Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
| ¨ | TRANSITION REPORT PURSUANT TO SECTIONS 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 001-32188
ORAGENICS, INC.
(Exact name of registrant as specified in its charter)
| Florida | 59-3410522 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(IRS Employer Identification No.) |
| 3000 Bayport Drive, Suite 685 Tampa, FL |
33607 | |
| (Address of Principal Executive Offices) | (Zip Code) |
813-286-7900
(Issuer’s Telephone Number, Including Area Code)
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
| Title of each class |
Name of each exchange on which registered | |
| None |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT:
Common stock, par value $.001 per share
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | x | |||
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2). Yes ¨ No x
The aggregate market value of the voting stock held by non-affiliates of the registrant, as of June 30, 2010 was approximately $17,209,815 based upon a last sales price of $7.00 as reported by the OTCBB.
As of March 23, 2011, there were 5,683,076 shares of the registrant’s Common Stock outstanding.
Table of Contents
| ii | ||||||||
| PART I | ||||||||
| ITEM 1. | 1 | |||||||
| ITEM 1A. | 22 | |||||||
| ITEM 2. | 38 | |||||||
| ITEM 3. | 38 | |||||||
| ITEM 4. | 38 | |||||||
| PART II | ||||||||
| ITEM 5. | 39 | |||||||
| ITEM 6. | 39 | |||||||
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
40 | ||||||
| ITEM 7A. | 52 | |||||||
| ITEM 8. | 53 | |||||||
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
53 | ||||||
| ITEM 9A | 53 | |||||||
| ITEM 9B. | 55 | |||||||
| PART III | ||||||||
| ITEM 10. | 56 | |||||||
| ITEM 11. | 59 | |||||||
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS |
68 | ||||||
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
69 | ||||||
| ITEM 14. | 71 | |||||||
| PART IV | ||||||||
| ITEM 15. | 72 | |||||||
| 73 | ||||||||
| F-2–F-3 | ||||||||
| F-4 | ||||||||
| F-5 | ||||||||
| F-6 | ||||||||
| F-7 | ||||||||
| F-8–F-24 | ||||||||
NOTE REGARDING REVERSE STOCK SPLIT
On September 24, 2010, we filed Articles of Amendment to our Amended and Restated Articles of Incorporation with the Secretary of State of the State of Florida to effect a reverse split of our common stock at a ratio of one for twenty. All historical share and per share amounts have been adjusted to reflect the resulting reverse stock split.
i
Table of Contents
FORWARD LOOKING STATEMENTS AND CERTAIN CONSIDERATIONS
This report, along with other documents that are publicly disseminated by us, contains or might contain forward-looking statements within the meaning of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). All statements included in this report and in any subsequent filings made by us with the SEC other than statements of historical fact, that address activities, events or developments that we or our management expect, believe or anticipate will or may occur in the future are forward-looking statements. These statements represent our reasonable judgment on the future based on various factors and using numerous assumptions and are subject to known and unknown risks, uncertainties and other factors that could cause our actual results and financial position to differ materially. We claim the protection of the safe harbor for forward-looking statements provided in the Private Securities Litigation Reform Act of 1995, Section 27A of the Securities Act and Section 21E of the Exchange Act. Examples of forward-looking statements include: (i) projections of revenue, earnings, capital structure and other financial items, (ii) statements of our plans and objectives, (iii) statements of expected future economic performance, and (iv) assumptions underlying statements regarding us or our business. Forward-looking statements can be identified by, among other things, the use of forward-looking language, such as “believes,” “expects,” “estimates,” “may,” “will,” “should,” “could,” “seeks,” “plans,” “intends,” “anticipates” or “scheduled to” or the negatives of those terms, or other variations of those terms or comparable language, or by discussions of strategy or other intentions.
Forward-looking statements are subject to known and unknown risks, uncertainties and other factors that could cause the actual results to differ materially from those contemplated by the statements. The forward-looking information is based on various factors and was derived using numerous assumptions. Important factors that could cause our actual results to be materially different from the forward-looking statements include the following risks and other factors discussed under the Item 1A “Risk Factors” in this Annual Report on Form 10-K. These factors include:
| • | Our inability to continue to raise capital, |
| • | We have incurred significant operating losses since our inception and cannot assure you that we will generate revenues or achieve profitability. |
| • | As a result of our lack of financial liquidity, our auditors have indicated there is substantial doubt about our ability to continue as a going concern. |
| • | If we fail to achieve positive cash flows from our operations and we fail to raise additional capital to meet our capital needs, we may need to significantly curtail operations. |
| • | If we raise additional capital it may be on terms that result in substantial dilution to our existing shareholders, |
| • | We are subject to extensive and costly regulation by the FDA, which must approve our product candidates in development and could restrict or delay the future commercialization of these product candidates. |
| • | We may be unable to achieve commercial viability and acceptance of our ProBiora3 products and proposed product candidates or increase sales of our ProBiora3 products. |
| • | Orders we receive for our consumer products may be subject to terms and conditions that could result in their cancellation or the return of products to us. |
| • | We may become dependent on a few large retail customers for sales of our consumer products. |
| • | We may be unable to successfully operate internationally. |
| • | We may be unable to improve upon, protect and/or enforce our intellectual property. |
| • | We may be unable to enter into strategic collaborations or partnerships for the development, commercialization, manufacturing and distribution of our proposed product candidates or maintain strategic collaborations or partnerships. |
ii
Table of Contents
| • | We may be adversely impacted by a continuing or worsening worldwide financial crises and its impact on consumers, retailers and equity and debt markets as well as our ability to obtain required additional funding to conduct our business. |
| • | We are subject to significant competition. |
| • | As a public company, we must implement additional and expensive finance and accounting systems, procedures and controls as we grow our business and organization to satisfy new reporting requirements, which will increase our costs and require additional management resources. |
| • | Success, timing and expenses of our expected clinical trials. |
| • | If we are unable to raise sufficient capital our license for our SMaRT™ Replacement Therapy and MU-1140 with the University of Florida Research Foundation could be terminated. |
We caution investors that actual results or business conditions may differ materially from those projected or suggested in forward-looking statements as a result of various factors including, but not limited to, those described above and in the Risk Factors section of this report. We cannot assure you that we have identified all the factors that create uncertainties. Moreover, new risks emerge from time to time and it is not possible for our management to predict all risks, nor can we assess the impact of all risks on our business or the extent to which any risk, or combination of risks, may cause actual results to differ from those contained in any forward-looking statements. Readers should not place undue reliance on forward-looking statements. Except as required by applicable law, we undertake no obligation to publicly release the result of any revision of these forward-looking statements to reflect events or circumstances after the date they are made or to reflect the occurrence of unanticipated events.
iii
Table of Contents
PART I
This description contains certain forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from the results discussed in the forward-looking statements as a result of certain of the risks set forth herein. We assume no obligation to update any forward-looking statements contained herein.
Overview
We are a biopharmaceutical company focused primarily on oral health products and novel antibiotics. Within oral health, we are developing our pharmaceutical product candidate, SMaRT Replacement Therapy, and we are also commercializing our oral probiotic blend, ProBiora3. Within antibiotics, we are developing our pharmaceutical product candidate, MU1140-S, and we intend to use our patented, novel organic chemistry platform to create additional antibiotics for therapeutic use.
Our SMaRT Replacement Therapy is designed to be a painless, one-time, five-minute topical treatment applied to the teeth that has the potential to offer lifelong protection against dental caries, or tooth decay. Dental diseases are the most prevalent chronic infectious diseases in the world, affecting up to 90% of schoolchildren and the vast majority of adults. In 2009, Popular Mechanics magazine named SMaRT Replacement Therapy as the “#1 New Biotech Breakthrough That Will Change Medicine.” In the United States alone, the annual cost to treat tooth decay is estimated to be $40 billion. SMaRT is based on the creation of a genetically modified strain of bacteria that colonizes in the oral cavity and replaces native decay-causing bacteria. We commenced a second Phase 1 clinical trial for our SMaRT Replacement Therapy, which we expect to conclude in the second half of 2011.
We have also developed and are commercializing a variety of products that contain the active ingredient ProBiora3, a patent-pending blend of oral probiotics that promotes fresher breath, whiter teeth and support overall oral health. The global probiotics market is expected to be $31.2 billion by 2014, representing a compound annual growth rate, or CAGR, of 12.6% from 2009. We have conducted extensive scientific studies on ProBiora3, in order to market our products under self-affirmed Generally Recognized As Safe status, or GRAS. We sell our ProBiora3 products through multiple distribution channels, and our customers include Walgreens, Kroger, and Garden of Life, among others.
While developing SMaRT Replacement Therapy, members of our scientific team discovered that the SMaRT bacterial strain produces MU1140, a molecule belonging to the novel class of antibiotics known as lantibiotics. MU1140 has proven active preclinically against Gram positive bacteria responsible for a number of healthcare-associated infections, HAIs. The direct cost to the U.S. healthcare system from HAIs is estimated to be up to $45 billion annually. We are in the process of scaling up production of our synthetic form of MU1140, or MU1140-S, and expect to commence preclinical testing during the first half of 2011 and to file an Investigational New Drug, or IND, application with the FDA in mid-2012. The key technology behind the production of MU1140-S is our Differentially Protected Orthogonal Lanthionine Technology platform, or DPOLT, which is a patented, novel organic chemistry platform that we believe will enable the first ever commercial scale, cost-effective production any of the 50 known lantibiotics. We intend to use DPOLT to create a pipeline of lantibiotics for therapeutic use.
Oragenics was founded in 1996 to commercialize the results of more than 30 years of research in oral biology by our principal founder and Chief Scientific Officer, Dr. Jeffrey Hillman. Dr. Hillman earned a DMD from Harvard School of Dental Medicine and a PhD in Molecular Genetics from Harvard University. He began his research career at the Harvard-affiliated Forsyth Institute in Boston, Massachusetts, where he introduced the concept of replacement therapy to prevent tooth decay by using a genetically modified strain of Streptococcus mutans, or S. mutans, to replace the decay-causing strains of S. mutans that are present on human teeth. He subsequently continued this research, now called SMaRT Replacement Therapy, at the University of Florida College of Dentistry. Under Dr. Hillman’s leadership, our scientific team has also developed other technologies such as ProBiora3, MU1140 and our DPOLT platform. Additionally, we are developing non-core technologies that originated from the discoveries of our scientific team, including LPT3-04, which is a potential weight loss product, and PCMAT, which is a biomarker discovery platform, both of which we believe could provide significant potential opportunities for us.
Our Product Portfolio
We are currently developing or commercializing three primary products or product candidates, including SMaRT Replacement Therapy, ProBiora3, and MU1140-S. Our product portfolio is protected by eight issued U.S. patents and eight filed U.S. patent applications, including patents exclusively licensed from the University of Florida Research Foundation, Inc., or UFRF. We are the exclusive worldwide licensee to the patents for SMaRT Replacement Therapy and MU1140, which are owned by the UFRF. We have retained worldwide commercialization rights to each of these products. Additionally, we believe that our SMaRT Replacement Therapy will qualify for a 12-year exclusivity period in the United States under the recently passed Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act.
1
Table of Contents
| Product/Candidate |
Description |
Application |
Status | |||
| SMaRT Replacement Therapy | Genetically modified strain of S. mutans that does not produce lactic acid | Tooth decay | Second Phase 1 clinical trial | |||
| ProBiora3 | Blend of three beneficial oral probiotic bacteria | Oral health, teeth whitening, breath freshening (humans, companion pets) | Commercial (GRAS) | |||
| MU1140-S | Member of lantibiotic class of antibiotics | Healthcare-associated infections | Preclinical testing | |||
SMaRT Replacement Therapy
SMaRT Replacement Therapy is designed to be a painless, one-time, five-minute topical treatment applied to the teeth that has the potential to offer lifelong protection against tooth decay caused by S. mutans, the principal cause of this disease. We have extensively and successfully tested the SMaRT strain for safety and efficacy in laboratory and animal models, and we are in the process of commencing a second Phase 1 clinical trial with an attenuated version of our SMaRT Replacement Therapy.
Market Opportunity
Dental diseases are the most prevalent chronic infectious diseases in the world, affecting up to 90% of schoolchildren and the vast majority of adults. Annual expenditures on the treatment of dental caries in the U.S. are estimated to be $40 billion a year according to the Dental, Oral and Craniofacial Data Resource Center. Tooth decay is characterized by the demineralization of enamel and dentin, eventually resulting in the destruction of the teeth. Dietary sugar is often misperceived as the cause of tooth decay; however, the immediate cause of tooth decay is lactic acid produced by microorganisms that metabolize sugar on the surface of the teeth. Studies suggest that of the approximately 700 oral microorganisms, S. mutans, a bacterium found in virtually all humans, is the principal causative agent in the development of tooth decay. Residing within dental plaque on the surface of teeth, S. mutans derives energy from carbohydrate metabolism as it converts dietary sugar to lactic acid which, in turn, promotes demineralization in enamel and dentin, eventually resulting in a cavity. The rate at which mineral is lost depends on several factors, most importantly the frequency and amount of sugar that is consumed.
Fluoride is used to reduce the effect of lactic acid-based demineralization of enamel and dentin. Despite the widespread use of fluoride in public water systems, toothpastes, dental treatments and sealants, and antiseptic mouth rinses, over 50% of 5-to-9-year-olds and almost 80% of 17-year-olds in the United States have at least one cavity or filling, according to the U.S. Surgeon General. In addition to non-compliance with the behavioral guidelines of the American Dental Association such as routine brushing and flossing, there are several factors that are likely to increase the incidence and frequency of tooth decay, including increasing consumption of both dietary sugar and bottled water. Bottled water generally does not contain fluoride, and thus does not impart any of the protective effects of fluoridated water from public systems. In 2008, U.S. consumers drank more bottled water than any other alcoholic or non-alcoholic beverage, with the exception of carbonated soft drinks, according to the Beverage Marketing Corporation.
Our Solution
Our replacement therapy technology is based on the creation of a genetically altered strain of S. mutans, called SMaRT, which does not produce lactic acid. Our SMaRT strain is engineered to have a selective colonization advantage over native S. mutans strains in that SMaRT produces minute amounts of a lantibiotic that kills off the native strains but leaves the SMaRT strain unharmed. Thus SMaRT Replacement Therapy can permanently replace native lactic acid-producing strains of S. mutans in the oral cavity, thereby potentially providing lifelong protection against the primary cause of tooth decay. The SMaRT strain has been extensively and successfully tested for safety and efficacy in laboratory and animal models.
2
Table of Contents
SMaRT Replacement Therapy is designed to be applied topically to the teeth by a dentist, pediatrician or primary care physician during a routine office visit. A suspension of the SMaRT strain is administered using a cotton-tipped swab during a single five-minute, pain-free treatment. Following treatment, the SMaRT strain should displace the native, decay-causing S. mutans strains over a six to twelve month period and permanently occupy the niche on the tooth surfaces normally occupied by native S. mutans.
Tooth decay is a largely preventable disease through implementation of an appropriate oral care hygiene program including brushing, flossing, irrigation, sealants and antiseptic mouth rinses. Nevertheless, tooth decay remains the most common chronic infectious disease in the world, which indicates that the lack of patient compliance with an overall oral care regimen remains a critical issue in tooth decay prevention. We believe that SMaRT Replacement Therapy addresses the issue of patient compliance by requiring only a one-time,five-minute treatment for the potential lifelong prevention of tooth decay.
Regulatory Status
We initiated our first Phase 1 clinical trial in April 2005, but we found it difficult to find subjects who fit the trial’s highly cautious inclusion and exclusion criteria, particularly with respect to the subjects’ lack of dentition. We concluded this trial early after enrolling only two of the 15 planned subjects. The FDA then recommended that we revise the protocol for the evaluation of ten healthy male subjects, ranging from 18 to 30 years old and with normal dentition, in an institutionalized setting. After we submitted additional information, the FDA issued a clinical hold letter in June 2007 for the proposed trial with the attenuated strain, citing the need for a plan with respect to serious adverse effects; a plan for the eradication of the attenuated strain in trial subjects’ offspring; and a required pregnancy test for female partners of subjects. We submitted additional protocols in response to the FDA’s concerns. In August 2007, the FDA issued a clinical hold letter with required revisions to the protocol for offspring of subjects. We submitted a response to the clinical hold letter in September 2007, and the FDA removed the clinical hold for our Phase 1 trial in the attenuated strain in October 2007.
We have commenced a second Phase 1 clinical trial of an attenuated version of our SMaRT Replacement Therapy, which will examine the safety and genetic stability of the SMaRT strain during administration to ten healthy adult male subjects over a two-week period. As a precautionary measure, this trial will use an attenuated version of the SMaRT strain that is dependent on D-alanine, which is a specific amino acid not normally found in the human diet. D-Alanine will be administered though a mouthwash provided to the patient group, and must be administered daily or the attenuated strain will perish in the oral cavity. We expect the second Phase 1 clinical trial of the attenuated strain, including a six-month follow-up examination of subjects, to be concluded in the second half of 2011. If the second Phase 1 trial of the attenuated strain is successful and if the FDA lifts the clinical hold on the IND for the non-attenuated version of the SMaRT strain, we anticipate that we would conduct a third Phase 1 trial using the non-attenuated SMaRT strain instead of the attenuated version.
The SMaRT strain has been extensively and successfully tested in the laboratory as well as in animal models, and has demonstrated the following:
| • | No lactic acid creation under any cultivation conditions tested; |
| • | Dramatically reduced ability to cause tooth decay; |
| • | Genetic stability as demonstrated in mixed culture and biofilm studies and in rodent model studies; |
| • | Production of a level of MU1140 that is comparable to its wild-type parent strain, which was previously shown to readily and persistently colonize the human oral cavity; and |
| • | Aggressive displacement of native, decay-causing strains of S. mutans and preemptive colonization of its niche on the teeth of laboratory rats. |
3
Table of Contents
In addition, during preclinical and early-stage clinical testing of our SMaRT Replacement Therapy, we observed the following:
| • | No adverse side effects in either acute or chronic testing in rodent models; |
| • | Colonization of the treated subjects following a five-minute application of SMaRT Replacement Therapy in our first Phase 1 study using the attenuated strain; and |
| • | No adverse side effects during our first Phase 1 study. |
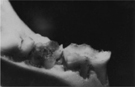
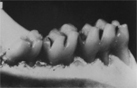
We conducted a preclinical study in which one group of rats was treated with the native S. mutans strain, and a second group of rats was treated with the SMaRT strain. Both groups of rats were subsequently fed a high-sugar diet for eight weeks. The group of rats treated with the SMaRT strain showed dramatically increased protection from tooth decay as compared to the group of rats treated with native S. mutans.
Rodent Teeth Treated with Native S. mutans (left) and SMaRT Strain (right)
|
|
Our Strategy
Our strategy is to develop our SMaRT Replacement Therapy through Phase 1 clinical trials. Upon the successful completion of Phase 1 trials, we intend to license our SMaRT Replacement Therapy to, or partner with, a major pharmaceutical company. We believe that the completion of Phase 1 trials would definitively establish clinical safety and therefore would represent a significant milestone in the development of SMaRT Replacement Therapy, which we anticipate would result in a substantial increase in the value of this technology. If we are unable to negotiate acceptable terms with a licensee or partner after Phase 1 trials have been completed, and assuming we are not required to undertake Phase 2 trials, we may consider pursuing Phase 3 clinical trials independently. However, we anticipate that we would need to partner with a major pharmaceutical company prior to marketing the product if our SMaRT Replacement Therapy ultimately achieves FDA approval. For our second Phase 1 clinical trial we have retained PRA International as the clinical research organization for clinical trials management services.
Manufacturing
The manufacturing methods for producing the SMaRT strain of S. mutans are standard Good Manufacturing Practice, or GMP, fermentation techniques. These techniques involve culturing bacteria in large vessels and harvesting them at saturation by centrifugation or filtration. The cells are then freeze dried or suspended in a pharmaceutical medium appropriate for application in the human oral cavity. These manufacturing methods are commonplace and readily available within the pharmaceutical industry. A single dose of our SMaRT Replacement Therapy contains approximately 10 billion S. mutans cells, which represents approximately 10 milliliters of fermentation product. The SMaRT strain grows readily in a variety of cultivation media and under a variety of common growth conditions including both aerobic and anaerobic incubations. The SMaRT strain can also utilize various carbon and nitrogen sources and is highly acid tolerant. There is no significant limitation to the manufacturing scale of our SMaRT strain other than the size of the containment vessel. For our first Phase 1 clinical trial, we engaged a contract manufacturer to produce an attenuated version our SMaRT strain, using a standard operating procedure provided by us that we believe is readily transferable to outside contract manufacturers with fermentation capabilities.
4
Table of Contents
ProBiora3
ProBiora3 is a proprietary blend of three naturally occurring strains of beneficial bacteria, including Streptococcus oralis, Streptococcus uberis, and Streptococcus rattus, which promote fresher breath, whiter teeth, and support overall oral health. We believe that ProBiora3 is the most comprehensive oral probiotic technology currently available in the oral care market. The scientific basis for the oral health and cosmetic benefits provided by these three strains of bacteria has been documented in numerous peer-reviewed publications over the last 30 years. We promote ProBiora3 as the active ingredient in our over-the-counter consumer branded products, including EvoraPlus, EvoraKids, Teddy’s Pride and the professional branded product, EvoraPro. EvoraPlus and EvoraKids are flavored probiotic tablets intended for twice-daily use by adults and children, respectively, after brushing their teeth. Teddy’s Pride is intended for companion pets such as cats and dogs, and comes in powder form, which is odorless and tasteless. The powder is intended to be sprinkled on a pet’s food once per day. EvoraPro is a professional strength product designed for the dental office channel. In addition to our house-branded products, we also market ProBiora3 as an active ingredient for private label products, as well as in bulk for licensing applications.
Market Opportunity
Probiotics are live microorganisms that confer a health benefit to their host when administered in sufficient amounts. The beneficial bacteria in a probiotic formulation help to maintain a healthy balance of bacteria in the body. Examples of common probiotic applications are yogurt containing live cultures, a-cidophilus capsules to improve digestion, and products for improved immune system and vaginal and urinary tract health. According to MarketsandMarkets, the global probiotics market is expected to reach $31.2 billion by 2014, representing a CAGR of 12.6% from 2009 to 2014. Probiotics products are relatively more common in Asia and Europe, with Europe accounting for nearly 42% and Asia accounting for 30% of the global market. The probiotics market in the United States, however, is emerging, and products that address gastrointestinal problems and other uses are rapidly becoming available, especially as dietary supplements and cultured foods and beverages. The Probiotic Foods & Beverages category currently represents over 75% of the overall probiotics market in the United States.
| • | Oral Care: The oral care market in the United States was $9.1 billion in 2008 and is expected to reach $10.9 billion by 2014, according to Packaged Facts. Packaged Facts segments this market into three comprehensive product categories: (i) Dental Preparations, which include toothpastes, tooth cleaners/whiteners, and denture products; (ii) Implements/Appliances, including toothbrushes, dental floss and irrigators; and (iii) Gum/Mouthwash/Breath Fresheners, which represented $2.4 billion of the market in 2009. |
| • | Companion Pets: In 2009, approximately 62% of U.S. households owned a pet, with an estimated 38.2 million and 45.6 million households owning cats and dogs, respectively, according to the American Pet Products Association, or APPA. The APPA also estimates that total 2010 U.S. pet industry expenditures were $47 billion, representing an increase of 4.3% from 2009. Within this market, approximately $10.4 billion was spent on Supplies/OTC Medicine, representing a 4.0% increase over 2009. |
Our Solution
ProBiora3 is a blend of three naturally occurring strains of bacteria for use in the promotion of oral health, including Streptococcus oralis strain KJ3SM, or S. oralis; Streptococcus uberis strain KJ2SM, or S. uberis; and Streptococcus rattus strain JH145SM, or S. rattus. In a healthy human oral cavity, S. oralis and S. uberis are commonly found in significant amounts, and conversely, the levels of bacteria associated with periodontal disease are usually quite low. The opposite situation prevails in periodontal disease sites, at which the beneficial bacteria S. oralis and S. uberis are usually undetectable. Our scientists have demonstrated that S. oralis and S. uberis produce hydrogen peroxide, which interferes with the growth of certain potentially harmful periodontal bacteria, and also gently and naturally whitens teeth. The third bacterial strain in our ProBiora3 blend, S. rattus, is able to establish and maintain a healthy balance of bacteria on the tooth surfaces by competing with certain other potentially harmful bacteria associated with tooth decay.
ProBiora3 has been extensively tested for safety and efficacy in the laboratory and in animal and human trials. In our pilot human study, a twice-daily administration of ProBiora3 was well tolerated by subjects and no safety issues were observed. ProBiora3 produced substantial decreases in the numbers of key potentially pathogenic bacteria associated with tooth decay and periodontal disease in young healthy adults.
5
Table of Contents
We market products containing ProBiora3 under our own house brand names, and have branded ProBiora3 as an active ingredient for licensing and private labeling. Our house brand products contain different ratios, or blends, of the three natural strains contained in ProBiora3, which vary depending on the intended use of the product. Our ProBiora3 products are designed for repetitive use in order to achieve the intended benefits, which we believe provides us with the potential for recurring revenues as consumers who continue to seek the benefits of our products will continue to make purchases. Our ProBiora3 products include:
| • | EvoraPlus: a product with equal weight of all three strains that is optimally designed for the general consumer market. EvoraPlus was initially launched in December 2008, but distribution was limited to sales through our own website. In March 2010, we obtained national distribution for EvoraPlus in the domestic mass retail channel with the addition of Walgreens as a vendor. During 2010 we continued to expand our distribution in the domestic mass retail channel, and we believe that EvoraPlus is currently available for stocking at over 11,000 retail stores. EvoraPlus is a mint-flavored probiotic tablet packaged in a 60-unit box with four 15-dose blister packs, representing a one-month supply. The intended use for EvoraPlus is for consumers to take one tablet twice per day after brushing their teeth. |
| • | EvoraKids: a product that has higher levels of S. rattus, which addresses dental health, but reduced levels of S. oralis and S. uberis since periodontal disease is not typically a pediatric concern. EvoraKids is a fruit-flavored probiotic tablet packaged in a 60-unit box with four 15-dose blister packs, representing a one-month supply. The intended use for EvoraKids is for consumers to take one tablet twice per day after brushing their teeth. We launched distribution of EvoraKids in January 2010. |
| • | Teddy’s Pride: a product that has higher levels of S. oralis and S. uberis, which address tooth staining and breath problems common to dogs and cats, but reduced levels of S. rattus since tooth decay is not typically a concern in companion pets. Teddy’s Pride comes in powder form, which is odorless and tasteless. The powder is intended to be sprinkled on a pet’s food once per day. It is sold in a jar containing a measuring scoop that provides the recommended dosage per application, representing a two-month supply. We launched Teddy’s Pride in October 2009. We anticipate continuing to use our Teddy’s Pride brand internationally and rebrand Teddy’s Pride for the domestic market as EvoraPet. |
| • | EvoraPro: a professional strength version of EvoraPlus that is designed for the dental office channel. EvoraPro is packaged as a ten-dose blister pack and is shrink-wrapped with one box of EvoraPlus. The intended use for EvoraPro is to take one tablet per day for ten days after a routine dental cleaning. EvoraPro can only be purchased from a professional dental office. EvoraPro was launched in early August 2010. |
Package and Delivery Transition
We are continually attentive to the needs of the market and ultimate consumers regarding the use of our ProBiora3 products and as such continue to seek ways to revise and improve on our product delivery mechanisms. For example, we are undergoing a change from a blister pack of 60 tablets to a bottled container of 30 tablets to be taken once daily (as opposed to twice daily) for our consumer ProBiora3 products based on our understanding of customer preferences. Such a change in delivery mechanisms or packaging will result in increased expenses while the change is being implemented. In December 2010, we setup reserves for inventory in the amount of $255,814 and sales returns in the amount of $105,588 to replace our existing inventory and customer held inventory as a result of this change.
Our Regulatory Strategy
We market ProBiora3 as a food ingredient utilizing self-affirmed Generally Recognized as Safe, or GRAS, status. GRAS is available for food products that are generally recognized as being safe for human use and do not claim to treat, prevent, or cure a disease. Furthermore, food products that make only cosmetic or structure-function claims are typically able to enter the market through what is known as self-affirmed GRAS status, which designates that we have performed all necessary research, including the formation of an expert panel to review safety concerns, and are prepared to use these findings to defend ProBiora3’s self-affirmed GRAS status. In 2008, we convened a panel, the members of which we believed to be qualified as experts by their scientific training and professional experience, to analyze and evaluate the safety data for ProBiora3. After review, the panel concluded that the safety data of ProBiora3 was sufficient to support our claim to self-affirmed GRAS status for human consumption. The same data dossier could be applied to support the safety for companion pet consumption of ProBiora3.
6
Table of Contents
Our marketing for ProBiora3 includes the cosmetic claims of teeth whitening and breath freshening, along with the general structure-function claim that ProBiora3 supports oral health. Regulations vary in markets outside the United States and it may be possible to assert other benefits including health and disease prevention claims associated with probiotic use, especially after independent clinical studies have been completed and appropriate regulatory fillings are approved. At present, we are aware of several independent academic studies that have been initiated on a variety of potential health and cosmetic benefits associated with ProBiora3 probiotic use by humans.
Sales, Marketing and Distribution
All of our house-branded ProBiora3 products have been launched and are available through various distribution channels. We have selected our distribution channels by focusing on our potential channel impact, as well as potential return on marketing expenditures.
| • | Mass Retail: The mass retail channel encompasses several sub-channels including large national retail stores, mass drugstore chains, independent drugstores, and grocery stores. In order to develop and manage this channel, we retained a team of independent manufacturers’ representatives with industry expertise and strong relationships with the buyers for many of the large national mass retailers. We worked with this team to identify the mass drugstore channel as the lead sub-channel in the overall mass retail channel. Mass drugstores are typically the first to adopt a new wellness-related product or technology, and once mass drugstores adopt such a product, other sub-channels typically follow. In March 2010 we received an initial order from Walgreens for EvoraPlus, and in April 2010, we received an initial order from Rite Aid for EvoraPlus. In addition to these mass drugstore chains, we received initial orders for EvoraPlus from a number of larger national and regional mass retailers, including GNC, Kroger, A&P Supermarkets, Pathmark, Albertsons Supermarkets, Sweetbay Supermarkets, Fred Meyer, Winn Dixie, Meijers, Harris Teeter and Hannaford Supermarkets, among others. While we were pleased with the level of initial success we achieved in establishing the mass retail distribution channel, the maintenance, continued use and expansion of this channel requires us to commit to expend capital resources on advertising and marketing campaigns. Because our available capital resources currently limit our ability to engage in significant advertising and marketing campaigns, we are undertaking an evaluation of the continued pursuit of the mass retail channel relative to other options. In connection with our ongoing evaluation of this channel in first quarter of 2011 we determined to pull our EvoraPlus product from the store shelves of Rite Aid. Also during the first quarter of 2011, GNC has refocused their strategy on selling higher margin GNC branded products and as such the decision was made to end distribution of our EvoraPlus product. Through this action we reduced our mass retail store presence from approximately 17,000 stores to just over 11,000 stores. |
| • | Direct-to-Consumer: The direct-to-consumer channel encompasses four sub-channels, including (i) Internet sales through our own websites; (ii) direct mail; (iii) direct-response television, or DRTV, which is usually initiated through an infomercial; and (iv) electronic-response television, or ERTV, which entails marketing through television shopping networks such as Home Shopping Network and QVC. |
| i. | Internet sales: We currently operate one corporate website through which we market our branded products direct to the consumer. An “Oragenics Store” provides the consumer with access to purchase our products. We will be initiating in the first quarter of 2011 an affiliate marketing program whereby we will pay external website operators click-through revenues when a customer visits our websites via an affiliate site and subsequently makes a purchase. |
| ii. | Direct mail: We are currently in discussions with one of the largest direct mailers of nutraceutical products in the United States and developing our own direct mail piece for our branded products. |
7
Table of Contents
| iii. | DRTV: We are developing a two-minute spot infomercial for Teddy’s Pride, our pet oral care product, that we expect to test on select networks and in select markets. The infomercial has been designed to promote a direct response from viewers. If tests prove successful and we are able to forecast a positive return on our marketing spend, we would anticipate expanding the geographic area and broadcasting frequency of the infomercial. |
| iv. | ERTV: We have been in discussions with both of the major domestic ERTV operators as well as companies that have established brands on their respective channels. We anticipate consummating one or more ERTV marketing opportunities by the end of second quarter 2011. |
| • | Professional Offices: The professional offices channel encompasses several sub-channels, including (i) the dental professional channel, which includes dentists, orthodontists and dental hygienists; (ii) the veterinarian professional channel; and (iii) the alternative medicine professional channel, which includes chiropractors, massage therapists and occupational therapists, among others. In August 2010, we launched EvoraPro, which is a product exclusively for the dental professional sub-channel. EvoraPro is an extra-strength, probiotic designed to be taken after dental cleaning or treatment. It is coupled with a box of EvoraPlus to be used once the EvoraPro has been exhausted. In January 2011 we entered into a distribution agreement with a leading distributor of products to the dental professional market with approximately 1,500 sales representatives. We have also established an affiliate marketing program through which dental professionals can earn recurring revenues from their patients’ subsequent purchases of EvoraPlus. If successful, we intend on following a similar plan to penetrate the other sub-channels in the professional offices channel. We would look to initiate a campaign in the veterinarian channel by the end of the second quarter 2011. |
| • | Private Label: The private label channel encompasses arrangements whereby we or third-party manufacturers market our products for resale under a third-party’s brand name. We typically establish private labeling arrangements in order to leverage an existing company’s brand equity and distribution channels. The first major private labeling agreement we consummated was with Garden of Life, which is a leading U.S. nutritional supplement products brand. Garden of Life has contracted to sell our EvoraPlus product under the brand name Probiotic Smile. Garden of Life sells exclusively in the health food channel, which includes many stores geographically disbursed around the United States. Oragenics has entered into agreements with Nutrahealth (US) and Pharmaforce (Denmark) for the distribution of product incorporating ProBiora3. Another notable private labeling sub-channel is the multi-level marketing, or MLM, channel. We have been in discussions with a number of large MLM companies regarding private labeling opportunities for our products. |
| • | International: Since the launch of our first product, EvoraPlus, we have entered into exclusive distribution agreements for our products internationally in various geographic locations. For example, we have executed distributorship agreements, with Natural Pharma International (Italy, Slovakia), Australian Pharmaceuticals Industries (Australia, New Zealand), Zooglobe (Poland), Benelux Cosmetics (Belgium, Netherlands, Luxembourg), Vetcom (Korea), Best Supplies (Taiwan), and Celgen. The international distributorship agreements we have entered into to date typically provide for exclusivity and require that the distributors provide upfront payment to us either by irrevocable letters of credit or wire transfers prior to our initiating production and as a result, we believe that we do not bear any credit risk with such agreements. We also require distributors to take possession of product at our manufacturing facility, which substantially reduces our inventory risk. We continually evaluate the effectiveness of these arrangements and may seek to terminate distributorship agreements in which limited purchase activity occurs. |
| • | Licensing/Bulk: The licensing/bulk channel encompasses the incorporation of ProBiora3 as an active ingredient into existing branded products. We have been in discussions with a number of companies regarding the licensing or bulk sale of ProBiora3. |
8
Table of Contents
Manufacturing
When produced, ProBiora3 comes in powder form. ProBiora3 is manufactured by separate fermentation of each of the three strains. The cells are recovered by centrifugation or filtration and freeze dried. Experimentally determined amounts of the resulting powders are blended with natural bulking agents to deliver the proper number of viable cells of each strain per unit weight. ProBiora3 for human use may be incorporated into various delivery vehicles; for example, in the case of EvoraPlus and EvoraKids, flavoring agents are added and the powder is pressed into tablets, which are sealed in blister packs. We are undergoing a change from blister pack tablets to one a day bottled product based on our understanding of customer preferences. In the case of Teddy’s Pride, the powder is not flavored and is simply added in bulk to a plastic container. Freeze-dried cells in ProBiora3-containing products are stable for up to 18 months after manufacture when kept in cool, dry conditions. The cells are revitalized when they come in contact with moisture, for example the saliva present in the oral cavity.
We have contracted with multiple manufacturers to: (i) produce our active ingredient, ProBiora3, (ii) blend and tablet EvoraPlus, EvoraKids, Teddy’s Pride and EvoraPro, and (iii) package our products. Each of our contract manufacturers has the ability to scale production as needed. With each manufacturer, we place orders for components or finished product to be produced for a fixed fee which we are expected to pay upon completion of the manufacturing process. Packaged probiotics products are shipped to us or to a destination specified by us, which is a central distribution center in the case of a mass retail customer or a private label distributor. We currently maintain an inventory of our products for Internet sales and other sales to distributors. We believe our arrangements with our contract manufacturers are satisfactory to meet our current and expected future needs. We have qualified and used at least two contract manufacturers for each step in our manufacturing process, although we do not have a long-term supply agreement or commitment with any of our manufacturers.
MU1140 and Other Lantibiotics
Our lantibiotic, MU1140, was discovered by Dr. Hillman in the course of developing SMaRT Replacement Therapy. MU1140 is a potent antibiotic that is naturally produced by the parent of the SMaRT strain, and we have produced a synthetic version of MU1140 known as MU1140-S. MU1140 is active against all Gram positive bacteria against which it has been tested, including those responsible for a variety of healthcare-associated infections, or HAIs. The key technology that enables our production of MU1140-S is our Differentially Protected Orthogonal Lanthionine Technology, or DPOLT, which is a patented, novel organic chemistry synthesis platform developed by our scientific team. We reported the successful, analytical scale synthesis of MU1140-S using DPOLT in October 2008, and thus achieved what we believe will lead to the first-ever synthetic route to commercial-scale production of lantibiotics.
Market Opportunity
The most common HAIs are caused by drug-resistant bacteria, including methicillin-resistant Staphylococcus aureus, or MRSA; vancomycin-resistant Enterococcus faecalis, or VRE; and Clostridium difficile, or C. diff. According to the Centers for Disease Control and Prevention, or CDC, HAIs are estimated to occur in approximately 5% of all acute-care hospitalizations, based on the 35 million patients admitted to 7,000 acute-care institutions in the United States, with an annual incidence of approximately 1.7 million cases, which result in 99,000 deaths. The CDC also estimates that the total direct medical cost to the U.S. healthcare system from HAIs is between $35.7 billion to $45 billion annually. HAIs are estimated to more than double the mortality and morbidity risks of any admitted patient in a U.S. hospital, which is the equivalent of 350,000 years of life lost annually. The critical care market for antibiotics is approximately $7 billion in the United States alone. Cubicin, a Gram positive lipopeptide antibiotic which was recently introduced by the biotechnology company Cubist, had 2010 sales of $600 million in the United States.
The need for novel antibiotics is increasing as a result of the growing resistance of target pathogens. In 2002, the CDC estimated that pathogenic bacteria resistant to known antibiotics cause between 6.3% and 89.1% of HAIs, and individual hospitals have resistance rates as high as 70% for many Gram positive infections. HAIs are not exclusively a problem in the United States as the rest of the world has also seen a dramatic rise in HAIs during the last decade. Vancomycin, which was introduced in 1956, has served as the last line of defense against certain life-threatening infections, and, more recently, Cubicin has also served in this capacity, but bacterial resistance to these drugs has been growing at an increasing rate. Novel antibiotics have become increasingly scarce as major pharmaceutical companies have focused more research and development resources on lifestyle drugs and fewer resources on specialty pharmaceuticals such as antibiotics. Between 1983 and 1987, 16 new antibiotics were approved by the FDA. Twenty years later, from 2003 to 2007, only five new antibiotics were approved, of which only two possessed a novel mechanism of action.
9
Table of Contents
Lantibiotics such as MU1140 are highly modified peptide antibiotics made by a small group of Gram positive bacterial species. Approximately 50 lantibiotics have been discovered since 1927 when the first lantibiotic, nisin, was discovered. Lantibiotics are known to be potent antibiotic agents, however, all attempts to investigate their usefulness have met with uniform failure due to the inability to produce sufficient pure amounts of any of these molecules to be able to test them as a therapeutic agent for the treatment of infectious diseases. Standard fermentation methods, such as those used to make a variety of other antibiotics, typically result in production of only minute amounts of the lantibiotic. In cases where large amounts of a lantibiotic are made, such as with nisin, the unique chemical structure of lantibiotics has prevented the necessary purification needed for clinical testing.
Our Solution
MU1140 has demonstrated activity against a wide variety of disease-causing Gram positive bacteria, including MRSA, VRE, C. diff., Mycobacterium tuberculosis, or M. tuberculosis, and anthrax. We have performed extensive preclinical testing on MU1140, which has demonstrated the molecule’s novel mechanism of action. In order to produce sufficient quantities for our clinical trials and commercialization efforts, we intend to use a synthetic version of MU1140, known as MU1140-S.
We created MU1140-S using our patented, novel organic chemistry synthesis platform known as DPOLT. We believe that DPOLT will enable large-scale, cost-effective production of clinical grade MU1140-S. We reported the successful, analytical scale synthesis of MU1140-S using DPOLT in October 2008, which we believe will lead to the first-ever synthetic route to commercial-scale production of a lantibiotic. In addition, we believe that DPOLT will allow us to synthetically produce any of the 50 known lantibiotics due to the shared chemical structure features of this class of molecule. We intend to use DPOLT to create a pipeline of lantibiotics for therapeutic use.
Regulatory Status
We have performed extensive preclinical testing using native MU1140, which demonstrated the following features:
| • | Bactericidal activity against Gram positive species and against both replicating and non-replicating M. tuberculosis; |
| • | Unusual chemical structure, which makes it extremely stable; |
| • | No immune response in a variety of animal models, even with the use of strong adjuvants and carriers; |
| • | Negligible toxicity when supra-therapeutic doses were tested in yeast, and fibroblast and kidney cell lines; |
In vivo efficacy in mouse and rat models, in which animals were infected intraperitoneally with MRSA (60xLD50) and MU1140 was administered intravenously at doses well below its maximum tolerated dose;
| • | Novel mechanism of action that involves binding to and abducting Lipid II, which is required for cell wall biosynthesis; |
| • | No spontaneous, genetically stable resistant mutants to MU1140; |
| • | Synergy with an aminoglycoside; and |
| • | Good pharmaceutical properties. |
We expect to conclude the preclinical testing of MU1140-S, including toxicity testing in rodent and non-rodent animal models, during the second half of 2011. Following successful completion of preclinical testing we would expect to file an Investigational New Drug, or IND, application with the FDA in early 2012. We estimate that, once commenced, the regulatory process will require at least four years of clinical testing and the application and FDA approval of a New Drug Application, or NDA, before MU1140-S would be commercially available. We have engaged Celerion (formerly known as MDS Pharma Services) on a fee-for-service basis to represent us in regulatory meetings with the FDA, and to perform the first-in-human trials with MU1140-S.
10
Table of Contents
Our Strategy
We intend to develop MU1140-S through Phase 1 clinical trials. If MU1140-S successfully completes Phase 1 trials, we believe that its value will substantially increase, and we would then seek to license MU1140-S to or partner with a major pharmaceutical company. If we are unable to consummate an acceptable licensing or partnership arrangement, we may pursue Phase 2 clinical trials independently.
Analysis of the 50 known lantibiotics suggests that there are possibly six to ten subclasses of lantibiotics as classified by known mechanisms of action, spectra of activity, or structural characteristics. In addition to MU1140-S, we intend to utilize DPOLT to synthesize additional lantibiotics of interest in the future.
Manufacturing
We have retained Almac Sciences, a leading contract manufacturer, to refine and scale-up GMP production of MU1140-S. Through this relationship, we expect to have access to sufficient amounts of MU1140-S during the first half of 2011 which will enable preliminary testing to demonstrate equivalence between the synthetic and native molecule.
Additional Areas of Development
As part of our past research efforts, we have identified and filed patent applications covering two technologies that we may seek to further develop internally or monetize through a sale, license, or partnership in the future. These areas include LPT3-04, our weight loss product, and PCMAT, our biomarker discovery platform.
Weight Loss Product (LPT3-04)
LPT3-04 is a naturally occurring compound, which is normally consumed in the human diet in small amounts. In the course of our SMaRT Replacement Therapy research, we discovered program that consumption of significantly larger amounts of LPT3-04 resulted in dose-dependent weight loss in experimental animal models. The mechanism of action appears to be induction of apoptosis, or programmed cell death, specifically in white fat cells. LPT3-04 consumption in the required amounts has been shown to be safe in humans. Anecdotally, weight loss has been observed in human volunteers. Due to the natural sweetness of LPT3-04 and the relatively large amounts of it that need to be consumed on a daily basis to achieve the desired weight loss effect, current product development efforts are focused on incorporating the compound into bars, milkshakes, and other food products. We expect to use these food products in a blinded placebo-controlled study to begin in 2011. We have submitted a patent application for the use of LPT3-04 for weight regulation with the United States Patent and Trademark Office, or U.S. PTO.
Biomarker Discovery Platform (PCMAT)
Our biomarker discovery platform is based on our Proteomics-based Change Mediated Antigen Technology, or PCMAT, and was discovered by members of our scientific team while searching for protein targets associated with the diagnosis of periodontal disease. This technology rapidly identifies proteins that are expressed when a cell undergoes any sort of change. Such proteins are excellent targets for medical diagnostics and therapeutic strategies. PCMAT is able to identify proteins shed from diseased tissues into bodily fluids such as blood, saliva and urine. We believe that PCMAT is faster, more cost-efficient and significantly more sensitive than competing technologies such as differential proteomics and microarrays. In addition, our technology uses the actual diseased host rather than an animal model, so that biomarkers that we discover are more likely to be of high clinical value. We have identified several widespread disease states that we intend to pursue. If we are able to discover protein targets with sufficient degrees of sensitivity and specificity, we intend to license these targets to major pharmaceutical or medical diagnostics companies.
Our In-Licensed Technology Agreements
SMaRT Replacement Therapy
We have exclusively licensed the intellectual property for our replacement therapy technology from the University of Florida Research Foundation, Inc., or the UFRF. The original license agreement was dated August 4, 1998 and was subsequently amended on September 15, 2000, July 10, 2002, September 25, 2002 and March 17, 2003. The amended license agreement provides us with an exclusive worldwide license to make, use and sell products and processes covered by Patent No. 5,607,672, entitled “Replacement Therapy for Dental Caries”, which was filed in the U.S. PTO on June 7, 1995 and made effective on March 4, 1997. The patent will expire on June 7, 2015. Our license is for the period of the patent, subject to the performance of terms and conditions contained therein. The patent covers the genetically altered strain of S. mutans which does not produce lactic acid, a pharmaceutical composition for administering the genetically altered strain and the method of preventing tooth decay by administering the strain. See “Our Intellectual Property.”
11
Table of Contents
We issued 29,997 shares of our common stock to the UFRF as partial consideration for the initial license.
MU1140
We have exclusively licensed the intellectual property for our MU1140 lantibiotic technology from the UFRF. The original license agreement was dated June 22, 2000 and was subsequently amended on September 15, 2000, July 10, 2002, September 25, 2002 and March 17, 2003. The amended license agreement provides us with an exclusive worldwide license to make, use and sell products and processes covered by Patent No. 5,932,469 entitled “Antimicrobial Polypeptide, Nucleic Acid and Methods of Use.” Our license is for the period of the patent, subject to the performance of terms and conditions contained therein. See “Our Intellectual Property.”
Additional Terms of License Agreements
In the amended license agreements for SMaRT Replacement Therapy and MU1140 the UFRF has reserved the right to use and sell products and services for research purposes only. The amended license agreements also provide the UFRF with a license, for research purposes only, to any improvements that we make to the products and processes covered by the patents.
We are obligated to pay 5% of the selling price of any products developed from the licensed technologies that we may sell as royalty to the UFRF. In addition, if we sublicense any rights granted by the amended license agreements, we are obligated to pay the UFRF 20% of all revenues received from the sublicenses, excluding monies received solely for development costs.
We are also obligated to make minimum annual royalty payments to the UFRF for the term of the amended license agreement in the amount of $50,000 by the end of each year for each license agreement. The minimum royalty payments are required to be paid in advance on a quarterly basis. For the SMaRT Replacement Therapy and MU1140 minimum royalty payments, we must pay the UFRF an aggregate of $100,000 which is required to be paid in equal quarterly installments of $25,000.
Under the terms of the amended license agreements, in each calendar year and in addition to the royalty payment obligations, we are obligated to spend, or cause to be spent, an aggregate of $1,000,000 on the research, development, and regulatory prosecution of our SMaRT Replacement Therapy and MU1140 technologies combined, until a product which is covered wholly or partially by the claims of the patent, or is manufactured using a process which is covered wholly or partially by the claims of the patent, is sold commercially. If we fail to make these minimum research and development expenditures, the UFRF may terminate our license agreement.
We must also pay all patent costs and expenses incurred by the UFRF for the preparation, filing, prosecution, issuance and maintenance of the patent.
We have agreed to indemnify and hold the UFRF harmless from any damages caused as a result of the production, manufacture, sale, use, lease, consumption or advertisement of the product.
We are required to maintain liability insurance coverage appropriate to the risk involved in marketing our products. Our liability insurance has been renewed through March 2012, however, there is no assurance that we can obtain continued coverage on reasonable terms.
The amended license agreements further provide that the U.S. government funded research grant No. RO1 DE04529 during the course of or under which the licensed inventions covered by the patent were conceived. As such the U.S. government is entitled, as a right, to a nonexclusive, nontransferable, irrevocable, paid-up license to practice or have practiced the inventions of such patents for governmental purposes.
In order to protect our license rights and their patents, we or the UFRF may have to file lawsuits and obtain injunctions. If we do that, we will have to spend large sums of money for attorney fees in order to obtain the injunctions. Even if we do obtain the injunctions, there is no assurance that those infringing on our licenses or the UFRF’s patents will comply with the injunctions. Further, we may not have adequate funds available to prosecute actions to protect or to defend the licenses and patents, in which case those infringing on the licenses and patents could continue to do so in the future.
12
Table of Contents
Government Regulations
The formulation, manufacturing, processing, packaging, labeling, advertising, distribution and sale of our products are subject to regulation by federal agencies, including, but not limited to the Food and Drug Administration, or FDA, and the Federal
Trade Commission, or FTC. These activities also are regulated by various agencies of the states, localities and foreign countries in which our products are sold. In particular, the FDA, under the Federal Food, Drug, and Cosmetic Act, or FDCA, regulates the safety, manufacturing, labeling and distribution of drugs, medical devices, food, and dietary supplements. In addition, the FTC has primary jurisdiction to regulate the advertising of drugs, medical devices, food and dietary supplements.
In foreign countries these same activities may be regulated by Ministries of Health, or other local regulatory agencies. The manner in which products sold in foreign countries are registered, how they are formulated, or what claims may be permitted may differ from similar products and practices in the United States.
FDA Regulation—Food
Under the FDCA, the FDA is responsible for ensuring that foods are safe, wholesome, and correctly labeled. The FDA enforces statutory prohibitions against misbranded and adulterated foods, and establishes safety standards for food processing and ingredients, manufacturing procedures for processed foods, and labeling standards for food products.
All facilities engaged in manufacturing, processing, packing or holding food for consumption in the United States must be registered with FDA before such activities begin. Those who manufacture, package, or hold food must comply with the Good Manufacturing Practices, or GMPs, for foods. The GMPs describe the methods, equipment, facilities, and controls for producing processed food, including requirements for personnel such as education, training and cleanliness requirements; proper maintenance and sanitization of buildings, facilities, and equipment; and processes and controls.
Acceptable claims for foods fall into three categories: health claims, structure/function claims and nutrient content claims. Health claims describe a relationship between a food, food component, or dietary ingredient and reducing the risk of a disease or health-related condition. The FDA authorizes these types of health claims based on an extensive review of the scientific literature, generally as a result of the submission of a health claim petition. Manufacturers also may make certain health claims based on “authoritative statements” from a scientific body of the U.S. Government or the National Academy of Sciences. Structure/function claims describe the role of a nutrient or dietary ingredient intended to affect or maintain normal structure or function of the body, and may characterize the means by which a nutrient or dietary ingredient acts to maintain such structure or function. Nutrient content claims expressly or by implication characterize the level of a nutrient in a food, by using terms such as “free,” “high” or “low.” The FDA’s regulations define the nutrient content claims that may be used and the requirements for making such claims.
Labels for food must not be false or misleading. Required information for labels includes the name of the food, the net quantity, the name and address of the manufacturer, packer or distributor, the ingredient list, and a Nutrition Facts label. In addition to the information required to be in a Nutrition Facts label, other nutrients must be included in the Nutrition Facts label if the nutrients are added as a nutrient supplement to the food, if the label makes a nutrition claim about them, or if advertising or product literature connects the nutrients to the food. The FDA considers information that is required or permitted in the Nutrition Facts label, on the front label or elsewhere on the package to be a nutrition content claim. In such cases, the package label must comply with the regulations for nutrient content claims.
Under the FDCA, any substance that is intentionally added to food is a food ingredient, which is subject to premarket review and approval by the FDA, unless the substance is Generally Recognized As Safe, or GRAS, which means that the substance is generally recognized, among qualified experts, as having been adequately shown to be safe under the conditions of its intended use, or unless the use of the substance is otherwise excluded from the definition of a food ingredient. Under FDA’s regulations, the use of a food substance may be GRAS either through scientific procedures that may be voluntarily submitted to the FDA, or, for a substance used in food before 1958, through experience based on common use in food. General recognition of safety through scientific procedures requires the same quantity and quality of scientific evidence as required to obtain approval of the substance as a food ingredient and ordinarily is based upon published studies, which may be corroborated by unpublished studies and other data and information. General recognition of safety through experience based on common use in foods requires a substantial history of consumption for food use by a significant number of consumers. To be considered “safe” for its intended use, there must be a reasonable certainty in the minds of competent scientists that the substance is not harmful under its intended conditions of use. The specific data and information that demonstrate safety depend on the characteristics of the substance, the estimated dietary intake, and the population that will consume the substance.
13
Table of Contents
Registered food facilities that manufacture, process, pack, or hold food for human or animal consumption in the United States are required to submit a report to the FDA’s Reportable Food Registry, or RFR, when there is a reasonable probability that the use of, or exposure to, an article of food will cause serious adverse health consequences or death. The RFR covers all foods regulated by FDA except infant formula and dietary supplements. Registered facilities must report as soon as practicable, but in no case later than 24 hours after it is determined that an article of food is a reportable food.
FDA Regulation—Dietary Supplements
The Dietary Supplement Health and Education Act of 1994, or DSHEA, amended the FDCA by establishing regulatory standards with respect to dietary supplements, and defining dietary supplements as a new category of food. Dietary supplements include vitamins, minerals, amino acids, nutritional supplements, herbs and botanicals intended for ingestion that are labeled as dietary supplements and are not represented for use as a conventional food or as a sole item of a meal or the diet. Under DSHEA, a firm that manufactures or distributes dietary supplements must determine that such products are safe and that any representations or claims made about the products are substantiated by adequate evidence to show that the claims are not false or misleading.
DSHEA does not require manufacturers or distributors to seek approval from the FDA before producing or selling a dietary supplement unless the supplement contains one or more ingredients that are considered to be a “new dietary ingredient.” A “new dietary ingredient” is one that was not marketed in the United States before October 15, 1994. The manufacturer or distributor of a dietary supplement that contains a “new dietary ingredient” must provide the FDA with information, including any citations to published articles, demonstrating why the ingredient is reasonably expected to be safe for use in a dietary supplement at least 75 days before the dietary supplement is introduced or delivered for introduction into interstate commerce. This requirement does not apply if the ingredient has been recognized as a food substance and is present in the food supply.
Because dietary supplements are foods, manufacturers of dietary supplements must register the facilities where the supplements are manufactured, processed, packed or held with the FDA before such activities begin. Those who manufacture, package or hold dietary supplements also must comply with GMPs for dietary supplements. According to the GMPs, dietary supplements must be prepared, packaged, labeled and held in compliance with specific requirements, including detailed quality control requirements, such as those for maintaining and cleaning facilities and instruments, hiring and training personnel and ensuring the appropriate manufacturing environment, testing requirements, recordkeeping requirements and handling of customer complaints. Anyone who manufactures, packages, labels or holds dietary supplements must evaluate and ensure the identity, purity, strength and composition of the products. FDA regulations also require that certain information appear on dietary supplement labels, including the name of the dietary supplement, the amount of the dietary supplement, nutrition labeling, a complete list of ingredients and the name and place of business of the manufacturer, packer or distributor. Manufacturers must ensure, and have substantiation showing, that claims made about dietary supplements are truthful and not misleading. Acceptable claims for dietary supplements are the same as those for conventional foods: health claims, structure/function claims and nutrient content claims. However, additional requirements apply to manufacturers of dietary supplements who make structure/function claims. Manufacturers of dietary supplements must notify the FDA of any structure/function claims made for a dietary supplement within 30 days of first marketing the product with the identified claims. A dietary supplement that includes a structure/function claim on its labeling is also required to bear a prescribed disclaimer: “This statement has not been evaluated by the Food and Drug Administration. This product is not intended to diagnose, treat, cure or prevent any disease.” The manufacturer, packer, or distributor of a dietary supplement must submit to the FDA any report it receives of a serious adverse event associated with the dietary supplement when used in the United States, accompanied by a copy of the label of the dietary supplement, no later than 15 business days after the report is received. A “serious adverse event” is an adverse event that results in death, a life-threatening experience, inpatient hospitalization, a persistent or significant disability or incapacity, a congenital anomaly or birth defect, or requires, based on a reasonable medical judgment, medical or surgical intervention to prevent such outcomes.
The FDA may take action to restrict use of a dietary supplement or to remove it from the marketplace if the agency believes the supplement presents a significant or unreasonable risk of illness or injury under conditions of use suggested in the labeling or under ordinary conditions of use. Under DSHEA, the FDA bears the burden of proof to show that a dietary supplement presents a significant or unreasonable risk of illness or injury. The FDA also may take enforcement action against a dietary supplement manufacturer or distributor for unlawful promotion of a dietary supplement, such as making claims that a supplement treats, prevents or cures a specific disease or condition. These claims would subject the dietary supplement to regulation as a drug product. If dietary supplements do not meet applicable requirements, the manufacturer may need to undertake a voluntary recall.
14
Table of Contents
FDA Regulation—Biological Products and New Drug Products
Under the FDCA all new drugs and biological products are subject to pre-market approval by the FDA. In contrast to chemically synthesized small molecular weight drugs, which have a well-defined structure and can be thoroughly characterized, biological products are generally derived from living material—human, animal, or microorganism—are complex in structure, and thus are usually not fully characterized. Biological products include blood-derived products, vaccines, in vivo diagnostic allergenic products, immunoglobulin products, products containing cells or microorganisms, and most protein products. Biological products subject to the Public Health Service, or PHS, Act also meet the definition of drugs under the FDCA, therefore both biological products and drugs are regulated under provisions of the FDCA. However, only biological products are licensed under the PHS Act. The overall development process for biological products is similar to that for drugs. The steps ordinarily required before a biological product or new drug may be marketed in the United States include:
| • | completion of preclinical studies according to Good Laboratory Practice, or GLP, regulations; |
| • | the submission of an IND application to the FDA, which must become effective before human clinical trials may commence; |
| • | performance of adequate and well-controlled human clinical trials according to Good Clinical Practices to establish the safety and efficacy of the proposed biological product or new drug for its intended use; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the product is manufactured, processed, packaged or held to assess compliance with GMPs; and |
| • | the submission to, and review and approval by, the FDA of a biologics license application, or BLA, or new drug application, or NDA, that includes satisfactory results of preclinical testing and clinical trials. |
Preclinical tests include laboratory evaluation of the product candidate, its formulation and stability, as well as animal studies. The FDA requires that preclinical tests be conducted in compliance with GLP regulations. The results of preclinical testing are submitted as part of an IND application to the FDA together with manufacturing information for the clinical supply, analytical data, the protocol for the initial clinical trials and any available clinical data or literature. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. FDA may also impose clinical holds at any time before or during studies due to safety concerns or non-compliance.
Clinical trials to support BLAs and NDAs involve the administration of the investigational product to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the efficacy criteria to be evaluated. Clinical trials are typically conducted in three sequential phases, but the phases may overlap.
In Phase 1 clinical trials, the biological or new drug product candidate is initially introduced into human subjects or patients and assessed for safety, dosage tolerance, absorption, metabolism, distribution and excretion, including any side effects associated with increasing doses.
Phase 2 clinical trials usually involve studies in a limited patient population to identify possible adverse effects and safety risks; preliminarily assess the efficacy of the product candidate in specific, targeted indications; and assess dosage tolerance and optimal dosage.
If a product candidate is found to be potentially effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken within an expanded patient population at multiple study sites to further demonstrate clinical efficacy and safety, further evaluate dosage and establish the risk-benefit ratio of the product and an adequate basis for product labeling.
15
Table of Contents
Phase 4, or post-marketing, trials may be mandated by the FDA or may be conducted voluntarily. Phase 4 trials are typically initiated to monitor the safety and efficacy of a biological product or new drug in its approved population and indication over a longer period of time, so that rare or long-term adverse effects can be detected over a much larger patient population and time than was possible during prior clinical trials. Alternatively, Phase 4 trials may be used to test a new method of product administration, or to investigate a product’s use in other indications. Adverse effects detected by Phase 4 trials may result in the withdrawal or restriction of a product.
If the required Phase 1, 2 and 3 clinical testing is completed successfully, the results of product development, preclinical studies and clinical trials, descriptions of the manufacturing process and other relevant information concerning the safety and effectiveness of the biological product or new drug candidate are submitted to the FDA in the form of a BLA or NDA. In most cases, the BLA or NDA must be accompanied by a substantial user fee. The FDA may deny a BLA or NDA if all applicable regulatory criteria are not satisfied or may require additional data, including clinical, toxicology, safety or manufacturing data. It can take several years for the FDA to approve a BLA or NDA once it is submitted, if at all, and the actual time required for any product candidate may vary substantially, depending upon the nature, complexity and novelty of the product candidate.
Before approving an application, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve a BLA or NDA unless it determines that the manufacturing processes and facilities are in compliance with GMP requirements.
If the FDA evaluations of the BLA or NDA and the manufacturing facilities are favorable, the FDA will issue an approval letter. If the FDA determines that it will not approve an NDA or BLA in its present form for one or more reasons, the FDA will issue a complete response letter. The complete response letter usually contains a number of conditions that must be met before FDA will approve the BLA or NDA. If the BLA or NDA does not meet the criteria for approval, the FDA may deny the application.
The required testing, data collection, analysis and compilation of an IND and a BLA or NDA are labor intensive and costly and may take a great deal of time. Tests may have to be redone or new tests performed in order to comply with FDA requirements. It can take considerable time (e.g. 5-10 years) and resources to achieve enrollment sufficient to commence such trials and complete Phase 2 or 3 clinical trials. Moreover, there is no guarantee a product will be approved.
FDA Regulation—Medical Devices
Medical devices also are subject to extensive regulation by the FDA. To be commercially distributed in the United States, devices that are not exempt from FDA’s premarket notification, or 510(k) procedures, or are pre-amendment devices, meaning they were on the market prior to May 28, 1976, must receive either 510(k) clearance or pre-market approval, or PMA, from the FDA prior to marketing. Devices are assigned to one of three classes depending on the controls the FDA deems necessary to ensure the safety and effectiveness of the devices. Devices deemed to pose the least risk are placed in Class I. A Class I device is 510(k) exempt unless the device is intended for a use which is of substantial importance in preventing impairment of human health, or the device presents a potential unreasonable risk of illness or injury. Class II devices require the manufacturer to submit a pre-market notification to FDA unless they are 510(k) exempt. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, devices deemed not substantially equivalent to a previously 510(k) cleared device and certain other devices are placed in Class III. Most Class III devices require approved PMAs before marketing, although some Class III devices can get to market through the 510(k) process.
To obtain 510(k) clearance, a manufacturer must submit a pre-market notification demonstrating that the proposed device is “substantially equivalent” to a “predicate device,” which is a previously 510(k) cleared Class I or Class II device, a pre-amendment Class III device for which the FDA has not yet called for PMA applications or a device that was in commercial distribution before May 28, 1976. To demonstrate substantial equivalence, the applicant must show that the device has the same intended use and the same technological characteristics as the predicate, or if the device has different technological characteristics than the predicate, the device does not raise new questions of safety and effectiveness, and is at least as safe and effective as the predicate. The FDA’s 510(k) clearance pathway usually takes from four to twelve months, but it can last longer. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require a PMA approval.
A product not eligible for 510(k) clearance must follow the PMA pathway, which requires proof that there is a reasonable assurance of a device’s safety and efficacy to the FDA’s satisfaction. The PMA pathway is much more costly and lengthy than the 510(k) pathway. A PMA application typically must provide extensive preclinical and clinical trial data and also information about the device and its components including, among other things, device design, manufacturing and labeling. As part of the PMA review, the FDA will typically inspect the manufacturer’s facilities for compliance with quality system regulation requirements, which impose elaborate testing, control, documentation and other quality assurance procedures. Upon acceptance by the FDA of what it considers a completed filing, the FDA commences an in-depth review of the PMA application, which typically takes from one to two years, but may last longer. Even after approval of a PMA, a new PMA or PMA supplement is required in the event of a modification affecting the safety or effectiveness of the device.
16
Table of Contents
FDA Regulation—Post-Market Requirements
Even if regulatory clearances or approvals for our product candidates are obtained, our products and the facilities manufacturing our products, including foods, will be subject to continued review and periodic inspections by the FDA. The FDA may perform these inspections at any time without advanced notice. For example, as a condition of approval of an NDA, the FDA may require us to engage in post-marketing testing and surveillance and to monitor the safety and efficacy of our products. Holders of an approved NDA, BLA, or PMA, or 510(k) clearance are subject to several post-market requirements, including the reporting of certain adverse events involving their products to the FDA, provision of updated safety and efficacy information, and compliance with requirements concerning the advertising and promotion of their products.
The FDA will inspect manufacturing facilities to confirm that the facilities comply with GMP requirements. To comply with GMP requirements, manufacturers must expend money, time and effort in the area of production and quality control to ensure full compliance. For example, manufacturers of biologic products must establish validated systems to ensure that products meet high standards of sterility, safety, purity, potency and identity, and must report to the FDA any deviations from GMP or any unexpected or unforeseeable event that may affect a product’s safety, purity, or potency. The regulations also impose documentation requirements and require manufacturers of drugs, biologics or devices to investigate and correct any unexplained discrepancy or the failure of a lot or unit to meet any of its specifications.
Failure to comply with the applicable FDA requirements may subject manufacturers and distributors to administrative or judicial sanctions. These sanctions could include, among other things, warning letters, product seizures, total or partial suspension of production or distribution, injunctions, civil money penalties, fines, restitution, disgorgement, or civil or criminal penalties. Further, the FDA has authority to issue mandatory recalls for medical devices and biologics, and we may need to undertake a voluntary recall for any of our products.
In addition to regulations enforced by the FDA, we are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other federal, state and local regulations. Our research and development activities involve the controlled use of hazardous materials, chemicals, biological materials and radioactive compounds.
FTC Regulation
The advertising of our products is subject to regulation by the FTC under the Federal Trade Commission Act, in addition to state and local regulation. The Federal Trade Commission Act prohibits unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce. The Federal Trade Commission Act also provides that the dissemination or the causing to be disseminated of any false advertisement pertaining to drugs or foods, which would include dietary supplements, is an unfair or deceptive act or practice. Under the FTC’s Substantiation Doctrine, an advertiser is required to have a “reasonable basis” for all objective product claims before the claims are made. Failure to adequately substantiate claims may be considered either deceptive or unfair practices. Pursuant to this FTC requirement we are required to have adequate substantiation for all advertising claims made for our products.
In recent years the FTC has initiated numerous investigations of dietary supplement and weight loss products and companies. We may be the subject of investigation in the future, and the FTC may impose limitations on our advertising of products. The FTC has a variety of processes and remedies available to it for enforcement, both administratively and judicially, including compulsory processes, cease and desist orders, and injunctions. FTC enforcement can result in orders requiring, among other things, limits on advertising, corrective advertising, consumer redress, divestiture of assets, rescission of contracts and such other relief as may be deemed necessary.
International Regulation
Our product candidates are subject to regulation in every country where they will be tested or used. Whether or not we obtain FDA approval for a product candidate, we must obtain the necessary approvals from the comparable regulatory authorities of foreign countries before we can commence testing or marketing of a product candidate in those countries. The requirements governing the conduct of clinical trials and the approval processes vary from country to country and the time required may be longer or shorter than that associated with FDA approval. As in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution, would apply to any product that is approved outside the United States.
17
Table of Contents
Future Legislation and Regulations
In the future we may be subject to additional laws or regulations by the FDA or other federal, state or foreign regulatory authorities, the repeal of laws or regulations, or more stringent interpretations of current laws or regulations. We are unable to predict the nature of such future laws, regulations, or interpretations, nor can we predict what effect additional governmental regulations or administrative orders, when and if promulgated, would have on our business in the future. For example, for dietary supplements, the FDA or other governmental regulatory bodies could require the reformulation of certain products to meet new standards, the recall or discontinuance of certain products not able to be reformulated, imposition of additional record keeping requirements, expanded documentation of the properties of certain products, expanded or different labeling and scientific substantiation. Any or all of such requirements could have a materially adverse effect on our business, financial condition, results of operations and cash flows.
Competition
Our industry is subject to rapid and intense technological change. Competition is intense among manufacturers of nutritional, non-prescription, and prescription pharmaceuticals. We face, and will continue to face, competition from nutraceutical, pharmaceutical, biopharmaceutical, medical device and biotechnology companies developing similar products and technologies both in the United States and abroad, as well as numerous academic and research institutions, governmental agencies and private organizations engaged in drug funding or research and discovery activities both in the United States and abroad. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for discovery, research and clinical development of technologies and products similar to ours. We also face competition from entities and healthcare providers using more traditional methods. We believe there are a substantial number of products under development by numerous nutraceutical, pharmaceutical, biopharmaceutical, medical device and biotechnology companies, and it is likely that other competitors will emerge.
Many of our existing and potential competitors are large, well established pharmaceutical, chemical or healthcare companies with considerably greater research and product development capabilities and financial, scientific, marketing and human resources than we have. As a result, these competitors may succeed in developing competing products earlier than we do; obtain patents that block or otherwise inhibit our ability to further develop and commercialize our product candidates; obtain approvals from the FDA or other regulatory agencies for products more rapidly than we do; or develop treatments or cures that are safer or more effective than those we propose to develop. These competitors may also devote greater resources to marketing or selling their products and may be better able to withstand price competition. In addition, these competitors may introduce or adapt more quickly to new technologies or scientific advances, which could render our technologies obsolete, and may introduce products or technologies that make the continued development, production, or marketing of our product candidates uneconomical. These competitors may also be more successful in negotiating third-party licensing or collaborative arrangements and may be able to take advantage of acquisitions or other strategic opportunities more readily than we can. These actions by competitors or potential competitors could materially affect our business, financial condition and results of operations. We cannot assure you that we will be able to compete successfully.
We have a limited ability to predict how competitive our products, technology platforms and replacement therapy will be in the market place. The competition we believe currently exists with respect to each of our products is as follows:
SMaRT Replacement Therapy
We are not aware of any direct competitors with respect to our licensed, patented replacement therapy technology. However, there may be several ways to disable or eradicate S. mutans. We know that certain companies and several academic and research institutions, such as the Forsyth Institute, the University of Alabama, and Guy’s Hospital of London, are developing and testing caries vaccines aimed at eradicating S. mutans. An alternative approach involves topical application of adhesion- blocking synthetic peptides that prevent S. mutans from attaching to the tooth surface. Products that result in the elimination of S. mutans from the natural ecosystem would require major studies to determine whether such eradication of naturally occurring bacteria might not create serious, unintended consequences. The problem with eradicating S. mutans is that it disrupts the natural ecosystem leaving a void for another pathogen potentially more harmful than S. mutans to dominate. We are not aware that any other company has filed an IND with the FDA to test their technology to address the matter. Any product based on our replacement therapy technology will compete against traditional oral care products used to combat tooth decay. These products include fluoride-based toothpastes as well as fluoride treatments and tooth sealants administered by dentists. These competitors could include, among others, Colgate, Procter & Gamble, Unilever, GlaxoSmithKline, and Dentsply.
18
Table of Contents
ProBiora3
Many companies sell probiotics that are principally designed for digestive health, vaginal and urinary tract health, and immune system support. Our product will not compete directly with the products of these companies. Recently, researchers at the University of Hiroshima in Japan have published studies indicating that Lactobacillus reuteri, or L. reuteri, a bacterial species isolated from the gastrointestinal tract, can reduce the levels of S. mutans in the mouth and may aid in the prevention of tooth decay. L. reuteri is widely used as a probiotic for other indications and recently has been promoted for dental health. We are aware of a probiotic product from BioGaia AB/Sunstar, containing a strain of L. reuteri, which is on the market today as GUM ® PerioBalance ® and is targeted to maintain oral health. Another probiotic bacteria for oral care, known as BLIS K12 probiotic, is commercially available from Frutarom, an Israeli company. BLIS K12 is promoted as a probiotic for bad breath and contains the bacterium, Streptococcus salivarius K12. This bacterium principally colonizes the cheek and tongue surfaces in the oral cavity, and as such is promoted only for its oral care activity as an aid for halitosis. As compared to all of these competitors, ProBiora3, with its unique blend of three proprietary probiotic strains, potentially has greater beneficial actions for maintaining oral health.
MU1140-S and Other Lantibiotics
MU1140-S will likely compete directly with antibiotic drugs such as vancomycin and newer drugs, including Cubicin (daptomycin) and Zyvox (linezolid). Given the growing resistance of target pathogens to even new antibiotics, we believe that there is ample room in the marketplace for additional antibiotics. We are aware of another mutacin peptide patented in the United States by the University of Laval in Quebec which patent expired on April 17, 2009 for failure to pay required maintenance fees. Management believes that the Laval peptide, if developed would infringe on the MU1140 patent. Many of our competitors are taking approaches to drug development differing from our approach, including using traditional screening of natural products; genomics to identify new targets, and combinatorial chemistry to generate new chemical structures. Competition in the pharmaceutical industry is based on drug safety, efficacy, ease of use, patient compliance, price, marketing, and distribution. Commercial success of MU1140-S technology will depend on our ability and/or the ability of our licensees and partners to compete effectively in all of these areas, against other companies with existing and pipeline antibiotics to be commercialized in the future. Producers of antibiotic products include many large, global pharmaceutical companies, who have much greater financial and technical resources than us.
Our Intellectual Property
We rely upon a combination of licenses, patents, trade secrets, know-how, and licensing opportunities to develop our business. Our future prospects depend on our ability to protect our intellectual property, particularly our patents. We also need to operate without infringing the proprietary rights of third parties.
License Agreements
We have exclusively licensed the intellectual property for our SMaRT Replacement Therapy and MU1140 technologies from the UFRF. The patents to which our exclusive license applies are U.S. Patent No. 5,607,672, “Replacement Therapy for Dental Caries,” and U.S. Patent No. 5,932,469, “Antimicrobial Polypeptide, Nucleic Acid and Methods of Use” (including derivative patents: 6,391,285, 6,475,771, 6,964,760 and 7,067,125) in addition to many corresponding international patents and applications. See “Our In-licensed Technology Agreements.”
Patents
We attempt to protect our technology and products through patents and patent applications. We have built a portfolio of patents and applications covering certain of our technologies. We have rights to eight issued U.S. patents and we have ten U.S. patent applications on file with the U.S. PTO (one of which is currently allowed) directed toward our products and technologies, including patents exclusively licensed from the UFRF. Our pending applications cover a range of technologies, including specific embodiments and applications for treatment of various medical indications, improved application methods and adjunctive utilization with other therapeutic modalities. We reassess the value of each patent at the time maintenance fees are due, and in cases where maintaining the patent is judged to be of no significant strategic value we decline to pay the fee. The patents and patent applications we have with respect to our products and technologies are set forth below:
| • | Consumer Products. We filed three U.S. patent applications on our probiotic technology (U.S. patent application serial number 10/567592, filed June 30, 2006 (this application was allowed on December 9, 2010, and the issue fee was paid on December 14, 2010, we expect the patent to issue shortly); U.S. patent application serial number 12/482,881, filed June 11, 2009; U.S. patent application serial number 13.017214, filed January 31, 2011). These applications were internationally filed as PCT/US04/025899 on August 10, 2004, and PCT/US09/047040, filed on June 11, 2009. We also filed a U.S. patent application entitled “Methods for Regulating Weight and Size of Animals” (U.S. patent application serial number 11/265, 414, filed November 2, 2005). This application was internationally filed as PCT/US05/39657 on November 2, 2005. A related application entitled “Methods of Treatment of Lipomas and Sarcomas” was filed as a PCT application on February 18, 2010 (PCT/US10/24562). |
19
Table of Contents
| • | Biomarker Discovery. In our Biomarker Discovery division we acquired the rights to our platform technology in November 2006 in connection with our acquisition of IviGene Corporation. We own patents and applications directed toward the identification and isolation of polynucleotides expressed during the process of infection: In Vivo Induced Antigen Technology-U.S. Patent No. 7,033,748, filed March 6, 2002, and U.S. patent application serial number 09/980,845; filed April 8, 2002 (internationally filed as PCT/US00/21340 on August 4, 2000); and 12/327,056, filed December 3, 2008; In Vivo Induced Genes of Mycobacterium tuberculosis, U.S. patent application serial number 12/293,497 filed on September 2, 2009 (internationally filed as PCT/US07/63850, on March 13, 2007); Compositions for Detection and Treatment of Colorectal Cancer, PCT/US09/050938, filed July 17, 2009, and filed in the U.S. as U.S. patent application serial number 13/054667 on March 2, 2011. |
| • | Antibiotics. In our Antibiotics division we have filed a patent application directed at the intellectual property surrounding the DPOLT solid/liquid phase peptide synthesis platform technology, as well as associated areas of lantibiotics technology, in the U.S. (Pat. No. 7,521,529 filed August 11, 2006; U.S. Pat. Appl. 12/413,551, filed March 28, 2009) and internationally (PCT/US06/31510 filed August 11, 2006; PCT/US10/028620 filed March 25, 2010). In addition, we have the exclusive license for our MU1140 lantibiotic technology from the UFRF. |
| • | Biologics. We have licensed our SMaRT Replacement Therapy technology, and the use of recombinant Streptococcus strains to combat dental caries, from the UFRF. |
We also have applications pending and/or allowed in Australia, Canada, China, Hong Kong, Israel, Japan, Mexico, New Zealand, South Africa, South Korea, as well as in the European Patent Office. Because of the differences in patent laws and laws concerning proprietary rights, the extent of protection provided by U.S. patents or proprietary rights owned by us may differ from that of their foreign counterparts.
The recently passed Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act provide a 12-year market exclusivity period for new biologics. We believe that our SMaRT Replacement Therapy technology would qualify for this exclusivity.
Trademarks
Our trademarks are of material importance to our business. We have developed many brand names and trademarks for our products. Accordingly, our future success may depend in part upon the goodwill associated with our brand names. We currently use the following unregistered trademarks: SMaRT Replacement Therapy™, MU1140™, IVIAT™ and CMAT™, LPT3-04™, KJ2™, KJ3™, JH145™, and DPOLT™. Oragenics™ is among our non-registered trademarks. We currently have pending with the U.S. PTO, an application for registration of the mark for PROBIORA™. We also hold U.S. trademark registrations for EVORAKID®, EVORAPRO®, EVORAPLUS ®, TEDDY’S PRIDE ® and PROBIORA3 ®. Finally, we hold a European Community trademark registration for PROBIORA3 ®.
We also have rights to use other names essential to our business. Federally registered trademarks have a perpetual life, as long as they are maintained and renewed on a timely basis and used properly as trademarks, subject to the rights of third parties to seek cancellation of the trademarks if they claim priority or confusion of usage. We regard our trademarks and other proprietary rights as valuable assets and believe they have significant value in marketing our products.
20
Table of Contents
Protection of Trade Secrets
We attempt to protect our trade secrets, including the processes, concepts, ideas and documentation associated with our technologies, through the use of confidentiality agreements and non-competition agreements with our current employees and with other parties to whom we have divulged such trade secrets. If our employees or other parties breach our confidentiality agreements and non-competition agreements or if these agreements are not sufficient to protect our technology or are found to be unenforceable, our competitors could acquire and use information that we consider to be our trade secrets and we may not be able to compete effectively. Most of our competitors have substantially greater financial, marketing, technical and manufacturing resources than we have and we may not be profitable if our competitors are also able to take advantage of our trade secrets.
Tax Credit
On November 1, 2010, we received notification that we were awarded federal grant funding for three of its therapeutic development programs under the Qualifying Therapeutic Discovery Project. The Qualifying Therapeutic Discovery Project, was recently enacted by Congress as part of the Patient Protection and Affordable Care Act of 2010, which was designed to provide grants or tax credits to qualified biotechnology companies that demonstrate the potential to either 1) develop new therapies to treat areas of unmet medical needs; 2) prevent, detect or treat chronic or acute diseases and conditions; 3) reduce long-term health care costs in United States; or 4) significantly advance the goal of curing cancer within the 30 year period beginning on May 21, 2010. We applied for funding on three of its programs: Prevention of Tooth Decay using Smart Replacement Therapy, Novel Antibiotics for the Treatment of Healthcare Associated Infections and Rapid and Sensitive Identification of Novel Diagnostic Biomarkers for Cancer and Infectious Diseases. We received a non-taxable cash grant award totaling $733,437 under the program. A payment of $371,219 was made to us in November 2010 and remaining grant award amount of $362,218 was received in February, 2011.
Research and Development Costs
We have spent $2,014,784 and $1,833,746 on research and development of our technologies in 2010 and in 2009, respectively.
Employees
We have 19 full-time and no part-time employees. We have three employees in research and development, nine employees in general and administrative and seven employees in sales, marketing and business development. We enjoy good relations with our employees. None of our employees is a member of any labor union, and we are not a party to any collective bargaining agreement.
Available Information
Our website is www.oragenics.com. On our website we make available at no cost our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished as soon as reasonably practicable after we electronically file such material with, or furnish them to, the United States Securities and Exchange Commission (“SEC”). The information contained on our website is not a part of this annual report on Form 10-K.
21
Table of Contents
An investment in our common stock involves a high degree of risk. You should carefully consider the risks described below before making an investment decision in our securities. These risk factors are effective as of the date of this Form 10-K and shall be deemed to be modified or superseded to the extent that a statement contained in our future filings modifies or replaces such statement. All of these risks may impair our business operations. The forward-looking statements in this Form 10-K involve risks and uncertainties and actual results may differ materially from the results we discuss in the forward-looking statements. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected. In that case, the trading price of our stock could decline, and you may lose all or part of your investment.
Risks Related to Our Business
We require additional financing to operate beyond June 2011, as well as complete the development of and to commercialize our SMaRT Replacement Therapy and MU1140-S product candidates and we do not know if additional financing will be available to us when and if needed, or, if available, on terms that we find acceptable, particularly given the current and potential future strain in the financial and credit markets.
We do not have sufficient capital to sustain our operations beyond June 2011. Our operations have required substantial capital funding since inception and we expect to continue to need substantial amounts to develop and commercialize our SMaRT Replacement Therapy and MU1140-S product candidates. We require additional funding and may be unable to raise capital on attractive terms, which would force us to significantly delay, scale back or discontinue the development or commercialization of our product candidates. Changing circumstances may cause us to use capital significantly faster than we currently anticipate, and we may incur higher expenses than currently expected because of circumstances beyond our control. If we are not able to raise additional capital and we are not generating positive cash flow from our ProBiora3 products and are unable to commercialize our product candidates, we may be unable to pursue further development of our product candidates, be forced to divest our product candidates prior to maximizing their potential value, be unable to maintain the licenses for our SMaRT Replacement Therapy and MU1140-S product candidates, or be forced to significantly scale back or cease our operations.
Other than the availability of $2,000,000 we are able to borrow under our Credit Facility with the Koski Family Limited Partnership (KFLP), we have no other committed sources of capital and do not know whether additional financing will be available to us when and if needed, or, if available, that the terms will be acceptable to us, particularly if the financial and credit markets continue to be constrained.
We may seek additional financing through public or private equity offerings or through arrangements with strategic third parties. If we raise additional financing by issuing equity securities, further dilution to existing stockholders may result. In addition, as a condition to providing additional financing to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. If we raise additional financing through arrangements with strategic third parties, we may be required to relinquish rights to or sell certain of our product candidates or products that we would not otherwise relinquish or sell.
We may also seek additional financing through long-term debt and lines of credit or through the issuance of debt securities. If we raise additional financing through borrowing or the issuance of debt securities, our debt service obligations may be significant. If we are unable to generate sufficient cash to meet these debt service obligations, we will need to use existing cash or liquidate assets in order to fund these obligations and to repay our debt, which could force us to delay or terminate our research, development and commercialization efforts.
We are dependent upon our Credit Facility with our largest shareholder, the Koski Family Limited Partnership (the “KFLP”) for funding.
On July 30, 2010 we entered into an unsecured revolving credit agreement (the “Credit Facility”) with our largest shareholder, the KFLP. Pursuant to the Credit Facility we were able to borrow up to $2,000,000 million from the KFLP at LIBOR plus 6.0%, subject to certain conditions precedent, including compliance with the Credit Facility and the receipt by the KFLP of a certificate of no adverse change from us in form and substance acceptable to the KFLP. On January 24, 2011 we entered into a First Amendment to the Credit Facility (the “First Amendment”) to increase the available borrowing from $2,000,000 to $2,500,000 and simultaneously therewith we drew on the Credit Facility as amended by the First Amendment to borrow the additional $500,000 in available funds. On February 4, 2011, the Company entered into a Second Amendment (the “Second Amendment”) to the Credit Facility. Under the Second Amendment, the due date of the amounts outstanding under the Credit Facility, as amended has been extended by one year from July 30, 2011 to July 30, 2012. The interest rate remained at LIBOR plus 6.0%. As a result of the Second Amendment, we were able to borrow up to an additional $2,500,000 from the KFLP which should be sufficient to enable us to continue our operations until June 2011. Future draws under the Credit Facility, as amended, are limited to $500,000 per month. There can be no assurance that there will be any adverse change in our business or that we will be able to draw funds under the Credit Facility. Any inability to draw upon, or delay in obtaining, funds under the Credit Facility when needed could have a significant adverse effect on our business and force us to cease operations.
22
Table of Contents
We have incurred significant losses since our inception and expect to continue to experience losses for the foreseeable future.
Since our inception, we have incurred operating losses and negative cash flow from operating activities. To achieve and maintain profitability, we must successfully develop, obtain regulatory approval for, manufacture, market and sell, or license, partner or sell the rights to, one or more of the product candidates we either license or own. Furthermore, our cash burn rate and expenses have increased significantly due to our recent commercialization initiatives with our ProBiora3 products. We expect to continue to incur losses for the foreseeable future as we expand our sales and marketing capabilities for our ProBiora3 products and continue our preclinical testing, clinical trials and research and development activities.
Net losses have totaled $7,805,165 and $5,519,348 for the years ended December 31, 2010, and 2009, respectively. We have experienced losses from operations during the last two years and have an accumulated deficit of $33,317,048 as of December 31, 2010. We have used cash in our operating activities of $6,448,434 and $5,799,481 for the years ended December 31, 2010 and 2009, respectively. Our accounts payable and accrued expenses have also increased due to operational changes instituted in connection with the launch of our consumer products and in connection with our abandoned public offering. We have a working capital deficit of $127,518 as of December 31, 2010 (a deficit of $603,175 when the current cash reserved for DPOLT research is excluded), and $2,564,147 ($114,147 capital when the current cash reserved for DPOLT research is excluded) as of December 31, 2009, respectively.
Our auditor has expressed substantial doubt about our ability to continue as a going concern.
In light of our recurring losses, accumulated deficit and negative cash flow, the report of our independent registered public accounting firm on our financial statements for the year ended December 31, 2010 contains an explanatory paragraph raising substantial doubt about our ability to continue as a going concern. Our financial statements do not include any adjustments that may be necessary in the event we are unable to continue as a going concern. If we are unable to establish to the satisfaction of our independent registered public accounting firm that the net proceeds from this offering will be sufficient to allow for the removal of this going concern qualification, we may need to significantly modify our operational plans for us to continue as a going concern.
Our success will depend on our ability to obtain regulatory approval of our SMaRT Replacement Therapy and MU1140-S product candidates and their successful commercialization.
Our SMaRT Replacement Therapy and MU1140-S product candidates have not received regulatory approval in any jurisdiction and they may never receive approval or, if approvals are obtained, may never be commercialized successfully. We have incurred and will continue to incur significant costs relating to the preclinical and clinical development of our SMaRT Replacement Therapy and MU1140-S product candidates. We are currently conducting a second Phase 1 clinical trial to examine the safety and genetic stability of an attenuated version of the SMaRT strain in humans. We do not know whether our planned and current clinical trials for our SMaRT Replacement Therapy product candidate will be completed on schedule, if at all. In addition, we do not know whether any of our clinical trials will be successful or can be completed within our current expected budget. For our MU1140-S product candidate, we have performed extensive preclinical testing using native MU1140 and expect to conclude the preclinical testing of MU1140-S, including toxicity testing in rodent and non-rodent animal models, during the second half of 2011. We intend to file an Investigational New Drug, or IND, application with the FDA in mid-2012. Even if we are able to conduct successful clinical trials or the required regulatory approvals are obtained, we may never be able to generate significant revenues from our SMaRT Replacement Therapy and MU1140-S product candidates. If our SMaRT Replacement Therapy or MU1140-S product candidates are unsuccessful, we may be unable to generate sufficient revenues to sustain and grow our business, and our business, financial condition and results of operations will be materially adversely affected.
23
Table of Contents
Our success will also depend on our ability to significantly increase sales of our ProBiora3 products which have only generated modest revenues to date.
Currently our sole source of product revenues is from sales of our ProBiora3 products, which began in late 2008 and have generated only modest revenues to date. Net sales of our ProBiora3 products were $1,128,895 and $366,801 for the years ended December 31, 2010 and 2009, respectively. If we are unable to generate significant revenues from our ProBiora3 products our business, financial condition and results of operations will be materially adversely affected.
Our financial results could vary significantly from quarter to quarter and are difficult to predict.
Our revenues and operating results could vary significantly from quarter to quarter due to a variety of factors, many of which are outside of our control. As a result, comparing our operating results on a period-to-period basis may not be meaningful. In addition, we may not be able to predict our future revenues or results of operations. We base our current and future expense levels on our internal operating plans and sales forecasts, and our operating costs are to a large extent fixed. As a result, we may not be able to reduce our costs sufficiently to compensate for an unexpected shortfall in revenues, and even a modest shortfall in revenues could disproportionately and adversely affect financial results for that quarter. Individual products represent meaningful portions of our revenues in any quarter. We may incur significant or unanticipated expenses associated with our research and development efforts of our product candidates under development. In addition to other risk factors discussed in this section, factors that may contribute to the variability of our quarterly results include:
| • | the timing of release of new products and services by us and our competitors, particularly those that may represent a significant portion of revenues in any given period; |
| • | the popularity of new products, and products released in prior periods; |
| • | changes in pricing policies by us or our competitors; |
| • | fluctuations in the size and rate of growth of overall consumer and retailer demand for our ProBiora3 products; |
| • | our success in entering new geographic markets; |
| • | decisions by us to incur additional expenses, such as increases in marketing or research and development; |
| • | the level of expenses associated with our clinical trials; |
| • | accounting rules governing recognition of revenues; |
| • | the amount we reserve against returns and allowances; and |
| • | the timing of compensation expense associated with equity compensation grants. |
As a result of these and other factors, our quarterly and annual operating results could be materially adversely affected. Moreover, our operating results may not meet announced guidance or the expectations of research analysts or investors, in which case the price of our common stock could decrease significantly.
Current and future economic and market conditions could materially adversely affect our revenues, expense levels and profitability.
The U.S. economy and the global economy are recovering from a severe recession. Factors such as uncertainties in consumer spending, a sustained regional and/or global economic downturn or slow recovery may reduce the demand for our ProBiora3 products. Furthermore, challenging economic conditions also may impair the ability of our customers to pay for our commercial products. Because consumer spending for our ProBiora3 products can generally be considered a discretionary purchase, we may experience a more negative impact on our business due to these conditions than other companies that do not depend on discretionary spending. If demand for our ProBiora3 products declines or our customers are otherwise unable to pay for our products, we may be required to offer extensive discounts or spend more on marketing than budgeted and our revenues, expense levels, and profitability will be materially adversely affected.
24
Table of Contents
Sales of our ProBiora3 products may be adversely affected by fluctuations in buying decisions of our retailers and consolidation among retailers.
Our ProBiora3 products are sold to national and regional retailers in the United States. Our revenues could be affected by fluctuations in the buying patterns of these customers, which may result from wholesale buying decisions, economic conditions and other factors. In addition, with the growing trend towards retail consolidation, we are increasingly dependent upon a limited number of leading retailers with greater bargaining strength. Such retailers have demanded, and may continue to demand, increased service and order accommodations as well as price and incremental promotional investment concessions. As a result, we may face pressure on our prices and experience increased expenses from promotions to meet these demands, which would reduce our profitability. We also may be negatively affected by changes in the policies of our customers such as inventory destocking, limitations on access to shelf space and other conditions.
Our agreements with large national mass retailers with respect to our ProBiora3 products may be delayed, terminated or reduced in scope with little or no notice, which could adversely impact our profitability.
Our agreements with large national mass retailers with respect to our ProBiora3 products may be terminated or reduced in scope with little or no notice. Cancellations may occur for a variety of reasons, including the failure of our products to satisfy safety requirements, and unexpected health consequences of our products. Agreements with national mass retailers may provide for rights of return that are unfavorable to us and may require us to adjust our future estimates of returns and allowances. We have vendor agreements whereby the vendors reserve the right to cancel a purchase order without penalty by providing notice to us on or before the given cancellation date and at any time if the completion or delivery date is not met. To the extent our capital resources continue to limit our ability to engage in significant advertising, we expect the number of mass retail stores that carry our consumer ProBiora3 products to continue to decline in the future.
We are subject to government regulation of the processing, formulation, packaging, labeling and advertising of our consumer products.
Under the Federal Food, Drug, and Cosmetic Act, or FDCA, companies that manufacture and distribute foods, such as our ProBiora3 products, are limited in the claims that they are permitted to make about nutritional support on the product label without FDA approval. Any failure by us to adhere to the labeling requirements could lead to the FDA’s requiring that our products be repackaged and relabeled, which would have a material adverse effect on our business. In addition, companies are responsible for the accuracy and truthfulness of, and must have substantiation for, any such statements. These claims must be truthful and not misleading. Statements must not claim to diagnose, mitigate, treat, cure or prevent a specific disease or class of disease. We are able to market our ProBiora3 products in reliance on the self-affirmed Generally Recognized As Safe, or GRAS, status of our active ingredient, ProBiora3. No governmental agency or other third party has made a determination as to whether or not ProBiora3 has achieved GRAS status. We make this determination based on independent scientific opinions that ProBiora3 is not harmful under its intended conditions of use. If the FDA, another regulatory authority or other third party denied our self-affirmed GRAS status for ProBiora3, we could face significant penalties or be required to undergo the regulatory approval process in order to market our probiotic products, and our business, financial condition and results of operations will be adversely affected. We cannot guarantee that in such a situation ProBiora3 would be approved.
The FDA’s Good Manufacturing Practices, or GMPs, describe the methods, equipment, facilities, and controls for producing processed food and set the minimum sanitary and processing requirements for producing safe and wholesome food. Those who manufacture, package, or hold human food must comply with the GMPs. If we or our suppliers fail to comply with the GMPs, the FDA may take enforcement action against us or our suppliers.
The processing, formulation, packaging, labeling and advertising of our probiotic products are subject to regulation by one or more federal agencies including the FDA, the Federal Trade Commission and the Environmental Protection Agency. Our activities are also subject to regulation by various agencies of the states and localities in which our probiotic products are sold. Any changes in the current regulatory environment could impose requirements that would limit our ability to market our probiotic products and make bringing new products to market more expensive. In addition, the adoption of new regulations or changes in the interpretation of existing regulations may result in significant compliance costs or discontinuation of product sales and may adversely affect our business, financial condition and results of operations. While our ProBiora3 products are categorized as foods, it is possible that the FDA or a state regulatory agency could classify these products as a cosmetic or a drug. If the products are classified as cosmetics rather than a food, we would be limited to making claims that the products cleanse and beautify, but we could not make structure or function claims. If our probiotic products are classified as drugs, we would not be able to market the ProBoira3 products without going through the drug approval process. Either of these events would limit our ability to effectively market our products and would adversely affect our financial condition and results of operations. If the FDA or a state regulatory agency viewed the products as cosmetics or drugs, they could claim that the products are misbranded and require that we repackage and relabel the products and impose civil and/or criminal penalties. Either or both of these situations could adversely affect our business and operations.
25
Table of Contents
If we undertake product recalls or incur liability claims with respect to our ProBiora3 products, such recalls or claims could increase our costs and adversely affect our reputation, revenues and operating income.
Our ProBiora3 products are designed for human and animal consumption and we may face product recalls or liability claims if the use of our products is alleged to have resulted in injury or death. ProBiora3 is classified as a food ingredient and is not subject to pre-market regulatory approval in the United States. Our ProBiora3 products contain ingredients that do not have long histories of human or animal consumption. Previously unknown adverse reactions resulting from consumption of these ingredients could occur. We may have to undertake various product recalls or be subject to liability claims, including, among others, that our ProBiora3 products include inadequate instructions for use or inadequate warnings concerning possible side effects and interactions with other substances. A product recall or liability claim against us could result in increased costs and could adversely affect our reputation with our customers, which, in turn, could have a material adverse effect on our business, financial condition and results of operations.
Product liability insurance is expensive, is subject to deductibles and coverage limitations, and may not be available in the future. While we currently maintain product liability insurance coverage, we cannot be sure that such coverage will be adequate to cover any incident or all incidents. In addition, we cannot be sure that we will be able to obtain or maintain insurance coverage at acceptable costs or in a sufficient amount, that our insurer will not disclaim coverage as to a future claim or that a product liability claim would not otherwise adversely affect our business, financial condition and results of operations. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. Uncertainties resulting from the initiation and continuation of product liability litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Product liability litigation and other related proceedings may also require significant management attention.
If we fail to comply with our obligations in the agreements with the University of Florida Research Foundation, Inc., under which we license our rights to SMaRT Replacement Therapy and MU1140, our licenses to these product candidates may be terminated and we will be unable to commercialize these products candidates.
We hold our SMaRT Replacement Therapy and MU1140 product candidates under licenses from the University of Florida Research Foundation, Inc., or UFRF. Under the terms of the licenses, we must spend at least $1,000,000 per year on development of those product candidates until the first commercial sale of products derived from those product candidates has occurred. In addition, we must pay $25,000 per quarter as minimum royalties to the UFRF under our license agreements. The UFRF may terminate our licenses to SMaRT Replacement Therapy and MU1140 if we breach our obligations to timely pay these amounts, to submit development reports as required under the license agreements or commit any other breach of any other covenants contained in the license agreements. There is no assurance that we will be able to comply with these conditions in a timely manner or at all. If our license agreements are terminated, we will be unable to commercialize these product candidates.
Until commercial sale of any products developed from these licensed product candidates takes place, we will not earn any revenues from these product candidates and will, therefore, need additional financing to fund our required royalty payments and development expenses, such as through the commercialization and sale of our ProBiora3 products, the sale of our debt or equity securities, or otherwise. There can be no assurance that we will achieve sufficient sales of our ProBiora3 products or be able to raise the capital necessary to meet our obligations under our licenses. If we are unable to meet our obligations under these licenses, we may lose the licenses to these product candidates and our business, financial condition and results of operations will be materially adversely affected.
We depend on third-party manufacturers for our ProBiora3 products. The loss of any manufacturer or any interruptions in the supply of our ProBiora3 products, would have a negative impact on our revenues and profitability.
We currently have no manufacturing facilities and are dependent upon establishing relationships with independent manufacturers to supply our product needs. We have contracted with multiple GMP-certified manufacturers to produce our active ingredient, ProBiora3, under GMPs. We believe our arrangements with our contract manufacturers have the capacity to meet our current and expected future manufacturing needs. Although we have qualified and used at least two contract manufacturers for each step in our manufacturing process, we do not have a long-term supply agreement or commitment with any of our manufacturers. If our manufacturers are unable or unwilling to produce our ProBiora products in sufficient quantities, or at all, at acceptable pricing in accordance with specifications we establish from time to time we would need to find alternative manufacturers that are qualified. If, in such instances, we are unsuccessful in obtaining alternative manufacturers, it could impair our ability to sell our ProBiora3 products and would have a negative impact on our revenues and profitability. In addition, competitors who own their manufacturing facilities may have an advantage over us with respect to pricing, availability of product and in other areas through their control of the manufacturing process.
26
Table of Contents
The risks associated with the numerous factors that could cause interruptions in the supply of our products, including manufacturing capacity limitations, regulatory inspections, changes in our sources for manufacturing, disputes with a manufacturer, our failure to timely locate and obtain replacement manufacturers as needed and conditions affecting the cost and availability of raw materials, are magnified when the suppliers are limited in number. Any interruption in the supply of finished products could hinder our ability to timely distribute our products and satisfy customer demand. If we are unable to obtain adequate product supplies to satisfy our customers’ orders, we may lose those orders, our customers may cancel other orders, and they may choose instead to stock and purchase competing products. This could result in a loss of our market share and negatively affect our revenues and profitability.
If our manufacturers in general fail to meet our requirements and the requirements of regulatory authorities, our revenues and profitability may be materially adversely affected.
We do not have the internal capability to manufacture our ProBiora3 products or our SMaRT Replacement Therapy and MU1140-S product candidates under GMPs, as required by the FDA and other regulatory agencies. In order to continue to develop our product candidates, apply for regulatory approvals for our SMaRT Replacement Therapy and MU1140-S product candidates, and commercialize our ProBiora3 products and other product candidates, we will need to contract with third parties that have, or otherwise develop, the necessary manufacturing capabilities.
There are a limited number of manufacturers that operate under GMPs that are capable of manufacturing our ProBiora3 products and SMaRT Replacement Therapy product candidate. Furthermore, manufacturing MU1140-S on a commercial scale has not yet been achieved, so there are additional technical skills needed for the manufacture of MU1140-S that will further limit the number of potential manufacturers. As such, if we are unable to enter into supply and processing contracts with any of these manufacturers or processors for our ProBiora3 products or our development stage product candidates, we may incur additional costs and delays in development and commercialization.
Problems with these manufacturing processes such as equipment malfunctions, facility contamination, labor problems, raw material shortages or contamination, natural disasters, power outages, terrorist activities, or disruptions in the operations of our suppliers and even minor deviations from normal procedures, could result in product defects or manufacturing failures that result in lot failures, product recalls, product liability claims and insufficient inventory.
If we are required to find an additional or alternative source of supply, there may be additional costs and delays in the development or commercialization of our product candidates. Additionally, the FDA and other regulatory agencies routinely inspect manufacturing facilities before approving a New Drug Application, or NDA, or Biologic License Application, or BLA, for a drug or biologic manufactured at those sites. If any of our manufacturers or processors fails to satisfy regulatory requirements, the approval and eventual commercialization of our commercial products and product candidates may be delayed.
All of our contract manufacturers must comply with the applicable GMPs, which include quality control and quality assurance requirements as well as the corresponding maintenance of records and documentation. If our contract manufacturers do not comply with the applicable GMPs and other FDA regulatory requirements, we may be subject to product liability claims, the availability of marketed products for sale could be reduced, our product commercialization could be delayed or subject to restrictions, we may be unable to meet demand for our products and may lose potential revenues and we could suffer delays in the progress of preclinical testing and clinical trials for products under development. We do not have control over our third-party manufacturers’ compliance with these regulations and standards. Moreover, while we may choose to manufacture ProBiora3 products ourselves in the future, we have no experience in the manufacture of pharmaceutical products for clinical trials or commercial purposes. If we decide to manufacture products, we would be subject to the regulatory requirements described above. In addition, we would require substantial additional capital and would be subject to delays or difficulties encountered in manufacturing pharmaceutical products. Regardless of the manufacturer of our products, we will be subject to continuing obligations regarding the submission of safety reports and other post-market information.
27
Table of Contents
We may be unable to find a method to produce MU1140-S in large-scale commercial quantities. If we cannot, we will be unable to generate revenues from sales of our MU1140-S product candidate.
Our antibiotic product candidate, MU1140-S, is a synthetic form of MU1140 produced by our strain of S. mutans. To date, it has been produced only in laboratory cultures. In March 2005 we successfully developed a methodology for producing MU1140 in quantities sufficient to undertake its preclinical testing. In addition, we developed the DPOLT synthetic chemistry methodology to allow large-scale commercial production of the MU1140-S antibiotic. However, this methodology may not be feasible for cost effective, large scale manufacture. If we are not able to utilize this methodology for large-scale manufacture, we will be unable to generate revenues from this product candidate and our business, financial condition and results of operations will be materially adversely affected. We have retained Almac Sciences to refine and scale-up GMP production of MU1140-S. The manufacturing of MU1140-S is a highly exacting and complex process. Manufacturing MU1140-S on a commercial scale has not yet been achieved so there are additional risks. Third-party manufacturers must have additional technical skills and must take multiple steps to attempt to control the manufacturing processes.
Our ProBiora3 products and our SMaRT Replacement Therapy and MU1140-S product candidates face various forms of competition from other products in the marketplace.
The pharmaceutical and biotechnology industries are characterized by intense competition, rapid product development and technological change. Most of the competition that our ProBiora3 products and our SMaRT Replacement Therapy and MU1140-S product candidates face comes from companies that are large, well established and have greater financial, marketing, sales and technological resources than we have. Our ProBiora3 products compete with a range of consumer and nutraceutical products. Our commercial success with SMaRT Replacement Therapy and MU1140-S will depend on our ability and the ability of any sub-licensees to compete effectively in marketing and product development areas including, but not limited to, sales and branding, drug safety, efficacy, ease of use, patient or customer compliance, price, marketing and distribution. There can be no assurance that competitors will not succeed in developing and marketing products that are more desirable or effective than our ProBiora3 products or the products developed from our product candidates or that would render our products obsolete and non-competitive. We anticipate that our SMaRT Replacement Therapy, if approved for the treatment of tooth decay, would compete with other companies attempting to develop technologies in the oral health care market, including vaccines.
We rely on the significant experience and specialized expertise of our senior management and scientific team; our Chief Executive Officer and President recently resigned and we currently do not have a Chief Executive Officer and President.
Our performance is substantially dependent on the continued services and on the performance of our senior management and scientific team, who have extensive experience and specialized expertise in our business. Our performance also depends on our ability to retain and motivate our other key employees. Our Chief Executive Officer and President recently resigned and our Board of Directors is engaged in a search for a new CEO and President. Until such time as a suitable candidate is identified and in place our lead contact executive is Brian Bohunicky, our Chief Financial Officer. Our failure to find and retain an experienced Chief Executive Officer and President could adversely affect the management of our business and our current financial condition could make it more difficult to attract qualified candidates. The loss of the services of our Chief Scientific Officer, Dr. Jeffrey Hillman, and our Chief Financial Officer, Brian Bohunicky, and any members of our scientific team, could harm our ability to develop and commercialize our product candidates. We have no key man life insurance policies. We have employment agreements with Dr. Hillman and Mr. Bohunicky. The term of each of these employment agreements is for an indefinite period and each employment agreement shall end when the employment relationship is terminated by either party.
We need to hire and retain additional qualified scientists and other highly skilled personnel to maintain and grow our business.
Our future success depends on our ability to identify, attract, hire, train, retain and motivate highly skilled technical, managerial and research personnel in all areas within our organization. We plan to continue to grow our business and will need to hire additional personnel to support this growth. We believe that there are only a limited number of individuals with the requisite skills to serve in many of our key positions, and we compete for key personnel with other biotechnology companies, as well as universities and research institutions. It is often difficult to hire and retain these persons, and we may be unable to replace key persons if they leave or be unable to fill new positions requiring key persons with appropriate experience. If we fail to attract, integrate and retain the necessary personnel, our ability to maintain and grow our business could suffer significantly.
28
Table of Contents
If our SMaRT Replacement Therapy and MU1140-S product candidates are shown to be ineffective or harmful in humans, we will be unable to generate revenues from these product candidates.
Before obtaining regulatory approvals for the commercial sale of our SMaRT Replacement Therapy or MU1140-S product candidates, we must demonstrate through preclinical testing and clinical trials that our products are safe and effective for use in humans. To date, the testing of our SMaRT Replacement Therapy product candidate has been undertaken solely in animals and a limited number of humans. Studies have proven our genetically altered strain of S. mutans to be effective in preventing tooth decay in animals. It is possible that our strain of S. mutans will be shown to be less effective in preventing tooth decay in humans in clinical trials. If our SMaRT Replacement Therapy product candidate is shown to be ineffective in preventing tooth decay in humans, we will be unable to commercialize and generate revenues from this product candidate. To date the testing of the antibiotic substance, MU1140, has been undertaken solely in the laboratory and in animals. We have not yet conducted human studies of MU1140-S. It is possible that when these studies are conducted, they will show that MU1140-S is ineffective or harmful in humans. If MU1140-S is shown to be ineffective or harmful in humans, we will be unable to commercialize and generate revenues from sales of this compound. If we are unable to generate revenues from our SMaRT Replacement Therapy and MU1140-S product candidates, our business, financial condition and results of operations will be materially adversely affected.
We intend to rely on third parties to pay the majority of the costs associated with obtaining regulatory approval for, and manufacturing and marketing of, our MU1140-S and SMaRT Replacement Therapy product candidates. If we are unable to obtain agreements with third parties to fund these costs, we will have to fund such costs ourselves or we may be unable to continue our operations.
Assuming the successful completion of Phase 1 trials for our MU1140-S and SMaRT Replacement Therapy product candidates, we intend to either license these product candidates to, or partner with, one or more major pharmaceutical companies prior to commercialization. If we do so, we intend for these licensees or partners to pay the costs associated with our remaining clinical trials and the manufacturing and marketing of our product candidates. If we are unable to license our product candidates or otherwise partner with third parties, we will have to fund the costs of our Phase 2 and Phase 3 trials ourselves. We will also have to establish our own manufacturing facilities and find our own distribution channels. This would greatly increase our future capital requirements and we cannot assure you that we would be able to obtain the necessary financing to pay these costs or that we will be generating significant revenues from any of our products, such as ProBiora3, sufficient to cover the associated costs. If we do not generate sufficient revenues to cover the associated costs or we cannot obtain financing on acceptable terms or at all, our business, financial condition and results of operations will be materially adversely affected.
Our dependence on collaborative arrangements with third parties subjects us to a number of risks. These collaborative arrangements may not be on terms favorable to us. Agreements with collaborative partners typically allow partners significant discretion in electing whether or not to pursue any of the planned activities. We cannot control the amount and timing of resources our collaborative partners may devote to products based on the collaboration, and our partners may choose to pursue alternative products. Our partners may not perform their obligations as expected. Business combinations or significant changes in a collaborative partner’s business strategy may adversely affect a partner’s willingness or ability to complete its obligations under the arrangement. Moreover, we could become involved in disputes with our partners, which could lead to delays or termination of the collaborations and time-consuming and expensive litigation or arbitration. Even if we fulfill our obligations under a collaborative agreement, our partner may be able to terminate the agreement under certain circumstances. If any collaborative partner were to terminate or breach our agreement with it, or otherwise fail to complete its obligations in a timely manner, our chances of successfully commercializing our product candidates would be materially and adversely affected.
We are subject to risks of doing business internationally as we attempt to expand our sales through international distributor relationships.
We recently entered into a number of international distributor agreements for our ProBiora3 products. As a result, we expect to increase our revenues from international sales. A number of factors can slow or prevent international sales, or substantially increase the cost of international sales, and we may encounter certain risks of doing business internationally including the following:
| • | reduced protection and enforcement for our intellectual property rights; |
| • | unexpected changes in, or impositions of, legislative or regulatory requirements that may limit our ability to sell our products and repatriate funds to the United States; |
29
Table of Contents
| • | political and economic instability; |
| • | fluctuations in foreign currency exchange rates; |
| • | difficulties in developing and maintaining distributor relationships in foreign countries; |
| • | difficulties in negotiating acceptable contractual terms and enforcing contractual obligations; |
| • | exposure to liabilities under the U.S. Foreign Corrupt Practices Act; |
| • | potential trade restrictions and exchange controls; |
| • | creditworthiness of foreign distributors, customer uncertainty and difficulty in foreign accounts receivable collection; and |
| • | the burden of complying with foreign laws. |
As we attempt to expand our sales internationally, our exposure to these risks could result in our inability to attain the anticipated benefits and our business could be adversely impacted. Our success will depend, in large part, on our ability to anticipate and effectively manage these and other risks associated with our international operations. However, any of these factors could adversely affect our international operations and, consequently, our operating results.
If our intellectual property rights do not adequately protect our products or product candidates, or if third parties claim we are infringing their intellectual property rights, others could compete against us more directly or we could be subject to significant litigation. Such results could prevent us from marketing our products or product candidates and hurt our profitability.
Our product portfolio is protected by eight issued U.S. patents and eight filed U.S. patent applications, including patents exclusively licensed from the University of Florida Research Foundation, Inc., or the UFRF. We are the exclusive worldwide licensee to the patents for SMaRT Replacement Therapy and MU1140, which are owned by the UFRF. Our success depends in part on our ability to obtain patents or rights to patents, protect trade secrets, operate without infringing upon the proprietary rights of others, and prevent others from infringing on our patents, trademarks and other intellectual property rights. We will be able to protect our intellectual property from unauthorized use by third parties only to the extent that it is covered by valid and enforceable patents, trademarks and licenses. Patent protection generally involves complex legal and factual questions and, therefore, enforceability of patent rights cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. Thus, any patents that we own or license from others may not provide adequate protection against competitors. In addition, any future patent applications may fail to result in patents being issued. Also, those patents that are issued may not provide us with adequate proprietary protection or competitive advantages against competitors with similar product candidates. Moreover, the laws of certain foreign countries do not protect intellectual property rights to the same extent as do the laws of the United States.
In addition to patents and trademarks, we rely on trade secrets and proprietary know-how. We seek protection of these rights, in part, through confidentiality and proprietary information agreements. These agreements may not provide meaningful protection or adequate remedies for violation of our rights in the event of unauthorized use or disclosure of confidential and proprietary information. Failure to protect our proprietary rights could seriously impair our competitive position.
In the event of an infringement or violation, we may face litigation and may be prevented from pursuing product development or commercialization. We may receive in the future, notice of claims of infringement of other parties’ proprietary rights. Infringement or other claims could be asserted or prosecuted against us in the future and it is possible that past or future assertions or prosecutions could harm our business.
30
Table of Contents
The FDA instituted numerous clinical holds with respect to our Phase 1 clinical trial program for our SMaRT Replacement Therapy product candidate. Although the clinical holds were lifted with respect to the attenuated version of the SMaRT strain, a clinical hold remains in effect for our IND for the non-attenuated SMaRT strain. If the FDA does not lift the clinical hold on this IND, we will be unable to conduct the clinical trials necessary to obtain marketing approval of our product candidate. The FDA’s concerns that resulted in these clinical holds may lead to the imposition of a new clinical hold or holds or the failure of our product candidates to obtain marketing approval.
In December 1998, the FDA imposed a clinical hold on the IND for our SMaRT strain. The FDA expressed concerns about the potential for DNA transfer into or from the SMaRT strain; the possible creation of an organism with increased ability to colonize and produce dental caries; and the potential for the transfer of the strain between humans. We met with the FDA to discuss proposed trials of an attenuated SMaRT strain to address the FDA’s concerns, and the FDA informed us that such trials required a separate IND. We submitted an IND for the attenuated strain in March 2003.
The FDA then imposed multiple clinical holds with respect to the Phase 1 clinical trials of the attenuated strain. Specifically, we received clinical hold letters in May 2003, February 2004, June 2007 and August 2007. In connection with the clinical hold letter in May 2003, the FDA requested that we provide preclinical data on eradication of the attenuated version of the SMaRT strain once it had been implanted and a plan for the eradication of the attenuated strain in the trial subjects and their spouse or partners. In an April 2004 meeting, the FDA suggested that we conduct two eradication trials: one in subjects without teeth, and, if successful, another trial in subjects with teeth. We submitted revised protocols in accordance with FDA’s suggestions, and in November 2004 the FDA lifted the clinical hold for the attenuated strain.
We initiated our first Phase 1 clinical trial in April 2005, but we found it difficult to find subjects who fit the FDA’s highly cautious inclusion and exclusion criteria, particularly with respect to the subjects’ lack of dentition. We concluded this trial early after enrolling only two of the 15 planned subjects. The FDA then recommended that we revise the protocol for the evaluation of ten healthy male subjects, ranging from 18 to 30 years old and with normal dentition, in an institutionalized setting. After we submitted additional information, the FDA issued another clinical hold letter in June 2007 for the proposed trial with the attenuated strain, citing the need for a plan with respect to serious adverse effects; a plan for the eradication of the attenuated strain in trial subjects’ offspring; and a required pregnancy test for female partners of subjects. We submitted additional protocols in response to the FDA’s concerns. In August 2007, the FDA issued another clinical hold letter with required revisions to the protocol for offspring of subjects. We submitted a response to the clinical hold letter in September 2007, and the FDA removed the clinical hold for our Phase 1 trial in the attenuated strain in October 2007.
We plan to discuss with the FDA whether the clinical hold for the non-attenuated SMaRT strain can be lifted after the completion of our second Phase 1 clinical trial using the attenuated strain, because we believe the results from the trial may address the FDA’s concerns with the non-attenuated SMaRT strain. However, there is no guarantee that our clinical trials for the attenuated strain will be successful, or that the FDA will ever lift the clinical hold on the non-attenuated SMaRT strain. If the FDA does not lift the clinical hold on the non-attenuated SMaRT strain, we cannot commence our anticipated third Phase 1 trial and we may not be able to conduct the clinical trials necessary to obtain marketing approval of the SMaRT strain.
The FDA may impose additional requirements that may significantly increase the time and expense of obtaining FDA approval. Although the FDA has removed the clinical holds with respect to our Phase 1 trials for the attenuated strain, if the FDA has further concerns regarding the safety or risks associated with our trials, the FDA could implement another clinical hold on our ongoing trial or future trials or raise concerns as a part of a future NDA review process for our SMaRT Replacement Therapy product candidate, which could delay or prevent marketing.
Our SMaRT Replacement Therapy and MU1140-S product candidates are subject to substantial government regulation, including the regulation of preclinical testing and clinical trials. If we are unable to obtain regulatory approval for our product candidates we will be unable to generate revenues.
The production and marketing of products which may be developed from our SMaRT Replacement Therapy and MU1140-S product candidates and our ongoing research and development, preclinical testing and clinical trial activities are subject to extensive regulation and review by numerous governmental authorities. Most of the product candidates we are developing must undergo rigorous preclinical testing and clinical trials and an extensive regulatory approval process before they can be marketed in the United States or internationally.
The FDA accepted our protocols to conduct Phase 1 human clinical trials of our SMaRT Replacement Therapy product candidate. We expect to file an IND application with the FDA in mid-2012 for our MU1140-S product candidate. If we fail to maintain regulatory approval for the clinical trials of our SMaRT Replacement Therapy, if the FDA fails to lift the clinical hold on our IND for the non-attenuated version of the SMaRT strain, or if we fail to obtain regulatory approval for our MU1140-S product candidate, we may have to cease further development. Clinical trials on our SMaRT Replacement Therapy and MU1140-S product candidates are expected to take several years to fully complete. The commencement or completion of preclinical studies or clinical trials can be delayed or prevented for a number of reasons, including:
| • | our belief that SMaRT Replacement Therapy is one of the first genetically modified bacterial strains for use in humans, which may cause the FDA to proceed with additional caution; |
31
Table of Contents
| • | findings in preclinical studies; |
| • | difficulties obtaining regulatory approval to commence a clinical trial or complying with conditions imposed by a regulatory authority regarding the scope or term of a clinical trial; |
| • | delays in reaching or failing to reach agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| • | insufficient or inadequate supply or quality of a product candidate or other materials necessary to conduct our clinical trials; |
| • | difficulties obtaining institutional review board, or IRB, approval to conduct a clinical trial at a prospective site; |
| • | challenges recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including the size and nature of patient population, proximity of patients to clinical sites, eligibility criteria for the trial, nature of the trial protocol, the availability of approved effective treatments for the relevant condition and competition from other clinical trial programs for similar indications; |
| • | severe or unexpected drug or biologic-related side effects experienced by patients in a clinical trial; and |
| • | difficulties retaining patients who have enrolled in a clinical trial but may be prone to withdraw due to rigors of the trial, lack of efficacy, side effects, or personal issues, or who are lost to further follow up. |
Clinical trials also may be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, or a data safety monitoring board, or DSMB, overseeing the clinical trial at issue, or other regulatory authorities due to a number of factors, including:
| • | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| • | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| • | unforeseen safety issues or lack of effectiveness; and |
| • | lack of adequate funding to continue the clinical trial. |
We cannot assure you that clinical trials will demonstrate the safety or effectiveness of our SMaRT Replacement Therapy or MU1140-S product candidates, or will otherwise satisfy regulatory requirements. Our preclinical studies or clinical trials may produce negative or inconclusive results, there may be inconsistencies between early clinical trial results and results obtained in later clinical trials. and we may decide, or regulators may require us, to conduct additional preclinical studies or clinical trials. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain FDA approval for their products. If we are unable to resolve the FDA’s concerns, we will not be able to obtain regulatory approval for these product candidates.
The pre-marketing approval process can be particularly expensive, uncertain and lengthy, and a number of products for which FDA or other governmental regulatory approval has been sought by other companies have never been approved for marketing. In addition to testing and approval procedures, extensive regulations also govern marketing, manufacturing, distribution, labeling, and record-keeping procedures. If we do not comply with applicable regulatory requirements, such violations could result in warning letters, non-approval, suspensions of regulatory approvals or ongoing clinical trials, civil penalties and criminal fines, product seizures and recalls, operating restrictions, injunctions, and criminal prosecution.
32
Table of Contents
We may encounter such delays and rejection of our product candidates by the FDA or other regulatory authority may also adversely affect our business. Such delays or rejection may be encountered due to, among other reasons, government or regulatory delays, lack of efficacy during clinical trials, unforeseen safety issues, or changes in regulatory policy during the period of product development. More stringent regulatory approval processes in product clearance and enforcement activities could result in our experiencing longer approval cycles, more uncertainty, greater risk, and higher expenses. Even if regulatory approval of a product is granted, this approval may entail limitations on uses for which the product may be labeled and promoted. It is possible, for example, that we may not receive FDA approval to market products based on our licensed, patented product candidates for different indications or to market updated products that represent extensions of our basic product candidates. In addition, we may not receive FDA approval to export our products based on our licensed, patented product candidates in the future, and countries to which products are to be exported may not approve them for import.
From time to time, legislative or regulatory proposals are introduced that could alter the review and approval process relating to our product candidates. It is possible that the applicable regulatory authority will issue additional regulations further restricting the sale of our product candidates. Any change in legislation or regulations that govern the review and approval process relating to our future product candidates could make it more difficult and costly to obtain approval for new products based on our product candidates, or to produce, market, and distribute such products if approved.
We cannot assure you that the market and consumers will accept our product candidates. If they do not, we will be unable to generate significant revenues from our product candidates and our business, financial condition and results of operations will be materially adversely affected.
The commercial success of our MU1140-S and SMaRT Replacement Therapy, ProBiora3, LPT3-04 and other product candidates will depend in part on market acceptance by consumers. Biopharmaceutical companies have received substantial support from the scientific community, regulatory agencies and many governmental officials in the United States and internationally. Future scientific developments, media coverage and political events may diminish such support. Public attitudes may be influenced by claims that health products are unsafe for consumption or pose unknown risks to the environment or to traditional social or economic practices. Securing governmental approvals for, and consumer confidence in, such products poses numerous challenges, particularly outside the United States. The market success of product candidates developed by biopharmaceutical companies could be delayed or impaired in certain geographical areas as a result. Products based on our product candidates may compete with a number of traditional dental therapies and drugs manufactured and marketed by major pharmaceutical companies and biotechnology companies. Market acceptance of products based on our product candidates will depend on a number of factors including potential advantage over alternative treatment methods. We can offer you no assurance that dentists, physicians, patients or the medical and dental communities in general will accept and utilize products developed from our product candidates. If they do not, we may be unable to generate significant revenues from our product candidates and our business, financial condition and results of operations will be materially adversely affected.
If our products do not receive favorable third-party reimbursement, or if new restrictive legislation is adopted, market acceptance of our products may be limited and we may not generate significant revenues.
Our ability to commercialize our products will depend in part on the extent to which appropriate reimbursement levels for the cost of our proposed formulations and products and related treatments are obtained by governmental authorities, private health insurers and other organizations, such as Health Maintenance Organizations, or HMOs. Reimbursement from third parties depends greatly on our ability to present data which demonstrate positive outcomes and reduced utilization of other products or services as well as cost data which show that treatment costs using the new product are equal to or less than what is currently covered for other products. If our products do not receive favorable third-party reimbursement and patients are unwilling or unable to pay for our products out-of-pocket, it could limit our revenues and harm our business.
The continuing efforts of government and insurance companies, health maintenance organizations and other payers of healthcare costs to contain or reduce costs of health care may affect our future revenues and profitability, and the future revenues and profitability of our potential customers, suppliers and collaborative partners and the availability of capital. For example, in certain foreign markets, pricing or profitability of prescription pharmaceuticals is subject to government control. In the United States, recent federal and state government initiatives have been directed at lowering the total cost of health care. In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Federal and state legislatures will likely continue to focus on health care reform, controlling the cost of prescription pharmaceuticals and on the reform of the Medicare and Medicaid systems. While we cannot predict whether any such legislative or regulatory proposals will be adopted, the announcement or adoption of such proposals could materially harm our business, financial condition and results of operations.
33
Table of Contents
Our ProBiora3 products are sold subject to a right of return to mass retailers. If our estimates for returned products are incorrect, there could be a materially adverse impact on our net revenues and results of operations.
Our ProBiora3 products may be sold to mass retailers with a right of return, which is a common practice in the mass retail channel. For example, a right of return may be granted when the shelf life has reached its expiration or the product has remained unsold for a period of time. We are required to estimate the amount of product that will ultimately be returned pursuant to our return policy and to record a related reserve at the time of sale. These amounts are deducted from our gross revenues to determine our net revenues. In order to reasonably estimate future returns, we will analyze both quantitative and qualitative information including, but not limited to, actual return rates by product, the level of product in the distribution channel, expected shelf life of the product, product demand, the introduction of competitive or generic products that may erode current demand, our new product launches and general economic and industry wide indicators. There are inherent limitations in estimating future product returns due to the time lapse between sale and actual return of the product. If we over- or under-estimate the amount of product that will ultimately be returned, there could be a material impact to our results of operations. We are just beginning our roll out of ProBiora3 products on a larger scale to mass retailers and we have limited data to determine the expected returns in future periods in situations when we may grant rights of return.
If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results or prevent fraud which could subject us to regulatory sanctions, harm our business and operating results and cause the trading price of our stock to decline.
Effective internal controls required under Section 404 of the Sarbanes-Oxley Act of 2002, or Sarbanes-Oxley Act, are necessary for us to provide reliable financial reports and effectively prevent fraud. If we cannot provide reliable financial reports or prevent fraud, our business, reputation and operating results could be harmed. We have discovered, and may in the future discover, areas of our internal controls that need improvement. For example, “material weaknesses” were identified in our year ended December 31, 2009 which means that there was “a significant deficiency, or a combination of significant deficiencies, that results in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected.” During the year ended December 31, 2010, while we made progress in remediating many of the material weaknesses we identified, we are under significant operational stress due to a lack of capital resources, and as such our continuing remediation efforts may be impacted. Until we can complete our remediation efforts including the re-staffing and training of our accounting personnel, we have a higher risk of deficiencies in our financial reporting. We cannot be certain that the measures we have taken or intend to take will ensure that we maintain adequate controls over our financial processes and reporting in the future. Any failure to implement required new or improved controls or difficulties encountered in their implementation could subject us to regulatory sanctions, harm our business and operating results or cause us to fail to meet our reporting obligations. Inferior internal controls could also harm our reputation and cause investors to lose confidence in our reported financial information, which could have a negative impact on the trading price of our stock.
Risks Related to Our Common Stock
The Koski Family Limited Partnership, or KFLP, together with members of the Koski family, have a controlling interest in our outstanding shares of common stock.
The KFLP, together with members of the Koski family, own approximately 56.6% of our outstanding shares of common stock. In addition, we have borrowed $2,500,000 under a Credit Facility provided by the KFLP and borrowings under the Credit Facility are subject to automatic conversion into any securities we may offer in an offering. Two members of the Koski family also serve on our Board of Directors. As a result, the Koski family will be able to affect the outcome of, or exert significant influence over, all matters requiring shareholder approval, including the election and removal of directors and any change in control. In particular, this concentration of ownership of our common stock could have the effect of delaying or preventing a change of control of us or otherwise discouraging or preventing a potential acquirer from attempting to obtain control of us. This, in turn, could have a negative effect on the market price of our common stock. It could also prevent our shareholders from realizing a premium over the market price for their shares of common stock. Moreover, the interests of this concentration of ownership may not always coincide with our interests or the interests of other shareholders, and, accordingly, the Koski family could cause us to enter into transactions or agreements that we would not otherwise consider.
34
Table of Contents
Certain provisions of our articles of incorporation, bylaws, executive employment agreements and stock option plan may prevent a change of control of our company that a shareholder may consider favorable.
Provisions of our articles of incorporation, bylaws, executive employment agreements and stock option plan may discourage, delay or prevent a change of control of our company that a shareholder may consider favorable. These provisions include:
| • | authorization of the issuance of “blank check” preferred stock that could be issued by our Board of Directors without shareholder approval and that may be substantially dilutive or contain preferences or rights objectionable to an acquiror; |
| • | the ability of our Board of Directors to amend the bylaws without shareholder approval; |
| • | vacancies on our Board may only be filled by the remaining directors and not our shareholders; |
| • | requirements that only our Board, our President or holders of more than 10% of our shares can call a special meeting of shareholders; |
| • | obligations to make certain payments under executive employment agreements in the event of a change of control and termination of employment; and |
| • | immediate vesting of all stock options. |
As a result, these provisions could discourage bids for our common stock at a premium and limit the price that investors are willing to pay in the future for shares of our common stock. However, in our articles of incorporation we expressly elected not to be governed by Sections 607.0901 and 607.0902 of the Florida Business Corporation Act, which are statutory anti-takeover provisions relating to affiliated transactions and control share acquisitions, respectively. As such, we do not have the protection of these statutes in connection with any unwanted takeover attempts which could have the effect of encouraging an attempted change of control without first negotiating with our Board of Directors.
Our common stock is not listed on a national U.S. securities exchange and the application of the “penny stock” rules could adversely affect the market price of our common stock as well as increase your transaction costs to sell those shares.
Our common stock trades on the OTC Bulletin Board which generally has significantly less liquidity than securities traded on a national securities exchange, not only in the number of shares that can be bought and sold, but also through delays in the timing of transactions, reduction in securities analyst and news media coverage, and lower market prices than might otherwise be obtained. As a result, purchasers of shares of our common stock may find it difficult to resell their shares at prices quoted in the market or at all. In addition, if at any time the trading price of our stock is below $5.00 per share it is subject to the SEC’s “penny stock” rules. Because the “penny stock” rules impose certain requirements on brokers, they may be less willing to execute transactions in our securities. Furthermore, because of the limited market and generally low volume of trading in our common stock, our common stock is more likely to be affected by broad market fluctuations, general market conditions, fluctuations in our operating results, changes in the market’s perception of our business, and announcements made by us, our competitors or parties with whom we have business relationships. Our ability to issue additional securities for financing or other purposes, or to otherwise arrange for any financing we may need in the future, may also be materially and adversely affected by the fact that our securities are not traded on a national securities exchange.
Our stock price has historically been volatile and the trading volume of our stock has been low.
Since our initial public offering in June 2003 and through March 23, 2011 our stock price has fluctuated from $90.00 to $1.00 per share. The trading price of our common stock has been, and may be, subject to wide fluctuations in response to a number of factors, many of which are beyond our control. These factors include:
| • | quarter-to-quarter variations in our operating results; |
35
Table of Contents
| • | our perceived funding needs; |
| • | the results of testing, technological innovations, or new commercial products by us or our competitors; |
| • | governmental regulations, rules, and orders; |
| • | our level of, and expected future use of, working capital; |
| • | general conditions in the healthcare, dentistry, or biopharmaceutical industries; |
| • | comments, research reports and/or earnings estimates by securities analysts; |
| • | developments concerning patents or other intellectual property rights; |
| • | litigation or public concern about the safety of our products; |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; |
| • | additions or departures of directors, officers and key personnel; |
| • | release of transfer restrictions on our outstanding shares of common stock or sales of additional shares of common stock; |
| • | potential litigation initiated against us; and |
| • | the additional sale of common stock by us in capital raising transactions. |
Historically, the daily trading volume of our common stock has been relatively low and on some days our stock does not trade at all. We cannot guarantee that an active public market for our common stock will be sustained or that the average trading volume will increase. In addition, the stock market in general has experienced significant price and volume fluctuations. Volatility in the market price for particular companies has often been unrelated or disproportionate to the operating performance of those companies. Broad market factors may seriously harm the market price of our common stock, regardless of our operating performance. In addition, securities class action litigation has often been initiated following periods of volatility in the market price of a company’s securities. A securities class action suit against us could result in substantial costs, potential liabilities, and the diversion of management’s attention and resources. To the extent our stock price fluctuates or remains low, it could impair our ability to raise capital through the offering of additional equity securities.
Trading on the OTC Bulletin Board may be volatile and sporadic, which could depress the market price of our common stock and make it difficult for our stockholders to resell their shares.
Our common stock is quoted on the OTC Bulletin Board service of the Financial Industry Regulatory Authority (FINRA). Trading in stock quoted on the OTC Bulletin Board is often thin and characterized by wide fluctuations in trading prices, due to many factors that may have little to do with our operations or business prospects. This volatility could depress the market price of our common stock for reasons unrelated to operating performance. Moreover, the OTC Bulletin Board is not a stock exchange, and trading of securities on the OTC Bulletin Board is often more sporadic than the trading of securities listed on a quotation system like Nasdaq or a stock exchange like Amex. Accordingly, shareholders may have difficulty reselling any of their shares.
As a result of our recent 1-for-20 reverse stock split, the liquidity of our common stock and market capitalization could be adversely affected.
On September 24, 2010, we filed articles of amendment to our Articles of Incorporation to effect a 1-for-20 reverse stock split. A reverse stock split is often viewed negatively by the market and, consequently, can lead to a decrease in our overall market capitalization. In addition, because of the resulting reverse split the number of shares of our common stock that are outstanding has been significantly reduced and, the liquidity of our common stock could be adversely affected and you may find it more difficult to purchase or sell shares of our common stock.
36
Table of Contents
The issuance of additional equity securities by us in the future could result in dilution to our existing shareholders.
Any issuance of additional equity securities by us in the future could result in dilution to our existing shareholders. Such issuances could be made at a price that reflects a discount or a premium to the then-current trading price of our common stock. In addition, our business strategy may include expansion through internal growth by acquiring complementary businesses, acquiring or licensing additional products or brands, or establishing strategic relationships with targeted customers and suppliers. In order to do so, or to finance the cost of our other activities, we may issue additional equity securities that could result in further dilution to our existing shareholders. Under our Credit Facility, amounts borrowed are subject to automatic conversion into equity securities upon the successful completion of an offering by the Company. In addition, the KFLP has a put right which authorizes the KFLP to put any unborrowed or undrawn available amounts to the Company in order for the KFLP to fully participate in any automatic conversion that may be triggered by a successful offering.
Future sales of our common stock may depress our stock price.
The market price of our common stock could decline as a result of sales of substantial amounts of our common stock in the public market, or the perception that these sales could occur. In addition, these factors could make it more difficult for us to raise funds through future offerings of common stock. As of March 23, 2011, there were 5,683,076 shares of our common stock outstanding, with another 306,388 shares of common stock issuable upon exercise of warrants to investors, 399,836 shares issuable upon exercise of options outstanding and an additional 192,165 shares available for option grants under our stock option plans. The issuance of shares of our common stock under our 2002 Stock Incentive Plan is covered by Form S-8 registration statements we filed with the Securities and Exchange Commission, or SEC, and upon exercise of the options, such shares may be resold into the market. We have also issued shares of common stock and warrants in connection with private placements. Such shares are available for resale as well as the shares of common stock issuable upon exercise of the warrants. We issued an aggregate of 1,050,813 shares of our common stock in the December 2009 Private Placement and July 2010 Financing Transaction which are deemed to be “restricted securities,” as that term is defined in Rule 144 promulgated under the Securities Act of 1933, as amended, or Securities Act, and may be resold pursuant to the provisions of Rule 144. The resale of shares acquired from us in private transactions could cause our stock price to decline significantly. In addition, from time to time, certain of our shareholders may be eligible to sell all or some of their restricted shares of common stock by means of ordinary brokerage transactions in the open market pursuant to Rule 144, subject to certain limitations. In general, pursuant to Rule 144, after satisfying a six-month holding period: (i) affiliated shareholders, or shareholders whose shares are aggregated, may, under certain circumstances, sell within any three-month period a number of securities which does not exceed the greater of 1% of the then-outstanding shares of common stock or the average weekly trading volume of the class during the four calendar weeks prior to such sale and (ii) non-affiliated shareholders may sell without such limitations, in each case provided we are current in our public reporting obligations. Rule 144 also permits the sale of securities by non-affiliates that have satisfied a one-year holding period without any limitation or restriction.
We could issue additional common stock, which might dilute the book value of our common stock.
Our board of directors has authority, without action or vote of our shareholders, to issue all or a part of our authorized but unissued shares. Such stock issuances could be made at a price that reflects a discount or a premium from the then-current trading price of our common stock. In addition, in order to raise capital, we may need to issue securities that are convertible into or exchangeable for a significant amount of our common stock. These issuances would dilute your percentage ownership interest, which would have the effect of reducing your influence on matters on which our shareholders vote, and might dilute the book value of our common stock. You may incur additional dilution if holders of stock options, whether currently outstanding or subsequently granted, exercise their options, or if warrant holders exercise their warrants to purchase shares of our common stock.
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain qualified members for our Board of Directors.
As a public company, we are already subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and the Sarbanes-Oxley Act. The requirements of these rules and regulations may increase our legal, accounting and financial compliance costs; may make some activities more difficult, time-consuming and costly; and may also place undue strain on our personnel, systems and resources. Our management and other personnel need to devote a substantial amount of time to comply with these requirements. Moreover, these rules and regulations increase our legal and financial compliance costs and make some activities more time-consuming and costly. These rules and regulations could also make it more difficult for us to attract and retain qualified persons to serve on our Board of Directors and Board committees or as executive officers.
37
Table of Contents
As a public company we are also required to assess our internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act and file periodic reports with the SEC. If we are unable to comply with these requirements in a timely manner, or if material weaknesses or significant deficiencies persist, the market price of our stock could decline and we could be subject to sanctions or regulatory investigations, which could harm our business. We must perform an assessment of our internal control over financial reporting to allow management to report on the effectiveness of our internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. Our compliance with Section 404 of the Sarbanes-Oxley Act requires that we incur substantial accounting expense and expend significant management efforts. If we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act or the SEC reporting requirements in a timely manner, or if we continue to note or identify deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources. As a smaller reporting company, we do not have to have our independent registered public accounting firm report on the effectiveness of our internal control over financial reporting. If in the future we no longer meet the requirements of a smaller reporting company, our independent registered public accounting firm will have to report on our internal controls over financial reporting. There can be no assurance that our independent registered public accounting firm will not identify material weaknesses which could have an adverse effect on our stock price.
In October 2009, we began leasing the office space located at 3000 Bayport Drive, Suite 685, Tampa, Florida 33607. This new location has become our principal executive office and is also being used for sales and marketing and some administrative matters. The office space is approximately 3,150 square feet and the annual lease cost is $63,312 which includes insurance, utilities and taxes. The lease term expires January 2013. Lease payments are capped during the term with the exception of taxes and insurance exceeding 3%. In addition to our Tampa location we continue to lease our research facility located at 13700 Progress Boulevard, Alachua, Florida 32615. This lease was renewed for a two-year period beginning December 2009 and expires November 2011. The facility is approximately 5,300 square feet of which approximately 60% is laboratory space and the remainder is office space and common areas. The 12-month lease costs for the year ended December 31, 2010 was $107,916 which includes insurance, taxes and utilities. Lease payments are capped during the term which expires in November 2011. We expect the location in Alachua, Florida to continue to be used primarily as our research and laboratory space as we seek to migrate more of the administrative and accounting functions to our Tampa, Florida office location. There were no leasehold improvements in 2010 and 2009. The Company terminated two lease agreements in August and October 2009 for office space located in Alachua and St. Petersburg, Florida, respectively.
We are not a party to any material legal proceedings and there are no material legal proceedings pending with respect to our property. We are not aware of any legal proceedings contemplated by any governmental authorities involving either us or our property. None of our Directors, officers or affiliates is an adverse party in any legal proceedings involving us, or has an interest in any proceeding which is adverse to us.
38
Table of Contents
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Our common stock has been quoted on the over-the counter (OTC) Bulletin Board under the ticker symbol “ORNI,” since December 2008. The following sets forth the high and low bid quotations reflected on the OTC Bulletin Board for the periods applicable in the last two fiscal years.
| Period |
2010 | 2009 | ||||||||||||||
| High | Low | High | Low | |||||||||||||
| First quarter |
$ | 20.40 | $ | 6.00 | $ | 9.00 | $ | 4.40 | ||||||||
| Second quarter |
$ | 14.60 | $ | 6.80 | $ | 8.20 | $ | 1.00 | ||||||||
| Third quarter |
$ | 9.60 | $ | 5.00 | $ | 10.20 | $ | 4.40 | ||||||||
| Fourth quarter |
$ | 8.50 | $ | 2.95 | $ | 6.40 | $ | 4.00 | ||||||||
On March 23, 2011, the closing bid price of the common stock, as reported by the OTC Bulletin Board, was $3.80. As of March 3, 2011, there were approximately 92 registered holders of our common stock according to Continental Stock Transfer. The number of record holders does not reflect the number of beneficial owners of the common stock for whom shares are held by banks, brokerage firms and others.
Dividends
To date, we have neither declared nor paid any dividends on our common stock nor do we anticipate that such dividends will be paid in the foreseeable future. Rather, we intend to retain any earnings to finance the growth and development of our business. Any payment of cash dividends on our common stock in the future will be dependent, among other things, upon our earnings, financial condition, capital requirements and other factors which the board of directors deems relevant. In addition, restrictive covenants contained in any financing agreements entered into in the future may preclude us from paying any dividends.
ITEM 6. SELECTED FINANCIAL DATA.
Not applicable.
39
Table of Contents
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following information should be read in conjunction with the Financial Statements, including the notes thereto, included elsewhere in this Form 10-K. This discussion contains certain forward-looking statements that involve risks and uncertainties. Our actual results and the timing of certain events could differ materially from those discussed in these forward-looking statements as a result of certain factors, including, but not limited to, those set forth herein and elsewhere in this Form 10-K.
Overview
We are a biopharmaceutical company focused primarily on oral health products and novel antibiotics. Within oral health, we are developing our pharmaceutical product candidate, SMaRT Replacement Therapy, and we are also commercializing our oral probiotic blend, ProBiora3. Within antibiotics, we are developing our pharmaceutical product candidate, MU1140-S, and we intend to use our patented, novel organic chemistry platform to create additional antibiotics for therapeutic use.
Our SMaRT Replacement Therapy product candidate is designed to be a painless, one-time, five-minute topical treatment applied to the teeth that has the potential to offer lifelong protection against dental caries, or tooth decay. Our SMaRT Replacement Therapy is based on the creation of a genetically modified strain of bacteria that colonizes in the oral cavity and replaces native bacteria that cause tooth decay. We commenced a second Phase 1 clinical trial for SMaRT Replacement Therapy which we expect to conclude in second half of 2011.
We have also developed and are commercializing a variety of products that contain the active ingredient ProBiora3, a patent pending blend of oral probiotics that promote fresher breath, whiter teeth and support overall oral health. We have conducted extensive scientific studies on ProBiora3 in order to market our products under self-affirmed Generally Recognized As Safe status, or GRAS. We sell our ProBiora3 products through multiple distribution channels.
While developing SMaRT Replacement Therapy, members of our scientific team discovered that the SMaRT bacterial strain produces MU1140, a molecule belonging to the novel class of antibiotics known as lantibiotics. MU1140 has proven active preclinically against Gram positive bacteria responsible for a number of HAIs. We are in the process of scaling up production of our synthetic form of MU1140, or MU1140-S, and expect to commence preclinical testing and to file an Investigational New Drug, or IND, application with the FDA in 2012 as our capital resources permit. The key technology behind the production of MU1140-S is our Differentially Protected Orthogonal Lanthionine Technology platform, or DPOLT, which is a patented, novel organic chemistry platform that we believe will enable the first ever commercial scale, cost-effective production of any of the 50 known lantibiotics. We intend to use DPOLT to create a pipeline of lantibiotics for therapeutic use. Additionally, we are developing non-core technologies that originated from the discoveries of our scientific team, including LPT3-04, which is a weight loss product, and PCMAT, which is a biomarker discovery platform, both of which we believe could provide significant potential opportunities for us.
We were incorporated in November 1996 and commenced operations in 1999. We consummated our initial public offering in June 2003. We have devoted substantially all of our resources to the commercialization of our ProBiora3 products as well as our discovery efforts comprising research and development, clinical trials for our product candidates, protection of our intellectual property and the general and administrative support of these operations. We have generated limited revenues from grants and ProBiora3 product sales through December 31, 2010, and have principally funded our operations through the sale of debt and equity securities, including the exercise of warrants issued in connection with these financing transactions. Prior to 2008 our revenues were derived solely from research grants. Since 2008, our revenues have also included sales of our ProBiora3 products, which we initiated in late 2008. Our net revenues were $1,308,910 for the year ended December 31, 2010 and $641,285 for the year ended December 31, 2009.
We have never been profitable and, as of December 31, 2010, we had an accumulated deficit of $33,317,048. We incurred net losses of $7,805,165 and $5,519,348 for the years ended December 31, 2010 and 2009, respectively. We expect to incur significant and increasing operating losses for the foreseeable future as we seek to advance our product candidates through preclinical testing and clinical trials to ultimately obtain regulatory approval and eventual commercialization. The report of our independent registered public accounting firm with respect to our financial statements appearing in our Form 10-K contains an explanatory paragraph stating that our operating losses and negative cash flows from operations since inception, and our need to raise additional financing and/or financial support prior to July 31, 2011 in order to continue to fund our operations, raise substantial doubt about our ability to continue as a going concern. Adequate additional funding may not be available to us on acceptable terms, or at all. We expect that research and development expenses will increase along with general and administrative costs, as we grow and operate our business. There can be no assurance that additional capital will be available to us on acceptable terms, if at all.
40
Table of Contents
Recent Developments and Trends
In December 2010, we setup reserves for inventory on hand in the amount of $255,814 and sales returns in the amount of $105,588 to replace our existing inventory and customer held inventory. The reserves resulted from a change in the packaging and delivery mechanisms for our consumer ProBiora products (EvoraPlus, EvoraKids and EvoraPro) from blister pack tablets to one a day tablets in bottled form. The changes resulted from our understanding of customer preferences. The changes in delivery mechanism and packaging are expected to result in increased expenses in future periods as the change is fully implemented.
On January 24, 2011, we entered into a First Amendment to the Credit Facility (the “First Amendment”) to our existing Credit Facility with the Koski Family Limited Partnership or KFLP, (the “KFLP”), our largest shareholder. The First Amendment increased the available borrowing on the Credit Facility from $2,000,000 to $2,500,000 and simultaneously with entering into the First Amendment we drew on the additional $500,000 in available funds.
On February 4, 2011, we entered into a Second Amendment (the “Second Amendment”) to the Credit Facility with the KFLP. As a result of the Second Amendment, we are able to borrow up to an additional $2,5000,000 from the KFLP. Future draws under the Credit Facility, as amended, are limited to $500,000 per month commencing no earlier than March 2011. Under the Second Amendment, the due date of the amounts outstanding under the existing Credit Facility, were extended by one year from July 30, 2011 to July 30, 2012. The interest rate remained at LIBOR plus 6.0%. The Second Amendment further provided for the automatic conversion of any amounts borrowed and outstanding under the Credit Facility into Company securities that may be issued by us in subsequent securities offerings. Any automatic conversion of amounts outstanding under the Credit Facility would be on the same terms of any such offering. In addition, the Second Amendment provided the KFLP with the right to put any undrawn available amounts under the Credit Facility, as amended, to us and thereby have a note issued to the KFLP. The KFLP can exercise its put right to the extent it desires to fully participate, through the automatic conversion provision, in any subsequent offering by us.
On March 15, 2011, we drew an additional $500,000 on the Credit Facility, as amended. We expect to continue to need to draw on the available borrowings under the Credit Facility each month while we seek to raise additional capital.
We have continued our efforts to broaden the distribution of our ProBiora3 products through the following business development activities:
Distribution Agreements:
| • | On February 11, 2011, we announced that EvoraPro, EvoraPlus and EvoraKids will be available to dental professionals across North America through a distribution agreement with Patterson Dental. |
| • | PharmaForce: On January 25, 2011, we announced that PharmaForce, ApS has been awarded rights to use our oral-care probiotic ingredient, ProBiora3. This agreement gives PharmaForce exclusive rights to incorporate its ProBiora3 oral care probiotics in adult oral care products in Denmark and Finland. |
| • | ZooGlobe: On January 24, 2011, we announced that Teddy’s Pride will be available in Poland through a distribution agreement with ZooGlobe, which offers distribution through regional representatives and wholesale companies and pet shops across Poland. |
| • | Schiaffino, Lasky & Associates, Inc: On November 29, 2010, we announced the Teddy’s Pride will be represented by SLA Brands providing pet product to distributors and retailers in the United States and Canada. |
Exhibitions:
| • | Yankee Dental Congress: From January 27-29, 2011, we exhibited EvoraPro to dental professionals at their Annual Conference & Exhibition. |
| • | Greater New York Dental Meeting: From November 28 through December 1, 2010, we exhibited EvoraPro to the dental community in attendance at the meeting. |
| • | Supply Side West: From October 19 – 23, 2010, we exhibited ProBiora3® to a wide array of thousands of executives from the food, supplement and cosmetic industries. |
41
Table of Contents
| • | American Dental Association: From October 9-12, 2010, we exhibited EvoraPro to dental professionals at their Annual Session and World Marketplace Exhibition. |
We are impacted by various trends and uncertainties, including the uncertainty associated with the availablity of sufficient capital resources to execute our plans and conduct our operations. In addition, while we were pleased with the level of initial success we achieved in establishing the mass retail distribution channel, the maintenance, continued use and expansion of this channel requires us to commit to expend capital resources on advertising and marketing campaigns. Because our available capital resources currently limit our ability to engage in significant advertising and marketing campaigns we are undertaking an evaluation of the continued pursuit of the mass retail channel relative to other options. In connection with our ongoing evaluation of this channel in first quarter of 2011 we determined to pull our EvoraPlus product from the store shelves of Rite Aid. Also during the first quarter of 2011, GNC has refocused their strategy on selling higher margin GNC branded products and as such the decision was made to end distribution of our EvoraPlus product. Through these actions our mass retail store presence was reduced from 17,000 stores approximately to just over 11,000 stores. To the extent our capital resources continue to limit our ability to engage in significant advertising, we expect the number of mass retail stores that carry our consumer ProBiora3 products to continue to decline in the future.
Financial Overview
Net Revenues
Our revenues prior to 2008 consisted exclusively of grant funding from government agencies under the National Science Foundation’s, or NSF, and National Institutes of Health’s, or NIH, Small Business Innovation Research, or SBIR, grants. Since the initial launch of our ProBiora3 products in late 2008, our net revenues for the year ended December 31, 2008 also included sales of our ProBiora3 products. Sales of our ProBiora3 products were $1,128,895, for the year ended December 31, 2010 and $366,801 for the year ended December 31, 2009, respectively. Because of our efforts to increase the distribution of our ProBiora3 products, we expect net revenues to continue to increase in the near future. However, our success will depend on a number of factors, including our marketing efforts related to our ProBiora3 products.
We expect that our revenues will fluctuate from quarter to quarter as a result of the volume of sales of our products and the amount of license fees, research and development reimbursements, milestone and other payments we may receive upon any license or strategic partnerships we may enter into in the future.
Cost of Goods Sold
Our cost of goods sold includes the production and manufacture of our ProBiora3 products, as well as shipping and processing expenses and scrap expense. Scrap expense represents product rework charges, inventory adjustments, inventory replacement reserves, and damaged inventory. We expect our costs of goods sold to increase as we expand our distribution and sales efforts for our ProBiora3 products.
Research and Development Expenses
Research and development consists of expenses incurred in connection with the discovery and development of our product candidates. These expenses consist primarily of employee-related expenses, which include salaries and benefits and attending science conferences; expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a substantial portion of our preclinical studies; the cost of acquiring and manufacturing clinical trial materials; facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities and equipment, and depreciation of fixed assets; license fees for and milestone payments related to in-licensed products and technology; stock-based compensation expense; and costs associated with non-clinical activities and regulatory approvals. We expense research and development costs as incurred.
We plan to increase our research and development expenses for the foreseeable future as we seek to advance the development of our SMaRT Replacement Therapy and MU1140-S product candidates, and to further advance our earlier stage research and development projects, such as LPT3-04, our potential weight loss product, and PCMAT, our biomarker discovery platform.
Prior to January 1, 2009, we did not track our internal research and development costs or our personnel and personnel-related costs on a project-by-project basis, instead, our research and development resources were allocated among all of our programs. Since January 1, 2009, we have tracked development expenses and personnel expense on a project-by-project basis and have allocated common expenses, such as scientific consultants and lab supplies, to each program based on the personnel resources allocated to each program.
42
Table of Contents
Our research and development expenses were $2,014,784 and $1,833,746 for the years ended December 31, 2010 and 2009, respectively. Our research and development expenses can be divided into (i) clinical research and development, and (ii) preclinical research and development activities. Clinical research and development costs consist of clinical trials, manufacturing services, regulatory activities and related personnel costs, and other costs such as rent, utilities, depreciation and stock-based compensation. Preclinical research and development costs consist of our research activities, preclinical studies, related personnel costs and laboratory supplies, and other costs such as rent, utilities, depreciation and stock-based compensation. While we are currently focused on advancing each of our product development programs, our future research and development expenses will depend on the clinical success of each product candidate, as well as ongoing assessments of each product candidate’s commercial potential. In addition, we cannot forecast with any degree of certainty which product candidates may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
We expect our research and development expenses to increase in the future as we continue the advancement of our clinical trials and preclinical product development programs. The lengthy process of completing clinical trials and seeking regulatory approval for our product candidates requires expenditure of substantial resources. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could cause a delay in generating product revenues and cause our research and development expenses to increase and, in turn, have a material adverse effect on our operations. Our current product development candidates are not expected to be commercially available before 2011.
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist principally of salaries and related costs for personnel in executive, finance, business development, marketing, information technology, legal and human resources functions. Other general and administrative expenses include facility costs not otherwise included in research and development expenses, patent filing, prosecution and defense costs and professional fees for legal, consulting, auditing and tax services.
We anticipate that our general and administrative expenses will increase for, among others, the following reasons:
| • | the sales and marketing of our ProBiora3 products; |
| • | to support our research and development activities, which, subject to available capital, we expect to expand as we continue the development of our product candidates; |
| • | the efforts we undertake from, time to time, to raise additional capital; and |
| • | the increased payroll, expanded infrastructure and higher consulting, legal, accounting and investor relations costs associated with being a public company. |
Other Income and (Expense)
Other income and expense includes gain or loss on sale of assets, local business taxes, extinguishment of payables, and loss from abandoned public offering, as well as interest income and expense. Interest income consists of interest earned on our cash and cash equivalents. The primary objective of our investment policy is capital preservation. Interest expense consists primarily of interest and costs associated with our loans payable.
Income Taxes
As of December 31, 2010 and 2009, we had federal and state net operating loss carryforwards and research and development tax credit carryforwards of approximately $30,150,000 and $23,130,000, respectively. Our net operating loss and research and development tax credit carryforwards will expire, if not used, by 2031. Our ability to utilize our net operating loss and tax credit carryforwards may be limited in the event a change in ownership, as defined in Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, has occurred or may occur in the future. The private placement transaction with the KFLP in June 2009 (the “June 2009 Private Placement”) constituted such an event and our historical loss carryfowards were limited. See “Tax Loss Carryforwards.” In each period since our inception, we have recorded a 100% valuation allowance for the full amount of our deferred tax asset, as the realization of the deferred tax asset is uncertain. As a result, we have not recorded any federal tax benefit in our statements of operations.
43
Table of Contents
Critical Accounting Estimates and Policies
Our discussion and analysis of our financial condition and results of operations are based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or U.S. GAAP. The preparation of financial statements in accordance with U.S. GAAP requires us to make estimates and assumptions that affect reported amounts and related disclosures. We consider an accounting estimate to be critical if it requires assumptions to be made that were uncertain at the time the estimate was made; and changes in the estimate or different estimates that could have been made could have a material impact on our results of operations or financial condition. The principal areas of estimation reflected in the financial statements are stock-based compensation, valuation of warrants, inventory obsolescence reserve, sales returns and allowances and allowance for doubtful accounts.
Revenue Recognition
We recognize revenues from the sales of product when title and risk of loss pass to the customer, which is generally when the product is shipped. Grant revenues are recognized as the reimbursable expenses are incurred over the life of the related grant. Grant revenues are deferred when reimbursable expenses have not been incurred. We record allowances for discounts and product returns at the time of sales as a reduction of revenues as such allowances can be reliably estimated based on historical experience or known trends. Product returns are limited to specific mass retail customers for expiration of shelf life or unsold product over a period of time. We maintain a return policy that allows our customers to return product within a specified period of time prior to and subsequent to the expiration date of the product. Our estimate of the provision for returns is analyzed quarterly and is based upon many factors, including industry data of product return rates, historical experience of actual returns, analysis of the level of inventory in the distribution channel, if any, and reorder rates. If the history or our product returns changes, the reserve will be adjusted. While we believe that the reserves we have established are reasonable and appropriate based upon current facts and circumstances, applying different judgments to the same facts and circumstances would result in the estimated amounts for sales returns and chargebacks to vary. Because our ProBiora3 products have only recently been introduced, we could experience different circumstances in the future and these differences could be material.
The Company has granted guaranteed rights of return on several mass retail customer accounts. At this time there are only two active mass retail customer accounts with guaranteed rights of return. Orders are processed and shipped on these accounts however the Company defers recognition of revenue until the customer provide notification to the Company that the product has sold to the end consumer. Once notification has been received and verified, the Company will record revenue in that accounting period.
Accounts Receivable
Accounts receivable are recorded at their net realizable value and consist of trade receivables from the sale of product to customers. We analyze accounts receivable on a monthly basis and determine the collectability based on the facts and circumstances relating to each customer. The company estimates their allowance for doubtful accounts based on sales trends and specific review of the creditworthiness of each customer.
Inventory
Inventories are stated at the lower of cost or market. Cost, which includes material, labor and overhead, is determined on a first-in, first-out basis. On a quarterly basis, we analyze our inventory levels and reserve for inventory that is expected to expire prior to being sold, inventory that has a cost basis in excess of its expected net realizable value, inventory in excess of expected sales requirements, or inventory that fails to meet commercial sale specifications. Expired inventory is disposed of and the related costs are written off to the reserve for inventory obsolescence. The inventory reserve at December 31, 2010 and 2009 was approximately $255,814 and $0, respectively.
Consigned Inventory
The Company has authorized a consignment inventory arrangement with one of its mass retail customers in March 2010. As of December 31, 2010, the Company has $64,999 of inventory on consignment located at the retailers stores and warehouses, which is included in our inventory reserve. Once consignment inventory has been sold by this customer, the customer notifies the Company of the sale and the Company records revenue in that accounting period.
44
Table of Contents
The Company authorizes the replenishment of consignment inventory based on orders placed by the customer. The Company is provided with weekly reports of consignment sales activity and balances.
Stock-Based Compensation
U.S. GAAP requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values as of the grant dates. Stock-based compensation expense is recorded over the requisite service period in which the grantee provides services to us, to the extent the options or warrants do not vest at the grant date and are subject to forfeiture.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards.
Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rate is recognized in operations in the period that includes the enactment date. Deferred tax assets are reduced to estimated amounts expected to be realized by the use of a valuation allowance. Based on our historical operating losses, a valuation allowance has been recognized for all deferred tax assets.
New Accounting Pronouncements
See Notes to Financial Statements – Item #1. Organization and Significant Accounting Policies: New Accounting Pronouncements.
Results of Operations:
| Years ended December 31 |
Three months ended December 31 |
|||||||||||||||
| 2010 | 2009 | 2010 | 2009 | |||||||||||||
| Revenue |
$ | 1,308,910 | $ | 641,285 | 298,157 | 275,443 | ||||||||||
| Cost of sales |
911,793 | 221,198 | 438,215 | 120,354 | ||||||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
2,014,784 | 1,833,746 | 641,536 | 426,230 | ||||||||||||
| Selling, general and administrative |
6,285,004 | 4,917,844 | 1,492,854 | 1,033,860 | ||||||||||||
| Total operating expenses |
8,299,788 | 6,751,590 | 2,134,390 | 1,460,090 | ||||||||||||
| Loss from operations |
(7,902,671 | ) | (6,331,503 | ) | (2,274,448 | ) | (1,305,001 | ) | ||||||||
| Other income (expense): |
||||||||||||||||
| Interest income |
3,657 | 922 | 521 | 126 | ||||||||||||
| Interest expense |
(33,859 | ) | (44,292 | ) | (28,866 | ) | (18,377 | ) | ||||||||
| Loss from abandoned public offering |
(603,012 | ) | — | (603,012 | ) | — | ||||||||||
| Gain (loss) on sale of property and equipment |
— | 22,743 | — | 11,469 | ||||||||||||
| Gain on extinguishment of payables |
— | 832,959 | — | 79,017 | ||||||||||||
| Local business tax |
(2,717 | ) | (177 | ) | (148 | ) | — | |||||||||
| Total other income, net |
(635,931 | ) | 812,155 | (631,505 | ) | 72,235 | ||||||||||
| Loss before income taxes |
(8,538,602 | ) | (5,519,348 | ) | (2,905,953 | ) | (1,232,766 | ) | ||||||||
| Income tax benefit |
733,437 | — | 733,437 | — | ||||||||||||
| Net Loss |
$ | (7,805,165 | ) | $ | (5,519,348 | ) | (2,172,516 | ) | (1,232,766 | ) | ||||||
45
Table of Contents
For the Three Months Ended December 31, 2010 and 2009
Net Revenues. We generated net revenues of $298,157 for the three months ended December 31, 2010 compared to $275,443 in the same period in 2009, a increase of $22,714. The increase was primarily attributable to ProBiora3 product sales and grant revenue, net of an increase in the sales return allowance.
Cost of Goods Sold. Cost of goods sold was $438,215 for the three months ended December 31, 2010 compared to $120,354 in the same period in 2009, an increase of $317,861. The increase was attributable to increased sales of our ProBiora3 products. Cost of goods sold includes the production and manufacturing costs of our ProBiora3 products sold of $116,484, shipping and processing expenses of $36,347, and scrap expense of $285,384. Scrap expenses represent product rework charges, inventory adjustments, inventory reserves of $255,814 associated with expected inventory replacement costs, and damaged inventory.
Research and Development. Research and development expenses were $641,536 for the three months ended December 31, 2010 compared to $426,230 in the same period in 2009, an increase of $215,306, or 50.5%. The increase was primarily attributed to an increase in clinical trial expense of $199,291 associated with the Phase 1 SMaRT clinical trial.
Selling, General and Administrative. Selling, general and administrative expenses were $1,492,854 for the three months ended December 31, 2010 compared to $1,033,860 in the same period in 2009, an increase of $458,994, or 44.4%. This increase was due to increases in advertising and marketing expenses of $355,423, salary and fringe costs of $227,052 as a result of additional staff and increases in compensation of existing personnel, non-employee director fees of $49,808, and an increase in travel expense of $13,138. The increase in selling, general and administrative expense was primarily offset by reduction in other expenses, including reductions in stock-based compensation expense of $50,593 and legal and professional support service fee savings of $63,572.
Other Income (Expense). Other income and (expense) was ($631,504) for the three months ended December 31, 2010 compared to $72,235 in the same period in 2009, a change of ($703,739). The decrease was primarily attributable to expenses of $603,012 we recognized in 2010 associated with the filing of a registration statement for our contemplated public offering (which we withdrew prior to the end of the year), as well as an increase in interest expense of $10,490. In addition, reductions in income from the prior period of $79,017 associated with the extinguishment of payables in connection with the June 2009 Private Placement and a $11,469 gain we realized in 2009 on the sale of assets.
For the Years Ended December 31, 2010 and 2009
Net Revenues. We generated net revenues of $1,308,910 for the year ended December 31, 2010 compared to $641,285 in the same period in 2009, an increase of $667,625. The increase in net revenues was primarily attributable to increased ProBiora3 product sales. The increase in net revenues also included a $99,398 decrease in grant revenues.
Cost of Goods Sold. Cost of goods sold was $911,793 for the year ended December 31, 2010 compared to $221,198 in the same period in 2009, an increase of $690,595. This increase was attributable to increased sales of our ProBiora3 products. Cost of goods sold also includes shipping and warehouse processing expenses of $145,049 and scrap expense of $394,667. Scrap expenses represent product rework charges, inventory adjustments, inventory reserves of $255,814 associated with expected inventory replacement costs, and damaged inventory.
Research and Development. Research and development expenses were $2,014,784 for the year ended December 31, 2010 compared to $1,833,746 in the same period in 2009, an increase of $181,038, or 9.9%. The increase was primarily attributable to an increase in clinical trial expense of $206,986, consulting expense of $94,509, and increases in patent expense of $47,859. The increase in research and development expense was offset by decreases in depreciation expense stemming from the full depreciation of certain lab equipment of $199,559.
Selling, General and Administrative. Selling, general and administrative expenses were $6,285,004 for the year ended December 31, 2010 compared to $4,917,844 in the same period in 2009, an increase of $1,367,160, or 27.8%. The increase was primarily attributable to increases in advertising and marketing expenses of $1,194,230, officer/staff salaries and benefits of $502,754, stock based compensation expense of $217,163, and non-employee director fees of $158,365. The increase in selling, general and administrative expense was offset by decreases in legal fees of $519,769 and consulting fees of $176,288.
46
Table of Contents
Other Income (Expense). Other income and expense was ($635,931) for the year ended December 31, 2010 compared to $812,155 in the same period in 2009, a change of $1,448,086. The decrease was primarily attributable to expenses of $603,012 we recognized in 2010 associated with the filing of a registration statement for our contemplated public offering (which we withdrew prior to the end of the year). In addition, the decrease in other income and expense for the period was impacted by a decrease in income from the prior period of $832,959 associated with the extinguishment of payables in connection with the June 2009 Private Placement and a $22,743 gain we realized in 2009 on the sale of assets.
Liquidity and Capital Resources
Since our inception, we have funded our operations primarily through the sale of equity securities in our initial public offering, the sale of equity securities and warrants in private placements, debt financing and grants. The following table sets forth the primary sources and uses of cash for each of the periods indicated:
| Years ended December 31, | ||||||||
| 2010 | 2009 | |||||||
| Net cash used in operating activities |
$ | (6,448,434 | ) | $ | (5,799,481 | ) | ||
| Net cash provided by (used in) investing activities |
(88,084 | ) | 30,927 | |||||
| Net cash provided by financing activities |
6,367,029 | 4,904,213 | ||||||
| Net decrease in cash and cash equivalents |
$ | (169,489 | ) | $ | (864,341 | ) | ||
During the years ended December 31, 2010 and 2009, our operating activities used cash of $6,448,434 and $5,799,481, respectively. The use of cash in all periods primarily resulted from our net losses adjusted for non-cash items and changes in operating assets and liabilities. We had negative working capital of $127,518 as of December 31, 2010 compared to positive working capital of $2,564,147 as of December 31, 2009.
During the years ended December 31, 2010 and 2009, our investing activities used cash of $88,084 and provided cash of $30,927, respectively. The cash provided and used in connection with investing activities primarily related to purchases and sales of equipment.
During the years ended December 31, 2010 and 2009, our financing activities provided cash of $6,367,029 and $4,904,213, respectively. The cash provided by financing activities in the year ended December 31, 2009 was primarily due to our debt and equity financings including the June 2009 Private Placement and the December 2009 Private Placement. The cash provided by financing activities in the year December 31, 2010 was primarily due to the release of restrictions on cash, borrowings under a note payable from a shareholder and proceeds from issuance of common stock, offset by reductions in long term notes payable.
Set forth below is a description of our various financing activities for the periods reflected in this report:
June 2008 Private Placement
In June 2008, we issued a total of 288,888 shares of restricted common stock and warrants to acquire 288,888 shares of common stock (the “June 2008 Warrants”) in a private placement to accredited investors, for net proceeds of $2,515,000 (the “June 2008 Private Placement”). The shares were sold at price per share of $9.00. The June 2008 Warrants were exercisable at $26.00 per share. The June 2008 Warrants expire May 30, 2013. Of the June 2008 Warrants, 161,000 were subsequently amended to reduce the exercise price in connection with the June 2009 Private Placement discussed below.
47
Table of Contents
June 2009 Private Placement
On June 29, 2009, we issued a total of 2,500,000 shares of restricted common stock and warrants to acquire 50,000 shares of common stock in a private placement to the Koski Family Limited Partnership, or KFLP, for total proceeds of $4,000,000 (the “June 2009 Private Placement”). The shares were sold at $1.60 per share. The warrants to purchase 50,000 shares of our common stock were exercisable at $2.00 per share and had a five year term. The consideration paid by the KFLP for the shares of common stock consisted of $4,000,000 as follows: $1,500,000 in cash at closing and $2,500,000 pursuant to a non-interest bearing promissory note providing for five consecutive monthly installment payments of $500,000 commencing July 31, 2009. In addition, pursuant to the securities purchase agreement (the “June 2009 Purchase Agreement”) with the KFLP, the KFLP also provided a secured loan of $1,000,000 to us. The loan was secured by substantially all of our assets, excluding receivables, and paid interest at the rate of prime plus 4.0% which was payable quarterly. This loan was subsequently repaid by us in connection with the December 2009 Private Placement described below. The principal of the loan was due in five years. As a result of the June 2009 Private Placement the Board of Directors believes there was a change of control, with the KFLP acquiring a controlling interest in our outstanding voting common stock.
We also agreed to provide the KFLP with certain registration rights in connection with any underwritten or other offering by us over the next five years. Specifically, we are obligated to register on behalf of the KFLP shares of common stock held by the KFLP equal to 15% of the total number of shares of common stock to be sold by us in a public offering subject to the discretion of the managing underwriter on the inclusion of shares in the offering to be sold by selling shareholders.
In addition to the above, as a further condition to the consummation of the transaction contemplated by the June 2009 Purchase Agreement, we were required to obtain satisfactory arrangements with three main creditors for reductions in the amounts payable by us to these creditors. As of June 29, 2009, these reductions amounted to $707,674 in the aggregate and were conditioned upon prompt payment of the remaining balances owed to such creditors after taking into account the agreed upon reductions. As of December 31, 2009, the amount of reductions arranged with our creditors totaled $832,959. These agreed upon reductions in payables have been fully reflected in our financial statements for the periods and reported under other income.
In connection with, and as a closing condition to the June 2009 Private Placement, the purchasers in the June 2008 Private Placement (including George Hawes, our largest shareholder prior to the June 2009 Private Placement), entered into a consent, waiver and mutual release agreement with us on June 25, 2009. In addition, the purchasers in the June 2008 Private Placement waived and relinquished any special rights they possessed pursuant to the agreements with us as part of the June 2008 Private Placement, including, but not limited to, (i) rights of first refusal, (ii) antidilution regarding future equity sales and (iii) covenants regarding secured lending by us. In connection with such consents, waivers and mutual releases, warrants to acquire 161,000 shares that were previously issued in connection with the June 2008 Private Placement were subject to the right of exchange for new replacement warrants to acquire the same number of shares under the same terms except for a change in the exercise price from $26.00 to $15.00. In addition, to the extent of any future underwritten registered offerings of our common stock, or the filing of any resale registration statement by us, in each case occurring within five years from the date of the consent, waiver and mutual release, the purchasers shall have the right to include an aggregate of up to 5.0% of the shares being registered in such offering or registration statement, subject to the discretion, in any underwritten primary offerings by us, of the underwriter on the inclusion of shares in the offering to be sold by selling shareholders.
December 2009 Private Placement
On December 30, 2009, we issued a total of 500,813 shares of restricted common stock in the initial closing of a private placement to accredited investors including the KFLP, our largest shareholder (the “December 2009 Private Placement”), for initial proceeds of $2,504,062. The shares were sold at $5.00 per share. The initial closing proceeds of $2,504,062 included the cancellation at closing of $54,062 in outstanding obligations we owed to Dr. Jeffrey Hillman, our Chief Scientific Officer, for compensation that had been deferred. Approximately half of the total investment, or $1,250,000, was made by the KFLP. In conjunction with, and as a condition to the initial closing of the December 2009 Private Placement, we also issued 200,000 shares of our common stock to the KFLP at $5.00 per share, which was the same price per share paid by the participating accredited investors, in exchange for the cancellation of the KFLP’s $1,000,000 secured promissory note we previously issued to the KFLP in connection with the June 2009 Private Placement.
Approximately $1,000,000 of the total proceeds from the December 2009 Private Placement were committed to further our development of the DPOLT synthetic chemistry platform, essential to the production of our lead antibiotic, MU1140, subject to the goals set forth by the two-year NSF SBIR Phase II grant that we received on February 15, 2008. Such allocation enabled us to be eligible to receive up to an additional $500,000 matching grant from the NSF, which grant was subsequently awarded in June 2010.
Contemporaneously with the initial closing of the December 2009 Private Placement, the KFLP also elected to exercise warrants it received as part of the June 2009 Private Placement to purchase 50,000 shares of our common stock. The warrants were exercised through the payment by the KFLP of the warrant exercise price of $2.00 per share.
48
Table of Contents
Additionally, Christine Koski and Robert Koski, as directors, each exercised previously issued options to purchase 5,000 shares of our common stock at the option exercise price of $2.00 per share. These options were granted to Christine Koski and Robert Koski when they became non-employee directors on June 30, 2009 in connection with our non-employee director compensation program.
On January 13, 2010, we completed the $3,004,062 private placement contemplated by the December 2009 Private Placement and issued another 100,000 shares of common stock at a price per share of $5.00 to the accredited investors for $500,000. Of this amount, the KFLP again participated in half of the remainder of the aggregate investment by acquiring 50,000 shares for $250,000.
May 2010 Note Financing
On May 28, 2010, we entered into an unsecured promissory note with a conversion provision (the “May 2010 Note”) to the KFLP pursuant to which we borrowed $1,000,000 from the KFLP. Interest on the May 2010 Note accrued at the rate of LIBOR plus 6.0% and the principal of the May 2010 Note, together with all accrued interest thereon, was due and payable the earlier of: (i) the closing date of a registered public offering of newly issued equity securities by us resulting in cash proceeds to us, other than in connection with employee option plans, or (ii) the May 24, 2011 maturity date; provided, however, that in the event we completed a subsequent private offering of equity securities prior to the May 24, 2011 maturity date, we could elect to convert the principal of the May 2010 Note into the same equity securities being sold in the private offering at the same price and terms to the KFLP.
July 2010 Financing Transaction
On July 5, 2010, we entered into a common stock purchase agreement (the “July 2010 Financing Transaction”) with the KFLP. At the closing of this financing transaction on July 30, 2010 we issued 250,000 shares of our common stock to the KFLP at a price of $8.00 per share. The $2,000,000 aggregate consideration paid by the KFLP consisted of (i) $1,000,000 cash and (ii) the exchange and cancellation of the outstanding May 2010 Note issued to the KFLP on May 28, 2010. Accrued interest on the May 2010 Note through closing was waived by the KFLP.
Concurrent with the July 2010 Financing Transaction and as part thereof, we entered into an unsecured revolving credit agreement (the “Credit Facility”) with the KFLP. Pursuant to the Credit Facility, we are able to borrow up to $2,000,000 from the KFLP at LIBOR plus 6.0%. The term of the Credit Facility is for 12 months commencing August 1, 2010. Our ability to draw on the Credit Facility is subject to (i) the receipt by the KFLP of a certificate of no adverse change from us in form and substance acceptable to the KFLP, (ii) the receipt by the KFLP of a revolving unsecured promissory note from us in the principal drawn down in the form attached to the Credit Facility and (iii) our compliance with the terms of the Credit Facility.
On September 13, 2010, we drew down on the Credit Facility in the amount of $1,000,000 and executed a revolving unsecured promissory note (the “September 2010 Promissory Note”) for such amount in favor of the KFLP. In addition, on November 8, 2010 we drew down on the remaining $1.0 million of available funds under the Credit Facility and executed another revolving unsecured promissory note (the “November 2010 Promissory Note”). The September 2010 Promissory Note and November 2010 Promissory Note each initially matured on July 30, 2011 until the Second Amendment discussed below, which extended the maturity date to July 30, 2012.
On January 24, 2011, we entered into a First Amendment to the Credit Facility (the “First Amendment”) to increase the available borrowing from $2,000,000 to $2,500,000 and simultaneously therewith we drew on the Credit Facility as amended by the First Amendment to borrow the additional $500,000 in available funds and executed another revolving unsecured promissory note (the “January 2011 Promissory Note”) initially due on July 30, 2011.
On February 4, 2011, we entered into a Second Amendment (the “Second Amendment”) to the Credit Facility with the KFLP. As a result of the Second Amendment, we are able to borrow up to an additional $2,500,000 from the KFLP. Future draws under the Credit Facility, as amended, are limited to $500,000 per month commencing no earlier than March 2011. Under the Second Amendment, the due date of the amounts then outstanding under the Credit Facility, (the September 2010 Promissory Note, November 2010 Promissory Note and January 2011 Promissory Note) were extended by one year from July 30, 2011 to July 30, 2012. The interest rate remained at LIBOR plus 6.0%. The Second Amendment further provided for the automatic conversion of any amounts borrowed and outstanding under the Credit Facility into securities that we may issue in subsequent securities offerings. Any automatic conversion of amounts outstanding under the Credit Facility would be on the same terms of any such offering. In addition, the Second Amendment provides the KFLP with the right to put any undrawn available amounts under the Credit Facility, as amended, to us and thereby have a note issued to the KFLP. The KFLP can exercise its put right to the extent it desires to fully participate, through the automatic conversion provision, in any subsequent offering by us.
49
Table of Contents
On March 15, 2011, we borrowed an additional $500,000 under the Credit Facility, as amended and executed a revolving unsecured promissory note (the “March 2011 Promissory Note”) in such amount. The March 2011 Promissory Note matures on July 30, 2012.
Other Financings
On March 17, 2009, we entered into a short-term note payable for $53,087 with an interest rate of 5.75% to finance product liability insurance. This note required principal and interest payments to be made evenly over a ten-month period and was repaid in full at December 31, 2009.
On April 15, 2009 we entered into a loan agreement with an accredited investor for a short-term note in the amount of $100,000. The note included an interest rate of 15% per annum and its maturity date was April 15, 2011. On August 21, 2009 we repaid this short-term note and outstanding accrued interest in full. In connection with this borrowing we also issued warrants to acquire 5,000 shares of our common stock at an exercise price of $10.00 per share to the investor and such warrants are exercisable for five years.
On May 4, 2009 and June 10, 2009, we borrowed $32,556 and $13,100, respectively, from Dr. Jeffery Hillman, our founder, Chief Scientific Officer and Director. These borrowings were to be repaid upon demand by Dr. Hillman, were unsecured and did not bear interest. The proceeds from these borrowings were used to purchase inventory for our Consumer Healthcare products division. On June 29, 2009 the aggregate amount of these obligations of $45,656 were repaid by us in full through the issuance of 22,828 shares of our common stock at a price of $2.00 per share, which was the closing price of our common stock on June 29, 2009.
On August 6, 2009 we entered into a short-term note payable for $70,025 with an interest rate of 5.75% to finance directors’ and officers’ liability insurance. This note required principal and interest payments to be made evenly over a ten-month period and was repaid in full on May 24, 2010 in accordance with its terms.
On March 17, 2010, we entered into a short-term note payable for $50,637 with an interest rate of 5.75% to finance product liability insurance. Payments on this note are made evenly based on a straight line amortization over a ten-month period with the final payment due on January 10, 2011.
On July 9, 2010, we entered into a non-interest bearing short-term note payable for $22,188 to finance a portion of our new enterprise resource planning system. Payments on this note began July 9, 2010 and are made quarterly with the final payment due on April 1, 2011.
On July 20, 2010 we entered into a short-term note payable for $63,835 with an interest rate of 5.75% to finance directors’ and officers’ liability insurance. Payments on this note begin on August 24, 2010 and are made evenly based upon a straight line amortization over a ten-month period with the final payment due on July 24, 2011.
On July 31, 2010, we entered into a short-term note payable for $85,185 bearing interest at 7.5% to finance a portion our new enterprise resource planning system. Principal and interest payments on this note begin August 31, 2010 and are made evenly based on a straight line amortization over a 17-month period with the final payment due on December 31, 2011.
On March 3, 2011, we entered into a short-term notes payable for $48,988 bearing interest at 5.48% to finance product liability insurance. Payments on this note are made evenly based on a straight line amortization over a ten-month period with the final payment due on January 10, 2012.
Grants
On February 15, 2008, we were awarded a two-year NSF SBIR Phase II grant to advance development of DPOLT. This federal grant supports studies focused on the synthesis and testing of our lead antibiotic, MU1140. The grant amount was $500,000 which we received $425,000 of these restricted funds. The balance of the grant was modified by the NSF revised budget and included in the subsequent grant awarded on June 10, 2010.
On September 1, 2009, we received a grant funding from the University of Florida under the prime grant with the Florida Citrus Production Advisory Council in the amount of $124,570. The purpose of the University of Florida grant is to identify disease-specific proteins expressed during citrus greening using our proprietary PCMAT biomarker technology.
50
Table of Contents
On June 10, 2010, we were awarded the matching $500,000 grant from the NSF to support the previously awarded SBIR Phase II grant for further development of our DPOLT platform. On June 17, 2010, we received $125,000 and on February 25, 2011 we received $125,000 related to this NSF awarded SBIR II Phase II grant for the company’s DPOLT platform. Proceeds from the financing are to be allocated to further the development of our DPOLT platform, essential to the production of our lead antibiotic, MU1140, subject to the goals set forth by the NSF SBIR Phase II grant received by us. The remainder of these grant funds are expected to be provided to us in $125,000 increments over 18 to 24 months from the grant dates.
Tax Credit
On November 1, 2010, we received notification that we were awarded federal grant funding for three of its therapeutic development programs under the Qualifying Therapeutic Discovery Project. The Qualifying Therapeutic Discovery Project, was recently enacted by Congress as part of the Patient Protection and Affordable Care Act of 2010, which was designed to provide grants or tax credits to qualified biotechnology companies that demonstrate the potential to either 1) develop new therapies to treat areas of unmet medical needs; 2) prevent, detect or treat chronic or acute diseases and conditions; 3) reduce long-term health care costs in United States; or 4) significantly advance the goal of curing cancer within the 30 year period beginning on May 21, 2010. We applied for funding on three of its programs: Prevention of Tooth Decay using Smart Replacement Therapy, Novel Antibiotics for the Treatment of Healthcare Associated Infections and Rapid and Sensitive Identification of Novel Diagnostic Biomarkers for Cancer and Infectious Diseases. We received a non-taxable cash grant award totaling $733,437 under the program. A payment of $371,219 was made to us in November 2010 and remaining grant award amount of $362,218 was received in February, 2011.
Future Capital Requirements
Our capital requirements for 2011 will depend on numerous factors, including the success of our commercialization efforts and of our research and development, the resources we devote to develop and support our technologies and the success of pursuing strategic licensing and funded product development relationships with external partners. Subject to our ability to generate revenues and cash flow from our ProBiora3 products and our ability to raise additional capital including through possible joint ventures and/or partnerships, we expect to incur substantial expenditures to further commercialize or develop each of our technologies including continued increases in costs related to research, preclinical testing and clinical studies, as well as significant costs associated with our capital raising efforts and being a public company. We will require substantial funds to conduct research and development and preclinical and Phase 1 clinical testing of our licensed, patented technologies and to develop sublicensing relationships for the Phase 2 and 3 clinical testing and manufacture and marketing of any products that are approved for commercial sale. Our plans include seeking both equity and debt financing, alliances or other partnership agreements with entities interested in our technologies, or other business transactions that would generate sufficient resources to ensure continuation of our operations and research and development programs.
In addition, the report of our independent registered public accounting firm with respect to our financial statements appearing in our Form 10-K contains an explanatory paragraph stating that our operating losses and negative cash flows from operations since inception, and our need to raise additional financing and/or financial support prior to December 31, 2011 in order to continue to fund our operations, raise substantial doubt about our ability to continue as a going concern. If we are unable to raise sufficient capital we will need to significantly modify our operational plans for us to continue as a going concern.
Our current available cash and cash equivalents are insufficient to satisfy our liquidity requirements. We believe our existing cash and cash equivalents together with the borrowings under our Credit Facility, as amended and grant funds will allow us to fund our operating plan through June 2011. We will need to raise capital through the additional sale of equity or debt securities. The sale of additional equity or debt securities may result in additional dilution to our shareholders. If we raise additional funds through the issuance of debt securities or preferred stock, these securities could have rights senior to those of our common stock and could contain covenants that would restrict our operations. We also require additional capital beyond our currently forecasted amounts, such as, for example, if we determine to proceed independently with a Phase 3 clinical trial for our SMaRT Replacement Therapy. Any such required additional capital may not be available on reasonable terms, if at all. If we were unable to obtain additional financing, we may be required to reduce the scope of, delay or eliminate some or all of our planned clinical testing, research and development and commercialization activities, which could harm our business.
Because of the numerous risks and uncertainties associated with sales of our ProBiora3 products as well as research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amounts of our working capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
| • | the cash flow generated from our ProBiora3 product sales; |
51
Table of Contents
| • | the number and characteristics of the product candidates we pursue; |
| • | the scope, progress, results and costs of researching and developing our product candidates, and conducting preclinical and clinical trials; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates; |
| • | the cost of commercialization activities for our ProBiora3 products and, if any of our product candidates are approved for sale, including marketing, sales and distribution costs; |
| • | the cost of manufacturing our ProBiora3 products and product candidates and any products we successfully commercialize; |
| • | our ability to establish strategic partnerships, licensing or other arrangements and the financial terms of such agreements; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; and |
| • | the timing, receipt and amount of sales of, or royalties on, our products and future products, if any. |
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements.
Tax Loss Carryforwards
As of December 31, 2010 and 2009, we have net operating loss carryforwards of approximately $30,150,000 and $23,126,000, respectively to offset future federal and state income taxes. We also have research and development and investment tax credit carryforwards of approximately $491,000 and $384,000 as of December 31,2010 and 2009, respectively, to offset future federal and state income taxes. Any greater than 50% change in ownership under Section 382 of the Internal Revenue Code, or the Code, places significant annual limitations on the use of such net operating loss carryforwards and we exceeded the 50% threshold when we consummated the June 2009 Private Placement transaction with the KFLP. As a result, our historical loss carryforwards through June 2009 will be limited to $172,000 per year over the next 20 years, or limited to an aggregate amount of up to $3,440,000 of such historical loss carryforwards over such period of time, and the remaining balance of our historical loss carryforwards prior to June 2009 will expire unused. Provided that there are no future ownership changes that would trigger the limitations on loss carryforwards provided under the Code, the operating losses we experience after the June 2009 Private Placement transaction are expected to add to our loss carryforwards and to be fully available to us.
At December 31, 2010 and 2009, we recorded a 100% valuation allowance against our deferred tax assets of approximately $11,769,000 and $8,717,000, respectively, as our management believes it is uncertain that they will be fully realized. If we determine in the future that we will be able to realize all or a portion of our net operating loss carryforwards, an adjustment to our net operating loss carryforwards would increase net income in the period in which we make such a determination.
Inflation
Inflation affects the cost of raw materials, goods and services that we use. In recent years, inflation has been modest. However, high energy costs and fluctuations in commodity prices can affect the cost of all raw materials and components. The competitive environment somewhat limits our ability to recover higher costs resulting from inflation by raising prices. Although we cannot precisely determine the effects of inflation on our business, it is management’s belief that the effects on revenues and operating results will not be significant. We do not believe that inflation has had a material impact on our results of operations for the periods presented, except with respect to payroll-related costs and other costs arising from or related to government imposed regulations.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Not applicable.
52
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
The Financial Statements are incorporated herein by reference to pages F-1 to F-23 at the end of this report and the supplementary data is not applicable.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
On March 11, 2011 the Company’s Audit Committee approved the appointment of Mayer Hoffman McCann P.C. as the Company’s new independent registered public accounting firm. The Company was notified that the shareholders of Kirkland, Russ, Murphy & Tapp, P.A. (“KRMT”), the independent registered public accounting firm engaged by the Company on November 2, 2009 became shareholders of Mayer Hoffman McCann P.C. pursuant to an asset purchase agreement effective November 1, 2010. KRMT now operates under the name Mayer Hoffman McCann P.C.
During the Company’s two most recent fiscal years ended December 31,2009 and through the date of the Company’s Report on Form 8-K, filed March 15, 2011, the Company did not consult with Mayer Hoffman McCann P.C. regarding any of the matters or reportable events set forth in Item 304 (a)(2) (i) and (ii) of Regulation S-K.
In connection with the audits of the Company’s financial statements for each of the fiscal years ended December 31, 2009 and through the date of the Form 8-K, there were (i) no disagreements between the Company and KRMT on any matters of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreements, if not resolved to the satisfaction of KRMT, would have caused KRMT to make reference to the subject matter of the disagreement in their reports on the Company’s financial statements for such years, or for any reporting period, since the Company’s last fiscal year end and (ii) no reportable events within the meaning set forth in Item 304(a)(1)(v) of Regulation S-K.
The Company provided KRMT a copy of the disclosures in its Form 8-K filing and requested that KRMT furnish it with a letter addressed to the Securities and Exchange Commission stating whether or not KRMT agreed with the Company’s statements made in the Form 8-K and, if not, stating the respects in which it did not agree. A copy of the letter dated March 11, 2011 furnished by KRMT in response to that request was filed as Exhibit 16.1 to the Form 8-K filed on March 15, 2011 and is incorporated as an exhibit to this Form 10-K.
ITEM 9A. CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures
Management’s evaluation of the effectiveness of the Company’s disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act was performed under the supervision and with the participation of our senior management, including our former Chief Executive Officer and Chief Financial Officer. The purpose of disclosure controls and procedures is to ensure that information required to be disclosed in the reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to management, including our former Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosures.
During 2010 and 2009 we disclosed and identified several material weaknesses in our internal controls. Since that time we have been working on remediation of the identified material weaknesses and have provided updates in our periodic reports. Management believes progress has been made during the year to remediate material weaknesses in the internal control over financial reporting. However, based on the continued existence of material weaknesses, our former Chief Executive Officer and Principal Financial Officer have concluded that, as of the year ended December 31, 2010, disclosure controls and procedures were not effective. Nevertheless, based on a number of factors, including the performance of additional procedures by management designed to ensure the reliability of our financial reporting, management believes that the financial statements in our Annual Report on December 31, 2010 Form 10-K fairly presented, in all material respects, our financial position, results of operations, and cash flows for the periods presented in conformity with GAAP.
53
Table of Contents
As previously disclosed and referenced above, the matters involving internal controls and procedures that our management identified and considered to be material weaknesses that have not yet been satisfactorily remediated are: (1) limited documentation of our system of internal control, (2) insufficient personnel to employ segregation of duties and (3) lack of formal written policies and procedures for accounting and financial reporting with respect to the requirements and application of U.S. GAAP and SEC disclosure requirements and related documentation. These deficiencies and weaknesses were largely attributable to the significant lack of available financial resources.
Management’s Remediation Initiatives
Although management has not fully remediated all the material weaknesses mentioned above, management believes progress has been made during the year ended December 31, 2010. For example, during the third quarter we have acquired a new business operating system to provided more data controls, improved business processes, process documentation and better financial reporting. The new system became operational during November, 2010. We also continued the engagement with a consulting firm specializing in Sarbanes-Oxley Section 404 compliance to assist us in the implementation of internal controls for financial reporting and disclosure and our remediation efforts. During the fourth quarter the consulting firm completed an evaluation of the treasury and payroll cycles. Following such evaluation, management implemented a remediation plan during the quarter and addressed the documentation and authority levels of our treasury and payroll controls. Management is currently working with the outside consulting firm to review and assess various facets of our information processing system, such as sales and billing, inventory, materials management and other procedures. We continue to evaluate and address these weaknesses to ensure adherence to our policies, completeness of reporting, segregation of incompatible duties and compliance with generally accepted accounting principles; and we intend to continue to monitor and evaluate these and other factors affecting our internal controls as our resources and available liquidity permit. Until such time, our internal controls over financial reporting may be subject to additional material weaknesses and deficiencies that we have not yet identified. Management is responsible for and is committed to achieving and maintaining a strong control environment, high ethical standards, and financial reporting integrity. This commitment continues to be communicated to, and reinforced with, our employees.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Because of the inherent limitations of internal control, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Changes in Internal Controls Over Financial Reporting
Except as indicated in the preceding paragraphs about management’s evaluation of disclosure controls and procedures and internal controls, our management, with the participation of our former Chief Executive Officer (our principal executive officer) and Chief Financial Officer, has concluded there were no other significant changes in our internal controls over financial reporting that occurred during our last fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Limitations on the Effectiveness of Controls
Our management, including our CFO, does not expect that our Disclosure Controls and internal controls will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management or board override of the control.
The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
54
Table of Contents
Principal Executive Officer and CFO Certifications
Appearing after the Signatures section of this report there are Certifications of the Principle Executive Officer and the CFO. The Certifications are required in accordance with Section 302 of the Sarbanes-Oxley Act of 2002 (the Section 302 Certifications). This Item of this report, which you are currently reading is the information concerning the Evaluation referred to in the Section 302 Certifications and this information should be read in conjunction with the Section 302 Certifications for a more complete understanding of the topics presented.
Management’s Report on Internal Control over Financial Reporting
The management of Oragenics, Inc. is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). The Company’s internal control over financial reporting is a process designed to provide reasonable assurance to the Company’s management and board of directors regarding the reliability of financial reporting and the preparation of the financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
The Company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. All internal control systems, no matter how well designed, have inherent limitations, including the possibility of human error and the circumvention of overriding controls. Accordingly, even effective internal control over financial reporting can provide only reasonable assurance with respect to financial statement preparation. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management, under the supervision of the former CEO and CFO, assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2010. In making this assessment, it used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework. Based on our assessment, we believe that, as of December 31, 2010, the Company’s internal control over financial reporting was not effective based on those criteria.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit us to provide only management’s report in this annual report.
The Company previously disclosed in Form 8-K dated August 30, 2010, the results of its Annual Meeting of Shareholders held on August 25, 2010 where our shareholders voted (i) to re-elect our directors and (ii) to approve an amendment to our Articles of Incorporation to effect a reverse stock split of our common stock. A reverse stock split of one-for-twenty subsequently became effective on September 24, 2010.
55
Table of Contents
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
Management
Our Board of Directors, executive officers and key employees are as follows:
| Name |
Age |
Position | ||||
| Dr. Frederick W. Telling |
|
59 — |
|
Chairman and Director President and Chief Executive Officer* | ||
| Dr. Jeffrey D. Hillman |
62 | Chief Scientific Officer and Director | ||||
| Robert C. Koski |
52 | Director | ||||
| Christine L. Koski |
53 | Director | ||||
| Charles L. Pope |
59 | Director | ||||
| Brian J. Bohunicky |
56 | Chief Financial Officer, lead executive (principal executive officer), Secretary and Treasurer | ||||
| Gerard “Gerry” V. David |
58 | Executive Vice President of Sales and Marketing | ||||
| Dr. Martin Handfield |
40 | Director of Research and Development | ||||
| * | See “Resignation of our President and Chief Executive Officer” below. |
Directors of the Company
Dr. Frederick W. Telling. Dr. Telling was elected Chairman of the Board of Directors on February 4, 2011. He has served as a Director since June 2010. Dr. Telling retired from Pfizer Inc. in June 2007 after 30 years of service. At Pfizer Dr. Telling served as its Corporate Vice President and Vice President of Corporate Strategic Planning and Policy since October 1994. Dr. Telling also serves as a director and member of the Compensation Committee and Audit Committee at Cell Therapeutics Inc. (NASDAQ: CTIC), a public company based in Seattle, Washington. Dr. Telling also serves on the boards of various civic and non-profit organizations. Dr. Telling holds a B.A. degree in History and Economics from Hamilton College and a MA degree in Industrial and Labor Relations and a PhD in Economics and Public Policy from Cornell University.
Dr. Telling brings to our Board an extensive array of business and industry experience as well as experience as a director of public companies.
Christine L. Koski. Ms. Koski has served as a Director since June 2009 and as the Chairperson of our Board of Directors from June 2009 until February 2011 when director Telling was appointed to succeed Ms. Koski. Ms. Koski also serves as President and CEO of nMetrics, LLC, a provider of web-based scheduling system software. Prior to joining nMetrics in September 2006, Ms. Koski founded Koski Consulting Group, Inc. in June 2001 to advise start-up companies in the areas of business strategy and marketing. In addition to her positions at nMetrics and Oragenics, Ms. Koski serves as a director at Sun Hydraulics Corporation (NASDAQ: SNHY), a manufacturer of high performance hydraulic valves and solutions, and Cheltec, Inc., a specialty chemical company. Ms. Koski is a managing partner of the Koski Family Limited Partnership, which beneficially owns a controlling interest in the Company. Ms. Koski is a member of the nonprofit National Association of Corporate Directors. Ms. Koski holds an Executive MBA degree from Southern Methodist University’s Cox School of Business and a B.S. degree in Chemistry from St. Lawrence University. Ms. Koski is the sister of our Director, Robert Koski.
Ms. Koski brings to the Board over a decade of experience as an executive officer and as a director of other privately held and public technology-based companies. Through her extensive executive management and board experience, Ms. Koski has developed the leadership, business judgment and consensus-building skills necessary to effectively execute her duties as director. Her strong expertise and background in management and marketing and track record as an accomplished executive have provided her with the business acumen and skills necessary to serve the company as it moves forward in commercializing its technology.
Dr. Jeffrey D. Hillman. Dr. Hillman has served as our Chief Scientific Officer and as a Director since November 1996, and as Chairman of our Board of Directors from November 1996 to December 2004. From 1992 through July 2008, Dr. Hillman was a Professor at the University of Florida College of Dentistry. Dr. Hillman received undergraduate training at the University of Chicago (Phi Beta Kappa), and holds a DMD degree ( cum laude ) from the Harvard School of Dental Medicine and a PhD from Harvard University Medical School. He is the inventor or co-inventor of various Oragenics’ technologies.
56
Table of Contents
Dr. Hillman, our founder and longest serving Board member, brings to our Board an extensive background spanning nearly 30 years in biotechnology research and development and a deep knowledge and understanding of Oragenics’ business, operation and employees.
Robert C. Koski. Mr. Koski has served as a Director since June 2009. Mr. Koski has practiced as an attorney with the Koski Firm, a sole proprietorship located in Atlanta, Georgia since 1992, where his practice includes litigation and tax law. Mr. Koski has also served as a partner in the Koski Family Limited Partnership, which beneficially owns a controlling interest in the Company, and as a director of the Koski Family Foundation since December 1996. Mr. Koski holds a B.A. degree in Philosophy and English from Colgate University, a JD from Emory School of Law and an LLM degree in Taxation and Litigation from Emory University. He is the brother of our Director, Christine Koski.
Mr. Koski brings to our Board over two decades of experience in the legal field as a practicing attorney. In addition to his legal experience, Mr. Koski’s educational background provides a foundation for leadership and consensus-building.
Charles L. Pope. Mr. Pope has served as a Director since June 2010. Mr. Pope currently serves as the Chief Financial Officer of Palm Bancorp, Inc. since June 2009. From September 2007 through June 2009, Mr. Pope served as the Chief Financial Officer of Aerosonic Inc., a manufacturer of aviation products. Mr. Pope served as the Chief Financial Officer of Reptron Inc., a manufacturer of electronic products, from March 2005 through June 2007. From March 2002 to February 2005, Mr. Pope served as Chief Financial Officer of SRI/Surgical Express, Inc. From February 2001 to March 2002 Mr. Pope served as Chief Financial Officer of Innovaro, Inc. (formerly UTEK Corporation). Prior to this time, Mr. Pope served as a Partner in the Audit and Financial Advisory Consulting Divisions and was a Partner in the Accounting and SEC Directorate at PricewaterhouseCoopers LLP. Mr. Pope serves on the board of directors of Inuvo, Inc. in Clearwater, Florida and Innovaro Inc. in Tampa, Florida, each of which are public companies. Mr. Pope holds a B.S. degree in Economics and Accounting from Auburn University and is a Certified Public Accountant in Florida.
Mr. Pope brings to our Board over three decades of experience in the finance and accounting fields. In addition, Mr. Pope also has experience serving as a director of public companies.
Resignation of Our President and Chief Executive Officer
On February 4, 2011 David Hirsch resigned as our President and Chief Executive Officer and director to pursue other opportunities. Our Board of Directors is engaged in a search to fill the vacancy. In the interim, Mr. Brian Bohunicky has been designated as our lead executive and therefore serves as our principal executive officer until such time as the vacancy is filled. During this period, the Board of Directors will provide direction to the company, with Robert Koski acting as its liaison with management.
On February 4, 2011, we entered into a separation and release agreement with our former Chief Executive Officer and President, Mr. David Hirsch. Mr. Hirsch’s compensation under the separation and release agreement includes the payment of severance in the amount of $112,500 over six months in accordance with the Company’s normal payroll practices. Mr. Hirsch was entitled to the immediate payment of his accrued vacation which totaled $10,961. Mr. Hirsch’s employee employment agreement was terminated in connection with the separation and release agreement.
Executive Management
Dr. Jeffrey D. Hillman. The biography of Dr. Hillman is included above under the section “Directors of the Company.”
Brian J. Bohunicky. Mr. Bohunicky has served as our Chief Financial Officer since June 2009 and as Secretary and Treasurer since August 2009. Since the resignation of Mr. David B. Hirsch on February 4, 2011. Mr. Bohunicky serves as our lead executive and principal executive officer. Mr. Bohunicky joined the Company in January 2009 as the Company Controller. Prior to joining the Company, Mr. Bohunicky was the Vice President and Controller of Idex Corporation’s (NYSE: IEX) Fire & Safety Segment from October 2002 to November 2008. Mr. Bohunicky holds a B.A. degree in Economics from Moravian College.
57
Table of Contents
Key Employees
Gerard “Gerry” V. David. Mr. David has served as our Executive Vice President of Sales and Marketing since September 2008. Prior to that time, he provided consulting services to Oragenics through his company, Certified Nutrition for Less, LLC. Mr. David served as President and Chief Operating Officer of Växa International in Tampa, Florida from March 2007 to July 2008. From August 2006 to February 2007 he served as Chief Operating Officer of Cyberwize in Sarasota, Florida. From March 2003 to July 2006, he served as President and Chief Operating Officer of Vitarich Laboratories, Inc. in Naples, Florida. Prior to his service at Vitarich Laboratories, Mr. David served as Chief Operating Officer of Oxyfresh. Mr. David attended Western Michigan University.
Dr. Martin Handfield. Dr. Handfield has served as our Director of Research and Development since January 2009. Prior to joining our Company, Dr. Handfield held a position as Tenured Associate Professor at the Center for Molecular Microbiology and the Department of Oral Biology at the University of Florida College of Dentistry, where he co-invented IVIAT and co-founded ivi Gene Corp. and Epicure Corp. to commercialize this and related technologies. Dr. Handfield holds a B.S. degree in Biochemistry, and a MS degree and PhD in Microbiology and Immunology from the Université Laval College of Medicine in Canada, and did postdoctoral training at the University of Florida under the mentorship of Dr. Hillman.
Board of Directors and Committees
Board of Directors
Our property, affairs and business are under the general management of our Board of Directors as provided by the laws of the State of Florida and our Bylaws.
The Board of Directors conducts its business through meetings of the full Board and through committees of the Board. The Board of Directors has appointed standing Audit, Compensation and Nominating Committees of the Board of Directors.
On June 4, 2010 our Board of Directors expanded the size of our Board by two additional seats in order to accommodate its appointment of Charles Pope and Dr. Frederick Telling to serve as additional non-employee, independent directors. The Board periodically reviews the size of the Board and recommends any changes it determines to be appropriate given our needs. Under our Bylaws, the number of members on the Board may be increased or decreased by resolution of the Board.
On February 4, 2011 Mr. David Hirsch resigned as President and Chief Executive Officer, and Dr. Frederick Telling was elected to succeed Christine Koski as Chairperson of the Board of Directors. As such, the Board currently consists of five members with one vacancy.
Director Independence
Since our securities are not listed on a national securities exchange or in an inter-dealer quotation system, we are not currently required to comply with director independence requirements. Notwithstanding the foregoing, historically we have determined director independence in accordance with the rules of a designated exchange. Accordingly, in determining whether our Directors are independent, we intend to comply with the rules of the NASDAQ Capital Market. We also expect to continue to comply with securities and other laws and regulations regarding the independence of directors, including those adopted under Section 301 of the Sarbanes-Oxley Act and Rule 10A-3 under the Securities and Exchange Act of 1934 with respect to the independence of Audit Committee members. The NASDAQ Capital Market listing standards define an “independent director” generally as a person, other than an officer of a company, who does not, in the view of the company’s Board of Directors, have a relationship with the company that would interfere with the director’s exercise of independent judgment. The Board has determined that each of the following directors, constituting a majority of the Board, is independent within the meaning of the NASDAQ Capital Market listing standards:
Frederick W. Telling
Christine L. Koski
Robert C. Koski
Charles L. Pope
58
Table of Contents
Such independence definition includes a series of objective tests, including that the director is not an employee of the company and has not engaged in various types of business dealings with the company. In addition, as further required by the NASDAQ listing standards, the Board has made a subjective determination as to each independent director that no relationships exist which, in the opinion of the Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Audit Committee Financial Expert
The audit committee is comprised of two non-employee, independent members of the board of directors, Mr. Charles Pope (chair) and Dr. Frederick Telling. The board of directors has determined that both of the audit committee members are able to read and understand fundamental financial statements. In addition, the board of directors has determined that Mr. Pope is an “audit committee financial expert” as that term is defined in Item 407(d)(5) of Regulation S-K promulgated under the Securities and Exchange Act of 1934.
Code of Ethics
We have adopted a code of ethics known as the Company Operating Principles, which is applicable to all of our directors and employees, including our principal executive officer and our principal financial officer. A copy of the Company Operating Principles can be found on our website at www.oragenics.com. Any future amendments to, or waivers from, the Company Operating Principles will be posted on our website.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934 requires the Company’s officers and Directors and any persons who beneficially own more than ten percent of the Company’s Common Stock to file reports of ownership and changes in ownership of such securities with the Securities and Exchange Commission Officers, Directors and beneficial owners of more than ten percent of the Common Stock are required by applicable regulations to furnish the Company with copies of all Section 16(a) forms they file. Based solely on its review of copies of forms furnished to the Company and written representations from the executive officers, directors and holders of ten percent or more of the Company’s Common Stock, the Company believes, all persons subject to the reporting requirements with regard to the Common Stock complied with the applicable filing requirements during 2010.
ITEM 11. EXECUTIVE COMPENSATION.
Compensation of Directors
Due primarily to our limited operating capital, our Director compensation program during the fiscal year ended December 31, 2009 consisted of a one-time option grant to acquire 5,000 shares of common stock in lieu of the payment of any meeting fees. Outside non-employee Directors are reimbursed for their expenses associated with travel to and from Board meetings and meetings with management. Certain fees previously earned by former non-employee Directors for attending Board and Committee meetings in the amount of $34,000 have been deferred instead of being paid.
On June 4, 2010, commensurate with the appointment of two new independent Directors, the Board approved changes to the standard Board compensation to be paid to non-employee Directors. Such changes primarily related to the reinstating of a cash fee component to the Director compensation program for non-employee directors. On March 11, 2011, the Board approved revisions to the Board compensation program. The revision consisted of the addition of one per board meeting fee for each non-employee director for any board meetings in excess of one per quarter. The Director compensation program consists of the following:
Cash Compensation
The Director compensation program changes provide that all non-employee Directors will receive an annual base fee for service on the Board of $24,000. In addition, the Chairperson of the Board and of our Audit Committee, Compensation Committee and Nominating Committee will also receive annual fees of $25,000, $20,000, $15,000 and $10,000, respectively. All non-employee Directors serving on committees (other than as the Chairperson) shall receive an annual fee of $5,000 in connection with such committee service. In addition all non-employee Directors shall receive a per board meeting fee of $2,000 for each board meeting in excess of one per quarter. All fees for Board service are to be paid quarterly in arrears.
59
Table of Contents
Equity Compensation
Equity compensation is to be issued to Directors upon joining our Board. Non-employee Directors receive a stock option for the purchase of 5,000 shares of our common stock at an exercise price per share equal to the fair market value per share on date they became a Director, which will immediately vest and be exercisable. As part of the Director compensation program, the Board may also make discretionary equity based awards from time to time under the Company’s existing Amended and Restated 2002 Stock Option and Incentive Plan, or 2002 Stock Incentive Plan.
Reimbursement of Expenses
Non-employee Directors are also reimbursed for expenses incurred in connection with their attendance at Board or committee meetings and reasonable out-of-pocket business expenses associated with their Board service.
Employee Directors
Consistent with past practice, the Director compensation program provides that employee Directors receive no additional compensation in connection with their board service.
The following table sets forth the compensation of our non-employee Directors in 2010.
| Name |
Fees earned or paid in cash(1) |
Option awards(2) |
All other compensation(3) |
Total | ||||||||||||
| Christine L. Koski |
$ | 29,500 | — | — | $ | 29,500 | ||||||||||
| Robert C. Koski |
$ | 19,500 | — | — | $ | 19,500 | ||||||||||
| Charles L. Pope |
$ | 28,125 | $ | 50,000 | — | $ | 78,125 | |||||||||
| Dr. Frederick W. Telling |
$ | 25,255 | $ | 50,000 | — | $ | 75,255 | |||||||||
| (1) | Amounts represent cash compensation paid to these Directors during 2010 in connection with their Board service. |
| (2) | The compensation amount reflected with respect to these awards represents the 2010 compensation expense associated with outstanding option grants to our non-employee directors. Upon joining our Board of Directors in June 2010, Mr. Pope and Dr. Telling as non-employee Directors were each granted options to acquire 5,000 shares of our common stock at $10.00 per share in accordance with our Director compensation program. The amounts reflected in the table with respect to these awards represent the 2010 compensation expense associated with such grants. The Company uses a Black-Scholes option pricing model to estimate the fair value of the stock option grant. The use of a valuation model requires the Company to make certain assumptions with respect to selected model inputs. The average expected life is based on the contractual term of the option and on the simplified approach provided by SAB 107. The risk-free interest rate is based on the U.S. Treasury zero-coupon issues equal to the expected life assumed at the date of the grant. |
| (3) | No other compensation was paid to the non-employee Directors except for reimbursement for travel expenses to Board meetings, which did not exceed $10,000 individually or in the aggregate for our non-employee Directors. |
60
Table of Contents
Executive compensation
Summary Compensation Table
The following table sets forth the aggregate compensation in 2009 and 2010 for services in all capacities paid or accrued by the Company to our most highly compensated officers and our former Principal Executive Officers (“PEO”) who earned more than $100,000 in total salary and bonus during the fiscal year ended December 31, 2010 (the “Named Executive Officers”).
| Name and principal position |
Year | Salary | Bonus | Option awards (5) |
All other compensation (6) |
Total | ||||||||||||||||||
| Dr. Jeffrey D. Hillman |
2010 | $ | 200,000 | — | — | $ | 6,000 | $ | 206,000 | |||||||||||||||
| Chief Scientific Officer (1) |
2009 | $ | 182,278 | — | $ | 318,205 | $ | 86,650 | $ | 587,133 | ||||||||||||||
| Brian J. Bohunicky |
2010 | $ | 200,000 | — | — | $ | 6,000 | $ | 206,000 | |||||||||||||||
| Chief Financial Officer and Principal Financial Officer (2) |
2009 | $ | 156,832 | — | $ | 195,750 | $ | 3,840 | $ | 356,422 | ||||||||||||||
| Gerard “Gerry” V. David |
2010 | $ | 180,000 | — | — | — | $ | 180,000 | ||||||||||||||||
| Executive Vice President Sales and Marketing |
2009 | $ | 161,250 | — | $ | 207,706 | — | $ | 368,956 | |||||||||||||||
| Dr. Martin Handfield |
2010 | $ | 171,000 | — | — | $ | 5,130 | $ | 176,130 | |||||||||||||||
| Director of Research and Development |
2009 | $ | 158,808 | — | $ | 136,281 | — | $ | 295,089 | |||||||||||||||
| Former PEOs |
||||||||||||||||||||||||
| David Hirsch (3) |
2010 | $ | 225,000 | — | — | $ | 6,750 | $ | 231,750 | |||||||||||||||
| 2009 | $ | 214,583 | $ | 100,000 | $ | 413,211 | $ | 9,417 | $ | 737,211 | ||||||||||||||
| Stanley Stein (4) |
2010 | — | — | — | — | — | ||||||||||||||||||
| 2009 | $ | 39,824 | — | — | $ | 120,000 | $ | 159,824 | ||||||||||||||||
| (1) | Effective December 1, 2009 Dr. Hillman’s annual salary was increased from $180,000 to $200,000. In addition, an amount of $81,250 in the other column reflects payments to Dr. Hillman in December 2009 for compensation and consulting fees that had previously been deferred. This amount net of applicable fees was paid through the issuance of common stock to Dr. Hillman as part of our December 2009 Private Placement . See “Certain Relationships and Related Transactions.” |
| (2) | Mr. Bohunicky joined our Company in January 2009 and became our Chief Financial Officer and Principal Financial Officer on June 29, 2009. Following the 2009 Private Placement Mr. Bohunicky’s annual compensation was increased by the Compensation Committee to $200,000. Included in Mr. Bohunicky’s salary for 2009 is $25,000 in compensation that had been deferred during a portion of the year which was paid to Mr. Bohunicky immediately following the 2009 Private Placement in 12,500 shares of our common stock at a price per share of $2.00. |
| (3) | On February 4, 2011 Mr. Hirsch resigned as our President and Chief Executive Officer and Director to pursue other opportunities. We entered into a separation and release agreement with Mr. Hirsch which provides for the payment of six months severance to Mr. Hirsch based upon his annual salary. |
| (4) | On March 18, 2009, Mr. Stein resigned as our President, Chief Executive Officer and Principal Executive Officer. We entered into a settlement and release agreement with Mr. Stein on August 31, 2009, pursuant to which we paid him $120,000 of severance which is included under “All Other Compensation.” |
61
Table of Contents
| (5) | The amounts included in this column do not reflect compensation actually received by the named executive officers. Instead the amounts in this column represent the aggregate grant date fair value computed in accordance with Financial Accounting Standards Board Accounting Standards Codification, Topic 718, Compensation-Stock Compensation (ASC 718). Under SEC rules relating to executive compensation disclosure, the amounts shown exclude the impact of estimated forfeitures related to service-based vesting conditions. Fair values relating to share grants have been determined under ASC 718 and were calculated using the common stock closing price on the date of grant and multiplying that price by the number of shares subject to the share grant. The equity-based compensation expense relating to the stock grants is recognized over the requisite service period of the grant. For option awards, we utilize the Black-Scholes option pricing model to determine the fair value on the date of the grant multiplied by the number of options subject to the option grants in accordance with ASC 718. The stock-based compensation expense relating to the stock option grants is recognized over the requisite service period of the grant. For information on the assumptions used to calculate the fair value of stock option grants, refer to Footnote 1, “Organization and Significant Accounting Policies” in our financial statements for the year ended December 31, 2010 included elsewhere in this prospectus. These amounts reflect our accounting expense for these awards, and do not necessarily correspond to the actual value that will be recognized by the executive officers. Amounts in this column for 2009 include $52,086, $41,455, $51,156 and $32,196 for Mr. Hirsch, Dr. Hillman, Mr. David and Dr. Handfield, respectively, which reflect the impact of the acceleration of the vesting of previously outstanding stock options by our Compensation Committee on August 13, 2009. The number of shares covered by the accelerated vesting were 21,666, 25,000, 15,166 and 13,000 for Mr. Hirsch, Dr. Hillman, Mr. David, and Dr. Handfield, respectively. A portion of stock option awards received by our executives were forfeited during 2010 due to not meeting performance criteria for shipping and invoicing of our ProBiora3 products. The amounts forfeited include $22,496 each for Mr. Hirsch, Dr,. Hillman, Mr. Bohunicky and Mr. David. All other terms of the prior option awards, including the share amounts covered by the options and exercise price remained the same. |
| (6) | Our Simple IRA retirement plan requires us to match employee contributions up to the first 3% of compensation earned and amounts presented also include our matching contribution and the amounts in this column represent such contributions. This column excludes certain payments for personal benefits for Mr. Hirsch and Dr. Hillman that do not exceed $10,000 individually or in the aggregate. |
Incentive Awards
The Compensation Committee believes that our future success depends, in large part, upon our ability to maintain a competitive position in attracting, retaining and motivating key personnel. The Compensation Committee utilizes the 2002 Stock Incentive Plan to provide incentives to employees. We do not have any long-term incentive plans that provide compensation intended to serve as incentives for performance other than options granted pursuant to our 2002 Stock Incentive Plan.
During the year ending December 31, 2010 there were no stock options awarded to or exercised by the named executive officers. In December 2009, options to purchase a total of 281,590 shares of our common stock which are subject to time vesting and performance vesting were awarded to our executive officers and employees. These option awards each have exercise prices of $5.40 per share, which was the closing price on the date the Compensation Committee granted the options. These option awards were made pursuant to individual award agreements substantially similar to the form of stock option agreement attached as an exhibit to our 2002 Stock Incentive Plan which has been previously filed with the SEC.
62
Table of Contents
Outstanding Equity Awards At Fiscal Year End
The following table provides information concerning unexercised options, stock that has not vested, and equity incentive plan awards outstanding as of December 31, 2010:
| Name |
Number of securities underlying unexercised options (#) exercisable |
Number of securities underlying unexercised options (#) unexercisable |
Equity incentive awards: number of securities underlying unexercised unearned options (#) |
Option exercise price ($) |
Option expiration date |
|||||||||||||||
| Dr. Jeffrey D. Hillman |
3,750 | — | 14.80 | 9/8/2011 | ||||||||||||||||
| 35,000 | — | 17.00 | 5/21/2018 | |||||||||||||||||
| 11,667 | (1) | 23,333 | (1) | 5.40 | 12/1/2019 | |||||||||||||||
| 5,000 | (2) | 5.40 | 12/1/2019 | |||||||||||||||||
| 2,084 | (3) | 5.40 | 12/1/2019 | |||||||||||||||||
| 5,000 | (4) | 5.40 | 12/1/2019 | |||||||||||||||||
| Brian J. Bohunicky |
8,333 | (1) | 16,667 | (1) | 5.40 | 12/1/2019 | ||||||||||||||
| 5,000 | (2) | 5.40 | 12/1/2019 | |||||||||||||||||
| 2,084 | (3) | 5.40 | 12/1/2019 | |||||||||||||||||
| Gerard “Gerry” V. David |
17,500 | 5.60 | 12/17/2018 | |||||||||||||||||
| 5,833 | (1) | 11,667 | (1) | 2,084 | (3) | 5.40 | 12/1/2019 | |||||||||||||
| 4,500 | (2) | 5.40 | 12/1/2019 | |||||||||||||||||
| Dr. Martin Handfield |
15,000 | 10.40 | 8/18/2018 | |||||||||||||||||
| 4,167 | (1) | 8,333 | (1) | 5.40 | 12/1/2019 | |||||||||||||||
| 4,275 | (2) | 5.40 | 12/1/2019 | |||||||||||||||||
| Former Officers: |
||||||||||||||||||||
| David B. Hirsch (5) |
25,000 | — | 9.80 | 5/30/2018 | ||||||||||||||||
| 16,667 | (1) | 33,000 | 5.40 | 12/1/2019 | ||||||||||||||||
| 5,625 | (2) | 5.40 | 12/1/2019 | |||||||||||||||||
| 2,084 | (3) | 5.40 | 12/1/2019 | |||||||||||||||||
| 5,000 | (4) | 5.40 | 12/1/2019 | |||||||||||||||||
| (1) | Represents awards that are time vested with each award vesting evenly on an annual basis over three years, subject to earlier vesting upon a change in control as defined in the award agreements. |
| (2) | Represents awards that vest upon the first calendar quarter in which we report a net profit in a Form 10-Q Report or Form 10-K Report. These awards expire on the earlier of (i) December 1, 2019 or (ii) such date we cease to be required to file quarterly or annual reports with the Securities and Exchange Commission. |
| (3) | Represents awards that vest upon our achieving certain performance goals tied to the shipment and invoicing of our ProBiora3 products with a third of these options expiring if we have not achieved the vesting performance targets by September 1, 2010, December 31, 2010 and March 31, 2011. Performance targets were not achieved on September 1, 2010 and December 1, 2010 and accordingly two thirds of these performance options have been forfeited. To the extent any of these option become vested and exercisable, they shall expire December 1, 2019. |
| (4) | Represents awards that are subject to vesting based on certain scientific performance milestones being achieved. These options expire and are void unless they become vested and exercisable on or before December 31, 2011. To the extent these options become vested and exercisable, they shall expire December 1, 2019. |
| (5) | Because of Mr. Hirsch’s resignation any awards that have vested (at the time of his resignation) must be exercised by May 5, 2011 or be forfeited. |
63
Table of Contents
Employment Contracts and Change in Control Arrangements
New 2010 Employment Agreements—Executive Officers
On March 11, 2010 our Compensation Committee met, approved and authorized new employment agreements with each of our executive officers. Each of the new employment agreements have substantially similar terms other than with respect to their annual compensation and title (the “2010 Employment Agreements”). As to our former President and Chief Executive Officer Mr. Hirsch and Dr. Hillman, the 2010 Employment Agreements replaced the existing employment agreements discussed above and as to Mr. Bohunicky, the 2010 Employment Agreement constitutes a new employment agreement between us and Mr. Bohunicky. The annual base salaries provided in the 2010 Employment Agreements are $225,000, $200,000 and $200,000 for Mr. Hirsch, Dr. Hillman and Mr. Bohunicky, respectively, and are payable in installments consistent with our normal payroll practices. The entering into of these 2010 Employment Agreements did not result in any change to any of the executive officers existing and previously disclosed annual base compensation. The executive officers are also eligible under the 2010 Employment Agreements to receive bonuses during the term at the discretion of the Compensation Committee and the Board of Directors.
New 2010 Employment Agreements—Key Employees
On March 11, 2010 our Compensation Committee met, approved and authorized new employment agreements with two of our key employees, Mr. Gerard David and Dr. Martin Handfield. As to Dr. Handfield, the 2010 Employment Agreements replaced an existing employment agreement, the 2010 Employment Agreement constitutes a new employment agreement between us and Mr. David. The annual base salaries provided in the 2010 Employment Agreements are $180,000 and $171,000 for Mr. David and Dr. Handfield, respectively, and are payable in installments consistent with our normal payroll practices. The entering into of these 2010 Employment Agreements did not result in any change to any of existing and previously disclosed annual base compensation. Mr. David and Dr. Handfield are also eligible under the 2010 Employment Agreements to receive bonuses during the term at the discretion of the Compensation Committee and the Board of Directors.
The 2010 Employment Agreements are terminable at any time by either party and if the executive officer or key employee is involuntarily terminated by us he shall receive his base salary and vacation pay each accrued through the date of termination, and any nonforfeitable benefits earned and payable to him under the terms of the employee handbook (which applies to all employees) and benefits available under any applicable incentive plan in which employee participates. In addition, if the executive officer or key employee’s separation from employment is not voluntary and without cause, we would be obligated to pay the executive officer or key employee six months of his annual base salary as severance and the executive shall be entitled to out placement service benefits. If the executive officer or key employee is terminated for cause, he shall be entitled to receive his base salary and accrued vacation due through the date of termination and any nonforfeitable benefits already earned and payable to the executive or key employee under the terms of the employee handbook or other applicable incentive plans maintained by us. Cause is defined in the 2010 Employment Agreements as any action that is illegal, immoral, or improper that reflects on the Company, the employee, or the ability of either to function optimally. If the executive officer or key employee voluntarily resigns, he shall be entitled to this base salary and accrued vacation due through the date of termination (including any mutually agreed upon notice period) and any nonforfeitable benefits already earned and payable to the executive officer or key employee under the terms of the employee handbook or other incentive plans maintained by us.
If the executive officer or key employee dies during the term of employment with us, the estate of the employee shall be paid the salary of the employee as it would have accrued over a period of thirty days after the executive officer’s death. We shall also extend the executive officer or key employee’s right to exercise vested stock options for six months provided such extension is permitted under the 2002 Stock Incentive Plan. In the event the executive officer or key employee becomes disabled (as defined in the then applicable short and long-term disability insurance policies) we shall pay to the executive officer or key employee his salary as it would have accrued over a period of 30 days after the executive or key employee became so disabled and we shall extend the executive officer or key employee’s right to exercise vested stock options for six months provided such extension is permitted under the 2002 Stock Incentive Plan.
The 2010 Employment Agreements also each include non-disclosure and Company ownership of development provisions, as well as a provision providing for the Company to defend and indemnify the executive or key employee if the executive or key employee is named as a defendant in any lawsuit regarding any action taken within the scope of employment.
64
Table of Contents
In the event of a change in control, any stock options or other awards granted (other than performance awards) under our 2002 Stock Incentive Plan shall become immediately vested in full and in the case of stock options exercisable in full. If the change in control results in an involuntary separation from employment of the executive officer or key employee within 180 days following a change in control, the executive officer or key employee would be entitled to (i) receive six months of salary and the extension of his benefits (excluding vacation time and paid time off) and (ii) exercise vested options for six months from the date of separation, provided said extension period is allowed under the 2002 Stock Incentive Plan. Under the 2010 Employment Agreements, “involuntary separation of employment” means (i) termination without cause, (ii) any reduction in responsibilities of office altering the status of the executive officer or key employee as an employee, or (iii) the duplication of the executive officer or key employees position by an equivalent executive in an acquiring entity and “change in control” means the sale of the entire company, or substantially all of its assets, or the sale of the business unit employing an individual which results in the termination of employment or subsequent transfer of the employment relationship to another legal entity, or entity, or single party acquiring more shares than are owned by the Koski Family Limited Partnership, including its members and their immediate families, including spouses and their children.
Dr. Jeffrey Hillman, Chief Scientific Officer
We previously had an employment agreement with Dr. Jeffrey Hillman, our Chief Scientific Officer which commenced on January 1, 2004 and provided for automatic one-year extensions after December 31, 2007. Under the terms of such employment agreement, we were obligated to pay compensation of $180,000. Dr. Hillman was also eligible for participation in incentive stock compensation plans. The employment agreement also provided for other benefits including the right to participate in fringe benefit plans, life and disability insurance plans, expense reimbursement and 20 days accumulating vacation/sick leave annually. If Dr. Hillman was terminated by the Company without cause (as defined in the agreement) or within 12 months following a change of control (as defined in the agreement), or if he left for good reason (as defined in the agreement), he would have been entitled to severance payments, at his then annual base salary and all stock options granted to the executive and any benefits under any benefit plans would have become immediately vested and exercisable. If Dr. Hillman voluntarily resigned he would have received no further compensation after the effective date of such resignation. The employment agreement also included non-disclosure and non-compete provisions, as well as salary payments for a three month period in the event of Dr. Hillman’s death or disability during the term of the agreement. Dr. Hillman was awarded options to acquire 35,000 shares of common stock under the 2002 Stock Incentive Plan on May 21, 2008. These options vested as follows: 10,000 shares immediately and the remaining 25,000 shares were scheduled to vest when the Company’s stock price reached certain levels as follows: 7,500 shares vest at $20.00 per share, 7,500 shares vest at $40.00 per share and 10,000 shares vest at $60.00 per share. On August 13, 2009 the Compensation Committee approved the accelerated vesting of the unvested, unexerciseable options.
On March 11, 2010 our Compensation Committee approved a new employment agreement with Dr. Hillman which replaced and superceded his prior employment agreement. See “New 2010 Employment Agreements—Executive Officers.” No changes were made during 2010 to Dr. Hillman’s employment agreement or compensation and no awards were made during 2010 to Dr. Hillman under our 2002 Stock Incentive Plan.
Brian Bohunicky, Chief Financial Officer, Secretary and Treasurer
Mr. Bohunicky joined the Company in January 2009 and became our Chief Financial Officer and Principal Financial Officer on June 29, 2009 following the June 2009 Private Placement. Also at this time, Mr. Bohunicky’s annual compensation was increased by the Compensation Committee to $200,000. Included in Mr. Bohunicky’s salary for 2009 is $25,000 in compensation that had been deferred during a portion of the year which was paid to Mr. Bohunicky immediately following the June 2009 Private Placement, in 12,500 shares of our common stock at a price per share of $2.00. During 2009 and the initial few months of 2010, Mr. Bohunicky’s employment with us was at will and was not subject to the terms of an employment agreement.
On March 11, 2010 our Compensation Committee approved a new employment agreement for Mr. Bohunicky. See “New 2010 Employment Agreements— Executive Officers.” No changes were made to Mr. Bohunicky’s employment agreement or compensation during 2010 and no awards were made during 2010 to Mr. Bohunicky under our 2002 Stock Incentive Plan. In addition, as a result of Mr. Bohunicky’s assuming the role of lead executive and principal executive officer following Mr. Hirsch’s resignation as our President and Chief Executive Officer, no changes were made to Mr. Bohunicky’s employment with us.
65
Table of Contents
On March 11, 2011 our Board of Directors and Compensation Committee approved an option award of 20,000 shares to Mr. Brian Bohunicky, our Chief Financial Officer under the Company’s Amended and Restated 2002 Stock Option and Incentive Plan, as amended (the “Plan”). These options vest equally over a three (3) year period from the date of grant and are exerciseable at $3.60 per share, the price our stock closed on March 11, 2011, the date of the grant. On that same date Mr. Bohunicky was also granted 10,000 shares of restricted common stock under the Plan half of which vests in six (6) months and the other half on the anniversary of the award. These restricted shares were granted at the price of $3.60, the closing price on March 11, 2011, the date of the award.
The Board also approved the payment of up to $15,000 to Mr. Bohunicky to reimburse him for relocation expenses he may incur in connection with his contemplated relocation to our primary corporate headquarters in Tampa, Florida.
Our former Chief Executive Officer and President, David Hirsh
On February 4, 2011 Mr. Hirsch resigned from the Company and we entered into a separation and release agreement with Mr. Hirsch that provided for the payment of six months severance. Mr. Hirsch began working for us as a consultant in April 2008 and became a full time employee in May 2008. In connection with Mr. Hirsch’s appointment, effective June 27, 2008, as our Chief Operating Officer, Mr. Hirsch entered into an employment agreement with us which was amended on July 15, 2008 when he also became our Chief Financial Officer upon the retirement of our former chief financial officer. Mr. Hirsch’s initial employment agreement was for one year, and was automatically extended for successive one-year renewal terms. Pursuant to his initial employment agreement, Mr. Hirsch initially received an annual salary of not less than $150,000 and was eligible to receive bonuses at the discretion of the Compensation Committee of the Board of Directors. Mr. Hirsch was granted stock options to acquire 25,000 shares of common stock under our Amended and Restated 2002 Stock Option and Incentive Plan, or 2002 Stock Incentive Plan. These options were scheduled to vest as follows: 3,333 shares vest immediately, 5,000 shares on the dates which the Company’s stock price equals or exceeds $20.00 per share, $40.00 per share and $60.00 per share respectively, and 6,666 shares on the date which the Company’s stock price equals or exceeds $100.00 per share.
Under the terms of his initial employment agreement, if Mr. Hirsch was involuntarily terminated he would receive his base salary accrued through the date of termination, and any nonforfeitable benefits earned and payable to him under the terms of the deferred compensation, incentive or other benefit plan, payable in accordance with the terms of the applicable plan. In addition, if Mr. Hirsch’s separation from employment was not voluntary, for cause or due to death or disability, the Company would be obligated to pay Mr. Hirsch a series of nine equal monthly payments equal to one-twelfth of his annual base salary in effect on the date of such termination as severance and any unvested options shall vest. If he was terminated for cause, he would be entitled to receive his base salary accrued through the date of termination and any nonforfeitable benefits already earned and payable to Mr. Hirsch under the terms of the deferred compensation or incentive plans maintained by the Company. If Mr. Hirsch voluntarily resigned, he would be entitled to this base salary accrued through termination and any nonforfeitable benefits already earned and payable to Mr. Hirsch under the terms of the deferred compensation or incentive plans maintained by the Company. In the event of a change in corporate control, the vesting of any stock options or other awards under the terms of the 2002 Stock Incentive Plan would become immediately vested in full and in the case of stock options, exercisable in full. If Mr. Hirsch is terminated within six months of a change in control, as such term is defined in his employment agreement, Mr. Hirsch would be entitled to receive, in lieu of the foregoing severance payment described above, a series of 24 equal monthly payments equal to one-twelfth of Mr. Hirsch’s annual base salary in effect at the time of a change in control. The initial employment agreement also included non-disclosure and non-compete provisions as well as a lump sum payment equal to the sum of the executive’s accrued base salary, unpaid amounts of any bonuses earned with respect to the fiscal year of the Company most recently ended and the death benefits payable under any retirement, deferred compensation or other employee benefit plan maintained by the Company in the event of Mr. Hirsch’s death during the term of the agreement.
Mr. Hirsch became our acting President and Chief Executive Officer effective March 18, 2009 upon the resignation of Stanley Stein. Mr. Hirsch also continued in his role as our Chief Financial Officer. Mr. Hirsch did not receive any adjustment in his compensation upon assuming the role of our acting President and Chief Executive Officer. On June 29, 2009, immediately following the June 2009 Private Placement, Mr. Hirsch became our President and Chief Executive Officer and relinquished his position as Chief Financial Officer to Mr. Bohunicky. In June 2009 Mr. Hirsch was awarded a bonus of $100,000 payable in 50,000 shares of our common stock at a price per share of $2.00. This bonus was paid to Mr. Hirsch in recognition of his efforts in guiding us through a significant adverse capital resource crisis. On August 13, 2009, the Compensation Committee approved acceleration of the vesting of the unvested, unexerciseable options awarded to Mr. Hirsch and approved an increase in his annual base salary to $225,000.
On March 11, 2010 our Compensation Committee approved a new employment agreement with Mr. Hirsch, which replaced and superceded his prior employment agreement. See “New 2010 Employment Agreements—Executive Officers.” No changes were made to Mr. Hirsch’s compensation during 2010.
66
Table of Contents
Our former Chief Executive Officer and President, Stanley Stein
On April 8, 2008, we entered into an employment agreement with our former Chief Executive Officer and President, Mr. Stanley Stein. Mr. Stein’s initial compensation arrangement was pursuant to an offer letter that provided for an annual rate of compensation of $175,000 and relocation expenses of $10,000. Mr. Stein also was compensated in the amount of $30,000 in connection with his initial services and was expected to receive an award of stock options under our 2002 Stock Incentive Plan. The initial term of the employment agreement was for one year and was subject to automatically being extended for successive one year renewal terms. Pursuant to the employment agreement, Mr. Stein received an annual salary of not less than $175,000 and was eligible to receive bonuses at the discretion of the Compensation Committee of the Board of Directors. Mr. Stein was granted stock options to acquire 37,500 shares of common stock under our 2002 Stock Incentive Plan. The options were subject to vesting as follows: 5,000 shares on April 9, 2008; 7,500 shares on the dates which the Company’s stock price equals or exceeds $20.00 per share, $40.00 per share and $60.00 per share respectively, and 10,000 shares on the date which the Company’s stock price equals or exceeds $100.00 per share. Mr. Stein resigned as President, Chief Executive Officer and Director effective March 18, 2009 and his employment agreement with us was terminated. In connection with Mr. Stein’s separation from employment he was to be paid his accrued compensation earned through the date of termination, which included an accrued bonus payment of $50,000 upon the occurrence of certain specified events. In addition, Mr. Stein was to be paid $1,500 for nine months to cover post-separation expenses. After separation from employment with us, Mr. Stein also became a consultant to the Company with his previously granted options continuing so long as Mr. Stein served as a consultant to the Company. On August 31, 2009, pursuant to a subsequent agreement with Mr. Stein, all continuing obligations and payments to Mr. Stein including his consulting agreement and options were terminated in exchange for a one time payment of $120,000. As a result of Mr. Stein’s resignation in March 2009, Mr. Hirsch was appointed to serve as our acting President and Chief Executive Officer.
67
Table of Contents
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The following table sets forth, as of March 24, 2011, certain information concerning the beneficial ownership of each class of our voting securities by: (i) each person known by us to own beneficially 5% or more of the outstanding shares of our common stock, (ii) each of our Directors and named executive officers, and (iii) all executive officers and Directors as a group.
The number of shares beneficially owned by each 5% shareholder, Director or named executive officer is determined under rules of the SEC and the information is not necessarily indicative of beneficial ownership for any other purpose. Under those rules, beneficial ownership includes any shares to which the individual has sole or shared voting power or investment power and also any shares that the individual has the right to acquire within 60 days after March 31, 2011 through the exercise of any stock option, warrant or other right, or conversion of any security. Unless otherwise indicated, each person has sole investment and voting power (or shares such power with his or her spouse) with respect to the shares set forth in the following table. The inclusion in the table below of any shares deemed beneficially owned does not constitute an admission of beneficial ownership of those shares.
| Name and address(1) |
Number of
shares beneficially owned |
Percentage
of ownership(2) |
||||||
| 5% shareholders |
||||||||
| Koski Family Limited Partnership(3) |
3,206,998 | 56.4 | % | |||||
| George T. Hawes(4) |
667,285 | 11.7 | % | |||||
| Directors and officers |
||||||||
| Jeffrey D. Hillman(5) |
283,364 | 5.0 | % | |||||
| Christine L. Koski(3)(6) |
2,234,666 | 39.3 | % | |||||
| Robert C. Koski(3)(7) |
2,282,666 | 40.2 | % | |||||
| Charles L. Pope(8) |
5,000 | * | ||||||
| Dr. Frederick W. Telling(8) |
5,000 | * | ||||||
| Brian J. Bohunicky(9) |
20,833 | * | ||||||
| All Directors and officers as a group (6 persons) |
3,041,528 | 53.59 | % | |||||
| * | less than one percent |
| (1) | Except as indicated, the address of the person named in the table is c/o Oragenics, Inc., 3000 Bayport Drive, Suite 685, Tampa, Florida 33607. |
| (2) | For each person and group included in this table, percentage ownership is calculated by dividing the number of shares beneficially owned by such person or group by the sum of 5,683,076 shares of common stock outstanding as of March 30, 2011, plus the number of shares of common stock that such person has the right to acquire within 60 days. |
| (3) | Based upon information provided by the Koski Family Limited Partnership, or KFLP, in the amendment to its Schedule 13D filing with the SEC on February 11, 2011, includes (i) 1,790,000 shares held directly by the KFLP, and (ii) 444,666 shares held by KFLP partner Christine Koski, 422,666 shares held by KFLP partner Robert Koski, 10,000 shares held by KFLP partner Koski Management, Inc. (solely owned by Beverly Koski), 469,666 shares held by KFLP partner, Thomas Koski and 70,000 shares held in trusts which Robert Koski serves as sole trustee (See Note 7 below). Christine L. Koski, Robert C. Koski, Thomas L. Koski and Beverly Koski (as sole owner of Koski Management, Inc.) share voting and investment powers as general partners of the KFLP. The address for the KFLP is 3525 Turtle Creek Boulevard, Unit 19-B, Dallas, Texas 75219. |
| (4) | Based upon information provided by Mr. Hawes in his Form 5, Form 4s and Schedule 13D filings with the SEC. The number of shares includes 539,397 shares owned directly, (as reflected on Form 4 filed October 13, 2010) and 127,888 shares issuable pursuant to currently exercisable warrants, and excludes 5,000 shares of common stock and warrants to purchase 5,250 shares of common stock owned by Mr. Hawes’ wife for which he disclaimed beneficial ownership. Mr. Hawes’ address as reflected in Schedule 13D/A is 390 Plandome Road, Suite 222, Manhasset, New York 11030. |
| (5) | Includes 202,845 shares held by the 2002 Jeffrey Hillman Trust, 39,283 shares held directly by Dr. Hillman and 50,417 shares pursuant to currently exercisable outstanding options, and excludes an aggregate of 35,417 shares able to be acquired pursuant to stock options which have not vested. Dr. Hillman disclaims beneficial ownership of 1,000 shares gifted to his minor grandchild. |
| (6) | In addition to the 1,790,000 shares reflected as being directly owned by the KFLP in Note 3, the share amounts include 444,666 shares owned directly by Ms. Koski (which includes 5,000 shares of our common stock acquired during 2009 upon exercise of Director options). |
68
Table of Contents
| (7) | In addition to the 1,790,000 shares reflected as directly owned by the KFLP in Note 3, the share amounts include: (i) 422,666 shares owned directly by Mr. Koski (which includes 5,000 shares of our common stock acquired during 2009 upon exercise of Director options) and (ii) 70,000 shares owned by trusts for which Mr. Koski serves as sole trustee as follows: the Robert Clayton Koski Trust for the benefit of Anthony James Hunter (10,000 shares); The Robert Clayton Koski Trust for the benefit of Hunter Buchanan Koski (25,000 shares); The Robert Clayton Koski Trust for the benefit of Clayton Ward Bennett (25,000 shares); and The Robert Clayton Koski Trust for the benefit of Robert Edward Koski (10,000 shares). Excludes 10,000 shares of common stock subject to vesting based upon a restricted stock award to Mr. Koski. |
| (8) | Includes 5,000 shares able to be acquired upon the exercise of currently exercisable stock options granted pursuant to our Director compensation program upon initially becoming Directors. |
| (9) | Includes 8,333 shares able to be acquired pursuant to currently exercisable options, and excludes an aggregate of 53,751 shares able to be acquired pursuant to stock options and restricted stock awards which have not vested. |
Equity Compensation Plan Information
We maintain an equity-based compensation plan—the Amended and Restated 2002 Stock Option and Incentive Plan (as amended, the “Incentive Plan”). A description of our equity based compensation plan can be found in Note 8 of the Notes to Financial Statements. The Incentive Plan has been approved by our shareholders. The following table sets forth the number of shares of our common stock subject to outstanding options and rights under the Incentive Plan, the weighted-average exercise price of outstanding options, and the number of shares remaining available for future award grants under the Incentive Plan as of December 31, 2010 (in thousands, except exercise price):
| Equity Compensation Plan Information | ||||||||||||
| (a) | (b) | (c) | ||||||||||
| Plan Category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights |
Weighted-average exercise price of outstanding options, warrants and rights |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||||
| Equity compensation plans approved by security holders |
379,836 | $ | 7.86 | 232,164 | ||||||||
| Equity compensation plans not approved by security holders(1) |
— | — | — | |||||||||
| Total |
379,836 | $ | 7.86 | 232,164 | ||||||||
| (1) | The Company does not have any equity compensation plans that have not been approved by security holders. The Company does have warrants to acquire 306,388 shares of common stock outstanding at a weighted average exercise price of $19.14 per share, the majority of which 288,888 were issued in connection with a private placement in June 2008. |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
The Audit Committee of the Board of Directors (or, to the extent applicable, our disinterested directors) is responsible for reviewing all transactions between the Company and any officer or Director of the Company or any entity in which an officer of Director has a material interest. Any such transactions must be on terms no less favorable than those that could be obtained on an arms-length basis from independent third parties.
69
Table of Contents
Consulting Fees
In December 2009, we paid Dr. Hillman, our director and Chief Scientific Officer, $55,000 for consulting services he provided to us in 2001 and 2002. No interest was accrued on this outstanding obligation. At the same time we paid Dr. Hillman $26,250 for salary deferred prior to 2008. Together these amounts, net of applicable taxes, totaled $54,062 were paid through the issuance of 216,250 shares of restricted common stock at a price per share of $0.25 in accordance with Dr. Hillman’s participation in the December 2009 Private Placement discussed below.
Financing Transactions
December 2009 Private Placement
On December 30, 2009, we issued a total of 500,813 shares of restricted common stock in the initial closing of a private placement to accredited investors including the Koski Family Limited Partnership, or KFLP, our largest shareholder (the “December 2009 Private Placement”), for initial proceeds of $2,504,062. The shares were sold at $5.00 per share. The initial closing proceeds of $2,504,062 included the cancellation at closing of $54,062 in outstanding obligations we owed to Dr. Jeffrey Hillman, our Chief Scientific Officer, for compensation that had been deferred. Approximately half of the total investment, or $1,250,000, was made by the KFLP. In conjunction with, and as a condition to the initial closing of the December 2009 Private Placement, we also issued 200,000 shares of our common stock to the KFLP at $5.00 per share, which was the same price per share paid by the participating accredited investors, in exchange for the cancellation of the KFLP’s $1,000,000 secured promissory note we previously issued to the KFLP in connection with a June 2009 private placement in which the KFLP initially acquired control of the Company (the “June 2009 Private Placement”).
Approximately $1,000,000 of the total proceeds from the December 2009 Private Placement were committed to further our development of the DPOLT synthetic chemistry platform, essential to the production of our lead antibiotic, MU1140, subject to the goals set forth by the two-year NSF SBIR Phase II grant that we received on February 15, 2008. Such allocation enabled us to be eligible to receive up to an additional $500,000 matching grant from the NSF, which grant was subsequently awarded in June 2010.
Contemporaneously with the initial closing of the December 2009 Private Placement, the KFLP also elected to exercise warrants it received as part of the June 2009 Private Placement to purchase 50,000 shares of our common stock. The warrants were exercised through the payment by the KFLP of the warrant exercise price of $2.00 per share. Additionally, Christine Koski and Robert Koski, as directors, each exercised previously issued options to purchase 5,000 shares of our common stock at the option exercise price of $2.00 per share. These options were granted to Christine Koski and Robert Koski when they became non-employee directors on June 30, 2009 in connection with our non-employee director compensation program.
On January 13, 2010, we completed the $3,004,062 private placement contemplated by the December 2009 Private Placement and issued another 100,000 shares of common stock at a price per share of $5.00 to the accredited investors for $500,000. Of this amount, the KFLP again participated in half of the remainder of the aggregate investment by acquiring 50,000 shares for $250,000.
May 2010 Note Financing
On May 28, 2010, we entered into an unsecured promissory note with a conversion provision (the “May 2010 Note”) to the KFLP pursuant to which we borrowed $1,000,000 from the KFLP. Interest on the May 2010 Note accrued at the rate of LIBOR plus 6.0% and the principal of the May 2010 Note, together with all accrued interest thereon, was due and payable the earlier of: (i) the closing date of a registered public offering of newly issued equity securities by us resulting in cash proceeds to us, other than in connection with employee option plans, or (ii) the May 24, 2011 maturity date; provided, however, that in the event we completed a subsequent private offering of equity securities prior to the May 24, 2011 maturity date, we could elect to convert the principal of the May 2010 Note into the same equity securities being sold in the private offering at the same price and terms to the KFLP.
July 2010 Financing Transaction
On July 5, 2010, we entered into a common stock purchase agreement (the “July 2010 Financing Transaction”) with the Koski Family Limited Partnership, or KFLP. At the closing of this financing transaction on July 30, 2010 we issued 250,000 shares of our common stock to the KFLP at a price of $8.00 per share. The $2,000,000 aggregate consideration paid by the KFLP consisted of (i) $1,000,000 cash and (ii) the exchange and cancellation of the outstanding May 2010 Note issued to the KFLP on May 28, 2010. Accrued interest on the May 2010 Note through closing was waived by the KFLP. Concurrent with the July 2010 Financing Transaction and as part thereof, we entered into an unsecured revolving credit agreement (the “Credit Facility”) with the KFLP. Pursuant to the Credit Facility, we are able to borrow up to $2,000,000 from the KFLP at LIBOR plus 6.0%. The term of the Credit Facility was initially for 12 months commencing August 1, 2010.
70
Table of Contents
On September 13, 2010, we drew down on the Credit Facility in the amount of $1,000,000 and executed a revolving unsecured promissory note (the “September 2010 Promissory Note”) for such amount in favor of the KFLP. In addition, on November 8, 2010 we drew down on the remaining $1,000,000 of available funds under the Credit Facility and executed another revolving unsecured promissory note (the “November 2010 Promissory Note”). The September 2010 Promissory Note and November 2010 Promissory Note each initially matured on July 30, 2011 until the Second Amendment discussed below was entered into by us, which extended the maturity date to July 30, 2012.
On January 24, 2011, we entered into a First Amendment to the Credit Facility (the “First Amendment”) to increase the available borrowing from $2,000,000 to $2,500,000 and simultaneously therewith we drew on the Credit Facility as amended by the First Amendment to borrow the additional $500,000 in available funds and executed another revolving unsecured promissory note (the “January 2011 Promissory Note”) initially due on July 30, 2011.
On February 4, 2011, we entered into a Second Amendment (the “Second Amendment”) to the Credit Facility with the KFLP. As a result of the Second Amendment, we are able to borrow up to an additional $2,500,000 from the KFLP. Future draws under the Credit Facility, as amended, are limited to $500,000 per month commencing no earlier than March 2011. Under the Second Amendment, the due date of the amounts then outstanding under the Credit Facility, (the September 2010 Promissory Note, November 2010 Promissory Note and January 2011 Promissory Note) were extended by one year from July 30, 2011 to July 30, 2012. The interest rate remained at LIBOR plus 6.0%. The Second Amendment further provided for the automatic conversion of any amounts borrowed and outstanding under the Credit Facility into securities that we may issue in subsequent securities offerings. Any automatic conversion of amounts outstanding under the Credit Facility would be on the same terms of any such offering. In addition, the Second Amendment provides the KFLP with the right to put any undrawn available amounts under the Credit Facility, as amended, to us and thereby have a note issued to the KFLP. The KFLP can exercise its put right to the extent it desires to fully participate, through the automatic conversion provision, in any subsequent offering by us.
On March 15, 2011, we borrowed an additional $500,000 under the Credit Facility, as amended and executed a revolving unsecured promissory note (the “March 2011 Promissory Note”) in such amount. The March 2011 Promissory Note matures on July 30, 2012.
Relationships
During 2010 we paid $226,760 (which included approximately $162,501 in costs reimbursements associated with maintaining our intellectual property) to a law firm that employs our director, Dr. Hillman’s daughter-in-law as a lawyer and from which we received intellectual property related legal services.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES.
Audit and Other Fees
The following table presents fees incurred for professional audit services rendered by our independent registered public accounting firms, Mayer Hoffman McCann P.C. (successor by acquisition to Kirkland, Russ, Murphy & Tapp, P.A.) and Kirkland, Russ, Murphy & Tapp, P.A. for the audits of our financial statements for the years ended December 31, 2010 and December 31, 2009, and quarterly reports on Form 10Q for 2010 and fees for other services rendered by Kirkland, Russ, Murphy & Tapp, P.A. and other accounting firms whom assisted on special projects during those periods.
| Type of Fees |
2010 | 2009 | ||||||
| Audit Fees (1) |
$ | 110,000 | $ | 124,625 | ||||
| Audit-Related Fees (2) |
89,721 | 37,037 | ||||||
| Tax Fees (3) |
3,500 | 3,100 | ||||||
| All Other Fees (4) |
36,180 | 14,821 | ||||||
| Total |
$ | 239,401 | $ | 179,583 | ||||
71
Table of Contents
| (1) | Audit Fees: These fees consist of aggregate fees billed or to be billed by Mayer Hoffman McCann P.C. and Kirkland, Russ, Murphy & Tapp, P.A. for professional services rendered in connection with their audits of the Company’s 2009 and 2008 financial statements, respectively, and the review of the financial statements included in the Company’s Quarterly Reports on Form 10-Q. |
| (2) | Audit-Related Fees: There were fees billed by Kirkland, Russ, Murphy & Tapp, P.A. and RSM McGladrey, Inc. and Edward Leiber, CPA, for assurance and related services that are reasonably related to the performance of the audit or review of the Company’s financial statements, registration statements and grant applications that are not reported above under the caption “Audit Fees.” |
| (3) | Tax Fees: There were fees billed by CBIZ Kirkland, Russ, Murphy & Tapp and Kirkland, Russ, Murphy & Tapp, P.A. for professional services for tax compliance and tax advice. |
| (4) | All Other Fees: There were fees billed by Taylor White Consulting firm in connection with the professional services associated with the Company’s compliance with the Sarbanes-Oxley Act of 2002 filings for small businesses and the valuation of the Company’s stock option awards in accordance with FASB standards. |
Pre-Approval Policies and Procedures
The Audit Committee approves in advance all audit and non-audit services to be performed by the Company’s independent registered public accounting firm. The Audit Committee considers whether the provision of any proposed non-audit services is consistent with the SEC’s rules on auditor independence and has pre-approved certain specified audit and non-audit services to be provided by Mayer Hoffman McCann P.C. for up to twelve (12) months from the date of the pre-approval. If there are any additional services to be provided, a request for pre-approval must be submitted by management to the Audit Committee for its consideration.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
(a) The documents filed as part of this report are as follows:
| 1. | The financial statements and accompanying report of independent registered public accounting firm are set forth immediately following the signature page of this report on pages F-1-F-21. |
| 2. | All financial statement schedules are omitted because they are inapplicable, not required or the information is included elsewhere in the financial statements or the notes thereto. |
| 3. | The exhibits required to be filed by this report or able to be incorporated by reference are listed in the “Exhibit Index” following the financial statements. |
(b) Other Exhibits
Exhibits required by Item 601 of Regulation S-K are submitted (or incorporated by reference) and listed in a separate section herein immediately following the “Exhibit Index” and are incorporated herein by reference by reference. No exhibits in addition to those previously filed or listed in item 15(a) (3) and filed herein.
(c) Not Applicable.
72
Table of Contents
Pursuant to the requirements of Section 13 and 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this amended report to be signed on its behalf by the undersigned, thereunto duly authorized.
Dated: March 30, 2011
| ORAGENICS, INC. | ||
| By: | /s/ Brian Bohunicky | |
| Brian Bohunicky, Chief Financial Officer, Principal Accounting Officer and Principal Executive Officer | ||
POWER OF ATTORNEY
Each of the undersigned officers and directors of Oragenics, Inc., hereby constitutes and appoints Jeffrey Hillman and Brian J. Bohunicky, their true and lawful attorney-in-fact and agent, for them and in their name, place and stead, in any and all capacities, to sign their name to any and all amendments to this Report on Form 10-K, and other related documents, and to cause the same to be filed with the Securities and Exchange Commission, granting unto said attorneys, full power and authority to do and perform any act and thing necessary and proper to be done in the premises, as fully to all intents and purposes as the undersigned could do if personally present, and the undersigned for himself hereby ratifies and confirms all that said attorney shall lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Brian J. Bohunicky Brian J. Bohunicky |
Principal Executive Officer, Chief Financial Officer (Principal Accounting Officer) | March 30, 2011 | ||
| /s/ Jeffrey D. Hillman Jeffrey D. Hillman |
Chief Scientific Officer and Director | March 30, 2011 | ||
| /s/ Christine L. Koski Christine L. Koski |
Director | March 30, 2011 | ||
| /s/ Robert C. Koski Robert C. Koski |
Director | March 30, 2011 | ||
| /s/ Frederick W. Telling Frederick W. Telling |
Chairman and Director | March 30, 2011 | ||
| /s/ Charles L. Pope Charles L. Pope |
Director | March 30, 2011 | ||
73
Table of Contents
Financial Statements
Years ended December 31, 2010 and 2009
Index
| Pg F-1 | ||||
| Report of Mayer Hoffman McCann P.C., Independent Registered Public Accounting Firm |
Pg F-2 | |||
| Report of Kirkland, Russ, Murphy & Tapp, P.A., Independent Registered Public Accounting Firm |
Pg F-3 | |||
| Audited Financial Statements |
||||
| Pg F-4 | ||||
| Pg F-5 | ||||
| Pg F-6 | ||||
| Pg F-7 | ||||
| Pg F-8 – F-24 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Shareholders of Oragenics, Inc.
We have audited the accompanying balance sheet of Oragenics, Inc. (the Company) as of December 31, 2010 and the related statements of operations, changes in shareholders’ equity (deficit), and cash flows for the year then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion of the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Oragenics, Inc. as of December 31, 2010, and the results of its operations and its cash flows for the year then ended in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming Oragenics, Inc. will continue as a going concern. As more fully described in Note 1, the Company has incurred recurring operating losses, negative operating cash flows and has an accumulated deficit. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
| March 25, 2011 | ||
| /s/ Mayer Hoffman McCann P.C. | ||
| Clearwater, Florida | Certified Public Accountants | |
F-2
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Shareholders of Oragenics, Inc.
We have audited the accompanying balance sheet of Oragenics, Inc. (the Company) as of December 31, 2009 and the related statements of operations, changes in shareholders’ equity (deficit), and cash flows for the year then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion of the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Oragenics, Inc. as of December 31, 2009, and the results of its operations and its cash flows for the year then ended in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming Oragenics, Inc. will continue as a going concern. As more fully described in Note 1, the Company has incurred recurring operating losses, negative operating cash flows and has an accumulated deficit. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
| March 29, 2010 | ||
| /s/ Kirkland, Russ, Murphy & Tapp, P.A. | ||
| Clearwater, Florida | Certified Public Accountants | |
F-3
Table of Contents
Balance Sheets
December 31, 2010 and 2009
| 2010 | 2009 | |||||||
| Assets | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 132,103 | 301,592 | |||||
| Restricted cash |
475,657 | 2,450,000 | ||||||
| Accounts receivables, net |
122,972 | 162,813 | ||||||
| Income tax receivable |
362,218 | — | ||||||
| Inventory, net |
266,628 | 132,112 | ||||||
| Prepaid expenses and other current assets |
139,883 | 80,839 | ||||||
| Total current assets |
1,499,461 | 3,127,356 | ||||||
| Property and equipment, net |
228,202 | 75,480 | ||||||
| Total assets |
$ | 1,727,663 | 3,202,836 | |||||
| Liabilities and Shareholders’ Equity (Deficit) | ||||||||
| Current liabilities: |
||||||||
| Accounts payable and accrued expenses |
$ | 1,514,885 | 478,111 | |||||
| Short term notes payable |
98,906 | 35,012 | ||||||
| Deferred revenue |
13,188 | 50,086 | ||||||
| Total current liabilities |
1,626,979 | 563,209 | ||||||
| Revolving note payable to shareholder |
2,000,000 | — | ||||||
| Total liabilities |
3,626,979 | 563,209 | ||||||
| Shareholders’ equity (deficit): |
||||||||
| Preferred stock, no par value; 20,000,000 shares authorized; none issued and outstanding |
— | — | ||||||
| Common stock, $0.001 par value; 15,000,000 shares authorized; 5,663,076 and 5,304,157 shares issued and outstanding at December 31, 2010 and December 31, 2009, respectively. |
5,663 | 5,304 | ||||||
| Additional paid-in capital |
31,412,069 | 28,146,206 | ||||||
| Accumulated deficit |
(33,317,048 | ) | (25,511,883 | ) | ||||
| Total shareholders’ equity (deficit) |
(1,899,316 | ) | 2,639,627 | |||||
| Total liabilities and shareholders’ equity (deficit) |
$ | 1,727,663 | 3,202,836 | |||||
See accompanying Reports of Independent Registered Public Accounting Firms and notes to the financial statements.
F-4
Table of Contents
Statements of Operations
For the Years Ended December 31, 2010 and 2009
| Year Ended December 31 | ||||||||
| 2010 | 2009 | |||||||
| Revenue, net |
$ | 1,308,910 | 641,285 | |||||
| Cost of sales |
911,793 | 221,198 | ||||||
| Operating expenses: |
||||||||
| Research and development |
2,014,784 | 1,833,746 | ||||||
| Selling, general and administration |
6,285,004 | 4,917,844 | ||||||
| Total operating expenses |
8,299,788 | 6,751,590 | ||||||
| Loss from operations |
(7,902,671 | ) | (6,331,503 | ) | ||||
| Other income (expense): |
||||||||
| Interest income |
3,657 | 922 | ||||||
| Interest expense |
(33,859 | ) | (44,292 | ) | ||||
| Loss from abandoned public offering |
(603,012 | ) | — | |||||
| Gain (loss) on sale of property and equipment |
— | 22,743 | ||||||
| Gain on extinguishment of payables |
— | 832,959 | ||||||
| Local business tax |
(2,717 | ) | (177 | ) | ||||
| Total other income, net |
(635,931 | ) | 812,155 | |||||
| Loss before income taxes |
(8,538,602 | ) | (5,519,348 | ) | ||||
| Income tax benefit |
733,437 | — | ||||||
| Net loss |
$ | (7,805,165 | ) | (5,519,348 | ) | |||
| Basic and diluted net loss per share |
$ | (1.42 | ) | (1.70 | ) | |||
| Shares used to compute basic and diluted net loss per share |
5,511,451 | 3,244,189 | ||||||
See accompanying Reports of Independent Registered Public Accounting Firms and notes to the financial statements.
F-5
Table of Contents
Statements of Changes in Shareholders’ Equity (Deficit)
For the Years Ended December 31, 2010 and 2009
| Common Stock | Additional Paid In Capital |
Accumulated Deficit |
Total Shareholders’ Equity (Deficit) |
|||||||||||||||||
| Shares | Amount | |||||||||||||||||||
| Balances at December 31, 2008 |
1,915,829 | $ | 1,916 | $ | 19,813,371 | $ | (19,992,535 | ) | $ | (177,248 | ) | |||||||||
| Exercise of common stock options and warrants |
60,000 | 60 | 119,940 | — | 120,000 | |||||||||||||||
| Issuance of common stock and warrants |
3,328,328 | 3,328 | 7,820,766 | — | 7,824,094 | |||||||||||||||
| Compensation expense relating to option issuances |
— | — | 392,129 | — | 392,129 | |||||||||||||||
| Net loss |
— | — | — | (5,519,348 | ) | (5,519,348 | ) | |||||||||||||
| Balances at December 31, 2009 |
5,304,157 | $ | 5,304 | $ | 28,146,206 | $ | (25,511,883 | ) | $ | 2,639,627 | ||||||||||
| Exercise of common stock options |
3,000 | 3 | 23,997 | — | 24,000 | |||||||||||||||
| Issuance of common stock, net of expenses |
356,000 | 356 | 2,574,644 | — | 2,575,000 | |||||||||||||||
| Compensation expense relating to option issuances |
— | — | 667,222 | — | 667,222 | |||||||||||||||
| Fractional shares cash payments from one for twenty reverse split of common stock |
(81 | ) | — | — | — | — | ||||||||||||||
| Net loss |
— | — | — | (7,805,165 | ) | (7,805,165 | ) | |||||||||||||
| Balances at December 31, 2010 |
5,663,076 | $ | 5,663 | $ | 31,412,069 | $ | (33,317,048 | ) | $ | (1,899,316 | ) | |||||||||
See accompanying Reports of Independent Registered Public Accounting Firms and notes to the financial statements.
F-6
Table of Contents
Statements of Cash Flows
For the Years Ended December 31, 2010 and 2009
| Year Ended December 31 | ||||||||
| 2010 | 2009 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss |
$ | (7,805,165 | ) | (5,519,348 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Non-cash bonus paid in common stock |
— | 100,000 | ||||||
| Non-cash services paid in common stock |
99,000 | 115,000 | ||||||
| Non-cash settlement of amounts owed to employees |
— | 113,439 | ||||||
| Depreciation and amortization |
42,735 | 239,760 | ||||||
| Stock-based compensation expense |
667,222 | 392,129 | ||||||
| Gain on extinguishment of payables |
— | (832,959 | ) | |||||
| (Gain) loss on sale of property and equipment |
— | (22,743 | ) | |||||
| Changes in operating assets and liabilities: |
||||||||
| Accounts receivable, net |
39,841 | (156,527 | ) | |||||
| Income tax receivable |
(362,218 | ) | ||||||
| Inventory, net |
(134,516 | ) | (120,298 | ) | ||||
| Prepaid expenses and other current assets |
4,791 | 128,939 | ||||||
| Accounts payable and accrued expenses |
1,036,774 | (286,959 | ) | |||||
| Deferred grant revenue |
(36,898 | ) | 50,086 | |||||
| Net cash used in operating activities |
(6,448,434 | ) | (5,799,481 | ) | ||||
| Cash flows from investing activities: |
||||||||
| Purchase of property and equipment, net |
(88,084 | ) | (9,073 | ) | ||||
| Proceeds from sale of property and equipment, net |
— | 40,000 | ||||||
| Net cash (used in) provided by investing activities |
(88,084 | ) | 30,927 | |||||
| Cash flows from financing activities: |
||||||||
| Borrowings under short term note payable |
— | 100,000 | ||||||
| Borrowings under note payable to shareholder |
1,000,000 | — | ||||||
| Borrowings under revolving note payable to shareholder |
2,000,000 | — | ||||||
| Borrowings under long term note payable |
— | 1,000,000 | ||||||
| Payments on short term notes payable |
(107,314 | ) | (215,787 | ) | ||||
| Net proceeds from issuance of common stock |
1,500,000 | 6,470,000 | ||||||
| Restricted cash from common stock issuance proceeds |
— | (2,450,000 | ) | |||||
| Restricted cash released from common stock proceeds |
1,974,343 | — | ||||||
| Net cash provided by financing activities |
6,367,029 | 4,904,213 | ||||||
| Net decrease in cash and cash equivalents |
(169,489 | ) | (864,341 | ) | ||||
| Cash and cash equivalents at beginning of year |
301,592 | 1,165,933 | ||||||
| Cash and cash equivalents at end of year |
$ | 132,103 | 301,592 | |||||
| Interest paid |
$ | 21,968 | 25,915 | |||||
| Non-cash investing and financing activities: |
||||||||
| Issuance of common stock to employees as settlement of amounts owed |
$ | — | 32,556 | |||||
| Borrowings under short term notes payable for prepaid expense |
$ | 63,835 | 123,112 | |||||
| Long-term note payable converted into common stock |
$ | — | 1,000,000 | |||||
| Common stock issued in exchange for cancellation of note payable to shareholder |
$ | 1,000,000 | — | |||||
| Borrowings under short term notes payable for purchase of property and equipment |
$ | 107,373 | — | |||||
See accompanying Reports of Independent Registered Public Accounting Firms and notes to the financial statements.
F-7
Table of Contents
Notes to Financial Statements
December 31, 2010 and 2009
1. Organization and Significant Accounting Policies
The Company
Oragenics, Inc. (formerly known as Oragen, Inc.) (the “Company” or “we”) was incorporated in November, 1996; however, operating activity did not commence until 1999. The Company is focused on the discovery, development and commercialization of a variety of technologies associated with oral health, broad spectrum antibiotics and other general health benefits.
Basis of Presentation
The accompanying financial statements of the Company have been prepared in accordance with accounting principles generally accepted in the United States (GAAP) including the assumption of a going concern basis which contemplates the realization of assets and the settlement of liabilities and commitments in the normal course of business.
The Company has incurred recurring losses and negative cash flows from operations since inception. To date the Company has not generated significant revenues from operations. The Company generated revenues of $1,308,910, incurred a net loss of $7,805,165 and used cash of $6,448,434 in its operating activities during the year ended December 31, 2010. As of December 31, 2010 the Company had an accumulated deficit of ($33,317,048) and cash flows from operations were negative throughout 2010. These factors raise substantial doubt about the Company’s ability to continue as a going concern.
During 2010, the Company’s primary source of debt and equity funding was provided by its largest shareholder, the Koski Family Limited Partnership, or KFLP. The Company expects to incur substantial expenditures to further develop each of its technologies. The Company believes the working capital at December 31, 2010, together with access to the recently amended Credit Facility with the KFLP will be sufficient to meet the business objectives as presently structured through June 2011. Management recognizes that the Company must generate additional capital resources or consider modifications to its technology development plans to enable it to continue as a going concern. Management’s plans include seeking financing, alliances or other partnership agreements with entities interested in the Company’s technologies, or other business transactions that would generate sufficient resources to assure continuation of the Company’s operations and research and development programs.
The Company intends to seek additional funding through sublicensing arrangements, joint venturing or partnering, sales of rights to technology, government grants and public or private financings. The Company’s future success depends on its ability to raise capital and ultimately generate revenue and attain profitability. The Company cannot be certain that additional capital, whether through selling additional debt or equity securities or obtaining a line of credit or other loan, will be available to it or, if available, will be on terms acceptable to the Company. If the Company issues additional securities to raise funds, these securities may have rights, preferences, or privileges senior to those of its common stock, and the Company’s current shareholders may experience dilution. If the Company is unable to obtain funds when needed or on acceptable terms, the Company may be required to curtail their current development programs, cut operating costs and forego future development and other opportunities. Without sufficient capital to fund their operations, the Company will be unable to continue as a going concern. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Reverse Stock Split
On September 24, 2010, we effected a 1-for-20 reverse stock split of all of our authorized, issued and outstanding shares of common stock (the “Reverse Stock Split”) by filing Articles of Amendment to Amended and Restated Articles of Incorporation with the Secretary of State of Florida. The par value of our common stock remained unchanged. The number of shares and per share amounts included in the financial statements and the accompanying notes have been adjusted to reflect the Reverse Stock Split retroactively. Unless otherwise indicated, all references to number of shares, per share amounts and earnings per share information contained in this report give effect to the Reverse Stock Split.
F-8
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
Recently Adopted Accounting Pronouncements
In January 2010, the FASB issued ASU 2010-06 “Improving Disclosures about Fair Value Measurements”, which is an update to ASC Topic 820, “Fair Value Measurement and Disclosure.” This Update establishes further disclosure requirements regarding transfers in and out of levels 1 and 2, and activity in level 3 fair value measurements. In addition, companies will be required to disclose quantitative information about the inputs used in determining fair values. ASU 2010-06 is effective for interim and annual reporting periods beginning after December 15, 2009, except for the new Level 3 disclosures, which become effective after December 15, 2010. The Company adopted ASU 2010-06 on January 1, 2010 and the adoption had no impact on the Company’s financial position or results of operations as it only amends required disclosures.
In February 2010, the FASB issued ASU 2010-09, which is an update to ASC Topic 855, “Subsequent Events.” This Update clarifies the date through which the Company is required to evaluate subsequent events. SEC filers will be required to evaluate subsequent events through the date that the financial statements are issued. ASU 2010-09 was effective upon issuance, and had no impact on the Company’s financial position or results of operations as it only amends required disclosures.
In April 2010, the FASB issued ASU No. 2010-17 “Milestone Method of Revenue Recognition- a consensus of the FASB emerging Issues Task Force” that recognizes the milestone method as an acceptable revenue recognition method for substantive milestones in research or development arrangements. This standard would require its provisions be met in order for an entity to recognize consideration that is contingent upon achievement of a substantive milestone as revenue in its entirety in the period in which the milestone is achieved. In addition, this ASU would require disclosure of certain information with respect to arrangements that contain milestones. ASU No. 2010-17 became effective on a prospective basis for milestones achieved in fiscal years or interim periods beginning on or after June 15, 2010. The adoption had no impact on the Company’s financial position or results of operations for the year ended December 31, 2010.
No other new accounting pronouncements issued or effective during 2010 have had or are expected to have had an impact on the Company’s financial statements.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. The principal areas of estimation reflected in the financial statements are stock based compensation, valuation of warrants, inventory obsolescence reserve, sales returns and allowances and allowance for doubtful accounts.
Fair Value of Financial Instruments
The fair value of the Company’s cash and cash equivalents, accounts payable and accrued expenses approximate their carrying values due to their short-term nature.
Cash and Cash Equivalents
Cash and cash equivalents consist of all cash balances and highly liquid investments with an original maturity of three months or less. The Company’s cash and cash equivalents are deposited in a financial institution and consist of demand deposits and overnight repurchase agreement investments and at times deposits are in excess of federally insured limits.
Restricted Cash
As of December 31, 2010, the Company had $475,657 of cash remaining that was restricted pursuant to the Common Stock Purchase Agreement dated December 30, 2009. The Company reserved and allocated $1,000,000 of the proceeds from the December 2009 Private Placement to the expenses incurred to further development of the Company’s DPOLT synthetic chemistry platform.
F-9
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
Accounts Receivable
Accounts receivable are recorded at their net realizable value and consist of trade receivables from the sale of product to customers. We analyze accounts receivable on a monthly basis and determine the collectability based on the facts and circumstances relating to each customer. The Company estimates their allowance for doubtful accounts based on sales trend and specific review of the creditworthiness of each customer. As of December 31, 2010 and 2009, the Company has recorded an allowance for doubtful accounts of approximately $149,000 and $5,000, respectively.
Inventory
Inventories are stated at the lower of cost or market. Cost, which includes material, labor and overhead, is determined on a first-in, first-out basis. On a quarterly basis, we analyze our inventory levels and reserve for inventory that is expected to expire prior to being sold, inventory that has a cost basis in excess of its expected net realizable value, inventory in excess of expected sales requirements, or inventory that fails to meet commercial sale specifications. Expired inventory is disposed of and the related costs are written off to the reserve for inventory obsolescence. The inventory reserve at December 31,2010 and 2009 was approximately $255,814 and $0, respectively.
Consigned Inventory
The Company has authorized a consignment inventory arrangement with one of its mass retail customers in March 2010. As of December 31, 2010, the Company has $64,999 of inventory on consignment located at the retailers stores and warehouses, which is included in our inventory reserve. Once consignment inventory has been sold by this customer, the customer notifies the Company of the sale and the Company records revenue in that accounting period. The Company authorizes the replenishment of consignment inventory based on orders placed by the customer. The Company is provided with weekly reports of consignment sales activity and balances.
Property and Equipment
Property and equipment is stated at cost less accumulated depreciation and amortization. Depreciation is provided on the straight-line method over the estimated useful lives of the assets (three to seven years). Leasehold improvements are amortized over the shorter of the estimated useful life or the lease term of the related asset (five years).
Business Segments
In accordance with GAAP, the Company is required to report segment information. As the Company only operates principally in one business segment, no additional reporting is required.
Stock-Based Compensation
GAAP requires all share-based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their fair values as of the grant date. Stock-based compensation expense is recorded over the requisite service period in which the grantee provides services to us, to the extent the options or warrants do not vest at the grant date and are subject to forfeiture.
Net Loss Per Share
During all periods presented, the Company had securities outstanding that could potentially dilute basic earnings per share in the future, but were excluded from the computation of diluted net loss per share, as their effect would have been antidilutive. Because the Company reported a net loss for all periods presented, shares associated with the stock options and warrants are not included because they are antidilutive. Basic and diluted net loss per share amounts are the same for the periods presented. Net loss per share is computed using the weighted average number of shares of common stock outstanding.
F-10
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
Revenue Recognition
We recognize revenues from the sales of product when title and risk of loss pass to the customer, which is generally when the product is shipped. Grant revenues are recognized as the reimbursable expenses are incurred over the life of the related grant. Grant revenues are deferred when reimbursable expenses have not been incurred.
We record allowances for discounts and product returns at the time of sales as a reduction of revenues as such allowances can be reliably estimated based on historical experience or known trends. Product returns are limited to specific mass retail customers for expiration of shelf life or unsold product over a period of time. We maintain a return policy that allows our customers to return product within a specified period of time prior to and subsequent to the expiration date of the product. Our estimate of the provision for returns is analyzed quarterly and is based upon many factors, including industry data of product return rates, historical experience of actual returns, analysis of the level of inventory in the distribution channel, if any, and reorder rates. If the history or our product returns changes, the reserve will be adjusted. While we believe that the reserves we have established are reasonable and appropriate based upon current facts and circumstances, applying different judgments to the same facts and circumstances would result in the estimated amounts for sales returns and chargeback’s to vary. Because our ProBiora3 products have only recently been introduced, we could experience different circumstances in the future and these differences could be material.
The Company has granted guaranteed rights of return on several mass retail customer accounts. At this time there are only two active mass retail customer accounts with guaranteed rights of return. Orders are processed and shipped on these accounts however the Company defers recognition of revenue until the customer provide notification to the Company that the product has sold to the end consumer. Once notification has been received and verified, the Company will record revenue in that accounting period.
Impairment of Long-Lived Assets
The Company periodically reviews their long-lived assets for impairment and reduces the carrying value to fair value whenever events or changes in circumstances indicate that the carrying value may not be recoverable. There were no impairment losses recorded during the years ended December 31, 2010 and 2009.
Advertising Expenses
The Company’s policy is to expense advertising and marketing costs as incurred. For the years ended December 31, 2010 and 2009, advertising and marketing expense was $1,615,268 and $421,038, respectively.
Research and Development Expenses
Research and development consists of expenses incurred in connection with the discovery and development of our product candidates. These expenses consist primarily of employee-related expenses, which include salaries and benefits and attending science conferences; expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a substantial portion of our preclinical studies; the cost of acquiring and manufacturing clinical trial materials; facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities and equipment, and depreciation of fixed assets; license fees for and milestone payments related to in-licensed products and technology; stock-based compensation expense; and costs associated with non-clinical activities and regulatory approvals. We expense research and development costs as incurred.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards.
Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rate is recognized in operations in the period that includes the enactment date. Deferred tax assets are reduced to estimated amounts expected to be realized by the use of a valuation allowance. Based on our historical operating losses, a valuation allowance has been recognized for all deferred tax assets.
F-11
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
In July 2006, the FASB issued guidance which clarifies accounting for uncertainty in income taxes recognized in an entity’s financial statements in accordance with GAAP and prescribes a recognition threshold and measurement attributes for financial statement disclosure of tax positions taken or expected to be taken on a tax return. Under GAAP, the impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. Additionally, GAAP provides guidance on derecognition, classification, interest and penalties, accounting for interim periods, disclosure and transition.
Concentrations
The Company is dependent on three key suppliers to provide probiotics, blending and packaging of it’s EvoraPlus, EvoraPlus Kids, EvoraPro, and Teddy’s Pride products. The majority of cost of sales are from these key suppliers. As of December 31, 2010 and 2009 our accounts payable and accrued expenses for these vendors totaled $107,980 and $10,526, respectively.
Abandoned Public Offering
On December 22, 2010, we withdrew the filing of a registration statement for our contemplated public offering. As of December 22, 2010 we had incurred $603,012 in expenses associated with the offering, respectively, which was charged to other income (expense).
2. Inventory, net
Inventory, net consists of the following as of December 31, 2010 and 2009:
| 2010 | 2009 | |||||||
| Finished goods |
$ | 368,102 | $ | 77,826 | ||||
| Consignment |
64,999 | — | ||||||
| Work-in-process |
71,996 | 27,286 | ||||||
| Raw materials |
17,346 | 27,000 | ||||||
| Total inventory |
522,442 | 132,112 | ||||||
| Less: inventory reserve |
(255,814 | ) | — | |||||
| $ | 266,628 | $ | 132,112 | |||||
F-12
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
3. Property and Equipment, net
Property and equipment, net consists of the following as of December 31, 2010 and 2009:
| 2010 | 2009 | |||||||
| Furniture and fixtures |
$ | 20,742 | $ | 17,109 | ||||
| Laboratory equipment |
758,766 | 804,279 | ||||||
| Leasehold improvements |
476,777 | 476,777 | ||||||
| Office and computer equipment |
271,245 | 33,908 | ||||||
| 1,527,530 | 1,332,073 | |||||||
| Accumulated depreciation and amortization |
(1,299,328 | ) | (1,256,593 | ) | ||||
| Property and equipment, net |
$ | 228,202 | $ | 75,480 | ||||
Depreciation and amortization expense for the years ending December 31, 2010 and 2009 were $42,735 and $239,760 respectively.
4. Related Party Transactions
At December 31, 2010 and 2009 deferred payments totaling $34,000 were owed to former directors in connection with their service on our Board and are included in the accompanying balance sheets in accounts payable and accrued expenses. These meeting fees are deferred until such time as management determines that we have sufficient funding to pay them to the former directors. The deferrals of payments to our former directors do not reduce our expenses, but serve to preserve our limited cash resources to the extent necessary to maintain our operations. These amounts are non-interest bearing.
5. Accounts Payable and Accrued Expenses
| 2010 | 2009 | |||||||
| Accounts payable trade |
$ | 745,569 | $ | 194,025 | ||||
| Legal fees |
256,478 | 107,656 | ||||||
| Vacation |
116,321 | 88,473 | ||||||
| Deferred compensation |
34,000 | 34,000 | ||||||
| Royalties payable |
25,000 | 25,000 | ||||||
| Interest |
30,268 | 18,377 | ||||||
| Consulting fees |
33,009 | 4,289 | ||||||
| Public offering costs |
149,163 | — | ||||||
| Sales return allowance |
121,728 | — | ||||||
| Other |
3,349 | 6,291 | ||||||
| Total accounts payable and accrued expenses |
$ | 1,514,885 | $ | 478,111 | ||||
Accounts payable and accrued expenses were $1,514,885 and $478,111 as of December 31, 2010 and 2009, respectively. Excluding accounts payable trade, legal fees represent the most significant expense totaling $256,478 as of December 31, 2010 and $107,656 as of December 31, 2009. In 2010, accrued legal fees are primarily for general counsel, public offering assistance and patent work. During 2009, the Company recorded $832,959 of extinguished accounts payable due to the reduction in payments owed to several creditors following the June 29, 2009 financing transaction.
F-13
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
6. Short Term Notes Payable
On July 9, 2010, we entered into a non-interest bearing short-term note payable for $22,188 to finance a portion of our new enterprise resource planning system. Payments on this note began July 9, 2010 and are made quarterly with the final payment due on April 1, 2011. At December 31, 2010, the balance due was $5,547.
On July 20, 2010, we entered into a short-term note payable for $63,835 with an interest rate of 5.75% to finance product liability insurance. Payments on this note begin on August 24, 2010 and are made evenly based upon a straight line amortization over ten-month period with the final payment due on May 24, 2011. At December 31, 2010, the balance due was $32,299.
On July 31, 2010, we entered into a short-term note payable for $85,185 bearing interest at 7.5% to finance a portion our new enterprise resource planning system. Principal and interest payments on this note begin August 31, 2010 and are made evenly based on a straight line amortization over a 17-month period with the final payment due on December 31, 2011. At December 31, 2010, the balance due was $61,060.
On August 6, 2009, the company entered into a short term note payable for $70,025 with an interest rate of 5.75% to finance directors and officers liability insurance. This note was paid in full during 2010.
7. Notes Payable to Shareholder
May 2010 Note Financing - On May 28, 2010, we entered into an unsecured promissory note with a conversion provision (the “May 2010 Note”) to the KFLP pursuant to which we borrowed $1,000,000 from the KFLP. Interest on the May 2010 Note accrued at the rate of LIBOR plus 6.0% and the principal of the May 2010 Note, together with all accrued interest thereon, was due and payable the earlier of: (i) the closing date of a registered public offering of newly issued equity securities by us resulting in cash proceeds to us, other than in connection with employee option plans, or (ii) the May 24, 2011 maturity date; provided, however, that in the event we completed a subsequent private offering of equity securities prior to the May 24, 2011 maturity date, we could elect to convert the principal of the May 2010 Note into the same equity securities being sold in the private offering at the same price and terms to the KFLP. The May 2010 Note was repaid through the issuance of common stock in connection with the July Financing Transaction. See Note 8 Shareholders’ Equity.
Credit Facility- On July 30, 2010, in connection with the July 2010 Financing Transaction, the Company entered into an unsecured revolving credit facility agreement with the KFLP (the “Credit Facility”) Pursuant to the Credit Facility, we are able to borrow up to $2,000,000 from the KFLP at LIBOR plus 6.0%. The term of the Credit Facility is for 12 months commencing August 1, 2010. Our continued ability to draw on the Credit Facility is subject to (i) the receipt by the KFLP of a certificate of no adverse change from us in form and substance acceptable to the KFLP, (ii) the receipt by the KFLP of a revolving unsecured promissory note from us in the principal drawn down in the form attached to the Credit Facility and (iii) our compliance with the terms of the Credit Facility. On September 13, 2010, the Company drew down on the Credit Facility in the amount of one million dollars $1,000,000 and executed a revolving unsecured promissory note (the “September 2010 Promissory Note”) for such amount in favor of the KFLP. In addition, on November 8, 2010 the Company drew down on the remaining $1,000,000 of available funds under the Credit Facility and executed another revolving unsecured promissory note (the “November 2010 Promissory Note”). The September 2010 Promissory Note and November 2010 Promissory Note each initially were to mature on July 30, 2011 prior to the amendments to the Credit Facility entered into after the end of the fiscal year which extended the maturity on the notes until July 30, 2012. See Note 14 Subsequent Events.
F-14
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
8. Shareholders’ Equity
Common Stock
At our 2009 annual shareholder meeting our proposal to amend the Company’s articles of incorporation to increase the authorized shares of common stock from 5,000,000 to 15,000,000 was approved by shareholders and the amendment to our articles of incorporation was filed with the Florida Department of State.
At our 2010 annual meeting our proposal to amend the Company’s articles of incorporation to consummate a reverse stock split of our authorized and outstanding shares of common stock was approved and thereafter our Board of Directors authorized a 1 for 20 reverse stock split of our authorized and outstanding shares of common stock and the Company filed an amendment to its articles of incorporation with the Florida Department of State to effect the reverse stock split.
June 2009 Private Placement
On June 29, 2009, we issued a total of 2,500,000 shares of restricted common stock and warrants to acquire 50,000 shares of common stock in a private placement to the KFLP, for total proceeds of $4,000,000 (the “June 2009 Private Placement”). The shares were sold at $1.60 per share. The warrants to purchase 50,000 shares of our common stock were exercisable at $2.00 per share and had a five year term. The consideration paid by the KFLP for the shares of common stock consisted of $4,000,000 as follows: $1,500,000 in cash at closing and $2,500,000 pursuant to a non-interest bearing promissory note providing for five consecutive monthly installment payments of $500,000 commencing July 31, 2009. In addition, pursuant to the securities purchase agreement (the “June 2009 Purchase Agreement”) with the KFLP, the KFLP also provided a secured convertible loan of $1,000,000 to the company. The loan was secured by substantially all of our assets, excluding receivables, and paid interest at the rate of prime plus 4.0% which was payable quarterly. This loan was subsequently repaid by us in connection with the December 2009 Private Placement described below. The principal of the loan was due in five years. As a result of the June 2009 Private Placement the Board of Directors believes there was a change of control, with the KFLP acquiring a controlling interest in our outstanding voting common stock.
We also agreed to provide the KFLP with certain registration rights in connection with any underwritten or other offering by us over the next five years. Specifically, we are obligated to register on behalf of the KFLP shares of common stock held by the KFLP equal to 15% of the total number of shares of common stock to be sold by us in a public offering subject to the discretion of the managing underwriter on the inclusion of shares in the offering to be sold by selling shareholders.
In connection with, and as a closing condition to the June 2009 Private Placement, the purchasers in the June 2008 Private Placement (including George Hawes, our largest shareholder prior to the June 2009 Private Placement), entered into a consent, waiver and mutual release agreement with us on June 25, 2009. In addition, the purchasers in the June 2008 Private Placement waived and relinquished any special rights they possessed pursuant to the agreements with us as part of the June 2008 Private Placement, including, but not limited to, (i) rights of first refusal, (ii) antidilution regarding future equity sales and (iii) covenants regarding secured lending by us. In connection with such consents, waivers and mutual releases, warrants to acquire 161,000 shares that were previously issued in connection with the June 2008 Private Placement were subject to the right of exchange for new replacement warrants to acquire the same number of shares under the same terms except for a change in the exercise price from $26.00 to $15.00. In addition, to the extent of any future underwritten registered offerings of our common stock, or the filing of any resale registration statement by us, in each case occurring within five years from the date of the consent, waiver and mutual release, the purchasers shall have the right to include an aggregate of up to 5.0% of the shares being registered in such offering or registration statement, subject to the discretion, in any underwritten primary offerings by us, of the underwriter on the inclusion of shares in the offering to be sold by selling shareholders.
In addition to the above, as a further condition to the consummation of the transaction contemplated by the June 2009 Purchase Agreement, we were required to obtain satisfactory arrangements with three main creditors for reductions in the amounts payable by us to these creditors. As of June 29, 2009, these reductions amounted to $707,674 in the aggregate and were conditioned upon prompt payment of the remaining balances owed to such creditors after taking into account the agreed upon reductions. As of December 31, 2009, the amount of reductions arranged with our creditors totaled $832,959. These agreed upon reductions in payables have been fully reflected in our 2009 financial statements for the periods and reported under other income.
F-15
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
December 2009 Private Placement
On December 30, 2009, we issued a total of 500,813 shares of restricted common stock in the initial closing of a private placement to accredited investors including the KFLP, our largest shareholder (the “December 2009 Private Placement”), for initial proceeds of $2,504,062. The shares were sold at $5.00 per share. The initial closing proceeds of $2,504,062 included the cancellation at closing of $54,062 in outstanding obligations we owed to Dr. Jeffrey Hillman, our Chief Scientific Officer, for compensation that had been deferred. Approximately half of the total investment, or $1,250,000, was made by the KFLP. In conjunction with, and as a condition to the initial closing of the December 2009 Private Placement, we also issued 200,000 shares of our common stock to the KFLP at $5.00 per share, which was the same price per share paid by the participating accredited investors, in exchange for the cancellation of the KFLP’s $1,000,000 secured promissory note we previously issued to the KFLP in connection with the June 2009 Private Placement. On January 13, 2010, we completed the $3,004,062 private placement contemplated by the December 2009 Private Placement and issued another 100,000 shares of common stock at a price per share of $5.00 to the accredited investors for $500,000. Of this amount, the KFLP again participated in half of the remainder of the aggregate investment by acquiring 50,000 shares for $250,000.
Approximately $1,000,000 of the total proceeds from the December 2009 Private Placement (See Note 1) were committed to further our development of the DPOLT synthetic chemistry platform, essential to the production of our lead antibiotic, MU1140, subject to the goals set forth by the two-year NSF SBIR Phase II grant that we received on February 15, 2008. Such allocation enabled us to be eligible to receive up to an additional $500,000 matching grant from the NSF, which grant was subsequently awarded in June 2010.
Contemporaneously with the initial closing of the December 2009 Private Placement, we issued 50,000 shares of common stock to the KFLP upon its exercise of warrants it received as part of the June 2009 Private Placement. The warrants were exercised through the payment by the KFLP of the warrant exercise price of $2.00 per share.
July 2010 Financing Transaction
In July 2010, we issued 250,000 shares of our common stock to the KFLP at a price of $8.00 per share in connection with a common stock purchase agreement (the “July 2010 Financing Transaction”) we entered into with the KFLP. The $2,000,000 aggregate consideration paid by the KFLP for the shares consisted of (i) $1,000,000 cash and (ii) the exchange and cancellation of the outstanding May 2010 Note issued to the KFLP on May 28, 2010. Accrued interest on the May 2010 Note through closing was waived by the KFLP.
Concurrent with the July 2010 Financing Transaction and as part thereof, the Company entered into the Credit Facility with the KFLP (see Note 7).
Other Share Issuances
In June 2010, we issued 6,000 shares to Athorn Clark partners (“Athorn”) at a price per share of $12.50 (based on the value of the services required to be provided by Athorn) in connection with an agreement for Athorn to provide media related services to us.
In September 2009, the Company issued 25,000 shares of restricted common stock to Media4Equity LLC (“M4E”) pursuant to an agreement with M4E effective September 3, 2009 whereby M4E will provide consulting services to us with respect to national media exposure of placements of print and radio features. The agreement also requires us to pay a monthly fee to M4E of $10,000 during the three year term of the agreement, subject to certain termination rights. The shares of common stock have a fair market value of $115,000 based on a price of $4.60 per share. This amount is included in selling, general and administrative expenses in the accompanying 2009 statement of operations.
In June 2009, following the closing of the June 2009 Private Placement, we issued shares of our common stock at a price per share of $2.00 (the closing price of our common stock on June 29, 2009) as follows: (i) 50,000 shares to our former chief executive officer for payment of a bonus, totaling $100,000 (ii) an aggregate of 29,687 shares to employees for deferred compensation we owed to them, (12,500 shares of which were issued to Mr. Brian Bohunicky, who was appointed as our Chief Financial Officer) and (iii) 22,828 shares to repay aggregate obligations of $45,656 owed to our Chief Scientific Officer, Dr. Hillman (these obligations arose due to borrowings on May 4, 2009 and June 10, 2009 and were used to purchase inventory, in the amounts of $32,556 and $13,100, respectively and were to be repaid upon demand, were unsecured and did not bear interest).
F-16
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
Warrants
In June 2008, we issued warrants to acquire 288,888 shares of common stock in connection with a private placement to accredited investors. These warrants are exercisable at $26.00 per share and expire May 30, 2013. In connection with the June 2009 Private Placement the exercise price of 161,000 of these warrant shares was amended from $26.00 to $15.00.
On April 15, 2009, the Company issued 5,000 warrants to an accredited investor to purchase common stock at an exercise price of $10.00 per share. These warrants were issued in connection with an April 15, 2009 bridge loan provided by the investor.
On September 14, 2009, the Company issued 12,500 warrants to Strategic Growth International to purchase common stock at an exercise price of $6.00 per share. These warrants were issued in connection with a contract to provide investor relations services.
The Company’s outstanding and exercisable warrants as of December 31, 2010 is presented below:
| Shares Underlying |
Exercise Price | Expiration Date | ||||||||
| 127,888 | $ | 26.00 | 5/30/2013 | |||||||
| 161,000 | 15.00 | 5/30/2013 | ||||||||
| 5,000 | 10.00 | 4/15/2014 | ||||||||
| 12,500 | 6.00 | 9/14/2012 | ||||||||
| 306,388 | ||||||||||
Stock Compensation Plan
The Company originally adopted the Oragenics, Inc. 2002 Stock Option and Incentive Plan (Plan) on September 17, 2002 (the “Original Plan”). The Original Plan was amended in May 2004 to increase the available shares from 50,000 to 75,000. In May 2006 the Company amended and restated the Original Plan (the “Amended and Restated Plan”) and simultaneously increased the shares available from 75,000 to 150,000. In April 2008 the Amended and Restated Plan was amended to increase the number of shares available from 150,000 to 250,000. In October 2009 the Amended and Restated Plan was amended to increase the shares available from 250,000 to 625,000. The Amended and Restated Plan together with the amendments are collectively referred to as the “Plan.” The Plan has been approved by our shareholders.
The purpose of the Plan is to advance the interests of the Company by affording certain employees and directors of the Company and key consultants and advisors an opportunity to acquire or increase their proprietary interests in the Company. The Plan authorizes the grant of stock options (incentive and non-statutory), stock appreciation rights and restricted stock. As of December 31, 2010 and 2009, the Company had not awarded any stock appreciation rights or restricted stock under the Plan.
As a result of option exercises under the Plan, an aggregate of 392,837 shares had been issued and the Company has an aggregate of 625,000 shares of common stock reserved for grants under the Plan. As of December 31, 2010 and 2009, there are 232,164 and 239,035 shares, respectively, available for future grants. The exercise price of each option shall be determined by the Board and an option’s maximum term is ten years.
During the years ended December 31, 2010 and 2009, the Company recognized a stock compensation expense of $667,222 and $392,129, respectively, in accordance with GAAP.
During 2010, there were 44,261 options forfeited due to lack by exercise of employees.
F-17
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
On August 13, 2009, the compensation committee approved the acceleration of the vesting of certain outstanding option awards, the vesting of which was tied to our share price reaching certain levels in the future. Option awards previously made to Mr. David Hirsch, our former President and Chief Executive Officer, Dr. Jeffrey Hillman, our Chief Science Officer and certain other Company employees were impacted by the accelerated vesting of these options (21,666 shares for Mr. Hirsch, 25,000 shares for Dr. Hillman, 28,166 for other Company employees). Following the acceleration of vesting by the compensation committee, Mr. Hirsch’s grant of options to acquire 25,000 shares of our common stock at $9.80 per share is fully vested and exercisable (including the 21,666 shares impacted by the acceleration of vesting), Dr. Hillman’s grant of options to acquire 35,000 shares of our common stock at $17.00 per share is fully vested and exercisable (including the 25,000 shares impacted by the acceleration of vesting). The options previously had a performance condition that was not probable. They are vested without any condition and a compensation expense of $177,800 was recognized at the modification date, no compensation expense was previously recognized. All other terms of the prior option awards, including the share amounts covered by the options and exercise prices remained the same.
On December 30th, 2009, we issued 5,000 shares to each of Christine L. Koski and Robert C. Koski upon the exercise of previously issued options under the Plan to purchase 5,000 shares of the common stock at an exercise price of $2.00 per share. These options were granted to both Christine and Robert Koski upon their appointment to our Board of Directors as non-employee directors on June 30, 2009.
In July 2010, we issued 3,000 shares to Mr. David McKeon (d/b/a Game On Consulting) at a price per share of $8.00 as part to a consulting agreement to provide services to the Company. The share price is based on value of the company’s common stock at the time the agreement was signed.
A summary of the status of the Company’s outstanding stock options as of December 31, 2010 and 2009 and changes during the periods ending on those dates is presented below:
| Options | Option Price Per Share |
Weighted Average Exercise Price |
||||||||||
| Outstanding at December 31, 2008 |
228,500 | $ | 5.60 - 85.00 | $ | 12.00 | |||||||
| Forfeited |
(132,250 | ) | 5.60 - 80.00 | 12.00 | ||||||||
| Granted |
299,717 | 2.00 - 6.00 | 5.40 | |||||||||
| Exercised |
(10,000 | ) | 2.00 - 2.00 | 2.00 | ||||||||
| Outstanding at December 31, 2009 |
385,967 | $ | 5.40 - 17.00 | $ | 17.00 | |||||||
| Forfeited |
(44,262 | ) | 5.40 - 14.00 | 6.77 | ||||||||
| Granted |
41,132 | 4.00 - 14.00 | 9.23 | |||||||||
| Exercised |
(3,000 | ) | 8.00 - 8.00 | 8.00 | ||||||||
| Outstanding at December 31, 2010 |
379,837 | $ | 5.40 - 17.00 | $ | 7.86 | |||||||
| Exercisable at the end of the year |
167,416 | $ | 5.60 - 17.00 | $ | 9.44 | |||||||
The range of exercise price for outstanding options at December 31, 2010 is $5.40 to $17.00 per share. The weighted-average per option fair value of options granted during 2010 and 2009 was $9.23 and $5.40, respectively, and the weighted average remaining contractual life of those options is 8.6 years. Options vest over a period of two to three years from respective grant dates and the options expire 10 years after the date of grant. The fair value of these options was estimated at the date of grant using the Black-Scholes option pricing model with the following weighted-average assumptions: weighted average risk-free interest rate of 3.15%; dividend yields of 0%; weighted-average volatility factors of the expected market price of the Company’s common stock of 146.0%; and an expected life of the option of ten years. Future compensation expense related to the outstanding options as of December 31, 2010 is $784,350.
F-18
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
As of the date of this filing there are 306,388 warrants and 379,836 stock options outstanding. The total number of outstanding warrants and unexercised stock options is 686,224. If all warrants and stock options were exercised, the total number of outstanding shares would be approximately 6,349,300.
9. Licenses
The Company has two license agreements with the University of Florida Research Foundation, Inc. (“UFRF”) for their technologies. The Company issued 29,997 shares of common stock as partial consideration in 1998. The license agreements provide for, among other things, the Company to make minimum annual research expenditures of $1,000,000 and to adhere to specific milestones. Beginning in 2005, the Company was required to pay minimum royalties on product sales of $50,000 annually per agreement. If the Company fails to perform certain of its obligations, UFRF may terminate the license agreements. The Company’s milestones are in compliance with UFRF and the Company had $25,000 of royalties payable to UFRF recorded in the accompanying balance sheets in accounts payable and accrued expenses at December 31, 2010 and 2009.
10. Retirement Plan
In January 2004, the Company established a defined contribution Simple Individual Retirement Arrangement (IRA) plan, replacing the previous plan that had been established in 2001. The new plan covers all employees and provides for a Company match of up to 3% of all employee contributions to the plan. Total matching contributions made by the Company in 2010 and 2009 were $40,197 and $24,718, respectively.
11. Income Taxes
The components of the provision for income taxes for the years ended December 31, 2010 and 2009 are as follows:
| 2010 | 2009 | |||||||
| Current |
$ | (733,437 | ) | — | ||||
| Deferred |
(3,502,706 | ) | (1,602,435 | ) | ||||
| Valuation Allowance |
3,502,706 | 1,602,435 | ||||||
| Total provision for income taxes |
$ | (733,437 | ) | — | ||||
F-19
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
At December 31, 2010 and 2009, the Company had temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and their respective income tax bases, as measured by enacted state and federal tax rates, as follows:
| 2010 | 2009 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforward |
$ | 11,345,514 | $ | 8,702,188 | ||||
| Bad debt reserve |
55,914 | — | ||||||
| Inventory reserve |
96,263 | — | ||||||
| Sales return allowance |
45,806 | — | ||||||
| Accrued vacation |
43,771 | — | ||||||
| Deferrals of compensation to Directors & Officers |
12,794 | 14,486 | ||||||
| Uniform capitalization (UNICAP) |
2,486 | — | ||||||
| Non-qualified stock compensation |
166,832 | — | ||||||
| Total deferred tax assets |
11,769,380 | 8,716,674 | ||||||
| Less valuation allowance |
(11,769,380 | ) | (8,716,674 | ) | ||||
| Total net deferred taxes |
$ | — | $ | — | ||||
The following is a reconciliation of tax computed at the statutory federal rate to the income tax benefit in the statements of operations for the years ended December 31, 2010 and 2009:
| 2010 | 2009 | |||||||
| Income tax benefit computed at statutory federal rate of 34% |
$ | (2,903,125 | ) | $ | (1,876,579 | ) | ||
| State income tax benefits, net of federal expense/benefit |
(309,951 | ) | (200,352 | ) | ||||
| Change in valuation allowance |
3,052,706 | 1,602,435 | ||||||
| Non-deductible expenses |
133,032 | 474,496 | ||||||
| Therapeutic discovery tax credit |
733,437 | — | ||||||
| Other |
27,338 | — | ||||||
| Total |
$ | 733,437 | $ | — | ||||
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies in making this assessment. Based upon the levels of historical taxable income and projections of future taxable income over which the deferred tax assets are deductible, the Company believes that it is more likely than not that it will not be able to realize the benefits of some of these deductible differences.
F-20
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
Accordingly, a valuation allowance of $11,769,380 and $8,716,674 has been provided in the accompanying financial statements as of December 31, 2010 and 2009, respectively. The 2010 net change in valuation allowance related to deferred tax assets was an increase of $3,052,706 primarily relating to net operating loss carryforwards.
At December 31, 2010, the Company has federal and state tax net operating loss carryforwards of approximately $30,150,000. The federal and state tax loss carryforward will expire through 2031, unless previously utilized. The Company also has federal research and development tax credit carryforwards of approximately $491,000. The federal tax credit carryforward will expire through 2021, unless previously utilized.
Pursuant to Internal Revenue Service Code Sections 382 and 383, use of the Company’s net operating losses and credit carryforwards are limited due to a cumulative change in ownership of more than 50% that occurred in 2009. As a result of the 50% change in ownership, the annual amount of pre-change net operating losses that may be used in periods subsequent to the change in ownership is approximately $172,000. The impact of this limitation is factored into management’s valuation allowance placed against the Company’s deferred tax assets.
For the years ended December 31, 2010 and 2009, the Company incurred $106,719 and $43,057, respectively, of additional unrecognized tax benefits that resulted in a decrease to the deferred tax asset valuation allowance, related to research and development credits. The entire amount of this unrecognized tax benefit, if recognized, would result in an increase to the deferred tax asset valuation allowance, and would not have an impact on the effective tax rate.
The Company files its income tax returns in the U.S. federal jurisdiction and in Florida. With few exceptions, the Company is no longer subject to federal or state income tax examinations by tax authorities for years before 2006.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
| Balance as of December 31, 2008 |
$ | 341,219 | ||
| Additions based on tax positions related to the current year |
43,057 | |||
| Additions for the tax positions of prior years |
— | |||
| Reductions for the tax positions of prior years |
— | |||
| Balance as of December 31, 2009 |
$ | 384,276 | ||
| Additions based on tax positions related to the current year |
106,719 | |||
| Additions for the tax positions of prior years |
— | |||
| Reductions for the tax positions of prior years |
— | |||
| Balance as of December 31, 2010 |
$ | 490,995 | ||
Included in the balance at December 31, 2010 and 2009, are $490,995 and $384,276, respectively, of tax positions for which there is uncertainty about the validity of certain credits. The disallowance of the credits would impact the amount of gross deferred tax assets reflected in the accompanying footnotes.
F-21
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
During the years 2010 and 2009 the Company did not recognize any interest and penalties. Due to the potential offset of the Company’s operating loss carryforward for any future activity, the amount attributed to interest and penalties would be immaterial.
On November 1, 2010, we received notification that we were awarded federal grant funding for three of its therapeutic development programs under the Qualifying Therapeutic Discovery Project. The Qualifying Therapeutic Discovery Project, was recently enacted by Congress as part of the Patient Protection and Affordable Care Act of 2010, which was designed to provide grants or tax credits to qualified biotechnology companies that demonstrate the potential to either 1) develop new therapies to treat areas of unmet medical needs; 2) prevent, detect or treat chronic or acute diseases and conditions; 3) reduce long-term health care costs in United States; or 4) significantly advance the goal of curing cancer within the 30 year period beginning on May 21, 2010. We applied for funding on three of its programs: Prevention of Tooth Decay using Smart Replacement Therapy, Novel Antibiotics for the Treatment of Healthcare Associated Infections and Rapid and Sensitive Identification of Novel Diagnostic Biomarkers for Cancer and Infectious Diseases. We received a non-taxable cash grant award totaling $733,437 under the program which was recorded as income tax benefits for the year. As of December 31, 2010, we had recorded income tax receivables totaling $362,218. A payment of $371,219 was made to us in November 2010 and remaining grant award amount of $362,218 was received in February, 2011.
12. Commitments and Contingencies
The Company’s Alachua facility is being leased from a real estate developer for a term of two years renewed in December 2009. Lease payments are capped during the term with the exception of taxes and insurance exceeding 3%. This operating lease agreement required the Company to pay a deposit of $6,400 and provides for monthly lease payments of $8,993, inclusive of utilities, insurance, sales taxes and real estate taxes. Rent expense under this lease was $107,916 and $97,187 for the years ended December 31, 2010 and 2009. On October 1, 2009 the Company leased office space for Corporate, Sales and Marketing personnel located in Tampa, FL. The lease is for approximately 3,150 square feet and is occupied by ten employees. The lease period for the office space is forty months in the amount of $5,276 per month inclusive of insurance, taxes and utilities. The lease expires on January 31, 2013. Rent expense under this lease was $63,312 and $15,828 for the year ended December 31, 2010 and 2009, respectively.
The Company terminated two lease agreements in August and October 2009 for office spaces which were located in Alachua and St. Petersburg, Florida, respectively.
Future annual minimum payments under all non-cancelable operating leases are as follows as of December 31, 2010:
| Year ended: | ||||
| 2011 |
161,773 | |||
| 2012 |
63,317 | |||
| 2013 |
5,276 | |||
| $ | 230,366 | |||
F-22
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
13. Unaudited Quarterly Financial Information
The quarterly interim financial information shown below has been prepared by the Company’s management and is unaudited. It should be read in conjunction with the audited financial statements appearing herein.
| 2010 | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
| Revenue |
$ | 341,483 | $ | 304,696 | $ | 364,574 | $ | 298,157 | ||||||||
| Total operating expenses |
1,890,997 | 2,186,467 | 2,087,934 | 2,134,390 | ||||||||||||
| Net loss |
(1,747,199 | ) | (2,010,222 | ) | (1,875,228 | ) | (2,172,516 | ) | ||||||||
| Loss per share: |
||||||||||||||||
| Basic and Diluted |
$ | (0.32 | ) | $ | (0.37 | ) | $ | (0.34 | ) | $ | (0.38 | ) | ||||
| 2009 | ||||||||||||||||
| First | Second | Third | Fourth | |||||||||||||
| Revenue |
$ | 124,272 | $ | 41,895 | $ | 199,675 | $ | 275,444 | ||||||||
| Total operating expenses |
2,092,819 | 1,605,328 | 1,593,353 | 1,460,091 | ||||||||||||
| Net loss |
(1,980,350 | ) | (869,048 | ) | (1,437,184 | ) | (1,232,766 | ) | ||||||||
| Loss per share: |
||||||||||||||||
| Basic and Diluted |
$ | (1.13 | ) | $ | (0.45 | ) | $ | (0.32 | ) | $ | (0.27 | ) | ||||
14. Subsequent Events
On January 24, 2011, the Company entered into a First Amendment to its Credit Facility with the KFLP (the “First Amendment”) to increase the available borrowing from $2,000,000 to $2,500,000 and simultaneously therewith the Company drew on the Credit Facility as amended by the First Amendment to borrow the additional $500,000 in available funds and executed another revolving unsecured promissory note (the “January 2011 Promissory Note”) initially due on July 30, 2011.
On February 4, 2011, the Company entered into a Second Amendment (the “Second Amendment”) to its Credit Facility with the KFLP. As a result of the Second Amendment, the Company will be able to borrow up to an additional $2,500,000 from the KFLP. Future draws under the Credit Facility, as amended, are limited to $500,000 per month commencing no earlier than March 2011. Under the Second Amendment, the due date of the amounts borrowed and outstanding under the Credit Facility, were extended by one year from July 30, 2011 to July 30, 2012. The interest rate remained at LIBOR plus 6.0%. The Second Amendment further provided for the automatic conversion of any amounts borrowed and outstanding under the Credit Facility into Company securities that may be issued by the Company in subsequent securities offerings. Any automatic conversion of amounts outstanding under the Credit Facility would be on the same terms of any such offering. In addition, the Second Amendment provides the KFLP with the right to put any undrawn available amounts under the Credit Facility, as amended, to the Company and thereby have a note issued to the KFLP. The KFLP can exercise its put right to the extent it desires to fully participate, through the automatic conversion provision, in any subsequent offering by the Company.
On February 4, 2011 David Hirsch resigned as our President and Chief Executive Officer and director to pursue other opportunities. Our Board of Directors is engaged in a search to fill the vacancy. In the interim, Mr. Brian Bohunicky has been designated as our lead executive and therefore serves as our principal executive officer until such time as the vacancy is filled. During this period, the Board of Directors will provide direction to the company, with Robert Koski acting as its liaison with management.
On February 4, 2011, we entered into a separation and release agreement with our former Chief Executive Officer and President, Mr. David Hirsch. Mr. Hirsch’s compensation under the separation and release agreement includes the payment of severance in the amount of $112,500 over six months in accordance with the Company’s normal payroll practices. Mr. Hirsch was entitled to the immediate payment of his accrued vacation which totaled $10,961. Mr. Hirsch’s employee employment agreement was terminated in connection with the separation and release agreement.
F-23
Table of Contents
Oragenics, Inc.
Notes to Financial Statements (continued)
December 31, 2010 and 2009
On March 11, 2011 our Board of Directors and Compensation Committee approved an option award of 20,000 shares to Mr. Brian Bohunicky, our Chief Financial Officer under the Company’s Amended and Restated 2002 Stock Option and Incentive Plan, as amended (the “Plan”). These options vest equally over a three (3) year period from the date of grant and are exerciseable at $3.60 per share, the price our stock closed on March 11, 2011, the date of the grant. On that same date Mr. Bohunicky was also granted 10,000 shares of restricted common stock under the Plan half of which vests in six (6) months and the other half on the anniversary of the award. These restricted shares were granted at the price of $3.60, the closing price on March 11, 2011, the date of the award.
The Board also approved the payment of up to $15,000 to Mr. Bohunicky to reimburse him for relocation expenses he may incur in connection with his contemplated relocation to our primary corporate headquarters in Tampa, Florida.
On March 11, 2011 our Board of Directors approved an award of 10,000 shares of restricted common stock under the Plan in connection with, and as recognition of, the time Mr. Koski is spending in his role as liaison between management and the Board during the vacancy in the Company’s position of Chief Executive Officer and President. Half of the shares of restricted common stock vest in six (6) months and the other half on the anniversary of the award. These restricted shares were granted at the price of $3.60 , the closing price on March 11, 2011, the date of the award.
On March 15 2011, we borrowed an additional $500,000 under the Credit Facility, as amended and executed a revolving unsecured promissory note (the “March 2011 Promissory Note”) in such amount. The March 2011 Promissory Note matures on July 30, 2012.
F-24
Table of Contents
| Incorporated by Reference | ||||||||||||
| Exhibit number |
Exhibit description |
Form |
File no. |
Exhibit |
Filing date |
Filed herewith | ||||||
| 3.1 | Amended and Restated Articles of Incorporation | SB-2 | 333-100568 | 3.3 | 10/16/02 | |||||||
| 3.2 | Articles of Amendment to Amended and Restated Articles of Incorporation | 8-K | 001-32188 | 10.2 | 10/30/09 | |||||||
| 3.3 | Articles of Amendment to Amended and Restated Articles of Incorporation | 8-K | 001-32188 | 3.1 | 9/27/10 | |||||||
| 3.4 | Bylaws | SB-2 | 333-100568 | 3.2 | 10/16/02 | |||||||
| 3.5 | First Amendment to Bylaws | 8-K | 001-32188 | 3.1 | 6/9/10 | |||||||
| 3.6 | Second Amendment to Bylaws | 8-K | 001-32188 | 3.1 | 8/24/10 | |||||||
| 4.1 | Specimen Stock Certificate | S-1/A | 333-169031 | 4.0 | 10/05/10 | |||||||
| 4.2 | Securities Purchase Agreement between George Hawes, William Matlack and Oragenics, Inc. dated June 12, 2008 (including form of June 2008 Warrant) | 8-K | 001-32188 | 10.1 | 6/16/08 | |||||||
| 4.3 | Securities Purchase Agreement dated June 29, 2009 by and between the Company and the Koski Family Limited Partnership (including the Form of the Promissory Note and Form of the Warrant) | 8-K | 001-32188 | 10.1 | 7/6/09 | |||||||
| 4.4 | Secured Promissory Note issued to the Koski Family Limited Partnership | 8-K | 001-32188 | 10.2 | 7/6/09 | |||||||
| 4.5 | Security Agreement between the Company and the Koski Family Limited Partnership | 8-K | 001-32188 | 10.3 | 7/6/09 | |||||||
| 10.1 | Exclusive License Agreement between the Company and the University of Florida Research Foundation, Inc. effective August 4, 1998 for Replacement Therapy for Dental Caries (the “Replacement Therapy License Agreement”) | SB-2 | 333-100568 | 10.1 | 10/16/02 | |||||||
| 10.2 | First Amendment to Replacement Therapy License Agreement dated September 15, 2000 | SB-2 | 333-100568 | 10.2 | 10/16/02 | |||||||
| 10.3 | Second Amendment to Replacement Therapy License Agreement dated June 2002 | SB-2 | 333-100568 | 10.3 | 10/16/02 | |||||||
| 10.4 | Third Amendment to Replacement Therapy License Agreement dated September 25, 2002 | SB-2 | 333-100568 | 10.4 | 10/16/02 | |||||||
| 10.5 | Fourth Amendment to Replacement Therapy License Agreement dated March 2003 | SB-2/A-3 | 333-100568 | 10.36 | 4/9/03 | |||||||
Table of Contents
| Incorporated by reference to | ||||||||||||
| Exhibit number |
Exhibit description |
Form |
File no. |
Exhibit |
Filing date |
Filed herewith | ||||||
| 10.6 | Standard Exclusive License Agreement with Sublicensing Terms between the Company and the University of Florida Research Foundation, Inc. effective June 22, 2000 (the “Antimicrobial Polypeptide License Agreement”) | SB-2 | 333-100568 | 10.5 | 10/16/02 | |||||||
| 10.7 | First Amendment to the Antimicrobial Polypeptide License Agreement dated September 15, 2000 | SB-2 | 333-100568 | 10.6 | 10/16/02 | |||||||
| 10.8 | Second Amendment to the Antimicrobial Polypeptide License Agreement dated June 10, 2002 | SB-2 | 333-100568 | 10.7 | 10/16/02 | |||||||
| 10.9 | Third Amendment to the Antimicrobial Polypeptide License Agreement dated September 25, 2002 | SB-2 | 333-100568 | 10.4 | 10/16/02 | |||||||
| 10.10 | Fourth Amendment to the Antimicrobial Polypeptide License Agreement dated March 2003 | SB-2/A-3 | 333-100568 | 10.36 | 4/9/03 | |||||||
| 10.11 | Amended and Restated 2002 Stock Option and Incentive Plan (including Form of Stock Option Agreement) | 10-QSB/A | 001-32188 | 10.1 | 9/29/06 | |||||||
| 10.12 | First Amendment to Amended and Restated 2002 Stock Option and Incentive Plan | 8-K | 001-32188 | 4.2 | 4/14/08 | |||||||
| 10.13 | Second Amendment to Amended and Restated 2002 Stock Option and Incentive Plan | 8-K | 001-32188 | 10.1 | 10/30/09 | |||||||
| 10.14 | Proprietary Information and Invention Agreement between the Company and Jeffrey D. Hillman | SB-2 | 333-100568 | 99.4 | 10/16/02 | |||||||
| 10.15 | Lease Agreement between the Company and Hawley-Wiggins LLC dated January 28, 2004; Subordination Agreement dated April 14, 2004; and First Amendment dated November 15, 2004 | 10-KSB | 001-32188 | 10.46 | 3/14/05 | |||||||
| 10.16 | Sublease Agreement between the Company and Astrazenca LP dated October 12, 2009 (3000 Bayport Drive, Suite 685, Tampa, FL 33607) | 10-K | 001-32188 | 10.17 | 3/31/10 | |||||||
| 10.17 | Lease Agreement between the Company and Hawley-Wiggins LLC dated October 23, 2009 (13700 Progress Blvd, Alachua, FL 32615) | 10-K | 001-32188 | 10.18 | 3/31/10 | |||||||
Table of Contents
| Incorporated by reference to | ||||||||||||
| Exhibit number |
Exhibit description |
Form |
File no. |
Exhibit |
Filing date |
Filed herewith | ||||||
| 10.18 | Common Stock Purchase Agreement dated December 30, 2009 | 10-K | 001-32188 | 10.19 | 3/3/10 | |||||||
| 10.19† | Executive Employment Agreement between the Company and Jeffrey D. Hillman, dated May 11, 2010 | 10-Q | 001-32188 | 10.2 | 5/14/10 | |||||||
| 10.20† | Executive Employment Agreement between the Company and Brian J. Bohunicky, dated May 10, 2010 | 10-Q | 001-32188 | 10.3 | 5/14/10 | |||||||
| 10.21 | Unsecured Promissory Note with Conversion Provisions issued to the Koski Family Limited Partnership, dated May 28, 2010 | 8-K | 001-32188 | 1.01 | 5/28/10 | |||||||
| 10.22 | Common Stock Purchase Agreement dated July 5, 2010, by and between Oragenics, Inc. and the Koski Family Limited Partnership | 8-K | 001-32188 | 10.1 | 7/7/10 | |||||||
| 10.23 | Revolving Credit Agreement dated July 30, 2010, by and between Oragenics, Inc. and the Koski Family Limited Partnership (including form of revolving unsecured promissory note). | 8-K | 001-32188 | 10.2 | 8/2/10 | |||||||
| 10.24 | Revolving Unsecured Promissory Note dated September 13, 2010 | 8-K | 001-32188 | 10.2 | 9/16/10 | |||||||
| 10.25 | Revolving Unsecured Promissory Note dated November 8, 2010 | 10-Q | 001-32188 | 10.4 | 11/12/10 | |||||||
| 10.26 | First Amendment to the Revolving Credit Agreement | 8-K | 001-32188 | 10.2 | 01/28/11 | |||||||
| 10.27 | Revolving Unsecured Promissory Note dated January 24, 2011 | 8-K | 001-32188 | 10.3 | 01/28/11 | |||||||
| 10.28 | Second Amendment to the Revolving Credit Agreement | 8-K | 001-32188 | 10.1 | 02/08/11 | |||||||
| 10.29 | Revolving Unsecured Promissory Note dated March 15, 2011 | 8-K | 001-32188 | 10.1 | 03/15/11 | |||||||
| 10.30 | Form of restricted stock agreement | 8-K | 001-32188 | 10.5 | 03/15/11 | |||||||
| 16.1 | Letter from Kirkland Russ, Murphy & Tapp P.A. to the U.S. Securities and Exchange Commission dated March 11, 2011 | 8-K | 001-32188 | 16.1 | 03/15/11 | |||||||
| 23.1 | Consent of Mayer Hoffman McCann P.C., an independent public accounting firm | X | ||||||||||
| 23.2 | Consent of Kirkland, Russ, Murphy & Tapp P.A., an independent public accounting firm | X | ||||||||||
| 24.1 | Powers of Attorney (included on signature page). | |||||||||||
| 31.1 | Certification of Principal Executive Officer pursuant to Rule 13a-14 and Rule 15d-14(a), promulgated under the Securities and Exchange Act of 1934, as amended. | X | ||||||||||
| 31.2 | Certification of Principal Financial Officer pursuant to Rule 13a-14 and Rule 15d-14(a), promulgated under the Securities and Exchange Act of 1934, as amended. | X | ||||||||||
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (Chief Executive Officer). | X | ||||||||||
| 32.2 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (Chief Financial Officer). | X | ||||||||||