Attached files
| file | filename |
|---|---|
| EX-21 - EX-21 - Cardiff Oncology, Inc. | a15-1864_1ex21.htm |
| EX-32.2 - EX-32.2 - Cardiff Oncology, Inc. | a15-1864_1ex32d2.htm |
| EX-10.7 - EX-10.7 - Cardiff Oncology, Inc. | a15-1864_1ex10d7.htm |
| EX-23.1 - EX-23.1 - Cardiff Oncology, Inc. | a15-1864_1ex23d1.htm |
| EX-31.1 - EX-31.1 - Cardiff Oncology, Inc. | a15-1864_1ex31d1.htm |
| EX-31.2 - EX-31.2 - Cardiff Oncology, Inc. | a15-1864_1ex31d2.htm |
| EX-10.8 - EX-10.8 - Cardiff Oncology, Inc. | a15-1864_1ex10d8.htm |
| EX-10.6 - EX-10.6 - Cardiff Oncology, Inc. | a15-1864_1ex10d6.htm |
| EX-32.1 - EX-32.1 - Cardiff Oncology, Inc. | a15-1864_1ex32d1.htm |
| EXCEL - IDEA: XBRL DOCUMENT - Cardiff Oncology, Inc. | Financial_Report.xls |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
x ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2014
o TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE TRANSITION PERIOD FROM TO
COMMISSION FILE NUMBER 000-54556
TROVAGENE INC.
(Name of small business issuer in its charter)
|
Delaware |
|
27-2004382 |
|
(State or other jurisdiction of |
|
(I.R.S. Employer |
|
incorporation or organization) |
|
Identification No.) |
11055 Flintkote Avenue, Suite B, San Diego, California 92121
(Address of principal executive offices) (Zip Code)
Issuer’s telephone Number: (858) 952-7570
Securities registered under Section 12(b) of the Exchange Act:
|
Title of each class |
|
Name of each exchange on which registered |
|
Units, each consisting of two shares of Common Stock and one Warrant to purchase one share of Common Stock |
|
The NASDAQ Capital Market |
|
|
|
|
|
Common Stock, $0.0001 par value |
|
The NASDAQ Capital Market |
|
|
|
|
|
Warrants to purchase Common Stock |
|
The NASDAQ Capital Market |
Securities registered under Section 12(g) of the Exchange Act: None.
Indicate by check mark is the issuer is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act Yes o No x
Indicate by check if the issuer is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes x No o
Indicate by check mark whether the issuer (1) filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the past 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if no disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer”, and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer o |
|
Accelerated filer x |
|
|
|
|
|
Non-accelerated filer o |
|
Smaller reporting company o |
|
(Do not check if a smaller reporting company) |
|
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of the voting and non-voting common equity held by non-affiliates based on a closing sale price of $3.50 per share which was the last sale price of the common stock as of June 30, 2014 was $63,974,477.
As of February 27, 2015, the issuer had 24,123,790 outstanding shares of Common Stock.
PART I
ITEM 1. BUSINESS
We are a molecular diagnostic company that focuses on the development and commercialization of a proprietary molecular diagnostic technology for use in disease detection and monitoring across a variety of medical disciplines. Our primary internal focus is to leverage our novel cell-free molecular diagnostic platform to facilitate improvements in the field of oncology, while our external focus includes entering into license agreements or collaborations to develop our technology in areas such as infectious disease, transplant medicine, and prenatal genetics.
We are leveraging our proprietary molecular diagnostic technology for the detection of cell-free DNA originating from diseased cell death that can be isolated and detected from urine, blood, and tissue samples to improve disease management. These genetic materials are also collectively referred to as “cell-free nucleic acids”, which result when cells in the body die and release their DNA contents into the bloodstream. The circulating fragments of genetic material are eventually filtered through the kidneys and therefore, can be detected and measured in urine. Cell-free nucleic acids can be used as genetic markers of disease. As such, the contents of urine or blood samples represent systemic liquid biopsies that can allow for simple, non-invasive or minimally-invasive sample collection methods.
Our fundamental cell-free molecular diagnostic platform, also known as our “Precision Cancer MonitoringSM” platform, (“PCM”) platform is protected by a strong intellectual property portfolio. We have developed significant intellectual property around cell-free nucleic acids in urine, the extraction of cell-free nucleic acids from urine, as well as novel assay designs, particularly our proprietary non-naturally occurring primers. Through this proprietary technology, we believe that we are at the forefront of a shift in the way diagnostic medicine is practiced, using simple, non-invasive or minimally invasive sampling and analysis of nucleic acids, which we believe will ultimately lead to more effective treatment monitoring, better management of serious illnesses such as cancer, and the ability to detect the recurrence of cancer earlier. As of February 27, 2015, our property intellectual property portfolio consists of over 50 issued patents and over 60 pending patent applications globally. Our patent estate includes the detection of cell-free nucleic acids that pass through the kidney into the urine, as well as their application in specific disease areas, including oncology, infectious disease, transplantation, and prenatal genetics.
We believe that our proprietary PCM platform is uniquely positioned to address a high unmet clinical need in field of oncology. Our PCM platform is designed to offer better cancer monitoring by tracking and analyzing levels of cell-free DNA from either urine or blood samples, and is intended to provide important clinical information beyond the current standard of care. Using urine as a sample, our cancer monitoring technology enables more frequent, non-invasive monitoring of oncogene mutation status, disease progression and disease recurrence. Our extensive research and development efforts were strengthened, due to investments to expand our intellectual property portfolio and were made commercially feasible following improved next-generation sequencing (“NGS”) technologies which are now available at a significantly lower cost. This combined with our extensive patent portfolio around cell-free DNA in urine gives us a competitive advantage to leverage an emerging trend toward monitoring cancer using cell-free DNA as a marker of disease status. Our proprietary sample preparation process forms the basis of our PCM platform. It includes novel technology for the extraction and isolation of cell-free DNA from either a urine or blood sample, proprietary non-naturally occuring primers to enrich the sample for mutant alleles, and the ability to sequence nucleic acids of interest using one of several leading gene sequencing technologies such as NGS or droplet digital PCR (“ddPCR”). We believe that our quantitative cell-free DNA detection and monitoring platform offers industry leading sensitivity, featuring single nucleic acid molecule detection.
Our PCM platform is poised to overcome a significant clinical dilemma in the area of cancer treatment. Recent scientific evidence supports the molecular basis of cancer, and has resulted in a paradigm shift in the way cancer is treated. Researchers and clinicians are now focused on specific oncogene mutations that are believed to be the drivers of cancer at the molecular level, and, as a result, there is a trend in the pharmaceutical research community toward developing targeted therapies. As such, there is a need for oncologists to have an ability to track the mutational status of their patients, including a given patient’s response to treatments designed to target driver oncogene mutations. Current monitoring tools such as imaging procedures, tissue biopsy, and circulating tumor cells are insufficient to meet the challenge of monitoring oncogene mutations. Imaging only provides a rough indication of tumor size, and is an important tool for surgeons, but provides little practical advice to oncologists regarding mutational status and appropriate treatment options, especially for molecular targeted therapies. Tissue biopsy usually involves a major surgical procedure and, in many cases, is not repeatable as there are limitations related to access for serial biopsies. In some cases, biopsies may not be available, significantly increasing the need to determine mutational status using an alternative method. In addition, tumor heterogeneity is important, as the surgeon may not obtain the proper tissue from the tumor sample. In the case of circulating tumor cells, which are typically measured using blood tests, there is very low sensitivity, and such tests are technically difficult and can be expensive.
Targeted drug therapies themselves are not without issues. Targeted therapies are typically very expensive and can have significant side effects. In order to measure effectiveness, repeated monitoring is needed and serial biopsies can be difficult to obtain. If resistance develops, fast and accurate detection of emerging or changing oncogene mutations is critical. Our PCM platform provides a novel solution using urine, a non-invasive, plentiful sample source, and we are continuing to build a growing body of evidence supporting the clinical utility of our technology to monitor cancer using cell-free DNA.
Our goal is to improve treatment outcomes for cancer patients using our proprietary technology to detect and quantitatively monitor cell-free DNA using urine or blood samples.
Developing a Market for Molecular Diagnostic Tests based on Liquid Biopsies using Cell-free DNA
We intend to develop and expand our cell-free molecular diagnostic technology into a pipeline of potentially groundbreaking commercial molecular detection and monitoring products. Our Clinical Laboratory Improvement Amendments (“CLIA”)-certified, College of American Pathologists (“CAP”)-accredited laboratory in San Diego will enable us to initially commercialize our testing services and launch our platform technology and associated innovative molecular monitoring tests. Urine-based cell-free molecular diagnostics can provide relevant information across multiple therapeutic and clinical areas, and may lead to improvements in patient management. We are focused on the oncology treatment market, and the opportunity to enable clinicians to track oncogene mutational status in cancer patients. Repeat testing is expected with most cancer patients, and there also exists a need to chronically monitor for the re-emergence of oncogenes in people that are cancer survivors.
In order to facilitate early availability and use of our products and technologies, in February 2012, we acquired the CLIA laboratory assets of MultiGEN Diagnostics, Inc., (“MultiGEN”), which included CLIA approval and licensing documentation, laboratory procedures, customer lists, and marketing materials. A CLIA lab is a clinical reference laboratory that can perform high complexity diagnostic assays (e.g. those requiring polymerase chain reaction, (“PCR”) amplification). Through this CLIA laboratory, we are able to offer laboratory developed tests (“LDTs”), in compliance with CLIA guidelines.
Targeting cell-free nucleic acid markers will allow for the development of genetic tests that use non-invasive and easy-to-obtain urine samples, rather than other more traditional, more invasive methods. These methods include imaging, blood testing, and bone marrow and tissue biopsies. We are exploring a broad range of clinical utilities where cell-free nucleic acid technology holds the potential to replace more complex, less robust existing technologies, which are based on circulating cells and nucleic acids in blood. We are developing more effective, non-invasive diagnostics, which align with the current industry shift toward highly personalized medicine. Urine-based cell-free nucleic acid molecular tests can make it easier to address important health problems, and may lead to significant advancements in patient care.
Our patented technology uses safe, non-invasive, cost effective, and simple urine collection, which can be applied to a broad range of testing including tumor mutation detection and monitoring, infectious disease monitoring, transplantation monitoring, and prenatal genetic diagnostics. We believe that our technology is ideally suited to be used in developing molecular diagnostic assays that will allow physicians to provide simple, non-invasive, and convenient screening and monitoring tests for their patients by identifying specific biomarkers involved in a disease process. Our novel urine-based assays can facilitate improved testing compliance, resulting in more effective use of targeted therapies, earlier detection of disease, and improvements in both patient outcomes and cost of care.
The material terms of certain of our clinical collaboration, research and development, and technology license agreements that we have entered into are as follows:
In September 2014, under the strategic partnership we established in March 2014, we entered into a Sponsored Research Agreement with Catholic Health Initiatives Center for Translational Research to conduct clinical studies to evaluate use of our PCM technology in the management of cancer patients. Under the terms of the agreement, we may pay our collaborator in the study approximately $151,000 for services provided. As of December 31, 2014 we have incurred approximately $30,000 related to this agreement.
In June 2014, we entered into a Sponsored Research Agreement with Dana Farber Cancer Institute to conduct a clinical study to evaluate use of our precision cancer monitoring technology in the management of lung cancer patients. Under the agreement, we may pay our collaborator in the study approximately $42,000 for services provided. As of December 31, 2014 we have incurred approximately $8,000 related to this agreement.
In June 2014, we entered into a Sponsored Research Agreement with Memorial Sloan Cancer Center to conduct a clinical study for the detection of oncogenic tumor mutations in the urine of lung cancer patients. Under the agreement with Memorial Sloan Kettering, we may pay our collaborator approximately $146,000 for services provided. As of December 31, 2014 we have incurred approximately $25,000 related to this agreement.
In May 2014, we entered into a Strategic Research Alliance with the Robert H. Lurie Comprehensive Cancer Center of Northwestern University to conduct one or more research agreements to evaluate use of our precision cancer monitoring technology in the management of cancer patients. Under the agreement, each party will be responsible for its own costs and obligations under each agreement. No services or costs have been incurred by us as of December 31, 2014.
In May 2014, we entered into a Patent Assignment and License Agreement, effective as of April 23, 2014, with GenSignia IP Ltd., a United Kingdom company, pursuant to which we assigned all of our miRNA patents, including methods of using miRNA for detection of in vivo cell death and detecting cell-free miRNA in urine and blood. Concurrent with the assignment, GenSignia granted to us an exclusive, world-wide, royalty-free, fully paid, perpetual license under the transferred patents in the urine field. Pursuant to the agreement, GenSignia will pay us a low single digit royalty on net sales and will pay an aggregate $6.5 million in milestone payments upon the achievement of up to $150 million in net sales. GenSignia shall be responsible for the preparation, filing and maintenance of all patents under the agreement. As of December 31, 2014, we had recorded $10,000 in license fee revenue related to the agreement. Costs have been incurred through December 31, 2014 and reimbursed by GenSignia.
In December 2013, we entered into a Clinical Trial Agreement with US Oncology Research LLC (“USOR”), pursuant to which USOR will provide the principal investigator and conduct a clinical study related to examining the utility of cell-free quantitative KRAS testing to monitor disease in patients with metastatic pancreatic cancer. Under the agreement, we committed to pay USOR approximately $270,000 for services provided. During the years ended December 31, 2014 and 2013, we incurred and recorded approximately $16,000 and $29,000 of research and development expense related to this agreement.
In August 2013, we entered into a Clinical Trial Agreement with the University of Southern California (“USC”), pursuant to which USC will provide the principal investigator and conduct a clinical study related to the genetic characterization of metastatic colorectal cancers. Under the agreement, we are committed to pay USC approximately $232,000 for services provided. During the years ended December 31, 2014 and 2013, we incurred approximately $38,000 and $0, respectively for expenses related to this agreement.
In June 2013, we entered into a Research Agreement with Illumina, Inc. pursuant to which the parties will work together to evaluate the potential for integrating our cell-free technology for isolating, extracting, and analyzing nucleic acids from urine with Illumina’s genetic analysis sequencing technology. The parties have agreed to share all results and reagents from the Research Plan. The agreement will terminate upon the earlier of 30 days after completion of the Research Plan or the one year anniversary of the agreement unless extended by mutual written agreement. In October 2014, the agreement was extended for an additional year to June 2015.
During 2012, we entered into research agreements with University of Texas MD Anderson Cancer Center (“MDACC”) to provide samples and evaluate methods used by us to identify pancreatic cancer mutations, as well as to measure the degree of concordance between the results of cell-free DNA mutation analysis from urine samples and tumor tissue. An amendment in 2013 increased the scope of the research agreements. We have committed to pay approximately $266,000 for the services performed by MDACC. As of December 31, 2014, 2013 and 2012, we have incurred and recorded approximately $124,000, $142,000 and $0, respectively of research and development expenses related to this agreement.
In December 2012, we entered into a sublicense agreement with Genoptix, Inc. for non-exclusive worldwide rights to develop and market laboratory testing services for nucleophosmin protein (“NPM1”) for the diagnosis and monitoring of patients with acute myeloid leukemia (“AML”),. Under this agreement, we have granted a license to certain NPM1 patents in exchange for a one time license fee of $100,000 due upon execution of the agreement and royalty payments on net revenues. During the years ended December 31, 2014, 2013 and 2012, we have recorded royalty and license fee revenues of approximately $30,000, $10,000 and $100,000, respectively.
In November 2012, we entered into a sublicense agreement with Duke University and Duke University Health Systems for non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. Under this agreement, we have granted a license to certain NPM1 patents in exchange for a one time license fee of $5,000 due upon execution of the agreement and royalty payments on net revenues. During the years ended December 31, 2014, 2013 and 2012, we have recorded $1,000, $0 and $5,000, respectively of royalty and license fee revenues related to this agreement.
In September 2012, we entered into a collaboration and license agreement with Strand Life Sciences related to the validation and commercial launch of a urine-based DNA test for Human Papillomavirus (“HPV”). Under this agreement, we have granted a license for use of our tests to Strand in exchange for royalty payments on net sales earned in the territory specified in the agreement. During the years ended December 31, 2014, 2013 and 2012, no royalties or license fees had been received under this agreement.
In September 2012, we entered into a sublicense agreement with Quest Diagnostics for non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. Under this agreement, we have granted a license to certain NPM1 patents in exchange for a one time license fee of $20,000 due upon execution of the agreement and royalty payments on net sales of Quest Diagnostics and its affiliates. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty and license revenues of approximately $26,000, $14,000 and $20,000, respectively.
In December 2011, we entered into an exclusive license agreement with Columbia University to license the patent rights to hairy cell leukemia biomarkers. In consideration of the license we paid $1,000 as an upfront license fee and agreed to make royalty payments as a single digit percentage of net sales if sales are made by us or a single digit royalty rate as a percentage on sublicense income received by us if sales are made by sublicensees. The license agreement shall continue until May 10, 2021, which is the date of the last to expire of the licensed patent rights covering the license product. The license agreement may also be terminated upon a material breach by any party or by us if we determine that it is not commercially or scientifically appropriate to further develop the license product rights. For the years ended December 31, 2014, 2013 and 2012, there has been no royalty expense recorded related to this agreement.
In October 2011, we entered into an exclusive license agreement with Gianluca Gaidano, Robert Foa and Davide Rossi for the patent rights to a specific gene mutation with respect to chronic lymphoblastic leukemia. In consideration of the license, we paid $1,000 as an upfront license fee and agreed to make royalty payments as a single digit percentage of net sales if sales are made by us or a single digit royalty rate as a percentage of sublicense income received by us if sales are made by sublicensees. We have an option to purchase the licensed patent rights in the event the licensor decides to sell such licensed patent rights. The license agreement shall continue until September 29, 2031, which is the date of the last to expire of the licensed patent rights covering the license product. The license agreement may also be terminated upon a material breach by any party or by us if it is determined that it is not commercially or scientifically appropriate to further develop the license product rights. For the years ended December 31, 2014, 2013, and 2012, there has been no royalty expense recorded related to this agreement.
In July 2011, we entered into a sublicense agreement with Fairview Health Services (“Fairview”) for the non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. Fairview paid an initial license fee of $10,000 upon execution of the agreement and will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. Fairview is obligated to pay a royalty with annual minimums of $1,000 each year. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty and license fee revenues of approximately $2,000, $1,000 and $2,000, respectively.
In February 2011, we entered into a sublicense agreement with MLL Münchner Leukämielabor (“MLL”) for the non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. MLL paid an initial license fee of $20,000 upon execution of the agreement and will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. MLL is obligated to pay a royalty with annual minimums of $15,000 for the first year and $20,000 thereafter. The term of the license ends on October 28, 2025, which is the date of expiration of the issued patent rights. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty and license fee revenues of approximately $81,000, $85,000 and $71,000, respectively.
In June 2010, we signed a sublicensing agreement with Skyline Diagnostics BV for the non-exclusive rights to develop, commercialize and market, research and diagnostic laboratory services for the stratification and monitoring of patients with AML. Skyline Diagnostics BV paid an initial licensing fee of $10,000 upon execution of the agreement and may make future payments to us upon the attainment of certain regulatory and commercial milestones. Skyline Diagnostics BV will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty and license revenues of approximately $0, $0 and $0, respectively. During those same periods, we did not record any license fee expenses.
In December 2008, we signed a sublicensing agreement with InVivoScribe Technologies, Inc. for the non-exclusive rights to develop and market lab testing services for NPM1 for the diagnosis and monitoring of patients with AML. InVivoScribe Technologies paid an initial licensing fee of $10,000 upon execution of the agreement. InVivoScribe Technologies will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on
October 28, 2025 which is the date of expiration of the issued patent rights. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty revenues of approximately $25,000, $25,000 and $27,000, respectively. During those same periods, we did not record any license fee expenses.
In August 2008, we signed a sublicensing agreement with LabCorp for the non-exclusive rights to develop and market lab testing services for NPM1, for the diagnosis and monitoring of patients with AML. LabCorp paid an initial licensing fee of $20,000 upon execution of the agreement. LabCorp will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends August 25, 2018. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty and license fee revenues of approximately $28,000, $20,000 and $5,000, respectively. During those same periods, we did not record any license fee expenses.
In October 2007, we signed a license agreement with ASURAGEN, Inc. for the co-exclusive rights to develop, manufacture and market, research and diagnostic products for the stratification and monitoring of patients with AML.ASURAGEN paid an initial licensing fee of $120,000 upon execution of the agreement and may make future payments to us upon the attainment of certain regulatory and commercial milestones. In June 2010, we signed amendment No. 1 to the co-exclusive license agreement. The amendment asserts that we may require a license from a third-party to perform laboratory testing services. ASURAGEN will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. In March 2013, we signed amendment No. 2 to the co-exclusive sublicense agreement with ASURAGEN. The amendment limited the field of use to research use only (RUO) kits. ASURAGEN was also granted a non-exclusive sublicense for NPM1 laboratory testing services. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty and license fee revenues of approximately $50,000, $50,000 and $50,000, respectively. During those same periods, we had no license fee expenses related to this agreement.
During August 2007, we signed a sublicensing agreement with IPSOGEN SAS, a leading molecular diagnostics company with operations in France and the United States for the co-exclusive rights to develop, manufacture and market, research and diagnostic products for the stratification and monitoring of patients with AML. Upon execution of this agreement, IPSOGEN paid an initial licensing fee of $120,000 and may make milestone payments upon the attainment of certain regulatory and commercial milestones. IPSOGEN will also pay us a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025, which is the date of expiration of the issued patent rights. In September 2010, we signed amendment No. 1 to the sublicensinging agreement. The amendment asserts that we may require a license from a third-party to perform laboratory testing services. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty, milestone and license fee revenues of approximately $60,000, $60,000 and $180,000, respectively. During those same periods, we had no license fee expenses.
In May 2006, we entered into a license agreement with Drs. Falini and Mecucci, wherein we obtained the exclusive rights for the genetic marker for AML with the intention to utilize these rights for the development of new diagnostic tools. In connection with this agreement, we paid $70,000 to Drs. Falini and Mecucci. In August 2010, we signed amendment No.1 to the license agreement with an obligation to pay royalties of 6% on royalty revenues and/or 10% of any sublicense income. During the years ended December 31, 2014, 2013 and 2012, we recorded royalty expenses of approximately $23,000, $30,000 and $24,000, respectively.
History
On April 26, 2002, we were incorporated in the State of Florida as Used Kar Parts, Inc. On July 2, 2004, we acquired Xenomics, a California corporation, which was in business to develop and commercialize urine-based molecular diagnostics technology. As part of the acquisition, our corporate name was changed to Xenomics, Inc. (“Xenomics”). In 2007, we changed our fiscal year end from January 31 to December 31. In January 2010, we re-domesticated our state of incorporation from Florida to Delaware and our name was changed to Trovagene, Inc. In June 2012, our common stock was listed on The NASDAQ Capital Market under the ticker symbol TROV.
The Basis for Our Urine-based Molecular Diagnostic Technology
Cell-free nucleic acids have been found in a variety of human bodily fluids, with the nucleic acids isolated from urine having been extensively characterized. Cell-free nucleic acids in urine have been proven to contain mutated DNA and other markers of disease, including microRNA. In contrast to other bodily fluids (e.g. blood plasma), urine allows for truly non-invasive collection of the sample, provides a larger sample size, and allows for frequent collection. Importantly, urine enables the collection of nucleic acid material from the systemic circulation over a period of time, and those DNA and RNA fragments remain stable in urine. These factors, combined with recently developed technologies to sequence, count, and track nucleic acids with low relative abundance, make the development of these non-invasive diagnostics commercially practical and scalable.
In the human body, about 1011 - 1012 cells die each day primarily as a consequence of natural physiological processes for tissue and organ maintenance, but also as a result of disease. Together, these dead and dying cells contain more than 1 gram of DNA, which is mostly degraded into short fragments by specific enzymes. A small proportion of these cell-free nucleic acids escapes complete degradation and appears in the bloodstream. Our scientists were the first to discover that circulating cell-free nucleic acids cross the kidney barrier and can be found in the urine as cell-free DNA. This simple yet remarkable discovery that genetic information from various cells throughout the body is present in urine enabled the development of new, non-invasive techniques for molecular diagnostics and genetic testing.
To unlock the full potential of cell-free nucleic acids, we have developed a proprietary method for the isolation of the short fragmented nucleic acids that pass through the kidneys, and proprietary “ultra-short” amplicon assays necessary for the efficient detection of cell-free nucleic acids, which can be analyzed at our San Diego-based CLIA laboratory.
Because of the small size of cell-free nucleic acids in urine, having an isolation method that efficiently captures short nucleic acids is critical. We have multiple methods (patents and pending patents) for the isolation of nucleic acids from bodily fluids, including urine. Many nucleic isolation methods are not properly suited for the isolation of cell-free nucleic acids in urine. For example, many DNA isolation kits only capture DNA greater than 200 base-pairs (“bp”) in length, with a few claiming 100 bp or longer. No manufacturer states that their product is suitable to capture DNA sequences shorter than 80 bp in length.
When compared to leading kits for the isolation of DNA from bodily fluids, we have observed by conducting internal studies, that our method is three to twenty times more efficient in isolating a 50 bp target. Our method is also suitable for the isolation of RNA, including miRNAs.
Paired with our cell-free nucleic acid isolation method, is our technology for detecting ultra-short amplicons (patents pending). By combining our proprietary nucleic acid isolation method with our ultra-short amplicon assays, we are able to detect at least six times more mutations in a urine-based cell-free DNA sample than any other PCR-based assay, according to our internal test data. We believe that these methods are also applicable to other small or fragmented nucleic acids, including cell-free DNA from blood and formalin-fixed, paraffin-embedded samples.
Determining DNA and RNA signatures using urine as a “systemic biopsy” may provide a more powerful and effective tool for following and uncovering both pre-clinical and clinical changes, which may include:
· monitoring cancer patients to determine therapeutic response or non-response and disease recurrence;
· following organ transplant status to watch for rejection;
· non-invasively securing samples for the clinical diagnosis of infectious diseases; and
· screening and testing expectant mothers, who’s fetus may be at risk for certain genetic abnormalities.
Currently, these clinical needs are addressed by the use of invasive blood and bone marrow tests, tissue biopsies, amniocentesis, as well as costly CT, MRI, and PET scans.
Urine is a relatively simple aqueous solution and, unlike plasma, contains few components that can attack and break down cell-free nucleic acid fragments. Since urine does not contain many cells, proteins and other contaminants, cell-free nucleic acid isolation is a procedure which can be easily automated for high throughput screening applications. Cell-free nucleic acid fragments can be accurately analyzed using conventional methods that are either in use or in development within many molecular genomics laboratories.
Our urine-based cell-free nucleic acid tests are based upon a proprietary method of nucleic acid isolation, followed by detection of specific genetic markers. These proven and well-established detection methods can be used to detect nucleic acids in blood, stool, and other specimen types. Using enhancements of these techniques, cell-free nucleic acid markers can be isolated from easily obtained urine specimens.
Our urine-based cell-free nucleic acid technology may be applied to the detection and monitoring of an extremely broad spectrum of medical conditions.
Characteristics of Urine-based Cell-free Nucleic Acid Testing
· The kidney acts as a filter, passing cell-free nucleic acids from complex, multicellular, multicomponent blood into urine, a much less complex aqueous environment.
· The collection procedure is non-invasive and does not require the involvement of trained medical staff.
· Urine as a sample type supports repeated testing when required and poses no discomfort for the patient.
· Cell-free nucleic acids in urine are stable at room temperature for extended periods of time with the addition of a simple preservative. Nucleic acids in blood and many other traditional samples are not.
· Sample processing and tests can often be easily automated.
· Isolation of cell-free markers from large sample volumes increases the sensitivity of the tests. This cannot be done as easily using blood or tissue specimens, which have inherent volume limitations.
· Blood or sputum samples for detection of infectious diseases may not be easily obtained from certain patients, including small children and the elderly. Urine specimens typically present minimal acquisition concerns.
· Blood and other bodily fluids can be highly infectious by nature, urine is not.
· Blood and other bodily fluids are legally considered biohazardous, urine is not.
Clinical Applications
We believe that our urine-based cell-free molecular diagnostic test will make it easier to address important health problems worldwide, and will lead to significant advances in personalized medicine for improved patient care. We intend to develop clinical evidence for our cancer monitoring tests in three distinct and potentially overlapping stages. Stage 1 studies are qualitative in nature and are designed to determine the mutational status of actionable biomarkers in urine especially when biopsy is not an option. These studies demonstrate concordance (agreement) of the oncogene mutation status between a urine sample and a tumor tissue sample. These studies are considered to have diagnostic value, and would prove that urine-based molecular test results match the tissue biopsy closely. The clinical utility of such a study would validate that mutational status of actionable biomarkers can be determined in urine when a biopsy is not an option. Stage 2 studies are quantitative in nature, and are designed to assess patient mutational status in urine longitudinally (over time) as an indicator of responsiveness to therapy and disease status of the patient. Demonstrated clinical utility includes quantitatively assessing mutation status in urine longitudinally as an indicator of responsiveness to therapy and disease status of the patient. Stage 3 studies are conducted with the goal to demonstrate improved patient outcomes and eventually could lead to changing medical guidelines and the clinical standard of care for managing certain cancers. Demonstrated clinical utility includes quantitatively assessing patient mutational status in urine longitudinally for mutational status as well as early detection of resistance to therapy as a decision tool for therapy selection. Generating data with our technology that supports better patient outcomes and more efficient use of healthcare resources is a key component of Stage 3.
We believe that there are several specific applications of our PCM platform technology with regard to helping oncologists monitor a patient’s mutational status, and thereby, optimize the treatment approach and improve outcomes. Our technology can be used to determinie a patient’s mutational status for the first time when a tissue biopsy is not feasible, or it can be used to monitor changes in mutational load over time to provide information that can be useful to direct treatment regimens. Should a patient have their tumor removed surgically, our technology can be used to broadly search for minimual residual disease, which can confirm a successful procedure, or can enable early detection of recurrent disease for improved patient management. Treatment-emergent mutations can also be a major problem, and may be drivers of resistance to first-line therapy. Examples of this include the emergent mutation EGFR T790M in lung cancer, or KRAS mutations in colorectal cancer. Because our platform uses a non-invasive, easy to obtain sample from the patient, the ability to monitor more accurately and more often with fewer barriers to doing so can provide us with key competitive advantages in the marketplace, particularly with regard to monitoring for treatment-emergent mutations.
Oncology
Urine may offer an alternative to blood-based tests such as circulating tumor cells, biopsy and imaging. By tracking mutations we can inform medical practice. Our initial pilot study was focused on the BRAF mutation because of its link to discreet cancers and associated treatments, as well as the KRAS mutation because of its broad applicability in many cancers. We are now developing
oncogene tracking tests using ddPCR and NGS for a variety of mutations seen in many cancer types. We believe the potential exists to expand the use of this latter technique across many cancer types for multiple mutations in test panels.
During 2014, we had over 15 ongoing clinical studies with leading cancer centers and pharmaceutical companies to demonstrate the qualitative and quantitative clinical utility of our tests. Clinical study sites include MD Anderson Cancer Center, USC Norris Cancer Center, US Oncology, pharmaceutical collaborators and other top cancer centers. In 2014, we signed clinical study collaboration agreements with Memorial Sloan Kettering Cancer Center, Catholic Health Initiatives Center for Translational Research, Dana Farber Cancer Insititute, The Robert H. Lurie Comprehensive Cancer Center of Northwestern University, City of Hope Comprehensive Care Center, and Genomac International Ltd, (also known as the Center for Applied Genomics of Solid Tumors, Genomac Research Institute).
Initial clinical results for KRAS and BRAF mutation assays
The MD Anderson Cancer Center clinical study is focused on detecting and monitoring BRAF and KRAS tumor mutations in cell-free DNA from urine in metastatic cancer patients. BRAF mutations are common in melanoma, thyroid, and other cancers. Within the U.S., it is estimated that nearly 730,000 patients have tumors with BRAF mutations. Several targeted therapies are either on the market or in development for BRAF-mutation positive cancers. Pancreatic cancer represents an additional diagnostic and treatment challenge. Each year, more than 43,000 new cases of pancreatic cancer are diagnosed, and 37,000 patients succumb to this disease. It is estimated that KRAS mutations occur in >90% of pancreatic cancers and 11%-17% of these patients do not express the CA19-9 marker, which makes their disease more difficult to track.
Initial results from the MD Anderson Cancer Center clinical study were published at the AACR-NCI-EROTC International Conference in October 2013. During the study, urine samples from metastatic cancer patients known to have BRAF V600E, KRAS G12D or KRAS G12V mutations were assessed. Our researchers analyzed the urine samples using our urine-based cell-free molecular diagnostic assays. Results demonstrated high concordance between urine and tissue mutational status. In addition, preliminary results indicate that cell-free BRAF V600E mutation monitoring in urine correlates with clinical response to therapy. The clinical study demonstrated that BRAF V600E mutations were detected in urine irrespective of the cancer type, and a multitude of different cancer types, including brain cancer (“glioblastoma”), were included in the initial study results. The BRAF V600E assay demonstrated 95% concordance vs. tissue biopsy (both detected and borderline), and also demonstrated that urine DNA can be used to detect DNA fragments from circulation that harbor tumor mutations. The following cancers were detected: non-small cell lung cancer, papillary thyroid carcinoma, melanoma, colorectal cancer, glioblastoma, adenocarcinoma of unknown primary, ovarian cancer, and appendiceal cancer. In addition, preliminary results indicate that cell-free BRAF V600E mutation monitoring in urine longitudinally correlates with clinical response to therapy.
The study also evaluated the feasibility of using massively parallel deep sequencing (i.e. NGS) to identify DNA mutations in the urine of metastatic cancer patients harboring known KRAS mutations. Leveraging proprietary enrichment methods, our researchers were able to detect mutant cell-free DNA in the urine of cancer patients with verified KRAS mutations.
CLIA validated BRAF mutation assay
In October 2013, our first urine test for cancer mutation monitoring was made available to clinicians through our CLIA laboratory. The robustness of our ultra-sensitive assay procedure has been demonstrated for the detection of the BRAF V600E mutation from cell-free DNA in urine. This mutation commonly occurs in melanoma. Of the more than 70,000 cases of melanoma diagnosed each year in the United States, up to 70 percent harbor a BRAF-type mutation and of those, 80 percent may be positive specifically for BRAF V600E. There are several approved targeted therapies for the treatment of BRAF-positive melanoma, making mutational status monitoring an area of clinical interest among treating physicians.
Our cell-free BRAF test is a LDT, designed to detect and monitor this mutation in metastatic cancer patients with biopsy-proven V600E BRAF mutation in their tumor. It is the first commercial assay within our cancer monitoring portfolio performed using a ddPCR platform. Using urine as a non-invasive, systemic sample, the cell-free BRAF test could help physicians monitor changes in mutation status for patients requiring therapy for cancers that have this mutation. For patients with difficult-to-biopsy metastatic tumors, urine-based mutation testing may also provide a viable alternative to gauge mutation status as part of the initial treatment workup.
In April, 2014, we announced the presentation of clinical study results at the American Association for Cancer Research (“AACR”) Annual Meeting. Of the 33 patients enrolled in the study, our BRAF V600E oncogene mutation assay was able to identify the mutation in 29 patients (88%) at least one time during the study, demonstrating a high level of concordance with tissue biopsy.
Longitudinal analysis was performed in 17 patients who had more than one urine-based test during the monitoring period. Of these patients, 13 (76%) showed a correlation between response to treatment and mutational status observed by the urine-based test. The results were presented by Filip Janku, M.D., Ph.D., University of Texas MD Anderson Cancer Center.
In June 2014, we announced that expanded clinical study results demonstrating the utility of our PCM platform were released at the 50th Annual Meeting of the American Society of Clinical Oncology (“ASCO”). Data from a study in multiple cancer types were published in the 2014 ASCO Annual Meeting Proceedings, a Journal of Clinical Oncology by Filip Janku, M.D., Ph.D., University of Texas MD Anderson Cancer. In this study, longitudinal analysis of sequential urine samples demonstrated a statistically significant correlation between changes in the amount of BRAF V600E mutation load and treatment response with targeted drug therapy (p=0.002), per RECIST 1.1 criteria. Results also demonstrated that patients with a decrease in BRAF V600E mutation load had a longer median time to treatment failure compared to those that did not (259 days vs. 61 days; p=0.002). Patients in the study had melanoma (n=7), non-small cell lung cancer (n=3), colorectal cancer (n=2), and other forms of cancer (n=5). Additionally, clinical results from a study in patients with histiocytic disease were presented by Eli Diamond, M.D., Memorial Sloan Kettering Cancer Center. In this study, the Company’s PCM technology demonstrated 93% concordance for identifying the BRAF V600E mutation, and confirmed the absence of the mutation in the six patients whose biopsies tested negative. Trovagene’s assay also detected the BRAF V600E mutation in two patients for whom tissue biopsy material was inadequate to determine mutational status, and these results were subsequently confirmed with follow-up biopsies. Our PCM platform showed 100% concordance in monitoring response to therapy in six study subjects who tested positive for the mutation and were treated with a BRAF inhibitor. Results from this study were published in clinical consensus guidelines for the diagnosis and treatment of patients with the histiocytic disease, Erdheim-Chester disease.
CLIA validated KRAS mutation assay
In March 2014, our urine based test for KRAS mutations became available to clinicians through our CLIA laboratory. This assay detects and monitors the seven most commonly encountered mutations of the KRAS oncogene, and is our first multiplexed oncogene mutation assay utilizing next-generation sequencing as a mutation detection platform. The robustness of our ultra-sensitive assays has been demonstrated for the detection of KRAS mutations from cell-free DNA in urine. This mutation commonly occurs in patients diagnosed with either colorectal cancer, pancreatic cancer, or lung cancer. Of the more than 1.1 million estimated cases of colorectal cancer in the United States, up to 40 percent are estimated to harbor KRAS mutations. In pancreatic cancer and lung cancer, approximately 90% and 15% of patients harbor KRAS mutations, respectively. Because of the prevalence of this mutation in several important cancer types, monitoring KRAS mutational status is an area of clinical interest among treating physicians.
The clinical study being conducted at the USC Norris Cancer Center is focused on mutation monitoring and the emergence of KRAS resistant mutations in colorectal cancer. With multiple targeted therapies for colorectal cancer on the market, detection of KRAS mutations in tissue has a direct impact on the initial treatment selection for these patients. The primary purpose of the collaborative study is to determine whether KRAS mutations can be evaluated in urine to monitor treatment response in patients that test either positive or negative for the mutation.
The US Oncology clinical study will test detection and monitoring of KRAS mutations in pancreatic cancer patients. In addition to the US Oncology Research affiliated community cancer care sites participating in this study, additional academic research institutions that specialize in oncology have also elected to participate. CT scans and CA19-9 blood levels are currently the only two methods available to clinicians to monitor metastatic pancreatic cancer tumor burden and response to therapy. However, approximately 11%-17% of patients will not display elevated CA 19-9, even with high tumor load. For patients that test negative for CA19-9, our method to follow disease status by detecting and monitoring KRAS mutations could be distinctly beneficial.
In November 2014, we presented clinical results at the EORTC-AACR-NCI International Symposium highlighting our ability to detect and quantitate KRAS mutations in blood and urine samples from patients with advanced colorectal cancer. Results showed a highly correlated response. Of the blinded retrospective plasma cell-free DNA samples evaluated, 95% displayed the KRAS mutation concordant with tumor tissue, and for evaluable urine samples in the study, 92% displayed the KRAS mutation concordant with tumor tissue. The majority of patients in the study underwent surgery and received neo-adjuvant or adjuvant therapy, and serial monitoring of KRAS mutations using our assay showed a clear correlation between blood and urine samples. An estimated analytical limit of detection of 7 copies per ~100,000 genome equivalents, or 0.0067% was observed in the study, demonstrating very high analytical sensitivity.
Research and Development of additional mutation assays
We have several programs to evaluate the detection and monitoring of EGFR mutational status in lung cancer patients. A focus of these studies is the emergence of the resistant mutation EGFR T790M in lung cancer patients, which can be important for therapeutic selection when this mutation type is or becomes present. Our collaborators for this program include Memorial Sloan Kettering Cancer Center, City of Hope Comprehensive Cancer Center, UC San Diego Moores Cancer Center, and Genomac Research Institute. In addition to our study collaborations with cancer centers for this indication, we also have an ongoing collaboration with a pharmaceutical company to determine and monitor EGFR T790 resistant mutations in lung cancer patients.
PIK3CA mutations are common in breast, colon and endometrial cancers. Within the U.S., nearly one million cancer patients are positive for these mutations. Among our platform applications in development, we are working on an assay for the detection and monitoring of PIK3CA oncogene mutations. Other mutation marker assays in development include: EGFR, NRAS, HER-2, and ALK rearrangements.
Infectious Disease
HPV-HR Detection Assay
Following the completion of a pilot clinical study with a urine-based DNA test for high-risk HPV, our first HPV-HR Detection assay became commercially available in March 2013. Initial data from the pilot study showed that our assay provided superior performance to the current leading HPV assay. Our HPV-HR Detection assay showed a sensitivity of 93.0% and specificity of 96.0% for the detection of HPV virus in a comparative study of 320 high-risk individuals.
In August 2014, we presented results from two clinical studies at the 29th International Papillomavirus conference for our urine-based diagnostic test for the detection of high risk strains of HPV. Results from both pilot studies consistently demonstrated that our urine-based HPV assay had sensitivity greater than 90% for identifying women with high grade cervical intraepithelial neoplasia (CIN2/3). Assay performance was comparable to traditional HPV testing with commercially available tests in patient-matched cervical samples. In one of the studies, urine collection was examined to establish standardization of urine as a clinical specimen for high-risk HPV testing.
Urine-based HPV testing offers a significant advantage over the traditional cervical swab sample, which can present a logistic, invasive, or privacy concern. A urine-based assay also makes both female and male carrier screening feasible.
Through licensing agreements, we are pursuing commercialization of our HPV-HR Detection test, particularly in those geographies where compliance with cervical cell sampling is problematic.
Prenatal Genetics
The combination of NGS or ddPCR with our proprietary cell-free nucleic acid technology would allow for truly non-invasive prenatal screening of aneuploidies and monogenic disorders. We may pursue the development of our technology for use in prenatal genetics through licensing agreements.
Transplant
Patients who receive solid organ or bone marrow transplants are at risk of rejection, particularly during the first few months following surgery. Non-invasive monitoring of transplant status could replace repeated biopsies and blood tests, while keeping both the patient and the physician informed about potential problems.
Changing the Molecular Diagnostic Paradigm
Diagnosis and detection of severe and life-threatening diseases are among the most important outcomes of the Human Genome Project (“HGP”). There are four requirements to realize the full benefit of the HGP in relation to cancer diagnostics; large catalogues of cancer mutations; affordable sequencing of patient samples; detection technologies capable of identifying and quantifying rare instances of mutations at affordable costs; and large, systemic samples that can be collected easily and frequently in order to monitor an individual’s cancer.
The first requirement has been met through the Sanger Centre’s Catalogue of Somatic Mutations in Cancer database, which has catalogued more than 233,000 mutations in more than 20,948 genes; and by the National Institutes of Health’s (“NIH”) The Cancer Genome Atlas, which has data on more than 20 cancer types and provides a host of tools for their analysis. The second requirement has been met through the dramatic and continuing decrease in the cost of both conventional sequencing and NGS. ddPCR, capable of detecting rare mutations among thousands of wild type molecules at a reasonable cost, fulfills the third requirement.
Our proprietary methods provide the fourth and final requirement, the provision of a large, systemic sample that allows the purification of cell-free nucleic acids in amounts necessary to detect rare mutations. Furthermore, the “liquid biopsy” provided by urine can be collected frequently, is truly non-invasive, and requires no specialized personnel to collect it.
Taken together, these developments will increase the effectiveness of cancer diagnostics, improve healthcare spending efficiency, and overall, enable better patient care. These developments have made the era of personalized precision medicine in cancer possible.
The Market
The global molecular diagnostics market is forecast to reach nearly $8.0 billion by 2018, a compound annual growth rate of 9.7% from 2013—2018. This molecular diagnostics market is segmented on the basis of application, technology, end user, product, and geography. Based on application, the market is further segmented into infectious diseases, oncology, genetics, blood screening, microbiology, and others. Infectious diseases secured the largest market share, whereas oncology was the fastest growing segment amongst the rest. The driving forces of the molecular diagnostics market include the rising incidences of infectious diseases, genetic disorders, and cancer, as well as technological advancements such as assay improvements, new diagnostic tests with novel clinical utility, and portability of equipment. The technology segment of the molecular diagnostics market comprises of polymerase chain reaction (“PCR”), Isothermal Nucleic Acid Amplification Technology, hybridization, DNA sequencing and NGS, microarray, and others. Among these, the PCR segment had the largest share in 2013 of the total molecular diagnostics technology market, whereas the microarray segment will be the fastest growing segment by 2018.
Based on products, the molecular diagnostics market is segmented into instruments, reagents, and services & software. Reagents occupy the largest market share and will also register the maximum growth rate in the forecasted period 2013-2018. These reagents include assays that detect and diagnose diseases and are also used as biomarkers that predict the biological properties of the potential drug compounds.
Based on end users, hospitals was the largest segment in 2013, whereas reference laboratories will grow at the highest CAGR between 2013 and 2018. Reference laboratories carry out complex, specialized, and obscure tests. The government regulations to cut down healthcare costs will lead to the rise in the reference laboratories segment. Therefore, reference laboratories are estimated to grow at highest CAGR.
North America accounted for the largest share in 2013 and is poised to grow at a high rate in the forecast period 2013 to 2018. The growth can be attributed to the rising infectious diseases, cancer prevalence, and genetic disorders that are further adding to the overall prevalence of chronic diseases. Europe was the second leading contributor to the molecular diagnostics market in 2013. However, the growth of this region is expected to be sluggish in the forecast period and is estimated to grow at a lower CAGR than North America, due to factors such as the uneven reimbursement policies and the European economic crisis. Asia is the most promising region for molecular diagnostics in the coming five years. It is expected to grow at a higher CAGR than North America and Europe over the forecast period. The high population base and improved purchasing power of patients are the major drivers of this market. Moreover, economic instability in some western countries enables companies to focus on the Asian region in order to meet their revenue targets.
Cell-free molecular diagnostics from urine and plasma provide relevant information that can lead to improvements in personalized patient management. Beyond cancer care and infectious diseases, new products that facilitate personalized care are also emerging in the areas of central nervous system diseases, diabetes, and autism. Most major pharmaceutical companies have active pharmacogenomic programs included in their clinical studies, anticipating the need to utilize diagnostic testing to stratify patients for clinical response. We believe that our broad intellectual property (“IP”) portfolio positions us to work within each of these markets, either alone or in partnership with other companies, to develop and market cell-free molecular diagnostic products, all of which we expect would address the large unmet market needs of simplicity, patient convenience and privacy, accuracy, and cost effectiveness. Such products could play key roles in their applications to improve testing compliance and as such, reduce morbidity and mortality. The use of urine as a sample should provide a paradigm shift in screening and monitoring practices as it provides an easier sample to acquire in a truly non-invasive fashion, with more nucleic acid targets present in the sample leading to greater sensitivity. We believe these modified screening practices will most likely meet with wide physician and patient acceptance in oncology, infectious disease, transplantation, and potentially, prenatal diagnostics.
Commercial Markets — Internal Focus
Oncology
Cancer mutation testing and monitoring is the priority area for our scientists and commercial personnel. Early data from ongoing clinical studies have shown that cell-free nucleic acid analysis may be useful for determining the presence or absence of actionable mutations, and for monitoring therapeutic response and recurrence in metastatic cancers. Such testing could serve to help physicians monitor ongoing response to therapy, identify signs of early progression, or see markers of resistance emerge prior to clinical presentation. Once therapy is completed, a simple urine test could be used to monitor for early signs of disease recurrence over time. The market for these tests—diagnosed cancer patients possessing mutations known to have clinical or therapeutic importance—is already established. Use of urine-based testing could be disruptive, and change the pattern of use of other cancer monitoring tools, including expensive imaging technologies, such as PET, CT and MRI scans.
According to the American Cancer Society’s (“ACS”) 2013 report, there are approximately 525,000 patients that die every year from cancer, not including cancers of the blood, bone marrow, or lymphatic system. Using this number as a proxy for metastatic cancers, it can be assumed that all of these patients are being treated within 12 months of death for their disease. Testing and monitoring these patients for response to therapy, progression while on therapy, or for markers of resistance to therapy (like T790M for lung cancer) would be a natural extension of our technology. The average lung, breast, or colon cancer patient receives between 18-21 radiographic imaging procedures (PET, CT, MRI, etc.) during the two years following their diagnosis. This averages to about 9-10 scans per patient per year. Use of a urine-based monitoring test at the start of therapy, at several time points during therapy, and at the completion of therapy would represent approximately six separate testing events that could occur within a 12 month period. At a reimbursed price of approximately $1,000 per test, the total available market (“TAM”) for treatment response monitoring in the U.S. could be worth more than $5.0 billion.
Once patients with cancer, primary or metastatic, have completed therapy, they will require monitoring for possible progression, and for the appearance of resistance markers, since many metastatic patients may remain on lower-dose “maintenance therapy” during the remainder of their lives, or until treatment is no longer considered an option. According to the ACS, as of 2012, there were over 11 million patients alive in the U.S. who have been treated for cancers that have metastatic potential, not including cancers of the blood, bone marrow, or lymphatic system. Use of a urine-based mutation monitoring test once a year at $1000 per test would equate to a TAM for recurrence monitoring in the U.S. at approximately $12 billion annually.
Both of these markets, treatment response and recurrence monitoring, are sizeable economic opportunities. Capturing 10% of the response monitoring market would produce revenues of ~$500 million, and 5% of the recurrence monitoring market would yield annual revenues of ~$600 million.
Beyond cancer patients being actively treated or monitored over time, cell-free nucleic acid testing may eventually emerge as a viable option for pre-cancerous screening. This was recently evaluated in a cancer clinical study at Thomas Jefferson University, funded jointly by the NIH and the National Cancer Institute (“NCI”). The study demonstrated that DNA fragments carrying a specific mutation (KRAS), and released from pre-cancerous colon polyps, can be detected in the urine of patients.
Studies have shown that cancer patients who have KRAS mutations do not respond successfully to treatment with anti-EGFR (epidermal growth factor receptor) drugs such as Erbitux, Iressa, Tarceva, Tykerb, and Vectibix.
These anti-EGFR agents, particularly Erbitux and Vectibix, are a mainstay of treatment for colorectal cancer. It has been estimated that 17-25% of all human cancers have been found to harbor KRAS mutations, with mutation rates as high as 59%-90% in pancreatic cancers and 35%-40% in colorectal cancers. These tumors have a low probability of responding to anti-EGFR drugs. By first testing for KRAS mutations, physicians will be able to better manage their patients and avoid costly treatments that are unlikely to have a positive clinical response.
Screening and monitoring for KRAS and other key biomarker mutations (i.e. BRAF, PIK3CA, EGFR, etc.) using urine-based tests would provide a simple, non-invasive, cost effective, and convenient testing alternative for physicians and patients. Specimens may even be collected in the patients’ home as required, or as requested by the physician.
Simple urine-based assays would likely lead to much improved personalized medicine for patients, resulting in the right drug being prescribed for the right disease at the right time. We believe this technology will lead to an improved quality of life for patients.
Drug Development and Monitoring of Therapeutic Outcomes
Cell-free DNA diagnostic technology has significant potential as a very simple, quick, non-invasive way of monitoring clinical responses to drugs in clinical development and evaluating patient-specific responses to already approved and marketed therapies. Specific target applications include, but are not limited to; the detection of metastasis following tumor surgery, monitoring of response and tumor progression during chemotherapy and/or radiation therapy, development of optimal hormonal and chemotherapeutic treatment protocols, and monitoring of transplantation patients on immunosuppressive drugs.
With cancer treatment today, it is often difficult to determine if a particular patient is responding to their current therapeutic regimen. Generally, patients are re-examined periodically to determine if a tumor has grown in size, reduced in size (i.e. partial response), disappeared (i.e. no sign of disease — complete response) or remained the same (stable disease). If the tumor has grown in size or remained the same, treatment may be adjusted. By measuring and monitoring tumor specific genetic markers in a patient’s urine pre-, peri- and post-chemotherapy, it may be possible to more quickly determine whether a patient is responding to therapy. Use of cell-free DNA diagnostics may permit more rapid and real-time therapeutic decisions on a patient-specific basis. About 1.6 million new cancer cases are diagnosed annually, and there are several hundred companies developing therapeutic agents in the United States alone. We believe this indicates a large potential application to use cell-free DNA diagnostic technology for both drug development and the monitoring of therapeutic outcomes.
One of the largest costs associated with development of a new therapy is the size of human clinical studies required to identify the cohort of responders, and the resulting statistical power required. By measuring specific genetic markers, it may be possible to pre-identify, and subsequently screen, for the most likely responders to the therapy, and to limit patient recruitment to this subset. This strategy could significantly reduce the cost to develop a drug, and can improve development timelines as well. We believe that there is significant commercial potential for our urine-based cell-free molecular diagnostic technology to be incorporated into these clinical trial protocols, and ultimately post-approval patient identification protocols.
Commercial Markets — External Focus
We will seek to license and/or partner with other companies who have vested interests or commercial strengths in the following areas in order to develop applicable diagnostic and/or monitoring tests using our cell-free molecular diagnostic technology.
Infectious Diseases — Human Papilloma Virus (“HPV”)
The rationale for screening HPV is that high-risk subtypes cause virtually all cases of cervical cancer. We have developed a urine-based HPV test capable of screening for known high-risk HPV types that are associated with the development of cervical cancer. Cervical cancer is the third most commonly diagnosed cancer, and the fourth leading cause of cancer deaths in females, worldwide. Deaths due to cervical cancer are a significant global problem, especially in the developing world where screening practices are far from adequate.
According to the American Cancer Society, India alone accounts for 27% (77,100) of total worldwide cervical cancer deaths. A recent clinical trial conducted in rural India found that a single round of HPV DNA testing was associated with about a 50% reduction in the risk of developing advanced cervical cancer and associated deaths. This compares with the United States, where better patient compliance and screening guidelines have reduced cervical cancer death rates to only 4,290 cases in 2011. The major drivers of poor screening in these developing regions are cultural acceptance, limited screening resources and funding, and poor cytology proficiency. Further exacerbating the compliance hurdles, is that the primary screening mechanism involves an invasive cervical scraping procedure (e.g. Pap smear). It is generally agreed that the early detection of cervical cancer leads to much higher cure rates, and lower rates of invasive disease.
Beyond women’s health and cervical cancer, HPV infection impacts the men who carry and help spread the virus. While not at risk for cervical cancer, men can experience clinical manifestations in the form of genital warts, as well as penile, anal, and oropharyngeal cancers. Determining male carrier status is an unmet medical need within the sexually-transmitted disease community. We intend to explore the viability of our urine-based HPV assay to be a potential screening test for both low-risk HPV types 6 and 11 (which cause up to 90% of all genital warts), as well as known high-risk HPV types that cause the development of cervical and other cancers. Knowing HPV carrier status may contribute to more stringent use of safe sex practices, and can prevent further spread of the disease during active infection periods. In addition to carrier screening, our test may also prove useful in monitoring patients with active HPV infection until resolution of the disease.
There is a tremendous unmet need for a new non-invasive, simple, private, and cost effective test to simplify the HPV screening process for patients, both male and female, and in turn improve compliance. We believe our urine-based HPV test can address these market needs.
Other areas beyond HPV detection and monitoring include those infectious diseases caused by viruses, bacteria, fungi, and parasites. Cell-free nucleic acid assays that detect molecular targets in such organisms can provide a quick, accurate, simple, and cost effective method for screening and monitoring disease. Specific areas of interest include testing for molecular targets from organisms that cause Lyme disease, JC Virus, valley fever, and various fungal infections. These organisms all tend to be difficult to identify with current technology, making differential diagnosis especially challenging, thus delaying the start of potentially curative anti-infective treatment.
Transplantation
According to government statistics, there are approximately 28,000 solid organ transplants performed in the U.S. annually. Post-transplant monitoring for organ rejection episodes requires a highly invasive tissue biopsy. Approximately 10 such biopsies are taken over a period of one year per patient. Because organ rejection is marked by the early death of cells, we believe that an early indication of rejection can be identified by measuring a unique series of genetic markers characteristic of the organ donor that can be easily detected in random urine specimens from the transplant recipient. Providing early evidence of tissue rejection is key to the administration and monitoring of immunosuppressive therapies used to fend off rejection. Given the annual number of transplants performed in the U.S. and the annual number of corresponding biopsies performed per patient, this would equate to a market opportunity in the U.S. of roughly 300,000 urine-based tests per year. Transplantation monitoring with our technology offers opportunities for partnering with companies developing drugs for controlling tissue rejection, developing cell transplantation, or developing novel transplantation technologies. This illustrates the breadth of commercial potential of our cell-free molecular testing platform technology, and we intend to leverage such potential applications to maximize shareholder value.
Ultra-sensitive Analytical and Detection System
As it relates to detection platforms, which are required for final assay analyses, we have potential to develop a new instrument that provides features that would be synergistic and complementary to our cell-free molecular diagnostic technology. In this regard, in August 2010 we acquired Etherogen, Inc. which owns the CMOS Sensor Detection Platform, and we may design a “next generation” version of this screening and detection device. The major differentiating features of this platform are simplicity, unsurpassed ultra-sensitive detection of nucleic acids and proteins without the need for target amplification or the resulting investments in amplification-related infrastructure or capital equipment, significantly heightened speed, and the ability to perform multi-analyte assays. We believe that such a platform would undoubtedly expand the user base for molecular diagnostics. Currently, the cost of adding these new testing modalities in hospitals can be daunting. These high costs include extensive capital equipment and infrastructure requirements (i.e. amplification technology, highly trained personnel, special facilities, etc.) that most hospitals cannot afford. Our platform may address cost efficiencies, and potentially could help overcome these adoption hurdles. Finalization of the system architecture, operating procedure, and software specifications for this platform are required, and system development will take place when resources are allocated to fund the project.
Technologies for the collection, shipment and storage of urine specimens, and cell-free nucleic acid extraction
Successful implementation of our cell-free nucleic acid technology in molecular testing is tightly linked to the availability of techniques and procedures for cell-free nucleic acid preservation, purification, and analysis. Our strategic plan includes the allocation of sufficient resources for the creation of robust, feasible, and inexpensive approaches to improve the efficiency of working with urine samples.
Instrumentation/System Platform
As part of our product offerings, we intend to provide various types of automation alternatives that will further enhance the acceptance and use of our urine-based assays incorporating our cell-free platform. In this regard, there are several alternatives that we will pursue. For example, in sample extraction, we will either develop applications for existing extraction systems that already exist in laboratories or recommend that they acquire instruments that can be used with our assays. An alternative will be to explore an OEM (original equipment manufacturer) arrangement with one of the instrument suppliers, which will allow us to private label the instrument thus supporting a complete system at the customer site.
Our Business Strategy
We plan to leverage our cell-free nucleic acid technology to develop and market, either independently or in conjunction with corporate partners, molecular diagnostic products in our core market, oncology, as well as other markets including infectious disease, transplantation, and prenatal diagnostics. Our marketing strategy includes approaches across multiple fronts. In the U.S. market, we have acquired a Clinical Laboratory Improvement Amendments of 1988 (“CLIA”) laboratory. At the late stages of development for
each product, while collecting clinical data for regulatory submissions, we intend to market the products as LDTs through our CLIA laboratory. CLIA laboratories can develop and offer their own in-house tests that receive reimbursement under the provisions of LDT rules.
Congress passed the Clinical Laboratory Improvement Amendments in 1988 to regulate development, evaluation, and use of LDTs. CLIA states that laboratories must demonstrate how well an LDT performs using certain performance standards. Laboratories that perform testing on human specimens for the diagnosis, prevention, or treatment of disease, or for the assessment of health, must comply with all applicable CLIA ‘88 regulations. These regulations, which were finalized in 2003, establish standards to help ensure the quality and accuracy of laboratory testing. While most common laboratory tests are commercial tests, manufactured and marketed to multiple laboratories, some new tests are developed, evaluated, and validated within one particular laboratory. These LDTs are used solely within that laboratory and are not distributed or sold to any other labs or health care facilities.
Because LDTs are not marketed to other labs or facilities, they do not require approval for marketing from the U.S. Food and Drug Administration (“FDA”) as do commercially developed and marketed tests. However, these types of tests must go through rigorous validation procedures and must meet several criteria before results can be used for decisions regarding patient care. These include demonstration of test accuracy, precision, sensitivity, and specificity.
We may pursue FDA review and approval for our products as clinical studies are completed. Assuming we receive FDA clearance or approval for our products, we plan to market such urine-based test kits through a U.S. commercial organization directly to national and regional CLIA medical testing laboratories. We also intend to complete business partnerships (out-license agreements) with diagnostic and pharmaceutical companies in the U.S., Europe, Asia Pacific and the rest of the world as appropriate given market conditions and opportunities. This strategy would provide both short term (license fees) and long term (royalty) revenue streams. Licensees of our technology will use our platform technology in the clinical development of their products, to monitor patients taking their marketed products (i.e. TNF inhibitors), and in certain situations to develop, market and sell our cell-free molecular diagnostic technology in predefined fields of use and geographic territories. We plan to become a fully vertically integrated business in which we develop, manufacture, register, market, and sell our products.
The major advantages of our cell-free nucleic acids tests, when commercially available, will be the ease of sample collection, anticipated higher levels of sensitivity and specificity, larger quantities of genetic material for analysis (allowing for the detection of oncogene mutations that are low in abundance), patient convenience, non-invasiveness, and the ability to provide more efficient and effective monitoring protocols. Our cell-free nucleic molecular diagnostic technology must be cost effective, and we believe the process to make, sell, and process our assays is relatively simple and suitable for automation.
During the last decade, medical laboratory operating margins have declined in the face of Medicare fee schedule reductions, managed care contracts, competitive bidding, and other cost containment measures. If our technology were commercially available today, reimbursement would be available under the current procedural terminology, or CPT codes, for molecular-based testing. We expect to market our tests through our CLIA laboratory directly to physicians, and we will work with public and private payers for appropriate reimbursement. We believe this strategy, coupled with strong clinical results supporting the use of our cell-free molecular diagnostic technology, will lead to broad market adoption of our technology.
Research and Development
As of December 31, 2014, we have 17 dedicated scientists who are located in our office in San Diego, CA. We plan to continue to grow our R&D organization to approximately 28 individuals that will represent a good mix of senior lead researchers and scientists (PhDs), laboratory associate scientists, and experts in clinical development and regulatory affairs of molecular diagnostics. We plan to rapidly introduce new products to the market that could be used as LDTs within our CLIA lab, and simultaneously continue funding and collaborating on the necessary clinical studies that can support the utility of our tests, and potentially support regulatory submissions for marketing approval or clearance depending upon the nature of the product. We currently have sufficient resources to complete these projects extending into 2016. We plan to seek additional funding as required to supplement current commercial and licensing revenue. Information and documentation systems infrastructure (e.g. design history files, firewalls, etc.) must be in place to support the confidentiality of multiple partnering programs, and the rigorous scientific and regulatory oversight needed for products in the in-vitro diagnostics markets. Research and development expenses for the years ended December 31, 2014, 2013 and 2012 were approximately $6.6 million, $3.9 million and $1.9 million, respectively.
Intellectual Property
We consider the protection of our proprietary technologies and products to be a critical element in the success of our business. As of February 27, 2015, our wholly owned and licensed intellectual property includes over 50 issued patents and over 60 pending patent applications in the U.S. and abroad. The pending applications include multiple international applications filed under the Patent Cooperation Treaty (“PCT applications”) that will be used as the basis of multiple additional patent applications.
One group of patents and patent applications includes 7 U.S. patents, with 30 counterpart patents in Japan, Hong Kong and Europe, including the major markets of the European countries. These patents are directed to the detection of nucleic acid sequences in urine and nucleic acid modifications and alterations in urine. This patent family includes claims directed to prenatal analysis of fetal DNA and determining the sex of a fetus and detecting diseases such as Down Syndrome caused by genetic alterations. Other patented claims are directed to detecting and monitoring cancer through urine-based testing; nucleic acid screening, and monitoring in cases of transplantation and infectious diseases, including infection by viruses and pathogens; and other potential diagnostic and genetic testing applications. Additional pending claims are directed to the preparation of cell-free nucleic acids, as well as detection of cell-free microRNA and short cell-free nucleic acid molecules. Members of this patent group expire between 2018 and 2026.
A second group is directed to detection of specific gene mutations and indicators of disease. These include NPM1 mutations, BRAF mutations, SF3B1 mutations, HPV, AML, and hairy cell leukemia. The detection includes analysis of cell-free nucleic acid molecules. The group includes U.S. patents 8,222,370 B1, 8,501,924 B1, and 8,642,261 B1, as well as 6 pending U.S. patent applications. There are also 25 pending non-U.S. patent applications, and one PCT application. Members of this patent group expire between 2025 and 2033.
A third group is directed to our molecular detection platform utilizing proprietary probe chemistry on optical detectors such as CMOS (complementary metal-oxide semiconductor). This platform technology utilizes a conjugated probe and optical detection of analytes in medical diagnostics. The group includes one issued Japanese patent and patents in the major markets of Europe, with pending applications in the US, Europe, and Hong Kong. Members of this patent group expire beginning in 2022.
Applications are also pending that are directed to detecting and monitoring mutations in histiocytosis, detecting mutations in disease over time, and synthesis and enrichment of short nucleic acid sequences.
Wherever possible, we seek to protect our inventions by filing U.S. patents as well as foreign counterpart applications in selected other countries. Because patent applications in the U.S. are maintained in secrecy for at least eighteen months after the applications are filed, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first to make the inventions covered by each of our issued or pending patent applications, or that we were the first to file for protection of inventions set forth in such patent applications. Our planned or potential products may be covered by third-party patents or other intellectual property rights, in which case continued development and marketing of the products would require a license. Required licenses may not be available to us on commercially acceptable terms, if at all. If we do not obtain these licenses, we could encounter delays in product introductions while we attempt to design around the patents, or could find that the development, manufacture or sale of products requiring such licenses are not possible.
We may rely on trade secrets to protect our technology. Trade secrets are difficult to protect. We seek to protect our proprietary technology and processes by entering into confidentiality agreements with our employees, certain consultants and contractors. These agreements may be breached, we may not have adequate remedies for any breach and our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees or our consultants or contractors use intellectual property owned by others in their work for us, disputes may also arise as to the rights in related or resulting know-how and inventions.
Manufacturing and Distribution
In 2015, we plan to continue introducing our LDTs into the marketplace through our CLIA licensed and CAP accredited laboratory. We source all reagents and consumables needed for our LDTs from third party vendors, and we currently do not manufacture reagents kits for use in our own lab, or to distribute to 3rd party labs.
We have established a sales and marketing organization to directly market our LDTs for oncogene mutations to end users in the oncology market segment. As of December 31, 2014 we had five employees dedicated to the sales and marketing of our LDTs in the U.S. market. We intend to add additional employees as needed to support the introduction of new LDTs planned for 2015.
Reimbursement
Medicare and other third-party payers will independently evaluate our technologies by, among other things, a cost/benefit analysis, assessing other available options and reviewing the published literature with respect to the results obtained from our clinical studies. Currently, CPT codes are available for molecular testing which we believe will allow our technologies to be billed following
completion of a test which has been prescribed (ordered) by a physician for a patient. We believe that the existence of current CPT codes with applicability to our tests will help facilitate Medicare’s reimbursement process, as well as that for third party insurance providers.
Reimbursement of our novel tests is a top priority, as physician and patient access to our technology is essential for widespread adoption of our products. To gain initial reimbursement, our qualitative tests will be billed and reimbursed under established Tier I codes for their respective mutation (ie BRAF, KRAS, EGFR, etc.). These are CPT codes from the American Medical Association (MoPath system), which should enable us to bill and obtain reimbursement for our tests without much issue. As we develop our tests and demonstrate novel clinical utility in cancer monitoring, supported by our high analytical sensitivity, quantitative performance over a large dynamic range, and clinical experience, we will pursue a Not Otherwise Classified (“NOC”) code for billing and reimbursement. Under these conditions, premium pricing is expected. Over time, we intend to pursue permanent CPT codes unique to our cancer monitoring diagnostics once sufficient value is being assigned under the NOC code system. We will engage with 3rd party payors including integrated healthcare networks and Medicare for reimbursement of our tests, with the goals of obtaining strong adoption of our tests, positive coverage decisions, and appropriate valuation of our tests on a widespread basis over time. In 2015, we plan to continue developing clinical evidence around the utility and performance of our testing platform, and interacting with payors for the reimbursement of our commercially available urine-based cell-free nucleic acid diagnostics.
Government Regulation
Regulation by governmental authorities in the United States and other countries will be a significant factor in the development, production and marketing of any products that we may develop. The nature and extent to which such regulation may apply will vary depending on the nature of any such products and the policy of each country. Virtually all of our potential products will require regulatory allowance or approval by governmental agencies prior to commercialization, except for the LDTs as mentioned above. We may submit and obtain FDA approval or clearance for some or all of our diagnostic products. Pursuing and receiving FDA approval or clearance may be vital to maximizing our customer base and revenue potential for our numerous products.
FDA clearance for our products may be obtained through submission of a 510(k) statement of equivalency. Another regulatory option, albeit more complicated and expensive, is to pursue FDA approval by submitting a Pre-Market Approval (“PMA”) application. A 510(k) submission requires that we show equivalency of results in a clinical study with parallel comparison against an existing and FDA-recognized reference method (predicate device).
The FDA also regulates the sale of certain reagents, including our potential reagents, used by laboratories under the LDT rules to perform tests. The FDA refers to such a reagent as an Analyte-Specific Reagent (“ASR”). ASR’s generally do not require FDA pre-market approval or clearance if they are (i) sold to clinical laboratories certified under the Clinical Laboratory Improvement Act to perform high complexity testing and (ii) are labeled in accordance with FDA requirements, including a statement that their analytical and performance characteristics have not been established. Prior to, or in lieu of FDA approval, we can sell our reagents to laboratories that meet the established criteria. The FDA also regulates all promotional materials and specifically prohibits medical and efficacy claims.
Assuming that FDA approval or clearance is received for our products, a number of other FDA requirements would apply to our manufacturing and distribution efforts. Medical device manufacturers must be registered and their products listed with the FDA, and certain adverse events, such as reagent failures, significant changes in quality control and other events requiring correction and/or replacement/removal of reagents must be documented and reported to the FDA. The FDA also regulates product labeling, promotion, and in some cases, advertising, of medical devices. As discussed above, we must comply with the FDA’s Quality System Regulation that establishes extensive requirements for design control, quality control, validation, and manufacturing. Thus, even with FDA approval or clearance, we must continue to be diligent in maintaining compliance with these various regulations, as failure to do so can lead to enforcement action. The FDA periodically inspects facilities to determine compliance with these and other requirements.
Competition
The medical diagnostic industry is characterized by rapidly evolving technology and intense competition. Our competitors include medical diagnostic companies, most of which have financial, technical, and marketing resources significantly greater than our resources. In addition, there are a significant number of biotechnology companies working on evolving technologies that may supplant our technology, or make it obsolete. Academic institutions, government agencies, and other public and private research organizations are also conducting research activities and seeking patent protection and may commercialize products on their own or through joint venture. We are aware of certain development projects for products to prevent or treat certain diseases targeted by us. The existence of these potential products or other products or treatments of which we are not aware, or products or treatments that may be developed in the future, may adversely affect the marketability of our products or product candidates.
We believe that direct competition in the area of cell-free DNA detection and analysis is precluded by our growing patent estate. However, there are other companies working in the area of cell-free nucleic acids and circulating tumor cell (“CTC”) collection and analysis in blood plasma that could compete in similar clinical areas - including disease detection, therapeutic response monitoring, and minimal disease detection. These companies include Johnson & Johnson (Veridex), Qiagen, Quest, Labcorp, Biocept, Exact Sciences, Boreal Genomics, Sysmex-Inostics, and numerous other smaller companies both in the R&D and early commercial development phases. However, we believe that the advantages of urine as a specimen (large amounts of cell-free nucleic acid material, ease of collection, continuous collection over time, and virtually no limit on sample size and frequency) position us favorably even among such competing companies.
Employees
As of February 27, 2015 we had 31 full-time employees.
ITEM 1A. RISK FACTORS
An investment in our securities involves a high degree of risk. An investor should carefully consider the risks described below as well as other information contained in this registration statement. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our securities could decline, and an investor may lose all or part of his or her investment.
Risks Related to Our Business
We are a development stage company and we may never earn a profit.
We are a development stage company and have incurred losses since we were formed. As of December 31, 2014, we have an accumulated total deficit of approximately $81.4 million. For the fiscal year ended December 31, 2014, we had a net loss and comprehensive loss attributable to common stockholders of approximately $14.3 million. To date, we have experienced negative cash flow from development of our cell-free molecular diagnostic technology. We have not generated any revenue from operations except for licensing, milestone and royalty income and expect to incur substantial net losses for the foreseeable future to further develop and commercialize the cell-free molecular diagnostic technology. We cannot predict the extent of these future net losses, or when we may attain profitability, if at all. If we are unable to generate significant revenue from the cell-free molecular diagnostic technology or attain profitability, we will not be able to sustain operations.
Because of the numerous risks and uncertainties associated with developing and commercializing our cell-free molecular diagnostic technology and any future tests, we are unable to predict the extent of any future losses or when we will become profitable, if ever. We may never become profitable and you may never receive a return on an investment in our common stock. An investor in our common stock must carefully consider the substantial challenges, risks and uncertainties inherent in the attempted development and commercialization of tests in the medical diagnostic industry. We may never successfully commercialize cell-free molecular diagnostic technology or any future tests, and our business may fail.
We will need to raise substantial additional capital to commercialize our cell-free molecular diagnostic technology, and our failure to obtain funding when needed may force us to delay, reduce or eliminate our product development programs or collaboration efforts.
As of February 27, 2015, our cash balance was approximately $46.0 million and our working capital was approximately $42.0 million. Due to our recurring losses from operations and the expectation that we will continue to incur losses in the future, we will be required to raise additional capital to complete the development and commercialization of our current product candidates. This amount will be sufficient to launch our products in the marketplace currently under development as LDTs. We have historically relied upon private and public sales of our equity to fund our operations. We currently have a $15.0 million loan payable. When we seek additional capital, we may seek to sell additional equity and/or debt securities or to obtain a credit facility, which we may not be able to do on favorable terms, or at all. Our ability to obtain additional financing will be subject to a number of factors, including market conditions, our operating performance and investor sentiment. If we are unable to raise additional capital when required or on acceptable terms, we may have to significantly delay, scale back or discontinue the development and/or commercialization of one or more of our product candidates, restrict our operations or obtain funds by entering into agreements on unattractive terms.
Our Loan and Security Agreement with Oxford Finance LLC, or Oxford, and Silicon Valley Bank, or SVB, contains certain covenants that could adversely affect our operations and, if an event of default were to occur, we could be forced to repay the outstanding indebtedness sooner than planned and possibly at a time when we do not have sufficient capital to meet this obligation. The occurrence of any of these events could cause a significant adverse impact on our business, prospects and stock price.
We have entered into a Loan and Security Agreement with Oxford and SVB for a term loan of $15 million. The term loan is secured by all of our assets, other than intellectual property. The Loan and Security Agreement contains affirmative and negative covenants that, among other things, restrict our ability to:
· incur additional indebtedness or guarantees;
· incur liens;
· make investments, loans and acquisitions;
· consolidate or merge;
· sell or assign any part of our business or property;
· engage in transactions with affiliates; and
· pay dividends.
The Loan and Security Agreement also includes events of default, including, among other things, payment defaults; breaches of representations, warranties or covenants; certain insolvency events; and the occurrence of certain material adverse changes. Upon the occurrence of an event of default and following any cure periods (if applicable), a default interest rate of an additional 5.0% per annum may be applied to the outstanding loan balances, and the lenders may declare all outstanding obligations immediately due and payable and take such other actions as set forth in the Loan and Security Agreement.
These terms of the Loan and Security Agreement could prevent us from taking certain actions without the consent of our lenders and, if an event of default should occur, we could be required to immediately repay the outstanding indebtedness. If we are unable to repay this debt, the lenders would be able to foreclose on the secured collateral, including our cash accounts, and take other remedies permitted under the Loan and Security Agreement. Even if we are able to repay the indebtedness on an event of default, the repayment of these sums may significantly reduce our working capital and impair our ability to operate as planned. The occurrence of any of these events could cause a significant adverse impact on our business, prospects and stock price.
Our ability to successfully commercialize our technology will depend largely upon the extent to which third-party payors reimburse our tests.
Physicians and patients may decide not to order our products unless third-party payors, such as managed care organizations as well as government payors such as Medicare and Medicaid pay a substantial portion of the test price.
Reimbursement by a third-party payor may depend on a number of factors, including a payor’s determination that our product candidates are:
· not experimental or investigational;
· effective;
· medically necessary;
· appropriate for the specific patient;
· cost-effective;
· supported by peer-reviewed publications; and
· included in clinical practice guidelines.
Market acceptance, sales of products based upon the cell-free molecular diagnostic technology, and our profitability may depend on reimbursement policies and health care reform measures. Several entities conduct technology assessments of medical tests and devices and provide the results of their assessments for informational purposes to other parties. These assessments may be used by third-party payors and health care providers as grounds to deny coverage for a test or procedure. The levels at which government authorities and third-party payors, such as private health insurers and health maintenance organizations, may reimburse the price patients pay for such products could affect whether we are able to commercialize our products. Our product candidates may receive negative assessments that may impact our ability to receive reimbursement of the test. Since each payor makes its own decision as to whether to establish a policy to reimburse our test, seeking these approvals may be a time-consuming and costly process. We cannot be sure that reimbursement in the U.S. or elsewhere will be available for any of our products in the future. If reimbursement is not available or is limited, we may not be able to commercialize our products.
If we are unable to obtain reimbursement approval from private payors and Medicare and Medicaid programs for our product candidates, or if the amount reimbursed is inadequate, our ability to generate revenues could be limited. Even if we are being reimbursed, insurers may withdraw their coverage policies or cancel their contracts with us at any time, stop paying for our test or reduce the payment rate for our test, which would reduce our revenue. Moreover, we may depend upon a limited number of third-party payors for a significant portion of our test revenues and if these or other third-party payors stop providing reimbursement or decrease the amount of reimbursement for our test, our revenues could decline.
Our business could be adversely impacted by adoption of new coding for molecular genetic tests.
If our technology were commercially available today, reimbursement would be available under the current procedural terminology, or CPT codes, for molecular-based testing. The American Medical Association CPT® Editorial Panel is continuing its process of establishing analyte specific billing codes to replace codes that describe procedures used in performing molecular testing. The adoption of analyte specific codes will allow payers to better determine tests being performed. This could lead to limited coverage decisions or payment denials.
The commercial success of our product candidates will depend upon the degree of market acceptance of these products among physicians, patients, health care payors and the medical community and on our ability to successfully market our product candidates.
The use of the cell-free molecular diagnostic technology has never been commercialized for any indication. Even if approved for sale by the appropriate regulatory authorities, physicians may not order diagnostic tests based upon the cell-free molecular diagnostic technology, in which event we may be unable to generate significant revenue or become profitable. Acceptance of the cell-free molecular diagnostic technology will depend on a number of factors including:
· acceptance of products based upon the cell-free molecular diagnostic technology by physicians and patients;
· successful integration into clinical practice;
· adequate reimbursement by third parties;
· cost effectiveness;
· potential advantages over alternative treatments; and
· relative convenience and ease of administration.
We will need to make leading physicians aware of the benefits of tests using our technology through published papers, presentations at scientific conferences and favorable results from our clinical studies. In addition, we will need to gain support from thought leaders who believe that testing a urine specimen for these molecular markers will provide superior performance. Ideally, we will need these individuals to publish support papers and articles which will be necessary to gain acceptance of our products. There is
no guarantee that we will be able to obtain this support. Our failure to be successful in these efforts would make it difficult for us to convince medical practitioners to order cell-free molecular diagnostic testsfor their patients and consequently our revenue and profitability will be limited.
We currently have limited experience in marketing products. If we are unable to establish marketing and sales capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to generate product revenue.
We have limited experience in marketing products. We intend to develop an in-house marketing organization and sales force, which will require significant capital expenditures, management resources and time. We will have to compete with other molecular diagnostic companies to recruit, hire, train and retain marketing and sales personnel.
If we are unable or decide not to establish internal sales, marketing and distribution capabilities, we will pursue collaborative arrangements regarding the sales and marketing of our product candidates or future products, however, we may not be able to establish or maintain such collaborative arrangements, or if we are able to do so, that they will have effective sales forces. Any revenue we receive will depend upon the efforts of such third parties, which may not be successful. We may have little or no control over the marketing and sales efforts of such third parties and our revenue from product sales may be lower than if we had commercialized our product candidates ourselves. We also face competition in our search for third parties to assist us with the sales and marketing efforts of our product candidates.
If our potential medical diagnostic tests are unable to compete effectively with current and future medical diagnostic tests targeting similar markets as our product candidates, our commercial opportunities will be reduced or eliminated.
The medical diagnostic industry is intensely competitive and characterized by rapid technological progress. In each of our potential product areas, we face significant competition from large biotechnology, medical diagnostic and other companies. The technologies associated with the molecular diagnostics industry are evolving rapidly and there is intense competition within such industry. Certain molecular diagnostics companies have established technologies that may be competitive to our product candidates and any future tests that we develop. Some of these tests may use different approaches or means to obtain diagnostic results, which could be more effective or less expensive than our tests for similar indications. Moreover, these and other future competitors have or may have considerably greater resources than we do in terms of technology, sales, marketing, commercialization and capital resources. These competitors may have substantial advantages over us in terms of research and development expertise, experience in clinical studies, experience in regulatory issues, brand name exposure and expertise in sales and marketing as well as in operating central laboratory services. Many of these organizations have financial, marketing and human resources greater than ours; therefore, there can be no assurance that we can successfully compete with present or potential competitors or that such competition will not have a materially adverse effect on our business, financial position or results of operations.
Since the cell-free molecular diagnostic technology is under development, we cannot predict the relative competitive position of any product based upon the cell-free molecular diagnostic technology. However, we expect that the following factors will determine our ability to compete effectively: safety and efficacy; product price; turnaround time; ease of administration; performance; reimbursement; and marketing and sales capability.
We believe that many of our competitors spend significantly more on research and development-related activities than we do. Our competitors may discover new diagnostic tools or develop existing technologies to compete with the cell-free molecular diagnostic technology. Our commercial opportunities will be reduced or eliminated if these competing products are more effective, are more convenient or are less expensive than our product candidates.
Our failure to obtain human urine samples from medical institutions for our clinical studies will adversely impact the development of our cell-free molecular diagnostic technology.
We will need to establish relationships with medical institutions in order to obtain urine specimens from patients who are testing positive for a relevant infectious disease or from patients that have been diagnosed with solid tumors. We must obtain a sufficient number in order to statistically prove the equivalency of the performance of our assays versus existing assays that are already on the market.
Cell-free nucleic acids in urine are stable at room temperature for extended periods of time with the addition of a simple preservative. Successful implementation of our cell-free nucleic acid technology in molecular testing is closely linked to the availability of techniques and procedures for cell-free nucleic acid preservation, purification, and analysis. In the event urine specimens are not adequately preserved, improperly stored, or contaminated, we may be delayed in pursuing our clinical studies, and we may incur additional costs associated with procuring new human urine samples.
If our clinical studies do not prove the superiority of our technologies and demonstrate clinical utility of our technology, we may never sell our product candidates and services.
The results of our clinical studies may not show that tests using our cell-free molecular diagnostic technology are superior to existing testing methods and demonstrate clinical utility. In that event, we will have to devote significant financial and other resources to further research and development, and commercialization of tests using our technologies will be delayed or may never occur. Our earlier clinical studies were small and included samples from high-risk patients. The results from these earlier studies may not be representative of the results we obtain from any future studies, including our next two clinical studies, which will include substantially more samples and a larger percentage of normal-risk patients.
We have limited experience in establishing strong business relationships with leading clinical reference laboratories to perform cell-free molecular diagnostic tests using our technologies which could limit our revenue growth.
A key step in our strategy is to sell diagnostic products that use our proprietary technologies to leading clinical reference laboratories that will perform cell-free molecular diagnostic tests. We have limited experience in establishing these business relationships. If we are unable to establish and maintain these business relationships, we will have limited ability to obtain revenues beyond the revenue we can generate from our limited in-house capacity to process tests.
We depend upon our officers, and if we are not able to retain them or recruit additional qualified personnel, the commercialization of our product candidates and any future tests that we develop could be delayed or negatively impacted.
Our success is largely dependent upon the continued contributions of our officers such as Dr. Antonius Schuh, Chief Executive Officer. Our success also depends in part on our ability to attract and retain highly qualified scientific, commercial and administrative personnel. In order to pursue our test development and commercialization strategies, we will need to attract and hire, or engage as consultants, additional personnel with specialized experience in a number of disciplines, including assay development, bioinformatics and statistics, laboratory and clinical operations, clinical affairs and studies, government regulation, sales and marketing, billing and reimbursement and information systems. There is intense competition for personnel in the fields in which we operate. If we are unable to attract new employees and retain existing employees, the development and commercialization of our product candidates and any future tests could be delayed or negatively impacted.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
We are a small company with 31 full-time employees as of February 27, 2015. Future growth will impose significant added responsibilities on members of management, including the need to identify, attract, retain, motivate and integrate highly skilled personnel. We may increase the number of employees in the future depending on the progress of our development of cell-free molecular diagnostic technology. Our future financial performance and our ability to commercialize cell-free molecular diagnostic testsand to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to:
· manage our clinical studies effectively;
· integrate additional management, administrative, manufacturing and regulatory personnel;
· maintain sufficient administrative, accounting and management information systems and controls; and
· hire and train additional qualified personnel.
We may not be able to accomplish these tasks, and our failure to accomplish any of them could harm our financial results.
If we do not receive regulatory approvals, we may not be able to develop and commercialize our cell-free molecular diagnostic technology.
We may need FDA approval to market products based on the cell-free molecular diagnostic technology for diagnostic uses in the United States and approvals from foreign regulatory authorities to market products on the cell-free molecular diagnostic technology outside the United States. We have not yet filed an application with the FDA to obtain approval to market any of our proposed products. If we fail to obtain regulatory approval for the marketing of products based on the cell-free molecular diagnostic technology, we will be unable to sell such product candidates and will not be able to sustain operations.
We believe the estimated molecular diagnostics market for many diseases in Europe is approximately as large as that of the United States. If we seek to market products or services such as a urine-based HPV HR Detection test in Europe, we need to receive a CE Mark. If we do not obtain a CE Mark for our urine-based HPV HR Detection test, we will be unable to sell this product in Europe and countries that recognize the CE Mark.
The regulatory review and approval process, which may include evaluation of preclinical studies and clinical studies of product candidates based on the cell-free molecular diagnostic technology, as well as the evaluation of manufacturing processes and contract manufacturers’ facilities, is lengthy, expensive and uncertain. Securing regulatory approval for products based upon the cell-free molecular diagnostic technology may require the submission of extensive preclinical and clinical data and supporting information to regulatory authorities to establish such product candidates’ safety and effectiveness for each indication. We have limited experience in filing and pursuing applications necessary to gain regulatory approvals.
Regulatory authorities generally have substantial discretion in the approval process and may either refuse to accept an application, or may decide after review of an application that the data submitted is insufficient to allow approval of any product based upon the cell-free molecular diagnostic technology. If regulatory authorities do not accept or approve our applications, they may require that we conduct additional clinical, preclinical or manufacturing studies and submit that data before regulatory authorities will reconsider such application. We may need to expend substantial resources to conduct further studies to obtain data that regulatory authorities believe is sufficient. Depending on the extent of these studies, approval of applications may be delayed by several years, or may require us to expend more resources than we may have available. It is also possible that additional studies may not suffice to make applications approvable. If any of these outcomes occur, we may be forced to abandon our applications for approval, which might cause us to cease operations.
If we do not comply with governmental regulations applicable to our CLIA-certified laboratory, we may not be able to continue our operations.
The establishment and operation of our laboratory is subject to regulation by numerous federal, state and local governmental authorities in the United States. The laboratory holds a CLIA certificate of compliance and is licensed by every state (other than the State of New York) and the District of Columbia, as required, which enables us to provide testing services to residents of almost every state. Failure to comply with state regulations or changes in state regulatory requirements could result in a substantial curtailment or even prohibition of the operations of our laboratory and could have a material adverse effect on our business. CLIA is a federal law that regulates clinical laboratories that perform testing on human specimens for the purpose of providing information for the diagnosis, prevention or treatment of disease. To renew CLIA certification, laboratories are subject to survey and inspection every two years. Moreover, CLIA inspectors may make unannounced inspections of these laboratories. If we were to lose our CLIA certification or our state licenses, whether as a result of a revocation, suspension or limitation, we would no longer be able to continue our testing operations which would have a material adverse effect on our business. Potential sanctions for violations of these statutes and regulations also include significant fines, the suspension or loss of various licenses, certificates and authorizations, or product suspension or recalls.
Changes in healthcare policy could subject us to additional regulatory requirements that may delay the commercialization of our tests and increase our costs.
The U.S. government and other governments have shown significant interest in pursuing healthcare reform. Any government-adopted reform measures could adversely impact the pricing of our diagnostic products and tests in the United States or internationally and the amount of reimbursement available from governmental agencies or other third party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors of health care services to contain or reduce health care costs may adversely affect our ability to set prices for our products and services which we believe are fair, and our ability to generate revenues and achieve and maintain profitability.
New laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, that relate to healthcare availability, methods of delivery or payment for products and services, or sales, marketing or pricing, may limit our potential revenue, and we may need to revise our research and development programs. The pricing and reimbursement environment may change in the future and become more challenging due to several reasons, including policies advanced by the current executive
administration in the United States, new healthcare legislation or fiscal challenges faced by government health administration authorities. Specifically, in both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably.
For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, (‘ PPACA”) has substantially changed the way health care is financed by both government health plans and private insurers. The PPACA contains a number of provisions that are expected to impact our business and operations in ways that may negatively affect our potential revenues in the future. While it is too early to predict all the specific effects the PPACA or any future healthcare reform legislation will have on our business, they could have a material adverse effect on our business and financial condition.
In September 2007, the Food and Drug Administration Amendments Act of 2007 was enacted, giving the FDA enhanced post-marketing authority, including the authority to require post-marketing studies and clinical studies, labeling changes based on new safety information, and compliance with risk evaluations and mitigation strategies approved by the FDA. The FDA’s exercise of this authority could result in delays or increased costs during product development, clinical studies and regulatory review, increased costs to assure compliance with post-approval regulatory requirements, and potential restrictions on the sale and/or distribution of approved products.
If the FDA were to begin regulating LDTs, or if we decide to market our products as a medical device rather than a LDT, we could be forced to delay commercialization of our current product candidates, experience significant delays in commercializing any future tests, incur substantial costs and time delays associated with meeting requirements for pre-market clearance or approval and/or experience decreased demand for or reimbursement of our test.
We intend to develop products that are considered to be medical devices and are subject to federal regulations including those covering Quality System Regulations (“QSR”) and Medical Device Reporting (“MDR”).
The QSR includes requirements related to the methods used in and the facilities and controls used for designing, purchasing, manufacturing, packaging, labeling, storing, installing and servicing of medical devices. Manufacturing facilities undergo FDA inspections to assure compliance with the QS requirements. The quality systems for FDA-regulated products are known as current good manufacturing practices (“cGMPs”) as described in the Code of Federal Regulations, part 820 (21 CFR part 820). Among the cGMP requirements are those requiring manufacturers to have sufficient appropriate personnel to implement required design controls and other portions of the QSR guidelines.
Design controls include procedures that describe the product design requirements (design goals) and compare actual output to these requirements, including documented Design Reviews. Required Design History Files (“DHFs”) for each device will document the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of the QSRs.
QSRs also include stipulation for control of all documents used in design and production, including history of any changes made. Production and process controls include stipulations to ensure products are in fact produced as specified by controlled documents resulting from the controlled design phase, using products and services purchased under controlled purchasing procedures.
Incidents in which a device may have caused or contributed to a death or serious injury must to be reported to FDA under the MDR program. In addition, certain malfunctions must also be reported. The MDR regulation is a mechanism for FDA and manufacturers to identify and monitor significant adverse events involving medical devices. The goals of the regulation are to detect and correct problems in a timely manner.
We may be required to participate in MDR through two routes. As a manufacturer of products for sale within the United States, we would be required to report to the FDA any deaths, serious injuries and malfunctions, and events requiring remedial action to prevent an unreasonable risk of substantial harm to the public health. Our CLIA lab offering services for sale is already currently required to report suspected medical device related deaths to both the FDA and the relevant manufacturers of products we purchase and use.
Clinical laboratory tests like our current product offerings are regulated in the United States under CLIA as well as by applicable state laws. Diagnostic kits that are sold and distributed through interstate commerce are regulated as medical devices by the FDA. Clinical laboratory tests that are developed and validated by a laboratory for its own use are called LDTs. Most LDTs currently are not subject to FDA regulation, although reagents or software provided by third parties and used to perform LDTs may be subject to regulation. We expect that, upon the commencement of commercialization, our product candidates will be an LDT and not
a diagnostic kit. As a result, we believe that our product candidates should not be subject to regulation under current FDA policies, however there is no assurance that it will not be subject to such regulation in the future. Further, if we decide to market our products as a diagnostic kit rather than a LDT, our products would be subject to FDA regulation as a medical device. The container we expect to provide for collection and transport of tumor samples from a pathology laboratory to our clinical reference laboratory may be a medical device subject to FDA regulation and while we expect that it will be exempt from pre-market review by FDA, there is no certainty in that respect.
We cannot provide any assurance that FDA regulation, including pre-market review, will not be required in the future for our LDT product candidates, either through new policies adopted by the FDA or new legislation enacted by Congress. It is possible that legislation will be enacted into law and may result in increased regulatory burdens for us to offer or continue to offer our product as a clinical laboratory service.
If pre-market review is required, our business could be negatively impacted until such review is completed and clearance to market or approval is obtained, and the FDA could require that we stop selling. If pre-market review of our LDTs is required by the FDA, there can be no assurance that our product offerings will be cleared or approved on a timely basis, if at all. Ongoing compliance with FDA regulations, such as the Quality System Regulation and Medical Device Reporting, would increase the cost of conducting our business, and subject us to inspection by the FDA and to the requirements of the FDA and penalties for failure to comply with these requirements. We may also decide voluntarily to pursue FDA pre-market review of our product offerings if we determine that doing so would be appropriate. Some competitors may develop competing tests cleared for marketing by the FDA. There may be a marketing differentiation or perception that an FDA-cleared test is more desirable than our product offerings, and that could discourage adoption and reimbursement of our test.
We may be required to conduct clinical studies and we may find it difficult to enroll patients in such clinical studies, which could delay or prevent clinical studies of our product candidates.
If the FDA decides to regulate our LDTs, it may require that we conduct extensive pre-market clinical studies prior to submitting a regulatory application for commercial sales. If we are required to conduct pre-market clinical studies, whether using retrospectively collected and banked samples or prospectively collected samples, delays in the commencement or completion of clinical studies could significantly increase our test development costs and delay commercialization. Many of the factors that may cause or lead to a delay in the commencement or completion of clinical studies may also ultimately lead to delay or denial of regulatory clearance or approval. The commencement and completion of clinical trials may be delayed by factors such as unforeseen safety issues, lack of effectiveness during clinical trials, inability to monitor patients adequately during or after testing, and slower than expected rates of patient recruitment.
Insufficient patient enrollment is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial. We may find it necessary to engage contract research organizations to perform data collection and analysis and other aspects of our clinical studies, which might increase the cost of the studies. We will also depend on clinical investigators, medical institutions and contract research organizations to perform the studies properly. If these parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality, completeness or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, FDA requirements or for other reasons, our clinical studies may have to be extended, delayed or terminated. Many of these factors would be beyond our control. We may not be able to enter into replacement arrangements without undue delays or considerable expenditures. If there are delays in testing as a result of the failure to perform by third parties, our research and development costs would increase, and we may not be able to obtain regulatory clearance or approval for our test. In addition, we may not be able to establish or maintain relationships with these parties on favorable terms, if at all. Each of these outcomes would harm our ability to market our test, or to become profitable.
In addition, in the event we are required to conduct clinical trials, it may be very expensive and difficult to design and implement due to the rigorous regulatory requirements to which clinical trials are subjected. Clinical trials are also time consuming, and we would be unable to provide certainty regarding when we might complete the clinical trial process.
If we are unable to protect our intellectual property effectively, we may be unable to prevent third parties from using our technologies, which would impair our competitive advantage.
We rely on patent protection as well as a combination of trademark, copyright and trade secret protection, and other contractual restrictions to protect our proprietary technologies, all of which provide limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. If we fail to protect our intellectual property, we will be unable to prevent third parties from using our technologies and they will be able to compete more effectively against us.
Our currently pending or future patent applications may not result in issued patents and any patents issued to us may be challenged, invalidated or held unenforceable. We may not be successful in defending challenges made in connection with our patents and patent applications.
In addition to our patents, we rely on contractual restrictions to protect our proprietary technology. We require our employees and third parties to sign confidentiality agreements and employees to also sign agreements assigning to us all intellectual property arising from their work for us. Nevertheless, we cannot guarantee that these measures will be effective in protecting our intellectual property rights.
The patents issued to us may not be broad enough to provide any meaningful protection one or more of our competitors may develop more effective technologies, designs or methods without infringing our intellectual property rights and one or more of our competitors may design around our proprietary technologies.
If we are not able to protect our proprietary technology, trade secrets and know-how, our competitors may use our inventions to develop competing products. We own certain patents relating to the cell-free molecular diagnostic technology. However, these patents may not protect us against our competitors, and patent litigation is very expensive. We may not have sufficient cash available to pursue any patent litigation to its conclusion because currently we do not generate revenues other than licensing, milestone and royalty income.
We cannot rely solely on our current patents to be successful. The standards that the U.S. Patent and Trademark Office and foreign patent office’s use to grant patents, and the standards that U.S. and foreign courts use to interpret patents, are not the same and are not always applied predictably or uniformly and can change, particularly as new technologies develop. As such, the degree of patent protection obtained in the U.S. may differ substantially from that obtained in various foreign countries. In some instances, patents have been issued in the U.S. while substantially less or no protection has been obtained in Europe or other countries.
We cannot be certain of the level of protection, if any, which will be provided by our patents if we attempt to enforce them and they are challenged in court where our competitors may raise defenses such as invalidity, unenforceability or possession of a valid license. In addition, the type and extent of any patent claims that may be issued to us in the future are uncertain. Any patents which are issued may not contain claims that will permit us to stop competitors from using similar technology.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or use, our cell-free molecular diagnostic technology.
Third parties may challenge the validity of our patents and other intellectual property rights, resulting in costly litigation or other time-consuming and expensive proceedings, which could deprive us of valuable rights. If we become involved in any intellectual property litigation, interference or other judicial or administrative proceedings, we will incur substantial expenses and the diversion of financial resources and technical and management personnel. An adverse determination may subject us to significant liabilities or require us to seek licenses that may not be available from third parties on commercially favorable terms, if at all. Further, if such claims are proven valid, through litigation or otherwise, we may be required to pay substantial financial damages, which can be tripled if the infringement is deemed willful, or be required to discontinue or significantly delay development, marketing, selling and licensing of the affected products and intellectual property rights. In our European patent that covers using microRNAs to detect in vivo cell death, an anonymous third party has recently filed an opposition against the claims in the patent. Oppositions against the patentability of claims in a European patent are considered by a panel of examiners at the European Patent Office, and we are considering the full range of options available for defending against the opposition.
Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours. Any such patent application may have priority over our patent applications and could further require us to obtain rights to issued patents covering such technologies. There may be third-party patents, patent applications and other intellectual property relevant to our potential products that may block or compete with our products or processes. If another party has filed a United States patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the United States Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our United States patent position with respect to such inventions. In addition, we cannot assure you that we would prevail in any of these suits or that the damages or other remedies if any, awarded against us would not be substantial. Claims of intellectual property infringement may require us to enter into
royalty or license agreements with third parties that may not be available on acceptable terms, if at all. We may also become subject to injunctions against the further development and use of our technology, which would have a material adverse effect on our business, financial condition and results of operations.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
The testing, manufacturing and marketing of medical diagnostic devices entails an inherent risk of product liability and personal injury claims.
To date, we have experienced no product liability or personal injury claims, but any such claims arising in the future could have a material adverse effect on our business, financial condition and results of operations. Potential product liability or personal injury claims may exceed the amount of our insurance coverage or may be excluded from coverage under the terms of our policy or limited by other claims under our umbrella insurance policy. Additionally, our existing insurance may not be renewed by us at a cost and level of coverage comparable to that presently in effect, if at all. In the event that we are held liable for a claim against which we are not insured or for damages exceeding the limits of our insurance coverage, such claim could have a material adverse effect on our cash flow and thus potentially a materially adverse effect on our business, financial condition and results of operations.
All of our diagnostic technology and services are performed at a single laboratory, and in the event this facility was to be affected by a termination of the lease or a man-made or natural disaster, our operations could be severely impaired.
We are performing all of our diagnostic services in our laboratory located in San Diego, California. Despite precautions taken by us, any future natural or man-made disaster at this laboratory, such as a fire, earthquake or terrorist activity, could cause substantial delays in our operations, damage or destroy our equipment and urine samples or cause us to incur additional expenses.
In addition, we are leasing the facilities where our lab operates. We are currently in compliance with all and any lease obligations, but should the lease terminate for any reason, or if at any time the lab is moved due to conditions outside our control, it could cause substantial delay in our diagnostics operations, damage or destroy our equipment and biological samples or cause us to incur additional expenses. In the event of an extended shutdown of our laboratory, we may be unable to perform our services in a timely manner or at all and therefore would be unable to operate in a commercially competitive manner. This could harm our operating results and financial condition.
Further, if we have to use a substitute laboratory while our facility was shut down, we could only use another facility with established state licensure and accreditation under CLIA. We may not be able to find another CLIA-certified facility and comply with applicable procedures, or find any such laboratory that would be willing to perform the tests for us on commercially reasonable terms. Additionally, any new laboratory opened by us would be subject to certification under CLIA and licensure by various states, which would take a significant amount of time and result in delays in our ability to continue our personalized medicine services operations.
Risks Related to Ownership of our Common Stock
If we discover material weaknesses and other deficiencies in our internal control and accounting procedures, our stock price could decline significantly and raising capital could be more difficult.
If we fail to comply with the rules under the Sarbanes-Oxley Act of 2002 related to disclosure controls and procedures, or, if we discover additional material weaknesses and other deficiencies in our internal control and accounting procedures, our stock price could decline significantly and raising capital could be more difficult. Moreover, effective internal controls are necessary for us to produce reliable financial reports and are important to helping prevent financial fraud. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock could drop significantly. In addition, we cannot be certain that additional material weaknesses or significant deficiencies in our internal controls will not be discovered in the future.
The rights of the holders of common stock may be impaired by the potential issuance of preferred stock.
Our certificate of incorporation gives our board of directors the right to create new series of preferred stock. As a result, the board of directors may, without stockholder approval, issue preferred stock with voting, dividend, conversion, liquidation or other rights which could adversely affect the voting power and equity interest of the holders of common stock. Preferred stock, which could be issued with the right to more than one vote per share, could be utilized as a method of discouraging, delaying or preventing a
change of control. The possible impact on takeover attempts could adversely affect the price of our common stock. Although we have no present intention to issue any additional shares of preferred stock or to create any new series of preferred stock and the certificate of designation relating to the Series A Convertible Preferred Stock restricts our ability to issue additional series of preferred stock, we may issue such shares in the future. Without the consent of the holders of the outstanding shares of Series A Convertible Preferred Stock we may not alter or change adversely the rights of the holders of the Series A Convertible Preferred Stock or increase the number of authorized shares of Series A Convertible Preferred Stock, create a class of stock which is senior to or on a parity with the Series A Convertible Preferred Stock, amend our certificate of incorporation in breach of these provisions or agree to any of the foregoing.
Our common stock price may be volatile and could fluctuate widely in price, which could result in substantial losses for investors.
The market price of our ordinary shares historically has been, and we expect will continue to be, subject to significant fluctuations over short periods of time. For example, the closing price of our common stock during the past 52-weeks has ranged from a low of $3.00 to a high of $6.74. These fluctuations may be due to various factors, many of which are beyond our control, including:
· technological innovations or new products and services by us or our competitors;
· clinical trial results relating to our tests or those of our competitors;
· announcements or press releases relating to the industry or to our own business or prospects;
· coverage and reimbursement decisions by third party payors, such as Medicare and other managed care organizations;
· regulation and oversight of our product candidates and services, including by the FDA, CMS and comparable ex-U.S. agency;
· FDA, CMS and comparable ex-U.S. agency regulation and oversight of our products and services;
· the establishment of partnerships with clinical reference laboratories;
· health care legislation;
· intellectual property disputes;
· additions or departures of key personnel;
· sales of our common stock;
· our ability to integrate operations, technology, products and services;
· our ability to execute our business plan;
· operating results below expectations;
· loss of any strategic relationship;
· industry developments;
· economic and other external factors; and
· period-to-period fluctuations in our financial results.
In addition, market fluctuations, as well as general political and economic conditions could adversely affect the market price of our securities. Because we are a development stage company with no revenue from operations to date, other than licensing, milestone and royalty income, you should consider any one of these factors to be material. Our stock price may fluctuate widely as a result of any of the foregoing.
Because certain of our stockholders control a significant number of shares of our common stock, they may have effective control over actions requiring stockholder approval.
As of February 27, 2015, our directors, executive officers and principal stockholders, and their respective affiliates, beneficially own approximately 29.3% of our outstanding shares of common stock. As a result, these stockholders, acting together, would have the ability to control the outcome of matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these stockholders, acting together, would have the ability to control the management and affairs of our company. Accordingly, this concentration of ownership might harm the market price of our common stock by:
· delaying, deferring or preventing a change in corporate control;
· impeding a merger, consolidation, takeover or other business combination involving us; or
· discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us.
We have not paid dividends on our common stock in the past and do not expect to pay dividends on our common stock for the foreseeable future. Any return on investment may be limited to the value of our common stock.
No cash dividends have been paid on our common stock. We expect that any income received from operations will be devoted to our future operations and growth. We do not expect to pay cash dividends on our common stock in the near future. Payment of dividends would depend upon our profitability at the time, cash available for those dividends, and other factors as our board of directors may consider relevant. If we do not pay dividends, our common stock may be less valuable because a return on an investor’s investment will only occur if our stock price appreciates. In addition, under the terms of our Loan and Security Agreement, we are precluded from paying cash dividends without the prior written consent of the lenders, and the terms of the Series A Convertible Preferred Stock prohibit us from paying dividends to the holders of our common stock so long as any dividends due on the Series A Convertible Preferred Stock remain unpaid. Investors in our common stock should not rely on an investment in our company if they require dividend income.
If securities or industry analysts do not publish research or reports about our business, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. We do not currently have and may never obtain research coverage by industry or financial analysts. If no or few analysts commence coverage of us, the trading price of our stock would likely decrease. Even if we do obtain analyst coverage, if one or more of the analysts who cover us downgrade our stock, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
Delaware law and our corporate charter and bylaws will contain anti-takeover provisions that could delay or discourage takeover attempts that stockholders may consider favorable.
Provisions in our certificate of incorporation and bylaws may have the effect of delaying or preventing a change of control or changes in our management. For example, our board of directors have the authority to issue up to 20,000,000 shares of preferred stock in one or more series and to fix the powers, preferences and rights of each series without stockholder approval. The ability to issue preferred stock could discourage unsolicited acquisition proposals or make it more difficult for a third party to gain control of our company, or otherwise could adversely affect the market price of our common stock. Our bylaws require that any stockholder proposals or nominations for election to our board of directors must meet specific advance notice requirements and procedures, which make it more difficult for our stockholders to make proposals or director nominations.
Furthermore, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law. These provisions may prohibit or restrict large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us. These provisions in our certificate of incorporation and bylaws and under Delaware law could discourage potential takeover attempts and could reduce the price that investors might be willing to pay for shares of our common stock in the future and result in our market price being lower than it would without these provisions.
A sale of a substantial number of shares of our common stock may cause the price of our common stock to decline and may impair our ability to raise capital in the future.
Our common stock is traded on The NASDAQ Capital Market and, despite certain increases of trading volume from time to time, there have been periods when it could be considered “thinly-traded,” meaning that the number of persons interested in purchasing our common stock at or near bid prices at any given time may be relatively small or non-existent. Finance transactions resulting in a large amount of newly issued shares that become readily tradable, or other events that cause current stockholders to sell shares, could place downward pressure on the trading price of our stock. In addition, the lack of a robust resale market may require a stockholder who desires to sell a large number of shares of common stock to sell the shares in increments over time to mitigate any adverse impact of the sales on the market price of our stock.
If our stockholders sell, or the market perceives that our stockholders intend to sell for various reasons, including the ending of restriction on resale, substantial amounts of our common stock in the public market, including shares issued upon the exercise of outstanding options or warrants, the market price of our common stock could fall. Sales of a substantial number of shares of our common stock may make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate. We may become involved in securities class action litigation that could divert management’s attention and harm our business.
We may be subject to stockholder litigation, thereby diverting our resources that may have a material effect on our profitability and results of operations.
As discussed in the preceding risk factors, the market for our common shares is characterized by significant price volatility, and we expect that our share price will continue to be at least as volatile for the indefinite future. In the past, plaintiffs have often initiated securities class action litigation against a company following periods of volatility in the market price of its securities. In addition, many companies have actions brought against them by stockholders relating to past transactions or other matters Any such actions could give rise to substantial damages, and thereby have a material adverse effect on our consolidated financial position, liquidity, or results of operations. Even if an action is not resolved against us, the uncertainty and expense associated with stockholder actions could harm our business, financial condition and reputation. Litigation can be costly, time-consuming and disruptive to business operations. The defense of lawsuits could also result in diversion of our management’s time and attention away from business operations, which could harm our business.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
We lease approximately 13,000 square feet of laboratory and office space in our headquarters in San Diego, California under a lease that expires in December 2017. We believe that our current facilities are adequate for our needs for the immediate future and that, should it be needed, suitable additional space will be available to accommodate expansion of our operations on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS
From time to time, we may become involved in litigation relating to claims arising out of its operations in the normal course of business. We are not involved in any pending legal proceeding or litigation and, to the best of our knowledge, no governmental authority is contemplating any proceeding to which we are a party or to which any of our properties is subject, which would reasonably be likely to have a material adverse effect on us.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock has been traded on The NASDAQ Capital Market (“NASDAQ”) under the symbol “TROV” since May 29, 2012.
Our common stock was traded over the counter on the pink sheets under the symbol TROV.PK from June 15, 2007 until May 30, 2012. From July 27, 2004 until June 14, 2007, our common stock was quoted on the OTC Bulletin Board under the symbol “XNOM.OB”. Prior to July 27, 2004, our common stock was quoted on the OTC Bulletin Board under the symbol “UKAR.OB” but never traded. The following table sets forth the range of high and low sales prices for our common stock on NASDAQ for the periods indicated since January 1, 2013.
The closing price of our common stock on NASDAQ on February 27, 2015 was $5.65 per share.
|
Fiscal 2014 |
|
High |
|
Low |
| ||
|
Fourth Quarter |
|
$ |
5.17 |
|
$ |
4.01 |
|
|
Third Quarter |
|
$ |
6.30 |
|
$ |
3.00 |
|
|
Second Quarter |
|
$ |
6.01 |
|
$ |
3.50 |
|
|
First Quarter |
|
$ |
6.74 |
|
$ |
5.13 |
|
|
Fiscal 2013 |
|
High |
|
Low |
| ||
|
Fourth Quarter |
|
$ |
8.50 |
|
$ |
4.81 |
|
|
Third Quarter |
|
$ |
10.27 |
|
$ |
6.61 |
|
|
Second Quarter |
|
$ |
7.23 |
|
$ |
5.27 |
|
|
First Quarter |
|
$ |
8.96 |
|
$ |
5.09 |
|
Number of Stockholders
As of February 27, 2015, we had approximately 65 stockholders of record of our common stock.
Dividend Policy
Historically, we have not paid any dividends to the holders of our common stock and we do not expect to pay any such dividends in the foreseeable future as we expect to retain our future earnings for use in the operation and expansion of our business. Pursuant to the terms of the Series A Convertible Preferred Stock, dividends cannot be paid to the holders of our common stock so long as any dividends due on the Series A Convertible Preferred Stock remain unpaid.
Corporate Performance Graph
COMPARISON OF CUMULATIVE TOTAL RETURN
Among the NASDAQ Stock Market (U.S.),
The NASDAQ Pharmaceutical Index, the Russell 3000 Index
and Trovagene, Inc.
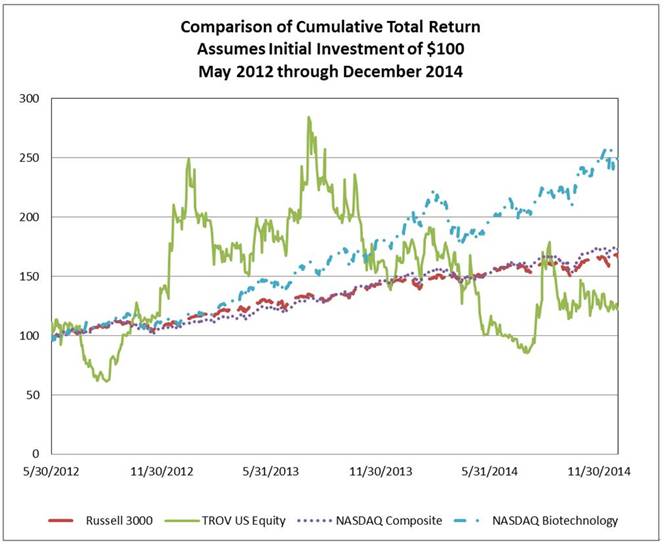
Equity Compensation Plan Information
The following table summarizes information about our equity compensation plans as of December 31, 2014:
|
Plan Category |
|
Number of |
|
Weighted- |
|
Number of |
| |
|
|
|
(a) |
|
(b) |
|
(c) |
| |
|
Equity Compensation Plans Approved by Stockholders |
|
4,784,306 |
|
$ |
4.74 |
|
1,371,832 |
|
|
Equity Compensation Plans Not Approved by Stockholders |
|
129,166 |
|
$ |
3.15 |
|
— |
|
|
Total |
|
4,913,472 |
|
$ |
4.66 |
|
1,371,832 |
|
ITEM 6. SELECTED FINANCIAL DATA
The following table sets forth our selected consolidated financial data and has been derived from our audited consolidated financial statements. Consolidated balance sheets as of December 31, 2014 and 2013, as well as consolidated statements of operations for the years ended December 31, 2014, 2013 and 2012, and the reports thereon are included elsewhere in this Annual Report on Form 10-K. The information should be read in conjunction with our audited consolidated financial statements and the notes to such statements, included below in Item 8, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Item 7. Historical results are not necessarily indicative of the results to be expected in the future.
|
|
|
Year ended December 31, |
| |||||||||||||
|
|
|
2014 |
|
2013 |
|
2012 |
|
2011 |
|
2010 |
| |||||
|
|
|
(in thousands, except for share and per share data) |
| |||||||||||||
|
Consolidated Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Revenues |
|
$ |
280 |
|
$ |
259 |
|
$ |
450 |
|
$ |
258 |
|
$ |
266 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Costs and Expenses: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cost of revenues |
|
15 |
|
— |
|
— |
|
— |
|
— |
| |||||
|
Research and development |
|
6,665 |
|
3,948 |
|
1,920 |
|
911 |
|
1,024 |
| |||||
|
Purchased in-process research and development |
|
— |
|
— |
|
— |
|
— |
|
2,667 |
| |||||
|
Selling and marketing |
|
2,735 |
|
1,530 |
|
506 |
|
— |
|
— |
| |||||
|
General and administrative |
|
5,810 |
|
5,472 |
|
2,873 |
|
2,324 |
|
1,954 |
| |||||
|
Total operating expenses |
|
15,225 |
|
10,950 |
|
5,299 |
|
3,235 |
|
5,645 |
| |||||
|
Loss from operations |
|
(14,945 |
) |
(10,691 |
) |
(4,849 |
) |
(2,977 |
) |
(5,379 |
) | |||||
|
Gain (loss) on disposal of equipment |
|
25 |
|
(23 |
) |
4 |
|
— |
|
— |
| |||||
|
Net interest (expense) income |
|
(831 |
) |
(13 |
) |
— |
|
(56 |
) |
(337 |
) | |||||
|
Gain (loss) on change in fair value of derivative instruments-warrants |
|
1,426 |
|
(1,084 |
) |
(6,271 |
) |
171 |
|
267 |
| |||||
|
Gain on extinguishment of debt |
|
— |
|
— |
|
— |
|
623 |
|
— |
| |||||
|
Net loss and comprehensive loss |
|
(14,325 |
) |
(11,811 |
) |
(11,566 |
) |
(2,239 |
) |
(5,449 |
) | |||||
|
Preferred stock dividend |
|
(23 |
) |
(30 |
) |
(38 |
) |
(38 |
) |
(38 |
) | |||||
|
Net loss and comprehensive loss attributable to common stockholders |
|
(14,348 |
) |
(11,841 |
) |
(11,604 |
) |
(2,277 |
) |
(5,487 |
) | |||||
|
| ||||||||||||||||
|
Net loss per common share - basic |
|
$ |
(0.76 |
) |
$ |
(0.70 |
) |
$ |
(0.89 |
) |
$ |
(0.23 |
) |
$ |
(0.77 |
) |
|
Net loss per common share - diluted |
|
$ |
(0.88 |
) |
$ |
(0.70 |
) |
$ |
(0.89 |
) |
$ |
(0.23 |
) |
$ |
(0.77 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Weighted average shares outstanding - basic* |
|
18,904,280 |
|
16,978,212 |
|
13,066,600 |
* |
9,711,519 |
* |
7,158,791 |
* | |||||
|
Weighted average shares outstanding - diluted |
|
19,071,112 |
|
16,978,212 |
|
13,066,600 |
* |
9,711,519 |
* |
7,158,791 |
* | |||||
(*) Weighted average shares outstanding reflects retroactive change of a one for 6 (1:6) reverse stock split effective on May 29, 2012
|
|
|
December 31, |
| |||||||||||||
|
|
|
2014 |
|
2013 |
|
2012 |
|
2011 |
|
2010 |
| |||||
|
|
|
($ in thousands) |
| |||||||||||||
|
Consolidated Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
| |||||
|
Cash and cash equivalents |
|
$ |
27,294 |
|
$ |
25,837 |
|
$ |
10,820 |
|
$ |
700 |
|
$ |
59 |
|
|
Working capital |
|
23,232 |
|
24,060 |
|
10,318 |
|
(588 |
) |
(3,137 |
) | |||||
|
Total assets |
|
28,897 |
|
27,156 |
|
11,665 |
|
1,039 |
|
512 |
| |||||
|
Total stockholders’ equity (deficit) |
|
$ |
8,350 |
|
$ |
20,392 |
|
$ |
2,169 |
|
$ |
(4,231 |
) |
$ |
(4,995 |
) |
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Forward-Looking Statements
The information in this report contains forward-looking statements. All statements other than statements of historical fact made in this report are forward looking. In particular, the statements herein regarding industry prospects and future results of operations or financial position are forward-looking statements. These forward-looking statements can be identified by the use of words such as “believes,” “estimates,” “could,” “possibly,” “probably,” anticipates,” “projects,” “expects,” “may,” “will,” or “should” or other variations or similar words. No assurances can be given that the future results anticipated by the forward-looking statements will be achieved. Forward-looking statements reflect management’s current expectations and are inherently uncertain. Our actual results may differ significantly from management’s expectations.
The following discussion and analysis should be read in conjunction with our financial statements, included herewith. This discussion should not be construed to imply that the results discussed herein will necessarily continue into the future, or that any conclusion reached herein will necessarily be indicative of actual operating results in the future. Such discussion represents only the best present assessment of our management.
Overview
We are focused on developing and commercializing our precision cancer monitoring technology, which can inform oncologists and guide treatment decisions by determining a tumor’s mutational status and enabling physicians to track therapeutic response and resistance over time.
We are expanding the body of clinical evidence supporting our urine-based cell-free molecular diagnostic platform through collaborations with major cancer treatment centers and integrated healthcare networks. We expect that the benefits of our precision cancer monitoring technology will become more apparent in terms of its clinical utility and impact on patient outcomes. Our intellectual property estate protecting our technology includes methods of extracting, purifying, preparing, and detecting cell-free DNA and RNA mutations in urine.
Through December 31, 2014, we have sustained a cumulative total deficit of $81,391,909. To date, we have generated minimal revenues and expect to incur additional losses to perform further research and development activities and commercial expansion. During 2014, we have advanced our business with the following activities:
· Presented clinical data at the 56th American Society of Hematology Annual Meeting and Exposition highlighting use of our PCM platform for the determination of mutational status and longitudinal monitoring of disease dynamics from both urine and plasma cell-free DNA.
· Presented study results at the EORTC-NCI-AACR International Symposium demonstrating high sensitivity and strong quantitative performance with our PCM platform for the detection and monitoring of KRAS mutations in circulating tumor DNA of colorectal cancer patients.
· Published results from a blinded prospective clinical study in Cancer Discovery demonstrating the ability to accurately determine mutational status and monitor treatment response to BRAF inhibitor therapy from urinary cell-free DNA.
· We secured $15.0 million in debt financing with Silicon Valley Bank and Oxford Finance to aid in funding our clinical programs, commercialization efforts, and continued expansion of our oncogene mutation portfolio for cancer monitoring research.
· Presented clinical data from an ongoing study demonstrating the utility of our PCM platform for the determination of oncogene mutational status in cell-free DNA obtained from a liquid biopsy at the ECD Global Alliance’s Second International Medical Symposium, in collaboration with the National Institutes of Health.
· Expanded clinical collaboration with the Memorial Sloan Kettering Cancer Center by initiating a new study evaluating our PCM platform for the detection and quantitative monitoring of EGFR Mutations in lung cancer patients.
· Entered into a strategic partnership with the Robert H. Lurie Comprehensive Cancer Center of Northwestern University and the Northwestern Medicine Developmental Therapeutics Institute to conduct a translational research program designed to assess the utility of our PCM platform in clinical practice.
· Entered into a clinical collaboration with Dana-Farber Cancer Institute to investigate the utility of quantitative urine-based mutation detection and the ability to monitor tumor mutation burden and treatment response over time in metastatic melanoma patients.
· Published study results in the 2014 ASCO Annual Meeting Proceedings demonstrating that oncogene mutation load in urinary cell-free DNA, as determined using our PCM platform, is significantly correlated with treatment response.
· Prepared multi-disciplinary clinical consensus guidelines for Erdheim-Chester disease which referenced the ability of our PCM platform to detect actionable oncogene mutation status and to monitor patients during treatment.
· Published data from ongoing clinical validation studies in Oncotarget demonstrating that our PCM platform is suitable to non-invasively determine oncogene mutation status in patients with malignant disease.
· Presented clinical data at American Association for Cancer Research annual meeting demonstrating the ability of our PCM platform to non-invasively track cancer patients’ response to therapy.
· Released first urine-based multiplexed oncogene mutation assay utilizing Next Generation Sequencing technology, for the detection and monitoring of seven mutations in the KRAS oncogene.
· Signed clinical study agreement with US Oncology Research to examine the utility of quantitative KRAS mutation detection and monitoring in metastatic pancreatic cancer patients.
Our product development and commercialization efforts are in their early stages, and we cannot make estimates of the costs or the time our development efforts will take to complete, or the timing and amount of revenues related to the sale of our tests and revenues related to our license agreements. The risk of completion of any program is high because of the many uncertainties involved in bringing new diagnostic products to market including the long duration of clinical testing, the specific performance of proposed products under stringent clinical trial protocols and/or CLIA requirements, the extended regulatory approval and review cycles, our ability to raise additional capital, the nature and timing of research and development expenses, and competing technologies being developed by organizations with significantly greater resources.
CRITICAL ACCOUNTING POLICIES
Financial Reporting Release No. 60 requires all companies to include a discussion of critical accounting policies or methods used in the preparation of financial statements. Our accounting policies are described in Item 8. Financial Statements—Note 2 Basis of Presentation and Summary of Significant Accounting Policies. The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates. We believe that the following discussion represents our critical accounting policies.
Revenue Recognition
Historically our revenues have been generated from royalty, license and milestones related to agreements we have with other healthcare companies, medical laboratories and biotechnology partners. In the future we will also have revenues from our diagnostics services.
We recognize revenues when persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable, and collection is reasonably assured.
Milestone, Royalty and License Revenues
We license and sublicense our patent rights to healthcare companies, medical laboratories and biotechnology partners. These agreements may involve multiple elements such as license fees, royalties and milestone payments. Revenue is recognized when the criteria described above have been met as well as the following:
· Up-front nonrefundable license fees pursuant to agreements under which we have no continuing performance obligations are recognized as revenues on the effective date of the agreement and when collection is reasonably assured.
· Minimum royalties are recognized as earned, and royalties in excess of minimum amounts are recognized upon receipt of payment when collection is assured.
· Milestone payments are recognized when both the milestone is achieved and the related payment is received.
Diagnostic Service Revenues
Diagnostic service revenue, which consists of fees for clinical laboratory tests may come from several sources, including commercial third-party payors, such as insurance companies and health maintenance organizations, government payors, such as Medicare and Medicaid in the United States, patient self-pay and, in some cases, from hospitals or referring laboratories who, in turn, bill third-party payors for testing.
Diagnostic services revenue will be recognized when the criteria described above has been met as well as upon cash collection until we can reliably estimate the amount that will be ultimately collected for our LDTs, at which time we will recognize revenues on an accrual basis.
We have not recognized any diagnostic service revenue to date.
Derivative Financial Instruments-Warrants
Our derivative liabilities are related to warrants issued in connection with financing transactions and are therefore not designated as hedging instruments. All derivatives are recorded on our balance sheet at fair value in accordance with current accounting guidelines for such complex financial instruments.
We have issued common stock warrants in connection with the execution of certain equity and debt financings. Such warrants are classified as derivative liabilities under the provisions of FASB ASC 815 Derivatives and Hedging (“ASC 815”) and are recorded at their fair market value as of each reporting period. Such warrants do not meet the exemption that a contract should not be considered a derivative instrument if it is (1) indexed to its own stock and (2) classified in stockholders’ equity. Changes in fair value of derivative liabilities are recorded in the consolidated statement of operations under the caption “Change in fair value of derivative instruments.”
The fair value of warrants is determined using the Black-Scholes option-pricing model using assumptions regarding volatility of our common share price, remaining life of the warrant, and risk-free interest rates at each period end. We thus use model-derived valuations where inputs are observable in active markets to determine the fair value and accordingly classify such warrants in Level 3 per ASC 820. At December 31, 2014 and 2013, the fair value of such warrants was $3,006,021 and $4,431,871, respectively, which are included in the derivative financial instruments’ liability on our balance sheet.
Cost of Revenue
Cost of revenue represents the cost of materials, personnel costs, costs associated with processing specimens including pathological review, quality control analyses, and delivery charges necessary to render an individualized test result. Costs associated with performing tests are recorded as the tests are processed.
Research and Development
Research and development costs, which include expenditures in connection with an in-house research and development laboratory, salaries and staff costs, application and filing for regulatory approval of proposed products, purchased in-process research and development, regulatory and scientific consulting fees, clinical samples as well as clinical collaborators and insurance, are accounted for in accordance with ASC Topic 730-10-55-2, Research and Development. Also, as prescribed by this guidance, patent filing and maintenance expenses are considered legal in nature and therefore classified as general and administrative expense. We are providing the following summary of our research and development expenses to supplement the more detailed discussions under results of operations. Costs are not allocated to projects as the majority of the costs relate to employees and facilities costs and we do not track employees’ hours by project or allocate facilities costs on a project basis.
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Salaries and staff costs |
|
$ |
3,465,211 |
|
$ |
2,063,474 |
|
$ |
950,861 |
|
|
Outside services, consultants and lab supplies |
|
2,435,917 |
|
1,301,190 |
|
594,342 |
| |||
|
Facilities |
|
628,535 |
|
466,138 |
|
352,920 |
| |||
|
Other |
|
135,243 |
|
116,787 |
|
22,175 |
| |||
|
Total Research and Development |
|
$ |
6,664,906 |
|
$ |
3,947,589 |
|
$ |
1,920,298 |
|
While certain of our research and development costs may have future benefits, our policy of expensing all research and development expenditures is predicated on the fact that we have no history of successful commercialization of molecular diagnostic products to base any estimate of the number of future periods that would be benefited.
ASC Topic 730, Research and Development requires that non-refundable advance payments for goods or services that will be used or rendered for future research and development activities should be deferred and capitalized. As the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided, the deferred amounts would be recognized as an expense. There are no non-refundable advance payments that are deferred and capitalized as of December 31, 2014 and 2013.
Stock-based Compensation
We rely heavily on incentive compensation in the form of stock options to recruit, retain and motivate directors, executive officers, employees and consultants. Incentive compensation in the form of stock options and warrants are designed to provide long-term incentives, develop and maintain an ownership stake and conserve cash. Stock-based compensation expense for employees and directors is recognized in the statement of operations based on estimated amounts, including the grant date fair value and the expected service period. For stock options, we estimate the grant date fair value using a Black-Scholes model. Share-based compensation recorded in our statement of operations is based on awards expected to ultimately vest and has been reduced for estimated forfeitures. We recognize the value of the awards on a straight-line basis over the awards’ requisite service periods. The requisite service period is generally the time over which our share-based awards vest.
We account for equity instruments granted to non-employees in accordance with ASC Topic 505-50 “Equity-Based Payment to Non-Employees” where the value of the share-based compensation is based upon the measurement date as determined at either: a) the date at which a performance commitment is reached, or b) at the date at which the necessary performance to earn the equity instruments is complete. Accordingly the fair value of these options is being “marked to market” quarterly until the measurement date is determined.
Fair value of financial instruments
Financial instruments consist of cash and cash equivalents, accounts receivable, accounts payable, debt and derivative liabilities. We have adopted FASB ASC 820 Fair Value Measurements and Disclosures (“ASC 820”) for financial assets and liabilities that are required to be measured at fair value, and non-financial assets and liabilities that are not required to be measured at fair value on a recurring basis. These financial instruments are stated at their respective historical carrying amounts which approximate to fair value due to their short term nature as they reflect current market interest rates. Debt is stated at its respective historical carrying amounts which approximates fair value as they reflect current market interest rates.
ASC 820 provides that the measurement of fair value requires the use of techniques based on observable and unobservable inputs. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect our market assumptions. The inputs create the following fair value hierarchy:
· Level 1 — Quoted prices for identical instruments in active markets.
· Level 2 — Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations where inputs are observable or where significant value drivers are observable.
· Level 3 — Instruments where significant value drivers are unobservable to third parties.
Off-Balance Sheet Arrangements
We do not believe that we have any off-balance sheet arrangements.
Inflation
It is our opinion that inflation has not had a material effect on our operations.
Recent Accounting Pronouncements
See Note 2 to the Notes to Financial Statements in Item 8 below for further discussion of recent accounting pronouncements.
Results of Operations
YEARS ENDED DECEMBER 31, 2014 AND 2013
Revenues
Our total revenues were $280,178 and $259,246 for the years ended December 31, 2014 and 2013, respectively. Total revenues consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
(Decrease)/Increase |
| |||
|
|
|
|
|
|
|
|
| |||
|
Royalty income |
|
$ |
270,178 |
|
$ |
259,246 |
|
$ |
10,932 |
|
|
License fees |
|
10,000 |
|
— |
|
10,000 |
| |||
|
Total revenues |
|
$ |
280,178 |
|
$ |
259,246 |
|
$ |
20,932 |
|
Royalty income increased by $10,932 in the year ended December 31, 2014 primarily as a result of certain licensees exceeding their minimum royalties in comparison to the same period of the prior year. In accordance with our revenue recognition policy, we do not record royalty revenues in excess of minimum royalty amounts until we have received the payment.
The $10,000 license fee earned in the year ended December 31, 2014 related to a licensing agreement signed in the second quarter of 2014. There were no license fees earned during the year ended December 31, 2013.
We expect our royalty income to fluctuate as the royalties are based on the portion of our partners’ revenues earned utilizing the patents they have licensed from us. Milestone and license fee revenues are difficult to predict and can vary significantly from period to period. In addition, we expect to have revenues from our diagnostics tests in future periods, but as the revenue recognition will initially be based on cash receipts, the timing of these revenues are also uncertain.
Cost of Revenues
Cost of revenues relates to the costs of our diagnostic services revenues and are recognized at the completion of testing. Gross margin on diagnostic tests are affected by test volumes, the timing of collections and overall reimbursement for the amount paid per test.
Research and Development Expenses
Research and development expenses increased by $2,717,317 to $6,664,906 for the year ended December 31, 2014 from $3,947,589 for the same period in 2013. Substantially all of the increase resulted from the expansion of our research and development efforts as we increased the average number of our internal research and development personnel from nine to 17, and purchased additional laboratory equipment to support the increase in clinical collaborations to support the commercialization of our tests. The clinical collaborations with external parties involve validation of our tests to detect certain types of cancer in urine samples.
Research and development expenses consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
Increase |
| |||
|
Salaries and staff costs |
|
$ |
2,669,203 |
|
1,514,009 |
|
$ |
1,155,194 |
| |
|
Stock-based compensation |
|
796,008 |
|
549,465 |
|
246,543 |
| |||
|
Outside services, consultants and lab supplies |
|
2,435,917 |
|
1,301,190 |
|
1,134,727 |
| |||
|
Facilities |
|
628,535 |
|
466,138 |
|
162,397 |
| |||
|
Travel and scientific conferences |
|
119,562 |
|
95,399 |
|
24,163 |
| |||
|
Other |
|
15,681 |
|
21,388 |
|
(5,707 |
) | |||
|
Total research and development |
|
$ |
6,664,906 |
|
$ |
3,947,589 |
|
$ |
2,717,317 |
|
To date our costs have related to validating our tests and supporting clinical collaborations. These costs are expected to increase as we expand current collaborations or enter into new collaborations and as we engage in research and development in areas other than detection of cancer in urine samples.
Selling and Marketing Expenses
Selling and marketing expenses increased by $1,204,743 to $2,734,903 for the year ended December 31, 2014, from $1,530,160 for the same period in 2013. The most significant increase was in outside services and related to reestablishing our corporate and product identity (“branding”), a commercial update to our website, and market research related to pricing and reimbursement of our tests. We have three commercially available tests as of December 31, 2014. We have increased our average internal headcount in this functional area from three to five, resulting in the increase in salaries and staff costs.
Selling and marketing expenses consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
Increase |
| |||
|
Salaries and staff costs |
|
$ |
1,139,855 |
|
734,060 |
|
405,795 |
| ||
|
Stock-based compensation |
|
145,240 |
|
100,189 |
|
45,051 |
| |||
|
Outside services |
|
902,181 |
|
274,625 |
|
627,556 |
| |||
|
Facilities and insurance |
|
115,713 |
|
98,081 |
|
17,632 |
| |||
|
Marketing |
|
258,658 |
|
193,701 |
|
64,957 |
| |||
|
Travel |
|
139,710 |
|
92,030 |
|
47,680 |
| |||
|
Other |
|
33,546 |
|
37,474 |
|
(3,928 |
) | |||
|
Total selling and marketing |
|
$ |
2,734,903 |
|
$ |
1,530,160 |
|
$ |
1,204,743 |
|
We expect our selling and marketing expenses to increase as we increase the number of tests we have commercially available and increase market acceptance of our tests.
General and Administrative Expenses
General and administrative expenses increased by $338,049 to $5,810,087 for the year ended December 31, 2014 from $5,472,038 for the same period in 2013. This increase was primarily due to an increase in legal fees related to filings and maintenance of patents. We have increased our average internal headcount in these functional areas from two to four, to support the growth in both research and development and sales and marketing, resulting in the increase in salaries and staff costs during the year ended December 31, 2014 as compared to the same period of the prior year. In addition, the costs associated with being a publicly traded company, such as additional costs for insurance, NASDAQ fees, and Sarbanes-Oxley compliance have added to our general and administrative expenses in comparison to the same period of the prior year. The overall increase was partially offset by decreases in stock-based compensation due to timing and quantity of stock-based awards.
General and administrative expenses consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
Increase/(Decrease) |
| |||
|
Salaries and staff costs |
|
$ |
773,353 |
|
663,190 |
|
110,163 |
| ||
|
Board of Directors’ (BOD) fees |
|
328,184 |
|
241,229 |
|
86,955 |
| |||
|
Stock-based compensation - employees |
|
691,128 |
|
927,404 |
|
(236,276 |
) | |||
|
Stock-based compensation - BOD |
|
437,820 |
|
601,097 |
|
(163,277 |
) | |||
|
Outside services |
|
1,472,448 |
|
1,394,796 |
|
77,652 |
| |||
|
Legal and accounting fees |
|
1,314,960 |
|
995,185 |
|
319,775 |
| |||
|
Facilities and insurance |
|
336,154 |
|
248,491 |
|
87,663 |
| |||
|
Travel |
|
208,651 |
|
220,691 |
|
(12,040 |
) | |||
|
Fees, licenses, taxes and other |
|
247,389 |
|
179,955 |
|
67,434 |
| |||
|
Total general and administrative |
|
$ |
5,810,087 |
|
$ |
5,472,038 |
|
$ |
338,049 |
|
We expect our general and administrative expenses to increase as we support the expansion of our sales and marketing of our diagnostic tests. In addition, at times, due to the use of options and warrants for compensation of services, stock based compensation expenses can vary significantly as the expense is based on assumptions in place at the measurement date of the award.
Interest Expense
Interest expense was $843,259 and 17,005 for the years ended December 31, 2014 and 2013, respectively. The increase results from the $15.0 million term loan we entered into in June 2014. We expect interest expense to continue to increase in comparison to the prior year as average debt outstanding will be higher and the term loan has interest only payments through July 2015.
Change in Fair Value of Derivative Instruments - Warrants
The change in fair value of derivative instruments resulted in a $1,425,850 gain in the year ended December 31, 2014 compared to a loss of $1,084,114 in the same period of the prior year. The gain was a result of the revaluation of the warrants based upon the change in our stock price from $5.74 at December 31, 2013 to $4.30 at December 31, 2014, and the changes in the expected term, volatility and risk free interest rates for the expected term. The increase in value was recorded as non-operating gain for the year ended December 31, 2014.
Net Loss
Net loss and per share amounts were as follows:
|
|
|
For the years ended December 31, |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
Net loss and comprehensive loss attributable to common shareholders |
|
$ |
(14,348,499 |
) |
$ |
(11,840,778 |
) |
|
Net loss per common share - basic |
|
$ |
(0.76 |
) |
$ |
(0.70 |
) |
|
Net loss per common share - diluted |
|
$ |
(0.88 |
) |
$ |
(0.70 |
) |
|
|
|
|
|
|
| ||
|
Weighted average shares outstanding - basic |
|
18,904,280 |
|
16,978,212 |
| ||
|
Weighted average shares outstanding - diluted |
|
19,071,112 |
|
16,978,212 |
| ||
The increase from a net loss and comprehensive loss attributable to common shareholders of $11,840,778 for the year ended December 31, 2013 to $14,348,999 for the year ended December 31, 2014 resulted from a slight increase in revenues, offset by increases in all operating expenses. The net loss per share - basic for the year ended December 31, 2014 increased by $0.06 to a net loss of $0.76 as a result of an overall increase in the net loss, slightly offset by an increase in weighted average shares outstanding during the year ended December 31, 2014 compared to the same period of the prior year. Weighted average shares outstanding - basic increased for the year ended December 31, 2014 due to 3.4 million shares issued as a result of shares sold through controlled equity offerings, and exercise of warrants and stock options during the year ended December 31, 2013, and approximately 13,000 shares issued as a result of the net exercise of warrants during the year ended December 31, 2014.
YEARS ENDED DECEMBER 31, 2013 AND 2012
Revenues
Our total revenues were $259,246 and $450,404 for the years ended December 31, 2013 and 2012, respectively. Total revenues consisted of the following:
|
|
|
Years ended December 31, |
| |||||||
|
|
|
2013 |
|
2012 |
|
(Decrease)/Increase |
| |||
|
|
|
|
|
|
|
|
| |||
|
Royalty income |
|
$ |
259,246 |
|
$ |
175,404 |
|
$ |
83,842 |
|
|
Milestone |
|
— |
|
150,000 |
|
(150,000 |
) | |||
|
License fees |
|
— |
|
125,000 |
|
(125,000 |
) | |||
|
Total revenues |
|
$ |
259,246 |
|
$ |
450,404 |
|
$ |
(191,158 |
) |
Royalty income increased by $83,842 in the year ended December 31, 2013, primarily as a result of more royalty bearing agreements in 2013 compared to the same period in 2012 and more royalty payments earned in excess of minimum royalty payments in the current year compared to the year ended December 31, 2012. In accordance with our revenue recognition policy, we do not record royalty revenues in excess of minimum royalty amounts until we have received the payment.
A milestone payment of $150,000 in 2012 was received upon achievement of a milestone with Ipsogen SAS during 2012.
License fees decreased by $125,000 in the year ended December 31, 2013 as there were more license agreements entered into during the year ended December 31, 2012 as compared to the same period in 2013.
Research and Development Expenses
Research and development expenses consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2013 |
|
2012 |
|
Increase |
| |||
|
Salaries and staff costs |
|
$ |
2,063,474 |
|
950,861 |
|
$ |
1,112,613 |
| |
|
Outside services, consultants and lab supplies |
|
1,301,190 |
|
594,342 |
|
706,848 |
| |||
|
Facilities |
|
466,138 |
|
352,920 |
|
113,218 |
| |||
|
Travel and scientific conferences |
|
95,400 |
|
— |
|
95,400 |
| |||
|
Other |
|
21,387 |
|
22,175 |
|
(788 |
) | |||
|
Total research and development |
|
$ |
3,947,589 |
|
$ |
1,920,298 |
|
$ |
2,027,291 |
|
Research and development expenses increased by $2,027,291 to $3,947,589 for the year ended December 31, 2013 from $1,920,298 for the same period in 2012. Substantially all of the increase resulted from the expansion of our research and development efforts as we began commercialization, increased the average number of our internal research and development personnel from four to nine, and purchase additional laboratory equipment to support the clinical collaborations we have entered into related to validating our tests to detect certain types of cancer in urine samples. We also established a clinical advisory board during the year ended December 31, 2013.
Selling and Marketing Expenses
Selling and marketing expenses consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2013 |
|
2012 |
|
Increase/(Decrease) |
| |||
|
Salaries and staff costs |
|
$ |
834,249 |
|
227,914 |
|
606,335 |
| ||
|
Outside services and consultants |
|
274,625 |
|
109,875 |
|
164,750 |
| |||
|
Facilities and insurance |
|
98,081 |
|
75,485 |
|
22,596 |
| |||
|
Marketing |
|
193,701 |
|
46,667 |
|
147,034 |
| |||
|
Travel |
|
92,030 |
|
4,360 |
|
87,670 |
| |||
|
Fees, licenses, taxes and other |
|
37,474 |
|
42,234 |
|
(4,760 |
) | |||
|
Total sales and marketing |
|
$ |
1,530,160 |
|
$ |
506,535 |
|
$ |
1,023,625 |
|
Selling and marketing expenses increased by $1,023,625 to $1,530,160 for the year ended December 31, 2013 from $ 506,535 for the same period in 2012. We have increased our average sales and marketing headcount from 2 to 3 and our marketing costs as we expand our efforts to inform the major cancer centers in the United States of our current and future product offerings. We expect these costs to increase as we continue to market and sell our tests.
General and Administrative Expenses
General and administrative expenses consisted of the following:
|
|
|
For the years ended December 31, |
| |||||||
|
|
|
2013 |
|
2012 |
|
Increase/(Decrease) |
| |||
|
Salaries and staff costs |
|
$ |
1,590,594 |
|
479,241 |
|
1,111,353 |
| ||
|
Outside services and Board of Directors’ fees |
|
2,237,122 |
|
1,442,352 |
|
794,770 |
| |||
|
Legal and accounting fees |
|
995,185 |
|
663,281 |
|
331,904 |
| |||
|
Facilities and insurance |
|
248,491 |
|
163,312 |
|
85,179 |
| |||
|
Travel |
|
220,691 |
|
80,166 |
|
140,525 |
| |||
|
Fees, licenses, taxes and other |
|
179,956 |
|
44,375 |
|
135,581 |
| |||
|
Total general and administrative |
|
$ |
5,472,039 |
|
$ |
2,872,727 |
|
$ |
2,599,312 |
|
General and administrative expenses increased by $2,599,312 to $5,472,039 for the year ended December 31, 2014 from $2,872,727 for the same period in 2013. This increase was primarily due to an increase in non-cash stock-based compensation expenses, 2013 bonus amounts and addition of personnel in comparison to the same period of the prior year. The increase in outside services and Board of Directors’ fees resulted from additional services received related to Sarbanes-Oxley compliance, investor relations efforts and equity financing services. Legal and accounting fees increased primarily as a result of legals costs related to filings and maintenance payments for our intellectual property portfolio.
We expect our general and administrative expenses to increase as we expand commercialization of our current diagnostic tests and future tests. In addition, at times, due to the use of options and warrants for compensation of services, stock based compensation expenses can vary significantly as the expense is based on assumptions in place at the measurement date of the award.
Interest Expense
There was $17,005 interest expense in the year ended December 31, 2013 compared to no interest expense in the same period of 2012. The increase results from the $1.0 million equipment line of credit agreement we entered into in June 2013.
Change in Fair Value of Derivative Instruments - Warrants
The change in fair value of derivative instruments resulted in a $1,084,114 loss in the year ended December 31, 2013 compared to a loss of $6,720,805 in the same period of the prior year. We issued securities that were accounted for as derivative liabilities at issuance during the years ended December 31, 2013 and 2012. On May 30, 2012 we closed an underwritten public offering that removed the condition that required the securities issued during the nine months ended September 30, 2012, as well as certain securities issued in prior periods, to be treated as derivative liabilities. Accordingly, the fair value of these securities of $3,317,463 was reclassified from a liability to additional paid in capital. During the quarter ended December 31, 2012, we issued additional securities that were accounted for as derivative liabilities at issuance. As of December 31, 2013, the remaining derivative liabilities were revalued to $4,431,871, resulting in a net decrease in value of $1,820,888 from December 31, 2012, based primarily upon changes in the fair value as a result of the underwritten public offering and the increase in the fair value of our common stock at December 31, 2013.
Net Loss
Net loss and per share amounts were as follows:
|
|
|
For the years ended December 31, |
| ||||
|
|
|
2013 |
|
2012 |
| ||
|
Net loss and comprehensive loss attributable to common shareholders |
|
$ |
(11,840,778 |
) |
$ |
(11,604,201 |
) |
|
Net loss per common share: basic and diluted |
|
$ |
(0.70 |
) |
$ |
(0.89 |
) |
|
Weighted average shares: basic and diluted |
|
16,978,212 |
|
13,066,600 |
| ||
The $236,577 increase in net loss and the $0.19 decrease in net loss per share in 2013 compared to 2012 reflected a slight decrease in revenues, more than offset by an increase in operating expenses and a loss from the change in fair value in derivative liabilities. Weighted average shares outstanding increased due to the issuance of 3.4 million shares of common stock during the year as a result of the public offering in July 2013, controlled equity offerings and exercise of warrants and stock options during the year ended December 31, 2013.
LIQUIDITY AND CAPITAL RESOURCES
As of December 31, 2014, we had $27,293,798 in cash and cash equivalents. Net cash used in operating activities for the year ended December 31, 2014 was $12,727,385, compared to $7,317,248 for the year ended December 31, 2013. Our use of cash was primarily a result of the net loss of $14,325,484 for the year ended December 31, 2014, adjusted for non-cash items related to stock-based compensation of $2,070,194, depreciation and amortization of $234,813 and the gain from the change in fair value of derivatives of $1,425,850. The changes in our operating assets and liabilities consisted of higher accounts payable and accrued expenses and a decrease in accounts receivable. At our current and anticipated level of operating loss, we expect to continue to incur an operating cash outflow for the next several years. As of December 31, 2014 and 2013, we had working capital of $23,231,596 and $24,059,854, respectively. The decrease in working capital is primarily due to the increase in the current portion of long-term debt.
Investing activities consisted of purchases for capital equipment that used $299,790 in cash during the year ended December 31, 2014, compared to $649,284 for the same period in 2013. We expect to invest approximately $985,000 in capital equipment over the next year. The investment will be predominantly for laboratory equipment.
Net cash provided by financing activities was $14,484,036 during the year ended December 31, 2014, compared to $22,983,688 during the year ended December 31, 2013. Financing activities during the year ended December 31, 2014 included $515,964 of repayments on equipment lines, offset by $15.0 million of new borrowings, while in 2013 financing activities included $18,829,644 from the sales of common stock, $3,638,080 from proceeds related to the exercise of warrants and options, and $515,964 from borrowings on equipment lines.
On February 11, 2015, we closed an underwritten public offering of 5,111,110 shares of our common stock. The offering price was $4.50 per share and net proceeds to us were approximately $21.3 million. As of February 27, 2015, our cash balance was approximately $46.0 million and our working capital was approximately $42.0 million. .
Our working capital requirements will depend upon numerous factors including but not limited to the nature, cost and timing of our research and development programs. We believe that we have sufficient cash and cash equivalents to fund our operations for at least the next twelve months. We do not anticipate that our existing working capital alone will be sufficient to fund our operations through the successful development and commercialization of products we develop. As a result, we will need to raise additional capital to fund our operations and continue to conduct activities to support our product development and commercialization. To date, our sources of cash have been primarily limited to the sale of equity securities and debentures and debt borrowings. We cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants that impact our ability to conduct business. If we are unable to raise additional capital when required or on acceptable terms, we may have to (i) significantly delay, scale back or discontinue the development and/or commercialization of one or more of product candidates; (ii) seek collaborators for product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; or (iii) relinquish or otherwise dispose of rights to technologies, product candidates or products that we would otherwise seek to develop or commercialize ourselves on unfavorable terms.
Public Offering and Controlled Equity Offering
On January 25, 2013 we filed a Form S-3 Registration Statement to offer and sell in one or more offerings, any combination of common stock, preferred stock, warrants, or units having an aggregate initial offering price not exceeding $150,000,000. The preferred stock, warrants, and units may be convertible or exercisable or exchangeable for common stock or preferred stock or other securities. This form was declared effective on February 4, 2013. In addition, in connection with the Form S-3, we entered into an agreement with Cantor Fitzgerald & Co. (“Agent”) on January 25, 2013 to issue and sell up to $30,000,000 of shares of common stock through them. As payment for their services, the Agent is entitled to a 3% commission on gross proceeds.
CONTRACTUAL OBLIGATIONS AND COMMITMENTS
The following table is a summary of contractual obligations for the periods indicated that existed as of December 31, 2014, and is based on information appearing in the notes to Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K.
|
|
|
Total |
|
Less than 1 |
|
1-2 Years |
|
3-5 Years |
|
More than 5 |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Operating leases |
|
$ |
1,119,505 |
|
360,775 |
|
371,324 |
|
387,406 |
|
$ |
— |
| |||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Research agreements (1) |
|
1,120,968 |
|
1,120,968 |
|
— |
|
— |
|
— |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Long-term debt (3) |
|
18,359,545 |
|
2,936,808 |
|
5,563,640 |
|
9,859,097 |
|
— |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Purchase obligations - major vendors (2) |
|
191,933 |
|
191,933 |
|
— |
|
— |
|
— |
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
Total obligations |
|
$ |
20,791,951 |
|
$ |
4,610,484 |
|
$ |
5,934,964 |
|
$ |
10,246,503 |
|
$ |
— |
|
(1) Payments under research agreements are based on the completion of activities as specified in the research agreement. The amounts in the table above assume the successful completion of the collaborative research activities contemplated in the agreements.
(2) Represents amounts that will become due upon future delivery of supplies and services from various suppliers under open purchase orders as of December 31, 2014
(3) Represents long-term debt and interest.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
Our cash and cash equivalent primary consists of securities issued by the U.S. government, deposits, and money market deposits managed by commercial banks. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk.
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of interest rates, particularly because our investments are in short-term money marketable funds. Due to the short-term duration of our investment portfolio and the relatively low risk profile of our investments, a sudden change in interest rates would not have a material effect on the fair market value of our portfolio, nor our operating results or cash flows.
Recently, there has been concern in the credit markets regarding the value of a variety of mortgage-backed and auction rate securities and the resulting effect on various securities markets. We do not hold any auction rate securities. We do not believe our cash, and cash equivalents investments have significant risk of default or illiquidity, however, we maintain significant amounts of cash and cash equivalents at one or more financial institutions that are in excess of federally insured limits. Given the current instability of financial institutions, we cannot provide assurance that we will not experience losses on these deposits.
Foreign Currency Risk
We have no operations outside the U.S. and do not hold any foreign currency denominated financial instruments.
Effects of Inflation
We do not believe that inflation and changing prices during the years ended December 31, 2014, 2013 and 2012 had a significant impact on our results of operations.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
All financial information required by this Item is attached hereto at the end of this report beginning on page F-1 and is hereby incorporated by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our chief executive officer and chief financial officer evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2014. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Securities Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Securities Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Based on that evaluation, as of December 31, 2014, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as such term is defined in Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
As of December 31, 2014, our management assessed the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework - 2013. Based on this assessment, our management concluded that, as of December 31, 2014, our internal control over financial reporting was effective based on those criteria.
The effectiveness of our internal control over financial reporting as of December 31, 2014 has been audited by BDO USA, LLP, an independent registered public accounting firm, as stated in their report which is included herein.
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Trovagene, Inc.
San Diego, California
We have audited Trovagene, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Trovagene, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Item 9A, Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Trovagene, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Trovagene, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2014 and our report dated March 12, 2015 expressed an unqualified opinion thereon.
/s/ BDO USA, LLP
San Diego, California
March 12, 2015
Changes in Internal Control
As required by Rule 13a-15(d) and Rule 15d-15(b) of the Exchange Act, our management, including our principal executive officer and our principal financial officer, conducted an evaluation of the internal control over financial reporting to determine whether any changes occurred during the three months ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. There was no change in our internal control over financial reporting during the three months ended December 31, 2014 that materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
EXECUTIVE OFFICERS, DIRECTORS AND KEY EMPLOYEES
The following table sets forth the names and ages of the members of our Board of Directors and our executive officers and the positions held by each as of February 27, 2015.
|
Name |
|
Age |
|
Position |
|
|
Thomas H. Adams, Ph.D. |
|
72 |
|
Chairman of the Board |
|
|
Antonius Schuh, Ph.D. |
|
51 |
|
Chief Executive Officer and Director |
|
|
Steve Zaniboni |
|
57 |
|
Chief Financial Officer |
|
|
John Brancaccio |
|
67 |
|
Director |
|
|
Gary S. Jacob, Ph.D. |
|
67 |
|
Director |
|
|
Dr. Paul Billings |
|
62 |
|
Director |
|
|
Dr. Stanley Tennant |
|
63 |
|
Director |
|
|
Dr. Rodney S. Markin |
|
58 |
|
Director |
|
|
Carl Feldbaum |
|
71 |
|
Director |
|
All directors hold office until the next annual meeting of stockholders and the election and qualification of their successors. Officers are elected annually by the board of directors and serve at the discretion of the board.
Executive Biographies
The principal occupations for the past five years (and, in some instances, for prior years) of each of our directors and executive officers are as follows:
Thomas H. Adams. Thomas H. Adams, Ph.D, has been our Chairman of the Board since April 2009. From June 2005 through 2011, Dr. Adams served as a director of IRIS International, Inc., a diagnostics company, and as Chief Technology Officer of IRIS from April 2006. Until November 2012, Dr. Adams was the Head of Iris Molecular Diagnostics since 2006 and the President of Iris Personalized Medicine since 2011. In November 2012, IRIS was acquired by Danaher Corporation. Dr. Adams served as Chairman and Chief Executive Officer of Leucadia Technologies, a privately held medical-device company, from 1998 to April 2006, when Leucadia was acquired by IRIS. In 1989, Dr. Adams founded Genta, Inc., a publicly held biotechnology company in the field of antisense technology, and served as its Chief Executive Officer until 1997. Dr. Adams founded Gen-Probe, Inc. in 1984 and served as its Chief Executive Officer and Chairman until its acquisition by Chugai Biopharmaceuticals, Inc. in 1989. Dr. Adams is currently a director of Synergy Pharmaceuticals Inc., a biotechnology company. Dr. Adams holds a Ph.D. in Biochemistry from the University of California, at Riverside. Dr. Adam’s executive leadership, particularly in the diagnostic field, and the extensive healthcare expertise he has developed qualifies Dr. Adams to serve as a director of our company.
Antonius Schuh. Antonius Schuh, Ph.D. joined us in October 2011 as our Chief Executive Officer and was elected as a Director in December 2011. Dr. Schuh co-founded Sorrento Therapeutics, Inc., a biopharmaceutical company developing monoclonal antibodies, in January 2006. He served as Chairman of the Board and Chief Executive Officer of Sorrento Therapeutics from November 2008 to April 2011. From April 2006 to September 2008, Dr. Schuh served as Chief Executive Officer of AviaraDx (now bioTheranostics, Inc., a bioMerieux company), a molecular diagnostic testing company that is focused on clinical applications in oncology. From March 2009 to January 2015 Dr. Schuh was a director of Diogenix, Inc., a privately held molecular diagnostic company, and since May 2009, he has served as a director of RScueRx, Inc., a privately held biotherapeutics company. Dr. Schuh is a certified pharmacist and earned his Ph.D. in pharmaceutical chemistry from the University of Bonn, Germany.
Stephen Zaniboni. Stephen Zaniboni joined us as Chief Financial Officer in January 2012. Prior to joining us, since June 2010, Mr. Zaniboni has served as Chief Financial Officer of Awarepoint Corporation, a leading provider of healthcare software. Prior to joining Awarepoint Corporation, Mr. Zaniboni served as Chief Financial Officer of XIFIN Inc., the leading provider of revenue cycle management for diagnostic service providers, from January 2009 through June 2010. Prior to joining XIFIN Inc., Mr. Zaniboni served as the Chief Financial Officer of Sorrento Therapeutics, Inc. from January 2006, and as a member of its board of directors from November 2008, through September 2009. From May 2006 to September 2008, Mr. Zaniboni served as Chief Financial Officer of AviaraDx (now bioTheranostics, a bioMerieux company), a molecular diagnostic testing cancer profiling company that is focused on developing and commercializing molecular diagnostic technologies with proven clinical utility. Mr. Zaniboni has also held various financial management positions at Aspect Medical Systems, Behring Diagnostics, and Boston Scientific. He was a practicing CPA with Arthur Andersen and holds a B.S. in accounting from Boston University and an M.B.A. from Boston College.
John Brancaccio. John Brancaccio, a retired CPA, has served as a director of our company since December 2005. Since April 2004, Mr. Brancaccio has been the Chief Financial Officer of Accelerated Technologies, Inc., an incubator for medical device companies.. Mr. Brancaccio is currently a director of Tamir Biotechnology, Inc. (formerly Alfacell Corporation) as well as a director of Synergy Pharmaceuticals Inc. and ContraVir Pharmaceuticals, Inc. Mr. Brancaccio’s chief financial officer experience provides him with valuable financial and accounting expertise which the Board believes qualifies him to serve as a director of our company.
Gary S. Jacob. Gary S. Jacob, Ph.D. has served as a director of our company since February 2009. Since July 2008, Dr. Jacob has been President, Chief Executive Officer and a Director of Synergy Pharmaceuticals Inc. and Chairman since September 2013. Dr. Jacob has been Chairman of ContraVir Pharmaceuticals, Inc. since May 2013. Prior to 1999, Dr. Jacob served as a Monsanto Science Fellow, specializing in the field of glycobiology, and from 1997 to 1998 was Director of Functional Genomics, Corporate Science & Technology, at Monsanto Company. Dr. Jacob earned a B.S. in Chemistry from the University of Missouri, and holds a Ph.D. in Biochemistry from the University of Wisconsin-Madison. Dr. Jacob’s broad management expertise in the pharmaceutical and biotechnology industries provides relevant experience in a number of strategic and operational areas and led to the Board’s conclusion that he should serve as a director of our company.
Dr. Paul Billings. Paul Billings, MD, PhD was appointed to our Board in October 2013 and has been a member of our Scientific Advisory Board since November 2012. Dr. Billings is a board certified internist and clinical geneticist, and currently is an Executive-in-Residence at the California Innovation Center of Johnson and Johnson, Inc. He is also the Medical Director of the IMPACT program at Thermo Fisher Scientifiic, Inc.(TFS) He recently finished serving as the first and only Chief Medical Officer at Life Technologies Corporation and the Genetic Sciences Division of TFS. Dr. Billings served as Chief Medical Officer at Life Technologies Corporation. Dr. Billings has extensive healthcare experience in many aspects of genomics and molecular medicine. In addition to serving as Chief Medical Officer at Life Technologies, he also serves on the Scientific Advisory Board of the Food and Drug Administration, the Genomic Medicine Advisory Committee at the Department of Veterans Affairs, and the National Academy of Sciences Institute of Medicine’s Roundtable on Genomics. In addition to Trovagene, he serves as an advisor or director for many companies including Omicia, BioScale, Applied Immunology, Aueon and PAX Neurosciences. Dr. Billings holds an M.D. from Harvard Medical School and a Ph.D. in immunology, also from Harvard University. Dr. Billings’ medical and managerial experience in the diagnostic field qualifies him to serve as a director of our company.
Dr. Stanley Tennant. Dr. Tennant has served as a director of our company since December 2010. Since 1983, Dr. Tennant has been a cardiologist in Greensboro, NC. He graduated from Wake Forest University School of Medicine in 1978 and completed postgraduatetraining in Internal Medicine and Cardiology at Vanderbilt University in 1983. Dr. Tennant’s practical experience in the healthcare field led to the Board’s conclusion that he should serve as a director of our company.
Dr. Rodney S. Markin. Dr. Markin has been a director of our company since February 2014. Dr. Markin is Chief Technology Officer and Associate Vice Chancellor for Business Development at the University of Nebraska Medical Center and a Professor of Pathology and Microbiology; David T. Purtilo Distinguished Professor Pathology and Microbiology and Courtesy Professor of Surgery. Dr. Markin is also a director on the Board of Children’s Hospital and Medical Center Foundation and on the Board of Trustees for Keck Graduate Institute and on the Board of the Make-A-Wish Foundation. The Board selected Dr. Markin to serve as a director because he has valuable executive experience in the healthcare business.
Carl Feldbaum. Carl Feldbaum has been a director of our company since December 2014. Mr. Feldbaum has been a director of Exelixis, Inc. since February 2007 and serves as a member of the Nominating and Governance Committee. In addition, Mr. Feldbaum is a member of the board of directors of Actelion, Ltd. and serves as Chairman of its Nominating and Governance Committee. Mr. Feldbaum previously served as a member of the board of directors of Connetics Corporation from 2005 until its acquisition by Stiefel Laboratories, Inc. in 2006. Mr. Feldbaum is a member of the board of directors of BIO Ventures for Global Health, a non-profit organization, where he has served as a member of the board of directors since its inception in 2004. Mr. Feldbaum is interim Chair and a member of the Board of the Life Sciences Foundation, a non-profit based in Oakland, California. Mr. Feldbaum is president emeritus of the Biotechnology Industry Organization (BIO), which represents more than 1,000
biotechnology companies, academic institutions and state biotechnology centers internationally. Mr. Feldbaum served as president of BIO from 1993 until his retirement in 2005. Prior to joining BIO, Mr. Feldbaum was chief of staff to Senator Arlen Specter of Pennsylvania. He also was president and founder of Palomar Corporation, a national security “think tank” in Washington, D.C. Before founding Palomar Corporation, Mr. Feldbaum was Assistant to the Secretary of Energy and served as the Inspector General for defense intelligence in the U.S. Department of Defense. Mr. Feldbaum received an A.B. in Biology from Princeton University and his J.D. from the University of Pennsylvania Law School. Mr. Feldbaum’s knowledge and experience with respect to the biotechnology, pharmaceutical and healthcare industries, his broad leadership experience resulting from service on various boards and as an executive officer and his knowledge and experience with policymaking, regulatory issues and other governmental matters qualifies him to serve as a director of our company.
Family Relationships
None.
Involvement in Certain Legal Proceedings
To our knowledge, during the last eleven years, none of our directors, executive officers (including those of our subsidiaries), promoters or control persons have:
· had a bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time;
· been convicted in a criminal proceeding or been subject to a pending criminal proceeding, excluding traffic violations and other minor offenses;
· been subject to any order, judgment or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities;
· been found by a court of competent jurisdiction (in a civil action), the Securities and Exchange Commission, or SEC, or the Commodities Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended or vacated; and
· been the subject to, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization, any registered entity, or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member.
Board Leadership Structure and Role in Risk Oversight
Since April 2009, we have separated the roles of Chairman of the Board and Chief Executive Officer (“CEO”). Although the separation of roles has been appropriate for us during that time period, in the view of the board of directors, the advisability of the separation of these roles depends upon the specific circumstances and dynamics of our leadership.
As Chairman of the Board, Dr. Adams serves as the primary liaison between the CEO and the independent directors and provides strategic input and counseling to the CEO. With input from other members of the board of directors, committee chairs and management, he presides over meetings of the board of directors. Dr. Adams has developed an extensive knowledge of our company, its challenges and opportunities and has a productive working relationship with our senior management team.
The board of directors, as a unified body and through committee participation, organizes the execution of its monitoring and oversight roles and does not expect its Chairman to organize those functions. Our primary rationale for separating the positions of Board Chairman and the CEO is the recognition of the time commitments and activities required to function effectively as Chairman and as the CEO of a company with a relatively flat management structure. The separation of roles has also permitted the board of directors to recruit senior executives into the CEO position with skills and experience that meet the board of director’s planning for the position who may not have extensive public company board experience.
The board of directors has three standing committees—Audit, Compensation and Corporate Governance/Nominating. The membership of each of the board committees is comprised of independent directors, with each of the committees having a separate chairman, each of whom is an independent director. Our non-management members of the board of directors meet in executive session at each board meeting.
Risk is inherent with every business, and how well a business manages risk can ultimately determine its success. Management is responsible for the day-to-day management of risks the company faces, while the board of directors, as a whole and through its committees, has responsibility for the oversight of risk management. In its risk oversight role, the board of directors has the responsibility to satisfy itself that the risk management processes designed and implemented by management are adequate and functioning as designed.
The board of directors believes that establishing the right “tone at the top” and that full and open communication between executive management and the board of directors are essential for effective risk management and oversight. Our CEO communicates frequently with members of the board to discuss strategy and challenges facing the company. Senior management usually attends our regular quarterly board meetings and is available to address any questions or concerns raised by the board of directors on risk management-related and any other matters. Each quarter, the board of directors receives presentations from senior management on matters involving our areas of operations.
Director Independence
Our board of directors has determined that a majority of the board consists of members who are currently “independent” as that term is defined under current listing standards of NASDAQ. The board of directors considers Messrs. Jacob, Billings, Tennant, Feldbaum, Markin and Brancaccio to be “independent.”
Audit Committee
The Audit Committee’s responsibilities include: (i) reviewing the independence, qualifications, services, fees, and performance of the independent registered public accountants, (ii) appointing, replacing and discharging the independent auditors, (iii) pre-approving the professional services provided by the independent auditors, (iv) reviewing the scope of the annual audit and reports and recommendations submitted by the independent auditors, and (v) reviewing our financial reporting and accounting policies, including any significant changes, with management and the independent auditors. The Audit Committee also prepares the Audit Committee report that is required pursuant to the rules of the SEC.
As of December 31, 2014, the Audit Committee consisted of John P. Brancaccio, chairman of the Audit Committee, Dr. Gary S. Jacob, and Dr. Thomas Adams. Our board of directors has determined that each of Mr. Brancaccio, Dr. Jacob and Dr. Adams is “independent” as that term is defined under applicable SEC and NASDAQ rules. Mr. Brancaccio is our audit committee financial expert. The board of directors has adopted a written charter setting forth the authority and responsibilities of the Audit Committee. As of January 1, 2015, the Audit Committee was reconstituted with Mr. Brancaccio remaining as chairman and Dr. Rodney Markin and Dr. Stanley Tennant joining the Committee.
Compensation Committee
The Compensation Committee has responsibility for assisting the board of directors in, among other things, evaluating and making recommendations regarding the compensation of the executive officers and directors of our company; assuring that the executive officers are compensated effectively in a manner consistent with our stated compensation strategy; producing an annual report on executive compensation in accordance with the rules and regulations promulgated by the SEC; periodically evaluating the terms and administration of our incentive plans and benefit programs and monitoring of compliance with the legal prohibition on loans to our directors and executive officers.
As of December 31, 2014, the Compensation Committee consisted of Dr. Stanley Tennant, chairman of the Compensation Committee, Dr. Gary S. Jacob and John P. Brancaccio. Our board of directors has determined that all of the members are “independent” under the current listing standards of NASDAQ. The board of directors has adopted a written charter setting forth the authority and responsibilities of the Compensation Committee. As of January 1, 2015, the Compensation Committee was reconstituted with Dr. Tennant remaining as chairman and Dr. Rodney Markin and Dr. Paul Billings joining Dr. Gary Jacob on the Committee.
Corporate Governance/Nominating Committee
The Corporate Governance/Nominating Committee has responsibility for assisting the board of directors in, among other things, effecting board organization, membership and function including identifying qualified board nominees; effecting the organization, membership and function of board committees including composition and recommendation of qualified candidates; establishment of and subsequent periodic evaluation of successor planning for the chief executive officer and other executive officers; development and evaluation of criteria for Board membership such as overall qualifications, term limits, age limits and independence; and oversight of compliance with the Corporate Governance Guidelines. The Corporate Governance/Nominating Committee shall
identify and evaluate the qualifications of all candidates for nomination for election as directors. Potential nominees are identified by the Board of Directors based on the criteria, skills and qualifications that have been recognized by the CorporateGovernance/Nominating Committee. While our nomination and corporate governance policy does not prescribe specific diversity standards, the Corporate Governance/Nominating Committee and its independent members seek to identify nominees that have a variety of perspectives, professional experience, education, difference in viewpoints and skills, and personal qualities that will result in a well-rounded Board of Directors.
As of December 31, 2014, the Corporate Governance/Nominating Committee consisted of John Brancaccio, chairman of the Corporate Governance/Nominating Committee, Thomas Adams and Stanley Tennant. The Board of Directors has determined that all of the members are “independent” under the current listing standards of NASDAQ. The Board of Directors has adopted a written charter setting forth the authority and responsibilities of the Corporate Governance/Nominating Committee. As of January 1, 2015, the Corporate Governance/Nominating Committee was reconstituted with Carl Feldbaum joining as chairman and Dr. Paul Billings and John Brancaccio joining as members.
Code of Ethics
We have adopted a formal Code of Business Conduct and Ethics applicable to all Board members, executive officers and employees. Our Code of Business Conduct and Ethics can be found on our website (www.trovagene.com).
Compliance With Section 16(A) of the Exchange Act
Section 16(a) of the Securities Exchange Act of 1934 requires our officers and directors, and persons who own more than ten percent of a registered class of our equity securities, to file reports of ownership and changes in ownership with the Securities and Exchange Commission. Officers, directors and greater than ten percent stockholders are required by SEC regulation to furnish us with copies of all Section 16(a) forms they file.
Based on a review of the copies of such forms received, we believe that during 2014, all filing requirements applicable to our officers, directors and greater than ten percent beneficial owners were complied with except that Antonius Schuh and Steve Zaniboni each filed one Form 4 late.
ITEM 11. EXECUTIVE COMPENSATION
Compensation Committee Report
Under the rules of the SEC, this Compensation Committee Report is not deemed to be incorporated by reference by any general statement incorporating this Annual Report by reference into any filings with the SEC.
The Compensation Committee has reviewed and discussed the following Compensation Discussion and Analysis with management. Based on this review and these discussions, the Compensation Committee recommended to the Board of Directors that the following Compensation Discussion and Analysis be included in this Annual Report on Form 10-K.
Submitted by the Compensation Committee
Dr. Stanley Tennant
John Brancaccio
Dr. Gary S. Jacob
Compensation Discussion and Analysis
Overview
We compete with many other medical diagnostic companies in seeking to attract and retain a skilled work force. To meet this challenge, we have developed our compensation structure to enable our management to make decisions regarding our compensation programs, to manage these programs, and to effectively communicate the goals of these programs to our employees and stockholders.
Our compensation philosophy is to offer our employees compensation and benefits that are competitive and that meet our goals of attracting, retaining and motivating highly skilled employees so that we can achieve our financial and strategic objectives.
Utilizing this philosophy, our compensation programs are designed to:
· be “market-based” and reflect the competitive environment for personnel;
· stress our “pay for performance” approach to managing pay levels;
· share risks and rewards with employees at all levels;
· be affordable, within the context of our operating expense model;
· align the interests of our employees with those of our stockholders;
· reflect our values; and
· be fairly and equitably administered.
In addition, as we administer our compensation programs, we plan to:
· evolve and modify our programs to reflect the competitive environment and our changing business needs;
· focus on simplicity, flexibility and choice wherever possible;
· openly communicate the details of our programs with our employees and managers to ensure that our programs and their goals are understood; and
· provide our managers and employees with the tools they need to administer our compensation programs.
Elements of Our Compensation Program
As a total rewards package, we design our compensation program to enable us to attract and retain talented personnel. The individual elements of our compensation program serve to satisfy this larger goal in specific ways as described below.
We design base pay to provide the essential reward for an employee’s work, and is required to be competitive in attracting talent. Once base pay levels are initially determined, increases in base pay are provided to recognize an employee’s specific performance achievements. Consistent with our compensation philosophy, we implement a “pay for performance” approach that provides higher levels of compensation to individual employees whose results merit greater rewards. Our managers typically make performance assessments throughout the year, and provide ongoing feedback to employees, provide resources and maximize individual and team performance levels.
We design equity-based compensation, including stock options, to ensure that we have the ability to retain talent over a longer period of time, and to provide optionees with a form of reward that aligns their interests with those of our stockholders.
We also utilize various forms of variable compensation, including cash bonuses that allow us to remain competitive with other companies while providing upside potential to those employees who achieve outstanding results.
Core benefits, such as our basic health benefits, are designed to provide a stable array of support to employees and their families.
The four key elements of our compensation structure are:
· base pay;
· variable pay;
· equity-based pay; and
· benefits.
Consistent with our compensation philosophy, we have structured each element of our rewards package as follows:
Base Pay
We create a set of base pay structures that are both affordable and competitive in relation to the market. We continuously monitor base pay levels within the market and make adjustments to our structures as needed. In general, an employee’s base pay level should reflect the employee’s overall sustained performance level and contribution to our company over time. We seek to structure the base pay for our top performers to be aggressive in relation to the market.
Variable Pay
We design our variable pay programs to be both affordable and competitive in relation to the market. We monitor the market and adjust our variable pay programs as needed. Our variable pay programs, such as our bonus program, are designed to motivate employees to achieve overall goals. Our programs are designed to avoid entitlements, to align actual payouts with the actual results achieved and to be easy to understand and administer.
Equity-Based Rewards
We design our equity programs to be both affordable and competitive in relation to the market. We monitor the market and applicable accounting, corporate, securities and tax laws and regulations and adjust our equity programs as needed. Stock options and other forms of equity compensation are designed to reflect and reward a high level of sustained individual performance over time. We design our equity programs to align employees’ interests with those of our stockholders.
Benefits Programs
We design our benefits programs to be both affordable and competitive in relation to the market while conforming with local laws and practices. We monitor the market, local laws and practices and adjust our benefits programs as needed. We design our benefits programs to provide an element of core benefits, and to the extent possible, offer options for additional benefits, be tax-effective for employees in each country and balance costs and cost sharing between us and our employees.
Our stock options typically have annual vesting over a three-year period and a term of ten years, in order to encourage a long-term perspective and to encourage key employees to remain with us. We also use performance based vesting in our option grants. Generally, vesting and exercise rights cease upon termination of employment. Prior to the exercise of an option, the holder has no rights as a stockholder with respect to the shares subject to such option, including voting rights and the right to receive dividends or dividend equivalents.
Timing of Equity Awards
Only the Compensation Committee may approve stock option grants to our executive officers. Stock options are generally granted at predetermined meetings of the Compensation Committee. On limited occasions, grants may occur upon unanimous written consent of the Compensation Committee, which occurs primarily for the purpose of approving a compensation package for newly hired or promoted executive. The exercise price of a newly granted option is the closing price of our common stock on the date of grant.
Executive Equity Ownership
We encourage our executives to hold a significant equity interest in our company. However, we do not have specific share retention and ownership guidelines for our executives.
Performance-Based Compensation and Financial Restatement
We have not considered or implemented a policy regarding retroactive adjustments to any cash or equity-based incentive compensation paid to our executives and other employees where such payments were predicated upon the achievement of certain financial results that were subsequently the subject of a financial restatement.
Severance and Change in Control Arrangements
Several of our executives have employment and other agreements which provide for severance payment arrangements and/or acceleration of stock option vesting that would be triggered by an acquisition or other change in control of our company. See “Employment Agreements” below for a description of the severance and change in control arrangements for our named executive officers.
Effect of Accounting and Tax Treatment on Compensation Decisions
In the review and establishment of our compensation programs, we consider the anticipated accounting and tax implications to us and our executives.
Section 162(m) of the Internal Revenue Code imposes a limit on the amount of compensation that we may deduct in any one year with respect to our chief executive officer and each of our next four most highly compensated executive officers, unless certain specific and detailed criteria are satisfied. Performance-based compensation, as defined in the Internal Revenue Code, is fully deductible if the programs are approved by stockholders and meet other requirements. We believe that grants of equity awards under our existing stock plans qualify as performance-based for purposes of satisfying the conditions of Section 162(m), thereby permitting us to receive a federal income tax deduction in connection with such awards. In general, we have determined that we will not seek to limit executive compensation so that it is deductible under Section 162(m). However, from time to time, we monitor whether it might be in our interests to structure our compensation programs to satisfy the requirements of Section 162(m). We seek to maintain flexibility in compensating our executives in a manner designed to promote our corporate goals and therefore our compensation committee has not adopted a policy requiring all compensation to be deductible. Our compensation committee will continue to assess the impact of Section 162(m) on our compensation practices and determine what further action, if any, is appropriate.
Role of Executives in Executive Compensation Decisions
Our board of directors and our Compensation Committee generally seek input from our Chief Executive Officer, Dr. Antonius Schuh, when discussing the performance of, and compensation levels for executives other than himself. The Compensation Committee also works with Dr. Schuh and our Chief Financial Officer evaluating the financial, accounting, tax and retention implications of our various compensation programs. Neither Dr. Schuh nor any of our other executives participates in deliberations relating to his or her compensation.
Role of Compensation Consultant
The Compensation Committee has the power to engage independent advisors to assist it in carrying out its responsibilities. For fiscal 2014, the Compensation Committee engaged Barney & Barney, LLC (“Barney& Barney”) as its independent executive and Board compensation consultant. Barney & Barney, who reports directly to the Compensation Committee and not to management, is independent from us, has not provided any services to us other than to the Compensation Committee, and receives compensation from us only for services provided to the Compensation Committee. The Compensation Committee assessed the independence of Barney & Barney pursuant to SEC rules and concluded that the work of Barney & Barney has not raised any conflict of interest.
Barney & Barney reviews and advises on all principal aspects of the executive and Board compensation program. Its main responsibilities are to:
· advise on alignment of pay and performance;
· review and advise on executive total compensation, including base salaries, short- and long-term incentives, associated performance goals, and retention and severance arrangements;
· advise on trends in executive compensation;
· advise on Board and Board committee compensation;
· provide recommendations regarding the composition of our peer group;
· analyze peer group proxy statements, compensation survey data, and other publicly available data (and apply its experience with other companies to this analysis); and
· perform any special projects requested by the Compensation Committee.
Barney & Barney has attended the Compensation Committee’s meetings, including executive sessions at which management is not present. Barney & Barney communicates regularly with the Compensation Committee’s Chair outside of Compensation Committee meetings, and also meets with management to gather information and review proposals. Barney & Barney is expected to remain the Compensation Committee’s independent consultant until determined otherwise by the Compensaton Committee or Barney & Barney.
Chief Executive Officer Compensation for Fiscal Year 2014
In 2011, we entered into an executive agreement with Antonius Schuh, Ph.D. in which he agreed to serve as our Chief Executive Officer. The term of the agreement is effective as of October 4, 2011 and continues until October 4, 2015 and is automatically renewed for successive one year periods at the end of each term. Dr. Schuh’s 2014 compensation was $385,000 per year. In December 2014, Dr. Schuh’s salary was increased to $470,000. Dr. Schuh is eligible to receive a cash bonus of up to 50% of his base salary per year based on meeting certain performance objectives and bonus criteria. Upon entering the agreement, Dr. Schuh was granted 633,333 non-qualified stock options which have an exercise price of $3.00 per share and vest annually in equal amounts over a period of four years. Dr. Schuh is also eligible to receive a realization bonus upon the occurrence of either of the following events, whichever occurs earlier;
(i) In the event that during the term of the agreement, for a period of 90 consecutive trading days, the market price of the common stock is $7.50 or more and the value of the common stock daily trading volume is $125,000 or more, we shall pay or issue Dr. Schuh a bonus in an amount of $3,466,466 in either cash or registered common stock or a combination thereof as mutually agreed by Dr. Schuh and us; or
(ii) In the event that during the term of the agreement, a change of control occurs where the per share enterprise value of our company equals or exceeds $7.50 per share, we shall pay Dr. Schuh a bonus in an amount determined by multiplying the enterprise value by 4.0%. In the event in a change of control the per share enterprise value exceeds a minimum of $14.40 per share, $22.80 per share or $30.00 per share, Dr.Schuh shall receive a bonus in an amount determined by multiplying the incremental enterprise value by 2.5%, 2.0% or 1.5%, respectively.
If the executive agreement is terminated by us for cause or as a result of Dr. Schuh’s death or permanent disability or if Dr. Schuh terminates his agreement voluntarily, Dr. Schuh shall receive a lump sum equal to (i) any portion of unpaid base compensation then due for periods prior to termination, (ii) any bonus or realization bonus earned but not yet paid through the date of termination and (iii) all expenses reasonably incurred by Dr. Schuh prior to date of termination. If the executive agreement is terminated by us without cause Dr. Schuh shall receive a severance payment equal to base compensation for three months if termination occurs ten months after the effective date of the agreement and six months if termination occurs subsequent to ten months from the effective date. If the executive agreement is terminated as a result of a change of control, Dr. Schuh shall receive a severance payment equal to base compensation for twelve months and all unvested stock options shall immediately vest and become fully exercisable for a period of six months following the date of termination.
2014 Bonus
On December 14, 2014, the Compensation Committee approved a bonus of $154,000 for Dr. Schuh, which was 40% of his base compensation. The Compensation Committee reviewed the following factors in determining the amount of the bonus awarded.
· Clinical development progress
· Business development progress
· Commercialization progress
· Financing of the company
Dr. Schuh’s employment agreement allows for an annual bonus equal to 50% of his base compensation. The Compensation Committee believed that Dr. Schuh did an outstanding job during 2014 in a challenging environment with limited resources.
In making its determination as to whether Dr. Schuh achieved his performance objectives for awarding 2014 bonus, the Compensation Committee looked at the above-mentioned performance objectives in totality and what the achievement of those performance objectives meant to us and our business. The Compensation Committee did not assign actual levels of achievement to each objective.
2015 Bonus Criteria
As of February 27, 2015, the Compensation Committee had not yet determined the performance criteria for Dr. Schuh’s 2015 bonus.
Compensation Risk Management
We have considered the risk associated with our compensation policies and practices for all employees, and we believe we have designed our compensation policies and practices in a manner that does not create incentives that could lead to excessive risk taking that would have a material adverse effect on us.
SUMMARY COMPENSATION TABLE
The following table provides certain summary information concerning compensation awarded to, earned by or paid to our Principal Executive Officer and the two other highest paid executive officers whose total annual salary and bonus exceeded $100,000 (collectively, the “named executive officers”) for fiscal year 2014.
|
Name & Principal Position |
|
Year |
|
Salary ($) |
|
Bonus ($) |
|
Option |
|
Total ($) |
|
|
Dr. Antonius Schuh, CEO (2) |
|
2014 |
|
385,000 |
|
154,000 |
|
399,999 |
|
938,999 |
|
|
|
|
2013 |
|
323,125 |
|
210,000 |
|
1,212,964 |
|
1,746,089 |
|
|
|
|
2012 |
|
275,000 |
|
137,500 |
|
468,916 |
|
881,416 |
|
|
Stephen Zaniboni, CFO (3) |
|
2014 |
|
242,000 |
|
96,800 |
|
263,400 |
|
602,200 |
|
|
|
|
2013 |
|
201,750 |
|
132,000 |
|
492,727 |
|
826,477 |
|
|
|
|
2012 |
|
199,333 |
|
80,000 |
|
107,331 |
|
386,664 |
|
|
Mark Erlander, CSO (4) |
|
2014 |
|
298,769 |
|
131,200 |
|
921,400 |
|
1,351,369 |
|
|
|
|
2013 |
|
169,692 |
|
100,000 |
|
1,366,187 |
|
1,635,879 |
|
(1) Amounts shown in this column do not reflect dollar amounts actually received by our named executive officers. Instead, these amounts represent the aggregate grant date fair value of stock option awards determined in accordance with FASB ASC Topic 718. The valuation assumptions used in determining 2014, 2013, and 2012 amounts are described in Note 6 to our financial statements included in our Annual Reports on Form 10-K for the fiscal years ended December 31, 2014, 2013, and 2012. Our named executive officers will only realize compensation to the extent the trading price of our common stock is greater than the exercise price of such stock options.
(2) Dr. Schuh was appointed CEO in October 2011.
(3) Mr. Zaniboni was appointed CFO in February 2012.
(4) Mr. Erlander was appointed CSO in January 2013.
OUTSTANDING EQUITY AWARDS AT FISCAL YEAR-END
The following table sets forth information for the named executive officers regarding the number of shares subject to both exercisable and unexercisable stock options, as well as the exercise prices and expiration dates thereof, as of December 31, 2014.
|
Name |
|
Number of |
|
Number of |
|
Weighted Average |
|
Option |
| |
|
Dr. Antonius Schuh (1) |
|
537,499 |
|
520,834 |
|
$ |
3.92 |
|
October 4, 2021-December 11, 2024 |
|
|
Stephen Zaniboni(2) |
|
110,834 |
|
225,833 |
|
$ |
4.46 |
|
February 1, 2022- December 11, 2024 |
|
|
Mark Erlander (3) |
|
85,000 |
|
490,000 |
|
$ |
5.12 |
|
September 13, 2022 — December 11, 2024 |
|
(1) The unexercisable options of 520,834 vest as follows: 158,334 on October 4, 2015; 50,000 each on June 24, 2015, 2016 and 2017 and 12,500 each on December 11, 2015, 2016 and 2017; 43,750 each on December 11, 2015, 2016, 2017 and 2018.
(2) The unexercisable options of 225,833 vest as follows: 41,666 on February 1, 2015 and 2016; 15,000 each on June 24, 2015, 2016 and 2017; 27,500 each on December 11, 2015, 2016, 2017 and 15,000 on December 11, 2018.
(3) The unexercisable options of 490,000 vest as follows: 1,666 on September 12, 2015; 3,334 on December 10, 2015; 50,000 each on January 28, 2015, 2016 and 2017; 40,000 each on December 11, 2015, 2016, 2017 and 15,000 on December 11, 2018; 50,000 each on July 16, 2015, 2016, 2017 and 2018.
DIRECTOR COMPENSATION
The following table sets forth summary information concerning the total compensation paid to our non-employee directors in 2014 for services to our company.
|
Name |
|
Fees Earned or Paid |
|
Option |
|
Total |
| |||
|
Thomas H. Adams(2) |
|
$ |
50,000 |
|
$ |
121,713 |
|
$ |
171,713 |
|
|
John P. Brancaccio(3) |
|
$ |
66,000 |
|
$ |
234,911 |
|
$ |
300,911 |
|
|
Gary S. Jacob(4) |
|
$ |
50,000 |
|
$ |
199,616 |
|
$ |
249,616 |
|
|
Stanley Tennant (5) |
|
$ |
52,000 |
|
$ |
184,395 |
|
$ |
236,395 |
|
|
Paul Billings (6) |
|
$ |
37,000 |
|
$ |
144,849 |
|
$ |
181,849 |
|
|
Rodney Markin (7) |
|
$ |
34,250 |
|
$ |
199,180 |
|
$ |
233,430 |
|
|
Carl Feldbaum (8) |
|
$ |
— |
|
$ |
103,200 |
|
$ |
103,200 |
|
(1) Amounts shown in this column do not reflect dollar amounts actually received by our non-employee directors. Instead, these amounts represent the aggregate grant date fair value of stock option awards determined in accordance with FASB ASC Topic 718. The valuation assumptions used in determining 2014 amounts are described in Note 6 to our financial statements included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2014. Our named executive officers will only realize compensation to the extent the trading price of our common stock is greater than the exercise price of such stock options.
(2) As of December 31, 2014, 342,849 stock options were outstanding, of which 324,565 were exercisable.
(3) As of December 31, 2014, 121,701 stock options were outstanding, of which 103,417 were exercisable.
(4) As of December 31, 2014, 123,672 stock options were outstanding, of which 105,388 were exercisable.
(5) As of December 31, 2014, 61,158 stock options were outstanding, of which 42,874 were exercisable.
(6) As of December 31, 2014, 51,230 stock options were outstanding, of which 23,000 were exercisable.
(7) As of December 31, 2014, 36,230 stock options were outstanding, of which none were exercisable.
(8) As of December 31, 2014, 24,000 stock options were outstanding, of which none were exercisable.
Employment Agreements
In January 2013, we entered into an employment agreement with Mark Erlander, Ph.D. in which he agreed to serve as Chief Scientific Officer. Dr. Erlander’s initial salary was $200,000 per year, increased to $260,000 and to $360,000 per year in 2014. Dr. Erlander is eligible to receive a cash bonus of up to 50% of his base salary per year at the discretion of the Compensation Committee based on goals mutually agreed upon by Dr. Erlander, the CEO and the Board of Directors. In connection with his employment, Dr. Erlander was granted a stock option to purchase 200,000 shares of common stock at an exercise price of $7.04. The option vests ratably over a four year period. If we terminate Dr. Erlander without cause, he is entitled to severance benefits equal to six months of his base salary.
During 2012, we entered into an executive agreement with Steve Zaniboni in which he agreed to serve as our Chief Financial Officer. The term of the agreement is effective as of February 1, 2012 and continues until February 1, 2013 and is automatically renewed for successive one year periods at the end to each term. Mr. Zaniboni’s initial compensation was $200,000 per year and was increased to $242,000 in 2013. In December 2014, Mr. Zaniboni’s salary was increased to $290,000. Mr. Zaniboni is eligible to receive a cash bonus of up to 50% of his base salary per year based on meeting certain performance objectives and bonus criteria. Upon entering the agreement, Mr. Zaniboni was granted 166,667 non-qualified stock options which have an exercise price of $3.60 per share and vest annually in equal amounts over a period of four years.
If the executive agreement is terminated by us for cause or as a result of Mr. Zaniboni’s death or permanent disability or if Mr. Zaniboni terminates his agreement voluntarily, Mr. Zaniboni shall receive a lump sum equal to (i) any portion of unpaid base compensation then due for periods prior to termination, (ii) any bonus or realization bonus earned but not yet paid through the date of termination and (iii) all expenses reasonably incurred by Mr. Zaniboni prior to date of termination. If the executive agreement is terminated by us without cause Mr. Zaniboni shall receive a severance payment equal to base compensation for three months if termination occurs ten months after the effective date of the agreement and six months if termination occurs subsequent to ten months from the effective date. If the executive agreement is terminated as a result of a change of control, Mr. Zaniboni shall receive a severance payment equal to base compensation for twelve months and all unvested stock options shall immediately vest and become fully exercisable for a period of six months following the date of termination.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding beneficial ownership of shares of our common stock as of
February 27, 2015 by (i) each person known to beneficially own more than 5% of our outstanding common stock, (ii) each of our directors, (iii) our named executive officers and (iv) all directors and executive officers as a group. Except as otherwise indicated, the persons named in the table have sole voting and investment power with respect to all shares beneficially owned, subject to community property laws, where applicable.
|
Name of Beneficial Owner |
|
Amount and nature of |
|
Percentage(2) |
| |
|
Executive officers and directors: |
|
|
|
|
|
|
|
Thomas Adams |
|
704,258 |
(3) |
2.9 |
|
|
|
Antonius Schuh |
|
537,499 |
(4) |
2.2 |
|
|
|
Paul Billings |
|
27,077 |
(4) |
* |
|
|
|
John Brancaccio |
|
138,604 |
(5) |
* |
|
|
|
Gary Jacob |
|
246,409 |
(6) |
1.0 |
|
|
|
Stanley Tennant |
|
308,157 |
(7) |
1.3 |
|
|
|
Rodney S. Markin |
|
12,077 |
(4) |
* |
|
|
|
Stephen Zaniboni |
|
152,501 |
(4) |
* |
|
|
|
Mark Erlander |
|
135,000 |
(4) |
* |
|
|
|
|
|
|
|
|
|
|
|
All Officers and Directors as a Group (9 persons) |
|
2,261,582 |
(8) |
8.8 |
|
|
|
5% or greater holders: |
|
|
|
|
|
|
|
Bridger Management, LLC |
|
2,587,301 |
(9) |
10.7 |
|
|
|
|
|
|
|
|
|
|
|
R. Merrill Hunter |
|
1,510,834 |
(10) |
6.1 |
|
|
|
Gabriele Cerrone |
|
1,569,404 |
(9) |
6.3 |
|
|
*less than 1%
(1) The address of each person is c/o Trovagene, Inc., 11055 Flintkote Avenue, Suite A, San Diego, CA 92121 unless otherwise indicated herein.
(2) The calculation in this column is based upon 24,123,790 shares of common stock outstanding on February 27, 2015. Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to the subject securities. Shares of common stock that are currently exercisable or exercisable within 60 days of February 27, 2015 are deemed to be beneficially owned by the person holding such securities for the purpose of computing the percentage beneficial ownership of such person, but are not treated as outstanding for the purpose of computing the percentage beneficial ownership of any other person.
(3) Includes (i) 332,086 shares of common stock issuable upon exercise of stock options and (ii) 45,686 shares of common stock issuable upon exercise of warrants.
(4) Consists of shares of common stock issuable upon exercise of stock options.
(5) Includes (i) 110,938 shares of common stock issuable upon exercise of stock options and (ii) 13,833 shares of common stock issuable upon exercise of warrants.
(6) Includes (i) 112,909 shares of common stock issuable upon exercise of stock options and (ii) 10,500 shares of common stock issuable upon exercise of warrants.
(7) Includes (i) 75,000 shares of common stock issuable upon exercise of warrants and (ii) 50,395 shares of common stock exercisable upon exercise of stock options.
(8) Includes (i) 1,470,482 shares of common stock issuable upon exercise of stock options and (ii) 145,019 shares of common stock issuable upon exercise of warrants.
(9) As per the Schedule 13G/A filed February 17, 2015.
(10) Includes 666,667 shares of common stock issuable upon exercise of warrants.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
On January 1, 2015, we entered into a consulting agreement with Tom Adams, our Chairman, pursuant to which Dr. Adams will provide consulting services to us in connection with applying our technology to infectious diseases. The agreement is for a term of one year and Dr. Adams shall be paid $9,500 per month for his services.
Any future transactions with officers, directors or 5% stockholders will be on terms no less favorable to us than could be obtained from independent parties. Any affiliated transactions must be approved by a majority of our independent and disinterested directors who have access to our counsel or independent legal counsel at our expense.
Board Determination of Independence
Our board of directors has determined that a majority of the board consists of members who are currently “independent” as that term is defined under current listing standards of NASDAQ.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Audit and Non-Audit Fees
The aggregate fees billed to us by BDO USA, LLP, our independent registered public account firm, for the indicated services for each of the last two fiscal years were as follows:
|
|
|
2014 |
|
2013 |
| ||
|
Audit fees (1) |
|
$ |
192,318 |
|
$ |
284,720 |
|
|
Tax fees (2) |
|
16,264 |
|
35,646 |
| ||
|
|
|
$ |
208,582 |
|
$ |
320,366 |
|
(1) Audit fees consist of fees for professional services performed by BDO USA, LLP for the audit and review of our financial statements included in SEC filings, and services that are normally provided in connection with regulatory filings or engagements.
(2) Tax fees consist of fees for professional services performed by BDO USA, LLP with respective to tax compliance.
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Auditors
Consistent with SEC policies and guidelines regarding audit independence, the Audit Committee is responsible for the pre-approval of all audit and permissible non-audit services provided by our principal accountants on a case-by-case basis. Our Audit Committee has established a policy regarding approval of all audit and permissible non-audit services provided by our principal accountants. No non-audit services were performed by our principal accountants during the fiscal years ended December 31, 2014 and 2013. Our Audit Committee pre-approves these services by category and service. Our Audit Committee has pre-approved all of the services provided by our principal accountants.
ITEM 15. EXHIBITS
|
Exhibit |
|
Description of Exhibit |
|
|
|
(a)(1)Financial Statements |
|
|
|
|
|
|
|
The financial statements required by this item are submitted in a separate section beginning on page F-1 of this annual report. |
|
b) Exhibits | ||
|
|
|
|
|
Exhibit |
|
Description |
|
1.2 |
|
Controlled Equity Offering SM Sales Agreement dated January 25, 2013 by and between Trovagene, Inc. and Cantor Fitzgerald & Co. (incorporated by reference to Exhibit 1.2 to Form S-3 filed on January 25, 2013). |
|
|
|
|
|
3.1 |
|
Amended and Restated Certificate of Incorporation of Trovagene, Inc. (incorporated by reference to Exhibit 3.1 to the Company’s Form 10-12G filed on November 25, 2011). |
|
|
|
|
|
3.2 |
|
Certificate of Amendment of Amended and Restated Certificate of Incorporation of Trovagene, Inc. (incorporated by reference to Appendix B to Trovagene, Inc.’s Proxy Statement on Schedule 14A filed March 20, 2012). |
|
|
|
|
|
3.3 |
|
By-Laws of Trovagene, Inc. (incorporated by reference to Exhibit 3.2 to the Company’s Form 10-12G filed on November 25, 2011). |
|
|
|
|
|
4.1 |
|
Form of Common Stock Certificate of Trovagene, Inc. (incorporated by reference to Exhibit 4.1 to the Company’s Form 10-12G filed on November 25, 2011). |
|
|
|
|
|
4.2 |
|
2004 Stock Option Plan (incorporated by reference to Exhibit 4.3 to the Company’s Current Report on Form 8-K filed on July 19, 2004)+ |
|
|
|
|
|
4.3 |
|
Form of Registration Rights Agreement (incorporated by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K, filed on April 16, 2012). |
|
|
|
|
|
4.4 |
|
Form of Warrant Agency Agreement by and between Trovagene, Inc. and Broadridge Corporate Issuer Solutions, Inc. and Form of Warrant Certificate (incorporated by reference to Exhibit 4.5 to Amendment No. 3 to Form S-1 filed on May 22, 2012). |
|
|
|
|
|
4.5 |
|
Form of Unit Agency Agreement by and between Trovagene, Inc. and Broadridge Corporate Issuer Solutions, Inc. (incorporated by reference to Exhibit 4.6 to Amendment No. 3 to Form S-1 filed on May 22, 2012). |
|
|
|
|
|
10.2 |
|
Executive Agreement between Trovagene, Inc. and Antonius Schuh dated October 4, 2011 (incorporated by reference to Exhibit 10.2 to the Company’s Form 10-12G filed on November 25, 2011).+ |
|
|
|
|
|
10.3 |
|
Summary of Terms of Lease Agreement dated as of October 28, 2009 between Trovagene, Inc. and BMR-Sorrento West LLC (incorporated by reference to Exhibit 10.3 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.4 |
|
Form of First Amendment to Standard Industrial Net Lease dated September 28, 2011 between Trovagene, Inc. and BMR-Sorrento West LLC (incorporated by reference to Exhibit 10.4 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
10.5 |
|
Form of Second Amendment to Standard Industrial Net Lease dated October 2011 between Trovagene, Inc. and BMR-Sorrento West LLC (incorporated by reference to Exhibit 10.5 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.6 |
|
Form of Third Amendment to Standard Industrial Net Lease dated October 22, 2012 between Trovagene, Inc. and BMR-Sorrento West , LP. |
|
|
|
|
|
10.7 |
|
Form of Fourth Amendment to Standard Industrial Net Lease dated December 2, 2013 between Trovagene, Inc. and BMR-Coast 9 LP. |
|
|
|
|
|
10.8 |
|
Form of Fifth Amendment to Standard Industrial Net Lease dated May 14, 2014 between Trovagene, Inc. and BMR-Coast 9 LP. |
|
|
|
|
|
10.9 |
|
Co-Exclusive Sublicense Agreement dated October 22, 2007 between Trovagene, Inc. and Asuragen, Inc. (incorporated by reference to Exhibit 10.6 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.10 |
|
Amendment to Co-Exclusive Sublicense Agreement dated June 1, 2010 between Trovagene, Inc. and Asuragen, Inc. (incorporated by reference to Exhibit 10.7 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.11 |
|
Sublicense Agreement dated as of August 27, 2007 between Trovagene, Inc. and Ipsogen SAS (incorporated by reference to Exhibit 10.8 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.12 |
|
Amendment to Co-Exclusive Sublicense Agreement dated as of September 1, 2010 between Trovagene, Inc. and Ipsogen SAS (incorporated by reference to Exhibit 10.9 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.13 |
|
Sublicense Agreement dated as of July 20, 2011 between Trovagene, Inc. and Fairview Health Services (incorporated by reference to Exhibit 10.11 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.14 |
|
Sublicense Agreement dated as of December 1, 2008 by and between Trovagene, Inc. and InVivoScribe Technologies, Inc. (incorporated by reference to Exhibit 10.13 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.15 |
|
Sublicense Agreement dated as of August 25, 2008 by and between Trovagene, Inc. and Laboratory Corporation of America Holdings. (incorporated by reference to Exhibit 10.14 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.16 |
|
Form of Sublicense Agreement effective as of February 8, 2011 by and between Trovagene, Inc. and MLL Munchner Leukamielabor GmbH. (incorporated by reference to Exhibit 10.15 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.17 |
|
Sublicense Agreement effective as of June 15, 2010 by and between Trovagene, Inc. and Skyline Diagnostics BV (incorporated by reference to Exhibit 10.16 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.18 |
|
Exclusive License Agreement effective as of December 12, 2011 by and between Columbia University and Trovagene, Inc. (incorporated by reference to Exhibit 10.20 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
10.19 |
|
Form of Exclusive License Agreement effective as of October 2011 by and between Gianluca Gaidano, Robert Foa and Davide Rossi and Trovagene, Inc. (incorporated by reference to Exhibit 10.21 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.20 |
|
Executive Agreement between Trovagene, Inc. and Steve Zaniboni dated February 1, 2012 (incorporated by reference to Exhibit 10.22 to the Company’s Form 10-12G/A filed on February 15, 2012). + |
|
|
|
|
|
10.21 |
|
Exclusive License Agreement effective as of May 2006 by and between Brunangelo Falini, Cristina Mecucci and Trovagene, Inc. (incorporated by reference to Exhibit 10.23 to the Company’s Form 10- |
|
|
|
12G/A filed on February 15, 2012). |
|
|
|
|
|
10.22 |
|
Form of First Amendment to Exclusive License Agreement effective as of August 2010 by and among Brunangelo Falini, Cristina Mecucci and Trovagene, Inc. (incorporated by reference to Exhibit 10.24 to the Company’s Form 10-12G/A filed on February 15, 2012). |
|
|
|
|
|
10.23 |
|
Form of Securities Purchase Agreement dated as of July 30, 2013 (incorporated by reference to Exhibit 10.1 to the Company’s Form 8-K filed on July 31, 2013). |
|
|
|
|
|
10.24 |
|
Research Agreement between Trovagene, Inc. and Illumina, Inc. dated June 20, 2013 (incorporated by reference to Exhibit 10.1 to the Company’s Form 10-Q filed on August 14, 2013)* |
|
|
|
|
|
14 |
|
Code of Business Conduct and Ethics Amended and Restated 2011 (incorporated by reference to Exhibit 14 to the Company’s Form 10-12G filed on November 25, 2011). |
|
|
|
|
|
21 |
|
List of Subsidiaries. |
|
|
|
|
|
23.1 |
|
Consent of BDO USA, LLP |
|
|
|
|
|
31.1 |
|
Certification of Chief Executive Officer required under Rule 13a-14(a)/15d-14(a) under the Exchange Act. |
|
|
|
|
|
31.2 |
|
Certification of Principal Financial Officer required under Rule 13a-14(a)/15d-14(a) under the Exchange Act. |
|
|
|
|
|
32.1 |
|
Certification of Chief Executive Officer pursuant to 18 U.S.C Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
|
32.2 |
|
Certification of Principal Financial Officer pursuant to 18 U.S.C Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
|
|
|
|
|
101 |
|
Financial statements from the annual report on Form 10-K of Trovagene for the year ended December 31, 2014, formatted in Extensible Business Reporting Language (XBRL): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations and Comprehensive Loss, (iii) the Consolidated Statement of Stockholders’ Equity (Deficiency) (iv) the Consolidated Statements of Cash Flows and (v) the Notes to Consolidated Financial Statements. |
|
|
|
+ Indicates a management contract or compensatory plan or arrangement *Portions of this exhibit were omitted and filed separately with the U.S. Securities and Exchange Commission pursuant to a request for confidential treatment. |
SIGNATURES
In accordance with Section 13 or 15(d) of the Exchange Act, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
TROVAGENE, INC. |
|
|
|
|
|
|
|
|
/s/ Dr. Antonius Schuh |
|
March 12, 2015 |
Chief Executive Officer |
In accordance with the Exchange Act, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
SIGNATURE |
|
TITLE |
|
DATE |
|
|
|
|
|
|
|
|
|
/s/ Dr. Antonius Schuh |
|
Chief Executive Officer and Director |
|
March 12, 2015 |
|
|
|
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Stephen Zaniboni |
|
Chief Financial Officer |
|
March 12, 2015 |
|
|
|
|
(Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Thomas H. Adams |
|
Chairman of the Board |
|
March 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ John P. Brancaccio |
|
Director |
|
March 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Gary S. Jacob |
|
Director |
|
March 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Paul Billings |
|
Director |
|
March 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Stanley Tennant |
|
Director |
|
March 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Rodney S. Markin |
|
Director |
|
March 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Carl Feldbaum |
|
Director |
|
March 12, 2015 |
|
TROVAGENE, INC.
Index to Consolidated Financial Statements
|
F-2 | |
|
|
|
|
Consolidated Balance Sheets as of December 31, 2014 and 2013 |
F-3 |
|
|
|
|
F-4 | |
|
|
|
|
F-5 | |
|
|
|
|
F-7 | |
|
|
|
|
F-8 |
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Trovagene, Inc.
San Diego, California
We have audited the accompanying consolidated balance sheets of Trovagene, Inc. and Subsidiaries’ (“Trovagene”) as of December 31, 2014 and 2013 and the related consolidated statements of operations and comprehensive loss, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Trovagene, Inc. and Subsidiaries at December 31, 2014 and 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Trovagene’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and our report dated March 12, 2015 expressed an unqualified opinion thereon.
|
/s/ BDO USA, LLP |
|
|
San Diego, California |
|
|
March 12, 2015 |
|
Trovagene, Inc. and Subsidiaries
|
|
|
December 31, |
|
December 31, |
| ||
|
|
|
2014 |
|
2013 |
| ||
|
|
|
|
|
|
| ||
|
Assets |
|
|
|
|
| ||
|
Current assets: |
|
|
|
|
| ||
|
Cash and cash equivalents |
|
$ |
27,293,798 |
|
25,836,937 |
| |
|
Accounts receivable |
|
56,694 |
|
78,994 |
| ||
|
Prepaid expenses and other assets |
|
369,259 |
|
152,789 |
| ||
|
Total current assets |
|
27,719,751 |
|
26,068,720 |
| ||
|
Property and equipment, net |
|
840,387 |
|
750,565 |
| ||
|
Other assets |
|
336,708 |
|
336,450 |
| ||
|
Total Assets |
|
$ |
28,896,846 |
|
27,155,735 |
| |
|
|
|
|
|
|
| ||
|
Liabilities and Stockholders’ Equity |
|
|
|
|
| ||
|
Current liabilities: |
|
|
|
|
| ||
|
Accounts payable |
|
747,799 |
|
286,608 |
| ||
|
Accrued expenses |
|
1,841,808 |
|
1,524,092 |
| ||
|
Current portion of long-term debt |
|
1,898,548 |
|
198,166 |
| ||
|
Total current liabilities |
|
4,488,155 |
|
2,008,866 |
| ||
|
Long-term debt, less current portion |
|
13,053,117 |
|
322,998 |
| ||
|
Derivative financial instruments |
|
3,006,021 |
|
4,431,871 |
| ||
|
Total liabilities |
|
20,547,293 |
|
6,763,735 |
| ||
|
|
|
|
|
|
| ||
|
Commitments and contingencies (Note 11) |
|
|
|
|
| ||
|
Stockholders’ equity |
|
|
|
|
| ||
|
|
|
|
|
|
| ||
|
Preferred stock, $0.001 par value, 20,000,000 shares authorized, 60,600 shares outstanding at December 31, 2014 and 2013, designated as Series A Convertible Preferred Stock with liquidation preference of $606,000 at December 31, 2014 and 2013 |
|
60 |
|
60 |
| ||
|
|
|
|
|
|
| ||
|
Common stock, $0.0001 par value, 150,000,000 shares authorized at December 31, 2014 and 2013; 18,915,793 and 18,902,782 issued and outstanding at December 31, 2014 and 2013, respectively |
|
1,891 |
|
1,890 |
| ||
|
|
|
|
|
|
| ||
|
Additional paid-in capital |
|
89,739,511 |
|
87,433,460 |
| ||
|
Deficit accumulated during |
|
(81,391,909 |
) |
(67,043,410 |
) | ||
|
Total stockholders’ equity |
|
8,349,553 |
|
20,392,000 |
| ||
|
Total Liabilities and Stockholders’ Equity |
|
$ |
28,896,846 |
|
$ |
27,155,735 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Trovagene, Inc. and Subsidiaries
Consolidated Statements of Operations and Comprehensive Loss
|
|
|
Year Ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Royalty income |
|
$ |
270,178 |
|
$ |
259,246 |
|
$ |
175,404 |
|
|
Milestone fees |
|
— |
|
— |
|
150,000 |
| |||
|
License fees |
|
10,000 |
|
— |
|
125,000 |
| |||
|
Total revenues |
|
280,178 |
|
259,246 |
|
450,404 |
| |||
|
Costs and expenses: |
|
|
|
|
|
|
| |||
|
Cost of revenue |
|
15,441 |
|
— |
|
— |
| |||
|
Research and development |
|
6,664,906 |
|
3,947,589 |
|
1,920,298 |
| |||
|
Selling and marketing |
|
2,734,903 |
|
1,530,160 |
|
506,535 |
| |||
|
General and administrative |
|
5,810,087 |
|
5,472,038 |
|
2,872,727 |
| |||
|
Total operating expenses |
|
15,225,337 |
|
10,949,787 |
|
5,299,560 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Loss from operations |
|
(14,945,159 |
) |
(10,690,541 |
) |
(4,849,156 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Interest income |
|
12,239 |
|
3,663 |
|
— |
| |||
|
Interest expense |
|
(843,259 |
) |
(17,005 |
) |
— |
| |||
|
Gain (loss) on disposal of equipment |
|
24,845 |
|
(22,941 |
) |
4,000 |
| |||
|
Gain (loss) from change in fair value of derivative instruments—warrants |
|
1,425,850 |
|
(1,084,114 |
) |
(6,720,805 |
) | |||
|
Net loss and comprehensive loss |
|
(14,325,484 |
) |
(11,810,938 |
) |
(11,565,961 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Preferred stock dividend |
|
(23,015 |
) |
(29,840 |
) |
(38,240 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Net loss and comprehensive loss attributable to common stockholders |
|
$ |
(14,348,499 |
) |
$ |
(11,840,778 |
) |
$ |
(11,604,201 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per common share - basic |
|
$ |
(0.76 |
) |
$ |
(0.70 |
) |
$ |
(0.89 |
) |
|
Net loss per common share - diluted |
|
$ |
(0.88 |
) |
$ |
(0.70 |
) |
$ |
(0.89 |
) |
|
|
|
|
|
|
|
|
| |||
|
Weighted average shares outstanding - basic |
|
18,904,280 |
|
16,978,212 |
|
13,066,600 |
| |||
|
Weighted average shares outstanding - diluted |
|
19,071,112 |
|
16,978,212 |
|
13,066,600 |
| |||
The accompanying notes are an integral part of these consolidated financial statements.
Trovagene, Inc. and Subsidiaries
Consolidated Statements of Stockholders’ Equity (Deficiency)
|
|
|
Preferred |
|
Preferred |
|
Common |
|
Common |
|
Additional |
|
Deficit |
|
Total |
| |||||
|
Balance, December 31, 2011 |
|
95,600 |
|
$ |
96 |
|
10,737,026 |
|
$ |
1,073 |
|
$ |
39,365,994 |
|
$ |
(43,598,431 |
) |
$ |
(4,231,268 |
) |
|
Units issued via registered underwritten direct public offering and private placement of units |
|
— |
|
— |
|
4,383,333 |
|
438 |
|
16,899,562 |
|
— |
|
16,900,000 |
| |||||
|
Fees and expenses related to financing transactions |
|
— |
|
— |
|
— |
|
— |
|
(1,576,452 |
) |
— |
|
(1,576,452 |
) | |||||
|
Derivative liability-fair value of warrants and price protected units issued |
|
— |
|
— |
|
— |
|
— |
|
(1,796,610 |
) |
— |
|
(1,796,610 |
) | |||||
|
Correction of error in derivative liability—fair value of warrants price protected units issued |
|
— |
|
— |
|
— |
|
— |
|
274,967 |
|
— |
|
274,967 |
| |||||
|
Warrants reclassified to additional paid in capital |
|
— |
|
— |
|
— |
|
— |
|
3,317,463 |
|
— |
|
3,317,463 |
| |||||
|
Issuance of common stock and warrant to shareholder as finder’s fees |
|
— |
|
— |
|
214,100 |
|
21 |
|
(21 |
) |
— |
|
— |
| |||||
|
Issuance of common stock in connection with Asset Purchase Agreement with MultiGen Diagnostics, Inc. |
|
— |
|
— |
|
125,000 |
|
13 |
|
187,487 |
|
— |
|
187,500 |
| |||||
|
Issuance of common stock in connection with consulting services |
|
— |
|
— |
|
9,916 |
|
1 |
|
22,380 |
|
— |
|
22,381 |
| |||||
|
Issuance of warrants in connection with advisory services |
|
— |
|
— |
|
— |
|
— |
|
142,508 |
|
— |
|
142,508 |
| |||||
|
Stock based compensation |
|
— |
|
— |
|
— |
|
— |
|
532,140 |
|
— |
|
532,140 |
| |||||
|
Issuance of common stock upon exercise of stock options |
|
— |
|
— |
|
200 |
|
— |
|
600 |
|
— |
|
600 |
| |||||
|
Issuance of common stock upon net exercise of warrant |
|
— |
|
— |
|
8,602 |
|
1 |
|
(1 |
) |
— |
|
— |
| |||||
|
Preferred stock dividend |
|
— |
|
— |
|
— |
|
|
|
|
|
(38,240 |
) |
(38,240 |
) | |||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(11,565,961 |
) |
(11,565,961 |
) | |||||
|
Balance, December 31, 2012 |
|
95,600 |
|
$ |
96 |
|
15,478,177 |
|
$ |
1,547 |
|
$ |
57,370,017 |
|
$ |
(55,202,632 |
) |
$ |
2,169,028 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Trovagene, Inc. and Subsidiaries
Consolidated Statements of Stockholders’ Equity (Deficiency)
|
|
|
Preferred |
|
Preferred |
|
Common |
|
Common |
|
Additional |
|
Deficit |
|
Total |
| |||||
|
Sale of common stock, net of expenses |
|
— |
|
— |
|
2,631,332 |
|
263 |
|
18,829,381 |
|
— |
|
18,829,644 |
| |||||
|
Issuance of warrants in connection with services |
|
— |
|
— |
|
— |
|
— |
|
198,791 |
|
— |
|
198,791 |
| |||||
|
Stock based compensation |
|
— |
|
— |
|
— |
|
— |
|
1,979,364 |
|
— |
|
1,979,364 |
| |||||
|
Derivative liability - Warrants reclassified to additional paid in capital |
|
— |
|
— |
|
— |
|
— |
|
5,417,871 |
|
— |
|
5,417,871 |
| |||||
|
Issuance of common stock upon conversion of preferred stock |
|
(35,000 |
) |
(36 |
) |
36,458 |
|
4 |
|
32 |
|
— |
|
— |
| |||||
|
Issuance of common stock upon net exercise of warrant |
|
— |
|
— |
|
7,284 |
|
1 |
|
(1 |
) |
— |
|
— |
| |||||
|
Issuance of common stock upon exercise of warrants |
|
— |
|
— |
|
715,743 |
|
72 |
|
3,599,759 |
|
— |
|
3,599,831 |
| |||||
|
Issuance of common stock upon net exercise of stock options |
|
— |
|
— |
|
22,955 |
|
2 |
|
(2 |
) |
— |
|
— |
| |||||
|
Issuance of common stock upon exercise of stock options |
|
— |
|
— |
|
10,833 |
|
1 |
|
38,248 |
|
— |
|
38,249 |
| |||||
|
Preferred stock dividend |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(29,840 |
) |
(29,840 |
) | |||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(11,810,938 |
) |
(11,810,938 |
) | |||||
|
Balance, December 31, 2013 |
|
60,600 |
|
$ |
60 |
|
18,902,782 |
|
$ |
1,890 |
|
$ |
87,433,460 |
|
$ |
(67,043,410 |
) |
$ |
20,392,000 |
|
|
Stock based compensation |
|
— |
|
— |
|
— |
|
— |
|
2,070,195 |
|
— |
|
2,070,195 |
| |||||
|
Issuance of warrant in connection with debt agreement |
|
— |
|
— |
|
— |
|
— |
|
235,857 |
|
— |
|
235,857 |
| |||||
|
Issuance of common stock upon net exercise of warrant |
|
— |
|
— |
|
13,011 |
|
1 |
|
(1 |
) |
— |
|
— |
| |||||
|
Preferred stock dividend |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(23,015 |
) |
(23,015 |
) | |||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(14,325,484 |
) |
(14,325,484 |
) | |||||
|
Balance, December 31, 2014 |
|
60,600 |
|
$ |
60 |
|
18,915,793 |
|
$ |
1,891 |
|
$ |
89,739,511 |
|
$ |
(81,391,909 |
) |
$ |
8,349,553 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Trovagene, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
|
|
|
Year |
|
Year |
|
Year |
| |||
|
|
|
|
|
|
|
|
| |||
|
Operating activities |
|
|
|
|
|
|
| |||
|
Net loss |
|
$ |
(14,325,484 |
) |
$ |
(11,810,938 |
) |
$ |
(11,565,961 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
| |||
|
Loss (gain) on disposal of fixed assets |
|
(24,845 |
) |
22,941 |
|
(4,000 |
) | |||
|
Depreciation and amortization |
|
234,813 |
|
130,520 |
|
41,842 |
| |||
|
Stock based compensation expense |
|
2,070,194 |
|
1,979,364 |
|
532,140 |
| |||
|
Amortization of debt costs |
|
248,799 |
|
— |
|
— |
| |||
|
Accretion of discount on debt |
|
57,117 |
|
— |
|
— |
| |||
|
Change in fair value of financial instruments |
|
(1,425,850 |
) |
1,084,114 |
|
6,720,805 |
| |||
|
Stock and warrant issued in connection with consulting services |
|
— |
|
198,791 |
|
164,889 |
| |||
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
| |||
|
Decrease (increase) in other assets |
|
(258 |
) |
25,631 |
|
— |
| |||
|
Decrease (increase) in accounts receivable |
|
22,300 |
|
89,387 |
|
(69,241 |
) | |||
|
(Increase) decrease in prepaid expenses |
|
(216,470 |
) |
(92,748 |
) |
(17,383 |
) | |||
|
Increase (decrease) in accounts payable and accrued expenses |
|
632,299 |
|
1,055,690 |
|
(737,752 |
) | |||
|
Net cash used in operating activities |
|
(12,727,385 |
) |
(7,317,248 |
) |
(4,934,661 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Investing activities: |
|
|
|
|
|
|
| |||
|
Capital expenditures |
|
(363,290 |
) |
(649,784 |
) |
(274,080 |
) | |||
|
Proceeds from disposals of capital equipment |
|
63,500 |
|
500 |
|
4,000 |
| |||
|
Net cash used in investing activities |
|
(299,790 |
) |
(649,284 |
) |
(270,080 |
) | |||
|
|
|
|
|
|
|
|
| |||
|
Financing activities |
|
|
|
|
|
|
| |||
|
Proceeds from sale of common stock, net of expenses |
|
— |
|
18,829,644 |
|
15,323,548 |
| |||
|
Proceeds from exercise of warrants |
|
— |
|
3,599,831 |
|
— |
| |||
|
Proceeds from exercise of options |
|
— |
|
38,249 |
|
600 |
| |||
|
Borrowings (repayments) under equipment line of credit |
|
(515,964 |
) |
515,964 |
|
— |
| |||
|
Borrowings under debt agreement |
|
15,000,000 |
|
— |
|
— |
| |||
|
Net cash provided by financing activities |
|
14,484,036 |
|
22,983,688 |
|
15,324,148 |
| |||
|
Net change in cash and cash equivalents |
|
1,456,861 |
|
15,017,156 |
|
10,119,407 |
| |||
|
Cash and cash equivalents—Beginning of period |
|
25,836,937 |
|
10,819,781 |
|
700,374 |
| |||
|
Cash and cash equivalents—End of period |
|
$ |
27,293,798 |
|
$ |
25,836,937 |
|
$ |
10,819,781 |
|
|
Supplementary disclosure of cash flow activity: |
|
|
|
|
|
|
| |||
|
Cash paid for taxes |
|
$ |
2,400 |
|
$ |
7,650 |
|
$ |
— |
|
|
Cash paid for interest |
|
$ |
425,256 |
|
$ |
9,459 |
|
$ |
— |
|
|
Supplemental disclosure of non-cash investing and financing activities: |
|
|
|
|
|
|
| |||
|
Warrants issued in connection with Loan and Security Agreement |
|
$ |
235,857 |
|
$ |
— |
|
$ |
— |
|
|
Issuance of 125,000 shares of common stock pursuant to Asset Purchase Agreement with Multigen Diagnostics, Inc. |
|
$ |
— |
|
$ |
— |
|
$ |
187,500 |
|
|
Reclassification of derivative financial instruments to additional paid in capital |
|
$ |
— |
|
$ |
(5,417,871 |
) |
$ |
(3,317,463 |
) |
|
Correction of error in derivative financial instruments |
|
$ |
— |
|
$ |
— |
|
$ |
(274,967 |
) |
|
Preferred stock dividends accrued |
|
$ |
23,015 |
|
$ |
29,840 |
|
$ |
38,240 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Trovagene, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
1. Business Overview and Liquidity
Trovagene, Inc. (“Trovagene” or the “Company”) is a molecular diagnostic company that focuses on the development and commercialization of a proprietary urine-based cell-free molecular diagnostic technology for use in disease detection and monitoring across a variety of medical disciplines. Trovagene’s primary internal focus is to leverage its novel urine-based molecular diagnostic platform to facilitate improvements in the field of oncology, while the Company’s external focus includes entering into collaborations to develop the Company’s technology in areas such as infectious disease, transplant medicine, and prenatal genetics. The Company’s goal is to improve treatment outcomes for cancer patients using its proprietary technology to detect and quantitatively monitor cell-free DNA in urine.
Underwritten Public Offering of Common Stock
On May 30, 2012, the Company completed an underwritten public offering in which an aggregate of 1,150,000 units, with each unit consisting of two shares of its common stock and one warrant to purchase one share of common stock were sold at a purchase price of $8.00 per unit. On June 13, 2012, the underwriters exercised their overallotment option in full for an additional 172,500 units. The Company raised a total of $9.1 million in net proceeds after deducting underwriting discounts and commissions of $0.7 million and offering expenses of $0.7 million. The units began trading on The NASDAQ Capital Market on May 30, 2012 under the symbol “TROVU”. The common stock and warrants became separately transferable upon the exercise in full of the underwriters’ overallotment. Each warrant has an exercise price of $5.32 per share, and expires five years from the closing of the offering. The warrants trade on The NASDAQ Capital Market under the symbol “TROVW”. Warrants issued in connection with the underwritten public offering and sale of units in May 2012 are not considered derivatives based on our analysis of the criteria of ASC 815, as the Company is not required to make any cash payments or net cash settlement to the registered holder in lieu of issuance of warrant shares.
To date, Trovagene’s efforts have been principally devoted to research and development, securing and protecting patents and raising capital. Through December 31, 2014, the Company has sustained cumulative net losses attributed to common stockholders of $81,391,909. The Company’s losses have resulted primarily from expenditures incurred in connection with research and development activities, stock based compensation expense, patent filing and maintenance expenses, outside accounting and legal services and regulatory, scientific and financial consulting fees, amortization and liquidated damages. To date, the Company has generated only limited revenue from operations and expects to incur additional losses to perform further research and development activities as well as selling and marketing expenses related to the diagnostic tests it has commercially available as of December 31, 2014.
Liquidity
The Company will need to continue to raise funds until it is able to generate revenues from operations sufficient to fund its development and commercial operations.
Cash used in operating activities was $12,727,385, $7,317,248 and $4,934,661 for the years ended December 31, 2014, 2013, and 2012, respectively. During the years ended December 31, 2014, 2013 and 2012, the Company incurred net loss and comprehensive loss attributable to common stockholders of $14,348,499, $11,840,778 and $11,604,201, respectively. The Company believes that it currently has adequate capital to continue operations for the subsequent one year business cycle. However, to carry the Company forward beyond the current business cycle until it can generate adequate cash flow from operations, additional cash resources will be necessary.
To date, Trovagene’s sources of cash have been primarily limited to the sale of debt and equity securities, as well as proceeds from warrant and option exercises. Net cash provided by financing activities for the years ended December 31, 2014, 2013 and 2012 was $14,484,036, $22,983,688 and $15,324,148, respectively. The Company cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that the Company can raise additional funds by issuing equity securities, the Company’s stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants that impact the Company’s ability to conduct its business.
If the Company is unable to raise additional capital when required or on acceptable terms, it may have to significantly delay, scale back or discontinue the development and/or commercialization of one or more of its product candidates. The Company may also be required to:
· Seek collaborators for product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; and
· Relinquish licenses or otherwise dispose of rights to technologies, product candidates or products that the Company would otherwise seek to develop or commercialize themselves, on unfavorable terms.
2. Basis of Presentation and Summary of Significant Accounting Policies
The accompanying consolidated financial statements of Trovagene, which include its wholly owned subsidiaries Xenomics, Inc., a California corporation, Xenomics Europa Ltd, (an inactive subsidiary formed in the United Kingdom and liquidated) and Etherogen, Inc., a Delaware corporation, have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). All intercompany balances and transactions have been eliminated.
On March 15, 2012, the Board of Directors and on April 27, 2012 a majority of the stockholders approved a proposal to amend the Company’s Amended and Restated Certificate of Incorporation to effect a reverse stock split of the Company’s issued and outstanding common stock at a ratio of not less than one-for-two and not greater than one-for-six at any time prior to April 27, 2013 at the discretion of the Board of Directors. On May 24, 2012, the Board of Directors approved a 1-for-6 reverse stock split of the Company’s issued and outstanding common stock effective on May 29, 2012. All the relevant information relating to number of shares and per share information contained in these consolidated financial statements has been retrospectively adjusted to reflect the reverse stock split for all periods presented.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents consist of operating accounts as of December 31, 2014 and 2013 on deposit with U.S. commercial banks. Cash equivalents are considered by the Company to be highly liquid investments purchased with original maturities of three months or less from the date of purchase. Cash and cash equivalents include money market accounts at December 31, 2014.
Concentration of credit risk
The Company maintains its cash in financial institutions, which at times may exceed the amount insured by the Federal Deposit Insurance Corporation (“FDIC”). All of the Company’s noninterest bearing cash balances were insured up to $250,000 at December 31, 2014 and December 31, 2013.
Revenues
We license and sublicense our patent rights to healthcare companies, medical laboratories and biotechnology partners. These agreements may involve multiple elements such as license fees, royalties and milestone payments. Revenue is recognized for each element when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable, and collection is reasonably assured.
· Up-front nonrefundable license fees pursuant to agreements under which we have no continuing performance obligations are recognized as revenues on the effective date of the agreement and when collection is reasonably assured.
· Minimum royalties are recognized as earned, and royalties in excess of minimum amounts are recognized upon receipt of payment when collection is assured.
· Milestone payments are recognized when both the milestone is achieved and the related payment is received.
Allowance for Doubtful Accounts
The Company reviews the collectability of accounts receivable based on an assessment of historic experience, current economic conditions, and other collection indicators. At December 31, 2014 and 2013, the Company has not recorded an allowance for doubtful accounts. When accounts are determined to be uncollectible, they are written off against the reserve balance and the reserve is reassessed. When payments are received on reserved accounts, they are applied to the individual’s account and the reserve is reassessed.
Derivative Financial Instruments—Warrants
The Company has issued common stock warrants in connection with the execution of certain equity financings. Such warrants are classified as derivative liabilities under the provisions of FASB ASC 815 Derivatives and Hedging (“ASC 815”) and are recorded at their fair market value as of each reporting period. Such warrants do not meet the exemption that a contract should not be considered a derivative instrument if it is (1) indexed to its own stock and (2) classified in stockholders’ equity. Changes in fair value of derivative liabilities are recorded in the consolidated statement of operations under the caption “Change in fair value of derivative instruments.”
The fair value of warrants is determined using the Black-Scholes option-pricing model using assumptions regarding volatility of Trovagene’s common share price, remaining life of the warrant, and risk-free interest rates at each period end. The Company thus uses model-derived valuations where inputs are observable in active markets to determine the fair value and accordingly classifies such warrants in Level 3 per ASC 820, Fair Value Measurements. At December 31, 2014 and 2013, the fair value of these warrants was $3,006,021 and $4,431,871, respectively, and were included in the derivative financial instruments liability on the balance sheet.
Stock-Based Compensation
ASC Topic 718 “Compensation—Stock Compensation” requires companies to measure the cost of employee services received in exchange for the award of equity instruments based on the estimated fair value of the award at the date of grant. The expense is recognized over the period during which an employee is required to provide services in exchange for the award. ASC Topic 718 did not change the way Trovagene accounts for non-employee stock-based compensation. Trovagene continues to account for shares of common stock, stock options and warrants issued to non-employees based on the fair value of stock, stock option or warrant, if that value is more reliably measurable than the fair value of the consideration or services received. The Company accounts for stock options issued and vesting to non-employees in accordance with ASC Topic 505-50 “Equity -Based Payment to Non-Employees” and accordingly the value of the stock compensation to non-employees is based upon the measurement date as determined at either a) the date at which a performance commitment is reached, or b) at the date at which the necessary performance to earn the equity instruments is complete. Accordingly the fair value of these options is being “marked to market” quarterly until the measurement date is determined.
Fair value of financial instruments
Financial instruments consist of cash and cash equivalents, accounts receivable, accounts payable, debt and derivative liabilities. We have adopted FASB ASC 820 Fair Value Measurements and Disclosures (“ASC 820”) for financial assets and liabilities that are required to be measured at fair value, and non-financial assets and liabilities that are not required to be measured at fair value on a recurring basis. These financial instruments are stated at their respective historical carrying amounts which approximate to fair value due to their short term nature as they reflect current market interest rates. Debt is stated at its respective historical carrying amounts which approximates fair value as they reflect current market interest rates.
In accordance with ASC subtopic 820-10, the Company measures certain assets and liabilities at fair value on a recurring basis using the three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. The three tiers include:
|
· |
|
Level 1 — |
|
Quoted prices for identical instruments in active markets. |
|
|
|
|
|
|
|
· |
|
Level 2 — |
|
Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations where inputs are observable or where significant value drivers are observable. |
|
|
|
|
|
|
|
· |
|
Level 3 — |
|
Instruments where significant value drivers are unobservable to third parties. |
Property, equipment and depreciation and amortization
Expenditures for additions, renewals and improvements are capitalized at cost. Depreciation and amortization is generally computed on a straight-line method based on the estimated useful lives of the related assets. Amortization of leasehold improvements is computed based on the shorter of the life of the asset or the term of the lease. The estimated useful lives of the major classes of depreciable assets are 3 to 5 years for lab equipment and furniture and fixtures. Expenditures for repairs and maintenance are charged to operations as incurred.
Impairment of Indefinite and Long-Lived Assets
The Company reviews its long and indefinite lived assets to determine if any event has occurred that may indicate its intangible assets with indefinite lives and other long-lived assets are potentially impaired. If indicators of impairment exist, the Company performs an impairment test to assess the recoverability of the affected assets by determining whether the carrying amount of such assets exceeds the undiscounted expected future cash flows. If the affected assets are not recoverable, the Company estimates the fair value of the assets and records an impairment loss if the carrying value of the assets exceeds the fair value. Factors that would indicate potential impairment include a significant decline in the Company’s stock price and market capitalization compared to its net book value, significant changes in the ability of a particular asset to generate positive cash flows, and significant changes in the Company’s strategic business objectives and utilization of a particular asset. The Company noted no indications of impairment for the years ended December 31, 2014, 2013, and 2012.
Income Taxes
Income taxes have been determined using the asset and liability approach of accounting for income taxes. Under this approach, deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. Deferred taxes result from differences between the financial statement and tax bases of Trovagene’s assets and liabilities and are adjusted for changes in tax rates and tax laws when changes are enacted. Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized. The assessment of whether or not a valuation allowance is required often requires significant judgment.
Contingencies
In the normal course of business, Trovagene is subject to loss contingencies, such as legal proceedings and claims arising out of its business, that cover a wide range of matters, including, among others, government investigations, shareholder lawsuits, product and environmental liability, and tax matters. In accordance with FASB ASC Topic 450, Accounting for Contingencies, Trovagene records such loss contingencies when it is probable that a liability has been incurred and the amount of loss can be reasonably estimated. Trovagene, in accordance with this guidance, does not recognize gain contingencies until realized.
Cost of Revenue
Cost of revenue represents the cost of materials, personnel costs, costs associated with processing specimens including pathological review, quality control analyses, and delivery charges necessary to render an individualized test result. Costs associated with performing tests are recorded as the tests are processed.
Research and Development
Research and development costs, which include expenditures in connection with an in-house research and development laboratory, salaries and staff costs, purchased in-process research and development, regulatory and scientific consulting fees, as well as contract research and insurance, are accounted for in accordance with ASC Topic 730-10-55-2, Research and Development. Also, as prescribed by this guidance, patent filing and maintenance expenses are considered legal in nature and therefore classified as general and administrative expense, if any.
While certain of our research and development costs may have future benefits, our policy of expensing all research and development expenditures is predicated on the fact that Trovagene has no history of successful commercialization of molecular diagnostic products to base any estimate of the number of future periods that would be benefited.
ASC Topic 730, Research and Development requires that non-refundable advance payments for goods or services that will be used or rendered for future research and development activities should be deferred and capitalized. As the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided, the deferred amounts are recognized as an expense. There were no non-refundable advance payments as of December 31, 2014 and 2013.
Net Loss Per Share
Basic and diluted net loss per share is presented in conformity with ASC Topic 260, Earnings per Share, for all periods presented. In accordance with this guidance, basic and diluted net loss per common share was determined by dividing net loss applicable to common stockholders by the weighted-average common shares outstanding during the period. Preferred dividends are included in income available to common stockholders in the computation of basic and diluted earnings per share. Shares used in calculating diluted net loss per common share exclude as antidilutive the following share equivalents:
|
|
|
December 31, |
| ||||
|
|
|
2014 |
|
2013 |
|
2012 |
|
|
Options to purchase Common Stock |
|
4,913,472 |
|
4,287,545 |
|
3,711,303 |
|
|
Warrants to purchase Common stock |
|
5,251,660 |
|
6,233,483 |
|
6,985,070 |
|
|
Series A Convertible Preferred Stock |
|
63,125 |
|
63,125 |
|
99,583 |
|
|
|
|
10,228,257 |
|
10,584,153 |
|
10,795,956 |
|
The following table summarizes the Company’s diluted net loss per share:
|
|
|
December 31, |
|
December 31, |
|
December 31, |
| |||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
Numerator: |
|
|
|
|
|
|
| |||
|
Net loss |
|
$ |
(14,348,499 |
) |
$ |
(11,840,778 |
) |
$ |
(11,604,201 |
) |
|
Adjustment for change in fair value of derivative instruments - warrants |
|
(2,422,337 |
) |
— |
|
— |
| |||
|
Net loss used for diluted loss per share |
|
$ |
(16,770,836 |
) |
$ |
(11,840,778 |
) |
$ |
(11,604,201 |
) |
|
|
|
|
|
|
|
|
| |||
|
Denominator: |
|
|
|
|
|
|
| |||
|
Weighted average shares used to compute basic loss per share |
|
18,904,280 |
|
16,978,212 |
|
13,066,600 |
| |||
|
Adjustments to reflect assumed exercise of warrants |
|
166,832 |
|
— |
|
— |
| |||
|
Weighted average shares used to compute diluted net loss per share |
|
19,071,112 |
|
16,978,212 |
|
13,066,600 |
| |||
|
|
|
|
|
|
|
|
| |||
|
Net loss per share diluted |
|
$ |
(0.88 |
) |
$ |
(0.70 |
) |
$ |
(0.89 |
) |
Recent Accounting Pronouncements
In January 2015, the Financial Accounting Standards Board (the FASB) issued an amendment to the accounting guidance related to the presentation of extraordinary items. The amendment simplifies the income statement presentation requirements in Subtopic 225-20 by eliminating the concept of extraordinary items. Extraordinary items are events and transactions that are distinguished by their unusual nature and by the infrequency of their occurrence. Eliminating the extraordinary classification simplifies income statement presentation by altogether removing the concept of extraordinary items from consideration. The amended guidance is effective prospectively for fiscal years beginning after December 15, 2015. The new guidance will not have an impact on the Company’s financial position, results of operations or cash flows.
In August 2014, the FASB issued an amendment to the accounting guidance related to the evaluation of an entity to continue as a going concern. The amendment establishes management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern in connection with preparing financial statements for each annual and interim reporting period. The amendment also gives guidance to determine whether to disclose information about relevant conditions and events when there is substantial doubt about an entity’s ability to continue as a going concern. The amended guidance is effective prospectively for fiscal years beginning after December 15, 2016. The new guidance will not have an impact on the Company’s financial position, results of operations or cash flows.
In June 2014, the FASB issued an accounting standards update that removes the financial reporting distinction between development stage entities and other reporting entities from GAAP. In addition, the amendments eliminate the requirements for development stage entities to: (1) present inception-to-date information in the statements of income, cash flows, and shareholder equity, (2) label the financial statements as those of a development stage entity, (3) disclose a description of the development stage activities in which the entity is engaged, and disclose in the first year in which the entity is no longer a development stage entity that in prior years it had been in the development stage. The guidance is effective prospectively for fiscal years, and interim periods within those years, beginning after December 15, 2014, with an option for early adoption. The Company elected early adoption, and does not believe the adoption of the standard had a material impact on its financial position, results of operations or related financial statement disclosures.
In May 2014, the FASB issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”). The standard provides companies with a single model for accounting for revenue arising from contracts with customers and supersedes current revenue recognition guidance, including industry-specific revenue guidance. The core principle of the model is to recognize revenue when control of the goods or services transfers to the customer, as opposed to recognizing revenue when the risks and rewards transfer to the customer under the existing revenue guidance. ASU 2014-09 is effective for annual reporting periods beginning after December 15, 2016. Early adoption is not permitted. The guidance permits companies to either apply the requirements retrospectively to all prior periods presented, or apply the requirements in the year of adoption, through a cumulative adjustment. The Company is in the process of evaluating the impact of adoption on its consolidated financial statements.
3. Asset Purchase
On February 1, 2012 the Company entered into an asset purchase agreement with MultiGen Diagnostics, Inc. The Company determined that the acquired asset did not meet the definition of a business, as defined in ASC 805, Business Combinations and was accounted for under ASC 350, Intangibles- Goodwill and Other. In connection with the acquisition, the Company issued 125,000 shares of restricted common stock to MultiGEN. In addition, up to an additional $3.7 million may be paid in a combination of common stock and cash to MultiGEN upon the achievement of specific sales and earnings targets. The Company is no longer offering these tests and has no plans to offer these tests. The Company does not anticipate any additional payments will become due. In addition, in connection with the acquisition, the Company entered into a Reagent Supply Agreement dated as of February 1, 2012 pursuant to which MultiGEN will supply and deliver reagents to be used in connection with a CLIA laboratory. The total purchase consideration was determined to be $187,500 which was paid in the Company’s common stock and allocated to an indefinite lived intangible asset related to the CLIA license.
Under ASC Topic 805, Business Combinations, the Company was required to assess the fair value of the assets acquired and the contingent consideration at the date of acquisition. Therefore, the Company assessed the fair value of the assets purchased and concluded that the purchase price would be allocated entirely to one intangible asset, a CLIA license. The contingent consideration of the $3.7 million milestone was determined to have no fair value by applying a weighted average probability on the achievement of the milestones developed during the valuation process. The Company assesses the fair value of the contingent consideration at each quarter and makes adjustments as necessary until the milestone dates have expired. As of December 31, 2014 and 2013, no adjustments to the fair value of the contingent consideration have been necessary, and therefore the fair value of the contingent consideration remains unchanged.
4. Property and Equipment
Fixed assets consist of laboratory, testing and computer equipment and fixtures stated at cost. Depreciation and amortization expense for the years ended December 31, 2014, 2013 and 2012 was $234,813, $130,520 and $41,842, respectively. Property and equipment consisted of the following:
|
|
|
As of December 31, |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
Furniture and office equipment |
|
$ |
365,955 |
|
$ |
236,645 |
|
|
Leasehold Improvements |
|
39,401 |
|
36,371 |
| ||
|
Laboratory equipment |
|
968,901 |
|
826,151 |
| ||
|
|
|
1,374,257 |
|
1,099,167 |
| ||
|
Less—accumulated depreciation and amortization |
|
(533,870 |
) |
(348,602 |
) | ||
|
Property and equipment, net |
|
$ |
840,387 |
|
$ |
750,565 |
|
5. Stockholders’ Equity (Deficiency)
All share and per share amounts have been restated to reflect the one-for-six reverse stock split effected on May 29, 2012 as described in Note 2.
Common Stock
During the year ended December 31, 2012, the Company closed five private placement financings which raised gross proceeds of $6,320,000. In total, the Company issued 1,738,333 shares of its common stock and warrants to purchase 1,738,333 shares of common stock (“units”).
The purchase price paid by the investors for 633,333 of the units sold in the period January 2012 through May 2012 was $3.00 each, determined by the price paid by investors in recent private placements. The warrants expire after eight years and are exercisable at $3.00 per share. The Company paid $96,500 in cash for a finder’s fee. Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, Trovagene determined that the warrants issued in connection with these private placements should be recorded as derivative liabilities at the time of issuance since they are all price protected however the completion of the underwritten public offering on May 30, 2012 removed the condition which required these warrants to be treated as derivative liabilities. Accordingly, the fair value of these warrants were marked to market through May 30, 2012 and then reclassified from a liability to additional paid in capital.
The purchase price paid by the investors for 1,105,000 of the units sold in the fourth quarter of 2012 was $4.00 each, determined by the market price on NASDAQ. The warrants expire after five years and are exercisable at $5.32 per share. The Company paid $24,989 in cash for a finder’s fee. Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, Trovagene has determined that the warrants issued in connection with these private placements should be recorded as derivative liabilities at the time of issuance since they are all price protected. The fair value of the warrants on the date of issuance was $1,031,281. However, the completion of the public offering in July 2013 removed the condition which required these warrants to be treated as derivative liabilities. Accordingly, the fair value of these warrants were marked to market through July 18, 2013 and then reclassified from a liability to additional paid in capital.
On May 30, 2012, the Company completed an underwritten public offering in which an aggregate of 1,150,000 units, with each unit consisting of two shares of its common stock and one warrant to purchase one share of common stock were sold at a purchase price of $8.00 per unit. On June 13, 2012 the underwriters exercised their overallotment option in full for an additional 172,500 units. In addition, the Company issued 92,000 warrants to selling agents at an exercise price of $7.00. The warrants were immediately exercisable and expire on May 29, 2017. Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, “Derivatives and Hedging - Contracts in an Entity’s Own Equity”, Trovagene determined that the warrants issued in connection with this transaction were not derivative liabilities. The Company raised a total of $9.1 million in net proceeds after deducting underwriting discounts and commissions of $0.7 million and offering expenses of $0.7 million.
During the year ended December 31, 2012, the Company issued in total 174,100 units to a selling agent, who is also a shareholder of the Company, consisting of 174,100 shares of common stock and warrants to purchase 174,100 shares of common stock. Of the total warrants issued, 30,450 issued in the period January 2012 through May 2012 have an exercise price of $3.00 per share, are immediately exercisable and expire December 31, 2018. The remaining 143,650 warrants issued in the fourth quarter of 2012, have an exercise price of $5.32, are immediately exercisable, and expire five years from the date of issuance. The units were issued as a finder’s fee in connection with certain private placements closed during the year ended December 31, 2012. The issuance of these units was treated as a non-compensatory cost of capital.
During the year ended December 31, 2012, the Company issued 40,000 units to Panetta Partners, Ltd., consisting of 40,000 shares of common stock and warrants to purchase 40,000 shares of common stock. Gabriele Cerrone, previously a member of the Board of Directors of Trovagene, is a director of Panetta Partners, Ltd. The warrants have an exercise price of $5.32, are immediately exercisable, and expire five years from the date of issuance. The units were issued for advisory services in connection with the private placements closed in the fourth quarter of 2012. The issuance of these units was treated as a non-compensatory cost of capital. In addition, the Company issued a warrant to purchase 50,000 shares of common stock to Panetta Partners, Ltd. at an exercise price of $4.14 for consulting services. The warrant was immediately exercisable and expires on December 10, 2017. The fair value of the warrant issued was determined using the Black-Scholes method. A total of approximately $133,000 was charged to general and administrative expenses in the Company’s consolidated statement of operations in 2012.
During the year ended December 31, 2012, the Company issued 125,000 shares of common stock in connection with an asset purchase agreement with MultiGen Diagnostics, Inc. See Note 3.
During the year ended December 31, 2012, the Company issued 9,916 shares of common stock in connection with consulting agreements. The fair value of the stock issued was determined using the Black-Scholes method. A total of $22,381 was charged to general and administrative expense in the Company’s consolidated statement of operations in 2012.
In December 2012, a Trovagene warrant holder exercised warrants to purchase 16,667 on a net exercise basis and received a total of 8,602 shares of common stock. The exercise price of the warrants was $3.00.
In December 2012, a Trovagene option holder exercised his option and purchased a total of 200 shares of common stock. Trovagene raised gross proceeds of $600 as a result of the option exercise. The purchase price paid by the option holder was $3.00.
On January 25, 2013, the Company filed a Form S-3 Registration Statement to offer and sell in one or more offerings, any combination of common stock, preferred stock, warrants, or units having an aggregate initial offering price not exceeding $150,000,000. The preferred stock, warrants, and units may be convertible or exercisable or exchangeable for common stock or preferred stock or other Trovagene securities. This form was declared effective on February 4, 2013. In addition, in connection with the Form S-3, the Company entered into an agreement with Cantor Fitzgerald & Co. (“Agent”) on January 25, 2013 to issue and sell up to $30,000,000 of shares of common stock through them. As payment for its services, the Agent is entitled to a 3% commission on gross proceeds. The Company has received gross proceeds of approximately $4.2 million from the sale of 488,476 shares of its common stock during the year ended December 31, 2013 under the agreement with the Agent. In addition, the Company has received gross proceeds of approximately $15.0 million from the sale of 2,142,857 shares of its common stock through a registered direct offering that occurred in July 2013.
During the year ended December 31, 2013, the Company issued a total of 3,424,605 shares of common stock. The Company sold 2,631,332 shares of Common Stock for net proceeds of $18,829,644. In addition, 36,458 shares were issued upon conversion of Series A Preferred Stock, 715,743 shares were issued upon exercise of 715,743 warrants for a weighted average price of $5.02 and 7,284 shares were issued upon net exercise of 12,745 warrants at an exercise price of $3.00. The remaining 33,788 shares include 22,955 shares that were issued upon net exercise of an option for 41,667 shares at an exercise price of $4.50 and the exercise of an option for 10,833 shares at a weighted average exercise price of $3.53.
During the year ended December 31, 2014, the Company issued a total of 13,011 shares of common stock upon the net exercise of warrants at a weighted average exercise price of $3.00.
Warrants
During the years ended December 31, 2014, 2013, and 2012, warrant activity was as follows:
|
|
|
Number |
|
Weighted |
|
Term |
| |
|
Warrants Outstanding 12/31/2011 |
|
3,601,474 |
|
$ |
3.18 |
|
1-8 years |
|
|
Granted |
|
3,416,934 |
|
$ |
4.88 |
|
|
|
|
Exercised |
|
(16,667 |
) |
|
|
|
| |
|
Expired |
|
(16,671 |
) |
$ |
3.00 |
|
|
|
|
Warrants Outstanding 12/31/2012 |
|
6,985,070 |
|
$ |
3.96 |
|
1 - 6 years |
|
|
Granted |
|
50,000 |
|
$ |
8.00 |
|
|
|
|
Exercised |
|
(728,488 |
) |
4.99 |
|
|
| |
|
Expired |
|
(73,099 |
) |
$ |
4.50 |
|
|
|
|
Warrants Outstanding 12/31/2013 |
|
6,233,483 |
|
$ |
3.87 |
|
1 -5 years |
|
|
Granted |
|
85,470 |
|
$ |
3.51 |
|
|
|
|
Exercised |
|
(36,666 |
) |
$ |
3.00 |
|
|
|
|
Expired |
|
(16,667 |
) |
$ |
10.80 |
|
|
|
|
Warrants Outstanding 12/31/2014 |
|
6,265,620 |
|
$ |
3.85 |
|
1 – 4 years |
|
The Company granted a total of 3,416,934 warrants during the year ended December 31, 2012. Of the total warrants granted, 713,784 were warrants that were price protected. These warrants had an exercise price of $3.00 per share and expire on December 31, 2018. The fair value of these warrants was estimated using the binomial option pricing model. The binomial model requires the input of variable inputs over time, including the expected stock price volatility, the expected price multiple at which warrant holders are likely to exercise their warrants and the expected forfeiture rate. The Company uses historical data to estimate forfeiture rate and expected stock price volatility within the binomial model. The risk-free rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve in effect at the date of grant for the expected term of the warrant. The completion of the underwritten public offering on May 30, 2012 removed the condition which required these warrants to be treated as derivative liabilities. Accordingly, the fair value of these warrants were marked to market through May 30, 2012 and then reclassified from a liability to additional paid in capital. See Note 7. In connection with underwritten public offering and sale of units in May 2012, 1,414,500 warrants were issued with exercise prices ranging from $5.32 to $7.00 and an expiration date five years after date of issuance. Warrants issued in connection with the underwritten public offering and sale of units are not considered derivatives based on Trovagene’s analysis of the criteria of ASC 815, as the Company is not required to make any cash payments or net cash settlement to the registered holder in lieu of issuance of warrant shares. An additional 1,288,650 warrants in the fourth quarter of 2012 were warrants that are price protected. These warrants have an exercise price of $5.32 per share and have an expiration date of five years from issuance. The fair value of these warrants was estimated using the binomial option pricing model. The binomial model requires the input of variable inputs over time, including the expected stock price volatility, the expected price multiple at which warrant holders are likely to exercise their warrants and the expected forfeiture rate. The Company uses historical data to estimate forfeiture rate and expected stock price volatility within the binomial model. The risk-free rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve in effect at the date of grant for the expected term of the warrant. See Note 7.
The Company issued a warrant to purchase 50,000 shares of common stock at an exercise price of $8.00 per share, during the year ended December 31, 2013. The warrants were issued in connection with an agreement to provide services related to investor and public relations materials and expire three years from date of grant. The estimated fair value of the warrant was determined on the date of grant using the Black-Scholes option valuation model using the following assumptions: a risk-free interest rate of 0.42%, dividend yield of 0%, expected volatility of 97% and expected term of three years. The resulting fair value of $198,791 was recorded as stock based compensation expense.
The Company issued warrants to purchase 85,470 shares of common stock at an exercise price of $3.51 per share, during the year ended December 31, 2014. The warrants were issued in connection with a $15.0 million debt agreement. The estimated fair value of the warrant was determined on the date of grant using the Black-Scholes option valuation model using the following assumptions: a risk-free interest rate of 2.53%, dividend yield of 0%, expected volatility of 73.8% and expected term of ten years. The resulting fair value of $235,857 was recorded as a debt discount and will be amortized to interest expense over the term of the loan using the effective interest method.
Series A Convertible Preferred Stock
The material terms of the Series A Convertible Preferred Stock consist of:
1) Dividends. Holders of the Series A Convertible Preferred Stock shall be entitled to receive cumulative dividends at the rate per share of 4% per annum, payable quarterly on March 31, June 30, September 30 and December 31, beginning with September 30, 2005. Dividends shall be payable, at the Company’s sole election, in cash or shares of common stock. As of December 31, 2014 and 2013, the Company had $244,055 and $221,040, respectively in accrued cumulative unpaid preferred stock dividends, included in Accrued Expenses in the Company’s consolidated balance sheets, and $23,015, $29,840, and $38,240, was recorded during the years ended December 31, 2014, 2013, and 2012, respectively.
2) Voting Rights. Shares of the Series A Convertible Preferred Stock shall have no voting rights. However, so long as any shares of Series A Convertible Preferred Stock are outstanding, the Company shall not, without the affirmative vote of the holders of the shares of Series A Convertible Preferred Stock then outstanding, (a) adversely change the powers, preferences or rights given to the Series A Convertible Preferred Stock, (b) authorize or create any class of stock senior or equal to the Series A Convertible Preferred Stock, (c) amend its articles of incorporation or other charter documents, so as to affect adversely any rights of the holders of Series A Convertible Preferred Stock or (d) increase the authorized number of shares of Series A Convertible Preferred Stock.
3) Liquidation. Upon any liquidation, dissolution or winding-up of the Company, the holders of the Series A Convertible Preferred Stock shall be entitled to receive an amount equal to the Stated Value per share, which is $10 per share plus any accrued and unpaid dividends.
4) Conversion Rights. Each share of Series A Convertible Preferred Stock shall be convertible at the option of the holder into that number of shares of common stock determined by dividing the Stated Value, currently $10 per share, by the conversion price, originally $2.15 per share.
5) Subsequent Equity Sales. The conversion price is subject to adjustment for dilutive issuances for a period of 12 months beginning upon registration of the common stock underlying the Series A Convertible Preferred Stock. The relevant registration statement became effective March 17, 2006 and during the following twelve month period the conversion price was adjusted to $9.60 per share.
6) Automatic Conversion. Beginning July 13, 2006, if the price of the common stock equals $25.80 per share for 20 consecutive trading days, and an average of 8,333 shares of common stock per day shall have been traded during the 20 trading days, the Company shall have the right to deliver a notice to the holders of the Series A Convertible Preferred Stock, to convert any portion of the shares of Series A Convertible Preferred Stock into shares of Common Stock at the conversion price. As of the date of these financial statements, such conditions have not been met.
During the year ended December 31, 2013, 35,000 shares of Series A Convertible Preferred Stock were converted into 36,458 shares of common stock, on a net converted basis. As of December 31, 2014 and 2013, and 2012, there were 60,600, 60,600, and 95,600 shares of Series A Convertible Preferred Stock outstanding.
6. Stock Option Plan
In June 2004 the Company adopted the Trovagene Stock Option Plan, as amended (the “2004 Plan”), under which up to 6,000,000 shares common stock were reserved for issuance to directors, eligible employees, including executive officers and consultants. The 2004 Plan provided for the grant of stock options to eligible recipients. Generally, vesting for options granted under the Plan was from three to four years, and options expird after a 10-year period. Options were granted at an exercise price not less than the fair market value at the date of grant. As of December 31, 2014 the 2004 Plan was expired. As of December 31, 2013, there were 1,788,921 shares available for issuance under the 2004 Plan.
During 2013, the Company issued 260,000 options over the authorized number of options in the 2004 Plan. As per ASC Topic 815-40, the options were accounted for as liabilities and recorded at fair value with the changes in fair value being recorded in the Company’s statement of operations. Stockholder approval was obtained on July 18, 2013 to increase the number of authorized shares in the Plan from 3,666,667 to 6,000,000. Accordingly, the options were remeasured as of the date of stockholder approval with the change recorded in stock based compensation expense and the $23,024 liability was reclassified to additional paid in capital.
In July 2013, an option to purchase 90,000 shares of common stock was granted to a Board Director for services provided outside of routine Board of Directors’ services. These options were immediately vested. The fair value of this option was approximately $500,000 and is included in general and administrative expenses.
The Trovagene Inc. 2014 Equity Incentive Plan, (the “2014 EIP’) authorizing up to 2,500,000 shares of common stock for issuance under the Plan, was approved by the Board of Directors in June 2014 and approved by the Shareholders at the September 17, 2014 Annual Shareholders’ Meeting. As of December 31, 2014, there were 1,371,832 shares available for issuance under the 2014 EIP.
Stock-based compensation has been recognized in operating results as follows:
|
|
|
Years ended December 31, |
| |||||||
|
|
|
2014 |
|
2013 |
|
2012 |
| |||
|
In research and development expenses |
|
$ |
796,008 |
|
$ |
549,465 |
|
$ |
136,148 |
|
|
In general and administrative expenses |
|
1,274,186 |
|
1,429,899 |
|
395,992 |
| |||
|
Total stock based compensation |
|
$ |
2,070,194 |
|
$ |
1,979,364 |
|
$ |
532,140 |
|
The estimated fair value of stock option awards was determined on the date of grant using the Black-Scholes option valuation model with the following assumptions during the years indicated below:
|
|
|
Years ended December 31, |
| ||||
|
|
|
2014 |
|
2013 |
|
2012 |
|
|
Risk-free interest rate |
|
1.42% - 2.1% |
|
.74% - 1.5% |
|
.62% - 1.04% |
|
|
Dividend yield |
|
0% |
|
0% |
|
0% |
|
|
Expected volatility |
|
81% - 86% |
|
82% - 100% |
|
90% - 97% |
|
|
Expected term (in years) |
|
5.0 yrs |
|
5.0 yrs |
|
5.0 yrs |
|
|
Stock price |
|
$3.00 - $6.74 |
|
$5.53 - $8.15 |
|
$0.50 - $4.87 |
|
Risk-free interest rate — Based on the daily yield curve rates for U.S. Treasury obligations with maturities that correspond to the expected term of the Company’s stock options.
Dividend yield — Trovagene has not paid any dividends on common stock since its inception and does not anticipate paying dividends on its common stock in the foreseeable future.
Expected volatility — Based on the historical volatility of a group of peer companies with attributes similar to Trovagene.
Expected term — The expected option term represents the period that stock-based awards are expected to be outstanding based on the simplified method provided in Staff Accounting Bulletin (“SAB”) No. 107, Share-Based Payment, (“SAB No. 107”), which averages an award’s weighted-average vesting period and expected term for “plain vanilla” share options. Under SAB No. 107, options are considered to be “plain vanilla” if they have the following basic characteristics: (i) granted “at-the-money”; (ii) exercisability is conditioned upon service through the vesting date; (iii) termination of service prior to vesting results in forfeiture; (iv) limited exercise period following termination of service; and (v) options are non-transferable and non-hedgeable.
Forfeitures — ASC Topic 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company estimates forfeitures based on its historical experience.
The weighted-average fair value per share of all options granted during the years ended December 31, 2014, 2013, and 2012, estimated as of the grant date using the Black-Scholes option valuation model was $2.19, $4.54, and $2.12, per share, respectively.
The unrecognized compensation cost related to non-vested stock options outstanding at December 31, 2014 and December 31, 2013 was $4,862,030 and $3,733,753, respectively. The weighted-average remaining contractual term at December 31, 2014 and 2013 for options outstanding and vested options was 3.0 and 6.7, respectively.
A summary of stock option activity and of changes in stock options outstanding is presented below:
|
|
|
Number of |
|
Weighted |
|
Intrinsic |
|
Weighted Average |
| ||
|
Balance outstanding, December 31, 2011 |
|
2,426,192 |
|
$ |
5.22 |
|
$ |
— |
|
6.7 years |
|
|
Granted |
|
1,294,668 |
|
$ |
3.74 |
|
|
|
|
| |
|
Exercised |
|
(200 |
) |
$ |
3.00 |
|
$ |
640 |
|
|
|
|
Forfeited |
|
(9,357 |
) |
$ |
3.00 |
|
|
|
|
| |
|
Balance outstanding, December 31, 2012 |
|
3,711,303 |
|
$ |
4.69 |
|
$ |
8,301,484 |
|
6.3 years |
|
|
Granted |
|
1,144,760 |
|
$ |
6.33 |
|
|
|
|
| |
|
Exercised |
|
(52,500 |
) |
$ |
4.30 |
|
$ |
275,492 |
|
|
|
|
Forfeited |
|
(516,018 |
) |
$ |
4.35 |
|
|
|
|
| |
|
Balance outstanding, December 31, 2013 |
|
4,287,545 |
|
$ |
5.18 |
|
$ |
5,896,329 |
|
6.7 years |
|
|
Granted |
|
1,410,038 |
|
$ |
4.42 |
|
|
|
|
| |
|
Forfeited |
|
(784,111 |
) |
$ |
7.08 |
|
|
|
|
| |
|
Balance outstanding, December 31, 2014 |
|
4,913,472 |
|
$ |
4.66 |
|
$ |
2,808,083 |
|
7.6 years |
|
|
Vested and exercisable, December 31, 2014 |
|
2,434,124 |
|
$ |
4.65 |
|
$ |
1,990,187 |
|
6.2 years |
|
ASC Topic 718 requires that cash flows resulting from tax deductions in excess of the cumulative compensation cost recognized for options exercised (excess tax benefits) be classified as cash inflows from financing activities and cash outflows from operating activities. Due to Trovagene’s accumulated deficit position, no tax benefits have been recognized in the cash flow statement.
7. Derivative Financial Instruments - Warrants
Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, Contracts in Entity’s Own Equity, Trovagene has determined that certain warrants issued in connection with the private placements must be recorded as derivative liabilities with a charge to additional paid in capital as they were issued with other equity instruments. In accordance with ASC Topic 815-40, the warrants are also being re-measured at each balance sheet date based on estimated fair value, and any resultant change in fair value is being recorded in the Company’s statement of operations. The Company estimates the fair value of (i) certain of these warrants using the Black-Scholes option pricing model and (ii) estimates the fair value of the price protected units using the Binomial option pricing model in order to determine the associated derivative instrument liability and change in fair value described above.
Warrants - Black-Scholes Option Pricing Model
The range of assumptions used to determine the fair value of the warrants valued using the Black-Scholes option pricing model during the periods indicated was:
|
|
|
Year ended |
|
Year ended |
|
Year ended |
|
|
Estimated fair value of Trovagene common stock |
|
$3.00 - $6.74 |
|
$5.74 - $7.18 |
|
$0.02 - $5.93 |
|
|
Expected warrant term |
|
4.0 years |
|
1 month 5.8 years |
|
10 months to 6 years |
|
|
Risk-free interest rate |
|
1.38% |
|
.03%-1.75% |
|
.06% - 1.54% |
|
|
Expected volatility |
|
86.4% |
|
82%-100% |
|
90%-97% |
|
|
Dividend yield |
|
0% |
|
0% |
|
0% |
|
Expected volatility is based on the volatility of a peer group of companies with attributes similar to Trovagene. The warrants have a transferability provision and based on guidance provided in SAB 107 for instruments issued with such a provision, Trovagene used the full contractual term as the expected term of the warrants. The risk free rate is based on the U.S. Treasury security rates consistent with the expected remaining term of the warrants at each balance sheet date.
The following table sets forth the components of changes in the Company’s derivative financial instruments liability balance, valued using the Black-Scholes option pricing method, for the periods indicated. For the period ending December 31, 2013, the derivative financial instruments liability in the table below only represents the Black-Scholes calculation whereas the loss recognized in the Statement of Operations represents both the Black-Scholes and the binomial methodology.
|
Date |
|
Description |
|
Number of |
|
Derivative |
| |
|
December 31, 2011 |
|
Balance of derivative financial instruments liability |
|
1,103,727 |
|
$ |
994,627 |
|
|
|
|
Expired warrants |
|
(16,667 |
) |
— |
| |
|
|
|
Change in fair value of warrants during the year recognized as a loss in the statement of operations |
|
— |
|
5,258,133 |
| |
|
December 31, 2012 |
|
Balance of derivative financial instruments liability |
|
1,087,060 |
|
6,252,760 |
| |
|
|
|
Expired warrants |
|
(73,099 |
) |
— |
| |
|
|
|
Change in fair value of warrants during the year recognized as a gain in the statement of operations |
|
— |
|
(1,820,889 |
) | |
|
December 31, 2013 |
|
Balance of derivative financial instruments liability |
|
1,013,961 |
|
$ |
4,431,871 |
|
|
|
|
Change in fair value of warrants during the year recognized as a gain in the statement of operations |
|
— |
|
(1,425,850 |
) | |
|
December 31, 2014 |
|
Balance of derivative financial instruments liability |
|
1,013,961 |
|
$ |
3,006,021 |
|
Warrants - Binomial Option Pricing Model
During the year ended 2011 and through May 2012, the Company issued 713,784 and 1,048,175 units, respectively, at $3.00 per unit. The units had a per unit price protection clause whereby from the date of issuance until the earlier of (i) thirty months from the final Closing or (ii) the closing date of a Subsequent Financing which generates within a one year period an amount equal to or in excess of $5,000,000, if the Company shall issue any Common Stock or Common Stock Equivalents, in a Subsequent Financing at an effective price per share less than the Per Unit Purchase Price, the Company shall issue to such the number of additional Units equal to (a) the Subscription Amount Investor at the Closing divided by the Discounted Purchase Price, less (b) the Units issued to such Investor at the Closing. Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, Contracts in Entity’s Own Equity, Trovagene has determined that these price protected units issued in connection with the private placements must be recorded as derivative liabilities with a charge to additional paid in capital. The price protected unit’s warrants had an exercise price of $3.00 per share and had expiration dates ranging from June 30, 2014 to December 31, 2018. The fair value of these price protected units was estimated using the binomial option pricing model. The binomial model requires the input of variable inputs over time, including the expected stock price volatility, the expected price multiple at which unit holders are likely to exercise their warrants and the expected forfeiture rate. The Company uses historical data to estimate forfeiture rate and expected stock price volatility within the binomial model. The risk-free rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve in effect at the date of grant for the expected term of the warrant. However, the completion of the underwritten public offering on May 30, 2012 removed the condition which required these warrants to be treated as derivative liabilities. Accordingly, the fair value of these warrants were marked to market through May 30, 2012 and then reclassified from a liability to additional paid in capital.
In addition, during the fourth quarter of 2012, the Company issued a total of 1,288,650 units at $4.00 per unit. The units had a per unit price protection clause whereby from the date of issuance until the earlier of (i) forty-eight months from the final Closing or (ii) the closing date of a Subsequent Financing which generates within a one year period an amount equal to or in excess of $10,000,000, if the Company shall issue any Common Stock or Common Stock Equivalents, in a Subsequent Financing at an effective price per share less than the Per Unit Purchase Price, the Company shall issue to such the number of additional Units equal to (a) the Subscription Amount Investor at the Closing divided by the Discounted Purchase Price, less (b) the Units issued to such Investor at the Closing. Based upon the Company’s analysis of the criteria contained in ASC Topic 815-40, Contracts in Entity’s Own Equity, Trovagene has determined that these price protected units issued in connection with the private placements must be recorded as derivative liabilities with a charge to additional paid in capital. The price protected unit’s warrants had an exercise price of $5.32 per share and had expiration dates five years from date of issuance. The fair value of these price protected units was estimated using the binomial option pricing model. The binomial model requires the input of variable inputs over time, including the expected stock price volatility, the expected price multiple at which unit holders are likely to exercise their warrants and the expected forfeiture rate. The Company uses historical data to estimate forfeiture rate and expected stock price volatility within the binomial model. The risk-free rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve in effect at the date of grant for the expected term of the warrant. However, the completion of the public offering in July 2013 removed the condition which required these warrants to be treated as derivative liabilities. Accordingly, the fair value of these warrants were marked to market through July 18, 2013 and then reclassified from a liability to additional paid in capital.
The fair value of the warrants granted during the year ended December 31, 2012 was estimated under the binomial method using the following weighted average assumptions:
|
|
|
2012 |
|
|
|
|
|
|
|
Range of risk-free interest rates |
|
0.53% to 1.61% |
|
|
Range of expected volatility |
|
90% -97% |
|
|
Expected fair value of the stock |
|
$0.25- $3.21 |
|
|
Expected warrant term |
|
2 years to 6.75 years |
|
The following table sets forth the components of changes in the Company’s derivative financial instruments liability balance, valued using the Binomial option pricing method, for the periods indicated:
|
Date |
|
Number of |
|
Derivative |
|
Change In |
|
Ending |
| |||
|
December 31, 2012 |
|
1,288,650 |
|
1,171,463 |
|
1,341,405 |
|
2,512,868 |
| |||
|
Reclassification of derivative liability to equity |
|
(1,288,650 |
) |
— |
|
(5,417,871 |
) |
(5,417,871 |
) | |||
|
Change in fair value of warrants during the year recognized as a loss in the statement of operations |
|
— |
|
— |
|
2,905,003 |
|
2,905,003 |
| |||
|
December 31, 2013 |
|
— |
|
$ |
1,171,463 |
|
$ |
(1,171,463 |
) |
$ |
— |
|
The weighted average remaining contractual term of all of the Company’s warrants outstanding at December 31, 2014 and 2013 was approximately 3.6 and 4.0 years, respectively.
At December 31, 2014 and 2013, the total fair value of the above warrants accounted for as derivative financial instruments, valued using the Black-Scholes option pricing model was $3,006,021 and $4,431,871, respectively, and is classified as derivative financial instruments liability on the balance sheet.
8. Fair Value Measurements
The following table presents the Company’s assets and liabilities that are measured and recognized at fair value on a recurring basis classified under the appropriate level of the fair value hierarchy as of December 31, 2014 and 2013:
|
|
|
Fair Value Measurements at |
| ||||||||||
|
|
|
December 31, 2014 |
| ||||||||||
|
|
|
Quoted Prices |
|
Significant |
|
Significant |
|
Total |
| ||||
|
Assets: |
|
|
|
|
|
|
|
|
| ||||
|
Money market fund (1) |
|
$ |
27,123,587 |
|
$ |
— |
|
$ |
— |
|
27,123,587 |
| |
|
Total Assets |
|
$ |
27,123,587 |
|
$ |
— |
|
$ |
— |
|
$ |
27,123,587 |
|
|
Liabilities: |
|
|
|
|
|
|
|
|
| ||||
|
Derivative liabilities related to warrants |
|
— |
|
$ |
— |
|
$ |
3,006,021 |
|
$ |
3,006,021 |
| |
|
Total Liabilities |
|
$ |
— |
|
$ |
— |
|
$ |
3,006,021 |
|
$ |
3,006,021 |
|
|
|
|
Fair Value Measurements at |
| ||||||||||
|
|
|
December 31, 2013 |
| ||||||||||
|
|
|
Quoted Prices |
|
Significant |
|
Significant |
|
Total |
| ||||
|
Assets: |
|
|
|
|
|
|
|
|
| ||||
|
Money market fund (1) |
|
$ |
25,703,330 |
|
$ |
— |
|
$ |
— |
|
25,703,330 |
| |
|
Total Assets |
|
$ |
25,703,330 |
|
$ |
— |
|
$ |
— |
|
$ |
25,703,330 |
|
|
Liabilities: |
|
|
|
|
|
|
|
|
| ||||
|
Derivative liabilities related to warrants |
|
— |
|
$ |
— |
|
$ |
4,431,871 |
|
$ |
4,431,871 |
| |
|
Total Liabilities |
|
$ |
— |
|
$ |
— |
|
$ |
4,431,871 |
|
$ |
4,431,871 |
|
(1) Included as a component of cash and cash equivalents on the accompanying consolidated balance sheet.
The following table sets forth a summary of changes in the fair value of the Company’s Level 3 liabilities for the years ended December 31, 2014 and 2013:
|
Description |
|
Balance at |
|
Unrealized |
|
Balance as of |
| |||
|
Derivative liabilities related to Warrants |
|
$ |
4,431,871 |
|
$ |
(1,425,850 |
) |
$ |
3,006,021 |
|
|
Description |
|
Balance at |
|
Fair Value of |
|
Unrealized |
|
Balance as of |
| |
|
Derivative liabilities related to Warrants |
|
$ |
8,765,628 |
|
(5,417,871 |
) |
1,084,114 |
|
4,431,871 |
|
The unrealized gains or losses on the derivative liabilities are recorded as a change in fair value of derivative liabilities in the Company’s statement of operations. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement. At each reporting period, the Company reviews the assets and liabilities that are subject to ASC Topic 815-40. At each reporting period, all assets and liabilities for which the fair value measurement is based on significant unobservable inputs or instruments which trade infrequently and therefore have little or no price transparency are classified as Level 3.
9. Debt
Loan and Security Agreement
In June 2014, the Company entered into a $15,000,000 loan and security agreement with two banks pursuant to which the lenders provided the Company a term loan, which was funded at closing. A portion of the proceeds were used to repay the existing equipment loan. The repayment with the same lender was accounted for as a debt extinguishment under ASC Topic 470-50, Debt. The interest rate on the new loan is 7.07% per annum. The Company will make interest only payments on the outstanding amount of the loan on a monthly basis through July 2015, after which equal monthly payments of principal and interest are due until the loan maturity date of July 1, 2018. The loan is secured by a security interest in all of the Company’s assets except intellectual property, which is subject to a negative pledge. In connection with the loan, the lenders received a warrant to purchase an aggregate 85,470 shares of the Company’s common stock at an exercise price of $3.51 per share exercisable for ten years from the date of issuance. The original value of the warrants, totaling $235,857, was recorded as debt discount and additional paid-in capital as the warrants were equity classified.
At the Company’s option, it may prepay all of the outstanding principal balance, subject to certain pre-payment fees ranging from 1% to 3% of the prepayment amount. In the event of a final payment of the loans under the loan agreement, either in the event of repayment of the loan at maturity or upon any prepayment, the Company is obligated to pay the amortized portion of the final fee of $1,050,000.
The Company is also subject to certain affirmative and negative covenants under the loan agreement, including limitations on its ability to: undergo certain change of control events; convey, sell, lease, license, transfer or otherwise dispose of any equipment financed by loans under the loan agreement; create, incur, assume, guarantee or be liable with respect to indebtedness, subject to certain exceptions; grant liens on any equipment financed under the loan agreement; and make or permit any payment on specified subordinated debt and pay dividends. In addition, under the loan agreement, subject to certain exceptions, the Company is required to maintain with the lender its primary operating, other deposit and securities accounts.
As of December 31, 2014, amounts due under the agreement include $1,898,548 in current liabilities and $13,053,117 in long-term liabilities, which includes $191,682 of accrued interest.
At December 31, 2014, Trovagene was in compliance with all covenants under the Loan and Security Agreement. .
The Company recorded $843,260 in interest expense related to the Loan and Security Agreement during the year ended December 31, 2014. Closing costs are being accreted over the life of the loan to interest expense.
Future maturities of long-term debt at December 31, 2014 are as follows:
|
2015 |
|
$ |
1,898,548 |
|
|
2016 |
|
4,790,628 |
| |
|
2017 |
|
5,140,519 |
| |
|
2018 |
|
3,170,305 |
| |
|
Total long-term obligations |
|
$ |
15,000,000 |
|
10. Income Taxes
At December 31, 2014, Trovagene has federal net operating loss carryforwards (NOLs) of approximately $49.3 million, which, if not used, expire beginning in 2020. Trovagene also has California NOLs of approximately $ 29.9 million which begin to expire in 2021 and New Jersey NOLs of $11.5 million which started to expire on January 31, 2013. Trovagene also has R&D credits available for federal purposes of approximately $698,000. The federal R&D credits will begin to expire January 31, 2025. The utilization of these NOLs and R&D tax credits is subject to limitations based on past and future changes in ownership of Trovagene pursuant to Internal Revenue Code Section 382. The Company has determined that ownership changes have occurred for Internal Revenue Code Section 382 purposes and therefore, the ability of the Company to utilize its NOLs is limited.
Signifcant components of the Company’s taxes and the rates as of December 31, are shown below:
Tax Rate Reconciliation
|
Tax computed at the federal statutory rate |
|
(4,871,000 |
) |
35 |
% |
|
State tax, net of federal tax benefit |
|
(891,000 |
) |
6 |
% |
|
Permanent Items |
|
(320,000 |
) |
2 |
% |
|
Valuation allowance increase (decrease) |
|
6,081,000 |
|
(43 |
)% |
|
Other |
|
1,000 |
|
— |
% |
|
Provision for income taxes |
|
— |
|
|
|
Significant components of the Company’s deferred tax assets as of December 31, are shown below:
|
|
|
Years ended |
| ||||
|
|
|
2014 |
|
2013 |
| ||
|
Deferred tax assets |
|
|
|
|
| ||
|
Tax loss carryforwards |
|
$ |
19,010,900 |
|
$ |
14,211,400 |
|
|
R&D credits and other tax credits |
|
698,000 |
|
235,300 |
| ||
|
Stock based compensation |
|
2,193,900 |
|
1,547,500 |
| ||
|
Other |
|
420,800 |
|
230,400 |
| ||
|
Total deferred tax assets |
|
22,323,600 |
|
16,224,600 |
| ||
|
Valuation allowance |
|
(22,323,600 |
) |
(16,224,600 |
) | ||
|
Net deferred tax asset |
|
$ |
— |
|
$ |
— |
|
The difference between the statutory rate of 35% on taxable income and the actual income tax rate of zero is a result of the full deferred tax asset valuation allowance. The valuation allowance increased by $6,099,000 and $4,398,700 during the years ended December 31, 2014 and 2013, respectively.
Trovagene records a valuation allowance against deferred tax assets to the extent that it is more likely than not that some portion, or all of, the deferred tax assets will not be realized.
ASC 740-10-30-7, Accounting for Income Taxes had no effect on Trovagene’s financial position, cash flows or results of operations upon adoption, as Trovagene does not have any unrecognized tax benefits. Trovagene’s practice is to recognize interest and/or penalties related to income tax matters in income tax expense and none have been incurred to date.
11. Commitments and Contingencies
Significant Research Agreements
During 2012, the Company entered into research agreements with University of Texas MD Anderson Cancer Center (“MDACC”) to provide samples and evaluate methods used by the Company in identification of pancreatic cancer mutations, as well as to measure the degree of concordance between results of cell-free DNA mutations analysis from urine samples and tumor tissue. During 2013, the agreements were amended to increase the scope of the agreements. Under these agreements, the Company has committed to pay approximately $266,000 for the services performed by MDACC. As of December 31, 2014, the Company has incurred and recorded approximately $124,000 of research and development expenses related to these agreements. There were approximately $142,000 of research and development expenses incurred during the year ended December 31, 2013.
In June 2013, the Company entered into a Research Agreement with Illumina, Inc. (“Illumina”) pursuant to which the parties will work together to evaluate the potential for integrating the Company’s cell-free technology for isolating, extracting and genetic analysis of nucleic acids from urine with Illumina’s genetic analysis sequencing technology (the “Research Plan”). The parties have agreed that all results and reagents from the Research Plan will be shared between the parties. The Agreement will terminate upon the earlier of 30 days after completion of the Research Plan or the one year anniversary of the Agreement unless extended by mutual written agreement. In October 2014, the agreement was extended for an additional year to June 2015.
In August 2013, the Company entered into a Clinical Trial Agreement with the University of Southern California (“USC”), pursuant to which USC will provide the principal investigator and conduct the clinical trial related to the genetic characterization of metastatic colorectal cancers. Under the agreement, the Company is committed to pay USC approximately $232,000 for services provided. As of December 31, 2013 the Company had not incurred any expense related to this agreement. As of December 31, 2014, the Company had incurred approximately $38,000 of research and development expenses.
In December 2013, the Company entered into a Clinical Trial Agreement with US Oncology Research LLC (“USOR”), pursuant to which USOR will provide the principal investigator and conduct the clinical trial related to the examining the utility of cell-free quantitative KRAS testing in disease monitoring in patients with metastatic pancreatic cancer. Under the agreement, the Company is committed to pay USOR approximately $270,000 for services provided. As of December 31, 2014 and 2013, the Company has incurred and recorded approximately $16,000 and $29,000 of research and development expense related to this agreement.
In October 2014, the Company entered into a Research Agreement with Genomac International, Ltd., pursuant to which Genomac will conduct a research study of minimally invasive molecular monitoring of efficiency of targeted biological therapy in advanced non-small lung cancer. Under the agreement, the Company is committed to pay Genomac approximately $175,000 for services provided. As of December 31, 2014, the Company has incurred and recorded approximately $90,000 of research and development expenses.
License Agreements
In May 2006, the Company entered into a license agreement with Drs. Falini and Mecucci, wherein it obtained the exclusive rights for the genetic marker for AML and intends to utilize these rights for the development of new diagnostic tools. In connection with this agreement, the Company paid $70,000 to Drs. Falini and Mecucci. In August 2010 the Company signed amendment No. 1 to the license agreement with an obligation to payroyalties of 6% of royalty revenues and/or 10% of any sublicense income. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty expenses of approximately $23,000, $30,000 and $24,000, respectively.
During August 2007, the Company signed a sublicensing agreement with IPSOGEN SAS, a leading molecular diagnostics company with operations in France and the United States for the co-exclusive rights to develop, manufacture and market, research and diagnostic products for the stratification and monitoring of patients with AML. Upon execution of this agreement, IPSOGEN paid an initial licensing fee of $120,000 and may make milestone payments upon the attainment of certain regulatory and commercial milestones. IPSOGEN will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. In September 2010, the Company signed amendment No. 1 to the sublicensing agreement. The amendment asserts that the Compay may require a license from a third-party to perform laboratory testing services. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty, milestone and license fee revenues of approximately $60,000, $60,000 and $180,000, respectively. During those same periods, the Company had no license fee expenses.
In October 2007, the Company signed a license agreement with ASURAGEN, Inc. for the co-exclusive rights to develop, manufacture and market, research and diagnostic products for the stratification and monitoring of patients with AML. ASURAGEN paid an initial licensing fee of $120,000 upon execution of the agreement and may make future payments to the Company upon the attainment of certain regulatory and commercial milestones. In June 2010, the Compay signed amendment No. 1 to the co-exclusive license agreement. The amendment asserts that the Company may require a license from a third-party to perform laboratory testing services. ASURAGEN will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. In March 2013, we signed amendment No. 2 to the co-exclusive sublicense agreement with ASURAGEN. The amendment limited the field of use to research use only (RUO) kits. ASURAGEN was also granted a non-exclusive sublicense for NPMI laboratory testing services. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty and license fee revenues of approximately $50,000, $50,000 and $50,000, respectively. During those same periods, the Company had no license fee expenses related to this agreement.
In August 2008, the Company signed a sublicensing agreement with LabCorp for the non-exclusive rights to develop and market lab testing services for NPM1, for the diagnosis and monitoring of patients with AML. LabCorp paid an initial licensing fee of $20,000 upon execution of the agreement. LabCorp will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends August 25, 2018. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty and license fee revenues of approximately $28,000, $20,000 and $5,000, respectively. During those same years, the Company has not recorded any license fee expenses.
In December 2008, the Company signed a sublicensing agreement with InVivoScribe Technologies, Inc. for the non-exclusive rights to develop and market lab testing services for NPM1 for the diagnosis and monitoring of patients with AML. InVivoScribe Technologies paid an initial licensing fee of $10,000 upon execution of the agreement. InVivoScribe Technologies will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. During the years ended December 31, 2014, 2013, and 2012, the Company recorded royalty and license fee revenues of approximately $25,000, $25,000 and $27,000, respectively. During those same periods, the Company has not recorded any license fee expenses.
In June 2010, the Company signed a sublicensing agreement with Skyline Diagnostics BV for the non-exclusive rights to develop, commercialize and market, research and diagnostic laboratory services for the stratification and monitoring of patients with AML. Skyline Diagnostics BV paid an initial licensing fee of $10,000 upon execution of the agreement and may make future payments to the Company upon the attainment of certain regulatory and commercial milestones. Skyline Diagnostics BV will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty and license fee revenues of approximately $0, $0 and $0, respectively. During those same periods, the Company has not recorded any license fee expenses.
In January 2011, the Company entered into an asset purchase agreement with TTFactor S.r.l.for a hybridoma able to produce a monoclonal antibody targeting the NPM1 biomarker for $10,000. In addition the Company agreed to pay the seller of the hybridoma for a period of seven years commencing with the first sale of the antibody, annual royalties on a country by country basis. In addition, the Company agreed to pay a percentage of all cash consideration received from licensees as an upfront license fee pursuant to any licenses of the product and a percentage of all cash consideration received from licensees as milestone payments. The agreement was terminated in 2013. During the years ended December 31, 2014, 2013, and 2012, there were no royalty expense, license fee or milestone payments recorded related to this agreement.
In February 2011, the Company entered into a sublicense agreement with MLL Münchner Leukämielabor, or MLL for non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. MLL paid an initial license fee of $20,000 upon execution of the agreement and will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. MLL is obligated to pay a royalty with annual minimums of $15,000 for the first year and $20,000 thereafter. The term of the license ends on October 28, 2025 which is the date of expiration of the issued patent rights. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty and license fee revenues of approximately $81,000, $85,000 and $71,000, respectively.
In July 2011, the Company entered into a sublicense agreement with Fairview Health Services (“Fairview”) for the non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. Fairview paid an initial license fee of $10,000 upon execution of the agreement and will also pay the Company a royalty on any net revenues during the term of the agreement, subject to certain minimums. Fairview is obligated to pay a royalty with annual minimums of $1,000 each year. During the years ended December 31, 2014, 2013, and 2012, the Company recorded royalty and license fee revenues of approximately $2,000, $1,000 and $2,000, respectively.
In October 2011, the Company entered into an exclusive license agreement with Gianluca Gaidano, Robert Foa and Davide Rossi for the patent rights to a specific gene mutation with respect to chronic lymphoblastic leukemia. In consideration of the license, the Company paid $1,000 as an upfront license fee and agreed to make royalty payments in the single digits on net sales if sales are made by the Company or a single digit royalty on sublicense income received by the Company if sales are made by sublicensees. The Company has an option to purchase the licensed patent rights in the event the licensor decides to sell such licensed patent rights. The license agreement shall continue until September 29, 2031 which is the date of the last to expire of the licensed patent rights covering the license product. The license agreement may also be terminated upon a material breach by any party or by the Company if it is determined that it is not commercially or scientifically appropriate to further develop the license product rights. During the years ended December 31, 2014, 2013 and 2012, no royalty expense has been recorded related to this agreement.
In December 2011, the Company entered into an exclusive license agreement with Columbia University to license the patent rights to hairy cell leukemia biomarkers. In consideration of the license, the Company paid $1,000 as an upfront license fee and agreed to make royalty payments in the single digits on net sales if sales are made by the Company or a single digit royalty on sublicense income received by the Company if sales are made by sublicensees. The license agreement shall continue until May 10, 2021 which is the date of the last to expire of the licensed patent rights covering the license product. The license agreement may also be terminated upon a material breach by any party or by the Company if we determine that it is not commercially or scientifically appropriate to further develop the license product rights. During the years ended December 31, 2014, 2013 and 2012, no royalty expense has been recorded related to this agreement.
In September 2012, the Company entered into a sublicense agreement with Quest Diagnostics for non-exclusive rights to develop and market laboratory testing services for NPM1 for the diagnosis and monitoring of patients with AML. Under this agreement, the Company has granted a license to certain NPM1 patents in exchange for a one time license fee of $20,000 due upon execution of the agreement and royalty payments on net sales of Quest Diagnostics and its affiliates. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty and license fee revenues of approximately $26,000, $14,000 and $20,000, respectively.
In September 2012, the Company entered into a collaboration and license agreement with Strand Life Sciences (“Strand”) related to the validation and commercial launch of a urine based DNA test for Human Papillomavirus. Under this agreement, the Company has granted a license for use of its tests to Strand in exchange for royalty payments on net sales earned in the territory specified in the agreement. During the years ended December 31, 2014, 2013 and 2012, the Company has recorded no royalty and license fee revenues related to this agreement.
In November 2012, the Company entered into a sublicense agreement with Duke University and Duke University Health Systems for non-exclusive rights to develop and market laboratory testing services for NPM1 for diagnosis and monitoring of patients with AML. Under this agreement, the Company has granted a license to certain NPM1 patents in exchange for a one time license fee of $5,000 due upon execution of the agreement and royalty payments on net revenues. During the years ended December 31, 2014, 2013 and 2012, the Company has recorded $1,000, $0 and $5,000, respectively of royalty and license fee revenues related to this agreement.
In December 2012, the Company entered into a sublicense agreement with Genoptix, Inc. for non-exclusive worldwide rights to develop and market laboratory testing services for NPM1 for diagnosis and monitoring of patients with AML. Under this agreement, the Company has granted a license to certain NPM1 patents in exchange for a one time license fee of $100,000 due upon execution of the agreement and royalty payments on net revenues. During the years ended December 31, 2014, 2013 and 2012, the Company recorded royalty and license fee revenues of approximately $30,000, $10,000 and $100,000, respectively.
Litigation
Trovagene does not believe that the Company has legal liabilities that are probable or reasonably possible that require either accrual or disclosure. From time to time, we may become involved in various lawsuits and legal proceedings which arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business.
We are not currently a party to any material legal proceedings.
Employment and Consulting Agreements
During 2011, the Company entered into an executive agreement with Antonius Schuh, Ph.D. in which he agreed to serve as Chief Executive Officer. The term of the agreement is effective as of October 4, 2011 and continues until October 4, 2015 and is automatically renewed for successive one year periods at the end to each term. In the event that during the term of the agreement, for a period of 90 consecutive trading days, the market price of the common stock is $7.50 or more and the value of the common stock daily trading volume is $125,000 or more, the Company shall pay or issue Dr. Schuh a bonus in an amount of $3,466,466 in either cash or registered common stock or a combination thereof as mutually agreed by Dr. Schuh the Company; or in the event that during the term of the agreement, a change of control occurs where the per share enterprise value of our company equals or exceeds $7.50 per share, the Company shall pay Dr. Schuh a bonus in an amount determined by multiplying the enterprise value by 4.0%. In the event in a change of control the per share enterprise value exceeds a minimum of $14.40 per share, $22.80 per share or $30.00 per share, Dr. Schuh shall receive a bonus in an amount determined by multiplying the incremental enterprise value by 2.5%, 2.0% or 1.5%, respectively.
If the executive agreement is terminated for cause or as a result of Dr. Schuh’s death or permanent disability or if Dr. Schuh terminates his agreement voluntarily, Dr. Schuh shall receive a lump sum equal to (i) any portion of unpaid base compensation then due for periods prior to termination, (ii) any bonus or realization bonus earned but not yet paid through the date of termination and
(iii) all expenses reasonably incurred by Dr. Schuh prior to date of termination. If the executive agreement is terminated without cause Dr. Schuh shall receive a severance payment equal to base compensation for three months if termination occurs ten months after the effective date of the agreement and six months if termination occurs subsequent to ten months from the effective date. If the executive agreement is terminated as a result of a change of control, Dr. Schuh shall receive a severance payment equal to base compensation for twelve months and all unvested stock options shall immediately vest and become fully exercisable for a period of six months following the date of termination.
During 2012, the Company entered into an executive agreement with Steve Zaniboni in which he agreed to serve as Chief Financial Officer. If the executive agreement is terminated by the Company for cause or as a result of Mr. Zaniboni’s death or permanent disability or if Mr. Zaniboni terminates his agreement voluntarily, Mr. Zaniboni shall receive a lump sum equal to (i) any portion of unpaid base compensation then due for periods prior to termination, (ii) any bonus or realization bonus earned but not yet paid through the date of termination and (iii) all expenses reasonably incurred by Mr. Zaniboni prior to date of termination. If the executive agreement is terminated by the Company without cause Mr. Zaniboni shall receive a severance payment equal to base compensation for three months if termination occurs ten months after the effective date of the agreement and six months if termination occurs subsequent to ten months from the effective date. If the executive agreement is terminated as a result of a change of control, Mr. Zaniboni shall receive a severance payment equal to base compensation for twelve months and all unvested stock options shall immediately vest and become fully exercisable for a period of six months following the date of termination.
Lease Agreements
The Company leases its facilities under a noncancelable operating lease that expires in 2017. Rent expense for the years ended December 31, 2014, 2013 and 2012 was $315,000, $294,000, and $270,000, and respectively. The Company is also a party to various operating lease agreements for office equipment.
Total annual commitments under current lease agreements for each of the years ended December 31, are as follows:
|
2015 |
|
360,775 |
| |
|
2016 |
|
371,324 |
| |
|
2017 |
|
381,026 |
| |
|
2018 |
|
6,380 |
| |
|
Total |
|
$ |
1,119,505 |
|
12. Employee Benefit Plan
The Company has a retirement savings plan under Section 401(k) of the Internal Revenue Code covering its employees. The plan allows employees to defer, up to the maximum allowed, a percentage of their income on a pre-tax basis through contributions to the plans, plus any employee of the age of 55 can participate in the caught-up dollars as allowed by IRS codes. The Company also has a Roth investment plan that is taken after taxes. The Company does not currently make matching contributions.
13. Related Party Transactions
Gabriele M. Cerrone, the Company’s former Co-Chairman, and former member of the Board of Directors, served as a consultant to the Company from June 27, 2005 until June 2008 and is affiliated with Panetta Partners Ltd. Transactions between the Company and Mr. Cerrone and Panetta Partners, Ltd. is disclosed in, Note 5, Stockholders’ Equity (Deficiency).
14. Selected Quarterly Financial Data (Unaudited)
The following is a summary of the quarterly results of operations of the Company for years ended December 31, 2014 and 2013:
|
|
|
Quarter Ended(1) |
| ||||||||||
|
|
|
March 31 |
|
June 30 |
|
September 30 |
|
December 31 |
| ||||
|
|
|
|
|
(Revised)(2) |
|
|
|
|
| ||||
|
|
|
(dollars in thousands, except per share data) |
| ||||||||||
|
2014 |
|
|
|
|
|
|
|
|
| ||||
|
Revenues |
|
$ |
111 |
|
$ |
56 |
|
$ |
57 |
|
$ |
56 |
|
|
Operating expenses |
|
3,371 |
|
3,297 |
|
3,997 |
|
4,560 |
| ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
Net loss and comprehensive loss attributable to common stockholders |
|
(3,198 |
) |
(1,079 |
) |
(5,382 |
) |
(4,689 |
) | ||||
|
Net loss per common share - basic |
|
$ |
(0.17 |
) |
$ |
(0.06 |
) |
$ |
(0.28 |
) |
$ |
(0.25 |
) |
|
Net loss per common share - diluted |
|
$ |
(0.17 |
) |
$ |
(0.17 |
) |
$ |
(0.28 |
) |
$ |
(0.25 |
) |
|
|
|
|
|
|
|
|
|
|
| ||||
|
Shares used in the calculation of net loss attributable to common stockholders - basic |
|
18,902,783 |
|
18,902,783 |
|
18,902,783 |
|
18,904,280 |
| ||||
|
Shares used in the calculation of net loss attributable to common stockholders - diluted |
|
18,902,783 |
|
19,232,760 |
|
18,902,783 |
|
19,071,112 |
| ||||
|
|
|
|
|
|
|
|
|
|
| ||||
|
2013 |
|
|
|
|
|
|
|
|
| ||||
|
Revenues |
|
$ |
119 |
|
$ |
49 |
|
$ |
44 |
|
$ |
47 |
|
|
Operating expenses |
|
2,509 |
|
2,423 |
|
3,121 |
|
2,897 |
| ||||
|
Net loss and comprehensive loss attributable to common stockholders |
|
(1,117 |
) |
(5,279 |
) |
(4,407 |
) |
(1,038 |
) | ||||
|
Net loss per common share - basic |
|
$ |
(0.07 |
) |
$ |
(0.34 |
) |
$ |
(0.25 |
) |
$ |
(0.05 |
) |
|
Net loss per common share - diluted |
|
$ |
(0.07 |
) |
$ |
(0.34 |
) |
$ |
(0.25 |
) |
$ |
(0.05 |
) |
|
|
|
|
|
|
|
|
|
|
| ||||
|
Shares used in the calculation of net loss attributable to common stockholders - basic |
|
15,510,340 |
|
15,583,957 |
|
17,870,703 |
|
18,900,781 |
| ||||
|
Shares used in the calculation of net loss attributable to common stockholders - diluted |
|
15,510,340 |
|
15,583,957 |
|
17,870,703 |
|
18,900,781 |
| ||||
(1) Basic and diluted EPS are computed independently for each of the periods presented. Accordingly, the sum of the quarterly EPS amount may not agree to the total for the year. (2) Second quarter fully diluted earnings per share have been adjusted for dilutive amounts related to the derivative liability. We believe that these changes are not material to the financial statements or related notes.
15. Subsequent Events
Public Offering and Controlled Equity Offering
On February 11, 2015, the Company closed an underwritten public offering of 5,111,110 shares of its common stock. The offering price was $4.50 per share and net proceeds to the Company were approximately $21.3 million. The effects of this offering would materially change the number of common shares outstanding at the end of the reporting period had the transaction occurred before December 31, 2014.
Related Party Consulting Agreement
On January 1, 2015, we entered into a consulting agreement with Tom Adams, our Chairman, pursuant to which Dr. Adams will provide consulting services to us in connection with applying our technology to infectious diseases. The agreement is for a term of one year and Dr. Adams shall be paid $9,500 per month for his services.