Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Timber Pharmaceuticals, Inc. | Financial_Report.xls |
| EX-31.1 - EX-31.1 - Timber Pharmaceuticals, Inc. | a2223386zex-31_1.htm |
| EX-4.3 - EX-4.3 - Timber Pharmaceuticals, Inc. | a2223386zex-4_3.htm |
| EX-23.1 - EX-23.1 - Timber Pharmaceuticals, Inc. | a2223386zex-23_1.htm |
| EX-21.1 - EX-21.1 - Timber Pharmaceuticals, Inc. | a2223386zex-21_1.htm |
| EX-32.1 - EX-32.1 - Timber Pharmaceuticals, Inc. | a2223386zex-32_1.htm |
| EX-10.4 - EX-10.4 - Timber Pharmaceuticals, Inc. | a2223386zex-10_4.htm |
| EX-10.15 - EX-10.15 - Timber Pharmaceuticals, Inc. | a2223386zex-10_15.htm |
| EX-4.2 - EX-4.2 - Timber Pharmaceuticals, Inc. | a2223386zex-4_2.htm |
Use these links to rapidly review the document
Table of Contents
FINANCIAL STATEMENTS
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark One) | ||
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2014 |
||
or |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the transition period from to |
||
Commission File No. 000-54871
BioPharmX Corporation
(Exact name of registrant as specified in its charter)
| Delaware (State or Other Jurisdiction of Incorporation or Organization) |
59-3843182 (I.R.S. Employer Identification No.) |
|
1098 Hamilton Court, Menlo Park, California (Address of principal executive offices) |
94025 (Zip Code) |
Registrant's telephone number, including area code: 650-889-5020
Securities registered pursuant to Section 12(b) of the Act: None.
Securities registered pursuant to Section 12(g) of the Act: Common Stock, $0.001 Par Value.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by checkmark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company.
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company ý |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
As of June 30, 2014, the last day of the Registrant's most recently completed second fiscal quarter, the aggregate market value of the shares of the Registrant's common stock held by non-affiliates was $1,300,000. Shares of the Registrant's common stock held by each executive officer and director and by each person who owns 10 percent or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates of the Registrant. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of March 24, 2015, there were outstanding 11,415,416 shares of the registrant's common stock, $.001 par value.
Documents incorporated by reference: None.
BioPharmX Corporation
Form 10-K
2
This Annual Report on Form 10-K, including the section entitled "Management's Discussion and Analysis of Financial Condition and Results of Operations," contains forward-looking statements regarding future events and our future results that are subject to the safe harbors created under the Securities Act of 1933, as amended (the Securities Act), and the Securities Exchange Act of 1934, as amended (the Exchange Act). Words such as "expect," "anticipate," "target," "goal," "project," "hope," "intend," "plan," "believe," "seek," "estimate," "continue," "may," "could," "should," "might," variations of such words and similar expressions are intended to identify such forward-looking statements. In addition, any statements other than statements of historical fact are forward-looking statements, including statements regarding overall trends, operating cost and revenue trends, liquidity and capital needs and other statements of expectations, beliefs, future plans and strategies, anticipated events or trends and similar expressions. We have based these forward-looking statements on our current expectations about future events. These statements are not guarantees of future performance and involve risks, uncertainties and assumptions that are difficult to predict. Our actual results may differ materially from those suggested by these forward-looking statements for various reasons. Given these risks, uncertainties and assumptions you are cautioned not to place undue reliance on forward-looking statements. The forward-looking statements included in this report are made only as of the date hereof. Except as required under federal securities laws and the rules and regulations of the Securities and Exchange Commission (SEC), we do not undertake, and specifically decline, any obligation to update any of these statements or to publicly announce the results of any revisions to any forward-looking statements after the distribution of this report, whether as a result of new information, future events, changes in assumptions or otherwise.
Unless the context otherwise requires, we use the terms "BioPharmX," "company," "we," "us" and "our" in this Annual Report on Form 10-K to refer to BioPharmX Corporation and its subsidiary.
3
Overview
BioPharmX Corporation is incorporated under the laws of the State of Delaware and originally incorporated on August 30, 2010 in Nevada under the name Thompson Designs, Inc. We have one wholly-owned subsidiary, BioPharmX, Inc., a Nevada corporation. Our headquarters are located at 1098 Hamilton Court, Menlo Park, California.
We are a specialty pharmaceutical company focused on utilizing our proprietary drug delivery technologies to develop and commercialize novel prescription and over-the-counter, or OTC, products that address large markets in women's health and dermatology. Our objective is to develop products that treat health or age-related conditions that: (1) are not presently being addressed or treated at all or (2) are currently treated with drug therapies or drug delivery approaches that are sub-optimal. Our strategy is designed to bring new products to market by identifying optimal delivery mechanisms and/or alternative applications for FDA-approved active pharmaceutical ingredients, or APIs, while, in appropriate circumstances, reducing the time, cost and risk typically associated with new product development by repurposing drugs with demonstrated safety profiles and taking advantage of the regulatory approval pathway under Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, or FDC Act. We believe the 505(b)(2) regulatory pathway may reduce drug development risk and could reduce the time and resources we spend during development. Our current platform technologies include innovative delivery mechanisms for molecular iodine, or I2, and antibiotics.
Our management team has experience in formulation development, intellectual property generation, clinical trial execution, regulatory strategy definition and commercialization of products through licensing as well as direct to consumer. Our business model is to outsource our manufacturing and at times commercialization activities in order to maintain our focus on technology sourcing, acquisitions, and strategic partner development to create new products to address unmet needs in well-defined global markets. Our current portfolio of product candidates targets significant market opportunities and includes two clinical stage product candidates: (1) BPX01, a topical antibiotic for the treatment of acne and (2) BPX03, a molecular iodine (I2) tablet for the treatment of benign breast pain associated with fibrocystic breast condition, or FBC, and cyclic mastalgia.
Since our inception, we have devoted our efforts to developing our product candidates including conducting preclinical and clinical trials and providing general and administrative support for these operations, and we commercially launched our breast health dietary supplement at the end of 2014. To date we have not generated any revenue from product sales. We have financed our operations primarily through the sale of equity securities and convertible debt securities from which we raised $9.6 million of net cash from our inception through December 31, 2014.
Share Exchange
On January 23, 2014, we (then operating as Thompson Designs, Inc.), BioPharmX, Inc. and stockholders of BioPharmX, Inc., who collectively owned 100% of BioPharmX, Inc., entered into and consummated transactions pursuant to a share exchange agreement, such transaction referred to as the Share Exchange, whereby we issued to the stockholders of BioPharmX, Inc. an aggregate of 7,025,000 shares of our common stock, in exchange for 100% of the shares of BioPharmX, Inc. held by stockholders. The shares of our common stock received by the stockholders of BioPharmX, Inc. in the Share Exchange constituted approximately 77.8% of our then issued and outstanding common stock, after giving effect to the issuance of shares pursuant to the share exchange agreement. As a result of the Share Exchange, BioPharmX, Inc. became our wholly-owned subsdidiary. For accounting purposes, the Share Exchange was treated as a reverse acquisition with BioPharmX, Inc. as the acquirer and us
4
as the acquired party, and as a result the historical financial statements prior to the Share Exchange included in this Annual Report on Form 10-K are the historical financial statements of BioPharmX, Inc. On March 3, 2014, we changed our name to BioPharmX Corporation. On April 25, 2014, we reincorporated from Nevada to Delaware.
Our Product Candidates
Our first commercial product, VI2OLET iodine, is a once-a-day OTC dietary supplement molecular iodine tablet that promotes overall breast health and is for the alleviation of benign breast pain associated with fibrocystic breast condition, or FBC. We launched VI2OLET iodine in December 2014 and are rolling out the product in drug store and retail chains throughout the United States. We are also developing a prescription molecular iodine tablet, BPX03, for the treatment of benign breast pain associated with FBC and cyclic mastalgia, intended for global distribution, where products such as ours may require a prescription due to regulatory requirements. We are preparing to conduct clinical studies under institutional review board, or IRB, oversight to inform the study design for our Phase 3 safety and efficacy study. We are planning to commence a Phase 3 clinical trial for BPX03 to support FDA and foreign regulatory requirements upon completion of the IRB studies and submission of our investigational new drug application, or IND, for BPX03. We shall seek approval only in those countries where we will seek to market the prescription product. It is our intent to commence a Phase 3 study in 2016.
We are also developing BPX01, a non-lipophilic, topical antibiotic for the treatment of acne. BPX01 utilizes a transepidermal delivery mechanism for minocycline and other APIs that we believe has the potential to kill p. acnes bacteria without the systemic side effects of orally-administered antibiotics. In addition, BPX01 has been shown in pre-clinical studies to possess anti-inflammatory properties, which reduce swelling and slow hyper-cornification. We are currently conducting an animal toxicity study, after which we expect to submit our IND to the FDA to initiate our first Phase 2a clinical trial of BPX01. We are also preparing to conduct a bridging safety study using oral minocycline as the comparator and a Phase 2 dose-finding clinical study for BPX01. We intend to pursue regulatory approval under Section 505(b)(2) of the FDC Act. We believe the 505(b)(2) regulatory pathway, which permits us to rely in part on FDA's prior findings of safety and/or efficacy for an approved product, may reduce the drug development risk and could reduce the time and resources we spend during development of BPX01.
Our product pipeline includes additional applications for the delivery of iodine, FDA-approved antibiotics, and biologics. Product candidates may be developed for delivery in oral, topical, inhalant and/or injectable forms depending on the platform technology employed and the underlying condition being treated.
Target Markets
We believe that the industry dynamics in the areas of women's health and dermatology represent significant opportunities for innovative new products to emerge as attractive solutions for unmet needs in multi-billion dollar therapeutic categories. In particular, we believe that both the women's health and dermatology markets are large specialty markets with significant global patient demand. We believe that our focus on these markets coupled with our proprietary platform technologies will enable us to develop and commercialize attractive products within these areas of women's health and dermatology.
5
Products and Pipeline
Overview
Our product portfolio has been developed using our proprietary drug delivery technologies including innovative delivery mechanisms for molecular iodine and antibiotics. We currently have one marketed product, VI2OLET iodine, and two clinical-stage product candidates, BPX01 and BPX03.
VI2OLET Iodine
Our first commercial product, VI2OLET iodine, is the only OTC molecular iodine dietary supplement that addresses cyclic breast discomfort and is clinically demonstrated to alleviate the symptoms associated with fibrocystic breast changes including tenderness, swelling and aches. Our patented molecular iodine (I2) formula is delivered to breast tissue and reduces the breast cell build-up that results in breast discomfort. Women who suffer from menstrual-related breast discomfort are recommended to take one tablet per day on an empty stomach for at least 60 days to realize initial symptom relief. They may take a second tablet every evening if they have more severe symptoms. Additionally, with consistent daily use, VI2OLET iodine has been shown to help maintain healthy breast tissue.
The product is currently available for sale in approximately 2,960 CVS retail pharmacy chains and 650 Vitamin Shoppe stores throughout the United States, as well as online through drugstore.com and walgreens.com.
The commercial launch of VI2OLET iodine is supported by an extensive consumer marketing program targeting women between the ages of 30 and 44. With a combination of brand and shopper marketing, both nationally and locally, we generate awareness, engagement, education, consideration and purchase interest.
BPX03
In addition to our VI2OLET iodine product, we are also developing BPX03, a prescription drug version of our molecular iodine (I2) tablet for the treatment of benign breast pain associated with FBC and cyclic mastalgia. We have licensed the patent rights to a set of iodine technologies. The licensors previously sponsored and completed Phase 1 and Phase 2 clinical studies. We intend to approach the FDA in 2016 for a pre-IND discussion regarding the study design for a Phase 3 clinical trial intended to commence in 2016 with a new IND submission.
BPX01
We are developing BPX01, a novel, topical formulation of minocycline. BPX01 delivers minocycline directly to the target sebaceous glands in the skin. We believe that our proprietary topical minocycline acne treatment is designed to have several advantages compared to both orally-administered and other topically-administered retinoid- and antibiotic-based solutions. Since BPX01 is not administered orally, its delivery route to the target site is not primarily through the bloodstream, and it therefore has the potential to lower the risk of systemic side effects common to orally-administered antibiotics. The gel form of BPX01, when applied topically, is designed to penetrate through the intercellular space among corneocytes in the stratum corneum to increase the delivery of the antibiotic at low dosages directly to the affected area. Unlike other topical lipophilic solutions formulated to ensure active pharmaceutical ingredient, or API, stability, BPX01 is non-lipophilic, which improves the aesthetic appearance and feel of the topical and is designed to allow the topical to be absorbed more quickly by the skin, and, we believe, without sacrificing long-term API stability.
6
Research and Development
Our core competency is providing the link between concept and commercialization through focused, practical product development based on innovative research. We employ highly-qualified scientists and consultants specializing in our various product development areas.
As a Silicon Valley-based company, we are located in a region with many strong biotechnology and pharmaceutical companies, which have drawn a high caliber of scientists and scientific support staff to the region. While there is intense competition for this type of personnel, we believe our location will enable us to expand our product development and consultant resources as our business grows. Our location also provides us with convenient access to local formulation resources and pre-clinical testing facilities.
Technology and Intellectual Property
Overview
Our success depends in large part upon our ability to obtain and maintain proprietary protection for our products and platform technologies. Our goal is to develop a strong intellectual property portfolio that enables us to capitalize on the research and development that we have performed to date and will perform in the future, particularly for each of the products in our development pipeline and each of the products marketed by us. We rely on a combination of patent, copyright, trademark and trade secret laws in the United States and other countries to obtain and maintain our intellectual property. We protect our intellectual property by, among other methods, filing for patent applications on inventions that are important to the development and conduct of our business with the United States Patent and Trademark Office and its foreign counterparts.
We also rely on a combination of non-disclosure, confidentiality, and other contractual restrictions to protect our technologies and intellectual property. We require our employees and consultants to execute confidentiality agreements in connection with their employment or consulting relationships with us. We also require them to agree to disclose and assign to us all inventions conceived in connection with the relationship.
Patents
Patent protection is an important aspect of our product development process and we are actively developing intellectual property in-house. In addition to an aggressive licensing strategy, we have several pending patent applications related to our novel iodine-based technologies for women's health and topical compositions for dermatological conditions. We have both United States provisional and utility patent applications pending. We also have pending international patent applications, which were filed according to the Patent Cooperation Treaty and which enable us to apply for patent protection for the described inventions in key individual countries in the future.
Our patent applications may not result in issued patents and we cannot assure you that any patents that issue will provide a competitive advantage. Moreover, any patents issued to us may be challenged by third parties as invalid or parties may independently develop similar or competing technology or design around our patents. We cannot be certain that the steps we have taken will prevent the misappropriation of our intellectual property, particularly in foreign countries where the laws may not protect our proprietary rights as fully as in the United States.
On March 1, 2013, we entered into a collaboration and license agreement with Iogen LLC, or Iogen, to license certain patents, formulations, and know-how relating to molecular iodine formulations. Our license is an exclusive, royalty-bearing license agreement with the right to enforce and sublicense. These patents have expiration dates between 2017 and 2029.
7
Strategic Alliances and Partnerships
We have entered into strategic alliances/partnerships with Iogen and NuTech Medical, Inc., or NuTech.
Iogen
We have executed collaboration and licensing agreements with Iogen, a biotechnology company with iodine-based solutions and associated intellectual property. Our molecular iodine dietary OTC product, VI2OLET iodine, and the development of our molecular iodine prescription product, BPX03, build upon this licensed technology and its associated intellectual property. Under the terms of the agreement, we received an exclusive worldwide perpetual irrevocable license to Iogen's patented technology relating to an oral iodine tablet. In consideration of the license granted under the agreement, we agreed to pay to Iogen a non-refundable license issue fee of $150,000, which we paid in full, and 30% of net profit associated with direct commercialization of an OTC iodine tablet product or 30% of net royalties received from any sub-licensee. For other products developed and commercialized using licensed technology and associated intellectual property covered by this agreement, including a prescription iodine tablet, we agreed to pay to Iogen a royalty of 3% of net sales for the first 12 months of commercialization and 2% of net sales thereafter.
NuTech
We have executed a collaboration and supply agreement with NuTech, a biologics company specializing in the spinal and orthopedics market. This agreement describes the collaboration between Nutech and us to develop products in the field of dermatology. Products and intellectual property developed under this agreement are exclusively owned by us and licensed to NuTech for use in indications outside of dermatology. In exchange for an exclusive license to NuTech's intellectual property in the field of dermatology, we will pay to NuTech a royalty of 3% of net sales on product sold in the field of dermatology. In exchange for granting NuTech an exclusive license to our intellectual property and intellectual property developed in collaboration with NuTech in indications outside of dermatology, we shall receive from NuTech a royalty of 3% of net sales on products sold by them.
Trademarks
We have applied for trademark protection for several trademarks in the United States. The United States Patent and Trademark Office has issued us a Notice of Acceptance of Statement of Use for the trademark "VIOLET" which means the mark will now register barring any extraordinary circumstances. We have received a Notice of Allowance from the United States Patent and Trademark Office for "BIOPHARMX," "VI2OLET," and "GET IT OFF YOUR CHEST." In the future, we may apply for trademark protection for one or more of these trademarks in key markets outside the United States.
Manufacturing, Supply and Production
Suppliers
The company has in place a commercial supply agreement with UPM Pharmaceuticals, or UPM, to manufacture and package its VI2OLET iodine tablets. As our volume grows, we will consider expanding to multiple suppliers to mitigate the risk of having a single source. Our joint development agreement with NuTech specifies that NuTech will supply materials for certain of our dermatological products.
8
Manufacturing
The company utilizes contract manufacturers to produce its products for commercial distribution. We have no plans to establish in-house manufacturing capabilities for large-scale production at this time.
UPM, an independent drug development and contract manufacturer serving the pharmaceutical and biotechnology industries and a division of Gregory Pharmaceutical Holdings, Inc., manufactures solid dose iodine supplement tablets for our VI2OLET iodine product. VI2OLET iodine is manufactured at UPM's 475,000 square-foot manufacturing facility in Bristol, Tennessee, under a commercial supply agreement. UPM provides high-quality drug development services including formulation development, the FDA's current good manufacturing practices, or cGMP clinical and commercial manufacturing, analytical methods development and stability testing. As our volume grows, we will consider expanding to multiple manufacturers to mitigate the risk of having a single source.
Marketing, Sales & Distribution
We plan to commercialize women's health and dermatology products in our pipeline into various channels, beginning with our VI2OLET iodine dietary supplement, which we launched in December 2014 and are currently rolling out in drug stores and retail chains throughout the United States.
Our product launch for VI2OLET iodine is supported by a marketing program, including in-store merchandising, a digital strategy focused on education and activation, public relations events and traditional media to drive awareness and a physician and pharmacist influencer program.
Customers
Potential customers for our products and product candidates include: pharmaceutical companies physician's practices, including OB-Gyn's, dermatologists and general practioners; and retail customers via retail sales channels and/or pharmacy outlets.
Competition
FBC and Cyclic Mastalgia
Our competitors, typically large pharmaceutical companies, vary from product to product. In the area of women's health, many companies sell iodine supplements, mostly for the purpose of delivering iodine with iodide salts to address hypothyroidism as iodine replacement therapy, as opposed to targeting breast tissue. We believe our competitive advantage is our solid dose proprietary formulation which delivers molecular iodine in a stable manner allowing the consumer to ingest orally and specifically to address breast symptoms. Addressing a condition that has long been neglected, we believe that VI2OLET iodine dietary supplement and, if approved, BPX03, are essentially new products in a new category.
9
The following figure presents a typical treatment algorithm for FBC given the current/limited options available to physicians.
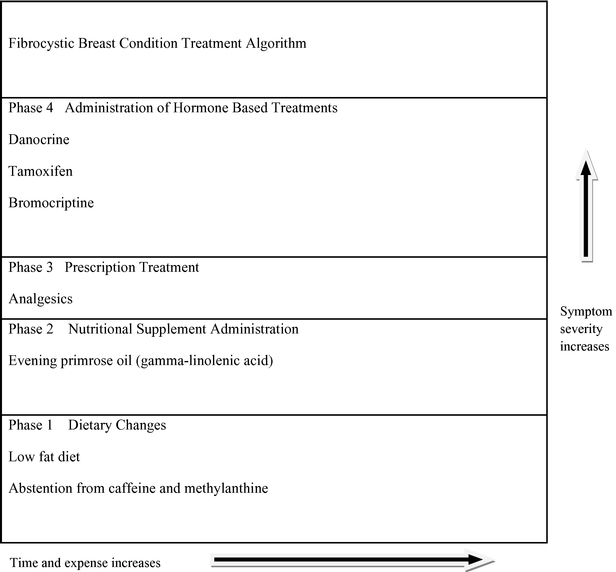
Some limitations of competitive approaches to addressing FBC and/or cyclic mastalgia include serious and sometimes dangerous side effects caused by prescription drugs and the temporary nature of relief provided by analgesics. Because optimal solutions do not exist, the majority of women choose to live with chronic pain.
Acne
While the acne market has a number of competitive products, BPX01 is being developed to combine the most successful oral approach for the treatment of moderate-severe acne without systemic side effects with a targeted topical antibiotic technology specifically designed to localize the delivery of the API. At the present time, there is no FDA-approved topical solution for this API that provides similar or equal clinical efficacy to that of oral treatments.
A number of approved prescription acne products currently exist in oral form such as isotretinoins, antibiotics, antimicrobials and contraceptives. These treatments are marketed by a number of large pharmaceutical and specialty pharmaceutical companies including, but not limited to, Valeant, Allergan, Pharmacia, Pfizer, Galderma S.A., Teva, and Bayer Healthcare. Additionally, there are also several
10
prescription acne products that exist in topical form such as antimicrobials, retinoids, or some combination of the two. The majority of these topical solutions are marketed by GlaxoSmithKline, Galderma S.A., Allergan, Valeant, and Mylan.
In addition to prescription acne therapies discussed above, there are numerous OTC products in the form of benzoyl peroxide and salicylic acid topical solutions available from various cosmetic and cosmeceutical companies such as Neutrogena, Clean & Clear, Aveeno, Proactiv, and Clearasil. Energy-based devices have also been widely used by dermatologists, such as intense pulsed light, or IPL, by Ellipse and combination of IPL and radiofrequency, or RF, devices, elos, by Syneron. Combination drug-device treatments such as photodynamic therapy, or PDT, with Blu-U by Dusa Pharmaceuticals, has been used off-label for treating acne, while the Blu-U light source without its PDT drug has been indicated for acne treatment.
Government Regulation
In the United States, foods (including dietary supplements), drugs (including biological products), medical devices, cosmetics, tobacco products, and radiation-emitting products are subject to extensive regulation by the FDA. The FDC Act and other federal and state statutes and regulations govern, among other things, the manufacture, distribution, and sale of these products. These laws and regulations prescribe criminal and civil penalties that can be assessed, and violation of these laws and regulations can result in enforcement action by the FDA and other regulatory agencies.
Regulation of Dietary Supplements
The formulation, manufacturing, packaging, labeling, advertising, distribution and sale (hereafter, "sale" or "sold" may be used to signify all of these activities) of dietary supplements are subject to regulation by one or more federal agencies, primarily the FDA and the Federal Trade Commission, or the FTC, and to a lesser extent the Consumer Product Safety Commission, or the CPSC.
Dietary supplements are also regulated by various governmental agencies for the states and localities in which product are sold. Among other matters, regulation by the FDA and the FTC is concerned with product safety, efficacy, and claims made with respect to a dietary supplement's ability to provide health related benefits. The FDA, under the FDC Act, regulates the formulation, manufacturing, packaging, labeling, distribution and sale of food, including dietary supplements. The FTC regulates the advertising of these products. The National Advertising Division, or NAD, of the Council of Better Business Bureaus oversees an industry sponsored, self-regulatory system that permits competitors to resolve disputes over advertising claims. The NAD has no enforcement authority of its own, but may refer matters that appear to violate the Federal Trade Commission Act, or FTC Act, or the FDC Act to the FTC or the FDA for further action, as appropriate.
Federal agencies, primarily the FDA and the FTC, have a variety of procedures and enforcement remedies available to them, including initiating investigations, issuing warning letters and cease and desist orders, requiring corrective labeling or advertising, requiring consumer redress (for example, requiring that a company offer to repurchase products previously sold to consumers), seeking injunctive relief or product seizures, imposing civil penalties or commencing criminal prosecution. In addition, certain state agencies have similar authority.
The Dietary Supplement Health and Education Act, or DSHEA, was enacted in 1994 and amended the FDC Act. DSHEA establishes a statutory class of dietary supplements, which includes vitamins, minerals, herbs, amino acids and other dietary ingredients for human use to supplement the diet. Dietary ingredients marketed in the U.S. before October 15, 1994 may be marketed without the submission of a new dietary ingredient, or NDI, premarket notification to the FDA. Dietary ingredients not marketed in the U.S. before October 15, 1994 may require the submission, at least 75 days before marketing, of an NDI notification containing information establishing that the ingredient is reasonably
11
expected to be safe for its intended use. Among other things, DSHEA prevents the FDA from regulating dietary ingredients in dietary supplements as food additives.
The FDA issued a draft guidance document in July 2011 that clarifies when the FDA believes a dietary ingredient is an NDI, when a manufacturer or distributor must submit an NDI premarket notification to the FDA, the evidence necessary to document the safety of an NDI and the methods for establishing the identity of an NDI. The FDA's interpretation of what constitutes an NDI is extremely broad and seems to imply that virtually every new dietary supplement requires a premarket notification. Although the industry has objected and questioned FDA's authority, it is unclear whether the FDA will make any changes to the draft guidance, and, if the agency does make changes, what changes will be made. In addition, the FDA may begin to take enforcement actions consistent with the interpretations in the draft guidance before issuing a final version.
The FDA's current good manufacturing practices, or cGMPs, regulations for dietary supplements apply to manufacturers and holders of finished dietary supplement products, including dietary supplements manufactured outside the U.S. that are imported for sale into the U.S. Among other things, the FDA's cGMPs: (a) require identity testing on all incoming dietary ingredients, (b) call for a scientifically valid system for ensuring finished products meet all specifications, (c) include requirements related to process controls, including statistical sampling of finished batches for testing and requirements for written procedures and (d) require extensive recordkeeping.
Under the Dietary Supplement and Nonprescription Drug Consumer Protection Act, FDA requires, among other things, that companies that manufacture or distribute nonprescription drugs or dietary supplements report serious adverse events associated with their products to the FDA and institute recordkeeping requirements for all adverse events. Based on serious adverse event (or other) information, the FDA or another agency may take actions against dietary supplements or dietary ingredients that in its determination present a significant or unreasonable risk of illness or injury, which could make it illegal to sell those products.
The FDA Food Safety Modernization Act, or FSMA, enacted January 4, 2011, amended the FDC Act to significantly enhance FDA's authority over various aspects of food regulation, including dietary supplements. Under FSMA, FDA may use the mandatory recall authority when the FDA determines there is a reasonable probability that a food is adulterated or misbranded and that the use of, or exposure to, the food will cause serious adverse health consequences or death to humans or animals. Also under FSMA, FDA has expanded access to records; the authority to suspend food facility registrations and require high risk imported food to be accompanied by a certification; stronger authority to administratively detain food; the authority to refuse admission of an imported food if it is from a foreign establishment to which a U.S. inspector is refusing entry for an inspection; and the requirement that importers verify that the foods they import meet domestic standards.
One of FSMA's more significant changes is the requirement of preventive controls for food facilities required to register with the FDA, except dietary supplement facilities in compliance with both cGMPs and the serious adverse event reporting requirements. Although dietary supplement facilities are exempt from the preventive controls requirements, dietary ingredient facilities might not qualify for the exemption. The FDA's proposed preventive controls regulations, issued in February 2013 and supplemented in September 2014, would require that facilities develop and implement preventive controls (including supplier controls) to assure that identified hazards are significantly minimized or prevented, monitor the effectiveness of the preventive controls, and maintain numerous records related to those controls. FSMA also requires that importers implement a foreign supplier verification program, or FSVP. The FDA's proposed FSVP regulations, issued in July 2013 and supplemented in September 2014, would require importers to implement supplier verification activities to ensure that the foods they import meet domestic standards, with a partial exemption that might or might not apply to
12
certain importers of dietary ingredients. When implemented, the FSVP requirements may affect the cost and the availability of dietary supplements and dietary ingredients.
The new FSMA requirements, as well as the FDA enforcement of the NDI draft guidance, can result in the detention and refusal of admission of imported products, the injunction of manufacturing of any dietary ingredients or dietary supplements until the FDA determines that such ingredients or products are in compliance, and the potential imposition of fees for re-inspection of noncompliant facilities.
The FDC Act, as amended by DSHEA, permits statements of nutritional support often referred to as "structure/function claims" to be included in labeling for dietary supplements without FDA pre-market approval. FDA regulation requires that FDA be notified of those statements within 30 days of marketing. Among other things, the statements may describe the role of a dietary ingredient intended to affect the structure or function of the body or characterize the documented mechanism of action by which a dietary ingredient maintains such structure or function, but may not expressly or implicitly represent that a dietary supplement will diagnose, cure, mitigate, treat, or prevent a disease. A company that uses a statement of nutritional support in labeling must possess information substantiating that the statement is truthful and not misleading. If the FDA determines that a particular statement of nutritional support is an unacceptable drug claim or an unauthorized version of a health claim, or if the FDA determines that a particular claim is not adequately supported by existing information or is otherwise false or misleading, the claim could not be used and any product bearing the claim could be subject to regulatory action.
The FTC and the FDA have pursued a coordinated effort to challenge the scientific substantiation for dietary supplement claims. Their efforts to date have focused on manufacturers and marketers as well as media outlets and have resulted in a significant number of investigations and enforcement actions, some resulting in civil penalties under the FTC Act of several million dollars. If the FTC and the FDA continue to focus on health related claims, including structure/function claims for dietary supplements, dietary supplements could be the subject of FTC and/or FDA inquiries, inquiries from the NAD and states Attorney Generals, as well as private class action lawsuits.
All states regulate foods and drugs under laws that generally parallel federal statutes. These products are also subject to state consumer health and safety regulations, such as California Safe Drinking Water and Toxic Enforcement Act of 1986, or Proposition 65. Violation of Proposition 65 may result in substantial monetary penalties.
FDA Regulation of Drugs
New Drug Approval Process
Pharmaceutical products are subject to extensive regulation by the FDA. The FDC Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending new drug applications, or NDAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical product development for a new product or certain changes to an approved product in the United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an investigational new drug application, or IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA
13
pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation, and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice, or GCP, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; as well as (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB's requirements, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage tolerance, and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the U.S. The NDA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product's pharmacology, chemistry, manufacture, and controls. The cost of preparing
14
and submitting an NDA is substantial. The submission of most NDAs is additionally subject to a substantial application user fee, currently exceeding $2,335,000, and the manufacturer and/or sponsor under an approved new drug application are also subject to annual product and establishment user fees, currently exceeding $110,000 per product and $569,000 per establishment. These fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency's threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of new drug applications. Most such applications for standard review drug products are reviewed within ten to twelve months; most applications for priority review drugs are reviewed in six to eight months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. For biologics, priority review is further limited to drugs intended to treat a serious or life-threatening disease relative to the currently approved products. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission.
The FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee—typically a panel that includes clinicians and other experts—for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with current good manufacturing practices, or cGMPs, is satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug's safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
15
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
The Best Pharmaceuticals for Children Act, or BPCA, provides NDA holders a six-month extension of any exclusivity—patent or non-patent—for a drug if certain conditions are met. Conditions for exclusivity include the FDA's determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
The Hatch-Waxman Amendments
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant's product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, pre-clinical or clinical tests to prove the safety or effectiveness of their drug product. Drugs approved in this way are commonly referred to as "generic equivalents" to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA's Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or
16
(iv) the listed patent is invalid or will not be infringed by the new product. The ANDA applicant may also elect to submit a section viii statement certifying that its proposed ANDA label does not contain (or carves out) any language regarding the patented method-of-use rather than certify to a listed method-of-use patent. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
A certification that the new product will not infringe the already approved product's listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the referenced product has expired.
Exclusivity
Upon NDA approval of a new chemical entity or NCE, which is a drug that contains no active moiety that has been approved by the FDA in any other NDA, that drug receives five years of marketing exclusivity during which FDA cannot receive any ANDA seeking approval of a generic version of that drug. Certain changes to a drug, such as the addition of a new indication to the package insert, are associated with a three-year period of exclusivity during which FDA cannot approve an ANDA for a generic drug that includes the change.
An ANDA may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, there may not be a Paragraph IV certification, and, thus, no ANDA may be filed before the expiration of the exclusivity period.
Patent Term Extension
After NDA approval, owners of relevant drug patents may apply for up to a five year patent extension. The allowable patent term extension is calculated as half of the drug's testing phase—the time between IND application and NDA submission—and all of the review phase—the time between NDA submission and approval up to a maximum of five years. The time can be shortened if the FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the U.S. Patent and Trademark Office must determine that approval of the drug covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA has not been submitted.
Section 505(b)(2) New Drug Applications
Most drug products obtain FDA marketing approval pursuant to an NDA or an ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2), or 505(b)(2), NDA,
17
which enables the applicant to rely, in part, on the FDA's previous approval of a similar product, or published literature, in support of its application.
505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. If the 505(b)(2) applicant can establish that reliance on the FDA's previous approval is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all, or some, of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. Thus approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, risk evaluation and mitigation strategies, or REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality-control, drug manufacture, packaging, and labeling procedures must continue to conform to cGMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Regulation Outside the United States
In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of drug products. Whether or not it obtains FDA approval for a
18
product, the company would need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trials or marketing of the product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
Regulation and Marketing Authorization in the European Union
The process governing approval of medicinal products in the European Union follows essentially the same lines as in the United States and, likewise, generally involves satisfactorily completing each of the following:
- •
- preclinical laboratory tests, animal studies and formulation studies all performed in accordance with the applicable E.U. Good
Laboratory Practice regulations;
- •
- submission to the relevant national authorities of a clinical trial application, or CTA, which must be approved before human clinical
trials may begin;
- •
- performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each proposed
indication;
- •
- submission to the relevant competent authorities of a marketing authorization application, or MAA, which includes the data supporting
safety and efficacy as well as detailed information on the manufacture and composition of the product in clinical development and proposed labelling;
- •
- satisfactory completion of an inspection by the relevant national authorities of the manufacturing facility or facilities, including
those of third parties, at which the product is produced to assess compliance with strictly enforced current cGMP;
- •
- potential audits of the non-clinical and clinical trial sites that generated the data in support of the MAA; and
- •
- review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product.
Preclinical Studies
Preclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animal studies, in order to assess the potential safety and efficacy of the product. The conduct of the preclinical tests and formulation of the compounds for testing must comply with the relevant E.U. regulations and requirements. The results of the preclinical tests, together with relevant manufacturing information and analytical data, are submitted as part of the CTA.
Clinical Trial Approval
Requirements for the conduct of clinical trials in the European Union including Good Clinical Practice, or GCP, are implemented in the Clinical Trials Directive 2001/20/EC and the GCP Directive 2005/28/EC. Pursuant to Directive 2001/20/EC and Directive 2005/28/EC, as amended, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, approval must be obtained from the competent national authority of an E.U. member state in which a study is planned to be conducted, or in multiple member states if the clinical trial is to be conducted in a number of member states. To this end, a CTA is
19
submitted, which must be supported by an investigational medicinal product dossier, or IMPD, and further supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and other applicable guidance documents. Furthermore, a clinical trial may only be started after a competent ethics committee has issued a favorable opinion on the clinical trial application in that country.
In April 2014, the E.U. legislator passed the new Clinical Trials Regulation, (EU) No 536/2014, which will replace the current Clinical Trials Directive 2001/20/EC. To ensure that the rules for clinical trials are identical throughout the European Union, the new E.U. clinical trials legislation was passed as a regulation that is directly applicable in all E.U. member states. All clinical trials performed in the European Union are required to be conducted in accordance with the Clinical Trials Directive 2001/20/EC until the new Clinical Trials Regulation (EU) No 536/2014 becomes applicable, which will be no earlier than May 28, 2016.
The new Regulation (EU) No 536/2014 aims to simplify and streamline the approval of clinical trial in the European Union. The main characteristics of the regulation include:
- •
- A streamlined application procedure via a single entry point, the E.U. portal.
- •
- A single set of documents to be prepared and submitted for the application as well as simplified reporting procedures that will spare
sponsors from submitting broadly identical information separately to various bodies and different member states.
- •
- A harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. Part I is
assessed jointly by all member states concerned. Part II is assessed separately by each member state concerned.
- •
- Strictly defined deadlines for the assessment of clinical trial application.
- •
- The involvement of the ethics committees in the assessment procedure in accordance with the national law of the member state concerned but within the overall timelines defined by the Regulation Regulation (EU) No 536/2014.
Marketing Authorization
Authorization to market a product in the member states of the European Union proceeds under one of four procedures: a centralized authorization procedure, a mutual recognition procedure, a decentralized procedure or a national procedure.
Centralized Authorization Procedure
The centralized procedure enables applicants to obtain a marketing authorization that is valid in all E.U. member states based on a single application. Certain medicinal products, including products developed by means of biotechnological processes, must undergo the centralized authorization procedure for marketing authorization, which, if granted by the European Commission, is automatically valid in all 28 E.U. member states. The EMA and the European Commission administer this centralized authorization procedure pursuant to Regulation (EC) No 726/2004.
Pursuant to Regulation (EC) No 726/2004, this procedure is mandatory for:
- •
- medicinal products developed by means of one of the following biotechnological processes:
- •
- recombinant DNA technology;
- •
- controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes including
transformed mammalian cells; and
- •
- hybridoma and monoclonal antibody methods;
20
- •
- advanced therapy medicinal products as defined in Article 2 of Regulation (EC) No. 1394/2007 on advanced therapy
medicinal products;
- •
- medicinal products for human use containing a new active substance that, on the date of effectiveness of this regulation, was not
authorized in the European Union, and for which the therapeutic indication is the treatment of any of the following diseases:
- •
- acquired immune deficiency syndrome;
- •
- cancer;
- •
- neurodegenerative disorder;
- •
- diabetes;
- •
- auto-immune diseases and other immune dysfunctions; and
- •
- viral diseases;
- •
- medicinal products that are designated as orphan medicinal products pursuant to Regulation (EC) No 141/2000.
The centralized authorization procedure is optional for other medicinal products if they contain a new active substance or if the applicant shows that the medicinal product concerned constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization is in the interest of patients in the European Union.
Administrative Procedure
Under the centralized authorization procedure, the EMA's Committee for Human Medicinal Products, or CHMP, serves as the scientific committee that renders opinions about the safety, efficacy and quality of medicinal products for human use on behalf of the EMA. The CHMP is composed of experts nominated by each member state's national authority for medicinal products, with expert appointed to act as Rapporteur for the co-ordination of the evaluation with the possible assistance of a further member of the Committee acting as a Co-Rapporteur. After approval, the Rapporteur(s) continue to monitor the product throughout its life cycle. The CHMP has 210 days to adopt an opinion as to whether a marketing authorization should be granted. The process usually takes longer in case additional information is requested, which triggers clock-stops in the procedural timelines. The process is complex and involves extensive consultation with the regulatory authorities of member states and a number of experts. When an application is submitted for a marketing authorization in respect of a drug that is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation, the applicant may pursuant to Article 14(9) Regulation (EC) No 726/2004 request an accelerated assessment procedure. If the CHMP accepts such request, the time-limit of 210 days will be reduced to 150 days but it is possible that the CHMP can revert to the standard time-limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment. Once the procedure is completed, a European Public Assessment Report, or EPAR, is produced. If the opinion is negative, information is given as to the grounds on which this conclusion was reached. After the adoption of the CHMP opinion, a decision on the MAA must be adopted by the European Commission, after consulting the E.U. member states, which in total can take more than 60 days.
Conditional Approval
In specific circumstances, E.U. legislation (Article 14(7) Regulation (EC) No 726/2004 and Regulation (EC) No 507/2006 on Conditional Marketing Authorisations for Medicinal Products for Human Use) enables applicants to obtain a conditional marketing authorization prior to obtaining the
21
comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted for product candidates (including medicines designated as orphan medicinal products) if (1) the risk-benefit balance of the product candidate is positive, (2) it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, (3) the product fulfills unmet medical needs and (4) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions and/or specific obligations. The timelines for the centralized procedure described above also apply with respect to the review by the CHMP of applications for a conditional marketing authorization.
Marketing Authorization under Exceptional Circumstances
Under Article 14(8) Regulation (EC) No 726/2004, products for which the applicant can demonstrate that comprehensive data (in line with the requirements laid down in Annex I of Directive 2001/83/EC, as amended) cannot be provided (due to specific reasons foreseen in the legislation) might be eligible for marketing authorization under exceptional circumstances. This type of authorization is reviewed annually to reassess the risk-benefit balance. The fulfillment of any specific procedures/obligations imposed as part of the marketing authorization under exceptional circumstances is aimed at the provision of information on the safe and effective use of the product and will normally not lead to the completion of a full dossier/approval.
Market Authorizations Granted by Authorities of E.U. Member States
In general, if the centralized procedure is not followed, there are three alternative procedures as pecribed in Directive 2001/83/EC:
- •
- The decentralized procedure allows applicants to file identical applications to several E.U. member states and receive simultaneous
national approvals based on the recognition by E.U. member states of an assessment by a reference member state.
- •
- The national procedure is only available for products intended to be authorized in a single E.U. member state.
- •
- A mutual recognition procedure similar to the decentralized procedure is available when a marketing authorization has already been obtained in at least one E.U. member state.
A marketing authorization may be granted only to an applicant established in the European Union.
Pediatric Studies
Prior to obtaining a marketing authorization in the European Union, applicants have to demonstrate compliance with all measures included in an EMA-approved Paediatric Investigation Plan, or PIP, covering all subsets of the paediatric population, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. The respective requirements for all marketing authorization procedures are set forth in Regulation (EC) No 1901/2006, which is referred to as the Pediatric Regulation. This requirement also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The Pediatric Committee of the EMA, or PDCO, may grant deferrals for
22
some medicines, allowing a company to delay development of the medicine in children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO may also grant waivers when development of a medicine in children is not needed or is not appropriate, such as for diseases that only affect the elderly population.
Before a marketing authorization application can be filed, or an existing marketing authorization can be amended, the EMA determines that companies actually comply with the agreed studies and measures listed in each relevant PIP.
Periods of Authorization and Renewals
A marketing authorization is valid for five years in principle and the marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the authorizing member state. To this end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the E.U. market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid (the so-called sunset clause).
Regulatory Data Protection
E.U. legislation also provides for a system of regulatory data and market exclusivity. According to Article 14(11) of Regulation (EC) No 726/2004, as amended, and Article 10(1) of Directive 2001/83/EC, as amended, upon receiving marketing authorization, new chemical entities approved on the basis of complete independent data package benefit from eight years of data exclusivity and an additional two years of market exclusivity. Data exclusivity prevents regulatory authorities in the European Union from referencing the innovator's data to assess a generic (abbreviated) application. During the additional two-year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator's data may be referenced, but no generic medicinal product can be marketed until the expiration of the market exclusivity. The overall ten-year period will be extended to a maximum of 11 years if, during the first eight years of those ten years, the marketing authorization holder, or MAH, obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the innovator is able to gain the period of data exclusivity, another company nevertheless could also market another version of the drug if such company obtained marketing authorization based on an MAA with a complete independent data package of pharmaceutical test, preclinical tests and clinical trials. However, products designated as orphan medicinal products enjoy, upon receiving marketing authorization, a period of ten years of orphan market exclusivity. Depending upon the timing and duration of the E.U. marketing authorization process, products may be eligible for up to five years' supplementary protection certificates, or SPCs, pursuant to Regulation (EC) No 469/2009. Such SPCs extend the rights under the basic patent for the drug.
23
Regulatory Requirements After a Marketing Authorization has been Obtained
If we obtain authorization for a medicinal product in the European Union, we will be required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products:
Pharmacovigilance and other requirements
We will, for example, have to comply with the E.U.'s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. Other requirements relate, for example, to the manufacturing of products and APIs in accordance with good manufacturing practice standards. E.U. regulators may conduct inspections to verify our compliance with applicable requirements, and we will have to continue to expend time, money and effort to remain compliant. Non-compliance with E.U. requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties in the European Union. Similarly, failure to comply with the E.U.'s requirements regarding the protection of individual personal data can also lead to significant penalties and sanctions. Individual E.U. member states may also impose various sanctions and penalties in case we do not comply with locally applicable requirements.
Manufacturing
The manufacturing of authorized drugs, for which a separate manufacturer's license is mandatory, must be conducted in strict compliance with the EMA's Good Manufacturing Practices, or GMP, requirements and comparable requirements of other regulatory bodies in the European Union, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and identity. The EMA enforces its current GMP requirements through mandatory registration of facilities and inspections of those facilities. The EMA may have a coordinating role for these inspections while the responsibility for carrying them out rests with the member states competent authority under whose responsibility the manufacturer falls. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and could subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
Marketing and Promotion
The marketing and promotion of authorized drugs, including industry-sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public, are strictly regulated in the European Union under Directive 2001/83/EC. The applicable regulations aim to ensure that information provided by holders of marketing authorizations regarding their products is truthful, balanced and accurately reflects the safety and efficacy claims authorized by the EMA or by the competent authority of the authorizing member state. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties.
Patent Term Extension
In order to compensate the patentee for delays in obtaining a marketing authorization for a patented product, a supplementary certificate, or SPC, may be granted extending the exclusivity period for that specific product by up to five years. Applications for SPCs must be made to the relevant patent office in each E.U. member state and the granted certificates are valid only in the member state of
24
grant. An application has to be made by the patent owner within six months of the first marketing authorization being granted in the European Union (assuming the patent in question has not expired, lapsed or been revoked) or within six months of the grant of the patent (if the marketing authorization is granted first). In the context of SPCs, the term "product" means the active ingredient or combination of active ingredients for a medicinal product and the term "patent" means a patent protecting such a product or a new manufacturing process or application for it. The duration of an SPC is calculated as the difference between the patent's filing date and the date of the first marketing authorization, minus five years, subject to a maximum term of five years.
A six month pediatric extension of an SPC may be obtained where the patentee has carried out an agreed pediatric investigation plan, the authorized product information includes information on the results of the studies and the product is authorized in all member states of the European Union.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of products approved by the FDA and other government authorities. Sales of products will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs in the United States such as Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the approved products for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. A payor's decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Third-party reimbursement may not be sufficient to maintain price levels high enough to realize an appropriate return on investment in product development.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of our drug candidate to currently available therapies (so called health technology assessment, or HTA) in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. E.U. member states may approve a specific price for a drug product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, there can be considerably pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various E.U. member states, and parallel distribution (arbitrage between low-priced and high-priced member states), can further reduce prices. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements.
25
Healthcare Law and Regulation
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with third-party payors and customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations. Such restrictions under applicable federal and state healthcare laws and regulations, include the following:
- •
- the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving
or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service,
for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid;
- •
- the federal False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or
entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an
obligation to pay money to the federal government;
- •
- the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing
a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
- •
- HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also
imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
- •
- the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making
any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services;
- •
- the federal transparency requirements under the Health Care Reform Law requires manufacturers of drugs, devices, biologics and medical
supplies to report to the Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals and physician ownership and
investment interests; and
- •
- analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers.
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Environmental, Health and Safety Matters
The manufacturing facilities of the third-parties that develop our product candidates are subject to extensive environmental, health and safety laws and regulations in a number of jurisdictions, governing, among other things: the use, storage, registration, handling, emission and disposal of chemicals, waste materials and sewage; chemicals, air, water and ground contamination; air emissions and the cleanup of
26
contaminated sites, including any contamination that results from spills due to our failure to properly dispose of chemicals, waste materials and sewage.
These laws, regulations and permits could potentially require the expenditure by us of significant amounts for compliance or remediation. If the third-party manufacturers fail to comply with such laws, regulations or permits, we may be subject to fines and other civil, administrative or criminal sanctions, including the revocation of permits and licenses necessary to continue our business activities. In addition, we may be required to pay damages or civil judgments in respect of third-party claims, including those relating to personal injury (including exposure to hazardous substances we use, store, handle, transport, manufacture or dispose of), property damage or contribution claims. Some environmental, health and safety laws allow for strict, joint and several liability for remediation costs, regardless of comparative fault. We may be identified as a responsible party under such laws. Such developments could have a material adverse effect on our business, financial condition and results of operations.
In addition, laws and regulations relating to environmental, health and safety matters are often subject to change. In the event of any changes or new laws or regulations, we could be subject to new compliance measures or to penalties for activities that were previously permitted.
Employees
As of December 31, 2014, we had 20 employees, all of which were full time. We had 12 employees in research and development. We had one employee located outside of the United States as of December 31, 2014. We also retain independent contractors to support our organization. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have not experienced any work stoppages and we consider our relations with our employees to be good.
Other Information
We are subject to the information requirements of the Exchange Act. Therefore, we file periodic reports, proxy statements and other information with the SEC. Such reports, proxy statements and other information may be obtained, free of charge, by visiting the Public Reference Room of the SEC at 100 F Street, NE, Washington, D.C. 20549 or by calling the SEC at 1-800-SEC-0330, by sending an electronic message to the SEC at publicinfo@sec.gov or by sending a fax to the SEC at 1-202-777-1027. In addition, the SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically.
Not applicable.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
Our principal executive office and laboratory is located at 1098 Hamilton Court, Menlo Park, California 94025, where the company occupies 10,800 sq. ft. of research and development and administration facilities that are nearby to external formulation, clinical and pre-clinical testing facilities. The lease expires in November 2016. We believe that our existing property is in good condition and suitable for our current needs.
27
We may become subject to legal proceedings, claims and litigation arising in the ordinary course of business. In addition, we may receive letters alleging infringement of patents or other intellectual property rights. We are not a party to any material legal proceedings, nor are we aware of any pending or threatened litigation that would have a material adverse effect on our business, operating results, cash flows or financial condition should such litigation be resolved unfavorably.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
28
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common shares currently trade on the OTCQB Marketplace under the symbol "BPMX." Except for one quotation dated February 14, 2013 of $0.15, there were no reported quotations for our common stock during 2013.
The following table sets forth, for each of the calendar periods indicated, the quarterly high and low bid prices for our common shares quoted on the OTCQB Marketplace. The prices in the table represent prices between dealers and do not include adjustments for retail mark-up, markdown or commission and may not represent actual transactions.
Period
|
High | Low | |||||
|---|---|---|---|---|---|---|---|
Fiscal Year Ended December 31, 2014: |
|||||||
First Quarter (from March 3, 2014) |
$ | 0.15 | $ | 0.15 | |||
Second Quarter |
$ | 0.15 | $ | 0.15 | |||
Third Quarter |
$ | 3.00 | $ | 0.15 | |||
Fourth Quarter |
$ | 3.50 | $ | 2.01 | |||
As of March 24, 2015, there were approximately 47 registered holders of record of our common shares, based upon information received from our stock transfer agent. However, this number does not include beneficial owners whose shares were held of record by nominees, including broker dealers. We believe that there are a significantly larger number of beneficial owners of our common shares than the number of record holders.
Transfer Agent and Registrar
The Transfer Agent for our capital stock is Empire Stock Transfer, located at 1859 Whitney Mesa Dr., Henderson, Nevada 89014.
Penny Stock Regulations
The SEC has adopted regulations which generally define "penny stock" to be an equity security that has a market price of less than $5.00 per share. Our common stock falls within the definition of penny stock and is subject to rules that impose additional sales practice requirements on broker-dealers who sell such securities to persons other than established customers and accredited investors (generally those with assets in excess of $1 million, or annual incomes exceeding $200,000 individually, or $300,000, together with their spouse).
For transactions covered by these rules, the broker-dealer must make a special suitability determination for the purchase of such securities and have received the purchaser's prior written consent to the transaction. Additionally, for any transaction, other than exempt transactions, involving a penny stock, the rules require the delivery, prior to the transaction, of a risk disclosure document mandated by the SEC relating to the penny stock market. The broker-dealer also must disclose the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and, if the broker-dealer is the sole market-maker, the broker-dealer must disclose this fact and the broker-dealer's presumed control over the market. Finally, monthly statements must be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks. Consequently, the "penny stock" rules may restrict the ability of broker-dealers to sell our common stock and may affect the ability of investors to sell their common stock in the secondary market.
29
Dividend Policy
We have not paid any cash dividends to our shareholders. Any future determination as to the declaration and payment of dividends on shares of our common stock will be made at the discretion of our board of directors out of funds legally available for such purpose. We are under no contractual obligations or restrictions to declare or pay dividends on our shares of common stock. In addition, we currently have no plans to pay such dividends.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table gives information about our common stock that may be issued upon the exercise or settlement of stock options and rights under all of our existing equity compensation plans as of December 31, 2014:
Plan Category
|
Number of Securities to be Issued Upon Exercise of Outstanding Options and Rights (Column a) |
Weighted- Average Exercise Price of Outstanding Options and Rights (Column b) ($) |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column a) (Column c) (3) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Equity compensation plans approved by security holders |
2,609,357 | $ | 0.82 | 1,163,000 | ||||||
Equity compensation plans not approved by security holders |
193,333 | (1) | — | — | ||||||
| | | | | | | | | | | |
Total |
2,802,690 | $ | 0.82 | 1,163,000 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- Shares to be issued for director services rendered by Ping Wang.
Unregistered Sales of Equity Securities
From September 3, 2014 to January 26, 2015, 15 individuals exercised stock options granted under the 2014 Equity Incentive Plan to purchase 767,748 shares of our common stock. These stock options were issued in exchange for services rendered to us in accordance with the terms of the 2014 Equity Incentive Plan. The weighted-average exercise price of the stock options exercised during this period was $0.29.
On November 10, 2014 we issued to Korea Investment Partners Company, Ltd., or KIP, 290,000 shares of our common stock, of which 96,667 shares vested immediately on issuance and 193,333 vest upon completion of a milestone, for director services rendered by Ping Wang. The shares have a fair value of $481,400 based on stock valuation at the date of issuance.
The foregoing issuances of the equity securities were effectuated pursuant to the exemption from the registration requirements of the Securities Act of 1933, as amended, or Securities Act, provided by Section 4(a)(2) of the Securities Act and/or Regulation D promulgated thereunder as a transaction not involving a public offering and are restricted shares as defined in the Securities Act. The Company did not engage in any general solicitation or advertising in connection with the foregoing issuances.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable.
30
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management's Discussion and Analysis of Financial Condition and Results of Operations, or MD&A, is intended to help the reader understand our results of operations and financial condition. MD&A is provided as a supplement to, and should be read in conjunction with, our audited financial statements and the accompanying notes to the financial statements and other disclosures included in this Annual Report on Form 10-K. Our financial statements have been prepared in accordance with U.S. generally accepted accounting principles and are presented in U.S. dollars.
Overview
We are a specialty pharmaceutical company focused on utilizing our proprietary drug delivery technologies to develop and commercialize novel prescription and over-the-counter, or OTC, products that address large markets in women's health and dermatology. Our objective is to develop products that treat health or age-related conditions that: (1) are not presently being addressed or treated at all or (2) are currently treated with drug therapies or drug delivery approaches that are sub-optimal. Our strategy is to bring new products to market by identifying optimal delivery mechanisms and/or alternative applications for FDA-approved active pharmaceutical ingredients, or APIs, and biological materials, while, in appropriate circumstances, reducing the time, cost and risk typically associated with new product development by taking advantage of the abbreviated regulatory pathway available for reformulated drugs that are bioequivalent to FDA-approved products. Our current platform technologies include innovative delivery mechanisms for molecular iodine, or I2, antibiotics and biologics.
Since our inception, we have devoted substantially all of our efforts to developing our product candidates including conducting preclinical and clinical trials and providing general and administrative support for these operations. We commercially launched our breast health supplement at the end of 2014, although to-date we have not generated any revenue from product sales and we are not dependent on sales to any one customer. We have financed our operations primarily through the sale of equity securities and convertible debt securities from which we raised $9.6 million of net cash from our inception through December 31, 2014.
Share Exchange
On January 23, 2014, we (then operating as Thompson Designs, Inc.), BioPharmX, Inc. and stockholders of BioPharmX, Inc., who collectively owned 100% of BioPharmX, Inc., entered into and consummated transactions pursuant to a share exchange agreement, such transaction referred to as the Share Exchange, whereby we issued to the stockholders of BioPharmX, Inc. an aggregate of 7,025,000 shares of our common stock, in exchange for 100% of the shares of BioPharmX, Inc. held by stockholders. The shares of our common stock received by the stockholders of BioPharmX, Inc. in the Share Exchange constituted approximately 77.8% of our then issued and outstanding common stock, after giving effect to the issuance of shares pursuant to the share exchange agreement. As a result of the Share Exchange, BioPharmX, Inc. became our wholly-owned subsdidiary. For accounting purposes, the Share Exchange was treated as a reverse acquisition with BioPharmX, Inc. as the acquirer and us as the acquired party, and as a result the historical financial statements prior to the Share Exchange included in this Annual Report on Form 10-K are the historical financial statements of BioPharmX, Inc. On March 3, 2014, we changed our name to BioPharmX Corporation. On April 25, 2014, we reincorporated from Nevada to Delaware.
31
Results of Operations
Fiscal Years Ended December 31, 2014 and 2013
Revenue
We did not recognize any revenue in the years ended December 31, 2014 and 2013. We shipped our first product to a retailer in December 2014. The product, the VI2OLET breast health tablet, is a new product in the dietary supplement field.
Research and Development Expenses
We expense both internal and external research and development expenses to operations as they are incurred.
| Year ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2014 | 2013 | Change | % | |||||||
| ($ in thousands) |
||||||||||
| $2,519 | $ | 671 | $ | 1,848 | 275 | % | ||||
Research and development expenses for the years ended December 31, 2014 and 2013 were $2.5 million and $671,000, respectively. The increase from year-to-year of $1.8 million is primarily due to a $1.0 million increase in employees' salaries and $198,000 due to stock compensation expense during 2014. In 2013, we were using primarily consultants who were converted to employees in early 2014. Additionally, costs increased $260,000 due to quality testing and one-time production costs related to producing our VI2OLET breast health tablet product. Laboratory expenses for on-going research and development on future products increased by $121,000. Overhead allocated to the research and development department increased by $170,000.
Research and development expenses for the year ended December 31, 2013 consisted primarily of employee and consultant compensation and non-employee stock compensation expense in the amount of $527,000 and laboratory supplies of $51,000.
As of December 31, 2014, we had 12 employees in research and development.
Sales and Marketing Expenses
We expense both sales and marketing expenses to operations as they are incurred. In the years shown, costs are related to establishing our corporate brand and efforts related to our VI2OLET breast health tablet.
| Year ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2014 | 2013 | Change | % | |||||||
| ($ in thousands) |
||||||||||
| $2,299 | $ | 132 | $ | 2,167 | 1,642 | % | ||||
Sales and marketing expenses for the years ended December 31, 2014 and 2013 were $2.3 million and $132,000, respectively. The increase from year-to-year of $2.2 million is primarily due to the ramp up in marketing and sales to launch our VI2OLET breast health tablet product. Sales and marketing compensation increased $392,000 and stock compensation increased $140,000 as a result of hiring people who had previously been consultants as employees in 2014. Outside agencies accounted for $769,000 of the increase from year-to-year to accomplish the marketing goals for our new product. Marketing costs to launch our new product increased by $496,000 and travel increased by $54,000 from 2014 to 2013. Allocated overhead, consisting of facilities, insurance and maintenance expenses, increased by $110,000.
32
Sales and marketing expenses for the year ended December 31, 2013 consisted primarily of consultant compensation and non-employee stock compensation expense in the amount of $93,000 and the cost of developing marketing strategy and material in the amount of $30,000.
As of December 31, 2014, we had 3 employees in sales and marketing.
General and Administrative Expenses
Our general and administrative expenses consist of the cost of our executive, finance, corporate development and other administrative functions.
| Year ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2014 | 2013 | Change | % | |||||||
| ($ in thousands) |
||||||||||
| $2,953 | $ | 711 | $ | 2,242 | 315 | % | ||||
General and administrative expenses for the years ended December 31, 2014 and 2013 were $3.0 million and $711,000, respectively. The increase from year-to-year of $2.2 million is primarily due to beginning in the year ended December 31, 2014 to pay our officers' salaries of $500,000, adding support staff, which resulted in $1.0 million in cash compensation, $160,000 in stock compensation to an investor for service as a director and other consulting services and $658,000 in stock compensation for employees and consultants. The cost of the reverse merger and overhead related to being a publicly-traded company increased costs by $318,000 including reporting, legal and audit expenses. Travel expense was up $63,000 from year to year. The remaining increase of $170,000 was due to allocated overhead and general office expenses.
General and administrative expenses for the year ended December 31, 2013 consisted primarily of compensation and benefits in the amount of $272,000, professional fees totaling $259,000 to our legal counsel and auditors, travel expense of $64,000, as well as other general and administrative expenses
As of December 31, 2014, we had 5 employees in general and administrative.
Loss from Operations
Loss from operations for the years ended December 31, 2014 and 2013 were $7.8 million and $1.5 million, respectively. The increase in the loss from year-to-year is due to ramping up research and development, production and launch of our first product and the costs related to our reverse merger and going public.
Net Loss
Net loss for the years ended December 31, 2014 and 2013 was $7.8 million and $1.6 million, respectively.
Inflation did not have a material impact on our operations for either of the periods. Other than the foregoing, management knows of no trends, demands, or uncertainties that are reasonably likely to have a material impact on our results of operations.
33
Capital Resources and Liquidity
A summary of the sources and uses of cash is as follows (in thousands):
| |
December 31 | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Net cash used in operating activities |
$ | (6,001 | ) | $ | (1,080 | ) | |
Net cash used in investing activities |
(263 | ) | (85 | ) | |||
Net cash provided by financing activities |
8,372 | 1,030 | |||||
| | | | | | | | |
Net increase (decrease) in cash |
$ | 2,108 | $ | (135 | ) | ||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Between September 2012 and March 2014, we issued 6% unsecured convertible notes to investors in the aggregate principal amount of $2.25 million. These notes had maturity dates from one to three years from the date of issuance, with principal and interest payable at maturity. The notes automatically converted in 2014 into shares of our common stock on the completion of our reverse acquisition by BioPharmX, Inc. in January 2014 and closing of a financing in the amount of at least $2 million at a conversion price per share equal to 80% of the per share offering price of such financing.
During the year ended December 31, 2014, we completed the private placement of shares of Series A preferred stock and warrants to purchase common stock. The private placement was consummated in a series of closings that occurred between April 2014 and November 2014. We sold to accredited investors and non-U.S. persons 4.2 million shares of Series A preferred stock at a per share price of $1.85 for net proceeds of approximately $7.3 million and issued to the investors, for no additional consideration, warrants to purchase in the aggregate 2.0 million shares of our common stock, at an exercise price of $3.70 per share pursuant to a series of subscription agreements.
Additionally, under the subscription agreement with one of the investors, that investor is committed to purchase an additional 1,081,081 shares of Series A preferred stock at a per share price of $1.85 upon the achievement of certain milestones which would raise another $2.0 million in gross proceeds. The milestones include our receiving revenues of $2.0 million for our VI2OLET product or uplisting our stock to NYSE or NASDAQ. Two of our majority common stockholders and this investor also entered into a voting agreement whereby these stockholders agreed to (i) vote in favor of any merger or sale of us which has been approved by the board of directors and holders of at least 50% of the then outstanding shares of Series A preferred stock, and (ii) irrevocably grant to such investor a proxy to vote in favor of such business combination transaction. These stockholders also agreed to sell their shares to a purchaser in a transaction approved by holders of at least 67% of shares of Series A preferred stock or 67% of shares of common stock and Series A preferred stock in the aggregate.
The following table summarizes total current assets, liabilities and working capital (in thousands).
| |
December 31, 2014 |
|||
|---|---|---|---|---|
Current assets |
$ | 2,520 | ||
Current liabilities |
1,367 | |||
| | | | | |
Working capital |
$ | 1,153 | ||
| | | | | |
| | | | | |
| | | | | |
Net cash used for operating activities for the year ended December 31, 2014 was $6.0 million. Cash used in operating activities was primarily due to a net loss for the year ended December 31, 2013 of $7.8 million which was partially offset by changes in operating assets and liabilities of $413,000, non-cash interest expense of $76,000, warrants issued for $99,000 and stock-based compensation of $1.2 million. Cash used in investing activities was primarily for acquisition of intellectual property and acquisition of fixed assets.
34
Net cash used for operating activities for the year ended December 31, 2013 was $1.1 million. Cash used in operating activities was primarily due to a net loss for the year ended December 31, 2013 of $1.6 million which was partially offset by changes in operating assets and liabilities of $371,000, non-cash interest expense of $74,000 and stock-based compensation of $58,000. Cash used in investing activities was primarily for the acquisition of intellectual property and the acquisition of fixed assets.
Net cash provided by financing activities for the years ended December 31, 2014 and 2013 was $8.4 million and $1.0 million, respectively. This consisted of $7.3 million from issuing Series A preferred stock in the year ended December 31, 2014 and $1.0 million in proceeds from issuing convertible notes for each of the years ended December 31, 2014 and 2013.
Subsequent Events
In March 2015, we amended certain warrants to reduce the exercise price of such warrants from $3.70 to $2.50 per share with a corresponding increase in the number of shares of common stock exercisable under the warrants so that the aggregate exercise value of such warrants remained the same. As of March 27, 2015, the holders had exercised such warrants for an aggregate of 397,996 shares of common stock for an aggregate cash exercise price of $994,990.
Going Concern
As reflected in the accompanying financial statements, those financial statements have been prepared assuming we will continue as a going concern. We have incurred recurring losses and negative cash flows from operations since inception. We have not generated revenues and have funded our operating losses through the issuance of convertible notes payable and Series A preferred stock. We have a limited operating history and our prospects are subject to risks, expenses and uncertainties frequently encountered by companies in the industry.
The significant risks described herein could have a significant negative impact on our financial viability and raise substantial doubt about our ability to continue as a going concern. We are working on our business model to increase working capital by managing our cash flow, securing financing and introducing our first product to market.
Risks include, but are not limited to, the uncertainty of availability of additional financing and the uncertainty of achieving future profitability. We intend to raise additional funds through the issuance of equity securities. There can be no assurance that such financing will be available or on terms which are favorable to us. Failure to generate sufficient cash flows from operations, raise additional capital or reduce certain discretionary spending could have a material adverse effect on our ability to achieve our intended business objectives. These factors raise substantial doubt about our ability to continue as a going concern. The financial statements do not contain any adjustments that might result from the outcome of this uncertainty.
As shown in the accompanying financial statements, we incurred a net loss of $7.8 million and $1.6 million during the years ended December 31, 2014 and 2013, respectively, and have an accumulated deficit of $9.5 million as of December 31, 2014. As of December 31, 2014, we had working capital of $1.2 million. While we believe that we have a plan to fund on-going operations, there is no assurance that our plan will be successfully implemented. We are experiencing the following risks and uncertainties in the business:
- •
- The discovery of key raw materials to formulate novel products depends on our ability to identify, negotiate and secure procurement of
such materials. This also depends on our ability to establish comprehensive and long term vendor contracts and relationships.
- •
- Our ability to compete and to achieve our product platform strategy depends on our ability to protect our proprietary discoveries and technologies. We currently rely on a combination of
35
- •
- Our continued operations are dependent upon our ability to identify, recruit and retain adequate management personnel and contractors
to perform certain jobs such as research and development, patent generation, regulatory affairs and general administrative functions. We require highly trained professionals of varying levels and
experience along with a flexible work force.
- •
- Our ability to generate income in the short-run will depend greatly on the rate of adoption and ability to establish a market for our
VI2OLET breast health tablet.
- •
- Research and development for novel prescription or OTC based products can be very extensive and lengthy in nature; and the clinical trial process with the Food and Drug Administration can require significant funding and time consuming patient studies. The competitive landscape could change significantly over the time period to complete targeted product development milestones. The current competition for our products could also turn into strategic partners or potential acquirers in the future.
copyrights, trademarks, trade secret laws and confidentiality agreements to protect our intellectual property rights. We also rely upon unpatented know-how and continuing technological innovation.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers, (ASU 2014-09), which converges the FASB and the International Accounting Standards Board standards on revenue recognition. Areas of revenue recognition that will be affected include, but are not limited to, transfer of control, variable consideration, allocation of transfer pricing, licenses, time value of money, contract costs and disclosures. This guidance is effective for the fiscal years and interim reporting periods beginning after December 15, 2016. The Company is currently evaluating the impact that the adoption of ASU 2014-09 will have on its consolidated financial statements and related disclosures.
On June 10, 2014, the FASB issued ASU 2014-10, Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation (ASU 2014-10), which eliminates the definition of a development stage entity, eliminates the development stage presentation and disclosure requirements under Accounting Standards Codification, or ASC, 915 Development Stage Entities, or ASC 915, and amends provisions of existing variable interest entity guidance under ASC 810 Consolidation. As a result of the changes, entities which meet the former definition of a development stage entity will no longer be required to: (1) present inception-to-date information in the statements of income, cash flows, and stockholder equity; (2) label the financial statements as those of a development stage entity; (3) disclose a description of the development stage activities in which the entity is engaged; and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. Furthermore, ASU 2014-10 clarifies disclosures about risks and uncertainties under ASC Topic 275, Risks and Uncertainties, that apply to companies that have not commenced planned principal operations. Finally, variable interest entity rules no longer contain an exception for development stage entities and, as a result, development stage entities will have to be evaluated for consolidation in the same manner as non-development stage entities.
Under ASU 2014-10, entities are no longer required to apply the presentation and disclosure provisions of ASC 915 during annual periods beginning after December 15, 2014. In addition, the revisions to the consolidation standards are effective for annual periods beginning after December 15, 2015 for public entities and are effective for annual periods beginning after December 15, 2016 for nonpublic entities. Early adoption is permitted for any annual reporting period or interim period for
36
which the entity's financial statements have not yet been issued (public business entities) or made available for issuance (other entities).
The Company has adopted ASU 2014-10 effective as of its issuance date. Adoption of this standard had no impact on its financial position, results of operations, or cash flows; however, the presentation of the consolidated financial statements and related disclosures in the notes to the consolidated financial statements has been changed to eliminate the disclosures that are no longer required.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements—Going Concern (ASU 2014-15). This standard includes guidance about management's responsibility to evaluate whether there is substantial doubt about an entity's ability to continue as a going concern within one year after the financial statements are issued. If conditions or events raise substantial doubt, the entity must disclose the conditions or events that raise substantial doubt about the entity's ability to continue as a going concern, management's evaluation of those conditions or events, and management's plans to mitigate the conditions or events. This update is effective for interim and annual reporting periods beginning after December 15, 2016. Early adoption is permitted. The Company is currently evaluating the impact that the adoption of ASU 2014-15 will have on its consolidated financial statements and related disclosures.
We have reviewed other recent accounting pronouncements and concluded they are either not applicable to the business, or no material effect is expected on the consolidated financial statements as a result of future adoption.
Critical Accounting Policies
Our consolidated financial statements and related financial information are based on the application of accounting principles generally accepted in the United States, or GAAP. GAAP requires the use of estimates, assumptions, judgments and subjective interpretations of accounting principles that have an impact on the assets, liabilities, revenues and expense amounts reported. These estimates can also affect supplemental information contained in our external disclosures including information regarding contingencies, risk and financial condition. We believe our use of estimates and underlying accounting assumptions adhere to GAAP and are consistently applied. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ materially from these estimates under different assumptions or conditions. We continue to monitor significant estimates made during the preparation of our financial statements.
Our significant accounting policies are summarized in Note 1 of our audited consolidated financial statements. While all these significant accounting policies impact our financial condition and results of operations, we view certain of these policies as critical. Policies determined to be critical are those policies that have the most significant impact on our financial statements and require management to use a greater degree of judgment and estimates. Actual results may differ from those estimates and such differences may be material to the financial statements. Our management believes that given current facts and circumstances, it is unlikely that applying any other reasonable judgments or estimate methodologies would have an effect on our results of operations, financial position or liquidity for the periods presented in this report.
37
We believe the following critical accounting policies and procedures, among others, affect our more significant judgments and estimates used in the preparation of our consolidated financial statements:
Revenue Recognition
We shipped our first product to a retailer in December 2014. The product, the VI2OLET breast health tablet, is a new product in the dietary supplement field. Revenue is recognized provided that persuasive evidence of a sales arrangement exists, the price is fixed or determinable, title and risk of loss has transferred, calculability of the resulting receivable is reasonably assured, there are no customer acceptance requirements and we do not have any significant post-shipment obligations. We recognize revenue on a sell through basis since we do not have the historical information to estimate product returns. As a result, we account for these product shipments using a deferred revenue recognition model. Under the deferred revenue recognition model, we do not recognize revenue upon product shipment. For these product shipments, we invoice the reseller, record deferred revenue at gross invoice sales price, and classify the cost basis of the product held by the wholesaler as a component of inventory. We recognize revenue when product is sold by the reseller to the end-user, on a first-in first-out (FIFO) basis.
Stock-based Compensation
We account for stock-based employee compensation arrangements which requires the recognition of compensation expense, using a fair-value based method for costs related to all employee share-based payments, including stock options. We estimate the fair value of share-based payment awards on the date of grant using an option pricing model. All option grants have been expensed on a straight-line basis over their vesting period. Equity instruments issued to nonemployees are recorded at their fair value on the measurement date and are subject to periodic adjustment as the underlying equity instruments vest.
For the years ended December 31, 2014 and 2013, stock-based compensation was $1.2 million and $58,000.
Off Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, financings, or other relationships with unconsolidated entities or other persons, also known as "special purpose entities," or SPEs.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our consolidated audited financial statements as of and for the fiscal years ended December 31, 2014 and 2013, together with the report of the independent registered public accounting firm thereon and the notes thereto, are presented beginning at page F-1.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
We changed our independent registered public accounting firm effective January 23, 2014 from Silberstein Ungar, PLLC ("SUPLLC") to Burr Pilger Mayer, Inc. Information regarding the change in the independent registered public accounting firm was disclosed in our Current Report on Form 8-K
38
filed with the SEC on January 27, 2014. There were no disagreements with SUPLLC or any reportable events requiring disclosure under Item 304(b) of Regulation S-K.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
As required by Rule 13a-15 under the Securities Exchange Act of 1934, we have carried out an evaluation of the effectiveness of our disclosure controls and procedures as of the end of the period covered by this annual report, December 31, 2014. This evaluation was carried out under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer.
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission's rules and forms. Disclosure controls and procedures include controls and procedures designed to ensure that information required to be disclosed in our company's reports filed under the Securities Exchange Act of 1934 is accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosure.
Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer has concluded that our disclosure controls and procedures were ineffective as of the end of the period covered by this annual report. This conclusion was based on the material weaknesses in our internal control over financial reporting further described below.
Management's Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934). Management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2014 based on criteria established in Internal Control-Integrated Framework 2013 issued by the Committee of Sponsoring Organizations of the Treadway Commission. As a result of this assessment, management concluded that, as of December 31, 2014, our internal control over financial reporting was not effective. Our management identified the following material weaknesses in our internal control over financial reporting, which are indicative of many small companies with small staff: (i) inadequate segregation of duties and ineffective risk assessment; and (ii) insufficient written policies and procedures for accounting and financial reporting with respect to the requirements and application of both US GAAP and Securities and Exchange Commission guidelines.
We plan to take steps to enhance and improve the design of our internal control over financial reporting. During the period covered by this annual report on Form 10-K, we have not remediated the material weaknesses identified above. To remediate such weaknesses, we plan to implement the following changes during our current fiscal year: (i) appoint additional qualified personnel to address inadequate segregation of duties and ineffective risk management; and (ii) adopt sufficient written policies and procedures for accounting and financial reporting. The remediation efforts set out in (i) and (ii) are largely dependent upon our securing additional financing to cover the costs of implementing the changes required. If we are unsuccessful in securing such funds, remediation efforts may be adversely affected in a material manner.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management's report was not subject to attestation by our registered public accounting firm because as a smaller reporting company we are not subject to Section 404(b) of the Sarbanes-Oxley Act of 2002.
Changes in Internal Control over Financial Reporting
No change in our system of internal control over financial reporting occurred during the fourth quarter of the year ended December 31, 2014 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
39
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
In connection with the Share Exchange, effective on January 21, 2014, James Pekarsky was appointed as Chairman of the board of directors, Chief Executive Officer and Chief Financial Officer, and Anja Krammer was appointed as a director and as President. Kade Thompson resigned as our sole director and officer at the same time. Kin F. Chan, Ph.D was appointed as our Executive Vice President of Research and Development effective February 17, 2014. Ping Wang was appointed as a director effective November 10, 2014. Michael Hubbard was appointed as a director effective January 22, 2015. Stephen Morlock was appointed as a director effective March 26, 2015.
The following table sets forth certain information as of March 27, 2015, concerning our directors and executive officers. All directors hold office until the next annual meeting of stockholders or until their respective successors are elected, except in the case of death, resignation or removal:
Name
|
Age | Position | ||
|---|---|---|---|---|
| James R. Pekarsky | 55 | Chief Executive Officer, Chief Financial Officer, and Chairman of the Board of Directors | ||
Anja Krammer |
47 |
President and Director |
||
Kin F. Chan, Ph.D. |
41 |
Executive Vice President of Research & Development |
||
Ping Wang |
38 |
Director |
||
Michael Hubbard |
63 |
Director |
||
Stephen Morlock |
61 |
Director |
Exeutive Officers
James R. Pekarsky, has been our Chief Executive Officer, Chief Financial Officer and Chairman of the Board since January 2014. Since September 2011, Mr. Pekarsky has served as Chief Executive Officer and Treasurer of BioPharmX, Inc. From November 2011 to August 2013, he served as Chief Financial Officer of Solar Power, Inc. From November 2007 to November 2011, Mr. Pekarsky was a consulting CFO to a variety of early-stage, start-up companies. Additionally, Mr. Pekarsky served as Chief Financial Officer of MoSys, Inc., from January 2006 to November 2007, AccelChip from December 2004 to December 2006 and Virage Logic from May 1999 to November 2003, where he helped lead the company's IPO in August 2000. Mr. Pekarsky also held general manager and senior operations positions at Mentor Graphics from January 1997 to May 1999, Advanced Molecular Systems from June 1995 to December 1996, Sclavo Diagnostics from November 1993 to May 1995 and Bio-Rad Laboratories from June 1989 to October 1993 where he resided abroad in Paris, Milan and London. Mr. Pekarsky holds a B.S. in accounting from Indiana University of Pennsylvania and an M.B.A. in finance from Golden Gate University. We believe that Mr. Pekarsky should serve on our board of directors due to his substantial leadership experience in senior finance, operations and general management roles in high tech and medical research companies, along with international business, strategic planning and thorough knowledge of public company requirements.
Anja Krammer has served as our President and a director since January 2014. Since September 2011 she has served as the President and Secretary of BioPharmX, Inc. Ms. Krammer previously served as Chief Marketing Officer/Founder of MBI, Inc., a management consulting firm from January 1998 to December 2013. While at MBI, Inc., Ms. Krammer also served as Vice President Global Marketing from April 2006 to August 2008 for Reliant Technologies, a venture backed startup in aesthetic medicine. From April 2004 to April 2006, Ms. Krammer served as Sr. Director of Strategic Marketing for Medtronic Corporation. From December 2000 to September 2001, Ms. Krammer was Vice
40
President, Solutions Marketing for Getronics Corporation, a global IT services company. From April 1999 to December 2000, Ms. Krammer served as Vice President, Indirect Channel Sales and Worldwide Industry Partnership Marketing, Itronix Division, Acterna Corporation, an optical communications company. Prior roles included, serving as Director of Worldwide Marketing and Communications for Tektronix Corporation in its Color Printing and Imaging Division from October 1997 to April 1999. From October 1995 to October 1997, Ms. Krammer was Director of Worldwide Sales and Marketing with KeyTronic Corporation, a computer equipment manufacturer. Ms. Krammer holds a BAIS degree with a focus on Marketing/Management from the University of South Carolina and an International Trade Certificate from the Sorbonne, University of Paris. We believe that Ms. Krammer should serve on our board of directors due to her experience in guiding healthcare and consumer enterprises in product development, sales and marketing management and commercialization strategies and her industry background in pharmaceuticals, medical devices, technology and consumer products.
Kin F. Chan, Ph.D. has served as Executive Vice President of Research & Development since February 2014. Prior to joining us, from April 2012 to January 2014, he was Vice President of Engineering at Demira, Inc., a biopharmaceutical company focusing on dermatology products. Prior to that he was the Managing Director of Advanced Research at Solta Medical, Inc. from 2003 to 2009, and was an optical R&D engineer at Ball Semiconductor, Inc. from 2000 to 2003. He was also the founder and President of Fourier Biotechnologies, LLC, which provides services in optical engineering and preclinical research, from 2009 to January 2014. Dr. Chan received his B.S., M.S., and Ph.D. in Electrical & Computer Engineering from the University of Texas at Austin.
Non-Employee Directors
Ping Wang has been a Principal of Korea Investment Partners, a venture capital investment firm, since May 2010. Prior to joining Korea Investment Partners in 2010, he worked at Great Pacific Financial Group from October 2007 to March 2010. Previously, Mr. Wang was an investment officer at Beijing Ancai Technology Venture Capital from May 2006 to October 2007, and earlier, worked at Matsuoka Industry Group as IT Manager from January 2000 to December 2002. He began his professional career as a software engineer at IBM from December 1998 to November 1999. Mr. Wang earned a B.S. degree in Computer Science at the University of Texas, Austin, and graduated from the MIT Sloan-Tsinghua joint program with an International MBA degree. We believe that Mr. Wang should serve on our board of directors due to his experience analyzing corporate performance as a venture capitalist and managing his firm's investments in private companies and knowledge of health care and pharmaceutical industries, important skills related to corporate finance, oversight of management and strategic positioning.
Michael Hubbard served as a senior audit partner at Deloitte & Touche LLP from August 2007 until retiring in June 2014 and also at PricewaterhouseCoopers LLP from September 1986 to July 2007. In these roles, he served private and publicly held clients across the life sciences, waste management, construction, and technology sectors, advising domestic and international issuer companies on complex transactions, including 19 IPOs and numerous follow-on equity and debt offerings. Mr. Hubbard holds a B.A. degree in Business Administration with a concentration in Accounting and an MBA degree from Washington State University. He is a licensed CPA in the states of Washington and California and is a certified practitioner of international financial reporting standards. We believe that Mr. Hubbard should serve on our board of directors due to his broad range of experience serving large public and private companies in the United States and internationally, including experience with the reporting requirements for complex transactions, including carve-outs and spin-offs; direct involvement with numerous SEC filings; and significant experience working with SEC staff, including the pre-clearance of accounting issues, responses to comments letters on periodic filings, and offering documents.
Stephen Morlock served as Executive Vice President and Chief Financial Officer at Otis Spunkmeyer, Inc. from May 1994 until his retirement in June 2004. He also served as Controller from
41
August 1992 to April 1994. Prior to that, he held various management positions in accounting, financial planning, and internal audit at Westinghouse Electric Supply Company from November 1977 to July 1992. Since his retirement in June 2004, Mr. Morlock has not been active in any business activities. Mr. Morlock holds a B.S. degree in Accounting from San Diego State University. We believe that Mr. Morlock should serve on our board of directors due to his extensive experience in the retail industry, including a variety of distribution channels, product merchandising, customer relationship management and brand name development, as well as his background in manufacturing capacity utilization and expansion, procurement and inventory management, compensation plan design and financial reporting.
All of our directors hold their positions on the board until our next annual meeting of the shareholders, and until their successors have been qualified after being elected or appointed. Officers serve at the discretion of the board of directors.
There are no family relationships among our directors and executive officers. There is no arrangement or understanding between or among our executive officers and directors pursuant to which any director or officer was or is to be selected as a director or officer, and there is no arrangement, plan or understanding as to whether non-management shareholders will exercise their voting rights to continue to elect the current board of directors.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors, executive officers and holders of more than 10% of our shares of common stock to file with the SEC initial reports of ownership and reports of changes in such ownership. SEC rules require such persons to furnish us with copies of all Section 16(a) reports that they file. Based on a review of these reports and on written representations from the reporting persons that no other reports were required, we believe that the applicable Section 16(a) reporting requirements were complied with for all of the transactions which occurred in the fiscal year ended December 31, 2014 with the following exceptions. In February 2014, a Form 3 for Ms. Krammer was not filed on a timely basis. In February 2014, a Form 3 for Dr. Chan was not filed on a timely basis. In February 2015, a Form 4 for Mr. Wang was not filed on a timely basis. In February 2015, a Form 4 for Mr. Hubbard was not filed on a timely basis.
Committees of the Board of Directors
We have standing nominating, compensation and audit committees. During 2014, our full board of directors performed the functions of these committees. We did not believe it was necessary for our board of directors to appoint such committees until recently because the volume of matters that came before our board of directors for consideration permitted the directors to give sufficient time and attention to such matters to be involved in all decision making. Additionally, because our common stock is not listed for trading on a national securities exchange, we were not required to have such committees.
Audit Committee Financial Expert
The board of directors has determined that Mr. Hubbard qualifies as an audit committee financial expert, as defined under Item 407(d)(5)(ii) of Regulation S-K.
Code of Ethics
In March 2015, we adopted a code of ethics that applies to all of our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer. The new code addresses, among other things, honesty and ethical conduct, conflicts of interest,
42
compliance with laws, regulations and policies, including disclosure requirements under the federal securities laws, confidentiality, trading on inside information and reporting of violations of the code.
Board Leadership Structure
Our Board recognizes that the leadership structure and combination or separation of the Chief Executive Officer and chairman roles is driven by our needs at any point in time. Currently, Mr. Pekarsky serves as our Chief Executive Officer and the Chairman of our Board, and Ms. Krammer serves as our President. We have no policy requiring the combination or separation of leadership roles and our governing documents do not mandate a particular structure. This has allowed, and will continue to allow, our Board the flexibility to establish the most appropriate structure for our company at any given time.
Non-Employee Director Compensation
In 2014, we did not pay any fees to, reimburse any expense of, make any equity awards or non-equity awards to, or pay any other compensation to any of our non-employee directors other than to KIP in connection with Ping Wang's services as a director. On November 10, 2014, in connection with Ping Wang's appointment to our board of directors, we issued to KIP 290,000 shares of our common stock, of which 96,667 shares vested immediately on issuance and 193,333 shares vest upon completion of a milestone in accordance with the terms of a subscription agreement entered into between us and KIP.
The compensation received by each of Mr. Pekarsky and Ms. Krammer as our employees is shown below in "Executive Compensation—Summary Compensation."
We do not have any standard policies, plans, or arrangements in place with respect to director compensation.
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation
The following is a summary of the compensation we paid to each of James Pekarsky, Anja Krammer, and Kin F. Chan, Ph.D. (our "Named Executive Officers"), for the two fiscal years ended December 31, 2014 and 2013 (in thousands).
Name and Principal Position
|
Year | Salary ($)(1) |
Totals ($) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| James R. Pekarsky(2) | 2014 | 250 | 250 | |||||||
CEO, CFO, Chairman of the Company; CEO and Director of BioPharmX, Inc. |
2013 | — | — | |||||||
| Anja Krammer(3) | 2014 | 250 | 250 | |||||||
President and Director of the Company; President and Director of BioPharmX, Inc. |
2013 | — | — | |||||||
| Kin F. Chan, Ph.D.(4) | 2014 | 197 | 197 | |||||||
Executive Vice President of Research & Development |
2013 | — | — | |||||||
| Kade Thompson(5) | 2014 | — | — | |||||||
CEO, CFO, Director of Thompson Design |
2013 | — | — | |||||||
- (1)
- Amounts
set forth in this column represent salary earned during fiscal 2014.
- (2)
- Mr. Pekarsky was appointed as our Chief Executive Officer, Chief Financial Officer and Chairman on January 21, 2014. Mr. Pekarsky has been the Chief Executive Officer and
43
Director of BioPharmX, Inc. since its inception. Mr. Pekarsky deferred a portion of his salary.
- (3)
- Ms. Krammer
was appointed as our Director and President on January 21, 2014. Ms. Krammer has been the President and Director of
BioPharmX, Inc. since its inception. Ms. Krammer deferred a portion of her salary.
- (4)
- Dr. Chan
was hired on February 17, 2014 as our Executive Vice President of Research & Development. Dr. Chan deferred a portion
of his salary.
- (5)
- Mr. Thompson resigned as Thompson Design Inc.'s sole director, Chief Executive Officer, President, Treasurer and Secretary on January 21, 2014, and received no compensation during the years ended December 31, 2013 and 2014.
Mr. Pekarsky's Employment Agreement
On January 21, 2014, we entered into an employment agreement with Mr. Pekarsky, pursuant to which Mr. Pekarsky was employed as our Chief Executive Officer and Chief Financial Officer for a term of four years with a one-year automatic renewal term. Mr. Pekarsky's employment agreement terminates immediately in the event of his death or disability or, in the event either we or Mr. Pekarsky delivers written notice of termination to the other party, on the fifteenth day following delivery of such notice of termination. In addition, we may immediately terminate Mr. Pekarsky's employment agreement in the event Mr. Pekarsky breaches such agreement or upon the occurrence of an event that would constitute cause (as defined in his employment agreement). Mr. Pekarsky's employment agreement provides for a base salary of $250,000 per year and an annual bonus if performance targets are met, which determination shall be made at the discretion of the board of directors. Mr. Pekarsky's employment agreement also provides that Mr. Pekarsky shall be subject to nondisclosure, noncompetition, and nonsolicitation covenants for specified periods following the termination of his employment with us.
If we terminate Mr. Pekarsky's employment without cause (as defined in his employment agreement) or if Mr. Pekarsky resigns for good reason (as defined in his employment agreement) within 12 months of a change in control (as defined in his employment agreement) and he delivers a customary release of claims, he would be entitled to: (i) an amount equal to four times his annual compensation; (ii) a continuation of company-paid health and group-term life insurance benefits applicable to him as of the change of control (or provision of benefits equivalent thereto) for 24 months; and (iii) 100% acceleration of his then unvested options, restricted stock awards, performance shares, stock appreciation rights, and, subject to limitations imposed by the applicable award agreement and Section 409A of the Code, restricted stock units, performance-based restricted stock units, and long-term cash incentives.
Ms. Krammer's Employment Agreement
On January 21, 2014, we entered into an employment agreement with Ms. Krammer, pursuant to which Ms. Krammer was employed as our President for a term of four years with a one-year automatic renewal term. Ms. Krammer's employment agreement terminates immediately in the event of her death or disability or, in the event either we or Ms. Krammer delivers written notice of termination to the other party, on the fifteenth day following delivery of such notice of termination. In addition, we may immediately terminate Ms. Krammer's employment agreement in the event Ms. Krammer breaches such agreement or upon the occurrence of an event that would constitute cause (as defined in her employment agreement). Ms. Krammer's employment agreement provides for a base salary of $250,000 per year and an annual bonus if performance targets are met, which determination shall be made at the discretion of the board of directors. Ms. Krammer's employment agreement also provides that
44
Ms. Krammer shall be subject to nondisclosure, noncompetition, and nonsolicitation covenants for specified periods following the termination of her employment with us.
If we terminate Ms. Krammer's employment without cause (as defined in her employment agreement) or if Ms. Krammer resigns for good reason (as defined in her employment agreement) within 12 months of a change in control (as defined in her employment agreement) and she delivers a customary release of claims, she would be entitled to: (i) an amount equal to four times her annual compensation; (ii) a continuation of company-paid health and group-term life insurance benefits applicable to her as of the change of control (or provision of benefits equivalent thereto) for 24 months; and (iii) 100% acceleration of her then unvested options, restricted stock awards, performance shares, stock appreciation rights, and, subject to limitations imposed by the applicable award agreement and Section 409A of the Code, restricted stock units, performance-based restricted stock units, and long-term cash incentives.
Dr. Chan's Employment Agreement
On February 17, 2014 we entered into an Employment Agreement with Dr. Chan, pursuant to which Dr. Chan was employed as Executive Vice President of Research & Development for a term of four years with a one-year automatic renewal term. Dr. Chan's employment agreement terminates immediately in the event of his death or disability or, in the event either we or Dr. Chan delivers written notice of termination to the other party, on the fifteenth day following delivery of such notice of termination. In addition, we may immediately terminate Dr. Chan's employment agreement in the event Dr. Chan breaches such agreement or upon the occurrence of an event that would constitute cause (as defined in his employment agreement). Dr. Chan's employment agreement provides for a base salary of $225,000 per year and an annual bonus if performance targets are met, which determination shall be made at the discretion of the board of directors. Dr. Chan's employment agreement also provides that Dr. Chan shall be subject to nondisclosure, noncompetition, and nonsolicitation covenants for specified periods following the termination of his employment with us.
If we terminate Dr. Chan's employment without cause (as defined in his employment agreement) or if Dr. Chan resigns for good reason (as defined in his employment agreement) within 12 months of a change in control (as defined in his employment agreement) and he delivers a customary release of claims, he would be entitled to: (i) an amount equal to two times his annual compensation; (ii) a continuation of company-paid health and group-term life insurance benefits applicable to him as of the change of control (or provision of benefits equivalent thereto) for 18 months; and (iii) 100% acceleration of his then unvested options, restricted stock awards, performance shares, stock appreciation rights, and, subject to limitations imposed by the applicable award agreement and Section 409A of the Code, restricted stock units, performance-based restricted stock units, and long-term cash incentives.
We have no plans that provide for the payment of retirement benefits, or benefits that will be paid primarily following retirement, including, but not limited to, tax qualified defined benefit plans, supplemental executive retirement plans, tax qualified defined contribution plans and non-qualified defined contribution plans.
Potential Payments upon Termination or Change of Control
The Named Executive Officers, which is defined as our principal executive officer and our next two most highly paid executive officers as of the end of the most recent fiscal year, are eligible to receive certain severance payments and benefits in connection with a termination of employment following a change in control of our company. Although we have entered into a written agreement with each of our named executive officers to provide such severance payments and benefits, we, or an
45
acquirer, may mutually agree with the Named Executive Officers to provide payments and benefits on terms that vary from those currently contemplated.
In the event of a qualifying termination of employment within 12 months of a change of control, the Named Executive Officers shall be eligible to receive the payments and benefits as described above in "Employment Agreements." The receipt of any termination based payments or benefits is subject to the Named Executive Officer executing (and not subsequently revoking) a waiver and release of claims in favor of our company or successor company.
Payments to each of Mr. Pekarsky and Ms. Krammer may be reduced in the event such payments are considered parachute payments within the meaning of Section 280G of the Code or in the event such payments trigger excise tax under Section 4999 of the Code.
Additional Narrative Disclosure
At December 31, 2014, none of our Named Executive Officers had outstanding stock options or any other equity awards.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth information regarding beneficial ownership of our common stock as of March 24, 2015:
- •
- each stockholder known by us to be the beneficial owner of more than 5% of our common stock;
- •
- each of our directors;
- •
- each of our Named Executive Officers; and
- •
- all of our directors and executive officers as a group.
We have determined beneficial ownership in accordance with the rules of the SEC, and thus this table reflects sole or shared voting or investment power with respect to our capital stock. Unless otherwise indicated below, to our knowledge, the persons and entities named in the table have sole voting and sole investment power with respect to all shares that they beneficially owned, subject to community property laws where applicable. We have deemed shares of our common stock subject to options and warrants that are currently exercisable or exercisable within 60 days of March 24, 2015 to be outstanding and to be beneficially owned by the person holding such option or warrant for the purpose of computing the percentage ownership of that person but have not treated them as outstanding for the purpose of computing the percentage ownership of any other person.
46
On March 24, 2015, there were 15,623,403 shares of common stock outstanding (including 4,207,987 shares of the Company's Series A preferred stock on an as-converted basis).
Name of Beneficial Owner(1)
|
Shares | Percentage | |||||
|---|---|---|---|---|---|---|---|
Directors and Executive Officers: |
|||||||
James R. Pekarsky(2) |
2,500,000 | 16.00 | % | ||||
Anja Krammer |
2,500,000 | 16.00 | % | ||||
Kin Chan |
1,200,000 | 7.68 | % | ||||
Michael Hubbard |
— | — | |||||
Stephen Morlock(3) |
191,082 | 1.22 | % | ||||
Ping Wang |
— | — | |||||
All executive officers and directors as a group (6 persons named above)(4) |
6,391,082 | 40.91 | % | ||||
5% or Greater Stockholders |
|||||||
KIP(5) |
1,101,101 | 7.05 | % | ||||
Hong Dong Ping(6) |
875,675 | 5.61 | % | ||||
Xiao Zheng(7) |
810,811 | 5.19 | % | ||||
- (1)
- Unless
otherwise noted, the address of each person is 1098 Hamilton Court, Menlo Park, CA 94025.
- (2)
- Consists
of 2,500,000 shares of our common stock held by The James Pekarsky Trust, of which Mr. Pekarsky is the sole beneficiary and trustee.
- (3)
- Consists
of 13,514 shares of Series A preferred stock (on an as-converted basis) and 177,568 shares of common stock held by the Stephen W. Morlock and Karen
R. Morlock TIEE UPT dated 04/21/03, of which Mr. and Ms. Morlock are co-trustees and co-beneficiaries.
- (4)
- Consists
of 13,514 shares of Series A preferred stock and 6,377,568 shares of our common stock held by all directors and executive officers as a group.
- (5)
- Consists
of (i) 290,000 shares of our common stock held by KIP, (ii) 540,541 shares of our Series A preferred stock held by KIP (on an
as-converted basis) and (iii) 270,270 shares of our common stock issuable to KIP upon exercise of a warrant that could be exercised within 60 days of March 24, 2015, thus
qualifying for inclusion in beneficial ownership. Although Ping Wang is a Principal of Korea Investment Partners Co., Ltd., which manages KIP, Mr. Wang does not have voting and
dispositive power over the shares issued to KIP.
- (6)
- Consists
of (i) 200,000 shares of our common stock held by Hong Dong Ping, (ii) 540,541 shares of Series A preferred stock (on an
as-converted basis) held by Mr. Ping and (iii) 135,134 shares of our common stock issuable to Mr. Ping upon exercise of a warrant that could be exercised within 60 days of
March 24, 2015, thus qualifying for inclusion in beneficial ownership.
- (7)
- Consists of 540,540 shares of Series A preferred stock (on an as-converted basis) held by Xiao Zheng and 270,270 shares of our common stock issuable to Ms. Zheng upon exercise of a warrant that could be exercised within 60 days of March 24, 2015, thus qualifying for inclusion in beneficial ownership.
47
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
We describe below transactions and series of similar transactions, since January 1, 2014, to which we were a party or will be a party, in which:
- •
- the amounts involved exceeded or will exceed the lesser of $120,000 or 1% of the average of our assets for the last two fiscal years;
and
- •
- any of our directors, executive officers, promoters or beneficial holders of more than 5% of any class of our capital stock had or will have a direct or indirect material interest.
Other than as described below, there have not been, nor are there any currently proposed, transactions or series of similar transactions to which we have been or will be a party other than compensation arrangements, which are described where required under "Executive Compensation."
Transactions with Founders
Our policy is that a contract or transaction either between us and a director, or between a director and another company in which he is financially interested is not necessarily void or voidable if the relationship or interest is disclosed or known to the board of directors and the stockholders are entitled to vote on the issue, or if it is fair and reasonable to our company.
Since the inception of BioPharmX, our founding executives, Mr. Pekarsky, Ms. Krammer and Dr. Chan, have made advances to cover short-term operating expenses. These advances are non-interest bearing. As of December 31, 2013 and 2014, related party payables of BioPharmX were $125,000 and $199,000, respectively, as noted in the table below.
| |
Year ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
James Pekarsky |
$ | 59,000 | $ | 112,000 | |||
Anja Krammer |
75,000 | 11,000 | |||||
Kin F. Chan |
65,000 | 2,000 | |||||
| | | | | | | | |
|
$ | 199,000 | $ | 125,000 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Share Exchange Agreement
On January 23, 2014, we entered into and consummated transactions pursuant to a share exchange agreement with BioPharmX, Inc., and the stockholders of BioPharmX, Inc. (consisting of Mr. Pekarsky, Ms. Krammer, Dr. Chan and Mr. Kevin Mszanowski (the "Founders")), whereby we issued to the Founders an aggregate of 7,025,000 shares of our common stock in exchange for 100% of the shares of BioPharmX, Inc. The shares of our common stock received by the Founders in the transaction constituted approximately 77.8% of our then issued and outstanding common stock giving effect to the issuance of shares pursuant to the share exchange agreement.
Subscription Agreements
From April 2014 through November 2014, we issued and sold to accredited investors an aggregate of 4,207,987 shares of Series A stock, at a purchase price of $1.85 per share, for aggregate consideration of $7.8 million pursuant to Subscription Agreements and issued warrants with an initial exercise price of $3.70 per share for an aggregate of 2,042,583 shares of common stock.
In connection with the Series A stock financing, (i) KIP acquired 540,541 shares of Series A stock from us for aggregate consideration of $1.0 million and warrants exercisable for an aggregate of
48
270,270 shares of common stock at $3.70 per share, (ii) Hong Dong Ping acquired 540,541 shares of Series A stock from us for aggregate consideration of $1.0 million and warrants exercisable for an aggregate of 270,270 shares of common stock at $3.70 per share and (iii) Xiao Zheng acquired 540,540 shares of Series A stock from us for aggregate consideration of $1.0 million and warrants exercisable for an aggregate of 270,270 shares of common stock at $3.70 per share. KIP, Hong Dong Ping and Xiao Zheng each hold more than 5% of our capital stock. Ping Wang, one of our directors, is an affiliate of KIP.
Investors also received registration rights pursuant to the subscription agreements.
Restricted Stock Purchase Agreement
On November 10, 2014 we entered into a restricted stock purchase agreement ("RSPA") with KIP. Pursuant to the RSPA, we issued and sold to KIP 290,000 shares of common stock with 96,667 shares vesting immediately and the remaining 193,333 shares vesting on the earlier of our achieving $2 million in revenues from VI2OLET or upon a "qualified listing", defined as uplisting to NYSE or NASDAQ within three years from the issuance of shares of Series A preferred stock, in exchange for Mr. Wang's, a principal of KIP, services as a director.
Investor Rights Agreements
We have entered into investor rights agreements with certain holders of our Series A preferred stock. These stockholders are entitled to rights with respect to the registration of their shares in connection with a public offering. The investor rights agreements will terminate upon receipt of approval of a qualified listing.
Voting Agreement
We are party to a voting agreement under which Mr. Pekarsky and Ms. Krammer have agreed to vote in a certain way on certain matters, including with respect to a merger or sale of us, or a sale of substantially all of our assets. Upon receipt of approval of a qualified listing, the voting agreement will terminate and Mr. Pekarsky and Ms. Krammer will no longer be required to vote in accordance with the agreement.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers. The indemnification agreements require us to indemnify our directors to the fullest extent permitted by Delaware law.
Employment Agreements
We have entered into employment agreements with Mr. Pekarsky, Ms. Krammer and Dr. Chan. For a description of these agreements, see "Executive Compensation."
Director Independence
We are not a "listed issuer" within the meaning of Item 407 of Regulation S-K and there are no applicable listing standards for determining the independence of our directors. Applying the definition of independence set forth in Rule 4200(a)(15) of The Nasdaq Stock Market, Inc., Mr. Hubbard, Mr. Wang and Mr. Morlock are considered independent directors. We have standing nominating, compensation and audit committees.
49
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following lists fees billed and to be billed by the auditors for the Company, for the years ended December 31, 2014 and 2013 (in thousands):
Financial Statements for the Year Ended December 31,
|
Audit Fees |
Audit Related Fees |
Tax Fees | Other Fees | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2014(1) |
$ | 110 | — | — | — | ||||||||
2013(1) |
$ | 58 | $ | 23 | — | — | |||||||
2013(2) |
$ | 4 | $ | 5 | $ | 1 | — | ||||||
- (1)
- These
services were provided by Burr Pilger Mayer, Inc., who was engaged on January 23, 2014.
- (2)
- These services were provided by Silberstein Ungar, PLLC, who was engaged through January 23, 2014.
- •
- Audit Fees. Represents fees for professional services
provided for the audit of our annual financial statements and review of our quarterly financial statements, and for audit services provided in connection with other statutory or regulatory filings.
- •
- Audit-Related Fees. Represents fees for assurance and
other services related to the audit of our financial statements.
- •
- Tax Fees. Represents fees for professional services
provided primarily for tax compliance and advice.
- •
- All Other Fees. Represents fees for products and services not otherwise included in the categories above.
We have a standing audit committee, and such committee may establish audit pre-approval policies and procedures.
50
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
- (a)
- Financial Statements and Schedules
The list of consolidated financial statements set forth in the accompanying Index to the Consolidated Financial Statements at page F-1 of this Annual Report on Form 10-K is incorporated herein by reference. Such consolidated financial statements and schedules are filed as part of this Annual Report on Form 10-K.
- (b)
- Exhibits
See the Exhibit Index immediately following the signature page of this Annual Report on Form 10-K.
51
BIOPHARMX CORPORATION
FINANCIAL STATEMENTS
Years ended December 31, 2014 and 2013
CONTENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To
the Board of Directors and Stockholders of
BioPharmX Corporation
We have audited the accompanying consolidated balance sheets of BioPharmX Corporation and its subsidiary (the "Company") as of December 31, 2014 and 2013, and the related consolidated statements of operations and comprehensive loss, convertible redeemable preferred stock and stockholders' deficit, and cash flows for each of the two years in the period ended December 31, 2014. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of the Company's internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of BioPharmX Corporation and its subsidiary as of December 31, 2014 and 2013, and the results of their operations and their cash flows for each of the two years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that BioPharmX Corporation and its subsidiary will continue as a going concern. As discussed in Note 3 to the consolidated financial statements, the Company's recurring losses from operations, available cash and accumulated deficit raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 3. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Burr Pilger Mayer, Inc.
San
Jose, California
March 30, 2015
F-2
BIOPHARMX CORPORATION
CONSOLIDATED BALANCE SHEETS
as of December 31, 2014 and 2013
(in thousands, except share and per share
data)
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
ASSETS |
|||||||
Current assets: |
|||||||
Cash |
$ | 2,111 | $ | 3 | |||
Accounts receivable |
2 | — | |||||
Inventory |
138 | — | |||||
Prepaid expenses and other current assets |
269 | 36 | |||||
| | | | | | | | |
Total current assets |
2,520 | 39 | |||||
Property and equipment, net |
235 | 32 | |||||
Intangible assets |
150 | 150 | |||||
Other assets |
50 | 150 | |||||
Restricted cash |
35 | — | |||||
| | | | | | | | |
Total assets |
$ | 2,990 | $ | 371 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
LIABILITIES, CONVERTIBLE REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS' DEFICIT |
|||||||
Current liabilities: |
|||||||
Accounts payable |
$ | 486 | $ | 229 | |||
Accrued liabilities |
426 | 198 | |||||
Payroll liabilities |
199 | 64 | |||||
Deferred rent |
51 | 65 | |||||
Deferred revenue |
6 | — | |||||
Related party payables |
199 | 125 | |||||
Convertible notes, short-term |
— | 90 | |||||
| | | | | | | | |
Total current liabilities |
1,367 | 771 | |||||
Convertible notes payable |
— | 938 | |||||
Other long-term liabilities |
— | 32 | |||||
| | | | | | | | |
Total liabilities |
1,367 | 1,741 | |||||
| | | | | | | | |
Commitments and contingencies (Note 11) |
|||||||
Series A convertible redeemable preferred stock, $0.001 par value; 10,000,000 shares authorized; 4,207,987 and no shares issued and outstanding at December 31, 2014, and 2013, respectively (liquidation preference of $7.9 million as of December 31, 2014) |
6,730 |
— |
|||||
| | | | | | | | |
Stockholders' deficit: |
|||||||
Common stock, $0.001 par value; 90,000,000 shares authorized; 11,375,311 and 7,025,000 shares issued and outstanding at December 31, 2014 and 2013, respectively |
11 | 7 | |||||
Additional paid-in capital |
4,372 | 306 | |||||
Accumulated deficit |
(9,490 | ) | (1,683 | ) | |||
| | | | | | | | |
Total stockholders' deficit |
(5,107 | ) | (1,370 | ) | |||
| | | | | | | | |
Total liabilities, convertible redeemable preferred stock and stockholders' deficit |
$ | 2,990 | $ | 371 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-3
BIOPHARMX CORPORATION
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
for the years ended December 31, 2014 and 2013
(in thousands, except share and per share data)
| |
Year ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Operating expenses: |
|||||||
Research and development |
$ | 2,519 | $ | 671 | |||
Sales and marketing |
2,299 | 132 | |||||
General and administrative |
2,953 | 711 | |||||
| | | | | | | | |
Total operating expenses |
7,771 | 1,514 | |||||
| | | | | | | | |
Loss from operations |
(7,771 | ) | (1,514 | ) | |||
Other income |
40 | — | |||||
Interest expense, net |
(76 | ) | (74 | ) | |||
| | | | | | | | |
Net and comprehensive loss |
(7,807 | ) | (1,588 | ) | |||
| | | | | | | | |
Accretion on Series A convertible redeemable preferred stock |
(163 | ) | — | ||||
Deemed dividend on Series A convertible redeemable preferred stock |
(159 | ) | — | ||||
| | | | | | | | |
Net loss available to common stockholders |
$ | (8,129 | ) | $ | (1,588 | ) | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Basic and diluted net loss available to common stockholders per share |
$ | (0.80 | ) | $ | (0.22 | ) | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Shares used in computing basic and diluted net loss per share |
10,217,000 | 7,119,000 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
BIOPHARMX CORPORATION
CONSOLIDATED STATEMENTS OF CONVERTIBLE REDEEMABLE PREFERRED
STOCK AND STOCKHOLDERS' DEFICIT
for the years ended
December 31, 2014 and 2013
(in thousands, except share and per share data)
| |
Series A Convertible Redeemable Preferred Stock |
|
|
|
|
|
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
|
|
|
|
|
||||||||||||||||||
| |
|
Common Stock | |
|
|
|||||||||||||||||||
| |
|
Additional Paid-in Capital |
Accumulated Deficit |
Total Stockholders' Deficit |
||||||||||||||||||||
| |
Shares | Amount |
|
Shares | Amount | |||||||||||||||||||
Balance at December 31, 2012 |
— | $ | — | 7,400,000 | $ | 7 | $ | 42 | $ | (95 | ) | $ | (46 | ) | ||||||||||
Repurchase of common stock |
— | — | (375,000 | ) | — | — | — | — | ||||||||||||||||
Stock-based compensation |
— | — | — | — | 58 | — | 58 | |||||||||||||||||
Issuance of convertible notes payable with beneficial conversion feature |
— | — | — | — | 206 | — | 206 | |||||||||||||||||
Net and comprehensive loss |
— | — | — | — | — | (1,588 | ) | (1,588 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 |
— | — | 7,025,000 | 7 | 306 | (1,683 | ) | (1,370 | ) | |||||||||||||||
Thompson Designs, Inc. common stock assumed in conjunction with Share Exchange |
— | — | 2,000,000 | 2 | (2 | ) | — | — | ||||||||||||||||
Issuance of convertible notes payable with beneficial conversion feature |
— | — | — | — | 204 | — | 204 | |||||||||||||||||
Issuance of common stock due to exercise of options and release of awards |
— | 824,310 | 1 | 98 | — | 99 | ||||||||||||||||||
Issuance of warrants to non-employees |
— | 204 | — | 204 | ||||||||||||||||||||
Conversion of convertible notes payable to common stock |
— | — | 1,526,001 | 1 | 1,846 | — | 1,847 | |||||||||||||||||
Stock-based compensation |
— | — | — | — | 1,193 | — | 1,193 | |||||||||||||||||
Issuance of preferred stock, related warrants and common stock |
4,207,987 | 6,408 | 845 | — | 845 | |||||||||||||||||||
Interest on preferred stock |
159 | (159 | ) | — | (159 | ) | ||||||||||||||||||
Accretion of stock issuance costs |
163 | (163 | ) | (163 | ) | |||||||||||||||||||
Net and comprehensive loss |
— | — | — | — | — | (7,807 | ) | (7,807 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2014 |
4,207,987 | $ | 6,730 | 11,375,311 | $ | 11 | $ | 4,372 | $ | (9,490 | ) | $ | (5,107 | ) | ||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-5
BIOPHARMX CORPORATION
CONSOLIDATED STATEMENTS OF CASH FLOWS
for the years ended December 31, 2014 and 2013
(in
thousands)
| |
Year ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Cash flows from operating activities: |
|||||||
Net loss |
$ | (7,807 | ) | $ | (1,588 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
|||||||
Stock-based compensation expense |
1,193 | 58 | |||||
Depreciation expense |
25 | 5 | |||||
Warrants issued for services provided |
99 | ||||||
Noncash interest expense |
76 | 74 | |||||
Changes in assets and liabilities: |
|||||||
Accounts receivable |
(2 | ) | — | ||||
Inventories |
(138 | ) | — | ||||
Prepaid expenses and other assets |
(133 | ) | (184 | ) | |||
Accounts payable |
257 | 446 | |||||
Accrued expenses |
214 | — | |||||
Payroll liabilities |
135 | — | |||||
Deferred revenue |
6 | — | |||||
Related party payables |
74 | 109 | |||||
| | | | | | | | |
Net cash used in operating activities |
(6,001 | ) | (1,080 | ) | |||
| | | | | | | | |
Cash flows from investing activities: |
|||||||
Change in restricted cash |
(35 | ) | — | ||||
Purchases of property and equipment |
(228 | ) | (25 | ) | |||
Purchase of intellectual property |
— | (60 | ) | ||||
| | | | | | | | |
Net cash used in investing activities |
(263 | ) | (85 | ) | |||
| | | | | | | | |
Cash flows from financing activities: |
|||||||
Proceeds from issuance of common stock |
99 | — | |||||
Repurchase of common stock |
— | — | |||||
Net proceeds from issuance of convertible redeemable preferred stock and common stock warrants |
7,253 | — | |||||
Proceeds from issuance of convertible notes payable |
1,020 | 1,030 | |||||
| | | | | | | | |
Net cash provided by financing activities |
8,372 | 1,030 | |||||
| | | | | | | | |
Net increase in cash |
2,108 | (135 | ) | ||||
Cash at beginning of year |
3 | 138 | |||||
| | | | | | | | |
Cash at end of year |
$ | 2,111 | $ | 3 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Non-cash investing and financing activities: |
|||||||
Intellectual assets purchase accrued |
$ | — | $ | 90 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Conversion of convertible notes payable to common stock |
$ | 1,847 | $ | — | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Fair value of beneficial conversion feature issued in connection with convertible notes payable |
$ | 204 | $ | 206 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Issuance of common stock warrants in connection with convertible notes payable |
$ | 105 | $ | — | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-6
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. FORMATION AND BUSINESS OF THE COMPANY
Description of Business
BioPharmX Corporation is incorporated under the laws of the state of Delaware and originally incorporated on August 30, 2010 in Nevada under the name Thompson Designs, Inc. We have only one wholly owned subsidiary, BioPharmX, Inc. a Nevada corporation.
The Company is a specialty pharmaceutical company focused on utilizing our proprietary drug delivery technologies to develop and commercialize novel prescription and over-the-counter, or OTC, products that address large markets in women's health and dermatology. The Company's objective is to develop products that treat health or age-related conditions that (1) are not presently being addressed or treated at all or (2) are currently treated with drug therapies or drug delivery approaches that are suboptimal. The Company's strategy is designed to bring new products to market by identifying optimal delivery mechanisms and/or alternative applications for FDA-approved active pharmaceutical ingredients, or APIs, while in appropriate circumstances, reducing the time, cost and risk typically associated with new product development by repurposing drugs with demonstrated safety profiles taking advantage of the regulatory approval pathway under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or FDC Act available for repurposed/reformulated drugs. The Company believes the 505(b)(2) regulatory pathway may reduce drug development risk and could reduce the time and resources we spend during development.
Share Exchange
On January 23, 2014, the Company (then operating as Thompson Designs, Inc.), BioPharmX, Inc. and stockholders of BioPharmX, Inc., who collectively owned 100% of BioPharmX, Inc., entered into and consummated transactions pursuant to a share exchange agreement, such transaction referred to as the Share Exchange, whereby the Company issued to the stockholders of BioPharmX, Inc. an aggregate of 7,025,000 shares of its common stock, in exchange for 100% of the shares of BioPharmX, Inc. held by stockholders. The shares of the Company's common stock received by the stockholders of BioPharmX, Inc. in the Share Exchange constituted approximately 77.8% of our then issued and outstanding common stock, after giving effect to the issuance of shares pursuant to the share exchange agreement. As a result of the Share Exchange, BioPharmX, Inc. became the Company's wholly-owned subsdidiary. For accounting purposes, the Share Exchange was treated as a reverse acquisition with BioPharmX, Inc. as the acquirer and the Company as the acquired party, and as a result the historical financial statements prior to the Share Exchange included in this Annual Report on Form 10-K are the historical financial statements of BioPharmX, Inc. On March 3, 2014, we changed the Company's name to BioPharmX Corporation. On April 25, 2014, the Company reincorporated from Nevada to Delaware.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America ("U.S. GAAP"). The accompanying financial statements include the accounts of BioPharmX and our wholly-owned subsidiary. All intercompany transactions have been eliminated in consolidation.
F-7
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Use of Estimates
The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosures. On an ongoing basis, management evaluates its significant accounting policies or estimates. We base our estimates on historical experience and on various market specific and other relevant assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates and such differences may be material to the financial statements.
Reclassification
Certain prior year amounts have been reclassified to conform to the current year presentation. The amounts for the prior periods have been reclassified to be consistent with the current year presentation and have no impact on previously reported total assets, total stockholders' deficit or net loss.
Fair Value of Financial Instruments
Carrying amounts of certain of the Company's financial instruments, including cash, prepaid and other current assets, accounts payable and accrued expenses and related party payables approximate fair value due to their short maturities. Based on borrowing rates currently available to the Company for loans with similar terms, the carrying value of the convertible notes payable approximates fair value.
Inventory
Inventory is stated at the lower of cost or market. Cost is determined using the standard cost method which approximates actual cost on a first-in, first-out basis. Market value is determined as the lower of replacement cost or net realizable value. The Company regularly reviews inventory quantities in consideration of actual loss experiences, projected future demand, and remaining shelf life to record a provision for excess and obsolete inventory when appropriate.
Shipping and Handling Costs
Shipping and handling costs are expensed as incurred and are included in cost of revenue.
Revenue Recognition
The Company shipped its first product to a retailer in December 2014. The product, the VI2OLET breast health tablet, is a new product in the dietary supplement field. Revenue is recognized provided that persuasive evidence of a sales arrangement exists, the price is fixed or determinable, title and risk of loss has transferred, calculability of the resulting receivable is reasonably assured, there are no customer acceptance requirements and we do not have any significant post-shipment obligations. The Company recognizes revenue on a sell through basis since we do not have the historical information to estimate product returns. As a result, the Company accounts for these product shipments using a deferred revenue recognition model. Under the deferred revenue recognition model, the Company does not recognize revenue upon product shipment. For these product shipments, the Company invoices the reseller, records deferred revenue at gross invoice sales price, and classifies the cost basis of the
F-8
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
product held by the wholesaler as a component of inventory. The Company recognizes revenue when product is sold by the reseller to the end-user, on a first-in first-out (FIFO) basis.
Property and Equipment
Property, plant and equipment is stated at cost less accumulated depreciation and amortization. Depreciation and amortization are recognized using the straight-line method. Repairs and maintenance costs are expensed as incurred. Estimated useful lives in years are as follows:
Description
|
Estimated Useful Life |
|
|---|---|---|
Furniture |
5 and 7 | |
Laboratory and manufacturing equipment |
5 | |
Computer & network equipment |
3 | |
Software |
3 |
Intangible Assets
Intangible assets related to in-process research and development (IPR&D) projects are considered to be indefinite-lived until the completion or abandonment of the associated research and development (R&D) efforts. During the period the assets are considered indefinite-lived, they will not be amortized but will be tested for impairment on an annual basis as well as between annual tests if we become aware of any events or changes that would indicate a reduction in the fair value of the IPR&D projects below their respective carrying amounts. If and when development is complete, which generally occurs if and when regulatory approval to market a product is obtained, the associated assets would be deemed finite-lived and would then be amortized based on their respective estimated useful lives at that point in time.
As of December 31, 2014 there have been no sales of VI2OLET. As such, the amortization period associated with the intangible asset has not commenced.
Intangible assets with finite useful lives are amortized over their estimated useful lives. Intangible assets with finite useful lives are reviewed for impairment when facts or circumstances suggest that the carrying value of these assets may not be recoverable.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. When such an event occurs, management determines whether there has been an impairment by comparing the anticipated undiscounted future net cash flows to the related asset's carrying value. If an asset is considered impaired, the asset is written down to fair value, which is determined based either on discounted cash flows or appraised value, depending on the nature of the asset. The Company has not identified any such impairment losses to date.
F-9
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Income Taxes
The Company accounts for income taxes using the liability method whereby deferred tax asset and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. Valuation allowances are established to reduce deferred tax assets when management estimates, based on available objective evidence, that it is more likely than not that the benefit will not be realized for the deferred tax assets.
The Company's policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. As of the date of adoption of accounting for uncertain tax positions there was no accrued interest or penalties associated with any unrecognized tax benefits, nor was any interest expense recognized during the year.
Comprehensive Loss
Comprehensive loss is the changes in equity of an enterprise, except those resulting from stockholder transactions. Accordingly, comprehensive loss includes certain changes in equity that are excluded from net loss. For the years ended December 31, 2014 and 2013, the Company's comprehensive loss is equal to the net loss. There were no components of other comprehensive loss for any of the periods presented.
Research and Development Expenses
Research and development expenses are expensed as incurred and consist primarily of personnel costs, including salaries, benefits and stock-based compensation, clinical studies performed by contract research organizations (CROs), materials and supplies and overhead allocations consisting of various and facilities-related costs.
Stock-Based Compensation
The Company recognizes stock-based compensation for awards granted to employees on a straight-line basis over the requisite service period, usually the vesting period, based on the grant-date fair value using the Black-Scholes pricing model.
The Company records stock-based compensation expense for awards granted to non-employees, excluding non-employee directors, based upon the estimated then-current fair value of the equity instrument using the Black-Scholes pricing model. The Company charges the value of the equity instrument to the Consolidated Statements of Operations and Comprehensive Loss over the term of the service agreement and the unvested shares underlying the option are subject to periodic revaluation over the remaining vesting period.
Advertising Expenses
The Company expenses the costs of advertising, including promotional expenses, as incurred. Advertising expenses were $68,000 and $7,000 in 2014 and 2013, respectively.
F-10
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Net Loss Per Share Attributable to BioPharmX Common Stockholders
The following table is a reconciliation of the numerator and denominator used in the calculation of basic and diluted net loss per share attributable to BioPharmX common stockholders:
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
Numerator: |
|||||||
Net loss attributable to BioPharmX stockholders (in thousands) |
$ | (8,129 | ) | $ | (1,588 | ) | |
Denominator: |
|||||||
Weighted-average shares of common stock outstanding used in the calculation of basic net loss per share attributable to BioPharmX common stockholders |
10,217,000 | 7,119,000 | |||||
Effect of dilutive securities: |
|||||||
Stock options and equivalents |
— | — | |||||
Convertible preferred stock |
— | — | |||||
| | | | | | | | |
Weighted-average shares of common stock outstanding used in the calculation of diluted net loss per share attributable to BioPharmX common stockholders |
10,217,000 | 7,119,000 | |||||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Basic net loss per share attributable to BioPharmX common stockholders is calculated based on the weighted-average number of shares of our common stock outstanding during the period. Diluted net loss per share attributable to BioPharmX common stockholders is calculated based on the weighted-average number of shares of our common stock outstanding and other dilutive securities outstanding during the period. The potential dilutive shares of our common stock resulting from the assumed exercise of outstanding stock options and the assumed conversion of convertible notes are determined under the treasury stock method.
The following outstanding shares of common stock equivalents were excluded from the computation of diluted net loss per share of common stock for the periods presented because including them would have been antidilutive:
| |
Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2014 | 2013 | |||||
Convertible redeemable preferred stock |
4,207,987 | — | |||||
Stock options and awards to purchase common stock |
2,802,690 | 2,606,000 | |||||
Common stock warrants |
2,702,537 | — | |||||
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers, (ASU 2014-09), which converges the FASB and the International Accounting Standards Board standards on revenue recognition. Areas of revenue recognition that will be affected include, but are not limited to, transfer of control, variable consideration, allocation of transfer pricing, licenses, time value of money, contract costs and
F-11
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
disclosures. This guidance is effective for the fiscal years and interim reporting periods beginning after December 15, 2016. The Company is currently evaluating the impact that the adoption of ASU 2014-09 will have on its consolidated financial statements and related disclosures.
On June 10, 2014, the FASB issued ASU 2014-10, Elimination of Certain Financial Reporting Requirements, Including an Amendment to Variable Interest Entities Guidance in Topic 810, Consolidation, (ASU 2014-10), which eliminates the definition of a development stage entity, eliminates the development stage presentation and disclosure requirements under Accounting Standards Codification, or ASC, 915 Development Stage Entities, or ASC 915, and amends provisions of existing variable interest entity guidance under ASC 810 Consolidation . As a result of the changes, entities which meet the former definition of a development stage entity will no longer be required to: (1) present inception-to-date information in the statements of income, cash flows, and stockholder equity; (2) label the financial statements as those of a development stage entity; (3) disclose a description of the development stage activities in which the entity is engaged; and (4) disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. Furthermore, ASU 2014-10 clarifies disclosures about risks and uncertainties under ASC Topic 275, Risks and Uncertainties, that apply to companies that have not commenced planned principal operations. Finally, variable interest entity rules no longer contain an exception for development stage entities and, as a result, development stage entities will have to be evaluated for consolidation in the same manner as non-development stage entities.
Under ASU 2014-10, entities are no longer required to apply the presentation and disclosure provisions of ASC 915 during annual periods beginning after December 15, 2014. In addition, the revisions to the consolidation standards are effective for annual periods beginning after December 15, 2015 for public entities and are effective for annual periods beginning after December 15, 2016 for nonpublic entities. Early adoption is permitted for any annual reporting period or interim period for which the entity's financial statements have not yet been issued (public business entities) or made available for issuance (other entities).
The Company has adopted ASU 2014-10 effective as of its issuance date. Adoption of this standard had no impact on its financial position, results of operations, or cash flows; however, the presentation of the consolidated financial statements and related disclosures in the notes to the consolidated financial statements has been changed to eliminate the disclosures that are no longer required.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements—Going Concern (ASU 2014-15). This standard includes guidance about management's responsibility to evaluate whether there is substantial doubt about an entity's ability to continue as a going concern within one year after the financial statements are issued. If conditions or events raise substantial doubt, the entity must disclose the conditions or events that raise substantial doubt about the entity's ability to continue as a going concern, management's evaluation of those conditions or events, and management's plans to mitigate the conditions or events. This update is effective for interim and annual reporting periods beginning after December 15, 2016. Early adoption is permitted. The Company is currently evaluating the impact that the adoption of ASU 2014-15 will have on its consolidated financial statements and related disclosures.
F-12
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
The Company has reviewed other recent accounting pronouncements and concluded they are either not applicable to the business, or no material effect is expected on the consolidated financial statements as a result of future adoption.
3. GOING CONCERN CONSIDERATIONS AND MANAGEMENT'S PLAN
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. The Company has incurred recurring losses and negative cash flows from operations since inception. The Company has not generated revenues and has funded its operating losses through the issuance of convertible notes payable and Series A convertible redeemable preferred stock (Series A). The Company has a limited operating history and its prospects are subject to risks, expenses and uncertainties frequently encountered by companies in the industry.
The significant risks and uncertainties described herein could have a significant negative impact on the financial viability of BioPharmX and raise substantial doubt about the Company's ability to continue as a going concern. Management is working on the Company's business model to increase working capital by managing its cash flow, securing financing and introducing its first product to market.
Risks include, but are not limited to, the uncertainty of availability of additional financing and the uncertainty of achieving future profitability. Management of the Company intends to raise additional funds through the issuance of equity securities. There can be no assurance that such financing will be available or on terms which are favorable to the Company. Failure to generate sufficient cash flows from operations, raise additional capital or reduce certain discretionary spending could have a material adverse effect on the Company's ability to achieve its intended business objectives. These factors raise substantial doubt about the Company's ability to continue as a going concern. The financial statements do not contain any adjustments that might result from the outcome of this uncertainty.
As shown in the accompanying consolidated financial statements, the Company incurred a net loss of $7.8 million and $1.6 million during the years ended December 31, 2014 and 2013, respectively, and has an accumulated deficit of $9.5 million as of December 31, 2014. As of December 31, 2014, the Company had working capital of $1.2 million. While management of the Company believes that it has a plan to fund on-going operations, there is no assurance that its plan will be successfully implemented. The Company is experiencing the following risks and uncertainties in the business:
The discovery of key raw materials to formulate novel products depends on the Company's ability to identify, negotiate and secure procurement of such materials. This also depends on the Company's ability to establish comprehensive and long term vendor contracts and relationships.
The Company's ability to compete and to achieve its product platform strategy depends on its ability to protect its proprietary discoveries and technologies. The Company currently relies on a combination of copyrights, trademarks, trade secret laws and confidentiality agreements to protect its intellectual property rights. The Company also relies upon unpatented know-how and continuing technological innovation.
The Company's continued operations are dependent upon its ability to identify, recruit and retain adequate management personnel and contractors to perform certain jobs such as research and development, patent generation, regulatory affairs and general administrative functions. The Company requires highly trained professionals of varying levels and experience along with a flexible work force.
F-13
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. GOING CONCERN CONSIDERATIONS AND MANAGEMENT'S PLAN (Continued)
The Company's ability to generate income in the short-run will depend greatly on the rate of adoption and ability to establish a market for the Company's VI2OLET breast health tablet.
Research and development for novel prescription or OTC based products can be very extensive and lengthy in nature; and the clinical trial process with the Food and Drug Administration can require significant funding and time consuming patient studies. The competitive landscape could change significantly over the time period to complete targeted product development milestones. The current competition for BioPharmX's products could also turn into strategic partners or potential acquirers in the future.
4. FAIR VALUE MEASUREMENTS
The Company recognizes and discloses the fair value of its assets and liabilities using a hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to valuations based upon unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to valuations based upon unobservable inputs that are significant to the valuation (Level 3 measurements). Each level of input has different levels of subjectivity and difficulty involved in determining fair value.
- •
- Level 1—Inputs used to measure fair value are unadjusted quoted prices that are available in active markets for the
identical assets or liabilities as of the reporting date. Therefore, determining fair value for Level 1 investments generally does not require significant judgment, and the estimation is not
difficult.
- •
- Level 2—Pricing is provided by third party sources of market information obtained through investment advisors. The
Company does not adjust for or apply any additional assumptions or estimates to the pricing information received from its advisors.
- •
- Level 3—Inputs used to measure fair value are unobservable inputs that are supported by little or no market activity and reflect the use of significant management judgment. These values are generally determined using pricing models for which the assumptions utilize management's estimates of market participant assumptions. The determination of fair value for Level 3 instruments involves the most management judgment and subjectivity.
As of December 31, 2014 and 2013, the Company held no assets or liabilities with instrument valuations measured on a recurring basis.
5. INVENTORY
Inventory at December 31, 2014 and 2013 consisted of the following (in thousands):
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
Work in Process |
$ | 135 | $ | — | |||
Finished Goods |
2 | — | |||||
Channel Inventory |
1 | — | |||||
| | | | | | | | |
|
$ | 138 | $ | — | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
F-14
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
6. PROPERTY AND EQUIPMENT
Property and equipment, net at December 31, 2014 and 2013 consisted of the following (in thousands):
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
Furniture |
$ | 18 | $ | 11 | |||
Lab equipment |
26 | 12 | |||||
Computers and equipment |
78 | 15 | |||||
Software |
144 | — | |||||
| | | | | | | | |
|
266 | 38 | |||||
Less: accumulated depreciation |
(31 | ) | (6 | ) | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
|
$ | 235 | $ | 32 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Depreciation expense for the years ended December 31, 2014 and 2013 was $25,000 and $5,000.
7. RESTRICTED CASH
The Company has restricted cash in the amount of $35,000 held by Bank of America in a money market account to secure the credit line of the Company's credit cards.
8. ACCRUED LIABILITIES
Accrued liabilities at December 31, 2014 and 2013 consisted of the following (in thousands):
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
Marketing |
$ | 267 | $ | — | |||
Legal |
80 | 89 | |||||
Production |
57 | — | |||||
Intellectual property |
— | 90 | |||||
Other |
22 | 19 | |||||
| | | | | | | | |
Total accrued liabilities |
$ | 426 | $ | 198 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
9. RELATED PARTY PAYABLES
Since inception, the founding executives of the Company have made advances to cover short-term operating expenses. Additionally, since the beginning of 2014 a portion of their compensation is being deferred and is included in this balance. These advances and deferred compensation are non-interest bearing. As of December 31, 2014 and 2013, related party payables were $199,000 and $125,000, respectively.
10. LONG-TERM OBLIGATIONS
Financing Arrangements
In September and November 2012, the Company issued convertible notes payable ("Notes") to two individuals, respectively, in exchange for $200,000 in cash. These Notes carry an interest rate of 6% per
F-15
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
10. LONG-TERM OBLIGATIONS (Continued)
annum and mature in September and November 2015, respectively, with principal and interest payable at maturity.
During the year ended December 31, 2013, the Company issued Notes to twelve individuals in exchange for $1.0 million in cash. These notes carry an interest rate of 6% per annum and mature between June 2014 and October 2016, with principal and interest payable at maturity.
During the first quarter of 2014, the Company issued convertible notes payable to thirteen individuals in exchange for $1.0 million in cash. The Notes carry an interest rate of 6% per annum and mature between January 2015 and March 2017, with principal and interest payable at maturity.
The Notes automatically convert into common stock upon the Company entering into a qualified preferred stock financing at 80% of the price per share at which such preferred stock is issued in such an offering. Additionally, there is a special conversion that at maturity, unless the Company repays all outstanding principal and interest, the Notes shall be automatically converted into a number of shares of common stock of the Company at 80% of the then fair market value per share.
As a result of this beneficial conversion feature, the Company recorded $204,000 and $206,000, as a debt discount during the years ended December 31, 2014 and 2013. The debt discount was amortized to interest expense over the term of the Notes using the effective interest rate method. The amortization expense related to the debt discount was $49,000 and $41,000 for the years ended December 31, 2014 and 2013.
During the year ended December 31, 2014, the Company completed a private placement (the "Private Placement") of shares of its Series A and warrants to purchase common stock ("Warrants"). The Private Placement was consummated in a series of closings that occurred between April 2014 and November 2014. The Company sold to accredited investors and non-U.S. persons 4.2 million shares of Series A at a per share price of $1.85 for total net proceeds of approximately $7.3 million and issued to the investors for no additional consideration the Warrants to purchase in the aggregate 2.0 million shares of the Company's common stock, at an exercise price of $3.70 per share pursuant to a series of subscription agreements. See Note 12.
As a result, upon the first Series A closing on April 11, 2014, the convertible notes payable balance, net of unamortized discounts, of $1.8 million was converted into 1,526,001 shares of common stock. The balance of the accrued interest was waived. As of December 31, 2014, there were no remaining outstanding convertible notes.
11. COMMITMENTS AND CONTINGENCIES
Lease Arrangements
On August 23, 2013, the Company signed a lease for 10,800 square feet of office and laboratory space in Menlo Park, California. The term of the lease is 39 months from the lease commencement
F-16
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. COMMITMENTS AND CONTINGENCIES (Continued)
date of September 1, 2013. Future minimum commitments under this lease are as follows (in thousands):
Years ending December 31,
|
|
|||
|---|---|---|---|---|
2015 |
$ | 288 | ||
2016 |
271 | |||
| | | | | |
Total |
$ | 559 | ||
| | | | | |
| | | | | |
| | | | | |
Legal Proceedings
We are not currently a party to any legal proceedings. We are not aware of any pending legal proceeding to which any of our officers, directors, or any beneficial holders of 5% or more of our voting securities are adverse to us or have a material interest adverse to us.
Indemnification
The Company enters into standard indemnification arrangements in the ordinary course of business. Pursuant to these arrangements, the Company indemnifies, holds harmless, and agrees to reimburse the indemnified parties for losses suffered or incurred by the indemnified party, in connection with any trade secret, copyright, patent or other intellectual property infringement claim by any third-party with respect to the Company's technology. The term of these indemnification agreements is generally perpetual. The maximum potential amount of future payments the Company could be required to make under these agreements is not determinable because it involves claims that may be made against the Company in the future, but have not yet been made.
The Company has entered into indemnification agreements with its directors and officers that may require the Company to indemnify its directors and officers against liabilities that may arise by reason of their status or service as directors or officers, other than liabilities arising from willful misconduct of the individual.
The Company has not incurred costs to defend lawsuits or settle claims related to these indemnification agreements. No liability associated with such indemnifications has been recorded to date.
License Agreement
In March 2013, the Company entered into an amended and restated collaboration and license agreement with Iogen LLC, which provides the Company the license the rights to label, market, and resell the finished inventory and ongoing manufacturing of the Iogen molecular iodine technology for future product formulation development and commercialization. New formulation patents developed by the Company will be the sole ownership of the Company. The agreement gives the Company a perpetual, fully paid-up, non-exclusive license under the licensed Intellectual Property (IP) rights to make, have made, use, sell, offer for sale and import products.
F-17
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. COMMITMENTS AND CONTINGENCIES (Continued)
Per terms of the license the Company is required to make future payments of:
- •
- Pay a fee for acquiring all finished inventory of the Iogen Product.
- •
- Pay a fee for the non-exclusive license to the IP.
- •
- Pay 30% of net profit associated with direct commercialization of an OTC product or 30% of net royalties received from any
sub-licensee.
- •
- Pay a royalty of 3% of net sales for the first 12 months of commercialization and 2% of net sales thereafter for other products
developed and commercialized under the license, including a prescription version of the iodine tablet.
- •
- Pay a fixed royalty fee for the protection and indemnification of licensed IP rights for the prescription product developed, marketed
and sold from newly developed formulations as long as the patents are valid and cover the prescription product.
- •
- Pay a fixed royalty fee for the protection and indemnification of licensed IP rights for the other products utilizing the molecular iodine technology developed, marketed and sold from newly developed formulations as long as the patents are valid and cover the prescription product.
The Company capitalized as Intangible Assets, in the year ended December 2013, the amount of $150,000 related to this agreement. No royalties have been paid as of December 31, 2014.
12. CONVERTIBLE REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS' EQUITY
Common Stock
On March 27, 2013, the Company terminated one of the founders and repurchased 375,000 shares for $18.
As described in Note 1, on January 23, 2014, the Company issued 7,025,000 shares of its common stock to BioPharmX, Inc. stockholders.
As described in Note 10, on April 11, 2014, the Company's convertible notes and eligible interest were converted to 1,526,001 shares of common stock upon the first closing of the offer and sale of Series A Preferred Stock.
During the year ended December 31, 2014, the Company issued 727,643 shares of common stock upon the exercise of stock options.
In November 2014, the Company issued 290,000 shares of common stock to Korea Investment Partners Expansion Platform Fund of which 96,667 vested immediately and 193,333 will vest upon completion of the $2 million investment outlined in the Series A subscription agreement. The unvested shares are not considered outstanding for financial reporting purposes.
At December 31, 2014, the Company has 11,375,311 shares of common stock currently issued and outstanding.
Series A Preferred Stock
The Company entered into Subscription Agreements for a private placement of shares of our Series A and Warrants with 47 accredited investors during 2014 whereby we sold an aggregate of
F-18
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. CONVERTIBLE REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS' EQUITY (Continued)
4,207,987 shares of Series A at a per share price of $1.85 for gross proceeds of $7.5 million and issued to the investors for no additional consideration the Warrants to purchase in the aggregate 2,042,583 shares of the Company's common stock, with an exercise price of $3.70 per share.
The Warrants with an allocated fair value of $845,000 were classified as additional paid-in capital. The Company determined the fair value using the Black-Scholes pricing model with the following assumptions: dividend rate of 0%, risk-free rate of 1.6% to 4.0%, contractual term of 5 years and expected volatility of 88.8%. These Warrants were immediately exercisable, and as of December 31, 2014, were all outstanding.
In connection with the Subscription Agreements, the Company, the majority shareholders of the Company and the Investors entered into Investor Rights Agreements (the Investor Rights Agreements) with the Investors, whereby the Investors were granted certain rights including: (i) right to receive copies of quarterly and annual reports of the Company, (ii) right of inspection of the Company's properties and records, (iii) right of participation in future securities offerings, (iv) tag-along rights in connection with sales of the Company's stock by a major shareholder, and (v) board of directors representation rights for the subscribers who purchased at least 500,000 shares of Series A and hold at least 30% of such shares (the "Qualified Subscribers"). The Company made certain covenants under the agreement including: (i) uplisting to NYSE or NASDAQ within three years from the issuance shares of Series A, and (ii) increase of the board of directors to five members including one member to be appointed by the Qualified Subscribers.
Significant terms of Series A are as follows:
- •
- Holders of the Series A are entitled to interest payment at the rate of 6% of the purchase price per annum. The Company has the
option to pay this interest in shares of common stock or in cash. As of December 31, 2014, $159,000 in interest has been accreted to the Series A. Holders of the Series A are
entitled to receive dividends on an as converted basis with the holders of the Company's common stock.
- •
- The holders of the Series A are entitled to vote together with the holders of the Company's common stock, with each such holder
of Series A entitled to the number of votes equal to the number of shares of the Company's common stock into which such Series A would be converted if converted on the record date for
the taking of a vote.
- •
- Each share of Series A is initially convertible, at any time at the sole option of the holder, into one share of the Company's
common stock, subject to future adjustments as provided for in the Series A Certificate. The Series A shall automatically convert into shares of the Company's common stock upon the
uplisting of the common stock to NYSE or NASDAQ within three years from the issuance of shares of Series A.
- •
- If the Company fails to effect the uplisting within three years from the issuance of shares of Series A, the holders will have the right to require the Company to redeem all or a portion of the then outstanding Series A at a price per share equal to the Series A liquidation preference.
F-19
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. CONVERTIBLE REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS' EQUITY (Continued)
Warrants
In addition to the Warrants issued in conjunction with the Subscription Agreements, the Company issued warrants on May 15, 2014, to a service provider for 316,395 shares of common stock at an exercise price of $2.035 per share, which were valued at $99,000 and expensed. The Company also issued to a qualified investor as a part of his convertible loan package for 343,559 shares of common stock at an exercise price of $1.85 per share, which was valued at $105,000. These warrants expire after five years. The Company determined the fair value using the Black-Scholes option pricing model with the following assumptions: dividend rate of 0%, risk-free rate of 1.6%, contractual term of 5 years and expected volatility of 88.8%. These Warrants were immediately exercisable, and as of December 31, 2014, were all outstanding.
Equity Incentive Plan
On January 23, 2014, the Company adopted the 2014 Equity Incentive Plan (the "2014 Plan") which permits the Company to grant stock options to directors, officers or employees of the Company or others to purchase shares of common stock of the Company through awards of incentive and nonqualified stock options ("Options"), stock ("Restricted Stock" or "Unrestricted Stock") and stock appreciation rights ("SARs"). Options previously issued under the BioPharmX, Inc. 2011 Equity Incentive Plan were cancelled, and options under the 2014 Plan were issued to replace all cancelled BioPharmX, Inc. options.
The Company currently has time-based options outstanding. The time-based options generally vest in two to four years and expire ten years from the date of grant. Total number of shares originally reserved and available for grant and issuance pursuant to this Plan was 2,700,000. Shares issued under the Plan will be drawn from authorized and unissued shares or shares now held or subsequently acquired by the Company. On November 7, 2014, the Company increased the stock available to the 2014 Equity Incentive Plan for options grants from 2,700,000 shares to 4,500,000 shares. At December 31, 2014, there were 1,163,000 shares available for grant under the Plan.
F-20
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. CONVERTIBLE REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS' EQUITY (Continued)
The following table summarizes the Company's stock option activities for the years ended December 31, 2014 and 2013:
| |
Shares | Weighted Average Exercise Price Per Share |
Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
|
|
(in thousands) | |||||||||
Outstanding as of January 1, 2013 |
1,150,000 | $ | 0.06 | ||||||||||
Granted |
1,456,000 | 0.40 | |||||||||||
Exercised |
— | — | |||||||||||
Cancelled |
— | — | |||||||||||
| | | | | | | | | | | | | | |
Outstanding as of December 31, 2013 |
2,606,000 | $ | 0.25 | ||||||||||
Granted |
891,000 | 1.85 | |||||||||||
Exercised |
(727,643 | ) | 0.14 | ||||||||||
Cancelled |
(160,000 | ) | 0.37 | ||||||||||
| | | | | | | | | | | | | | |
Outstanding as of December 31, 2014 |
2,609,357 | $ | 0.82 | 8.52 | $ | 5,686 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Vested and exercisable |
972,462 | $ | 0.39 | 7.71 | $ | 2,542 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Vested and expected to vest |
2,490,074 | $ | 0.80 | 8.49 | $ | 5,478 | |||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The weighted average grant date fair values of the stock options granted during the years ended December 31, 2014 and 2013 were $1.10 and $0.28 per share, respectively.
The following table summarizes significant ranges of outstanding and exercisable options as of December 31, 2014 (in thousands, except contractual life and exercise price):
| |
|
Options Outstanding | Options Vested and Exercisable |
|
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Range of Exercise Price
|
Number Outstanding |
Weighted Average Remaining Contractual Life (in Years) |
Weighted Average Exercise Price |
Number Vested and Exercisable |
Weighted Average Exercise Price |
|
||||||||||||||
| $0.05 - $0.35 | 1,550,857 | 7.68 | $ | 0.21 | 797,462 | $ | 0.17 | ||||||||||||||
| $1.00 | 167,500 | 6.76 | $ | 1.00 | 100,000 | $ | 1.00 | ||||||||||||||
| $1.85 | 891,000 | 9.79 | $ | 1.85 | 75,000 | $ | 1.85 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| 2,609,357 | 8.52 | $ | 0.82 | 972,462 | $ | 0.39 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | |
The total intrinsic value of employee stock options exercised during the years ended December 31, 2014 and 2013 was $676,000 and zero, respectively.
As of December 31, 2013, total compensation costs related to unvested, but not yet recognized, stock-based awards was $2,573,000, net of estimated forfeitures. This cost will be amortized on a
F-21
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. CONVERTIBLE REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS' EQUITY (Continued)
straight-line basis over a weighted average remaining period of 3.29 years and will be adjusted for subsequent changes in estimated forfeitures.
13. STOCK-BASED COMPENSATION.
The following table summarizes the stock-based compensation expenses included in our Statement of Operations and Comprehensive Loss for the years ended (in thousands):
| |
2014 | 2013 | |||||
|---|---|---|---|---|---|---|---|
Research and development |
$ | 228 | $ | 30 | |||
Sales and marketing |
147 | 7 | |||||
General and administrative expenses |
818 | 21 | |||||
| | | | | | | | |
Stock-based compensation expense, net of tax |
$ | 1,193 | $ | 58 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The Company estimates the fair value of time-based stock options, if any, granted using the Black-Scholes pricing model. The fair value is then amortized on a straight-line basis over the requisite service periods of the awards, which is generally the vesting period. Time-based and performance-based options, if any, typically have a ten-year life from date of grant and vesting periods of two to four years.
Valuation Assumptions
The fair value of stock-based awards to employees is calculated through the use of the Black-Scholes pricing model, even though such model was developed to estimate the fair value of freely tradable, fully transferable options without vesting restrictions, which differ significantly from the Company's stock option awards. This model also requires subjective assumptions, including future stock price volatility and expected time to exercise, which greatly affect the calculated values.
Expected Term
The expected term represents the period that the Company's stock-based awards are expected to be outstanding. For awards granted subject only to service vesting requirements, the Company utilizes the simplified method for estimating the expected term of the stock-based award, instead of historical exercise data.
Expected Volatility
The Company uses the historical volatility of the price of the common shares of selected public companies in the biotechnology sector.
Expected Dividend
The Company has never paid dividends on its common shares and currently does not intend to do so and, accordingly, the dividend yield percentage is zero for all periods.
F-22
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
13. STOCK-BASED COMPENSATION. (Continued)
Risk-Free Interest Rate
The Company bases the risk-free interest rate used in the Black-Scholes-Merton valuation method upon the implied yield curve currently available on U.S. Treasury zero-coupon issues with a remaining term equal to the expected term used as the assumption in the model.
We used the following assumptions to calculate the estimated fair value of the awards for the years ended:
| |
2014 | 2013 | |||
|---|---|---|---|---|---|
Expected volatility |
82.2 | % | 82.1% | ||
Expected term in years |
6.0 | 5.51 - 6.08 | |||
Risk-free interest rate |
1.74 | % | 0.61% - 1.62% | ||
Expected dividend yield |
— | % | —% | ||
14. EMPLOYEE BENEFIT PLAN
The Company sponsors a 401(k) defined contribution plan for its employees. This plan provides for tax-deferred salary deductions for all employees. Employee contributions are voluntary. Employees may contribute up to 100% of their annual compensation to this plan, as limited by an annual maximum amount as determined by the Internal Revenue Service. The Company may match employee contributions in amounts to be determined at the Company's sole discretion. The Company has made no contributions to the plan for the years ended December 31, 2014 and 2013.
15. INCOME TAXES
No federal income taxes were provided in the years ended December 31, 2014 and 2013 due to the Company's net losses. State minimum income and franchise taxes are included in general and administrative expenses and were immaterial for the periods presented.
At December 31, 2014, the Company had available federal net operating loss ("NOL") carry-forwards of approximately $7.6 million which will begin to expire in 2031 and California state NOL carry-forwards of approximately $7.6 million which will begin to expire in 2021. At December 31, 2014 and 2013, the net deferred tax assets of approximately $3.2 million and $594,000, respectively, generated primarily by NOL carry-forwards, have been fully reserved due to the uncertainty surrounding the realization of such benefits. The net valuation allowance increased by approximately $2.6 million and $563,000 during the years ended December 31, 2014 and 2013, respectively.
Current tax laws impose substantial restrictions on the utilization of net operating loss and credit carry-forwards in the event of an "ownership change," as defined by the Internal Revenue Code. If there should be an ownership change, the Company's ability to utilize its carry-forwards could be limited.
As of December 31, 2014 and 2013, the Company did not have any material unrecognized tax benefits. The tax years from 2010 to 2014 remain open for examination by the federal and state authorities.
F-23
BIOPHARMX CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
16. SUBSEQUENT EVENTS
In March 2015, we amended certain warrants to reduce the exercise price of such warrants from $3.70 to $2.50 per share with a corresponding increase in the number of shares of common stock exercisable under the warrants so that the aggregate exercise value of such warrants remained the same. As of March 27, 2015, the holders had exercised such warrants for an aggregate of 397,996 shares of common stock for an aggregate cash exercise price of $994,990.
F-24
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized, in Menlo Park, State of California, on this 30th day of March, 2015.
| BioPharmX Corporation | ||||
By: |
/s/ JAMES PEKARSKY Name: James Pekarsky Title: Chief Executive Officer, Chief Financial Officer and Director (Principal Executive Officer, Principal Financial Officer and Principal Accounting Officer) |
|||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints James Pekarsky and Anja Krammer or either of them his or her true and lawful attorney-in-fact and agents, each with the full power of substitution and re-substitution, for such person in such person's name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might do or could do in person, hereby ratifying and confirming all that each of said attorneys-in-fact and agents, or his substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ JAMES PEKARSKY James Pekarsky |
Chief Executive Officer, Chief Financial Officer and Chairman of the Board of Directors (Principal Executive Officer, Principal Financial Officer and Principal Accounting Officer) | March 30, 2015 | ||
/s/ ANJA KRAMMER Anja Krammer |
President and Director |
March 30, 2015 |
S-1
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ PING WANG Ping Wang |
Director | March 30, 2015 | ||
/s/ MICHAEL HUBBARD Michael Hubbard |
Director |
March 30, 2015 |
||
/s/ STEPHEN MORLOCK Stephen Morlock |
Director |
March 30, 2015 |
S-2
| |
|
|
Incorporated by Reference | |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
|
Filed Herewith |
|||||||||||
| Description of Document | Form | File No. | Filing Date | Exhibit | ||||||||||
| 2.1 | Form of Share Exchange Agreement dated January 23, 2014 by and among Thompson Designs, Inc., BioPharmX, Inc. and BioPharmX, Inc. Stockholders | 8-K | 000-54871 | 1/27/2014 | 2.1 | |||||||||
| 3.1 | Certificate of Incorporation | S-8 | 333-201708 | 1/26/2015 | 4.01 | |||||||||
| 3.2 | Bylaws | S-8 | 333-201708 | 1/26/2015 | 4.02 | |||||||||
| 4.1 | Specimen Stock Certificate | S-8 | 333-201708 | 1/26/2015 | 4.03 | |||||||||
| 4.2 | Certificate of Designations, Preferences and Rights of Series A Preferred Stock | X | ||||||||||||
| 4.3 | Certificate of Validation of Certificate of Designations, Preferences and Rights of Series A Preferred Stock filed with the Delaware Secretary of State on March 17, 2015 | X | ||||||||||||
| 10.1 | Form of Stock Purchase Agreement between Kade Thompson and BioPharmX, Inc. | 8-K | 000-54871 | 1/27/2014 | 10.1 | |||||||||
| 10.2 | * | Form of Employment Agreement between James Pekarsky and Thompson Designs, Inc. | 8-K | 000-54871 | 1/27/2014 | 10.2 | ||||||||
| 10.3 | * | Form of Employment Agreement between Anja Krammer and Thompson Designs, Inc. | 8-K | 000-54871 | 1/27/2014 | 10.3 | ||||||||
| 10.4 | * | Employment Agreement between Kin F. Chan, Ph.D. and the Company | X | |||||||||||
| 10.5 | Amended and Restated Collaboration and License Agreement dated March 1, 2013 between BioPharmX, Inc. and Iogen LLC | 8-K | 000-54871 | 1/27/2014 | 10.4 | |||||||||
| 10.6 | Collaboration and Supply Agreement dated October 22, 2013 between BioPharmX, Inc. and Nutech Medical, Inc. | 8-K | 000-54871 | 1/27/2014 | 10.5 | |||||||||
| 10.7 | Lease Agreement dated August 23, 2013 between Prologis, L.P. and BioPharmX, Inc. | 8-K | 000-54871 | 1/27/2014 | 10.6 | |||||||||
| 10.8 | * | 2014 Equity Incentive Plan | 8-K | 000-54871 | 1/27/2014 | 10.7 | ||||||||
| 10.9 | * | Form of 2014 Equity Incentive Plan award agreement | S-8 | 333-201708 | 1/26/2015 | 4.05 | ||||||||
| 10.10 | Form of Securities Purchase Agreement | 8-K | 000-54871 | 1/27/2014 | 10.8 | |||||||||
| 10.11 | Form of Subscription Agreement | 10-K | 000-54871 | 3/31/14 | 10.11 | |||||||||
| 10.12 | Investor Rights Agreement between the Company, Senior Management of the Company and the parties to Subscription Agreements | 10-K | 000-54871 | 3/31/14 | 10.12 | |||||||||
| |
|
|
Incorporated by Reference | |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exhibit Number |
|
|
Filed Herewith |
|||||||||||
| Description of Document | Form | File No. | Filing Date | Exhibit | ||||||||||
| 10.13 | Promissory Note, dated December 21, 2012 between Thompson Designs, Inc. and Kade Thompson | 10-K | 000-54871 | 12/31/12 | 10.1 | |||||||||
| 10.14 | Voting Agreement, dated October 24, 2014, between KIP Overseas Platform Expansion Fund, James Pekarsky and Anja Krammer | 8-K | 000-54871 | 11/12/2014 | 10.3 | |||||||||
| 10.15 | Subscription Agreement, dated October 24, 2014, between the Company and KIP Overseas Expansion Platform Fund | X | ||||||||||||
| 16.1 | Letter of Silberstein Ungar, PLLC to the SEC dated January 23, 2014 | 8-K | 000-54871 | 1/27/2014 | 16.1 | |||||||||
| 21.1 | Subsidiaries of the Registrant | X | ||||||||||||
| 23.1 | Consent of Burr Pilger Mayer, Inc., independent registered public accounting firm | X | ||||||||||||
| 24.1 | Power of attorney. Reference is made to the signature page hereto. | X | ||||||||||||
| 31.1 | Certification of Principal Executive Officer and Principal Financial Officer Required Under Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended | X | ||||||||||||
| 32.1 | Certification of Chief Executive Officer and Chief Financial Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. §1350 | X | ||||||||||||
| 101.INS | XBRL Instance Document | X | ||||||||||||
| 101.SCH | XBRL Taxonomy Schema Linkbase Document | X | ||||||||||||
| 101.CAL | XBRL Taxonomy Calculation Linkbase Document | X | ||||||||||||
| 101.DEF | XBRL Taxonomy Definition Linkbase Document | X | ||||||||||||
| 101.LAB | XBRL Taxonomy Labels Linkbase Document | X | ||||||||||||
| 101.PRE | XBRL Taxonomy Presentation Linkbase Document | X | ||||||||||||
- *
- Indicates a management contract, compensatory plan or arrangement