Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Celldex Therapeutics, Inc. | Financial_Report.xls |
| EX-32 - EX-32 - Celldex Therapeutics, Inc. | a2207736zex-32.htm |
| EX-21.0 - EX-21.0 - Celldex Therapeutics, Inc. | a2207736zex-21_0.htm |
| EX-23.1 - EX-23.1 - Celldex Therapeutics, Inc. | a2207736zex-23_1.htm |
| EX-31.2 - EX-31.2 - Celldex Therapeutics, Inc. | a2207736zex-31_2.htm |
| EX-31.1 - EX-31.1 - Celldex Therapeutics, Inc. | a2207736zex-31_1.htm |
Use these links to rapidly review the document
TABLE OF CONTENTS
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark one) | ||
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2011 |
||
or |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
Commission File Number 000-15006
CELLDEX THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 13-3191702 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
119 Fourth Avenue, Needham, Massachusetts 02494
(Address of principal executive offices) (Zip Code)
Registrant's telephone number, including area code: (781) 433-0771
Securities registered pursuant to Section 12(b) of the Act:
| Title of Class: | Name of Each Exchange on Which Registered: | |
|---|---|---|
| Common Stock, par value $.001 | NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this Chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of "large accelerated filer," "accelerated filer," and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer o | Accelerated filer ý | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller Reporting Company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the registrant's common stock held by non-affiliates as of June 30, 2011 was $156.1 million. Exclusion of shares held by any person should not be construed to indicate that such person possesses the power, direct or indirect, to direct or cause the actions of the management or policies of the registrant, or that such person is controlled by or under common control with the registrant.
The number of shares of common stock outstanding at February 29, 2012 was 57,160,636 shares.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive Proxy Statement for our 2012 Annual Meeting of Stockholders are incorporated by reference into Part III of this Report.
CELLDEX THERAPEUTICS, INC.
ANNUAL REPORT ON FORM 10-K
YEAR ENDED DECEMBER 31, 2011
i
Safe Harbor Statement under the Private Securities Litigation Reform Act of 1995: This Annual Report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements include statements with respect to our beliefs, plans, objectives, goals, expectations, anticipations, assumptions, estimates, intentions and future performance, and involve known and unknown risks, uncertainties and other factors, which may be beyond our control, and which may cause our actual results, performance or achievements to be materially different from future results, performance or achievements expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be forward-looking statements. You can identify these forward-looking statements through our use of words such as "may," "will," "can," "anticipate," "assume," "should," "indicate," "would," "believe," "contemplate," "expect," "seek," "estimate," "continue," "plan," "point to," "project," "predict," "could," "intend," "target," "potential" and other similar words and expressions of the future.
There are a number of important factors that could cause the actual results to differ materially from those expressed in any forward-looking statement made by us. These factors include, but are not limited to:
- •
- our ability to raise sufficient capital to fund our clinical studies to meet our long-term liquidity needs, on
terms acceptable to us, or at all;
- •
- our ability to successfully complete research and further development, including animal, preclinical and clinical studies,
and commercialization of rindopepimut, CDX-011, CDX-1127, and other drug candidates and the growth of the markets for those drug candidates;
- •
- our ability to manage multiple clinical trials for a variety of drug candidates at different stages of development,
including our Phase 3 trial for rindopepimut;
- •
- the cost, timing, scope and results of ongoing safety and efficacy trials of rindopepimut, CDX-011,
CDX-1127 and other preclinical and clinical testing;
- •
- our ability to fund and complete the development and commercialization of rindopepimut for North America internally and to
find a strategic partner to commercialize rindopepimut outside of North America;
- •
- the ability to negotiate strategic partnerships, where appropriate, for our lead programs, including CDX-011
and CDX-1127, as well as for our non-core programs;
- •
- the strategies and business plans of our partners, such as GlaxoSmithKline's plans with respect to Rotarix®
and Vaccine Technologies' plans concerning the CholeraGarde® (Peru-15) and ETEC E. coli vaccines, which are not within our control, and our ability to maintain strong, mutually
beneficial relationships with these partners;
- •
- our ability to adapt our APC Targeting Technology™ to develop new, safe and effective vaccines against
oncology and infectious disease indications;
- •
- our ability to develop technological capabilities and expand our focus to broader markets for vaccines;
- •
- the availability, cost, delivery and quality of clinical and commercial grade materials produced by our own manufacturing
facility or supplied by contract manufacturers and partners;
- •
- the availability, cost, delivery and quality of clinical management services provided by our clinical research
organization partners;
- •
- the timing, cost and uncertainty of obtaining regulatory approvals for our drug candidates;
ii
- •
- our ability to develop and commercialize products before competitors that are superior to the alternatives developed by
such competitors;
- •
- the validity of our patents and our ability to avoid intellectual property litigation, which can be costly and divert
management time and attention; and
- •
- the factors listed under "Risk Factors" in this Annual Report on Form 10-K.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference into this report. We have no obligation, and expressly disclaim any obligation, to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise. We have expressed our expectations, beliefs and projections in good faith and we believe they have a reasonable basis. However, we cannot assure you that our expectations, beliefs or projections will result or be achieved or accomplished.
iii
Overview
Celldex Therapeutics, Inc., which we refer to as "Celldex," "we," "us," "our" or the "Company," is a biopharmaceutical company focused on the development and commercialization of several immunotherapy technologies for the treatment of cancer and other difficult-to-treat diseases.
Our lead drug candidate, rindopepimut (CDX-110), is an immunotherapeutic vaccine that targets the tumor-specific molecule, epidermal growth factor receptor variant III (EGFRvIII). EGFRvIII is a mutated form of the epidermal growth factor receptor (EGFR) that is only expressed in cancer cells and not in normal tissue and can directly contribute to cancer cell growth. EGFRvIII has been shown by polymerase chain reaction (PCR) analysis to be expressed in approximately 31% of glioblastoma (GB) tumors, also referred to as glioblastoma multiforme (GBM), the most common and aggressive form of brain cancer. Based on results from three prior Phase 2 trials, in December 2011, we initiated ACT IV, a pivotal, randomized, double-blind, controlled Phase 3 study of rindopepimut in patients with surgically resected EGFRvIII-positive GB. In December 2011, we also initiated ReACT, a Phase 2 study of rindopepimut in combination with Avastin® in patients with recurrent EGFRvIII-positive GB.
Our other lead drug candidates include CDX-011 and CDX-1127. CDX-011 is an antibody-drug conjugate (ADC) that consists of a fully-human monoclonal antibody, CR011, linked to a potent cell-killing drug, monomethyl-auristatin E (MMAE). The CR011 antibody specifically targets glycoprotein NMB (GPNMB) that is expressed in a variety of human cancers including breast cancer. In December 2011, we completed enrollment of EMERGE, a randomized Phase 2b study of CDX-011 in patients with heavily pre-treated, advanced, GPNMB-positive breast cancer and expect to present results at an appropriate scientific conference in the first half of 2012.
CDX-1127 is a fully human monoclonal antibody that targets CD27. CD27 is a co-stimulatory molecule on T cells and is over-expressed in certain lymphomas and leukemias. CDX-1127 is an agonist antibody designed to have two potential therapeutic mechanisms. CDX-1127 has been shown to activate immune cells that can target and eliminate cancerous cells in tumor bearing mice and to directly kill or inhibit the growth of CD27 expressing lymphomas and leukemias in vitro and in vivo. In November 2011, we initiated an open label, dose-escalating Phase 1 study of CDX-1127 in patients with selected malignant solid tumors or hematologic cancers.
We have additional clinical and preclinical programs, including CDX-1401, an APC Targeting Technology™ program, CDX-301, an immune cell mobilizing agent and dendritic cell growth factor and CDX-1135, a molecule that inhibits a part of the immune system called the complement system.
Generally our strategy is to develop and demonstrate proof-of-concept for our drug candidates before leveraging their value through partnerships or, in appropriate situations, continuing late stage development through commercialization ourselves. Demonstrating proof-of-concept for a drug candidate generally involves bringing it through Phase 1 clinical trials and one or more Phase 2 clinical trials so that we are able to demonstrate, based on human trials, good safety data for the drug candidate and some data indicating its effectiveness. We thus leverage the value of our technology portfolio through corporate, governmental and non-governmental partnerships. This approach allows us to maximize the overall value of our technology and product portfolio while best ensuring the expeditious development of each individual product. Our current collaborations include the commercialization of an oral human rotavirus vaccine. We are exploring potential development and commercialization collaborations for certain drug candidates such as CDX-011 and CDX-1127. Furthermore, while we plan to retain the rights to develop and commercialize rindopepimut in North America, we are exploring potential partnership opportunities to commercialize rindopepimut outside
1
of North America. Our drug candidates address market opportunities for which we believe current therapies are inadequate or non-existent.
Our products are derived from a broad set of complementary technologies which have the ability to utilize the human immune system and enable the creation of therapeutic agents. We are using these technologies to develop targeted immunotherapeutics comprised of antibodies, adjuvants and monotherapies and antibody-drug conjugates that prevent or treat cancer and other diseases that modify undesirable activity by the body's own proteins or cells. A number of our immunotherapeutic and antibody-drug conjugate drug candidates are in various stages of clinical trials. We expect that a large percentage of our research and development expenses will be incurred in support of our current and future clinical trial programs.
The following table includes the programs that we currently believe are material to our business:
Product (generic)
|
Indication/Field | Partner | Status | |||
|---|---|---|---|---|---|---|
CLINICAL |
||||||
CDX-110 |
Front-line Glioblastoma | — | Phase 3 | |||
CDX-110 |
Recurrent Glioblastoma | — | Phase 2 | |||
CDX-011 |
Metastatic breast cancer and melanoma | — | Phase 2b | |||
CDX-1127 |
Lymphoma/leukemia and solid tumors | — | Phase 1 | |||
CDX-1401 |
Multiple solid tumors | — | Phase 1/2 | |||
CDX-301 |
Cancer, autoimmune disease and transplant | — | Phase 1 | |||
PRECLINICAL |
||||||
CDX-1135 |
Renal disease | — | Preclinical | |||
CDX-014 |
Ovarian and renal cancer | — | Preclinical | |||
MARKETED PRODUCTS |
||||||
Rotarix |
Rotavirus infection | GlaxoSmithKline | Marketed |
Using our expertise in immunology, we are building business franchises in major disease areas including oncology. We have pursued some of these opportunities independently in a highly focused manner. In other cases, we have leveraged the financial support and development capabilities of corporate and public sector partners to bring our development projects to fruition. The research we have pursued over the past several years has matured into what we believe is an exciting and diverse portfolio of drug candidates.
Our success has depended and will continue to depend upon many factors, including our ability, and that of our licensees and collaborators, to successfully develop, obtain regulatory approval for and commercialize our drug candidates. Commercial sales are currently generated by GlaxoSmithKline, which is marketing Rotarix. We have had no commercial revenues from sales of our human therapeutic or other human vaccine products and we have had a history of operating losses. It is possible that we may not be able to successfully develop, obtain regulatory approval for or commercialize our drug candidates, and we are subject to a number of risks that you should be aware of before investing in us. These risks are described more fully in "Item 1A. Risk Factors."
We are a Delaware corporation organized in 1983. Our website is located at http://www.celldextherapeutics.com. On our website, investors can obtain, free of charge, a copy of our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and
2
other reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act of 1934, as amended, as soon as reasonably practicable after we file such material electronically with, or furnish it to, the Securities and Exchange Commission (SEC). None of the information posted on our website is incorporated by reference into this Annual Report.
Clinical Development Programs
Rindopepimut
Our lead clinical development program, rindopepimut, is a peptide-based immunotherapy that targets the tumor specific molecule called EGFRvIII, a functional variant of the naturally expressed EGFR, a protein which has been well validated as a target for cancer therapy. Unlike EGFR, EGFRvIII is not present in normal tissues and has been shown to be a transforming oncogene that can directly contribute to cancer cell growth. EGFRvIII is commonly present in glioblastoma or GB, also commonly referred to as glioblastoma multiforme or GBM, the most common and aggressive form of brain cancer. The rindopepimut vaccine is composed of the EGFRvIII peptide linked to a carrier protein called Keyhole Limpet Hemocyanin (KLH) and administered together with the adjuvant GM-CSF. The Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have both granted orphan drug designation for rindopepimut for the treatment of EGFRvIII expressing GB and the FDA has also granted Fast Track designation.
In April 2008, we and Pfizer Inc. (Pfizer) entered into a License and Development Agreement (the "Pfizer Agreement") under which Pfizer was granted an exclusive worldwide license to rindopepimut. The Pfizer Agreement provided for reimbursement by Pfizer of all costs incurred by us in connection with the collaboration since the effective date. In November 2010, the Pfizer Agreement was terminated (the "Pfizer Termination") and all rights to rindopepimut were returned to us. Pfizer did not provide a reason for termination. Effective with the Pfizer Termination, Pfizer is no longer funding the development of rindopepimut.
The Phase 1 (VICTORI) and Phase 2a (ACTIVATE) studies of EGFRvIII immunotherapy were led by collaborating investigators at the Brain Center at Duke Comprehensive Cancer Center in Durham, North Carolina and at M.D. Anderson Cancer Center in Houston, Texas and enrolled 14 and 18 evaluable GB patients, respectively. An extension of the Phase 2a study (ACT II) at the same two institutions evaluated 22 additional GB patients treated in combination with maintenance temozolomide (TMZ) (the current standard of care).
We initiated a Phase 2b/3 randomized study (ACT III) of rindopepimut combined with standard of care, TMZ, versus standard of care alone in patients with GB in over 30 sites throughout the United States. In December 2008, we announced an amendment to convert the ACT III study to a single-arm Phase 2 clinical trial in which all patients were to receive rindopepimut in combination with temozolomide. The decision, which followed the recommendation of the Independent Data Monitoring Committee, was based on the observation that the majority of patients randomized to the control (standard of care) arm withdrew from this open-label study after being randomized to the control arm. Patients participating in the control arm of the study were offered the option to receive treatment with rindopepimut. Under this amendment, the ACT III study provided for a multi-center, non-randomized dataset for rindopepimut in patients with newly diagnosed GB.
In November 2010, we announced complete data for the primary endpoint of the 65 patients enrolled to receive rindopepimut in combination with maintenance TMZ in the ACT III study. The data showed that 43 of 65 patients (66%) were progression-free at 5.5 months after entry into the study. Taking into account the 3 to 3.5 months required to complete pre-study chemoradiation and enter into the study, the 5.5 month time point in ACT III corresponds to approximately 8.5 months after diagnosis. The ACTIVATE and ACT II trials, which were conducted in two leading centers,
3
reported progression-free rates at 8.5 months after diagnosis of 70% and 80%, respectively, and similar results were seen in the ACT III trial, which enrolled patients in over 25 centers in the United States.
The following table summarizes the progression free survival (PFS) and overall survival (OS) rates from clinical trials of rindopepimut as compared to matched historical controls and the standard of care (SOC).
| |
Median PFS from diagnosis (months) |
Median OS from diagnosis (months) |
OS at 24 months | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
ACT III (n=65) |
12.3 | (1) | 24.6 | 52 | % | |||||
ACT II (n=22) |
15.3 | 24.4 | 50 | % | ||||||
ACTIVATE (n=18) |
14.2 | 24.6 | 50 | % | ||||||
Matched historical control (n=17)(2) |
6.4 | 15.2 | 6 | % | ||||||
Standard of care radiation/TMZ (n=287)(3) |
6.9 | 14.6 | 27 | % | ||||||
- (1)
- Change
in median PFS not statistically significant from ACTIVATE and ACT II.
- (2)
- Sampson,
et al. J. Clin. Oncol. 2010 Nov 1, 28(31), 4722-9. Historical controls were treated at M.D. Anderson and matched for eligibility
(EGFRvIII-positive, karnofsky performance status (KPS) greater-than or equal to 80%, complete resection, radiation/temozolomide (TMZ) and without progression through ~ 3 months
post-diagnosis).
- (3)
- Stupp, et al. N. Engl. J. Med. 2005, 352, 987-96.
Importantly, rindopepimut showed a similar benefit in patients whether or not they expressed an active DNA repair gene (MGMT) that has been shown to limit the benefit from TMZ treatment. In ACT III, the number of patients who were expected to be resistant to the TMZ chemotherapy appeared to do better with vaccination than the numbers observed in the historical data. Patients who have an active DNA repair gene, MGMT (unmethylated), generally have a worse outcome, presumably because they do not gain much benefit from TMZ as reported in the literature. Patients with a methylated MGMT promoter in their tumor do not express MGMT and have a more favorable outcome to TMZ treatment. Patients with methylated tumors (n=25) that were treated with the rindopepimut regimen experienced a median PFS of 17.5 months, which compares favorably with the published data from the SOC of radiation plus TMZ (R+TMZ) of 10.3 months. Those with unmethylated tumors (n=40) treated with the rindopepimut regimen experienced a PFS of 11.2 months, which compared favorably to the PFS with SOC of 5.3 months in this patient population. Thus, rindopepimut would appear to benefit both methylated and unmethylated MGMT patients.
In December 2011, we initiated ACT IV, a pivotal, randomized, double-blind, controlled Phase 3 study of rindopepimut in patients with surgically resected, EGFRvIII-positive GB. Patients will be randomized after the completion of surgery and standard chemoradiation. The treatment regime includes a vaccine priming phase post-radiation followed by an adjuvant temozolomide phase and a vaccine maintenance therapy phase. Patients will be treated until disease progression or intolerance to therapy. The primary objective of the study is to determine whether rindopepimut plus adjuvant GM-CSF improves the overall survival of patients with newly diagnosed EGFRvIII-positive GB after Gross Total Resection (GTR) when compared to treatment with TMZ and a control injection of KLH. KLH is a component of rindopepimut and was selected due to its ability to generate a similar injection site reaction to that observed with the rindopepimut vaccine. ACT IV will enroll up to 440 patients at over 150 centers worldwide to recruit approximately 374 patients with GTR to be included in the primary analysis. Our targeted patient accrual is 24 months and another 18 to 24 months of follow-up. We expect it will cost over $50 million to complete this Phase 3 study.
In December 2011, we also initiated ReACT, a Phase 2 study of rindopepimut in combination with Avastin® in patients with recurrent EGFRvIII-positive GB. ReACT will enroll approximately 95
4
patients in a first or second relapse of GB following receipt of standard therapy and will be conducted at approximately 20 sites across the United States. Approximately 70 patients who have yet to receive Avastin will be randomized to receive either rindopepimut and Avastin or a control injection of KLH and Avastin in a blinded fashion. Another 25 patients who are refractory to Avastin having received Avastin in either the frontline or recurrent setting with subsequent progression will receive rindopepimut plus Avastin in a single treatment arm. We expect preliminary data from this study to be available in mid-2013.
In addition, researchers at Stanford University are conducting a pilot trial of rindopepimut in pediatric patients with pontine glioma in an investigator sponsored trial. Patient screening is ongoing for this trial.
Glembatumumab Vedotin (CDX-011)
CDX-011 is an antibody-drug conjugate (ADC) that consists of a fully-human monoclonal antibody, CR011, linked to a potent cell-killing drug, monomethyl-auristatin E (MMAE). The CR011 antibody specifically targets glycoprotein NMB (GPNMB) that is expressed in a variety of human cancers including breast cancer and melanoma. The ADC technology, comprised of MMAE and a stable linker system for attaching it to CR011, was licensed from Seattle Genetics, Inc. The ADC is designed to be stable in the bloodstream. Following intravenous administration, CDX-011 targets and binds to GPNMB and upon internalization into the targeted cell, CDX-011 is designed to release MMAE from CR011 to produce a cell-killing effect. The FDA has granted Fast Track designation to CDX-011 for the treatment of advanced, refractory/resistant GPNMB-expressing breast cancer.
Treatment of Breast Cancer: In June 2008, an open-label, multi-center Phase 1/2 study was initiated of CDX-011 administered intravenously once every three weeks to patients with locally advanced or metastatic breast cancer who had received prior therapy (median of seven prior regimens). The study began with a bridging phase to confirm the maximum tolerated dose (MTD) and then expanded into a Phase 2 open-label, multi-center study.
The study confirmed the safety of CDX-011 at the pre-defined maximum dose level (1.88 mg/kg) in 6 patients. An additional 28 patients were enrolled as an expanded Phase 2 cohort (for a total of 34 treated patients at 1.88 mg/kg, the Phase 2 dose) to evaluate the PFS rate at 12 weeks. As previously seen in melanoma patients, the 1.88 mg/kg dose was well tolerated in this patient population with the most common adverse events of rash, alopecia, and fatigue. The primary activity endpoint, which called for at least 5 of 25 (20%) patients in the Phase 2 study portion to be progression-free at 12 weeks, was met as 9 of 26 (35%) evaluable patients were progression-free at 12 weeks.
For all patients treated at the maximum dose level, tumor shrinkage was seen in 62% (16/26) and median PFS was 9.1 weeks. A subset of 10 patients had "triple negative disease," a more aggressive breast cancer subtype that carries a high risk of relapse and reduced survival as well as limited therapeutic options due to lack of over-expression of HER2/neu, estrogen and progesterone receptors. In these patients, 78% (7/9) had some tumor shrinkage, 12-week PFS rate was 70% (7/10), and median PFS was 17.9 weeks. Tumor samples from a subset of patients across all dose groups were analyzed for GPNMB expression. The tumor samples from most patients showed evidence of stromal and/or tumor cell expression of GPNMB. The most common treatment-related toxicities were fatigue, rash, nausea, alopecia (hair loss), neutropenia and vomiting.
In December 2011, we completed enrollment of EMERGE, a randomized, multi-center Phase 2b study of CDX-011 in patients with heavily pre-treated, advanced, GPNMB-positive breast cancer. Patients were randomized (2:1) to receive either CDX-011 or single-agent "Investigator's Choice" chemotherapy. Patients randomized to receive Investigator's Choice are allowed to cross over to CDX-011 following disease progression. Activity endpoints include response rate (RR) and PFS. We expect that a significant portion of the enrolled patients will have triple-negative disease, since GPNMB
5
is frequently expressed in this patient population. We expect to present results at the 2012 American Society of Clinical Oncology (ASCO) meeting in June 2012.
In 2009, Formatech, Inc., a third party contract manufacturer ("Formatech") was engaged by us for the aseptic filling of one lot of our CDX-011 product candidate being used in our ongoing Phase 2b study. The CDX-011 lot from Formatech has passed all of the sterility testing performed during drug release and in subsequent stability studies. At the end of January 2012, we were notified by the FDA that because significant Good Manufacturing Practice (GMP) violations were uncovered during inspection of Formatech, our Phase 2b study for CDX-011 was being placed on partial clinical hold. The FDA uncovered these findings during their inspections of the Formatech facility between August to October 2010 and July to August 2011. These inspections began approximately one year after the CDX-011 drug product was filled at Formatech. Specifically, the FDA requested that no new patients be treated with CDX-011. However, patients already undergoing treatment with CDX-011 could continue treatment using vials of CDX-011 from the lot filled by Formatech, after such patients were informed of the potential risk and reconsented to continued participation in the study. Since the Phase 2b trial completed accrual of patients in December 2011 and there are currently no other ongoing open studies with CDX-011, the only patients that are affected by the partial hold are in the Investigator's Choice (control arm) of the study who currently are not receiving CDX-011 and may be eligible to cross over at the time of progression and receive CDX-011 under the study protocol. Currently there are eight patients remaining on the control arm.
We have initiated discussions with the FDA regarding our proposal to utilize vials of CDX-011 that were filled by a different contract manufacturer. Although the FDA has stated that no new patients may receive CDX-011 and that no patients may cross over to receive CDX-011, we have asked the FDA to reconsider allowing patients currently on the control arm to cross over to CDX-011 after stability testing and confirmation that the product filled by the other contract manufacturer is acceptable for continued use. If the FDA agrees to our proposal concerning use of the alternative CDX-011 for the eligible cross-over patients, we believe that we should have sufficient clinical supply of CDX-011 to treat these cross-over patients. If we are not able to treat the eight remaining cross-over patients with CDX-011, patients may withdraw from the control arm study upon learning that they will not be allowed to cross over to CDX-011 following disease progression. However, the primary analyses for the study are entirely based upon the primary randomization and do not include the cross over results. Based on our discussions with our clinical investigators, we do not believe that a high proportion of patients will withdraw from the control arm prior to progression. We do not believe that this partial hold will significantly impact analysis of the Phase 2b data for purposes of determining next steps in our future development of CDX-011.
In addition, the FDA has agreed in concept that we could reprocess the remaining available vials of CDX-011 manufactured at Formatech at another cGMP contract manufacturer. The FDA's final decision regarding the acceptability of this reprocessing will be made upon review of data concerning the stability and sterility of the reprocessed vials of CDX-011. If we are unsuccessful at reprocessing the available drug product or if FDA does not approve the use of these reprocessed vials, we will need to manufacture new drug product for subsequent clinical studies for CDX-011, which may cause a delay in the initiation of a subsequent trial with CDX-011.
Treatment of Metastatic Melanoma: In 2009, we completed enrollment of 117 patients in a Phase 1/2 open-label, multi-center, dose escalation study to evaluate the safety, tolerability and pharmacokinetics of CDX-011 for patients with un-resectable Stage III or Stage IV melanoma who had failed no more than one prior line of cytotoxic therapy. The MTD was determined to be 1.88 mg/kg administered intravenously (IV) once every three weeks. The study achieved its primary activity objective with an ORR in the Phase 2 cohort of 15% (5/34). Median PFS was 3.9 months. CDX-011 was generally well tolerated, with the most frequent treatment-related adverse events being rash, fatigue, alopecia (hair loss), pruritus, diarrhea and neuropathy. In the subset of patients with tumor
6
biopsies, high levels of tumor expression of GPNMB appeared to correlate with favorable outcome. In the seven patients whose tumors were found to express high amounts of GPNMB, and who were treated at the maximum tolerated doses across all dosing schedules, median PFS was 4.9 months. The development of rash, which may be associated with the presence of GPNMB in the skin also seemed to correlate with greater PFS.
We intend to initially focus our resources on advancing CDX-011 in breast cancer while pursuing further development of CDX-011 in melanoma through collaborations and investigator sponsored studies.
CDX-1127
CDX-1127 is a human monoclonal antibody that targets CD27, a member of the tumor necrosis factor (TNF) receptor superfamily. We have entered into license agreements with the University of Southampton, UK for intellectual property related to uses of anti-CD27 antibodies and with Medarex for access to the UltiMab technology to develop and commercialize human antibodies to CD27. CD27 acts downstream from CD40 and may provide a novel way to regulate the immune responses. CD27 is a co-stimulatory molecule on T cells and is over-expressed in certain lymphomas and leukemias. CDX-1127 is an agonist antibody designed to have two potential therapeutic mechanisms. CDX-1127 has been shown to activate immune cells that can target and eliminate cancerous cells in tumor bearing mice and to directly kill or inhibit the growth of CD27 expressing lymphomas and leukemias in vitro and in vivo. Both mechanisms have been seen even at low doses in appropriate preclinical models.
In November 2011, we initiated an open label, dose-escalating Phase 1 study of CDX-1127 in patients with selected malignant solid tumors or hematologic cancers at multiple clinical sites in the United States. The Phase 1 study is designed to test five escalating doses of CDX-1127 to determine a Phase 2 dose for further development based on safety, tolerability, potential activity and immunogenicity. The study will accrue approximately 30 patients in each of the two arms, either selected refractory/relapsed solid tumors or lymphomas/leukemias known to express CD27. Patients will have received all appropriate prior therapies for their specific disease. The trial design incorporates both single dosing and multiple dosing regimens at each dose level. We expect to complete enrollment in this study by the end of 2012.
CDX-1401
CDX-1401, developed from our APC Targeting Technology, is a fusion protein consisting of a fully human monoclonal antibody with specificity for the dendritic cell receptor, DEC-205, linked to the NY-ESO-1 tumor antigen. In humans, NY-ESO-1 has been detected in 20 – 30% of all melanoma, lung, esophageal, liver, gastric, prostate, ovarian and bladder cancers, thus representing a broad opportunity. This product is intended to selectively deliver the NY-ESO-1 antigen to dendritic cells for generating robust immune responses against cancer cells expressing NY-ESO-1. We are developing CDX-1401 for the treatment of malignant melanoma and a variety of solid tumors which express the proprietary cancer antigen NY-ESO-1, which we licensed from the Ludwig Institute for Cancer Research in 2006. Preclinical studies have shown that CDX-1401 is effective for activation of human T cell responses against NY-ESO-1.
In September 2009, we initiated enrollment in a dose-escalating, multi-center, Phase 1/2 study aimed at determining the optimal dose for further development based on the safety, tolerability, and immunogenicity of the CDX-1401 vaccine. The trial will evaluate three different doses of the vaccine in combination with TLR agonists poly-ICLC or Hiltonol™ and/or R848 or resiquimod.
In October 2010, we announced interim data for the first 20 patients enrolled in the study, 35% of whom had confirmed NY-ESO-1 expression. The interim data showed that six patients maintained stable disease and were eligible for multiple cycles of the treatment regimen, including 4 patients who
7
have received 3 or more cycles (6 weeks of treatment followed by a 6 week rest), with stable disease of up to 11.5+ months. The treatment was well tolerated and there were no dose-limiting toxicities. Robust anti-NY-ESO-1 immunity was induced with the majority of the patients developing anti-NY-ESO-1 antibody responses and 39% of the patients having increases in NY-ESO-1 specific T cell responses, including both CD4 and CD8 responses. Importantly, the T cell responses were directed against multiple regions of the NY-ESO-1 antigen.
In February 2012, we completed enrollment in the Phase 1 portion of the study and expect to report data at an appropriate scientific conference in the second half of 2012.
CDX-301
CDX-301 is a FMS-like tyrosine kinase 3 ligand (Flt3L) stem cell mobilizer and dendritic cell growth factor. We licensed CDX-301 from Amgen in March 2009. CDX-301 is a potent hematopoietic cytokine that stimulates the expansion and differentiation of hematopoietic progenitor and stem cells. CDX-301 has demonstrated a unique capacity to increase the number of circulating dendritic cells in both laboratory and clinical studies. In addition, CDX-301 has shown impressive results in models of cancer, infectious diseases and inflammatory/autoimmune diseases. We believe CDX-301 may hold significant opportunity for synergistic development in combination with other proprietary molecules in our portfolio.
In January 2012, we initiated a dose-escalating Phase 1 study of CDX-301 in approximately 30 healthy subjects in collaboration with Rockefeller University. The Phase 1 study will evaluate seven different dosing regimens of CDX-301 to determine the appropriate dose for further development based on safety, tolerability, and biological activity.
Preclinical Programs
CDX-1135
CDX-1135 is a molecule that inhibits a part of the human immune system called the complement system. The complement system is a series of proteins that are important initiators of the body's acute inflammatory response against disease, infection and injury. Excessive complement activation also plays a role in some persistent inflammatory conditions. CDX-1135 is a soluble form of naturally occurring Complement Receptor 1 that has been shown to inhibit the activation of the complement cascade in animal models and in human clinical trials. In preclinical studies, CDX-1135 has been shown to inhibit both the classical and alternative pathways of complement activation.
Dense Deposit Disease (DDD) is a rare and devastating disease that is caused by uncontrolled activation of the alternative pathway of complement and leads to progressive kidney damage in children. There is currently no treatment for patients with DDD and about half progress to end-stage renal disease within 10 years. Because DDD recurs in virtually all patients who receive a kidney transplant, transplantation is not a viable option for these patients. In animal models of DDD, CDX-1135 treatment showed evidence of reversal of kidney damage. We believe that regulating the complement system could have therapeutic and prophylactic applications in DDD and several other acute and chronic conditions, including organ transplantation, multiple sclerosis, rheumatoid arthritis, age-related macular degeneration (AMD), atypical Hemolytic Uremic Syndrome (aHUS), Paroxysmal Nocturnal Hemaglobinuria (PNH), and myasthenia gravis.
8
CDX-1135 is currently being studied in DDD under an investigator sponsored IND and initial experience indicates that CDX-1135 limits complement abnormalities. In 2011, we completed process development activities and manufactured GMP clinical drug product. Based on discussion to date with the FDA, we are currently planning to initiate a Phase 2 pilot study of CDX-1135 in a small number of DDD patients to determine the appropriate dose and regimen for further clinical development based on safety, tolerability and biological activity in 2012.
CDX-014
CDX-014 is a fully-human monoclonal ADC that targets TIM-1, a molecule that is highly expressed on renal and ovarian cancers with minimal expression in normal tissues. The antibody, CDX-014, is linked to a potent chemotherapeutic, monomethyl auristatin E (MMAE), using Seattle Genetics' proprietary technology. The ADC is designed to be stable in the bloodstream, but to release MMAE upon internalization into TIM-1-expressing tumor cells, resulting in a targeted cell-killing effect. CDX-014 has shown potent activity in preclinical models of ovarian and renal cancer.
Development Strategy
Precision Targeted Immunotherapy Platform:
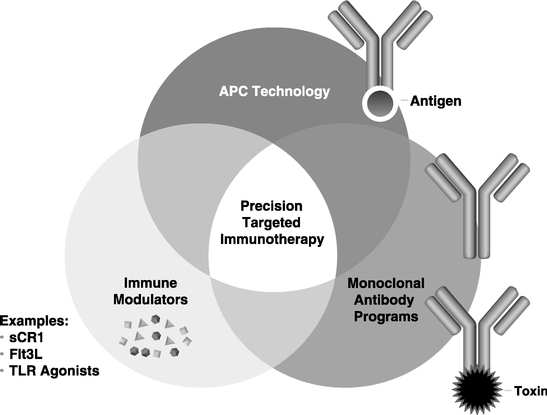
We believe there is tremendous untapped potential in immunotherapy that can be exploited through the right combination of therapeutic agents. Immunotherapy approaches have encountered difficulties when following standard drug development. The mechanisms of action are complex, activity is generally not dependent on highest tolerated dose and patient response is highly variable. Our understanding of the immune system, cancer's effect on immune mediated mechanisms, and the impact of conventional therapies on the immune system provides a new rationale for combining therapies that may lead to significant clinical responses. The concept of Precision Targeted Immunotherapy is to exploit this knowledge and the availability of good, tested products that may not be sufficiently effective
9
to be commercialized as a monotherapy, but which we believe may be very effective in combination approaches. Our goal is to develop products that maximize the efficacy of immunotherapy regimens through combinations of therapeutic agents. These therapeutic agents include:
Therapeutic Antibodies: These programs are based on the well validated approach to using antibodies that target cancer and other diseases directly, or interfere with critical interactions between the patient and the disease. Our antibody programs include antibody-drug conjugates (ADCs) that are designed to deliver potent cytotoxic molecules to cancer cells, and traditional unmodified antibody approaches. Our current programs are based on fully human sequence antibodies to minimize patient reactivity against the drug. In addition, we have access through a Research and Commercialization Agreement with Medarex (now a subsidiary of Bristol-Myers Squibb) to the UltiMAb® Technology for generating fully human monoclonal antibodies. Under this agreement, we can exercise up to ten separate licenses to develop and commercialize therapeutic antibody products, either alone or through collaboration with our licensing partners.
Our APC Targeting Technology™: This is a new class of vaccines based on our proprietary antibody-targeted vaccine technology that is used to generate an immune response against cancer or other diseases. Our APC Targeting Technology™ uses human monoclonal antibodies linked to disease associated antigens to efficiently deliver the attached antigens to immune cells known as antigen presenting cells, or APCs. This technology has been designed to allow us to take advantage of many important characteristics of human monoclonal antibodies, including their long circulating half-life, well known safety profile, and standardized manufacturing procedures. We believe that our APC Targeting Technology™ provides significant manufacturing, regulatory and other practical advantages over patient specific and other immune-based treatments and can substantially reduce the dosage and cost currently required in conventional immunotherapies. Preclinical studies have demonstrated that APC Targeting Technology™ is more effective than conventional non-targeted vaccines. We have developed several proprietary monoclonal antibodies that can independently be developed to generate new product opportunities. Our CDX-1401 program is in clinical development utilizing the APC technology.
Immune System Modulators: Immune system modulators include drugs that activate or suppress specific parts of the immune system. Currently we are combining our APC technology drug candidates with molecules known as Toll-Like Receptor (TLR) agonists that can activate patients' innate and adaptive immunity. We are also developing an immune cell growth factor called FMS-like tyrosine kinase 3 ligand (FLT3-L or CDX-301) designed to expand immune cells and stem cells. In addition, we are investigating the activity of a complement inhibitor (CDX-1135) that suppresses inflammatory reactions. These agents further support our Precision Targeted Immunotherapy Platform.
Antibody-Drug Conjugates (ADC): ADCs are monoclonal antibodies that are linked to potent cell-killing drugs. Our ADCs utilize fully-human monoclonal antibodies that internalize within target cells after binding to their cell-surface receptors. Enzymes present inside the cell cause the cell-killing drug to be released from the monoclonal antibody, allowing it to have the desired activity. A key component of our ADCs is the linker that attaches the drug to the monoclonal antibody. When the ADC is internalized within the target cell, the drug is released, thereby minimizing toxicity to normal tissues. Our CDX-011 program is in clinical development with the ADC technology.
Our strategy is to utilize our expertise to design and develop targeted immunotherapeutics that have significant and growing market potential; to establish governmental and corporate alliances to fund development; and to commercialize our products either through corporate partners or, in appropriate circumstances, through our own direct selling efforts. Our goal is to demonstrate clinical proof-of-concept for each product, and then seek partners to help see those products which we cannot develop ourselves through to commercialization. This approach allows us to maximize the overall value of our technology and product portfolios while best ensuring the expeditious development of each individual product.
10
Factors that may significantly harm our commercial success, and ultimately the market price of our common stock, include but are not limited to, announcements of technological innovations or new commercial products by our competitors, disclosure of unsuccessful results of clinical testing or regulatory proceedings and governmental approvals, adverse developments in patent or other proprietary rights, public concern about the safety of products developed by us and general economic and market conditions. See "Item 1A. Risk Factors."
Partnerships
Our strategy is to develop and demonstrate proof-of-concept for our drug candidates before leveraging their value through partnerships or, in appropriate situations, continuing late stage development through commercialization ourselves. We are exploring potential development and commercialization collaborations for certain product candidates such as CDX-011 and CDX-1127. Furthermore, while we plan to retain the rights to develop and commercialize rindopepimut in North America, we are exploring potential partnership opportunities to commercialize rindopepimut outside of North America.
We have entered into collaborative partnership agreements with pharmaceutical and other companies and organizations that provide financial and other resources, including capabilities in research, development, manufacturing, and sales and marketing, to support our research and development programs. We depend on these relationships and may enter into more of them in the future. Some of our partners have substantial responsibility to commercialize a product and to make decisions about the amount and timing of resources that are devoted to developing and commercializing a product. As a result, we do not have complete control over how resources are used toward some of our products.
Partnership agreements may terminate without benefit to us if the underlying products are not fully developed. If we fail to meet our obligations under these agreements, they could terminate and we might need to enter into relationships with other collaborators and to spend additional time, money, and other valuable resources in the process.
We cannot predict whether our collaborators will continue their development efforts or, if they do, whether their efforts will achieve success. Many of our collaborators face the same kinds of risks and uncertainties in their business that we face. A delay or setback to a partner will, at a minimum, delay the commercialization of any affected products, and may ultimately prevent it. Moreover, any partner could breach its agreement with us or otherwise not use best efforts to promote our products. A partner may choose to pursue alternative technologies or products that compete with our technologies or products. In either case, if a partner failed to successfully develop one of our products, we would need to find another partner. Our ability to do so would depend upon our legal right to do so at the time and whether the product remained commercially viable.
GlaxoSmithKline plc (Glaxo)
Rotavirus is a major cause of diarrhea and vomiting in infants and children. In 1997, we licensed our oral rotavirus strain to Glaxo and Glaxo assumed responsibility for all subsequent clinical trials and all other development activities. Glaxo gained approval for its rotavirus vaccine, Rotarix, in Mexico in July 2004, which represented the first in a series of worldwide approvals and commercial launches for the product leading up to the approval in Europe in 2006 and in the U.S. in 2008. We licensed-in our rotavirus strain in 1995 and owe a license fee of 30% to Cincinnati Children's Hospital Medical Center (CCH) on net royalties received from Glaxo. We are obligated to maintain a license with CCH with respect to the Glaxo agreement. The term of the Glaxo agreement is through the expiration of the last of the relevant patents covered by the agreement, although Glaxo may terminate the agreement upon
11
90 days prior written notice. The last relevant patent is scheduled to expire in December 2012. No additional milestone payments are due from Glaxo under the agreement.
In May 2005, we entered into an agreement whereby an affiliate of Paul Royalty Fund II, L.P. (PRF) purchased a 70% interest in the milestone payments and net royalties that we will receive on the development and worldwide sales of Rotarix. We have received a total of $60 million in milestone payments under the PRF agreement. No additional milestone payments are due from PRF under the agreement. The PRF agreement terminates in December 2012, unless otherwise extended. We would retain approximately 65% of the royalties on worldwide sales of Rotarix if PRF receives 2.45 times the aggregate cash payments of $60 million it made to us prior to December 2012. We do not expect this to occur.
Vaccine Technologies, Inc. (VTI)
In January 2009, we entered into a license agreement with VTI under which we granted a worldwide exclusive license to VTI to develop and commercialize our CholeraGarde and ETEC vaccine programs. We may receive milestones payments of up to $0.8 million and royalties in the low to mid teens with respect to development and commercialization of the technology licensed to VTI.
TopoTarget A/S (TopoTarget)
Under our April 2008 agreement (TopoTarget Agreement) with TopoTarget, we could receive up to $6 million in either potential commercial milestone payments related to future net sales of Belinostat or 10% of any sublicense income received by TopoTarget (TopoTarget Payments). We have no financial and operational responsibility for the clinical development of Belinostat under the TopoTarget Agreement. In February 2010, TopoTarget entered into a co-development and commercialization agreement for Belinostat with Spectrum Pharmaceuticals, Inc. resulting in our receipt of $3 million of the TopoTarget Payments.
Research Collaboration and License Agreements
We have entered into license agreements whereby we have received licenses or options to license technology, specified patents and/or patent applications. These license and collaboration agreements generally provide for royalty payments equal to specified percentages of product sales, annual license maintenance fees, continuing patent prosecution costs and potential future milestone payments to third parties upon the achievement of certain development, regulatory and/or commercial milestones.
Medarex, Inc., a subsidiary of Bristol-Myers Squibb (Medarex)
We and Medarex, a former related party, have entered into the following agreements, each of which was approved by a majority of its independent directors who did not have an interest in the transaction. These agreements include:
- •
- An Assignment and License Agreement, as amended, (Assignment and License Agreement) that provides for the assignment of
certain patent and other intellectual property rights and a license to certain Medarex technology related to the Company's APC Targeting Technology™ and an anti-mannose
receptor product; and
- •
- A Research and Commercialization Agreement, as amended, (Research and Commercialization Agreement) that provides us with certain rights to obtain exclusive commercial licenses to proprietary monoclonal antibodies raised against certain antigens utilizing the Medarex UltiMAb technology platform for generating antibodies.
Under the terms of the Assignment and License Agreement, we may be required to pay royalties in the low-single digits on any net product sale of a Licensed Royalty-Bearing Product or Anti-Mannose
12
Product to Medarex until the later of (i) the expiration of the last to expire applicable patent and (ii) the tenth anniversary of the first commercial sale of such licensed product. Under the terms of the Research and Commercialization Agreement, we may be required to pay milestones of up to $7.0 million upon obtaining first approval for commercial sale in a first indication of a product containing a licensed antibody and royalty payments in the low- to mid-single digits on any net product sales to Medarex with respect to the development of any products containing such licensed antibodies until the later of (i) the expiration of the last to expire applicable patent and (ii) the tenth anniversary of the first commercial sale of such licensed product. In September 2010, we exercised an option under our Research and Commercialization Agreement, whereby we licensed from Medarex access to the UltiMab technology to develop and commercialize human antibodies to CD27, including CDX-1127.
Rockefeller University (Rockefeller)
In November 2005, we and Rockefeller entered into a license agreement for the exclusive worldwide rights to human DEC-205 receptor, with the right to sublicense the technology. The license grant is exclusive except that Rockefeller may use and permit other nonprofit organizations to use the human DEC-205 receptor patent rights for educational and research purposes. We may be required to pay milestones of up to $3.9 million upon obtaining first approval for commercial sale in a first indication of a product targeting the licensed receptor and royalty payments in the low- to mid-single digits on any net product sales to Rockefeller with respect to development and commercialization of the human DEC-205 receptor.
Duke University Brain Tumor Cancer Center (Duke)
In September 2006, we and Duke entered into a license agreement that gave us access and reference to the clinical data generated by Duke and its collaborators in order for us to generate our own filing with the FDA relating to rindopepimut. We may be required to pay milestones of up to $1.0 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Duke with respect to development and commercialization of rindopepimut.
Ludwig Institute for Cancer Research (Ludwig)
In October 2006, we and Ludwig entered into an agreement for the nonexclusive rights to certain cancer tumor targets for use in combination with our APC Targeting Technology. The term of the agreement is for ten years. We may be required to pay milestones of up to $1.0 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Ludwig with respect to development and commercialization of the technology licensed from Ludwig.
Alteris Therapeutics, Inc. (Alteris)
In October 2005, we completed the acquisition of the assets of Alteris, including the EGFRvIII molecule. We may be required to pay Alteris up to $5.0 million upon obtaining the first approval for commercial sale of a product containing EGFRvIII, including rindopepimut.
Thomas Jefferson University (TJU)
In connection with our acquisition of the assets of Alteris, we obtained the rights to two exclusive license agreements with TJU dated February 2003 related to the EGFRvIII tumor antigen. Under these licenses, we may be required to pay milestones of up to $3.0 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to TJU with respect to development and commercialization of rindopepimut.
13
3M Company
In June 2008, we and 3M Company entered into a license agreement for the exclusive worldwide rights to access 3M Company's proprietary Immune Response Modifier, Resiquimod™, (and additional Toll-Like Receptor 7/8 agonists (TLR)) for clinical study with our proprietary APC Targeting Technology, for use as vaccine adjuvants, with the right to sublicense the technology. We may be required to pay milestones of up to $3.8 million upon obtaining first approval for commercial sale of each product using this vaccine adjuvant and royalty payments in the low-single digits on any net product sales to 3M Company with respect to development and commercialization of the technology licensed from 3M Company.
University of Southampton, UK (Southampton)
In November 2008, we entered into a license agreement with Southampton to develop human antibodies towards CD27, a potentially important target for immunotherapy of various cancers. We may be required to pay milestones of up to approximately $1.4 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Southampton with respect to development and commercialization of CDX-1127.
Amgen Inc. (Amgen)
In March 2009, we entered into a license agreement with Amgen to expand our Precision Targeted Immunotherapy Platform by acquiring exclusive rights to FMS-like tyrosine kinase 3 ligand (Flt3L or CDX-301) and CD40 ligand (CD40L). CDX-301 and CD40L are immune modulating molecules that increase the numbers and activity of immune cells that control immune responses. We may be required to pay milestones of up to $1.3 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Amgen with respect to development and commercialization of the technology licensed from Amgen, including CDX-301.
Amgen Fremont (formerly Abgenix)
In connection with our acquisition of CuraGen Corporation (CuraGen) in 2009, we assumed the license agreement between CuraGen and Amgen Fremont (successor in-interest to Abgenix) to develop fully-human monoclonal antibody therapeutics. In May 2009, an amendment to the license agreement (Amgen Amendment) was entered into related to CuraGen's exclusive rights to develop and commercialize CDX-011, CDX-014 and 10 other licensed antigens. Under the Amgen Amendment, CuraGen and Amgen Fremont agreed to modify the terms of their existing cross-license of antigens whereby our amended license would be fully paid-up and royalty-free (except for any potentially required payments to the original licensor of CDX-011).
Seattle Genetics, Inc. (Seattle Genetics)
In connection with our acquisition of CuraGen, we assumed the license agreement between CuraGen and Seattle Genetics whereby CuraGen acquired the rights to proprietary ADC technology for use with its proprietary antibodies for the potential treatment of cancer. We may be required to pay milestones of up to $7.5 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the mid-single digits on any net product sales to Seattle Genetics with respect to development and commercialization of the ADC technology, including CDX-011.
Competition
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Many of the products that we are attempting to develop and commercialize will be competing with existing therapies. In addition, a number of companies are
14
pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. We face competition from pharmaceutical and biotechnology companies both in the United States and abroad. Our competitors may utilize discovery technologies and techniques or partner with collaborators in order to develop products more rapidly or successfully than us or our collaborators are able to do. Many of our competitors, particularly large pharmaceutical companies, have substantially greater financial, technical and human resources than we do. In addition, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies and may establish exclusive collaborative or licensing relationships with our competitors.
We face intense competition in our development activities. We face competition from many companies in the United States and abroad, including a number of large pharmaceutical companies, firms specialized in the development and production of vaccines, adjuvants and vaccine and immunotherapeutic delivery systems and major universities and research institutions. The competitors for which we are aware have initiated a Phase 3 study or have obtained marketing approval for a potentially competitive drug include Alexion, Agenus, Baxter, BMS, Dendreon, Eli Lilly, GlaxoSmithKline, ImmunoGen, Merck, Pfizer, Roche, Sanofi-Aventis, Seattle Genetics, and Takeda. Many other companies are developing or commercializing products in areas that we have targeted for product development. Some of these products use therapeutic approaches that may compete directly with our drug candidates. Most of our competitors possess substantially greater financial, technical and human resources than we possess. In addition, many of our competitors have significantly greater experience than we have in conducting preclinical and nonclinical testing and human clinical trials of drug candidates, scaling up manufacturing operations and obtaining regulatory approvals of drugs and manufacturing facilities. Accordingly, our competitors may succeed in obtaining regulatory approval for drugs more rapidly than we do. If we obtain regulatory approval and launch commercial sales of our drug candidates, we also will compete with respect to manufacturing efficiency and sales and marketing capabilities, areas in which we currently have limited experience.
We also face competition from pharmaceutical and biotechnology companies, academic institutions, government agencies and private research organizations in recruiting and retaining highly qualified scientific personnel and consultants and in the development and acquisition of technologies. Moreover, technology controlled by third parties that may be advantageous to our business may be acquired or licensed by our competitors, thereby preventing us from obtaining technology on commercially reasonable terms, if at all. We will also compete for the services of third parties that may have already developed or acquired internal biotechnology capabilities or made commercial arrangements with other biopharmaceutical companies to target the diseases on which we have focused both in the U.S. and outside of the U.S.
Our competitive position will also depend upon our ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary products or processes and secure sufficient capital resources for the often lengthy period between technological conception and commercial sales. We will require substantial capital resources to complete development of some or all of our products, obtain the necessary regulatory approvals and successfully manufacture and market our products. In order to secure capital resources, we anticipate having to sell additional capital stock, which would dilute existing stockholders. We may also attempt to obtain funds through research grants and agreements with commercial collaborators. However, these types of fundings are uncertain because they are at the discretion of the organizations and companies that control the funds. As a result, we may not receive any funds from grants or collaborations. Alternatively, we may borrow funds from commercial lenders, likely at high interest rates, which would increase the risk of any investment in us.
15
Manufacturing
We have limited experience in large scale manufacturing and we have relied upon collaborators or contractors to manufacture some of our proposed products for both clinical and commercial purposes to date. We have established our own manufacturing facility in Fall River, Massachusetts, to produce antibodies, vaccines and other products that we may develop at scale for clinical trials. In 2010, we completed renovations at our Fall River, MA manufacturing facility which increased our capacity by installing a 1000L bioreactor and made improvement to the facility to be able to manufacture in compliance with European Medicines Agency (EMEA) regulations. Implementing EMEA requirements along with FDA Good Manufacturing Practices (GMP) will allow us to distribute potential products to clinical sites in both the US and EU. In order for us to establish a commercial manufacturing facility, we will require substantial additional funds and will be required to hire and retain significant additional personnel and comply with the extensive GMP regulations applicable to such facility. The commercial manufacturing facility would also need to be licensed for the production of antibodies, vaccines and other products by the FDA. We intend to establish manufacturing arrangements with contract manufacturers that comply with the FDA's requirements and other regulatory standards, although there can be no assurance that we will be able to do so.
While we believe that there is currently sufficient capacity worldwide for the production of our potential products by our collaborators or through contract manufacturers, establishing long-term relationships with contract manufacturers and securing multiple sources for the necessary quantities of clinical and commercial materials required can be a challenge. Qualifying the initial source of clinical and ultimately commercial material is a time consuming and expensive process due to the highly regulated nature of the pharmaceutical/biotech industry. These costs are hopefully mitigated in the economies of scale realized in commercial manufacture and product sale. The key difficulty in qualifying more than one source for each product is the duplicated time and expense in doing so without the potential to mitigate these costs if the secondary source is never utilized.
We use rindopepimut drug product that was manufactured by Pfizer in the ACT IV and ReACT clinical studies. We plan to establish a relationship with a contract manufacturer to support future clinical trials and for the commercial manufacturing of rindopepimut. To date, we have utilized contract manufacturers for the manufacture of clinical trial supplies of CDX-011. We manufacture clinical materials of CDX-1127, CDX-1401, CDX-1135 and CDX-301 in our Fall River facility for our current and planned Phase 1 and Phase 2 clinical trials. Manufacture of the rotavirus vaccine is the responsibility of Glaxo, which has received from us a worldwide exclusive license to commercialize this vaccine.
The manufacturing processes for our other vaccine and immunotherapeutic delivery systems and vaccines utilize known technologies. We believe that the products we currently have under development can be scaled up to permit manufacture in commercial quantities. However, there can be no assurance that we will not encounter difficulties in scaling up the manufacturing processes.
Use of third party manufacturers limits our control over and ability to monitor the manufacturing process. As a result, we may not be able to detect a variety of problems that may arise and may face additional costs in the process of interfacing with and monitoring the progress of our contract manufacturers. If third party manufacturers fail to meet our manufacturing needs in an acceptable manner, we would face delays and additional costs while we develop internal manufacturing capabilities or find alternative third party manufacturers. It may not be possible to have multiple third party manufacturers ready to supply us with needed material at all or without incurring significant costs.
16
Marketing
Under the terms of existing and future partnership agreements, we rely and expect to continue to rely on the efforts of our collaborators, including Glaxo, for the sale and marketing of our products. There can be no assurance that our collaborators will develop and market vaccine products incorporating our technologies, or, if marketed, that such efforts will be successful. The failure of our collaborators to successfully market products would harm our business.
We have retained, and in the future intend to retain, marketing rights to some of our drug candidates, including vaccine and immunotherapeutic delivery systems and vaccine candidates, in selected geographic areas and for specified indications. We intend to seek marketing and distribution agreements and/or co-promotion agreements for the distribution of our products in these geographic areas and for these indications. We believe that these arrangements could enable us to generate greater financial return than might be obtained from early stage licensing and collaboration agreements. We currently have no marketing and sales staff and limited experience relating to marketing and distribution of commercial products, including vaccines. If we determine in the future to engage in direct marketing of our products, we will be required to recruit an experienced marketing group, develop a supporting distribution capability and incur significant additional expenditures. There can be no assurance that we will be able to establish a successful marketing force. We may choose or find it necessary to enter into strategic partnerships on uncertain, but potentially unfavorable, terms to sell, market and distribute our products. Any delay in the marketing or distribution of our products, whether it results from problems with internal capabilities or with a collaborative relationship, could harm the value of an investment in us.
Patents, Licenses and Proprietary Rights
In general, our intellectual property strategy is to protect our technology by filing patent applications and obtaining patent rights covering our own technology, both in the United States and in foreign countries that we consider important to our business. In addition, we have acquired and will seek to acquire as needed or desired, exclusive rights of others through assignment or license to complement our portfolio of patent rights. We also rely on trade secrets, unpatented know-how and technological expertise and innovation to develop and maintain our competitive position.
Patents
The successful development and marketing of products by us will depend in part on our ability to create and maintain intellectual property, including patent rights. We are the owner or exclusive licensee to proprietary patent positions in the areas of vaccine technologies, antibody technologies and complement inhibitor technology. Although we continue to pursue patent protection for our products, no assurance can be given that any pending application will issue as a patent, that any issued patent will have a scope that will be of commercial benefit, or that we will be able to successfully enforce our patent position against infringers. We routinely review our patent portfolio and adjust our strategies for prosecution and maintenance of individual cases according to a number of factors including program priorities, stage of development, and patent term.
We own or license rights under more than 400 granted patents and national and regional patent applications around the world covering inventions relating to our business. The key patents and patent applications owned by us or licensed to us that we consider important to our business include the following (the indicated and estimated patent expiry dates do not include any possible Patent Term Extensions or Supplementary Protection Certificates, if these may be secured in due course):
- •
- Patents for the technology used in rindopepimut have expiration dates through 2014 in the United States and through 2015 in the United Kingdom, Germany and France. We also have rights under patent applications around the world relating to uses of rindopepimut which are
17
- •
- Our patent portfolio for CDX-011 includes an issued patent in Europe and pending patent applications in the
U.S. and Japan. If issued and maintained to full term in due course, these would have estimated patent expiry dates in 2025. In addition, patent rights relating to the toxin and conjugation technology
used in CDX-011 have been licensed from Seattle Genetics.
- •
- We have licensed pending patent applications in the U.S., Europe and Japan relating to the technology used in
CDX-1127. If issued and maintained to full term in due course, these would have estimated patent expiry dates in 2027. U.S. and international patent applications have also been filed
which, if issued in the main designated territories and maintained to full term in due course, would have estimated patent expiry dates in 2031.
- •
- We have pending patent application relating to the technology used in CDX-1401 which, if issued and maintained
to full term in due course, would have estimated patent expiry dates in 2028.
- •
- Patents for the technology used in CDX-1135 have expiration dates that range from 2013 to 2016. Further patent
applications are also pending.
- •
- Patents for the technology used in CDX-301 have current expiration dates that range from 2016 in the major
European territories to 2020 in the US.
- •
- Our patent portfolio for CDX-014 includes pending patent applications in the U.S., Europe and Japan. If issued
and maintained to full term in due course, these would have estimated patent expiry dates in 2024.
- •
- Patents for the technology used in the cholera and typhoid vaccines expire between 2013 and 2016. Our patent portfolio for
ETEC includes pending patent applications around the world which, if issued and maintained to full term in due course, would have estimated patent expiry dates in 2028.
- •
- A U.S. patent for our rotavirus strain that we licensed to Glaxo has an expiration date in December 2012.
currently pending. If issued and maintained to full term in a form which covers commercial use of rindopepimut, the latter filings could potentially provide additional patent protection for the relevant use in the relevant territories to 2026. We also have rights to an international patent application relating to methods of manufacture and formulation of rindopepimut, which, if issued in the main designated territories in a form which covers manufacture and/or formulation rindopepimut and maintained to full term in due course, would have estimated patent expiry dates in 2030.
There can be no assurance that patent applications owned by or licensed to us will result in granted patents or that, if granted, the resultant patents will afford protection against competitors with similar technology. It is also possible that third parties may obtain patents or other proprietary rights that may be necessary or useful to us. In cases where third parties are first to invent a particular product or technology, it is possible that those parties will obtain patents that will be sufficiently broad to prevent us from using important technology or from further developing or commercializing important vaccine and immunotherapeutic systems and vaccine candidates. If licenses from third parties are necessary but cannot be obtained, commercialization of the covered products might be delayed or prevented. Even if these licenses can be obtained, they would probably require us to pay ongoing royalties and other costs, which could be substantial.
Although a patent has a statutory presumption of validity in the United States, the issuance of a patent is not conclusive as to validity or as to the enforceable scope of the patent claims. The validity or enforceability of a patent after its issuance by the Patent and Trademark Office can be challenged in litigation. As a business that uses a substantial amount of intellectual property, we face a heightened
18
risk of intellectual property litigation. If the outcome of the litigation is adverse to the owner of the patent, third parties may then be able to use the invention covered by the patent without authorization or payment. There can be no assurance that our issued patents or any patents subsequently issued to or licensed by us will not be successfully challenged in the future. In addition, there can be no assurance that our patents will not be infringed or that the coverage of our patents will not be successfully avoided by competitors through design innovation.
We are aware that others, including universities and companies, have filed patent applications and have been granted patents in the United States and other countries which claim subject matter potentially useful or necessary to the commercialization of our products. The ultimate scope and validity of existing or future patents which have been or may be granted to third parties, and the availability and cost of acquiring rights in those patents necessary to the manufacture, use or sale of our products presently cannot be determined by us.
Third parties may have or may obtain valid and enforceable patents or proprietary rights that could block us from developing products using our technology, including:
- •
- certain patents and applications in the United States and Europe owned by Sanofi-Aventis, which relate to antibody-antigen
conjugates and methods of their use for eliciting an immune response against the antigen;
- •
- certain patents and applications in the United States and foreign countries covering particular antigens and antigenic
fragments targeted by our current vaccine drug candidates, including CDX-1401;
- •
- certain patents and pending applications related to particular receptors and other molecules on dendritic cells and
macrophages that may be useful for generating monoclonal antibodies and can be employed in our APC Targeting Technology;
- •
- two United States patents and related foreign patents and applications covering methods of diagnosing gliomas by detecting
the presence of the EGFRvIII (tumor specific splice variant) protein;
- •
- a United States patent relating to certain uses of GM-CSF;
- •
- a United States patent owned by Genentech, Inc., relating to the production of recombinant antibodies in host
cells;
- •
- a United States patent owned by GlaxoSmithKline plc related to methods of culturing cells under certain conditions;
and
- •
- certain patents held by third parties relating to antibody expression in particular types of host cells.
In addition to the patents referred to in the previous paragraphs, there may be other patent applications and issued patents belonging to competitors that may require us to alter our vaccine candidates and vaccine and immunotherapeutic delivery systems, pay licensing fees or cease some of our activities. If our drug candidates conflict with patents that have been or may be granted to competitors, universities or others, the patent owners could bring legal action against us claiming damages and seeking to enjoin manufacturing and marketing of the patented products. If any of these actions is successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to manufacture or market the affected products. There can be no assurance that we would prevail in any such action or that any license required under any such third party patent would be made available on acceptable terms or at all. We believe that there may be significant litigation in the biotechnology and vaccine industries regarding patent and other intellectual property rights. If we become involved in that litigation, we could consume substantial resources.
19
Licenses
We have entered into several significant license agreements relating to technology that is being developed by us and/or our collaborators. In general, these institutions have granted us an exclusive worldwide license (with right to sublicense) to make, use and sell products embodying the licensed technology, subject to the reservation by the licensor of a non-exclusive right to use the technologies for non-commercial research purposes. Generally, the term of each license is through the expiration of the last of the patents issued with respect to the technologies covered by the license. We have generally agreed to use reasonable efforts to develop and commercialize licensed products and to achieve specified milestones and pay license fees, milestone payments and royalties based on the net sales of the licensed products or to pay a percentage of sublicense income. If we breach our obligations, the licensor has the right to terminate the license, and, in some cases, convert the license to a non-exclusive license. Generally, we control and are responsible for the cost of defending the patent rights of the technologies that we license.
Proprietary Rights
We also rely on unpatented technology, trade secrets and confidential information, and no assurance can be given that others will not independently develop substantially equivalent information and techniques or otherwise gain access to our know-how and information, or that we can meaningfully protect our rights in such unpatented technology, trade secrets and information. We require each of our employees, consultants and advisors to execute a confidentiality agreement at the commencement of an employment or consulting relationship with us. The agreements generally provide that all inventions conceived by the individual in the course of employment or in providing services to us and all confidential information developed by, or made known to, the individual during the term of the relationship shall be the exclusive property of us and shall be kept confidential and not disclosed to third parties except in limited specified circumstances. There can be no assurance, however, that these agreements will provide meaningful protection for our information in the event of unauthorized use or disclosure of such confidential information.
Government Regulation
Our activities and products are significantly regulated by a number of governmental entities, including the FDA in the United States and by comparable authorities in other countries. These entities regulate, among other things, the manufacture, testing, safety, effectiveness, labeling, documentation, advertising and sale of our products. We must obtain regulatory approval for a product in all of these areas before we can commercialize the product. Product development within this regulatory framework takes a number of years and involves the expenditure of substantial resources. Many products that initially appear promising ultimately do not reach the market because they are found to be unsafe or ineffective when tested. Our inability to commercialize a product would impair our ability to earn future revenues.
FDA Approval Process
In the United States, vaccines and immunotherapeutics for human use are subject to FDA approval as "biologics" under the Public Health Service Act and "drugs" under the Federal Food, Drug and Cosmetic Act. The steps required before a new product can be commercialized include: preclinical studies in animals, clinical trials in humans to determine safety and efficacy and FDA approval of the product for commercial sale.
Data obtained at any stage of testing is susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. Moreover, during the regulatory process, new or changed drug approval policies may cause unanticipated delays or rejection of our product. We may not obtain
20
necessary regulatory approvals within a reasonable period of time, if at all, or avoid delays or other problems in testing our products. Moreover, even if we received regulatory approval for a product, the approval may require limitations on use, which could restrict the size of the potential market for the product.
The FDA provides that human clinical trials may begin thirty (30) days after receipt and review of an IND application, unless the FDA requests additional information or changes to the study protocol within that period. An IND must be sponsored and filed for each of our proposed products. Authorization to conduct a clinical trial in no way assures that the FDA will ultimately approve the product. Clinical trials are usually conducted in three sequential phases. In a Phase 1 trial, the product is given to a small number of healthy volunteers to test for safety (adverse effects). Phase 2 trials are conducted on a limited group of the target patient population; safety, optimal dosage and efficacy are studied. A Phase 3 trial is performed in a large patient population over a wide geographic area to provide evidence for the safety of the product and to prove and confirm efficacy. The FDA has ongoing oversight over all these trials and can order a temporary or permanent discontinuation if warranted. Such an action could materially harm us. Clinical tests are critical to the success of our products but are subject to unforeseen and uncontrollable delay, including delay in enrollment of patients. Any delay in clinical trials could delay our commercialization of a product.
A product's safety and effectiveness in one test is not necessarily indicative of its safety and effectiveness in another test. Moreover, we may not discover all potential problems with a product even after completing testing on it. Some of our products and technologies have undergone only preclinical testing. As a result, we do not know whether they are safe or effective for humans. Also, regulatory authorities may decide, contrary to our findings, that a product is unsafe or not as effective in actual use as its test results indicated. This could prevent the product's widespread use, require its withdrawal from the market or expose us to liability.
The results of the clinical trials and all supporting data are submitted to the FDA for approval. A Biologics License Application (BLA) is submitted for a biologic product; a New Drug Application (NDA) for a drug product. The interval between IND filing and BLA/NDA filing is usually at least several years due to the length of the clinical trials, and the BLA/NDA review process can take over a year. During this time the FDA may request further testing or additional trials or may turn down the application. Even with approval, the FDA frequently requires post-marketing safety studies (known as Phase 4 trials) to be performed.
The FDA requires that the manufacturing facility that produces a licensed product meet specified standards, undergo an inspection and obtain an establishment license prior to commercial marketing. Subsequent discovery of previously unknown problems with a product or its manufacturing process may result in restrictions on the product or the manufacturer, including withdrawal of the product from the market. Failure to comply with the applicable regulatory requirements can result in fines, suspensions of regulatory approvals, product recalls, operating restrictions and criminal prosecution.
Expedited Review and Approval
The FDA has various programs, including fast track, priority review, and accelerated approval, that are intended to expedite or simplify the process for reviewing drugs, and/or provide for approval on the basis of surrogate endpoints. Generally, drugs that may be eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments, however, these programs do not affect the standards for approval. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform post-marketing clinical trials.
21
Orphan Drug
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for seven years.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. Approval by the FDA does not ensure approval by the regulatory bodies of other countries.
Under European Union regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology and optional for those which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under the decentralized procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessments report each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states. As in the United States, we may apply for designation of our products as orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including a 10-year market exclusivity period for the approved indication, but not for the same drug, unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan-designated product.
Our collaborators are also subject to all of the above-described regulations in connection with the commercialization of products utilizing our technology.
22
Other Regulatory Processes
We are subject to a variety of financial disclosure and securities trading regulations as a public company in the U.S., including laws relating to the oversight activities of the SEC and the regulations of the NASDAQ Global Market, on which our shares are traded. We are also subject to regulation under other federal laws and regulation under state and local laws, including laws relating to occupational safety, laboratory practices, environmental regulations, and hazardous substance control.
Employees
As of December 31, 2011, we employed 103 employees, 14 of whom have Ph.D. and/or M.D. degrees. Of these employees, 86 were engaged in or directly support research and development activities. We believe that our employee relations are good. We believe that our future success will depend in large part on our ability to attract and retain experienced and skilled employees.
You should consider carefully these risk factors together with all of the information included or incorporated by reference in this Annual Report in addition to our financial statements and the notes to our financial statements. This section includes forward-looking statements.
The following is a discussion of the risk factors that we believe are material to us at this time. These risks and uncertainties are not the only ones facing us and there may be additional matters that we are unaware of or that we currently consider immaterial. All of these could adversely affect our business, results of operations, financial condition and cash flows.
Risks Related to our Business
We expect to incur future losses and we may never become profitable.
We have incurred operating losses of $43.4 million, $6.5 million and $36.9 million during 2011, 2010 and 2009, respectively, and expect to incur an operating loss in 2012. We believe that operating losses will continue beyond 2012 because we are planning to incur significant costs associated with the clinical development and manufacturing commercial supply of rindopepimut to prepare for the potential launch of rindopepimut. In addition, we are planning to incur significant costs in the clinical development of CDX-011, CDX-1127, CDX-1401, CDX-301 and CDX-1135. Our net losses have had and will continue to have an adverse effect on, among other things, our stockholders' equity, total assets and working capital. We expect that losses will fluctuate from quarter to quarter and year to year, and that such fluctuations may be substantial. We cannot predict when we will become profitable, if at all.
Our long term success depends heavily on our ability to fund and complete research and development activities for, and to commercialize, our lead drug candidate, rindopepimut, which we are developing internally.
While in the past we have typically focused on developing and demonstrating proof-of-concept for our product candidates by bringing such candidates through Phase 1 and one or more Phase 2 clincial trials, and then leveraging their value through partnerships, we have decided to fund and complete the research and development activities for rindopepimut ourselves. We plan to commercialize rindopepimut ourselves in North America and to find a partner to commercialize rindopepimut outside of North America. Therefore, we must allocate a significant portion of our time, personnel and financial resources to the development of rindopepimut. We initiated ACT IV, our pivotal Phase 3 clinical trial of rindopepimut, in December 2011. While we are targeting two years for patient accrual, it could take up to three years to enroll all the patients, and another 18 to 24 months of follow-up, at a
23
cost of over $50 million to complete this Phase 3 study. Our management team lacks significant experience in completing Phase 3 clinical trials and bringing a drug through commercialization. If we face delays, difficulties or unanticipated costs in completing the development of rindopepimut, we will need substantial additional financing. Further, even if we complete the development of rindopepimut and gain marketing approvals from the FDA and comparable foreign regulatory authorities in a timely manner, we cannot be sure that rindopepimut will be commercially successful in the pharmaceutical market. If the results of clinical trials of rindopepimut, the anticipated or actual timing of marketing approvals for rindopepimut, or the market acceptance of rindopepimut, if approved, do not meet the expectations of investors or public market analysts, the market price of our common stock would likely decline.
We may be unable to manage one Phase 3 clinical trial or multiple late stage clinical trials for a variety of product candidates simultaneously.
As our current clinical trials progress, we may need to manage multiple late stage clinical trials simultaneously in order to continue developing all of our current products. Our management team does not have significant experience in completing late stage clinical trials and the management of late stage clinical trials is more complex and time consuming than early stage trials. Typically, early stage trials involve several hundred patients in no more than 10-30 clinical sites. Late stage (Phase 3) trials may involve up to several thousand patients in up to several hundred clinical sites and may require facilities in several countries. Therefore, the project management required to supervise and control such an extensive program is substantially larger than early stage programs. As the need for these resources is not known until some months before the trials begin, it is necessary to recruit large numbers of experienced and talented individuals very quickly. If the labor market does not allow this team to be recruited quickly, the sponsor is faced with a decision to delay the program or to initiate it with inadequate management resources. This may result in recruitment of inappropriate patients, inadequate monitoring of clinical investigators and inappropriate handling of data or data analysis. Consequently it is possible that conclusions of efficacy or safety may not be acceptable to permit filing of a BLA or NDA for any one of the above reasons or a combination of several.
We will need additional capital to fund our operations, including the development, manufacture and potential commercialization of our drug candidates. If we do not have or cannot raise additional capital when needed, we will be unable to develop and ultimately commercialize our drug candidates successfully.
We expect to incur significant costs as we develop our drug candidates. In particular, the continuing development and commercialization of rindopepimut requires additional capital beyond our current resources. As of December 31, 2011, we had cash, cash equivalents and marketable securities of $53.3 million, which, at that time, we believed would support expected operations for more than 12 months.
During the next twelve months, we may take further steps to raise additional capital to fund our long-term liquidity needs. Our capital raising activities may include, but may not be limited to, one or more of the following:
- •
- licensing of drug candidates with existing or new collaborative partners;
- •
- possible business combinations;
- •
- issuance of debt; or
- •
- issuance of common stock or other securities via private placements or public offerings.
While we may continue to seek capital through a number of means, there can be no assurance that additional financing will be available on acceptable terms, if at all, and our negotiating position in
24
capital-raising efforts may worsen as existing resources are used. There is also no assurance that we will be able to enter into further collaborative relationships. Additional equity financing may be dilutive to our stockholders; debt financing, if available, may involve significant cash payment obligations and covenants that restrict our ability to operate as a business; and licensing or strategic collaborations may result in royalties or other terms which reduce our economic potential from products under development. If we are unable to raise the funds necessary to meet our long-term liquidity needs, we may have to delay or discontinue the development of one or more programs, discontinue or delay on-going or anticipated clinical trials, license out programs earlier than expected, raise funds at significant discount or on other unfavorable terms, if at all, or sell all or part of our business.
In January 2011, we entered into a financing facility with Cantor Fitzgerald & Co., providing for the sale of up to 5 million shares of our common stock from time to time into the open market at prevailing prices. During the year ended December 31, 2011, we sold 575,000 shares of common stock under the Cantor Agreement and raised $2.2 million in net proceeds, after deducting commission and offering expenses. As of December 31, 2011, we had 4,425,000 shares available to be sold under the Cantor Agreement. In January 2012, we sold 2,450,000 shares of common stock under the Cantor Agreement and raised $8.5 million in net proceeds. Under the terms of the Cantor Agreement, we will have the ability to sell up to 1,975,000 shares of our common stock upon the expiration or earlier waiver of our 90-day lock-up with the underwriters of our recent offering in February 2012. We may or may not sell additional shares under this facility, depending on the volume and price of our common stock, as well as our capital needs and potential alternative sources of capital. If we actively sell shares under this facility, a significant number of shares of common stock could be issued in a short period of time, although we would attempt to structure the volume and price thresholds in a way that minimizes market impact. Notwithstanding these control efforts, these sales, or the perceived risk of dilution from potential sales of stock through this facility, may depress our stock price, cause holders of our common stock to sell their shares, or encourage short selling by market participants, which could contribute to a decline in our stock price. A decline in our stock price might impede our ability to raise capital through the issuance of additional shares of common stock or other equity securities, and may cause our stockholders to lose part or all of the value of their investment in our stock.
In February 2012, we issued 10,500,000 shares of our common stock in an underwritten public offering. The net proceeds to us were $37.7 million, after deducting underwriting fees and estimated offering expenses. We have granted the underwriters a 30-day option to purchase up to an aggregate of 1,575,000 additional shares of common stock to cover overallotments, if any.
We rely on third parties to plan, conduct and monitor our clinical tests, and their failure to perform as required would interfere with our product development.
We rely on third parties to conduct a significant portion of our clinical development activities. These activities include clinical patient recruitment and observation, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management. We conduct project management and medical and safety monitoring in-house for some of our programs and rely on third parties for the remainder of our clinical development activities. If any of these third parties fails to perform as we expect or if their work fails to meet regulatory standards, our testing could be delayed, cancelled or rendered ineffective.
The significant third parties who we currently rely on for clinical development activities include Novella Clinical Inc. (Novella) for ACT IV. If Novella is unable to perform in a quality and timely manner, and at a feasible cost, ACT IV will face delays.
25
We may enter into collaboration agreements for the licensing, development and ultimate commercialization of some of our drug candidates including, where appropriate, for our lead drug candidates. In such cases, we will depend greatly on our third party collaborators to license, develop and commercialize such drug candidates, and they may not meet our expectations.
We are exploring potential co-development and commercialization partnerships for certain products, including rindopepimut for commercialization outside of North America, CDX-011 and CDX-1127. The process of identifying collaborators and negotiating collaboration agreements for the licensing, development and ultimate commercialization of some of our drug candidates may cause delays and increased costs. We may not be able to enter into collaboration agreements on terms favorable to us. Furthermore some of those agreements may give substantial responsibility over our drug candidates to the collaborator. Some collaborators may be unable or unwilling to devote sufficient resources to develop our drug candidates as their agreements require. They often face business risks similar to ours, and this could interfere with their efforts. Also, collaborators may choose to devote their resources to products that compete with ours. If a collaborator does not successfully develop any one of our products, we will need to find another collaborator to do so. The success of our search for a new collaborator will depend on our legal right to do so at the time and whether the product remains commercially viable.
If we enter into collaboration agreements for one or more of our lead drug candidates, the success of such drug candidates will depend in great part upon our and our collaborators' success in promoting them as superior to other treatment alternatives. We believe that our drug candidates can be proven to offer disease prevention and treatment with notable advantages over drugs in terms of patient compliance and effectiveness. However, there can be no assurance that we will be able to prove these advantages or that the advantages will be sufficient to support the successful commercialization of our drug candidates.
We may face delays, difficulties or unanticipated costs in establishing sales, distribution and manufacturing capabilities for our commercially ready products.
Our current plan is to retain, rather than license to a third party, all rights to rindopepimut in North America (and to explore partnership opportunities to commercialize rindopepimut outside of North America) and our APC Targeting Technology programs. As a result, we will have full responsibility for commercialization of these products if and when they are approved for sale. We currently lack the marketing, sales and distribution capabilities that we will need to carry out this strategy. To market any of our products directly, we must develop a substantial marketing and sales force with technical expertise and a supporting distribution capability. We have little expertise in this area, and we may not succeed. We may find it necessary to enter into strategic partnerships on uncertain but potentially unfavorable terms to sell, market and distribute our products when they are approved for sale.
Some of our products are difficult to manufacture, especially in large quantities, and we have not yet developed commercial scale manufacturing processes for any of our products. We do not currently plan to develop internal manufacturing capabilities to produce any of our products at commercial scale if they are approved for sale. To the extent that we choose to market and distribute these products ourselves, this strategy will make us dependent on other companies to produce our products in adequate quantities, in compliance with regulatory requirements, and at a competitive cost. We may not find third parties capable of meeting those manufacturing needs.
Our drug candidates are subject to extensive regulatory scrutiny.
All of our drug candidates are at various stages of development and commercialization and our activities and drug candidates are significantly regulated by a number of governmental entities,
26
including the FDA in the United States and by comparable authorities in other countries. These entities regulate, among other things, the manufacture, testing, safety, effectiveness, labeling, documentation, advertising and sale of drugs and drug candidates. We or our partners must obtain regulatory approval for a drug candidate in all of these areas before we can commercialize the drug candidate. Product development within this regulatory framework takes a number of years and involves the expenditure of substantial resources. This process typically requires extensive preclinical and clinical testing, which may take longer or cost more than we anticipate, and may prove unsuccessful due to numerous factors. Many drug candidates that initially appear promising ultimately do not reach the market because they are found to be unsafe or ineffective when tested. Companies in the pharmaceutical, biotechnology and vaccines industries have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier trials. Our inability to commercialize a drug candidate would impair our ability to earn future revenues.
If our products do not pass required tests for safety and effectiveness, we will not be able to derive commercial revenue from them.
In order to succeed, we will need to derive commercial revenue from the products we have under development. The FDA has not approved our rindopepimut, CDX-011 or CDX-1127 drug candidates or any of our other products for sale to date. Our drug candidates are in various stages of preclinical and clinical testing. Preclinical tests are performed at an early stage of a product's development and provide information about a product's safety and effectiveness on laboratory animals. Preclinical tests can last years. If a product passes its preclinical tests satisfactorily, and we determine that further development is warranted, we would file an IND application for the product with the FDA, and if the FDA gives its approval we would begin Phase 1 clinical tests. Phase 1 testing generally lasts between 6 and 24 months. If Phase 1 test results are satisfactory and the FDA gives its approval, we can begin Phase 2 clinical tests. Phase 2 testing generally lasts between 6 and 36 months. If Phase 2 test results are satisfactory and the FDA gives its approval, we can begin Phase 3 pivotal studies. Phase 3 studies generally last between 12 and 48 months. Once clinical testing is completed and a new drug application is filed with the FDA, it may take more than a year to receive FDA approval.
In all cases we must show that a pharmaceutical product is both safe and effective before the FDA, or drug approval agencies of other countries where we intend to sell the product, will approve it for sale. Our research and testing programs must comply with drug approval requirements both in the United States and in other countries, since we are developing our lead products with the intention to, or could later decide to, commercialize them both in the U.S. and abroad. A product may fail for safety or effectiveness at any stage of the testing process. A major risk we face is the possibility that none of our products under development will come through the testing process to final approval for sale, with the result that we cannot derive any commercial revenue from them after investing significant amounts of capital in multiple stages of preclinical and clinical testing.
Product testing is critical to the success of our products but subject to delay or cancellation if we have difficulty enrolling patients.
As our portfolio of potential products moves from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, we will need to enroll an increasing number of patients with the appropriate characteristics. At times we have experienced difficulty enrolling patients and we may experience more difficulty as the scale of our clinical testing program increases. The factors that affect our ability to enroll patients are largely uncontrollable and include principally the following:
- •
- the nature of the clinical test;
- •
- the size of the patient population;
27
- •
- patients' willingness to receive a placebo or less effective treatment on the control arm of a clinical study;
- •
- the distance between patients and clinical test sites; and
- •
- the eligibility criteria for the trial.
If we cannot enroll patients as needed, our costs may increase or it could force us to delay or terminate testing for a product.
We may have delays in completing our clinical trials and we may not complete them at all.
We have not completed the clinical trials necessary to obtain FDA approval to market rindopepimut, CDX-011 or any of our other products in development. We initiated a Phase 3 study of rindopepimut in December 2011 but we have not initiated Phase 3 studies for CDX-011 or any of our other products in development. Our management lacks significant experience in completing Phase 3 trials and bringing a drug through commercialization. Our rindopepimut Phase 3 trial, CDX-011 Phase 2b studies and planned clinical trials for other products in development may be delayed or terminated as a result of many factors, including the following:
- •
- difficulty in enrolling patients in our clinical trials;
- •
- patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons
- •
- failure by regulators to authorize us to commence a clinical trial;
- •
- suspension or termination by regulators of clinical research for many reasons, including concerns about patient safety or
failure of our contract manufacturers to comply with cGMP requirements;
- •
- delays or failure of the FDA to remove the partial clinical hold on our CDX-011 studies;
- •
- treatment candidates demonstrating a lack of efficacy during clinical trials;
- •
- inability to continue to fund clinical trials or to find a partner to fund the clinical trials;
- •
- competition with ongoing clinical trials and scheduling conflicts with participating clinicians; and
- •
- delays in completing data collection and analysis for clinical trials.
Any delay or failure to complete clinical trials and obtain FDA approval for our drug candidates could have a material adverse effect on our cost to develop and commercialize, and our ability to generate revenue from, a particular drug candidate.
Any delay in obtaining regulatory approval would have an adverse impact on our ability to earn future revenues.
It is possible that none of the drug candidates that we develop will obtain the regulatory approvals necessary for us to begin commercializing them. The time required to obtain FDA and other approvals is unpredictable but often can take years following the commencement of clinical trials, depending upon the nature of the drug candidate. Any analysis we perform of data from clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues from the particular drug candidate including, but not limited to, loss of patent term during the approval period. Furthermore, if we, or our partners, do not reach the market with our products before our competitors offer products for the same or similar uses, or if
28
we, or our partners, are not effective in marketing our products, our revenues from product sales, if any, will be reduced.
We face intense competition in our development activities. We face competition from many companies in the United States and abroad, including a number of large pharmaceutical companies, firms specialized in the development and production of vaccines, adjuvants and vaccine and immunotherapeutic delivery systems and major universities and research institutions. The competitors for which we are aware have initiated a Phase 3 study or have obtained marketing approval for a potentially competitive drug include Alexion, Agenus, Baxter, BMS, Dendreon, Eli Lilly, GlaxoSmithKline, ImmunoGen, Merck, Pfizer, Roche, Sanofi-Aventis, Seattle Genetics, and Takeda. Most of our competitors have substantially greater resources, more extensive experience in conducting preclinical studies and clinical testing and obtaining regulatory approvals for their products, greater operating experience, greater research and development and marketing capabilities and greater production capabilities than those of ours. These companies might succeed in obtaining regulatory approval for competitive products more rapidly than we can for our products, especially if we experience any delay in obtaining required regulatory approvals.
Failure to comply with applicable regulatory requirements would adversely impact our operations.
Even after receiving regulatory approval, our products would be subject to extensive regulatory requirements, and our failure to comply with applicable regulatory requirements will adversely impact our operations. In the United States, the FDA requires that the manufacturing facility that produces a product meet specified standards, undergo an inspection and obtain an establishment license prior to commercial marketing. Subsequent discovery of previously unknown problems with a product or its manufacturing process may result in restrictions on the product or the manufacturer, including withdrawal of the product from the market. Failure to comply with the applicable regulatory requirements can result in fines, suspensions of regulatory approvals, product recalls, operating restrictions and criminal prosecution.
We depend greatly on the intellectual capabilities and experience of our key executives and scientists and the loss of any of them could affect our ability to develop our products.
The loss of Anthony S. Marucci, our President and Chief Executive Officer, or other key members of our staff, including Avery W. Catlin, our Chief Financial Officer, Dr. Thomas Davis, our Chief Medical Officer, Dr. Tibor Keler, our Chief Scientific Officer or Ronald Pepin, our Chief Business Officer, could harm us. We entered into employment agreements with Messrs. Marucci, Catlin, Davis, Keler and Pepin although an employment agreement as a practical matter does not guarantee retention of an employee. We also depend on our scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to us. In addition, we believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific, managerial and marketing personnel, particularly as we expand our activities in clinical trials, the regulatory approval process and sales and manufacturing. We routinely enter into consulting agreements with our scientific and clinical collaborators and advisors, key opinion leaders and heads of academic departments in the ordinary course of our business. We also enter into contractual agreements with physicians and institutions who recruit patients into our clinical trials on our behalf in the ordinary course of our business. Notwithstanding these arrangements, we face significant competition for this type of personnel from other companies, research and academic institutions, government entities and other organizations. We cannot predict our success in hiring or retaining the personnel we require for continued growth.
29
We rely on contract manufacturers over whom we have limited control. Should the cost, delivery and quality of clinical and commercial grade materials supplied by contract manufacturers vary to our disadvantage, our business operations could suffer significant harm.
We have limited experience in large scale manufacturing at our Fall River facility. We rely on sourcing from third-party manufacturers for suitable quantities of some of our clinical and commercial grade materials and certain filling and packaging essential to preclinical and clinical studies currently underway and to planned clinical trials in addition to those currently being conducted by third parties or us. The inability to have suitable quality and quantities of these essential materials produced in a timely manner would result in significant delays in the clinical development and commercialization of products, which could adversely affect our business, financial condition and results of operations.
For example, one lot of our CDX-011 product candidate being used in our on-going Phase 2b study was aseptically filled in 2009 by Formatech, a third party contract manufacturer. The CDX-011 lot from Formatech has passed all of the sterility testing performed during drug release and in subsequent stability studies. At the end of January 2012, we were notified by the FDA that because significant Good Manufacturing Practice (GMP) violations were uncovered during inspection of Formatech, our Phase 2b study for CDX-011 was being placed on partial clinical hold. The FDA uncovered these findings during their inspections of the Formatech facility between August to October 2010 and July to August 2011. These inspections began approximately one year after the CDX-011 drug product was filled at Formatech. Specifically, the FDA requested that no new patients be treated with CDX-011. However, patients already undergoing treatment with CDX-011 could continue treatment using vials of CDX-011 from the lot filled by Formatech, after such patients were informed of the potential risk and reconsented to continued participation in the study. Since the Phase 2b trial completed accrual of patients in December 2011 and there are currently no other ongoing open studies with CDX-011, the only patients that are affected by the partial hold are the eight patients remaining in the control arm of the study, who currently are not receiving CDX-011, and may be eligible to cross-over at the time of progression and receive CDX-011 under the study protocol.
We have initiated discussions with the FDA regarding our proposal to utilize vials of CDX-011 that were filled by a different contract manufacturer. Although the FDA has stated that no new patients may receive CDX-011 and that no patients may cross-over to receive CDX-011, we have asked the FDA to reconsider allowing patients currently on the control arm to cross-over to CDX-011 after stability testing and confirmation that the product filled by the other contract manufacturer is acceptable for continued use. If the FDA agrees to our proposal concerning use of the alternative CDX-011 for the eligible cross-over patients, we believe that we should have sufficient clinical supply of CDX-011 to treat these cross-over patients. If we are not able to treat the eight remaining cross-over patients with CDX-011, patients may withdraw from the control arm study upon learning that they will not be allowed to cross-over to CDX-011 following disease progression. However, the primary analyses for the study are entirely based upon the primary randomization and do not include the cross-over results. Based on our discussions with our clinical investigators, we do not believe that a high proportion of patients will withdraw from the control arm prior to progression. We do not believe that this partial hold will significantly impact analysis of the Phase 2b data for purposes of determining next steps in our future development of CDX-011.
30
In addition, the FDA has agreed in concept that we could reprocess the remaining available vials of CDX-011 manufactured at Formatech at another cGMP contract manufacturer. The FDA's final decision regarding the acceptability of this reprocessing will be made upon review of data concerning the stability and sterility of the reprocessed vials of CDX-011. If we are unsuccessful at reprocessing the available drug product or if FDA does not approve the use of these reprocessed vials, we will need to manufacture new drug product for subsequent clinical studies for CDX-011, which may cause a delay in the initiation of a subsequent trial with CDX-011. We also rely on collaborators and contract manufacturers to manufacture proposed products in both clinical and commercial quantities in the future. Our leading vaccine candidates require specialized manufacturing capabilities and processes.
We may face difficulty in securing commitments from U.S. and foreign contract manufacturers as these manufacturers could be unwilling or unable to accommodate our needs. Relying on foreign manufacturers involves peculiar and increased risks, including the risk relating to the difficulty foreign manufacturers may face in complying with GMP requirements as a result of language barriers, lack of familiarity with GMP or the FDA regulatory process or other causes, economic or political instability in or affecting the home countries of our foreign manufacturers, shipping delays, potential changes in foreign regulatory laws governing the sales of our product supplies, fluctuations in foreign currency exchange rates and the imposition or application of trade restrictions.
There can be no assurances that we will be able to enter into long-term arrangements with third party manufacturers on acceptable terms, or at all. Further, contract manufacturers must also be able to meet our timetable and requirements, and must operate in compliance with GMP; failure to do so could result in, among other things, the disruption of product supplies. As noted above, non-U.S. contract manufacturers may face special challenges in complying with GMP requirements, and although we are not currently dependent on non-U.S. collaborators or contract manufacturers, we may choose or be required to rely on non-U.S. sources in the future as we seek to develop stable supplies of increasing quantities of materials for ongoing clinical trials of larger scale. Our dependence upon third parties for the manufacture of our products may adversely affect our profit margins and our ability to develop and deliver products on a timely and competitive basis.
The significant third parties who we currently rely on for sourcing of suitable quantities of some of our clinical and commercial grade materials include Biosyn, Bayer and Sanofi for our rindopepimut drug candidate. If we or our third-party manufacturers are unable to produce drug material in suitable quantities of appropriate quality, in a timely manner, and at a feasible cost, our clinical tests will face delays.
Certain factors could negatively affect the demand for and sales of Rotarix®, which would have a material adverse affect on our revenues.
We have licensed a rotavirus strain to Glaxo for the purposes of Glaxo developing and commercializing their Rotarix® vaccine worldwide. The term of the Glaxo agreement is through the expiration of the last of the relevant patents covered by the agreement, although Glaxo may terminate the agreement upon 90 days prior written notice. The last relevant patent is scheduled to expire in December 2012. Glaxo gained approval for Rotarix® in Mexico in July 2004, in the European Union in February 2006 and in the United States in April 2008. In May 2005, we entered into an agreement whereby an affiliate of PRF purchased a 70% interest in the net royalties we receive on worldwide sales of Rotarix®. In addition, we retain upside participation in the worldwide net royalties from Rotarix® once, and if, PRF receives an agreed upon return on capital invested (2.45 times PRF's aggregate cash payments to us of $60 million). The PRF agreement terminates in December 2012,
31
unless otherwise extended. The following are potential factors, among others, that may negatively affect the demand for Rotarix®:
- •
- competitors in the pharmaceuticals, biotechnology and vaccines market have greater financial and management resources, and
significantly more experience in bringing products to market, and may develop, manufacture and market products that are more effective or less expensive than Rotarix®;
- •
- Rotarix® could be replaced by a novel product and may become obsolete;
- •
- Glaxo may be unable to prevent third parties from infringing upon their proprietary rights related to
Rotarix®;
- •
- users may not accept such a recently approved product without years of proven history; and
- •
- we are dependent on Glaxo for the manufacturing, testing, acquisition of regulatory approvals, marketing, distribution and commercialization of Rotarix®.
Any of these factors could have a material adverse effect on the sales of Rotarix® and our revenues. However, any decline in revenue would not impact our net income because any royalty revenue we receive from sales of Rotarix® is offset by a corresponding royalty expense that we pay to PRF.
Other factors could affect the demand for and sales of Rotarix and any other products that we may commercialize in the future.
In general, other factors that could affect the demand for and sales and profitability of our products include, but are not limited to:
- •
- the timing of regulatory approval, if any, of competitive products;
- •
- our, Glaxo's, or any other of our partners' pricing decisions, as applicable, including a decision to increase or decrease
the price of a product, and the pricing decisions of our competitors;
- •
- government and third-party payer reimbursement and coverage decisions that affect the utilization of our products and
competing products;
- •
- negative safety or efficacy data from new clinical studies conducted either in the U.S. or internationally by any party
could cause the sales of our products to decrease or a product to be recalled;
- •
- the degree of patent protection afforded our products by patents granted to or licensed by us and by the outcome of
litigation involving our or any of our licensor's patents;
- •
- the outcome of litigation involving patents of other companies concerning our products or processes related to production
and formulation of those products or uses of those products;
- •
- the increasing use and development of alternate therapies;
- •
- the rate of market penetration by competing products; and
- •
- the termination of, or change in, existing arrangements with our partners.
Any of these factors could have a material adverse effect on Glaxo's sales of Rotarix and on the sales of any other products that we may commercialize in the future.
32
We face the risk of product liability claims, which could exceed our insurance coverage, and produce recalls, each of which could deplete our cash resources.
As a participant in the pharmaceutical, biotechnology and vaccines industries, we are exposed to the risk of product liability claims alleging that use of our drug candidates caused an injury or harm. These claims can arise at any point in the development, testing, manufacture, marketing or sale of our drug candidates and may be made directly by patients involved in clinical trials of our products, by consumers or healthcare providers or by individuals, organizations or companies selling our products. Product liability claims can be expensive to defend, even if the drug or drug candidate did not actually cause the alleged injury or harm.
Insurance covering product liability claims becomes increasingly expensive as a drug candidate moves through the development pipeline to commercialization. Under our license agreements, we are required to maintain clinical trial liability insurance coverage up to $14 million. However, there can be no assurance that such insurance coverage is or will continue to be adequate or available to us at a cost acceptable to us or at all. We may choose or find it necessary under our collaborative agreements to increase our insurance coverage in the future. We may not be able to secure greater or broader product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any liability for damages resulting from a product liability claim could exceed the amount of our coverage, require us to pay a substantial monetary award from our own cash resources and have a material adverse effect on our business, financial condition and results of operations. Moreover, a product recall, if required, could generate substantial negative publicity about our products and business and inhibit or prevent commercialization of other products and drug candidates.
In addition, some of our licensing and other agreements with third parties require or might require us to maintain product liability insurance. If we cannot maintain acceptable amounts of coverage on commercially reasonable terms in accordance with the terms set forth in these agreements, the corresponding agreements would be subject to termination, which could have a material adverse impact on our operations.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them.
Because we rely on third parties to develop our products, we must share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties. We also conduct joint research and development programs which may require us to share trade secrets under the terms of research and development partnership or similar agreements. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. A competitor's discovery of our trade secrets would impair our competitive position.
33
We may not be able to successfully integrate newly-acquired technology with our existing technology or to modify our technologies to create new vaccines.
As part of our acquisition of technology assets from entities such as 3M Company and Amgen, we have acquired access to Resiquimod™ (a TLR 7/8 agonist) and Flt3L, which may improve the immunogenicity of our vaccines. If we are able to integrate these licensed assets with our vaccine technologies, we believe these assets will give our vaccines a competitive advantage. However, if we are unable to successfully integrate licensed assets, or other technologies which we have acquired or may acquire in the future, with our existing technologies and potential products currently under development, we may be unable to realize any benefit from our acquisition of these assets, or other technologies which we have acquired or may acquire in the future and may face the loss of our investment of financial resources and time in the integration process.
We believe that our vaccine technology portfolio may offer opportunities to develop vaccines that treat a variety of oncology, inflammatory and infectious diseases by stimulating a patient's immune system against those disease organisms. If our vaccine technology portfolio cannot be used to create effective vaccines against a variety of disease organisms, we may lose all or portions of our investment in development efforts for new vaccine candidates.
We license technology from other companies to develop products, and those companies could influence research and development or restrict our use of it.
Companies that license technologies to us that we use in our research and development programs may require us to achieve milestones or devote minimum amounts of resources to develop products using those technologies. They may also require us to make significant royalty and milestone payments, including a percentage of any sublicensing income, as well as payments to reimburse them for patent costs. The number and variety of our research and development programs require us to establish priorities and to allocate available resources among competing programs. From time to time we may choose to slow down or cease our efforts on particular products. If in doing so we fail to fully perform our obligations under a license, the licensor can terminate the licenses or permit our competitors to use the technology. Moreover, we may lose our right to market and sell any products based on the licensed technology.
We have many competitors in our field and they may develop technologies that make ours obsolete.
Biotechnology, pharmaceuticals and therapeutics are rapidly evolving fields in which scientific and technological developments are expected to continue at a rapid pace. We have many competitors in the U.S. and abroad. The competitors for which we are aware have initiated a Phase 3 study or have obtained marketing approval for a potentially competitive drug include Alexion, Agenus, Baxter, BMS, Dendreon, Eli Lilly, GlaxoSmithKline, ImmunoGen, Merck, Pfizer, Roche, Sanofi-Aventis, Seattle Genetics, and Takeda. Our success depends upon our ability to develop and maintain a competitive position in the product categories and technologies on which we focus. Many of our competitors have greater capabilities, experience and financial resources than we do. Competition is intense and is expected to increase as new products enter the market and new technologies become available. Our competitors may:
- •
- develop technologies and products that are more effective than ours, making ours obsolete or otherwise noncompetitive;
- •
- obtain regulatory approval for products more rapidly or effectively than us; and
- •
- obtain patent protection or other intellectual property rights that would block our ability to develop competitive products.
34
We rely on patents, patent applications and other intellectual property protections to protect our technology and trade secrets; which are expensive and may not provide sufficient protection.
Our success depends in part on our ability to obtain and maintain patent protection for technologies that we use. Biotechnology patents involve complex legal, scientific and factual questions and are highly uncertain. To date, there is no consistent policy regarding the breadth of claims allowed in biotechnology patents, particularly in regard to patents for technologies for human uses like those we use in our business. We cannot predict whether the patents we seek will issue. If they do issue, a competitor may challenge them and limit their scope. Moreover, our patents may not afford effective protection against competitors with similar technology. A successful challenge to any one of our patents could result in a third party's ability to use the technology covered by the patent. We also face the risk that others will infringe, avoid or circumvent our patents. Technology that we license from others is subject to similar risks and this could harm our ability to use that technology. If we, or a company that licenses technology to us, were not the first creator of an invention that we use, our use of the underlying product or technology will face restrictions, including elimination.
If we must defend against suits brought against us or prosecute suits against others involving intellectual property rights, we will incur substantial costs. In addition to any potential liability for significant monetary damages, a decision against us may require us to obtain licenses to patents or other intellectual property rights of others on potentially unfavorable terms. If those licenses from third parties are necessary but we cannot acquire them, we would attempt to design around the relevant technology, which would cause higher development costs and delays, and may ultimately prove impracticable.
Our business requires us to use hazardous materials, which increases our exposure to dangerous and costly accidents.
Our research and development activities involve the use of hazardous chemicals, biological materials and radioactive compounds. Although we believe that our safety procedures for handling and disposing of hazardous materials comply with the standards prescribed by applicable laws and regulations, we cannot completely eliminate the risk of accidental contamination or injury from these materials. In the event of an accident, an injured party will likely sue us for any resulting damages with potentially significant liability. The ongoing cost of complying with environmental laws and regulations is significant and may increase in the future.
Health care reform and restrictions on reimbursement may limit our returns on potential products.
Because our strategy ultimately depends on the commercial success of our products, we assume, among other things, that end users of our products will be able to pay for them. In the United States and other countries, in most cases, the volume of sales of products like those we are developing depends on the availability of reimbursement from third-party payors, including national health care agencies, private health insurance plans and health maintenance organizations. Third-party payors increasingly challenge the prices charged for medical products and services. Accordingly, if we succeed in bringing products to market, and reimbursement is not available or is insufficient, we could be prevented from successfully commercializing our potential products.
The health care industry in the United States and in Europe is undergoing fundamental changes as a result of political, economic and regulatory influences. Reforms proposed from time to time include mandated basic health care benefits, controls on health care spending, the establishment of governmental controls over the cost of therapies, creation of large medical services and products purchasing groups and fundamental changes to the health care delivery system. We anticipate ongoing review and assessment of health care delivery systems and methods of payment in the United States and other countries. We cannot predict whether any particular reform initiatives will result or, if
35
adopted, what their impact on us will be. However, we expect that adoption of any reform proposed will impair our ability to market products at acceptable prices and that uncertainty concerning future government regulation of consumer healthcare purchasing and insurance may result in difficulties for drug development companies, like Celldex, in raising capital.
Changes in laws affecting the health care industry could adversely affect our business.
In the U.S., there have been numerous proposals considered at the federal and state levels for comprehensive reforms of health care and its cost, and it is likely that federal and state legislatures and health agencies will continue to focus on health care reform in the future. Congress has considered legislation to reform the U.S. health care system by expanding health insurance coverage, reducing health care costs and making other changes. While health care reform may increase the number of patients who have insurance coverage for our products, it may also include cost containment measures that adversely affect reimbursement for our products. Congress has also considered legislation to change the Medicare reimbursement system for outpatient drugs, increase the amount of rebates that manufacturers pay for coverage of their drugs by Medicaid programs and facilitate the importation of lower-cost prescription drugs that are marketed outside the U.S. Some states are also considering legislation that would control the prices of drugs, and state Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and requiring prior authorization by the state program for use of any drug for which supplemental rebates are not being paid. Managed care organizations continue to seek price discounts and, in some cases, to impose restrictions on the coverage of particular drugs. Government efforts to reduce Medicaid expenses may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products.
We and our collaborators and partners operate in a highly regulated industry. As a result, governmental actions may adversely affect our business, operations or financial condition, including:
- •
- new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to
health care availability, method of delivery and payment for health care products and services;
- •
- changes in the FDA and foreign regulatory approval processes that may delay or prevent the approval of new products and
result in lost market opportunity;
- •
- changes in FDA and foreign regulations that may require additional safety monitoring, labeling changes, restrictions on
product distribution or use, or other measures after the introduction of our products to market, which could increase our costs of doing business, adversely affect the future permitted uses of
approved products, or otherwise adversely affect the market for our products;
- •
- new laws, regulations and judicial decisions affecting pricing or marketing practices; and
- •
- changes in the tax laws relating to our operations.
The enactment in the U.S. of health care reform, possible legislation which could ease the entry of competing follow-on biologics in the marketplace, new legislation or implementation of existing statutory provisions on importation of lower-cost competing drugs from other jurisdictions, and legislation on comparative effectiveness research are examples of previously enacted and possible future changes in laws that could adversely affect our business. In addition, the Food and Drug Administration Amendments Act of 2007 included new authorization for the FDA to require post-market safety monitoring, along with an expanded clinical trials registry and clinical trials results database, and expanded authority for the FDA to impose civil monetary penalties on companies that fail to meet certain commitments.
36
If physicians, patients and third-party payors do not accept any future drugs that we may develop, we may be unable to generate significant revenue, if any.
Even if our drug candidates as well as any drug candidates that we may develop or acquire in the future obtain regulatory approval, they may not gain market acceptance among physicians, patients and health care payors. Physicians may elect not to recommend these drugs for a variety of reasons including:
- •
- timing of market introduction of competitive drugs;
- •
- lower demonstrated clinical safety and efficacy compared to other drugs;
- •
- lack of cost-effectiveness;
- •
- lack of availability of reimbursement from third-party payors;
- •
- convenience and ease of administration;
- •
- prevalence and severity of adverse side effects;
- •
- other potential advantages of alternative treatment methods; and
- •
- ineffective marketing and distribution support.
If any drugs that we develop fail to achieve market acceptance, we would not be able to generate sufficient revenue from product sales to maintain or grow our business.
Risks Related to our Capital Stock
Our history of losses and uncertainty of future profitability make our common stock a highly speculative investment.
We have had no commercial revenue to date from sales of our human therapeutic or vaccine products and cannot predict when we will have commercial revenue from such sales. We had an accumulated deficit of $205 million as of December 31, 2011. We expect to spend substantial funds to continue the research and development testing of our products that we have in the preclinical and clinical testing stages of development that have not been partnered.
In anticipation of FDA approval of these products, we will need to make substantial investments to establish sales, marketing, quality control, and regulatory compliance capabilities. These investments will increase if and when any of these products receive FDA approval. We cannot predict how quickly our lead products will progress through the regulatory approval process. As a result, we may continue to lose money for several years.
We cannot be certain that we will achieve or sustain profitability in the future. Failure to achieve profitability could diminish our ability to sustain operations, pay dividends on our common stock, obtain additional required funds and make required payments on our present or future indebtedness.
Our share price has been and could remain volatile.
The market price of our common stock has historically experienced and may continue to experience significant volatility. From January 2011 through December 2011, the market price of our common stock has fluctuated from a high of $4.46 per share in the second quarter of 2011, to a low of $2.11 per share in the fourth quarter of 2011. Our progress in developing and commercializing our products, the impact of government regulations on our products and industry, the potential sale of a large volume of our common stock by stockholders, our quarterly operating results, changes in general conditions in the economy or the financial markets and other developments affecting us or our competitors could cause the market price of our common stock to fluctuate substantially with
37
significant market losses. If our stockholders sell a substantial number of shares of common stock, especially if those sales are made during a short period of time, those sales could adversely affect the market price of our common stock and could impair our ability to raise capital. In addition, in recent years, the stock market has experienced significant price and volume fluctuations. This volatility has affected the market prices of securities issued by many companies for reasons unrelated to their operating performance and may adversely affect the price of our common stock. In addition, we could be subject to a securities class action litigation as a result of volatility in the price of our stock, which could result in substantial costs and diversion of management's attention and resources and could harm our stock price, business, prospects, results of operations and financial condition.
The restrictive covenants contained in our credit agreement may limit our activities.
On December 30, 2010, we entered into a Loan and Security Agreement (the "Loan Agreement") with MidCap Financial, LLC (MidCap) pursuant to which we borrowed $10 million (the "Term Loan") from MidCap. In March 2011, we amended the Loan Agreement and borrowed an additional $5 million from General Electric Capital Corporation (GECC) (collectively with MidCap, the "Lenders") to increase the amount owed under the Term Loan to $15 million. Our obligations under the Term Loan are secured by a first priority lien upon and security interest in substantially all of our existing and after-acquired assets, excluding our intellectual property assets (the "Collateral"). Under the Term Loan, we are subject to specified affirmative covenants customary for loans of this type, including but not limited to the obligations to maintain good standing, provide various notices to the Lenders, deliver financial statements to the Lenders, maintain adequate insurance, promptly discharge all taxes, protect our intellectual property and protect the Collateral. We are also subject to certain negative covenants customary for loans of this type, including but not limited to prohibitions against certain mergers and consolidations, certain management and ownership changes constituting a "change of control," and the imposition of additional liens on Collateral or other of our assets, as well as prohibitions against additional indebtedness, certain dispositions of property, changes in our business, name or location, payment of dividends, prepayment of certain other indebtedness, certain investments or acquisitions, and certain transactions with affiliates, in each case subject to certain customary exceptions, including exceptions that allow us to enter into non-exclusive and/or exclusive licenses and similar agreements providing for the use of our intellectual property in collaboration with third parties provided certain conditions are met.
Failure to comply with the restrictive covenants in our Term Loan could accelerate the repayment of any debt outstanding under the Term Loan. Additionally, as a result of these restrictive covenants, we may be at a disadvantage compared to our competitors that have greater operating and financing flexibility than we do.
Our ability to use our net operating loss carryforwards will be subject to limitation and, under certain circumstances, may be eliminated.
Utilization of our net operating loss (NOL) and research and development credit (R&D credit) carryforwards may be subject to substantial annual limitation due to ownership change limitations that have occurred previously or that could occur in the future provided by Section 382 of the Internal Revenue Code of 1986 (Section 382), as well as similar state provisions. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50 percentage points over a three-year period.
In October 2007, June 2009 and in December 2009, we experienced a change in ownership as defined by Section 382 of the Internal Revenue Code. Historically, we have raised capital through the issuance of capital stock on several occasions which, combined with shareholders' subsequent disposition of those shares, has resulted in three changes of control, as defined by Section 382. As a
38
result of the ownership change in October 2007, utilization of its Federal NOLs is subject to an annual limitation. Any unused annual limitation may be carried over to later years, and the amount of the limitation may, under certain circumstances, be subject to adjustment if the fair value of the our net assets are determined to be below or in excess of the tax basis of such assets at the time of the ownership change, and such unrealized loss or gain is recognized during the five-year period after the ownership change. Subsequent ownership changes, as defined in Section 382, could further limit the amount of net operating loss carryforwards and research and development credits that can be utilized annually to offset future taxable income.
We have not undertaken a study to assess whether an ownership change or multiple ownership changes has occurred for (i) AVANT or CuraGen prior to our acquisitions, (ii) the Company on the state level, (iii) the Company since December 2009, or (iv) R&D credits. If there has been an ownership change at any time since its formation, utilization of NOL or tax credit carryforwards would be subject to an annual limitation under Section 382.
Refer to Note 15, "Income Taxes," in the accompanying notes to the consolidated financial statements for additional discussion on income taxes.
Item 1B. UNRESOLVED STAFF COMMENTS
None.
Our significant leased properties are described below.
Property Location
|
Approximate Square Feet | Use | Lease Expiration Date | ||||
|---|---|---|---|---|---|---|---|
| Needham, Massachusetts | 35,200 | Office Headquarters and Laboratory | April 2017 | ||||
| Fall River, Massachusetts | 23,400 | Manufacturing Facility | December 2017(1) | ||||
| Phillipsburg, New Jersey | 19,400 | Office and Laboratory | August 2016(2) | ||||
| New Haven, Connecticut | 3,000 | Office | January 2013(3) | ||||
- (1)
- Lease
includes two renewal options of five years each. Lease also includes provision for early termination in December 2015 upon prior notice of one year.
- (2)
- Lease
includes one renewal option of five years.
- (3)
- Lease includes one renewal option of three years.
We are not currently a party to any material legal proceedings.
Item 4. MINE SAFETY DISCLOSURES
Not applicable.
39
Item 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock currently trades on the Nasdaq Global Market (NASDAQ) under the symbol "CLDX". The following table sets forth for the periods indicated the high and low sale prices per share for our common stock, as reported by NASDAQ.
Fiscal Period
|
High | Low | |||||
|---|---|---|---|---|---|---|---|
Year Ended December 31, 2010 |
|||||||
First Quarter |
$ | 6.48 | $ | 4.35 | |||
Second Quarter |
9.49 | 4.53 | |||||
Third Quarter |
5.59 | 2.91 | |||||
Fourth Quarter |
4.98 | 3.90 | |||||
Year Ended December 31, 2011 |
|||||||
First Quarter |
$ | 4.22 | $ | 3.45 | |||
Second Quarter |
4.46 | 3.03 | |||||
Third Quarter |
3.73 | 2.29 | |||||
Fourth Quarter |
3.21 | 2.11 | |||||
As of February 29, 2012, there were approximately 562 shareholders of record of our common stock. On February 29, 2012 the closing price of our common stock, as reported by NASDAQ, was $3.79 per share. We have not paid any dividends on our common stock since our inception and do not intend to pay any dividends in the foreseeable future.
Equity Compensation Plan Information
The following table provides information as of December 31, 2011 regarding shares of our common stock that may be issued under our existing equity compensation plans, including our 2008 Stock Option and Incentive Plan (the "2008 Plan") and our 2004 Employee Stock Purchase Plan (the "2004 ESPP Plan").
| |
Equity Compensation Plan Information | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
Number of securities to be issued upon exercise of outstanding options and rights(1) |
Weighted Average exercise price of outstanding options and rights |
Number of securities remaining available for future issuance under equity compensation plan (excluding securities referenced in column (a)) |
|||||||
Equity compensation plans approved by security holders(2) |
4,459,034 | (3) | $ | 6.08 | 997,493 | (4) | ||||
- (1)
- Does
not include any Restricted Stock as such shares are already reflected in our outstanding shares.
- (2)
- Consists
of the 2008 Plan, 2004 ESPP Plan, Celldex Research's 2005 Equity Incentive Plan and CuraGen's 2007 Stock Plan.
- (3)
- Does
not include purchase rights accruing under the 2004 ESPP Plan because the purchase price (and therefore the number of shares to be purchased) will not
be determined until the end of the purchase period.
- (4)
- Includes shares available for future issuance under the 2008 Plan and the 2004 ESPP Plan.
40
CELLDEX THEAPEUTICS, INC., NASDAQ MARKET INDEX—U.S. AND
PEER GROUP INDICES
The graph below compares the cumulative total stockholder return on the common stock for the period from December 31, 2006 through December 31, 2011, with the cumulative return on (i) NASDAQ Market Index—U.S. Companies and (ii) NASDAQ Pharmaceutical Index. The comparison assumes investment of $100 on December 31, 2006 in our common stock and in each of the indices and, in each case, assumes reinvestment of all dividends. The points on the graph are as of December 31 of the year indicated.
| |
2006 | 2007 | 2008 | 2009 | 2010 | 2011 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Celldex Therapeutics, Inc. |
$ | 100 | $ | 37 | $ | 49 | $ | 29 | $ | 26 | $ | 16 | |||||||
NASDAQ Stock Market (U.S.) Index |
$ | 100 | $ | 108 | $ | 66 | $ | 95 | $ | 113 | $ | 114 | |||||||
NASDAQ Pharmaceutical Stock Index |
$ | 100 | $ | 105 | $ | 98 | $ | 110 | $ | 119 | $ | 128 | |||||||
Item 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data are derived from our financial statements. The consolidated statement of operations data for the years ended December 31, 2011, 2010 and 2009 and the consolidated balance sheet data as of December 31, 2011 and 2010 have been derived from our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K. This data should be read in conjunction with our audited consolidated financial statements and related notes which are included elsewhere in this Annual Report on Form 10-K, and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included in Item 7 below.
On October 1, 2009, our acquisition of CuraGen became effective. The CuraGen acquisition was accounted for using the acquisition method of accounting and was treated as our acquisition of CuraGen. Accordingly, the financial information presented below for periods prior to October 1, 2009 reflects the financial position and the results of operations of us alone, and for periods from October 1, 2009 forward the combined financial position and combined results of operations of us and CuraGen.
On March 7, 2008, the AVANT merger became effective. The AVANT merger was accounted for using the purchase method of accounting and was treated as our acquisition of AVANT. Accordingly, the financial information presented below for periods prior to March 8, 2008 reflects the financial position and the results of operations of us alone, and for periods from March 8, 2008 forward the
41
combined financial position and combined results of operations of us and AVANT. All amounts are in thousands except per share data.
CONSOLIDATED STATEMENTS OF OPERATIONS DATA
| |
2011 | 2010 | 2009 | 2008 | 2007 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
REVENUE: |
||||||||||||||||
Product Development and Licensing Agreements |
$ | 110 | $ | 40,187 | $ | 5,662 | $ | 3,716 | $ | 466 | ||||||
Contracts and Grants |
36 | 220 | 1,802 | 533 | 940 | |||||||||||
Product Royalties |
9,119 | 6,386 | 7,716 | 3,207 | — | |||||||||||
Total Revenue |
9,265 | 46,793 | 15,180 | 7,456 | 1,406 | |||||||||||
OPERATING EXPENSE: |
||||||||||||||||
Research and Development |
32,439 | 27,650 | 26,169 | 22,636 | 9,892 | |||||||||||
Royalty |
9,119 | 12,077 | 8,397 | 3,711 | — | |||||||||||
Charge for In-Process Research and Development(2) |
— | — | — | 14,756 | — | |||||||||||
Other Operating Expense |
11,106 | 13,521 | 17,464 | 15,109 | 7,022 | |||||||||||
Total Operating Expense |
52,664 | 53,248 | 52,030 | 56,212 | 16,914 | |||||||||||
Operating Loss |
(43,399 | ) | (6,455 | ) | (36,850 | ) | (48,756 | ) | (15,508 | ) | ||||||
Investment and Other Income, Net |
396 | 5,259 | 248 | 1,411 | 435 | |||||||||||
Interest Expense |
(1,796 | ) | (1,337 | ) | (452 | ) | (156 | ) | — | |||||||
Net Loss Before Income Taxes |
(44,799 | ) | (2,533 | ) | (37,054 | ) | (47,501 | ) | (15,073 | ) | ||||||
Income Tax Benefit |
— | — | 529 | — | — | |||||||||||
Net Loss |
$ | (44,799 | ) | $ | (2,533 | ) | $ | (36,525 | ) | $ | (47,501 | ) | $ | (15,073 | ) | |
Basic and Diluted Net Loss Per Common Share |
$ | (1.13 | ) | $ | (0.08 | ) | $ | (1.84 | ) | $ | (3.34 | ) | $ | (1.81 | ) | |
Shares Used in Calculating Basic and Diluted Net Loss Per Common Share(1) |
39,501 | 31,868 | 19,823 | 14,217 | 8,309 | |||||||||||
- (1)
- Weighted
average common shares outstanding for 2007 have been adjusted to reflect the AVANT merger and a reverse stock split of
1-for-12 effective March 7, 2008.
- (2)
- The 2008 amount arose as a result of the merger between AVANT and us.
CONSOLIDATED BALANCE SHEET DATA
| |
2011 | 2010 | 2009 | 2008 | 2007 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Working Capital |
$ | 40,386 | $ | 42,739 | $ | 69,569 | $ | 32,975 | $ | (4,438 | ) | |||||
Total Assets |
97,994 | 109,943 | 140,364 | 69,793 | 9,375 | |||||||||||
Long Term Liabilities |
14,974 | 14,480 | 52,190 | 37,558 | 370 | |||||||||||
Accumulated Deficit |
(205,006 | ) | (160,207 | ) | (157,674 | ) | (121,149 | ) | (73,648 | ) | ||||||
Total Stockholders' Equity (Deficit) |
68,722 | 75,255 | 73,767 | 18,134 | (1,132 | ) | ||||||||||
42
Item 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
OVERVIEW
We are a biopharmaceutical company focused on the development and commercialization of several immunotherapy technologies for the treatment of cancer and other difficult-to-treat diseases.
Our lead drug candidate, rindopepimut (CDX-110), is an immunotherapeutic vaccine that targets the tumor-specific molecule, epidermal growth factor receptor variant III (EGFRvIII). EGFRvIII is a mutated form of the epidermal growth factor receptor (EGFR) that is only expressed in cancer cells and not in normal tissue and can directly contribute to cancer cell growth. EGFRvIII has been shown by polymerase chain reaction (PCR) analysis to be expressed in approximately 31% of glioblastoma (GB) tumors, also referred to as glioblastoma multiforme (GBM), the most common and aggressive form of brain cancer. Based on results from three prior Phase 2 trials, in December 2011, we initiated ACT IV, a pivotal, randomized, double-blind, controlled Phase 3 study of rindopepimut in patients with surgically resected EGFRvIII-positive GB. In December 2011, we also initiated ReACT, a Phase 2 study of rindopepimut in combination with Avastin® in patients with recurrent EGFRvIII-positive GB.
Our other lead drug candidates include CDX-011 and CDX-1127. CDX-011 is an antibody-drug conjugate (ADC) that consists of a fully-human monoclonal antibody, CR011, linked to a potent cell-killing drug, monomethyl-auristatin E (MMAE). The CR011 antibody specifically targets glycoprotein NMB (GPNMB) that is expressed in a variety of human cancers including breast cancer. In December 2011, we completed enrollment of EMERGE, a randomized Phase 2b study of CDX-011 in patients with heavily pre-treated, advanced, GPNMB-positive breast cancer and expect to present results at an appropriate scientific conference in the first half of 2012.
CDX-1127 is a fully human monoclonal antibody that targets CD27. CD27 is a co-stimulatory molecule on T cells and is over-expressed in certain lymphomas and leukemias. CDX-1127 is an agonist antibody designed to have two potential therapeutic mechanisms. CDX-1127 has been shown to activate immune cells that can target and eliminate cancerous cells in tumor bearing mice and to directly kill or inhibit the growth of CD27 expressing lymphomas and leukemias in vitro and in vivo. In November 2011, we initiated an open label, dose-escalating Phase 1 study of CDX-1127 in patients with selected malignant solid tumors or hematologic cancers.
We have additional clinical and preclinical programs, including CDX-1401, an APC Targeting Technology™ program, CDX-301, an immune cell mobilizing agent and dendritic cell growth factor and CDX-1135, a molecule that inhibits a part of the immune system called the complement system.
Generally our strategy is to develop and demonstrate proof-of-concept for our drug candidates before leveraging their value through partnerships or, in appropriate situations, continuing late stage development through commercialization ourselves. Demonstrating proof-of-concept for a drug candidate generally involves bringing it through Phase 1 clinical trials and one or more Phase 2 clinical trials so that we are able to demonstrate, based on human trials, good safety data for the drug candidate and some data indicating its effectiveness. We thus leverage the value of our technology portfolio through corporate, governmental and non-governmental partnerships. This approach allows us to maximize the overall value of our technology and product portfolio while best ensuring the expeditious development of each individual product. Our current collaborations include the commercialization of an oral human rotavirus vaccine. We are exploring potential development and commercialization collaborations for certain drug candidates such as CDX-011 and CDX-1127. Furthermore, while we plan to retain the rights to develop and commercialize rindopepimut in North America, we are exploring potential partnership opportunities to commercialize rindopepimut outside of North America. Our drug candidates address market opportunities for which we believe current therapies are inadequate or non-existent.
43
Our products are derived from a broad set of complementary technologies which have the ability to utilize the human immune system and enable the creation of therapeutic agents. We are using these technologies to develop targeted immunotherapeutics comprised of antibodies, adjuvants and monotherapies and antibody-drug conjugates that prevent or treat cancer and other diseases that modify undesirable activity by the body's own proteins or cells. A number of our immunotherapeutic and antibody-drug conjugate drug candidates are in various stages of clinical trials. We expect that a large percentage of our research and development expenses will be incurred in support of our current and future clinical trial programs.
The following table includes the programs that we currently believe are material to our business:
Product (generic)
|
Indication/Field | Partner | Status | |||
|---|---|---|---|---|---|---|
CLINICAL |
||||||
CDX-110 |
Front-line Glioblastoma | — | Phase 3 | |||
CDX-110 |
Recurrent Glioblastoma | — | Phase 2 | |||
CDX-011 |
Metastatic breast cancer and melanoma | — | Phase 2b | |||
CDX-1127 |
Lymphoma/leukemia and solid tumors | — | Phase 1 | |||
CDX-1401 |
Multiple solid tumors | — | Phase 1/2 | |||
CDX-301 |
Cancer, autoimmune disease and transplant | — | Phase 1 | |||
PRECLINICAL |
||||||
CDX-1135 |
Renal disease | — | Preclinical | |||
CDX-014 |
Ovarian and renal cancer | — | Preclinical | |||
MARKETED PRODUCTS |
||||||
Rotarix |
Rotavirus infection | GlaxoSmithKline | Marketed |
The expenditures that will be necessary to execute our business plan are subject to numerous uncertainties. Completion of clinical trials may take several years or more, and the length of time generally varies substantially according to the type, complexity, novelty and intended use of a product candidate. It is not unusual for the clinical development of these types of product candidates to each take five years or more, and for total development costs to exceed $100 million for each product candidate. Our estimates that clinical trials of the type we generally conduct are typically completed over the following timelines:
Clinical Phase
|
Estimated Completion Period |
|
|---|---|---|
Phase 1 |
1 - 2 Years | |
Phase 2 |
1 - 5 Years | |
Phase 3 |
1 - 5 Years |
The duration and the cost of clinical trials may vary significantly over the life of a project as a result of differences arising during the clinical trial protocol, including, among others, the following:
- •
- the number of patients that ultimately participate in the trial;
- •
- the duration of patient follow-up that seems appropriate in view of results;
- •
- the number of clinical sites included in the trials;
44
- •
- the length of time required to enroll suitable patient subjects; and
- •
- the efficacy and safety profile of the product candidate.
We test potential product candidates in numerous preclinical studies for safety, toxicology and immunogenicity. We may then conduct multiple clinical trials for each product candidate. As we obtain results from trials, we may elect to discontinue or delay clinical trials for certain product candidates in order to focus our resources on more promising product candidates.
An element of our business strategy is to pursue the research and development of a broad portfolio of product candidates. This is intended to allow us to diversify the risks associated with our research and development expenditures. As a result, we believe our future capital requirements and our future financial success are not substantially dependent on any one product candidate. To the extent we are unable to maintain a broad range of product candidates, our dependence on the success of one or a few product candidates increases.
Regulatory approval is required before we can market our product candidates as therapeutic or vaccine products. In order to proceed to subsequent clinical trial stages and to ultimately achieve regulatory approval, the regulatory agency must conclude that our clinical data is safe and effective. Historically, the results from preclinical testing and early clinical trials (through Phase 2) have often not been predictive of results obtained in later clinical trials. A number of new drugs, biologics and vaccines have shown promising results in early clinical trials, but subsequently failed to establish sufficient safety and efficacy data to obtain necessary regulatory approvals.
Furthermore, our business strategy includes the option of entering into collaborative arrangements with third parties to complete the development and commercialization of our product candidates. In the event that third parties take over the clinical trial process for one of our product candidates, the estimated completion date would largely be under control of that third party rather than us. We cannot forecast with any degree of certainty which proprietary products, if any, will be subject to future collaborative arrangements, in whole or in part, and how such arrangements would affect our development plan or capital requirements. Our programs may also benefit from subsidies, grants, contracts or government or agency-sponsored studies that could reduce our development costs.
As a result of the uncertainties discussed above, among others, it is difficult to accurately estimate the duration and completion costs of our research and development projects or when, if ever, and to what extent we will receive cash inflows from the commercialization and sale of a product. Our inability to complete our research and development projects in a timely manner or our failure to enter into collaborative agreements, when appropriate, could significantly increase our capital requirements and could adversely impact our liquidity. These uncertainties could force us to seek additional, external sources of financing from time to time in order to continue with our business strategy. Our inability to raise additional capital, or to do so on terms reasonably acceptable to us, would jeopardize the future success of our business.
During the past five years through December 31, 2011, we incurred an aggregate of $118.8 million in research and development expenses. The following table indicates the amount incurred for each of our significant research programs and for other identified research and development activities during the years ended December 31, 2011, 2010 and 2009. The amounts disclosed in the following table
45
reflect direct research and development costs, license fees associated with the underlying technology and an allocation of indirect research and development costs to each program.
| |
Year Ended December 31, 2011 |
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
|||||||||
Rindopepimut |
$ | 8,366 | $ | 1,718 | $ | 3,249 | ||||
CDX-011 |
4,917 | 4,104 | 1,098 | |||||||
CDX-1127 |
5,965 | 4,967 | 1,308 | |||||||
CDX-1401 |
2,464 | 2,899 | 4,293 | |||||||
CDX-301 |
1,112 | 4,345 | 2,424 | |||||||
CDX-1135 |
5,524 | 839 | 473 | |||||||
CDX-014 |
481 | 130 | 8 | |||||||
CDX-1307 |
1,440 | 4,067 | 6,510 | |||||||
Other Programs |
2,170 | 4,581 | 6,806 | |||||||
Total R&D Expense |
$ | 32,439 | $ | 27,650 | $ | 26,169 | ||||
Clinical Development Programs
Rindopepimut
Our lead clinical development program, rindopepimut, is a peptide-based immunotherapy that targets the tumor specific molecule called EGFRvIII, a functional variant of the naturally EGFR, a protein which has been well validated as a target for cancer therapy. Unlike EGFR, EGFRvIII is not present in normal tissues and has been shown to be a transforming oncogene that can directly contribute to cancer cell growth. EGFRvIII is commonly present in glioblastoma or GB, also commonly referred to as glioblastoma multiforme or GBM, the most common and aggressive form of brain cancer. The rindopepimut vaccine is composed of the EGFRvIII peptide linked to a carrier protein called Keyhole Limpet Hemocyanin (KLH) and administered together with the adjuvant GM-CSF. The Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have both granted orphan drug designation for rindopepimut for the treatment of EGFRvIII expressing GB and the FDA has also granted Fast Track designation.
In April 2008, we and Pfizer Inc. (Pfizer) entered into a License and Development Agreement (the "Pfizer Agreement") under which Pfizer was granted an exclusive worldwide license to rindopepimut. The Pfizer Agreement provided for reimbursement by Pfizer of all costs incurred by us in connection with the collaboration since the effective date. In November 2010, the Pfizer Agreement was terminated (the "Pfizer Termination") and all rights to rindopepimut were returned to us. Pfizer did not provide a reason for termination. Effective with the Pfizer Termination, Pfizer is no longer funding the development of rindopepimut.
The Phase 1 (VICTORI) and Phase 2a (ACTIVATE) studies of EGFRvIII immunotherapy were led by collaborating investigators at the Brain Center at Duke Comprehensive Cancer Center in Durham, North Carolina and at M.D. Anderson Cancer Center in Houston, Texas and enrolled 14 and 18 evaluable GB patients, respectively. An extension of the Phase 2a study (ACT II) at the same two institutions evaluated 22 additional GB patients treated in combination with maintenance temozolomide (TMZ) (the current standard of care).
We initiated a Phase 2b/3 randomized study (ACT III) of rindopepimut combined with standard of care, TMZ, versus standard of care alone in patients with GB in over 30 sites throughout the United States. In December 2008, we announced an amendment to convert the ACT III study to a single-arm Phase 2 clinical trial in which all patients were to receive rindopepimut in combination with
46
temozolomide. The decision, which followed the recommendation of the Independent Data Monitoring Committee, was based on the observation that the majority of patients randomized to the control (standard of care) arm withdrew from this open-label study after being randomized to the control arm. Patients participating in the control arm of the study were offered the option to receive treatment with rindopepimut. Under this amendment, the ACT III study provided for a multi-center, non-randomized dataset for rindopepimut in patients with newly diagnosed GB.
In November 2010, we announced complete data for the primary endpoint of the 65 patients enrolled to receive rindopepimut in combination with maintenance TMZ in the ACT III study. The data showed that 43 of 65 patients (66%) were progression-free at 5.5 months after entry into the study. Taking into account the 3 to 3.5 months required to complete pre-study chemoradiation and enter into the study, the 5.5 month time point in ACT III corresponds to approximately 8.5 months after diagnosis. The ACTIVATE and ACT II trials, which were conducted in two leading centers, reported progression-free rates at 8.5 months after diagnosis of 70% and 80%, respectively, and similar results were seen in the ACT III trial, which enrolled patients in over 25 centers in the United States.
The following table summarizes the progression free survival (PFS) and overall survival (OS) rates from clinical trials of rindopepimut as compared to matched historical controls and the standard of care (SOC).
| |
Median PFS from diagnosis (months) |
Median OS from diagnosis (months) |
OS at 24 months | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
ACT III (n=65) |
12.3 | (1) | 24.6 | 52 | % | |||||
ACT II (n=22) |
15.3 | 24.4 | 50 | % | ||||||
ACTIVATE (n=18) |
14.2 | 24.6 | 50 | % | ||||||
Matched historical control (n=17)(2) |
6.4 | 15.2 | 6 | % | ||||||
Standard of care radiation/TMZ (n=287)(3) |
6.9 | 14.6 | 27 | % | ||||||
- (1)
- Change
in median PFS not statistically significant from ACTIVATE and ACT II.
- (2)
- Sampson,
et al. J. Clin. Oncol. 2010 Nov 1, 28(31), 4722-9. Historical controls were treated at M.D. Anderson and matched for eligibility
(EGFRvIII-positive, karnofsky performance status (KPS) greater-than or equal to 80%, complete resection, radiation/ temozolomide (TMZ) and without progression through ~ 3 months
post-diagnosis).
- (3)
- Stupp, et al. N. Engl. J. Med. 2005, 352, 987-96.
Importantly, rindopepimut showed a similar benefit in patients whether or not they expressed an active DNA repair gene (MGMT) that has been shown to limit the benefit from TMZ treatment. In ACT III, the number of patients who were expected to be resistant to the TMZ chemotherapy appeared to do better with vaccination than the numbers observed in the historical data. Patients who have an active DNA repair gene, MGMT (unmethylated), generally have a worse outcome, presumably because they do not gain much benefit from TMZ as reported in the literature. Patients with a methylated MGMT promoter in their tumor do not express MGMT and have a more favorable outcome to TMZ treatment. Patients with methylated tumors (n=25) that were treated with the rindopepimut regimen experienced a median PFS of 17.5 months, which compares favorably with the published data from the SOC of radiation plus TMZ (R+TMZ) of 10.3 months. Those with unmethylated tumors (n=40) treated with the rindopepimut regimen experienced a PFS of 11.2 months, which compared favorably to the PFS with SOC of 5.3 months in this patient population. Thus, rindopepimut would appear to benefit both methylated and unmethylated MGMT patients.
In December 2011, we initiated ACT IV, a pivotal, randomized, double-blind, controlled Phase 3 study of rindopepimut in patients with surgically resected, EGFRvIII-positive GB. Patients will be randomized after the completion of surgery and standard chemoradiation. The treatment regime
47
includes a vaccine priming phase post-radiation followed by an adjuvant temozolomide phase and a vaccine maintenance therapy phase. Patients will be treated until disease progression or intolerance to therapy. The primary objective of the study is to determine whether rindopepimut plus adjuvant GM-CSF improves the overall survival of patients with newly diagnosed EGFRvIII-positive GB after Gross Total Resection (GTR) when compared to treatment with TMZ and a control injection of KLH. KLH is a component of rindopepimut and was selected due to its ability to generate a similar injection site reaction to that observed with the rindopepimut vaccine. ACT IV will enroll up to 440 patients at over 150 centers worldwide to recruit approximately 374 patients with GTR to be included in the primary analysis. Our targeted patient accrual is 24 months and another 18 to 24 months of follow-up. We expect it will cost over $50 million to complete this Phase 3 study.
In December 2011, we also initiated ReACT, a Phase 2 study of rindopepimut in combination with Avastin® in patients with recurrent EGFRvIII-positive GB. ReACT will enroll approximately 95 patients in a first or second relapse of GB following receipt of standard therapy and will be conducted at approximately 20 sites across the United States. Approximately 70 patients who have yet to receive Avastin will be randomized to receive either rindopepimut and Avastin or a control injection of KLH and Avastin in a blinded fashion. Another 25 patients who are refractory to Avastin having received Avastin in either the frontline or recurrent setting with subsequent progression will receive rindopepimut plus Avastin in a single treatment arm. We expect preliminary data from this study to be available in mid-2013.
In addition, researchers at Stanford University are conducting a pilot trial of rindopepimut in pediatric patients with pontine glioma in an investigator sponsored trial. Patient screening is ongoing for this trial.
Glembatumumab Vedotin (CDX-011)
CDX-011 is an antibody-drug conjugate (ADC) that consists of a fully- human monoclonal antibody, CR011, linked to a potent cell-killing drug, monomethyl-auristatin E (MMAE). The CR011 antibody specifically targets glycoprotein NMB (GPNMB) that is expressed in a variety of human cancers including breast cancer and melanoma. The ADC technology, comprised of MMAE and a stable linker system for attaching it to CR011, was licensed from Seattle Genetics, Inc. The ADC is designed to be stable in the bloodstream. Following intravenous administration, CDX-011 targets and binds to GPNMB and upon internalization into the targeted cell, CDX-011 is designed to release MMAE from CR011 to produce a cell-killing effect. The FDA has granted Fast Track designation to CDX-011 for the treatment of advanced, refractory/resistant GPNMB-expressing breast cancer.
Treatment of Breast Cancer: In June 2008, an open-label, multi-center Phase 1/2 study was initiated of CDX-011 administered intravenously once every three weeks to patients with locally advanced or metastatic breast cancer who had received prior therapy (median of seven prior regimens). The study began with a bridging phase to confirm the maximum tolerated dose (MTD) and then expanded into a Phase 2 open-label, multi-center study.
The study confirmed the safety of CDX-011 at the pre-defined maximum dose level (1.88 mg/kg) in 6 patients. An additional 28 patients were enrolled as an expanded Phase 2 cohort (for a total of 34 treated patients at 1.88 mg/kg, the Phase 2 dose) to evaluate the PFS rate at 12 weeks. As previously seen in melanoma patients, the 1.88 mg/kg dose was well tolerated in this patient population with the most common adverse events of rash, alopecia, and fatigue. The primary activity endpoint, which called for at least 5 of 25 (20%) patients in the Phase 2 study portion to be progression-free at 12 weeks, was met as 9 of 26 (35%) evaluable patients were progression-free at 12 weeks.
For all patients treated at the maximum dose level, tumor shrinkage was seen in 62% (16/26) and median PFS was 9.1 weeks. A subset of 10 patients had "triple negative disease," a more aggressive breast cancer subtype that carries a high risk of relapse and reduced survival as well as limited
48
therapeutic options due to lack of over-expression of HER2/neu, estrogen and progesterone receptors. In these patients, 78% (7/9) had some tumor shrinkage, 12-week PFS rate was 70% (7/10), and median PFS was 17.9 weeks. Tumor samples from a subset of patients across all dose groups were analyzed for GPNMB expression. The tumor samples from most patients showed evidence of stromal and/or tumor cell expression of GPNMB. The most common treatment-related toxicities were fatigue, rash, nausea, alopecia (hair loss), neutropenia and vomiting.
In December 2011, we completed enrollment of EMERGE, a randomized, multi-center Phase 2b study of CDX-011 in patients with heavily pre-treated, advanced, GPNMB-positive breast cancer. Patients were randomized (2:1) to receive either CDX-011 or single-agent "Investigator's Choice" chemotherapy. Patients randomized to receive Investigator's Choice are allowed to cross-over to CDX-011 following disease progression. Activity endpoints include response rate (RR) and PFS. We expect that a significant portion of the enrolled patients will have triple-negative disease, since GPNMB is frequently expressed in this patient population. We expect to present results at the 2012 American Society of Clinical Oncology (ASCO) meeting in June 2012.
In 2009, Formatech, Inc., a third party contract manufacturer ("Formatech") was engaged by us for the aseptic filling of one lot of our CDX-011 product candidate being used in our ongoing Phase 2b study. The CDX-011 lot from Formatech has passed all of the sterility testing performed during drug release and in subsequent stability studies. At the end of January 2012, we were notified by the FDA that because significant Good Manufacturing Practice (GMP) violations were uncovered during inspection of Formatech, our Phase 2b study for CDX-011 was being placed on partial clinical hold. The FDA uncovered these findings during their inspections of the Formatech facility between August to October 2010 and July to August 2011. These inspections began approximately one year after the CDX-011 drug product was filled at Formatech. Specifically, the FDA requested that no new patients be treated with CDX-011. However, patients already undergoing treatment with CDX-011 could continue treatment using vials of CDX-011 from the lot filled by Formatech, after such patients were informed of the potential risk and reconsented to continued participation in the study. Since the Phase 2b trial completed accrual of patients in December 2011 and there are currently no other ongoing open studies with CDX-011, the only patients that are affected by the partial hold are in the Investigator's Choice (control arm) of the study who currently are not receiving CDX-011 and may be eligible to cross over at the time of progression and receive CDX-011 under the study protocol. Currently there are eight patients remaining on the control arm.
We have initiated discussions with the FDA regarding our proposal to utilize vials of CDX-011 that were filled by a different contract manufacturer. Although the FDA has stated that no new patients may receive CDX-011 and that no patients may cross over to receive CDX-011, we have asked the FDA to reconsider allowing patients currently on the control arm to cross over to CDX-011 after stability testing and confirmation that the product filled by the other contract manufacturer is acceptable for continued use. If the FDA agrees to our proposal concerning use of the alternative CDX-011 for the eligible cross-over patients, we believe that we should have sufficient clinical supply of CDX-011 to treat these cross-over patients. If we are not able to treat the eight remaining cross-over patients with CDX-011, patients may withdraw from the control arm study upon learning that they will not be allowed to cross over to CDX-011 following disease progression. However, the primary analyses for the study are entirely based upon the primary randomization and do not include the cross over results. Based on our discussions with our clinical investigators, we do not believe that a high proportion of patients will withdraw from the control arm prior to progression. We do not believe that this partial hold will significantly impact analysis of the Phase 2b data for purposes of determining next steps in our future development of CDX-011.
In addition, the FDA has agreed in concept that we could reprocess the remaining available vials of CDX-011 manufactured at Formatech at another cGMP contract manufacturer. The FDA's final decision regarding the acceptability of this reprocessing will be made upon review of data concerning
49
the stability and sterility of the reprocessed vials of CDX-011. If we are unsuccessful at reprocessing the available drug product or if FDA does not approve the use of these reprocessed vials, we will need to manufacture new drug product for subsequent clinical studies for CDX-011, which may cause a delay in the initiation of a subsequent trial with CDX-011.
Treatment of Metastatic Melanoma: In 2009, we completed enrollment of 117 patients in a Phase 1/2 open-label, multi-center, dose escalation study to evaluate the safety, tolerability and pharmacokinetics of CDX-011 for patients with un-resectable Stage III or Stage IV melanoma who had failed no more than one prior line of cytotoxic therapy. The MTD was determined to be 1.88 mg/kg administered intravenously (IV) once every three weeks. The study achieved its primary activity objective with an ORR in the Phase 2 cohort of 15% (5/34). Median PFS was 3.9 months. CDX-011 was generally well tolerated, with the most frequent treatment-related adverse events being rash, fatigue, alopecia (hair loss), pruritus, diarrhea and neuropathy. In the subset of patients with tumor biopsies, high levels of tumor expression of GPNMB appeared to correlate with favorable outcome. In the seven patients whose tumors were found to express high amounts of GPNMB, and who were treated at the maximum tolerated doses across all dosing schedules, median PFS was 4.9 months. The development of rash, which may be associated with the presence of GPNMB in the skin also seemed to correlate with greater PFS.
We intend to initially focus our resources on advancing CDX-011 in breast cancer while pursuing further development of CDX-011 in melanoma through collaborations and investigator sponsored studies.
CDX-1127
CDX-1127 is a human monoclonal antibody that targets CD27, a member of the tumor necrosis factor (TNF) receptor superfamily. We have entered into license agreements with the University of Southampton, UK for intellectual property related to uses of anti-CD27 antibodies and with Medarex for access to the UltiMab technology to develop and commercialize human antibodies to CD27. CD27 acts downstream from CD40 and may provide a novel way to regulate the immune responses. CD27 is a co-stimulatory molecule on T cells and is over-expressed in certain lymphomas and leukemias. CDX-1127 is an agonist antibody designed to have two potential therapeutic mechanisms. CDX-1127 has been shown to activate immune cells that can target and eliminate cancerous cells in tumor bearing mice and to directly kill or inhibit the growth of CD27 expressing lymphomas and leukemias in vitro and in vivo. Both mechanisms have been seen even at low doses in appropriate preclinical models.
In November 2011, we initiated an open label, dose-escalating Phase 1 study of CDX-1127 in patients with selected malignant solid tumors or hematologic cancers at multiple clinical sites in the United States. The Phase 1 study is designed to test five escalating doses of CDX-1127 to determine a Phase 2 dose for further development based on safety, tolerability, potential activity and immunogenicity. The study will accrue approximately 30 patients in each of the two arms, either selected refractory/relapsed solid tumors or lymphomas/leukemias known to express CD27. Patients will have received all appropriate prior therapies for their specific disease. The trial design incorporates both single dosing and multiple dosing regimens at each dose level. We expect to complete enrollment in this study by the end of 2012.
CDX-1401
CDX-1401, developed from our APC Targeting Technology, is a fusion protein consisting of a fully human monoclonal antibody with specificity for the dendritic cell receptor, DEC-205, linked to the NY-ESO-1 tumor antigen. In humans, NY-ESO-1 has been detected in 20 – 30% of all melanoma, lung, esophageal, liver, gastric, prostate, ovarian and bladder cancers, thus representing a broad opportunity. This product is intended to selectively deliver the NY-ESO-1 antigen to dendritic cells for
50
generating robust immune responses against cancer cells expressing NY-ESO-1. We are developing CDX-1401 for the treatment of malignant melanoma and a variety of solid tumors which express the proprietary cancer antigen NY-ESO-1, which we licensed from the Ludwig Institute for Cancer Research in 2006. Preclinical studies have shown that CDX-1401 is effective for activation of human T cell responses against NY-ESO-1.
In September 2009, we initiated enrollment in a dose-escalating, multi-center, Phase 1/2 study aimed at determining the optimal dose for further development based on the safety, tolerability, and immunogenicity of the CDX-1401 vaccine. The trial will evaluate three different doses of the vaccine in combination with TLR agonists poly-ICLC or Hiltonol™ and/or R848 or resiquimod.
In October 2010, we announced interim data for the first 20 patients enrolled in the study, 35% of whom had confirmed NY-ESO-1 expression. The interim data showed that six patients maintained stable disease and were eligible for multiple cycles of the treatment regimen, including 4 patients who have received 3 or more cycles (6 weeks of treatment followed by a 6 week rest), with stable disease of up to 11.5+ months. The treatment was well tolerated and there were no dose-limiting toxicities. Robust anti-NY-ESO-1 immunity was induced with the majority of the patients developing anti-NY-ESO-1 antibody responses and 39% of the patients having increases in NY-ESO-1 specific T cell responses, including both CD4 and CD8 responses. Importantly, the T cell responses were directed against multiple regions of the NY-ESO-1 antigen.
In February 2012, we completed enrollment in the Phase 1 portion of the study and expect to report data at an appropriate scientific conference in the second half of 2012.
CDX-301
CDX-301 is a FMS-like tyrosine kinase 3 ligand (Flt3L) stem cell mobilizer and dendritic cell growth factor. We licensed CDX-301 from Amgen in March 2009. CDX-301 is a potent hematopoietic cytokine that stimulates the expansion and differentiation of hematopoietic progenitor and stem cells. CDX-301 has demonstrated a unique capacity to increase the number of circulating dendritic cells in both laboratory and clinical studies. In addition, CDX-301 has shown impressive results in models of cancer, infectious diseases and inflammatory/autoimmune diseases. We believe CDX-301 may hold significant opportunity for synergistic development in combination with other proprietary molecules in our portfolio.
In January 2012, we initiated a dose-escalating Phase 1 study of CDX-301 in approximately 30 healthy subjects in collaboration with Rockefeller University. The Phase 1 study will evaluate seven different dosing regimens of CDX-301 to determine the appropriate dose for further development based on safety, tolerability, and biological activity.
Preclinical Programs
CDX-1135
CDX-1135 is a molecule that inhibits a part of the human immune system called the complement system. The complement system is a series of proteins that are important initiators of the body's acute inflammatory response against disease, infection and injury. Excessive complement activation also plays a role in some persistent inflammatory conditions. CDX-1135 is a soluble form of naturally occurring Complement Receptor 1 that has been shown to inhibit the activation of the complement cascade in animal models and in human clinical trials. In preclinical studies, CDX-1135 has been shown to inhibit both the classical and alternative pathways of complement activation.
51
Dense Deposit Disease (DDD) is a rare and devastating disease that is caused by uncontrolled activation of the alternative pathway of complement and leads to progressive kidney damage in children. There is currently no treatment for patients with DDD and about half progress to end-stage renal disease within 10 years. Because DDD recurs in virtually all patients who receive a kidney transplant, transplantation is not a viable option for these patients. In animal models of DDD, CDX-1135 treatment showed evidence of reversal of kidney damage. We believe that regulating the complement system could have therapeutic and prophylactic applications in DDD and several other acute and chronic conditions, including organ transplantation, multiple sclerosis, rheumatoid arthritis, age-related macular degeneration (AMD), atypical Hemolytic Uremic Syndrome (aHUS), Paroxysmal Nocturnal Hemaglobinuria (PNH), and myasthenia gravis.
CDX-1135 is currently being studied in DDD under an investigator sponsored IND and initial experience indicates that CDX-1135 limits complement abnormalities. In 2011, we completed process development activities and manufactured GMP clinical drug product. Based on discussion to date with the FDA, we are currently planning to initiate a Phase 2 pilot study of CDX-1135 in a small number of DDD patients to determine the appropriate dose and regimen for further clinical development based on safety, tolerability and biological activity in 2012.
CDX-014
CDX-014 is a fully-human monoclonal ADC that targets TIM-1, a molecule that is highly expressed on renal and ovarian cancers with minimal expression in normal tissues. The antibody, CDX-014, is linked to a potent chemotherapeutic, monomethyl auristatin E (MMAE), using Seattle Genetics' proprietary technology. The ADC is designed to be stable in the bloodstream, but to release MMAE upon internalization into TIM-1-expressing tumor cells, resulting in a targeted cell-killing effect. CDX-014 has shown potent activity in preclinical models of ovarian and renal cancer.
Marketed Products
Rotavirus Vaccine
Rotavirus is a major cause of diarrhea and vomiting in infants and children. In 1997, we licensed our oral rotavirus strain to Glaxo and Glaxo assumed responsibility for all subsequent clinical trials and all other development activities. Glaxo gained approval for its rotavirus vaccine, Rotarix, in Mexico in July 2004, which represented the first in a series of worldwide approvals and commercial launches for the product leading up to the approval in Europe in 2006 and in the U.S. in 2008. We licensed-in our rotavirus strain in 1995 and owe a license fee of 30% to Cincinnati Children's Hospital Medical Center (CCH) on net royalties received from Glaxo. We are obligated to maintain a license with CCH with respect to the Glaxo agreement. The term of the Glaxo agreement is through the expiration of the last of the relevant patents covered by the agreement, although Glaxo may terminate the agreement upon 90 days prior written notice. The last relevant patent is scheduled to expire in December 2012. No additional milestone payments are due from Glaxo under the agreement.
In May 2005, we entered into an agreement whereby an affiliate of PRF purchased a 70% interest in the milestone payments and net royalties that we will receive on the development and worldwide sales of Rotarix. We have received a total of $60 million in milestone payments under the PRF agreement. No additional milestone payments are due from PRF under the agreement. The PRF agreement terminates in December 2012, unless otherwise extended. We would retain approximately 65% of the royalties on worldwide sales of Rotarix if PRF receives 2.45 times the aggregate cash payments of $60 million it made to us prior to December 2012. We do not expect this to occur.
52
CRITICAL ACCOUNTING POLICIES
Our significant accounting policies are described in Note 2 to the consolidated financial statements included in Item 8 of this Form 10-K. We believe our most critical accounting policies include accounting for business combinations, revenue recognition, impairment of long-lived assets, research and development expenses and stock-based compensation expense.
The methods, estimates and judgments we use in applying our most critical accounting policies have a significant impact on the results we report in our consolidated financial statements. We evaluate our estimates and judgments on an on-going basis. We base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances. Our experience and assumptions form the basis for our judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what we anticipate and different assumptions or estimates about the future could materially change our reported results. We believe the following accounting policies are the most critical to us in that they are important to the portrayal of our financial statements and they require our most difficult, subjective or complex judgments in the preparation of our consolidated financial statements:
Business Combinations
We account for business combinations under the acquisition method of accounting. We record the fair value of the consideration transferred to acquire a business to the tangible assets and identifiable intangible assets acquired and liabilities assumed on the basis of their fair values at the date of acquisition. We assess the fair value of assets, including intangible assets such as IPR&D, using a variety of methods including present-value models. Each asset is measured at fair value from the perspective of a market participant. The method used to estimate the fair values of IPR&D assets incorporates significant assumptions regarding the estimates a market participant would make in order to evaluate an asset, including a market participant's assumptions regarding the probability of completing IPR&D projects, which would require obtaining regulatory approval for marketing of the associated drug candidate; a market participant's estimates regarding the timing of and the expected costs to complete IPR&D projects; a market participant's estimates of future cash flows from potential product sales; and the appropriate discount rates for a market participant. Transaction costs and restructuring costs associated with the transaction are expensed as incurred.
IPR&D assets acquired in a business combination initially are recorded at fair value and accounted for as indefinite-lived intangible assets. These assets are maintained on our consolidated balance sheets until either the project underlying them is completed or the assets become impaired. If a project is completed, the carrying value of the related intangible asset is amortized over the remaining estimated life of the asset beginning in the period in which the project is completed. If a project becomes impaired or is abandoned, the carrying value of the related intangible asset is written down to its fair value and an impairment charge is taken in the period in which the impairment occurs. IPR&D assets are tested for impairment on an annual basis during the third quarter, or earlier if impairment indicators are present. We performed an annual impairment test of the IPR&D assets as of July 1, 2011 and concluded that the IPR&D assets were not impaired.
Intangible assets acquired in a business combination with a finite life are recorded at fair value and amortized over the greater of economic consumption or on a straight-line basis over their estimated useful life.
The difference between the purchase price and the fair value of assets acquired and liabilities assumed in a business combination is recorded to goodwill. Goodwill is evaluated for impairment on an annual basis during the third quarter, or earlier if impairment indicators are present. We performed an annual impairment test of the goodwill asset as of July 1, 2011 and concluded that the goodwill asset was not impaired.
53
Revenue Recognition
We recognizes revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the seller's price to the buyer is fixed or determinable; and collectability is reasonably assured.
We have entered into and in the future may enter into biopharmaceutical product development agreements with collaborative partners for the research and development of therapeutic drug products. The terms of the agreements may include nonrefundable signing and licensing fees, funding for research, development and manufacturing, milestone payments and royalties on any product sales derived from collaborations. These multiple element arrangements are analyzed to determine whether the deliverables can be separated or whether they must be accounted for as a single unit of accounting. In accounting for these transactions, we allocate revenue to the various elements based on their fair value. The fair value of a revenue generating element can be based on current selling prices offered by us or another party for current products or the our best estimate of a selling price for future products. Revenue allocated to an individual element is recognized when all other revenue recognition criteria are met for that element.
These collaborative and other agreements may contain milestone payments. Revenues from milestones, if they are considered substantive, are recognized upon successful accomplishment of the milestones. Determining whether a milestone is substantive involves judgment, including an assessment of our involvement in achieving the milestones and whether the amount of the payment is commensurate to our performance. If not considered substantive, milestones are initially deferred and recognized over the remaining performance obligation.
Payments received to fund certain research activities are recognized as revenue in the period in which the research activities are performed. Revenue from contracts and grants is recognized as the services are performed and recorded as effort is expended on the contracted work and billed to the government or our contractual partner. Payments received in advance that are related to future performance are deferred and recognized as revenue when the research projects are performed.
Product royalty revenue consists of payments received from licensees for a portion of sales proceeds from products that utilize our licensed technologies and are recognized when the amount of and basis for such royalty payments are reported to us in accurate and appropriate form and in accordance with the related license agreement.
Impairment of Long-Lived Assets
We evaluate the recoverability of our long-lived assets, including property and equipment, and intangible assets when circumstances indicate that an event of impairment may have occurred. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written-down to their estimated fair values.
Research and Development Expenses
Research and development costs, including internal and contract research costs, are expensed as incurred. Research and development expenses consist mainly of clinical trial costs, manufacturing of clinical material, toxicology and other studies, personnel costs, depreciation, license fees and funding of outside research.
Clinical trial expenses include expenses associated with clinical research organizations (CRO). The invoicing from CROs for services rendered can lag several months. We accrue the cost of services rendered in connection with CRO activities based on our estimate of site management, monitoring
54
costs, and project management costs. We maintain regular communication with our CROs to gauge the reasonableness of our estimates. Differences between actual clinical trial expenses and estimated clinical trial expenses recorded have not been material and are adjusted for in the period in which they become known.
Stock-Based Compensation Expense
We record stock-based compensation expense for all stock-based awards made to employees and directors based on the estimated fair values of the stock-based awards expected to vest at the grant date and is adjusted, if necessary, to reflect actual forfeitures. Compensation expense for all stock-based awards to employees and directors is recognized using the straight-line method over the term of vesting or performance.
We record stock-based compensation expense for stock options granted to non-employees based on the fair value of the stock options which is re-measured over the vesting term resulting in periodic adjustments to stock-based compensation expense.
RESULTS OF OPERATIONS
Year Ended December 31, 2011 compared with Year Ended December 31, 2010
| |
Year Ended December 31, | |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) $ |
Increase/ (Decrease) % |
|||||||||||
| |
2011 | 2010 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Revenue: |
|||||||||||||
Product Development and Licensing Agreements |
$ | 110 | $ | 40,187 | $ | (40,077 | ) | (100 | )% | ||||
Contracts and Grants |
36 | 220 | (184 | ) | (84 | )% | |||||||
Product Royalties |
9,119 | 6,386 | 2,733 | 43 | % | ||||||||
Total Revenue |
$ | 9,265 | $ | 46,793 | $ | (37,528 | ) | (80 | )% | ||||
Operating Expense: |
|||||||||||||
Research and Development |
32,439 | 27,650 | 4,789 | 17 | % | ||||||||
Royalty |
9,119 | 12,077 | (2,958 | ) | (24 | )% | |||||||
Gain on Sale of Assets |
(50 | ) | (50 | ) | — | — | |||||||
General and Administrative |
9,243 | 10,428 | (1,185 | ) | (11 | )% | |||||||
Amortization of Acquired Intangible Assets |
1,913 | 3,143 | (1,230 | ) | (39 | )% | |||||||
Total Operating Expense |
52,664 | 53,248 | (584 | ) | (1 | )% | |||||||
Operating Loss |
(43,399 | ) | (6,455 | ) | 36,944 | 572 | % | ||||||
Investment and Other Income, Net |
396 | 5,259 | (4,863 | ) | (92 | )% | |||||||
Interest Expense |
(1,796 | ) | (1,337 | ) | 459 | 34 | % | ||||||
Net Loss |
$ | (44,799 | ) | $ | (2,533 | ) | $ | 42,266 | 1,669 | % | |||
Net Loss
The $42.3 million increase in net loss for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily the result of a decrease in product development and licensing agreement revenue.
55
Revenue
The $40.1 million decrease in product development and licensing agreement revenue for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to the termination of the Pfizer Agreement which resulted in us recognizing the remaining deferred revenue of $35.6 million during the year ended December 31, 2010. The $0.2 million decrease in contracts and grants revenue for the year ended December 31, 2011 compared to the year ended December 31, 2010 was due to a decrease in revenue related to our vaccine development work on Rockefeller's DCVax-001 (CDX-2401) program. The $2.7 million increase in product royalty revenue for the year ended December 31, 2011 compared to the year ended December 31, 2010 was related to our retained interests in Rotarix net royalties which were not sold to PRF and which is equal to the amount payable to CCH and recognized in royalty expense by us. We expect that royalty revenue related to the Glaxo agreement will end during the year ended December 31, 2013 as the term of the Glaxo agreement is through the expiration of the last of the relevant patents covered by the agreement and the last relevant patent is scheduled to expire in December 2012.
Research and Development Expense
Research and development expenses consist primarily of (i) personnel expenses, (ii) laboratory supply expenses relating to the development of our technology, (iii) facility expenses, and (iv) product development expenses associated with our drug candidates as follows:
| |
Year Ended December 31, | |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) $ |
Increase/ (Decrease) % |
|||||||||||
| |
2011 | 2010 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Personnel |
$ | 12,715 | $ | 12,204 | $ | 511 | 4 | % | |||||
Laboratory Supplies |
1,920 | 1,779 | 141 | 8 | % | ||||||||
Facility |
4,674 | 4,962 | (288 | ) | (6 | )% | |||||||
Product Development |
10,044 | 5,832 | 4,212 | 72 | % | ||||||||
Personnel expenses primarily include salary, benefits, stock-based compensation and payroll taxes. The $0.5 million increase in personnel expenses for the year ended December 31, 2011 compared to the year ended December 31, 2010 was due to higher headcount. We expect personnel expenses to increase over the next twelve months as we plan to continue to increase our headcount, primarily in clinical research personnel, to support our Phase 3 study of rindopepimut.
Laboratory supply expenses include laboratory materials and supplies, services, and other related expenses incurred in the development of our technology. The $0.1 million increase in laboratory supply expenses for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to the renovations completed during the year ended December 31, 2010 at our Fall River, MA manufacturing facility during which manufacturing activities ceased. We expect supply expenses to increase over the next twelve months as a result of increased research and development activities, although there may be fluctuations on a quarterly basis.
Facility expenses include depreciation, amortization, utilities, rent, maintenance, and other related expenses incurred at our facilities. The $0.3 million decrease in facility expenses for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to lower depreciation and amortization expenses. We expect facility expenses to remain relatively consistent over the next twelve months, although there may be fluctuations on a quarterly basis.
Product development expenses include clinical investigator site fees, external trial monitoring costs, data accumulation costs, contracted research and outside clinical drug product manufacturing. The $4.2 million increase in product development expenses for the year ended December 31, 2011
56
compared to the year ended December 31, 2010 was primarily due to an increase in clinical trial costs of $4.6 million primarily due to our rindopepimut and CDX-011 programs. We expect product development expenses to increase over the next twelve months primarily due to the increase in clinical trial expenses related to rindopepimut, although there may be fluctuations on a quarterly basis.
Royalty Expense
Royalty expenses include product royalty and sublicense royalty fees on our out-licensed programs. The $3.0 million decrease in royalty expenses for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to the termination of the Pfizer Agreement which resulted in us recognizing the remaining deferred sublicense fees of $5.1 million, partially offset by an increase in Rotarix related royalty fees. Our retained interests in Rotarix net royalties which were not sold to PRF are recorded as product royalty revenue and a corresponding amount that is payable to CCH is recorded as royalty expense. We expect royalty expense related to the Glaxo agreement will end during the year ended December 31, 2013 as the term of the Glaxo agreement is through the expiration of the last of the relevant patents covered by the agreement and the last relevant patent is scheduled to expire in December 2012.
General and Administrative Expense
The $1.2 million decrease in general and administrative expenses for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due a decrease in legal, patent and other professional services. We expect general and administrative expense to remain relatively consistent over the next twelve months, although there may be fluctuations on a quarterly basis.
Amortization Expense
The $1.2 million decrease in amortization expenses for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to the TopoTarget intangible asset becoming fully amortized during the year ended December 31, 2011. We recorded amortization expense related to the TopoTarget Agreement of $0.3 million and $1.7 million for the years ended December 31, 2011 and 2010, respectively. We expect amortization expense of acquired intangible assets to decrease over the next twelve months as a result of intangible assets becoming fully amortized during 2011.
Investment and Other Income, Net
The $4.9 million decrease in investment and other income, net for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to $3.0 million received in connection with the TopoTarget Agreement and $1.7 million received related to the IRS Qualifying Therapeutic Discovery Grants during the year ended December 31, 2010. We anticipate investment income to increase over the next twelve months due to higher cash and investment balances resulting from fundraising efforts in January and February 2012. In January 2012, we sold 2,450,000 shares of common stock under the Cantor Agreement and raised $8.5 million in net proceeds. In February 2012, we issued 10,500,000 shares of our common stock in an underwritten public offering. The net proceeds to us were $37.7 million, after deducting underwriting fees and estimated offering expenses. We have granted the underwriters a 30-day option to purchase up to an aggregate of 1,575,000 additional shares of common stock to cover overallotments, if any.
Interest Expense
The $0.5 million increase in interest expense for the year ended December 31, 2011 compared to the year ended December 31, 2010 was primarily due to our Term Loan. On December 30, 2010, we
57
entered into a Loan and Security Agreement (the "Loan Agreement") with MidCap Financial, LLC (MidCap) pursuant to which we borrowed $10 million (the "Term Loan") from MidCap. In March 2011, we amended the Loan Agreement and borrowed an additional $5 million from General Electric Capital Corporation (GECC) (collectively with MidCap, the "Lenders") to increase the amount owed under the Term Loan to $15 million. The Term Loan accrues interest at a fixed annual interest rate equal to the greater of (i) the sum of (A) the LIBOR Rate (as defined in the Loan Agreement) plus (B) 6.25%; or (ii) a minimum rate of 9.50%. We anticipate interest expense to remain relatively consistent over the next twelve months.
Year Ended December 31, 2010 compared with Year Ended December 31, 2009
| |
Year Ended December 31, | |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) $ |
Increase/ (Decrease) % |
|||||||||||
| |
2010 | 2009 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Revenue: |
|||||||||||||
Product Development and Licensing Agreements |
$ | 40,187 | $ | 5,662 | $ | 34,525 | 610 | % | |||||
Contracts and Grants |
220 | 1,802 | (1,582 | ) | (88 | )% | |||||||
Product Royalties |
6,386 | 7,716 | (1,330 | ) | (17 | )% | |||||||
Total Revenue |
$ | 46,793 | $ | 15,180 | $ | 31,613 | 208 | % | |||||
Operating Expense: |
|||||||||||||
Research and Development |
27,650 | 26,169 | 1,481 | 6 | % | ||||||||
Royalty |
12,077 | 8,397 | 3,680 | 44 | % | ||||||||
Gain on Sale of Assets |
(50 | ) | (604 | ) | 554 | 92 | % | ||||||
General and Administrative |
10,428 | 17,119 | (6,691 | ) | (39 | )% | |||||||
Amortization of Acquired Intangible Assets |
3,143 | 949 | 2,194 | 231 | % | ||||||||
Total Operating Expense |
53,248 | 52,030 | 1,218 | 2 | % | ||||||||
Operating Loss |
(6,455 | ) | (36,850 | ) | (30,395 | ) | (82 | )% | |||||
Investment and Other Income, Net |
5,259 | 248 | 5,011 | 2,021 | % | ||||||||
Interest Expense |
(1,337 | ) | (452 | ) | 885 | 196 | % | ||||||
Net Loss Before Income Taxes |
(2,533 | ) | (37,054 | ) | (34,521 | ) | (93 | )% | |||||
Income Tax Benefit |
— | 529 | (529 | ) | (100 | )% | |||||||
Net Loss |
$ | (2,533 | ) | $ | (36,525 | ) | $ | (33,992 | ) | (93 | )% | ||
Net Loss
The $34.0 million decrease in net loss for the year ended December 31, 2010 compared to the year ended December 31, 2009 was primarily the result of an increase in product development and licensing agreement revenue and other income and a decrease in general and administrative expense. This impact was partially offset by a decrease in contracts and grants and product royalty revenue and increased research and development, royalty and amortization expense on acquired intangible assets.
Revenue
The $34.5 million increase in product development and licensing agreement revenue for the year ended December 31, 2010 was primarily due to the Pfizer Termination which resulted in us recognizing the remaining deferred revenue of $35.6 million related to the Pfizer Agreement. The $1.6 million decrease in contracts and grants revenue for the year ended December 31, 2010 was due to a decrease in revenue related to our vaccine development work on Rockefeller's DCVax-001 (CDX-2401) program. The $1.3 million decrease in product royalty revenue for the year ended December 31, 2010 was
58
related to our retained interests in Rotarix net royalties which were not sold to PRF and which is equal to the amount payable to CCH and recognized in royalty expense by us. In late March 2010, the FDA temporarily suspended the use of Rotarix in the United States as a precautionary measure following the discovery of PCV-1 DNA material in the vaccine. In May 2010, the FDA determined that healthcare practitioners could resume the use of Rotarix. Our royalties from sales of Rotarix were negatively impacted during the year ended December 31, 2010 by the FDA's decision to temporarily suspend the use of Rotarix from March 2010 through May 2010 and that negative impact from the temporary suspension may extend into the future as well. However, our net loss and cash position were not impacted even though our royalty revenue was impacted because there was an offsetting impact on our royalty expense.
Research and Development Expense
Research and development expenses consist primarily of (i) personnel expenses, (ii) laboratory supply expenses relating to the development of our technology, (iii) facility expenses, and (iv) product development expenses associated with our drug candidates as follows:
| |
Year Ended December 31, | |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Increase/ (Decrease) $ |
Increase/ (Decrease) % |
|||||||||||
| |
2010 | 2009 | |||||||||||
| |
(In thousands) |
|
|||||||||||
Personnel |
$ | 12,204 | $ | 11,108 | $ | 1,096 | 10 | % | |||||
Laboratory Supplies |
1,779 | 2,517 | (738 | ) | (29 | )% | |||||||
Facility |
4,962 | 4,782 | 180 | 4 | % | ||||||||
Product Development |
5,832 | 5,758 | 74 | 1 | % | ||||||||
Personnel expenses primarily include salary, benefits, stock-based compensation and payroll taxes. The $1.1 million increase in personnel expenses for the year ended December 31, 2010 was due to higher headcount, partially offset by $0.9 million in severance expense during the year ended December 31, 2009 related to the CuraGen acquisition.
Laboratory supply expenses include laboratory materials and supplies, services, and other related expenses incurred in the development of our technology. The $0.7 million decrease in laboratory supply expenses was primarily due to the renovations completed during the year ended December 31, 2010 at our Fall River, MA manufacturing facility during which manufacturing activities ceased.
Facility expenses include depreciation, amortization, utilities, rent, maintenance, and other related expenses incurred at our facilities. The $0.2 million increase in facility expenses for the year ended December 31, 2010 was primarily due to higher depreciation and amortization expenses.
Product development expenses include clinical investigator site fees, external trial monitoring costs, data accumulation costs, contracted research and outside clinical drug product manufacturing. The $0.1 million increase in product development expenses for the year ended December 31, 2010 was primarily due to an increase in product development costs related to our CDX-011 program.
Royalty Expense
Royalty expenses include product royalty and sublicense royalty fees on our out-licensed programs. The $3.7 million increase in royalty expenses for the year ended December 31, 2010 was primarily due to the Pfizer Termination which resulted in us recognizing the remaining deferred sublicense fees related to the Pfizer Agreement of $5.1 million, partially offset by a decrease in Rotarix related royalty fees. Our retained interests in Rotarix net royalties which were not sold to PRF are recorded as product royalty revenue and a corresponding amount that is payable to CCH is recorded as royalty expense.
59
General and Administrative Expense
The $6.7 million decrease in general and administrative expenses for the year ended December 31, 2010 was primarily due to $3.3 million in severance expense incurred during the year ended December 31, 2009 related to the CuraGen acquisition and a decrease of $2.1 million in consultant and legal expense primarily related to costs incurred in connection with the CuraGen acquisition in 2009. The decrease was also a result of $0.7 million in severance expense, including related non-cash stock-based compensation expense, incurred during the year ended December 31, 2009 related to our former SVP, Business Development.
Amortization Expense
The $2.2 million increase in amortization expenses for the year ended December 31, 2010 was primarily due to intangible assets acquired in connection with the CuraGen acquisition including the TopoTarget Agreement. In February 2010, TopoTarget entered into a co-development and commercialization agreement for Belinostat with Spectrum Pharmaceuticals, Inc. which resulted in our receipt of $3.0 million which we recorded as Other Income for the year ended December 31, 2010. We recorded amortization expense related to the TopoTarget Agreement of $1.7 million for the year ended December 31, 2010.
Investment and Other Income, Net
The $5.0 million increase in investment and other income, net for the year ended December 31, 2010 was primarily due to $3.0 million received in connection with the TopoTarget Agreement and $1.7 million in other income related to the receipt of IRS Qualifying Therapeutic Discovery Grants.
Interest Expense
The $0.9 million increase in interest expense for the year ended December 31, 2010 was primarily due to the CuraGen Debt we assumed in connection with the CuraGen acquisition.
Income Tax Benefit
The $0.5 million decrease in income tax benefit for the year ended December 31, 2010 was due to non-cash tax consequences as a result of the CuraGen acquisition.
LIQUIDITY AND CAPITAL RESOURCES
At December 31, 2011, our principal sources of liquidity consisted of cash, cash equivalents and marketable securities of $53.3 million. Our working capital at December 31, 2011 was $40.4 million. At December 31, 2011, our Term Loan balance was $15.1 million. We incurred a loss of $44.8 million for the year ended December 31, 2011. Net cash used in operations for the year ended December 31, 2011 was $35.7 million. In January 2012, we sold 2,450,000 shares of common stock under the Cantor Agreement and raised $8.5 million in net proceeds. In February 2012, we issued 10,500,000 shares of our common stock in an underwritten public offering. The net proceeds to us were $37.7 million, after deducting underwriting fees and estimated offering expenses. We have granted the underwriters a 30-day option to purchase up to an aggregate of 1,575,000 additional shares of common stock to cover overallotments, if any. We believe that the cash, cash equivalents and marketable securities at December 31, 2011, the $8.5 million raised under the Cantor Agreement in January 2012, the $37.7 million raised in the underwritten offering in February and interest income on invested funds, are sufficient to meet estimated working capital requirements and fund planned operations for at least the next twelve months.
60
Our cash equivalents are highly liquid investments with a maturity of three months or less at the date of purchase and consist primarily of investments in money market mutual funds with commercial banks and financial institutions. We maintain cash balances with financial institutions in excess of insured limits. We do not anticipate any losses with respect to such cash balances. We invest our excess cash balances in marketable securities including municipal bond securities, U.S. government agency securities, and high-grade corporate bonds that meet high credit quality standards, as specified in our investment policy. Our investment policy seeks to manage these assets to achieve our goals of preserving principal and maintaining adequate liquidity.
The use of our cash flows for operations has primarily consisted of salaries and wages for our employees, facility and facility-related costs for our offices, laboratories and manufacturing facility, fees paid in connection with preclinical studies, clinical studies, contract manufacturing, laboratory supplies and services, consulting, legal and other professional fees. To date, the primary sources of cash flows from operations have been payments received from our collaborative partners and from government entities. The timing of any new collaboration agreements, government contracts or grants and any payments under these agreements, contracts or grants cannot be easily predicted and may vary significantly from quarter to quarter.
We raised net proceeds of $35.9 million during the year ended December 31, 2011 and $46.2 million during the two months ended February 29, 2012 from the sale of our common stock. During the next twelve months, we may take further steps to raise additional capital to fund our long-term liquidity needs. Our capital raising activities may include, but may not be limited to, one or more of the following: the licensing of technology programs with existing or new collaborative partners, possible business combinations, issuance of debt, or the issuance of common stock or other securities via private placements or public offerings. While we may continue to seek capital through a number of means, there can be no assurance that additional financing will be available on acceptable terms, if at all, and our negotiating position in capital-raising efforts may worsen as existing resources are used. There is also no assurance that we will be able to enter into further collaborative relationships. Additional equity financing may be dilutive to our stockholders; debt financing, if available, may involve significant cash payment obligations and covenants that restrict our ability to operate as a business; and licensing or strategic collaborations may result in royalties or other terms which reduce our economic potential from products under development. If we are unable to raise the funds necessary to meet our long-term liquidity needs, we may have to delay or discontinue the development of one or more programs, discontinue or delay on-going or anticipated clinical trials, license out programs earlier than expected, raise funds at significant discount or on other unfavorable terms, if at all, or sell all or part of our business.
Operating Activities
Net cash used in operating activities was $35.7 million for the year ended December 31, 2011 compared to $30.4 million for the year ended December 31, 2010. The increase in net cash used in operating activities was primarily due to changes in working capital and decreases during the year ended December 31, 2011 in other income and the related amortization of intangible assets resulting from the $3.0 million received in connection with the TopoTarget Agreement during the year ended December 31, 2010. We expect that cash used in operating activities will increase over the next twelve months primarily related to costs incurred on our rindopepimut program.
We have incurred and will continue to incur significant costs in the area of research and development, including preclinical and clinical trials, as our drug candidates are developed. We plan to spend significant amounts to progress our current drug candidates through the clinical trial and commercialization process as well as to develop additional drug candidates. As our drug candidates progress through the clinical trial process, we may be obligated to make significant milestone payments.
61
Investing Activities
Net cash used in investing activities was $2.2 million for the year ended December 31, 2011 compared to $16.2 million for the year ended December 31, 2010. The decrease in net cash used in investing activities was primarily due to net purchases of marketable securities for the year ended December 31, 2011 of $1.7 million as compared to $14.1 million for the year ended December 31, 2010. We expect that cash provided by investing activities will increase over the next twelve months as we fund our operations through the net proceeds from the sale and maturity of marketable securities, cash provided by financing activities and/or new partnerships.
Financing Activities
Net cash provided by financing activities was $28.4 million for the year ended December 31, 2011 compared to $10.8 million for the year ended December 31, 2010. The increase in net cash provided by financing activities was primarily due to the $35.9 million in net proceeds we received through the sale of 11,500,000 shares of our common stock in an underwritten public offering in May 2011 and the sale of 575,000 shares of common stock under the Cantor Agreement during the year ended December 31, 2011. In February 2011, we paid $12.5 million to satisfy all outstanding principal related to the CuraGen Debt. In March 2011, we amended the Loan Agreement and borrowed an additional $5 million from GECC to increase the amount owed under the Term Loan to $15 million.
Equity Offerings
In April 2010, we filed a shelf registration statement with the Securities and Exchange Commission to register for sale any combination of the types of securities described in the shelf registration statement up to a dollar amount of $150 million. The shelf registration became effective on April 22, 2010.
On January 6, 2011, we entered into a controlled equity offering sales agreement (the "Cantor Agreement") with Cantor Fitzgerald & Co. (Cantor) pursuant to which we may issue and sell up to 5,000,000 shares of our common stock from time to time through Cantor, acting as agent. We agreed to pay Cantor a commission of up to 5% of the gross proceeds from each sale and to reimburse Cantor for certain expenses incurred in connection with entering into the Cantor Agreement. The Cantor Agreement terminates upon the sale of all 5,000,000 shares or upon ten day notice by either Cantor or us. During the year ended December 31, 2011, we sold 575,000 shares of common stock under the Cantor Agreement and raised $2.2 million in net proceeds, after deducting commission and offering expenses. In January 2012, we sold 2,450,000 shares of common stock under the Cantor Agreement and raised $8.5 million in net proceeds. Under the terms of the Cantor Agreement, we will have the ability to sell up to 1,975,000 shares of our common stock upon the expiration or earlier waiver of our 90-day lock-up with the underwriters of our recent offering in February 2012.
In May 2011, we issued 11,500,000 shares of our common stock in an underwritten public offering, including the underwriter's exercise of their full over-allotment option to purchase an additional 1,500,000 shares of common stock. The net proceeds to us were $33.7 million, after deducting underwriting fees and offering expenses.
In February 2012, we issued 10,500,000 shares of our common stock in an underwritten public offering. The net proceeds to us were $37.7 million, after deducting underwriting fees and estimated offering expenses. We have granted the underwriters a 30-day option to purchase up to an aggregate of 1,575,000 additional shares of common stock to cover overallotments, if any.
62
Term Loan
On December 30, 2010, we entered the Loan Agreement with MidCap pursuant to which we borrowed $10 million from MidCap. In March 2011, we amended the Loan Agreement and borrowed an additional $5 million from GECC to increase the amount owed under the Term Loan to $15 million. No additional advances are available under the Loan Agreement. The Term Loan accrues interest at a fixed annual interest rate equal to the greater of (i) the sum of (A) the LIBOR Rate (as defined in the Loan Agreement) plus (B) 6.25%; or (ii) a minimum rate of 9.50%. In September 2011, we exercised an option to extend the interest-only period by 6 months from October 1, 2011 to April 1, 2012. In March 2012, we amended the Loan Agreement to extend the maturity date from December 2013 to December 2014 in return for an upfront fee of $25,000 and an additional fee of $37,500 due upon repayment of the Term Loan in full.
Interest on the Term Loan is payable monthly and principal is due, as amended, in 34 equal consecutive monthly installments commencing on April 1, 2012. All unpaid principal and accrued interest with respect to the Term Loan is due and payable on the earlier of (A) December 30, 2014 or (B) the date that the Term Loan otherwise becomes due and payable under the terms of the Loan Agreement. We may prepay all, but not less than all, of the Term Loan subject to a prepayment premium of 1% in year three and 2% in year two of the original principal amount of the Term Loan. There is no prepayment premium if the loan is paid off early in year four. We are also obligated to make a payment of $0.5 million (Payment Fee) upon the earlier of (A) December 30, 2013 or (B) upon repayment of the Term Loan in full prior to December 30, 2013.
Our obligations under the Loan Agreement are secured by a first priority lien upon and security interest in substantially all of our existing and after-acquired assets, excluding our intellectual property assets. Under the Loan Agreement, we are subject to specified affirmative and negative covenants customary for financings of this type. The Loan Agreement provides that, upon the occurrence of certain specified events of default customary for financings of this type, our obligations under the Loan Agreement may be automatically accelerated, whereupon our obligations under the Loan Agreement shall be immediately due and payable. At December 31, 2011, we believe we are in compliance with the Loan Agreement.
AGGREGATE CONTRACTUAL OBLIGATIONS
We have entered into license agreements whereby we have received licenses or options to license technology, specified patents and/or patent applications. These license and collaboration agreements generally provide for royalty payments equal to specified percentages of product sales, annual license maintenance fees, continuing patent prosecution costs and potential future milestone payments to third parties upon the achievement of certain development, regulatory and/or commercial milestones. Because the achievement of these milestones had not occurred as of December 31, 2011, such contingencies have not been recorded in our financial statements. We expect to incur approximately $0.7 million of milestone payments in 2012.
The following table summarizes our contractual obligations (not including contingent royalty and milestone payments as described above) at December 31, 2011 and the effect such obligations and commercial commitments are expected to have on our liquidity and cash flow in future years. These
63
obligations, commitments and supporting arrangements represent expected payments based on current operating forecasts, which are subject to change:
| |
Total | 2012 | 2013 - 2014 | 2015 - 2016 | Thereafter | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
|||||||||||||||
Contractual obligations: |
||||||||||||||||
Operating lease obligations(1) |
$ | 13,181 | $ | 2,405 | $ | 4,859 | $ | 4,903 | $ | 1,014 | ||||||
Other contractual obligations(2) |
18,090 | 18,090 | — | — | — | |||||||||||
Other long-term liabilities(3) |
527 | 51 | 118 | 129 | 229 | |||||||||||
Term Loan(4) |
15,450 | 3,971 | 11,479 | — | — | |||||||||||
Total contractual obligations |
$ | 47,248 | $ | 24,517 | $ | 16,456 | $ | 5,032 | $ | 1,243 | ||||||
- (1)
- Such
amounts primarily consist of payments for our facility leases and do not assume the exercise of renewal terms or early termination provisions.
- (2)
- Such
amounts primarily consist of research and development commitments with third parties. Our obligation to pay certain of these amounts may increase or be
reduced based on certain future events.
- (3)
- Such
amounts include the outstanding balance on a loan payable which accrues interest at 5.5% and is payable monthly.
- (4)
- Such amounts include the outstanding balance at December 31, 2011 on the Term Loan along with the Payment Fee. The Term Loan accrues interest at a fixed annual interest rate equal to the greater of (i) the sum of (A) the LIBOR Rate (as defined in the Loan Agreement) plus (B) 6.25%; or (ii) a minimum rate of 9.50%. As described above, we amended our Term Loan in March 2012 and currently all amounts borrowed under the Term Loan mature in December 2014.
RECENT ACCOUNTING PRONOUNCEMENTS
Refer to Note 2, "Summary of Significant Accounting Policies," in the accompanying notes to the consolidated financial statements for a discussion of recent accounting pronouncements.
OFF-BALANCE SHEET ARRANGEMENTS
None.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We own financial instruments that are sensitive to market risk as part of our investment portfolio. Our investment portfolio is used to preserve our capital until it is used to fund operations, including our research and development activities. None of these market-risk sensitive instruments are held for trading purposes. We invest our cash primarily in money market mutual funds. These investments are evaluated quarterly to determine the fair value of the portfolio. From time to time, we invest our excess cash balances in marketable securities including municipal bond securities, U.S. government agency securities, and high-grade corporate bonds that meet high credit quality standards, as specified in our investment policy. Our investment policy seeks to manage these assets to achieve our goals of preserving principal and maintaining adequate liquidity. Because of the short-term nature of these investments, we do not believe we have material exposure due to market risk. The impact to our financial position and results of operations from likely changes in interest rates is not material.
We do not utilize derivative financial instruments. The carrying amounts reflected in the consolidated balance sheet of cash and cash equivalents, accounts receivables and accounts payable approximates fair value at December 31, 2011 due to the short-term maturities of these instruments.
64
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Celldex Therapeutics, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations and comprehensive loss, of stockholders' equity, and of cash flows, present fairly, in all material respects, the financial position of Celldex Therapeutics, Inc. and its subsidiaries at December 31, 2011 and December 31, 2010, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2011 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Annual Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Boston,
Massachusetts
March 8, 2012
65
CELLDEX THERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
| |
December 31, 2011 | December 31, 2010 | |||||
|---|---|---|---|---|---|---|---|
ASSETS |
|||||||
Current Assets: |
|||||||
Cash and Cash Equivalents |
$ | 11,899 | $ | 21,287 | |||
Marketable Securities |
41,413 | 39,811 | |||||
Accounts and Other Receivables |
170 | 324 | |||||
Prepaid and Other Current Assets |
1,202 | 1,525 | |||||
Total Current Assets |
54,684 | 62,947 | |||||
Property and Equipment, Net |
9,093 | 10,832 | |||||
Intangible Assets, Net |
24,923 | 26,836 | |||||
Other Assets |
329 | 363 | |||||
Goodwill |
8,965 | 8,965 | |||||
Total Assets |
$ | 97,994 | $ | 109,943 | |||
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||||
Current Liabilities: |
|||||||
Accounts Payable |
$ | 935 | $ | 931 | |||
Accrued Expenses |
7,008 | 4,936 | |||||
Current Portion of Long-Term Liabilities |
219 | 818 | |||||
Current Portion of Term Loan |
6,136 | 1,111 | |||||
Convertible Subordinated Debt |
— | 12,412 | |||||
Total Current Liabilities |
14,298 | 20,208 | |||||
Term Loan, less Current Portion |
9,008 | 8,889 | |||||
Other Long-Term Liabilities |
5,966 | 5,591 | |||||
Total Liabilities |
29,272 | 34,688 | |||||
Commitments and Contingent Liabilities (Notes 14 and 16) |
|||||||
Stockholders' Equity: |
|||||||
Convertible Preferred Stock, $.01 Par Value; 3,000,000 Shares Authorized; No Shares Issued and Outstanding at December 31, 2011 and 2010 |
— | — | |||||
Common Stock, $.001 Par Value; 297,000,000 Shares Authorized; 44,210,636 and 32,055,382 Shares Issued and Outstanding at December 31, 2011 and 2010, respectively |
44 | 32 | |||||
Additional Paid-In Capital |
271,032 | 232,679 | |||||
Accumulated Other Comprehensive Income |
2,652 | 2,751 | |||||
Accumulated Deficit |
(205,006 | ) | (160,207 | ) | |||
Total Stockholders' Equity |
68,722 | 75,255 | |||||
Total Liabilities and Stockholders' Equity |
$ | 97,994 | $ | 109,943 | |||
The accompanying notes are an integral part of the consolidated financial statements.
66
CELLDEX THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except per share amounts)
| |
Year Ended December 31, 2011 |
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
REVENUE: |
||||||||||
Product Development and Licensing Agreements |
$ | 110 | $ | 40,187 | $ | 5,662 | ||||
Contracts and Grants |
36 | 220 | 1,802 | |||||||
Product Royalties |
9,119 | 6,386 | 7,716 | |||||||
Total Revenue |
9,265 | 46,793 | 15,180 | |||||||
OPERATING EXPENSE: |
||||||||||
Research and Development |
32,439 | 27,650 | 26,169 | |||||||
Royalty |
9,119 | 12,077 | 8,397 | |||||||
Gain on Sale of Assets |
(50 | ) | (50 | ) | (604 | ) | ||||
General and Administrative |
9,243 | 10,428 | 17,119 | |||||||
Amortization of Acquired Intangible Assets |
1,913 | 3,143 | 949 | |||||||
Total Operating Expense |
52,664 | 53,248 | 52,030 | |||||||
Operating Loss |
(43,399 | ) | (6,455 | ) | (36,850 | ) | ||||
Investment and Other Income, Net |
396 | 5,259 | 248 | |||||||
Interest Expense |
(1,796 | ) | (1,337 | ) | (452 | ) | ||||
Net Loss Before Income Taxes |
(44,799 | ) | (2,533 | ) | (37,054 | ) | ||||
Income Tax Benefit |
— | — | 529 | |||||||
Net Loss |
$ | (44,799 | ) | $ | (2,533 | ) | $ | (36,525 | ) | |
Basic and Diluted Net Loss Per Common Share (See Note 2) |
$ | (1.13 | ) | $ | (0.08 | ) | $ | (1.84 | ) | |
Shares Used in Calculating Basic and Diluted Net Loss per Share (See Note 2) |
39,501 | 31,868 | 19,823 | |||||||
COMPREHENSIVE LOSS: |
||||||||||
Net Loss |
$ | (44,799 | ) | $ | (2,533 | ) | $ | (36,525 | ) | |
Other Comprehensive (Loss) Income: |
||||||||||
Foreign Currency Translation Adjustments |
(9 | ) | 2 | (12 | ) | |||||
Unrealized (Loss) Gain on Marketable Securities |
(90 | ) | 203 | (48 | ) | |||||
Comprehensive Loss |
$ | (44,898 | ) | $ | (2,328 | ) | $ | (36,585 | ) | |
The accompanying notes are an integral part of the consolidated financial statements.
67
CELLDEX THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(In thousands, except share amounts)
| |
Common Stock Shares |
Common Stock Par Value |
Additional Paid-In Capital |
Accumulated Other Comprehensive Income |
Accumulated Deficit |
Total Stockholders' Equity |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Balance at December 31, 2008 |
15,789,756 | $ | 16 | $ | 136,661 | $ | 2,606 | $ | (121,149 | ) | $ | 18,134 | |||||||
Shares Issued under Stock Option and Employee Stock Purchase Plans |
172,592 | — | 917 | — | — | 917 | |||||||||||||
Shares Issued in Connection with the CuraGen acquisition |
15,722,713 | 16 | 88,227 | — | — | 88,243 | |||||||||||||
Share-Based Compensation |
— | — | 3,058 | — | — | 3,058 | |||||||||||||
Foreign Currency Translation Adjustments |
— | — | — | (12 | ) | — | (12 | ) | |||||||||||
Unrealized Losses on Marketable Securities |
— | — | — | (48 | ) | — | (48 | ) | |||||||||||
Net Loss |
— | — | — | — | (36,525 | ) | (36,525 | ) | |||||||||||
Balance at December 31, 2009 |
31,685,061 | 32 | 228,863 | 2,546 | (157,674 | ) | 73,767 | ||||||||||||
Shares Issued under Stock Option and Employee Stock Purchase Plans |
370,321 | — | 1,014 | — | — | 1,014 | |||||||||||||
Share-Based Compensation |
— | — | 2,802 | — | — | 2,802 | |||||||||||||
Foreign Currency Translation Adjustments |
— | — | — | 2 | — | 2 | |||||||||||||
Unrealized Gains on Marketable Securities |
— | — | — | 203 | — | 203 | |||||||||||||
Net Loss |
— | — | — | — | (2,533 | ) | (2,533 | ) | |||||||||||
Balance at December 31, 2010 |
32,055,382 | 32 | 232,679 | 2,751 | (160,207 | ) | 75,255 | ||||||||||||
Shares Issued under Stock Option and Employee Stock Purchase Plans |
80,254 | — | 173 | — | — | 173 | |||||||||||||
Shares Issued in Connection with Cantor Agreement |
575,000 | 1 | 2,154 | — | — | 2,155 | |||||||||||||
Shares Issued in Underwritten Offering |
11,500,000 | 11 | 33,684 | — | — | 33,695 | |||||||||||||
Share-Based Compensation |
— | — | 2,342 | — | — | 2,342 | |||||||||||||
Foreign Currency Translation Adjustments |
— | — | — | (9 | ) | — | (9 | ) | |||||||||||
Unrealized Losses on Marketable Securities |
— | — | — | (90 | ) | — | (90 | ) | |||||||||||
Net Loss |
— | — | — | — | (44,799 | ) | (44,799 | ) | |||||||||||
Balance at December 31, 2011 |
44,210,636 | $ | 44 | $ | 271,032 | $ | 2,652 | $ | (205,006 | ) | $ | 68,722 | |||||||
The accompanying notes are an integral part of the consolidated financial statements.
68
CELLDEX THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| |
Year Ended December 31, 2011 |
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cash Flows From Operating Activities: |
||||||||||
Net Loss |
$ | (44,799 | ) | $ | (2,533 | ) | $ | (36,525 | ) | |
Adjustments to Reconcile Net Loss to Cash Used in Operating Activities: |
||||||||||
Depreciation and Amortization |
2,248 | 2,718 | 2,583 | |||||||
Amortization of Intangible Assets |
1,913 | 3,143 | 949 | |||||||
Amortization and Premium of Marketable Securities |
17 | (14 | ) | — | ||||||
Realized Loss (Gain) on Sales and Maturities of Marketable Securities |
5 | (4 | ) | 24 | ||||||
(Gain) Loss on Sale or Disposal of Assets |
(58 | ) | (38 | ) | (556 | ) | ||||
Stock-Based Compensation Expense |
2,342 | 2,802 | 3,058 | |||||||
Non-Cash Interest Expense |
307 | 728 | 181 | |||||||
Non-Cash Tax Benefit |
— | — | (529 | ) | ||||||
Changes in Operating Assets and Liabilities, Net of Acquisitions: |
||||||||||
Accounts and Other Receivables |
154 | 220 | 1,283 | |||||||
Prepaid and Other Current Assets |
241 | (546 | ) | 743 | ||||||
Other Assets |
34 | 5,592 | 722 | |||||||
Accounts Payable and Accrued Expenses |
2,076 | (1,193 | ) | (2,463 | ) | |||||
Deferred Revenue |
— | (39,382 | ) | (2,038 | ) | |||||
Other Liabilities |
(138 | ) | (1,865 | ) | 2,699 | |||||
Net Cash Used in Operating Activities |
(35,658 | ) | (30,372 | ) | (29,869 | ) | ||||
Cash Flows From Investing Activities: |
||||||||||
Cash Acquired in the CuraGen acquisition |
— | — | 51,654 | |||||||
Sales and Maturities of Marketable Securities |
51,003 | 42,383 | 2,674 | |||||||
Purchases of Marketable Securities |
(52,717 | ) | (56,522 | ) | (9,559 | ) | ||||
Acquisition of Property and Equipment |
(509 | ) | (2,100 | ) | (528 | ) | ||||
Proceeds from Sale or Disposal of Assets |
68 | 77 | 850 | |||||||
Net Cash (Used in) Provided by Investing Activities |
(2,155 | ) | (16,162 | ) | 45,091 | |||||
Cash Flows From Financing Activities: |
||||||||||
Net Proceeds from Stock Issuances |
36,023 | 1,014 | 668 | |||||||
Related Party Loan Due to Medarex |
— | — | (2,957 | ) | ||||||
Issuance of Term Loan |
5,000 | 10,000 | — | |||||||
Payment of Convertible Subordinated Debt |
(12,503 | ) | — | — | ||||||
Payment of Other Liabilities |
(86 | ) | (197 | ) | (176 | ) | ||||
Net Cash Provided by (Used in) Financing Activities |
28,434 | 10,817 | (2,465 | ) | ||||||
Effect of Exchange Rate Changes on Cash and Cash Equivalents |
(9 | ) | 2 | (12 | ) | |||||
Net (Decrease) Increase in Cash and Cash Equivalents |
(9,388 | ) | (35,715 | ) | 12,745 | |||||
Cash and Cash Equivalents at Beginning of Period |
21,287 | 57,002 | 44,257 | |||||||
Cash and Cash Equivalents at End of Period |
$ | 11,899 | $ | 21,287 | $ | 57,002 | ||||
Supplemental Disclosure of Non-Cash Flow Information |
||||||||||
Shares Issued to Executive Officers |
$ | — | $ | — | $ | 250 | ||||
Supplemental Disclosure of Cash Flow Information |
||||||||||
Cash Paid for Interest |
$ | 1,560 | $ | 604 | $ | 157 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
69
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(1) NATURE OF BUSINESS AND OVERVIEW
Celldex Therapeutics, Inc. (the "Company" or "Celldex") is a biopharmaceutical company focused on the development and commercialization of several immunotherapy technologies for the treatment of cancer and other difficult-to-treat diseases. The Company's lead drug candidates include rindopepimut (CDX-110), an immunotherapeutic vaccine in a pivotal Phase 3 study for the treatment of glioblastoma, CDX-011, an antibody-drug conjugate in a randomized Phase 2b study for the treatment of advanced breast cancer and CDX-1127, a therapeutic human antibody in a Phase 1 study for cancer indications. The Company has additional clinical and preclinical programs, including CDX-1401, an APC Targeting Technology™ program, CDX-301, an immune cell mobilizing agent, and CDX-1135, a molecule that inhibits a part of the immune system called the complement system. The Company's collaboration with GlaxoSmithKline (Glaxo) resulted in the commercialization of Rotarix, an oral human rotavirus vaccine.
At December 31, 2011, the Company had cash, cash equivalents and marketable securities of $53.3 million; working capital of $40.4 million; and a Term Loan balance of $15.1 million. The Company incurred a loss of $44.8 million for the year ended December 31, 2011. Net cash used in operations for the year ended December 31, 2011 was $35.7 million. In January 2012, the Company sold 2,450,000 shares of common stock under the Cantor Agreement and raised $8.5 million in net proceeds. In February 2012, the Company issued 10,500,000 shares of its common stock in an underwritten public offering. The net proceeds to the Company were $37.7 million, after deducting underwriting fees and estimated offering expenses. The Company has granted the underwriters a 30-day option to purchase up to an aggregate of 1,575,000 additional shares of common stock to cover overallotments, if any. The Company believes that the cash, cash equivalents and marketable securities at December 31, 2011, the $8.5 million raised under the Cantor Agreement in January and the $37.7 million raised in the underwritten public offering in February and interest income on invested funds, will be sufficient to meet estimated working capital requirements and fund planned operations for at least the next twelve months.
The Company raised net proceeds of $35.9 million during the year ended December 31, 2011 and $46.2 million during the two months ended February 29, 2012 from the sale of its common stock. During the next twelve months, the Company may take further steps to raise additional capital to meet its long-term liquidity needs. These capital raising activities may include, but may not be limited to, one or more of the following: the licensing of technology programs with existing or new collaborative partners, possible business combinations, issuance of debt, or the issuance of common stock or other securities via private placements or public offerings. While the Company continues to seek capital through a number of means, there can be no assurance that additional financing will be available on acceptable terms, if at all, and the Company's negotiating position in capital-raising efforts may worsen as existing resources are used. There is also no assurance that the Company will be able to enter into further collaborative relationships. Additional equity financing may be dilutive to the Company's stockholders; debt financing, if available, may involve significant cash payment obligations and covenants that restrict the Company's ability to operate as a business; and licensing or strategic collaborations may result in royalties or other terms that reduce the Company's economic potential from products under development. If the Company is unable to raise the funds necessary to meet its long-term liquidity needs, it may have to delay or discontinue the development of one or more programs, discontinue or delay on-going or anticipated clinical trials, license out programs earlier than expected, raise funds at significant discount or on other unfavorable terms, if at all, or sell all or a part of the Company.
70
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements reflect the operations of the Company and its wholly-owned subsidiary. All intercompany balances and transactions have been eliminated in consolidation. The Company operates in one segment, which is the business of development, manufacturing and commercialization of novel therapeutics for human health care.
Use of Estimates
The preparation of the consolidated financial statements in conformity with accounting principles generally accepted in the United States of America (U.S. GAAP) requires management to make estimates and use assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the dates of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with a maturity date of 90 days or less at the date of purchase to be cash equivalents. Cash equivalents consist principally of money market funds and debt securities.
Marketable Securities
The Company invests its excess cash balances in marketable securities including municipal bond securities, U.S. government agency securities, and high-grade corporate bonds. The Company classifies all of its marketable securities as current assets on the consolidated balance sheets because they are available-for-sale and available to fund current operations. Marketable securities are stated at fair value with their unrealized gains and losses included as a component of accumulated other comprehensive income (loss), which is a separate component of stockholders' equity, until such gains and losses are realized. If a decline in the fair value is considered other-than-temporary, based on available evidence, the unrealized loss is transferred from other comprehensive income (loss) to the consolidated statements of operations. Realized gains and losses are determined on the specific identification method and are included in investment and other income, net.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk primarily consist of cash, cash equivalents, marketable securities and accounts receivable. The Company invests its cash, cash equivalents and marketable securities in debt instruments and interest bearing accounts at major financial institutions in excess of insured limits. The Company mitigates credit risk by limiting the investment type and maturity to securities that preserve capital, maintain liquidity and have a high credit quality. The Company has not historically experienced credit losses from its accounts receivable and therefore has not established an allowance for doubtful accounts.
Revenue from Glaxo and Pfizer represented 98% and none for the year ended December 31, 2011, 14% and 85% for the year ended December 31, 2010 and 52% and 34% for the year ended December 31, 2009 of total Company revenue, respectively.
71
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Fair Value Measurements
The Company has certain assets and liabilities that are measured at fair value in the financial statements. The Company seeks to maximize the use of observable inputs (market data obtained from sources independent from the Company) and to minimize the use of unobservable inputs (the Company's assumptions about how market participants would price assets and liabilities) when measuring the fair value of its assets and liabilities. These assets and liabilities are classified into one of three levels of the following fair value hierarchy as defined by U.S. GAAP:
Level 1: Observable inputs such as quoted prices in active markets for identical assets or liabilities. An active market for an asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis.
Level 2: Observable inputs other than Level 1 prices, such as quoted prices in active markets for similar assets or liabilities and quoted prices for identical assets or liabilities in markets that are not active.
Level 3: Unobservable inputs based on the Company's assessment of the assumptions that market participants would use in pricing the asset or liability.
Property and Equipment
Property and equipment is stated at cost and depreciated over the estimated useful lives of the related assets using the straight-line method. Laboratory equipment and office furniture and equipment are depreciated over five years and computer equipment is depreciated over three years. Manufacturing equipment is amortized over seven to ten years. Leasehold improvements are amortized over the shorter of the estimated useful life or the non-cancelable term of the related lease, including any renewals that are reasonably assured of occurring. Property and equipment under construction is classified as construction in progress and is depreciated or amortized only after the asset is placed in service. Expenditures for maintenance and repairs are charged to expense whereas the costs of significant improvements which extend the life of the underlying asset are capitalized. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are eliminated and any resulting gain or loss is reflected in the Company's consolidated statements of operations.
The treatment of costs to construct property and equipment depends on the nature of the costs and the stage of construction. Costs incurred in the project planning, design, construction and installation phases are capitalized as part of the cost of the asset. The Company stops capitalizing these costs when the asset is substantially complete and ready for its intended use. For manufacturing property and equipment, the Company also capitalizes the cost of validating these assets for the underlying manufacturing process. The Company completes the capitalization of validation costs when the asset is substantially complete and ready for its intended use. Costs capitalized include incremental labor and fringe benefits, and direct consultancy services.
Business Combinations
The Company records the fair value of the consideration transferred to acquire a business to the tangible assets and identifiable intangible assets acquired and liabilities assumed on the basis of their fair values at the date of acquisition. The Company assesses the fair value of assets, including
72
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
intangible assets such as in-process research and development (IPR&D), using a variety of methods including present value models. Each asset is measured at fair value from the perspective of a market participant. The method used to estimate the fair values of IPR&D assets incorporates significant assumptions regarding the estimates a market participant would make in order to evaluate an asset, including a market participant's assumptions regarding the probability of completing IPR&D projects, which would require obtaining regulatory approval for marketing of the associated drug candidate; a market participant's estimates regarding the timing of and the expected costs to complete IPR&D projects; a market participant's estimates of future cash flows from potential product sales; and the appropriate discount rates for a market participant. Transaction costs and restructuring costs associated with the transaction are expensed as incurred.
Intangible Assets
IPR&D assets acquired in a business combination initially are recorded at fair value and accounted for as indefinite-lived intangible assets. These assets are maintained on the Company's consolidated balance sheets until either the project underlying them is completed or the assets become impaired. If a project is completed, the carrying value of the related intangible asset is amortized over the remaining estimated life of the asset beginning in the period in which the project is completed. If a project becomes impaired or is abandoned, the carrying value of the related intangible asset is written down to its fair value and an impairment charge is taken in the period in which the impairment occurs. IPR&D assets will be tested for impairment on an annual basis during the third quarter, or earlier if impairment indicators are present.
Intangible assets acquired in a business combination with a finite life are recorded at fair value and amortized over the greater of economic consumption or on a straight-line basis over their estimated useful life.
Goodwill
The difference between the purchase price and the fair value of assets acquired and liabilities assumed in a business combination is allocated to goodwill. Goodwill is evaluated for impairment on an annual basis during the third quarter, or earlier if impairment indicators are present.
Impairment of Long-Lived Assets
The Company evaluates the recoverability of its long-lived assets, including property and equipment, and intangible assets when circumstances indicate that an event of impairment may have occurred. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written-down to their estimated fair values.
Revenue Recognition
The Company recognizes revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the seller's price to the buyer is fixed or determinable; and collectability is reasonably assured.
73
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
The Company has entered into and in the future may enter into biopharmaceutical product development agreements with collaborative partners for the research and development of therapeutic drug products. The terms of the agreements may include nonrefundable signing and licensing fees, funding for research, development and manufacturing, milestone payments and royalties on any product sales derived from collaborations. These multiple element arrangements are analyzed to determine whether the deliverables can be separated or whether they must be accounted for as a single unit of accounting. In accounting for these transactions, the Company allocates revenue to the various elements based on their fair value. The fair value of a revenue generating element can be based on current selling prices offered by the Company or another party for current products or the Company's best estimate of a selling price for future products. Revenue allocated to an individual element is recognized when all other revenue recognition criteria are met for that element.
These collaborative and other agreements may contain milestone payments. Revenues from milestones, if they are considered substantive, are recognized upon successful accomplishment of the milestones. Determining whether a milestone is substantive involves judgment, including an assessment of the Company's involvement in achieving the milestones and whether the amount of the payment is commensurate to the Company's performance. If not considered substantive, milestones are initially deferred and recognized over the remaining performance obligation.
Payments received to fund certain research activities are recognized as revenue in the period in which the research activities are performed. Revenue from contracts and grants is recognized as the services are performed and recorded as effort is expended on the contracted work and billed to the government or the Company's contractual partner. Payments received in advance that are related to future performance are deferred and recognized as revenue when the research projects are performed. During the year ended December 31, 2010, the Company recorded $1.7 million to other income related to IRS Qualifying Therapeutic Discovery Grants because the grant arrangement was not part of the Company's on-going operations.
Product royalty revenue consists of payments received from licensees for a portion of sales proceeds from products that utilize the Company's licensed technologies and are recognized when the amount of and basis for such royalty payments are reported to the Company in accurate and appropriate form and in accordance with the related license agreement.
Research and Development Expenses
Research and development costs, including internal and contract research costs, are expensed as incurred. Research and development expenses consist mainly of clinical trial costs, manufacturing of clinical material, toxicology and other studies, personnel costs, depreciation, license fees and funding of outside research.
Clinical trial expenses include expenses associated with clinical research organizations (CRO). The invoicing from CROs for services rendered can lag several months. We accrue the cost of services rendered in connection with CRO activities based on our estimate of site management, monitoring costs, and project management costs. We maintain regular communication with our CROs to gauge the reasonableness of our estimates. Differences between actual clinical trial expenses and estimated clinical trial expenses recorded have not been material and are adjusted for in the period in which they become known.
74
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Patent Costs
Patent costs are expensed as incurred. Certain patent costs are reimbursed by the Company's product development and licensing partners. Any reimbursed patent costs are recorded as product development and licensing agreement revenues in the Company's financial statements.
Stock-Based Compensation
The Company records stock-based compensation expense for all stock-based awards made to employees and directors based on the estimated fair values of the stock-based awards expected to vest at the grant date and is adjusted, if necessary, to reflect actual forfeitures. Compensation expense for all stock-based awards to employees and directors is recognized using the straight-line method over the term of vesting or performance.
The Company records stock-based compensation expense for stock options granted to non-employees based on the fair value of the stock options which is re-measured over the vesting term resulting in periodic adjustments to stock-based compensation expense.
Foreign Currency Translation
Net unrealized gains and losses resulting from foreign currency translation are included in other comprehensive income (loss). At December 31, 2011 and December 31, 2010, accumulated other comprehensive income includes a net unrealized gain related to foreign currency translation of $2.6 million. In 2011, the Company's foreign subsidiary voluntarily liquidated in order to consolidate the Company's foreign operations into Celldex Therapeutics, Inc.
Income Taxes
The Company uses the asset and liability method to account for income taxes, including the recognition of deferred tax assets and deferred tax liabilities for the anticipated future tax consequences attributable to differences between financial statement amounts and their respective tax basis. Quarterly, the Company reviews its deferred tax assets for recovery. A valuation allowance is established when the Company believes that it is more likely than not that its deferred tax assets will not be realized. Changes in valuation allowances from period to period are included in the Company's tax provision in the period of change.
The Company records uncertain tax positions in the financial statements only if it is more likely than not that the uncertain tax position will be sustained upon examination by the taxing authorities. The Company records interest and penalties related to uncertain tax positions in income tax expense.
Comprehensive Loss
Comprehensive loss is comprised of net loss and certain changes in stockholders' equity that are excluded from net loss. The Company includes foreign currency translation adjustments and unrealized gains and losses on marketable securities in other comprehensive loss. In December 2011, the Company adopted a new U.S. GAAP accounting standard which requires an entity to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income, or in two separate but consecutive statements. This new standard eliminates the option to present components of other
75
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(2) SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
comprehensive income as part of the statement of equity. The adoption of this new standard did not have a material effect on the Company's operating results or financial position. The consolidated statements of operations and comprehensive loss reflect total comprehensive loss for the years ended December 31, 2011, 2010 and 2009. There were no significant reclasses to income during the years ended December 31, 2011, 2010 and 2009.
Net Loss Per Share
Basic net loss per common share is based upon the weighted-average number of common shares outstanding during the period, excluding restricted stock that has been issued but is not yet vested. Diluted net loss per common share is based upon the weighted-average number of common shares outstanding during the period plus additional weighted-average potentially dilutive common shares outstanding during the period when the effect is dilutive. The potentially dilutive common shares that have not been included in the net loss per common share calculations because the effect would have been anti-dilutive are as follows:
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
Stock options |
4,459,034 | 4,019,982 | 3,576,159 | |||||||
Convertible debt |
— | 353,563 | 353,563 | |||||||
Restricted stock |
6,000 | 9,338 | 16,000 | |||||||
|
4,465,034 | 4,382,883 | 3,945,722 | |||||||
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (FASB) or other standard setting bodies that are adopted by the Company as of the specified effective date. Unless otherwise discussed, the Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on the Company's financial position or results of operations upon adoption.
In September 2011, the FASB amended the guidance on the annual testing of goodwill for impairment. The amended guidance will allow companies to assess qualitative factors to determine if it is more-likely-than-not that goodwill might be impaired and whether it is necessary to perform the two-step goodwill impairment test required under current accounting standards. This guidance will be effective for the Company's fiscal year ending December 30, 2012. The Company does not expect the adoption of this new standard to have a material effect on its operating results or financial position.
In May 2011, the FASB issued a new accounting standard which clarifies the application of certain existing fair value measurement guidance and expands the disclosures for fair value measurements that are estimated using significant unobservable (Level 3) inputs. This new standard is effective on a prospective basis for annual and interim reporting periods beginning on or after December 15, 2011. The adoption of this standard is not expected to have a material impact on the Company's operating results or financial position.
76
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(3) BUSINESS COMBINATIONS
Acquisition of CuraGen Corporation (CuraGen)
On October 1, 2009, the Company acquired CuraGen, a former publicly-traded company. Following the CuraGen acquisition, the financial statements reflect the financial position, results of operation and cash flows of the combined companies. In connection with the CuraGen acquisition, effective October 1, 2009, the Company (i) issued 15,722,713 shares of common stock of the Company, or 0.2739 shares, in exchange for each share of outstanding CuraGen common stock, plus cash in lieu of fractional shares (the "CuraGen Exchange Ratio"), (ii) assumed the obligations under CuraGen's 2007 Stock Plan (the "CuraGen 2007 Plan") and each outstanding option to purchase common stock (a "CuraGen Stock Option") granted under the CuraGen 2007 Plan and (iii) assumed the $12.5 million in 4% convertible subordinated debt due February 15, 2011 (the "CuraGen Debt").
The transaction was accounted for under the acquisition method of accounting. The acquisition-date fair value of the consideration transferred consisted of the fair value of the Company's common stock issued of $85.4 million and fair value of CuraGen Stock Options that were attributed to precombination service of $2.9 million. Of the CuraGen Stock Options assumed, all but 1%, were immediately vested upon closing in accordance with the terms of the stock option agreements and employment agreements.
The Company recorded the fair value of acquired assets and liabilities to net tangible assets, intangible assets, goodwill and a severance obligation. The difference between the aggregate consideration transferred and the fair value of assets acquired and liabilities assumed was recorded to goodwill. This goodwill relates to synergies from the CuraGen acquisition and a deferred tax liability related to acquired IPR&D intangible assets. None of the goodwill is expected to be deductible for income tax purposes. The following table summarizes the fair values of the assets acquired and liabilities assumed at the acquisition date (in thousands):
Cash and cash equivalents |
$ | 51,654 | ||
Marketable securities |
18,638 | |||
Identifiable intangible assets: |
||||
IPR&D |
11,800 | |||
Amgen Amendment |
14,500 | |||
TopoTarget Agreement |
2,400 | |||
Other current and long-term assets |
756 | |||
Goodwill |
8,965 | |||
CuraGen Debt |
(11,503 | ) | ||
Net deferred tax liability |
(5,190 | ) | ||
Other assumed liabilities |
(3,778 | ) | ||
Total |
$ | 88,242 | ||
The values assigned to the intangible assets acquired, including the IPR&D, were determined based on fair market value using a risk adjusted discounted cash flow approach. The values assigned to IPR&D related to the development of CDX-011. At the date of acquisition, CDX-011 had not yet reached technological feasibility nor did it have any alternative future use. In December 2011, the Company completed enrollment in a randomized Phase 2b controlled study for CDX-011 in patients with heavily pre-treated, advanced breast cancer. The Company expects to incur approximately
77
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(3) BUSINESS COMBINATIONS (Continued)
$1.4 million in 2012 on the Phase 2b study. Estimated revenues from CDX-011 are expected to be generated by 2016. The remaining acquired intangible assets arising from the acquisition are being amortized on a straight line basis over their estimated lives. The net deferred tax liability primarily relates to the temporary differences associated with the IPR&D intangible assets, which are not deductible for tax purposes.
In connection with the CuraGen acquisition, effective October 1, 2009, the Company, CuraGen, and The Bank of New York Mellon (the "Trustee") amended the CuraGen Debt to provide that the CuraGen Debt shall be convertible into 353,563 shares of the Company's common stock at the rate of 28.27823 shares of the Company's common stock per $1,000 principal amount of notes, or $35.36 per share. The initial carrying value of the CuraGen Debt was accreted ratably, over the term of the CuraGen Debt, to $12.5 million due at maturity. Interest expense on the CuraGen Debt was $0.2 million, $1.2 million and $0.3 million for the years ended December 31, 2011, 2010 and 2009 and included $0.1 million, $0.7 million and $0.2 million in discount accretion, respectively. In February 2011, the Company paid the Trustee $12.8 million to satisfy all outstanding principal and accrued interest related to the CuraGen Debt.
CuraGen employees who did not receive offers of employment were terminated upon the consummation of the CuraGen acquisition. These employees were eligible for severance payments upon termination of employment under certain circumstances, including following the CuraGen acquisition. U.S. GAAP requires severance obligations that are incurred by the acquiree for the benefit of the acquirer to be recognized as an expense in the post-combination period. Because the offer of employment was at the option of the Company, the Company has deemed the CuraGen Severance to be at its benefit. Accordingly, the Company recorded $3.3 million and $0.9 million in CuraGen severance expenses to general and administrative and research and development, respectively, in the consolidated statements of operations for the year ended December 31, 2009. In addition, the Company incurred $2.9 million in acquisition-related expenses including investment banking, legal, accounting, and valuation services in the consolidated statements of operations for the year ended December 31, 2009.
(4) FAIR VALUE MEASUREMENTS
The following tables set forth the Company's financial assets subject to fair value measurements:
| |
As of December 31, 2011 |
Level 1 | Level 2 | Level 3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||||||||
Money market funds and cash equivalents |
$ | 11,038 | $ | 11,038 | — | — | |||||||
Marketable securities |
$ | 41,413 | — | $ | 41,413 | — | |||||||
|
$ | 52,451 | $ | 11,038 | $ | 41,413 | — | ||||||
78
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(4) FAIR VALUE MEASUREMENTS (Continued)
| |
As of December 31, 2010 |
Level 1 | Level 2 | Level 3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||||||||
Money market funds and cash equivalents |
$ | 10,975 | $ | 10,975 | — | — | |||||||
Marketable securities |
$ | 39,811 | — | $ | 39,811 | — | |||||||
|
$ | 50,786 | $ | 10,975 | $ | 39,811 | — | ||||||
There have been no transfers of assets or liabilities between the fair value measurement classifications. The Company's financial instruments consist mainly of cash and cash equivalents, marketable securities, short-term accounts receivable, accounts payable and debt obligations. The Company values its marketable securities utilizing independent pricing services which normally drive security prices from recently reported trades for identical or similar securities, making adjustments based on significant observable transactions. At each balance sheet date, observable market inputs may include trade information, broker or dealer quotes, bids, offers or a combination of these data sources. Short-term accounts receivable and accounts payable are reflected in the accompanying consolidated financial statements at cost, which approximates fair value due to the short-term nature of these instruments. Based on current market interest rates available to the Company for long-term liabilities with similar terms and maturities, the Company believes the fair value approximates the carrying value of the principal portion of the Term Loan and note payable at December 31, 2011.
(5) MARKETABLE SECURITIES
A summary of marketable securities is shown below:
| |
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||||||||
December 31, 2011 |
|||||||||||||
Marketable securities |
|||||||||||||
U.S. government and municipal obligations |
|||||||||||||
Maturing in one year or less |
$ | 19,993 | $ | 20 | $ | — | $ | 20,013 | |||||
Maturing after one year through three years |
10,808 | 122 | 6 | 10,924 | |||||||||
Total U.S. government and municipal obligations |
$ | 30,801 | $ | 142 | $ | 6 | $ | 30,937 | |||||
Corporate debt securities |
|||||||||||||
Maturing in one year or less |
$ | 5,817 | $ | 3 | $ | 4 | $ | 5,816 | |||||
Maturing after one year through three years |
4,730 | 2 | 72 | 4,660 | |||||||||
Total corporate debt securities |
$ | 10,547 | $ | 5 | $ | 76 | $ | 10,476 | |||||
Total marketable securities |
$ | 41,348 | $ | 147 | $ | 82 | $ | 41,413 | |||||
79
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(5) MARKETABLE SECURITIES (Continued)
| |
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Value |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||||||||
December 31, 2010 |
|||||||||||||
Marketable securities |
|||||||||||||
U.S. government and municipal obligations |
|||||||||||||
Maturing in one year or less |
$ | 14,836 | $ | 35 | $ | — | $ | 14,871 | |||||
Maturing after one year through three years |
11,428 | 103 | — | 11,531 | |||||||||
Total U.S. government and municipal obligations |
$ | 26,264 | $ | 138 | $ | — | $ | 26,402 | |||||
Corporate debt securities |
|||||||||||||
Maturing in one year or less |
$ | 11,798 | $ | 18 | $ | 2 | $ | 11,814 | |||||
Maturing after one year through three years |
1,594 | 1 | — | 1,595 | |||||||||
Total corporate debt securities |
$ | 13,392 | $ | 19 | $ | 2 | $ | 13,409 | |||||
Total marketable securities |
$ | 39,656 | $ | 157 | $ | 2 | $ | 39,811 | |||||
The marketable securities held by the Company were high investment grade and there were no marketable securities that the Company considered to be other-than-temporarily impaired as of December 31, 2011.
(6) PROPERTY AND EQUIPMENT, NET
Property and equipment include the following:
| |
December 31, 2011 | December 31, 2010 | |||||
|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||
Laboratory Equipment |
$ | 2,630 | $ | 2,942 | |||
Manufacturing Equipment |
1,999 | 1,418 | |||||
Office Furniture and Equipment |
1,314 | 1,299 | |||||
Leasehold Improvements |
13,280 | 13,244 | |||||
Construction in Progress |
240 | 703 | |||||
Total Property and Equipment |
19,463 | 19,606 | |||||
Less Accumulated Depreciation and Amortization |
(10,370 | ) | (8,774 | ) | |||
|
$ | 9,093 | $ | 10,832 | |||
Depreciation and amortization expense related to property and equipment was $2.2 million, $2.7 million and $2.6 million for the years ended December 31, 2011, 2010 and 2009, respectively.
80
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(7) INTANGIBLE ASSETS AND GOODWILL
Intangible assets, net of accumulated amortization, and goodwill are as follows:
| |
December 31, 2011 | December 31, 2010 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Estimated Life |
Cost | Accumulated Amortization |
Net | Cost | Accumulated Amortization |
Net | ||||||||||||||
| |
|
(In thousands) |
|||||||||||||||||||
Intangible Assets: |
|||||||||||||||||||||
IPR&D |
Indefinite | $ | 11,800 | $ | — | $ | 11,800 | $ | 11,800 | $ | — | $ | 11,800 | ||||||||
Amgen Amendment |
16 years | 14,500 | (2,018 | ) | 12,482 | 14,500 | (1,121 | ) | 13,379 | ||||||||||||
TopoTarget Agreement |
— | 2,400 | (2,400 | ) | — | 2,400 | (2,057 | ) | 343 | ||||||||||||
Core Technology |
4.5 – 11 years | 1,948 | (1,307 | ) | 641 | 1,948 | (1,040 | ) | 908 | ||||||||||||
Strategic Partner Agreement |
— | 630 | (630 | ) | — | 630 | (224 | ) | 406 | ||||||||||||
Total Intangible Assets |
$ | 31,278 | $ | (6,355 | ) | $ | 24,923 | $ | 31,278 | $ | (4,442 | ) | $ | 26,836 | |||||||
Goodwill |
Indefinite | $ | 8,965 | — | $ | 8,965 | $ | 8,965 | — | $ | 8,965 | ||||||||||
The estimated fair value attributed to the April 2008 agreement (TopoTarget Agreement) between the Company (as a successor to CuraGen) and TopoTarget A/S (TopoTarget) relates to the Company's rights under the TopoTarget Agreement to receive up to $6 million in either potential commercial milestone payments related to future net sales of Belinostat or 10% of any sublicense income received by TopoTarget (TopoTarget Payments). In February 2010, TopoTarget entered into a co-development and commercialization agreement for Belinostat with Spectrum Pharmaceuticals, Inc. which resulted in the Company's receipt of $3.0 million of the TopoTarget Payments. The Company recorded this cash receipt as Other Income for the year ended December 31, 2010.
During the year ended December 31, 2011, the Company recorded an impairment loss of $0.3 million in Strategic Partnership Agreement to amortization of intangible asset expense due to the Company's termination of rights to intellectual property underlying that Strategic Partnership Agreement. During the year ended December 31, 2010, the Company wrote-off $0.2 million in Core Technology to amortization of intangible asset expense. In January 2009, the Company entered into a purchase agreement with Lohmann Animal Health International ("LAHI") to sell its poultry vaccines assets to LAHI. Under the purchase agreement, LAHI paid an upfront fee of $0.8 million and agreed to pay potential milestone payments. During the year ended December 31, 2009, the Company recorded a gain of $0.6 million related to the LAHI Agreement based on the upfront fee less the net book value of the related asset.
Amortization expense for intangible assets was $1.9 million, $3.1 million and $0.9 million for the years ended December 31, 2011, 2010 and 2009, respectively. The estimated future amortization expense of intangible assets as of December 31, 2011, for the next five years is as follows (in thousands):
2012 |
$ | 1,089 | ||
2013 |
1,014 | |||
2014 |
1,014 | |||
2015 |
1,014 | |||
2016 |
994 |
81
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(8) ACCRUED EXPENSES
Accrued expenses include the following:
| |
December 31, 2011 |
December 31, 2010 |
|||||
|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||
Accrued Royalty and License Fees |
$ | 1,179 | $ | 826 | |||
Accrued Payroll and Employee Benefits |
2,145 | 1,925 | |||||
Accrued Research and Development Contract Costs |
3,035 | 1,218 | |||||
Accrued Professional Fees |
297 | 449 | |||||
Other Accrued Expenses |
352 | 518 | |||||
|
$ | 7,008 | $ | 4,936 | |||
(9) OTHER LONG-TERM LIABILITIES
Other long-term liabilities include the following:
| |
December 31, 2011 | December 31, 2010 | |||||
|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||
Deferred Rent |
$ | 435 | $ | 450 | |||
Severance |
— | 685 | |||||
Net Deferred Tax Liability |
4,661 | 4,661 | |||||
Deferred Income from Sale of Tax Benefits |
510 | — | |||||
Loan Payable |
527 | 581 | |||||
Other |
52 | 32 | |||||
Total |
6,185 | 6,409 | |||||
Less Current Portion |
(219 | ) | (818 | ) | |||
Long-Term Portion |
$ | 5,966 | $ | 5,591 | |||
Sale of Tax Benefits
In January 2011, the Company received approval from the New Jersey Economic Development Authority and agreed to sell New Jersey tax benefits worth $0.6 million (consisting of R&D tax credits) to an independent third party for $0.5 million. Under the agreement, the Company must maintain a base of operations in New Jersey for five years or the tax benefits must be paid back on a pro-rata basis based on the number of years completed. The Company is recognizing the $0.5 million over five years starting in 2012.
Loan Payable
In December 2003, the Company entered into a lease with the Massachusetts Development Finance Agency whereby the Company received a loan to finance the build-out of its manufacturing facility in Fall River, Massachusetts. Principal and interest payments on the loan are due monthly using
82
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(9) OTHER LONG-TERM LIABILITIES (Continued)
an amortization period of 15 years and interest accrues at a rate of 5.5% per annum. The Company is obligated to repay the following principal amounts for the loan as follows (in thousands):
2012 |
$ | 51 | ||
2013 |
58 | |||
2014 |
60 | |||
2015 |
63 | |||
2016 |
66 | |||
Thereafter |
229 | |||
Total |
$ | 527 | ||
(10) TERM LOAN
In December 2010, the Company entered into a Loan and Security Agreement (the "Loan Agreement") with MidCap Financial, LLC (MidCap) pursuant to which the Company borrowed $10 million (the "Term Loan") from MidCap. In March 2011, as the Company had anticipated, the Company amended the Loan Agreement and borrowed an additional $5 million from General Electric Capital Corporation (GECC) (collectively with MidCap, the "Lenders") to increase the amount owed under the Term Loan to $15 million. No additional advances are available under the Loan Agreement. The Term Loan accrues interest at a fixed annual interest rate equal to the greater of (i) the sum of (A) the LIBOR Rate (as defined in the Loan Agreement) plus (B) 6.25%; or (ii) a minimum rate of 9.50%. In September 2011, the Company exercised an option to extend the interest-only period by 6 months from October 1, 2011 to April 1, 2012. In March 2012, the Company amended the Loan Agreement to extend the maturity date from December 2013 to December 2014 in return for an upfront fee of $25,000 and an additional fee of $37,500 due upon repayment of the Term Loan in full.
Interest on the Term Loan is payable monthly and principal is due, as amended, in 34 equal consecutive monthly installments commencing on April 1, 2012. All unpaid principal and accrued interest with respect to the Term Loan is due and payable on the earlier of (A) December 30, 2014 or (B) the date that the Term Loan otherwise becomes due and payable under the terms of the Loan Agreement. The Company may prepay all, but not less than all, of the Term Loan subject to a prepayment premium of 1% in year three and 2% in year two of the original principal amount of the Term Loan. There is no prepayment premium if the loan is paid off early in year four. The Company is also obligated to make a payment fee of $0.5 million (the "Payment Fee") upon the earlier of (A) December 30, 2013 or (B) upon repayment of the Term Loan in full prior to December 30, 2013. The Company is accreting the Payment Fee ratably over the original term of the Term Loan to interest expense.
The obligations of the Company under the Loan Agreement are secured by a first priority lien upon and security interest in substantially all of the Company's existing and after-acquired assets, excluding its intellectual property assets. Under the Loan Agreement, the Company is subject to specified affirmative and negative covenants customary for financings of this type. The Loan Agreement provides that, upon the occurrence of certain specified events of default customary for financings of this type, the Company's obligations under the Loan Agreement may be automatically accelerated, whereupon the Company's obligations under the Loan Agreement shall be immediately due and payable. At December 31, 2011, the Company believes it is in compliance with the Loan Agreement.
83
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(10) TERM LOAN (Continued)
At December 31, 2011 and 2010, the Company had $0.2 million in capitalized deferred financing costs incurred in connection with the Term Loan and is amortizing these costs over the original term of the Term Loan to interest expense. Interest expense on the Term Loan including the accretion of the Payment Fee and amortization of the deferred financing costs was $1.6 million for the year ended December 31, 2011.
The Company is obligated to repay the following principal amounts for the Term Loan (including Payment Fee) as follows (in thousands):
2012 |
$ | 3,971 | ||
2013 |
5,744 | |||
2014 |
5,735 | |||
2015 |
— | |||
2016 |
— | |||
Thereafter |
— | |||
Total |
$ | 15,450 | ||
(11) STOCKHOLDERS' EQUITY
Common Stock
In April 2010, the Company filed a shelf registration statement with the Securities and Exchange Commission to register for sale any combination of the types of securities described in the shelf registration statement up to a dollar amount of $150 million. The shelf registration became effective on April 22, 2010.
In January 2011, the Company entered into a controlled equity offering sales agreement (the "Cantor Agreement") with Cantor Fitzgerald & Co. (Cantor) pursuant to which the Company may issue and sell up to 5,000,000 shares of its common stock from time to time through Cantor, acting as agent. The Company agreed to pay Cantor a commission of up to 5% of the gross proceeds from each sale and to reimburse Cantor for certain expenses incurred in connection with entering into the Cantor Agreement. The Cantor Agreement terminates upon the sale of all 5,000,000 shares or upon ten day notice by either Cantor or the Company. During the year ended December 31, 2011, the Company sold 575,000 shares of common stock under the Cantor Agreement and raised $2.2 million in net proceeds, after deducting commission and offering expenses. In January 2012, the Company sold 2,450,000 shares of common stock under the Cantor Agreement and raised $8.5 million in net proceeds. Under the terms of the Cantor Agreement, the Company will have the ability to sell up to 1,975,000 shares of its common stock upon the expiration or earlier waiver of the 90-day lock-up with the underwriters of the Company's recent offering in February 2012.
In May 2011, the Company issued 11,500,000 shares of its common stock in an underwritten public offering, including the underwriter's exercise of their full over-allotment option to purchase an additional 1,500,000 shares of common stock. The net proceeds to the Company were $33.7 million, after deducting underwriting fees and offering expenses.
84
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(11) STOCKHOLDERS' EQUITY (Continued)
In February 2012, the Company issued 10,500,000 shares of its common stock in an underwritten public offering. The net proceeds to the Company were $37.7 million, after deducting underwriting fees and estimated offering expenses. The Company has granted the underwriters a 30-day option to purchase up to an aggregate of 1,575,000 additional shares of common stock to cover overallotments, if any.
Convertible Preferred Stock
At December 31, 2011, the Company had authorized 3,000,000 shares of preferred stock all of which have been designated Class C Preferred Stock including 350,000 shares which have been designated Series C-1 Junior Participating Cumulative Preferred Stock (the "Series C-1 Preferred Stock").
Shareholder Rights Plan
The Company's Board has adopted a Shareholder Rights Plan, as set forth in the Shareholder Rights Agreement, as amended, between the Company and Computershare Trust Company, N.A., as Rights Agent (the "Rights Agreement"). Pursuant to the terms of the Rights Agreement, the Board declared a dividend distribution of one Preferred Stock Purchase Right (a "Right") for each outstanding share of the Company's common stock. Each Right, which expires in November 2014, entitles their holder to purchase from the Company one ten-thousandth of a share (a "Unit") of Series C-1 Preferred Stock at a cash exercise price of $35.00 per Unit, subject to adjustment. The Rights will trade separately from the common stock and will become exercisable only when a person or group has acquired 15% or more of the outstanding common stock or upon the commencement by a person or group of a tender offer that would result in such person or group acquiring 15% or more of the outstanding common stock other than as a result of repurchases of stock by the Company or certain inadvertent actions by a shareholder. In the event a person or group acquires 15% or more of the outstanding common stock each holder of a Right (except for any such person or group) would be entitled to receive upon exercise sufficient Units of Series C-1 Preferred Stock to equal a value of two times the exercise price of the Right. In the event the Company is acquired in a merger or other business combination transaction or if 50% or more of the Company's assets or earning power is sold, each holder of a Right (except for any such person or group described above) would receive upon exercise common stock of the acquiring company with a value equal to two times the exercise price of the Right.
85
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(12) STOCK-BASED COMPENSATION
The Company has the following stock-based compensation plans: the 2004 Employee Stock Purchase Plan (the "2004 ESPP Plan"), the 2008 Stock Option and Incentive Plan (the "2008 Plan"), Celldex Research's 2005 Equity Incentive Plan (the "Celldex Research 2005 Plan") and the CuraGen 2007 Plan. There are no shares available for future grant under the Celldex Research 2005 Plan and CuraGen 2007 Plan.
Employee Stock Purchase Plan
At December 31, 2011, a total of 62,500 shares of common stock are reserved for issuance under the 2004 ESPP Plan. Under the 2004 ESPP Plan, each participating employee may purchase up to 250 shares of common stock per year, through payroll deductions, at a purchase price equal to 85% of the lower of the fair market value of the common stock at either the beginning of the offering period or the applicable exercise date. During the years ended December 31, 2011 and 2010, the Company issued 6,627 and 5,897 shares under the 2004 ESPP Plan, respectively. At December 31, 2011, 44,382 shares were available for issuance under the 2004 ESPP Plan.
Employee Stock Option and Incentive Plan
The 2008 Plan permits the granting of incentive stock options (intended to qualify as such under Section 422A of the Internal Revenue Code of 1986, as amended), non-qualified stock options, stock appreciation rights, performance share units, restricted stock and other awards of restricted stock in lieu of cash bonuses to employees, consultants and non-employee directors.
At December 31, 2011, the 2008 Plan allowed for a maximum of 3,900,000 shares of common stock to be issued for grants of Stock Options and other Awards made prior to March 7, 2018 and grants of Incentive Stock Options made prior to October 20, 2017. The Company's board of directors determines the term of each option, option price, and number of shares for which each option is granted and the rate at which each option vests. Options generally vest over a period not to exceed four years. The term of each option cannot exceed ten years (five years for options granted to holders of more than 10% of the voting stock of the Company) and the exercise price of stock options cannot be less than the fair market value of the common stock at the date of grant (110% of fair market value for incentive stock options granted to holders of more than 10% of the voting stock of the Company). Vesting of all employee and non-employee director stock option awards is accelerated upon a change in control as defined in the 2008 Plan.
86
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(12) STOCK-BASED COMPENSATION (Continued)
A summary of stock option activity for the year ended December 31, 2011 is as follows:
| |
Shares | Weighted Average Exercise Price Per Share |
Weighted Average Remaining Contractual Term (In Years) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Options Outstanding at December 31, 2010 |
4,019,982 | $ | 6.93 | 6.6 | ||||||
Granted |
942,884 | $ | 2.90 | |||||||
Exercised |
(61,627 | ) | $ | 2.50 | ||||||
Canceled |
(442,205 | ) | $ | 7.51 | ||||||
Options Outstanding at December 31, 2011 |
4,459,034 | $ | 6.08 | 6.9 | ||||||
Options Vested and Expected to Vest at December 31, 2011 |
4,412,254 | $ | 6.11 | 6.9 | ||||||
Options Exercisable at December 31, 2011 |
2,818,895 | $ | 7.16 | 5.9 | ||||||
Shares Available for Grant under the 2008 Plan |
953,111 | |||||||||
The total intrinsic value of stock options exercised during the years ended December 31, 2011, 2010 and 2009 was $0.03 million, $1.0 million and $0.3 million, respectively. The weighted average grant-date fair value of stock options granted during the years ended December 31, 2011, 2010 and 2009 was $1.83, $2.82 and $5.29, respectively. The total fair value of stock options vested during the years ended December 31, 2011, 2010 and 2009 was $2.6 million, $2.5 million and $2.6 million, respectively.
The aggregate intrinsic value of stock options outstanding at December 31, 2011 was $0.03 million. The aggregate intrinsic value of stock options vested and expected to vest at December 31, 2011 was $0.03 million. As of December 31, 2011, total compensation cost related to non-vested employee and non-employee director stock options not yet recognized was approximately $3.3 million, net of estimated forfeitures, which is expected to be recognized as expense over a weighted average period of 2.5 years.
Restricted Stock
A summary of restricted stock activity under the 2008 Plan for the year ended December 31, 2011 is as follows:
| |
Shares | Weighted Average Grant Date Fair Value (per share) |
|||||
|---|---|---|---|---|---|---|---|
Outstanding and unvested at December 31, 2010 |
9,338 | $ | 3.96 | ||||
Granted |
12,000 | $ | 3.24 | ||||
Vested |
(15,338 | ) | $ | 3.68 | |||
Canceled |
— | — | |||||
Outstanding and unvested at December 31, 2011 |
6,000 | $ | 3.24 | ||||
87
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(12) STOCK-BASED COMPENSATION (Continued)
Valuation and Expenses Information
Stock-based compensation expense for the years ended December 31, 2011, 2010 and 2009 was recorded as follows:
| |
2011 | 2010 | 2009 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
|||||||||
Research and development |
$ | 1,412 | $ | 1,625 | $ | 1,383 | ||||
General and administrative |
930 | 1,177 | 1,675 | |||||||
Total stock-based compensation expense |
$ | 2,342 | $ | 2,802 | $ | 3,058 | ||||
The fair values of employee and non-employee director stock options granted during the years ended December 31, 2011, 2010 and 2009 were valued using the Black-Scholes option-pricing model with the following assumptions:
| |
Year Ended December 31, 2011 |
Year Ended December 31, 2010 |
Year Ended December 31, 2009 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
Expected stock price volatility |
68 – 70 | % | 65 – 67 | % | 65 – 68 | % | ||||
Expected option term |
6.0 Years | 6.2 Years | 5.5 – 6.3 Years | |||||||
Risk-free interest rate |
1.4 – 2.9 | % | 1.8 – 3.2 | % | 1.8 – 3.4 | % | ||||
Expected dividend yield |
None | None | None | |||||||
(13) SIGNIFICANT REVENUE ARRANGEMENTS
A summary of the Company's significant revenue contracts and arrangements follows:
GlaxoSmithKline plc (Glaxo) and Paul Royalty Fund II, L.P. (PRF)
In 1997, the Company entered into an agreement with Glaxo to collaborate on the development and commercialization of the Company's oral rotavirus strain and Glaxo assumed responsibility for all subsequent clinical trials and all other development activities. The Company's licensed-in the rotavirus strain that was used to develop Glaxo's Rotarix rotavirus vaccine in 1995 and owes a license fee of 30% to Cincinnati Children's Hospital Medical Center (CCH) on net royalties received from Glaxo. The Company is obligated to maintain a license with CCH with respect to the Glaxo agreement. The term of the Glaxo agreement is through the expiration of the last of the relevant patents covered by the agreement, although Glaxo may terminate the agreement upon 90 days prior written notice. The last relevant patent is scheduled to expire in December 2012. No additional milestone payments are due from Glaxo under the agreement.
In May 2005, the Company entered into an agreement whereby an affiliate of PRF purchased a 70% interest in the milestone payments and net royalties the Company will receive on worldwide sales of Rotarix. We have received a total of $60 million in milestone payments under the PRF agreement. No additional milestone payments are due from PRF under the agreement. The PRF agreement terminates in December 2012, unless otherwise extended. The Company's retained interests in Rotarix net royalties which were not sold to PRF are recorded as product royalty revenue and a corresponding amount that is payable to CCH is recorded as royalty expense. Product royalty revenue and royalty expense related to the Company's retained interest in Rotarix was $9.1 million, $6.4 million and $7.7 million for the years ended December 31, 2011, 2010 and 2009, respectively.
88
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(13) SIGNIFICANT REVENUE ARRANGEMENTS (Continued)
Pfizer Inc. (Pfizer)
In April 2008, the Company and Pfizer entered into a License and Development Agreement (the "Pfizer Agreement") under which Pfizer was granted an exclusive worldwide license to rindopepimut. Under the Pfizer Agreement, Pfizer made an upfront payment to the Company of $40 million and made a $10 million equity investment in the Company. The Pfizer Agreement also provided for reimbursement by Pfizer of all costs incurred by the Company in connection with the collaboration since the effective date.
The Company had determined that its performance obligations under this collaboration should be accounted for as a single unit of accounting. The Company's deliverables under this collaboration primarily included an exclusive license to rindopepimut, research and development services as required under the collaboration and participation in the joint clinical development committee. The Company had estimated that its expected performance period under the collaboration would be 9.5 years based on an assessment of the period over which the Company would have met its performance obligations under the collaboration. The $40 million up-front payment and research and development reimbursements were initially recorded as deferred revenue and recognized as revenue over this 9.5 year period.
In November 2010, the Pfizer Agreement was terminated (the "Pfizer Termination") and all rights to rindopepimut were returned to the Company. Pfizer did not provide a reason for termination. As a result of the Pfizer Termination, the Company recognized the remaining deferred revenue related to the Pfizer Agreement to product development and licensing agreement revenue during the year ended December 31, 2010. The Company recorded $39.9 million and $5.2 million in product development and licensing agreement revenue under the Pfizer Agreement during the years ended December 31, 2010 and 2009, respectively. The Company incurred and invoiced Pfizer reimbursable costs related to the Pfizer collaboration of $0.8 million and $3.2 million for the years ended December 31, 2010 and 2009, respectively. Effective with the Pfizer Termination, Pfizer is no longer funding the development of rindopepimut.
In connection with the Pfizer Agreement, the Company paid a total of $6.9 million in sublicense fees to Duke University and Thomas Jefferson University. The Company recorded these deferred sublicense fees to other assets in the consolidated balance sheets and was amortizing them to royalty expense over the 9.5-year performance period. As a result of the Pfizer Termination, the Company recognized the remaining deferred costs related to the Pfizer Agreement to royalty expense during year ended December 31, 2010. The Company recorded $5.7 million and $0.7 million in royalty expense related to these deferred sublicense fees during the years ended December 31, 2010 and 2009, respectively.
Rockefeller University (Rockefeller)
The Company has provided research and development support to Rockefeller on the development of their vaccine, DCVax-001, which the Company refers to as CDX-2401, aimed at providing protection from infection with HIV, the virus known to cause AIDS. Payments to the Company are made on a time and materials basis. The Company recorded grant revenue from Rockefeller of $0.2 million and $1.8 million for the years ended December 31, 2010 and 2009, respectively.
89
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(14) COLLABORATION AGREEMENTS
The Company has entered into license agreements whereby the Company has received licenses or options to license technology, specified patents or patent applications. The Company's licensing and development collaboration agreements generally provide for royalty payments equal to specified percentages of product sales, annual license maintenance fees, continuing patent prosecution costs and potential future milestone payments to third parties upon the achievement of certain developmental, regulatory and/or commercial milestones. Nonrefundable license fee expense was $1.4 million, $1.0 million and $0.7 million for the years ended December 31, 2011, 2010 and 2009, respectively.
Medarex, Inc., a subsidiary of Bristol-Myers Squibb (Medarex)
Medarex, a former related party, and the Company have entered into the following agreements, each of which was approved by a majority of its independent directors who did not have an interest in the transaction. These agreements include:
- •
- An Assignment and License Agreement, as amended, (Assignment and License Agreement) that provides for the assignment of
certain patent and other intellectual property rights and a license to certain Medarex technology related to the Company's APC Targeting Technology™ and an anti-mannose
receptor product; and
- •
- A Research and Commercialization Agreement, as amended, (Research and Commercialization Agreement) that provides the Company with certain rights to obtain exclusive commercial licenses to proprietary monoclonal antibodies raised against certain antigens utilizing the Medarex UltiMAb technology platform for generating antibodies.
Under the terms of the Assignment and License Agreement, the Company may be required to pay royalties in the low-single digits on any net product sale of a Licensed Royalty-Bearing Product or Anti-Mannose Product to Medarex until the later of (i) the expiration of the last to expire applicable patent and (ii) the tenth anniversary of the first commercial sale of such licensed product. Under the terms of the Research and Commercialization Agreement, the Company may be required to pay milestones of up to $7.0 million upon obtaining first approval for commercial sale in a first indication of a product containing a licensed antibody and royalty payments in the low- to mid-single digits on any net product sales to Medarex with respect to the development of any products containing such licensed antibodies until the later of (i) the expiration of the last to expire applicable patent and (ii) the tenth anniversary of the first commercial sale of such licensed product. In September 2010, the Company exercised an option under the Research and Commercialization Agreement, whereby it licensed from Medarex access to the UltiMab technology to develop and commercialize human antibodies to CD27, including CDX-1127.
Rockefeller University (Rockefeller)
In November 2005, the Company and Rockefeller entered into a license agreement for the exclusive worldwide rights to human DEC-205 receptor, with the right to sublicense the technology. The license grant is exclusive except that Rockefeller may use and permit other nonprofit organizations to use the human DEC-205 receptor patent rights for educational and research purposes. The Company may be required to pay milestones of up to $3.9 million upon obtaining first approval for commercial sale in a first indication of a product targeting the licensed receptor and royalty payments in the low- to mid-single digits on any net product sales to Rockefeller with respect to development and commercialization of the human DEC-205 receptor.
90
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(14) COLLABORATION AGREEMENTS (Continued)
Duke University Brain Tumor Cancer Center (Duke)
In September 2006, the Company and Duke entered into a license agreement that gave the Company access and reference to the clinical data generated by Duke and its collaborators in order for the Company to generate its own filing with the FDA relating to rindopepimut. The Company may be required to pay milestone of up to $1.0 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Duke with respect to development and commercialization of rindopepimut.
Ludwig Institute for Cancer Research (Ludwig)
In October 2006, the Company and Ludwig entered into an agreement for the nonexclusive rights to certain cancer tumor targets for use in combination with the Company's APC Targeting Technology. The term of the agreement is for ten years. The Company may be required to pay milestones of up to $1.0 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Ludwig with respect to development and commercialization of the technology licensed from Ludwig.
Alteris Therapeutics, Inc. (Alteris)
In October 2005, the Company completed the acquisition of the assets of Alteris, including the EGFRvIII molecule. The Company may be required to pay Alteris up to $5.0 million upon obtaining the first approval for commercial sale of a product containing EGFRvIII, including rindopepimut.
Thomas Jefferson University (TJU)
In connection with our acquisition of the assets of Alteris, the Company obtained the rights to two exclusive license agreements with TJU dated February 2003 related to the EGFRvIII tumor antigen. Under Under these licenses, the Company may be required to pay milestones of up to $3.0 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to TJU with respect to development and commercialization of rindopepimut.
3M Company
In June 2008, the Company and 3M Company entered into a license agreement for the exclusive worldwide rights to access 3M Company's proprietary Immune Response Modifier, Resiquimod™, (and additional Toll-Like Receptor 7/8 agonists (TLR)) for clinical study with the Company's proprietary APC Targeting Technology, for use as vaccine adjuvants, with the right to sublicense the technology. The Company may be required to pay milestones of up to $3.8 million upon obtaining first approval for commercial sale of each product using this vaccine adjuvant and royalty payments in the low-single digits on any net product sales to 3M Company with respect to development and commercialization of the technology licensed from 3M Company.
University of Southampton, UK (Southampton)
In November 2008, the Company entered into a license agreement with Southampton to develop human antibodies towards CD27, a potentially important target for immunotherapy of various cancers.
91
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(14) COLLABORATION AGREEMENTS (Continued)
The Company may be required to pay milestones of up to approximately $1.4 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Southampton with respect to development and commercialization of CDX-1127
Amgen Inc. (Amgen)
In March 2009, the Company entered into a license agreement with Amgen to expand its Precision Targeted Immunotherapy Platform by acquiring exclusive rights to CDX-301 and CD40 ligand (CD40L). CDX-301 and CD40L are immune modulating molecules that increase the numbers and activity of immune cells that control immune responses. The Company may be required to pay milestones of up to $1.3 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the low-single digits on any net product sales to Amgen with respect to development and commercialization of the technology licensed from Amgen, including CDX-301.
Seattle Genetics, Inc. (Seattle Genetics)
In connection with the CuraGen acquisition, the Company assumed the license agreement between CuraGen and Seattle Genetics whereby CuraGen acquired the rights to proprietary antibody-drug conjugate (ADC) technology for use with the Company's proprietary antibodies for the potential treatment of cancer. The Company may be required to pay milestones of up to $7.5 million upon obtaining first approval for commercial sale in a first indication and royalty payments in the mid-single digits on any net product sales to Seattle Genetics with respect to development and commercialization of the ADC technology, including CDX-011.
(15) INCOME TAXES
The components of income tax expense attributable to continuing operations consist of the following:
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2011 | 2010 | 2009 | |||||||
| |
(In thousands) |
|||||||||
Income tax benefit (provision): |
||||||||||
Federal |
$ | 16,204 | $ | 1,512 | $ | 12,750 | ||||
State |
3,131 | 779 | (1,757 | ) | ||||||
Foreign |
84 | 107 | 126 | |||||||
Expiration of Net Operating Losses and Research & Development Tax Credits |
(411 | ) | (13,924 | ) | (3,992 | ) | ||||
|
19,008 | (11,526 | ) | 7,127 | ||||||
Deferred tax valuation allowance |
(19,008 | ) | 11,526 | (6,598 | ) | |||||
|
$ | — | $ | — | $ | 529 | ||||
Included in the state tax provision above for the year ended December 31, 2009 is the effect of a rate decrease on the deferred tax asset and liabilities offset by a $0.5 million tax benefit due to non-cash tax consequences of the CuraGen acquisition.
92
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(15) INCOME TAXES (Continued)
A reconciliation between the amount of reported income tax and the amount computed using the U.S. Statutory rate of 34% follows:
| |
2011 | 2010 | 2009 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands) |
|||||||||
Pre-tax book income (loss) |
$ | (44,799 | ) | $ | (2,533 | ) | $ | (37,054 | ) | |
Loss at Statutory Rates |
(15,213 | ) | (838 | ) | (12,571 | ) | ||||
Research and Development Credits |
(1,736 | ) | (1,498 | ) | (1,456 | ) | ||||
State Taxes |
(3,131 | ) | (779 | ) | 1,757 | |||||
Other |
661 | 717 | 1,151 | |||||||
Expiration of Net Operating Losses and Research & Development Tax Credits |
411 | 13,924 | 3,992 | |||||||
Change in Valuation Allowance |
19,008 | (11,526 | ) | 6,598 | ||||||
Income tax (benefit) provision |
$ | — | $ | — | $ | (529 | ) | |||
Deferred tax assets and liabilities are recognized based on temporary differences between the financial reporting and tax basis of assets and liabilities using future expected enacted rates. A valuation allowance is recorded against deferred tax assets if it is more likely than not that some or all of the deferred tax assets will not be realized.
The principal components of the deferred tax assets and liabilities at December 31, 2011 and 2010, respectively, are as follows:
| |
December 31, 2011 |
December 31, 2010 |
|||||
|---|---|---|---|---|---|---|---|
| |
(In thousands) |
||||||
Gross Deferred Tax Assets |
|||||||
Net Operating Loss Carryforwards |
$ | 58,802 | $ | 50,228 | |||
Tax Credit Carryforwards |
20,285 | 17,998 | |||||
Deferred Expenses |
31,002 | 23,705 | |||||
Stock-based Compensation |
3,184 | 3,066 | |||||
Fixed Assets |
2,029 | 1,957 | |||||
Accrued Expenses and Other |
197 | 311 | |||||
|
115,499 | 97,265 | |||||
Gross Deferred Tax Liabilities |
|||||||
Other Acquired Intangibles |
(4,878 | ) | (5,615 | ) | |||
IPR&D Intangibles |
(4,661 | ) | (4,661 | ) | |||
Deferred License Costs and Other |
— | (37 | ) | ||||
|
(9,539 | ) | (10,313 | ) | |||
Total Deferred Tax Assets and Liabilities |
105,960 | 86,952 | |||||
Deferred Tax Assets Valuation Allowance |
(110,621 | ) | (91,613 | ) | |||
Net Deferred Tax Asset (Liability) |
$ | (4,661 | ) | $ | (4,661 | ) | |
93
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(15) INCOME TAXES (Continued)
The net deferred tax liability of $4.7 million at December 31, 2011 and 2010 relates to the temporary differences associated with the IPR&D intangible assets acquired in the CuraGen acquisition, which are not deductible for tax purposes.
As of December 31, 2011, the Company had the following federal net operating loss (NOL) carryforwards:
- •
- Prior to the merger of the Company and AVANT, $33.0 million was generated by the Company which expire at various
dates starting in 2023 and going through 2028;
- •
- Prior to the merger of the Company and AVANT, $132.4 million, net of expirations and utilization, was generated by
AVANT which expire at various dates starting in 2012 and going through 2028. NOLs of $0.8 million were utilized in 2009. $13.0 million and $12.1 million in NOLs expired in 2011
and 2010, respectively;
- •
- Following the merger of the Company and AVANT, $73.9 million was generated by the combined company which expire at
various dates starting in 2028 and going through 2031; and
- •
- Prior to its acquisition by the Company, $518.3 million was generated by CuraGen.
In general, an ownership change, as defined by Section 382 of the Internal Revenue Code, results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50 percentage points over a three-year period. Such ownership changes can significantly limit the amount of NOL carryforwards that may be utilized in future periods. The Company currently expects that it is not more likely than not that the CuraGen loss carryforwards may be utilized and, as such, no related asset has been recorded for such losses. The Company has not completed an analysis of losses generated by AVANT, however, the Company believes it is remote that $92 million of the AVANT loss carryforwards may be utilized in future periods and there may be substantial limitations on the Company's ability to use the remaining losses of $40.4 million. Following the merger of the Company and AVANT, the Company experienced changes in ownership as defined by Section 382 in June 2009 and December 2009. Further, prior to the AVANT merger, the Company as a stand alone company experienced a change in ownership in October 2007. As a result of the ownership change in October 2007, utilization of the Company's NOLs prior to October 2007 is subject to an annual limitation of $4.5 million on $28.3 million of NOLs generated before that date. As a result of the ownership changes in June 2009 and December 2009, there is an annual limitation amount of $6.0 million on $67.7 million NOLs. Any unused annual limitation may be carried over to later years, and the amount of the limitation may, under certain circumstances, be subject to adjustment if the fair value of the Company's net assets are determined to be below or in excess of the tax basis of such assets at the time of the ownership change, and such unrealized loss or gain is recognized during the five-year period after the ownership change.
Similar to the AVANT and CuraGen NOL carryforwards above, the Company believes that it is not more likely than not that federal and state research and development credits ("R&D credit") of $20.8 million and $14.4 million, respectively, will be utilized in the future periods. Further, the Company's ability to use the state NOL carryforwards of approximately $88.7 million and the remaining federal and state R&D credit carryforwards of approximately $14.0 million and $9.2 million, respectively, may be substantially limited. These state NOLs and federal and state credits expire at various dates starting in 2012 going through 2031. The Company has not yet completed a study of these
94
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(15) INCOME TAXES (Continued)
credits to substantiate the amounts. Until a study is completed, no amounts are being presented as an uncertain tax position.
Subsequent ownership changes, as defined in Section 382, could further limit the amount of net operating loss carryforwards and research and development credits that can be utilized annually to offset future taxable income.
The Company applies the authoritative guidance on account for and disclosure of uncertainty in income tax positions which requires the Company to determine whether an income tax position of the Company is more likely than not to be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position. For income tax positions meeting the more likely than not threshold, the tax amount recognized in the financial statements is reduced to the largest benefit that has a greater than fifty perfect likelihood of being realized upon the ultimate settlement with the relevant taxing authority. At December 31, 2011 and 2010, we had no unrecognized tax benefits. A full valuation allowance has been provided against our deferred tax assets and liabilities and, if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. Thus, there would be no impact to the consolidated balance sheet or statement of operations if an adjustment were required.
Massachusetts, New Jersey and Connecticut are the three states in which the Company primarily operates or has operated and has income tax nexus. The Company completed an examination by the Internal Revenue Service with respect to 2008 which resulted in no change to our 2008 tax return. The Company is not currently under examination by any other jurisdictions for any tax year.
The Company has evaluated the positive and negative evidence bearing upon the realizability of its net deferred tax assets, which are comprised principally of net operating loss carryforwards, capitalized R&D expenditures and R&D tax credit carryforwards. The Company has determined that it is more likely than not that it will not recognize the benefits of federal and state deferred tax assets and, as a result, a full valuation allowance was maintained at December 31, 2011 against the Company's net deferred tax assets.
(16) COMMITMENTS AND CONTINGENCIES
The Company has facility and equipment leases that expire at various dates through 2017. Certain of these facility leases contain renewal options, early termination provisions, and provisions that escalate the base rent payments and require the Company to pay common area maintenance costs (CAM) during the lease term. The Company entered into a letter of credit facility with a national U.S. financial institution which is collateralized by a security deposit for the leased facility in Phillipsburg, New Jersey. The Company recorded restricted cash related to this security deposit of $0.2 million to other assets in the consolidated balance sheets at December 31, 2011 and December 31, 2010.
95
CELLDEX THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
(16) COMMITMENTS AND CONTINGENCIES (Continued)
The following obligations for base rent and CAM costs under facility and other non-cancelable operating leases as of December 31, 2011 do not include the exercise of renewal terms or early termination provisions (in thousands):
2012 |
$ | 2,405 | ||
2013 |
2,414 | |||
2014 |
2,445 | |||
2015 |
2,503 | |||
2016 |
2,400 | |||
Thereafter |
1,014 | |||
Total minimum lease payments |
$ | 13,181 | ||
The Company's total rent and CAM expense for all facility leases was $2.5 million for the years ended December 31, 2011, 2010 and 2009.
(17) RETIREMENT SAVINGS PLAN
The Company maintains a 401(k) Plan which is available to substantially all employees. Under the terms of the 401(k) Plan, participants may elect to contribute up to 15% of their compensation, or the statutory prescribed limits. The Company may make 50% matching contributions on up to 4% of a participant's annual salary. Benefit expense for the 401(k) Plan was $0.2 million, $0.2 million and $0.1 million for the years ended December 31, 2011, 2010 and 2009, respectively.
(18) SELECTED QUARTERLY FINANCIAL DATA (Unaudited)
2011
|
Q1 2011 | Q2 2011 | Q3 2011 | Q4 2011 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands, except per share amounts) |
||||||||||||
Total revenue |
$ | 2,516 | $ | 1,952 | $ | 2,363 | $ | 2,433 | |||||
Net loss |
(10,059 | ) | (10,236 | ) | (11,772 | ) | (12,732 | ) | |||||
Basic and diluted net loss per common share |
(0.31 | ) | (0.27 | ) | (0.27 | ) | (0.29 | ) | |||||
2010
|
Q1 2010 | Q2 2010 | Q3 2010 | Q4 2010 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
(In thousands, except per share amounts) |
||||||||||||
Total revenue |
$ | 3,713 | $ | 2,951 | $ | 2,408 | $ | 37,721 | |||||
Net (loss) income |
(6,582 | ) | (9,525 | ) | (9,087 | ) | 22,661 | ||||||
Basic net (loss) income per common share |
(0.21 | ) | (0.30 | ) | (0.28 | ) | 0.71 | ||||||
Diluted net (loss) income per common share |
(0.21 | ) | (0.30 | ) | (0.28 | ) | 0.70 | ||||||
96
Item 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
Item 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
As of December 31, 2011, we evaluated, with the participation of our Chief Executive Officer and Chief Financial Officer, the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the "Exchange Act")). Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31, 2011. Our disclosure controls and procedures are designed to provide reasonable assurance that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within time periods specified by the SEC's rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Management's Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act as the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of our financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
- •
- pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and
dispositions of assets;
- •
- provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in
accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with the authorizations of management and directors; and
- •
- provide reasonable assurance regarding the prevention or timely detection of unauthorized acquisition, use or disposition of assets that could have a material effect on our financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework provided in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2011.
The effectiveness of our internal control over financial reporting as of December 31, 2011 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
97
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the three months ended December 31, 2011 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item 10 will be included in the definitive Proxy Statement for our 2012 Annual Meeting of Stockholders, or the 2012 Proxy Statement, under "Information Regarding the Current Directors and Executive Officers of Celldex Therapeutic, Inc.," "Section 16(a) Beneficial Ownership Reporting Compliance," "Code of Business Conduct and Ethics" and "The Board of Directors and Its Committees" and is incorporated herein by reference. If the 2012 Proxy Statement is not filed with the SEC within 120 days after the end of our most recent fiscal year, we will provide such information by means of an amendment to this Annual Report on Form 10-K.
Item 11. EXECUTIVE COMPENSATION
The information required by this Item 11 will be included in the 2012 Proxy Statement under "Executive Compensation," and "Compensation Committee Interlocks and Insider Participation," and is incorporated herein by reference. If the 2012 Proxy Statement is not filed with the SEC within 120 days after the end of our most recent fiscal year, we will provide such information by means of an amendment to this Annual Report on Form 10-K.
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item 12 will be included in the 2012 Proxy Statement under "Security Ownership of Certain Beneficial Owners and Management" and "Equity Compensation Plan Information" and is incorporated herein by reference. If the 2012 Proxy Statement is not filed with the SEC within 120 days after the end of our most recent fiscal year, we will provide such information by means of an amendment to this Annual Report on Form 10-K.
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item 13 will be included in the 2012 Proxy Statement under "Election of Directors" and "Approval of Related Person Transactions and Transactions with Related Persons" and is incorporated herein by reference. If the 2012 Proxy Statement is not filed with the SEC within 120 days after the end of our most recent fiscal year, we will provide such information by means of an amendment to this Annual Report on Form 10-K.
Item 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this Item 14 will be included in the 2012 Proxy Statement under "Independent Registered Public Accounting Firm" and is incorporated herein by reference. If the 2012 Proxy Statement is not filed with the SEC within 120 days after the end of our most recent fiscal year, we will provide such information by means of an amendment to this Annual Report on Form 10-K.
98
Item 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
- (A)
- The
following documents are filed as part of this Form 10-K:
- (1)
- Financial Statements:
The Financial Statements and Supplementary Data are included in Part II Item 8 of this report.
- (2)
- Financial Statement Schedules:
Schedules are omitted since the required information is not applicable or is not present in amounts sufficient to require submission of the schedule, or because the information required is included in the Consolidated Financial Statements or Notes thereto.
- (3)
- Exhibits:
| |
|
Incorporated by Reference to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Description | Form and SEC File No. |
Exhibit No. |
SEC Filing Date |
|||||
| Plan of Acquisition, Reorganization, Arrangement, Liquidation or Succession | |||||||||
| 2.1 | Agreement and Plan of Merger, dated as of October 19, 2007, by and among AVANT, Celldex Merger Corporation, and Celldex Therapeutics, Inc. | 8-K (000-15006) |
2.1 | 10/22/07 | |||||
2.2 |
Agreement and Plan of Merger, dated as of May 28, 2009, by and among Celldex Therapeutics, Inc., CuraGen Corporation and Cottrell Merger Sub, Inc. |
8-K (000-15006) |
2.1 |
5/29/09 |
|||||
Articles of Incorporation and By-Laws |
|||||||||
| 3.1 | Third Restated Certificate of Incorporation | S-4 (333-59215) |
3.1 | 7/16/98 | |||||
3.2 |
Certificate of Amendment of Third Restated Certificate of Incorporation |
S-4 (333-59215) |
3.1 |
7/16/98 |
|||||
3.3 |
Second Certificate of Amendment of Third Restated Certificate of Incorporation |
S-4 (333-59215) |
3.2 |
7/16/98 |
|||||
3.4 |
Third Certificate of Amendment of Third Restated Certificate of Incorporation |
10-Q (000-15006) |
3.1 |
5/10/02 |
|||||
3.5 |
Fourth Certificate of Amendment of Third Restated Certificate of Incorporation |
8-K (000-15006) |
3.1 |
3/11/08 |
|||||
3.6 |
Fifth Certificate of Amendment of Third Restated Certificate of Incorporation |
8-K (000-15006) |
3.2 |
3/11/08 |
|||||
3.7 |
Amended and Restated By-Laws as of March 14, 2007 |
10-K (000-15006) |
3.5 |
3/18/08 |
|||||
Instruments Defining the Rights of Security Holders |
|||||||||
| 4.1 | Specimen of Common Stock Certificate | S-3 (000-15006) |
4.17 | 4/5/10 | |||||
4.2 |
Shareholder Rights Agreement dated November 5, 2004 |
8-A (000-15006) |
4.1 |
11/8/04 |
|||||
99
| |
|
Incorporated by Reference to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Description | Form and SEC File No. |
Exhibit No. |
SEC Filing Date |
|||||
| 4.3 | Amendment No. 1 to Shareholder Rights Agreement dated October 19, 2007 | 8-A/A (000-15006) |
10.1 | 10/22/07 | |||||
4.4 |
Amendment No. 2 to Shareholder Rights Agreement dated March 7, 2008 |
8-A/A (000-15006) |
10.1 |
3/7/08 |
|||||
4.5 |
Certificate of Designations, Preferences and Rights of a Series of Preferred Stock classifying and designating the Series C-1 Junior Participating Cumulative Preferred Stock |
8-A (000-15006) |
3.1 |
11/8/04 |
|||||
Material Contracts—Leases |
|||||||||
| 10.1 | Commercial Lease Agreement of May 1, 1996 between the Company and Fourth Avenue Ventures Limited Partnership | 10-Q/A (000-15006) |
10.11 | 8/23/96 | |||||
10.2 |
Extension of Lease Agreement of May 1, 1997 between the Company and DIV Needham 53 LLC (successor in interest to Fourth Avenue Ventures Limited Partnership) dated as of August 23, 2001 |
10-K (000-15006) |
10.9 |
3/27/02 |
|||||
10.3 |
First Amendment to Lease by and between the Company and DIV Needham 53 LLC dated November 29, 2005 |
10-K (000-15006) |
10.40 |
3/16/06 |
|||||
*10.4 |
Lease Agreement, by and between the Company and the Massachusetts Development Finance Agency, dated as of December 22, 2003 |
10-Q (000-15006) |
10.1 |
4/30/04 |
|||||
10.5 |
First Amendment to Lease between Massachusetts Development Finance Agency and the Company dated March 17, 2005 |
10-K/A (000-15006) |
10.6 |
12/23/10 |
|||||
10.6 |
Second Amendment to Lease by and between the Company and the Massachusetts Development Finance Agency dated as of November 4, 2005 |
10-K (000-15006) |
10.41 |
3/16/06 |
|||||
10.7 |
Third Amendment to Lease between Massachusetts Development Finance Agency and the Company dated December 20, 2006 |
10-K/A (000-15006) |
10.7 |
12/23/10 |
|||||
10.8 |
Fifth Amendment to Lease between Massachusetts Development Finance Agency and the Company dated October 3, 2008 |
10-K/A (000-15006) |
10.8 |
12/23/10 |
|||||
10.9 |
Sixth Amendment to Lease between Massachusetts Development Finance Agency and the Company dated August 20, 2009 |
10-K/A (000-15006) |
10.9 |
12/23/10 |
|||||
10.10 |
Seventh Amendment to Lease by and between the Company and the Massachusetts Development Finance Agency dated as of June 22, 2010 |
10-Q (000-15006) |
10.1 |
8/5/10 |
|||||
10.11 |
Lease Agreement dated as of October 21, 2005 by and between Phillipsburg Associates, L.P. and the Company. |
S-4 (333-148291) |
10.10 |
1/18/08 |
|||||
100
| |
|
Incorporated by Reference to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Description | Form and SEC File No. |
Exhibit No. |
SEC Filing Date |
|||||
| 10.12 | First Amendment to Lease between Phillipsburg Associates, L.P. and the Company dated October 11, 2010 | 10-Q/A (000-15006) |
10.1 | 12/23/10 | |||||
10.13 |
Subordination, Non-Disturbance and Attornment Agreement between Bank of America and the Company dated October 11, 2010 |
10-Q/A (000-15006) |
10.2 |
12/23/10 |
|||||
Material Contracts—License, Collaboration, Supply and Distribution Agreements |
|||||||||
| *10.14 | License Agreement between the Company and SmithKline Beecham PLC dated as of December 1, 1997 | 10-K (000-15006) |
10.20 | 3/28/00 | |||||
10.15 |
Amendment Agreement, dated January 9, 2003, between the Company and SmithKline Beecham PLC |
10-K/A (000-15006) |
10.21 |
9/12/03 |
|||||
10.16 |
License and Clinical Trials Agreement, effective as of February 27, 1995, between the Company and the James N. Gamble Institute of Medical Research |
10-K/A (000-15006) |
10.23 |
9/12/03 |
|||||
10.17 |
Amendment Agreement between Cincinnati Children's Hospital Medical Center and the Company dated November 17, 2003 |
10-K/A (000-15006) |
10.10 |
12/23/10 |
|||||
10.18 |
License Agreement, dated as of November 25, 1988, by and among The Johns Hopkins University, Brigham and Women's Hospital and the Company |
10-K/A (000-15006) |
10.28 |
9/12/03 |
|||||
10.19 |
Purchase Agreement, dated as of May 16, 2005, by and between the Company and PRF Vaccine Holdings LLC |
8-K (000-15006) |
10.1 |
5/18/05 |
|||||
10.20 |
Amendment Agreement to Purchase Agreement between the Company and PRF Vaccine Holdings LLC, dated as of March 14, 2006 |
8-K (000-15006) |
10.1 |
3/15/06 |
|||||
*10.21 |
Exclusive License Agreement dated February 1, 2003 by and between Thomas Jefferson University and the Company |
S-4 (333-148291) |
10.1 |
1/18/08 |
|||||
*10.22 |
Amendment to License Agreement between Thomas Jefferson University and the Company dated March 27, 2008 |
10-K/A (000-15006) |
10.12 |
12/23/10 |
|||||
*10.23 |
License Agreement dated as of November 1, 2005 by and between The Rockefeller University and the Company |
S-4 (333-148291) |
10.2 |
1/18/08 |
|||||
*10.24 |
License Agreement dated September 1, 2006 by and between Duke University and the Company |
S-4 (333-148291) |
10.3 |
1/18/08 |
|||||
10.25 |
Amendment to License Agreement between Duke University and the Company dated April 2, 2008 |
10-K/A (000-15006) |
10.5 |
12/23/10 |
|||||
*10.26 |
License Agreement between Duke University, The Johns Hopkins University and the Company dated December 31, 2003 |
10-K/A (000-15006) |
10.11 |
12/23/10 |
|||||
101
| |
|
Incorporated by Reference to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Description | Form and SEC File No. |
Exhibit No. |
SEC Filing Date |
|||||
| *10.27 | Amendment to License Agreement between Duke University, The Johns Hopkins University and the Company dated April 2, 2008 | 10-K/A (000-15006) |
10.13 | 12/23/10 | |||||
*10.28 |
Assignment and License Agreement, as amended, dated April 6, 2004 by and among Medarex, Inc., GenPharm International, Inc. and the Company |
S-4 (333-148291) |
10.4 |
1/18/08 |
|||||
*10.29 |
Research and Commercialization Agreement, as amended, dated as of April 6, 2004 by and among Medarex, Inc., GenPharm International, Inc. and the Company |
S-4 (333-148291) |
10.5 |
1/18/08 |
|||||
*10.30 |
Supply Agreement dated August 18, 2006 by and between the Company and Biosyn |
S-4 (333-148291) |
10.9 |
1/18/08 |
|||||
*10.31 |
Research Collaboration and Commercialization Agreement effective October 20, 2006 between the Company and the Ludwig Institute for Cancer Research |
10-K (000-15006) |
10.45 |
3/2/09 |
|||||
*10.32 |
Vaccine Adjuvant License and Collaboration Agreement dated on May 30, 2008 between the Company and 3M Innovation Properties Company |
10-K (000-15006) |
10.46 |
3/2/09 |
|||||
*10.33 |
Exclusive Patent and Know-How License Agreement dated as of November 5, 2008 between the Company and the University of Southampton |
10-K (000-15006) |
10.47 |
3/2/09 |
|||||
*10.34 |
License and Assignment Agreement, between Amgen Inc. and the Company dated March 16, 2009 |
10-K/A (000-15006) |
10.1 |
12/23/10 |
|||||
*10.35 |
Collaboration Agreement dated June 18, 2004 between Seattle Genetics and CuraGen |
10-K (000-15006) |
10.27 |
3/12/10 |
|||||
*10.36 |
Second Restated Collaboration Agreement dated April 12, 2004 and amended October 19, 2004 between Abgenix Inc. and CuraGen |
10-K (000-15006) |
10.28 |
3/12/10 |
|||||
10.37 |
Amgen Letter Agreement, by and between CuraGen and Amgen Fremont, Inc. dated May 2, 2009 |
10-K (000-15006) |
10.29 |
3/12/10 |
|||||
*10.38 |
Transfer and Termination Agreement, dated as of April 21, 2008 by and between TopoTarget A/S and CuraGen |
10-K (000-15006) |
10.30 |
3/12/10 |
|||||
*10.39 |
License Agreement between Medarex and Company dated September 17, 2010 |
10-Q/A (000-15006) |
10.3 |
12/23/10 |
|||||
10.40 |
Master Services Agreement dated March 29, 2010 by and between the Company and Prologue Research International, Inc. (Prologue) |
10-Q (000-15006) |
10.2 |
11/3/11 |
|||||
10.41 |
Amendment to Master Services Agreement dated July 6, 2011 by and between the Company and Novella Clinical Inc. (formerly known as Prologue) |
10-Q (000-15006) |
10.3 |
11/3/11 |
|||||
102
| |
|
Incorporated by Reference to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Description | Form and SEC File No. |
Exhibit No. |
SEC Filing Date |
|||||
| Material Contracts—Stock Purchase, Financing and Credit Agreements | |||||||||
| 10.42 | Loan and Security Agreement, dated as of December 30, 2010, by and among Celldex Therapeutics, Inc., Celldex Research Corporation and MidCap Financial, LLC. | 8-K (000-15006) |
10.1.1 | 1/6/11 | |||||
10.43 |
Promissory Note issued by Celldex Therapeutics, Inc. and Celldex Research Corporation to MidCap Financial, LLC. |
8-K (000-15006) |
10.1.2 |
1/6/11 |
|||||
10.44 |
Joinder and First Loan Modification Agreement, dated as of March 7, 2011, by and among Celldex Therapeutics, Inc., Celldex Research Corporation, MidCap Funding V, LLC and General Electric Capital Corporation. |
10-K (000-15006) |
10.53 |
3/9/11 |
|||||
10.45 |
Promissory Note issued by Celldex Therapeutics, Inc. and Celldex Research Corporation to General Electric Capital Corporation. |
10-K (000-15006) |
10.54 |
3/9/11 |
|||||
10.46 |
Second Loan Modification Agreement, dated as of March 2, 2012, by and among Celldex Therapeutics, Inc., Celldex Research Corporation, MidCap Funding V, LLC and General Electric Capital Corporation. |
8-K (000-15006) |
10.1 |
3/7/12 |
|||||
10.47 |
Sales Agreement, dated January 6, 2011, between Celldex Therapeutics, Inc. and Cantor Fitzgerald & Co. |
8-K (000-15006) |
10.1.3 |
1/6/11 |
|||||
Material Contracts—Management Contracts and Compensatory Plans |
|||||||||
†10.48 |
2008 Stock Option and Incentive Plan, as amended and restated |
10-K (000-15006) |
10.34 |
3/12/10 |
|||||
†10.49 |
2004 Employee Stock Purchase Plan, as amended and restated |
10-K (000-15006) |
10.35 |
3/12/10 |
|||||
†10.50 |
Employment Agreement, dated January 6, 2009, by and between the Company and Avery W. Catlin |
8-K (000-15006) |
10.1 |
1/8/09 |
|||||
†10.51 |
Employment Agreement, dated January 6, 2009, by and between the Company and Thomas Davis, MD |
8-K (000-15006) |
10.2 |
1/8/09 |
|||||
†10.52 |
Employment Agreement, dated January 6, 2009, by and between the Company and Tibor Keler, Ph.D. |
8-K (000-15006) |
10.3 |
1/8/09 |
|||||
†10.53 |
Amended and Restated Employment Agreement, dated January 6, 2009, by and between the Company and Anthony S. Marucci. |
8-K (000-15006) |
10.4 |
1/8/09 |
|||||
†10.54 |
Amended and Restated Employment Agreement, dated July 1, 2011, by and between the Company and Ronald A. Pepin, Ph.D. |
8-K (000-15006) |
10.1 |
7/6/11 |
|||||
†10.55 |
Form of Stock Option Agreement |
8-K (000-15006) |
10.1 |
1/25/10 |
|||||
103
| |
|
Incorporated by Reference to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Description | Form and SEC File No. |
Exhibit No. |
SEC Filing Date |
|||||
| †10.56 | CuraGen 2007 Stock Incentive Plan, amended and restated | 10-K (000-15006) |
10.41 | 3/12/10 | |||||
†10.57 |
Form of Restricted Stock Award |
10-K (000-15006) |
10.42 |
3/12/10 |
|||||
21.0 |
List of Subsidiaries |
Filed herewith |
|||||||
23.1 |
Consent of PricewaterhouseCoopers LLP, an Independent Registered Public Accounting Firm |
Filed herewith |
|||||||
31.1 |
Certification of President and Chief Executive Officer |
Filed herewith |
|||||||
31.2 |
Certification of Senior Vice President and Chief Financial Officer |
Filed herewith |
|||||||
32 |
Section 1350 Certifications |
Furnished herewith |
|||||||
+101 |
XBRL Instance Document |
||||||||
+102 |
XBRL Taxonomy Extension Schema Document |
||||||||
+103 |
XBRL Taxonomy Extension Calculation Linkbase Document |
||||||||
+104 |
XBRL Taxonomy Extension Definition Linkbase Document |
||||||||
+105 |
XBRL Taxonomy Extension Label Linkbase Document |
||||||||
+106 |
XBRL Taxonomy Extension Presentation Linkbase Document |
||||||||
- *
- Confidential
treatment has been requested for certain provisions of this Exhibit pursuant to Rule 24b-2 promulgated under the Securities
Exchange Act of 1934, as amended.
- †
- Indicates
a management contract or compensation plan, contract or arrangement.
- +
- The XBRL information is being furnished and not filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and is not incorporated by reference into any registration statement under the Securities Act of 1933, as amended.
104
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| |
|
|
||
|---|---|---|---|---|
|
CELLDEX THERAPEUTICS, INC. | |||
|
By: |
/s/ ANTHONY S. MARUCCI |
||
Date |
Anthony S. Marucci | |||
March 8, 2012 |
President and Chief Executive Officer | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ ANTHONY S. MARUCCI Anthony S. Marucci |
President, Chief Executive Officer, and Director (Principal Executive Officer) |
March 8, 2012 | ||
/s/ AVERY W. CATLIN Avery W. Catlin |
Senior Vice President, Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) |
March 8, 2012 |
||
/s/ LARRY ELLBERGER Larry Ellberger |
Director, Chairman of the Board of Directors |
March 8, 2012 |
||
/s/ HERBERT J. CONRAD Herbert J. Conrad |
Director |
March 8, 2012 |
||
/s/ GEORGE O. ELSTON George O. Elston |
Director |
March 8, 2012 |
||
/s/ HARRY H. PENNER, JR. Harry H. Penner, Jr. |
Director |
March 8, 2012 |
||
/s/ TIMOTHY M. SHANNON, M.D. Timothy M. Shannon, M.D. |
Director |
March 8, 2012 |
||
/s/ KAREN L. SHOOS Karen L. Shoos |
Director |
March 8, 2012 |
105