Attached files
| file | filename |
|---|---|
| EX-23.1 - EX-23.1 - Jaguar Health, Inc. | a2221465zex-23_1.htm |
| EX-10.23 - EX-10.23 - Jaguar Health, Inc. | a2221465zex-10_23.htm |
| EX-4.1 - EX-4.1 - Jaguar Health, Inc. | a2221465zex-4_1.htm |
| EX-10.24 - EX-10.24 - Jaguar Health, Inc. | a2221465zex-10_24.htm |
| EX-10.22 - EX-10.22 - Jaguar Health, Inc. | a2221465zex-10_22.htm |
Use these links to rapidly review the document
TABLE OF CONTENTS
Index to Financial Statements
As filed with the Securities and Exchange Commission on October 10, 2014.
Registration No. 333-198383
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
Amendment No. 2
To
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
JAGUAR ANIMAL HEALTH, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
2834 (Primary Standard Industrial Classification Code Number) |
46-2956775 (I.R.S. Employer Identification Number) |
185 Berry Street, Suite 1300
San Francisco, California 94107
(415) 371-8300
(Address, including zip code, and telephone number, including area code, of registrant's principal executive office)
Lisa A. Conte
Chief Executive Officer and President
Jaguar Animal Health, Inc.
185 Berry Street, Suite 1300
San Francisco, California 94107
(415) 371-8300
(Name, address, including zip code, and telephone number, including area code, of agent for service)
| Copies to: | ||
Donald C. Reinke Marianne C. Sarrazin Reed Smith LLP 101 Second Street, Suite 1800 San Francisco, California 94105 (415) 543-8700 |
Divakar Gupta John T. McKenna Cooley LLP 1114 Avenue of the Americas New York, New York 10036 (212) 479-6000 |
|
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer ý (Do not check if a smaller reporting company) |
Smaller reporting company o |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
SUBJECT TO COMPLETION DATED OCTOBER 10, 2014
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting offers to buy these securities in any jurisdiction where the offer or sale is not permitted.
PRELIMINARY PROSPECTUS
Shares

Common Stock
$ per share
This is the initial public offering of Jaguar Animal Health, Inc. We are offering shares of common stock. Prior to this offering, there has been no public market for our common stock. We estimate that the initial public offering price will be between $ and $ per share.
We have applied for listing of our common stock on The NASDAQ Capital Market under the symbol "JAGX."
We are an "emerging growth company" as defined by the Jumpstart Our Business Startups Act of 2012 and, as such, we have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
Investing in our common stock involves a high degree of risk. See "Risk Factors" beginning on page 11.
| |
Per Share |
Total | |||||
|---|---|---|---|---|---|---|---|
Initial public offering price |
$ | $ | |||||
Underwriting discounts and commissions(1) |
$ | $ | |||||
Proceeds, before expenses to us |
$ | $ | |||||
- (1)
- We refer you to "Underwriting" for additional information regarding underwriter compensation.
We have granted the underwriters a 30-day option to purchase a total of up to additional shares of common stock.
The underwriters expect to deliver shares of common stock to purchasers on or about , 2014.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| BMO Capital Markets | Guggenheim Securities |
Roth Capital Partners
The date of this prospectus is , 2014.

Until , 2014 (25 days after the commencement of this offering), all dealers that buy, sell or trade shares of our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the obligation of dealers to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
We have not, and the underwriters have not, authorized anyone to provide any information or to make any representations other than those contained in this prospectus or in any free writing prospectus prepared by or on behalf of us or to which we have referred you. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. This prospectus is an offer to sell only the shares offered hereby, but only under the circumstances and in the jurisdictions where it is lawful to do so. The information contained in this prospectus or in any applicable free writing prospectus is current only as of its date, regardless of its time of delivery or any sale of shares of our common stock. Our business, financial condition, results of operations and prospects may have changed since that date.
i
For investors outside the United States: we have not and the underwriters have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of securities and the distribution of this prospectus outside the United States.
Jaguar Animal Health, our logo, Canalevia and Neonorm are our trademarks that are used in this prospectus. This prospectus also includes trademarks, tradenames and service marks that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this prospectus appear without the © and ™ symbols, but those references are not intended to indicate that we will not assert, to the fullest extent under applicable law, our rights or that the applicable owner will not assert its rights, to these trademarks and tradenames.
ii
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should read this entire prospectus carefully, especially the section in this prospectus titled "Risk Factors" and our financial statements and related notes appearing elsewhere in this prospectus, before making an investment decision.
As used in this prospectus, references to "Jaguar," "we," "us" or "our" refer to Jaguar Animal Health, Inc.
Our Company
We are an animal health company focused on developing and commercializing first-in-class gastrointestinal products for companion and production animals. Canalevia is our lead prescription drug product candidate for the treatment of various forms of watery diarrhea in dogs. We expect to announce data from our proof-of-concept study of Canalevia for general acute watery diarrhea in dogs in the fourth quarter of 2014. We also expect to initiate filing of a rolling new animal drug application, or NADA, for Canalevia for chemotherapy-induced diarrhea, or CID, in dogs, by the end of 2014. Canalevia is a canine-specific formulation of crofelemer, an active pharmaceutical ingredient isolated and purified from the Croton lechleri tree. A human-specific formulation of crofelemer, Fulyzaq, was approved by the U.S. Food and Drug Administration, or FDA, in 2012 for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. Members of our management team developed crofelemer, including while at Napo Pharmaceuticals, Inc., or Napo. Neonorm is our lead non-prescription product to address the symptoms of watery diarrhea, or scours. We recently launched Neonorm in the United States, for preweaned dairy calves under the brand name Neonorm Calf and expect to launch additional formulations of Neonorm for other animal species beginning in 2015. Neonorm is a botanical extract also derived from the Croton lechleri tree. Canalevia and Neonorm are distinct products that are formulated to address specific species and market channels. We have filed eight investigational new animal drug applications, or INADs, with the FDA and intend to develop species-specific formulations of Neonorm in six additional target species.
Diarrhea is one of the most common reasons for veterinary office visits for dogs and is the second most common reason for visits to the veterinary emergency room, yet there are no FDA-approved anti-secretory products for the treatment of diarrhea. We estimate that in the United States, veterinarians see approximately six million annual cases of acute and chronic watery diarrhea in dogs, approximately two-thirds of which are acute watery diarrhea. We believe Canalevia will be effective in treating watery diarrhea because it acts at the last physiological step, conserved across mammalian species, in the manifestation of watery diarrhea, regardless of cause, by normalizing ion and water flow in the intestinal lumen. We are first seeking a minor use, minor species, or MUMS, designation for Canalevia for CID in dogs to shorten the timeframe to commercialization. If we receive conditional approval pursuant to MUMS designation, we expect to commercialize Canalevia for CID in dogs in early 2016. We are also enrolling a placebo-controlled proof-of-concept study of approximately 240 dogs with multiple preselected and distinct types of watery diarrhea. We are conducting this study to support full approval of Canalevia for CID, as well as protocol concurrence discussions with the FDA regarding expansion of labeled indications of watery diarrhea beyond CID to include general acute watery diarrhea. We plan to market Canalevia, if approved, through a focused direct sales force and to complement our internal efforts with distribution partners.
According to the Dairy 2007 study conducted by the United States Department of Agriculture, or USDA, almost one in four preweaned dairy heifer, or female, calves suffers from diarrhea or other digestive problems. The preweaning period is generally the first 60 days after birth. Scours, diarrhea or other digestive problems are responsible for more than half of all preweaned heifer calf deaths, and
1
result in supportive care and treatment costs, impaired weight gain and long-term reduction in milk production. We believe the incidence rate of scours and its corresponding financial impact represent a large opportunity and that Neonorm has the potential to effectively meet this need. In our clinical study completed in May 2014, Neonorm demonstrated a statistically significant reduction in the severity of watery diarrhea, reduced morbidity and mortality, and improved weight gain as compared to placebo in newborn dairy calves with scours.
We recently launched Neonorm for preweaned dairy calves under the brand name Neonorm Calf. Our commercialization activities are initially focusing on large commercial dairy operations and will include active ongoing education and outreach to dairy industry key opinion leaders, such as academics involved in dairy cattle research or who advise the dairy cattle industry, as well as veterinarians. We intend to augment these commercialization efforts by working with regional distributors to leverage the geographic concentration of the dairy market in the United States as well as national distributors to provide wider geographic access to our products. In August 2014, we entered into our first regional distribution agreement for the Upper Midwest region, and in September 2014, entered into an agreement with a national master distributor, who also distributes prescription products for the companion animal market. We estimate that the commercial launch will cost approximately $1.0 million. We expect the ongoing launch of Neonorm to drive awareness among veterinarians regarding the utility of our first-in-class anti-secretory Croton lechleri-derived products, including Canalevia.
We have an exclusive worldwide license to Napo's intellectual property rights and technology related to our products and product candidates, including rights to its library of over 2,300 medicinal plants, for all veterinary treatment uses and indications for all species of animals. This license includes rights to Canalevia, Neonorm and other distinct prescription drug product candidates and non-prescription products in our pipeline along with the corresponding existing pre-clinical and clinical data packages.
Our management team has significant experience in gastrointestinal and animal health product development. This experience includes the development of crofelemer for human use, from discovery and preclinical and clinical toxicity studies, including the existing animal studies to be used for Canalevia regulatory approvals, through human clinical development. Our team also includes individuals who have prior animal health experience at major pharmaceutical companies, including Ciba-Geigy Corp., now Novartis International AG, SmithKline Beecham Corporation, now GlaxoSmithKline LLC, the animal health group of Pfizer Inc., now Zoetis Inc., and Vétoquinol S.A.
2
Product Pipeline
We are developing a pipeline of prescription drug product candidates and non-prescription products to address unmet needs in animal health. Our pipeline currently includes prescription drug product candidates for eight indications across multiple species, and non-prescription products targeting seven species.
Prescription Drug Product Candidates
| Product Candidates |
Species |
Indication |
Recent Developments(1) |
Anticipated Near-Term Milestones |
||||
|---|---|---|---|---|---|---|---|---|
| Canalevia |
Dogs | CID | • INAD filed in November 2013(2) • Scheduled MUMS designation / pre-NADA meeting |
• Initiate rolling NADA filing with the FDA in fourth quarter of 2014 | ||||
| Dogs | General acute watery diarrhea |
• INAD filed in February 2014 • Initiated proof-of-concept study in June 2014 |
• Proof-of-concept data in fourth quarter of 2014 • Top line pivotal efficacy data in 2015 |
|||||
| Horses | Acute colitis | • INAD filed in February 2014 • Initiated hamster C. difficile study in April 2014 |
• Safety data in fourth quarter of 2014 • Proof-of-concept data in second half of 2015 • Apply for MUMS designation in second half of 2015 |
|||||
| Species-specific formulations of crofelemer |
Horses | Colonic and gastric ulcers(3) | • Proof-of-concept data in second half of 2015 | |||||
| |
Cats | General acute watery diarrhea |
• INAD filed in February 2014 | • Safety data in first half of 2015 • Top line pivotal efficacy data in 2015 |
||||
| Virend (topical) | Cats |
Herpes virus | • INAD filed in July 2014 | • Proof-of-concept data in first half of 2015 • Top line pivotal efficacy data in 2015 |
||||
| Dogs |
Obesity-related metabolic dysfunction | • INAD filed September 2014 | ||||||
| Species-specific formulations of NP-500 |
Horses |
Metabolic syndrome | • INAD filed in March 2014 | |||||
| Cats |
Type II diabetes | • INAD filed in March 2014 | ||||||
- (1)
- Each INAD was filed by us unless otherwise noted.
- (2)
- Initially filed by Napo; transferred to us in March 2014
- (3)
- In combination with omeprazole.
| Products |
Species |
Use |
Recent Developments |
Anticipated Near-Term Milestones |
||||
|---|---|---|---|---|---|---|---|---|
| Neonorm Calf | Dairy calves |
For scours in preweaned dairy calves |
• Commercial launch in September 2014 | • Field study data by end of 2014 | ||||
| Horse foals | Normalize stool formation | • Completed pilot formulation in April 2014 | • Safety and palatability data in 2014 • Efficacy data in first half of 2015 • Commercial launch in 2015 |
|||||
| Species-specific formulations of Neonorm |
Adult horses | Normalize stool formation | • Completed pilot formulation in April 2014 | • Safety and efficacy data in first half of 2015 • Commercial launch in 2015 |
||||
| Sheep and other farm animals |
Normalize stool formation |
• Initiated international market research in New Zealand in May 2014 | • Initiate proof-of-concept studies in various species based on market research | |||||
3
Novel Mechanism of Action
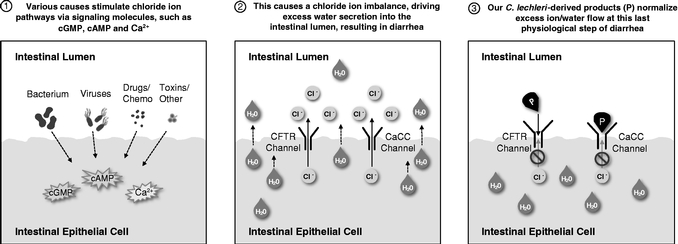
Our gastrointestinal products and product candidates act by normalizing the flow of ions and water in the intestinal lumen, the dysregulation of which is the last step common to the manifestation of watery diarrhea. As a result, we believe that our products and product candidates may be effective in addressing watery diarrhea, regardless of cause. In addition, the channels that regulate this ion and water flow, including channels known as CFTR and CaCC (the sites of action of our gastrointestinal products), are generally present in mammals. We therefore expect that the clinical benefit shown in humans and preweaned dairy calves will be confirmed in multiple other species, including dogs. Accordingly, we believe we can bring to market multiple products for a range of species that are first-in-class and effective in preventing the debilitating and devastating ramifications of watery diarrhea in companion and production animals. The following diagram illustrates the mechanism of action of our gastrointestinal products and product candidates, which normalize chloride and water flow and transit time of fluids within the intestinal lumen.
Business Strategy
Our goal is to become a leading animal health company with first-in-class products that address unmet medical needs in both the companion and production animal markets. To accomplish this goal, we plan to:
- •
- Leverage our significant gastrointestinal knowledge, experience and intellectual property
portfolio to develop a line of products addressing watery diarrhea for both companion and production animals. In addition to Canalevia for dogs and Neonorm for preweaned dairy
calves, we are developing formulations of these products across multiple animal species and market channels.
- •
- Establish commercial capabilities, including third-party sales and distribution networks and our
own targeted commercial efforts, through the launch of Neonorm. We recently launched Neonorm in the United States under the brand name Neonorm Calf. We intend to establish a
focused direct sales force for both the companion and production animal markets, as well as partner with leading distributors to commercialize our products.
- •
- Launch Canalevia and our other product candidates for companion animals, if approved, leveraging the commercial capabilities and brand awareness we are currently building. We believe the ongoing Neonorm launch will allow us to establish sales and marketing capabilities in advance of the planned launch of Canalevia in 2016 for both CID (early 2016) and general acute watery diarrhea (2016) in dogs, to build corporate brand identity awareness, and to establish distributor relationships relevant to both our non-prescription and prescription drug product lines.
4
- •
- Identify market needs that can be readily accessed and develop species-specific products by leveraging our broad intellectual property portfolio, deep pipeline and extensive botanical library. In addition to our gastrointestinal product pipeline, we are also developing products such as Virend for feline herpes and NP-500 for Type II diabetes and metabolic syndrome, both of which have been through Phase 2 human clinical testing. We have exclusive worldwide rights to a library of over 2,300 medicinal plants for all veterinary treatment uses and indications for all species of animals.
Risks Related to Our Business
Our business, and our ability to execute our business strategy, is subject to a number of risks as more fully described in the section titled "Risk Factors." These risks include, among others, the following:
- •
- We have a limited operating history, have not yet generated any material revenues, expect to continue to incur significant
research and development and other expenses, and may never become profitable. Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going
concern.
- •
- We have never generated any material revenue from operations and may need to raise additional capital to achieve our
goals.
- •
- We are substantially dependent on the success of our current lead prescription drug product candidate, Canalevia, and
non-prescription product, Neonorm, and cannot be certain that necessary approvals will be received or that these products will be successfully commercialized.
- •
- We are dependent upon our license agreement with Napo, and if this agreement is terminated, we will be unable to
commercialize our products and our business will be harmed.
- •
- The results of earlier studies may not be predictive of the results of our pivotal trials or other future studies, and we
may be unable to obtain any necessary regulatory approvals for our existing or future prescription drug product candidates under applicable regulatory requirements.
- •
- Development of prescription drug products, and to a lesser extent, non-prescription products, for the animal health market
is inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials, or dosage or formulation studies, would harm our business and prospects.
- •
- Even if we obtain any required regulatory approvals for our current or future prescription drug product candidates, they
may never achieve market acceptance or commercial success.
- •
- We are dependent upon contract manufacturers for supplies of our current prescription drug product candidates and
non-prescription products and, in the short term, intend to rely on contract manufacturers for commercial quantities of any of our commercialized products.
- •
- If we are not successful in identifying, developing and commercializing additional prescription drug product candidates and non-prescription products, our ability to expand our business and achieve our strategic objectives would be impaired.
We were founded in San Francisco, California as a Delaware corporation on June 6, 2013. Napo formed our company to develop and commercialize animal health products. As of December 31, 2013, we were a wholly-owned subsidiary of Napo, and as of September 30, 2014, we are a majority-owned subsidiary of Napo. Upon the closing of this offering, we will no longer be majority-owned by Napo. See "Certain Relationships and Related Person Transactions—Transactions with Napo" and "—Napo Arrangements" for information regarding our transactions with Napo.
5
Our executive offices are located at 185 Berry Street, Suite 1300, San Francisco, California 94107, and our telephone number is (415) 371-8300. Our website address is www.jaguaranimalhealth.com. The information contained on, or that can be accessed through, our website is not part of, and is not incorporated by reference into this prospectus and should not be considered to be part of this prospectus.
Implications of Being an Emerging Growth Company
We qualify as an "emerging growth company" as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from specified disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
- •
- being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim
financial statements, with correspondingly reduced "Management's Discussion and Analysis of Financial Condition and Results of Operations" disclosure;
- •
- not being required to comply with the auditor attestation requirements in the assessment of our internal control over
financial reporting;
- •
- reduced disclosure obligations regarding executive compensation; and
- •
- exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved.
We can take advantage of these provisions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we were to generate more than $1.0 billion in annual revenues, have more than $700.0 million in market value of our capital stock held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. As an emerging growth company, we may choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of some reduced reporting burdens in this prospectus. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock.
6
Common stock offered by us |
shares (or shares if the underwriters exercise their option to purchase additional shares in full) | |
Common stock to be outstanding after this offering |
shares (or shares if the underwriters exercise their option to purchase additional shares in full) |
|
Option to purchase additional shares |
We have granted the underwriters a 30-day option to purchase up to additional shares of our common stock to cover over-allotments, if any. |
|
Use of proceeds |
We intend to use the net proceeds from this offering for further development work for Canalevia and our other prescription drug products, for studies and commercial activities related to Neonorm, for formulation costs and establishing manufacturing capabilities, and for other research and product development activities, working capital and general corporate purposes. See "Use of Proceeds" for a more detailed description of the intended use of proceeds from this offering. |
|
Risk factors |
See "Risk Factors" and other information included in this prospectus for a discussion of factors that you should consider carefully before deciding to invest in our common stock. |
|
Proposed NASDAQ Capital Market symbol |
"JAGX" |
The number of shares of common stock to be outstanding after this offering is based on shares of common stock outstanding as of June 30, 2014, and excludes:
- •
- 311,498 shares of common stock issuable upon exercise of outstanding warrants as of June 30, 2014 with an exercise
price of $1.6854 per share;
- •
- 25,000 shares of common stock issuable upon exercise of an outstanding warrant as of June 30, 2014 with an exercise
price equal to 90% of the initial public offering price;
- •
- 50,000 shares of our common stock issuable upon exercise of outstanding warrants issued after June 30, 2014 with an
exercise price equal to 80% of the initial public offering price;
- •
- 1,129,673 shares issuable upon exercise of outstanding options as of June 30, 2014 with a weighted-average exercise
price of $1.77 per share;
- •
- 118,953 shares issuable upon vesting of outstanding restricted stock unit awards as of June 30, 2014;
- •
- 22,674 shares of common stock reserved for future issuance under our 2013 Equity Incentive Plan as of June 30,
2014; and
- •
- 500,000 shares of common stock reserved for future issuance under our 2014 Stock Incentive Plan, which will become effective in connection with this offering, as well as any automatic increases in the shares of common stock reserved for future issuance under the 2014 Stock Incentive Plan.
7
Unless otherwise indicated, the information in this prospectus assumes the following:
- •
- the filing of our amended and restated certificate of incorporation and the adoption of our amended and restated bylaws,
which will be in effect as of the closing of this offering;
- •
- the conversion of all outstanding shares of Series A preferred stock into 3,015,902 shares of common stock on a
one-for-one basis upon the closing of this offering;
- •
- the issuance of shares of common stock upon the conversion of convertible promissory notes in the
aggregate principal amount of $450,000 (which includes $150,000 aggregate principal amount of notes issued in July 2014) upon the closing of this offering at a conversion price equal to 80% of the
assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, and which shares will be
unregistered;
- •
- no conversion into shares of common stock of up to $1,000,000 aggregate principal amount of borrowings under our standby
letter of credit entered into in August 2014;
- •
- no exercise of outstanding options or warrants, or issuance of shares upon the vesting of restricted stock units; and
- •
- no exercise by the underwriters of their option to purchase additional shares of common stock.
Recent Developments
Subsequent to June 30, 2014, we completed the following transactions and issuances of securities.
Convertible Promissory Notes
In July 2014, we issued convertible promissory notes in the aggregate principal amount of $150,000. Upon the closing of this offering, these notes will convert into shares of common stock at a conversion rate equal to 80% of the initial public offering price per share, and which shares will be unregistered.
Standby Line of Credit and Warrant Issuance
In August 2014, we entered into a standby line of credit with an individual, who is an accredited investor, for up to $1.0 million pursuant to a Line of Credit Loan Agreement dated August 26, 2014. The minimum amount of any drawdown is $250,000, the lender has no obligation to fund more than once every 10 calendar days, we must provide 15 business days prior notice for any drawdown and may not drawdown funds after March 31, 2015. Outstanding borrowings bear interest at a rate of 3.0% per annum, and all borrowings are due in full on the one-year anniversary of our first drawdown. Following closing of this offering, outstanding principal amounts borrowed under the standby line of credit may be converted, at the option of the lender, into shares of our common stock at a conversion price equal to 80% of the initial public offering price per share. In connection with the entry into the standby line of credit, we issued the lender a warrant to purchase 50,000 shares of our common stock at an exercise price equal to 80% of the initial public offering price per share, which expires in August 2016.
8
Summary Selected Financial Data
The following tables set forth a summary of our selected historical financial data as of and for the periods ended on the dates indicated. We have derived the statements of comprehensive loss data for the period from June 6, 2013 (inception) through December 31, 2013 from our audited financial statements included elsewhere in this prospectus. We have derived the statements of comprehensive loss data for the period from June 6, 2013 (inception) through June 30, 2014 and for the six months ended June 30, 2014, and the balance sheet data as of June 30, 2014 from our unaudited interim financial statements appearing elsewhere in this prospectus. The unaudited interim financial statements have been prepared on the same basis as our audited financial statements and, in our opinion, reflect all adjustments, consisting only of normal and recurring adjustments, which we consider necessary for a fair presentation of our financial position as of June 30, 2014. You should read this data together with our financial statements and related notes appearing elsewhere in this prospectus and the sections in this prospectus titled "Selected Financial Data" and "Management's Discussion and Analysis of Financial Condition and Results of Operations." The historical results are not necessarily indicative of the results to be expected for any future periods and the results for the six months ended June 30, 2014 should not be considered indicative of results expected for the full year 2014.
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
Period from June 6, 2013 (inception) through June 30, 2014 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
(unaudited) |
|||||||
Statements of Comprehensive Loss Data: |
||||||||||
Operating expenses: |
||||||||||
General and administrative expense |
$ | 458,473 | $ | 1,740,515 | $ | 2,198,988 | ||||
Research and development expense |
324,479 | 2,149,555 | 2,474,034 | |||||||
| | | | | | | | | | | |
Total operating expenses |
782,952 | 3,890,070 | 4,673,022 | |||||||
Loss from operations |
(782,952 |
) |
(3,890,070 |
) |
(4,673,022 |
) |
||||
Interest expense, net |
(18,251 | ) | (20,164 | ) | (38,415 | ) | ||||
| | | | | | | | | | | |
Net loss and comprehensive loss |
$ | (801,203 | ) | $ | (3,910,234 | ) | $ | (4,711,437 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Accretion of redeemable convertible preferred stock |
— | (285,009 | ) | (285,009 | ) | |||||
| | | | | | | | | | | |
Net loss attributable to common stockholders |
$ | (801,203 | ) | $ | (4,195,234 | ) | $ | (4,996,446 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share attributable to common stockholders, basic and diluted(1) |
$ | (0.20 | ) | $ | (0.99 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted-average common shares outstanding, basic and diluted(1) |
4,000,000 | 4,250,929 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro forma net loss per share, basic and diluted(1) |
$ | (0.20 | ) | $ | (0.67 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro forma weighted-average number of common shares outstanding, basic and diluted(1) |
4,000,000 | 6,304,461 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- See Notes 2 and 12 to our financial statements for a description of the method used to compute basic and diluted net loss per share and pro forma net loss per share.
9
| |
As of June 30, 2014 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Actual | Pro Forma(1) | Pro Forma, As Adjusted(2)(3) |
|||||||
| |
|
(unaudited) |
|
|||||||
Balance Sheet Data: |
||||||||||
Cash and cash equivalents |
$ | 4,281,698 | $ | $ | ||||||
Total assets |
6,217,115 | |||||||||
Total liabilities |
3,175,169 | |||||||||
Convertible promissory notes |
231,250 | |||||||||
Redeemable convertible preferred stock |
6,943,250 | |||||||||
Total stockholders' (deficit) |
(3,901,304 | ) | ||||||||
- (1)
- Pro forma column reflects (i) the conversion of all outstanding shares of Series A preferred stock into shares of common stock upon the closing of this offering; (ii) the issuance of shares of common stock upon the conversion of convertible promissory notes in the aggregate principal amount of $450,000 (which includes $150,000 aggregate principal amount of notes issued in July 2014) upon the closing of this offering at a conversion price equal to 80% of the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus; and (iii) the filing and effectiveness of our amended and restated certificate of incorporation upon the closing of this offering.
- (2)
- Pro forma as adjusted column further reflects the sale of shares of common stock that we are offering at an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
- (3)
- A $1.00 increase (decrease) in the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) each of cash and cash equivalents, total assets and total stockholders' equity (deficit) on a pro forma as adjusted basis by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. An increase (decrease) of 1,000,000 shares in the number of shares offered by us would increase (decrease) each of cash and cash equivalents, total assets, and total stockholders' equity (deficit) on a pro forma as adjusted basis by approximately $ million, assuming the assumed initial public offering price remains the same, and after deducting underwriting discounts and commissions. The pro forma as adjusted information discussed above is illustrative only and will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing.
10
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as the other information in this prospectus, including our financial statements and the related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations," before deciding whether to invest in our common stock. The occurrence of any of the events or developments described below could harm our business, financial condition, results of operations and prospects. In such an event, the market price of our common stock could decline, and you may lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may harm our business, financial condition, results of operations and prospects.
We have a limited operating history, expect to incur further losses as we grow and may be unable to achieve or sustain profitability. Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
Since formation in June 2013, our operations have been primarily limited to the research and development of our lead prescription drug product candidate, Canalevia, to treat various forms of watery diarrhea in dogs, and our lead non-prescription product, Neonorm, to address symptoms of scours in preweaned dairy calves. As a result, we have no meaningful historical operations upon which to evaluate our business and prospects and have not yet demonstrated an ability to broadly commercialize any of our products, obtain any required marketing approval for any of our prescription drug product candidates or successfully overcome the risks and uncertainties frequently encountered by companies in emerging fields such as the animal health industry. We also have not generated any material revenue to date, and expect to continue to incur significant research and development and other expenses. Our net loss and comprehensive loss for the period from June 6, 2013 (inception) through December 31, 2013 was $801,203 and for the six months ended June 30, 2014 was $3,910,234. As of June 30, 2014, we had a total stockholders' deficit of $3,901,304. We expect to continue to incur losses for the foreseeable future, which will increase significantly from historical levels as we expand our product development activities, seek necessary approvals for our product candidates, conduct species-specific formulation studies for our non-prescription products and begin commercialization activities. Even if we succeed in developing and broadly commercializing one or more of our products or product candidates, we expect to continue to incur losses for the foreseeable future, and we may never become profitable. If we fail to achieve or maintain profitability, then we may be unable to continue our operations at planned levels and be forced to reduce or cease operations.
Our auditors have included an explanatory paragraph in their audit report on our financial statements for the year ended December 31, 2013, regarding our assessment of substantial doubt about our ability to continue as a going concern. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. We believe that the successful completion of this offering will eliminate the doubt and enable us to continue as a going concern. However, if we are unable to successfully complete this offering, we will need to obtain alternate financing or create operational plans to continue as a going concern.
We have never generated any material revenue from operations and may not generate any material revenue from our operations in the foreseeable future.
We are an animal health company focused on developing and commercializing prescription drug and non-prescription products for companion and production animals. Since formation in June 2013, we have not generated any material revenue from operations. There is no guarantee that our commercial launch of Neonorm for preweaned dairy calves in the United States will be successful or that we will be able to sell any products in the future. Further, in order to commercialize our
11
prescription drug product candidates, we must receive regulatory approval from the FDA in the United States and other regulatory agencies in various jurisdictions. We have not yet received any regulatory approvals for our prescription drug product candidates. In addition, certain of our non-prescription products, such as Neonorm, may be subject to regulatory approval outside the United States prior to commercialization. Accordingly, until and unless we receive any necessary regulatory approvals, we cannot market or sell our products. Moreover, even if we receive the necessary approvals, we may not be successful in generating revenue from sales of our products as we do not have any meaningful experience marketing or distributing our products. Accordingly, we may never generate any material revenue from our operations.
We expect to incur significant additional costs as we begin commercialization efforts for Neonorm, and undertake the clinical trials necessary to obtain regulatory approvals for Canalevia, which will increase our losses.
We recently commenced sales of Neonorm for preweaned dairy calves in the United States under the brand name Neonorm Calf. We will need to continue to invest in developing our internal and third-party sales and distribution network and outreach efforts to key opinion leaders in the dairy industry, including veterinarians. We will also need to conduct clinical trials for Canalevia in order to obtain necessary initial regulatory approvals and subsequently broaden Canalevia to additional indications and additional species. We will also need to conduct species-specific testing with Neonorm to expand to additional animal populations.
We are actively identifying additional products for development and commercialization, and will continue to expend substantial resources for the foreseeable future to develop Canalevia and Neonorm and develop products from the library of over 2,300 medicinal plants that we have licensed. These expenditures will include costs associated with:
- •
- identifying additional potential prescription drug product candidates and non-prescription products;
- •
- formulation studies;
- •
- conducting pilot, pivotal and toxicology studies;
- •
- completing other research and development activities;
- •
- payments to technology licensors;
- •
- maintaining our intellectual property;
- •
- obtaining necessary regulatory approvals;
- •
- establishing commercial manufacturing and supply capabilities; and
- •
- sales, marketing and distribution of our commercialized products.
We also may incur unanticipated costs in connection with developing and commercializing our products. Because the outcome of our development activities and commercialization efforts is inherently uncertain, the actual amounts necessary to successfully complete the development and commercialization of our current or future products and product candidates may be greater than we anticipate.
Because we anticipate incurring significant costs for the foreseeable future, if we are not successful in broadly commercializing any of our current or future products or product candidates or raising additional funding to pursue our research and development efforts, we may never realize the benefit of our development efforts and our business may be harmed.
12
We may need to raise additional capital to achieve our business goals and such funding may not be available to us on acceptable terms, or at all, which would force us to delay, limit, reduce or terminate one or more of our product development programs or future commercialization efforts.
We believe the net proceeds from this offering, together with our existing cash and cash equivalents, will be sufficient to fund our operating plan through the planned commercial launch of Neonorm for preweaned dairy calves and anticipated commercial launch of Canalevia for CID in dogs, as well as for general acute watery diarrhea in dogs. However, we may experience unexpected events that require us to seek additional funds sooner than planned through public or private equity or debt financings or other sources such as strategic collaborations. We do not expect that the net proceeds from this offering will be sufficient to complete the development of all the current products in our pipeline, or any additional products we may identify. We may need to raise additional capital to fund these activities. Other than our standby line of credit, we have no current agreements or arrangements with respect to any such financings or collaborations, and any such financings or collaborations may result in dilution to our stockholders, the imposition of debt covenants and repayment obligations or other restrictions that may harm our business or the value of our common stock. We may also seek from time to time to raise additional capital based upon favorable market conditions or strategic considerations such as potential acquisitions.
Our future capital requirements depend on many factors, including, but not limited to:
- •
- the scope, progress, results and costs of researching and developing our current and future prescription drug product
candidates and non-prescription products;
- •
- the timing of, and the costs involved in, obtaining any regulatory approvals for our current and any future products;
- •
- the number and characteristics of the products we pursue;
- •
- the cost of manufacturing our current and future products and any products we successfully commercialize;
- •
- the cost of commercialization activities for Neonorm and Canalevia, if approved, including sales, marketing and
distribution costs;
- •
- the expenses needed to attract and retain skilled personnel;
- •
- the costs associated with being a public company;
- •
- our ability to establish and maintain strategic collaborations, distribution or other arrangements and the financial terms
of such agreements; and
- •
- the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation.
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate one or more of our product development programs or future commercialization efforts.
We are substantially dependent on the success of Canalevia and Neonorm and cannot be certain that Canalevia will be approved or that we can successfully commercialize these products.
We currently do not have regulatory approval for any of our prescription drug product candidates, including Canalevia. Our current efforts are primarily focused on the commercial launch of Neonorm in the United States, and development efforts related to Canalevia for CID in dogs. We are also focused on expanding Canalevia's proposed indications to cover general acute watery diarrhea in dogs
13
and full FDA approval for CID for dogs. Accordingly, our near-term prospects, including our ability to generate material product revenue, obtain any new financing if needed to fund our business and operations or enter into potential strategic transactions, will depend heavily on the success of Neonorm and, if approved, Canalevia.
Substantial time and capital resources have been previously devoted by third parties in the development of crofelemer, the active pharmaceutical ingredient, or API, in Canalevia, and the botanical extract used in Neonorm. Both crofelemer and the botanical extract used in Neonorm were originally developed at Shaman Pharmaceuticals, Inc., or Shaman, by certain members of our management team, including Lisa A. Conte, our Chief Executive Officer and President, and Steven R. King, Ph.D., our Executive Vice President, Sustainable Supply, Ethnobotanical Research and Intellectual Property and Secretary. Shaman spent significant development resources before voluntarily filing for bankruptcy in 2001 pursuant to Chapter 11 of the U.S. Bankruptcy Code. The rights to crofelemer and the botanical extract used in Neonorm, as well as other intellectual property rights, were subsequently acquired by Napo from Shaman in 2001 pursuant to a court approved sale of assets. Ms. Conte founded Napo in 2001 and is the current interim chief executive officer of Napo and a member of its board of directors. While at Napo, certain members of our management team, including Ms. Conte and Dr. King, continued the development of crofelemer. In 2005, Napo entered into license agreements with Glenmark Pharmaceuticals Ltd. and Luye Pharma Group Limited for rights to various human indications of crofelemer in certain territories as defined in the respective license agreements with these licensees. Subsequently, after expending significant sums developing crofelemer, including trial design and on-going patient enrollment in the final pivotal Phase 3 trial for crofelemer for non-infectious diarrhea in adults with HIV/AIDS on antiretroviral therapy, in late 2008, Napo entered into a collaboration agreement with Salix Pharmaceuticals, Inc., or Salix, for development and commercialization rights to certain indications worldwide and certain rights in North America, Europe, and Japan, to crofelemer for human use. In January 2014, we entered into the Napo License Agreement pursuant to which we acquired an exclusive worldwide license to Napo's intellectual property rights and technology, including crofelemer and the botanical extract used in Neonorm, for all veterinary treatment uses and indications for all species of animals. In February 2014, most of the executive officers of Napo, and substantially all Napo's employees, became our employees. If we are not successful in the development and commercialization of Neonorm and Canalevia, our business and our prospects will be harmed.
The successful development and commercialization of Neonorm and, if approved, Canalevia will depend on a number of factors, including the following:
- •
- the successful completion of the pivotal trials and toxicology studies for Canalevia, which may take significantly longer
than we currently anticipate and will depend, in part, upon the satisfactory performance of third-party contractors;
- •
- our ability to demonstrate to the satisfaction of the FDA and any other regulatory bodies, the safety and efficacy of
Canalevia;
- •
- our ability and that of our contract manufacturers to manufacture supplies of Neonorm and Canalevia and to develop,
validate and maintain viable commercial manufacturing processes that are compliant with current good manufacturing practices, or cGMP, if required;
- •
- the success of Neonorm field studies and acceptance of their results by dairy producers;
- •
- our ability to successfully launch Neonorm, whether alone or in collaboration with others;
- •
- our ability to successfully launch Canalevia assuming approval is obtained, whether alone or in collaboration with others;
14
- •
- the availability, perceived advantages, relative cost, relative safety and relative efficacy of our prescription drug
product candidates and non-prescription products compared to alternative and competing treatments;
- •
- the acceptance of our prescription drug product candidates and non-prescription products as safe and effective by
veterinarians, animal owners and the animal health community;
- •
- our ability to achieve and maintain compliance with all regulatory requirements applicable to our business; and
- •
- our ability to obtain and enforce our intellectual property rights and obtain marketing exclusivity for our prescription drug product candidates and non-prescription products, and avoid or prevail in any third-party patent interference, patent infringement claims or administrative patent proceedings initiated by third parties or the U.S. Patent and Trademark Office, or USPTO.
Many of these factors are beyond our control. Accordingly, we may not be successful in developing or commercializing Neonorm, Canalevia or any of our other potential products. If we are unsuccessful or are significantly delayed in developing and commercializing Neonorm, Canalevia or any of our other potential products, our business and prospects will be harmed and you may lose all or a portion of the value of your investment in our common stock.
If we are not successful in identifying, licensing, developing and commercializing additional product candidates and products, our ability to expand our business and achieve our strategic objectives could be impaired.
Although a substantial amount of our efforts are focused on the commercial launch of Neonorm and the continued development and potential approval of Canalevia, a key element of our strategy is to identify, develop and commercialize a portfolio of products to serve the animal health market. Most of our potential products are based on our knowledge of medicinal plants. Our current focus is primarily on product candidates and products for animals whose active pharmaceutical ingredient or botanical extract has been successfully commercialized or demonstrated to be safe and effective in human trials. In some instances, we may be unable to further develop these potential products because of perceived regulatory and commercial risks. Even if we successfully identify potential products, we may still fail to yield products for development and commercialization for many reasons, including the following:
- •
- competitors may develop alternatives that render our potential products obsolete;
- •
- potential products we seek to develop may be covered by third-party patents or other exclusive rights;
- •
- a potential product may on further study be shown to have harmful side effects in animals or other characteristics that
indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria;
- •
- a potential product may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and
- •
- a potential product may not be accepted as safe and effective by veterinarians, animal owners, key opinion leaders and other decision-makers in the animal health market.
While we are developing species-specific formulations, including flavors, methods of administration, new patents and other strategies with respect to our current potential products, we may be unable to prevent competitors from developing substantially similar products and bringing those products to market earlier than we can. If such competing products achieve regulatory approval and commercialization prior to our potential products, our competitive position may be impaired. If we fail
15
to develop and successfully commercialize other potential products, our business and future prospects may be harmed and we will be more vulnerable to any problems that we encounter in developing and commercializing our current potential products.
Our animal health products face significant competition from other pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The development and commercialization of animal health products is highly competitive and our success depends on our ability to compete effectively with other products in the market. We expect to compete with the animal health divisions of major pharmaceutical and biotechnology companies such as Merck Animal Health, Merial Limited, Elanco Animal Health, Bayer Animal Health GmbH, Novartis Animal Health Inc. and Boehringer Ingelheim Animal Health, as well as specialty animal health medicines companies such as Zoetis Inc., Phibro Animal Health Corporation and, in Europe, Virbac S.A., Vétoquinol S.A., Ceva Animal Health S.A. and Dechra Pharmaceuticals PLC. We are also aware of several early-stage companies that are developing products for use in the animal health market, including Aratana Therapeutics, Inc., Kindred Biosciences, Inc., Parnell Pharmaceuticals Holdings Ltd and ImmuCell Corporation. We also compete with academic institutions, governmental agencies and private organizations that are conducting research in the field of animal health products.
Although there are currently no FDA-approved anti-secretory products to treat watery diarrhea in dogs, we anticipate that Canalevia, if approved, will face competition from various products, including products approved for use in humans that are used extra-label in animals. Extra-label use is the use of an approved drug outside of its cleared or approved indications in the animal context. All of our potential products could also face competition from new products in development. These and other potential competing products may benefit from greater brand recognition and brand loyalty than our products and product candidates may achieve.
Many of our competitors and potential competitors have substantially more financial, technical and human resources than we do. Many also have more experience in the development, manufacture, regulation and worldwide commercialization of animal health products, including animal prescription drugs and non-prescription products.
For these reasons, we cannot be certain that we and our products can compete effectively.
We may be unable to obtain, or obtain on a timely basis, regulatory approval for our existing or future prescription drug product candidates under applicable regulatory requirements, which would harm our operating results.
The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of animal health products are subject to extensive regulation. We are usually not permitted to market our prescription drug product candidates in the United States until we receive approval of an NADA from the FDA. To gain approval to market an animal prescription drug for a particular species, we must provide the FDA with efficacy data from pivotal trials that adequately demonstrate that our prescription drug product candidates are safe and effective in the target species (e.g., dogs, cats or horses) for the intended indications. In addition, we must provide manufacturing data evidencing that we can produce our product candidates in accordance with cGMP. For the FDA, we must also provide data from toxicology studies, also called target animal safety studies, and in some cases environmental impact data. In addition to our internal activities, we will partially rely on contract research organizations, or CROs, and other third parties to conduct our toxicology studies and for certain other development activities. The results of toxicology studies and other initial development activities, and of any previous studies in humans or animals conducted by us or third parties, may not be predictive of future results of pivotal trials or other future studies, and failure can occur at any time during the conduct of pivotal trials and other development activities by us or our CROs. Our pivotal trials may fail
16
to show the desired safety or efficacy of our prescription drug product candidates despite promising initial data or the results in previous human or animal studies conducted by others, and success of a prescription drug product candidate in prior animal studies, or in the treatment of humans, does not ensure success in subsequent studies. Clinical trials in humans and pivotal trials in animals sometimes fail to show a benefit even for drugs that are effective because of statistical limitations in the design of the trials or other statistical anomalies. Therefore, even if our studies and other development activities are completed as planned, the results may not be sufficient to obtain a required regulatory approval for a product candidate.
Regulatory authorities can delay, limit or deny approval of any of our prescription drug product candidates for many reasons, including:
- •
- if they disagree with our interpretation of data from our pivotal studies or other development efforts;
- •
- if we are unable to demonstrate to their satisfaction that our product candidate is safe and effective for the target
indication;
- •
- if they require additional studies or change their approval policies or regulations;
- •
- if they do not approve of the formulation, labeling or the specifications of our current and future product candidates;
and
- •
- if they fail to approve the manufacturing processes of our third-party contract manufacturers.
Further, even if we receive a required approval, such approval may be for a more limited indication than we originally requested, and the regulatory authority may not approve the labeling that we believe is necessary or desirable for successful commercialization.
Any delay or failure in obtaining any necessary regulatory approval for the intended indications of our product candidates would delay or prevent commercialization of such product candidates and would harm our business and our operating results.
The results of our earlier studies of Neonorm may not be predictive of the results in any future species-specific formulation studies, and we may not be successful in our efforts to develop or commercialize line extensions of Neonorm.
Our product pipeline includes a number of species-specific formulations of Neonorm, our lead non-prescription product. The results of our dairy calf studies and other initial development activities and of any previous studies in humans or animals conducted by us or third parties may not be predictive of future results of these formulation studies. Failure can occur at any time during the conduct of these trials and other development activities. Even if our species-specific formulation studies and other development activities are completed as planned, the results may not be sufficient to pursue a particular line extension for Neonorm. Further, even if we obtain promising results from our species-specific formulation studies, we may not successfully commercialize any line extension. Because line extensions are developed for a particular species market, we may not be able to leverage our experience from the commercial launch of Neonorm Calf in new animal species markets. If we are not successful in developing and successfully commercializing these line extension products, we may not be able to grow our revenue and our business may be harmed.
Development of prescription drug products is inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials would harm our business and prospects.
Development of prescription drug products for animals remains an inherently lengthy, expensive and uncertain process, and our development activities may not be successful. We do not know whether
17
our current or planned pivotal trials for any of our product candidates, will begin or conclude on time, and they may be delayed or discontinued for a variety of reasons, including if we are unable to:
- •
- address any safety concerns that arise during the course of the studies;
- •
- complete the studies due to deviations from the study protocols or the occurrence of adverse events;
- •
- add new study sites;
- •
- address any conflicts with new or existing laws or regulations; or
- •
- reach agreement on acceptable terms with study sites, which can be subject to extensive negotiation and may vary significantly among different sites.
Further, we may not be successful in developing species-specific formulations for Neonorm, and Neonorm may be subject to the same regulatory regime as prescription drug products in jurisdictions outside the United States. Any delays in completing our development efforts will increase our costs, delay our development efforts and approval process and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may harm our business, financial condition and prospects. In addition, factors that may cause a delay in the commencement or completion of our development efforts may also ultimately lead to the denial of regulatory approval of our product candidates which, as described above, would harm our business and prospects.
We will partially rely on third parties to conduct our development activities. If these third parties do not successfully carry out their contractual duties, we may be unable to obtain regulatory approvals or commercialize our current or future product candidates on a timely basis, or at all.
We will partially rely upon CROs to conduct our toxicology studies and for other development activities. We intend to rely on CROs to conduct one or more of our planned pivotal trials. These CROs are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs or manage the risks associated with their activities on our behalf. We are responsible for ensuring that each of our studies is conducted in accordance with the development plans and trial protocols presented to regulatory authorities. Any deviations by our CROs may adversely affect our ability to obtain regulatory approvals, subject us to penalties or harm our credibility with regulators. The FDA and foreign regulatory authorities also require us and our CROs to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, or good laboratory practices, or GLPs, for conducting, monitoring, recording and reporting the results of our studies to ensure that the data and results are scientifically valid and accurate.
Agreements with CROs generally allow the CROs to terminate in certain circumstances with little or no advance notice. These agreements generally will require our CROs to reasonably cooperate with us at our expense for an orderly winding down of the CROs' services under the agreements. If the CROs conducting our studies do not comply with their contractual duties or obligations, or if they experience work stoppages, do not meet expected deadlines, or if the quality or accuracy of the data they obtain is compromised, we may need to secure new arrangements with alternative CROs, which could be difficult and costly. In such event, our studies also may need to be extended, delayed or terminated as a result, or may need to be repeated. If any of the foregoing were to occur, regulatory approval, if required, and commercialization of our product candidates may be delayed and we may be required to expend substantial additional resources.
18
Even if we obtain regulatory approval for Canalevia or our other product candidates, they may never achieve market acceptance. Further, even if we are successful in commercially launching Neonorm, it may not achieve commercial success.
If we obtain necessary regulatory approvals for Canalevia or our other product candidates, such products may still not achieve market acceptance and may not be commercially successful. Market acceptance of Canalevia, Neonorm and any of our other products depends on a number of factors, including:
- •
- the safety of our products as demonstrated in our target animal studies;
- •
- the indications for which our products are approved or marketed;
- •
- the potential and perceived advantages over alternative treatments or products, including generic medicines and competing
products currently prescribed by veterinarians, and products approved for use in humans that are used extra-label in animals;
- •
- the acceptance by veterinarians, companion animal owners and production animal owners, including in the dairy industry, of
our products as safe and effective;
- •
- the cost in relation to alternative treatments and willingness on the part of veterinarians and animal owners to pay for
our products;
- •
- the prevalence and severity of any adverse side effects of our products;
- •
- the relative convenience and ease of administration of our products; and
- •
- the effectiveness of our sales, marketing and distribution efforts.
Any failure by Canalevia, Neonorm or any of our other products to achieve market acceptance or commercial success would harm our financial condition and results of operations.
The dairy industry is subject to conditions beyond our control and the occurrence of any such conditions may harm our business and impact the demand for our products.
The demand for production animal health products, such as Neonorm Calf, is heavily dependent on factors that affect the dairy market that are beyond our control, including the following, any of which may harm our business:
- •
- cost containment measures within the dairy industry, in response to international, national and local general economic
conditions, which may affect the market adoption of our products;
- •
- state and federal government policies, including government-funded programs or subsidies whose discontinuance or
modification could erode the demand for our products;
- •
- a decline in demand for dairy products due to changes in consumer diets away from dairy products, which could adversely
affect the demand for production animal health products;
- •
- adverse weather conditions and natural disasters, such as floods, droughts, and pestilence, which can lower dairy yields;
and
- •
- disease or other conditions beyond our control.
Animal products, like human products, are subject to unanticipated post-approval safety or efficacy concerns, which may harm our business and reputation.
The success of our commercialization efforts will depend upon the perceived safety and effectiveness of animal health products, in general, and of our products, in particular. Unanticipated safety or efficacy concerns can subsequently arise with respect to approved prescription drug products,
19
or non-prescription products, such as Neonorm, which may result in product recalls or withdrawals or suspension of sales, as well as product liability and other claims. Any safety or efficacy concerns, or recalls, withdrawals or suspensions of sales of our products, or human products derived from Croton lechleri, if any, could harm our reputation and business, regardless of whether such concerns or actions are justified.
Future federal and state legislation may result in increased exposure to product liability claims, which could result in substantial losses.
Under current federal and state laws, companion and production animals are generally considered to be the personal property of their owners and, as such, the owners' recovery for product liability claims involving their companion and production animals may be limited to the replacement value of the animal. Companion animal owners and their advocates, however, have filed lawsuits from time to time seeking non-economic damages such as pain and suffering and emotional distress for harm to their companion animals based on theories applicable to personal injuries to humans. If new legislation is passed to allow recovery for such non-economic damages, or if precedents are set allowing for such recovery, we could be exposed to increased product liability claims that could result in substantial losses to us if successful. In addition, some horses can be worth millions of dollars or more, and product liability for horses may be very high. While we currently have product liability insurance, such insurance may not be sufficient to cover any future product liability claims against us.
If we fail to retain current members of our senior management, or to identify, attract, integrate and retain additional key personnel, our business will be harmed.
Our success depends on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Lisa A. Conte, our President and Chief Executive Officer, and Serge Martinod, D.V.M., Ph.D., our Chief Veterinary Officer. The loss of services of any of our key personnel would cause a disruption in our ability to develop our current or future product pipeline and commercialize our products and product candidates. Although we have offer letters with these key members of senior management, such agreements do not prohibit them from leaving our employ at any time. We currently do not maintain "key man" life insurance on any of our senior management team. The loss of Ms. Conte, Dr. Martinod or other members of our current senior management could adversely affect the timing or outcomes of our current and planned studies, as well as the prospects for commercializing our products.
In addition, competition for qualified personnel in the animal health field is intense, because there are a limited number of individuals who are trained or experienced in the field. Further, our headquarters are located in San Francisco, California, and the dairy and agriculture industries are not prevalent in urban areas such as San Francisco. We will need to hire additional personnel as we expand our product development and commercialization activities. Even if we are successful in hiring qualified individuals, as we are a growing organization, we do not have a track record for integrating and retaining individuals. If we are not successful in identifying, attracting, integrating or retaining qualified personnel on acceptable terms, or at all, our business will be harmed.
We are dependent on two suppliers for the raw material used to produce the active pharmaceutical ingredient in Canalevia and the botanical extract in Neonorm. The termination of either of these contracts would result in a disruption to product development and our business will be harmed.
The raw material used to manufacture Canalevia and Neonorm is crude plant latex, or CPL, derived from the Croton lechleri tree, which is found in countries in South America, principally Peru. The ability of our contract suppliers to harvest CPL is governed by the terms of their respective agreements with local government authorities. Although CPL is available from multiple suppliers, we
20
only have contracts with two suppliers to obtain CPL and arrange the shipment to our contract manufacturer. Accordingly, if our contract suppliers do not or are unable to comply with the terms of our respective agreements, and we are not able to negotiate new agreements with alternate suppliers on terms that we deem commercially reasonable, it may harm our business and prospects. The countries from which we obtain CPL could change their laws and regulations regarding the export of the natural products or impose or increase taxes or duties payable by exporters of such products. Restrictions could be imposed on the harvesting of the natural products or additional requirements could be implemented for the replanting and regeneration of the raw material. Such events could have a significant impact on our cost and ability to produce Canalevia, Neonorm and anticipated line extensions.
We are dependent upon third-party contract manufacturers, both for the supply of the active pharmaceutical ingredient in Canalevia and the botanical extract in Neonorm, as well as for the supply of finished products for commercialization.
To date, the CPL, API, botanical extract and some finished products that we have used in our studies and trials were obtained from Napo. We have also contracted with third parties for the formulation of API and botanical extract into finished products for our studies. We have entered into memorandums of understanding with Indena S.p.A. for the manufacture of CPL received from our suppliers into the API in Canalevia to support our regulatory filings, as well as the botanical extract in Neonorm and agreed to negotiate a commercial supply agreement. Indena S.p.A. has never manufactured either such ingredient to commercial scale. We also plan to contract with different third parties for the formulation and supply of finished products, which we will use in our planned studies and commercialization efforts. However, we have not entered into any definitive agreements with any third parties for the supply of commercial quantities of finished products.
We also intend to use a portion of the net proceeds from this offering to develop our own manufacturing capability for the API in Canalevia. However, we do not yet have such capability, and we cannot be certain that we will have sufficient funds to develop this facility, and we may not be able to successfully manufacture the API in Canalevia. If we are not able to develop our own manufacturing capability, we will be dependent upon our contract manufacturer for the supply of the API in Canalevia. We currently have approximately 2,000 kg of the botanical extract used in Neonorm. However, we will require additional quantities of the botanical extract if our commercial launch of Neonorm is successful. If we are not successful in reaching agreements with third parties on terms that we consider commercially reasonable for manufacturing and formulation, or if our contract manufacturer and formulator are not able to produce sufficient quantities or quality of API, botanical extract or finished product under their agreements, it could delay our plans and harm our business prospects.
The facilities used by our third-party contractors are subject to inspections, including by the FDA, and other regulators, as applicable. We also depend on our third-party contractors to comply with cGMP. If our third-party contractors do not maintain compliance with these strict regulatory requirements, we and they will not be able to secure or maintain regulatory approval for their facilities, which would have an adverse effect on our operations. In addition, in some cases, we also are dependent on our third-party contractors to produce supplies in conformity to our specifications and maintain quality control and quality assurance practices and not to employ disqualified personnel. If the FDA or a comparable foreign regulatory authority does not approve the facilities of our third-party contractors if so required, or if it withdraws any such approval in the future, we may need to find alternative manufacturing or formulation facilities, which could result in delays in our ability to develop or commercialize our products, if at all. We and our third-party contractors also may be subject to penalties and sanctions from the FDA and other regulatory authorities for any violations of applicable regulatory requirements. The USDA and the European Medicines Agency, or the EMA, employ different regulatory standards than the FDA, so we may require multiple manufacturing processes and
21
facilities for the same product candidate or any approved product. We are also exposed to risk if our third-party contractors do not comply with the negotiated terms of our agreements, or if they suffer damage or destruction to their facilities or equipment.
If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our current or future products and product candidates, if approved, and generate product revenue.
We currently have limited sales, marketing or distribution capabilities, and prior to our recent launch of Neonorm for preweaned dairy calves, had no experience in the sale, marketing and distribution of animal health products. There are significant risks involved in building and managing a sales organization, including our potential inability to attract, hire, retain and motivate qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively oversee a geographically-dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities and entry into adequate arrangements with distributors would adversely impact the commercialization of Neonorm, and Canalevia, if approved. If we are not successful in commercializing Neonorm, Canalevia or any of our other line extension products, either on our own or through one or more distributors, we may never generate significant revenue and may continue to incur significant losses, which would harm our financial condition and results of operations.
Changes in distribution channels for animal prescription drugs may make it more difficult or expensive to distribute our prescription drug products.
In the United States, animal owners typically purchase their animal prescription drugs from their local veterinarians who also prescribe such drugs. There is a trend, however, toward increased purchases of animal prescription drugs from Internet-based retailers, "big-box" retail stores and other over-the-counter distribution channels, which follows an emerging shift in recent years away from the traditional veterinarian distribution channel. It is also possible that animal owners may come to rely increasingly on Internet-based animal health information rather than on their veterinarians. We currently expect to market our animal prescription drugs directly to veterinarians, so any reduced reliance on veterinarians by animal owners could harm our business and prospects by making it more difficult or expensive for us to distribute our prescription drug products. Animal owners also may substitute human health products for animal prescription drugs if the human health products are less expensive or more readily available, which could also harm our business.
Legislation has been or may be proposed in various states that would require veterinarians to provide animal owners with written prescriptions and disclosures that the animal owner has the right to fill the prescriptions through other means. If enacted, such legislation could lead to a reduction in the number of animal owners who purchase their animal pharmaceuticals directly from veterinarians, which also could harm our business.
Consolidation of our customers could negatively affect the pricing of our products.
Veterinarians will be our primary customers for our prescription drug products, as well as, to some extent, our non-prescription products, such as Neonorm. In recent years, there has been a trend towards the consolidation of veterinary clinics and animal hospitals. If this trend continues, these large clinics and hospitals could attempt to leverage their buying power to obtain favorable pricing from us and other animal health product companies. Any downward pressure on the prices of any of our products could harm our operating results and financial condition.
22
We will need to increase the size of our organization and may not successfully manage our growth.
As of September 30, 2014, we have 17 employees. Our ability to manage our growth effectively will require us to hire, train, retain, manage and motivate additional employees and to implement and improve our operational, financial and management systems. These demands also may require the hiring of additional senior management personnel or the development of additional expertise by our senior management personnel. If we fail to expand and enhance our operational, financial and management systems in conjunction with our potential future growth, it could harm our business and operating results.
Our research and development relies on evaluations in animals, which is controversial and may become subject to bans or additional regulations.
The evaluation of our products and product candidates in target animals is required to develop, formulate and commercialize our products and product candidates. Although our animal testing will be subject to GLPs and GCPs, as applicable, animal testing in the human pharmaceutical industry and in other industries continues to be the subject of controversy and adverse publicity. Some organizations and individuals have sought to ban animal testing or encourage the adoption of additional regulations applicable to animal testing. To the extent that such bans or regulations are imposed, our research and development activities, and by extension our operating results and financial condition, could be harmed. In addition, negative publicity about animal practices by us or in our industry could harm our reputation among potential customers.
If approved, our prescription drug product candidates may be marketed in the United States only in the target animals and for the indications for which they are approved, and if we want to expand the approved animals or indications, we will need to obtain additional approvals, which may not be granted.
If our prescription drug product candidates are approved by regulatory authorities, we may market or advertise them only in the specific species and for treatment of the specific indications for which they were approved, which could limit use of the products by veterinarians and animal owners. We intend to develop, promote and commercialize approved products for other animals and new treatment indications in the future, but we cannot be certain whether or at what additional time and expense we will be able to do so. If we do not obtain marketing approvals for other species or for new indications, our ability to expand our business may be harmed.
Under the Animal Medicinal Drug Use Clarification Act of 1994, veterinarians are permitted to prescribe extra-label uses of certain approved animal drugs and approved human drugs for animals under certain conditions. While veterinarians may in the future prescribe and use human-approved products or our products for extra-label uses, we may not promote our products for extra-label uses. If the FDA determines that any of our marketing activities constitute promotion of an extra-label use, we could be subject to regulatory enforcement, including seizure of any misbranded or mislabeled drugs, and civil or criminal penalties, any of which could have an adverse impact on our reputation and expose us to potential liability. We will continue to spend resources ensuring that our promotional claims for our products and product candidates remain compliant with applicable FDA laws and regulations, including materials we post or link to on our website. For example, in 2012, our Chief Executive Officer received an "untitled letter" from the FDA while at Napo regarding preapproval promotion statements constituting misbranding of crofelemer, which was then an investigational drug. These statements were included in archived press releases included on Napo's website. Napo was required to expend time and resources to revise its website to remove the links in order to address the concerns raised in the FDA's letter.
23
If our prescription drug product candidates are approved by regulatory authorities, the misuse or extra-label use of such products may harm our reputation or result in financial or other damages.
If our prescription drug product candidates are approved by regulatory authorities, there may be increased risk of product liability if veterinarians, animal owners or others attempt to use such products extra-label, including the use of our products in species (including humans) for which they have not been approved. Furthermore, the use of an approved drug for indications other than those indications for which such products have been approved may not be effective, which could harm our reputation and lead to an increased risk of litigation. If we are deemed by a governmental or regulatory agency to have engaged in the promotion of any approved product for extra-label use, such agency could request that we modify our training or promotional materials and practices and we could be subject to significant fines and penalties, and the imposition of these sanctions could also affect our reputation and position within the industry. Any of these events could harm our reputation and our operating results.
We may not obtain or maintain the benefits associated with MUMS designation, including market exclusivity.
Although we requested MUMS designation for Canalevia for CID in dogs, we may not be granted MUMS designation. Even if granted, we may not receive or maintain the benefits associated with MUMS designation. As the sponsor, we are allowed under FDA regulations to apply for MUMS designation of our product candidate prior to its approval. MUMS designation is a status similar to "orphan drug" status for human drugs. If we are granted MUMS designation, we are eligible for incentives to support the approval or conditional approval of the designated use. This designation does not allow us to commercialize a product until such time as we obtain approval or conditional approval of the product.
If Canalevia receives MUMS designation for the identified particular intended use, we will be eligible to obtain seven years of exclusive marketing rights upon approval (or conditional approval) of Canalevia for that intended use and become eligible for grants to defray the cost of our clinical work. Each designation that is granted must be unique, i.e., only one designation can be granted for a particular API in a particular dosage form for a particular intended use. The intended use includes both the target species and the disease or condition to be treated.
Even if granted, at some point, we could lose MUMS designation. The basis for a lost designation can include but is not limited to, our failure to engage with due diligence in moving forward with a non-conditional approval, or a competing product has received MUMS designation prior to our product candidate for the same indication or species. In addition, MUMS designation may be withdrawn for a variety of reasons such as where the FDA determines that the request for designation was materially defective, or if the manufacturer is unable to assure sufficient quantity of the prescription drug product to meet the needs of animals with the rare disease or condition. If this designation is lost, it could have a negative impact on the product and our company, which includes but is not limited to, market exclusivity pursuant to MUMS designation, or eligibility for grants as a result of MUMS designation.
The market for our products and the animal health market as a whole, is uncertain and may be smaller than we anticipate, which could lead to lower revenue and harm our operating results.
It is very difficult to estimate the commercial potential of any of our products because of the emerging nature of our industry as a whole. The animal health market continues to evolve and it is difficult to predict the market potential for our products. The market will depend on important factors such as safety and efficacy compared to other available treatments, changing standards of care, preferences of veterinarians, the willingness of companion and production animal owners to pay for such products, and the availability of competitive alternatives that may emerge either during the product development process or after commercial introduction. If the market potential for our products
24
is less than we anticipate due to one or more of these factors, it could negatively impact our business, financial condition and results of operations. Further, the willingness of companion and production animal owners to pay for our products may be less than we anticipate, and may be negatively affected by overall economic conditions. The current penetration of animal insurance in the United States is low, animal owners are likely to have to pay out-of-pocket, and such owners may not be willing or able to pay for our products.
Our largest stockholder, Napo, controls a significant percentage of our common stock, and its interests may conflict with those of our other stockholders.
Upon the closing of this offering, Napo will beneficially own in the aggregate % of our common stock. This concentration of ownership gives Napo significant influence over the way we are managed and the direction of our business. In addition, because we and Napo are party to a license agreement, Napo's interests as the licensor of our technology may be different from ours or those of our other stockholders. As a result, the interests of Napo with respect to matters potentially or actually involving or affecting us, such as future acquisitions, licenses, financings and other corporate opportunities and attempts to acquire us, may conflict with the interests of our other stockholders. Further, Napo has pledged its interests in our common stock as security for certain of its monetary obligations. Accordingly, Napo's ability to take action with respect to these shares may be limited by its agreements with its secured creditor, which may conflict with your interests or those of our other stockholders. In addition, our Chief Executive Officer is also the interim chief executive officer of Napo and her duties as interim chief executive officer of Napo may conflict with her duties as our Chief Executive Officer, and the resolution of these conflicts may not always be in our or your best interest.
Napo's principal business currently consists of, among other activities, the management of its intellectual property portfolio, including rights under license agreements with respect to such intellectual property. Napo has limited assets, and its primary sources of revenues in recent years have been license fees, warrant exercises, equity and debt investments and, since late 2013, the receipt of royalties pursuant to its license agreements, which have been limited to date. If Napo fails to generate sufficient revenues to cover its operating costs, it could revise its business strategy in ways that could affect its relationship with our company. For example, it could decide to divest its assets, including its stock in our company. Napo's interests in managing its business, including its ownership in our company, may conflict with your interests.
We may engage in future acquisitions that increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities and subject us to other risks.
We may evaluate various strategic transactions, including licensing or acquiring complementary products, technologies or businesses. Any potential acquisitions may entail numerous risks, including increased operating expenses and cash requirements, assimilation of operations and products, retention of key employees, diversion of our management's attention and uncertainties in our ability to maintain key business relationships of the acquired entities. In addition, if we undertake acquisitions, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition opportunities and this inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
Certain of the countries in which we plan to commercialize our products in the future are developing countries, some of which have potentially unstable political and economic climates.
We may commercialize our products in jurisdictions that are developing and emerging countries. This may expose us to the impact of political or economic upheaval, and we could be subject to unforeseen administrative or fiscal burdens. At present, we are not insured against the political and economic risks of operating in these countries. Any significant changes to the political or economic climate in any of the developing countries in which we operate or plan to sell products either now or in the future may have a substantial adverse effect on our business, financial condition, trading performance and prospects.
25
Fluctuations in the exchange rate of foreign currencies could result in currency transactions losses.
As we expand our operations, we expect to be exposed to risks associated with foreign currency exchange rates. We anticipate that we will commercialize Neonorm for preweaned dairy calves and its line extensions, as well as possibly Canalevia and its line extensions in jurisdictions outside the United States. As a result, we will also be further affected by fluctuations in exchange rates in the future to the extent that sales are denominated in currencies other than U.S. dollars. We do not currently employ any hedging or other strategies to minimize this risk, although we may seek to do so in the future.
Risks Related to Intellectual Property
We are dependent upon our license agreement with Napo and if the agreement is terminated for any reason our business will be harmed.
In January 2014, we entered into a license agreement with Napo, or the Napo License Agreement, which we amended and restated in August 2014. Pursuant to the Napo License Agreement, we acquired an exclusive worldwide license to Napo's intellectual property rights and technology, including rights to its library of over 2,300 medicinal plants, for all veterinary treatment uses and indications for all species of animals. Under the terms of the Napo License Agreement, we are responsible for, and shall ensure, the development and commercialization of products that contain or are derived from the licensed Napo technology worldwide in the field of veterinary treatment uses and indications for all species of animals. In consideration for the license, we are obligated to pay a one-time non-refundable license fee and royalties. Napo has the right to terminate the Napo License Agreement upon our uncured material breach of the agreement or if we declare bankruptcy. If the Napo License Agreement is terminated for any reason, our business will be harmed.
Napo has also entered into secured financing agreements with certain lenders, for whom Nantucket Investments Limited is acting as collateral agent. The security includes certain assets, including the intellectual property and technology licensed to us pursuant to the Napo License Agreement and Napo's shares of our common stock. Although Napo and Nantucket Investments Limited, on behalf of the secured lenders, have entered into a non-disturbance agreement with respect to the Napo License Agreement, in the event of a bankruptcy of Napo or foreclosure action with respect to Napo's assets, there can be no guarantee that the bankruptcy trustee or any other party to such action will not attempt to interfere with or terminate the Napo License Agreement or otherwise require its terms to be changed, which could harm our business. Under the terms of the Napo License Agreement, certain events, such as an acquisition of Napo or a sale by Napo of all of the intellectual property and technology licensed to us pursuant to the Napo License Agreement, should result in a fully-paid up license to us of all of such intellectual property and technology. If for any reason, Napo ceases to be the owner of the intellectual property and technology licensed to us pursuant to the Napo License Agreement in such a manner that did not result in a fully-paid up license provided for therein, the owner of such intellectual property and technology could attempt to interfere with or terminate the Napo License Agreement or otherwise attempt to renegotiate the arrangement, which would harm our business.
If Napo experiences financial difficulties and becomes unable to pay its liabilities when due and declares bankruptcy, its creditors could attempt to assert claims against Napo relating to the formation of our company and the grant of an exclusive license to us.
Napo formed our company in June 2013, and in January 2014, we entered into the Napo License Agreement. Napo currently has no commercial operations and its potential sources of revenue are limited to the third parties who have licensed or may license Napo's intellectual property and technology, or collaborate with Napo in the future. Napo has been involved in litigation with Salix and has expended significant resources in the litigation. At the time of the formation of our company and
26
the date of the Napo License Agreement, Napo's liabilities exceeded its assets on a balance sheet prepared in conformity with U.S. generally accepted accounting principles. Napo has been able to pay its liabilities when due but if Napo experiences financial difficulties and becomes unable to pay its liabilities when due, or declares bankruptcy, a creditor, trustee in bankruptcy, or other representative of a Napo bankruptcy estate could attempt to assert claims against us relating to our formation and Napo's grant of an exclusive license to us. One theory such a party could use to challenge our formation and the license grant is that of fraudulent conveyance. This theory is used by creditors to challenge the transfer of assets made with actual intent to hinder, delay, or defraud creditors, or where a financially distressed entity transfers assets without receiving reasonably equivalent value in exchange, provided such litigation is brought within the applicable statute of limitations. Although we do not believe that our formation or Napo's grant of the license was a fraudulent conveyance, litigation based on such theory, if successful, could result in a court order setting aside the license for the benefit of the creditor pursuing the litigation or all creditors of Napo should it occur in the context of a Napo bankruptcy. Even if unsuccessful, any such action would divert management's attention, potentially be costly to defend and could harm our business.
We currently do not own any issued patents, most of our intellectual property is licensed from Napo and we cannot be certain that our patent strategy will be effective to enhance marketing exclusivity.
The patent prosecution process is expensive and time-consuming, and we may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of inventions made in the course of development and commercialization activities in time to obtain patent protection on them. Moreover, in some circumstances, we may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology that we license from third parties. In particular, we are dependent upon Napo and its licensees to file, prosecute and maintain the intellectual property we license pursuant to the Napo License Agreement. The patents and patent applications we licensed from Napo, or the Napo Patents, which cover both human and veterinary uses, are also licensed by Napo to Salix for certain fields of human use. Under the terms of the collaboration agreement between Salix and Napo, or the Salix Collaboration Agreement, Napo and Salix agreed on who has the first right and responsibility to file, prosecute and maintain the Napo Patents. As a result, under the Napo License Agreement, we only have the right to maintain any issued patents within the Napo Patents that are not maintained in accordance with the rights and responsibilities of the parties under the Salix Collaboration Agreement. There are three issued Napo Patents in the United States that cover, collectively, enteric protected formulations of proanthocyanidin polymers isolated from Croton spp. and methods of treating watery diarrhea using the enteric protected formulations for both human and veterinary uses.
Napo has also licensed its Croton lechleri related intellectual property to Salix, Glenmark Pharmaceuticals Ltd. and Luye Pharma Group Limited to develop and commercialize crofelemer for human indications in various geographies. In May 2011, Napo filed a lawsuit against Salix in the Supreme Court of the State of New York, County of New York, alleging, among other items, that Salix had breached its collaboration agreement with Napo. By orders entered in December 2013 and January 2014, the court granted Salix's motion for partial summary judgment and narrowed the issues for trial. In February 2014, the jury rendered its verdict, concluding that Salix had complied with its contractual obligations in commercializing Fulyzaq in the United States, and had not breached the collaboration agreement. In May 2014, Napo filed a notice of appeal from the court's partial summary judgment ruling as well as from certain court rulings and the judgment entered in February 2014. That appeal is pending. Fulyzaq is dependent upon intellectual property protection from the Napo Patents. Salix currently markets Fulyzaq in the United States for human use and has listed the three issued Napo Patents in the FDA's Orange Book for Fulyzaq. We rely on these issued Napo Patents as intellectual property protection for our prescription drug product candidates and non-prescription products.
27
Pending patent applications within Napo Patents either may not be relevant to veterinary indications and/or may not issue as patents. If any patent application within the Napo Patents is not filed or prosecuted as provided in the Salix Collaboration Agreement, including due to a lack of financial resources, and we are not able to file and prosecute such patent application within the Napo Patents, our business may be harmed. Also, under the Salix Collaboration Agreement, Napo and Salix have agreed on who has the first right to enforce the Napo Patents against potential infringers. In addition, as between Napo and us, Napo has the first right to enforce the Napo Patents against potential infringers. If we are not the party who enforces the Napo Patents, we will receive no proceeds from such enforcement action. In each case, such proceeds are subject to reimbursement of costs and expenses incurred by the other party in connection with such action. If our current or future licensors fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced or eliminated.
We currently do not own any issued patents. We have filed three provisional patent applications in the veterinary field, of which we control the filing, prosecution and maintenance; however, patents based on any patent applications we may submit may never be issued. We have an exclusive worldwide license from Napo to various issued patents and pending patent applications in the field of animal health. The strength of patents in the field of animal health involves complex legal and scientific questions and can be uncertain. Even if patents do successfully issue, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents, if issued, and the patents we have licensed may not adequately protect our intellectual property or prevent others from designing around their claims. If we cannot obtain issued patents or the patents we have licensed are not maintained or their scope is significantly narrowed, our business and prospects would be harmed.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of any patent applications and the enforcement or defense of any patents that issue. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a "first-to-invent" system to a "first-to-file" system. Under a "first-to-file" system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO has developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, became effective on March 16, 2013. Among some of the other changes to the patent laws are changes that limit where a patentee may file a patent infringement suit and that provide opportunities for third parties to challenge any issued patent in the USPTO. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that issue, all of which could harm our business and financial condition.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable
28
rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering prescription drug product candidates and non-prescription products, our competitors might be able to enter the market, which would harm our business.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of our current or future products and product candidates.
Our research, development and commercialization activities may infringe or otherwise violate or be claimed to infringe or otherwise violate patents owned or controlled by other parties. There may be patents already issued of which we are unaware that might be infringed by one of our current or future prescription drug product candidates or non-prescription products. Moreover, it is also possible that patents may exist that we are aware of, but that we do not believe are relevant to our current or future prescription drug product candidates or non-prescription products, which could nevertheless be found to block our freedom to market these products. Because patent applications can take many years to issue and may be confidential for 18 months or more after filing, there may be applications now pending of which we are unaware and which may later result in issued patents that may be infringed by our current or future prescription drug product candidates or non-prescription products. We cannot be certain that our current or future prescription drug product candidates or non-prescription products will not infringe these or other existing or future third-party patents. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents.
To the extent we become subject to future third-party claims against us or our collaborators, we could incur substantial expenses and, if any such claims are successful, we could be liable to pay substantial damages, including treble damages and attorney's fees if we or our collaborators are found to be willfully infringing a third party's patents. If a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the prescription drug or non-prescription product that is the subject of the suit. Even if we are successful in defending such claims, infringement and other intellectual property claims can be expensive and time-consuming to litigate and divert management's attention from our business and operations. As a result of or in order to avoid potential patent infringement claims, we or our collaborators may be compelled to seek a license from a third party for which we would be required to pay license fees or royalties, or both. Moreover, these licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain such a license, the rights may be nonexclusive, which could allow our competitors access to the same intellectual property. Any of these events could harm our business and prospects.
There has been substantial litigation regarding patents and other intellectual property rights in the field of therapeutics, as well as patent challenge proceedings, including interference, derivation and administrative law proceedings before the USPTO, and oppositions and other comparable proceedings in foreign jurisdictions. Under U.S. patent reform laws, new procedures, including inter partes review and post-grant review, were implemented as of September 16, 2012, with post-grant review available for patents issued on applications filed on or after March 16, 2013, and the implementation of such reform laws presents uncertainty regarding the outcome of any challenges to our future patents, if any, and to patents we have in licensed. In addition to possible infringement claims against us, we may be subject to third-party pre-issuance submission of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review, or other patent office proceedings or litigation in the United States or elsewhere, challenging our patent rights or the patent rights of others.
29
For applications filed before March 16, 2013 or patents issuing from such applications, if third parties have prepared and filed patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings in the USPTO to determine the priority of invention. Because patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to either file patent applications on or invent any of the inventions claimed in our patent applications. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal court necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. We may also become involved in opposition or similar proceedings in patent offices in other jurisdictions regarding our intellectual property rights with respect to our prescription drug or non-prescription products and technology. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our future patent rights, if any, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
Our proprietary position depends upon patents that are formulation or method-of-use patents, which do not prevent a competitor from using the same drug candidate for another use.
Composition-of-matter patents on the API in prescription drug products are generally considered to be the strongest form of intellectual property protection because such patents provide protection without regard to any particular method of use or manufacture or formulation of the API used. The composition-of-matter patents for crofelemer, the API in Canalevia, have expired, and we have licensed from Napo patents and applications covering formulations and methods of use for crofelemer and the botanical extract in Neonorm.
Method-of-use patents protect the use of a product for the specified method and formulation patents cover formulations of the API or botanical extract. These types of patents do not prevent a competitor from developing or marketing an identical product for an indication that is outside the scope of the patented method or from developing a different formulation that is outside the scope of the patented formulation. Moreover, with respect to method-of-use patents, even if competitors do not actively promote their product for our targeted indications or uses for which we may obtain patents, veterinarians may recommend that animal owners use these products extra-label, or animal owners may do so themselves. Although extra-label use may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
If our efforts to protect intellectual property are not adequate, we may not be able to compete effectively in our markets.
We intend to rely upon a combination of regulatory exclusivity periods, patents, trade secret protection, confidentiality agreements, and license agreements to protect the intellectual property related to our current prescription drug product candidates and non-prescription products and our development programs.
If the breadth or strength of protection provided by any patents, patent applications or future patents we may own, license, or pursue with respect to any of our current or future product candidates or products is threatened, it could threaten our ability to commercialize any of our current or future product candidates or products. Further, if we encounter delays in our development efforts, the period of time during which we could market any of our current or future product candidates or products under any patent protection we obtain would be reduced.
30
Given the amount of time required for the development, testing and regulatory review of new product candidates or products, patents protecting such candidates might expire before or shortly after such product candidates or products are commercialized. Patent term extension have been applied for US 7,323,195 and US 7,341,744 to account for regulatory delays in obtaining human marketing approval for crofelemer, however, only one patent may be extended per marketed compound. If such extensions are received, then US 7,323,195 may be extended to June 2021 or US 7,341,744 may be extended to December 2020. However, the applicable authorities, including the USPTO and the FDA, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to patents, or may grant more limited extensions than requested. If this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Even where laws provide protection or we are able to obtain patents, costly and time-consuming litigation may be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have.
If we are unable to prevent disclosure of our trade secrets or other confidential information to third parties, our competitive position may be impaired.
We also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or for which we have not filed patent applications, processes for which patents are difficult to enforce and other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us, and endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or had access to our proprietary information, or that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. If we are unable to prevent disclosure of our intellectual property to third parties, we may not be able to maintain a competitive advantage in our market, which would harm our business.
Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, and erode our competitive position in our market.
We may be involved in lawsuits to protect or enforce any future patents issued to us, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe any patents that may issue to us, or any patents that we may license. To counter infringement or unauthorized use of any patents we may obtain, we may be required to file infringement claims or request that our licensor file an infringement claim, which can be expensive and time-consuming to litigate. In addition, if we or one of our future collaborators were to initiate legal proceedings against a third party to enforce a patent covering our current product candidates, or one of our future products, the defendant could counterclaim that the patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or lack of statutory
31
subject matter. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant material information from the USPTO, or made a materially misleading statement, during prosecution. Third parties may also raise similar validity claims before the USPTO in post-grant proceedings such as ex parte reexaminations, inter partes review, or post-grant review, or oppositions or similar proceedings outside the United States, in parallel with litigation or even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. We cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. For the patents and patent applications that we have licensed, we may have limited or no right to participate in the defense of any licensed patents against challenge by a third party. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of any future patent protection on our current or future product candidates. Such a loss of patent protection could harm our business.
Litigation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be unsuccessful, it could have an adverse effect on the price of our common stock. Finally, we may not be able to prevent, alone or with the support of our licensors, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other animal health product companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the animal health industry involves both technological and legal complexity. Therefore, obtaining and enforcing patents is costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and implemented wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce patents that we have licensed or that we might obtain in the future.
We may not be able to protect our intellectual property rights throughout the world, which could impair our business.
Filing, prosecuting and defending patents on prescription drug products, product candidates and non-prescription products throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we may obtain patent protection, but where patent enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and any future patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
32
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to animal health products, which could make it difficult for us to stop the infringement of our future patents, if any, or patents we have in licensed, or marketing of competing products in violation of our proprietary rights generally. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. Proceedings to enforce our future patent rights, if any, in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Our business could be harmed if we fail to obtain certain registered trademarks in the United States or in other countries.
In October 2014, our trademark applications for Canalevia and Neonorm were approved for publication. Although we have filed a trademark application for our company name and our logo in the United States, our applications have not been granted and the corresponding marks have not been registered in the United States. We have not filed for these or other trademarks in any other countries. During trademark registration proceedings, we may receive rejections. If so, we will have an opportunity to respond, but we may be unable to overcome such rejections. In addition, the USPTO and comparable agencies in many foreign jurisdictions may permit third parties to oppose pending trademark applications and to seek to cancel registered trademarks. If opposition or cancellation proceedings are filed against any of our trademark applications or any registered trademarks, our trademarks may not survive such proceedings. Moreover, any name we propose to use with our prescription drug product candidates in the United States, including Canalevia, must be approved by the FDA, regardless of whether we have registered or applied to register as a trademark. The FDA typically conducts a review of proposed prescription drug product names, including an evaluation of potential for confusion with other product names. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology, pharmaceutical or animal health companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise improperly used or disclosed confidential information of these third parties or our employees' former employers. Litigation may be necessary to defend against any such claims. Even if we are successful in defending against any such claims, such litigation could result in substantial cost and be a distraction to our management and employees.
Risks Related to Government Regulation
Even if we receive any required regulatory approvals for our current or future prescription drug product candidates and non-prescription products, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.
If the FDA or any other regulatory body approves any of our current or future prescription drug product candidates, or if necessary, our non-prescription products, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and
33
recordkeeping for the product may be subject to extensive and ongoing regulatory requirements. These requirements include, but are not limited to, submissions of safety and other post-marketing information and reports, establishment registration, and product listing, as well as continued compliance with cGMP, GLP and GCP for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our contract manufacturers or manufacturing processes, or failure to comply with regulatory requirements, must be reported in many instances to the FDA and may result in, among other things:
- •
- restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, revised
labeling, or voluntary or involuntary product recalls;
- •
- fines, warning letters or holds on target animal studies;
- •
- refusal by the FDA, or other regulators to approve pending applications or supplements to approved applications filed by
us or our strategic collaborators related to the unknown problems, or suspension or revocation of the problematic product's license approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; and
- •
- injunctions or the imposition of civil or criminal penalties.
The FDA or other regulatory agency's policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates or require certain changes to the labeling or additional clinical work concerning safety and efficacy of the product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would harm our business. In addition, failure to comply with these regulatory requirements could result in significant penalties.
In addition, from time to time, we may enter into consulting and other financial arrangements with veterinarians, who prescribe or recommend our products, once approved. As a result, we may be subject to state, federal and foreign healthcare and/or veterinary medicine laws, including but not limited to anti-kickback laws. If our financial relationships with veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
The FDA issuing protocol concurrences for our pivotal studies does not guarantee ultimate approval of our NADA.
We intend to seek protocol concurrences with the FDA for the pivotal trial of Canalevia that we plan to conduct for general acute watery diarrhea in dogs and for future pivotal trials in other indications. A pivotal study protocol is submitted to the FDA voluntarily by a drug sponsor for purposes of obtaining FDA review of the protocol. Prior FDA review of the protocol for a pivotal study makes it more likely that the study will generate information the sponsor needs to demonstrate whether the drug is safe and effective for its intended use. It creates an expectation by the sponsor that the FDA should not later alter its perspectives on these issues unless public or animal health concerns appear that were not recognized at the time of protocol assessment. Even if the FDA issues a protocol concurrence, ultimate approval of an NADA by the FDA is not guaranteed because a final determination that the agreed-upon protocol satisfies a specific objective, such as the demonstration of efficacy, or supports an approval decision, will be based on a complete review of all the data submitted to the FDA. Even if we were to obtain protocol concurrence such concurrence does not guarantee that the results of the study will support a particular finding or approval of the new drug.
34
Any of our current or future prescription drug product candidates or non-prescription products may cause or contribute to adverse medical events that we would be required to report to regulatory authorities and, if we fail to do so, we could be subject to sanctions that would harm our business.
If we are successful in commercializing any of our current or future prescription drug product candidates or non-prescription products, at least certain regulatory authorities will require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the regulatory authorities could take action including, but not limited to, criminal prosecution, seizure of our products or delay in approval or clearance of future products.
Legislative or regulatory reforms with respect to animal health may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our current or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress or other jurisdictions in which we intend to operate that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, the FDA and other regulations and guidance are often revised or reinterpreted by the FDA and such other regulators in ways that may significantly affect our business and our products and product candidates. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of any of our current or future products and product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
- •
- changes to manufacturing methods;
- •
- new requirements related to approval to enter the market;
- •
- recall, replacement, or discontinuance of certain products; and
- •
- additional record keeping.
Each of these would likely entail substantial time and cost and could harm our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
We do not believe that our non-prescription products are subject to regulation by regulatory agencies in the United States, but there is a risk that regulatory bodies may disagree with our interpretation, or may redefine the scope of its regulatory reach in the future, which would result in additional expense and could delay or prevent the commercialization of these products.
The FDA retains jurisdiction over all prescription drug products however, in many instances, the Federal Trade Commission will exercise primary or concurrent jurisdiction with FDA on non-prescription products as to post marketing claims made regarding the product. On April 22, 1996 the FDA published a statement in the Federal Register, 61 FR 17706, that it does not believe that the Dietary Supplement and Health Education Act applies to animal supplement products. Therefore, animal supplements that fall within the FDA definition of an animal drug, food or food additive are
35
regulated by the FDA. The Federal Food Drug and Cosmetic Act defines food as "articles used for food or drink for man or other animals." The guidance and regulations providing insight into this definition further clarify that any "article that is intended to be used as an animal feed ingredient, to become part of an ingredient or feed or added to an animal's drinking water" is deemed to be a feed within the definition. Our non-prescription products are not added to food, are not ingredients in food nor are they added to any animal's drinking water and therefore, our non-prescription products do not fall within the definition of a food. In light of the pronouncement by the FDA that the Dietary Supplement and Health Education Act was not intended to apply to animals, the FDA seeks to regulate such supplements as food or food additives depending on the intended use of the product. The intended use is demonstrated by how the article is included in a food, or added to the animals' intake (i.e., through its drinking water). If the intended use of the product does not fall within the proscribed use making the product a food, it cannot be regulated as a food. There is no intent to make our non-prescription products a component of an animal food, either directly or indirectly. A feed additive is a product that is added to a feed for any reason including the top dressing of an already prepared feed. Some additives, such as certain forage, are deemed to be Generally Recognized as Safe, or GRAS, and therefore, not subject to a feed Additive Petition approval prior to use. However, the substances deemed GRAS are generally those that are recognized as providing nutrients as a food does. We do not believe that our non-prescription products fit within this framework either. Finally, a new animal drug refers to drugs intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in animals. Our non-prescription products are not intended to diagnose, cure, mitigate, treat or prevent disease and therefore, do not fit within the definition of an animal drug. We do not believe that our non-prescription products fit the definition of an animal drug, food or food additive and therefore are not regulated by the FDA at this time.
However, despite many such unregulated animal supplements currently on the market, the FDA may choose in the future to exercise jurisdiction over animal supplement products in which case, we may be subject to unknown regulations thereby inhibiting our ability to launch or to continue marketing our non-prescription products. In the past the FDA has redefined or attempted to redefine some non-prescription non-feed products as falling within the definition of drug, feed or feed additive and therefore subjected those products to the relevant regulations. Should the FDA assert regulatory authority over our non-prescription products, we would take commercially reasonable steps to address the FDA's concerns, potentially including but not limited to, seeking registration for such products, reformulating such products to further distance such products from regulatory control, or ceasing sale of such products. Further, the Animal and Plant Health Inspection Service, an agency of the USDA, may at some point choose to exercise jurisdiction over certain non-prescription products that are not intended for production animals. We do not believe we are currently subject to such regulation, but could be in the future. If the FDA or other regulatory agencies, such as the USDA, try to regulate our non-prescription products, we could be required to seek regulatory approval for our non-prescription products, which would result in additional expense and could delay or prevent the commercialization of these products.
Risks Related to this Offering and Our Common Stock
The price of our common stock could be subject to volatility related or unrelated to our operations, and purchasers of our common stock could incur substantial losses.
If a market for our common stock develops following this offering, the trading price of our common stock could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include those discussed previously in this "Risk Factors" section of this prospectus and others, such as:
- •
- delays in the commercialization of Neonorm, Canalevia or our other current or future prescription drug product candidates and non-prescription products;
36
- •
- any delays in, or suspension or failure of, our current and future studies;
- •
- announcements of regulatory approval or disapproval of any of our current or future product candidates or of regulatory
actions affecting us or our industry;
- •
- manufacturing and supply issues that affect product candidate or product supply for our studies or commercialization
efforts;
- •
- quarterly variations in our results of operations or those of our competitors;
- •
- changes in our earnings estimates or recommendations by securities analysts;
- •
- announcements by us or our competitors of new prescription drug products or product candidates or non-prescription
products, significant contracts, commercial relationships, acquisitions or capital commitments;
- •
- announcements relating to future development or license agreements including termination of such agreements;
- •
- adverse developments with respect to our intellectual property rights or those of our principal collaborators;
- •
- commencement of litigation involving us or our competitors;
- •
- any major changes in our board of directors or management;
- •
- new legislation in the United States relating to the prescription, sale, distribution or pricing of animal health
products;
- •
- product liability claims, other litigation or public concern about the safety of our prescription drug product candidates
and non-prescription products or any such future products;
- •
- market conditions in the animal health industry, in general, or in the animal health sector, in particular, including
performance of our competitors; and
- •
- general economic conditions in the United States and abroad.
In addition, the stock market, in general, or the market for stocks in our industry, in particular, may experience broad market fluctuations, which may adversely affect the market price or liquidity of our common stock. Any sudden decline in the market price of our common stock could trigger securities class-action lawsuits against us. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the time and attention of our management would be diverted from our business and operations. We also could be subject to damages claims if we are found to be at fault in connection with a decline in our stock price.
No active market for our common stock exists or may develop, and you may not be able to resell your common stock at or above the initial public offering price.
Prior to this offering, there has been no public market for shares of our common stock. We and the representatives of the underwriters determined the initial public offering price of our common stock by arm's-length negotiations, and the initial public offering price does not necessarily reflect the price at which investors in the market will be willing to buy and sell our shares following this offering. If no active trading market for our common stock develops or is sustained following this offering, you may be unable to sell your shares when you wish to sell them or at a price that you consider attractive or satisfactory. The lack of an active market may also adversely affect our ability to raise capital by selling securities in the future, or impair our ability to license or acquire other product candidates, businesses or technologies using our shares as consideration.
37
Purchasers in this offering will experience immediate and substantial dilution in the book value of their investment.
The initial public offering price of our common stock is substantially higher than the pro forma net tangible book value per share of our common stock before giving effect to this offering. Accordingly, if you purchase our common stock in this offering, you will incur immediate dilution of approximately $ per share, representing the difference between the initial public offering price of $ per share and our pro forma as adjusted net tangible book value per share as of , 2014. In addition, following this offering, purchasers in this offering will have contributed approximately % of the total gross consideration paid by stockholders to us to purchase shares of our common stock through , 2014, but will own only approximately % of the shares of common stock outstanding immediately after this offering. Furthermore, if the underwriters exercise their option to purchase additional shares of our common stock or our outstanding stock options are exercised, you will experience further dilution. For a further description of the dilution that you will experience immediately after this offering, see the section in this prospectus titled "Dilution."
If securities or industry analysts do not publish research or reports about our company, or if they issue an adverse or misleading opinions regarding us or our stock, our stock price and trading volume could decline.
We do not currently have research coverage by securities and industry analysts, and if no significant coverage is initiated or maintained following this offering, the market price for our stock may be adversely affected. Our stock price also may decline if any analyst who covers us issues an adverse or erroneous opinion regarding us, our business model, our intellectual property or our stock performance, or if our animal studies and operating results fail to meet analysts' expectations. If one or more analysts cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline and possibly adversely affect our ability to engage in future financings.
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after the expiration or termination of the lock-up and other legal restrictions on resale discussed in this prospectus, the trading price of our common stock could decline. Based upon the number of shares outstanding as of , upon the closing of this offering, we will have outstanding a total of shares of common stock. Of these shares, approximately shares, plus any shares sold upon exercise of the underwriters' option to purchase additional shares of our common stock, will be freely tradable in the public market immediately following this offering.
The lock-up agreements pertaining to this offering will expire 180 days from the date of this prospectus. After the lock-up agreements expire, up to an additional shares of common stock will be eligible for sale in the public market, of which shares are held by directors, executive officers and other affiliates and will be subject to vesting schedules or volume limitations under Rule 144 under the Securities Act of 1933, as amended, or the Securities Act. The representatives of the underwriters may, in their sole, joint discretion, permit our officers, directors and other stockholders who are subject to lock-up agreements to sell shares even prior to the expiration of the lock-up agreements. In addition, shares of common stock that are subject to outstanding options under our 2013 Equity Incentive Plan will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, the lock-up agreements and Rule 144 and Rule 701 under the Securities Act. The sale or possible sale of these additional shares may adversely affect the trading price of our common stock.
38
We will have broad discretion to use the net proceeds from this offering, and may use them in ways that do not enhance our operating results or the market price of our common stock.
Our management will have broad discretion regarding the use of the net proceeds from this offering, and we could spend the net proceeds in ways our stockholders may not agree with or that do not yield a favorable return, if at all. We intend to use the net proceeds from this offering for the research and development of our prescription drug and non-prescription products and product candidates, manufacturing, marketing, distribution and commercialization of any products and other general corporate and working capital purposes. We may also use a portion of the net proceeds to acquire additional product candidates or complementary assets or businesses; however, we currently have no agreements or commitments to complete any such transaction. Our use of these proceeds may differ substantially from our current plans. If we do not invest or apply the net proceeds from this offering in ways that improve our operating results or our prospects, our stock price could decline.
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our amended and restated certificate of incorporation and amended and restated bylaws that will be in effect upon the closing of this offering will contain provisions that could delay or prevent changes in control or changes in our management without the consent of our board of directors. We expect these provisions to include the following:
- •
- a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the
membership of a majority of our board of directors;
- •
- no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director
candidates;
- •
- the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board
of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors;
- •
- the ability of our board of directors to alter our bylaws without obtaining stockholder approval;
- •
- the required approval of the holders of at least two-thirds of the shares entitled to vote at an election of directors to
adopt, amend or repeal our bylaws or repeal the provisions of our amended and restated certificate of incorporation regarding the election and removal of directors;
- •
- a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or
special meeting of our stockholders;
- •
- the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the
chief executive officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of
directors; and
- •
- advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders' meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer's own slate of directors or otherwise attempting to obtain control of us.
These provisions could inhibit or prevent possible transactions that some stockholders may consider attractive.
39
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation generally may not engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
Our amended and restated bylaws designate the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain actions and proceedings that may be initiated by our stockholders, which could limit our stockholders' ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our amended and restated bylaws that will be in effect upon the closing of this offering provide that, unless we consent in writing to an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law, or (iv) any action asserting a claim that is governed by the internal affairs doctrine. Any person purchasing or otherwise acquiring any interest in any shares of our capital stock shall be deemed to have notice of and to have consented to this provision of our amended and restated bylaws. This choice-of-forum provision may limit our stockholders' ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits. Alternatively, if a court were to find this provision of our amended and restated bylaws inapplicable or unenforceable with respect to one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could harm our business and financial condition.
We do not intend to pay dividends on our common stock, and your ability to achieve a return on your investment will depend on appreciation in the market price of our common stock.
As described in the section titled "Dividend Policy" in this prospectus, we currently intend to invest our future earnings, if any, to fund our growth and not to pay any cash dividends on our common stock. Because we do not intend to pay dividends, your ability to receive a return on your investment will depend on any future appreciation in the market price of our common stock. We cannot be certain that our common stock will appreciate in price.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Upon the closing of this offering, based on shares outstanding as of June 30, 2014, our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates will beneficially own in the aggregate approximately % of our outstanding shares of common stock. As a result of their stock ownership, these stockholders may have the ability to influence our management and policies, and will be able to significantly affect the outcome of matters requiring stockholder approval such as elections of directors, amendments of our organizational documents or approvals of any merger, sale of assets or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders.
As a newly public company, we will incur significant additional costs, and our management will be required to devote substantial management time and attention to our public reporting obligations.
As a privately-held company, we have not been required to comply with public reporting, corporate governance and financial accounting practices and policies required of a publicly-traded company. As a
40
publicly-traded company, we will incur significant additional legal, accounting and other expenses compared to historical levels. In addition, new and changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act and the rules and regulations thereunder, as well as under the Sarbanes-Oxley Act, the JOBS Act and the rules and regulations of the U.S. Securities and Exchange Commission, or the SEC, and The NASDAQ Capital Market, may result in an increase in our costs and the time that our board of directors and management must devote to our compliance with these rules and regulations. We expect these rules and regulations to substantially increase our legal and financial compliance costs and to divert management time and attention from our product development and other business activities.
The Sarbanes-Oxley Act requires, among other things, that we assess the effectiveness of our internal control over financial reporting annually and the effectiveness of our disclosure controls and procedures quarterly. In particular, Section 404 of the Sarbanes-Oxley Act, or Section 404, requires us to perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on, and our independent registered public accounting firm potentially to attest to, the effectiveness of our internal control over financial reporting. We will need to expend time and resources on documenting our internal control over financial reporting so that we are in a position to perform such evaluation when required. As an "emerging growth company," we expect to avail ourselves of the exemption from the requirement that our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting under Section 404. However, we may no longer avail ourselves of this exemption when we cease to be an "emerging growth company." When our independent registered public accounting firm is required to undertake an assessment of our internal control over financial reporting, the cost of our compliance with Section 404 will correspondingly increase. Our compliance with applicable provisions of Section 404 will require that we incur substantial accounting expense and expend significant management time on compliance-related issues as we implement additional corporate governance practices and comply with reporting requirements. Moreover, if we are not able to comply with the requirements of Section 404 applicable to us in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources.
We are an "emerging growth company" and we cannot be certain if the reduced disclosure requirements applicable to "emerging growth companies" will make our common stock less attractive to investors.
We are an "emerging growth company," as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not "emerging growth companies." In particular, while we are an "emerging growth company" (i) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (ii) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (iii) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved. In addition, the JOBS Act provides that an emerging growth company can delay its adoption of any new or revised accounting standards, but we have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. In addition, investors may find our common stock less attractive if we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline and/or become more volatile.
41
We may remain an "emerging growth company" until as late as December 31, 2019 (the fiscal year-end following the fifth anniversary of the closing of this offering), although we may cease to be an "emerging growth company" earlier under certain circumstances, including (i) if the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of any June 30, in which case we would cease to be an "emerging growth company" as of December 31 of such year, (ii) if our gross revenue exceeds $1.0 billion in any fiscal year or (iii) if we issue more than $1.0 billion of non-convertible debt over a three-year period.
42
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements. All statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations and financial position, business strategy, prospective products, product approvals, research and development costs, timing of receipt of clinical trial, field study and other study data, and likelihood of success, commercialization plans and timing, other plans and objectives of management for future operations, and future results of current and anticipated products are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as "may," "will," "should," "expect," "plan," "aim," "anticipate," "could," "intend," "target," "project," "contemplate," "believe," "estimate," "predict," "potential" or "continue" or the negative of these terms or other similar expressions. The forward-looking statements in this prospectus are only predictions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our business, financial condition and results of operations. These forward-looking statements speak only as of the date of this prospectus and are subject to a number of risks, uncertainties and assumptions described under the sections in this prospectus titled "Risk Factors" and "Management's Discussion and Analysis of Financial Condition and Results of Operations" and elsewhere in this prospectus. Forward-looking statements are subject to inherent risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in a dynamic industry and economy. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties that we may face. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
Unless otherwise indicated, information contained in this prospectus concerning our industry and the markets in which we operate, including our general expectations and market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. In addition, assumptions and estimates of our and our industry's future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in "Risk Factors." These and other factors could cause our future performance to differ materially from our assumptions and estimates. See "Special Note Regarding Forward-Looking Statements."
43
We estimate that the net proceeds from our issuance and sale of shares of common stock in this offering will be approximately $ million, assuming an initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. If the underwriters exercise their option to purchase additional shares from us in full, we estimate that the net proceeds from this offering will be approximately $ million, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
A $1.00 increase (decrease) in the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) the net proceeds from this offering by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. An increase (decrease) by 1,000,000 shares in the number of shares offered by us would increase (decrease) the net proceeds to us from this offering by approximately $ million, assuming that the assumed initial public offering price remains the same, and after deducting underwriting discounts and commissions. We do not expect that a change in the initial public offering price or the number of shares by these amounts would have a material effect on our anticipated uses of the net proceeds from this offering, although it may accelerate the time at which we will need to seek additional capital.
We anticipate that we will use the net proceeds from this offering as follows:
- •
- approximately $4.2 million for clinical studies and regulatory approval costs related to Canalevia for CID
($0.3 million) and general acute watery diarrhea ($3.9 million) in dogs;
- •
- approximately $7.0 million for clinical studies and regulatory approval costs related to the other prescription
drug products in our pipeline, namely species-specific formulations of crofelemer for general acute watery diarrhea in cats ($1.7 million), acute colitis in horses ($1.3 million),
gastric and colonic ulcers in horses ($2.0 million) and the herpes virus in cats ($0.9 million), as well as proof-of-concept studies for NP-500 ($1.1 million);
- •
- approximately $3.0 million for studies and commercial activities inside and outside the United States related to
Neonorm for scours in preweaned dairy calves ($2.1 million) and for the equine field ($0.9 million);
- •
- approximately $2.7 million for the formulation costs associated with developing species-specific formulations of
our products and for other third-party costs;
- •
- approximately $5.0 million for establishing manufacturing capability, including the technology transfer and
manufacturing and support equipment at third-party manufacturing facilities, and including approximately $3.7 million of fees payable to Indena S.p.A. pursuant to our memorandums of
understanding; and
- •
- the remaining funds will be utilized for working capital and general corporate purposes.
These expected uses of the net proceeds from this offering represents our intentions based upon our current financial condition, results of operations, business plans and conditions. As of the date of this prospectus, we cannot predict with certainty all of the particular uses for the net proceeds to be received upon the closing of this offering or the amounts that we will actually spend on the uses set forth above. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors. As a result, our management will retain broad discretion over the allocation of the net proceeds from this offering.
We may also use a portion of the net proceeds from this offering for the acquisition of, or investment in, complementary business, products or technologies, although we have no present commitments or agreements for any specific acquisitions or investments. Pending our use of the net proceeds from this offering, we intend to invest the net proceeds in a variety of capital preservation investments, including short-term, investment grade, interest bearing instruments and U.S. government securities.
44
We have never declared or paid any cash dividends on our capital stock. We intend to retain future earnings, if any, to finance the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future. Any future determination related to dividend policy will be made at the discretion of our board of directors after considering our financial condition, results of operations, capital requirements, business prospects and other factors the board of directors deems relevant, and subject to the restrictions contained in any future financing instruments.
45
The following table sets forth our cash and cash equivalents and capitalization as of June 30, 2014, as follows:
- •
- on an actual basis;
- •
- on a pro forma basis to give effect to (i) the conversion of all outstanding shares of Series A preferred
stock into 3,015,902 shares of common stock upon the closing of this offering; (ii) the issuance of shares of common stock upon the conversion of convertible promissory notes
in
the aggregate principal amount of $450,000 (which includes $150,000 aggregate principal amount of notes issued in July 2014) upon the closing of this offering at a conversion price equal to 80% of the
assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, and which shares will be
unregistered; and
(iii) the filing and effectiveness of our amended and restated certificate of incorporation upon the closing of this offering; and
- •
- on a pro forma as adjusted basis to give further effect to the sale of shares of common stock in this offering at the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
You should read this information in conjunction with our financial statements and related notes appearing elsewhere in this prospectus and the sections in this prospectus titled "Selected Financial Data" and "Management's Discussion and Analysis of Financial Condition and Results of Operations."
| |
As of June 30, 2014 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Actual | Pro Forma | Pro Forma As Adjusted(1) |
|||||||
| |
(unaudited) |
|||||||||
Cash and cash equivalents |
$ | 4,281,698 | $ | $ | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Convertible promissory notes |
$ | 231,250 | $ | $ | ||||||
| | | | | | | | | | | |
Series A redeemable convertible preferred stock, par value $0.0001 per share: 3,017,488 shares authorized, 3,015,902 shares issued and outstanding, actual; no shares authorized, issued and outstanding, pro forma and pro forma as adjusted |
6,943,250 | |||||||||
Stockholders' equity (deficit): |
||||||||||
Common stock, par value $0.0001 per share: 10,000,000 shares authorized, 4,311,498 shares issued and outstanding, actual; shares authorized, shares issued and outstanding, pro forma; shares issued and outstanding, pro forma as adjusted |
431 | |||||||||
Additional paid-in capital |
809,702 | |||||||||
Deficit accumulated during the development stage |
(4,711,437 | ) | ||||||||
| | | | | | | | | | | |
Total stockholders' (deficit) |
(3,901,304 | ) | ||||||||
| | | | | | | | | | | |
Total capitalization |
$ | 3,273,196 | $ | $ | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- A $1.00 increase (decrease) in the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) each of cash and cash equivalents, additional paid-in capital, total stockholders' equity (deficit) and total capitalization by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
46
An increase (decrease) of 1,000,000 shares in the number of shares offered by us would increase (decrease) cash and cash equivalents, additional paid-in capital, total stockholders' equity (deficit) and total capitalization by approximately $ million, assuming the assumed initial public offering price remains the same, and after deducting underwriting discounts and commissions. The pro forma as adjusted information discussed above is illustrative only and will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing.
- •
- 311,498 shares of common stock issuable upon exercise of outstanding warrants as of June 30, 2014 at an exercise
price of $1.6854 per share; and
- •
- 25,000 shares of common stock issuable upon exercise of an outstanding warrant as of June 30, 2014 with an exercise
price equal to 90% of the initial public offering price;
- •
- 50,000 shares of our common stock issuable upon exercise of outstanding warrants issued after June 30, 2014 with an
exercise price equal to 80% of the initial public offering price;
- •
- 1,129,673 shares issuable upon exercise of outstanding options as of June 30, 2014 with a weighted-average exercise
price of $1.77 per share;
- •
- 118,953 shares issuable upon vesting of outstanding restricted stock unit awards as of June 30, 2014;
- •
- 22,674 shares of common stock reserved for future issuance under our 2013 Equity Incentive Plan as of June 30,
2014; and
- •
- 500,000 shares of common stock reserved for future issuance under our 2014 Stock Incentive Plan, which will become effective in connection with this offering, as well as any automatic increases in the shares of common stock reserved for future issuance under the 2014 Stock Incentive Plan.
The outstanding share information in the table above is based on 4,311,498 shares of common stock outstanding as of June 30, 2014, and excludes:
47
If you invest in our common stock in this offering, your ownership interest will be immediately diluted to the extent of the difference between the initial public offering price per share of our common stock and the pro forma as adjusted net tangible book value per share of our common stock immediately after this offering.
As of June 30, 2014, our historical net tangible book value was $ , or $ per share of common stock. Our historical net tangible book value per share represents the amount of our total tangible assets less total liabilities divided by the number of shares of common stock outstanding as of June 30, 2014.
Our pro forma net tangible book value as of June 30, 2014 was $ or $ per share of common stock, after giving effect to (i) the conversion of all outstanding shares of Series A preferred stock into 3,015,902 shares of common stock upon the closing of this offering; (ii) the issuance of shares of common stock upon the conversion of convertible promissory notes in the aggregate principal amount of $450,000 (which includes $150,000 aggregate principal amount of notes issued in July 2014) upon the closing of this offering at a conversion price equal to 80% of the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, and which shares will be unregistered; and (iii) the filing and effectiveness of our amended and restated certificate of incorporation upon the closing of this offering.
After giving further effect to the sale of the shares of common stock in this offering at the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, our pro forma as adjusted net tangible book value as of June 30, 2014 would have been approximately $ , or $ per share. This amount represents an immediate increase in pro forma net tangible book value of $ per share to our existing stockholders, and an immediate dilution in pro forma net tangible book value of approximately $ per share to new investors purchasing shares of common stock in this offering.
Dilution per share to new investors is determined by subtracting pro forma as adjusted net tangible book value per share after this offering from the assumed initial public offering price per share paid by new investors. The following table illustrates this dilution:
Assumed initial public offering price per share |
$ | ||||||
Historical net tangible book value per share as of June 30, 2014 |
$ | ||||||
Increase attributable to conversion of all outstanding shares of Series A preferred stock and convertible promissory notes |
|||||||
| | | | | | | | |
Pro forma net tangible book value per share as of June 30, 2014 |
|||||||
Increase in net tangible book value per share attributable to new investors |
|||||||
| | | | | | | | |
Pro forma as adjusted net tangible book value per share after this offering |
|||||||
| | | | | | | | |
Dilution per share to new investors |
$ | ||||||
| | | | | | | | |
| | | | | | | | |
If the underwriters exercise their option to purchase additional shares in full, the pro forma as adjusted net tangible book value will increase to $ per share, representing an immediate dilution of $ per share to new investors, assuming that the initial public offering price will be $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus.
A $1.00 increase (decrease) in the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) our pro forma as adjusted net tangible book value per share after this offering by $ per share and the dilution to new investors by $ per share, assuming the number of shares
48
offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. An increase (decrease) of 1,000,000 shares in the number of shares offered by us would increase (decrease) the pro forma as adjusted net tangible book value by $ per share and the dilution to new investors by $ per share, assuming the assumed initial public offering price remains the same and after deducting underwriting discounts and commissions. The pro forma as adjusted information discussed above is illustrative only and will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing.
The following table summarizes, on a pro forma as adjusted basis as of June 30, 2014, the differences between the number of shares of common stock purchased from us, the total consideration and the average price per share paid by existing stockholders and by investors participating in this offering, before deducting underwriting discounts and commissions and estimated offering expenses payable by us, at an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus.
| |
Shares Purchased | Total Consideration | |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Average Price Per Share |
|||||||||||||||
| |
Number | Percent | Amount | Percent | ||||||||||||
Existing stockholders |
% | $ | % | $ | ||||||||||||
New investors |
% | % | ||||||||||||||
| | | | | | | | | | | | | | | | | |
Total |
100 | % | $ | 100 | % | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
The number of shares of common stock to be outstanding after this offering excludes:
- •
- 311,498 shares of common stock issuable upon exercise of outstanding warrants as of June 30, 2014 with an exercise
price of $1.6854 per share; and
- •
- 25,000 shares of common stock issuable upon exercise of an outstanding warrant as of June 30, 2014 with an exercise
price equal to 90% of the initial public offering price;
- •
- 50,000 shares of our common stock issuable upon exercise of outstanding warrants issued after June 30, 2014 with an
exercise price equal to 80% of the initial public offering price;
- •
- 1,129,673 shares issuable upon exercise of outstanding options as of June 30, 2014 with a weighted-average exercise
price of $1.77 per share;
- •
- 118,953 shares issuable upon vesting of outstanding restricted stock unit awards as of June 30, 2014;
- •
- 22,674 shares of common stock reserved for future issuance under our 2013 Equity Incentive Plan as of June 30,
2014; and
- •
- 500,000 shares of common stock reserved for future issuance under our 2014 Stock Incentive Plan, which will become effective in connection with this offering, as well as any automatic increases in the shares of common stock reserved for future issuance under the 2014 Stock Incentive Plan.
To the extent any of these outstanding options are exercised, there will be further dilution to new investors. If all of such outstanding options had been exercised as of June 30, 2014, the pro forma as adjusted net tangible book value after this offering would be $ per share, and total dilution to new investors would be $ per share.
If the underwriters exercise their option to purchase additional shares of common stock in full:
- •
- the percentage of shares of common stock held by existing stockholders will decrease to approximately % of
the total number of shares of common stock outstanding after this offering; and
- •
- the number of shares held by new investors will increase to , or approximately % of the total number of shares of common stock outstanding after this offering.
49
You should read the following selected financial data together with our financial statements and related notes appearing elsewhere in this prospectus and the section in this prospectus titled "Management's Discussion and Analysis of Financial Condition and Results of Operations." Napo formed our company to develop and commercialize animal health products. As of December 31, 2013, we were a wholly-owned subsidiary of Napo, and as of June 30, 2014, we are a majority-owned subsidiary of Napo.
The following tables set forth our selected statements of comprehensive loss data since inception in June 2013 and our selected balance sheet data as of December 31, 2013 and June 30, 2014. We are a development stage company. Data for the period from June 6, 2013 (inception) through and as of December 31, 2013 is derived from our audited financial statements included elsewhere in this prospectus. Data for the period from June 6, 2013 (inception) through June 30, 2014 and for the six months ended and as of June 30, 2014 is derived from our unaudited financial statements appearing elsewhere in this prospectus. The unaudited interim financial statements have been prepared on the same basis as our audited financial statements and, in our opinion, reflect all adjustments, consisting only of normal and recurring adjustments, which we consider necessary for a fair presentation of our financial position as of June 30, 2014. The historical results are not necessarily indicative of the results to be expected for any future periods, and the results for the six months ended June 30, 2014 should not be considered indicative of results expected for the full year 2014.
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
Period from June 6, 2013 (inception) through June 30, 2014 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
(unaudited) |
|||||||
Statements of Comprehensive Loss Data: |
||||||||||
Operating expenses: |
||||||||||
General and administrative expense |
$ | 458,473 | $ | 1,740,515 | $ | 2,198,988 | ||||
Research and development expense |
324,479 | 2,149,555 | 2,474,034 | |||||||
| | | | | | | | | | | |
Total operating expenses |
782,952 | 3,890,070 | 4,673,022 | |||||||
Loss from operations |
(782,952 |
) |
(3,890,070 |
) |
(4,673,022 |
) |
||||
Interest expense, net |
(18,251 | ) | (20,164 | ) | (38,415 | ) | ||||
| | | | | | | | | | | |
Net loss and comprehensive loss |
$ | (801,203 | ) | $ | (3,910,234 | ) | $ | (4,711,437 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Accretion of redeemable convertible preferred stock |
— | (285,009 | ) | (285,009 | ) | |||||
| | | | | | | | | | | |
Net loss attributable to common stockholders |
$ | (801,203 | ) | $ | (4,195,243 | ) | $ | (4,996,446 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share attributable to common stockholders, basic and diluted(1) |
$ | (0.20 | ) | $ | (0.99 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted-average common shares outstanding, basic and diluted(1) |
4,000,000 | 4,250,929 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro forma net loss per share, basic and diluted(1) |
$ | (0.20 | ) | $ | (0.67 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro forma weighted-average number of common shares(1) |
4,000,000 | 6,304,461 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
- (1)
- See Notes 2 and 12 to our financial statements for a description of the method used to compute basic and diluted net loss per share and pro forma net loss per share.
| |
As of December 31, 2013 |
As of June 30, 2014 |
|||||
|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
|||||
Balance Sheet Data: |
|||||||
Cash and cash equivalents |
$ | 185,367 | $ | 4,281,698 | |||
Total assets |
289,261 | 6,217,115 | |||||
Total liabilities |
724,114 | 3,175,169 | |||||
Convertible promissory notes |
519,486 | 231,250 | |||||
Redeemable convertible preferred stock |
— | 6,943,250 | |||||
Total stockholders' (deficit) |
(434,853 | ) | (3,901,304 | ) | |||
50
MANAGEMENT'S DISCUSSION
AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes and other financial information included elsewhere in this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. You should review the "Risk Factors" section of this prospectus for a discussion of important factors that could cause our actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are an animal health company focused on developing and commercializing first-in-class gastrointestinal products for companion and production animals. Canalevia is our lead prescription drug product candidate for the treatment of various forms of watery diarrhea in dogs. We expect to announce data from our field efficacy trial of Canalevia for general acute watery diarrhea in dogs in the fourth quarter of 2014. We also expect to initiate filing of a rolling new animal drug application, or NADA, for Canalevia for chemotherapy-induced diarrhea, or CID, in dogs, by the end of 2014. Canalevia is a canine-specific formulation of crofelemer, an active pharmaceutical ingredient isolated and purified from the Croton lechleri tree. A human-specific formulation of crofelemer, Fulyzaq, was approved by the U.S. Food and Drug Administration, or FDA, in 2012 for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. Members of our management team developed crofelemer, including while at Napo Pharmaceuticals, Inc., or Napo. Neonorm is our lead non-prescription product to address the symptoms of watery diarrhea, or scours. We recently launched Neonorm in the United States for preweaned dairy calves under the brand name Neonorm Calf and expect to launch additional formulations of Neonorm for other animal species beginning in 2015. Neonorm is a botanical extract also derived from the Croton lechleri tree. Canalevia and Neonorm are distinct products that are formulated to address specific species and market channels. We have filed eight investigational new animal drug applications, or INADs, with the FDA and intend to develop species-specific formulations of Neonorm in six additional target species.
Since inception, we have been primarily focused on designing protocols for studies of Canalevia to treat multiple preselected and distinct types of watery diarrhea in dogs and for Neonorm for scours in preweaned dairy calves. We have also conducted a clinical study of Neonorm for scours in preweaned dairy calves. A portion of our activities has also been focused on other efforts associated with being a newly formed company, including securing necessary intellectual property, recruiting management and key employees and initial financing activities.
In January 2014, we entered into the Napo License Agreement, pursuant to which we acquired an exclusive worldwide license to Napo's intellectual property rights and technology, including rights to its library of over 2,300 medicinal plants, for all veterinary treatment uses and indications for all species of animals. Under the Napo License Agreement, Napo also assigned to us equipment, inventory and granted us a right to cross-reference any regulatory submissions or drug-matter files for which Napo has rights and access.
In consideration for the license from Napo, we are obligated to pay a one-time non-refundable license fee of $2,000,000, less an option fee of $100,000 we paid in July 2013. This license fee payment will be deferred until the combined net sales of one or more products we commercialize exceed $2,000,000 and can be paid in our common stock at our option. For products derived from Croton lechleri, we will owe Napo a 2% royalty on annual net sales of all products that are prescription drugs (such as Canalevia and any line extensions) approved by the FDA or the equivalent regulatory agency in another country, and, 1% of net sales of non-prescription products (such as Neonorm and any line extensions) that do not require pre-marketing approval from the FDA or the equivalent regulatory
51
agency in another country. Following the closing of this offering, we will not owe Napo any royalties on sales of non-Croton lechleri products.
Financial Operations Overview
We were incorporated in June 2013 in Delaware and are considered a development stage company. Napo formed our company to develop and commercialize animal health products. Prior to our incorporation, the only activities of Napo related to animal health were limited to the retention of consultants to evaluate potential strategic alternatives. As of December 31, 2013, we were a wholly-owned subsidiary of Napo, and as of June 30, 2014, we are a majority-owned subsidiary of Napo. Upon the closing of this offering, we will no longer be majority-owned by Napo.
We have presented our financial statements on a standalone basis without predecessor or carve-out financial information because we do not have a "predecessor" within the meaning of Rule 405 of Regulation C under the Securities Act. We did not succeed to a major portion of the business or assets of Napo, nor a separately identifiable line of business of Napo. Prior to our formation, Napo's operations did not include an animal health business and we were formed for the purpose of developing and commercializing products in the animal health field. Napo's business was focused on the development of human-specific formulations of its product, as well as licensing activities related to its intellectual property. Since early 2011, Napo's business activities have been limited to activities related to the licensing of its intellectual property, which we did not acquire or succeed to, and there were no predecessor operations of the animal health business in Napo prior to our formation. For these reasons, we did not succeed to substantially all of the business of Napo nor a separately identifiable line of business of Napo.
In July 2013, we entered into an employee leasing and overhead allocation agreement with Napo, or the Service Agreement. The term of the Service Agreement was from July 1, 2013 through February 28, 2014. Pursuant to the Service Agreement, Napo provided us the services of certain Napo employees, and on March 1, 2014, these employees joined our company. In addition, we also agreed to pay Napo for a portion of its overhead costs during the term of the agreement. We agreed to pay Napo $71,811 per month (consisting of $65,811 for employee services and $6,000 for overhead costs) for the months from July 2013 through February 2014 as follows: (1) for the period from July 2013 through November 2013, in 4,000,000 shares of common stock and (2) for the period from December 2013 through February 2014, in cash.
We have not generated any material revenue to date and expect to continue to incur significant research and development and other expenses. Our net loss for the period from June 6, 2013 (inception) through December 31, 2013 and for the six months ended June 30, 2014 was $801,203 and $3,910,234, respectively. As of June 30, 2014, we had a total stockholders' deficit of $3,901,304. We expect to continue to incur losses for the foreseeable future as we expand our product development activities, seek necessary approvals for our product candidates, conduct species-specific formulation studies for our non-prescription products, establish API manufacturing capabilities and begin commercialization activities.
Operating Expenses
The majority of our operating expenses to date have been for research and development activities related to Canalevia and Neonorm and for costs associated with our formation, including legal, recruiting, travel and financing activities. During 2013, we did not incur any stock-based compensation expense. For the six months ended June 30, 2014, operating expenses include $90,952 of stock-based compensation expense.
Research and Development Expense
Research and development costs are expensed as incurred. Research and development expense consists primarily of third-party consultant fees, expenses attributable to services received from Napo
52
under the Service Agreement and expenses related to our clinical studies. Beginning January 1, 2014, research and development expense also includes personnel-related costs, including salaries and benefits, and other operational costs related to our research and development activities, including costs of studies, raw material acquisition costs, contract manufacturers and service providers, regulatory, professional and consulting fees, and travel costs.
We typically use our employee and infrastructure resources across multiple development programs. We track outsourced development costs by prescription drug product candidate and non-prescription product but do not allocate personnel or other internal costs related to development to specific programs or development compounds.
The timing and amount of our research and development expenses will depend largely upon the outcomes of current and future trials for our prescription drug product candidates as well as the related regulatory requirements, the outcomes of current and future species-specific formulation studies for our non-prescription products, manufacturing costs and any costs associated with the advancement of our line extension programs. We cannot determine with certainty the duration and completion costs of the current or future development activities.
The duration, costs and timing of trials, formulation studies and development of our prescription drug and non-prescription products will depend on a variety of factors, including:
- •
- the scope, rate of progress, and expense of our ongoing, as well as any additional, clinical trials, formulation studies
and other research and development activities;
- •
- future clinical trial and formulation study results;
- •
- potential changes in government regulations; and
- •
- the timing and receipt of any regulatory approvals.
A change in the outcome of any of these variables with respect to the development of a prescription drug product candidate or non-prescription product could mean a significant change in the costs and timing associated with our development activities.
We expect research and development expense to increase significantly as we add personnel, commence additional clinical studies and other activities to develop our prescription drug product candidates and non-prescription products. Over the next two years, we anticipate spending approximately $13 million for the research and development of Neonorm for scours in preweaned dairy calves and Canalevia for CID and general acute watery diarrhea in dogs, as well as the research and development of other prescription drug products for horses and cats and non-prescription products for multiple animal species.
General and Administrative Expense
General and administrative expense consists of personnel-related costs, including salaries and benefits, and also includes expenses attributable to services received from Napo under the Service Agreement, rent and other facilities costs and professional and consulting fees for legal, accounting, tax services and other general business services. During 2013, we did not incur any stock-based compensation expense. For the six months ended June 30, 2014, general and administrative expense includes $58,051 of stock-based compensation expense.
We expect general and administrative expense to increase significantly as we incur operating costs related to being a public company, including building our corporate infrastructure.
Interest (Expense) Income, Net
Interest (expense) income, net consists primarily of interest expense related to our convertible promissory notes issued from July to September 2013, which converted to common stock in February 2014. It also includes interest expense and the amortization of a beneficial conversion feature related to convertible promissory notes issued in June 2014.
53
Results of Operations
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
|||||
|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
|||||
Operating expenses: |
|||||||
General and administrative expense |
$ | 458,473 | $ | 1,740,515 | |||
Research and development expense |
324,479 | 2,149,555 | |||||
| | | | | | | | |
Total operating expenses |
782,952 | 3,890,070 | |||||
| | | | | | | | |
Loss from operations |
(782,952 | ) | (3,890,070 | ) | |||
Interest (expense) income, net |
(18,251 | ) | (20,164 | ) | |||
| | | | | | | | |
Net loss and comprehensive loss |
$ | (801,203 | ) | $ | (3,910,234 | ) | |
| | | | | | | | |
| | | | | | | | |
General and Administrative Expense
The following table presents the components of general and administrative expense for the periods indicated:
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
|||||
|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
|||||
Personnel and related benefits |
$ | — | $ | 655,431 | |||
Accounting fees |
— | 144,850 | |||||
Third-party consulting fees and Napo service fees |
391,493 | 254,174 | |||||
Legal fees |
4,780 | 227,092 | |||||
Other expenses |
62,200 | 400,917 | |||||
Stock-based compensation |
— | 58,051 | |||||
| | | | | | | | |
Total |
$ | 458,473 | $ | 1,740,515 | |||
| | | | | | | | |
| | | | | | | | |
General and administrative expense for 2013 primarily consists of third-party consulting fees and services provided by Napo personnel pursuant to the Service Agreement related to fundraising, corporate organization and administrative services, as well as Napo overhead allocation expense. Legal fees were related to general corporate activities. Other expenses included costs related to marketing studies, business development consultants and travel.
General and administrative expense for the six months ended June 30, 2014 primarily consists of salaries and related benefits for employees, third-party consulting fees and two months of services provided by Napo personnel pursuant to the Service Agreement, as well as Napo overhead allocation expense and legal costs related to intellectual property development and general corporate activities. In March 2014, upon the conclusion of the Service Agreement with Napo, four Napo employees joined us as our employees.
54
Research and Development Expense
The following table presents the components of research and development expense for the periods indicated:
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
|||||
|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
|||||
Personnel and related benefits |
$ | — | $ | 407,554 | |||
Third-party consulting and Napo service fees |
136,274 | 104,594 | |||||
Materials expense |
— | 1,196,425 | |||||
Studies, formulation and assay costs |
159,048 | 118,291 | |||||
Other |
29,157 | 133,924 | |||||
Supply |
— | 155,866 | |||||
Stock-based compensation |
— | 32,901 | |||||
| | | | | | | | |
Total |
$ | 324,479 | $ | 2,149,555 | |||
| | | | | | | | |
| | | | | | | | |
Research and development expense for 2013 includes expenses associated with services provided by Napo employees, raw material supply costs and manufacturing-related activities. We also retained third-party consultants in connection with our application for MUMS designation for Canalevia for CID in dogs, and the development of a protocol for a study of Neonorm in preweaned dairy calves. Study and assay costs include costs of a study of Neonorm in preweaned dairy calves conducted at a veterinary school.
Research and development expense for the six months ended June 30, 2014 primarily consists of materials to be used in studies and pre-commercial manufacturing that were transferred to our company as part of the Napo License Agreement, and expensed. Research and development expenses also include payroll and related benefits for research and development personnel, the costs of a study of Neonorm in preweaned dairy calves, services provided by Napo personnel before they became employees of our company in March 2014, consultants, and manufacturing and raw material supply costs and related activities.
Liquidity and Capital Resources
Since inception, we have not generated any material revenue and we have funded our operations primarily through the issuance of equity securities and convertible promissory notes. We have incurred increasing losses and negative cash flow from operations, and as of June 30, 2014, we had an accumulated deficit of $4,711,437. We anticipate that we will continue to incur losses for the next several years due to expenses relating to:
- •
- trials of our products and product candidates;
- •
- toxicology studies for our product candidates;
- •
- establishing manufacturing capabilities; and
- •
- commercialization of one or more of our prescription drug product candidates, if approved, and commercialization of our non-prescription products.
As of June 30, 2014, we had cash and cash equivalents of $4,281,698. In April and May 2014, we received aggregate gross proceeds of $1,777,338 from the issuance of 790,911 shares of Series A preferred stock, and in June and July 2014, we issued an aggregate of $450,000 principal amount of convertible promissory notes. In August 2014, we entered into a standby line of credit with an individual, who is an accredited investor, for up to $1,000,000.
Our auditors have included an explanatory paragraph in their audit report on our financial statements for the year ended December 31, 2013, regarding our assessment of substantial doubt about our ability to continue as a going concern. Our financial statements do not include any adjustments that
55
may result from the outcome of this uncertainty. We believe that the successful completion of this offering will eliminate the doubt and enable us to continue as a going concern. However, if we are unable to successfully complete this offering, we will need to obtain alternate financing or create operational plans to continue as a going concern.
We believe the net proceeds from this offering, together with our existing cash and cash equivalents, will be sufficient to fund our operating plan through the next 24 months and through the commercial launch of Neonorm for preweaned dairy calves and anticipated commercial launch of Canalevia for CID in dogs, as well as for general acute watery diarrhea in dogs. However, our operating plan may change due to many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations. Such financing may result in dilution to stockholders, imposition of debt covenants and repayment obligations or other restrictions that may affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
We expect that we will increase our expenditures following the closing of this offering once we have additional capital on hand in order to continue our efforts to develop animal health products, commercially launch Neonorm and continue development of Canalevia in the near term. We currently estimate that the commercial launch of Neonorm will cost approximately $1.0 million, and have agreed to pay Indena S.p.A. fees of approximately $3.7 million under memorandums of understanding relating to the establishment of our commercial manufacturing arrangement. The exact amounts and timing of any expenditures may vary significantly from our current intentions.
Cash Flows
The following table shows a summary of cash flows for the periods set forth below:
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
||||
|---|---|---|---|---|---|---|
| |
|
(unaudited) |
||||
Cash used in operating activities |
$ | (334,839 | ) | $(1,776,865 | ) | |
Cash used in investing activities |
— | (55,149 | ) | |||
Cash provided by financing activities |
520,206 | 5,928,345 | ||||
Cash Used in Operating Activities
During the period from June 6, 2013 (inception) through December 31, 2013, cash used in operating activities was the result of our net loss of $801,203 offset by the issuance of common stock to Napo for services $359,055, further offset by changes in operating assets and liabilities of $104,628.
During the six months ended June 30, 2014, cash used in operating activities was the result of our net loss of $3,910,234 and changes in operating assets and liabilities of $906,293, both of which were primarily offset by the expense of certain materials received from Napo of $1,082,626.
Cash Used in Investing Activities
During the period from June 6, 2013 to December 31, 2013, we did not have any cash provided by or used in investing activities. In the six months ended June 30, 2014, cash used in investing activities primarily consisted of purchases of manufacturing-related equipment.
Cash Provided by Financing Activities
During the period from June 6, 2013 through December 31, 2013, cash provided by financing activities primarily consisted of the gross proceeds from the issuance of convertible promissory notes and warrants to purchase common stock. On February 4, 2014, the convertible notes issued in 2013 were converted in full in exchange for an aggregate of 311,498 shares of common stock at a conversion
56
price of $1.6854, which was equal to 75% of the price per share paid by the purchasers of Series A preferred stock.
During the six months ended June 30, 2014, cash provided by financing activities consisted of net proceeds of $6,658,241 from the issuance of 3,015,902 shares of Series A preferred stock and $300,000 from the issuance of convertible promissory notes due June 1, 2015.
Description of Indebtedness
Standby Line of Credit
In August 2014, we entered into a standby line of credit with an individual, who is an accredited investor, for up to $1.0 million pursuant to a Line of Credit Loan Agreement dated August 26, 2014. The minimum amount of any drawdown is $250,000, the lender has no obligation to fund more than once every 10 calendar days, we must provide 15 business days prior notice for any drawdown and may not draw down funds after March 31, 2015. Outstanding borrowings bear interest at a rate of 3.0% per annum, and all borrowings are due in full on the one-year anniversary of our first drawdown. Following the closing of this offering, outstanding principal amounts borrowed under the standby line of credit may be converted, at the option of the lender, into shares of our common stock at a conversion price equal to 80% of the initial public offering price per share. In connection with the entry into the standby line of credit, we issued the lender a warrant to purchase 50,000 shares of our common stock at an exercise price equal to 80% of the initial public offering price per share, which expires in August 2016.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in the use of any off-balance sheet arrangements, such as structured finance entities, special purpose entities or variable interest entities.
Commitments and Contingencies
The following table summarizes our contractual obligations as of December 31, 2013:
| |
Payments Due by Period | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Less Than 1 Year |
1 to 3 Years |
3 to 5 Years |
More Than 5 Years |
|||||||||||
Convertible promissory notes(1) |
$ | 525,000 | $ | 525,000 | $ | — | $ | — | $ | — | ||||||
Total |
$ | 525,000 | $ | 525,000 | $ | — | $ | — | $ | — | ||||||
- (1)
- In February 2014, these convertible notes were converted in full in exchange for an aggregate of 311,498 shares of common stock at a conversion price of $1.6854 per share.
57
The following table summarizes our contractual obligations as of June 30, 2014:
| |
Payments Due by Period | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Total | Less Than 1 Year |
1 to 3 Years |
3 to 5 Years |
More Than 5 Years |
|||||||||||
Napo License Agreement(1) |
$ | 1,900,000 | $ | — | $ | 1,900,000 | $ | — | $ | — | ||||||
Convertible promissory notes(2) |
300,000 | 300,000 | — | — | — | |||||||||||
Sublease(3) |
138,715 | 138,715 | — | |||||||||||||
Total(4) |
$ | 2,338,715 | $ | 438,715 | $ | 1,900,000 | $ | — | $ | — | ||||||
- (1)
- The Napo License Agreement obligates us to pay (i) license fees upon achievement of a cumulative level of $2,000,000 of product sales, (ii) royalties on net sales of products utilizing licensed technology and (iii) milestone payments. Royalties are dependent on future product sales and are not reflected in the table above, as they are not estimable. Milestone payments are not payable if we complete this initial public offering prior to December 31, 2015 and are not reflected in the table above.
- (2)
- Does not include an additional $150,000 aggregate principal amount of notes issued in July 2014.
- (3)
- Represents future lease payments for our San Francisco, California headquarters.
- (4)
- Does not include approximately $3.7 million of payments potentially payable to Indena S.p.A. pursuant to our memorandums of understanding regarding establishing a manufacturing arrangement. Does not include any amounts that may become payable under our August 2014 $1.0 million standby line of credit.
The contractual commitment amounts in the table above are associated with agreements that are enforceable and legally binding. We also enter into agreements in the normal course of business with contract research organizations for clinical trials and with vendors for preclinical studies and other services and products for operating purposes, which are generally cancelable at any time by us with advance written notice. The amounts due under these agreements are not included in the above tables.
Quantitative and Qualitative Disclosures about Market Risk
Interest Rate Fluctuation Risk
Our cash and cash equivalents as of June 30, 2014 were held in a cash account. Upon completion of this offering, the proceeds from the sale of shares of our common stock will be placed in interest bearing accounts. As a result, our primary exposure to market risk for our cash will be interest income sensitivity, which is affected by changes in the general level of U.S. interest rates. However, because our cash is held in bank accounts, a sudden change in the interest rates associated with our cash and cash equivalents balances would not be expected to have a material impact on our financial condition or results of operations.
We do not have any foreign currency or derivative financial instruments.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles, or U.S. GAAP, requires the use of estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and related disclosures in the financial statements. Critical accounting policies are those accounting policies that may be material due to the levels of subjectivity and judgment necessary to account for highly uncertain matters or the susceptibility of such matters to change, and that have a material impact on financial condition or operating performance. While we base our estimates and judgments on our experience and on various other factors that we believe to be reasonable under the circumstances, actual results may differ from these estimates under different assumptions or conditions. We believe the following critical accounting policies used in the preparation of our financial statements require significant judgments and estimates.
58
For additional information relating to these and other accounting policies, see Note 2 to our audited financial statements, appearing elsewhere in this prospectus.
Accrued Research and Development Expenses
As part of the process of preparing our financial statements, we are required to estimate accrued research and development expenses. Estimated accrued expenses include fees paid to vendors and clinical sites in connection with our clinical trials and studies. We review new and open contracts and communicate with applicable internal and vendor personnel to identify services that have been performed on our behalf and estimate the level of service performed and the associated costs incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost for accrued expenses. The majority of our service providers invoice us monthly in arrears for services performed or as milestones are achieved in relation to our contract manufacturers. We make estimates of our accrued expenses as of each reporting date.
We base our accrued expenses related to clinical trials and studies on our estimates of the services received and efforts expended pursuant to contracts with vendors, our internal resources, and payments to clinical sites based on enrollment projections. The financial terms of the vendor agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of animals and the completion of development milestones. We estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the related expense accrual accordingly on a prospective basis. If we do not identify costs that have been incurred or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates. To date, we have not made any material adjustments to our estimates of accrued research and development expenses or the level of services performed in any reporting period presented.
Accounting for Stock-Based Compensation
During 2013, we did not issue any stock awards to employees, directors or consultants and did not incur any stock based compensation expense. Beginning in the second quarter of 2014, we awarded options and restricted stock units. We measure stock-based awards granted to employees and directors at fair value on the date of grant and recognize the corresponding compensation expense of the awards, net of estimated forfeitures, over the requisite service periods, which correspond to the vesting periods of the awards.
Key Assumptions. Our Black-Scholes-Merton option-pricing model requires the input of highly subjective assumptions, including the fair value of the underlying common stock, the expected volatility of the price of our common stock, the expected term of the option, risk-free interest rates and the expected dividend yield of our common stock. These estimates involve inherent uncertainties and the application of management's judgment. If factors change and different assumptions are used, our stock-based compensation expense could be materially different in the future. These assumptions are estimated as follows:
- •
- Fair value of our common stock—Because the common stock is not yet publicly traded, we estimated the fair
value of our common stock, as discussed in "Common Stock Valuations" below. Upon the closing of this public offering, our common stock will be valued by reference to the publicly-traded price of our
common stock.
- •
- Expected volatility—As we do not have any trading history for our common stock, the expected stock price volatility for our common stock was estimated by taking the average historic price volatility for industry peers based on daily price observations for common stock values over a period equivalent to the expected term of our stock option grants. We did not rely on implied volatilities of traded options in our industry peers' common stock because the
59
- •
- Expected term—The expected term represents the period that our stock-based awards are expected to be
outstanding. It is based on the "simplified method" for developing the estimate of the expected life of a "plain vanilla" stock option. Under this approach, the expected term is presumed to be the
midpoint between the average vesting date and the end of the contractual term for each vesting tranche. We intend to continue to apply this process until a sufficient amount of historical exercise
activity is available to be able to reliably estimate the expected term.
- •
- Risk-free interest rate—The risk-free interest rate is based on the yields of U.S. Treasury securities with
maturities similar to the expected term of the options for each option group.
- •
- Dividend yield—We have never declared or paid any cash dividends and do not presently plan to pay cash dividends in the foreseeable future. Consequently, we used an expected dividend yield of zero.
volume of activity was relatively low. We intend to continue to consistently apply this process using the same or similar public companies until a sufficient amount of historical information regarding the volatility of our own common stock share price becomes available.
Common Stock Valuations. The fair value of the common stock underlying our stock options was determined by our board of directors, which intended all options granted to be exercisable at a price per share not less than the per share fair value of our common stock underlying those options on the date of grant. The valuations of our common stock were determined in accordance with the guidelines outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. The assumptions we used in the valuation model are highly complex and subjective. We base our assumptions on future expectations combined with management judgment. In the absence of a public trading market, our board of directors, with input from management, exercised significant judgment and considered numerous objective and subjective factors to determine the fair value of our common stock as of the date of each option grant and stock award. These judgments and factors will not be necessary to determine the fair value of new awards once the underlying shares begin trading. For now we included the following factors:
- •
- the prices, rights, preferences and privileges of our Series A preferred stock relative to those of our common
stock;
- •
- lack of marketability of our common stock;
- •
- our actual operating and financial performance;
- •
- current business conditions and projections;
- •
- hiring of key personnel and the experience of our management;
- •
- our stage of development;
- •
- illiquidity of share-based awards involving securities in a private company;
- •
- the U.S. capital market conditions; and
- •
- likelihood of achieving a liquidity event, such as this offering or a merger or acquisition of our company given prevailing market conditions.
Following the closing of this initial public offering, the fair value per share of our common stock for purposes of determining stock-based compensation will be the closing price of our common stock as reported on The NASDAQ Stock Market on the applicable grant date.
Income Taxes
As of December 31, 2013, we had net operating loss carryforwards for federal and state income tax purposes of $788,486, which will begin to expire in 2033, subject to limitations. Our management has evaluated the factors bearing upon the realizability of our deferred tax assets, which are comprised
60
principally of net operating loss carryforwards. Our management concluded that, due to the uncertainty of realizing any tax benefits as of December 31, 2013, a valuation allowance was necessary to fully offset our deferred tax assets. We have evaluated our uncertain tax positions and determined that we have no liabilities from unrecognized tax benefits and therefore we have not incurred any penalties or interest.
Recently Issued Accounting Pronouncements
In June 2014, the Financial Accounting Standards Board, or FASB, issued authoritative guidance that eliminates the distinction of a development stage entity and certain related disclosure requirements, including the elimination of inception-to-date information on the statements of operations, cash flows and stockholders' equity. The amendments will be effective prospectively for annual reporting periods beginning after December 15, 2014, and interim periods within those annual periods, however early adoption is permitted. We did not implement early adoption of this standard. The adoption of this guidance will have no impact on our financial condition, results of operations or cash flows.
In June 2014, the FASB issued authoritative guidance that requires that a performance target that affects vesting and that could be achieved after the requisite service period be treated as a performance condition. The performance target should not be reflected in estimating the grant-date fair value of the award. Compensation cost should be recognized in the period in which it becomes probable that the performance target will be achieved and should represent the compensation cost attributable to the period(s) for which the requisite service has already been rendered. If the performance target becomes probable of being achieved before the end of the requisite service period, the remaining unrecognized compensation cost should be recognized prospectively over the remaining requisite service period. The total amount of compensation cost recognized during and after the requisite service period should reflect the number of awards that are expected to vest and should be adjusted to reflect those awards that ultimately vest. The requisite service period ends when the employee can cease rendering service and still be eligible to vest in the award if the performance target is achieved. This guidance will be effective for annual periods (and interim periods within those annual periods) beginning after December 15, 2015. We will implement this guidance for all interim and annual periods beginning after December 15, 2015. The adoption of this guidance is not expected to have an impact on our financial condition, results of operations or cash flows.
In May 2014, the FASB issued Accounting Standards Update, or ASU, No. 2014-09, "Revenue from Contracts with Customers." The objective of ASU 2014-19 is to establish a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and will supersede most of the existing revenue recognition guidance, including industry-specific guidance. The core principle of the new standard is that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard is effective for annual reporting periods beginning after December 15, 2016 and allows for prospective or retrospective application. We are evaluating this pronouncement and do not believe it will have a material effect on our financial statements.
JOBS Act
In April 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period, and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.
61
Overview
We are an animal health company focused on developing and commercializing first-in-class gastrointestinal products for companion and production animals. Canalevia is our lead prescription drug product candidate for the treatment of various forms of watery diarrhea in dogs. We expect to announce data from our proof-of-concept study of Canalevia for general acute watery diarrhea in dogs in the fourth quarter of 2014. We also expect to initiate filing of a rolling new animal drug application, or NADA, for Canalevia for chemotherapy-induced diarrhea, or CID, in dogs, by the end of 2014. Canalevia is a canine-specific formulation of crofelemer, an active pharmaceutical ingredient isolated and purified from the Croton lechleri tree. A human-specific formulation of crofelemer, Fulyzaq, was approved by the U.S. Food and Drug Administration, or FDA, in 2012 for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. Members of our management team developed crofelemer, including while at Napo Pharmaceuticals, Inc., or Napo. Neonorm is our lead non-prescription product to address the symptoms of watery diarrhea, or scours. We recently launched Neonorm in the United States for preweaned dairy calves under the brand name Neonorm Calf and expect to launch additional formulations of Neonorm for other animal species beginning in 2015. Neonorm is a botanical extract also derived from the Croton lechleri tree. Canalevia and Neonorm are distinct products that are formulated to address specific species and market channels. We have filed eight investigational new animal drug applications, or INADs, with the FDA and intend to develop species-specific formulations of Neonorm in six additional target species.
Diarrhea is one of the most common reasons for veterinary office visits for dogs and is the second most common reason for visits to the veterinary emergency room, yet there are no FDA-approved anti-secretory products for the treatment of diarrhea. We estimate that in the United States, veterinarians see approximately six million annual cases of acute and chronic watery diarrhea in dogs, approximately two-thirds of which are acute watery diarrhea. We believe Canalevia will be effective in treating watery diarrhea because it acts at the last physiological step, conserved across mammalian species, in the manifestation of watery diarrhea, regardless of cause, by normalizing ion and water flow in the intestinal lumen. We are first seeking a minor use, minor species, or MUMS, designation for Canalevia for CID in dogs to shorten the timeframe to commercialization. If we receive conditional approval pursuant to MUMS designation, we expect to commercialize Canalevia for CID in dogs in early 2016. We are also enrolling a placebo-controlled proof-of-concept study of approximately 240 treated dogs with multiple preselected and distinct types of watery diarrhea. We are conducting this study to support full approval of Canalevia for CID, as well as protocol concurrence discussions with the FDA regarding expansion of labeled indications of watery diarrhea beyond CID, to include general acute watery diarrhea. We plan to market Canalevia, if approved, through our focused direct sales force and to complement our internal efforts with distribution partners.
According to the Dairy 2007 study conducted by the United States Department of Agriculture, or USDA, almost one in four preweaned dairy heifer, or female, calves suffers from diarrhea or other digestive problems. The preweaning period is generally the first 60 days after birth. Scours, diarrhea or other digestive problems are responsible for more than half of all preweaned heifer calf deaths, and result in impaired weight gain and long-term reduction in milk production. We believe the incidence rate of scours and its corresponding financial impact represent a large opportunity and that Neonorm has the potential to effectively meet this need. In our clinical study completed in May 2014, Neonorm demonstrated a statistically significant reduction in the severity of watery diarrhea, reduced morbidity and mortality, and improved weight gain as compared to placebo in newborn dairy calves with scours.
We recently launched Neonorm for preweaned dairy calves in the United States. Our commercialization activities are initially focusing on large commercial dairy operations and will include active ongoing education and outreach to dairy industry key opinion leaders, such as academics
62
involved in dairy cattle research or who advise the dairy cattle industry, as well as veterinarians. We intend to augment these commercialization efforts by working with regional distributors to leverage the geographic concentration of the dairy market in the United States as well as national distributors to provide wider geographic access to our products. In August 2014, we entered into our first regional distribution agreement for the Upper Midwest region and, together with this partner, launched Neonorm at the 2014 World Dairy Expo, and in September 2014, entered into an agreement with a national master distributor, who also distributes prescription products for the companion animal market. We estimate that the commercial launch will cost approximately $1.0 million. We expect the ongoing launch of Neonorm to drive awareness among veterinarians regarding the utility of our first-in-class anti-secretory Croton lechleri-derived products, including Canalevia.
We have an exclusive worldwide license to Napo's intellectual property rights and technology related to our products and product candidates, including rights to its library of over 2,300 medicinal plants, for all veterinary treatment uses and indications for all species of animals. This includes rights to Canalevia, Neonorm and other distinct prescription drug product candidates in our pipeline along with the corresponding existing pre-clinical and clinical data packages.
Our management team has significant experience in gastrointestinal and animal health product development. This experience includes the development of crofelemer for human use, from discovery and preclinical and clinical toxicity studies, including the existing animal studies to be used for Canalevia regulatory approvals, through human clinical development. Our team also includes individuals who have prior animal health experience at major pharmaceutical companies including Ciba-Geigy Corp., now Novartis International AG, SmithKline Beecham Corporation, now GlaxoSmithKline LLC, the animal health group of Pfizer Inc., now Zoetis Inc., and Vétoquinol S.A.
Product Pipeline
We are developing a pipeline of prescription drug product candidates and non-prescription products to address unmet needs in animal health. Our pipeline currently includes prescription drug
63
product candidates for eight indications across multiple species, and non-prescription products targeting seven species.
Prescription Drug Product Candidates
| Product Candidates |
Species |
Indication |
Recent Developments(1) |
Anticipated Near-Term Milestones |
||||
|---|---|---|---|---|---|---|---|---|
| Canalevia |
Dogs | CID | • INAD filed in November 2013(2) • Scheduled MUMS designation / pre-NADA meeting |
• Initiate rolling NADA filing with the FDA in fourth quarter of 2014 | ||||
| Dogs | General acute watery diarrhea |
• INAD filed in February 2014 • Initiated proof-of-concept study in June 2014 |
• Proof-of-concept data in fourth quarter of 2014 • Top line pivotal efficacy data in 2015 |
|||||
| Horses | Acute colitis | • INAD filed in February 2014 • Initiated hamster C. difficile study in April 2014 |
• Safety data in fourth quarter of 2014 • Proof-of-concept data in second half of 2015 • Apply for MUMS designation in second half of 2015 |
|||||
| Species-specific formulations of crofelemer |
Horses | Colonic and gastric ulcers(3) | • Proof-of-concept data in second half of 2015 | |||||
| |
Cats | General acute watery diarrhea |
• INAD filed in February 2014 | • Safety data in first half of 2015 • Top line pivotal efficacy data in 2015 |
||||
| Virend (topical) | Cats |
Herpes virus | • INAD filed in July 2014 | • Proof-of-concept data in first half of 2015 • Top line pivotal efficacy data in 2015 |
||||
| Dogs |
Obesity-related metabolic dysfunction | • INAD filed September 2014 | ||||||
| Species-specific formulations of NP-500 |
Horses |
Metabolic syndrome | • INAD filed in March 2014 | |||||
| Cats |
Type II diabetes | • INAD filed in March 2014 | ||||||
- (1)
- Each INAD was filed by us unless otherwise noted.
- (2)
- Initially filed by Napo; transferred to us in March 2014
- (3)
- In combination with omeprazole.
| Products |
Species |
Use |
Recent Developments |
Anticipated Near-Term Milestones |
||||
|---|---|---|---|---|---|---|---|---|
| Neonorm Calf | Dairy calves |
For scours in preweaned dairy calves |
• Commercial launch in September 2014 | • Field study data by end of 2014 | ||||
| Horse foals | Normalize stool formation | • Completed pilot formulation in April 2014 | • Safety and palatability data in 2014 • Efficacy data in first half of 2015 • Commercial launch in 2015 |
|||||
| Species-specific formulations of Neonorm |
Adult horses | Normalize stool formation | • Completed pilot formulation in April 2014 | • Safety and efficacy data in first half of 2015 • Commercial launch in 2015 |
||||
| Sheep and other farm animals |
Normalize stool formation |
• Initiated international market research in New Zealand in May 2014 | • Initiate proof-of-concept studies in various species based on market research | |||||
Canalevia is our lead prescription drug product candidate for CID and general acute watery diarrhea in dogs. Neonorm is our lead non-prescription product for preweaned dairy calves with scours.
64
Both Canalevia and Neonorm are derived from the Croton lechleri tree and act at the same last step in a physiological pathway generally present in mammals. However, they are distinct products based on species-specific formulations of such derivatives and have distinct chemical compositions as well as different levels of purification. Canalevia is a canine-specific formulation of crofelemer, an active pharmaceutical ingredient that is an isolated and purified compound. Neonorm is a formulation of a botanical extract that is less refined than crofelemer and includes many chemical constituents.
We are developing Canalevia as a prescription drug product and Neonorm as a non-prescription product due to differences between the companion and production animal markets. Companion animal owners generally visit veterinarians, who prescribe a product to treat a disease or condition. We believe the ability to make a disease treatment claim is important in this market, and such a claim is only possible with FDA approval as a prescription product. In contrast, dairy farm and other production animal owners generally make purchasing decisions based on a product's ability to demonstrate an economic benefit from health endpoints, such as weight gain. We believe that data from our clinical study of Neonorm demonstrates such an economic benefit and do not believe the ability to make disease treatment claims will be needed to commercialize the product.
We are initially pursuing conditional FDA approval for Canalevia for CID in dogs pursuant to MUMS designation, and are conducting studies to broaden the Canalevia label to include general acute watery diarrhea in dogs. A MUMS designation is a status similar to the orphan drug designation in humans. In the case of major animal species such as dogs, cats and horses, MUMS designations are typically limited to drugs that are used to treat a small number of animals each year. For dogs and cats that number is no more than 70,000 and 120,000 animals, respectively. MUMS designation can potentially expedite the process of drug review and approval. In addition, a sponsor of a MUMS drug can apply for conditional approval, which allows the sponsor to make the drug commercially available before collecting all necessary effectiveness data, but after proving the drug is safe and showing that there is a reasonable expectation of effectiveness.
We also plan to expand our gastrointestinal product line to other animals by developing species-specific formulations and expect to seek protocol concurrences with the FDA where appropriate. For example, we have planned trials to develop formulations of crofelemer for watery diarrhea in cats and acute severe colitis in adult horses. We also plan to develop specific formulations of Neonorm for foals and adult horses, as well as sheep and other farm animals. A protocol concurrence in animal drug development means that the FDA agrees that the design and analyses proposed in a protocol are acceptable to support regulatory approval of the product candidate with respect to effectiveness of the indication studied and will not change its view of these matters, unless public or animal health concerns arise that were not recognized at the time of concurrence or we change the protocol.
We have also licensed intellectual property from Napo to develop prescription drug product candidates for diabetes and metabolic syndrome for dogs, cats and horses, as well as a topical herpes product for cats. Similar to our lead prescription drug product candidate, these products were tested in animals for safety to support their development for use in humans. We are leveraging the data and knowledge gained during the development of human therapeutics into veterinary applications.
Business Strategy
Our goal is to become a leading animal health company with first-in-class products that address unmet medical needs in both the companion and production animal markets. To accomplish this goal, we plan to:
65
Leverage our significant gastrointestinal knowledge, experience and intellectual property portfolio to develop a line of products addressing watery diarrhea for both companion and production animals.
Our management team collectively has over 100 years of experience in the development of gastrointestinal prescription drug and non-prescription products. This experience covers all aspects of product development, including discovery, pre-clinical and clinical development and regulatory strategy. In addition to our near-term development efforts advancing Canalevia for dogs and Neonorm for preweaned dairy calves, we are developing formulations of these products to address the unmet medical need for the treatment of watery diarrhea across multiple animal species and market channels. Our products are designed with a thorough understanding of not only species-specific health issues, but also market practices, the economics of current treatment strategies, competitive dynamics, government initiatives, such as concern for extensive antibiotic usage, and effective channels for new product introductions. Many of our products are being formulated into separate and distinct gastrointestinal products accounting for multiple specific species, markets and regulatory dynamics.
Establish commercial capabilities, including third-party sales and distribution networks and our own targeted commercial efforts, through the launch of Neonorm.
We recently launched Neonorm in the United States under the brand name Neonorm Calf. We intend to establish a focused direct sales force, initially for the production animal markets and have already hired our first sales representative. We will direct our sales and marketing efforts on educational activities and outreach to key opinion leaders and decision makers at targeted regional and global accounts and also plan to partner with leading distributors to commercialize our products. We expect that our current and future distribution partners will have the presence, name recognition, reputation and reach in the veterinary markets and in both key urban and rural centers, as appropriate. We believe this overall approach is scalable and transferable as we expand our commercialization efforts to companion animals, as well as when we expand internationally.
Launch Canalevia and our other product candidates for companion animals, if approved, leveraging the commercial capabilities and brand awareness we are currently building.
We expect to launch Canalevia in 2016 for both CID and general acute watery diarrhea in dogs, leveraging the sales and marketing capabilities established from our launch of Neonorm. As our focus shifts to companion animals, our direct sales force will also increasingly target high prescribing veterinarians for companion animals. We believe the third-party sales and distribution networks we establish in connection with our launch of Neonorm will be highly relevant for the companion animal market as well. In addition, while we believe Neonorm addresses a smaller market opportunity than our companion animal product candidates, it is a first-in-class product with the same novel mechanism of action as Canalevia. As such, Neonorm provides a scientific and promotional foundation that we believe we can leverage for our companion animal drug development and launch events.
Identify market needs that can be readily accessed and develop species-specific products by leveraging our broad intellectual property portfolio, deep pipeline and extensive botanical library.
In addition to our gastrointestinal product development efforts, we are developing products such as Virend for feline herpes and NP-500 for Type II diabetes and metabolic syndrome. Both of these product candidates have been through Phase 2 human clinical testing. In addition, we have exclusive worldwide rights to Napo's library of over 2,300 medicinal plants for veterinary use in all species. We believe we have the product candidates and expertise to address many unmet animal health needs for both companion and production animals. We believe our extensive library of medicinal plants will enable us to develop first-in-class products that address significant health issues and concerns of many markets and geographies.
66
Products in Development
Market Background—Watery Diarrhea
We believe there is an unmet medical need for the treatment of watery diarrhea. The devastating dehydration that often occurs as a result of watery diarrhea in animals, including dogs, horses and preweaned dairy calves, can manifest quickly, have long-term health implications and result in death. Other than the FDA-approved human formulation of crofelemer, there are no approved anti-secretory agents that directly address the water loss associated with watery diarrhea. Current treatments for watery diarrhea include oral rehydration solution, or ORS, anti-motility agents, absorbents and antibiotics. However, each of these approaches has known limitations. While ORS replaces the water loss associated with diarrhea, it can often extend the duration and severity of diarrhea. Anti-motility agents work by the mechanism of constipation, or temporarily paralyzing normal intestinal contractions, or peristaltic activity. These agents are contraindicated for chronic use and are therefore inappropriate for certain conditions, such as chronic CID. Anti-motility agents can also cause pain, cramping, and rebound diarrhea. Absorbents simply attempt to absorb the toxin in the gut, often causing additional pain and cramping, and do not directly address the water loss. Antibiotics attempt to treat the infectious agent releasing the toxin, but do not directly address water loss and carry a risk of altering gut flora, which alteration itself can cause diarrhea. Antibiotics usage has also come under increased scrutiny by the FDA due to problems associated with antibiotic resistance.
We believe that an ideal treatment for watery diarrhea would directly address water loss without causing constipation, affecting normal peristaltic activity or altering normal body absorption of other drugs or normal physiological function of the gut. We believe addressing water loss associated with watery diarrhea will improve the quality of life of dogs and provide attendant benefits to the dog owner, improve the health and productivity of dairy cattle and provide similar health and economic benefits in multiple other species. Our gastrointestinal products and product candidates act by normalizing the flow of ions and water in the intestinal lumen, the dysregulation of which is the last step common to the manifestation of watery diarrhea. As a result, we believe that our products and product candidates may be effective in addressing watery diarrhea, regardless of cause. In addition, the channels that regulate this ion and water flow, including channels known as CFTR and CaCC (the sites of action of our gastrointestinal products), are generally present in mammals. We therefore expect that the clinical benefit shown in humans and preweaned dairy calves will be confirmed in multiple other species, including dogs. Accordingly, we believe we can bring to market multiple products among multiple species that are first-in-class and effective in preventing the debilitating and devastating ramifications of watery diarrhea in animals.
The following diagram illustrates the mechanism of action of our gastrointestinal products, which normalize chloride and water flow and transit time of fluids within the intestinal lumen.
67
Canalevia—Chemotherapy-Induced Diarrhea in Dogs
Overview
Canalevia is an oral, twice daily, chewable, beef-flavored formulation of crofelemer that we are developing for the treatment of CID in dogs. Canalevia is enteric coated for targeted release of crofelemer, the active pharmaceutical ingredient, or API, in Canalevia, in the intestine. We are seeking MUMS designation for Canalevia for CID in dogs to shorten the timeframe to commercialization. We expect to initiate filing of a rolling NADA by the end of 2014. If we receive conditional approval from the FDA for this indication, we expect to launch Canalevia for CID in dogs by early 2016. Under MUMS designation, we would be required to initiate a pivotal study in the five years following conditional approval to generate the data required for full approval. We expect to meet this requirement with data generated from our ongoing placebo-controlled proof-of-concept study of approximately 240 dogs that we commenced in May 2014. We are conducting this study to support full approval of Canalevia for CID, as well as labeled indications beyond CID, such as general acute watery diarrhea in dogs.
Market Opportunity
We believe there is a significant unmet medical need for the treatment of CID in dogs. There is currently no FDA-approved anti-secretory product to treat CID in dogs. We estimate that there are over 230,000 dogs receiving chemotherapy treatment for cancer each year in the United States, with over 25% suffering from CID. Severe diarrhea is a frequent side effect of the most commonly administered chemotherapy drugs. Similar to the effects in humans, we believe that if left untreated, CID in dogs can result in:
- •
- fluid and electrolyte losses, which can cause dehydration, electrolyte imbalance and renal insufficiency;
- •
- nutritional deficiencies from alteration of gastrointestinal transit and digestion; and
- •
- increased risk of infectious complication.
Efficacy of the underlying cancer treatment may also be jeopardized if CID severity requires reductions in the absorption, frequency and/or dosage of chemotherapy. From the dog owner's perspective, there are significant practical implications of CID in dogs that may affect living arrangements, as well as the cost, time and attention required to clean and care for the dog and its surroundings on a daily basis. Veterinarians sometimes prescribe human drugs in an effort to treat CID in dogs, but do not have the benefit of clinical support with respect to efficacy or dosing. In addition, administering a potentially unpalatable human formulation is often difficult and may lead to further uncertainty of the amount actually ingested by the dog.
Our Solution
We believe that Canalevia is an ideal treatment for CID in dogs because of its demonstrated novel anti-secretory mechanism of action. Canalevia acts locally in the gut and is minimally absorbed systemically. It does not alter gastrointestinal motility, has no significant effects on normally functioning intestinal ion channels and electrolyte or fluid transport, and has no side effects different from placebo. These features are further augmented by its lack of effects on the absorption and/or metabolism of co-administered chemotherapy drugs, orally or by other routes of administration. Canalevia acts by normalizing the flow of excess ions and water in the intestinal lumen. The flow of excess ions and water into the intestinal lumen is the last step common to the manifestation of watery diarrhea. As a result, we believe Canalevia may be effective in the treatment of watery diarrhea, regardless of cause, including CID.
68
Human formulations of crofelemer have been studied and found effective in human patients with various types of watery diarrhea, including traveler's diarrhea, HIV-related diarrhea and other acute infectious diarrheas, including cholera. Crofelemer has been clinically demonstrated to have a safety profile not different from placebo in humans and several animal species, including dogs.
Clinical Data
Canalevia is a canine-specific formulation of crofelemer. A human-specific formulation of crofelemer, Fulyzaq, was approved by the FDA in 2012 for the symptomatic relief of noninfectious diarrhea in adults with HIV/AIDS on antiretroviral therapy. A number of clinical studies of crofelemer were conducted by Napo in dogs in support of this approval that included dose toxicity studies. Safety was established by conducting a series of toxicity studies involving a total of 32 dogs. Dosage levels varied within and across the studies: two single dose acute toxicity studies were conducted on four dogs each; two seven-day repeat administration studies were conducted on four dogs each; one 30-day repeat administration study was conducted on four dogs; and one nine-month repeat administration study on eight dogs. The toxicology studies in dogs showed minimal to no adverse effects following dosing up to approximately 50 times the anticipated efficacious dose. The clinical studies previously conducted in dogs also included multiple dose studies. We believe these studies will meet FDA requirements for a pivotal safety package and will support our anticipated dosing of Canalevia in dogs.
In multiple third-party human clinical trials involving approximately 2,400 patients, enteric-coated crofelemer showed statistically significant results relative to placebo in normalizing stool formation and improvements in other endpoints related to treating watery diarrhea. In these trials, the "p" values were statistical calculations to determine whether the effects of crofelemer were significant in comparison to placebo based on pre-specified statistical targets. Depending on the trial design, we specified that any result less than p=0.05 would be significant. In a pivotal trial in support of approval for human use, crofelemer demonstrated significant benefit in the chronic indication of diarrhea in adults with HIV/AIDS on anti-retroviral therapy, achieving highly significant results (p=0.0096) in the primary endpoint measuring frequency of diarrhea.
In addition to the pivotal trial in HIV/AIDS associated diarrhea, human clinical trials included double-blind, placebo-controlled chronic and acute studies, across different human patient populations, and included safety studies in pediatric patients as young as three months of age. For example, in a 3-day treatment study of approximately 100 adult human patients with acute watery diarrhea of multiple and/or unknown etiologies, crofelemer achieved clinical success in 79% of the patients, compared to 28% receiving placebo (p<0.05). Clinical success was defined as the complete cessation of diarrhea for 12 hours or two consecutive normal stools within 48 hours of first dose. Crofelemer also achieved statistical significance across each of the seven other endpoints measured in that study, including a 96% reduction in watery stools from baseline, compared to 54% for placebo (p<0.05) and an 89% reduction in urgency compared to 43% for placebo (p<0.05). Across the diseases and human patient populations studied to date with crofelemer, there have been no drug related serious adverse events or safety profile different from placebo.
Next Steps and Commercialization Plans
We are seeking MUMS designation for Canalevia for the treatment of CID in dogs. MUMS designation provides an opportunity to shorten the time to commercialization. We are relying on previously conducted toxicology studies in dogs that were required for FDA approval of the human formulation of crofelemer to provide required safety data. We have established a safety database that we believe meets the qualifications for MUMS designation and initiation of a rolling NADA filing for Canalevia for CID in dogs. Our initial meeting with the FDA to commence the rolling NADA process is scheduled for late October 2014. At this meeting, we expect to reach agreement on the timing for submissions of the technical sections of the NADA filing. We anticipate initiating the formal submission
69
of the rolling NADA soon thereafter. If we receive conditional approval with MUMS designation, we could begin sales of Canalevia for this indication by early 2016. With conditional approval under MUMS designation, we would be required to initiate a pivotal study in the five years following such conditional approval to generate the data required for full FDA approval. We expect to meet this requirement with data generated from our ongoing placebo-controlled proof-of-concept study of approximately 240 dogs with multiple preselected and distinct types of watery diarrhea that we commenced in May 2014. We are conducting this study in connection with our planned expansion of Canalevia's labeled indication to general acute watery diarrhea in dogs.
We plan to market Canalevia, if conditionally approved by the FDA, through a focused direct sales force and to complement our internal efforts with a distribution partner.
Canalevia—Expansion to General Acute Watery Diarrhea in Dogs
Overview
We are also developing Canalevia for general acute watery diarrhea in dogs, regardless of cause. According to the American Veterinary Medical Association, there were approximately 70 million dogs in the United States in 2012. In May 2014, we commenced a placebo-controlled proof-of-concept study of approximately 240 dogs with multiple preselected and distinct types of watery diarrhea. We anticipate having data from this study in support of this general acute watery diarrhea indication in the fourth quarter of 2014. Crofelemer, the API in Canalevia, demonstrated efficacy in numerous human clinical trials of acute watery diarrhea induced by various infectious pathogens, including E. coli, V. cholera and non-specific pathogens (e.g., Traveler's). Following oral dosing for two or three days, crofelemer, together with ORS, produced significant reduction in watery diarrhea, as demonstrated by the reduction of watery stool passage as well as reduced duration of diarrhea, urgency and dehydration.
Market Opportunity
Diarrhea is one of the most common reasons for veterinary office visits for dogs and the second most common reason for visits to the veterinary emergency room, yet there are no FDA-approved anti-secretory agents to treat the indication. We estimate that veterinarians see approximately six million annual cases of acute and chronic watery diarrhea in dogs in the United States, approximately two-thirds of which are acute watery diarrhea.
Veterinarians typically treat watery diarrhea in dogs with antibiotics, probiotics, dietary restrictions and products approved and formulated for humans, such as Imodium and other anti-motility agents, as well as binding agents that absorb water such as Kaopectate and Pepto-Bismol. None of these treatment options address the water loss associated with watery diarrhea. Further, because none of the human products are FDA approved for animal use, veterinarians do not have the benefit of clinical support with respect to efficacy or dosing. Moreover, administering a potentially unpalatable human formulation is often difficult and may lead to further uncertainty of the amount actually ingested by the dog.
We believe that Canalevia is an ideal treatment for general acute watery diarrhea in dogs because of its demonstrated novel anti-secretory mechanism of action. If approved for use in general acute watery diarrhea in dogs, Canalevia will be the only FDA-approved anti-secretory agent to treat diarrhea in dogs.
Next Steps
In May 2014, we initiated a multicenter placebo-controlled proof-of-concept study of approximately 240 dogs with acute watery diarrhea. We are conducting this study in veterinary hospitals to provide a controlled environment.
70
The study is a double-blind, block-randomized format that will compare five distinct treatment groups based on the cause of diarrhea with each of five related placebo groups, as well as a global analysis. The study design, enrollment criteria, endpoints, and powering assumptions have been developed in conjunction with the team that conducted over a dozen clinical trials with crofelemer in support of the FDA approval of the human formulation, and our veterinary experts.
The study is enrolling dogs presenting with general acute watery diarrhea for less than three days. A thorough clinical examination of each dog is conducted to determine if the cause of diarrhea is chemotherapy, bacterial infection, pancreatitis, dietary indiscretion or Giardia infection. Each of these five causes is considered a subgroup and dogs with any other cause of diarrhea are being excluded from the study. Enrolled dogs are hospitalized at a clinic for four days and treated according to their weight classification with 2-4 mg/kg of Canalevia (enteric-coated micro-granules; dosage range due to the size and weight variability of dog species) or an enteric-coated placebo twice a day for three days. In addition, all enrolled dogs are treated according to the "standard of care" for diarrhea, including oral or intravenous fluids for rehydration and disease-specific medications such as fenbendazole for Giardia infection, pain control for pancreatitis and an anti-emetic for vomiting (Maropitant citrate).
The endpoints being evaluated are feces consistency, a comprehensive gastrointestinal score, duration of diarrhea, frequency of defecation, appetite, attitude and body temperature. Dogs are examined twice a day for four days and assessed using two validated scoring systems: the Nestle Purina Fecal and the Waltham Fecal Scoring systems. Feces samples are taken once a day to establish or confirm the cause of diarrhea and to measure the dry matter content of the stool. Blood chemistry analyses are also being studied.
The protocol for this study is based on our experience and success in previous human and dairy calf studies evaluating Croton lechleri derivatives and their effect on watery diarrhea. The design provides for the evaluation of data by subgroup, and as a whole, across a range of relevant clinical endpoints. As a result, we believe the study will enable us to confirm the clinical benefits of Canalevia for all or some of these subgroups of general acute watery diarrhea and allow us to move forward with a pivotal clinical trial. We expect to seek protocol concurrence from the FDA and anticipate commercial launch for Canalevia for this indication in 2016.
We expect to announce top line results from this proof-of-concept study by the fourth quarter of 2014.
Crofelemer—Equine Line Extension
We intend to develop a species-specific formulation of crofelemer to treat acute colitis in horses. We believe colitis affects thousands of horses in the United States each year. Acute colitis can cause sudden, massive fluid loss and severe electrolyte imbalances that can result in death in a matter of hours. Acute colitis often occurs when salmonella and C. difficile, bacteria that are normally present in the gut, are activated by stress, or when the bacteria N. risticii is ingested, causing Potomac horse fever. A 2009 Compendium Equine article reported fatality rates of 32% to 60% for salmonellosis and 15% to 35% for Potomac horse fever. Stress (e.g., shipping, changes in daily routines, illness, hospitalization, racing), recent diet changes, recent antimicrobial administration and non-steroidal anti-inflammatory drug therapy can also put horses at risk for acute colitis. The current standard of care includes hospitalization, intubation and intravenous fluids, with little opportunity to culture stools to determine the exact source of the disease. We believe treatment of acute colitis in high-value race and performance horses with crofelemer represents a premium niche market opportunity.
We are currently evaluating crofelemer in hamsters, a validated gastrointestinal model for adult horses, for treatment of acute colitis resulting from C. difficile. If the results are positive, we intend to seek MUMS designation for our product for treatment of acute colitis in adult horses, which may shorten the timeframe to commercialization. If approved, we believe we could launch an equine formulation of crofelemer in early 2016.
71
We are also developing a formulation of a Croton lechleri-derived product in combination with omeprazole as a total intestinal tract health product for horses. Ulcers are lesions of the lining of the digestive tract and are very common in horses used for many competitive activities including racing, dressage, show jumping, endurance events, and western performance. We believe that because Croton lechleri-derived products have been shown to act locally in the gut and have traditional use for ulcers, this equine formulation of a Croton lechleri-derived product in combination with omeprazole has the potential to address both gastric and colonic ulcers in horses, as well as diarrhea. There are currently no marketed FDA-approved treatments for colonic ulcers in horses, because physiological factors such as pH, bile, etc. render many drugs ineffective. According to a 2005 study, 54% of performance horses have both colonic and gastric ulcers and 97% of performance horses have either a gastric or a colonic ulcer. We believe that many owners give their horses daily doses of omeprazole to prevent ulcers, which practice can cost up to $50 per day. We believe a product treating both gastric and colonic ulcers, as well as diarrhea, would represent a significant advance in the management of gastrointestinal disease in horses. We anticipate that this product will capitalize on our work with targeted delivery in the gastrointestinal tract of other mammals.
Crofelemer—Cats
According to the American Veterinary Medical Association, there were approximately 74 million cats in the United States in 2012. We estimate that veterinarians see approximately 2.9 million annual cases of acute and chronic watery diarrhea in cats, approximately two-thirds of which are acute watery diarrhea. Veterinarians typically treat watery diarrhea in cats with the same treatments used for dogs, namely antibiotics, probiotics, dietary restrictions and products approved and formulated for humans, such as Imodium and other anti-motility agents, as well as binding agents that absorb water such as Kaopectate and Pepto-Bismol.
We are currently developing a species-specific formulation of crofelemer for cats and we intend to begin studies in cats in 2015. If data is positive and we receive FDA approval, we anticipate commercial launch in 2017.
Neonorm—Scours in Preweaned Dairy Calves
Overview
This formulation of Neonorm is an enteric-coated tablet designed to be orally administered to preweaned dairy calves twice daily for three days. In our clinical study completed in May 2014, Neonorm demonstrated a statistically significant reduction in morbidity, as well as reduced mortality and improved weight gain as compared to placebo in newborn dairy calves with scours. We recently launched Neonorm for preweaned dairy calves in the United States under the brand name Neonorm Calf. We do not believe that Neonorm fits the definition of an animal drug, as it does not contain an active pharmaceutical ingredient, nor is it a food or food additive. Thus, we do not believe that it is regulated by the FDA at this time. To support the commercial launch, we are also conducting field studies of Neonorm involving approximately 1,300 preweaned dairy calves in total at the following leading veterinary academic institutions: Cornell University, Tufts University and the University of California at Davis. We expect to announce this data by the end of 2014. Our commercialization activities are initially focused on large commercial dairy operations, and include active ongoing education and outreach to dairy industry key opinion leaders in the dairy industry, such as academics involved in dairy cattle research or who advise the dairy cattle industry, as well as veterinarians. We intend to augment these commercialization efforts by working with regional distributors to leverage the geographic concentration of the dairy market. In August 2014, we entered into our first regional distribution agreement for the Upper Midwest region, and together with this partner, launched Neonorm Calf at the 2014 World Dairy Expo. In September 2014, we entered into an agreement with a
72
national master distributor, who also distributes prescription products for the companion animal market.
Scours Market Opportunity
Scours refers to watery diarrhea in production animals, including dairy calves, which results from infectious agents that cause the secretion of ions and water into the intestinal lumen. Animals with scours may experience severe dehydration and electrolyte imbalance, which can lead to renal insufficiency, nutritional deficiencies, lower production in dairy cattle and even death. Current treatments include fluid and electrolyte replacement, continuous milk feeding, antibiotics (for calves with systemic involvement (e.g., fever) with an increased risk of bacteremia), non-steroidal anti-inflammatory drug therapy and vaccines.
According to the USDA, there are approximately 9.2 million milk-producing dairy cows in the United States. We estimate from USDA sources that there were over 11 million dairy calves born in 2013. Dairy cows are continuously bred, both to maintain lactation and to produce dairy calves to maintain the herd. Dairy heifer calves are separated from their mothers shortly after birth and raised on commercial milk replacers until weaned at about 60 days of age. Male dairy calves are typically sold into the beef industry.
Almost one in four, or 23.9%, of dairy heifer calves had diarrhea or other digestive problems according to the USDA Dairy 2007 study. Scours, diarrhea or other digestive problems are responsible for more than half of all preweaned heifer calf deaths, and result in supportive care and treatment costs, impaired weight gain and long-term reduction in milk production. Of dairy farm operations surveyed in the Dairy 2007 study, 62.1% used antibiotics for diarrhea or other digestive problems, including preweaned heifer calves not reporting diseases or disorders. Of preweaned heifer calves that were affected by diarrhea or other digestive problems, almost three-fourths, or 74.5%, were treated with an antibiotic.
Our Solution
We believe Neonorm is an ideal solution to address scours in dairy calves. Neonorm has been formulated and clinically tested to improve gut health by specifically addressing the normalization of stool formation and ion and water flow in the intestinal lumen of newborn dairy calves with scours. Like Canalevia, Neonorm acts locally in the gut and is minimally absorbed systemically. It does not alter gastrointestinal motility, has no significant effects on normally functioning intestinal ion channels and electrolyte or fluid transport, and has no side effects different from placebo. As a result, stool formation is normalized in a short period of time, weight loss is mitigated, supportive care costs and rehydration therapies such as ORS are reduced, and the risk of mortality is minimized.
Clinical Data
Overview. Neonorm demonstrated a statistically significant reduction in the severity of watery diarrhea and reduced daily incidence of watery diarrhea in a double-blind, randomized, placebo-controlled challenge study in newborn dairy calves with scours completed in May 2014. Neonorm also showed improvements in average daily weight gain and mortality. Scours-associated financial losses to the dairy industry arise not only from dairy calf mortality and impaired growth, but also from costs associated with veterinary care, medications and incremental labor to treat the sick dairy calves. The lifetime productivity for dairy cattle is influenced by early development and weight gain. Dairy calves with impaired preweaned growth may produce less milk over their lifetime. We believe our results demonstrate that the use of Neonorm in calves with scours can improve the economic return to dairy producers.
73
Study Protocol. The study enrolled 39 healthy newborn dairy calves, randomized into two groups. The calves were all challenged with enterotoxigenic E. coli, the most common bacterial cause of scours in dairy calves, in a controlled clinical setting. Clinical signs of watery diarrhea generally occurred 12 hours after the challenge. The first dose was administered to all calves at 12 hours. Additional doses were administered every 12 hours until hour 72 for a total of 6 doses. Twenty calves received Neonorm and 19 calves received placebo. Consistent with standard industry practice, calves with watery diarrhea were treated for dehydration with oral rehydration therapy or intravenous fluid. We examined the calves twice daily for 10 days as well as at days 15 and 25 for fecal consistency, dehydration, appetite, attitude and other adverse health disorders. In addition, all calves were weighed on the first day of the study and 25 days later. For all measurements except weight, we believe that days 1 through 8 to 10 following the E. coli challenge (i.e., 4.5 and 6.5 days after treatment is concluded) represent the most relevant timeframe to evaluate the treatment effect of Neonorm or placebo. After that period, other digestive ailments unrelated to the challenge may occur during the preweaning development of the calves. While most calves that do not die will eventually return to normal stool formulation after suffering from scours, studies have shown that the weight loss as a result of scours has a detrimental effect on lifetime milk productivity of the dairy cow. Thus, we believe resolving scours in the first ten days after onset can have positive economic impacts.
The study's goal was to evaluate the severity and incidence of diarrhea, mortality and weight gain. In this study, the "p" values were statistical calculations to determine whether the effects of Neonorm were significant in comparison to placebo based on pre-specified statistical targets. We specified that any result less than p=0.05 would be significant. The fecal scoring chart used in the study was the University of Wisconsin Calf Health Scoring Chart, modified to track a subset of the most severe, and potentially fatal, watery diarrhea, which we scored as 4, as set out in the following chart.
Modified University of Wisconsin Calf Health Scoring Chart
| |
Score |
Calf Feces Description |
Potential Treatment |
|||
|---|---|---|---|---|---|---|
|
0 |
Normal, formed pasty feces |
|
|||
Normal |
None | |||||
|
1 |
Semi-formed pasty feces |
||||
Moderate Diarrhea |
2 |
Loose, watery feces but stays on top of bedding |
Oral electrolytes |
|||
|
3 |
Watery feces with mucus, sifts through bedding |
Oral electrolytes, |
|||
Severe Diarrhea |
intravenous fluids, and | |||||
|
4 |
Diarrhea with blood |
antibiotics |
Reduced Fecal Consistency Score. Neonorm significantly decreased the severity of watery diarrhea over the course of the 25-day period (p=0.0133) and increased the speed of improvement in watery diarrhea, as shown by the decrease in average daily fecal scores. Quantitative analysis of fecal samples collected through day 10 further supported these results. Neonorm significantly increased the average fecal dry matter content, an objective measure of fecal consistency, compared to placebo (p=0.03). The multivariate analyses used each fecal consistency score, or fecal dry matter measure, data point collected for each animal in calculating statistical significance. The following chart sets out the average fecal consistency score over 25 days.
74
Average Daily Calf Fecal Scores
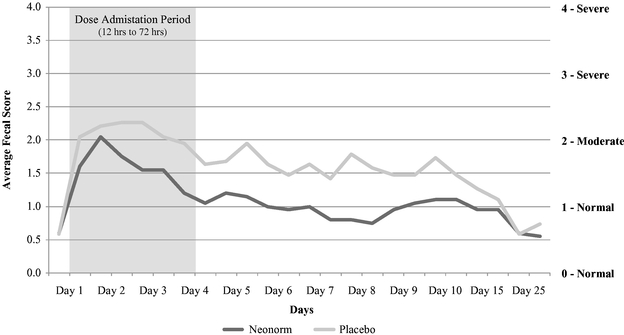
Reduced Daily Incidence of Watery Diarrhea. Neonorm decreased the daily incidence of watery diarrhea, which was defined as an average daily fecal consistency score of two or greater, over the 25-day period (p=0.0545). On day eight, there were no calves that had been administered Neonorm with an average daily fecal score of two or greater, whereas 37% of calves administered placebo had an average daily fecal score of two or greater.
Daily Incidence of Watery Diarrhea
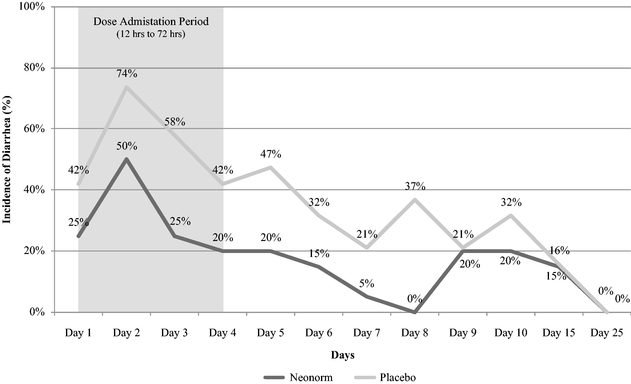
75
Proactive and Reactive Use. We believe the data support both the proactive and reactive use of Neonorm. In order to standardize the treatments, the first dose was administered 12 hours after the challenge with the bacteria. At that stage, some calves showed signs of watery diarrhea and others were at an earlier phase of the disease pathogenesis. We anticipate that Neonorm would be used in a comparable manner in dairy operations to dose sick calves reactively, and dose other calves proactively before development of clinical symptoms. We are further evaluating proactive use of Neonorm in field studies on commercial dairy farms.
Duration, Mortality and Weight Gain. Calves administered Neonorm showed a reduced average duration of watery diarrhea as compared to placebo. While this study was not powered for statistical significance, we plan to use our observations in this study to power our planned field studies to seek statistical significance on these endpoints. The following chart shows the average duration of watery diarrhea, as defined by a fecal score of two or greater, and severe watery diarrhea, as defined by a fecal score of three or greater, of calves administered Neonorm as compared to placebo. Measurements were taken twice daily and each case of watery or severe diarrhea counted for one half day of duration. Calves that died during the study were measured based on their most recently available fecal score until day 25.
| |
Average Duration of Watery Diarrhea (Score 2 and above) |
Average Duration of Severe Watery Diarrhea (Score 3 and 4) |
|||||
|---|---|---|---|---|---|---|---|
Administered |
3.03 days | 1.10 days | |||||
Placebo |
5.16 days | 2.42 days | |||||
After 25 days, only one calf died in the group administered Neonorm as compared to four calves in the placebo group. In addition, the calves administered Neonorm gained an average of 217g/day compared to 175g/day for the control group, indicating a trend of increased weight gain for calves receiving Neonorm during this period (41g difference per day). This translated to a weight gain of 11.9 pounds over 25 days for calves administered Neonorm, compared to 9.7 pounds for those receiving placebo, which is a relative improvement of approximately 24% during the period. While this study was not powered for statistical significance for these endpoints, we plan to use our observations in this study to power our planned field studies to seek statistical significance on weight gain. The lifetime productivity for dairy cattle is influenced by early development and weight gain. Preweaning nutrition has a significant effect on mammary gland development, the timing of puberty and the age at which the dairy cow first produces milk.
To support our commercial launch, we are also conducting field studies of Neonorm involving approximately 1,300 preweaned dairy calves in total at the following leading veterinary academic institutions: Cornell University, Tufts University and the University of California at Davis. We anticipate announcing data from these studies by the end of 2014. If the results confirm our existing data, and based on current industry cost standards, we estimate that Neonorm could save approximately $110 on average per treated dairy calf presenting with scours, accounting for costs to replace the dairy calf, costs of supportive care and improvement in future milk production. We believe that dairy farm operators currently target approximately $3 of expected savings for every $1 spent on animal health products. We believe our study demonstrates the potential for Neonorm to be a novel first-in-class product that provides health and economic benefits to the dairy industry.
Commercialization Plans
We recently launched Neonorm in the United States under the brand name Neonorm Calf. In July 2014, we commenced initial launch activities and met with key opinion leaders at a dairy industry conference, and in August 2014, we entered into our first regional distribution agreement for the Upper Midwest region. In September 2014, together with this distribution partner, we launched Neonorm Calf at the World Dairy Expo, which launch focused on key dairy states including Wisconsin,
76
Minnesota and Iowa. In September 2014, we entered into an agreement with a national master distributor, who also distributes prescription products for the companion animal market. We expect to launch Neonorm Calf nationwide by the end of 2014. According to the USDA, ten states account for approximately 75% of the U.S. dairy market, with three primary geographic regions: the North East, the Upper Midwest and California, as illustrated by the following map.
©Reprinted by permission from the March 25, 2013 issue of Hoard's Dairyman Magazine, Fort Atkinson, Wis.
Workforce Needs in Veterinary Medicine estimates that there are approximately 1,000 dairy veterinarians engaged in the food-animal industry. We believe a focused direct sales force initially targeting large commercial dairy operations, and potentially in conjunction with regional distribution partners, can be effective in reaching this market. We plan to establish an active ongoing industry education and outreach program. We expect to publish our clinical data in peer reviewed journals and to present at conferences attended by members of the dairy industry.
Neonorm Line Extensions
We believe that due to Neonorm's mechanism of action and our data in preweaned dairy calves, we will be able to develop and commercialize species-specific formulations of Neonorm for the estimated approximately 22 million beef calves in the United States, and multiple other animal species, such as horses, goats and sheep. Published sources indicate that approximately 2.4% of beef calves younger than three weeks old suffer from diarrhea. We believe that there is an opportunity to target large-scale commercial livestock operations, first in the United States, and later, internationally. In less developed nations, where not only dairy and beef cattle but also buffalo, goat and sheep provide livelihoods for local populations, reducing losses related to diarrhea can provide significant monetary, social and health benefits. Today, these groups are already accessed by distributors with whom we intend to work to extend the reach of Neonorm and line extension products.
We are planning studies of an equine formulation of Neonorm both for foals that have not been weaned and that are experiencing watery diarrhea, as well as adult horses with episodic diarrhea.
77
Published studies estimate that there were 9.2 million horses in the United States in 2005. We estimate that over 10% of these horses experience diarrhea. Diarrhea is among the most common clinical complaints in foals. Often, diarrhea occurs in the first 30 days of the foal's life, both from infections and non-infectious causes, such as lactose intolerance and overfeeding. Some cases are severe and life threatening. A majority of foals will exhibit diarrhea at some point within the first two months of life. In adult horses, episodic diarrhea is mostly associated with diseases of the large intestine and damage to the colon or disturbance of colonic function. Typically, diarrhea in horses is treated with fluid replenishment and electrolytes, deworming agents and antibiotics, and intestinal protectants and absorbents, as well as anti-motility agents. There are no anti-secretory products approved by the FDA for veterinary use.
Other Product Candidates and Development
We have planned multiple clinical studies over the next 12 to 18 months, to expand Canalevia and Neonorm to additional species. We believe that we will be successful because:
- •
- we have existing safety and efficacy data for our products and product candidates in dogs, dairy calves and/or humans;
- •
- each of these products works through the normalization of ion and water flow into the intestinal lumen; and
- •
- this physiological pathway is generally present in mammals.
Additionally, we will be initiating clinical studies for Virend and NP-500, both of which have been through Phase 2 human clinical testing by third parties. NP-500 is isolated and purified from a plant indigenous to the southwestern United States, and in traditional medicine, the plant was brewed as a tea and used for the treatment of diabetes and other various illnesses. We are currently developing species-specific formulations of NP-500 to treat obesity-related metabolic dysfunction in dogs, Type II diabetes in cats and metabolic syndrome in horses, and have filed three INADs for these indications.
According to a 2012 national survey of veterinarians, approximately 21% of dogs in the United States are obese. Studies show that obesity is more common in elderly dogs, as well as in neutered dogs. Obesity-related metabolic dysfunction manifests in altered lipid profiles, insulin resistance and mild hypertension, which could decrease a dog's lifespan. There are currently no FDA-approved products for the treatment of metabolic syndrome or insulin resistance in dogs. In cats, the prevalence of obesity-related diabetes or Type II diabetes is high and increasing. In horses, insulin resistance is associated with an equine metabolic syndrome characterized by obesity, regional adiposity and hypertriglyceridaemia. It is also known to be a risk factor for laminitis. Various studies report the prevalence of insulin resistance as 10% and 28% in horses and ponies, respectively. There are also currently no FDA-approved products for the treatment of metabolic syndrome in horses.
We anticipate that our development activities will benefit from centralized activities, including shared use of the manufacturing and regulatory documentation for chemistry, manufacturing and controls, or CMC. We also anticipate being able to enter into combined clinical research agreements and activities with companion animal clinical trial sites for dogs and cats.
Sales and Distribution
In September 2014, we launched Neonorm for preweaned dairy calves under the brand name Neonorm Calf in the Upper Midwest region, and we expect to launch Neonorm nationwide by the end of 2014 and Canalevia in 2016. We intend to establish a focused direct sales force for both the production and companion animal markets, and we have already hired our first sales representative for Neonorm Calf. We will focus our sales and marketing efforts on educational activities and outreach to key opinion leaders and decision makers at key regional and global accounts for production animals
78
and high prescriber veterinarians targets for companion animals. In August 2014, we entered into our first regional distribution agreement for the Upper Midwest region, and in September 2014, entered into an agreement with a national master distributor, who also distributes prescription products for the companion animal market. We plan to partner with other leading distributors to deliver our products to customers. We expect that our current and future partners will have the presence, name recognition, reputation and reach in the veterinary markets and in both key urban and rural centers, as appropriate. We believe this overall approach is scalable and transferable as we expand our commercialization efforts, as well as when we expand internationally including to resource-constrained countries where food safety issues are emerging global challenges.
Manufacturing
The manufacture of our Croton lechleri-derived products starts with the sustainable harvest of CPL. CPL, which is found in countries in South America, including Peru, is plentiful and naturally regenerating in the rain forest. Our contract suppliers in Peru obtain the raw materials and arrange the shipment of CPL to our third-party contract manufacturer. CPL will also be shipped to us for manufacturing after we establish our own API manufacturing capability.
Our third-party contract manufacturer will process CPL into both crofelemer, the API in Canalevia, and the botanical extract used in Neonorm. This manufacturing process uses exclusive Napo intellectual property licensed pursuant to the Napo License Agreement. Canalevia will be manufactured by the same process used to manufacture the API that was used in the animal safety studies and the human studies in support of the approval of Fulyzaq. Napo has also licensed this intellectual property to third parties in connection with its licenses related to the development and commercialization of crofelemer for human use. While we believe these third parties have developed their own proprietary manufacturing specifications pursuant to their license agreements, such third-party intellectual property is unknown to us, is not licensed to us pursuant to the Napo License Agreement, and is not part of the intellectual property that we intend to use for the manufacture of API in our licensed field of use. Similarly, the manufacture of Neonorm depends only on technology licensed from Napo. The license grant specifically excludes intellectual property rights developed pursuant to a prior collaboration agreement between Napo and Glenmark Pharmaceuticals, Ltd., the manufacturer of the API in Fulyzaq. In May 2014 and June 2014, we entered into binding memorandums of understanding with Indena S.p.A. to negotiate a definitive commercial manufacturing agreement for the manufacture of the API in Canalevia and the botanical extract in Neonorm. We have furnished equipment to Indena S.p.A. for use in a facility that will be dedicated to the manufacture of crofelemer and the botanical extract. Although we have not yet entered into the commercial manufacturing agreement, we currently have approximately 2,000 kg of the botanical extract in Neonorm, a quantity we believe is sufficient to meet or exceed expected volume requirements for approximately 12 months following our recent commercial launch of Neonorm and Indena S.p.A. has agreed to supply us with two pilot lots (approximately 60 kg) of botanical extract, as well as the API in Canalevia (approximately 3 kg) and data to support our anticipated regulatory filings.
Pursuant to the memorandums of understanding, we agreed to pay Indena S.p.A. the following fees in connection with the establishment of our manufacturing arrangement:
- •
- a start-up fee equal to €500,000, payable in two equal installments, the first of which will be payable
following the closing of this offering, the second of which will be payable upon installation and qualification of the manufacturing equipment we furnished to Indena;
- •
- fees associated with the technology transfer and manufacturing process adaptation equal to (i) €430,000 for API and (ii) €190,000 for the botanical extract, each of which are payable in two equal installments, the first of which have already been paid, and the second of which are payable within five days of delivery of the final study reports on batch testing;
79
- •
- if construction of a dedicated suite is required, initial fees of €100,000 for engineering, which will be
credited as a deposit towards construction costs, plus 100% of the construction costs; and
- •
- deliverables fees equal to €500,000, €250,000 of which is payable upon acceptance of the first GMP batch of API, and €250,000 of which is payable upon acceptance of the CMC necessary for our regulatory filings; with the understanding that these fees may be credited against payments agreed to under the future commercial manufacturing agreement.
We expect to use a portion of the proceeds of this offering for payment of these fees. In June 2014, as contemplated by the memorandums of understanding, we also issued Indena S.p.A. a warrant to acquire 25,000 shares our common stock at an exercise price per share equal to 90% of the initial public offering price, which expires in June 2019.
Utilizing proceeds from this offering, we intend to develop our own manufacturing capabilities for crofelemer to ensure, together with our contract manufacturer, a long-term supply of the Croton lechleri derivatives used in our prescription drug product candidates and non-prescription products. We plan to develop our own manufacturing capabilities in the United States and believe we will need an approximately 10,000 square foot facility. We anticipate that this size of facility will enable us to produce approximately 600 kg annually of the API in our prescription drug product candidates.
We also plan to enter into agreements with third parties for the formulation of the API and botanical extracts into finished products to be used for planned studies and commercialization. We are currently negotiating a commercial agreement with a third-party who currently provides formulated Canalevia for our ongoing general acute watery diarrhea proof-of-concept study in approximately 240 dogs.
The facilities of our third-party contract manufacturers that will manufacture our API and botanical extract, as well as formulate our finished products, comply with cGMP and other relevant manufacturing requirements.
Competition
The animal health industry is dominated by large independent companies such as Zoetis Inc., a stand alone animal health company that was spun out from Pfizer, Inc. in 2013, as well as subsidiaries of large pharmaceutical companies, including Novartis Animal Health Inc., a subsidiary of Novartis International AG., Merck Animal Health, the animal health division of Merck & Co., Inc., Merial Limited, the animal health division of Sanofi S.A., Elanco Animal Health, the animal health division of Eli Lilly and Company, Bayer Animal Health GmbH, a subsidiary of Bayer AG, and Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH. There are also animal health companies based in Europe, including Vétoquinol S.A., Virbac S.A., Dechra Pharmaceuticals PLC and Ceva Animal Health S.A.
Additionally, smaller animal health companies, such as Aratana Therapeutics, Inc., Kindred Biosciences, Inc., Phibro Animal Health Corporation and Parnell Pharmaceuticals Holdings Ltd, recently completed initial public offerings of their stock in the United States and may choose to develop competitive products. We believe that the large human pharmaceutical companies may also decide to spin out their animal health subsidiaries into stand alone companies.
Although there are currently no FDA-approved anti-secretory products to treat watery diarrhea in dogs, we anticipate that Canalevia, if approved, will face competition from various products, including products approved for use in humans that are used extra-label in animals. We are aware that veterinarians typically treat watery diarrhea in dogs with antibiotics, probiotics, dietary restrictions and products approved and formulated for humans, such as Imodium and other anti-motility agents, as well as binding agents that absorb water, such as Kaopectate and Pepto-Bismol. None of these treatment
80
options address the water loss associated with watery diarrhea. We are not aware of any veterinarians prescribing Fulyzaq extra-label for use in dogs, and the indication of Fulyzaq is for a disease that does not occur in dogs. Further, because none of the human products are FDA approved for animal use, veterinarians, although allowed to dispense human products for animal use, do not have the benefit of clinical support with respect to efficacy or dosing. Moreover, administering a potentially unpalatable human formulation is often difficult and may lead to further uncertainty of the amount actually ingested by the dog. However, this practice may continue and Canalevia may face competition from these products. Canalevia could also potentially face competition from Fulyzaq were veterinarians to prescribe it extra-label. Extra-label use is the use of an approved drug outside of its cleared or approved indications in the animal context. All of our potential products could also face competition from new products in development. These and other potential competing products may benefit from greater brand recognition and brand loyalty than our products and product candidates may achieve.
Intellectual Property
Napo License Agreement
In January 2014, we entered into the Napo License Agreement, which we amended and restated in August 2014, pursuant to which we acquired an exclusive, sublicensable, transferable, worldwide license to intellectual property rights of Napo and its affiliates to research, develop, formulate, make, have made, use, have used, market, offer for sale, sell, have sold, and import, and to otherwise exploit products of Napo and its other affiliates for all veterinary treatment uses and indications for all species of animals. The license grant specifically excludes intellectual property rights developed pursuant to a prior collaboration agreement between Napo and Glenmark Pharmaceuticals, Ltd., the manufacturer of the API in Fulyzaq. Under the Napo License Agreement, Napo also assigned to us certain raw materials and equipment and granted us a right of reference to the entirety of the information included in the human approved new drug application of crofelemer.
Under the terms of the Napo License Agreement, we are responsible for, and shall ensure, the development and commercialization of products that contains or are derived from the licensed Napo technology (collectively referred to herein as the Products) worldwide in the field of veterinary treatment uses and indications for all species of animals.
In consideration for the license, we are obligated to pay a one-time non-refundable license fee of $2,000,000, less the option fee of $100,000 paid in July 2013 pursuant to a term sheet we signed with Napo. This license fee payment will be deferred until the combined net sales of one or more Products with Napo technology exceed $2,000,000 and can be paid, at our option, in shares of our common stock.
Pursuant to the Napo License Agreement, if this offering is not completed before December 31, 2015 or if we do not receive net proceeds of at least $10,000,000 from this offering, we may owe Napo (x) milestone payments of up to $3,000,000 per Product derived from Croton lechleri and milestone payments of up to $150,000 per non-Croton lechleri Product, (y) an 8% royalty on cumulative annual net sales of all such Products derived from Croton lechleri up to a specified amount of net sales and a 10% royalty on cumulative annual net sales of all such Products above such specified amount of net sales, and (z) a 2% royalty on annual net sales of all non-Croton lechleri Products that are prescription drugs approved by the FDA or the equivalent regulatory agency in another country, and a 1% royalty on annual net sales of non-prescription non-Croton lechleri Products that do not require pre-marketing approval from the FDA or the equivalent regulatory agency in another country. If this offering closes before such milestones are achieved or such sales occur and before December 31, 2015 and we receive net proceeds of at least $10,000,000 from this offering, we will not owe Napo such milestone payments or such royalties on non-Croton lechleri Products and for Products derived from Croton lechleri, we will owe Napo a 2% royalty on annual net sales of all Products that are prescription drugs (such as Canalevia and any line extensions) approved by the FDA or the equivalent regulatory agency in
81
another country, and a 1% royalty of annual net sales of non-prescription Products (such as Neonorm and any line extensions) that do not require pre-marketing approval from the FDA or the equivalent regulatory agency in another country.
The royalty term expires on a country-by-country and Product-by-Product basis on the later of: (i) 10 years from the first sale of a Product in such country, on an animal by animal basis; and (ii) the first date on which there is no longer (A) a valid claim within the licensed patent rights covering the use, manufacture or sale of such Product, or (B) any data exclusivity with respect to such Product in such country conferred by the applicable regulatory authority, and in each case of (A) and (B), a competitive product has been introduced into the market in such country. The royalties payable to Napo are subject to reduction, capped at a specified percentage, for any third-party payments made to obtain a license or other rights to issued patents that might present a commercial obstacle to the development, manufacture, use, or sale of a Product in a country. Additionally, if the royalty term for a Product is ongoing post-expiration of the last valid claim within the licensed patent rights that covers such product in any given country, then the royalties we owe Napo will be reduced by a specified percentage until expiration of the royalty term for such Product in such country. Upon the expiration of each royalty term, on a country-by-country and Product-by-Product basis, the license grants shall be fully paid up and we will have perpetual non-exclusive licenses for such Products in such countries. At any time during the term of the agreement, if Napo sells all of its assets relating to the use, production or exploitation of Croton lechleri derivative products to a third party, all of the rights granted to us relating to Croton lechleri derivative products under the license shall become exclusive in the field of veterinary treatment uses and indications for all species of animals, perpetual, fully paid-up, royalty-free and irrevocable, with the right to grant sublicenses.
Under the terms of the Napo License Agreement, we own all rights, title and interest in our intellectual property and any joint intellectual property developed under the license. We granted Napo a non-exclusive, paid-up, irrevocable worldwide license to our intellectual property developed under the Napo License Agreement for use outside the veterinary field, and an exclusive, paid-up worldwide license to any joint intellectual property developed under the Napo License Agreement outside the veterinary field. We agreed to defend, indemnify and hold Napo, its affiliates, and its officers, directors, employees, consultants and contractors harmless from and against any losses, costs, damages, liabilities, fees and expenses arising out of any third-party claim related to our gross negligence or willful misconduct, breach of our representations, warranties or covenants or the manufacture, sale or use of the Product or Products, in each case, unless such third-party claim is subject to indemnification by Napo. Napo agreed to defend, indemnify and hold us, our affiliates, and our officers, directors, employees, consultants and contractors harmless from and against any losses, costs, damages, liabilities, fees and expenses arising out of any third-party claim related to Napo's, its affiliate's or its licensees' (except for us) gross negligence or willful misconduct, or Napo's breach of its representations, warranties or covenants.
We may terminate the Napo License Agreement upon Napo's uncured material breach, bankruptcy or at will after certain notification periods. Napo may terminate the Napo License Agreement upon our uncured material breach or bankruptcy after certain notification periods.
Patent Portfolio
Under the Napo License Agreement, we have exclusive rights in the veterinary field to an international patent family related to International Patent Application WO1998/16111. The patents and patent applications in this family are directed to enteric protected formulations of proanthocyanidin polymers isolated from Croton spp. (such as crofelemer and Neonorm), and methods of treating watery diarrhea using the enteric protected formulations for both human and veterinary uses. As such, the patents and patent applications of this family cover certain formulations of crofelemer, including Canalevia, as well as the standardized botanical extract in Neonorm, and methods of treating diarrhea
82
using these formulations. There are three U.S. patents and a pending U.S. patent application in this family, including, US 7,323,195, which has a term until at least June 7, 2018, US 7,341,744, which has a term until at least January 11, 2018, and US 8,574,634, which has a term until at least February 4, 2018. The term of one of US 7,323,195 or US 7,341,744 may be extended to June 2021 and December 2020, respectively, to account for regulatory delay in obtaining human marketing approval for crofelemer (such potential extensions have been filed for). Patent protection for enteric protected formulations of crofelemer and methods of use has also been obtained outside the United States, including in Europe, Australia, Canada, India, Japan, Korea, Mexico, New Zealand and Taiwan, with terms extending until at least October 14, 2017 in these jurisdictions. In particular, European patent EP 0 935 417 and Japanese patent no. 4195728 provide protection for enteric protected formulations of crofelemer and the standardized botanical extract in Neonorm in Europe and Japan, respectively, with terms that extend until at least October 14, 2017.
The patents and patent applications we licensed from Napo, or the Napo Patents, are also licensed by Napo to Salix Pharmaceuticals, Inc., or Salix, for certain fields of human use. Under the terms of the collaboration agreement between Salix and Napo, or the Salix Collaboration Agreement, Napo and Salix have agreed on who has the first right and responsibility to file, prosecute and maintain the Napo Patents. As a result, under the Napo License Agreement, we only have the right to maintain any issued patents within the Napo Patents that are not maintained in accordance with the rights and responsibilities of the parties under the Salix Collaboration Agreement. US 7,323,195; US 7,341,744; and US 8,574,634 are issued Napo Patents. Salix has licensed rights only to human use in certain territories and for certain indications, and currently markets crofelemer (Fulyzaq) for human use and has listed US 7,323,195; US 7,341,744; and US 8,574,634 in the FDA's Orange Book for Fulyzaq. We rely on these issued Napo Patents as intellectual property protection for our veterinary prescription drug product candidates and non-prescription products. Pending patent applications within Napo Patents either may not be relevant to veterinary indications and/or may not issue as patents. Similarly, under the Salix Collaboration Agreement, Napo and Salix agreed on who has the first right to enforce the Napo Patents against potential infringers, even in our field of use. In addition, as between Napo and us, Napo has the first right to enforce the Napo Patents against potential infringers. If we are not the party who enforces the Napo Patents, we will receive no proceeds from such enforcement action. In each case, such proceeds are subject to reimbursement of costs and expenses incurred by the other party in connection with such action.
We have filed three provisional patent applications in the United States relating to veterinary uses of Croton proanthocyanidin polymer compositions, including crofelemer and Neonorm. These applications are directed to treatment of watery diarrhea in newborn and young animals, including methods of improving mortality and weight gain in newborn animals, treatment of stress-induced diarrhea in animals, and treatment of watery diarrhea caused by salmonella in animals. Patents that may issue based upon applications filed claiming benefit of these provisional patent applications should have terms that extend until at least May 2035.
Trademarks
We plan to market our products under a trademark or trademarks we select and we will own all rights, title and interest, including all goodwill, associated with such trademarks.
Government Regulation
The development, approval and sale of animal health products are governed by the laws and regulations of each country in which we intend to seek approval, where necessary, to market and subsequently sell our prescription drug and non-prescription products. To comply with these regulatory requirements, we are establishing processes and resources to provide oversight of the development,
83
approval processes and launch of our products and to position those products in order to gain market share in each respective market.
United States
Certain federal regulatory agencies are charged with oversight and regulatory authority of animal health products in the United States. These agencies, depending on the product and its intended use may include the FDA, the USDA and the Environmental Protection Agency. In addition, the Drug Enforcement Administration regulates animal therapeutics that are classified as controlled substances. In addition, the Federal Trade Commission may in the case of non-prescription products, regulate the marketing and advertising claims being made.
The approval of prescription drugs intended for animal use is regulated by the FDA, Center for Veterinary Medicine, or CVM. The CVM consists of six offices that work together to, in part, approve new drugs for commercialization and thereafter monitor those commercialized drugs once in the market. The Office of New Animal Drug Evaluation, or ONADE, is the lead office for reviewing novel drug candidates. We, as the sponsor of a novel drug candidates commence the development and approval process by initiating communication with the ONADE and opening an INAD file. As part of this process, we will also schedule a discussion of the novel drug's development plan in order to obtain agreement from the CVM for the number, type and design of studies needed to obtain FDA approval of the novel drug.
As required by the FDA, new animal drug products must obtain marketing approval through the NADA process. Under the Administrative New Animal Drug Application, or Administrative NADA, process, a sponsor can engage in a phased submission of the required technical sections of an NADA, known as a rolling NADA, as opposed to submitting the entire application at once with a standard NADA. The requirements for all NADAs are the same regardless of whether a sponsor chooses the rolling NADA or the standard NADA submission. Under the phased review, once all technical sections have been submitted and reviewed, the sponsor submits an Administrative NADA to reflect that all technical sections of the NADA have been submitted and reviewed, each such technical section meets the requirements for approval and the CVM has issued technical section complete letters for each technical section. The phased review and Administrative NADA allow a drug sponsor to engage with the FDA as to each technical section to ensure that each section meets all requirements prior to submission of the application for approval. Phasing of NADA submissions is a voluntary process.
Once the tasks set forth in the development plan have been completed, including the clinical work as well as the chemistry and manufacturing work (feasibility, validation and stability of the drug inclusive), we, as the novel drug sponsor will need to provide to the FDA through the application process, information as to the safety and efficacy of the drug candidate, and, if needed, a human food safety study. This food study is only required for drugs intended for use in production animals, and we currently have no plans to develop drugs for production animals. Additionally, the application will contain a module on CMC, which describes the plan for manufacturing the drug including the API, the final formulation, where it will be made, how it will be made, how the drug will be packaged, how it can be stored, the conditions required for storage and how long it can be stored before expiry. A major part of the CMC section is the analysis we employ to ensure that the manufactured drug is of a high quality, is consistently manufactured under cGMP and is stable. Other significant components to the application we have to complete before receiving drug approval includes a draft label that will list specific information such as dosing information, intended use, warnings, directions for use, and other information as required by the regulations. The package insert that will contain information on studies, warnings, drug interactions, intended use and dosing is considered part of the label in addition to that which is adhering to the container itself. The CVM ensures that the labeling provides all the necessary information to use the drug safely and effectively, and that it clearly discloses the risks associated with the drug.
84
MUMS Designation
The Minor Use and Minor Species Animal Health Act, or MUMS Act, became effective in August 2004. The purpose of the MUMS Act was twofold: first, to encourage the development and availability of more animal drugs which are intended to be used in a major species defined as dogs, cats, cattle, horses, chickens, turkeys and pigs to treat diseases which occur infrequently or in limited geographic areas, therefore having an impact on a smaller number of animals on a yearly basis; and second, to encourage the development and availability of animal drugs for use in minor species (defined as all animals other than humans that are not one of the major species). In order to be considered a MUMS product, the drug sponsor must seek the MUMS designation by working with the CVM. The MUMS designation is modeled on the orphan drug designation for human drug development and has certain financial incentives available to encourage MUMS drug development such as the availability of grants to help with the cost of the MUMS drug development. Also, drug developers of MUMS drugs are eligible to apply for a waiver of the user fees once the MUMS designation has been given by the Office of Minor Use Minor Species. Once a drug has received a MUMS designation, the drug sponsor may seek conditional approval of the drug product. MUMS is a designation that is requested by the applicant to the CVM for drug products where the intended use fits within MUMS designation. We believe that we qualify for MUMS designation for Canalevia as a minor use in a major species because the estimated total number of dogs in the United States affected by CID is less than 70,000. Once designation has been granted, then the company must submit safety and acceptable evidence of efficacy data as well as CMC data similar to the requirements for an NADA. However, with a MUMS product, in order to encourage development of MUMS drugs, the FDA allows interspecies data extrapolation in order to minimize the amount of new research needed to support efficacy and safety. After the filing of an NADA and the review of the application, the FDA through the CVM can then grant a conditional approval. This approval allows for the commercialization of the product, while collecting the remaining efficacy data required for a non-conditional approval of the drug. The sponsor has up to five years to collect this efficacy data. Following submission, review and approval of the pivotal field effectiveness study, the FDA may grant a full NADA approval. Ideally, MUMS designation helps move the product forward in development; however, it may not shorten the time to full commercialization. A sponsor that gains approval or conditional approval for a designated new animal drug receives a seven-year marketing exclusivity.
Protocol Concurrence
We intend to seek protocol concurrences with the FDA for the planned pivotal trial of Canalevia that we plan to conduct for general acute watery diarrhea in dogs and for future pivotal trials in other indications. A protocol is submitted to the FDA voluntarily by a drug sponsor. The FDA review of the protocol for a pivotal study makes it more likely that the study will generate information the sponsor needs to demonstrate whether the drug is safe and effective for its intended use. It creates an expectation by the sponsor that the FDA should not later alter its perspectives on these issues unless public or animal health concerns appear that were not recognized at the time of protocol assessment. Even if FDA issues a protocol concurrence, ultimate approval of an NADA by the FDA is not guaranteed because a final determination that the agreed-upon protocol satisfies a specific objective, such as the demonstration of efficacy, or supports an approval decision, will be based on a complete review of all the data submitted to the FDA. Even if we were to obtain protocol concurrence, such concurrence does not guarantee that the results of the study will support a particular finding or approval of the new drug.
Marketing Exclusivity
We are currently planning on seeking MUMS designation for our prescription drug products and if we receive such a designation, we are entitled to a seven-year marketing exclusivity, which means that we will face no competition from another sponsor marketing the same drug in the same dosage form
85
for the same intended use. If we were to lose such designation or not receive such designation but our application as a New Animal Drug is found to be a new chemical entity that meets the criteria described by the FDA, we would be entitled to a five-year marketing exclusivity. In order to receive this five-year exclusivity, the FDA would have to find in its approval of our application that our NADA contains an API not previously approved in another application, that the application itself is an original application, not a supplemental application, and that our application included the following studies: one or more investigations to demonstrate substantial evidence of effectiveness of the drug for which we are seeking approval; animal safety studies and human food safety studies (where applicable). If the NADA is seeking approval of a drug for which we have received conditional approval, we, upon approval would still be entitled to a five-year marketing exclusivity provided it meets the criteria as set forth above. If however, our NADA is for a drug for which the FDA has determined that the drug contains an API that has previously been approved, regardless of whether the original approval was for use in humans or not, we may only be entitled to a three-year marketing exclusivity provided that the NADA is an original, not supplemental, application and contains both safety and efficacy studies demonstrating the safety and efficacy of the drug which is the subject of the application.
European Union
The European Union, or EU, definition of a veterinary medicinal product closely matches the definition of an animal drug in the United States. In the EU, a company can market a veterinary medicinal product only after a marketing authorization has been issued by an EU member state, (i.e., approval on a country-by-country basis) or by the EU Commission through the European Medicines Agency, or the EMA. Before the EU member state or the EU Commission issues marketing authorization, we must submit a marketing authorization application, known as the dossier. The dossier includes data from studies showing the product's quality, safety, and efficacy and is similar to an NADA filed with the FDA.
For an animal drug, the Committee for Medicinal Products for Veterinary Use, or CVMP, is responsible for the scientific evaluation. Experts from all EU member states are on the CVMP. The Rapporteur, or lead reviewer on the dossier, prepares an overview of the committee's scientific evaluation, called the CVMP Assessment Report.
The CVMP Assessment Report:
- •
- summarizes the data submitted by the company on the product's quality, safety, and efficacy;
- •
- explains the assessment done by the CVMP to support the committee's recommendation to the EU Commission to issue a
marketing authorization; and
- •
- is the basis for the European Public Assessment Report published on the EMA's website.
Labeling
The FDA plays a significant role in regulating the labeling, advertising and promotion of animal drugs. This is also true of regulatory agencies in the EU and other territories. In addition, advertising and promotion of animal health products is controlled by regulations in many countries. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and approved by the applicable agency. We will conduct a review of advertising and promotional material for compliance with the local and regional requirements in the markets where we eventually may sell our product candidates.
Our non-prescription products will be labeled in accordance with the health guidelines outlined by the National Animal Supplements Council, an industry organization that sets industry standards for certain non-prescription animal products, including but not limited to product labeling.
86
Other Regulatory Considerations
We believe regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to the products we currently are developing because our prescription drug product candidates are not intended for use in production animals, with the exception of horses, which qualify as food animals in Europe and Canada; and our non-prescription products are not regulated by section 201(g) of the Federal Food, Drug, and Cosmetic Act, which the FDA is authorized to administer.
Our prescription drug product candidates currently in development, if approved, may eventually face generic competition in the United States and in the EU after the period of exclusivity has expired. In the United States, a generic animal drug may be approved pursuant to an Abbreviated New Animal Drug Application, or ANADA. With an ANADA, a generic applicant is not subject to the submission of new clinical and safety data but instead must only show that the proposed generic product is a copy of the novel drug product, and bioequivalent to the approved novel product. However, if our product candidates are the first approved by the FDA or the EMA as applicable for use in animals, they will be eligible for a five-year marketing exclusivity in the United States and 10 years in the EU thereby prohibiting generic entry into the market. If the product has MUMS designation it has a seven-year marketing exclusivity.
In addition to the foregoing, we may be subject to state, federal and foreign healthcare and/or veterinary medicine laws, including but not limited to anti-kickback laws, as we may from time to time enter consulting and other financial arrangements with veterinarians, who may prescribe or recommend our products. If our financial relationships with veterinarians are found to be in violation of such laws that apply to us, we may be subject to penalties.
Employees
As of September 30, 2014, we had 17 employees. Of our employees, five hold D.V.M. or Ph.D. degrees and eight of our employees are engaged in research and development activities. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Description of Properties
Our corporate headquarters are located in San Francisco, California, where we rent approximately 3,125 square feet of office space from Napo. Napo's lease agreement expires in June 2015. In June 2014, Napo assigned the lease to our company. We believe that our existing facilities are adequate for our near-term needs. We believe that suitable additional or alternative space would be available if required in the future on commercially reasonable terms.
Legal Proceedings
From time to time, we may become involved in litigation relating to claims arising from the ordinary course of business. There are currently no claims or actions pending against us, the ultimate disposition of which could have a material adverse effect on our results of operations, financial condition or cash flows.
87
The following table lists our executive officers and directors and their respective ages and positions as of September 30, 2014:
Name
|
Age | Position
|
|||
|---|---|---|---|---|---|
| Lisa A. Conte | 55 | Chief Executive Officer, President and Director | |||
| Steven R. King, Ph.D. | 56 | Executive Vice President, Sustainable Supply, Ethnobotanical Research and Intellectual Property and Secretary | |||
| Serge Martinod, D.V.M., Ph.D. | 58 | Chief Veterinary Officer | |||
| John A. Kallassy | 50 | Executive Vice President, Chief Financial Officer, Chief Operating Officer and Treasurer | |||
| James J. Bochnowski(1)(2)(3) | 70 | Chairman of the Board of Directors | |||
| Jiahao Qiu(1)(3) | 29 | Director | |||
| Zhi Yang, Ph.D.(1)(2) | 58 | Director | |||
- (1)
- Member of the audit committee.
- (2)
- Member of the compensation committee.
- (3)
- Member of the nominating committee.
Executive Officers
Lisa A. Conte. Ms. Conte has served as our President, Chief Executive Officer and a member of our board of directors since she founded the company in June 2013. From 2001 to 2014, Ms. Conte served as the Chief Executive Officer of Napo Pharmaceuticals, Inc., a biopharmaceutical company she founded in November 2001. In 1989, Ms. Conte founded Shaman Pharmaceuticals, Inc., a natural product pharmaceutical company, which declared bankruptcy in 2001. Additionally, Ms. Conte is Napo Pharmaceutical's current Interim Chief Executive Officer and has served as a member of its board of directors since 2001. Ms. Conte is also currently a member of the board of directors of Healing Forest Conservatory, a California not-for-profit public benefit corporation. Ms. Conte holds an M.S. in Physiology and Pharmacology from the University of California, San Diego, and an M.B.A. and A.B. in Biochemistry from Dartmouth College.
We believe Ms. Conte is qualified to serve on our board of directors due to her extensive knowledge of our company and experience with our product and product candidates, as well as her experience managing and raising capital for public and private companies.
Steven R. King, Ph.D. Dr. King has served as our Executive Vice President of Sustainable Supply, Ethnobotanical Research and Intellectual Property since March 2014 and as our Secretary since September 2014. From 2002 to 2014, Dr. King served as the Senior Vice President of Sustainable Supply, Ethnobotanical Research and Intellectual Property at Napo Pharmaceuticals, Inc. Prior to that, Dr. King served as the Vice President of Ethnobotany and Conservation at Shaman Pharmaceuticals, Inc. Dr. King has been recognized by the International Natural Products and Conservation Community for the creation and dissemination of research on the long-term sustainable harvest and management of Croton lechleri, the widespread source of crofelemer. Dr. King is currently a member of the board of directors of Healing Forest Conservatory, a California not-for-profit public benefit corporation. Dr. King holds a Ph.D. in Biology from the Institute of Economic Botany of the New York Botanical Garden and an M.S. in Biology from the City University of New York.
Serge Martinod, D.V.M, Ph.D. Dr. Martinod has served as our Chief Veterinary Officer since June 2013. Since January 2013, Dr. Martinod has also served as the Chief Executive Officer, Chief Scientific Officer and a member of the board of directors of TNG Pharmaceuticals, Inc., an animal
88
pharmaceutical company. From 2012 to 2013, Dr. Martinod served as a research consultant for Napo Pharmaceuticals, Inc. From 2008 to 2012, Dr. Martinod served as Chief Scientific Officer of ArcaNatura LLC, an animal health company he co-founded. Dr. Martinod holds an M.B.A. from the University of Nebraska, a Ph.D. in Biology from the University of Science and Medicine in Grenoble, France and a D.V.M. from the National Veterinary School in Lyon, France.
John A. Kallassy. Mr. Kallassy has served as our Executive Vice President and Chief Operating Officer since February 2014, and as our Chief Financial Officer and Treasurer since September 2014. From 2012 to 2014, Mr. Kallassy was the President and co-founder of I-Bankers Direct, LLC, an investment bank and currently serves on its board of directors. From 2005 to 2011, Mr. Kallassy was the Chief Executive Officer of Zargis Medical Corp., a medical device company, whose assets were acquired by 3M Company in 2011, and as Chief Financial Officer of Speedus Corp., a then-NASDAQ listed affiliate of Zargis Medical Corp. Mr. Kallassy holds an M.B.A. from Cornell University and a B.S. in Biology from the State University of New York at Brockport.
Non-Employee Directors
James J. Bochnowski. Mr. Bochnowski has served as a member of our board of directors since February 2014 and as Chairman of our board since June 2014. Since 1988, Mr. Bochnowski has served as the founder and Managing Member of Delphi Ventures, a venture capital firm. In 1993, Mr. Bochnowski co-founded Technology Venture Investors. Mr. Bochnowski holds an M.B.A. from Harvard University Graduate School of Business and a B.S. in Aeronautics and Astronautics from Massachusetts Institute of Technology.
We believe Mr. Bochnowski is qualified to serve on our board of directors due to his significant experience with venture capital backed healthcare companies and experience as both an executive officer and member of the board of directors of numerous companies.
Jiahao Qiu. Mr. Qiu has served as a member of our board of directors since February 2014. Since 2010, Mr. Qiu has served as a Principal of BioVeda China Fund, a life science investment firm. From 2009 to 2010, he served as an interpreter for the Delegation of the European Union to China. Mr. Qiu holds a B.S. in Biotechnology from the Jiao Tong University in Shanghai, China.
We believe Mr. Qiu is qualified to serve on our board of directors due to his experience with evaluating, managing and investing in life science portfolio companies for BioVeda China Fund.
Zhi Yang, Ph.D. Dr. Yang has served as a member of our board of directors since February 2014. Since 2005, Dr. Yang has served as the Chairman, Managing Partner and Founder of BioVeda China Fund, a life science investment firm. Dr. Yang is currently an advisor to the China Health and Medical Development Foundation, under China's Ministry of Health. Dr. Yang holds a Ph.D. in Molecular Biology and Biochemistry, as well as an M.A. in Cellular and Developmental Biology, both from Harvard University.
We believe Dr. Yang is qualified to serve on our board of directors due to his significant experience as a founder, investor and member of the board of directors of numerous life sciences companies, as well as his life sciences background and education.
Family Relationships
There are no family relationships among any of our directors or executive officers.
89
Corporate Governance
Board Composition and Risk Oversight
Our business and affairs are managed under the direction of our board of directors, which currently consists of four members. Certain members of our board of directors were elected pursuant to the provisions of a voting agreement among certain of our major stockholders, as amended. See "Certain Relationships and Related Persons Transactions—Voting Agreement" for more information regarding the voting agreement.
Our board of directors consists of four members. Three of the four directors that will comprise our board of directors upon the closing of this offering are independent within the meaning of the independent director rules of the NASDAQ Stock Market, LLC, or NASDAQ. All of the current directors were initially elected to our board of directors pursuant to a voting agreement that will terminate automatically by its terms upon the closing of this offering, or appointed by the then members of the board.
Upon closing of this offering, our board of directors will be divided into three classes of directors. At each annual meeting of stockholders, a class of directors will be elected for a three-year term to succeed the class whose term is then expiring. The terms of the directors will expire upon the election and qualification of successor directors at the annual meeting of stockholders to be held during the years 2015 for the Class I directors, 2016 for the Class II directors and 2017 for the Class III directors.
The Class I directors will be Ms. Conte and Mr. Bochnowski.
The Class II director will be Mr. Qiu.
The Class III director will be Dr. Yang.
We expect that any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors. The division of our board of directors into three classes with staggered three-year terms may delay or prevent a change of our management or a change in control.
Our board of directors has an active role, as a whole and when it establishes committees, will at the committee level, in overseeing the management of our risks. Our board of directors is responsible for general oversight of risks and regular review of information regarding our risks, including credit risks, liquidity risks and operational risks. Our compensation and nominating committees will be responsible for overseeing the management of risks relating to our executive compensation plans and arrangements and the risks associated with the independence of our board of directors and potential conflicts of interest. Our audit committee will be responsible for overseeing the management of our risks relating to accounting matters and financial reporting. While each committee is responsible for evaluating certain risks and overseeing the management of such risks, our entire board of directors will be regularly informed through discussions from committee members about such risks. Our board of directors believes its administration of risk oversight function has not affected our board of directors' leadership structure.
Director Independence
Upon the closing of this offering, we anticipate that our common stock will be listed on The NASDAQ Capital Market. Under the NASDAQ rules, independent directors must comprise a majority of a listed company's board of directors within a specified period of the closing of this offering. In addition, NASDAQ rules require that, subject to specified exceptions, each member of a listed company's audit, compensation and nominating committee be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Under the NASDAQ rules, a director will only qualify as an
90
"independent director" if, in the opinion of that company's board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
To be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, our board of directors, or any other board committee (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
In July 2014, our board of directors undertook a review of its composition, the composition of its committees and the independence of our directors and considered whether any director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our board of directors has determined that three of our four directors that will be seated upon the closing of this offering does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is "independent" as that term is defined under the NASDAQ rules. Our board of directors also determined that Mr. Bochnowski (chairperson), Mr. Qiu and Dr. Yang, who will comprise our audit committee, and Mr. Bochnowski (chairperson) and Dr. Yang, who will comprise our compensation committee and Mr. Bochnowski (chairperson) and Mr. Qiu, who will comprise our nominating committee, satisfy the independence standards for those committees established by applicable SEC rules and the NASDAQ rules and listing standards.
In making this determination, our board of directors considered the relationships that each non-employee director has with us and all other facts and circumstances our board of directors deemed relevant in determining independence, including the beneficial ownership of our capital stock by each non-employee director.
Board Committees
Upon the closing of this offering, our board of directors will establish an audit committee, a compensation committee and a nominating committee.
Audit Committee
The members of our audit committee will be Mr. Bochnowski, Mr. Qiu and Dr. Yang. Mr. Bochnowski will be the chairperson of the audit committee. Upon the closing of this offering, our audit committee's responsibilities will include:
- •
- appointing, approving the compensation of, and assessing the independence of our registered public accounting firm;
- •
- overseeing the work of our independent registered public accounting firm, including through the receipt and consideration
of reports from that firm;
- •
- reviewing and discussing with management and our independent registered public accounting firm our annual and quarterly
financial statements and related disclosures;
- •
- monitoring our internal control over financial reporting, disclosure controls and procedures and code of conduct;
- •
- discussing our risk management policies;
91
- •
- establishing policies regarding hiring employees from our independent registered public accounting firm and procedures for
the receipt and retention of accounting related complaints and concerns;
- •
- reviewing and approving or ratifying any related person transactions; and
- •
- preparing the audit committee report required by SEC rules.
All audit and non-audit services, other than de minimis non-audit services, to be provided to us by our independent registered public accounting firm must be approved in advance by our audit committee.
Our board of directors has determined that each of Mr. Bochnowski, Mr. Qiu and Dr. Yang is an independent director under NASDAQ rules and under Rule 10A-3. All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and NASDAQ. Our board of directors has determined that Mr. Bochnowski is an "audit committee financial expert," as defined by applicable SEC rules, and has the requisite financial sophistication as defined under the applicable NASDAQ rules and regulations.
Compensation Committee
The members of our compensation committee will be Mr. Bochnowski and Dr. Yang. Mr. Bochnowski will be the chairperson of the compensation committee. Upon the closing of this offering, our compensation committee's responsibilities will include:
- •
- determining, or making recommendations to our board of directors with respect to, the compensation of our Chief Executive
Officer;
- •
- determining, or making recommendations to our board of directors with respect to, the compensation of our other executive
officers;
- •
- overseeing and administering our cash and equity incentive plans;
- •
- reviewing and making recommendations to our board of directors with respect to director compensation;
- •
- reviewing and discussing at least annually with management our "Compensation Discussion and Analysis" disclosure if and to
the extent then required by SEC rules; and
- •
- preparing the compensation committee report and necessary disclosure in our annual proxy statement in accordance with applicable SEC rules.
Our board has determined that each of Mr. Bochnowski and Dr. Yang is independent under the applicable NASDAQ rules and regulations, is a "non-employee director" as defined in Rule 16b-3 promulgated under the Exchange Act, and is an "outside director" as that term is defined in Section 162(m) of the Internal Revenue Code of 1986, as amended.
Nominating Committee
The members of our nominating committee will be Mr. Bochnowski and Mr. Qiu. Mr. Bochnowski will be the chairperson of the nominating committee. Upon the closing of this offering, our nominating committee's responsibilities will include:
- •
- identifying individuals qualified to become members of our board of directors;
- •
- recommending to our board of directors the persons to be nominated for election as directors and to each of the committees
of our board of directors; and
- •
- overseeing an annual evaluation of our board of directors.
92
Code of Ethics and Conduct
We have adopted a written code of ethics and conduct that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions that will become effective upon the closing of this offering. Following the closing of this offering, a current copy of the code will be available on our website at www.jaguaranimalhealth.com. We intend to disclose future amendments to certain provisions of our code of business conduct and ethics, or waivers of such provisions on our website to the extent required by applicable rules and exchange requirements. The inclusion of our website address in this prospectus does not incorporate by reference the information on or accessible through our website into this prospectus.
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee has ever been an officer or employee of our company. None of our executive officers currently serves, or in the past year has served, as a member of the board of directors or compensation committee or other board committee performing equivalent functions of any entity that has one or more of its executive officers serving on our board of directors or compensation committee.
Limitation of Liability and Indemnification
Our amended and restated certificate of incorporation and amended and restated bylaws that will become effective upon the closing of this offering contain provisions that limit the personal liability of our directors for monetary damages to the fullest extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for:
- •
- any breach of the director's duty of loyalty to us or our stockholders;
- •
- any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law;
- •
- unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the
Delaware General Corporation Law, or DGCL; or
- •
- any transaction from which the director derived an improper personal benefit.
Such limitation of liability does not apply to liabilities arising under federal securities laws and does not affect the availability of equitable remedies, such as injunctive relief or rescission.
Our amended and restated certificate of incorporation that will become effective upon the closing of this offering provides that we indemnify our directors to the fullest extent permitted by Delaware law. In addition, our amended and restated bylaws that will become effective prior to the closing of this offering, provide that we indemnify our directors and officers to the fullest extent permitted by Delaware law. Our amended and restated bylaws that will become effective upon the closing of this offering also provide that we shall advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity, regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law. We have entered and expect to continue to enter into agreements to indemnify our directors, executive officers and other employees as determined by our board of directors. With certain exceptions, these agreements provide for indemnification for related expenses including, among others, attorneys' fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding. We believe that these bylaw provisions and
93
indemnification agreements are necessary to attract and retain qualified persons as directors and officers. We also maintain directors' and officers' liability insurance.
The limitation of liability and indemnification provisions in our amended and restated certificate of incorporation and amended and restated bylaws that will become effective upon the closing of this offering, and our indemnification agreements, may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duty of care. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Furthermore, a stockholder's investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers. There is no pending litigation or proceeding involving any of our directors, officers or employees for which indemnification is sought, and we are not aware of any threatened litigation that may result in claims for indemnification.
Board Leadership Structure
Our amended and restated bylaws and corporate governance guidelines provide our board of directors with flexibility in its discretion to combine or separate the positions of chairman of the board and chief executive officer. As a general policy, our board of directors believes that separation of the positions of chairman and chief executive officer reinforces the independence of the board of directors from management, creates an environment that encourages objective oversight of management's performance and enhances the effectiveness of the board of directors as a whole. We expect and intend the positions of chairman of the board and chief executive officer to be held by two individuals in the future.
Director Compensation
During 2013, we did not pay any compensation to our directors for their service on our board of directors. We currently do not pay our directors any cash compensation for their services on our board of directors.
We intend to make annual equity grants to directors serving on our board who are not employees nor serving as designees of our investors, along with an additional equity grant to the chairman of our board of directors. We may in the future determine to make additional equity grants or pay other equity compensation for service on our board of directors.
In June 2014, we granted Mr. Bochnowski, our Chairman of the Board, a stock option to acquire 59,116 shares of common stock at an exercise price of $3.22 per share, which expires 10 years after the grant date. The option vests as follows: 25% vests on March 2, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option is vested in full on June 2, 2017.
94
Summary Compensation Table
In 2013, we paid Dr. Martinod, our Chief Veterinary Officer, $40,000 pursuant to a consulting agreement. We did not pay any other compensation to any other executive officer in 2013. In 2013, we paid Napo $394,866 for services provided by its employees, which includes services provided by our executive officers, pursuant to the Service Agreement, in the amounts of $137,080 for Lisa A. Conte, $63,650 for Charles O. Thompson (our former Chief Financial Officer) and $21,865 for Steven R. King. See "Management's Discussion and Analysis of Financial Condition and Results of Operations—Financial Operations Overview" for further information regarding the Service Agreement.
Outstanding Equity Awards at Fiscal Year-End
We did not have any outstanding equity awards at December 31, 2013.
Executive Employment Agreements
Lisa A. Conte
In March 2014, we entered into an offer letter with Ms. Conte to serve as our Chief Executive Officer, effective March 1, 2014, in an at-will capacity. Under this offer letter, Ms. Conte's annual base salary is $400,000, she is eligible for an annual target bonus of 30% of her base salary, and she is eligible to participate in the employee benefit plans that we offer to our other employees. In April 2014, Ms. Conte was granted a stock option to purchase 240,575 shares of common stock at an exercise price of $1.69 per share. The option has a 10 year term and vests as follows: 25% vests on January 1, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option is vested in full on April 1, 2017. On June 2, 2014, Ms. Conte was granted 26,731 restricted stock units, or RSUs. Assuming the closing of this offering and compliance with the other terms of the RSU award agreement, 50% of the shares of common stock underlying the RSUs will vest and be issuable on January 1, 2016, and the remaining 50% will vest and be issuable on July 1, 2017. In the event of a change in control, as defined in the Jaguar Animal Health, Inc. 2013 Equity Incentive Plan, or the 2013 Plan, the vesting of all outstanding awards granted to Ms. Conte under the 2013 Plan will accelerate if Ms. Conte's service with us is terminated without cause within twelve months of the change in control.
Serge Martinod, D.V.M., Ph.D.
In January 2014, we entered into an offer letter with Dr. Martinod to serve as our Chief Veterinary Officer in an at-will capacity, effective as upon the closing of our first sale of Series A preferred stock on February 5, 2014. Under the offer letter, Dr. Martinod's annual base salary is $220,000, he is eligible for an annual target bonus of 30% of his base salary, and will be eligible to participate in the employee benefit plans we offer to our other employees. In April 2014, Dr. Martinod was granted a stock option to purchase 120,287 shares of common stock at an exercise price of $1.69 per share. The option has a 10-year term and vests as follows: 25% vests on January 1, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option is vested in full on April 1, 2017. In June 2014, Dr. Martinod was granted 13,366 RSUs. Assuming the closing of this offering and compliance with the other terms of the RSU award agreement, 50% of the shares of common stock underlying the RSUs will vest and be issuable on January 1, 2016, and the remaining 50% will vest and be issuable on July 1, 2017. In the event of a change in control, as defined in the 2013 Plan, the vesting of all outstanding awards granted to Dr. Martinod under the 2013 Plan will accelerate if Dr. Martinod's service with us is terminated without cause within twelve months of the change in control.
95
Steven R. King, Ph.D.
In February 2014, we entered into an offer letter with Dr. King to serve as our Executive Vice President, Sustainable Supply, Ethnobotanical Research and Intellectual Property, effective March 1, 2014, in an at-will capacity. Under the offer letter, Dr. King's annual base salary of $255,000, he is eligible for an annual target bonus of 30% of his base salary, and he is eligible to participate in the employee benefit plans we offer to our other employees. In April 2014, Dr. King was granted a stock option to purchase 140,335 shares of common stock at an exercise price of $1.69 per share. The option has a 10-year term and vests as follows: 25% vests on January 1, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option is vested in full on April 1, 2017. In June 2014, Dr. King was granted 15,593 RSUs. Assuming the closing of this offering and compliance with the other terms of the RSU award agreement, 50% of the shares of common stock underlying the RSUs will vest and be issuable on January 1, 2016, and the remaining 50% will vest and be issuable on July 1, 2017. In the event of a change in control, as defined in the 2013 Plan, the vesting of all outstanding awards granted to Dr. King under the 2013 Plan will accelerate if Dr. King's service with us is terminated without cause within twelve months of the change in control.
John A. Kallassy
In January 2014, we entered into an offer letter with Mr. Kallassy to serve as our Executive Vice President and Chief Operating Officer, effective as upon the closing of our first sale of Series A preferred stock on February 5, 2014. Effective as of September 19, 2014, we entered into a new offer letter with Mr. Kallassy in connection with his appointment to serve as our Chief Financial Officer. Under the current offer letter, Mr. Kallassy's annual base salary is $245,000, and he is eligible for an annual target bonus of 30% of his base salary and is eligible to participate in the employee benefit plans that we offer to our other employees. In April 2014, Mr. Kallassy was granted a stock option to purchase 120,287 shares of common stock at an exercise price of $1.69 per share. The option has a 10-year term and vests as follows: 25% vests on January 1, 2015, 9 months after the grant date, with the remainder vesting equally over the next 27 months such that the option is vested in full on April 1, 2017. In June 2014, Mr. Kallassy was granted 13,366 RSUs. Assuming the closing of this offering and compliance with the other terms of the RSU award agreement, 50% of the shares of common stock underlying the RSUs will vest and be issuable on January 1, 2016, and the remaining 50% will vest and be issuable on July 1, 2017. We also agreed that Mr. Kallassy is eligible for the grant of an additional 2,227 RSUs, as well as an option to purchase an additional 20,048 shares of common stock, subject to approval by our board of directors. In the event of a change in control, as defined in the 2013 Plan, the vesting of all outstanding awards granted to Mr. Kallassy under the 2013 Plan will accelerate if Mr. Kallassy's service with us is terminated without cause within twelve months of the change in control.
Employee Benefit Plans
2014 Stock Incentive Plan
In July 2014, our board of directors adopted the Jaguar Animal Health, Inc. 2014 Stock Incentive Plan, or the 2014 Plan, and our stockholders are expected to approve the 2014 Plan before the completion of this offering. The 2014 Plan will be effective on the business day immediately before the effective date of the registration statement of which this prospectus forms a part. The 2014 Plan provides for the grant of incentive stock options to our eligible employees, and for the grant of nonstatutory stock options, restricted stock, and RSUs to eligible employees, directors and consultants.
96
Authorized Shares
We have reserved 500,000 shares of our common stock for issuance pursuant to the 2014 Plan. In addition, the number of shares will automatically increase on January 1st of each year, for a period of not more than five years, beginning on January 1st of the year following the year in which the Plan became effective and ending no later than January 1, 2019, in an amount equal to 2% of the total number of shares of common stock outstanding on December 31st of the preceding calendar year. The board of directors may act prior to January 1st of any given year, at its discretion, to provide for no increase in shares or to add a lesser number of shares than provided for in the prior sentence.
If a stock award expires without having been exercised in full, or, with respect to restricted stock and RSUs, a stock award is forfeited, the shares that were subject to those stock awards will become available for future grant or sale under the 2014 Plan (unless the 2014 Plan has terminated). If unvested shares of restricted stock or RSUs are repurchased by the company or are forfeited to the company, such shares will become available for future awards under the 2014 Plan.
Plan Administration
Our board of directors or one or more committees appointed by our board of directors will administer the 2014 Plan. In the case of awards intended to qualify as "performance-based compensation" within the meaning of Section 162(m) of the Code, the committee will consist of two or more "outside directors" within the meaning of Section 162(m) of the Code. In addition, if we determine it is desirable to qualify transactions under the 2014 Plan as exempt under Rule 16b-3, such transactions will be structured to satisfy the requirements for exemption under Rule 16b-3. Subject to the provisions of the 2014 Plan, the committee has the power to administer the 2014 Plan, including but not limited to, the power to interpret the terms of the 2014 Plan and awards granted under it, to create, amend and revoke rules relating to the 2014 Plan, including creating sub-plans, and to determine the terms of the awards, including the exercise price, the number of shares subject to each such award, the exercisability of the awards and the form of consideration, if any, payable upon exercise.
Options
Both incentive stock options qualifying under Section 422 of the Code and non-statutory stock options may be granted under the 2014 Plan. The exercise price of options granted under the 2014 Plan must at least be equal to the fair market value of the common stock on the date of grant. The term of an incentive stock option may not exceed ten years, except that with respect to any participant who owns more than 10% of the voting power of all classes of our outstanding stock, the term must not exceed five years and the exercise price must equal at least 110% of the fair market value on the grant date. For nonstatutory stock options the exercise price must equal at least 100% of the fair market value. The committee will determine the methods of payment of the exercise price of an option, which may include cash, shares or other property acceptable to the committee, as well as other types of consideration permitted by applicable law. After the termination of service of an employee, director or consultant, he or she may exercise the vested portion of his or her option for the period of time stated in his or her award agreement, except in the case of an employee terminated for cause (as defined in the 2014 Plan) the option will terminate upon his or her termination from service. Generally, if termination is due to death or disability, the vested portion of the option will remain exercisable for 12 months. In all other cases, the vested portion of the option generally will remain exercisable for three months following the termination of service. An option may not be exercised after expiration of its term. However, if the exercise of an option is prevented by applicable law the exercise period may be extended under certain circumstances. Subject to the provisions of the 2014 Plan, the committee determines the other terms of options.
97
Restricted Stock
Restricted stock awards may be granted under the 2014 Plan. Restricted stock awards are grants of shares of our common stock that vest in accordance with terms and conditions established by the committee. The committee will determine the number of shares of restricted stock granted to any employee, director or consultant and, subject to the provisions of the 2014 Plan, will determine the terms and conditions of such awards. The committee may impose whatever conditions to vesting it determines to be appropriate (for example, the committee may set restrictions based on the achievement of specific performance goals or continued service to us); provided, however, that the committee, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed. Recipients of restricted stock awards generally will have voting and dividend rights with respect to such shares upon grant without regard to vesting, unless the committee provides otherwise. Shares of restricted stock that do not vest are subject to our right of repurchase or forfeiture.
RSUs
Awards of RSUs may be granted under the 2014 Plan. An RSU is the right to receive a share of common stock at a future date. The committee determines the terms and conditions of RSUs, including the vesting criteria (which may include accomplishing specified performance criteria or continued service to us) and the form and timing of payment. Notwithstanding the foregoing, the committee, in its sole discretion, may accelerate the time at which RSUs will vest.
Non-Transferability of Awards
Unless the committee provides otherwise, stock awards issued under the 2014 Plan are not transferrable other than by will or the laws of descent and distribution, and only the recipient of an award may exercise an award during his or her lifetime, although a recipient may designate a beneficiary to exercise an award after death.
Certain Adjustments
In the event of certain changes in the capitalization, to prevent diminution or enlargement of the benefits or potential benefits available under the 2014 Plan, the committee will adjust the number and class of shares that may be delivered under the 2014 Plan and/or the number, class and price of shares covered by each outstanding award, and the numerical share limits set forth in the 2014 Plan. In the event of the proposed liquidation or dissolution, the committee will notify participants as soon as practicable and all awards will terminate immediately prior to the consummation of such proposed transaction.
Merger or Change in Control
The 2014 Plan provides that in the event of a merger or change in control, as defined under the 2014 Plan, each outstanding award will be treated as the committee determines, including (i) the assumption, continuation or substitution of the stock awards by the successor corporation or its parent or subsidiary, (ii) the acceleration of vesting for any unvested portion of the stock awards, or (iii) the cash-out of the stock awards.
Amendment; Termination
The committee has the authority to amend, suspend or terminate the 2014 Plan provided such action does not impair the existing rights of any participant.
98
2013 Equity Incentive Plan, as Amended
In November 2013, our board of directors adopted the 2013 Plan, effective November 1, 2013. The 2013 Plan was approved by our stockholders in November 2013. The 2013 Plan is intended to provide incentives to attract, retain and motivate eligible persons whose present and potential contributions are important to the success of our company by offering them an opportunity to participate in our company's future performance through awards of options, restricted stock, RSUs and stock bonuses.
A total of 1,271,300 shares of common stock have been approved by the board of directors and our shareholders for issuance under the 2013 Plan. The 2013 Plan will be terminated in connection with this offering, and accordingly, no shares will be available for issuance under the 2013 Plan following the completion of this offering. The 2013 Plan will continue to govern outstanding awards granted thereunder. As of July 31, 2014, options to purchase 1,129,673 shares of our common stock and RSUs covering 118,953 shares remained outstanding under the 2013 Plan.
The 2013 Plan and outstanding stock awards will be administered by the compensation committee of our board of directors, or the committee, or our board of directors, acting as the committee. The committee has the authority to select grantees and set the terms of awards under the 2013 Plan, to construe and interpret the Plan and to make all other determinations necessary or advisable for the administration of the Plan. Grantees are selected in the discretion of the committee.
Awards under the Plan are evidenced by a written award agreement that contains the terms and conditions of the award. Awards granted under the 2013 Plan are generally not transferable other than by will or the laws of descent and distribution, or as otherwise provided in an award agreement.
The exercise price for options granted under the 2013 Plan may not be less than the fair market value of our common stock on the grant date. Under the 2013 Plan, fair market value will be determined by the board of directors in good faith.
In the event of certain corporate transactions, the vesting of outstanding stock awards under the 2013 Plan granted to non-executive employees will accelerate such that the stock awards are fully vested upon the corporate transaction.
Our board of directors may terminate the 2013 Plan at any time and also has the right to alter or amend the 2013 Plan or any part of the 2013 Plan from time to time. However, no change can be made to a granted option, if it would impair the rights of such grantee, without the consent of the grantee.
99
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
The following includes a summary of transactions since inception, June 6, 2013, to which we have been a party in which the amount involved exceeded or will exceed $120,000 and in which any of our directors, executive officers or beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest. Compensation arrangements for our directors and executive officers are described elsewhere in this prospectus.
Transactions with Napo
Formation
We were founded in San Francisco, California as a Delaware corporation on June 6, 2013. Napo formed our company to develop and commercialize animal health products. In connection with our formation, we issued 4,000,000 shares of common stock to Napo, pursuant to a stock purchase agreement, for $400 in cash and services to be provided by Napo to our company pursuant to the Service Agreement discussed below. As of December 31, 2013, we were a wholly-owned subsidiary of Napo and as of June 30, 2014, we were a majority-owned subsidiary of Napo.
Napo Service Agreement
Effective July 1, 2013, we entered into an Employee Leasing and Overhead Allocation Agreement with Napo, or the Service Agreement. The term of the agreement was from July 1, 2013 through February 28, 2014. In connection with the Service Agreement, Napo provided us with the services of Napo employees and we agreed to pay Napo for a portion of Napo's overhead costs including rent. For additional information relating to the Service Agreement, see "Management's Discussion and Analysis of Financial Condition and Results of Operations—Financial Operations Overview."
Napo License Agreement
In January 2014, we entered into the Napo License Agreement, pursuant to the term sheet for which we paid Napo a $100,000 option fee, and agreed to make royalty and milestone payments to Napo based on sales of our products. Lisa A. Conte, our Chief Executive Officer, President and member of our board of directors is also the interim chief executive officer and serves on the board of directors of Napo. Ms. Conte intends to continue to act as interim chief executive officer of Napo until a suitable chief executive officer for Napo activities is recruited and approved by Napo's board of directors. For additional information relating to the Napo License Agreement, see "Business—Intellectual Property—Napo License."
In connection with the entry into certain financing arrangements in October 2014, which we refer to as the Nantucket Financing Arrangements, Napo and Nantucket Investments Limited, or Nantucket, on behalf of Napo's secured lenders, entered into a non-disturbance agreement with respect to the Napo License Agreement. The non-disturbance agreement provides that we are a third party beneficiary of such agreement and also provides, among other items, that notwithstanding any transfer of or sale or other disposition by Nantucket of the intellectual property and technology licensed to us pursuant to the Napo License Agreement, including without limitation, in connection with any enforcement of the Nantucket Financing Arrangements, transfer in lieu of enforcement or by operation of law, the intellectual property and technology licensed to us pursuant to the Napo License Agreement shall remain subject to the Napo License Agreement, the Napo License Agreement shall survive in accordance with its terms, and our rights under the Napo License Agreement shall not be terminated unless we fail to make payments thereunder within the time periods required.
100
Napo Arrangements
Lease
Our corporate headquarters are located in San Francisco, California, where we rent approximately 3,125 square feet of office space. Since our formation in June 2013, we have shared premises with Napo pursuant to its lease. See "Napo Service Agreement" above. Since March 2014, we have made the rent payments under Napo's lease. The lease was assigned to us in June 2014 and expires in June 2015.
Napo Beneficial Ownership
The following table sets forth information with respect to beneficial ownership of Napo common stock by the current members of our board of directors and our executive officers. The column titled "Percentage of Shares Beneficially Owned" is based on a total of 108,452,786 shares of Napo common stock outstanding as of June 30, 2014.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to Napo common stock. Shares of Napo common stock subject to options or warrants that are currently exercisable or vested, or exercisable or subject to vesting within 60 days after June 30, 2014 are considered outstanding and beneficially owned by the person holding the options or warrants for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person.
Name of Beneficial Owner
|
Number of Shares Beneficially Owned |
Percentage of Shares Beneficially Owned |
|||||
|---|---|---|---|---|---|---|---|
James J. Bochnowski(1) |
8,067,505 | 7.4 | % | ||||
Lisa A. Conte(2) |
2,697,770 | 2.4 | |||||
Jiahao Qiu |
— | — | |||||
Zhi Yang, Ph.D.(3) |
2,151,174 | 2.0 | |||||
Steven R. King, Ph.D.(4) |
797,175 | * | |||||
John A. Kallassy |
— | — | |||||
- *
- Less than 1%
- (1)
- Includes (i) 7,522,051 shares of Napo common stock and (ii) warrants to purchase 545,454 shares of Napo common stock, all of which are held by the Bochnowski Family Trust. Mr. Bochnowski, a member of our board of directors, is a co-trustee and beneficiary of such trust, and shares voting and investment control over such shares with his spouse.
- (2)
- Includes (i) 981,122 shares of Napo common stock and (ii) a fully-vested option to purchase 1,716,648 shares of Napo common stock. In addition, Ms. Conte holds RSUs for an aggregate of 7,300,134 shares of Napo common stock (3,475,734 of which were issued prior to 2011; and 3,824,400 of which were issued post 2011). Ms. Conte, our Chief Executive Officer, President and a member of our board of directors, is the interim chief executive officer of Napo and a member of Napo's board of directors.
- (3)
- Includes (i) 30,828 shares of Napo common stock held by Mr. Yang; (ii) 65,309 shares of Napo common stock held by BioVeda China Limited, an entity affiliated with BioVeda China Fund; and (iii) 2,055,037 shares of Napo common stock held by BioVeda China LP, an entity affiliated with BioVeda China Fund. Mr. Yang, a member of our board of directors, is the Founder and Managing Partner of BioVeda China Fund, and may be deemed to beneficially own such shares.
- (4)
- Includes (i) 337,460 shares of Napo common stock and (ii) a fully-vested option to purchase 459,715 shares of Napo common stock. In addition, Dr. King holds RSUs for an aggregate of 2,042,098 shares of Napo common stock (1,073,273 of which were issued
101
prior to 2011; and 968,825 of which were issued post 2011). Dr. King, our Executive Vice President of Sustainable Supply, Ethnobotanical Research and Intellectual Property, held an office in the same capacity at Napo.
Assuming satisfaction of the service requirements, Napo's RSU awards granted post 2011 will vest and the shares will be issued when: (i) the performance criteria set out in the award agreement are met (which include (A) the repayment in full by Napo of certain debts owed to third parties and (B) Napo's successful resolution of the litigation against Salix) and (ii) there is a Napo liquidity event (such as a merger, an asset sale or a liquidation or dissolution). Napo's RSU awards granted prior to 2011 will vest and the shares will be issued when there is a Napo liquidity event. For all Napo RSUs, the vesting and issuance criteria must be satisfied by December 31, 2018 or the Napo RSUs will lapse.
Financings
Note and Warrant Financings
In July 2013, pursuant to a note and warrant purchase agreement dated July 8, 2013, we issued a convertible promissory note in the aggregate principal amount of $100,000 and warrants to purchase 59,333 shares of common stock to Sichuan Biopharma, an entity affiliated with BioVeda China Fund, or BVCF, a significant stockholder. Dr. Zhi Yang, a member of our board of directors, is the Chairman, Founder and Managing Partner of BVCF, and Mr. Jiahao Qiu, a member of our board of directors, is an employee of BVCF. In September 2013, we also issued a convertible promissory note in the aggregate principal amount of $250,000 and warrants to purchase 148,333 shares of common stock to the Bochnowski Family Trust pursuant to the same purchase agreement. Mr. Bochnowski, a member of our board of directors, is a co-trustee and beneficiary of the Bochnowski Family Trust. The warrants have an exercise price of $1.6854 per share and expire February 5, 2019, the fifth anniversary of the initial closing of the sale of Series A preferred stock. The warrants are outstanding and have not been exercised as of the date of this prospectus.
In January 2014, the noteholders acknowledged and agreed that all principal under the notes would convert in full into shares of common stock at $1.6854 per share immediately prior to the initial closing of the sale of Series A preferred stock and we subsequently issued 148,333 shares of common stock to the Bochnowski Family Trust and 59,333 shares of common stock to Sichuan Biopharma in February 2014.
In June 2014, pursuant to a convertible note purchase agreement, we issued convertible promissory notes in the aggregate principal amount of $300,000 to two accredited investors, including a convertible promissory note for $200,000 to the Bochnowski Family Trust. Upon the closing of this offering, all of these convertible promissory notes will convert into shares of common stock at a conversion price equal to 80% of the initial public offering price per share. Such shares of common stock will be unregistered.
Series A Financing
In February 2014, we entered into a Series A preferred stock purchase Agreement with Kunlun Pharmaceuticals Ltd., or Kunlun, pursuant to which we issued 2,224,991 shares of Series A preferred stock at a price per share of $2.2472 for aggregate gross proceeds of $5,000,000. Kunlun is a wholly-owned subsidiary of BVCF. Dr. Zhi Yang, a member of our board of directors, is the Chairman, Founder and Managing Partner of BVCF. Mr. Jiahao Qiu, a member of our board of directors, is an employee of BVCF.
In April and May 2014, pursuant to a series of joinder agreements to the Series A preferred stock purchase agreement, we issued 790,911 shares of Series A preferred stock to eight accredited investors at a price per share of $2.2472 for aggregate gross proceeds of $1,777,338. The Bochnowski Family
102
Trust purchased 222,499 shares of Series A preferred stock for $500,000. Mr. Bochnowski, a member of our board of directors, is a co-trustee and beneficiary of the Bochnowski Family Trust.
Investors' Rights Agreement
In February 2014, we entered into an investors' rights agreement with the holders of Series A preferred stock. This agreement provides for certain rights relating to the registration of their shares of common stock issuable upon conversion of their Series A preferred stock, a right of first refusal to purchase future securities sold by us and certain additional covenants made by us. The right of first refusal and certain additional covenants will terminate upon the closing of this offering. The registration rights will terminate upon the later to occur of (a) for any particular holder with registration rights, at such time following this offering when all securities held by that stockholder subject to registration rights may be sold pursuant to Rule 144 under the Securities Act of 1933, as amended, or the Securities Act, during any 90-day period, or (b) five years following the closing of this offering. See "Description of Securities—Registration Rights" for additional information.
Voting Agreement
In February 2014, we entered into a voting agreement with the holders of Series A preferred stock and common stock. Pursuant to the voting agreement, the following directors were each elected to serve as members on our board of directors: Lisa A. Conte and James J. Bochnowski (as representatives of holders of common stock, as designated by a majority of common stockholders), and Zhi Yang and Jiahao Qiu (as representatives of the holders of Series A preferred stock).
The voting agreement will terminate upon the closing of this offering, and members previously elected to our board of directors pursuant to this agreement will continue to serve as directors until they resign, are removed or their successors are duly elected by holders of our common stock. The composition of our board of directors after this offering is described in more detail under "Management—Board of Directors and Executive Officers."
Right of First Refusal and Co-Sale Agreement
In February 2014, we entered into a right of first refusal and co-sale agreement with the holders of our Series A preferred stock and common stock. The right of first refusal, right of co-sale and certain additional covenants will terminate upon the closing of this offering.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors, and intend to enter into such agreements with our officers prior to the closing of this offering. These agreements, among other things, require us or will require us to indemnify each director to the fullest extent permitted by Delaware law, including indemnification of expenses such as expenses, judgments, penalties, fines and amounts paid in settlement to the extent legally permitted incurred by the director in any action or proceeding, including any action or proceeding by or in right of us, arising out of the person's services as a director.
Other Transactions
We have granted stock options and/or RSUs to our executive officers. For a description of these options and RSUs, see the section of this prospectus titled "Management—Executive Compensation." We have also granted stock options to certain members of our board of directors. For a description of these stock options, see the section of this prospectus titled "Management—Director Compensation."
103
Policies and Procedures for Related Person Transactions
Our board of directors has adopted a written related person transaction policy, to be effective upon the closing of this offering, setting forth the policies and procedures for the review and approval or ratification of related-person transactions. This policy will cover, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person had or will have a direct or indirect material interest, including, without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm's length transaction and the extent of the related person's interest in the transaction. All of the transactions described in this section occurred prior to the adoption of this policy.
104
The following table sets forth certain information with respect to the beneficial ownership of our common stock as of September 30, 2014 by:
- •
- each person, or group of affiliated persons, who is known by us to be the beneficial owner of more than 5% of our
outstanding common stock;
- •
- each of our directors;
- •
- each of our executive officers; and
- •
- all of our executive officers and directors as a group.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to our common stock. Shares of our common stock subject to options, warrants or RSUs that are currently exercisable or vested, or exercisable or subject to vesting within 60 days after September 30, 2014 are considered outstanding and beneficially owned by the person holding the options, warrants, or RSUs for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Except as otherwise noted, the persons and entities in this table have sole voting and investing power with respect to all of the shares of our common stock beneficially owned by them, subject to community property laws, where applicable. The information is not necessarily indicative of beneficial ownership for any other purpose, including for purposes of Section 13(d) and Section 13(g) of the Securities Act.
The column titled "Percentage of Shares Beneficially Owned—Before Offering" is based on a total of 7,327,400 shares of common stock outstanding as of September 30, 2014 and includes the conversion of all outstanding shares of Series A preferred stock into 3,015,902 shares of common stock. The total shares of common stock outstanding may be adjusted for the purpose of calculating the percentage ownership of a person that has options, warrants or RSUs that are currently exercisable or vested, or exercisable or subject to vesting within 60 days after September 30, 2014 but not for the purpose of recalculating the percentage ownership of any other person. It does not include the conversion of all outstanding convertible promissory notes into shares of common stock, assuming an initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, upon the closing of this offering. The column titled "Percentage of Shares Beneficially Owned—After Offering" is based on shares of common stock to be outstanding after this offering, including the shares of common stock that we are selling in this offering and the conversion of all outstanding convertible promissory notes into shares of common stock based upon a conversion price equal to 80% of the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, upon the closing of this offering.
105
Except as otherwise set forth below, the address of each beneficial owner listed in the table below is c/o Jaguar Animal Health, Inc., 185 Berry Street, Suite 1300, San Francisco, California 94107.
| |
|
Percentage of Shares Beneficially Owned |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Name and Address of Beneficial Owner
|
Number of Shares Beneficially Owned |
Before Offering |
After Offering |
|||||||
5% Stockholders |
||||||||||
Napo Pharmaceuticals, Inc.(1) |
4,000,000 | 54.6 | % | % | ||||||
Entities affiliated with BVCF(2) |
2,343,657 | 31.7 | % | % | ||||||
Executive Officers and Directors |
||||||||||
James J. Bochnowski(3) |
519,165 | 6.9 | % | % | ||||||
Lisa A. Conte |
— | — | — | |||||||
Jiahao Qiu |
— | — | — | |||||||
Zhi Yang, Ph.D.(4) |
2,343,657 | 31.7 | % | % | ||||||
Steven R. King, Ph.D. |
— | — | — | |||||||
Serge Martinod, D.V.M., Ph.D. |
— | — | — | |||||||
John A. Kallassy |
— | — | — | |||||||
All executive officers and directors as a group (7 persons) |
2,862,822 | 38.0 | % | % | ||||||
- (1)
- Lisa A. Conte, our Chief Executive Officer, is the interim chief executive officer of Napo. Napo's five-person board of directors, consisting of Lisa A. Conte, Richard W. Fields, Joshua Mailman, Gregory Stock and Thomas Van Dyck, has ownership and control of the shares of common stock held by Napo. Certain members of our board of directors, as well as certain of our executive officers and employees beneficially own common stock in Napo. As a group, our executive officers and directors (7 persons total), collectively beneficially own 12.7% of the issued and outstanding common stock of Napo, including the Bochnowski Family Trust, which holds 7.4%. Mr. Bochnowski, a member of our board of directors, is a co-trustee and beneficiary of such trust and shares voting and investment control over such shares with his spouse. See "Certain Relationships and Related Persons Transactions—Napo Arrangements—Napo Beneficial Ownership."
- (2)
- Includes (i) 2,224,991 shares of common stock held by Kunlun Pharmaceuticals, Ltd., and (ii) 59,333 shares of common stock and warrants to purchase 59,333 shares of common stock held by Sichuan Biopharma. Kunlun Pharmaceuticals, Ltd. is a wholly-owned subsidiary of BioVeda China Fund, or BVCF, and Sichuan Biopharma is an investment vehicle of BVCF's management company. Accordingly, BVCF may be deemed to beneficially own all shares held by Kunlun Pharmaceuticals, Ltd. and Sichuan Biopharma. BVCF's principal business address is Suite 2606, Tower 1, New Richport Center, 763 Mengzi Road, Huangpu District, Shanghai 200023, China.
- (3)
- Includes (i) 370,832 shares of common stock and (ii) warrants to purchase 148,333 shares of common stock, all of which are held by the Bochnowski Family Trust. Does not include shares of common stock issuable upon the closing of this offering upon the conversion of convertible promissory notes in the aggregate principal amount of $200,000, held by the Bochnowski Family Trust. Mr. Bochnowski is a co-trustee and beneficiary of such trust and shares voting and investment control over such shares with his spouse.
- (4)
- Represents 2,343,657 shares of common stock beneficially held by BVCF. Dr. Yang is the Chairman, Founder and Managing Partner of BVCF and he may be deemed to beneficially own all the shares held by BVCF.
106
General
The following is a summary of the rights of our common stock and preferred stock and of certain provisions of our amended and restated certificate of incorporation and amended and restated bylaws, as they will be in effect upon the closing of this offering. This summary is not complete. For more detailed information, please see the amended and restated certificate of incorporation and amended and restated bylaws which are filed as exhibits to the registration statement of which this prospectus is a part.
Immediately upon the closing of this offering, our authorized capital stock will consist of shares, all with a par value of $0.0001 per share, of which shares are designated as common stock.
Upon the closing of this offering, all the outstanding shares of Series A preferred stock will convert into an aggregate of 3,015,902 shares of common stock. Additionally, warrants to purchase an aggregate of 386,498 shares of common stock, if not exercised, will remain outstanding upon the closing of the offering.
Common Stock
Based on (i) 4,311,498 shares of common stock outstanding as of June 30, 2014; (ii) the conversion of all outstanding shares of Series A preferred stock into 3,015,902 shares of common stock upon the closing of this offering; (iii) the issuance of shares of common stock upon the conversion of convertible promissory notes in the aggregate principal amount of $450,000 upon the closing of this offering at a conversion price equal to 80% of the initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, and which shares will be unregistered; and (v) no exercise of outstanding options or warrants, or issuance of shares upon vesting of RSUs, there will be shares of common stock outstanding upon the closing of this offering.
As of June 30, 2014, assuming the conversion of all outstanding shares of Series A preferred stock into common stock upon the closing of this offering, we had 11 record holders of common stock.
As of June 30, 2014, there were 311,498 shares of common stock subject to outstanding warrants with an exercise price of $1.6854 per share and 25,000 shares of common stock subject to outstanding warrants with an exercise price equal to 90% of the initial public offering price. Subsequent to June 30, 2014, we issued a warrant to purchase 50,000 shares of common stock with an exercise price equal to 80% of the initial public offering price.
As of June 30, 2014, there were outstanding options to purchase 1,129,673 shares of common stock with a weighted-average exercise price of $1.77 per share and outstanding RSUs for 118,953 shares of common stock.
Voting Rights
The holders of our common stock are entitled to one vote per share on all matters to be voted on by our stockholders. Subject to preferences that may be applicable to any outstanding shares of preferred stock, holders of common stock are entitled to receive ratably such dividends as may be declared by our board of directors out of funds legally available for that purpose. In the event of our liquidation, dissolution or winding up, the holders of common stock are entitled to share ratably in all assets remaining after the payment of liabilities, subject to the prior distribution rights of preferred stock then outstanding. Holders of common stock have no preemptive, conversion or subscription rights. There are no redemption or sinking fund provisions applicable to the common stock.
107
Dividends
Subject to preferences that may be applicable to any outstanding preferred stock, holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds. For more information, see the section titled "Dividend Policy."
Liquidation
In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any then outstanding shares of preferred stock.
Rights and Preferences
Holders of common stock have no preemptive, conversion, subscription or other rights, and there are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences and privileges of the holders of common stock are subject to and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate in the future.
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued pursuant to this offering, when paid for, will be fully paid and nonassessable.
Warrants
As of June 30, 2014, we had outstanding warrants to purchase an aggregate of 386,498 shares of common stock, 311,498 of which are exercisable at a price of $1.6854 per share, and expire on February 5, 2019, 25,000 of which are exercisable at a price per share equal to 90% of the initial public offering price, and expire June 26, 2019. Subsequent to June 30, 2014, we issued a warrant to purchase 50,000 shares of our common stock at an exercise price per share equal to 80% of the initial public offering price per share, which expires August 26, 2016. These warrants, if not exercised, will remain outstanding following the closing of this offering.
Registration Rights
Pursuant to the investor rights agreement entered into in February 2014 described above under "—Investor Rights Agreement," the current holders of Series A preferred stock have certain registration rights with respect to their shares of common stock, including shares of common stock issuable upon conversion thereof and shares of common stock issued as a dividend or other distribution with respect to or in exchange for or in replacement of the foregoing shares, as described below.
Demand Registration Rights
If at any time beginning 180 days after this offering, the holders of at least 20% of the registrable securities request in writing that we effect a registration with respect to their shares in an offering with an anticipated aggregate offering price of at least $10.0 million, we may be required to register their shares. We are obligated to effect at most four registrations for the holders of registrable securities in response to these demand registration rights. If the holders requesting registration intend to distribute their shares by means of an underwriting, the managing underwriter of such offering will have the right to limit the numbers of shares to be underwritten for reasons related to the marketing of the shares.
108
Piggyback Registration Rights
If we propose to register any shares of our common stock under the Securities Act, subject to certain exceptions, the holders of registrable securities will be entitled to notice of the registration and to include their shares of registrable securities in the registration. If such demand is made by the holders of registrable securities, we must use commercially reasonable efforts to include such holders' shares in the registration. If our proposed registration involves an underwriting, the managing underwriter of such offering will have the right to limit the number of shares to be underwritten for reasons related to the marketing of the shares.
Form S-3 Registration Rights
After this offering, if we become entitled under the Securities Act to register our shares on Form S-3 a holder of registrable securities requests in writing that we register their shares for public resale on Form S-3 in an offering with an anticipated aggregate offering price of at least $1.0 million, we will be required to use commercially reasonable efforts to effect such registration; provided, however, that we will not be required to effect such a registration if, within the preceding 12 months, we have already effected two registrations on Form S-3 for the holders of registrable securities.
Expenses
All expenses incurred in connection with the registration will be borne by us, except for if a demand registration is withdrawn under certain conditions. These expenses may include all registration and filing fees, printing expenses, fees and disbursements of our counsel, reasonable fees and disbursements of a counsel for the selling securityholders, blue sky fees and expenses and the expenses of any regular and special audits incident to the registration.
Termination of Registration Rights
The registration rights terminate upon the later of (i) with respect to the registration rights of an individual holder, when the holder can sell all of such holder's registrable securities in compliance with Rule 144 of the Securities Act within a ninety day period and (ii) five years after the effective date of the registration statement of which this prospectus is a part.
Anti-Takeover Effects of Delaware Law and Our Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Delaware Law
Certain provisions of Delaware law and our amended and restated certificate of incorporation and amended and restated bylaws that will become effective upon the closing of this offering contain provisions that could have the effect of delaying, deferring or discouraging another party from acquiring control of us. These provisions, which are summarized below, are expected to discourage certain types of coercive takeover practices and inadequate takeover bids. These provisions are also designed in part to encourage anyone seeking to acquire control of us to negotiate with our board of directors. We believe that the advantages gained by protecting our ability to negotiate with any unsolicited and potentially unfriendly acquirer outweigh the disadvantages of discouraging such proposals, including those priced above the then-current market value of our common stock, because, among other reasons, the negotiation of such proposals could improve their terms.
109
Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Our amended and restated certificate of incorporation and amended and restated bylaws to become effective in connection with this offering include provisions that:
- •
- require that any action to be taken by our stockholders be effected at a duly called annual or special meeting and not by
written consent;
- •
- specify that special meetings of our stockholders can be called only by our board of directors, the chairman of our board
of directors, the chief executive officer or the president;
- •
- establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our
stockholders, including proposed nominations of persons for election to our board of directors;
- •
- provide that directors may be removed only for cause;
- •
- provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though
less than a quorum;
- •
- establish that our board of directors is divided into three classes, Class I, Class II and Class III,
with each class serving staggered terms;
- •
- specify that no stockholder is permitted to cumulate votes at any election of our board of directors; and
- •
- require a super majority of the stockholders and a majority of the board of directors to amend certain of the above-mentioned provisions.
Exclusive Jurisdiction
Under the provisions of our amended and restated certificate of incorporation to become effective upon the closing of this offering, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for: (i) any derivative action or proceeding brought on behalf of us; (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees or agents to us or our stockholders; (iii) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law or our amended and restated certificate of incorporation or amended and restated bylaws; or (iv) any action asserting a claim against us governed by the internal affairs doctrine. The enforceability of similar choice of forum provisions in other companies' certificates of incorporation has been challenged in legal proceedings, and it is possible that, in connection with any action, a court could find the choice of forum provisions contained in our amended and restated certificate of incorporation to be inapplicable or unenforceable in such action.
Delaware Anti-Takeover Statute
We are subject to the provisions of Section 203 of the Delaware General Corporation Law regulating corporate takeovers. In general, Section 203 prohibits a publicly-held Delaware corporation from engaging, under certain circumstances, in a business combination with an interested stockholder for a period of three years following the date the person became an interested stockholder unless:
- •
- prior to the date of the transaction, our board of directors of the corporation approved either the business combination
or the transaction which resulted in the stockholder becoming an interested stockholder;
- •
- upon the closing of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the
110
- •
- at or subsequent to the date of the transaction, the business combination is approved by our board of directors of the corporation and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock which is not owned by the interested stockholder.
corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding, but not for determining the outstanding voting stock owned by the interested stockholder, (1) shares owned by persons who are directors and also officers, and (2) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
Generally, a business combination includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. An interested stockholder is a person who, together with affiliates and associates, owns or, within three years prior to the determination of interested stockholder status, did own 15% or more of a corporation's outstanding voting stock. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage business combinations or other attempts that might result in the payment of a premium over the market price for the shares of common stock held by our stockholders.
The provisions of Delaware law and our restated certificate of incorporation and amended and restated bylaws to become effective upon the closing of this offering could have the effect of discouraging others from attempting hostile takeovers and, as a consequence, they may also inhibit temporary fluctuations in the market price of our common stock that often result from actual or rumored takeover attempts. These provisions may also have the effect of preventing changes in our management. It is possible that these provisions could make it more difficult to accomplish transactions that stockholders may otherwise deem to be in their best interests.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare Trust Company N.A. The transfer agent and registrar's address is 250 Royall St., Canton, MA 02021. The transfer agent's telephone number is (800) 962-4284.
Listing
We have applied to have our common stock listed on The NASDAQ Capital Market under the symbol "JAGX."
111
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no public market for our common stock, and although we expect that our common stock will be approved for listing on The NASDAQ Capital Market, we cannot assure you that there will be an active public market for our common stock following this offering. We cannot predict what effect, if any, sales of our shares in the public market or the availability of shares for sale will have on the market price of our common stock. Future sales of substantial amounts of common stock in the public market, including shares issued upon exercise of outstanding options, or the perception that such sales may occur, however, could adversely affect the market price of our common stock and also could adversely affect our future ability to raise capital through the sale of our common stock or other equity-related securities at times and prices we believe appropriate.
Upon the closing of this offering, based on our shares outstanding as of June 30, 2014 and after giving effect to (i) the conversion of all outstanding shares of Series A preferred stock into an aggregate of 3,015,902 shares of common stock upon the closing of this offering; (ii) the conversion of all outstanding convertible promissory notes into shares of common stock at a conversion price of 80% of the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus; and (iii) the issuance of shares of common stock being offered hereby, shares of common stock will be outstanding. All of the shares of common stock to be sold in this offering will be freely tradable without restriction or further registration under the Securities Act unless held by our "affiliates," as that term is defined in Rule 144 under the Securities Act. The remaining outstanding shares of our common stock will be deemed "restricted securities" as that term is defined under Rule 144. Restricted securities may be sold in the public market only if their offer and sale is registered under the Securities Act or if the offer and sale of those securities qualify for an exemption from registration, including exemptions provided by Rules 144 and 701 under the Securities Act, which are summarized below.
As a result of the lock-up agreements and market stand-off provisions described below and the provisions of Rules 144 or 701, the shares of common stock that will be deemed "restricted securities" will be available for sale in the public market following the closing of this offering as follows:
- •
- no shares will be eligible for sale on the date of this prospectus; and
- •
- approximately shares will be eligible for sale upon expiration of the lock-up agreements and market stand-off provisions described below, beginning more than 180 days after the date of this prospectus, subject in some cases to applicable volume limitations under Rule 144.
We may issue shares of common stock from time to time for a variety of corporate purposes, including in capital-raising activities through future public offerings or private placements, in connection with exercise of stock options, vesting of restricted stock units and other issuances relating to our employee benefit plans and as consideration for future acquisitions, investments or other purposes. The number of shares of common stock that we may issue may be significant, depending on the events surrounding such issuances. In some cases, the shares we issue may be freely tradable without restriction or further registration under the Securities Act; in other cases, we may grant registration rights covering the shares issued in connection with these issuances, in which case the holders of common stock will have the right, under certain circumstances, to cause us to register any resale of such shares to the public.
Lock-up Agreements
We and each of our directors and executive officers and holders of substantially all of our outstanding capital stock have agreed that, without the prior written consent of BMO Capital Markets Corp. and Guggenheim Securities, LLC, on behalf of the underwriters, we and they will not, subject to
112
limited exceptions that are described in "Underwriting" below, during the period ending 180 days after the date of this prospectus:
- •
- offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell,
grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, directly or indirectly, any shares of our common stock or any securities convertible into or exercisable or
exchangeable for common stock; or
- •
- enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of our common stock,
whether any transaction described above is to be settled by delivery of our common stock or such other securities, in cash or otherwise.
Upon the expiration of the applicable lock-up periods, substantially all of the shares subject to such lock-up restrictions will become eligible for sale, subject to the limitations discussed above.
Rule 144
In general, under Rule 144, beginning 90 days after the date of this prospectus, a person who is not our affiliate and has not been our affiliate for purposes of the Securities Act at any time during the preceding three months will be entitled to sell any shares of common stock that such person has beneficially owned for at least six months, including the holding period of any prior owner other than one of our affiliates, subject only to the availability of current public information about us. Sales of common stock by any such person would be subject to the availability of current public information about us if the shares to be sold were beneficially owned by such person for less than one year.
In addition, under Rule 144, a person may sell shares of common stock acquired from us immediately upon the closing of this offering, without regard to the registration requirements of the Securities Act or the availability of public information about us, if:
- •
- the person is not our affiliate and has not been our affiliate at any time during the preceding three months; and
- •
- the person has beneficially owned the shares to be sold for at least one year, including the holding period of any prior owner other than one of our affiliates.
Beginning 90 days after the date of this prospectus, our affiliates who have beneficially owned shares of common stock for at least six months, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell within any three-month period a number of shares that does not exceed the greater of:
- •
- 1% of the number of shares of our common stock then outstanding, which will equal approximately shares
immediately after this offering; and
- •
- the average weekly trading volume in our common stock on The NASDAQ Capital Market during the four calendar weeks preceding the date of filing of a notice on Form 144 with respect to the sale.
Sales under Rule 144 by our affiliates are also subject to manner of sale provisions and notice requirements and to the availability of current public information about us. To the extent that shares were acquired from one of our affiliates, a person's holding period for the purpose of effecting a sale under Rule 144 would commence on the date of transfer from the affiliate.
113
Rule 701
In general, under Rule 701, any of an issuer's employees, directors, officers, consultants or advisors who purchases shares from the issuer in connection with a compensatory stock or option plan or other written agreement before the effective date of a registration statement under the Securities Act is entitled to sell such shares 90 days after such effective date in reliance on Rule 144. An affiliate of the issuer can resell shares in reliance on Rule 144 without having to comply with the holding period requirement, and non-affiliates of the issuer can resell shares in reliance on Rule 144 without having to comply with the current public information and holding period requirements.
The Securities and Exchange Commission has indicated that Rule 701 will apply to typical stock options granted by an issuer before it becomes subject to the reporting requirements of the Exchange Act, along with the shares acquired upon exercise of such options, including exercises after an issuer becomes subject to the reporting requirements of the Exchange Act.
As of September 30, 2014, no shares of our outstanding common stock had been issued in reliance on Rule 701. If options are exercised or shares are issued upon the vesting of RSUs, any such shares will be subject to lock-up agreements as discussed above, and, as a result, these shares will only become eligible for sale at the earlier of the expiration of the lock-up period or upon obtaining the consent of BMO Capital Markets Corp. and Guggenheim Securities, LLC, on behalf of the underwriters to release all or any portion of these shares from the lock-up agreements.
Equity Plan Awards
As of June 30, 2014, we had options to purchase 1,129,673 shares of common stock with a weighted-average exercise price of $1.77 per share and RSUs for 118,953 shares of common stock outstanding. We intend to file one or more registration statements on Form S-8 under the Securities Act to register the offer and sale of all shares of common stock subject to outstanding stock options and RSUs and all shares issuable under our stock plans. We expect to file the registration statement covering these shares after the date of this prospectus, which will permit the resale of such shares by persons who are non-affiliates of ours in the public market without restriction under the Securities Act, subject, with respect to certain of the shares, to the provisions of the lock-up agreements and market stand-off provisions described above.
Warrants
See "Description of Capital Stock—Warrants" for additional information. Such shares issued upon exercise of the warrants may be able to be sold after the expiration of the lock-up period described above subject to the requirements of Rule 144 described above.
Registration Rights
Upon the closing of this offering, the holders of approximately shares of common stock, will be eligible to exercise certain rights to cause us to register their shares of common stock for resale under the Securities Act, subject to various conditions and limitations. These registration rights are described under the caption "Description of Capital Stock—Registration Rights." Upon the effectiveness of a registration statement covering these shares, the shares would become freely tradable without restriction under the Securities Act.
114
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS OF COMMON STOCK
The following is a general discussion of the material U.S. federal income tax consequences applicable to a non-U.S. holder (as defined below) with respect to the acquisition, ownership and disposition of our common stock. This discussion is limited to non-U.S. holders who purchase our common stock issued pursuant to this offering for cash and who hold our common stock as a "capital asset" within the meaning of Section 1221 of the Internal Revenue Code of 1986, as amended, or the Code (generally, property held for investment). This discussion is based upon the applicable provisions of the Code, applicable U.S. Treasury regulations promulgated thereunder, or the Treasury Regulations, and administrative and judicial interpretations thereof, promulgated thereunder, all as in effect on the date hereof, and all of which are subject to change, possibly on a retroactive basis. Any such changes could alter the tax consequences to non-U.S. holders described herein. This discussion is not a complete analysis of all of the potential U.S. federal income tax consequences applicable to a non-U.S. holder, and does not address all of the U.S. federal income tax consequences that may be relevant to a particular non-U.S. holder in light of such non-U.S. holder's particular circumstances or the U.S. federal income tax consequences applicable to non-U.S. holders that are subject to special rules, such as United States expatriates, banks, financial institutions, insurance companies, regulated investment companies, real estate investment trusts, controlled foreign corporations, passive foreign investment companies, corporations that accumulate earnings to avoid U.S. federal income tax, brokers, dealers or traders in securities, commodities or currencies, partnerships or other pass-through entities (or investors in such entities), tax-exempt organizations, tax-qualified retirement plans, persons subject to the alternative minimum tax, and non-U.S. holders that hold our common stock as part of a straddle, hedge, conversion transaction or other integrated investment. In addition, this discussion does not describe any state or local income, estate or other tax consequences of holding and disposing of our common stock.
As used in this discussion, the term "non-U.S. holder" means any beneficial owner of our common stock that is, for U.S. federal income tax purposes, neither a partnership nor any of the following:
- •
- an individual citizen or resident of the United States;
- •
- a corporation or other entity taxable as a corporation created or organized under the laws of the United States or any
political subdivision thereof;
- •
- an estate, the income of which is subject to U.S. federal income tax regardless of its source; or
- •
- a trust if (i) a United States court is able to exercise primary supervision over the administration of the trust and one or more United States persons have authority to control all substantial decisions of the trust or (ii) the trust has a valid election in effect under applicable Treasury Regulations to be treated as a United States person.
If any entity classified as a partnership for U.S. federal income tax purposes holds our common stock, the tax treatment of a partner in such partnership generally will depend on the status of the partner and the activities of the partnership. Partnerships and their partners should consult their tax advisors as to the tax consequences to them of the acquisition, ownership and disposition of our common stock.
THE FOLLOWING DISCUSSION IS FOR GENERAL INFORMATION ONLY AND IS NOT TAX ADVICE. PROSPECTIVE INVESTORS ARE URGED TO CONSULT THEIR TAX ADVISORS REGARDING THE PARTICULAR U.S. FEDERAL, STATE, LOCAL AND FOREIGN TAX CONSEQUENCES TO THEM OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF OUR COMMON STOCK.
115
Distributions on Common Stock
Distributions on our common stock generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will be treated first as a tax-free return of a capital to the extent of the non-U.S. holder's adjusted tax basis in the common stock below zero, and thereafter as capital gain, subject to the tax treatment described under "—Sale, Exchange or Other Disposition of Our Common Stock," below.
Subject to the discussions below regarding backup withholding and FATCA, the gross amount of dividends paid to a non-U.S. holder of our common stock that are not effectively connected with a U.S. trade or business conducted by such non-U.S. holder generally will be subject to U.S. federal withholding tax at a rate of 30%, or such lower rate specified by an applicable income tax treaty if we have received proper certification as to the application of such treaty. If a non-U.S. holder holds our common stock in connection with the conduct of a trade or business within the United States, and dividends paid on our common stock are effectively connected with such non-U.S. holder's U.S. trade or business (and, if under an applicable income tax treaty, such dividends are attributable to a permanent establishment or fixed base maintained by the non-U.S. holder within the United States), such non-U.S. holder generally will be subject to U.S. federal income tax at ordinary U.S. federal income tax rates (on a net income basis), and such dividends will not be subject to the U.S. federal withholding tax described above. In the case of a non-U.S. holder that is a corporation, such non-U.S. holder may also be subject to a 30% "branch profits tax" unless such corporate non-U.S. holder qualifies for a lower rate under an applicable income tax treaty.
In general, to claim the benefit of any applicable income tax treaty or an exemption from U.S. federal withholding because the income is effectively connected with the conduct of a trade or business within the United States, a non-U.S. holder must provide a properly executed Internal Revenue Service, or IRS, Form W-8BEN for treaty benefits or IRS Form W-8ECI for effectively connected income (or such successor form as the IRS designates), before the distributions are made. These forms must be updated periodically. If you are a non-U.S. holder, you may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. Non-U.S. holders should consult their tax advisers regarding their entitlement to benefits under an applicable income tax treaty and the specific manner of claiming the benefits of such treaty.
Sale, Exchange or Other Disposition of Common Stock
Subject to the discussions below regarding backup withholding and FATCA, a non-U.S. holder generally will not be subject to U.S. federal income tax on any gain realized upon the sale, exchange or other disposition (collectively, a "disposition") of our common stock, unless:
- •
- the gain is effectively connected with the non-U.S. holder's conduct of a trade or business within the United States, and
if an income tax treaty applies, is attributable to a permanent establishment maintained by the non-U.S. holder within the United States;
- •
- the non-U.S. holder is an individual who is present in the United States for 183 days or more during the taxable
year of the disposition and certain other requirements are met; or
- •
- we are or have been a U.S. real property holding corporation, or USRPHC, for U.S. federal income tax purposes at any time within the shorter of (i) the five-year period ending on the date of the disposition of our common stock or (ii) the non-U.S. holder's holding period for our common stock.
If the gain is described in the first bullet point above, the non-U.S. holder generally will be subject to U.S. federal income tax on a net income basis with respect to such gain in the same manner as if
116
such non-U.S. holder were a United States person. In addition, if the non-U.S. holder is a corporation for U.S. federal income tax purposes, such gain may be subject to a 30% branch profits tax unless such corporate non-U.S. holder qualifies for a lower rate under an applicable income tax treaty.
A non-U.S. holder described in the second bullet point above generally will be subject to U.S. federal income tax with respect to such gain at a flat 30% rate (or such lower rate specified by an applicable income tax treaty), which may be offset by U.S. source capital losses of the non-U.S. holder during the taxable year of disposition (even though the individual is not considered a resident of the United States), provided that the non-U.S. holder has timely filed U.S. federal income tax returns with respect to such losses.
With respect to the third bullet point above, we believe that we are not currently, and we do not anticipate becoming, a USRPHC. However, because the determination of whether we are a USRPHC depends on the fair market value of our U.S. real property interests relative to the fair market value of our other business assets and our non-U.S. real property interests, there can be no assurance that we will not become a USRPHC in the future. In general, a corporation is a USRPHC if the fair market value of its "United States real property interests" (as defined in the Code) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests and its other assets used or held for use in a trade or business. Even if we are or become a USRPHC, a non-U.S. holder would not be subject to U.S. federal income tax on a sale, exchange or other taxable disposition of shares of our common stock by reason of our status as a USRPHC so long as (i) shares of our common stock continue to be regularly traded on an established securities market (within the meaning of Section 897(c)(3) of the Code) during the calendar year in which such disposition occurs and (ii) such non-U.S. holder does not own and is not deemed to own (directly, indirectly or constructively) more than 5% of the shares of our common stock at any time during the shorter of the five-year period ending on the date of the disposition of our common stock or the non-U.S. holder's holding period for our common stock. If gain on the disposition of our common stock were subject to taxation under the third bullet point above, the non-U.S. holder generally would be subject to U.S. federal income tax with respect to such gain in the same manner as gain that is effectively connected with the conduct of a U.S. trade or business (as described above), except that the branch profits tax generally would not apply.
Information Reporting and Backup Withholding
In general, a non-U.S. holder will be required to comply with certain certification procedures to establish that such holder is not a United States person in order to avoid backup withholding with respect to dividends or the proceeds from disposition of common stock. In addition, we are required to report annually to the IRS the amount of any dividends paid to a non-U.S. holder, regardless of whether we actually withheld any tax. Copies of the information returns reporting such dividends and the amount withheld may also be made available to the tax authorities in the country in which the non-U.S. holder resides under the provisions of an applicable income tax treaty.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit against a non-U.S. holder's U.S. federal income tax liability, provided the required information is timely furnished to the IRS.
Foreign Accounts Tax Compliance Act
Under the Foreign Account Tax Compliance Act, as modified by Treasury Regulations and subject to any official interpretations thereof, any applicable intergovernmental agreement between the United States and a non-U.S. government to implement these rules and improve international tax compliance, or any fiscal or regulatory legislation or rules adopted pursuant to any such agreement (collectively, "FATCA"), after June 30, 2014, withholding at a rate of 30% will be required on dividends in respect of, and, after December 31, 2016, gross proceeds from the disposition of, our common stock held by or
117
through certain foreign financial institutions (including investment funds), unless such institution enters into an agreement with the Secretary of the Treasury to report, on an annual basis, information with respect to interests in, and accounts maintained by, the institution to the extent such interests or accounts are held by certain United States persons and by certain non-U.S. entities that are wholly or partially owned by United States persons and to withhold on certain payments. An intergovernmental agreement between the United States and an applicable foreign country, or future Treasury Regulations or other guidance, may modify these requirements. Accordingly, the entity through which our common stock is held will affect the determination of whether such withholding is required. Similarly, dividends in respect of, and gross proceeds from the sale of, our common stock held by an investor that is a non-financial non-U.S. entity that does not qualify under certain exemptions will be subject to withholding at a rate of 30%, unless such entity either (i) certifies to us that such entity does not have any "substantial United States owners" or (ii) provides certain information regarding the entity's "substantial United States owners," which we will provide to Secretary of the Treasury. We will not pay any additional amounts to holders in respect of any amounts withheld. Prospective investors are urged to consult their tax advisors regarding the possible implications of FATCA on their investment in our common stock.
118
BMO Capital Markets Corp. and Guggenheim Securities, LLC are acting as representatives of each of the underwriters named below. Subject to the terms and conditions of an underwriting agreement among us and the underwriters, we have agreed to sell to the underwriters, and each underwriter has agreed, severally and not jointly, to purchase from us, the number of shares of common stock shown opposite its name in the table below:
Underwriter
|
Number of Shares | |||
|---|---|---|---|---|
BMO Capital Markets Corp. |
||||
Guggenheim Securities, LLC |
||||
Roth Capital Partners, LLC |
||||
| | | | | |
Total |
||||
| | | | | |
| | | | | |
The underwriting agreement provides that the obligations of the underwriters are subject to certain conditions precedent including approval of certain legal matters by their counsel. The underwriting agreement provides that the underwriters will purchase all of the shares of common stock if any of them are purchased. If an underwriter defaults, the underwriting agreement provides that the purchase commitments of the non-defaulting underwriters may be increased or the underwriting agreement may be terminated. We have agreed to indemnify the underwriters and certain of their controlling persons against certain liabilities, including liabilities under the Securities Act, and to contribute to payments that the underwriters may be required to make in respect of those liabilities.
The underwriters are offering the shares of common stock subject to their acceptance of the shares of common stock from us and subject to prior sale. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part. In addition, the underwriters have advised us that they do not intend sales to discretionary accounts to exceed 5% of the total number of shares of common stock being offered.
Option to Purchase Additional Shares
We have granted to the underwriters an option, exercisable for 30 days from the date of this prospectus, to purchase, from time to time, in whole or in part, up to an aggregate of additional shares from us at the public offering price set forth on the cover page of this prospectus, less underwriting discounts and commissions. The underwriters may exercise this option solely for the purpose of covering over-allotments, if any, made in connection with the offering of the shares of common stock offered by this prospectus. If the underwriters exercise this option, each underwriter will be obligated, subject to specified conditions, to purchase a number of additional shares proportionate to that underwriter's initial purchase commitment as indicated in the table above.
Commission and Expenses
The underwriters have advised us that they propose to offer the shares of common stock to the public at the initial public offering price set forth on the cover page of this prospectus and to certain dealers, which may include the underwriters, at that price less a concession not in excess of $ per share of common stock. The underwriters may allow, and certain dealers may re-allow, a discount from the concession not in excess of $ per share of common stock to certain brokers and dealers. After the offering, the offering price, concession and reallowance to dealers may be varied from time to time by the representative.
119
The following table shows the public offering price, the underwriting discounts and comissions and proceeds, before expenses, to us in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the underwriters' option to purchase additional shares.
| |
Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Per Share | Without Option to Purchase Additional Shares |
With Exercise of Option to Purchase Additional Shares in Full |
|||||||
Public offering price |
$ | $ | $ | |||||||
Underwriting discounts and commissions paid by us |
$ | $ | $ | |||||||
| | | | | | | | | | | |
Proceeds to us, before expenses |
$ | $ | $ | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
We estimate expenses payable by us in connection with this offering, other than the underwriting discounts and commissions referred to above, will be approximately $ , which includes legal, accounting and printing costs and various other fees associated with the registration and listing of our common stock. We have also agreed to reimburse the underwriters for certain of their expenses in an amount up to $20,000.
No Sales of Similar Securities
We, our executive officers, directors and holders of substantially all of our outstanding capital stock have agreed, subject to specified exceptions, not to directly or indirectly, for a period of 180 days after the date of this prospectus, without the prior written consent of the representatives of the underwriters:
- •
- sell, offer, contract or grant any option to sell (including any short sale), pledge, transfer, establish an open "put
equivalent position" within the meaning of Rule 16a-l(h) under the Exchange Act; or
- •
- otherwise dispose of any shares of common stock, options or warrants to acquire shares of common stock, or securities
exchangeable or exercisable for or convertible into shares of common stock, currently or hereafter owned either of record or beneficially; or
- •
- publicly announce an intention to do any of the foregoing or make any demand or exercise any right with respect to the registration of any shares of our common stock or any security convertible into or exchangeable for our common stock.
However, in the case of our officers, directors and stockholders, these lock-up restrictions will not apply to, subject to certain restrictions:
- •
- bona fide gifts made by the holder;
- •
- the surrender or forfeiture of shares of common stock to us to satisfy tax withholding obligations upon exercise or
vesting of stock options or equity awards;
- •
- transfers of common stock or any security convertible into or exercisable for common stock to an immediate family member,
an immediate family member of a domestic partner or a trust for the benefit of the undersigned, a domestic partner or an immediate family member;
- •
- transfers of shares of common stock or any security convertible into or exercisable for common stock to any corporation, partnership, limited liability company or other entity all of the beneficial ownership interests of which are held exclusively by the holder, a domestic
120
- •
- transfers of shares of common stock or any security convertible into or exercisable for common stock upon death by will or
intestate succession;
- •
- distributions of shares of common stock or securities convertible into or exercisable for common stock to members,
partners or stockholders of the holder;
- •
- securities transferred to one or more affiliates of the holder and distributions of securities to partners, members or
stockholders of the holder;
- •
- transactions relating to securities purchased in open market transactions after the date of this prospectus;
- •
- entry into a trading plan established pursuant to Rule 10b5-1 under the Exchange Act, provided that such plan does
not provide for any sales or other dispositions of shares of common stock during the 180-day restricted period;
- •
- any shares purchased by the holder in this offering; or
- •
- securities transferred pursuant to a bona fide third-party tender offer, merger, consolidation or other similar transaction made to all holders of our common stock and involving a change of control of the company.
partner and/or one or more family members of the holder or the holder's domestic partner in a transaction not involving a disposition for value;
The representatives may, in their sole, joint discretion and at any time or from time to time before the termination of the 180-day period release all or any portion of the securities subject to lock-up agreements. There are no existing agreements between the underwriters and any of our stockholders who will execute a lock-up agreement, providing consent to the sale of shares prior to the expiration of the lock-up period.
Listing
We have applied to list our common stock on The NASDAQ Capital Market under the symbol "JAGX." In order to meet the requirements for listing on that exchange, the underwriters have undertaken to sell a minimum number of shares to a minimum number of beneficial owners as required by that exchange.
Prior to this offering, there has been no public market for our common stock. The initial public offering price will be determined through negotiations between us and the representative. In determining the initial public offering price, we and the representative expect to consider a number of factors, including:
- •
- the information set forth in this prospectus and otherwise available to the underwriters;
- •
- our prospects for future earnings;
- •
- our prospects and the history and prospects of the industry in which we compete;
- •
- an assessment of our management;
- •
- the present state of our development;
- •
- the general condition of the securities markets at the time of this offering; and
- •
- the recent market prices of, and demand for, publicly traded common stock of generally comparable companies.
121
We and the underwriters cannot assure you that an active trading market for the shares will develop or that shares will trade in the public market at or above the initial public offering price after this offering.
Stabilization
The underwriters have advised us that they may engage in short sale transactions, stabilizing transactions, syndicate covering transactions or the imposition of penalty bids in connection with this offering. These activities may have the effect of stabilizing or maintaining the market price of the common stock at a level above that which might otherwise prevail in the open market. Establishing short sales positions may involve either "covered" short sales or "naked" short sales. "Covered" short sales are sales made in an amount not greater than the underwriters' option to purchase additional shares of our common stock in this offering. The underwriters may close out any covered short position by either exercising their option to purchase additional shares of our common stock or purchasing shares of our common stock in the open market. In determining the source of shares to close out the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the option to purchase additional shares. "Naked" short sales are sales in excess of the option to purchase additional shares of our common stock. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of the shares of our common stock in the open market after pricing that could adversely affect investors who purchase in this offering. A penalty bid is an arrangement permitting the underwriters to reclaim the selling concession otherwise accruing to a syndicate member in connection with the offering if the common stock originally sold by such syndicate member are purchased in a syndicate covering transaction and therefore have not been effectively placed by such syndicate member.
Similar to other purchase transactions, the underwriter's purchases to cover the syndicate short sales may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. The underwriters may conduct these transactions on the NASDAQ Stock Market, in the over-the-counter market or otherwise.
Neither we, nor any of the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common stock. The underwriters are not obligated to engage in these activities and, if commenced, any of the activities may be discontinued at any time.
Electronic Distribution
A prospectus in electronic format may be made available by e-mail or through online services maintained by one or more of the underwriters or their affiliates. In those cases, prospective investors may view offering terms online and may be allowed to place orders online. The underwriters may agree with us to allocate a specific number of shares of common stock for sale to online brokerage account holders. Any such allocation for online distributions will be made by the underwriters on the same basis as other allocations. Other than the prospectus in electronic format, the information on the underwriters' web sites and any information contained in any other web site maintained by any of the underwriters is not part of this prospectus, has not been approved and/or endorsed by us or the underwriters and should not be relied upon by investors.
122
Other Activities and Relationships
The underwriters and certain of their affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The underwriters and certain of their affiliates may in the future perform, various commercial and investment banking and financial advisory services for us and our affiliates, for which they received or will receive customary fees and expenses.
In the ordinary course of their various business activities, the underwriters and their respective affiliates, officers, directors and employees may purchase, sell or hold a broad array of investments and actively trade securities, derivatives, loans, commodities, currencies, credit default swaps and other financial instruments for their own account and for the accounts of their customers, and such investment and trading activities may involve or relate to our securities.
Disclaimers about Non-U.S. Jurisdictions
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a "Relevant Member State") an offer to the public of any shares of our common stock may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any shares of our common stock may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
- (a)
- to
any legal entity which is a qualified investor as defined in the Prospectus Directive;
- (b)
- to
fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons
(other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer;
or
- (c)
- in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares of our common stock shall result in a requirement for the publication by us or any underwriter of a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an "offer to the public" in relation to any shares of our common stock in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any shares of our common stock to be offered so as to enable an investor to decide to purchase any shares of our common stock, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State, the expression "Prospectus Directive" means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State), and includes any relevant implementing measure in the Relevant Member State, and the expression "2010 PD Amending Directive" means Directive 2010/73/EU.
United Kingdom
Each underwriter has represented and agreed that:
- (a)
- it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000 (the "FSMA"))
123
- (b)
- it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares of our common stock in, from or otherwise involving the United Kingdom.
received by it in connection with the issue or sale of the shares of our common stock in circumstances in which Section 21(1) of the FSMA does not apply to us; and
Canada
The common shares may be sold only to purchasers purchasing as principal that are both "accredited investors" as defined in National Instrument 45-106 Prospectus and Registration Exemptions and "permitted clients" as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the common shares must be made in accordance with an exemption from the prospectus requirements and in compliance with the registration requirements of applicable securities laws.
Notice to Residents of Germany
Each person who is in possession of this prospectus is aware of the fact that no German securities prospectus (wertpapierprospekt) within the meaning of the securities prospectus act (wertpapier-prospektgesetz, the "act") of the federal republic of Germany has been or will be published with respect to the shares of our common stock. In particular, each underwriter has represented that it has not engaged and has agreed that it will not engage in a public offering in the federal republic of Germany (ôffertliches angebot) within the meaning of the act with respect to any of the shares of our common stock otherwise than in accordance with the act and all other applicable legal and regulatory requirements.
Hong Kong
The common shares may not be offered or sold in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), or (ii) to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a "prospectus" within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to common shares which are or are intended to be disposed of only to persons outside Hong Kong or only to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
Notice to Residents of the Netherlands
The offering of the shares of our common stock is not a public offering in The Netherlands. The shares of our common stock may not be offered or sold to individuals or legal entities in The Netherlands unless (i) a prospectus relating to the offer is available to the public, which has been approved by the Dutch Authority for the Financial Markets (Autoriteit Financiële Markten) or by the competent supervisory authority of another state that is a member of the European Union or party to the Agreement on the European Economic Area, as amended or (ii) an exception or exemption applies to the offer pursuant to Article 5:3 of The Netherlands Financial Supervision Act (Wet op het financieel toezicht) or Article 53 paragraph 2 or 3 of the Exemption Regulation of the Financial
124
Supervision Act, for instance due to the offer targeting exclusively "qualified investors" (gekwalificeerde beleggers) within the meaning of Article 1:1 of The Netherlands Financial Supervision Act.
Notice to Residents of Japan
The underwriters will not offer or sell any of the shares of our common stock directly or indirectly in Japan or to, or for the benefit of, any Japanese person or to others, for re-offering or re-sale directly or indirectly in Japan or to any Japanese person, except in each case pursuant to an exemption from the registration requirements of, and otherwise in compliance with, the Financial Instruments and Exchange Law of Japan and any other applicable laws and regulations of Japan. For purposes of this paragraph, "Japanese person" means any person resident in Japan, including any corporation or other entity organized under the laws of Japan.
Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the common shares may not be circulated or distributed, nor may the common shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the "SFA"), (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with conditions set forth in the SFA.
Where the common shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
- (a)
- a
corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the
entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or
- (b)
- a
trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who
is an accredited investor, shares, debentures and units of shares and debentures of that corporation or the beneficiaries' rights and interest (howsoever described) in that trust shall not be
transferred within six months after that corporation or that trust has acquired the common shares pursuant to an offer made under Section 275 of the SFA except:
- (1)
- to
an institutional investor (for corporations, under Section 274 of the SFA) or to a relevant person defined in Section 275o(2) of the SFA,
or to any person pursuant to an offer that is made on terms that such shares, debentures and units of shares and debentures of that corporation or such rights and interest in that trust are acquired
at a consideration of not less than S$200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other
assets, and further for corporations, in accordance with the conditions specified in Section 275 of the SFA;
- (2)
- where
no consideration is or will be given for the transfer; or
- (3)
- where the transfer is by operation of law.
125
Switzerland
The common shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (the "SIX") or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the common shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the common shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of common shares will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of common shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes ("CISA"). Accordingly, no public distribution, offering or advertising, as defined in CISA, its implementing ordinances and notices, and no distribution to any non-qualified investor, as defined in CISA, its implementing ordinances and notices, shall be undertaken in or from Switzerland, and the investor protection afforded to acquirers of interests in collective investment schemes under CISA does not extend to acquirers of common shares.
United Arab Emirates
This offering has not been approved or licensed by the Central Bank of the United Arab Emirates (the "UAE"), Securities and Commodities Authority of the UAE and/or any other relevant licensing authority in the UAE including any licensing authority incorporated under the laws and regulations of any of the free zones established and operating in the territory of the UAE, in particular the Dubai Financial Services Authority ("DFSA"), a regulatory authority of the Dubai International Financial Centre ("DIFC"). The offering does not constitute a public offer of securities in the UAE, DIFC and/or any other free zone in accordance with the Commercial Companies Law, Federal Law No 8 of 1984 (as amended), DFSA Offered Securities Rules and NASDAQ Dubai Listing Rules, accordingly, or otherwise. The common shares may not be offered to the public in the UAE and/or any of the free zones.
The common shares may be offered and issued only to a limited number of investors in the UAE or any of its free zones who qualify as sophisticated investors under the relevant laws and regulations of the UAE or the free zone concerned.
France
This prospectus (including any amendment, supplement or replacement thereto) is not being distributed in the context of a public offering in France within the meaning of Article L. 411-1 of the French Monetary and Financial Code (Code monétaire et financier).
This prospectus has not been and will not be submitted to the French Autorité des marchés financiers (the "AMF") for approval in France and accordingly may not and will not be distributed to the public in France.
Pursuant to Article 211-3 of the AMF General Regulation, French residents are hereby informed that:
- 1.
- the transaction does not require a prospectus to be submitted for approval to the AMF;
126
- 2.
- persons
or entities referred to in Point 2°, Section II of Article L.411-2 of the Monetary and Financial Code may take part in the
transaction solely for their own account, as provided in Articles D. 411-1, D. 734-1, D. 744-1, D. 754-1 and D. 764-1 of the Monetary and Financial Code; and
- 3.
- the financial instruments thus acquired cannot be distributed directly or indirectly to the public otherwise than in accordance with Articles L. 411-1, L. 411-2, L. 412-1 and L. 621-8 to L. 621-8-3 of the Monetary and Financial Code.
This prospectus is not to be further distributed or reproduced (in whole or in part) in France by the recipients of this prospectus. This prospectus has been distributed on the understanding that such recipients will only participate in the issue or sale of our common stock for their own account and undertake not to transfer, directly or indirectly, our common stock to the public in France, other than in compliance with all applicable laws and regulations and in particular with Articles L. 411-1 and L. 411-2 of the French Monetary and Financial Code.
127
The validity of the securities being offered by this prospectus will be passed upon for us by Reed Smith LLP, San Francisco, California. Cooley LLP, New York, New York, is representing the underwriters in connection with this offering.
The financial statements as of December 31, 2013 and for the period from June 6, 2013 (inception) through December 31, 2013 included in this prospectus and the registration statement, have been so included in reliance on the report of BDO USA, LLP, an independent registered public accounting firm (the report on the financial statements contains an explanatory paragraph regarding the Company's ability to continue as a going concern), appearing elsewhere herein and in the registration statement, given on the authority of said firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC under the Securities Act a registration statement on Form S-1 with respect to the common stock offered by this prospectus. This prospectus, which constitutes part of the registration statement, does not contain all the information set forth in the registration statement or the exhibits and schedules which are part of the registration statement, portions of which are omitted as permitted by the rules and regulations of the SEC. Statements made in this prospectus regarding the contents of any contract or other document are summaries of the material terms of the contract or document. With respect to each contract or document filed as an exhibit to the registration statement, please see the copy of the contract or document that has been filed. For further information pertaining to us and the common stock offered by this prospectus, reference is made to the registration statement, including the exhibits and schedules thereto, copies of which may be inspected without charge at the public reference facilities of the SEC at 100 F Street, NE., Room 1580, Washington, D.C. 20549, as may the other reports, statements and information we file with the SEC. Copies of all or any portion of the registration statement may be obtained from the SEC at prescribed rates. Information on the public reference facilities may be obtained by calling the SEC at 1-800-SEC-0330. In addition, the SEC maintains a website that contains reports, proxy and information statements and other information that is filed through the SEC's EDGAR System. The website can be accessed at http.//www.sec.gov.
As a result of this offering, we will become subject to the information and reporting requirements of the Exchange Act and, in accordance with this law, will file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements, and other information will be available for inspection and copying at the SEC's public reference facilities and the website of the SEC referred to above. We also maintain a website at www.jaguaranimalhealth.com. After the closing of this offering, you may access our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act with the SEC free of charge at our website as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. The information contained in, or that can be accessed through, our website is not incorporated by reference into this prospectus.
128
Jaguar Animal Health, Inc.
(A Development Stage Company)
F-1
Report of Independent Registered Public Accounting Firm
Board
of Directors and Stockholders
Jaguar Animal Health, Inc.
San Francisco, CA
We have audited the accompanying balance sheet of Jaguar Animal Health, Inc., a development stage company (the "Company") and majority-owned subsidiary of Napo Pharmaceuticals, Inc. as of December 31, 2013 and the related statements of comprehensive loss, changes in common stock, convertible preferred stock and stockholders' deficit and cash flows for the period from June 6, 2013 (inception) through December 31, 2013. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As described in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to this matter.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Jaguar Animal Health, Inc. as of December 31, 2013, and the results of its operations and its cash flows for the period from June 6, 2013 (inception) through December 31, 2013, in conformity with accounting principles generally accepted in the United States of America.
/s/ BDO USA, LLP
San Francisco, California
June 16, 2014
F-2
Jaguar Animal Health, Inc.
(A Development Stage Company)
Balance Sheets
| |
December 31, 2013 |
June 30, 2014 |
Pro Forma as of June 30, 2014 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
(unaudited) |
|||||||
Assets |
||||||||||
Cash and cash equivalents |
$ | 185,367 | $ | 4,281,698 | ||||||
Deferred offering costs |
— | 1,029,896 | ||||||||
Prepaid license fee |
100,000 | — | ||||||||
Prepaid expenses |
— | 32,998 | ||||||||
Deferred finance charge |
3,894 | — | ||||||||
| | | | | | | | | | | |
Total current assets |
289,261 | 5,344,592 | ||||||||
Equipment |
— | 872,523 | ||||||||
| | | | | | | | | | | |
Total assets |
$ | 289,261 | $ | 6,217,115 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Liabilities, Convertible Preferred Stock and Stockholders' (Deficit) |
||||||||||
Accounts payable |
$ | 8,995 | $ | 202,191 | ||||||
Due to parent |
116,383 | 63,055 | ||||||||
Convertible notes payable |
519,486 | 231,250 | ||||||||
Accrued expenses |
79,250 | 778,673 | ||||||||
| | | | | | | | | | | |
Total current liabilities |
724,114 | 1,275,169 | ||||||||
License fee payable |
— | 1,900,000 | ||||||||
| | | | | | | | | | | |
Total current and long term liabilities |
724,114 | 3,175,169 | ||||||||
| | | | | | | | | | | |
Commitments and Contingencies (Note 6) |
||||||||||
Series A redeemable convertible preferred stock; $0.0001 par value, zero and 3,017,488 shares authorized at December 31, 2013 and June 30, 2014 (unaudited), respectively; 3,015,902 shares issued and outstanding at June 30, 2014 (unaudited); (liquidation preference of $6,777,338 at June 30, 2014 (unaudited)); no shares issued or outstanding pro forma at June 30, 2014 (unaudited) |
— | 6,943,250 | $ | — | ||||||
Stockholders' (Deficit) Equity |
||||||||||
Common stock: $0.0001 par value, 10,000,000 shares authorized at December 31, 2013 and June 30, 2014 (unaudited); 4,000,000 and 4,311,498 shares issued and outstanding at December 31, 2013 and June 30, 2014 (unaudited), respectively; 7,327,400 shares issued and outstanding pro forma at June 30, 2014 (unaudited) |
400 | 431 | 733 | |||||||
Additional paid-in capital |
365,950 | 809,702 | 7,467,641 | |||||||
Deficit accumulated during the development stage |
(801,203 | ) | (4,711,437 | ) | (4,711,437 | ) | ||||
| | | | | | | | | | | |
Total Stockholders' (Deficit) Equity |
(434,853 | ) | (3,901,304 | ) | $ | 2,756,937 | ||||
Total liabilities, convertible preferred stock and stockholders' (deficit) |
$ | 289,261 | $ | 6,217,115 | ||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements
F-3
Jaguar Animal Health, Inc.
(A Development Stage Company)
Statements of Comprehensive Loss
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
Cumulative Period from June 6, 2013 (inception) through June 30, 2014 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
(unaudited) |
|||||||
Operating expenses |
||||||||||
General and administrative expense |
$ | 458,473 | $ | 1,740,515 | $ | 2,198,988 | ||||
Research and development expense |
324,479 | 2,149,555 | 2,474,034 | |||||||
| | | | | | | | | | | |
Total operating expenses |
782,952 | 3,890,070 | 4,673,022 | |||||||
| | | | | | | | | | | |
Loss from operations |
(782,952 | ) | (3,890,070 | ) | (4,673,022 | ) | ||||
Interest expense, net |
(18,251 | ) | (20,164 | ) | (38,415 | ) | ||||
| | | | | | | | | | | |
Net loss and comprehensive loss |
$ | (801,203 | ) | $ | (3,910,234 | ) | $ | (4,711,437 | ) | |
Accretion of redeemable convertible preferred stock |
— | (285,009 | ) | (285,009 | ) | |||||
| | | | | | | | | | | |
Net loss attributable to common stockholders |
$ | (801,203 | ) | $ | (4,195,243 | ) | $ | (4,996,446 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net loss per share attributable to common stockholders, basic and diluted |
$ | (0.20 | ) | $ | (0.99 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted-average common shares outstanding, basic and diluted |
4,000,000 | 4,250,929 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro forma net loss per share attributable to common stockholders, basic and diluted (unaudited) |
$ | (0.20 | ) | $ | (0.67 | ) | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Weighted-average shares used in computing pro forma net loss per share attributable to common stockholders, basic and diluted (unaudited) |
4,000,000 | 6,304,461 | ||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements
F-4
Jaguar Animal Health, Inc.
(A Development Stage Company)
Statement of Changes in Common Stock, Convertible
Preferred Stock and Stockholders' (Deficit)
|
Series A Convertible Preferred Stock |
|
Common Stock | Additional Paid-in Capital |
Deficit Accumulated During the Development Stage |
Total Stockholders' (Deficit) |
||||||||||||||||||
|
Shares | Amount | Shares | Amount | ||||||||||||||||||||
Balances at June 6, 2013 (inception) |
— | $ | — | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||
Issuance of common stock to parent for services |
— | — | 4,000,000 | 400 | 359,055 | — | 359,455 | |||||||||||||||||
Issuance of common stock warrants |
— | — | — | — | 6,895 | — | 6,895 | |||||||||||||||||
Net loss and comprehensive loss |
— | — | — | — | — | (801,203 | ) | (801,203 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balances at December 31, 2013 |
— | — | 4,000,000 | 400 | 365,950 | (801,203 | ) | (434,853 | ) | |||||||||||||||
Stock-based compensation |
— | — | — | — | 90,952 | — | 90,952 | |||||||||||||||||
Conversion of notes payable into common stock |
— | — | 311,498 | 31 | 524,969 | — | 525,000 | |||||||||||||||||
Issuance of redeemable convertible preferred stock, net |
3,015,902 | 6,658,241 | — | — | — | — | — | |||||||||||||||||
Beneficial conversion feature on issuance of convertible promissory notes |
— | — | — | — | 75,000 | — | 75,000 | |||||||||||||||||
Warrants issue in connection with transfer agreement |
— | — | — | — | 37,840 | — | 37,840 | |||||||||||||||||
Deemed dividends on redeemable convertible preferred stock |
— | 269,237 | — | — | (269,237 | ) | — | (269,237 | ) | |||||||||||||||
Accretion of issuance costs to liquidity amount |
— | 15,772 | — | — | (15,772 | ) | — | (15,772 | ) | |||||||||||||||
Net loss and comprehensive loss |
— | — | — | — | — | (3,910,234 | ) | (3,910,234 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balances at June 30, 2014 (unaudited) |
3,015,902 | $ | 6,943,250 | 4,311,498 | $ | 431 | $ | 809,702 | $ | (4,711,437 | ) | $ | (3,901,304 | ) | ||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements
F-5
Jaguar Animal Health, Inc.
(A Development Stage Company)
Statements of Cash Flows
| |
Period from June 6, 2013 (inception) through December 31, 2013 |
Six Months Ended June 30, 2014 |
Cumulative Period from June 6, 2013 (inception) through June 30, 2014 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
(unaudited) |
|||||||
Cash Flows from Operating Activities |
||||||||||
Net loss |
$ | (801,203 | ) | $ | (3,910,234 | ) | $ | (4,711,437 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||
Materials cost in connection with license activity |
— | 1,082,626 | 1,082,626 | |||||||
Stock issued to parent for services |
359,055 | — | 359,055 | |||||||
Warrants issued in connection with transfer agreement |
— | 37,840 | 37,840 | |||||||
Stock-based compensation |
— | 90,952 | 90,952 | |||||||
Amortization of beneficial conversion feature |
— | 6,250 | 6,250 | |||||||
Accretion of debt discount |
1,381 | 5,514 | 6,895 | |||||||
Amortization of deferred finance charge |
1,300 | 3,894 | 5,194 | |||||||
Changes in assets and liabilities: |
||||||||||
Prepaid license fee |
(100,000 | ) | 100,000 | — | ||||||
Prepaid expenses |
— | (32,998 | ) | (32,998 | ) | |||||
Due to parent |
116,383 | (53,328 | ) | 63,055 | ||||||
Accounts payable |
8,995 | 193,196 | 202,191 | |||||||
Accrued expenses |
79,250 | 699,423 | 778,673 | |||||||
| | | | | | | | | | | |
Total Cash Used in Operating Activities |
(334,839 | ) | (1,776,865 | ) | (2,111,704 | ) | ||||
| | | | | | | | | | | |
Cash Flows from Investing Activities |
||||||||||
Purchase of equipment |
— | (55,149 | ) | (55,149 | ) | |||||
| | | | | | | | | | | |
Total Cash Used in Investing Activities |
— | (55,149 | ) | (55,149 | ) | |||||
| | | | | | | | | | | |
Cash Flows from Financing Activities |
||||||||||
Proceeds from issuance of redeemable convertible preferred stock, net |
— | 6,658,241 | 6,658,241 | |||||||
Proceeds from issuance of redeemable convertible notes payable, net |
519,806 | 300,000 | 819,806 | |||||||
Deferred offering costs |
— | (1,029,896 | ) | (1,029,896 | ) | |||||
Proceeds from issuance of common stock to parent |
400 | — | 400 | |||||||
| | | | | | | | | | | |
Total Cash Provided by Financing Activities |
520,206 | 5,928,345 | 6,448,551 | |||||||
| | | | | | | | | | | |
Net increase in cash and cash equivalents |
185,367 | 4,096,331 | 4,281,698 | |||||||
Cash and cash equivalents, beginning of period |
— | 185,367 | — | |||||||
| | | | | | | | | | | |
Cash and cash equivalents, end of period |
$ | 185,367 | $ | 4,281,698 | $ | 4,281,698 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Supplemental Schedule of Non-Cash Financing and Investing Activities |
||||||||||
Equipment recorded in connection with license agreement |
— | $ | 817,374 | $ | 817,374 | |||||
| | | | | | | | | | | |
Notes payable converted into common stock |
— | $ | 525,000 | $ | 525,000 | |||||
| | | | | | | | | | | |
Accretion of redeemable convertible preferred stock |
— | $ | 285,009 | $ | 285,009 | |||||
| | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements
F-6
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements
1. Organization and Business
Jaguar Animal Health, Inc. ("Jaguar" or the "Company") was incorporated on June 6, 2013 (inception) in Delaware. The Company, a majority-owned subsidiary of Napo Pharmaceuticals, Inc. ("Napo" or the "Parent"), was formed to develop and commercialize gastrointestinal products for companion and production animals. The Company is an animal health company in the development-stage whose activities since inception have consisted principally of raising capital, recruiting management, and performing research and development. All losses accumulated since inception are considered part of the Company's development-stage activities. The Company operates in one segment and is headquartered in San Francisco, California.
The following series of transactions between Jaguar and Napo were executed in order to separate the Company's business from Napo:
On June 11, 2013, Jaguar issued 4,000,000 shares of common stock to Napo in exchange for cash and services. On July 1, 2013, Jaguar entered into an employee leasing and overhead agreement (the "Service Agreement") with Napo, under which Napo agreed to provide the Company with the services of certain Napo employees for research and development and the general administrative functions of the Company. On January 27, 2014, Jaguar executed an intellectual property license agreement with Napo pursuant to which Napo transferred fixed assets and development materials, and licensed intellectual property and technology to Jaguar. On February 28, 2014, the Service Agreement terminated and the associated employees became employees of Jaguar effective March 1, 2014. Included in the statement of comprehensive loss from the period of June 6, 2013 (inception) through June 30, 2014 (unaudited) are general and administrative expense of $459,432 and research and development expense of $115,056 that were charged to Jaguar by Napo for the services of certain employees and overhead allocations. See Note 8 for additional information regarding the capital contributions and Notes 3 and 4 for the Service Agreement and license agreement details, respectively.
Liquidity
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. The Company has incurred recurring operating losses since inception and has an accumulated deficit of $4,711,437 as of June 30, 2014 (unaudited). The Company expects to incur substantial losses in future periods. Further, the Company's future operations are dependent on the success of the Company's ongoing development and commercialization efforts. There is no assurance that profitable operations, if ever achieved, could be sustained on a continuing basis.
The Company plans to finance its operations and capital funding needs through equity and/or debt financing as well as revenue from future product sales. However, there can be no assurance that additional funding will be available to the Company on acceptable terms on a timely basis, if at all, or that the Company will generate sufficient cash from operations to adequately fund operating needs or ultimately achieve profitability. If the Company is unable to obtain an adequate level of financing needed for the long-term development and commercialization of its products, the Company will need to curtail planned activities and reduce costs. Doing so will likely have an adverse effect on the Company's ability to execute on its business plan. These matters raise substantial doubt about the ability of the Company to continue in existence as a going concern. The accompanying financial statements do not include any adjustments that might result from the outcome of these uncertainties.
F-7
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies
Basis of Presentation
The financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America ("U.S. GAAP").
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires the Company's management to make judgments, assumptions and estimates that affect the amounts reported in its financial statements and the accompanying notes. Actual results could differ materially from those estimates.
Unaudited Interim Financial Information
The accompanying balance sheet at June 30, 2014, statements of comprehensive loss, changes in common stock, convertible preferred stock and stockholders' (deficit) and cash flows for the six months ended June 30, 2014 and the cumulative period from June 6, 2013 (inception) through June 30, 2014, are unaudited. The interim unaudited financial statements have been prepared on the same basis as the audited financial statements and, in the opinion of management, reflect all adjustments, which include only normal recurring adjustments, necessary for the fair presentation of the Company's financial position as of June 30, 2014 and the results of its operations and its cash flows for the six months ended June 30, 2014 and the cumulative period from June 6, 2013 (inception) through June 30, 2014. The financial data and other information disclosed in these notes related to the six months ended June 30, 2014 and the cumulative period from June 6, 2013 (inception) through June 30, 2014 are unaudited. The results for the six months ended June 30, 2014 are not necessarily indicative of the results to be expected for the year ending December 31, 2014, any other interim periods, or any future year or period.
Unaudited Pro Forma Information
The Company is filing a registration statement on Form S-1 with the U.S. Securities and Exchange Commission in connection with a proposed initial public offering ("IPO") of its common stock. If the IPO is consummated, the Company's outstanding convertible preferred stock will convert into shares of common stock of the Company on a one-for-one basis. The accompanying unaudited pro forma balance sheet as of June 30, 2014, statements of comprehensive loss for the six months ended June 30, 2014 and statements of comprehensive loss for the period from June 6, 2013 (inception) through December 31, 2013 have been prepared to give effect to the conversion of all outstanding shares of the Company's redeemable convertible preferred stock into 3,015,902 shares of the Company's common stock, as if such conversion had occurred at the beginning of the period, or the issuance date if later, and the reclassification of the redeemable convertible preferred stock to common stock and additional paid-in capital upon the closing of the IPO.
Deferred Offering Costs
Deferred offering costs, consisting of legal, accounting and filing fees related to the Company's proposed IPO are capitalized. The deferred offering costs will be offset against IPO proceeds upon the effectiveness of the offering. In the event the offering is terminated, deferred offering costs will be expensed.
F-8
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
Concentration of Credit Risk and Cash and Cash Equivalents
The financial instrument that potentially subjects the Company to a concentration of credit risk is cash and cash equivalents that are held at a financial institution of high credit standing. The Company considers all highly liquid instruments with an original maturity of three months or less to be cash equivalents. Cash is generally in excess of FDIC insurance limits. Therefore, the Company is exposed to credit risk in the event that the balances exceed FDIC insurance limits. The carrying value of cash and cash equivalents approximates estimated market value at December 31, 2013 and June 30, 2014 (unaudited).
Fair Value Measurements
Certain assets and liabilities are carried at fair value under U.S. GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. A fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last is considered unobservable, is used to measure fair value:
| Level 1: | Valuations for assets and liabilities traded in active markets from readily available pricing sources such as quoted prices in active markets for identical assets or liabilities. | |
Level 2: |
Observable inputs (other than Level 1 quoted prices) such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data. |
|
Level 3: |
Unobservable inputs that are supported by little or no market activity and that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. |
The carrying amount of the Company's financial instruments, including cash and cash equivalents, accounts payable, accrued expenses and amounts due to parent, approximate fair value due to the short maturities of these financial instruments.
As of December 31, 2013 and June 30, 2014 (unaudited), the Company does not have any financial assets or liabilities that are measured at fair value on a recurring basis.
Property and Equipment
Equipment is stated at cost, less accumulated depreciation. Equipment begins to be depreciated when it is placed into service. Depreciation will be calculated using the straight-line method over the estimated useful lives of 3 to 10 years.
Expenditures for repairs and maintenance of assets are charged to expense as incurred. Costs of major additions and betterments are capitalized and depreciated on a straight-line basis over their useful lives. Upon retirement or sale, the cost and related accumulated depreciation of assets disposed of are removed from the accounts and any resulting gain or loss is included in income (loss) from operations.
F-9
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
Long-Lived Assets
The Company regularly reviews the carrying value and estimated lives of all of its long-lived assets, including property and equipment to determine whether indicators of impairment may exist that warrant adjustments to carrying values or estimated useful lives. The determinants used for this evaluation include management's estimate of the asset's ability to generate positive income from operations and positive cash flow in future periods as well as the strategic significance of the assets to the Company's business objectives.
Should an impairment exist, the impairment loss would be measured based on the excess of the carrying amount over the asset's fair value. The Company has not recognized any impairment losses through December 31, 2013.
Research and Development Expense
Research and development expense consists of expenses incurred in performing research and development activities including related salaries, clinical trial and related drug and non-drug product costs, contract services and other outside service expenses. Research and development expense is charged to operating expense in the period incurred.
Stock-Based Compensation
The Company's equity incentive plan (see Note 10) provides for the grant of stock options, restricted stock and restricted stock unit awards.
The Company measures stock awards granted to employees and directors at fair value on the date of grant and recognizes the corresponding compensation expense of the awards, net of estimated forfeitures, over the requisite service periods, which correspond to the vesting periods of the awards. Generally, the Company issues stock awards with only service-based vesting conditions, and records compensation expense for these awards using the straight-line method.
The Company values its shares of common stock by taking into consideration its most recently available valuation of common stock performed by management and the board of directors, as well as additional factors that may have changed since the date of the most recent contemporaneous valuations through the date of grant.
In the absence of a valuation, the fair value of the Company's common stock underlying stock awards is determined by its board of directors, with assistance from management, based upon information available at the time of grant. Given the absence of a public trading market for its common stock, and in accordance with the "American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation," the Company's board of directors exercises its reasonable judgment and considers numerous objective and subjective factors to determine the best estimate of the fair value of its common stock at each grant date.
Income Taxes
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the financial statements or in the Company's tax returns. Deferred taxes are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse.
F-10
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. The Company assesses the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent it believes, based upon the weight of available evidence, that it is more likely than not that all or a portion of deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.
The Company accounts for uncertainty in income taxes recognized in the financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate, as well as the related net interest and penalties.
Comprehensive Loss
Comprehensive loss is defined as changes in stockholders' (deficit) exclusive of transactions with owners (such as capital contributions and distributions). For the period from June 6, 2013 (inception) through December 31, 2013, the six months ended June 30, 2014 (unaudited), and the cumulative period from June 6, 2013 (inception) through June 30, 2014 (unaudited), there was no difference between net loss and comprehensive loss.
Segment Data
The Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions. The Company is a development stage animal health company focused on developing and commercializing prescription and non-prescription products for companion and production animals.
Basic and Diluted Net Loss Per Common Share
Basic net loss per common share is computed by dividing net loss attributable to common stockholders for the period by the weighted-average number of common shares outstanding during the period. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders for the period by the weighted-average number of common shares, including potential dilutive shares of common stock assuming the dilutive effect of potential dilutive securities. For periods in which the Company reports a net loss, diluted net loss per common share is the same as basic net loss per common share, because their impact would be anti-dilutive to the calculation of net loss per common share. Diluted net loss per common share is the same as basic net loss per common share for the period from June 6, 2013 (inception) through December 31, 2013 and the six months ended June 30, 2014 (unaudited).
F-11
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
Revenue Recognition
The Company is a development-stage enterprise and has not generated any revenue since inception.
Recent Accounting Pronouncements
In June 2014, the Financial Accounting Standards Board ("FASB") issued authoritative guidance which eliminates the distinction of a development stage entity and certain related disclosure requirements, including the elimination of inception-to-date information on the statements of operations, cash flows and stockholders' equity. The amendments will be effective prospectively for annual reporting periods beginning after December 15, 2014, and interim periods within those annual periods, however early adoption is permitted. The Company did not implement early adoption of this standard. The adoption of this guidance will have no impact on the Company's financial condition, results of operations or cash flows.
In June 2014, the FASB issued authoritative guidance which requires that a performance target that affects vesting and that could be achieved after the requisite service period be treated as a performance condition. The performance target should not be reflected in estimating the grant-date fair value of the award. Compensation cost should be recognized in the period in which it becomes probable that the performance target will be achieved and should represent the compensation cost attributable to the period(s) for which the requisite service has already been rendered. If the performance target becomes probable of being achieved before the end of the requisite service period, the remaining unrecognized compensation cost should be recognized prospectively over the remaining requisite service period. The total amount of compensation cost recognized during and after the requisite service period should reflect the number of awards that are expected to vest and should be adjusted to reflect those awards that ultimately vest. The requisite service period ends when the employee can cease rendering service and still be eligible to vest in the award if the performance target is achieved. This guidance will be effective for annual periods (and interim periods within those annual periods) beginning after December 15, 2015. The Company will implement this guidance for all interim and annual periods beginning after December 15, 2015. The adoption of this guidance is not expected to have an impact on the Company's financial condition, results of operations or cash flows.
In May 2014, the FASB issued Accounting Standards Update ("ASU") No. 2014-09, "Revenue from Contracts with Customers." The objective of ASU 2014-19 is to establish a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and will supersede most of the existing revenue recognition guidance, including industry-specific guidance. The core principle of the new standard is that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard is effective for annual reporting periods beginning after December 15, 2016 and allows for prospective or retrospective application. The Company is evaluating and does not believe this pronouncement will have a material effect on its financial statements.
3. Employee Leasing and Overhead Allocation Agreement
Effective July 1, 2013, the Company entered into an employee leasing and overhead allocation agreement (the "Service Agreement") with its parent, Napo. The term of the Service Agreement was from July 1, 2013 through February 28, 2014. In connection with the Service Agreement, Napo
F-12
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
provided the Company with the services of Napo employees. The Service Agreement also stipulated that Jaguar would pay for a portion of Napo's overhead costs. The Company agreed to pay Napo $71,811 per month (consisting of $38,938 for executive compensation, $26,873 for employee services, and $6,000 for overhead costs) for the months from July 2013 through February 2014 as follows: (1) for the period from July 2013 through November 2013, in 4,000,000 shares of common stock and (2) for the period from December 2013 through February 2014, in cash. Included in due to parent on the accompanying balance sheet at December 31, 2013 is $71,811 related to the amount due for December 2013. Commencing March 1, 2014, certain Napo employees became employees of the Company and all overhead costs related to the animal health business will be paid by the Company.
General and administrative expense recognized under the Service Agreement was $344,574 and $114,858 for the period from June 6, 2013 (inception) through December 31, 2013 and the six months ended June 30, 2014 (unaudited), respectively.
Research and development expense recognized under the Service Agreement was $86,292 and $28,764 for the period from June 6, 2013 (inception) through December 31, 2013 and the six months ended June 30, 2014 (unaudited), respectively.
4. License Agreement
On July 11, 2013, Jaguar entered into an option to license Napo's intellectual property and technology (the "Option Agreement"). Under the Option Agreement, upon the payment of $100,000 in July 2013, the Company obtained an option for a period of two years to execute an exclusive worldwide license to Napo's intellectual property and technology to use for the Company's animal health business. The option price is creditable against future license fees to be paid to Napo under the License Agreement (as defined below). As such, $100,000 is included on the balance sheet as a prepaid license fee at December 31, 2013.
In January 2014, the Company exercised its option and entered into a license agreement (the "License Agreement") with Napo for an exclusive worldwide license to Napo's intellectual property and technology to permit the Company to develop, formulate, manufacture, market, use, offer for sale, sell, import, export, commercialize and distribute products for veterinary treatment uses and indications for all species of animals. The Company is obligated to pay a one-time non-refundable license fee of $2,000,000, less the option fee of $100,000. At the Company's option, the license fee can be paid in common stock. This license fee will be payable to Napo when the Company's cumulative net sales exceed $2,000,000. Milestone payments may also be due to Napo aggregating $3,150,000 based on regulatory approvals of various veterinary products. In addition to the milestone payments, the Company will owe Napo an 8% royalty on annual net sales of products derived from Croton lechleri, up to $30,000,000 and then, a royalty of 10% on annual net sales of $30,000,000 or more. Additionally, if any other products are developed, the Company will owe Napo a 2% royalty on annual net sales of pharmaceutical prescription products that are not derived from Croton lechleri and a 1% royalty on annual net sales of nonprescription products that are not derived from Croton lechleri. The royalty term expires at the longer of 10 years from the first sale of each individual product or when there is no longer a valid patent claim covering any of the products and a competitive product has entered the market. However, in the event of an IPO of at least $10,000,000 prior to December 31, 2015, the royalty shall be reduced to 2% of annual net sales of its prescription products derived from Croton lechleri and 1% of net sales of its nonprescription products derived from Croton lechleri and no milestone payment will be due and no royalties will be owed on any additional products developed.
F-13
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
In addition to receiving a License Agreement to Napo's intellectual property and technology, the License also transferred to the Company certain materials and equipment. Materials transferred from Napo have been included in research and development expense on the statements of comprehensive loss. Equipment of $817,374 related to the License is included on the balance sheet at June 30, 2014 (unaudited) at the cost paid by Napo, which approximates fair value. As of June 30, 2014, the equipment has not been placed into service. The Company will begin depreciating the equipment on a straight-line basis over its estimated life of 10 years at the time it is placed into service.
Jaguar has agreed under the License Agreement to defend, indemnify and hold Napo, its affiliates, and the officers, directors, employees, consultants and contractors of Napo harmless from and against any losses, costs, damages, liabilities, fees and expenses arising out of any third-party claim related to Jaguar's gross negligence, breach of covenants or the manufacture, sale or use of the product or products.
5. Accrued Expenses
Accrued expenses at December 31, 2013 and June 30, 2014 (unaudited) consist of the following:
| |
December 31, 2013 |
June 30, 2014 |
|||||
|---|---|---|---|---|---|---|---|
| |
|
(unaudited) |
|||||
Accrued legal costs |
$ | — | $ | 542,376 | |||
Accrued printing costs |
— | 125,000 | |||||
Due to veterinary school of medicine |
$ | 45,000 | — | ||||
Accrued consulting fees |
17,683 | 18,000 | |||||
Accrued interest |
15,671 | 740 | |||||
Accrued vacation |
— | 64,725 | |||||
Other |
896 | 27,832 | |||||
| | | | | | | | |
|
$ | 79,250 | $ | 778,673 | |||
| | | | | | | | |
| | | | | | | | |
6. Commitments and Contingencies
In 2013, a veterinary school of medicine began a field study for the Company. The Company agreed to make payments to the school totaling $190,000. The total expense for the period from June 6, 2013 (inception) to December 31, 2013 was $145,000 and for the six months ended June 30, 2014 was $45,000 (unaudited).
Since March 1, 2014, the date the Service Agreement terminated, the Company paid Napo $33,897 (unaudited) for rent related to the office space utilized by the Company for the months of March, April and May, 2014. Effective on June 1, 2014, the Company assumed the existing sublease from Napo. The term of the sublease is from June 1, 2014 through June 30, 2015 (unaudited).
F-14
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
Effective June 26, 2014 the Company entered into a technology transfer and commercial manufacturing agreement (the "Transfer Agreement") with a contract manufacturer in Italy (the "Manufacturer"), whereby the Company and the Manufacturer will cooperate to develop and refine the manufacturing process for the Company's prescription and non-prescription products. Pursuant to the Transfer Agreement, the Company will make prepayments to the Manufacturer as follows: (1) a start-up fee of €500,000, €250,000 of which is to be paid at the earlier to occur of September 15, 2014 or the closing date of an initial public offering and €250,000 of which is to be paid at the time of installation and qualification of the Company's equipment at their facility, (2) related to the technology transfer, €620,000, €310,000 of which is to be paid within five days of the signature of the Transfer Agreement and €310,000 of which is to be paid after the delivery of a final study report, and (3) for design of a portion of the Manufacturer's facility, €100,000 to be paid within five days of the signature of the Transfer Agreement. Additionally, the Transfer Agreement stipulates that the Company will pay the Manufacturer an aggregate of €500,000 upon the delivery of agreed-upon levels of satisfactory product. Further, the Company issued the Manufacturer warrants to purchase 25,000 shares of common stock with an exercise price of 90% of the initial public offering price.
7. Convertible Promissory Notes and Common Stock Warrants
From July through September 2013, the Company issued four convertible promissory notes (collectively the "Notes") for gross aggregate proceeds of $525,000 to various third-party lenders. The Notes bear interest at 8% per annum. The Notes automatically mature and the entire outstanding principal amount, together with accrued interest, are due and payable in cash at the earlier of July 8, 2015 (the "Maturity Date") or ten business days after the date of consummation of the initial closing of a first equity round of financing.
If the Company consummates a first equity round of financing prior to the Maturity Date with a pre-money valuation of greater than $3,000,000, principal and accrued interest may, at the option of each noteholder, be converted into shares of common stock at 75% of the purchase price paid by such equity investors. If the Company consummates a first equity round of financing prior to the Maturity Date with a pre-money valuation of $3,000,000 or less, principal and accrued interest may, at the option of each noteholder, be converted into shares of common stock at the same purchase price paid by such equity investors.
In connection with the Notes, the Company issued to the noteholders warrants which became exercisable to purchase an aggregate of 311,498 shares of common stock as of the issuance of the first equity round of financing (the "Warrants"). The Warrants are fully exercisable from the initial date of the first equity round of financing and have a five-year term subsequent to that date. If the Company consummates a first equity round of financing with a pre-money valuation of greater than $3,000,000, the exercise price of the Warrants is 75% of the purchase price paid by such equity investors. If the Company consummates a first equity round of financing with a pre-money valuation of $3,000,000 or less, the exercise price of the Warrants will be the same purchase price paid by such equity investors.
In February 2014, the Company closed its first equity round of financing and sold 2,224,991 shares of Series A convertible preferred stock at a price of $2.2472 per share. The pre-money valuation was in excess of $3,000,000 setting the exercise price of the Warrants at 75% of the purchase price paid by the investors, or $1.6854 per share. As such, the fair value of the Warrants, $6,895, was recorded as equity in February 2014. The Warrants were valued at $6,895 using the Black-Scholes model with the following assumptions: strike price of $0.18, exercise price of $1.6854, term of five years, volatility of 64%,
F-15
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
dividend yield of 0%, and risk-free interest rate of 1.82%. Based on the fair value of the Warrants, the Company deemed it appropriate to use the residual value of the total proceeds from the issuance of the Notes and Warrants to record the Notes on the balance sheet as of issuance of the Notes. Thus, the amount recorded, in the aggregate, for the Notes on issuance was $518,105, net. The debt discount of $6,895 is recorded as interest expense over the five-year term of the Warrants. Interest expense recorded from the period from June 6, 2013 (inception) through December 31, 2013 was $1,381. At December 31, 2013, the net amount of the Notes is $519,486.
In February 2014, in connection with the first equity round of financing and issuance of the Series A convertible preferred stock, the noteholders exercised their option to convert their Notes into 311,498 shares of common stock and accrued interest was paid in cash to the noteholders. As such, the Notes are classified as current on the accompanying balance sheet as of December 31, 2013. The accreted interest expense related to the discount on the Notes was $1,443 for the period from January 1, 2014 to the conversion date of the Notes. Upon conversion, the entire remaining debt discount of $4,071 was recorded as interest expense.
On June 2, 2014, pursuant to a convertible note purchase agreement, the Company issued convertible promissory notes in the aggregate principal amount of $300,000 to two accredited investors, including a convertible promissory note for $200,000 to the same board member to which Series A preferred stock was sold. These notes bear interest at 3% per annum and automatically mature on June 1, 2015. Accrued interest shall be paid in cash upon maturity. Upon the closing of an initial public offering, the outstanding principal amount shall automatically convert into common stock at 80% of the price in the initial public offering. If the Company has not consummated an initial public offering on or before June 1, 2015, then the principal then outstanding will automatically convert at 80% of the Company's next preferred stock financing. The Company has analyzed the beneficial nature of the conversion terms and determined that a beneficial conversion feature ("BCF") exists because the effective conversion price was less than the fair value at the time of the issuance. The Company calculated the value of the BCF using the intrinsic method. A BCF of $75,000 has been recorded as a discount to the notes payable and to additional paid-in capital. For the six months ended June 30, 2014 (unaudited), the Company has amortized $6,250 of the beneficial conversion feature which has also been recorded as interest expense.
In connection with the Transfer Agreement (Note 6) the Company issued fully vested and immediately exercisable warrants to the Manufacturer to purchase 25,000 shares of common stock at 90% of the IPO price, for a period of five years. The fair value of the warrants, $37,840, was recorded as research and development expense and additional paid-in capital in June 2014. The warrants were valued using the Black-Scholes model with the following assumptions: stock price of $3.22, exercise price of $2.90, term of five years, volatility of 49%, dividend yield of 0%, and risk-free interest rate of 1.64%.
F-16
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
8. Redeemable Convertible Preferred Stock
The following is a summary of the Company's Series A redeemable convertible preferred stock at June 30, 2014 (unaudited):
Preferred shares authorized |
3,017,488 | |
Issuance dates |
February, April and May 2014 | |
Preferred shares issued and outstanding |
3,015,902 | |
Redemption value/liquidation preference |
$9,020,637/$6,777,338 | |
Carrying value |
$6,943,250 |
The differences between the respective redemption values/liquidation preference and carrying values are being accreted over the period from the date of issuance to the earliest possible redemption date, February 2017.
Costs incurred in connection with the issuance of Series A redeemable convertible preferred stock (the "Preferred Stock") during the six months ended June 30, 2014 (unaudited) were $119,097 which have been recorded as a reduction to the carrying amounts of Preferred Stock and are being accreted to the carrying value of the applicable preferred stock to the redemption date.
The rights, preferences, and privileges of the Preferred Stock are as follows:
Voting—Except as provided by law, the holders of the Preferred Stock vote together and not as a separate class. On any matter presented to the stockholders of the Company for their action, each holder of outstanding shares of Preferred Stock is entitled to cast the number of votes equal to the number of whole shares of common stock into which the shares of Preferred Stock held by such holder could be converted as of the record date. The holders of Preferred Stock shall be entitled to vote on all matters on which the common stock shall be entitled to vote. Holders of Preferred Stock shall be entitled to notice of any stockholders' meeting in accordance with the bylaws of the Company. Fractional votes shall not, however, be permitted and any fractional voting rights shall be disregarded.
Dividends—The holders of Preferred Stock are entitled to receive cumulative dividends at an annual rate of 8% of the Preferred Stock original issue price of $2.2472 per share, which dividends accrue daily in arrears, whether or not such dividends are declared by the Company's board of directors (the "Accruing Dividends"). The dividends are only payable when declared by the board of directors, out of any funds legally available. No such dividends have been declared or paid through June 30, 2014 (unaudited). Dividends in arrears as of June 30, 2014 totaled $166,936 (unaudited).
Liquidation Rights—In the event of any voluntary or involuntary liquidation, dissolution or winding-up of the Company, either voluntary or involuntary, the holders of Preferred Stock then outstanding shall be entitled to receive, prior and in preference to any distribution of any of the assets of the Company to the holders of common stock by reason of their ownership of such stock, an amount equal to $2.2472 per share of Preferred Stock, plus any declared but unpaid dividends. If, upon liquidation, dissolution or winding up of the Company, the assets of the corporation legally available for distribution to the holders of the Preferred Stock are insufficient to permit the payment in full of the liquidation preference above, then the entire assets of the Company legally available for distribution shall be distributed with equal priority and pro rata among the holders of Preferred Stock in proportion to the full amounts they would otherwise be entitled to receive.
F-17
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
After the payment or setting aside for payment to the holders of Preferred Stock of the full amounts of the liquidation preferences, the entire remaining assets of the Company legally available for distribution after satisfaction of the liquidation preferences of the Preferred Stock shall be distributed to the holders of Preferred Stock and common stock, pro rata based upon the number of shares held by each such holder.
Conversion—Each share of Preferred Stock is convertible into shares of common stock a conversion price initially equal to $2.2472 per share and is subject to adjustment as set forth in the Company's certificate of incorporation, as amended and restated. Conversion price adjustments may occur if there are new issuances of common stock at less than the conversion price or in the event of an IPO. At June 30, 2014 (unaudited), no such events have occurred and as such, no conversion adjustment has been recorded. At June 30, 2014 (unaudited) the shares of Preferred Stock were convertible into shares of common stock on a 1-for-1 basis. The shares of Preferred Stock are convertible into shares of common stock, at the option of the holder, at any time after the date of issuance. Further, upon the closing of an IPO (1) within six months of the initial Preferred Stock closing at a price per share at least three times the original issue price or (2) after six months at a price per share at least five times the original issue price, and which in both (1) and (2) results in at least $25,000,000 of gross proceeds to the Company, all outstanding shares of Preferred Stock shall automatically convert into shares of common stock. Each share of Preferred Stock is convertible into shares of common stock at the applicable conversion rate then in effect at the time of conversion, which is calculated by dividing the original issue price by the respective conversion price. Any shares of Preferred Stock that are converted into common stock will be canceled and cannot be reissued by the Company.
Redemption—Upon certain change in control events that are outside the Company's control, including liquidation, sale or transfer of control of the Company, the holders of the Preferred Stock can cause its redemption. If the Company fails to complete an initial public offering of its common stock by February 2017, the holders of a majority of the shares of the Preferred Stock may thereafter request redemption at a price equal to the Preferred Stock original issue price per share, plus, in lieu of any Accruing Dividends, 10% percent of the Preferred Stock original issue price per share for each 12 month period after the date of the initial closing, on a compounded basis, commencing not more than 60 days after receipt by the Company of the written notice requesting redemption. The Company has recorded cumulative deemed dividends for the preferred return of $269,237 as of June 30, 2014 (unaudited).
The Preferred Stock has been classified outside of stockholders' (deficit) in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities.
9. Common Stock
The Company's certificate of incorporation, as amended and restated, authorizes the Company to issue 10,000,000 shares of common stock $0.0001 par value.
The holders of common stock are entitled to one vote for each share of common stock held at all meetings of stockholders. The number of authorized shares of common stock may be increased or decreased by the affirmative vote of the holders of shares of capital stock of the Company representing a majority of the votes represented by all shares (including Preferred Stock) entitled to vote.
F-18
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
In June 2013, the Company issued 4,000,000 shares of common stock to its parent, Napo, for total cash consideration of $400 and services to be performed by the parent from July 1, 2013 through November 30, 2013 (see Note 3).
In February 2014, holders of certain convertible promissory notes exercised their option to convert the notes into 311,498 shares of the Company's common stock (see Note 7).
10. Stock-Based Awards (Unaudited)
2013 Equity Incentive Plan
Effective November 1, 2013, the Company's board of directors and sole stockholder adopted the Jaguar Animal Health, Inc. 2013 Equity Incentive Plan (the "2013 Plan"). The 2013 Plan allows the Company's board of directors to grant stock options, restricted stock awards and restricted stock unit awards to employees, officers, directors and consultants of the Company. As of December 31, 2013, the Company had reserved 450,000 shares of its common stock for issuance under the 2013 Plan. In April 2014, the board of directors amended the 2013 Plan to increase the shares reserved for issuance to 1,171,177 shares. As of December 31, 2013 no stock awards have been granted under the 2013 Plan.
Stock Options
During the six months ended June 30, 2014, the Company granted stock options for the purchase of 1,129,673 shares of common stock to employees and a director. The vesting conditions of these awards are time-based, and the awards all vest 25% after 9 months and monthly thereafter for the next 27 months. Awards expire after 10 years.
The Company grants stock options with exercise prices equal to the fair value of its common stock as of the date of grant. The process of estimating the fair value of stock awards and recognizing stock-based compensation cost over their requisite service period involves significant assumptions and judgments. The Company estimates the fair value of stock option awards on the date of grant using the Black-Scholes option-valuation model for the remaining awards, which requires it to use certain assumptions regarding: (i) the expected volatility in the market price of its common stock; (ii) dividend yield; (iii) risk-free interest rates; and (iv) the period of time employees are expected to hold the award prior to exercise (referred to as the expected holding period). As a result, if the Company revises its assumptions and estimates, stock compensation expense could change materially for future grants. The Company classifies stock compensation expense in the statement of comprehensive loss in the same manner in which the recipient's payroll costs are classified.
The relevant data used to determine the weighted-average value of $1.03 of the stock option grants is as follows, presented on a weighted average basis:
| |
Six Months Ended June 30, 2014 |
|||
|---|---|---|---|---|
Risk free interest rate |
2 | % | ||
Expected term (in years) |
5.81 | |||
Expected volatility |
63 | % | ||
Expected dividend yield |
0 | % | ||
F-19
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
The following table summarizes stock option activity for the six months ended June 30, 2014:
| |
Shares issuable under options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term (in years) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Outstanding as of December 31, 2013 |
— | $ | — | |||||||
Granted |
1,129,673 | 1.77 | ||||||||
| | | | | | | | | | | |
Outstanding as of June 30, 2014 |
1,129,673 | 1.77 | 9.8 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Options expected to vest as of June 30, 2014 |
1,129,673 | 1.77 | 9.8 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Options exercisable as of June 30, 2014 |
— | |||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
Restricted Stock Units
The Company's 2013 Plan provides for the award of restricted stock units ("RSUs") with time-based and liquidity-event based vesting. Unvested RSUs may not be sold or transferred by the holder. These restrictions lapse according to the vesting.
During the six months ended June 30, 2014, the Company granted 118,953 RSUs. These shares vest upon the occurrence of both a liquidity event and satisfaction of the service-based requirement. The time-based vesting provides that 50% of the RSU will vest on January 1, 2016 and the remaining 50% will vest on July 1, 2017. Because the liquidity condition is not met until the occurrence of a qualifying liquidity event (an IPO or change of control), the Company have not recorded any expense to date relating to the RSU grants. In connection with the IPO, the Company will begin recording stock compensation expense based on the grant date fair value of the RSUs using the straight-line method, net of estimated forfeitures. If the IPO had occurred on June 30, 2014, the Company would have recorded $10,640 of stock-based compensation expense on that date related to RSUs and would have had an additional $372,389 in unamortized stock-based compensation expense related to RSUs.
The Company did not issue any RSUs prior to December 31, 2013.
Stock-Based Compensation
The Company recognizes compensation expense for only the portion of the awards that are expected to vest. The Company recorded stock-based compensation expense related to stock options of $58,051 and $32,901 to general and administrative expense and research and development expense, respectively.
As of June 30, 2014, the Company had $1,072,953 of unrecognized stock-based compensation expense for options outstanding, which is expected to be recognized over a weighted average period of 2.8 years.
11. Related Party Transactions
The Company is a majority-owned subsidiary of Napo. The Company has total outstanding liabilities to Napo in the amount of $116,383 as of December 31, 2013 and $63,055 as of June 30, 2014
F-20
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
(unaudited). Additionally, Lisa A. Conte, Chief Executive Officer of the Company, is also the interim Chief Executive Officer of Napo Pharmaceuticals, Inc.
A member of the board of directors of the Company purchased 222,499 shares of the Company's preferred stock during the six months ended June 30, 2014 (unaudited). The Company also issued a convertible promissory note for $200,000 to the same board member to whom the preferred stock was sold.
12. Net Loss Per Share Attributable to Common Stockholders
Basic net loss per share is calculated by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted net loss per share is computed by dividing net loss by the weighted average number of common shares and common share equivalents outstanding for the period. Common stock equivalents are only included when their effect is dilutive. The Company's potentially dilutive securities which include convertible preferred stock and warrants have been excluded from the computation of diluted net loss per share as they would be anti-dilutive. For all periods presented, there is no difference in the number of shares used to compute basic and diluted shares outstanding due to the Company's net loss position.
The following table sets forth the outstanding potentially dilutive securities that have been excluded in the calculation of diluted net loss per shares.
| |
June 30, 2014 | |||
|---|---|---|---|---|
| |
(unaudited) |
|||
Convertible preferred stock |
3,015,902 | |||
Warrants to purchase common stock |
336,498 | |||
| | | | | |
Total |
3,352,400 | |||
| | | | | |
| | | | | |
Unaudited Pro Forma Net Loss Per Share
The following table summarizes the unaudited pro forma net loss per share attributable to common stockholders:
| |
June 30, 2014 | |||
|---|---|---|---|---|
| |
(unaudited) |
|||
Numerator: |
||||
Net loss attributable to common stockholders |
$ | (4,195,243 | ) | |
Denominator: |
||||
Weighted-average common shares outstanding basic and diluted |
4,250,929 | |||
Pro forma adjustments to reflect assumed conversion of preferred stock |
2,053,532 | |||
| | | | | |
Shares used to compute pro forma net loss per share, basic and diluted |
6,304,461 | |||
| | | | | |
| | | | | |
Pro forma basic and diluted net loss per share attributable to common stockholders |
$ | (0.67 | ) | |
| | | | | |
| | | | | |
As of December 31, 2013, there were 4,000,000 shares of common stock outstanding, therefore the pro forma net loss per share, basic and diluted, for the period from June 6, 2013 (inception) through December 31, 2013 is $0.20 per share, and is equal to the net loss per share, basic and diluted, for the period from June 6, 2013 (inception) through December 31, 2013.
F-21
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
13. Income Taxes
The Company has not recorded a provision for income taxes in the statement of comprehensive loss for the period from June 6, 2013 (inception) through December 31, 2013. The Company had a net comprehensive loss for the period from June 6, 2013 (inception) through December 31, 2013, of $801,203.
Due to continued losses for the six months ended June 30, 2014 (unaudited), and a full valuation allowance, the Company has not recorded a provision for income taxes for the six months ended June 30, 2014 (unaudited).
The components of the provision for income taxes for the period from June 6, 2013 (inception) through December 31, 2013 are as follows:
| |
December 31, 2013 |
|||
|---|---|---|---|---|
Current: |
||||
Federal |
$ | — | ||
State |
$ | — | ||
Foreign |
$ | — | ||
| | | | | |
Total current |
$ | — | ||
Deferred: |
||||
Federal |
$ | (273,843 | ) | |
State |
$ | (48,002 | ) | |
Foreign |
$ | — | ||
| | | | | |
Total deferred |
$ | (321,845 | ) | |
Less: valuation allowance |
$ | 321,845 | ||
| | | | | |
Total provision for income taxes |
$ | — | ||
| | | | | |
| | | | | |
The Company's effective tax rate for the period from June 6, 2013 (inception) through December 31, 2013, differed from the federal statutory rate as follows:
| |
December 31, 2013 |
|||
|---|---|---|---|---|
Statutory rate |
(34.0 | )% | ||
State taxes |
(6.0 | )% | ||
Tax credits |
(0.7 | )% | ||
Non-deductible interest expense |
0.7 | % | ||
Valuation allowance |
40.0 | % | ||
| | | | | |
Effective tax rate |
0.0 | % | ||
| | | | | |
| | | | | |
F-22
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
Net deferred tax assets as of December 31, 2013 consists of the following:
| |
December 31, 2013 |
|||
|---|---|---|---|---|
Non-current deferred tax assets: |
||||
Net operating losses |
$ | 314,089 | ||
Tax credits |
7,756 | |||
| | | | | |
|
321,845 | |||
Valuation allowance |
(321,845 | ) | ||
| | | | | |
Net non-current deferred tax assets |
$ | — | ||
| | | | | |
| | | | | |
A valuation allowance is provided when it is more likely than not that the deferred tax assets will not be realized. The Company has established a valuation allowance to offset net deferred tax assets as of December 31, 2013, due to the uncertainty of realizing future tax benefits from its net operating loss carryforwards and other deferred tax assets.
As of December 31, 2013, the Company had federal and California net operating loss carryovers of $788,486 and $788,486, respectively. The federal and California net operating losses will expire in 2033.
As of December 31, 2013, the Company had federal and California research credit carryovers of $5,758 and $3,028, respectively. The federal research credits will begin to expire in 2033. The California research credits carry forward indefinitely.
The Tax Reform Act of 1986, as amended, limits the use of net operating loss and tax credit carryforward in certain situations where changes occur in the stock ownership of a company. In the event the Company has a change in ownership in the future, as defined by the tax law, utilization of the carryforwards could be limited.
A reconciliation of the beginning and ending amounts of unrecognized tax benefits in 2013 is as follows:
| |
December 31, 2013 |
|||
|---|---|---|---|---|
Beginning balance |
$ | — | ||
Change for tax positions |
— | |||
| | | | | |
Ending balance |
$ | — | ||
| | | | | |
| | | | | |
There are no liabilities from unrecognized tax benefits included in the Company's balance sheet as of December 31, 2013, and therefore the Company has not incurred any penalties or interest.
The Company files income tax returns in the United States and California, where the statute of limitations are 3 years and 4 years, respectively. The Company remains open for audit by the United States Internal Revenue Service and California state tax jurisdictions since inception. The Company is not currently under examination by income tax authorities in federal or state jurisdictions.
F-23
Jaguar Animal Health, Inc.
(A Development Stage Company)
Notes to Financial Statements (continued)
14. Subsequent Events
The Company completed an evaluation of the impact of subsequent events through October 10, 2014, the date these financial statements were issued. The following capital transaction has occurred. The effect of this transaction has not been included in the financial statements.
On July 16, 2014, pursuant to a convertible note purchase agreement, the Company issued a convertible promissory note in the principal amount of $150,000 to an accredited investor. This note bears an annual interest rate of 3%. Upon the closing of an IPO, these notes will convert into shares of common stock at a conversion rate equal to 80% of the IPO price per share.
In July 2014, the Company adopted the Jaguar Animal Health, Inc. 2014 Stock Incentive Plan ("2014 Plan"). The 2014 Plan provides for the grant of incentive stock options to eligible employees, and for the grant of nonstatutory stock options, restricted stock, and RSUs to eligible employees, directors and consultants. The Company has reserved 500,000 shares of common stock for issuance pursuant to the 2014 Plan.
In August 2014, the Company entered into a standby line of credit with an accredited investor for up to $1,000,000 pursuant to a Line of Credit and Loan Agreement dated August 26, 2014. The minimum amount of any drawdown is $250,000, the lender has no obligation to fund more than once every 10 calendar days, and the Company must provide 15 business days prior notice for any drawdown and may not draw down funds after March 31, 2015. Outstanding borrowings bear interest at a rate of 3.0% per annum, and all borrowings are due in full on the one-year anniversary of the first drawdown. In the event of closing of an IPO, outstanding principal amounts borrowed under the standby line of credit may be converted, at the option of the lender, into shares of common stock at a conversion price equal to 80% of the IPO price. In connection with the entry into the standby line of credit, the Company issued the lender a fully vested warrant to purchase 50,000 shares of common stock at an exercise price equal to 80% of the IPO price, which expires in August 2016. If an IPO is not consummated prior to August 26, 2015, the exercise price of the warrants will be $2.25 per share.
F-24
Shares
Common Stock
| BMO Capital Markets | Guggenheim Securities |
Roth Capital Partners
The date of this prospectus is , 2014.
Part II—INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION.
The following table sets forth an itemized statement of the expenses (excluding underwriting discounts and commissions) that are payable by us in connection with the registration, offer and sale of the common stock described in this registration statement. With the exception of the SEC registration fee, the FINRA filing fee and The NASDAQ Capital Market listing fee, the amounts set forth below are estimates.
| |
Amount to be Paid |
|||
|---|---|---|---|---|
SEC registration fee |
9,016 | |||
FINRA filing fee |
11,000 | |||
NASDAQ Capital Market listing fee |
50,000 | |||
Accounting fees and expenses |
* | |||
Legal fees and expenses |
* | |||
Printing and related expenses |
* | |||
Transfer agent and registrar fees |
* | |||
Miscellaneous |
* | |||
| | | | | |
Total |
$ | * | ||
| | | | | |
| | | | | |
- *
- To be filed by amendment.
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS.
Section 102(b)(7) of the DGCL authorizes a corporation in its certificate of incorporation to eliminate or limit personal liability of directors of the corporation for violations of the directors' fiduciary duty of care. However, directors remain liable for breaches of duties of loyalty, failing to act in good faith, engaging in intentional misconduct, knowingly violating a law, paying a dividend or approving a stock repurchase which was illegal under DGCL Section 174 or obtaining an improper personal benefit. In addition, equitable remedies for breach of fiduciary duty of care, such as injunction or recession, are available.
Our current certificate of incorporation eliminates the personal liability of the members of our board of directors to the fullest extent permitted by the DGCL. Any repeal or modification of that provision by the stockholders of the corporation will not adversely affect any right or protection of a director of the corporation existing at the time of such repeal or modification.
Section 145 of the DGCL provides that a corporation has the power to indemnify a director, officer, employee or agent of the corporation, or a person serving at the request of the corporation for another corporation, partnership, joint venture, trust or other enterprise in related capacities against expenses (including attorneys' fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with an action, suit or proceeding to which he was or is a party or is threatened to be made a party to any threatened, ending or completed action, suit or proceeding by reason of such position, if such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, in any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful, except that, in the case of actions brought by or in the right of the corporation, no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or other adjudicating court determines that, despite the adjudication of liability but in view of all of the circumstances of the
II-1
case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
Our current bylaws provide for indemnification of our officers and directors to the fullest extent permitted by the DGCL.
We have entered into indemnification agreements with each of our directors, and intend to enter into such agreements with each of our officers prior to this offering, pursuant to which we agreed, to the maximum extent permitted by applicable law and subject to the specified terms and conditions set forth in each agreement, to indemnify a director or officer who acts on our behalf and is made or threatened to be made a party to any action or proceeding against expenses, judgments, fines and amounts paid in settlement that are incurred by such officer or director in connection with the action or proceeding. The indemnification provisions apply whether the action was instituted by a third party or by us.
We have purchased and maintain insurance on behalf of our officers and directors that provides coverage for expenses and liabilities incurred by them in their capacities as officers and directors.
In any underwriting agreement we enter into in connection with the sale of common stock being registered hereby, the underwriters will agree to indemnify, under certain conditions, us, our directors, our officers and persons who control us within the meaning of the Securities Act against certain liabilities.
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES.
Since June 6, 2013 (inception), we have issued and sold the following securities without registration under the Securities Act:
- (1)
- In
June 2013, pursuant to a founder stock purchase agreement, we issued 4,000,000 shares of common stock to Napo Pharmaceuticals, Inc. for $400.
- (2)
- From
July through September 2013, pursuant to a note and warrant purchase agreement dated July 8, 2013, we issued convertible promissory notes in the
aggregate principal amount of $525,000 and warrants to purchase 311,498 shares of common stock at an exercise price of $1.6854 per share, which warrants expire February 5, 2019, to four
accredited investors. On February 4, 2014, these noteholders converted the notes in full for an aggregate of 311,498 shares of common stock.
- (3)
- In
February 2014, we issued an aggregate of 2,224,991 shares of Series A preferred stock for aggregate gross proceeds of $5,000,000 to Kunlun
Pharmaceuticals, Ltd., an accredited investor.
- (4)
- In
April 2014, we granted stock options to purchase 1,070,557 shares of common stock under our 2013 Equity Incentive Plan, with an exercise price of $1.69
per share to our executive officers and employees.
- (5)
- In
April 2014, we issued 585,321 shares of Series A preferred stock for aggregate gross proceeds of $1,315,337, to six accredited investors.
- (6)
- In
May 2014, we issued an aggregate of 205,590 shares of Series A preferred stock for aggregate gross proceeds of $462,002, to two accredited
investors.
- (7)
- In June 2014, we granted stock options to purchase 59,116 shares of common stock under our 2013 Equity Incentive Plan, which options have an exercise price of $3.22 per share to a member of our board of directors.
II-2
- (8)
- In
June 2014, we granted 118,953 restricted stock unit awards under our 2013 Equity Incentive Plan to our executive officers and employees.
- (9)
- In
June 2014, pursuant to a convertible note purchase agreement dated June 2, 2014, we issued convertible promissory notes in the aggregate principal
amount of $300,000, to two accredited investors.
- (10)
- In
June 2014, we issued a warrant to purchase 25,000 shares of common stock at an exercise price equal to 90% of the initial public offering price per
share, to a contract manufacturer.
- (11)
- In
July 2014, pursuant to a convertible note purchase agreement dated June 2, 2014, we issued a convertible promissory note in the aggregate
principal amount of $150,000, to an accredited investor.
- (12)
- In August 2014, we entered into a standby line of credit with an individual, who is an accredited investor, for up to $1,000,000. Following closing of this offering, outstanding principal amounts borrowed under the standby line of credit may be converted, at the option of the lender, into shares of our common stock at a conversion price equal to 80% of the initial public offering price per share. In connection with the entry into the standby line of credit, we issued the lender a warrant to purchase 50,000 shares of our common stock at an exercise price equal to 80% of the initial public offering price per share, which expires in August 2016. If an IPO is not consummated prior to August 26, 2015, the exercise price of the warrants will be $2.25 per share.
The offers, sales, and issuances of the securities described in paragraphs (1)-(3), (5), (6) and (9)-(12) above were deemed to be exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act, Regulation D or Regulation S promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited or sophisticated person and had adequate access, through employment, business or other relationships, to information about us.
The offers, sales and issuances of the securities described in paragraphs (4), (7) and (8) above were deemed to be exempt from registration under the Securities Act under Rule 701 promulgated under the Securities Act as offers and sale of securities pursuant to certain compensatory benefit plans and contracts relating to compensation in compliance with Rule 701.
II-3
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
- (a)
- Exhibits. The following exhibits are included herein or incorporated herein by reference.
| Exhibit No. | Description | ||
|---|---|---|---|
| 1.1 | ** | Form of Underwriting Agreement. | |
3.1 |
* |
Amended and Restated Certificate of Incorporation, as amended and as currently in effect. |
|
3.2 |
* |
First Certificate of Amendment to the Amended and Restated Certificate of Incorporation, as currently in effect. |
|
3.3 |
** |
Form of Second Amended and Restated Certificate of Incorporation, to be effective upon the closing of this offering. |
|
3.4 |
* |
Bylaws as currently in effect. |
|
3.5 |
* |
Form of Amended and Restated Bylaws, to be effective upon the closing of this offering. |
|
4.1 |
Specimen Common Stock Certificate of Jaguar Animal Health, Inc. |
||
4.2 |
* |
Investor Rights Agreement by and between Jaguar Animal Health, Inc. and certain of its stockholders, dated February 5, 2014. |
|
5.1 |
** |
Opinion of Reed Smith LLP. |
|
10.1 |
* |
Form of Indemnification Agreement by and between Jaguar Animal Health, Inc. and its directors and officers. |
|
10.2 |
* |
Jaguar Animal Health, Inc. 2013 Equity Incentive Plan. |
|
10.3 |
* |
Form of Notice of Grant of Stock Option and Stock Option Agreement under the 2013 Equity Incentive Plan. |
|
10.4 |
* |
Form of Notice of Grant of Restricted Stock Units and Restricted Stock Unit Agreement under the 2013 Equity Incentive Plan. |
|
10.5 |
* |
Jaguar Animal Health, Inc. 2014 Stock Incentive Plan. |
|
10.6 |
* |
Form of Notice of Grant of Stock Option and Stock Option Agreement under the 2014 Stock Incentive Plan. |
|
10.7 |
* |
Form of Notice of Grant of Restricted Stock and Restricted Stock Agreement under the 2014 Stock Incentive Plan. |
|
10.8 |
* |
Form of Notice of Grant of Restricted Stock Units and Restricted Stock Unit Agreement under the 2014 Stock Incentive Plan. |
|
10.9 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Lisa A. Conte, dated March 1, 2014. |
|
10.10 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Serge Martinod, D.V.M., Ph.D., dated January 28, 2014. |
|
10.11 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Steven R. King, Ph.D., dated February 28, 2014. |
|
10.12 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Charles O. Thompson, dated February 28, 2014. |
|
10.13 |
* |
Amended and Restated License Agreement by and between Jaguar Animal Health, Inc. and Napo Pharmaceuticals, Inc., dated August 6, 2014. |
|
10.14 |
* |
Employee Leasing and Overhead Allocation Agreement by and between Jaguar Animal Health, Inc. and Napo Pharmaceuticals, Inc., dated July 1, 2013. |
|
II-4
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.15 | * | Assignment of Sublease and Landlord Consent by and between Jaguar Animal Health, Inc. and Napo Pharmaceuticals, Inc., dated June 1, 2014. | |
10.16 |
* |
Form of Common Stock Warrant that expires February 5, 2019. |
|
10.17 |
* |
Form of Common Stock Warrant issued to Indena S.p.A. that expires June 26, 2019. |
|
10.18 |
* |
Form of Convertible Note Purchase Agreement dated June 2, 2014 by and between Jaguar Animal Health, Inc. and certain of its Investors. |
|
10.19 |
* |
Form of Convertible Promissory Note. |
|
10.20 |
* |
Line of Credit Loan Agreement by and between Jaguar Animal Health, Inc. and Joshua Mailman, dated August 26, 2014. |
|
10.21 |
* |
Form of Common Stock Warrant issued to Joshua Mailman that expires August 26, 2016. |
|
10.22 |
Offer Letter by and between Jaguar Animal Health, Inc. and John A. Kallassy, dated as of September 19, 2014. |
||
10.23 |
Non-Disturbance Letter Agreement by and between Napo Pharmaceuticals, Inc. and Nantucket Investments Limited, as Administrative Agent and Collateral Agent, dated October 10, 2014. |
||
10.24 |
Separation Letter Agreement by and between Jaguar Animal Health, Inc. and Charles O. Thompson, dated October 2, 2014. |
||
23.1 |
Consent of Independent Registered Public Accounting Firm. |
||
23.2 |
** |
Consent of Reed Smith LLP (included in Exhibit 5.1). |
|
24.1 |
* |
Power of Attorney (included on page II-7 of the original filing of this registration statement on Form S-1). |
|
- *
- Previously filed.
- **
- To be filed by amendment.
II-5
- (b)
- Financial Statement Schedules. See page F-1.
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
The undersigned registrant hereby undertakes that:
(a) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(b) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
II-6
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this Amendment No. 2 to the Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of San Francisco, State of California, on October 10, 2014.
| JAGUAR ANIMAL HEALTH, INC. | ||||||
By: |
/s/ LISA A. CONTE |
|||||
| Name: | Lisa A. Conte | |||||
| Title: | Chief Executive Officer and President | |||||
Pursuant to the requirements of the Securities Act of 1933, this Amendment No. 2 to the Registration Statement has been signed by the following persons in the capacities and on the date indicated.
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ LISA A. CONTE Lisa A. Conte |
Chief Executive Officer, President and Director (Principal Executive Officer) | October 10, 2014 | ||
/s/ JOHN A. KALLASSY John A. Kallassy |
Chief Financial Officer, Chief Operating Officer and Treasurer (Principal Financial and Accounting Officer) |
October 10, 2014 |
||
* James J. Bochnowski |
Chairman of the Board |
October 10, 2014 |
||
* Jiahao Qiu |
Director |
October 10, 2014 |
||
* Zhi Yang, Ph.D. |
Director |
October 10, 2014 |
| *By: | /s/ LISA A. CONTE Lisa A. Conte, Attorney-in-Fact |
II-7
| Exhibit No. | Description | ||
|---|---|---|---|
| 1.1 | ** | Form of Underwriting Agreement. | |
3.1 |
* |
Amended and Restated Certificate of Incorporation, as amended and as currently in effect. |
|
3.2 |
* |
First Certificate of Amendment to the Amended and Restated Certificate of Incorporation, as currently in effect. |
|
3.3 |
** |
Form of Second Amended and Restated Certificate of Incorporation, to be effective upon the closing of this offering. |
|
3.4 |
* |
Bylaws as currently in effect. |
|
3.5 |
* |
Form of Amended and Restated Bylaws, to be effective upon the closing of this offering. |
|
4.1 |
Specimen Common Stock Certificate of Jaguar Animal Health, Inc. |
||
4.2 |
* |
Investor Rights Agreement by and between Jaguar Animal Health, Inc. and certain of its stockholders, dated February 5, 2014. |
|
5.1 |
** |
Opinion of Reed Smith LLP. |
|
10.1 |
* |
Form of Indemnification Agreement by and between Jaguar Animal Health, Inc. and its directors and officers. |
|
10.2 |
* |
Jaguar Animal Health, Inc. 2013 Equity Incentive Plan. |
|
10.3 |
* |
Form of Notice of Grant of Stock Option and Stock Option Agreement under the 2013 Equity Incentive Plan. |
|
10.4 |
* |
Form of Notice of Grant of Restricted Stock Units and Restricted Stock Unit Agreement under the 2013 Equity Incentive Plan. |
|
10.5 |
* |
Jaguar Animal Health, Inc. 2014 Stock Incentive Plan. |
|
10.6 |
* |
Form of Notice of Grant of Stock Option and Stock Option Agreement under the 2014 Stock Incentive Plan. |
|
10.7 |
* |
Form of Notice of Grant of Restricted Stock and Restricted Stock Agreement under the 2014 Stock Incentive Plan. |
|
10.8 |
* |
Form of Notice of Grant of Restricted Stock Units and Restricted Stock Unit Agreement under the 2014 Stock Incentive Plan. |
|
10.9 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Lisa A. Conte, dated March 1, 2014. |
|
10.10 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Serge Martinod, D.V.M., Ph.D., dated January 28, 2014. |
|
10.11 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Steven R. King, Ph.D., dated February 28, 2014. |
|
10.12 |
* |
Offer Letter by and between Jaguar Animal Health, Inc. and Charles O. Thompson, dated February 28, 2014. |
|
10.13 |
* |
Amended and Restated License Agreement by and between Jaguar Animal Health, Inc. and Napo Pharmaceuticals, Inc., dated August 6, 2014. |
|
10.14 |
* |
Employee Leasing and Overhead Allocation Agreement by and between Jaguar Animal Health, Inc. and Napo Pharmaceuticals, Inc., dated July 1, 2013. |
|
10.15 |
* |
Assignment of Sublease and Landlord Consent by and between Jaguar Animal Health, Inc. and Napo Pharmaceuticals, Inc., dated June 1, 2014. |
|
II-8
| Exhibit No. | Description | ||
|---|---|---|---|
| 10.16 | * | Form of Common Stock Warrant that expires February 5, 2019. | |
10.17 |
* |
Form of Common Stock Warrant issued to Indena S.p.A. that expires June 26, 2019. |
|
10.18 |
* |
Form of Convertible Note Purchase Agreement dated June 2, 2014 by and between Jaguar Animal Health, Inc. and certain of its Investors. |
|
10.19 |
* |
Form of Convertible Promissory Note. |
|
10.20 |
* |
Line of Credit Loan Agreement by and between Jaguar Animal Health, Inc. and Joshua Mailman, dated August 26, 2014. |
|
10.21 |
* |
Form of Common Stock Warrant issued to Joshua Mailman that expires August 26, 2016. |
|
10.22 |
Offer Letter by and between Jaguar Animal Health, Inc. and John A. Kallassy, dated as of September 19, 2014. |
||
10.23 |
Non-Disturbance Letter Agreement by and between Napo Pharmaceuticals, Inc. and Nantucket Investments Limited, as Administrative Agent and Collateral Agent, dated October 10, 2014. |
||
10.24 |
Separation Letter Agreement by and between Jaguar Animal Health, Inc. and Charles O. Thompson, dated October 2, 2014. |
||
23.1 |
Consent of Independent Registered Public Accounting Firm. |
||
23.2 |
** |
Consent of Reed Smith LLP (included in Exhibit 5.1). |
|
24.1 |
* |
Power of Attorney (included on page II-7 of the original filing of this registration statement on Form S-1). |
|
- *
- Previously filed.
- **
- To be filed by amendment.
II-9