Attached files
| file | filename |
|---|---|
| EX-23.2 - EXHIBIT 23.2 - Vaxart, Inc. | ex23-2.htm |
| EX-23.1 - EXHIBIT 23.1 - Vaxart, Inc. | ex23-1.htm |
| EX-32.2 - EXHIBIT 32.2 - Vaxart, Inc. | ex32-2.htm |
| EX-21.1 - EXHIBIT 21.1 - Vaxart, Inc. | ex21-1.htm |
| EX-23.1A - EXHIBIT 23.1A - Vaxart, Inc. | ex23-1a.htm |
| EX-31.1 - EXHIBIT 31.1 - Vaxart, Inc. | ex31-1.htm |
| EX-31.2 - EXHIBIT 31.2 - Vaxart, Inc. | ex31-2.htm |
| EX-32.1 - EXHIBIT 32.1 - Vaxart, Inc. | ex32-1.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES |
|
|
EXCHANGE ACT OF 1934 |
||
|
For the fiscal year ended June 30, 2015 |
||
|
OR |
||
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES |
|
|
EXCHANGE ACT OF 1934 |
For the transition period from ____________ to ____________
Commission file number: 001-35285
Biota Pharmaceuticals Inc.
(Exact name of Registrant as specified in its charter)
|
Delaware |
|
59-1212264 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification Number) |
|
2500 Northwinds Parkway, Suite 100, Alpharetta, GA |
30009 | |
|
(Address of Principal Executive Offices) |
(Zip Code) |
(678) 221 3343
(Registrant’s telephone number, including area code)
Securities registered pursuant to section 12(b) of the Act:
|
Title of each class |
Name of each exchange on which registered | |
|
Common Stock, par value $.10 per share |
The Nasdaq Stock Market LLC NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☑
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☑ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer”, ”accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ Accelerated filer ☑ Non-accelerated filer ☐ Smaller reporting company ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☑
The aggregate market value of the common stock held by non-affiliates of the registrant, based on the closing price on December 31, 2014 was approximately $78.1 million.
Number of shares of Common Stock outstanding as of September 9, 2015: 38,609,086. The common stock is listed on the NASDAQ Global Select Market (trading symbol “BOTA”)
Documents incorporated by reference:
Portions of the definitive Proxy Statement with respect to the 2015 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days after the close of the fiscal year are incorporated by reference into Part III of this report.
TABLE OF CONTENTS
|
Page | |||
|
Item1 |
Business |
4 | |
|
Item1A |
Risk Factors |
23 | |
|
Item 1B |
Unresolved Staff Comments |
40 | |
|
Item2 |
Properties |
40 | |
|
Item3 |
Legal Proceedings |
41 | |
|
Item4 |
Mine Safety Disclosures |
41 | |
|
Item5 |
Market for the Registrant’s Common Equity, Related Stockholders’ Matters, and Issuer Purchases of Equity Securities |
42 | |
|
Item6 |
Select Financial Data |
44 | |
|
Item7 |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
45 | |
|
Item7A |
Quantitative and Qualitative Disclosures about Market Risk |
54 | |
|
Item8 |
Financial Statements and Supplementary Data |
55 | |
|
Item9 |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
55 | |
|
Item9A |
Controls and Procedures |
56 | |
|
Item9B |
Other Information |
56 | |
|
Item10 |
Directors, Executives Officers and Corporate Governance |
57 | |
|
Item11 |
Executive Compensation |
57 | |
|
Item12 |
Security Ownership of Certain Beneficial Owners; and Management; and Related Stockholder Matters |
57 | |
|
Item13 |
Certain Relationships, Related Transactions, and Director Independence |
57 | |
|
Item14 |
Principal Accounting Fees and Services |
57 | |
|
Item15 |
Exhibits; Financial Statement Schedules |
58 | |
|
Signatures |
59 | ||
|
ITEM 1. |
BUSINESS |
PART I
SPECIAL NOTE ON FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the Securities Exchange Act of 1934, as amended. These forward-looking statements are principally contained in the sections entitled “Item 1-Business”, “Item 2-Properties” and “Item 7-Management’s Discussion and Analysis of Financial Condition and Results of Operations”, but may appear elsewhere. All statements other than those of historical facts contained herein are forward looking statements, which reflect our current expectations and assumptions about the future. Forward looking statements involve known and unknown risks and uncertainties that may cause actual future results, performance, achievements or events to be materially different from any results, performance, achievements or events expressed or implied by the forward-looking statements. In general, you can identify forward-looking statements by terms such as, but not limited to, “may,” “will,” “should,” “could,” “would,” “expect,” “plan,” “intend,” “anticipate,” “believe,” “estimate,” “project,” “predict,” “forecast,” “potential,” “continue,” “target,” “likely” or “possible,” as well as the negative of such expressions, and similar expressions intended to identify forward-looking statements. These forward-looking statements include, but are not limited to, statements relating to:
|
● |
our anticipated timing to fully enroll and report top line-data from our Phase2b SPIRITUS clinical trial; |
|
● |
our anticipated timing to complete the Phase 1 single ascending dose clinical trial for BTA585 and report top-line data; |
|
● |
our planned timing of the initiation of the Phase 1 multiple ascending dose clinical trial for BTA585 and report top-line data; |
|
● |
our planned timing of the initiation of the Phase 2a challenge study for BTA585; |
|
● |
our planned timing of the initiation of the Phase 2 clinical trial for BTA074 and reporting top line-data; |
|
● |
our preclinical respiratory syncytial virus (“RSV”) non-fusion inhibitor that may complement BTA585; |
|
● |
our plans to select a lead candidate from our RSV non-fusion inhibitor program; |
|
● |
our plans to out-license the rights to laninamivir octanoate (“LANI”) outside of Japan in concert with Daiichi Sankyo Company Ltd. (“Daiichi Sankyo”); |
|
● |
our anticipation that royalty revenue from the net sales of Relenza® may decrease in fiscal 2016 due to the expiration of the composition of matter patents for Relenza® in multiple countries and the outcome of the pending patent application in the U.S.; |
|
● |
our anticipation that we will generally incur net losses from operations in the future due to our intention to continue to support the preclinical and clinical development of our product candidates; |
|
● |
our future financing requirements, the factors that may influence the timing and amount of those requirements and our ability to fund them; |
|
● |
the number of months that our current cash, cash equivalents and anticipated future proceeds from existing royalty-bearing licenses and other existing license and collaboration agreements will allow us to operate; and |
|
● |
our plan to continue to finance our operations with our existing cash, cash equivalents and proceeds from existing or potential future royalty-bearing licenses, government contracts, or collaborative research and development arrangements, or through future equity and/or forms of asset or debt financings or other financing vehicles. |
These forward looking statements are subject to key risks and uncertainties including, without limitation: the U.S. Food and Drug Administration (“FDA”) or similar foreign regulatory agency, a data safety monitoring board, an institutional review board delaying, or we limiting, suspending or terminating the clinical development of any of our clinical development programs at any time for a lack of safety, tolerability, biologic activity, commercial viability, regulatory or manufacturing issues, or any other reason whatsoever; the safety or efficacy data from ongoing or future preclinical studies of any of our product candidates not supporting the clinical development of that product candidate; our capacity to successfully enroll, manage and conduct several simultaneous clinical trials on a worldwide timely basis; our ability to comply with applicable government regulations in various countries and regions in which we are conducting, or expect to conduct, clinical trials; our ability to manufacture and maintain sufficient quantities of preclinical and clinical trial material on hand to conduct and complete our preclinical studies or clinical trials on a timely basis; our ability, or that of our clinical research organizations or clinical investigators, to enroll a sufficient number of patients in our clinical trials on a timely basis; our ability to retain and recruit sufficient staff, including key executive management and employees, to manage our business; our ability to secure, manage and retain qualified third-party clinical research, preclinical research, data management, contract manufacturing and other similar vendors who we outsource many of our activities to and rely on to assist us in the design, development and implementation of the development of our product candidates; our third-party contract research, data management and manufacturing organizations fulfilling their contractual obligations on a timely basis or otherwise performing satisfactorily in the future; GlaxoSmithKline (“GSK”) or Daiichi Sankyo continuing to generate net sales from Relenza® and Inavir®, respectively, and otherwise continuing to fulfill their obligations under our royalty-bearing license agreements with them in the future; our ability to maintain, protect or defend our proprietary intellectual property rights from unauthorized use by others, or not infringe on the intellectual property rights of others; our ability to successfully manage our expenses, operating results and financial position in line with our plans and expectations; the condition of the financial equity and debt markets and our ability to raise sufficient funding in such markets; changes in general economic business or competitive conditions related to our industry or product candidates; and other statements contained elsewhere in this Annual Report on Form 10-K and the risk factors described in or referred to in greater detail in the “Risk Factors” section of this Form 10-K. There may be events in the future that we are unable to predict accurately, or over which we have no control. You should read this Form 10-K, as well as the documents that we reference herein and that have been filed or incorporated by reference as exhibits, completely and with the understanding that our actual future results may be materially different from our expectations. Our business, financial condition, results of operations, and prospects may change. We undertake no obligation to update these forward-looking statements, unless we are required by law. We qualify all of the information presented in this Form 10-K, and particularly our forward-looking statements, by these cautionary statements.
Biota is a registered trademark of Biota Pharmaceuticals, Inc., Relenza® is a registered trademark of GlaxoSmithKline plc, Inavir® is a registered trademark of Daiichi Sankyo Company, Ltd, and TwinCaps® is a registered trademark of Hovione FarmaCiencia SA.
References to “we,” “us,” and “our” refer to Biota Pharmaceuticals, Inc. and its subsidiaries.
Our Business
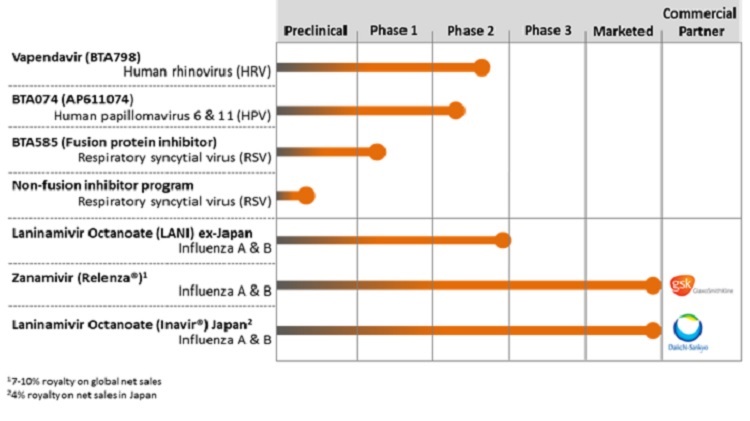
We are focused on the discovery and development of direct-acting antivirals to treat infections that affect a significant number of patients globally. We have four product candidates in clinical development that address viral infections that have limited therapeutic options. vapendavir, an oral treatment for human rhinovirus (“HRV”) infections in moderate-to-severe asthmatics, currently being evaluated in our ongoing Phase 2b SPIRITUS trial; BTA074, a Phase 2 topical antiviral treatment for genital warts caused by human papillomavirus (“HPV”) types 6 & 11; BTA585, an oral fusion (“F”) protein inhibitor in Phase 1 development for the treatment of RSV-A and RSV-B infections; and laninamivir octanoate, a one-time, inhaled treatment in Phase 2 development for influenza A and B infections. We also have preclinical RSV non-fusion inhibitor program that we believe complements our F-protein inhibitor BTA585.
Background
We have historically focused our research and drug development capabilities on discovering and developing small molecule compounds that can prevent or treat infectious diseases. Infectious diseases are caused by pathogens that are present in the environment, such as viruses and bacteria, which enter the body through various means and overwhelm its natural defenses and cause an infection. The severity of an infectious disease varies depending on the nature of the infectious pathogen, as well as the degree to which the body’s immune system or available therapies can prevent or fight the infection. The market for anti-infective drugs generally can be divided into three general categories: antiviral, antibacterial and antifungal. We are currently focused on developing antiviral compounds.
The use of antiviral drugs has led to a significant reduction in the morbidity and mortality associated with infectious diseases. However, for many infectious diseases, current treatment options, to the extent any such treatment options are currently available, are associated with suboptimal treatment outcomes, significant toxicities, tolerability issues or adverse side effects, the emergence of drug resistant pathogens, complex dosing schedules, and inconvenient methods of administration. These sub-optimal characteristics of many existing treatment options often lead to patients prematurely discontinuing treatment or not fully complying with treatment dosing schedules, resulting in a treatment failure. A patient’s failure to comply fully with a recommended dosing schedule can also both accelerate and exacerbate the emergence of drug-resistant strains. In recent years, the increasing prevalence of drug-resistant pathogens has created ongoing treatment challenges with respect to many infectious diseases. The ability of viruses to adapt rapidly to existing or new treatments through genetic mutations allows new strains to develop that may be resistant to currently available drugs. In recent years, the increasing prevalence of drug-resistant pathogens has created ongoing treatment challenges with respect to many infectious diseases.
Our Pipeline
The following chart summarizes key information regarding our antiviral product candidates:
|
|
Human Rhinovirus (“HRV”), Asthma and Chronic Obstructive Pulmonary Disease (“COPD”) |
HRV is a non-enveloped, single-stranded viruses that belong to the Picornaviridiae family. Currently more than 100 distinct serotypes of HRV are classified into three species, HRV-A, HRV-B, and HRV-C. Primary market research conducted by the IMS Consulting Group on our behalf with pulmonologists, internists and general practitioners indicated that adult asthma and chronic obstructive pulmonary disease (“COPD”) patients experience four to six colds per year. Asthma is a common disease with underlying inflammation of the airways that affects an estimated 300 million people worldwide and 26 million people in the United States. A 2015 study commissioned by us with the IMS Consulting Group indicated that there were 10.5 million people in the U.S. categorized as having moderate to severe asthma based on its research of primary and secondary market sources dating between 2008 and 2015.1 Acute asthma exacerbations are a major healthcare burden, accounting for almost half of the total healthcare costs associated with asthma, and also have a major impact on the quality of life and in some cases can cause death. Recent studies in adults with asthma have documented an association between respiratory tract infection and worsening asthma symptoms, decline in lung function (disease progression), and exacerbations. Respiratory viruses, and in particular HRV, are a significant cause of exacerbations. In a 2014 study of asthma patients with cold-like symptoms, 63% of the patients had respiratory viruses that were detected by qPCR (quantitative PCR) and the majority of those samples (68%) contained HRV.
Exacerbations are important sequelae of HRV infection in asthma patients and their prevention has historically been the focus of asthma drug development. Poor asthma control and use of asthma reliever bronchodilator medications have been linked with an increased risk of death and asthma exacerbations can be fatal. In recent years asthma treatment guidelines have also focused on asthma control as an important goal. Asthma control is defined by a global assessment of symptoms, use of rescue medications, lung function, and patient-reported functioning and activity limitations. The Asthma Control Questionnaire (“ACQ-6”) is a patient reported outcome (“PRO”) tool often used to measure a drug’s therapeutic impact on the worsening of asthma symptoms. In general, a well-controlled asthma patient has an ACQ score of ≤ 0.75 – 1.0 and a patient with uncontrolled asthma has an ACQ score of ≥ 1.50. An improvement in ACQ score of ≥ 0.5 is generally considered indicative of a clinically meaningful change. Although there are several FDA approved drugs for the treatment of asthma, none are directed at respiratory viruses, including HRV.
COPD is the most common chronic respiratory condition in adults whose prevalence is expected to continue to increase in the future. Currently, the World Health Organization (“WHO”) estimates that 64 million people have moderate to severe COPD worldwide. In the U.S. there are an estimated 28 million individuals over the age of 40 with COPD, with an annual average growth rate of 1.9%. Further, of the estimated 28 million COPD patients in the U.S., approximately 13 million are classified as having moderate to severe/very severe COPD.
Similar to asthma, HRV is the most common virus detected during exacerbations of COPD. In COPD patients, colds often precede exacerbation symptoms. In a published experimental challenge study, COPD patients with an HRV infection showed more severe and prolonged lower respiratory symptoms, airway obstruction, and neutrophilic airway inflammation than subjects without COPD. In addition, a recent natural exposure study in COPD patients demonstrated that HRV prevalence and viral load at exacerbation presentation were significantly higher compared to a period when the patient was not experiencing an exacerbation. Further, the HRV viral load was elevated in COPD patients that presented to the clinic, consistent with the experimental challenge study, suggesting that viral replication may be ongoing, and antiviral therapy may be an effective treatment modality to prevent or reduce the severity of exacerbations.
There are currently no direct antiviral drugs approved for the treatment of HRV. As such, there remains a significant unmet medical need to identify treatments that can reduce the impact that HRV infection has on the frequency of exacerbations and loss of control, prevent viral transmission, lessen the severity and duration of cold-like HRV symptoms and minimize secondary bacterial infections in asthma and COPD patients.
|
|
Vapendavir (BTA798) |
We are developing vapendavir (BTA798), a potent antiviral capsid binder that is designed to bind to a highly conserved pocket in the HRV capsid and interfere with receptor binding and/or related early steps in the infectious cycle. Vapendavir is a potent inhibitor of picornaviruses and has been shown to inhibit the replication of a wide range of HRV serotypes and the replication of a majority of recent HRV clinical isolates in tissue culture assays. The median EC50 value for vapendavir against the 100 HRV serotypes is 5.8 ng/mL (15.2 nM). Vapendavir has also demonstrated antiviral activity against other clinically relevant enteroviruses (“EV”) including EV- 71 and poliovirus types 1, 2 and 3.
Vapendavir (BTA798) Clinical Trials
Phase 2b SPIRITUS Trial. The ongoing Phase 2b SPIRITUS clinical trial of vapendavir is being conducted at approximately 65 sites in North America and Central Europe with a goal of enrolling approximately 190 laboratory-confirmed HRV-infected patients for the intent to treat-infected (“ITT-I”) population. Patients aged 18-70 years of age that have an established history of moderate-to-severe asthma and a history of losing asthma control as a result of an upper respiratory tract infection will be eligible to be enrolled in the trial.
The following diagram summarizes the design of the multi-center, 1:1:1 randomized, double-blind, placebo-controlled dose-ranging SPIRITUS Trial:
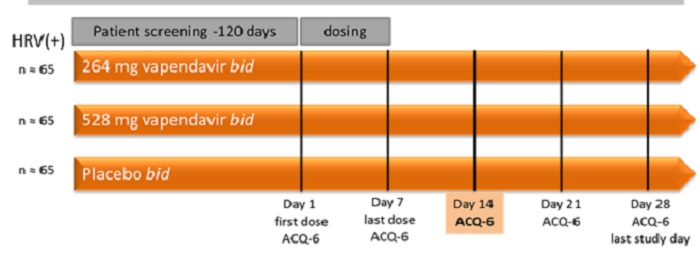
The primary endpoint of SPIRITUS is the least square mean change from baseline (day 1) to study day 14 in ACQ-6 total score.
The secondary endpoints of this study are focused on safety and tolerability, lung function assessments such as forced expiratory volume in one second (“FEV1”), incidence of asthma exacerbations, assessments of the severity and duration of cold symptoms as measured by the Wisconsin Upper Respiratory Symptom Survey-21 (“WURSS-21”), and virology assessments such as changes in viral load. The primary efficacy analysis population will be the ITT-I population defined as all subjects with confirmed HRV infection (by either the eSensor® Respiratory Viral Panel (GenMark) or RT-PCR on any of Study Days 1, 3, 5, or 7).
Phase 1 Bioavailability Trial. In 2014, we completed enrollment in a Phase 1 study entitled ‘A Randomized, Single-Center, Open-Label, Two-Period, Two-Sequence, Crossover, Comparative Study to Compare the Oral Bioavailability of Single Doses of Two Vapendavir Drug Product Formulations in Healthy Volunteers’ designed to establish the systemic exposure profile of a single dose of a vapendavir free-base tablet formulation compared to the exposure profile following a single dose of two vapendavir phosphate salt capsules, which was the formulation used in prior clinical trials. Thirty-six subjects received a single dose of the capsule formulation or the tablet formulation in Period 1 and, after a washout, crossed over to receive a single dose of the other formulation in Period 2 of the study, according to their sequence treatment assignment. No serious adverse events occurred during the study. The pharmacokinetics (“PK”) results demonstrated that the free-base tablet formulation achieved approximately 60% of the mean bioavailability as the phosphate salt capsule formulation. The half-life and time to peak concentration (“Tmax”) were comparable between the two formulations. Based on these PK findings additional formulation activities with the free-base tablet were initiated to improve its bioavailability and the phosphate salt form of vapendavir was selected for use in the Phase 2b SPIRITUS trial.
Phase 1 Drug-Drug Interaction Trial. In 2014, we also completed a drug-drug interaction study entitled ‘A Phase 1, Randomized, Open-Label Study to Evaluate the Effect of Vapendavir (BTA798) on the Pharmacokinetics of Orally Administered Midazolam, a CYP3A4 Substrate, in Healthy Male and Female Volunteers’. This study was designed to assess the effect of vapendavir on the PK profile of midazolam, a CYP3A4 substrate. Additionally, the effect of midazolam on the PK profile of vapendavir, the PK profile differences of vapendavir in males and females, and the safety profile of vapendavir were assessed. Twelve (12) male and 12 female subjects aged 18 to 55 years were randomized to receive one of two oral doses of vapendavir and midazolam. Of the 24 subjects randomized, 22 completed all study visits. No serious adverse events (“SAEs”) occurred during the study. The results of the study confirmed vapendavir’s pharmacokinetic profile as established in prior clinical trials and established that vapendavir is a weak to moderate inducer of CYP3A4, which suggests that vapendavir may be used to treat asthma and COPD patients receiving multiple background medications.
Phase 2. In 2012, we completed a 300-patient, multicenter, randomized, double-blind, placebo-controlled study of vapendavir in adults with mild to moderate asthma that had a symptomatic HRV infection. The primary objective of the study was to determine the efficacy of vapendavir on symptoms of presumptive HRV infection in asthmatic adults, as measured by the WURSS-21 severity scores. Vapendavir was dosed twice daily for six days., The study was conducted over two HRV seasons (18 months), with an estimated 1200 individuals screened in order to randomize 300 subjects, 155 in the vapendavir arm and 145 in the placebo group. The trial successfully met its primary endpoint, which was a reduction of cold symptoms based on the WURSS-21 severity score averaged over days two through day four. The mean daily reduction in WURSS-21 severity score averaged over days two to four was significantly greater in the vapendavir treated group compared to the placebo group (least square mean difference: -4.01, p = 0.020). Vapendavir was generally tolerated and most treatment-related adverse events were of mild intensity, with moderate treatment-related events reported in 2.3% of subjects. No SAE’s occurred during the study.
Phase 2 HRV39 Challenge Study. In 2009, we completed a Phase 2a placebo-controlled, double-blind, randomized, parallel group trial to determine the potential of 25 mg, 100 mg and 400 mg of vapendavir, when dosed twice daily for 10 days, to prevent experimental HRV39 infection (challenge design) in 41 healthy volunteers. Subjects that received 400 mg of vapendavir achieved a statistically significant reduction compared to placebo in mean viral load on days two to five inclusive. Vapendavir was generally well tolerated, and the overall incidence of adverse events was low, not dose dependent, and was similar to placebo. There was one SAE of neutropenic sepsis in a subject in the 100 mg arm of the trial.
Phase 1 Single Ascending and Multiple Ascending Trials. In 2006, we completed Phase 1, placebo-controlled, single and multiple ascending oral dose, safety, tolerability and pharmacokinetic studies in 56 healthy volunteers. Single oral doses of 25 mg, 50 mg, 100 mg, 200 mg, 400 mg, 800 mg or 1600 mg of vapendavir were evaluated. Vapendavir was generally well tolerated and there were no dose limiting toxicities or trends in adverse events or laboratory parameters observed, with the incidence and nature of adverse events similar between placebo recipients and all dosing groups of vapendavir. In a subsequent multiple ascending dose trial evaluating 200 and 400 mg of vapendavir, administered either once a day (“QD”) or twice a day (“BID”) for seven or eight consecutive days, vapendavir was well tolerated. There were no SAE’s and there were no dose limiting toxicities or clinically relevant changes in vital signs, ECG or laboratory parameters observed.
Human Papillomavirus (“HPV”)
HPVs are small non-enveloped, double stranded DNA viruses that infect mucosal or cutaneous squamous epithelia, where they may cause benign or malignant hyperproliferation of the skin and mucosa. HPV is the most common cause of sexually transmitted infections and the disease burden includes skin warts, genital warts, cervical and other anogenital dysplasias and carcinomas, oropharyngeal cancer and recurrent respiratory papillomatosis (“RRP”). HPV is the most common sexually transmitted infection. Over 40 distinct types can infect the genital tract. Approximately 90% of infections caused by HPV’s are asymptomatic and resolve spontaneously within two years. However, persistent infection with some HPV types can cause cancer and other benign diseases. Of the 13 HPV types designated as human carcinogens, types 16 and 18 account for 70% of cervical cancers worldwide. Among non-carcinogenic types, types, HPV 6 and 11 are responsible for 90% of anogenital warts.
Genital warts also referred to as anogenital warts or condyloma is the most commonly identified pathology caused by genital HPVs. Genital warts are sexually transmitted, with a high rate of transmission and significant psychosocial morbidity. Genital warts are one of the most common viral sexually transmitted disease (“STD”) worldwide. It is one of the most frequent STDs diagnosed among genitourinary medicine (“GUM”) clinics and accounts for more frequent visits to general practitioners or GUM clinics than those for genital herpes. In 2013, the Centers for Disease Control and Prevention estimated that in the U.S. there were >400,000 visits to physicians’ offices related to genital warts.
HPV types 6 and 11 are also associated with RRP, a condition of tumors or wart-like lesions of the upper respiratory tract, particularly the larynx. In the U.S., the incidence of RRP is two per 100,000 adults and four per 100,000 children. Juvenile-onset RRP (“JORRP”) is usually diagnosed between the ages of one and four years, is equally prevalent in both sexes, and is believed to be acquired by newborns from their mothers during labor. Adult-onset RRP (“AORRP”) has a broad peak of occurrence between ages 20 and 40 years, and is most frequent in males. Afflicted infants and children with JORRP present with difficulty breathing or swallowing and therefore the lesions can become life-threatening, while adults usually present with hoarseness, chronic coughing or breathing problems. A small percentage of afflicted patients go on to have systemic disseminated disease which can invade the lungs and become potentially lethal. Typically, RRP lesions have to be removed surgically. On average, in the U. S., children undergo 19.7 surgical procedures over their lifetime, with a mean frequency of 4.4 procedures per year. In 20 percent of JORRP and AORRP patients the disease will be more aggressive and can require more than 40 surgical procedures during a lifetime. It is estimated that 15,000 surgical procedures due to RRP are performed per year in the U. S., at a total cost of $150 million, and lifetime costs per individual patient can reach up to $470,000.
Currently, no approved HPV-specific direct acting anti-viral drugs exist to treat genital warts or RRP. Existing treatments for genital warts can be divided broadly into two categories: provider-administered ablative/cytodestructive therapies (including cryotherapy, laser ablation, and trichloroacetic acid) and patient-administered topical therapies, such as podophyllotoxin, sinecatechins, and imiquimod. Imiquimod directly activates innate immune cells through Toll-like receptor 7, resulting in production of cytokines. Treatment choice depends on the morphology, number, and distribution of warts and patient preference. Significant failure and relapse rates, often as much as 20-30% or more have been reported for all of these existing treatments. Further, all existing therapies are associated with local skin reactions including itching, burning, erosions and pain. There are no cures for RRP, however current maintenance treatments include CO2 laser surgery, pulse dye laser surgery, endoscopic microdebriders, and intralesional injection with cidofovir. The high reoccurrence rates make the current therapies less than optimal for patients suffering with RRP. Therefore, despite the existence of marketed prophylactic vaccines, effective therapies against pathologies caused by HPV6 and HPV11 are still needed.
|
BTA074 (AP611074) |
BTA074 is in development for the treatment of genital warts, as well as the orphan indication of RRP. BTA074 is a potent and selective inhibitor of the interaction between two viral proteins from HPV6 and HPV11, E1 and E2, an interaction that is an essential step for HPV DNA replication and thus, viral production and pathogenesis. This inhibition results from the binding of BTA074 to the E2 protein (Kd=168 nM). BTA074 is a first-in-class directing acting antiviral specific to HPV and possesses new mechanism of action that can be exploited to treat infections caused by HPV types 6 & 11. BTA074 was selected for clinical development among more than 1200 unique compounds tested. BTA074 was developed by combining chemo-informatics modeling and in cellulo screening of E1/E2 protein-protein interactions. These studies showed that BTA074 inhibits the HPV6 and HPV11 E1/E2 interaction or HPV DNA replication in cellulo with an IC50 of 0.5-1 “M. Moreover, BTA074 is highly selective for low-risk types HPV 6 and HPV 11, since it does not inhibit replication of HPV 18 or E1/E2 protein interactions of other HPVs.
BTA074 (AP611074) Clinical Trials
Phase 2. We plan to initiate a Phase 2 trial in late 2015 to further validate BTA074 favorable local skin tolerability profile and antiviral activity. The trial is designed as a double-blind placebo controlled, randomized, Phase 2 study the primarily objective of which is to assess the safety, tolerability, pharmacokinetics and efficacy of twice daily topical treatments of BTA074 5% gel for up to 16 weeks in approximately 210 genital warts patients. The primary objective of the trial is to evaluate the safety and tolerability in genital warts patients with special focus on local skin reactions. A primary efficacy endpoint is to determine the complete clearance rate for baseline genital warts lesions after twice daily application of BTA074 5% gel or placebo from baseline week 0 visit to the completion of the treatment.
Phase 2a. In 2013, a Phase 2a clinical trial of BTA074 5% gel was completed. The six-week, Phase 2a study in 24 subjects (sixteen active; eight placebo) demonstrated that twice daily application of 100 mg BTA074 5% gel had an excellent local skin tolerability profile and resulted in high patient compliance and no patient drop-outs or treatment interruptions. Further, treatment with BTA074 produced a 56% overall response rate and a -38% change in mean baseline wart area.
Phase 1b. In 2013, a Phase 1b multicenter, double-blind, randomized, placebo-controlled study in eight genital warts subjects (six active; two placebo) was completed. 100 mg BTA074 5% gel was applied topically twice daily for seven days to the infected area. No adverse events were reported during this study and no clinically relevant findings were observed in clinical examination, laboratory parameters, vital signs or electrocardiogram (“ECG”) parameters.
Phase 1. In 2011, a Phase 1 single center, double-blind, randomized, placebo-controlled study in eight healthy male volunteers (six active; two placebo) was completed. The study drug dose was 250 mg BTA074 5% gel applied on 25 cm² skin, corresponding to 12.5 mg BTA074/application. BTA074 5% gel was applied twice daily for seven days. Repeated topical applications of BTA074 on the back of healthy volunteers were well tolerated. One subject receiving BTA074 5% gel and one subject receiving placebo experienced mild isolated erythema on pre-dose day five.
|
|
Respiratory Syncytial Virus (“RSV”) |
RSV, a member of the Paramyxoviridae family of viruses, is a major cause of acute upper and lower respiratory tract infections in infants, young children, and adults. Datamonitor estimates that approximately 18 million people are infected annually with RSV in the seven major markets worldwide, including over nine million children under the age of four, five and half million elderly, and three million adults with underlying disease. About 900,000 of these individuals are hospitalized for their RSV infection. These infections are particularly problematic in infants, as approximately 91,000 are hospitalized with RSV infection in the U.S. in any given year. RSV infections are also responsible for 40 to 50% of hospitalizations for pediatric bronchiolitis and 25% of hospitalizations for pediatric pneumonia. In addition to pediatric patients, elderly patients with cardiac or pulmonary conditions and adults that have received a bone marrow transplant are at an increased risk for severe RSV infection. The overall magnitude of hospitalizations makes RSV a costly disease, although mortality is low.
To date, only three drugs have been approved to either prevent or treat RSV infections. Ribavirin is used to treat serious RSV infections in infants with severe bronchiolitis and in immunocompromised patients. However, its use is restricted due to highly variable efficacy and toxicity risks. In fact, current American Academy of Pediatrics guidelines for the treatment of bronchiolitis in children do not recommend the routine use of ribavirin to treat RSV infection due to lack of clinical evidence supporting its use. Antibody-based products RespiGam® (no longer available) and Synagis® (palivizumab) were designed, developed and approved to prevent, not treat, RSV infections in high risk premature infants. Due to the high cost of treatment with Synagis®, its use is limited in many hospitals. There remains a significant unmet need for a safe and effective treatment for RSV in all at-risk populations.
|
|
BTA585 |
Our lead compound, BTA585, is a potent, non-cytotoxic and selective inhibitor of the RSV F protein. Data from studies investigating the mechanism of BTA585 anti-viral activity, including analysis of RSV resistance mutants, support the conclusion that BTA585 inhibits the function of the RSV protein. Therefore, BTA585 exerts its antiviral activity by interfering with the earliest stage of infection by inhibiting the attachment and/or fusion of the virus to the host cell. BTA585 is equally active against both RSV A and B subtypes but has no known activity against other pathogenic viruses. When tested against a panel of RSV clinical isolates, BTA585 was found to be highly potent with an average EC50 =95.6nM.
In September 2015, we presented at the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy Meeting in Washington, D.C. from a number of in vivo studies designed to assess the antiviral activity of BTA585 prior to and during experimental RSV infection in a cotton rat model. These studies demonstrated a dose-dependent decrease in virus titers in lung tissue. Similarly, a highly significant dose-dependent decrease in RSV mRNA in lung tissue was also observed in the cotton rat model.
BTA585 Clinical Trials
Phase 1 Single Ascending Dose (“SAD”) Clinical Trial
In August 2015, we commenced dosing in a 50-subject, randomized, placebo-controlled, Phase 1 single ascending dose (“SAD”) clinical trial to evaluate the safety and PK of BTA585 in healthy volunteers. The Phase 1 SAD trial has five dose level cohorts ranging from 50 mg to 500 mg and includes an evaluation of the effect of food on the PK of BTA585.
Non-Fusion RSV Inhibitors
In addition to BTA585, we have identified a series of potent RSV non-fusion inhibitors that we intend to further develop and believe could be useful as a stand-alone treatment or potentially in combination therapy with BTA585 for the treatment of patients infected with RSV. Our goal is to select a lead candidate from this program by mid-year 2016.
|
|
Influenza |
Influenza, or the flu, is an acute viral infection caused by an influenza virus. There are three types of influenza – A, B and C. Influenza A viruses are further typed into subtypes according to the different kinds and combinations of virus surface proteins. Among the many subtypes of influenza A viruses, currently influenza A(H1N1)pmd09 and influenza A(H3N2) are the most common subtypes circulating among humans. Type C influenza cases occur much less frequently than types A and B.
Influenza is characterized by a sudden onset of high fever, headache, muscle and joint pain, severe malaise (feeling unwell), cough (usually dry), sore throat and a runny nose. Most people generally recover from fever and most of the other symptoms within a week without requiring medical attention. However, influenza can cause severe illness, hospitalization or death in people at high risk, which include the very young, the elderly and the chronically ill. The time from infection to illness, known as the incubation period, is generally about two days. Influenza epidemics generally occur annually during the autumn and winter in temperate regions. According to the WHO, annual epidemics result in about three to five million cases of severe illness and about 250,000 to 500,000 deaths worldwide. Most deaths associated with influenza in industrialized countries occur among people ages 65 or older.
Controlling influenza virus infections continues to be a major public health challenge. Despite increasingly widespread vaccination, influenza remains a significant burden that can give rise to a potential crisis, even in communities with advanced health care. Data from the CDC suggest that each year 5-20% of the U.S. population (16-63 million according to 2012 U.S. population estimates) suffer from influenza, and approximately 200,000 people in the U.S. are hospitalized each year for respiratory and heart conditions associated with influenza infections. In pandemic influenza outbreaks, vaccines can only be developed following identification of the pandemic strain, which results in vaccines not being available immediately. These limitations of vaccination emphasize the importance of and need for antiviral drugs to treat and prevent influenza.
The market opportunity for antivirals to prevent or treat influenza, and their utilization in the seasonal influenza market, is often difficult to project given the year-to-year variability in the circulating strain of influenza, the severity of influenza illness, and the length of the influenza season. Further, if there is a year in which a pandemic occurs, the variability in the market potential is magnified. In addition to this seasonal influenza market, government stockpiling of antivirals to prevent or treat influenza have historically contributed significantly to the overall market opportunity.
|
|
Laninamivir Octanoate (“LANI”) |
In 2003, we cross-licensed intellectual property related to a new class of inhaled long acting neuraminidase inhibitors (“NI’s”) with Daiichi Sankyo. The lead product from this collaboration is LANI, also known as CS-8958, a second-generation octanoyl ester pro-drug of laninamivir. LANI has been shown to have in vitro neuraminidase-inhibitory activity against various influenza A and B viruses, including subtypes N1 to N9 and oseltamivir-resistant viruses, and it has also been found to be effective against a swine origin H1N1 strain. Moreover, LANI has long-lasting antiviral activity. Preclinical studies in mice have demonstrated that after intranasal administration, it was rapidly converted to its active metabolite, laninamivir, which was retained in the lungs where it had a long half-life of approximately 40 hours. Further, a single intranasal dose of LANI exhibited efficacy similar to that of repeated doses of zanamivir or oseltamivir phosphate.
LANI was successfully developed by Daiichi Sankyo in Japan and since 2010 has been marketed there as Inavir® for the treatment of influenza A and B infections. In December 2013, Inavir® was approved for use in the post-exposure prevention of influenza. Since 2009, we have been developing LANI under an IND in the U.S. for the treatment of influenza A and B.
|
|
LANI Clinical Trials |
Phase 2. In June 2013, we commenced enrollment in a multi-national, randomized, double blind, placebo controlled, parallel arm Phase 2 clinical trial that compared the safety and efficacy of 40 mg and 80 mg of LANI with placebo, all delivered by a TwinCaps® inhaler in adults with symptomatic influenza A or B infection. The trial, which we refer to as “IGLOO”, was designed to enroll 636 subjects, randomized equally across the three treatment arms. The primary endpoint of the IGLOO trial was the reduction in the median time to alleviation (reported to be mild or absent for greater than 24 hours) of seven influenza symptoms (headache, feeling feverish, body aches and pains, fatigue, cough, sore throat and nasal congestion) plus fever (<38oC), compared to placebo. Symptom data were collected through the Flu-iiQTM patient-recorded outcomes (“PRO”) questionnaire. Secondary end points included quantitative changes in virus shedding, evaluating whether the use of LANI reduces the incidence of secondary bacterial infections compared to placebo, the development of resistance by phenotypic and genotypic analyses, and the impact of treatment with LANI on the quality of life.
On August 1, 2014, we announced top-line data from the IGLOO trial. We enrolled a total of 639 patients across 12 countries in the northern and southern hemispheres from June 2013 to April 2014. Of the 639 patients enrolled, 248, or 39%, had PCR confirmed influenza A or B virus and were included in the intent-to-treat efficacy analyses. Approximately 75% and 19% of the influenza-confirmed patients were infected with influenza A H1N1 2009 and H3N2, respectively, while 6% were infected with influenza B. As compared to placebo, neither the 40 mg nor the 80 mg cohort achieved a statistically significant reduction in the median time to alleviation of the seven influenza symptoms, which was the primary efficacy endpoint. The median time to alleviation of the seven influenza symptoms was 102.3 hours for the 40 mg cohort and 103.2 hours for the 80 mg cohort, as compared to 104.1 hours for the placebo cohort.
Although neither the 40 mg nor the 80 mg LANI cohorts achieved a statistically significant difference compared to placebo for the entire seven symptom primary endpoint, notable effects were seen in individual symptoms, the subset of systemic symptoms (headache, feeling feverish, body aches and pains and fatigue) and a number of key secondary endpoints. Subjects in the 40 mg cohort reported alleviation of the subset of systemic symptoms significantly earlier as compared to placebo (median time 59 hours and 79 hours, respectively, p=0.029). Patients in the 40 mg cohort also reported a significant reduction in the number of days in which all seven symptoms were severe (p=0.02) and in secondary bacterial infections as compared to placebo (0% compared to 7.8% of placebo recipients; p=0.013). Patients in the 40 mg (p<0.001) cohort demonstrated a statistically significant reduction in viral shedding on day 3 of the study compared to placebo as quantified by qRT-PCR. In addition, a statistically significant proportion of patients in both the 40 mg (p=0.002) and 80 mg (p=0.02) cohorts were culture negative on day 3 of the study as compared to placebo. The nature and extent of adverse events were similar in the three cohorts, with diarrhea (3.1% vs. 0.9%), headache (1.4% vs. 0.5%), gastritis (1.4% vs. 0%), urinary tract infection (1.4% vs. 0%), and sinusitis (1.2% vs. 0.9%) being the most common adverse events that occurred more frequently in the treatment cohorts as compared to placebo. The incidence of serious adverse events was low and balanced across the three cohorts.
We and Daiichi Sankyo intend to collectively pursue a license agreement, or other similar transaction, with regional or global third-party pharmaceutical or biopharmaceutical companies that have greater clinical development, manufacturing and commercialization capabilities than we do that we believe could advance the development and/or commercialization of LANI regionally or globally.
Phase 1 Clinical Trials. In 2014, we completed two Phase 1 clinical trials of LANI; one to evaluate its safety and pharmacokinetics in patients with chronic asthma and the other being a QT/QTc study to evaluate the effect of therapeutic (40 mg) and supra-therapeutic (240 mg) doses of laninamivir octanoate on the QT-interval.
The safety profile of 40 mg and 80 mg doses of LANI by inhalation via the TwinCaps® DPI in adults with mild or moderate chronic asthma demonstrated that inhaled LANI was well tolerated. Treatment with inhaled LANI at therapeutic antiviral doses did not result in significant changes in lung function as measured by spirometry. These results suggest that inhaled LANI can be safely administered to asthma patients. Top-line results of the QT/QTc study indicate LANI was not associated with any clinically significant ECG changes, no serious adverse events (“SAEs”) occurred, no subjects terminated the study or the study treatment due to an adverse event and a low proportion of subjects experienced adverse events during the study treatments. Between these two recent Phase 1safety studies, a total of 211 subjects participated and, of these, 118 received inhaled LANI at doses ranging from 40 mg to 240 mg.
Prior to initiating the Phase 2 IGLOO trial in June 2013, we completed three other Phase 1 trials of inhaled LANI. These trials provided safety and pharmacokinetic data at single doses of LANI ranging from five mg to 40 mg in healthy volunteers aged 18 to 77 years, and at multiple doses up to 40 mg (twice daily for three days or twice weekly for six weeks) in healthy volunteers aged 20 to 47 years. A total of 94 subjects were enrolled in these studies, with 70 of those receiving LANI. In healthy adult volunteers, LANI was generally well tolerated at single doses up to 120 mg and at multiple doses up to 40 mg administered twice daily for three days or twice weekly for six weeks.
Our Strategy
We are focused on the discovery and development of direct-acting antivirals to treat infections that affect a significant number of patients globally for which there are limited therapeutic options. In the near-term we intend to employ the following strategy:
|
● |
Focus Our Resources on the Development of our Clinical Stage Antiviral Product Candidates. We plan to focus our resources on vapendavir, an oral treatment for HRV infections in moderate-to-severe asthmatics; BTA074, a novel topical treatment for genital warts caused by HPV types 6 & 11; and BTA585, an oral fusion inhibitor in development for the treatment of RSV infections. |
More specifically, over the next 12 months we intend to:
|
■ |
Initiate a Phase 2 clinical trial for BTA074 in patients with genital warts late 2015; |
|
■ |
Complete and report top-line data from a Phase 1 single ascending dose clinical trial for BTA585 in healthy volunteers in the fourth quarter of 2015; |
|
■ |
Complete and report top-line data from a Phase 1 multiple ascending dose clinical trial for BTA585 in healthy volunteers in the first quarter of 2016; |
|
■ |
Complete the Phase 2b SPIRITUS trial of vapendavir in patients with moderate-to-severe asthma and report top-line data in mid-2016; |
|
■ |
Initiate a Phase 2a RSV challenge study in healthy volunteers for BTA585 in the second quarter of 2016; and |
|
■ |
Continue process development and formulation activities (adult and pediatric) for BTA585 and BTA074. | |
|
● |
Evaluate our RSV preclinical candidates and select a lead candidate for IND-enabling studies. We intend to develop a series of potent RSV non-fusion inhibitors that we believe could be used as a stand-alone treatment or potentially in combination therapy. Our goal is to select a lead candidate from this series by mid-year 2016. |
|
● |
Seek to out-license LANI. We, in concert with Daiichi Sankyo, intend to pursue a license agreement, or other similar transaction, with larger third-party regional or global pharmaceutical or biopharmaceutical companies that have greater clinical development, manufacturing and commercialization capabilities than we do that we believe could advance the development and/or commercialization of LANI. |
Research and Development
Our research and development expense in fiscal 2015, 2014 and 2013 was $19.8 million, $17.5 million and $19.2 million, respectively. In fiscal 2016, we plan to focus our research and development resources primarily on (i) the clinical development of vapendavir, BTA074, and BTA585, (ii) continue process development and formulation activities for vapendavir, BTA074, and BTA585, and (iii) conduct screening, lead-optimization, and preclinical studies on several series of RSV non-fusion inhibitors.
Our basic research and discovery activities, including medicinal chemistry, virology, and cell culture assays have historically been conducted internally by our research staff. In March 2015, we closed our laboratory facilities in Melbourne, Australian. We now use third party research firms and consultants more extensively to conduct these activities under our management. We do not have any future plans to build laboratory facilities or hire significant staff to conduct research, discovery and certain development activities. To the extent we continue these activities in the near-future, we anticipate that we will outsource them and rely on third-party research firms and consultants to a greater extent than we did in the past.
Sales and Marketing
We currently do not have any commercialization or sales and marketing capabilities, and we have no near term plans to invest in or build such capabilities internally. At this time, we anticipate partnering, collaborating with or licensing certain rights to our development programs to other larger pharmaceutical or biopharmaceutical companies in the future to support the late stage development and commercialization of our product candidates.
Manufacturing
We currently do not own or operate any facilities in which we can formulate, manufacture, fill or package our product candidates. We currently rely on single group of contract manufacturers to produce our drug substance and to fill and package the materials required to conduct clinical trials under current good manufacturing practices, (“cGMP”). If an existing contract manufacture fails to deliver on schedule, or at all, or fails to manufacture our material is accordance with their or our specifications and/or FDA regulations, it could significantly delay or interrupt the development or commercialization of our product candidates and affect our operating results and estimated development timelines. We have used contract manufacturers to produce all of the clinical trial material used in the preclinical studies and clinical trials we have conducted to-date.
Competition
The pharmaceutical and biotechnology industries are intensely competitive. Many companies, including biotechnology, chemical and pharmaceutical companies, are actively engaged in activities similar to ours, including research and the development of product candidates for the treatment of infectious diseases. Many of these companies have substantially greater financial and other resources, larger research and development staffs, and more extensive marketing and manufacturing capabilities than we do. In addition, some of them have considerably more experience in preclinical testing, conducting clinical trials and other regulatory approval procedures. There are also academic institutions, governmental agencies and other research organizations that are conducting research in areas in of infectious disease which we are working. We expect to encounter significant direct competition for any of the product candidates we plan to develop. Companies that complete clinical trials obtain required regulatory approvals and commence commercial sales of their products before their competitors may achieve a significant competitive advantage.
Currently, there are no approved direct-acting antiviral drugs to treat HRV infections. However, our vapendavir product candidate, if successfully developed, would indirectly compete with drugs approved to reduce the incidence of exacerbations or improve lung function in patients with asthma and COPD, such as fluticasone propionate (Advair®), tiotoprium bromide (Spiriva®), fluticasone furoate/vilanterol (Breo Ellipta®), and roflumilast (Daliresp®). In addition to these approved drugs, there are compounds in the clinical development stage, such as inhaled ß-interferon, that if successfully developed for the treatment of HRV infections could compete with vapendavir.
Currently there no approved HPV-specific directing acting anti-viral drugs to treat genital warts or RRP. Treatments for genital warts can be divided broadly into two categories: provider-administered ablative/cytodestructive therapies (including cryotherapy, laser ablation, and trichloroacetic acid) and patient-administered topical therapies such as podophyllotoxin (Condylox; Wartec), sinecatechins (Veregen®), and imiquimod (Zyclara®, Aldara®). There are no cures for RRP, however current maintenance treatments include CO2 laser surgery, pulse dye laser surgery, endoscopic microdebriders, and intralesional injection with cidofovir. We anticipate that BTA074, if successfully developed, would directly compete with the patient-applied topical treatments for genital warts and could become first-line therapy for RRP. We believe key differentiating features of BTA074 could be its mechanism of action, favorable local skin tolerability, efficacy, and lower reoccurrence rate. Three prophylactic vaccines, primarily designed to prevent cervical, vulvar, vaginal, and anal cancers, are currently marketed: a bivalent HPV16/18 vaccine (Cervarix®; GSK), quadrivalent HPV16/18/6/11 (Gardasil®, Merck) and the 9-valent HPV 6/11/16/18/33/52/58 (Gardasil®9; Merck). Gardasil® 9 is indicated for females aged 9 through 26 and males aged 9 through 15, to prevent various HPV related cancers and genital warts in both sexes. Gardasil®, Gardasil® 9, and Cervarix® are not known to exhibit a therapeutic effect on existing HPV lesions.
Effective treatments of RSV infections in pediatrics, the elderly, and the immunocompromised are very limited. Currently, only Virazole® (ribavirin) is indicated for the treatment of hospitalized infants and young children with severe lower respiratory tract infections due to RSV. We are aware that the following compounds are under development: Gilead’s GS-5806, Johnson and Johnson’s JJ-53718678 (ALS-8176), Ark Biosciences’ AK0529 and Teva Pharmaceutical’s MDT-637. The only approved drug for the prevention of RSV infections in high risk infants is MedImmune’s palivizumab (Synagis®), a monoclonal antibody.There are several vaccines and antibody products designed to prevent RSV infections in clinical development. Among the clinical stage product candidates in development are Novavax’s RSV F vaccine, GSK’s GSK3003898A vaccine, GSK’s GSK3003898A vaccine, Bavarian Nordic’s BN® RSV vaccine, MedImmune’s MEDI ÄM2-2 vaccine, MedImmune’s monoclonal antibody MEDI8897, and Regeneron’s monoclonal antibody REGN2222C.
The pharmaceutical market for products that prevent or treat influenza is competitive. Key competitive advantages for LANI may include its single administration treatment regimen, its resistance profile, and to-date, its reported safety profile. A number of NIs are currently available in the U.S. and/or other counties, including Japan, for the prevention and/or treatment of influenza. These include oseltamivir phosphate from Hoffmann-La Roche Ltd. (“Roche”), which is marketed as Tamiflu®, zanamivir from GSK, which is marketed as Relenza®, laninamivir octanoate from Daiichi Sankyo, which is marketed as Inavir®, and peramivir marketed as Rapivab® or Rapiacta® by CSL Limited and Shionogi & Co. Ltd., respectively. We anticipate that LANI, if ever successfully developed and approved, would compete directly or indirectly with drugs that will be “generic” by the time LANI may be approved for sale. Generic drugs are drugs whose patent protection has expired, which generally have an average selling price substantially lower than drugs protected by patents and intellectual property rights. Unless a patented drug can sufficiently differentiate itself from a competing generic drug in a meaningful manner, the existence of generic competition in any indication will generally impose significant pricing pressure on competing patented drugs.
Intellectual Property Rights and Patents
Patents and other proprietary intellectual rights are crucial in our business and industry, and establishing and maintaining these rights are essential to justify the cost to develop and commercialize any of our product candidates and products. We have sought, and intend to continue to seek, viable and strategic intellectual property rights, including, but not limited to, patent protection for our inventions, and intend to rely upon patents, trade secrets, confidential information, know-how, trademarks, improvements in our technological innovations and licensing opportunities to develop and maintain a competitive advantage for our products and product candidates. In order to protect our intellectual property rights, we typically require employees, consultants, collaborators, advisors, potential partners, service providers and contractors to enter into confidentiality agreements with us, generally stating that they will not disclose our confidential information to third parties for a certain period of time, and will otherwise not use our confidential information for anyone’s benefit but ours.
The patent positions of biotechnology and pharmaceutical companies are highly uncertain and involve complex legal and factual questions. Therefore, the patentability of subject matter we claim in our patent applications, the breadth of the claims ultimately granted, or their enforceability cannot be predicted. For this reason, we may not have or be able to obtain or maintain worldwide patent protection for any or all of our products and product candidates, and our intellectual property rights may not be protected or legally enforceable in all countries throughout the world. In some cases we may rely upon data exclusivity or similar exclusivities, although there is no guarantee that such exclusivity will be available or obtained in any jurisdiction. Further, as the publication of discoveries in the scientific and/or patent literature often lags behind the actual discoveries, we cannot be certain that we or our licensors were the first to make the inventions described in our patent applications or that we or our licensors were the first to file patent applications for such inventions.
Pursuant to the terms of the Uruguay Round Agreements Act, patents filed on or after June 8, 1995 in the U. S. have a term of 20 years from the date of filing, regardless of the period of time it may take for the patent to ultimately issue. This may shorten the period of patent protection afforded to our products as patent applications in the biopharmaceutical sector often take considerable time to issue. Under the Drug Price Competition and Patent Term Restoration Act of 1984, a sponsor may obtain marketing exclusivity for a period of time following FDA approval of certain drug applications, regardless of patent status, if the drug is a new chemical entity or if new clinical studies were used to support the marketing application for the drug.
Zanamivir, a neuraminidase inhibitor approved for the treatment and prevention of influenza A and B, is marketed worldwide as Relenza® by GSK. Most of our Relenza® patents have expired and the only substantial remaining intellectual property related to the Relenza® patent portfolio is scheduled to expire in July 2019 in Japan. On May 12, 2015, we filed a request for rehearing with the U.S. Patent and Trademark Office, Patent Trial and Appeal Board (“PTAB”) in relation to the pending patent application No. 08/737,141 related to Relenza IP in the U.S. On June 23, 2015 the PTAB denied our request for a rehearing. We reported on September 11, 2015, that we have filed an another appeal in relation to the pending patent application No. 08/737,141 related to Relenza® to the United States Court of Appeals for the Federal Circuit. While we cannot determine the duration or the outcome of this appeal process, or how long this patent application will remain pending, however we do believe that if this most recent appeal is unsuccessful, it is unlikely that the patent claims will be ever issued and we will receive no further royalties. If the patent claims are ultimately issued, we would be eligible to receive royalties from net sales of Relenza® in the U.S. for an additional 17 years from the date of allowance.
LANI, a long acting NI for the treatment and prevention of influenza A and B, is currently marketed as Inavir® in Japan by Daiichi-Sankyo. The patent relating to the structure of LANI expires in 2017 in the U.S., the EU and Japan. The patent relating to hydrates and the crystalline form of LANI actually used in the product expires in 2021 (not including extensions) in the U.S. and EU and in 2024 in Japan. In February 2015, a patent containing claims relevant to the manufacture of Inavir® was issued in Japan and expires in December 2029. The dry-powder inhaler device patent portfolio, which includes TwinCaps®, is owned by Hovione International Limited (“Hovione”) and is exclusively licensed to us and Daiichi Sankyo worldwide for the prevention and treatment of influenza and other influenza-like viral infections. These patents will expire in 2029 in the U.S., and in 2027 in the EU and Japan.
Vapendavir is an oral antiviral capsid binder we are developing to treat HRV infections. We exclusively own the vapendavir patent portfolio, and issued claims under this portfolio will begin to expire in some countries in December 2021, not including extensions. Claims filed in recent patent applications related to a free-base form of vapendavir, if allowed, would extend coverage until 2034, without extensions, however we cannot make any assurance that these claims will be allowed.
BTA074 is a direct-acting antiviral we are developing as a treatment for genital warts and RRP caused by HPV 6 and 11. The patent containing composition of matter claims expires in the U.S. in 2029 without extensions. Pending U.S. patent applications related to pharmaceutical compositions and methods of synthesis of BTA074 if allowed, would extend coverage until 2033, without extensions, however we cannot make any assurance that these claims will be allowed.
We also own a patent portfolio focused on developing several series of oral antivirals for RSV. Our RSV patent portfolio is comprised of a number of patent filings directed to several compound series, with the earliest projected expiries of such patents ranging from late-2024 to late-2031. Issued patent claims covering the BTA585 composition of matter will begin to expire in 2031 without extensions.
Patent Term Restoration/Extension and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval for the intended use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term, or extension, of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. Subject to certain limitations, the patent term restoration period is generally one-half the time between the effective date of an investigational new drug (“IND”) and the submission date of a new drug application (“NDA”) plus the time between the submission date of an NDA and the approval of that application, up to a total of five years. Only one patent applicable to an approved drug is eligible for the extension. The application for such extension must be submitted prior to the expiration of the patent and within 60 days of the drug’s approval. The United States Patent and Trademark Office (“USPTO”), in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, we may apply for restoration of patent term for one or more of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the Federal Drug, Food and Cosmetic Act (“FDCA”) can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA. The FDCA provides a five-year period of non-patent data exclusivity within the U.S. to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an Abbreviated New Drug Application (“ANDA”), or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness. We cannot assure you that we will be able to take advantage of either the patent term extension or marketing exclusivity provisions of this law.
Pediatric exclusivity is another type of exclusivity available in the U.S. Pediatric exclusivity, if granted, provides an additional six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or the patent term, may be granted based on the voluntary completion of a pediatric study in accordance with a FDA request for such a study. The current pediatric exclusivity provision was reauthorized in September 2007 as part of the Food and Drug Administration Amendments Act.
Licenses and Agreements
|
|
GSK |
In 1990, we entered into a royalty-bearing research and license agreement with GSK for the development and commercialization of zanamivir, a NI marketed by GSK as Relenza® to prevent and treat influenza. Under the terms of the agreement, we licensed zanamivir to GSK on an exclusive, worldwide basis and are entitled to receive royalty payments of 7% of GSK's annual net sales of Relenza® in the U.S., Europe, Japan and certain other countries and 10% in Australia, New Zealand, South Africa and Indonesia to the extent that the underlying patents in those respective countries do not expire. Most of our Relenza® patents have expired and the only substantial remaining intellectual property related to the Relenza® patent portfolio is scheduled to expire in July 2019 in Japan. GSK has recently verified that we will continue to receive royalties on the net sales of Relenza® in the U.S. beyond December 2014 to the extent that U.S. Patent Application No. 08/737,141 remains pending.
|
|
Daiichi Sankyo |
In 2003, we entered into collaboration and license agreement with Daiichi Sankyo related to the development of second generation long acting NIs, including LANI. Under the collaboration and license agreement, we and Daiichi Sankyo cross-licensed the right to develop, make, use, sell or offer for sale, or import products based on our respective intellectual property related to our long acting NIs. A primary focus of the agreement was for the parties to collectively seek third-party licensees that could develop and commercialize the related long-acting NIs on a worldwide basis. In the event that the related intellectual property was out-licensed to a third party, we would share equally with Daiichi Sankyo in any future royalties, license fees, milestones or other payments received from such a licensee. Further, although it was the intention of the parties to seek a third-party licensee or licensees worldwide, the parties retained the right to market or co-market related products in the U.S. and other markets outside of Japan, and any sales made by either party in the U.S. would result in the selling party paying the other party a royalty rate that was half of the royalty rate paid by any other third-party licensee. To date, there have been no third-party licenses granted pursuant to this agreement; therefore a royalty rate on net sales outside of Japan has not been established.
In March 2009, we entered into a commercialization agreement with Daiichi Sankyo, pursuant to which Daiichi Sankyo obtained exclusive marketing rights in Japan for long acting NI’s, including LANI, covered by the 2003 collaboration and license agreement between the parties. In consideration for these rights, Daiichi Sankyo agreed to pay us a royalty rate equal to 4% on net sales in Japan. In September 2010, LANI (Inavir®) was approved for sale by the Japanese Ministry of Health and Welfare for the treatment of influenza in adults and children. Accordingly, under this agreement, we currently receive a 4% royalty on net sales of Inavir® in Japan and are eligible to earn sales milestone payments.
|
|
Hovione |
On January 25, 2007, we and Daiichi Sankyo collectively entered into an exclusive license agreement with Hovione for the use of its proprietary dry-powder inhaler technology for prevention and treatment of influenza and other influenza-like viral infections with LANI or any other long-acting NI selected by us or Daiichi Sankyo during the term of the agreement. Under the terms of the agreement, in the event we sublicense LANI administered by the dry-powder inhaler to a third-party, we will owe Hovione a sublicense fee and a royalty on net sales. In the event we or Daiichi Sankyo commercialize LANI administered by the dry-powder inhaler outside of Japan, the terms, conditions, and any royalty rate due to Hovione have not yet been determined. The license agreement terminates with expiration of the last patent claim covering the dry-powder inhaler intellectual property used. These claims are currently scheduled to expire in 2029 in the U.S., and in 2027 in the EU and Japan.
Regulatory Matters
|
|
Overview |
The preclinical and clinical testing, manufacture, labeling, storage, distribution, promotion, sale, export, reporting and record-keeping of drug products and product candidates is subject to extensive regulation by numerous governmental authorities in the U.S., principally the FDA and corresponding state agencies, and similar regulatory authorities in other countries.
Non-compliance with applicable regulatory requirements can result in, among other things, total or partial suspension of the clinical development, manufacturing and marketing of a product or product candidate, the refusal of the FDA or similar regulatory authorities in other countries to grant marketing approval, the withdrawal of marketing approvals, fines, injunctions, seizure of products and criminal prosecution.
|
|
U.S. Regulatory Approval |
Pursuant to FDA regulations, we are required to successfully undertake a long and rigorous development process before any of our product candidates can be approved and marketed or sold in the U.S. This regulatory process typically includes the following steps:
|
● |
the successful completion of satisfactory preclinical studies under the FDA’s good laboratory practices (“GLP”) regulations; |
|
● |
the submission and acceptance of an IND that must be reviewed and accepted by the FDA and become effective before human clinical trials may begin; |
|
● |
the approval of an Institutional Review Board (“IRB”) at each site or location where we plan to conduct a clinical trial to protect the welfare and rights of human subjects in clinical trials; |
|
● |
the successful completion of a series of adequate and well-controlled human clinical trials to establish the safety, potency, efficacy and purity of any product candidate for its intended use, which conform to the FDA’s good clinical practice (“GCP”) regulations; |
|
● |
the development and demonstration of manufacturing processes that conform to FDA-mandated current Good Manufacturing Practices (“cGMPs”); and |
|
● |
the submission to, and review and approval by, the FDA of a NDA prior to any commercial sale or shipment of a product. |
Successfully completing this development process requires a substantial amount of time, risk and financial resources. We cannot assure you that this process will be completed for any of our product candidates, or will result in the granting of an approval for any of our product candidates on a timely basis, if at all, or that we will have sufficient financial resources to see the process for any of our product candidates through to completion.
|
|
Preclinical Studies |
Preclinical studies generally include laboratory, or in vitro, evaluation of a product candidate, its chemistry, formulation, stability and toxicity, as well as certain in vivo animal studies to assess its potential safety and biologic activity. We must submit the results of these preclinical studies, together with other information, including manufacturing records, analytical data and proposed clinical trial protocols, to the FDA as part of an IND, which must be reviewed by the FDA and become effective before we may begin any human clinical trials. An IND generally becomes effective approximately 30 days after receipt by the FDA, unless the FDA, within this 30-day time period, raises material concerns or questions about the intended conduct of the proposed trials and imposes what is referred to as a clinical hold or partial clinical hold. If one or more of our product candidates is placed on clinical hold, we may be required to resolve any outstanding issues to the satisfaction of the FDA before we can begin, or continue, clinical trials of such product candidates.
Certain preclinical studies must be conducted in compliance with the FDA’s GLP regulations and the U.S. Department of Agriculture’s Animal Welfare Act. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring such studies to be conducted again. Preclinical studies supportive of an IND generally take a year or more to complete, and there is no guarantee that an IND based on those studies will become effective, thus allowing human clinical testing to begin.
|
|
Clinical Trials |
The clinical trial phase of drug development occurs after a successful IND submission, and involves the activities necessary to demonstrate the safety, tolerability, biologic activity, efficacy and dosage of an investigational new drug substance in humans, as well as the ability to produce the drug substance in accordance with the FDA’s cGMP requirements. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial and the parameters to be used in assessing the safety and the activity or efficacy of the product candidate. Each clinical trial protocol must be submitted to the FDA under the IND prior to beginning the trial. Each trial, and the related clinical protocol, must be reviewed, approved and conducted under the auspices of an IRB and, with limited exceptions, requires the patient’s informed consent to participate in the trial. Sponsors, investigators, and IRBs also must satisfy extensive GCPs, including regulations and guidelines for obtaining informed consent from the study subjects, complying with the protocol and investigational plan, adequately monitoring the clinical trial, and reporting any SAEs on a timely basis.
Clinical trials to support a NDA for marketing approval are typically conducted in three sequential phases: Phase 1, 2 and 3. Data from these activities are compiled in a NDA for submission to the FDA requesting approval to market the drug. These phases may be compressed, may overlap, or may be omitted in some circumstances. The FDA may also require sponsors to conduct Phase 4 clinical trials after market approval to study certain safety issues or other patient populations.
|
● |
Phase 1: After an IND becomes effective, Phase 1 human clinical trials can begin. A product candidate is typically introduced either into healthy human subjects or in certain cases, patients with the medical condition for which the product candidate is intended to be used. Generally, the purpose of a Phase 1 trial is to assess a product candidate’s safety and the ability of the human body to tolerate it at different dose levels. Absorption, metabolism, distribution and pharmacokinetic trials are also generally performed at this stage. Phase 1 trials typically evaluate these aspects of the investigational drug in both single and multiple doses. |
|
● |
Phase 2: During Phase 2 clinical trials, a product candidate is generally studied in an exploratory trial or trials in a limited number of patients with the disease or medical condition for which it is intended to be used in order to (i) further identify any possible adverse side effects and safety risks, (ii) assess the preliminary or potential effectiveness or biologic activity of the product candidate for specific targeted diseases or medical conditions, and (iii) assess dose tolerance and determine the optimal dose for a subsequent Phase 2 or Phase 3 trial. Phase 2 trials generally involve patients who are divided into one or more groups that will get one of several dose levels of the product candidate, and a control group that is not treated with the product candidate but either receives a placebo or a drug already on the market for the same indication. Typically, two or more Phase 2 studies will be conducted for a product candidate prior to advancing to Phase 3. |
|
● |
Phase 3: If and when one or more Phase 2 trials demonstrate that a specific dose or range of doses of a product candidate is potentially effective and has an acceptable safety and tolerability profile, one or more Phase 3 trials may be undertaken to further demonstrate or confirm the clinical efficacy and safety of the investigational drug in an expanded patient population, with the goal of evaluating its overall risk-benefit relationship. Phase 3 trials are generally designed to reach a specific goal or end point, the achievement of which is intended to demonstrate the product candidate’s clinical efficacy. The successful demonstration of clinical efficacy and safety in one or more Phase 3 trials is typically a prerequisite to the filing of a NDA for a product candidate. |
The sponsor of a clinical-stage development program may request an “end-of-Phase 2 Meeting” with the FDA to assess the safety of the dose regimen to be studied in a Phase 3 clinical trial, to evaluate the planned design of a Phase 3 trial, and to identify any additional information that will be needed to support an NDA. If a Phase 3 clinical trial has been the subject of discussion at an end-of-Phase 2 Meeting, the sponsor may be eligible for a Special Protocol Assessment (“SPA”), a process by which the FDA, at the request of the sponsor, will evaluate the trial protocol and issues relating to the protocol to assess whether it is deemed to be adequate to meet the scientific and regulatory requirements identified by the sponsor. If the FDA and the sponsor reach agreement on the design and size of a Phase 3 clinical trial intended to form the primary basis of an efficacy claim in an NDA, the FDA may reduce the understanding to writing. The SPA, however, is not a guarantee of product approval by the FDA, or approval of any permissible claims about the product.
Throughout the various phases of clinical development, samples of the product candidate made in different batches are tested for stability to establish any shelf life constraints. In addition, large-scale production protocols and written standard operating procedures for each aspect of commercial manufacture and testing must be developed and validated.
Phase 1, 2, and 3 testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical development and may, at its discretion, reevaluate, alter, suspend, or terminate further evaluation or trials based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA, the sponsor, a data safety monitoring board or an IRB may suspend or terminate a clinical trial at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health or safety risk. The FDA can also request additional clinical trials be conducted as a condition to product approval or advancement to the next stage of development. Additionally, new government requirements may be established that could delay or prevent regulatory approval of product candidates under development.
Clinical trials performed outside the U.S. under an IND must meet the same requirements that apply to studies conducted in the U.S. The FDA may also accept a foreign clinical study not conducted under an IND if the study is well-designed, well-conducted, performed by qualified investigators, and conforms to the ethical principles contained in the Declaration of Helsinki, or with the laws and regulations of the country in which the research was conducted, whichever provides greater protection of the human subjects.
Certain information about clinical trials, including a description of the study, participation criteria, location of study sites, and contact information, is required to be sent to the National Institutes of Health, (“NIH”) for inclusion in a publicly-accessible database that is available at www.clinicaltrials.gov. Sponsors also are subject to certain state laws imposing requirements to make publicly available certain information on clinical trial results. In addition, the Food and Drug Administration Amendments Act of 2007 directed the FDA to issue regulations that will require sponsors to submit to the NIH the results of all controlled clinical studies, other than Phase 1 studies.
|
|
New Drug Applications (“NDA”) |
If and when we believe that all the requisite clinical trials for a product candidate have been completed with satisfactory and supporting clinical, toxicology, safety and manufacturing-related data, we must submit an NDA to the FDA in order to obtain approval for the marketing and sale of a product candidate in the U.S. Among many other items, an NDA typically includes the results of all preclinical and toxicology studies and human clinical trials and a description of the manufacturing process and quality control methods. The FDA must approve the NDA prior to the marketing and sale of the related product. The FDA may deny or reject an NDA if it believes all applicable regulatory criteria are not satisfied, or it may require additional data, including clinical, toxicology, safety or manufacturing data prior to approval. The FDA has 60 days from its receipt of an NDA to review the application to ensure that it is sufficiently complete for a substantive review before accepting it for filing. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be amended with any additional information requested. The FDA may also refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee.
An NDA can receive either standard or priority review. A product candidate representing a potentially significant improvement in the treatment, prevention or diagnosis of a life threatening or serious disease may receive a priority review. In addition, product candidates studied for their safety and effectiveness in treating serious or life-threatening illnesses that provide meaningful therapeutic benefit over existing treatments may also receive accelerated approval on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. Priority review and accelerated approval do not change the standards for approval, but may expedite the approval process.
If the results of the FDA’s evaluation of the NDA and inspection of manufacturing facilities are favorable, the FDA may issue an approval letter. An approval letter authorizes the commercial marketing of the drug with specific prescribing information for a specific indication. As a condition of NDA approval, the FDA may require post-approval testing, including Phase 4 trials, and surveillance to monitor the drug’s safety or efficacy and may impose other conditions, including labeling or distribution restrictions which can materially impact the potential market and profitability of the drug. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
If the FDA determines that it cannot approve the NDA in its present form, it generally issues what is referred to as a complete response letter. A complete response letter will describe all of the specific deficiencies that the agency has identified in an application that must be met in order to secure final approval of the NDA. If and when those conditions are met to the FDA’s satisfaction, the FDA will typically re-review the application and possibly issue an approval letter. However, even after submitting this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. It can take several years for the FDA to approve a NDA once it is submitted, and the actual time required for any product candidate to be approved may vary substantially, depending upon the nature, complexity and novelty of the product candidate.
We cannot assure you that the FDA, or any other similar regulatory authority in another country, will grant approval for any of our product candidates on a timely basis, if at all. Success in preclinical or early-stage clinical trials does not assure success in later stage clinical trials. Data obtained from preclinical and clinical activities is not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval.
|
|
Post-Approval Regulations |
If and when a product candidate receives regulatory approval to be marketed and sold, the approval is typically limited to a specific clinical indication or use. Further, even after regulatory approval is obtained, subsequent discovery of previously unknown safety problems with a product may result in restrictions on its use, or even complete withdrawal of the product from the market. Any FDA-approved products manufactured or distributed by us are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse events or experiences. Further, drug manufacturers and their subcontractors are required to register their establishments with the FDA and state agencies, and are subject to periodic inspections by the FDA and state agencies for compliance with cGMP regulations, which impose rigorous procedural and documentation requirements upon us and our contract manufacturers. We cannot be certain that we, or our present or future contract manufacturers or suppliers, will be able to comply with cGMP regulations and other FDA regulatory requirements. Failure to comply with these requirements may result in, among other things, total or partial suspension of production activities for our current and future product candidates, failure of the FDA to grant approval for the marketing of such product candidates, and withdrawal, suspension, or revocation of marketing approvals.
If the FDA approves one or more of our product candidates, we and our contract manufacturers must provide the FDA with certain updated safety, efficacy and manufacturing information. Product changes, as well as certain changes in the manufacturing process or facilities where the manufacturing occurs or other post-approval changes may necessitate additional FDA review and approval. We rely, and expect to continue to rely, on third parties for the formulation and manufacture of clinical and commercial quantities of our products. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
The labeling, advertising, promotion, marketing and distribution of an approved drug or biologic product must also comply with FDA and Federal Trade Commission, (“FTC”) requirements which include, among others, standards and regulations for direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing the company to correct deviations from regulatory standards and enforcement actions that can include seizures, fines, injunctions and criminal prosecution.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products that have been commercialized, and in some circumstances the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
From time to time, legislation is drafted and later introduced and passed that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will change or what the impact of such changes, if any, may be. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad, or the impact such changes could have on our business.
|
|
Other U.S. Health Care Laws and Compliance Requirements |
In the U.S., our activities are subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of HHS (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act (“HIPAA”) and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Under the Veterans Health Care Act, (“VHCA”), drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including the U.S. Department of Veterans Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Recent legislative changes purport to require that discounted prices be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing a product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities or register their sales representatives, as well as prohibiting pharmacies and other health care entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing, and prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
|
|
Foreign Regulation |
In addition to regulations in the U.S., we are subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates to the extent we choose to develop these product candidates or sell any products outside of the U.S. Whether or not we obtain FDA approval for a product, we must obtain similar approval by comparable regulatory authorities in foreign countries before we can commence clinical trials or the marketing of a product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
EU member states require both regulatory clearances by the national competent authority and a favorable ethics committee opinion prior to the commencement of a clinical trial. Under the EU regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all EU member states. The centralized procedure is compulsory for medicines produced by certain biotechnological processes, products with a new active substance indicated for the treatment of certain diseases and products designated as orphan medicinal products and optional for those products which are highly innovative or for which a centralized process is in the interest of patients. The decentralized procedure of approval provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state, known as the reference member state. Under the decentralized approval procedure, an applicant submits an application, or dossier, and related materials (draft summary of product characteristics, draft labeling and package leaflet) to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states.
|
|
Pharmaceutical Coverage, Pricing and Reimbursement |
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical products for which we may obtain regulatory approval to market and sell. In the U.S. and other countries, revenue from any products for which we receive regulatory approval to sell will depend considerably on the availability of reimbursement from third-party payers. Third-party payers include government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payer will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payer will pay for the product. Third-party payers may limit coverage to specific products on an approved list, or formulary, which might not include all of the FDA-approved products for a particular indication. Third-party payers are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, which would be in addition to the costs required to obtain FDA approvals. Our products may not be considered medically necessary or cost-effective. A payer’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in developing a product.
In 2003, the U.S. government enacted legislation providing a prescription drug benefit for Medicare recipients, which became effective at the beginning of 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through prescription drug plans operating pursuant to this legislation. These plans will likely negotiate discounted prices for our products. In March 2010, the Patient Protection and Affordable Care Act became law in the U.S., which substantially changed the way healthcare is financed by both governmental and private insurers. We anticipate that this legislation will result in additional downward pressure on the price, if any, that we may receive for any approved product. Federal, state and local governments in the U.S. continue to consider legislation to limit the growth of health care costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceutical products, including the product candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the EU, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of our particular drug products to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any products for which we receive regulatory approval to sell may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the U.S. has increased, and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Employees
As of June 30, 2015, we had 19 full-time employee equivalents, eleven of whom were engaged in research and development, and eight of whom were engaged in corporate, administration, finance, and business development activities. All of our employees have entered into non-disclosure agreements with us regarding our intellectual property, trade secrets and other confidential information. None of our employees are represented by a labor union or covered by a collective bargaining agreement, nor have we experienced any work stoppages. We believe that we maintain satisfactory relations with our employees.
Available Information
Our website address is www.biotapharma.com. Please note that this website address is provided as an inactive textual reference only. We make available free of charge through our website our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. The information provided on our website is not part of this report, and is therefore not incorporated by reference unless such information is otherwise specifically referenced elsewhere in this report.
|
ITEM 1A. |
RISK FACTORS |
You should carefully consider the following discussion of risks, together with the other information contained in this Form 10-K. The occurrence of any of the following risks could materially harm our business, our financial condition, our ability to raise additional capital in the future, or ever become profitable. In that event, the market price per share of our common stock could decline and you could lose a portion or all of your investment in our common stock.
RISKS RELATED TO THE DEVELOPMENT OF OUR PRODUCT CANDIDATES
Our success depends largely upon our ability to advance our product candidates through the various stages of drug development. If we are unable to successfully advance or develop our product candidates, our business will be materially harmed.
Even though we generate royalty revenue from our two commercialized influenza products, all of our remaining product candidates are in early stages of development and their commercial viability remains subject to the successful outcome of current and future preclinical studies, clinical trials, manufacturing processes, regulatory approvals and the risks generally inherent in the development of pharmaceutical product candidates. Failure to advance the development of one or more of our product candidates may have a material adverse effect on our business. The long-term success of our business ultimately depends upon our ability to advance the development of our product candidates through preclinical studies and clinical trials, appropriately formulate and consistently manufacture them in accordance with strict specifications and regulations, obtain approval of our product candidates for sale by the FDA or similar regulatory authorities in other countries, and ultimately have our product candidates successfully commercialized by us or a strategic partner or licensee. We cannot assure you that the results of our ongoing or future research, preclinical studies or clinical trials will support or justify the continued development of our product candidates, or that we will ultimately receive approval from the FDA, or similar regulatory authorities in other countries, to advance the development of our product candidates.
Our product candidates must satisfy rigorous regulatory standards of safety, efficacy and manufacturing before we can advance or complete their development and before they can be approved for sale by the FDA or similar regulatory authorities in other countries. To satisfy these standards, we must engage in expensive and lengthy studies and clinical trials, develop acceptable and cost effective manufacturing processes, and obtain regulatory approval of our product candidates. Despite these efforts, our product candidates may not:
|
● |
demonstrate clinically meaningful therapeutic or other medical benefits as compared to a patient receiving no treatment or over existing drugs or other product candidates in development to treat the same patient population; |
|
● |
be shown to be safe and effective in current and future preclinical studies or clinical trials; |
|
● |
have the desired therapeutic or medical effects; |
|
● |
be tolerable or free from undesirable or unexpected side effects; |
|
● |
meet applicable regulatory standards; |
|
● |
be capable of being appropriately formulated and manufactured in commercially suitable quantities or scale and at an acceptable cost; or |
|
● |
be successfully commercialized by us or by our licensees or collaborators. |
Even if we demonstrate favorable results in preclinical studies and early-stage clinical trials, we cannot assure you that the results of late-stage clinical trials will be sufficient to support the continued development of our product candidates. Many, if not most companies in the pharmaceutical and biopharmaceutical industries have experienced significant delays, setbacks and failures in all stages of development, including late-stage clinical trials, even after achieving promising results in preclinical testing or early-stage clinical trials. Accordingly, results from completed preclinical studies and early-stage clinical trials of our product candidates may not be predictive of the results we may obtain in future late-stage trials. Furthermore, even if the data collected from preclinical studies and clinical trials involving any of our product candidates demonstrate a satisfactory safety, tolerability and efficacy profile, such results may not be sufficient to obtain regulatory approval from the FDA in the U.S., or other similar regulatory agencies in other jurisdictions, which is required to market and sell the product.
Clinical trials are risky, lengthy and expensive. We incur substantial expense for, and devote significant time and resources to, preclinical testing and clinical trials, yet cannot be certain that these tests and trials will demonstrate that a product candidate is effective and well tolerated, or will ever support its approval and commercial sale. For example, clinical trials require adequate supplies of clinical trial material and sufficient patient enrollment to power the study. Delays in patient enrollment can result in increased costs and longer development times. Even if we, or a licensee or collaborator, if applicable, successfully complete clinical trials for our product candidates, we or they might not file the required regulatory submissions in a timely manner and may not receive marketing approval for the product candidate. We cannot assure you that any of our product candidates will successfully progress further through the drug development process, or ultimately will result in an approved and commercially viable product.
If the actual or perceived therapeutic benefits, or the safety or tolerability profile of any of our product candidates are not equal to or superior to other competing treatments approved for sale or in clinical development, we may terminate the development of any of our product candidates at any time, and our business prospects and potential profitability could be harmed.
We are aware of a number of companies marketing or developing various classes of anti-infective product candidates or products for the treatment of patients infected with HRV, RSV, HPV and influenza that are either approved for sale or further advanced in clinical development than ours, such that their time to approval and commercialization may be shorter than that for our product candidates.
Currently, there are no approved direct-acting antiviral drugs to treat HRV infections. However, if ever approved, our vapendavir product candidate would indirectly compete with drugs approved to reduce the incidence of exacerbations or improve lung function in patients with asthma and COPD, such as fluticasone propionate (Advair®), tiotoprium bromide (Spiriva®), fluticasone furoate/vilanterol (Breo Ellipta®), and roflumilast (Daliresp®). In addition to these approved drugs, there are compounds at the clinical development stage, such as inhaled ß-interferon, that if successfully developed for the treatment of HRV infections could compete with vapendavir in the future.
Currently, there is no HPV-specific direct acting antiviral drugs available to treat genital warts or RRP. Treatments for genital warts can be divided broadly into two categories: provider-administered ablative/cytodestructive therapies (including cryotherapy, laser ablation, and trichloroacetic acid) and patient-administered topical therapies such as podophyllotoxin (Condylox; Wartec), sinecatechins (Veregen®), and imiquimod (Zyclara®, Aldara®). There are no cures for RRP, however current maintenance treatments include CO2 laser surgery, pulse dye laser surgery, endoscopic microdebriders, and intralesional injection with cidofovir. The high reoccurrence rates make the current therapies less than optimal for patients suffering with genital warts or RRP. We anticipate that BTA074, if successfully developed and commercialized, would directly compete with the patient-applied topical treatments for condylomaand could become first-line therapy for the treatment of RRP. We believe key differentiating features of BTA074 could be its mechanism of action, favorable local skin tolerability, efficacy, and a lower reoccurrence rate. Three prophylactic vaccines, primarily designed to prevent cervical, vulvar, vaginal, and anal cancers, are currently marketed: a bivalent HPV16/18 vaccine (Cervarix®; GSK), quadrivalent HPV16/18/6/11 (Gardasil®, Merck) and the 9-valent HPV 6/11/16/18/33/52/58 (Gardasil®9; Merck). Gardasil®9 is indicated for females aged 9 through 26 and males aged 9 through 15, to prevent various HPV related cancers and genital warts in both sexes. Gardasil®, Gardasil®9, and Cervarix® are not known to exhibit a therapeutic effect on existing HPV lesions.
Effective treatments of RSV infections in pediatrics, the elderly, and the immunocompromised are very limited. Currently, only Virazole® (ribavirin) is indicated for the treatment of hospitalized infants and young children with severe lower respiratory tract infections due to RSV. We are aware that the following compounds are under development: Gilead’s GS-5806, Johnson and Johnson’s JJ-53718678 (ALS-8176), Ark Biosciences’ AK0529 and Teva Pharmaceutical’s MDT-637. The only approved drug for the prevention of RSV infections in high risk infants is MedImmune’s palivizumab (Synagis®), monoclonal antibody. There are several vaccines and antibody products designed to prevent RSV infections in clinical development. Among the clinical stage product candidates in development are Novavax’s RSV F vaccine, GSK’s GSK3003898A vaccine, GSK’s GSK3003893A vaccine, Bavarian Nordic’s BN® RSV vaccine, MedImmune’s MEDI ÄM2-2 vaccine, MedImmune’s monoclonal antibody MEDI8897, and Regeneron’s monoclonal antibody REGN2222C.
The pharmaceutical market for products that prevent or treat influenza is competitive. Key competitive advantages for LANI may include its single administration treatment regimen, its antiviral resistance profile, and to-date, its reported safety profile. A number of NIs are currently available in the U.S. and/or other counties, including Japan, for the prevention and/or treatment of influenza. These include oseltamivir phosphate from Roche Ltd., which is marketed as Tamiflu®, zanamivir from GSK, which is marketed as Relenza®, LANI from Daiichi Sankyo, which is marketed as Inavir®, and peramivir marketed as Rapivab® or Rapiacta® by CSL Limited and Shionogi & Co. Ltd., respectively. We anticipate that LANI, if ever successfully developed and approved, would compete directly or indirectly with drugs that will be “generic” by the time LANI may be approved for sale. Generic drugs are drugs whose patent protection has expired, which generally have an average selling price substantially lower than drugs protected by patents and intellectual property rights. Unless a patented drug can sufficiently differentiate itself from a competing generic drug in a meaningful manner, the existence of generic competition in any indication will generally impose significant pricing pressure on competing patented drugs.
If at any time we believe that any of our product candidates may not provide meaningful or differentiated therapeutic benefits, perceived or real, equal to or better than our competitor’s products or product candidates, or we believe our product candidates may not have as favorable a safety or tolerability profile as potentially competitive compounds, we may delay or terminate the future development of any of our product candidates. We cannot provide any assurance that the future development of any or our product candidates will demonstrate any meaningful therapeutic benefits over potentially competitive compounds currently approved for sale or in development, or an acceptable safety or tolerability profile sufficient to justify its continued development.
We plan to conduct a Phase 2 clinical trial for BTA074 in Argentina and possibly in Chile for the treatment of genital warts, the treatment period of which will likely extend beyond that evaluated in preclinical studies. If Argentine National Administration of Drugs, Foods and Medical Devices (“ANMAT”) and/or the Chilean regulatory agency were to delay, request a change of scope, or institute a clinical hold for our planned Phase 2 trial, the trail could be substantially delayed and our business could be materially harmed.
We intend to evaluate BTA074 for up to 16 weeks in our planned Phase 2 trial, which we anticipate will begin in the fourth quarter of 2015. Sixteen (16) week preclinical toxicology studies have not yet been completed for BTA074. Typically the duration of preclinical studies match the duration of planned human clinical studies. Our currently planned Phase 2 trial for BTA074 has been approved by ANMAT, however if we were unable complete the planned Phase 2 trial in Argentina or Chile, we will likely have a significant delay in the development of BTA074 due to the time required to complete the 16-week toxicology studies and potentially conducting the Phase 2 trial in other countries. The time to complete and the additional costs associated with these activities could significantly harm our business and the value of BTA074.
Our product candidates may exhibit undesirable side effects when used alone or in combination with other approved pharmaceutical products, which may delay or preclude their development or regulatory approval, or limit their use if ever approved.
Throughout the drug development process, we must continually demonstrate the activity, safety and tolerability of our product candidates in order to obtain regulatory approval to further advance their clinical development, or to eventually market them. Even if our product candidates demonstrate adequate biologic activity and clear clinical benefit, any unacceptable side effects or adverse events, when administered alone or in the presence of other pharmaceutical products, may outweigh these potential benefits. We may observe adverse or serious adverse events or drug-drug interactions in preclinical studies or clinical trials of our product candidates, which could result in the delay or termination of their development, prevent regulatory approval, or limit their market acceptance if they are ultimately approved.
If the results from preclinical studies or clinical trials of our product candidates, including those that are subject to existing or future license or collaboration agreements, are unfavorable, we could be delayed or precluded from the further development or commercialization of our product candidates, which could materially harm our business.
In order to further advance the development of, and ultimately receive marketing approval to sell our product candidates, we must conduct extensive preclinical studies and clinical trials to demonstrate their safety and efficacy to the satisfaction of the FDA or similar regulatory authorities in other countries, as the case may be. Preclinical studies and clinical trials are expensive, complex, can take many years to complete, and have highly uncertain outcomes. Delays, setbacks, or failures can and do occur at any time, and in any phase of preclinical or clinical testing, and can result from concerns about safety, tolerability, toxicity, a lack of demonstrated biologic activity or improved efficacy over similar products that have been approved for sale or are in more advanced stages of development, poor study or trial design, and issues related to the formulation or manufacturing process of the materials used to conduct the trials. The results of prior preclinical studies or early-stage clinical trials are not predictive of the results we may observe in late-stage clinical trials. In many cases, product candidates in clinical development may fail to show the desired tolerability, safety and efficacy characteristics, despite having favorably demonstrated such characteristics in preclinical studies or early-stage clinical trials.
In addition, we may experience numerous unforeseen events during, or as a result of, preclinical studies and the clinical trial process, which could delay or impede our ability to advance the development of, receive marketing approval for, or commercialize our product candidates, including, but not limited to:
|
● |
communications with the FDA, or similar regulatory authorities in different countries, regarding the scope or design of a trial or trials, or placing the development of a product candidate on clinical hold or delaying the next phase of development until questions or issues are satisfactorily resolved, including preforming additional studies to answer their queries; |
|
● |
regulatory authorities or IRB’s not authorizing us to commence or conduct a clinical trial at a prospective trial site; |
|
● |
enrollment in our clinical trials being delayed, or proceeding at a slower pace than we expected, because we have difficulty recruiting participants or participants drop out of our clinical trials at a higher rate than we anticipated; |
|
● |
our third-party contractors, upon whom we rely to conduct preclinical studies, clinical trials and the manufacturing of our clinical trial materials, failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
|
● |
having to suspend or ultimately terminate a clinical trial if participants are being exposed to unacceptable health or safety risks; |
|
● |
regulatory authorities or IRBs requiring that we hold, suspend or terminate our preclinical studies and clinical trials for various reasons, including non-compliance with regulatory requirements; and |
|
● |
the supply or quality of material necessary to conduct our preclinical studies or clinical trials being insufficient, inadequate or unavailable. |
Even if the data collected from preclinical studies or clinical trials involving our product candidates demonstrate a satisfactory tolerability, safety and efficacy profile, such results may not be sufficient to support the submission of an NDA to obtain regulatory approval from the FDA in the U.S., or other similar regulatory authorities in other foreign jurisdictions, which is required for us to market and sell our product candidates.
Most of our product candidates are generally being developed to treat seasonal respiratory infections, which could cause their clinical development to be more complex, take longer and cost more to complete than product candidates intended for non-seasonal infections.
HRV, RSV and influenza are respiratory infections that generally occur more frequently in certain months of the year in a particular geography. Accordingly, it is more efficient to conduct clinical trials in patients with these respiratory infections during the months in which the infections are more prevalent, and these trials generally cannot be as efficiently conducted year-round in any one region of the world. The seasonality in the incidence of these respiratory infections may require us to conduct additional clinical trials in both the northern and southern hemispheres in order to fully enroll these trials on a timely basis. Seasonality or variability in the incidence of these infections increases the complexity of our trial designs, exposes us to additional regulatory oversight in more countries, and generally increases the cost and time to conduct these trials.
If third-party contract manufacturers, upon whom we rely to formulate and manufacture our product candidates, do not perform, fail to manufacture according to our specifications, or fail to comply with strict government regulations, our preclinical studies or clinical trials could be adversely affected and the development of our product candidates could be delayed or terminated, or we could incur significant additional expenses.
We do not currently own any manufacturing facilities. We have historically used third-party contract manufacturers and we intend to continue to rely on third-party contractors for the foreseeable future, to formulate, manufacture, fill and package our product candidates. Our reliance on these third-party contract manufacturers, which in some cases are sole sourced, exposes us to a number of risks, any of which could delay or prevent the completion of our preclinical studies or clinical trials, or the regulatory approval or commercialization of our product candidates, result in higher costs or deprive us of potential product revenues in the future. Some of these risks include, but are not limited to:
|
● |
our contract manufacturers failing to develop an acceptable formulation to support late-stage clinical trials for, or the commercialization of, our product candidates; |
|
● |
our contract manufacturers failing to manufacture our product candidates according to their own standards, our specifications, cGMPs or regulatory guidelines, or otherwise manufacturing material that we or regulatory authorities deem to be unsuitable for our clinical trials or commercial use; |
|
● |
our contract manufacturers being unable to increase the scale of or the capacity for, or reformulate the form of our product candidates, which may cause us to experience a shortage in supply, or cause the cost to manufacture our product candidates to increase. We cannot assure you that our contract manufacturers will be able to manufacture our product candidates at a suitable commercial scale, or that we will be able to find alternative manufacturers acceptable to us that can do so; |
|
● |
our contract manufacturers placing a priority on the manufacture of other customers’ or their own products, rather than ours; |
|
● |
our contract manufacturers failing to perform as agreed or exiting from the contract manufacturing business; and |
|
● |
our contract manufacturers’ plants being closed as a result of regulatory sanctions or a natural disaster. |
Manufacturers of pharmaceutical drug products are subject to ongoing periodic inspections by the FDA, the U.S. Drug Enforcement Administration (“DEA”) and corresponding state and other foreign agencies to ensure strict compliance with FDA-mandated cGMPs, other government regulations and corresponding foreign standards. We do not have control over our third-party contract manufacturers’ compliance with these regulations and standards and accordingly, failure by our third-party manufacturers, or us, to comply with applicable regulations could result in sanctions being imposed on us or the manufacturer, which could significantly and adversely affect our business.
In the event that we need to change our third-party contract manufacturers, our preclinical studies, our clinical trials or the commercialization of our product candidates could be delayed, adversely affected or terminated, or such a change may result in the need for us to incur significantly higher costs, which could materially harm our business.
Due to various regulatory restrictions in the U.S. and many other countries, as well as potential capacity constraints on manufacturing that occur from time-to-time in our industry, various steps in the manufacture of our product candidates are sole-sourced to certain contract manufacturers. In accordance with cGMPs, changing manufacturers may require the re-validation of manufacturing processes and procedures, and may require further preclinical studies or clinical trials to show comparability between the materials produced by different manufacturers. Changing our current or future contract manufacturers may be difficult, if not impossible for us, and could be extremely costly if we do make such a change, which could result in our inability to manufacture our product candidates for an extended period of time and a delay in the development of our product candidates. Further, in order to maintain our development timelines in the event of a change in a third-party contract manufacturer, we may incur significantly higher costs to manufacture our product candidates.
If third-party vendors, upon whom we rely to conduct our preclinical studies or clinical trials, do not perform or fail to comply with strict regulations, these studies or trials may be delayed, terminated, or fail, or we could incur significant additional expenses, which could materially harm our business.
We have limited resources dedicated to designing, conducting and managing our preclinical studies and clinical trials. We have historically relied on, and intend to continue to rely on, third parties, including clinical research organizations, consultants and principal investigators, to assist us in designing, managing, conducting, monitoring and analyzing the data from our preclinical studies and clinical trials. We rely on these vendors and individuals to perform many facets of the clinical development process on our behalf, including conducting preclinical studies, the recruitment of sites and patients for participation in our clinical trials, maintenance of good relations with the clinical sites, and ensuring that these sites are conducting our trials in compliance with the trial protocol and applicable regulations. If these third parties fail to perform satisfactorily, or do not adequately fulfill their obligations under the terms of our agreements with them, we may not be able to enter into alternative arrangements without undue delay or additional expenditures, and therefore the preclinical studies and clinical trials of our product candidates may be delayed or prove unsuccessful.
Further, the FDA, or similar regulatory authorities in other countries, may inspect some of the clinical sites participating in our clinical trials or our third-party vendors’ sites to determine if our clinical trials are being conducted according to GCP or similar regulations. If we or a regulatory authority determine that our third-party vendors are not in compliance with, or have not conducted our clinical trials according to applicable regulations, we may be forced to exclude certain data from the results of the trial, or delay, repeat or terminate such clinical trials.
We have limited capacity for managing clinical trials, which could delay or impair our ability to initiate or complete clinical trials of our product candidates on a timely basis and materially harm our business.
We have limited capacity to recruit and manage all of the clinical trials necessary to obtain approval for our product candidates by the FDA or similar regulatory authorities in other countries. By contrast, larger pharmaceutical and biopharmaceutical companies often have substantial staff or departments with extensive experience in conducting clinical trials with multiple product candidates across multiple indications and obtaining regulatory approval in various countries. In addition, these companies may have greater financial resources to compete for the same clinical investigators, sites and patients that we are attempting to recruit for our clinical trials. As a result, we may be at a competitive disadvantage that could delay the initiation, recruitment, timing and completion of our clinical trials and obtaining of marketing approvals, if achieved at all, for our product candidates.
If we are unable to attract or retain key employees, advisors or consultants, we may be unable to successfully develop our product candidates in a timely manner, if at all, or otherwise manage our business effectively.
We have increasingly adopted an operating model that relies on the outsourcing of a number of key responsibilities and activities to third-party vendors, such as contract research and manufacturing organizations, in order to advance the development of our product candidates. Therefore, our success depends in part on our ability to retain highly qualified key management, personnel to develop, implement and execute our business strategy and operations, and oversee the activities of our vendors, as well as any academic and corporate advisors or consultants that may assist us in this regard. We are currently highly dependent upon the efforts of our small management team to accomplish this. In order to advance the development of our product candidates, we need to retain and be able to recruit certain key personnel, consultants or advisors with experience in a number of disciplines, including but not limited to, research and development, product development, clinical trials, medical affairs, government regulation approval of pharmaceutical products, quality control and assurance, formulation and manufacturing, business development, accounting, finance, human resources and information systems. We may not be able to continue to do so in the future on acceptable terms, if at all. If we lose any key personnel, or are unable to retain qualified key personnel, directors, advisors or consultants, the development of our product candidates could be delayed or terminated and our business may be harmed.
Our Phase 2 IGLOO clinical trial of LANI did not achieve its primary efficacy endpoint, coupled with the termination of our contract with BARDA and the inability to reach a revised equitable agreement with Daiichi Sankyo and Hovione with respect to our commercial rights for LANI outside of Japan, has impaired the value of this program and our ability to further develop or commercialize LANI, or license it to a third-party, which could materially harm our financial condition, prospects and business.
On May 7, 2014 HHS/ASPR/BARDA notified us of its decision to terminate the contract for the convenience of the U.S. Government for the development of LANI. On August 1, 2014, we announced top-line data from the Phase 2 IGLOO trial. As compared to placebo, neither the 40 mg or 80 mg cohort achieved a statistically significant reduction in the median time to alleviation of influenza symptoms as measured by the Flu-iiQ patient-recorded outcome questionnaire, which was the primary endpoint of the study. Pursuant to our collaboration and license agreement with Daiichi Sankyo, if a third-party licensee other than Biota or Daiichi Sankyo develops and commercializes LANI in territories outside Japan, we and Daiichi Sankyo will share all licensing revenue equally. The agreement does not, however, specifically address the respective rights or obligations of, or any consideration between, the parties in the event that either we or Daiichi Sankyo directly market LANI in territories outside Japan.
The failure to achieve the primary endpoint of the Phase 2 IGLOO study, the lack of sufficient capital resources to continue to independently fund the future development and commercialization of LANI and the collaboration and licensing arrangement we have with Daiichi Sankyo may have impaired the value of the program and our ability to continue the development of LANI or for us and our partner Daiichi Sankyo to find a third-party licensee or collaborator that is willing to further develop LANI. If we and Daiichi Sankyo license the rights to develop and commercialize LANI outside of Japan to a third party, we may be required to give up a significant portion of the commercial value of LANI to this third party licensee. Further, we would be required be contractually required to share such proceeds equally with Daiichi Sankyo. We cannot assure you that LANI can be developed further or otherwise monetized by us at all in the future.
Our industry is highly competitive and subject to rapid technological changes. As a result, we may be unable to compete successfully or develop innovative or differentiated products, which could harm our business.
Our industry is highly competitive and characterized by rapid technological change. Key competitive factors in our industry include, among others, the ability to successfully advance the development of a product candidate through preclinical and clinical trials; the efficacy, toxicology, tolerability, safety, resistance or cross-resistance, interaction or dosing profile of a product or product candidate; the timing and scope of marketing approvals, if ever achieved; reimbursement rates for and the average selling price of competing products and pharmaceutical products in general; the availability of raw materials and qualified contract manufacturing and manufacturing capacity to produce our product candidates; relative manufacturing costs; establishing, maintaining and protecting our intellectual property and patent rights; and sales and marketing capabilities.
Developing pharmaceutical product candidates is a highly competitive, expensive and risky activity with a long business cycle. Many organizations, including the large pharmaceutical and biopharmaceutical companies that have existing products on the market or in clinical development that may compete with our product candidates, have substantially more resources than we have, as well as much greater capabilities and experience than we have in research and discovery, designing and conducting preclinical studies and clinical trials, operating in a highly regulated environment, formulating and manufacturing drug substances, products and devices, and marketing and sales. Our competitors may be more successful than we are in obtaining regulatory approvals for their product candidates and achieving broad market acceptance once they are approved. Our competitors’ products or product candidates may be more effective, have fewer adverse effects, be more convenient to administer, have a more favorable resistance profile, or be more effectively marketed and sold than any product we, or our potential future licensees or collaborators, may develop or commercialize. New drugs or classes of drugs from competitors may render our product candidates obsolete or non-competitive before we are able to successfully develop them or, if approved, before we can recover the expenses of developing and commercializing them. We anticipate that we or our potential future licensees or collaborators will face intense and increasing competition as new drugs and drug classes enter the market and advanced technologies or new drug targets become available. If our product candidates do not demonstrate any meaningful competitive advantages over existing products, or new products or product candidates, we may terminate the development or commercialization of our product candidates at any time.
These competitors, either alone or with their collaborators, may succeed in developing product candidates or products that are more effective, safer, less expensive or easier to administer than ours. Accordingly, our competitors may succeed in obtaining regulatory approval for their product candidates more rapidly than we can. Companies that can complete clinical trials, obtain required marketing approvals and commercialize their products before their competitors do so may achieve a significant competitive advantage, including certain patent and marketing exclusivity rights that could delay the ability of competitors to market certain products.
We also face, and expect that we will continue to face, intense competition from other companies in a number of other areas, including (i) attracting larger pharmaceutical and biopharmaceutical companies to enter into collaborative arrangements with us to acquire, license or co-develop our product candidates, (ii) identifying and obtaining additional clinical-stage development programs to bolster our pipeline, (iii) attracting investigators and clinical sites capable of conducting our clinical trials, and (iv) recruiting patients to participate in our clinical trials. We cannot assure you that product candidates resulting from our research and development efforts, or from joint efforts with our potential future licensees or collaborators, will be able to compete successfully with our competitors’ existing products or product candidates in development.
We may be unable to successfully develop a product candidate that is the subject of an existing or future license agreement or collaboration if our licensee or collaborator does not perform or fulfill its contractual obligations, delays the development of our product candidate, or terminates our agreement.
We expect to continue to enter into and rely on license and collaboration agreements in the future, or other similar business arrangements with third parties, to further develop and/or commercialize some or all of our existing and future product candidates. Such licensees or collaborators may not perform as agreed upon or anticipated, may fail to comply with strict regulations, or may elect to delay or terminate their efforts in developing or commercializing our product candidates even though we have met our obligations under the arrangement.
A majority of the potential revenue from existing and any future licenses and collaborations we may enter into will likely consist of contingent milestone payments, such as payments received for achieving development or regulatory milestones, and royalties payable on the sales of approved products. Milestone and royalty revenues that we may receive under these licenses and collaborations will depend primarily upon our licensee’s or collaborator’s ability to successfully develop and commercialize our product candidates. In addition, our licensees or collaborators may decide to enter into arrangements with third parties to commercialize products developed under our existing or future collaborations using our technologies, which could reduce the milestone and royalty revenue that we may receive, if any. In many cases, we will not be directly or closely involved in the development or commercialization of our product candidates that are subject to licenses or collaborations and, accordingly, we will depend largely on our licensees or collaborators to develop or commercialize our product candidates. Our licensees or collaborators may fail to develop or effectively commercialize our product candidates because they:
|
● |
do not allocate the necessary resources due to internal constraints, such as limited personnel with the requisite scientific expertise, limited capital resources, or the belief that other product candidates or internal programs may have a higher likelihood of obtaining regulatory approval, or may potentially generate a greater return on investment; |
|
● |
do not have sufficient resources necessary to fully support the product candidate through clinical development, regulatory approval and commercialization; |
|
● |
are unable to obtain the necessary regulatory approvals; or |
|
● |
prioritize other programs or otherwise diminish their support for developing and/or marketing our product candidate or product due to a change in management, business operations or strategy. |
Should any of these events occur, we may not realize the full potential or intended benefit of our license or collaboration arrangements, and our results of operations may be adversely affected. In addition, a licensee or collaborator may decide to pursue the development of a competitive product candidate developed outside of our agreement with them. Conflicts may also arise if there is a dispute about the progress of, or other activities related to, the clinical development or commercialization of a product candidate, the achievement and payment of a milestone amount, the ownership of intellectual property that is developed during the course of the arrangement, or other license agreement terms. If a licensee or collaborator fails to develop or effectively commercialize our product candidates for any of these reasons, we may not be able to replace them with another third-party willing to develop and commercialize our product candidates under similar terms, if at all. Similarly, we may disagree with a licensee or collaborator as to which party owns newly or jointly-developed intellectual property. Should an agreement be revised or terminated as a result of a dispute and before we have realized the anticipated benefits of the arrangement, we may not be able to obtain certain development support or revenues that we anticipated receiving. We may also be unable to obtain, on terms acceptable to us, a license from such collaboration partner to any of its intellectual property that may be necessary or useful for us to continue to develop and commercialize the product candidate. We cannot assure you that any product candidates will emerge from any existing or future license or collaboration agreements we may enter into for any of our product candidates.
We may be unable to successfully integrate the operations of Anaconda Pharma, which could increase our cost of doing business or harm our operations.
We previously reported the closing of the Anaconda Pharma acquisition in June 2015. Anaconda Pharma was a privately-held biotechnology company whose lead candidate was BTA074, a patented, direct-acting antiviral with activity against HPV types 6 and 11. We may incur additional costs and consume a significant amount of management’s time to integrate operations, including the development plans for BTA074 and the financial, information and legal records, in a timely manner, if at all, which could result in us incurring additional costs. Further we may be exposed to additional claims or unrecorded liabilities in the future for acts preceding the acquisition. The agreement calls for the escrow of shares for a one year period to cover any unknown claims or liabilities. If a third party claim or liability arise that exceeds the escrow amount or becomes known after the one year escrow period we may be liable for these prior acts, which would harm our business. Additionally, in the event we are liable for a claim, or liability that requires the payment of cash, we may have to use our own cash to pay such claim or liability, which would deplete our cash resources, or sell our common stock to raise the funds to pay the claim or liability.
Our ability to conduct research and discovery activities to support our current antiviral pipeline or the discovery of future product candidates may be impaired as a result of our decision to close our research facility in Melbourne Australia, which could result in increased costs and time to outsource these activities and potentially impair our ability to supplement our pipeline and harm our business prospects.
Our basic research and discovery activities, including medicinal chemistry, virology, and cell culture assays have historically been conducted internally by our research staff in in our laboratory facility in Melbourne, Australia. Upon the closing in March 2015 we now use third party research firms and consultants to conduct these activities under our management. We currently do not have any plans to build laboratory facilities or hire significant staff to conduct research, discovery and certain development activities. To the extent we continue these activities in the near-future, we anticipate that we will outsource them and rely more on third-party research firms and consultants, which may require us to incur higher costs and the lengthen the time it takes to conduct activities. There can be no assurance that our research efforts will be rewarded and that will continue to develop future preclinical candidates, which ultimately may harm our business.
RISKS RELATED TO COMMERCIAL MATTERS
We have a history of incurring net losses and we may never achieve profitability.
We have a history of incurring net losses, some of which have been significant. We expect to incur additional net losses in the near-term, and these losses would likely increase as our research and development efforts progress to later stage activities. To become profitable, we, or our licensees or collaborators if applicable, must successfully manufacture and develop product candidates, receive regulatory approval, successfully commercialize and/or enter into profitable agreements with other parties and maintain existing and/or obtain additional intellectual property rights. It could be several years, if ever, before we receive significant revenues from any future license agreements or revenues directly from the sale of any of our product candidates.
Royalty revenues from Relenza® and Inavir® are unpredictable and subject to the seasonal incidence and severity of influenza, which could adversely affect our results of operations and financial condition.
We currently earn royalty revenue from the net sales of Relenza® and Inavir®, which are marketed by our licensees. Although the royalty rates paid to us by our licensees are fixed at a proportion of our licensees’ net sales of these products, our periodic and annual revenues from these royalties have historically been variable and subject to fluctuation based on the seasonal incidence and severity of influenza. In addition, returns of products to our licensees that were sold in prior years are taken into account in the calculation of net sales for purposes of determining the royalty revenue we receive and the amount of such returns are generally unpredictable. We cannot predict with any certainty what our royalty revenues are likely to be in any given year. Further, most of our Relenza® patents have expired and the only substantial remaining intellectual property related to the Relenza® patent portfolio, which is solely owned by us and exclusively licensed to GSK, is scheduled to expire in July 2019 in Japan. GSK has verified that we will continue to receive royalties on the net sales of Relenza® in the U.S. beyond December 2014 to the extent that U.S. Patent Application No. 08/737,141 remains pending.
If safety, tolerability, resistance, drug-drug interactions, or efficacy concerns should arise with Relenza® or Inavir®, our future royalty revenue may be reduced, which would adversely affect our financial condition and business.
We currently earn royalty revenue from Relenza® and Inavir®, which are marketed by our licensees. Data supporting the marketing approvals and forming the basis for the safety warnings in the product labels for these products were obtained in controlled clinical trials of limited duration in limited patient populations and, in some cases, from post-approval use. As these marketed products are used over longer periods of time and by more patients, some with underlying health problems or taking other medicines, new issues such as safety, tolerability, resistance or drug-drug interaction issues could arise, which may require our licensees to provide additional warnings or contraindications on their product labels, or otherwise narrow the approved indications. Further, additional information from ongoing research or clinical trials of these products that raise any doubts or concerns about their efficacy may arise. If serious safety, tolerability, resistance, drug-drug interaction, efficacy, or any other concerns or issues arise with respect to these marketed products, sales of these products could be impaired, limited or abandoned by our licensees or by regulatory authorities, in which case our royalty revenue would decrease.
If government and third-party payers fail to provide adequate reimbursement or coverage for our products or those that are developed through licenses or collaborations, our revenues and potential for profitability may be harmed.
In the U.S. and most foreign markets, product revenues or related royalty revenue, and therefore the inherent value of our products, will depend largely upon the reimbursement rates established by third-party payers for such products. Third-party payers include government health administration authorities, managed-care organizations, private health insurers and other similar organizations. Third-party payers are increasingly examining the cost effectiveness of medical products, services and pharmaceutical drugs and challenging the price of these products and services. In addition, significant uncertainty exists as to the reimbursement status, if any, of newly approved pharmaceutical products. Further, the comparative effectiveness of new products over existing therapies and the assessment of other non-clinical outcomes are increasingly being considered in the decision by payers to establish reimbursement rates. We, or our licensees or collaborators if applicable, may also be required to conduct post-marketing clinical trials in order to demonstrate the cost-effectiveness of our products. Such studies may require us to commit a significant amount of management time and financial resources. We cannot assure you that any products we or our licensees or collaborators may successfully develop will be reimbursed in part, or at all, by any third-party payers in any country.
Many governments continue to propose legislation designed to expand the coverage, yet reduce the cost, of healthcare, including pharmaceutical products. In many foreign markets, governmental agencies control the pricing of prescription drugs. In the U.S., significant changes in federal health care policy were approved over the past several years and continue to evolve, and will likely result in reduced reimbursement rates for many pharmaceutical products in the future. We expect that there will continue to be federal and state proposals to implement increased government control over reimbursement rates of pharmaceutical products. In addition, we expect that increasing emphasis on managed care and government intervention in the U.S. healthcare system will continue to put downward pressure on the pricing of pharmaceutical products there. Government cost control initiatives could decrease the price that we or our licensees or collaborators may receive for any of our products that may be approved for sale in the future, which would limit our revenues and profitability. Legislation and regulations affecting the pricing of pharmaceutical products may change before our product candidates are approved for sale, which could further limit or eliminate their reimbursement rates. Further, social and patient activist groups, whose goal it is to reduce the cost of healthcare, and in particular the price of pharmaceutical products, may also place downward pressure on the price of these products, which could result in decreased prices of our products.
If any product candidates that we develop independently, or through licensees or collaborators if applicable, are approved but do not gain meaningful acceptance in their intended markets, we are not likely to generate significant revenues.
Even if our product candidates are successfully developed and we or a licensee or collaborator obtain the requisite regulatory approvals to market them in the future, they may not gain market acceptance or broad utilization among physicians, patients or third-party payers. The degree of market acceptance that any of our products may achieve will depend on a number of factors, including:
|
● |
the efficacy or perceived clinical benefit of the product, if any, relative to existing therapies; |
|
● |
the timing of market approval and the existing market for competitive drugs, including the presence of generic drugs; |
|
● |
the level of reimbursement provided by third-party payers to cover the cost of the product to patients; |
|
● |
the net cost of the product to the user or third-party payer; |
|
● |
the convenience and ease of administration of the product; |
|
● |
the product’s potential advantages over existing or alternative therapies; |
|
● |
the actual or perceived safety of similar classes of products; |
|
● |
the actual or perceived existence, incidence and severity of adverse effects; |
|
● |
the effectiveness of sales, marketing and distribution capabilities; and |
|
● |
the scope of the product label approved by the FDA or similar regulatory agencies in other jurisdictions. |
There can be no assurance that physicians will choose to prescribe or administer our products, if approved, to the intended patient population. If our products do not achieve meaningful market acceptance, or if the market for our products proves to be smaller than anticipated, we may never generate significant revenues.
If we fail to enter into or maintain collaborations or other sales, marketing and distribution arrangements with third parties to commercialize our product candidates, or otherwise fail to establish marketing and sales capabilities in the future, we may not be able to successfully commercialize our products.
We currently have no infrastructure to support the commercialization of any of our product candidates, and have little, if any, experience in the commercialization of pharmaceutical products. Therefore, if we successfully develop any product candidate, and it is ultimately approved for sale, our future profitability will depend largely on our ability to access, arrange or develop suitable marketing and sales capabilities. We anticipate that we will need to establish relationships with other companies, through license, collaboration, commercialization or similar marketing and sales agreements, to successfully commercialize and market our product candidates in the U.S. and other countries around the world. To the extent that we enter into these types of agreements with other companies to sell, promote or market our products in the U.S. or abroad, our product revenues, which may be in the form of indirect revenue, a royalty, or a split of profits, may depend largely on the efforts of the other party, which may not be successful. In the event we decide to develop our own sales force and marketing capabilities, this may result in us incurring significant upfront costs to do so before we may generate any significant product revenues. We may not be able to attract and retain qualified third parties or marketing or sales personnel, or be able to establish marketing capabilities or an effective sales force.
Currency fluctuations and changes in exchange rates could increase our costs or lower our revenues.
We collect and pay a portion of our revenue and expenses in currencies other than the U.S. dollar. Fluctuations in foreign currency exchange rates can affect our operating results. We retain the majority of our cash and cash equivalents in U.S. dollars and utilize foreign currency accounts for collection and payment of revenues and expenses. Any significant foreign exchange rate fluctuations could adversely affect our financial position and results of operations.
Unless we reach an agreement with Daiichi Sankyo and Hovione with respect to commercial rights to LANI outside of Japan, disputes between us and these parties may occur and could adversely affect our business prospects.
Pursuant to our 2003 license and collaboration agreement with Daiichi Sankyo, if the parties agree to license the commercial rights to LANI in territories outside Japan to a third-party licensee, we share all licensing revenue equally with Daiichi Sankyo. The agreement does not, however, specifically address the respective rights or obligations of, or any consideration between, the parties in the event that either we or Daiichi Sankyo directly market LANI in territories outside Japan and a license has not been granted to a third-party licensee anywhere in the world. Further, the license agreement that we and Daiichi Sankyo collectively entered into with Hovione for use of the TwinCaps® dry powder inhaler provides us and Daiichi Sankyo with the exclusive right to import, export, make, use, and distribute for sale the drug product comprised of LANI and the TwinCaps® dry powder inhaler (“drug product”) worldwide in the field of preventing and/or treating influenza infections. The contract specifies what consideration is payable to Hovione in the event drug product is marketed by a third-party licensee other than us or Daiichi Sankyo outside of Japan. The agreement does not, however, specifically address the respective rights or obligations of, or any consideration between, the parties in the event that either we or Daiichi Sankyo directly market drug product in territories outside Japan.
The consideration potentially payable to Daiichi Sankyo under our license agreement with it, if any, or to Hovione under our license with it, related to direct sales of LANI we may generate in territories outside of Japan is uncertain. If we fail to reach a mutually acceptable commercial agreement in the future with either Daiichi Sankyo, Hovione, or both with respect to the development and marketing of LANI or drug product of Japan, disputes could result, which could further result in arbitration, litigation or other legal proceedings, or delay our ability to generate significant revenue from the sale of such products outside Japan. Such proceedings can be expensive and consume a significant amount of our management’s time. We cannot assure you we will reach a satisfactory commercial agreement with Daiichi Sankyo or Hovione in the future.
Our employees, representatives or agents may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could expose us to financial, reputational or other harm.
Our employees, representatives or agents may engage in any fraud or other improper activities, including but not limited to:
|
● |
complying with FDA regulations or similar regulations of similar regulatory authorities in other countries; |
|
● |
providing accurate information to the FDA or similar regulatory authorities in other countries; |
|
● |
complying with manufacturing standards we or the FDA have established; |
|
● |
complying with federal and state healthcare fraud and abuse laws and regulations or similar laws and regulations established and enforced by comparable foreign regulatory authorities; |
|
● |
complying with the provisions of the Foreign Corrupt Practices Act; or |
|
● |
reporting financial information or clinical or preclinical data accurately. |
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent these activities may not be effective in controlling unknown or unmanaged risks or losses, or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
Laws and regulations governing international operations may preclude us from developing, manufacturing and selling certain product candidates outside of the United States and require us to develop and implement costly compliance programs.
Because we have subsidiaries and conduct business outside of the U.S., we must comply with numerous laws and regulations in each jurisdiction in which we operate. The creation and implementation of international business practices compliance programs is costly and such programs are difficult to enforce, particularly where reliance on third parties is required.
The Foreign Corrupt Practices Act (“FCPA”) includes provisions that prohibits any U.S. individual or business from paying, offering, authorizing payment or offering anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the U.S. to comply with certain accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the company, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. The anti-bribery provisions of the FCPA are enforced primarily by the Department of Justice, while the SEC is involved with enforcement of the books and records provisions of the FCPA.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with the conduct of clinical studies and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the U.S., or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. Our operations outside of the U.S. require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the U.S., which could limit our growth potential and increase our development costs.
The failure to comply with laws governing international business practices may result in substantial penalties, including suspension or debarment from government contracting. Violation of the FCPA can result in significant civil and criminal penalties. Indictment alone under the FCPA can lead to suspension of the right to do business with the U.S. Government until the pending claims are resolved. Conviction for a violation of the FCPA can result in long-term disqualification as a government contractor. The termination of a government contract or relationship as a result of our failure to satisfy any of our obligations under laws governing international business practices could have a negative impact on our operations and harm our reputation and ability to procure government contracts. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA's accounting provisions.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
If we are unable to adequately protect or expand our intellectual property related to our products, or current or future product candidates, our business prospects could be materially harmed.
Our business success depends in part on our ability to:
|
● |
obtain, maintain and protect our intellectual property rights; |
|
● |
protect our trade secrets; and |
|
● |
prevent others from infringing on our proprietary rights or patents. |
We can protect our proprietary intellectual property rights from unauthorized use by third parties only to the extent that our proprietary rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. The patent position of pharmaceutical and biopharmaceutical companies involves complex legal and factual questions, and, therefore, we cannot predict with certainty whether we will be able to ultimately enforce our patents or proprietary rights, or avoid infringing on the patents or proprietary rights of others. Any issued patents that we own or otherwise have rights to may be challenged, invalidated or circumvented, and may not provide us with the protection against competitors that we anticipate.
The degree of future protection for our proprietary intellectual property rights is uncertain because issued patents and other legal means of establishing proprietary rights afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. Our future patent position will be influenced by the following factors:
|
● |
we, or our licensors, may not have been the first to discover the inventions covered by each of our or our licensors’ pending patent applications and issued patents, and we may have to engage in expensive and protracted interference proceedings to determine priority of invention; |
|
● |
our, or our licensors’, pending patent applications may be denied and may not result in issued patents; |
|
● |
our, or our licensors’, issued patents may not provide a basis for commercially viable products, may not provide us with any competitive advantages, or may be challenged by third parties; and |
|
● |
third parties may develop intellectual property that circumvents our or our licensors’ patent claims or design competitive intellectual property and ultimately product candidates that fall outside the scope of our or our licensors’ patents. |
Due to the extensive time required for the development, testing and regulatory review and approval of a product candidate, it is possible that before a product candidate of ours may be approved for sale and commercialized, our relevant patent rights may expire, or such patent rights may remain in force for only a short period following marketing approval. We currently rely on certain patents to provide us and our licensees with exclusive rights for certain of our products. When all patents underlying a license expire, our revenue from that license may cease, and there can be no assurance that we will be able to replace it with revenue from new or existing licenses.
Zanamivir, a neuraminidase inhibitor approved for the treatment and prevention of influenza A and B, is marketed worldwide as Relenza® by GSK. Most of our Relenza® patents have expired and the only substantial remaining intellectual property related to the Relenza® patent portfolio, which is solely owned by us and exclusively licensed to GSK, is scheduled to expire in July 2019 in Japan. On May 12, 2015, we filed a request for rehearing from the PTAB in relation to the pending patent application No. 08/737,141 related to Relenza IP in the U.S. On June 23, 2015, the PTAB denied our request for a rehearing. We reported on September 11, 2015, that we have filed another appeal in relation to the pending patent application No. 08/737,141 related to Relenza® to the United States Court of Appeals for the Federal Circuit. We cannot determine the duration or the outcome of this appeal process, or how long this patent application will remain pending, however we do believe that if this most recent appeal is unsuccessful, it is unlikely that the patent claims will be ever issued and we will receive no further royalties. If the patent claims are ultimately issued, we would be eligible to receive royalties from net sales of Relenza® in the U.S. for an additional 17 years from the date of allowance.
LANI, a long acting NI for the treatment and prevention of influenza A and B, is currently marketed as Inavir® in Japan by Daiichi-Sankyo. The patent relating to the structure of LANI expires in 2017 in the U.S., the EU and Japan. The patent relating to hydrates and the crystalline form of LANI actually used in the product expires in 2021 (not including extensions) in the U.S. and EU and in 2024 in Japan. In February 2015, a patent containing claims relevant to the manufacture of Inavir® was issued in Japan and expires in December 2029. The dry-powder inhaler device patent portfolio, which includes TwinCaps®, is owned by Hovione International Limited (“Hovione”) and is exclusively licensed to us and Daiichi Sankyo worldwide for the prevention and treatment of influenza and other influenza-like viral infections. These patents will expire in 2029 in the U.S., and in 2027 in the EU and Japan.
Vapendavir is an oral direct acting antiviral we are developing to treat HRV infections. We exclusively own the vapendavir patent portfolio, and issued claims under this portfolio will begin to expire in some countries in December 2021, not including extensions. Claims filed in recent patent applications related to a free-base form of vapendavir, if allowed, would extend coverage until 2034, without extensions, however we cannot make any assurance that these claims will be allowed.
BTA074 is a direct-acting antiviral we are developing as a topical treatment for genital warts caused by HPV 6 and 11. The patent containing composition of matter claims expires in the U.S. in 2029 without extensions. Pending U.S. patent applications related to pharmaceutical compositions and methods of synthesis of BTA074 if allowed, would extend coverage until 2033, without extensions, however we cannot make any assurance that these claims will be allowed.
We also own a patent portfolio focused on developing oral antivirals for RSV. Our RSV patent portfolio is comprised of a number of patent filings directed to several compound series, with the earliest projected expiries of such patents ranging from late-2024 to late-2031. Issued patent claims covering the BTA585 composition of matter will begin to expire in 2031 without extensions.
Patent rights may not provide us with adequate proprietary protection or competitive advantages against competitors with or developing similar technologies or approaches to ours. The laws of certain foreign countries do not protect intellectual property rights to the same extent as do the laws of the U.S., and certain countries may lack adequate rules and procedures for defending our intellectual property rights. For example, we may not be able to prevent a third party from infringing our patents in a country that does not recognize or enforce patent rights, or that imposes compulsory licenses on or restricts the prices of drugs. Changes in either patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. We may need to in-license certain technologies to successfully develop and commercialize our product candidates. We may not develop or obtain rights to products or processes that are patentable. Even if we or our licensors do obtain patents, such patents may not adequately protect the products or technologies licensed, or may otherwise be limited in scope. In addition, we may not have total control over the patent prosecution of subject matter that we license from others. Accordingly, we may be unable to exercise the same degree of control over this intellectual property as we would over our own. Others may challenge, seek to invalidate, infringe or circumvent any pending or issued patents we own or license, and rights we receive under those issued patents may not provide competitive advantages to us. We cannot assure you of the degree of protection that will be afforded by any of our issued or pending patents, or those licensed by us.
We cannot be sure that any patents will be issued from the patent applications we own or have licensed or, should any patents issue, that we will be provided with adequate protection against potentially competitive products. Furthermore, we cannot be sure that patents issued or licensed to us will be of any commercial value, or that private parties or competitors will not successfully challenge these patents or circumvent our patent position in the U.S. or abroad. In the absence of adequate patent protection, our business may be adversely affected by competitors who develop comparable technology or products.
If a third-party claims we are infringing on its intellectual property rights, we could incur significant expenses, or be prevented from further developing or commercializing our product candidates, which could materially harm our business.
Our success will also depend on our ability to operate without infringing the patents and other proprietary intellectual property rights of third parties. This is generally referred to as having the “freedom to operate.” The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. The defense and prosecution of intellectual property claims, interference proceedings and related legal and administrative proceedings, both in the U.S. and internationally, involve complex legal and factual questions. As a result, such proceedings are lengthy, costly and time-consuming, and their outcome is highly uncertain. We may become involved in protracted and expensive litigation in order to determine the enforceability, scope and validity of the proprietary rights of others, or to determine whether we have the freedom to operate with respect to the intellectual property rights of others.
Patent applications in the U.S. are, in most cases, maintained in secrecy until approximately 18 months after the patent application is filed. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made. Therefore, patent applications relating to product candidates similar to ours may have already been filed by others without our knowledge. In the event that a third party has also filed a patent application covering our product candidate or other claims, we may have to participate in an adversarial proceeding, known as an interference proceeding, in the USPTO or similar proceedings in other countries, to determine the priority of invention. In the event an infringement claim is brought against us, we may be required to pay substantial legal fees and other expenses to defend such a claim and, if we are unsuccessful in defending the claim, we may be prevented from pursuing the development and commercialization of a product candidate and may be subject to injunctions and/or damage awards.
In the future, the USPTO or a foreign patent office may grant patent rights to our product candidates or other claims to third parties. Subject to the issuance of these future patents, the claims of which will be unknown until issued, we may need to obtain a license or sublicense to these rights in order to have the appropriate freedom to further develop or commercialize them. Any required licenses may not be available to us on acceptable terms, if at all. If we need to obtain such licenses or sublicenses, but are unable to do so, we could encounter delays in the development of our product candidates, or be prevented from developing, manufacturing and commercializing our product candidates at all. If it is determined that we have infringed an issued patent and do not have the freedom to operate, we could be subject to injunctions, and/or compelled to pay significant damages, including punitive damages. In cases where we have in-licensed intellectual property, our failure to comply with the terms and conditions of such agreements could harm our business.
It is becoming common for third parties to challenge patent claims on any successfully developed product candidate or approved drug. If we or our licensees or collaborators become involved in any patent litigation, interference or other legal proceedings, we could incur substantial expense, and the efforts and attention of our technical and management personnel could be significantly diverted. A negative outcome of such litigation or proceedings may expose us to the loss of our proprietary position or to significant liabilities, or require us to seek licenses that may not be available from third parties on commercially acceptable terms, if at all. We may be restricted or prevented from developing, manufacturing and selling our product candidates in the event of an adverse determination in a judicial or administrative proceeding, or if we fail to obtain necessary licenses.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information and may not adequately protect our intellectual property.
We also rely on trade secrets to protect our technology, especially where we do not believe patent protection is obtainable, or prior to us filing patent applications on any inventions we may make. However, trade secrets are difficult to protect. In order to protect our proprietary technology and processes, we also rely in part on confidentiality and intellectual property assignment agreements with our corporate and academic partners, employees, consultants, outside scientific collaborators and sponsored researchers and other advisors. These agreements may not effectively prevent disclosure of confidential information or result in the effective assignment to us of intellectual property, and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information or other breaches of these agreements. In addition, others may independently discover our trade secrets and proprietary information, and in such case we may not be able to assert any trade secret rights against such party. Enforcing a claim that a third party illegally obtained and is using our trade secret is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the U.S. may be less willing to protect trade secrets. Costly and time-consuming litigation could be necessary to seek to enforce and determine the scope of our proprietary rights, and our failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submissions, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary fee payments and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our product candidates, our competitive position would be adversely affected.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees, including our senior management, were previously employed at other biotechnology or pharmaceutical companies. These employees typically executed proprietary rights, non-disclosure and non-competition agreements in connection with their previous employment. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee's former employer. We are not aware of any threatened or pending claims related to these matters, but in the future litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
RISKS RELATED TO OWNING OUR COMMON STOCK
Our revenue, expenses and results of operations may be subject to significant fluctuations, which will make it difficult to compare our operating results from period to period.
Our revenues have historically been highly variable. Royalty revenues we earn are derived from the net sales of products used for the treatment and/or prevention of influenza. Influenza as a disease is seasonal and highly unpredictable, and sales of these products to treat influenza fluctuate in line with the nature and extent of the incidence and severity of influenza each season. Payments potentially due to us under our existing or any future collaborative arrangements, including any milestone and royalty payments, are generally intermittent in nature and are subject to significant fluctuation in both timing and amount, or may never be earned or paid at all. In addition, the returns of products to our licensees are taken into account in the calculation of net sales for purposes of calculating the royalty revenue we receive and the amount of such returns are in general unpredictable. Further, in May 2014 our contract with BARDA was terminated, such that we do not anticipate any future revenue, or cost of revenue, associated with that contract. Accordingly, our quarterly and annual revenue may be highly variable, and comparisons to previous periods may be difficult to make, especially due to the cancellation of our BARDA contract which was a cost plus margin contract that we earned revenue from and had cost of sale expense. Our historical and current revenues may not be indicative of our ability to achieve additional payment-generating milestones or royalties in the future, or vice versa. We expect that our operating results will also vary significantly from quarter-to-quarter and year-to-year as a result of the initiation and success or failure of preclinical studies or clinical trials we undertake, the timing of the formulation and manufacture of our product candidates, or other development-related factors and activities, as well as any business or corporate development activities we may undertake. Accordingly, our revenues, expenses and results of operations for any period, particularly over the next several quarters, may not be comparable to the revenues, expenses or results of operations for any other period.
The reporting requirements of being a company that is publicly traded on the NASDAQ Global Select Market (NASDAQ) increases our overall operating costs and subject us to further increased costs and regulatory risk that may negatively impact our business or our ability to raise capital in the future.
As a company that is publicly-traded on NASDAQ, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), and the listing requirements of NASDAQ. Further, Section 404 of the Sarbanes-Oxley Act requires that we maintain effective internal control over financial reporting and disclosure controls and procedures. In particular, management must perform system and process evaluation and testing of our internal control over financial reporting to assess the effectiveness of our internal control over financial reporting and our independent auditor must perform its own assessment on our internal control over financial reporting. This testing is expensive and requires the attention of our limited management resources. The various financial reporting, legal, corporate governance and other obligations associated with being a company that is publicly traded on NASDAQ in the U.S. require us to incur significant expenditures and place additional demands and requirements on our board of directors, executive officers, and other administrative, operational and financial personnel and resources. If we are unable to comply with these requirements in a timely and effective manner, we and/or our executive officers may be subject to sanctions by the SEC. We expect that we will continue to incur additional expenses as a result of being a company that is publicly traded on NASDAQ.
The price of our common stock price has been highly volatile, and your investment in us could suffer a decline in value.
The market price of our common stock has been and is likely to continue to be highly volatile and could be subject to wide fluctuations in response to various factors and events, including but not limited to:
|
● |
our ability to successfully advance our product candidates through preclinical and clinical development; |
|
● |
disclosure of any favorable or unfavorable data from our preclinical studies or clinical trials, or other regulatory developments concerning our preclinical studies or clinical trials, the formulation and manufacturing of our product candidates, or those of our competitors; |
|
● |
the approval or commercialization of new products by us or our competitors, and the disclosure thereof; |
|
● |
novel scientific innovations by us or our competitors; |
|
● |
rumors relating to us or our competitors; |
|
● |
public concern about the safety or tolerability of our products, product candidates, or similar classes of compounds; |
|
● |
litigation to which we may become subject; |
|
● |
actual or anticipated variations in our quarterly or annual revenue or operating results; |
|
● |
changes in general conditions or trends in the biotechnology and pharmaceutical industries; |
|
● |
changes in drug reimbursement rates or government policies related to such reimbursement; |
|
● |
significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
|
● |
new regulatory legislation adopted in the U.S. or abroad; |
|
● |
changes in patent legislation in the U.S. or abroad; |
|
● |
our failure to achieve or meet equity research analysts’ expectations or their estimates of our business or prospects, or a change in their recommendations concerning us, the value of our common stock or our industry in general; |
|
● |
termination or delay in any of our existing or future license or collaboration arrangements; |
|
● |
future sales of equity or debt securities, or the perception that such future sales may occur; |
|
● |
the loss of our eligibility to have shares of our common stock traded on the NASDAQ Market or other listed markets due to our failure to maintain minimum listing standards; |
|
● |
changes in accounting principles or a restatement of previously reported financial results; |
|
● |
failure to comply with the periodic reporting requirements of publicly-owned companies under the Exchange Act and the Sarbanes-Oxley Act; and |
|
● |
conditions in the economy generally and the capital markets in particular. |
In addition, the stock market in general, and more specifically NASDAQ, which our common stock is traded, and the market for smaller biotechnology stocks in particular have historically experienced significant price and volume fluctuations. Volatility in the market price for a particular biotechnology company’s stock has often been unrelated or disproportionate to the operating performance of that company. Market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. Due to this volatility, you may be unable to sell your shares of our common stock at or above the price you paid and you could lose all or part of your investment in us.
In order to develop our product candidates and support our operations beyond 12 months from June 30, 2015, we may need to raise additional capital. Such capital may not be available to us on acceptable terms, if at all, which could materially harm our financial condition, business and business prospects.
We believe that our existing cash, cash equivalents and investments of $65.5 million and our accounts receivable balance as of June 30, 2015, along with the anticipated proceeds from our existing royalty-bearing licenses for Relenza® and Inavir® will enable us to operate for a period of at least 12 months from June 30, 2015. This estimate assumes that we pursue our current strategy and continue the development of our existing product candidates. This estimate does not include the impact of any other significant transaction or change in our strategy or development plans in the near-future. We currently do not have any commitments for additional future funding, nor do we anticipate that we will generate any significant incremental revenue from the sale of any of our product candidates in the foreseeable future. Therefore, in order to meet our anticipated liquidity needs beyond twelve months to continue the development of our product candidates, or possibly sooner in the event we enter into other transactions, change our strategy or accelerate our development plans, we may need to secure additional capital. In the event we need to raise additional capital we expect to raise it primarily through the sale of our common stock or other equity securities, as well as through proceeds from future licensing agreements, strategic collaborations, forms of asset and debt financing, or any other financing vehicle we may enter into in the future. Funds from these sources may not be available to us on acceptable terms, if at all, and our failure to raise such funds could have a material adverse impact on our future business strategy, plans, financial condition and results of operations. If adequate capital is not available to us on acceptable terms in the future, we may be required to delay, reduce the scope of, or eliminate one or more of our research and development programs, or delay or curtail our preclinical studies and clinical trials. If additional capital is not available to us on acceptable terms, we may also need to obtain funds through license or collaborative arrangements, pursuant to which we would likely relinquish potentially valuable rights to certain of our product candidates that we might otherwise choose to develop or commercialize independently, or be forced to enter into such arrangements earlier than we would prefer, which would likely result in less favorable transaction terms. Additional equity financings may be dilutive to holders of our common stock, and debt financing, if available, may involve significant payment obligations and restrictive covenants that restrict how we operate our business.
The timing and extent of our future financing needs are uncertain and will depend on many factors, some of which are very difficult to predict or may be beyond our control, including:
|
● |
the variability of future royalty revenue we may receive under our existing royalty-bearing license agreements; |
|
● |
the development timelines and plans for our product candidates, including any changes to those timelines, plans or our strategy; |
|
● |
the variability, timing and costs associated with conducting clinical trials for our product candidates, the rate of enrollment in such clinical trials, and the results of these clinical trials: |
|
● |
the variability, timing and costs associated with conducting preclinical studies, and the results of these studies; |
|
● |
the cost of scaling up, formulating and manufacturing preclinical and clinical trial materials to evaluate our product candidates; |
|
● |
whether we receive regulatory approval to advance the clinical development of our product candidates in a timely manner, if at all; |
|
● |
the cost and time to obtain regulatory approvals required to advance the development of our product candidates; |
|
● |
the scope and size of our research and development efforts; |
|
● |
the terms and timing of any collaborative, licensing and other arrangements that we may establish in the future; |
|
● |
the cost to maintain a corporate infrastructure to support being a company that is publicly traded in the U.S. on NASDAQ; and |
|
● |
the cost of filing, prosecuting, and enforcing patent and other intellectual property claims. |
Future issuances of shares of our common stock may cause our stock price to decline, even if our business is doing well.
The sale and issuance of additional shares of our common stock, or the perception that such future sales could occur, including any sales by our directors, executive officers, and other insiders or their affiliates, could materially and adversely affect the market price of our common stock and impair our ability to raise capital through the sale of additional equity securities at a price we deem appropriate.
If we raise additional capital in the future, your level of ownership in us could be diluted or we could be required to relinquish certain rights.
Any issuance of securities we may undertake in the future to raise additional capital could cause the price of our common stock to decline, or require us to issue shares at a price that is lower than that paid by holders of our common stock in the past, which would result in those newly issued shares being dilutive. Further, if we obtain funds through a debt financing or through the issuance of debt or preferred securities, these securities would likely have rights senior to your rights as a common stockholder, which could impair the value of our common stock. The terms of any debt financing we enter into may include covenants that limit our flexibility in conducting our business. We also could be required to seek funds through arrangements with collaborators or others, which might require us to relinquish valuable rights to our intellectual property or product candidates that we would have otherwise retained.
We do not anticipate paying cash dividends in the foreseeable future, and accordingly, you must rely on appreciation in the price of our common stock for any return on your investment in us.
We anticipate that we will retain our earnings, if any, for future growth and therefore do not anticipate paying cash dividends in the foreseeable future. As a result, our common stock will likely only provide a return to stockholders in the event there is appreciation in its price.
Our certificate of incorporation, our bylaws, and the laws of Delaware contain provisions that could discourage, delay or prevent a change in our control or in our management.
Certain provisions of our restated certificate of incorporation, our bylaws and the laws of Delaware, the state in which we are incorporated, may discourage, delay or prevent a change in control of us or a change in our directors or management that stockholders may consider favorable. These provisions:
|
● |
allow the authorized number of directors to be changed only by resolution of our Board of Directors; |
|
● |
provide that our stockholders may remove our directors only for cause; |
|
● |
authorize our Board of Directors to issue without stockholder approval, up to 5,000,000 shares of preferred stock, the rights of which will be determined at the discretion of the Board of Directors that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our Board of Directors; |
|
● |
establish advance notice requirements for stockholder nominations to our Board of Directors or for stockholder proposals that can be acted on at stockholder meetings; |
|
● |
limit who may call stockholder meetings; and |
|
● |
contain a fair price provision. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights of our common stock, from merging or combining with us for a prescribed period of time. These provisions could discourage proxy contests and make it more difficult for you and other stockholders to remove and elect directors and take other corporate actions. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock.
We may be subject to securities litigation, which is expensive and could divert management attention.
The market price of our common stock has been and may continue to be volatile, and in the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management's attention from other business concerns, which could seriously harm our business.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable rating, about our business, our stock price and trading volume could decline.
The trading market for our common stock is influenced by independent research and reports that securities or industry analysts publish about us or our business from time to time. There can be no assurance that analysts will continue to cover us or provide favorable ratings. If any analysts who cover us downgrade our stock, change their opinion of our stock or disseminate negative information regarding our business, our share price may decline. If any analysts cease coverage of our company, or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our share price or trading volume to decline.
RISKS RELATED TO OTHER ASPECTS OF OUR BUSINESS
If a product liability claim is successfully brought against us for uninsured liabilities, or such claim exceeds our insurance coverage, we could be forced to pay substantial damage awards that could materially harm our business.
The use of any of our existing or future product candidates in clinical trials and the sale of any approved pharmaceutical products may expose us to significant product liability claims. We currently have product liability insurance coverage for our ongoing clinical trials in the amount of $15 million. Further, we also require clinical research and manufacturing organizations that assist us in the conduct of our clinical trials or manufacture materials used in these trials to carry product liability insurance against such claims. This insurance coverage may not protect us against any or all of the product liability claims that may be brought against us in the future. We may not be able to acquire or maintain adequate product liability insurance coverage at a commercially reasonable cost or in sufficient amounts or scope to protect us against potential losses. In the event a product liability claim is brought against us, we may be required to pay legal and other expenses to defend the claim, as well as uncovered damage awards resulting from a claim brought successfully against us. In the event any of our product candidates are approved for sale by the FDA or similar regulatory authorities in other countries and commercialized, we may need to substantially increase the amount of our product liability coverage. Defending any product liability claim or claims could require us to expend significant financial and managerial resources, which could have an adverse effect on our business.
Our ability to use our net operating loss carry forwards to reduce taxable income generated in the future could be substantially limited or eliminated.
Our ability to use our net operating losses in the U.S., Australia, France and the United Kingdom is subject to limitations and re-assessment due to ownership changes that have occurred, or that could occur in the future. Depending on the actual amount of any limitation on our ability to use our net operating loss carry forwards, a significant portion of our future taxable income could be taxable. Additionally, tax law limitations may result in our net operating losses expiring before we have the ability to use them. In addition, financing and acquisition transactions that we may enter into in the future could significantly limit or eliminate our ability to realize any value from our net operating losses.
Our internal computer systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our drug development programs.
Despite the implementation of security measures, our internal computer systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing clinical trials for any of our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breaches were to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
|
ITEM 1B. |
UNRESOLVED STAFF COMMENTS |
None.
|
ITEM 2. |
PROPERTIES |
We have entered into an operating lease for office space in Alpharetta, Georgia through September 2019. The total annual rent expense under this lease is approximately $0.1 million. We do not own any real property. We believe that our facilities are adequate for our current business as a conducted, as well as our expected business for the foreseeable future.
|
ITEM 3. |
LEGAL PROCEEDINGS |
We may from time to time become subject to various claims and legal actions during the ordinary course of our business. We are not party to any legal proceedings at the date of filing of this Annual Report on Form 10-K.
|
ITEM 4. |
MINE SAFETY DISCLOSURES |
Not applicable.
PART II
|
ITEM 5. |
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock trades on the NASDAQ Global Select Market under the symbol “BOTA.” On September 9, 2015, we had 9,575 common stockholders of record. This figure does not represent the actual number of beneficial owners of common stock because shares are generally held in “street name” by securities dealers and others for the benefit of individual owners who may vote the shares.
The following table shows the range of high and low sales prices for our common stock for each completed fiscal quarter since June 30, 2013.
|
2014 |
||||||||
|
High |
Low |
|||||||
|
First Quarter (July 2013 to September 2013) |
$ | 4.44 | $ | 3.20 | ||||
|
Second Quarter (October 2013 to December 2013) |
4.30 | 3.75 | ||||||
|
Third Quarter (January 2014 to March 2014) |
7.07 | 4.17 | ||||||
|
Fourth Quarter (April 2014 to June 2014) |
6.17 | 2.36 | ||||||
|
2015 |
||||||||
|
High |
Low |
|||||||
|
First Quarter (July 2014 to September 2014) |
$ | 3.44 | $ | 2.10 | ||||
|
Second Quarter (October 2014 to December 2014) |
2.59 | 2.15 | ||||||
|
Third Quarter (January 2015 to March 2015) |
3.00 | 2.20 | ||||||
|
Fourth Quarter (April 2015 to June 2015) |
2.53 | 1.94 | ||||||
Securities Authorized for Issuance under Equity Compensation Plans
For certain information concerning securities authorized for issuance under our 2007 Omnibus Equity and Incentive Plan, see Item 12 – Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Dividend Policy
We have not paid or declared any dividends on our common stock in either of the two most recent fiscal years, and we do not anticipate paying any cash dividends in the foreseeable future. We currently intend to retain any earnings we may generate to fund our product development, operations and future growth. Any future determination to pay a dividend will be at the sole discretion of our Board of Directors, and will depend upon a number of factors, including our results of operations, capital requirements, financial condition, future prospects, contractual arrangements, restrictions imposed by applicable law, any limitations on payments of dividends present in any debt arrangements we may enter into in the future and other factors our Board of Directors may deem relevant.
Comparative Stock Performance
The following graph assumes $100 invested on June 30, 2010 into Biota Pharmaceuticals, Inc., Nasdaq stock market index and Nasdaq Biotech index and related information should not be deemed “soliciting material” or to be “filed” with the Securities and Exchange Commission, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933, as amended or the Exchange Act, except to the extent that we specifically incorporate it by reference into such filing.
|
6/30/2010 |
6/30/2011 |
6/30/2012 |
6/30/2013 |
6/30/2014 |
6/30/2015 | |
|
Biota Pharmaceuticals (1) |
$100 |
$99 |
$29 |
$63 |
$52 |
$38 |
|
Nasdaq Stock Market |
$100 |
$131 |
$139 |
$161 |
$209 |
$236 |
|
Nasdaq Biotech Index |
$100 |
$139 |
$167 |
$227 |
$336 |
$425 |
Assumes $100 invested on June 30, 2010.
|
(1) |
Nabi Pharmaceuticals, Inc. stock performance from June 30, 2010 to November 7, 2012. On November 8, 2012, Biota Holdings Limited completed a reverse merger with Nabi Pharmaceuticals, Inc. and renamed the resulting company Biota Pharmaceuticals, Inc. |
Issuer Purchases of Equity Securities
There were no stock repurchases or other purchases of equity securities by the Company during the fourth quarter ended June 30, 2015.
|
ITEM 6. |
SELECTED FINANCIAL DATA |
The following derived consolidated financial data as of June 30, 2015, 2014 and 2013 are from our audited consolidated financial statements appearing elsewhere in this Annual Report. The following consolidated financial data for June 30, 2012 and 2011 are derived from our audited consolidated financial statements not included in this Annual Report. This data should be read in conjunction with our audited consolidated financial statements and related notes, which are included elsewhere in this Annual Report on Form 10-K, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Item 7 below.
|
Years Ended June 30, |
||||||||||||||||||||
|
2015 |
2014 |
2013 |
2012 |
2011 |
||||||||||||||||
|
(in millions, except share and per share data) |
||||||||||||||||||||
|
Statement of Operations Data: |
||||||||||||||||||||
|
Revenues |
$ | 24.6 | $ | 68.7 | $ | 33.6 | $ | 20.4 | $ | 12.5 | ||||||||||
|
Operating expense: |
||||||||||||||||||||
|
Cost of revenue |
3.6 | 51.1 | 20.4 | 9.9 | 2.5 | |||||||||||||||
|
Research and development |
19.8 |
17.5 | 19.2 | 24.1 | 33.5 | |||||||||||||||
|
In-process research and development |
17.6 | - | - | - | - | |||||||||||||||
|
General and administrative |
9.4 | 10.2 | 18.0 | 9.4 | 7.0 | |||||||||||||||
|
Foreign exchange (gain) loss |
(6.5 |
) |
1.4 | (1.9 | ) | (0.1 |
) |
- | ||||||||||||
|
Loss on disposal of assets |
0.2 | - | - | - | - | |||||||||||||||
|
Total operating expense |
44.1 | 80.2 | 55.7 | 43.3 | 43.0 | |||||||||||||||
|
Operating loss |
(19.5 |
) |
(11.5 |
) |
(22.1 |
) |
(22.9 |
) |
(30.5 |
) | ||||||||||
|
Total non-operating income, net |
0.3 | 0.2 | 13.3 | 3.2 | 4.3 | |||||||||||||||
|
Income tax benefit (expense) |
0.1 | 0.3 | (0.1 |
) |
0.5 | 0.8 | ||||||||||||||
|
Net loss |
(19.1 |
) |
(11.0 |
) |
(8.9 |
) |
(19.2 |
) |
(25.4 |
) | ||||||||||
|
Net loss per common share: |
||||||||||||||||||||
|
Basic and Diluted |
$ | (0.54 |
) |
$ | (0.35 |
) |
$ | (0.32 |
) |
$ | (0.85 | ) | $ | (1.12 |
) | |||||
|
Weighted average number of shares used in per common share calculations: |
||||||||||||||||||||
|
Basic |
35,360,841 | 31,347,888 | 28,217,515 | 22,713,566 | 22,567,958 | |||||||||||||||
|
Diluted |
35,360,841 | 31,347,888 | 28,217,515 | 22,713,566 | 22,567,958 | |||||||||||||||
|
As of June 30, |
||||||||||||||||||||
|
2015 |
2014 |
2013 |
2012 |
2011 |
||||||||||||||||
|
(in millions) |
||||||||||||||||||||
|
Balance Sheet Data: |
||||||||||||||||||||
|
Cash, cash equivalents and investments |
$ | 65.5 | $ | 91.7 | $ | 66.8 | $ | 53.8 | $ | 74.2 | ||||||||||
|
Total assets |
79.4 | 114.0 | 85.8 | 69.3 | 88.5 | |||||||||||||||
|
Total liabilities |
9.9 | 27.1 | 17.8 | 10.0 | 7.1 | |||||||||||||||
|
Total stockholders’ equity |
$ | 69.5 | $ | 86.9 | $ | 68.0 | $ | 59.3 | $ | 81.4 | ||||||||||
|
ITEM 7. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read this discussion together with the audited financial statements, related notes and other financial information included elsewhere in this Form 10-K. The following discussion contains assumptions, estimates and other forward-looking statements that involve a number of risks and uncertainties, including those discussed under “Risk Factors,” “Special Note on Forward-Looking Statements” and elsewhere in this Form 10-K. These risks could cause our actual results to differ materially from those anticipated in these forward-looking statements.
References to “we,” “us,” and “our” refer to Biota Pharmaceuticals, Inc. and its consolidated subsidiaries. References to “Notes” refer to the Notes to Consolidated Financial Statements included herein (refer to Item 8).
Overview
We are focused on the discovery and development of direct-acting antivirals to treat infections that affect a significant number of patients globally. We have four product candidates in clinical development that address viral infections that have limited therapeutic options. vapendavir, an oral treatment for human rhinovirus (“HRV”) infections in moderate-to-severe asthmatics, currently being evaluated in our ongoing Phase 2b SPIRITUS trial; BTA074, a Phase 2 topical antiviral treatment for genital warts caused by human papillomavirus (“HPV”) types 6 & 11; BTA585, an oral fusion (“F”) protein inhibitor in Phase 1 development for the treatment of RSV-A and RSV-B infections; and laninamivir octanoate (“LANI”), a one-time, inhaled treatment in Phase 2 development for influenza A and B infections. We also have preclinical RSV non-fusion inhibitor program that we believe complements our F-protein inhibitor BTA585.
Although several of our influenza product candidates have been successfully developed and commercialized to-date by other larger pharmaceutical companies under license, collaboration or commercialization agreements with us, we have not independently developed or received regulatory approval for any product candidate, and we do not currently have any sales, marketing or commercial capabilities. Therefore, it is possible that we may not derive any significant product revenues from any product candidates that we are developing now, or may develop in the future. We expect to incur losses for the foreseeable future as we intend to support the clinical and preclinical development of our product candidates. Also, due to the termination of our contract with BARDA, we anticipate that we will not generate any revenue from service and cost of revenue in the near future as compared to recent historical levels.
We plan to continue to finance our operations with (i) our existing cash, cash equivalents, and investments (ii) proceeds from existing or potential future royalty-bearing licenses, collaborative research and development arrangements, (iii) future equity and/or forms of asset and debt financing or (iv) other financing arrangements. Our ability to continue to support our operations is dependent, in the near-term, upon our successful management of our cash resources, our continuing to receive royalty revenue under our existing licenses, our ability to enter into future collaboration, license or commercialization agreements, the successful development of our product candidates, our ability to execute future financings, if needed, and ultimately, upon the approval of our products for sale and achievement of positive cash flows from operations on a consistent basis. There can be no assurance that additional capital or funds will be available on terms acceptable to us, if at all, or that we will be able to enter into collaboration, license or commercialization agreements in the future, or that we will ever generate significant product revenue and become operationally profitable on a consistent basis.
Recent Corporate Developments
BTA585 Phase 1 Trial Ongoing. In August 2015, we reported that we have commenced dosing in a 50-subject, randomized, placebo-controlled, Phase 1 single ascending dose (“SAD”) clinical trial to evaluate the safety and pharmacokinetics (“PK”) of BTA585 in healthy volunteers. BTA585, a potent inhibitor of viral entry into cells, is an orally bioavailable compound in clinical development for the treatment of acute RSV infections in children, the elderly and immunocompromised patients. The ongoing Phase 1 SAD clinical trial has five dose level cohorts ranging from 50 mg to 500 mg and will include an evaluation of the effect of food on the plasma PK of BTA585. Following a safety assessment of the initial dose level cohorts of the SAD trial, we plan to begin dosing in a Phase 1 multiple ascending dose (“MAD”) clinical trial in the fourth quarter of calendar year 2015. We expect to report Phase 1 SAD data in fourth quarter of calendar year 2015 and MAD data in the first quarter of calendar year 2016.
Initiation of Phase 2 Trial with BTA074 Planned for Q4, 2015. In June 2015, we reported closing the acquisition of Anaconda Pharma. Anaconda Pharma was a privately-held Paris-based biotechnology company, whose lead candidate, BTA074 (AP611074), is a novel, direct-acting antiviral with activity against HPV types 6 and 11. BTA074 is in development for the treatment of genital warts, or also referred to as anogenital warts and condyloma, as well as recurrent respiratory papillomatosis (“RRP”). Prior to the acquisition, Anaconda Pharma had completed a Phase 2a clinical trial, which demonstrated a 38% reduction in the total genital warts area after six weeks of treatment with BTA074 5% gel while exhibiting a favorable local skin tolerability profile. We plan on initiating a double-blind placebo-controlled, randomized, Phase 2 study to assess the safety, tolerability, pharmacokinetics and efficacy of twice daily up to 16 weeks topical applications of BTA074 (5% gel) in approximately 210 adult genital warts patients in the fourth quarter of calendar year 2015.
Vapendavir Phase 2b SPIRITUS Trial Ongoing. The multi-center, randomized, double-blind, placebo-controlled dose-ranging SPIRITUS trial designed and powered to equally randomize approximately 190 laboratory-confirmed HRV infected patients across three treatment arms. The primary endpoint of the trial is the change from baseline to study day 14 in asthma symptoms and lung function as measured by the asthma control questionnaire-6 total score. Key secondary endpoints include safety and tolerability, specific lung function assessments such as forced expiratory volume in one second, forced vital capacity, peak expiratory flow, daily ß2-agonist use and the incidence of moderate and severe asthma exacerbations. Based upon the number of patients screened to date, we anticipate top-line data from this trial to be available in mid-2016.
Relenza® Related Intellectual Property Status. On September 11, 2015, we reported that on August 21, 2015 we filed another appeal in relation to the pending patent application No. 08/737,141 related to Relenza® to the United States Court of Appeals for the Federal Circuit. On March 19, 2015, we reported that the United States Patent Trial and Appeal Board (“USPTAB”) had issued a decision rejecting the previous appeal affirming the Examiner’s prima facie case of obviousness rejection under 35 U.S.C. 103(a). On May 12, 2015, we filed a request for rehearing under 37 C.F.R. § 41.50 (b)(2) with the USPATB. On June 23, 2015 the USPATB denied the Company’s request for a rehearing.
BARDA Contract Termination. On September 11, 2015, we reported that we have resolved all outstanding claims and collected all payments due from the Biomedical Advanced Research and Development Authority (“BARDA”) associated with the termination of its contract in May 2014.
Financial Operations Overview
Revenue. We have historically generated revenue primarily from royalty payments, license fees, milestone payments, payments for services performed pursuant to contracts, such as the terminated BARDA contract, and certain early-stage research and development activities pursuant to collaborations with other entities. Revenues are earned when the underlying service is rendered and all contingencies have been satisfied. Revenue for royalties is recognized when the net sales of the underlying product by the relevant third party, including actual or estimated returns within the royalty period based on agreement, are determinable. In fiscal 2016, we anticipate revenue from services to decrease to zero due to the termination of our contract with BARDA for the clinical advancement of laninamivir octanoate, also we expect our royalty revenues will be lower than in fiscal 2015, due to most of our Relenza® patents having expired with the only substantial remaining intellectual property related to the Relenza® patent portfolio scheduled to expire in July 2019 in Japan. GSK has verified that we will continue to receive royalties on the net sales of Relenza® in the U.S. beyond December 2014 to the extent that U.S. Patent Application No. 08/737,141 remains pending. We are unable at this time to determine the duration or final outcome of this appeal process, or how long this patent application will remain pending, and how long we might continue to receive royalty revenue from net sales of Relenza® in the U.S. during our 2016 fiscal year.
Cost of Revenue. Cost of revenue represents expenses incurred by us in performing services and activities pursuant to government contracts or grants for which we record related revenue and expense on the gross basis of accounting. Cost of revenue expense, which historically related to the terminated BARDA contract, includes, but is not limited to, the cost of third-party service providers incurred in connection with conducting external preclinical studies and treating patients enrolled in clinical trials and monitoring, accumulating and evaluating the related clinical data; salaries and personnel-related expenses for our internal staff allocated to a contract or grant, including benefits; and, the cost to develop, formulate and manufacture product candidates directly allocated to the specific contract. Cost of Revenue expenses are expensed as incurred. In fiscal 2016, we expect our cost of revenue to decrease to zero from our 2015 levels due to the termination of our contract with BARDA.
Research and Development Expense. Research and development expense represents the cost of activities associated with the discovery, preclinical development, and clinical development of our product candidates other than those captured under Cost of revenue. These costs include, but are not limited to, fees paid to third-party service providers in connection with conducting external preclinical studies and clinical trials, monitoring, accumulating and evaluating the related preclinical and clinical data; salaries and personnel-related expenses for our internal staff, including benefits and share-based compensation; the cost to develop, formulate and manufacture product candidates; legal fees associated with patents and intellectual property related to our product candidates; external research and chemistry, consulting fees; license expenses and sponsored research fees paid to third parties; and outsourced cost of specialized information systems to evaluate and monitor our programs, depreciation and laboratory facility costs. Research and development costs are expensed as incurred.
We anticipate that our research and development expense will increase in fiscal 2016, as compared to 2015 based on our plans for the ongoing clinical development of vapendavir Phase 2 clinical trial in patients with moderate and severe asthma with a presumptive HRV infection, the advancement of our RSV compound, BTA585 into Phase 1 and Phase 2 clinical trials, for the treatment of RSV and the initiation of a Phase 2 clinical trial for BTA-074 for the treatment of genital warts. Due to the early stage nature of our programs, our future research and development expense may be highly variable in future periods depending on the results and timing of these activities. From time-to-time, we will make determinations as to how much funding or resources to direct to these programs in response to their scientific, clinical and regulatory status, anticipated market opportunity and the availability of capital to fund our programs.
A discussion of the risks and uncertainties associated with the development of our existing or future product candidates, is set forth in the “Risk Factors” section of this Form 10-K.
In-Process Research and Development (“IPR&D”) Expense.
IPR&D expense and other charges represent impairments and other costs associated with product candidates under development that have not received regulatory approval for marketing at the time of acquisition. IPR&D acquired through an asset acquisition is written off at the acquisition date if the assets have no alternative future use. IPR&D acquired in a business combination is capitalized as indefinite-lived intangible assets (irrespective of whether these assets have an alternative future use) until completion or abandonment of the related research and development activities. Costs associated with the development of acquired IPR&D assets are expensed as incurred.
In fiscal 2015, we recorded charge of $17.6 million primarily due to the write-off of an IPR&D asset related to acquisition of Anaconda Pharma. The IPR&D project is BTA074, a patented, direct-acting antiviral in development for the treatment of genital warts, as well as the orphan disease RRP, both of which are caused by HPV types 6 and 11. The transaction also includes additional contingent financial consideration of up to $30.0 million, which is based on the successful achievement of certain future clinical and regulatory milestones, plus a royalty that was deemed not probable based on the current status of the BTA074 compound. If and when these contingent considerations are probable the effect of a change in estimate will be accounted in the period of change by recording a cumulative catch-up adjustment to retroactively apply the new estimate in accordance with generally accepted accounting principles in the U.S (“U.S. GAAP”)
General and Administrative Expense. General and administrative expense reflects the costs incurred to manage and support our research and development activities, operations, contracts and grants, and status as a publicly-traded company. General and administrative expense consists primarily of salaries and personnel-related expenses, including share-based compensation for personnel in executive, finance, accounting, information technology, business development and human resources functions. Other significant costs include professional fees for legal, auditing, tax, and consulting services, insurance premiums, other expenses incurred as a result of being a company that is publicly traded, and depreciation and facility expenses. In fiscal 2016, we anticipate our general and administrative expense to slightly decrease from our 2015 levels as result of previous integration activities and reductions in personnel.
Foreign Exchange (Gain) or Loss. Foreign exchange (gain) or loss primarily relates to translation of foreign currency balances in our subsidiaries that have a different functional currency than the reporting currency of the parent per ASC 830, Foreign Currency Matters. In April 2015, we changed the function currency of our subsidiaries to the U.S. dollar. Due to this change in fiscal 2016, we expect our foreign exchange (gain) or loss to be minimal in comparison to historical levels.
Other Income (Expense). Other income (expense) has historically consisted of the proceeds from the gain or loss on the disposal of equipment and research and development tax grants. Interest income consists of interest earned on our cash, cash equivalents, and short-term and long-term investments.
Critical Accounting Policies and Estimates
This discussion and analysis of our current financial condition and historical results of operations are based on our audited financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of our financial statements requires us to make estimates and judgments with respect to the selection and application of accounting policies that affect the reported amounts of assets, liabilities, revenues and expenses, and the disclosures of contingent assets and liabilities. We believe the following critical accounting policies are important in understanding our financial statements and operating results.
Use of Estimates. The preparation of our financial statements in conformance with U. S. GAAP requires us to make estimates and judgments with respect to the selection and application of accounting policies that affect the reported amounts of assets, liabilities, revenues and expenses, and the disclosures of contingent assets and liabilities. We base our estimates on historical experience, current economic and industry conditions, and various other factors that we believe to be reasonable at the time, the results of which form the basis for making judgments about the carrying values of certain assets and liabilities. Actual future results may differ from these estimates under different assumptions or conditions.
Revenue Recognition. Revenue from royalties is recognized when the net sales of the underlying product by the relevant third-party licensee, including actual or estimated returns within the royalty period based on agreement, are determinable. Revenue from services performed pursuant to a contract or grant is generally recognized as revenue when earned, typically when the underlying services or activities are rendered. Revenue from collaborative research and development activities typically consists of fees for services, or payments when specific milestones are met and match underlying activities occurring during the term of the arrangement.
Accrued Expenses. The preparation of our financial statements requires us to estimate expenses that we believe have been incurred, but for which we have not yet received invoices from our vendors and for employee services that we have not yet made payment. This process primarily involves identifying services and activities that have been performed by third-party vendors on our behalf and estimating the level to which they have been performed and the associated cost incurred for such service as of each balance sheet date. Examples of expenses for which we generally accrue based on estimates include fees for services, such as those provided by clinical research and data management organizations and investigators in conjunction with the conduct of our clinical trials, research organizations that perform preclinical studies, and fees owed to contract manufacturers in connection with the formulation or manufacture of materials for our preclinical studies and clinical trials. In order to estimate costs incurred to-date and evaluate the adequacy of a related accrued liability, we monitor and analyze the progress and related activities, under the terms of the underlying contract or agreement, any invoices received and the budgeted costs. We make these estimates based upon the facts and circumstances known to us at the time and in accordance with U.S. GAAP.
Share-Based Compensation. We use the Black-Scholes method to estimate the value of stock options granted to employees and directors. Our forfeiture rate is based on historical experience as well as anticipated turnover and other qualitative and quantitative factors, which may change over time. Also, we have used in the past a lattice model with a Monte Carlo simulation to value the grants of market stock units (“MSUs”). This valuation methodology utilizes several key assumptions, including the average closing stock price on the grant date, expected volatility of the Company’s stock price, risk-free rates of return and expected dividend yield. There may be adjustments to future periods if actual forfeitures differ from current estimates. Our time-based awards are issued with graded vesting. The compensation cost of these graded vesting awards is recognized using the straight-line method.
In-Process Research and Development (“IPR&D”) Expense.
IPR&D expense and other charges represent impairments and other costs associated with product candidates under development that have not received regulatory approval for marketing at the time of acquisition. IPR&D acquired through an asset acquisition is written off at the acquisition date if the assets have no alternative future use. IPR&D acquired in a business combination is capitalized as indefinite-lived intangible assets (irrespective of whether these assets have an alternative future use) until completion or abandonment of the related research and development activities. Costs associated with the development of acquired IPR&D assets are expensed as incurred.
Recent Accounting Pronouncements
In August 2014, the Financial Accounting Standard Board issued Accounting Standards Update No. 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which defines management’s responsibility to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosures if there is substantial doubt about its ability to continue as a going concern. The pronouncement is effective for annual reporting periods ending after December 15, 2016 with early adoption permitted. The adoption of this guidance is not expected to have a material impact on our financial statements.
Results of Operations
|
|
Fiscal Years Ended June 30, 2015 and 2014 |
Summary. For the fiscal year ended June 30, 2015, we reported a net loss of $19.1 million as compared to net loss of $11.0 million in 2014. The $8.1 million increase in net loss from the prior year was primarily due to the non-recurring $17.6 million IPR&D expense recorded during the three month period ended June 30, 2015 related to the acquisition of Anaconda Pharma, a $44.1 million decrease in revenues primarily due to the cancellation of the BARDA contract, a $2.3 million increase in research and development expense related to an increase in preclinical, clinical and manufacturing costs for the vapendavir program, and the IND-enabling studies for BTA585, a $0.2 million reduction in income tax benefits and a $0.2 million loss on disposal of assets, offset in part by a $47.5 million decrease in cost of revenue related to the cancellation of the BARDA contract, a $7.9 million change in foreign exchange from a loss to a gain, a $0.8 million decrease in general and administrative expense and a $0.1 increase in other income. Basic and diluted net loss per share was $0.54 for the twelve month period ended June 30, 2015, as compared to a basic and diluted net loss per share of $0.35 in the same period of 2014.
We expect to incur losses for the foreseeable future as we intend to support the clinical and preclinical development of our product candidates. Also, due to the recent termination of our contract with BARDA, we anticipate that our revenue from services and cost of revenue will be zero in the near future as compared to recent historical levels.
Revenue. Revenue decreased to $24.6 million for the fiscal year ended 2015 from $68.7 million in 2014. The following table summarizes the key components of our revenue in 2015 and 2014:
|
(in millions) |
||||||||
|
Twelve Months Ended June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
Royalty revenue – Relenza® |
$ | 11.4 | $ | 10.6 | ||||
|
– Inavir® |
4.8 | 4.5 | ||||||
|
Revenues from contract services, grants and collaborations |
8.4 | 53.6 | ||||||
|
Total revenue |
$ | 24.6 | $ | 68.7 | ||||
Royalty revenue from net sales of Relenza® increased primarily due to a rise in government stockpiling orders, offset in part by lower seasonal sales of Relenza®. Royalty revenue from Inavir® increased slightly due to higher seasonal sales in Japan. Revenues from contract services, grants and collaborations decreased due to a reduction in contract service revenue related to the cancellation of our contract with BARDA in May 2014 for the convenience of the U.S. Government offset in part by contract revenue from BARDA related close-out activities.
Cost of Revenue. Cost of revenue decreased to $3.6 million in fiscal year ended 2015 from $51.1 million in 2014. The following table summarizes the components of our cost of revenue in 2015 and 2014:
|
Twelve Months Ended June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
(in millions) |
||||||||
|
Direct preclinical, clinical and product development expenses |
$ | 3.3 | $ | 44.6 | ||||
|
Salaries, benefits and share-based compensation expenses |
0.2 | 5.9 | ||||||
|
Other expenses |
0.1 | 0.6 | ||||||
|
Total cost of revenue expense |
$ | 3.6 | $ | 51.1 | ||||
Direct preclinical, clinical and product development expenses decreased due to reduced third-party clinical costs incurred associated with the development of LANI as a result of the BARDA contract terminating in May 2014, offset in part by contract revenue from BARDA related close-out activities. Salaries, benefits and share-based compensation expenses decreased primarily as a result of the cost of certain personnel no longer being allocated to work under the BARDA contract. Other expenses decreased due to reduction in miscellaneous costs as a result of the termination of the BARDA contract.
Research and Development Expense. Research and development expense increased to $19.8 million in fiscal year ended 2015 from $17.5 million in 2014. The following table summarizes the components of our research and development expense for 2015 and 2014.
|
Twelve Months Ended June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
(in millions) |
||||||||
|
Direct preclinical, clinical and product development expenses |
$ | 11.1 | $ | 5.1 | ||||
|
Salaries, benefits and share-based compensation expenses |
5.8 | 7.1 | ||||||
|
Other expenses |
0.8 | 2.0 | ||||||
|
Depreciation and facility related expenses |
2.1 | 3.3 | ||||||
|
Total research and development expense |
$ | 19.8 | $ | 17.5 | ||||
Direct preclinical, clinical and product development expenses increased largely due to the initiation of our Phase 2 SPIRITUS clinical trial of vapendavir in February 2015 and IND-enabling studies associated with BTA585, our lead RSV compound, which were completed in June 2015. Salaries, benefits and share-based compensation expenses decreased primarily due to reductions in personnel working on other research and development activities. Other expenses decreased due to lower research and intellectual patent filing costs on other product candidates. Depreciation and facility-related expenses decreased as a result of the closure of our Melbourne, Australia research facility in March 2015.
In-Process Research and Development Expense. IPR&D was $17.6 million in our fiscal year ended 2015 which was related to the acquisition of Anaconda Pharma in June 2015. We accounted for the acquisition as an asset acquisition of IPR&D with no alternative future use, and therefore expensed the total consideration paid for the acquisition. IPR&D expenses also included $1.0 million of transaction costs we incurred directly related to the Anaconda Pharma acquisition. No IPR&D or related transaction expenses were incurred in the previous year.
General and Administrative Expense. General and administrative expense decreased to $9.4 million in fiscal year ended 2015 from $10.2 million in 2014. The following table summarizes the components of our general and administrative expense in 2015 and 2014.
|
Twelve Months Ended June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
(in millions) |
||||||||
|
Salaries, benefits and share-based compensation expenses |
$ | 5.8 | $ | 5.3 | ||||
|
Professional and legal fees expenses |
0.8 | 1.6 | ||||||
|
Other expenses |
2.8 | 3.3 | ||||||
|
Total general and administrative expense |
$ | 9.4 | $ | 10.2 | ||||
Salaries, benefits and share-based compensation expenses increased largely due to a higher share-based compensation, incentive compensation expenses and staff benefits. Professional and legal fees expenses decreased primarily due to lower ongoing professional fees as result of integration of the Company’s administrative functions to one location. Other expenses decreased due to lower administrative expenses as a result of the Company’s previous integration and restructuring efforts.
Foreign Exchange (Gain) Loss. Foreign exchange changed from a loss to a gain primarily due to the appreciation of the U.S. dollar as compared to the Australian dollar during our 2015 fiscal year and the related translation of those foreign currency balances and transactions in our subsidiaries that have a different functional currency than the U.S. reporting currency on our statement of operations. The vast majority of our cash holdings in our foreign subsidiaries are held in the U.S. dollar. We also translate all of the assets and liabilities of our non-U.S. subsidiaries at the period-end exchange rate and the net effect of these translation adjustments is shown on our condensed consolidated balance sheet as a component of stockholders’ equity.
|
|
Fiscal Years Ended June 30, 2014 and 2013 |
Summary. For the fiscal year ended June 30, 2014, we reported a net loss of $11.0 million, as compared to a $8.9 million net loss in 2013. The $2.1 million increase in net loss in 2014 was the result of a $31.4 million increase in cost of revenue, a $3.3 million change from a foreign exchange gain to a loss, non-recurring other income of $12.0 million in 2013 as a result of a gain on merger and a research and development tax credit and $1.1 million decrease in interest income, offset in part by a $35.1 million increase in revenue, a $7.8 million decrease in general and administrative expenses, a $2.4 million decrease in research and development expense and a $0.4 million increase in income tax benefit. Basic and diluted net loss per share were $0.35 for the year ended June 30, 2014, as compared to a basic and diluted net loss per share of $0.32 in 2013.
Revenue. Revenue increased to $68.7 million for the fiscal year ended 2014 from $33.6 million in 2013. The following table summarizes the key components of our revenue for 2014 and 2013.
|
(in millions) |
||||||||
|
Twelve Months Ended June 30, |
||||||||
|
2014 |
2013 |
|||||||
|
Royalty revenue – Relenza® |
$ | 10.6 | $ | 2.6 | ||||
|
– Inavir® |
4.5 | 4.2 | ||||||
|
Commercial milestone – Inavir® |
- | 2.8 | ||||||
|
Revenues from contract services, grants and collaborations |
53.6 | 24.0 | ||||||
|
Total revenue |
$ | 68.7 | $ | 33.6 | ||||
Royalty revenue from net sales of Relenza® increased primarily due to a government stock pile order and higher gross commercial sales of Relenza®. Royalty revenue from Inavir® increased due to higher seasonal sales. A non-recurring commercial milestone was earned in 2013 due to the net sales of Inavir® reaching a certain threshold. Revenues from contract services increased primarily due to the increased reimbursements received as a result of the clinical and manufacturing advancements of the LANI program under the BARDA contract that was terminated in May 2014, offset by a slight decrease in other grant revenue.
Cost of Revenue. Cost of revenue increased to $51.1 million in the fiscal year ended 2014 from $19.7 million in 2013.The following table summarizes the components of our cost of revenue in 2014 and 2013.
|
Twelve Months Ended June 30, |
||||||||
|
2014 |
2013 |
|||||||
|
(in millions) |
||||||||
|
Direct preclinical, clinical and product development expenses |
$ | 44.6 | $ | 14.5 | ||||
|
Salaries, benefits and share-based compensation expenses |
5.9 | 4.5 | ||||||
|
Other expenses |
0.6 | 0.7 | ||||||
|
Total cost of revenue expense |
$ | 51.1 | $ | 19.7 | ||||
Direct preclinical, clinical and product development expenses increased primarily due to the Phase 2 IGLOO trial and three other Phase 1 and Phase 2 trials of laninamivir octanoate and related manufacturing activities being conducted in 2014 under the BARDA contract, which was terminated in May 2014. Salaries, benefits and share-based compensation expenses increased principally due to severance benefits of $1.6 million being recorded as a result of the restructuring related to the termination of the BARDA contract. Other expenses decreased due to lower administration expenses as a result of the termination of the BARDA contract.
Research and Development Expense. Research and development expense decreased to $17.5 million in the fiscal year ended 2014 from $19.9 million in 2013. The following table summarizes the components of our research and development expense for 2014 and 2013.
|
Twelve Months Ended June 30, |
||||||||
|
2014 |
2013 |
|||||||
|
(in millions) |
||||||||
|
Direct preclinical, clinical and product development expenses |
$ | 5.1 | $ | 3.7 | ||||
|
Salaries, benefits and share-based compensation expenses |
7.1 | 9.3 | ||||||
|
Other expenses |
2.0 | 3.3 | ||||||
|
Depreciation and facility related expenses |
3.3 | 3.6 | ||||||
|
Total research and development expense |
$ | 17.5 | $ | 19.9 | ||||
Direct preclinical, clinical and product development expenses increased due to an increase in direct clinical expenses associated with our vapendavir and RSV program, offset in part by a decrease in cost of other research and development programs. Salaries, benefits and share-based compensation expenses slightly decreased due to a reduction in research personnel, offset in part by non-recurring severance benefits recorded as a result of restructurings in the previous years. Other expenses decreased due to a reduce number of research programs. Depreciation and facility expenses decreased to a reduction in research facilities.
General and Administrative Expense. General and administrative expense decreased to $10.2 million in fiscal year ended 2014 from $18.0 million in 2013. The following table summarizes the components of our general and administrative expense in 2014 and 2013.
|
Twelve Months Ended June 30, |
||||||||
|
2014 |
2013 |
|||||||
|
(in millions) |
||||||||
|
Salaries, benefits and share-based compensation expenses |
$ | 5.3 | $ | 9.8 | ||||
|
Professional and legal fees expenses |
1.6 | 3.8 | ||||||
|
Other expenses |
3.3 | 4.4 | ||||||
|
Total general and administrative expense |
$ | 10.2 | $ | 18.0 | ||||
Salaries, benefits and share-based compensation expenses decreased largely due to a reduction in ongoing compensation expenses as result of integration of the company’s administrative functions and severance benefits and merger expenses that were incurred in 2013. Professional and legal fees expenses decreased primarily due to non-recurring merger expenses that we incurred in 2013, as well as lower ongoing professional fees as result of integration of our administrative functions. Other expenses decreased due to lower administrative expenses as a result of our integration efforts.
Foreign Exchange (Gain) Loss. Foreign exchange changed from a gain to loss due to an increase in volatility of the U.S. dollar exchange rate to the Australian dollar during the 2014 fiscal year related to a decrease in the value of the U.S. dollar as compared to the Australian dollar and the related translation of foreign currency transactions in our subsidiaries that have a different functional currency than the reporting currency of the parent on our income statement. We also translate all of assets and liabilities of our non-U.S. subsidiaries at the period-end exchange rate and the net effect of these translation adjustments is shown in condensed consolidated balance sheet as a component of stockholders’ equity.
Other Income (Expense). Other income decreased primarily due to a non-recurring gain on merger, as well as the receipt of an Australian research and development credit in 2013 and lower interest income due to lower available interest rates, as well as a higher amount of U.S. dollar cash balances as compared to previous year.
Liquidity and Capital Resources
|
|
Sources of Liquidity |
Since our inception in 1965 through June 30, 2015 we have funded our operations primarily with public offerings of equity securities and license fees, royalties, research agreements, government contracts and grants. In March 2011, we were awarded a contract by BARDA for the late-stage development of LANI on a cost-plus-fixed-fee basis, the total of which is not to exceed $231.2 million. On May 7, 2014, the HHS office of the ASPR and BARDA notified us of its decision to terminate the contract for the development of laninamivir octanoate for the convenience of the U.S. Government.
At June 30, 2015, our cash, cash equivalents and investments were $65.5 million. Our cash and cash equivalents are generally held in a variety of interest-bearing short-term deposits with large U.S. banks, and our investments have an average maturity of less than two years.
|
|
Cash Flows |
For the year ended June 30, 2015, cash and cash equivalents decreased by $29.1 million and $7.9 million for the effects of exchange rate movements on cash and cash equivalents. This decrease was primarily the result of cash used for operating activities, the purchase of Anaconda Pharma and the purchase of additional investment securities during the period.
Net cash used in operating activities was $9.6 million, which reflected our net loss for the period of $19.1 million and a net decrease in operating liabilities of $17.8 million, offset in part by non-cash charges totaling $21 million for depreciation and amortization, share-based compensation and an IPR&D expense, and by an increase in net operating assets of $6.3 million.
Our net loss resulted largely from our funding of research and development activities including basic research, conducting preclinical studies, manufacturing and formulation of our product candidates, and ongoing general and administrative activities, offset in part by revenue from services, royalties and other revenue from grants and collaborations. The net change in operating assets and liabilities reflects the decrease in accounts receivable and accounts payable and accrued expenses largely due to payment of revenue billed under the contract with BARDA and the corresponding payments to the BARDA vendors that performed those services, offset in part by higher receivables related to royalty revenue from GSK.
Net cash used in investing activities during 2015 was $19.4 million, which reflects net investments of $10.8 million in short and long-term investments and $8.9 million used for the purchase of Anaconda Pharma, offset in part by the net receipt of $0.3 million from the sale of property and equipment.
|
|
Funding Requirements |
Our future funding requirements are difficult to determine and will depend on a number of factors, including:
|
● |
the variability of future royalty revenue we may receive from existing royalty-bearing license agreements; |
|
● |
the development timelines and plans for our product candidates, including any changes to those timelines, plans or our strategy; |
|
● |
the variability, timing and costs associated with conducting clinical trials for our product candidates, the rate of enrollment in such clinical trials, and the results of these clinical trials: |
|
● |
the variability, timing and costs associated with conducting preclinical studies, and the results of these studies; |
|
● |
the cost of scaling up, formulating and manufacturing preclinical and clinical trial materials to evaluate our product candidates; |
|
● |
whether we receive regulatory approval to advance or begin the clinical development of our product candidates in a timely manner, if at all; |
|
● |
the cost and time to obtain regulatory approvals required to advance the development of our product candidates; |
|
● |
the scope and size of our research and development efforts; |
|
● |
the size and cost of the general and administrative function we need to manage our operations, including the infrastructure to support being a publicly-traded company; and |
|
● |
the cost of filing, prosecuting, and enforcing patent and other intellectual property claims. |
Based on our current strategy and operating plan, and considering the potential costs associated with advancing the preclinical and clinical development of our product candidates, we believe that our existing cash, cash equivalents and investments of approximately $65.5 million, as well as our accounts receivables as of June 30, 2015, along with the anticipated proceeds from existing royalty-bearing licenses will enable us to operate for a period of at least 12 months ending June 30, 2016.
We currently do not have any commitments for future funding, nor do we anticipate that we will generate significant revenue, aside from revenue from existing royalty-bearing arrangements. Therefore, in order to meet our anticipated liquidity needs beyond 12 months to support the development of our product candidates, or possibly sooner in the event we enter into other transactions or revise our strategy or development plans, we may need to raise or secure additional capital. If we do, we would expect to do so primarily through the sale of additional common stock or other equity securities, as well as through proceeds from future licensing agreements, strategic collaborations, forms of asset or debt financing, or any other financing arrangement. Funds from these sources may not be available to us on acceptable terms, if at all, and our failure to raise such funds could have a material adverse impact on our future business strategy and plans, financial condition and results of operations. If adequate funds are not available to us on acceptable terms in the future, we may be required to delay, reduce the scope of, or eliminate one or more, if not all, of our research and development programs, or delay or curtail preclinical studies and clinical trials, or reduce our internal cost structure. If additional capital is not available to us on acceptable terms, we may need to obtain funds through license agreements, or collaborative or partner arrangements pursuant to which we will likely relinquish rights to certain product candidates that we might otherwise choose to develop or commercialize independently, or be forced to enter into such arrangements earlier than we would prefer, which would likely result in less favorable transaction terms. Additional equity financings may be dilutive to holders of our common stock, and debt financing, if available, may involve significant payment obligations and restrictive covenants that restrict how we operate our business.
Off-Balance Sheet Arrangements
At June 30, 2015, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. We are, therefore, not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in such relationships.
|
|
Contractual Obligations and Commitments |
We have entered into an operating lease for our corporate office in Alpharetta, Georgia through September, 2019. The total annual rent expense under these leases is approximately $0.1 million. As of June 30, 2015, future payments under this non-cancellable operating leases and purchase obligations are as follows (in millions):
|
Payments Due By Period |
||||||||||||||||||||
|
Total |
Less than |
1-3 Years |
4-5 Years |
After |
||||||||||||||||
|
Operating leases |
$ | 0.5 | $ | 0.1 | $ | 0.4 | $ | — | $ | — | ||||||||||
|
Purchase obligations |
1.8 | 1.8 | — | — | — | |||||||||||||||
|
Total contractual obligations |
$ | 2.3 | $ | 1.9 | $ | 0.4 | $ | — | $ | — | ||||||||||
The above contractual obligations table does not include any amounts or payments related to development, regulatory, or commercialization milestones on our product candidates, as the payments are contingent on the achievement of these milestones, which have not occurred.
|
ITEM 7A. |
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
|
|
Interest Rate Risk |
Our exposure to interest rate risk is currently confined to interest earnings, as our cash, cash equivalents and short and long term investments are invested in highly liquid money market funds, short-term bank deposits, U.S. agency securities, U.S. treasury securities, certificates of deposit and AA/ Aa grade bond securities. The primary objective of our investment activities is to preserve our capital to fund operations. We do not use derivative financial instruments to manage interest rate risk. If a 10% change in interest rates were to have occurred on June 30, 2015, this change would not have had a material effect on future earnings or cash flows.
Our exposure to credit risk is managed through our investment policy that specifies credit quality standards for our cash, cash equivalents and investments, which limits the amount of credit exposure to any single party or industry. We place any excess cash not needed to fund operations with high credit investments in order to limit the amount of credit exposure.
|
|
Foreign Currency Exchange Rate Risk |
We report our financial results in U.S. dollars; however we conduct business in other foreign countries. In April 2015, we changed our subsidiaries functional currency to the U.S. dollar.
We generated a portion of our revenue and collection of those receivables in foreign currencies. Similarly, we incur costs in foreign currencies and are subject to fluctuations in the exchange rate of the U.S. dollar against major foreign currencies, including the Euro, British Pound, Japanese Yen and Australian dollar, which can result in foreign currency exchange gains and losses that may significantly impact our financial results. Continued currency exposure to fluctuation in these exchange rates could result in financial results that are not comparable from quarter-to-quarter, or year-to-year. Where appropriate, we hold cash reserves in currencies in which those reserves are anticipated to be expended, but a majority of cash holdings are in the U.S. dollar. If a 10% change or more in currency rates were to have occurred on June 30, 2015, this change would not have had a material effect on future earnings or cash flows.
|
ITEM 8. |
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The information required by this Item is included in our Financial Statements and Supplementary Data listed in Item 15 of Part IV of this Annual Report on Form 10-K.
|
ITEM 9. |
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
|
ITEM 9A. |
CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
Our management, including our Chief Executive Officer and Chief Financial Officer, have evaluated the effectiveness of our “disclosure controls and procedures” (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934,) as amended (the “Exchange Act”)) of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, management and our Chief Executive Officer have concluded that our disclosure controls and procedures are effective as of the end of the period covered by this report.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rules
13a-15(f) or 15d-15(f)) under the Exchange Act as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the Company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
|
● |
Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Company’s assets; |
|
● |
Provide reasonable assurance that transactions are recorded as necessary to permit the preparation of financial statements in accordance with generally accepted accounting principles, and that the receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and, |
|
● |
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements. |
Our management, including our Chief Executive Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all error and all fraud. A control system, no matter how well conceived, operated, tested and monitored, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our company have been detected.
Our management, including our Chief Executive Officer, assessed the effectiveness of our internal control over financial reporting as of the end of the fiscal year covered by this Annual Report on Form 10-K. In making this assessment, management used the criteria set forth in Internal Control-Integrated 2013 Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, (“COSO”). Based on their assessment, management has concluded that, as of June 30, 2015, our internal control over financial reporting is effective based on the COSO criteria.
The effectiveness of our internal control over financial reporting as of June 30, 2015 has been audited by PricewaterhouseCoopers, LLP, an independent registered public accounting firm, as stated in their report, which is included in this Annual Report on Form 10-K.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the fourth quarter of 2015 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
|
ITEM 9B. |
OTHER INFORMATION |
None.
PART III
|
ITEM 10. |
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Incorporated by reference to the sections labeled “Proposal 1 Election of Directors,” “Executive Officers,” and “Corporate Governance” in our definitive proxy statement to be filed in connection with our 2015 annual meeting of stockholders.
Code of Ethics
We have adopted a code of ethics for our directors, officers and employees, which is available on our website at www.biotapharma.com in the Investor section under “Corporate Governance.” If we make any substantive amendments to the code of ethics or grant any waiver from a provision of the code of ethics to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website. The information on, or that can be accessed from, our website is not incorporated by reference into this Annual Report.
|
ITEM 11. |
EXECUTIVE COMPENSATION |
Incorporated by reference to the sections labeled “Executive Compensation,” “Compensation of Directors” and “Compensation Committee Report” in our definitive proxy statement to be filed in connection with our 2015 annual meeting of stockholders.
|
ITEM 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT, AND RELATED STOCKHOLDER MATTERS |
Incorporated by reference to the sections labeled “Principal Stockholders,” and “Executive Compensation” in our definitive proxy statement to be filed in connection with our 2015 annual meeting of stockholders.
|
ITEM 13. |
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Incorporated by reference to the sections labeled “Certain Relationships and Related Transactions” and “Corporate Governance” in our definitive proxy statement to be filed in connection with our 2015 annual meeting of stockholders.
|
ITEM 14. |
PRINCIPAL ACCOUNTING FEES AND SERVICES |
Incorporated by reference to the section labeled “Independent Registered Public Accountants” in our definitive proxy statement to be filed in connection with our 2015 annual meeting of stockholders.
PART IV
|
ITEM 15. |
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(a)(1) Financial Statements
The following documents are included on pages F-1 through F-29 attached hereto and are filed as part of this annual report on Form 10-K.
|
Report of Independent Registered Public Accounting Firm |
|
F-1, F-2 |
|
Consolidated Balance Sheets as of June 30, 2015 and 2014 |
|
F-3 |
|
Consolidated Statements of Operations and Comprehensive Loss for the Years Ended June 30, 2015, 2014 and 2013 |
|
F-4 |
|
Consolidated Statements of Stockholders’ Equity for the Years Ended June 30, 2015, 2014 and 2013 |
|
F-5 |
|
Consolidated Statements of Cash Flows for the Years Ended June 30, 2015, 2014 and 2013 |
|
F-6 |
|
Notes to Financial Statements |
|
F-7 |
(a)(2) Financial Statements Schedules
Not applicable
(a)(3) List of Exhibits Required by Item 601 of Regulation S-K
See Item 15(b) below.
(b) Exhibits
The exhibits which are filed or furnished with this report or which are incorporated herein by reference are set forth in the Exhibit Index hereto.
SIGNATURES
Pursuant to the requirements of the Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Alpharetta, Georgia on this 11th day of September 2015.
Biota Pharmaceuticals, Inc.
|
|
By: |
/s/ Joseph M. Patti, PhD |
|
|
|
|
|
|
|
Joseph M. Patti |
|
|
|
President and Chief Executive Officer |
Pursuant to the requirements the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date | ||||||
|
| ||||||||||
|
/s/ Joseph M. Patti, PhD
Joseph M. Patti |
|
President, Chief Executive Officer and Director (Principal Executive Officer) |
|
September 11, 2015 | ||||||
|
/s/ Russell H. Plumb
Russell H. Plumb |
|
Executive Chairmen (Principal Financial Officer) |
|
September 11, 2015 | ||||||
|
/s/ Peter Azzarello
Peter Azzarello |
|
Vice President of Finance and Chief Accounting Officer (Principal Accounting Officer) |
|
September 11, 2015 | ||||||
|
/s/ James Fox, PhD
James Fox |
|
Lead Director |
|
September 11, 2015 | ||||||
|
| ||||||||||
|
/s/ Geoffrey Cox, PhD
Geoffrey Cox |
|
Director |
|
September 11, 2015 | ||||||
|
| ||||||||||
|
/s/ John Richard
John Richard |
|
Director |
|
September 11, 2015 | ||||||
|
| ||||||||||
|
/s/ Anne VanLent
Anne VanLent |
|
Director |
|
September 11, 2015 | ||||||
|
| ||||||||||
|
/s/ Michael Dougherty
Michael Dougherty |
|
Director |
|
September 11, 2015 | ||||||
|
ITEM 8. |
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Biota Pharmaceuticals, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations and comprehensive loss, of stockholders’ equity and of cash flows present fairly, in all material respects, the financial position of Biota Pharmaceuticals, Inc. and its subsidiaries at June 30, 2015 and 2014, and the results of their operations and their cash flows for each of the two years in the period ended June 30, 2015 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of June 30, 2015, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Atlanta, Georgia
September 11, 2015
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Biota Pharmaceuticals, Inc.
In our opinion, the accompanying consolidated statements of operations and comprehensive loss, of stockholders' equity, and of cash flows present fairly, in all material respects, the results of operations and cash flows of Biota Pharmaceuticals, Inc. for the year ended June 30, 2013 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit. We conducted our audit of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
|
|
||
|
/s/ PricewaterhouseCoopers Melbourne Melbourne, Australia September 27, 2013 |
||
Biota Pharmaceuticals, Inc.
Consolidated Balance Sheets
(in millions), except share data
|
As of June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
ASSETS |
||||||||
|
Current assets: |
||||||||
|
Cash and cash equivalents |
$ | 44.7 | $ | 81.7 | ||||
|
Contract receivable (BARDA) |
— | 17.8 | ||||||
|
Other accounts receivable, net of allowance |
12.6 | 0.9 | ||||||
|
Short-term investments |
12.9 | — | ||||||
|
Prepaid expenses and other assets |
0.6 | 0.7 | ||||||
|
Total current assets |
70.8 | 101.1 | ||||||
|
Non-current assets: |
||||||||
|
Long-term investments |
7.9 | 10.0 | ||||||
|
Property and equipment, net |
0.2 | 2.0 | ||||||
|
Deferred tax asset |
0.5 | 0.9 | ||||||
|
Total non-current assets |
8.6 | 12.9 | ||||||
|
Total assets |
$ | 79.4 | $ | 114.0 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
|
Current liabilities: |
||||||||
|
Contract payable (BARDA) |
$ | 1.0 | $ | 18.6 | ||||
|
Accounts payable |
1.9 | 2.8 | ||||||
|
Accrued expenses and other current liabilities |
5.3 | 3.4 | ||||||
|
Accrued severance obligations |
0.1 | 1.2 | ||||||
|
Deferred tax liability |
0.5 | 0.9 | ||||||
|
Short-term note payable |
0.2 | — | ||||||
|
Total current liabilities |
9.0 | 26.9 | ||||||
|
Long-term note payable, net of current portion |
0.8 | — | ||||||
|
Other liabilities, net of current portion |
0.1 | 0.2 | ||||||
|
Total liabilities |
$ | 9.9 | $ | 27.1 | ||||
| Commitments and contingencies | ||||||||
|
Stockholders’ equity: |
— | — | ||||||
|
Common stock, $0.10 par value; 200,000,000 shares authorized; 38,609,086 shares and 35,100,961 shares issued and outstanding at June 30, 2015 and June 30, 2014, respectively |
3.9 | 3.5 | ||||||
|
Common stock-treasury |
— | — | ||||||
|
Additional paid-in capital |
155.6 | 146.4 | ||||||
|
Accumulated other comprehensive income |
18.9 | 26.8 | ||||||
|
Accumulated deficit |
(108.9 |
) |
(89.8 |
) | ||||
|
Total stockholders’ equity |
69.5 | 86.9 | ||||||
|
Total liabilities and stockholders’ equity |
$ | 79.4 | $ | 114.0 | ||||
Biota Pharmaceuticals, Inc.
Consolidated Statements of Operations and Comprehensive Loss
(in millions, except share data)
|
Years Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Revenue: |
||||||||||||
|
Royalty revenue and milestones |
$ | 16.1 | $ | 15.1 | $ | 9.6 | ||||||
|
Revenue from services |
8.4 | 53.5 | 23.2 | |||||||||
|
Other |
0.1 | 0.1 | 0.8 | |||||||||
|
Total revenue |
24.6 | 68.7 | 33.6 | |||||||||
|
Operating expense (income): |
||||||||||||
|
Cost of revenue |
3.6 | 51.1 | 19.7 | |||||||||
|
Research and development |
19.8 |
17.5 | 19.9 | |||||||||
|
In-process research and development (IPR&D) |
17.6 | — | — | |||||||||
|
General and administrative |
9.4 | 10.2 | 18.0 | |||||||||
|
Foreign exchange (gain) loss |
(6.5 |
) |
1.4 | (1.9 |
) | |||||||
|
Loss on disposal of assets |
0.2 | — | — | |||||||||
|
Total operating expense |
44.1 | 80.2 | 55.7 | |||||||||
|
Loss from operations |
(19.5 |
) |
(11.5 |
) |
(22.1 |
) | ||||||
|
Other income: |
||||||||||||
|
Gain recorded on merger |
— | — | 7.6 | |||||||||
|
Research and development credit |
— | — | 4.4 | |||||||||
|
Other income |
0.3 | 0.2 | 1.3 | |||||||||
|
Total other income |
0.3 | 0.2 | 13.3 | |||||||||
|
Loss before tax |
(19.2 |
) |
(11.3 |
) |
(8.8 |
) | ||||||
|
Income tax benefit (expense) |
0.1 | 0.3 | (0.1 |
) | ||||||||
|
Net loss |
$ | (19.1 |
) |
$ | (11.0 |
) |
$ | (8.9 |
) | |||
|
Basic and diluted loss per share |
(0.54 |
) |
(0.35 |
) |
(0.32 |
) | ||||||
|
Basic and diluted weighted average shares outstanding |
35,360,841 | 31,347,888 | 28,217,515 | |||||||||
|
Comprehensive loss: |
||||||||||||
|
Net loss |
(19.1 |
) |
(11.0 |
) |
(8.9 |
) | ||||||
|
Exchange differences on translation of foreign operations |
(7.8 | ) | 1.5 | (4.2 |
) | |||||||
|
Change in fair value of available for sale investments |
(0.1 | ) | — | — | ||||||||
|
Total comprehensive loss |
$ | (27.0 |
) |
$ | (9.5 |
) |
$ | (13.1 |
) | |||
See accompanying notes to the financial statements
Biota Pharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
(in millions, except share data)
|
Common Stock |
Treasury Shares |
Accumulated |
||||||||||||||||||||||||||||||
|
Shares |
Amount |
Additional Paid-in Capital |
Shares |
Amount |
Accumulated Deficit |
Other Comprehensive Income |
Total Stockholders’ Equity |
|||||||||||||||||||||||||
|
Balances at June 30, 2012 |
182,350,316 | 100.4 | 0.7 | (1,816,178 | ) | (1.4 | ) | $ | (69.9 | ) | 29.5 | $ | 59.3 | |||||||||||||||||||
|
Comprehensive loss |
||||||||||||||||||||||||||||||||
|
Exchange differences on translation of foreign operations |
- | - | - | - | - | - | (4.2 | ) | (4.2 | ) | ||||||||||||||||||||||
|
Net loss |
- | - | - | - | - | (8.9 | ) | - | (8.9 | ) | ||||||||||||||||||||||
|
Total Comprehensive loss |
(13.1 | ) | ||||||||||||||||||||||||||||||
|
New shares issued on exercise of options |
413,335 | 0.4 | (0.4 | ) | - | - | - | - | - | |||||||||||||||||||||||
|
New shares issued on vesting of options on merger |
4,639,104 | 1.1 | (1.1 | ) | - | - | - | - | - | |||||||||||||||||||||||
|
Acquisition of Nabi Biopharmaceuticals |
(153,398,048 | ) | (98.5 | ) | 233.4 | (4,051,183 | ) | (115.7 | ) | - | - | 19.2 | ||||||||||||||||||||
|
Retirement of treasury shares |
(5,867,361 | ) | (0.6 | ) | (116.5 | ) | 5,867,361 | 117.1 | - | - | - | |||||||||||||||||||||
|
Restricted stock units issued, net |
214,983 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
Retirement of common stock |
(3 | ) | - | - | - | - | - | - | - | |||||||||||||||||||||||
|
Share-based compensation |
- | - | 2.6 | - | - | - | - | 2.6 | ||||||||||||||||||||||||
|
Balances at June 30, 2013 |
28,352,326 | $ | 2.8 | $ | 118.7 | - | - | $ | (78.8 | ) | $ | 25.3 | $ | 68.0 | ||||||||||||||||||
|
Comprehensive income |
||||||||||||||||||||||||||||||||
|
Exchange differences on translation of foreign operations |
- | - | - | - | - | - | 1.5 | 1.5 | ||||||||||||||||||||||||
|
Net loss |
- | - | - | - | - | (11.0 | ) | - | (11.0 | ) | ||||||||||||||||||||||
|
Total Comprehensive loss |
(9.5 | ) | ||||||||||||||||||||||||||||||
|
Common stock issued |
6,685,985 | 0.7 | 26.1 | - | - | - | - | 26.8 | ||||||||||||||||||||||||
|
Restricted stock units issued, net |
62,650 | - | 0.2 | - | - | - | - | 0.2 | ||||||||||||||||||||||||
|
Share-based compensation |
- | - | 1.4 | - | - | - | - | 1.4 | ||||||||||||||||||||||||
|
Balances at June 30, 2014 |
35,100,961 | $ | 3.5 | $ | 146.4 | - | - | $ | (89.8 | ) | $ | 26.8 | $ | 86.9 | ||||||||||||||||||
|
Comprehensive loss |
||||||||||||||||||||||||||||||||
|
Exchange differences on translation of foreign operations |
- | - | - | - | - | - | (7.8 | ) | (7.8 | ) | ||||||||||||||||||||||
|
Change in fair value of investments |
- | - | - | - | - | - | (0.1 | ) | (0.1 | ) | ||||||||||||||||||||||
|
Net loss |
- | - | - | - | - | (19.1 | ) | - | (19.1 | ) | ||||||||||||||||||||||
|
Total Comprehensive loss |
(27.0 | ) | ||||||||||||||||||||||||||||||
|
Common stock issued |
3,500,000 | 0.4 | 7.1 | - | - | - | - | 7.5 | ||||||||||||||||||||||||
|
Restricted stock units issued, net |
8,125 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
Share-based compensation |
- | - | 2.1 | - | - | - | - | 2.1 | ||||||||||||||||||||||||
|
Balances at June 30, 2015 |
38,609,086 | $ | 3.9 | $ | 155.6 | - | - | $ | (108.9 | ) | $ | 18.9 | $ | 69.5 | ||||||||||||||||||
See accompanying notes to the financial statements
Biota Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(in millions, except share data)
|
Years Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Cash flows from operating activities provided by/(used in): |
||||||||||||
|
Net loss |
$ | (19.1 |
) |
$ | (11.0 |
) |
$ | (8.9 |
) | |||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
|
Depreciation and amortization |
1.1 | 2.4 | 3.0 | |||||||||
|
Share-based compensation |
2.1 | 1.7 | 2.6 | |||||||||
|
Loss (gain) recorded on disposal of assets |
0.2 | — | — | |||||||||
|
Acquisition of IPR&D |
17.6 | — | — | |||||||||
|
Gain recorded on merger |
— | — | (7.6 |
) | ||||||||
|
Deferred income taxes |
— | — | 1.5 |
| ||||||||
|
Change in operating assets and liabilities (net of liabilities acquired): |
||||||||||||
|
Accounts receivable |
6.2 | (7.1 |
) |
(4.2 |
) | |||||||
|
Prepaid expenses and other assets |
0.1 | 1.4 | (0.6 |
) | ||||||||
|
Deferred revenue |
— | (0.3 |
) |
(0.1 |
) | |||||||
|
Accounts payable and accrued expenses and other liabilities |
(15.8 |
) |
10.6 | 2.8 | ||||||||
|
Accrued severance obligations |
(2.0 |
) |
(1.0 |
) |
(2.4 |
) | ||||||
|
Net cash used in operating activities |
(9.6 |
) |
(3.3 |
) |
(13.9 |
) | ||||||
|
Cash flows from investing activities: |
||||||||||||
|
Acquisition of asset, net of cash acquired |
(8.9 | ) | — | — | ||||||||
|
Cash acquired on merger |
— | — | 32.7 | |||||||||
|
Purchases of short and long-term investments |
(17.7 |
) |
(10.0 |
) |
— | |||||||
|
Call redemption of long-term investments |
6.9 | — | — | |||||||||
|
Proceeds from sale of property and equipment |
0.4 | — | — | |||||||||
|
Purchases of property and equipment |
(0.1 |
) |
(0.1 |
) |
(1.0 |
) | ||||||
|
Net cash (used in) provided by investing activities |
(19.4 |
) |
(10.1 |
) |
31.7 | |||||||
|
Cash flows from financing activities: |
||||||||||||
|
Repayments on note payable |
(0.1 | ) | — | — | ||||||||
|
Issuance of common stock |
— | 26.8 | — | |||||||||
|
Net cash (used in) provided by financing activities |
(0.1 | ) | 26.8 | — | ||||||||
|
Net (decrease) increase in cash and cash equivalents |
(29.1 |
) |
13.4 | 17.8 |
| |||||||
|
Cash and cash equivalents at beginning of period |
81.7 | 66.8 | 53.8 | |||||||||
|
Effects of exchange rate movements on cash and cash equivalents |
(7.9 |
) |
1.5 | (4.8 |
) | |||||||
|
Cash and cash equivalents at end of period |
$ | 44.7 | $ | 81.7 | $ | 66.8 | ||||||
|
Supplemental cash flow disclosure: |
||||||||||||
|
Proceeds from issuance of common stock on merger |
$ | — | $ | — | $ | 27.0 | ||||||
|
Proceeds to settle accrued severance obligations and other accrued liabilities on merger |
— | — | 5.7 | |||||||||
|
Cash acquired on merger |
$ | — | $ | — | $ | 32.7 | ||||||
|
Supplemental cash flow disclosure of non-cash transaction: |
||||||||||||
|
Asset acquired through issuance of common stock |
$ | 7.5 | $ | — | $ | — | ||||||
See accompanying notes to the financial statements
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
|
(1) |
Company Overview |
Biota Pharmaceuticals, Inc., together with its wholly owned subsidiaries (“Biota”, or the “Company”) is a biopharmaceutical company focused on the discovery and development of direct-acting antivirals to treat infections that affect a significant number of patients globally. The Company is focused on the discovery and development of direct-acting antivirals to treat infections that affect a significant number of patients globally. The Company has four product candidates in clinical development that address viral infections that have limited therapeutic options. vapendavir, an oral treatment for human rhinovirus (“HRV”) infections in moderate-to-severe asthmatics, currently being evaluated in an ongoing Phase 2b SPIRITUS trial; BTA074, a Phase 2 topical antiviral treatment for genital warts caused by human papillomavirus (“HPV”) types 6 & 11; BTA585, an oral fusion (“F”) protein inhibitor in Phase 1 development for the treatment of RSV-A and RSV-B infections; and laninamivir octanoate (“LANI”), a one-time, inhaled treatment in Phase 2 development for influenza A and B infections. The Company also has a preclinical RSV non-fusion inhibitor program that the Company believes complements its F-protein inhibitor BTA585. The Company has been incorporated in the state of Delaware since 1969 and its corporate headquarters are located in Alpharetta, Georgia.
Although several of the Company’s influenza product candidates have been successfully developed and commercialized to-date by other larger pharmaceutical companies under collaboration, license or commercialization agreements with the Company, it has not independently developed or received regulatory approval for any product candidate, and the Company does not currently have any sales, marketing or commercial capabilities. Therefore, it is possible that the Company may not successfully derive any significant product revenues from any product candidates that it is developing now, or may develop in the future. The Company expects to incur losses for the foreseeable future as it intends to support the clinical and preclinical development of its product candidates.
The Company plans to continue to finance its operations with (i) existing cash, cash equivalents and investments, (ii) proceeds from existing or potential future royalty-bearing licenses or collaborative research and development arrangements, (iii) future equity and/or asset or debt financings, or (iv) other financing arrangements. The Company’s ability to continue to support its operations is dependent, in the near-term, upon managing its cash resources, continuing to receive royalty revenue under existing licenses, entering into future collaboration, license or commercialization agreements, the successful development of its product candidates, executing future financings and ultimately, upon the approval of its products for sale and achieving positive cash flows from operations on a consistent basis. There can be no assurance that additional capital or funds will be available on terms acceptable to the Company, if at all, that the Company will be able to enter into collaboration, license or commercialization agreements in the future, or that the Company will ever generate significant product revenue and become operationally profitable on a consistent basis.
|
|
|
(2) |
Summary of Significant Accounting Policies |
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements of Biota Pharmaceuticals, Inc. and its wholly owned subsidiaries have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”). All intercompany balances and transactions have been eliminated in consolidation. The Company’s fiscal year ends on June 30.
Use of Estimates
The preparation of the consolidated financial statements requires management of the Company to make a number of estimates and assumptions relating to the reported amount of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the period. Significant items subject to such estimates and assumptions include accruals and obligations, tangible and intangible assets and deferred income taxes. Actual results could differ from those estimates.
Fair Value of Financial Instruments
Financial instruments include cash and cash equivalents, investments, accounts receivable, accounts payable, note payable and accrued liabilities. The carrying amounts of those financial instruments are considered to be representative of their respective fair values because of the short-term nature of those investments.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Cash Equivalents and Investments
Cash equivalents consist of short-term, highly liquid investments with original maturities of 90 or fewer days when purchased. Investments with original maturities between 90 and 365 days when purchased are considered to be short-term investments. Investments with original maturities over 365 days when purchased are considered to be long-term investments. The Company has classified its entire investment portfolio as available-for-sale. These securities are recorded as cash equivalents, short-term or long-term investments. Short-term and long-term investments are carried at the fair value based upon observable inputs based on quoted market prices. The amortized cost of securities is adjusted for amortization of premiums and accretion of discounts to maturity. Amortization and accretion are included in interest income, net, and any realized gains and losses are also included in interest income, net. All unrealized gains and losses are reported in other comprehensive loss. The cost basis of all securities sold is based on the specific identification method. Available-for-sale securities as of June 30, 2015 consisted primarily of U.S. treasury securities, U.S. government agency securities, corporate notes and certificates of deposit.
Concentration of Credit Risk and Other Risks and Uncertainties
Cash, cash equivalents and short- and long-term investments consist of financial instruments that potentially subject the Company to concentrations of credit risk to the extent recorded on the balance sheets. The Company believes that it has established guidelines for investment of its excess cash that maintain principal and liquidity through its policies on concentration, diversification, investment maturity, and investment grade.
Receivables
Accounts receivable are recorded at the invoiced amount. An allowance for returns is estimated based on historical information patterns and sales and return information provided by the partner. An allowance for doubtful accounts is estimated based on probable credit losses in the existing accounts receivable and returns allowed under the Company’s contract based on a combination of default history, aging analysis and any specific, known troubled accounts. The allowance is determined based on a review of individual accounts for collectability, generally focusing on those that are past due. The current year expense to adjust receivables for returns and doubtful accounts, if any, is recorded in the consolidated statements of operations.
Property and Equipment
Fixed assets are recorded at acquisition cost, net of accumulated depreciation and impairment. Depreciation on tangible and intangible property and equipment is calculated using the straight-line method over the estimated useful lives of the assets. The estimated useful life of machinery, equipment, software and fixtures is three to five years. Leasehold improvements are amortized using the straight-line method over the shorter of the remaining lease term or estimated useful life of the asset. Maintenance and repairs are charged to operations as incurred.
Leased Assets
The Company accounts for its leases at their inception as either an operating or capital lease, depending on certain defined criteria. All of the Company’s leases in effect at June 30, 2015 and 2014 are considered operating leases. The costs of operating leases are charged to the consolidated statement of operations on a straight-line basis over the lease term. The difference between cash payments and straight line rent expense is recorded as deferred rent liability. The balance of deferred rent liabilities is classified in the balance sheet as other liabilities. Additionally, any incentives the Company receives are treated as a reduction of expenses over the term of the agreement. Leasehold improvements provided by the landlord are capitalized at cost and amortized over the lesser of their expected useful life or the life of the lease, without assuming renewal features, if any, are exercised.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Impairment of Assets
The Company reviews its tangible and intangible assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition is less than the carrying amount of the asset. If the evaluation indicates that the carrying value of an asset is not recoverable from its undiscounted cash flows, an impairment loss is measured by comparing the carrying value of the asset to its fair value.
Foreign Currency
Functional and reporting currency. Items included in the Company’s consolidated financial statements are measured using the currency of the primary economic environment in which the entity operates, referred to as the functional currency. The Company operates in several jurisdictions with functional currencies including the Euro, the Australian dollar, British Pound, and the U.S. dollar. The consolidated financial statements are presented in U.S. dollars. Effective April 1, 2015, the Company changed its subsidiaries functional currency to the U.S. dollar based on significant changes in economic facts and circumstances indicated clearly that the functional currency had changed.
Transactions and balances. Foreign currency transactions are translated into the functional currency using the exchange rates prevailing at the dates of the related transactions. Foreign exchange gains and losses resulting from the settlement of such transactions, as well as from the translation at year-end exchange rates of monetary assets and liabilities denominated in foreign currencies, are recognized in the consolidated statements of operations.
The results and financial position of any operations that have a functional currency different from the U.S. dollar are translated into U.S. dollar amounts. Assets and liabilities are translated into U.S. dollars at exchange rates in effect at the balance sheet date. Income and expense items are translated at average rates for the period. All resulting exchange differences are recognized as accumulated other comprehensive income, a separate component of stockholders’ equity. On consolidation, exchange differences arising from the translation of any net investment in foreign entities are recorded in stockholders’ equity as part of accumulated other comprehensive income, net of related taxes.
Patent Expense
Legal fees incurred for patent application costs for product candidates have been charged to expense and reported in research and development expense.
Share-Based Compensation Expense
Share-based compensation expense relates to stock options, restricted stock units or other equity-based grants. The fair market value of stock options is determined at the grant date using the Black-Scholes option pricing model based on the date the grant is issued. The fair market value of restricted stock units or other equity-based grants are also determined at the grant date, based on the closing price of the Company’s common stock on that date. The value of the awards that are ultimately expected to vest is recognized, net of forfeitures, as an expense on a straight-line basis over the employee's requisite service period. The Company uses the lattice model with a Monte Carlo simulation to value the grants of market stock units (“MSUs”). This valuation methodology utilizes several key assumptions, including the average closing stock price on the grant date, expected volatility of the Company’s stock price, risk-free rates of return and expected dividend yield.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Income Taxes
The Company applies ASC 740 – Income Taxes, which established financial accounting and reporting requirements for the effects of income taxes that result from the Company’s activities during the current and preceding years. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, and operating losses and tax credit carry forwards. Deferred tax assets and liabilities are measured using enacted statutory tax rates expected to apply to taxable income in the jurisdictions and years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
Where the Company determines that it is more likely than not that some portion or all of the deferred tax assets will not be realized in the future, the deferred tax assets are reduced by a valuation allowance. The valuation allowance is sufficient to reduce the deferred tax assets to the amount that the Company determines is more likely than not to be realized.
Revenue Recognition
Revenue consists primarily of royalty payments, license fees, milestone payments and payments for services performed pursuant to contracts. Revenue from royalties is recognized when the net sales of the underlying product by the relevant third party, including actual or estimated returns within the royalty period based on agreement, are determinable. The Company receives estimates of the amount of royalty revenue from its licensees on a quarterly basis. Revenue from services performed pursuant to contracts or grants is recognized when earned, typically when the underlying services or activities are rendered. The Company analyzes cost reimbursable grants and contracts to determine whether it should report such reimbursements as revenue, or as an offset to the related research and development expenses incurred. For costs incurred and revenues generated from third parties where the Company is deemed to be the principal participant, such as the previous BARDA contract, it recognizes revenue and costs using the gross basis of accounting. Revenue for collaborative research and development activities typically consists of fees for services, or payments when specific milestones are met and match underlying activities occurring during the term of the arrangement.
For milestones that are deemed substantive, the Company recognizes the contingent revenue when: (i) the milestones have been achieved; (ii) no further performance obligations with respect to the milestones exist; and (iii) collection is reasonably assured. A milestone is considered substantive if all of the following conditions are met: (i) the milestone is non-refundable; (ii) achievement of the milestone was not reasonably assured at the inception of the arrangement; (iii) substantive effort is involved to achieve the milestone; and (iv) the amount of the milestone appears reasonable in relation to the effort expended with the other milestones in the arrangement and the related risk associated with achievement of the milestone. If a milestone is deemed not to be substantive, the Company recognizes the portion of the milestone payment as revenue that correlates to activities already performed; the remaining portion of the milestone payment is deferred and recognized as revenue as the Company completes its performance obligations.
Cost of Revenue
Cost of revenue represents expenses incurred by the Company in performing services and activities pursuant to government contracts or grants for which it records related revenue and expense on the gross basis of accounting. Cost of revenue expense, which relates to the terminated BARDA contract, includes, but is not limited to, the cost of third-party service providers incurred in connection with conducting external preclinical studies and clinical trials, monitoring, accumulating and evaluating the related preclinical and clinical data; salaries and personnel-related expenses for our internal staff allocated to the contract or grant, including benefits; and, the cost to develop, formulate and manufacture product candidates directly allocated to the specific contract.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Research and Development Expense
Research and development expense represents the cost of activities associated with the discovery, preclinical development, and clinical development of the Company’s product candidates other than those captured under cost of revenue. These costs include, but are not limited to, fees paid to third-party service providers in connection with conducting external preclinical studies and clinical trials, monitoring, accumulating and evaluating the related preclinical and clinical data; salaries and personnel-related expenses for our internal staff, including benefits and share-based compensation; the cost to develop, formulate and manufacture product candidates; legal fees associated with patents and intellectual property related to our product candidates; external research and chemistry, consulting fees; license expenses and sponsored research fees paid to third parties; and outsourced cost of specialized information systems to evaluate and monitor our programs, depreciation and laboratory facility costs. Research and development expenses are expensed as incurred.
In-Process Research and Development (“IPR&D”) Expense
IPR&D expense and other charges represent impairments and other costs associated with product candidates under development that have not received regulatory approval for marketing at the time of acquisition. IPR&D acquired through an asset acquisition is written off at the acquisition date if the assets have no alternative future use. IPR&D acquired in a business combination is capitalized as indefinite-lived intangible assets (irrespective of whether these assets have an alternative future use) until completion or abandonment of the related research and development activities. Costs associated with the development of acquired IPR&D assets are expensed as incurred.
General and Administrative Expense
General and administrative expense reflects the costs incurred to manage and support our research and development activities, operations, contracts and grants, and status as a publicly-traded company. General and administrative expense consists primarily of salaries and personnel-related expenses, including share-based compensation for personnel in executive, finance, accounting, information technology, business development and human resources functions. Other significant costs include professional fees for legal, auditing, tax, and consulting services, insurance premiums, other expenses incurred as a result of being a company that is publicly traded, and depreciation and facility expenses.
Total Comprehensive Income
Comprehensive income is defined as the total change in stockholders’ equity during the period other than from transactions with stockholders, and for the Company, includes net income, unrealized gains and loss from available for sale securities and cumulative translation foreign currency adjustments.
Limited Suppliers
The Company may rely on single-source third-party suppliers and contract manufacturers to formulate or manufacture its product candidates pursuant to FDA current good manufacturing practices (“cGMP”) requirements. The failure of a single-source supplier or single-source contract manufacturer to produce and deliver specific candidates on a timely basis, or at all, could delay or interrupt the development process and affect the Company’s operating results.
Recent Accounting Standards
In August 2014, the Financial Accounting Standard Board issued Accounting Standards Update No. 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which defines management’s responsibility to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosures if there is substantial doubt about its ability to continue as a going concern. The pronouncement is effective for annual reporting periods ending after December 15, 2016 with early adoption permitted. The adoption of this guidance is not expected to have a material impact on the Company’s financial statements.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
|
(3) |
Stock Purchase Agreement |
On June 3, 2015, the Company completed the acquisition of Anaconda Pharma pursuant to a stock purchase agreement, dated February 25, 2015, the (“agreement”). Under the terms of the agreement, at closing all of Anaconda Pharma's outstanding shares were acquired for 3.5 million shares of the Company’s common stock and $8.0 million in cash, subject to certain closing and post-closing adjustments. The transaction also includes additional contingent financial consideration of up to $30.0 million, which is based on the successful achievement of certain future clinical and regulatory milestones, plus a royalty. If and when these contingent considerations are probable the effect of a change in estimate will be accounted in the period of change by recording a cumulative catch-up adjustment to retroactively apply the new estimate in accordance with U.S. GAAP.
The fair value of the issuance of 3.5 million of Biota’s common stock in the acquisition of Anaconda Pharma's was $7.5 million or $2.14 per share, based on the volume weighted average price on June 3, 2015. The estimated purchase price paid at closing was calculated as follows:
|
(in millions) |
||||
|
Fair value of Biota common stock issued |
$ | 7.5 | ||
|
Estimated transaction and exit costs |
1.0 | |||
|
Cash consideration issued |
8.0 | |||
|
Total purchase price |
$ | 16.5 |
The Company had also incurred $1.0 million of transaction costs directly related to the Anaconda Pharma acquisition, which includes expenditures for advisory, legal, fairness opinion, accounting and other similar services.
The total estimated purchase price was allocated to the tangible and intangible assets acquired and liabilities assumed in connection with the transaction, based on their estimated fair values. The purchase price allocation is preliminary and additional adjustments may occur. Anaconda Pharma was a development stage enterprise, therefore the acquisition was not considered to be a business combination as it did not meet the definition of a business The Company determined that the acquired assets can only be used for a specific and intended purpose and have no alternative future use after taking into consideration further research and development, regulatory and marketing approval efforts required in order to reach technological feasibility. Accordingly, the acquisition was accounted for as a purchase of in-process research and development (“IPR&D”) assets with no alternative future use and the entire amount was charged to IPR&D expense as of the acquisition date. Further, the contingent financial consideration will be recognized if and when the contingency is resolved and becomes payable.
The preliminary allocation of the total purchase price, as shown above, to the acquired tangible and intangible assets and assumed liabilities of Anaconda Pharma based on their fair values as of the acquisition date are as follows:
|
(in millions) |
||||
|
Cash and cash equivalents |
$ | 0.3 | ||
|
Prepaid expenses and other current assets |
0.2 | |||
|
Notes payable |
(1.0 |
) | ||
|
Accounts payable |
(0.4 |
) | ||
|
Accrued expenses |
(0.2 |
) | ||
|
Sub-total net fair value of acquired assets and liabilities |
(1.1 |
) | ||
|
In-process research and development |
17.6 | |||
|
Total purchase price |
$ | 16.5 |
The IPR&D project is BTA074, a patented, direct-acting antiviral in development for the treatment of genital warts, as well as the orphan disease recurrent respiratory papillomatosis, both of which are caused by HPV types 6 and 11. The accounting fair value of BTA074 IPR&D was $17.6 million.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Further, the Company assumed an interest-free loan of $1.0 million with a French local development authority for previous research and development activities related to BTA074. As of June 30, 2015, $1 million was outstanding under this note payable.
Future minimum payments due under notes payable as of June 30, 2015 are as follows:
|
Year ending June 30, |
||||
|
2016 |
$ | 0.2 | ||
|
2017 |
0.4 | |||
|
2018 |
0.4 | |||
|
2019 |
- | |||
|
2020 |
- | |||
|
Total future payments |
$ | 1.0 |
|
(4) |
Short-Term Financial Instruments |
Financial Assets (in millions)
|
As of June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
Financial assets: |
||||||||
|
Cash and cash equivalents |
$ | 44.7 | $ | 81.7 | ||||
| Accounts receivable, net of allowance | 12.6 | 18.7 | ||||||
|
Short-term investments |
12.9 | - | ||||||
|
Total current financial assets |
70.2 | 100.4 | ||||||
|
Financial liabilities: |
||||||||
|
Accounts payable and current accrued liabilities |
8.3 | 26.0 | ||||||
|
Short-term note payable |
0.2 | - | ||||||
|
Total current financial liabilities |
8.5 | 26.0 | ||||||
|
Net financial assets |
$ | 61.7 | $ | 74.4 | ||||
The carrying value of the cash and cash equivalents, accounts receivable, short-term note payable and accounts payable approximates fair value because of their short-term nature. The Company regularly reviews all financial assets for impairment. There were no impairments recognized in 2015 and 2014.
|
|
|
(5) |
Fair Value Measurements |
A fair value hierarchy has been established which requires the Company to maximize the use of observable inputs, where available, and minimize the use of unobservable inputs when measuring fair value. The fair value hierarchy describes three levels of inputs that may be used to measure fair value:
|
Level 1 |
Quoted prices in active markets for identical assets or liabilities. | |
|
Level 2 |
Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. | |
|
Level 3 |
Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
The following table sets forth the financial assets and liabilities that were measured at fair value on a recurring basis at June 30, 2015, by level within the fair value hierarchy. The assets and liabilities measured at fair value are classified in their entirety based on the lowest level of input that is significant to the fair value measurement.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
The Company’s long-term investments have been classified as Level 2, which have been initially valued at the transaction price and subsequently revalued, at the end of each reporting period, utilizing a third party pricing service. The pricing service utilizes industry standard valuation models and observable market inputs to determine value that include surveying the bond dealer community, obtaining benchmark quotes, incorporating relevant trade data, and updating spreads daily. There have been no transfers of assets or liabilities between the fair value measurement classifications.
|
Quoted Prices in |
||||||||||||||||
|
Active Markets for |
Significant Other |
Significant |
||||||||||||||
|
(in millions) |
Identical Assets |
Observable Inputs |
Unobservable |
|||||||||||||
|
June 30, 2015 |
Total |
(Level 1) |
(Level 2) |
Inputs (Level 3) |
||||||||||||
|
Cash equivalents |
$ | 6.3 | $ | 6.3 | $ | — | $ | — | ||||||||
|
Short-term investments available-for-sale |
12.9 | 12.9 | — | — | ||||||||||||
|
Long-term investments available-for-sale |
7.9 | — | 7.9 | — | ||||||||||||
|
Total |
$ | 27.1 | $ | 19.2 | $ | 7.9 | $ | — | ||||||||
|
Quoted Prices in |
||||||||||||||||
|
Active Markets for |
Significant Other |
Significant |
||||||||||||||
|
Identical Assets |
Observable Inputs |
Unobservable |
||||||||||||||
|
June 30, 2014 |
Total |
(Level 1) |
(Level 2) |
Inputs (Level 3) |
||||||||||||
|
Cash equivalents |
$ | 36.9 | $ | 36.9 | $ | — | $ | — | ||||||||
|
Long-term investments available-for-sale |
10.0 | — | 10.0 | — | ||||||||||||
|
Total |
$ | 46.9 | $ | 36.9 | $ | 10.0 | $ | — | ||||||||
Cash equivalents consist primarily of money market funds. Long-term investments consist of U.S. agency securities and U.S. Treasury securities, classified as available-for-sale and have maturities greater than 365 days from the date of acquisition.
The Company has had no realized gains or losses from the sale of investments for the twelve months ended June 30, 2015. The following table shows the unrealized gains and losses and fair values for those investments as of June 30, 2015 and June 30, 2014 aggregated by major security type:
|
(in millions) |
Unrealized |
Unrealized |
||||||||||||||
|
June 30, 2015 |
At Cost |
Gains |
(Losses) |
At Fair Value |
||||||||||||
|
Money market funds |
$ | 6.3 | $ | — | $ | — | $ | 6.3 | ||||||||
|
Debt securities of U.S. government agencies |
6.5 | — | — | 6.5 | ||||||||||||
|
U.S. Treasury securities |
9.6 | — | (0.1 | ) | 9.5 | |||||||||||
|
Corporate notes |
2.9 | — | — | 2.9 | ||||||||||||
|
Certificates of deposit |
1.9 | — | — | 1.9 | ||||||||||||
|
Total |
$ | 27.2 | $ | — | $ | (0.1 | ) | $ | 27.1 | |||||||
|
Unrealized |
Unrealized |
|||||||||||||||
|
June 30, 2014 |
At Cost |
Gains |
(Losses) |
At Fair Value |
||||||||||||
|
Money market funds |
$ | 36.9 | $ | — | $ | — | $ | 36.9 | ||||||||
|
Debt securities of U.S. government agencies |
4.9 | — | — | 4.9 | ||||||||||||
|
U.S. Treasury securities |
5.1 | — | — | 5.1 | ||||||||||||
|
Total |
$ | 46.9 | — | — | 46.9 | |||||||||||
As of June 30, 2014, the Company had investments in an unrealized loss position below material disclosure thresholds in the table above. The Company has determined that the unrealized losses on these investments are temporary in nature and expects the security to mature at its stated maturity principal. All available-for-sale securities held at June 30, 2015 will mature in over a two year period. The fair value of cash, accounts receivable, accounts payable and accrued liabilities approximate their carrying value because of the short-term nature of these financial instruments respectively, respectively, at June 30, 2015 and June 30, 2014. The fair value of our short and long term note payable, which is measured using Level 2 inputs, approximates book value, at June 30, 2015.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
|
(6) |
Property and Equipment |
Property and equipment consist of the following (in millions):
|
As of June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
Property and equipment |
$ | 0.2 | $ | 20.8 | ||||
|
Leasehold improvements |
0.2 | 6.9 | ||||||
|
Total Property and equipment |
0.4 | 27.7 | ||||||
| Accumulated depreciation | (0.2 | ) | (25.7 | ) | ||||
| Property and equipment, net | $ | 0.2 | $ | 2.0 | ||||
Depreciation and amortization expense was $1.1 million, $2.4 million and $3.0 million for the fiscal years ended June 30, 2015, 2014 and 2013, respectively.
|
|
|
(7) |
Accrued and Other Current Liabilities |
Accrued and other current liabilities consist of the following (in millions):
|
As of June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
Professional fees |
$ | 0.8 | $ | 1.0 | ||||
|
Salary and related costs |
1.6 | 0.4 | ||||||
|
Research and development services |
1.7 | 0.8 | ||||||
|
Other accrued expenses |
1.2 | 1.2 | ||||||
|
Total accrued expenses and other liabilities |
$ | 5.3 | $ | 3.4 | ||||
|
(8) |
Stockholders’ Equity |
In January 2014, the Company closed a public offering in which it sold approximately 6.7 million shares of its common stock at a purchase price of $4.30 per share. The net proceeds to the Company from the sale of these shares after underwriting discounts, commissions and other offering expenses were approximately $26.8 million.
|
|
|
(9) |
Commitments and Contingent Liabilities |
Operating Leases
The Company has one operating lease. The lease at 2500 Northwinds Parkway is for the Company’s corporate headquarters in Alpharetta, Georgia. The lease commenced in April, 2013 and expires in September, 2018. The lease includes an escalating base rent schedule as well as a seven month rent holiday and a tenant incentive towards leasehold improvements of approximately $0.1 million which are being recognized as a reduction in rent expense on a straight line basis over the term of the lease.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
Future minimum lease payments, in millions, under non-cancellable operating leases (with initial or remaining lease terms in excess of one year) as of June 30, 2015 are:
|
(in millions) |
||||
|
2016 |
$ | 0.1 | ||
|
2017 |
0.2 | |||
|
2018 |
0.2 | |||
|
Thereafter |
0.0 | |||
|
Total minimum lease payments |
$ | 0.5 |
Rent expense was $0.7 million, $0.7 million and $1.1 million for the fiscal years ended June 30, 2015, 2014 and 2013, respectively.
|
|
|
(10) |
Income Taxes |
For financial reporting purposes, income before taxes includes the following components (in millions):
|
Years Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
United States |
$ | (26.7 | ) | $ | (8.3 | ) | $ | 6.5 | ||||
|
Foreign |
7.5 | (3.0 |
) |
(15.3 |
) | |||||||
|
Total |
$ | (19.2 | ) | $ | (11.3 |
) |
$ | (8.8 |
) | |||
The expense (benefit) for income taxes is comprised of:
|
Years Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Current: |
||||||||||||
|
Federal |
$ | - | $ | - | $ | - | ||||||
|
State |
- | - | - | |||||||||
|
Foreign |
(0.1 | ) | (0.3 | ) | 0.1 |
| ||||||
| (0.1 | ) | (0.3 | ) | 0.1 |
| |||||||
|
Deferred: |
||||||||||||
|
Federal |
- | - | - | |||||||||
|
State |
- | - | - | |||||||||
|
Foreign |
- | - | - | |||||||||
| - | - | - | ||||||||||
|
Total tax (benefit) expense |
$ | (0.1 | ) | $ | (0.3 | ) | $ | 0.1 |
| |||
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
The reconciliation between the company's effective tax rate and the statutory rate is as follows:
|
Years Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Income tax (benefit) expense at federal statutory rate |
$ | (6.7 | ) | $ | (4.0 |
) |
$ | (3.3 |
) | |||
|
State and local income taxes, net of federal benefit |
(1.0 | ) | (0.3 |
) |
0.3 | |||||||
|
Foreign tax rate differential |
(0.4 | ) | 0.4 | 1.3 | ||||||||
|
In-process research and development |
6.9 | - | - | |||||||||
|
Change in valuation allowance |
0.8 | 4.1 | 4.2 | |||||||||
|
Merger related items |
0.4 | 0.1 | (3.0 |
) | ||||||||
|
Research and development expenses |
- | (0.1 |
) |
1.1 | ||||||||
|
Research and development tax credits |
(0.1 | ) | (0.3 |
) |
(1.1 |
) | ||||||
| Foreign Tax Credit | (0.2 | ) | - | - | ||||||||
|
Employee stock options |
- | - | 0.5 | |||||||||
|
Other |
0.2 | (0.2 |
) |
0.1 | ||||||||
|
Income tax (benefit) expense |
$ | (0.1 | ) | $ | (0.3 |
) |
$ | 0.1 | ||||
The following table includes deferred tax assets and liabilities as of June 30, 2015 and 2014:
|
As of June 30, |
||||||||
|
2015 |
2014 |
|||||||
|
Deferred tax assets: |
||||||||
|
Foreign net operating loss carryforwards |
$ | 19.8 | $ | 18.2 | ||||
|
US federal and state loss carryforwards |
5.4 | 2.8 | ||||||
|
Amortization |
0.6 | 0.8 | ||||||
|
Depreciation |
- | 0.6 | ||||||
|
Accrued compensated-related costs |
1.4 | 0.8 | ||||||
|
Other |
0.4 | 4.3 | ||||||
|
Subtotal |
27.6 | 27.5 | ||||||
|
Less valuation allowance |
(27.1 | ) | (26.4 | ) | ||||
|
Total net deferred tax asset |
0.5 | 1.1 | ||||||
|
Unearned Income |
(0.5 |
) |
(1.1 |
) | ||||
|
Net deferred tax assets |
$ | - | $ | - | ||||
|
Current net deferred tax liability |
(0.5 | ) | (0.9 |
) | ||||
|
Noncurrent net deferred tax asset |
0.5 | 0.9 | ||||||
|
Net deferred taxes |
$ | - | $ | - | ||||
Significant components of deferred income taxes reflect the net tax effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting and tax purposes. As of June 30, 2015 a full valuation allowance has been established, as the Company has determined that the realization of its deferred tax assets is not more likely than not. The Company recorded $27.1 million and $26.4 million of valuation allowance as of June 30, 2015 and 2014, respectively.
As of June 30, 2015, the Company has $13.8 million of gross U.S. federal net operating loss carryforwards that expire at various dates through 2034. Under IRC section 382, certain significant changes in ownership may restrict the future utilization of its U.S. tax loss carryforwards. As of June 30, 2015, the Company also has accumulated tax losses of $34.7 million for Australia, $25 million for the United Kingdom and $13.2 million for France available for carry forward against future earnings, which under relevant tax laws do not expire but may not be available under certain circumstances.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
As of June 30, 2015, the Company’s foreign subsidiaries have no positive accumulated earnings. As such, no federal or state income taxes have been provided on the losses of its foreign subsidiaries under ASC 740. If in the future there are positive earnings generated from the Company’s foreign subsidiaries, the Company will evaluate whether to record any applicable federal and state income taxes on such earnings.
Uncertain Tax Positions
The Company files income tax returns in the U.S, Australia, France and the United Kingdom, as well as with various U.S. states. The Company is subject to tax audits in all jurisdictions for which we file income tax returns. Tax audits by their very nature are often complex and can require several years to complete. There are currently no tax audits that have commenced with respect to income tax returns in any jurisdiction.
Under the tax statute of limitations applicable to the Internal Revenue Code, we are no longer subject to U.S. federal income tax examinations by the Internal Revenue Service for years before 2012. Under the statute of limitations applicable to most state income tax laws, the Company is no longer subject to state income tax examinations by tax authorities for years before 2011 in states in which we have filed income tax returns. Certain states may take the position that the Company is subject to income tax in such states even though the Company has not filed income tax returns in such states and, depending on the varying state income tax statutes and administrative practices, the statute of limitations in such states may extend to years before 2009. The Company began foreign operations in 1985. The Company is subject to foreign tax examinations by tax authorities for all years of operations.
The Company does not have any unrecognized tax benefits as of June 30, 2015.
|
(11) |
Share-Based Compensation |
For the twelve months ended June 30, 2015, 2014 and 2013, the Company recorded share-based compensation expense related to grants from equity incentive plans of $2.1 million, $1.7 million and $2.6 million, respectively. No income tax benefit was recognized in the statements of operations and no share-based compensation expense was capitalized as part of any assets for the twelve months ended June 30, 2015 and 2014.
Stock Options. The fair value of each stock option award was estimated at its respective date of grant using the Black-Scholes option-pricing model with the following weighted average assumptions:
|
For the Twelve Month Period Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Risk-free interest rate |
1.70 |
% |
1.50 |
% |
1.50 |
% | ||||||
|
Dividend yield |
— | — | — | |||||||||
|
Expected volatility |
.82 | .79 | .79 | |||||||||
|
Expected life of options (years) |
5.9 | 5.9 | 6.0 | |||||||||
|
Fair value of options granted |
$ | 1.65 | $ | 2.47 | $ | 2.39 | ||||||
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
The risk-free rate interest rate is based on the expected life of the option and the corresponding U.S. Treasury bond, which in most cases is the U.S. five year Treasury bond. The expected term of stock options granted is derived from actual and expected option behavior and represents the period of time that options granted are expected to be outstanding. The Company uses historical data to estimate option exercise patterns and future employee terminations to determine expected life and forfeitures. Expected volatility is based on the historical volatility of the Company’s publicly traded common stock. The following table summarizes the stock option activity for the year end:
|
Weighted Average |
Weighted-Average |
Aggregate Intrinsic |
||||||||||||||
|
Number of |
Exercise Price |
Remaining Contractual |
Value |
|||||||||||||
|
Stock Options |
Per Option |
Term |
($0000) | |||||||||||||
|
Balance at June 30, 2014 |
2,463,369 | $ | 9.09 | |||||||||||||
|
Granted |
1,432,000 | 2.42 | ||||||||||||||
|
Exercised |
— | — | ||||||||||||||
|
Forfeited or expired |
(531,252 | ) | 13.37 | |||||||||||||
|
Balance at June 30, 2015 |
3,364,117 | $ | 5.58 | 7.9 | $ | - | ||||||||||
The total intrinsic value of stock options exercised during the twelve month period ended June 30, 2015 was zero, and no cash proceeds were received by the Company. Further, no actual tax benefits were realized, as the Company currently records a full valuation allowance for all tax benefits due to uncertainties with respect to its ability to generate sufficient taxable income in the future.
The following tables summarize information relating to outstanding and exercisable stock options as of June 30, 2015:
|
June 30, 2015 |
|||||||||||||||||||||||
|
Outstanding |
Exercisable | ||||||||||||||||||||||
|
Weighted Average |
|
||||||||||||||||||||||
|
Number of |
Remaining |
Weighted Average |
Number of |
Weighted Average |
|||||||||||||||||||
| Exercise Prices |
Shares |
Contractual Life |
Exercise Price |
Shares |
Exercise Price |
||||||||||||||||||
|
(In Years) |
|||||||||||||||||||||||
| $2.07 | — | $2.40 | 272,000 | 8.62 | $ | 2.29 | 5,000 | $ | 2.20 | ||||||||||||||
| $2.45 | — | $2.45 | 1,035,000 | 9.25 | 2.45 | — | — | ||||||||||||||||
| $2.47 | — | $4.05 | 605,000 | 8.58 | 3.18 | 248,750 | 3.47 | ||||||||||||||||
| $4.07 | — | $75.81 | 1,452,117 | 6.55 | 9.42 | 975,338 | 11.91 | ||||||||||||||||
| 3,364,117 | 7.91 | $ | 5.58 | 1,229,088 | $ | 10.16 | |||||||||||||||||
Restricted Stock Awards. A summary of the Company’s outstanding restricted stock activity for the twelve months ended June 30, 2015 is as follows:
|
Weighted-Average |
||||||||
|
Grant Date |
||||||||
|
Shares |
Fair Value |
|||||||
|
Outstanding at June 30, 2014 |
8,750 | $ | 3.93 | |||||
|
Granted |
— | — | ||||||
|
Forfeited |
— | — | ||||||
|
Outstanding at June 30, 2015 |
8,750 | $ | 3.93 | |||||
Restricted Stock Units and Market Stock Units (MSUs). A summary of the Company’s outstanding restricted stock and market stock unit (MSU) activity for the twelve months ended June 30, 2015 is as follows:
|
Shares |
Weighted |
|||||||
|
Outstanding at June 30, 2014 |
189,427 | $ | 3.94 | |||||
|
Awarded |
47,500 | 2.41 | ||||||
|
Released |
(80,144 | ) | 3.94 | |||||
|
Forfeited |
(60,202 | ) | 3.87 | |||||
|
Unvested at June 30, 2015 |
96,581 | $ | 3.23 | |||||
In December 2013, the Company awarded 108,133 MSUs to employees that can vest on January 1, 2017. The vesting of these awards is subject to the respective employee’s continued employment through this settlement period. The number of MSUs granted represents the target number of units that are eligible to be earned based on the attainment of certain market-based criteria involving the Company’s stock price. The number of MSUs actually earned is calculated upon the vesting of the award. Participants may ultimately earn between 0% and 250% of the target number of units granted based on actual stock performance. Accordingly, additional MSUs may be issued or currently outstanding MSUs may be cancelled upon final determination of the number of awards earned. Compensation expense, including the effect of forfeitures, is recognized over the applicable service period.
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
The Company values grants of MSUs using a lattice model with a Monte Carlo simulation. This valuation methodology utilizes several key assumptions, including the 20-day average closing stock price on the grant date, expected volatility of the Company’s stock price, risk-free rates of return and expected dividend yield. The assumptions used in the Company’s valuation of the MSU’s are summarized as follows:
|
For the Twelve Month Period Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Expected dividend yield |
— | 0.00% | — | |||||||||
|
Expected stock price volatility |
— | 0.86% | — | |||||||||
|
Risk-free interest rate |
— | 0.64% | — | |||||||||
|
20-day trading average stock price on grant date |
— | $3.98 | — | |||||||||
|
Weighted-average per share grant date fair value |
— | $7.69 | — | |||||||||
As of June 30, 2015 there was $3.2 million of unrecognized share-based compensation expense related to all unvested share-based awards, not discounted for future forfeitures. This balance is expected to be recognized over a weighted-average period of two years.
|
|
|
(12) |
Retirement Benefits |
The Company contributed $0.4 million, $0.8 million and $1.1 million for the fiscal years ended June 30, 2015, 2014 and 2013, respectively, toward standard defined contribution plans for employees. Contributions by the Company for non-U.S. employees can be up to nine percent of an employee’s salary during fiscal year ending June 30, 2015 and up to four per cent of an employee’s salary for U.S. employees.
|
|
|
(13) |
Net Loss per Share |
Basic and diluted loss per share has been computed based on net loss and the weighted-average number of common shares outstanding during the applicable period. For diluted net loss per share, common stock equivalents (shares of common stock issuable upon the exercise of stock options and warrants) are excluded from the calculation of diluted net loss per share as their inclusion would be anti-dilutive. The Company has excluded all options to purchase common stock in periods indicating a loss, as their effect is anti-dilutive.
The following table sets forth the computation of historical basic and diluted net loss per share.
|
Year Ended |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Net loss (in millions) |
$ | (19.1 | ) | $ | (11.0 | ) | $ | (8.9 | ) | |||
|
Weighted average shares outstanding |
35,367,117 | 31,347,888 | 28,217,515 | |||||||||
|
Shares used to compute diluted earnings per share |
35,367,117 | 31,347,888 | 28,217,515 | |||||||||
|
Basic loss per share |
$ | (0.54 | ) | $ | (0.35 | ) | $ | (0.32 | ) | |||
|
Diluted loss per share |
$ | (0.54 | ) | $ | (0.35 | ) | $ | (0.32 | ) | |||
|
Number of antidilutive stock options excluded from computation |
3,604,737 | 2,725,441 | 1,828,668 | |||||||||
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
|
(14) |
Licenses, Royalty Collaborative and Contractual Arrangements |
Royalty agreements
The Company entered into a royalty-bearing research and license agreement with GSK in 1990 for the development and commercialization of zanamivir, a neuraminidase inhibitor (“NI”) marketed by GSK as Relenza® to treat influenza. Under the terms of the agreement, the Company licensed zanamivir to GSK on an exclusive, worldwide basis and is entitled to receive royalty payments of 7% of GSK's annual net sales of Relenza® in the U.S., Europe, Japan and certain other countries as well as 10% of GSK's annual net sales of Relenza® in Australia, New Zealand, South Africa and Indonesia. Most of the Company’s Relenza® patents have expired and the only substantial remaining intellectual property related to the Relenza® patent portfolio, which is solely owned by the Company and exclusively licensed to GSK, is scheduled to expire in July 2019 in Japan. On May 12, 2015, the Company filed a request for rehearing with the U.S. Patent and Trademark Office, Patent Trial and Appeal Board (“PTAB”) in relation to the pending patent application No. 08/737,141 related to Relenza intellectual property in the U.S. On June 23, 2015 the PTAB denied the Company’s request for a rehearing. The Company reported on September 11, 2015, that it filed an appeal in relation to the pending patent application No. 08/737,141 related to Relenza® to the United States Court of Appeals for the Federal Circuit. While the Company cannot determine the duration or the outcome of this appeal process, or how long this patent application will remain pending, if the patent claims are ultimately issued, the Company would be eligible to receive royalties from net sales of Relenza® in the U.S. for an additional 17 years from the date of allowance. If the patents claims are ultimately not issued, the Company will not receive any further royalties on U.S. sales.
The Company also generates royalty revenue from the sale of Inavir® in Japan, pursuant to a collaboration and license agreement that the Company entered into with Daiichi Sankyo in 2009. In September 2010, laninamivir octanoate was approved for sale by the Japanese Ministry of Health and Welfare for the treatment of influenza in adults and children, which Daiichi Sankyo markets as Inavir®. Under the agreement, the Company currently receives a 4% royalty on net sales of Inavir® in Japan and is eligible to earn sales milestone payments. Under the collaboration and license agreement, the Company and Daiichi Sankyo have cross-licensed the world-wide rights to develop and commercialize the related intellectual property, and have agreed to share equally in any royalties, license fees, or milestone or other payments received from any third party licenses outside of Japan. Patents on the composition of matter for laninamivir octanoate in Japan generally expire in 2024.
Collaborative and contract arrangements
In March 2011, the Company’s wholly owned subsidiary, Biota Scientific Management Pty Ltd., was awarded a contract by BARDA for the late-stage development of LANI on a cost-plus-fixed-fee basis, the total of which was not to exceed $231.2 million. BARDA is part of the U.S. Office of the Assistant Secretary for Preparedness and Response ("ASPR") within the U.S. Department of Health and Human Services ("HHS"). The BARDA contract was designed to fund and provide the Company with all technical and clinical data and U.S. based manufacturing to support the filing of a U.S. new drug application (“NDA”) with the FDA for LANI. The performance period of the BARDA contract commenced on March 31, 2011, and was intended to continue for five years. On May 7, 2014 HHS/ASPR/BARDA notified the Company of its decision to terminate the contract for the development of LANI for the convenience of the U.S. Government. The Company completed and finalized all activities related to the settlement and close out this contract in June 2015. The Company was considered an active participant in the BARDA contract, with exposure to significant risks and rewards of commercialization relating to the development of LANI. Therefore, revenues from and costs associated with the contract are recorded and recognized on a gross basis in the consolidated statement of operations.
The following tables summarize the key components of the Company’s revenues (in millions):
|
Years Ended June 30, |
||||||||||||
|
2015 |
2014 |
2013 |
||||||||||
|
Royalty revenue – Relenza® |
$ | 11.4 | $ | 10.6 | $ | 2.6 | ||||||
|
– Inavir® |
4.8 | 4.5 | 4.2 | |||||||||
|
Commercial milestone – Inavir® |
- | - | 2.8 | |||||||||
|
Revenue from services |
8.4 | 53.6 | 24.0 | |||||||||
|
Total revenue |
$ | 24.6 | $ | 68.7 | $ | 33.6 | ||||||
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
|
(15) |
Restructuring Charges |
The Company recognizes restructuring charges when a plan that materially changes the scope of its business or the manner in which that business is conducted is adopted and communicated to the impacted parties, and the expenses have been incurred or are reasonably estimable.
Fiscal 2014 Restructuring Activity
In the fourth quarter of fiscal 2014, the Company announced a restructuring as result of the termination for convenience of the BARDA contract. These restructuring activities were completed in fiscal 2015. The Company recorded $2.1 million in restructuring charges during fiscal 2014, which comprised of severance and other employee related benefits of which $0.9 million was recorded in cost of revenue, $1.0 million in research and development and $0.2 million in general and administrative. The remaining severance and other employment costs of approximately $2.0 million were paid in fiscal 2015.
Following is a reconciliation of the beginning and ending balances of the restructuring liability:
|
Balance at |
Balance at |
|||||||||||||||
|
June 30, |
June 30, |
|||||||||||||||
|
Fiscal 2014 Restructuring Plans: |
2014 |
Provision |
Payments |
2015 |
||||||||||||
|
Severance and employment costs |
2.0 | — | (2.0 |
) |
$ | — | ||||||||||
|
Total restructuring costs |
$ | 2.0 | $ | — | $ | (2.0 |
) |
$ | — | |||||||
|
(16) |
Research and Development Credit |
In fiscal 2013, an application for a claim of $4.4 million was made by the Company’s subsidiary, Biota Holdings Limited, under the Australian Government’s Research and Development tax incentive when Biota Holdings Limited submitted its tax return for its fiscal year ended June 30, 2012. This amount was recorded as a contingent asset as of June 30, 2012. On November 7, 2012, Biota Holdings Limited received cash for this claim. Although the credit is administered by the Australian government, it is not linked to the level of taxable income and is effectively a government grant. As such, the Company obtained an immediate benefit and therefore, the entire amount has been recognized within non-operating income in the consolidated statement of operations for the year ending June 30, 2013. For fiscal 2014 and 2015, the Company did not receive a research and development credit as its revenue has exceeded the qualifying revenue threshold.
|
(17) |
Quarterly Financial Information (Unaudited) |
The table below sets forth summary unaudited consolidated quarterly financial information for the years ended June 30, 2015 and 2014 (in millions):
|
Quarter Ended |
||||||||||||||||
|
6/30/2015 |
3/31/2015 |
12/31/2014 |
9/30/2014 |
|||||||||||||
|
Revenues |
$ | 4.1 | $ | 5.9 | $ | 13.9 | $ | 0.7 | ||||||||
|
Operating expenses |
24.1 | 4.8 | 7.5 | 7.7 | ||||||||||||
|
Net (loss) income |
(19.9 | ) | 1.2 | 6.5 | (6.9 | ) | ||||||||||
|
Net (loss) income per share (1): |
||||||||||||||||
|
Basic |
$ | (.55 | ) | $ | 0.02 | $ | 0.19 | $ | (0.20 | ) | ||||||
|
Diluted |
$ | (.55 | ) | $ | 0.02 | $ | 0.19 | $ | (0.20 | ) | ||||||
Biota Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
|
Quarter Ended |
||||||||||||||||
|
6/30/2014 |
3/31/2014 |
12/31/2013 |
9/30/2013 |
|||||||||||||
|
Revenues |
$ | 8.5 | $ | 29.5 | $ | 18.5 | $ | 12.2 | ||||||||
|
Operating expenses |
19.0 |
26.3 | 18.6 | 16.3 | ||||||||||||
|
Net (loss) income |
(10.2 | ) | 3.2 | (0.1 |
) |
(3.9 | ) | |||||||||
|
Net (loss) per share (1): |
||||||||||||||||
|
Basic |
$ | (0.29 | ) | $ | 0.09 | $ | 0.00 | $ | (0.15 | ) | ||||||
|
Diluted |
$ | (0.29 | ) | $ | 0.09 | $ | 0.00 | $ | (0.15 | ) | ||||||
|
|
(1) |
Due to the use of the weighted average shares outstanding for each quarter for computing earnings per share, the sum of the quarterly per share amounts may not equal the per share amount for the year. |
EXHIBIT INDEX
|
Filed |
Incorporation by Reference | |||||||||
|
Exhibit |
Exhibit Title |
with this Form |
Form |
File No. |
Date Filed | |||||
|
2.1 |
Merger Implementation Agreement, dated April 22, 2012, between Nabi Biopharmaceuticals and Biota Holdings Limited |
8-K |
001-35285-12773718 |
04/23/12 | ||||||
|
2.2 |
Amendment Deed, dated August 6, 2012, to the Merger Implementation Agreement, dated April 22, 2012, between Nabi Biopharmaceuticals and Biota Holdings Limited |
8-K |
001-35285-121016660 |
08/08/12 | ||||||
|
2.3 |
Amendment Deed, dated September 17, 2012, to the Merger Implementation Agreement, dated April 22, 2012, as amended by the Merger Implementation Agreement Amendment dated August 6, 2012, between Nabi Biopharmaceuticals and Biota Holdings Limited |
8-K |
001-35285-121096040 |
09/18/12 | ||||||
|
2.4 |
Stock Purchase Agreement, dated February 25, 2015, among Biota Pharmaceuticals, Inc., each of the shareholders of Anaconda Pharma party thereto and the Holder Representative thereunder |
10-Q |
001-35285-15847337 |
05/08/15 | ||||||
|
3.1 |
Composite Certificate of Incorporation of Biota Pharmaceuticals, Inc. |
10-Q |
001-35285-13592912 |
02/11/13 | ||||||
|
3.2 |
By-Laws of Biota Pharmaceuticals, Inc. |
10-Q |
001-35285-13592912 |
02/11/13 | ||||||
|
4.1 |
Form of Common Stock Certificate |
10-K |
000-04829-08651814 |
03/15/07 | ||||||
|
10.1† |
Collaboration and License Agreement, dated September 29, 2003, between Biota Holdings Limited and Sankyo Co., Ltd. |
10-Q |
001-35285-13834721 |
05/10/13 | ||||||
|
10.2† |
Amendment #1 to Collaboration and License Agreement, dated June 30, 2005, between Biota Holdings Limited, Biota Scientific Management Pty. Ltd. and Sankyo Company, Ltd. |
10-Q |
001-35285-13834721 |
05/10/13 | ||||||
|
10.3 |
Amendment #2 to Collaboration and License Agreement, dated March 27, 2009, between Biota Holdings Limited, Biota Scientific Management Pty. Ltd. and Daiichi Sankyo Company, Limited. |
10-Q |
001-35285-13834721 |
05/10/13 | ||||||
|
10.4† |
Commercialization Agreement, dated March 27, 2009, between Biota Holdings Limited, Biota Scientific Management Pty. Ltd and Daiichi Sankyo Company, Ltd. |
10-Q |
001-35285-13834721 |
05/10/13 | ||||||
|
10.5† |
Contract, dated March 31, 2011, between Biota Scientific Management Pty. Ltd. and Office of Biomedical Advanced Research and Development Authority within the Office of the Assistant Secretary for preparedness and Response at the U.S. Department of Health and Human Services. |
10-Q |
001-35285-13834721 |
05/10/13 | ||||||
|
10.6† |
Research and License Agreement, dated February 21, 1990, by and among Biota Scientific Management Pty. Ltd., Biota Holdings Limited, Glaxo Australia Pty. Ltd. and Glaxo Group Limited. |
10-Q |
001-35285-13834721 |
5/10/13 | ||||||
|
10.7 |
Form of Indemnification Agreement for Directors and Executive Officers |
8-K |
001-35285-13817036 |
05-06-13 | ||||||
|
10.9+ |
Employment Agreement, dated as of October 1, 2014, between Biota Pharmaceuticals, Inc., and Russell H. Plumb |
10-Q |
001-35285-15584221 |
02/06/15 | ||||||
|
10.10+ |
Amended Executive Employment Agreement, dated as of October 1, 2014, between Biota Pharmaceuticals, Inc., and Joseph M. Patti |
10-Q |
001-35285-15584221 |
02/06/15 | ||||||
|
10.11+ |
Form Non-Plan Stock Units Agreement |
8-K |
001-35285-121206005 |
11/14/12 | ||||||
|
10.12+ |
Form of Letter Agreement for Stock Option Grant |
8-K |
001-35285-121206005 |
11/14/12 | ||||||
|
10.13+ |
2007 Omnibus Equity and Incentive Plan |
DEF 14A |
000-04829-07763351 |
04/12/07 | ||||||
|
10.14+ |
Executive Employment Agreement, dated as of November 26, 2013, between Biota Pharmaceuticals, Inc., and Peter Azzarello |
8-K |
001-35285-131247987 |
11/27/13 | ||||||
|
10.15+ |
Form of Employee Stock Option Agreement under the 2007 Omnibus Equity and Incentive Plan |
8-K |
001-35285-131266832 |
12/10/13 | ||||||
|
10.16+ |
Form of Market-Based Stock Unit Award Agreement under the 2007 Omnibus Equity and Incentive Plan |
8-K |
001-35285-131266832 |
12/10/13 |
|
16.2 |
Letter from PricewaterhouseCoopers dated April 29, 2014 |
8-K |
001-35285-14795541 |
4/30/14 | ||||||
|
21.1 |
List of Subsidiaries |
X |
||||||||
|
23.1 |
Consent of PricewaterhouseCoopers LLP. |
X |
||||||||
|
23.1a |
Consent of PricewaterhouseCoopers. |
X |
||||||||
|
23.2 |
Consent of IMS Consulting Group |
X |
||||||||
|
31.1* |
Certification of Principal Executive Officer Required Under Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended |
X |
||||||||
|
31.2* |
Certification of Principal Financial Officer Required Under Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended |
X |
||||||||
| 32.1* | Certification of Principal Executive Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. §1350 | X | ||||||||
| 32.2* | Certification of Principal Financial Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. §1350 | X | ||||||||
|
101** |
XBRL Instance Document |
X |
||||||||
|
101** |
XBRL Taxonomy Extension Schema Document |
X |
||||||||
|
101** |
XBRL Taxonomy Calculation Document |
X |
||||||||
|
101** |
XBRL Taxonomy Definition Linkbase Document |
X |
||||||||
|
101** |
XBRL Taxonomy Label Linkbase Document |
X |
||||||||
|
101** |
XBRL Taxonomy Presentation Linkbase Document |
X |
+ Indicates a management contract or compensatory plan or arrangement in which any director or named executive officer participates.
† Confidential treatment has been granted with respect to certain portions of this exhibit.
* This certification is being furnished solely to accompany this annual report pursuant to 18 U.S.C. Section 1350, and is not being filed for purposes of Section 18 of the Securities Exchange Act of 1934 and is not to be incorporated by reference into any filing of Biota Pharmaceuticals, Inc., whether made before or after the date hereof, regardless of any general incorporation language in such filing.
** Furnished, not filed.
F-26