaluation, we concluded that our internal control over financial reporting was effective as of December 31, 2015.
Our independent registered public accounting firm, Ernst & Young LLP, has audited our Consolidated Financial Statements included in this Annual Report on Form 10-K and have issued a report on our internal control over financial reporting as of December 31, 2015. Their report on the audit of internal control over financial reporting appears below.
(c) Changes in Internal Control over Financial Reporting
Our management, including our Chief Executive Officer and Chief Financial Officer, has evaluated any changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2015, and has concluded that there was no change during such quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
107
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Gilead Sciences, Inc.
We have audited Gilead Sciences, Inc.'s internal control over financial reporting as of December 31, 2015, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Gilead Sciences, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Gilead Sciences, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2015, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2015 consolidated financial statements of Gilead Sciences, Inc. and our report dated February 24, 2016 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
Redwood City, California
February 24, 2016
108
ITEM 9B. | OTHER INFORMATION |
Not applicable.
PART III
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this Item concerning our directors and executive officers is incorporated by reference to the sections of our Definitive Proxy Statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A in connection with our 2016 Annual Meeting of Stockholders (the Proxy Statement) under the headings “Nominees,” “Board Committees and Meetings,” “Executive Officers,” and “Section 16(a) Beneficial Ownership Reporting Compliance.”
Our written Code of Ethics applies to all of our directors and employees, including our executive officers, including without limitation our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions. The Code of Ethics is available on our website at http://www.gilead.com in the Investors section under “Corporate Governance.” Changes to or waivers of the Code of Ethics will be disclosed on the same website. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding any amendment to, or waiver of, any provision of the Code of Ethics by disclosing such information on the same website.
ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item is incorporated by reference to the sections of the Proxy Statement under the headings “Executive Compensation,” “Compensation Committee Interlocks and Insider Participation,” “Compensation Committee Report,” and “Compensation of Non-Employee Board Members.”
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item is incorporated by reference to the sections of the Proxy Statement under the headings “Security Ownership of Certain Beneficial Owners and Management” and “Securities Authorized for Issuance under Equity Compensation Plans.”
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item is incorporated by reference to the sections of the Proxy Statement under the headings “Nominees,” and “Certain Relationships and Related Party Transactions.”
ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item is incorporated by reference to the section of the Proxy Statement under the heading “Principal Accountant Fees and Services.”
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) The following documents are filed as part of this Annual Report on Form 10-K:
(1) Index list to Consolidated Financial Statements:
Audited Consolidated Financial Statements | |
(2) Schedule II is included on page 106 of this report. All other schedules are omitted because they are not required or the required information is included in the financial statements or notes thereto.
(3) Exhibits.
The following exhibits are filed herewith or incorporated by reference:
109
ITEM 15. | EXHIBITS |
Exhibit Footnote | Exhibit Number | Description of Document | ||
(1) | 1.1 | Underwriting Agreement, dated September 9, 2015, among Registrant and Merrill Lynch, Pierce, Fenner & Smith Incorporated and J.P. Morgan Securities LLC, as representatives of the several underwriters listed in Schedule 1 thereto | ||
†(2) | 2.1 | Agreement and Plan of Merger among Registrant, Merger Sub and Pharmasset, Inc., dated as of November 21, 2011 | ||
(3) | 3.1 | Restated Certificate of Incorporation of Registrant | ||
(4) | 3.2 | Amended and Restated Bylaws of Registrant | ||
4.1 | Reference is made to Exhibit 3.1 and Exhibit 3.2 | |||
(5) | 4.2 | Indenture related to the Convertible Senior Notes due 2016 (2016 Notes), between Registrant and Wells Fargo Bank, National Association, as trustee (including form of 1.625% Convertible Senior Note due 2016), dated July 30, 2010 | ||
(6) | 4.3 | Indenture related to Senior Notes, dated as of March 30, 2011, between Registrant and Wells Fargo, National Association, as Trustee | ||
(6) | 4.4 | First Supplemental Indenture related to Senior Notes, dated as of March 30, 2011, between Registrant and Wells Fargo, National Association, as Trustee (including form of Senior Notes) | ||
(7) | 4.5 | Second Supplemental Indenture related to Senior Notes, dated as of December 13, 2011, between Registrant and Wells Fargo, National Association, as Trustee (including Form of 2014 Note, Form of 2016 Note, Form of 2021 Note, Form of 2041 Note) | ||
(8) | 4.6 | Third Supplemental Indenture related to Senior Notes, dated as of March 7, 2014, between Registrant and Wells Fargo, National Association, as Trustee (including Form of 2019 Note, Form of 2024 Note, Form of 2044 Note) | ||
(9) | 4.7 | Fourth Supplemental Indenture related to Senior Notes, dated as of November 17, 2014, between Registrant and Wells Fargo, National Association, as Trustee (including Form of 2020 Note, Form of 2025 Note, Form of 2045 Note) | ||
(1) | 4.8 | Fifth Supplemental Indenture, dated as of September 14, 2015, between Registrant and Wells Fargo Bank, National Association, as Trustee (including Form of 2018 Note, Form of 2020 Note, Form of 2022 Note, Form of 2026 Note, Form of 2035 Note and Form of 2046 Note) | ||
(10) | 10.1 | Confirmation of OTC Convertible Note Hedge related to 2016 Notes, dated July 26, 2010, between Registrant and Goldman, Sachs & Co. | ||
(10) | 10.2 | Confirmation of OTC Convertible Note Hedge related to 2016 Notes, dated July 26, 2010, between Registrant and JPMorgan Chase Bank, National Association | ||
(10) | 10.3 | Confirmation of OTC Warrant Transaction, dated July 26, 2010, between Registrant and Goldman, Sachs & Co. for warrants expiring in 2016 | ||
(10) | 10.4 | Confirmation of OTC Warrant Transaction, dated July 26, 2010, between Registrant and JPMorgan Chase Bank, National Association for warrants expiring in 2016 | ||
(11) | 10.5 | Confirmation of OTC Additional Convertible Note Hedge related to 2016 Notes, dated August 5, 2010, between Registrant and Goldman, Sachs & Co. | ||
(11) | 10.6 | Confirmation of OTC Additional Convertible Note Hedge related to 2016 Notes, dated August 5, 2010, between Registrant and JPMorgan Chase Bank, National Association | ||
(11) | 10.7 | Confirmation of OTC Additional Warrant Transaction, dated August 5, 2010, between Registrant and Goldman, Sachs & Co. for warrants expiring in 2016 | ||
(11) | 10.8 | Confirmation of OTC Additional Warrant Transaction, dated August 5, 2010, between Registrant and JPMorgan Chase Bank, National Association for warrants expiring in 2016 | ||
(11) | 10.9 | Amendment to Confirmation of OTC Convertible Note Hedge related to 2016 Notes, dated August 30, 2010, between Registrant and Goldman, Sachs & Co. | ||
(11) | 10.10 | Amendment to Confirmation of OTC Convertible Note Hedge related to 2016 Notes, dated August 30, 2010, between Registrant and JPMorgan Chase Bank, National Association | ||
(11) | 10.11 | Amendment to Confirmation of OTC Additional Convertible Note Hedge related to 2016 Notes, dated August 30, 2010, between Registrant and Goldman, Sachs & Co. | ||
(11) | 10.12 | Amendment to Confirmation of OTC Additional Convertible Note Hedge related to 2016 Notes, dated August 30, 2010, between Registrant and JPMorgan Chase Bank, National Association | ||
(12) | 10.13 | Amendment to Base Warrants (2016), dated May 8, 2015, between Registrant and Goldman, Sachs & Co. | ||
(12) | 10.14 | Amendment to Base Warrants (2016), dated May 8, 2015, between Registrant and JPMorgan Chase Bank, National Association | ||
(13) | 10.15 | 5-Year Revolving Credit Facility Credit Agreement among Registrant and Gilead Biopharmaceutics Ireland UC (formerly Gilead Biopharmaceutics Ireland Corporation), as Borrowers, Bank of America, N.A., as Administrative Agent, Swing Line Lender and L/C Issuer, certain other lenders parties thereto, Barclays Capital, as Syndication Agent, and Goldman Sachs Bank USA, JPMorgan Chase Bank, N.A., Royal Bank of Canada and Wells Fargo Bank, N.A., as Co-Documentation Agents, dated as of January 12, 2012 | ||
(13) | 10.16 | Parent Guaranty Agreement (5-Year Revolving Credit Facility), dated as of January 12, 2012, by Registrant | ||
*(3) | 10.17 | Gilead Sciences, Inc. 2004 Equity Incentive Plan, as amended through May 8, 2013 | ||
110
*(14) | 10.18 | Form of employee stock option agreement used under 2004 Equity Incentive Plan (for grants prior to February 2008) | ||
*(15) | 10.19 | Form of employee stock option agreement used under 2004 Equity Incentive Plan (for grants made February 2008 through April 2009) | ||
*(16) | 10.20 | Form of employee stock option agreement used under 2004 Equity Incentive Plan (for grants commencing in May 2009) | ||
*(17) | 10.21 | Form of employee stock option agreement used under 2004 Equity Incentive Plan (for grants commencing in February 2010) | ||
*(18) | 10.22 | Form of employee stock option agreement used under 2004 Equity Incentive Plan (for 2011 and subsequent year grants) | ||
*(15) | 10.23 | Form of non-employee director stock option agreement used under 2004 Equity Incentive Plan (for grants prior to 2008) | ||
*(15) | 10.24 | Form of non-employee director option agreement used under 2004 Equity Incentive Plan (for initial grants made in 2008) | ||
*(15) | 10.25 | Form of non-employee director option agreement used under 2004 Equity Incentive Plan (for annual grants made in May 2008 and through May 2012) | ||
*(16) | 10.26 | Form of non-employee director option agreement used under 2004 Equity Incentive Plan (for annual grants commencing in May 2009 and through May 2012) | ||
*(19) | 10.27 | Form of non-employee director option agreement used under 2004 Equity Incentive Plan (for annual grants made in May 2013) | ||
*(19) | 10.28 | Form of non-employee director option agreement (non-U.S.) used under 2004 Equity Incentive Plan (for annual grants made in May 2013) | ||
*(20) | 10.29 | Form of non-employee director option agreement used under 2004 Equity Incentive Plan (for annual grants made in and after May 2014) | ||
*(21) | 10.30 | Form of restricted stock unit issuance agreement used under 2004 Equity Incentive Plan (for annual grants to non-employee directors in May 2012) | ||
*(16) | 10.31 | Form of restricted stock award agreement used under 2004 Equity Incentive Plan (for annual grants to certain non-employee directors prior to May 2012) | ||
*(19) | 10.32 | Form of restricted stock unit issuance agreement used under 2004 Equity Incentive Plan (for annual grants to non-employee directors commencing in May 2013) | ||
*(20) | 10.33 | Form of restricted stock unit issuance agreement used under 2004 Equity Incentive Plan (for annual grants to non-employee directors commencing in and after May 2014) | ||
*(19) | 10.34 | Form of restricted stock unit issuance agreement (non-U.S.) used under 2004 Equity Incentive Plan (for annual grants to non-employee directors commencing in May 2013) | ||
*(16) | 10.35 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for grants to certain executive officers made in 2009) | ||
*(17) | 10.36 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for grants to certain executive officers made in 2010) | ||
*(18) | 10.37 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for grants to certain executive officers made in 2011) | ||
*(19) | 10.38 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for grants to certain executive officers made in 2012) | ||
*(22) | 10.39 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for TSR Goals in 2013 and 2014) | ||
*(23) | 10.40 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for Revenue Goals in 2013 and 2014) | ||
*(24) | 10.41 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for TSR Goals - Non-US in 2015) | ||
*(24) | 10.42 | Form of performance share award agreement used under the 2004 Equity Incentive Plan (for Revenue Goals - Non-US in 2015) | ||
*(25) | 10.43 | Form of restricted stock unit issuance agreement used under the 2004 Equity Incentive Plan (for grants to certain executive officers made prior to May 2009) | ||
*(16) | 10.44 | Form of restricted stock unit issuance agreement used under the 2004 Equity Incentive Plan (for grants to certain executive officers commencing in May 2009) | ||
*(26) | 10.45 | Form of restricted stock unit issuance agreement used under the 2004 Equity Incentive Plan (service-based vesting for certain executive officers commencing in November 2009) | ||
*(18) | 10.46 | Form of restricted stock unit issuance agreement used under the 2004 Equity Incentive Plan (service-based vesting for certain executive officers commencing in 2011) | ||
*(27) | 10.47 | Gilead Sciences, Inc. Employee Stock Purchase Plan, restated on January 22, 2015 | ||
*(28) | 10.48 | Gilead Sciences, Inc. Deferred Compensation Plan-Basic Plan Document | ||
*(26) | 10.49 | Gilead Sciences, Inc. Deferred Compensation Plan-Adoption Agreement | ||
*(28) | 10.50 | Addendum to the Gilead Sciences, Inc. Deferred Compensation Plan | ||
*(29) | 10.51 | Gilead Sciences, Inc. 2005 Deferred Compensation Plan, as amended and restated on October 23, 2008 | ||
*(22) | 10.52 | Gilead Sciences, Inc. Severance Plan, as amended on January 26, 2012 | ||
111
* | 10.53 | Gilead Sciences, Inc. Corporate Bonus Plan, amended on November 4, 2015 | ||
*(30) | 10.54 | Amended and Restated Gilead Sciences, Inc. Code Section 162(m) Bonus Plan | ||
*(31) | 10.55 | 2016 Base Salaries for the Named Executive Officers | ||
*(32) | 10.56 | Offer Letter dated April 16, 2008 between Registrant and Robin Washington | ||
*(33) | 10.57 | Form of Indemnity Agreement entered into between Registrant and its directors and executive officers | ||
*(34) | 10.58 | Form of Employee Proprietary Information and Invention Agreement entered into between Registrant and certain of its officers and key employees | ||
*(17) | 10.59 | Form of Employee Proprietary Information and Invention Agreement entered into between Registrant and certain of its officers and key employees (revised in September 2006) | ||
+ (35) | 10.60 | Amended and Restated Collaboration Agreement by and among Registrant, Gilead Holdings, LLC, Bristol-Myers Squibb Company, E.R. Squibb & Sons, L.L.C., and Bristol-Myers Squibb & Gilead Sciences, LLC, dated September 28, 2006 | ||
+ (15) | 10.61 | Commercialization Agreement by and between Gilead Sciences Ireland UC (formerly Gilead Sciences Limited) and Bristol-Myers Squibb Company, dated December 10, 2007 | ||
+ (36) | 10.62 | Amendment Agreement, dated October 25, 1993, between Registrant, the Institute of Organic Chemistry and Biochemistry (IOCB) and Rega Stichting v.z.w. (REGA), together with the following exhibits: the License Agreement, dated December 15, 1991, between Registrant, IOCB and REGA (the 1991 License Agreement), the License Agreement, dated October 15, 1992, between Registrant, IOCB and REGA (the October 1992 License Agreement) and the License Agreement, dated December 1, 1992, between Registrant, IOCB and REGA (the December 1992 License Agreement) | ||
+ (37) | 10.63 | Amendment Agreement between Registrant and IOCB/REGA, dated December 27, 2000 amending the 1991 License Agreement and the December 1992 License Agreement | ||
+ (35) | 10.64 | Sixth Amendment Agreement to the License Agreement, between IOCB/REGA and Registrant, dated August 18, 2006 amending the October 1992 License Agreement and the December 1992 License Agreement | ||
+ (38) | 10.65 | Seventh Amendment Agreement to the License Agreement, between IOCB/REGA and Registrant dated July 1, 2013 amending the October 1992 License Agreement and the December 1992 License Agreement | ||
+ (39) | 10.66 | Exclusive License Agreement between Registrant (as successor to Triangle Pharmaceuticals, Inc.), Glaxo Group Limited, The Wellcome Foundation Limited, Glaxo Wellcome Inc. and Emory University, dated May 6, 1999 | ||
+ (40) | 10.67 | Royalty Sale Agreement by and among Registrant, Emory University and Investors Trust & Custodial Services (Ireland) Limited, solely in its capacity as Trustee of Royalty Pharma, dated July 18, 2005 | ||
+ (40) | 10.68 | Amended and Restated License Agreement between Registrant, Emory University and Investors Trust & Custodial Services (Ireland) Limited, solely in its capacity as Trustee of Royalty Pharma, dated July 21, 2005 | ||
+ (41) | 10.69 | License Agreement between Japan Tobacco Inc. and Registrant, dated March 22, 2005 | ||
+ (42) | 10.70 | First Amendment to License Agreement between Japan Tobacco Inc. and Registrant, dated May 19, 2005 | ||
+ (42) | 10.71 | Second Amendment to License Agreement between Japan Tobacco Inc. and Registrant, dated May 17, 2010 | ||
+(12) | 10.72 | Third Amendment (Revised) to License Agreement between Japan Tobacco Inc. and Registrant, dated June 10, 2015 | ||
+ (42) | 10.73 | Fourth Amendment to License Agreement between Japan Tobacco Inc. and Registrant, dated July 5, 2011 | ||
+(43) | 10.74 | Amendment to License Agreement between Japan Tobacco Inc. and Registrant, dated October 10, 2013 | ||
+(44) | 10.75 | Fifth Amendment to License Agreement between Japan Tobacco Inc. and Registrant, dated September 29, 2014 | ||
+(45) | 10.76 | Amended and Restated Collaboration Agreement by and among Registrant, Gilead Sciences Ireland UC (formerly Gilead Sciences Limited) and Janssen R&D Ireland, dated December 23, 2014 | ||
+(46) | 10.77 | Master Clinical and Commercial Supply Agreement between Gilead World Markets, Limited, Registrant and Patheon Inc., dated January 1, 2003 | ||
+(47) | 10.78 | Restated and Amended Toll Manufacturing Agreement between Gilead Sciences Ireland UC (formerly Gilead Sciences Limited), Registrant and Takeda GmbH (formerly Nycomed GmbH and Altana Pharma Oranienburg GmbH), dated November 7, 2005 | ||
21.1 | Subsidiaries of Registrant | |||
23.1 | Consent of Independent Registered Public Accounting Firm | |||
31.1 | Certification of Chief Executive Officer, as required by Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended | |||
31.2 | Certification of Chief Financial Officer, as required by Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as amended | |||
32.1** | Certifications of Chief Executive Officer and Chief Financial Officer, as required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350) | |||
101*** | The following materials from Registrant's Annual Report on Form 10-K for the year ended December 31, 2015, formatted in Extensible Business Reporting Language (XBRL) includes: (i) Consolidated Balance Sheets at December 31, 2015 and 2014, (ii) Consolidated Statements of Income for the years ended December 31, 2015, 2014 and 2013, (iii) Consolidated Statements of Comprehensive Income for the years ended December 31, 2015, 2014 and 2013, (iv) Consolidated Statements of Stockholders' Equity for the years ended December 31, 2015, 2014 and 2013 (v) Consolidated Statements of Cash Flows for years ended December 31, 2015, 2014 and 2013, and (vi) Notes to Consolidated Financial Statements. | |||
112
(1) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on September 14, 2015, and incorporated herein by reference. |
(2) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on November 25, 2011, and incorporated herein by reference. |
(3) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on May 8, 2014, and incorporated herein by reference. |
(4) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on December 23, 2015, and incorporated herein by reference. |
(5) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on August 2, 2010, and incorporated herein by reference. |
(6) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on April 1, 2011, and incorporated herein by reference. |
(7) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on December 13, 2011, and incorporated herein by reference. |
(8) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on March 7, 2014, and incorporated herein by reference. |
(9) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on November 17, 2014, and incorporated herein by reference. |
(10) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2010, and incorporated herein by reference. |
(11) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2010, and incorporated herein by reference. |
(12) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2015, and incorporated herein by reference. |
(13) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on January 17, 2012, and incorporated herein by reference. |
(14) | Filed as an exhibit to Registrant's Current Report on Form 8-K/A filed on February 22, 2006, and incorporated herein by reference. |
(15) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2007, and incorporated herein by reference. |
(16) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2009, and incorporated herein by reference. |
(17) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2009, and incorporated herein by reference. |
(18) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2011, and incorporated herein by reference. |
(19) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, and incorporated herein by reference |
(20) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2014, and incorporated herein by reference. |
(21) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2012, and incorporated herein by reference. |
(22) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2012, and incorporated herein by reference. |
(23) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2013, and incorporated herein by reference. |
(24) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2015, and incorporated herein by reference. |
(25) | Filed as an exhibit to Registrant's Current Report on Form 8-K first filed on December 19, 2007, and incorporated herein by reference. |
(26) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, and incorporated herein by reference. |
(27) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on May 8, 2015, and incorporated herein by reference. |
(28) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2001, and incorporated herein by reference. |
(29) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2008, and incorporated herein by reference. |
(30) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on May 13, 2013, and incorporated herein by reference. |
(31) | Filed as an exhibit to Registrant's Current Report on Form 8-K filed on February 3, 2016, and incorporated herein by reference. |
(32) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended June 30, 2008, and incorporated herein by reference. |
(33) | Filed as an exhibit to Registrant's Registration Statement on Form S-1 (No. 33-55680), as amended, and incorporated herein by reference. |
(34) | Filed as an exhibit to Registrant's Registration Statement on Form S-8 (No. 333-102912) filed on January 31, 2003, and incorporated herein by reference. |
(35) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2006, and incorporated herein by reference. |
(36) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended March 31, 1994, and incorporated herein by reference. |
(37) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2000, and incorporated herein by reference. |
(38) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2013, and incorporated herein by reference. |
(39) | Filed as an exhibit to Triangle Pharmaceuticals, Inc.'s Quarterly Report on Form 10-Q/A filed on November 3, 1999, and incorporated herein by reference. |
(40) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2005, and incorporated herein by reference. |
(41) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended March 31, 2005, and incorporated herein by reference. |
(42) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2011, and incorporated herein by reference. |
(43) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2013, and incorporated herein by reference. |
(44) | Filed as an exhibit to Registrant's Quarterly Report on Form 10-Q for the quarter ended September 30, 2014, and incorporated herein by reference. |
(45) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2014, and incorporated herein by reference. |
(46) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2003, and incorporated herein by reference. |
(47) | Filed as an exhibit to Registrant's Annual Report on Form 10-K for the fiscal year ended December 31, 2005, and incorporated herein by reference. |
† | The Agreement and Plan of Merger (the Pharmasset Merger Agreement) contains representations and warranties of Registrant, Merger Sub and Pharmasset, Inc. made solely to each other as of specific dates. Those representations and warranties were made solely for purposes of the Pharmasset Merger Agreement and may be subject to important qualifications and limitations agreed to by Registrant, Merger Sub and Pharmasset, Inc. Moreover, some of those representations and warranties may not be accurate or complete as of any specified date, may be subject to a standard of materiality provided for in the Pharmasset Merger Agreement and have been used for the purpose of allocating risk among Registrant, Merger Sub and Pharmasset, Inc. rather than establishing matters as facts. |
* | Management contract or compensatory plan or arrangement. |
** | This certification accompanies the Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing. |
*** | XBRL information is filed herewith. |
+ | Certain confidential portions of this Exhibit were omitted by means of marking such portions with an asterisk (the Mark). This Exhibit has been filed separately with the Secretary of the Securities and Exchange Commission without the Mark pursuant to Registrant's Application Requesting Confidential Treatment under Rule 24b-2 under the Securities Exchange Act of 1934, as amended. |
113
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
GILEAD SCIENCES, INC. | |
By: | /S/ JOHN C. MARTIN |
John C. Martin, Ph.D. Chairman and Chief Executive Officer | |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints John C. Martin and Brett A. Pletcher, and each of them, as his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place, and stead, in any and all capacities, to sign any and all amendments to this Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that all said attorneys-in-fact and agents, or any of them or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
114
Signature | Title | Date | ||
/S/ JOHN C. MARTIN | Chairman and Chief Executive Officer | February 24, 2016 | ||
John C. Martin, Ph.D. | (Principal Executive Officer) | |||
/S/ ROBIN L. WASHINGTON | Executive Vice President and Chief Financial Officer | February 24, 2016 | ||
Robin L. Washington | (Principal Financial and Accounting Officer) | |||
/S/ JOHN F. COGAN | Director | February 24, 2016 | ||
John F. Cogan | ||||
/S/ ETIENNE F. DAVIGNON | Director | February 24, 2016 | ||
Etienne F. Davignon | ||||
/S/ CARLA A. HILLS | Director | February 24, 2016 | ||
Carla A. Hills | ||||
/S/ KEVIN E. LOFTON | Director | February 24, 2016 | ||
Kevin E. Lofton | ||||
/S/ JOHN W. MADIGAN | Director | February 24, 2016 | ||
John W. Madigan | ||||
/S/ JOHN F. MILLIGAN | Director | February 24, 2016 | ||
John F. Milligan | ||||
/S/ NICHOLAS G. MOORE | Director | February 24, 2016 | ||
Nicholas G. Moore | ||||
/S/ RICHARD J. WHITLEY | Director | February 24, 2016 | ||
Richard J. Whitley | ||||
/S/ GAYLE E. WILSON | Director | February 24, 2016 | ||
Gayle E. Wilson | ||||
/S/ PER WOLD-OLSEN | Director | February 24, 2016 | ||
Per Wold-Olsen | ||||
115