Attached files
| file | filename |
|---|---|
| EX-23.1 - EX-23.1 - PLx Pharma Inc. | a2218793zex-23_1.htm |
Use these links to rapidly review the document
Table of Contents
TABLE OF CONTENTS 2
As filed with the Securities and Exchange Commission on March 6, 2014
Registration Number 333-193780
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 2
TO
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
DIPEXIUM PHARMACEUTICALS, LLC
(to be converted to Dipexium Pharmaceuticals, Inc.)
(Exact Name of Registrant as Specified in its Charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
2834 (Primary Standard Industrial Classification Code Number) |
27-1707962 (I.R.S. Employer Identification No.) |
74 Broad Street
New York, New York 10004
Phone: (212) 422-5717
Fax: (212) 269-6441
(Address, Including Zip Code, and Telephone Number, Including Area Code, of Registrant's Principal Executive Offices)
David P. Luci, Esq.
President and Chief Executive Officer
74 Broad Street
New York, New York 10004
Phone: (212) 422-5717
Fax: (212) 269-6441
(Name, Address, Including Zip Code, and Telephone Number, Including Area Code, of Agent for Service)
| with copies to: | ||
Lawrence A. Rosenbloom, Esq. Ellenoff Grossman & Schole LLP 1345 Avenue of the Americas, 11th Floor New York, NY 10105 Phone: (212) 370-1300 Fax: (212) 370-7889 |
Ivan K. Blumenthal, Esq. Mintz Levin Cohn Ferris Glovsky and Popeo, P.C. Chrysler Center, 666 Third Avenue New York, NY 10017 Phone: (212) 935-3000 Fax: (212) 983-3115 |
|
Approximate date of commencement of proposed sale to public:
As soon as practicable after the effective date hereof.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company ý |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission, acting pursuant to Section 8(a), may determine.
Dipexium Pharmaceuticals, LLC, the registrant whose name appears on the cover of this registration statement, is a Delaware limited liability company. Immediately prior to the effectiveness of this registration statement, Dipexium Pharmaceuticals, LLC intends to convert into a Delaware corporation pursuant to a statutory conversion and change its name to Dipexium Pharmaceuticals, Inc. As a result of the corporate conversion:
- •
- the Class A membership interests of Dipexium Pharmaceuticals, LLC will become shares of common stock of
Dipexium Pharmaceuticals, Inc. pursuant to a conversion ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each Class A membership interest of Dipexium
Pharmaceuticals, LLC previously held. Accordingly, 767,911 outstanding Class A membership interests of Dipexium Pharmaceuticals, LLC issued and outstanding immediately prior to
the corporate conversion will be converted automatically into 5,375,377 shares of Dipexium Pharmaceuticals, Inc.; and
- •
- all of the outstanding warrants to purchase Class A membership interests of Dipexium Pharmaceuticals, LLC
will become warrants to purchase shares of common stock of Dipexium Pharmaceuticals, Inc. in a ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each
Class A membership interest of Dipexium Pharmaceuticals, LLC underlying such warrants, with the effect that warrants to purchase 4,900 Class A membership interests of Dipexium
Pharmaceuticals, LLC outstanding immediately prior to the corporate conversion will automatically convert into warrants to purchase 34,300 shares of Dipexium Pharmaceuticals, Inc. upon
consummation of the corporate conversion; and
- •
- the exercise price of all of the outstanding warrants will be adjusted in the same ratio as the seven-for-one conversion ratio noted above such that all of our outstanding warrants to purchase Class A membership interests of Dipexium Pharmaceuticals, LLC which are currently exercisable at $60 per Class A membership interest will automatically be adjusted such that the new exercise price for the outstanding warrants upon consummating the corporate conversion will be $8.57 per share, subject to certain adjustments noted in each of the warrants.
The financial statements and summary historical financial data included in this registration statement are those of Dipexium Pharmaceuticals, LLC and do not give effect to the corporate conversion. All share and warrant amounts and related prices reflected in the accompanying prospectus give effect to the corporate conversion, however such amounts appearing in Part II of the accompanying registration statement do not give effect to the corporate conversion.
The information contained in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to Completion, Dated March 6, 2014
2,307,693 Shares

Common Stock
$ per share
This is the initial public offering of common stock of Dipexium Pharmaceuticals, Inc. We are offering 2,307,693 shares of our common stock in this offering. Prior to this offering, there has been no public market for our common stock.
We currently expect the initial public offering price per share to be between $12.00 and $14.00.
We have applied to have our common stock listed on The NASDAQ Capital Market under the symbol "DPRX."
We are an "emerging growth company" under the federal securities laws and have elected to comply with certain reduced public company reporting requirements.
Investing in our common stock involves a high degree of risk. See "Risk Factors" beginning on page 10.
| |
Per Share | Total | |||||
|---|---|---|---|---|---|---|---|
Public offering price |
$ | $ | |||||
Discounts and commissions to underwriters(1) |
$ | $ | |||||
Proceeds to us, before expenses |
$ | $ | |||||
- (1)
- See "Underwriting" beginning on page 86 for additional information regarding underwriting compensation.
We have granted a 30-day option to the representative of the underwriters to purchase up to 346,154 additional shares of common stock solely to cover over-allotments, if any.
The underwriters expect to deliver the shares to purchasers on or about , 2014 through the book-entry facilities of The Depository Trust Company.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Oppenheimer & Co. | ||
| Feltl and Company |
The date of this prospectus is , 2014
You should rely only on the information contained in this prospectus. We have not authorized any other person to provide you with information different from or in addition to that contained in this prospectus. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offer to sell these securities in any jurisdiction where an offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
In this prospectus, we rely on and refer to information and statistics regarding our industry. We obtained this statistical, market and other industry data and forecasts from publicly available information. While we believe that the statistical data, market data and other industry data and forecasts are reliable, we have not independently verified the data.
Locilex™ is a trademark of Dipexium Pharmaceuticals, LLC. All other trademarks used herein are the property of their respective owners.
This summary highlights certain information appearing elsewhere in this prospectus. Because it is only a summary, it does not contain all of the information that you should consider before investing in shares of our common stock and it is qualified in its entirety by, and should be read in conjunction with, the more detailed information appearing elsewhere in this prospectus. Before you decide to invest in our common stock, you should read the entire prospectus carefully, including "Risk Factors" beginning on page 9 and the financial statements and related notes included in this prospectus.
Immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, we will undertake a corporate conversion pursuant to which Dipexium Pharmaceuticals, Inc. will succeed to the business of Dipexium Pharmaceuticals, LLC and the holders of membership interests of Dipexium Pharmaceuticals, LLC will become stockholders of Dipexium Pharmaceuticals, Inc. In this prospectus, we refer to this transaction as the "corporate conversion." References in this prospectus to our capitalization and other matters pertaining to our common equity relate to the capitalization and common equity of Dipexium Pharmaceuticals, LLC after giving effect to the corporate conversion. However, the financial statements and summary historical financial data included in this prospectus are those of Dipexium Pharmaceuticals, LLC and do not give effect to the corporate conversion.
Unless the context indicates otherwise, as used in this prospectus, the terms "Dipexium," "we," "us," "our," "our company" and "our business" refer, prior to the corporate conversion discussed herein, to Dipexium Pharmaceuticals, LLC, and after the corporate conversion to Dipexium Pharmaceuticals, Inc.
Our Company
We are a late stage pharmaceutical company focused on the development and commercialization of Locilex™ (pexiganan acetate cream 1%), a novel, first-in-class, broad spectrum, topical antibiotic. Locilex™ is a chemically synthesized, 22-amino acid peptide isolated from the skin of the African Clawed Frog. Its novel mechanism of action kills microbial targets through disruption of bacterial cell membrane permeability. Locilex™ is initially being targeted for the treatment of mild infections of diabetic foot ulcers (or Mild DFI). In 2011, the market for diabetic foot infection therapeutics worldwide was $1.46 billion. We recently received a U.S. patent on the formulation of Locilex™ that expires in June 2032. Our primary objective is to establish Locilex™ as the standard of care for the treatment of patients with Mild DFI. Thereafter, our growth strategy includes potentially expanding the indications for Locilex™ to include moderate infections of diabetic foot ulcers (or Moderate DFI) and certain other mild or moderate skin and skin structure infections in superficial wounds.
We believe that we have a clear clinical and regulatory pathway with the potential for near term United States Food and Drug Administration (or FDA) approval of Locilex™. We believe that Locilex™ may be approved by the FDA within 24 months of commencing the Phase 3 enrollment. We have reached agreement with the FDA through a special protocol assessment (or SPA) for our Phase 3 program. We intend to conduct and complete two pivotal Phase 3, double blind, placebo-controlled, superiority studies. We expect to begin enrollment in these studies in the second quarter of 2014, with top line data from the Phase 3 studies anticipated in the first quarter of 2015. Concurrently, we also intend to conduct two separate Phase 1 skin irritation and skin sensitization studies, which are expected to commence in the first quarter of 2014. If the data from our Phase 3 studies are sufficient to meet the primary endpoints, we expect to submit our new drug application (or NDA) for Locilex™ in the second half of 2015. We expect to receive a response from the FDA within six months of our NDA submission.
1
According to the Infectious Disease Society of America (or IDSA), diabetic foot infections (or DFI) may be classified by their clinical severity as mild, moderate, or severe. Forty-seven percent of DFI patients present at the mild stage, 34% of DFI patients present at the moderate stage and 18% of DFI patients present at the severe stage. At the mild stage, patients can typically be treated on an outpatient basis and amputation risk is minimal (2 to 3% in Mild DFI). When not managed effectively, the potential for Mild DFI to progress to a limb- or life-threatening infection increases dramatically. Published research suggests amputation rates increase in Moderate DFI and severe infections of diabetic foot ulcers (or Severe DFI) to approximately 45% and 75%, respectively. Similarly, the hospitalization rate for Mild DFI patients is approximately 10%, increasing in Moderate and Severe DFI to approximately 55% and 85%, respectively. Thus, DFI are a major cause of patient morbidity, a substantial burden to the healthcare system, and a source of high financial costs.
Systemic antibiotics currently prescribed off-label to treat Mild DFI generate resistant pathogens which create infections that are more difficult to treat. Such antibiotics are also associated with toxic side effects in patients who typically have some degree of compromised liver and kidney function. We believe that a topical preparation like Locilex™, which is locally administered on the open wound and skin, offers significant advantages over systemic treatments. Currently, there are no products specifically approved by the FDA for the treatment of Mild DFI, nor are there any topical antibiotics currently approved for any severity of DFI. As such, we believe that Locilex™ has the potential to be the first topical antibiotic approved for the treatment of DFI, as well as the first product of any kind to be labeled specifically for the treatment of Mild DFI.
We believe the key attributes of Locilex™ are:
- •
- it has not generated resistant bacteria systemically;
- •
- it has not generated cross resistance with other antibiotics;
- •
- it has demonstrated activity against a broad spectrum of pathogens, including difficult to treat gram negative, and
anaerobic bacteria;
- •
- it has not been systemically absorbed; and
- •
- it has not caused any significant safety issues in over 500 patients treated.
These attributes lead us to believe that Locilex™ has the potential to be positioned as the standard of care to treat patients with Mild DFI. In addition, Locilex™ data generated to date support the potential use of Locilex™ to treat a broad array of mild or moderate skin and skin structure infections in superficial wounds.
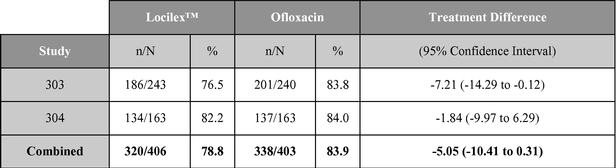
As reported in published research, Locilex™ previously has been evaluated in two large-scale clinical studies in patients with Mild or Moderate DFI. Each of these trials was a randomized, active-controlled, double blind, multi-center trial designed to establish equivalence of Locilex™ to ofloxacin, a systemic antibiotic. Across both trials, 835 patients received twice-daily Locilex™ cream plus placebo pill or twice-daily oral ofloxacin plus placebo cream for 14 to 28 days. The primary outcome was the clinical improvement of infection in response to antimicrobial treatment. Secondary outcomes included eradication of the wound pathogens, healing of the ulcer, development of antibiotic resistance to a study drug, and safety.
Although one study (Study 303) did not demonstrate equivalence, the second study (Study 304) and the combined data for the two trials did demonstrate equivalent results for topical Locilex™ and oral ofloxacin in clinical improvement rates and overall microbiological eradication rates. Guidelines for establishing equivalence for these results were a 95% confidence interval with a lower bound delta of -15%, for clinical effectiveness, and a lower bound delta of -20%, for overall microbiological effectiveness. In each case, the confidence interval must cross zero. Additionally, there were no statistically significant differences in wound assessment scores between patients treated with Locilex™ and ofloxacin. Wound assessment scores decreased at the end of treatment visit in both studies for both treatment arms, and they decreased further at the follow-up visit. No serious adverse events were
2
determined to have been related to either Locilex™ or ofloxacin. Moreover, the overall incidence of serious adverse events unrelated to the study medication (12% Locilex™ to 9% ofloxacin), including worsening cellulitis (2% to 4%) and amputation (2% to 3%), did not differ significantly between treatment arms. Bacterial resistance to ofloxacin emerged in some patients in the control arm, but no significant resistance to Locilex™ emerged among patients in the treatment arm.
A summary table of the primary clinical outcome and secondary microbiological outcome are shown below:
Clinical outcome (% cure or improved)
Microbiological outcome (% responders)
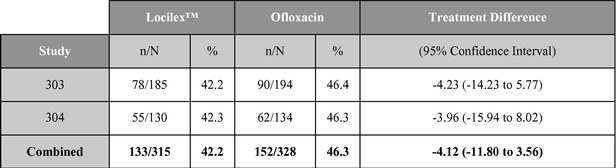
Source: Lipsky, et al. Clinical Infectious Disease. 2008; 47(12): 1537-45.
We have conducted microbiology studies that highlight the sensitivity of resistant bacteria, including methicillin-resistant staphylococcus aureus (or MRSA), vancomycin-resistant enterococcus (or VRE), extended-spectrum b-lactamase (or ESBL) and multi-drug resistant (or MDR) bacteria, to pexiganan, the active pharmaceutical ingredient (or API) in Locilex™. Due to the increased global prevalence of resistant bacteria, Locilex™ may provide an important therapeutic advance.
We hold U.S. rights to a patent covering our proprietary formulation of Locilex™ and the method of using it for the treatment of skin and wound infections. This patent was granted in September 2013 and expires in the U.S. in June 2032. In addition, we have filed a Patent Cooperation Treaty (or PCT) application that will allow us to seek corresponding protection outside of the U.S., including in Europe, Japan, China, Australia, and Korea, as well as in other PCT jurisdictions. We also hold an exclusive sublicense to the composition-of-matter patent covering the pexiganan technology, which expires in June 2016, not including any patent term extension that we expect to seek under The Drug Price Competition and Patent Term Restoration Act of 1984 (or the Hatch-Waxman Act).
We have contracted with third party vendors with respect to all key elements of our clinical and regulatory program, including vendors to: (i) conduct the Phase 3 and Phase 1 clinical trials for Locilex™; (ii) manufacture the API; (iii) formulate the finished product; and (iv) label and package the product. We believe these key relationships will help drive our clinical and regulatory program for Locilex™ in a timely and efficient manner.
3
Locilex™ was originally sponsored by Magainin Pharmaceuticals, Inc. (or Magainin), which engaged in the FDA review process for a prior formulation of Locilex™. In its 1999 non-approvable letter, the FDA identified two cGMP manufacturing issues. The first issue concerned the stability of the product. Examination of the product over time showed evidence of water separation from the cream matrix. The second issue related to the purity level of the API in the product. The prior source of the API yielded a purity level as low as 95%. We acquired the worldwide rights to pexiganan, the API in Locilex™, from a third party in April 2010. These rights included the prior formulation and all of the clinical and preclinical data generated by Magainin in its FDA review process. This includes data from over 1,000 evaluable patients, including 835 in large-scale, randomized, active-controlled, double blind multi-center clinical studies as compared to a systemic quinolone standard of care. After acquiring the rights to Locilex™, we developed a detailed product development plan to arrive at an optimized formulation to address these issues to the satisfaction of the FDA. In October 2013, we submitted our manufacturing data, including data from the cGMP batch as well as 18-month stability data on our non-cGMP batch, to the FDA, and in December 2013 the FDA indicated in written communications with us that Locilex™'s stability and purity levels are acceptable for use in our upcoming Phase 3 studies. We believe we have corrected the manufacturing problems encountered by the prior sponsor.
Our Strategy
Our primary objective is to establish Locilex™ as the standard of care to treat patients with Mild DFI. The key elements of our strategy are as follows:
- •
- Complete the Phase 3 program for
Locilex™;
- •
- Obtain FDA approval of Locilex™ for Mild
DFI;
- •
- Commercially launch Locilex™ in the U.S.;
- •
- Expand Locilex™'s FDA-approved uses;
and
- •
- Commence clinical and regulatory activities in Europe.
We will rely on our strong management team, board of directors and scientific advisory board to execute our strategy. The individuals on our management team, board of directors and scientific advisory board will contribute their significant development and regulatory experience to the development and commercialization of Locilex™.
Corporate Conversion
We currently operate as a limited liability company, organized in the State of Delaware under the name Dipexium Pharmaceuticals, LLC. Immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, Dipexium Pharmaceuticals, LLC will convert from a Delaware limited liability company to a Delaware corporation and will be renamed Dipexium Pharmaceuticals, Inc. As a result of the corporate conversion:
- •
- the Class A membership interests of Dipexium Pharmaceuticals, LLC will become shares of common stock of
Dipexium Pharmaceuticals, Inc. pursuant to a conversion ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each Class A membership interest of Dipexium
Pharmaceuticals, LLC previously held. Accordingly, 767,911 Class A membership interests of Dipexium Pharmaceuticals, LLC issued and outstanding immediately prior to the corporate
conversion will be converted automatically into 5,375,377 shares of Dipexium Pharmaceuticals, Inc.;
- •
- all of the outstanding warrants to purchase Class A membership interests of Dipexium Pharmaceuticals, LLC will become warrants to purchase shares of common stock of Dipexium Pharmaceuticals, Inc. in a ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each Class A membership interest of Dipexium Pharmaceuticals, LLC underlying such
4
- •
- the exercise price of all of the outstanding warrants will be adjusted in the same ratio as the seven-for-one conversion ratio noted above such that all of our outstanding warrants to purchase Class A membership interests of Dipexium Pharmaceuticals, LLC which are currently exercisable at $60 per Class A membership interest will automatically be adjusted such that the new exercise price for the outstanding warrants upon consummating the corporate conversion will be $8.57 per share, subject to certain adjustments noted in each of the warrants.
warrants, with the effect that warrants to purchase 4,900 Class A membership interests of Dipexium Pharmaceuticals, LLC outstanding immediately prior to the corporate conversion will automatically convert into warrants to purchase 34,300 shares of Dipexium Pharmaceuticals, Inc. upon consummation of the corporate conversion; and
The purpose of the corporate conversion is to reorganize our corporate structure so that our company will continue as a corporation rather than a limited liability company following this offering, and so that our existing investors will own our common stock rather than equity interests in a limited liability company. For further information regarding the corporate conversion, see "Corporate Conversion."
Risks Associated with our Business
Our business and ability to execute our business strategy are subject to a number of risks of which you should be aware before you decide to buy our common stock. In particular, you should consider the following risks, which are discussed more fully in the section entitled "Risk Factors" in this prospectus:
- •
- We are heavily dependent on attaining regulatory approval for and, if approved, successfully commercializing
Locilex™. Locilex™ is our only product candidate. As such, all of our resources and efforts have been and are expected for the foreseeable future to be
dedicated to the development and commercialization of Locilex™. As such, we are subject to the risk of dependency on this sole product, and if our efforts fail to gain FDA or other
regulatory approvals for Locilex™ (for example, for use in treating mild or moderate skin and skin structure infections in superficial wounds), our viability would be materially impacted
unless we were able to develop or acquire other product candidates.
- •
- The regulatory approval process for Locilex™ may be lengthy and is inherently
unpredictable. Although we anticipate certain time frames for our regulatory pathway for Locilex™, the approval process of the FDA and comparable foreign regulatory
authorities may be lengthy and time consuming. The process also is inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for Locilex™, our business will
be substantially harmed.
- •
- Manufacturing issues may prevent Locilex™ from receiving regulatory
approval. Although we believe that we have successfully worked with our third-party vendors to resolve the manufacturing issues
encountered by Locilex™'s prior sponsor and previously identified by the FDA, to the extent that such issues are not resolved, regulatory approval for Locilex™ may be delayed
or withheld, and we may not be able to meet the developmental milestones necessary to continue our business.
- •
- Even if Locilex™ is approved, the market may not accept it as a viable treatment
option. Even if Locilex™ is approved, there is a risk that it may not be accepted in the marketplace for a variety of reasons. If we are unable to generate revenue
from sales of Locilex™, our business will be substantially harmed.
- •
- We have a very limited operating history, have not generated revenues to date, and have incurred significant losses since our inception. We were formed in 2010, and we have not yet generated any revenues from product sales or otherwise. As such, you have limited data on which to evaluate our past performance and ability to progress our business plan forward. Moreover, we do not expect to generate revenues, and we expect to incur losses, for the foreseeable future and may never achieve or maintain profitability.
5
- •
- We rely on single source third-party contract manufacturing and contract research organizations to manufacture and supply
Locilex™ for us and conduct our clinical trials. We are a small organization with limited full time employees, and thus we rely and will continue to rely on key
third party vendors to, among other functions, manufacture, formulate and package Locilex™ and conduct our upcoming clinical trials. If one of our suppliers, manufacturers or vendors fails
to perform adequately or fulfill our needs, or if our agreements with these parties are terminated, we may be required to incur significant costs and devote significant efforts to find new suppliers
or manufacturers. We may also face delays in the development and commercialization of Locilex™ or other product candidates that we may develop or acquire in the future.
- •
- We may have a need for funding beyond this offering to reach profitability. Even following this
offering, if we are unable to raise capital if and when needed, we could be forced to delay, reduce or eliminate one or more aspects of our development or commercial activities.
- •
- We are subject to risks associated with clinical trials and regulatory oversight. The conduct of
clinical trials is inherently subject to numerous risks and uncertainties, some of which will be beyond our control. If clinical trials of Locilex™, or any other product candidate that we
may develop in the future, fail to demonstrate safety and efficacy to the satisfaction of the FDA or similar regulatory authorities outside the U.S. or do not otherwise produce positive results, we
may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of Locilex™ or any other product candidate we may
develop in the future. In addition, the pharmaceutical industry is intensely regulated. Compliance with such regulations is costly and time consuming, and our failure to comply with such regulations
would have a material adverse effect on our ability to implement our business plans.
- •
- Our future viability depends on the strength of our intellectual property. Due to the exclusivity
that intellectual property protection can afford, our commercial success depends to a material extent on our ability to maintain adequate protection for our intellectual property for
Locilex™ (and any other product candidates we may develop in the future) in the U.S. and other countries. We consider our U.S. Patent No. 8,530,409, relating to our new proprietary
formulation and methods of use for Locilex™, to be particularly important to our company. If we are unable to protect U.S. Patent No. 8,530,409 or other intellectual property rights
for Locilex™, or our intellectual property for any future product candidates, it may materially and adversely affect our ability to market and generate sales of the product.
- •
- Our current and future viability depends on our existing management and our ability to attract and retain other key employees. Given their history with our company, we are materially reliant on our current management, including David P. Luci and Robert J. DeLuccia. The loss of their services would have a materially adverse impact on our ability to progress our business plan. Moreover, as we grow, we will be required to attract and retain other key employees. Our inability to do so could also have a material adverse impact on our company.
Corporate Information
We were organized originally as a limited liability company under the laws of the State of Delaware in January 2010 and will convert to a corporation immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. Our principal executive office is located at 74 Broad Street, New York, New York 10004, and our phone number is (212) 422-5717.
Implications of Being an Emerging Growth Company
We qualify as an "emerging growth company" as defined in the Jumpstart Our Business Startups Act of 2012 (or the JOBS Act). As a result, we are permitted to, and intend to, rely on exemptions from
6
certain disclosure requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
- •
- being permitted to present only two years of audited financial statements and only two years of related "Management's
Discussion and Analysis of Financial Condition and Results of Operations" in this prospectus;
- •
- not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of
2002, as amended (or the Sarbanes-Oxley Act);
- •
- reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration
statements; and
- •
- exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an emerging growth company to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this extended transition period.
We will remain an emerging growth company until the earliest to occur of: (i) our reporting $1 billion or more in annual gross revenues; (ii) the end of fiscal year 2018; (iii) our issuance, in a three year period, of more than $1 billion in non-convertible debt; and (iv) the end of the fiscal year in which the market value of our common stock held by non-affiliates exceeded $700 million on the last business day of our second fiscal quarter.
7
| Common stock offered by us | 2,307,693 shares | |
Common stock to be outstanding after this offering |
7,683,070 shares (or 8,029,224 shares if the underwriters exercise their over-allotment option in full). |
|
Over-allotment option |
We have granted the underwriters a 30-day option to purchase up to an additional 346,154 shares of our common stock at the initial public offering price to cover over-allotments, if any. |
|
Use of proceeds |
We expect to use the proceeds received from the offering to further develop Locilex™ including, without limitation, conducting our pivotal Phase 3 clinical studies, commencing preliminary commercialization activities, expanding uses of Locilex™, as well as for working capital and general corporate purposes. See the "Use of Proceeds" section in this prospectus for a more complete description of the intended use of proceeds from this offering. |
|
Concentration of ownership |
Upon completion of this offering, our executive officers and directors will beneficially own, in the aggregate, approximately 52.1% of the outstanding shares of our common stock. |
|
Proposed NASDAQ Capital Market symbol |
"DPRX" |
|
Risk Factors |
Investing in our common stock involves a high degree of risk. See "Risk Factors" beginning on page 9 and the other information in this prospectus for a discussion of the factors you should consider carefully before you decide to invest in our common stock. |
All information in this prospectus assumes the underwriters do not exercise their over-allotment option. The total number of shares of our common stock outstanding assuming our corporate conversion took place on March 6, 2014 is 5,375,377 and excludes the following (numbers presented on a post-conversion basis):
- •
- 70,000 shares of common stock issuable to members of our board of directors which are currently unvested and, in each
case, vest over a period of time for service on the board of directors;
- •
- 34,300 shares of common stock issuable upon conversion of warrants issued to investors in prior financings, in each case,
with a conversion price equal to $8.57 per share;
- •
- 1,536,614 shares of our common stock (which is equal to 20% of our issued and outstanding common stock immediately after the consummation this offering) reserved for future issuance under our 2013 Equity Incentive Plan, which will become effective as of the closing of this offering.
Summary Financial Information
The following summary financial data as of and for the years ended December 31, 2013 and 2012, have been derived from our audited financial statements included in this prospectus. The financial data set forth below should be read in conjunction with "Management's Discussion and Analysis of
8
Financial Condition and Results of Operations" and the financial statements and notes thereto included elsewhere in this prospectus.
| |
Year Ended December 31, 2013 |
Year Ended December 31, 2012 |
||||||
|---|---|---|---|---|---|---|---|---|
| |
(in thousands) |
|||||||
Statement of Operations Data: |
||||||||
OPERATING EXPENSES: |
||||||||
Research and Development |
$ | 1,730 | $ | 845 | ||||
General and Administrative |
2,284 | 999 | ||||||
TOTAL OPERATING EXPENSES |
4,014 | 1,844 | ||||||
LOSS FROM OPERATIONS |
4,014 | 1,844 | ||||||
NET LOSS |
4,014 | $ | 1,844 | |||||
| |
As of December 31, 2013 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Actual | Pro Forma, Warrant Exchange(1) |
Pro Forma, Corporate Conversion(2) |
Pro Forma, As Adjusted(3) |
||||||||||
| |
(in thousands) |
|||||||||||||
Balance Sheet Data: |
||||||||||||||
Current Assets |
$ |
3,907 |
3,907 |
3,907 |
31,105 |
|||||||||
Total Assets |
4,030 | 4,030 | 4,030 | 31,105 | ||||||||||
Total Liabilities |
824 | 824 | 824 | 701 | ||||||||||
Total Members' Equity |
3,206 | 3,206 | — | — | ||||||||||
Shareholders' Equity(4) |
— | 3,206 | 30,404 | |||||||||||
Total Liabilities and Members' Equity/Shareholders' Equity |
4,030 | 4,030 | 4,030 | 31,105 | ||||||||||
- (1)
- The
December 31, 2013 pro forma (warrant exchange) balance sheet data reflects the February 2014 issuance of an aggregate of 23,719 Class A
membership interests to investors in our prior financings in exchange for the redemption of outstanding warrants to purchase an aggregate of 89,900 Class A membership interests in our company.
As the warrants and membership interests are both classified as equity instruments, the exchange resulted in no impact to Total Members' Equity.
- (2)
- The
December 31, 2013 pro forma (corporate conversion) balance sheet data reflects our conversion from a limited liability company to a corporation,
and, in connection therewith, the conversion of 767,911 Class A membership interests in our company issued and outstanding immediately prior to the conversion into an aggregate of
5,375,377 shares of common stock.
- (3)
- The
December 31, 2013 pro forma as adjusted balance sheet data reflects the sale of shares of our common stock in this offering, assuming an initial
public offering price of $13.00 per share (the mid-point of the price range set forth on the cover page of this prospectus), after deducting estimated underwriting discounts and commissions and
estimated offering expenses payable by us.
- (4)
- Reflects the conversion of Members' Equity following our conversion from a limited liability company to a corporation.
9
Any investment in our securities involves a high degree of risk. You should carefully consider the risks described below, which we believe represent certain of the material risks to our business, together with the information contained elsewhere in this prospectus, before you make a decision to invest in our shares of common stock. If any of the following events occur, our business, financial condition and operating results may be materially adversely affected. In that event, the trading price of our securities could decline and you could lose all or part of your investment.
Risks Relating to Our Business and Industry
We have a very limited operating history and are expected to incur significant operating losses during the early stage of our corporate development.
We were organized on January 14, 2010 and we acquired the rights to our product candidate Locilex™ in April 2010. Accordingly, we have a limited operating history. We have not sold our product candidate because it is currently investigational in nature and requires completion of additional clinical and non-clinical trials and studies in order to obtain regulatory approval in the U.S. and abroad. Therefore, our historical financial information consists only of an audit of our financial results at and for the years ended December 31, 2012 and 2013. This is very limited historical financial information upon which to base an evaluation of our performance. We are an emerging company, and thus our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operation, particularly in the pharmaceutical industry. We have generated cumulative losses of approximately $8.7 million since inception, and we expect to continue to incur losses until Locilex™ is approved by the FDA and foreign regulatory authorities. Even if regulatory approval is obtained, there is a risk that we will not be able to generate material sales of Locilex™, which would cause us to continue to incur losses. We thus expect to incur substantial operating expenses over the next several years as our product development and marketing activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain.
We have never generated revenue, may never generate revenue, are not profitable and may never become profitable.
We expect to incur substantial losses and negative operating cash flow for the foreseeable future, and we may never achieve or maintain profitability. Even if we are able to launch Locilex™, we expect to incur substantial losses for the foreseeable future and may never become profitable.
As we have no operating revenue, we also expect to experience negative cash flow for the foreseeable future as we continue to fund our operating losses and capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability would negatively impact the value of your securities and potentially require us to shut down our business, which would result in the loss of your investment.
Given our lack of revenue and cash flow, we may need to raise additional capital, which may be unavailable to us or, even if consummated, may cause dilution or place significant restrictions on our ability to operate.
Since we will be unable to generate sufficient, if any, revenue or cash flow to fund our operations for the foreseeable future, we may need to seek additional equity or debt financing to provide the capital required to maintain or expand our operations. We may also need additional funding to continue the
10
development of Locilex™, increase our sales and marketing capabilities, promote brand identity, or develop or acquire complementary companies, technologies and assets, as well as for working capital requirements and other operating and general corporate purposes. Moreover, the regulatory compliance arising out of being a publicly registered company will dramatically increase our costs.
We do not currently have any arrangements or credit facilities in place as a source of funds, and there can be no assurance that we will be able to raise sufficient additional capital if needed on acceptable terms, or at all. If such financing is not available on satisfactory terms, or is not available at all, we may be required to delay, scale back or eliminate the development of business opportunities and our ability to achieve our business objectives, our competitiveness, and our operations and financial condition may be materially adversely affected. Our inability to fund our business could thus lead to the loss of your investment.
If we raise additional capital by issuing equity securities, the percentage ownership of our existing stockholders may be reduced, and accordingly these stockholders may experience substantial dilution. We may also issue equity securities that provide for rights, preferences and privileges senior to those of our common stock. Given our need for cash and that equity issuance is the most common type of fundraising for companies like ours, the risk of dilution is particularly significant for stockholders of our company.
Debt financing, if obtained, may involve agreements that include liens on our assets and covenants limiting or restricting our ability to take specific actions, such as incurring additional debt. Debt financing would also be required to be repaid regardless of our operating results.
If we raise additional funds through collaborations and licensing arrangements, we may be required to relinquish some rights to Locilex™, or to grant licenses on terms that are not favorable to us.
We have no experience as a company in obtaining regulatory approval for, or commercializing, any product candidate.
As a company, we have never obtained regulatory approval for, or commercialized, any product candidate. It is possible that the FDA may refuse to accept our planned NDA for Locilex™ for substantive review, or may conclude after review of our data that our application is insufficient to obtain regulatory approval of Locilex™ or any future product candidates. If the FDA does not accept or approve our planned NDA for Locilex™, it may require that we conduct additional clinical, preclinical or manufacturing validation studies, which may be costly, and submit that data before it will reconsider our applications. Depending on the extent of these or any other FDA required studies, approval of any NDA or application that we submit may be significantly delayed, possibly for several years, or may require us to expend more resources than we have available. Any delay in obtaining, or an inability to obtain, regulatory approvals would prevent us from commercializing Locilex™, generating revenues and achieving and sustaining profitability. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA to approve any NDA we submit. If any of these outcomes occur, we may be forced to abandon our planned NDA for Locilex™, which would materially adversely affect our business and could potentially cause us to cease operations. We face similar risks for any approval in a foreign jurisdiction.
Our current and future operations substantially depend on our management team and our ability to hire other key personnel, the loss of any of whom could disrupt our business operations.
Our business depends and will continue to depend in substantial part on the continued service of David P. Luci and Robert J. DeLuccia. Upon the consummation of this offering, Messrs. Luci and DeLuccia will be parties to employment agreements for an initial three year term of service, subject to automatic one year renewals absent termination with at least six months notice. Such agreements may also be terminated by us with or without Cause (as defined in the employment agreements) and by each of Messrs. Luci or DeLuccia voluntarily or with Good Reason (as defined in the employment
11
agreements). The loss of the services of either of these individuals would significantly impede implementation and execution of our business strategy and may result in the failure to reach our goals. We do not carry key person life insurance on any of our management, which would leave our company uncompensated for the loss of any of our management.
Our future viability and ability to achieve sales and profit will also depend on our ability to attract, retain and motivate highly qualified personnel in the diverse areas required for continuing our operations. There is a risk that we will be unable to attract, train or retain qualified personnel, both near term or in the future, and our failure to do so may severely damage our prospects.
Our independent registered public accounting firm, in their audit report related to our financial statements for the fiscal year ended December 31, 2013, expressed substantial doubt about our ability to continue as a going concern.
As a result of our continued losses, our independent registered public accounting firm has included an explanatory paragraph in its report on our financial statements for the fiscal year ended December 31, 2013, expressing substantial doubt as to our ability to continue as a going concern. The inclusion of a going concern explanatory paragraph in the report of our independent registered public accounting firm may make it more difficult for us to secure additional financing or enter into strategic relationships on terms acceptable to us, if at all, and may materially and adversely affect the terms of any financing that we might obtain.
There is a risk that Locilex™ will not receive regulatory approval, and without regulatory approval we will not be able to market Locilex™.
Our business currently depends entirely on the successful development and commercialization of Locilex™. Our ability to generate revenue related to product sales, if ever, will depend on the successful development and regulatory approval of Locilex™ for the treatment of Mild DFI.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. The development of a product candidate and issues relating to its approval and marketing are subject to extensive regulation by the FDA in the U.S., the European Medicines Agency (or EMA) in Europe and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the U.S. or Europe until we receive approval of a NDA from the FDA or a Marketing Authorisation Application (or MAA) from the EMA, respectively.
NDAs and MAAs must include extensive data and supporting information to establish the product candidate's safety and effectiveness for each desired indication. NDAs and MAAs must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of a NDA or a MAA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA and the EMA review processes can take years to complete and approval is never guaranteed. If we submit a NDA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA. Regulators of other jurisdictions, such as the EMA, have their own procedures for approval of product candidates.
Even if a product is approved, the FDA or the EMA, as the case may be, may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the U.S. and Europe also have requirements for approval of drug candidates with which we must comply prior to marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country.
12
In addition, delays in approvals or rejections of marketing applications in the U.S., Europe or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, preclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding Locilex™ or other product candidates we may develop or acquire in the future. Also, regulatory approval for Locilex™ or other product candidates we may develop or acquire in the future may be withdrawn.
Before we submit a NDA to the FDA or a MAA to the EMA for Locilex™ for Mild DFI, we must successfully complete two Phase 3 trials and two Phase 1 trials. We cannot predict whether our future trials and studies will be successful or whether regulators will agree with our conclusions regarding the preclinical studies and clinical trials we have conducted to date.
If we are unable to obtain approval from the FDA, the EMA or other regulatory agencies for Locilex™ and any other product candidates we may develop or acquire in the future, or if, subsequent to approval, we are unable to successfully commercialize Locilex™ or our other product candidates we may develop or acquire in the future, we will not be able to generate sufficient revenue to become profitable or to continue our operations.
Locilex™'s prior sponsor encountered certain difficulties in manufacturing Locilex™ on a commercial scale, and there is a risk that we will continue to experience manufacturing issues that may prevent Locilex™ from receiving regulatory approval.
In the late 1990s, a prior formulation of Locilex™ was tested by Magainin, Locilex™'s prior sponsor, with over 1,000 human subjects exposed to such prior formulation of Locilex™, including 835 evaluable patients in two Phase 3 clinical trials. The FDA Advisory Committee reviewing Locilex™ at the time unanimously approved the safety of the product, but did not approve its efficacy and recommended an additional Phase 3 placebo controlled trial. In its 1999 non-approvable letter, the FDA identified certain cGMP manufacturing deficiencies, namely stability and quality control issues, and questions regarding the comparability of the product used in the Phase 3 program versus that which was produced at commercial scale.
Since we acquired rights to Locilex™, we have worked with our third-party vendors to address the stability and purity concerns previously articulated by the FDA. However, we cannot guarantee that such deficiencies will be deemed resolved to the FDA's satisfaction, that the FDA or other regulatory agencies will not identify new manufacturing concerns, or that, once regulatory approval is granted, our manufacturers will be able to comply with applicable regulations to maintain the quality of our product. If we are unable to resolve any outstanding manufacturing issues, the cost and timing for achieving regulatory approval could materially increase and could prevent us from meeting the developmental milestones necessary to maintain the viability of our business.
Even if Locilex™ gains regulatory approval, it may never achieve market acceptance or any level of commercial success. Our failure to achieve market acceptance will prevent or delay our ability to generate material revenues.
Our future financial performance will depend, to a large extent, upon the introduction and physician and patient acceptance of our product candidate, Locilex™. Even if approved for marketing by the necessary regulatory authorities, Locilex™ may not achieve market acceptance or reimbursement by Medicare, Medicaid or third party payors.
The degree of market acceptance for Locilex™ will depend upon a number of factors, including:
- •
- regulatory clearance of marketing claims for the uses that we are developing;
- •
- demonstration of the advantages, safety and efficacy for Locilex™;
13
- •
- pricing and reimbursement policies of government and third-party payors such as insurance companies, health maintenance
organizations and other health plan administrators;
- •
- our ability to attract corporate partners, including pharmaceutical companies, to assist in commercializing our
formulation of Locilex™; and
- •
- our ability to timely and effectively manufacture and market Locilex™, either on our own or through third parties.
Physicians, various other healthcare providers, patients, payors or the medical community in general may be unwilling to accept, utilize or recommend Locilex™. If we are unable to obtain regulatory approval, or are unable (either on our own or through third parties) to manufacture, commercialize and market Locilex™ or any future product candidates we may develop or acquire when planned, we may not achieve any market acceptance or generate revenue, which could cause our business to fail.
Our failure to complete or meet key milestones relating to the development of Locilex™ or other product candidates we may develop or acquire in the future would significantly impair the viability of our company.
In order to be commercially viable, we must successfully research, develop, obtain regulatory approval for, manufacture, introduce, market and distribute Locilex™ and, if applicable, any future product candidates we may develop. With respect to Locilex™ in particular, we must meet a number of critical developmental milestones, including:
- •
- demonstration, through clinical trials, that Locilex™ is safe and effective;
and
- •
- establishment of a viable Good Manufacturing Process capable of potential scale-up.
The estimated required capital and time-frames necessary to achieve these developmental milestones as described in this prospectus or as we may state from time to time is subject to inherent risks, many of which may be beyond our control. As such, we may not be able to achieve these or similar milestones for Locilex™ or any product candidate we may develop or acquire in the future. Our failure to meet these or other critical milestones would adversely affect the viability of our company.
Conducting and completing the clinical trials necessary for FDA approval is costly and subject to intense regulatory scrutiny as well as the risk of failing to meet the primary endpoints of such trials. We will not be able to commercialize and sell Locilex™ without completing such trials.
In order to conduct clinical trials that are necessary to obtain approval by the FDA to market Locilex™ or any other product candidate we may develop or acquire in the future, it is necessary to receive clearance from the FDA to conduct such clinical trials. We must conduct two Phase 3 and two Phase 1 clinical trials for Locilex™. We have not commenced these trials as of the date of this prospectus, and we may not be able to commence, enroll sufficient patients in, properly conduct or complete such trials. If we can not achieve all of these goals, our viability will be materially impaired.
In addition, the FDA can halt clinical trials at any time during the conduct of clinical trials for safety reasons or because we or our clinical investigators did not follow the FDA's requirements for conducting clinical trials. If we commence our clinical trials and trials are permanently halted by the FDA, we would not be able to achieve any revenue from such product as it is illegal to sell any drug for human consumption or use without FDA approval. If our trials are temporarily halted by the FDA, the cost and timing for potentially achieving regulatory approval could be materially increased.
Moreover, there is a risk that our clinical trials will fail to meet their primary endpoints, which would make them unacceptable in having Locilex™ approved by the FDA. If this were to occur, the announcement of such an event would very likely cause our public stock price to decrease, perhaps significantly, and such event would otherwise materially and adversely affect our business, results of operations and viability.
14
Locilex™ may have undesirable side effects which may delay or prevent marketing approval, or, if approval is received, require it to be taken off the market, require it to include safety warnings or otherwise limit sales of the product.
Unforeseen side effects from Locilex™ could arise either during clinical development or, if approved, after Locilex™ has been marketed. This could cause regulatory approvals for, or market acceptance of, Locilex™ harder and more costly to obtain.
To date, no serious adverse events have been attributed to Locilex™, though approximately 12% of patients in prior clinical trials experienced serious adverse events unrelated to Locilex™. The results of our planned or any future clinical trials may show that Locilex™ causes undesirable or unacceptable side effects, which could interrupt, delay or halt clinical trials, and result in delay of, or failure to obtain, marketing approval from the FDA and other regulatory authorities, or result in marketing approval from the FDA and other regulatory authorities with restrictive label warnings.
If Locilex™ receives marketing approval and we or others later identify undesirable or unacceptable side effects caused by the use of Locilex™:
- •
- regulatory authorities may withdraw their approval of the product, which would force us to remove Locilex™
from the market;
- •
- regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field
alerts to physicians and pharmacies;
- •
- we may be required to change instructions regarding the way the product is administered, conduct additional clinical
trials or change the labeling of the product;
- •
- we may be subject to limitations on how we may promote the product;
- •
- sales of the product may decrease significantly;
- •
- we may be subject to litigation or product liability claims; and
- •
- our reputation may suffer.
Any of these events could prevent us or our potential future collaborators from achieving or maintaining market acceptance of Locilex™ or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of Locilex™.
We currently have no marketing and sales organization and have no experience as a company in marketing pharmaceutical products. If we are unable to establish our own marketing and sales capabilities, or enter into agreements with third parties, to market and sell our products after they are approved, we may not be able to generate product revenues.
We do not have a sales organization for the marketing, sales and distribution of any pharmaceutical products. In order to commercialize Locilex™ or any other product candidate we may develop or acquire in the future, we must develop these capabilities on our own or make arrangements with third parties for the marketing, sales and distribution of our products. The establishment and development of our own sales force would be expensive and time consuming and could delay any product launch, and we cannot be certain that we would be able to successfully develop this capability. As a result, we may seek one or more licensing partners to handle some or all of the sales and marketing of Locilex™ in the U.S. and elsewhere. There also may be certain markets within the U.S. for Locilex™ for which we may seek a co-promotion arrangement. However, we may not be able to enter into arrangements with third parties to sell Locilex™ on favorable terms or at all. In the event we are unable to develop our own marketing and sales force or collaborate with a third-party marketing and sales organization, we would not be able to commercialize Locilex™ or any other product candidates that we develop, which would negatively impact our ability to generate product revenues. Furthermore, whether we
15
commercialize products on our own or rely on a third party to do so, our ability to generate revenue will be dependent on the effectiveness of the sales force. In addition, to the extent we rely on third parties to commercialize our approved products, we will likely receive less revenues than if we commercialized these products ourselves.
We rely heavily on third parties for conducting clinical trials, marketing and distributing Locilex™.
We presently are party to, and expect that we will be required to enter into, agreements with commercial partners to perform clinical trials for us and to engage in sales, marketing and distribution efforts for Locilex™ or other product candidates we may acquire in the future. We may be unable to establish or maintain third-party relationships on a commercially reasonable basis, if at all. In addition, these third parties may have similar or more established relationships with our competitors or other larger customers. Moreover, the loss for any reason of one or more of these key partners could have a significant and adverse impact on our business. If we are unable to obtain or retain third party sales and marketing vendors on commercially acceptable terms, we may not be able to commercialize Locilex™ as planned and we may experience delays in or suspension of our marketing launch of Locilex™. The same could apply to other product candidates we may develop or acquire in the future. Our dependence upon third parties may adversely affect our ability to generate profits or acceptable profit margins and our ability to develop and deliver such products on a timely and competitive basis.
We rely on single source third-party contract manufacturing organizations to manufacture and supply Locilex™ for us. If one of our suppliers or manufacturers fails to perform adequately or fulfill our needs, or if these agreements are terminated by the third parties, we may be required to incur significant costs and devote significant efforts to find new suppliers or manufacturers. We may also face delays in the development and commercialization of Locilex™ or other product candidates that we may develop or acquire in the future.
We currently have limited experience in, and we do not own facilities for, manufacturing Locilex™ or any other product candidate we may develop or acquire in the future. We rely upon single source third-party contract manufacturing organizations to manufacture and supply large quantities of our product candidates. We currently utilize PolyPeptide Laboratories, Inc. for the bulk manufacturing of Locilex™ and DPT Laboratories, Inc. for the formulation of Locilex™. In addition, we are using Almac Group Limited to label the 15 gram tubes of Locilex™ and distribute as we direct.
The manufacture of pharmaceutical products in compliance with current good manufacturing practice (or cGMP) regulations requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, including difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, or shortages of qualified personnel. If our manufacturers were to encounter any of these difficulties or otherwise fail to comply with their obligations to us or under applicable regulations, our ability to provide study materials in our preclinical studies and clinical trials would be jeopardized. Any delay or interruption in the supply of clinical trial materials could delay the completion of our clinical trials, increase the costs associated with maintaining our clinical trial programs and, depending upon the period of delay, require us to commence new trials at significant additional expense or terminate the studies and trials completely.
Our suppliers and manufacturers for Locilex™ must comply with cGMP requirements enforced by the FDA through its facilities inspection program. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. Manufacturers of our component materials may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. The FDA or similar foreign regulatory agencies at any time may also implement new standards, or change their interpretation and enforcement of
16
existing standards for manufacture, packaging or testing of products. We have little control over our manufacturers' compliance with these regulations and standards. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any product supplied is compromised due to our manufacturers' failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our products, and we may be held liable for any injuries sustained as a result. Any of these factors could cause a delay of clinical trials, regulatory submissions, approvals or commercialization of Locilex™ or any product candidates we may develop or acquire in the future or entail higher costs or impair our reputation.
Our current agreements with our suppliers do not provide for the entire supply of the bulk drug necessary for full-scale commercialization. In the event that we and our suppliers cannot agree to the terms and conditions for them to provide some or all of our bulk drug clinical and commercial supply needs, or if any single-source supplier terminates the agreement in response to a breach by us, we would not be able to manufacture the bulk drug on a commercial scale until a qualified alternative supplier is identified, which could also delay the development of, and impair our ability to commercialize, Locilex™ or any product candidates we may develop or acquire in the future.
The number of third-party suppliers with the necessary manufacturing and regulatory expertise and facilities for our company is limited, and it could be expensive and take a significant amount of time to arrange for alternative suppliers, which could have a material adverse effect on our business. New suppliers of any bulk drug would be required to qualify under applicable regulatory requirements and would need to have sufficient rights under applicable intellectual property laws to the method of manufacturing such ingredients. Obtaining the necessary FDA approvals or other qualifications under applicable regulatory requirements and ensuring non-infringement of third-party intellectual property rights could result in a significant interruption of supply and could require the new manufacturer to bear significant additional costs which may be passed on to us.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
As of March 6, 2014, we had seven employees and consultants, of which two are full-time employees and five are part-time employees or consultants. We will need to expand our managerial, operational, financial and other resources in order to manage our operations and clinical trials, continue our development activities and commercialize Locilex™ and any other product candidates we may develop or acquire in the future. Our management and personnel, systems and facilities currently in place are not adequate to support this future growth. Our need to effectively execute our business strategy requires that we:
- •
- manage our clinical trials effectively, including two Phase 3 clinical trials and two Phase 1 clinical
trials for Locilex™. The Phase 3 clinical trials are expected to be conducted at up to 40 trial sites in the U.S. and are expected to be managed on our behalf by RRD
International, LLC;
- •
- manage our internal development efforts effectively while carrying out our contractual obligations to licensors,
contractors, collaborators, government agencies and other third parties;
- •
- continue to improve our operational, financial and management controls, reporting systems and procedures;
and
- •
- identify, recruit, maintain, motivate and integrate additional employees.
If we are unable to expand our managerial, operational, financial and other resources to the extent required to manage our development and commercialization activities, our business will be materially adversely affected.
17
We are exposed to product liability, non-clinical and clinical liability risks which could place a substantial financial burden upon us, should lawsuits be filed against us.
Our business exposes us to potential product liability and other liability risks that are inherent in the testing, manufacturing and marketing of pharmaceutical formulations and products. We expect that such claims are likely to be asserted against us at some point. In addition, the use in our clinical trials of pharmaceutical formulations and products and the subsequent sale of these formulations or products by us or our potential collaborators may cause us to bear a portion of or all product liability risks. We currently have $3 million in insurance coverage relating to personal injury, product liability, medical expenses, and office premises. However, any claim under such insurance policies may be subject to certain exceptions, and may not be honored fully, in part, in a timely manner, or at all, and may not cover the full extent of liability we may actually face. Therefore, a successful liability claim or series of claims brought against us could have a material adverse effect on our business, financial condition and results of operations.
We and our management are parties to a lawsuit which, if adversely decided against, could impact our rights to Locilex™.
In April 2010, we acquired the worldwide rights to develop pexiganan, the active pharmaceutical ingredient in Locilex™, from Genaera Liquidating Trust, which was put in place to liquidate the assets of Genaera Corporation. In June 2012, we, along with our two senior executives and several other unrelated defendants, were sued in the Federal District Court for the Eastern District of Pennsylvania by a former shareholder of Genaera Corporation and purported to be on behalf of other Genaera Corporation shareholders, alleging, in pertinent part, that our company's acquisition of the rights to pexiganan (the active ingredient in Locilex™, and which rights included the rights to the prior formulation of Locilex™) was for what was alleged to be inadequate consideration, and as a result, it was alleged that we and our senior executives aided and abetted a breach of fiduciary duty by Genaera Corporation and the Genaera Liquidating Trust to the former shareholders of Genaera Corporation. It was also alleged that we and our senior executives aided and abetted a breach of the duty of the trustee at common law and under a certain trust agreement which was alleged to exist and which was executed by Argyce LLC (or Argyce), as trustee. The agreement called for Argyce to create the Genaera Liquidating Trust pursuant to which Argyce apparently was appointed to liquidate the assets formerly held by Genaera Corporation. One of these assets was pexiganan, which we acquired via public auction conducted by Argyce on behalf of the Genaera Liquidating Trust.
As of the date of this prospectus, the case against our company and our senior executives has been dismissed with prejudice, and a subsequent motion to reconsider such dismissal has been denied. Prior to the dismissal there was no request or action to seek class certification by the plaintiff though it was purportedly filed on behalf of other former Genaera Corporation shareholders. Plaintiff has appealed the dismissal of the suit as well as the denial of the motion to reconsider. The appeal has been filed in the United States Court of Appeals for the Third Circuit. As of the date of this prospectus, briefing is underway in the appellate court.
If the appellate court reverses the decision of the underlying federal court, a civil case would be reinstated and if we were to lose such case, our rights to the prior formulation of Locilex™ could be lost, which may impair the commercial viability of our product or the timeline to potential regulatory approval. If we were required to settle the case, we may lose certain rights to Locilex™ or be required to pay damages, which could have a material adverse effect on our company, our business plans and results of operations.
18
We may form strategic alliances in the future with respect to our Locilex™ development program and we may not realize the benefits of such alliances.
We may form one or more strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with one or more third parties with respect to our Locilex™ drug development program that we believe will complement or augment our existing business. For example, we may attempt to find a partner for licensing, development and/or commercialization of Locilex™ in one or more Asian territories, or other unpartnered areas of the world. We routinely engage, and are engaged, in partnering discussions with a range of pharmaceutical and biotechnology companies and could enter into new collaborations at any time. We face significant competition in seeking appropriate strategic partners, and the negotiation process to secure appropriate terms is time-consuming and complex. Any delays in identifying suitable development partners and entering into agreements to develop our product candidates could also delay the commercialization of our product candidates, which may reduce their competitiveness even if they reach the market.
In addition, we may be unable to establish such a strategic partnership for any future product candidates and programs on terms that are acceptable to us, or at all. This may be because our product candidates and programs may be deemed to be at too early of a stage of development for collaborative effort, our research and development pipeline may be viewed as insufficient, and/or third parties may not view our product candidates and programs as having sufficient potential for commercialization. Even if we are able to enter into a strategic alliance or license arrangement with one or more third parties, there is a risk that the collaboration will not achieve any measure of success, or that any future partner will commit sufficient resources to the development, regulatory approval, and commercialization effort for such products, or that such alliances will result in us achieving revenues that justify such transactions.
Trends toward managed healthcare and downward price pressures on medical products and services may limit our ability to profitably sell Locilex™ or other product candidates we may develop or acquire in the future.
Lower prices for pharmaceutical products may result from:
- •
- third-party payors' increasing challenges to the prices charged for medical products and services;
- •
- the trend toward managed healthcare in the U.S. and the concurrent growth of HMOs and similar organizations that can
control or significantly influence the purchase of healthcare services and products; and
- •
- legislative proposals to reform healthcare or reduce government insurance programs.
The cost containment measures that healthcare providers are instituting, including practice protocols and guidelines and clinical pathways, and the effect of any healthcare reform, could limit our ability to profitably sell Locilex™ or any other products that we may develop or acquire which obtain FDA approval. Moreover, any future legislation or regulation, if any, relating to the healthcare industry or third-party coverage and reimbursement, may adverse impact our ability to generate sales revenue and cause our business to suffer.
We will compete with larger and better capitalized companies, and competitors in the drug development or pharmaceutical industries may develop competing products which outperform or supplant Locilex™.
Competition in the worldwide pharmaceutical industry, including the sector for antibiotic agents that we will operate in, is intense. Pharmaceutical companies and/or other technology companies have developed (and are currently marketing in competition with us), have sought to develop and may in
19
the future seek to develop or acquire and market other products that may successfully treat skin or wound infections which do and may compete with Locilex™. Competitors have developed and may in the future develop similar or different products which may become more accepted by the marketplace or which may supplant Locilex™ entirely. In addition, many of our current competitors are, and future competitors may be, significantly larger and better financed than we are, thus giving them a significant advantage. With respect to Mild, Moderate or Severe DFI specifically, such competitors include very large international organizations such as Pfizer, Inc., Eli Lilly and Company, Johnson & Johnson and GlaxoSmithKline plc. We may be unable to respond to competitive forces presently in the marketplace (including competition from larger companies), which would severely impact our business.
If ultimate users of Locilex™ or any other product candidates we may develop or acquire in the future are unable to obtain adequate reimbursement from third-party payors, or if new restrictive legislation is adopted, market acceptance of our proposed products may be limited and we may not achieve material revenues.
The continuing efforts of government and insurance companies, health maintenance organizations and other payors of healthcare costs to contain or reduce costs of healthcare may affect our future revenues and profitability, and the future revenues and profitability of our potential customers, suppliers and collaborative partners and the availability of capital. For example, in the U.S., given recent federal and state government initiatives directed at lowering the total cost of healthcare, the U.S. Congress and state legislatures will likely continue to focus on healthcare reform, the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid systems. While we cannot predict whether any such legislative or regulatory proposals will be adopted, the announcement or adoption of such proposals and related laws, rules and regulations could materially harm our business, financial conditions results of operations or stock price. Moreover, the passage of the Patient Protection and Affordable Care Act in 2010, and efforts to amend or repeal such law, has created significant uncertainty relating to the scope of government regulation of healthcare and related legal and regulatory requirements, which could have an adverse impact on sales of our products.
In addition, our ability to commercialize Locilex™ or any future product candidates will depend in part on the extent to which appropriate reimbursement levels for the cost of such products and related treatments are adopted by third party payors, such as by governmental health programs, private health insurers and other organizations, such as HMOs. Consumers and third-party payors are increasingly challenging the prices charged for drugs and healthcare services.
Even if we obtain FDA approval of Locilex™, or any other product candidate we may develop or acquire in the future, we may never obtain approval or commercialize our products outside of the U.S., which would limit our ability to realize their full market potential. If foreign approval is obtained, there are risks in conducting business in international markets.
In order to market Locilex™ or any other products we may develop or acquire outside of the U.S., we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and require additional preclinical studies or clinical trials which would be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of Locilex™ or any future products in those countries. Satisfying these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. In addition, our failure to obtain regulatory approval in the U.S. or any foreign country may delay or
20
have negative effects on the process for regulatory approval in other countries. We do not have any product candidates approved for sale in the U.S. or any foreign country and we do not have experience as a company in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in a foreign country or to obtain and maintain required approvals, our potential market for Locilex™ or other products will be reduced and our ability to realize the full market potential of our products will be harmed.
If approved for commercialization in a foreign country, we intend to enter into agreements with third parties to market Locilex™ whenever it may be approved and wherever we have the right to market it. Consequently, we expect that we will be subject to additional risks relating to entering into international business relationships, including:
- •
- lack of adequate protection from intellectual property rights in foreign countries, which could occur if we do not have
issued patents in force in such foreign countries covering our products, their methods of use and methods of manufacture;
- •
- the potential for so-called parallel importing, which is what happens when a local seller, faced with high or higher local
prices (for instance, because the goods have patent protection in such country), opts to import goods from a foreign market (with low or lower prices) rather than buy them locally;
- •
- unexpected changes in tariffs, trade barriers and regulatory requirements;
- •
- economic weakness, including inflation, or political instability in particular foreign economies and markets;
- •
- compliance with laws for employees traveling abroad;
- •
- foreign taxes, including withholding of payroll taxes;
- •
- foreign currency fluctuations, which could result in increased operating expenses and reduced revenues;
- •
- workforce uncertainty in countries where labor unrest is more common than in the U.S.;
- •
- production shortages resulting from any events affecting the API and/or finished drug product supply or manufacturing
capabilities abroad;
- •
- business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters including
earthquakes, typhoons, floods and fires; and
- •
- failure to comply with Office of Foreign Asset Control rules and regulations and the Foreign Corrupt Practices Act.
These and other risks may materially adversely affect our ability to attain or sustain revenue from international markets.
Our independent registered public accounting firm has previously identified material weaknesses in our financial reporting process in their audit of our 2012 financial statements, and we may experience such weaknesses in the future.
As of December 31, 2012, our independent registered public accounting firm had identified two material weaknesses in our internal controls over financial reporting. Specifically, our independent registered public accounting firm identified material weaknesses with respect to:
- •
- our financial statement closing process, in that our current process did not include an effective review to identify all
journal entries that needed to be recorded; and
- •
- an inadequate segregation of duties by management in the financial reporting area.
21
During the quarter ended December 31, 2013, we took the following steps toward remediating the material weaknesses previously identified:
- •
- We retained a new Chief Accounting Officer, who is currently consulting with us but with whom we have executed an
employment agreement which shall be effective upon closing this offering, and
- •
- We have hired an external consultant with the requisite accounting expertise to assist in the proper recording of accounting transactions and to assist with our financial closing process.
We intend to implement additional measures to avoid material weaknesses and improve our periodic financial statement reporting process to:
- •
- limit access to the accounting and information systems and related data to strengthen our segregation of duties; and
- •
- implement procedures and controls in the financial statement closing process to improve the accuracy and timeliness of the preparation of quarterly and annual financial statements.
While our independent registered public accounting firm did not note any material weaknesses in our internal controls in connection with its audit of our 2013 financial statements, we believe that as we become a publicly-reporting company, we will be required to further augment our internal financial and reporting controls to avoid or remediate material weaknesses or significant deficiencies in such controls as they may arise. However, there is a risk we will be unable to successfully implement our plans relating to our financial and disclosure controls or remediate internal control issues should they arise. Our failure to successfully implement our plans or remediate any control issues could cause us to fail to meet our reporting obligations, to produce timely and reliable financial information, and to effectively prevent fraud. Additionally, such failure, or other weaknesses or deficiencies that we may experience in our financial reporting or disclosure controls or similar processes, could cause investors to lose confidence in our reported financial information, which could have a negative impact on our financial condition and stock price.
Failure to achieve and maintain effective internal controls could have a material adverse effect on our business.
Effective internal controls are necessary for us to safeguard our assets and provide reliable financial reports. If we cannot provide reliable financial reports, our operating results could be harmed. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
While we continue to evaluate and improve our internal controls, we are a small company with limited staff, and we cannot be certain that the measures we implement will ensure that we design, undertake and maintain adequate controls over our financial processes and reporting in the future. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations.
Failure to achieve and maintain an effective internal control environment could cause investors to lose confidence in our reported financial information, which could have a material adverse effect on our stock price. See "– Our independent registered public accounting firm has previously identified material weaknesses in our financial reporting process in their audit of our 2012 financial statements, and we may experience such weaknesses in the future." In addition, if our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
22
Our internal computer systems, or those of our contract research organization and other key vendors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development program.
Our internal computer systems and those of our contract research organization and other key vendors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs. For example, the loss of clinical trial data from completed or ongoing clinical trials for Locilex™ could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of Locilex™ could be delayed.
Our employees and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee or consultant fraud or other misconduct. Misconduct by our employees or consultants could include intentional failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing standards, comply with federal and state healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee and consultant misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Business disruptions could seriously harm our future revenues and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or manmade disasters or business interruptions. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
We may engage in strategic transactions that could impact our liquidity, increase our expenses and present significant distractions to our management.
From time to time, we may consider strategic transactions, such as acquisitions of companies, asset purchases, and out-licensing or in-licensing of products, product candidates or technologies. Additional potential transactions that we may consider include a variety of different business arrangements, including spin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and investments. Any such transaction may require us to incur non-recurring or other charges, may increase our near- and long-term expenditures and may pose significant integration
23
challenges or disrupt our management or business, which could adversely affect our operations and financial results. For example, these transactions may entail numerous operational and financial risks, including:
- •
- exposure to unknown liabilities;
- •
- disruption of our business and diversion of our management's time and attention in order to develop acquired products,
product candidates or technologies;
- •
- incurrence of substantial debt or dilutive issuances of equity securities to pay for acquisitions;
- •
- higher-than-expected acquisition and integration costs;
- •
- write-downs of assets or goodwill or impairment charges;
- •
- increased amortization expenses;
- •
- difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and
personnel;
- •
- impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and
ownership; and
- •
- inability to retain key employees of any acquired businesses.
Accordingly, although there can be no assurance that we will undertake or successfully complete any transactions of the nature described above, any transactions that we do complete may be subject to the foregoing or other risks, could have a material adverse effect on our business, results of operations, financial condition and prospects.
Risks Relating to Our Intellectual Property
Our ability to achieve commercial success depends to a material extent on our ability to maintain adequate intellectual property protection for our products and technology. If we are unable to obtain and maintain adequate intellectual property rights for Locilex™, it may materially and adversely affect our ability to market and generate sales of the product.
Due to the exclusivity that intellectual property protection can afford, our commercial success depends to a material extent on our ability to obtain and maintain adequate intellectual property protection for Locilex™ (and any other products we may develop in the future) in the U.S. and other countries. As of February 1, 2014, our patent estate included a U.S. patent (U.S. Patent No. 8,530,409), which has corresponding applications pending before the United States Patent and Trademark Office (or USPTO) and Patent Cooperation Treaty (or PCT). We also have an exclusive sublicense from Scripps Research Institute (or Scripps) , the inventor of the pexiganan technology, to a U.S. patent (U.S. Patent No. 5,912,231), directed to pexiganan, the API used in Locilex.™ We consider our U.S. Patent No. 8,530,409, which relates to our new, proprietary formulation of Locilex™ and methods of using it to treat skin or wound infection, to be particularly important to our company primarily due to its substantially longer patent term coverage, its novel attributes as a topical formulation, its potentially broader scope of coverage and its opportunity for foreign patent protection. While we currently have pending a foreign PCT application corresponding to U.S. Patent No. 8,530,409, no assurance can be given that any foreign patents will issue, or that even if any such patents were to issue, such patents would provide meaningful protection for Locilex™.
Our patent estate related to Locilex™ is critical to our commercial viability. There is a risk that our pending patent applications may not result in issued patents, and that any of our issued patents will not include claims that are sufficiently broad to provide adequate protection for Locilex™, including meaningful protection from our competitors. Additionally, the success of an application for the patent term extension of our licensed patent will require the cooperation of the licensor, which cooperation cannot be guaranteed. We will be able to protect our proprietary rights from unauthorized use by
24
third parties only to the extent that Locilex™ (and any other products we may develop in the future) is covered by valid and enforceable patents that are of sufficient scope to effectively prevent competitive products or are effectively maintained as trade secrets within our organization. If third parties disclose or misappropriate or design around our proprietary rights, it may materially and adversely impact our position in the market.
We apply for patents covering both our technologies and product candidates, as we deem appropriate. However, we may fail to apply for patents on important technologies or improvements in our technologies in a timely fashion, or at all. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from using our technologies or from developing competing products and technologies. Moreover, the patent positions of numerous biotechnology and pharmaceutical companies are highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. As a result, the validity and enforceability of our patents cannot be predicted with certainty. In addition, no assurances can be given that:
- •
- we were the first to make the inventions covered by each of our issued patents and pending patent applications;
- •
- we were the first to file patent applications for these inventions;
- •
- others will not independently develop similar or alternative technologies or duplicate any of our technologies by
inventing around our claims;
- •
- a third party will not challenge our proprietary rights, and if challenged that a court will hold that our patents are
valid and enforceable;
- •
- any patents issued to us or our collaboration partners will cover our product as ultimately developed, or provide us with
any competitive advantages, or will not be challenged by third parties;
- •
- we will develop additional proprietary technologies that are patentable;
or
- •
- the patents of others will not have an adverse effect on our business.
In addition, there are numerous recent changes to the patent laws and proposed changes to the rules of the USPTO which may have a significant impact on our ability to protect our technology and enforce our intellectual property rights. For example, on September 16, 2011, President Obama signed the Leahy-Smith America Invents Act which codifies several significant changes to the U.S. patent laws, including, among other things, changing from a "first to invent' to a "first inventor to file" system, limiting where a patentee may file a patent suit, eventually eliminating interference proceedings while maintaining derivation actions, and creating a set of procedures to challenge patents in the USPTO after they have issued. The effects of these changes are currently uncertain as the USPTO has just implemented regulations related to these changes and the courts have yet to address many of these provisions in the context of a dispute. Furthermore, we have not assessed the applicability of the act and new regulations on the specific patents discussed herein. The U.S. Supreme Court has also issued decisions, the full impact of which is not yet known. For example, on March 20, 2012 in Mayo Collaborative Services, DBA Mayo Medical Laboratories, et al. v. Prometheus Laboratories, Inc., the Court held that several claims drawn to measuring drug metabolite levels from patient samples and correlating them to drug doses were not patentable subject matter. The decision appears to impact diagnostics patents that merely apply a law of nature via a series of routine steps and it has created uncertainty around the ability to patent certain biomarker-related method claims. Additionally, on June 13, 2013 in Association for Molecular Pathology v. Myriad Genetics, Inc., the Court held that claims to isolated genomic DNA are not patentable, but claims to complementary DNA (or cDNA) molecules were held to be valid. The effect of the decision on patents for other isolated natural products is uncertain.
25
We are dependent on a third party to maintain the patent exclusivity for the API in Locilex™.
We hold an exclusive, worldwide sublicense to the composition-of-matter patent, U.S. Patent No. 5,912,231, which expires in June 2016, excluding any patent term restoration that we may seek under the Hatch-Waxman Act. Our rights to practice the pexiganan technology are derived through a license agreement between Scripps and Multiple Peptide Systems Inc. (or MPS). MPS subsequently sublicensed the pexiganan technology to the prior sponsor of the pexiganan clinical and regulatory program. On October 1, 1996, both the license agreement and sublicense agreement were amended by the parties to confirm that the license and sublicense were fully-paid and royalty-free with no further economic obligations for the practice of the pexiganan technology.
We will need to enlist the cooperation of Scripps should we decide to apply for a five-year patent term extension of U.S. Patent No. 5,912,231 under the Hatch Waxman Act, and such cooperation is not guaranteed. Although U.S. Patent 5,912,231 supplements our existing intellectual property portfolio, we are chiefly reliant on our U.S. Patent 8,530,409, which covers the novel formulation and method of use for Locilex™ and provides for substantially longer patent coverage (until June 2032) than U.S. Patent 5,912,231. As such, we have not yet engaged in any discussions with Scripps regarding a possible patent term extension for U.S. Patent No. 5,912,231. If and when we decide to apply for an extension, we will bear the costs of preparing and filing the application, but Scripps, as the owner of U.S. Patent No. 5,912,231, will submit the application. Accordingly, we will need to work with Scripps throughout the application process to facilitate its approval. An inability to extend the patent past 2016 may impair our competitive position if other companies use pexiganan as an API to develop a product that, once approved by the FDA, competes with Locilex™.
We may become subject to third parties' claims alleging infringement of their patents and proprietary rights or seeking to invalidate our patents or proprietary rights, or we may need to become involved in lawsuits to protect or enforce our patents, which could be costly, time consuming, delay or prevent the development and commercialization of our product candidates, or put our patents and other proprietary rights at risk.
Litigation relating to infringement or misappropriation of patent and other intellectual property rights in the pharmaceutical and biotechnology industries is common. We may become subject to third-party claims in the future relating to our technologies, processes, formulations, methods, or products that would cause us to incur substantial expenses and which, if successful, could cause us to pay substantial damages and attorney's fees, if we are found to be infringing a third party's patent rights. We may also become subject to claims that we have misappropriated the trade secrets of others. These risk are exacerbated by the fact that the validity and breadth of claims covered in pharmaceutical patents is, in most instances, uncertain and highly complex. We would be particularly at risk if any such claims relate to our key U.S. Patent No. 8,530,409 covering our particular formulation of and method of use for Locilex™.
Furthermore, if a patent infringement suit is brought against us relating to Locilex™ (or any other products we may develop or acquire in the future), our research, development, manufacturing or sales activities relating to Locilex™ or the product candidate that is the subject of the suit may be delayed or terminated. As a result of patent infringement claims, or in order to avoid potential infringement claims, we or our collaborators may choose to seek, or be required to seek, a license from the third party, which would be likely to include a requirement to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if a license can be obtained on acceptable terms, the rights may be nonexclusive, which would give our competitors access to the same intellectual property rights. If we are unable to enter into a license on acceptable terms, we or our collaborators could be prevented from commercializing Locilex™ (or any other products we may develop or acquire in the future), or forced to modify such product candidates, or to cease some aspect of our business operations, which could harm our business significantly.
26
In addition, competitors may infringe our patents, or misappropriate or violate our other intellectual property rights. To counter infringement or unauthorized use, we may find it necessary to file infringement or other claims to protect our intellectual property rights. In addition, in any infringement proceeding brought by us against a third party to enforce our rights, a court may decide that a patent of ours is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the basis that our patents do not cover the technology in question. An adverse result in any such litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly, which could open us up to additional competition and have a material adverse effect on our business.
The cost to us of any patent litigation or other proceedings, even if resolved in our favor, could be substantial. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings, and such litigation could impair our ability to raise funding for our company. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and, if securities analysts or investors perceive these results to be negative, there could be a substantial adverse effect on the price of our common stock. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also require significant time and attention of management and technical staff, which may materially and adversely impact our financial position and results of operations. Furthermore, because of the substantial amount of discovery required in connection with any intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
As a result of any such litigation, we may also be required to: (i) cease selling, making, importing, incorporating or using one or more or all of our products that incorporate intellectual property of others, which would adversely affect our revenue; or (ii) redesign our products, which would be costly and time-consuming.
Restrictions on our patent rights relating to Locilex™ may limit our ability to prevent third parties from competing against us.
Assuming FDA approval, our ability to market and sell Locilex™ will depend, in part, on our ability to obtain and maintain patent protection for Locilex™ (or any products we may develop in the future), preserve our trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others. The U.S. patent that we sublicense from Scripps (U.S. Patent No. 5,912,231), which is directed to the composition of matter of pexiganan, expires in 2016. The foreign patents corresponding to U.S. Patent No. 5,912,231 expired in 2009. As a result, we have no foreign patent protection for the pexiganan API. We have recently been issued a U.S. patent, U.S. Patent No. 8,530,409, covering our new formulation of Locilex™ as well as a method of using this new formulation to treat skin or wound infections. The U.S. Patent No. 8,530,409 claims are directed to very specific formulations of the pexiganan API, and their methods of use to treat skin or wound infections. As a result, U.S. Patent No. 8,530,409 would not prevent third party competitors from creating, making and marketing alternative formulations of pexiganan, including topical formulations, that fall outside the scope of the U.S. Patent No. 8,530,409 claims. There can be no assurance that any such alternative formulations will not be equally effective as Locilex.TM Introduction of any such competitive product could have a material adverse effect on sales of Locilex™. Moreover, even if these competitors do not actively promote their product for our targeted indication, physicians may prescribe these products "off-label." Although off-label prescriptions may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
27
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on Locilex™ or any product candidates we may develop or acquire in the future throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and furthermore, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the U.S. These products may compete with our future products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
If our Locilex™ trademark is not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered trademark, Locilex™, may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to this trademark, which we need to build name recognition by potential partners or customers in our markets of interest. Over the long term, if we are unable to establish name recognition based on our trademark, then we may not be able to compete effectively and our business may be adversely affected.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patent protection for Locilex™, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants that obligate them to assign their inventions to us. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult,
28
expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S., including in foreign jurisdictions, are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
Risks Relating to Our Securities and this Offering
There is no existing market for our common stock and we do not know if one will develop to provide you with adequate liquidity.
Prior to this offering, there has not been a public market for our common stock. We cannot assure you that an active trading market for our common stock will develop following this offering, or if it does develop, it may not be maintained. You may not be able to sell your shares quickly or at the market price if trading in our common stock is not active. The initial public offering price for the shares will be determined by negotiations between us and representatives of the underwriters and may not be indicative of prices that will prevail in the trading market.
The market price of our common stock may be highly volatile, and you could lose all or part of your investment.
The trading price of our common stock is likely to be volatile. This volatility may prevent you from being able to sell your shares at or above the price you paid for your shares. Our stock price could be subject to wide fluctuations in response to a variety of factors, which include:
- •
- any adverse results or delays in commencement or completion of our planned Phase 3 clinical trials for
Locilex™;
- •
- any adverse results or delays in the commencement or completion of our planned Phase 1 clinical trials for
Locilex™;
- •
- any delay in preparing or filing our NDA for Locilex™ and any adverse development or perceived adverse
development with respect to the FDA's review of the NDA, including without limitation the FDA's issuance of a "refusal to file" letter or a request for additional information;
- •
- changes in laws or regulations applicable to Locilex™ or any future product candidates, including but not
limited to clinical trial requirements for approvals;
- •
- unanticipated serious safety concerns related to the use of Locilex™ or any future product candidates;
- •
- a decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial;
- •
- our inability to obtain adequate product supply for Locilex™ or any future product candidate, or the inability
to do so at acceptable prices;
- •
- adverse regulatory decisions;
- •
- the introduction of new products or technologies offered by us or our competitors;
- •
- the effectiveness of our or our potential partners' commercialization efforts;
- •
- the inability to effectively manage our growth;
- •
- actual or anticipated variations in quarterly operating results;
- •
- our failure to meet or exceed the estimates and projections of the investment community;
29
- •
- the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community;
- •
- the overall performance of the U.S. equity markets and general political and economic conditions;
- •
- developments concerning our sources of manufacturing supply and any commercialization partners;
- •
- announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our
competitors;
- •
- disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to
obtain patent protection for our technologies;
- •
- additions or departures of key scientific or other consultants or management personnel;
- •
- adverse market reaction to any indebtedness we may incur or securities we may issue in the future;
- •
- sales of our common stock by our stockholders in the future;
- •
- significant lawsuits, including patent or stockholder litigation;
- •
- changes in the market valuations of similar companies;
- •
- the trading volume of our common stock;
- •
- effects of natural or man-made catastrophic events or other business interruptions;
and
- •
- other events or factors, many of which are beyond our control.
In addition, the stock market in general, and the stock of biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
The NASDAQ Capital Market may not list our securities, which could limit investors' ability to make transactions in our securities and subject us to additional trading restrictions.
We anticipate that our securities will be listed on The NASDAQ Capital Market, a national securities exchange, upon consummation of this offering. Although, after giving effect to this offering, we expect to meet, on a pro forma basis, The NASDAQ Capital Market's minimum initial listing standards, which generally mandate that we meet certain requirements relating to stockholders' equity, market capitalization, aggregate market value of publicly held shares and distribution requirements, we cannot assure you that we will be able to meet those initial listing requirements. If The NASDAQ Capital Market does not list our securities for trading on its exchange, we could face significant material adverse consequences, including:
- •
- a limited availability of market quotations for our securities;
- •
- reduced liquidity with respect to our securities;
- •
- a determination that our shares of common stock are "penny stock" which will require brokers trading in our shares of
common stock to adhere to more stringent rules, possibly resulting in a reduced level of trading activity in the secondary trading market for our shares of common stock;
- •
- a limited amount of news and analyst coverage for our company; and
- •
- a decreased ability to issue additional securities or obtain additional financing in the future.
30
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as "covered securities." Assuming our common stock will be listed on The NASDAQ Capital Market, our common stock will be covered securities. Although the states are preempted from regulating the sale of our securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Furthermore, if we were no longer listed on The NASDAQ Capital Market, our common stock would not be covered securities and we would be subject to regulation in each state in which we offer our securities.
Our failure to meet the continued listing requirements of The NASDAQ Capital Market could result in a de-listing of our common stock.
If after listing we fail to satisfy the continued listing requirements of The NASDAQ Capital Market, such as the corporate governance requirements or the minimum closing bid price requirement, the NASDAQ Stock Market (or NASDAQ) may take steps to de-list our common stock. Such a de-listing would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a de-listing, we would take actions to restore our compliance with NASDAQ's listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the NASDAQ minimum bid price requirement or prevent future non-compliance with NASDAQ's listing requirements.
If our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The Securities and Exchange Commission (or SEC) has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not obtain or retain a listing on The NASDAQ Capital Market and if the price of our common stock is less than $5.00, our common stock will be deemed a penny stock. The penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser's written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks; and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
There can be no assurance that we will ever provide liquidity to our investors through a sale of our company.
While acquisitions of pharmaceutical companies like ours are not uncommon, potential investors are cautioned that no assurances can be given that any form of merger, combination, or sale of our company will take place following this offering, or that any merger, combination, or sale, even if consummated, would provide liquidity or a profit for our investors following this offering. You should not invest in our company with the expectation that we will be able to sell the business in order to provide liquidity or a profit for our investors.
31
The financial and operational projections that we may make from time to time are subject to inherent risks.
The projections that we provide herein or our management may provide from time to time (including, but not limited to, those relating to potential peak sales amounts, clinical and regulatory timelines, production and supply matters, commercial launch dates, and other financial or operational matters) reflect numerous assumptions made by management, including assumptions with respect to our specific as well as general business, regulatory, economic, market and financial conditions and other matters, all of which are difficult to predict and many of which are beyond our control. Accordingly, there is a risk that the assumptions made in preparing the projections, or the projections themselves, will prove inaccurate. There may be differences between actual and projected results, and actual results may be materially different from than those contained in the projections. The inclusion of the projections in this prospectus should not be regarded as an indication that we, our management, the underwriters or their respective representatives considered or consider the projections to be a guaranteed prediction of future events, and the projections should not be relied upon as such.
Messrs. Luci and DeLuccia will hold a significant concentration of our common stock following this offering, which could limit the ability of our other stockholders to influence the direction of our company.
As calculated by the SEC rules of beneficial ownership, David P. Luci and Robert J. DeLuccia, each executive officers and directors of our company, own 34.2% and 34.1%, respectively, of our common stock on an as converted basis as of March 6, 2014, prior to giving effect to this offering. Accordingly, they collectively may have the ability to significantly influence or determine the election of all of our directors or the outcome of most corporate actions requiring stockholder approval such as: (i) a merger or a sale of our company, (ii) a sale of all or substantially all of our assets, and (iii) amendments to our articles of incorporation and bylaws. This concentration of voting power and control could have a significant effect in delaying, deferring or preventing an action that might otherwise be beneficial to our other stockholders and be disadvantageous to our stockholders with interests different from those individuals. These individuals also have significant control over our business as officers and directors of our company. There is a risk that they may exercise this ability in a manner that advances their best interests and not necessarily those of our other stockholders.
In making your investment decision, you should understand that we and the underwriters have not authorized any other party to provide you with information concerning us or this offering.
You should carefully evaluate all of the information in this prospectus before investing in our company. We may receive media coverage regarding our company, including coverage that is not directly attributable to statements made by our officers, that incorrectly reports on statements made by our officers or employees, or that is misleading as a result of omitting information provided by us, our officers or employees. We and the underwriters have not authorized any other party to provide you with information concerning us or this offering, and you should not rely on this information in making an investment decision.
Future sales by our stockholders may adversely affect our stock price and our ability to raise funds in new stock offerings.
Sales of our common stock in the public market following this offering could lower the market price of our common stock. Sales may also make it more difficult for us to sell equity securities or equity-related securities in the future at a time and price that our management deems acceptable or at all. Assuming our corporate conversion, of the 5,375,377 shares of common stock outstanding as of March 6, 2014, no shares are, or will be, freely tradable without restriction immediately after the consummation of this offering, but approximately 1,370,131 of these shares, representing shares not
32
held by our "affiliates," generally may be resold under SEC Rule 144 beginning 180 days from the effectiveness of the registration statement of which this prospectus forms a part.
Furthermore, assuming the corporate conversion and the completion of this offering had occurred on March 6, 2014, warrants exercisable for up to 34,300 shares of our common stock at an exercise price of $8.57 per share were outstanding. The exercise of any of these warrants would result in additional dilution, and the sale of the 34,300 shares issuable upon exercise of outstanding warrants could also lower the market price of our common stock.
You will experience immediate and substantial dilution as a result of this offering and may experience additional dilution in the future.
You will incur immediate and substantial dilution as a result of this offering. After giving effect to the sale by us of 2,307,693 shares of common stock offered in this offering at an assumed public offering price of $13.00 per share (which represents the mid-point of the estimated range of the initial public offering price shown on the front cover of this prospectus), and after deducting underwriting commissions and estimated offering expenses payable by us, investors in this offering can expect an immediate dilution of $9.06 per share, or 69.7%, at the assumed public offering price. In addition, in the past, we issued warrants to acquire shares of common stock. Additionally, to the extent that these warrants, or options we will grant to our officers, directors and employees, are ultimately exercised, you will sustain future dilution. We may also acquire or license other technologies or finance strategic alliances by issuing equity, which may result in additional dilution to our stockholders.
We will incur significant increased costs as a result of operating as a public company and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and NASDAQ, has imposed various requirements on public companies. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, we anticipate that compliance with these rules and regulations will increase our legal, accounting and financial compliance costs substantially. In addition, these rules and regulations may make our activities related to legal, accounting and financial compliance more difficult, time-consuming and costly and may also place undue strain on our personnel, systems and resources. If these requirements divert the attention of our management and personnel from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to incur substantial costs to maintain our current levels of such coverage. We estimate the additional costs we may incur to respond to these requirements to range from $500,000 to $1.0 million annually, although unforeseen circumstances could increase actual costs.
An investment in our company may involve tax implications, and you are encouraged to consult your own advisors as neither we nor any related party is offering any tax assurances or guidance regarding our company or your investment.
The formation of our company and our financings, as well as an investment in our company generally, involves complex federal, state and local income tax considerations. Neither the Internal Revenue Service nor any State or local taxing authority has reviewed the transactions described herein, and may take different positions than the ones contemplated by management. You are strongly urged to consult your own tax and other advisors prior to investing, as neither we nor any of our officers, directors or related parties is offering you tax or similar advice, nor are any such persons making any representations and warrants regarding such matters.
33
Our management will have broad discretion in how we use the net proceeds of this offering.
Our management will have considerable discretion over the use of proceeds from this offering. You will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used in a manner which you may consider most appropriate. Furthermore, you will have no direct say on how our management allocates the net proceeds of this offering. Until the net proceeds are used, they may be placed in investments that do not produce significant income or that may lose value.
Our ability to use our net operating loss carry-forwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, referred to as the Internal Revenue Code, if a corporation undergoes an "ownership change" (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation's ability to use its pre-change net operating loss carry-forwards and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited. We believe that, with this offering, taken together with our private placements within a three-year period and other transactions that have occurred over the past three years, we may have triggered an "ownership change" limitation. We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership, including as a result of the completion of this offering when it is taken together with other transactions we may consummate in the succeeding three-year period. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carry-forwards to offset U.S. federal taxable income may be subject to limitations, which potentially could result in increased future tax liability to us.
As an "emerging growth company" under applicable law, we will be subject to lessened disclosure requirements, which could leave our stockholders without information or rights available to stockholders of more mature companies.
For as long as we remain an "emerging growth company" as defined in the Jumpstart Our Business Startups Act of 2012 (which we refer to herein as the JOBS Act), we have elected to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not "emerging growth companies" including, but not limited to:
- •
- not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act;
- •
- taking advantage of an extension of time to comply with new or revised financial accounting standards;
- •
- reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements;
and
- •
- exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved.
We expect to take advantage of these reporting exemptions until we are no longer an "emerging growth company." Because of these lessened regulatory requirements, our stockholders would be left without information or rights available to stockholders of more mature companies.
34
Because we have elected to use the extended transition period for complying with new or revised accounting standards for an "emerging growth company" our financial statements may not be comparable to companies that comply with public company effective dates.
We have elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates, and thus investors may have difficulty evaluating or comparing our business, performance or prospects in comparison to other public companies, which may have a negative impact on the value and liquidity of our common stock.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they change their recommendations regarding our common stock adversely, the price of our common stock and trading volume could decline.
The trading market for our common stock may be influenced by the research and reports that securities or industry analysts may publish about us, our business, our market or our competitors. If any of the analysts who may cover us change their recommendation regarding our common stock adversely, or provide more favorable relative recommendations about our competitors, the price of our common stock would likely decline. If any analyst who may cover us was to cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause the price of our common stock or trading volume to decline.
Anti-takeover provisions in our charter documents and Delaware law could discourage, delay or prevent a change in control of our company and may affect the trading price of our common stock.
Upon the completion of our anticipated corporate conversion, we will be a Delaware corporation. The anti-takeover provisions of the Delaware General Corporation Law may discourage, delay or prevent a change in control by prohibiting us from engaging in a business combination with an interested stockholder for a period of three years after the person becomes an interested stockholder, even if a change in control would be beneficial to our existing stockholders. In addition, our certificate of incorporation and bylaws may discourage, delay or prevent a change in our management or control over us that stockholders may consider favorable. Our certificate of incorporation and bylaws will:
- •
- provide that vacancies on our board of directors, including newly created directorships, may be filled only by a majority
vote of directors then in office;
- •
- not provide stockholders with the ability to take action by written consent;
- •
- provide that special meetings of stockholders may only be called by our Chairman and/or President, our board of directors
or a super-majority (662/3%) of our stockholders;
- •
- place restrictive requirements (including advance notification of stockholder nominations and proposals) on how special
meetings of stockholders may be called by our stockholders;
- •
- not provide stockholders with the ability to cumulate their votes; and
- •
- provide that only a super-majority of our stockholders (662/3%) may amend our bylaws.
We do not expect to pay dividends for the foreseeable future.
We do not expect to pay dividends on our common stock offered in this transaction for the foreseeable future. Accordingly, any potential investor who anticipates the need for current dividends should not purchase our securities.
35
Cautionary Note Regarding Forward-Looking Statements
This prospectus contains "forward-looking statements" within the meaning of the federal securities laws, and that involve significant risks and uncertainties. Words such as "may," "should," "could," "would," "predicts," "potential," "continue," "expects," "anticipates," "future," "intends," "plans," "believes," "estimates," and similar expressions, as well as statements in future tense, identify forward-looking statements. Forward-looking statements should not be read as a guarantee of future performance or results and may not be accurate indications of when such performance or results will be achieved. Forward-looking statements are based on information we have when those statements are made or management's good faith belief as of that time with respect to future events, and are subject to significant risks and uncertainties that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. Important factors that could cause such differences include, but are not limited to:
- •
- risks and uncertainties associated with our research and development activities, including our clinical trials;
- •
- our dependence on Locilex™ as our only product;
- •
- our ability to raise capital when needed;
- •
- the timing of and our ability to achieve U.S. or international regulatory approvals for Locilex™ or any other
product candidates we may develop;
- •
- our dependence on others to conduct clinical research of, and to manufacture and market, Locilex™;
- •
- the terms of future licensing arrangements, and whether we can enter into such arrangements at all;
- •
- risks associated with the timing and receipt of licensing and milestone revenues, if any;
- •
- our ability to gain market acceptance for Locilex™ or any other product candidates we may develop;
- •
- our ability to maintain or protect the validity of our patents and other intellectual property, including in connection
with pending or future litigation against us;
- •
- our ability to secure registration for our current and future patent applications;
- •
- our ability to extend our licensed composition of matter patent No. 5,912,231 under the Hatch-Waxman Act with the
cooperation of Scripps;
- •
- our estimates of the size of the prospective markets in which we may offer Locilex™;
- •
- our ability to continue as a going concern;
- •
- our expectations regarding minimizing our development risk;
- •
- our ability to establish new relationships and maintain current relationships;
- •
- our ability to attract and retain key personnel; and
- •
- acceptance of our business model by investors.
The foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or risk factors that we are faced with. Forward-looking statements necessarily involve risks and uncertainties, and our actual results could differ materially from those anticipated in the forward-looking statements due to a number of factors, including those set forth above under "Risk Factors" and elsewhere in this prospectus. The factors set forth above under "Risk Factors" and other cautionary statements made in this prospectus should be read and understood as
36
being applicable to all related forward-looking statements wherever they appear in this prospectus. The forward-looking statements contained in this prospectus represent our judgment as of the date of this prospectus. We caution readers not to place undue reliance on such statements. Except as required by law, we undertake no obligation to update publicly any forward-looking statements for any reason, even if new information becomes available or other events occur in the future. All subsequent written and oral forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the cautionary statements contained above and throughout this prospectus.
37
We estimate that the net proceeds from the sale of the shares of common stock we are offering will be approximately $27.2 million based on an assumed offering price of $13.00 per share (which represents the mid-point of the estimated range of the initial public offering price shown on the front cover of this prospectus). If the underwriters fully exercise the over-allotment option, the net proceeds of the shares we sell will be approximately $31.4 million. "Net proceeds" is what we expect to receive after deducting the underwriting discount and commission and estimated offering expenses payable by us.
Each $1.00 increase (decrease) in the assumed offering price of $13.00 would increase (decrease) the net proceeds to us from this offering by approximately $2.1 million, after deducting estimated underwriting discount and commission and estimated offering expenses payable by us, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same. Each increase of 100,000 shares in the number of shares offered by us at the assumed public offering price would increase the net proceeds to us in this offering by approximately $1.2 million. Similarly, each decrease of 100,000 shares in the number of shares offered by us at the assumed public offering price would decrease the net proceeds to us from this offering by approximately $1.2 million. A change in the offering price or the number of shares by these amounts could have a material effect on our uses of the proceeds from this offering, and it may impact the amount of time prior to which we will need to seek additional capital.
We intend to use the net proceeds of this offering primarily to continue clinical testing and commercialization of Locilex™ and for working capital and other general corporate purposes.
We anticipate an approximate allocation of the use of net proceeds as follows:
| Use of Net Proceeds | $ (in millions)* |
% | ||||||
|---|---|---|---|---|---|---|---|---|
Completion of clinical trials and NDA submission for Locilex™ |
12.5 | 46 | ||||||
Preliminary commercialization expenses in anticipation of FDA approval of Locilex™ |
7.5 | 27 | ||||||
Expansion of approved uses of Locilex™ |
1.5 | 6 | ||||||
Working capital and other general corporate purposes |
5.7 | 21 | ||||||
Total |
27.2 | 100 | ||||||
- *
- Assuming the over-allotment option is not exercised.
While we expect to use the net proceeds for the purposes described above, the amounts and timing of our actual expenditures will depend upon numerous factors, including the ongoing status and results of the Locilex™ clinical trials. The expected net proceeds from the sale of the shares offered hereby, if added to our current cash and cash equivalents is anticipated to be sufficient to fund our operations through the end of 2016. In the event that our plans change, our assumptions change or prove to be inaccurate, or the net proceeds of this offering are less than as set forth herein or otherwise prove to be insufficient, it may be necessary or advisable to reallocate proceeds or curtail expansion activities, or we may be required to seek additional financing or curtail our operations. As a result of the foregoing, our success will be affected by our discretion and judgment with respect to the application and allocation of the net proceeds of this offering.
Pending their use, we plan to invest the net proceeds from this offering in short- and intermediate-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the U.S. government.
38
We have never declared or paid any cash dividends on our equity interests and we do not anticipate paying any cash dividends in the foreseeable future. The payment of dividends, if any, in the future is within the discretion of our board of directors and will depend on our earnings, capital requirements and financial condition and other relevant facts. We currently intend to retain all future earnings, if any, to finance the development and growth of our business.
Immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, we will convert Dipexium Pharmaceuticals, LLC from a Delaware limited liability company to a Delaware corporation. As a result of the corporate conversion:
- •
- the Class A membership interests of Dipexium Pharmaceuticals, LLC will become shares of common stock of
Dipexium Pharmaceuticals, Inc. pursuant to a conversion ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each Class A membership interest of Dipexium
Pharmaceuticals, LLC previously held. Accordingly, 767,911 Class A membership interests of Dipexium Pharmaceuticals, LLC issued and outstanding immediately prior to the corporate
conversion will be converted automatically into 5,375,377 shares of Dipexium Pharmaceuticals, Inc.; and
- •
- all of the outstanding warrants to purchase Class A membership interests of Dipexium Pharmaceuticals, LLC
will become warrants to purchase shares of common stock of Dipexium Pharmaceuticals, Inc. in a ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each
Class A membership interest of Dipexium Pharmaceuticals, LLC underlying such warrants, with the effect that warrants to purchase 4,900 Class A membership interests of Dipexium
Pharmaceuticals, LLC outstanding immediately prior to the corporate conversion will automatically convert into warrants to purchase 34,300 shares of Dipexium Pharmaceuticals, Inc. upon
consummation of the corporate conversion; and
- •
- the exercise price of all of the outstanding warrants will be adjusted in the same ratio as the seven-for-one conversion ratio noted above such that all of our outstanding warrants to purchase Class A membership interests of Dipexium Pharmaceuticals, LLC which are currently exercisable at $60 per Class A membership interest will automatically be adjusted such that the new exercise price for the outstanding warrants upon consummating the corporate conversion will be $8.57 per share, subject to certain adjustments noted in each of the warrants.
In connection with the corporate conversion, Dipexium Pharmaceuticals, Inc. will continue to hold all property of Dipexium Pharmaceuticals, LLC and will assume all of the debts and obligations of Dipexium Pharmaceuticals, LLC. Dipexium Pharmaceuticals, Inc. will be governed by a certificate of incorporation filed with the Delaware Secretary of State and bylaws, the material portions of which are described in "Description of Capital Stock." On the effective date of the corporate conversion, the members of the board of directors of Dipexium Pharmaceuticals, LLC will become the members of the board of directors of Dipexium Pharmaceuticals, Inc. and the officers of Dipexium Pharmaceuticals, LLC will become the officers of Dipexium Pharmaceuticals, Inc. The purpose of the corporate conversion is to reorganize our corporate structure so that our company will continue is a corporation rather than a limited liability company following this offering, and so that our existing investors will own our common stock rather than equity interests in a limited liability company. In order to consummate the corporate conversion, a certificate of conversion will be filed with the Secretary of State of the State of Delaware.
39
The following table sets forth our cash and equivalents and capitalization as of December 31, 2013:
- •
- on an actual basis;
- •
- on a pro forma basis to give effect to the February 2014 issuance of an aggregate of 23,719 Class A membership
interests to investors in our prior financings in exchange for the redemption of outstanding warrants to purchase an aggregate of 89,900 Class A membership interests in our company;
- •
- on a pro forma basis to give further effect to our corporate conversion from a limited liability company to a corporation
and, in connection therewith, the conversion of 767,911 outstanding Class A membership interests in our company issued and outstanding immediately prior to the conversion into an aggregate of
5,375,377 shares of common stock; and
- •
- on a pro forma as adjusted basis to additionally give effect to the sale of shares of our common stock in this offering, assuming an initial public offering price of $13.00 per share (the mid-point of the price range set forth on the cover page of this prospectus), after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
You should read the information in this table together with our financial statements and accompanying notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" appearing elsewhere in this prospectus.
| |
As of December 31, 2013 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Actual | Pro Forma, Warrant Exchange(1) |
Pro Forma, Corporate Conversion(1) |
Pro Forma As Adjusted(1) |
||||||||||
| |
(in thousands, except share data) (Unaudited) |
|||||||||||||
Cash and equivalents |
$ | 3,861 | $ | 3,861 | $ | 3,861 | $ | 31,059 | ||||||
Class A membership interests, 735,588 issued and outstanding, actual; 767,911 issued and outstanding, pro forma (warrant exchange); none, pro forma (corporate conversion) and pro forma as adjusted |
11,944 | 11,944 | — | — | ||||||||||
Shareholders' equity (deficit) |
||||||||||||||
Common Stock, $0.001 par value, no shares authorized, issued and outstanding, actual; no shares authorized, issued and outstanding, pro forma (warrant exchange); 30,000,000 shares authorized, 5,375,377 shares issued and outstanding, pro forma (corporate conversion); 30,000,000 shares authorized, 7,683,070 shares issued and outstanding, pro forma as adjusted |
– | 5 | 8 | |||||||||||
Additional paid-in capital |
– | 11,939 | 30,396 | |||||||||||
Deficit accumulated during the development stage |
(8,738 | ) | (8,738 | ) | (8,738 | ) | — | |||||||
Total shareholders' equity (deficit) |
3,206 | 3,206 | 3,206 | 30,404 | ||||||||||
Total capitalization |
$ | 3,206 | 3,206 | 3,206 | 30,404 | |||||||||
- (1)
- The pro forma and pro forma as adjusted information is illustrative only and following the completion of this offering will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing.
40
If you purchase shares of our common stock in this offering, your interest will be diluted immediately to the extent of the difference between the assumed public offering price of $13.00 per share (the mid-point of the range appearing on the front cover of this prospectus) and the as adjusted net tangible book value per share of our common stock immediately upon the consummation of this offering.
On a pro forma basis, after giving effect to the exchange of 89,900 outstanding warrants into an aggregate of 23,719 Class A membership interests in February 2014 and the effect of our corporate conversion from a limited liability company to a corporation resulting in the conversion of 767,911 Class A membership interests issued and outstanding immediately prior to the conversion into an aggregate of 5,375,377 shares of our common stock, our net tangible book value as of December 31, 2013 was approximately $3.1 million, or approximately $0.57 per share. Net tangible book value per share represents our total tangible assets less total liabilities, divided by the number of shares of common stock outstanding as of December 31, 2013.
Net tangible book value dilution per share to new investors represents the difference between the amount per share paid by purchasers in this offering and the as adjusted net tangible book value per share of common stock immediately after completion of this offering. After giving effect to our sale of 2,307,693 shares of common stock in this offering at an assumed public offering price of $13.00 per share, and after deducting underwriters' commissions and estimated offering expenses, our as adjusted net tangible book value as of December 31, 2013 would have been $30.3 million, or $3.94 per share. This represents an immediate increase in net tangible book value of $3.37 per share to existing stockholders and an immediate dilution in net tangible book value of $9.06 per share to purchasers of shares in this offering, as illustrated in the following table:
Assumed public offering price per share |
$ | 13.00 | |||||
Net tangible book value per share as of December 31, 2013, after giving effect to the warrant exchange in February 2014 and our corporate conversion |
$ | 0.57 | |||||
Increase in net tangible book value per share attributable to new investors |
$ | 3.37 | |||||
Adjusted net tangible book value per share as of December 31, 2013, after giving effect to the offering |
$ | 3.94 | |||||
Dilution per share to new investors in the offering |
$ | 9.06 |
The above discussion and tables do not include the following:
- •
- 70,000 shares of common stock issuable to members of our board of directors which are currently unvested and, in each
case, vest over a period of time for service on the board of directors;
- •
- 34,300 shares of common stock issuable upon conversion of warrants issued to investors in prior financings, in each case,
with a conversion price equal to $8.57 per share; or
- •
- 1,536,614 shares of our common stock (which is equal to 20% of our issued and outstanding common stock immediately after the consummation of this offering) reserved for future issuance under our 2013 Equity Incentive Plan, which will become effective as of the closing of this offering.
The above discussion and tables assume that our 767,911 Class A membership interests currently outstanding are converted into 5,375,377 shares of common stock following the conversion of Dipexium Pharmaceuticals, LLC into Dipexium Pharmaceuticals, Inc.
41
Management's Discussion and Analysis of Financial Condition
and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with the section titled "Summary Financial Information" and the financial statements and related notes thereto included elsewhere in this prospectus. This discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those discussed below. Period-to-period comparisons of our operating results should not be relied upon as predictive of future performance. Factors that could cause or contribute to such differences include, but are not limited to, those identified below and those discussed in the section titled "Risk Factors" included elsewhere in this prospectus.
Overview
We are a late stage pharmaceutical company focused on the development and commercialization of Locilex™ (pexiganan acetate cream 1%), a novel, first-in-class, broad spectrum, topical antibiotic. We were formed in January 2010, and we acquired the worldwide rights to pexiganan, the active pharmaceutical ingredient in Locilex™ from a third party in April 2010. We thus have a very limited history of operations. We have not generated any revenues to date, having focused all of our effort on research and development activities.
Results of Operations
Year Ended December 31, 2012 Compared to Year Ended December 31, 2013
Summary Table
The following table presents a summary of the changes in our results of operations for the year ended December 31, 2012, compared with the year ended December 31, 2013:
| |
Years Ended December 31, |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Percentage Increase |
|||||||||
| |
2013 | 2012 | ||||||||
| |
(in thousands) |
|
||||||||
Research and Development Expenses |
$ | 1,730 | $ | 845 | 104.8 | % | ||||
General and Administrative Expenses |
$ | 2,284 | $ | 999 | 128.6 | % | ||||
Total Expenses |
$ | 4,014 | $ | 1,844 | 117.7 | % | ||||
Net Loss |
$ | 4,014 | $ | 1,844 | 117.7 | % | ||||
Research and Development Expenses
Research and development expenses were $0.8 million for the year ended December 31, 2012, and $1.7 million for the year ended December 31, 2013, an increase of 104.8%. Research and development expenses increased primarily as a result of the increased consulting costs of $0.5 million associated with the planned clinical trials and increased manufacturing costs for validation of stability study of $0.4 million.
General and Administrative Expenses
General and administrative expenses were $1.0 million for the year ended December 31, 2012, and $2.3 million for the year ended December 31, 2013, an increase of $1.3 million, or 128.6%. The increase in general and administrative expenses is primarily attributable to an increase in share based compensation expenses to the board of directors of $0.9 million as a result of new members to the
42
board of directors and accelerated vesting due to certain director resignations, increased professional fees of $0.3 million as a result of increased accounting and consulting work and an increase in executive compensation related expenses of $0.1 million.
Net Loss
Net loss was $1.8 million for the year ended December 31, 2012, compared to $4.0 million for the year ended December 31, 2013, an increase of $2.2 million, or 117.7%, primarily due to increases in research and development expenses and general and administrative expenses for the reasons stated above.
Net Cash Used in Operating Activities
Net cash used in operating activities was $1.5 million for the year ended December 31, 2012. The net loss for this period was greater than the net cash used in operating activities by $0.3 million, which was primarily attributable to share based executive compensation of $0.2 million, share based compensation to the board of directors and vendors of $0.3 million, which was paid in restricted Class A membership interests, and an increase in accounts payable and accrued expenses of $0.2 million.
Net cash used in operating activities was $2.2 million for the year ended December 31, 2013. The net loss for this period was greater than the net cash used in operating activities by $1.8 million, which was primarily attributable to share based compensation of $1.1 million, share based vendor payments of $0.1 million, executive compensation of $0.1 million, which was paid in restricted Class A membership interests, and an increase in accounts payable and accrued expenses of $0.5 million.
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the year ended December 31, 2012 was $2.0 million, which was attributable to aggregate net proceeds of $0.5 million received from the private placement of our Class A membership interests on March 30, 2012 and net proceeds of $1.5 million received from the private placement of our Class A membership interests on November 21, 2012.
Net cash provided by financing activities for the year ended December 31, 2013 was $4.6 million, which was attributable to aggregate net proceeds of $0.9 million received from the private placement of our Class A membership interests on February 13, 2013, net proceeds of $2.8 million received from the private placement of our Class A membership interests on July 12, 2013, and net proceeds of $0.9 million received from the private placement of our Class A membership interests that closed on November 15, 2013.
Liquidity and Capital Resources
Overview
We have generated no revenue from operations and we have incurred cumulative losses of approximately $8.7 million since inception. We have funded our operations primarily from equity issuances and a Federal government grant under the Qualified Therapeutic Discovery Act. We received net cash proceeds of approximately $9.7 million from equity financings closed between July 2010 and November 2013, and received approximately $0.3 million in additional funding from the U.S. federal government under the Qualified Therapeutic Discovery Act program. All of our equity financings were consummated at $7.14 per Class A membership interest with 50% warrant coverage at 120% of the issue price ($8.57, or $60 per interest on a pre-conversion basis).
Based upon our lack of revenue expected for 2014, together with the planned expenditures, management currently believes that current cash will be insufficient to fund our research and development expenses and general and administrative expenses beyond the end of 2014. Upon
43
completion of this offering, the expected net proceeds from this offering, added to our current cash and cash equivalents, is anticipated to be sufficient to fund our operations through the end of 2016.
Furthermore, if our assumptions underlying our anticipated timing for the completion of our clinical and regulatory program and our anticipated expenses prove to be wrong, we may have to raise additional capital sooner than anticipated. Because of numerous risks and uncertainties associated with the research, development and future commercialization of our product candidate, we are unable to estimate with certainty the amounts of increased capital outlays and operating expenditures associated with our anticipated clinical trials and development activities. Our current estimates may be subject to change as circumstances regarding requirements further develop. We may decide to raise capital through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. We do not have any existing commitments for future external funding. We may seek to sell additional equity or debt securities or obtain a bank credit facility if available. The sale of additional equity or debt securities, if convertible, could result in dilution to our stockholders. The incurrence of indebtedness would result in increased fixed obligations and could also result in covenants that would restrict our operations or other financing alternatives.
Our ability to continue as a going concern may be dependent on our ability to raise additional capital, to fund our research and development and commercial programs and meet our obligations on a timely basis. If we are unable to successfully raise sufficient additional capital we may not have sufficient cash flow and liquidity to fund our business operations, forcing us to delay, discontinue or prevent product development and clinical trial activities or the approval of any of our potential products or curtail our activities and, ultimately, potentially cease operations. Even if we are able to raise additional capital, such financings may only be available on unattractive terms, or could result in significant dilution of stockholders' interests and, in such event, the value and potential future market price of our common stock may decline. In addition, the incurrence of indebtedness would result in increased fixed obligations and could result in covenants that would restrict our operations or other financing alternatives.
As of December 31, 2013, we had working capital of $3.1 million, consisting primarily of $3.9 million of cash and cash equivalents, offset by $0.8 million of accounts payable and accrued expenses.
Recent Accounting Pronouncements
The Financial Accounting Standards Board has issued certain accounting pronouncements as of December 31, 2013 that will become effective in subsequent periods; however, we do not believe that any of those pronouncements would have significantly affected our financial accounting measurements or disclosures had they been in effect during 2013 or that they will have a significant impact on us at the time they become effective.
Critical Accounting Policies and Estimates
On April 5, 2012, the JOBS Act was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for qualifying public companies. As an "emerging growth company," we may, under Section 7(a)(2)(B) of the Securities Act of 1933 (or Securities Act), delay adoption of new or revised accounting standards applicable to public companies until such standards would otherwise apply to private companies. We may take advantage of this extended transition period until the first to occur of the date that we (i) are no longer an "emerging growth company" or (ii) affirmatively and irrevocably opt out of this extended transition period. We have elected to take advantage of the benefits of this extended transition period. Our financial statements may therefore not be comparable to those of companies that comply with such new or revised accounting standards. Until the date that we are no longer an "emerging growth company" or affirmatively and irrevocably opt out of the exemption provided by Securities Act Section 7(a)(2)(B), upon issuance of a new or revised accounting standard that applies to our financial statements and that has a different effective date for public and private companies, we will disclose the date on which
44
adoption is required for non-emerging growth companies and the date on which we will adopt the recently issued accounting standard. Our management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on our historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates.
Share Based Compensation
We account for the cost of services performed by directors received in exchange for an award of Class A membership interests based upon the grant date fair value of the award. In accordance with the Accounting Standards Codification, we recognize compensation expense, net of estimated forfeitures, on a straight-line basis over the vesting period.
We account for the cost of services performed by vendors in exchange for an award of membership interests based upon the grant date fair value of the award or fair value of the services rendered, whichever is more readily determinable. In accordance with the Accounting Standards Codification, we recognize the expense in the same period and in the same manner as if we had paid cash for the services.
Off-Balance Sheet Arrangements
We did not have, during the periods presented, and we are currently not party to, any off-balance sheet arrangements.
45
Overview
We are a late stage pharmaceutical company focused on the development and commercialization of Locilex™ (pexiganan acetate cream 1%), a novel, first-in-class, broad spectrum, topical antibiotic. Locilex™ is a chemically synthesized, 22-amino acid peptide isolated from the skin of the African Clawed Frog. Its novel mechanism of action kills microbial targets through disruption of bacterial cell membrane permeability. Locilex™ is initially being targeted for the treatment of mild infections of diabetic foot ulcers (or Mild DFI). In 2011, the market for diabetic foot infection therapeutics worldwide was $1.46 billion. Our primary objective is to establish Locilex™ as the standard of care for the treatment of patients with Mild DFI. Thereafter, our growth strategy includes potentially expanding the indications for Locilex™ to include moderate infections of diabetic foot ulcers (or Moderate DFI) and certain other mild or moderate skin and skin structure infections in superficial wounds.
We believe that we have a clear clinical and regulatory pathway with the potential for near term United States Food and Drug Administration (or FDA) approval of Locilex™. We believe that Locilex™ may be approved by the FDA within 24 months of commencing the Phase 3 enrollment. We have reached agreement with the FDA through a special protocol assessment (or SPA) for our Phase 3 program. We intend to conduct and complete two pivotal Phase 3, double blind, placebo-controlled superiority studies. We expect to begin enrollment in these studies in the second quarter of 2014, with top line data from the Phase 3 studies anticipated in the first quarter of 2015. Concurrently, we also intend to conduct two separate Phase 1 skin irritation and skin sensitization studies, which are expected to commence in the first quarter of 2014. If the data from our Phase 3 studies are sufficient to meet the primary endpoints, we expect to submit our new drug application (or NDA) for Locilex™ to the FDA in the second half of 2015. We expect to receive a response from the FDA within six months of our NDA submission.
According to the Infectious Disease Society of America (or IDSA), diabetic foot infections (or DFI) may be classified by their clinical severity as mild, moderate, or severe. Forty-seven percent of DFI patients present at the mild stage, 34% of DFI patients present at the moderate stage and 18% of DFI patients present at the severe stage. At the mild stage, patients can typically be treated on an outpatient basis and amputation risk is minimal (2 to 3% in Mild DFI). When not managed effectively, the potential for Mild DFI to progress to a limb- or life-threatening infection increases dramatically. Published research suggests amputation rates increase in Moderate DFI and severe infections of diabetic foot ulcers (or Severe DFI) to approximately 45% and 75%, respectively. Similarly, the hospitalization rate for Mild DFI patients is approximately 10%, increasing in Moderate and Severe DFI to approximately 55% and 85%, respectively. Thus, DFI are a major cause of patient morbidity, a substantial burden to the healthcare system, and a source of high financial costs.
Systemic antibiotics currently prescribed off-label to treat Mild DFI generate resistant pathogens which create infections that are more difficult to treat. Such antibiotics are also associated with toxic side effects in patients who typically have some degree of compromised liver and kidney function. We believe that a topical preparation like Locilex™, which is locally administered on the open wound and skin, offers significant advantages over systemic treatments. As a topical antibiotic, Locilex™ affects only the area to which it is applied, and unlike systemic antibiotics, it does not spread to the entire body through the bloodstream, which is host to a number of different bacteria other than bacteria causing the skin infection. Topical antibiotics are also delivered to the infected area at a higher concentration than those antibiotics that are delivered systemically. As a result, we believe that topical antibiotics are less likely to develop bacterial resistance, both because the higher antibiotic concentration more effectively kills the infection-causing bacteria before they develop resistance, and because other bacteria in the bloodstream are not exposed, and therefore not given the chance to generate resistance.
46
Currently, there are no products specifically approved by the FDA for the treatment of Mild DFI, nor are there any topical antibiotics currently approved for any severity of DFI. As such, we believe that Locilex™ has the potential to be the first topical antibiotic approved for the treatment of DFI, as well as the first product of any kind to be labeled specifically for the treatment of Mild DFI.
We believe that the key attributes of Locilex™ are: (i) it has not generated resistant bacteria systemically; (ii) it has not generated cross resistance with other antibiotics; (iii) it has demonstrated activity against a broad spectrum of pathogens, including difficult to treat gram negative, and anaerobic bacteria; (iv) it has not been systemically absorbed; and (v) it has not caused any significant safety issues in over 500 patients treated. These attributes lead us to believe that Locilex™ has the potential to be positioned as the standard of care to treat patients with Mild DFI. In addition, data generated to date support the potential use of Locilex™ to treat a broad array of mild or moderate skin and skin structure infections in superficial wounds.
As reported in published research, Locilex™ has previously demonstrated statistical non-inferiority to a systemic antibiotic in a large-scale, randomized, active-controlled double blind, multi-center clinical studies in patients with Mild or Moderate DFI. We have conducted microbiology studies that highlight the sensitivity of resistant bacteria, including methicillin-resistant staphylococcus aureus (or MRSA), vancomycin-resistant enterococcus (or VRE), extended-spectrum b-lactamase (or ESBL) and multi-drug resistant (or MDR) bacteria, to pexiganan, the active pharmaceutical ingredient (or API) in Locilex™. Due to the increased global prevalence of resistant bacteria in all types of skin infections, Locilex™ may provide an important therapeutic advance.
We have contracted with third party vendors with respect to all key elements of our clinical and regulatory program, including vendors to: (i) conduct the Phase 3 and Phase 1 clinical trials for Locilex™; (ii) manufacture the API; (iii) formulate the finished product; and (iv) label and package the product. We believe these key relationships will help drive our clinical and regulatory program for Locilex™ in a timely and efficient manner.
Locilex™ was originally sponsored by Magainin Pharmaceuticals, Inc. (or Magainin), which engaged in the FDA review process during 1998 and 1999, ultimately receiving a non-approvable letter based upon two manufacturing issues. We acquired the worldwide rights to pexiganan, the API in Locilex™, from a third party in April 2010. These rights included the prior formulation and all of the clinical and preclinical data generated by Magainin in its FDA review process. This includes data from over 1,000 evaluable patients, including 835 in large-scale, randomized, active-controlled, double blind, multi-center clinical studies as compared to a systemic quinolone standard of care as more fully described below. We believe we have corrected the manufacturing problems encountered by the prior sponsor. See "History of Locilex™" below.
Our Strategy
Our primary objective is to establish Locilex™ as the standard of care to treat patients with Mild DFI. The key elements of our strategy are as follows:
- •
- Complete the Phase 3 program for
Locilex™. As a result of our SPA for Locilex™, we believe the clinical pathway for Locilex™ is
clear. Working with our key third party vendors, we expect to commence our Phase 3 program in the second quarter of 2014 and expect to report top line data in the first quarter of 2015.
- •
- Obtain FDA approval of Locilex™ for Mild
DFI. If our Phase 3 trials meet their primary endpoints, we plan to submit our NDA for Locilex™. We believe that
Locilex™ may be approved by the FDA within 24 months of commencing the Phase 3 program.
- •
- Commercially launch Locilex™ in the U.S. We plan to utilize a small specialty sales force to launch Locilex™, if it is approved, in the U.S., initially targeting podiatrists and potentially expanding to other specialty healthcare providers.
47
- •
- Expand Locilex™'s FDA-approved uses. If we are
able to obtain FDA approval in Mild DFI, we will consider obtaining additional FDA approvals that will enable us to expand the Locilex™
label to include patients with Moderate DFI and certain other mild or moderate skin and skin structure infections in superficial wounds.
- •
- Commence clinical and regulatory activities in Europe. We plan to finalize our European Union (or E.U.) clinical and regulatory strategy and work toward filing a clinical trial authorization application (or CTA) in one or more E.U. member states promptly following the completion of our Phase 3 program in the U.S.
We will rely on our strong management team, board of directors and scientific advisory board to execute our strategy. The individuals on our management team, board of directors and scientific advisory board will contribute their significant development and regulatory experience to the development and commercialization of Locilex™.
The Antibiotics Market
The widespread use of antibiotics has led to the development of resistant strains of bacteria, which limits the effectiveness of existing drugs, leading the World Health Organization to state in 2011 that antibiotic resistance is one of the greatest threats to human health.
Antibiotic resistance is primarily caused by genetic mutations in bacteria or when bacteria acquire resistance genes from other bacteria. In addition to mutated bacteria being resistant to the drug used for treatment, many bacterial strains can also be cross-resistant, meaning that the use of a particular treatment to address one kind of bacteria can result in resistance to other types of antibiotics. As a result, the effectiveness of many antibiotics has declined, limiting physicians' options to treat serious infections and creating a global health issue. According to the U.S. Centers for Disease Control and Prevention (or CDC), each year in the U.S., at least two million people acquire serious infections with bacteria that are resistant to one or more of the antibiotics designed to treat those infections. Antibiotic resistance has a significant impact on mortality and contributes heavily to healthcare system costs worldwide. In a 2013 report, the CDC estimates antibiotic resistance contributes to as much as $20 billion in direct healthcare costs and $35 billion in indirect costs (lost productivity) to the U.S. economy.
In addition to resistance issues, current antibiotic therapies have other limitations, including serious side effects. These side effects may include: severe allergic reaction, decreased blood pressure, nausea and vomiting, suppression of platelets, pain and inflammation at the site of injection, muscle, renal and oto-toxicities, optic and peripheral neuropathies and headaches. Some of these side effects may be significant enough to require that therapy be discontinued or not used. Furthermore, some systemic treatments require clinicians to closely monitor diabetic patients' blood glucose levels and other parameters, increasing the expense and inconvenience of treatment. As such, we believe that there is a need for new antibiotics that have improved potency and pharmacokinetics, effectiveness against resistant bacterial strains, improved side effect profiles and more flexible administration formulations. Topical formulations having no systemic absorption can complement systemic antibiotics with no drug-drug interaction in patients with localized skin infections.
Market Opportunity for Diabetic Foot Infection
Based upon our analysis of data from industry sources, in 2011, the markets for DFI therapeutics in the U.S. and worldwide were $1.01 billion and $1.46 billion, respectively. According to the CDC, there were approximately 26 million people with diabetes in the U.S. (or approximately 8% of the population) in 2010. Approximately 1.9 million adults were diagnosed with diabetes in 2010, and based on long-term trends, the number of newly diagnosed cases is expected to increase annually. In published data, results of a meta-analysis of studies that followed patients with diabetes for periods of time between 12 weeks and four years, researchers reported that the incidence of foot ulceration in
48
diabetics was between 8% and 17%. In another three-year study of patients with diabetes, the cumulative incidence of diabetic foot ulcers (or DFUs) was approximately 6%. The majority (approximately 61%) of DFUs are clinically infected at the time the patient initially presents. In addition, the incidence of infection recurrence after complete healing approaches 32%.
Foot infection is the most frequent diabetic complication requiring hospitalization and the most common precipitating event leading to lower limb amputation. Approximately 1.35 million patients are diagnosed with DFI each year in the U.S., of which approximately 650,000 patients are diagnosed with Mild DFI and approximately 470,000 patients are diagnosed with Moderate DFI. According to published research, 47% of DFI patients present at the mild stage, 34% of DFI patients present at the moderate stage and 18% of DFI patients present at the severe stage. At the mild stage, patients can typically be treated on an outpatient basis and amputation risk is minimal (from 2 to 3% in Mild DFI). When not managed effectively, the potential for Mild DFI to progress to a limb- or life-threatening infection increases dramatically. Published research suggests amputation rates increase in Moderate and Severe DFI to approximately 45% and 75%, respectively. Similarly, the hospitalization rate for Mild DFI patients is approximately 10%, increasing in Moderate and Severe DFI to approximately 55% and 85%, respectively. Thus, DFI are a major cause of patient morbidity, a substantial burden to the healthcare system, and a source of high financial costs.
The goal of any antibiotic regimen is to treat patients with the least invasive therapy for the shortest amount of time, such that infection control can be achieved while minimizing the risk of antibiotic resistance. According to published research, there has long been interest in treating Mild DFI, the extent of which are confined to superficial skin and skin structures, with topical antibacterial agents. In Mild DFI, assuming equivalent or greater efficacy, we believe that a topical treatment such as Locilex™ could hold significant advantages over intravenous and oral therapies by avoiding systemic adverse events, providing increased target site concentration, and most importantly, avoiding patient exposure to systemic antibiotic resistance.
While various systemic antibiotics have been approved to treat more severe levels of DFI, according to the 2012 IDSA Clinical Practice Guideline for the Diagnosis and Treatment of Diabetic Foot Infections, there is no standard of care in Mild DFI. In the absence of any approved treatment for Mild DFI, oral antibiotics, such as quinolones, cephalosporins, or penicillins, are considered to be acceptable for off-label use. We are not aware of any ongoing clinical trials evaluating the use of these oral antibiotics for the treatment of Mild DFI. The IDSA does not recommend the use of any currently FDA approved topical antibiotics for the treatment of Mild DFI. In infected DFUs, currently available prescription wound healing products are either contraindicated, indicated for use only after establishing infection control, or indicated for use as an adjunct, and not a substitute for, antibiotics.
All classes of systemic antibiotics are associated with antibiotic resistance. Furthermore, side effects associated with a variety of systemic therapies include: gastrointestinal infections, liver and renal side effects, serious and sometimes fatal hypersensitivity reactions, central nervous system effects, QT interval prolongation (effects on heartbeat), myelosuppression (effects on bone marrow), and black box warnings for tendinopathy and tendon rupture. Some of these side effects can be exacerbated by drug-drug interactions in diabetic patients who often are on concurrent medications. Diabetics taking a hypoglycemic agent or insulin must be particularly mindful of blood glucose disturbances that can occur with systemic quinolone antibiotic therapy.
In addition to the absence of FDA-approved therapeutics labeled for the treatment of Mild DFI, research published in 2012 stated that no approved topical antibiotic products had demonstrated sufficient activity to recommend for off-label treatment in DFI of any level of severity. However, in a large-scale, randomized, active-controlled, double blind, multi-center clinical trial, topical Locilex™ was noted to be as effective as an oral quinolone in treating Mild or Moderate DFI.
49
Based upon discussions with our scientific advisors, the following chart highlights the key attributes of products which we believe are the most commonly used products to treat Mild or Moderate DFI:
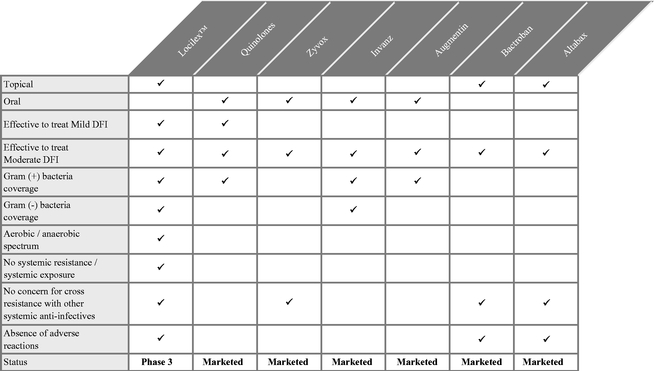
Mechanism of Action
We believe that the particular chemical attributes of Locilex™ will differentiate it from other antibiotic agents. Our API, pexiganan acetate, is a new chemical entity that is a chemically synthesized 22-amino acid analog of Magainin II, a natural host defense peptide isolated from skin secretions of the African Clawed Frog. Magainin II is one of a group of peptides that are considered to be the first line of defense against microbial invasion in this host. The peptides are simple linear structures that have a common feature of being basic peptides, which are able to form an amphipathic a-helical structure. This basic a-helical structure is the basis for the peptides' antimicrobial activity as inferred by studies in artificial lipid bilayers. Our API binds to artificial membranes, assumes an a-helical structure, and induces membrane lysis similar to Magainin II. In models of membrane interactions, the peptides exist as a random conformation in aqueous solution and an amphipathic a-helical structure is induced upon an initial ionic interaction with the microbial membrane. This interaction is not mediated by any membrane linked receptor. After ionic interaction, it is postulated that the hydrophobic side of the amphipathic a-helix of the lytic peptide is incorporated into the bilayer. The peptides could interact to form a raft-like structure in the membrane ultimately leading to the disruption of the membrane. In the model, the peptide raft will then realign perpendicular to the membrane surface to form a discrete pore structure with the helical peptides forming the sides of the pore like staves in a barrel. Although membrane disruption as a proposed mechanism of action is shared by other antimicrobial agents (e.g. polymyxin and amphotericin), the distinguishing characteristic of our API is its ability to cause membrane disruption in a broad spectrum of microbes including gram-negative bacteria, gram-positive bacteria, and fungi.
50
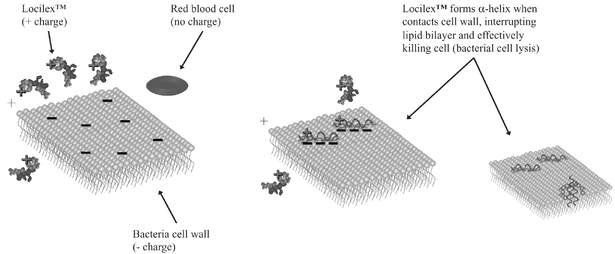
Clinical Development Strategy
Drug Development Strategy
We plan to concurrently conduct two Phase 3 clinical trials of Locilex™ for the treatment of Mild DFI. We have an SPA with the FDA relating to the trial design for our Phase 3 studies. The SPA calls for us to conduct two placebo-controlled Phase 3 studies, but does not require us to demonstrate equivalence to oral antibiotics. We expect to begin enrollment in these studies in the second quarter of 2014, with top line data anticipated in the first quarter of 2015. Both trials will be large-scale, randomized, active-controlled double blind, multi-center trials designed to establish the superiority of Locilex™ over placebo-cream in the treatment of Mild DFI. We plan to conduct our trials across approximately 40 clinical sites in the U.S. Each trial will be comprised of 180 patients randomized 1:1 (statistical power 90%; P=0.05) to receive standard wound care (off-loading, debridement) plus twice-daily Locilex™ cream or placebo-cream for 14 days. The primary endpoint of the trial will be clinical success, defined as resolution of infection per the clinical judgment of each treating physician using the 2012 DFI treatment guidelines. Microbiological success, defined as complete microbiological response, will be assessed as a secondary endpoint. The primary and secondary endpoints will be assessed for each patient at day 28. Furthermore, the FDA agreed that data from the Locilex™ arm of the two prior studies conducted by Locilex™'s prior sponsor can be used to supplement our safety database.
Concurrently, we also intend to initiate Phase 1 skin irritation and skin sensitization studies, which are expected to commence in the first quarter of 2014. The first Phase 1 study, for skin irritation, will evaluate 30 healthy volunteers, and the second Phase 1 study, for skin sensitization, will evaluate 200 healthy volunteers.
We have selected Research Pharmaceutical Services, Inc. as the contract research organization to conduct our Phase 3 studies for Locilex™. This work will be performed under the auspices of RRD International, LLC, which will manage all aspects of our scientific, clinical and regulatory development.
Previous Phase 3 Clinical Trial Results: Studies 303 and 304
Locilex™ was previously investigated in two Phase 3 clinical trials in patients with DFI, referred to as Studies 303 and 304, conducted by the prior sponsor. Each trial was a large-scale, randomized, active-controlled, double blind, multi-center trial designed to establish equivalence of Locilex™ to systemic ofloxacin. Across both trials, 835 patients received twice-daily Locilex™ cream plus placebo pill or twice-daily oral ofloxacin plus placebo cream for 14 to 28 days. The primary outcome was the clinical improvement of infection in response to antimicrobial treatment. Secondary outcomes
51
included eradication of the wound pathogens, healing of the ulcer, development of antibiotic resistance to a study drug, and safety.
Although one study (Study 303) did not demonstrate equivalence, the second study (Study 304) and the combined data for the two trials did demonstrate equivalent results for topical Locilex™ and oral ofloxacin in clinical improvement rates and overall microbiological eradication rates. Guidelines for establishing equivalence for these results were a 95% confidence interval with a lower bound delta of -15%, for clinical effectiveness, and a lower bound delta of -20%, for overall microbiological effectiveness. In each case, the confidence interval must cross zero. Additionally, there were no statistically significant differences in wound assessment scores between patients treated with Locilex™ and ofloxacin. Wound assessment scores decreased at the end of treatment visit in both studies for both treatment arms, and they decreased further at the follow-up visit. No serious adverse events were determined to have been related to either Locilex™ or ofloxacin. Moreover, the overall incidence of serious adverse events unrelated to the study medication (12% Locilex™ to 9% ofloxacin), including worsening cellulitis (2% to 4%) and amputation (2% to 3%), did not differ significantly between treatment arms. Bacterial resistance to ofloxacin emerged in some patients in the control arm, but no significant resistance to Locilex™ emerged among patients in the treatment arm.
A summary table of the primary clinical outcome, secondary microbiological outcome and wound assessment scores are shown below:
Clinical outcome (% cure or improved)
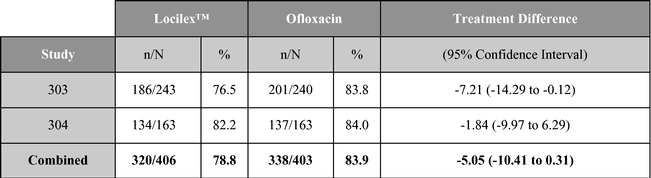
Microbiological outcome (% responders)
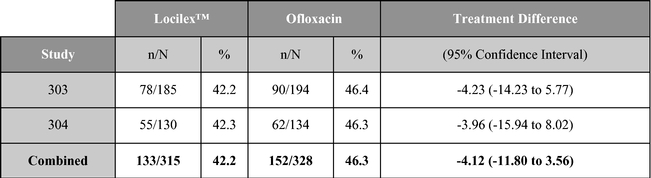
52
Wound assessment scores
| |
Locilex™ | Ofloxacin | |
|
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Change from baseline | |
Change from baseline | P | ||||||||||||||||||||
| |
Baseline value |
Baseline value |
|||||||||||||||||||||||
Study(s) and variable
|
EOT | Follow-up | EOT | Follow-up | EOT | Follow-up | |||||||||||||||||||
303 |
|||||||||||||||||||||||||
Total wound score, mean value |
25.5 | -7.6 | -8.5 | 24.2 | -8.0 | -8.7 | .61 | .78 | |||||||||||||||||
Wound infection score, mean value |
7.1 | -4.5 | -5.1 | 6.9 | -5.0 | -5.3 | .13 | .55 | |||||||||||||||||
Wound area, median mm2 |
131.5 | -61.9 | 67.8 | 117.3 | -58.3 | -64.4 | .53 | .57 | |||||||||||||||||
Wound depth, median mm |
3.0 | -1.0 | -1.5 | 3.0 | -1.0 | -1.5 | .30 | .48 | |||||||||||||||||
304 |
|||||||||||||||||||||||||
Total wound score, mean value |
26.2 | -8.6 | -9.0 | 25.7 | -8.5 | -8.9 | .92 | .87 | |||||||||||||||||
Wound infection score, mean value |
6.9 | -4.5 | -4.7 | 7.5 | -5.1 | -5.2 | .11 | .24 | |||||||||||||||||
Wound area, median mm2 |
146.9 | -63.0 | -64.7 | 160.8 | -70.2 | -86.4 | .17 | .03 | |||||||||||||||||
Wound depth, median mm |
3.0 | -1.0 | -1.5 | 3.0 | -1.0 | -2.0 | .42 | .58 | |||||||||||||||||
303 and 304 |
|||||||||||||||||||||||||
Total wound score, mean value |
25.8 | -8.0 | -8.7 | 24.8 | -8.2 | -8.7 | .75 | .91 | |||||||||||||||||
Wound infection score, mean value |
7.0 | -4.5 | -5.0 | 7.2 | -5.1 | -5.3 | .03 | .22 | |||||||||||||||||
Wound area, median mm2 |
138.0 | -62.6 | 67.1 | 138.7 | -65.8 | -72.1 | .17 | .07 | |||||||||||||||||
Wound depth, median mm |
3.0 | -1.0 | -1.0 | 3.0 | -1.0 | -2.0 | .20 | .36 | |||||||||||||||||
Source: Lipsky, et al. Clinical Infectious Disease. 2008; 47(12): 1537-45.
Microbiology Study Results
JMI Laboratories, an independent microbiology testing laboratory, conducted a minimum inhibitory concentration (or MIC) study and a minimum bactericidal concentration (or MBC) study on our behalf, in part, to respond to the FDA's request for information on the sensitivity of certain resistant bacteria to Locilex™. The results demonstrate that resistant bacteria, including MRSA, MDR pathogens and VRE, are particularly sensitive to the API in Locilex™. These results were deemed satisfactory by the FDA in February 2012. Many of the resistant isolates included in these two microbiology studies are included in the definition of "Qualifying Pathogens" under the U.S. Federal Government's GAIN Act (Generating Antibiotic Incentives Now) signed into law by President Obama in July 2012. See "– Government Regulation and Product Approval" below.
Most if not all of resistant bacteria tested in the MIC and MBC studies are commonly involved with mild or moderate skin and skin structure infections in superficial wounds. Therefore, we believe these data suggest that Locilex™ has potential as a standard of care for mild or moderate skin and skin structure infections in superficial wounds.
Manufacturing and Supply
We utilize three contract manufacturers (or CMOs) to produce Locilex™. Our manufacturing supply chain for Locilex™ starts with PolyPeptide Laboratories, Inc. which manufactures pexiganan, the API in Locilex™. At our direction, Polypeptide Laboratories delivers the API to DPT Laboratories, Inc., which formulates the API into a cream formulation on our behalf. DPT Laboratories then delivers the formulated product to Almac Group Limited, which labels, packages, and delivers the finished goods as we request.
In the late 1990s, our prior sponsor engaged in an FDA review process for a prior formulation of Locilex™. In its 1999 non-approvable letter, the FDA identified two cGMP manufacturing issues. The first issue concerned the stability of the product. Examination of the product over time showed evidence of water separation from the cream matrix. The second issue related to the purity level of the API in the product. The prior source of the API yielded a purity level as low as 95%.
53
After acquiring the rights to Locilex™, we developed a detailed product development plan to arrive at an optimized formulation to address these issues to the satisfaction of the FDA. We believe that the changes we made to the formulation have resolved the previously observed product separation. We have manufactured our initial commercial scale 30 kg batch of Locilex™ in accordance with current good manufacturing practices (or cGMPs), and this batch is currently subject to ongoing stability testing. Our stability testing is conducted in compliance with the ICH Guideline Q1A(R2): International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartate Guideline, Stability Testing of New Drug Substances and Products, Current Step 4 version, Dated 6 February 2003. The purpose of the stability testing is to provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors such as temperature, humidity, and light, and to establish a re-test period for the drug substance or a shelf life for the drug product and recommended storage conditions. We have achieved one-month stability data on our cGMP batch that we intend to use in our clinical studies in the first quarter of 2014. Our CMOs have confirmed that no stability problems have been observed to date. At the present time, the stability data supports a shelf life of at least 36 months for Locilex™.
We also believe our peptide is highly purified. The impurity levels of the API used in our commercial scale batch of Locilex™ has been confirmed to be 0.6% for the API used in our cGMP batch.
In October 2013, we submitted our manufacturing data, including data from the cGMP batch as well as 18-month stability data on our non-cGMP batch, to the FDA, and in December 2013 the FDA indicated in written communications with us that Locilex™'s stability and purity levels are acceptable for use in our upcoming Phase 3 studies. As a result of the aforementioned activities, we believe we have resolved the stability and purity concerns previously articulated by the FDA.
Commercial Plans
We intend to market and sell Locilex™, if approved, through a small, specialty sales force primarily to podiatrists at extended care facilities, wound care clinics, high volume group podiatry practices, and other treatment centers that specialize in the care of patients with DFI. Initially, we intend to hire a senior national sales officer, a senior marketing officer, and either directly or through a contract sales organization, a small team of fully dedicated Locilex™ sales representatives. Based on current data from the Bureau of Labor Statistics, we estimate that there are approximately 13,700 podiatrists in the U.S. Our marketing efforts may also include other specialty healthcare providers.
Intellectual Property
We hold rights to a U.S. patent covering our proprietary formulation of Locilex™ and the method of using it for the treatment of skin and wound infections (U.S. Patent Number 8,530,409). This patent was granted in September 2013 and expires in the U.S. in June 2032. The patent incorporates discoveries made by Dow Pharmaceutical Sciences, Inc. (later acquired by Valeant Pharmaceuticals International, Inc.). The application which gave rise to U.S. Patent No. 8,520,409 was assigned to us in June 2013. In addition, we have filed a Patent Cooperation Treaty (or PCT) application claiming priority to U.S. Patent No. 8,520,409 that will allow us to seek corresponding protection outside of the U.S., including in Europe, Japan, China, Australia, and Korea, as well as in other PCT jurisdictions.
In addition to this patent, we hold an exclusive sublicense to the composition-of-matter patent covering the pexiganan technology (U.S. Patent No. 5,912,231) which expires in June 2016, not including any patent term extension that we expect to seek under the Drug Price Competition and Patent Term Restoration Act of 1984 (or the Hatch-Waxman Act). We acquired this sublicense when we acquired the rights to Locilex™ in April 2010. Our rights to practice the pexiganan technology are originally derived through a license agreement between Scripps Research Institute (or Scripps), the inventor of the pexiganan technology, and Multiple Peptide Systems, Inc. (or MPS). MPS then
54
sublicensed the pexiganan technology to the prior sponsor of the pexiganan program. In October 1996, both of the license and sublicense agreements were amended by Scripps, MPS and the prior sponsor of Locilex™ to confirm that the license and sublicense were fully-paid, royalty free and of indefinite duration, with no further economic obligations for the practice of the pexiganan technology.
Although U.S. Patent 5,912,231 supplements our existing intellectual property portfolio, we are chiefly reliant on our U.S. Patent 8,530,409, which covers the novel formulation and method of use for Locilex™ and provides for substantially longer patent coverage than U.S. Patent 5,912,231, and that U.S. Patent 8,530,409's attributes as a topical formulation, its potentially broader scope of coverage and opportunity for foreign patent protection offer greater benefits to us than U.S. Patent 5,912,231. As such, we have not yet engaged in any discussions with Scripps regarding a possible patent term extension for U.S. Patent No. 5,912,231. If and when we decide to apply for an extension, we will we will need to work with Scripps throughout the application process to facilitate its approval.
Competition
The pharmaceutical and biotechnology industry is characterized by intense competition, rapid product development and technological change. Competition is intense among manufacturers of prescription pharmaceuticals and other product areas where we may develop and market products in the future. Most of our competitors are large, well established pharmaceutical or healthcare companies with considerably greater financial, marketing, sales and technical resources than are available to us. Additionally, many of our competitors have research and development capabilities that may allow such competitors to develop new or improved products that may compete with Locilex™. Our product could be rendered obsolete or made uneconomical by the development of new products to treat Mild DFI or other acute bacterial skin infections.
Our business, financial condition and results of operation could be materially adversely affected by any one or more of such developments. We cannot assure you that we will be able to compete successfully against current or future competitors or that competition will not have a material adverse effect on our business, financial condition and results of operations. Academic institutions, governmental agencies and other public and private research organizations are also conducting research activities and seeking patent protection and may commercialize products on their own or with the assistance of major healthcare companies for indications for which we are developing Locilex™. We are aware of certain development projects for products to treat or prevent certain diseases targeted by us. The existence of these potential products or other products or treatments of which we are not aware, or products or treatments that may be developed in the future, may adversely affect the marketability of Locilex™.
Even if Locilex™ receives regulatory approval, of which there can be no assurance, our competitors' drugs may be more effective, more effectively marketed and sold, or less costly than Locilex™, and may render our product obsolete or non-competitive before we can recover the expenses of developing and commercializing Locilex™.
Although there are currently no products approved specifically to treat Mild DFI, we anticipate that, if approved, Locilex™ will compete with other anti-infective products that are marketed for the general treatment of diabetic foot ulcers. These include systemic products that have been generally accepted by the ISDA to treat DFI at other levels of severity such as linezolid (marketed by Pfizer, Inc. as Zyvox), piperacillin/tazobactam (marketed by Pfizer, Inc. as Zosyn), ertapenam (marketed by Merck & Co. as Invanz); generic products recommended by the IDSA for off-label use in the treatment of all severity levels of DFI such as dicloxacillin, clindamycin, cephalexin, ofloxacin, trimethoprim, amoxicillin-clavulanate, and others; as well as antimicrobial wound care dressings such as medicinal grade honey and antiseptics such as silver and iodine. Furthermore, we are aware of other development stage topical or systemic products that may represent significant competition if approved. These include, but are not limited to nemonoxacin/TG-873870 (under development by
55
TaiGen Biotechnology Co., Ltd.), Dermacyn (under development by Oculus Innovative Sciences, Inc.), and Cogenzia (under development by Innocoll, Inc.).
In addition, some of our competitors have greater experience than we do in conducting preclinical and clinical trials and obtaining FDA and other regulatory approvals. Accordingly, our competitors may succeed in obtaining FDA or other regulatory approvals for drug candidates more rapidly than we do. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before their competitors may achieve a significant competitive advantage. Locilex™ therefore may not be commercially competitive with existing products or products under development. With respect to Mild, Moderate or Severe DFI specifically, such competitors include very large international organizations such as Pfizer, Inc., Eli Lilly and Company, Johnson & Johnson and GlaxoSmithKline plc.
History of Locilex™
In August 1992, the prior sponsor of Locilex™, Magainin, submitted an initial IND for the prior formulation of Locilex™ to study broad spectrum anti-infective activity for the treatment of superficial and complicated dermatological infections. Another IND was submitted in November 1993 to cover a new indication for the treatment of DFI. In the late 1990s, the prior sponsor tested the prior formulation of Locilex™, with over 1,000 human subjects exposed without safety concerns, to such prior formulation of Locilex™, including 835 evaluable patients in two Phase 3 clinical trials. The Phase 3 trial results showed that such prior formulation of topical Locilex™ had an approximate 80% response rate measured as resolution or improvement in infection in patients who under today's standards would be considered to have Mild or Moderate DFI. See "– Clinical Development Strategy – Phase 3 Clinical Trial Results: Studies 303 and 304" above.
The FDA Advisory Committee reviewing Locilex™ at the time unanimously approved the safety of the product, but did not approve its efficacy and recommended an additional Phase 3 placebo controlled trial. In its 1999 non-approvable letter, the FDA identified certain cGMP manufacturing deficiencies, namely stability and quality control issues, and questions regarding the comparability of the product used in the Phase 3 program versus that which was produced at commercial scale. We believe that these hurdles and a lack of financing ultimately caused Magainin (later renamed Genaera Corporation) to deprioritize the product within their product pipeline. MacroChem Corporation (or MacroChem) licensed the technology in late 2007, after several years of attempting to remediate the manufacturing deficiencies. In February 2009, MacroChem was acquired by Access Pharmaceuticals, Inc. (or Access), which focused on oncology and oncology supportive care product candidates. Rights to Locilex™ reverted to Genaera Liquidating Trust (established to sell the drug related assets of Genaera Corporation in liquidation) when Access failed to start a Phase 3 trial by the two-year anniversary of the effective date of the license agreement, triggering a termination right for Genaera Liquidating Trust in December 2009.
In April 2010, we acquired the worldwide rights to pexiganan, the API in Locilex™, and the prior formulation of the product and all related assets after participating in a public auction for our product conducted by Genaera Liquidating Trust. During the period between the FDA non-approval letter received in July 1999 and the second half of 2006, SmithKline Beecham Corporation (now part of GlaxoSmithKline plc) held the exclusive distribution rights to Locilex™ in the U.S.
In March 2011, we exercised our exclusive option to buy out the downstream, success-based milestones and royalty obligations related to Locilex™ and currently own 100% of our product candidate.
Government Regulation and Product Approval
Governmental authorities in the U.S., at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling,
56
packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Our product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the U.S. and by the European Medicines Agency (or EMA) through the Marketing Authorisation Application (or MAA) process before they may be legally marketed in Europe. Our product candidates will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Government Regulation
NDA Approval Processes
In the U.S., the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (or FDCA) and implementing regulations. Failure to comply with the applicable U.S. requirements at any time during the product development process or approval process, or after approval, may subject an applicant to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include:
- •
- refusal to approve pending applications;
- •
- withdrawal of an approval;
- •
- imposition of a clinical hold;
- •
- warning letters;
- •
- product seizures;
- •
- total or partial suspension of production or distribution; or
- •
- injunctions, fines, disgorgement, or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the U.S. generally involves the following:
- •
- completion of nonclinical laboratory tests, animal studies and formulation studies conducted according to Good Laboratory
Practices (or GLPs) or other applicable regulations;
- •
- submission to the FDA of an investigational new drug application (or IND), which must become effective before human
clinical trials may begin;
- •
- performance of adequate and well-controlled human clinical trials according to Good Clinical Practices (or GCPs) to
establish the safety and efficacy of the proposed drug for its intended use;
- •
- submission to the FDA of an NDA;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced
to assess compliance with current cGMPs to assure that the facilities, methods and controls are adequate to preserve the drug's identity, strength, quality and purity;
and
- •
- FDA review and approval of the NDA.
Once a pharmaceutical candidate is identified for development, it enters the preclinical or nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the nonclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some nonclinical testing may continue even after the IND is submitted. In addition to including the results of the nonclinical studies, the IND will also include a protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness
57
criteria to be evaluated if the first phase lends itself to an efficacy determination. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the IND on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND, and may affect one or more specific studies or all studies conducted under the IND.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCPs. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject selection and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and progress reports detailing the status of the clinical trials must be submitted to the FDA annually. Sponsors also must timely report to FDA serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigation brochure, or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the drug. An institutional review board, or IRB, at each institution participating in the clinical trial must review and approve the protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each research subject or the subject's legal representative, monitor the study until completed and otherwise comply with IRB regulations.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
- •
- Phase 1. The drug is initially introduced into
healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some products for severe or life-threatening diseases, such as
cancer, especially when the product may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
- •
- Phase 2. Clinical trials are performed on a limited
patient population intended to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage
tolerance and optimal dosage.
- •
- Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product and provide an adequate basis for product labeling.
Human clinical trials are inherently uncertain and Phase 1, Phase 2 and Phase 3 testing may not be successfully completed. The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements or if the drug has been associated with unexpected serious harm to patients.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to the submission of an IND, at the end of Phase 2 and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support the approval of the new drug. If a Phase 2 clinical trial is the subject of discussion at the end of Phase 2 meeting with the FDA, a sponsor may be able to request a SPA, the purpose of which is to reach agreement with the FDA on the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
58
According to published guidance on the SPA process, a sponsor which meets the prerequisites may make a specific request for a SPA and provide information regarding the design and size of the proposed clinical trial. The FDA is supposed to evaluate the protocol within 45 days of the request to assess whether the proposed trial is adequate, and that evaluation may result in discussions and a request for additional information. A SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the testing began.
Concurrent with clinical trials, sponsors usually complete additional animal safety studies and also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing commercial quantities of the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug and the manufacturer must develop methods for testing the quality, purity and potency of the drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its proposed shelf-life.
The results of product development, nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of user fees, but a waiver of such fees may be obtained under specified circumstances. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. It may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in-depth review. NDAs receive either standard or priority review. A drug representing a significant improvement in treatment, prevention or diagnosis of disease may receive priority review. The FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The FDA may refer the NDA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured and tested. We anticipate that our NDA submission, which may technically be classified as an amended NDA (a so-called Class II resubmission), will address the manufacturing concerns previously articulated by the FDA regarding the prior formulation of Locilex™. Should our NDA be accepted for review, the FDA is supposed to respond within six months of submission.
Recent Changes to the Regulatory Landscape for Anti-Infective Drugs
The analytic approach of the FDA's Anti-Infective Drugs Division has undergone evolution in recent years, primarily driven by concerns that increasingly less effective antibiotics may have been approved in the last 10 to 15 years. The impact of these changes was a rethinking of how antibiotic efficacy is measured in clinical trials, and a review of the statistical tools used to analyze the data. In March 2009, the FDA published a draft guidance entitled "Guidance for Industry Community-Acquired Bacterial Pneumonia: Developing Drugs for Treatment" and in August 2010, it published draft
59
guidance (subsequently published as final guidance in October 2013) entitled "Guidance for Industry Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment" (or 2010 Guidance). The purpose of this guidance was to address many of the uncertainties regarding what the FDA expected from sponsors and clinical trials for the indications of acute bacterial skin and skin structure infections and community-acquired bacterial pneumonia.
The FDA asked sponsors to include additional measurements in their evaluation of efficacy that the FDA believes are more objective and less susceptible to interpretation by investigators. Non-inferiority comparisons of drugs are the standards for antibiotics, and non-inferiority margins are the margins used in the statistical analysis comparing two treatment arms in a study. These are the statistical margins or rules used to distinguish the degree of potential difference between two antibiotics in a study. In September 2010, one month after issuing the 2010 Guidance, the FDA approved the first antibiotic NDA reviewed pursuant to these new endpoints and non-inferiority margins. The clinical protocol that was reviewed by the FDA in support of our SPA with the FDA includes provisions that are consistent with the 2010 Guidance, as well as the FDA's final guidance published in October 2013.
Expedited Review and Approval
The FDA has various programs, including Fast Track, priority review, and accelerated approval, which are intended to expedite or simplify the process for reviewing drugs, and/or provide for the approval of a drug on the basis of a surrogate endpoint. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will be shortened. Generally, drugs that are eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious or life-threatening diseases or conditions and fill unmet medical needs. Priority review is designed to give drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of ten months.
Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track designated drug and expedite review of the application for a drug designated for priority review. Accelerated approval, which is described in Subpart H of 21 CFR Part 314, provides for an earlier approval for a new drug that is intended to treat a serious or life-threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint. A surrogate endpoint is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome. As a condition of approval, the FDA may require that a sponsor of a product candidate receiving accelerated approval perform post-marketing clinical trials.
In the Food and Drug Administration Safety and Innovation Act (or FDASIA), which was signed into law in July 2012, Congress encouraged the FDA to utilize innovative and flexible approaches to the assessment of products under accelerated approval. The law required the FDA to issue related draft guidance within a year after the law's enactment and also promulgate confirming regulatory changes. In June 2013, the FDA published a draft Guidance for Industry entitled, "Expedited Programs for Serious Conditions – Drugs and Biologics" which provides guidance on FDA programs that are intended to facilitate and expedite development and review of new drugs as well as threshold criteria generally applicable to concluding that a drug is a candidate for these expedited development and review programs. In addition to the Fast Track, accelerated approval and priority review programs discussed above, the FDA also provided guidance on a new program for Breakthrough Therapy designation. A request for Breakthrough Therapy designation should be submitted concurrently with, or as an amendment to an IND. FDA has already granted this designation to around 30 new drugs and recently approved a couple of Breakthrough Therapy designated drug.
60
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our drug candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product's approval date. The patent term restoration period is generally one-half the time between the effective date of an IND, and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for extension must be made prior to expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (or ANDA) or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
FDASIA includes the Generating Antibiotic Incentives Now Act (or GAIN Act), which is intended to provide incentives for the development of new qualified infectious disease products. A new drug that is designated as a qualified infectious disease product after a request by the sponsor that is made before an NDA is submitted will be eligible, if approved, for an additional five years of exclusivity beyond any period of exclusivity to which it would have previously been eligible. In addition, a qualified infectious disease product will receive priority review and Fast Track designation. Qualified infectious disease products are defined as antibacterial or antifungal drugs intended to treat serious or life-threatening infections that are resistant to treatment, or that treat qualifying resistant pathogens identified by the FDA. Examples of pathogens that may be designated as a qualifying pathogen include MRSA, VRE and multi-drug resistant gram-negative bacteria. We believe that Locilex™ will qualify for this designation because it has already shown promising results in treating several qualifying pathogens in preclinical studies and clinical trials, and we have applied for "qualified infectious disease product" status under the GAIN Act. On January 14, 2014, we received notification from the FDA that it had denied our request for such status, primarily because there is no clinical evidence that Mild DFI is a serious condition. The FDA invited us to reapply either now if such evidence exists or in the future if such evidence becomes available or if we would like to target another clinical indication. Accordingly, we intend to re-apply for "qualified infectious disease
61
product" status in the first quarter of 2014 based on clinical evidence from the prior Studies 303 and 304 that demonstrates that Locilex™ successfully treats resistant bacteria, including MRSA, multi-drug resistant bacteria and VRE, in patients with Moderate DFI, which we believe is widely accepted as a serious condition.
Pediatric Exclusivity and Pediatric Use
Under the Best Pharmaceuticals for Children Act (BPCA) certain drugs may obtain an additional six months of exclusivity, if the sponsor submits information requested in writing by the FDA (or Written Request) relating to the use of the active moiety of the drug in children. The FDA may not issue a Written Request for studies on unapproved or approved indications or where it determines that information relating to the use of a drug in a pediatric population, or part of the pediatric population, may not produce health benefits in that population.
We have not received a Written Request for such pediatric studies, although we may ask the FDA to issue a Written Request for such studies in the future. To receive the six-month pediatric market exclusivity, we would have to receive a Written Request from the FDA, conduct the requested studies in accordance with a written agreement with the FDA or, if there is no written agreement, in accordance with commonly accepted scientific principles, and submit reports of the studies. A Written Request may include studies for indications that are not currently in the labeling if the FDA determines that such information will benefit the public health. The FDA will accept the reports upon its determination that the studies were conducted in accordance with and are responsive to the original Written Request or commonly accepted scientific principles, as appropriate, and that the reports comply with the FDA's filing requirements.
In addition, the Pediatric Research Equity Act (or PREA) requires all applications (or supplements to an application) submitted under section 505 of the FDCA (21 U.S.C. Section 355) for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration to contain a pediatric assessment unless the applicant has obtained a waiver or deferral. It also authorizes the FDA to require holders of approved NDAs for marketed drugs to conduct pediatric studies under certain circumstances. In general, PREA applies only to those drugs developed for diseases and/or conditions that occur in both the adult and pediatric populations. Products intended for pediatric-specific indications will be subject to the requirements of PREA only if they are initially developed for a subset of the relevant pediatric population.
As part of FDASIA, Congress reauthorized both BPCA and PREA, which were slated to expire on September 30, 2012, and made both laws permanent.
Post-approval Requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Any drug products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things:
- •
- record-keeping requirements;
- •
- reporting of adverse experiences with the drug;
62
- •
- providing the FDA with updated safety and efficacy information;
- •
- drug sampling and distribution requirements;
- •
- notifying the FDA and gaining its approval of specified manufacturing or labeling changes;
and
- •
- complying with FDA promotion and advertising requirements.
Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMP and other laws.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be.
Regulation Outside of the U.S.
In addition to regulations in the U.S., we will be subject to regulations of other countries governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of countries outside of the U.S. before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the E.U., before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under E.U. regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology or those medicines intended to treat AIDS, cancer, neurodegenerative disorders or diabetes and optional for those medicines which are highly innovative, provides for the grant of a single marketing authorization that is valid for all E.U. member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessments report, each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
Reimbursement
Sales of our products will depend, in part, on the extent to which the costs of our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on
63
reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (or MMA) imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor's product could adversely affect the sales of our product candidates. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (or collectively, the ACA), enacted in March 2010, is expected to have a significant impact on the health care industry. ACA is expected to expand coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, the ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the impact of the ACA on pharmaceutical companies, as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet occurred. In addition, some members of the U.S. Congress have been seeking to overturn at least portions of the legislation and we expect they will continue to review and assess this legislation and alternative health care reform proposals. Any legal challenges to the ACA, as well as Congressional efforts to repeal the ACA, add to the uncertainty of the legislative changes enacted as part of the ACA.
64
In addition, in some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the E.U. provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the E.U. do not follow price structures of the U.S. and generally tend to be significantly lower.
Scientific Advisory Board
In December 2013, we formally established a Scientific Advisory Board to advise our management regarding our clinical and regulatory development programs and other customary matters. Our scientific advisors are experts in various areas of medicine including DFI, mild and moderate skin and skin structure infections in superficial wounds and podiatry. We believe the advice of our scientific advisors was integral to the quality of our clinical trial protocol for our Phase 3 program and the resulting SPA. Our Scientific Advisory Board is comprised of the following individuals:
- •
- Dr. Jonathan Wilkin. Founding Director (retired)
of the Division of Dermatology and Dental Products at the FDA. Remains active in regulatory matters after over 12 years of FDA service, which included membership on the FDA's Dermatology Drugs
Advisory Committee.
- •
- Dr. Benjamin Lipsky. Emeritus Professor of
Medicine, University of Washington; Visiting Professor, Infectious Diseases, University of Geneva. Teaching Associate, Green Templeton College, University of Oxford. Head of the International Working
Group on the Diabetic Foot (or IWGDF) and lead author of the Diabetic Foot Infection Treatment Guidelines, published in June 2012.
- •
- Dr. David Armstrong. Professor of Surgery and
Director, Southern Arizona Limb Salvage Alliance (or SALSA), University of Arizona College of Medicine. Co-Sponsor of annual Diabetic Foot Global Conference (or DFCon).
- •
- Dr. Warren Joseph. Adjunct Clinical Associate
Professor, Dr. William Scholl College of Podiatric Medicine, Rosland Franklin University of Medicine and Science. Managing Editor of the Journal of the American Podiatric Medical
Association.
- •
- Dr. Michael Zasloff. Original inventor of the "magainin peptides" which include pexiganan, while he was a research scientist at the National Institutes of Health (or NIH) in 1987. Co-Founder of Magainin, the original owner of Locilex™. Current Professor, Departments of Surgery & Pediatrics; and Director, Surgical Immunology, Georgetown University School of Medicine.
We will continue to rely upon our scientific advisors in various aspects of our product development program including, without limitation, assisting with the publication in the future of the clinical data generated in our Phase 3 program in coordination with us.
Our Management
Our management team has extensive experience in leading the development of innovative therapeutics and significant expertise in operational, financial and corporate development functions. Our co-founder, President and Chief Executive Officer, David P. Luci, Esq., has managed multiple drug development companies including Bioenvision, Inc. (sold to Genzyme Corporation for $345 million in 2007), MacroChem (sold to Access in 2009), Access and our company. Our co-founder and Executive Chairman, Robert J. DeLuccia, has extensive product development, sales and marketing experience as
65
well as experience in management and operation of pharmaceutical and biotechnology companies of various sizes. Collectively, Messrs. Luci and DeLuccia have over 50 years of combined experience in the pharmaceutical and biotechnology sectors.
Employees
As of March 6, 2014, we have a total of seven employees and consultants, of which two are full-time employees and five are part-time employees or consultants. We believe our relationships with our employees and consultants are satisfactory. We have never experienced employment-related work stoppages and consider that we maintain good relations with our personnel. In addition, to complement our internal expertise, we have contracts with medical and scientific consultants, manufacturers, laboratories, and contract research organizations that specialize in various aspects of drug development including clinical development, preclinical development, manufacturing and regulatory affairs.
Description of Office Facility
We maintain one facility of approximately 80 square feet for administrative offices at 74 Broad Street, New York, New York 10004 in a month to month tenancy. Adjacent space may be available for expansion which we believe would accommodate our growth in the foreseeable future. We believe that our existing properties are suitable for the conduct of our business and adequate to meet our current needs.
Corporate Information
We were organized originally as a limited liability company under the laws of the State of Delaware in 2010 as Dipexium Pharmaceuticals, LLC, and we plan to convert to a corporation under the name Dipexium Pharmaceuticals, Inc. in connection with this offering. Our principal executive office is located at 74 Broad Street, New York, New York 10004, and our phone number is (212) 422-5717.
Web Availability
We intend to make our annual reports on Form 10-K for the years ended at or after December 31, 2014 and other reports that we file with the Securities and Exchange Commission (or SEC) as well as certain of our corporate governance policies, including the charters for the audit, compensation and nominating and corporate governance committees of our board of directors and our code of ethics available free of charge through our website, www.dipexiumpharmaceuticals.com. We will also provide to any person without charge, upon request, a copy of any of the foregoing materials. Any such request must be made in writing to us at: Dipexium Pharmaceuticals, Inc., 74 Broad Street, New York, New York 10004, Attn: Investor Relations.
Legal Proceedings
We and our key employees, David P. Luci and Robert J. DeLuccia, were three of some 30 defendants in a lawsuit filed by a former stockholder of Genaera Corporation, which was the predecessor of the Genaera Liquidating Trust, the party which sold us the worldwide rights to pexiganan, the API in our product Locilex™, in April 8, 2010.
The complaint was filed on June 8, 2012 in the United States District Court for the Eastern District of Pennsylvania (Civil Action No. 12-3265) by Alan W. Schmidt, individually and on behalf of former Genaera Corporation shareholders. Among others, the suit was filed against us, as well as John A. Skolas and Argyce LLC (or Argyce), who were responsible for the administration of the Genaera Liquidating Trust and who sold pexiganan to us via a public auction. The defendants listed in the
66
complaint included several individuals and companies formerly associated with Genaera Corporation, Genaera Liquidating Trust and/or Argyce, as well as against our company and Messrs. Luci and DeLuccia, as individuals. Also included in the defendant group were several other pharmaceutical companies that were involved in acquiring the former drug-related assets of Genaera Corporation and their related institutional and personal investors.
The complaint alleged, among other things, that our company and Messrs. Luci and DeLuccia aided and abetted a breach of fiduciary duty alleged to have been committed by the Argyce and the Genaera Liquidating Trust as well as former individuals associated with Genaera Corporation before it went into liquidation via a majority vote of its shareholders. Plaintiff claimed that the defendants, including our company and Messrs. Luci and DeLuccia, aided and abetted a breach of the duties of the trustee under common law and under a certain trust agreement allegedly signed between Argyce, as trustee, and the Genaera Liquidating Trust. With regard to the claims made against our company and Messrs. Luci and DeLuccia, the plaintiff alleged, in pertinent part, that our company's acquisition of the pexiganan rights (including the rights to the prior formulation of Locilex™) was for alleged inadequate consideration, and that we and our management aided and abetted a breach of fiduciary duty by the defendants who were formerly associated with Genaera Corporation and/or the Genaera Liquidating Trust. Plaintiff also claimed that we and Messrs. Luci and DeLuccia aided and abetted a breach of the duty of the trustee at common law and under a certain trust agreement alleged to exist and signed by Argyce, as trustee, and Genaera Liquidating Trust pursuant to which Argyce apparently was appointed to liquidate the assets formerly held by Genaera Corporation.
We and Messrs. Luci and DeLuccia filed a motion to dismiss the complaint within the proscribed time period. All of the other defendants in this litigation also filed motions to dismiss, and a court order granting each and every motion to dismiss, with prejudice, without leave to refile, was granted by the court on August 12, 2013. Plaintiff's lawyers then filed a Motion for Reconsideration which was also denied by the court on September 25, 2013. Plaintiff has filed an appeal in the United States Court of Appeals for the Third Circuit seeking reversal of the court's orders dismissing the complaint and denying the motion to reconsider. Appellate court briefing is underway and expected to be concluded in mid-2014. We and Messrs. Luci and DeLuccia continue to believe these claims have no merit and we intend to vigorously defend our position in the appellate court.
67
The following table sets forth the names of our directors and executive officers along with their respective ages and positions:
| Name | Age | Title | ||||
|---|---|---|---|---|---|---|
Robert J. DeLuccia(1) |
68 | Executive Chairman and Director | ||||
David P. Luci, Esq.(1) |
47 | President, Chief Executive Officer, Secretary and Director | ||||
William J. McSherry, Jr., Esq.(2)(4) |
65 | Director | ||||
Dr. Jack H. Dean(3)(4) |
72 | Director | ||||
Christopher Coughlin(2)(3) |
61 | Director | ||||
Thomas Harrison(3) |
66 | Director | ||||
Michael Duffy, Esq.(2)(4) |
44 | Director | ||||
David Garrett(1) |
38 | Vice President, Finance and Corporate Development | ||||
Robert G. Shawah, CPA(1) |
47 | Chief Accounting Officer and Treasurer | ||||
- (1)
- Messrs.
DeLuccia, Luci, Garrett and Shawah will assume their respective officer positions upon the consummation of our corporate conversion.
- (2)
- Audit
Committee Appointee
- (3)
- Nominating
and Corporate Governance Committee Appointee
- (4)
- Member, Compensation Committee
All directors hold office for one-year terms until the election and qualification of their successors. Officers are appointed by our board of directors and serve at the discretion of the board, subject to applicable employment agreements.
No director, officer, affiliate or promoter of our company has, within the past ten years, filed any bankruptcy petition, been convicted in or been the subject of any pending criminal proceedings, or is any such person the subject or any order, judgment or decree involving the violation of any state or federal securities laws.
The following is a description of the business experience of each director and executive officer of our company:
Robert J. DeLuccia. Mr. DeLuccia serves as a director of our company and is one of the two co-founders and managing partners of our company, which was formed in 2010. Upon the consummation of our corporate conversion, he will serve as our Executive Chairman. From 2004 to 2009, Mr. DeLuccia served in several capacities at MacroChem, a development-stage, publicly traded pharmaceutical company using topical drug delivery technology for products in dermatology, podiatry, urology and cancer, including as Chairman, President and Chief Executive Officer, and as director. Prior to joining MacroChem, Mr. DeLuccia served as President and Chief Executive Officer of Immunomedics, Inc., a publicly-traded biopharmaceutical company focused on antibody-based therapeutic products and diagnostic imaging for cancer and infectious diseases. Mr. DeLuccia also served as President of Sterling Winthrop, Inc. (or Sterling Winthrop) (as an independent corporation and then as subsidiary of Eastman Kodak), and subsequently, upon acquisition, the U.S. subsidiary of Sanofi-Aventis (or Sanofi) and currently serves as a member of the board of directors of IBEX Technologies Inc., which manufactures and markets proprietary enzymes (heparinases and chondroitinases) for use in pharmaceutical research and Heparinase I, used in many leading hemostasis monitoring devices. Mr. DeLuccia began his career as a pharmaceutical sales representative for Pfizer, Inc. (or Pfizer) and progressed to Director of Marketing, Pfizer Laboratories Division and to Vice President Marketing and Sales Operations for Pfizer's Roerig Division. Mr. DeLuccia received a Bachelor of Business Administration with a concentration in Marketing and a Master's Degree in Business Administration, both from Iona College.
68
David P. Luci. Mr. Luci is a director of our company and is one of the two co-founders and managing partners of our company, which was formed in 2010. Upon the consummation of our corporate conversion, he will serve as our President, Chief Executive Officer and Secretary. Prior to co-founding our company, from February 2009 to January 2010, Mr. Luci served as a member of the board of directors of Access, where he also served as Chairman of the Audit Committee and Chairman of the Compensation Committee as well as serving in a consulting capacity following the acquisition of MacroChem. From December 2007 through February 2009, Mr. Luci served as a member of the board of directors and President of MacroChem. Prior to that, Mr. Luci served as Executive Vice President, Chief Financial Officer, General Counsel and Corporate Secretary of Bioenvision, Inc. (or Bioenvision), an international biopharmaceutical company focused upon the development, marketing and commercialization of oncology products and product candidates. Mr. Luci created and managed Bioenvision's principal executive offices located in New York as well as its satellite office located in Tokyo, Japan. Mr. Luci was instrumental in creating Bioenvision's international commercial enterprise; managed the worldwide development of Evoltra (clofarabine) as a member of the product's Joint Steering Committee in conjunction with senior executives of Bioenvision's partner, Genzyme Corporation; and orchestrated, structured and negotiated the sale of Bioenvision in 2007 to Genzyme Corporation for $345 million. Mr. Luci began his career with Ernst & Whinney LLP (now Ernst &Young LLP) in New York as a certified public accountant working in the Healthcare Practice Group. He later practiced corporate law at Paul Hastings LLP in New York, where his practice encompassed all aspects of public and private mergers and acquisitions, corporate finance, restructurings and private equity transactions, with a core focus in the healthcare industry. Mr. Luci graduated from Bucknell University with a degree as a Bachelor of Science in Business Administration with a concentration in Accounting and graduated from Albany Law School of Union University where he served as Managing Editor of the Journal of Science & Technology. Mr. Luci became a certified public accountant in the State of Pennsylvania in 1990 (inactive) and is a member of the New York State Bar Association.
William J. McSherry, Jr., Esq. Mr. McSherry has served as a director of our company since October 2010. Mr. McSherry has served as a partner at Eaton & Van Winkle LLP in New York since 2014 in the firm's litigation department, where he specializes in the areas of securities, mergers and acquisitions, financial institutions, derivatives, structured finance, insurance, reinsurance, life sciences, real estate, product liability and trademark law. Mr. McSherry previously served as a partner and the New York Chair of Crowell & Moring's Litigation Group from 2006 to 2014, during which he conducted numerous trials and arbitrations throughout the U.S. Prior to joining Crowell & Moring LLP, Mr. McSherry served as a partner at Arent Fox LLP in New York from 2000 to 2006. Mr. McSherry received his B.A. from Fordham College in 1969 and received a J.D. from Harvard Law School in 1973. Mr. McSherry is an active member of the American Bar Association, the New York State Bar Association, the U.S. Supreme Court Historical Society and the Association of the Bar of the City of New York, where he also served as a member of several committees: State Courts of Superior Jurisdiction (from 1980 to 1983), Arbitration and ADR (from 1987 to 1989) and Sports Law (from 1999 to 2001). Mr. McSherry is also a member of the New York State Bar Association. Mr. McSherry has published articles on numerous litigation related topics as well as a chapter on derivatives litigation in a publication entitled "Derivatives and Risk Management."
Jack H. Dean, Ph.D., Sc.D. (Hon.), DABT, Fellow ATS. Dr. Dean has been a director of our company since October 2010. From January 2006 to the present, Dr. Dean has served as an advisor to the Executive Vice President of Drug Development for Sanofi, consulting on drug development strategy, drug safety issues and immunotoxicology through his company Drug Development Advisors, LLC where he serves as President. He is also a research professor in the departments of Medical Pharmacology and Pharmacology/Toxicology, Colleges of Medicine and Pharmacy, at University of Arizona in Tucson. Prior to January 2006, Dr. Dean served as the President, U.S. Science and Medical Affairs (R&D), Sanofi in Malvern, Pennsylvania and the Global Director of Preclinical Development for Sanofi. During his tenure at Sanofi and legacy companies over an 18 year period, he was involved
69
with the registration of eight NDAs for the U.S. and global market including Plavix, Avapro, Avalide, Ambien CR, and Eloxatin. He joined Sterling Winthrop in 1988, as Director of the Department of Toxicology and was appointed Vice President, Drug Safety worldwide in 1989. In addition, Dr. Dean served as Director of the Sterling Winthrop Research Center in Alnwick, England from 1990 to 1992. Dr. Dean was appointed Executive Vice President, Drug Development, in 1992 where he managed Non-Clinical and Clinical Development, and Regulatory Affairs. Before joining Sterling Winthrop, Dr. Dean headed the Department of Cellular and Molecular Toxicology, Chemical Industry Institute of Toxicology, Research Triangle Park, NC from 1982 to 1988. Prior to 1982, he headed the Immunotoxicology Section, National Institute of Environmental Health Services and National Toxicology Program, NIH in Research Triangle Park. From 1972 to 1979, Dr. Dean was in the Department of Immunology at Litton Bionetics (Dept. Director from 1975 to 1979) doing research in tumor immunology. Dr. Dean holds a B.S. in microbiology and an M.S. in medical microbiology from California State University at Long Beach. He earned a Ph.D. in molecular biology and minor in biochemistry in 1972 from the College of Medicine, University of Arizona. Dr. Dean held adjunct professorships at the University of North Carolina, Chapel Hill and Duke University from 1981 to 1988.
Christopher J. Coughlin. Mr. Coughlin has served as a director of our company since November 2012. Mr. Coughlin has served as Executive Vice President and Chief Financial Officer of Tyco International Ltd. (or Tyco), a global business with positions in residential and commercial security, fire protection and industrial products and services, from March 2005 until December 2010 and from that time until September 2012 he served as an advisor to the Chairman and Chief Executive Officer of Tyco. Prior to joining Tyco, Mr. Coughlin served as Chief Operating Officer at Interpublic Group of Companies, Inc., a leading global advertising and marketing services holding company. Previously, he was Chief Financial Officer of Pharmacia Corporation (or Pharmacia). Mr. Coughlin has significant experience in major acquisition, divestiture, as well as other types of transactions. This experience includes major acquisitions such as Pharmacia's acquisition of Monsanto Company (or Monsanto), and the subsequent spin-off of Monsanto, numerous licensing and co-development deals, the acquisition of Sugen by Pharmacia, as well as the eventual sale of Pharmacia to Pfizer. He more recently managed the spin-offs of Covidien Ltd. and Tyco Electronics to the shareholders of Tyco, and the acquisition of Brinks (Broadview) Security. Before joining Pharmacia, he served as President of Nabisco International in 1997 after joining the company as Vice President and Chief Financial Officer in 1996. Prior to that Mr. Coughlin held a number of management positions at Sterling Drug Inc. (later Sterling Winthrop), including Corporate Audit Director, Vice President of International Finance and Vice President of Worldwide Consumer Healthcare. In 1992, he was named Chief Financial Officer of Sterling Winthrop. Mr. Coughlin began his career with Arthur Young & Company (now Ernst & Young LLP). He currently serves on the Board of Directors for Dun & Bradstreet, Inc., Forest Laboratories, Inc., and Covidien Ltd. He has previously served on the Board of Directors of each of Monsanto, Interpublic Group of Companies, Inc. and Perrigo Company. Mr. Coughlin holds a Bachelor's Degree in Accounting from Boston College.
Thomas L. Harrison, LH.D. Mr. Harrison has served as a director of our company since November 2012. He has worked at the Omnicom Group since 1992 and currently serves as Chairman (Emeritus) of Diversified Agency Services, a division of the Omnicom Group. Prior to joining the Omnicom Group by acquisition of his company in 1992, Mr. Harrison was the founder and Chairman of the Harrison & Star Group, a healthcare advertising agency. Mr. Harrison began his career in 1974 as a pharmaceutical sales representative at Pfizer before continuing on to found and/or manage several healthcare advertising agencies. Currently, Mr. Harrison is a member of the Executive Committee of the Montefiore Hospital, New York, and is a Fellow of the New York Academy of Medicine. He also currently serves as a board member of Morgans Hotel Group and as a governor of the New York Academy of Sciences. He previously served as a Board Member of ePocrates, Inc., a healthcare information company, and the New York Chapter of the Arthritis Foundation. Mr. Harrison holds
70
advanced degrees in biology and physiology earning an undergraduate degree from West Virginia University in 1972 and an Honorary Doctorate from West Virginia University in 2007.
Michael E. Duffy, Esq. Mr. Duffy has served as a director of our company since February 2013. Mr. Duffy currently serves as the managing partner of the law firm, Duffy & Duffy, PLLC, is a highly experienced civil litigator with a passion for the law and a desire to represent victims of medical malpractice, where he has practiced since 1996. Mr. Duffy concentrates his practice on catastrophically injured victims and wrongful death claims involving medical malpractice and personal injury. Throughout his career, Mr. Duffy has been repeatedly named in Best Lawyers in America. Mr. Duffy received a J.D. from St. John's University and a Bachelor of Science degree in Business Administration from St. John's University. Mr. Duffy was admitted to the New York State Bar in 1995.
David Garrett. Mr. Garrett will serve as our Vice President, Finance and Corporate Development upon the consummation of our corporate conversion. Beginning in January 2012 and presently, Mr. Garrett serves as Managing Partner of Aumoe Partners, LLC (or Aumoe Partners), a financial advisory firm which he founded. He will join our company as a full time employee upon the completion of this offering. Aumoe Partners has served as a financial and business development consultant to our company. Prior to founding Aumoe Partners, Mr. Garrett served as Director, Healthcare Equity Sales and Capital Markets at Canaccord Genuity, Inc. from July 2008 to November 2011. From 1999 to 2008, Mr. Garrett served as an equity analyst covering the biotechnology and specialty pharmaceuticals industries at Scudder Kemper Investments, Wachovia Securities, UBS Securities, and Fortis Securities. Over the course of his career, Mr. Garrett has assisted over 45 emerging biotechnology and medical technology companies in initial public offerings, secondary public offerings and private placements of public equity that collectively have raised over $2.9 billion. Mr. Garrett received a Bachelor's Degree in Economics from the University of Wisconsin, Madison.
Robert G. Shawah, CPA. Mr. Shawah will serve as our Chief Accounting Officer and Treasurer upon the consummation of our corporate conversion. From 2005 to 2013, Mr. Shawah served as a Vice President of Baldwin Pearson & Co., Inc. focusing on structuring transactions in the commercial and industrial real estate market in Fairfield County, Connecticut, as well as financial reporting responsibility. From 1997 to 2005, he served Sales and Financial Engineer for CC1 Inc., a private New Hampshire firm that designed and manufactured camera-based technical equipment for the printing industry. Prior to 1997, Mr. Shawah held financial management positions at Victorinox/Swiss Army Brands and Grace Cocoa, a division of W.R. Grace. His responsibilities at these firms included accounting, financial reporting, and foreign currency transactions. Mr. Shawah is a certified public accountant in the Commonwealth of Pennsylvania (inactive) and spent the first five years of his career in the audit division of Arthur Andersen LLP.
Committees of the Board of Directors
Our board of directors has established an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. The Compensation Committee became effective as of November 12, 2013, and the Audit Committee and Nominating and Corporate Governance Committee will become effective as of the consummation of this offering. Each of our board committees acts pursuant to a separate written charter adopted by our board of directors.
The Compensation Committee is currently comprised of William J. McSherry, Esq. (Chairman), Dr. Jack H. Dean and Michael Duffy, Esq. Messrs. McSherry, and Duffy are non-employee directors under applicable SEC rules, and are "outside" directors under Internal Revenue Code Section 162(m). Dr. Dean and Messrs. McSherry and Duffy are each independent under applicable SEC and NASDAQ rules and regulations.
The Audit Committee will be comprised of Christopher Coughlin (Chairman), William J. McSherry, Esq. and Michael Duffy, Esq. Our board has determined that Mr. Coughlin, the Chairman of the
71
Audit and Finance Committee, is an "audit committee financial expert," under applicable SEC rules and regulations. The Audit Committee's responsibilities and duties are among other things to engage the independent auditors, review the audit fees, supervise matters relating to audit functions and review and set internal policies and procedure regarding audits, accounting and other financial controls. Messrs. Coughlin, McSherry and Duffy are each independent under applicable SEC and NASDAQ rules and regulations.
The Nominating and Corporate Governance Committee will be comprised of Dr. Jack H. Dean (Chairman) and Messrs. Thomas Harrison and Christopher Coughlin. The committee members are independent under applicable NASDAQ rules and regulations. The Nominating and Corporate Governance Committee is responsible for, among other things, considering potential board members, making recommendations to the full board as to nominees for election to the board, assessing the effectiveness of the board and implementing our corporate governance guidelines. Dr. Dean and Messrs. Harrison and Coughlin are each independent under applicable SEC and NASDAQ rules and regulations.
Our board of directors may at any time or from time to time appoint certain other committees in its sole discretion as it deems necessary or appropriate to carry out its functions.
Scientific Advisory Board
In December 2013, we formally established a Scientific Advisory Board to advise our management regarding our clinical and regulatory development programs and other customary matters. Our scientific advisors are experts in various areas of medicine including DFI, mild and moderate skin and skin structure infections in superficial wounds and podiatry. We believe the advice of our scientific advisors was integral to the quality of our clinical trial protocol for our Phase 3 program and the resulting SPA. Our Scientific Advisory Board is comprised of the following individuals:
- •
- Dr. Jonathan Wilkin. Founding Director (retired)
of the Division of Dermatology and Dental Products at the FDA. Remains active in regulatory matters after over 12 years of FDA service, which included membership on the FDA's Dermatology Drugs
Advisory Committee.
- •
- Dr. Benjamin Lipsky. Emeritus Professor of
Medicine, University of Washington; Visiting Professor, Infectious Diseases, University of Geneva. Teaching Associate, Green Templeton College, University of Oxford. Head of the IWGDF and lead author
of the Diabetic Foot Infection Treatment Guidelines, published in June 2012.
- •
- Dr. David Armstrong. Professor of Surgery and
Director, SALSA, University of Arizona College of Medicine. Co-Sponsor of the annual DFCon.
- •
- Dr. Warren Joseph. Adjunct Clinical Associate
Professor, Dr. William Scholl College of Podiatric Medicine, Rosland Franklin University of Medicine and Science. Managing Editor of the Journal of the American Podiatric Medical
Association.
- •
- Dr. Michael Zasloff. Original inventor of the "magainin peptides" which include pexiganan, while he was a research scientist at the NIH in 1987. Co-Founder of Magainin, the original owner of Locilex™. Current Professor, Departments of Surgery & Pediatrics; and Director, Surgical Immunology, Georgetown University School of Medicine.
We will continue to rely upon our scientific advisors in various aspects of our product development program including, without limitation, assisting with the publication in the future of the clinical data generated in our Phase 3 program in coordination with us.
72
The following table sets forth the aggregate compensation paid to our Chief Executive Officer and each of our other executive officers whose aggregate salary and bonus exceeded $100,000 for services rendered in all capacities for the fiscal years ended December 31, 2013, 2012 and 2011.
Summary Compensation Table
| Name and Principal Position | Year | Salary ($) |
Stock Awards ($) |
Option Awards ($) |
All Other Compensation(1) |
Total ($) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Robert J. DeLuccia, Executive Chairman(2) |
2013 | $ | 219,250 | $ | 57,750 | – | $12,000 | $ | 289,000 | |||||||||
|
2012 | 168,000 | 84,000 | – | 12,000 | 264,000 | ||||||||||||
|
2011 | 126,000 | 126,000 | – | 12,000 | 264,000 | ||||||||||||
David P. Luci, President & Chief Executive Officer(2) |
2013 | $ | 219,250 | $ | 57,750 | – | $22,000 | $ | 299,000 | |||||||||
|
2012 | 168,000 | 84,000 | – | 22,000 | 274,000 | ||||||||||||
|
2011 | 126,000 | 126,000 | – | 22,000 | 274,000 | ||||||||||||
- (1)
- Amounts
reported consist of amounts paid or reimbursed for health insurance for each of the named executives.
- (2)
- David P. Luci and Robert J. DeLuccia served as our Managing Partners prior to our corporate conversion. Each of Messrs. Luci and DeLuccia executed an amended and restated employment agreement in January 2014 that will become effective upon consummation of this offering.
In December 2013, we issued 45 Class A membership interests to each of Messrs. Luci and DeLuccia in connection with the terms of their pre-offering employment agreements.
In September 2013, we issued 120 Class A membership interests to each of Messrs. Luci and DeLuccia in connection with the terms of their pre-offering employment agreements.
In January 2013, we issued 240 Class A membership interests to each of Messrs. Luci and DeLuccia in connection with the terms of their pre-offering employment agreements.
In January 2011, we issued 360 Class A membership interests in accordance with their pre-offering employment agreements with Messrs. Luci and DeLuccia for services rendered in 2011.
Outstanding Equity Awards at Fiscal Year-End
There were no outstanding stock awards held by any of our executive officers at December 31, 2013.
Compensation Pursuant to Agreements and Plans
Employment Agreements
David P. Luci, Esq., President, Chief Executive Officer and Secretary. In February 2014, we entered into an amended and restated employment agreement with Mr. Luci which will become effective at the closing of this offering. Pursuant to the terms of this employment agreement, Mr. Luci will be paid an annual base salary of $395,000 and will be considered for an annual bonus of up to 45% of the annual base salary at the discretion of the Compensation Committee of our board of directors. In addition, pursuant to the employment agreement, Mr. Luci will receive options to purchase shares of our common stock following the closing of this offering (which is equal to 3.5% of the post-offering common stock outstanding) at price equal to the price paid by investors in this offering. The options shall be issued pursuant to our 2013 Equity Incentive Plan and shall vest in thirty-six (36) equal monthly installments beginning on the one month anniversary of the effective date of the agreement, subject to accelerated vesting upon a change of control of our company. We will also pay Mr. Luci a one-time cash bonus equal to $100,000 upon consummation of this offering.
73
Our agreement with Mr. Luci has a three year term and is subject to automatic one year renewals unless we terminate the agreement on no less than six months notice. Our agreement with Mr. Luci may be terminated by us with or without Cause (as defined in the agreement) or by Mr. Luci voluntarily or with Good Reason (as defined in the agreement). If we terminate Mr. Luci's agreement without Cause, or if he terminates the agreement with Good Reason (which includes a change in control of our company), we will be required to pay Mr. Luci a severance package which includes, among other items, a lump sum payment equal to 24 months of his annual compensation and an acceleration of all unvested equity awards.
Mr. Luci's employment agreement contains customary confidentiality and intellectual property covenants and one-year post-termination non-competition and non-solicitation covenants.
Robert J. DeLuccia, Executive Chairman. In February 2014, we entered into an amended and restated employment agreement with Mr. DeLuccia which will become effective at the closing of this offering. Pursuant to the terms of this employment agreement, Mr. DeLuccia will be paid an annual base salary of $395,000 and will be considered for an annual bonus of up to 30% of the annual base salary at the discretion of the Compensation Committee of our board of directors. In addition, pursuant to the employment agreement, Mr. DeLuccia will receive options to purchase shares of our common stock following the closing of this offering (which is equal to 3.5% of the post-offering common stock outstanding) at price equal to the price paid by investors in this offering. The options shall be issued pursuant to our 2013 Equity Incentive Plan and shall vest in thirty-six (36) equal monthly installments beginning on the one month anniversary of the effective date of the agreement, subject to accelerated vesting upon a change of control of our company.
Our agreement with Mr. DeLuccia has a three year term and is subject to automatic one year renewals unless we terminate the agreement on no less than six months notice. Our agreement with Mr. DeLuccia may be terminated by us with or without Cause (as defined in the agreement) or by Mr. DeLuccia voluntarily or with Good Reason (as defined in the agreement). If we terminate Mr. DeLuccia's agreement without Cause, or if he terminates the agreement with Good Reason (including a change in control of our company), we will be required to pay Mr. DeLuccia a severance package which includes, among other items, a lump sum payment equal to 18 months of his annual compensation and an acceleration of all unvested equity awards.
Mr. DeLuccia's employment agreement contains customary confidentiality and intellectual property covenants and one-year post-termination non-competition and non-solicitation covenants.
David Garrett, Vice President, Finance and Corporate Development. In February 2014, we entered into an Employment Agreement with Mr. Garrett which will become effective at the closing of this offering. Pursuant to the terms of this employment agreement, Mr. Garrett shall be paid an annual base salary in the amount of $265,000 and will be considered for an annual bonus of up to 25% of the annual base salary at the discretion of the Compensation Committee of our board of directors. In addition, pursuant to the terms of the Employment Agreement, Mr. Garrett will receive options to purchase shares of our common stock following the closing of this offering (which is equal to 2.0% of the post-offering common stock outstanding) at price equal to the price paid by investors in this offering. The options shall be issued pursuant to our 2013 Equity Incentive Plan and shall vest in thirty-six (36) equal monthly installments beginning on the one month anniversary of the effective date of the agreement, subject to accelerated vesting upon a change of control of our company. We will also pay Mr. Garrett a one-time cash bonus equal to $50,000 upon consummation of this offering.
Our agreement with Mr. Garrett has a three year term and is subject to automatic one year renewals unless we terminate the agreement on no less than six months notice. Our agreement with Mr. Garrett may be terminated by us with or without Cause (as defined in the agreement) or by Mr. Garrett voluntarily or with Good Reason (as defined in the agreement). If we terminate Mr. Garrett's agreement without Cause, or if he terminates the agreement with Good Reason
74
(including a change in control of our company), we will be required to pay Mr. Garrett a severance package which includes, among other items, a lump sum payment equal to 12 months of his base salary and an acceleration of all unvested equity awards.
Mr. Garrett's employment agreement contains customary confidentiality and intellectual property covenants and one-year post-termination non-competition and non-solicitation covenants.
Robert G. Shawah, CPA, Chief Accounting Officer and Treasurer. In February 2014, we entered into an Employment Agreement with Mr. Shawah which will become effective at the closing of this offering. Pursuant to the terms of this employment agreement, Mr. Shawah shall be employed for the first year of his agreement with us for 60% of his working time, for which he will be paid an annual base salary in the amount of $165,000 and will be considered for an annual bonus of up to 25% of the annual base salary at the discretion of the Compensation Committee of our board of directors. We will reevaluate Mr. Shawah's time commitment to our company after the first year of his employment. In addition, pursuant to the terms of the Employment Agreement, Mr. Shawah will receive options to purchase shares of our common stock following the closing of this offering (which is equal to 1% of the post-offering common stock outstanding) at price equal to the price paid by investors in this offering. The options shall be issued pursuant to our 2013 Equity Incentive Plan and shall vest in thirty-six (36) equal monthly installments beginning on the one month anniversary of the effective date of the agreement, subject to accelerated vesting upon a change of control of our company. We will also pay Mr. Shawah a one-time cash bonus equal to $15,000 upon consummation of this offering.
Our agreement with Mr. Shawah has a three year term and is subject to automatic one year renewals unless we terminate the agreement on no less than six months notice. Our agreement with Mr. Shawah may be terminated by us with or without Cause (as defined in the agreement) or by Mr. Shawah voluntarily or with Good Reason (as defined in the agreement). If we terminate Mr. Shawah's agreement without Cause, or if he terminates the agreement with Good Reason (including a change of control of our company), we will be required to pay Mr. Shawah a severance package which includes, among other items, a lump sum payment equal to 12 months of his base salary and an acceleration of all unvested equity awards.
Mr. Shawah's employment agreement contains customary confidentiality and intellectual property covenants and one-year post-termination non-competition and non-solicitation covenants.
Compensation of Directors
Director Compensation Table – 2013
The table below represents the compensation paid to our outside directors during the year ended December 31, 2013. Messrs. DeLuccia and Luci, our directors who also serve as executive officers of our company, receive no compensation for acting in their capacities as directors of our company.
| Name | Fees earned or Paid in Cash ($) |
Stock Awards ($)(1) |
Option Awards ($) |
All Other Compensation ($) |
Total ($) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Jack H. Dean |
$ | 500 | $ | 33,350 | – | – | $ | 33,850 | |||||||
William J. McSherry, Jr., Esq. |
500 | 33,350 | – | – | 33,850 | ||||||||||
Christopher Coughlin |
500 | 50,000 | – | – | 50,500 | ||||||||||
Thomas Harrison |
500 | 50,000 | – | – | 50,500 | ||||||||||
Michael Duffy, Esq. |
500 | – | – | – | 500 | ||||||||||
- (1)
- The value listed represents the fair value of the shares of common stock based upon the price of the most recent financing ($8.57 per share on a post-conversion basis). Such awards were originally issued a Class A membership interests in our company.
75
2013 Equity Incentive Plan
On November 12, 2013, our board of directors adopted a 2013 Equity Incentive Plan for our company, which plan will be effective as of the completion of our corporate conversion and the closing of this offering. The holders of majority of our outstanding Class A membership interests approved such plan on November 20, 2013. An aggregate number of shares of our common stock equal to 20% of our issued and outstanding common stock immediately after the consummation of this offering are reserved for issuance under our 2013 Equity Incentive Plan. No awards have been granted as of the date of this prospectus under our 2013 Equity Incentive Plan. However, at the closing of this offering:
- (i)
- Mr. Luci
will receive options to purchase shares of our common stock equal to 3.5% of our common stock outstanding immediately after the consummation
of this offering;
- (ii)
- Mr. DeLuccia
will receive options to purchase shares of our common stock equal to 3.5% of our common stock outstanding immediately after the
consummation of this offering;
- (iii)
- Mr. Garrett
will receive options to purchase shares of our common stock equal to 2% of our common stock outstanding immediately after the
consummation of this offering; and
- (iv)
- Mr. Shawah will receive options to purchase shares of our common stock equal to 1% of our common stock outstanding immediately after the consummation of this offering.
The foregoing options shall be issued pursuant to our 2013 Equity Incentive Plan and shall vest ratably over a period of three years (on the first, second and third anniversaries of the agreement) subject to accelerated vesting upon a change of control of our company.
The purpose of our 2013 Equity Incentive Plan is to attract and retain directors, officers, consultants, advisors and employees whose services are considered valuable, to encourage a sense of proprietorship and to stimulate an active interest of such persons in our development and financial achievements. The 2013 Equity Incentive Plan will be administered by the Compensation Committee of our board of directors or by the full board, which may determine, among other things, the (a) terms and conditions of any option or stock purchase right granted, including the exercise price and the vesting schedule, (b) persons who are to receive options and stock purchase rights and (c) the number of shares to be subject to each option and stock purchase right. The 2013 Equity Incentive Plan will provide for the grant of (i) "incentive" options (qualified under section 422 of the Internal Revenue Code of 1986, as amended) to employees of our company and (ii) non-qualified options to directors and consultants of our company.
In connection with the administration of our 2013 Equity Incentive Plan, our Compensation Committee will:
- •
- determine which employees and other persons will be granted awards under our 2013 Equity Incentive Plan;
- •
- grant the awards to those selected to participate;
- •
- determine the exercise price for options; and
- •
- prescribe any limitations, restrictions and conditions upon any awards, including the vesting conditions of awards.
Any grant of awards to any of directors under our 2013 Equity Incentive Plan must be approved by the Compensation Committee of our board of directors. In addition, our Compensation Committee will: (i) interpret our 2013 Equity Incentive Plan; and (ii) make all other determinations and take all other action that may be necessary or advisable to implement and administer our 2013 Equity Incentive Plan.
76
The 2013 Equity Incentive Plan provides that in the event of a change of control event, the Compensation Committee or our board of directors shall have the discretion to determine whether and to what extent to accelerate the vesting, exercise or payment of an award.
In addition, our board of directors may amend our 2013 Equity Incentive Plan at any time. However, without stockholder approval, our 2013 Equity Incentive Plan may not be amended in a manner that would:
- •
- increase the number of shares that may be issued under our 2013 Equity Incentive Plan;
- •
- materially modify the requirements for eligibility for participation in our 2013 Equity Incentive Plan;
- •
- materially increase the benefits to participants provided by our 2013 Equity Incentive Plan; or
- •
- otherwise disqualify our 2013 Equity Incentive Plan for coverage under Rule 16b-3 promulgated under the Securities Exchange Act of 1934, as amended.
Awards previously granted under our 2013 Equity Incentive Plan may not be impaired or affected by any amendment of our 2013 Equity Incentive Plan, without the consent of the affected grantees.
77
Based solely upon information made available to us, the following table sets forth information as of March 6, 2014 regarding the beneficial ownership of our common stock after giving effect to our anticipated corporate conversion by:
- •
- each person known by us to be the beneficial owner of more than 5% of our outstanding shares of common stock;
- •
- each of our named executive officers and directors; and
- •
- all our executive officers and directors as a group.
The percentage ownership information shown in the table is based upon 5,375,377 shares of common stock outstanding after giving effect to our anticipated corporate conversion. In addition, the number of shares and percentage of shares beneficially owned after the offering gives effect to the issuance by us of 2,307,693 shares of common stock in this offering assuming an initial public offering price of $13.00 per share (the mid-point of the price range set forth on the cover page of this prospectus). The percentage ownership information assumes no exercise of the underwriters' over-allotment option.
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Except as otherwise indicated, each person or entity named in the table has sole voting and investment power with respect to all shares of our capital shown as beneficially owned, subject to applicable community property laws.
In computing the number and percentage of shares beneficially owned by a person, shares that may be acquired by such person (for example, upon the exercise of options or warrants) within 60 days of the date of this prospectus are counted as outstanding, while these shares are not counted as outstanding for computing the percentage ownership of any other person.
The address of each holder listed below, except as otherwise indicated, is c/o Dipexium Pharmaceuticals, Inc., 74 Broad Street, New York, New York 10004.
| Name of Beneficial Owner | Shares of Common Beneficially Stock Owned(1) |
Percent of Common Stock Beneficially Owned Before Offering(1) |
Percent of Common Stock Beneficially Owned After Offering(1) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
David P. Luci(2) |
1,837,626 | 34.2 | % | 23.9 | % | ||||||
Robert J. DeLuccia(3) |
1,834,826 | 34.1 | % | 23.9 | % | ||||||
William J. McSherry, Jr. |
21,924 | * | * | ||||||||
Dr. Jack H. Dean |
43,484 | * | * | ||||||||
Christopher Coughlin(4) |
86,226 | 1.6 | % | 1.1 | % | ||||||
Thomas Harrison(5) |
125,832 | 2.3 | % | 1.6 | % | ||||||
Michael Duffy, Esq.(6) |
47,404 | * | * | ||||||||
David Garrett(7) |
7,924 | * | * | ||||||||
Robert G. Shawah(8) |
– | – | – | ||||||||
All directors and executive officers as a group (9 persons) |
4,005,246 | 74.5 | % | 52.1 | % | ||||||
- *
- Less
than 1%.
- (1)
- Percentage
ownership is based on 5,375,377 shares of our common stock outstanding prior to this offering and 7,683,070 shares of our common stock
outstanding after this offering, assuming in each case the consummation of our anticipated corporate conversion.
- (2)
- This amount includes 7,462 shares of common stock held by Donna Luci, the wife of Mr. Luci. Mr. Luci disclaims beneficial ownership of the shares held by Donna Luci. This amount also includes an additional 2,800 shares of common stock beneficially owned by certain family members of Mr. Luci to which Mr. Luci maintains voting control. Mr. Luci disclaims beneficial ownership of the shares of common stock held by such family members and maintains no pecuniary
78
interest therein. Excludes unvested options to purchase shares of our common stock equal to 3.5% of our outstanding common stock after this offering to be issued to Mr. Luci under our 2013 Equity Incentive Plan at the closing of this offering.
- (3)
- This
amount includes 17,640 shares of common stock held by Rosemary DeLuccia, Mr. DeLuccia's wife, and 5,600 shares for which Mr. DeLuccia
transferred beneficial ownership to certain other family members but maintains voting control. Mr. DeLuccia disclaims beneficial ownership of the shares of common stock held by such family
members and maintains no pecuniary interest therein. Excludes unvested options to purchase shares of our common stock equal to 3.5% of our outstanding common stock after this offering to be issued to
Mr. DeLuccia under our 2013 Equity Incentive Plan at the closing of this offering.
- (4)
- Excludes
21,000 shares of our common stock issuable for board services rendered which vest as follows: (i) 7,000 shares vest on November 21,
2014; (ii) 7,000 shares vest on November 21, 2015; and (iii) 7,000 shares vest on November 21, 2016 (all of these excluded shares vest immediately upon a change of control
of our company).
- (5)
- Excludes
21,000 shares of our common stock issuable for board services rendered which vest as follows: (i) 7,000 shares vest on February 13,
2015; (ii) 7,000 shares vest on February 13, 2016; and (iii) 7,000 shares vest on February 13, 2017 (all of these excluded shares vest immediately upon a change of control
of our company).
- (6)
- Excludes
20,000 shares of our common stock issuable for board services rendered which vest as follows: (i) 7,000 shares vest on February 13,
2015; (iii) 7,000 shares vest on February 13, 2016; and (iii) 7,000 shares vest on February 13, 2017 (all of these excluded shares vest immediately upon a change of control
of our company).
- (7)
- Excludes
unvested options to purchase shares of our common stock equal to 2% of our outstanding common stock after this offering to be issued to
Mr. Garrett under our 2013 Equity Incentive Plan at the closing of this offering.
- (8)
- Excludes unvested options to purchase shares of our common stock equal to 1% of our outstanding common stock after this offering to be issued to Mr. Shawah under our 2013 Equity Incentive Plan at the closing of this offering.
The above table does not include any shares that our officers and directors or their affiliates or related parties may purchase in this offering.
79
Certain Relationships and Related Party Transactions
On occasion we may engage in certain related party transactions. All prior related party transactions were approved by a majority of the disinterested directors. Upon the consummation of offering, our policy is that all related party transactions will be reviewed and approved by the Audit Committee of our board of directors prior to our entering into any related party transactions.
On November 12, 2013, our board of directors elected Robert J. DeLuccia as Executive Chairman and David P. Luci as President and Chief Executive Officer, in each case, to be pursuant to an amended and restated employment agreement effective upon the closing of this offering. Such amended and restated employment agreements were approved by the Compensation Committee of our board of directors in January 2014. See "Executive Compensation – Compensation Pursuant to Agreements and Plans – Employment Agreements."
Mr. David Garrett will serve as our Vice President, Finance and Corporate Development as of the closing of this offering. Mr. Garrett is the owner of Aumoe Partners, which we engaged in January 2012 to perform certain financial advisory services. We paid Aumoe Partners $43,375 and $77,500 in fees for services rendered in 2012 and the year ended December 31, 2013, respectively.
Statement of Policy
All future transactions between us and our officers, directors or five percent stockholders, and respective affiliates will be on terms no less favorable than could be obtained from unaffiliated third parties and will be approved by a majority of our independent directors who do not have an interest in the transactions and who had access, at our expense, to our legal counsel or independent legal counsel.
To the best of our knowledge, during the past three fiscal years, other than as set forth above, there were no material transactions, or series of similar transactions, or any currently proposed transactions, or series of similar transactions, to which we were or are to be a party, in which the amount involved exceeds $120,000, and in which any director or executive officer, or any security holder who is known by us to own of record or beneficially more than 5% of any class of our common stock, or any member of the immediate family of any of the foregoing persons, has an interest (other than compensation to our officers and directors in the ordinary course of business).
80
Our certificate of incorporation to be adopted at the time of our anticipated corporate conversion authorizes the issuance of 30,000,000 shares of common stock, $0.001 par value per share. As of March 6, 2014, there were 5,375,377 shares of our common stock issued and outstanding (on an as converted basis after giving effect to the anticipated conversion of each Class A membership interest of Dipexium Pharmaceuticals, LLC into seven shares of common stock of Dipexium Pharmaceuticals, Inc.) and held of record by approximately 100 stockholders.
The following is a summary of our capital stock upon consummation of our anticipated corporate conversion and the consummation of this offering. This summary does not purport to be complete and is qualified in its entirety by the provisions of our certificate of incorporation and bylaws to be in effect upon consummation of this offering, copies of which have been filed as exhibits to the registration statement of which this prospectus is a part.
Common Stock
Holders of our common stock are entitled to one vote for each share held on all matters submitted to a vote of stockholders and are not entitled to cumulative voting rights.
Holders of our common stock are entitled to receive ratably such dividends, if any, as may be declared by our board of directors out of funds legally available therefor, subject to any preferential distribution rights of third parties (of which there are presently none). Upon our liquidation, dissolution or winding up, the holders of our common stock are entitled to receive ratably our net assets available after the payment of all debts and other liabilities.
Holders of our common stock have no preemptive, subscription, redemption or conversion rights. There are no redemption or sinking fund provisions applicable to the common stock. All of the outstanding shares of our common stock are fully-paid and nonassessable. The rights, preferences and privileges of holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of any indebtedness of our company.
Preferred Stock
Our certificate of incorporation that will be in effect following consummation of this offering does not permit our board of directors to designate and issue shares of preferred stock.
Warrants
As of March 6, 2014, warrants for the issuance of 34,300 shares of our common stock were outstanding, all of which are exercisable at an exercise price of $8.57 per share, after taking into effect the anticipated conversion of each Class A membership interest of Dipexium Pharmaceuticals, LLC into seven shares of common stock of Dipexium Pharmaceuticals, Inc. See "Corporate Conversion." All of the warrants are exercisable through various dates expiring between July 23, 2015 and November 15, 2018. The exercise price and the number of warrant shares purchasable upon the exercise of the warrants are subject to adjustment upon the occurrence of certain events, including stock dividends, stock splits, combinations and reclassifications of our capital stock. The warrants also contain a "cashless exercise" provision.
Registration Rights
Each of our investors in our previous private placements are party to an Investor Rights Agreement affording them certain "piggy back" registration rights with respect to their Class A membership interests in our company and Class A membership interests underlying warrants held by such investors. This is comprised of 1,492,233 shares of our common stock on a post-conversion basis and shares of common stock (on a post-conversion basis) underlying warrants to purchase 34,300 shares.
81
We refer to all of such Class A membership interests as "registrable securities." If at any time when there is not an effective registration statement covering all of the registrable securities, we determine to prepare and file with the SEC a registration statement relating to an offering for our own account or the account of others under the Securities Act of any of our equity securities (other than on Form S-4 or Form S-8), we are required to send to each holder of registrable securities written notice of such determination and, if within seven business days after receipt of such notice, any such holder shall so request in writing (which request shall specify the registrable securities intended to be disposed of by the holder), we will cause the registration under the Securities Act of all registrable securities which we have been so requested to register by the holder; provided, however, that, in connection with any underwritten public offering of our securities, we maintain the right to not register all or any portion of the registrable securities if it is determined that such registration would materially and adversely affect such underwritten public offering. We have exercised such rights in connection with this offering and therefore are not including any such shares in the registration statement of which this prospectus forms a part.
Transfer Agent and Registrar
The transfer agent and registrar of our common stock is VStock Transfer LLC.
Delaware Law and Certain Charter and By-Law Provisions
Delaware Anti-Takeover Law. Upon our corporate conversion, we will be subject to Section 203 of the Delaware General Corporation Law. Section 203 generally prohibits a public Delaware corporation from engaging in a "business combination" with an "interested stockholder" for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
- •
- prior to the date of the transaction, the board of directors of the corporation approved either the business combination
or the transaction which resulted in the stockholder becoming an interested stockholder;
- •
- upon consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested
stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding specified shares;
or
- •
- at or subsequent to the date of the transaction, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock which is not owned by the interested stockholder.
Section 203 defines a "business combination" to include:
- •
- any merger or consolidation involving the corporation and the interested stockholder;
- •
- any sale, lease, exchange, mortgage, pledge, transfer or other disposition of 10% or more of the assets of the corporation
to or with the interested stockholder;
- •
- subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the
corporation to the interested stockholder;
- •
- subject to exceptions, any transaction involving the corporation that has the effect of increasing the proportionate share
of the stock of any class or series of the corporation beneficially owned by the interested stockholder; or
- •
- the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
In general, Section 203 defines an "interested stockholder" as any person that is:
- •
- the owner of 15% or more of the outstanding voting stock of the corporation;
82
- •
- an affiliate or associate of the corporation who was the owner of 15% or more of the outstanding voting stock of the
corporation at any time within three years immediately prior to the relevant date; or
- •
- the affiliates and associates of the above.
Under specific circumstances, Section 203 makes it more difficult for an "interested stockholder" to effect various business combinations with a corporation for a three-year period, although the stockholders may, by adopting an amendment to the corporation's certificate of incorporation or bylaws, elect not to be governed by this section, effective 12 months after adoption.
Our certificate of incorporation and bylaws do not exclude us from the restrictions of Section 203. We anticipate that the provisions of Section 203 might encourage companies interested in acquiring us to negotiate in advance with our board of directors since the stockholder approval requirement would be avoided if a majority of the directors then in office approve either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder.
Certificate of Incorporation and Bylaws. Our certificate of incorporation and bylaws contain provisions that could have the effect of discouraging potential acquisition proposals or tender offers or delaying or preventing a change of control of our company. These provisions are as follows:
- •
- they do not permit our stockholders to act by written consent. As a result, any action by our stockholders must be taken
at a duly called annual or special meeting of stockholders;
- •
- they provide that special meetings of stockholders may be called only by the board of directors, our Chairman of the board
of directors, one of our executive officers, or at the request in writing by stockholders of record owning at least sixty-six and two thirds (662/3%) percent of the issued and
outstanding voting shares of common stock; and
- •
- they do not include a provision for cumulative voting in the election of directors. Under cumulative voting, a minority stockholder holding a sufficient number of shares may be able to ensure the election of one or more directors. The absence of cumulative voting may have the effect of limiting the ability of minority stockholders to effect changes in our board of directors.
Elimination of Monetary Liability for Officers and Directors
Our certificate of incorporation incorporates certain provisions permitted under the Delaware General Corporation Law relating to the liability of directors. The provisions eliminate a director's liability for monetary damages for a breach of fiduciary duty, including gross negligence, except in circumstances involving certain wrongful acts, such as the breach of director's duty of loyalty or acts or omissions, which involve intentional misconduct or a knowing violation of law. These provisions do not eliminate a director's duty of care. Moreover, these provisions do not apply to claims against a director for certain violations of law, including knowing violations of federal securities law. Our certificate of incorporation also contains provisions to indemnify the directors, officers, employees or other agents to the fullest extent permitted by the Delaware General Corporation Law. We believe that these provisions will assist us in attracting and retaining qualified individual to serve as directors.
Indemnification of Officers and Directors
Our certificate of incorporation also contains provisions to indemnify the directors, officers, employees or other agents to the fullest extent permitted by the Delaware General Corporation Law. These provisions may have the practical effect in certain cases of eliminating the ability of shareholders to collect monetary damages from directors. We are also a party to indemnification agreements with each of our directors. We believe that these provisions will assist us in attracting or retaining qualified individuals to serve as our directors.
Disclosure of Commission Position on Indemnification for Securities Act Liabilities
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
83
Shares Eligible For Future Sale
Immediately prior to this offering, there was no public market for our common stock. Future sales of substantial amounts of our common stock in the public market could adversely affect prevailing market prices. Furthermore, since only a limited number of shares will be available for sale shortly after this offering because of contractual and legal restrictions on resale described below, sales of substantial amounts of common stock in the public market after the restrictions lapse could adversely affect the prevailing market price for our common stock as well as our ability to raise equity capital in the future.
After giving effect to our anticipated corporate conversion and the closing of this offering, approximately 7,683,070 shares of common stock will be outstanding, assuming an initial public offering price of $13.00 per share (the mid-point of the price range set forth on the cover page of this prospectus) and further assuming no exercise of the underwriters' over-allotment option. All of the shares sold in this offering will be freely tradable unless held by an affiliate of ours. The remaining 5,375,377 shares of common stock outstanding after this offering and our corporate conversion will be restricted as a result of securities laws or lock-up agreements. These remaining shares will generally become available for sale in the public market as follows: approximately 1,370,131 restricted shares held by non-affiliates will be eligible for sale under Rule 144 or Rule 701 upon expiration of lock-up agreements at least 180 days after the date of this offering.
Rule 144
In general, under Rule 144 as currently in effect, beginning 90 days after the effective date of the registration statement of which this prospectus is a part, any person who is not an affiliate of ours and has held their shares for at least six months, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, may sell shares without restriction, provided current public information about us is available. In addition, under Rule 144, any person who is not an affiliate of ours and has held their shares for at least one year, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of shares immediately upon the closing of this offering without regard to whether current public information about us is available. Beginning 90 days after the effective date of the registration statement of which this prospectus is a part, a person who is an affiliate of ours and who has beneficially owned restricted securities for at least six months, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of restricted shares within any three-month period that does not exceed the greater of:
- •
- 1% of the number of shares of our common stock then outstanding, which will equal approximately 76,831 shares immediately
after this offering; and
- •
- the average weekly trading volume of our common stock on The NASDAQ Capital Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale.
Sales of restricted shares under Rule 144 held by our affiliates are also subject to requirements regarding the manner of sale, notice and the availability of current public information about us. Rule 144 also provides that affiliates relying on Rule 144 to sell shares of our common stock that are not restricted shares must nonetheless comply with the same restrictions applicable to restricted shares, other than the holding period requirement. Notwithstanding the availability of Rule 144, the holders of 5,257,847 of our restricted shares have entered into lock-up agreements as described below and their restricted shares will become eligible for sale at the expiration of the restrictions set forth in those agreements.
84
Rule 701
Under Rule 701, shares of our common stock acquired upon the exercise of currently outstanding options or pursuant to other rights granted under our stock plans may be resold, by:
- •
- persons other than affiliates, beginning 90 days after the effective date of the registration statement of which
this prospectus is a part, subject only to the manner-of-sale provisions of Rule 144; and
- •
- our affiliates, beginning 90 days after the effective date of the registration statement of which this prospectus is a part, subject to the manner-of-sale and volume limitations, current public information and filing requirements of Rule 144, in each case, without compliance with the six-month holding period requirement of Rule 144.
Lock-up Agreements
We, our executive officers, directors and other certain holders of an aggregate of 5,257,847 shares of our capital stock and securities convertible into or exchangeable for our capital stock have agreed that, subject to certain exceptions, for a period of 180 days after the date of this prospectus, we and they will not, without the prior written consent of Oppenheimer & Co. Inc., dispose of or hedge any shares or any securities convertible into or exchangeable for shares of our capital stock. Oppenheimer & Co. Inc. may, in its discretion, release any of the securities subject to these lock-up agreements at any time.
Registration Rights
Upon completion of this offering, the holders of 1,492,233 shares of our common stock on a post-conversion basis and the holders of our warrants to purchase 34,300 shares of common stock on a post-conversion basis will be entitled to certain "piggy back" rights with respect to the registration of their shares under the Securities Act, subject to the lock-up arrangement described above. See "Description of Securities – Registration Rights."
Equity Incentive Plans
We intend to file a registration statements on Form S-8 under the Securities Act after the closing of this offering to register the shares of our common stock that are issuable pursuant to our 2013 Equity Incentive Plan. The registration statements are expected to be filed and become effective as soon as practicable after the completion of this offering. Accordingly, shares registered under the registration statements will be available for sale in the open market following their effective dates, subject to Rule 144 volume limitations and the lock-up arrangement described above, if applicable.
85
We have entered into an underwriting agreement with the underwriters named below. Oppenheimer & Co. Inc. is acting as representative of the underwriters.
The underwriting agreement provides for the purchase of a specific number of shares of common stock by each of the underwriters. The underwriters' obligations are several, which means that each underwriter is required to purchase a specified number of shares, but is not responsible for the commitment of any other underwriter to purchase shares. Subject to the terms and conditions of the underwriting agreement, each underwriter has severally agreed to purchase the number of shares of common stock set forth opposite its name below:
| Underwriter | Number of Shares | ||
|---|---|---|---|
Oppenheimer & Co. Inc. |
|||
Feltl & Company, Inc. |
|||
Total |
|||
The underwriters have agreed to purchase all of the shares offered by this prospectus (other than those covered by the over-allotment option described below) if any are purchased. Under the underwriting agreement, if an underwriter defaults in its commitment to purchase shares, the commitments of non-defaulting underwriters may be increased or the underwriting agreement may be terminated, depending on the circumstances. Certain of our officers and directors or their affiliates or related parties have indicated that they may have an interest in purchasing shares in this offering, which would reduce the number of shares sold to the general public. However, because indications of interest are not binding agreements or commitments to purchase, these persons or entities may determine not to purchase any shares in this offering.
The underwriters are offering the shares subject to various conditions and may reject all or part of any order. The representative has advised us that the underwriters propose to offer the shares directly to the public at the public offering price that appears on the cover page of this prospectus. In addition, the representative may offer some of the shares to other securities dealers at such price less a concession of $ per share. The underwriters may also allow, and such dealers may reallow, a concession not in excess of $ per share to other dealers. After the shares are released for sale to the public, the representative may change the offering price and other selling terms at various times.
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 30 days after the date of this prospectus, permits the underwriters to purchase a maximum of 346,154 additional shares from us to cover over-allotments. If the underwriters exercise all or part of this option, they will purchase shares covered by the option at the initial public offering price that appears on the cover page of this prospectus, less the underwriting discount. If this option is exercised in full, the total price to public will be $ , the total proceeds to us will be $ . The underwriters have severally agreed that, to the extent the over-allotment option is exercised, they will each purchase a number of additional shares proportionate to the underwriter's initial amount reflected in the foregoing table.
The following table provides information regarding the amount of the discount to be paid to the underwriters by us:
| |
Per Share | Total Without Exercise of Over-Allotment Option |
Total With Full Exercise of Over-Allotment Option |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Public offering price |
$ | $ | $ | ||||||||
Underwriting discounts and commissions |
$ | $ | $ | ||||||||
Proceeds, before expenses, to us |
$ | $ | $ | ||||||||
86
Trout Capital LLC, a FINRA member, acted as our financial advisor in connection with this offering. The expenses of this offering include a fee to Trout Capital LLC equal to three percent (3%) of the underwriting discount for services rendered as our financial advisor in connection with this offering. This fee will be payable in its entirety out of the underwriting discount and will not be an expense in addition to the underwriting discount or a deduction to the gross proceeds of this offering.
We estimate that our total expenses of the offering, excluding the underwriting discounts and commissions, will be approximately $ , which includes $125,000 that we have agreed to reimburse Oppenheimer & Co. Inc. for the fees and expenses incurred by them in connection with this offering.
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act of 1933.
We, our officers and directors and other stockholders have agreed to a 180 day "lock up" with respect to 5,257,847 shares of common stock and other of our securities that they beneficially own, including securities that are convertible into shares of common stock and securities that are exchangeable or exercisable for shares of common stock. This means that, subject to certain exceptions, for a period of 180 days following the date of this prospectus, we and such persons may not offer, sell, pledge or otherwise dispose of these securities without the prior written consent of Oppenheimer & Co. Inc.
The representative has informed us that they do not expect discretionary sales by the underwriters to exceed five percent of the shares offered by this prospectus.
Rules of the Securities and Exchange Commission may limit the ability of the underwriters to bid for or purchase shares before the distribution of the shares is completed. However, the underwriters may engage in the following activities in accordance with the rules:
- •
- Stabilizing transactions – The representative may make bids or purchases for the purpose of pegging,
fixing or maintaining the price of the shares, so long as stabilizing bids do not exceed a specified maximum.
- •
- Over-allotments and syndicate covering transactions – The underwriters may sell more shares of our
common stock in connection with this offering than the number of shares than they have committed to purchase. This over-allotment creates a short position for the underwriters. This short sales
position may involve either "covered" short sales or "naked" short sales. Covered short sales are short sales made in an amount not greater than the underwriters' over-allotment option to purchase
additional shares in this offering described above. The underwriters may close out any covered short position either by exercising their over-allotment option or by purchasing shares in the open
market. To determine how they will close the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market, as compared to
the price at which they may purchase shares through the over-allotment option. Naked short sales are short sales in excess of the over-allotment option. The underwriters must close out any naked short
position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that, in the open market after pricing, there may be downward
pressure on the price of the shares that could adversely affect investors who purchase shares in this offering.
- •
- Penalty bids – If the representative purchases shares in the open market in a stabilizing transaction or syndicate covering transaction, they may reclaim a selling concession from the underwriters and selling group members who sold those shares as part of this offering.
Similar to other purchase transactions, the underwriters' purchases to cover the syndicate short sales or to stabilize the market price of our common stock may have the effect of raising or maintaining the market price of our common stock or preventing or mitigating a decline in the market price of our common stock. As a result, the price of the shares of our common stock may be higher than the price that might otherwise exist in the open market. The imposition of a penalty bid might also have an effect on the price of the shares if it discourages resales of the shares.
87
Neither we nor the underwriters makes any representation or prediction as to the effect that the transactions described above may have on the price of the shares. These transactions may occur on The NASDAQ Capital Market or otherwise. If such transactions are commenced, they may be discontinued without notice at any time.
Electronic Delivery of Preliminary Prospectus: A prospectus in electronic format may be delivered to potential investors by one or more of the underwriters participating in this offering. The prospectus in electronic format will be identical to the paper version of such preliminary prospectus. Other than the prospectus in electronic format, the information on any underwriter's web site and any information contained in any other web site maintained by an underwriter is not part of the prospectus or the registration statement of which this prospectus forms a part
Determination of Offering Price
Prior to the offering, there has not been a public market for our common stock. Consequently, the public offering price for our common stock has been determined by negotiations between us and Oppenheimer & Co. Inc., the representative of the several underwriters for this offering. Among the factors to be considered in these negotiations were the prevailing market conditions, our financial information, market valuations of other companies that we and the representative believe to be comparable to us, estimates of our business potential, the present state of our development and other factors deemed relevant.
We offer no assurances that the public offering price will correspond to the price at which the common stock will trade in the public market subsequent to the offering or that an active trading market for the common stock will develop and continue after the offering.
Notice to Non-U.S. Investors
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive, or a Relevant Member State, an offer to the public of any securities which are the subject of the offering contemplated by this prospectus may not be made in that Relevant Member State except that an offer to the public in that Relevant Member State of any securities may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
(a) to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
(b) to any legal entity which has two or more of (i) an average of at least 250 employees during the last financial year; (ii) a total balance sheet of more than €43,000,000 and (iii) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts;
(c) by the representative to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive); or
(d) in any other circumstances falling within Article 3(2) of the Prospectus Directive,
provided, that no such offer of securities shall result in a requirement for the publication by us or any representative of a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an "offer to the public" in relation to any securities in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any securities to be offered so as to enable an investor to decide to purchase any securities, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State and the expression "Prospectus Directive" means Directive 2003/71/EC and includes any relevant implementing measure in each Relevant Member State.
88
Each underwriter has represented, warranted and agreed that:
(a) it has only communicated or caused to be communicated and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of section 21 of the Financial Services and Markets Act 2000, or the FSMA, received by it in connection with the issue or sale of any securities in circumstances in which section 21(1) of the FSMA does not apply to us; and
(b) it has complied with and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the securities in, from or otherwise involving the United Kingdom.
In the State of Israel, the common stock offered hereby may not be offered to any person or entity other than the following:
(a) a fund for joint investments in trust (i.e., mutual fund), as such term is defined in the Law for Joint Investments in Trust, 5754-1994, or a management company of such a fund;
(b) a provident fund as defined in Section 47(a)(2) of the Income Tax Ordinance of the State of Israel, or a management company of such a fund;
(c) an insurer, as defined in the Law for Oversight of Insurance Transactions, 5741-1981, (d) a banking entity or satellite entity, as such terms are defined in the Banking Law (Licensing), 5741-1981, other than a joint services company, acting for their own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(d) a company that is licensed as a portfolio manager, as such term is defined in Section 8(b) of the Law for the Regulation of Investment Advisors and Portfolio Managers, 5755-1995, acting on its own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(e) a company that is licensed as an investment advisor, as such term is defined in Section 7(c) of the Law for the Regulation of Investment Advisors and Portfolio Managers, 5755-1995, acting on its own account;
(f) a company that is a member of the Tel Aviv Stock Exchange, acting on its own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(g) an underwriter fulfilling the conditions of Section 56(c) of the Securities Law, 5728-1968;
(h) a venture capital fund (defined as an entity primarily involved in investments in companies which, at the time of investment, (i) are primarily engaged in research and development or manufacture of new technological products or processes and (ii) involve above-average risk);
(i) an entity primarily engaged in capital markets activities in which all of the equity owners meet one or more of the above criteria; and
(j) an entity, other than an entity formed for the purpose of purchasing common stock in this offering, in which the shareholders equity (including pursuant to foreign accounting rules, international accounting regulations and U.S. generally accepted accounting rules, as defined in the Securities Law Regulations (Preparation of Annual Financial Statements), 1993) is in excess of NIS 250 million.
Any offeree of the common stock offered hereby in the State of Israel shall be required to submit written confirmation that it falls within the scope of one of the above criteria. This prospectus will not be distributed or directed to investors in the State of Israel who do not fall within one of the above criteria.
89
CohnReznick LLP, our independent registered public accounting firm, has audited our balance sheets as of December 31, 2013 and 2012, and the related statements of operations, changes in members' equity and cash flows for each of the years in the two-year period ended December 31, 2013 and the period from January 14, 2010 (inception) through December 31, 2013, as set forth in their report, which includes an explanatory paragraph relating to our ability to continue as a going concern. We have included our financial statements in this prospectus and in this registration statement in reliance on CohnReznick's report given on their authority as experts in accounting and auditing.
Ellenoff Grossman & Schole LLP, New York, New York, will pass upon the validity of the securities offered hereby. A partner of Ellenoff Grossman & Schole LLP is also a shareholder of our company. Certain matters are being passed on for the underwriters by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., New York, New York.
Where You Can Find More Information
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of our common stock offered by this prospectus. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement, some of which is contained in exhibits to the registration statement as permitted by the rules and regulations of the SEC. For further information with respect to us and our common stock, we refer you to the registration statement, including the exhibits filed as a part of the registration statement. Statements contained in this prospectus concerning the contents of any contract or any other document is not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, please see the copy of the contract or document that has been filed. Each statement is this prospectus relating to a contract or document filed as an exhibit is qualified in all respects by the filed exhibit. You may obtain copies of this information by mail from the Public Reference Section of the SEC, 100 F Street, N.E., Room 1580, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy statements and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
As a result of this offering, we will become subject to the information and reporting requirements of the Securities Exchange Act of 1934, as amended, and, in accordance with this law, will file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information will be available for inspection and copying at the SEC's public reference facilities and the website of the SEC referred to above.
We also maintain a website at www.dipexiumpharmaceuticals.com. Upon completion of this offering, you may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC. Information contained on our website is not a part of this prospectus and the inclusion of our website address in this prospectus is an inactive textual reference only.
90
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Contents
F-1
Report of Independent Registered Public Accounting Firm
The
Board of Directors and Members
Dipexium Pharmaceuticals, LLC
We have audited the accompanying balance sheets of Dipexium Pharmaceuticals, LLC as of December 31, 2013 and 2012, and the related statements of operations, changes in members' equity and cash flows for each of the years in the two-year period ended December 31, 2013 and the period from January 14, 2010 (date of inception) through December 31, 2013. Dipexium Pharmaceuticals, LLC's management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Dipexium Pharmaceuticals, LLC as of December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2013 and the period from January 14, 2010 (date of inception) through December 31, 2013, in conformity with accounting principles generally accepted in the United States of America.
The financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in the Note 1 to the financial statements, the Company has incurred losses since inception and has no revenue. Those conditions raise substantial doubt about its ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/
CohnReznick LLP
Roseland,
New Jersey
February 5, 2014
F-2
DIPEXIUM PHARMACEUTICALS, LLC
(A DEVELOPMENT STAGE COMPANY)
BALANCE SHEETS
December 31, 2013 and 2012
| |
2013 | 2012 | |||||
|---|---|---|---|---|---|---|---|
ASSETS |
|||||||
CURRENT ASSETS |
|||||||
Cash |
$ | 3,861,145 | $ | 1,457,115 | |||
Prepaid Expenses |
46,094 | 6,498 | |||||
| | | | | | | | |
TOTAL CURRENT ASSETS |
3,907,239 | 1,463,613 | |||||
| | | | | | | | |
OTHER ASSETS |
|||||||
Deferred Initial Public Offering costs |
122,826 | — | |||||
Security Deposit |
— | 3,670 | |||||
| | | | | | | | |
TOTAL OTHER ASSETS |
122,826 | 3,670 | |||||
| | | | | | | | |
TOTAL ASSETS |
$ | 4,030,065 | $ | 1,467,283 | |||
| | | | | | | | |
| | | | | | | | |
LIABILITIES AND MEMBERS' EQUITY |
|||||||
CURRENT LIABILITIES |
|||||||
Accounts Payable and Accrued Expenses |
$ | 823,834 | $ | 162,176 | |||
| | | | | | | | |
TOTAL LIABILITIES |
823,834 | 162,176 | |||||
| | | | | | | | |
COMMITMENTS AND CONTINGENCIES |
|||||||
MEMBERS' EQUITY |
|||||||
Members' Equity |
3,206,231 | 1,305,107 | |||||
| | | | | | | | |
TOTAL MEMBERS' EQUITY |
3,206,231 | 1,305,107 | |||||
| | | | | | | | |
TOTAL LIABILITIES AND MEMBERS' EQUITY |
$ | 4,030,065 | $ | 1,467,283 | |||
| | | | | | | | |
| | | | | | | | |
See notes to financial statements.
F-3
DIPEXIUM PHARMACEUTICALS, LLC
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF OPERATIONS
Years Ended December 31, 2013 and 2012 and the Cumulative Period from January 14, 2010 (inception) to December 31, 2013
| |
Years Ended December 31, | Cumulative Period from January 14, 2010 (inception) to December 31, 2013 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2013 | 2012 | ||||||||
REVENUES |
$ | — | $ | — | $ | — | ||||
| | | | | | | | | | | |
EXPENSES |
||||||||||
OPERATING EXPENSES |
||||||||||
Research and Development Expenses |
1,729,540 | 844,630 | 3,823,364 | |||||||
Acquired In-process Research and Development |
— | — | 272,500 | |||||||
General and Administrative Expenses |
2,284,080 | 999,356 | 4,641,900 | |||||||
| | | | | | | | | | | |
TOTAL OPERATING EXPENSES |
4,013,620 | 1,843,986 | 8,737,764 | |||||||
| | | | | | | | | | | |
LOSS FROM OPERATIONS |
(4,013,620 | ) | (1,843,986 | ) | (8,737,764 | ) | ||||
| | | | | | | | | | | |
NET LOSS |
$ | (4,013,620 | ) | $ | (1,843,986 | ) | $ | (8,737,764 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro Forma C Corporation Information (Unaudited)-See Note 12 |
||||||||||
Historical loss from operations before income taxes |
$ | (4,013,620 | ) | $ | (1,843,986 | ) | $ | (8,737,764 | ) | |
Pro forma provision (benefit) for income taxes |
— | — | — | |||||||
| | | | | | | | | | | |
Pro forma net loss |
$ | (4,013,620 | ) | $ | (1,843,986 | ) | $ | (8,737,764 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Pro forma net loss per common share basic and diluted |
$ | (0.87 | ) | $ | (0.45 | ) | ||||
Weighted average pro forma shares outstanding basic and diluted |
4,623,861 | 4,056,287 | ||||||||
See notes to financial statements.
F-4
DIPEXIUM PHARMACEUTICALS, LLC
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF CHANGES IN MEMBERS' EQUITY
Cumulative Period from January 14, 2010 (inception) to December 31, 2013
| |
Class A Membership Interests |
Membership Interests Amount |
Total Members' Equity |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Balance at January 14, 2010 |
— | $ | — | $ | — | |||||
Initial Investment |
500,000 | 250,000 | 250,000 | |||||||
Private Placement Offerings at $50 per unit, net of issuance costs |
28,400 | 1,408,950 | 1,408,950 | |||||||
Executive Compensation settled with Membership Interests |
2,208 | 110,400 | 110,400 | |||||||
Share-Based Compensation |
13,889 | 13,889 | ||||||||
Net Loss |
(915,665 | ) | (915,665 | ) | ||||||
| | | | | | | | | | | |
Balance at December 31, 2010 |
530,608 | 867,574 | 867,574 | |||||||
Private Placement Offerings at $50 per unit, net of issuance costs |
28,750 | 1,430,000 | 1,430,000 | |||||||
Executive Compensation settled with Membership Interests |
5,040 | 252,000 | 252,000 | |||||||
Share-Based Compensation |
126,390 | 126,390 | ||||||||
Issuance of Restricted Membership Interest Awards |
1,334 | |||||||||
Net Loss |
(1,964,494 | ) | (1,964,494 | ) | ||||||
| | | | | | | | | | | |
Balance at December 31, 2011 |
565,732 | 711,470 | 711,470 | |||||||
Private Placement Offerings at $50 per unit, net of issuance costs |
40,100 | 1,982,346 | 1,982,346 | |||||||
Executive Compensation settled with Membership Interests |
3,360 | 168,000 | 168,000 | |||||||
Share-Based Compensation |
215,277 | 215,277 | ||||||||
Issuance of Restricted Membership Interest Awards |
4,001 | |||||||||
Share-Based Payments to Vendors |
1,440 | 72,000 | 72,000 | |||||||
Net Loss |
(1,843,986 | ) | (1,843,986 | ) | ||||||
| | | | | | | | | | | |
Balance at December 31, 2012 |
614,633 | 1,305,107 | 1,305,107 | |||||||
Private Placement Offerings at $50 per unit, net of issuance costs |
92,350 | 4,598,300 | 4,598,300 | |||||||
Executive Compensation settled with Membership Interests |
2,310 | 115,500 | 115,500 | |||||||
Share-Based Compensation |
1,077,777 | 1,077,777 | ||||||||
Issuance of Restricted Membership Interest Awards |
22,665 | |||||||||
Share-Based Payments to Vendors |
123,167 | 123,167 | ||||||||
Issuance of Restricted Membership Interest to Vendors |
3,630 | — | — | |||||||
Net Loss |
(4,013,620 | ) | (4,013,620 | ) | ||||||
| | | | | | | | | | | |
Balance at December 31, 2013 |
735,588 | $ | 3,206,231 | $ | 3,206,231 | |||||
| | | | | | | | | | | |
| | | | | | | | | | | |
See notes to financial statements.
F-5
DIPEXIUM PHARMACEUTICALS, LLC
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF CASH FLOWS
Years Ended December 31, 2013 and 2012 and the Cumulative Period from January 14, 2010 (inception) to December 31, 2013
| |
Years Ended December 31, | Cumulative Period from January 14, 2010 (inception) to December 31, 2013 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2013 | 2012 | ||||||||
Operating Activities: |
||||||||||
Net Loss |
$ | (4,013,620 | ) | $ | (1,843,986 | ) | $ | (8,737,764 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||
Share Based Compensation |
1,077,777 | 215,277 | 1,433,332 | |||||||
Share Based Payments to Vendors |
123,167 | 72,000 | 195,167 | |||||||
Executive compensation settled with Membership Interests |
115,500 | 168,000 | 645,900 | |||||||
(Increase) / Decrease In: |
||||||||||
Prepaid Expenses |
(39,596 | ) | 13,600 | (46,094 | ) | |||||
Security Deposit |
3,670 | — | — | |||||||
Accounts Payable and Accrued Expenses |
538,832 | (168,717 | ) | 701,008 | ||||||
| | | | | | | | | | | |
Net Cash Used In Operating Activities |
(2,194,270 | ) | (1,543,826 | ) | (5,808,451 | ) | ||||
| | | | | | | | | | | |
Financing Activities: |
||||||||||
Proceeds from Private Placement Offerings, net of issuance costs |
4,598,300 | 1,982,346 | 9,669,596 | |||||||
| | | | | | | | | | | |
Net Cash Provided By Financing Activities |
4,598,300 | 1,982,346 | 9,669,596 | |||||||
| | | | | | | | | | | |
Net Increase In Cash |
2,404,030 | 438,520 | 3,861,145 | |||||||
Cash at Beginning of Period |
1,457,115 | 1,018,595 | — | |||||||
| | | | | | | | | | | |
Cash at End of Period |
$ | 3,861,145 | $ | 1,457,115 | $ | 3,861,145 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
SUPPLEMENTAL DISCLOSURE: |
||||||||||
NONCASH FINANCING ACTIVITY |
||||||||||
Deferred Initial Public Offering Costs |
$ | 122,826 | $ | — | $ | 122,826 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
See notes to financial statements.
F-6
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements
NOTE 1 — NATURE OF OPERATIONS
Business:
Dipexium Pharmaceuticals, LLC, a Delaware limited liability company (the "Company"), is a biopharmaceutical company addressing the need for new antibiotics to treat infectious diseases. The Company was formed on January 14, 2010. The Company is a late-stage pharmaceutical company focused on the development and commercialization of Locilex™ (pexiganan acetate cream 1%).
The Company has experienced net losses and negative cash flows from operations since inception and expects these conditions to continue for the foreseeable future. The Company has needed to raise capital from sales of its securities to sustain operations.
There can be no assurance that the Company's research and development will be successfully completed or that any Company product will be approved or commercially viable. The Company is subject to risks common to companies in the biotechnology industry including, but not limited to, dependence on collaborative arrangements, development by the Company or its competitors of new technological innovations, dependence on key personnel, protection of proprietary technology, and compliance with Food and Drug Administration ("FDA") and other governmental regulations and approval requirements.
Basis of Presentation:
The Company's primary activities since formation have been organizational activities, including recruiting personnel, acquiring rights to a pharmaceutical compound, performing business and financial planning, performing research and development activities and raising funds through issuances of Class A Membership Interests and warrants to purchase Class A Membership Interests. The Company has not generated any revenues and, accordingly, the Company is considered to be in its development stage.
The Company's financial statements have been prepared on a going concern basis which contemplates the realization of assets and the settlement of liabilities and commitments through the ordinary course of business. The Company incurred net losses of $4,013,620 and $1,843,986, for the years ended December 31, 2013 and 2012, respectively, and net losses of $8,737,764 for the period from January 14, 2010 (inception) to December 31, 2013. The Company has total members' equity of $3,206,231 and $1,305,107 as of December 31, 2013 and 2012, respectively. Management believes that the Company will continue to incur losses for the foreseeable future and will need additional resources to sustain its operations until it can achieve profitability and positive cash flows, if ever. Management plans to seek additional equity financing for the Company, but cannot assure that such financing will be available on acceptable terms, or at all. These matters raise substantial doubt about the Company's ability to continue as a going concern. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
NOTE 2 — SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States ("GAAP") requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
F-7
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 2 — SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Federal Income Taxes
The Company is not a taxpaying entity for Federal income tax purposes and, therefore, no income tax expense has been recorded in the financial statements. Income of the Company is taxed to the members in their respective income tax returns.
Concentration of Credit Risk
The Company maintains its cash balance in one financial institution. The balance is insured up to the maximum allowable by the Federal Deposit Insurance Company ("FDIC"). The Company has not experienced any losses in such accounts and does not believe it is exposed to any significant risk of loss on cash. At times, the cash balance may exceed the maximum limit of the FDIC.
Guaranteed Payments to Members
Guaranteed payments to members of the Company that are designated to represent reasonable compensation for services rendered are accounted for as Company expenses rather than an allocation of the Company's net income.
Research and Development
The Company follows the policy of expensing its research and development costs in the period in which they are incurred in accordance with Accounting Standards Codification ("ASC") 730, "Accounting for Research and Development Costs". The Company incurred net research and development expenses in the amount of $1,729,540 and $844,630 for the years ended December 31, 2013, and 2012, respectively, and $3,823,364 for the cumulative period from January 14, 2010 (inception) to December 31, 2013.
Share-Based Compensation
The Company accounts for the cost of services performed by officers and directors received in exchange for an award of Company membership interests based on the grant-date fair value of the award. In accordance with the ASC, the Company recognizes compensation expense, net of estimated forfeitures, on a straight-line basis over the service period.
Share-Based Payments to Vendors
The Company accounts for the cost of services performed by vendors in exchange for an award of Company membership interests based on the grant-date fair value of the award or fair value of the services rendered; whichever is more readily determinable. The Company recognizes the expense in the same period and in the same manner as if the Company had paid cash for the services.
Deferred Initial Public Offering Costs
The Company has incurred legal and accounting costs relating to a proposed initial public offering. These specific incremental costs directly attributable to the proposed offering of equity securities have been capitalized in other noncurrent assets and will be applied against the proceeds of the public offering when effective.
F-8
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 3 — ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses at December 31, 2013 and 2012 are as follows:
| |
2013 | 2012 | |||||
|---|---|---|---|---|---|---|---|
Accrued compensation expense |
$ | 247,845 | $ | 121,153 | |||
Accrued research and development |
367,065 | 7,710 | |||||
Accrued professional fees |
160,370 | 9,400 | |||||
Other accounts payable and accrued expenses |
48,554 | 23,913 | |||||
| | | | | | | | |
Total |
$ | 823,834 | $ | 162,176 | |||
| | | | | | | | |
| | | | | | | | |
NOTE 4 — ASSET PURCHASE
On April 8, 2010, the Company purchased the intellectual property and other rights (the "Asset") relating to the compound pexiganan from the Genaera Liquidating Trust, Argyce, LLC (the "Trust"). On March 21, 2011, the Company exercised its right to purchase the Trust's rights to milestone payments and royalties, thereby acquiring all worldwide interests to the Asset. The Company paid a total of $272,500, in the aggregate, for all of the interests in and to the Asset.
In accordance with ASC 805, the Company has defined the Asset purchase as "in-process research and development", as the assets, including the drug compound and the associated patents and other intangible assets, are in early stage development and will require significant additional investments in research and development to realize their full value.
The transaction was treated as an asset acquisition as it was determined that the Asset purchased did not meet the definition of a business. The Company determined that the acquired Asset can only be economically used for the specific and intended purpose and has no alternative future use taking into consideration future research and development, regulatory and marketing approval efforts required in order to reach technological feasibility. Accordingly, the entire purchase consideration was immediately expensed as acquired in-process research and development.
NOTE 5 — EXECUTIVE COMPENSATION
Executive compensation is paid to two executives pursuant to employment agreements effective July 23, 2010. The agreements stipulate that the executives will receive a base salary of $252,000 per annum, of which one-half is payable with the issuance of Class A Membership Interests of the Company at their most recent offering price (currently $50 per share), until such time as the Company has raised aggregate proceeds of at least $2,000,000. This threshold was reached on March 11, 2011; however, both executives signed temporary waivers until January 2012, at which time the executives revoked one-half of each waiver, and then subsequently reinstated the waivers in full. The Company issued 2,310 and 3,360 Class A Membership Interests to the two executives for the years ended December 31, 2013 and 2012, respectively, and 12,918 Class A Membership Interests for the cumulative period from January 14, 2010 (inception) to December 31, 2013.
The agreements also stipulate that the executives will receive five weeks of vacation per annum. Any unused weeks at the end of the year are payable to the executives in cash. Accordingly, compensation expense representing accrued vacation pay has been recorded in the amount of $51,692 and $35,861 for the years ended December 31, 2013 and 2012, respectively, and $172,845 for the cumulative period from January 14, 2010 (inception) to December 31, 2013.
F-9
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 6 — ISSUANCE OF MEMBERSHIP INTERESTS
On July 23, 2010, the Company entered into a securities purchase agreement with 27 investors for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 28,400 units, comprised of 28,400 Class A Membership Interests and warrants to purchase up to 14,200 additional Class A Membership Interests for gross proceeds of $1,420,000. Each warrant, exercisable for five years from July 23, 2010, has an exercise price of $60 per Class A Membership Interest.
On March 20, 2011, the Company entered into a securities purchase agreement with 22 investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 13,400 units, comprised of 13,400 Class A Membership Interests and warrants to purchase up to 6,700 additional Class A Membership Interests for gross proceeds of $670,000. Each warrant, exercisable for five years from March 20, 2011, has an exercise price of $60 per Class A Membership Interest.
On October 14, 2011, the Company entered into a securities purchase agreement with 23 investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 15,350 units, comprised of 15,350 Class A Membership Interests and warrants to purchase up to 7,675 additional Class A Membership Interests for gross proceeds of $767,500. Each warrant, exercisable for five years from October 14, 2011, has an exercise price of $60 per Class A Membership Interest.
On March 30, 2012, the Company entered into a securities purchase agreement with 16 investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 10,150 units, comprised of 10,150 Class A Membership Interests and warrants to purchase up to 5,075 additional Class A Membership Interests for gross proceeds of $507,500. Each warrant, exercisable for five years from March 30, 2012, has an exercise price of $60 per Class A Membership Interest.
On November 21, 2012, the Company entered into a securities purchase agreement with 21 investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 29,950 units, comprised of 29,950 Class A Membership Interests and warrants to purchase up to 14,975 additional Class A Membership Interests for gross proceeds of $1,497,500. Each warrant, exercisable for five years from November 21, 2012, has an exercise price of $60 per Class A Membership Interest.
On February 13, 2013, the Company entered into a securities purchase agreement with seven investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is
F-10
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 6 — ISSUANCE OF MEMBERSHIP INTERESTS (Continued)
comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 18,000 units, comprised of 18,000 Class A Membership Interests and warrants to purchase up to 9,000 additional Class A Membership Interests for gross proceeds of $900,000. Each warrant, exercisable for five years from February 13, 2013, has an exercise price of $60 per membership interest.
On July 12, 2013, the Company entered into a securities purchase agreement with 28 investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 55,500 units, comprised of 55,500 Class A Membership Interests and warrants to purchase up to 27,750 additional Class A Membership Interests for gross proceeds of $2,775,000. Each warrant, exercisable for five years from July 12, 2013, has an exercise price of $60 per Class A Membership Interest.
On November 15, 2013, the Company entered into a securities purchase agreement with 25 investors, for the private placement of the Company's Class A Membership Interests and warrants to purchase its Class A Membership Interests, at a purchase price of $50 per unit. Each unit is comprised of one Class A Membership Interest and a warrant to purchase one-half of the total Class A Membership Interests purchased. The Company issued and sold an aggregate of 18,850 units, comprised of 18,850 Class A Membership Interests and warrants to purchase up to 9,425 additional Class A Membership Interests for gross proceeds of $942,500. Each warrant, exercisable for five years from November 15, 2013, has an exercise price of $60 per Class A Membership Interest.
NOTE 7 — SHARE-BASED COMPENSATION
The Company grants restricted Class A Membership Interests awards to board members in exchange for services. These membership interests awards vest over a period of three or four years, with the first year beginning on the date the member joined the board. Accelerated vesting will occur upon a change of control or other business combination. The fair value of the membership interests granted is equal to the per-membership interest value of the most recent private placement ($50 per membership interest). In December 2013, the Company received resignation letters from six members of the board of directors and the continuing members of the board of directors authorized accelerated vesting of their Class A Membership Interests. The Company issued 20,333 Class A Membership Interests in connection with both the accelerated and annual vesting of the awards and recorded compensation expense in the amount of $693,750 associated with this accelerated vesting. Total compensation expense in the amount of $1,077,777 and $215,277 has been recorded as director fees for the years ended December 31, 2013 and 2012, respectively, and $1,433,332 for the cumulative period period from January 14, 2010 (inception) to December 31, 2013.
During 2012, Class A Membership Interests vested in full for one board member by board resolution when this board member decided to leave the board of directors. The following table summarizes the
F-11
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 7 — SHARE-BASED COMPENSATION (Continued)
non-vested Class A Membership Interests and the associated activity for the years ended December 31, 2013 and 2012.
| |
Number of Class A Membership Interests |
|||
|---|---|---|---|---|
Nonvested at January 1, 2012 |
8,666 | |||
Granted |
12,000 | |||
Vested |
(4,668 | ) | ||
| | | | | |
Nonvested at December 31, 2012 |
15,998 | |||
Granted |
16,000 | |||
Vested |
(20,998 | ) | ||
| | | | | |
Nonvested at December 31, 2013 |
11,000 | |||
| | | | | |
| | | | | |
The vested amount in 2012 includes 667 Membership Interests that were not issued until 2013.
In December 2013, 1,000 Membership Interests were issued to a member of board of directors which did not vest until February 2014.
As of December 31, 2013, there was $466,667 of total unrecognized compensation cost related to nonvested share-based compensation arrangements. That cost is expected to be recognized over a period of 3.25 years.
In November 2013, the board of directors adopted the 2013 Equity Incentive Plan. The plan will be effective as of the completion of the corporate conversion and the closing of the initial public offering. The Equity Incentive Plan reserves an aggregate number of common shares equal to 20% of the issued and outstanding common stock. The purpose of the plan is to attract and retain directors, officers, and employees whose services are considered valuable to the Company. No awards have been granted under this plan as of December 31, 2013.
NOTE 8 — SHARE-BASED PAYMENTS TO VENDORS
During 2012, the Company granted 1,440 Class A Membership Interests to two vendors in exchange for consulting services. The fair value of the Class A Membership Interests granted is equal to the value of the most recent private placement ($50 per share). Consulting expense in the amount of $72,000 has been recorded for the year ended December 31, 2012.
In January 2013, the Company granted an additional 218 Class A Membership Interests to one vendor in exchange for consulting services. The fair value of the Class A Membership Interests granted is equal to the value of the most-recent private placement ($50 per Class A Membership Interest). Consulting expense in the amount of $10,900 was recorded for the year ended December 31, 2013.
In August 2013, the Company engaged a consultant for investor related services for a 12 month term. The Company agreed to pay for the services rendered in cash and a grant of 2,000 Class A Membership Interests which will vest one year after the effective date of the agreement. The Company will expense the fair value of the Class A Membership Interests ($50 per Class A Membership Interest based on the most recent private placement and to be remeasured based on the market value at each reporting date) over the term of the agreement and when the cash is paid. Consulting expense in the amount of $101,666 was recorded for the year ended December 31, 2013.
F-12
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 8 — SHARE-BASED PAYMENTS TO VENDORS (Continued)
In October 2013, the Company engaged a consultant for product development work for a three month term completed in December 2013. The Company agreed to pay for the services rendered in cash and a grant of 1,412 Class A Membership Interests. The Company expensed the fair value of the Class A Membership Interests ($50 per Class A Membership Interest based on the most recent private placement). Consulting expense in the amount of $353,000, which includes cash payments and the membership interest grant, was recorded for the year ended December 31, 2013.
NOTE 9 — LEASE OF OFFICE SPACE
On December 27, 2011, the Company entered into a one year lease agreement with monthly payments of $1,835. The lease expired on January 1, 2013, and the Company did not renew the lease.
NOTE 10 — LEGAL MATTERS
The Company, as well as its two executives, were three of some thirty defendants in a lawsuit filed by a former stockholder of Genaera Corporation, which was the predecessor of the Trust, the party which sold the Company the worldwide rights to pexiganan, the active ingredient of the product Locilex™ in April 8, 2010. The complaint was filed on June 8, 2012 in the United States District Court for the Eastern District of Pennsylvania (Civil Action No. 12-3265) by Alan W. Schmidt, individually and on behalf of former Genaera Corporation shareholders. Among others, the suit was filed against the Company, as well as John A. Skolas and Argyce LCC, who were responsible for the administration of the Trust and who sold pexiganan to the Company via a public auction. The defendants listed in the complaint included several individuals and companies formerly associated with Genera Corporation, the Trust and/or Argyce LLC, as well as against the Company and its two executives, as individuals. Also included in the defendant group were several other pharmaceutical companies that were involved in acquiring the former drug-related assets of the Genaera Corporation and their related institutional and personal investors.
The complaint alleged, among other things, the Company and its two executives aided and abetted a breach of fiduciary duty alleged to have been committed by Argyce, LLC, the trustee of the Trust, and the Trust as well as former individuals associated with Genaera Corporation before it went into liquidation via a majority vote of its stockholders. Plaintiff claimed that the defendants, aided and abetted a breach of the duties of the trustee under common law and under a certain trust agreement allegedly signed between Argyce, LLC, as the trustee, and the Trust. With regard to the claims made against the Company and two executives, the plaintiff alleged, in pertinent part, that the Company's acquisition of the pexiganan rights was for alleged inadequate consideration, and that the Company and its management aided and abetted a breach of fiduciary duty by the Genaera Corporation defendants who were formerly associated with Genaera Corporation and/or the Trust.
The Company and its two executives filed a motion to dismiss the complaint within the prescribed time period. All of the other defendants in this litigation also filed motions to dismiss, and a court order granting each and every motion to dismiss, with prejudice, without leave to refile, was granted by the court on August 12, 2013. Plaintiff's lawyers then filed a Motion for Reconsideration which was also denied by the court on September 25, 2013. Plaintiff has filed an appeal in the United States Third Circuit Court of Appeals seeking reversal of the court's orders dismissing the complaint and denying the motion to reconsider. Appellate court briefing is underway and expected to be concluded in mid-2014. The Company and its two executives continue to believe these claims have no merit and
F-13
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 10 — LEGAL MATTERS (Continued)
will vigorously defend their position in the appellate court. The Company believes that this legal matter will not have a material adverse effect on the Company's financial position.
NOTE 11 — RELATED PARTY TRANSACTIONS
The individual to be employed as the Company's Vice President, Finance and Corporate Development as of the closing ofthe proposed initial public offering, is the owner of Aumoe Partners, LLC ("Aumoe"), which was engaged in January 2012 to perform certain financial advisory services.
The Company incurred $77,500 and $43,375 in fees for services rendered by Aumoe for the years ended December 31, 2013 and 2012, respectively, and $120,875 for the cumulative period from January 14, 2010 (inception) to December 31, 2013.
NOTE 12 — PRO FORMA INCOME TAXES AND LOSS PER SHARE (unaudited)
Immediately prior to the effectiveness of the Company's registration statement on Form S-1, the Company will convert into a Delaware C-corporation and will be subject to federal and state income taxes. Accordingly, a pro forma income tax provision has been disclosed as if the Company was a corporation for all periods presented. Based on the Company's history of generating operating losses and its anticipation of operating losses continuing in the foreseeable future, the Company has determined that it would not have been more likely than not that the tax benefits from these net operating losses would be realized and a full valuation allowance against all deferred tax assets would be recorded on a pro forma basis. Therefore, for the purposes of the pro forma tax provision, we have applied a 0% combined federal and state income rate.
A pro forma net loss per common share has been disclosed for the years ended December 31, 2013 and 2012, assuming that a 7 to 1 conversion ratio will be used to convert the Class A Membership Interests into shares of common stock at the time of the proposed initial public offering. Pro forma basic net income or loss per common share is computed by dividing net income or loss available to common shareholders by the weighted average number of common shares outstanding during the periods.
NOTE 13 — RECENT ACCOUNTING PRONOUCEMENTS
In February 2013, the Financial Accounting Standards Board issued amendments to the accounting guidance for presentation of comprehensive income to improve the reporting of reclassifications out of accumulated other comprehensive income. The amendments do not change the current requirements for reporting net income or other comprehensive income, but do require an entity to provide information about the amounts reclassified out of accumulated other comprehensive income by component. In addition, an entity is required to present, either on the face of the statement where the net income is presented or in the notes, significant amounts reclassified out of accumulated other comprehensive income by the respective line items of net income loss but only if the amount reclassified is required under GAAP to be reclassified to net income in its entirety in the same reporting period. For other amounts that are not required under GAAP to be reclassified in their entirety to net income, an entity is required to cross-reference to other disclosures required under GAAP that provide additional detail about these amounts. These amendments are effective prospectively for reporting periods beginning after December 15, 2012. The adoption of this guidance did not have any impact on the financial statements.
F-14
DIPEXIUM PHARMACEUTICALS, LLC
(A Development Stage Company)
Notes to Financial Statements (Continued)
NOTE 14 — SUBSEQUENT EVENTS
In January 2014, the Company entered into a Product Development Agreement with RRD International, LLC ("RRD") with an initial estimated contract value of $1.9 million. Pursuant to the agreement, RRD will provide the Company with certain strategic product development services including the development, planning and execution of a fully integrated product development strategy and implementation of that strategy either directly or with third party vendors. For services to be rendered in calendar year 2014, the Company has agreed to compensate RRD for an estimated $380,000 of the services rendered in January and February 2014 in Class A Membership Interests, with the balance to be paid in cash as services are performed. Accordingly, the Company will issue approximately 7,600 Class A Membership Interests ($50 per Class A Membership Interest based on the most recent private placement) in the first quarter of 2014. Terms of services to be provided after 2014, if any, will be negotiated at a later date. The Company will recognize the costs incurred in connection with this agreement as research and development expenses as services are rendered.
In order to simplify the Company's capital structure in advance of the initial public offering, in February 2014, the Company issued an aggregate of 23,719 Class A Membership Interests to investors in its prior financings in exchange for 89,900 outstanding warrants with an exercise price of $60 per Class A Membership Interest or $8.57 on a 7 to 1 as converted basis. The exchange was effectively a cashless exercise in which 23,719 Class A Membership Interests issued were calculated using an implied intrinsic value of the warrants at $2.93 per share on a 7 to 1 as converted basis and an estimated issuance price of $11.50 per share. As the warrants and membership interests are both classified as equity instruments, the warrant exercise resulted in an immaterial reclassification within members' equity.
The Company has evaluated subsequent events through February 5, 2014, the date the financial statements were available to be issued.
F-15
Shares
Common Stock
PROSPECTUS
, 2014
Oppenheimer & Co. |
||
| Feltl and Company |
You should rely only on the information contained in this prospectus. No dealer, salesperson or other person is authorized to give information that is not contained in this prospectus. This prospectus is not an offer to sell nor is it seeking an offer to buy these securities in any jurisdiction where the offer or sale is not permitted. The information contained in this prospectus is correct only as of the date of this prospectus, regardless of the time of the delivery of this prospectus or any sale of these securities.
Through and including , 2014 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer's obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
Part II
Information Not Required In Prospectus
Item 13. Other Expenses of Issuance and Distribution
Expenses of the Registrant in connection with the issuance and distribution of the securities being registered, are estimated as follows:
SEC Registration Fee |
$ | 4,443.60 | |||
FINRA Registration Fee |
$ | 5,675.00 | |||
NASDAQ Listing Fee |
$ | 50,000.00 | |||
Printing and Engraving Expenses |
$ | 100,000.00 | |||
Legal Fees and Expenses |
$ | 325,000.00 | |||
Accounting Fees and Expenses |
$ | 130,000.00 | |||
Transfer Agent Fees and Expenses |
$ | 2,000.00 | |||
Miscellaneous Costs |
$ | 85,000.00 | |||
Total |
$ | 702,118.60 | |||
Item 14. Indemnification of Directors and Officers
Section 145 of the Delaware General Corporation Law empowers a Delaware corporation to indemnify our officers and directors and certain other persons to the extent and under the circumstances set forth therein.
The Registrant's certificate of incorporation and by-laws provide for indemnification of officers and directors of the Registrant and certain other persons against liabilities and expenses incurred by any of them to the fullest extent permitted by the Delaware General Corporation Law, subject to certain stated conditions.
The above discussion of the Registrant's certificate of incorporation, by-laws, and Section 145 of the Delaware General Corporation Law is not intended to be exhaustive and is qualified in its entirety by such certificate of incorporation, by-laws and statute.
Item 15. Recent Sales of Unregistered Securities
In February 2014, we issued 1,000 Class A membership interests to Mr. Michael Duffy, one of our board members, representing the first one-fourth of his board interests which had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In February 2014, we issued 7,604 Class A membership interests to RRD International, LLC as compensation for certain services rendered. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In February 2014, we issued an aggregate of 23,719 Class A membership interests to investors in our prior financings in exchange for the redemption of outstanding warrants to purchase an aggregate of 89,900 Class A membership interests. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In December 2013, we issued 1,412 Class A membership interests to RRD International, LLC as compensation for certain services rendered. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
II-1
In December 2013, we issued 2,000 Class A membership interests to PG Advisers, LLC as compensation for certain investor relations and related consulting services rendered. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In December 2013, we issued 16,333 Class A membership interests to departing board members Steven D. Hurd, John Terrana, Barry Kagan, Russell L. Jenkins, Ellen Zaroff and John Leone, constituting the unvested portion of their board shares which our board of directors agreed to accelerate the vesting in recognition of their ongoing efforts and dedication to the board of directors. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In December 2013, we issued 315 Class A membership interests to each of Messrs. Luci and DeLuccia in connection with the terms of their employment agreements. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In December 2013, we issued 1,000 Class A membership interests to each of Messrs. Christopher Coughlin, Thomas Harrison and John Leone, who are or were board members, representing the first one-fourth of their respective board shares which had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In December 2013, we issued 667 Class A membership interests to each of Dr. Jack H. Dean and Messrs. William J. McSherry, Jr. and Steven D. Hurd, who are or were board members, representing the last one-third of their respective board interests which had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In November 2013, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 18,850 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 9,425 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $942,500. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In September 2013, we issued 840 Class A membership interests to each of Messrs. Luci and DeLuccia in connection with the terms of their employment agreements. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In August 2013, we issued 677 Class A membership interests to each of Messrs. Steven D. Hurd and John Terrana, who were board members, representing issuance of a portion of their respective board interests that had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In July 2013, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 55,500 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 27,750 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $2,775,000. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In February 2013, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 18,000 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 9,000 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $900,000. The issuance of our securities in settlement
II-2
of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In January 2013, we issued 1,680 Class A membership interests to each of Messrs. Luci and DeLuccia in connection with the terms of their employment agreements. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In February 2013, we issued 218 Class A membership interests to a consultant to our company for certain services rendered. The price used to issue the interests was the price used in the most recent financing ($50 per Class A membership interest). The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In January 2013, we issued 1,332 Class A membership interests to each of Dr. Jack H. Dean and Mr. William J. McSherry, Jr., two of our board members, representing the second one-third of their respective board interests which had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In November 2012, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 29,950 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 14,975 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $1,497,500. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In March 2012, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 10,150 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 5,075 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $507,500. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
From March 2012 to June 2012, we issued a total of 1,440 Class A membership interests to a consultant to our company for services rendered. The price used to issue the interests was the price used in the most recent financing ($50 per Class A membership interest). The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In June 2012, we issued the remaining 1,333 Class A membership interests of Ambassador Milos Koterec, one of our former board members, constituting the unvested portion of his board interests which our board of directors agreed to accelerate the vesting in recognition of his ongoing efforts and dedication to the board of directors. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In March 2012, we issued 667 Class A membership interests to Mr. John Terrana, one of our former board members, representing the first one-third of his board interests which had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In January 2012, we issued 667 Class A membership interests to each of Dr. Jack H. Dean, Mr. William J. McSherry, Jr. and Ambassador Milos Koterec, who are or were board members, representing issuance of the first one-third of their board interests which had then vested. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
II-3
In October 2011, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 15,350 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 7,675 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $767,500. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In March 2011, we entered into Securities Purchase Agreements with accredited investors whereby we agreed to sell 13,400 Class A membership interests for an issue price of $50 per interest, and agreed to issue warrants to purchase an additional 6,700 Class A membership interests at an exercise price of $60 per interest, for gross proceeds of $670,000. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
In January 2011, we issued 2,520 Class A membership interests in accordance with the employment agreements then in effect with Messrs. Luci and DeLuccia for services rendered in 2011. The issuance of our securities in settlement of these accounts was made pursuant to Section 4(a)(2) and Rule 506(b) of the Securities Act of 1933, as amended.
The abovementioned issuances of unregistered securities do not reflect the conversion of each Class A membership interest of Dipexium Pharmaceuticals, LLC into seven shares of common stock of Dipexium Pharmaceuticals, Inc., which is anticipated to take place prior to the effectiveness of this registration statement.
The following is a list of exhibits filed as a part of this registration statement:
| Exhibit Number |
Description of Document | ||
|---|---|---|---|
| 1.1 | Underwriting Agreement.* | ||
2.1 |
Asset Purchase Agreement, dated April 8, 2010, by and between the registrant and Genaera Liquidating Trust.* |
||
2.2 |
Form of Certificate of Conversion pursuant to which the registrant will be reorganized into a corporation.* |
||
3.1 |
Certificate of Formation of Dipexium Pharmaceuticals, LLC.* |
||
3.2 |
Certificate of Incorporation of Dipexium Pharmaceuticals, Inc.* |
||
3.3 |
Bylaws of Dipexium Pharmaceuticals, Inc.* |
||
5.1 |
Opinion of Ellenoff Grossman & Schole LLP.* |
||
10.1 |
2013 Equity Incentive Plan.* |
||
10.2 |
Form of Director Indemnification Agreement.* |
||
10.3 |
Amended and Restated Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and David P. Luci.* |
||
10.4 |
Amended and Restated Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and Robert J. DeLuccia.* |
||
10.5 |
Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and David Garrett.* |
||
10.6 |
Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and Robert G. Shawah.* |
||
II-4
| Exhibit Number |
Description of Document | ||
|---|---|---|---|
| 10.7 | Form of Securities Purchase Agreement, dated July 23, 2010, by and between the registrant and each of the named purchasers.* | ||
10.8 |
Form of Securities Purchase Agreement, dated March 11, 2011, by and between the registrant and each of the named purchasers.* |
||
10.9 |
Form of Securities Purchase Agreement, dated October 14, 2011, by and between the registrant and each of the named purchasers.* |
||
10.10 |
Form of Securities Purchase Agreement, dated March 30, 2012, by and between the registrant and each of the named purchasers.* |
||
10.11 |
Form of Securities Purchase Agreement, dated November 21, 2012, by and between the registrant and each of the named purchasers.* |
||
10.12 |
Form of Securities Purchase Agreement, dated February 13, 2013, by and between the registrant and each of the named purchasers.* |
||
10.13 |
Form of Securities Purchase Agreement, dated July 12, 2013, by and between the registrant and each of the named purchasers.* |
||
10.14 |
Form of Warrant, dated July 23, 2010, issued by the registrant to certain purchasers.* |
||
10.15 |
Form of Warrant, dated March 11, 2011, issued by the registrant to certain purchasers.* |
||
10.16 |
Form of Warrant, dated October 14, 2011, issued by the registrant to certain purchasers.* |
||
10.17 |
Form of Warrant, dated March 30, 2012, issued by the registrant to certain purchasers.* |
||
10.18 |
Form of Warrant, dated November 21, 2012, issued by the registrant to certain purchasers.* |
||
10.19 |
Form of Warrant, dated February 13, 2013, issued by the registrant to certain purchasers.* |
||
10.20 |
Form of Warrant, dated July 12, 2013, issued by the registrant to certain purchasers.* |
||
10.21 |
Form of Investor Rights Agreement, dated July 23, 2010, between the registrant and certain purchasers.* |
||
10.22 |
Form of Investor Rights Agreement Joinder, between the registrant and certain purchasers.* |
||
10.23 |
Master Services Agreement, dated August 23, 2010, between the registrant and RRD International, LLC.*† |
||
10.24 |
Bill of Sale and Assignment Agreement, dated March 21, 2011, between the registrant and Genaera Liquidating Trust.* |
||
10.25 |
Research and Development Agreement, dated December 8, 2011, between the registrant and DPT Laboratories, Inc.* |
||
10.26 |
Laboratory Services Agreement, dated May 22, 2012, between the registrant and Covance Laboratories Inc.*† |
||
10.27 |
Master Services Agreement, dated September 3, 2013, between the registrant and PolyPeptide Laboratories, Inc.* |
||
10.28 |
Quality Agreement, dated September 3, 2013, between registrant and PolyPeptide Laboratories, Inc.* |
||
10.29 |
Master Agreement for the Provision of Pharmaceutical Support Services, dated October 9, 2013, between registrant and Almac Group Limited.* |
||
II-5
| Exhibit Number |
Description of Document | ||
|---|---|---|---|
| 10.30 | Master Services Agreement, dated October 25, 2013, between the registrant and Research Pharmaceutical Sciences, Inc.* | ||
10.31 |
License Agreement, dated October 20, 1988, by and between Scripps Research and Clinic Foundation and Multiple Peptide Systems.* |
||
10.32 |
Second Amendment to License Agreement, dated September 24, 1996, by and between Scripps Research and Clinic Foundation and Multiple Peptide Systems.* |
||
10.33 |
Agreement, dated November 4, 1988, by and between Multiple Peptide Systems and Magainin Sciences Inc.* |
||
10.34 |
Second Amendment to Agreement, dated September 24, 1996, by and between Multiple Peptide Systems and Magainin Pharmaceuticals Inc.* |
||
10.35 |
Product Development Agreement, dated January 1, 2014, between the registrant and RRD International, LLC.* |
||
23.1 |
Consent of CohnReznick LLP. |
||
23.2 |
Consent of Ellenoff Grossman & Schole LLP (included in Exhibit 5.1).* |
||
- *
- Previously
filed.
- †
- Confidential treatment is requested for certain portions of this exhibit pursuant to 17 CFR Sections 200.8(6)(4) and 240.246.2.
II-6
The undersigned registrant hereby undertakes:
1. For purposes of determining any liability under the Securities Act of 1933, as amended (the "Securities Act") the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
2. For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
3. To provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
II-7
Pursuant to the requirements of the Securities Act, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in New York, New York on March 6, 2014.
| DIPEXIUM PHARMACEUTICALS, LLC | ||||||
By: |
/s/ DAVID P. LUCI |
|||||
| Name: | David P. Luci, Esq. | |||||
| Title: | Managing Partner | |||||
Pursuant to the requirements of the Securities Act, this registration statement has been signed below by the following persons in the capacities and on the dates indicated.
Person
|
Capacity
|
Date
|
||
|---|---|---|---|---|
| /s/ ROBERT J. DELUCCIA* Robert J. DeLuccia |
Managing Partner and Director | March 6, 2014 | ||
/s/ DAVID P. LUCI David P. Luci, Esq. |
Managing Partner and Director (Principal Executive, Financial and Accounting Officer) |
March 6, 2014 |
||
/s/ CHRISTOPHER COUGHLIN* Christopher Coughlin |
Director |
March 6, 2014 |
||
/s/ JACK H. DEAN* Dr. Jack H. Dean |
Director |
March 6, 2014 |
||
/s/ MICHAEL DUFFY* Michael Duffy, Esq. |
Director |
March 6, 2014 |
||
/s/ THOMAS HARRISON* Thomas Harrison |
Director |
March 6, 2014 |
||
/s/ WILLIAM J. MCSHERRY* William J. McSherry, Jr., Esq. |
Director |
March 6, 2014 |
| *By: | /s/ DAVID P. LUCI David P. Luci, Esq. Attorney-in-Fact |
S-1
| Exhibit Number |
Description of Document | ||
|---|---|---|---|
| 1.1 | Underwriting Agreement.* | ||
2.1 |
Asset Purchase Agreement, dated April 8, 2010, by and between the registrant and Genaera Liquidating Trust.* |
||
2.2 |
Form of Certificate of Conversion pursuant to which the registrant will be reorganized into a corporation.* |
||
3.1 |
Certificate of Formation of Dipexium Pharmaceuticals, LLC.* |
||
3.2 |
Certificate of Incorporation of Dipexium Pharmaceuticals, Inc.* |
||
3.3 |
Bylaws of Dipexium Pharmaceuticals, Inc.* |
||
5.1 |
Opinion of Ellenoff Grossman & Schole LLP.* |
||
10.1 |
2013 Equity Incentive Plan.* |
||
10.2 |
Form of Director Indemnification Agreement.* |
||
10.3 |
Amended and Restated Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and David P. Luci.* |
||
10.4 |
Amended and Restated Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and Robert J. DeLuccia.* |
||
10.5 |
Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and David Garrett.* |
||
10.6 |
Employment Agreement, dated February 3, 2014, between Dipexium Pharmaceuticals, Inc. and Robert G. Shawah.* |
||
10.7 |
Form of Securities Purchase Agreement, dated July 23, 2010, by and between the registrant and each of the named purchasers.* |
||
10.8 |
Form of Securities Purchase Agreement, dated March 11, 2011, by and between the registrant and each of the named purchasers.* |
||
10.9 |
Form of Securities Purchase Agreement, dated October 14, 2011, by and between the registrant and each of the named purchasers.* |
||
10.10 |
Form of Securities Purchase Agreement, dated March 30, 2012, by and between the registrant and each of the named purchasers.* |
||
10.11 |
Form of Securities Purchase Agreement, dated November 21, 2012, by and between the registrant and each of the named purchasers.* |
||
10.12 |
Form of Securities Purchase Agreement, dated February 13, 2013, by and between the registrant and each of the named purchasers.* |
||
10.13 |
Form of Securities Purchase Agreement, dated July 12, 2013, by and between the registrant and each of the named purchasers.* |
||
10.14 |
Form of Warrant, dated July 23, 2010, issued by the registrant to certain purchasers.* |
||
10.15 |
Form of Warrant, dated March 11, 2011, issued by the registrant to certain purchasers.* |
||
10.16 |
Form of Warrant, dated October 14, 2011, issued by the registrant to certain purchasers.* |
||
10.17 |
Form of Warrant, dated March 30, 2012, issued by the registrant to certain purchasers.* |
||
10.18 |
Form of Warrant, dated November 21, 2012, issued by the registrant to certain purchasers.* |
||
10.19 |
Form of Warrant, dated February 13, 2013, issued by the registrant to certain purchasers.* |
||
| Exhibit Number |
Description of Document | ||
|---|---|---|---|
| 10.20 | Form of Warrant, dated July 12, 2013, issued by the registrant to certain purchasers.* | ||
10.21 |
Form of Investor Rights Agreement, dated July 23, 2010, between the registrant and certain purchasers.* |
||
10.22 |
Form of Investor Rights Agreement Joinder, between the registrant and certain purchasers.* |
||
10.23 |
Master Services Agreement, dated August 23, 2010, between the registrant and RRD International, LLC.*† |
||
10.24 |
Bill of Sale and Assignment Agreement, dated March 21, 2011, between the registrant and Genaera Liquidating Trust.* |
||
10.25 |
Research and Development Agreement, dated December 8, 2011, between the registrant and DPT Laboratories, Inc.* |
||
10.26 |
Laboratory Services Agreement, dated May 22, 2012, between the registrant and Covance Laboratories Inc.*† |
||
10.27 |
Master Services Agreement, dated September 3, 2013, between the registrant and PolyPeptide Laboratories, Inc.* |
||
10.28 |
Quality Agreement, dated September 3, 2013, between registrant and PolyPeptide Laboratories, Inc.* |
||
10.29 |
Master Agreement for the Provision of Pharmaceutical Support Services, dated October 9, 2013, between registrant and Almac Group Limited.* |
||
10.30 |
Master Services Agreement, dated October 25, 2013, between the registrant and Research Pharmaceutical Sciences, Inc.* |
||
10.31 |
License Agreement, dated October 20, 1988, by and between Scripps Research and Clinic Foundation and Multiple Peptide Systems.* |
||
10.32 |
Second Amendment to License Agreement, dated September 24, 1996, by and between Scripps Research and Clinic Foundation and Multiple Peptide Systems.* |
||
10.33 |
Agreement, dated November 4, 1988, by and between Multiple Peptide Systems and Magainin Sciences Inc.* |
||
10.34 |
Second Amendment to Agreement, dated September 24, 1996, by and between Multiple Peptide Systems and Magainin Pharmaceuticals Inc.* |
||
10.35 |
Product Development Agreement, dated January 1, 2014, between the registrant and RRD International, LLC.* |
||
23.1 |
Consent of CohnReznick LLP. |
||
23.2 |
Consent of Ellenoff Grossman & Schole LLP (included in Exhibit 5.1).* |
||
- *
- Previously
filed.
- †
- Confidential treatment is requested for certain portions of this exhibit pursuant to 17 CFR Sections 200.8(6)(4) and 240.246.2.