Attached files
| file | filename |
|---|---|
| 8-K - 8-K - Radius Health, Inc. | a14-7553_18k.htm |
Exhibit 10.1
CLINICAL TRIAL SERVICES AGREEMENT AMENDMENT NO. 1 TO WORK STATEMENT NB-3
RADIUS HEALTH, INC., a Delaware corporation (“Radius”) and NORDIC BIOSCIENCE CLINICAL DEVELOPMENT VII A/S, a Danish corporation (“NB”) that is a wholly-owned subsidiary of Nordic Bioscience Clinical Development A/S entered into a Clinical Trial Services Agreement dated March 29, 2011 (“Agreement”) and Work Statement NB-3 under the Agreement (“Work Statement NB-3”) as of February 21, 2013 (“Effective Date”)
Pursuant to Section 2.3, 2.11 and 11.7 of the Agreement, the parties wish to enter into this Amendment No. 1 to Work Statement NB-3 (“Amendment No. 1”) effective as of February 28, 2014 (“Amendment Date”). Capitalized terms used in this Amendment No. 3 and not defined herein are used with the meanings ascribed to them in the Agreement and Work Statement NB-3.
NOW THEREFORE, in consideration of the mutual promises contained in the Agreement and for other good and valuable consideration the receipt and adequacy of which each of the parties does hereby acknowledge, the parties hereby agree to the terms of this Amendment No. 1 to Work Statement NB-3 as follows:
1. Addition of Period 2 to NB-3. (a) At Radius’ request, NB will contract with up to 28 CCBR and non-CCBR Sites to enroll patients in a Period 2 extension of the clinical study that is the subject of Work Statement NB-3 entitled: BA058-05-005 “An Extension Study to Evaluate 24 Months of Standard-of-Care Osteoporosis Management Following Completion of 18 Months of BA058 or Placebo Treatment in Protocol BA058-05-00” (Attachment 3 to the Agreement). Radius wishes to provide for payment to NB for Services during Period 2 under this Amendment No. 1.
(b) Attachment A to Work Statement NB-3 is amended to add Study Assumptions for Period 2 immediately following the Payment Schedule table for Period 1 (Attachment 1 to the Agreement).
(c) Attachment B to Work Statement NB-3 is amended to add Payment Schedule for Period 2 immediately following the final paragraph (Attachment 2 to the Agreement).
This Amendment No. 1 to Work Statement NB-3 contains the following Attachments, each of which is made a part hereof:
Attachment 1 — Specifications/Key Assumptions/Services/Division of
Responsibilities/Timeline Specifications
Attachment 2 — Budgets, Fees, Pass-through Costs, and Payment Schedule
Attachment 3 — Protocol
2. Ratification. Except to the extent expressly amended by this Amendment No. 1, all of the terms, provisions and conditions of the Agreement and Work Statement NB-3 are hereby ratified and confirmed and shall remain in full force and effect. The term “Work Statement NB-3”, as used in the Agreement, shall henceforth be deemed to be a reference to Work Statement NB-3 as amended by this Amendment No. 1.
3. General. This Amendment No. 1 may be executed in counterparts, each of which will be deemed an original with all such counterparts together constituting one instrument.
IN WITNESS WHEREOF the parties have caused this Amendment No. 1 to be executed by their respective duly authorized officers, and have duly delivered and executed this Amendment No. 1 under seal as of the Amendment Date.
|
RADIUS HEALTH, INC. |
|
NORDIC BIOSCIENCE CLINICAL DEVELOPMENT VII A/S |
|
|
|
|
|
/s/ Robert E. Ward |
|
/s/ Jeppe R. Andersen |
|
By: Robert E. Ward |
|
By: Jeppe R. Andersen |
|
Title: Chief Executive Officer |
|
Title: Chief Executive Officer |
|
|
|
|
|
Notice Address |
|
Notice Address |
|
Radius Health, Inc. |
|
Nordic Bioscience Clinical Development VII A/S |
|
201 Broadway, 6th Floor |
|
Herlev Hovedgade 207 |
|
Cambridge, MA 02139 |
|
2730 Herlev |
|
USA |
|
Denmark |
|
Attn: President |
|
Attn: Clinical Trial Leader & Medical Advisor / Clinical Studies |
|
Phone: 01.617.444.1834 |
|
Phone: 45.4452.5251 |
|
Fax: 01.617.551.4701 |
|
Fax: 45.4452.525 |
Amendment No. 1 to Work Statement NB-3
Attachment 1
Specifications/Key Assumptions/Services/Division of Responsibilities/Timeline Specifications
Study Assumptions for Period 2
Radius Health, Inc.
Protocol: BA058-05-005, “An Extension Study to Evaluate 24 Months of Standard-of-Care Osteoporosis Management Following Completion of 18 Months of BA058 or Placebo Treatment in Protocol BA058-05-00”
|
Protocol Number: BA058-05-005 |
|
Number of Sites: 28 |
|
Argentina |
|
1 |
|
Brazil |
|
5 |
|
Czech Republic |
|
3 |
|
Denmark |
|
3 |
|
Estonia |
|
2 |
|
Hong Kong |
|
1 |
|
Lithuania |
|
1 |
|
Poland |
|
6 |
|
Romania |
|
1 |
|
USA |
|
5 |
Clinical Trial Timeline and Budget
Page 1 of 2
RADIUS
BA058-05-005 Amendment 1 (Period 2)
Protocol 05 February 2013
Cost Proposal 02 April 2013
|
Sponsor: |
|
RADIUS |
|
|
|
|
Protocol ID: |
|
BA058-05-005 Amendment 1 (Period 2) |
|
|
|
|
Development Phase: |
|
III |
|
|
|
|
Disease: |
|
Osteoporosis |
|
|
|
|
Total # of Randomized and completed subjects |
|
925 |
|
|
|
|
Nordic Start Study Activity |
|
1-Mar-13 |
|
|
|
|
Expected Date of FPFV: |
|
1-May-13 |
|
|
|
|
Expected Length of Recruitment (months): |
|
18 |
|
|
|
|
Treatment duration (months) |
|
18 |
|
|
|
|
Follow-up duration (months) |
|
1 |
|
|
|
|
Close Out (months) |
|
Transferred from period 1 |
|
|
|
|
Duration of Nordic Involvement |
|
37 |
|
|
|
|
Number of visits per patient: |
|
2.5 |
|
|
|
|
Number of Countries: |
|
10 |
|
Ar, Br, Dk, Cz, Ee, Li, Po, Ro, HK, USA |
|
|
Number of Sites: |
|
28 |
|
Ar x 1, Br x 5, Dk x 3, Cz x 3, Ee x 2, Li x 1, Po x 6, Ro x 1, HK x 1, USA x 5 |
|
|
Total Budget |
|
EURO |
|
|
|
|
Clinic Fee |
|
1,802,138 |
|
925 subjects |
|
|
CRO Activities (Nordic Bioscience) |
|
1,165,500 |
|
|
|
|
Central Lab Fee (Synarc Lab) |
|
0 |
|
No lab tests to be performed |
|
|
EDC system |
|
|
|
Pass through. Estimated to be below 100,000 Euro |
|
|
Calcium and D |
|
|
|
Pass through. Estimated to be 15 Euro per month per subject |
|
|
Alendronate |
|
|
|
Pass through. Estimated to be 30 Euro per month per subject |
|
|
Total Budget (Euro) |
|
2,967,638 |
|
|
|
|
|
|
USD |
|
|
|
|
Central Imaging Reading (Synarc Imaging) |
|
527,740 |
|
Scalable and invoiced as such + shipment estimated 41,617 USD |
|
|
Total Budget (USD) |
|
527,740 |
|
|
|
Page 2 of 2
|
Pass through Cost |
|
EURO |
|
|
|
|
Monitoring Travel Expenses & Accommodations/ other travels |
|
Included |
|
|
|
|
Translation |
|
Included |
|
|
|
|
Investigator Meeting |
|
NA |
|
|
|
|
Alendronate and calcium/ D supllement |
|
Not included |
|
|
|
|
Image shipments |
|
Not included |
|
|
|
|
Submission Fee to ERC and CA |
|
Not included |
|
|
|
|
EDC system |
|
Not included |
|
|
|
|
Data Monitoring Committe |
|
Not included |
|
|
|
|
Patient insurance |
|
Not included |
|
|
|
|
Medical writing |
|
Not included |
|
|
|
|
External advisory Board |
|
Not included |
|
|
|
|
Statistical Data analysis and Clinical Study Report |
|
Not included |
|
|
|
|
Payment schedule |
|
Euro |
|
|
|
|
|
|
|
|
|
|
|
Upfront |
|
222,573 |
|
|
|
|
1,243 per enrolled subject |
|
1,149,775 |
|
|
|
|
60,514 per month for 19 months |
|
1,149,766 |
|
|
|
|
Rest |
|
445,524 |
|
|
|
|
|
|
2,967,638 |
|
|
|
Amendment No. 1 to Work Statement NB-3
Attachment B
Budgets, Fees, Pass-through Costs, and Payment Schedule*
For Period 2
Protocol: BA058-05-005, “An Extension Study to Evaluate 24 Months of Standard-of-Care Osteoporosis Management Following Completion of 18 Months of BA058 or Placebo Treatment in Protocol BA058-05-00”
The Cash budget is Euro 2,967,638 and USD 527,740
The Bonus Equity Payment Amount budget is Euro 2,967,638 and USD 527,740
The Cash budget will be paid as follows:
· 7.5% (Euro 222,573) upon execution.
· 39% (Euro 1,149,775) of the Euro cash budget will be paid during enrollment at Euro 1,243 per randomized patients at all non-US sites (the SITES).
· The USD budget will be paid according to invoices received from Synarc Inc.
· 39% (Euro 1,149,766) of the Euro cash budget will be paid on a monthly basis over 19 months starting after patient randomization is completed with Euro 60,514 per month.
· The USD budget will be paid according to invoices received from Synarc Inc.
· 15% (Euro 445,524) of the Euro cash budget will be paid when the database is locked and transferred to and accepted by Radius.
Pass through costs will be invoiced on a monthly basis.
The Equity budget will be paid in concert with the cash payment after the same model as for Work Statement NB-1, NB-2 and NB-3 (Period 1) under an amended Stock Issuance Agreement modeled on the Amended and Restated Stock Issuance Agreement entered into by the parties as of May 16, 2011.
The pricing specified in this Budget is calculated based upon 925 subjects randomized and completed but will be adjusted for the number of completed patients greater or less than 925 subjects as follows (i) on a fully pro rata basis for the Clinic Fee and the Central Imaging Reading Fee; and (ii) by an amount of euro 487 per subject for the CRO Activities Fee. Should greater than 925 patients be randomized into the study, Euro 1,243 per patient shall be paid at the time of randomization of the additional patients. Thereafter, at the time of completion of patient randomization for the study, the monthly payments over the remaining 19 months of clinic activities shall be adjusted to reflect the actual number of randomized patients but also factoring in the actual drop-out rate at the time of completion of patient randomization and preserving a 15% final payment amount when the database is locked and transferred to Radius. The rate of drop-outs will be monitored over the 19 month period and appropriate adjustments will be made on a quarterly basis to the monthly payment amount as well as the final payment amount to reflect a higher or lower drop-out rate following completion of patient randomization according to following table.
|
Dropout time |
|
Payment (if no radiology performed) |
|
Payment (if radiology performed) |
|
|
|
|
|
|
|
After Visit 3, before Visit 4 |
|
EUR 643 (33% of the total Clinic Fee) |
|
EUR 1,123.5 (58% of the total Clinic Fee) |
|
|
|
|
|
|
|
After Visit 4, before Visit 5 |
|
EUR 1,052 (54% of the total Clinic Fee) |
|
EUR 1,535.8 (78.88% of the total Clinic Fee) |
|
|
|
|
|
|
|
After Visit 5 |
|
n/a radiology is mandatory |
|
EUR 1,948 (100% of the total Clinic Fee)* |
*The additional payment for radiology shall not be owed for any patient that is lost to follow-up and no radiology is completed
The Cash Budget and the Bonus Equity Payment Amount shall be reduced by an amount of euro 1,948 per subject for Clinic Activities not performed at the SITES for any patients enrolled in the United States. Such reduction to be applied in pro rata installments to monthly payments after patient randomization is completed. Otherwise, all pricing will be adjusted on a pro rata fashion to reflect the actual study activities completed by the study subjects.

CLINICAL STUDY PROTOCOL
An Extension Study to Evaluate 24 Months of Standard-of-Care Osteoporosis Management Following Completion of 18 Months of BA058 or Placebo Treatment in Protocol BA058-05-003
This study will be conducted according to the protocol and in compliance with Good Clinical Practice, the ethical principles stated in the Declaration of Helsinki, and other applicable regulatory requirements.
|
Protocol Number: |
Protocol BA058-05-005 |
|
Protocol Date (Version): |
Original (23 July 2012) Amendment 1, Version 1 (13 February 2013) |
|
EudraCT Number |
2012-002216-10 |
|
IND Number: |
73,176 |
|
Study Sponsor: |
Radius Health, Inc. 201 Broadway, 6th Floor Cambridge, MA 02139, USA Tel: 617.551.4700. Fax: 617.551.4701 |
|
Sponsor Medical Monitor: |
Louis Brenner, MD Chief Medical Officer, Radius Health, Inc. Tel: 617.551.4006. Fax: 617.551.4701. Email: lbrenner@radiuspharm.com |
|
Study Safety Officer |
Bente Juel Riis, MD Medical Advisor, Nordic Bioscience A/S Tel: +45 22 90 13 17. Fax: +41 91 970 2988 Email: bjr@nordicbioscience.com |
|
Contract Research Organization (CRO): |
Nordic Bioscience A/S Herlev Hovedgade 207 2730 Herlev, Denmark Tel: +45 4452 5252. Fax: +45 4452 5251 |
Disclosure Statement
This document contains information that is confidential and proprietary to Radius Health, Incorporated (RADIUS). This information is being provided to you solely for the purpose of evaluation and/or conducting a clinical trial for RADIUS. You may disclose the contents of this document only to study personnel under your supervision and/or to your institutional review board(s) or ethics committee(s) who need to know the contents for this purpose and who have been advised on the confidential nature of the document.
PROTOCOL SYNOPSIS
Title: An Extension Study to Evaluate 24 Months of Standard-of-Care Osteoporosis Management Following Completion of 18 Months of BA058 or Placebo Treatment in Protocol BA058-05-003
Protocol Number: BA058-05-005
Test Drug: BA058 Injection
Study Objectives:
The primary objective of this study is to collect clinical information regarding six months of treatment with alendronate, in subjects who have previously received 18 months of blinded treatment with BA058 Injection or Placebo in Study BA058-05-003. Safety data will be obtained via clinical, laboratory and radiologic assessments. Following the initial six months of treatment in the study, subjects will then enter the long-term observational phase of the study during which subjects will continue to receive alendronate treatment for an additional 18 months (for a total of 24 months).
The specific objectives of this study are to:
· Provide additional information on safety in study subjects receiving six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide information on the vertebral fracture rate in subjects receiving six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide additional information on non-vertebral fractures and BMD change associated with six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide additional information on BMD change and osteoporosis status associated with 24 months of treatment with alendronate after 18 months of treatment with BA058 Injection 80 µg/Placebo.
The analysis performed at six months of this Extension Study will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. The analyses performed on data in this study will be descriptive in nature. Full details of the statistical procedures to be used will be provided in the Statistical Analysis Plan.
Study Population:
Subjects with postmenopausal osteoporosis who completed the End-of-Treatment Visit (Visit 9) for Study BA058-05-003 and were previously randomized to either blinded BA058 Injection 80 µg or blinded Placebo are eligible for inclusion into this Extension Study provided that they fulfill the Inclusion/Exclusion criteria described below.
Inclusion/Exclusion Criteria
Otherwise healthy ambulatory postmenopausal women who participated in, and who completed 18 months of treatment with either blinded BA058 Injection 80 µg or blinded Placebo in Study BA058-05-003, are scheduled to complete or have completed the End-of-Treatment visit (Visit 9 in Study BA058-05-003), and who have provided a new written informed consent for the Extension Study, are eligible for enrollment into this study. Participants must be no more than 40 days from Visit 9 in Study BA058-05-003 to be eligible for this study. The physical examinations and clinical laboratory measurements from the End-of-Treatment visit from Protocol BA058-05-003 (Visit 9) of the BA058-
05-003 study will provide baseline data for this Extension Study. In addition, the subjects must, in the opinion of the Investigator, be appropriate candidates for further osteoporosis management.
Subjects will not be enrolled if they experienced a treatment-related SAE as assessed by the Investigator, or if they were withdrawn from Study BA058-05-003 for any reason. Subjects who are not candidates for alendronate treatment will receive standard-of-care management determined to be appropriate by the Investigator. Specific inclusion and exclusion criteria are described in Section 4.1 and Section 4.2, respectively.
Study Design and Methodology:
Number of Subjects
All subjects who were randomized to the BA058 Injection 80 µg/Placebo arms in Study BA058-05-003, and who completed 18 months of treatment will be offered the opportunity to participate in this study. There will, therefore, be a potential maximum of 1,600 subjects eligible to be enrolled in this study.
Design
This study will be an open-label extension of Study BA058-05-003. The purpose of the study is to provide longer term safety data, fracture data and BMD data after six months of treatment with alendronate, in otherwise healthy ambulatory postmenopausal women with severe osteoporosis who have previously received 18 months of blinded treatment with BA058 Injection or Placebo. The analysis performed at six months will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. In addition, this study will examine changes in osteoporosis status after 24 months of treatment with alendronate in otherwise healthy ambulatory women with severe osteoporosis who have previously received 18 months of blinded treatment with BA058 Injection/Placebo. The analysis performed at 24 months will be descriptive in nature.
Subjects randomized to BA058 Injection 80 µg/Placebo in Study BA058-05-003 and who are candidates for ongoing osteoporosis care, will receive six months of treatment with oral alendronate at a total dose of 70 mg once per week for 24 months. All subjects will undergo protocol specified procedures (Section 7.0, Appendix 14.1 and 14.2) including BMD and fracture assessment. The study design is presented in Figure 1, below.
Figure 1: Protocol BA058-05-005 Study Design
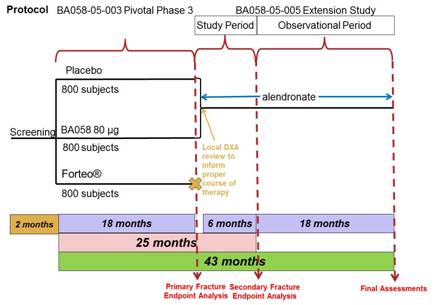
In this study, the Follow-up Visit from the 18 month study (Visit 10 from Study BA058-05-003) will serve as the Day 1 Visit (Visit 1) for this six month Extension Study (Study BA058-05-005).
Following the initial six months of treatment, subjects will enter the long-term observational phase of this study during which the subjects will continue to receive alendronate treatment for an additional 18 months. During the long-term follow-up of this study, subjects will continue to undergo study related procedures as outlined in Section 14.1 and Section 14.2.
All subjects will continue to take calcium and vitamin D supplementation throughout the Extension Study.
Study Visits
At the End-of-Treatment Visit (Visit 9) for Study BA058-05-003, the possibility of participating in the Extension Study will be discussed with subjects randomized to BA058 80 µg/Placebo. This Extension Study will be comprised of standard-of-care osteoporosis management, including 24 months of treatment with alendronate. In the month between Visit 9 and Visit 10 (between months 18 and 19 of Study BA058-05-003), the Investigator will consider the results of the assessments performed at Visit 9, including a local review of BMD, and determine if alendronate is appropriate for the subject, as part of this Extension Study.
At the Follow-up (Visit 10 for Protocol BA058-05-003, Day 1 for Protocol BA058-05-005) subjects; who were randomized to BA058 Injection 80 µg/Placebo, who fulfill the inclusion/exclusion criteria (Section 4.1 and Section 4.2), and who have agreed to participate in the Extension Study; will sign the Informed Consent Form and be enrolled in the study.
Alendronate is the recommended osteoporosis treatment for this Extension Study. Subjects who have been determined by the Investigator to be candidates for alendronate therapy will receive open-label oral alendronate treatment at a total dose of 70 mg once per week for 24 months. Subjects will be instructed to take their first dose of alendronate for Study BA058-05-005 in the morning, on the day following their Day 1 visit. Following the initial six months of treatment in this study, subjects will
enter the long-term observational phase of this study, during which subjects will continue to receive alendronate treatment for an additional 18 months.
All subjects will have clinic visits for study related procedures at Day 1, Month 3, Month 6, Month 12, Month 18 and Month 24. For the purpose of this study one month is equal to 30 days.
Statistical Considerations:
Exploratory statistical analyses will assess longer term safety, fracture incidence (including vertebral and non-vertebral fracture), and BMD change following treatment with alendronate for six months after the completion of a subject’s participation of 18 months in study BA058-05-003.
The analyses performed at six months will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. At this time-point, subjects will be analyzed based on the randomization assignment in the BA058-05-003 study. Other analyses performed on the data in this study will be descriptive in nature.
Fractures and BMD Analyses
All specified endpoints will be summarized by treatment group and study period using standard descriptive statistics (n, mean, SD, median, minimum, maximum or n and %, as appropriate). The fracture incidence; either clinically or radiologically determined, based on clinical events or protocol-directed vertebral x-rays at Month 6 of this Extension Study; will be tabulated. In addition, BMD results from the six months of treatment with alendronate will also be tabulated based on the treatment arm they were randomized to in the BA058-05-003 study, with additional tabular categories for results from the entire contiguous 24 months from baseline of study BA058-05-003 through the end of Study BA058-05-005, as well as the results during the 18 months of study BA058-05-003, for subjects who eventually enter study BA058-05-005. These descriptive analyses will be conducted on all subjects with baseline and post-baseline data.
Safety Analysis
Data will be summarized and tabulated based on the enrolled population for this Extension Study. All subjects enrolled in the Extension Study will be included in the safety analysis that will be performed on the following parameters:
· Incidence and severity of AEs.
· Pathological changes in hematology, chemistry and urinalysis data based on normal ranges supplied by the clinical laboratory, if applicable.
Safety assessments for changes in physical examination, vital signs, ECG, and laboratory tests will be descriptively summarized by treatment and study periods. Concomitant medication classes will be categorized using World Health Organization (WHO) drug dictionary and summarized by number and percent of subjects using each class by treatment group. All treatment emergent adverse events (TEAEs) will be coded for system organ class (SOC) and preferred term (PT) using MedDRA and the number (%) of subjects experiencing each AE (SOC/PT) will be summarized by treatment, relationship to treatment, and severity. All serious adverse events (SAE) will be listed and the number (%) of subjects with an SAE presented by treatment group.
Procedures and Assessments
Fractures and BMD
The End-of-Treatment (Visit 9) evaluations for vertebral fracture assessment, non-vertebral fracture assessment and BMD from Study BA058-05-003 will serve as the baseline evaluations in this study. The Day 1 assessment will be concurrent with the Follow-up Visit (Visit 10) for Study BA058-05-003. Subjects will return to the clinic for assessment of BMD at spine, hip and wrist (for those
subjects who had wrist DXAs performed in Study BA058-05-003) at Month 6 and at Month 24. Clinical and radiographic assessments for fractures will be performed at Month 6 and Month 24, and bone marker assessments of anabolism (PINP, bone-specific alkaline phosphatase and osteocalcin) and resorption (CTX) will be performed at Day 1 and Month 6.
Safety
Safety evaluations performed will include physical examinations, vital signs, 12-lead ECGs, clinical laboratory tests, and monitoring and recording of adverse events.
Complete details of the study assessments are provided in Section 7.0, in the Schedule of Visits and Procedures (Appendix 14.1) and in the Suggested Schedule of Events and Procedures by Study Visit (Appendix 14.2).
Treatments Administered
Alendronate sodium (Fosamax®, Merck & Co., Inc., or other approved generic manufacturer) 70 mg tablets for oral administration contain 91.35 mg of alendronate monosodium salt trihydrate which is the molar equivalent of 70 mg free acid and excipients. Alendronate should be stored in a well-closed container at room temperature, 15-30ºC. The alendronate may be generic substitutable approved versions which contain different inactive ingredients, but the amount of active free alendronate must be equivalent to 70 mg. Alendronate for Europe, Hong Kong and the US will be sourced centrally; alendronate for South America will be sourced locally by the medical center and reimbursed by the Sponsor.
Calcium (500—1000 mg) and vitamin D (400—800 IU) supplements will be sourced locally by the medical center and provided to the subjects at the expense of the Sponsor. Subjects will continue to take calcium and vitamin D as they did in Study BA058-05-003.
Duration of Subject Participation:
Participation in the initial phase of this study will be approximately six months from enrollment to completion of the six month study evaluations. Participation for both the initial and observational phases of the study will be approximately 24 months. In combination with Study BA058-05-003, subjects will participate in this clinical postmenopausal osteoporosis program for 43 to 44 months. The first visit of Study BA058-05-005 will be concurrent with Visit 10 of Study BA058-05-003.
TABLE OF CONTENTS
|
PROTOCOL SYNOPSIS |
9 | |||
|
|
| |||
|
Table of Contents |
14 | |||
|
|
| |||
|
List of Abbreviations |
17 | |||
|
|
| |||
|
1.0 |
introduction |
19 | ||
|
|
1.1 |
Background Information |
19 | |
|
|
1.2 |
Drug Under Study |
20 | |
|
|
|
1.2.1 |
Efficacy of Alendronate |
20 |
|
|
|
1.2.2 |
Safety of Alendronate Sodium |
20 |
|
|
1.3 |
Study Rationale and Selection of Doses |
21 | |
|
|
|
1.3.1 |
Study Rationale |
21 |
|
|
|
1.3.2 |
Study Design |
22 |
|
|
|
1.3.3 |
Study Population |
22 |
|
|
|
1.3.4 |
Selection of Endpoints |
22 |
|
|
|
1.3.5 |
Selection of Dose |
22 |
|
|
|
| ||
|
2.0 |
STUDY OBJECTIVES |
23 | ||
|
|
|
| ||
|
3.0 |
INVESTIGATIONAL PLAN |
23 | ||
|
|
3.1 |
Overall Design and Study Plan |
23 | |
|
|
|
3.1.1 |
Treatment Period |
24 |
|
|
|
| ||
|
4.0 |
SELECTION OF STUDY POPULATION |
25 | ||
|
|
4.1 |
Number of Subjects |
25 | |
|
|
4.2 |
Inclusion Criteria |
25 | |
|
|
4.3 |
Exclusion Criteria |
25 | |
|
|
4.4 |
Withdrawal of Subjects from the Study |
26 | |
|
|
4.5 |
Temporary Suspension of Treatment |
26 | |
|
|
4.6 |
Replacement of Subjects |
26 | |
|
|
|
| ||
|
5.0 |
STUDY TREATMENTS |
27 | ||
|
|
5.1 |
Study Medications |
27 | |
|
|
|
5.1.1 |
Alendronate |
27 |
|
|
|
5.1.1.1 |
Restrictions on Alendronate Use |
27 |
|
|
|
5.1.2 |
Calcium and Vitamin D Supplements |
27 |
|
|
5.2 |
Packaging, Labeling and Storage |
27 | |
|
|
|
5.2.1 |
Storage |
27 |
|
|
5.3 |
Treatment Assignment |
27 | |||
|
|
5.4 |
Study Medication Administration |
28 | |||
|
|
|
5.4.1 |
Alendronate Administration |
28 | ||
|
|
5.5 |
Treatment Compliance |
28 | |||
|
|
5.6 |
Unblinding of Study Medication |
28 | |||
|
|
|
| ||||
|
6.0 |
CONCOMITANT MEDICATIONS |
28 | ||||
|
|
6.1 |
Concomitant Medications |
28 | |||
|
|
6.2 |
Prohibited Medications |
29 | |||
|
|
|
| ||||
|
|
7.0 |
STUDY ASSESSMENTS |
29 | |||
|
|
7.1 |
Clinical Procedures/Assessments |
29 | |||
|
|
|
7.1.1 |
Informed Consent |
29 | ||
|
|
|
7.1.2 |
Recent Health Status |
30 | ||
|
|
|
7.1.3 |
Vital Signs |
30 | ||
|
|
|
7.1.4 |
Height and Weight |
30 | ||
|
|
|
7.1.5 |
Orthostatic Blood Pressure and Heart Rate |
30 | ||
|
|
|
7.1.6 |
Electrocardiogram |
30 | ||
|
|
|
7.1.7 |
Clinical Laboratory Evaluations |
30 | ||
|
|
|
7.1.8 |
Clinical Chemistry and Urinalysis (Dipstick) |
31 | ||
|
|
|
7.1.9 |
Hematology |
31 | ||
|
|
|
7.1.10 |
Coagulation |
32 | ||
|
|
|
7.1.11 |
24-Hour Urine Collection |
32 | ||
|
|
|
7.1.12 |
Bone Mineral Density |
32 | ||
|
|
|
7.1.13 |
Serum Markers of Bone Metabolism |
32 | ||
|
|
|
7.1.14 |
Clinical and Radiologic Evaluation of Fractures |
33 | ||
|
|
|
7.1.15 |
BA058 Antibody Assessments |
33 | ||
|
|
|
7.1.16 |
Subject Diaries |
33 | ||
|
|
|
7.1.17 |
Activity and Diet |
33 | ||
|
|
|
| ||||
|
8.0 |
ADVERSE EVENTS AND SAFETY EVALUATION |
33 | ||||
|
|
8.1 |
Definitions, Documentation, and Reporting |
34 | |||
|
|
|
8.1.1 |
Adverse Event Definition |
34 | ||
|
|
|
8.1.2 |
Serious Adverse Event Definition |
34 | ||
|
|
8.2 |
Monitoring of Adverse Events and Period of Observation |
35 | |||
|
|
8.3 |
Procedures for Recording and Reporting AEs and SAEs |
35 | |||
|
|
8.4 |
Rules for Suspension of the Study |
36 | |||
|
|
|
| ||||
|
9.0 |
Statistical Procedures |
37 | ||||
|
|
9.1 |
Sample Size |
37 | |||
|
|
9.2 |
Randomization, Stratification and Blinding |
37 | |||
|
|
9.3 |
Populations for Analysis |
37 | |||
|
|
|
9.3.1 |
Safety Population |
37 | ||
|
|
9.4 |
Procedures for Handling Missing, Unused, and Spurious Data |
37 | |||
|
|
9.5 |
Statistical Methods |
38 | |
|
|
|
9.5.1 |
Statistical Considerations |
38 |
|
|
|
9.5.2 |
Baseline Comparisons |
38 |
|
|
|
9.5.3 |
Fractures and BMD Analysis |
38 |
|
|
|
9.5.4 |
Safety Analysis |
38 |
|
|
|
9.5.5 |
Procedures for Reporting Deviations to Original Statistical Analysis Plan |
39 |
|
|
9.6 |
Data Oversight |
39 | |
|
|
|
9.6.1 |
Central Review of Radiographs and DXA Scans |
39 |
|
|
|
| ||
|
10.0 |
ADMINISTRATIVE REQUIREMENTS |
39 | ||
|
|
10.1 |
Good Clinical Practice |
39 | |
|
|
10.2 |
Ethical Considerations |
39 | |
|
|
10.3 |
Subject Information and Informed Consent |
40 | |
|
|
10.4 |
Protocol Compliance |
40 | |
|
|
10.5 |
Case Report Form Completion |
40 | |
|
|
10.6 |
Source Documents |
41 | |
|
|
10.7 |
Study Monitoring |
41 | |
|
|
10.8 |
On-Site Audits |
41 | |
|
|
10.9 |
Drug Accountability |
41 | |
|
|
10.10 |
Record Retention |
42 | |
|
|
10.11 |
Study Termination |
42 | |
|
|
10.12 |
Liability and Insurance |
42 | |
|
|
|
| ||
|
11.0 |
USE OF INFORMATION AND PUBLICATION OF STUDY FINDINGS |
43 | ||
|
|
11.1 |
Use of Information |
43 | |
|
|
11.2 |
Publication |
43 | |
|
|
|
| ||
|
12.0 |
INVESTIGATOR AGREEMENT |
44 | ||
|
|
|
| ||
|
13.0 |
References |
45 | ||
|
|
|
| ||
|
14.0 |
APPENDICES |
47 | ||
|
|
14.1 |
Schedule of Visits and Procedures |
48 | |
|
|
14.2 |
Suggested Schedule of Events and Procedures by Study Visit |
48 | |
|
|
14.3 |
Eastern Cooperative Oncology Group (ECOG) Common Toxicity Criteria |
57 | |
LIST OF ABBREVIATIONS
|
Abbreviation |
|
Term |
|
°C |
|
Degree Celsius |
|
°F |
|
Degree Fahrenheit |
|
µg |
|
Microgram |
|
µmol |
|
Micromole |
|
AE |
|
Adverse event |
|
ALT |
|
Alanine aminotransferase |
|
AST |
|
Aspartate aminotransferase |
|
BMD |
|
Bone mineral density |
|
BMI |
|
Body mass index |
|
bpm |
|
Beats per minute |
|
BSAP |
|
Bone-specific alkaline phosphatase |
|
BUN |
|
Blood urea nitrogen |
|
cm |
|
Centimeter |
|
CPK |
|
Creatine phosphokinase |
|
CRF |
|
Case report form |
|
CRO |
|
Contract research organization |
|
CTX |
|
C-telopeptides of type 1 collagen crosslinks (serum) |
|
DXA |
|
Dual energy x-ray absorptiometry |
|
ECG |
|
Electrocardiogram |
|
eCRF |
|
Electronic case report form |
|
FDA |
|
Food and Drug Administration |
|
g |
|
Gram |
|
GCP |
|
Good clinical practice |
|
GGT |
|
Gamma-glutamyltranspeptidase |
|
GLP |
|
Good laboratory practice |
|
GMP |
|
Good manufacturing practice |
|
ICH |
|
International Conference on Harmonization |
|
IEC |
|
Independent ethics committee |
|
IRB |
|
Institutional review board |
|
ITT |
|
Intent-to-treat |
|
IU |
|
International unit |
|
IV |
|
Intravenous |
|
IVRS |
|
Interactive voice response system |
|
kg |
|
Kilogram |
|
L |
|
Liter |
|
LDH |
|
Lactate dehydrogenase |
|
MCH |
|
Mean corpuscular hemoglobin |
|
MCHC |
|
Mean corpuscular hemoglobin concentration |
|
Abbreviation |
|
Term |
|
MCV |
|
Mean corpuscular volume |
|
MedDRA |
|
Medical dictionary for regulatory activities |
|
µL |
|
Microliter |
|
mg |
|
Milligram |
|
mL |
|
Milliliter |
|
mmHg |
|
Millimeter of mercury |
|
msec |
|
Millisecond |
|
NPO |
|
Nothing by mouth |
|
ng |
|
Nanogram |
|
ONJ |
|
Osteonecrosis of the jaw |
|
PA |
|
Posterior-anterior |
|
PD |
|
Pharmacodynamic |
|
pg |
|
Picogram |
|
PINP |
|
N-terminal propeptide of type I procollagen |
|
PK |
|
Pharmacokinetic |
|
PT |
|
Prothrombin time |
|
PTH |
|
Parathyroid hormone |
|
PTHrP |
|
Parathyroid hormone related peptide |
|
PTT |
|
Partial thromboplastin time |
|
PUBs |
|
Upper gastrointestinal perforations, ulcers and bleeds |
|
QT |
|
Total depolarization and repolarization time |
|
QTc |
|
Total depolarization and repolarization time corrected with heart rate |
|
RBC |
|
Red blood cell |
|
SAE |
|
Serious adverse event |
|
SC |
|
Subcutaneous |
|
SD |
|
Standard deviation |
|
SERMs |
|
Selective estrogen receptor modulators |
|
SOC |
|
System organ class |
|
SOP |
|
Standard operating procedure |
|
TEAEs |
|
Treatment emergent adverse events |
|
ULN |
|
Upper Limit of Normal |
|
WBC |
|
White blood cells |
|
WHO |
|
World Health Organization |
1.0 INTRODUCTION
1.1 Background Information
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue which leads to enhanced fragility and increased risk of fractures (Rizzoli, 2001). It is estimated that over 200 million people worldwide have osteoporosis (Reginster, 2006) and osteoporosis causes more than 8.9 million fractures worldwide, of which more than 4.5 million occur in the Americas and Europe (WHO Scientific Group, 2007). The vast majority of osteoporotic fractures occur in elderly women and incidence increases markedly with age. Most fractures occur at the spine, wrist and hip. Of these, hip fractures carry the highest morbidity and mortality. In 1990, the total number of hip fractures in men and women was estimated to be 1.26 million worldwide, and it is estimated that this number will increase to 3.6 million by 2025 and to 4.5 million by 2050 (Gullberg, 1997).
Subject enrolled in this Extension Study will have completed 18 months of treatment with BA058 Injection 80 µg/Placebo. BA058 is a synthetic 34 amino acid analog of parathyroid hormone related peptide(PTHrP), with molecular modifications of specific amino acids, and is under clinical development for the prevention of fractures in postmenopausal women with severe osteoporosis who are at a risk for fracture. BA058 shows particular potential for reversing bone loss at both the spine and the hip, the site of the most debilitating osteoporotic fractures in elderly women. BA058 is a synthetic analog of PTHrP (1-34) designed to give a greater anabolic effect than human parathyroid hormone (hPTH). Initial in vitro and in vivo studies identified BA058 as displaying bone anabolic properties without a significant hypercalcemic effect. In humans, BA058 has different pharmacokinetics (PK) and pharmacodynamics (PD) properties than hPTH(1-34) and has been shown in a Phase 2 study (BA058-05-002) to have similar or greater efficacy in restoring bone mineral density (BMD) in individuals with osteoporosis than hPTH(1-34). Overall, BA058 has been well tolerated in previous studies.
This is an open-label extension of Study BA058-05-003. Enrollment requires previous participation in, and successful completion of, 18 months of treatment with BA058 Injection 80 µg/Placebo in Study BA058-05-003. The purpose of this extension is to accumulate longer-term safety, fracture, and BMD data in subjects who receive six months of standard-of-care osteoporosis treatment, including treatment with alendronate, following 18 months of treatment with blinded BA058/Placebo treatment. Following the initial six months of treatment in this study, subjects will then enter the long-term observational phase of this study during which the subjects will continue to receive alendronate treatment for an additional 18 months. The analyses performed at six months will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. Other analyses performed on the data in this study will be descriptive in nature. Full details of the statistical procedures to be used will be provided in the Statistical Analysis Plan. Alendronate, a bisphosphonate, is approved and marketed world-wide for the treatment and prevention of osteoporosis in postmenopausal women.
1.2 Drug Under Study
1.2.1 Efficacy of Alendronate
Alendronate is a bisphosphonate that acts as a specific inhibitor of osteoclast-mediated bone resorption. Bisphosphonates are synthetic analogs of pyrophosphate that bind to the hydroxyapatite found in bone. At the cellular level, alendronate shows preferential localization to sites of bone resorption, specifically under osteoclasts. The osteoclasts adhere normally to the bone surface but lack the ruffled border that is indicative of active resorption. Alendronate does not interfere with osteoclast recruitment or attachment, but it does inhibit osteoclast activity. (Fosamax Package Insert)
Bisphosphonates including alendronate are widely used to treat osteoporosis. In animal models, minipigs treated with alendronate exhibited a direct correlation between cancellous bone volume and bone strength (Lefage 1995). In primates, treatment with alendronate increased the strength of cancellous bone between 44 and 100% (the effect was dose dependent) when compared to vehicle, and also increased bone mass (Balena 1993). In dogs, this increase in bone mass occurred without causing abnormalities in bone modeling of bone structure (Balena, 1996).
In postmenopausal women, alendronate has been demonstrated to increase bone mineral density, decrease bone turnover and reduce the risk of fracture among women with osteoporosis (Tucci, 1996; Devogelaer, 1996; Liberman, 1995). The therapeutic effects on bone density, remodeling and fracture prevention persist following daily treatment at an oral dose of 10 mg for up to 10 years (Bone, 2004). Studies have demonstrated that sequential treatment of osteoporosis with one year of treatment with PTH followed by one year of treatment with alendronate resulted in an increase in vertebral bone density that was considerably greater than previously reported for alendronate alone (Rittmaster, 2000). In subjects receiving PTH(1-84) followed by alendronate, there were significant increases in BMD, in particular trabecular spine, when compared to PTH(1-84) followed by placebo (31% vs. 14%, p<0.001) (Black, 2005).
1.2.2 Safety of Alendronate Sodium
According to the US package insert for Fosamax® (alendronate sodium), in studies of up to five years duration, adverse experiences usually were mild and generally did not require discontinuation of therapy. In a three-year, placebo-controlled, double blind study in which 196 subjects were treated with 10 mg/day, discontinuation due to any adverse experience occurred in 4.1% of subjects treated with alendronate, and 6% of 397 subjects treated with placebo. The most frequently reported adverse event (occurring in >2% of subjects treated with alendronate) in this study were abdominal pain, musculoskeletal pain, nausea, dyspepsia, constipation, diarrhea, flatulence, headache and acid regurgitation.
Alendronate may cause local irritation of the upper gastrointestinal mucosa. Esophageal adverse experiences, such as esophagitis, esophageal ulcers and esophageal erosions occasionally with bleeding and rarely followed by esophageal stricture or perforation have been reported. Osteonecrosis of the jaw (ONJ), which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing, has been reported in subjects taking alendronate. For subjects requiring dental procedures, discontinuation of alendronate therapy may reduce the risk for ONJ.
Atypical, low-energy, or low trauma fractures of the femoral shaft have been reported in bisphosphonate-treated patients. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Causality has not been established as these fractures also occur in osteoporotic patients who have not been treated with bisphosphonates.
Atypical femur fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture.
Any patient with a history of bisphosphonate exposure who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patients presenting with an atypical fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of bisphosphonate therapy should be considered, pending a risk/benefit assessment, on an individual basis.
According to the Summary of Product Characteristics for alendronate from the EMA, the following adverse experiences have been reported in alendronate treated subject during clinical trials and/or post-marketing use:
Common: Headache, abdominal pain, dyspepsia, constipation, diarrhea, flatulence, esophageal ulcer, dysphagia, abdominal distension, acid regurgitation and musculoskeletal pain.
Uncommon: Nausea, vomiting, gastritis, esophagitis, esophageal erosions, melena, rash, pruritus and erythema.
Rare: Hypersensitivity reactions including urticarial and angioedema, symptomatic hypocalcemia (often in association with predisposing conditions), uveitis, scleritis, episcleritis, esophageal stricture, oropharyngeal ulceration, upper gastrointestinal perforations, ulcers and bleeds (PUBs), rash with photosensitivity, osteonecrosis of the jaw, atypical subtrochanteric and diaphyseal femoral fractures and transient symptoms as in an acute-phase response (myalgia, malaise and rarely, fever), typically associated with initiation of treatment.
1.3 Study Rationale and Selection of Doses
1.3.1 Study Rationale
The purpose of the study is to provide longer term safety data, fracture data and BMD data after six months of treatment with alendronate, in otherwise healthy ambulatory postmenopausal women with severe osteoporosis who have previously received 18 months of blinded treatment with BA058 Injection or Placebo. Following the initial six months of treatment in this study, subjects will enter the long-term observational phase of the study during which the subjects will continue to receive alendronate for an additional 18 months.
1.3.2 Study Design
Subjects randomized to BA058 Injection 80 µg/Placebo, who have completed 18 months of treatment in Protocol BA058-05-003 and, who meet the Inclusion/Exclusion criteria (Sections 4.2 and 4.3) are eligible to participate in this study. Subjects originally randomized to BA058 Injection 80 µg/Placebo in Study BA058-05-003 and who are candidates for ongoing osteoporosis care, will receive 24 months of daily open-label alendronate treatment at a total dose of 70 mg/week.
1.3.3 Study Population
The study population in this protocol is comprised of otherwise healthy ambulatory postmenopausal women who:
1. have participated in Study BA058-05-003,
2. were randomized to either BA058 Injection 80 µg/Placebo,
3. have completed the End-of-Treatment Visit (Visit 9 in Study BA058-05-003), and
4. have provided a new written informed consent for this protocol.
Subjects will not be enrolled if they experienced treatment-related SAE or were withdrawn from Study BA058-05-003 for any reason.
1.3.4 Selection of Endpoints
The fracture incidence; either clinically or radiologically determined, based on clinical events or protocol-directed vertebral x-rays at Month 6 of this Extension Study; will be tabulated. In addition, BMD results from the six months of treatment with alendronate will also be tabulated, with additional tabular categories for results from the entire contiguous 24 months from baseline of study BA058-05-003 through the end of Study BA058-05-005, as well as the results during the 18 months of study BA058-05-003, for subjects who eventually enter study BA058-05-005. Bone formation (PINP, osteocalcin, BSAP) and resorption (CTX) markers will also be assessed. Clinical incidence of any fracture and radiologic incidence of vertebral fracture will also be evaluated at Month 24. The End-of-Treatment (Visit 9) evaluations for BMD, vertebral fracture, and non-vertebral fracture assessments from BA058-05-003 will serve as the baseline evaluations in this study.
Subjects will be monitored for safety events and will have safety assessments performed at each study visit. At Month 6 and Month 24, BMD by DXA, as well as clinical and radiologic assessment of the spine for assessment of fractures will be performed. Bone formation and resorption markers will also be assessed at Day 1 and Month 6. Further details of these assessments are in Section 7.0, and in Appendix 14.1 and 14.2.
1.3.5 Selection of Dose
The dose of alendronate (70 mg per week, oral) selected for this study is based upon the recommended daily dose in the product’s prescribing information.
All enrolled subjects will also continue to receive calcium (500-1000 mg) and vitamin D (400-800 IU) supplementation.
2.0 STUDY OBJECTIVES
The primary objective of this study is to evaluate data obtained following six months of treatment with alendronate, in subjects who have previously received 18 months of blinded treatment with BA058 Injection 80 µg/Placebo. Safety will be evaluated with clinical, laboratory and radiologic assessment. The analysis at six months will be based on the treatment that subjects were randomized to in the BA058-05-003 study. Following the initial six months of treatment in this study, subjects will then enter the long-term observational phase of the study during which the subjects will continue to receive alendronate treatment for an additional 18 months.
The specific objectives of this study are to:
· Provide additional information on safety in study subjects receiving six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide information on the vertebral fracture rate in subjects receiving six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide additional information on non-vertebral fractures and BMD change associated with six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide additional information on BMD change and osteoporosis status associated with 24 months of treatment with alendronate after 18 months of BA058 Injection 80 µg/Placebo.
The analysis performed at six months will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. Additional analyses performed on the data in this study will be descriptive in nature. Full details of the statistical procedures to be used will be provided in the Statistical Analysis Plan.
3.0 INVESTIGATIONAL PLAN
3.1 Overall Design and Study Plan
This study is an open-label extension of Study BA058-05-003. Subjects and Investigators who participate in Study BA058-05-005 will remain blinded to prior treatment assignment as part of BA058-05-003. At the End-of-Treatment visit (Visit 9) for Study BA058-05-003, the possibility of participating in the Extension Study will be discussed with subjects randomized to BA058 80 µg/Placebo. The Extension Study will be comprised of an initial six months of treatment with alendronate. In the month between Visit 9 and Visit 10, the Investigator will review the results of the assessments performed at Visit 9, including a local interpretation of BMD, and determine if alendronate is appropriate for the subject. All subjects will continue to receive vitamin D and calcium supplementation as they did in Study BA058-05-003. The study design is presented in Figure 2, below.
Figure 2: Protocol BA058-05-005 Study Design
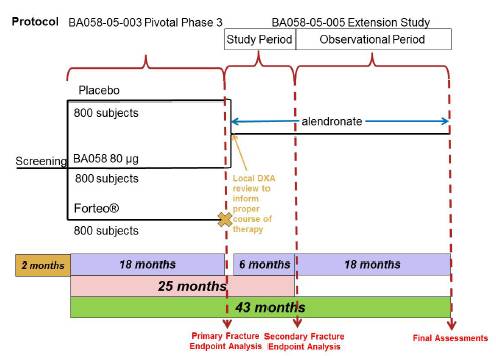
Participation for both the initial and observational phases of the will be approximately 24 months. There are a total of six clinic visits during the course of the study.
A brief summary of the study is provided below. For a summary of the study assessments to be performed, refer to Section 7.0 (Study Assessments) and to the Schedule of Visits and Procedures (Appendix 14.1). A more detailed description of the study procedures on a by-visit basis is provided in Appendix 14.2 (Suggested Schedule of Events and Procedures by Study Visit). A suggested order of procedures is also provided in this schedule.
3.1.1 Treatment Period
Subjects will enter into Study BA058-05-005 on Day 1, and Day 1 will also serve as Visit 10 (the Follow-up Visit) for Study BA058-05-003. The Informed Consent must be signed prior to undergoing any BA058-05-005 study related procedures, and may be signed at either Visit 9 or Visit 10 of Study BA058-05-003. Subjects who received BA058 Injection 80 µg/Placebo in Study BA058-05-003 will receive six months of open-label oral alendronate treatment as part of this study (BA058-05-005). Following the initial six months of treatment in this study, subjects will then enter the long-term observational phase of this study during which the subjects will continue to receive osteoporosis care for an additional 18 months.
If determined by the Investigator to be appropriate, treatment will be by oral administration of alendronate at a total dose of 70 mg once per week. Subjects will be given a weekly diary card to record missed doses of medication including calcium and vitamin D.
A total of six clinic visits are scheduled during the study (Day 1, Month 3, Month 6, Month 12, Month 18 and Month 24).
Subjects will be instructed to take their first dose of study drug for Study BA058-05-005 in the morning on the day following their Day 1 visit (Day 2 of this study). Study subjects will continue calcium and vitamin D supplementation during this study as was administered during BA058-05-003 (Section 6.1).
At Month 3, subjects will return to the clinic for medication resupply, subject diary review and questioning as to their use of concomitant medications and the occurrence of adverse events.
At the Month 6 visit ECG, and safety labs will be performed. Vertebral fractures will be determined clinically and via protocol directed x-ray evaluation; non-vertebral fractures will be determined clinically. In addition, subjects will undergo a DXA of the hip and spine (and wrist, if the subject was enrolled in the wrist DXA sub-study in Study BA058-05-003), and have samples drawn for bone markers and anti-BA058 antibodies. Procedures are to be performed as described in Section 7.0, Appendix 14.1 and Appendix 14.2.
At Months 12 and 18, subjects will return to the clinic for medication resupply, subject diary review and questioning as to their use of concomitant medications and occurrence of adverse events.
At Month 24, subjects will return to the clinic and will undergo clinical and radiologic fracture assessments and have DXA of the hip and spine (and wrist, if the subject was enrolled in the wrist DXA sub-study in Study BA058-05-003). Any adverse event or clinical laboratory abnormality recorded at the Month 6 Visit will be monitored until it has resolved, become chronic or stable.
4.0 SELECTION OF STUDY POPULATION
4.1 Number of Subjects
Subjects who completed 18 months of treatment with either BA058 Injection 80 µg/Placebo in Study BA058-05-003 will be given the opportunity to participate in the Extension Study at all participating centers. Based on randomization to the BA058 Injection 80 µg/Placebo arms in Study BA058-05-003, up to 1,600 subjects may be entered into this study.
The specific inclusion and exclusion criteria for enrolling subjects in this study are presented below in Sections 4.2 and 4.3, respectively. Exceptions to these criteria should occur infrequently and should be discussed in advance and approved by the Sponsor Medical Monitor.
4.2 Inclusion Criteria
Subjects must meet all of the following criteria to be eligible to participate in this study:
1. The subject was enrolled, randomized to BA058 Injection 80 µg/Placebo and completed 18-months of blinded treatment within Study BA058-05-003.
2. The subject is no more than 40 days from Visit 9 in Study BA058-05-003.
3. The subject has read, understood, and signed the written informed consent form for the Extension Study.
4.3 Exclusion Criteria
Subjects with any of the following characteristics are not eligible to participate in the study:
1. Subjects who were withdrawn from Study BA058-05-003 for any reason.
2. Subjects who experienced a treatment-related SAE during Study BA058-05-003.
4.4 Withdrawal of Subjects from the Study
Subjects will be informed that they have the right to withdraw from the study at any time for any reason without prejudice to their medical care.
Consistent with the prior protocol, BA058-05-003, the Investigator must withdraw subjects from the study prior at any time in the study for the following reasons:
· Treatment-related SAEs;
· Refusal of treatment;
· Refusal or inability to complete study procedures;
· Lost to follow-up.
The Investigator should exercise his/her best judgment and also has the right to withdraw subjects from the study during the study for any of the following reasons:
· ECOG Grade 3 or 4 adverse events [Refer to Appendix14.3];
· A complex of adverse events which, in the judgment of the Investigator justifies treatment cessation;
· Serious intercurrent illness;
· Non-compliance;
· Protocol violations;
· Administrative reasons.
If a subject is withdrawn or discontinued from the study, the reason for withdrawal is to be recorded in the source documents and on the case report form. All subjects withdrawn prior to completing the study should be encouraged to complete the Month 6 or Month 24 Visit (depending on the length of time on study) including any outstanding radiologic assessment or BMD assessment by DXA.
4.5 Temporary Suspension of Treatment
The Investigator has the right to suspend treatment with alendronate without withdrawal of the subject from the study. Reasons for temporary suspension of treatment may include a medical reason unrelated to an adverse event (e.g., a planned procedure), or important social or administrative events. The reason for the suspension of treatment is to be documented in the electronic case report form (eCRF) and in source documents.
When treatment with alendronate is restarted, the subject should resume treatment with the next scheduled dose (as if treatment had not been interrupted).
4.6 Replacement of Subjects
Subjects who have been enrolled into the study and subsequently withdraw or drop out of the study will not be replaced.
5.0 STUDY TREATMENTS
5.1 Study Medications
Alendronate will be sourced locally. Calcium and vitamin D will be provided by the study centers, similar to their provision in Study BA058-05-003.
5.1.1 Alendronate
Alendronate will be sourced centrally for Europe, Hong Kong and the US, and will be sourced locally for South America at the expense of the Sponsor.
Subjects will receive total weekly dose of oral alendronate at a dose of 70 mg once per week beginning on Day 2 for 24 months. Additional provisions for dosing of alendronate should be followed based on the prescribing information. Alendronate provided will be in the approved, marketed formulation. The alendronate may be generic substitutable approved versions which contain different inactive ingredients, but the amount of active free alendronate must be equivalent to 70 mg per week.
5.1.1.1 Restrictions on Alendronate Use
Subjects should not receive alendronate if they have the following conditions/limitations:
· Abnormalities of the esophagus and other factors which delay espophageal emptying such as stricture or achalasia.
· Inability to stand or sit upright for at least 30 minutes.
· Hypocalcemia.
· Known history of hypersensitivity to alendronate, alendronate excipients, or related compounds.
5.1.2 Calcium and Vitamin D Supplements
Calcium and vitamin D supplements will be sourced locally and provided by the sites at the expense of the Sponsor.
5.2 Packaging, Labeling and Storage
Centrally supplied alendronate will not be repackaged for the study, but will be over-labeled according to local regulatory requirements as necessary.
Calcium and vitamin D supplements will not be relabeled for the study.
5.2.1 Storage
Alendronate must be kept in a secure, limited-access storage area until dispensed for use to a study subject. Alendronate sodium should be stored in the container provided at room temperature, 15-30ºC (59-86ºF).
Calcium and vitamin D supplements may be stored at room temperature.
5.3 Treatment Assignment
All subjects who participate will continue to be identified by the same 7-digit subject number that was assigned upon enrollment into Study BA058-05-003 throughout the study and on the eCRF.
5.4 Study Medication Administration
5.4.1 Alendronate Administration
Alendronate must be taken with water only (not mineral water) at least 30 minutes before the first food, beverage or medicinal product (including antacids, calcium supplements and vitamins) of the day. Other beverages (including mineral water), food and some medicinal products are likely to reduce the absorption of alendronate.
The following instructions should be followed exactly in order to minimize the risk of esophageal irritation and related adverse reactions.
· Alendronate should only be swallowed after getting up for the day with a full glass of water (not less than 200 mL or 7 fl. oz.).
· Subjects should only swallow alendronate whole. Subjects should not crush or chew the tablet or allow the tablet to dissolve in their mouths because of a potential for oropharyngeal ulceration.
· Subjects should not lie down until after their first food of the day.
· Subjects should not lie down for at least 30 minutes after taking alendronate.
· Alendronate should not be taken at bedtime or before arising for the day.
At the Month 3, Month 6, Month 12 and Month 18 visits, the unused alendronate tablets are to be returned to the clinic for counting and the subject will be dispensed additional alendronate. At the Month 24 visit, all unused alendronate tablets are to be returned to the study site.
5.5 Treatment Compliance
The study site personnel will perform drug accountability at each clinic visit and review each subject diary (refer to Section 7.1.16). Accountability will be documented on the appropriate forms and subjects will be re-trained on administration as appropriate. All doses of study medication are to be self-administered.
If a subject does not administer or take all study medication including vitamin D or calcium, the reason for the missed dosing is to be recorded in source documents and on the eCRF.
Returned, unused alendronate will be accounted for by the study site and destroyed as appropriate.
5.6 Unblinding of Study Medication
Not applicable.
6.0 CONCOMITANT MEDICATIONS
6.1 Concomitant Medications
Vitamin D and calcium supplements are required to be administered daily from Day 1 (continuing from Protocol BA058-05-003) until the Month 6 Visit. Vitamin D and calcium supplements will be administered in the following doses: 400-800 IU/day (Vitamin D) and 500-100mg/day (calcium), or at a dose to be determined by the Investigator and agreed upon by the Sponsor Medical Monitor according to the subjects need. The doses and schedule of Vitamin D and calcium supplements, which are part of the study medication protocol, should
be adhered to and not be changed other than for medical necessity. The supplements should be taken in the evening with or without food or as otherwise instructed by the Investigator.
For any required concomitant medication, such as statins or antihypertensives, the subject must be on a stable dose at study entry and every effort should be made to maintain a stable dose during study participation.
The occasional use of over-the-counter medications at approved doses (e.g., ibuprofen or acetaminophen) for headache or minor discomfort is allowed. Occasional short term (<3 months) use of corticosteroids for seasonal allergies or asthma is also allowed. These are to be recorded on the appropriate case report form. Subjects should not take any other medications, including over-the-counter medications, herbal medications, or mega-doses of vitamins during the study without prior approval of the Investigator.
If it becomes necessary for a subject to take any other medication during the study, the specific medication(s) and indication(s) must be discussed with the Investigator. All concomitant medications taken during the course of the study must be recorded in the Subject’s medical record or source document and transcribed into the case report form.
6.2 Prohibited Medications
Subjects who require treatment during the course of the study with either an anticonvulsant (phenobarbital, phenytoin, carbamazepin or primidone) or chronic treatment with any form of heparin will be discontinued. Estrogens given as HRT are allowed at entry into the study but cannot be initiated during the study except for local low dose vaginal estrogen.
Drugs that may compromise renal function such as non-steroidal anti-inflammatory drugs should be used with caution.
7.0 STUDY ASSESSMENTS
Subjects randomized to BA058 Injection 80 µg/Placebo in Study BA058-05-003 will receive alendronate at a dose of 70 mg once per week for a total of 24 months.
The assessments performed at each study visit are displayed in the Schedule of Visits and Procedures in Appendix 14.1. Appendix 14.2 provides a more detailed schedule of the study procedures by study visit with a suggested order of procedure conduct. Exact procedures for centrifuging, storage, and shipping of laboratory samples will be detailed in a separate document. The actual time of each blood collection will be recorded on the appropriate source documents and in the eCRF.
Study-specific assessments are to be conducted only after the subject has provided written informed consent to participate in this study. The study assessments are described in more detail in Section 7.1 below.
7.1 Clinical Procedures/Assessments
7.1.1 Informed Consent
At the End-of-Treatment Visit (Visit 9) for Study BA058-05-003, the possibility of participating in the Extension Study will be discussed with the subjects randomized to BA058 80 µg/Placebo. The Informed Consent must be signed prior to undergoing any
BA058-05-005 study related procedures, and may be signed at either Visit 9 or Visit 10 of Study BA058-05-003.
7.1.2 Recent Health Status
The subject’s health status will be updated from their last visit in Study BA058-05-003, as necessary. Any changes in health status should be recorded as an adverse event, as appropriate.
The physical examination from the End-of-Treatment visit (Visit 9) of Study BA058-05-003 will be the baseline for this study (Day 1).
Interim or symptom-directed physical examinations may be performed at the discretion of the Investigator, if necessary, to evaluate adverse events or clinical laboratory abnormalities.
7.1.3 Vital Signs
Blood pressure, body temperature (ºC), pulse (bpm) and respiration rate (breaths per minute) are to be measured and recorded at each study visit (Day 1, Month 3 and Month 6, Month 12, Month 18 and Month 24). Only the Day 1 blood pressure assessments need be conducted as an orthostatic measurement (See Section 7.1.5).
7.1.4 Height and Weight
Height and weight are to be measured at each study visit (Day 1, Month 3, Month 12, Month 18 and Month 24). Height is to be measured in the standing position using a medical stadiometer.
7.1.5 Orthostatic Blood Pressure and Heart Rate
The Day 1 orthostatic blood pressure measurement for Study BA058-05-005 will serve as the Visit 10 orthostatic blood pressure for Study BA058-05-003. Blood pressure (mmHg; measured in the same arm at each visit) and pulse rate (bpm) will be measured after five minutes in the supine position. Immediately following this measurement, blood pressure will be measured again after three minutes in the standing position.
7.1.6 Electrocardiogram
A twelve-lead supine electrocardiograms (ECGs) will be performed and the following ECG parameters will be recorded: rhythm, heart rate, PR interval, QRS duration and QT/QTc.
The Day 1 ECG measurement for Study BA058-05-005 will serve as the Visit 10 ECG measurement for Study BA058-05-003. An ECG will also be obtained at Month 6.
7.1.7 Clinical Laboratory Evaluations
Clinical laboratory evaluations will be performed by a central laboratory. Prior to starting the study, the Sponsor (or its designee) will provide each Investigator with copies of the appropriate laboratory certifications and normal ranges for all laboratory parameters to be performed by that laboratory.
The blood and urinalysis samples are to be obtained under fasting conditions (NPO for 8 hours; water is acceptable) in the morning of each scheduled study visits on Day 1 and Month 6.
All clinically significant laboratory abnormities indicating an adverse event will be followed up by repeat testing and further investigated as necessary, according to the judgment of the Investigator.
7.1.8 Clinical Chemistry and Urinalysis (Dipstick)
Clinical chemistry and dipstick urinalysis will be performed on Day 1 and at Month 6. Urinalysis will be performed using samples freshly voided during the clinic visit. If there are positive findings noted on the dipstick, a urine microscopic examination will be performed. The following tests will be performed:
|
Serum Chemistry |
· |
Sodium |
|
|
· |
Potassium |
|
|
· |
Chloride |
|
|
· |
Inorganic phosphorus |
|
|
· |
Albumin |
|
|
· |
Total protein |
|
|
· |
Glucose |
|
|
· |
Blood urea nitrogen (BUN) |
|
|
· |
Creatinine |
|
|
· |
Uric acid |
|
|
· |
Aspartate aminotransferase (AST) |
|
|
· |
Alanine aminotransferase (ALT) |
|
|
· |
Gamma-glutamyltranspeptidase (GGT) |
|
|
· |
Creatine phosphokinase (CPK) |
|
|
· |
Alkaline phosphatase |
|
|
· |
Total bilirubin |
|
|
· |
Lactate dehydrogenase (LDH) |
|
|
· |
Cholesterol |
|
|
· |
Triglycerides |
|
|
· |
Total calcium |
|
Urinalysis |
· |
pH |
|
|
· |
Glucose |
|
|
· |
Protein |
|
|
· |
Ketones |
|
|
· |
Bilirubin |
|
|
· |
Blood |
|
|
· |
Urobilinogen |
|
|
· |
Specific gravity |
|
|
· |
Nitrite |
|
|
· |
Leukocytes |
7.1.9 Hematology
Hematology testing will be performed on Day 1 and at Month 6. The following tests will be performed:
|
Hematology: |
· |
Hemoglobin |
|
|
· |
Hematocrit |
|
|
· |
WBC count with differential in absolute counts |
|
|
· |
RBC count |
|
|
· |
Mean corpuscular volume (MCV) |
|
|
· |
Mean corpuscular hemoglobin concentration (MCHC) |
|
|
· |
Mean corpuscular hemoglobin (MCH) |
|
|
· |
Platelet count |
7.1.10 Coagulation
Coagulation testing will be performed on Day 1 and at Month 6. The following tests will be performed:
|
|
· |
Prothrombin time (PT) |
|
|
· |
Partial thromboplastin time (PTT) |
7.1.11 24-Hour Urine Collection
The 24-hour urine collection is to be begun the day before the Day 1 and Month 6 visits. If a sample was not able to be collected on the day prior to the Day 1 visit (i.e., if the subject had not yet signed the ICF for study participation), a 24-hour urine sample must be collected on the day prior to the Month 3 visit. Subjects are to be instructed to begin the urine collection by discarding the first morning void (~6 a.m.) the day prior to the scheduled clinic visit and to then collect their urine for 24 hours. A final void is to be collected at the end of the 24-hour period and the urine collection transported to the clinic by the subject. The 24-hour urinalysis will be used to measure urinary calcium and urinary creatinine.
7.1.12 Bone Mineral Density
All subjects will have bone mineral density measurements (BMD) taken via DXA at Month 6 and at Month 24. The End-of-Treatment (Visit 9) bone mineral density tests for Study BA058-05-003 will serve as the baseline BMD measurements for Study BA058-05-005.
DXAs will be performed on the hip (femoral neck) and spine (L1-4). The spinal DXA is to be taken in the postero-anterior (PA) projection with any subsequent spinal DXA to be taken in the same projection. Subjects who underwent wrist DXAs in Study BA058-05-003 will also have wrist DXAs performed at Month 6 and Month 24. The same side of the hip and wrist that were used in Study BA058-05-003 must be used for the DXA scan, and the same scanner should be used throughout the study, when possible.
7.1.13 Serum Markers of Bone Metabolism
Blood samples to measure bone markers will be taken on Day 1 and at Month 6. The results of the bone markers will be reported in the same subset of subjects reported on for Study BA058-05-003.
The following markers of bone formation will be measured:
· Serum N-terminal propeptide of type I procollagen (PINP);
· Serum bone-specific alkaline phosphatase (BSAP);
· Serum osteocalcin.
The following marker of bone resorption will be measured:
· Serum C-telopeptides of type 1 collagen crosslinks (CTX).
7.1.14 Clinical and Radiologic Evaluation of Fractures
Subjects will undergo protocol directed antero-posterior and lateral radiographs of the lumbar and thoracic spines at Month 6 and Month 24. The End-of-Treatment (Visit 9) clinical and radiological evaluation of fractures for Study BA058-05-003 will serve as the baseline assessments for Study BA058-05-005. Subjects will also be clinically evaluated for non-vertebral fractures (wrist, hip, rib, etc.) that occur de novo during the Treatment Period.
All radiographs will be viewed and assessed centrally by a blinded, independent assessor (radiologist) on the basis of existing baseline and study-acquired vertebral deformity. Fractures will be assessed according to the severity scale of Genant (1993). A second blinded radiologist will confirm the assessment of the first reviewer for all subject radiographs in which an incident fracture has been identified. In the case of any disagreement, a third consensus assessment will be made to adjudicate the incident fracture.
Fractures identified during the study will not be recorded as AEs unless the subject is hospitalized, the fracture is complicated, or the Investigator considers the fracture to be unrelated to the subject’s underlying osteoporosis. All fractures (vertebral and non-vertebral) will be identified and evaluated as part of the disease assessment and will be documented in the case report forms and source documents.
7.1.15 BA058 Antibody Assessments
The occurrence of anti-drug antibodies will be assessed at the completion of the initial six months of the study. Serum samples will be drawn at Month 6. Any subject who tests positive, or has previously tested positive for antibodies subjects will be retested at six month intervals under a separate Safety Surveillance protocol. Exact procedures for collection, preparation, storage, and shipping of these samples will be detailed in a separate document. The actual time and date of each blood collection will be recorded on the appropriate source document and in the eCRF.
7.1.16 Subject Diaries
A weekly diary will be completed by the subject beginning on the Day 1 visit and continuing until the last day of Month 24. This diary will capture missed doses of vitamin D, calcium and alendronate. The weekly diary will be reviewed at each study visit.
7.1.17 Activity and Diet
Subjects who qualify for enrollment in the study will have no restrictions placed on their usual level of activity or on their usual diet, unless directed by the treating physician for medically justified reasons.
8.0 ADVERSE EVENTS AND SAFETY EVALUATION
Timely, accurate, and complete reporting and analysis of safety information from clinical studies are crucial for the protection of subjects, Investigators and the Sponsor, and is mandated by Regulatory Agencies worldwide. All clinical trials sponsored by RADIUS will be conducted in accordance with Standard Operating Procedures (SOPs) that have been established to conform to regulatory requirements worldwide to ensure appropriate reporting of safety information.
8.1 Definitions, Documentation, and Reporting
8.1.1 Adverse Event Definition
An adverse event (AE) is any untoward medical occurrence in a subject administered a pharmaceutical product, which does not necessarily have a causal relationship with the treatment. An AE can be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of the study drug, whether or not it is considered to be study drug related. This includes any newly occurring event or previous condition that has increased in severity or frequency since the administration of study drug.
8.1.2 Serious Adverse Event Definition
A serious adverse event (SAE) is any adverse event, occurring at any dose and regardless of causality that:
· Results in death.
· Is life-threatening. Life-threatening means that the subject was at immediate risk of death from the reaction as it occurred, i.e., it does not include a reaction which hypothetically might have caused death had it occurred in a more severe form.
· Requires in-patient hospitalization or prolongation of existing hospitalization. Hospitalization admissions and/or surgical operations scheduled to occur during the study period, but planned prior to study entry are not considered AEs if the illness or disease existed before the subject was enrolled in the trial. Provided that the illness/disease did not deteriorate in an unexpected manner during the trial (e.g., surgery performed earlier than planned).
· Results in persistent or significant disability/incapacity. Disability is defined as a substantial disruption of a person’s ability to conduct normal life functions.
· Is a congenital anomaly/birth defect. This includes any anomaly detected at or after birth, or any anomaly that results in fetal loss.
· Is an important medical event. An important medical event is an event that may not result in death, be life-threatening, or require hospitalization, but may be considered an SAE when, based upon appropriate medical judgment, it may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed in the definitions for SAEs. Examples of such medical events include allergic bronchospasm requiring intensive treatment in an emergency room or at home, blood dyscrasias or convulsions that do not result in in-patient hospitalization, or the development of drug dependency or drug abuse.
Clarification should be made between the terms “serious” and “severe” since they are not synonymous. The term “severe” is often used to describe the intensity (synonym: severity) of a specific event (as in mild, moderate, or severe myocardial infarction); the event itself, however, may be of relatively minor medical significance (such as a severe headache). This is not the same as “serious,” which is based on subject/event outcome or action criteria described above and are usually associated with events that pose a threat to a subject’s life or functioning. A severe adverse event does not necessarily need to be considered serious. For example, persistent nausea of several hours duration may be considered severe nausea but not
an SAE. On the other hand, a stroke resulting in only a minor degree of disability may be considered mild, but would be defined as an SAE based on the above noted criteria. Seriousness (not severity) serves as a guide for defining regulatory reporting obligations.
8.2 Monitoring of Adverse Events and Period of Observation
All AEs will be monitored until they are resolved or have become chronic or stable. AEs and SAEs will be recorded on the case report forms starting from the time of subject entry from Day 1 of the study until the final study visit (Month 24). Any SAEs that occur at any time after completion of the study, which the Investigator considers to be related to study drug, must be reported to the Sponsor or its designee.
8.3 Procedures for Recording and Reporting AEs and SAEs
All adverse events spontaneously reported by the subject and/or in response to an open question from study personnel or revealed by observation, physical examination or other diagnostic procedures must be recorded in the source document and on the appropriate page of the case report form. Any clinically relevant deterioration in laboratory assessments or other clinical findings is considered an adverse event and must be recorded on the appropriate pages of the case report form. When possible, signs and symptoms indicating a common underlying pathology should be noted as one comprehensive event.
All SAEs that occur during the course of the study, as defined by the protocol, must be reported by the Investigator to the Study Safety Officer by completing and transmitting the SAE Form within one working day from the point in time when the Investigator becomes aware of the SAE. In addition, all SAEs including all deaths, which occur up to and including 30 days after administration of the last dose of study drug, must be reported to the Study Safety Officer within one working day. All SAEs and deaths must be reported whether or not considered causally related to the study drug. SAE forms will be provided to the study site. The information collected will include a minimum of the following: Subject number, a narrative description of the event, and an assessment by the Investigator as to the intensity of the event, and relatedness to study drug. Follow-up information on the SAE may be requested by the CRO, the Study Safety Officer or the Sponsor Medical Monitor. Contact information for reporting SAEs to the Study Safety Officer is provided on the SAE form.
It is the responsibility of the Investigator to promptly notify the Institutional Review Board (IRB)/Independent Ethics Committee (IEC) of all serious adverse drug reactions involving risk to human subjects in accordance with the requirements of the IRB/IEC. An unexpected event is one that is not reported in the Investigator’s Brochure.
Planned hospital admissions or surgical procedures for an illness or disease that existed before the subject was enrolled in the trial or before study drug was given are not to be considered AEs unless they occur at a time other than the planned date.
Fractures identified during the study are not to be recorded as AEs unless the subject is hospitalized, the fracture is complicated, or the Investigator considers the fracture to be unrelated to the subject’s underlying osteoporosis. All fractures will be identified and evaluated as part of the disease assessment and will be documented in the case report forms and source documents.
For both serious and non-serious adverse events, the Investigator must determine the intensity of the event and the relationship of the event to study drug administration. Intensity for each AE will be defined according to the following criteria:
|
Intensity |
|
Definition |
|
Mild |
|
Awareness of sign or symptom, but easily tolerated. |
|
Moderate |
|
Discomfort enough to cause interference with normal daily activities. |
|
Severe |
|
Inability to perform normal daily activities |
If the intensity of an adverse event changes within a day, the maximum intensity should be recorded. If the intensity changes over a longer period of time, the changes should be recorded as separate events (having separate onset and stop dates for each intensity).
Relationship to study drug administration will be determined by the Investigator according to the following criteria:
|
Relationship |
|
Definition |
|
None |
|
No relationship between the event and the administration of study drug. The event is related to other etiologies, such as concomitant medications or subject’s clinical state. |
|
Unlikely |
|
The current state of knowledge indicates that a relationship to study drug is unlikely or the temporal relationship is such that study drug would not have had any reasonable association with the observed event. |
|
Possible |
|
A reaction that follows a plausible temporal sequence from administration of the study drug and follows a known response pattern to the suspected study drug. The reaction might have been produced by the subject’s clinical state or other modes of therapy administered to the subject. |
|
Probable |
|
A reaction that follows a plausible temporal sequence from administration of the study drug and follows a known response pattern to the suspected study drug. The reaction cannot be reasonably explained by the known characteristics of the subject’s clinical state or other modes of therapy administered to the subject. |
For the purpose of safety analyses, all AEs that are classified with a relationship to study medication administration of possible or probable will be considered treatment-related events.
8.4 Rules for Suspension of the Study
As this is an extension study using standard-of-care management of osteoporosis, including products approved for treatment, it is not anticipated that the study will need to be suspended, and therefore, suspension rules are not assigned. In the event that the prior study (Study BA058-05-003) is suspended, the circumstances of the Study BA058-05-003 suspension will be considered to determine if this study, Study BA058-05-005, should be suspended as well.
9.0 STATISTICAL PROCEDURES
The primary objective of this study is to evaluate data obtained following six months of standard-of-care osteoporosis treatment, including treatment with alendronate, in subjects who have previously received 18 months of blinded treatment with BA058 Injection 80 µg/Placebo. Safety data will be obtained with clinical, laboratory and radiologic assessment. Following the initial six months of treatment in this study, subjects will then enter the long-term observational phase of this study during which the subjects will continue to receive alendronate treatment for an additional 18 months.
The specific objectives of this study are to:
· Provide additional information on safety in study subjects receiving six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide information on the vertebral fracture rate of subjects receiving six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide additional information on non-vertebral fractures and BMD change associated with six months of treatment with alendronate following 18 months of treatment with BA058 Injection 80 µg/Placebo.
· Provide additional information on BMD change and osteoporosis status associated with 24 months of treatment with alendronate after 18 months of treatment with BA058 Injection 80 µg/Placebo.
The analysis performed at six months will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. Additional analyses performed on data in this study will be descriptive in nature. Full details of the statistical procedures to be used will be provided in the Statistical Analysis Plan.
9.1 Sample Size
As this is an extension study, no formal sample size analysis was performed for this study. Study data will be tabulated and summarized.
9.2 Randomization, Stratification and Blinding
Osteoporosis treatment will be open label and no randomization is required.
9.3 Populations for Analysis
All analyses and data summaries will be presented for the Safety Population.
9.3.1 Safety Population
The Safety Population is comprised of all subjects who qualify on the basis of Study BA058-05-003 and provide informed consent to enroll in this Extension Study.
9.4 Procedures for Handling Missing, Unused, and Spurious Data
All available data will be included in the data listings and tabulations. Where appropriate, imputations of values for missing data will be performed using last observation carried
forward and specified in the Statistical Analysis Plan. All data recorded on the CRF will be included in the data listings that will accompany the clinical study report.
9.5 Statistical Methods
9.5.1 Statistical Considerations
Statistical analysis will focus on longer term safety, fracture incidence, including vertebral fracture and BMD change following six months of standard-of-care osteoporosis treatment in subjects who have previously received 18 months of blinded treatment with BA058 Injection 80 µg/Placebo. The analyses will be performed based on the treatment arm they were randomized to in the BA058-05-003 study. The analyses performed on data collected at Month 12 and Month 24 will be descriptive in nature and will be summarized descriptively with number, percentage (for categorical variables), mean, standard deviation, median, minimum and maximum (for continuous variables).
The analyses performed at six months will be used as a follow-up to the 18 month fracture endpoint for Study BA058-05-003. At this time-point, subjects will be analyzed based upon the randomization assignment in the BA058-05-003 study.
9.5.2 Baseline Comparisons
Baseline characteristics, medical history, physical examination, vital signs and ECG, will be summarized using standard descriptive statistics.
9.5.3 Fractures and BMD Analysis
All specified endpoints will be summarized by treatment group and study period using standard descriptive statistics (n, mean, SD, median, minimum, maximum, or n and %, as appropriate). The fracture incidence and BMD results from the additional six months of treatment with alendronate will be tabulated based on the treatment arm they were randomized to in the BA058-05-003 study, with additional tabular categories for results from the entire contiguous 24 months from the baseline of Study BA058-05-003, for subjects who eventually enter study BA058-05-005. These descriptive analyses will be conducted on all subjects with baseline and post-baseline data. The analysis performed at six months will be used a follow-up to the 18 month fracture endpoint for Study BA058-05-003. The data collected at Month 12 and Month 24 will be tabulated for each efficacy endpoint (vertebral fracture, non-vertebral fracture, and spine, femoral neck and forearm BMD). This information will be summarized according to original treatment assignment, osteoporosis medications taken between Month 6 and Month 24, and other patient characteristics.
9.5.4 Safety Analysis
Data will be summarized and tabulated based on the enrolled population for this Extension Study. All subjects enrolled in the Extension Study will be included in the safety analysis that will be performed on the following parameters:
· Incidence and severity of AEs;
· Pathological changes in hematology, chemistry and urinalysis data based on normal ranges supplied by the clinical laboratory, if applicable;
Safety assessments for changes in physical examination, vital signs, ECG, and laboratory tests will be descriptively summarized by treatment and study periods. The results of anti-
BA058 testing will be summarized. Concomitant medication classes will be categorized using World Health Organization (WHO) drug dictionary and summarized by number and percent of subjects using each class by treatment group. All treatment emergent adverse events (TEAEs) will be coded for system organ class (SOC) and preferred term (PT) using MedDRA and the number (%) of subjects experiencing each AE (SOC/PT) will be summarized by treatment, relationship to treatment, and severity. All serious adverse events (SAE) will be listed and the number (%) of subjects with an SAE presented by treatment group.
9.5.5 Procedures for Reporting Deviations to Original Statistical Analysis Plan
All deviations from the original statistical analysis plan will be provided in the final clinical study report.
9.6 Data Oversight
9.6.1 Central Review of Radiographs and DXA Scans
All radiographs will be viewed and assessed by a blinded, independent assessor (radiologist) on the basis of existing baseline and study-acquired vertebral deformity, and fractures will be assessed according to the method of Genant. A second blinded radiologist will review the assessment of the first reviewer for all subject radiographs in which an incident fracture has been identified. In the case of any disagreement, a third consensus assessment will be made to adjudicate the incident fracture. All study DXA scans will also be evaluated centrally by a blinded independent reviewer. The primary objective of the independent review is to provide objective data to determine the treatment benefit as demonstrated on the pertinent radiologic and clinical data associated with this study.
10.0 ADMINISTRATIVE REQUIREMENTS
10.1 Good Clinical Practice
This study will be conducted in accordance with the International Conference on Harmonization (ICH) for Good Clinical Practice (GCP) and the appropriate regulatory requirements. The Investigator will be thoroughly familiar with the appropriate use of the study medication as described in the protocol and the Investigator’s Brochure. Essential clinical documents will be maintained to demonstrate the validity of the study and the integrity of the data collected. The Investigator/institution should establish master files at the beginning of the study which will be maintained and updated during the study and retained thereafter according to the appropriate regulations.
10.2 Ethical Considerations
The study will be conducted in accordance with ethical principles founded in the Declaration of Helsinki. The Institutional Review Board (IRB)/Independent Ethics Committee (IEC) will review all appropriate study documentation in order to safeguard the rights, safety and well-being of the subjects. The study can only be conducted at study sites where IRB/IEC approval has been obtained. The protocol, informed consent form, Investigator’s Brochure, advertisements (if applicable), and all other forms of information given to subjects will be provided to the IRB/IEC by the Investigator. In addition, reports on the progress of the study will be submitted to the IRB/IEC by the Investigator at the appropriate intervals.
10.3 Subject Information and Informed Consent
Each subject (or a legally authorized representative) must give written informed consent prior to any new study-specific procedures being conducted. It is the responsibility of the Investigator to ensure written informed consent is obtained from each subject participating in this study after an explanation of the objectives, methods, discomforts and potential risks of the study has been provided. The Investigator (or study personnel) must also explain to each subject that he/she is free to refuse participation in the study or to withdraw from it at any time. Each subject will also be told that his/her records may be examined by competent authorities and authorized persons but that personal information will be treated as strictly confidential and will not be publicly available.
The informed consent form must be in accordance with the Declaration of Helsinki, ICH and GCP guidelines, and be approved by the Sponsor and the IRB/IEC. State or local laws may require additional information. Each subject (or his/her legally authorized representative) must sign and be given a copy of the informed consent form. Each subject’s signed informed consent form must be maintained by the Investigator and be readily available for review by the Sponsor (or its designee) or the Regulatory Authorities.
10.4 Protocol Compliance
The Investigator will conduct this study in compliance with the protocol provided by the Sponsor and given approval/favorable opinion by the IRB/IEC and the appropriate Regulatory Authority(ies). Changes to the protocol will not be made without agreement of the Sponsor Medical Monitor. All changes to the protocol will require IRB/IEC approval prior to implementation, except when necessary to eliminate an immediate hazard to study subjects or when the change involves only logistical or administrative aspects of the study (e.g., change in Sponsor Medical Monitor or telephone number). The IRB/IEC may provide, if applicable regulations permit, expedited review and approval/favorable opinion for minor changes in ongoing studies. The Sponsor will submit all protocol changes to the appropriate Regulatory Authority in accordance with the governing regulations.
In situations requiring a departure from the protocol, the Investigator or other physician in attendance will contact the Sponsor Medical Monitor by telephone, e-mail or fax. If possible, this contact will be made before implementing any departure from the protocol. In all cases, contact with the Sponsor Medical Monitor must be made as soon as possible in order to review the situation and agree on an appropriate course of action. The case report form and source document will describe any departure from the protocol and the circumstances requiring it.
10.5 Case Report Form Completion
eCRFs will be developed to collect information obtained during this study. It is the Investigator’s responsibility to ensure that the e-CRFs are completed for each subject enrolled in this study and for the accuracy, completeness, legibility and timeliness of the data reported in each e-CRF. Data for subjects who are screened but not enrolled into the study because they do not meet study criteria or do not complete all screening procedures, should be recorded in the e-CRF.
eCRFs will be completed and any corrections of data will be made according to procedures provided by the Sponsor (or designee).
10.6 Source Documents
Source documents are defined as original documents, data and records. This may include hospital records, clinical and office charts, laboratory data/information, work sheets, subjects’ diaries or evaluation checklists, pharmacy dispensing and other records, recorded data from automated instruments, microfiches, photographic negatives, microfilm or magnetic media, ECG printouts, and/or x-rays.
The Investigator(s)/institution(s) will permit trial-related monitoring, audits, IRB/IEC review, and regulatory inspection(s), providing direct access to source data documents.
10.7 Study Monitoring
The Sponsor (or its designee) will ensure that the study is monitored in accordance with ICH-GCP Guidelines. Monitoring is the act of overseeing the progress of a clinical trial and of ensuring that it is conducted, recorded, and reported in accordance with the protocol, standard operating procedures, Good Clinical Practice, and the applicable regulatory requirements and that the study data are accurate, complete and verifiable from source data. All study documentation and other source data will be made available to the Sponsor (or its designee), the IRB and to Regulatory Authorities for inspection upon request.
10.8 On-Site Audits
Representatives of the IRB or the Sponsor (or designee) may visit the study site to carry out an audit of the study in compliance with regulatory guidelines and company policy. Such audits will require access to all study records including source documents, CRFs, and other study documents. Direct access to these study records must be guaranteed by the Investigator, who must provide support for these activities at all times.
Similar auditing procedures may also be conducted by agents of any Regulatory Authority reviewing the results of this study. The Investigator/institution should immediately notify the Sponsor if they have been contacted by a Regulatory Authority concerning an upcoming inspection.
10.9 Drug Accountability
Accountability for the study medication at the study site is the responsibility of the Investigator. Drug accountability will be performed only on alendronate, calcium and vitamin D. The Investigator will ensure that the study medication is used only in accordance with this protocol. Where allowed, the Investigator may choose to assign some of the study medication accountability responsibilities to qualified study personnel.
Study medication accountability records indicating the delivery date to the study site, inventory at the study site and dispensing/use will be maintained. These records will adequately document that the study medications were dispensed and returned as specified in the protocol. Accountability records will include dates, quantities, and subject numbers. The Sponsor (or its designee) will review study medication accountability records at the study site on an ongoing basis during the study. All used and unused study medication must be inventoried, accounted for, and approved by the Sponsor (or its designee) prior to destruction. If the site is not capable of study drug disposal/destruction, the Sponsor will arrange for an alternative method. Records of disposal must be maintained with the study records.
10.10 Record Retention
The Investigator will maintain all study records according to ICH/GCP and applicable regulatory requirements. Essential documents must be retained for two years after the final marketing approval in an ICH region or at least two years have elapsed since the discontinuation of clinical development of the study medication. It is the responsibility of the Sponsor to inform the Investigator of when these documents can be destroyed. In addition, all subject medical records and other source documentation will be kept for the maximum time permitted by the hospital, institution or medical practice.
The Investigator/institution will take measures to prevent accidental or premature destruction of these documents. If the responsible Investigator retires, relocates, or for other reasons withdraws from the responsibility of keeping the study records, custody must be transferred to a person who will accept the responsibility. The Sponsor must be notified in writing of the name and address of the new custodian.
10.11 Study Termination
This study may be terminated at any time by the Sponsor if there is sufficient reasonable cause. Circumstances that may warrant termination include, but are not limited to:
· Determination of unexpected, significant, or unacceptable risk to subjects.
· Failure of enrollment
· Administrative reasons
· Plans to modify, suspend or discontinue the development of the study drug.
In addition, individual study sites may be terminated from study participation for reasons including, but not limited to the following:
· Failure to enter subjects at an acceptable rate.
· Insufficient adherence to protocol requirements.
· Incomplete and/or non-evaluable data.
In all cases, the terminating parties will provide written notification documenting the reason for study termination to all the relevant parties.
Should the study or an individual site be prematurely closed, all study materials (completed, partially completed, and blank CRFs, study drug, etc.) must be returned to the Sponsor (or its designee).
10.12 Liability and Insurance
The Sponsor has subscribed to an insurance policy covering, in its terms and provisions, its legal liability for injuries caused to participating persons and arising out of this research performed strictly in accordance with the scientific protocol as well as with applicable law and professional standards.
11.0 USE OF INFORMATION AND PUBLICATION OF STUDY FINDINGS
11.1 Use of Information
All information regarding BA058 supplied by the Sponsor (or its designee) to the Investigator is privileged and confidential information. The Investigator agrees to use this information to accomplish the study and will not use it for other purposes without prior consent from the Sponsor.
The information developed during the conduct of this clinical study is also considered confidential and will be used by the Sponsor in connection with the development of BA058. This information may be disclosed as deemed necessary by the Sponsor to other clinical Investigators, other pharmaceutical companies, and to Regulatory Authorities. To allow for the use of the information derived from this study and to ensure complete and thorough analysis, the Investigator is obligated to provide the Sponsor (or its designee) with complete study results and all data developed in this study and to allow direct access to source data/documents for study-related monitoring, audits, IRB/IEC review, and regulatory inspection.
11.2 Publication
One publication of the study data is planned. A Publications Committee composed of Investigators participating in the study and representatives from the Sponsor as appropriate will be formed to oversee the publication of the study results, which will reflect the experience of all participating study centers.
Subsequently, individual Investigators may publish results from the study in compliance with their agreement with the Sponsor. A pre-publication manuscript must be provided to the Sponsor at least 30 days prior to the submission of the manuscript to a publisher. Similarly, the Sponsor will provide any company prepared manuscript to the Investigators for review at least 30 days prior to submission to a publisher.
The Investigator shall comply with the policy of the Sponsor regarding confidential or proprietary information in any such paper and agrees to withhold publication of same for an additional 60 days in order to permit the Sponsor to obtain patent or other proprietary rights protection, if the Sponsor deems it necessary.
12.0 INVESTIGATOR AGREEMENT
To be completed by the Investigator
I have read Protocol BA058-05-005: “An Extension Study to Evaluate 24 Months of Standard-of-Care Osteoporosis Management Following Completion of 18 Months of BA058 or Placebo Treatment in Protocol BA058-05-003”.
I agree to conduct the study as detailed herein and in compliance with ICH Guidelines for Good Clinical Practice and applicable regulatory requirements and to inform all who assist me in the conduct of this study of their responsibilities and obligations.
The signature below constitutes my agreement to the contents of this protocol.
|
|
|
|
|
Signature of Principal Investigator |
|
Date |
|
|
|
|
|
|
|
|
|
Principal Investigator (print) |
|
|
Signature of Sponsor’s Medical Officer (where applicable)
|
|
|
|
|
Louis Brenner, MD |
|
Date |
13.0 REFERENCES
Balena R, Toolan BC, Shea M, Markatos A, Myers ER, Lee SC, Opas EE, Seedor JG, Klein H, Frankenfield D, Quartuccio H, Fiovanti C, Clair J, Brown E, Hayes WC, Rodan GA. The effects of 2-year treatment with the aminobisphosphonate alendronate on bone metabolism, bone histomorphometry, and bone strength in ovariectomized nonhuman primates. J Clin Invest 1993; 92:2577-2586.
Balena R, Markatos A, Seedor JG, Gentile M, Stark C, Peter CP, Rodan GA. Long-term safety of the aminobisphosphonate alendronate in adult dogs. II Histomorphometric analysis of the L5 vertebrae. J Pharmacol Exp Ther 1996; 276(1):277-83.
Black DM, Bilezikian JP, Ensrud KE, Greenspan SL, Palermo L, Hue T, Lang TF, McGowan JA, Rosen CJ. One year of alendronate after one year of parathyroid hormone (1-84) for osteoporosis. N Engl J Med 2005; 353(6):555-565.
Bone HG, Hosking D, Devogelaer JP, Tucci JR, Emkey RD, Tonito RP, Rodriguez-Portales JA, Downs RW, Grupta J, Santora AC, Liberman UA, Alendronate Phase III Osteoporosis Treatment Study Group. Ten years’ experience with alendronate for osteoporosis in postmenopausal women. N Eng J Med 2004; 350(12):1189-99.
Devogelaer JP, Broll H, Correa-Rotter R, Coming DC, De Deuxchaisnes CN, Geusens P, Hosking D, Jaeger P, Kaufman JM, Leite M, Leon J, Liberman U, Menkes CJ, Meunier PJ, Reid I, Rodriguez J, Romanowicz A, Seeman E, Vermeulen A, Hirsch LJ, Lombardi A, Plezia K, Santora AC, Yates AJ, Yuan W. Oral alendronate induces progressive increases in bone mass of the spine, hip and total body over 3 years in postmenopausal women with osteoporosis. Bone 1996; 18(2):141-50.
EMEA. Guideline on the evaluation of medicinal products in the treatment of primary osteoporosis. 2007.
FDA. Guidelines for preclinical and clinical evaluation of agents used in the prevention or treatment of postmenopausal osteoporosis. 1994.
FDA. Draft guidance: Development of parathyroid hormone for the prevention and treatment of osteoporosis. 2000.
Fosamax® Prescribing Information. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2012.
Genant HK, Wu CY, van KC, and Nevitt MC. Vertebral fracture assessment using a semiquantitative technique. J Bone Miner Res 1993; 8:1137-1148.
Guidance for Industry. E6 Good Clinical Practice: Consolidated Guidance. U.S.Department of Health and Human Services, Food and Drug Administration. April 1996.
Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407-413.
Lefage M-H, Balena R, Battle MA, Shea M, Seedor JG, Klein H, Hayes WC, Rodan GA. Comparison of alendronate and sodium fluoride effects on cencellous and cortical bone in minipigs. J Clin Invest 1995; 95:2127-2133.
Liberman UA, Weiss SR, Broll J, Minne HW, Quan H, Bell NH, Rodriguez-Portales J, Downs Jr. RW, Dequeker J, Favus M, Seeman E, Recker RR, Capizzi T, Santora AC, Lombardi A, Shah RV, Hirsch LJ, Karpe DB. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. N Engl J Med 1995; 333(22):1437-43.
Reginster JY, Burlet N. Osteoporosis: still increasing prevalence. Bone 2006; 38(Suppl 1):S4-9.
Rittmaster RS, Bolognese M, Ettinger MP, Hanley DA, Hodsman AB, Kendler DL, Rosen CJ. Enhancement of bone mass in osteoporotic women with parathyroid hormone followed by alendronate. J Clin Endocrinol Metab 2000; 85(6):2129-34.
Rizzoli R, Bonjour JP, and Ferrari SL. Osteoporosis, genetics and hormones. J Mol Endocrinol 2001; 26:79-94.
Tucci JR, Tonino RP, Emkey RD, Peverly CA, Kher U, Santora AC 2nd. Effect of three years of oral alendroneate treatment in postmenopausal women with osteoporosis. Am J Med 1996; 101(5):488-501.
WHO Scientific Group on the assessment of osteoporosis at primary health care level. World Health Organization Summary Meeting Report 2007.
World Medical Association Declaration of Helsinki. The World Medical Association, Inc. 2008.
|
|
Visit 1 2 3 4 5 6 Procedure Study Day/Month: Visit 10 003/ Visit 11 005 Month 3 Month 6 Month 12 Month 18 Month 24 Day 1 90 180 360 540 720 Visit Window (Days) N/A ± 5 ± 14 ± 14 ± 14 ± 14 Informed consent X Review of entrance criteria X Recent health status X X X X X X Vital signs, weight and height measurements2 X X X X X X Electrocardiogram X X Urinalysis (dipstick) 3 X X Chemistry blood collection4 X X Hematology blood collection5 X X Coagulation blood collection5 X X PTH(1-84) X 25-hydroxy vitamin D level X 1,25-dihydroxy vitamin D level X Serum markers of bone metabolism 5 X X BA058 antibody levels X 24-hour urine collection (for calcium:creatinine and creatinine clearance)6 X X7 X Clinical and radiologic (spine, lumbar and thoracic vertebrae) fracture X X assessments Bone mineral density of hip and spine by DXA8 X X Bone mineral density of wrist by DXA9 X X Calcium and vitamin D supplements Daily Alendronate administration (if applicable) Dosing as per prescribing information Study medication resupply (if applicable) X X X X Subject diary review10 X X X X X Document adverse events and concomitant medications Daily |
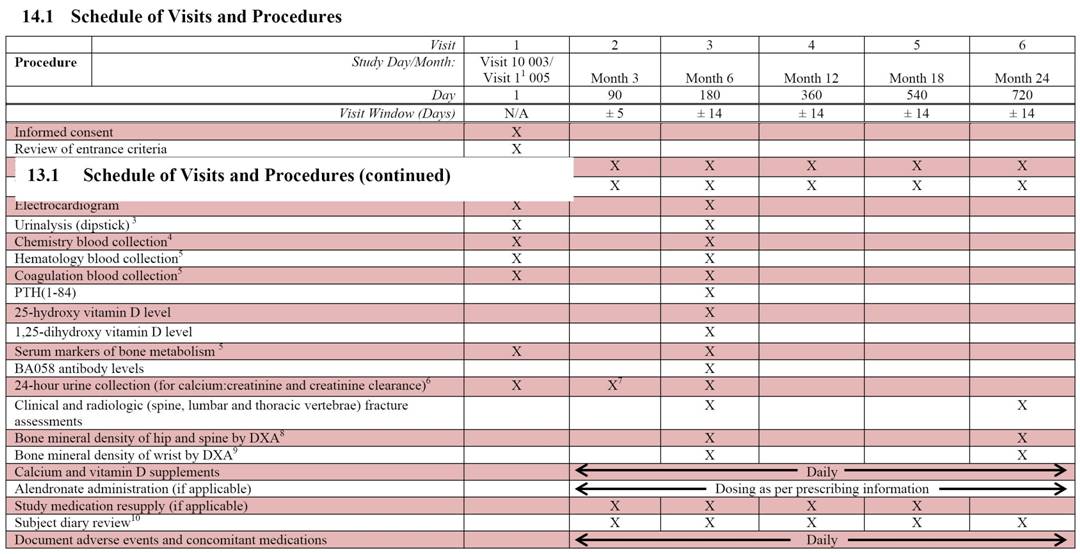
14.2 Suggested Schedule of Events and Procedures by Study Visit
The purpose of this guide is to provide more detailed instructions for the study procedures listed in Appendix 14.1. This guide presents the procedures in a suggested sequence of performance at each study visit. Further information may be found within the protocol and in other study reference manuals (e.g., ECG, clinical laboratory sample processing).
Of note:
· Blood and urinalysis samples are to be obtained under fasting conditions (NPO. for 8 hours; water is acceptable) in the morning of Day 1 and Month 6.
· DXA Scans: Always use the same study-validated machine; preferably the same technician.
· The 24-hour urine collection will be started at home the day before the clinic visit where the collection is required. Subjects will be instructed to discard the first morning void and begin the collection at least 24 hours before their clinic visit the following day. They will collect all urine for 24 hours with a final void before coming to the clinic. Routine urinalyses are to be performed using samples freshly voided during the clinic visit. Subjects should receive a reminder to initiate their 24-hour urine 2 days before their scheduled visit.
· Alendronate for Europe, Hong Kong and the US will be sourced centrally; alendronate for South America will be sourced locally by the medical center and reimbursed by the Sponsor.
· Subjects will be instructed to take the calcium and vitamin D supplements daily (in the evening with or without food or as otherwise instructed by the Investigator) until discharge from the study. This is required until the end of Month 24.
Definitions of Common Procedures:
The terms used in the by-visit schedule that follows are further defined below.
Recent health status (document any changes from last visit)
· Question subject regarding any new health issues
· Question subject regarding any new adverse events
· Question subject regarding any new concomitant medications
· Question subject regarding any new issues related to ability to continue with study
Pulse, respiration and temperature:
· Pulse rate (beats/minute) taken after approximately five minutes in the supine position.
· Respiration rate (breaths/minute).
· Body temperature (°C).
Weight and height measurements:
· Weight (kg).
· Height (cm) standing measurements are to be performed using the same medical stadiometer and standardized procedures each time.
Orthostatic blood pressure:
· Orthostatic blood pressure (mmHg) (measured in same arm each time/each visit) is measured after five minutes in the supine position followed by a measurement taken after 3 minutes in the standing position. Only the blood pressure assessment on Day 1 (Visit 10) needs to be orthostatic.
ECG
· Twelve-lead supine electrocardiogram
· Print hard copy for reading by qualified study personnel
· More than one ECG may be performed per time-point.
24 hour urine collection
· Subject to discard first morning void (suggest 6 a.m.) on day before clinic visit
· Subject to collect urine for approximately 24 hours
· Subject to collect final void at end of collection and bring collection to clinic.
· Process for calcium and creatinine
Urinalysis
· Obtain under fasting conditions (NPO. except water for 8 hours)
· Routine urinalysis is to be performed using a sample freshly voided during the clinic visit (microscopic examination if positive dipstick).
Review study medication administration procedures with subject
· Alendronate should be taken daily, preferably at the same time each morning/day of the week
Scheduling and instructions for next clinic visit
· Schedule visit
· Remind subject of any fasting requirements
· Provide urine collection instructions and materials as necessary
· Remind subjects to complete the diaries until the end of the study
Vitamin D and calcium supplements
· Vitamin D and calcium supplements are required throughout the study. Only those supplements supplied as part of study medication may be used and are to be used at the daily recommended dose (see Section 5.1.2).
· Supplements should be taken in the evening, with or without food as instructed by the Investigator.
· At each study visit, assess the subject’s supply and resupply as necessary.
· Drug usage reconciliation is to be performed when a new supply is provided.
Visit 10 for Study BA058-05-003
Day 1 Visit for Study BA058-05-005
Day 1
|
VISIT |
|
ACTIVITIES |
|
Day 1
Day 1 Visit for Study BA058-05-005
Visit 10 for Study BA058-05-003 |
|
Written informed consent must be obtained Recent health status · Document any changes since End-of-Treatment visit (Visit 9) from Study BA058-05-003 Study staff will receive your prior days 24-hour urine sample (if a 24-hour urine sample was not collected prior to Day 1, the subject must begin a 24-hour urine sample on the day prior to the Month 3 visit Subject diary review · Review study medication diary (calcium and vitamin D)/dispense new diary if necessary · Record deviations in dosing or any AEs in source documents and CRFs · Collect diaries and enter data into CRF Vital signs, weight and height measurement Orthostatic blood pressure ECG Blood collection: fasting conditions (NPO except water for 8 hours) · Chemistry · Hematology · Coagulation (PT and PTT) · Serum markers of bone metabolism, where applicable · PINP · bone-specific alkaline phosphatase · serum osteocalcin · serum CTX · Urinalysis (Dipstick) Study medication · Dispense three month supply of alendronate · Assess subject’s supply of calcium and vitamin D supplements; resupply as necessary · Instruct subject to take daily until they are discharged from the study Scheduling and instructions for next clinic visit · Remind subject to take study medication as instructed · 24-hour urine collection: If subjects did not provide a 24-hour urine sample for Visit 1, dispense urine collection container and instruct subjects to perform 24-hour urine collection beginning the morning 24 hours prior to their next scheduled visit (Month 3) · Remind subject to record study medication use |
Month 3 Visit for Study BA058-05-005
Day 90 (±5 days)
|
VISIT |
|
Activities |
|
Month 3 |
|
Recent health status · Document any changes since previous visit Study staff will receive the prior days 24 hour urine sample, if applicable Vital signs, height and weight measurement Subject diary review · Review study medication diary/dispense new diary if necessary · Record deviations in dosing or any AEs in source documents and CRFs. · Collect diaries and enter data into CRF Study medication · Dispense three month supply of alendronate · Assess subject’s supply of calcium and vitamin D supplements; resupply as necessary, instruct subject to take daily until they are discharged from the study Scheduling and instructions for next clinic visit · 24-hour urine collection: Dispense urine collection container and instruct subjects to perform 24-hour urine collection beginning the morning 24 hours prior to their next scheduled visit (Month 6) · Remind subject to take study medication as instructed · Remind subject to record study medication use |
Month 6 Visit for Study BA058-05-005
Day 180 (±14 Days)
|
VISIT |
|
Activities |
|
Month 6 |
|
Physical Examination Recent Health Status · Document any changes from last visit Collect 24 hour urine sample from subject Study staff will receive your prior days 24-hour urine sample · Review diary of study medication · Collect diary and enter data into CRF, record dosing deviations or any AEs in source documents and CRFs Vital signs, weight and height measurement ECG Blood collection: fasting conditions (NPO except water for 8 hours) · Chemistry · Hematology · Coagulation (PT and PTT) · Serum markers of bone metabolism, where applicable · PINP · bone-specific alkaline phosphatase · serum osteocalcin · serum CTX · BA058 antibody levels · Urinalysis (Dipstick) Clinical and radiologic fracture evaluations · Obtain antero-posterior and lateral radiographs of the lumbar and thoracic vertebrae · Document any non-vertebral fractures Bone mineral density · Perform DXA of spine (L1-L4), hip (femoral neck), and wrist (distal 1/3 radius), where applicable. Study Medication · Dispense six month supply of alendronate · Assess subject’s supply of calcium and vitamin D supplements, resupply as necessary Scheduling and instructions for next clinic visit · Remind subject to take study medication as instructed · Remind subject to record study medication use |
Month 12 Visit for Study BA058-05-005
Day 360 (±5 days)
|
VISIT |
|
Activities |
|
Month 12 |
|
Recent health status · Document any changes since previous visit Vital signs, height and weight measurement Subject diary review · Review study medication diary/dispense new diary if necessary · Record deviations in dosing or any AEs in source documents and CRFs. · Collect diaries and enter data into CRF Study medication · Dispense six month supply of alendronate · Assess subject’s supply of calcium and vitamin D supplements; resupply as necessary, instruct subject to take daily until they are discharged from the study Scheduling and instructions for next clinic visit · Remind subject to take study medication as instructed · Remind subject to record study medication use |
Month 18 Visit for Study BA058-05-005
Day 540 (±5 days)
|
VISIT |
|
Activities |
|
Month 18 |
|
Recent health status · Document any changes since previous visit Vital signs, height and weight measurement Subject diary review · Review study medication diary/dispense new diary if necessary · Record deviations in dosing or any AEs in source documents and CRFs. · Collect diaries and enter data into CRF Study medication · Dispense six month supply of aalendronate · Assess subject’s supply of calcium and vitamin D supplements; resupply as necessary, instruct subject to take daily until they are discharged from the study Scheduling and instructions for next clinic visit · Remind subject to take study medication as instructed · Remind subject to record study medication use |
Month 24 Visit for Study BA058-05-005
Day 720 (±5 days)
|
VISIT |
|
Activities |
|
Month 24 |
|
Recent health status · Document any changes since previous visit Vital signs, height and weight measurement Subject diary review · Review study medication diary · Record deviations in dosing or any AEs in source documents and CRFs. · Collect diaries and enter data into CRF Clinical and radiologic fracture evaluations · Obtain antero-posterior and lateral radiographs of the lumbar and thoracic vertebrae · Document any non-vertebral fractures Bone mineral density · Perform DXA of spine (L1-L4), hip (femoral neck), and wrist (distal 1/3 radius), where applicable. Study medication · Collect unused study medication Discharge subject from study · Subject is terminated from the study unless adverse events require further follow through Discuss continuing treatment options · Subjects will receive standard-of-care management according to their physician |
14.3 Eastern Cooperative Oncology Group (ECOG) Common Toxicity Criteria
|
Category |
|
Grade 0 |
|
Grade 1 |
|
Grade 2 |
|
Grade 3 |
|
Grade 4 |
|
Haematology |
|
|
|
|
|
|
|
|
|
|
|
WBC (x109/L) |
|
4 |
|
3.0 - 3.9 |
|
2.0 - 2.9 |
|
1.0 - 1.9 |
|
< 1.0 |
|
Platelets (x109/L) |
|
WNL |
|
75.0 - normal |
|
50.0 - 74.9 |
|
25.0 - 49.9 |
|
< 25.0 |
|
Haemoglobin (g/L); (mmol/L) |
|
WNL |
|
100.0 - normal; 6.2 - normal |
|
80.0 - 99.0; 5.0 - 6.1 |
|
65.0 - 79.0 4.0 - 4.9 |
|
< 65.0 < 4.0 |
|
Granulocytes/ Bands (x109/L) |
|
2 |
|
1.5 - 1.9 |
|
1.0 - 1.4 |
|
0.5 - 0.9 |
|
< 0.5 |
|
Lymphocytes (x109/L) |
|
2 |
|
1.5 - 1.9 |
|
1.0 - 1.4 |
|
0.5 - 0.9 |
|
< 0.5 |
|
Haemorrhage |
|
none |
|
mild, no transfusion |
|
gross, |
|
gross, |
|
massive, |
|
Coagulation |
|
|
|
|
|
|
|
|
|
|
|
Fibrinogen |
|
WNL |
|
0.99 - 0.75 x N |
|
0.74 - 0.50 x N |
|
0.49 - 0.25 x N |
|
< 0.25 x N |
|
Prothrombin time(quick) |
|
WNL |
|
1.01 - 1.25 x N |
|
1.26 - 1.50 x N |
|
1.51 - 2.00 x N |
|
> 2.00 x N |
|
Partial thromboplastin time |
|
WNL |
|
1.01 - 1.66 x N |
|
1.67 - 2.33 x N |
|
2.34 - 3.00 x N |
|
> 3.00 x N |
|
Metabolic |
|
|
|
|
|
|
|
|
|
|
|
Hyperglycaemia (mmol/L) |
|
< 6.4 |
|
6.4 - 8.9 |
|
9.0 - 13.9 |
|
14.0 - 27.8 |
|
> 27.8 or ketoacidosis |
|
Hypoglycaemia (mmol/L) |
|
> 3.6 |
|
3.6 - 3.1 |
|
3.0 - 2.3 |
|
2.2 - 1.7 |
|
< 1.7 |
|
Amylase |
|
WNL |
|
< 1.5 x N |
|
1.5 - 2.0 x N |
|
2.1 - 5.0 N |
|
> 5.0 x N |
|
Hypercalcaemia (mmol/L) |
|
< 2.65 |
|
2.65 - 2.88 |
|
2.89 - 3.13 |
|
3.14 - 3.36 |
|
> 3.37 |
|
Hypocalcaemia (mmol/L) |
|
> 2.10 |
|
2.10 - 1.94 |
|
1.93 - 1.74 |
|
1.73 - 1.52 |
|
< 1.51 |
|
Hypomagnesaemia (mmol/L) |
|
> 0.58 |
|
0.58 - 0.48 |
|
0.47 - 0.36 |
|
0.35 - 0.24 |
|
< 0.23 |
|
Gastrointestinal |
|
|
|
|
|
|
|
|
|
|
|
Nausea |
|
none |
|
able to eat reasonable intake |
|
intake significantly decreased but can eat |
|
no significant intake |
|
— |
|
Vomiting |
|
none |
|
1 episode |
|
2 - 5 episodes |
|
6 - 10 episodes |
|
> 10 episodes in 24 hrs or requiring parenteral support |
|
Diarrhoea |
|
none |
|
increase of 2 - 3 stools/day over pre-Rx |
|
increase of 4 - 6 stools/day, or nocturnal stools, or moderate cramping |
|
increase of 7 - 9 stools/day, or incontinence, or severe cramping |
|
increase of > 10 stools/day or grossly bloody diarrhoea, or need for parenteral support |
|
Stomatitis |
|
none |
|
painless ulcers, erythema, or mild soreness |
|
painful erythema, oedema, or ulcers but can eat solids |
|
painful erythema, oedema, or ulcers and cannot eat solids |
|
requires parenteral or enteral support for alimentation |
|
Liver |
|
|
|
|
|
|
|
|
|
|
|
Bilirubin (N = 17 µmol/L) |
|
WNL |
|
— |
|
< 1.5 x N |
|
1.5 - 3.0 x N |
|
> 3.0 x N |
|
Transaminase (SGOT, SGPT) |
|
WNL |
|
2.5 x N |
|
2.6 - 5.0 x N |
|
5.1 - 20.0 x N |
|
> 20.0 x N |
|
Alkaline phosphatase or 5-nucleotidase |
|
WNL |
|
< 2.5 x N |
|
2.6 - 5.0 x N |
|
5.1 - 20.0 x N |
|
> 20.0 x N |
|
Category |
|
Grade 0 |
|
Grade 1 |
|
Grade 2 |
|
Grade 3 |
|
Grade 4 |
|
Liver- clinical |
|
No change from baseline |
|
— |
|
— |
|
precoma |
|
hepatic coma |
|
Kidney, bladder |
|
|
|
|
|
|
|
|
|
|
|
Creatinine |
|
WNL |
|
< 1.5 x N |
|
1.5 - 3.0 x N |
|
3.1 - 6.0 x N |
|
> 6.0 x N |
|
Proteinuria |
|
No change |
|
1 (+) or |
|
2 - 3 (+) or |
|
4 (+) or |
|
nephrotic syndrome |
|
Haematuria |
|
Negative |
|
microscopic only |
|
gross, |
|
gross and clots |
|
requires transfusion |
|
Weight gain/ loss |
|
< 5.0 % |
|
5.0 - 9.9 % |
|
10.0 - 19.9 % |
|
20.00% |
|
— |
|
Pulmonary |
|
|
|
|
|
|
|
|
|
|
|
Pulmonary |
|
none or no change |
|
asymptomatic, with abnormality in PFTs |
|
dyspnoea on significant exertion |
|
dyspnoea at normal level of activity |
|
dyspnoea at rest |
|
Cardiac |
|
|
|
|
|
|
|
|
|
|
|
Cardiac arrhythmias |
|
none |
|
asymptomatic, transient, requiring no therapy |
|
recurrent or persistent, no therapy required |
|
requires treatment |
|
requires monitoring; or hypotension, or ventricular tachycardia or fibrillation |
|
Cardiac function |
|
none |
|
asymptomatic, decline of resting ejection fraction by less than 20 % of baseline value |
|
asymptomatic, decline of resting ejection fraction by more than 20 % of baseline value |
|
mild CHF, |
|
severe or |
|
Cardiac ischaemia |
|
none |
|
non-specific T- wave flattening |
|
asymptomatic, ST and T wave changes suggesting ischaemia |
|
angina without evidence of infraction |
|
acute myocardial infarction |
|
Cardiac- pericardial |
|
none |
|
asymptomatic effusion, no intervention required |
|
pericarditis (rub, chest pain, ECG changes) |
|
symptomatic effusion; drainage required |
|
tamponade; drainage urgently required |
|
Hypertension |
|
none or no change |
|
asymptomatic, transient increase by greater than 20 mmHg (D) or to > 150/100 if previously WNL. |
|
recurrent or persistent increase by greater than 20 mmHG (D) or to > 150/100 if previously WNL. |
|
requires therapy |
|
hypertensive crisis |
|
Hypotension |
|
none or no change |
|
changes requiring no therapy (including transient orthostatic hypotension) |
|
requires fluid replacement or other therapy but not hospitalisation |
|
requires therapy and hospitalisation; resolves within 48 hrs of stopping the agent |
|
requires therapy and hospitalisation for > 48 hrs after stopping the agent |
|
Neurologic |
|
|
|
|
|
|
|
|
|
|
|
Neuro: sensory |
|
none or no change |
|
mild paraesthesias; |
|
mild or moderate objective sensory loss moderate paraesthesias |
|
severe objective sensory loss or paraesthesias that interfere with function |
|
— |
|
Category |
|
Grade 0 |
|
Grade 1 |
|
Grade 2 |
|
Grade 3 |
|
Grade 4 |
|
Neuro: motor |
|
none or no change |
|
subjective weakness; |
|
mild objective weakness without significant impairment of function |
|
objective weakness with impairment of function |
|
paralysis |
|
Neuro: cortical |
|
none |
|
mild somnolence or agitation |
|
moderate somnolence or agitation |
|
severe somnolence, (>50 % waking hours), agitation, confusion, disorientation or hallucinations |
|
coma, seizures, |
|
Neuro: cerebellar |
|
none |
|
slight incoordination, dysdiadochokinesia |
|
intention tremor, dysmetria, slurred speech, nystagmus |
|
locomotor ataxia |
|
cerebellar necrosis |
|
Neuro: mood |
|
no change |
|
mild anxiety or depression |
|
moderate anxiety or depression |
|
severe anxiety or depression |
|
suicidal ideation |
|
Neuro: headache |
|
none |
|
mild |
|
moderate or severe |
|
unrelenting and severe |
|
— |
|
Neuro: constipation |
|
none or no change |
|
mild |
|
moderate |
|
severe |
|
ileus > 96 hrs |
|
Neuro: hearing |
|
none or no change |
|
asymptomatic, hearing loss on audiometry only |
|
tinnitus |
|
hearing loss interfering with function but correctable with hearing aid |
|
deafness not correctable |
|
Neuro: vision |
|
none or no change |
|
— |
|
— |
|
symptomatic subtotal loss of vision |
|
blindness |
|
Pain |
|
|
|
|
|
|
|
|
|
|
|
Pain |
|
none |
|
mild |
|
moderate |
|
severe |
|
reg. narcotics |
|
Skin |
|
|
|
|
|
|
|
|
|
|
|
Skin |
|
none or no change |
|
scattered macular or papular eruption or erythema that is asymptomatic |
|
scattered macular or papular eruption or erythema with pruritus or other associated symptoms |
|
generalised symptomatic macular, papular or vesicular eruption |
|
exfoliative dermatitis or ulcerating dermatitis |
|
Alopecia |
|
|
|
|
|
|
|
|
|
|
|
Alopecia |
|
no loss |
|
mild hair loss |
|
pronounced or total hair loss |
|
— |
|
— |
|
Allergy |
|
|
|
|
|
|
|
|
|
|
|
Allergy |
|
none |
|
transient rash, |
|
urticaria, |
|
serum sickness, bronchospasm requiring parenteral medication |
|
anaphylaxis |
|
Local |
|
|
|
|
|
|
|
|
|
|
|
Local |
|
none |
|
pain |
|
pain and swelling with inflammation or phlebitis |
|
ulceration |
|
plastic surgery indicated |
|
Fever of unknown origin |
|
|
|
|
|
|
|
|
|
|
|
Fever of unknown origin |
|
none |
|
37.1 - 38.0o C |
|
38.1 - 40.0o C |
|
> 40.0oC (> 104o F) for less than 24hrs |
|
> 40.0o C (> 104o F) for more than 24 hrs or accompanied by hypotension |
|
Infection |
|
|
|
|
|
|
|
|
|
|
|
Infection |
|
none |
|
mild |
|
moderate |
|
severe |
|
life-threatening |
|
Category |
|
Grade 0 |
|
Grade 1 |
|
Grade 2 |
|
Grade 3 |
|
Grade 4 |
|
Additional events |
|
|
|
|
|
|
|
|
|
|
|
Asthenia |
|
analogous to Karnofsky index (WHO grading) |
|
|
|
|
|
|
|
|
|
Chills |
|
analogous to fever |
|
|
|
|
|
|
|
|
|
Peripheral oedema |
|
analogous to weight gain |
|
|
|
|
|
|
|
|
|
Anorexia |
|
analogous to weight loss |
|
|
|
|
|
|
|
|
(1)The procedures for the Follow-up visit (Visit 10) for Study BA058-05-003 will serve as the procedures performed at Day 1 (for Study BA058-05-005). The consent form will need to be signed if it was not signed during the End-of-Treatment Visit (Visit 9) of Study BA058-05-003.
(2) Vital signs (blood pressure, pulse rate, body temperature, and respiration rate) are to be recorded at each study visit. Only the blood pressure assessment on Day 1 (Visit 10) needs to be orthostatic. Height is to be measured at each visit in the standing position using a medical stadiometer. Weight is to be measured at each visit. Orthostatic blood pressure is to be measured initially after 5 minutes in the supine position and then again after standing for three minutes.
(3) All routine urinalysis will be performed on a sample freshly voided during the clinic visit.
(4) These blood samples are to be obtained under fasting conditions (N.P.O. for 8 hours; water is acceptable) in the morning of each scheduled study visit.
(5) Includes blood samples for PINP, bone-specific alkaline phosphatase, serum osteocalcin and CTX.
(6) Twenty-four hour urine collection will be used for urinary calcium and urinary creatinine measurements. Subjects will discard the 1st void and begin a 24-hour urine collection the day prior to the clinic visit.
(7) A 24-hour urine collection will be collected at Month 3 only if a sample was not collected for the Day 1 (Visit 10).
(8) Each DXA for a given subject should be performed on the same machine, and if available, preferably by the same technician
(9) Each DXA for a given subject should be performed on the same machine, and if available, preferably by the same technician. Only subjects who had wrist DXA assessments in Study BA058-05-003 will have wrist DXAs performed.
(10) The subjects will maintain a diary throughout the study to record missed doses of medication (including supplements) on a weekly basis; the diaries are to be reviewed with the subject at each study visit.