Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - DELCATH SYSTEMS, INC. | Financial_Report.xls |
| EX-32.2 - EXHIBIT 32.2 - DELCATH SYSTEMS, INC. | ex32_2.htm |
| EX-32.1 - EXHIBIT 32.1 - DELCATH SYSTEMS, INC. | ex32_1.htm |
| EX-23.1 - EXHIBIT 23.1 - DELCATH SYSTEMS, INC. | ex23_1.htm |
| EX-31.1 - EXHIBIT 31.1 - DELCATH SYSTEMS, INC. | ex31_1.htm |
| EX-31.2 - EXHIBIT 31.2 - DELCATH SYSTEMS, INC. | ex31_2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| ☒ | Annual report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 for the fiscal year ended December 31, 2014 |
| ☐ | Transition report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 for the transition period from ____________ to ____________ |
Commission file number: 001-16133
DELCATH SYSTEMS, INC.
|
Delaware
|
06-1245881
|
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
|
|
1301 Avenue of the Americas, 43rd Floor New York, NY
|
10019
|
|
|
(Address of principal executive offices)
|
(Zip Code)
|
212-489-2100
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class
|
Name of Each Exchange on Which Registered
|
|
|
Common Stock, par value $0.01 per share
|
The NASDAQ Stock Market LLC
|
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act.
Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ☐ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐
|
Accelerated filer ☐
|
|
Non-accelerated filer ☒ (Do not check if smaller reporting company)
|
Smaller reporting company ☐
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ☐ No ☒
The aggregate market value of the voting common stock held by non-affiliates of the registrant, based on the closing sale price on The NASDAQ Capital Market of $2.62 per share, was $24,619,516 as of June 30, 2014.
At March 11, 2015, the registrant had outstanding 12,169,464 shares of par value $0.01 Common Stock.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for its 2015 Annual Meeting of Stockholders are incorporated by reference into Part III (Items 10, 11, 12, 13 and 14) of this Annual Report on Form 10-K. The definitive Proxy Statement will be filed with the Securities and Exchange Commission within 120 days after the close of the fiscal year covered by this Annual Report on Form 10-K.
|
Page
|
||
|
PART I
|
||
|
Item 1.
|
1
|
|
|
Item 1A.
|
16
|
|
|
Item 1B.
|
31
|
|
|
Item 2.
|
31
|
|
|
Item 3.
|
32
|
|
|
Item 4.
|
33
|
|
|
PART II
|
||
|
Item 5.
|
34
|
|
|
Item 6.
|
36
|
|
|
Item 7.
|
36
|
|
|
Item 7A.
|
40
|
|
|
Item 8.
|
42
|
|
|
Item 9.
|
43
|
|
|
Item 9A.
|
43
|
|
|
Item 9B.
|
43
|
|
|
PART III
|
||
|
Item 10.
|
44
|
|
|
Item 11.
|
44
|
|
|
Item 12.
|
44
|
|
|
Item 13.
|
44
|
|
|
Item 14.
|
44
|
|
|
PART IV
|
||
|
Item 15.
|
45
|
|
|
46
|
||
Disclosure Regarding Forward-Looking Statements
This Annual Report on Form 10-K for the period ended December 31, 2014 contains certain “forward-looking statements” within the meaning of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995 with respect to our business, financial condition, liquidity and results of operations. Words such as “anticipates,” “expects,” “intends,” “plans,” “predicts,” “believes,” “seeks,” “estimates,” “could,” “would,” “will,” “may,” “can,” “continue,” “potential,” “should,” and the negative of these terms or other comparable terminology often identify forward-looking statements. Statements in this Annual Report on Form 10-K for the period ending December 31, 2014 that are not historical facts are hereby identified as “forward-looking statements” for the purpose of the safe harbor provided by Section 21E of the Exchange Act and Section 27A of the Securities Act. These forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties that could cause actual results to differ materially from the results contemplated by the forward-looking statements, including the risks discussed in this Annual Report on Form 10-K for the fiscal year ended December 31, 2014 in Item 1A under “Risk Factors” as well as in Item 7A “Quantitative and Qualitative Disclosures About Market Risk,” our Quarterly Report on Form 10-Q for the period ended September 30, 2014 in Part II, Item 1A under “Risk Factors” as well as in Part I, Item 3 “Quantitative and Qualitative Disclosures About Market Risk” and the risks detailed from time to time in our future SEC reports. These forward-looking statements include, but are not limited to, statements about:
| ● | our estimates regarding sufficiency of our cash resources, anticipated capital requirements and our need for additional financing; |
| ● | the commencement of future clinical trials and the results and timing of those clinical trials; |
| ● | our ability to successfully commercialize CHEMOSAT/Melphalan/HDS, generate revenue and successfully obtain reimbursement for the procedure and System; |
| ● | the progress and results of our research and development programs; |
| ● | submission and timing of applications for regulatory approval and approval thereof; |
| ● | our ability to successfully source certain components of the system and enter into supplier contracts; |
| ● | our ability to successfully manufacture CHEMOSAT/Melphalan/HDS; |
| ● | our ability to successfully negotiate and enter into agreements with distribution, strategic and corporate partners; and |
| ● | our estimates of potential market opportunities and our ability to successfully realize these opportunities. |
Many of the important factors that will determine these results are beyond our ability to control or predict. You are cautioned not to put undue reliance on any forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. Except as otherwise required by law, we do not assume any obligation to publicly update or release any revisions to these forward-looking statements to reflect events or circumstances after the date of this Annual Report on Form 10-K or to reflect the occurrence of unanticipated events.
Unless the context otherwise requires, all references in this Annual Report on Form 10-K to the “Company”, “Delcath”, “Delcath Systems”, “we”, “our”, and “us” refers to Delcath Systems, Inc., a Delaware corporation, incorporated in August 1988, and all entities included in our consolidated financial statements. Our corporate offices are located at 1301 Avenue of the Americas, 43rd Floor, New York, New York 10019. Our telephone number is (212) 489-2100.
Company Overview
Delcath Systems, Inc. is a late-stage clinical development company with early commercial activity in Europe focused on cancers of the liver. We are a specialty pharmaceutical and medical device company developing our proprietary product—Melphalan Hydrochloride for Injection for use with the Delcath Hepatic Delivery System (Melphalan/HDS). In Europe, our proprietary system to deliver and filter melphalan hydrochloride is marketed as a device under the trade name Delcath Hepatic CHEMOSAT® Delivery System for Melphalan (CHEMOSAT).
Our primary focus is on the execution of our clinical development program (CDP) in ocular melanoma liver metastases (mOM), intrahepatic cholangiocarncinoma (ICC), hepatocellular carcinoma (HCC or primary liver), and certain other cancers that are metastatic to the liver. We believe the disease states we are investigating represent a multi-billion dollar global market opportunity and a clear unmet medical need.
Our clinical development program for CHEMOSAT/Melphalan/HDS is comprised of: a planned Global Phase 3 clinical trial investigating overall survival in ocular melanoma liver metastases, and a Global Phase 2 clinical trial investigating Melphalan/HDS with and without sorafenib in HCC, opened for enrollment in the fall of 2014. We expect to expand the Global Phase 2 HCC trial to include a cohort of patients with ICC. Our CDP also includes sponsorship of select investigator initiated trials (IITs) in HCC and colorectal cancer liver metastases (mCRC) and the establishment of a commercial registry for CHEMOSAT non-clinical commercial cases performed in Europe.
The direction and focus of our CDP for CHEMOSAT/Melphalan/HDS is informed by our prior clinical development program, which was conducted between 2004 and 2010. This prior program included a Phase 3 trial in 93 patients that demonstrated efficacy for Melphalan/HDS in ocular melanoma liver metastases, and a Phase 2 multi-histology trial in 56 patients that also provided an efficacy signal for Melphalan/HDS in HCC. Our CDP is also informed by non-clinical, commercial CHEMOSAT cases performed on over 100 patients in Europe, and prior regulatory experience with the FDA. Experience gained from this research, development, early European commercial and U.S. regulatory activity has led to the implementation of several safety improvements to both our product and the associated medical procedure.
In the United States, Melphalan/HDS is considered a combination drug and device product, and is regulated as a drug by the FDA. The FDA has granted us five orphan drug designations, including two orphan designations for the use of the drug melphalan for the treatment of patients with ocular melanoma liver metastases and HCC. Melphalan/HDS has not been approved for sale in the United States.
In Europe, the current version of our CHEMOSAT product is regulated as a Class IIb medical device and received its CE Mark in 2012. We are in an early phase of commercializing CHEMOSAT in select markets in the European Union (United Kingdom and Germany) where we believe the prospect of securing adequate reimbursement for the procedure is strongest.
Currently there are few effective treatment options for certain cancers in the liver. Traditional treatment options include surgery, chemotherapy, liver transplant, radiation therapy, interventional radiology techniques, and isolated hepatic perfusion. We believe that CHEMOSAT/Melphalan/HDS represents a potentially important advancement in regional therapy for primary liver cancer and certain other cancers metastatic to the liver. We believe that CHEMOSAT/Melphalan/HDS is uniquely positioned to treat the entire liver either as a standalone therapy or as a complement to other therapies.
Cancers in the Liver – A Significant Unmet Need
Cancers of the liver remain a major unmet medical need globally. According to GLOBOCAN and American Cancer Society (ACS) Facts & Figures 2008, approximately 1.2 million patients globally are diagnosed each year with primary liver cancer or cancer that has metastasized to the liver. According to the American Cancer Society’s (ACS) Cancer Facts & Figures 2013 report, cancer is the second leading cause of death in the United States, with an estimated 580,350 deaths and 1,660,290 new cases expected to be diagnosed in 2013. Cancer is one of the leading causes of death worldwide, accounting for approximately 8.2 million deaths and 14.1 million new cases in 2012 according to GLOBOCAN. The financial burden of cancer is enormous for patients, their families and society. The National Institutes of Health (NIH) estimates that the overall costs of cancer in 2008 were $201 billion: $77 billion for direct medical costs (total of all health expenditures) and $124 billion for indirect mortality costs (cost of lost productivity due to premature death). The liver is often the life-limiting organ for cancer patients and one of the leading causes of cancer death. Patient prognosis is generally poor once cancer has spread to the liver.
Liver Cancers—Incidence and Mortality
There are two types of liver cancers: primary liver cancer and metastatic liver disease. Primary liver cancer (hepatocellular carcinoma or HCC, including intrahepatic bile duct cancers or ICC) originates in the liver or biliary tissue and is particularly prevalent in populations where the primary risk factors for the disease, such as hepatitis-B, hepatitis-C, high levels of alcohol consumption, aflatoxin, cigarette smoking and exposure to industrial pollutants, are present. Metastatic liver disease, also called liver metastasis, or secondary liver cancer, is characterized by microscopic cancer cell clusters that detach from the primary site of disease and travel via the blood stream and lymphatic system into the liver, where they grow into new tumors. These metastases often continue to grow even after the primary cancer in another part of the body has been removed. Given the vital biological functions of the liver, including processing nutrients from food and filtering toxins from the blood, it is not uncommon for metastases to settle in the liver. In many cases patients die not as a result of their primary cancer, but from the tumors that metastasize to their liver. In the United States, metastatic liver disease is more prevalent than primary liver cancer.
Ocular Melanoma
Ocular melanoma is one of the cancer histologies with a high likelihood of metastasizing to the liver. We estimate that up to 8,600 cases of ocular melanoma are diagnosed in the U.S. and Europe annually, and that approximately 55% of these patients will develop metastatic disease. Of metastatic cases of ocular melanoma, we estimate that approximately 90% of patients will development liver involvement. Once ocular melanoma has spread to the liver, current evidence suggests median overall survival for these patients is generally six to eight months. Currently there is no standard of care for patients with ocular melanoma liver metastases. As a result, we estimate that up to 4,300 patients with ocular melanoma liver metastases in the U.S. and Europe may be eligible for treatment with the Melphalan/HDS.
Hepatocellular Carcinoma (HCC) and Intrahepatic Cholangiocarcinoma (ICC)
Hepatobiliary cancers---including HCC and ICC---are among the most prevalent and lethal forms of cancer. According to GLOBOCAN, an estimated 76,000 new cases of primary liver cancers are diagnosed in the U.S. and Europe annually. Approximately 90% of these patients are diagnosed with HCC. Excluding patients who are eligible for surgical resection or certain focal treatments, we estimate that approximately 15,000 patients with HCC in the U.S. and Europe may be eligible for treatment with Melphalan/HDS. We estimate that an additional 6,500 patients diagnosed with ICC may also be eligible for treatment with Melphalan/HDS. According to the ACS, the overall five-year survival rate for liver cancer patients in the U.S is approximately 15% compared to 68% for all cancer combined. Globally, with 782,000 new cases in 2012, HCC was the fifth most common cancer in men and the ninth in women according to GLOBOCAN. GLOBOCAN estimates indicate that HCC was responsible for 746,000 deaths in 2012 (9.1% of the total cancer deaths), making it the second most common cause of death from cancer worldwide.
The prognosis for primary liver cancer is very poor, as indicated by an overall ratio of mortality to incidence of 0.95. The American Cancer Society’s Cancer Facts & Figures 2013 outlines the treatment options for HCC as follows: “Early stage HCC can sometimes be successfully treated with surgery in patients with sufficient healthy liver tissue; liver transplantation may also be an option. Surgical treatment of early stage HCC is often limited by pre-existing liver disease that has damaged the portion of the liver not affected by cancer. Patients whose tumors cannot be surgically removed may choose ablation (tumor destruction) or embolization, a procedure that cuts off blood flow to the tumor. Fewer treatment options exist for patients diagnosed at an advanced stage of the disease. sorafenib (Nexavar) is a targeted drug approved for the treatment of HCC in patients who are not candidates for surgery.”
In HCC, the impact of systemic chemotherapy has been very limited, primarily due to the poor liver function of presenting patients. Modest results have been reported in Phase II trials with various agents such as doxorubicin, gemcitabine, and capecitabine. A Phase III open label study compared advanced HCC patients who received doxorubicin or FOLFOX4 (5-fluorouracil and leucovirin plus oxaliplatin), and found no statistical difference in median overall survival. For HCC patients who are not eligible for surgery or TACE, the only current FDA-approved chemotherapy option is sorafenib. Sorafenib, taken orally, is a small molecule, multikinase inhibitor with activity against Raf-1, B-raf, VEGFR2 and PDGFR- proteins and signaling pathways shown to be involved in the pathogenesis of HCC. Data from two randomized Phase III clinical trials (the SHARP trial and the Asian trial) in patients with unresectable advanced HCC with Child-Pugh A score reported very modest response rates (2%), but demonstrated statistically significant survival advantages favoring sorafenib. Phase III trials evaluating the efficacy of sorafenib alone versus a combination of sorafenib plus doxorubicin, or sorafenib plus capecitabine and oxaliplatin (SECOX) are currently ongoing.
Despite its approval and widespread use as the first line treatment for unresectable HCC, there are challenges and limitations associated with sorafenib. High rates of dermatologic side effects, such as hand-foot skin reaction (HSFR) were reported in the SHARP and Asian Phase III trials. Acute diarrhea has also been described as an early and common side effect of sorafenib treatment. More recently, there have been reports of pancreatic atrophy associated with long-term sorafenib therapy, possibly due to its overall anti-angiogenic activity. Resistance to sorafenib despite initial responses has also been reported. This has spurned efforts to develop other chemotherapeutics, most of which target the multiple signaling pathways and molecules involved in HCC. Notable among these are small molecule agents such as brivanib (targeting VEGFR, FGFR), ARQ 197 or tivantinib (targeting c-MET), XL184 or cabozantinib (targeting c-MET and VEGFR), and ABT-869 or linifanib (targeting VEGFR, PDGFR, c-KIT, FLT-3), and monoclonal antibody/biologics such as ramucirumab (anti-VEGFR2), all of which are currently in Phase III trials. Other agents currently in HCC or metastatic liver cancer Phase II trials include axitinib, cediranib, and oranitinib (all targeting VEGFR), lapatinib, and gefitinib (both targeting EGFR), selumetinib (targeting MEK), and belinostat (targeting histone deacetylase). In addition to these agents, MDX-1106 (also known as BMS-936558 or nivolumab), an antibody that targets the PD-1 immuno-inhibitory receptor, is in a Phase I trial for advanced HCC.
About CHEMOSAT/Melphalan/HDS
CHEMOSAT/Melphalan/HDS administers concentrated regional chemotherapy to the liver. This “whole organ” therapy is performed by isolating the circulatory system of the liver, infusing the liver with chemotherapeutic agent, and then filtering the blood prior to returning it to the patient. During the procedure, known as percutaneous hepatic perfusion (PHP), three catheters are placed percutaneously through standard interventional radiology techniques. The catheters temporarily isolate the liver from the body’s circulatory system, allow administration of the chemotherapeutic agent melphalan hydrochloride directly to the liver, and collect blood exiting the liver for filtration by our proprietary filters. The filters absorb chemotherapeutic agent in the blood, thereby reducing systemic exposure to the drug and related toxic side effects, before the filtered blood is returned to the patient’s circulatory system.
The PHP procedure is performed in an interventional radiology suite in approximately two to three hours. Patients remain in an intensive care or step-down unit overnight for observation following the procedure. Treatment with CHEMOSAT/Melphalan/HDS is repeatable, and a new disposable CHEMOSAT/Melphalan/HDS is used for each treatment. In early clinical trials patients received an average of three procedures in four to eight week intervals. Patients treated in both clinical and non-clinical settings have received up to six treatments. In the United States, melphalan hydrochloride for injection will be included with the system. In Europe, the system is sold separately and used in conjunction with melphalan hydrochloride commercially available from a third party.
Prior Clinical Development
Our Phase 3 clinical trial and multi-arm Phase 2 clinical trial of the Melphalan/HDS with melphalan in patients with liver cancers are summarized below. The Phase 3 and Phase 2 clinical trials were subject to the terms and conditions of the Cooperative Research and Development Agreement (CRADA), between the Company and the National Cancer Institute (NCI). The Phase 3 trial was conducted under an FDA Special Protocol Assessment (SPA) and was conducted at centers throughout the United States.
Phase 3—Melanoma Metastases Trial
The most advanced application for which Melphalan/HDS was evaluated is for the treatment of metastatic melanoma in the liver. In February 2010, we concluded a randomized Phase 3 multi-center study for patients with unresectable metastatic ocular or cutaneous melanoma exclusively or predominantly in the liver. In the trial, patients were randomly assigned to receive PHP treatments with melphalan using the Melphalan/HDS, or to a control group providing best alternative care (BAC). Patients assigned to the PHP arm were eligible to receive up to six cycles of treatment at approximately four to eight week intervals. Patients randomized to the BAC arm were permitted to cross-over into the PHP arm at radiographic documentation of hepatic disease progression. A majority of the BAC patients did in fact cross over to the PHP arm. Secondary objectives of the study were to determine the response rate, safety, tolerability and overall survival.
On April 21, 2010, we announced that our randomized Phase 3 clinical trial of PHP with melphalan using Melphalan/HDS for patients with unresectable metastatic ocular and cutaneous melanoma in the liver had successfully achieved the study’s primary endpoint of extended hepatic progression-free survival, or hPFS. An updated summary of the results was presented at the European Multidisciplinary Cancer Congress organized by the European Cancer Organization (ECCO) and the European Society of Medical Oncology (ESMO) in September 2011. Data submitted in October 2012 to the FDA in Delcath’s New Drug Application (NDA) comparing treatment with the PHP with melphalan (the treatment group) to BAC (the control group), showed that patients in the PHP arm had a statistically significant longer median hPFS of 7.0 months compared to 1.7 months in the BAC control group, according to the Independent Review Committee (IRC) assessment. This reflects a 4-fold increase of hPFS over that of the BAC arm, with 50% reduction in the risk of progression and/or death in the PHP treatment arm compared to the BAC control arm. Authors of this study submitted these results for publication in a leading peer-reviewed journal in February 2015.
Phase 2 Multi-Histology, Unresectable Hepatic Tumor Trial
Also in 2010, we concluded a separate multi-arm Phase 2 clinical trial of PHP with melphalan using an early version of the Melphalan/HDS in patients with primary and metastatic liver cancers, stratified into four arms: neuroendocrine tumors (carcinoid and pancreatic islet cell tumors), ocular or cutaneous melanoma, metastatic colorectal adenocarcinoma (mCRC), and HCC. In the metastatic neuroendocrine (mNET) cohort (n=24), the objective tumor response rate was 42%, with 66% of patients achieving hepatic tumor shrinkage and durable disease stabilization. In the mCRC cohort, there was inconclusive efficacy possibly due to advanced disease status of the patients. Similar safety profiles were seen across all tumor types studied in the trial.
Phase 2 Multi-Histology Clinical Trial - HCC Cohort
In the HCC cohort (n=8) of our Phase 2 Multi-Histology trial, a positive signal in hepatic malignancies was observed in 5 patients. Among these patients, one patient received four treatments, achieved a partial response lasting 12.22 months, and survived 20.47 months. Three other patients with stable disease received 3-4 treatments, with hepatic progression free survival (hPFS) ranging 3.45 to 8.15 months, and overall survival (OS) ranging 5.26 to 19.88 months. There was no evidence of extrahepatic disease progression. The observed duration of hPFS and OS in this limited number of patients exceeded that generally associated with this patient population. We believe these results constitute a promising signal that warrants further clinical investigation.
Risks associated with the CHEMOSAT/Melphalan/HDS Procedure
As with many cancer therapies, treatment with CHEMOSAT/Melphalan/HDS is associated with toxic side effects and certain risks, some of which are potentially life threatening. In our Phase 2 and 3 clinical trials using early versions of CHEMOSAT/Melphalan/HDS and treatment protocol, the integrated safety population of patients treated with CHEMOSAT/Melphalan/HDS showed these risks to include: a 4.1% incidence of deaths due to adverse reactions; 4% incidence of stroke; 2% incidence of myocardial infarction in the setting of an incomplete cardiac risk assessment; a ≥ 70% incidence of grade 4 bone marrow suppression with a median time of recovery of greater than 1 week; and 8% incidence of febrile neutropenia, along with the additive risk of hepatic injury, severe hemorrhage, and gastrointestinal perforation. In this integrated safety population, deaths due to certain adverse reactions did not occur again during the clinical trials following the adoption of related protocol amendments.
Procedure and Product Refinements
The trials that comprised this integrated safety population used early versions of the device and procedure. As a consequence of these identified risks and experience gained in non-clinical, commercial usage in Europe, we have continued to develop and refine both the CHEMOSAT/Melphalan/HDS and the PHP procedure. The procedure refinements have included modifications to the pre-, peri- and post-procedure patient management and monitoring, as well as the use of the following: prophylactic administration of proton pump inhibitors, prophylactic platelet transfusions, prophylactic hydration at key pre-treatment intervals, use of vasopressor agents coupled with continuous monitoring for maintenance of blood pressure and prophylactic administration of growth factors to reduce risk of serious myelosuppression. In addition, in 2012, we introduced the Generation Two version of the CHEMOSAT system, which offered improved hemofiltration and other product enhancements.
Reports from treating physicians in both Europe and the U.S. using the Generation Two CHEMOSAT/Melphalan/HDS in a non-clinical, commercial setting have suggested that these product improvements and procedure refinements have improved the safety profile.
Clinical Development Program
The focus of our CDP is to generate clinical data for the CHEMOSAT/Melphalan/HDS in various disease states and validate the safety profile of the current version of the product and treatment procedure. The program also seeks to address the requirements contained in the FDA’s Complete Response Letter (CRL) received in September 2013, which was issued in response to our New Drug Application which we submitted in 2012 seeking an indication in ocular melanoma liver metastases. We believe that the improvements we have made to CHEMOSAT/Melphalan/HDS and to the PHP procedure have addressed the severe toxicity and procedure-related risks observed during the previous Phase 2 and 3 clinical trials. The CDP is also designed to support clinical adoption of and reimbursement for CHEMOSAT in Europe, and to support regulatory approvals in various jurisdictions, including the U.S.
Global Phase 3 Ocular Melanoma Trial
Ocular melanoma is one of the cancer histologies with a high likelihood of metastasizing to the liver. Once ocular melanoma has spread to the liver, current evidence suggests median overall survival for these patients is generally six to eight months. According to the ACS and other international health agencies, approximately 8,600 cases of ocular melanoma are diagnosed annually in the U.S. and Europe. Over half of these patients will develop metastatic disease and approximately 90% of these patients will have liver involvement. As a result, we believe that up to 4,300 patients eligible for treatment with the CHEMOSAT/Melphalan/HDS. There currently is no standard of care for the treatment of ocular melanoma metastatic to the liver. Melphalan Hydrochloride has been granted orphan drug status by FDA for treatment of patients with ocular melanoma.
We are advancing plans to initiate a pivotal Phase 3 overall survival (OS) clinical trial in ocular melanoma that is metastatic to the liver for resubmission of our NDA to the FDA. Based on the strength of the efficacy data in this disease observed in our previous Phase 3 clinical trial and the reports of an improved safety profile from over 100 patients treated in a non-clinical trial setting in Europe, we are confident that this program can address the concerns raised by the FDA in its CRL. We are working with the relevant Health Authorities in Europe and the U.S. to initiate this trial. We believe that ocular melanoma liver metastases represent a high unmet medical need, and that pursuit of an indication in this disease state may be the fastest path to potential approval of the Melphalan/HDS in the U.S.
Phase 2 Hepatocellular Carcinoma (HCC) & Intrahepatic Cholangiocarcinoma (ICC) Program
Based on third party research, we estimate that approximately 15,000 of the 76,000 patients diagnosed annually in the U.S. and Europe could be eligible candidates for treatment with the Melphalan/HDS. The FDA has granted orphan drug status to Melphalan Hydrochloride for treatment of patients with unresectable HCC. We believe that there is a large unmet medical need in first line therapy for patients with HCC, with sorafenib the only currently approved systemic therapy in the U.S., Europe and certain Asian markets.
ICC is the second most common primary liver tumor and accounts for 3% of all gastrointestinal cancers and 15% of HCC cases diagnosed in the U.S. and Europe annually. Outside of resection, which is the only cure for ICC, there is currently no standard of care (SOC). Based on third party research we believe that 90% of ICC patients are not candidates for surgical resection, and that approximately 20-30% of these may be candidates for certain focal interventions. We estimate that approximately 6,500 ICC patients in the U.S. and Europe annually could be candidates for treatment with Melphalan/HDS, which we believe represents a significant market opportunity. We intend to pursue an orphan drug designation from the FDA for Melphalan Hydrochloride for the treatment of patients with ICC.
In 2014 we initiated a new clinical trial program in Europe and the U.S., with the goal of obtaining an efficacy and safety signal for Melphalan/HDS in the treatment of HCC and ICC. Due to differences in treatment practice patterns between Europe and the U.S., we established separate European and U.S. trial protocols for the HCC Phase 2 program with different inclusion and exclusion patient selection criteria:
| o | Protocol 201 – Conducted in the U.S., this trial will assess the safety and efficacy of Melphalan/HDS followed by sorafenib. The trial will evaluate overall response rate via modified Response Evaluation Criteria in Solid Tumors (mRECIST), progression free survival, characterize the systemic exposure of melphalan and assess patient quality of life. The Moffitt Cancer Center opened for enrollment in this trial October 2014, and we expect to add additional centers in the U.S. in 2015. |
| o | Protocol 202 – conducted in Europe, this trial will assess the safety and efficacy of Melphalan/HDS without sorafenib. The trial will also evaluate overall response rate via (mRECIST) criteria, progression free survival, characterize the systemic exposure of melphalan and assess patient quality of life. Three hospitals in Germany have opened for enrollment --- Goethe University Hospital, Hannover Medical School Hospital and Jena University Hospital. We intend to open additional centers in Germany and the U.K., subject to the applicable authorizations and approvals including ethics committee approval at participating hospitals. |
| o | ICC Cohort – In 2015 we expect to expand Protocol 202 to include a cohort of patients with ICC. We expect the trial for this cohort will be conducted at the same centers participating in the Phase 2 HCC trial. |
Clinical trials are long, expensive and highly uncertain processes and failure can unexpectedly occur at any stage of clinical development. The start or end of a clinical trial is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparator treatment or required prior therapy.
European Investigator Initiated Trials
In addition to the clinical trials in our CDP, we are supporting data generation in other areas. We are currently supporting two Investigator Initiated Trials (IITs) in Europe– one in colorectal carcinoma metastatic to the liver (mCRC) at Leiden University Medical Center in The Netherlands, and another in HCC at Goethe University Hospital in Frankfurt Germany. Both of these trials have opened for enrollment. We continue to evaluate other IITs as suitable opportunities present in Europe. We believe IITs will serve to build clinical experience at key cancer centers, and will help support efforts to obtain full reimbursement in Europe.
European Clinical Data Generation
We are also initiating a prospective registry in Europe to collect data from cases performed in a commercial setting. This registry will gather data in multiple tumor types from commercial cases performed by participating cancer centers. A prospective registry is an organized system that uses observational study methods to collect defined clinical data under normal conditions of use to evaluate specified outcomes for a population defined by a particular disease, condition, or exposure. We intend to collect essential patient safety and efficacy information beginning with treatment centers in the Netherlands. Registry data is non-randomized, and as such cannot be used for either registration approval, promotional or competitive claims. However, we believe the Patient Registry will provide a valuable data repository from a commercial setting that can be used to support clinical adoption and reimbursement in Europe.
Recent Data Presentations
In October 2014, three abstracts detailing the clinical experiences at three leading cancer hospitals using CHEMOSAT/Melphalan HDS to perform percutaneous hepatic perfusion (PHP) were presented at the European Society of Surgical Oncology (ESSO) congress. The three presentations were:
| · | A Single Institution Experience with Percutaneous Hepatic Perfusion for Unresectable Ocular Melanoma and Sarcoma in the Liver, presented by Dr. Jonathan Zager of the Moffitt Cancer Center in Tampa, FL. Dr. Zager reported that among 13 patients treated at Moffitt a 67% positive response rate was observed, with one partial response and one complete response. |
| · | Percutaneous Hepatic Perfusion with Melphalan in Treating Unresectable Liver Metastases from Colorectal Cancer and Uveal (Ocular) Melanoma, presented by Dr. Neal de Leede of Leiden University Medical Centre (LUMC) in the Netherlands. Dr. de Leede reported that among 11 patients treated at LUMC an 80% partial response rate was observed. |
| · | Initial United Kingdom Experience with Melphalan Percutaneous Hepatic Perfusion (PHP) For Treatment of Inoperable Ocular Melanoma Metastases, presented by Dr. Brian Steadman of the University Hospital Southampton (UHS) in the United Kingdom. Dr. Steadman reported that in 19 patients treated at UHS, a 63% positive response was observed with 47% having a partial response and 16% having a complete response. |
These response rates were achieved with a range of one to six treatments. All authors concluded that in their opinion CHEMOSAT or Melphalan/HDS is a safe and effective procedure for selected patients. These abstracts, as submitted, can be downloaded from the ECCO-ESSO website. A link to this site is available on www.delcath.com.
Market Access & Commercial Clinical Adoption
European Union
With continued economic and reimbursement challenges in certain European markets, in 2014 our immediate market access and clinical adoptions efforts were focused on the key target markets of Germany and the United Kingdom, which represent a majority of the total potential liver cancer market (primary and metastatic) in the EU and where progress in securing reimbursement for CHEMOSAT treatments offers the best near-term opportunities. We also continue to support clinical adoption of CHEMOSAT in the Netherlands, Spain, France and Italy. We employ a combination of direct and indirect sales channels to market and sell CHEMOSAT in these markets. In 2014, we phased out a medical science liaison consultant program as part of our cost reduction and restructuring efforts, and have integrated these capabilities into existing resources. Our European Headquarters is in Galway, Ireland.
Since its February 2012 launch, CHEMOSAT has been used to perform more than 160 treatments. During 2014, 79 CHEMOSAT commercial treatments were performed, with 34 of these representing retreatments. This represents a nearly 100% increase in treatments and a nearly 200% increase in retreatments over 2013.
Since launching CHEMOSAT in Europe, treatments have been performed at 19 leading European cancer centers. Physicians in Europe have used CHEMOSAT to treat patients with a variety of cancers in the liver primarily ocular melanoma liver metastases, and other tumor types, including hepatocellular carcinoma, cholangiocarcinoma, and liver metastases from colorectal cancer, breast, and cutaneous melanoma.
European Reimbursement
A critical driver of utilization growth for CHEMOSAT in Europe is the expansion of reimbursement mechanisms for the procedure in our priority markets. In Europe, there is no centralized pan-European medical device reimbursement body. Reimbursement is administered on a regional and national basis. Medical devices are typically reimbursed under Diagnosis Related Groups (DRG) as part of a procedure. Prior to obtaining permanent DRG reimbursement codes, in certain jurisdictions, the Company is actively seeking interim reimbursement from existing mechanisms that include specific interim reimbursement schemes, new technology payment programs as well as existing DRG codes. In most EU countries, the government provides healthcare and controls reimbursement levels. Since the EU has no jurisdiction over patient reimbursement or pricing matters in its member states, the methodologies for determining reimbursement rates and the actual rates may vary by country.
Germany
In February 2015, we announced that the Institut fϋr das Entgeltsystem im Krankenhaus (InEk), the German federal reimbursement agency, again granted Value 4 coverage status for the treatment of patients with liver metastases with CHEMOSAT. The InEk determines three status levels for medical procedures submitted for its review: Value 1 (mandated reimbursement), Value 2 (declined for reimbursement), and Value 4 (negotiated reimbursement). The InEk may also decline to make a determination regarding an application. Under the Neue Untersuchungs und Behandlungsmethoden (NUB) reimbursement scheme, Value 4 Status, while not mandating reimbursement, allows participating cancer centers to negotiate a budget to fund reimbursement coverage for CHEMOSAT procedure with insurers serving their region. The InEk first established NUB Value 4 status for CHEMOSAT procedures in 2013, and repeated this assessment in 2014. The NUB is an annual process, and participating centers in Germany are required to apply each year for subsequent coverage under the NUB scheme.
Separately, throughout 2014 physicians and patients in Germany submitted and received approvals for Individual Funding Requests (IFRs) granting reimbursement for the treatment of liver metastases with CHEMOSAT. IFRs are case-by-case appeals for reimbursement made to the patient’s insurance carrier (“sickness funds”). While each IFR is evaluated independently, the majority of these applications were approved during the year. Of those IFRs that were initially rejected, subsequent appeals over-ruled most of these rejections and allowed treatments to be funded. IFR approvals have covered a range of sickness funds across a number of regions in Germany including ocular melanoma, cutaneous melanoma, intrahepatic cholangiocarcinoma, pancreatic cancer and sarcoma; and some were granted for multiple treatments of the same patients. We expect that IFRs will continue to be the main reimbursement vehicle in the German market in 2015.
The German Radiology Society resubmitted its application for ZE (Zusatzentgeld) for CHEMOSAT in March 2014, but did not affect the relevant DRG codes in 2014. ZE is a national interim reimbursement code granted by the InEk until a specific DRG code can be created. A ZE code is dependent on having enough financial data related to the procedure to establish cost averages, and our efforts are focused on ensuring that treatment and cost data from specific hospitals are provided to the InEk to support a future ZE application.
United Kingdom
In the United Kingdom, though Delcath and our participating cancer centers identified existing Healthcare Resource Groups (HRG) code(s), we have been advised that hospitals have not used it for coverage of CHEMOSAT related costs. We continue to work with the HRG organization that decides on new HRG codes toward receipt of a dedicated and permanent reimbursement code in the future.
Throughout 2014, we supported efforts to seek a block fund grant through the Commissioning Through Evaluation (CTE) process, which may ultimately provide funding for up to 50-75 ocular melanoma patients to be treated utilizing CHEMOSAT at two or three centers in the U.K. This process has been driven by our partner centers and their clinical community, with the centers applying for funding for a limited number of patients with ocular melanoma. In the fourth quarter of 2014, Aintree University Hospital in Liverpool was activated with the intention of it becoming one of these CTE centers. The British healthcare system continues to evolve however, and ongoing changes to the CTE process and funding streams have resulted in delays that made the award and timing of any block grant funding difficult to predict. Our current expectation is for the process to be completed by the end of the second quarter 2015 with the funding, if any, becoming available in the third quarter of 2015. The entire CTE funding mechanism is a new process and the ongoing policy changes in the National Health Service (NHS) make it difficult to predict the likelihood of success in the near term.
In May 2014, the National Institute for Clinical Excellence (NICE), a non-departmental public body that provides guidance and advice to improve health and social care in the UK, completed a clinical review of CHEMOSAT. The NICE review indicated that as the current body of evidence on the safety and efficacy of PHP with CHEMOSAT for primary or metastatic liver cancer is limited, the procedure should be performed within the context of research by clinicians with specific training in its use and techniques. NICE stated that this research may take the form of observational studies. With UK participation in our Phase 2 HCC trial beginning in 2015, we believe the data generated from these studies will help provide supporting clinical data and address the concerns raised by NICE relative to survival, quality of life and adverse events. NICE may decide to conduct a Technology Appraisal of CHEMOSAT thereafter, the outcome of which could influence the long-term reimbursement status.
Public patients will continue to be treated in the UK through clinical trials and potentially the CTE process. Private patients will continue to be treated through the established private treatment pathway such as private insurance coverage or self-pay.
Other European Markets
Permanent reimbursement coverage in remaining EU markets will require additional time to secure. In the interim period, we are seeking payment through various avenues, including new technology programs. In France, we plan to present our Phase 3 trial data to the French healthcare authorities assuming publication in 2015 to set the foundation for a potential DRG code in 2016.
For France, Spain and the Netherlands, publication of the Phase 3 trial manuscript is a key component of the reimbursement process. The Phase 3 trial manuscript has been submitted and we expect publication in 2015.
Distribution Partners
As a result of the Company’s strategy to prioritize resources on the key direct markets of Germany and the United Kingdom, the Company expects that its distribution strategy will continue to play a lesser role in its current commercial activities. In Spain, the Company has determined that there was no benefit to continuing with an indirect model and therefore terminated its relationship with its distributor in Spain and is now represented in Spain through a sales agency.
Regulatory Status
Our products are subject to extensive and rigorous government regulation by foreign regulatory agencies and the FDA. Foreign regulatory agencies, the FDA and comparable regulatory agencies in state and local jurisdictions impose extensive requirements upon the clinical development, pre-market clearance and approval, manufacturing, labeling, marketing, advertising and promotion, pricing, storage and distribution of pharmaceutical and medical device products. Failure to comply with applicable foreign regulatory agency or FDA requirements may result in Warning Letters, fines, civil or criminal penalties, suspension or delays in clinical development, recall or seizure of products, partial or total suspension of production or withdrawal of a product from the market.
U.S. Regulatory History
In August 2012, we submitted our New Drug Application (NDA) for the Melblez Kit under Section 505(b)(2) of the Federal Food Drug Cosmetic Act (FFDCA) seeking an indication for the percutaneous intra-arterial administration of melphalan for use in the treatment of patients with metastatic melanoma in the liver, and subsequently amended the indication to ocular melanoma metastatic to the liver. Data submitted to the FDA used the early clinical trial versions of the system along with early clinical procedure techniques. Our NDA was accepted for filing by the Food and Drug Administration (FDA) on October 15, 2012, and was designated for standard review with an initial Prescription Drug User Fee Act (PDUFA) goal date of June 15, 2013. On April 3, 2013, the FDA extended its PDUFA goal date to September 13, 2013.
On May 2, 2013 the Company announced that an Oncologic Drug Advisory Committee (ODAC) panel convened by the FDA voted 16 to 0, with no abstentions, that the benefits of treatment with the Melblez Kit do not outweigh the risks associated with the procedure using the early clinical trial versions of the system. A significant portion of FDA’s presentation to the ODAC panel was focused on the FDA’s assessment of treatment related risks, including the analysis of treatment-related deaths that occurred during clinical trials. Five deaths (4.1%) in the Phase 2 and Phase 3 clinical trials were considered by the treating principal investigators to be treatment-related and resulted from adverse events. In the FDA’s presentation at ODAC, FDA disagreed with this assessment of procedure related deaths and added three additional deaths, for a total of a 7% percent death rate, in the combined Phase 2 and Phase 3 programs. Two deaths related to hepatic failure and one death related to myelosuppression, were described. Upon being advised of the FDA’s assessment of these deaths, we requested that the cases be re-reviewed by the treating principal investigators. After this review, the treating principal investigators continue to be convinced that these patients died of disease progression.
The FDA also expressed concerns about hypotension (low blood pressure) during the procedure, length of hospital stay, as well as risks of stroke, heart attack, renal failure, and bone marrow suppression. We believe that the protocol amendments and other procedure refinements instituted during clinical trials and subsequently in commercial, non-clinical usage in Europe, including changes to the way blood pressure is managed and monitored, may help address these procedure related risks. Collection of adequate safety data on all aspects of the procedure is a major focus of the clinical trials planned in our CDP.
Briefing materials presented to the ODAC by both the FDA and Delcath are available on our website at http://delcath.com/clinical-research/clinical-bibliography.
Complete Response Letter
On September 12, 2013, the FDA issued a complete response letter (CRL) regarding our NDA for Melblez Kit. The FDA issues a CRL after the review of a file has been completed and questions remain that preclude approval of the NDA in its current form. The FDA comments included, but were not limited to, a statement that Delcath must perform another "well-controlled randomized trial(s) to establish the safety and efficacy of Melblez Kit using overall survival as the primary efficacy outcome measure," and which "demonstrates that the clinical benefits of Melblez Kit outweigh its risks." The FDA also requires that the additional clinical trial(s) be conducted using the product the Company intends to market. In November 2013 Delcath and FDA participated in a meeting to discuss and clarify certain clinical, clinical pharmacology, Human Factors and product quality components of the CRL. We are working to incorporate the requirements referenced in the CRL into our clinical development program.
United States Regulatory Environment
In the United States, the FDA regulates drug and device products under the Federal Food, Drug, and Cosmetic Act (FFDCA), and it’s implementing regulations. The Delcath Melphalan/HDS is subject to regulation as a combination product, which means it is composed of both a drug product and device product. If marketed individually, each component would therefore be subject to different regulatory pathways and reviewed by different centers within the FDA. A combination product, however, is assigned to a center that will have primary jurisdiction over its pre-market review and regulation based on a determination of its primary mode of action, which is the single mode of action that provides the most important therapeutic action. In the case of the Melphalan/HDS, the primary mode of action is attributable to the drug component of the product, which means that the Center for Drug Evaluation and Research (CDER), has primary jurisdiction over its pre-market development and review.
The process required by the FDA before drug product candidates may be marketed in the United States generally involves the following:
| o | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin and must be updated annually; |
| o | completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA’s Good Laboratory Practice, or GLP, regulations; |
| o | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; |
| o | submission to the FDA of an NDA after completion of all pivotal clinical trials; |
| o | a determination by the FDA within 60 days of its receipt of an NDA to file the NDA for review; |
| o | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the product is produced and tested to assess compliance with current good manufacturing practice, or cGMP, regulations; and |
| o | FDA review and approval of an NDA prior to any commercial marketing or sale of the drug in the United States. |
The development and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product will be granted on a timely basis, if at all.
The results of preclinical tests (which include laboratory evaluation as well as GLP studies to evaluate toxicity in animals) for a particular product candidate, together with related manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the proposed clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. IND submissions may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive good clinical practice regulations and regulations for informed consent and privacy of individually identifiable information. Similar requirements to the U.S. IND are required in the EEA and other jurisdictions in which we may conduct clinical trials.
Clinical Trials
For purposes of NDA submission and approval, clinical trials are typically conducted in the following sequential phases, which may overlap:
| o | Phase I Clinical Trials. Studies are initially conducted in a limited population to test the product candidate for safety, dose tolerance, absorption, distribution, metabolism and excretion, typically in healthy humans, but in some cases in patients. |
| o | Phase 2 Clinical Trials. Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, explore the initial efficacy of the product for specific targeted indications and to determine dose range or pharmacodynamics. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| o | Phase 3 Clinical Trials. These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, provide substantial evidence of clinical efficacy and further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial centers. |
| o | Phase IV Clinical Trials. The FDA may approve an NDA for a product candidate, but require that the sponsor conduct additional clinical trials to further assess the drug after NDA approval under a post-approval commitment. In addition, a sponsor may decide to conduct additional clinical trials after the FDA has approved an NDA. Post-approval trials are typically referred to as Phase IV clinical trials. |
Sponsors of clinical trials may submit proposals for the design, execution, and analysis for their pivotal trials under a Special Protocol Assessment (SPA). A SPA is an evaluation by the FDA of a protocol with the goal of reaching an agreement that the Phase 3 trial protocol design, clinical endpoints, and statistical analyses are acceptable to support regulatory approval of the drug product candidate with respect to effectiveness for the indication studied. Under a SPA, the FDA agrees to not later alter its position with respect to adequacy of the design, execution or analyses of the clinical trial intended to form the primary basis of an effectiveness claim in an NDA, without the sponsor’s agreement, unless the FDA identifies a substantial scientific issue essential to determining the safety or efficacy of the drug after testing begins. Prior to initiating our Phase 3 clinical trial, we submitted a proposal for the design, execution and analysis under a SPA, and we conducted our Phase 3 trial under a SPA.
New Drug Applications
The results of drug development, preclinical studies and clinical trials are submitted to the FDA as part of a New Drug Application (NDA). NDAs also must contain extensive chemistry, manufacturing and control information. An NDA must be accompanied by a significant user fee, which may be waived in certain circumstances. Once the submission has been accepted for filing, the FDA’s goal is to review applications within ten months of submission or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months from submission. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. For new oncology products, the FDA will often solicit an opinion from an Oncologic Drugs Advisory Committee (ODAC), a panel of expert authorities knowledgeable in the fields of general oncology, pediatric oncology, hematologic oncology, immunologic oncology, biostatistics, and other related professions. The ODAC panel reviews and evaluates data concerning the safety and effectiveness of marketed and investigational human drug products for use in the treatment of cancer, and makes appropriate recommendations to the Commissioner of Food and Drugs. The FDA is not bound by the recommendation of an advisory committee. The FDA may deny approval of an NDA by issuing a Complete Response Letter (CRL) if the applicable regulatory criteria are not satisfied. A CRL may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Approval may be contingent on a Risk Evaluation and Mitigation Strategy (REMS) that limits the labeling, distribution or promotion of a drug product. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, and surveillance programs to monitor the safety effects of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
There are three primary regulatory pathways for a New Drug Application under Section 505 of The Federal Food Drug and Cosmetics Act (FFDCA): Section 505 (b)(1), Section 505 (b)(2) and Section 505(j). A Section 505 (b)(1) application is used for approval of a new drug (for clinical use) whose active ingredients have not been previously approved. A Section 505 (b)(2) application is used for a new drug that relies on data not developed by the applicant. Section 505(b)(2) of the FFDCA was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act. This statutory provision permits the approval of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The Hatch-Waxman Act permits the applicant to rely in part upon the FDA’s findings of safety and effectiveness for previously approved products. Section 505(j) application, also known as an abbreviated NDA, is used for a generic version of a drug that has already been approved.
Orphan Drug Exclusivity
Some jurisdictions, including the United States, may designate drugs for relatively small patient populations as orphan drugs. Pursuant to the Orphan Drug Act, the FDA grants orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. The orphan designation is granted for a combination of a drug entity and an indication and therefore it can be granted for an existing drug with a new (orphan) indication. Applications are made to the Office of Orphan Products Development at the FDA and a decision or request for more information is rendered in 60 days. NDAs for designated orphan drugs are exempt from user fees, obtain additional clinical protocol assistance, are eligible for tax credits up to 50% of research and development costs, and are granted a seven-year period of exclusivity upon approval. The FDA cannot approve the same drug for the same condition during this period of exclusivity, except in certain circumstances where a new product demonstrates superiority to the original treatment. Exclusivity begins on the date that the marketing application is approved by the FDA for the designated orphan drug, and an orphan designation does not limit the use of that drug in other applications outside the approved designation in either a commercial or investigational setting.
The FDA has granted Delcath five orphan drug designations. In November 2008, the FDA granted Delcath two orphan drug designations for the drug melphalan for the treatment of patients with cutaneous melanoma as well as patients with ocular melanoma. In May 2009, the FDA granted Delcath an additional orphan drug designation of the drug melphalan for the treatment of patients with neuroendocrine tumors. In August 2009, the FDA granted Delcath an orphan drug designation of the drug doxorubicin for the treatment of patients with primary liver cancer. In October 2013, the FDA granted Delcath orphan drug designation of the drug melphalan for the treatment of HCC.
Other Regulatory Requirements
Products manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping, annual product quality review and reporting requirements. Adverse event experience with the product must be reported to the FDA in a timely fashion and pharmacovigilance programs to proactively look for these adverse events are mandated by the FDA. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Following such inspections, the FDA may issue notices on Form 483 and Untitled Letters or Warning Letters that could cause us or our third-party manufacturers to modify certain activities. A Form 483 Notice, if issued at the conclusion of an FDA inspection, can list conditions the FDA investigators believe may have violated cGMP or other FDA regulations or guidelines. In addition to Form 483 Notices and Untitled Letters or Warning Letters, failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If we or our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may require us to recall our products from distribution or withdraw any potential approvals of an NDA for that product.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. Failure to comply with these requirements can result in adverse publicity, Warning Letters, corrective advertising and potential civil and criminal penalties.
Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, in particular in oncology. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use.
European Regulatory Environment
In the EEA, the CHEMOSAT system is subject to regulation as a medical device. The EEA is composed of the 28 Member States of the European Union plus Norway, Iceland, Liechtenstein, Switzerland and Turkey. Under the EU Medical Devices Directive (Directive No 93/42/ECC of 14 June 1993, as last amended), drug delivery products such as the CHEMOSAT system is governed by the EU laws on pharmaceutical products only if they are (i) placed on the market in such a way that the device and the pharmaceutical product form a single integral unit which is intended exclusively for use in the given combination, and (ii) the product is not reusable. In such cases, the drug delivery product is governed by the EU Code on Medicinal Products for Human Use (Directive 2001/83/EC, as last amended), while the essential requirements of the EU Medical Devices Directive apply to the safety and performance-related device features of the product. Because we do not intend to place the CHEMOSAT system on the EEA market as a single integral unit with melphalan, the product is governed solely by the EU Medical Devices Directive, while the separately marketed drug is governed by the EU Code relating to Medicinal Products for Human Use and other EU legislation applicable to drugs for human use.
Before we may commercialize a medical device in the EEA, we must comply with the essential requirements of the EU Medical Devices Directive. Compliance with these requirements entitles a manufacturer to affix a CE conformity mark, without which the products cannot be commercialized in the EEA. To demonstrate compliance with the essential requirements and obtain the right to affix the CE conformity mark, medical device manufacturers must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. In April 2011, we obtained authorization to affix a CE Mark for the Generation One CHEMOSAT system and began European commercialization with this version of the CHEMOSAT system in early 2012. In April 2012, the Company obtained authorization to affix a CE Mark for the Generation Two CHEMOSAT system, and since this time all procedures in Europe have been performed with this version of the system
The Medical Devices Directive establishes a classification system placing devices into Class I, IIa, IIb, or III, depending on the risks and characteristics of the medical device. For certain types of low risk medical devices (i.e., Class I devices which are non-sterile and do not have a measuring function), the manufacturer may issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the essential requirements of the EU Medical Devices Directives. Other devices are subject to a conformity assessment procedure requiring the intervention of a Notified Body, which is an organization designated by a Member State of the EEA to conduct conformity assessments.
CHEMOSAT is regulated as a Class IIb medical device. As a Class IIb medical device, the Notified Body is not required to carry out an examination of the product’s design dossier as part of its conformity assessment prior to commercialization. The Company must continue to comply with the essential requirements of the EU Medical Devices Directive (Directive 93/42 EC) and is subject to a conformity assessment procedure requiring the intervention of a Notified Body. The conformity assessment procedure for Class IIb medical devices requires the manufacturer to apply for the assessment of its quality system for the design, manufacture and inspection of its medical devices by a Notified Body. The Notified Body will audit the system to determine whether it conforms to the provisions of the Medical Devices Directive. If the Notified Body’s assessment is favorable it will issue a Full Quality Assurance Certificate, which enables the manufacturer to draw a Declaration of Conformity and affix the CE mark to the medical devices covered by the assessment. Thereafter, the Notified Body will carry out periodic audits to ensure that the approved quality system is applied by the manufacturer.
A manufacturer without a registered place of business in a Member State of the European Union which places a medical device on the market under its own name must designate an authorized representative established in the European Union who can act before, and be addressed by, the Competent Authorities on the manufacturer’s behalf with regard to the manufacturer’s obligations under the EU Medical Devices Directive. We appointed such a representative prior to establishing our infrastructure in the EEA and expect that we will not need a third party representative in the future.
In the EEA, we must also comply with the Medical Device Vigilance System, which is designed to improve the protection of health and safety of patients, users and others by reducing the likelihood of recurrence of incidents related to the use of a medical device. Under this system, incidents are defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient, or user or of other persons or to a serious deterioration in their state of health. When a medical device is suspected to be a contributory cause of an incident, its manufacturer or authorized representative in the European Union must report it to the Competent Authority of the Member State where the incident occurred. Incidents are generally investigated by the manufacturer. The manufacturer’s investigation is monitored by the Competent Authority, which may intervene, or initiate an independent investigation if considered appropriate. An investigation may conclude in the adoption of a Field Safety Corrective Action (FSCA). An FSCA is an action taken by a manufacturer to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market. An FSCA may include device recall, modification exchange and destruction. FSCAs must be notified by the manufacturer or its authorized representative to its customers and/or the end users of the medical device via a Field Safety Notice.
In the EEA, the off-label promotion of a pharmaceutical product is strictly prohibited under the EU Community Code on Medicinal Products, which provides that all information provided within the context of the promotion of a drug must comply with the information contained in its approved summary of product characteristics. Our product instructions and indication reference the chemotherapeutic agent melphalan hydrochloride. However, no melphalan labels in the EEA reference our product, and the labels vary from country to country with respect to the approved indication of the drug and its mode of administration. In the exercise of their professional judgment in the practice of medicine, physicians are generally allowed, under certain conditions, to use or prescribe a product in ways not approved by regulatory authorities. Physicians intending to use our device must obtain melphalan separately for use with the CHEMOSAT system and must use melphalan independently at their discretion.
In the EEA, the advertising and promotion of our products is also subject to EEA Member States laws implementing the EU Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising and Directive 2005/29/EC on unfair commercial practices, as well as other EEA Member State legislation governing the advertising and promotion of medical devices. These laws may further limit or restrict the advertising and promotion of our products to the general public and may also impose limitations on our promotional activities with health care professionals.
Failure to comply with the EEA Member State laws implementing the Medical Devices Directive, with the EU and EEA Member State laws on the promotion of medicinal products or with other applicable regulatory requirements can result in enforcement action by the EEA Member State authorities, which may include any of the following: fines, imprisonment, orders forfeiting products or prohibiting or suspending their supply to the market, or requiring the manufacturer to issue public warnings, or to conduct a product recall.
The European Commission reviewed the medical devices legislative framework in 2012 with the aim of simplifying it and ensuring a more uniform application of the provisions contained in the medical devices directives across the EEA. We do not believe the adopted regulatory changes will impact our business at this time, though future changes to the medical device legislation may adversely affect our business, financial condition and results of operations or restrict our operations.
Other International Regulations
The CHEMOSAT device has received registrations in the following countries: Australia, New Zealand, Argentina, Taiwan, and Singapore. With limited resources and our attention focused on European commercial and clinical adoption efforts, pursuing other markets at this time is not practical. We will continue to evaluate commercial opportunities in these and other markets when resources are available and at an appropriate time.
Competition
The healthcare industry is characterized by extensive research, rapid technological progress and significant competition from numerous healthcare companies and academic institutions. Competition in the cancer treatment industry is intense. We believe that the primary competitive factors for products addressing cancer include safety, efficacy, ease of use, reliability and price. We also believe that physician relationships, especially relationships with leaders in the medical and surgical oncology communities, are important competitive factors. We also believe that the current global economic conditions and new healthcare reforms could put competitive pressure on us, including reduced selling prices and potential reimbursement rates, and overall procedure rates. Certain markets in Europe are experiencing the effects of continued economic weakness, which is affecting healthcare budgets and reimbursement.
The CHEMOSAT/Melphalan/HDS competes with all forms of liver cancer treatments, including surgery, systemic chemotherapy, focal therapies and palliative care. In the disease states we are targeting there are also numerous clinical trials sponsored by third-parties, which can compete for potential patients in the near term and may ultimately lead to new competitive therapies.
For ocular melanoma liver metastases, there are currently no approved or effective treatment options, and patients are generally treated with a variety of focal and regional techniques. There are numerous companies developing and marketing devices for the performance of focal therapies, including Covidian, Biocompatibles, Merit, CeleNova, SirTex, AngioDynamics, and many others.
For HCC, sorafenib (Nexavar, Onyx Pharmaceuticals) remains the only targeted drug approved for the treatment of HCC in patients who are not candidates for surgery.
Several therapies have been recently approved for unresectable or metastatic cutaneous melanoma, which may encompass liver metastases. Dabrafenib (Tafinlar™, GlaxoSmithKline), is indicated as single agent for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation, and in combination with trametinib in unresectable or metastatic melanoma with BRAF V600E or V600K mutations. Furthermore, trametinib (MEKINIST™, GlaxoSmithKline) is indicated as single agent (in addition to in combination with dabrafinib) for treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations. Previously approved melanoma therapies such as the biologic ipilimumab (Yervoy™, Bristol Myers Squibb) and the B-RAF targeted drug vemurafenib (Zelboraf™, Genentech) may also make up the competitive landscape for the treatment of metastatic liver disease.
Many of these treatments are approved in Europe and other global markets.
Many of our competitors have substantially greater financial, technological, research and development, marketing and personnel resources. In addition, some of our competitors have considerable experience in conducting clinical trials, regulatory, manufacturing and commercialization capabilities. Our competitors may develop alternative treatment methods, or achieve earlier product development, in which case the likelihood of us achieving meaningful revenues or profitability will be substantially reduced.
Manufacturing and Quality Assurance
We manufacture certain components including our proprietary filter media, and assemble and package the CHEMOSAT/Melphalan/HDS at our facility in Queensbury, New York. We have established our European headquarters and distribution facility in Galway, Ireland where we intend to conduct final manufacturing and assembly in the future. Delcath currently utilizes third-parties to manufacture some components of the CHEMOSAT/Melphalan/HDS. The CHEMOSAT/Melphalan/HDS and its components must be manufactured and sterilized in accordance with approved manufacturing and pre-determined performance specifications. In addition, certain components will require sterilization prior to distribution and Delcath relies on third-party vendors to perform the sterilization process.
We are committed to providing high quality products to our customers. To honor this commitment, Delcath has implemented updated quality systems throughout our organization. Delcath’s quality system starts with the initial product specification and continues through the design of the product, component specification process and the manufacturing, sale and servicing of the product. These systems are designed to enable us to satisfy the various international quality system regulations including those of the FDA with respect to products sold in the United States and those established by the International Standards Organization (ISO) with respect to products sold in the EEA. The Company is required to maintain ISO 13485 certification for medical devices to be sold in the EEA, which requires, among other items, an implemented quality system that applies to component quality, supplier control, product design and manufacturing operations. On February 17, 2011, we announced that we had achieved ISO 13485 certification for our Queensbury manufacturing facility. On December 28, 2011, we announced that we had achieved ISO 13485 certification for our Galway, Ireland facility.
Intellectual Property and Other Rights
Our success depends in part on our ability to obtain patents and trademarks, maintain trade secret and know-how protection, enforce our proprietary rights against infringers, and operate without infringing on the proprietary rights of third parties. Because of the length of time and expense associated with developing new products and bringing them through the regulatory approval process, the healthcare industry places considerable emphasis on obtaining patent protection and maintaining trade secret protection for new technologies, products, processes, know-how, and methods. The Company currently holds seven United States patents, ten foreign patents with patent validity in 15 countries, four pending United States patent applications, and two pending foreign patent applications.
When appropriate, the Company actively pursues protection of our proprietary products, technologies, processes, and methods by filing United States and international patent and trademark applications. We seek to make patent improvements that we identify through research and development, manufacturing, and clinical use of the CHEMOSAT/Melphalan/HDS that will enable us to expand our platform beyond the treatment of cancers in the liver. There can be no assurance that pending patent applications will result in the issuance of patents, that patents issued to or licensed by us will not be challenged or circumvented by competitors, or that these patents will be found to be valid or sufficiently broad to protect our technology or provide us with a competitive advantage.
To maintain our proprietary position, we also rely on trade secrets and proprietary technological experience to protect proprietary manufacturing processes, technology, and know-how relating to our business. We rely, in part, on confidentiality agreements with our marketing partners, employees, advisors, vendors and consultants to protect our trade secrets and proprietary technological expertise. In addition, we also seek to maintain our trade secrets through maintenance of the physical security of the premises where our trade secrets are located. There can be no assurance that these agreements will not be breached, that we will have adequate remedies for any breach, that others will not independently develop equivalent proprietary information or that third parties will not otherwise gain access to our trade secrets and proprietary knowledge.
Certain of our United States and foreign patents have already expired and other patents relating to the CHEMOSAT/Melphalan/HDS will expire in 2016. In certain circumstances, United States patent law allows for the extension of a patent’s duration for a period of up to five years after FDA approval. The Company intends to seek extension for one of our patents after FDA approval if it has not expired prior to the date of approval. In addition to our proprietary protections, the FDA has granted Delcath five orphan drug designations that provide us a seven-year period of exclusive marketing beginning on the date that our NDA is approved by the FDA for the designated orphan drug. While the exclusivity only applies to the indication for which the drug has been approved, the Company believes that it will provide us with added protection once commercialization of an orphan drug designated product begins.
There has been and continues to be substantial litigation regarding patent and other intellectual property rights in the pharmaceutical and medical device areas. If a third party asserts a claim against Delcath, the Company may be forced to expend significant time and money defending such actions and an adverse determination in any patent litigation could subject us to significant liabilities to third parties, require us to redesign our product, require us to seek licenses from third parties, and, if licenses are not available, prevent us from manufacturing, selling or using our system. Additionally, Delcath plans to enforce its intellectual property rights vigorously and may find it necessary to initiate litigation to enforce our patent rights or to protect our trade secrets or know-how. Patent litigation can be costly and time consuming and there can be no assurance that the outcome will be favorable to us.
Employees
During 2014 we reduced our workforce by over 30% in order to increase efficiencies and focus available financial resources on its clinical development program and European commercialization. The Company believes that these actions will help preserve our ability to achieve our objectives for 2015. As of December 31, 2014, the Company had 25 full-time employees. None of our employees is represented by a union and we believe relationships with our employees are good.
Management Transition
In September 2014, the Company announced the reorganization of the Company's leadership under which Dr. Jennifer Simpson was appointed interim President and CEO. Dr. Simpson served as interim Co-President and interim Co-CEO of Delcath since September 2013. Dr. Graham Miao, who had served as interim Co-President, interim Co-CEO and CFO, left the Company at the end of September to pursue other opportunities. Barbra Keck, Vice President, Controller & Principal Accounting Officer assumed the responsibilities of Principal Financial Officer. Dr. Roger G. Stoll, who has been a member of the Delcath Board of Directors since 2008, was appointed to the newly created position of Executive Chairman. He succeeds Gabriel Leung, who stepped down from his position as Chairman of the Board.
In December 2014, we announced the appointment of Dr. Dennis H. Langer, William D. Rueckert and Dr. Marco Taglietti to the Company's Board of Directors. Dr. Langer will serve as a Class III director with his term expiring at the 2015 annual meeting; and Mr. Rueckert and Dr. Taglietti will both serve as Class I directors with terms expiring at the 2016 annual meeting. Concurrent with these additions, the Company also announced the resignations from the Board of Laura A. Brege, Tasos G. Konidaris, and Gabriel Leung. All such appointments and resignations were effective as of December 11, 2014.
Available Information
Delcath maintains a website at www.delcath.com. The Company makes available, free of charge on our website, our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after the Company electronically files those reports with, or furnishes them to, the Securities and Exchange Commission, or the SEC. The Company is not including the information contained at www.delcath.com or at any other internet address as part of, or incorporating by reference into, this Annual Report on Form 10-K.
Risks Related to Our Business and Financial Condition
Drug development is an inherently uncertain process with a high risk of failure at every stage of development. We received a complete response letter from the FDA regarding our Melblez Kit system, which precludes approval of our existing NDA.
Preclinical testing and clinical trials are long, expensive and highly uncertain processes and failure can unexpectedly occur at any stage of clinical development. Drug development is very risky and it takes several years to complete clinical trials. The start or end of a clinical trial is often delayed or halted due to changing regulatory requirements, manufacturing challenges, required clinical trial administrative actions, slower than anticipated patient enrollment, changing standards of care, availability or prevalence of use of a comparator treatment or required prior therapy, clinical outcomes including insufficient efficacy, safety concerns, or our own financial constraints.
In September 2013, the FDA issued a complete response letter (CRL) with respect to our NDA seeking an indication or ocular melanoma liver metastases for our Melblez Kit system. A CRL is issued by the FDA when the review of a file is completed and questions remain that precludes approval of the NDA in its current form. The FDA comments in the CRL included, but were not limited to, a statement that Delcath must perform additional “well-controlled randomized trial(s) to establish the safety and efficacy of Melblez Kit using overall survival as the primary efficacy outcome measure” and which “demonstrates that the clinical benefits of Melblez Kit outweigh its risks.” The FDA also requires that the additional clinical trial(s) be conducted using the product the Company intends to market. Prior to conducting additional clinical trials, we must satisfy certain other requirements of the CRL, including, but not limited to, product quality testing and human factors.
As a part of the regulatory process of obtaining marketing clearance for Melphalan/HDS, we will conduct and participate in numerous clinical trials with a variety of study designs, patient populations and trial endpoints. In 2014, we initiated a Phase 2 clinical trial for HCC in both the United States and Europe. In 2015, we expect to expand the Phase 2 clinical trial for HCC to include a cohort of patients with ICC. The trial for this cohort will be conducted at the same centers participating in the Phase 2 HCC trial. Additionally, we are advancing plans to initiate a pivotal Phase 3 overall survival clinical trial in ocular melanoma liver metastases. Our ability to initiate this trial is subject to FDA clearance of our trial protocol and the satisfaction of certain requirements the CRL. Unfavorable or inconsistent clinical data from clinical trials, including the Phase 2 clinical trial for HCC or the market's or FDA's perception of this clinical data, may adversely impact our ability to obtain approval, and the financial condition and results of operations. Additionally, even if the results of our Phase 2 clinical trial for HCC are positive, there is a substantial risk that it will fail to have positive results in Phase 3 clinical trials with regard to efficacy, safety or other clinical outcomes and may never obtain regulatory approval.
We do not expect to generate significant revenue for the foreseeable future.
Our entire focus has been on developing, commercializing, and obtaining regulatory authorizations and approvals of CHEMOSAT/Melphalan/HDS and currently we have only developed this system for the treatment of cancers in the liver. If CHEMOSAT/Melphalan/HDS for the treatment of cancers in the liver fails as a commercial product, we have no other products to sell. In addition, since CHEMOSAT is currently only authorized for marketing in the EEA and limited other jurisdictions, if we are unsuccessful in commercializing the product in the EEA and if Melphalan/HDS is not approved in the United States and elsewhere, we will have no means of generating revenue. In September 2013, the FDA issued a CRL with respect to our NDA for our Melblez Kit system. A CRL is issued by the FDA when the review of a file is completed and questions remain that precludes approval of the NDA in its then current form. Accordingly, we do not expect to realize any revenues from product sales in the United States in the next several years, if at all. As a result, our revenue sources are, and will remain, extremely limited until our product candidates are approved by the FDA or other additional foreign regulatory agencies and successfully marketed. CHEMOSAT/Melphalan/HDS may not be successful in clinical trials, approved by the FDA or other additional foreign regulatory agency or marketed at any time in the foreseeable future or at all.
Continuing losses may exhaust our capital resources.
As of December 31, 2014, we had $20.5 million in cash and cash equivalents. We have had minimal revenue to date, and we have a substantial accumulated deficit, recurring operating losses and negative cash flow. For the years ended December 31, 2014, 2013, and 2012, we incurred net losses of approximately $17.4 million, $30.3 million and $51.9 million, respectively, and we expect to continue to incur losses in 2015. To date, we have funded our operations through a combination of private placements and public offerings of our securities. If we continue to incur losses, we may exhaust our capital resources, and as a result may be unable to complete our clinical trials, product development, regulatory approval process and commercialization of CHEMOSAT/Melphalan/HDS or any other versions of the system.
If we cannot raise additional capital, our potential to generate future revenues will be significantly limited since we may not be able to commercialize CHEMOSAT/Melphalan/HDS, complete our HCC clinical trial or conduct future development and clinical trials.
We will require additional financing to complete our clinical trial program or seek other approvals, to conduct future development and clinical trials and to commercialize our product in the EEA and any other markets where we receive approval for our system. In addition, we are obligated to make payments under long-term research and development obligations and lease agreements. If financing is unavailable to make the required payments under these agreements, we could be subject to legal liability and our ability to complete our development projects or our clinical trials could be impaired. We do not know if additional financing will be available when needed at all or on acceptable terms. If we are unable to obtain additional financing as needed, we may not be able to commercialize CHEMOSAT/Melphalan/HDS commercially, obtain regulatory approvals or complete our development projects or our clinical trials.
Our liquidity and capital requirements will depend on numerous factors, including:
| o | clinical studies, including a Phase 2 clinical trial to establish proof of concept in HCC and ICC and a Phase 3 clinical trial to investigate overall survival in ocular melanoma liver metastases; |
| o | the timing and costs of our various U.S. and foreign regulatory filings, obtaining approvals and complying with regulations; |
| o | the timing and costs associated with developing our manufacturing operations; |
| o | the timing of product commercialization activities, including marketing and distribution arrangements overseas; |
| o | the timing and costs involved in preparing, filing, prosecuting, defending and enforcing intellectual property rights; and |
| o | the impact of competing technological and market developments. |
In February 2015, we completed the sale of approximately 2.4 million shares of our common stock and the issuance of warrants to purchase approximately 1.1 million shares of our common stock pursuant to an underwriting agreement. We received proceeds of approximately $2.8 million, with net cash proceeds after related expenses from this transaction of approximately $2.4 million. The shares and warrants were issued pursuant to an effective registration statement on Form S-3. Form S-3 limits the aggregate market value of securities that we are permitted to offer in any 12 month-period under Form S-3 to one-third of our public float. Given the offering in February 2015, other sales under our at the market equity offering program during the relevant 12 month-period, and our current aggregate market value of securities, we are at the applicable limit under Form S-3. As a result, unless the market value of our securities increases our ability to raise capital may be impaired and we currently are unable to utilize the Form S-3 or access our at the market equity offering program.
Insufficient funds may require us to curtail or stop our commercialization activities, submissions or ongoing activities for regulatory approval, research and development and clinical trials, which will significantly limit our potential to generate future revenues.
Risks Related to FDA and Foreign Regulatory Approval
Our failure to obtain, or delays in obtaining, regulatory approvals may have a material adverse effect on our business, financial condition and results of operations.
CHEMOSAT/Melphalan/HDS is subject to extensive and rigorous government regulation by the FDA and other foreign regulatory agencies. The FDA regulates the research, development, pre-clinical and clinical testing, manufacture, safety, effectiveness, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import and export of pharmaceutical and medical device products. Failure to comply with FDA and other applicable regulatory requirements may, either before or after product approval, subject us to administrative or judicially imposed sanctions.
In the United States, the FDA regulates drug and device products under the FFDCA, and its implementing regulations. Melphalan/HDS is subject to regulation by the FDA as a combination product, which means it is composed of both a drug product and device product. If marketed individually, each component would therefore be subject to different regulatory pathways and reviewed by different centers within the FDA. A combination product, however, is assigned to a center that will have primary jurisdiction over its pre-market review and regulation based on a determination of the product’s primary mode of action, which is the single mode of action that provides the most important therapeutic action. In the case of Melphalan/HDS, the primary mode of action is attributable to the drug component of the product, which means that the CDER has primary jurisdiction over its pre-market development and review.
We are not permitted to market Melphalan/HDS in the United States unless and until we obtain regulatory approval from the FDA. To market the product in the United States, we must submit to the FDA and obtain FDA approval of an NDA. An NDA must be supported by extensive clinical and preclinical data, as well as extensive information regarding chemistry, manufacturing and controls, or CMC, to demonstrate the safety and effectiveness of the applicable product candidate. Regulatory approval of an NDA is not guaranteed. The number and types of preclinical studies and clinical trials that will be required varies depending on the product candidate, the disease or condition that the product candidate is designed to target and the regulations applicable to any particular product candidate. Despite the time and expense associated with preclinical and clinical studies, failure can occur at any stage, and we could encounter problems that cause us to repeat or perform additional preclinical studies, CMC studies or clinical trials. The FDA and similar foreign authorities could delay, limit or deny approval of a product candidate for many reasons, including because they:
| o | may not deem a product candidate to be adequately safe and effective; |
| o | may not find the data from preclinical studies, CMC studies and clinical trials to be sufficient to support a claim of safety and efficacy; |
| o | may interpret data from preclinical studies, CMC studies and clinical trials significantly differently than we do; |
| o | may not approve the manufacturing processes or facilities associated with our product candidates; |
| o | may change approval policies (including with respect to our product candidates’ class of drugs) or adopt new regulations; or |
| o | may not accept a submission due to, among other reasons, the content or formatting of the submission. |
Undesirable side effects caused by any product candidate that we develop could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications or cause us to evaluate the future of our development programs. The regulatory review and approval process is lengthy, expensive and inherently uncertain. As part of the U.S. Prescription Drug User Fee Act, the FDA has a goal to review and act on a percentage of all submissions in a given time frame. In August 2012, we submitted the Melblez Kit system NDA seeking an indication for ocular melanoma liver metastases. In September 2013, the FDA issued a CRL. A CRL is issued by the FDA when the review of a file is completed and questions remain that precludes approval of the NDA in its current form. The FDA comments in the CRL included, but were not limited to, a statement that Delcath must perform additional “well-controlled randomized trial(s) to establish the safety and efficacy of Melblez Kit using overall survival as the primary efficacy outcome measure” and which “demonstrates that the clinical benefits of Melblez Kit outweigh its risks.” The FDA also requires that the additional clinical trial(s) be conducted using the product the Company intends to market. Prior to conducting additional clinical trials, we must satisfy certain other requirements of the CRL, including, but not limited to, product quality testing and human factors. However, even if we complete clinical trials and satisfy all the requirements of the CRL, we may not obtain regulatory approval from the FDA. Continued failure to obtain, or additional delays in obtaining, regulatory approvals may:
| o | adversely affect the commercialization of the current version of CHEMOSAT/Melphalan/HDS or any products that we develop in the future; |
| o | impose additional costs on us; |
| o | diminish any competitive advantages that may be attained; and |
| o | adversely affect our ability to generate revenues. |
We have obtained the right to affix the CE Mark for the Delcath Hepatic CHEMOSAT Delivery System as a medical device for the delivery of melphalan. Since we may only promote the device within this specific indication, if physicians are unwilling to obtain melphalan separately for use with CHEMOSAT, our ability to commercialize CHEMOSAT in the EEA will be significantly limited.
In the EEA, CHEMOSAT is regulated as a Class IIb medical device indicated for the intra-arterial administration of a chemotherapeutic agent, melphalan hydrochloride, to the liver with additional extracorporeal filtration of the venous blood return. Our ability to market and promote CHEMOSAT is limited to this approved indication. To the extent that our promotion of CHEMOSAT is found to be outside the scope of our approved indication, we may be subject to fines or other regulatory action, limiting our ability to commercialize CHEMOSAT in the EEA.
We are limited to marketing CHEMOSAT in the EEA as a medical device for the delivery of melphalan. If physicians are unwilling to obtain melphalan separately for use with CHEMOSAT, our ability to commercialize CHEMOSAT in the EEA will be significantly limited. Our product instructions and indication reference the chemotherapeutic agent melphalan. However, no melphalan labels in the EEA reference our product, and the labels vary from country to country with respect to the approved indication of the drug and its mode of administration. As a result, the delivery of melphalan with our device may not be within the applicable label with respect to some indications in some Member States of the EEA where the drugs are authorized for marketing. In the exercise of their professional judgment in the practice of medicine, physicians are generally allowed, under certain conditions, to use or prescribe a product in ways not approved by regulatory authorities. Physicians intending to use our device must obtain melphalan separately for use with CHEMOSAT and must use melphalan independently at their discretion. If physicians are unwilling to obtain melphalan separately from our product and/or to prescribe the use of melphalan independently, our sales opportunities in the EEA will be significantly impaired.
While we have obtained the right to affix the CE Mark, we will be subject to significant ongoing regulatory obligations and oversight in the EEA and in any other country where we receive marketing authorization or approval.
In April 2012, we obtained the required certification from our European Notified Body, enabling us to complete an EC Declaration of Conformity with the essential requirements of the EU Medical Devices Directive and affix the CE Mark to the Generation Two CHEMOSAT system. In order to maintain the right to affix the CE Mark in the EEA, we are subject to compliance obligations, and any material changes to the approved product, such as manufacturing changes, product improvements or revised labeling, may require further regulatory review. Additionally, we are subject to ongoing audits by our European Notified Body, and the right to affix the CE Mark to the Generation Two CHEMOSAT system may be withdrawn for a number of reasons, including the later discovery of previously unknown problems with the product.
To the extent that CHEMOSAT/ Melphalan/HDS is approved by the FDA or any other regulatory agency, we will be subject to similar ongoing regulatory obligations and oversight in those countries where we obtain approval. For example, we may be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or requirements for potentially costly post-marketing testing, including Phase IV clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. In addition, if the FDA approves a product candidate, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs, good clinical practices, or GCPs, and good laboratory practices, which are regulations and guidelines enforced by the FDA for all products in clinical development, for any clinical trials that we conduct post-approval. In addition, post-marketing requirements for CHEMOSAT/Melphalan/HDS may include implementation of a risk evaluation and mitigation strategies (REMS) program to ensure that the benefits of the product outweigh its risks. A REMS may include a Medication Guide, a patient package insert, a communication plan to healthcare professionals and/or other elements to assure safe use of the product.
Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| o | refusals or delays in the approval of applications or supplements to approved applications; |
| o | refusal of a regulatory authority to review pending market approval applications or supplements to approved applications; |
| o | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market or voluntary or mandatory product recalls or seizures; |
| o | fines, Warning Letters or holds on clinical trials; |
| o | import or export restrictions; |
| o | injunctions or the imposition of civil or criminal penalties; |
| o | restrictions on product administration, requirements for additional clinical trials or changes to product labeling or REMS programs; or |
| o | recommendations by regulatory authorities against entering into governmental contracts with us. |
If we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would have a material adverse effect on our business, results of operations, financial condition and prospects.
The development and approval process in the United States will take many years, require substantial resources and may never lead to the approval of Melphalan/HDS by the FDA for use in the United States.
We cannot sell or market Melphalan/HDS with melphalan or other chemotherapeutic agents in the United States without prior FDA approval of an NDA for Melphalan/HDS. Although melphalan and other drugs have been approved by the FDA for use as chemotherapeutic agents, regulatory approval is required in the United States for the combined medical device component and drug component and the specific indication, dose and route of administration of melphalan or other chemotherapeutic agent used in our system. We are seeking approval of Melphalan/HDS for a substantially higher dose of melphalan than prior approved doses of melphalan and such other drugs. We must obtain separate regulatory approvals for Melphalan/HDS with melphalan and every other chemotherapeutic agent or other compound used with our system that we intend to market, and all the manufacturing facilities used to manufacture components or assemble our system must be inspected and meet legal requirements. Securing regulatory approval requires the submission of extensive pre-clinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish to the FDA’s satisfaction the product’s safety, efficacy, potency and purity for each intended use. The pre-clinical testing and clinical trials of Melphalan/HDS with melphalan or any other chemotherapeutic agent or compound we use in our system must comply with the regulations of the FDA and other federal, state and local government authorities in the United States. Clinical development is a long, expensive and uncertain process and is subject to delays. We may encounter delays or rejections for various reasons, including our inability to enroll enough patients to complete our clinical trials. Moreover, approval policies or regulations may change. If we do not obtain and maintain regulatory approval for our system and our use of melphalan or other chemotherapeutic agents, the value of our company, our results of operations and our ability to raise additional capital will be harmed.
In August 2012, we submitted a NDA seeking an indication for ocular melanoma liver metastases for our Melblez Kit system. In September 2013, the FDA issued a complete response letter (CRL). A CRL is issued by the FDA when the review of a file is completed and questions remain that precludes approval of the NDA in its current form. The FDA comments in the CRL included a statement that Delcath must perform additional well-controlled randomized trials to establish the safety and efficacy of Melblez Kit using overall survival as the primary efficacy outcome measure and which demonstrates that the clinical benefits of Melblez Kit outweigh its risks. Failure to obtain FDA approval will have a material adverse effect on our business, financial condition and results of operations.
Even if we obtain regulatory approval for the Melblez Kit system in the United States, our ability to market the Melblez Kit system would be limited to those uses that are approved.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. If the FDA approves an application for the Melblez Kit, our ability to market and promote the Melblez Kit system would be limited to the approved indication, so even with FDA approval, the Melblez Kit system may only be promoted in this limited market. Physicians may prescribe legally available drugs for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, including oncology. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use, and FDA approval may otherwise limit our sales practices and our ability to promote, sell and distribute the product. Thus, we may only market the Melblez Kit system, if approved by the FDA, for its approved indication and we could be subject to enforcement action for off-label marketing.
Further, if there are any modifications to the product, including changes in indications, labeling or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. Failure to comply with these requirements can result in adverse publicity, Warning Letters, corrective advertising and potential civil and criminal penalties.
If future clinical trials are unsuccessful, significantly delayed or not completed, we may not be able to market Melphalan/HDS for other indications.
The clinical trial data on our product is limited to specific types of liver cancer. In 2010, we concluded a Phase 3 clinical trial of Melphalan/HDS in patients with metastatic ocular and cutaneous melanoma to the liver and also completed a multi-arm Phase 2 clinical trial of Melphalan/HDS in patients with primary and metastatic melanoma stratified into four arms.
In 2014, we initiated a Phase 2 clinical trial for HCC in both the United States and Europe. In 2015, we expect to expand the Phase 2 clinical trial for HCC to include a cohort of patients with ICC. Additionally, we are advancing plans to initiate a pivotal Phase 3 overall survival clinical trial in ocular melanoma liver metastases, subject to FDA clearance of our trial protocol and the satisfaction of certain requirements contained in the CRL.
It may take several years to complete the testing of Melphalan/HDS for use in the treatment of these indications, and failure can occur at any stage of development, for many reasons, including:
| o | any pre-clinical or clinical test may fail to produce results satisfactory to the FDA or foreign regulatory authorities; |
| o | pre-clinical or clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval; |
| o | negative or inconclusive results from a pre-clinical study or clinical trial or adverse medical events during a clinical trial could cause a pre-clinical study or clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are successful; |
| o | the FDA or foreign regulatory authorities can place a clinical hold on a trial if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury; |
| o | we may encounter delays or rejections based on changes in regulatory agency policies during the period in which we are developing a system or the period required for review of any application for regulatory agency approval; |
| o | our clinical trials may not demonstrate the safety and efficacy of any system or result in marketable products; |
| o | the FDA or foreign regulatory authorities may request additional clinical trials, including an additional Phase 3 trial, relating to our NDA submissions; |
| o | the FDA or foreign regulatory authorities may change its approval policies or adopt new regulations that may negatively affect or delay our ability to bring a system to market or require additional clinical trials; and |
| o | a system may not be approved for all the requested indications. |
The failure or delay of clinical trials could cause an increase in the cost of product development, delay filing of an application for marketing approval or cause us to cease the development of Melphalan/HDS for other indications. If we are unable to develop Melphalan/HDS for other indications the future growth of our business could be negatively impacted. In addition, we have limited clinical data relating to the effectiveness of Melphalan/HDS in certain types of cancer. Such limited data could slow the adoption of CHEMOSAT/ Melphalan/HDS, significantly reduce our ability to commercialize CHEMOSAT/ Melphalan/HDS.
We rely on third parties to conduct certain elements of the clinical trials for CHEMOSAT/Melphalan/HDS, and if they do not perform their obligations to us, we may not be able to obtain regulatory approvals for our system.
We design the clinical trials for Melphalan/HDS, but we rely on academic institutions, corporate partners, contract research organizations and other third parties to assist us in managing, monitoring and otherwise carrying out these trials. We rely heavily on these parties for the execution of our clinical studies and control only certain aspects of their activities. Accordingly, we may have less control over the timing and other aspects of these clinical trials than if we conducted them entirely on our own. We rely upon third parties to conduct monitoring and data collection of our ongoing and future clinical trials, including our Phase 2 HCC clinical trial with an ICC cohort and our planned Phase 3 ocular melanoma trial. Although we rely on these third parties to manage the data from these clinical trials, we are responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with GCPs for conducting, recording and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the trial participants are adequately protected. The FDA enforces these GCP regulations through periodic inspections of trial sponsors, principal investigators and trial sites. Our reliance on third parties does not relieve us of these responsibilities and requirements, and if we or the third parties upon whom we rely for our clinical trials fail to comply with the applicable GCPs, the data generated in our clinical trials may be deemed unreliable and the FDA or other foreign regulatory agencies may require us to perform additional trials before approving our marketing application. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply or complied with GCPs. In addition, our clinical trials must be conducted with product that complies with the FDA’s cGMP requirements. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process, and we may fail to obtain regulatory approval for Melphalan/HDS if these requirements are not met.
Purchasers of CHEMOSAT in the EEA may not receive third-party reimbursement or such reimbursement may be inadequate. Without adequate reimbursement, we may not be able to successfully commercialize CHEMOSAT in the EEA.
We have obtained the right to affix the CE Mark for CHEMOSAT, and we intend to seek third-party or government reimbursement within those countries in the EEA where we expect to market and sell CHEMOSAT. In Germany, we have received approval for Value 4 status reimbursement. Value 4 status does not mandate reimbursement, but allows participating cancer centers to negotiate reimbursement coverage for the CHEMOSAT procedure with all insurers serving their region. Consequently, we may not be able to obtain reimbursement, and any reimbursement obtained may not be for the full amount sought. In countries where we are able to obtain reimbursement, local policy could limit our ability to obtain adequate and consistent reimbursement and limit other sales opportunities in those countries. In the United Kingdom, we began seeking a block fund grant in 2014. Ongoing changes to the process and funding streams have resulted in delays that made the award and timing of any block grant funding difficult to predict. Accordingly, we may not receive the grant in a timely manner or at all.
In other countries, until we obtain government reimbursement, we will rely on private payors or local pre-approved funds where available. New technology payment programs may provide interim funding, but there are no assurances that we will qualify for such funding. Even if we do qualify, the amount and the duration of this funding may be limited. There are also no assurances that third-party payors or government health agencies of members states of the EEA will reimburse the product’s use in the long term or at all. For example, throughout 2014, physicians and patients in Germany submitted and received approvals for Individual Funding Requests (IFRs) granting reimbursement for the treatment of liver metastases with CHEMOSAT. We expect that IFRs will continue to be the main reimbursement vehicle in the German market in 2015. Further, each country has its own protocols regarding reimbursement, so successfully obtaining third party or government health agency reimbursement in one country does not necessarily translate to similar reimbursement in other EEA countries. Physicians, hospitals and other health care providers may be reluctant to purchase CHEMOSAT if they do not receive substantial reimbursement for the cost of using our product from third-party payors or government entities. The lack of adequate reimbursement may significantly limit sales opportunities in the EEA.
The success of our products may be harmed if the government, private health insurers and other third-party payers do not provide sufficient coverage or reimbursement.
Our ability to commercialize our systems successfully will depend in part on the extent to which reimbursement for the costs of such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payors. CHEMOSAT/Melphalan/HDS is currently not approved by the FDA or any other regulatory body outside the EEA. Medicare, Medicaid, private health insurance plans and their foreign equivalents will not reimburse the use of Melphalan/HDS since the product is currently not approved outside the EEA. We will seek reimbursement by third-party payors of the cost of Melphalan/HDS after its use is approved, but there are no assurances that adequate third-party coverage will be available for us to establish and maintain price levels sufficient for us to realize an appropriate return on our investment in developing new therapies. Government, private health insurers and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new therapeutic products approved for marketing. Accordingly, even if coverage and reimbursement are provided by government, private health insurers and third-party payors for uses of our products, market acceptance of these products would be adversely affected if the reimbursement available proves to be unprofitable for healthcare providers.
Implementation of healthcare reforms in the United States and in significant overseas markets may limit the ability to commercialize CHEMOSAT/ Melphalan/HDS and the demand for CHEMOSAT/ Melphalan/HDS. Healthcare providers may respond to such cost-containment pressures by choosing lower cost products or other therapies. In March 2010, the Patient Protection and Affordable Care Act and Health Care and Education Reconciliation Act of 2010 were enacted into law in the United States, which included a number of provisions aimed at improving quality and decreasing costs. It is uncertain what consequences these provisions will have on our efforts to commercialize CHEMOSAT/Melphalan/HDS.
CHEMOSAT/ Melphalan/HDS may not achieve sufficient acceptance by the medical community to sustain our business.
The commercial success of CHEMOSAT/Melphalan/HDS will depend upon their acceptance by the medical community and third-party payers as clinically useful, cost effective and safe. Acceptance by the medical community may depend on the extent to which leaders in the scientific and medical communities publish scientific papers in reputable academic journals. If testing and clinical practice do not confirm the safety and efficacy of CHEMOSAT/Melphalan/HDS or even if further testing and clinical practice produce positive results but the medical community does not view these favorably, CHEMOSAT/Melphalan/HDS as effective and desirable, our efforts to market CHEMOSAT/Melphalan/HDS may fail, which would have an adverse effect on our business, financial condition and results of operations.
Consolidation in the healthcare industry could lead to demands for price concessions.
The cost of healthcare has risen significantly over the past decade and numerous initiatives and reforms initiated by legislators, regulators and third-party payors to curb these costs have resulted in a consolidation trend in the medical device industry. Group purchasing organizations, independent delivery networks and large single accounts in the United States and foreign markets may result in a consolidation of purchasing decisions for potential healthcare provider customers. We expect that market demand, government regulation, third-party reimbursement policies and societal pressures will continue to change the worldwide healthcare industry, resulting in further business consolidations and alliances which may exert further downward pressure on the price of CHEMOSAT/Melphalan/HDS and adversely impact our business, financial condition and results of operations.
Further, third-party payors may deny reimbursement if they determine that CHEMOSAT/Melphalan/HDS is not used in accordance with established payor protocols regarding cost effective treatment methods or is used outside its approved indication or for forms of cancer or with drugs not specifically approved by the FDA or other foreign regulatory bodies in the future. Without reimbursement, physicians, hospitals and other health care providers will be less likely to purchase CHEMOSAT/Melphalan/HDS, thereby harming our results of operations.
We may not realize the expected benefits from our restructuring and optimization initiatives; our long-term expense reduction programs may result in an increase in short-term expense; and our efforts may lead to additional unintended consequences.
In early 2013, we announced a plan to increase efficiencies and reduce cash utilization. To achieve the program's goals, we broadened our workforce restructuring actions throughout 2013. As a result of the restructuring program and attrition, we reduced our workforce by approximately 60% in 2013 and an additional 32% in 2014. In addition, we have reduced expenses incurred with outside consultants. In furtherance of our plan, we entered into two sublease agreements to sublease our office space at our corporate headquarters and relocated our corporate headquarters to a new location. The subleases and subsequent relocation represent a significant decrease in total square footage and ongoing facility expenses. These measures could have unintended consequences, such as distraction of our management and employees, business disruption, attrition beyond our planned reduction in workforce and reduced employee productivity. We may be unable to attract or retain key personnel. Attrition beyond our planned reduction in workforce or a material decrease in employee morale or productivity could negatively affect our business and results of operations. In addition, headcount reductions may subject us to the risk of litigation, which could result in substantial cost. These measures, or other expense reduction measures we take in the future, may not result in the expected cost savings.
If we engage in acquisitions, reorganizations or business combinations, we will incur a variety of risks that could adversely affect our business operations or our stockholders.
We may consider strategic alternatives, such as acquiring businesses, technologies or products or entering into a business combination with another company. If we do pursue such a strategy, we could, among other things:
| o | issue equity securities that would dilute our current stockholders’ percentage ownership; |
| o | incur substantial debt that may place strains on our operations; |
| o | spend substantial operational, financial and management resources in integrating new businesses, personnel intellectual property, technologies and products; |
| o | assume substantial actual or contingent liabilities; |
| o | reprioritize our programs and even cease development and commercialization of CHEMOSAT/Melphalan/HDS; |
| o | suffer the loss of key personnel, or |
| o | merge with, or otherwise enter into a business combination with, another company in which our stockholders would receive cash or shares of the other company or a combination of both on terms that certain of our stockholders may not deem desirable. |
Although we intend to evaluate and consider different strategic alternatives, we have no agreements or understandings with respect to any acquisition, reorganization or business combination at this time.
Risks Related to Manufacturing, Commercialization and Market Acceptance of CHEMOSAT/Melphalan/HDS
There is only one approved third-party manufacturer of melphalan in the EEA. If this manufacturer fails to provide end-users with adequate supplies of melphalan or fails to comply with the requirements of regulatory authorities, we may be unable to successfully commercialize our product in the EEA.
Under the regulatory scheme in the EEA, CHEMOSAT is approved for marketing as a device only, and doctors will separately obtain melphalan for use with CHEMOSAT. Although melphalan has been approved in the EEA for over a decade, we are aware that there is currently only one approved manufacturer of melphalan in the EEA, with whom we have no supply arrangements or other affiliation, and therefore we will not have any control over the quality, availability, price or labeling of melphalan in that market. As a result, there may not be sufficient supply of melphalan for use with our system, and any adverse change in the sole manufacturer’s commercial operations or regulatory approval status may seriously impair our sales opportunities in the EEA. Additionally, melphalan is not available in certain foreign countries outside the EEA where we may seek to market CHEMOSAT. If supply of melphalan remains limited or unavailable, we will be unable to commercialize our product in these markets, thereby limiting future sales opportunities.
We purchase components for CHEMOSAT/ Melphalan/HDS from third parties, some of which are sole-source suppliers.
The components of CHEMOSAT/Melphalan/HDS, including catheters, filters, introducers and chemotherapy agents, must be manufactured and assembled in accordance with approved manufacturing and predetermined performance specifications and must meet cGMP and quality systems requirements. Some states also have similar regulations. Many of the components of CHEMOSAT/Melphalan/HDS are manufactured by sole-source suppliers that may have proprietary manufacturing processes. If Delcath or any of our suppliers fails to meet those regulatory obligations, we may be forced to suspend or terminate our clinical trials, and, once a product is approved for marketing, the manufacture, assembly or distribution thereof. Further, if we need to find a new source of supply, we may face long interruptions in obtaining necessary components for CHEMOSAT/Melphalan/HDS, in obtaining FDA or foreign regulatory agency approval of these components and in establishing the manufacturing process, which could jeopardize our ability to supply CHEMOSAT/Melphalan/HDS to the market.
All of the manufacturers of the components for CHEMOSAT/Melphalan/HDS must comply with a number of FDA and International Organization for Standardization, or ISO, and foreign regulatory agency requirements and regulations. If we or one of our suppliers fails to meet such requirements, we may need to change suppliers. If we are unable to successfully change suppliers, the successful completion of some of our future clinical trials and/or commercialization of CHEMOSAT/Melphalan/HDS could be jeopardized. CHEMOSAT/Melphalan/HDS and its components must be manufactured and sterilized with approved manufacturing and pre-determined performance specifications. Certain components will require sterilization prior to distribution and Delcath relies on third-party vendors to perform the sterilization process. A third-party vendor's failure to properly sterilize a component may cause manufacturing or assembly delays.
If we cannot maintain or enter into acceptable arrangements for the production of melphalan and other chemotherapeutic agents we will be unable to successfully commercialize the Delcath system in the United States or complete our Phase 2 clinical trial for HCC in the U.S., our planned global Phase 3 in ocular melanoma liver metastases or any future clinical trials.
We have entered into a manufacturing and supply agreement with Synerx Pharma, LLC, or Synerx, and Bioniche Teoranta, or Bioniche, an affiliate of Mylan, Inc., for the supply of our branded melphalan for injection. The agreement with Synerx and Bioniche currently represents our sole source of branded melphalan in the United States. We intend to use the melphalan supplied by Synerx and Bioniche to conduct our planned Phase 2 clinical trial for HCC and ICC in the United States and our planned global Phase 3 trial for ocular melanoma liver metatases. We may pursue agreements with additional contract manufacturers to produce melphalan and other chemotherapeutic agents that we will use in the future for our clinical trial program and the commercialization of CHEMOSAT/Melphalan/HDS, as well as for labeling and finishing services. We may not be able to enter into such arrangements on acceptable terms or at all. To manufacture melphalan or other chemotherapeutic agents on our own, we would first have to develop a manufacturing facility that complies with FDA requirements and regulations for the production of melphalan and each other chemotherapeutic agent we choose to manufacture for our system. Developing these resources would be an expensive and lengthy process and would have a material adverse effect on our revenues and profitability. If we are unable to obtain sufficient melphalan and labeling services on acceptable terms, if we should encounter delays or difficulties in our relationships with our current and future suppliers or if our current and future suppliers of melphalan do not comply with applicable regulations for the manufacturing and production of melphalan, our business, financial condition and results of operations may be materially harmed.
If we cannot successfully manufacture CHEMOSAT/Melphalan/HDS, our ability to develop and commercialize the system would be impaired.
We manufacture CHEMOSAT/Melphalan/HDS for distribution worldwide in our Queensbury, NY facility. We have a limited manufacturing history and we may not be able to manufacture the system in sufficient commercial quantities, in a cost-effective manner or in compliance with the regulatory requirements applicable to such manufacturing. Additionally, we may have difficulty obtaining components for the system from our third-party suppliers in a timely manner or at all which may adversely affect our ability to deliver CHEMOSAT/Melphalan/HDS to purchasers.
In addition to limiting sales opportunities, delays in manufacturing CHEMOSAT/Melphalan/HDS may adversely affect our ability to obtain regulatory approval in other jurisdictions. Our ability to conduct timely clinical trials in the United States and abroad depends on our ability to manufacture the system, including sourcing the chemotherapeutic agents or other compounds through third parties in accordance with FDA and other regulatory requirements. If we are unable to manufacture CHEMOSAT/Melphalan/HDS in a timely manner, we may not be able to conduct the clinical trials required to obtain regulatory approval and commercialize our product.
If our Queensbury, NY facility fails to maintain compliance with ISO 13485, a comprehensive management system for the design and manufacture of medical devices, and FDA cGMP or fails to pass facility inspection or audits, our ability to manufacture at the facility could be limited or terminated. In the future, we may manufacture and assemble CHEMOSAT/Melphalan/HDS in the EEA, and any facilities in the EEA would have to obtain and maintain similar approvals or certifications of compliance.
We do not have written contracts with all of our suppliers for the manufacture of components for CHEMOSAT/Melphalan/HDS.
We do not have written contracts with all our suppliers for the manufacture of components for CHEMOSAT/Melphalan/HDS. If we are unable to obtain an adequate supply of the necessary components or negotiate acceptable terms, we may not be able to manufacture the system in commercial quantities or in a cost-effective manner, and commercialization of CHEMOSAT/Melphalan/HDS in the EEA may be delayed. In addition, certain components are available from only a limited number of sources. Components of CHEMOSAT/Melphalan/HDS are currently manufactured for us in small quantities and we may require significantly greater quantities to further commercialize the product. We may not be able to find alternate sources of comparable components. If we are unable to obtain adequate supplies of components from our existing suppliers or need to switch to an alternate supplier and obtain FDA or other regulatory agency approval of that supplier, commercialization of CHEMOSAT/Melphalan/HDS may be delayed.
We have limited experience in marketing and commercializing our products, and as a result, we may not be successful in commercializing CHEMOSAT in the EEA.
We have not previously sold, marketed or distributed any products and have limited experience in building a sales and marketing organization and in entering into and managing relationships with third-party distributors. Even though we have obtained the right to affix the CE Mark, we currently have limited sales, marketing, commercial or distribution capabilities in any countries in the EEA. In order to pursue our strategy to commercialize CHEMOSAT in the EEA, we must acquire or internally develop a sales, marketing and distribution infrastructure and/or enter into strategic alliances to perform these services. The development of sales, marketing and distribution infrastructure is difficult, time consuming and requires substantial financial and other resources. If we cannot successfully develop the infrastructure to market and commercialize CHEMOSAT, our ability to generate revenues in the EEA may be harmed, and we may not generate sufficient revenue to sustain our business or we may be required to enter into strategic alliances to have such activities carried out on our behalf, which may not be on favorable terms.
Competition for sales and marketing personnel is intense, and we may not be successful in attracting or retaining such personnel. Our inability to attract and retain skilled sales and marketing personnel or to reach an agreement with a third party could adversely affect our business, financial condition and results of operations. Further, since our marketing strategy in the EEA includes establishing a network of third-party distributors, we must enter into collaborative arrangements with these third-party distributors. We may not be able to enter into such arrangements on reasonable terms or at all
Even if we receive FDA or other foreign regulatory approvals, we may be unsuccessful in commercializing CHEMOSAT/Melphalan/HDS in markets outside the EEA, because of inadequate infrastructure or an ineffective commercialization strategy.
Outside the EEA, even if we obtain regulatory approval from the FDA or other foreign regulatory agencies, our ability to commercialize CHEMOSAT/Melphalan/HDS may be limited due to our inexperience in developing a sales, marketing and distribution infrastructure. If we are unable to develop this infrastructure in the United States or elsewhere or to collaborate with an alliance partner to market our products in the United States or foreign countries, particularly in Asia, our efforts to commercialize CHEMOSAT/Melphalan/HDS or any other product outside of the EEA may be less successful.
Even if we are successful in commercializing CHEMOSAT/Melphalan/HDS in the EEA, we may not be successful in the United States and other foreign countries. Each country requires a different commercialization strategy, so our EEA strategy may not translate to other markets. Without a successful commercialization strategy tailored for each market, our efforts to promote and market CHEMOSAT in each of our target markets may fail in any or all of those markets.
Our plan to use collaborative arrangements with third parties to help finance and to market and sell CHEMOSAT/Melphalan/HDS may not be successful.
We may be unable to enter into collaborative agreements without additional clinical data or unable to continue a collaborative agreement as a result of unsuccessful future clinical trials. Additionally, we may face competition in our search for alliances. As a result, we may not be able to enter into any additional alliances on acceptable terms, if at all. Our collaborative relationships may never result in the successful development or commercialization of CHEMOSAT/Melphalan/HDS or any other product. The success of any collaboration will depend upon our ability to perform our obligations under any agreements as well as factors beyond our control, such as the commitment of our collaborators and the timely performance of their obligations. The terms of any such collaboration may permit our collaborators to abandon the alliance at any time for any reason or prevent us from terminating arrangements with collaborators who do not perform in accordance with our expectations or our collaborators may breach their agreements with us. In addition, any third parties with which we collaborate may have significant control over important aspects of the development and commercialization of our products, including research and development, market identification, marketing methods, pricing, composition of sales force and promotional activities. We are not able to control or influence the amount and timing of resources that any collaborator may devote to our research and development programs or the commercialization, marketing or distribution of our products. We may not be able to prevent any collaborators from pursuing alternative technologies or products that could result in the development of products that compete with CHEMOSAT/Melphalan/HDS or the withdrawal of their support for our products. The failure of any such collaboration could have a material adverse effect on our business.
If we fail to overcome the challenges inherent in international operations, our business and results of operations may be materially adversely affected.
Currently we have only received authorization to market CHEMOSAT in the EEA, and intend to seek similar authorization or approvals in other foreign countries. As a result, we expect international sales of our products to account for a significant portion of our revenue, which exposes us to risks inherent in international operations. To accommodate our international sales, we will need to further invest financial and management resources to develop an international infrastructure that will meet the needs of our customers. Accordingly, we will face additional risks resulting from our international operations including:
| o | difficulties in enforcing agreements and collecting receivables in a timely manner through the legal systems of many countries outside the United States; |
| o | the failure to fulfill foreign regulatory requirements to market our products on a timely basis or at all; |
| o | availability of, and changes in, reimbursement within prevailing foreign healthcare payment systems; |
| o | difficulties in managing foreign relationships and operations, including any relationships that we establish with foreign sales or marketing employees and agents; |
| o | limited protection for intellectual property rights in some countries; |
| o | fluctuations in currency exchange rates; |
| o | the possibility that foreign countries may impose additional withholding taxes or otherwise tax our foreign income, impose tariffs or adopt other restrictions on foreign trade; |
| o | the possibility of any material shipping delays; |
| o | significant changes in the political, regulatory, safety or economic conditions in a country or region; |
| o | protectionist laws and business practices that favor local competitors; and |
| o | trade restrictions, including the imposition of, or significant changes to, the level of tariffs, customs duties and export quotas. |
If we fail to overcome the challenges we encounter in our international operations, our business and results of operations may be materially adversely affected.
CHEMOSAT has been used a limited number of times in a clinical setting in the EEA, so market acceptance of our product will depend on EEA healthcare professionals’ efforts to learn about our product.
Since all of our prior clinical studies were conducted in the United States and CHEMOSAT has had limited use in a clinical setting in the EEA, physicians in the EEA have no clinical experience with our product. As a result, CHEMOSAT may not gain significant market acceptance among physicians, hospitals, patients and healthcare payors in the EEA until healthcare professionals are properly educated about the procedure. Market acceptance of CHEMOSAT in the EEA will depend upon a variety of factors including:
| o | whether our future clinical trials demonstrate significantly improved patient outcomes; |
| o | our ability to educate and train physicians to perform the procedure and drive acceptance of the use of CHEMOSAT; |
| o | our ability to obtain adequate reimbursement and convince healthcare payors that use of CHEMOSAT results in reduced treatment costs and improved outcomes for patients; |
| o | whether CHEMOSAT replaces and/or complements treatment methods in which many hospitals have made a significant investment; and |
| o | whether doctors and hospitals are willing to replace their existing technology with a new medical technology until the new technology’s value has been demonstrated. |
We intend to establish clinical training and centers of excellence to educate and train physicians and healthcare payors in the EEA, but the key opinion thought leadership required for initial market acceptance within the healthcare arena may take time to develop. Without effort from healthcare professionals to become educated about our product, the market may not accept CHEMOSAT and our efforts to commercialize CHEMOSAT in the EEA may be unsuccessful.
Similar considerations apply in any other market where we receive approval. Successful commercialization of CHEMOSAT in these markets will depend on market acceptance by healthcare professionals.
Rapid technological developments in treatment methods for liver cancer and competition with other forms of liver cancer treatments could affect our ability to achieve meaningful revenues or profit.
Competition in the cancer treatment industry is intense. CHEMOSAT/Melphalan/HDS competes with all forms of liver cancer treatments that are alternatives to the “gold standard” treatment of surgical resection. Many of our competitors have substantially greater resources and considerable experience in conducting clinical trials and obtaining regulatory approvals. If these competitors develop more effective or more affordable products or treatment methods, or achieve earlier product development, our revenues or profitability will be substantially reduced.
Our ability to develop CHEMOSAT/Melphalan/HDS for other indications could affect our orphan drug exclusivity. In November 2008, the FDA granted Delcath two orphan drug designations for the drug melphalan for the treatment of patients with cutaneous melanoma as well as patients with ocular melanoma. In May 2009, the FDA granted Delcath an additional orphan drug designation of the drug melphalan for the treatment of patients with neuroendocrine tumors. In August 2009, the FDA granted Delcath an orphan drug designation of the drug doxorubicin for the treatment of patients with primary liver cancer. In October 2013, the FDA granted Delcath orphan drug designation of the drug melphalan for the treatment of HCC. If CHEMOSAT/Melphalan/HDS is approved for an indication different than the indications for which we have received orphan drug designations, we will not obtain orphan drug exclusivity, which could increase our competition.
The loss of key personnel could adversely affect our business.
The loss of a member of our senior executive staff could harm our business. Competition for experienced personnel is intense. If we cannot retain our current personnel or attract additional experienced personnel, our ability to compete could be adversely affected.
We have been named as a party to a purported stockholder class action and stockholder derivative complaint, and we may be named in additional litigation, all of which will require significant management time and attention, result in substantial legal expenses and may result in an unfavorable outcome, which could have a material adverse effect on us.
A purported class action lawsuit has been filed against us on behalf of certain purchasers of our common stock. The complaint includes allegations that we violated federal securities laws by, among other things, knowingly making false and misleading statements or omissions regarding our NDA for our Melblez Kit, thereby artificially inflating the price of our common stock. The complaint seeks compensatory damages, equitable relief, and reasonable attorneys’ fees, expert fees and other costs. In addition, stockholder derivative actions have been initiated against us and certain of our directors and officers. These complaints purport to seek relief on behalf of the company to remedy alleged breaches of fiduciary duty and other misconduct by the defendants. Our insurance coverage and assets may be insufficient to cover any damage awards or settlement arrangements we may enter into in connection with such claims. Any such payments or settlement arrangements in this current litigation or any future litigation could have material adverse effects on our business, operating results or financial condition. Even if the plaintiffs' claims are not successful, this or future litigation could result in substantial costs and significantly and adversely impact our reputation and divert management's attention and resources, which could have a material adverse effect on our business, operating results or financial condition. In addition, such lawsuits may make it more difficult for us to finance our operations.
Risks Related to Patents, Trade Secrets and Proprietary Rights
Our success depends in part on our ability to obtain patents, maintain trade secret protection, operate without infringing on the proprietary rights of third parties and commercialize CHEMOSAT/Melphalan/HDS prior to the expiration of our patent protection.
Our patent portfolio consists of seven U.S. patents, one pending Patent Cooperation Treaty application, 22 issued foreign counterpart patents and four pending foreign counterpart patent applications. Certain of our U.S., European and other foreign patents have already expired and other U.S. patents relating to CHEMOSAT/Melphalan/HDS have expired in 2013 and will continue to expire through 2016.
Due to the uncertainty of the patent prosecution process, there are no guarantees that any of our pending patent applications will result in the issuance of a patent. Even if we are successful in obtaining a patent, there is no assurance that it will be upheld if later challenged or will provide significant protection or commercial advantage. Because of the length of time and expense associated with bringing new medical drugs and devices to the market, the healthcare industry has traditionally placed considerable emphasis on patent and trade secret protection for significant new technologies. Other parties may challenge patents, patent claims or patent applications licensed or issued to us or may design around technologies we have patented, licensed or developed.
Companies in the medical drug/device industry may use intellectual property infringement litigation to gain a competitive advantage. In the United States, patent applications filed in recent years are confidential for 18 months, while older applications are not publicly available until the patent issues. As a result, avoiding patent infringement may be difficult. Litigation may be necessary to enforce any patents issued or assigned to us or to determine the scope and validity of third-party proprietary rights. Litigation could be costly and could divert our attention from our business. There are no guarantees that we will receive a favorable outcome in any such litigation. If a third party claims that we infringed its patents, any of the following may occur:
| o | we may become liable for substantial damages for past infringement if a court decides that our technologies infringe upon a competitor’s patent; |
| o | a court may prohibit us from selling or licensing our product without a license from the patent holder, which may not be available on commercially acceptable terms or at all, or which may require us to pay substantial royalties or grant cross-licenses to our patents; and |
| o | we may have to redesign our product so that it does not infringe upon others’ patent rights, which may not be possible or could require substantial funds or time. |
If others file patent applications with respect to inventions for which we already have patents issued to us or have patent applications pending, we may be forced to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention, which could also be costly and could divert our attention from our business. If a third party violates our intellectual property rights, we may be unable to enforce our rights because of our limited resources. Use of our limited funds to enforce or to defend our intellectual property rights or to defend against legal proceedings alleging infringement of third party proprietary rights may also affect our financial condition adversely.
Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before CHEMOSAT/Melphalan/HDS or any other product can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantages of the patent. Not all of our U.S. patent rights have corresponding patent rights effective in Europe or other foreign jurisdictions.
Similar considerations apply in any other country where we are prosecuting patent applications, have been issued patents, or have decided not to pursue patent protection relating to our technology. The laws of foreign countries may not protect our intellectual property rights to the same extent as do laws of the United States.
We rely solely on trade secret protection for important proprietary technologies in the EEA.
We presently only have a validly issued patent with claims related to certain features of the current version of CHEMOSAT/Melphalan/HDS in the United States and other parts of CHEMOSAT/Melphalan/HDS are protected by trade secret. Outside the United States, we have no patent protection for CHEMOSAT/Melphalan/HDS and rely on trade secret protection. Without patent protection in the EEA, CHEMOSAT will only be covered by trade secret protection. Unlike patents, trade secrets are only recognized under applicable law if they are kept secret by restricting their disclosure to third parties. We protect our trade secrets and proprietary knowledge in part through confidentiality agreements with employees, consultants and other parties. However, certain consultants and third parties with whom we have business relationships, and to whom in some cases we have disclosed trade secrets and other proprietary knowledge, may also provide services to other parties in the medical device industry, including companies, universities and research organizations that are developing competing products. In addition, some of our former employees who were exposed to certain of our trade secrets and other proprietary knowledge in the course of their employment may seek employment with, and become employed by, our competitors. We cannot be assured that consultants, employees and other third parties with whom we have entered into confidentiality agreements will not breach the terms of such agreements by improperly using or disclosing our trade secrets or other proprietary knowledge or that we will have adequate remedies for any such breach.
Trade secret protection does not prevent independent discovery of the technology or proprietary information or use of the same. Competitors may independently duplicate or exceed our technology in whole or in part. If we are not successful in maintaining the confidentiality of our technology, the loss of trade secret protection or know-how relating to CHEMOSAT/Melphalan/HDS will significantly impair our ability to commercialize CHEMOSAT in the EEA, and our value and results of operations will be harmed. In particular, we rely on trade secret protection for the filter media, which is a key component of our system.
Similar considerations apply in any other foreign country where we receive approval. Since we do not have valid issued patents for the current version of CHEMOSAT/Melphalan/HDS in these countries, our ability to successfully commercialize CHEMOSAT/Melphalan/HDS will depend on our ability to maintain trade secret protection in these markets.
Risks Related to Products Liability
We may be the subject of product liability claims or product recalls, and we may be unable to maintain insurance adequate to cover potential liabilities.
Our business exposes us to potential liability risks that may arise from clinical trials and the testing, manufacture, marketing, sale and use of CHEMOSAT/Melphalan/HDS. In addition, because CHEMOSAT/Melphalan/HDS is intended for use in patients with cancer, there is an increased risk of death among the patients treated with our system which may increase the risk of product liability lawsuits related to clinical trials or commercial sales. We may be subject to claims against us even if the injury is due to the actions of others. For example, if the medical personnel that use our system on patients are not properly trained or are negligent in the use of our system, the patient may be injured through the use of our system, which may subject us to claims. Were such a claim asserted we would likely incur substantial legal and related expenses even if we prevail on the merits. Claims for damages, whether or not successful, could cause delays in clinical trials and result in the loss of physician endorsement, adverse publicity and/or limit our ability to market and sell the system, resulting in loss of revenue. In addition, it may be necessary for us to recall products that do not meet approved specifications, which would also result in adverse publicity, as well as resulting in costs connected to the recall and loss of revenue. A successful products liability claim or product recall would have a material adverse effect on our business, financial condition and results of operations. We currently carry product liability and clinical trial insurance coverage, but it may be insufficient to cover one or more large claims.
Risks Related to Our Common Stock
The market price of our common stock has been, and may continue to be volatile and fluctuate significantly, which could result in substantial losses for investors.
The trading price for our common stock has been, and we expect it to continue to be, volatile. The price at which our common stock trades depends upon a number of factors, including our historical and anticipated operating results, our financial situation, announcements of technological innovations or new products by us or our competitors, our ability or inability to raise the additional capital we may need and the terms on which we raise it, and general market and economic conditions. Some of these factors are beyond our control. Broad market fluctuations may lower the market price of our common stock and affect the volume of trading in our stock, regardless of our financial condition, results of operations, business or prospect. Among the factors that may cause the market price of our common stock to fluctuate are the risks described in this “Risk Factors” section and other factors, including:
| o | fluctuations in our quarterly operating results or the operating results of our competitors; |
| o | variance in our financial performance from the expectations of investors; |
| o | changes in the estimation of the future size and growth rate of our markets; |
| o | changes in accounting principles or changes in interpretations of existing principles, which could affect our financial results; |
| o | failure of our products to achieve or maintain market acceptance or commercial success; |
| o | conditions and trends in the markets we serve; |
| o | changes in general economic, industry and market conditions; |
| o | success of competitive products and services; |
| o | changes in market valuations or earnings of our competitors; |
| o | changes in our pricing policies or the pricing policies of our competitors; |
| o | announcements of significant new products, contracts, acquisitions or strategic alliances by us or our competitors; |
| o | changes in legislation or regulatory policies, practices or actions; |
| o | the commencement or outcome of litigation involving our company, our general industry or both; |
| o | recruitment or departure of key personnel; |
| o | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| o | actual or expected sales of our common stock by our stockholders; and |
| o | the trading volume of our common stock. |
In addition, the stock markets, in general, the NASDAQ Capital Market and the market for pharmaceutical companies in particular, may experience a loss of investor confidence. Such loss of investor confidence may result in extreme price and volume fluctuations in our common stock that are unrelated or disproportionate to the operating performance of our business, financial condition or results of operations. These broad market and industry factors may materially harm the market price of our common stock and expose us to securities class action litigation. Such litigation, even if unsuccessful, could be costly to defend and divert management’s attention and resources, which could further materially harm our financial condition and results of operations.
Our warrants contain anti-dilution provisions that, if triggered, could cause dilution to our existing stockholders.
The warrants issued in our May 2012 and February 2015 offerings are subject to an exercise price adjustment upon certain equity issuances below $0.82 per share or $1.38 per share, respectively (as may be further adjusted). In addition to the potential dilutive effect of these provisions, there is the potential that a large number of the shares may be sold in the public market at any given time, which could place additional downward pressure on the trading price of our common stock.
Anti-takeover provisions in our Certificate of Incorporation and By-laws may reduce the likelihood of a potential change of control, or make it more difficult for our stockholders to replace management.
Certain provisions of our Certificate of Incorporation and By-laws could have the effect of making it more difficult for our stockholders to replace management at a time when a substantial number of our stockholders might favor a change in management. These provisions include:
| o | providing for a staggered board; and |
| o | authorizing the board of directors to fill vacant directorships or increase the size of our board of directors. |
Furthermore, our board of directors has the authority to issue up to 10,000,000 shares of preferred stock in one or more series and to determine the rights and preferences of the shares of any such series without stockholder approval. Any series of preferred stock is likely to be senior to the common stock with respect to dividends, liquidation rights and, possibly, voting rights. Our board’s ability to issue preferred stock may have the effect of discouraging unsolicited acquisition proposals, thus adversely affecting the market price of our common stock.
Our common stock is listed on The NASDAQ Capital Market and if we do not maintain compliance with NASDAQ Marketplace Rules our common stock may be delisted from the NASDAQ Capital Market.
To keep our listing on The NASDAQ Capital Market, we are required to maintain: (i) a minimum bid price of $1.00 per share, (ii) a certain public float, (iii) a certain number of round lot shareholders and (iv) one of the following: a net income from continuing operations (in the latest fiscal year or two of the three last fiscal years) of at least $500,000, a market value of listed securities of at least $35 million or a stockholders’ equity of at least $2.5 million. On June 13, 2013, we were notified by the NASDAQ Listing Qualifications Department that we do not comply with the $1.00 minimum bid threshold as our common stock has traded below the $1.00 minimum bid price for 30 consecutive business days. We were automatically provided with a 180-calendar day period within which to regain compliance and we subsequently qualified for an additional 180-day grace period. To regain compliance, our common stock was required to close at or above the $1.00 minimum bid price for at least 10 consecutive days or more at the discretion of NASDAQ. On February 24, 2014, we obtained shareholder approval of an amendment to our Certificate of Incorporation to effect a reverse stock split of our common stock at a specific ratio within a range from 1-for-8 to 1-for-16, inclusive, on or prior to December 31, 2014. In April 2014, we filed a Certificate of Amendment to our Amended and Restated Certificate of Incorporation with the Secretary of State of the State of Delaware, which has effected a reverse stock split of our common stock at a ratio of 1-to-16. Following the completion of the reverse stock split, we resumed compliance with the $1.00 minimum bid threshold; however, we may fail to comply with the requirement in the future.
We are also required to maintain certain corporate governance requirements. In the event that in the future we are notified that we no longer comply with NASDAQ’s corporate governance requirements, and we fail to regain compliance within the applicable cure period, our common stock could be delisted from The NASDAQ Capital Market.
If our common stock is delisted, trading of the stock will most likely take place on an over-the-counter market established for unlisted securities, such as the Pink Sheets or the OTC Bulletin Board. An investor is likely to find it less convenient to sell, or to obtain accurate quotations in seeking to buy, our common stock on an over-the-counter market, and many investors may not buy or sell our common stock due to difficulty in accessing over-the-counter markets, or due to policies preventing them from trading in securities not listed on a national exchange or other reasons. For these reasons and others, delisting would adversely affect the liquidity, trading volume and price of our common stock, causing the value of an investment in us to decrease and having an adverse effect on our business, financial condition and results of operations, including our ability to attract and retain qualified executives and employees and to raise capital.
If our common stock is delisted from The NASDAQ Capital Market, we may be subject to the risks relating to penny stocks.
If our common stock were to be delisted from trading on The NASDAQ Capital Market and the trading price of the common stock were below $5.00 per share on the date the common stock were delisted, trading in our common stock would also be subject to the requirements of certain rules promulgated under the Exchange Act. These rules require additional disclosure by broker-dealers in connection with any trades involving a stock defined as a “penny stock” and impose various sales practice requirements on broker-dealers who sell penny stocks to persons other than established customers and accredited investors, generally institutions. These additional requirements may discourage broker-dealers from effecting transactions in securities that are classified as penny stocks, which could severely limit the market price and liquidity of such securities and the ability of purchasers to sell such securities in the secondary market. A penny stock is defined generally as any non-exchange listed equity security that has a market price of less than $5.00 per share, subject to certain exceptions.
We have never declared or paid any dividends to the holders of our common stock and we do not expect to pay cash dividends in the foreseeable future.
We currently intend to retain all earnings for use in connection with the expansion of our business and for general corporate purposes. Our board of directors will have the sole discretion in determining whether to declare and pay dividends in the future. The declaration of dividends will depend on our profitability, financial condition, cash requirements, future prospects and other factors deemed relevant by our board of directors. Our ability to pay cash dividends in the future could be limited or prohibited by the terms of financing agreements that we may enter into or by the terms of any preferred stock that we may authorize and issue. We do not expect to pay dividends in the foreseeable future. As a result, holders of our common stock must rely on stock appreciation for any return on their investment.
The issuance of additional stock in connection with acquisitions or otherwise will dilute all other stockholdings.
We are not restricted from issuing additional shares of our common stock, or from issuing securities that are convertible into or exchangeable for, or that represent the right to receive, common stock. As of December 31, 2014, we had an aggregate of 160.3 million shares of common stock authorized but unissued.. Subject to certain volume limitations imposed by The NASDAQ Capital Market, we may issue all of these shares without any action or approval by our shareholders. We have established an at the market equity offering program, and we may issue shares under this program without any action or approval by our shareholders. We may expand our business through complementary or strategic business combinations or acquisitions of other companies and assets, and we may issue shares of common stock in connection with those transactions. The market price of our common stock could decline as a result of our issuance of a large number of shares of common stock, particularly if the per share consideration we receive for the stock we issue is less than the per share book value of our common stock or if we are not expected to be able to generate earnings with the proceeds of the issuance that are as great as the earnings per share we are generating before we issue the additional shares. In addition, any shares issued in connection with these activities, the exercise of stock options or otherwise would dilute the percentage ownership held by our investors. We cannot predict the size of future issuances or the effect, if any, that they may have on the market price of our common stock.
In February 2015, we completed the sale of approximately 2.4 million shares of our common stock and the issuance of warrants to purchase approximately 1.1 million shares of our common stock pursuant to an underwriting agreement. We received proceeds of approximately $2.8 million, with net cash proceeds after related expenses from this transaction of approximately $2.4 million. The shares and warrants were issued pursuant to an effective registration statement on Form S-3. Form S-3 limits the aggregate market value of securities that we are permitted to offer in any 12 month-period under Form S-3 to one-third of our public float. Given the offering in February 2015, other sales under our at the market equity offering program during the relevant 12 month-period, and our current aggregate market value of securities, we are at the applicable limit under Form S-3. As a result, unless the market value of our securities increases our ability to raise capital may be impaired and we currently are unable to utilize the Form S-3 or access our at the market equity offering program.
None.
Our corporate offices currently occupy 5,818 square feet of office space at 1301 Avenue of the Americas, New York, New York under a license agreement that expires in May 2016. The Company leases three additional spaces in the United States including approximately 18,000 square feet at Suites 2 and 3 Country Club Road and 6,000 square feet at 95-97 Park Road in Queensbury, New York, as well as 17,320 square feet of office space at 810 Seventh Avenue, New York, New York. The lease agreements expire in June 2015, October 2015, and March 2021 respectively. The Company has subleased the office space at 810 Seventh Avenue. See Note 12 to the Company’s audited financial statements contained in this Annual Report on Form 10-K for more details. Delcath owns a building containing approximately 10,320 square feet at 566 Queensbury Avenue in Queensbury, NY. These facilities house manufacturing, quality assurance and quality control, research and development, and office space. The Company also owns approximately six acres of land at 10, 12 and 14 Park Road in Queensbury, New York. In addition, Delcath Systems Limited leases a facility for office and manufacturing containing approximately 19,200 square feet at 19 Mervue, Industrial Park in Galway, Ireland under a lease agreement that expires August 2, 2021, but can be terminated after the fifth year (August 2016). The Company has sublet 5,662 square feet of this facility. The Company believes substantially all of our property and equipment is in good condition and that we have sufficient capacity to meet our current operational needs.
In re Delcath Systems, Inc. Securities Litigation, United States District Court for the Southern District of New York (Case No. 13-cv-3116)
On May 8, 2013, a purported stockholder of the Company filed a putative class action complaint in the United States District Court for the Southern District of New York, captioned Bryan Green, individually and on behalf of all others similar situated, v. Delcath Systems, Inc., et al. (“Green”), Case No. 1:13-cv-03116-LGS. On June 14, 2013, a substantially similar complaint was filed in the United States District Court for the Southern District of New York, captioned Joseph Connico, individually and on behalf of all others similarly situated, v. Delcath Systems, Inc., et al. (“Connico”), Case No. 1:13-cv-04131-LGS.
At a hearing on August 2, 2013, the Court consolidated the Green and Connico actions under the caption In re Delcath Systems, Inc. Securities Litigation, No. 13-cv-3116, appointed Lead Plaintiff, Delcath Investor Group, and approved Pomerantz Grossman Hufford Dahlstrom & Gross LLP as Lead Plaintiff’s choice of counsel.
On September 18, 2013, Lead Plaintiff filed a consolidated amended complaint, naming the Company and Eamonn P. Hobbs as defendants (the “Defendants”). The consolidated amended complaint asserts that Defendants violated Sections 10(b) and 20(a) of the Securities Exchange Act of 1934 by allegedly making false and misleading statements or omissions regarding the Company’s New Drug Application for its Melblez Kit (Melblez (melphalan) for Injection for use with the Delcath Hepatic Delivery System), for the treatment of patients with unresectable metastatic ocular melanoma in the liver. The putative class period alleged in the amended complaint is April 21, 2010 through and including September 13, 2013. Lead Plaintiff seeks compensatory damages, equitable relief, and reasonable attorneys’ fees, expert fees and other costs. On October 31, 2013, Defendants filed their motion to dismiss, which was subsequently denied on June 27, 2014. On July 25, 2014, Defendants filed their respective answers to Lead Plaintiff’s consolidated amended complaint. On July 29, 2014, the Court held a scheduling conference setting forth a case management plan. The parties are proceeding with discovery. On October 15, 2014, Lead Plaintiff served Defendants with a Motion for Class Certification to which Defendants served an opposition on December 16, 2014. On February 4, 2015, Lead Plaintiff served Defendants with a reply in support of the Motion for Class Certification and the parties filed all class certification pleadings with the Court.
The Company believes that the In re Delcath Systems, Inc. Securities Litigation action lacks merit and intends to defend the case vigorously.
In re Delcath Systems, Inc. Derivative Shareholder Litigation, United States District Court for the Southern District of New York (Lead Case No. 1:13-cv-03494-LGS)
On May 23, 2013, purported stockholders of the Company filed a shareholder derivative lawsuit in the United States District Court for the Southern District of New York, captioned Vincent J. Orlando and Carol Orlando, derivatively on behalf of Delcath Systems, Inc. v. Harold S. Koplewicz, et al. (“Orlando”), Case No. 1:13-cv-03494-LGS. On June 11, 2013, a substantially similar complaint was filed in the United States District Court for the Southern District of New York, captioned Howard Warsett, derivatively on behalf of Delcath Systems, Inc. v. Harold S. Koplewicz, et al. (“Warsett”), Case No. 1:13-cv-04002-LGS. On July 19, 2013, another substantially similar complaint was filed in the United States District Court for the Southern District of New York, captioned Patricia Griesi, derivative on behalf of nominal defendant Delcath Systems, Inc. v. Harold S. Koplewicz, et al. (“Griesi”), Case No. 13 cv 5024. In all three cases, Harold S. Koplewicz, Laura A. Brege, Tasos G. Konidaris, Eamonn P. Hobbs, Douglas G. Watson, Laura A. Philips, Roger G. Stoll, and Gabriel Leung were named as defendants (the“Individual Defendants”), and the Company was named as a nominal defendant.
All three complaints assert claims for breach of fiduciary duty for disseminating false and misleading information, breach of fiduciary duty for failing to properly oversee and manage the company, and gross mismanagement for making false and misleading statements or failing to disclose material information regarding (i) the Company’s New Drug Application for its Melblez Kit (Melblez (melphalan) for Injection for use with the Delcath Hepatic Delivery System), for the treatment of patients with unresectable metastatic ocular melanoma, and (ii) the status of the Company’s manufacturing facilities. In addition, the Orlando complaint further asserts claims for contribution and indemnification, abuse of control, and waste of corporate assets, while the Warsett complaint asserts an additional claim for unjust enrichment. The Griesi complaint also asserts additional claims for breach of fiduciary duties for failing to maintain internal controls, unjust enrichment, abuse of control, and violations of Section 14(a) of the Securities Exchange Act of 1934. The relevant time period alleged in the Orlando action is April 21, 2010 through the present, and the relevant time period alleged in the Warsett action is April 10, 2010 through the present. The relevant time period alleged in Griesi is April 21, 2010 through May 2, 2013. The Orlando, Warsett, and Griesi plaintiffs seek damages as well as reasonable costs and attorneys’ fees. The Griesi plaintiffs also seek corporate governance reforms and improvements and restitution.
On June 25, 2013, the Court consolidated the Orlando and Warsett actions with the caption In re Delcath Systems, Inc. Derivative Shareholder Litigation, Lead Case No. 1:13-cv-03494-LGS (“Consolidated Derivative Case”). On August 1, 2013, the Court consolidated the Griesi action under the caption In re Delcath Systems, Inc. Derivative Shareholder Litigation, Lead Case No. 1:13-cv-03494-LGS. At a hearing on August 2, 2013, the Court entered an order approving Federman & Sherwood as lead counsel. The Court stayed the Consolidated Derivative Case, pending resolution of an anticipated motion to dismiss in In re Delcath Systems, Inc. Securities Litigation, United States District Court for the Southern District of New York, No. 13-cv-3116.
On September 12, 2014, Plaintiffs Vincent Orlando and Carol Orlando filed a Verified Amended Consolidated Shareholder Derivative Complaint (the “Amended Complaint”) in the Consolidated Derivative Case. The Amended Complaint is brought against the Individual Defendants, and names the Company as a nominal defendant (collectively, the “Defendants”). The Amended Complaint alleges breaches of fiduciary duty against the Individual Defendants for disseminating false and misleading information and for failing to properly oversee and manage the company. In addition, the Amended Complaint alleges claims for gross mismanagement, contribution and indemnification, abuse of control, and waste of corporate assets. The relevant time period alleged in the Amended Complaint is April 21, 2010 through the present. The Plaintiffs in the Amended Complaint seek damages as well as reasonable costs and attorneys’ fees. On October 27, 2014, Defendants served Plaintiffs with their Motion to Dismiss the Amended Complaint, and the motion is fully briefed.
The Individual Defendants in the Consolidated Derivative Case deny any wrongdoing, believe the claims are baseless, and will defend accordingly.
Howard D. Weinstein, derivatively on behalf of Delcath Systems, Inc. v. Harold S. Koplewicz, et al., Supreme Court of the State of New York County of New York (Case No. 652030/2013)
On June 7, 2013, a purported stockholder of the Company filed a shareholder derivative lawsuit in the Supreme Court of the State of New York County of New York, captioned Howard D. Weinstein, derivatively on behalf of Delcath Systems, Inc. v. Harold S. Koplewicz, et al., (“Weinstein”) Case No. 652030/2013. The action named Harold S. Koplewicz, Laura A. Brege, Tasos G. Konidaris, Eamonn P. Hobbs, Douglas G. Watson, Laura A. Philips, Roger G. Stoll, and Gabriel Leung as individual defendants (the “Individual Defendants”), as well as the Company, as a nominal defendant.
The complaint asserts claims for breach of fiduciary duty for disseminating false and misleading information, breach of fiduciary duty for failing to properly oversee and manage the company, gross mismanagement, contribution and indemnification, abuse of control, and waste of corporate assets in connection with allegations that the Individual Defendants made false and misleading statements or failed to disclose material information regarding (i) the Company’s New Drug Application for its Melblez Kit (Melblez (melphalan) for Injection for use with the Delcath Hepatic Delivery System), for the treatment of patients with unresectable metastatic ocular melanoma, and (ii) the status of the Company’s manufacturing facilities. The relevant time period alleged is April 21, 2010 through the present. The plaintiff seeks damages, as well as reasonable costs and attorneys’ fees.
In July 2014, the parties in the Weinstein matter agreed to stipulate to stay the proceeding until the federal district court rules on the anticipated motion to dismiss in In re Delcath Systems, Inc. Derivative Shareholder Litigation, United States District Court for the Southern District of New York (Lead Case No. 1:13-cv-03494-LGS).
The Individual Defendants in the Weinstein matter deny any wrongdoing, believe the claims are baseless, and will defend accordingly.
Part II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Our common stock is traded on The NASDAQ Capital Market under the symbol “DCTH”.
The following table sets forth the high and low last reported sales prices of our common stock for the fiscal quarters indicated as reported on The NASDAQ Capital Market:
Common Stock Price Range
|
2014
|
||||||||
|
High
|
Low
|
|||||||
|
Quarter ended March 31, 2014
|
$
|
6.24
|
$
|
4.16
|
||||
|
Quarter ended June 30, 2014
|
4.88
|
2.53
|
||||||
|
Quarter ended September 30, 2014
|
2.68
|
1.93
|
||||||
|
Quarter ended December 31, 2014
|
1.96
|
1.11
|
||||||
|
2013
|
||||||||
|
High
|
Low
|
|||||||
|
Quarter ended March 31, 2013
|
$
|
34.08
|
$
|
20.80
|
||||
|
Quarter ended June 30, 2013
|
30.56
|
5.92
|
||||||
|
Quarter ended September 30, 2013
|
6.88
|
4.74
|
||||||
|
Quarter ended December 31, 2013
|
9.44
|
3.68
|
||||||
On March 11, 2015 there were 27 stockholders of record of our common stock.
Dividend Policy
The Company has never declared or paid cash dividends on our common stock and has no intention to do so in the foreseeable future.
Recent Sales of Unregistered Securities
The Company did not sell any equity securities that were not registered under the Securities Act of 1933, as amended, in the years ended December 31, 2014, 2013 and 2012.
Performance Graph
The graph below matches the cumulative 5-Year total return of holders of Delcath Systems Inc.'s common stock with the cumulative total returns of the NASDAQ Composite index and a customized peer group of fifteen companies that includes: Acura Pharmaceuticals Inc., Adamis Pharmaceuticals Corp., Alkermes PLC, Aradigm Corp., Columbia Laboratories Inc., Delcath Systems Inc., Flamel Technologies SA, Generex Biotechnology Corp., Hospira Inc., Insite Vision Inc., Intellipharmaceutics International Inc., Novadel Pharma Inc., Petmed Express Inc., Psivida Corp. and Valeant Pharmaceuticals International Inc. The graph assumes that the value of the investment in our common stock, in each index, and in the peer group (including reinvestment of dividends) was $100 on 12/31/2009 and tracks it through 12/31/2014.
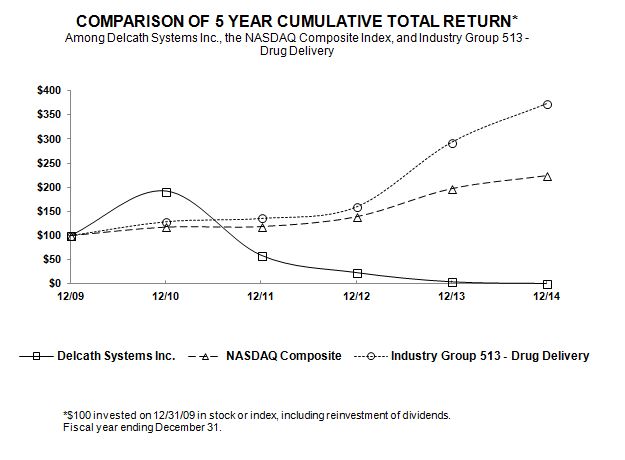
|
12/09
|
12/10
|
12/11
|
12/12
|
12/13
|
12/14
|
|||||||||||||||||||
|
Delcath Systems Inc.
|
100.00
|
191.78
|
59.69
|
24.07
|
4.99
|
1.48
|
||||||||||||||||||
|
NASDAQ Composite
|
100.00
|
117.61
|
118.70
|
139.00
|
196.83
|
223.74
|
||||||||||||||||||
|
Industry Group 513 - Drug Delivery
|
100.00
|
128.72
|
135.61
|
159.21
|
292.88
|
372.90
|
|
12/09
|
12/10
|
12/11
|
12/12
|
12/13
|
12/14
|
|||||||||||||||||||
|
Delcath Systems Inc.
|
91.78
|
%
|
-68.88
|
%
|
-59.67
|
%
|
-79.27
|
%
|
-70.34
|
%
|
||||||||||||||
|
NASDAQ Composite
|
17.61
|
%
|
0.92
|
%
|
17.11
|
%
|
41.60
|
%
|
13.67
|
%
|
||||||||||||||
|
Industry Group 513 - Drug Delivery
|
28.72
|
%
|
5.35
|
%
|
17.40
|
%
|
83.96
|
%
|
27.32
|
%
|
The stock price performance included in this graph is not necessarily indicative of future stock price performance.
The selected financial data set forth below should be read together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and related notes included in this Annual Report on Form 10-K.
The selected financial data set forth below as of December 31, 2014, 2013, 2012, 2011, and 2010 and for the years ended December 31, 2014, 2013, 2012, 2011, and 2010 are derived from our audited financial statements included in this Annual Report on Form 10-K. All other selected financial data set forth below is derived from our audited financial statements not included in this Annual Report on Form 10-K. Our historical results are not necessarily indicative of our results of operations to be expected in the future.
|
Year Ended December 31,
|
||||||||||||||||||||
|
(Dollars in thousands)
|
2014
|
2013
|
2012
|
2011
|
2010
|
|||||||||||||||
|
Statement of Operations Data
|
||||||||||||||||||||
|
Total revenue
|
$
|
1,069
|
$
|
790
|
$
|
346
|
$
|
-
|
$
|
-
|
||||||||||
|
Costs and expenses
|
20,082
|
33,345
|
54,178
|
46,456
|
30,743
|
|||||||||||||||
|
Operating loss
|
19,304
|
33,019
|
53,871
|
46,456
|
30,743
|
|||||||||||||||
|
Net loss
|
17,381
|
30,324
|
51,868
|
30,885
|
46,684
|
|||||||||||||||
|
Loss per share
|
(1.84
|
)
|
(1.81
|
)
|
(13.54
|
)
|
(0.68
|
)
|
(1.20
|
)
|
||||||||||
|
Year Ended December 31,
|
||||||||||||||||||||
|
(Dollars in thousands)
|
2014
|
2013
|
2012
|
2011
|
2010
|
|||||||||||||||
|
Balance Sheet Data
|
||||||||||||||||||||
|
Current assets
|
$
|
21,966
|
$
|
34,028
|
$
|
26,432
|
$
|
31,988
|
$
|
48,898
|
||||||||||
|
Total assets
|
23,764
|
37,097
|
30,474
|
35,241
|
50,578
|
|||||||||||||||
|
Current liabilities
|
4,576
|
6,632
|
10,156
|
8,837
|
21,197
|
|||||||||||||||
|
Stockholder’s equity
|
18,145
|
30,099
|
20,009
|
26,104
|
29,081
|
|||||||||||||||
Overview
Delcath Systems, Inc. is a late-stage clinical development company with early commercial activity in Europe focused on cancers of the liver. We are a specialty pharmaceutical and medical device company developing our proprietary product—Melphalan Hydrochloride for Injection for use with the Delcath Hepatic Delivery System (Melphalan/HDS). In Europe, our proprietary system to deliver and filter melphalan hydrochloride is marketed as a device under the trade name Delcath Hepatic CHEMOSAT® Delivery System for Melphalan (CHEMOSAT).
Our primary focus is on the execution of our clinical development program (CDP) in ocular melanoma liver metastases (mOM), intrahepatic cholangiocarncinoma (ICC), hepatocellular carcinoma (HCC or primary liver), and certain other cancers that are metastatic to the liver. We believe the disease states we are investigating represent a multi-billion dollar global market opportunity and a clear unmet medical need.
In the United States, Melphalan/HDS is considered a combination drug and device product, and is regulated as a drug by the FDA. The FDA has granted us five orphan drug designations, including two orphan designations for the use of the drug melphalan for the treatment of patients with ocular melanoma liver metastases and HCC. Melphalan/HDS has not been approved for sale in the United States.
In Europe, the current version of our CHEMOSAT product is regulated as a Class IIb medical device and received its CE Mark in 2012. We are in an early phase of commercializing CHEMOSAT in select markets in the European Union (United Kingdom and Germany) where we believe the prospect of securing adequate reimbursement for the procedure is strongest.
Currently there are few effective treatment options for certain cancers in the liver. Traditional treatment options include surgery, chemotherapy, liver transplant, radiation therapy, interventional radiology techniques, and isolated hepatic perfusion. We believe that CHEMOSAT/Melphalan/HDS represents a potentially important advancement in regional therapy for primary liver cancer and certain other cancers metastatic to the liver. We believe that CHEMOSAT/Melphalan/HDS is uniquely positioned to treat the entire liver either as a standalone therapy or as a complement to other therapies.
Liquidity and Capital Resources
The Company’s future results are subject to substantial risks and uncertainties. Delcath has operated at a loss for its entire history and anticipates that losses will continue over the coming year. There can be no assurance that Delcath will ever generate significant revenues or achieve profitability. The Company expects to use cash, cash equivalents and investment proceeds to fund its operating activities. Delcath’s future liquidity and capital requirements will depend on numerous factors, including the progress of clinical trials and research and product development programs, obtaining approvals and complying with regulations; the timing and effectiveness of product commercialization activities, including marketing arrangements; the timing and costs involved in preparing, filing, prosecuting, defending and enforcing intellectual property rights; and the effect of competing technological and market developments.
At December 31, 2014, the Company had cash and cash equivalents totaling $20.5 million, as compared to cash and cash equivalents totaling $31.2 million at December 31, 2013. During the year ended December 31, 2014, the Company used $15.6 million of cash in its operating activities, which compares to $34.1 million used for operating activities during the comparable twelve month period in 2013. The decrease of $18.5 million is primarily driven by a reduction in regulatory and clinical costs related to an NDA submission made in 2012, a decrease in severance expenses following significant restructuring efforts in 2013, a reduction in compensation related expenses as the Company further reduced the number of employees from 37 employees at December 31, 2013 to 25 employees at December 31, 2014, and continued efforts to improve efficiency in the Company’s organization and operations. The Company believes it has sufficient capital to fund its operating activities into the first half of 2016.
Because Delcath’s business does not generate positive cash flow from operating activities, the Company will need to raise additional capital in order to fund clinical trial research and support development efforts relating to Ocular Melanoma liver metastases, ICC, HCC or other indications, and to fully commercialize the product. The Company believes it will be able to raise additional capital in the event it is in its best interest to do so. The Company anticipates raising such additional capital by either borrowing money, selling shares of Delcath’s capital stock, or entering into strategic alliances with appropriate partners. To the extent additional capital is not available when needed, the Company may be forced to abandon some or all of its development and commercialization efforts, which would have a material adverse effect on the prospects of our business. Further, the Company’s assumptions relating to its cash requirements may differ materially from its actual requirements because of a number of factors, including significant unforeseen delays in the regulatory approval process, changes in the timing, scope, focus and direction of clinical trials and costs related to commercializing the product.
The Company has funded its operations through a combination of private placements of its securities, public offerings in 2000, 2003, 2009, 2010, 2011, 2012, 2013, and 2015, registered direct offerings in 2007, 2009 and 2013, and “at the market” equity offering programs initiated in 2012 and 2013. For a detailed discussion of the Company’s various sales of securities and the “at the market” equity offering program see Note 10 to the Company’s audited financial statements contained in this Annual Report on Form 10-K.
As of December 31, 2014, the Company had two active registration statements.
On March 13, 2013, the Company filed a registration statement on Form S-3 with the SEC and also entered into a new sales agreement (the “March 2013 Sales Agreement”) with Cowen and Company, LLC to sell shares of the Company’s common stock, par value $.01 per share, having aggregate sales proceeds of $50,000,000, from time to time, through an “at the market” equity offering program under which Cowen and Company, LLC will act as sales agent. The registration statement became effective on May 1, 2013 (333-187230). As of December 31, 2014, Delcath had approximately $39.9 million available under this registration statement and intends to use this for its “at the market” equity offering program.
In December 2011, the Company filed a registration statement on Form S-3 with the SEC, which allowed the Company to offer and sell, from time to time in one or more offerings, up to $100,000,000 of common stock, preferred stock, warrants, debt securities and stock purchase contracts as it deemed prudent or necessary to raise capital at a later date. The registration statement became effective on February 13, 2012 (333-178819). The Company used this registration statement for its May 2012 public offering detailed in Note 10 to the Company’s audited financial statements contained in this Annual Report on Form 10-K. The Company subsequently filed a new shelf registration statement on Form S-3 with the SEC which became effective on October 9, 2012 (333-183675). This new shelf replaces the shelf registration filed in December 2011 and allows the Company to offer and sell, from time to time in one or more offerings, up to $100,000,000 of common stock, preferred stock, warrants, debt securities and stock purchase contracts as it deems prudent or necessary to raise capital at a later date. The Company used this registration statement for its Common Stock Purchase Agreement with Terrapin Opportunity, L.P. and registered direct offering in October 2013 detailed in Note 10 to the Company’s audited financial statements contained in this Annual Report on Form 10-K. As of December 31, 2014, Delcath had approximately $80.4 million available under this registration statement, of which approximately $4.6 million is reserved for the potential issuance of shares upon the exercise of warrants.
Form S-3 limits the aggregate market value of securities that we are permitted to offer in any 12 month-period under Form S-3 to one-third of our public float. Given the offering in February 2015, other sales under our at the market equity offering program during the relevant 12 month-period, and our current aggregate market value of securities, we are at the applicable limit under Form S-3. As a result, unless the market value of our securities increases our ability to raise capital may be impaired and we currently are unable to utilize the Form S-3 or access our at the market equity offering program.
The Company intends to use the net proceeds from any future offerings for general corporate purposes, including, but not limited to, funding clinical trials, obtaining regulatory approvals, commercialization of its products, capital expenditures and working capital.
On February 24, 2014, shareholders of the Company approved, through a shareholder vote, an amendment to the Company’s Amended and Restated Certificate of Incorporation authorizing the Board of Directors to effect a reverse stock split of Delcath’s common stock. The reverse stock split became effective on April 8, 2014 at which time Delcath’s common stock began trading on the NASDAQ Stock Exchange on a one-for-sixteen (1:16) split-adjusted basis. All owners of record as of the close of the NASDAQ market on April 8, 2014 received one share of Delcath common stock in exchange for sixteen issued and outstanding shares of Delcath common stock. No fractional shares were issued in connection with the reverse stock split. All fractional shares created by the one-for-sixteen exchange were rounded up to the next whole share. The reverse stock split had no impact on the number of common shares authorized or the par value per share of Delcath common stock, which remain 170,000,000 and $0.01, respectively. All current and prior period amounts related to shares, share prices and earnings per share, presented in these Consolidated Financial Statements and the accompanying Notes have been restated to give retrospective presentation for the reverse stock split.
Contractual Obligations, Commercial Commitments and Off-Balance Sheet Arrangements
The Company is obligated to make future payments under various operating lease agreements. The following table provides a summary of significant contractual obligations at December 31, 2014:
|
Payments Due by Period
|
||||||||||||||||||||
|
(in millions)
|
Total
|
Less than 1 year
|
1-3 years
|
3-5 years
|
More than 5 years
|
|||||||||||||||
|
Operating Activities:
|
||||||||||||||||||||
|
Operating Leases
|
$
|
2.2
|
$
|
0.9
|
$
|
0.9
|
$
|
0.4
|
$
|
-
|
||||||||||
Our operating lease obligations at December 31, 2014 include: the annual rent for our office space at 1301 Avenue of the Americas, New York, New York, which will expire in May 2016; the annual rent under the lease for our office space at 810 Seventh Avenue, New York, New York, which will expire in March 2021 and of which a certain amount of expense has been offset by two sub-leases; the annual rent under the leases for our facilities in Queensbury, New York, which expire in June 2015 and October 2015; and the annual rent for our facility in Galway, Ireland, which will expire in August 2021, but can be terminated after the fifth year (August 2016) upon not less than six months notice and of which a certain amount of expense has been offset by a sub-lease. See Part I, Item 2, “Properties” and Notes 8 and 12 to the Company’s audited financial statements contained in this Annual Report on Form 10-K.
Future Capital Needs; Additional Future Funding
Our future results are subject to substantial risks and uncertainties. The Company has operated at a loss for its entire history and there can be no assurance that it will ever achieve consistent profitability. The Company believes that it has adequate resources to fund operations through 2015 and anticipates that additional working capital will be required to continue our operations. There can be no assurance that such working capital will be available on acceptable terms, if at all.
Results of Operations for the Year Ended December 31, 2014; Comparisons of Results of the Years Ended December 31, 2013 and 2012
Revenue
The Company recorded approximately $1.1 million in total revenue during the year ended December 31, 2014. During the same period in 2013, Delcath recorded $0.8 million in total revenue, of which $0.3 million is related to the recognition of previously deferred revenue as a result of satisfying certain requirements of the Company’s agreement with Chi-Fu Trading Co. Ltd. The remainder of the revenue is related to product sales. During the same period in 2012, Delcath recorded $0.3 million in revenue related to product sales.
Cost of Goods Sold
During the year ended December 31, 2014, the Company recognized cost of goods sold of approximately $0.3 million related to product revenue of $1.1 million.
During the year ended December 31, 2013, the Company recognized cost of goods sold of approximately $0.5 million related to product revenue of $0.5 million. Due to adjustments in the anticipated use of inventory, the Company recorded $0.3 million cost for expired, obsolete and slow-moving inventory during the year ended December 31, 2013.
During the year ended December 31, 2012, the Company recognized cost of goods sold of approximately $39,000 related to product revenue of $0.4 million. As discussed in Note 3 to the Company’s audited financial statements contained in this Annual Report on Form 10-K, the Company did not recognize any cost of goods sold associated with the revenue or deferred revenue reported in the second or third quarters of 2012 because a portion of the Company’s inventory was purchased prior to obtaining authorization to affix the CE Mark to its Generation Two CHEMOSAT system in April 2012, including components used in the kits sold during those periods.
As Delcath continues progress with clinical adoption, the Company expects to see a certain amount of volatility in both the average selling price and gross margin for the next several years. This volatility will be related to several factors, including: adjustments to volume forecasts; the expected use of third party distributors whose purchase prices will be lower than direct-to-customer prices; the gradual increase in cost of goods sold as the Company exhausts raw materials that were purchased and expensed in prior periods and begins to recognize the actual costs of materials, labor and overhead; and an improvement in efficiencies as the Company increases its production of CHEMOSAT.
Operating Expenses
Selling, General and Administrative Expenses
For the year ended December 31, 2014, selling, general and administrative expenses decreased to $15.8 million from $20.7 million for the year ended December 31, 2013.The decrease reflects a reduction in severance and compensation related expenses following the Company’s significant workforce restructurings throughout 2013. As discussed further in Note 8, in 2014 the Company recognized $1.3 million in expenses related to vacating its 810 Seventh Avenue office space, offsetting a portion of the decrease in year over year expenses.
For the year ended December 31, 2013, selling, general and administrative expenses decreased to $20.7 million from $28.0 million for the year ended December 31, 2012. The decrease reflects the Company’s efforts to increase organizational efficiencies, including workforce restructurings initiated early in 2013 and efforts to streamline its European operations to focus on key direct markets. During the first half of 2012, the Company incurred certain expenses related to the early stages of its European commercial activities, including creating the appropriate subsidiaries, and the hiring of staff for sales and support positions across Europe.
Research and Development Expenses
For the year ended December 31, 2014, research and development expenses decreased to $4.3 million from $12.7 million for the year ended December 31, 2013. The decrease is primarily due to a significant reduction in regulatory and clinical expenses related to the Company’s NDA submission to the FDA, as well as a reduction in severance and compensation related expenses following the Company’s significant workforce restructurings throughout 2013.
For the year ended December 31, 2013, research and development expenses decreased to $12.7 million from $26.2 million for the year ended December 31, 2012. The decrease is primarily due to a significant reduction in regulatory and clinical expenses related to the Company’s NDA submission to the FDA.
Interest Income
Interest income is from a money market account and interest earned on operating accounts. For the year ended December 31, 2014, the Company had interest income of $5,232 as compared to interest income of $19,777 for the same period in 2013.
For the year ended December 31, 2013, the Company had interest income of $19,777 as compared to interest income of $19,358 for the same period in 2012.
Other Expense and Interest Expense
Other expense is primarily related to currency gains and losses. Interest expense is related to the commitment fee paid in 2012 upon entering into a Loan and Security Agreement with Silicon Valley Bank (SVB) and an ongoing Revolving Line Facility Fee as required by the agreement with SVB as discussed in Note 11 to the Company’s audited financial statements contained in this Annual Report on Form 10-K.
Net Loss
The Company had a net loss for the year ended December 31, 2014 of $17.4 million, a decrease of $12.9 million, or 42.6%, compared to the net loss for the same period in 2013. This decrease is primarily due to a $13.3 million decrease in operating expenses. The decrease in operating expenses reflects a significant decrease in severance and compensation related expenses following the Company’s workforce restructurings throughout 2013 as well as a reduction in costs related to the Company’s NDA submission.
The Company had a net loss for the year ended December 31, 2013 of $30.3 million, a decrease of $21.5 million, or 41.4%, compared to the net loss for the same period in 2012. This decrease is primarily due to a $20.8 million decrease in operating expenses. The decrease in operating expenses reflects a significant decrease in costs related to the Company’s NDA submission and overall operations.
Application of Critical Accounting Policies
The Company’s financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (GAAP). Certain accounting policies have a significant impact on amounts reported in the financial statements. A summary of those significant accounting policies can be found in Note 3 to the Company’s audited financial statements contained in this Annual Report on Form 10-K. During 2012, Delcath transitioned from a development stage company to a commercialization organization.
The Company considers the valuation allowance for the deferred tax assets to be a significant accounting estimate. In applying ASC 740 management estimates future taxable income from operations and tax planning strategies in determining if it is more likely than not that the Company will realize the benefits of its deferred tax assets. Management believes the Company does not have any uncertain tax positions.
The Company has adopted the provisions of ASC 718, which establishes accounting for equity instruments exchanged for employee services. Under the provisions of ASC 718, share-based compensation is measured at the grant date, based upon the fair value of the award, and is recognized as an expense over the option holders’ requisite service period (generally the vesting period of the equity grant). The Company expenses its share-based compensation under the ratable method, which treats each vesting tranche as if it were an individual grant.
The Company has adopted the provisions of ASC 505-50, which establishes accounting for equity-based payments to non-employees. Measurement of compensation cost related to common shares issued to non-employees for services is based on the value of the services provided or the fair value of the shares issued. Each transaction is reviewed to determine the more reliably measurable basis for the valuation. The measurement of non-employee stock-based compensation is subject to periodic adjustment as the underlying equity instrument vests. Non-employee stock-based compensation charges are amortized over the vesting period or period of performance of the services.
The Company has adopted the provisions of ASC 820, which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
ASC 820 emphasizes that fair value is a market-based measurement, not an entity-specific measurement. Therefore, a fair value measurement should be determined based on the assumptions that market participants would use in pricing the asset or liability. As a basis for considering market participant assumptions in fair value measurements, ASC 820 establishes a fair value hierarchy that distinguishes between market participant assumptions based on market data obtained from sources independent of the reporting entity (observable inputs that are classified within Levels 1 and 2 of the hierarchy) and the reporting entity’s own assumptions about market participant assumptions (unobservable inputs classified within Level 3 of the hierarchy).
Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities that the Company has the ability to access. Level 2 inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 inputs may include quoted prices for similar assets and liabilities in active markets, as well as inputs that are observable for the asset or liability (other than quoted prices), such as interest rates, foreign exchange rates, and yield curves that are observable at commonly quoted intervals. Level 3 inputs are unobservable inputs for the asset or liability which are typically based on an entity’s own assumptions, as there is little, if any, related market activity. In instances where the determination of the fair value measurement is based on inputs from different levels of the fair value hierarchy, the level in the fair value hierarchy within which the entire fair value measurement falls is based on the lowest level input that is significant to the fair value measurement in its entirety. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment, and considers factors specific to the asset or liability. See Note 9 to the Company’s audited financial statements contained in this Annual Report on Form 10-K for assets and liabilities the Company has evaluated under ASC 820.
The Company may be exposed to market risk through changes in market interest rates that could affect the interest earned on its cash balances.
The Company measures all derivatives, including certain derivatives embedded in contracts, at fair value and recognizes them on the balance sheet as an asset or a liability, depending on the Company’s rights and obligations under the applicable derivative contract.
In October 2013, the Company completed the sale of 1.3 million shares of its common stock and the issuance of warrants to purchase approximately 0.6 million common shares (the “2013 Warrants”) pursuant to a placement agency agreement. The Company received proceeds of $7.5 million, with net cash proceeds after related expenses from this transaction of approximately $6.9 million. Of those proceeds, the Company allocated an estimated fair value of $1.9 million to the 2013 Warrants. The 2013 Warrants became exercisable on April 30, 2014. At December 31, 2014, the 2013 Warrants were exercisable at $7.04 per share with approximately 0.6 million warrants outstanding. The 2013 Warrants have a five-year term. The shares and warrants were issued pursuant to an effective registration statement on Form S-3. There were no 2013 Warrants exercised during the year ended December 31, 2014.
In May 2012, the Company completed the sale of 1.0 million shares of its common stock and the issuance of warrants to purchase 0.3 million common shares (the “2012 Warrants”) pursuant to an underwriting agreement. The Company received proceeds of $21.5 million, with net cash proceeds after related expenses from this transaction of approximately $21.1 million. Of those proceeds, the Company allocated an estimated fair value of $3.4 million to the 2012 Warrants. As required by the 2012 Warrant agreement, the exercise price of the warrants was adjusted following the Company’s October 2014 sale of common stock. At December 31, 2014, the 2012 Warrants were exercisable at $1.75 per share with approximately 0.3 million warrants outstanding. The 2012 Warrants have a three-year term. The shares and warrants were issued pursuant to an effective registration statement on Form S-3. During the year ended December 31, 2014, 14,000 2012 Warrants were exercised for net proceeds of approximately $34,000.
In June 2009, the Company completed the sale of 0.1 million shares of its common stock and the issuance of warrants to purchase 0.1 million common shares (the “2009 Warrants”) pursuant to a subscription agreement with a single investor. The Company received proceeds of $3.0 million, with net cash proceeds after related expenses from this transaction of approximately $2.7 million. Of those proceeds, the Company allocated an estimated fair value of $2.2 million to the 2009 Warrants. The shares and warrants were issued pursuant to an effective registration statement on Form S-3. During the year ended December 31, 2014, 35,000 2009 Warrants were exercised for net proceeds of approximately $0.1 million. The 2009 Warrants had a five-year term which expired on June 15, 2014. The remaining liability after warrant exercises was credited to pre-tax derivative instrument income as of June 30, 2014.
For the year ended December 31, 2014, the Company recorded pre-tax derivative warrant income of $1.9 million. The resulting derivative warrant liabilities totaled $0.2 million at December 31, 2014. In the event of a hypothetical 10% increase in the market price of our common shares on which the December 31, 2014 valuation was based, the value of the derivative liability would have increased by approximately $40,000. Management expects that the warrants will either be exercised or expire worthless. The fair value of the warrants at December 31, 2014 was determined by using an option pricing model with the following assumptions:
|
2013 Warrants
|
2012 Warrants
|
|||||||
|
Expected volatility
|
89.89
|
%
|
49.23
|
%
|
||||
|
Risk-free interest rates
|
1.38
|
%
|
0.12
|
%
|
||||
|
Expected life (in years)
|
3.83
|
0.41
|
||||||
Subsequent to December 31, 2014, the Company completed the sale of 2.5 million shares of its common stock and the issuance of warrants to purchase 1.1 million common shares (the “2015 Warrants”) pursuant to an underwriting agreement. The Company received proceeds of $2.6 million, with net cash proceeds after related expenses from this transaction expected to be approximately $2.4 million. Of those proceeds, the Company allocated an estimated fair value of $0.8 million to the 2015 Warrants. The 2015 Warrants have a five-year term. The shares and warrants were issued pursuant to an effective registration statement on Form S-3.
|
Report of Ernst & Young LLP - Independent Registered Public Accounting Firm
|
F-1
|
|
Consolidated Balance Sheets at December 31, 2014 and 2013
|
F-2
|
|
Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2014, 2013, and 2012
|
F-3
|
|
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2014, 2013, and 2012
|
F-4
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013, and 2012
|
F-5
|
|
Notes to Consolidated Financial Statements
|
F-6 – F-19
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Delcath Systems, Inc.
We have audited the accompanying consolidated balance sheets of Delcath Systems, Inc., as of December 31, 2014 and 2013, and the related consolidated statements of operations and comprehensive loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Delcath Systems, Inc. at December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
|
/s/ Ernst & Young LLP
|
|
MetroPark, NJ
|
|
March 11, 2015
|
DELCATH SYSTEMS, INC.
Consolidated Balance Sheets as of December 31, 2014 and 2013
(in thousands, except share data)
|
December 31,
2014
|
December 31,
2013
|
|||||||
|
Assets:
|
||||||||
|
Current assets
|
||||||||
|
Cash and cash equivalents
|
$
|
20,469
|
$
|
31,249
|
||||
|
Accounts receivables, net
|
174
|
349
|
||||||
|
Inventories
|
349
|
719
|
||||||
|
Prepaid expenses and other current assets
|
974
|
1,711
|
||||||
|
Total current assets
|
21,966
|
34,028
|
||||||
|
Property, plant and equipment, net
|
1,798
|
3,069
|
||||||
|
Total assets
|
$
|
23,764
|
$
|
37,097
|
||||
|
Liabilities and Stockholders’ Equity:
|
||||||||
|
Current liabilities
|
||||||||
|
Accounts payable
|
$
|
748
|
$
|
582
|
||||
|
Accrued expenses
|
3,603
|
3,740
|
||||||
|
Warrant liability
|
225
|
2,310
|
||||||
|
Total current liabilities
|
4,576
|
6,632
|
||||||
|
Other non-current liabilities
|
1,043
|
366
|
||||||
|
Total liabilities
|
5,619
|
6,998
|
||||||
|
Commitments and contingencies (Note 12)
|
–
|
–
|
||||||
|
Stockholders’ equity
|
||||||||
|
Preferred stock, $.01 par value; 10,000,000 shares authorized; no shares issued and outstanding at December 31, 2014 and 2013
|
–
|
–
|
||||||
|
Common stock, $.01 par value; 170,000,000 shares authorized; 9,740,394 and 8,402,922 shares issued and 9,708,841 and 8,401,165 outstanding at December 31, 2014 and December 31, 2013, respectively*
|
97
|
84
|
||||||
|
Additional paid-in capital
|
264,592
|
259,102
|
||||||
|
Accumulated deficit
|
(246,513
|
)
|
(229,132
|
)
|
||||
|
Treasury stock, at cost; 1,757 shares at December 31, 2014 and December 31, 2013*
|
(51
|
)
|
(51
|
)
|
||||
|
Accumulated other comprehensive income
|
20
|
96
|
||||||
|
Total stockholders’ equity
|
18,145
|
30,099
|
||||||
|
Total liabilities and stockholders’ equity
|
$
|
23,764
|
$
|
37,097
|
||||
*Reflects a one-for-sixteen (1:16) reverse stock split effected on April 8, 2014.
See Accompanying Notes to these Consolidated Financial Statements.
DELCATH SYSTEMS, INC.
Consolidated Statements of Operations and Comprehensive Loss
for the Years Ended December 31, 2014, 2013 and 2012
(in thousands, except share and per share data)
|
Year ended December 31,
|
||||||||||||
|
2014
|
2013
|
2012
|
||||||||||
|
Revenue
|
$
|
1,069
|
$
|
490
|
$
|
346
|
||||||
|
Other revenues
|
—
|
300
|
—
|
|||||||||
|
Total revenue
|
1,069
|
790
|
346
|
|||||||||
|
Costs of goods sold
|
(291
|
)
|
(464
|
)
|
(39
|
)
|
||||||
|
Gross profit
|
778
|
326
|
307
|
|||||||||
|
Operating expenses
|
||||||||||||
|
Selling, general and administrative
|
$
|
15,783
|
$
|
20,657
|
$
|
27,963
|
||||||
|
Research and development
|
4,299
|
12,688
|
26,215
|
|||||||||
|
Total operating expenses
|
20,082
|
33,345
|
54,178
|
|||||||||
|
Operating loss
|
(19,304
|
)
|
(33,019
|
)
|
(53,871
|
)
|
||||||
|
Change in fair value of warrant liability, net
|
1,942
|
2,756
|
2,159
|
|||||||||
|
Interest income
|
5
|
20
|
19
|
|||||||||
|
Other expense and interest expense
|
(24
|
)
|
(81
|
)
|
(175
|
)
|
||||||
|
Net Loss
|
$
|
(17,381
|
)
|
$
|
(30,324
|
)
|
$
|
(51,868
|
)
|
|||
|
Common Share data:
|
||||||||||||
|
Basic loss per share*
|
$
|
(1.84
|
)
|
$
|
(4.81
|
)
|
$
|
(13.54
|
)
|
|||
|
Diluted loss per share*
|
$
|
(1.84
|
)
|
$
|
(5.10
|
)
|
$
|
(13.54
|
)
|
|||
|
Weighted average number of basic common shares outstanding*
|
9,452,050
|
6,300,614
|
3,829,721
|
|||||||||
|
Weighted average number of diluted common shares outstanding*
|
9,452,050
|
6,569,011
|
3,829,721
|
|||||||||
|
Other comprehensive income (loss):
|
||||||||||||
|
Foreign currency translation adjustments
|
$
|
(76
|
)
|
$
|
59
|
$
|
37
|
|||||
|
Comprehensive loss
|
$
|
(17,457
|
)
|
$
|
(30,265
|
)
|
$
|
(51,831
|
)
|
|||
*Reflects a one-for-sixteen (1:16) reverse stock split effected on April 8, 2014.
See Accompanying Notes to these Consolidated Financial Statements.
DELCATH SYSTEMS, INC.
Consolidated Statements of Stockholders’ Equity
for the Years Ended December 31, 2014, 2013 and 2012
(in thousands, except share data)
|
Common Stock Issued
$0.01 Par Value |
In Treasury
|
|||||||||||||||||||||||||||||||
|
# of Shares
|
Amount
|
# of
Shares
|
Amount
|
Additional
Paid-in
Capital
|
Accumulated
deficit
|
Accumulated
Other
Comprehensive
(loss)
income
|
Total
Stockholders'
Equity
|
|||||||||||||||||||||||||
|
Balance at December 31, 2011
|
3,023,374
|
$
|
30
|
(1,757
|
)
|
$
|
(51
|
)
|
$
|
173,065
|
$
|
(146,940
|
)
|
$
|
-
|
$
|
26,104
|
|||||||||||||||
|
Compensation expense for issuance of stock options
|
-
|
-
|
-
|
-
|
2,807
|
-
|
-
|
2,807
|
||||||||||||||||||||||||
|
Compensation expense for issuance of restricted stock
|
25,543
|
-
|
-
|
-
|
1,018
|
-
|
-
|
1,018
|
||||||||||||||||||||||||
|
Sale of common stock, net of expenses
|
1,576,704
|
16
|
-
|
-
|
37,231
|
-
|
-
|
37,247
|
||||||||||||||||||||||||
|
Exercise of warrants
|
185,967
|
2
|
-
|
-
|
4,432
|
-
|
-
|
4,434
|
||||||||||||||||||||||||
|
Fair value of warrants exercised
|
-
|
-
|
-
|
-
|
908
|
-
|
-
|
908
|
||||||||||||||||||||||||
|
Fair value of warrants issued classified as a liability
|
-
|
-
|
-
|
-
|
(678
|
)
|
-
|
-
|
(678
|
)
|
||||||||||||||||||||||
|
Foreign currency translation
|
-
|
-
|
-
|
-
|
-
|
-
|
37
|
37
|
||||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
(51,868
|
)
|
-
|
(51,868
|
)
|
||||||||||||||||||||||
|
Balance at December 31, 2012
|
4,811,588
|
$
|
48
|
(1,757
|
)
|
$
|
(51
|
)
|
$
|
218,783
|
$
|
(198,808
|
)
|
$
|
37
|
$
|
20,009
|
|||||||||||||||
|
Compensation expense for issuance of stock options
|
-
|
-
|
-
|
-
|
174
|
-
|
-
|
174
|
||||||||||||||||||||||||
|
Compensation expense for issuance of restricted stock
|
8,604
|
-
|
-
|
-
|
117
|
-
|
-
|
117
|
||||||||||||||||||||||||
|
Sale of common stock, net of expenses
|
3,570,061
|
36
|
-
|
-
|
41,424
|
-
|
-
|
41,460
|
||||||||||||||||||||||||
|
Exercise of warrants
|
12,669
|
-
|
-
|
-
|
243
|
-
|
-
|
243
|
||||||||||||||||||||||||
|
Fair value of warrants exercised
|
-
|
-
|
-
|
-
|
218
|
-
|
-
|
218
|
||||||||||||||||||||||||
|
Fair value of warrants issued classified as liability
|
-
|
-
|
-
|
-
|
(1,857
|
)
|
-
|
-
|
(1,857
|
)
|
||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
(30,324
|
)
|
(30,324
|
)
|
|||||||||||||||||||||||
|
Foreign currency translation
|
-
|
-
|
-
|
-
|
-
|
-
|
59
|
59
|
||||||||||||||||||||||||
|
Balance at December 31, 2013
|
8,402,922
|
$
|
84
|
(1,757
|
)
|
$
|
(51
|
)
|
$
|
259,102
|
$
|
(229,132
|
)
|
$
|
96
|
$
|
30,099
|
|
||||||||||||||
|
Compensation expense for issuance of stock options
|
-
|
-
|
-
|
-
|
391
|
-
|
-
|
391
|
||||||||||||||||||||||||
|
Compensation expense for issuance of restricted stock
|
28,256
|
-
|
-
|
-
|
100
|
-
|
-
|
100
|
||||||||||||||||||||||||
|
Sale of common stock, net of expenses
|
1,260,079
|
13
|
-
|
-
|
4,730
|
-
|
-
|
4,743
|
||||||||||||||||||||||||
|
Exercise of warrants
|
49,137
|
-
|
-
|
-
|
126
|
-
|
-
|
126
|
||||||||||||||||||||||||
|
Fair value of warrants exercised
|
-
|
-
|
-
|
-
|
143
|
-
|
-
|
143
|
||||||||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
-
|
-
|
(17,381
|
)
|
-
|
(17,381
|
)
|
||||||||||||||||||||||
|
Foreign currency translation
|
-
|
-
|
-
|
-
|
-
|
-
|
(76
|
)
|
(76
|
)
|
||||||||||||||||||||||
|
Balance at December 31, 2014
|
9,740,394
|
$
|
97
|
(1,757
|
)
|
$
|
(51
|
)
|
$
|
264,592
|
$
|
(246,513
|
)
|
$
|
20
|
$
|
18,145
|
|
||||||||||||||
See Accompanying Notes to these Consolidated Financial Statements.
DELCATH SYSTEMS, INC.
Consolidated Statements of Cash Flows
for the Years Ended December 31, 2014, 2013, and 2012 (in thousands)
|
Year ended December 31,
|
||||||||||||
|
2014
|
2013
|
2012
|
||||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net loss
|
$
|
(17,381
|
)
|
$
|
(30,324
|
)
|
$
|
(51,868
|
)
|
|||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Stock option compensation expense
|
391
|
174
|
2,807
|
|||||||||
|
Restricted stock compensation expense
|
100
|
117
|
1,018
|
|||||||||
|
Depreciation expense
|
958
|
1,126
|
1,331
|
|||||||||
|
Provision for inventory obsolescence
|
118
|
—
|
—
|
|||||||||
|
Loss on disposal of equipment
|
282
|
5
|
—
|
|||||||||
|
Warrant liability fair value adjustment
|
(1,942
|
)
|
(2,756
|
)
|
(2,159
|
)
|
||||||
|
Non-cash interest income
|
—
|
(1
|
)
|
2
|
||||||||
|
Changes in assets and liabilities:
|
||||||||||||
|
Decrease (increase) in prepaid expenses and other current assets
|
718
|
(235
|
)
|
(228
|
)
|
|||||||
|
Decrease (increase) in accounts receivable
|
161
|
(211
|
)
|
(144
|
)
|
|||||||
|
Decrease (increase) in inventories
|
237
|
391
|
(1,105
|
)
|
||||||||
|
Increase (decrease) in accounts payable and accrued expenses
|
132
|
(2,445
|
)
|
331
|
||||||||
|
Increase in other non-current liabilities
|
674
|
57
|
9
|
|||||||||
|
Net cash used in operating activities
|
(15,552
|
)
|
(34,102
|
)
|
(50,006
|
)
|
||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Purchase of property, plant, and equipment
|
(44
|
)
|
(142
|
)
|
(2,120
|
)
|
||||||
|
Proceeds from sales of property, plant and equipment
|
37
|
—
|
—
|
|||||||||
|
Proceeds from maturities of short-term investments
|
—
|
—
|
4,980
|
|||||||||
|
Net cash (used in) provided by investing activities
|
(7
|
)
|
(142
|
)
|
2,860
|
|||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Net proceeds from sale of stock and exercise of stock options and warrants
|
4,869
|
41,702
|
45,058
|
|||||||||
|
Net cash provided by financing activities
|
4,869
|
41,702
|
45,058
|
|||||||||
|
Foreign currency effects on cash
|
(90
|
)
|
65
|
37
|
||||||||
|
(Decrease) increase in cash and cash equivalents
|
(10,780
|
)
|
7,523
|
(2,051
|
)
|
|||||||
|
Cash and cash equivalents at beginning of period
|
31,249
|
23,726
|
25,777
|
|||||||||
|
Cash and cash equivalents at end of period
|
$
|
20,469
|
$
|
31,249
|
$
|
23,726
|
||||||
|
Supplemental non-cash activities:
|
||||||||||||
|
Fair value of warrants issued
|
$
|
—
|
$
|
1,857
|
$
|
4,055
|
||||||
|
Fair value of warrants exercised
|
$
|
143
|
$
|
218
|
$
|
908
|
||||||
See Accompanying Notes to these Consolidated Financial Statements.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
|
(1)
|
Description of Business
|
Delcath Systems, Inc. is a late-stage clinical development company with early commercial activity in Europe focused on cancers of the liver. We are a specialty pharmaceutical and medical device company developing our proprietary product—Melphalan Hydrochloride for Injection for use with the Delcath Hepatic Delivery System (Melphalan/HDS). In Europe, our proprietary system to deliver and filter melphalan hydrochloride is marketed as a device under the trade name Delcath Hepatic CHEMOSAT® Delivery System for Melphalan (CHEMOSAT).
Our primary focus is on the execution of our clinical development program (CDP) in ocular melanoma liver metastases (mOM), intrahepatic cholangiocarncinoma (ICC), hepatocellular carcinoma (HCC or primary liver), and certain other cancers that are metastatic to the liver. We believe the disease states we are investigating represent a multi-billion dollar global market opportunity and a clear unmet medical need.
The Company has incurred losses since inception. The Company anticipates incurring additional losses until such time, if ever, that it can generate significant sales. Management believes that its capital resources are adequate to fund operations into the first half of 2016, but anticipates that additional working capital will be required to continue operations. To the extent additional capital is not available when needed, the Company may be forced to abandon some or all of its development and commercialization efforts, which would have a material adverse effect on the prospects of the business. Operations of the Company are subject to certain risks and uncertainties, including, among others, uncertainty of product development and clinical trial results; uncertainty regarding regulatory approval; technological uncertainty; uncertainty regarding patents and proprietary rights; comprehensive government regulations; limited commercial manufacturing, marketing or sales experience; and dependence on key personnel.
| (2) | Basis of Consolidated Financial Statement Presentation |
The accounting and financial reporting policies of the Company conform to generally accepted accounting principles in the United States of America (GAAP). The preparation of consolidated financial statements in conformity with GAAP requires management to make assumptions and estimates that impact the amounts reported in the Company’s consolidated financial statements. The consolidated financial statements include the accounts of all entities controlled by Delcath. All significant inter-company accounts and transactions are eliminated.
|
(3)
|
Summary of Significant Accounting Policies
|
Use of Estimates
The Company bases its estimates and judgments on historical experience and on various other assumptions that it believes are reasonable under the circumstances. The amounts of assets and liabilities reported in the Company’s consolidated balance sheets and the amount of expenses reported for each of its periods presented are affected by estimates and assumptions, which are used for, but not limited to, the accounting for derivative instrument liabilities, stock-based compensation, valuation of inventory, impairment of long-lived assets, income taxes and operating expense accruals. Such assumptions and estimates are subject to change in the future as additional information becomes available or as circumstances are modified. Actual results could differ from these estimates.
Cash Equivalents and Concentrations of Credit Risk
The Company considers investments with original maturities of three months or less at date of acquisition to be cash equivalents. The Company has deposits that exceed amounts insured by the Federal Deposit Insurance Corporation (FDIC), however, the Company does not consider this a significant concentration of credit risk based on the strength of the financial institution.
Accounts Receivable
Accounts receivable, principally trade, are generally due within 30 days and are stated at amounts due from customers. As the Company’s commercial activities expand, collections and payments from customers will be monitored and a provision for estimated credit losses will be created based upon historical experience and specific customer collection issues that may be identified.
Inventories
Inventories are valued at the lower of cost or market value using the first-in, first-out method. The reported net value of inventory includes finished saleable products, work-in-process, and raw materials that will be sold or used in future periods. The Company reserves for expired, obsolete, and slow-moving inventory.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
Prior to obtaining authorization to affix the CE Mark to its Generation Two CHEMOSAT System in April 2012, the Company expensed all of its inventory costs as research and development. Inventory as of December 31, 2014 includes finished goods and components that have been purchased since April 2012. Therefore, to the extent that materials expensed prior to April 2012 are used in manufacturing finished goods for sale, the Company’s cost of goods sold will be adjusted accordingly.
Property, Plant and Equipment
Property, plant and equipment are recorded at cost, less accumulated depreciation. The Company provides for depreciation on a straight line basis over the estimated useful lives of the assets which range from three to seven years. Leasehold improvements will be amortized over the shorter of the lease term or the estimated useful life of the related assets when they are placed into service. Maintenance and repairs are charged to operations as incurred. Expenditures which substantially increase the useful lives of the related assets are capitalized.
Derivative Instrument Liability
The Company accounts for derivative instruments in accordance with ASC 815, which establishes accounting and reporting standards for derivative instruments and hedging activities, including certain derivative instruments embedded in other financial instruments or contracts and requires recognition of all derivatives on the balance sheet at fair value, regardless of the hedging relationship designation. Accounting for changes in the fair value of the derivative instruments depends on whether the derivatives qualify as hedge relationships and the types of relationships designated are based on the exposures hedged. At December 31, 2014 and 2013, the Company did not have any derivative instruments that were designated as hedges.
Fair Value Measurements
The Company adopted ASC 820, which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements. ASC 820 applies to reported balances that are required or permitted to be measured at fair value under existing accounting pronouncements; accordingly, the standard does not require any new fair value measurements of reported balances.
ASC 820 emphasizes that fair value is a market-based measurement, not an entity-specific measurement. Therefore, a fair value measurement should be determined based on the assumptions that market participants would use in pricing the asset or liability. As a basis for considering market participant assumptions in fair value measurements, ASC 820 establishes a fair value hierarchy that distinguishes between market participant assumptions based on market data obtained from sources independent of the reporting entity (observable inputs that are classified within Levels 1 and 2 of the hierarchy) and the reporting entity’s own assumptions about market participant assumptions (unobservable inputs classified within Level 3 of the hierarchy).
| · | Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities that the Company has the ability to access. |
| · | Level 2 inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 inputs may include quoted prices for similar assets and liabilities in active markets, as well as inputs that are observable for the asset or liability (other than quoted prices), such as interest rates, foreign exchange rates, and yield curves that are observable at commonly quoted intervals. |
| · | Level 3 inputs are unobservable inputs for the asset or liability, which is typically based on an entity’s own assumptions, as there is little, if any, related market activity. |
In instances where the determination of the fair value measurement is based on inputs from different levels of the fair value hierarchy, the level in the fair value hierarchy within which the entire fair value measurement falls is based on the lowest level input that is significant to the fair value measurement in its entirety. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment, and considers factors specific to the asset or liability.
Revenue Recognition
Revenue from product sales is generally recognized when all of the following criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred; product price is fixed or determinable; and collection of the resulting receivable is reasonably assured. When obligations or contingencies remain after the products are shipped, such as training and certifying the treatment centers, revenue is deferred until the obligations or contingencies are satisfied.
Selling, General and Administrative
Selling, general and administrative costs include personnel costs and related expenses for the Company’s sales, marketing, general management and administrative staff, recruitment, costs related to the Company’s commercialization efforts in Europe, professional service fees, professional license fees, business development and certain general legal activities. All such costs are charged to expense when incurred.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
Research and Development
Research and development costs include the costs of materials used for clinical trials and R&D, personnel costs associated with device and pharmaceutical R&D, clinical affairs, medical affairs, medical science liaisons, and regulatory affairs, costs of outside services and applicable indirect costs incurred in the development of the Company’s proprietary drug delivery system. All such costs are charged to expense when incurred.
Stock Based Compensation
The Company accounts for its share-based compensation in accordance with the provisions of ASC 718, which establishes accounting for equity instruments exchanged for employee services and ASC 505-50, which establishes accounting for equity-based payments to non-employees. Under the provisions of ASC 718, share-based compensation is measured at the grant date, based upon the fair value of the award, and is recognized as an expense over the option holders’ requisite service period (generally the vesting period of the equity grant). The Company is required to record compensation cost for all share-based payments granted to employees based upon the grant date fair value, estimated in accordance with the provisions of ASC 718. Under the provisions of ASC 505-50, measurement of compensation cost related to common shares issued to non-employees for services is based on the value of the services provided or the fair value of the shares issued. The measurement of non-employee stock-based compensation is subject to periodic adjustment as the underlying equity instrument vests. The Company expensed its share-based compensation for share-based payments granted under the accelerated method, which treats each vesting tranche as if it were an individual grant.
The Company periodically grants stock options for a fixed number of shares of common stock to its employees, directors and non-employee contractors, with an exercise price greater than or equal to the fair market value of Delcath’s common stock at the date of the grant. The Company estimates the fair value of stock options using an option pricing model. Key inputs used to estimate the fair value of stock options include the exercise price of the award, the expected post-vesting option life, the expected volatility of Delcath’s stock over the option’s expected term, the risk-free interest rate over the option’s expected term, and Delcath’s expected annual dividend yield. Estimates of fair value are not intended to predict actual future events or the value ultimately realized by persons who receive equity awards.
Income Taxes
The Company accounts for income taxes following the asset and liability method in accordance with the ASC 740 “Income Taxes.” Under such method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the consolidated financial statement carrying amounts of existing assets and liabilities and their respective tax bases. The Company applies the accounting guidance issued to address the accounting for uncertain tax positions. This guidance clarifies the accounting for income taxes, by prescribing a minimum recognition threshold a tax position is required to meet before being recognized in the financial statements as well as provides guidance on derecognition, measurement, classification, interest and penalties, accounting in interim periods, disclosure and transition. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years that the asset is expected to be recovered or the liability settled. See Note 13 for additional information.
Net Loss per Common Share
Basic net loss per share is determined by dividing net loss by the weighted average shares of common stock outstanding during the period. Diluted net loss per share is determined by dividing net loss by diluted weighted average shares outstanding. Diluted weighted average shares reflects the dilutive effect, if any, of potentially dilutive common shares, such as stock options and warrants calculated using the treasury stock method. In periods with reported net operating losses, all common stock options and warrants are deemed anti-dilutive such that basic net loss per share and diluted net loss per share are equal. However, in certain periods in which the exercise price of the warrants was less than the last reported sales price of Delcath’s common stock on the final trading day of the period and there is a gain recorded pursuant to the change in fair value of the warrant derivative liability, the impact of gains related to the mark-to-market adjustment of the warrants outstanding at the end of the period is reversed and the treasury stock method is used to determine diluted earnings per share.
The calculation of net loss and the number of shares used to compute basic and diluted earnings per share for the years ended December 31, 2014, 2013, and 2012 are as follows:
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
|
(in thousands, except share data)
|
2014
|
2013
|
2012
|
|||||||||
|
Net loss – basic
|
$
|
(17,381
|
)
|
$
|
(30,324
|
)
|
$
|
(51,868
|
)
|
|||
|
Adjust for gain on warrant derivative liability
|
—
|
(1,835
|
)
|
—
|
||||||||
|
Net loss - diluted
|
(17,381
|
)
|
(32,159
|
)
|
(51,868
|
)
|
||||||
|
Net loss per share – basic
|
(1.84
|
)
|
(4.81
|
)
|
(13.54
|
)
|
||||||
|
Weighted average shares outstanding – basic
|
9,452,050
|
6,300,614
|
3,829,721
|
|||||||||
|
Potentially dilutive warrant exercises
|
—
|
268,397
|
—
|
|||||||||
|
Weighted average shares outstanding – diluted
|
9,452,050
|
6,569,011
|
3,829,721
|
|||||||||
|
Net loss per share – diluted
|
(1.84
|
)
|
(5.10
|
)
|
(13.54
|
)
|
||||||
For the years ended December 31, 2014, 2013 and 2012 the following potentially dilutive securities were excluded from the computation of diluted earnings per share (EPS) because their effects would be antidilutive.
Shares excluded from the computation of diluted EPS:
|
|
2014
|
2013
|
2012
|
|||||||||
|
Stock options
|
281,735
|
252,158
|
299,242
|
|||||||||
|
Unvested restricted shares
|
29,799
|
20,347
|
31,267
|
|||||||||
|
Warrants
|
850,138
|
589,500
|
352,664
|
|||||||||
|
Total
|
1,161,672
|
862,005
|
683,173
|
|||||||||
Segment Information
The Company currently operates in one business segment, which is the development and commercialization of CHEMOSAT/Melphalan/HDS. A single management team that reports to the interim CEO and President comprehensively manages the business. Accordingly, the Company does not have separately reportable segments.
Foreign Currency and Currency Translation
Transactions that are denominated in a foreign currency are remeasured into the functional currency at the current exchange rate on the date of the transaction. Any foreign currency-denominated monetary assets and liabilities are subsequently remeasured at current exchange rates, with gains or losses recognized as foreign exchange (losses)/gains in the statement of operations.
The assets and liabilities of the Company’s international subsidiaries are translated from their functional currencies into United States dollars at exchange rates prevailing at the balance sheet date. Average rates of exchange during the period are used to translate the statement of operations, while historical rates of exchange are used to translate any equity transactions.
Translation adjustments arising on consolidation due to differences between average rates and balance sheet rates, as well as unrealized foreign exchange gains or losses arising from translation of intercompany loans that are of a long-term-investment nature, are recorded in other comprehensive income.
Recent Accounting Pronouncements
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”) that updates the principles for recognizing revenue. The core principle of the guidance is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 also amends the required disclosures of the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. ASU 2014-09 is effective for annual reporting periods beginning after December 15, 2016, including interim periods within that reporting period. The Company expects to adopt this guidance when effective, and the potential impact on its financial statements is not currently estimable.
In June 2014, the FASB issued ASU 2014-12, Accounting for Share-Based Payments When the Terms of an Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period (“ASU 2014-12”). ASU 2014-12 requires that a performance target that affects vesting and that could be achieved after the requisite service period be treated as a performance condition. As such, the performance target should not be reflected in estimating the grant-date fair value of the award. ASU 2014-12 is effective for annual reporting periods beginning after December 15, 2015, with early adoption permitted. The Company expects to adopt this guidance when effective, and does not anticipate that this guidance will materially impact its consolidated financial statements.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements — Going Concern, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (“ASU 2014-15”). ASU 2014-15 requires management to assess an entity’s ability to continue as a going concern by incorporating and expanding upon certain principles that are currently in U.S. auditing standards. Specifically, the ASU (1) provides a definition of the term substantial doubt, (2) requires an evaluation every reporting period including interim periods, (3) provides principles for considering the mitigating effect of management’s plans, (4) requires certain disclosures when substantial doubt is alleviated as a result of consideration of management’s plans, (5) requires an express statement and other disclosures when substantial doubt is not alleviated, and (6) requires an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). This standard is effective for the fiscal years ending after December 15, 2016, and for annual periods and interim periods thereafter. Early application is permitted. The Company is currently evaluating the accounting transition and disclosure requirements of the standard and cannot currently estimate the financial statement impact of adoption.
| (4) | Inventories |
Inventories consist of:
|
(in thousands)
|
December 31, 2014
|
December 31, 2013
|
||||||
|
Raw materials
|
$
|
203
|
$
|
249
|
||||
|
Work-in-process
|
63
|
364
|
||||||
|
Finished goods
|
83
|
106
|
||||||
|
Total inventory
|
$
|
349
|
$
|
719
|
||||
| (5) | Prepaid Expenses and Other Current Assets |
Prepaid expenses and other current assets include the following:
|
(in thousands)
|
December 31, 2014
|
December 31, 2013
|
||||||
|
Insurance premiums
|
$
|
591
|
$
|
407
|
||||
|
Professional fees
|
—
|
377
|
||||||
|
Income tax credits receivable
|
—
|
326
|
||||||
|
Kits for clinical use
|
161
|
287
|
||||||
|
Other1
|
222
|
314
|
||||||
|
Total prepaid and other current assets
|
$
|
974
|
$
|
1,711
|
||||
1 Other consists of various prepaid expenses and other current assets, with no individual item accounting for more than 5% at December 31, 2014 and 2013.
| (6) | Property, Plant, and Equipment |
Property, plant, and equipment consists of:
|
(in thousands)
|
December 31, 2014
|
December 31, 2013
|
||||||
|
Leaseholds
|
$
|
1,629
|
$
|
1,749
|
||||
|
Enterprise hardware and software
|
1,573
|
2,143
|
||||||
|
Furniture
|
361
|
957
|
||||||
|
Equipment
|
1,380
|
1,552
|
||||||
|
Buildings and land
|
603
|
603
|
||||||
|
Property, plant and equipment, gross
|
5,546
|
7,004
|
||||||
|
Accumulated depreciation
|
(3,748
|
)
|
(3,935
|
)
|
||||
|
Property, plant and equipment, net
|
$
|
1,798
|
$
|
3,069
|
||||
Depreciation expense for the years ended December 31, 2014, 2013, and 2012 was $1.0 million, $1.1 million, and $1.3 million, respectively.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
|
(7)
|
Current Accrued Expenses
|
Current accrued expenses include the following:
|
(in thousands)
|
December 31, 2014
|
December 31, 2013
|
||||||
|
Compensation, excluding taxes
|
$
|
2,187
|
$
|
1,866
|
||||
|
Professional fees
|
341
|
360
|
||||||
|
Short-term portion of lease restructuring
|
239
|
—
|
||||||
|
Deferred rent
|
58
|
485
|
||||||
|
Other1
|
778
|
1,029
|
||||||
|
Total accrued liabilities
|
$
|
3,603
|
$
|
3,740
|
||||
1 Other consists of various accrued expenses, with no individual item accounting for more than 5% of current liabilities at December 31, 2014 and 2013.
| (8) | Restructuring Expenses |
During 2013, the Company implemented workforce restructurings to reduce operating costs, better focus its organizational structure, increase efficiency and concentrate financial resources on its clinical development program and European commercialization activity. This resulted in a total reduction in the Company’s workforce by 50 employees. During the second and third quarters of 2014, the Company implemented additional workforce restructurings that resulted in a total reduction in the Company’s workforce by eight employees. As a result of termination benefits provided to these 58 employees the Company has incurred a total restructuring charge of approximately $5.3 million for employee related expenses, of which $4.0 million was incurred in 2013 and $1.3 million was incurred in 2014. At December 31, 2014, the remaining restructuring reserve of approximately $0.8 million is included in Accrued expenses on the Consolidated Balance Sheets.
In order to help reduce operating costs and more appropriately align its office space with the reduced size of its workforce, during the quarter ended June 30, 2014, the Company implemented a plan to vacate and sub-lease office space at its 810 Seventh Avenue office. On May 22, 2014, the Company entered into a sub-lease agreement (“Sub-lease #1”) for approximately one-half of the office space at this location (“Suite 3500”), and had vacated and relinquished the premises to the sub-tenant as of June 30, 2014. Pursuant to Sub-lease #1, the sub-lease term commenced on July 1, 2014 and will expire on March 30, 2021, concurrent with the expiration of the Lease Agreement between Delcath and the landlord dated February 5, 2010 and subsequently modified by a Lease Modification, Extension and Additional Space Agreement between Delcath and the landlord dated September 27, 2010 (together the “Prime Lease”). At June 30, 2014, the Company’s future rent obligations for Suite 3500 under the Prime Lease over the remaining lease term of 81 months totaled approximately $3.6 million. Under Sub-lease #1 the Company will receive future sub-lease rental receipts totaling approximately $2.6 million, resulting in net future cash outflows of approximately $1.0 million. In accordance with ASC 420, Exit or Disposal Cost Obligations, the Company calculated the fair value of the remaining net cash flow liability for Suite 3500 and recorded a lease restructuring reserve of approximately $0.9 million as of June 30, 2014. Additionally, during the quarter ended June 30, 2014, the Company recorded contract termination costs related to Sub-lease #1 of approximately $150,000 and wrote off approximately $50,000 of unamortized leasehold improvements related to Suite 3500. The expenses related to this lease restructuring were recorded in Selling, general and administrative on the Consolidated Statements of Operations.
On August 18, 2014, the Company entered into a sub-lease agreement (“Sub-lease #2”) with a third party for the remaining one-half of office space at its 810 Seventh Avenue office “(Suite 3505”). On September 24, 2014, the Company received written consent from the landlord of the building with respect to Sub-lease #2, and had vacated and relinquished the premises to the sub-tenant as of September 30, 2014. Pursuant to Sub-lease #2, the sub-lease term commenced on October 1, 2014 and will expire concurrently with the expiration of the Prime Lease on March 30, 2021. At September 30, 2014, the Company’s future rent obligations for Suite 3505 under the Prime Lease over the remaining lease term of 78 months totaled approximately $3.4 million. Under Sub-lease #2 the Company will receive future sub-lease rental receipts totaling approximately $2.6 million, resulting in net future cash outflows of approximately $0.8 million. In accordance with ASC 420, Exit or Disposal Cost Obligations, the Company calculated the fair value of the remaining net cash flow liability for Suite 3505 and recorded a lease restructuring reserve of approximately $0.7 million as of September 30, 2014. Additionally, during the quarter ended September 30, 2014, the Company recorded contract termination costs related to Sub-lease #2 of approximately $150,000. The expenses related to this lease restructuring were recorded in Selling, General and Administrative expenses on the Consolidated Statements of Operations.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
As of December 31, 2014, the total remaining lease restructuring liability for its leased office space at 810 Seventh Ave was approximately $1.3 million, of which approximately $0.3 million and $1.0 million were included in Accrued expenses and Other non-current liabilities on the Consolidated Balance Sheets, respectively.
|
(in thousands)
|
Employee
Costs
|
Operating
Lease
|
||||||
|
Reserve balance as of December 31, 2013
|
$
|
2,019
|
$
|
–
|
||||
|
Charges
|
1,324
|
1,557
|
||||||
|
Payments / utilizations
|
(2,519
|
)
|
(275
|
)
|
||||
|
Reserve balance as of December 31, 2014
|
$
|
824
|
$
|
1,282
|
||||
| (9) | Assets and Liabilities Measured at Fair Value |
Derivative Financial Instruments
As disclosed in Note 10, the Company allocated part of the proceeds of public offerings in 2009, 2012, and 2013 of the company’s common stock to warrants issued in connection with those transactions. The valuation of the warrants is determined using an option pricing model. This model uses inputs such as the underlying price of the shares issued when the warrant is exercised, volatility, risk free interest rate and expected life of the instrument. The Company has determined that the warrant derivative liability should be classified within Level 3 of the fair-value hierarchy by evaluating each input for the option pricing model against the fair-value hierarchy criteria and using the lowest level of input as the basis for the fair-value classification as called for in ASC 820. There are six inputs: closing price of Delcath stock on the day of evaluation; the exercise price of the warrants; the remaining term of the warrants; the volatility of Delcath’s stock over that term; annual rate of dividends; and the riskless rate of return. Of those inputs, the exercise price of the warrants and the remaining term are readily observable in the warrant agreements. The annual rate of dividends is based on the Company’s historical practice of not granting dividends. The closing price of Delcath stock would fall under Level 1 of the fair-value hierarchy as it is a quoted price in an active market (ASC 820-10). The riskless rate of return is a Level 2 input as defined in ASC 820-10, while the historical volatility is a Level 3 input as defined in ASC 820. Since the lowest level input is a Level 3, Delcath determined the warrant derivative liability is most appropriately classified within Level 3 of the fair value hierarchy.
Money Market Funds
The Company has determined that the inputs associated with the fair value determination are based on quoted prices (unadjusted) and as a result the investments are classified within Level 1 of the fair value hierarchy.
The table below presents the Company’s assets and liabilities measured at fair value on a recurring basis as of December 31, 2014 and 2013, aggregated by the level in the fair value hierarchy within which those measurements fall.
Assets and Liabilities Measured at Fair Value on a Recurring Basis
|
Level 1
|
Level 2
|
Level 3
|
Balance at
December 31,
|
|||||||||||||||||||||||||||||
|
(in thousands)
|
2014
|
2013
|
2014
|
2013
|
2014
|
2013
|
2014
|
2013
|
||||||||||||||||||||||||
|
Assets
|
||||||||||||||||||||||||||||||||
|
Money market funds
|
$
|
1,943
|
$
|
1,956
|
—
|
—
|
—
|
—
|
$
|
1,943
|
$
|
1,956
|
||||||||||||||||||||
|
Liabilities
|
||||||||||||||||||||||||||||||||
|
Derivative instrument liabilities
|
—
|
—
|
—
|
—
|
$
|
225
|
$
|
2,310
|
$
|
225
|
$
|
2,310
|
||||||||||||||||||||
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
Fair Value Measurements Using Significant Unobservable
Inputs (Level 3)
|
(in thousands)
|
Derivative
|
|||
|
Balance at January 1, 2012
|
$
|
2,439
|
||
|
Total change in the fair value of the liability included in earnings
|
(2,159
|
)
|
||
|
Fair value of warrants issued
|
4,055
|
|||
|
Fair value of warrants exercised
|
(908
|
)
|
||
|
Balance at December 31, 2012
|
$
|
3,427
|
||
|
Total change in the fair value of the liability included in earnings
|
(2,756
|
)
|
||
|
Fair value of warrants exercised
|
1,639
|
|||
|
Balance at December 31, 2013
|
$
|
2,310
|
||
|
Total change in the fair value of the liability included in earnings
|
(1,942
|
)
|
||
|
Fair value of warrants exercised
|
(143
|
)
|
||
|
Balance at December 31, 2014
|
$
|
225
|
||
For the year ended December 31, 2014, the Company recorded pre-tax derivative instrument income of $1.9 million. The resulting derivative instrument liabilities totaled $0.2 million at December 31, 2014. Management expects that the Warrants will either be exercised or expire worthless. The fair value of the Warrants at December 31, 2014 was determined by using an option pricing model assuming the following:
|
2013 Warrants
|
2012 Warrants
|
|||||||
|
Expected volatility
|
89.89
|
%
|
49.23
|
%
|
||||
|
Risk-free interest rates
|
1.38
|
%
|
0.12
|
%
|
||||
|
Expected life (in years)
|
3.83
|
0.41
|
||||||
|
(10)
|
Stockholders’ Equity
|
Stock Issuances
Reverse Stock Split
On February 24, 2014, shareholders of the Company approved, through a shareholder vote, an amendment to the Company’s Amended and Restated Certificate of Incorporation authorizing the Board of Directors to effect a reverse stock split of Delcath’s common stock. The reverse stock split became effective on April 8, 2014 at which time Delcath’s common stock began trading on the NASDAQ Stock Exchange on a one-for-sixteen (1:16) split-adjusted basis. All owners of record as of the close of the NASDAQ market on April 8, 2014 received one issued and outstanding share of Delcath common stock in exchange for sixteen issued and outstanding shares of Delcath common stock. No fractional shares were issued in connection with the reverse stock split. All fractional shares created by the one-for-sixteen exchange were rounded up to the next whole share. The reverse stock split had no impact on the number of common shares authorized or the par value per share of Delcath common stock, which remain 170,000,000 and $0.01, respectively. All current and prior period amounts related to shares, share prices and earnings per share, presented in these Consolidated Financial Statements and the accompanying Notes have been restated to give retrospective presentation for the reverse stock split.
At-the-Market (“ATM”) Programs
In December 2011, the Company entered into an agreement with Cowen and Company, LLC (“Cowen”) to sell shares of its common stock, par value $.01 per share, from time to time, through an ATM equity offering program having aggregate sales proceeds of $39.8 million, under which Cowen would act as sales agent. During the first quarter of 2013, the Company sold approximately 0.9 million shares of its common stock under this ATM program for proceeds of approximately $20.9 million, with net cash proceeds after related expenses of approximately $20.8 million, and successfully completed the program. As of March 31, 2013, there were no shares of common stock of the Company remaining for sale under this ATM program.
In March 2013, the Company entered into a new agreement with Cowen to sell shares of the Company’s common stock, par value $.01 per share, from time to time, through an ATM equity offering program having aggregate sales proceeds of $50.0 million, under which Cowen will act as sales agent. During the year ended December 31, 2013, the Company sold approximately 1.0 million shares of its common stock under this ATM program for proceeds of approximately $5.0 million, with net cash proceeds after related expenses of approximately $4.8 million. During the year ended December 31, 2014 the Company sold an additional 1.0 million shares of its common stock under this ATM program for proceeds of approximately $4.8 million, with net cash proceeds after related expenses of approximately $4.7 million. The shares were issued pursuant to an effective registration statement on Form S-3 (333-187230). The net proceeds will be used for general corporate purposes, including, but not limited to, commercialization of its products, obtaining regulatory approvals, funding of clinical trials, capital expenditures and working capital. As of December 31, 2014, the Company has approximately $39.9 million available under the program subject to market conditions and certain limitations.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
In December 2012, the Company entered into a two-year agreement with Terrapin Opportunity, L.P. (“Terrapin”) for a CEFF program. Under the agreement Terrapin committed to purchase up to $35.0 million of Delcath common stock over a 24-month term. Since inception, the Company has sold approximately 0.5 million shares of its common stock through the program for total proceeds of approximately $11.1 million, with net cash proceeds after related expenses of approximately $10.8 million. The shares were issued pursuant to an effective registration statement on Form S-3 (333-183675). The net proceeds have been used for general corporate purposes including, but not limited to, commercialization of its products, funding of clinical trials, obtaining regulatory approvals, capital expenditures and general working capital needs. The agreement expired in December 2014.
The Company currently has two active registration statements on Forms S-3. Form S-3 limits the aggregate market value of securities that Delcath is permitted to offer in any 12 month-period under Form S-3 to one-third of its public float. Given the Company’s offering in February 2015, other sales under the market equity offering program during the relevant 12 month-period, and Delcath’s current aggregate market value of securities, the Company is at the applicable limit under Form S-3. As a result, unless the market value of Delcath’s securities increases the Company’s ability to raise capital may be impaired and Delcath is currently unable to utilize the Form S-3 or access its at the market equity offering program.
Stock Incentive Plans
The Company established the 2004 Stock Incentive Plan and the 2009 Stock Incentive Plan (collectively, the “Plans”) under which 187,500, and 406,250 shares, respectively, were reserved for the issuance of stock options, stock appreciation rights, restricted stock, stock grants and other equity awards. A stock option grant allows the holder of the option to purchase a share of the Company’s common stock in the future at a stated price. The Plans are administered by the Compensation and Stock Option Committee of the board of directors which determines the individuals to whom awards shall be granted as well as the type, terms and conditions of each award, the option price and the duration of each award.
Options granted under the Plans vest as determined by the Company’s Compensation and Stock Option Committee and expire over varying terms, but not more than ten years from the date of grant. Stock option activity for 2014, 2013, and 2012 is as follows:
|
Number of
Options
|
Exercise Price
per Share
|
Weighted Average
Exercise Price
|
Weighted
Average
Remaining
Life (Years)
|
|||||||||||||
|
Outstanding at January 1, 2012
|
258,069
|
$
|
19.68-248.64
|
$
|
81.45
|
6.38
|
||||||||||
|
Granted
|
75,437
|
22.88-73.60
|
60.80
|
|||||||||||||
|
Expired
|
(26,251
|
)
|
30.08-72.32
|
76.90
|
||||||||||||
|
Forfeited
|
(8,013
|
)
|
36.16-146.88
|
80.87
|
||||||||||||
|
Outstanding at December 31, 2012
|
299,242
|
$
|
19.68-248.64
|
$
|
76.66
|
6.88
|
||||||||||
|
Granted
|
130,328
|
4.80-34.08
|
17.41
|
|||||||||||||
|
Expired
|
(16,874
|
)
|
6.08-197.44
|
63.50
|
||||||||||||
|
Forfeited
|
(160,538
|
)
|
19.68-29.92
|
24.47
|
||||||||||||
|
Outstanding at December 31, 2013
|
252,158
|
$
|
4.80 – 248.64
|
$
|
57.90
|
7.36
|
||||||||||
|
Granted
|
165,000
|
1.24 – 2.42
|
1.78
|
|||||||||||||
|
Expired
|
(45,666
|
)
|
4.80 – 110.40
|
47.54
|
||||||||||||
|
Forfeited
|
(89,757
|
)
|
4.80 – 248.64
|
67.58
|
||||||||||||
|
Outstanding at December 31, 2014
|
281,735
|
$
|
1.24 – 245.12
|
$
|
23.63
|
8.83
|
||||||||||
|
Exercisable at December 31, 2014
|
55,634
|
$
|
21.12 – 245.12
|
$
|
99.23
|
6.08
|
||||||||||
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
For the years ended December 31, 2014, 2013 and 2012 the Company recognized compensation expense related to stock option grants of approximately $0.4 million, $0.2 million and $2.8 million, respectively.
The estimated fair value of each option award granted was determined on the date of grant using an option pricing model with the following assumptions for option grants during the years ended December 31, 2014, 2013 and 2012:
|
Year Ended December 31,
|
||||||||||||
|
2014
|
2013
|
2012
|
||||||||||
|
Weighted average risk-free interest rate
|
1.53
|
%
|
1.30
|
%
|
1.11
|
%
|
||||||
|
Weighted average expected volatility
|
96.68
|
%
|
92.31
|
%
|
79.89
|
%
|
||||||
|
Expected volatility
|
95.47%-98.14
|
%
|
86.16%-97.21
|
%
|
77.37%-84.81
|
%
|
||||||
|
Dividend yield
|
0.00
|
%
|
0.00
|
%
|
0.00
|
%
|
||||||
|
Weighted average expected option term (in years)
|
4.5
|
5.66
|
6.17
|
|||||||||
|
Weighted average grant date fair value
|
$
|
1.78
|
$
|
17.41
|
$
|
60.80
|
||||||
No dividend yield was assumed because the Company has never paid a cash dividend on its common stock and does not expect to pay dividends in the foreseeable future. Volatilities were developed using the Company’s historical volatility. The risk-free interest rate was developed using the U.S. Treasury yield for periods equal to the expected life of the stock options on the grant date. The expected option term for grants made during 2014, 2013 and the second half of 2012 is based on actual historical results. The expected option term for grants made prior to that was developed based on the mid-point between the vesting date and the expiration date of each respective grant as permitted under ASC 718. This method of determining the expected holding period was utilized because the Company did not have sufficient historical experience from which to estimate the period.
A summary of the Company’s non-vested options to purchase shares as of December 31, 2014 and changes during the year ended December 31, 2014 and December 31, 2013 are presented below:
|
Non-Vested Options
|
||||||||
|
Number of
Options
|
Weighted
Average
Exercise Price
|
|||||||
|
Non-vested at January 1, 2013
|
105,143
|
$
|
74.85
|
|||||
|
Granted
|
130,328
|
17.41
|
||||||
|
Vested
|
(42,893
|
)
|
85.99
|
|||||
|
Forfeited
|
(73,968
|
)
|
53.93
|
|||||
|
Non-vested at December 31, 2013
|
118,610
|
$
|
20.76
|
|||||
|
Granted
|
165,000
|
1.78
|
||||||
|
Vested
|
(46,737
|
)
|
26.35
|
|||||
|
Forfeited
|
(10,772
|
)
|
35.92
|
|||||
|
Non-vested at December 31, 2014
|
226,101
|
$
|
5.03
|
|||||
Additional compensation expense of $0.2 million, relating to the unvested portion of stock options granted, is expected to be recognized over a remaining average period of 0.89 years.
The aggregate intrinsic value of options outstanding and options exercisable at December 31, 2014 is $0. The aggregate intrinsic value represents the total pretax intrinsic value, based on options with an exercise price less than the Company’s closing stock price of $1.21 as of December 31, 2014, which would have been received by the option holders had those option holders exercised their options as of that date.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
A summary of the Company’s restricted stock activity as of December 31, 2014 and changes during the year ended December 31, 2014 and December 31, 2013 are presented below:
|
Restricted Stock Activity
|
||||||||
|
Number of Shares
|
Weighted Average
Grant Date Fair
Value
|
|||||||
|
Non-vested at January 1, 2013
|
31,267
|
$
|
52.02
|
|||||
|
Granted
|
17,258
|
6.73
|
||||||
|
Vested
|
(19,582
|
)
|
41.14
|
|||||
|
Forfeited
|
(8,596
|
)
|
69.16
|
|||||
|
Non-vested at December 31, 2013
|
20,347
|
$
|
16.84
|
|||||
|
Granted
|
62,504
|
2.60
|
||||||
|
Vested
|
(18,815
|
)
|
13.60
|
|||||
|
Forfeited
|
(34,237
|
)
|
3.40
|
|||||
|
Non-vested at December 31, 2014
|
29,799
|
$
|
4.46
|
|||||
For the years ended December 31, 2014, 2013 and 2012 the Company recognized compensation expense related to restricted stock grants of approximately $0.01 million, $0.01 million and $1.0 million, respectively. Additional compensation expense of $0.04 million relating to the unvested portion of restricted stock granted is expected to be recognized over a remaining average period of 0.5 years.
Warrants
The Company allocated part of the proceeds of public offerings in 2009, 2012 and 2013 of the Company’s common stock to warrants issued in connection with those transactions. A summary of warrant activity is as follows:
|
Warrants
|
Exercise Price
per Share
|
Weighted
Average
Exercise
Price
|
Weighted
Average
Remaining Life
(Years)
|
|||||||||||||
|
Outstanding at January 1, 2012
|
157,063
|
$
|
55.04 – 57.60
|
$
|
56.10
|
1.45
|
||||||||||
|
Issued
|
407,695
|
26.40
|
||||||||||||||
|
Exercised
|
(185,967
|
)
|
23.84
|
|||||||||||||
|
Expired
|
(26,127
|
)
|
23.84
|
|||||||||||||
|
Outstanding at December 31, 2012
|
352,664
|
$
|
19.20
|
$
|
19.20
|
2.24
|
||||||||||
|
Issued
|
589,500
|
7.04
|
||||||||||||||
|
Exercised
|
(12,669
|
)
|
19.20
|
|||||||||||||
|
Expired
|
–
|
–
|
||||||||||||||
|
Outstanding at December 31, 2013
|
929,495
|
$
|
2.56 – 7.04
|
$
|
5.40
|
3.51
|
||||||||||
|
Issued
|
–
|
–
|
||||||||||||||
|
Exercised
|
(49,139
|
)
|
2.56
|
|||||||||||||
|
Expired
|
(30,218
|
)
|
2.56
|
|||||||||||||
|
Outstanding at December 31, 2014
|
850,138
|
$
|
1.75 – 7.04
|
$
|
5.42
|
2.78
|
||||||||||
| (11) | Loan and Security Agreement |
In April 2012, the Company entered into a four-year Loan and Security Agreement (the “Credit Agreement“) with Silicon Valley Bank (“SVB”), as lender. The Credit Agreement consists of a revolving credit facility in an amount equal to the lesser of $20,000,000 and the Company’s Borrowing Base (as defined in the Credit Agreement). In order to draw down on the facility, the Company will need to have at least the greater of (i) $15,000,000 in cash and cash equivalents in its account with SVB plus the amount of all outstanding obligations of the Company owed to SVB and (ii) trailing 3 months Cash Burn (as defined in the Credit Agreement) plus the amount of all outstanding obligations of the Company owed to SVB. At December 31, 2014, the Company had not used any of the available funds.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
| (12) | Commitments |
Operating Leases
In February 2010, the Company entered into an agreement to lease (Initial Lease) 8,629 square feet of office space in New York, New York with an option to expand an additional 8,629 square feet. The term of the Initial Lease began in March, 2010. In September 2010, the Company exercised its option right under the Initial Lease and entered into an agreement to lease (Lease Amendment) an additional 8,629 square feet of office space. The term of the Lease Amendment began in January 2011 and will expire in March 2021. In addition, the Lease Amendment extends the term of the Initial Lease to March 2021. The Initial Lease and the Lease Amendment provide for annual rent of $961,000 in 2012, $970,000 in 2013, $996,000 in 2014 and 2015, $1.0 million in 2016, and $1.1 million in 2017-2020. As discussed in Note 8, the Company has sub-leased this office space.
In August 2011, Delcath Systems Limited entered into an agreement of lease for an office and manufacturing facility located in the city of Galway, Ireland. This facility is approximately 19,200 square feet and is intended to be the location of Delcath’s European headquarters. The Lease is for a term of ten years, commencing August 2, 2011; although Delcath Limited has the option to terminate the Lease after the fifth year upon not less than six months notice. The Lease provides for fixed annual lease amounts payable in advance in equal quarterly installments. The annual lease amounts, which escalate annually, are as follows (USD conversions are based on the December 31, 2014 conversion rate): Year 1 – €106,051 ($128,322), Year 2 – €134,974 ($163,319), Year 3 – €159,077 ($192,483) and Years 4 and 5 – €183,179 ($221,647). Annual lease amounts in years 6 through 10 are subject to adjustment based upon the percentage increase in the consumer price index as published by the Ireland Central Statistics Office. Delcath Limited is also required to pay for customary building operating expenses. Delcath Limited’s payment obligations and performance of the Lease are guaranteed by Delcath.
In May 2012, the Company entered into an agreement to purchase 10,320 square feet located at 566 Queensbury Avenue, Queensbury, NY (the “Facility”), which was previously leased. The purchase price for the Facility was $440,000 as stated in the initial lease agreement, which commenced on September 1, 2009.
In June 2012, the Company entered into an agreement to lease 18,000 square feet at Suites 2 and 3 Country Club Road, Queensbury, NY for a three year period. This amends the initial lease at 2 Country Club Road, which commenced in November 2010. The location houses a portion of the Company’s research and manufacturing operations. The lease provides for annual base rent of $216,000, as well as the payment of customary building operating expenses and real estate taxes.
In September 2014, the Company entered into a license agreement to lease approximately 5,818 square feet of office space at 1301 Avenue of the Americas, New York, New York. The term began on October 1, 2014 and is effective through May 20, 2016. The agreement provides for total annual base rent of $200,000.
In October 2014, the Company entered into a lease agreement for 95-97 Park Road in Queensbury, NY, agreeing to lease the 6,000 square feet at that location. The term began on November 1, 2014 and is effective for a one year period. The agreement provides for total annual base rent of $45,900.
Future minimum lease payments under all operating leases at December 31, 2014 are as follows:
|
(in thousands)
|
Future Lease Payment
|
|||
|
2015
|
$
|
869
|
||
|
2016
|
426
|
|||
|
2017
|
252
|
|||
|
2018
|
260
|
|||
|
2019
|
239
|
|||
|
$
|
2,046
|
|||
Rent expense totaled approximately $2.3 million, $1.5 million and $1.5 million, for the years ended December 31, 2014, 2013 and 2012, respectively. Of the $2.3 million in expense for 2014, approximately $1.3 million is related to restructuring charges discussed in Note 8.
Letters of Credit
Under the terms of the lease agreement for office space in New York City, the Company is required to maintain a letter of credit in the amount of $881,297. Under the terms of the license agreement for office space at 1301 Avenue of the Americas, the Company is required to maintain a letter of credit in the amount of $200,000. The letters of credit expire in February 2016 if not renewed by the Company.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
| (13) | Income Taxes |
The provision for income taxes differs from the amount computed by applying the statutory rate as follows:
|
Year Ended December 31,
|
||||||||||||
|
(in thousands)
|
2014
|
2013
|
2012
|
|||||||||
|
Income taxes using U.S. federal statutory rate
|
$
|
(5,926
|
)
|
$
|
(10,309
|
)
|
$
|
(17,621
|
)
|
|||
|
Amortization of gain on IP migration
|
767
|
781
|
754
|
|||||||||
|
State income taxes, net of federal benefit
|
2,215
|
(1,390
|
)
|
(4,299
|
)
|
|||||||
|
Foreign rate differential
|
1,069
|
2,761
|
3,716
|
|||||||||
|
Valuation allowance
|
886
|
7,683
|
17,561
|
|||||||||
|
Derivative charge
|
(660
|
)
|
(937
|
)
|
(734
|
)
|
||||||
|
Stock option exercises and cancellations
|
1,748
|
1,589
|
310
|
|||||||||
|
Research and development credits
|
(125
|
)
|
(1,090
|
)
|
326
|
|
||||||
|
Other
|
26
|
912
|
(13
|
)
|
||||||||
|
Total
|
$
|
–
|
$
|
–
|
$
|
–
|
||||||
Significant components of the Company’s deferred tax assets are as follows:
|
(in thousands)
|
2014
|
2013
|
||||||
|
Deferred tax assets:
|
||||||||
|
Employee compensation accruals
|
$
|
1,866
|
$
|
4,077
|
||||
|
Accrued liabilities
|
1,356
|
870
|
||||||
|
Research tax credits
|
3,597
|
3,472
|
||||||
|
Other
|
45
|
79
|
||||||
|
Net operating losses
|
74,359
|
71,874
|
||||||
|
Total deferred tax assets
|
81,223
|
80,372
|
||||||
|
Deferred tax liability:
|
||||||||
|
Total deferred tax liabilities
|
–
|
–
|
||||||
|
Valuation allowance
|
81,223
|
80,372
|
||||||
|
Net deferred tax assets
|
$
|
–
|
$
|
–
|
||||
As of December 31, 2014 and December 31, 2013, the Company had net operating loss carryforwards for U.S. federal income tax purposes of approximately $171.6 million and $158.4 million, respectively. A portion of the federal amount, $8.6 million, is subject to an annual limitation of approximately $123,000 as a result of a change in the Company’s ownership through May 2003, as defined by Federal Internal Revenue Code Section 382 and the related income tax regulations. As a result of the limitation, approximately $163.0 million is available to offset future federal taxable income which will expire between 2018 and 2034. As of December 31, 2014 and December 31, 2013, the Company had net operating loss carryforwards for state and city income tax purposes of approximately $242.9 million and $268.8 million, respectively, which expire through 2034. As of December 31, 2014 and December 31, 2013, the Company had a net operating loss carryforward for foreign income tax purposes of $21.8 million and $19.0 million, respectively, which have indefinite carryforward periods. As of December 31, 2014 and December 31, 2013, the Company had federal research and development tax credit carryforward of approximately $3.6 million and $3.5 million, respectively, which expire through 2034.
The Company has a tax benefit of approximately $1.0 million related to the exercise of non-qualified stock options. Pursuant to ASC 718, the benefit will be recognized and recorded to additional paid-in capital when the benefit is realized through the reduction of taxes payable. Management has established a 100% valuation allowance against the deferred tax assets as management does not believe it is more likely than not that these assets will be realized. The Company’s valuation allowance increased by approximately $0.9 million, $7.7 million, $17.6 million, $20.5 million, and $15.0 million in 2014, 2013, 2012, 2011, and 2010, respectively.
DELCATH SYSTEMS, INC.
Notes to Consolidated Financial Statements
for the Years Ending December 31, 2014, 2013 and 2012
The Company complies with the provisions of ASC 740-10 in accounting for its uncertain tax positions. ASC 740-10 addresses the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740-10, the Company may recognize the tax benefit from an uncertain tax position only if it is more likely that not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The Company has determined that the Company has no significant uncertain tax positions requiring recognition under ASC 740-10.
The Company is subject to income tax in the U.S., as well as various state and international jurisdictions. The Company has not been audited by the U.S. Internal Revenue Service, international tax authorities, or any states in connection with income taxes. The Company’s New York State tax returns have been subject to annual desk reviews which have resulted in insignificant adjustments to the related franchise tax liabilities and credits. The Company’s tax years generally remain open to examination for all federal, state and foreign tax matters until its net operating loss carryforwards are utilized and the applicable statutes of limitation have expired. The federal and state tax authorities can generally reduce a net operating loss (but not create taxable income) for a period outside the statute of limitations in order to determine the correct amount of net operating loss which may be allowed as a deduction against income for a period within the statute of limitations.
Delcath recognizes interest accrued related to unrecognized tax benefits and penalties, if incurred, as a component of income tax expense.
| (14) | Quarterly Financial Data (Unaudited) |
Set forth below is selected quarterly financial data for each of the quarters in the years ended December 31, 2014 and 2013.
|
2014 Quarters Ended
|
||||||||||||||||
|
(in thousands except per share amounts)
|
March 31
|
June 30
|
September 30
|
December 31
|
||||||||||||
|
Operating loss
|
$
|
(5,059
|
)
|
(5,904
|
)
|
(5,054
|
)
|
(3,285
|
)
|
|||||||
|
Change in fair value of warrant liability, net
|
(205
|
)
|
1,297
|
519
|
330
|
|||||||||||
|
Net loss
|
(5,278
|
)
|
(4,600
|
)
|
(4,558
|
)
|
(2,945
|
)
|
||||||||
|
Basic loss per share
|
(0.57
|
)
|
(0.49
|
)
|
(0.48
|
)
|
(0.31
|
)
|
||||||||
|
Diluted loss per share
|
(0.57
|
)
|
(0.52
|
)
|
(0.48
|
)
|
(0.31
|
)
|
||||||||
|
2013 Quarters Ended
|
||||||||||||||||
|
(in thousands except per share amounts)
|
March 31
|
June 30
|
September 30
|
December 31
|
||||||||||||
|
Operating loss
|
$
|
(10,202
|
)
|
(10,587
|
)
|
(6,702
|
)
|
(5,526
|
)
|
|||||||
|
Change in fair value of warrant liability, net
|
(2,272
|
)
|
5,115
|
(497
|
)
|
410
|
||||||||||
|
Net loss
|
(12,845
|
)
|
(5,482
|
)
|
(7,206
|
)
|
(4,792
|
)
|
||||||||
|
Basic and diluted loss per share
|
(2.40
|
)
|
(0.91
|
)
|
(1.15
|
)
|
(0.63
|
)
|
||||||||
| (15) | Subsequent Events |
On February 11, 2015, the Company entered into an underwriting agreement (the “Underwriting Agreement”) with Roth Capital Partners, LLC, as Underwriter (the “Underwriter”). The Underwriting Agreement provided for the sale to the Underwriter of 2,460,000 shares of the Company’s common stock, par value $0.01 per share (the “Common Stock”), and warrants (the “Warrants”) to initially purchase up to an aggregate of 1,107,000 shares of the Common Stock (the “Offering”). The Offering closed on February 17, 2015. The Warrants are exercisable beginning on the date six months after the date of issuance and will expire five years from the date of issuance. The exercise price of the Warrants is $1.38 per share of Common Stock, subject to certain adjustments. The net proceeds to the Company from the offering are approximately $2.4 million after underwriting discounts and commissions and other estimated offering expenses payable by the Company, and excluding any proceeds the Company may receive upon exercise of the warrants to be issued in the Offering. As a result, the exercise price of the 2012 Warrants has been adjusted to $0.82.
None.
Evaluation of Disclosure Controls and Procedures
The Company’s management, with the participation of its interim Chief Executive Officer, evaluated the effectiveness of the design and operation of its disclosure controls and procedures (as defined in Rule 13a-15(e) or 15d-15(e) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”)). Based on that evaluation, Delcath’s interim Chief Executive Officer concluded that the Company’s disclosure controls and procedures as of December 31, 2014 (the end of the period covered by this Annual Report on Form 10-K), have been designed and are functioning effectively to provide reasonable assurance that the information required to be disclosed by the Company in its reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and that such information is accumulated and communicated to the Company’s management, including the interim Chief Executive Officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control Over Financial Reporting
There were no changes to the Company’s internal control over financial reporting that occurred during the fourth fiscal quarter ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, its internal control over financial reporting.
Management’s Annual Report on Internal Control over Financial Reporting
Delcath’s management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the Company’s board of directors, management and other personnel, to provide reasonable assurance regarding reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company:
| · | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and |
| · | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the consolidated financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Delcath’s management assessed the effectiveness of its internal control over financial reporting as of December 31, 2014. In making this assessment, it used the criteria set forth in Internal Control-Integrated Framework (1992) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on such assessment, management has concluded that, as of December 31, 2014, the Company’s internal control over financial reporting was effective based on those criteria.
None.
PART III
Except for the information about our Code of Ethics below, the information required by this Item 10 is incorporated by reference from our definitive proxy statement for our 2015 Annual Meeting of Stockholders (the “Proxy Statement”).
We maintain a Code of Business Conduct and Ethics (Code) that applies to all employees, including our principal executive officer, principal financial officer, principal accounting officer, controller and persons performing similar functions, and including our independent directors, who are not employees of the Company, with regard to their Delcath-related activities. The Code incorporates guidelines designed to deter wrongdoing and to promote honest and ethical conduct and compliance with applicable laws, rules and regulations. The Code also incorporates our expectations of our employees that enable us to provide accurate and timely disclosure in our filings with the SEC and other public communications. In addition, the Code incorporates guidelines pertaining to topics such as complying with applicable laws, rules, and regulations; insider trading; reporting Code violations; and maintaining accountability for adherence to the Code. The full text of our Code is published on our web site at http://delcath.com/investors/governance. We intend to disclose future amendments to certain provisions of our Code, or waivers of such provisions granted to our principal executive officer, principal financial officer, principal accounting officer or controller and persons performing similar functions on our web site.
The information required for this Item is incorporated by reference from our Proxy Statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The information required for this Item is incorporated by reference from our Proxy Statement.
The information required for this Item is incorporated by reference from our Proxy Statement.
The information required for this Item is incorporated by reference from our Proxy Statement.
PART IV
The following documents are filed as part of this Annual Report on Form 10-K:
1. Consolidated Financial Statements: The following Consolidated Financial Statements and Supplementary Data of Delcath and the Report of Independent Registered Public Accounting Firm included in Part II, Item 8:
Consolidated Balance Sheets at December 31, 2014 and 2013
Consolidated Statements of Comprehensive Loss for the years ended December 31, 2014, 2013, and 2012
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2014, 2013, and 2012
Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013, and 2012
Notes to Consolidated Financial Statements
2. Exhibits: The exhibits listed in the accompanying Exhibit Index are filed or incorporated by reference as part of this Annual Report on Form 10-K.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
DELCATH SYSTEMS, INC.
|
|
|
/s/ Jennifer K. Simpson
|
|
|
Jennifer K. Simpson
|
|
|
Interim President and Chief Executive Officer
|
|
|
(Principal Executive Officer)
|
|
|
Dated: March 11, 2015
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/Jennifer K. Simpson
|
Interim President and Chief Executive Officer
|
March 11, 2015
|
||
|
Jennifer K. Simpson
|
(Principal Executive Officer)
|
|||
|
/s/Barbra C. Keck
|
VP, Controller
|
March 11, 2015
|
||
|
Barbra C. Keck
|
(Principal Financial Officer and Principal Accounting Officer)
|
|||
|
/s/ Roger Stoll
|
Executive Chairman of the Board
|
March 11, 2015
|
||
|
Roger Stoll
|
||||
|
/s/ Harold S. Koplewicz, M.D.
|
Director
|
March 11, 2015
|
||
|
Harold S. Koplewicz, M.D.
|
||||
|
/s/ Dennis Langer
|
Director
|
March 11, 2015
|
||
|
Dennis Langer
|
||||
|
/s/ Laura Philips
|
Director
|
March 11, 2015
|
||
|
Laura Philips, Ph.D.
|
||||
|
/s/ William Rueckert
|
Director
|
March 11, 2015
|
||
|
William Rueckert
|
||||
|
/s/ Marco Taglietti
|
Director
|
March 11, 2015
|
||
|
Marco Taglietti
|
Exhibit Index
|
Exhibit
No.
|
Description
|
|
|
3.1
|
Amended and Restated Certificate of Incorporation of the Company, as amended to June 30, 2005 (incorporated by reference to Exhibit 3.1 to Company’s Current Report on Form 8-K filed June 5, 2006 (Commission File No. 001-16133).
|
|
|
3.2
|
Certificate of Amendment to the Amended and Restated Certificate of Incorporation of Delcath Systems, Inc. (incorporated by reference to Exhibit 3.1 to Company’s Current Report on Form 8-K filed April 8, 2014 (Commission File No. 001-16133).
|
|
|
3.3
|
Amended and Restated By-Laws of the Company (incorporated by reference to Exhibit 3.2 to Amendment No. 1 to Company’s Registration Statement on Form SB-2 (Registration No. 333-39470)).
|
|
|
4.2
|
Form of Warrant to Purchase Shares of Common Stock dated June 15, 2009 (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed June 10, 2009 (Commission File No,. 001-16133)).
|
|
|
4.3
|
Form of Warrant to Purchase Shares of Common Stock dated May 31, 2012 (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed May 31, 2012 (Commission File No,. 001-16133)).
|
|
|
4.4
|
Form of Warrant to Purchase Shares of Common Stock dated October 28, 2013 (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed October 28, 2013 (Commission File No,. 001-16133)).
|
|
|
4.5
|
Form of Warrant to Purchase Shares of Common Stock dated February 17, 2015 (incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K filed February 17, 2015 (Commission File No. 001-16133)).
|
|
|
10.1
|
*
|
2009 Stock Incentive Plan (incorporated by reference to Appendix B to the Company’s definitive Proxy Statement dated April 30, 2009 (Commission File No. 001-16133)).
|
|
10.2
|
Form of Indemnification Agreement dated April 8, 2009 between the Company and members of the Company’s Board of Directors (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed April 10, 2009 (Commission File No. 001-16133)).
|
|
|
10.3
|
*
|
Employment Agreement dated as of July 1, 2009 between the Company and Eamonn P. Hobbs (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed July 7, 2009 (Commission File No. 001-16133)).
|
|
10.4
|
Lease between SLG 810 Seventh Lessee LLC and the Company dated as of February 5, 2010 (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 (Commission File No. 001-16133)).
|
|
|
10.5
|
Research and Distribution Agreement between CHIFU Trading Co Ltd and the Company dated as of February 9, 2010 (incorporated by reference to Exhibit 10.6 to the Company’s Quarterly Report on Form 10-Q/A for the quarter ended March 31, 2010 (Commission File No. 001-161233)).
|
|
|
10.6
|
Amended and Restated Supply Agreement between B. Braun Medical Inc and the Company dated as of May 4, 2010 (incorporated by reference to Exhibit 10.7 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2010 (Commission File No. 001-16133)).
|
|
|
10.7
|
Lease Modification, Extension and Additional Space Agreement between SLG 810 Seventh Lessee LLC and the Company dated as of September 27, 2010 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed September 30, 2010 (Commission File No. 001-16133)).
|
|
|
10.8
|
†
|
License, Supply and Contract Manufacturing Agreement between Synerx Pharma, LLC and Bioniche Teoranta and the Company dated as of October 13, 2010.
|
|
10.9
|
*
|
Form of Restricted Stock Agreement under the Company's 2009 Stock Incentive Plan (incorporated by reference to Exhibit 10.3 to the Company's Current Report on Form 8-K filed December 20, 2010 (Commission File No. 001-16133)).
|
|
10.10
|
Form of Restricted Stock Agreement (Non-Employee Directors) under the Company's 2009 Stock Incentive Plan (incorporated by reference to Exhibit 10.4 to the Company's Current Report on Form 8-K filed December 20, 2010 (Commission File No. 001-16133)).
|
|
|
10.11
|
Form of Restricted Stock Agreement (Consultants) under the Company's 2009 Stock Incentive Plan (incorporated by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed December 20, 2010 (Commission File No. 001-16133)).
|
|
|
10.12
|
*
|
Form of Non-Statutory Stock Option Grant Letter under the Company's 2009 Stock Incentive Plan (incorporated by reference to Exhibit 10.6 to the Company's Current Report on Form 8-K filed December 20, 2010 (Commission File No. 001-16133)).
|
|
10.13
|
Form of Non-Statutory Stock Option Grant Letter (Non-Employee Directors) under the Company's 2009 Stock Incentive Plan (incorporated by reference to Exhibit 10.7 to the Company's Current Report on Form 8-K filed December 20, 2010 (Commission File No. 001-16133)).
|
|
10.14
|
Form of Non-Statutory Stock Option Grant Letter (Consultants) under the Company's 2009 Stock Incentive Plan (incorporated by reference to Exhibit 10.8 to the Company's Current Report on Form 8-K filed December 20, 2010 (Commission File No. 001-16133)).
|
|
|
10.15
|
*
|
Interim Agreement, dated July 6, 2011, by and between Delcath Systems, Inc. and Eamonn Hobbs (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed July 11, 2011 (Commission File No. 001-16133)).
|
|
10.16
|
*
|
Second Interim Agreement between Delcath Systems, Inc. and Eamonn Hobbs, dated August 8, 2011 (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed August 11, 2011 (Commission File No. 001-16133)).
|
|
10.17
|
*
|
Employment Agreement between Delcath Systems, Inc. and Eamonn Hobbs, dated August 10, 2011 (incorporated by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed August 11, 2011 (Commission File No. 001-16133)).
|
|
10.18
|
*
|
Employment Offer Letter between Delcath Systems, Inc. and Graham Miao, Ph.D., dated August 31, 2011 (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed September 26, 2011 (Commission File No. 001-16133)).
|
|
10.19
|
Form of Employee Confidentiality and Restrictive Covenant Agreement (incorporated by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed September 26, 2011 (Commission File No. 001-16133)).
|
|
|
10.20
|
Lease Agreement, dated August 2, 2011 (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 (Commission File No. 001-16133)).
|
|
|
10.21
|
Loan and Security Agreement dated April 20, 2012 between Silicon Valley Bank and Delcath Systems, Inc.1
|
|
|
10.22
|
Employment Agreement between Delcath Systems, Inc. and Krishna Kandarpa, MD, Ph.D., dated July 16, 2012 (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed July 19, 2012 (Commission File No. 001-16133))
|
|
|
10.23
|
First Amendment to Research and Distribution Agreement between Delcath Systems, Inc. and CHI-FU Trading Co., Ltd., dated January 26, 2013 (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed January 30, 2013 (Commission File No. 001-16133))
|
|
|
10.24
|
Sublease betaween Delcath Systems, Inc. and SLG 810 Seventh Lessee LLC, dated May 22, 2014. (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed May 28, 2014 (Commission File No. 001-16133))
|
|
|
10.25
|
Sublease Agreement between Delcath Systems, Inc. and ICV Partners, LLC dated August 18, 2014 (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed September 30, 2014 (Commission File No. 001-16133))
|
|
|
10.26
|
License Agreement between Delcath Systems, Inc. and Dresdner Kleinwort Group Holdings, LLC dated September 23, 2014 (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed September 30, 2014 (Commission File No. 001-16133))
|
|
**
|
Consent of Ernst & Young LLP
|
|
**
|
Certification by Principal executive officer Pursuant to Rule 13a 14.
|
|
|
**
|
Certification by Principal financial officer Pursuant to Rule 13a 14.
|
|
|
**
|
Certification of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
**
|
Certification of VP, Controller Pursuant to 18 U.S.C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
| † | Portions of this exhibit have been redacted and are subject to a confidential treatment request filed with the Secretary of the Securities and Exchange Commission pursuant to Rule 24b-2 under the Securities Exchange Act of 1934, as amended. |
| * | Indicates management contract or compensatory plan or arrangement. |
| ** | Filed herewith. |
| 1 | (incorporated by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2012). |
49