Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - KERYX BIOPHARMACEUTICALS INC | Financial_Report.xls |
| EX-32.1 - EX-32.1 - KERYX BIOPHARMACEUTICALS INC | d846100dex321.htm |
| EX-23.1 - EX-23.1 - KERYX BIOPHARMACEUTICALS INC | d846100dex231.htm |
| EX-32.2 - EX-32.2 - KERYX BIOPHARMACEUTICALS INC | d846100dex322.htm |
| EX-31.1 - EX-31.1 - KERYX BIOPHARMACEUTICALS INC | d846100dex311.htm |
| EX-31.2 - EX-31.2 - KERYX BIOPHARMACEUTICALS INC | d846100dex312.htm |
| EX-21.1 - EX-21.1 - KERYX BIOPHARMACEUTICALS INC | d846100dex211.htm |
| EX-10.16 - EX-10.16 - KERYX BIOPHARMACEUTICALS INC | d846100dex1016.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014.
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number 000-30929
KERYX BIOPHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 13-4087132 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
| 750 Lexington Avenue New York, New York |
10022 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (212) 531-5965
Securities registered pursuant to Section 12(b) of the Act:
| Common Stock, Par Value $0.001 Per Share | Nasdaq Capital Market | |
| (Title of Class) | (Name of Each Exchange on Which Registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act). (Check one):
| Large accelerated filer | x | Accelerated filer | ¨ | |||
| Non-accelerated filer | ¨ | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of voting common stock held by non-affiliates of the registrant (assuming, for purposes of this calculation, without conceding, that all executive officers and directors are “affiliates”) was $1,378,888,655 as of June 30, 2014, based on the closing sale price of such stock as reported on the Nasdaq Capital Market.
There were 103,596,337 shares of the registrant’s common stock outstanding as of February 17, 2015.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for the 2015 Annual Meeting of Stockholders are incorporated by reference in Part III of this Annual Report on Form 10-K.
Table of Contents
KERYX BIOPHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2014
This Annual Report on Form 10-K contains trademarks and trade names of Keryx Biopharmaceuticals, Inc., including our name and logo. All other trademarks, service marks, and trade names referenced in this Annual Report on Form 10-K are the property of their respective owners.
Table of Contents
SPECIAL CAUTIONARY NOTICE REGARDING FORWARD-LOOKING STATEMENTS
Certain matters discussed in this report, including matters discussed under the caption “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” may constitute forward-looking statements for purposes of the Securities Act of 1933, as amended, or the Securities Act, and the Securities Exchange Act of 1934, as amended, or the Exchange Act, and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from the future results, performance or achievements expressed or implied by such forward-looking statements. The words “anticipate,” “believe,” “estimate,” “may,” “expect” and similar expressions are generally intended to identify forward-looking statements. Our actual results may differ materially from the results anticipated in these forward-looking statements due to a variety of factors, including, without limitation, those discussed under the captions “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this report, as well as other factors which may be identified from time to time in our other filings with the Securities and Exchange Commission, or the SEC, or in the documents where such forward-looking statements appear. All written or oral forward-looking statements attributable to us are expressly qualified in their entirety by these cautionary statements. Such forward-looking statements include, but are not limited to, statements about our:
| • | estimates regarding market size and projected growth, as well as our expectation of market acceptance of Auryxia; |
| • | expectations for increases or decreases in expenses; |
| • | expectations for the pre-clinical and clinical development, including our pending Marketing Authorization Application with the European Medicines Agency, manufacturing, regulatory approval, and commercialization (including market acceptance) of AuryxiaTM (ferric citrate) or any other products that we may acquire or in-license; |
| • | expectations for incurring capital expenditures to expand our research and development and manufacturing capabilities; |
| • | expectations regarding our ability to successfully market Riona® through our Japanese partner, Japan Tobacco, Inc. and Torii Pharmaceutical Co., Ltd.; |
| • | expectations regarding our ability to successfully develop ferric citrate for the treatment of iron deficiency anemia in non-dialysis chronic kidney disease patients; |
| • | expectations that the European Medicines Agency will concur with our interpretation of our registration studies in End Stage Renal Disease and non-dialysis dependent chronic kidney disease, supportive data, conduct of such studies, or any other part of our Marketing Authorization Application submission; |
| • | expectations for generating revenue or becoming profitable on a sustained basis; |
| • | expectations of the scope of patent protection with respect to Auryxia; |
| • | expectations or ability to enter into marketing and other partnership agreements; |
| • | expectations or ability to enter into product acquisition and in-licensing transactions; |
| • | expectations or ability to build our own commercial infrastructure to manufacture, market and sell our drug candidate; |
| • | estimates of the sufficiency of our existing cash and cash equivalents to finance our operating requirements, including expectations regarding the value and liquidity of our investments; |
| • | expected losses; and |
| • | expectations for future capital requirements. |
1
Table of Contents
The forward-looking statements contained in this report reflect our views and assumptions only as of the date that this report is signed. Except as required by law, we assume no responsibility for updating any forward-looking statements.
We qualify all of our forward-looking statements by these cautionary statements. In addition, with respect to all of our forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995.
2
Table of Contents
Unless the context requires otherwise, references in this report to “Keryx,” “Company,” “we,” “us” and “our” and similar designations refer to Keryx Biopharmaceuticals, Inc. and our subsidiaries.
| ITEM 1. | BUSINESS. |
| OVERVIEW |
We are a biopharmaceutical company focused on bringing innovative therapies to market for patients with renal disease. Our first product, AuryxiaTM (ferric citrate), an oral, absorbable iron-based compound, received marketing approval from the U.S. Food and Drug Administration, or FDA, in September 2014 for the control of serum phosphorus levels in patients with chronic kidney disease, or CKD, on dialysis. The U.S. approval of Auryxia was based on data from our Phase 3 registration program, in which Auryxia effectively reduced serum phosphorus levels to well within the National Kidney Foundation Kidney Disease Outcomes Quality Initiative, or KDOQI, guidelines range of 3.5 to 5.5 mg/dL. In addition to the effects on serum phosphorus levels, Auryxia’s pharmacodynamic properties resulted in increased ferritin, iron and transferrin saturation, or TSAT, whereas these parameters remained relatively constant in patients treated with active control (Renvela® and/or PhosLo®). The most common adverse events for Auryxia treated patients were gastrointestinal-related, including diarrhea, nausea, constipation, vomiting and cough.
We launched Auryxia in the U.S. in late December 2014. Auryxia is being marketed in the U.S. through our specialty salesforce and commercial infrastructure. We currently have 60 sales representatives in the field calling on approximately 5,000 target nephrologists.
Our Japanese partner, Japan Tobacco Inc. or JT, and Torii Pharmaceutical Co. Ltd., or Torii, received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare as an oral treatment for the improvement of hyperphosphatemia in patients with CKD, including dialysis and non-dialysis dependent CKD, or NDD-CKD, in January 2014. JT’s subsidiary, Torii, launched the product under the brand name Riona® in May 2014.
We have also submitted, in March 2014, a Marketing Authorization Application, or MAA, with the European Medicines Agency, or EMA, for the approval of Auryxia in patients with CKD, including dialysis and NDD-CKD. Also in March 2014, the EMA validated our MAA, confirming that the submission is sufficiently complete to begin the formal review process.
In September 2014, we announced the initiation of a pivotal Phase 3 study of Auryxia for the treatment of iron deficiency anemia, or IDA, in patients with Stage 3-5 NDD-CKD. This study’s primary endpoint is the between group comparison of the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any point during the 16-week randomized period. In our completed 12-week Phase 2 study of Auryxia for the management of elevated serum phosphorus levels and iron deficiency in subjects with Stage 3 to 5 NDD-CKD, a post-hoc analysis of this endpoint demonstrated that the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any time point during the study was 40% in the Auryxia arm vs. 15% in the placebo arm (p-value <0.001). Secondary endpoints in the Phase 3 study include change from baseline to the end of the randomized period for hemoglobin, ferritin, TSAT and serum phosphorus.
Currently, our only drug product is Auryxia. We may engage in business development activities that include seeking strategic relationships for Auryxia, as well as evaluating other compounds and companies for in-licensing or acquisition. We have also generated, and expect to continue to generate, revenue from the sublicensing of rights to Auryxia in Japan to our Japanese partner, JT and Torii.
3
Table of Contents
OUR STRATEGY
Our mission is to create long-term stockholder value by bringing differentiated products to market that provide meaningful benefits to patients and their healthcare providers. Our strategy to achieve this mission is to:
| • | continue building our infrastructure to commercialize Auryxia and any future drug candidate(s), either alone or in partnership depending on the path we believe will provide maximum stockholder value; |
| • | identify and explore licensing, partnership and other business development opportunities for Auryxia, and any drug candidates we may in-license or acquire; |
| • | seek to acquire or in-license medically important drug candidates in clinical development; and |
| • | utilize our clinical development capabilities to manage and progress any future drug candidates through the clinical development and regulatory processes to approval. |
CORPORATE INFORMATION
We were incorporated in Delaware in October 1998 and commenced operations in November 1999. Our executive offices are located at 750 Lexington Avenue, New York, New York 10022. Our telephone number is 212-531-5965, and our e-mail address is info@keryx.com.
We maintain a website with the address www.keryx.com. We make available free of charge through our Internet website our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and any amendments to these reports, as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the SEC. We are not including the information on our website as a part of, nor incorporating it by reference into, this report. You may read and copy any materials we file at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549 on official business days during the hours of 10:00 a.m. to 3:00 p.m. Please call the SEC at 1-800-SEC-0330 for information on the Public Reference Room. Additionally, the SEC maintains a website that contains annual, quarterly, and current reports, proxy statements, and other information that issuers (including us) file electronically with the SEC. The SEC’s website address is http://www.sec.gov.
LEAD PRODUCT
Auryxia (ferric citrate)
Overview
Auryxia (ferric citrate) is an oral, ferric iron-based compound that has the capacity to bind to phosphate in the gastrointestinal tract and form non-absorbable complexes and can potentially also treat iron deficiency anemia. The FDA approved Auryxia for the control of serum phosphorus levels in patients with CKD on dialysis in September 2014. The U.S. approval of Auryxia was based on data from our Phase 3 registration program. In the Phase 3 clinical trials, which were conducted under a Special Protocol Assessment, or SPA, agreement with the FDA, Auryxia effectively reduced serum phosphorus levels to well within the KDOQI guidelines range of 3.5 to 5.5 mg/dL. In addition to the effects on serum phosphorus levels, Auryxia’s pharmacodynamic properties resulted in increased ferritin, iron and TSAT; whereas these parameters remained relatively constant in patients treated with active control (Renvela® and/or Phoslo®). The most common adverse events for Auryxia treated patients were gastrointestinal-related, including diarrhea, nausea, constipation, vomiting and cough. In July 2014, we announced the publication of results from the long-term pivotal Phase 3 study of Auryxia in the Journal of the American Society of Nephrology.
Auryxia is being marketed in the U.S. through our specialty salesforce and commercial infrastructure. We currently have 60 sales representatives in the field calling on approximately 5,000 target nephrologists. We have also established account management and medical affairs teams who are responsible for working with the dialysis centers where ESRD patients receive dialysis treatment. In addition, we have a small team of national
4
Table of Contents
account managers who are primarily responsible for working with insurance plans, health maintenance organizations and other payers to secure reimbursement and formulary status for Auryxia. We are currently undergoing formulary reviews with Medicare Part D insurance plans and as of January 1, 2015 have been placed on formulary with four of those plans. We have also created the Keryx Patient Plus program to assist with patient accessibility to Auryxia. Beginning at our reimbursement hub, personalized service is provided through dedicated regional case managers to provide the renal care team with comprehensive insurance and reimbursement support for their patients. Keryx Patient Plus offers benefit verification, co-pay assistance for eligible commercial patients, no-cost drug program for those who qualify, and a short-term prescription bridge program that may assist those already on Auryxia who are in danger of suffering a break in coverage.
In July 2014, we completed a long-term, open-label extension, or OLE, study for Auryxia in dialysis-dependent CKD patients. Patients who had participated in and successfully completed the long-term pivotal Phase 3 study were eligible for enrollment in the 48-week OLE study, providing for cumulative exposure to Auryxia of up to two years. Patients in the OLE study (n=168) were titrated to achieve and maintain serum phosphorus levels within a range of 3.5 to 5.5 mg/dL, with a maximum daily dose of 12 grams per day of Auryxia. The safety profile observed in the OLE study was consistent with that seen in the long-term pivotal Phase 3 study and there were no clinically meaningful changes in liver enzymes or aluminum levels over the course of the study.
We are also developing Auryxia as a treatment for IDA in patients with Stage 3-5 NDD-CKD and announced the initiation of a pivotal Phase 3 study for that indication in September 2014. This study’s primary endpoint is the between group comparison of the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any point during the 16-week Randomized Period. In our completed 12-week Phase 2 study in NDD-CKD, a post-hoc analysis of this endpoint demonstrated that the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any time point during the study was 40% in the Auryxia arm vs. 15% in the placebo arm (p-value <0.001). Secondary endpoints in the Phase 3 study include change from baseline to end of Randomized Period for hemoglobin, ferritin, TSAT and serum phosphorus.
In October 2014, we announced that the U.S. Patent and Trademark Office issued U.S. Patent No. 8,846,976. The patent, which expires in 2024, claims a method of treating hyperphosphatemia comprised of administering a therapeutically effective amount of an orally administrable form of Auryxia to a subject, wherein the orally administrable form is prepared from an Auryxia active pharmaceutical ingredient having an intrinsic dissolution rate of at least 1.88/mg/cm2/min. In addition, U.S. Patent No. 8,846,976 contains claims directed to the FDA approved dosing and daily administration of Auryxia. This newly issued patent further enhances our key patent family, which includes U.S. Patent Nos. 7,767,851, 8,299,298, 8,338,642, 8,609,896, 8,754,257 and 8,754,258, which expire in 2024, and U.S. Patent No. 8,093,423, which expires in 2026, before patent term extension. Each of these patents contains composition and method of use claims covering Auryxia.
Our Japanese partner, JT and Torii, received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare as an oral treatment for the improvement of hyperphosphatemia in patients with CKD, including dialysis and NDD-CKD, in January 2014. JT’s subsidiary, Torii, launched the product under the brand name Riona® in May 2014. Under the license agreement with JT and Torii, we received a non-refundable payment of $10.0 million in February 2014 for the achievement of the marketing approval milestone. We also receive royalty payments based on a tiered double-digit percentage of net sales of Riona® in Japan escalating up to the mid-teens, as well as up to an additional $55.0 million upon the achievement of certain annual net sales milestones. In October 2014, following the regulatory approval of Auryxia in Japan earlier this year, the Japan Patent office granted patent term extensions for patents #4964585 and #4173553, which extended the terms of these patents in Japan to November 2025 and November 2022, respectively.
We have also submitted, in March 2014, a MAA with the EMA for the approval of Auryxia in patients with CKD, including dialysis and NDD-CKD. Also in March 2014, the EMA validated our MAA, confirming that the submission is sufficiently complete to begin the formal review process.
5
Table of Contents
Market Opportunity
In the U.S., there were approximately 450,000 adult patients with End Stage Renal Disease, or ESRD, who required dialysis in 2012. Managing ESRD is complex as many metabolic factors, such as iron and phosphorus, are out of balance. Phosphate retention and the resulting hyperphosphatemia in dialysis patients are typically associated with increased risk for heart and bone disease, and death. The majority of ESRD patients require chronic treatment with phosphate-binding agents to lower and maintain serum phosphorus at acceptable levels. In addition, iron can be severely depleted in dialysis patients and they are therefore often treated with intravenous iron, or IV iron, erythropoiesis-stimulating agents, or ESAs, and other medications.
In addition, it is estimated that more than 10% of the U.S. adult population is affected by NDD-CKD, a condition generally characterized by greater than 50% reduction of normal kidney function. In addition, elevated levels of serum phosphorus become more prevalent in Stage 3 to 5 NDD-CKD patients. Several studies have shown that higher serum phosphorus concentrations may be associated with increased mortality and morbidity in CKD, however, no phosphate binders are currently FDA approved for NDD-CKD.
IDA is extremely prevalent in the NDD-CKD population and is associated with fatigue, lethargy, decreased quality of life and is also believed to be associated with cardiovascular complications, hospitalizations, and increased mortality. Based on data contained in a 2009 publication in the Journal of the American Society of Nephrology, it is estimated that over 1.5 million adults with NDD-CKD in the U.S. alone are also afflicted with IDA. To combat this anemia, iron replacement therapy is essential to increase iron stores, which is reflected in ferritin and TSAT levels, and raise hemoglobin levels. Currently available oral iron supplements are associated with limited efficacy and dose-limiting tolerability issues. No oral iron agents are currently FDA approved to treat IDA in NDD-CKD. ESAs and IV iron are not frequently administered in NDD-CKD due to both the FDA boxed warning label of potential cardiovascular risk for ESAs and logistical complications associated with administering IV medicines in office settings. Consequently, the NDD-CKD patient population remains underserved.
CKD on Dialysis: Phase 3 Registration Clinical Program – Short-Term Study
Study Design
The Phase 3 short-term study was a multicenter, randomized, open-label trial with a two-week washout period, following which patients were randomized 1:1:1 to receive a fixed dose of Auryxia of either 1 gram, 6 grams or 8 grams per day for a treatment period of 28 days. Auryxia was administered using a 1 gram oral tablet formulation, and the fixed-dose arms of 1 gram, 6 grams and 8 grams per day represented 1, 6 and 8 pills per day, respectively. One hundred fifty-four dialysis patients were enrolled into the study. The ITT group included 146 patients, representing all patients who took at least one dose of Auryxia and provided a Baseline (at the end of washout) and at least one post-Baseline efficacy assessment. Efficacy assessments were taken weekly starting at Baseline and subsequently at days 7, 14, 21 and 28.
Study Results
The primary endpoint of the study was to determine whether there was a dose response in the change in serum phosphorus from Baseline to Day 28 in the ITT group, using a regression analysis to evaluate this objective. The study met the primary endpoint, with the regression analysis indicating a highly statistically significant dose response (p<0.0001). Additional efficacy results are as follows:
| Mean Serum Phosphorus (mg/dL) ITT (n=146) |
1g/day (n=50) |
6g/day (n=51) |
8g/day (n=45) |
|||||||||
| Baseline (End of Washout) |
7.3 | 7.6 | 7.5 | |||||||||
| Day 28 (End of Treatment) |
7.4 | 5.6 | 5.3 | |||||||||
| Change from Baseline at Day 28 P-Value |
0.1 |
|
-2.0 <0.0001 |
|
|
-2.2 <0.0001 |
| |||||
| % Change from Baseline at Day 28 |
0.5 | % | -25.7 | % | -29.6 | % | ||||||
6
Table of Contents
In addition, a statistically significant dose response increase in serum bicarbonate was observed in the study, indicating the potential ability of Auryxia to address metabolic acidosis. Metabolic acidosis is a condition that occurs in many dialysis patients when the kidneys do not remove sufficient acid from the body, leading to low blood pH. The consequences of metabolic acidosis can be severe. The inability to manage metabolic acidosis is believed to be a drawback for some of the currently marketed phosphate binders.
Importantly, no clinically meaningful change in serum calcium was observed in the study. Additionally, a statistically significant dose response reduction in calcium-phosphorus product was also observed in the study. Elevated levels of serum calcium, or hypercalcemia, and high levels of calcium-phosphorus product, both of which are believed to be drawbacks from the use of some of the currently marketed phosphate binders, increase the risk of soft tissue calcification and may contribute to the substantial morbidity and mortality seen in patients with ESRD.
Certain iron parameters, including ferritin and TSAT, were measured in the study. Over the 28-day treatment period, modest upward trends in ferritin and TSAT levels were observed in the 6 grams/day and 8 grams/day dose groups.
No serious adverse events were deemed to be drug-related by the Data Safety Monitoring Committee in this clinical study.
CKD on Dialysis: Phase 3 Registration Clinical Program – Long-Term Study
In January 2013, we announced successful top-line results from the long-term Phase 3 study of Auryxia for the treatment of hyperphosphatemia in patients with CKD on dialysis. In this study, conducted pursuant to a SPA agreement with the FDA, Auryxia met the study’s primary endpoint, described below, demonstrating a highly statistically significant change in serum phosphorus versus placebo over the four-week Efficacy Assessment Period of the study. In addition, Auryxia met the key pre-defined secondary endpoints of increasing ferritin and TSAT and reducing the use of IV iron and ESAs versus the active control group (Renvela® [sevelamer carbonate] and/or Phoslo® [calcium acetate]) over the 52-week Safety Assessment Period of the study. This long-term study was the final component of our Phase 3 registration program.
Study Design
This Phase 3 long-term study was a multicenter, randomized, open-label, safety and efficacy clinical trial in 441 CKD patients on hemodialysis or peritoneal dialysis. The study consisted of a 2-week washout period followed by a 52-week Safety Assessment Period in which subjects were randomized 2:1 to receive either Auryxia or an active control (Renvela® [sevelamer carbonate] and/or Phoslo® [calcium acetate]). The 52-week Safety Assessment Period was followed by a 4-week Efficacy Assessment Period. During the Efficacy Assessment Period, only those subjects randomized to treatment with Auryxia during the Safety Assessment Period and completed the Safety Assessment Period were randomized in a 1:1 ratio to either continue treatment with Auryxia or switch to placebo for a 4-week treatment period. Subjects were titrated during the study to achieve serum phosphorus levels that ranged between 3.5 to 5.5 mg/dL.
The primary objectives of this study were to determine the long-term safety of Auryxia in subjects with CKD undergoing either hemodialysis or peritoneal dialysis, and the efficacy of Auryxia following 52 weeks of treatment in a four-week, randomized, open-label, placebo-controlled Efficacy Assessment Period. Auryxia was administered using a 1 gram oral tablet formulation.
Oral iron therapy was not permitted during the course of the study. IV iron therapy was not permitted if a subject’s serum ferritin level was greater than 1000 ng/mL or TSAT was greater than 30%. The use of ESAs was at the physician’s discretion.
7
Table of Contents
Efficacy results were as follows:
Primary Efficacy Endpoint
The primary efficacy endpoint of this trial was the mean change in serum phosphorus from baseline (Week 52) to end of the four-week Efficacy Assessment Period (Week 56) versus placebo in the Intent-to-Treat, or ITT, group. The ITT group included 182 subjects, representing all subjects who took at least one dose of Auryxia or placebo in the Efficacy Assessment Period and provided at least one post-baseline efficacy assessment.
Auryxia met the primary efficacy endpoint with a highly statistically significant result (p<0.0001).
| Mean Serum Phosphorus (mg/dL) |
Placebo (n=91) |
Auryxia (n=91) |
||||||
| Baseline (Week 52) |
5.4 | 5.1 | ||||||
| End of Treatment1 (Week 56) |
7.2 | 4.9 | ||||||
| Mean Change from Baseline at Week 56 |
1.8 | -0.2 | ||||||
| Least Squares (LS) Mean Difference from Placebo2 |
-2.2 | |||||||
| p-value2 |
p<0.0001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | The LS Mean treatment difference and p-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
Key Secondary Efficacy Endpoints Related to Serum Phosphorus
During the 52-week Safety Assessment Period, Auryxia maintained serum phosphorus in the normal range, with highly statistically significant changes in mean serum phosphorus concentration at Weeks 12, 24, 36, 48, and 52 as compared to baseline (Day 0).
| n=281 |
Baseline | Week | ||||||||||||||||||||||
| 12 | 24 | 36 | 48 | 52 | ||||||||||||||||||||
| Auryxia Mean Serum Phosphorus (mg/dL)1 |
7.4 | 5.4 | 5.3 | 5.2 | 5.3 | 5.4 | ||||||||||||||||||
| Mean Change from Baseline |
-2.0 | -2.1 | -2.2 | -2.1 | -2.0 | |||||||||||||||||||
| % Change from Baseline |
-27.0 | % | -28.4 | % | -29.7 | % | -28.4 | % | -27.0 | % | ||||||||||||||
| p-value |
<0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |||||||||||||||||||
| 1 | Last observation carried forward was used for missing data. |
In addition, as agreed to with the EMA, the treatment difference between Auryxia and Renvela® (sevelamer carbonate) at Week 12 of the Safety Assessment Period in terms of change from baseline (Day 0) in serum phosphorus was analyzed. Auryxia successfully achieved the non-inferiority endpoint versus Renvela®.
Key Secondary Efficacy Endpoints Related to Iron
The objectives of the key iron-related secondary endpoints, which were all pre-specified in the statistical analysis plan in a sequential strategy to control overall Type I error rate, were to corroborate prior data which suggested that Auryxia may increase iron storage parameters and reduce the need for IV iron and/or ESAs as compared to the active control group. Auryxia met all the key pre-defined secondary efficacy endpoints related
8
Table of Contents
to iron with statistically significant treatment differences versus the active control group (Renvela® [sevelamer carbonate] and/or Phoslo® [calcium acetate]), as follows:
Mean Change in Ferritin
Auryxia demonstrated a statistically significant treatment difference versus the active control group in mean change in serum ferritin from baseline (Day 0) to Week 52.
| Mean Ferritin (ng/mL)1 |
Active Controls (n=137) |
Auryxia (n=253) |
||||||
| Baseline (Day 0) |
610 | 593 | ||||||
| Week 12 |
649 | 750 | ||||||
| Week 24 |
651 | 847 | ||||||
| Week 36 |
633 | 864 | ||||||
| Week 48 |
622 | 886 | ||||||
| Week 52 |
632 | 895 | ||||||
| Mean Change from Baseline at Week 52 % Change from Baseline |
|
22 3.6 |
% |
|
302 50.9 |
% | ||
| LS Mean Difference from Active Control Group at Week 522 |
274 | |||||||
| p-value2 |
p<0.0001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | The LS Mean treatment difference and p-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
Mean Change in TSAT
Auryxia demonstrated a statistically significant treatment difference versus the active control group in mean change in TSAT from baseline (Day 0) to Week 52.
| Mean TSAT (%)1 |
Active Controls (n=137) |
Auryxia (n=252) |
||||||
| Baseline (Day 0) |
31 | 31 | ||||||
| Week 12 |
31 | 40 | ||||||
| Week 24 |
31 | 40 | ||||||
| Week 36 |
31 | 40 | ||||||
| Week 48 |
29 | 41 | ||||||
| Week 52 |
30 | 39 | ||||||
| Mean Change from Baseline at Week 52 % Change from Baseline |
|
-1 -3.2 |
% |
|
8 25.8 |
% | ||
| LS Mean Difference from Active Control Group at Week 522 |
9 | |||||||
| p-value2 |
p<0.0001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | The LS Mean treatment difference and p-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
Cumulative IV iron Use
Each subject’s average cumulative IV iron intake was calculated over the 52-week Safety Assessment Period. The ITT group consisted of 271 subjects and 138 subjects for the Auryxia and active control groups, respectively. Auryxia demonstrated a 51% decrease in median IV iron intake as compared to the active control group (median 1.87 mg/day for Auryxia versus 3.83 mg/day for active control, p<0.0001).
9
Table of Contents
Cumulative Erythropoiesis-Stimulating Agent (ESA) Use
Each subject’s average cumulative ESA intake was calculated over the 52-week Safety Assessment Period. The ITT group consisted of 273 subjects and 141 subjects for the Auryxia and active control groups, respectively. Auryxia demonstrated a 24% decrease in median ESA intake as compared to the active control group (median 756 units/day for Auryxia versus 993 units/day for active control, p<0.05).
Mean Change in Hemoglobin
Auryxia demonstrated a statistically significant treatment difference versus the active control group in mean change in hemoglobin from baseline (Day 0) to Week 52.
| Mean Hemoglobin (g/dL)1 |
Active Controls (n=133) |
Auryxia (n=248) |
||||||
| Baseline (Day 0) |
11.7 | 11.6 | ||||||
| Week 52 |
11.2 | 11.4 | ||||||
| Mean Change from Baseline at Week 52 |
-0.6 | -0.2 | ||||||
| LS Mean Difference from Active Control Group at Week 522 |
0.3 | |||||||
| p-value2 |
p<0.05 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | The LS Mean treatment difference and p-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
Safety and Tolerability Profile
For reference, subjects previously intolerant to Renvela® (sevelamer carbonate) and/or Phoslo® (calcium acetate) were ineligible to participate in this study. Based on an analysis of safety data, the side-effect profile of Auryxia and the active control group appeared similar, with the most common adverse events gastrointestinal-related. The most common gastrointestinal adverse events were: diarrhea, including soft stools (26% Auryxia vs. 14% Active Control), nausea (14% Auryxia vs. 14% Active Control), feces discoloration (17% Auryxia vs. 0% Active Control), vomiting (9% Auryxia vs. 15% Active Control) and constipation (8% Auryxia vs. 5% Active Control). Adverse events were generally characterized as mild to moderate in nature.
The overall serious adverse event rates in the study were 39% Auryxia vs. 49% Active Control. Importantly, there were no clinically meaningful or statistically significant differences between Auryxia and the active control group in serum calcium levels, aluminum levels and liver enzymes, as measured by alanine transaminase, or ALT, and aspartate transaminase, or AST.
CKD on Dialysis: Open-Label Safety Extension Study
In July 2014, we completed the long-term, open-label extension, or OLE, study for Auryxia in dialysis-dependent CKD patients. Patients who had participated in and successfully completed the long-term pivotal Phase 3 study were eligible for enrollment in the 48-week OLE study, providing for cumulative exposure to Auryxia of up to two years. Patients in the OLE study (n=168) were titrated to achieve and maintain serum phosphorus levels within a range of 3.5 to 5.5 mg/dL, with a maximum daily dose of 12 grams per day of Auryxia. The safety profile observed in the OLE study was consistent with that seen in the long-term pivotal Phase 3 study and there were no clinically meaningful changes in liver enzymes or aluminum levels over the course of the study.
10
Table of Contents
NDD-CKD: Phase 3 Clinical Study
In September 2014, we announced the initiation of a pivotal Phase 3 study of Auryxia for the treatment of iron deficiency anemia in patients with Stage 3-5 NDD-CKD. The placebo-controlled study is expected to 230 patients and the study’s primary endpoint is the between group comparison of the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any point during the 16-week Randomized Period. In our completed 12-week Phase 2 study in NDD-CKD, a post-hoc analysis of this endpoint demonstrated that the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any time point during the study was 40% in the Auryxia arm vs. 15% in the placebo arm (p-value <0.001). Secondary endpoints in the Phase 3 study include change from baseline to end of Randomized Period for hemoglobin, ferritin, TSAT and serum phosphorus.
NDD-CKD: Phase 2 Clinical Study
In November 2013, we announced successful top-line results from the U.S.-based Phase 2 study of Auryxia in managing serum phosphorus and iron deficiency anemia in patients with Stage 3 to 5 NDD-CKD. In this study, Auryxia met both co-primary endpoints, demonstrating highly statistically significant changes in serum phosphorus and TSAT versus placebo over the 12-week treatment period. In addition, Auryxia met the key secondary endpoints of increasing ferritin and hemoglobin, and decreasing fibroblast growth factor-23, or FGF-23, versus placebo.
Study Design
This Phase 2 study was a multicenter, randomized, double-blind, placebo-controlled clinical trial in subjects with stage 3 to 5 NDD-CKD, with elevated serum phosphorus ³4.0 mg/dL and iron deficiency anemia. The study consisted of a 2-week washout period (for subjects on a phosphate binder at screening) followed by a 12-week treatment period in which subjects were randomized 1:1 to receive either Auryxia or placebo. One hundred forty-nine (149) subjects were randomized into the study from 20 sites in the United States.
The use of IV iron and ESAs were not permitted within 8 weeks and 4 weeks prior to the study, respectively, and not permitted during the course of the study. Oral iron therapy was also not permitted during the course of the study.
Efficacy results were as follows:
Co-Primary and Key Secondary Endpoints
Auryxia met both co-primary and all key secondary endpoints with highly statistically significant results. The Intent-to Treat (ITT) group included 141 subjects, representing all subjects who took at least one dose of Auryxia or placebo and provided at least one post-baseline efficacy assessment. The co-primary efficacy endpoints of this trial were the mean changes in serum phosphorus and TSAT from baseline to the end of the 12-week treatment period versus placebo in the ITT group.
| Mean Serum Phosphorus (mg/dL) |
Placebo (n=69) |
Auryxia (n=72) |
||||||
| Baseline |
4.7 | 4.5 | ||||||
| End of Treatment1 (Week 12) |
4.4 | 3.9 | ||||||
| Treatment Difference p-value2 |
p<0.001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | P-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
11
Table of Contents
| TSAT (%) |
Placebo (n=69) |
Auryxia (n=72) |
||||||
| Baseline |
21 | 22 | ||||||
| End of Treatment1 (Week 12) |
20 | 32 | ||||||
| Treatment Difference p-value2 |
p<0.001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | P-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
The key secondary endpoints of the study were the mean changes in ferritin, hemoglobin and FGF-23 from baseline to the end of the 12-week treatment period versus placebo in the ITT group.
| Mean Ferritin (ng/mL) |
Placebo (n=69) |
Auryxia (n=72) |
||||||
| Baseline |
110 | 116 | ||||||
| End of Treatment1 (Week 12) |
106 | 189 | ||||||
| Treatment Difference p-value2 |
p<0.001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | P-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
| Mean Hemoglobin (g/dL) |
Placebo (n=69) |
Auryxia (n=72) |
||||||
| Baseline |
10.6 | 10.5 | ||||||
| End of Treatment1 (Week 12) |
10.4 | 11.0 | ||||||
| Treatment Difference p-value2 |
p<0.001 | |||||||
| 1 | Last observation carried forward was used for missing data. |
| 2 | P-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
| Mean Intact FGF-23 (pg/mL) |
Placebo (n=60) |
Auryxia (n=63) |
||||||
| Baseline |
263 | 319 | ||||||
| End of Treatment1 (Week 12) |
293 | 200 | ||||||
| Treatment Difference p-value2 |
P=0.017 | |||||||
| 1 | Last observation carried forward was used for missing data. Intact FGF-23 was assessed at baseline, Week 6 and Week 12. |
| 2 | P-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
| Mean C-Terminal FGF-23 (pg/mL) |
Placebo (n=60) |
Auryxia (n=63) |
||||||
| Baseline |
511 | 468 | ||||||
| End of Treatment1 (Week 12) |
579 | 316 | ||||||
| Treatment Difference p-value2 |
p<0.001 | |||||||
| 1 | Last observation carried forward was used for missing data. C-Terminal FGF-23 was assessed at baseline, Week 6 and Week 12. |
| 2 | P-value is created via an ANCOVA model with treatment as the fixed effect and baseline as the covariate. |
Auryxia was also highly statistically significant in its mean changes at Week 12 versus baseline for all the above-mentioned co-primary and key secondary endpoints.
12
Table of Contents
Treatment Failures
Patients were discontinued from the study if they had hemoglobin measurements <9.0 g/dL on two consecutive visits or serum phosphorus measurements ³6.0 mg/dL on two consecutive visits following randomization. Treatment Failures in the study were as follows:
| Treatment Failures n (%) |
Placebo (n=74) |
Auryxia (n=75) |
||||||
| Hemoglobin <9.0 g/dL |
9 | (12%) | 1 | (1%) | ||||
| Serum Phosphorus ³6.0 mg/dL |
2 | (3%) | 0 | (0%) | ||||
Safety and Tolerability Profile
The safety population in the study included all randomized patients who took at least one dose of study drug. We believe that Auryxia appeared to be safe and well-tolerated in this Phase 2 study, with discontinuation rates of 19% and 32% in the Auryxia and placebo groups, respectively, including Treatment Failures. There were no study discontinuations due to hypophosphatemia in the study.
Serious adverse events occurred in six Auryxia subjects (8%) versus nine placebo subjects (12%). Two deaths were recorded in the study, both from the placebo group. There were no clinically meaningful or statistically significant differences in serum calcium levels and liver enzymes as measured by ALT and AST.
COSTS AND TIME TO COMPLETE PRODUCT DEVELOPMENT
The information below provides estimates regarding the costs associated with the completion of the current development phase and our current estimated range of the time that will be necessary to complete such development for Auryxia, which is currently our only product candidate. We also direct your attention to the risk factors which could significantly affect our ability to meet these cost and time estimates found in this report in Item 1A under the heading “Risks Associated with Our Product Development Efforts.”
| Product candidate |
Target indication | Development status | Expected completion of phase |
Estimated cost to complete phase | ||||
| Auryxia (ferric citrate) | Hyperphosphatemia in CKD | EU MAA submitted and under review |
2H 2015 | $1 - $3 million | ||||
|
|
|
|
|
| ||||
| Auryxia (ferric citrate) | Iron deficiency anemia in NDD-CKD |
Phase 3 ongoing | 1Q 2016 | $6 - $8 million |
Completion dates and costs in the above table are estimates and are subject to the uncertainties associated with regulatory submissions, clinical trials and the related requirements of development. In the cases where the requirements for regulatory submissions, clinical trials and development programs have not been fully defined, or are dependent on the success of other trials, we cannot estimate trial completion or cost with any certainty. The actual spending on each trial during the year is also dependent on our ability to fund such clinical trials. We therefore direct your attention to Item 7 under the heading “Liquidity and Capital Resources.”
INTELLECTUAL PROPERTY AND PATENTS
General
Patents and other proprietary rights are very important to the development of our business. We will be able to protect our proprietary technologies from unauthorized use by third parties only to the extent that our proprietary rights are covered by valid and enforceable patents supported by regulatory exclusivity, or are effectively maintained as trade secrets. It is our intention to seek and maintain patent and trade secret protection for our drug candidates and our proprietary technologies. As part of our business strategy, our policy is to
13
Table of Contents
actively file patent applications in the U.S. and, when appropriate, internationally to cover methods of use, processes of manufacture, new chemical compounds, pharmaceutical compositions, dosing of the compounds and compositions, and improvements in each of these areas. We also rely on trade secret information, technical know-how, innovation and agreements with third parties to continuously expand and protect our competitive position. We have a number of patents and patent applications related to our compounds and other technology, but we cannot guarantee the scope of protection of the issued patents, or that such patents will survive a validity or enforceability challenge, or that any of the pending patent applications will issue as patents.
Generally, patent applications in the U.S. are maintained in secrecy for a period of 18 months or more. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we are not certain that we were the first to make the inventions covered by each of our pending patent applications or that we were the first to file those patent applications. The patent positions of biotechnology and pharmaceutical companies are highly uncertain and involve complex legal and factual questions. Therefore, we cannot predict the breadth of claims allowed in biotechnology and pharmaceutical patents, or their enforceability. To date, there has been no consistent policy regarding the breadth of claims allowed in biotechnology patents. Third parties or competitors may challenge or circumvent our patents or patent applications, if issued. If our competitors prepare and file patent applications in the U.S. that claim technology also claimed by us, we may have to participate in interference or derivation proceedings in front of the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost, even if the eventual outcome is favorable to us. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that before we commercialize any of our products, any related patent may expire or remain in existence for only a short period following commercialization, thus reducing any advantage of the patent. However, the life of a patent covering a product that has been subject to regulatory approval may have the ability to be extended through the Patent Term Extension program available under 35 U.S.C. § 156, although any such extension could still be minimal.
If a patent is issued to a third party containing one or more preclusive or conflicting claims, and those claims are ultimately determined to be valid and enforceable, we may be required to obtain a license under such patent or to develop or obtain alternative technology. In the event of a litigation involving a third party claim, an adverse outcome in the litigation could subject us to significant liabilities to such third party, require us to seek a license for the disputed rights from such third party, and/or require us to cease use of the technology. Further, our breach of an existing license or failure to obtain a license to technology required to commercialize our products may seriously harm our business. We also may need to commence litigation to enforce any patents issued to us or to determine the scope and validity of third-party proprietary rights. Litigation would involve substantial costs.
Pursuant to our license for Auryxia (ferric citrate) with Panion & BF Biotech, Inc., or Panion, we have the exclusive commercial rights to a series of patent applications worldwide, excluding certain Asian-Pacific countries. These patents and patent applications include claims directed to compositions of matter, pharmaceutical compositions, methods of treatment, as well as methods for the manufacture of Auryxia. We have also filed a patent application directed to formulations of certain ferric citrate drug products.
The patent rights that we own or have licensed relating to Auryxia are limited in ways that may affect our ability to exclude third parties from competing against us. In particular:
| • | Composition of matter patents can provide protection for pharmaceutical products to the extent that the specifically covered compositions are key, non-interchangeable components of the pharmaceutical product. The first composition of matter and method patent relating to Auryxia in the United States (U.S. Patent No. 5,753,706) expires in February 2017. We licensed additional composition of matter and method of use patents expiring in 2024 with independent claims covering forms of ferric citrate (the active pharmaceutical ingredient, or API, of Auryxia), pharmaceutical compositions that include the API, pharmaceutical compositions having ferric citrate in an amount effective to reduce serum phosphate levels, and methods of treating hyperphosphatemia and metabolic acidosis. |
14
Table of Contents
| • | Our method of use patents, including U.S. Patent Nos. 7,767,851, 8,299,298 and 8,338,642 and (which expire in 2024), and U.S. Patent No. 8,093,423 (which expires in 2026) only protect the product when used or sold for the claimed methods. However, these types of patents do not limit a competitor from making and marketing a product that is identical to our product that is labeled for an indication that is outside of our patented methods. |
| • | We have filed applications under the Patent Term Extension provisions of 35 U.S.C. § 156 on the above mentioned patents for delays caused by FDA regulatory review. If granted we can utilize the patent term extension on one of these patents, however, we cannot assure you that we can obtain any extension of the term of these patents. If obtained, the maximum term of extension available under 35 U.S.C. § 156 would extend the term of the chosen patent by no more than five years. Upon expiration of these patents, competitors who obtain the requisite regulatory approval may potentially offer products with the same composition and/or method of use as our product, so long as the competitors do not infringe any other patents that we may hold. |
| • | Our pending patent applications may not issue as patents and may not issue in all countries in which we develop, manufacture or potentially sell our product(s) or in countries where others develop, manufacture and potentially sell products using our technologies. Moreover, our pending patent applications, if issued as patents, may not provide additional protection for our product. |
In October 2014, we announced that the U.S. Patent and Trademark Office issued U.S. Patent No. 8,846,976. The patent, which expires in 2024, claims a method of treating hyperphosphatemia comprised of administering a therapeutically effective amount of an orally administrable form of Auryxia to a subject, wherein the orally administrable form is prepared from an active pharmaceutical ingredient having an intrinsic dissolution rate of at least 1.88/mg/cm2/min. In addition, U.S. Patent No. 8,846,976 contains claims directed to the FDA approved dosing and daily administration of Auryxia. This newly issued patent further enhances our key patent family, which includes U.S. Patent Nos. 7,767,851, 8,299,298, 8,338,642, 8,609,896, 8,754,257 and 8,754,258, which expire in 2024, and U.S. Patent No. 8,093,423, which expires in 2026, before patent term extension. Each of these patents contains composition and method of use claims covering Auryxia.
In October 2014, following the regulatory approval of Auryxia in Japan earlier this year, the Japan Patent office granted patent term extensions for patents #4964585 and #4173553, which extended the terms of these patents in Japan to November 2025 and November 2022, respectively.
Obtaining proof of direct infringement by a competitor for a method of use patent requires us to demonstrate that the competitors make and market a product for the patented use(s). Alternatively we can prove that our competitors induce or contribute others in engaging in direct infringement. Proving that a competitor contributes to, or induces, infringement of a patented method by another has additional proof requirements. For example, proving inducement of infringement requires proof of intent by the competitor. If we are required to defend ourselves against claims or to protect our own proprietary rights against others, it could result in substantial costs to us and the distraction of our management. An adverse ruling in any litigation or administrative proceeding could prevent us from marketing and selling Auryxia, increase the risk that a generic version of Auryxia could enter the market to compete with Auryxia, limit our development and commercialization of Auryxia, or otherwise harm our competitive position and result in additional significant costs. In addition, any successful claim of infringement asserted against us could subject us to monetary damages or injunction, which could prevent us from making or selling Auryxia. We also may be required to obtain licenses to use the relevant technology. Such licenses may not be available on commercially reasonable terms, if at all.
Moreover, physicians may prescribe a competitive identical product for indications other than the one for which the product has been approved, or off-label indications, that are covered by the applicable patents. Although such off-label prescriptions may directly infringe or contribute to or induce infringement of method of use patents, such infringement is difficult to prevent.
15
Table of Contents
In addition, any limitations of our patent protection described above may adversely affect the value of our product candidate and may inhibit our ability to obtain a corporate partner at terms acceptable to us, if at all.
Other Intellectual Property Rights
We depend upon trademarks, trade secrets, know-how and continuing technological advances to develop and maintain our competitive position. To maintain the confidentiality of trade secrets and proprietary information, we require our employees, scientific advisors, consultants and collaborators, upon commencement of a relationship with us, to execute confidentiality agreements and, in the case of parties other than our research and development collaborators, to agree to assign their inventions to us. These agreements are designed to protect our proprietary information and to grant us ownership of technologies that are developed in connection with their relationship with us. These agreements may not, however, provide protection for our trade secrets in the event of unauthorized disclosure of such information.
In addition to patent protection, we may utilize orphan drug regulations, pediatric exclusivity or other provisions of the Food, Drug and Cosmetic Act of 1938, as amended, or FDCA, such as new chemical entity exclusivity or new formulation exclusivity, to provide market exclusivity for a drug candidate. Orphan drug regulations provide incentives to pharmaceutical and biotechnology companies to develop and manufacture drugs for the treatment of rare diseases, currently defined as diseases that exist in fewer than 200,000 individuals in the U.S., or, diseases that affect more than 200,000 individuals in the U.S. but that the sponsor does not realistically anticipate will generate a net profit. Under these provisions, a manufacturer of a designated orphan drug can seek tax benefits, and the holder of the first FDA approval of a designated orphan product will be granted a seven-year period of marketing exclusivity for such FDA-approved orphan product. In the U.S., the FDA has the authority to grant additional data protection for approved drugs where the sponsor conducts specified testing in pediatric or adolescent populations. If granted, this pediatric exclusivity may provide an additional six months which are added to the term of data protection as well as to the term of a relevant patent, to the extent these protections have not already expired. We may also seek to utilize market exclusivities in other territories, such as in the EU. We cannot assure that our drug candidate, Auryxia, or any drug candidates we may acquire or in-license, will obtain such orphan drug designation, pediatric exclusivity, new chemical entity exclusivity or any other market exclusivity in the U.S., EU or any other territory, or that we will be the first to receive the respective regulatory approval for such drugs so as to be eligible for any market exclusivity protection.
LICENSING AGREEMENTS AND COLLABORATIONS
We have formed strategic alliances with a number of companies for the development, manufacture and commercialization of Auryxia. Our current key strategic alliances are discussed below.
Panion & BF Biotech, Inc.
In November 2005, we entered into a license agreement with Panion & BF Biotech, Inc. (“Panion”). Under the license agreement, we acquired the exclusive worldwide rights, excluding certain Asian-Pacific countries, for the development and marketing of Auryxia. To date, we have paid an aggregate of $9.6 million to Panion and Panion is eligible to receive one additional milestone payment of $2.0 million upon our successful achievement of European marketing approval, in addition to royalty payments based on a mid-single digit percentage of net sales of Auryxia.
The license agreement terminates upon the expiration of our obligations to pay royalties thereunder. In addition, we may terminate the license agreement (i) in its entirety or (ii) with respect to one or more countries of the territory covered by the agreement, in either case upon 90 days’ notice. We and Panion also have the right to terminate the license agreement upon the occurrence of a breach of a material provision of the license agreement and certain insolvency events.
16
Table of Contents
Japan Tobacco Inc. and Torii Pharmaceutical Co., Ltd.
In September 2007, we entered into a Sublicense Agreement with JT and Torii, JT’s pharmaceutical business subsidiary, under which JT and Torii obtained the exclusive sublicense rights for the development and commercialization of ferric citrate in Japan, which is being developed and marketed in the U.S. under the trade name Auryxia. JT and Torii are responsible for the future development and commercialization costs in Japan. Effective as of June 8, 2009, we entered into an Amended and Restated Sublicense Agreement (the “Revised Agreement”) with JT and Torii, which, among other things, provided for the elimination of all significant on-going obligations under the sublicense agreement.
In January 2014, JT and Torii received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare. Ferric citrate, launched in May 2014 and being marketed in Japan by JT’s subsidiary, Torii Pharmaceutical Co., Ltd., under the brand name Riona®, is indicated as an oral treatment for the improvement of hyperphosphatemia in patients with CKD. Under the terms of the license agreement with JT and Torii, we receive royalty payments based on a tiered double-digit percentage of net sales of Riona® in Japan escalating up to the mid-teens, as well as up to an additional $55.0 million upon the achievement of certain annual net sales milestones. In accordance with our revenue recognition policy, royalty revenues are recognized in the quarter that JT and Torii provide their written report and related information to us regarding sales of Riona®, which generally will be one quarter following the quarter in which the underlying sales by JT and Torii occurred. We record the associated mid-single digit percentage of net sales royalty expense due Panion, the licensor of Auryxia, in the same period as the royalty revenue from JT and Torii is recorded.
The sublicense terminates upon the expiration of all underlying patent rights. Also, JT and Torii may terminate the sublicense agreement with or without cause upon at least six months prior written notice to us. Additionally, either party may terminate the sublicense agreement for cause upon 60 days’ prior written notice after the breach of any material provision of the sublicense agreement, or after certain insolvency events.
COMPETITION
The pharmaceutical and biotechnology industries are highly competitive. Our competitors include pharmaceutical companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies that are active in different but related fields represent substantial competition for us. Many of our competitors have significantly greater capital resources, larger research and development staffs and facilities and greater experience in drug development, regulation, manufacturing and marketing than we do. These organizations also compete with us to recruit qualified personnel, attract partners for joint ventures or other collaborations, and license technologies that are competitive with ours. To compete successfully in this industry we must identify novel and unique drugs or methods of treatment and then complete the development of those drugs as treatments in advance of our competitors.
Aluminum-type phosphate binders were widely used in the past. However, the systemic absorption of aluminum from these agents and the potential toxicity associated with their use no longer make this type of binder a viable long-term treatment option.
Calcium-type phosphate binders are commonly used to bind dietary phosphate; however, they promote positive net calcium balance and an increased risk of metastatic calcification in many patients, especially in those patients taking vitamin D analogs and those with adynamic bone disease. Calcification of the cardiovascular system is believed to represent a significant risk factor for morbidity and mortality in patients with CKD.
Non-calcium-based, non-absorbed phosphate binders, including sevelamer hydrochloride and sevelamer carbonate are among the most prescribed phosphate binders in the U.S. Compared to the calcium-type binders, fewer coronary and aortic calcifications have been documented, however, there is a risk of metabolic acidosis with sevelamer hydrochloride, as well as the potential for gastrointestinal problems, and sevelamer can affect concomitant vitamin K and vitamin D treatment.
17
Table of Contents
Lanthanum-type phosphate binders are another alternative. Lanthanum is a rare earth element and is minimally absorbed in the gastrointestinal tract. Lower level tissue deposition, particularly in bone and liver, has been observed in animals. However, the long-term, potentially harmful, effects due to the accumulation of lanthanum in these tissues have not been clearly determined.
The need for alternative phosphate-binding agents has long been recognized, especially given the increasing prevalence of ESRD and shortcomings with current therapies available to such patients.
Auryxia, currently our only drug product, which we launched in December 2014, is competing with existing therapies. In addition, other companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting with Auryxia, including the treatment of hyperphosphatemia and iron deficiency anemia. Other companies have products or drug candidates in various stages of pre-clinical or clinical development to treat diseases for which we are also seeking to acquire and develop drug products. Some of these potential competing drugs are further advanced in development than Auryxia and other potential drug candidates we may acquire or in-license, and may be commercialized earlier. Additional information can be found under Item 1A - Risk Factors – Other Risks Related to Our Business within this report.
SUPPLY AND MANUFACTURING
We have limited experience in manufacturing products for clinical or commercial purposes. We intend to continue, in whole or in part, to use third parties to manufacture and analytically test our drug, Auryxia, for use in clinical trials and for sales.
We believe that we have established contract manufacturing relationships for the supply of Auryxia to ensure that we will have sufficient material for clinical trials and the ongoing commercial launch. In addition, we are establishing the basis for long-term commercial production capabilities to supply the potential expanded demand for Auryxia in future years. As with any supply program, obtaining raw materials of the required quality and quantity cannot be guaranteed and we cannot ensure that we will be successful in this endeavor.
As we continue to build inventory for the expanded commercialization of Auryxia, we intend to engage additional suppliers to produce Auryxia under current Good Manufacturing Practice, or cGMP, regulations. Our third-party manufacturers have a limited number of facilities in which Auryxia can be produced and will have limited experience in manufacturing Auryxia in quantities sufficient for commercialization. Our third-party manufacturers will have other clients and may have other priorities that could affect their ability to perform the work satisfactorily and/or on a timely basis. Both of these occurrences would be beyond our control.
We expect to similarly rely on contract manufacturing relationships for any products that we may in-license or acquire in the future. However, there can be no assurance that we will be able to successfully contract with such manufacturers on terms acceptable to us, or at all.
Contract manufacturers are subject to ongoing periodic and unannounced inspections by the FDA, the Drug Enforcement Administration and corresponding state and foreign government agencies to ensure strict compliance with cGMP and other state and federal regulations and corresponding foreign standards. Any of our contractors in Europe face similar challenges from the numerous European Union and member state regulatory agencies and authorized bodies. We do not have control over third-party manufacturers’ compliance with these regulations and standards, other than through contractual obligations and periodic auditing. If they are deemed out of compliance with cGMPs, approvals could be delayed, product recalls could result, inventory could be destroyed, production could be stopped and supplies could be delayed or otherwise disrupted.
If we need to change manufacturers after commercialization, the FDA and corresponding foreign regulatory agencies must approve these new manufacturers in advance, which will involve testing and additional inspections to ensure compliance with FDA regulations and standards and may require significant lead times and delay, and
18
Table of Contents
disruption of supply. Furthermore, switching manufacturers may be difficult because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer quickly or on terms acceptable to us, or at all.
GOVERNMENT AND INDUSTRY REGULATION
Numerous governmental authorities, principally the FDA and corresponding state and foreign regulatory agencies, impose substantial regulations upon the clinical development, manufacture and marketing of Auryxia and any future drug candidate(s), as well as our ongoing research and development activities. Before marketing in the U.S., any drug that we develop must undergo rigorous pre-clinical testing and clinical trials and an extensive regulatory approval process implemented by the FDA under the FDCA. The FDA regulates, among other things, the pre-clinical and clinical testing, safety, efficacy, approval, manufacturing, testing, packaging, labeling, storage, recordkeeping, distribution, adverse event reporting, advertising, promotion, and the import and export of biopharmaceutical products.
The regulatory review and approval process is lengthy, expensive and uncertain. We are required to submit extensive pre-clinical, clinical and manufacturing data and supporting information to the FDA for each indication for use to establish a drug candidate’s safety and efficacy before we can secure FDA approval to market or sell a product in the U.S. The approval process can take many years, requires the expenditure of substantial resources, and can include ongoing requirements for post-market surveillance and possibly post-marketing studies. Before commencing clinical trials in humans, we must submit an IND to the FDA containing, among other things, pre-clinical data, chemistry, manufacturing and control information, and an investigative plan. Our submission of an IND may not result in FDA authorization to commence a clinical trial.
The FDA may permit expedited development, evaluation, and marketing of new therapies intended to treat persons with serious or life-threatening conditions for which there is an unmet medical need under its fast track drug development programs. A sponsor can apply for fast track designation at the time of submission of an IND, or at any time prior to receiving marketing approval of the new drug application, or NDA. To receive Fast Track designation, an applicant must demonstrate:
| • | that the drug is intended to treat a serious or life-threatening condition; and |
| • | that the drug demonstrates the potential to address unmet medical needs. |
The FDA must respond to a request for fast track designation within 60 calendar days of receipt of the request. If the FDA determines that the conditions for fast track designation have been met, the FDA will provide a designation letter stating that fast track designation has been granted for development of the drug product for use in treating serious or life-threatening conditions, and informing the sponsor that development studies must show that the product fulfills unmet medical needs. A fast track designation applies to the product coupled with the specific indication for which it is being studied, but not to a product alone.
If the fast track request is incomplete, or the drug development program fails to meet the criteria for fast track designation, the FDA will issue a nondesignation determination. If the sponsor submits a subsequent request for consideration, the FDA will respond to that request within 60 calendar days of receipt of the subsequent request.
Over the course of drug development, a product in a fast track development program must continue to meet the criteria for fast track designation. Sponsors of products in fast track drug development programs must be in regular contact with the reviewing division of the FDA to ensure that the evidence necessary to support marketing approval will be developed and presented in a format conducive to an efficient review. Sponsors of products in fast track drug development programs ordinarily are eligible for priority review of a completed application in six months or less, if the application submission is supported by clinical data, and also may be permitted to submit portions of a NDA to the FDA for review before the complete application is submitted.
19
Table of Contents
Sponsors of drugs designated as fast track also may seek approval under the FDA’s accelerated approval regulations. Under this authority, the FDA may grant marketing approval for a new drug product on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely, based on epidemiologic, therapeutic, pathophysiologic, or other evidence, to predict clinical benefit or an effect on a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, taking into account the threat posed by the condition and whether the drug provides a meaningful advantage over available therapies. Accelerated approval based on a surrogate endpoint or an effect on a clinical endpoint other than survival or irreversible morbidity will be subject to the requirement that the applicant study the drug further to verify and describe its clinical benefit where there is uncertainty as to the relation of the surrogate endpoint to clinical benefit or uncertainty as to the relation of the observed clinical benefit to ultimate outcome. When required to be conducted, such post-marketing studies must also be adequate and well-controlled. The applicant must carry out any such post-marketing studies with due diligence. Failure to conduct such studies, or conducting such studies that do not establish the required safety and efficacy may result in revocation of the original approval. Many companies who have been granted the right to utilize an accelerated approval approach have failed to obtain approval. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch or subsequent marketing of the product.
Clinical testing must meet requirements for institutional review board oversight, informed consent and good clinical practices, and must be conducted pursuant to an IND, unless exempted.
For purposes of NDA approval, clinical trials are typically conducted in the following sequential phases:
| • | Phase 1: The drug is administered to a small group of humans, either healthy volunteers or patients, to test for safety, dosage tolerance, absorption, metabolism, excretion, and clinical pharmacology. In the case of some products for severe or life-threatening diseases, the initial human testing is often conducted in patients having the specific disease. |
| • | Phase 2: Studies are conducted on a larger number of patients to assess the efficacy of the product, to ascertain dose tolerance and the optimal dose range, and to gather additional data relating to safety and potential adverse events. |
| • | Phase 3: Studies further evaluate dosage, and establish safety and efficacy in an expanded patient population. Generally, at least two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval. |
| • | Phase 4: The FDA may require Phase 4 post-marketing studies to find out more about the drug’s long-term risks, benefits, and optimal use, or to test the drug in different populations. |
The length of time necessary to complete clinical trials varies significantly and may be difficult to predict. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Additional factors that can cause delay or termination of our clinical trials, or that may increase the costs of these trials, include:
| • | slow patient enrollment due to the nature of the clinical trial plan, the proximity of patients to clinical sites, the eligibility criteria for participation in the study, competing clinical trials or other factors; |
| • | inadequately trained or insufficient personnel at the study site to assist in overseeing and monitoring clinical trials or delays in approvals from a study site’s review board; |
| • | longer treatment time required to demonstrate efficacy or determine the appropriate product dose; |
| • | insufficient supply of the drug candidates; |
| • | high drop-out rate in the clinical trial; |
| • | adverse medical events or side effects in treated patients; and |
| • | lack of efficacy of the drug candidates. |
20
Table of Contents
In addition, the FDA, equivalent foreign regulatory authority, or a data safety monitoring committee for a trial may place a clinical trial on hold or terminate it if it concludes that subjects are being exposed to an unacceptable health risk, or for futility. Any drug is likely to produce some toxicity or undesirable side effects in animals and in humans when administered at sufficiently high doses and/or for a sufficiently long period of time. Unacceptable toxicity or side effects may occur at any dose level at any time in the course of studies in animals designed to identify unacceptable effects of a drug candidate, known as toxicological studies, or clinical trials of drug candidates. The appearance of any unacceptable toxicity or side effect could cause us or regulatory authorities to interrupt, limit, delay or abort the development of any of our drug candidates and could ultimately prevent approval by the FDA or foreign regulatory authorities for any or all targeted indications.
Sponsors of drugs may apply for a Special Protocol Assessment (SPA) from the FDA. Through the SPA process, the FDA provides official evaluation and written guidance on the design and size of proposed protocols that are intended to form the basis for a new drug application. However, final marketing approval depends on the results of efficacy, the adverse event profile and an evaluation of the benefit/risk of treatment demonstrated in Phase 3 trials. The SPA agreement may only be changed through a written agreement between the sponsor and the FDA, or if the FDA becomes aware of a substantial scientific issue essential to product safety or efficacy.
Before receiving FDA approval to market a product, we must demonstrate that the product is safe and effective for its intended use by submitting to the FDA an NDA containing the pre-clinical and clinical data that have been accumulated, together with chemistry, manufacturing and controls specifications and information, and proposed labeling, among other things. The FDA may refuse to accept an NDA for filing if certain content criteria are not met and, even after accepting an NDA, the FDA may often require additional information, including clinical data, before issuing approval to market a product. Whether or not the FDA requests additional information, there is no assurance that the NDA will be approved.
It is also becoming more common for the FDA to request a Risk Evaluation and Mitigation Strategy, or REMS, as part of a NDA. A REMS plan may contain post-market obligations of the sponsor to, among other things, train prescribing physicians, monitor off-label drug use, and conduct sufficient Phase 4 follow-up studies and registries to ensure the continued safe use of the drug.
As part of the approval process, the FDA must inspect and approve each manufacturing facility. Among the conditions of approval is the requirement that a manufacturer’s quality control and manufacturing procedures conform to current good manufacturing practices (cGMPs), which are established under FDA regulations. Manufacturers must expend significant time, money and effort to ensure continued compliance, and, in addition to preapproval inspections, the FDA conducts periodic inspections to evaluate continued compliance with cGMP and other requirements. It may be difficult for our manufacturers or us to comply with applicable cGMPs, as interpreted by the FDA, and other FDA regulatory requirements. If we, or our contract manufacturers, fail to comply, then the FDA may not allow us to market products that have been affected by the failure to comply.
If the FDA grants approval, the approval will be limited to those disease states, conditions and patient populations for which the product is deemed by the FDA to be safe and effective, as determined by the FDA’s review of the clinical studies and other data submitted in the NDA. Further, a product may be marketed only in those dosage forms and for those indications approved in the NDA. Certain changes to an approved NDA, including, with certain exceptions, any significant changes to manufacturing, drug product, or labeling, require approval of a supplemental application before the drug may be marketed as changed. Any products that we manufacture or distribute pursuant to FDA approvals are subject to continuing monitoring and regulation by the FDA, including compliance with cGMPs and the reporting of field alerts and adverse reactions and experiences with the drugs. The nature of marketing claims that the FDA will permit us to make in the labeling and advertising of our products will be limited to those specified in FDA approved labeling, and the promotion and advertising of our products will be subject to comprehensive monitoring, review and regulation by the FDA. Drugs whose review was accelerated may carry additional restrictions on marketing activities, including the requirement that all promotional materials are pre-submitted to the FDA. Claims exceeding or deemed
21
Table of Contents
inconsistent with those contained in approved labeling, or deemed to be false or misleading, will be deemed by FDA to constitute a violation of the FDCA. Violations of the FDCA or regulatory requirements at any time during the product development process, approval process, or marketing and sale following approval may result in recalls, warning letters or enforcement actions, including withdrawal of approval, seizure of products, injunctions, fines and/or civil or criminal penalties. In addition, there may be instances in which the U.S. Department of Justice or Office of Inspector General at the U.S. Department of Health & Human Services pursues an enforcement action against our company or our contract manufacturers due to manufacturing or marketing activities or due to alleged kickbacks to health care professionals or false claims to the government if we are able to obtain reimbursement for our product. Any agency enforcement action could have a material adverse effect on our business.
Should we wish to market our products outside the U.S., we must receive marketing authorization from the appropriate foreign regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. At present, companies may apply for foreign marketing authorizations at a national level. However, within the European Union, registration procedures are available to companies wishing to market a product in more than one European Union member state. Typically, if the regulatory authority is satisfied that a company has presented adequate evidence of safety, quality and efficacy, then the regulatory authority will grant a marketing authorization. This foreign regulatory approval process, however, involves risks similar or identical to the risks associated with FDA approval discussed above, and therefore we cannot guarantee that we will be able to obtain the appropriate marketing authorization for any product in any particular country.
Failure to comply with applicable federal, state and foreign laws and regulations would likely have a material adverse effect on our business. In addition, federal, state and foreign laws and regulations regarding the manufacture and sale of new drugs are subject to future changes. We cannot predict the likelihood, nature, effect or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
RESEARCH AND DEVELOPMENT
Company sponsored research and development expenses totaled $20.0 million in 2012, $34.7 million in 2013 and $51.5 million in 2014. Research and development expenses consist primarily of salaries and related personnel costs (including stock-based compensation expense), fees paid to consultants and outside service providers for clinical and laboratory development, manufacturing, including pre-FDA approval inventory, facilities-related and other expenses relating to the design, development, manufacture, testing, and enhancement of our drug candidates and technologies, as well as expenses related to in-licensing of new product candidates. See “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—Overview.”
EMPLOYEES
As of February 17, 2015, we had 155 full and part-time employees. None of our employees are represented by a collective bargaining agreement, and we have never experienced a work stoppage. We consider our relations with our employees to be good.
| ITEM 1A. | RISK FACTORS. |
You should carefully consider the following risks and uncertainties. If any of the following occurs, our business, financial condition and/or operating results could be materially harmed. These factors could cause the trading price of our common stock to decline, and you could lose all or part of your investment.
22
Table of Contents
Risks related to our business and industry
We have a limited operating history and have incurred substantial operating losses since our inception. We expect to continue to incur losses in the future and may never become profitable.
We have a limited operating history. You should consider our prospects in light of the risks and difficulties frequently encountered by early stage companies. In addition, we have incurred substantial operating losses since our inception and expect to continue to incur operating losses for the foreseeable future and may never become profitable. As of December 31, 2014, we had an accumulated deficit of $550.9 million. As we continue our research and development and initial commercial efforts, we will incur increasing losses. We may continue to incur substantial operating losses even after we begin to generate revenues from our drug, Auryxia. Our ability to achieve profitability depends on a number of factors, including our ability to complete our development efforts, obtain additional regulatory approvals for our drug, successfully complete any post approval regulatory obligations and successfully manufacture and commercialize our drug.
We are highly dependent on the commercial success of Auryxia in the U.S. for the foreseeable future; we may be unable to attain profitability and positive cash flow from operations.
In September 2014, the FDA approved Auryxia (formerly known as Zerenex) for the control of serum phosphorus levels in patients with CKD on dialysis. The commercial success of Auryxia will depend on a number of factors, including:
| • | the effectiveness of Auryxia as a treatment for adult patients with CKD on dialysis; |
| • | the size of the treatable patient population; |
| • | the effectiveness of the sales, managed markets and marketing efforts by us and our competitors; |
| • | the adoption of Auryxia by physicians, which depends on whether physicians view it as a safe and effective treatment to lower serum phosphorus levels in patients with CKD on dialysis; |
| • | our ability to both secure and maintain adequate reimbursement for, and optimize patient access to, Auryxia by providing third party payers with a strong value proposition based on the existing burden of illness associated with CKD patients on dialysis and the benefits of Auryxia; |
| • | the occurrence of any side effects, adverse reactions or misuse, or any unfavorable publicity in these areas, associated with Auryxia; |
| • | our ability to obtain and maintain strong intellectual property protection for Auryxia; |
| • | the development or commercialization of competing products or therapies for the control of serum phosphorus levels in patients with CKD on dialysis; and |
| • | our ability to identify reliable suppliers and successfully manufacture Auryxia. |
Our revenues from the commercialization of Auryxia are subject to these and other factors, and therefore may be unpredictable from quarter-to-quarter. Ultimately, we may never generate sufficient revenues from Auryxia to reach or maintain profitability or sustain our anticipated levels of operations.
Auryxia may cause undesirable side effects or have other properties that could limit its commercial potential.
The most commonly reported adverse reactions in the clinical trials that supported the approval of Auryxia in the U.S. were diarrhea (21%), nausea (11%), constipation (8%), vomiting (7%) and cough (6%). Gastrointestinal adverse reactions were the most common reason for discontinuing Auryxia (14%) in clinical trials. If we or others identify previously unknown side effects, if known side effects are more frequent or severe than in the past, if we or others detect unexpected safety signals for Auryxia or any products perceived to be similar to Auryxia, or if any of the foregoing are perceived to have occurred, then in any of these circumstances:
| • | sales of Auryxia may be impaired; |
23
Table of Contents
| • | regulatory approvals for Auryxia may be restricted or withdrawn; |
| • | we may decide to, or be required to, send drug warnings or safety alerts to physicians, pharmacists and hospitals, or may decide to conduct a product recall; |
| • | reformulation of the product, additional nonclinical or clinical studies, changes in labeling or changes to or reapprovals of manufacturing facilities may be required; |
| • | we may be precluded from pursuing additional development opportunities to enhance the clinical profile of Auryxia within its indicated populations, as well as be precluded from studying Auryxia in additional indications and populations and in new formulations; and |
| • | government investigations or lawsuits, including class action suits, may be brought against us. |
Any of the above occurrences would harm or prevent sales of Auryxia, likely increase our expenses and impair our ability to successfully commercialize Auryxia.
Furthermore, as we explore development opportunities to enhance the clinical profile of Auryxia, any clinical trials conducted, if successful, may expand the patient populations treated with Auryxia within or outside of its current indications or patient populations, which could result in the identification of previously unknown side effects, increased frequency or severity of known side effects, or detection of unexpected safety signals. In addition, now that Auryxia will soon be commercially available, it will be used in a wider population and in less rigorously controlled environments than in clinical studies. As a result, regulatory authorities, healthcare practitioners, third party payers or patients may perceive or conclude that the use of Auryxia is associated with serious adverse effects, undermining our commercialization efforts.
We rely on third parties to manufacture and analytically test our drug. If these third parties do not successfully manufacture and test our drug, our business will be harmed.
We have limited experience in manufacturing products for clinical or commercial purposes. We intend to continue, in whole or in part, to use third parties to manufacture and analytically test our drug for use in clinical trials and for future commercial distribution. We may not be able to enter into future contract agreements with these third-parties on terms acceptable to us, if at all.
Our ability to conduct clinical trials, manufacture and commercialize our drug will depend on the ability of such third parties to manufacture our drug on a large scale at a competitive cost and in accordance with current Good Manufacturing Practice regulations, (or cGMPs), and other regulatory requirements, including requirements from federal, state and local environmental and safety regulatory agencies and foreign regulatory requirements, if applicable. Significant scale-up of manufacturing may result in unanticipated technical challenges and will require validation studies that are subject to FDA inspection. Scale-up/technology transfer activities can be complex, and insufficient process knowledge can result in a poorly scaled up process with inadequate process control. A lack of process control can lead to increased deviations during the manufacturing process, out of specification test results, batch rejection and the possible distribution of drug products that do not conform to predetermined specifications. In addition, a variety of factors can affect a contract manufacturer’s qualifications to produce acceptable product, including deficiencies in the contractor’s quality unit, lack of training, a shortage of qualified personnel, capacity constraints and changes in the contractor’s commercial or quality related priorities. Any of these difficulties, if they occur, and are not overcome to the satisfaction of the FDA or other regulatory agency, could lead to significant delays and possibly the termination of the development program for our drug, particularly given that some of the third parties we intend to employ in the manufacturing process are single source providers. These risks become more acute as we scale up for commercial quantities, where a reliable source of active pharmaceutical ingredient (or API) and a qualified contract manufacturer become critical to commercial success. For example, given the large quantity of materials required for Auryxia production and the large quantities of Auryxia that will be required for commercial success, the commercial viability of Auryxia will also depend on adequate supply of starting materials that meet quality, quantity and cost
24
Table of Contents
standards and the ability of our contract manufacturers to produce the API and finished drug product on a commercial scale. Failure to achieve this level of supply can jeopardize and prevent the successful commercialization of the product. Moreover, issues that may arise in our scale-up/technology transfer of Auryxia can lead to significant delays in our development and commercial timelines.
Our third-party manufacturers may not perform as required under the terms of our supply agreement or quality agreement, or may not remain in the contract manufacturing business for the time required by us to successfully manufacture and distribute our drug. In addition, our contract manufacturers will be subject to ongoing periodic and unannounced inspections by the FDA and corresponding foreign governmental agencies to ensure strict compliance with cGMPs, as well as other governmental regulations and corresponding foreign standards. While we periodically audit our contractors for adherence to regulatory requirements, and are ultimately held responsible for their regulatory compliance, we cannot assure you that unforeseen changes at these contractors will not occur that could change their regulatory standing. The same issues apply to contract analytical services which we use for quality, impurity and release testing of our drug. We are required by law to establish adequate oversight and control over raw materials, components and finished products furnished by our third-party manufacturers, which we establish by contract, supplier qualification and periodic audits, but unforeseen circumstances could affect our third-party manufacturers’ compliance with applicable regulations and standards. As we continue to scale up production, we continue to develop analytical tools for Auryxia drug substance and drug product testing. Failure to develop effective analytical tools could result in regulatory or technical delay or could jeopardize our ability to obtain FDA approval. Moreover, even with effective analytical methods available, there is no assurance that we will be able to analyze all the raw materials and qualify all impurities to the satisfaction of the FDA, possibly requiring additional analytical studies, analytical method development, or preclinical studies, which could significantly delay our ability to receive regulatory approvals for our drug. Additionally, changes in the analytical specifications required by the FDA or other regulatory authority, such as United States Pharmacopeial Convention standards, from time to time, could delay our ability to receive regulatory approvals for our drug or our commercial efforts. Switching or engaging multiple third-party contractors to produce our drug substance or drug product may be difficult and time consuming because the number of potential manufacturers may be limited and the process by which multiple manufacturers make the drug substance or drug product must meet established specifications at each manufacturing facility. It may be difficult and time consuming for us to find and engage replacement or multiple manufacturers quickly and on terms acceptable to us, if at all. For Auryxia, the loss of any of our drug substance or drug product manufacturers would result in significant additional costs and delays in our development program. Moreover, if we need to add or change manufacturers after commercialization, the FDA and corresponding foreign regulatory agencies must approve any new manufacturers in advance, which will involve additional inspections to ensure compliance with FDA and foreign regulations and standards.
If we do not establish or maintain manufacturing, drug development and marketing arrangements with third parties, we may be unable to commercialize our products.
We do not possess all of the capabilities to fully commercialize our product on our own. From time to time, we may need to contract with additional third parties, or renew or revise contracts with existing third parties, to:
| • | manufacture our drug; |
| • | assist us in developing, testing and obtaining regulatory approval for and commercializing our compound and technologies; and |
| • | market and distribute our drug. |
We can provide no assurance that we will be able to successfully enter into agreements with such third parties on terms that are acceptable to us, if at all. If we are unable to successfully contract with third parties for these services when needed, or if existing arrangements for these services are terminated, whether or not through our actions, or if such third parties do not fully perform under these arrangements, we may have to delay, scale back or end one or more of our drug development programs or seek to develop or commercialize our product
25
Table of Contents
independently, which could result in significant delays. Furthermore, such failure could result in the termination of license rights to our product. If these manufacturing, development or marketing agreements take the form of a partnership or strategic alliance, such arrangements may provide our collaborators with significant discretion in determining the efforts and resources that they will apply to the development and commercialization of our product. We cannot predict the form or scope that any such collaboration might take, and we may pursue other strategic alternatives if terms or proposed collaborations are not attractive. To the extent that we rely on third parties to research, develop or commercialize our product, we are unable to control whether such product will be scientifically or commercially successful. Additionally, if these third parties fail to perform their obligations under our agreements with them or fail to perform their work in a satisfactory manner, in spite of our efforts to monitor and ensure the quality of such work, we may face delays in achieving the business or regulatory milestones required for commercialization of our current drug and any future drug candidate.
We will incur significant liability if it is determined that we are promoting any “off-label” use of Auryxia.
Physicians are permitted to prescribe drug products for uses that are not described in the product’s labeling and that differ from those approved by the FDA or other applicable regulatory agencies. Such “off-label” uses are common across medical specialties. Although the FDA and other regulatory agencies do not regulate a physician’s choice of treatments, the FDA and other regulatory agencies do restrict communications on the subject of off-label use. Companies are not permitted to promote drugs for off-label uses or promote drugs using marketing claims that are not otherwise consistent with the FDA-approved labeling, including comparative or superiority claims that are not consistent with the FDA-approved labeling or supported by substantial evidence. Accordingly, we may not promote Auryxia in the U.S. for use in any indications other than for the control of serum phosphorus levels in patients with CKD on dialysis and all promotional claims must be consistent with the FDA-approved labeling for Auryxia. The FDA and other regulatory and enforcement authorities actively enforce laws and regulations prohibiting promotion of off-label uses and the promotion of products for which marketing approval has not been obtained as well as the false advertising or misleading promotion of drugs. A company that is found to have improperly promoted off-label uses or to have otherwise engaged in false or misleading promotion of drugs will be subject to significant liability, including civil and administrative remedies as well as criminal sanctions.
Notwithstanding the regulatory restrictions on off-label promotion, the FDA and other regulatory authorities allow companies to engage in truthful, non-misleading, and non-promotional scientific exchange concerning their products in certain circumstances. We intend to engage in medical education activities and communicate with healthcare providers in compliance with all applicable laws, regulatory guidance and industry best practices. Although we believe we have put in place a robust compliance program designed to ensure that all such activities are performed in a legal and compliant manner, Auryxia is our first commercial product, so our implementation of our compliance program in connection with commercialization activities is still relatively new.
If we fail to comply with healthcare regulations, we could face substantial penalties and our business, operations and financial condition could be adversely affected.
As a manufacturer of pharmaceuticals, even though we do not (and do not expect in the future to) control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payers, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We are subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. The regulations include:
| • | federal healthcare program anti-kickback laws, which prohibit, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid; |
26
Table of Contents
| • | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent, and which may apply to entities like us which provide coding and billing advice to customers; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| • | the Federal Food, Drug, and Cosmetic Act, which among other things, strictly regulates drug product marketing, prohibits manufacturers from marketing drug products for off-label use and regulates the distribution of drug samples; |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts; |
| • | the federal Foreign Corrupt Practices Act which prohibits corporations and individuals from paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity; and |
| • | the federal Physician Payments Sunshine Act, which was passed as part of the Patient Protection and Affordable Care Act of 2010, and similar state laws in certain states, that require pharmaceutical and medical device companies to monitor and report certain payments and transfers of value made to physicians and teaching hospitals. |
If our operations are found to be in violation of any of the laws described above or any other laws, rules or regulations that apply to us, we will be subject to penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results.
In preparation for the commercial launch of Auryxia, we assembled an experienced compliance team who compiled a program based on industry best practices that is designed to ensure that our commercialization of Auryxia complies with all applicable laws, regulations and industry standards. We also hire, manage and incentivize our employees around a culture of compliance, trust, respect and ownership. Because our program is relatively new and the requirements in this area are constantly evolving, we cannot be certain that our program will eliminate all areas of potential exposure. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business, as well as damage our business or reputation. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security, fraud and reporting laws may prove costly.
If our competitors develop and market products that are less expensive, more effective or safer than our drug product, or our drug product does not achieve market acceptance vis-à-vis existing treatments, our commercial opportunities may be reduced or eliminated.
The pharmaceutical industry is highly competitive. Our competitors include pharmaceutical companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies that are active in different but related fields represent substantial competition for us. Many of our competitors have significantly greater capital resources, larger research and development staffs and facilities and
27
Table of Contents
greater experience in drug development, regulation, manufacturing and marketing than we do. These organizations also compete with us to recruit qualified personnel, attract partners for joint ventures or other collaborations, and license technologies that are competitive with ours. As a result, our competitors may be able to more easily develop technologies and products that could render our drug product obsolete or noncompetitive. To compete successfully in this industry we must identify novel and unique drugs or methods of treatment and then complete the development of those drugs as treatments in advance of our competitors.
Auryxia will compete in the U.S. with other FDA approved phosphate binders such as Renagel® (sevelamer hydrochloride) and Renvela® (sevelamer carbonate), both marketed by Genzyme Corporation (a wholly-owned subsidiary of Sanofi), or Genzyme, PhosLo® (calcium acetate), marketed by Fresenius Medical Care, Fosrenol® (lanthanum carbonate), marketed by Shire Pharmaceuticals Group plc, and Velphoro® (sucroferric oxyhydroxide), marketed by Fresenius Medical Care North America, as well as over-the-counter calcium carbonate products such as TUMS® and metal-based options such as aluminum and magnesium. Our strategy to compete against these existing treatments depends in part on physicians and patients accepting that Auryxia is differentiated in the marketplace versus these FDA approved phosphate binders. In addition, we may have to compete against existing treatments on price, which becomes more challenging as generic versions of these existing treatments come to market. For example, an authorized generic of Renvela® was launched in the U.S. in April 2014 by Impax Laboratories, Inc., or Impax, under a settlement agreement with Genzyme whereby Genzyme agreed to grant Impax a license to sell a one-time allotment of a specified number of bottles of an authorized generic version of Renvela® tablets. Impax is also pursuing approval of its pending Abbreviated New Drug Application, or ANDA, for generic Renvela® with the FDA. In addition, a generic formulation of PhosLo® manufactured by Roxane Laboratories, Inc. was launched in the U.S. in October 2008. In addition, upon the expiration of its core patents, generic formulations of Fosrenol® may be launched. These generic formulations could have a further material effect on the pricing of phosphate binders.
Furthermore, our commercial opportunities may be reduced or eliminated if our competitors develop and market products that are less expensive, more effective or safer than our drug product. Other companies have drug candidates in various stages of pre-clinical or clinical development to treat diseases for which we are also seeking to acquire and develop drug products. Even if we are successful in developing effective drugs, our product(s) may not compete successfully with products produced by our competitors.
If we lose our key personnel or are unable to attract and retain additional personnel, our operations could be disrupted and our business could be harmed.
As of February 17, 2015, we had 155 full and part-time employees. To successfully develop and commercialize our drug and any drug candidates we may in-license or acquire, we must be able to attract and retain highly skilled personnel. Our limited resources may hinder our efforts to attract and retain highly skilled personnel. In addition, if we lose the services of our current personnel our ability to continue to execute on our business plan could be materially impaired. Although we have employment agreements with Ron Bentsur and Greg Madison, these agreements do not prevent them from terminating their employment with us.
In January 2015, we announced the transitioning of the role of Chief Executive Officer from Ron Bentsur to Greg Madison. Mr. Madison joined Keryx in February 2014 as Executive Vice President and Chief Operating Officer to transition Keryx from a development-stage organization into a fully integrated commercial entity, bringing to Keryx a wealth of relevant expertise in both the phosphate binder and iron deficiency anemia markets. Mr. Madison has been appointed President of Keryx and will work with Mr. Bentsur to ensure a successful leadership transition by the end of May, when Mr. Bentsur’s contract expires.
In February 2015, we announced a planned consolidation of our finance and accounting function into our Boston office and that our Chief Financial Officer, James Oliviero, will be leaving Keryx by October 2015. Mr. Oliviero has been with Keryx for twelve years and has served as the Chief Financial Officer since 2009. We
28
Table of Contents
have commenced a search for a new Chief Financial Officer who will be based in our Boston office. Mr. Oliviero will continue to manage our finance and accounting team during the remainder of his tenure and will assist in the transition of his duties to the new Chief Financial Officer.
Risks associated with our product development efforts
If we do not receive regulatory approvals to market our drug product in a timely manner, or at all, our business will be materially harmed and our stock price may be adversely affected.
We are developing Auryxia, an oral, ferric iron-based compound that has the capacity to bind to phosphate and form non-absorbable complexes. In May 2011, we announced positive Scientific Advice from the EMA for the development of Auryxia for the management and control of serum phosphorus in CKD patients undergoing dialysis, and in NDD-CKD patients. The Scientific Advice from the EMA indicates that our successful Phase 3 program in dialysis in the U.S., in conjunction with safety data generated from other clinical studies with Auryxia, will be considered sufficient to support a MAA to the EMA for the indication in CKD patients on dialysis. The Scientific Advice also provided us with a regulatory path forward in the NDD-CKD setting in Europe. As a result, we believe that since our Phase 3 program in dialysis, and Phase 2 study in NDD-CKD, in the U.S. were successful, we will not need to conduct any additional clinical trials to assess the safety or efficacy of Auryxia in order to obtain European approval in CKD, including the dialysis and NDD-CKD settings. Accordingly, in March 2014, we submitted a MAA with the EMA for both dialysis and NDD-CKD, which was validated by the EMA in March 2014. Scientific Advice is legally non-binding and is based on the current scientific knowledge, which may be subject to future changes. Many companies which have been provided with positive Scientific Advice by the EMA have ultimately failed to obtain approval of an MAA for their drugs. Additionally, even if the primary endpoint in a Phase 3, or other pivotal, clinical trial is achieved, the Scientific Advice does not guarantee approval. The EMA may raise issues of safety, study conduct, bias, deviation from the protocol, statistical power and analyses, patient demographics, patient completion rates, changes in scientific or medical parameters or internal inconsistencies in the data prior to making its final decision, which may delay or prevent EMA approval of Auryxia.
Obtaining approval of an MAA by the EMA is highly uncertain and like many product candidates, we may fail to obtain the approval even though our MAA has been validated by the EMA. The MAA review processes are extensive, lengthy, expensive and uncertain, and the EMA may delay, limit or deny approval of Auryxia for many reasons, including:
| • | we may not be able to demonstrate to the satisfaction of the regulatory authority that Auryxia is safe and effective for any indication; |
| • | the data arising from the clinical trials, including the Phase 3 results for dialysis patients and our Phase 2 results for NDD-CKD, the development program or the MAA for Auryxia may not be satisfactory to the EMA; |
| • | the EMA may disagree with the number, design, size, conduct or implementation of our clinical trials or conclude that the data fails to meet statistical or clinical significance; |
| • | the EMA may not find the data from preclinical and clinical studies sufficient to demonstrate that Auryxia’s clinical and other benefits outweigh its safety risks; |
| • | the EMA may disagree with our interpretation of data from preclinical studies or clinical trials, and may reject conclusions from preclinical studies or clinical trials, or determine that primary or secondary endpoints from clinical trials were not met, or reject safety conclusions from such studies; |
| • | the EMA may not accept data generated at one or more of our clinical trial sites; |
| • | the EMA may determine that we did not properly oversee our clinical trials or follow the regulatory authority’s advice or recommendations in conducting our clinical trials; |
29
Table of Contents
| • | an advisory committee, if convened by the EMA, may recommend against approval of our application or may recommend that the respective regulatory authority require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions, or even if an advisory committee, if convened, makes a favorable recommendation, the respective regulatory authority may still not approve Auryxia; |
| • | data and analyses submitted to the EMA in response to questions raised during the review processes may not be satisfactory to the regulatory authority, and this may lead to significant delays in the approval of Auryxia or to the rejection of the Auryxia MAA; and |
| • | the EMA may identify deficiencies in the chemistry, manufacturing and controls, or CMC, sections of our MAA, our manufacturing processes, facilities or analytical methods or those of our third party contract manufacturers, and this may lead to significant delays in the approval of Auryxia or to the rejection of the Auryxia MAA. |
Additionally, our March 2014 MAA submission to the EMA was our first MAA filing in Europe. During the regulatory review process, regulatory agencies will typically ask questions of drug sponsors, such as the Day 120 questions which we received from the EMA for which we submitted our responses in January 2015. We will endeavor to answer all such questions in a timely and complete fashion; however, we cannot assure you that our answers to such questions will be complete and to the satisfaction of the regulatory agencies. If certain questions asked have not been fully and satisfactorily answered by us, approval of our filings may be delayed, or the filings may be rejected.
Accordingly, we may not receive the regulatory approvals needed to market Auryxia. Any failure or delay in completion of the development program or the EMA review processes would delay or foreclose commercialization of Auryxia and severely harm our business and financial condition.
If we are unable to successfully complete our clinical trial programs, or if such clinical trials take longer to complete than we project, our ability to execute our current business strategy will be adversely affected.
Whether or not and how quickly we complete our clinical trials is dependent in part upon the rate at which we are able to engage clinical trial sites and, thereafter, the rate of enrollment of patients, and the rate we collect, clean, lock and analyze the clinical trial database. Patient enrollment is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the study, the existence of competitive clinical trials, and whether existing or new drugs are approved for the indication we are studying. We are aware that other companies are currently conducting or planning clinical trials that seek to enroll patients with the same disease that we are studying. If we experience delays in identifying and contracting with sites and/or in patient enrollment in our clinical trial programs, we may incur additional costs and delays in our development programs, and may not be able to complete our clinical trials in a cost-effective or timely manner or at all. In addition, conducting multi-national studies adds another level of complexity and risk. As a result, we may be subject to events affecting countries outside the U.S.
Negative or inconclusive results from the clinical trials we conduct, such as the ongoing Phase 3 study of Auryxia for the treatment of iron deficiency anemia in patients with NDD-CKD, or unanticipated adverse medical events could cause us to have to repeat or terminate the clinical trials. For example, in May 2012, we abandoned our development efforts and terminated our license for KRX-0401 (perifosine) following negative results from the Phase 3 trial. We may also opt to change the delivery method, formulation or dosage which could affect efficacy results for the drug. Accordingly, we may not be able to complete our current or future clinical trials within an acceptable time frame, if at all.
30
Table of Contents
Pre-clinical testing and clinical development are long, expensive and uncertain processes. If our drug does not receive the necessary regulatory approvals, we will be unable to commercialize our drug, Auryxia in Europe.
We have not received, and may never receive, regulatory approval for the commercial sale of Auryxia by the EMA. We may need to conduct significant additional research and human testing before we receive product approval with the EMA or with regulatory authorities of other countries. Pre-clinical testing and clinical development are long, expensive and uncertain processes. Satisfaction of regulatory requirements typically depends on the nature, complexity and novelty of the product. It requires the expenditure of substantial resources. Data obtained from pre-clinical and clinical tests can be interpreted in different ways, which could delay, limit or prevent regulatory approval. The EMA or a regulatory authority of another country, as applicable, may pose additional questions or request further toxicological, drug-drug interaction, pre-clinical or clinical data or substantiation. For example, while Auryxia is a Generally Recognized as Safe, or GRAS, substance in the U.S., and the EMA has not requested that we conduct a two-year carcinogenicity study in animals, there is no assurance that the EMA or some other regulatory authority will not ask us to conduct such a study in order to obtain regulatory approval. In addition, the EMA has not requested us to conduct reproductive toxicity, genotoxicity and single-dose toxicity studies and we are referencing such studies from the published scientific literature in our regulatory submissions. However, we can provide no assurance that the EMA will not ask us to conduct additional studies. We received Day 120 questions from the EMA on our MAA. We provided our responses to the EMA’s Day 120 questions in January 2015; however, we cannot assure you that we have answered these questions to the EMA’s satisfaction or that the EMA will not have additional questions as part of the MAA review. Consequently, it may take us many years to complete the testing of our drug and failure can occur at any stage of this process. Negative, inconclusive, or insufficient results or medical events during a pre-clinical or clinical trial could cause us to delay or terminate our development efforts. Furthermore, interim results of preclinical or clinical studies do not necessarily predict their final results, and acceptable results in early studies might not be obtained in later studies.
Safety signals detected during clinical studies and pre-clinical animal studies, such as the gastrointestinal bleeding and liver toxicities that have been seen in some high-dose Auryxia canine studies, may require us to perform additional safety studies or analyses, which could delay the development of the drug or lead to a decision to discontinue development of the drug. We have submitted to the EMA data from our short-term and long-term rat and canine pre-clinical studies for Auryxia. While the EMA has reviewed the data from these studies and we have conducted our Phase 3 clinical program for CKD patients on dialysis, and Phase 2 study in NDD CKD patients, we can provide no assurance that the EMA will not raise any safety concerns in the future from these studies. Drug candidates in the later stages of clinical development may fail to show the desired traits of safety and efficacy despite positive results in earlier clinical testing. Moreover, the risk remains that the safety and efficacy data from our pivotal Phase 3 program for dialysis dependent CKD patients may be insufficiently persuasive for the approval of the drug, or may raise safety concerns that may prevent approval of the drug, for the indication sought. The risk also remains that a clinical program conducted by one of our partners may raise efficacy or safety concerns that may prevent approval of the drug. In addition, qualitative, quantitative and statistical interpretation of any of the prior pre-clinical and clinical safety and efficacy data of our drug may be viewed as flawed by the EMA or any other regulatory agency. In addition, there can be no assurance that safety and/or efficacy concerns from the prior data were not overlooked or misinterpreted by us or our consultants, which in subsequent, larger studies might appear and prevent approval of such drug candidate. In addition, top-line results reported on completed clinical trials, such as those from our long-term open label extension, or OLE, study for Auryxia in dialysis-dependent CKD patients, are based on a preliminary analysis of then available data (both safety and efficacy) and there is the risk that such findings and conclusions could change following a more comprehensive review of the data by a regulatory authority. For example, in January 2013, we announced successful top-line results from our long-term Phase 3 study of Auryxia for the treatment of elevated serum phosphorus levels, or hyperphosphatemia, in patients with ESRD on dialysis. Updated results from the study were presented in June 2013 at the World Congress of Nephrology. We can provide no assurance that our findings and conclusions from our long-term Phase 3 study of Auryxia or from our long-term OLE study for Auryxia in dialysis-dependent CKD patients will not change following a more comprehensive review of the data by a regulatory authority.
31
Table of Contents
Clinical trials have a high risk of failure. A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after achieving what appeared to be promising results in earlier trials. We experienced such a setback with our Phase 3 KRX-0401 (perifosine) results in April 2012, and we can provide no assurance that we will not experience such setbacks with Auryxia or any other drug candidate we develop. If we experience delays in the testing or approval process for our existing drug or if we need to perform more or larger clinical trials than originally planned, our financial results and the commercial prospects for our drug may be materially impaired. In addition, we have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approval. Accordingly, we may encounter unforeseen problems and delays in the approval process. Although we engage, from time to time, clinical research organizations with experience in conducting regulatory trials, errors in the conduct, monitoring, data capture and analysis, and/or auditing could potentially invalidate the results.
Because all of our proprietary technologies are licensed or sublicensed to us by third parties, termination of these license rights would prevent us from developing and commercializing Auryxia.
We do not own our drug, Auryxia. We have licensed and sublicensed the rights, patent or otherwise, to Auryxia from a third party, Panion & BF Biotech, Inc., or Panion, who in turn licenses certain rights to Auryxia from one of the inventors of Auryxia. The license agreement with Panion requires us to meet development milestones and imposes development and commercialization due diligence requirements on us. In addition, under the agreement, we must pay royalties based on a mid-single digit percentage of net sales of product resulting from the licensed technologies (including Auryxia) and pay the patent filing, prosecution and maintenance costs related to the license. If we do not meet our obligations in a timely manner or if we otherwise breach the terms of our license agreement (including upon certain insolvency events), Panion could terminate the agreement, and we would lose the rights to Auryxia. In addition, if Panion breaches its agreement with the inventor from whom it licenses rights to Auryxia, Panion could lose its license, which could impair or delay our ability to develop and commercialize Auryxia. From time to time, we may have disagreements with our licensors or collaborators, or they and/or we may have disagreements with the original inventors, regarding the terms of our agreements or ownership of proprietary rights, which could lead to delays in the research, development and commercialization of our current drug and any future drug candidate, could require or result in litigation or arbitration, which would be time-consuming and expensive, or could lead to the termination of a license, or force us to negotiate a revised or new license agreement on terms less favorable than the original. In addition, in the event that the owners and/or licensors of the rights we license were to enter into bankruptcy or similar proceedings, we could potentially lose our rights to our drug or drug candidates or our rights could otherwise be adversely affected, which could prevent us from developing or commercializing our drugs. Finally, our rights to develop and commercialize Auryxia, whether ourselves or with third parties, are subject to and limited by the terms and conditions of our licenses to Auryxia and the licenses and sublicenses we grant to others.
Our reliance on third parties, such as clinical research organizations, or CROs, may result in delays in completing, or a failure to complete, clinical trials if such CROs fail to perform under our agreements with them.
In the course of product development, we engage CROs and other vendors to conduct and manage clinical studies and to assist us in guiding our products through the FDA review and approval process. If the CROs or applicable vendors fail to perform their obligations under our agreements with them or fail to perform clinical trials in a satisfactory or timely manner, we may face significant delays in completing our clinical trials, submitting our regulatory filings, or approval, as well as the commercialization of one or more drug candidates. Furthermore, any loss or delay in obtaining contracts with such entities may also delay the completion of our clinical trials and the market approval of drug candidate(s).
32
Table of Contents
Other risks related to our business.
Any acquisitions we make may require a significant amount of our available cash and may not be scientifically or commercially successful.
As part of our business strategy, we may effect acquisitions to obtain additional businesses, products, technologies, capabilities and personnel. If we make one or more significant acquisitions in which the consideration includes cash, we may be required to use a substantial portion of our available cash.
Acquisitions involve a number of operational risks, including:
| • | difficulty and expense of assimilating the operations, technology and personnel of the acquired business; |
| • | our inability to retain the management, key personnel and other employees of the acquired business; |
| • | our inability to maintain the acquired company’s relationship with key third parties, such as alliance partners; |
| • | exposure to legal claims for activities of the acquired business prior to the acquisition; |
| • | the diversion of our management’s attention from our core business; and |
| • | the potential impairment of goodwill and write-off of in-process research and development costs, adversely affecting our reported results of operations. |
The status of reimbursement from third-party payors for newly approved health care drugs is uncertain and failure to obtain adequate reimbursement could limit our ability to generate revenue.
Our ability to commercialize pharmaceutical products may depend, in part, on the extent to which reimbursement for the products will be available from:
| • | government and health administration authorities; |
| • | private health insurers; |
| • | managed care programs; and |
| • | other third-party payors. |
Significant uncertainty exists as to the coverage and reimbursement status of newly approved health care products, as well as the timing of coverage and reimbursement decisions by third-party payors. Third-party payors, including Medicare and Medicaid, are challenging the prices charged for medical products and services. Government and other third-party payors increasingly are attempting to contain health care costs by limiting both coverage and the level of reimbursement for new drugs and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has not granted labeling approval. In 2003, Congress passed the Medicare Prescription Drug, Improvement and Modernization Act of 2003, which for the first time established prescription drug coverage for Medicare beneficiaries, under Medicare Part D. Under this program, beneficiaries purchase insurance coverage from private insurance companies to cover the cost of their prescription drugs. However, third-party insurance coverage may not be available to patients for our product. If government and other third-party payors do not provide adequate coverage and reimbursement levels for our product, its market acceptance may be significantly reduced.
Health care reform measures could adversely affect our business.
The business prospects and financial condition of pharmaceutical and biotechnology companies are affected by the efforts of governmental and third-party payors to contain or reduce the costs of health care. In the U.S. and in foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and
33
Table of Contents
regulatory proposals aimed at changing the health care system, such as proposals relating to the pricing of healthcare products and services in the U.S. or internationally, the reimportation of drugs into the U.S. from other countries (where they are then sold at a lower price), and the amount of reimbursement available from governmental agencies or other third party payors. For example, drug manufacturers are required to have a national rebate agreement with the Department of Health and Human Services, or HHS, in order to obtain state Medicaid coverage, which requires manufacturers to pay a rebate on drugs dispensed to Medicaid patients. On January 27, 2012, the Centers for Medicare and Medicaid Services, or CMS, issued a proposed regulation covering the calculation of Average Manufacturer Price, or AMP, which is the key variable in the calculation of these rebates.
Furthermore, in the U.S., health care reform legislation titled the Patient Protection and Affordable Care Act, or PPACA, was signed into law in March 2010. The impact of this legislation on our business is inherently difficult to predict as many of the details regarding the implementation of this legislation have not been determined. In a decision issued on June 29, 2012, the United States Supreme Court upheld the majority of PPACA. The Court’s decision allows implementation of key provisions impacting drug and device manufacturers to go forward. This includes PPACA changes to the Medicare Part D Program (including closing the “donut hole”), Medicaid Drug Rebate Program (including the definition of AMP), and expansion of the 340B Drug Discount Program. The decision also allows the FDA and CMS to continue with implementation efforts, including related to the Biologics Price Competition and Innovation Act and the Physician Payments Sunshine Act, both of which were enacted as part of the PPACA. Regulations to implement PPACA could result in a decrease in our stock price or limit our ability to raise capital or to obtain strategic partnerships or licenses. Government-financed comparative efficacy research could also result in new practice guidelines, labeling or reimbursement policies that discourages use of our product.
For example, in July 2010, CMS released its final rule to implement a bundled prospective payment system for end-stage renal disease facilities as required by the Medicare Improvements for Patients and Providers Act, or MIPPA. The final rule delayed the inclusion of oral medications without intravenous equivalents, such as phosphate binders, in the bundle until January 1, 2014; however, on January 3, 2013, the United States Congress passed legislation known as the American Taxpayer Relief Act of 2012, which, among other things, delayed by two years the implementation of oral-only end-stage renal disease related drugs, including phosphate binders, in the bundled ESRD prospective payment system, until January 1, 2016. In April 2014, the United States Congress passed legislation known as Protecting Access to Medicare Act of 2014, which, among other things, delays by eight years the implementation of oral-only ESRD related drugs, including phosphate binders, in the bundled ESRD prospective payment system, until January 1, 2025. If phosphate binders are included in the bundle beginning in 2025, or earlier, separate Medicare reimbursement will no longer be available for phosphate binders, as it is today under Medicare Part D. While it is too early to project the impact bundling may have on the phosphate binder industry, the impact could potentially cause dramatic price reductions for phosphate binders, which could significantly reduce the commercial potential of Auryxia.
On September 27, 2007, the Food and Drug Administration Amendments Act of 2007 was enacted, giving the FDA enhanced post-market authority, including the authority to require post-marketing studies and post-marketing clinical trials related to serious risks, labeling changes based on new safety information, and compliance with risk evaluation and mitigation strategies approved by the FDA. The FDA’s exercise of this authority may result in delays or increased costs during the period of product development, clinical trials and regulatory review and approval, which may also increase costs related to complying with new post-approval regulatory requirements, and increase potential FDA restrictions on the sale or distribution of approved products. On July 9, 2012, the Food and Drug Administration Safety and Innovation Act was enacted to, among other things, renew the drug user fee program, expand the FDA’s inspection records access and require manufacturers to establish appropriate oversight and controls over their suppliers and the supply chain, including raw material suppliers and contract manufacturers, as a part of cGMP compliance. On November 27, 2013, the Drug Quality and Security Act, which includes the Drug Supply Chain Security Act, was signed into law to, among other things, build an electronic, interoperable system to identify and trace certain prescription drugs as they are
34
Table of Contents
distributed in the United States. Requirements for the tracing of products through the pharmaceutical distribution supply chain took effect on January 1, 2015 for manufacturers and building internal systems to ensure compliance with this law will require dedication of resources. In addition, this law requires engaging in transactions only with authorized trading partners and could limit our pool of available trading partners.
We face product liability risks and may not be able to obtain adequate insurance.
The use of our drug or future drug candidates in clinical trials, and the future sale of any approved drug and new technology, exposes us to liability claims. Although we are not aware of any historical or anticipated product liability claims against us, if we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to cease clinical trials of our drug product or limit commercialization of any approved product.
We have expanded our insurance coverage to include the commercial sale of Auryxia; however, insurance coverage is becoming increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost. We also may not be able to obtain additional insurance coverage that will be adequate to cover product liability risks that may arise. Regardless of merit or eventual outcome, product liability claims may result in:
| • | decreased demand for a product; |
| • | injury to our reputation; |
| • | our inability to continue to develop a drug candidate; |
| • | withdrawal of clinical trial volunteers; and |
| • | loss of revenues. |
Consequently, a product liability claim or product recall may result in losses that could be material to our business.
Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, our proprietary business information and that of our suppliers and business partners, as well as personally identifiable information of Auryxia patients, clinical trial participants and employees. We also have outsourced elements of our information technology structure, and as a result, we are managing independent vendor relationships with third parties who may or could have access to our confidential information. Similarly, our business partners and other third party providers possess certain of our sensitive data. The secure maintenance of this information is critical to our operations and business strategy. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. We, our partners, vendors and other third party providers could be susceptible to third party attacks on our, and their, information security systems, which attacks are of ever increasing levels of sophistication and are made by groups and individuals with a wide range of motives and expertise, including criminal groups. Any such breach could compromise our, and their, networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, disrupt our operations, and damage our reputation, any of which could adversely affect our business.
Risks related to our financial condition
Our existing capital resources may not be adequate to finance our operating cash requirements for the length of time that we have estimated.
We currently expect that our existing capital resources combined with future anticipated cash flows will be sufficient to execute our business plan. The actual amount of cash that we will need to operate is subject to many
35
Table of Contents
factors, including, but not limited to, the timing and expenditures associated with the build-up of inventory and capacity expansion, the timing and expenditures associated with the regulatory review process for our EU MAA filing, the timing and expenditures associated with commercial activities related to Auryxia, and the timing, design and conduct of clinical trials for Auryxia. As a result of these factors, we may need to seek additional financings to provide the cash necessary to execute our current operations, including beyond the initial commercialization of Auryxia, and to develop any drug candidates we may in-license or acquire.
Our forecast of the period of time through which our existing capital resources will be adequate to support our current operations is a forward-looking statement that involves risks and uncertainties. The actual amount of funds we will need to operate is subject to many factors, some of which are beyond our control. These factors include, but are not limited to, the following:
| • | the timing and expenditures associated with the build-up of inventory and capacity expansion; |
| • | the timing and expenditures associated with the regulatory review process for our EU MAA filing; |
| • | the timing and expenditures associated with commercial activities related to Auryxia; |
| • | the timing, design and conduct of, and results from, clinical trials for Auryxia; |
| • | the timing of expenses associated with manufacturing and product development of Auryxia and those proprietary drug candidates that may be in-licensed, partnered or acquired; |
| • | the timing of the in-licensing, partnering and acquisition of new product opportunities; |
| • | the progress of the development efforts of parties with whom we have entered, or may enter, into research and development agreements; |
| • | our ability to achieve our milestones under our licensing arrangement; |
| • | the timing and expenses associated with capital expenditures to expand our manufacturing capabilities; |
| • | the timing and expenses associated with building our own commercial infrastructure to manufacture, market and sell our drug and those that may be in-licensed, partnered or acquired; |
| • | the costs involved in prosecuting and enforcing patent claims and other intellectual property rights or defending against claims of infringement initiated by third parties in respect of their intellectual property rights; and |
| • | the timing and magnitude of cash received from product sales. |
If our cash is insufficient to meet future operating requirements, we will have to raise additional funds. If we are unable to obtain additional funds on terms favorable to us or at all, we may be required to cease or reduce our operating activities or sell or license to third parties some or all of our intellectual property. If we raise additional funds by selling additional shares of our capital stock, the ownership interests of our stockholders will be diluted. If we need to raise additional funds through the sale or license of our intellectual property, we may be unable to do so on terms favorable to us, if at all.
Risks related to our intellectual property and third-party contracts
If we are unable to adequately protect our intellectual property, third parties may be able to use our intellectual property, which could adversely affect our ability to compete in the market.
Our commercial success will depend in part on our ability, and the ability of our licensors, to obtain and maintain patent protection on our drug product and technologies, and to successfully defend these patents against third-party challenges. The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date. Accordingly, the patents we use may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products. Furthermore,
36
Table of Contents
others may independently develop similar or alternative drug products or technologies or design around our patented drug product and technologies which may have an adverse effect on our business. If our competitors prepare and file patent applications in the U.S. that claim technology also claimed by us, we may have to participate in interference or derivation proceedings in front of the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial cost, even if the eventual outcome is favorable to us. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that any related patent may expire prior to, or remain in existence for only a short period following, commercialization, thus reducing any advantage of the patent. The patents we use may be challenged or invalidated or may fail to provide us with any competitive advantage. As many of the patents we use are licensed or sublicensed from third parties, we may not be able to enforce such licensed patents against third party infringers without the cooperation of the patent owner and the licensor, which may not be forthcoming. In addition, we may not be successful or timely in obtaining any patents for which we submit applications.
Additionally, the laws of foreign countries may not protect our intellectual property rights to the same extent as do the laws of the U.S. In addition, in jurisdictions outside the U.S. where we own or license patent rights, we may be unable to prevent unlicensed parties from selling or importing products or technologies derived elsewhere using our proprietary technology.
We also rely on trade secrets and know-how to protect our intellectual property where we believe patent protection is not appropriate or obtainable. Trade secrets are difficult to protect. While we require our employees, licensees, collaborators and consultants to enter into confidentiality agreements, this may not be sufficient to adequately protect our trade secrets or other proprietary information. In addition, we share ownership and publication rights to data relating to our drug product and technologies with our research collaborators and scientific advisors. If we cannot maintain the confidentiality of this information, our ability to receive patent protection or protect our trade secrets or other proprietary information will be at risk.
The intellectual property that we own or have licensed relating to our drug, Auryxia, is limited, which could adversely affect our ability to compete in the market and adversely affect the value of Auryxia.
The patent rights that we own or have licensed relating to Auryxia are limited in ways that may affect our ability to exclude third parties from competing against us. In particular:
| • | Composition of matter patents can provide protection for pharmaceutical products to the extent that the specifically covered compositions are key, non-interchangeable components of the pharmaceutical product. The first composition of matter and method patent relating to Auryxia in the United States (U.S. Patent No. 5,753,706) expires in February 2017. We licensed additional composition of matter and method of use patents expiring in 2024 with independent claims covering forms of ferric citrate (the active pharmaceutical ingredient, or API, of Auryxia), pharmaceutical compositions that include the API, pharmaceutical compositions having ferric citrate in an amount effective to reduce serum phosphate levels, and methods of treating hyperphosphatemia and metabolic acidosis. |
| • | Our method of use patents, including U.S. Patent Nos. 7,767,851, 8,299,298 and 8,338,642 and (which expire in 2024), and U.S. Patent No. 8,093,423 (which expires in 2026) only protect the product when used or sold for the claimed methods. However, these types of patents do not limit a competitor from making and marketing a product that is identical to our product that is labeled for an indication that is outside of our patented methods. |
| • | We have filed applications under the Patent Term Extension provisions of 35 U.S.C. § 156 on the above mentioned patents for delays caused by FDA regulatory review. If granted we can utilize the patent term extension on one of these patents, however, we cannot assure you that we can obtain any extension of the term of these patents. If obtained, the maximum term of extension available under 35 U.S.C. § 156 would extend the term of the chosen patent by no more than five years. Upon expiration of these patents, competitors who obtain the requisite regulatory approval may potentially offer products with the same composition and/or method of use as our product, so long as the competitors do not infringe any other patents that we may hold. |
37
Table of Contents
| • | Our pending patent applications may not issue as patents and may not issue in all countries in which we develop, manufacture or potentially sell our product(s) or in countries where others develop, manufacture and potentially sell products using our technologies. Moreover, our pending patent applications, if issued as patents, may not provide additional protection for our product. |
Obtaining proof of direct infringement by a competitor for a method of use patent requires us to demonstrate that the competitors make and market a product for the patented use(s). Alternatively we can prove that our competitors induce or contribute others in engaging in direct infringement. Proving that a competitor contributes to, or induces, infringement of a patented method by another has additional proof requirements. For example, proving inducement of infringement requires proof of intent by the competitor. If we are required to defend ourselves against claims or to protect our own proprietary rights against others, it could result in substantial costs to us and the distraction of our management. An adverse ruling in any litigation or administrative proceeding could prevent us from marketing and selling Auryxia, increase the risk that a generic version of Auryxia could enter the market to compete with Auryxia, limit our development and commercialization of Auryxia, or otherwise harm our competitive position and result in additional significant costs. In addition, any successful claim of infringement asserted against us could subject us to monetary damages or injunction, which could prevent us from making or selling Auryxia. We also may be required to obtain licenses to use the relevant technology. Such licenses may not be available on commercially reasonable terms, if at all.
Moreover, physicians may prescribe a competitive identical product for indications other than the one for which the product has been approved, or off-label indications, that are covered by the applicable patents. Although such off-label prescriptions may directly infringe or contribute to or induce infringement of method of use patents, such infringement is difficult to prevent.
In addition, any limitations of our patent protection described above may adversely affect the value of our drug product and may inhibit our ability to obtain a corporate partner at terms acceptable to us, if at all.
In addition to patent protection, we may utilize pediatric exclusivity or other provisions of the Food, Drug and Cosmetic Act of 1938, as amended, or FDCA, such as new chemical entity exclusivity, or NCE, or new formulation exclusivity, to provide market exclusivity for a drug candidate.
In the U.S., the FDA has the authority to grant additional data protection for approved drugs where the sponsor conducts specified testing in pediatric or adolescent populations. If granted, this pediatric exclusivity may provide an additional six months which are added to the term of data protection as well as to the term of a relevant patent, to the extent these protections have not already expired.
The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a New Chemical Entity, or NCE. A drug is an NCE if the FDA has not previously approved any other new drug containing the same active moiety, which consists of the molecule(s) or ion(s) responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application (for example, for new indications, dosages, or strengths of an existing drug). This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or tentative approval of a full ANDA; however, an applicant submitting a full ANDA would be required to conduct sufficient studies to demonstrate that their generic product is bioequivalent to Auryxia.
38
Table of Contents
We may also seek to utilize market exclusivities in other territories, such as in the EU.
We cannot assure that Auryxia or any drug candidates we may acquire or in-license, will obtain such pediatric exclusivity, NCE exclusivity or any other market exclusivity in the U.S., EU or any other territory, or that we will be the first to receive the respective regulatory approval for such drugs so as to be eligible for any market exclusivity protection. We also cannot assure that Auryxia or any drug candidates we may acquire or in-license will obtain patent term extension.
Litigation or third-party claims could require us to spend substantial time and money defending such claims and adversely affect our ability to develop and commercialize our product.
We may be forced to initiate litigation to enforce our contractual and intellectual property rights, or we may be sued by third parties asserting claims based on contract, tort or intellectual property infringement. In addition, third parties may have or may obtain patents in the future and claim that Auryxia or any other technologies infringe their patents. If we are required to defend against suits brought by third parties, or if we sue third parties to protect our rights, we may be required to pay substantial litigation costs, and our management’s attention may be diverted from operating our business. In addition, any legal action against our licensor or us that seeks damages or an injunction of our commercial activities relating to Auryxia or other technologies could subject us to monetary liability, a temporary or permanent injunction preventing the development, marketing and sale of Auryxia or such technologies, and/or require our licensor or us to obtain a license to continue to use Auryxia or other technologies. We cannot predict whether our licensor or we would prevail in any of these types of actions or that any required license would be made available on commercially acceptable terms, if at all.
Risks Related to Our Common Stock
Future sales or other issuances of our common stock could depress the market for our common stock.
Sales of a substantial number of shares of our common stock, or the perception by the market that those sales could occur, could cause the market price of our common stock to decline or could make it more difficult for us to raise funds through the sale of equity in the future.
On January 21, 2015, we announced the pricing of an underwritten public offering in which we sold 10,541,667 shares of our common stock at a price of $12.00 per share for gross proceeds of approximately $126.5 million. Net proceeds from this offering were approximately $118.3 million, net of underwriting discounts and offering expenses of approximately $8.2 million. The shares were sold under Registration Statements (Nos. 333-201605 and 333-201639) on Form S-3 and Form S-3MEF, respectively, filed by us with the Securities and Exchange Commission.
We may need to seek additional financing to provide cash necessary to execute our current operations, including beyond the initial commercialization of Auryxia, and to develop any drug candidates we may in-license or acquire. Future issuances of common stock could depress the market for our common stock.
If we make one or more significant acquisitions in which the consideration includes stock or other securities, our stockholders’ holdings may be significantly diluted. In addition, stockholders’ holdings may also be diluted if we enter into arrangements with third parties permitting us to issue shares of common stock in lieu of certain cash payments upon the achievement of milestones.
Our stock price can be volatile, which increases the risk of litigation, and may result in a significant decline in the value of your investment.
The trading price of our common stock is likely to be highly volatile and subject to wide fluctuations in price in response to various factors, many of which are beyond our control. These factors include:
| • | announcements of technological innovations by us or our competitors; |
39
Table of Contents
| • | introductions or announcements of new products by us or our competitors; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments involving us or our competitors; |
| • | changes in financial estimates by securities analysts; |
| • | actual or anticipated variations in quarterly or annual operating results; |
| • | developments relating to the marketing, safety and efficacy of our drug product, and regulatory filings and approvals for us or our competitors; |
| • | expectations regarding our financial condition; |
| • | expiration or termination of licenses, research contracts or other collaboration agreements; |
| • | expectations or investor speculation regarding the strength of our intellectual property position, or the availability of other forms of regulatory exclusivity; |
| • | conditions or trends in the regulatory climate and the biotechnology and pharmaceutical industries; |
| • | changes in the market valuations of similar companies; |
| • | negative comments and sentiment in the media; and |
| • | additions or departures of key personnel. |
In addition, equity markets in general, and the market for biotechnology and life sciences companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of companies traded in those markets. These broad market and industry factors may materially affect the market price of our common stock, regardless of our development and operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation has often been instituted against that company. For example, in the past, we have been the subject of a putative stockholders securities class action alleging misstatements or omissions in relation to our clinical trials for KRX-0401 (perifosine), which we abandoned in May 2012 following negative Phase 3 results. Any litigation instituted against us could cause us to incur substantial costs to defend such claims and divert management’s attention and resources, which could seriously harm our business.
Certain anti-takeover provisions in our charter documents and Delaware law could make a third-party acquisition of us difficult. This could limit the price investors might be willing to pay in the future for our common stock.
Provisions in our amended and restated certificate of incorporation and bylaws could have the effect of making it more difficult for a third party to acquire, or of discouraging a third party from attempting to acquire, or control us. These factors could limit the price that certain investors might be willing to pay in the future for shares of our common stock. Our amended and restated certificate of incorporation allows us to issue preferred stock without the approval of our stockholders. The issuance of preferred stock could decrease the amount of earnings and assets available for distribution to the holders of our common stock or could adversely affect the rights and powers, including voting rights, of such holders. In certain circumstances, such issuance could have the effect of decreasing the market price of our common stock. Our amended and restated bylaws eliminate the right of stockholders to call a special meeting of stockholders, which could make it more difficult for stockholders to effect certain corporate actions. Any of these provisions could also have the effect of delaying or preventing a change in control.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS. |
None.
40
Table of Contents
| ITEM 2. | PROPERTIES. |
Our corporate and executive office is located in New York, New York. Our New York facility consists of approximately 18,500 square feet of leased space at 750 Lexington Avenue, New York, New York 10022, with a lease term through September 30, 2016. We were a party to an office sharing agreement with a third-party for a portion of our leased space through September 29, 2014.
In March 2014, we entered into a sublease for approximately 10,395 square feet of leased office space in Boston, Massachusetts, with a term through December 31, 2015.
| ITEM 3. | LEGAL PROCEEDINGS. |
We, and our subsidiaries, are not a party to, and our property is not the subject of, any material pending legal proceedings, except as stated below.
On February 1, 2013, a lawsuit was filed against us and our chief executive officer on behalf of a putative class of all of our shareholders (other than the defendants) who acquired our shares between June 1, 2009 and April 1, 2012. Smith v. Keryx Biopharmaceuticals, Inc., et al., Case No. 1:13-CV-0755-TPG (S.D.N.Y.). On February 26, 2013, a substantially similar lawsuit was filed against us and our chief executive officer as well as our chief financial officer. Park v. Keryx Biopharmaceuticals, Inc., et al., Case No. 1:13-CV-1307-TPG (S.D.N.Y.). On June 10, 2013, the Court entered an Order consolidating the two lawsuits and appointing a lead plaintiff. The case was styled In re Keryx Biopharmaceuticals, Inc. Securities Litigation, Case No. 1:13-CV-0755-KBF (S.D.N.Y.). On July 10, 2013, the lead plaintiff filed a Consolidated Amended Complaint that, in substance, repeated the claims alleged in the consolidated lawsuits. The Consolidated Amended Complaint asserted claims against (i) us for alleged violations of Section 10(b) of the Securities Exchange Act of 1934 (“Exchange Act”) and Rule 10b-5 promulgated thereunder and (ii) our chief executive officer for alleged violations of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5. The claims in the Consolidated Amended Complaint were premised on general allegations that we and the individual defendant participated directly or indirectly in the preparation and/or issuance of purportedly false and misleading earnings reports, SEC filings, press releases, and other public statements, which allegedly caused our stock to trade at artificially inflated prices. On August 26, 2013, we filed a motion to dismiss the Consolidated Amended Complaint. On February 14, 2014, the Court entered an Opinion and Order granting the motion to dismiss. The Court entered Judgment for the Defendants on February 24, 2014. The lead plaintiff did not appeal the Judgment and this matter is now concluded.
| ITEM 4. | MINE SAFETY DISCLOSURES. |
Not applicable
41
Table of Contents
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market Information
Our common stock is listed on the Nasdaq Capital Market and trades under the symbol “KERX.”
The following table sets forth the high and low closing sale prices of our common stock for the periods indicated.
| High | Low | |||||||
| Fiscal Year Ended December 31, 2014 |
||||||||
| Fourth Quarter |
$ | 17.20 | $ | 13.57 | ||||
| Third Quarter |
$ | 18.19 | $ | 12.71 | ||||
| Second Quarter |
$ | 17.03 | $ | 12.11 | ||||
| First Quarter |
$ | 17.04 | $ | 12.16 | ||||
| High | Low | |||||||
| Fiscal Year Ended December 31, 2013 |
||||||||
| Fourth Quarter |
$ | 14.68 | $ | 8.76 | ||||
| Third Quarter |
$ | 10.22 | $ | 7.87 | ||||
| Second Quarter |
$ | 8.75 | $ | 6.92 | ||||
| First Quarter |
$ | 9.08 | $ | 2.73 | ||||
Holders
The number of record holders of our common stock as of February 13, 2015 was 51.
Dividends
We have never declared or paid any cash dividends on our common stock and do not anticipate paying any cash dividends in the foreseeable future. Any future determination to pay dividends will be at the discretion of our board of directors.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information as of December 31, 2014, regarding the securities authorized for issuance under our equity compensation plans, consisting of the 1999 Stock Option Plan, as amended, 2004 Long-Term Incentive Plan, 2007 Incentive Plan, 2009 CEO Incentive Plan and 2013 Incentive Plan, as amended.
42
Table of Contents
| Equity Compensation Plan Information |
||||||||||||
| Plan Category |
Number of securities to be issued upon exercise of outstanding options |
Weighted-average exercise price of outstanding options |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders |
4,532,426 | $ | 10.51 | 5,528,526 | ||||||||
| Equity compensation plans not approved by security holders |
600,000 | 0.35 | — | |||||||||
|
|
|
|
|
|||||||||
| Total |
5,132,426 | $ | 9.32 | 5,528,526 | ||||||||
|
|
|
|
|
|||||||||
For information about all of our equity compensation plans, see Note 11 to our Consolidated Financial Statements included in this report.
COMMON STOCK PERFORMANCE GRAPH
The following graph compares the cumulative total stockholder return on our common stock for the period from December 31, 2009 through December 31, 2014, with the cumulative total return over such period on (i) the U.S. Index of The Nasdaq Stock Market and (ii) the Biotechnology Index of The Nasdaq Stock Market. The graph assumes an investment of $100 on December 31, 2009, in our common stock (at the closing market price) and in each of the indices listed above, and assumes the reinvestment of all dividends. Measurement points are December 31 of each year.
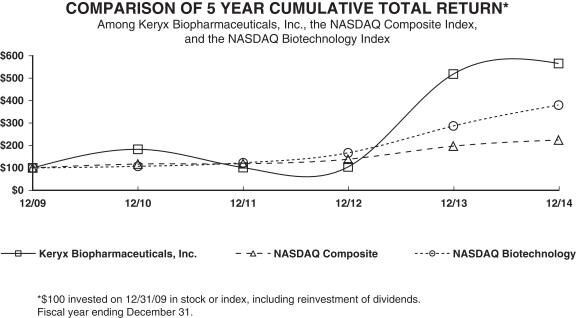
43
Table of Contents
| ITEM 6. | SELECTED FINANCIAL DATA. |
The following Statement of Operations Data for the years ended December 31, 2014, 2013, 2012, 2011 and 2010, and Balance Sheet Data as of December 31, 2014, 2013, 2012, 2011 and 2010, as set forth below are derived from our audited consolidated financial statements. This financial data should be read in conjunction with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8. Financial Statements and Supplementary Data.”
| Years ended December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| License revenue |
$ | 10,825 | $ | 7,000 | $ | — | $ | 5,000 | $ | — | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| License expenses |
495 | — | — | — | — | |||||||||||||||
| Research and development |
51,502 | 34,734 | 20,031 | 27,012 | 14,964 | |||||||||||||||
| Selling, general and administrative |
70,057 | 19,349 | 7,048 | 6,737 | 6,251 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
122,054 | 54,083 | 27,079 | 33,749 | 21,215 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Operating loss |
(111,229 | ) | (47,083 | ) | (27,079 | ) | (28,749 | ) | (21,215 | ) | ||||||||||
| Other income (expense): |
||||||||||||||||||||
| Interest and other income, net |
411 | 351 | 1,719 | 380 | 764 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from continuing operations before income taxes |
(110,818 | ) | (46,732 | ) | (25,360 | ) | (28,369 | ) | (20,451 | ) | ||||||||||
| Income taxes |
(700 | ) | — | — | — | — | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from continuing operations |
(111,518 | ) | (46,732 | ) | (25,360 | ) | (28,369 | ) | (20,451 | ) | ||||||||||
| Gain from discontinued operations |
— | — | — | 246 | 120 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss before extraordinary gain |
(111,518 | ) | (46,732 | ) | (25,360 | ) | (28,123 | ) | (20,331 | ) | ||||||||||
| Extraordinary gain |
— | — | 2,639 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (111,518 | ) | $ | (46,732 | ) | $ | (22,721 | ) | $ | (28,123 | ) | $ | (20,331 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted loss per common share: |
||||||||||||||||||||
| Continuing operations |
$ | (1.23 | ) | $ | (0.58 | ) | $ | (0.36 | ) | $ | (0.42 | ) | $ | (0.34 | ) | |||||
| Discontinued operations |
— | — | — | — | * | — | * | |||||||||||||
| Extraordinary gain |
— | — | 0.04 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted loss per common share |
$ | (1.23 | ) | $ | (0.58 | ) | $ | (0.32 | ) | $ | (0.42 | ) | $ | (0.34 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| * Amount less than one cent per share. |
| |||||||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents, interest receivable and short-term investment securities |
$ | 85,840 | $ | 55,696 | $ | 14,677 | $ | 39,470 | $ | 28,512 | ||||||||||
| Working capital |
69,285 | 41,600 | 7,068 | 30,237 | 22,520 | |||||||||||||||
| Total assets |
103,628 | 60,766 | 18,569 | 43,488 | 32,114 | |||||||||||||||
| Deferred tax liability |
700 | — | — | — | — | |||||||||||||||
| Other liabilities |
133 | 38 | 36 | 35 | 35 | |||||||||||||||
| Contingent equity rights |
— | — | — | 2,639 | 2,639 | |||||||||||||||
| Total stockholders’ equity |
73,484 | 45,400 | 10,494 | 31,047 | 23,248 | |||||||||||||||
44
Table of Contents
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following discussion and analysis contains forward-looking statements about our plans and expectations of what may happen in the future. Forward-looking statements are based on a number of assumptions and estimates that are inherently subject to significant risks and uncertainties, and our results could differ materially from the results anticipated by our forward-looking statements as a result of many known or unknown factors, including, but not limited to, those factors discussed in “Item 1A. Risk Factors.” See also the “Special Cautionary Notice Regarding Forward-Looking Statements” set forth at the beginning of this report.
You should read the following discussion and analysis in conjunction with “Item 6. Selected Financial Data,” “Item 8. Financial Statements and Supplementary Data,” and our consolidated financial statements beginning on page F-1 of this report.
Overview
We are a biopharmaceutical company focused on bringing innovative therapies to market for patients with renal disease. Our first product, AuryxiaTM (ferric citrate), an oral, absorbable iron-based compound, received marketing approval from the U.S. Food and Drug Administration, or FDA, in September 2014 for the control of serum phosphorus levels in patients with chronic kidney disease, or CKD, on dialysis.
Auryxia received marketing approval from the U.S. FDA in September 2014 for the control of serum phosphorus levels in patients with CKD on dialysis. The U.S. approval of Auryxia was based on data from our Phase 3 registration program, in which Auryxia effectively reduced serum phosphorus levels to well within the KDOQI guidelines range of 3.5 to 5.5 mg/dL. In addition to the effects on serum phosphorus levels, Auryxia’s pharmacodynamic properties resulted in increased ferritin, iron and TSAT, whereas these parameters remained relatively constant in patients treated with active control (Renvela® and/or PhosLo®). The most common adverse events for Auryxia treated patients were gastrointestinal-related, including diarrhea, nausea, constipation, vomiting and cough.
We launched Auryxia in the U.S. in late December 2014. Auryxia is being marketed in the U.S. through our specialty salesforce and commercial infrastructure. We currently have 60 sales representatives in the field calling on approximately 5,000 target nephrologists.
Our Japanese partner, JT and Torii, received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare as an oral treatment for the improvement of hyperphosphatemia in patients with CKD, including dialysis and NDD-CKD, in January 2014. JT’s subsidiary, Torii, launched the product under the brand name Riona® in May 2014. Under the license agreement with JT and Torii, we received a non-refundable payment of $10.0 million in February 2014 for the achievement of the marketing approval milestone. We also receive royalty payments based on a tiered double-digit percentage of net sales of Riona® in Japan escalating up to the mid-teens, as well as up to an additional $55.0 million upon the achievement of certain annual net sales milestones.
We have also submitted, in March 2014, a MAA with the EMA for the approval of Auryxia in patients with CKD, including dialysis and NDD-CKD. Also in March 2014, the EMA validated our MAA, confirming that the submission is sufficiently complete to begin the formal review process.
In September 2014, we announced the initiation of a pivotal Phase 3 study of Auryxia for the treatment of IDA in patients with Stage 3-5 NDD-CKD. This study’s primary endpoint is the between group comparison of the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any point during the 16-week randomized period. In our completed 12-week Phase 2 study of Auryxia for the management of elevated serum phosphorus levels and iron deficiency in subjects with Stage 3 to 5 NDD-CKD, a post-hoc analysis of this
45
Table of Contents
endpoint demonstrated that the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any time point during the study was 40% in the Auryxia arm vs. 15% in the placebo arm (p-value <0.001). Secondary endpoints in the Phase 3 study include change from baseline to the end of the randomized period for hemoglobin, ferritin, TSAT and serum phosphorus.
Currently, our only drug product is Auryxia. We may engage in business development activities that include seeking strategic relationships for Auryxia, as well as evaluating other compounds and companies for in-licensing or acquisition. We have also generated, and expect to continue to generate, revenue from the sublicensing of rights to Auryxia in Japan to JT and Torii.
In April 2012 we announced that our Phase 3 trial for KRX-0401 (perifosine) for refractory advanced colorectal cancer did not meet the primary endpoint of improving overall survival versus capecitabine and placebo. Following these results, we abandoned our development efforts and terminated our license relating to the KRX-0401 (perifosine) drug candidate, and re-focused our development efforts on our drug candidate, Auryxia.
We have devoted substantially all of our efforts to the identification, in-licensing, development and partnering of drug candidates, as well as pre-commercial/commercial activities related to Auryxia, and have incurred negative cash flow from operations each year since our inception. We have spent, and expect to continue to spend, substantial amounts in connection with implementing our business strategy, including our product development efforts, our clinical trials, commercial, partnership and licensing activities. Prior to the launch of Auryxia in December 2014, we have not commercialized any drug, and we may not become profitable. Our ability to achieve profitability depends on a number of factors, including our ability to complete our development efforts, obtain additional regulatory approvals for our drug, successfully complete any post-approval regulatory obligations and successfully manufacture and commercialize our drug. We may continue to incur substantial operating losses even after we begin to generate revenues from our drug.
Our license revenues consist of license fees and milestone payments arising from our agreement with JT and Torii. We recognize license revenue in accordance with the revenue recognition guidance of the Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or the Codification. We analyze each element of our licensing agreement to determine the appropriate revenue recognition. The terms of the license agreement may include payment to us of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. We recognize revenue from upfront payments over the period of significant involvement under the related agreements unless the fee is in exchange for products delivered or services rendered that represent the culmination of a separate earnings process and no further performance obligation exists under the contract. We recognize milestone payments as revenue upon the achievement of specified milestones only if (1) the milestone payment is non-refundable, (2) substantive effort is involved in achieving the milestone, (3) the amount of the milestone is reasonable in relation to the effort expended or the risk associated with the achievement of the milestone, and (4) the milestone is at risk for both parties. If any of these conditions are not met, we defer the milestone payment and recognize it as revenue over the estimated period of performance under the contract.
For arrangements for which royalty revenue information becomes available and collectability is reasonably assured, we recognize revenue during the applicable period earned. When collectability is reasonably assured but a reasonable estimate of royalty revenue cannot be made, the royalty revenue is recognized in the quarter that the licensee provides the written report and related information to us. Based on our agreement with JT and Torii, and in accordance with our revenue recognition policy, royalty revenues are recognized in the quarter that JT and Torii provide their written report and related information to us regarding sales of Riona®, which generally will be one quarter following the quarter in which the underlying sales by JT and Torii occurred.
Our commercial launch of Auryxia occurred in December 2014. We sell product to a limited number of major wholesalers, our Distributors, as well as certain pharmacies, or collectively, our Customers. Our
46
Table of Contents
Distributors resell the product to retail pharmacies for purposes of their reselling the product to fill patient prescriptions. In accordance with GAAP, until we have the ability to reliably estimate returns of Auryxia from our Customers, revenue will be recognized based on the resale of Auryxia for the purposes of filling patient prescriptions, and not based on sales from us to such Customers. Consistent with industry practice, once we achieve sufficient history such that we can reliably estimate returns based on sales to our Customers, we anticipate that our revenues will be recognized based on sales to our Customers. We currently defer Auryxia revenue recognition until the earlier of the product being resold for purposes of filling patient prescriptions and the expiration of the right of return (twelve months after the expiration date of the product). The deferred revenue is recorded net of discounts, rebates, and chargebacks. We also defer the related cost of product sales and record such amounts as finished goods inventory held by others, which is included in inventory on our balance sheet, until revenue related to such product sales is recognized.
Our license expenses consist of royalty and other expenses due to the licensor of Auryxia related to our license agreement with JT and Torii. With regard to royalty expense, such expense is directly related to the royalty revenue received from JT and Torii and is recognized in the same period as the revenue is recorded. Other expenses are recognized in the period they are incurred.
Our research and development expenses consist primarily of salaries and related personnel costs, including stock-based compensation, fees paid to consultants and outside service providers for clinical and laboratory development, manufacturing, including pre-approval inventory build-up, regulatory, facilities-related and other expenses relating to the design, development, manufacture, testing, and enhancement of our drug candidates and technologies, as well as expenses related to in-licensing of new product candidates. We expense our research and development costs as they are incurred. Research and development expenses for the years ended December 31, 2014, 2013 and 2012 were $51.5 million, $34.7 million and $20.0 million, respectively.
The following table sets forth the research and development expenses per project, for the periods presented.
| Years ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Auryxia (ferric citrate) |
$ | 44,735 | $ | 32,001 | $ | 15,494 | ||||||
| Other |
670 | 1,017 | 641 | |||||||||
| Terminated programs (primarily KRX-0401) |
(282 | ) | (631 | ) | 3,234 | |||||||
| Stock-based compensation expense |
6,379 | 2,347 | 662 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 51,502 | $ | 34,734 | $ | 20,031 | ||||||
|
|
|
|
|
|
|
|||||||
Our selling, general and administrative expenses consist primarily of salaries and related expenses, including stock-based compensation, for executive, finance, sales, marketing and other administrative personnel, recruitment expenses, professional fees and other corporate expenses, including investor relations, legal activities, pre-commercial/commercial activities and facilities-related expenses.
Our results of operations include stock-based compensation expense as a result of the grants of stock options and restricted stock. Compensation expense for awards of options and restricted stock granted to employees and directors represents the fair value of the award recorded over the respective vesting periods of the individual awards. The expense is included in the respective categories of expense in the consolidated statements of operations. We expect to continue to incur significant stock-based compensation expenses.
For awards of options and restricted stock to consultants and other third-parties, compensation expense is determined at the “measurement date.” The expense is recognized over the vesting period of the award. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record compensation expense based on the fair value of the award at the reporting date. The awards to consultants and other third-parties are then revalued, or the total compensation is recalculated based on the then current fair
47
Table of Contents
value, at each subsequent reporting date. This results in a change to the amount previously recorded in respect of the equity award grant, and additional expense or a reversal of expense may be recorded in subsequent periods based on changes in the assumptions used to calculate fair value, such as changes in market price, until the measurement date is reached and the compensation expense is finalized.
In addition, certain options and restricted stock issued to employees, consultants and other third-parties vest upon the achievement of certain milestones, therefore the total expense is uncertain until the milestone is met.
Even though our trials demonstrated that Auryxia is effective in the control of serum phosphorus levels in patients with CKD on dialysis, there is no guarantee that we will be able to record meaningful commercial sales of Auryxia in the future or become profitable. In addition, we expect losses to continue as we continue to fund the development and commercialization of Auryxia, including, but not limited to, supplemental new drug application submissions, MAA submission review, building of inventory, commercial activities, ongoing and additional clinical trials, and the potential acquisition and development of additional drug candidates in the future. As we continue our development efforts, we may enter into additional third-party collaborative agreements and may incur additional expenses, such as licensing fees and milestone payments. As a result, our quarterly results may fluctuate and a quarter-by-quarter comparison of our operating results may not be a meaningful indication of our future performance.
Corporate
In January 2014, we formed a subsidiary in the United Kingdom, Keryx Biopharma UK Ltd., related to the submission of our MAA in Europe.
In March 2014, we entered into a sublease for approximately 10,395 square feet of leased office space in Boston, Massachusetts, with a term through December 31, 2015.
In January 2015, we announced the transitioning of the role of Chief Executive Officer from Ron Bentsur to Greg Madison. Mr. Madison joined Keryx in February 2014 as Executive Vice President and Chief Operating Officer to transition Keryx from a development-stage organization into a fully integrated commercial entity, bringing to Keryx a wealth of relevant expertise in both the phosphate binder and iron deficiency anemia markets. Mr. Madison has been appointed President of Keryx and will work with Mr. Bentsur to ensure a successful leadership transition by the end of May, when Mr. Bentsur’s contract expires.
In February 2015, we announced a planned consolidation of our finance and accounting function into our Boston office and that our Chief Financial Officer, James Oliviero, will be leaving Keryx by October 2015. Mr. Oliviero has been with Keryx for twelve years and has served as the Chief Financial Officer since 2009. We have commenced a search for a new Chief Financial Officer who will be based in our Boston office. Mr. Oliviero will continue to manage our finance and accounting team during the remainder of his tenure and will assist in the transition of his duties to the new Chief Financial Officer.
RESULTS OF OPERATIONS
Years Ended December 31, 2014 and 2013
License Revenue. License revenue for the year ended December 31, 2014 was $10.8 million due to the recognition of a $10.0 million non-refundable milestone payment in January 2014 related to JT and Torii’s achievement of marketing approval in Japan and $0.8 million of royalty payments from sales of Riona® in Japan. License revenue for the year ended December 31, 2013 was $7.0 million due to the recognition of a non-refundable milestone payment received in January 2013 from JT and Torii following their filing of their NDA with the Japanese Ministry of Health, Labour and Welfare for marketing approval of Auryxia in Japan. We receive royalty payments based on a tiered double-digit percentage of net sales of Riona® in Japan escalating up to the mid-teens for sales made by Torii. We may also receive up to an additional $55 million of milestone payments upon the achievement of certain annual net sales milestones.
48
Table of Contents
License Expenses. For the year ended December 31, 2014, we recognized $0.5 million in license expenses related to royalties due to the licensor of Auryxia relating to sales of Riona® in Japan. There were no license expenses for the year ended December 31, 2013. We owe a mid-single digit percentage of net sales royalty to the licensor of Auryxia associated with net sales of Riona® in Japan.
Research and Development Expenses. Research and development expenses increased by $16.8 million to $51.5 million for the year ended December 31, 2014, as compared to $34.7 million for the year ended December 31, 2013. The increase in research and development expenses was due primarily to a $12.7 million increase in research and development expenses related to our Auryxia program, including costs associated with the manufacturing of pre-approval inventory and the submission of our MAA filing. The year ended December 31, 2014, includes a $2.0 million one-time milestone payment to Panion, the licensor of Auryxia, for JT and Torii’s achievement of the Japanese marketing approval milestone in January 2014 and a $3.0 million one-time milestone payment to Panion for our achievement of U.S. FDA approval of Auryxia in September 2014. Stock-based compensation expense increased by $4.0 million to $6.4 million for the year ended December 31, 2014, as compared to $2.3 million for the year ended December 31, 2013, primarily related to $4.6 million of expense due to the vesting of milestone-based stock options and restricted shares upon the FDA approval. We expect our research and development expenses for 2015 to decrease due to the capitalization of inventory, which prior to FDA approval had been expensed, expected completion of our MAA filing and a decrease in stock-based compensation expense.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased by $50.7 million to $70.1 million for the year ended December 31, 2014, as compared to $19.3 million for the year ended December 31, 2013. The increase was primarily related to a $24.3 million increase in pre-commercial/commercial activities, including associated personnel costs, in preparation for the commercialization of Auryxia and increased stock compensation expense. Stock-based compensation expense increased by $17.0 million to $20.6 million for the year ended December 31, 2014, as compared to $3.6 million for the year ended December 31, 2013, primarily related to $11.0 million of expense due to the vesting of milestone-based stock options and restricted shares upon the FDA approval and first commercial sale of Auryxia, as well as to increased selling, general and administrative personnel and the recording of the fair value of equity awards granted, which are expensed over the vesting periods of the individual awards. We expect our selling, general and administrative costs to increase in 2015 related to a full year of commercialization of Auryxia, partially offset by a decrease in stock-based compensation expense.
Interest and Other Income, Net. Interest and other income, net, increased by $60,000 to $411,000 for the year ended December 31, 2014, as compared to $351,000 for the year ended December 31, 2013. The increase was due to a higher level of invested funds in our investment portfolio following our January 2014 public offering.
Income Taxes. For the year ended December 31, 2014, we recognized $0.7 million in income tax expense related to the recording of a deferred tax liability associated with capitalized goodwill, an indefinite-lived intangible asset that is being amortized for tax purposes. Indefinite-lived intangibles are non-monetary assets which are not amortized under GAAP since there is no foreseeable limit to the cash flows provided by them. Our lack of earnings history and the uncertainty surrounding our ability to generate taxable income prior to the reversal or expiration of such deferred tax liability were the primary factors considered by management when recording the deferred tax liability. There was no income tax expense for the year ended December 31, 2013. We continue to maintain a full valuation allowance against our net deferred tax assets.
Years Ended December 31, 2013 and 2012
License Revenue. License revenue for the year ended December 31, 2013 was $7.0 million due to the recognition of a non-refundable milestone payment received in January 2013 from JT and Torii following their filing of their NDA with the Japanese Ministry of Health, Labour and Welfare for marketing approval of Auryxia in Japan for the treatment of hyperphosphatemia in patients with CKD. There was no license revenue for the year ended December 31, 2012.
49
Table of Contents
License Expenses. There were no license expenses for the years ended December 31, 2013 and 2012.
Research and Development Expenses. Research and development expenses increased by $14.7 million to $34.7 million for the year ended December 31, 2013, as compared to $20.0 for the year ended December 31, 2012. The increase in research and development expenses was due primarily to a $16.5 million increase in research and development expenses related to our Auryxia program, including costs associated with the filing of our NDA, preparation of our MAA submission, and manufacturing of pre-launch inventory, an increase in $1.7 million in stock-based compensation expenses (primarily due to the recording of $1.2 million in stock-based compensation expense associated with the vesting of 17,500 stock options and 100,000 shares of restricted stock in October 2013 upon the filing acceptance of our NDA for Auryxia), partially offset by a $3.9 million decrease in research and development expenses related to KRX-0401, which license agreement was terminated in May 2012. The year ended December 31, 2013, included a $1.0 million one-time milestone payment to Panion & BF Biotech, Inc., the licensor of Auryxia, related to our submission of the NDA in August 2013.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased by $12.3 million to $19.3 million for the year ended December 31, 2013, as compared to $7.0 million for the year ended December 31, 2012. The increase was primarily related to a $7.2 million increase in pre-commercial activities as we scaled-up our operations and infrastructure to prepare to commercialize Auryxia, a $2.1 million increase in stock-based compensation expense (primarily due to the recording of $1.6 million of stock-based compensation expense associated with the vesting of 150,000 shares of restricted stock in October 2013 upon the filing acceptance of our NDA for Auryxia), and a $1.5 million increase in legal fees.
Interest and Other Income, Net. Interest and other income, net, decreased by $1.4 million to $0.4 million for the year ended December 31, 2013, as compared to $1.7 million for the year ended December 31, 2012. During the year ended December 31, 2012, we were awarded $1.5 million in compensatory damages, net of fees and legal expenses, related to a statement of claim we filed with FINRA against an SEC registered broker-dealer for damages arising from that broker-dealer’s recommendations and purchases of certain securities for our cash management account.
Income Taxes. There were no income tax expenses for the years ended December 31, 2013 and 2012.
Extraordinary Gain. For the year ended December 31, 2012, we recorded a non-cash extraordinary gain of $2.6 million related to a write-off of the contingent equity rights liability following the termination of the license agreement for KRX-0401.
LIQUIDITY AND CAPITAL RESOURCES
Our major sources of cash have been proceeds from various public and private offerings of our common stock, option and warrant exercises, interest income, and from the upfront and milestone payments from our Sublicense Agreement with JT and Torii and miscellaneous payments from our other prior licensing activities. The commercial launch of our first product, Auryxia, occurred in December 2014. Even though we are commercializing Auryxia, we may not become profitable. Our ability to achieve profitability depends on a number of factors, including our ability to complete our development efforts, obtain additional regulatory approvals for our drug, successfully complete any post-approval regulatory obligations and successfully manufacture and commercialize our drug alone or in partnership. We may continue to incur substantial operating losses even after we begin to generate meaningful revenues from our drug.
As of December 31, 2014, we had $85.8 million in cash, cash equivalents, short-term investments and interest receivable, an increase of $30.1 million from December 31, 2013. On January 21, 2015, we announced the pricing of an underwritten public offering in which we sold 10,541,667 shares of our common stock at a price of $12.00 per share for gross proceeds of approximately $126.5 million. Net proceeds from this offering were
50
Table of Contents
approximately $118.3 million, net of underwriting discounts and offering expenses of approximately $8.2 million. The shares were sold under Registration Statements (Nos. 333-201605 and 333-201639) on Form S-3 and Form S-3MEF, respectively, filed by us with the Securities and Exchange Commission.
On January 22, 2014, we announced the pricing of an underwritten public offering in which we sold 7,935,000 shares of our common stock at a price of $14.50 per share for gross proceeds of approximately $115.1 million. Net proceeds from this offering were approximately $107.5 million, net of underwriting discounts and offering expenses of approximately $7.5 million. The shares were sold under a Registration Statement (No. 333-190353) on Form S-3, filed by us with the SEC.
In January 2014, our Japanese partner, JT and Torii, received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare. Ferric citrate, launched in May 2014 and being marketed in Japan by JT’s subsidiary, Torii, under the brand name Riona®, is indicated as an oral treatment for the improvement of hyperphosphatemia in patients with CKD, including dialysis and NDD-CKD. Under the license agreement with JT and Torii, we received a non-refundable payment of $10.0 million in February 2014 for the achievement of the marketing approval milestone. We also receive royalty payments based on a tiered double-digit percentage of net sales of Riona® in Japan escalating up to the mid-teens, as well as up to an additional $55.0 million upon the achievement of certain annual net sales milestones. We owe a mid-single digit percentage of net sales royalty to the licensor of Auryxia associated with net sales of Riona® in Japan.
We currently expect that our existing capital resources combined with future anticipated cash flows will be sufficient to execute our business plan. The actual amount of cash that we will need to operate is subject to many factors, including, but not limited to, the timing and expenditures associated with the build-up of inventory and capacity expansion, the timing and expenditures associated with the regulatory review process for our EU MAA filing, the timing and expenditures associated with commercial activities related to Auryxia, and the timing, design and conduct of clinical trials for Auryxia. As a result of these factors, we may need to seek additional financings to provide the cash necessary to execute our current operations, including beyond the initial commercialization of Auryxia, and to develop any drug candidates we may in-license or acquire.
Net cash used in operating activities for the year ended December 31, 2014 was $81.0 million, as compared to $34.3 million for the year ended December 31, 2013. This increase in net cash used in operating activities was primarily related to increased Auryxia development and pre-commercial/commercial expenditures, including the manufacturing of inventory.
For the year ended December 31, 2014, net cash used in investing activities was $13.0 million, as compared to $0.3 million for the year ended December 31, 2013. The increase in net cash used in investing activities was primarily due to our investments in held-to-maturity short-term securities following our public offering of common stock in January 2014.
For the year ended December 31, 2014, net cash provided by financing activities was $112.6 million as compared to $75.7 million for the year ended December 31, 2013. The increase was primarily related to $107.5 million of net proceeds received from our public offering of common stock in January 2014, as compared to $74.8 million of net proceeds received from our public offering of common stock in January 2013.
OFF-BALANCE SHEET ARRANGEMENTS
We have not entered into any transactions with unconsolidated entities whereby we have financial guarantees, subordinated retained interests, derivative instruments or other contingent arrangements that expose us to material continuing risks, contingent liabilities, or any other obligations under a variable interest in an unconsolidated entity that provides us with financing, liquidity, market risk or credit risk support, or engages in leasing, hedging, or research and development services on our behalf.
51
Table of Contents
OBLIGATIONS AND COMMITMENTS
As of December 31, 2014, we have known contractual obligations, commitments and contingencies of $46.4 million. Of this amount, $13.9 million relates to selling, general and administrative agreements primarily associated with the launch and commercialization of Auryxia, of which $10.0 million is due within the next year, $4.4 million relates to research and development agreements (relating to our Auryxia clinical and regulatory programs), of which $4.1 million is due within the next year, and $25.7 million relates to various third-party contract manufacturing agreements for the production and packaging of Auryxia drug substance and drug product, of which $24.7 million is due within the next year. The additional $2.4 million relates to our operating lease obligations.
| (in thousands) |
Payment due by period | |||||||||||||||||||
| Contractual obligations | Total | Less than 1 year |
1-3 years |
3-5 years |
More than 5 years |
|||||||||||||||
| Selling, general and administrative agreements |
$ | 13,938 | $ | 10,031 | $ | 3,807 | $ | 100 | $ | — | ||||||||||
| Research and development agreements |
4,406 | 4,073 | 333 | — | — | |||||||||||||||
| Manufacturing agreements |
25,712 | 24,663 | 1,049 | — | — | |||||||||||||||
| Operating leases |
2,375 | 1,521 | 854 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 46,431 | $ | 40,288 | $ | 6,043 | $ | 100 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
Our lease on our corporate and executive office located in New York City extends through September 30, 2016. In March 2014, we entered into a sublease for approximately 10,395 square feet of leased office space in Boston, Massachusetts, with a term through December 31, 2015.
We have undertaken to make a contingent milestone payment to the licensor of Auryxia of $2.0 million, which will be due upon the regulatory approval of Auryxia in Europe, which is included in research and development agreements in the table above. In addition the licensor will be due royalty payments based on a mid-single digit percentage of net sales of Auryxia.
CRITICAL ACCOUNTING POLICIES
The discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amount of assets and liabilities and related disclosure of contingent assets and liabilities at the date of our consolidated financial statements and the reported amounts of revenues and expenses during the applicable period. Actual results may differ from these estimates under different assumptions or conditions.
We define critical accounting policies as those that are reflective of significant judgments and uncertainties and which may potentially result in materially different results under different assumptions and conditions. In applying these critical accounting policies, our management uses its judgment to determine the appropriate assumptions to be used in making certain estimates. These estimates are subject to an inherent degree of uncertainty. Our critical accounting policies include the following:
Stock Compensation. We have granted stock options and restricted stock to employees, directors and consultants, as well as warrants to other third parties. For employee and director grants, the value of each option award is estimated on the date of grant using the Black-Scholes option-pricing model. The Black-Scholes model takes into account volatility in the price of our stock, the risk-free interest rate, the estimated life of the option, the closing market price of our stock and the exercise price. We base our estimates of our stock price volatility on the historical volatility of our common stock and our assessment of future volatility; however, these estimates are neither predictive nor indicative of the future performance of our stock. For purposes of the calculation, we assumed that no dividends would be paid during the life of the options and warrants. The estimates utilized in the
52
Table of Contents
Black-Scholes calculation involve inherent uncertainties and the application of management judgment. In addition, we are required to estimate the expected forfeiture rate and only recognize expense for those equity awards expected to vest. As a result, if other assumptions had been used, our recorded stock-based compensation expense could have been materially different from that reported. In addition, because some of the options and warrants issued to employees, consultants and other third-parties vest upon the achievement of certain milestones, the total expense is uncertain.
Total compensation expense for options and restricted stock issued to consultants is determined at the “measurement date.” The expense is recognized over the vesting period for the options and restricted stock. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record stock-based compensation expense based on the fair value of the equity awards at the reporting date. These equity awards are then revalued, or the total compensation is recalculated based on the then current fair value, at each subsequent reporting date. This results in a change to the amount previously recorded in respect of the equity award grant, and additional expense or a reversal of expense may be recorded in subsequent periods based on changes in the assumptions used to calculate fair value, such as changes in market price, until the measurement date is reached and the compensation expense is finalized.
Accruals for Clinical Research Organization and Clinical Site Costs. We make estimates of costs incurred in relation to external clinical research organizations, or CROs, and clinical site costs. We analyze the progress of clinical trials, including levels of patient enrollment, invoices received and contracted costs when evaluating the adequacy of the amount expensed and the related prepaid asset and accrued liability. Significant judgments and estimates must be made and used in determining the accrued balance and expense in any accounting period. We review and accrue CRO expenses and clinical trial study expenses based on work performed and rely upon estimates of those costs applicable to the stage of completion of a study. Accrued CRO costs are subject to revisions as such trials progress to completion. Revisions are charged to expense in the period in which the facts that give rise to the revision become known. With respect to clinical site costs, the financial terms of these agreements are subject to negotiation and vary from contract to contract. Payments under these contracts may be uneven, and depend on factors such as the achievement of certain events, the successful recruitment of patients, and the completion of portions of the clinical trial or similar conditions. The objective of our policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical site costs are recognized based on our estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
Revenue. We recognize license revenue in accordance with the revenue recognition guidance of the Codification. We analyze each element of our licensing agreement to determine the appropriate revenue recognition. The terms of the license agreement may include payment to us of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. We recognize revenue from upfront payments over the period of significant involvement under the related agreements unless the fee is in exchange for products delivered or services rendered that represent the culmination of a separate earnings process and no further performance obligation exists under the contract. We recognize milestone payments as revenue upon the achievement of specified milestones only if (1) the milestone payment is non-refundable, (2) substantive effort is involved in achieving the milestone, (3) the amount of the milestone is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (4) the milestone is at risk for both parties. If any of these conditions are not met, we defer the milestone payment and recognize it as revenue over the estimated period of performance under the contract.
For arrangements for which royalty revenue information becomes available and collectability is reasonably assured, we recognize revenue during the applicable period earned. When collectability is reasonably assured but a reasonable estimate of royalty revenue cannot be made, the royalty revenue is recognized in the quarter that the licensee provides the written report and related information to us.
We recognize other revenues at the time such fees and payments are earned.
53
Table of Contents
Our commercial launch of Auryxia occurred in December 2014. We sell product to a limited number of major wholesalers, our Distributors, as well as certain pharmacies, or collectively, our Customers. Our Distributors resell the product to retail pharmacies for purposes of their reselling the product to fill patient prescriptions. In accordance with GAAP, until we have the ability to reliably estimate returns of Auryxia from our Customers, revenue will be recognized based on the resale of Auryxia for the purposes of filling patient prescriptions, and not based on sales from us to such Customers. Consistent with industry practice, once we achieve sufficient history such that we can reliably estimate returns based on sales to our Customers, we anticipate that our revenues will be recognized based on sales to our Customers. We currently defer Auryxia revenue recognition until the earlier of the product being resold for purposes of filling patient prescriptions and the expiration of the right of return (twelve months after the expiration date of the product). The deferred revenue is recorded net of discounts, rebates, and chargebacks. We also defer the related cost of product sales and record such amounts as finished goods inventory held by others, which is included in inventory on our consolidated balance sheet, until revenue related to such product sales is recognized.
Inventory. Inventories are stated at the lower of cost or estimated realizable value. We determine the cost of our inventories, which include amounts related to materials, third-party contract manufacturing and packaging services, and manufacturing overhead, on a first-in, first-out basis. We capitalize inventory costs at our suppliers when, based on management’s judgment, the realization of future economic benefit is probable at each given supplier. We received FDA approval for Auryxia on September 5, 2014, and on that date began capitalizing inventory purchases of saleable product from certain suppliers. Prior to FDA approval, all saleable product purchased from such suppliers were included as a component of research and development expense.
Accounts Receivable, Allowances for Doubtful Accounts and Cash Discounts. We extend credit to our customers for product sales resulting in accounts receivable. Customer accounts are monitored for past due amounts. Past due accounts receivable, determined to be uncollectible, are written off against the allowance for doubtful accounts. Allowances for doubtful accounts are estimated based upon past due amounts, historical losses and existing economic factors, and are adjusted periodically. We offer cash discounts to our customers, generally 2% of the sales price, as an incentive for prompt payment. The estimate of cash discounts is recorded at the time of sale. We account for the cash discounts by reducing revenue and accounts receivable by the amount of the discounts we expect our customers to take. The accounts receivable are reported in the consolidated balance sheets, net of the allowances for doubtful accounts and cash discounts. There was no allowance for doubtful accounts at December 31, 2014 and 2013.
Accounting Related to Goodwill. As of December 31, 2014, there was approximately $3.2 million of goodwill on our consolidated balance sheet. Goodwill is reviewed for impairment annually, or when events arise that could indicate that an impairment exists. We test for goodwill impairment using a two-step process. The first step compares the fair value of the reporting unit with the unit’s carrying value, including goodwill. When the carrying value of the reporting unit is greater than fair value, the unit’s goodwill may be impaired, and the second step must be completed to measure the amount of the goodwill impairment charge, if any. In the second step, the implied fair value of the reporting unit’s goodwill is compared with the carrying amount of the unit’s goodwill. If the carrying amount is greater than the implied fair value, the carrying value of the goodwill must be written down to its implied fair value.
We are required to perform impairment tests annually, at December 31, and whenever events or changes in circumstances suggest that the carrying value of an asset may not be recoverable. For all of our acquisitions, various analyses, assumptions and estimates were made at the time of each acquisition that were used to determine the valuation of goodwill and intangibles. In future years, the possibility exists that changes in forecasts and estimates from those used at the acquisition date could result in impairment indicators.
Accounting For Income Taxes. In preparing our consolidated financial statements, we are required to estimate our income taxes in each of the jurisdictions in which we operate. This process involves management estimation of our actual current tax exposure and assessment of temporary differences resulting from differing
54
Table of Contents
treatment of items for tax and accounting purposes. These differences result in deferred tax assets and liabilities. We must then assess the likelihood that our deferred tax assets will be recovered from future taxable income and, to the extent we believe that recovery is not likely, we must establish a valuation allowance. To the extent we establish a valuation allowance or increase this allowance in a period, we must include an expense within the tax provision in the consolidated statement of operations. Significant management judgment is required in determining our provision for income taxes, our deferred tax assets and liabilities and any valuation allowance recorded against our net deferred tax assets. We have fully offset our deferred tax assets with a valuation allowance. Our lack of earnings history and the uncertainty surrounding our ability to generate taxable income prior to the reversal or expiration of such deferred tax assets were the primary factors considered by management in maintaining the valuation allowance.
For the year ended December 31, 2014, we recognized $0.7 million in income tax expense related to the recording of a deferred tax liability associated with capitalized goodwill, an indefinite-lived intangible asset that is being amortized for tax purposes. Indefinite-lived intangibles are non-monetary assets which are not amortized under GAAP since there is no foreseeable limit to the cash flows provided by them. Our lack of earnings history and the uncertainty surrounding our ability to generate taxable income prior to the reversal or expiration of such deferred tax liability were the primary factors considered by management when recording the deferred tax liability.
RECENTLY ISSUED ACCOUNTING STANDARDS
In August 2014, the Financial Accounting Standards Board issued a new standard, Accounting Standards Update No. 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. This new standard will explicitly require management to assess an entity’s ability to continue as a going concern and to provide footnote disclosures in certain cases. Currently there is no guidance in GAAP about management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern. The new standard applies to all entities and provides an explicit requirement that management assesses and discloses going concern uncertainties. Previous guidance in auditing standards required auditors to evaluate going concern. The new standard will be effective for all entities in the first annual period ending after December 15, 2016, which is December 31, 2016 for calendar year-end entities. Earlier application is permitted.
In May 2014, the Financial Accounting Standards Board issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2016 and allows for adoption using a full retrospective method, or a modified retrospective method. We are currently assessing the method of adoption and the expected impact the new standard has on our financial position and results of operations.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK. |
The primary objective of our investment activities is to preserve principal while maximizing our income from investments and minimizing our market risk. We currently invest in government and investment-grade corporate debt in accordance with our investment policy, which we may change from time to time. The securities in which we invest have market risk. This means that a change in prevailing interest rates, and/or credit risk, may cause the fair value of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the prevailing interest rate later rises, the fair value of our investment will probably decline. As of December 31, 2014, our portfolio of financial instruments consists of cash equivalents and short-term interest bearing securities, including government debt and money market funds. The
55
Table of Contents
average duration of all of our held-to-maturity investments held as of December 31, 2014, was less than 12 months. Due to the short-term nature of these financial instruments, we believe there is no material exposure to interest rate risk, and/or credit risk, arising from our portfolio of financial instruments.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
Our consolidated financial statements and the notes thereto, included in Part IV, Item 15(a), part 1, are incorporated by reference into this Item 8.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES. |
Evaluation of Disclosure Controls and Procedures. As of December 31, 2014, management carried out, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act). Our disclosure controls and procedures are designed to provide reasonable assurance that information we are required to disclose in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in applicable rules and forms. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2014, our disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting. Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) or Rule 15d-15(f) under the Exchange Act). Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2014. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO, in Internal Control-Integrated Framework (2013). Our management has concluded that, as of December 31, 2014, our internal control over financial reporting was effective based on these criteria. UHY LLP, our independent registered public accounting firm, has audited the accompanying consolidated balance sheets as of December 31, 2014 and 2013, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the years in the three-year period ended December 31, 2014, included in this annual report on page F-1. UHY LLP has issued an attestation report on our internal control over financial reporting as of December 31, 2014, which is found below.
Changes in Internal Control Over Financial Reporting. There were no changes in our internal control over financial reporting during the quarter ended December 31, 2014, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on the Effectiveness of Controls. Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within our company have been detected.
56
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Stockholders of Keryx Biopharmaceuticals, Inc.
We have audited Keryx Biopharmaceuticals, Inc.’s (the “Company”) internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in Part II, Item 9A of this Form 10-K. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Keryx Biopharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets and the related consolidated statements of operations, stockholders’ equity, and cash flows of Keryx Biopharmaceuticals, Inc., and our report dated February 27, 2015, expressed an unqualified opinion thereon.
/s/ UHY LLP
New York, New York
February 27, 2015
| ITEM 9B. | OTHER INFORMATION. |
None.
57
Table of Contents
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2015 Annual Meeting of Stockholders.
| ITEM 11. | EXECUTIVE COMPENSATION. |
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2015 Annual Meeting of Stockholders.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2015 Annual Meeting of Stockholders.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE. |
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2015 Annual Meeting of Stockholders.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES. |
The information required by this Item is incorporated herein by reference from our Proxy Statement for our 2015 Annual Meeting of Stockholders.
58
Table of Contents
| ITEM 15. | EXHIBITS and FINANCIAL STATEMENT SCHEDULES. |
| (a) | 1. Consolidated Financial Statements |
The following consolidated financial statements of Keryx Biopharmaceuticals, Inc. are filed as part of this report.
2. Consolidated Financial Statement Schedules
All schedules are omitted as the information required is inapplicable or the information is presented in the consolidated financial statements or the related notes.
3. Exhibits
| Exhibit Number |
Exhibit Description | |
| 3.1 | Amended and Restated Certificate of Incorporation of Keryx Biopharmaceuticals, Inc., filed as Exhibit 3.1 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2004, filed on August 12, 2004 (File No. 000-30929), and incorporated herein by reference. | |
| 3.2 | Amended and Restated Bylaws of Keryx Biopharmaceuticals, Inc., filed as Exhibit 3.2 to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2001, filed on March 26, 2002 (File No. 000-30929), and incorporated herein by reference. | |
| 3.3 | Amendment to Amended and Restated Certificate of Incorporation of Keryx Biopharmaceuticals, Inc., dated July 24, 2007, filed as Exhibit 3.3 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2007, filed on August 9, 2007 and incorporated herein by reference. | |
| 3.4 | Amendment Number 3 to Amended and Restated Certificate of Incorporation of Keryx Biopharmaceuticals, Inc. dated June 18, 2013, filed as Exhibit 3.4 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, filed on August 2, 2013 and incorporated herein by reference. | |
| 4.1 | Specimen Common Stock Certificate, filed as Exhibit 4.1 to the Registrant’s First Amendment to the Registration Statement on Form S-1 filed on June 30, 2000 (File No. 333-37402), and incorporated herein by reference. | |
| 10.1† | 1999 Stock Option Plan, as amended, filed as Exhibit 10.2 to the Registrant’s Quarterly Report of Form 10-Q for the quarter ended March 31, 2003 filed on May 15, 2003 (File No. 000-30929) and incorporated herein by reference. | |
59
Table of Contents
| 10.2† | Keryx Biopharmaceuticals, Inc. 2004 Long-Term Incentive Plan, filed with the Registrant’s Definitive Proxy Statement for the Annual Meeting of Stockholders on June 10, 2004, filed on April 29, 2004, and incorporated herein by reference. | |
| 10.3! | License Agreement between Keryx Biopharmaceuticals, Inc. and Panion & BF Biotech, Inc. dated as of November 7, 2005, filed as Exhibit 10.21 to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2005, filed on March 8, 2006, and incorporated herein by reference. | |
| 10.4† | Amendment to the Keryx Biopharmaceuticals, Inc. 2004 Long-Term Incentive Plan dated April 11, 2006, filed as Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2006, filed on August 9, 2006, and incorporated herein by reference. | |
| 10.5† | 2007 Incentive Plan, filed as Annex D to the Registrant’s Definitive Proxy Statement on Schedule 14A (File No. 000-30929) filed on April 30, 2007, and incorporated herein by reference. | |
| 10.6† | Keryx Biopharmaceuticals, Inc. 2013 Incentive Plan, filed with the Registrant’s Definitive Proxy Statement for the Annual Meeting of Stockholders on June 18, 2013, filed on April 30, 2013, and incorporated herein by reference. | |
| 10.7† | Amendment to Keryx Biopharmaceuticals, Inc. 2013 Incentive Plan, filed with the Registrant’s Definitive Proxy Statement for the Special Meeting of Stockholders on November 17, 2014, filed on October 10, 20072014, and incorporated herein by reference. | |
| 10.8! | Amended and Restated License Agreement by and between Panion & BF Biotech, Inc. and Keryx Biopharmaceuticals, Inc. dated March 17, 2008, filed as Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, filed on May 12, 2008, and incorporated herein by reference. | |
| 10.9! | First Amendment to Amended and Restated License Agreement by and between Panion & BF Biotech, Inc. and Keryx Biopharmaceuticals, Inc. dated March 17, 2008, filed as Exhibit 10.16 to the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2008, filed on March 31, 2009, and incorporated herein by reference. | |
| 10.10! | Amended and Restated Sub-License Agreement dated June 8, 2009, by and between Keryx Biopharmaceuticals, Inc., Japan Tobacco, Inc. and Japan Torii Pharmaceutical Co. Ltd., filed as Exhibit 10.1 to the Registrant’s quarterly report on Form 10-Q for the quarter ended June 30, 2009, filed on August 8, 2009, and incorporated herein by reference. | |
| 10.11! | License Termination and Technology Transfer Agreement dated May 4, 2012, among AOI Pharma, Inc., Keryx Biopharmaceuticals, Inc., AEterna Zentaris GmbH, filed as Exhibit 10.1 to the Registrant’s quarterly report on Form 10-Q for the quarter ended March 31, 2012, filed on May 9, 2012, and incorporated herein by reference. | |
| 10.12† | Employment Agreement with Ron Bentsur dated September 14, 2009, filed as Exhibit 10.1 to the Registrant’s Form 8-K filed on September 16, 2009, and incorporated herein by reference. | |
| 10.13† | First Amendment to Employment Agreement with Ron Bentsur dated January 13, 2012, filed as Exhibit 10.1 to the Registrant’s Form 8-K filed on January 19, 2012, and incorporated herein by reference. | |
| 10.14† | Second Amendment to Employment Agreement with Ron Bentsur dated June 11, 2013, filed as Exhibit 10.1 to the Registrant’s Form 8-K filed on June 13, 2013, and incorporated herein by reference. | |
| 10.15† | Third Amended and Restated Directors Equity Compensation Plan, filed as Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2014, filed on August 7, 2014, and incorporated herein by reference. | |
60
Table of Contents
| 10.16† | Employment Agreement with James F. Oliviero, dated February 26, 2015. | |
| 10.17! | Manufacturing Services Agreement by and between Keryx Biopharmaceuticals, Inc. and Norwich Pharmaceuticals, Inc. dated January 17, 2014, filed as Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014, filed on November 6, 2014, and incorporated herein by reference. | |
| 10.18! | First Addendum to Manufacturing Services Agreement by and between Keryx Biopharmaceuticals, Inc. and Norwich Pharmaceuticals, Inc. dated October 24, 2014, filed as Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2014, filed on November 6, 2014, and incorporated herein by reference. | |
| 21.1 | List of subsidiaries of Keryx Biopharmaceuticals, Inc. | |
| 23.1 | Consent of UHY LLP. | |
| 24.1 | Power of Attorney of Director and Officers of Keryx Biopharmaceuticals, Inc. (included herein). | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 101 | The following financial information from Keryx Biopharmaceuticals, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2014, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Stockholders’ Equity, (iv) Consolidated Statements of Cash Flows, (v) the Notes to Consolidated Financial Statements. | |
| ! | Confidential treatment has been granted with respect to the omitted portions of this exhibit. |
| † | Indicates management contract or compensatory plan or arrangement. |
61
Table of Contents
Keryx Biopharmaceuticals, Inc.
Consolidated Financial Statements as of December 31, 2014
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Stockholders of Keryx Biopharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Keryx Biopharmaceuticals, Inc. (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2014. The Company’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Keryx Biopharmaceuticals, Inc. as of December 31, 2014 and 2013, and the consolidated results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated February 27, 2015, expressed an unqualified opinion thereon.
/s/ UHY LLP
New York, New York
February 27, 2015
F-1
Table of Contents
Keryx Biopharmaceuticals, Inc.
Consolidated Balance Sheets as of December 31,
(in thousands, except share and per share amounts)
| 2014 | 2013 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 74,284 | $ | 55,696 | ||||
| Short-term investment securities |
11,508 | — | ||||||
| Interest receivable |
48 | — | ||||||
| Inventory |
7,830 | — | ||||||
| Accounts receivable, net |
834 | — | ||||||
| Other current assets |
4,092 | 1,232 | ||||||
|
|
|
|
|
|||||
| Total current assets |
98,596 | 56,928 | ||||||
| Property, plant and equipment, net |
1,532 | 349 | ||||||
| Goodwill |
3,208 | 3,208 | ||||||
| Other assets, net |
292 | 281 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 103,628 | $ | 60,766 | ||||
|
|
|
|
|
|||||
| Liabilities and stockholders’ equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable and accrued expenses |
$ | 24,146 | $ | 14,004 | ||||
| Accrued compensation and related liabilities |
4,751 | 1,324 | ||||||
| Deferred revenue |
414 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
29,311 | 15,328 | ||||||
| Deferred tax liability |
700 | — | ||||||
| Other liabilities |
133 | 38 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
30,144 | 15,366 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Notes 14 and 15) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value per share (5,000,000 shares authorized, no shares issued and outstanding) |
— | — | ||||||
| Common stock, $0.001 par value per share (130,000,000 shares authorized, 92,758,789 and 82,723,145 shares issued, 92,678,841 and 82,643,197 shares outstanding at December 31, 2014 and 2013, respectively) |
93 | 83 | ||||||
| Additional paid-in capital |
624,606 | 485,014 | ||||||
| Treasury stock, at cost, 79,948 shares at December 31, 2014 and 2013, respectively |
(357 | ) | (357 | ) | ||||
| Accumulated deficit |
(550,858 | ) | (439,340 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
73,484 | 45,400 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 103,628 | $ | 60,766 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of the consolidated financial statements.
F-2
Table of Contents
Keryx Biopharmaceuticals, Inc.
Consolidated Statements of Operations for the Year Ended December 31,
(in thousands, except share and per share amounts)
| 2014 | 2013 | 2012 | ||||||||||
| License revenue |
$ | 10,825 | $ | 7,000 | $ | — | ||||||
| Operating expenses: |
||||||||||||
| License expenses |
495 | — | — | |||||||||
| Research and development |
51,502 | 34,734 | 20,031 | |||||||||
| Selling, general and administrative |
70,057 | 19,349 | 7,048 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
122,054 | 54,083 | 27,079 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating loss |
(111,229 | ) | (47,083 | ) | (27,079 | ) | ||||||
| Interest and other income, net |
411 | 351 | 1,719 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes and extraordinary gain |
(110,818 | ) | (46,732 | ) | (25,360 | ) | ||||||
| Income taxes |
700 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss before extraordinary gain |
(111,518 | ) | (46,732 | ) | (25,360 | ) | ||||||
| Extraordinary gain |
— | — | 2,639 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (111,518 | ) | $ | (46,732 | ) | $ | (22,721 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per common share: |
||||||||||||
| Loss before extraordinary gain |
$ | (1.23 | ) | $ | (0.58 | ) | $ | (0.36 | ) | |||
| Extraordinary gain |
— | — | 0.04 | |||||||||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per common share |
$ | (1.23 | ) | $ | (0.58 | ) | $ | (0.32 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average shares used in computing basic and diluted net loss per common share |
91,000,902 | 81,009,561 | 71,633,464 | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-3
Table of Contents
Keryx Biopharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
for the Years Ended December 31, 2014, 2013 and 2012
(in thousands, except share amounts)
| Common stock | Additional paid-in capital |
Treasury stock | Accumulated deficit |
Total | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
| Balance at January 1, 2012 |
71,102,899 | $ | 71 | $ | 401,220 | 79,948 | $ | (357 | ) | $ | (369,887 | ) | $ | 31,047 | ||||||||||||||
| Issuance of restricted stock |
997,300 | 1 | — | — | — | — | 1 | |||||||||||||||||||||
| Forfeiture of restricted stock |
(97,250 | ) | (— | )* | — | — | — | — | (— | )* | ||||||||||||||||||
| Compensation in respect of options and restricted stock granted to employees, directors and third-parties |
— | — | 2,167 | — | — | — | 2,167 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (22,721 | ) | (22,721 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2012 |
72,002,949 | $ | 72 | $ | 403,387 | 79,948 | $ | (357 | ) | $ | (392,608 | ) | $ | 10,494 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Issuance of common stock in public offering (net of offering costs of $5,640) |
9,469,100 | 10 | 74,743 | — | — | — | 74,753 | |||||||||||||||||||||
| Issuance of restricted stock |
831,020 | 1 | — | — | — | — | 1 | |||||||||||||||||||||
| Forfeiture of restricted stock |
(23,737 | ) | (— | )* | — | — | — | — | (— | )* | ||||||||||||||||||
| Issuance of common stock in connection with the exercise of options |
443,813 | — | * | 931 | — | — | — | 931 | ||||||||||||||||||||
| Compensation in respect of options and restricted stock granted to employees, directors and third-parties |
— | — | 5,953 | |
— |
|
— | — | 5,953 | |||||||||||||||||||
| Net loss |
— | — | — | — | — | (46,732 | ) | (46,732 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2013 |
82,723,145 | $ | 83 | $ | 485,014 | 79,948 | $ | (357 | ) | $ | (439,340 | ) | $ | 45,400 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Issuance of common stock in public offering (net of offering costs of $7,525) |
7,935,000 | 8 | 107,524 | — | — | — | 107,532 | |||||||||||||||||||||
| Issuance of restricted stock |
1,451,558 | 1 | — | — | — | — | 1 | |||||||||||||||||||||
| Forfeiture of restricted stock |
(88,859 | ) | (— | )* | — | — | — | — | (— | )* | ||||||||||||||||||
| Issuance of common stock in connection with the exercise of options |
737,945 | 1 | 5,053 | — | — | — | 5,054 | |||||||||||||||||||||
| Compensation in respect of options and restricted stock granted to employees, directors and third-parties |
— | — | 27,015 | — | — | — | 27,015 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (111,518 | ) | (111,518 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2014 |
92,758,789 | $ | 93 | $ | 624,606 | 79,948 | $ | (357 | ) | $ | (550,858 | ) | $ | 73,484 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| * | Amount less than one thousand dollars. |
The accompanying notes are an integral part of the consolidated financial statements.
F-4
Table of Contents
Keryx Biopharmaceuticals, Inc.
Consolidated Statements of Cash Flows for the Year Ended December 31,
(in thousands)
| 2014 | 2013 | 2012 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES |
||||||||||||
| Net loss |
$ | (111,518 | ) | $ | (46,732 | ) | $ | (22,721 | ) | |||
| Extraordinary gain |
— | — | 2,639 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss before extraordinary gain |
(111,518 | ) | (46,732 | ) | (25,360 | ) | ||||||
| Adjustments to reconcile loss to cash flows used in operating activities: |
||||||||||||
| Stock compensation expense |
26,957 | 5,953 | 2,167 | |||||||||
| Depreciation and amortization |
306 | 54 | 35 | |||||||||
| Deferred income taxes |
700 | — | — | |||||||||
| Changes in assets and liabilities: |
||||||||||||
| (Increase) decrease in other current assets |
(2,860 | ) | (802 | ) | 104 | |||||||
| Increase in accounts receivable, net |
(834 | ) | — | — | ||||||||
| (Increase) decrease in accrued interest receivable |
(48 | ) | — | 7 | ||||||||
| Increase in inventory |
(7,771 | ) | — | — | ||||||||
| (Increase) decrease in other assets |
(11 | ) | (83 | ) | 11 | |||||||
| Increase (decrease) in accounts payable and accrued expenses |
10,142 | 6,792 | (1,658 | ) | ||||||||
| Increase (decrease) in accrued compensation and related liabilities |
3,427 | 497 | (70 | ) | ||||||||
| Increase in deferred revenue |
414 | — | — | |||||||||
| Increase in other liabilities |
95 | 2 | 1 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(81,001 | ) | (34,319 | ) | (24,763 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| CASH FLOWS FROM INVESTING ACTIVITIES |
||||||||||||
| Purchases of property, plant and equipment |
(1,489 | ) | (346 | ) | (24 | ) | ||||||
| Investment in held-to-maturity short-term securities |
(49,771 | ) | (24,403 | ) | (11,263 | ) | ||||||
| Proceeds from maturity of held-to-maturity short-term securities |
38,263 | 24,403 | 15,475 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) provided by investing activities |
(12,997 | ) | (346 | ) | 4,188 | |||||||
|
|
|
|
|
|
|
|||||||
| CASH FLOWS FROM FINANCING ACTIVITIES |
||||||||||||
| Gross proceeds from public offerings |
115,057 | 80,393 | — | |||||||||
| Offering costs related to public offerings |
(7,525 | ) | (5,640 | ) | — | |||||||
| Proceeds from exercise of options |
5,054 | 931 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
112,586 | 75,684 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS |
18,588 | 41,019 | (20,575 | ) | ||||||||
| Cash and cash equivalents at beginning of year |
55,696 | 14,677 | 35,252 | |||||||||
|
|
|
|
|
|
|
|||||||
| CASH AND CASH EQUIVALENTS AT END OF YEAR |
$ | 74,284 | $ | 55,696 | $ | 14,677 | ||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-5
Table of Contents
Keryx Biopharmaceuticals, Inc.
Notes to the Consolidated Financial Statements
Unless the context requires otherwise, references in this report to “Keryx,” “Company,” “we,” “us” and “our” refer to Keryx Biopharmaceuticals, Inc. and our subsidiaries.
NOTE 1 - ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
DESCRIPTION OF BUSINESS
We are a biopharmaceutical company focused on bringing innovative therapies to market for patients with renal disease. Our first product, AuryxiaTM (ferric citrate), an oral, absorbable iron-based compound, received marketing approval from the U.S. Food and Drug Administration, or FDA, in September 2014 for the control of serum phosphorus levels in patients with chronic kidney disease (“CKD”) on dialysis. Auryxia, which was launched in December 2014, is being marketed in the U.S. through our specialty salesforce and commercial infrastructure. We currently have 60 sales representatives in the field calling on approximately 5,000 target nephrologists.
Our Japanese partner, Japan Tobacco Inc. (“JT”) and Torii Pharmaceutical Co. Ltd. (“Torii”), received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare as an oral treatment for the improvement of hyperphosphatemia in patients with CKD, including dialysis and non-dialysis dependent CKD (“NDD-CKD”), in January 2014. JT’s subsidiary, Torii, launched the product under the brand name Riona® in May 2014.
We have also submitted, in March 2014, a Marketing Authorization Application (“MAA”) with the European Medicines Agency (“EMA”) for the approval of Auryxia in patients with CKD, including dialysis and NDD-CKD. Also in March 2014, the EMA validated our MAA, confirming that the submission is sufficiently complete to begin the formal review process.
In September 2014, we announced the initiation of a pivotal Phase 3 study of Auryxia for the treatment of iron deficiency anemia (“IDA”) in patients with Stage 3-5 NDD-CKD. This study’s primary endpoint is the between group comparison of the proportion of patients achieving a 1 g/dL or greater increase in hemoglobin at any point during the 16-week randomized period.
Currently, our only drug product is Auryxia. We may engage in business development activities that include seeking strategic relationships for Auryxia, as well as evaluating other compounds and companies for in-licensing or acquisition. To date, we have not recognized revenue on any prescription sales from Auryxia or any other drug product. We have generated, and expect to continue to generate, revenue from the sublicensing of rights to Auryxia in Japan to our Japanese partner, JT and Torii.
We own a 100% interest in each of ACCESS Oncology, Inc. (“ACCESS Oncology”), Neryx Biopharmaceuticals, Inc., and Accumin Diagnostics, Inc. (“ADI”), all inactive U.S. corporations incorporated in the State of Delaware. Most of our biopharmaceutical development and substantially all of our administrative operations during 2014, 2013 and 2012 were conducted in the U.S.
LIQUIDITY AND CAPITAL RESOURCES
Except for 2009, we have incurred substantial operating losses since our inception, and expect to continue to incur operating losses for the foreseeable future and may never become profitable. As of December 31, 2014, we have an accumulated deficit of $550.9 million.
Our major sources of cash have been proceeds from various public and private offerings of our common stock, option and warrant exercises, interest income, and from the upfront and milestone payments from our
F-6
Table of Contents
Sublicense Agreement with JT and Torii and miscellaneous payments from our other prior licensing activities. Prior to the launch of Auryxia in December 2014, we have not commercialized any drug, and we may not become profitable. Our ability to achieve profitability depends on a number of factors, including our ability to complete our development efforts, obtain additional regulatory approvals for our drug, successfully complete any post-approval regulatory obligations and successfully manufacture and commercialize our drug alone or in partnership. We may continue to incur substantial operating losses even after we begin to generate revenues from our drug.
In January 2015, we raised approximately $118.3 million, net of underwriting discounts and offering expenses of approximately $8.2 million, in an underwritten public offering. The shares were sold under Registration Statements (Nos. 333-201605 and 333-201639) on Form S-3 and Form S-3MEF, respectively, filed by us with the Securities and Exchange Commission.
We currently expect that our existing capital resources combined with future anticipated cash flows will be sufficient to execute our business plan. The actual amount of cash that we will need to operate is subject to many factors, including, but not limited to, the timing and expenditures associated with the build-up of inventory and capacity expansion, the timing and expenditures associated with the regulatory review process for our EU MAA filing, the timing and expenditures associated with commercial activities related to Auryxia, and the timing, design and conduct of clinical trials for Auryxia. As a result of these factors, we may need to seek additional financings to provide the cash necessary to execute our current operations, including beyond the initial commercialization of Auryxia, and to develop any drug candidates we may in-license or acquire.
Our common stock is listed on the Nasdaq Capital Market and trades under the symbol “KERX.”
CORPORATE
In January 2014, we formed a subsidiary in the United Kingdom, Keryx Biopharma UK Ltd., related to the submission of our MAA in Europe.
In March 2014, we entered into a sublease for approximately 10,395 square feet of leased office space in Boston, Massachusetts, with a term through December 31, 2015.
In January 2015, we announced the transitioning of the role of Chief Executive Officer from Ron Bentsur to Greg Madison. Mr. Madison joined Keryx in February 2014 as Executive Vice President and Chief Operating Officer to transition Keryx from a development-stage organization into a fully integrated commercial entity, bringing to Keryx a wealth of relevant expertise in both the phosphate binder and iron deficiency anemia markets. Mr. Madison has been appointed President of Keryx and will work with Mr. Bentsur to ensure a successful leadership transition by the end of May, when Mr. Bentsur’s contract expires.
In February 2015, we announced a planned consolidation of our finance and accounting function into our Boston office and that our Chief Financial Officer, James Oliviero, will be leaving Keryx by October 2015. Mr. Oliviero has been with Keryx for twelve years and has served as the Chief Financial Officer since 2009. We have commenced a search for a new Chief Financial Officer who will be based in our Boston office. Mr. Oliviero will continue to manage our finance and accounting team during the remainder of his tenure and will assist in the transition of his duties to the new Chief Financial Officer.
RECENTLY ISSUED ACCOUNTING STANDARDS
In August 2014, the Financial Accounting Standards Board issued a new standard, Accounting Standards Update No. 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. This new standard will explicitly require management to assess an entity’s ability to continue as a going concern and to provide footnote disclosures in certain cases. Currently there is no guidance in GAAP about management’s
F-7
Table of Contents
responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern. The new standard applies to all entities and provides an explicit requirement that management assesses and discloses going concern uncertainties. Previous guidance in auditing standards required auditors to evaluate going concern. The new standard will be effective for all entities in the first annual period ending after December 15, 2016, which is December 31, 2016 for calendar year-end entities. Earlier application is permitted.
In May 2014, the Financial Accounting Standards Board issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2016 and allows for adoption using a full retrospective method, or a modified retrospective method. We are currently assessing the method of adoption and the expected impact the new standard has on our financial position and results of operations.
PRINCIPLES OF CONSOLIDATION
The consolidated financial statements include our financial statements and those of our wholly-owned subsidiaries. Intercompany transactions and balances have been eliminated in consolidation.
USE OF ESTIMATES
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (“GAAP”) requires management to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of these consolidated financial statements and the reported amounts of revenues and expenses during the applicable reporting period. Actual results could differ from those estimates. Such differences could be material to these consolidated financial statements.
CASH AND CASH EQUIVALENTS
We treat liquid investments with original maturities of three months or less when purchased as cash and cash equivalents.
INVESTMENT SECURITIES
We classify our short-term debt securities as held-to-maturity. Held-to-maturity securities are those securities in which we have the ability and intent to hold the security until maturity. Held-to-maturity securities are recorded at amortized cost, adjusted for the amortization or accretion of premiums or discounts. Premiums and discounts are amortized or accreted over the life of the related held-to-maturity security as an adjustment to yield using the effective interest method. Available-for-sale investment securities are recorded at fair value. Other-than-temporary impairment charges are included in interest and other income, net, and unrealized gains (losses), if determined to be temporary, are included in accumulated other comprehensive income (loss) in stockholders’ equity.
INVENTORY
Inventories are stated at the lower of cost or estimated realizable value. We determine the cost of our inventories, which include amounts related to materials, third-party contract manufacturing and packaging services, and manufacturing overhead, on a first-in, first-out basis. We capitalize inventory costs at our suppliers when, based on management’s judgment, the realization of future economic benefit is probable at each given
F-8
Table of Contents
supplier. We received FDA approval for Auryxia on September 5, 2014, and on that date began capitalizing inventory purchases of saleable product from certain suppliers. Prior to FDA approval, all saleable product purchased from such suppliers were included as a component of research and development expense.
ACCOUNTS RECEIVABLE, NET
We extend credit to our customers for product sales resulting in accounts receivable. Customer accounts are monitored for past due amounts. Past due accounts receivable, determined to be uncollectible, are written off against the allowance for doubtful accounts. Allowances for doubtful accounts are estimated based upon past due amounts, historical losses and existing economic factors, and are adjusted periodically. We offer cash discounts to our customers, generally 2% of the sales price, as an incentive for prompt payment. The estimate of cash discounts is recorded at the time of sale. We account for the cash discounts by reducing revenue and accounts receivable by the amount of the discounts we expect our customers to take. The accounts receivable are reported in the consolidated balance sheets, net of the allowances for doubtful accounts and cash discounts. There was no allowance for doubtful accounts at December 31, 2014 and 2013.
PROPERTY, PLANT AND EQUIPMENT
Property, plant and equipment are stated at historical cost. Depreciation is computed using the straight-line method over the estimated useful lives of the assets:
| Estimated useful life (years) |
||||
| Office furniture and equipment |
3-7 | |||
| Computers, software and related equipment |
3 | |||
Leasehold improvements are amortized over the shorter of their useful life or the remaining term of the lease exclusive of renewal options.
PATENT COSTS
We expense patent maintenance costs as incurred. We have classified our patent expenses in selling, general and administrative.
REVENUE RECOGNITION
We recognize license revenue in accordance with the revenue recognition guidance of the FASB Accounting Standards Codification (the “Codification”). We analyze each element of our licensing agreement to determine the appropriate revenue recognition. The terms of the license agreement may include payments to us of non-refundable up-front license fees, milestone payments if specified objectives are achieved, and/or royalties on product sales. We recognize revenue from upfront payments over the period of significant involvement under the related agreements unless the fee is in exchange for products delivered or services rendered that represent the culmination of a separate earnings process and no further performance obligation exists under the contract. We recognize milestone payments as revenue upon the achievement of specified milestones only if (1) the milestone payment is non-refundable, (2) substantive effort is involved in achieving the milestone, (3) the amount of the milestone is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (4) the milestone is at risk for both parties. If any of these conditions are not met, we defer the milestone payment and recognize it as revenue over the estimated period of performance under the contract.
For arrangements for which royalty revenue information becomes available and collectability is reasonably assured, we recognize revenue during the applicable period earned. When collectability is reasonably assured but a reasonable estimate of royalty revenue cannot be made, the royalty revenue is recognized in the quarter that the licensee provides the written report and related information to us.
F-9
Table of Contents
Our commercial launch of Auryxia occurred in December 2014. We sell product to a limited number of major wholesalers, our Distributors, as well as certain pharmacies, or collectively, our Customers. Our Distributors resell the product to retail pharmacies for purposes of their reselling the product to fill patient prescriptions. In accordance with GAAP, until we have the ability to reliably estimate returns of Auryxia from our Customers, revenue will be recognized based on the resale of Auryxia for the purposes of filling patient prescriptions, and not based on sales from us to such Customers. Consistent with industry practice, once we achieve sufficient history such that we can reliably estimate returns based on sales to our Customers, we anticipate that our revenues will be recognized based on sales to our Customers. We currently defer Auryxia revenue recognition until the earlier of the product being resold for purposes of filling patient prescriptions and the expiration of the right of return (twelve months after the expiration date of the product). The deferred revenue is recorded net of discounts, rebates, and chargebacks. We also defer the related cost of product sales and record such amounts as finished goods inventory held by others, which is included in inventory on our consolidated balance sheet, until revenue related to such product sales is recognized.
LICENSE EXPENSES
License expenses include royalty and other expenses due to the licensor of Auryxia related to our license agreement with JT and Torii. With regard to royalty expense, such expense is directly related to the royalty revenue received from JT and Torii and is recognized in the same period as the revenue is recorded. Other expenses are recognized in the period they are incurred.
RESEARCH AND DEVELOPMENT COSTS
Research and development costs are expensed as incurred. Pre-approval inventory expenditures are recorded as research and development expense as incurred. The capitalization of inventory for our product candidate(s) commence when it is probable that the product will be approved for commercial marketing. Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and amortized over the period that the goods are delivered or the related services are performed, subject to an assessment of recoverability. We make estimates of costs incurred in relation to external clinical research organizations (“CROs”) and clinical site costs. We analyze the progress of clinical trials, including levels of patient enrollment, invoices received and contracted costs when evaluating the adequacy of the amount expensed and the related prepaid asset and accrued liability. Significant judgments and estimates must be made and used in determining the accrued balance and expense in any accounting period. We review and accrue CRO expenses and clinical trial study expenses based on work performed and rely upon estimates of those costs applicable to the stage of completion of a study. Accrued CRO costs are subject to revisions as such trials progress to completion. Revisions are charged to expense in the period in which the facts that give rise to the revision become known. With respect to clinical site costs, the financial terms of these agreements are subject to negotiation and vary from contract to contract. Payments under these contracts may be uneven, and depend on factors such as the achievement of certain events, the successful recruitment of patients, the completion of portions of the clinical trial or similar conditions. The objective of our policy is to match the recording of expenses in our consolidated financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical site costs are recognized based on our estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
INCOME TAXES
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, operating losses and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The
F-10
Table of Contents
effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. If the likelihood of realizing the deferred tax assets or liability is less than “more likely than not,” a valuation allowance is then created.
We, and our subsidiaries, file income tax returns in the U.S. federal jurisdiction and in various states. Our subsidiary, Keryx Biopharma UK Ltd., files annual returns and accounts in the United Kingdom. We have tax net operating loss carryforwards that are subject to examination for a number of years beyond the year in which they were generated for tax purposes. Since a portion of these net operating loss carryforwards may be utilized in the future, many of these net operating loss carryforwards will remain subject to examination.
We are continuing our practice of recognizing interest and penalties related to uncertain income tax positions in income tax expense.
STOCK - BASED COMPENSATION
We recognize all share-based payments to employees and to non-employee directors for service on our board of directors as compensation expense in the consolidated financial statements based on the grant date fair values of such payments. Stock-based compensation expense recognized each period is based on the value of the portion of share-based payment awards that is ultimately expected to vest during the period. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
For share-based payments to consultants and other third-parties, compensation expense is determined at the “measurement date.” The expense is recognized over the vesting period of the award. Until the measurement date is reached, the total amount of compensation expense remains uncertain. We record compensation expense based on the fair value of the award at the reporting date. The awards to consultants and other third-parties are then revalued, or the total compensation is recalculated based on the then current fair value, at each subsequent reporting date.
BASIC AND DILUTED NET LOSS PER COMMON SHARE
Basic net loss per share is computed by dividing the losses allocable to common stockholders by the weighted average number of shares of common stock outstanding for the period. Diluted net loss per share does not reflect the effect of shares of common stock to be issued upon the exercise of stock options and warrants, as their inclusion would be anti-dilutive. The options outstanding as of December 31, 2014, 2013 and 2012, which are not included in the computation of net loss per share amounts, were 5,132,426, 3,845,370 and 3,401,671, respectively. No warrants were outstanding during each of these periods.
SEGMENT REPORTING
We operate in only one reportable segment: the Products segment.
ACQUISITIONS
We adopted Accounting Standards Codification (“ASC”) Topic 805, Business Combinations, as of January 1, 2009. The adoption of ASC Topic 805 was effective on a prospective basis. Prior to the adoption of ASC Topic 805, we accounted for acquired businesses using the purchase method of accounting which required that the assets acquired and liabilities assumed be recorded at the date of acquisition at their respective fair values. Our consolidated financial statements and results of operations through 2008 reflected an acquired business after the completion of the acquisition and were not retroactively restated. The cost to acquire a business, including transaction costs, was allocated to the underlying net assets of the acquired business in proportion to their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired was recorded as goodwill. Any excess of the net assets acquired over the purchase price represented negative goodwill.
F-11
Table of Contents
IMPAIRMENT
Long lived assets are reviewed for an impairment loss when circumstances indicate that the carrying value of long-lived tangible and intangible assets with finite lives may not be recoverable. Management’s policy in determining whether an impairment indicator exists, a triggering event, comprises measurable operating performance criteria as well as qualitative measures. If an analysis is necessitated by the occurrence of a triggering event, we make certain assumptions in determining the impairment amount. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to future undiscounted cash flows expected to be generated by the asset or used in its disposal. If the carrying amount of an asset exceeds its estimated future undiscounted cash flows, an impairment charge is recognized.
Goodwill is reviewed for impairment annually, or when events arise that could indicate that an impairment exists. We test for goodwill impairment using a two-step process. The first step compares the fair value of the reporting unit with the unit’s carrying value, including goodwill. When the carrying value of the reporting unit is greater than fair value, the unit’s goodwill may be impaired, and the second step must be completed to measure the amount of the goodwill impairment charge, if any. In the second step, the implied fair value of the reporting unit’s goodwill is compared with the carrying amount of the unit’s goodwill. If the carrying amount is greater than the implied fair value, the carrying value of the goodwill must be written down to its implied fair value. As of December 31, 2012, 2013 and 2014, management conducted its annual assessments of goodwill and concluded that there were no impairments. We will continue to perform impairment tests annually, at December 31, and whenever events or changes in circumstances suggest that the carrying value of an asset may not be recoverable.
CONCENTRATIONS OF CREDIT RISK
We do not have significant off-balance-sheet risk or credit risk concentrations. We maintain our cash and cash equivalents and held-to-maturity investments, when applicable, with multiple financial institutions that invest in investment-grade securities with average maturities of less than twelve months. See Note 3 – Investment Securities and Note 4 – Fair Value Measurements.
NOTE 2 – CASH AND CASH EQUIVALENTS
| (in thousands) |
December 31, 2014 |
December 31, 2013 |
||||||
| Money market funds |
$ | 69,591 | $ | 29,904 | ||||
| Checking and bank deposits |
4,693 | 25,792 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 74,284 | $ | 55,696 | ||||
|
|
|
|
|
|||||
A significant portion of our cash is maintained in Federal Deposit Insurance Corporation (“FDIC”) insured accounts at credit qualified financial institutions. At times, such amounts may exceed the FDIC insurance limits. At December 31, 2014, uninsured cash balances totaled approximately $73.8 million.
NOTE 3 - INVESTMENT SECURITIES
We record our investments as either held-to-maturity or available-for-sale. Held-to-maturity investments are recorded at amortized cost. Available-for-sale investment securities are recorded at fair value (see Note 4 – Fair Value Measurements). Other-than-temporary impairment charges are included in interest and other income, net, and unrealized gains (losses), if determined to be temporary, are included in accumulated other comprehensive income (loss) in stockholders’ equity.
F-12
Table of Contents
The following tables summarize our investment securities at December 31, 2014:
| December 31, 2014 | ||||||||||||||||
| (in thousands) |
Amortized cost, as adjusted |
Gross unrealized holding gains |
Gross unrealized holding losses |
Estimated fair value |
||||||||||||
| Short-term investments (held-to-maturity): |
||||||||||||||||
| Obligations of domestic governmental agencies (mature January 2015) |
$ | 11,508 | $ | — | $ | — | $ | 11,508 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total short-term investment securities |
$ | 11,508 | $ | — | $ | — | $ | 11,508 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
We were not invested in investment securities at December 31, 2013.
NOTE 4 – FAIR VALUE MEASUREMENTS
We measure certain financial assets and liabilities at fair value on a recurring basis in the financial statements. The hierarchy ranks the quality and reliability of inputs, or assumptions, used in the determination of fair value and requires financial assets and liabilities carried at fair value to be classified and disclosed in one of the following three categories:
| • | Level 1 – quoted prices in active markets for identical assets and liabilities; |
| • | Level 2 – inputs other than Level 1 quoted prices that are directly or indirectly observable; and |
| • | Level 3 – unobservable inputs that are not corroborated by market data. |
We review investment securities for impairment and to determine the classification of the impairment as temporary or other-than-temporary. Losses are recognized in our consolidated statement of operations when a decline in fair value is determined to be other-than-temporary. We review our investments on an ongoing basis for indications of possible impairment. Once identified, the determination of whether the impairment is temporary or other-than-temporary requires significant judgment.
The following table provides the fair value measurements of applicable financial assets as of December 31, 2014 and 2013:
| Financial assets at fair value as of December 31, 2014 |
||||||||||||
| (in thousands) |
Level 1 | Level 2 | Level 3 | |||||||||
| Money market funds (1) |
$ | 69,591 | $ | — | $ | — | ||||||
| Obligations of domestic governmental agencies (held-to-maturity) (2) |
11,508 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 81,099 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Financial assets at fair value as of December 31, 2013 |
||||||||||||
| (in thousands) |
Level 1 | Level 2 | Level 3 | |||||||||
| Money market funds (1) |
$ | 29,904 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 29,904 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| (1) | Included in cash and cash equivalents on our consolidated balance sheet. The carrying amount of money market funds approximates fair value. |
| (2) | Amortized cost approximates fair value. |
F-13
Table of Contents
NOTE 5 - INVENTORY
Upon approval of Auryxia on September 5, 2014 by the FDA, we began capitalizing our purchases of saleable inventory of Auryxia from suppliers. Inventories consist of the following (in thousands):
| December 31, 2014 |
December 31, 2013 |
|||||||
| Raw materials |
$ | 111 | $ | — | ||||
| Work in process |
7,263 | — | ||||||
| Finished goods |
409 | — | ||||||
| Finished goods inventory held by others |
47 | — | ||||||
|
|
|
|
|
|||||
| Total inventory |
$ | 7,830 | $ | — | ||||
|
|
|
|
|
|||||
NOTE 6 - PROPERTY, PLANT AND EQUIPMENT
| (in thousands) |
December 31, 2014 |
December 31, 2013 |
||||||
| Leasehold improvements |
$ | 39 | $ | 32 | ||||
| Office furniture and equipment |
853 | 556 | ||||||
| Computers, software and related equipment |
1,823 | 638 | ||||||
|
|
|
|
|
|||||
| 2,715 | 1,226 | |||||||
| Accumulated depreciation and amortization |
(1,183 | ) | (877 | ) | ||||
|
|
|
|
|
|||||
| Net book value |
$ | 1,532 | $ | 349 | ||||
|
|
|
|
|
|||||
The following table summarizes depreciation expense for the years ended December 31, 2014, 2013 and 2012.
| For the year ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Depreciation expense: |
||||||||||||
| Research and development |
$ | 56 | $ | 32 | $ | 21 | ||||||
| Selling, general and administrative |
250 | 22 | 14 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 306 | $ | 54 | $ | 35 | ||||||
|
|
|
|
|
|
|
|||||||
NOTE 7 – GOODWILL
On April 6, 2006, ADI, our wholly-owned subsidiary, completed the acquisition of AccuminTM, a novel, patent protected, diagnostic for the direct measurement of total, intact urinary albumin, from AusAm Biotechnologies, Inc. The purchase price of Accumin was $4.0 million. We accounted for the ADI transaction as a purchase. The excess of the purchase price over the net assets acquired in the ADI transaction represented goodwill of approximately $3.2 million, which was allocated to our Products segment based on the proposed synergies with our then existing drug pipeline activities. In September 2008, we terminated our license agreement related to the ADI product.
F-14
Table of Contents
NOTE 8 - OTHER ASSETS
| (in thousands) |
December 31, 2014 |
December 31, 2013 |
||||||
| Patents |
$ | 352 | $ | 352 | ||||
| Deposits |
279 | 163 | ||||||
| Deferred registration fees |
13 | 118 | ||||||
|
|
|
|
|
|||||
| 644 | 633 | |||||||
| Accumulated amortization |
(352 | ) | (352 | ) | ||||
|
|
|
|
|
|||||
| $ | 292 | $ | 281 | |||||
|
|
|
|
|
|||||
There were no amortization expenses for the years ended December 31, 2014, 2013 and 2012. We do not expect to record amortization expenses going forward, as all definite-lived intangible assets are fully amortized.
NOTE 9 - LICENSE AGREEMENTS
In November 2005, we entered into a license agreement with Panion & BF Biotech, Inc. (“Panion”). Under the license agreement, we acquired the exclusive worldwide rights, excluding certain Asian-Pacific countries, for the development and marketing of Auryxia. To date, we have paid an aggregate of $9.6 million to Panion, including the $3.0 million milestone payment paid upon the FDA approval of Auryxia, and Panion is eligible to receive one additional milestone payment of $2.0 million upon our successful achievement of European marketing approval, in addition to royalty payments based on a mid-single digit percentage of net sales of Auryxia.
In September 2007, we entered into a Sublicense Agreement with JT and Torii, JT’s pharmaceutical business subsidiary, under which JT and Torii obtained the exclusive sublicense rights for the development and commercialization of ferric citrate in Japan, which is being marketed in the U.S. under the trade name Auryxia. JT and Torii are responsible for the future development and commercialization costs in Japan. Effective as of June 8, 2009, we entered into an Amended and Restated Sublicense Agreement (the “Revised Agreement”) with JT and Torii, which, among other things, provided for the elimination of all significant on-going obligations under the sublicense agreement.
In January 2013, JT and Torii filed its new drug application (“NDA”) with the Japanese Ministry of Health, Labour and Welfare for marketing approval of ferric citrate in Japan for the treatment of hyperphosphatemia in patients with CKD. Under the terms of the license agreement with JT and Torii, we received a non-refundable milestone payment of $7.0 million in January 2013 for the achievement of the NDA filing milestone. As a result, we recorded license revenue of $7.0 million in accordance with our revenue recognition policy, which is included in the year ended December 31, 2013.
In January 2014, JT and Torii received manufacturing and marketing approval of ferric citrate from the Japanese Ministry of Health, Labour and Welfare. Ferric citrate, launched in May 2014 and being marketed in Japan by JT’s subsidiary, Torii Pharmaceutical Co., Ltd., under the brand name Riona®, is indicated as an oral treatment for the improvement of hyperphosphatemia in patients with CKD. Under the terms of the license agreement with JT and Torii, we received a non-refundable payment of $10.0 million in February 2014 for the achievement of the marketing approval milestone. As a result, we recorded license revenue of $10.0 million in accordance with our revenue recognition policy, which is included in the year ended December 31, 2014. We also receive royalty payments based on a tiered double-digit percentage of net sales of Riona® in Japan escalating up to the mid-teens, as well as up to an additional $55.0 million upon the achievement of certain annual net sales milestones. In accordance with our revenue recognition policy, royalty revenues are recognized in the quarter that JT and Torii provide their written report and related information to us regarding sales of Riona®, which generally will be one quarter following the quarter in which the underlying sales by JT and Torii occurred. For the year ended December 31, 2014, we recorded $0.8 million in license revenue related to royalties earned on net
F-15
Table of Contents
sales of Riona® in Japan. We record the associated mid-single digit percentage of net sales royalty expense due Panion, the licensor of Auryxia, in the same period as the royalty revenue from JT and Torii is recorded. For the year ended December 31, 2014, we recorded $0.5 million in license expenses related to royalties due to the licensor of Auryxia relating to sales of Riona® in Japan.
On April 2, 2012, we reported that the Phase 3 “X-PECT” (Xeloda® + Perifosine Evaluation in Colorectal cancer Treatment) clinical trial evaluating perifosine (KRX-0401) + capecitabine (Xeloda) in patients with refractory advanced colorectal cancer did not meet the primary endpoint of improving overall survival versus capecitabine + placebo. On May 4, 2012, we executed a License Termination and Technology Transfer Agreement with Aeterna Zentaris GmbH (“Zentaris”), whereby the license agreement for KRX-0401 (perifosine) was terminated, and in exchange for the transfer of the U.S. Investigational New Drug Application, development data, intellectual property and contracts to Zentaris, we will receive a royalty on future net sales, if any, of perifosine in the U.S., Canada and Mexico. Zentaris has assumed all costs related to the Perifosine program going forward.
NOTE 10 – CONTINGENT EQUITY RIGHTS
On February 5, 2004, we acquired ACCESS Oncology, a related party, for a purchase price of approximately $19.5 million. The purchase price included our assumption of certain liabilities of ACCESS Oncology equal to approximately $8.7 million, the issuance of shares of our common stock valued at approximately $6.3 million, contingent equity rights valued at approximately $4.0 million and transaction costs of approximately $0.5 million.
At the effective date of the merger, each share of ACCESS Oncology common stock, including shares issuable upon the exercise of options exercised before March 1, 2004, and upon the exercise of outstanding warrants, was converted into the right to share in the contingent equity rights pro rata with the other holders of ACCESS Oncology common stock. Pursuant to the merger agreement, 623,145 shares of our common stock valued at approximately $6.3 million have been issued to the former preferred stockholders of ACCESS Oncology. An additional 4,433 shares of our common stock are issuable to those preferred stockholders of ACCESS Oncology who have yet to provide the necessary documentation to receive shares of our common stock.
On December 16, 2009, we announced the initiation of a Phase 3 registration trial of KRX-0401 (perifosine) for the treatment of patients with relapsed / refractory multiple myeloma. The achievement of this event triggered contingent milestone stock consideration payable to the former stockholders of ACCESS Oncology in the amount of an aggregate of 500,000 shares of our common stock valued at $1.4 million.
Due to the termination of the license for KRX-0401 in May 2012, we were no longer committed to pay to the former stockholders of ACCESS Oncology, Inc. certain contingent equity rights (up to 2,872,422 shares of our common stock). For the year ended December 31, 2012, we recognized a non-cash extraordinary gain of $2.6 million relating to the write-off of the contingent equity rights liability.
NOTE 11 - STOCKHOLDERS’ EQUITY
Preferred Stock
Our amended and restated certificate of incorporation authorizes the issuance of up to 5,000,000 shares of preferred stock, $0.001 par value, with rights senior to those of our common stock.
Common Stock
On June 18, 2013, at the 2013 Annual Meeting of Stockholders, the Company’s stockholders approved an amendment to the Company’s amended and restated certificate of incorporation increasing the shares of authorized common stock from 95,000,000 shares to 130,000,000 shares $0.001 par value common stock. The number of authorized shares of preferred stock remains unchanged at 5,000,000 shares.
F-16
Table of Contents
On January 22, 2014, we announced the pricing of an underwritten public offering in which we sold 7,935,000 shares of our common stock at a price of $14.50 per share for gross proceeds of approximately $115.1 million. Net proceeds from this offering were approximately $107.5 million, net of underwriting discounts and offering expenses of approximately $7.5 million. The shares were sold under a Registration Statement (No. 333-190353) on Form S-3, filed by us with the Securities and Exchange Commission.
On January 21, 2015, we announced the pricing of an underwritten public offering in which we sold 10,541,667 shares of our common stock at a price of $12.00 per share for gross proceeds of approximately $126.5 million. Net proceeds from this offering were approximately $118.3 million, net of underwriting discounts and offering expenses of approximately $8.2 million. The shares were sold under Registration Statements (Nos. 333-201605 and 333-201639) on Form S-3 and Form S-3MEF, respectively, filed by us with the Securities and Exchange Commission.
Treasury Stock
As of December 31, 2014 and 2013, we held a total of 79,948 shares of our common stock in treasury, at a total cost of $0.4 million.
Equity Incentive Plans
We have in effect the following stock option and incentive plans.
a. The 1999 Stock Option Plan was adopted in November 1999. Under the 1999 Stock Option Plan, our board of directors could grant stock-based awards to directors, consultants and employees. The plan authorizes grants to purchase up to 4,230,000 shares of authorized but unissued common stock. The plan limits the term of each option, to no more than 25 years from the date of the grant, unless otherwise authorized by the board. The plan permits the board of directors or a committee appointed by the board to administer the plan. The administrator has the authority, in its discretion, to determine the terms and conditions of any option granted to a service provider, including the vesting schedule. As of December 31, 2014, no additional shares of our common stock may be issued under the 1999 Stock Option Plan.
b. The 2004 Long-Term Incentive Plan was adopted in June 2004 by our stockholders. Under the 2004 Long-Term Incentive Plan, the compensation committee of our board of directors is authorized to grant stock-based awards to directors, consultants and employees. The 2004 plan authorizes grants to purchase up to 4,000,000 shares of authorized but unissued common stock. The plan limits the term of each option to no more than 10 years from the date of grant. As of December 31, 2014, no additional shares of our common stock may be issued under the 2004 Long-Term Incentive Plan.
c. The 2007 Incentive Plan was adopted in June 2007 by our stockholders. Under the 2007 Incentive Plan, the compensation committee of our board of directors is authorized to grant stock-based awards to directors, consultants, employees and officers. The 2007 Incentive Plan authorizes grants to purchase up to 6,000,000 shares of authorized but unissued common stock. The plan limits the term of each option to no more than 10 years from the date of grant. As of December 31, 2014, up to an additional 11,921 shares may be issued under the 2007 Incentive Plan.
d. The 2009 CEO Incentive Plan was adopted in May 2009. Under the 2009 CEO Incentive Plan, our board of directors granted an option to Ron Bentsur, our Chief Executive Officer, to purchase up to 600,000 shares of authorized but unissued common stock. The option has a term of 10 years from the date of grant. As of December 31, 2014, the option is fully vested and exercisable.
e. The 2013 Incentive Plan was adopted in June 2013 by our stockholders at our 2013 Annual Meeting of Stockholders. The 2013 Incentive plan was amended by our stockholders at a special meeting of our stockholders
F-17
Table of Contents
in November 2014, which increased the number of authorized shares issuable thereunder from 3,500,000 to 9,500,000. Under the 2013 Incentive Plan, the compensation committee of the Company’s board of directors is authorized to grant stock-based awards to directors, officers, employees and consultants. The plan limits the term of each option to no more than 10 years from the date of their grant. As of December 31, 2014, up to an additional 5,516,605 shares may be issued under the 2013 Incentive Plan.
Total shares available for the issuance of stock options or other stock-based awards under our stock option and incentive plans were 5,528,526 shares at December 31, 2014.
Stock Options
The following table summarizes stock option activity for all plans for the years ended December 31, 2014, 2013 and 2012:
| Number of shares |
Weighted- average exercise price |
Weighted- average Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at January 1, 2012 |
3,517,000 | 6.40 | $ | 2,139,130 | ||||||||||||
|
|
|
|||||||||||||||
| Granted |
521,500 | 2.36 | ||||||||||||||
| Exercised |
— | — | $ | — | ||||||||||||
|
|
|
|||||||||||||||
| Forfeited |
(266,621 | ) | 7.77 | |||||||||||||
| Expired |
(370,208 | ) | 11.02 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2012 |
3,401,671 | 5.17 | $ | 2,373,509 | ||||||||||||
|
|
|
|||||||||||||||
| Granted |
932,366 | 6.29 | ||||||||||||||
| Exercised |
(443,813 | ) | 2.10 | $ | 4,614,741 | |||||||||||
|
|
|
|||||||||||||||
| Forfeited |
(44,854 | ) | 8.70 | |||||||||||||
| Expired |
— | — | ||||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2013 |
3,845,370 | 5.75 | $ | 28,361,438 | ||||||||||||
|
|
|
|
|
|||||||||||||
| Granted |
2,264,550 | 14.69 | ||||||||||||||
| Exercised |
(737,945 | ) | 6.85 | $ | 6,252,223 | |||||||||||
|
|
|
|||||||||||||||
| Forfeited |
(239,549 | ) | 10.38 | |||||||||||||
| Expired |
— | — | ||||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2014 |
5,132,426 | $ | 9.32 | 7.0 | $ | 26,916,823 | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Vested and expected to vest at December 31, 2014 |
5,046,754 | $ | 9.25 | 6.9 | $ | 26,784,040 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Exercisable at December 31, 2014 |
2,575,056 | $ | 5.29 | 4.8 | $ | 22,953,150 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The following table summarizes information about stock options outstanding at December 31, 2014:
| Options outstanding | Options exercisable | |||||||||||||||||||
| Range of exercise prices |
Number outstanding |
Weighted- average remaining contractual life (years) |
Weighted- average exercise price |
Number exercisable |
Weighted- average exercise price |
|||||||||||||||
| $0.10 - $ 3.00 |
1,417,063 | 5.6 | $ | 1.44 | 1,243,990 | $ | 1.29 | |||||||||||||
| 3.70 - 8.56 |
818,900 | 6.5 | 5.72 | 683,482 | 5.29 | |||||||||||||||
| 9.34 - 17.23 |
2,896,463 | 7.7 | 14.20 | 647,584 | 12.98 | |||||||||||||||
|
|
|
|
|
|||||||||||||||||
| $0.10 - $ 17.23 |
5,132,426 | 7.0 | $ | 9.32 | 2,575,056 | $ | 5.29 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
F-18
Table of Contents
Upon the exercise of stock options, we issue new shares of our common stock. As of December 31, 2014, 125,000 options issued to employees are unvested, milestone-based options.
Restricted Stock
Certain employees, directors and consultants have been awarded restricted stock under our equity incentive plans. The time-vesting restricted stock grants vest primarily over a period of three to four years. The following table summarizes restricted share activity for the years ended December 31, 2014, 2013 and 2012:
| Number of Shares |
Weighted Average Grant Date Fair Value |
Aggregate Intrinsic Value |
||||||||||
| Outstanding at January 1, 2012 |
621,581 | $ | 3.16 | $ | 1,572,600 | |||||||
|
|
|
|||||||||||
| Granted |
997,300 | 1.94 | ||||||||||
| Vested |
(339,954 | ) | 2.91 | $ | 965,496 | |||||||
|
|
|
|||||||||||
| Forfeited |
(97,250 | ) | 2.33 | |||||||||
|
|
|
|||||||||||
| Outstanding at December 31, 2012 |
1,181,677 | 2.27 | $ | 3,095,994 | ||||||||
|
|
|
|||||||||||
| Granted |
831,020 | 7.68 | ||||||||||
| Vested |
(568,030 | ) | 2.43 | $ | 4,612,275 | |||||||
|
|
|
|||||||||||
| Forfeited |
(23,737 | ) | 8.52 | |||||||||
|
|
|
|||||||||||
| Outstanding at December 31, 2013 |
1,420,930 | 5.27 | $ | 18,401,044 | ||||||||
|
|
|
|||||||||||
| Granted |
1,451,558 | 14.38 | ||||||||||
| Vested |
(1,856,682 | ) | 8.78 | $ | 28,608,133 | |||||||
|
|
|
|||||||||||
| Forfeited |
(88,859 | ) | 8.22 | |||||||||
|
|
|
|||||||||||
| Outstanding at December 31, 2014 |
926,947 | $ | 12.22 | $ | 13,116,300 | |||||||
|
|
|
|
|
|
|
|||||||
As of December 31, 2014, 75,000 shares of restricted stock issued to employees are unvested, milestone-based shares.
On September 14, 2009, we entered in an employment agreement with Ron Bentsur, our Chief Executive Officer, which was amended on January 13, 2012, and further amended on September 11, 2013. The agreement, as amended, terminates on May 20, 2015, subject to certain early termination events. As of December 31, 2014, Mr. Bentsur has been granted a total of 1,250,000 shares of restricted stock based on the achievement of certain milestone awards described in his employment agreement. In addition, as of December 31, 2014, Mr. Bentsur has the opportunity to earn certain milestone awards as follows:
| • | 100,000 shares of restricted stock will be granted to Mr. Bentsur upon each event of our outlicensing Auryxia in a foreign market, other than Japan, resulting in a greater than $10 million non-refundable cash payment to us with a gross deal value to us of at least $50 million. Such restricted stock will vest in equal installments over each of the first three anniversaries of the date of grant, provided that Mr. Bentsur remains an employee during such vesting period. |
As per his employment agreement, in December 2014, 500,000 shares of fully vested common stock were granted to Mr. Bentsur, upon the first commercial sale of Auryxia in the U.S. off an approved NDA. In addition, upon reaching the same milestone, 266,666 shares of restricted stock previously issued to Mr. Bentsur were vested. We recorded $10.1 million of stock-based compensation expense associated with the granting and vesting of the 766,666 shares of restricted stock in December 2014, which is included in selling, general and administrative expenses in the year ended December 31, 2014.
F-19
Table of Contents
Stock-Based Compensation
The following tables summarize stock-based compensation expense information about equity incentive grants for the years ended December 31, 2014, 2013 and 2012:
| For the year ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Research and development expenses |
$ | 6,379 | $ | 2,347 | $ | 662 | ||||||
| Selling, general and administrative expenses |
20,578 | 3,606 | 1,505 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 26,957 | $ | 5,953 | $ | 2,167 | |||||||
|
|
|
|
|
|
|
|||||||
| For the year ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Stock-based compensation expense associated with restricted stock |
$ | 20,031 | $ | 3,859 | $ | 967 | ||||||
| Stock-based compensation expense associated with stock options |
6,926 | 2,094 | 1,200 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 26,957 | $ | 5,953 | $ | 2,167 | |||||||
|
|
|
|
|
|
|
|||||||
Stock-based compensation costs capitalized as part of inventory were immaterial for the year ended December 31, 2014.
The twelve months ended December 31, 2014, included $4.6 million and $11.0 million of stock-based compensation in research and development and selling, general and administrative expense, respectively, related to the vesting of milestone-based stock options and restricted shares upon the FDA approval and first commercial sale of Auryxia.
The fair value of stock options granted is estimated at the date of grant using the Black-Scholes pricing model. The expected term of options granted is derived from historical data and the expected vesting period. Expected volatility is based on the historical volatility of our common stock. The risk-free interest rate is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. We have assumed no expected dividend yield, as dividends have never been paid to stock or option holders and will not be paid for the foreseeable future.
| Black-Scholes Option Valuation Assumptions |
2014 | 2013 | 2012 | |||||||||
| Risk-free interest rates |
1.9% | 0.7% | 0.6% | |||||||||
| Dividend yield |
— | — | — | |||||||||
| Volatility |
103.0% | 102.0% | 106.3% | |||||||||
| Weighted-average expected term |
6.0 years | 3.8 years | 4.0 years | |||||||||
The weighted average grant date fair value of options granted was $11.81, $4.28 and $1.73 per option for the years ended December 31, 2014, 2013 and 2012, respectively. We used historical information to estimate forfeitures within the valuation model. As of December 31, 2014, there was $20.6 million and $8.1 million of total unrecognized compensation cost related to non-vested stock options and restricted stock, respectively, which is expected to be recognized over weighted-average periods of 2.3 years and 2.2 years, respectively. These amounts do not include, as of December 31, 2014, 125,000 options outstanding and 75,000 shares of restricted stock outstanding which are milestone-based and vest upon certain corporate milestones, such as change in control. Stock-based compensation will be measured and recorded if and when it is probable that the milestone will occur.
NOTE 12 – INCOME TAXES
We account for income taxes under the asset and liability method. Deferred tax assets and liabilities are determined based on differences between the financial reporting and tax basis of assets and liabilities and are
F-20
Table of Contents
measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is established when necessary to reduce deferred tax assets to the amount expected to be realized. In determining the need for a valuation allowance, management reviews both positive and negative evidence, including current and historical results of operations, future income projections and the overall prospects of our business. Based upon management’s assessment of all available evidence, we believe that it is more-likely-than-not that the deferred tax assets will not be realizable; and therefore, a full valuation allowance is established. The valuation allowance for deferred tax assets was $205.0 million and $162.8 million as of December 31, 2014 and 2013, respectively.
As of December 31, 2014, we have U.S. net operating loss carryforwards (“NOL’s”) of approximately $509.0 million, of which approximately $77.6 million were derived from certain stock option exercises and any such benefit realized will be credited to additional paid in capital. For income tax purposes, these NOL’s will expire in the years 2019 through 2034. Due to our various equity transactions, the utilization of certain NOL’s could be subject to annual limitations imposed by Internal Revenue Code Section 382 relating to the change of control provision and/or the separate return limitation year losses limitation.
For the year ended December 31, 2014, we recognized $0.7 million in income tax expense related to the recording of a deferred tax liability associated with capitalized goodwill, an indefinite-lived intangible asset that is being amortized for tax purposes. Indefinite-lived intangibles are non-monetary assets which are not amortized under GAAP since there is no foreseeable limit to the cash flows provided by them. Our lack of earnings history and the uncertainty surrounding our ability to generate taxable income prior to the reversal or expiration of such deferred tax liability were the primary factors considered by management when recording the deferred tax liability. There was no income tax expense for the year ended December 31, 2013.
The income tax provision consists of the following:
| (in thousands) |
December 31, 2014 |
December 31, 2013 |
||||||
| Current: |
||||||||
| Federal |
$ | — | $ | — | ||||
| State |
— | — | ||||||
|
|
|
|
|
|||||
| Total current |
— | — | ||||||
|
|
|
|
|
|||||
| Deferred: |
||||||||
| Federal |
640 | — | ||||||
| State |
60 | — | ||||||
|
|
|
|
|
|||||
| Total deferred |
700 | — | ||||||
|
|
|
|
|
|||||
| Total income taxes |
$ | 700 | $ | — | ||||
|
|
|
|
|
|||||
F-21
Table of Contents
Income tax expense differed from amounts computed by applying the US federal income tax rate of 34% to pretax loss as follows:
| For the year ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Loss before income taxes, as reported in the consolidated statements of operations |
$ | (110,818 | ) | $ | (46,732 | ) | $ | (25,360 | ) | |||
| Computed “expected” tax (benefit) expense |
(37,678 | ) | (15,889 | ) | (8,622 | ) | ||||||
| Increase (decrease) in income taxes resulting from: |
||||||||||||
| Expected (benefit) expense from state & local taxes |
(3,594 | ) | (1,523 | ) | (827 | ) | ||||||
| Stock compensation |
(7,178 | ) | (1,842 | ) | 905 | |||||||
| Deferred impact rate change |
— | — | — | |||||||||
| Permanent differences |
97 | 66 | (196 | ) | ||||||||
| Impact of state NOL carryforward change |
6,726 | — | — | |||||||||
| Prior year true-up |
70 | (409 | ) | (6,450 | ) | |||||||
| Change in the balance of the valuation allowance for deferred tax assets allocated to income tax expense |
42,257 | 19,597 | 15,190 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 700 | $ | — | $ | — | |||||||
|
|
|
|
|
|
|
|||||||
The significant components of deferred income tax expense (benefit) attributable to loss from operations are as follows:
| For the year ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Deferred tax (benefit) expense |
$ | (41,557 | ) | $ | (19,597 | ) | $ | (15,190 | ) | |||
| Federal deferred tax benefit relating to the exercise of stock options |
(— | ) | (— | ) | (— | ) | ||||||
| Increase (decrease) in the valuation allowance for deferred tax asset |
42,257 | 19,597 | 15,190 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 700 | $ | — | $ | — | |||||||
|
|
|
|
|
|
|
|||||||
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets and deferred tax liabilities at December 31, 2014 and 2013 are presented below.
| (in thousands) |
December 31, 2014 |
December 31, 2013 |
||||||
| Deferred tax assets (liabilities): |
||||||||
| Net operating loss carryforwards |
$ | 182,864 | $ | 149,510 | ||||
| Stock-based compensation expense |
11,379 | 7,022 | ||||||
| Unrealized / realized loss on securities |
1,052 | 1,164 | ||||||
| Capitalized Inventory |
7,106 | 3,061 | ||||||
| Research and development |
2,087 | 2,088 | ||||||
| Intangible assets due to different amortization methods |
(321 | ) | (135 | ) | ||||
| Accrued expenses |
— | 53 | ||||||
| Deferred revenue |
154 | — | ||||||
| Other temporary differences |
9 | 11 | ||||||
|
|
|
|
|
|||||
| Net deferred tax asset, excluding valuation allowance |
204,330 | 162,774 | ||||||
| Less valuation allowance |
(205,030 | ) | (162,774 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax (liabilities) assets |
$ | (700 | ) | $ | — | |||
|
|
|
|
|
|||||
F-22
Table of Contents
We file income tax returns in the U.S federal and various state and local jurisdictions. For federal and state income tax purposes, the 2011, 2012 and 2013 tax years remain open for examination under the normal three year statute of limitations. The statute of limitations for income tax audits in the U.S. will commence upon utilization of net operating losses and will expire three years from the filing of the tax return.
There was no accrual for uncertain tax positions or for interest and penalties related to uncertain tax positions for 2014, 2013 and 2012. We do not believe that there will be a material change in our unrecognized tax positions over the next twelve months. All of the unrecognized tax benefits, if recognized, would be offset by the valuation allowance.
NOTE 13 – INTEREST AND OTHER INCOME, NET
The components of interest and other income, net are as follows:
| For the year ended December 31, | ||||||||||||
| (in thousands) |
2014 | 2013 | 2012 | |||||||||
| Interest income |
$ | 290 | $ | 190 | $ | 52 | ||||||
| Other income |
121 | 161 | 166 | |||||||||
| Compensatory damage award, net |
— | — | 1,501 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 411 | $ | 351 | $ | 1,719 | |||||||
|
|
|
|
|
|
|
|||||||
In 2012, we recorded other income due to the award of $1.5 million in compensatory damages, net of fees and legal expenses, relating to the statement of claim we filed with the Financial Institution Regulatory Authority against an SEC registered broker-dealer for damages arising from that broker-dealer’s recommendations and purchases of auction rate securities for our cash management account.
NOTE 14 – COMMITMENTS AND CONTINGENCIES
As of December 31, 2014, we have known contractual obligations, commitments and contingencies of $46.4 million. Of this amount, $13.9 million relates to selling, general and administrative agreements primarily associated with the launch and commercialization of Auryxia, of which $10.0 million is due within the next year, $4.4 million relates to research and development agreements (relating to our Auryxia clinical and regulatory programs), of which $4.1 million is due within the next year, and $25.7 million relates to various third-party contract manufacturing agreements for the production and packaging of Auryxia drug substance and drug product, of which $24.7 million is due within the next year. The additional $2.4 million relates to our operating lease obligations.
| (in thousands) |
Payment due by period | |||||||||||||||||||
| Total | Less than 1 year |
1-3 years |
3-5 years |
More than 5 years |
||||||||||||||||
| Contractual obligations |
||||||||||||||||||||
| Selling, general and administrative agreements |
$ | 13,938 | $ | 10,031 | $ | 3,807 | $ | 100 | $ | — | ||||||||||
| Research and development agreements |
4,406 | 4,073 | 333 | — | — | |||||||||||||||
| Manufacturing agreements |
25,712 | 24,663 | 1,049 | — | — | |||||||||||||||
| Operating leases |
2,375 | 1,521 | 854 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
$ | 46,431 | $ | 40,288 | $ | 6,043 | $ | 100 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
Leases
In March and September 2013, we extended our lease on our corporate and executive office located in New York City, adding approximately 6,800 square feet of additional leased space and extending its term through September 30, 2016. We also executed an amendment to our office sharing agreement with a third party for a portion of our leased space, which term ended on September 29, 2014.
F-23
Table of Contents
In March 2014, we entered into a sublease for approximately 10,395 square feet of leased office space in Boston, Massachusetts, with a term through December 31, 2015.
Total rental expense was approximately $1.6 million, $0.7 million and $0.5 million for the years ended December 31, 2014, 2013, and 2012, respectively. We recognized sublet income of $0.1 million, $0.2 million and $0.2 million for the years ended December 31, 2014, 2013, and 2012, respectively, related to the office sharing agreement which ended in September 2014.
Future minimum lease commitments as of December 31, 2014, in the aggregate total approximately $2.4 million through September 2016. The following table shows future minimum lease commitments, which include our office leases in New York and Boston, by period as of December 31, 2014.
Royalty and Contingent Milestone Payments
Under the license agreement with Panion, we acquired the exclusive worldwide rights, excluding certain Asian-Pacific countries, for the development and marketing of Auryxia. To date, we have paid an aggregate of $9.6 million to Panion, including the $3.0 million milestone payment paid upon the FDA approval of Auryxia, and Panion is eligible to receive one additional milestone payment of $2.0 million upon our successful achievement of European marketing approval, in addition to royalty payments based on a mid-single digit percentage of net sales of Auryxia. The $2.0 million contingent milestone payment is included in research and development agreements in the table above.
NOTE 15 – LITIGATION
In October 2009, we filed a statement of claim with the Financial Institution Regulatory Authority, or FINRA, to commence an arbitration proceeding against an SEC registered broker-dealer. In this arbitration proceeding, we sought damages arising from that broker-dealer’s recommendations and purchases of auction rate securities for our cash management account. On May 7, 2012, we received the arbitrators’ award, which required the broker-dealer to pay us compensatory damages in the amount of approximately $1.8 million. In June 2012, we received the award, which amounted to, after fees and legal expenses, approximately $1.5 million.
On February 1, 2013, a lawsuit was filed against us and our chief executive officer on behalf of a putative class of all of our shareholders (other than the defendants) who acquired our shares between June 1, 2009 and April 1, 2012. Smith v. Keryx Biopharmaceuticals, Inc., et al., Case No. 1:13-CV-0755-TPG (S.D.N.Y.). On February 26, 2013, a substantially similar lawsuit was filed against us and our chief executive officer as well as our chief financial officer. Park v. Keryx Biopharmaceuticals, Inc., et al., Case No. 1:13-CV-1307-TPG (S.D.N.Y.). On June 10, 2013, the Court entered an Order consolidating the two lawsuits and appointing a lead plaintiff. The case was styled In re Keryx Biopharmaceuticals, Inc. Securities Litigation, Case No. 1:13-CV-0755-KBF (S.D.N.Y.). On July 10, 2013, the lead plaintiff filed a Consolidated Amended Complaint that, in substance, repeated the claims alleged in the consolidated lawsuits. The Consolidated Amended Complaint asserted claims against (i) us for alleged violations of Section 10(b) of the Securities Exchange Act of 1934 (“Exchange Act”) and Rule 10b-5 promulgated thereunder and (ii) our chief executive officer for alleged violations of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5. The claims in the Consolidated Amended Complaint were premised on general allegations that we and the individual defendant participated directly or indirectly in the preparation and/or issuance of purportedly false and misleading earnings reports, SEC filings, press releases, and other public statements, which allegedly caused our stock to trade at artificially inflated prices. On August 26, 2013, we filed a motion to dismiss the Consolidated Amended Complaint. On February 14, 2014, the Court entered an Opinion and Order granting the motion to dismiss. The Court entered Judgment for the Defendants on February 24, 2014. The lead plaintiff did not appeal the Judgment and this matter is now concluded.
F-24
Table of Contents
NOTE 16 – QUARTERLY CONSOLIDATED FINANCIAL DATA (UNAUDITED)
| 2014 | ||||||||||||||||
| Mar. 31 | June 30 | Sept. 30 | Dec. 31 | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenue: |
||||||||||||||||
| License revenue |
$ | 10,000 | $ | — | $ | 256 | $ | 569 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating expenses: |
||||||||||||||||
| License expenses: |
— | — | 154 | 341 | ||||||||||||
| Research and development |
16,359 | 10,275 | 19,053 | 5,815 | ||||||||||||
| Selling, general and administrative |
7,292 | 12,268 | 16,447 | 34,050 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
23,651 | 22,543 | 35,654 | 40,206 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating loss |
(13,651 | ) | (22,543 | ) | (35,398 | ) | (39,637 | ) | ||||||||
| Other income |
||||||||||||||||
| Interest and other income, net |
121 | 129 | 109 | 52 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss before income taxes |
(13,530 | ) | (22,414 | ) | (35,289 | ) | (39,585 | ) | ||||||||
| Income taxes |
— | — | — | 700 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (13,530 | ) | $ | (22,414 | ) | $ | (35,289 | ) | $ | (40,285 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted net loss per common share* |
$ | (0.15 | ) | $ | (0.24 | ) | $ | (0.38 | ) | $ | (0.44 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| 2013 | ||||||||||||||||
| Mar. 31 | June 30 | Sept. 30 | Dec. 31 | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenue: |
||||||||||||||||
| License revenue |
$ | 7,000 | $ | — | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating expenses: |
||||||||||||||||
| License expenses: |
— | — | — | — | ||||||||||||
| Research and development |
6,430 | 7,177 | 10,670 | 10,457 | ||||||||||||
| Selling, general and administrative |
2,728 | 4,277 | 5,062 | 7,282 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
9,158 | 11,454 | 15,732 | 17,739 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating loss |
(2,158 | ) | (11,454 | ) | (15,732 | ) | (17,739 | ) | ||||||||
| Other income |
||||||||||||||||
| Interest and other income, net |
103 | 96 | 81 | 71 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss before income taxes Income taxes |
|
(2,055 — |
)
|
|
(11,358 — |
)
|
|
(15,651 — |
)
|
|
(17,668 — |
)
| ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (2,055 | ) | $ | (11,358 | ) | $ | (15,651 | ) | $ | (17,668 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted net loss per common share* |
$ | (0.03 | ) | $ | (0.14 | ) | $ | (0.19 | ) | $ | (0.21 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| * | The aggregate of quarterly computed basic and diluted net loss per common share may not agree with the annual amount due to rounding. |
NOTE 17 – SUBSEQUENT EVENTS
On January 21, 2015, we announced the pricing of an underwritten public offering in which we sold 10,541,667 shares of our common stock at a price of $12.00 per share for gross proceeds of approximately $126.5 million. Net proceeds from this offering were approximately $118.3 million, net of underwriting discounts and offering expenses of approximately $8.2 million. The shares were sold under Registration Statements (Nos. 333-201605 and 333-201639) on Form S-3 and Form S-3MEF, respectively, filed by us with the Securities and Exchange Commission.
F-25
Table of Contents
In January 2015, we announced the transitioning of the role of Chief Executive Officer from Ron Bentsur to our current Chief Operating Officer, Greg Madison. Mr. Madison joined Keryx in February 2014 as Chief Operating Officer to transition the Company from a development-stage organization into a fully integrated commercial entity, bringing to Keryx a wealth of relevant expertise in both the phosphate binder and iron deficiency anemia markets. Mr. Madison has been appointed President of Keryx and will work with Mr. Bentsur to ensure a successful leadership transition by the end of May, when Mr. Bentsur’s contract expires.
In February 2015, we announced a planned consolidation of our finance and accounting function into our Boston office and that our Chief Financial Officer, James Oliviero, will be leaving Keryx by October 2015. Mr. Oliviero has been with Keryx for twelve years and has served as the Chief Financial Officer since 2009. We have commenced a search for a new Chief Financial Officer who will be based in our Boston office. Mr. Oliviero will continue to manage our finance and accounting team during the remainder of his tenure and will assist in the transition of his duties to the new Chief Financial Officer.
F-26
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: February 27, 2015
| KERYX BIOPHARMACEUTICALS, INC. | ||
| By: | /s/ Ron Bentsur | |
| Ron Bentsur Chief Executive Officer and Director | ||
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Ron Bentsur and James F. Oliviero, his true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution, for him and his name, place and stead, in any and all capacities, to sign any or all amendments to this annual report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the SEC, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent or any of his substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Form 10-K has been signed by the following persons on behalf of the Registrant on February 27, 2015, and in the capacities indicated:
| Signatures |
Title | |
| /s/ Ron Bentsur Ron Bentsur |
Chief Executive Officer and Director (principal executive officer) | |
| /s/ James F. Oliviero James F. Oliviero, CFA |
Chief Financial Officer (principal financial and accounting officer) | |
| /s/ Michael P. Tarnok Michael P. Tarnok |
Chairman of the Board of Directors | |
| /s/ Kevin Cameron Kevin Cameron |
Director | |
| /s/ Joseph Feczko, M.D Joseph Feczko, M.D |
Director | |
| /s/ Senator Wyche Fowler, Jr. Senator Wyche Fowler, Jr. |
Director | |
| /s/ Jack Kaye Jack Kaye |
Director | |
| /s/ Daniel P. Regan Daniel P. Regan |
Director | |
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Exhibit Description | |
| 10.16† | Employment Agreement with James F. Oliviero, dated February 26, 2015. | |
| 21.1 | List of Subsidiaries. | |
| 23.1 | Consent of UHY LLP. | |
| 24.1 | Power of Attorney of Director and Officers of Keryx Biopharmaceuticals, Inc. (included herein). | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated February 27, 2015. | |
| 101 | The following financial information from Keryx Biopharmaceuticals, Inc.’s Annual Report on Form 10-K for the year ended December 31, 2014, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Stockholders’ Equity, (iv) Consolidated Statements of Cash Flows, and (v) the Notes to Consolidated Financial Statements. | |
| † | Indicates management contract or compensatory plan or arrangement. |