Attached files
| file | filename |
|---|---|
| 8-K - 8-K - Radius Health, Inc. | a14-11420_18k.htm |
| EX-99.2 - EX-99.2 - Radius Health, Inc. | a14-11420_1ex99d2.htm |
| EX-10.1 - EX-10.1 - Radius Health, Inc. | a14-11420_1ex10d1.htm |
Exhibit 99.1
BUSINESS
OVERVIEW
We are a science-driven biopharmaceutical company focused on developing novel differentiated therapeutics for patients with osteoporosis as well as other serious endocrine-mediated diseases. Our lead product candidate is abaloparatide (BA058), a bone anabolic for the treatment of osteoporosis delivered via subcutaneous injection, which we refer to as Abaloparatide-SC. We are currently in Phase 3 development of Abaloparatide-SC and expect to announce top-line data from this study in late 2014. If the results are positive, we plan to submit a new drug application, or NDA, in the United States, and a marketing authorization application, or MAA, in Europe, in mid-2015. We hold worldwide commercialization rights to Abaloparatide-SC, other than in Japan, and with a favorable regulatory outcome, we anticipate our first commercial sales of Abaloparatide-SC will take place in 2016. We are leveraging our investment in Abaloparatide-SC to develop Abaloparatide-TD. We expect this line extension will provide improved patient convenience by enabling administration of abaloparatide through a short-wear-time transdermal patch. We have recently completed a successful Phase 2 proof of concept study.
Our current clinical product portfolio also includes a novel oral agent, RAD1901, a selective estrogen receptor down-regulator/degrader, or SERD. We are developing RAD1901 at higher doses for the treatment of breast cancer brain metastases, or BCBM, and at lower doses as a selective estrogen-receptor modulator, or SERM, for the treatment of vasomotor symptoms such as hot flashes. In 2014, we expect to commence a Phase 1 clinical trial to evaluate RAD1901 for the treatment of BCBM, and we previously completed a successful Phase 2 clinical trial of RAD1901 for the treatment of vasomotor symptoms.
Abaloparatide
Abaloparatide is a novel synthetic peptide analog of parathyroid hormone-related protein, or PTHrP, that we are developing as a bone anabolic treatment for osteoporosis. Osteoporosis is a disease that affects nearly 10 million people in the United States, with an additional approximately 43 million people at increased risk for the disease. It is characterized by low bone mass and structural deterioration of bone tissue, which leads to greater fragility and an increase in fracture risk. Anabolic agents, like Forteo (teriparatide), are used to increase bone mineral density, or BMD, and to reduce the risk of fracture. We believe abaloparatide has the potential to increase BMD and bone quality to a greater degree and at a faster rate than other approved drugs for the treatment of osteoporosis. We are developing two formulations of abaloparatide:
· Abaloparatide-SC is an injectable subcutaneous formulation of abaloparatide. In August 2009, we announced positive Phase 2 data that showed Abaloparatide-SC produced faster and greater BMD increases at the spine and the hip with substantially less hypercalcemia than Forteo (teriparatide), which is the only approved subcutaneous injectable anabolic agent for the treatment of osteoporosis in the United States. A subsequent Phase 2 clinical
trial announced in January 2014 also confirmed the results of our first clinical trial by demonstrating that Abaloparatide-SC produces BMD increases from baseline in the spine and hip that are comparable to our earlier Phase 2 trial. In April 2011, we commenced a Phase 3 clinical trial of Abaloparatide-SC. Enrollment was completed in March 2013, and we expect to announce top-line data at the end of the fourth quarter of 2014. Assuming a favorable outcome, we plan to use the results from this Phase 3 clinical trial to support a new drug application, or NDA, with the U.S. Food and Drug Administration, or FDA, and believe we could obtain approval of the NDA in 2016.
· Abaloparatide-TD is a line extension of Abaloparatide SC in the form of a convenient, short-wear-time (approximately five minutes) transdermal patch. In a recent Phase 2 clinical trial, Abaloparatide-TD demonstrated a statistically significant mean percent increase from baseline in BMD as compared to placebo at the lumbar spine and at the hip. These results demonstrated a clear proof of concept by achieving a dose dependent increase in BMD. Following additional formulation development work, we intend to advance an optimized Abaloparatide-TD product in additional clinical studies and to a Phase 3 bridging study and to subsequently submit for approval. We hold worldwide commercialization rights to Abaloparatide-TD technology.
RAD1901
RAD1901 is a SERD that we believe crosses the blood-brain barrier and that we are evaluating for the treatment of BCBM. RAD1901 has been shown to bind with good selectivity to the estrogen receptor and to have both estrogen-like and estrogen-antagonistic effects in different tissues. In many cancers, hormones, like estrogen, stimulate tumor growth and a desired therapeutic goal is to block this estrogen-dependent growth while inducing apoptosis of the cancer cells. SERDs are an emerging class of endocrine therapies that directly induce estrogen-receptor, or ER, degradation, enabling them to remove the estrogen growth signal in ER-dependent tumors without allowing ligand-independent resistance to develop. There is currently only one SERD, Faslodex (fulvestrant), approved for the treatment of hormone-receptor positive metastatic breast cancer; however, for patients with brain metastases (BCBM), there are no approved targeted therapies that cross the blood-brain barrier. We believe there is a significant opportunity for RAD1901 to be the first ER-targeted therapy that crosses the blood-brain barrier to more effectively treat ER-positive BCBM and potentially reduce both intracranial and extracranial BCBM tumors. We intend to commence a Phase 1 clinical trial in 2014 to evaluate high-dose RAD1901 for the treatment of BCBM. In March 2014, we submitted to the FDA an application for orphan medicinal product designation of RAD1901 for the treatment of BCBM.
We are also developing RAD1901 at lower doses as a selective estrogen receptor modulator, or SERM, for the treatment of vasomotor symptoms. Historically, hormone replacement therapy, or HRT, with estrogen or progesterone was considered the most efficacious approach to relieving menopausal symptoms such as hot flashes. However, because of the concerns about the potential long-term risks and contraindications associated with HRT, we believe a significant need exists
for new therapeutic treatment options to treat vasomotor symptoms. In a Phase 2 proof of concept study, RAD1901 at lower doses demonstrated a reduction in the frequency and severity of moderate and severe hot flashes. We believe RAD1901 is an attractive candidate for advancement into Phase 2b development as a treatment for vasomotor symptoms.
Additional information regarding our clinical trials, their designs and the results of previously completed clinical trials is contained in or incorporated by reference into our public filings with the Securities and Exchange Commission. The U.S. National Institutes of Health also provides a database of human clinical trials, that includes our Phase 2 or later clinical trials, which can be found at www.clinicaltrials.gov. The information contained in, or that can be accessed through, this website is not part of, and is not incorporated into, this filing and should not be considered part of this filing.
OUR OPPORTUNITY
Osteoporosis
Osteoporosis is a disease characterized by low bone mass and structural deterioration of bone tissue, which leads to greater fragility and an increase in fracture risk. All bones become more fragile and susceptible to fracture as the disease progresses. People tend to be unaware that their bones are getting weaker, and a person with osteoporosis can fracture a bone from even a minor fall. The debilitating effects of osteoporosis have substantial costs. Loss of mobility, admission to nursing homes and dependence on caregivers are all common consequences of osteoporosis. The prevalence of osteoporosis is growing and, according to the National Osteoporosis Foundation, or NOF, is significantly under-recognized and under-treated in the population. While the aging of the population is a primary driver of an increase in cases, osteoporosis is also increasing from the use of drugs that induce bone loss, such as chronic use of glucocorticoids and aromatase inhibitors that are increasingly used for breast cancer and the hormone therapies used for prostate cancer.
The NOF has estimated that 10 million people in the United States, composed of eight million women and two million men, already have osteoporosis, and another approximately 43 million have low bone mass placing them at increased risk for osteoporosis. In addition, the NOF has estimated that osteoporosis is responsible for more than two million fractures in the United States each year resulting in an estimated $19 billion in costs annually. The NOF expects that the number of fractures in the United States due to osteoporosis will rise to three million by 2025, resulting in an estimated $25.3 billion in costs each year. Worldwide, osteoporosis affects an estimated 200 million women according to the International Osteoporosis Foundation, or IOF, and causes more than 8.9 million fractures annually, which is equivalent to an osteoporotic fracture occurring approximately every three seconds. The IOF has estimated that 1.6 million hip fractures occur worldwide each year, and by 2050 this number could reach between 4.5 million
and 6.3 million. The IOF estimates that in Europe alone, the annual cost of osteoporotic fractures could surpass €76 billion by 2050.
There are two main types of osteoporosis drugs currently available in the United States, anti-resorptive agents and anabolic agents. Anti-resorptive agents act to prevent further bone loss by inhibiting the breakdown of bone, whereas anabolic agents stimulate bone formation to build new, high-quality bone. According to industry sources, sales of these drugs in the United States, Japan and the five major markets in Europe exceeded $6 billion in 2011. We believe there is a large unmet need in the market for osteoporosis treatment because existing therapies have shortcomings in efficacy, tolerability and convenience. For example, one current standard of care, bisphosphonates, an anti-resorptive agent, has been associated with infrequent but serious adverse events, such as osteonecrosis of the jaw and atypical fractures, especially of long bones. These side effects, although uncommon, have created increasing concern with physicians and patients. Many physicians are seeking alternatives to bisphosphonates. The two primary alternatives to bisphosphonates that are approved for the treatment of osteoporosis, Lilly’s Forteo and Amgen’s Prolia, had reported sales of approximately $1.2 billion and $700 million, respectively, in 2013. Forteo, a 34 amino acid recombinant peptide of human parathyroid hormone, is the only anabolic drug approved in the United States for the treatment of osteoporosis. We believe there is a significant opportunity for an anabolic agent that is able to increase BMD to a greater degree and at a faster rate than other approved drugs for the treatment of osteoporosis with added advantages in convenience and safety.
Our Solution — Abaloparatide
Abaloparatide is a novel synthetic peptide analog of PTHrP that we are developing as a bone anabolic treatment for osteoporosis. PTHrP, unlike parathyroid hormone, is critical in the formation of the skeleton, is involved in the regulation of bone formation and is able to rebuild bone with low associated risk of inducing the presence of too much calcium in the blood, known as hypercalcemia, as a side effect. We believe that abaloparatide is the most advanced PTHrP analog in clinical development for the treatment of osteoporosis and that it can provide the following advantages over other current standard of care treatments for osteoporosis:
· improved efficacy — greater bone build at hip and spine;
· faster benefit for building bone;
· shorter treatment duration;
· less hypercalcemia;
· no additional safety risks; and
· no refrigeration required in use.
Abaloparatide-SC. In August 2009, we announced positive Phase 2 data that showed Abaloparatide-SC produced faster and greater BMD increases at the spine and the hip with substantially less hypercalcemia than Forteo, the only approved anabolic agent for the treatment
of osteoporosis in the United States. Specifically, our study demonstrated that total hip BMD showed a more than five-fold benefit of Abaloparatide-SC at a dose of 80µg over Forteo after 24 weeks. Abaloparatide-SC at 80µg increased mean lumbar spine BMD by 6.7% at 24 weeks, compared to 5.5% with Forteo, and by 12.9% at 48 weeks, compared to 8.6% with Forteo. A subsequent Phase 2 clinical trial announced in January 2014 also confirmed the results of the first clinical trial by demonstrating that Abaloparatide-SC produces increases in BMD from baseline in the spine and hip that are comparable to the earlier Phase 2 trial. In April 2011, we began dosing patients in a pivotal, multinational Phase 3 clinical trial designed to show that Abaloparatide-SC prevents new vertebral fracture compared to placebo. We completed enrollment in this Phase 3 clinical study in March 2013 and expect to report top-line 18-month fracture data at the end of the fourth quarter of 2014. If the data from this Phase 3 clinical trial are positive, we plan to submit the NDA and the MAA for Abaloparatide-SC in mid-2015. We expect commercial launch to follow in 2016 in the United States, if and when the FDA approves the NDA for Abaloparatide-SC, and in the EU if and when an MAA for Abaloparatide-SC is approved.
Abaloparatide-TD. Abaloparatide-TD is a convenient, short-wear-time transdermal patch formulation of abaloparatide with Phase 2 clinical results suggesting efficacy, safety and tolerability in the treatment of osteoporosis. In January 2014, we reported positive results from a Phase 2 clinical trial of Abaloparatide-TD which showed at each dose there was a statistically significant mean percent increase from baseline in BMD, as compared to placebo, at the lumbar spine. Additionally, at the 100 µg and 150 µg doses, there was a statistically significant mean percent increase from baseline in BMD, as compared to placebo, at the hip. In addition, there was a consistent dose effect observed with increasing doses of Abaloparatide-TD, with a statistically significant dosing trend seen for changes in both spine and total hip BMD. As a result, we believe that by offering an alternative to daily injections, Abaloparatide-TD, if approved, could further improve patient outcomes by increasing patient acceptance.
We currently plan to optimize Abaloparatide-TD to achieve efficacy comparable to that of Abaloparatide-SC. If Abaloparatide-SC is approved by the FDA, we believe that we will only need to conduct a single non-inferiority Phase 3 clinical trial comparing the change in lumbar spine BMD at 12 months for patients dosed with Abaloparatide-TD to patients dosed with Abaloparatide-SC to show that the effect of Abaloparatide-TD treatment is comparable to that of Abaloparatide-SC. If our clinical trials of Abaloparatide-SC and Abaloparatide-TD are successful, we expect to seek marketing approval of Abaloparatide-TD as a line extension of Abaloparatide-SC. The FDA approval of Abaloparatide-TD, and the timing of any such approval, is dependent upon the approval of Abaloparatide-SC.
Metastatic Breast Cancer
According to the World Health Organization, breast cancer is the second most common cancer in the world and the most prevalent cancer in women, accounting for 16% of all female cancers.
The major cause of death from breast cancer is metastases, most commonly to the bone, liver, lung and brain. About 5% of patients have distant metastases at the time of diagnoses, and these patients have a 5-year survival rate of only 24%, compared with a greater than 98% survival rate for patients with only local disease. Importantly, even patients without metastases at diagnosis are at risk for developing metastases over time.
Approximately, 70% of breast cancers express the estrogen receptor, or ER, and depend on estrogen signaling for growth and survival. There are three main classes of therapies for ER-positive tumors available: aromatase inhibitors, or AIs; SERMs; and SERDs. AIs, which block the generation of estrogen, and SERMs, which selectively inhibit an ER’s ability to bind estrogen, both block ER-dependent signaling but leave functional ERs present on breast cancer cells. For this reason, although these classes of drugs are effective as adjuvants for breast cancer, patients’ tumors often acquire resistance to them by developing the ability to signal through the ER in a ligand-independent manner. SERDs, in contrast, are an emerging class of endocrine therapies that directly induce ER degradation. Therefore, these agents should be able to treat ER-dependent tumors without allowing ligand-independent resistance to develop, and they should also be able to act on AI- and SERM-resistant ER-positive tumors. Symptomatic BCBM occur in 10% to 16% of patients with metastatic breast cancer and are associated with particularly high morbidity and mortality. Current standard of care involves a combination of whole-brain radiotherapy, surgery or stereotactic radiosurgery, depending on the clinical context. These treatments are often only marginally effective, and are themselves associated with significant morbidity and mortality.
There is currently only one SERD approved for the treatment of ER-positive metastatic breast cancer, but there are no approved targeted therapies that cross the blood-brain barrier and can treat patients with ER-positive BCBM. We believe a significant opportunity exists for a SERD that can cross the blood-brain barrier to more effectively treat ER-positive BCBM and potentially delay or eliminate the need for radiation or surgery.
Our Solution — RAD1901
We are developing RAD1901 as a high-dose SERD in an oral formulation in Phase 1 clinical development for the treatment of BCBM. RAD1901 has been shown to bind with good selectivity to the estrogen hormone receptor and to have both estrogen-like and estrogen-antagonist effects in different tissues. In cell culture, RAD1901 does not stimulate replication of breast cancer cells, and antagonizes the stimulating effects of estrogen on cell proliferation. Furthermore, in breast cancer cell lines a dose dependent down regulation of ER, has been observed, a process we have shown to involve proteosomal mediated degradation pathway. In a model of breast cancer in which human breast cancer cells are implanted in mice and allowed to establish tumors in response to estrogen treatment, we have shown that treatment with RAD1901 results in decreased tumor growth. Studies with RAD1901 have established the pharmacokinetic profile, including demonstration of good oral bioavailability and the ability of RAD1901 to cross
the blood-brain barrier, with therapeutic levels detectable in the brain. We believe that RAD1901 could offer the following advantages over other current standard of care treatments for BCBM:
· ability to penetrate the blood-brain barrier;
· oral administration; and
· treatment of hormone-resistant breast cancers.
In late 2014, we intend to advance the development of high-dose RAD1901 for the treatment of ER-positive BCBM with the initiation of a Phase 1b clinical trial.
Vasomotor symptoms
Vasomotor symptoms, such as hot flashes and night sweats, are common during menopause, with up to 85% of women experiencing them during the menopause transition, for a median duration of four years. In 2010, approximately 11.5 million women in the United States were in the 45 to 49 year age range upon entering perimenopause/menopause. In addition, most women receiving systemic therapy for breast cancer suffer hot flashes, often with more severe or prolonged symptoms than women experiencing natural menopause. These symptoms can disrupt sleep and interfere with quality of life. An estimated two million women go through menopause every year in the United States, with a total population of 50 million postmenopausal women.
Historically, hormone replacement therapy, or HRT, with estrogen and/or progesterone was considered the most efficacious approach to relieving menopausal symptoms such as hot flashes. However, data from the Women’s Health Initiative, or WHI, identified increased risks for malignancy and cardiovascular disease associated with estrogen therapy. Sales of HRT declined substantially after the release of the initial WHI data, but HRT remains the current standard of care for many women suffering from hot flashes. However, due to concerns about the potential long-term risks and contraindications associated with HRT, we believe that there is a significant need for new therapeutic options to treat vasomotor symptoms.
Our Solution — RAD1901
We are developing RAD1901 as a low dose SERM in an oral formulation for the treatment of vasomotor symptoms. We conducted a Phase 2 proof of concept study in 100 healthy perimenopausal women using four doses of RAD1901 — 10 mg, 25 mg, 50 mg and 100 mg — and placebo. While a classic dose-response effect was not demonstrated, at the 10 mg dose level RAD1901 achieved a statistically significant reduction in the frequency of moderate and severe hot flashes both by linear trend test and by comparison to placebo and in overall hot flashes at either the two-, three- or four-week time-points. A similar reduction in composite score — frequency × severity of hot flashes — was identified at all time-points, with a statistically significant difference from placebo achieved at the two-, three- or four-week time-points.
We anticipate our next clinical study will be a Phase 2b study conducted in approximately 200 perimenopausal women experiencing a high frequency of hot flashes at baseline. The main study endpoints would be an assessment of the change in the frequency and severity of moderate and severe hot flashes.
OUR STRATEGY
Our goal is to become a leading provider of therapeutics for osteoporosis and other serious endocrine-mediated diseases. To achieve this goal we plan to:
· Advance the development and obtain regulatory approval of Abaloparatide-SC. We are evaluating Abaloparatide-SC in an ongoing Phase 3 clinical trial for the treatment of osteoporosis and expect to report top-line 18-month fracture data at the end of the fourth quarter of 2014. If the results are positive, we plan to submit an NDA for Abaloparatide-SC in the United States, and an MAA in the European Union, in mid-2015.
· Extend the lifecycle of abaloparatide through the continued development of Abaloparatide-TD. We are developing Abaloparatide-TD as a short-wear-time transdermal patch and we anticipate, pending a favorable regulatory outcome, commercial launch two to three years after the first commercial sale of Abaloparatide-SC. If Abaloparatide-SC is approved by the FDA, we believe that we will only need to conduct a single non-inferiority Phase 3 clinical trial comparing the change in BMD for patients dosed with Abaloparatide-TD as compared to patients dosed with Abaloparatide-SC. If our clinical trials of Abaloparatide-SC and Abaloparatide-TD are successful, we expect to seek marketing approval of Abaloparatide-TD as a line extension of Abaloparatide-SC.
· Establish internal sales and marketing capabilities to commercialize our product candidates in the United States. We currently plan to commercialize any of our approved product candidates by developing an internal sales force focused on specialists in core strategic markets in the United States. We believe that we can effectively target those markets using a sales force of approximately 150 representatives and that by doing so we can achieve a greater return on our product investment than if we license our products to third parties for sale. We plan to expand the use of our products to primary care physicians through selective co-promotion partnerships. Our management team has experience commercializing products in these core strategic markets, and understands the relevant sales, marketing and reimbursement requirements.
· Selectively pursue collaborations to commercialize our product candidates outside the United States. We intend to seek to enter into one or more collaborations for the commercialization of our approved product candidates in strategic markets in Europe and in other countries worldwide.
· Continue to expand our product portfolio. We plan to leverage our drug development expertise to discover and develop additional product candidates focused on serious endocrine-related diseases and conditions, including the development of RAD1901 as a potential treatment for ER-positive BCBM. We may also consider opportunistically expanding our product portfolio through in-licensing, acquisitions or partnerships.
OUR PRODUCT CANDIDATES
The following table identifies the product candidates in our current product portfolio, their proposed indication and stage of development:

Abaloparatide
Overview
Abaloparatide is a novel synthetic peptide analog of parathyroid hormone-related protein, or PTHrP, that we are developing as a bone anabolic treatment for osteoporosis. PTHrP, unlike PTH, is critical in the formation of the skeleton, is involved in the regulation of bone formation and is able to rebuild bone with low associated risk of inducing the presence of too much calcium in the blood, known as hypercalcemia, as a side effect. Human PTHrP (a protein of 139 to 173 amino acids) is different from PTH (a protein of 84 amino acids) in its structure and role. In 2009, the medical journal, Nature Chemical Biology, published the results of a study
indicating that PTH (which primarily regulates calcium homeostasis and bone resorption) and PTHrP activate the same parathyroid hormone receptor, or PTHR1, but produce divergent effects in bone due to differences in receptor conformation selectivity, receptor localization and downstream cell signaling. Forteo is a 34 amino acid recombinant peptide of human parathyroid hormone. We believe that abaloparatide is the most advanced PTHrP analog in clinical development for the treatment of osteoporosis. We acquired and maintain exclusive worldwide rights, excluding Japan, to certain patents, data and technical information related to abaloparatide through a license agreement with an affiliate of Ipsen Pharma SAS, or Ipsen.
We are developing abaloparatide for the prevention of fractures in postmenopausal women at risk of fracture from severe osteoporosis. Recognizing both the therapeutic potential of abaloparatide in this indication as well as the drawbacks inherent in self-injection therapies in this population, we are also developing Abaloparatide-TD for transdermal administration of the product using a microneedle technology from 3M. We plan to develop and register Abaloparatide-SC as our lead product, with Abaloparatide-TD as a line extension that provides greater patient convenience. We believe the ability of Abaloparatide-TD to capitalize on the more extensive fracture study data of Abaloparatide-SC will allow the patch product to be accelerated through later-phase development without requiring its own fracture study.
Abaloparatide-SC
We are developing Abaloparatide-SC as a once daily sub-cutaneous injection of abaloparatide for the treatment of osteoporosis. We have completed two Phase 2 clinical trials of Abaloparatide-SC. We announced results from our first Phase 2 clinical trial in August 2009, which showed that Abaloparatide-SC produced faster and greater BMD increases at the spine and the hip with substantially less hypercalcemia than Forteo, the only approved anabolic agent for the treatment of osteoporosis in the United States. Specifically, our study demonstrated that total hip BMD showed a more than five-fold benefit of Abaloparatide-SC at a dose of 80µg over Forteo after 24 weeks. Abaloparatide-SC at 80µg increased mean lumbar spine BMD by 6.7% at 24 weeks, compared to 5.5% with Forteo, and by 12.9% at 48 weeks, compared to 8.6% with Forteo. Abaloparatide-SC at 80µg increased mean femoral neck BMD by 3.1% at 24 weeks, compared to 1.1% with Forteo, and 0.8% with a placebo, and by 4.1% at 48 weeks, compared to 2.2% with Forteo. At 24 weeks, 66% of Abaloparatide-SC treated patients had increases in BMD at the lumbar spine, total hip and femoral neck, compared with 29% of placebo treated patients (p=0.0015) and 40% of Forteo treated patients (p=0.0250). In January 2014, we reported positive data from a second Phase 2 clinical trial of abaloparatide. Consistent with our Phase 2 clinical trial of Abaloparatide-SC completed in 2009, our second clinical trial demonstrated that patients who received a 80 µg dose of Abaloparatide-SC experienced increases in BMD from baseline in the lumbar spine (5.8% increase from baseline), total hip (2.7% increase from baseline) and femoral neck (2.7% increase from baseline and 3.1% increase net placebo). In addition to the BMD results, these study results add to the safety data from the prior Phase 2 clinical study with Abaloparatide-SC, which demonstrated that abaloparatide is generally safe and well tolerated.
In April 2011, we commenced our ongoing Phase 3 clinical trial of Abaloparatide-SC, which completed enrollment in March 2013 with 2,463 subjects. The trial was designed to enroll 2,400 subjects that would be randomized equally to receive daily doses of one of the following: 80 µg of abaloparatide, a matching placebo, or the approved dose of 20 µg of Forteo for 18 months. The trial is designed to test our belief that abaloparatide is superior to placebo for the prevention of vertebral fracture and to Forteo for greater BMD improvement at major skeletal sites and for a lower occurrence of hypercalcemia. We believe the trial will also show that BMD gains for abaloparatide patients will occur earlier than for Forteo patients. In 2012 we participated in a Type A meeting with the Division of Reproductive and Urologic Products of the FDA and discussed the Abaloparatide-SC single pivotal placebo-controlled, comparative Phase 3 fracture study. The FDA indicated that it wanted us to provide additional feedback on the design of our ongoing Phase 3 clinical trial so that the data would be adequate for submission of an NDA for the treatment of osteoporosis. Following this meeting, we amended our protocol to incorporate changes in response to our discussions with the FDA, which included the addition of a 6-month extension study during which patients will receive an approved alendronate therapy in order to obtain 24-month fracture data. The FDA determination of the approvability of any NDA is made based on their independent assessment of the totality of the data submitted. Based on our discussions with the FDA, we believe that a successful, single pivotal placebo-controlled, comparative Phase 3 fracture study will be sufficient to support approval of Abaloparatide-SC for the treatment of osteoporosis in the United States. We believe that the use of a single pivotal placebo-controlled comparative Phase 3 fracture study is consistent with the approach taken with Forteo and Prolia, which were each approved by the FDA for the treatment of osteoporosis in the United States on the basis of a single pivotal placebo-controlled Phase 3 fracture study.
Before we submit an NDA to the FDA for Abaloparatide-SC as a treatment for osteoporosis, we must complete our pivotal Phase 3 clinical trial based upon 18-month fracture data, a carcinogenicity study in rats, and bone quality studies in rats and monkeys. We also may need to complete several additional studies including, a thorough QT Phase 1 study, a Phase 1 pharmacokinetic, or PK, study in renal patients, a Phase 1 absolute bioavailability PK study and several drug interaction studies. Assuming we do not encounter any unforeseen delays during the course of developing Abaloparatide-SC, we expect to present top-line 18-month fracture data at the end of the fourth quarter of 2014. If the data from this Phase 3 clinical trial are positive, and we have completed the other necessary trials, we plan to submit the NDA for
Abaloparatide-SC in mid-2015. We expect commercial launch to follow in the United States, if and when the FDA approves the NDA for Abaloparatide-SC.
We understand that Phase 3 clinical trials with similar size, design and endpoints as our Phase 3 clinical trial have been sufficient to support registration with the European Medicines Agency, or the EMA, for other bone anabolic drugs used to treat women with osteoporosis in the European Union, or the EU. In December 2012, we met with the Swedish Medical Products Agency, or the MPA, to review the design and the overall progress of the Phase 3 clinical trial. The MPA
confirmed that the program, based on the current single pivotal trial design, could support the submission and potential approval of an MAA in the EU, depending on the results of the Phase 3 clinical trial.
Abaloparatide-TD
We are developing Abaloparatide-TD as a line extension of Abaloparatide-SC in a short-wear-time transdermal patch formulation. In January 2014, we reported positive data from our Phase 2 clinical trial of Abaloparatide-TD. The results demonstrated that for each Abaloparatide-TD dose there was a statistically significant mean percent increase from baseline in BMD at the lumbar spine, as compared to placebo. For the 100 µg and 150 µg Abaloparatide-TD doses, there was also a statistically significant mean percent increase from baseline in BMD at the hip, as compared to placebo. The highest Abaloparatide-TD dose of 150 µg produced increases in BMD from baseline in the lumbar spine and total hip of +2.9% and +1.5%, respectively, compared to changes in the placebo group of +0.04% and -0.02%, respectively. In addition, there was a consistent dose effect seen with increasing doses of Abaloparatide-TD, with a statistically significant dosing trend seen for changes in both spine and total hip BMD. Further, the overall tolerability and safety profile was acceptable, there were no clinically significant signs of anti-abaloparatide antibodies, and patient ratings of patch adhesion and local skin response to the transdermal patch technology were also acceptable.
In order to further enhance BMD efficacy, we currently plan to modify the pharmacokinetic profile of Abaloparatide-TD to more closely resemble that of Abaloparatide-SC. If Abaloparatide-SC is already approved by the FDA, we believe that we will only need to conduct a single non-inferiority Phase 3 clinical trial comparing the change in lumbar spine BMD at 12 months for patients dosed with Abaloparatide-TD to patients dosed with Abaloparatide-SC to confirm that the effect of Abaloparatide-TD treatment is comparable to that of Abaloparatide-SC. If our clinical trials of Abaloparatide-SC and Abaloparatide-TD are successful, we expect to seek marketing approval of Abaloparatide-TD as a line extension of Abaloparatide-SC. The FDA approval of Abaloparatide-TD, and the timing of any such approval, is dependent upon the approval of Abaloparatide-SC.
Clinical Development
Pivotal Phase 3 Clinical Trial of Abaloparatide-SC
In April 2011, we commenced our ongoing Phase 3 trial, which completed enrollment in March 2013. The trial completed enrollment with 2,463 patients at 28 medical centers in 10 countries in the United States, Europe, Latin America and Asia. Patients in the trial are randomized equally to receive daily doses of one of the following for 18 months: 80 µg of abaloparatide; a matching placebo or the approved dose of 20 µg of Forteo.
On February 15, 2012, we received a letter from the FDA stating that, after internal consideration, the agency believes that a minimum of 24-month fracture data are necessary for approval of new products for the treatment of postmenopausal osteoporosis. Our ongoing Abaloparatide-SC pivotal Phase 3 clinical study is designed to produce fracture data based on an 18-month primary endpoint. The FDA’s letter solicited a meeting to review the status of our Phase 3 clinical study and discuss options for fulfilling the FDA’s new request for 24-month fracture data in the context of the ongoing Phase 3 study. We subsequently met with the FDA on March 21, 2012 to discuss satisfying the 24-month data request while preserving the current 18-month primary endpoint. Based upon our discussion with the FDA, we believe that continued use of the 18-month primary endpoint will be acceptable, provided that our NDA includes the 24-month fracture data derived from a 6-month extension of the abaloparatide 80 µg and placebo groups in our Phase 3 study that will receive an approved alendronate (generic Fosamax) therapy for osteoporosis management. We intend to submit the NDA with the 24-month fracture data.
Study population — The study enrolled otherwise healthy ambulatory women aged 50 to 85 (inclusive) who had been postmenopausal for at least five years, met the study entry criteria and had provided written informed consent. Osteoporosis is defined as when a patient’s t-score is less than or equal to -2.5, meaning that the patient has a BMD that is two and a one-half standard deviations below the mean BMD of an ethnically matched thirty year old man or woman, as applicable. The women enrolled in the study have a BMD t-score < -2.5 and >-5.0 at the lumbar spine or hip (femoral neck) by dual-energy X-ray absorptiometry, or DXA, and radiological evidence of two or more mild or one or more moderate lumbar or thoracic vertebral fractures, or history of low trauma forearm, humerus, sacral, pelvic, hip, femoral or tibial fracture within the past five years. Postmenopausal women older than 65 who met the above fracture criteria but had a t-score of < -2.0 and >-5.0 could also be enrolled. Women older than 65 who did not meet the fracture criteria could also be enrolled if their t-score was < -3.0 and >-5.0. All patients were to be in good general health as determined by medical history, physical examination (including vital signs) and clinical laboratory testing. We believe this study population contains a patient population reflective of the type of severe osteoporosis patients that specialists will treat in their practices.
Study design

The 2,463 eligible patients were randomized equally to receive one of the following for 18 months:
· abaloparatide at a dose of 80 µg;
· a matching placebo; or
· Forteo at a dose of 20 µg.
The study drug was blinded to patients and medical personnel until the randomization process was completed. Treatment with abaloparatide at a dose of 80 µg or placebo will remain blinded to all parties throughout the study. Forteo comes as a proprietary prefilled drug and device combination that cannot be repackaged. Therefore, its identity cannot be blinded to treating physicians and patients once use begins. Study medication will be self-administered daily by subcutaneous injection for a maximum of 18 months. All enrolled patients will also receive calcium and vitamin D supplementation from the time of enrollment until the end of the treatment period. It will be recommended to patients that they also continue these supplements through the one month follow-up period.
Primary efficacy endpoints — The primary efficacy endpoint will be the number of patients treated with Abaloparatide-SC that show new vertebral fractures at end-of-treatment when compared to placebo as evaluated by a blinded assessor according to a standardized graded scale of severity of the vertebral deformity. The sample size per treatment arm provides 90% power at
a two-sided alpha to detect a superiority difference on vertebral fracture incidence between placebo patients and those who receive Abaloparatide-SC at a dose of 80 µg.
Secondary efficacy endpoints — Secondary efficacy parameters will also include reduction in the incidence of non-vertebral fractures to the wrist, hip and rib, for example, and reduction in moderate and severe vertebral fractures from baseline to end-of-treatment. Other secondary efficacy endpoints will include changes in BMD of the spine, hip, femoral neck and wrist from baseline to end-of-treatment as assessed by DXA and as compared to Forteo, as well as the number of hypercalcemic events in Abaloparatide-SC treated patients when compared to Forteo at end-of-treatment.
Additional secondary endpoints will include change in standing height and changes in serum bone formation markers across treatment, such as P1NP, osteocalcin and bone-specific alkaline phosphatase.
Extension study design.
Each of the abaloparatide 80 µg and placebo groups in our Phase 3 study will be eligible to continue in an extension study and will receive an approved alendronate (generic Fosamax) therapy for osteoporosis management. A key endpoint of the extension study will be the reduction in new vertebral fractures at up to 24 months in all randomized patients, including abaloparatide-treated and placebo-treated patients who are treated with alendronate at the end of treatment.

Safety outcomes — Safety evaluations to be performed will include physical examinations, vital signs, 12-lead electrocardiograms, or ECGs, clinical laboratory tests and monitoring and recording of adverse events. Specific safety assessments will include post-dose (four hours) determination of serum calcium, determination of creatinine clearance, post-dose ECG assessments at selected visits and assessments of postural hypotension (60 minutes post-dose) at selected clinic visits.
Bone biopsy of the iliac crest will be performed in a subset of patients receiving abaloparatide at a dose of 80 µg and placebo for assessment of bone quality and quantitative bone histomorphometry which is the quantitative study of the microscopic organization and structure of the bone tissue, and will be read blinded to treatment by an independent blinded assessor. Renal safety will be further evaluated in a subset of approximately 100 patients in each treatment group by renal computed tomography, or CT, scan.
Overall study safety is being monitored by an independent DSMB.
Abaloparatide-SC Phase 2 Clinical Trial
We conducted a randomized, placebo-controlled, parallel group dose-finding Phase 2 study (Study BA058-05-002) in the United States, Argentina, India and the United Kingdom. A total of 270 patients (mean age: 65 years) entered the pretreatment period, 222 patients were randomized, and 221 patients received study treatment and were analyzed in the intent-to-treat, or ITT, population with 55 continuing into an additional 24 weeks of treatment. A total of 155 patients were included in the efficacy population (per protocol) in the initial 24 weeks of treatment. The purpose of the study was to evaluate the safety and efficacy of daily injections of Abaloparatide-SC in women with osteoporosis. Postmenopausal women between the ages of 55 and 85 (inclusive) who had a BMD t-score < -2.5 at the lumbar spine or hip (femoral neck) by DXA or a BMD t-score < -2 and a prior low trauma fracture or an additional risk factor were candidates for this study. The study evaluated the effects of Abaloparatide-SC at multiple doses (placebo, 20 µg, 40 µg and 80 µg) on recovery of BMD, a marker of fracture risk, and on biomarkers of anabolic and resorptive activity in bone. The study also included a Forteo treatment arm for reference. After the initial 24 weeks of treatment, eligible patients were offered a second 24 weeks of their assigned treatment. Safety was assessed throughout the study and reported on at both 24 weeks and 48 weeks. Abaloparatide-SC and placebo were self-administered using a prefilled cartridge in a pen-injector device. Forteo was self-administered as the marketed product at the approved dose of 20 µg per day by subcutaneous injection. Four weeks prior to start of treatment, patients began taking calcium and vitamin D supplements that continued throughout the study.
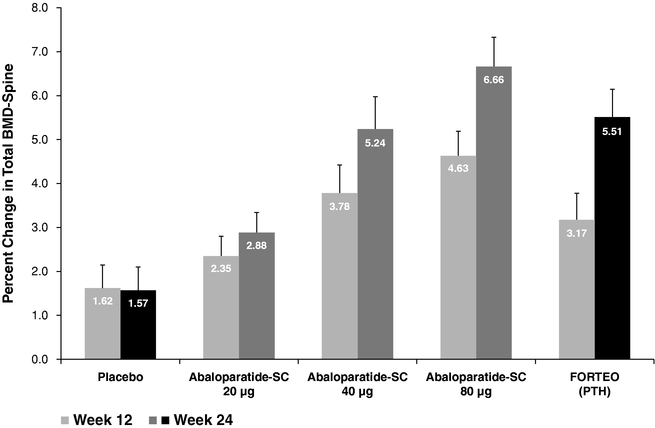
Initial 24 weeks of treatment — The following tables depict the percent change in total BMD-spine and BMD-hip at 12 and 24 weeks for each of arm of the trial.
In the ITT population, the mean percent change from baseline at week 12 in lumbar spine BMD (active treatment — placebo) for Abaloparatide-SC 40 µg and 80 µg groups were statistically significant (p = 0.0013 and p < 0.001, respectively). The difference was not statistically significant in the Abaloparatide-SC 20 µg group, in the placebo group or in the Forteo group (p = 0.055). At week 24, the mean percent change from baseline continued to increase and was statistically significantly proportional to dose (p < 0.001) as shown in Figure A below. Again, the mean gain in total spine BMD was statistically significant for Abaloparatide-SC 40 µg (p <
0.001) and 80 µg (p < 0.001) groups. The mean BMD gain at week 24 was also statistically significant for the Forteo group (p < 0.001). The difference was not statistically significant in the Abaloparatide-SC 20 µg group or in the placebo group. The response of lumbar spine BMD to Abaloparatide-SC was dose dependent, and the 80 µg Abaloparatide-SC dose produced a larger percentage increase in BMD at the lumbar spine than the approved 20 µg Forteo dose.
Figure A — Mean Standard Error of the Mean (SEM) Percent Change from Baseline at weeks 12 and 24 in Total Spine BMD (ITT Population,N =221)
An even greater proportional response in BMD was elicited in the hip region. By week 24, mean percent changes in total hip BMD were 0.4%, 1.4%, 2.0% and 2.6% for the placebo, abaloparatide at a dose of 20 µg, abaloparatide at a dose of 40 µg, and abaloparatide at a dose of 80 µg groups, respectively. Mean percent change in the Forteo (0.5%) group was similar to placebo as shown in Figure B below. The change in total hip BMD showed a dose response to Abaloparatide-SC and a more than five-fold benefit of abaloparatide at a dose of 80 µg over Forteo. A similar relative benefit of abaloparatide at a dose of 80 µg over Forteo was seen in all regions of the hip.
Figure B — Mean (SEM) Percent Change from Baseline at weeks 12 and 24 in Total Hip BMD (ITT Population, N=221)

Abaloparatide-SC also induced a dose-dependent rise in major markers of bone anabolic activity, including P1NP, bone specific alkaline phosphatase, or BSAP, and osteocalcin. The response to Forteo was somewhat greater for anabolic markers and bone resorption markers (C-telopeptides of type I collagen crosslinks, or CTX, and N-telopeptides of type I collagen crosslinks, or NTX), consistent with published data, suggesting a close of the anabolic window and attenuation in the anabolic benefit of continued Forteo administration. While elevated over baseline, the Abaloparatide-SC patient group maintained lower levels of resorption markers (CTX) throughout the study period as compared to Forteo. We believe abaloparatide may demonstrate a lengthening of the anabolic window as compared to Forteo.
Abaloparatide-SC was well tolerated at all doses and safety events were consistent with usual medical events in a study population of this age and gender. The safety profile was also similar to that of Forteo and there were no treatment-related serious adverse events, or SAE’s. Adverse events were reported by 74% of patients in the first six months of treatment, with a similar incidence across all treatment groups. The majority of on-treatment events were mild-to-moderate in severity and there were no deaths reported. Treatment-related treatment emergent adverse events were reported in approximately 30% of patients, with similar incidence across all
treatment groups. Seven subjects discontinued due to adverse events: one in the abaloparatide 20 µg group, one in the abaloparatide 40 µg group, three in the abaloparatide 80 µg group and two in the Forteo group. Eight patients (four percent) experienced at least one SAE and the incidence of such events was similar across treatment groups. Five SAEs, unrelated to treatment, were reported in three patients. Local tolerance at the injection site was similar across treatment groups and fewer than 20% of subjects reported any symptoms, such as redness at the injection site across the many months of injections.
The level of calcium in the blood, known as serum calcium levels, were monitored throughout the study and clinically significant elevated levels (greater than or equal to 10.5 milligrams per deciliter, or mg/dL) were observed in 40% of the Forteo group while also observed in four percent, 12%, 19% and 18% of the placebo, Abaloparatide-SC at doses of 20 µg, 40 µg and 80 µg groups, respectively. Most elevations were noted at the four-hour post-injection time point.
Blood pressure was assessed throughout the study for postural change. Postural changes in blood pressure (predetermined level of change in systolic or diastolic from lying to standing) were reported in seven patients, including 0%, 5%, 2%, 2% and 7% of patients in the placebo, Abaloparatide-SC 20 µg, 40 µg, 80 µg and Forteo groups, respectively. Pre-dose postural changes in blood pressure were similar across treatment groups. There were no clinically meaningful differences in ECG parameters between the placebo and active treatment groups.
Sixteen patients had low titer antibodies against abaloparatide after 24 weeks of treatment. Of these, five were in the abaloparatide 20 µg group, six were in the abaloparatide 40 µg group and five were in the abaloparatide 80 µg group. There were no associated safety events or attenuation of treatment efficacy. One antibody- positive patient in the Abaloparatide-SC 40 µg group was found to have possible evidence of neutralizing activity using an in vitro assay at 24 weeks without evidence of attenuation of drug efficacy; the patient achieved a 9.3% gain in total spine BMD at the week 24 assessment.
Extended 24 weeks of treatment — Patients who completed the initial 24 weeks of treatment and continued to meet eligibility criteria were offered participation in the 24-week extension study in which they would continue their assigned treatment. On completion of the regulatory process to approve the study extension, 69 patients remained eligible and 55 participated, including 13, 10, 7, 11 and 14 patients in Abaloparatide-SC 20 µg, 40 µg, 80 µg, placebo and Forteo groups, respectively. Forty-eight patients completed the extended treatment period.
BMD continued to increase during the extended 24 weeks of treatment, with the largest percent increases in total spine BMD, femoral neck BMD and total hip BMD observed in the Abaloparatide-SC 80 µg group, as shown in Figure C below. By week 48, mean percent changes in spine BMD were 0.7%, 5.1%, 9.8% and 12.9% for the placebo, Abaloparatide-SC 20 µg, Abaloparatide-SC 40 µg and Abaloparatide-SC 80 µg, groups, respectively, while mean percent change from baseline in the Forteo group was 8.6%. At week 48, the mean femoral neck BMD in
the Abaloparatide-SC 80 µg group gained 4.1% compared to the mean of the Forteo group at 2.2%. The total gain in hip BMD was 0.7%, 2.0%, 2.1% and 2.7% for the placebo, Abaloparatide-SC 20 µg, Abaloparatide-SC 40 µg and Abaloparatide-SC 80 µg groups, respectively, compared to 1.3% for the Forteo group.
Figure C — Mean (SEM) Percent Change from Baseline at weeks 12, 24 and 48 in Total Spine BMD (Extension Population N=55)

No treatment-related SAEs or deaths were reported during this time period. Two patients discontinued treatment, one for bilateral femoral hernias (Abaloparatide-SC 80 µg) and one for moderate syncope (Abaloparatide-SC 40 µg). Study-related adverse events occurred in a similar proportion of patients in each treatment group across the 52-week study period and the majority of events were mild or moderate in severity. The profile of events was not different during the second 24 weeks of study treatment.
Non-Head-to-Head Comparison of Abaloparatide-SC and Amgen Anti-sclerostin Antibody Phase 2 Study Results
Our Abaloparatide-SC Phase 2 clinical study used substantially similar patient inclusion and exclusion criteria as a study completed by Amgen of the use of a human anti-sclerostin antibody, romosozumab or AMG 785, for the treatment of osteoporosis. A comparison of the 6-month and 12-month spine BMD results of the AMG 785 study at the 210 mg once-monthly subcutaneous dosing regimen, including both patients treated with AMG 785 and a control group of patients treated with Forteo, and our Abaloparatide-SC study at the 80 mcg single daily subcutaneous
dose are set forth in the following table. While we believe the comparison is useful in evaluating the results of our Phase 2 clinical study of Abaloparatide-SC, the Abaloparatide-SC and AMG 785 studies were separate trials conducted at different sites, and we have not conducted a head-to-head comparison of the drugs in a single clinical trial. Results of an actual head-to-head comparison study may differ significantly from those set forth in the following table. In addition, because the Abaloparatide-SC and AMG 785 studies were separate studies and because the Abaloparatide-SC Phase 2 clinical study involved a lesser number of patients, differences between the results of the two studies may not be statistically or clinically meaningful.
|
|
|
Abaloparatide-SC |
|
AMG 785 |
| ||||
|
Product |
|
Abaloparatide |
|
Forteo |
|
AMG 785 |
|
Forteo |
|
|
Dose |
|
80 mcg |
|
20 mcg |
|
210 mg |
|
20 mcg |
|
|
Dosing frequency |
|
Daily |
|
Daily |
|
Monthly |
|
Daily |
|
|
No. of Injections per dose |
|
1 |
|
1 |
|
3 |
|
1 |
|
|
Type of Injection |
|
Self |
|
Self |
|
Physician |
|
Self |
|
|
Spine Mean Percent BMD Change from Baseline — 24 weeks/6 months |
|
+6.7 |
% |
+5.5 |
% |
+8.2 |
% |
+4.8 |
% |
|
Spine Mean Percent BMD Change from Baseline — 48 weeks/12 months |
|
+12.9 |
% |
+8.6 |
% |
+11.3 |
% |
+7.1 |
% |
|
Femoral Neck Mean Percent BMD Change from Baseline — 48 weeks/12 months |
|
+4.1 |
% |
+2.2 |
% |
+3.7 |
% |
+1.1 |
% |
(1) Abaloparatide-SC Study n=221 (24 weeks) and n=55 (48 weeks), 5 arms
(2) AMG 785 Study n=419 (12 months), 9 arms
Abaloparatide-SC Phase 1 Clinical Trials
We have completed three Phase 1 clinical trials of Abaloparatide-SC. Together with our Phase 2 clinical trials and ongoing Phase 3 clinical trial, over 1,300 patients have received the drug. The
results of our Phase 1 clinical trials suggest that Abaloparatide-SC is safe and well tolerated at doses of up to 100 µg administered once daily. These studies also demonstrated that abaloparatide was 100% bioavailable, meaning it was absorbed completely, when administered subcutaneously, and that it was rapidly cleared from the circulation.
Abaloparatide-TD Phase 2 Clinical Trials
We conducted a randomized, double-blind, placebo-controlled, Phase 2 clinical trial of abaloparatide administered via a coated transdermal microarray delivery system in healthy postmenopausal women with osteoporosis. This study was conducted in nine centers in the United States, Denmark, Poland and Estonia. The primary objective of this study was to determine the clinical safety and efficacy of Abaloparatide-TD as assessed by changes in BMD when compared to a transdermal placebo and Abaloparatide-SC. Postmenopausal women between the ages of 55 and 85 (inclusive) who had a BMD t-score < -2.5 at the lumbar spine or hip (femoral neck) by DXA or a BMD t-score < -2 and a prior low trauma fracture or an additional risk factor were candidates for this study. Abaloparatide-TD was administered via a spring-loaded applicator and Abaloparatide-SC was administered by a multi-use pen injector into which a multi-dose glass cartridge was inserted. Four weeks prior to the start of treatment, subjects began taking calcium and vitamin D supplements which were continued throughout the study. The study drug was to be administered once daily for a total of six months.
A total of 372 subjects were screened and 250 were randomized to treatment in one of five treatment regimens: transdermal placebo, Abaloparatide-TD at doses of 50 µg, 100 µg, and 150 µg or Abaloparatide-SC at a dose of 80 µg. Two hundred and forty-nine subjects were included in the safety population and 231 subjects were included in the modified intent-to-treat, or mITT, population.
In the mITT population, the mean percent change from baseline in total spine BMD after six months of treatment increased with Abaloparatide-TD dose (0.04%, 1.87%, 2.33% and 2.95% in the placebo, Abaloparatide 50 µg, 100 µg and 150 µg groups, respectively). The test for a dose response was statistically significant (<0.0001). The mean differences (active treatment — placebo) of the percent change from baseline in total spine BMD at six months were 1.83%, 2.29% and 2.91% in the Abaloparatide-TD 50 µg, 100 µg, and 150 µg groups, respectively. The results for all Abaloparatide-TD dose groups were statistically significantly better than placebo (p=0.0066, 0.0005, and <0.0001, respectively).
Figure D — Mean (SEM) Percent Change from Baseline at Six Months in Total Spine BMD

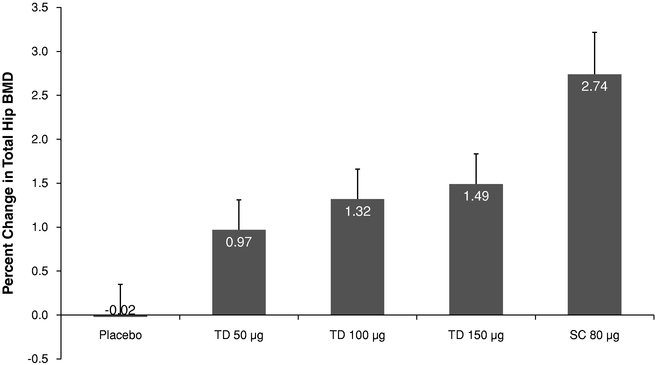
Similar to the findings in the spine, the mean percent change from baseline in total hip BMD after six months of treatment also increased with Abaloparatide-TD dose (-0.02% and 0.97%, 1.32% and 1.49% in the placebo, abaloparatide 50 µg, 100 µg and 150 µg groups. The mean differences (active treatment — placebo) of the percent change from baseline in total hip BMD at six months were 0.99%, 1.33% and 1.51% in the abaloparatide 50 µg, 100 µg, and 150 µg groups, respectively; the results for the 100 µg and 150 µg Abaloparatide-TD dose groups were statistically significantly better than placebo (p=0.0056 and 0.0018, respectively).
Figure E — Mean (SEM) Percent Change from Baseline at Six Months in Total Analyzable Hip BMD
Analysis of adverse events was performed on treatment-emergent adverse events, or TEAEs. There were no apparent differences between the TEAE profiles across the five treatment groups. Overall, nasopharyngitis, headache, and influenza were the most frequently reported TEAEs. There were 9 serious TEAEs reported, one in the placebo group, two in the 100 µg group, two in the 150 µg group and four in the Abaloparatide-SC group. No subjects died during the course of this study. All of the events were consistent with medical events in women with postmenopausal osteoporosis, and none of the events were considered to be related to treatment with study medication.
Assessment of local tolerance consisted of daily self-evaluation by the subject of any dermal reaction for two months during the course of the study using scales that ranged from 0 to 3 or 6, with 0 indicating no effect. In general, the types of symptoms reported were similar across the treatment groups, with dermal response and swelling being the effects most frequently reported. In an initial analysis, detectable antibodies against abaloparatide were noted in a subset of patients. However, these antibodies were of low titer, and there was no evidence of an effect on safety or attenuation of treatment efficacy.
Abaloparatide-TD Phase 1 Clinical Trials
We have completed three Phase 1 clinical trials that collectively evaluated the safety, PK, time course of delivery and dose ranging. Abaloparatide-TD was characterized by a rapid release of abaloparatide with a faster time to reach peak concentration as well as more rapid elimination in plasma compared to Abaloparatide-SC. Peak transdermal drug levels were consistent with Abaloparatide-SC. An optimal wear time of five minutes or less was identified as well as effective sites of application. Abaloparatide-TD showed an increase in the bone-formation marker P1NP in serum after seven days of exposure, consistent with bone-building activity, and was shown to be safe and well tolerated in all doses studied.
Preclinical Pharmacology of Abaloparatide
We have completed several preclinical studies of abaloparatide, and the following has been shown:
· Abaloparatide is a potent selective agonist of the human PTH type 1 receptor (PTHR1), with binding selectivity for the RG vs R0 receptor conformation compared to PTH(1-34) and greater selectivity than PTHrP(1-34);
· In models of calcium mobilization, abaloparatide has significantly less calcium mobilizing activity at higher doses than the native PTHrP(1-34), and less activity than PTH(1-34);
· Abaloparatide-SC stimulates the formation of normal, well-organized bone and restores BMD in ovariectomized (OVX), osteopenic rats and primates. Mechanical testing of bones from OVX rats after treatment with Abaloparatide-SC revealed a significant increase in femur and vertebral bone strength. Similar studies in rats with Abaloparatide-TD show comparable restoration of bone;
· Abaloparatide-SC was generally well tolerated over a wide range of doses in two species, rats and primates, for up to six months and nine months, respectively; and
· Safety pharmacology studies demonstrated no respiratory, gastroenterologic, hematologic, renal or central nervous system effects.
A two-year subcutaneous injection carcinogenicity study of abaloparatide in Fischer 344 albino rats was conducted to assess the carcinogenic potential of abaloparatide. The study was conducted according to the provisions set forth in Guidance ICH-S1A, ICH-S1B, and ICH-S1C(R2), and the design was accepted by the FDA on July 15, 2009. This study evaluated three abaloparatide dose levels. The doses were selected based upon findings and tolerance in completed long-term rat toxicology studies and the anticipated tolerance over a two-year dosing period. Furthermore, the doses represent an exposure multiple over maximum clinical doses. The study included a cohort of rats being dosed with a daily subcutaneous injection of PTH(1-34)as a positive control, as it was anticipated that osteosarcomas would be observed with this treatment, as previously published for both rhPTH(1-34) and rhPTH(1-84) in similar 2-year rat
carcinogenicity studies. The positive control served to provide confirmation of the sensitivity of the model. A preliminary unaudited analysis of histopathology data revealed osteosarcomas in our carcinogenicity study in both the abaloparatide and PTH(1-34) treated groups, with similar frequency between abaloparatide and PTH(1-34) when comparing comparable exposure multiples to the human therapeutic dose.
We are also conducting one preclinical bone quality study in OVX rats with 12 months of daily Abaloparatide-SC dosing and a second preclinical bone quality study in adult OVX monkeys for 16 months. The primary objective of these studies is to determine the long-term treatment effects of Abaloparatide-SC on bone quality. Effects on bone mass, both cortical bone and cancellous bone, will be assessed by BMD and peripheral quantitative computed tomography, and bone strength will be determined by biomechanical testing. The mechanisms by which abaloparatide affects bone will be assessed by evaluation of biomarkers of bone turnover and histomorphometric indices of bone turnover. Preliminary data from the 12-month rat study has shown marked, dose dependent increases in BMD following abaloparatide treatment, increases in bone formation markers, but not bone resorption, and an increase in bone strength.
Preliminary results from the 16-month monkey OVX study have also shown significant BMD gains, together with increases in bone strength.
RAD1901
In June 2006, we exclusively licensed the worldwide rights (except for Japan) to RAD1901 from Eisai. We are developing RAD1901 as a SERD in Phase 1 clinical development for the treatment of BCBM. We are also developing RAD1901, which at lower doses acts as a SERM, in an oral formulation as a treatment for vasomotor symptoms, commonly known as hot flashes or hot flushes. We currently intend to advance the development of RAD1901 for the treatment of ER-positive BCBM with the initiation of a Phase 1b clinical trial in late 2014. We currently intend to seek potential collaborations with third parties in order to advance the development of RAD1901 for the treatment of vasomotor symptoms. We anticipate the next clinical study would be a Phase 2b study conducted in approximately 200 perimenopausal women experiencing a high frequency of hot flashes at baseline. The main study endpoints would be an assessment of the change in the frequency and severity of moderate and severe hot flashes.
Pharmacologic Characteristics
RAD1901 has been shown to bind with good selectivity to the ER alpha, or ERα, and to have both estrogen-like and estrogen antagonist effects in different tissues. RAD1901 has also been shown to have estrogen-like behavioral effects in an animal model of partner preference and to reduce vasomotor signs in an animal model of menopausal hot flashes. In bone, RAD1901 protects against gonadectomy-induced bone loss. RAD1901 does not stimulate the endometrium, as shown in short and long term animal models, where changes in uterine weight, uterine epithelial thickness and C3 gene expression are measured, all of which are sensitive indicators.
In studies in which an estrogen is used to stimulate the endometrium, RAD1901 antagonizes this estrogen-mediated stimulation of the endometrium. In cell culture, RAD1901 does not stimulate replication of breast cancer cells, and antagonizes the stimulating effects of estrogen on cell proliferation. Furthermore, in breast cancer cell lines a dose dependent down regulation of ERα is observed, a process we have shown to involve proteosomal mediated degradation pathway. In a model of breast cancer, in which human breast cancer cells are implanted in mice and allowed to establish tumors in response to estrogen treatment, we have shown that treatment with RAD1901 results in decreased tumor growth.
Pharmacokinetic studies with RAD1901 have established the PK profile including demonstration of good oral bioavailability and the ability of RAD1901 to cross the blood-brain barrier, with pharmacological levels detectable in the brain. We believe that RAD1901 is a promising agent for development in the treatment both vasomotor symptoms and a range of estrogen-driven cancers.
Clinical Development Program
Phase 2 Study — Vasomotor Symptoms
A Phase 2 proof of concept study was conducted in 100 healthy perimenopausal women using four doses of RAD1901 (10 mg, 25 mg, 50 mg and 100 mg) and placebo. The primary study outcome was reduction in the frequency and severity of moderate and severe hot flashes. While a classic dose-response effect was not demonstrated, efficacy was determined to occur at the 10 mg dose level which achieved a statistically significant reduction in the frequency of moderate and severe hot flashes both by linear trend test and by comparison to placebo and in overall (mild-moderate-severe) hot flashes at either the two-, three- or four-week time-points. A similar reduction in composite score (frequency × severity of hot flashes) was identified at all time-points, with a statistically significant difference from placebo achieved at the two-, three- or four-week time-points. Numerical reductions in mean severity and mean daily severity were observed, but did not reach statistical significance. We believe RAD1901 is an attractive candidate for advancement to Phase 3 development as a treatment for vasomotor symptoms.
No SAEs were reported during the course of the study. Overall, 69% of patients had an adverse event, generally mild or moderate in severity, with some evidence of dose dependency, and events were most commonly gastrointestinal symptoms and headaches. Three severe adverse events occurred, one in a placebo patient, none of which were considered treatment related. Two patients discontinued treatment due to an adverse event, neither in relation to the 10 mg dose.
Phase 1 Study — Vasomotor Symptoms
We have conducted Phase 1 safety, PK and bioavailability studies of RAD1901 in 80 healthy postmenopausal women over a range of doses. Bioavailability was determined to be approximately 10%. Food effect was also investigated and the presence of food was determined
to increase absorption and delay clearance of RAD1901. RAD1901 was generally well tolerated at all dose levels tested. All study-related adverse events were of mild intensity, with some increase in frequency at the higher doses in the multiple dose group, most commonly gastrointestinal symptoms and headaches. There were no SAEs observed.
RAD140
RAD140 is a nonsteroidal selective androgen receptor modulator, or SARM, that resulted from an internal drug discovery program that began in 2005. RAD140 has demonstrated potent anabolic activity on muscle and bone in preclinical studies and has completed 28-day preclinical toxicology studies in both rats and monkeys. Because of its high anabolic efficacy, receptor selectivity, potent oral activity and long duration half-life, we believe that RAD140 has clinical potential in a number of indications where the increase in lean muscle mass and/or bone density is beneficial, such as treating the weight loss due to cancer cachexia, muscle frailty and osteoporosis, and also in the treatment of breast cancer.
We may choose to advance the RAD140 program internally or to collaborate with third parties for its further development and commercialization. Therefore, the date of any FDA approval of RAD140, if ever, cannot be predicted at this time. As a result of the uncertainties around the development strategy for RAD140, we are unable to determine the duration and costs to complete current or future clinical stages of our RAD140 product candidate.
Manufacturing
We do not own or operate manufacturing facilities for the production of any of our product candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. The active pharmaceutical ingredient, or API, of abaloparatide is manufactured on a contract basis by Lonza Group Ltd., or Lonza, using a solid phase peptide synthesis assembly process, and purification by high pressure liquid chromatography. Abaloparatide-SC is supplied as a liquid in a multi-dose cartridge for use in a pen delivery device. The multi-dose cartridges are manufactured by Vetter. Abaloparatide-TD is manufactured by 3M based on their patented microneedle technology to administer drugs through the skin, as an alternative to subcutaneous injection. The API of RAD1901 is manufactured for us on a contract basis by Irix Pharmaceuticals, Inc.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Our contract manufacturing organizations manufacture our product candidates under current Good Manufacturing Practice (cGMP) conditions. cGMP is a regulatory standard for the production of pharmaceuticals that will be used in humans.
Intellectual Property
As of December 31, 2013, we owned five issued United States patents, as well as ten pending U.S. patent applications and 45 pending foreign patent applications in Europe and 15 other jurisdictions, and 12 granted foreign patents. As of December 31, 2013, we had licenses to nine U.S. patents as well as numerous foreign counterparts to many of these patents and patent applications.
We strive to protect the proprietary technology that we believe is important to our business, including seeking and maintaining patents intended to cover our product candidates and compositions, their methods of use and processes for their manufacture and any other inventions that are commercially important to the development of our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our success will significantly depend on our ability to obtain and maintain patent and other proprietary protection for commercially important technology and inventions and know-how related to our business, defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on know-how and continuing technological innovation to develop and maintain our proprietary position.
Abaloparatide
We acquired and maintain exclusive worldwide rights, excluding Japan, to certain patents, data and technical information related to abaloparatide through a license agreement with an affiliate of Ipsen. Composition of matter of abaloparatide is claimed in the United States (U.S. Patent No. 5,969,095), Europe, Australia, Canada, China, Hong Kong, South Korea, New Zealand, Poland, Russia, Singapore, Mexico, Hungary, and Taiwan. These cases have a normal patent expiration date of 2016 absent any U.S. patent term extension under the Hatch-Waxman Act. The Phase 3 clinical dosage of abaloparatide by the subcutaneous route for use in treating osteoporosis is covered by Patent No. 7,803,770 until 2028 (statutory term extended with 175 days of patent term adjustment due to delays in patent prosecution by the United States Patent and Trademark Office, or USPTO) in the United States (absent any patent term extension under the Hatch-Waxman Act). The intended therapeutic formulation for Abaloparatide-SC is covered by Patent No. 8,148,333 until 2027 (statutory term extended with 36 days of patent term adjustment due to delays in patent prosecution by the USPTO) in the United States (absent any patent term extension under the Hatch-Waxman Act). Related cases granted in China, Australia, Singapore, Japan, Mexico, New Zealand, and Ukraine, and currently pending in Europe, China, Australia, Canada, Brazil, Singapore, South Korea, India, Israel, Norway, Russia, and Hong Kong will have a normal un-extended patent expiration date of 2027. Patent applications which cover various aspects of abaloparatide for microneedle application are pending in the United
States, Australia, Brazil, Canada, China, Europe, Israel, India, Japan, Korea, Mexico, New Zealand, Russia, Singapore, and Ukraine. Any claims that might issue from these applications will have a normal expiration date no earlier than 2032.
RAD1901
We exclusively license the worldwide rights, except for Japan, to RAD1901 from Eisai. US Patent No. 7,612,114 (statutory term extended to 2026 with 967 days of patent term adjustment absent any Hatch-Waxman patent term extension) and US Patent No. 8,399,520 (statutory term expires 2023) cover RAD1901 as a composition of matter as well as the use of RAD1901 for treatment of estrogen-dependent breast cancer. Corresponding cases issued in Australia and Canada and pending in India and Europe will have a normal expiration date in 2023. Patent applications covering methods of using RAD1901 for the treatment of vasomotor symptoms are pending in the United States (published as US 2010/0105733A1) and Canada, and granted in Europe; any issued claims will have a normal expiration in 2027. Patent applications covering a dosage form have been filed in the United States, Europe, Canada and Mexico, and any claims that might issue from these applications will have a normal expiration date no earlier than 2031.
RAD140
The composition of matter of, and methods of using, RAD140 is covered by US Patent No. 8,067,448 (effective filing date February 19, 2009, and a statutory term extended to September 25, 2029, with 281 days of patent term adjustment due to delays by the USPTO) and U.S. Patent No. 8,268,872 (effective filing date February 19, 2009 with term understood to be extended with 232 days of patent term adjustments). Related patents have been granted in Australia, Japan and Mexico and additional patent applications are pending in the United States and numerous additional countries worldwide. Any patents issued from these filings will have a normal expiration in 2029 absent any extensions.
There can be no assurance that an issued patent will remain valid and enforceable in a court of law through the entire patent term. Should the validity of a patent be challenged, the legal process associated with defending the patent can be costly and time consuming. Issued patents can be subject to oppositions, interferences and other third party challenges that can result in the revocation of the patent or that can limit patent claims such that patent coverage lacks sufficient breadth to protect subject matter that is commercially relevant. Competitors may be able to circumvent our patents. Development and commercialization of pharmaceutical products can be subject to substantial delays and it is possible that at the time of commercialization any patent covering the product has expired or will be in force for only a short period of time following commercialization. We cannot predict with any certainty if any third party U.S. or foreign patent rights, or other proprietary rights, will be deemed infringed by the use of our technology. Nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. Should we need to defend ourselves and our partners against any such claims,
substantial costs may be incurred. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability to develop or commercialize some or all of our products in the U.S. and abroad, and could result in the award of substantial damages. In the event of a claim of infringement, we or our partners may be required to obtain one or more licenses from a third party. There can be no assurance that we can obtain a license on a reasonable basis should we deem it necessary to obtain rights to an alternative technology that meets our needs. The failure to obtain a license may have a material adverse effect on our business, results of operations and financial condition.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that we can meaningfully protect our trade secrets on a continuing basis. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to our trade secrets.
It is our policy to require our employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from us to execute confidentiality agreements upon the commencement of employment or consulting relationships. These agreements provide that all confidential information developed or made known to these individuals during the course of the individual’s relationship with the company is to be kept confidential and is not to be disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee shall be the property of the company. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Our success will depend in part on our ability to obtain and maintain patent protection, preserve trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others, both in the U.S. and other territories worldwide.
Competition
The development and commercialization of new products to treat the targeted indications of our product candidates is highly competitive, and our products, if approved, will face considerable competition from major pharmaceutical, biotechnology and specialty pharmaceutical companies, including Amgen, UCB, Merck & Co, Novartis, Lilly, Genentech, Warner Chilcott, Asahi Kasei and Zosano, that are seeking to develop products for similar indications. Many of our competitors have substantially more resources than we do, including financial, manufacturing, marketing, research and drug development resources. In addition, many of these companies have longer operating histories and more experience than us in preclinical and clinical development, manufacturing, regulatory and global commercialization.
Within the osteoporosis market, Lilly launched Forteo in December 2002 as the first-to-market anabolic or bone-building agent for the treatment of osteoporosis. In April 2012, UCB and Amgen started a Phase 3 clinical trial program for their anti-sclerostin antibody for the treatment of osteoporosis. We are also aware that Zosano is developing a transdermal form of PTH(1-34) that would compete with Abaloparatide-TD.
RAD1901 for the treatment of BCBM will face competition from SERDs, CNS-penetrant anti-cancer agents and from chemotherapy derivatives. RAD1901 will also face competition from other therapeutics in development for the treatment of hot flashes. We cannot assure you that our current product candidates, if approved, will be able to compete effectively against these, or any other competing therapeutics that may become available on the market.
COLLABORATIONS AND LICENSE AGREEMENTS
Nordic Bioscience
Abaloparatide-SC Phase 3 Clinical Trial — We entered into a letter of intent with Nordic Bioscience Clinical Development VII A/S, or Nordic, on September 3, 2010, pursuant to which we funded preparatory work by Nordic in respect of a Phase 3 clinical study of Abaloparatide-SC, which is being conducted at centers operated by the Center for Clinical and Basic Research, or CCBR, as well as other medical centers. CCBR is a leading global clinical research organization, or CRO, with extensive experience in global osteoporosis registration studies. The letter of intent was extended on December 15, 2010 and on January 31, 2011. Pursuant to the letter of intent and the two extensions, we funded an aggregate $1.5 million of preparatory work by Nordic during 2010 and funded an additional $750,000 of preparatory work by Nordic during 2011. On March 29, 2011, we and Nordic entered into a Clinical Trial Services Agreement, a Work Statement NB-1, or the Work Statement NB-1, under such Clinical Trial Services Agreement and a related Stock Issuance Agreement. Pursuant to the Work Statement NB-1, Nordic is managing the Phase 3 clinical trial, or the Phase 3 Clinical Trial, of Abaloparatide-SC and Nordic will be compensated for such services in a combination of cash and shares of stock.
The Clinical Trial Services Agreement has a five-year term unless it is sooner terminated. The Clinical Trial Services Agreement or any Work Statement may be terminated by mutual agreement of the parties at any time. Either party may also terminate any Work Statement upon a material breach by the other party with respect to such Work Statement unless such other party cures the alleged breach within the notice period specified in the Clinical Trial Services Agreement or if not capable of being cured within such period the party alleged to be in breach commences efforts to cure and diligently proceeds to cure. Termination of any Work Statement does not result in termination of the Clinical Trial Services Agreement or any other Work Statements, which remain in force until terminated. Either party may also terminate a Work Statement if force majeure conditions have prevented performance by the other party for more than a specified period of time. We may also terminate a Work Statement with notice to Nordic
if authorization and approval to perform any clinical study that is the subject of such Work Statement is withdrawn by the FDA or other toxicological test results support termination of the clinical study relating to such Work Statement for reasons of safety or if the emergence of any adverse event or side effect in the clinical study relating to such Work Statement is of such magnitude or incidence in our opinion as to support termination.
The Clinical Trial Services Agreement contains customary risk allocation clauses with each party indemnifying the other in respect of third-party claims arising out of or resulting from: (1) the negligence or intentional misconduct of such party, its employees, agents or representatives in performing its obligations under the Clinical Trial Services Agreement or any Work Statement; and (2) any breach by such party of its representations and warranties under the Clinical Trial Services Agreement. Risk Factors
We have agreed to indemnify Nordic in respect of third-party claims for product liability or personal injury arising from or relating to our products or our use of any deliverables. In addition, we separately provide indemnification to the investigative sites performing services pursuant to Work Statement NB-1 in respect of third-party claims of injury, illness or adverse side effects to a patient in the study that is the subject of Work Statement NB-1 that are attributable to the Radius study drug under indemnification letters with such investigative sites. The Clinical Trial Services Agreement contains other customary clauses and terms as are common in similar agreements in the industry.
In December 2011, we entered into an amendment to the Work Statement NB-1, or the First Amendment. Pursuant to the original terms of the Work Statement NB-1, the study was to be conducted in 10 countries at a specified number of sites within each country. The terms of the First Amendment (1) provided for two additional countries (the United States and India) in which the trial will be conducted, (2) specified a certain number of sites within each such additional country for the conduct of the study and (3) amended various terms and provisions of the Work Statement NB-1 to reflect the addition of such countries and sites within the study’s parameters. Payments to be made by us to Nordic under the First Amendment in connection with the conduct of the study in such additional countries are denominated in both euros and U.S. dollars and total up to both €717,700 ($988,919) and $289,663, respectively, for the 15 additional study sites in India contemplated by the First Amendment and up to both €1.2 million ($1.7 million) and $143,369, respectively, for the five additional study sites in the United States contemplated by the First Amendment.
In June 2012, we entered into a second amendment to the Work Statement NB-1, or the Second Amendment. Pursuant to the original terms of the Work Statement NB-1, as amended by the First Amendment, the study was to be conducted in 12 countries at a specified number of sites within each country. The terms of the Second Amendment (1) increased the overall number of sites by adding sites in Europe, Brazil and Argentina and removing other sites, (2) specified a certain number of sites within each country for the conduct of the study, and (3) amended
various terms and provisions of the Work Statement NB-1 to reflect additional services provided at existing sites and the addition of the new study sites within the study’s parameters. The Second Amendment also provided that cash payments to Nordic under the Clinical Trial Services Agreement as well as the payment of shares of stock under the related Stock Issuance Agreement each be reduced by an amount of €11,941 ($16,454) per subject for any subjects enrolled in India or the United States. Such reductions are applied in pro rata monthly installments. Payments to be made by us to Nordic under the Second Amendment in connection with the extra services provided at existing sites and the conduct of the study at the new study sites are denominated in both euros and U.S. dollars and total €3.7 million ($5.1 million) and $205,540, respectively.
In March 2014, we entered into a fourth amendment to the Work Statement NB-1, or the Fourth Amendment. Pursuant to the terms of the Fourth Amendment, we agreed to pay to Nordic an additional performance incentive, or a Performance Incentive Payment, of $500,000 for every 50 patients that, subsequent to March 28, 2014, complete all end-of-study procedures, up to a maximum aggregate amount of additional payments equal to $5.0 million. Any Performance Incentive Payment will be paid in cash in the event an underwritten initial public offering of shares of our common stock, or an IPO, is consummated prior to May 31, 2014. Should an IPO not be consummated by us prior to May 31, 2014, any Performance Incentive Payments will be paid instead through the issuance to Nordic of shares of our capital stock under the same model for equity-based compensation that is contemplated by our existing outstanding work statements under the Clinical Trial Services Agreement.
The Work Statement NB-1, as amended on December 9, 2011, June 18, 2012 and March 28, 2014, provides for a total of up to approximately €41.2 million ($56.7 million) of euro-denominated payments and a total of up to approximately $3.2 million of U.S. dollar-denominated payments over the course of the Phase 3 Clinical Trial, plus aggregate Performance Incentive Payments of up to $5.0 million. These payments may be adjusted based upon actual sites opened, work performed or number of patients enrolled.
Pursuant to the Work Statement NB-1, we are required to make certain per patient payments denominated in both euros and U.S. dollars for each patient enrolled in the Phase 3 Clinical Trial followed by monthly payments for the duration of the study and final payments in two equal euro-denominated installments and two equal U.S. dollar-denominated installments. Changes to the Clinical Study schedule may alter the timing, but not the aggregate amounts of the payments.
Pursuant to the Stock Issuance Agreement, Nordic agreed to purchase the equivalent of €371,864 of Series A-5 Convertible Preferred Stock at $8.142 per share, and 64,430 shares of Series A-5 Convertible Preferred Stock were sold to Nordic on May 17, 2011 for proceeds of $525,154. These shares were exchanged in the Merger for an aggregate of 6,443 shares of our Series A-5. Convertible Preferred Stock, or Series A-5.
The Stock Issuance Agreement provides that Nordic is entitled to receive quarterly stock dividends, payable in shares of our Series A-6 Convertible Preferred Stock, or Series A-6, or shares of common stock if our preferred stock has been converted in accordance with its amended certificate of incorporation, having an aggregate value, under the Work Statement NB-1 of up to €36.8 million ($50.7 million) (the “Nordic Accruing Dividend”). In the event Nordic sells the shares of Series A-5 or in the event the shares of Series A-5 are converted into common stock in accordance with our amended certificate of incorporation, this right to receive the Nordic Accruing Dividend will terminate, but a right to receive an equivalent number of shares of Series A-6 or common stock, as applicable, will remain with Nordic as a contractual right under the Stock Issuance Agreement.
The Nordic Accruing Dividend related to the Phase 3 Clinical Trial is determined based upon the estimated period that will be required to complete the Phase 3 Clinical Trial. On the last business day of each calendar quarter (each, an “Accrual Date”), beginning with the quarter ended June 30, 2011, the Company has a liability, under the Work Statement NB-1 to issue shares of Series A-6 (or common stock, after the conversion of the Company’s preferred stock into common stock) to Nordic that is referred to as the “Applicable Quarterly Amount” and is equal to €36.8 million ($50.7 million) (subject to adjustment in accordance with the applicable provisions of the Second Amendment relating to consideration payable for patients enrolled in India and the U.S.) minus the aggregate value of any prior Nordic Accruing Dividend accrued divided by the number of calendar quarters it will take to complete the Phase 3 Clinical Trial. To calculate the aggregate number of shares due to Nordic in each calendar quarter, we convert the portion of €36.8 million ($50.7 million) to accrue in such calendar quarter into U.S. dollars using the simple average of the exchange rate for buying U.S. dollars with euros for all Mondays in such calendar quarter. We then calculate the aggregate number of shares to accrue in such calendar quarter by dividing the U.S. dollar equivalent of the Applicable Quarterly Amount, by the greater of (1) the fair market value of our common stock as of the applicable Accrual Date or (2) $81.42 and rounding down the resulting quotient to the nearest whole number. Such shares due to Nordic are to be issued when declared or paid by our Board of Directors, who are required to do so upon Nordic’s request, or upon an event of sale. As of December 31, 2013, 438,124 shares of Series A-6 were due to Nordic under Work Statement NB-1, as amended, or, after the automatic conversion into common stock of our preferred stock, 1,921,596 shares of our common stock. In December 2013, Nordic requested that all shares of Series A-6 accrued as of December 31, 2013 under Work Statement NB-1 be issued. Accordingly, our Board of Directors declared a dividend to Nordic of all 438,124 shares accrued under Work Statement NB-1 on December 31, 2013.
On March 28, 2014, we entered into Amendment No. 2 to the Amended and Restated Stock Issuance Agreement entered into by the parties as of May 16, 2011, or the Second Stock Issuance Agreement Amendment, with Nordic. The Second Stock Issuance Agreement Amendment required that our board of directors declare, as soon as reasonably practical, a stock dividend of
29 shares of our Series A-6 for each share of our outstanding Series A-5, all of which are held by Nordic, for a total of 186,847 shares of Series A-6, in full satisfaction of all stock dividends payable in 2014 under the terms of the Stock Issuance Agreement in relation to Work Statement NB-1 and Work Statement NB-3, excluding any Performance Incentive Payments payable in stock. In March 2014, Nordic requested that all 186,847 shares of Series A-6 be issued. Accordingly, our Board of Directors declared a dividend to Nordic of all 186,847 shares on March 31, 2014. The Second Stock Issuance Agreement Amendment provides further that in the event an IPO occurs prior to May 31, 2014, any payments owed by us to Nordic in relation to Work Statement NB-1 and Work Statement NB-3 for all periods of time after 2014, excluding any Performance Incentive Payments, will be changed from the right to receive stock to the right to receive a total cash payment from us of $4.3 million payable in ten equal monthly installments of $430,000 beginning on March 31, 2015. The Second Stock Issuance Agreement Amendment also stipulates that all consideration to be paid to Nordic pursuant to the Stock Issuance Agreement at any time after the consummation of an IPO be payable in cash.
Abaloparatide-SC Phase 3 Clinical Extension Study — In February 2013, we entered into a Work Statement NB-3 (the “Work Statement NB-3”) under the Clinical Trial Services Agreement and the related Stock Issuance Agreement. Pursuant to Work Statement NB-3, Nordic will perform an extension study to evaluate six months of standard-of-care osteoporosis management following the completion of the 18-month Abaloparatide-SC Phase 3 Clinical Trial, or the Extension Study, and will be compensated for such services in a combination of cash and shares of stock. Under the terms of a Letter of Intent that we entered into with Nordic on October 22, 2012 setting forth the parties’ obligations to negotiate in good faith to enter into Work Statement NB-3, we were required to make an initial payment of €806,468 ($1.1 million).
In March 2014, we entered into an amendment to the Work Statement NB-3, or the NB-3 Amendment. The NB-3 Amendment was effective as of February 28, 2014 and provides that Nordic will perform a Period 2 extension study, or the Second Extension, to evaluate an additional eighteen months of standard-of-care osteoporosis management following the Period 1 extension of six months upon completion of the Phase 3 clinical study of our Abaloparatide-SC product. Payments in cash to be made by us to Nordic under the NB-3 Amendment are denominated in both euros and U.S. dollars and total up to approximately €3.0 million ($4.1 million) and $527,740, respectively. In addition, we agreed to issue to Nordic shares of our series A-6 convertible preferred stock having a value of up to the sum of approximately €3.0 million ($4.1 million) and $527,740 as additional payment for the services to be provided under the NB-3 Amendment, with the issuance of such shares to be made pursuant to the terms of an Amendment No. 2, entered into by us with Nordic on March 28, 2014, to the Amended and Restated Stock Issuance Agreement entered into by the parties as of May 16, 2011, or the Second Stock Issuance Agreement Amendment.
Payments in cash to be made to Nordic under the Work Statement NB-3, as amended by the NB-3 Amendment, are denominated in both euros and U.S. dollars and total up to €7.5 million
($10.3 million) and $1.1 million, respectively. In addition, the Company will issue to Nordic, shares of our Series A-6 having a value of up to €7.5 million ($10.3 million) and $0.8 million, as additional payment for services to be provided under the Work Statement NB-3 and the Clinical Trial Services Agreement.
The Stock Issuance Agreement provides that, beginning with the quarter ended March 31, 2013, Nordic was entitled to receive quarterly stock dividends in connection with services performed under the Work Statement NB-3, payable in shares of Series A-6 or shares of common stock if our preferred stock has been automatically converted into common stock in accordance with our amended certificate of incorporation, having an aggregate value of up to €7.5 million ($10.3 million) and $0.8 million. In the event Nordic sells the shares of Series A-5 or in the event the shares of Series A-5 are converted into common stock in accordance with our amended certificate of incorporation, this right to receive the Nordic Accruing Dividend will terminate, but a right to receive an equivalent number of shares of Series A-6 or common stock, as applicable, will remain with Nordic as a contractual right under the Stock Issuance Agreement.
The Nordic Accruing Dividend related to the Extension Study is determined based upon the estimated period that will be required to complete the Extension Study. On each Accrual Date, beginning with the quarter ended March 31, 2013, we will recognize a liability to issue shares of Series A-6 to Nordic with an Applicable Quarterly Amount value equal to €7.5 million ($10.3 million) and $0.8 million minus the aggregate value of any previously accrued Nordic Accruing Dividend related to the Extension Study divided by the number of calendar quarters it will take to complete the Extension Study. We calculate the aggregate number of shares of Series A-6 to accrue in such calendar quarter by dividing such Applicable Quarterly Amount, by the greater of (1) the fair market value of our common stock as of the applicable Accrual Date or (2) $81.42 and rounding down the resulting quotient to the nearest whole number. Such shares due to Nordic are to be issued when declared or paid by our Board of Directors, who are required to do so upon Nordic’s request, or upon an event of sale. As of December 31, 2013, 25,772 shares of Series A-6 were due to Nordic under Work Statement NB-3, or, after the automatic conversion into common stock of our preferred stock, 113,035 shares of our common stock. In December 2013, Nordic requested that all shares of Series A-6 accrued as of December 31, 2013 under Work Statement NB-3 be issued. Accordingly, our Board of Directors declared a dividend to Nordic of all 25,772 shares accrued under Work Statement NB-3 on December 31, 2013.
On March 28, 2014, we entered into Amendment No. 2 to the Amended and Restated Stock Issuance Agreement entered into by the parties as of May 16, 2011, or the Second Stock Issuance Agreement Amendment, with Nordic. The Second Stock Issuance Agreement Amendment required that our board of directors declare, as soon as reasonably practical, a stock dividend of 29 shares of our Series A-6 for each share of our outstanding Series A-5, all of which are held by Nordic, for a total of 186,847 shares of Series A-6, in full satisfaction of all stock dividends payable in 2014 under the terms of the Stock Issuance Agreement in relation to Work Statement NB-1 and Work Statement NB-3, excluding any Performance Incentive Payments payable in
stock. In March 2014, Nordic requested that all 186,847 shares of Series A-6 be issued. Accordingly, our Board of Directors declared a dividend to Nordic of all 186,847 shares on March 31, 2014. The Second Stock Issuance Agreement Amendment provides further that in the event an IPO occurs prior to May 31, 2014, any payments owed by us to Nordic in relation to Work Statement NB-1 and Work Statement NB-3 for all periods of time after 2014, excluding any Performance Incentive Payments, will be changed from the right to receive stock to the right to receive a total cash payment from us of $4.3 million payable in ten equal monthly installments of $430,000 beginning on March 31, 2015. The Second Stock Issuance Agreement Amendment also stipulates that all consideration to be paid to Nordic pursuant to the Stock Issuance Agreement at any time after the consummation of an IPO be payable in cash.
On December 6, 2013, we entered into a Letter of Intent, or the Letter of Intent, with Nordic, which provided that we and Nordic would continue to negotiate the definitive terms of the NB-3 Amendment. Pursuant to the Letter of Intent, we were required to make an initial payment of €222,573 ($0.3 million) and agreed to commence payment of the cash compensation due in consideration of the services being provided by Nordic under the NB-3 Amendment. The Letter of Intent terminated in accordance with its terms on February 28, 2014 (pursuant to an extension mutually agreed to by us and Nordic).
Abaloparatide-TD Phase 2 Clinical Trial — On July 26, 2012, we entered into a Letter of Intent (the “Letter of Intent”) with Nordic, which provides that the Company and Nordic will, subject to compliance by the Company with certain requirements of our amended certificate of incorporation and applicable securities laws, negotiate in good faith to enter into (1) a Work Statement NB-2 (the “Work Statement NB-2”), a draft of which is attached to the Letter of Intent, and (2) an amendment to the Amended and Restated Stock Issuance Agreement.
In February 2013, we executed the final Work Statement NB-2 under the Clinical Trial Services Agreement and the related Stock Issuance Agreement. Pursuant to the Work Statement NB-2, Nordic will provide clinical trial services relating to the Phase 2 Clinical Trial and will be compensated for such services in a combination of cash and shares of stock. Payments in cash to be made by us to Nordic under the Work Statement NB-2 are denominated in both euros and U.S. dollars and total up to €3.6 million ($5.0 million) and $0.3 million, respectively. In addition, we will issue to Nordic shares of our Series A-6 stock having a value of up to $2.9 million, as additional payment for services to be provided under the Work Statement NB-2 and the Clinical Trial Services Agreement.
The Stock Issuance Agreement provides that Nordic is entitled to receive quarterly stock dividends in connection with services performed under Work Statement NB-2, payable in shares of Series A-6, or shares of common stock if our preferred stock has been automatically converted in accordance with its amended certificate of incorporation. In the event Nordic sells the shares of Series A-5 or in the event the shares of Series A-5 are converted into common stock in accordance with our amended certificate of incorporation, this right to receive the Nordic
Accruing Dividend will terminate, but a right to receive an equivalent number of shares of Series A-6 or common stock, as applicable, will remain with Nordic as a contractual right under the Stock Issuance Agreement.
The Nordic Accruing Dividend related to the Phase 2 Clinical Trial is determined based upon the estimated period that will be required to complete the Phase 2 Clinical Trial. On each Accrual Date, beginning with the quarter ended December 31, 2012, we will recognize a liability to issue shares of Series A-6 to Nordic with an Applicable Quarterly Amount value equal to up to $2.9 million minus the aggregate value of any prior Nordic Accruing Dividend related to the Phase 2 Clinical Study divided by the number of calendar quarters it will take to complete the Phase 2 Clinical Study. We calculate the aggregate number of shares of Series A-6 to accrue in such calendar quarter by dividing such Applicable Quarterly Amount, by the greater of (1) the fair market value of our common stock as of the applicable Accrual Date or (2) $81.42 and rounding down the resulting quotient to the nearest whole number. Such shares due to Nordic are to be issued when declared or paid by our Board of Directors, who are required to do so upon Nordic’s request, or upon an event of sale. As of December 31, 2013, 32,215 shares of Series A-6 were due to Nordic under Work Statement NB-2, or, after the automatic conversion into common stock of our preferred stock, 141,293 shares of common stock. In December 2013 Nordic requested that all shares of Series A-6 accrued as of December 31, 2013 under Work Statement NB-2 be issued. Accordingly, our Board of Directors declared a dividend to Nordic of all 32,215 shares accrued under Work Statement NB-2 on December 31, 2013.
3M
In December 2008, we entered into a Feasibility Agreement with 3M whereby 3M assessed the feasibility of developing an Abaloparatide-TD product and supplying the product for preclinical studies in an animal model. Upon successful completion of the feasibility study, during June 2009, we entered into a Development and Clinical Supplies Agreement with 3M under which 3M is responsible to develop an Abaloparatide-TD product and manufacture clinical and toxicology supplies of such patch product for preclinical, Phase 1 and Phase 2 studies on an exclusive basis during the term of the agreement. In December 2012, we entered into an amendment to the Development and Clinical Supplies Agreement in which 3M agreed to develop and manufacture clinical and toxicology supplies for the Phase 3 Abaloparatide-TD clinical study. In addition, 3M agreed that it will not use jointly owned intellectual property developed during and resulting from its work with Radius on Abaloparatide-TD in relation to any other PTH or PTHrP analogue or derivative. We hold exclusive worldwide rights to this use of transdermal technology.
We pay 3M for services delivered pursuant to the Development and Clinical Supplies Agreement on a fee for service or a fee for deliverable basis as specified in the Development and Clinical Supplies Agreement. The Feasibility Agreement expired on or around September 2009. We have paid 3M approximately $15.0 million, in the aggregate, through December 31, 2013 in respect to
services and deliverables delivered pursuant to the Feasibility Agreement and the Development and Clinical Supplies Agreement.
The Development and Clinical Supplies Agreement, as amended, provides for services through December 31, 2017, unless it is sooner terminated. Either party may terminate the Development and Clinical Supplies Agreement upon a material breach by the other party unless such other party cures the alleged breach within the notice period specified in the Development and Clinical Supplies Agreement. The Development and Clinical Supplies Agreement contains customary risk allocation clauses with 3M indemnifying us in respect of third-party claims arising from any personal injury to the extent that such claim results from 3M’s breach of warranty with respect to Abaloparatide-TD meeting applicable specifications; and us indemnifying 3M in respect of third-party claims arising with from our or our agent’s use, testing or clinical studies of Abaloparatide-TD. The Development and Clinical Supplies Agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Ipsen Pharma
In September 2005, we entered into a License Agreement with Ipsen, as amended in September 2007 and May 2011, under which we exclusively licensed certain Ipsen compound technology and related patents covering abaloparatide to research, develop, manufacture and commercialize certain compounds and related products in all countries, except Japan (where we do not hold commercialization rights) and France (where our commercialization rights are subject to certain co-marketing and co-promotion rights retained by Ipsen). Ipsen also granted us an exclusive right and license under the Ipsen compound technology and related patents to make and have made compounds or product in Japan. Ipsen also granted us an exclusive right and license under certain Ipsen formulation technology and related patents solely for purposes of enabling us to develop, manufacture and commercialize compounds and products covered by the compound technology license in all countries, except Japan (where we do not hold commercialization rights) and France (where our commercialization rights are subject to certain co-marketing and co-promotion rights retained by Ipsen). With respect to France, if Ipsen exercises its co-marketing and co-promotion rights then Ipsen may elect to receive a percentage of the aggregate revenue from the sale of products by both parties in France (subject to a mid-double digit percentage cap) and Ipsen shall bear a corresponding percentage of the costs and expenses incurred by both parties with respect to such marketing and promotion efforts in France; Ipsen shall also pay us a mid-single digit royalty on Ipsen’s allocable portion of aggregate revenue from the sale of products by both parties in France. Specifically, we licensed US Patent No. 5,969,095 (effective filing date March 29, 1996, statutory term expires March 29, 2016) entitled “Analogs of Parathyroid Hormone,” US Patent No. 6,544,949, (effective filing date March 29, 1996, statutory term ends March 29, 2016) entitled “Analogs of Parathyroid Hormone” and the corresponding foreign patents and continuing patent applications.
In addition, we have rights to joint intellectual property including rights to US Patent No. 7,803,770 (effective filing date October 3, 2007, statutory term extended to March 26, 2028 with 175 days of patent term adjustment due to delays in patent prosecution by USPTO), US Patent No. 8,148,333 (effective filing date October 3, 2007, statutory term extended to November 8, 2027 with 36 days of patent term adjustment due to delays in patent prosecution by the USPTO) and related patents and patent applications both in the United States and worldwide that cover the method of treating osteoporosis using the Phase 3 clinical dosage strength and form.
As consideration for the rights to abaloparatide licensed to us by Ipsen, we paid Ipsen a non-refundable, non-creditable initial license fee of $250,000. The License Agreement requires us to make payments to Ipsen upon the achievement of certain development milestones in the range of $750,000 and upon the achievement of certain development, regulatory and commercial milestones in the range of €10.0 million to €36.0 million ($13.8 million to $49.6 million), and we have, as of December 31, 2013, paid $750,000 in milestone payments and issued 17,326 shares of series A-1 convertible preferred stock to Ipsen on May 17, 2011 in lieu of a €1.0 million cash payment due to Ipsen upon initiation of the first abaloparatide Phase 3 clinical study. If we or our sublicensees commercialize a product that includes the compound licensed from Ipsen or any analog thereof, we will be obligated to pay to Ipsen a fixed five percent royalty based on net sales of the product on a country-by-country basis until the later of the last to expire of the licensed patents or for a period of 10 years after the first commercial sale in such country.
The date of the last to expire of the abaloparatide patents, barring any extension thereof, is expected to be March 26, 2028. In the event that we sublicense the rights licensed from Ipsen to a third party, we are obligated to pay Ipsen a percentage of certain payments received from such sublicensee (in lieu of milestone payments not achieved at the time of such sublicense). The applicable percentage is in the low double digit range. In addition, if we or our sublicensees commercialize a product that includes a compound discovered by us based on or derived from confidential Ipsen know-how, we will be obligated to pay to Ipsen a fixed low single digit royalty on net sales of such product on a country-by-country basis until the later of the last to expire of our patents that cover such product or for a period of 10 years after the first commercial sale of such product in such country. The License Agreement expires on a country by country basis on the later of (1) the date the last remaining valid claim in the licensed patents expires, in that country; or (2) a period of 10 years after the first commercial sale of the licensed products in such country, unless it is sooner terminated.
The License Agreement may be terminated by us with prior notice to Ipsen. The License Agreement may be terminated by Ipsen upon notice to us with immediate effect, if we, in any country of the world, bring an action or proceeding seeking to have any Ipsen patent right declared invalid or unenforceable. The License Agreement can also be terminated by Ipsen if we fail to use reasonable commercial efforts to develop the licensed product for sale and commercialization in those countries within the territory where it is commercially reasonable to
do so as contemplated by the License Agreement, or fail to use reasonable commercial efforts to perform our obligations under the latest revised version of the development plan approved by the joint steering committee, or fail to use reasonable commercial efforts to launch and sell one licensed product in those countries within the territory where it is commercially reasonable to do so. Either party may also terminate the License Agreement upon a material breach by the other party unless such other party cures the alleged breach within the notice period specified in the License Agreement. Ipsen may terminate the License Agreement in the event that the License Agreement is assigned or sublicensed or in the event that a third party acquires us or in the event that we acquire control over a PTH or a PTHrP compound that is in clinical development or is commercially available in the territory and that, following such assignment, sublicense, acquisition, or acquisition of control by us, such assignee, sublicensee, acquirer or we fail to meet the timetable under the latest revised version of the development plan approved by the joint steering committee under the License Agreement. Any failure to meet such timetable for purposes of such termination clause is deemed a material breach by us.
The License Agreement contains customary risk allocation clauses with each party indemnifying the other in respect of third-party claims arising out of or resulting from: (1) the gross negligence or willful misconduct of such party, its affiliates, licensees, distributors or contractors; (2) any breach by such party of its representations and warranties or any other provision of the License Agreement or any related agreement; (3) the manufacture on behalf of such party of any licensed product or compound; (4) (in the case of Ipsen) the use, development, handling or commercialization of any licensed compound, licensed product or the Ipsen formulation technology by or on behalf of Ipsen or any of its affiliates, licensees, distributors or contractors; and (5) (in our case) the making, use, development, handling or commercialization of any licensed compound or any licensed product by or on our behalf or any of our affiliates, licensees or contractors. The License Agreement contains other customary clauses and terms as are common in similar agreements in the industry. The License Agreement was amended on September 12, 2007 and May 11, 2011.
In January 2006, we entered into a Pharmaceutical Development Agreement as contemplated by the License Agreement with Ipsen. The Pharmaceutical Development Agreement, as amended in July 2007, February 2009, June 2010 and December 2011, provides for the supply of quantities of licensed product for use in certain clinical trials. Beaufour Ipsen Industrie SAS, a subsidiary of Ipsen, is responsible for the supply of Abaloparatide-SC in liquid form in a multi-dose cartridge for use in a pen delivery device. The multi-dose cartridges are manufactured for Beaufour Ipsen Industrie SAS by Vetter under a separate agreement between those parties, and abaloparatide API is manufactured by Lonza for us and is delivered to Vetter for vialing in the multi-dose cartridges. The Pharmaceutical Development Agreement expires upon the completion of the work plan entered into under the Pharmaceutical Development Agreement unless it is sooner terminated. The Pharmaceutical Development Agreement shall automatically terminate upon termination of the Ipsen license Agreement. We may terminate the Pharmaceutical
Development Agreement at any time and for any reason with a specified prior notice period to Ipsen. Either party may terminate the Pharmaceutical Development Agreement upon a material breach by the other party with respect to the Pharmaceutical Development Agreement or the Ipsen License Agreement unless such other party cures the alleged breach within the notice period specified in the Development and Manufacturing Services Agreement. The Pharmaceutical Development Agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Eisai
In June 2006, we exclusively licensed the worldwide (except Japan) rights to research, develop, manufacture and commercialize RAD1901 and related products from Eisai. Specifically, we licensed the patent application that subsequently issued as US Patent No. 7,612,114 (effective filing date December 25, 2003, statutory term extended to August 18, 2026 with 967 days of patent term adjustment due to delays by the USPTO) entitled “Selective Estrogen Receptor Modulator,” the corresponding foreign patent applications and continuing patent applications. As consideration for the rights to RAD1901, we paid Eisai an initial license fee of $0.5 million. In connection with the License Agreement, we have agreed to pay Eisai certain fees in the range of $1.0 million to $20.0 million (inclusive of the $0.5 million initial license fee), payable upon the achievement of certain clinical and regulatory milestones.
Should a product covered by the licensed technology be commercialized, we will be obligated to pay to Eisai royalties in a variable mid-single digit range based on net sales of the product on a country-by-country basis until the later of the last to expire of the licensed patents or the expiration of data protection clauses covering such product in such country; the royalty rate shall then be subject to reduction and the royalty obligation will expire at such time as sales of lawful generic version of such product account for more than a specified minimum percentage of the total sales of all products that contain the licensed compound. The latest valid claim to expire, barring any extension thereof, is expected on August 18, 2026.
We were also granted the right to sublicense with prior written approval from Eisai, and subject to a right of first negotiation held by Eisai if we seek to grant sublicenses limited to particular Asian countries. If we sublicense the licensed technology to a third party, we will be obligated to pay Eisai, in addition to the milestones referenced above, a fixed low double digit percentage of certain fees we receive from such sublicensee and royalties in low single digit range based on net sales of the sublicensee. The license agreement expires on a country by country basis on the later of (1) date the last remaining valid claim in the licensed patents expires, lapses or is invalidated in that country, the product is not covered by data protection clauses, and the sales of lawful generic version of the product account for more than a specified percentage of the total sales of all pharmaceutical products containing the licensed compound in that country; or (2) a period of 10 years after the first commercial sale of the licensed products in such country, unless it is sooner terminated.
The license agreement may be terminated by us with respect to the entire territory with prior notice to Eisai if we reasonably determine that the medical/scientific, technical, regulatory or commercial profile of the licensed product does not justify continued development or marketing. The license agreement can also be terminated by Eisai on a country by country basis at any time prior to the date on which we have submitted for either an FDA NDA approval or an EMA marketing approval with respect to a licensed product, upon prior written notice to us if Eisai makes a good faith determination that we have not used commercially reasonable efforts to develop the licensed product in the territory having reference to prevailing principles and time scales associated with the development, clinical testing and government approval of products of a like nature to such licensed product, unless such default is cured within the period specified in the license agreement or if not capable of being cured within such period we commence efforts to cure and make diligent efforts to do so. Either party may also terminate the license agreement upon a material breach by the other party unless such other party cures the alleged breach within the notice period specified in the license agreement. Either party may also terminate the license agreement upon the bankruptcy or insolvency of the other party. Eisai may also terminate the license agreement with prior notice if we are acquired by, or if we transfer all of our pharmaceutical business assets (or an essential part of such assets) or more than 50% of our voting stock to, any third-party person or organization, or otherwise come under the control of, such a person or organization, whether resulting from merger, acquisition, consolidation or otherwise in the event that Eisai reasonably determines that the person or organization assuming control of us is not able to perform the license agreement with the same degree of skill and diligence that we would use, such determination being made with reference to the following criteria with respect to the person or organization assuming control of us: (1) whether such person or organization has the financial resources to assume our obligations with respect to development and commercialization of products; (2) whether such person or organization has personnel with skill and experience adequate to assume our obligations with respect to development and commercialization of products at the stage of development and commercialization as of the date of such change; and (3) whether such person or organization expressly assumes all obligations imposed on us by the license agreement and agrees to dedicate personnel and financial resources to the development and commercialization of the licensed product that are at least as great as those provided by us. Eisai shall further have the right to terminate if the acquiring person or organization: (a) has any material and active litigations with Eisai; (b) is a certain type of pharmaceutical company; or (c) is a hostile takeover bidder against us which has not been approved by our board of directors as constituted immediately prior to such change of control.
The license agreement contains customary risk allocation clauses with each party indemnifying the other in respect of third-party claims arising out of or resulting from: (1) the negligence, reckless or intentional acts or omissions of such party, its affiliates, and licensees; (2) any breach by such party of its representations and warranties; and (3) any personal injury arising out of the labeling, packaging, package insert, other materials or promotional claims with respect to any
licensed product by such party or its affiliates, licensees or distributors in the territory (in our case) or Japan (in the case of Eisai). The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Lonza
In October 2007, we entered into a Development and Manufacturing Services Agreement with Lonza. We and Lonza have entered into a series of Work Orders pursuant to the Development and Manufacturing Services Agreement pursuant to which Lonza has performed pharmaceutical development and manufacturing services for our abaloparatide product. We pay Lonza for services rendered and deliverables delivered pursuant to these work orders on a fee for service basis as specified in the applicable work statement. The Development and Manufacturing Services Agreement will expire on December 31, 2015 unless it is sooner terminated, and is subject to renewal by us for successive multiple-year terms with notice to Lonza.
The Development and Manufacturing Services Agreement or any Work Order may be terminated by either party upon a material breach by the other party with respect to the Development and Manufacturing Services Agreement unless such other party cures the alleged breach within the notice period specified in the Development and Manufacturing Services Agreement. Either party may also terminate a Work Order if force majeure conditions have prevented performance by the other party for more than a specified period of time with respect to such Work Order. Termination of any Work Order for force majeure shall not result in termination of the Development and Manufacturing Services Agreement or any other Work Orders, which shall remain in force until terminated. Either party may also terminate the Development and Manufacturing Services Agreement upon the bankruptcy or insolvency of the other party. We may also terminate the Development and Manufacturing Services Agreement or any Work Order with prior notice to Lonza for convenience. We may also terminate the Development and Manufacturing Services Agreement or any Work Order if we reasonably determine that Lonza is or will be unable to perform the applicable services in accordance with the agreed upon timeframe and budget set forth in the applicable Work Order, or if Lonza fails to obtain or maintain any material governmental licenses or approvals required in connection with such services.
The Development and Manufacturing Services Agreement contains customary risk allocation clauses with each party indemnifying the other in respect of third-party claims arising out of or resulting from: (i) the negligence or willful misconduct of such party, its affiliates and their respective officers, directors, employees and agents in performing its obligations under the Developing and Manufacturing Services Agreement; and (ii) any breach by such party of its representations and warranties under the Development and Manufacturing Services Agreement. We have agreed to indemnify Lonza in respect of third-party claims arising from or relating to the use of our product.
On December 23, 2011, we entered into Work Order No. 4, or Work Order No. 4, under that certain Development and Manufacturing Services Agreement with Lonza. Pursuant to Work Order No. 4, Lonza agreed to perform activities required for our filing of an NDA in the United States with the FDA and similar applications required by the EMA and other authorities, excluding authorities in Japan, for abaloparatide, including production of three validation batches. These activities will provide for full process qualification and all required documentation necessary for regulatory submissions of the NDA to the FDA and the NDA equivalents to such other authorities. The total compensation payable to Lonza from us for services performed under Work Order No. 4 is up to €363.5 thousand plus up to €1.1 million ($500.9 thousand plus up to $1.5 million), for the regulatory qualification and validation campaigns.
Charles River Laboratories
In March 2004, we entered into a Laboratory Services and Confidentiality Agreement with Charles River Laboratories, Inc., or CRLI, and amended this agreement on November 7, 2008. We have entered into a series of letter agreements with CRLI pursuant to this Laboratory Services and Confidentiality Agreement, covering the performance of certain testing and analytical services concerning our product candidates. We pay CRLI for services rendered and deliverables delivered pursuant to these letter agreements on a fee for service basis. We are permitted to terminate any on-going study under the Laboratory Services and Confidentiality Agreement at any time with the specified prior notice to CRLI and subject to the payment of applicable study costs and fees. Either party may terminate the Laboratory Services and Confidentiality Agreement at any time with the specified prior notice to the other party and subject to the completion of any then on-going studies and the payment by us of any fees for such studies. Either party may also terminate the Laboratory Services and Confidentiality Agreement upon a material breach by the other party unless such other party cures the alleged breach within the notice period specified in the Laboratory Services and Confidentiality Agreement.
The Laboratory Services and Confidentiality Agreement contains customary risk allocation clauses with each party indemnifying the other in respect of third-party claims arising out of or in connection with the negligence or willful misconduct of such party. We also agreed to indemnify CRLI in respect of third-party claims arising out of or in connection with the manufacture, distribution, use, sale or other disposition by us, or any of our distributors, customers, sublicensees or representatives, of any of our products or processes and/or any other substances which are produced, purified, tested or vialed by CRLI. We also agreed to indemnify CRLI against any and all liability that may be incurred as the result of any contact by us or our employees with CRLI’s animals, tissues or specimens during visits to CRLI or after delivery of any samples/specimens to us. The Laboratory Services and Confidentiality Agreement contains other customary clauses and terms as are common in similar agreements in the industry.
GOVERNMENT REGULATION
United States — FDA process
The research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing, among other things, of our products are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Failure to comply with the applicable United States requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, and/or criminal prosecution. We expect abaloparatide, RAD1901 and RAD140 will each be subject to review by the FDA as a drug under NDA standards though we currently only have an active IND application in relation to abaloparatide in the United States. We anticipate filing an investigational new drug application for RAD1901 with the FDA in 2014, as previous studies of RAD1901 were performed outside the United States.
Approval process. None of our drugs may be marketed in the United States until the drug has received FDA approval. The steps required to be completed before a drug may be marketed in the United States include:
· preclinical laboratory tests, animal studies, and formulation studies, all performed in accordance with the FDA’s Good Laboratory Practice, or GLP, regulations;
· submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may begin and must be updated annually;
· adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each indication to FDA’s satisfaction;
· submission to the FDA of an NDA;
· satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with cGMP regulations; and
· FDA review and approval of the NDA.
Preclinical tests include laboratory evaluation of product chemistry, toxicity, and formulation, as well as animal studies. The conduct of the preclinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application, which must become effective before human clinical trials may begin. An IND application will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in the IND application. In such a case, the IND application sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed.
We cannot be sure that submission of an IND application will result in the FDA allowing clinical trials to begin.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND application.
Clinical trials necessary for product approval are typically conducted in three sequential phases, but the Phases may overlap. The study protocol and informed consent information for study subjects in clinical trials must also be approved by an Institutional Review Board, or IRB, for each institution where the trials will be conducted, and each IRB must monitor the study until completion. Study subjects must provide informed consent and sign an informed consent form before participating in a clinical trial. Clinical testing also must satisfy extensive good clinical practice, or GCP, regulations and regulations for informed consent and privacy of individually identifiable information. Phase 1 usually involves the initial introduction of the investigational drug into people to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness.
Phase 1 studies are usually conducted in healthy individuals and are not intended to treat disease or illness. However, Phase 1b studies are conducted in healthy volunteers or in patients diagnosed with the disease or condition for which the study drug is intended, who demonstrate some biomarker, surrogate, or possibly clinical outcome that could be considered for “proof of concept.” Proof of concept in a Phase 1b study typically confirms the hypothesis that the current prediction of biomarker, or outcome benefit is compatible with the mechanism of action.
Phase 2 usually involves trials in a limited patient population to (1) evaluate dosage tolerance and appropriate dosage; (2) identify possible adverse effects and safety risks; and (3) evaluate preliminarily the efficacy of the drug for specific indications. Several different doses of the drug may be looked at in Phase 2 to see which dose has the desired effects. Patients are monitored for side effects and for any improvement in their illness, symptoms, or both. Phase 3 trials usually further evaluate clinical efficacy and test further for safety by using the drug in its final form in an expanded patient population.
A Phase 3 trial usually compares how well the study drug works compared with an inactive placebo and/or another approved medication. One group of patients may receive the new drug being tested, while another group of patients may receive the comparator drug (already approved drug for the disease being studied), or placebo.
There can be no assurance that Phase 1, Phase 2 or Phase 3 testing will be completed successfully within any specified period of time, if at all. Furthermore, we or the FDA or an IRB
(with respect to a particular study site) may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The FDCA permits FDA and the IND application sponsor to agree in writing on the design and size of clinical studies intended to form the primary basis of an effectiveness claim in an NDA. This process is known as a Special Protocol Assessment, or SPA. Under an SPA, the FDA agrees to not alter its position with respect to adequacy of the design, execution or analyses of the clinical trial intended to form the primary basis of an effectiveness claim in an NDA without the sponsor’s agreement, unless the FDA identifies a substantial scientific issue essential to determining the safety or efficacy of the drug after testing begins.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. The testing and approval process requires substantial time, effort and financial resources. The FDA reviews the application and may deem it to be inadequate, and companies cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but it typically follows such recommendations.
The FDA has various programs, including fast track, breakthrough designation, priority review and accelerated approval, that are intended to expedite or simplify the process for reviewing drugs and/or provide for approval on the basis of surrogate endpoints. Generally, drugs that may be eligible for one or more of these programs are those intended to treat serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that provide meaningful benefit over existing treatments. From time to time, we anticipate applying for orphan designation and/or breakthrough designation for programs that we believe meet the applicable FDA criteria. A company cannot be sure that any of its drugs will qualify for any of these programs, or even if a drug does qualify, that the review time will be reduced.
Before approving an NDA, the FDA usually will inspect the facility or the facilities at which the drug is manufactured and will not approve the product unless the manufacturing is in compliance with cGMP regulations. If the NDA and the manufacturing facilities are deemed acceptable by the FDA, it may issue an approval letter, or in some cases, an approvable letter followed by an approval letter. Both letters usually contain a number of conditions that must be met in order to secure final approval of the NDA. When and if those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter. The approval letter authorizes commercial marketing of the drug for specific indications. As a condition of NDA approval, the FDA may require post-marketing testing and surveillance to monitor the drug’s safety or efficacy, or
impose other conditions. Approval may also be contingent on a Risk Evaluation and Mitigation Strategy, or REMS, that limits the labeling, distribution or promotion of a drug product. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market.
After approval, certain changes to the approved product, such as adding new indications, making certain manufacturing changes or making certain additional labeling claims, are subject to further FDA review and approval. Before a company can market products for additional indications, it must obtain additional approvals from the FDA. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. A company cannot be sure that any additional approval for new indications for any product candidate will be approved on a timely basis, or at all.
Post-approval requirements. Often times, even after a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. If such post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA are required to: (1) report certain adverse reactions to the FDA, (2) comply with certain requirements concerning advertising and promotional labeling for their products, and (3) continue to have quality control and manufacturing procedures conform to cGMP regulations after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of ongoing compliance with cGMP regulations. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. We have used and intend to continue to use third-party manufacturers to produce our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product after approval may result in restrictions on a product, including withdrawal of the product from the market.
Hatch-Waxman Act. Under the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, Congress created an abbreviated FDA review process for generic versions of pioneer (brand name) drug products. In considering whether to approve such a generic drug product, the FDA requires that an Abbreviated New Drug Application, or ANDA, applicant demonstrate, among other things, that the proposed generic drug product’s active ingredient is the same as that of the reference product, that any impurities in the proposed product do not affect the product’s safety or effectiveness, and that its manufacturing processes and methods ensure the consistent potency and purity of its proposed product.
The Hatch-Waxman Act provides five years of data exclusivity for new chemical entities which prevents the FDA from accepting ANDAs and 505(b)(2) applications containing the protected
active ingredient. We expect to be eligible for five years of data exclusivity following any FDA approval of Abaloparatide-SC.
The Hatch-Waxman Act also provides three years of exclusivity for applications containing the results of new clinical investigations (other than bioavailability studies) essential to the FDA’s approval of new uses of approved products, such as new indications, delivery mechanisms, dosage forms, strengths or conditions of use. For example, if Abaloparatide-SC is approved for commercialization and we are successful in performing a clinical trial of Abaloparatide-TD that provides a new basis for approval (a different delivery mechanism) it is possible that we may become eligible for an additional three-year period of data exclusivity which protects against the approval of ANDAs and 505(b)(2) applications for the protected use but will not prohibit the FDA from accepting or approving ANDAs or 505(b)(2) applications for other products containing the same active ingredient.
The Hatch-Waxman Act requires NDA applicants and NDA holders to provide certain information about patents related to the drug for listing in the FDA’s list of Approved Drug Products with Therapeutic Equivalence Evaluations (commonly known as the Orange Book). ANDA and 505(b)(2) applicants must then certify regarding each of the patents listed with the FDA for the reference product. A certification that a listed patent is invalid or will not be infringed by the marketing of the applicant’s product is called a “Paragraph IV certification.” If the ANDA or 505(b)(2) applicant provides such a notification of patent invalidity or non-infringement, then the FDA may accept the ANDA or 505(b)(2) application beginning four years after approval of the NDA. If an ANDA or 505(b)(2) application containing a Paragraph IV certification is submitted to the FDA and accepted as a reviewable filing by the agency, the ANDA or 505(b)(2) applicant then must provide, within 20 days, notice to the NDA holder and patent owner stating that the application has been submitted and providing the factual and legal basis for the applicant’s opinion that the patent is invalid or not infringed. The NDA holder or patent owner then may file suit against the ANDA or 505(b)(2) applicant for patent infringement. If this is done within 45 days of receiving notice of the Paragraph IV certification, a one-time 30-month stay of the FDA’s ability to approve the ANDA or 505(b)(2) application is triggered. The 30-month stay begins at the end of the NDA holder’s data exclusivity period, or, if data exclusivity has expired, on the date that the patent holder is notified of the submission of the ANDA. The FDA may approve the proposed product before the expiration of the 30-month stay if a court finds the patent invalid or not infringed or if the court shortens the period because the parties have failed to cooperate in expediting the litigation.
European Union — EMA process
In the EU, medicinal products are authorized following a similar demanding process as that required in the United States and applications are based on the ICH Common Technical Document. Prior to submitting a European Marketing Authorization Application, or MAA, it is necessary to gain approval of a detailed Pediatric Investigation Plan, or PIP, with the European
Medicines Agency’s Pediatric Committee, or PDCO. After gaining PIP approval, medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures.
Centralized procedure. Under the centralized procedure, after the EMA issues an opinion, the European Commission issues a single marketing authorization valid across the EU, as well as Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human medicines that are: derived from biotechnology processes, such as genetic engineering, contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions, and officially designated orphan medicines. For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the EMA, as long as the medicine concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health.
National authorization procedures. There are also two other possible routes to authorize medicinal products in several countries, which are available for products that fall outside the scope of the centralized procedure:
· Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one EU country of a medicinal product that has not yet been authorized in any EU country and that does not fall within the mandatory scope of the centralized procedure.
· Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one EU Member State, in accordance with the national procedures of that country. Thereafter, further marketing authorizations can be sought from other EU countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization.
In light of the fact that there is no policy at the EU level governing pricing and reimbursement, the 27 EU Member States each have developed their own, often varying, approaches. In many EU Member States, pricing negotiations must take place between the holder of the marketing authorization and the competent national authorities before the product is sold in their market with the holder of the marketing authorization required to provide evidence demonstrating the pharmaco-economic superiority of its product in comparison with directly and indirectly competing products. We have reviewed our development program, proposed Phase 3 study design, and overall non-clinical and clinical data package and believe they support future regulatory approval of Abaloparatide-SC in the EU. In December 2012, we met with the Swedish Medical Products Agency, or MPA, to review the design and the overall progress of the Phase 3 study. The MPA confirmed that the program, based on the current single pivotal trial design, could support the submission and potential approval of an MAA in the EU, depending on the results of the Phase 3 study.
Good manufacturing practices. Like the FDA, the EMA, the competent authorities of the EU Member States and other regulatory agencies regulate and inspect equipment, facilities and processes used in the manufacturing of pharmaceutical and biologic products prior to approving a product. If, after receiving clearance from regulatory agencies, a company makes a material change in manufacturing equipment, location, or process, additional regulatory review and approval may be required. Once we or our partners commercialize products, we will be required to comply with cGMP, and product-specific regulations enforced by, the European Commission, the EMA and the competent authorities of EU Member States following product approval. Also like the FDA, the EMA, the competent authorities of the EU Member States and other regulatory agencies also conduct regular, periodic visits to re-inspect equipment, facilities, and processes following the initial approval of a product. If, as a result of these inspections, it is determined that our or our partners’ equipment, facilities, or processes do not comply with applicable regulations and conditions of product approval, regulatory agencies may seek civil, criminal or administrative sanctions and/or remedies against us, including the suspension of our manufacturing operations or the withdrawal of our product from the market.
Data and Market Exclusivity. Similar to the United States, there is a process for generic versions of innovator drug products in the EU. Abridged applications for the authorization of generic versions of drugs authorized by EMA can be submitted to the EMA through a centralized procedure referencing the innovator’s data and demonstrating bioequivalence to the reference product, among other things.
New medicinal products in the EU can receive eight years of data exclusivity coupled with two years of market exclusivity, and a potential one year extension, if the marketing authorizations holder obtains an authorization for one or more new therapeutic indications that demonstrates “significant clinical benefit” in comparison with existing therapies; this system is usually referred to as “8+2+1”. We expect to be eligible for at least ten years of market exclusivity following any approval of Abaloparatide-SC.
Abridged applications cannot rely on an innovator’s data until after expiry of the 8 year date exclusivity term; applications for a generic product can be filed but the product cannot be marketed until the end of the market exclusivity term.
Other international markets — drug approval process
In some international markets (e.g., China or Japan), although data generated in United States or EU trials may be submitted in support of a marketing authorization application, additional clinical trials conducted in the host territory, or studying people of the ethnicity of the host territory, may be required prior to the filing or approval of marketing applications within the country.
Pricing and reimbursement
In the United States and internationally, sales of products that we market in the future, and our ability to generate revenues on such sales, are dependent, in significant part, on the availability and level of reimbursement from third-party payers such as state and federal governments, managed care providers and private insurance plans. Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and also the out-of-pocket obligations of member patients for such products. In addition, particularly in the United States and increasingly in other countries, we may be required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our products that are reimbursed by such entities. It is possible that future legislation in the United States and other jurisdictions could be enacted which could potentially impact the reimbursement rates for the products we are developing and may develop in the future and also could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market.
There is no legislation at the EU level governing the pricing and reimbursement of medicinal products in the EU. As a result, the competent authorities of each of the 27 EU Member States have adopted individual strategies regulating the pricing and reimbursement of medicinal products in their territory. These strategies often vary widely in nature, scope and application. However, a major element that they have in common is an increased move towards reduction in the reimbursement price of medicinal products, a reduction in the number and type of products selected for reimbursement and an increased preference for generic products over innovative products. These efforts have mostly been executed through these countries’ existing price-control methodologies. The government of the UK announced the phase-out of its established Pharmaceutical Pricing Reimbursement Scheme approach in January 2014 and the adoption of a new value-based pricing approach. Under this approach, in a complete departure from established methodologies, reimbursement levels of each drug will be explicitly based on an assessment of value, looking at the benefits for the patient, unmet need, therapeutic innovation, and benefit to society as a whole. It is increasingly common in many EU Member States for Marketing Authorization Holders to be required to demonstrate the pharmaco-economic superiority of their products as compared to products already subject to pricing and reimbursement in specific countries. In order for drugs to be evaluated positively under such criteria, pharmaceutical companies may need to re-examine, and consider altering, a number of traditional functions relating to the selection, study, and management of drugs, whether currently marketed, under development, or being evaluated as candidates for research and/or development.
Future legislation, including the current versions being considered at the federal level in the United States and at the national level in EU Member States, or regulatory actions implementing recent or future legislation may have a significant effect on our business. Our ability to
successfully commercialize products depends in part on the extent to which reimbursement for the costs of our products and related treatments will be available in the United States and worldwide from government health administration authorities, private health insurers and other organizations. Substantial uncertainty exists as to the reimbursement status of newly approved healthcare products by third-party payers.
Sales and Marketing
The FDA regulates all advertising and promotion activities for products under its jurisdiction both prior to and after approval. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, and often reflect a physician’s belief that the off-label use is the best treatment for the patients. The FDA does not regulate the behavior of physicians in their choice of treatments, but FDA regulations do impose stringent restrictions on manufacturers’ communications regarding off-label uses. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
We may also be subject to various federal and state laws pertaining to healthcare “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations and very few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. Moreover, recent healthcare reform legislation has strengthened these laws. For example, the recently enacted PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes, so that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, PPACA permits the government to assert that a claim that includes items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment, to third-party payers (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal and civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal healthcare programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions
and penalties also can be imposed upon executive officers and employees, including criminal sanctions against executive officers under the so-called “responsible corporate officer” doctrine, even in situations where the executive officer did not intend to violate the law and was unaware of any wrongdoing.
Given the significant penalties and fines that can be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the government were to allege or convict us or our executive officers of violating these laws, our business could be harmed. In addition, private individuals have the ability to bring similar actions. Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. Further, there are an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state authorities.
Similar rigid restrictions are imposed on the promotion and marketing of medicinal products in the EU and other countries. Laws (including those governing promotion, marketing and anti-kickback provisions), industry regulations and professional codes of conduct often are strictly enforced. Even in those countries where we are not directly responsible for the promotion and marketing of our products, inappropriate activity by our international distribution partners can have adverse implications for us.
Other laws and regulatory processes
We are subject to a variety of financial disclosure and securities trading regulations as a public company in the United States, including laws relating to the oversight activities of the SEC and, following the listing of our capital stock on the NASDAQ Global Market, we will be subject to the regulations of the NASDAQ Global Market. In addition, the Financial Accounting Standards Board, or FASB, the SEC and other bodies that have jurisdiction over the form and content of our accounts, our financial statements and other public disclosure are constantly discussing and interpreting proposals and existing pronouncements designed to ensure that companies best display relevant and transparent information relating to their respective businesses.
Our international operations are subject to compliance with the Foreign Corrupt Practices Act, or the FCPA, which prohibits corporations and individuals from paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to
otherwise influence a person working in an official capacity. We also may be implicated under the FCPA for activities by our partners, collaborators, CROs, vendors or other agents.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights or acquisitions may be subject to national or supranational antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
EMPLOYEES
As of March 31, 2014, we employed 11 full-time employees and two part-time employees, four of whom held Ph.D. or M.D. degrees. Eight of our employees were engaged in research and development activities and five were engaged in support administration, including business development and finance. We intend to use CROs and other third parties to perform our clinical studies and manufacturing.
LEGAL PROCEEDINGS
We are not currently involved in any material legal proceedings.