Attached files
| file | filename |
|---|---|
| 8-K - FORM 8-K - SYNAGEVA BIOPHARMA CORP | d275119d8k.htm |
| EX-10.1 - AMENDED AND RESTATED REGISTRATION RIGHTS AGREEMENT - SYNAGEVA BIOPHARMA CORP | d275119dex101.htm |
| EX-99.2 - UNAUDITED FINANCIAL STATEMENTS OF PRIVATE SYNAGEVA - SYNAGEVA BIOPHARMA CORP | d275119dex992.htm |
Exhibit 99.1
SYNAGEVA’S BUSINESS
Overview
Synageva BioPharma Corp. (“Synageva”) is a clinical stage biopharmaceutical company focused on the discovery, development and commercialization of therapeutic products for patients with life-threatening rare diseases and unmet medical needs. Synageva’s management team is experienced in the development and commercialization of drugs for diseases with small patient populations, including clinical and translational research, working with payors to establish reimbursement and designing and building commercial organizations to reach highly specialized physicians to facilitate patient identification. The rare disease field has smaller patient populations and a close community of experts. Members of the Synageva management team have long-standing relationships with many of these experts based on prior leadership roles on most of the novel protein therapeutics marketed for ultra-rare diseases, including Cerezyme, Myozyme, Fabrazyme, Aldurazyme, and Soliris. Based on these relationships, these experts are working with Synageva on clinical development initiatives to advance SBC-102 and its other pipeline programs, which are a prerequisite to commercial success.
Product development in the rare disease space has several potential advantages over other areas of pharmaceutical development. Often rare diseases have no or very limited therapeutic alternatives and thus represent significant unmet medical need. Many rare diseases are a result of specific genetic mutations that impact the expression or function of a specific protein. As a result, animal models of these diseases are very relevant, and the translation of a product’s effects in animal models to effects in human patients has been very high. In addition, the genetic basis of many of these rare disorders can aid in a definitive diagnosis through the identification of specific mutations in human patients, and the care of these patients is usually handled by a small number of specialized physicians. The rapidly evolving field of human genetics and DNA sequencing technology is expected to further facilitate efforts to identify patients in the future. All of these factors can help provide comfort to regulatory agencies, and thus clinical development timelines for rare diseases are often significantly shorter and less expensive than for other more common diseases.
One factor that has been a significant limitation to the development of treatments for some of these rare diseases has been the ability to manufacture and supply proteins with the necessary post-translational modifications required for their uptake in appropriate cells to correct the protein deficiency using existing conventional protein manufacturing systems. Synageva believes that its proprietary protein production platform has the ability to address some of these difficulties and potentially provides a unique opportunity for Synageva in the rare disease space.
SBC-102, recombinant human lysosomal acid lipase (“LAL”), is Synageva’s most advanced program in the rare disease space. Synageva believes that this lead program will offer patients and health care practitioners a unique therapy to treat LAL Deficiency, which is a rare, devastating genetic disease that causes significant morbidity and mortality. There are currently no approved treatments for LAL Deficiency. LAL Deficiency is an autosomal recessive lysosomal storage disorder that is caused by a marked decrease or almost complete absence of the LAL enzyme. Although a single disease, LAL Deficiency presents as a clinical continuum with two major phenotypes, early onset LAL Deficiency, frequently referred to as Wolman Disease, and late onset LAL Deficiency, frequently referred to as CESD. The marked reduction of LAL activity in patients with LAL Deficiency leads to the accumulation of lipids in various tissues and cell types. This leads to a range of abnormalities, including enlargement of the liver and spleen, severe liver dysfunction, liver fibrosis, cirrhosis, and ultimately hepatic failure as well as severe malabsorption. Early onset LAL Deficiency is characterized by malabsorption, significant growth failure, and hepatic failure and is usually fatal within the first year of life. In late onset LAL Deficiency, liver involvement and type II hyperlipidemia (high cholesterol and triglycerides) dominate the clinical picture. Sandro Muntoni, an Italian physician who performed a genotyping study in Western Europe (published in 2007 in Arteriosclerosis, Thrombosis, and Vascular Biology) (the “Muntoni Study”), concluded that approximately 25 individuals per million lives are affected by the genetic abnormality associated with LAL Deficiency. This incidence rate is comparable to Gaucher Disease, Fabry Disease and Pompe Disease, which are also lysosomal storage disorders. In addition, as evidenced in a research article by Livia Pisciotta, et al., published in Mol Genetic Metab in 2009 (the “Pisciotta Article”), cases of late onset LAL Deficiency with at least one copy of a common genetic mutation that is associated with a majority of published cases of late onset LAL Deficiency have been reported from a number of different countries.
As a therapeutic approach, the medical value and long-term safety of enzyme replacement therapy established initially for Gaucher Disease has now been demonstrated for other lysosomal storage disorders, including Pompe Disease and Fabry Disease and MPS I, II and VI. In addition, it is generally accepted that preclinical proof of concept in the relevant genetic model for enzyme replacement therapies for lysosomal storage disorders is highly predictive of clinical effectiveness. Published examples of preclinical efficacy data in an animal model of a lysosomal storage disorder predicting clinical efficacy for products that have been subsequently approved include articles by Schull, et al., in Medical Sciences in 1994 regarding Aldurazyme (the “Schull Article”), Byers, et al., in Pediatrics Research in 2000 regarding Naglazyme (the “Byers Article”), Kikuchi, et al., in The Journal of Clinical Investigation in 1998 regarding Myozyme (the “Kikuchi Article”), and Ioannou, et al., in The American Journal of Human Genetics in 2001 regarding Fabrazyme (the “Ioannou Article”). In a rat model of LAL Deficiency that demonstrates many of the abnormalities of the human disease, including elevated liver enzymes, organomegaly, and growth failure leading to rapidly premature death, SBC-102 has demonstrated significant efficacy.
SBC-102 has been granted orphan drug designations (“Orphan Drug Designations”) by the U.S. Food and Drug Administration (“FDA”) and European Medicines Agency (“EMA”) and a Fast Track Designation by the FDA, and Synageva has received regulatory clearance in the U.S. and EU to conduct clinical trials in patients with LAL Deficiency. In February 2011, Synageva initiated Phase I/II clinical trials for SBC-102 and in May 2011 began enrolling patients into its Phase I/II studies to evaluate the safety and tolerability of SBC-102 in adult patients with liver dysfunction due to late onset LAL Deficiency and its Phase I/II study in children with growth failure due to early onset LAL Deficiency. SBC-102 is currently being dosed in humans as part of its global clinical development plan. Synageva also plans to initiate a global trial to evaluate the long-term safety and efficacy of SBC-102 in both adults and children with the late onset form of LAL Deficiency. Additionally, Synageva has initiated natural history studies in approximately 20 countries. These studies will be used to investigate and characterize key aspects of the clinical course of LAL Deficiency to inform the evaluation and care of affected patients and to provide a reference for pivotal studies of SBC-102 by identifying appropriate clinical endpoints.
In addition to the patients with LAL Deficiency enrolled in clinical trials, an infant with the disease has received treatment with SBC-102 on a compassionate use basis. The infant, who presented with growth failure, anemia and progressively increasing serum transaminases prior to beginning treatment, has now received weekly infusions for 4.5 months. The infant continues to receive SBC-102, and is demonstrating substantial improvements in growth rate, liver function tests (reduction in serum transaminases), and other disease-related abnormalities consistent with Synageva’s preclinical data for SBC-102.
In addition to SBC-102, Synageva is also progressing protein therapeutic programs for other rare diseases, which are at various stages of preclinical development. These include two enzyme replacement therapies for other lysosomal storage disorders and two programs for other rare life-threatening conditions. These protein therapeutic programs were selected based on scientific rationale, unmet medical need within the patient population, potential to substantially impact disease course, and strategic alignment with Synageva’s corporate and commercial efforts, including a potentially significant commercial opportunity. Synageva believes its other programs, SBC-103, 104, 105, and 106, also have the potential to present patients and health care practitioners with effective therapies to treat the rare and devastating diseases targeted by these programs. Like LAL Deficiency, these diseases are characterized by significant morbidity and mortality, currently have high unmet medical need and are conditions in which protein replacement treatment has the potential to have a meaningful impact on disease progression.
The most advanced of these additional programs is SBC-103, an enzyme replacement therapy for MPS IIIB. This enzyme is a recombinant form of human NAGLU. There is currently no approved therapy available for MPS IIIB. Similar to LAL Deficiency, MPS IIIB is an autosomal recessive lysosomal storage disease that may affect as many as eight individuals per million lives. While initially appearing unaffected, children born with MPS IIIB usually present with a slowing of development and/or behavioral problems around two years of age, followed by progressive intellectual decline and immobility with complete dependency on care providers. The life-span of an affected child does not usually extend beyond late teens to early twenties. Preliminary characterization of the enzyme produced using Synageva’s production platform demonstrates favorable uptake properties compared to previously published attempts to produce this enzyme using standard cell culture based approaches. Synageva has not yet received approval to market this product, and is not currently commercializing any other products.
Synageva’s business focus on products for rare diseases was established in 2008 with the appointment of Sanj K. Patel as President and Chief Executive Officer and his redirection of the company. This change represented a substantial shift in the business strategy of the original company, AviGenics, inc., which was founded in 1996 and was focused on the development of a novel protein production technology. Today, Synageva’s protein therapeutics are produced using this proprietary expression system based on over 15 years of research and clinical development. Synageva’s proprietary expression technology is highly capital efficient, thus allowing the rapid initiation and simultaneous advancement of multiple programs, enabling Synageva to cost-effectively perform initial investigations of programs in additional disease areas. The proprietary expression system has allowed Synageva to create the current pipeline of product candidates and provides the ongoing opportunity to renew and expand its portfolio with additional products at a rate that far outpaces what would be possible using existing protein expression technologies.
Synageva’s Strategy
Synageva’s goal is to develop and market therapies for patients with life-threatening rare diseases. Key elements of its strategy include:
| • | Advance SBC-102 toward regulatory approval and successful commercialization for the treatment of LAL Deficiency. No treatments are approved for this severe and life-threatening lysosomal storage disorder, and Synageva’s most important near-term objective is to advance its first-mover SBC-102 program toward regulatory approval and successful commercialization. Synageva has established a medical affairs effort to assist in identifying and enrolling patients and to build upon the existing connections it has within the physician community. This group will take on medical education support upon SBC-102 marketing approval. Synageva has also established an initial commercial team that, in addition to supporting the efforts of medical affairs, is focused on laying the groundwork necessary for the successful launch of an ultra-orphan product. Upon approval, Synageva expects to market SBC-102 globally with initial focus on the U.S., EU, Japan, and Brazil. |
| • | Continue to develop other pipeline programs. Synageva believes that it is important to maintain a diverse pipeline of product candidates to sustain future growth. Synageva plans to advance current pipeline programs and in addition has the ability to efficiently add new research programs targeting other rare life-threatening conditions. |
| • | Enter into strategic partnerships to generate capital and supplement Synageva’s internal resources. When establishing strategic collaborations, Synageva seeks to leverage its expertise in both product development for rare diseases and its production technology. Synageva will consider collaborations in rare diseases at appropriate stages in the drug development process. In addition, in selective instances Synageva may also consider collaborations to provide access to the company’s proprietary expression platform to other companies interested in the development of products outside of Synageva’s areas of interest. |
In order to achieve these strategic objectives, Synageva has, and will remain, focused on hiring and retaining a highly skilled management team that has extensive experience and specific skill sets relating to the selection, development and commercialization of therapies for life-threatening rare diseases. Members of the current management team have prior leadership responsibility for most of the protein therapeutics approved for ultra-rare diseases to date. Synageva intends to continue its efforts to build and expand this team as it aggressively grows its business. This strategy is intended to allow Synageva to build medical and financial value for patients and stockholders, respectively, by capitalizing on its translational and drug development capabilities and commercial expertise in rare diseases and by leveraging its robust manufacturing platform and supply capabilities.
SBC-102
Disease Overview
LAL Deficiency is an autosomal recessive lysosomal storage disorder caused by a marked decrease in the activity of the native LAL enzyme, which plays a key role in the degradation and metabolism of cholesteryl esters and triglycerides. The significant reduction of LAL activity in these patients leads to the accumulation of these lipids in various tissues and cell types. The organ most affected by the disease is the liver, leading to substantial enlargement and dysfunction with progression to liver failure. Other affected organs include the spleen, as well as the small intestine, leading to profound malabsorption. Similar to other lysosomal storage diseases such as Pompe Disease, LAL Deficiency is a single disease that presents as a clinical continuum with two major phenotypes, early onset LAL Deficiency, frequently referred to as Wolman Disease, and late onset LAL Deficiency, frequently referred to as CESD. Early onset LAL Deficiency is characterized by malabsorption, growth failure, and hepatic failure and is usually fatal within the first year of life. In late onset LAL Deficiency, liver involvement and type II hyperlipidemia dominate the clinical picture. As described above, the Muntoni Study concluded that approximately 25 individuals per million lives are affected by known genetic abnormalities that lead to LAL Deficiency. In addition, as evidenced in the Pisciotta Article, cases of late onset LAL Deficiency with at least one copy of a common genetic mutation that is associated with a majority of published cases of late onset LAL Deficiency have been reported from a number of different countries. This incidence rate is comparable to that of Gaucher Disease, Fabry Disease, and Pompe Disease, which are other lysosomal storage disorders for which enzyme replacement therapies have been approved and successfully marketed. There are currently no approved treatments for LAL Deficiency.
SBC-102 has demonstrated efficacy in a highly relevant disease model and it is generally accepted that proof of concept for enzyme replacement therapies for lysosomal storage disorders is highly predictive of clinical effectiveness. For most other diseases, preclinical models are recognized as having limited value in assessing the probability of clinical efficacy in humans. The situation is different in enzyme replacement therapies for lysosomal storage disorders. In these storage diseases, where animal models have been developed, the missing protein performs a similar function in the animal as it does in humans and replacement of the missing enzyme corrects the disease-related abnormalities. Published examples of preclinical efficacy data in an animal model of a lysosomal storage disorder predicting clinical efficacy for products that have been subsequently approved include the Schull Article, the Byers Article, the Kikuchi Article, and the Ioannou Article. SBC-102 has also demonstrated such efficacy in a highly relevant in vivo disease model and is currently being dosed in humans as part of its global clinical development plan. In addition, the medical value and long-term safety of enzyme replacement therapy first established for Gaucher Disease has now been extended to a broader range of disorders, including Pompe Disease, Fabry Disease, and MPS I, II, and VI.
SBC-102 has been granted Orphan Drug Designations by the FDA and EMA, and Synageva has received regulatory clearance in the U.S. and EU to conduct clinical trials in patients with late and early onset LAL Deficiency. Additionally, due to the severity of LAL Deficiency, the FDA granted SBC-102 Fast Track Designation which allows for an expedited regulatory review for the product. In February 2011, Synageva initiated Phase I/II clinical trials for SBC-102 and in May 2011 began enrolling patients in two Phase I/II studies to evaluate the safety and tolerability of SBC-102 in adult patients with liver dysfunction due to late onset LAL Deficiency and in children with growth failure due to early onset LAL Deficiency. Synageva also plans to initiate a global trial to evaluate the long-term safety and efficacy of SBC-102 in both adults and children with the late onset form of LAL Deficiency. Additionally, Synageva has initiated natural history studies in approximately 20 countries for LAL Deficiency. These studies will be used to investigate and characterize key aspects of the clinical course of the disease to inform the evaluation and care of affected patients and to provide a reference for pivotal studies of SBC-102 by identifying appropriate clinical endpoints.
Early Onset LAL Deficiency
According to an article by Meikle, PJ, et al., in the Journal of the American Medical Association in 1999, early onset LAL Deficiency presents shortly after birth with predominant gastrointestinal and liver involvement and has an estimated incidence of approximately two individuals per million lives. This form of the disease is the most rapidly fatal of LAL Deficiency and is characterized by growth failure, malabsorption, steatorrhea, and liver enlargement. These patients usually die within the first year of life. In this form of LAL Deficiency, growth failure, which is caused by a number of mechanisms, including a marked reduction in the capacity of the gastrointestinal tract to absorb nutrients, is the predominant clinical feature and a key contributor to the early mortality. Hepatic involvement, as evidenced by liver enlargement and elevation of transaminases, is also common and is very similar to that described in other patients across the LAL Deficiency continuum.
In the absence of approved therapies for LAL Deficiency, supportive therapies are used in an attempt to mitigate some of the effects of this rapidly fatal disease. Although some stabilization of the clinical condition has been described with nutritional support, these interventions are not believed to substantially modify the outcome in affected patients. As there is presently no effective treatment (including enzyme replacement therapy) for LAL Deficiency, patients with early onset LAL Deficiency are sometimes offered experimental therapy with hematopoietic stem cell transplantation. Based on information, presented in a chapter by Assmann, G. and Seedorf, U. in the The Metabolic and Molecular Bases of Inherited Disease edited by A. Beaudet et al., early onset LAL Deficiency remains almost universally fatal.
Late Onset LAL Deficiency
Late onset LAL Deficiency presents with predominant liver involvement and type II hyperlipidemia (high cholesterol and triglycerides), and is the more common form of LAL Deficiency with an estimated incidence of 25 individuals per million lives. The liver and spleen are the most severely affected organs with marked organ enlargement, elevation of transaminases and severe liver fibrosis progressing to cirrhosis. Cardiovascular involvement is characterized by dyslipidemia (high cholesterol, high triglycerides and low high-density lipoprotein) with early onset vascular disease due to accumulation of lipid deposits in artery walls. The presentation of late onset LAL Deficiency is highly variable with some patients going undiagnosed until complications manifest in late adulthood, while others can present with liver dysfunction in early childhood. Late onset LAL Deficiency is associated with significant ill health and while the natural history is not well described, there is evidence that life expectancy is reduced with premature death due to liver, cardiovascular and vascular complications including strokes. This evidence includes case reports described by Beaudet, A, et al., in The Journal of Pediatrics in 1977, Cagel, et al., in American Journal of Medical Genetics in 1986, Elleder, et al., in Journal of Hepatology in 2000, and Riva, et al., in Digestive and Liver Disease in 2008.
Although no approved therapies are available for treatment of LAL Deficiency, palliative care is sometimes used to try to mitigate some of the effects of the disease. These treatments are mainly focused on control of plasma lipid levels through diets that exclude foods rich in cholesterol and triglycerides and suppression of cholesterol synthesis and apolipoprotein B production through administration of statins and other lipid lowering therapies. As described in the medical literature, including in the case study by Di Bisceglie, A., et al., published in Hepatology, Volume 11, Issue 5, 1990, although some improvement may be seen in serum lipid levels, the underlying disease manifestations persist and disease progression still occurs. As the disease progresses, this can lead to a need for liver transplantation or death.
SBC-102 Pharmacology
SBC-102 is a recombinant human LAL. This enzyme is responsible for the metabolism of cholesteryl esters and triglycerides that are delivered to lysosomes by a variety of routes, including low-density lipoprotein receptor mediated endocytosis. SBC-102 is produced by recombinant DNA technology in egg white (“EW”) using Synageva’s proprietary protein manufacturing platform. The protein contains glycan structures which are specifically recognized and internalized via receptors into key target cells. LAL Deficiency has parallels with Gaucher Disease, as both require effective macrophage targeting. Unlike certain other approved enzyme replacement therapies, however, SBC-102 does not require additional processes during manufacturing to either modify glycan synthesis or remove terminal glycans to allow for the correct glycan ligands for macrophage uptake. In addition, levels of mannose-6-phosphate that enable efficient uptake into other cell types are higher in SBC-102 than described in other forms of recombinant LAL produced using cell culture based manufacturing platforms. In November 2010, Synageva reported data from preclinical studies of SBC-102 demonstrating uptake and localization to lysosomes into key cell types, including macrophages and fibroblasts.
Preclinical Development
Synageva reported data from preclinical studies of SBC-102 at the American Society of Human Genetics meeting in November 2010, which demonstrated SBC-102’s efficacy in a disease model of LAL Deficiency. SBC-102 reduced lipid substrate levels in diseased tissues and corrected disease-related abnormalities associated with LAL enzyme deficiency, including growth failure and
liver pathology. The liver histology and gross pathology pictures shown below in Figure 1 are representative of the data presented. The photomicrograph of the untreated liver on the left shows marked disruption of liver structure due to the abnormal accumulation of lipids in the Kupffer cells. The significant expansion of these cells results in the pale, mottled appearance of the tissue. There is also damage to the tissue around the periportal tract resulting in fibrosis. The tissue abnormalities are also seen in the picture of the whole untreated liver on the left. It is enlarged and pale in color due to the accumulated lipids. In the photomicrograph of the SBC-102 treated liver on the lower right, the pathological abnormalities have been corrected and normal liver architecture has been restored. This is demonstrated by the smooth, uniform appearance of the tissue. The picture of the whole SBC-102 treated liver on the right is consistent with the histological findings with a normal appearance due to the reduction in the lipid accumulation.
|
| ||
| Untreated liver histology and gross pathology showing tissue abnormality due to lipid accumulation |
SBC-102 treated liver histology and whole organ showing normalization of tissue structure | |
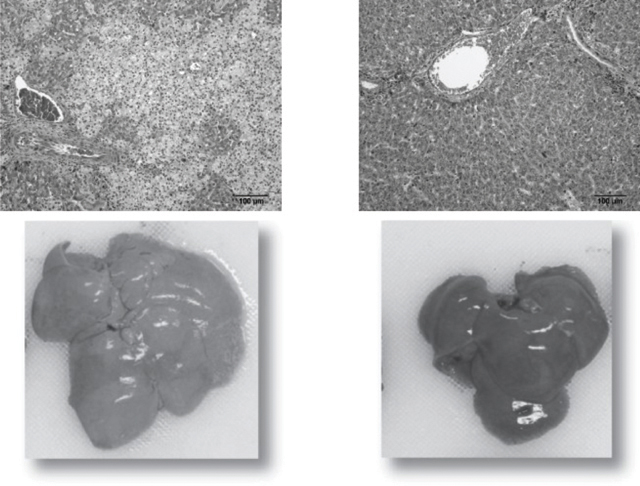
Figure 1. Effect of SBC-102 on liver morphology
Additional data was reported at the European Society for the Study of Liver Disease meeting in March 2011 and the European Society for Pediatric Gastroenterology, Hepatology and Nutrition meeting in May 2011. These studies reported that weekly (“qw”) and every other week (“qow”) administration of SBC-102 improved growth, decreased LAL substrate content in affected organs and normalized liver pathology in association with decreases in liver size and in serum transaminases and reversal of liver histopathologic findings. As shown in the figures below, detailed dose response analysis in a LAL deficient animal model has established a range of effective qw and qow doses supporting dose selection in clinical studies. These data demonstrate that doses equal to or greater than 1 mg/kg qow are highly effective in reducing lipid accumulation in the liver, which is the organ predominantly affected in late onset LAL Deficiency. For growth failure, which is characteristic of patients with early onset LAL Deficiency, maximum efficacy is seen at doses equal to or greater than 3 mg/kg qw.
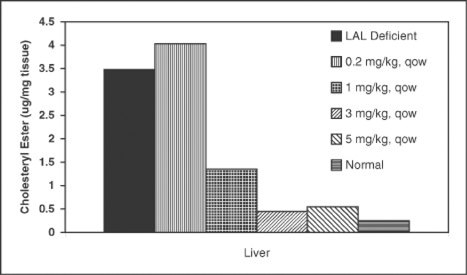
Figure 2. SBC-102 dose dependent reduction of cholesteryl esters from the liver in a LAL deficient animal model
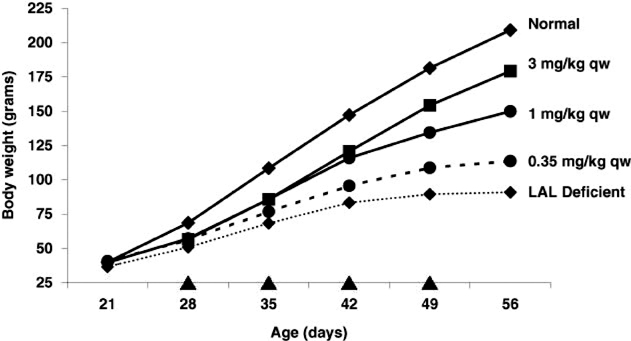
Figure 3. SBC-102 dose dependent restoration of growth in a LAL deficient animal model, qw dosing
These studies establish proof of concept for SBC-102 as an enzyme replacement therapy for LAL Deficiency. In contrast to preclinical testing of most other experimental therapies, it is generally accepted that efficacy for this class of therapies in lysosomal storage disorder disease models is highly predictive of clinical effectiveness. In these storage diseases, where animal models have been developed, the missing protein performs a similar function in the animal as it does in humans and replacement of the missing enzyme corrects the disease-related abnormalities. Published examples of preclinical efficacy data in an animal model of a lysosomal storage disorder predicting clinical efficacy for products that have been subsequently approved include the Schull Article, the Byers Article, the Kikuchi Article, and the Ioannou Article.
Synageva has also conducted toxicology studies to support initiation of human dosing. There were no meaningful toxicological findings in four-week repeat dose toxicology studies in the rat and monkey administered intravenous (“IV”) infusions of SBC-102 at doses up to 50 mg/kg once weekly. In a six-month repeated dose toxicity study in monkeys administered once-weekly IV infusions of SBC-102 at doses of 3 mg/kg, 10 mg/kg or 30 mg/kg (five males and five females per dose group), or placebo infusions (five males, five females), SBC-102 was well tolerated in monkeys at dose levels up to 30mg/kg per dose, which suggests a lack of safety concerns for long-term dosing with SBC-102.
Clinical Development
Synageva is pursuing a development strategy for SBC-102 that includes clinical trials in patients with both early and late onset LAL Deficiency. The overall goals of the program are to assess safety and tolerability in a broad population of patients, including infants, children, and adults, and to demonstrate clinically meaningful effects on the medical complications of LAL Deficiency. Consistent with study protocols for marketed products for other rare diseases such as Fabrazyme and Myozyme, Synageva anticipates that the patient numbers required for the SBC-102 clinical program will be small. Synageva has gained Fast Track Designation for the Biologic License Application (“BLA”) for SBC-102 that it intends to file with the FDA if clinical trials are successful.
In December 2010, Synageva filed an IND application with the FDA for late onset LAL Deficiency and submitted clinical trial applications for both late and early onset LAL Deficiency with the UK Medicines and Healthcare Products Regulatory Agency in January 2011. Clinical studies for these indications have now been initiated and enrollment of patients has begun.
Unlike most common diseases where clinical familiarity exists, many aspects of the clinical presentation, disease progression (including mortality and key morbidities) and response to treatment are poorly understood for rare diseases. Two key factors are responsible for these differences. First, the rarity of the disease often limits a physician’s clinical experience to a single case. Secondly, the historical absence of any effective therapy reduces the interest and research funding available to support coordinated investigation of the disease. The development of a new potential therapy requires accurate knowledge of the natural course of a disease to support patient diagnosis, endpoint selection and to provide historical data on mortality and morbidity. These are required to inform the evaluation and care of affected patients and to provide a reference for efficacy studies of enzyme replacement or other novel therapies. In order to generate this historical context for LAL Deficiency in support of the SBC-102 development program, Synageva is currently running two clinical study protocols in approximately 20 countries requiring case record review of patients with early and late onset LAL Deficiency. These natural history studies will be used to investigate and characterize key aspects of the clinical course of the disease, to inform the evaluation and care of affected patients and to provide a reference for efficacy studies of SBC-102 by identifying appropriate clinical endpoints.
In addition to the patients with LAL Deficiency enrolled in clinical trials, an infant with the disease has received treatment with SBC-102 on a compassionate use basis due to the life-threatening nature of the disease. Prior to treatment, the infant exhibited the classic symptoms of Wolman Disease (early onset LAL Deficiency), including anemia, increasingly abnormal liver function tests, and growth failure. The infant began receiving SBC-102 in April 2011 at the age of four months and the patient has so far received 4.5 months of treatment with no complications and continues to receive treatment with SBC-102. The infant is demonstrating substantial improvements in growth rate, liver function tests (reduction in serum transaminases), and other disease-related abnormalities consistent with Synageva’s preclinical data for SBC-102. It is anticipated that this patient will be enrolled into an extension study as part of the clinical trial program.
The current and planned clinical trials for SBC-102 are:
| • | Phase I/II Open Label Dose Escalation Study in Adult Patients with Liver Dysfunction Due to LAL Deficiency. In May 2011, Synageva initiated a four week study to evaluate the safety and tolerability of SBC-102 administered weekly in patients with liver dysfunction due to late onset LAL Deficiency. This study will also characterize the pharmacokinetics of SBC-102 delivered by IV infusion. The study is expected to enroll a total of approximately nine patients at multiple centers in the U.S. and Europe. |
| • | Phase I/II O pen Label Repeat Dose Escalation Study in Children with Growth Failure Due to LAL Deficiency. In 2011, Synageva also initiated a 16 week study to evaluate the safety and tolerability of SBC-102 in children with growth failure due to early onset LAL Deficiency. This study will also determine the effect of SBC-102 on growth and explore pharmacokinetics of SBC-102 and change in pharmacodynamics markers in this population. This study is expected to enroll a total of approximately eight patients at multiple centers in the U.S. and Europe. U.S. sites are expected to join this study once the adult safety and tolerability study has been reviewed by the FDA. As early onset LAL Deficiency has nearly a zero percent survival rate at one year of age, enrollment in this study is contingent on timely identification of newly diagnosed cases. |
| • | Planned Study in Subjects with Liver Dysfunction due to LAL Deficiency. Synageva is planning to initiate a trial to evaluate the safety and efficacy of SBC-102 in both adults and children with late onset LAL Deficiency. In this study, Synageva plans to investigate two doses of SBC-102 to define dosing requirements for approval and commercialization. The study is expected to enroll patients at multiple centers in the U.S. and Europe. Currently, Synageva expects to initiate this study in the second half of 2012. |
Synageva has currently enrolled a total of four patients in its Phase I/II studies of SBC-102. In addition to these trials, extension studies are planned for all patients enrolled into the SBC-102 development program in order to support requirements for long-term safety and to provide patients ongoing access to drug product. Patients in these extension studies will continue on therapy until BLA approval of SBC-102, if received.
Regulatory
SBC-102 has been granted Orphan Drug Designation by the FDA, and Synageva has received regulatory clearance in the U.S. to conduct clinical trials in adult patients with LAL Deficiency. U.S. Orphan Drug Designation is granted to a product that treats a rare disease, a condition that affects fewer than 200,000 Americans. As a result of the Orphan Drug Designation, Synageva is eligible to receive a number of benefits, including access to grant funding for clinical trials, tax credits, waiver of the FDA filing and registration fees, and seven years of market exclusivity if approval is received. Additionally, due to the severity of LAL Deficiency, the FDA granted SBC-102 Fast Track Designation which allows for an expedited regulatory review for the product.
SBC-102 has also been granted Orphan Drug Designation by the EMA, and Synageva has received regulatory clearance in key countries in the EU to conduct clinical trials in patients with late and early onset LAL Deficiency. The EMA’s Orphan Drug Designation is given to therapies that treat rare diseases, defined as conditions that affect no more than five in 10,000 persons in the EU. As a result of the EMA Orphan Drug Designation, Synageva is eligible to receive access to protocol assistance, direct access to centralized marketing authorization, up to 10 years of marketing exclusivity if approval is received, fee reductions or exemptions, and other national incentives. Under the EU Pediatric Regulations established in 2007, SBC-102 may be eligible for an additional two years of market exclusivity.
Commercialization
LAL Deficiency is a lysosomal storage disorder, and like other storage diseases such as Gaucher Disease, Fabry Disease, and Pompe Disease, affects a very small patient population hallmarked by unmet medical need, substantial morbidity and increased risk of mortality. As indicated in the table below, Cerezyme, Fabrazyme, Myozyme and Soliris are ultra-orphan drugs that provide precedent for the ability to commercialize a breakthrough treatment for an ultra-rare medical condition. Members of Synageva’s management team have previously held leadership roles in the successful development and commercialization for all four of these precedent products.
| Drug | SBC-102 | Cerezyme | Fabrazyme | Myozyme | Soliris | |||||
| Indication |
LAL Deficiency | Type I Gaucher Disease |
Fabry Disease |
Pompe Disease |
PNH | |||||
| Estimated Prevalence |
1 in 40,000 | 1 in 59,000 to 86,000 |
1 in 40,000 to 476,000 |
1 in 40,000 to 146,000 |
1 in ~77,000 | |||||
| Reported 2010 Revenue ($M) |
Significant Unmet Need |
$719.6 | $188.2 | $411.8 | $540.9 | |||||
Table 1. A sampling of ultra-orphan products and their associated incidence and revenue
(The revenue numbers above are not necessarily predictive of the commercial potential for SBC-102)
LAL Deficiency is an ultra-rare disorder that falls within the scope of metabolic specialists and hepatologists. Liver complications such as fibrosis, cirrhosis and liver failure dominate the late onset form, and patients may resemble those with other, more common diseases such as non-alcoholic fatty liver disease or non-alcoholic steatohepatitis. Similar to other lysosomal storage disorders, increased disease awareness and improvements in diagnosis supported by the patient and physician communities are critical for identifying patients and facilitating treatment. Synageva is in the process of engaging the physician and patient communities to establish a registry that will encourage involvement of all parties to raise awareness of and interest in LAL Deficiency. These include metabolic, hepatic, and lipid physician specialists and the LAL Solace, National Organization for Rare Disorders, Eurordis and CLIMB patient groups.
The diagnosis of late onset LAL Deficiency patients is anticipated to begin with the hepatologists’ clinical diagnosis aided by the use of differentiating biochemical markers, including abnormal lipid profile and confirmed by a simple blood test for the LAL enzyme. Synageva makes available to individuals information regarding which laboratories perform these tests and the procedures required for submitting a specimen. Adrenal calcification noted during investigations for gastrointestinal symptoms and growth failure allows for the rapid diagnosis of early onset LAL Deficiency patients. Presently, early onset patients usually receive care in a tertiary academic medical center.
Synageva’s commercial strategy for SBC-102 focuses on four imperatives: (i) facilitating diagnosis, (ii) generating prescriptions, (iii) supporting treatment, and (iv) ensuring coverage. By aligning resources against these imperatives, Synageva believes that its commercial footprint will be efficient and scaled to a highly specialized market niche. Similar to other companies with such specialized call points, Synageva’s current plans for resourcing the commercialization effort for SBC-102 include a small number of highly specialized field-based representatives supported by a specialized organization in-house. The past experience of members of Synageva’s management team in leading commercial efforts for ultra-rare products highlights the value of recruiting professionals who have experience with these specialized and focused physician and patient communities. The planned commercial organization is being developed to initially address the needs and opportunities for the North American, European, Latin American, and Asia Pacific regions.
Other Programs
In addition to SBC-102, Synageva is also progressing protein therapeutic programs for other rare diseases, which are at different stages of preclinical development. These include two enzyme replacement therapies for other lysosomal storage disorders and two programs for other rare life-threatening conditions. These protein therapeutic programs are selected based on scientific rationale, unmet medical need within the patient population, potential to substantially impact disease course, and strategic alignment with Synageva’s corporate and commercial efforts, including a potentially significant commercial opportunity.
Synageva believes its other programs, SBC-103 through 106, also have the potential to present patients and health care practitioners with effective therapies to treat the rare and devastating diseases targeted by these programs, which, like LAL Deficiency, are characterized by significant morbidity and mortality. The most advanced of these additional programs is SBC-103, an enzyme replacement therapy for MPS IIIB. This enzyme is a recombinant form of NAGLU. Similar to LAL Deficiency, MPS IIIB is an autosomal recessive lysosomal storage disease that may affect as many as eight individuals per million lives. While initially appearing unaffected, children born with MPS IIIB usually present with a slowing of development and/or behavioral problems around two years of age, followed by progressive intellectual decline and immobility with complete dependency on care providers. The life-span of an affected child does not usually extend beyond late teens to early twenties. Recent systematic studies using mutation based approaches have also revealed that MPS IIIB can demonstrate a large variability in the course of the disease, with a substantial number of additional patients with an attenuated phenotype with longer lasting, stable intellectual disability followed by regression later in life. Preliminary characterization of the enzyme produced using Synageva’s production platform demonstrates favorable uptake properties compared to previously published attempts to produce this enzyme using cell culture based approaches. There is currently no approved therapy available for MPS IIIB.
The following table describes Synageva’s product candidate pipeline:
| Program | SBC-102 (rhLAL) |
SBC-103 (rhNAGLU) |
SBC-104 | SBC-105 | SBC-106 | |||||
| Therapeutic |
Recombinant Lysosomal Acid Lipase |
Recombinant a-N-acetyl- |
Extra Cellular Protein |
Enzyme Replacement Therapy |
Enzyme Replacement Therapy | |||||
| Disease |
LAL Deficiency (LSD) |
MPS IIIB/ Sanfilippo B (LSD) |
Severe Genetic Condition |
Severe Metabolic Disorder |
Severe Genetic Condition | |||||
| Development Status |
Clinical | Preclinical | Preclinical | Preclinical | Preclinical | |||||
| Regulatory Opportunity |
Orphan Designation Granted U.S. Granted EU Fast Track |
Qualify for Orphan Designation & Potential Fast Track Designation |
Qualify for Orphan Designation & Potential Fast Track Designation |
Qualify for Orphan Designation & Potential Fast Track Designation |
Qualify for Orphan Designation & Potential Fast Track Designation |
Table 2. Synageva’s pipeline programs.
SBC-104 is an extracellular protein that targets a severe, rare genetic condition. There are no approved therapies for this disease, which is devastatingly debilitating and frequently results in early death. SBC-104 is being developed as a protein replacement therapy.
SBC-105 is an enzyme replacement therapy being developed to treat a severe, rare metabolic disorder. Synageva believes this program has opportunity in a number of related rare diseases with similar underlying biology.
SBC-106 is a protein therapy that targets a severe and rare genetic condition.
Synageva’s Expression Platform
Overview
In mid-2008, Sanj K. Patel was appointed President, Chief Executive Officer and Director of Synageva, after nearly a decade at Genzyme Corporation where he led, at different times, both Clinical and Commercial Operations for Genzyme’s rare disease franchise. Synageva’s business focus on products for rare diseases was established in 2008 with Mr. Patel’s appointment and his redirection of the company. This change represented a substantial shift in the business strategy of the original company, AviGenics, inc., which was founded in 1996 to develop a technology for expressing protein therapeutics in EW with the intention of creating follow-on biologics, including both biosimilar and biobetter products. With the change in business strategy, the expression platform was switched to a focus on the manufacture of protein therapeutics for rare diseases with unmet medical need.
Synageva’s proprietary expression platform remains a key element of Synageva’s business, and is an important contributor to Synageva’s current pipeline of rare disease therapeutics. Synageva’s expression platform is an integrated approach using recombinant DNA technology for the creation, optimization and commercial production of protein therapeutics. This mature platform, encompassing over 15 years of research and clinical development, is distinct from cell culture based approaches for protein therapeutic manufacturing and has a number of potential advantages which include:
| • | Reduced capital investment; |
| • | Very high and competitive protein expression levels; |
| • | Flexibility and consistency during scale-up; and |
| • | Human-like glycosylation patterns that can be tailored for the application. |
Synageva believes that this system will avoid the scale-up and inconsistency issues often associated with cell culture-based manufacturing of many types of therapeutic proteins. The ability to manufacture its products in-house and the important advantages of its manufacturing platform is a key element of Synageva’s business. This capability decreases the financial requirements for progressing a protein therapeutic into clinical development and allows Synageva to retain control over manufacturing of the commercial product.
Biology
The foundation of Synageva’s platform is an integrated system of proprietary vectors and methods that allow the generation of transgenic lines of hens which produce high levels of therapeutic protein in EW, a protein friendly matrix. Expression is achieved using Synageva’s proprietary vectors which allow targeted expression in EW-producing cells, and results in high expression levels of therapeutic proteins. The EW matrix facilitates bulk storage of unpurified EW prior to purification for prolonged periods and is one of a number of manufacturing advantages of the Synageva platform over traditional protein manufacturing technologies. In contrast to mammalian cell culture based approaches which utilize immortalized cell lines with high genetic and epigenetic instability, Synageva’s proprietary vectors allow incorporation of the gene of interest into the genome of normal cells of an avian (Gallus) with selective expression of the resulting protein in the oviduct tissues and secretion into EW. The importance of this cellular environment for therapeutic protein expression is highlighted by the tight consistency of post-translational modification, including glycosylation, seen in proteins manufactured using the Synageva platform compared to cell culture produced material. Furthermore, Synageva’s expression system yields consistent expression levels and quality of protein within production lines and through multiple generations. Synageva believes that its platform is the only approach for creating transgenic hens that produce a therapeutic protein which has been used successfully in clinical trials and which can enable commercial scale manufacturing.
Synageva’s proprietary technology allows for the production of proteins with glycosylation patterns that are suitable for many different diseases without a requirement for additional processes either during manufacturing (inhibition specific glycosylation enzymes) or post purification (enzymatic removal of terminal sugars) to modify terminal glycan structures impacting biodistribution. Furthermore, unlike some alternative expression platforms, Synageva’s expression system produces proteins with ‘human like’ glycan structures and does not incorporate non-human sugars into glycans.
Manufacturing and Supply
Synageva has demonstrated the platform’s ability to successfully produce a wide array of therapeutic proteins, including therapeutic enzymes, cytokines, monoclonal antibodies and fusion proteins. Furthermore, from 2004 to 2008, AviGenics, inc. manufactured investigational products using its expression system to supply Phase I and Phase II multinational clinical studies run in the U.S., EU, and India, which included more than 250 patients. The regulatory clearance for these studies included detailed considerations related to the manufacturing platform and there is good regulatory familiarity with the expression system. The largest study conducted was a multinational Phase II study of glycosylated recombinant human granulocyte colony stimulating factor (“G-CSF”) in 189 breast cancer patients with chemotherapy induced neutropenia. The overall safety and efficacy profile was comparable to the characteristics of Neupogen® (filgrastim) and no patient dosed with recombinant human G-CSF showed evidence of G-CSF antibody seroconversion during extended follow-up out to 12 months after initiation of dosing.
Finally, this platform is covered by a comprehensive intellectual property portfolio owned or exclusively licensed by Synageva and, as Synageva believes, provides expanded freedom to operate compared to other systems.
Cytovance Biologics LLC (“Cytovance”) is currently Synageva’s sole supplier of clinical trial material for SBC-102 from protein provided by Synageva and has provided manufacturing services to Synageva since December 2010. Synageva’s agreement with Cytovance will remain in effect until Cytovance completes the projects mutually agreed to by the parties in the agreement. Synageva may terminate its agreement with Cytovance upon 60 days’ written notice, but Cytovance may only terminate the agreement in the event of a material uncured breach by Synageva.
Patents and Proprietary Rights
Synageva actively seeks to aggressively protect the proprietary technology that is important to its business, including pursuing patents that cover its product candidates and compositions, their methods of use and the processes for their manufacture, as well as any other relevant inventions and improvements that are commercially important to the development of its business. Synageva also relies on trade secrets and other know-how that may be important to the development of its business.
Synageva’s patent portfolio is currently composed of 78 issued patents and 64 patent applications in the major territories, including the U.S., Europe, Brazil, Japan, China, Canada, India, and Australia, and includes patents and patent applications that Synageva owns as well as licenses from other parties. These patents and patent applications cover various aspects of Synageva’s manufacturing expression platform, product candidate pipeline and other product candidates that Synageva is no longer developing. Patents covering aspects of Synageva’s manufacturing expression platform will expire between 2016 and 2026. If they were to issue, current patent applications covering the expression platform will expire between 2018 and 2032 and current patent applications covering SBC-102 will expire between 2031 and 2032.
In addition, Synageva’s own pending applications contain claims directed to compositions and improved methods for expression of therapeutic proteins, with expirations between 2018 and 2032. Synageva continues to develop new intellectual property based on ongoing research to improve and enhance its expression platform.
University of Georgia Research Foundation
Synageva’s patent rights include the patent rights owned by University of Georgia Research Foundation (“UGARF”) subject to an exclusive, worldwide, sublicensable license granted to Synageva to develop, make, use and commercialize the avian transgenesis technology. Under the license, which was initially executed in 1996 and amended in 2008, Synageva paid UGARF an upfront license fee and issued shares of Synageva common stock in return for worldwide exclusive rights under the license. Additionally, UGARF is eligible to receive low single digit royalties on net sales of products covered under the license. This license agreement covers 21 patents and 18 patent applications worldwide that are the basis of Synageva’s expression platform. The UGARF license is effective until the last to expire of the licensed patents. Patents exclusively licensed from UGARF covering key aspects of Synageva’s expression platform will expire between 2018 and 2021. UGARF can terminate the license or, at UGARF’s discretion, convert the license into a non-exclusive license, if Synageva materially breaches the agreement, makes any materially false reports to UGARF, or fails to pay any required consideration under the agreement. Synageva has the right to terminate the agreement upon 60 days’ prior written notice to UGARF.
University of Minnesota
In 2009, Synageva entered into an exclusive license agreement, with the right to grant sublicenses, with the University of Minnesota that relates to compositions and methods useful for generating transgenic Gallus. In exchange for the license, which is effective until the last to expire of the licensed patents, Synageva paid the University of Minnesota an upfront license fee. In addition, University of Minnesota is entitled to minimal annual royalties which are creditable against low single digit royalties on net sales of products covered under the license. The 18 patents included in this license agreement expire by 2016. The University of Minnesota may terminate the agreement for Synageva’s failure to timely cure any material breach or failure to perform any obligations under the agreement. Synageva may terminate the agreement at any time after the third anniversary of the agreement.
Pangenix
The patents included in Synageva’s non-exclusive, sublicensable license agreement entered into with Pangenix in 2000 also cover compositions and methods useful for generating transgenic Gallus. In exchange for the license granted by Pangenix, which is effective until the last to expire of the licensed patents, Pangenix received an upfront license fee and is entitled to receive minimum annual royalties creditable against low single digit royalties on net sales of products covered under the license. The patents included in this license agreement are set to expire in 2015. Pangenix may terminate the agreement for Synageva’s failure to timely cure any material breach or failure to perform its obligations under the agreement. Synageva may terminate upon 60 days’ notice to Pangenix.
While there can be no assurance that patent applications relating to Synageva’s product candidates will ultimately issue or what the scope of the claims of such patent applications will cover if they were to issue, Synageva expects to rely heavily on orphan drug exclusivity for its product candidates, including SBC-102, which grants seven years of marketing exclusivity under the Federal Food, Drug, and Cosmetic Act, and up to 10 years of marketing exclusivity in Europe. In addition, Synageva continues to aggressively pursue intellectual property protection for its product candidates in the form of patent applications that have been and will continue to be filed in the U.S. and internationally.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which Synageva files, the patent term is 20 years from the date of filing the non-provisional application. In the U.S., a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent. Additional patent extension can be granted for time spent in development and regulatory review in certain jurisdictions as well.
Sales and Marketing
Synageva is currently developing marketing, sales and distribution capabilities to support commercialization for SBC-102. Similar to other companies with such specialized call points, Synageva’s current plans for commercialization resources include limited field-based customer-facing representatives supported by a specialized in-house organization to address the needs and opportunities for the North American, European, Latin American, and Asia Pacific regions.
With respect to pipeline products and other product candidates, Synageva may elect to utilize its own commercial capabilities to market and sell a product for which it obtains regulatory approval.
Competition
Synageva’s industry is highly competitive and subject to rapid and significant technological change. Synageva’s potential competitors include large pharmaceutical and biotechnology companies and specialty pharmaceutical companies, academic institutions, government agencies, and research institutions. The market for enzyme replacement therapies is becoming increasingly competitive. However, Synageva’s products, upon approval, will be focused, at least initially, on specific orphan markets characterized by high unmet medical need. Key competitive factors affecting the commercial success of Synageva’s product candidates are likely to be efficacy, safety and tolerability profile, reliability and durability of response, convenience of dosing, and price and reimbursement.
Government Regulation
The preclinical studies and clinical testing, manufacture, labeling, storage, record keeping, advertising, promotion, export, and marketing, among other things, of Synageva’s product candidates and future products, are subject to extensive regulation by governmental authorities in the U.S. and other countries. In the U.S., pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act and other laws, including, in the case of biologics, the Public Health Service Act. Synageva expects SBC-102 to be regulated by the FDA as a biologic. Biologics require the submission of a BLA and approval by the FDA prior to being marketed in the U.S. Manufacturers of biologics may also be subject to state regulation. Failure to comply with FDA requirements, both before and after product approval, may subject Synageva and/or its partners, contract manufacturers, and suppliers to administrative or judicial sanctions, including FDA refusal to approve applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, fines and/or criminal prosecution.
The steps required before a biologic may be approved for marketing of an indication in the U.S. generally include:
| • | preclinical laboratory tests and animal tests; |
| • | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may commence; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product; |
| • | submission to the FDA of a BLA or supplemental BLA; |
| • | FDA pre-approval inspection of product manufacturers; and |
| • | FDA review and approval of the BLA or supplemental BLA. |
Preclinical studies include laboratory evaluation, as well as animal studies to assess the potential safety and efficacy of the product candidate. Preclinical safety tests must be conducted in compliance with FDA regulations regarding good laboratory practices. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may be commenced. The IND will automatically become effective 30 days after receipt by the FDA, unless the FDA before that time raises concerns about the drug candidate or the conduct of the trials as outlined in the IND. The IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. Submission of an IND does not guarantee FDA authorization to commence clinical trials.
Clinical trials involve the administration of the investigational product to healthy volunteers or to patients, under the supervision of qualified principal investigators. Each clinical study at each clinical site must be reviewed and approved by an independent institutional review board prior to the recruitment of subjects.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap and different trials may be initiated with the same drug candidate within the same phase of development in similar or differing patient populations. Phase I studies may be conducted in a limited number of patients, but are usually conducted in healthy volunteer subjects. The drug is usually tested for safety and, as appropriate, for absorption, metabolism, distribution, excretion, pharmaco-dynamics and pharmaco-kinetics.
Phase II usually involves studies in a larger, but still limited, patient population to evaluate preliminarily the efficacy of the drug candidate for specific, targeted indications to determine dosage tolerance and optimal dosage and to identify possible short-term adverse effects and safety risks.
Phase III trials are undertaken to further evaluate clinical efficacy of a specific endpoint and to test further for safety within an expanded patient population at geographically dispersed clinical study sites. Phase I, Phase II, or Phase III testing might not be completed successfully within any specific time period, if at all, with respect to any of Synageva’s product candidates. Results from one trial are not necessarily predictive of results from later trials. Furthermore, the FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture and composition of the product, are submitted to the FDA as part of a BLA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee Act, as amended, the fees payable to the FDA for reviewing a BLA, as well as annual fees for commercial manufacturing establishments and for approved products, can be substantial. The BLA review fee alone can exceed $500,000, subject to certain limited deferrals, waivers, and reductions that may be available. Each BLA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within 45 to 60 days following submission of the application. If found complete, the FDA will “accept” the BLA, thus triggering a full review of the application. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission. The FDA’s established goal is to review 90% of priority BLA applications in six months and 90% of standard BLA applications in 10 months, whereupon a review decision is to be made. The FDA, however, may not approve a drug within these established goals and its review goals are subject to change from time to time. Further, the outcome of the review, even if generally favorable, may not be an actual approval but an “action letter” that describes additional work that must be done before the application can be approved. Before approving a BLA, the FDA may inspect the facilities at which the product is manufactured and will not approve the product unless compliance with cGMP is satisfactory. The FDA may deny approval of a BLA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any application may include many delays or never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, may require that warning statements be included in the product labeling, may require that additional studies be conducted following approval as a condition of the approval, and may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. Marketing a product for other indicated uses or making certain manufacturing or other changes requires FDA review and approval of a BLA supplement or new BLA. Further post-marketing testing and surveillance to monitor the safety or efficacy of a product is required. Also, product approvals may be withdrawn if compliance with regulatory standards is not maintained or if safety or manufacturing problems occur following initial marketing. In addition, new government requirements may be established that could delay or prevent regulatory approval of Synageva’s product candidates under development.
As part of the newly enacted Patient Protection and Affordable Care Act of 2010, Public Law No. 111-148, under the subtitle of Biologics Price Competition and Innovation Act of 2009 (“BPCI”), a statutory pathway has been created for licensure, or approval, of biological products that are biosimilar to, and possibly interchangeable with, earlier biological products licensed under the Public Health Service Act. Also under the BPCI, innovator manufacturers of original reference biological products are granted 12 years of exclusive use before biosimilars can be approved for marketing in the U.S. The objectives of the BPCI are conceptually similar to those of the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the “Hatch-Waxman Act,” which established abbreviated pathways for the approval of drug products. The implementation of an abbreviated approval pathway for biological products is under the direction of the FDA and is currently being developed. In late 2010, the FDA held a hearing to receive comments from a broad group of stakeholders regarding the implementation of the BCPI. The approval of a biologic product biosimilar to one of Synageva’s products could have a material adverse impact on its business as it may be significantly less costly to bring to market and may be priced significantly lower than Synageva’s products.
Both before and after the FDA approves a product, the manufacturer and the holder or holders of the BLA for the product are subject to comprehensive regulatory oversight. For example, quality control and manufacturing procedures must conform, on an ongoing basis, to cGMP requirements, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to spend time, money and effort to maintain cGMP compliance.
Orphan Drug Act
The Orphan Drug Act provides incentives to manufacturers to develop and market drugs for rare diseases and conditions affecting fewer than 200,000 persons in the U.S. at the time of application for Orphan Drug Designation. The first developer to receive FDA marketing approval for an orphan drug is entitled to a seven year exclusive marketing period in the U.S. for that product. However, a drug that the FDA considers to be clinically superior to, or different from, another approved orphan drug, even though for the same indication, may also obtain approval in the U.S. during the seven year exclusive marketing period. In addition, holders of exclusivity for orphan drugs are expected to assure the availability of sufficient quantities of their orphan drugs to meet the needs of patients. Failure to do so could result in the withdrawal of marketing exclusivity for the drug.
Legislation similar to the Orphan Drug Act has been enacted in other countries outside the U.S., including the EU. The orphan legislation in the EU is available for therapies addressing chronic debilitating or life-threatening conditions that affect five or fewer out of 10,000 persons or are financially not viable to develop. The market exclusivity period is for 10 years, although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product is sufficiently profitable not to justify maintenance of market exclusivity. The market exclusivity may be extended to 12 years if sponsors complete a pediatric investigation plan agreed upon with the relevant committee of the EMA.
Foreign Regulation
In addition to regulations in the U.S., Synageva is subject to a variety of foreign regulatory requirements governing human clinical trials and marketing approval for drugs. The foreign regulatory approval process includes all of the risks associated with FDA approval set forth above, as well as additional country-specific regulations. Whether or not Synageva obtains FDA approval for a product, it must obtain approval of a product by the comparable regulatory authorities of foreign countries before it can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from country to country.
Corporate Information
Synageva was incorporated in Delaware in 1996. Prior to August 2008, Synageva was named AviGenics, inc. Synageva’s address is 128 Spring Street, Suite 520, Lexington, Massachusetts 02421. Synageva’s telephone number is (781) 357-9900. Synageva’s website address is www.synageva.com. Information contained in, and that can be accessed through, Synageva’s website is not incorporated into and does not form a part of this joint proxy statement/prospectus.
Employees
As of June 30, 2011, Synageva had 72 full-time employees. Approximately 51 were primarily engaged in research and development activities and 21 were primarily engaged in general, administrative, and pre-commercial activities. None of Synageva’s employees are subject to a collective bargaining agreement, and Synageva believes its employee relations to be good.
Property and Facilities
Synageva’s corporate headquarters and research laboratories are located in Lexington, Massachusetts, where Synageva leases and occupies approximately 11,000 rentable square feet of office and laboratory space. The lease expires on August 31, 2013 with an option to extend the lease by an additional two years. Synageva’s technical and manufacturing operations are located in and around Athens, Georgia, where Synageva leases and occupies approximately 53,000 rentable square feet of office and laboratory space in three different buildings. The first facility lease expires June 30, 2012, the second facility lease expires April 10, 2013, and the third facility lease expires June 30, 2013. Synageva believes that its facilities are suitable and adequate for its needs.
Legal Proceedings
Synageva is not currently engaged in any material legal proceedings.
SYNAGEVA’S MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This discussion of Synageva’s financial condition and results of operations should be read together with the consolidated financial statements and notes contained elsewhere in this joint proxy statement/prospectus. Certain statements in this section and other sections are forward-looking. While Synageva believes these statements are accurate, its business is dependent on many factors, some of which are discussed in the sections entitled “Risk Factors” and “Synageva’s Business”. Many of these factors are beyond Synageva’s control and any of these and other factors could cause actual results to differ materially from the forward-looking statements made in this joint proxy statement/prospectus. See the section entitled “Risk Factors” for further information regarding these factors. Synageva undertakes no obligation to release publicly the results of any revisions to the statements contained in this report to reflect events or circumstances that occur subsequent to the date of this joint proxy statement/prospectus.
Overview
Synageva is a clinical stage biopharmaceutical company focused on the discovery, development, and commercialization of therapeutic products for patients with life-threatening rare diseases and unmet medical needs. Synageva’s management team is experienced in the development and commercialization of drugs for diseases with small patient populations, including clinical and translational research, working with payors to establish reimbursement, and designing and building commercial organizations to reach highly specialized physicians to facilitate patient identification. Synageva has several protein therapeutics in development, including two enzyme replacement therapies for lysosomal storage disorders and two programs for life-threatening genetic conditions for which there are currently no approved treatments. Its lead program, SBC-102, is a recombinant human LAL currently under clinical investigation in the U.S. and EU for the treatment of patients with LAL Deficiency, which is a rare, devastating disease that causes significant morbidity and mortality. SBC-102 has been granted Orphan Drug Designations by FDA and the EMA and a Fast Track Designation by the FDA. Synageva has not yet received approval to market this product and is not currently commercializing any other products.
Financial Operations Overview
General
Synageva’s future operating results will depend on the progress of drug candidates currently in its research and development pipeline. The results of Synageva’s operations will vary significantly from year to year and quarter to quarter and will depend largely on, among other factors, the cost and outcome of any preclinical development or clinical trials then being conducted.
Synageva, on a stand-alone basis, anticipates that its existing cash and cash equivalents as of June 30, 2011 should enable it to maintain current and planned operations into the first half of 2012. Synageva’s ability to continue funding its planned operations through and beyond the first half of 2012 is dependent on its ability to manage its expenses and its ability to raise additional funds through additional equity or debt financings, other sources of financing or corporate collaborations.
A discussion of certain risks and uncertainties that could affect Synageva’s liquidity, capital requirements and ability to raise additional funds is set forth under the section entitled “Risk Factors” in this joint proxy statement/prospectus.
Revenue
Synageva does not expect to generate any revenue from the direct sale of products for several years, if ever. Substantially all of its revenues to date have been derived from grant revenue and license and collaboration fees.
Synageva’s only source of significant revenues and/or cash flows from operations for the foreseeable future will be up-front license payments and funded research and development that it may receive under new collaboration agreements, if any. Synageva’s ability to enter into new collaborations and its receipt of additional payments under its existing collaborations cannot be assured, nor can it predict the timing of any such arrangements or payments, as the case may be.
Research and Development
Synageva expenses research and development costs as incurred. Research and development expense consists of costs incurred to discover, research and develop drug candidates, including personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs, outside consulting services and other external costs.
General and Administrative
General and administrative expense consists primarily of salaries, stock-based compensation expense and other related costs for personnel in executive, finance, accounting, business development, legal, information technology, corporate communications and human resource functions. Synageva also expenses patent costs and expenses associated with maintaining its intellectual property as incurred. Other costs include facility costs not otherwise included in research and development expense, insurance, and professional fees for legal and accounting services.
Results of Operations
Six Months Ended June 30, 2011 and June 30, 2010
Revenues
Total revenues increased approximately $26,000, or 9%, to $301,000 for the six months ended June 30, 2011 as compared to $275,000 for the comparable period in 2010. In May 2010, Synageva was awarded a two-year National Institute of Health grant (the “NIH Grant”) totaling $747,000. Grant revenues for the six months ended June 30, 2011 and June 30, 2010 were $196,000 and $132,000, respectively. Grant revenues are recognized in the period in which Synageva has incurred the expenditures in compliance with the specific restrictions of the NIH Grant. At June 30, 2011, Synageva had one ongoing collaboration. In accordance with the proportional performance method of revenue recognition, due to the timing of milestone payments, revenue is limited to amounts billed or cash paid. Collaboration revenues for the six months ended June 30, 2011 and June 30, 2010 were $105,000 and $143,000, respectively.
Research and Development Expenses
Research and development expenses are summarized as follows:
| Six Months Ended June 30, |
||||||||||||||||
| 2011 | 2010 | $ Change | % Change | |||||||||||||
| Compensation and benefits-related |
$ | 2,652,732 | $ | 1,629,679 | $ | 1,023,053 | 63 | % | ||||||||
| Clinical trials and manufacturing |
2,768,870 | 229,044 | 2,539,826 | 1,109 | % | |||||||||||
| External services |
1,369,144 | 655,855 | 713,289 | 109 | % | |||||||||||
| Facilities related |
972,295 | 724,726 | 247,569 | 34 | % | |||||||||||
| Non-cash stock-based compensation |
58,316 | 20,031 | 38,285 | 191 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total research and development expense |
$ | 7,821,357 | $ | 3,259,335 | $ | 4,562,022 | 140 | % | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Synageva’s research and development expenses increased by approximately $4,562,000, or 140%, to $7,821,000 for the six months ended June 30, 2011 as compared to $3,259,000 for the comparable period in 2010. The increase is primarily attributable to increased spending related to Synageva’s lead program SBC-102 as the program moved forward into clinical trials. The most significant costs included expenses associated with SBC-102 clinical trials, outsourced contract manufacturing activities, and toxicology work. Higher compensation expense mainly related to higher personnel expense, as Synageva hired additional staff between 2010 and 2011 to move the SBC-102 program forward. As part of these activities, Synageva also incurred additional facilities expense resulting from the opening of its new corporate headquarters and laboratory facilities in Lexington, Massachusetts. Synageva expects these expenses to continue to increase as clinical development for SBC-102 continues.
General and Administrative Expenses
General and administrative expenses increased by approximately $1,411,000, or 76%, to $3,278, 000 for the six months ended June 30, 2011 as compared to $1,868,000 for the comparable period in 2010. This increase was primarily due to increases in external spending for legal, accounting, and professional fees of approximately $1,036,000, which includes merger-related transaction costs, and employee-related compensation expense increases of approximately $367,000 due to a growth in Synageva’s headcount between 2010 and 2011.
Years Ended December 31, 2010 and December 31, 2009
Revenues
Total revenues increased by approximately $434,000, or 268%, to $595,000 for the year ended December 31, 2010, as compared to $162,000 for the year ended December 31, 2009. The increase in revenues was the result of higher grant revenues and higher collaboration revenues as compared to the prior year. In March 2009 and March 2010, Synageva entered into collaboration agreements with a third party to develop antibodies using Synageva’s proprietary expression platform. Collaboration revenues are the result of this collaboration. Grant revenues of $315,000 for the year ended December 31, 2010 relate to the NIH Grant.
Research and Development Expenses
Research and development expenses are summarized as follows:
| Year
Ended December 31, |
||||||||||||||||
| 2010 | 2009 | $ Change | % Change | |||||||||||||
| Compensation and benefits-related |
$ | 3,673,908 | $ | 2,674,911 | $ | 998,997 | 37 | % | ||||||||
| Clinical trials and manufacturing |
1,378,820 | 1,890,728 | (511,908 | ) | -27 | % | ||||||||||
| External services |
3,285,987 | 809,760 | 2,476,227 | 306 | % | |||||||||||
| Facilities related |
1,482,553 | 1,181,695 | 300,858 | 25 | % | |||||||||||
| Non-cash stock-based compensation |
44,541 | 25,938 | 18,603 | 72 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total research and development expense |
$ | 9,865,809 | $ | 6,583,032 | $ | 3,282,777 | 50 | % | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Synageva’s research and development expenses increased by approximately $3,283,000, or 50%, to $9,866,000 for the year ended December 31, 2010 as compared to $6,583,000 for the year ended December 31, 2009. The increase is primarily attributable to increased spending related to Synageva’s lead program SBC-102 and related preclinical and clinical start-up costs as the program moved toward the clinic. The increased costs included external services for preclinical work such as toxicology studies and lab research, as well as higher compensation expense after Synageva hired additional staff to move the SBC-102 program forward. As part of these activities, Synageva also incurred additional facilities expense resulting from the opening of its corporate headquarters and laboratory facilities in Lexington, Massachusetts. Synageva expects these expenses to continue to increase as clinical development for SBC-102 continues.
General and Administrative Expenses
General and administrative expenses increased by approximately $10,000 to $3,853,000 for the year ended December 31, 2010 as compared to $3,843,000 for the comparative prior year period. This increase was primarily due to higher facilities related expense of approximately $207,000 resulting from the opening of Synageva’s Lexington, Massachusetts corporate headquarters, partially offset by lower compensation spending of $197,000.
Other Income
Other income increased by approximately $2,213,000 to $2,295,000 for the year ended December 31, 2010 as compared to $82,000 for the year ended December 31, 2009. The increase was due to the receipt of a tax grant related to the approval of Synageva’s applications for the Qualifying Therapeutic Discovery Project program during 2010.
Interest Income
Interest income decreased by approximately $18,000 to $4,000 for the year ended December 31, 2010 as compared to the prior year period. The decrease was primarily due to lower average investment balances and lower interest rates.
Interest Expense
Interest expense decreased by approximately $1,260,000 to $0 for the year ended December 31, 2010 as compared to the prior year period. The decrease was due to the repayment of an outstanding loan in October 2009. Interest amounts for the year ended December 31, 2009 included interest charges while the loan was outstanding in addition to an early payment penalty of $223,000.
Liquidity and Capital Resources
Sources of Liquidity
Synageva has financed its operations to date primarily through license fees, contingent cash payments and research and development funding from its collaborators, government grants and licensors, the private placement of its equity securities, and debt financings.
As of June 30, 2011, Synageva’s principal sources of liquidity consisted of cash and cash equivalents of approximately $17,904,000. Synageva’s cash and cash equivalents are highly liquid investments with a maturity of three months or less at date of purchase and consist of investments in money market funds. Synageva maintains cash balances with financial institutions in excess of insured limits.
Cash Flows
The use of Synageva’s cash flows for operations has primarily consisted of salaries and wages for its employees, facility and facility-related costs for its office, laboratory, and manufacturing facilities, fees paid in connection with clinical studies, preclinical studies, laboratory supplies, consulting fees, and legal fees. Synageva expects that costs associated with clinical studies will increase in future periods assuming that SBC-102 advances into further stages of clinical testing and its other preclinical candidates reach clinical trials.
Net cash used in operating activities was $9,169,000 for the six-month period ended June 30, 2011 as compared to net cash used of $4,397,000 for the six-month period ended June 30, 2010 and $10,550,000 for the year ended December 31, 2010 as compared to net cash used of $13,585,000 for the year ended December 31, 2009. Net cash used in operating activities during the six-month period ended June 30, 2011 and the year ended December 31, 2010 was primarily the result of Synageva’s net loss for the six-month period ended June 30, 2011 of $11,005,000 and for the year ended December 31, 2010 of $10,824,000. In addition, changes in certain operating assets and liabilities affected operating cash during the six-month period ended June 30, 2011, including a decrease of $455,000 in Synageva’s prepaid expense and other current assets due to receipt of grant funds earned in 2010, an increase in accrued expenses of $795,000 as a result of increased spending and non-cash items of $217,000 for depreciation and amortization of fixed assets, $156,000 for stock-based compensation, and $199,000 due to the revaluation of outstanding preferred stock warrants. Changes in certain operating assets and liabilities affected operating cash during the year ended December 31, 2010, including an increase of $889,000 in Synageva’s prepaid expense and other current assets due to receivable balances associated with grant funds earned in 2010, increased accrued expenses of $373,000 and accounts payable of $230,000 due to increased spending on toxicology studies and non-cash items of $384,000 for depreciation and amortization of fixed assets, $204,000 for stock-based compensation, and a decrease of $20,000 due to the revaluation of outstanding preferred stock warrants.
Net cash used by operating activities during the six-month period ended June 30, 2010 was primarily the result of Synageva’s net loss for the period of $4,833,000. Net cash used by operating activities during the year ended December 31, 2009 was primarily the result of Synageva’s net loss for the period of $11,420,000. In addition, changes in certain operating assets and liabilities affected operating cash during the six-month period ended June 30, 2010 and the year ended December 31, 2009, respectively.
Synageva expects to continue to use cash in operations as it continues to seek to advance its orphan drug programs through clinical development and preclinical testing. In addition, in the future Synageva may owe royalties and other contingent payments to its licensors based on the achievement of developmental milestones, product sales, and other specified objectives.
Investing activities used cash of approximately $214,000 and $169,000 for the six-month periods ended June 30, 2011 and June 30, 2010, respectively, and $585,000 and $163,000 for the years ended December 31, 2010 and December 31, 2009, respectively, resulting from the purchases of property and equipment during such periods.
Financing activities provided cash of approximately $12,573,000 for the six-month period ended June 30, 2011 primarily resulting from the proceeds from an issuance of convertible notes. In March 2011, Synageva issued $12.5 million of convertible notes as the first tranche of a $25 million convertible note offering. There were no financing activities during the six-month period ended June 30, 2010 other than minimal proceeds from the issuance of common stock. Financing activities resulted in no cash provided for the year ended December 31, 2010 as issuance costs related to preferred stock offerings were offset by the issuance of common stock. Financing activities for the year ended December 31, 2009 of $36,905,000 were the result of the issuance of convertible preferred stock and convertible notes totaling $44,666,000 partially offset by payments on debt and capital leases of $7,766,000.
Funding Requirements
Synageva has incurred significant losses since its inception. As of June 30, 2011, Synageva had an accumulated deficit of approximately $101,539,000. Synageva will require substantial funds to continue its research and development programs and to fulfill its planned operating goals. In particular, Synageva’s currently planned operating and capital requirements include the need for working capital to support its research and development activities for SBC-102 and other preclinical candidates that it is seeking to develop, and to fund its general and administrative costs and expenses.
Synageva’s ability to finance itself and to generate revenues will depend heavily on its ability to obtain favorable results in the ongoing clinical trials of SBC-102 and to successfully develop and commercialize SBC-102. Synageva has no current source of significant research funding revenue. Synageva expects that its only source of cash flows from operations for the foreseeable future will be grant revenue and collaboration revenue, if any.
Synageva may not be able to successfully enter into or continue any corporate collaborations and the timing, amount, and likelihood of it receiving payments under such collaborations is highly uncertain. As a result, Synageva cannot assure that it will attain any further revenue under any collaborations or licensing arrangements.
Synageva anticipates that existing cash and cash equivalents as of June 30, 2011 should enable it to maintain current and planned operations into the first half of 2012. Synageva’s future capital requirements, however, may vary from what it currently expects. There are a number of factors that may adversely affect Synageva’s planned future capital requirements and accelerate its need for additional financing, many of which are beyond its control, including the following:
| • | unanticipated costs in its research and development programs; |
| • | the timing, receipt and amount of payments, if any, from current and potential future collaborators; |
| • | the timing and amount of payments due to licensors of patent rights and technology used in its drug candidates; and |
| • | unplanned costs to prepare, file, prosecute, maintain and enforce patent claims and other patent-related costs, including litigation costs and technology license fees. |
Synageva may seek additional funding through private financings of debt or equity. The fundraising environment for emerging life science companies in general is highly volatile. Due to this and various other factors, including currently adverse general market conditions and the early-stage status of Synageva’s development pipeline, additional funding may not be available to Synageva on acceptable terms, if at all. In addition, the terms of any financing may be dilutive or otherwise adversely affect other rights of the Synageva stockholders. Synageva also expects to seek additional funds through arrangements with collaborators, licensees or other third parties. These arrangements would generally require Synageva to relinquish or encumber rights to some of its technologies or drug candidates, and Synageva may not be able to enter into such arrangements on acceptable terms, if at all. If Synageva is unable to obtain additional funding on a timely basis, whether through sales of debt or equity or through third-party collaboration or license arrangements, Synageva may be required to curtail or terminate some or all of its development programs, including some or all of its drug candidates.
Off-Balance Sheet Arrangements
Synageva has no off-balance sheet arrangements as of June 30, 2011.
Inflation
Synageva believes that inflation has not had a significant impact on its revenue and results of operations since inception.
Critical Accounting Policies and Estimates
Use of Estimates
The methods, estimates, and judgments Synageva uses in applying its most critical accounting policies have a significant impact on the results reported in its consolidated financial statements. Synageva evaluates its estimates and judgments on an ongoing basis. Synageva bases its estimates on historical experience and on assumptions that it believes to be reasonable under the circumstances. Such estimates and judgments include revenue recognition, research and development costs and related accrued expenses, the carrying value of property and equipment, stock based compensation, and certain liabilities, including, but not limited to a warrant liability. Synageva’s experiences and assumptions form the basis for its judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what Synageva anticipates, and different assumptions or estimates about the future could materially change its reported results.
Synageva believes the following accounting policies are most critical because they are important to the portrayal of its consolidated financial statements and require its most difficult, subjective or complex judgments in the preparation of its consolidated financial statements.
Principles of Consolidation
Synageva’s consolidated financial statements include the accounts of Synageva and its wholly owned subsidiary, Synageva BioPharma Limited, an entity incorporated in the UK. Synageva’s subsidiary commenced operations in May 2011. All significant intercompany accounts and transactions have been eliminated in consolidation.
Revenue Recognition
Synageva’s business strategy includes entering into collaborative agreements with biotechnology and pharmaceutical companies. Revenue under such collaborations may include the receipt of non-refundable license fees, payments based on achievement of development objectives, reimbursement of research and development costs and royalties. Synageva follows the provisions of ASC 605.
In March 2009 and March 2010, Synageva entered into collaboration agreements with a third party to develop antibodies using Synageva’s proprietary expression platform. The collaboration agreements included up-front payments to Synageva totaling $200,000, potential future milestone payments totaling $400,000, and potential option payments. Cumulative revenues earned through June 30, 2011 for the two agreements total approximately $385,000. As of June 30, 2011, one of these collaborations has been completed and one is ongoing.
Synageva recognizes revenue using the proportional performance method, provided that it can reasonably estimate the level of effort required to complete its performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Revenue recognized under the proportional performance method is determined by multiplying the total payments
under an arrangement, excluding royalties and payments contingent upon achievement of development objectives, by the ratio of level of effort incurred to date to estimated total level of effort required to complete Synageva’s performance obligations under such arrangement. Payments contingent upon achievement of development objectives are included once the contingency is removed. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the proportional performance method, as of each reporting period.
Significant management judgment is required in determining the level of effort required under an arrangement and the period over which Synageva is expected to complete its performance obligations under such arrangement.
Reimbursement of costs is recognized as revenue, provided that Synageva has determined that it is acting primarily as a principal in the transaction according to the provisions outlined in ASC 605, the amounts are determinable and collection of the related receivable is reasonably assured.
Synageva recognizes revenues from grants in the period in which it incurred the expenditures, in compliance with the specific restrictions of the grants.
Research and Development
Research and development costs are charged to operations when incurred and include personnel and facility-related expenses, outside contracted services including clinical trial costs, manufacturing and process development costs, research costs, outside consulting services and other external costs. Clinical development costs are a significant component of research and development expenses. Synageva contracts with third parties that perform various clinical trial activities on its behalf in the ongoing development of its product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flow.
Accrued Expenses Related to Research and Development Programs
As part of the process of preparing its consolidated financial statements, Synageva estimates accrued expenses. This process involves identifying yet to be invoiced services which have been performed on Synageva’s behalf and estimating the level of service performed and the associated cost incurred for such service as of each balance sheet date in its consolidated financial statements. Examples of estimated accrued expenses include:
| • | consulting fees; |
| • | contract clinical service fees; |
| • | fees paid to data management organizations and investigators in conjunction with clinical trials; and |
| • | fees paid to contract manufacturers in conjunction with the production of clinical materials. |
In connection with such service fees, Synageva’s estimates are most affected by its understanding of the status, timing and billing of services provided. Although Synageva’s service providers generally invoice it in arrears for services performed, contract agreements may contain prepayment provisions. Synageva records payments made under those provisions in a prepaid account and offsets those amounts when it subsequently incurs costs related to those prepayments. Contracts vary significantly in length and payments thereunder may be for a fixed amount, a variable amount based on actual costs incurred, capped at certain limits, or a combination of any of these elements. Activity levels are monitored through communication with vendors, including detailed invoice and task completion reviews, analysis of expenses against budgeted amounts, and pre-approval of any changes in scope of the services being performed. In the event that Synageva does not identify certain costs which have begun to incur, or it under- or over-estimates the level of services performed or the costs of such services in a given period, its reported expenses for such period could be too low or too high. The date on which certain services commence, the level of services performed on or before a given date, and the cost of such services are often determined based on subjective judgments. There have been no changes in Synageva’s estimation methodology for accruing contract services fees that had a material effect on its net losses for any of the periods presented herein.
Stock-Based Compensation
Effective January 1, 2006, Synageva adopted the Statement of Financial Accounting Standards No. 123 (revised 2004), Share-Based Payment, which generally requires that such transactions be accounted for using a fair-value-based method. Synageva measures compensation cost for stock-based compensation at fair value, including estimated forfeitures, and recognizes the expense as compensation expense using the straight-line method over the period that the recipient is required to provide service in exchange for the award, which generally is the vesting period.
Synageva uses the Black-Scholes option pricing model to measure the fair value of stock options. This model requires significant estimates related to the fair value of its common stock, the option’s expected life, future stock price volatility of the underlying equity security, and the risk-free interest rate for the expected term of the option. In determining the amount of expense to be recorded, Synageva is also required to estimate forfeiture rates for awards, based on the probability that employees will complete
the required service period. Synageva estimates the forfeiture rate based on historical experience. If actual forfeitures differ significantly from Synageva’s estimates, additional adjustments to compensation expense may be required in future periods. Ultimately, the actual expense recognized over the vesting period will only be for those shares that vest. Future changes in such assumptions could result in Synageva recording varying levels of stock-based compensation expenses for stock-based compensation awards granted in the future.
Synageva records stock-based compensation expense for stock options granted to non-employees based on the fair value of the stock options which is re-measured over the vesting term resulting in periodic adjustments to stock-based compensation expense.
Synageva recognized stock-based compensation expense on all stock option awards for the six months ended June 30, 2011 and June 30, 2010 in the following categories:
| Six Months Ended June 30, |
||||||||
| 2011 | 2010 | |||||||
| Research and development |
$ | 58,316 | $ | 20,031 | ||||
| General and administrative |
97,234 | 79,504 | ||||||
|
|
|
|
|
|||||
| $ | 155,550 | $ | 99,535 | |||||
|
|
|
|
|
|||||
Synageva has granted and expects that it may grant additional options in 2011 that could increase the amount of stock-based compensation recognized. The amount of the incremental employee stock-based compensation expense attributable to 2011 employee stock options to be granted will depend primarily on the number of stock options granted, the fair market value of Synageva’s common stock at the respective grant dates, and the specific terms of the stock options.
As of June 30, 2011, the total unrecognized compensation cost related to unvested stock options was approximately $1,623,000. The cost is expected to be recognized over a weighted average period of approximately 3.4 years.
Recently Issued and Proposed Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board issued an amendment to the accounting for multiple-deliverable revenue arrangements. This amendment provides guidance on whether multiple deliverables exist, how the arrangements should be separated, and how the consideration paid should be allocated. As a result of this amendment, entities may be able to separate multiple-deliverable arrangements in more circumstances than under existing accounting guidance. This guidance amends the requirement to establish the fair value of undelivered products and services based on objective evidence and instead provides for separate revenue recognition based upon management’s best estimate of the selling price for an undelivered item when there is no other means to determine the fair value of that undelivered item. The existing guidance previously required that the fair value of the undelivered item reflect the price of the item either sold in a separate transaction between unrelated third parties or the price charged for each item when the item is sold separately by the vendor. If the fair value of all of the elements in the arrangement was not determinable, then revenue was deferred until all of the items were delivered or fair value was determined. This amendment will be effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. Synageva adopted the provisions of this guidance on a prospective basis on January 1, 2011, the effect of which will be dependent upon the terms of any new or materially modified arrangements subsequent to such adoption.
In April 2010, the Financial Accounting Standards Board issued ASU 2010-17, Revenue Recognition—Milestone Method (Topic 605): Milestone Method of Revenue Recognition (“ASU 2010-17”). ASU 2010-17 provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. Consideration that is contingent on achievement of a milestone in its entirety may be recognized as revenue in the period in which the milestone is achieved only if the milestone is judged to meet certain criteria to be considered substantive. The new rule was adopted by Synageva on January 1, 2011. Adoption of this new standard did not materially affect Synageva’s financial statements.
In May 2011, the Financial Accounting Standards Board issued an amendment to the accounting guidance for fair value to develop common requirements between GAAP and International Financial Reporting Standards. The amendment changes the wording used to describe many of the requirements in GAAP for measuring fair value and for disclosing information about fair value measurements, including (i) application of the highest and best use and valuation premise concepts solely for nonfinancial assets and liabilities, (ii) measuring the fair value of an instrument classified in a reporting entity’s shareholders’ equity, and (iii) disclosing quantitative information about unobservable inputs used in the fair value measurement within Level 3 of the fair value hierarchy. For public entities, the amendment is effective for interim and annual periods beginning after December 15, 2011. Early application is not permitted. Synageva is currently evaluating the disclosure requirements related to providing quantitative information about unobservable inputs used to measure the fair value of the warrant liability.
In June 2011, the Financial Accounting Standards Board issued an amendment to the accounting guidance for presentation of comprehensive income. Under the amended guidance, a company may present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. In either case, an entity is required to present each component of net income along with total net income, each component of other comprehensive income along with a total for other comprehensive income, and a total amount for comprehensive income. Regardless of choice in presentation, the entity is required to present on the face of the financial statements reclassification adjustments for items that are reclassified from other comprehensive income to net income in the statement(s) where the components of net income and the components of other comprehensive income are presented. For public entities, the amendment is effective for fiscal years, and interim periods within those years, beginning after December 15, 2011, and shall be applied retrospectively. Early adoption is permitted. Other than a change in presentation, the adoption of this update is not expected to have a material impact on Synageva’s consolidated financial statements.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT OF THE COMBINED COMPANY FOLLOWING THE MERGER
The following table sets forth information as of September 21, 2011, regarding the beneficial ownership of the combined company upon completion of the Merger by (i) each person known by the management of Trimeris, Inc. (“Trimeris”) and Synageva that is expected to become the beneficial owner of 5% of the common stock of the combined company upon completion of the Merger, (ii) each director and named executive officer of the combined company, and (iii) all directors and named executive officers of the combined company as a group. Information with respect to beneficial ownership is based solely on a review of Trimeris’ capital stock transfer records and on publicly available filings made with the SEC by or on behalf of the stockholders listed below and on Synageva’s stock ledger. Unless otherwise indicated in the footnotes, the address for each listed stockholder is: c/o Synageva BioPharma Corp., 128 Spring Street, Suite 520, Lexington, Massachusetts 02421.
Percentage of beneficial ownership is calculated based on 22,399,649 shares of Trimeris common stock outstanding as of September 21, 2011 and 31,225,776 shares of Synageva capital stock outstanding as of September 21, 2011 (which includes 524,153 shares of Synageva common stock, the conversion of all 25,997,351 shares of Synageva preferred stock at a ratio of 1:1, and the conversion of all of Synageva’s convertible notes into 4,999,987 shares of Synageva preferred stock and thereafter into common stock at a ratio of 1:1). The percent of common stock of the combined company is based on 87,901,307 shares of common stock of the combined company outstanding upon completion of the Merger and assumes that the exchange ratio, as such ratio is calculated pursuant to the formula set forth in the merger agreement, dated June 13, 2011 between Triemeris, Syangeva and Tesla Merger Sub, Inc., to be used in connection with the Merger is approximately 2.078 shares of Trimeris common stock for each share of Synageva capital stock (without giving effect to the proposed reverse stock split described elsewhere in this joint proxy statement/prospectus). Shares of Trimeris common stock subject to stock options that are currently exercisable or exercisable within 60 days after September 21, 2011 are treated as outstanding and beneficially owned by the holder of such options for the purpose of computing the percentage ownership of the combined company’s common stock of such holder, but are not treated as outstanding for the purpose of computing the percentage ownership of the combined company’s common stock of any other stockholder. Shares of Synageva common stock subject to options or warrants that are currently exercisable or exercisable within 60 days of September 21, 2011 are treated as outstanding and beneficially owned by the holder of such options for the purpose of computing the percentage ownership of the combined company’s common stock of such holder, but are not treated as outstanding for the purpose of computing the percentage ownership of the combined company’s common stock of any other stockholder. Unless otherwise indicated and subject to the voting agreements between certain stockholders of Trimeris with Synageva and certain stockholders of Synageva with Trimers, Trimeris and Synageva believe that each of the persons named in this table has sole voting and investment power with respect to all shares shown as beneficially owned by them.
| Name and Address of Beneficial Owner |
Amount of Beneficial Ownership |
Percent of Class |
||||||
| 5% Stockholders |
||||||||
| Baker Bros. Advisors LLC(1) |
31,841,391 | 36.2 | % | |||||
| Tisch Investors(2) |
11,634,007 | 13.2 | % | |||||
| New Leaf Ventures II, L.P.(3) |
8,619,685 | 9.8 | % | |||||
| Tullis Dickerson Affiliates(4) |
7,972,911 | 9.1 | % | |||||
| HealthCor Management, L.P.(5) |
4,413,657 | 5.0 | % | |||||
| Named Executive Officers and Directors: |
||||||||
| Srini Akkaraju(6) |
8,619,685 | 9.8 | % | |||||
| Felix J. Baker(7) |
31,951,403 | 36.3 | % | |||||
| Stephen R. Biggar(8) |
510 | * | ||||||
| Carsten Boess |
0 | * | ||||||
| Stephen R. Davis(9) |
80,000 | * | ||||||
| Mark Goldberg(10) |
61,556 | * | ||||||
| Eric Grinstead(11) |
216,240 | * | ||||||
| Thomas R. Malley(12) |
236,665 | * | ||||||
| Sanj K. Patel(13) |
1,979,999 | 2.2 | % | |||||
| Barry Quart(14) |
76,250 | * | ||||||
| Anthony G. Quinn(15) |
301,742 | * | ||||||
| Robyn Samuels |
0 | * | ||||||
| All executive officers and directors as a group (12 persons) |
43,524,050 | 48.1 | % |
| * | Less than 1%. | |
| (1) | Baker Bros. beneficially owns 3,515,475 shares of Trimeris common stock through the following limited partnerships: FBB Associates; Baker Tisch Investments, L.P.; Baker Bros. Investments, L.P.; Baker Bros. Investments II, L.P.; 667, L.P.; Baker Biotech Fund II (A), L.P.; Baker Brothers Life Sciences, L.P.; and 14159, L.P. Baker Bros. beneficially owns 13,631,336 shares of Synageva preferred stock through the following entities: FBB Associates; Baker Tisch Investments, L.P.; Baker Bros. Investments, L.P.; Baker Bros. Investments II, L.P., 667, L.P.; Baker Brothers Life Sciences, L.P. and 14159, L.P., including 2,352,166 shares of Synageva preferred stock issuable pursuant to notes convertible within 60 days of September 21, 2011. Felix J. Baker has voting and investment power over these shares. The address of Baker Bros. is 667 Madison Avenue, 21st Floor, New York, New York 10065. | |
| (2) | Includes 1,399,663 shares of Synageva preferred stock beneficially owned by Andrew H. Tisch; 1,447,545 shares of Synageva preferred stock beneficially owned by Daniel R. Tisch; 1,447,544 shares of Synageva preferred stock beneficially owned by James S. Tisch; and 1,303,904 shares of Synageva preferred stock beneficially owned by Thomas J. Tisch, which amounts include 223,331, 223,331, 223,330 and 223,331 shares of Synageva preferred stock issuable pursuant to notes convertible within 60 days of September 21, 2011, respectively. The address of Andrew H. Tisch, James S. Tisch, and Thomas J. Tisch is 667 Madison Avenue, New York, NY 10065, and the address of Daniel R. Tisch is c/o TowerView LLC, 500 Park Avenue, New York, N.Y. 10022. | |
| (3) | Includes 948,068 shares of Synageva preferred stock issuable pursuant to notes convertible within 60 days of September 21, 2011. The shares are directly held by New Leaf Ventures II, L.P. (“NLV II”) and indirectly held by New Leaf Venture Management II, L.P. (“NLV Venture Management”), the sole general partner of NLV II, New Leaf Venture Management II, L.L.C. (“NLV Management”), the sole general partner of NLV Venture Management, and the individual managers of NLV Management. The individual managers of NLV Management filing jointly with NLV II, NLV Venture Management and NLV Management are Philippe O. Chambon, James Niedel, Vijay Lathi, Jeani Delagardelle, Ronald Hunt, and Srinivas Akkaraju and each has shared investment and voting power over the securities held by NLV II. The address of NLV II is 2500 Sand Hill Road, Suite 203, Menlo Park, CA 94025. | |
| (4) | Tullis Dickerson Affiliates (“Tullis Dickerson”) beneficially owns 3,836,820 shares of Synageva preferred stock owned by the following: TD Javelin-Capital Fund II, L.P., TD Lighthouse Capital Fund, L.P. and Tullis-Dickerson Capital Focus II, L.P., including 469,984 shares of Synageva preferred stock issuable pursuant to notes convertible within 60 days of September 21, 2011. James Tullis has investment and voting power over the securities held by Tullis Dickerson. The address for Tullis Dickerson is One Stamford Plaza, 263 Tresser Boulevard, Stamford, CT 06901. | |
| (5) | Information in the table and this footnote is based solely upon information contained in a Schedule 13-D/A filed with the SEC on March 16, 2010 (which filing constitutes Amendment No. 9 to a Schedule 13-D filed with the SEC on August 8, 2007) by HealthCor. HealthCor beneficially owns 4,413,657 shares as investment manager of HealthCor L.P., HealthCor Offshore Master Fund, L.P. and HealthCor Hybrid Offshore Master Fund, L.P. HealthCor Associates, LLC is the general partner of HealthCor and Arthur Cohen and Joseph Healy, as the managers of HealthCor Associates, LLC, have voting and investment power over the shares held by the HealthCor Funds. The address of the HealthCor Funds is Carnegie Hall Tower, 152 West 57th Street, 47th Floor, New York, New York 10019. | |
| (6) | Dr. Akkaraju has shared voting and investment power over the 4,148,068 shares of Synageva preferred stock beneficially owned by NLV II. |
| (7) | Felix J. Baker has shared voting and investment power over 3,515,475 shares of Trimeris common stock and 13,631,336 shares of Synageva preferred stock indirectly owned through the following limited partnerships: FBB Associates; Baker Tisch Investments, L.P.; Baker Bros. Investments, L.P.; Baker Bros. Investments II, L.P.; 667, L.P.; Baker Biotech Fund II (A), L.P.; Baker Brothers Life Sciences, L.P.; and 14159, L.P. Additionally, Felix J. Baker owns 110,000 stock options to purchase Trimeris common stock exercisable within 60 days of September 21, 2011. This also includes 6 options to purchase Synageva common stock exercisable within 60 days of September 21, 2011. | |
| (8) | Includes 510 shares of Trimeris common stock owned by Dr. Biggar as of September 21, 2011. Excludes shares of Trimeris common stock beneficially owned by Dr. Felix Baker and Baker Bros., as to which Dr. Biggar disclaims beneficial ownership. | |
| (9) | Consists of 80,000 stock options exercisable within 60 days of September 21, 2011. The address for Mr. Davis is: c/o Trimeris, Inc., 2530 Meridian Parkway, 2nd Floor, Durham, North Carolina 27713. | |
| (10) | Includes 29,623 options to purchase Synageva common stock exercisable within 60 days of September 21, 2011. | |
| (11) | Includes 104,062 options to purchase Synageva common stock exercisable within 60 days of September 21, 2011. | |
| (12) | Includes 32,206 options to purchase Synageva common stock exercisable within 60 days of September 21, 2011 and 3,972 shares of Synageva preferred stock issuable pursuant to notes convertible within 60 days of September 21, 2011. | |
| (13) | Includes 949,645 options to purchase Synageva common stock exercisable within 60 days of September 21, 2011. Includes 694 shares of Synageva preferred stock issuable pursuant to notes convertible within 60 days of September 21, 2011. | |
| (14) | Consists of 76,250 stock options exercisable within 60 days of September 21, 2011. | |
| (15) | Includes 33,625 options to purchase Synageva common stock exercisable within 60 days of September 21, 2011. | |