Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 000-29089
Agenus Inc.
(exact name of registrant as specified in its charter)
| Delaware | 06-1562417 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
3 Forbes Road, Lexington, Massachusetts 02421
(Address of principal executive offices, including zip code)
Registrant’s telephone number, including area code:
(781) 674-4400
Securities registered pursuant to Section 12(b) of the Act:
| Common Stock, $.01 Par Value | The NASDAQ Capital Market | |
| (Title of each class) | (Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulations S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer þ Non-accelerated filer ¨ Smaller reporting company ¨
(Do not check if a smaller
reporting company)
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of voting stock held by non-affiliates of the registrant as of June 30, 2010 was: $67.8 million. There were 112,653,700 shares of the registrant’s Common Stock outstanding as of March 1, 2011.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for the registrant’s 2011 Annual Meeting of Stockholders, which definitive proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year end of December 31, 2010, are incorporated by reference into Part III of this Annual Report on Form 10-K.
Table of Contents
| Page | ||||||
| PART I | ||||||
| ITEM 1. |
3 | |||||
| 3 | ||||||
| 4 | ||||||
| 10 | ||||||
| 11 | ||||||
| 13 | ||||||
| 13 | ||||||
| 13 | ||||||
| 14 | ||||||
| ITEM 1A. |
14 | |||||
| ITEM 1B. |
30 | |||||
| ITEM 2. |
30 | |||||
| ITEM 3. |
30 | |||||
| ITEM 4. |
31 | |||||
| 31 | ||||||
| PART II | ||||||
| ITEM 5. |
33 | |||||
| ITEM 6. |
35 | |||||
| ITEM 7. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
37 | ||||
| ITEM 7A. |
48 | |||||
| ITEM 8. |
49 | |||||
| ITEM 9. |
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
80 | ||||
| ITEM 9A. |
80 | |||||
| ITEM 9B. |
82 | |||||
| PART III | ||||||
| ITEM 10. |
82 | |||||
| ITEM 11. |
82 | |||||
| ITEM 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
82 | ||||
| ITEM 13. |
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
82 | ||||
| ITEM 14. |
82 | |||||
| PART IV | ||||||
| ITEM 15. |
83 | |||||
Table of Contents
Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K and other written and oral statements the Company makes from time to time contain certain “forward-looking” statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. You can identify these forward-looking statements by the fact they use words such as “could,” “expect,” “anticipate,” “estimate,” “target,” “may,” “project,” “guidance,” “intend,” “plan,” “believe,” “will,” “potential,” “opportunity,” “future” and other words and terms of similar meaning and expression in connection with any discussion of future operating or financial performance. One can also identify forward-looking statements by the fact that they do not relate strictly to historical or current facts. Such forward-looking statements are based on current expectations and involve inherent risks and uncertainties, including factors that could delay, divert or change any of them, and could cause actual outcomes to differ materially from current expectations. These statements are likely to relate to, among other things, our business strategy, our future research and development, our product development efforts, our ability to commercialize our product candidates, our sales and marketing activities in Russia, our prospects for initiating partnerships or collaborations, the timing of the introduction of our products, the effect of new accounting pronouncements, uncertainty regarding our future operating results and our profitability, anticipated sources of funds as well as our plans, objectives, expectations, and intentions. The Company has included important factors in the cautionary statements included in this Annual Report, particularly under “Item 1A. Risk Factors,” that the Company believes could cause actual results to differ materially from any forward-looking statement.
Although the Company believes it has been prudent in its plans and assumptions, no assurance can be given that any goal or plan set forth in forward-looking statements can be achieved and readers are cautioned not to place undue reliance on such statements, which speak only as of the date made. The Company undertakes no obligation to release publicly any revisions to forward-looking statements as a result of new information, future events or otherwise.
We have included more detailed descriptions of these risks and uncertainties and other risks and uncertainties applicable to our business in Item 1A. “Risk Factors” of this Annual Report on Form 10-K. We encourage you to read those descriptions carefully. We caution investors not to place significant reliance on forward-looking statements contained in this document; such statements need to be evaluated in light of all the information contained in this document. Furthermore, the statements speak only as of the date of this document, and we undertake no obligation to update or revise these statements.
Oncophage® and Stimulon® are registered trademarks of Agenus Inc. and its subsidiaries. All rights reserved.
2
Table of Contents
PART I
| Item 1. | Business |
Overview
Agenus Inc., including its subsidiaries, referred to in this Annual Report on Form 10-K as “Agenus,” the “Company,” “we,” “us,” and “our,” is a biotechnology company focused on the development and commercialization of technologies to treat cancers and infectious diseases, primarily based on immunological approaches. As of January 6, 2011, we changed our name from Antigenics Inc. to Agenus Inc. to more accurately reflect our existing product pipeline, which has expanded over the years beyond antigen-based vaccines, as well as to highlight our business strategy as we seek partnering opportunities to grow and diversify our business. In conjunction with this name change, our autologous cancer immunotherapies have been named the Prophage Series of cancer vaccines (vitespen; HSPPC-96). The name Oncophage® vaccine will be retained in the adjuvant renal cell carcinoma indication as part of the Prophage Series. AG-707 was renamed HerpV.
Some of our key assets are highlighted below:
| • | The Prophage Series of cancer vaccines: The Prophage Series of cancer vaccines is based on our core heat shock protein technology. We believe that the collective results from our clinical trials to date indicate a favorable safety profile and signals of efficacy in multiple cancer types. In a registry following patients from a large randomized Phase 3 trial in non-metastatic renal cell carcinoma (RCC; kidney cancer), patients at intermediate risk of recurrence who were in the treatment arm demonstrated an approximately 46 percent lower risk of death compared with those in the control arm (n = 362; P < 0.05; hazard ratio = 0.54). This product is approved for sale in this indication in Russia. Phase 2 trials are underway testing the Prophage Series vaccines G-100 and G-200 in newly diagnosed and recurrent glioma, respectively. Although promising results have been observed to date there can be no assurances that we will successfully complete all clinical trials or obtain regulatory approvals for these products. Additional trials are under evaluation in metastatic RCC and metastatic melanoma in combination with potentially synergistic therapies, as well as in pediatric neurological tumors. |
| • | QS-21 Stimulon® adjuvant (“QS-21”): QS-21 is an adjuvant, or a substance added to a vaccine or other immunotherapy that is intended to enhance immune response. The key licensees of QS-21 are GlaxoSmithKline (GSK) and JANSSEN Alzheimer Immunotherapy. There are 14 vaccines containing QS-21 in clinical development, including four in Phase 3 testing for malaria, melanoma, non-small cell lung cancer and shingles. The first products containing QS-21 are expected to be launched in the 2013-2014 timeframe, and we are entitled to royalties for at least 10 years post-launch. However, there is no guarantee that we will be able to collect royalties in the future. The pipeline of product candidates containing QS-21 is extraordinarily diverse, encompassing prophylactic as well as therapeutic vaccines for infectious diseases, multiple cancer types, and Alzheimer’s disease. We do not incur clinical development costs for these products and are generally reimbursed for any related expenses by our licensees. |
| • | HerpV: HerpV is a therapeutic vaccine for the treatment of genital herpes, which is based on our HSP technology. It has completed Phase 1 testing, where it was shown to elicit both CD4 and CD8 positive T cell responses—a first of its kind finding in genital herpes treatment. Because the product contains multiple antigens derived from the herpes simplex 2 virus (HSV-2), it may be applicable to a broader patient population and may have potential in managing outbreaks and disease transmission. We consider this to be a platform technology, since with the integration of heat shock proteins with antigenic peptides we could potentially create therapeutic vaccines for many infectious diseases. We are currently seeking partners to advance HerpV and the platform technology into further development. |
3
Table of Contents
In addition to our internal development efforts, we are actively pursuing multiple partnering opportunities. We are seeking regional and/or global partners for select products in our portfolio, including Oncophage, the Prophage G-Series vaccines, G-100 and G-200, and HerpV. We are also exploring a variety of in-licensing opportunities that would be complementary to our existing business while expanding our product pipeline. Our business activities have included product research and development, intellectual property prosecution, manufacturing, regulatory and clinical affairs, corporate finance and development, market development, business development, and support of our collaborations. Research and development expenses for the years ended December 31, 2010, 2009, and 2008, were $12.9 million, $16.9 million, and $20.7 million, respectively.
On March 3, 2011, we were notified by the Listing Qualifications Staff of Nasdaq indicating that we are not in compliance with the Nasdaq Marketplace Rule 5550(a)(2) (the “Bid Price Requirement”) because the bid price for our common stock had closed below the minimum $1.00 per share requirement for 30 consecutive business days. In accordance with Nasdaq Marketplace Rule 5810(c)(3)(A), we have been provided 180 calendar days, or until August 30, 2011, to regain compliance with the Bid Price Requirement.
Our Products and Technologies Under Development
Heat Shock Protein Technology
Heat shock proteins, also known as HSPs, are also called stress proteins, as their expression is increased when cells experience various stresses like extremes of temperature (hot or cold) and oxygen deprivation. HSPs are present in all cells in all life forms from bacteria to mammals, and their structure and function are similar across these diverse life forms. Under normal conditions, HSPs play a major role in protein folding and transport of protein fragments called peptides within a cell, and are thus also known as “chaperones.” Antigenic peptides, those portions of a protein that stimulate immune responses when recognized by the immune cells, are also transported by these chaperones. Because HSPs interact with and bind many cellular proteins and peptides, they chaperone a broad array of antigenic peptides to facilitate their recognition by the immune system. Thus, HSPs play an integral role in capturing and presenting the antigenic “fingerprint” of a cell to a host’s immune system.
Although HSPs are normally found inside cells, they also provide important danger signals when found outside of cells. Detection of HSPs outside of cells is indicative that cell death has occurred. This may have been caused by disease, mutation, or injury, whereby a cell’s contents are spilled into body tissue. These HSPs send powerful “danger signals” to the immune system that initiate a cascade of events capable of generating a targeted immune response against the infection or disease-related cell death.
Combined, these functions of HSPs form the basis of our technology. The “chaperoning” nature of HSPs allows us to produce vaccines containing the antigenic fingerprint of a given disease. In the case of cancer, the vaccines are patient-specific, consisting of heat shock protein-peptide complexes, also known as HSPPCs, purified from a patient’s tumor cells. These HSPPCs, when injected into the skin, are expected to stimulate a powerful cellular immune response potentially capable of targeting and killing the cancer cells from which these complexes were derived. Because cancer is a highly variable disease from one patient to another, due to rapid mutation of cancer cells, we believe that a patient-specific vaccination approach is required to generate a more robust and targeted immune response against the disease.
For certain diseases, such as genital herpes, we do not believe that a personalized vaccination approach is required, since the pathogen does not vary as greatly from patient to patient as do cancer cells. For example, in our HerpV product candidate for the treatment of genital herpes, we complex, or bind, several defined antigenic herpes peptides to an HSP (Hsc70) that we genetically engineer, creating an HSPPC. This HSPPC, when injected into the skin, is designed to elicit a cellular immune response to the synthetic peptides carried by the HSP.
The Prophage Series of Cancer Vaccines
The Prophage Series of cancer vaccines describes our portfolio of patient-specific HSP-based therapeutic cancer vaccines, including the R-Series candidates in RCC, M-Series candidates in melanoma, G-Series
4
Table of Contents
candidates in glioma, and NP-Series candidate in pediatric neurological tumors. The first product derived from the R-Series (R-100, registered in Russia as Oncophage), represents the only approved treatment for adjuvant or non-metastatic kidney cancer patients at intermediate risk for disease recurrence. In 2008, we submitted a marketing authorization application (“MAA”) to the European Medicines Agency (“EMA”) requesting approval for Oncophage in earlier-stage, localized kidney cancer under the conditional authorization provision. After its review, the Committee for Medicinal Products for Human Use (“CHMP”) of the EMA adopted a negative opinion on our application and subsequently we withdrew our application. In a registry following patients from our large randomized Phase 3 trial in non-metastatic RCC, patients at intermediate risk of recurrence who were in the treatment arm demonstrated an approximately 46 percent lower risk of death compared with those in the control arm (n = 362; P < 0.05; hazard ratio = 0.54). Phase 2 trials testing the Prophage Series candidates G-100 and G-200 are underway in both newly diagnosed and recurrent glioma, respectively, where promising data has been generated to date. Additional trials are planned in metastatic RCC (R-200) and metastatic melanoma (M-200) in combination with potentially synergistic therapies, as well as in pediatric neurological tumors (NP-150).
Each Prophage Series vaccine candidate is made from a patient’s tumor tissue. After a surgeon removes a patient’s tumor, the majority of that tumor tissue is frozen and shipped to our manufacturing facility. Using a proprietary manufacturing process that takes approximately eight to 10 hours per individual patient lot, we isolate the HSPPCs from the tumor tissue. Through this isolation process, the HSPPCs are extracted, purified, and sterile-filtered from the tumor tissue, then formulated in solution and packaged in standard single-injection vials. After the performance of quality control testing, including sterility testing, we ship the frozen vaccine back to the hospital or clinic for administration. Medical professionals administer the vaccine by injecting the product into the skin.
Although we believe that our technology is applicable to all cancer types, our initial focus with the Prophage Series vaccines is on cancers that have limited or no available treatment options and in cancers that typically yield sufficient quantities of tumor tissue from the surgical procedure to allow for manufacture.
Since our first patient was enrolled in a clinical trial studying a Prophage Series vaccine in 1997, we have treated more than 850 cancer patients in our clinical trials. Because our vaccines are derived from the patient’s own tumor, they are unlike the majority of approved therapies and as such, they may experience a long regulatory review process and high development costs, either of which could delay or prevent our commercialization efforts. For additional information regarding regulatory risks and uncertainties, please read the risks identified under “Risk Factors.”
We believe that the collective results from our clinical trials thus far show that the vaccine candidates that have been clinically evaluated have a favorable safety profile. We also believe that available results from clinical trials suggest that treatment with the Prophage Series vaccines can generate immunological and anti-tumor responses.
Phase 3 Renal Cell Carcinoma Program
Renal cell carcinoma is the most common type of kidney cancer. The American Cancer Society estimated that there would be 58,240 new cases of kidney cancer and 13,040 people would die from the disease in the United States in 2010. The Kidney Cancer Research Bureau, a Russian non-profit, non-government research organization, estimated that in 2008, approximately 16,000 Russians would be diagnosed with kidney cancer and approximately 50% of those diagnosed would die of the disease.
We initiated a Phase 3, multicenter, international trial for non-metastatic RCC into which the first patient was randomized in February 2001. As announced on March 24, 2006, the trial did not reach statistical significance in its primary endpoint of recurrence-free survival in the total patient population, though a positive trend was observed. During the protocol design process in 1999 and 2000, key opinion leaders were consulted,
5
Table of Contents
and the non-metastatic RCC patient population designated for enrollment in the trial was thought to be a relatively uniform group. In 2006, the Eastern Cooperative Oncology Group (“ECOG”) initiated a trial in adjuvant RCC with sorafenib and sunitinib that stratified their patient population into intermediate-risk, high-risk, and very high-risk recurrence categories. Using these ECOG defined criteria, analysis of the intermediate risk patients (362 of the 604 eligible patients in the trial) in the trial showed a statistically significant difference in recurrence-free survival in favor of the Oncophage arm. In part because the intermediate-risk category was not prospectively delineated prior to the trial’s initiation, the Food & Drug Administration (“FDA”) has indicated that, by itself, part I of our Phase 3 clinical trial in renal cell carcinoma is not sufficient to support a biologics license application (“BLA”) filing.
We opened a subsequent protocol that continued to follow patients from this trial in the format of a registry in order to collect overall survival information, as well as investigator reports of disease recurrence. The registry, which is expected to provide additional data on the effectiveness of Oncophage, followed patients until March 2010, an additional three years from closure of the initial trial, providing more than five years of data collection following the enrollment of the last patient in the trial. Final analysis of this data is in process. At the 2009 American Society of Clinical Oncology (“ASCO”) annual meeting, we announced results of an interim analysis from the ongoing global patient survival registry, which showed that patients with kidney cancer at intermediate risk of disease recurrence demonstrated an approximately 46 percent lower risk of death when treated with Oncophage cancer vaccine after surgery compared with no treatment (n = 362; P < 0.05; hazard ratio = 0.54).
In addition to the patient registry, patient enrollment has commenced into a small study in non-metastatic RCC to assess immune response in the intermediate-risk patient population. The results of this study, continued data collection through the survival registry, and ongoing analysis are uncertain, and may not positively affect the acceptability of the overall results of the trial and, even if clinically meaningful, may not meet the requirements of the FDA or other regulatory authorities for submission and approval of a marketing application or similar applications for product approval outside the United States.
In April 2008, the Russian Ministry of Public Health issued a registration certificate for the use of Oncophage for the treatment of kidney cancer patients at intermediate risk for disease recurrence. Because, among other things, we have limited resources and minimal sales and marketing experience, commercialization of Oncophage has been slow, and only modest sales of Oncophage in Russia have occurred during the year ended December 31, 2010. The Russian registration was our first product approval from a regulatory authority, and the first approval of a patient-specific therapeutic cancer vaccine in a major market. Since this approval we have been focusing our efforts in Russia on securing one or more distribution and/or partnering arrangements and related commercialization activities. The amount of any future revenue generated from the sale of Oncophage in Russia will depend on our ability to successfully execute on these efforts and identify and obtain adequate reimbursement, as well as decisions of physicians and patients, among other factors. Furthermore, we may experience significant delays in the receipt of payment for Oncophage, or an inability to collect payments at all.
In October 2008, we announced the submission of a MAA to the EMA requesting conditional authorization of Oncophage in earlier-stage, localized kidney cancer. On November 20, 2009, we announced that the CHMP of the EMA formally adopted a negative opinion on this MAA. Subsequently we withdrew our application and have been evaluating a variety of options to potentially bring Oncophage to patients and physicians globally. Although we are no longer in active discussions with a potential partner for the European market, we continue to actively seek partnership discussions for multiple products generated from our portfolio of Prophage Series vaccines.
Glioma
Glioma is a cancer affecting the central nervous system that begins in glial cells (connective tissue cells that surround and support nerve cells). Malignant glioma is currently a fatal disease. The American Cancer Society estimated that 22,020 new cases of the brain and other nervous system cancers would be diagnosed during 2010 in the U.S., and that about 13,140 people would die from these tumors.
6
Table of Contents
A Phase 2 clinical trial with Prophage Series G-200 in recurrent, high-grade glioma is currently ongoing. This study is being led by the Brain Tumor Research Center at the University of California, San Francisco (“UCSF”), with grants from the American Brain Tumor Association and the National Cancer Institute Special Programs of Research Excellence. Phase 1 results, presented at the Society for Neuro-Oncology Annual Meeting Conference in November 2008, showed that vaccination following brain cancer surgery increased overall median survival to approximately 10.5 months, with four patients surviving beyond 12 months and one patient surviving almost 2.5 years. The study also showed that all 12 treated patients demonstrated a significant immune response after vaccination with G-200 ( P < 0.001) and that patients with minimal residual disease at time of first vaccination (n = 7) were more likely to survive beyond nine months compared with patients with significant residual disease.
The study, which is designed to enroll approximately 50 patients, has expanded to include New York-Presbyterian Hospital/Columbia University Medical Center and University Hospitals/Case Western Reserve. Interim data was presented at the Society for Neuro-Oncology meeting in October 2009, which showed a median survival of 10.1 months in the first 20 patients treated with G-200, and that to date six patients (30 percent) had survived at or beyond 12 months. This early data shows an improvement in overall survival over the previous long-standing historical median survival of 6.5 months, and is also slightly favorable to the recently reported median survival of 9.2 months with Avastin® (bevacizumab) in patients with recurrent high-grade glioma. In May 2010, data presented at the International Conference on Brain Tumor Research and Therapy suggested that vaccination with this candidate may improve overall survival in patients with recurrent high-grade glioma. An overall median survival of 44 weeks after tumor resection was observed. Approximately 70% of the evaluable patients survived beyond 36 weeks, and 41% survived up to or longer than one year. Additional data from this trial will be reported by mid-2011. UCSF also initiated an additional Phase 2 clinical trial in newly diagnosed glioma testing Prophage Series G-100 in combination with Temodar® (temozolomide). This trial is currently enrolling, with a target of 50 patients. Based on promising trends to date, this trial is being expanded to include up to 10 clinical sites.
Other Clinical Trials
Initial clinical trials of Prophage Series vaccines were aimed at assessing feasibility, safety and preliminary efficacy; select studies measured immune response. A series of small, single-arm trials were performed in various solid tumor types, including RCC, melanoma, colorectal cancer, gastric cancer, pancreatic cancer, and non-small cell lung cancer. A single Phase 1 trial was conducted in non-Hodgkin’s lymphoma, and a non-registrational Phase 3 trial was conducted in metastatic melanoma.
Collectively, results across all trials provided evidence of manufacturing and logistical feasibility as well as an initial demonstration of safety and signals of efficacy, which included patients who had complete disappearance (a complete response), substantial shrinkage (partial response), minor shrinkage (minor response), or no change in the size (disease stabilization) of tumor lesions. Median overall survival results exceeded historical controls that were relevant at the time when the studies were performed. Additionally, tumor-specific T-cell responses were noted in studies where they were measured: melanoma and colorectal cancer. In the Phase 3 metastatic melanoma trial, earlier-stage patients who received at least 10 doses of the vaccine showed a survival benefit over patients in the control arm.
Manufacturing
Commercial and clinical supplies of Oncophage and other vaccine candidates deriving from the Prophage Series are manufactured in our Lexington, Massachusetts facility. We estimate that the facility’s current capacity for these products is approximately 10,000 patient courses per year, expandable to approximately 200,000 patient courses per year, by building-out currently available space, adding second and third shifts, and automating various functions. On average, it takes eight to 10 hours of direct processing time to manufacture a patient batch of vaccine.
7
Table of Contents
After manufacturing, Prophage Series vaccines are tested and released by our quality systems staff. The quality control organization performs a series of release assays designed to ensure that the product meets all applicable specifications. Our quality assurance staff also reviews manufacturing and quality control records prior to batch release in an effort to assure conformance with current Good Manufacturing Practices, also known as cGMP, as mandated by the FDA and foreign regulatory agencies.
Our manufacturing staff is rigorously trained and routinely evaluated for conformance to manufacturing procedures and quality standards. This oversight is intended to ensure compliance with FDA and foreign regulations and to provide consistent vaccine output. Our quality control and quality assurance staff is similarly trained and evaluated as part of our effort to ensure consistency in the testing and release of the product, as well as consistency in materials, equipment, and facilities.
Preclinical Activities
We continue with product characterization efforts to better define the complex structure of the Prophage Series vaccines. These efforts are made more challenging by the autologous nature of the products. In addition, we are developing methods that will assess the intensity of immunological responses following vaccination with these vaccines. We expect to continue these efforts during 2011. In addition, we are currently planning to study the Prophage Series vaccine candidates in combination with potentially synergistic therapies in later-stage cancers.
QS-21
QS-21 Stimulon® adjuvant is an adjuvant, or a substance added to a vaccine or other immunotherapy that is intended to enhance immune response. The key licensees of QS-21 are GSK and JANSSEN Alzheimer Immunotherapy. There are 14 vaccines containing QS-21 in clinical development, including four in Phase 3 testing for malaria, melanoma, non-small cell lung cancer and shingles. The first products containing QS-21 are expected to be launched in the 2013-2014 timeframe, and we are entitled to royalties for at least 10 years post-launch. The pipeline of product candidates containing QS-21 is diverse, encompassing prophylactic as well as therapeutic vaccines for infectious diseases, multiple cancer types, and Alzheimer’s disease. The Company does not incur clinical development costs for these products and is generally reimbursed for any related expenses by its licensees.
QS-21 is best known for its ability to stimulate antibody, or humoral, immune response, and has also been shown to activate cellular immunity. A natural product, QS-21 is a triterpene glycoside, or saponin, a natural compound purified from the bark of a South American tree called Quillaja saponaria. It is sufficiently characterized with a known molecular structure, thus distinguishing it from other adjuvant candidates, which are typically emulsions, polymers, or biologicals. QS-21 has been tested in approximately 185 clinical trials involving, in the aggregate, nearly 14,000 subjects in a variety of cancer indications, infectious diseases, and other disorders. These studies have been carried out by academic institutions and pharmaceutical companies in the United States and internationally. A number of these studies have shown QS-21 to be significantly more effective in stimulating antibody responses than aluminum hydroxide or aluminum phosphate, the adjuvants most commonly used in approved vaccines in the United States today.
Partnered QS-21 Programs
A number of pharmaceutical and biotechnology companies have licensed QS-21 from us for use in vaccines to treat a wide variety of human diseases. Companies with QS-21 programs include GSK and JANSSEN Alzheimer Immunotherapy. In return for rights to use QS-21, these companies have generally agreed to pay us license fees, manufacturing payments, milestone payments, and royalties on product sales for a minimum of 10 years after commercial launch. In addition to our corporate licensing arrangements, we have developed a number of academic collaborations to test new vaccine concepts and products containing QS-21. There are 14 vaccines currently in clinical development that contain QS-21.
8
Table of Contents
GSK. In July 2006, we entered into a license agreement and a supply agreement with GSK for the use of QS-21. On January 16, 2009, we entered into an Amended and Restated Manufacturing Technology Transfer and Supply Agreement (the “Amended GSK supply agreement”) under which GSK has the right to manufacture all of its requirements of commercial grade QS-21. GSK is obligated to supply us (or our affiliates, licensees, or customers) certain quantities of commercial grade QS-21 for a stated period of time. To date, we have received $10.5 million of a potential $15.3 million in upfront and milestone payments related to these agreements. We are entitled to receive low single-digit royalties on net sales for a period of at least 10 years after the first commercial sale of a resulting GSK product. The agreements may be terminated by either party upon a material breach if the breach is not cured within the time specified in the agreement. The termination or expiration of the GSK license agreement does not relieve either party from any obligation which accrued prior to the termination or expiration. Among other provisions, the milestone payment obligations survive termination or expiration for any reason, and the license rights granted to GSK survive expiration of the GSK license agreement. The license rights and payment obligations of GSK under the Amended GSK supply agreement survive termination or expiration, except that GSK’s license rights and future royalty obligations do not survive if we terminate due to GSK’s material breach unless we elect otherwise.
We understand that QS-21 is a key component included in several of GSK’s proprietary adjuvant systems and that a number of GSK’s vaccine candidates currently under development are formulated using adjuvant systems containing QS-21. GSK has initiated Phase 3 studies evaluating its investigational MAGE-A3 Antigen-Specific Cancer Immunotherapeutic containing QS-21 in non-small cell lung cancer and melanoma. GSK has also initiated Phase 3 clinical trials in malaria and shingles.
Elan/JANSSEN Alzheimer’s Immunotherapy. Elan Pharmaceuticals, Inc. and/or its affiliates (“Elan”) had a commercial license for the use of QS-21 in the research and commercialization of Elan’s Alzheimer’s disease vaccine candidate that contains QS-21 (“Licensed Product”). Effective September 14, 2009, we entered into an Amended and Restated License Agreement (“Amended License Agreement”) with Elan, and on September 17, 2009, the Amended License Agreement was assigned to JANSSEN Alzheimer Immunotherapy, a subsidiary of Johnson & Johnson. Under the terms of the Amended License Agreement, JANSSEN Alzheimer Immunotherapy has the right to develop, make, have made, use, sell, offer for sale, import, and have sold, the Licensed Product. In addition, JANSSEN Alzheimer Immunotherapy has the right to manufacture all of its requirements of QS-21 for use in the Licensed Product and we have no further supply obligations. Assuming all benchmarks are met under this agreement, we could receive up to $11.5 million in future milestone payments; $1.5 million has been received as of December 31, 2010. Furthermore, under the terms of the Amended License Agreement, we are entitled to receive middle single-digit royalties on net sales of the Licensed Product for a period of at least 10 years after the first commercial sale of such product, if any. Expiration or termination of the Amended License Agreement is without prejudice to any rights that accrued to the benefit of the parties prior to the date of such expiration or termination. Upon expiration of the Amended License Agreement, JANSSEN Alzheimer Immunotherapy will have a royalty-free license. Upon early termination of the Amended License Agreement, JANSSEN Alzheimer Immunotherapy’s license rights terminate and future payment obligations do not accrue.
Manufacturing
Except in the case of GSK and JANSSEN Alzheimer Immunotherapy, we have retained worldwide manufacturing rights for QS-21. We have the right to subcontract manufacturing for QS-21 and we have a supply agreement with a contract manufacturer for the production of QS-21 through September 2012. In addition, under the terms of our agreement with GSK, GSK is contractually committed to supply certain quantities of commercial grade QS-21 to us and our licensees in the future.
HerpV
HerpV is an investigational therapeutic vaccine candidate directed at the virus that causes genital herpes (herpes simplex virus-2, or HSV-2) and is the first potential off-the-shelf application of our HSP technology.
9
Table of Contents
HerpV is a multivalent vaccine containing multiple synthetic HSV-2 peptides, which means that it may be applicable to a broader patient population and may have potential in managing outbreaks and disease transmission.
A 2005-2008 study of the Centers for Disease Control and Prevention estimates 16.2% of people 14 to 49 years of age in the U.S. have HSV-2 infection. The World Health Organization estimated in 2003 that approximately 23.6 million people aged 15 to 49 worldwide are infected each year with HSV-2. Genital herpes is currently treated with palliative topical drugs or antiviral agents that reduce further replication of the virus during the period of treatment.
Based on the results of completed toxicology studies and other preclinical activities, we submitted to the FDA an investigational new drug application (“IND”) for HerpV during the second quarter of 2005. In October 2005, we initiated a multicenter Phase 1 clinical trial of HerpV in genital herpes. In this four-arm, phase 1 study, 35 HSV-2 seropositive patients received HerpV with QS-21, HerpV alone, QS-21 alone, or placebo. The vaccine was well tolerated, with injection site pain as the most common reported adverse event. All patients who were evaluable for immune response and received HerpV with QS-21 showed a statistically significant CD4+ T cell response (100%; 7/7) to HSV-2 antigens as detected by IFNy Elispot, and the majority of those patients demonstrated a CD8+ T cell response (63%; 5/8). This study is the first to demonstrate that HSPs complexed to viral antigens induce an antigen-specific T cell response in humans.
We believe this is a first of its kind finding in genital herpes treatment. We consider HerpV to be part of a platform technology, since with the integration of heat shock proteins with antigenic peptides, we could potentially create therapeutic vaccines for many infectious diseases. We hope to advance HerpV and the platform technology in development through a partnership, and we are actively pursuing licensing discussions.
Intellectual Property Portfolio
We seek to protect our technologies through a combination of patents, trade secrets and know-how and currently have exclusive rights, through outright ownership or through exclusive licenses, to 73 issued United States patents and 117 issued foreign patents. We also have exclusive rights to 4 pending United States patent applications and 29 pending foreign patent applications. While we have patent coverage in Russia for Oncophage, we may not have rights in other territories where we may pursue regulatory approval for Prophage Series vaccine candidates.
Our issued patents include those that cover our core technologies including HSPs for the treatment of cancers and infectious disease, and saponin adjuvants.
The issued patents that cover the Prophage Series vaccines expire at various dates between 2015 and 2024. The issued patents to HerpV expire at various dates between 2014 and 2017. Our patent to purified QS-21 expired in most territories in 2008. Additional protection for QS-21 in combination with other agents is provided by our other issued patents which expire between 2016 and 2019.
Various patents and patent applications have been exclusively licensed to us by the following entities:
Mount Sinai School of Medicine
In November 1994, we entered into a patent license agreement with the Mount Sinai School of Medicine (the “Mount Sinai Agreement”). Through the Mount Sinai Agreement, we obtained an exclusive, worldwide license to patent rights relating to the heat shock protein technology that resulted from the research and development performed by Dr. Pramod Srivastava, our founding scientist and a former member of our Board of Directors. We agreed to pay Mount Sinai a royalty on the net sales of products covered by the licensed patent rights and also provided Mount Sinai with a 0.45% equity interest in the Company (approximately 62,000 shares)
10
Table of Contents
valued at approximately $90,000 at the time of issuance. The term of the Mount Sinai Agreement ends when the last of the licensed patents expires (2018) or becomes no longer valid. If we fail to pay royalties that are due under the agreement, Mount Sinai may issue written notice to us. If we continue to fail to pay royalties after 60 days from receipt of the written notice, Mount Sinai can terminate the agreement. The Mount Sinai Agreement requires us to use due diligence to make the products covered by the licensed patent rights commercially available, including a requirement for us to use best efforts to reach a number of developmental milestones, which have been achieved. If we fail to comply with the due diligence provisions of the agreement, Mount Sinai could take actions to convert our exclusive license to a non-exclusive license after six months written notice. The Mount Sinai Agreement does not contain any milestone payment provisions.
Fordham University
During 1995, Dr. Srivastava moved his research to Fordham University (“Fordham”). We entered into a sponsored research and technology license agreement with Fordham in March 1995 (the “Fordham Agreement”) relating to the continued development of the heat shock protein technology and agreed to make payments to Fordham to sponsor Dr. Srivastava’s research. Through the Fordham Agreement, we obtained an exclusive, perpetual, worldwide license to all of the intellectual property, including all the patent rights, which resulted from the research and development performed by Dr. Srivastava at Fordham. We also agreed to pay Fordham a royalty on the net sales of products covered by the Fordham Agreement through the last expiration date on the patents under the agreement (2018) or when the patents become no longer valid. The agreement does not contain any milestone payment provisions or any diligence provisions. Dr. Srivastava moved his research to the University of Connecticut Health Center (“UConn”) during 1997 and, accordingly, the parts of the agreement related to payments for sponsored research at Fordham terminated in mid-1997. During the term of this agreement, we paid Fordham approximately $2.4 million.
University of Connecticut
In May 2001, we entered into a license agreement with UConn which was amended in March 2003 and June 2009. Through the license agreement, we obtained an exclusive worldwide license to patent rights resulting from inventions discovered under a research agreement that was effective from February 1998 until December 2006. The term of the license agreement ends when the last of the licensed patents expires (2022) or becomes no longer valid. UConn may terminate the agreement: (1) if, after 30 days written notice for breach, we continue to fail to make any payments due under the license agreement, or (2) we cease to carry on our business related to the patent rights or if we initiate or conduct actions in order to declare bankruptcy. We may terminate the agreement upon 90 days written notice. The license agreement contains aggregate milestone payments of approximately $1.2 million for each product we develop covered by the licensed patent rights. These milestone payments are contingent upon regulatory filings, regulatory approvals, and commercial sales of products. We have also agreed to pay UConn a royalty on the net sales of products covered by the license agreement as well as annual license maintenance fees beginning in May 2006. Royalties otherwise due on the net sales of products covered by the license agreement may be credited against the annual license maintenance fee obligations. Under the March 2003 amendment, we agreed to pay UConn an upfront payment and to make future payments for each patent or patent application with respect to which we exercised our option under the research agreement. As of December 31, 2010, we have paid approximately $340,000 to UConn under the license agreement. The license agreement gives us complete discretion over the commercialization of products covered by the licensed patent rights but also requires us to use commercially reasonable diligent efforts to introduce commercial products within and outside the United States. If we fail to meet these diligence requirements, UConn may be able to terminate the license agreement.
Governmental authorities in the United States and other countries extensively regulate the preclinical and clinical testing, manufacturing, labeling, storage, record keeping, advertising, promotion, export, marketing and
11
Table of Contents
distribution, among other things, of our investigational product candidates. In the United States, the FDA under the Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal statutes and regulations, subject pharmaceutical products to rigorous review.
In order to obtain approval of a new product from the FDA, we must, among other requirements, submit proof of safety and efficacy as well as detailed information on the manufacture and composition of the product. In most cases, this proof entails extensive preclinical, clinical, and laboratory tests. Before approving a new drug or marketing application, the FDA may also conduct pre-licensing inspections of the company, its contract research organizations and/or its clinical trial sites to ensure that clinical, safety, quality control, and other regulated activities are compliant with Good Clinical Practices, or GCP, or Good Laboratory Practices, or GLP, for specific non-clinical toxicology studies. The FDA may also require confirmatory trials, post-marketing testing, and extra surveillance to monitor the effects of approved products, or place conditions on any approvals that could restrict the commercial applications of these products. Once approved, the labeling, advertising, promotion, marketing, and distribution of a drug or biologic product must be in compliance with FDA regulatory requirements.
In Phase 1 clinical trials, the sponsor tests the product in a small number of patients or healthy volunteers, primarily for safety at one or more doses. Phase 1 trials in cancer are often conducted with patients who have end-stage or metastatic cancer. In Phase 2, in addition to safety, the sponsor evaluates the efficacy of the product in a patient population somewhat larger than Phase 1 trials. Phase 3 trials typically involve additional testing for safety and clinical efficacy in an expanded population at geographically dispersed test sites. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time.
The sponsor must submit to the FDA the results of preclinical and clinical testing, together with, among other things, detailed information on the manufacture and composition of the product, in the form of a new drug application or, in the case of biologics, like the Prophage Series vaccines, a BLA. In a process that can take a year or more, the FDA reviews this application and, when and if it decides that adequate data is available to show that the new compound is both safe and effective for a particular indication and that other applicable requirements have been met, approves the drug or biologic for marketing.
Whether or not we have obtained FDA approval, we must generally obtain approval of a product by comparable regulatory authorities of international jurisdictions prior to the commencement of marketing the product in those jurisdictions. We are also subject to cGMP, GCP, and GLP compliance obligations, and are subject to inspection by international regulatory authorities. International requirements may in some circumstances be more rigorous than U.S. requirements and may require additional investment in manufacturing process development, non-clinical studies, clinical studies, and record keeping that are not required for U.S. regulatory compliance or approval. The time required to obtain this approval may be longer or shorter than that required for FDA approval and can also require significant resources in time, money, and labor.
Under the laws of the United States, the countries of the European Union, and other nations, we and the institutions where we sponsor research are subject to obligations to ensure the protection of personal information of human subjects participating in our clinical trials. We have instituted procedures that we believe will enable us to comply with these requirements and the contractual requirements of our data sources. The laws and regulations in this area are evolving, and further regulation, if adopted, could affect the timing and the cost of future clinical development activities.
We are also subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other current and potential future federal, state, or local regulations. Our research and development activities involve the controlled use of hazardous materials, chemicals, biological materials, various radioactive compounds, and for some experiments we use recombinant DNA. We believe that our procedures comply with the standards prescribed by local, state, and federal regulations; however, the risk of injury or accidental contamination cannot be completely eliminated. We conduct our activities in compliance with the National Institutes of Health Guidelines for Recombinant DNA Research.
12
Table of Contents
Competition in the pharmaceutical and biotechnology industries is intense. Many pharmaceutical or biotechnology companies have products on the market and are actively engaged in the research and development of products for the treatment of cancer and infectious diseases. In addition, many competitors focus on immunotherapy as a treatment for cancer and infectious diseases. In particular, some of these companies are developing cancer vaccines produced from a patient’s own cells or tissue. Others are focusing on developing heat shock protein products. Prior to regulatory approval, we may compete for access to patients with other products in clinical development, with products approved for use in the indications we are studying, or with off-label use of products in the indications we are studying. In addition, we compete for funding, access to licenses, personnel, and third-party collaborations. Many competitors have substantially greater financial, manufacturing, marketing, sales, distribution, and technical resources, and more experience in research and development, clinical trials, and regulatory matters, than we do. Competing companies developing or acquiring rights to more efficacious therapeutic products for the same diseases we are targeting, or which offer significantly lower costs of treatment, could render our products noncompetitive or obsolete.
Academic institutions, governmental agencies, and other public and private research institutions conduct significant amounts of research in biotechnology, medicinal chemistry, and pharmacology. These entities have become increasingly active in seeking patent protection and licensing revenues for their research results. They also compete with us in recruiting and retaining skilled scientific talent.
We are aware of certain programs and products under development by other companies that may compete with our programs and products. Several of these companies have products that utilize similar technologies and/or patient-specific medicine techniques, such as Dendreon and Accentia, as well as Immuncell-LC, ICT-107, DC-Vax and CDX-110, being developed by Innocell Corp, ImmunoCellular Therapeutics, Northwest Biotherapeutics and Celldex, respectively, for treatment of patients with newly diagnosed glioma.
We are aware of at least one saponin adjuvant which claims to be identical to QS-21. OPT-821 was developed by Optimer Pharmaceuticals and is being used in ongoing cancer vaccine trials. Several other vaccine adjuvants are in development and could compete with QS-21 for inclusion in vaccines in development. These adjuvants include, but are not limited to, oligonucleotides, under development by Pfizer, Idera, Juvaris, and Dynavax, MF59 under development by Novartis, IC31, under development by Intercell, and MPL, under development by GSK. In addition, at least one company, CSL Limited, as well as academic institutions, are developing saponin adjuvants, including derivatives and synthetic formulations.
The existence of products developed by these and other competitors, or other products of which we are not aware or which other companies may develop in the future, may adversely affect the marketability of products we develop.
As of February 25, 2011, we had approximately 56 employees, of whom 7 were Ph.D.s and 2 were MDs. None of our employees are subject to a collective bargaining agreement. We believe that we have good relations with our employees.
Antigenics L.L.C. was formed as a Delaware limited liability company in 1994 and was converted to Antigenics Inc., a Delaware corporation, in February 2000 in conjunction with our initial public offering of common stock. As of January 6, 2011, we changed our name from Antigenics Inc. to Agenus Inc. to more accurately reflect our existing product pipeline, which has expanded over the years beyond antigen-based vaccines, as well as to highlight our business strategy as we seek partnering opportunities to grow and diversify our business.
13
Table of Contents
Availability of Periodic SEC Reports
Our Internet website address is www.agenusbio.com. We make available free of charge through our website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (“Securities Exchange Act”) as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the Securities and Exchange Commission (the “SEC”). The contents of our website are not part of, or incorporated into, this document.
| Item 1A. | Risk Factors |
Our future operating results could differ materially from the results described in this Annual Report on Form 10-K due to the risks and uncertainties described below. We cannot assure investors that our assumptions and expectations will prove to be correct. Important factors could cause our actual results to differ materially from those indicated or implied by forward-looking statements. See “Note Regarding Forward-Looking Statements” on page 2 of this Annual Report on Form 10-K. Factors that could cause or contribute to such differences include those factors discussed below.
Risks Related to our Business
If we incur operating losses for longer than we expect, or we are not able to raise additional capital, we may be unable to continue our operations, or we may become insolvent.
From our inception through December 31, 2010, we have incurred net losses totaling $584.4 million. Our net losses for the years ended December 31, 2010, 2009, and 2008, were $21.9 million, $30.3 million, and $30.8 million, respectively. We expect to incur significant losses over the next several years as we continue research and clinical development of our technologies, apply for regulatory approvals, and pursue partnering opportunities, commercialization, and related activities. Furthermore, our ability to generate cash from operations is dependent on the success of our licensees and collaborative partners, as well as the likelihood and timing of new strategic licensing and partnering relationships and/or successful commercialization of Oncophage and our various product candidates. If we incur operating losses for longer than we expect and/or we are unable to raise additional capital, we may become insolvent and be unable to continue our operations.
On December 31, 2010, we had $19.8 million in cash and cash equivalents. We believe that, based on our current plans and activities, our working capital resources at December 31, 2010, combined with anticipated revenues, and the estimated proceeds from our license, supply, and collaborative agreements, will be sufficient to satisfy our liquidity requirements through 2011. We expect to attempt to raise additional funds in advance of depleting our current funds. For the year ended December 31, 2010, our average monthly cash used in operating activities was $1.2 million. We do not anticipate significant capital expenditures during 2011.
We are required to maintain effective registration statements in connection with certain private placement agreements. If we are unable to keep the registration statements continuously effective in accordance with the terms of the private placement agreements, or do not maintain our listing on Nasdaq or any electronic bulletin board, we are subject to liquidated damages penalties of up to a maximum of 10% of the aggregate purchase price paid by the original investors, or up to $3.8 million.
Since our inception, we have financed our operations primarily through the sale of equity and convertible notes, interest income earned on cash, cash equivalents, and short-term investment balances, and debt provided through secured lines of credit. In order to finance future operations, we will be required to raise additional funds in the capital markets, through arrangements with collaborative partners, or from other sources. During February 2010, we entered into an At the Market Sales Agreement with McNicoll, Lewis & Vlak LLC and Wm Smith & Co (the “Sales Agents”) under which we may sell an aggregate of up to 20 million shares of our common stock from time to time through the Sales Agents. To date we have sold approximately 7.0 million shares of our common stock under this agreement for net proceeds, after expenses, of $8.8 million.
14
Table of Contents
Additional financing may not be available on favorable terms, or at all. If we are unable to raise additional funds when we need them, we will be required to delay, reduce, or eliminate some or all of our development, commercialization and clinical trial programs. We also may be forced to license or sell technologies to others under agreements that allocate to third parties substantial portions of the potential value of these technologies. We may also be unable to continue our operations, or we may become insolvent.
The weakness of the United States economy and the global economy may have a material adverse effect on our liquidity and financial condition, particularly if our ability to raise additional funds is impaired. The ability of potential patients and/or health care payers to pay for our products could also be adversely impacted, thereby limiting our potential revenue. In addition, any negative impacts from any further deterioration in the credit markets and related financial crisis on our collaborative partners could limit potential revenue from our product candidates.
We have significant debt, and we may not be able to make interest or principal payments when due.
At the option of the holders, our 8% senior secured convertible notes due August 2014 (the “2006 Notes”) can be converted into an interest in one of our wholly-owned subsidiaries that holds the rights or patents to QS-21 and HerpV. If converted into an interest of this subsidiary, the ownership interest in the subsidiary will be determined by multiplying the quotient of the conversion amount divided by $25.0 million, by 30%. If the holders elect not to convert into the subsidiary, then at the maturity of the 2006 Notes, we may elect to repay the then outstanding balance, $34.7 million at December 31, 2010, in cash or in common stock, subject to certain limitations. In no event will any of the note holders be obligated to accept equity that would result in them owning in excess of 9.99% of our outstanding common stock at any given time in connection with any conversion, redemption, or repayment of these notes. The 2006 Notes are secured by the equity of the subsidiary that holds the rights or patents to QS-21 and HerpV.
Our ability to satisfy our obligations will depend upon our future performance, which is subject to many factors, including the factors identified in this “Risk Factors” section and other factors beyond our control. If we are not able to generate sufficient cash flow from operations in the future to service our indebtedness, we may be required, among other things, to:
| • | seek additional financing in the debt or equity markets; |
| • | refinance or restructure all or a portion of our indebtedness; |
| • | sell, out-license, or otherwise dispose of assets; and/or |
| • | reduce or delay planned expenditures on research and development and/or commercialization activities. |
Such measures might not be sufficient to enable us to make principal and interest payments. In addition, any such financing, refinancing, or sale of assets might not be available on economically favorable terms, if at all.
To date, we have had negative cash flows from operations. For the years ended December 31, 2010, 2009, and 2008, net cash used in operating activities was $14.8 million, $24.2 million, and $28.9 million, respectively.
Several factors could prevent the successful commercialization of Oncophage in Russia. In addition, we do not expect to generate significant revenue from sales of Oncophage in Russia in the near term.
In April 2008, the Russian Ministry of Public Health issued a registration certificate for the use of Oncophage for the treatment of kidney cancer patients at intermediate risk for disease recurrence and, in September 2008, the FDA granted the necessary permission to allow for the export of Oncophage from the United States to Russia. The Russian registration was our first product approval from a regulatory authority.
15
Table of Contents
Since approval, modest sales have occurred in Russia. Complexities unique to the logistics of this product may delay shipments and limit our ability to move commercial product in an efficient manner without incident. We currently do not have a business presence outside of the United States and rely on third parties to conduct our Oncophage operations in Russia. The reimbursement system in Russia is changing rapidly and has experienced serious funding and administrative problems in its national and regional reimbursement programs. If we are unable to obtain local distribution arrangements including favorable pricing and payment terms, and/or develop appropriate logistical processes for distribution of Oncophage, our commercialization efforts would be adversely affected.
To date we have not been able to secure government reimbursement and there appears to be a limited private-pay market in Russia. If we are unsuccessful in obtaining substantial reimbursement for Oncophage from national or regional funds, we will have to rely on private-pay for the foreseeable future, which may limit or prevent our sales efforts because the ability and willingness of patients to pay is unclear and many patients will not be capable of paying for Oncophage by themselves. Because we have limited resources and minimal sales and marketing experience, successful commercialization of Oncophage may not materialize. Furthermore, we may experience significant delays in the receipt of payment for Oncophage, or an inability to collect payments at all.
If we fail to obtain adequate levels of reimbursement for our product candidates there may be no commercially viable market for these products, or the commercial potential of these products may be significantly limited.
Public and private insurance programs may determine that they will not cover our or our collaborative partners’ product candidates. In Russia, Europe, and other countries outside the United States, government-sponsored health care systems typically pay a substantial share of health care costs, and they may regulate reimbursement levels of products to control costs. If we or our collaborative partners are unsuccessful in obtaining substantial reimbursement for our product candidates from national or regional funds, we will have to rely on private-pay, which may delay or prevent our launch efforts, because the ability and willingness of patients to pay for our products is unclear.
It is possible that there will be substantial delays in obtaining coverage of our product candidates, or the product candidates of our licensees or collaborative partners, if at all, and that, if coverage is obtained, there may be significant restrictions on the circumstances in which there would be reimbursement. We are unable to predict what impact any future regulation or third-party payer initiatives relating to reimbursement will have on our sales.
If we fail to comply with regulatory requirements in the countries in which we conduct our business, if these regulatory requirements change, or if we experience unanticipated regulatory problems, our commercial launch of our Prophage Series product candidates could be prevented or delayed, or our product candidates could be subjected to restrictions, or be withdrawn from the market, or some other action may be taken that may be adverse to our business.
Regulatory authorities generally approve products for particular indications. If an approval is for a limited indication, this limitation reduces the size of the potential market for that product. Product approvals, once granted, are subject to continual review and periodic inspections by regulatory authorities. Later discovery of previously unknown problems or safety issues and/or failure to comply with applicable regulatory requirements can result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal of the government to renew marketing applications, complete withdrawal of a marketing application, and/or criminal prosecution. Such regulatory enforcement could have a direct and negative impact on the product for which approval is granted, but also could have a negative impact on the approval of any pending applications for marketing approval of new drugs or supplements to approved applications.
16
Table of Contents
In addition, our operations and marketing practices are subject to regulation and scrutiny by the United States government, as well as governments of any other countries in which we do business or conduct activities. Because we are a company operating in a highly regulated industry, regulatory authorities could take enforcement action against us in connection with our business and marketing activities for various reasons.
For example, our marketing and sales, labeling, and promotional activities in Russia are subject to local regulations. If we fail to comply with regulations prohibiting the promotion of products for non-approved indications or products for which marketing approval has not been granted, regulatory authorities could bring enforcement actions against us that could inhibit our marketing capabilities, as well as result in penalties. In addition, the United States Foreign Corrupt Practices Act prohibits U.S. companies and their representatives from offering, promising, authorizing, or making payments to foreign officials for the purpose of obtaining or retaining business abroad. Failure to comply with domestic or foreign laws, knowingly or unknowingly, could result in various adverse consequences, including possible delay in approval or refusal to approve a product, recalls, seizures, withdrawal of an approved product from the market, exclusion from government health care programs, imposition of significant fines, injunctions, and/or the imposition of civil or criminal sanctions against us and/or our officers or employees.
From time to time, new legislation is passed into law that could significantly change the statutory provisions governing the approval, manufacturing, and marketing of products regulated by the FDA and other global health authorities. Additionally, regulations and guidance are often revised or reinterpreted by health agencies in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative changes will be enacted, or whether regulations, guidance, or interpretations will change, and what the impact of such changes, if any, may be.
We may not be able to make the Prophage Series of cancer vaccines available in countries other than Russia or in indications other than renal cell carcinoma.
The Prophage Series R-100 is currently only approved for marketing in Russia as Oncophage for the adjuvant treatment of kidney cancer patients at intermediate risk for disease recurrence. The probability and timing of submissions and/or approval in any jurisdiction or indication for this product is uncertain.
In 2008, we submitted a marketing authorization application (“MAA”), to the European Medicines Agency (“EMA”), requesting conditional authorization of Oncophage in earlier-stage, localized kidney cancer. After its review, the Committee for Medicinal Products for Human Use (“CHMP”) of the EMA adopted a negative opinion on our MAA and subsequently we withdrew our application. If we continue to pursue a marketing authorization application for Oncophage with the EMA, there is a high level of uncertainty regarding the probability and timing of a favorable outcome. In addition, even if we continue this pursuit, Oncophage may not achieve approval in Europe because we may not successfully address issues associated with post-hoc analysis, subgroup analysis, lack of immunological data, product characterization, or other issues that may be of concern to the EMA.
The FDA has indicated that our Phase 3 clinical trials of Prophage Series R-100 (Oncophage) and M-200 cannot, by themselves, support biologics license application (“BLA”) filings in the studies’ indications (renal cell carcinoma and metastatic melanoma). The signals and trends observed in the Phase 3 renal cell carcinoma and melanoma trials are based on data analysis of subgroups of patients, some of which were not pre-specified. While the subgroup data might be suggestive of treatment effect, under current regulatory guidelines the results cannot be expected, alone, to support registration or approval in the United States, and our existing data may not support registration or approval in other territories outside of Russia, including in Europe. Due to our lack of resources, our ability to perform additional studies may be limited. Furthermore, studies may take years to complete and may fail to support regulatory filings for many reasons. In addition, our Prophage Series vaccines are a novel class of patient-specific (derived from the patient’s own tumor) oncology therapies, and the FDA and foreign regulatory agencies, including the EMA, which is responsible for product approvals in Europe, and Health
17
Table of Contents
Canada, which is responsible for product approvals in Canada, have relatively little experience in reviewing these types of therapies. Therefore, Prophage Series product candidates may experience a long regulatory review process and high development costs, either of which could delay or prevent our commercialization efforts.
Risks associated with doing business internationally could negatively affect our business.
The Prophage Series vaccine R-100 is currently only approved for sale in Russia as Oncophage. Russia is an evolving market and regulatory, legal, and commercial structures are less predictable than in more mature markets. This unpredictability, as well as potential geopolitical instability in the Russian region, could negatively impact the regulatory and/or commercial environment there, which in turn could have an adverse effect on our business.
In addition, various other risks associated with foreign operations may impact our success. Possible risks include fluctuations in the value of foreign and domestic currencies, disruptions in the import, export, and transportation of patient tumors and our product, the product and service needs of foreign customers, difficulties in building and managing foreign relationships, the performance of our licensees or collaborators, and unexpected regulatory, economic, or political changes in foreign markets.
Our financial position, results of operations, and cash flows can be affected by fluctuations in foreign currency exchange rates, primarily for the euro and the ruble. Movement in foreign currency exchange rates could cause revenue or clinical trial costs to vary significantly in the future and may affect period-to-period comparisons of our operating results. Historically, we have not hedged our exposure to these fluctuations in exchange rates.
Our commercial and international operations experience and resources are limited and need to be developed or acquired. If we fail to do so, our revenues may be limited or nonexistent. In addition, we may be required to incur significant costs and devote significant efforts to augment our existing capabilities.
As we have limited experience with commercial and international operations, it may be difficult to accurately estimate our costs. We currently do not have employees, manufacturing, or business operations facilities outside of the United States and we will rely significantly on consultants, partners, and other third parties to conduct our sales, marketing, and distribution operations. If these third parties are unable to fulfill their obligations this could have a material adverse effect on our commercialization efforts. If in the future we elect to perform sales, marketing, and distribution functions ourselves, we will face a number of additional risks, including the need to recruit experienced marketing and sales personnel, or incur significant expenditures. In addition, we may need to compete with other companies that have more experienced and better-funded operations. Where we have licensed our products to third-party collaborators or licensees, we will be dependent on their commercial operations, sales and marketing expertise and resources, and any revenues we receive from those products will depend primarily on the sales and marketing efforts of others.
Our competitors in the biotechnology and pharmaceutical industries may have superior products, manufacturing capability, selling and marketing expertise and/or financial and other resources.
Our products and the products in development by our collaborative partners may fail because of intense competition from major pharmaceutical companies and specialized biotechnology companies engaged in the development of product candidates directed at cancer, infectious diseases and degenerative disorders. Several of these companies have products that utilize technologies similar to our Prophage Series and/or patient-specific or other vaccine based techniques, such as Dendreon and Accentia, as well as Immuncell-LC, ICT-107, DC-Vax and CDX-110, being developed by Innocell Corp, ImmunoCellular Therapeutics, Northwest Biotherapeutics and Celldex, respectively, for treatment of patients with newly diagnosed glioma.
There is no guarantee that we will be able to compete with potential future products being developed by our competitors. For example, Oncophage may compete with therapies currently in development for non-metastatic
18
Table of Contents
renal cell carcinoma, such as Wilex AG’s Rencarex (WX-G250), which is in Phase 3 clinical trials. Additionally, sorafenib and sunitinib, which are approved for advanced renal cell carcinoma, are being studied in non-metastatic renal cell carcinoma, and other products that have been developed for metastatic renal cell carcinoma, such as temsirolimus, bevacizumab and pazopanib, may also be developed for non-metastatic renal cell carcinoma. As our Prophage Series vaccines are potentially developed in other indications, they will face additional competition in those indications. In addition, for our Prophage Series vaccines, and all of our product candidates, prior to regulatory approval, we may compete for access to patients with other products in clinical development, with products approved for use in the indications we are studying, or with off-label use of products in the indications we are studying. We anticipate that we will face increased competition in the future as new companies enter markets we seek to address and scientific developments surrounding immunotherapy and other traditional cancer therapies continue to accelerate.
Our patent to purified QS-21 expired in most territories in 2008. Additional protection for our QS-21 proprietary adjuvant in combination with other agents is provided by our other patents. Our license and manufacturing agreements for QS-21 typically provide royalties for at least 10 years after commercial launch independent of patent expiry. However, there is no guarantee that we will be able to collect royalties in the future.
We are aware of compounds that claim to be identical to QS-21 that are being used in clinical trials. Several other vaccine adjuvants are in development and could compete with QS-21 for inclusion in vaccines in development. These adjuvants include, but are not limited to, oligonucleotides, under development by Pfizer, Idera, Juvaris, and Dynavax, MF59 under development by Novartis, IC31, under development by Intercell, and MPL, under development by GSK. In addition, at least one company, CSL Limited, as well as academic institutions, are developing saponin adjuvants, including derivatives and synthetic formulations.
Many of our competitors, including large pharmaceutical companies, have greater financial and human resources and more experience than we do. Our competitors may:
| • | commercialize their product candidates sooner than we commercialize our own; |
| • | develop safer or more effective therapeutic drugs or preventive vaccines and other therapeutic products; |
| • | implement more effective approaches to sales and marketing and capture some of our potential market share; |
| • | establish superior intellectual property positions; |
| • | discover technologies that may result in medical insights or breakthroughs, which render our drugs or vaccines obsolete, possibly before they generate any revenue; or |
| • | adversely affect our ability to recruit patients for our clinical trials. |
Manufacturing problems may cause product launch delays, unanticipated costs, or loss of revenue streams.
If the demand for our Prophage Series vaccines is substantially greater than we anticipate, our capacity may not be able to meet product demand. In addition, higher manufacturing loads may result in higher manufacturing failure rates as the operation becomes more complex. We currently manufacture our Prophage Series vaccines in our Lexington, Massachusetts facility and we intend to continue using this facility to satisfy all demands for product. While we believe we will be able to cover all demands in the near term, there is no guarantee that we will be able to meet all future or unanticipated increases in demand, and a failure to do so could adversely affect our business. Such demand may also limit our ability to manufacture product in support of clinical trials, and this could cause a delay or failure in our Prophage Series programs. Manufacturing of Prophage Series vaccines is complex, and various factors could cause delays or an inability to supply vaccine. Deviations in the processes controlling manufacture could result in production failures.
19
Table of Contents
We can also manufacture other clinical products in our own manufacturing facility. Our manufacturing facility has support areas that it shares with the Prophage Series manufacturing areas. As we seek to make Prophage Series vaccines available in other territories, the applicable regulatory bodies may require us to make our manufacturing facility a single product facility. In such an instance, we would no longer have the ability to manufacture products such as HerpV in our current facility. In order to prepare additional HerpV to support future clinical trials, we would then have to manufacture or have manufactured this product in an appropriate alternative facility.
Currently, we do not manufacture QS-21 in our own manufacturing facility, and we have given two QS-21 licensees who have the most advanced QS-21 programs the right to manufacture QS-21 themselves or through third-party manufacturers. If these licensees are unable to successfully manufacture or have manufactured QS-21, the commercialization of the product candidates being developed by such licensees could be delayed or prevented, and we could lose important potential future revenue streams. We currently outsource the manufacture of QS-21 under an agreement that expires in 2012. If we are not able to renew this agreement we may have to identify an alternative manufacturing source or the investment of substantial funds would be required to develop our own manufacturing facility. We or our currently contracted suppliers may never have the ability to manufacture commercial grade QS-21.
We currently rely upon and expect to continue to rely upon third parties, potentially including our collaborators or licensees, to produce materials required to support our product candidates, preclinical studies, clinical trials, and commercial efforts. A number of factors could cause production interruptions at our manufacturing facility or at our contract manufacturers or suppliers, including equipment malfunctions, labor or employment retention problems, natural disasters, power outages, terrorist activities, or disruptions in the operations of our suppliers. Alternatively, there is the possibility we may have excess manufacturing capacity if product candidates do not progress as planned.
There are a limited number of contract manufacturers or suppliers that are capable of manufacturing our product candidates or the materials used in their manufacture. If we are unable to do so ourselves or to arrange for third-party manufacturing or supply of these product candidates or materials, or to do so on commercially reasonable terms, we may not be able to complete development of these product candidates or commercialize them ourselves or through our collaborative partners or licensees. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured products ourselves, including reliance on the third party for regulatory compliance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control, and the possibility of termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
Manufacturing is also subject to extensive government regulation. Regulatory authorities must approve the facilities in which human health care products are produced. In addition, facilities are subject to ongoing inspections, and minor changes in manufacturing processes may require additional regulatory approvals, either of which could cause us to incur significant additional costs and lose revenue.
The drug development and approval process is uncertain, time-consuming, and expensive.
Clinical development, including preclinical testing and the process of obtaining and maintaining regulatory approvals for new therapeutic products, is lengthy, expensive, and uncertain. It also can vary substantially based on the type, complexity, and novelty of the product. We must provide regulatory authorities with manufacturing, product characterization, and preclinical and clinical data demonstrating that our product candidates are safe and effective before they can be approved for commercial sale. It may take us many years to complete our testing, and failure can occur at any stage of testing. Interim results of preclinical studies or clinical trials do not necessarily predict their final results, and acceptable results in early studies might not be seen in later studies. Any preclinical or clinical test may fail to produce results satisfactory to regulatory authorities for many reasons, including but not limited to insufficient product characterization, poor study structure conduct or statistical
20
Table of Contents
analysis planning, failure to enroll a sufficient number of patients or failure to prospectively identify the most appropriate patient eligibility criteria, and collectability of data. Preclinical and clinical data can be interpreted in different ways, which could delay, limit, or prevent regulatory approval. Negative or inconclusive results from a preclinical study or clinical trial, adverse medical events during a clinical trial, or safety issues resulting from products of the same class of drug could require a preclinical study or clinical trial to be repeated or cause a program to be terminated, even if other studies or trials relating to the program are successful. As of December 31, 2010, we have spent approximately 16 years and $281.9 million on our research and development program in heat shock proteins for cancer.
The timing and success of a clinical trial is dependent on enrolling sufficient patients in a timely manner, avoiding serious or significant adverse patient reactions, and demonstrating efficacy of the product candidate in order to support a favorable risk versus benefit profile, among other considerations. The timing and success of our clinical trials, in particular, are also dependent on clinical sites and regulatory authorities accepting each trial’s protocol, statistical analysis plan, product characterization tests, and clinical data. In addition, regulatory authorities may request additional information or data that is not readily available. Delays in our ability to respond to such requests would delay, and failure to adequately address concerns would prevent, our commercialization efforts.
Our existing Oncophage data may not support registration or approval in territories outside of Russia, including in the U.S. or Europe. Any additional studies may take years to complete and may fail to support regulatory filings for many reasons. In October 2008, we submitted a MAA to the EMA, requesting conditional authorization of Oncophage in earlier-stage, localized kidney cancer. After its review the CHMP of the EMA adopted a negative opinion on this MAA and subsequently we withdrew our application. We are currently evaluating our options to determine whether and how to proceed with Oncophage in renal cell carcinoma. If we continue to pursue a MAA for Oncophage with the EMA, there is a high level of uncertainty regarding the probability and timing of a favorable outcome. In addition, even if we continue this pursuit, Oncophage may not achieve approval in Europe. Additionally, the FDA has indicated that our Phase 3 clinical trials of Prophage Series R-100 (Oncophage) and M-200 cannot, by themselves, support BLA filings in the studies’ indications (renal cell carcinoma and metastatic melanoma). The signals and trends observed in these Phase 3 renal cell carcinoma and melanoma trials are based on data analysis of subgroups of patients, some of which were not pre-specified. While the subgroup data might be suggestive of treatment effect, under current regulatory guidelines the results cannot be expected, alone, to support registration or approval in the United States. Furthermore, regulatory authorities, including the FDA and the EMA, may have varying opinions of our product characterization, preclinical and clinical trial data for our other product candidates, which could delay, limit, or prevent regulatory approval or clearance. Delays or difficulties in obtaining regulatory approvals or clearances for our product candidates may:
| • | adversely affect the marketing of any products we or our licensees or collaborators develop; |
| • | impose significant additional costs on us or our licensees or collaborators; |
| • | diminish any competitive advantages that we or our licensees or collaborators may attain; |
| • | limit our ability to receive royalties and generate revenue and profits; and |
| • | adversely affect our business prospects and ability to obtain financing. |
Delays or failures in our receiving regulatory approval for our product candidates in a timely manner may result in us having to incur additional development expense and subject us to having to secure additional financing. As a result, we will not be able to commercialize them in the timeframe anticipated, and our business will suffer.
21
Table of Contents
New data from our research and development activities and/or resource considerations could modify our strategy and result in the need to adjust our projections of timelines and costs of programs.
Because we are focused on novel technologies, our research and development activities, including our nonclinical studies and clinical trials, involve the ongoing discovery of new facts and the generation of new data, based on which we determine next steps for a relevant program. These developments can occur with varying frequency and constitute the basis on which our business is conducted. We need to make determinations on an ongoing basis as to which of these facts or data will influence timelines and costs of programs. We may not always be able to make such judgments accurately, which may increase the costs we incur attempting to commercialize our product candidates. We monitor the likelihood of success of our initiatives and we may need to discontinue funding of such activities if they do not prove to be commercially feasible, due to our limited resources.
We may need to successfully address a number of technological challenges in order to complete development of our product candidates. Moreover, these product candidates may not be effective in treating any disease or may prove to have undesirable or unintended side effects, toxicities, or other characteristics that may preclude our obtaining regulatory approvals or prevent or limit commercial use.
Failure to enter into significant licensing, distribution and/or collaboration agreements may hinder our efforts to develop and commercialize our product candidates and will increase our need to rely on other financing mechanisms, such as sales of debt or equity securities, to fund our operations.
We have been engaged in efforts to enter into licensing, distribution and/or collaborative agreements with one or more pharmaceutical or biotechnology companies to assist us with development and/or commercialization of our product candidates. If we are successful in entering into such agreements, we may not be able to negotiate agreements with economic terms similar to those negotiated by other companies. We may not, for example, obtain significant upfront payments or substantial royalty rates. If we fail to enter into any such agreements, our efforts to develop and/or commercialize our products or product candidates may be undermined. In addition, if we do not raise funds through any such agreements, we will need to rely on other financing mechanisms, such as sales of debt or equity securities, to fund our operations.
While we have been pursuing these business development efforts for several years, we have not entered into a substantial agreement relating to the potential development or commercialization of Oncophage or any of the other Prophage Series vaccines. Due to the announcements in March 2006 that part I of our Phase 3 trial in renal cell carcinoma did not achieve its primary endpoint in the intent to treat population, and in November 2009 that the CHMP adopted a negative opinion on our MAA, and because companies may be skeptical regarding the potential success of a patient-specific product candidate, many companies may be unwilling to commit to an agreement prior to receipt of additional clinical data, if at all.
In addition, we would consider license and/or co-development opportunities to advance HerpV. This product is at an early stage and collaborative partners or licensees may defer discussions until results from early clinical trials become available, or they may not engage in such discussions at all. Further clinical development of HerpV will require a partner to support its advancement.
Because we rely on collaborators and licensees for the development and commercialization of some of our product candidate programs, these programs may not prove successful, and/or we may not receive significant payments from such parties.
Part of our strategy is to develop and commercialize some of our product candidates by continuing our existing arrangements with academic and corporate collaborators and licensees and by entering into new collaborations. Our success depends on our ability to negotiate such agreements and on the success of the other parties in performing research, preclinical and clinical testing, completing regulatory applications, and commercializing product candidates. For example, the development of Prophage G Series is currently dependent
22
Table of Contents
in large part on the efforts of our institutional collaborators, such as the Brain Tumor Research Center at the University of California, San Francisco, which is conducting Phase 2 clinical trials of Prophage Series G-100 and G-200 for the treatment of glioma. In addition, substantially all product candidates containing QS-21 depend on the success of our collaborative partners or licensees, and the Company’s relationships with these third parties. Such product candidates depend on the successful and adequate manufacture and/or supply of QS-21, and our collaborators and licensees successfully enrolling patients and completing clinical trials, being committed to dedicating the resources to advance these product candidates, obtaining regulatory approvals, and successfully commercializing product candidates.
These development activities may fail to produce marketable products due to unsuccessful results or abandonment of these programs, failure to enter into future collaborations or license agreements, or the inability to manufacture product supply requirements for our collaborators and licensees. For example, an undisclosed infectious disease Phase 3 program has been discontinued by one of our collaborators, and in August 2006, Pharmexa A/S announced a decision to cease dosing patients in their Phase 2 clinical trial of their HER-2 Protein AutoVac™ breast cancer vaccine containing our QS-21 adjuvant, after it was determined that the trial was unlikely to meet its primary endpoint. Several of our agreements also require us to transfer important rights and regulatory compliance responsibilities to our collaborators and licensees. As a result of collaborative agreements, we will not control the nature, timing, or cost of bringing these product candidates to market. Our collaborators and licensees could choose not to devote resources to these arrangements or, under certain circumstances, may terminate these arrangements early. They may cease pursuing product candidates or elect to collaborate with different companies. In addition, these collaborators and licensees, outside of their arrangements with us, may develop technologies or products that are competitive with those that we are developing. From time to time, we may also become involved in disputes with our collaborators or licensees. Such disputes could result in the incurrence of significant expense, or the termination of collaborations. We may be unable to fulfill all of our obligations to our collaborators, which may result in the termination of collaborations. As a result of these factors, our strategic collaborations may not yield revenue. Furthermore, we may be unable to enter into new collaborations or enter into new collaborations on favorable terms. Failure to generate significant revenue from collaborations would increase our need to fund our operations through sales of debt or equity securities and would negatively affect our business prospects.
If we are unable to purify heat shock proteins we may have difficulty successfully initiating or completing our clinical trials, and, even if we do successfully complete our clinical trials, generating sizable market potential.
Depending on the type and stage of cancer and the patient population, our ability to successfully develop and commercialize the Prophage Series vaccines for a particular cancer depends in part on our ability to purify heat shock proteins from that type of cancer. If we experience difficulties in purifying heat shock proteins for a sufficiently large number of patients in our clinical trials, we may face delays in enrolling sufficient patients and subsequently utilize more internal resources to satisfy enrolment requirements. Manufacturing failures may also lower the probability of a successful analysis of the data from clinical trials and, ultimately, the ability to obtain regulatory approvals. We have successfully manufactured product across many different cancer types, however, the success rate per indication has varied. We have evolved our manufacturing processes to better accommodate a wider range of tumor types. Our current manufacturing technologies have been successful in manufacturing product from approximately 92% of the RCC tumors received and approximately 90% of the tumors received for patients enrolled in our ongoing clinical trials in glioma. We expect to continue to devote resources to allow for a better evaluation of tumor characteristics and screening methods in an attempt to increase manufacturing success rates.
We may encounter problems with other types of cancer or patients, such as pediatric patients, as we expand our research. If we cannot overcome these problems, the number of cancer types that our heat shock protein product candidates could treat would be limited. In addition, if we commercialize our heat shock protein product candidates, we may not be able to replicate past manufacturing success rates and we may face claims from patients for whom we are unable to produce a vaccine.
23
Table of Contents
If we fail to sustain and further build our intellectual property rights, competitors will be able to take advantage of our research and development efforts to develop competing products.
If we are not able to protect our proprietary technology, trade secrets, and know-how, our competitors may use our inventions to develop competing products. We currently have exclusive rights to 73 issued United States patents and 117 issued foreign patents. We also have exclusive rights to 4 pending United States patent applications and 29 pending foreign patent applications. However, our patents may not protect us against our competitors. Our patent positions, and those of other pharmaceutical and biotechnology companies, are generally uncertain and involve complex legal, scientific, and factual questions. The standards which the United States Patent and Trademark Office uses to grant patents, and the standards which courts use to interpret patents, are not always applied predictably or uniformly and can change, particularly as new technologies develop. Consequently, the level of protection, if any, that will be provided by our patents if we attempt to enforce them, and they are challenged, is uncertain. In addition, the type and extent of patent claims that will be issued to us in the future is uncertain. Any patents that are issued may not contain claims that permit us to stop competitors from using similar technology.
Furthermore, the product development timeline for biotechnology products is lengthy and it is possible that our issued patents covering our product candidates in the United States and other jurisdictions may expire prior to commercial launch. In addition, because our patent on QS-21 composition of matter has already expired in virtually all territories, we are limited to protecting certain combinations of QS-21 with other adjuvants or formulations of QS-21 with other agents, e.g., excipients that improve performance of the compound. However, there is no guarantee that a third party would necessarily choose to use QS-21 in combination with such adjuvants or formulate it with the excipients covered by our patents. We are aware of at least one other party that makes a synthetic version of QS-21, claimed by such party to be equivalent in activity to natural QS-21, and has also developed derivatives of QS-21 which have shown biological activity.
In addition to our patented technology, we also rely on unpatented technology, trade secrets, and confidential information. We may not be able to effectively protect our rights to this technology or information. Other parties may independently develop substantially equivalent information and techniques or otherwise gain access to or disclose our technology. We generally require each of our employees, consultants, collaborators, and certain contractors to execute a confidentiality agreement at the commencement of an employment, consulting, collaborative, or contractual relationship with us. However, these agreements may not provide effective protection of our technology or information, or in the event of unauthorized use or disclosure, they may not provide adequate remedies.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, and we may be unable to protect our rights in, or to use, our technology.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. We may become a party to patent litigation or other proceedings regarding intellectual property rights.
If we choose to go to court to stop someone else from using the inventions claimed in our patents, that individual or company has the right to ask a court to rule that our patents are invalid and should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources even if we were successful in stopping the infringement of our patents. In addition, there is a risk that the court will decide that our patents are not valid and that we do not have the right to stop the other party from using the claimed inventions. There is also the risk that, even if the validity of our patents is upheld, the court will refuse to stop the other party on the grounds that such other party’s activities do not infringe our patents.
We may not have rights under some patents or patent applications related to some of our existing and proposed products or processes. Third parties may own or control these patents and patent applications in the United States and abroad. Therefore, in some cases, such as those described below, in order to develop, use,
24
Table of Contents
manufacture, sell, or import some of our existing or proposed products, or develop or use some of our existing or proposed processes, we or our collaborators may choose to seek, or be required to seek, licenses under third-party patents issued in the United States and abroad, or those that might issue from United States and foreign patent applications. In such an event, we likely would be required to pay license fees or royalties or both to the licensor. If licenses are not available to us on acceptable terms, we or our collaborators may not be able to exploit these products or processes.
Furthermore, a third party may claim that we are using inventions covered by such third-party’s patents or other intellectual property rights and may go to court to stop us from engaging in our normal operations and activities. These lawsuits are expensive. Some of our competitors may be able to sustain the cost of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. There is a risk that a court would decide that we are infringing the third-party’s patents and would order us to stop the activities covered by the patents. In addition, there is a risk that a court will order us to pay the other party substantial damages for having violated the other party’s patents. The biotechnology industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. Moreover, patent holders sometimes send communications to a number of companies in related fields suggesting possible infringement, and we, like a number of biotechnology companies, have received such communications in the past and may receive others in the future. If we are sued for patent infringement, we would need to demonstrate that our products either do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid, which we may not be able to do. Proving invalidity, in particular, is difficult, since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
If patent litigation or other proceeding is resolved against us, we or our licensees or collaborators may be enjoined from using, manufacturing, selling, or importing our products or processes without a license from the other party, and we may be held liable for significant damages. We may not be able to obtain any required licenses on commercially acceptable terms or at all.
Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to enter into collaborations with other entities, obtain financing, or compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time and other resources.
If we fail to comply with our obligations in our intellectual property licenses with third parties, we could lose license rights that are important to our business.
We are a party to various license agreements under which we receive the right to practice and use important third-party patent rights and we may enter into additional licenses in the future. Our existing licenses impose, and we expect future licenses will impose, various diligence, milestone payment, royalty, insurance, and other obligations on us. If we fail to comply with these obligations, the licensor may have the right to terminate the license, in which event we might not be able to market any product that is covered by the licensed patents.
If we fail to retain the services of, and/or maintain positive relations with, key individuals and our employees, we may not be able to achieve our strategic and operational objectives.
Garo H. Armen, Ph.D., the Chairman of our Board of Directors and our Chief Executive Officer, co-founded the Company in 1994 with Pramod K. Srivastava, Ph.D., and has been and continues to be integral to building our company and developing our technology. If Dr. Armen severed his relationship with Agenus, our business may be adversely impacted.
Effective December 1, 2005, we entered into an employment agreement with Dr. Armen. Subject to the earlier termination as provided in the agreement, the agreement had an original term of one year and is
25
Table of Contents
automatically extended thereafter for successive terms of one year each, unless either party provides notice to the other at least ninety days prior to the expiration of the original or any extension term. Dr. Armen plays an important role in our day-to-day activities. We do not carry key employee insurance policies for Dr. Armen or any other employee.
Dr. Srivastava currently has a consulting agreement with us pursuant to which he is retained to provide advice and services to Agenus from time to time. This agreement has an initial term ending March 31, 2011.
We also rely greatly on engaging and retaining other highly trained and experienced senior management and scientific and operations personnel and consultants. The competition for these and other qualified personnel in the biotechnology field is intense. In order to reduce our expenses, we have eliminated certain employee benefits, restructured our business, and reduced staffing levels. This restructuring has eliminated any redundancy in skills and capabilities in key areas. If we are not able to attract and retain qualified personnel, we may not be able to achieve our strategic and operational objectives.
We may face litigation that could result in substantial damages and may divert management’s time and attention from our business.
Agenus, our Chairman and Chief Executive Officer, Garo H. Armen, Ph.D., and two investment banking firms that served as underwriters in our initial public offering were named as defendants in a federal civil class action lawsuit in the United States District Court for the Southern District of New York. Substantially similar actions were filed concerning the initial public offerings for more than 300 different issuers, and the cases were coordinated for pre-trial purposes as In re Initial Public Offering Securities Litigation, 21 MC 92. The suit alleges that the brokerage arms of the investment banking firms charged secret excessive commissions to certain of their customers in return for allocations of our stock in the offering. The suit also alleges that shares of our stock were allocated to certain of the investment banking firms’ customers based upon agreements by such customers to purchase additional shares of our stock in the secondary market. The parties have reached a global settlement of the litigation. Under the settlement, the insurers will pay the full amount of settlement share allocated to the defendants, and the defendants will bear no financial liability. Agenus and the other defendants will receive complete dismissals from the case. In October 2009, the Court entered an order granting final approval of the settlement, and subsequently judgment was entered. Various objectors have filed appeals. If for any reason the settlement does not become effective, we believe we have meritorious defenses to the claims and intend to defend the action vigorously. We are unable to predict the likelihood of an unfavorable outcome or estimate our potential liability, if any.
In addition, we may currently be, or may become involved in additional litigation. Any such litigation could be expensive in terms of out-of-pocket costs and management time, and the outcome of any such litigation is uncertain.
Our directors and officers insurance policies provide $30.0 million of coverage. This insurance coverage may not be sufficient to cover us for future claims.
Product liability and other claims against us may reduce demand for our products and/or result in substantial damages.
We face an inherent risk of product liability exposure related to testing our product candidates in human clinical trials and commercial sales of Oncophage in Russia, and may face even greater risks if we sell Oncophage in other territories and/or sell our other product candidates commercially. An individual may bring a product liability claim against us if Oncophage or one of our product candidates causes, or merely appears to have caused, an injury. Product liability claims may result in:
| • | decreased demand for Oncophage or our product candidates; |
| • | regulatory investigations; |
| • | injury to our reputation; |
26
Table of Contents
| • | withdrawal of clinical trial volunteers; |
| • | costs of related litigation; and |
| • | substantial monetary awards to plaintiffs. |
We manufacture the Prophage Series vaccines from a patient’s cancer cells, and medical professionals must inject the vaccines into the same patient from which they were manufactured. A patient may sue us if a hospital, a shipping company, or we fail to deliver the removed cancer tissue or that patient’s Prophage Series vaccine. We anticipate that the logistics of shipping will become more complex if the number of patients we treat increases and that shipments of tumor and/or Prophage Series vaccines may be lost, delayed, or damaged. Additionally, complexities unique to the logistics of commercial products may delay shipments and limit our ability to move commercial product in an efficient manner without incident. To date, we have obtained transportation insurance coverage for commercial Oncophage being shipped to Russia. We do not have any other insurance that covers loss of or damage to the Prophage Series vaccines or tumor material, and we do not know whether such insurance will be available to us at a reasonable price or at all. We have limited product liability coverage for use of our product candidates. Our product liability policy provides $10.0 million aggregate coverage and $10.0 million per occurrence coverage. This limited insurance coverage may be insufficient to fully cover us for future claims.
If we do not comply with environmental laws and regulations, we may incur significant costs and potential disruption to our business.
We use hazardous, infectious, and radioactive materials, and recombinant DNA in our operations, which have the potential of being harmful to human health and safety or the environment. We store these hazardous (flammable, corrosive, toxic), infectious, and radioactive materials, and various wastes resulting from their use, at our facilities pending use and ultimate disposal. We are subject to a variety of federal, state, and local laws and regulations governing use, generation, storage, handling, and disposal of these materials. We may incur significant costs complying with both current and future environmental health and safety laws and regulations. In particular, we are subject to regulation by the Occupational Safety and Health Administration, the Environmental Protection Agency, the Drug Enforcement Agency, the Department of Transportation, the Centers for Disease Control and Prevention, the National Institutes of Health, the International Air Transportation Association, and various state and local agencies. At any time, one or more of the aforementioned agencies could adopt regulations that may affect our operations. We are also subject to regulation under the Toxic Substances Control Act and the Resource Conservation Development programs.
Although we believe that our current procedures and programs for handling, storage, and disposal of these materials comply with federal, state, and local laws and regulations, we cannot eliminate the risk of accidents involving contamination from these materials. Although we have limited pollution liability coverage ($2.0 million) and a workers’ compensation liability policy, we could be held liable for resulting damages in the event of an accident or accidental release, and such damages could be substantially in excess of any available insurance coverage and could substantially disrupt our business.
Risks Related to our Common Stock
The unaffiliated holders of certain convertible securities have the right to convert such securities into a substantial percentage of our outstanding common stock.
According to publicly filed documents, Mr. Brad M. Kelley beneficially owns 5,546,240 shares of our outstanding common stock and 31,620 shares of our series A convertible preferred stock. The shares of preferred stock are currently convertible at any time into 2,000,000 shares of common stock at an initial conversion price of $15.81, are non-voting, and carry a 2.5% annual dividend yield. If Mr. Kelley had converted all of the shares of preferred stock on December 31, 2010, he would have held approximately 7% of our outstanding common stock. We currently have a right of first refusal agreement with Mr. Kelley that provides us with limited rights to purchase certain of Mr. Kelley’s shares if he proposes to sell them to a third party.
27
Table of Contents
Mr. Kelley’s substantial ownership position provides him with the ability to substantially influence the outcome of matters submitted to our stockholders for approval. Furthermore, collectively, Mr. Kelley and Garo Armen, our CEO, control approximately 11% of our outstanding common stock as of December 31, 2010, providing ability, if they vote in the same manner, to determine the outcome of matters submitted to a stockholder vote. If Mr. Kelley were to convert all of his preferred stock into common stock, the combined total would increase to 12%. Additional purchases of our common stock by Mr. Kelley also would increase both his percentage of outstanding voting rights and the percentage combined with our CEO. While Mr. Kelley’s shares of preferred stock do not carry voting rights, the shares of common stock issuable upon conversion carry the same voting rights as other shares of common stock.
Our stock may be delisted from the Nasdaq Capital Market, which could affect its market price and liquidity.
Our common stock is currently listed on the Nasdaq Capital Market under the symbol “AGEN.” In the event that we fail to satisfy any of the listing requirements, our common stock may be put under review or removed from the listing on the Nasdaq Capital Market.
On March 3, 2011, we were notified by the Listing Qualifications Staff of Nasdaq (the “Staff”) indicating that we are not in compliance with the Nasdaq Marketplace Rule 5550(a)(2) (the “Bid Price Requirement”) because the bid price for our common stock had closed below the minimum $1.00 per share requirement for 30 consecutive business days. In accordance with Nasdaq Marketplace Rule 5810(c)(3)(A), we have been provided 180 calendar days, or until August 30, 2011, to regain compliance with the Bid Price Requirement.
If compliance is not demonstrated within the applicable compliance period, the Staff will notify us that our securities will be delisted from the Nasdaq Capital Market. However, we may appeal the Staff’s determination to delist our securities to a Hearings Panel. During any appeal process, shares of our common stock would continue to trade on the Nasdaq Capital Market. There can be no assurance that we will meet the requirements for continued listing on the Nasdaq Capital Market or whether any appeal would be granted by the Hearings Panel.
Provisions in our organizational documents could prevent or frustrate attempts by stockholders to replace our current management.
Our certificate of incorporation and bylaws contain provisions that could make it more difficult for a third party to acquire us without the consent of our Board of Directors. Our certificate of incorporation provides for a staggered board and removal of directors only for cause. Accordingly, stockholders may elect only a minority of our Board at any annual meeting, which may have the effect of delaying or preventing changes in management. In addition, under our certificate of incorporation, our Board of Directors may issue additional shares of preferred stock and determine the terms of those shares of stock without any further action by our stockholders. Our issuance of additional preferred stock could make it more difficult for a third party to acquire a majority of our outstanding voting stock and thereby effect a change in the composition of our Board of Directors. Our certificate of incorporation also provides that our stockholders may not take action by written consent. Our bylaws require advance notice of stockholder proposals and director nominations and permit only our President or a majority of the Board of Directors to call a special stockholder meeting. These provisions may have the effect of preventing or hindering attempts by our stockholders to replace our current management. In addition, Delaware law prohibits a corporation from engaging in a business combination with any holder of 15% or more of its capital stock until the holder has held the stock for three years unless, among other possibilities, the board of directors approves the transaction. Our Board of Directors may use this provision to prevent changes in our management. Also, under applicable Delaware law, our Board of Directors may adopt additional anti-takeover measures in the future.
28
Table of Contents
Our stock has historically had low trading volume, and its public trading price has been volatile.
Between our initial public offering on February 4, 2000 and December 31, 2010, and for the year ended December 31, 2010, the closing price of our common stock has fluctuated between $0.30 and $52.63 per share and $0.60 and $1.38 per share, respectively. The average daily trading volume for the year ended December 31, 2010 was approximately 1,103,000 shares. The market may experience significant price and volume fluctuations that are often unrelated to the operating performance of individual companies. In addition to general market volatility, many factors may have a significant adverse effect on the market price of our stock, including:
| • | continuing operating losses, which we expect over the next several years as we continue our development activities; |
| • | announcements of decisions made by public officials; |
| • | results of our preclinical studies and clinical trials; |
| • | announcements of new collaboration agreements with strategic partners or developments by our existing collaborative partners; |
| • | announcements of technological innovations, new commercial products, failures of products, or progress toward commercialization by our competitors or peers; |
| • | developments concerning proprietary rights, including patent and litigation matters; |
| • | publicity regarding actual or potential results with respect to product candidates under development; and |
| • | quarterly fluctuations in our financial results. |
The sale of a significant number of shares could cause the market price of our stock to decline.
The sale by us or the resale by stockholders of a significant number of shares of our common stock could cause the market price of our common stock to decline. As of December 31, 2010, we had approximately 111,625,000 shares of common stock outstanding. All of these shares are eligible for sale on the Nasdaq Capital Market, although certain of the shares are subject to sales volume and other limitations. We have filed registration statements to permit the sale of approximately 25,437,000 shares of common stock under our equity incentive plan and certain equity plans that we assumed in our acquisitions of Aquila Biopharmaceuticals, Inc. and Aronex Pharmaceuticals, Inc. We have also filed registration statements to permit the sale of 1,000,000 shares of common stock under our employee stock purchase plan, to permit the sale of 450,000 shares of common stock under our Directors’ Deferred Compensation Plan, to permit the sale of 49,643,966 shares of common stock pursuant to various private placement agreements. As of December 31, 2010, an aggregate of 39.3 million shares remain available for sale under these registration statements. The market price of our common stock may decrease based on the expectation of such sales.
As of December 31, 2010, options to purchase 7,272,850 shares of our common stock with a weighted average exercise price per share of $2.24 were outstanding. Many of these options are subject to vesting that generally occurs over a period of up to four years following the date of grant. As of December 31, 2010, we have 513,449 nonvested shares outstanding.
Because we are a small public company we believe we have been disproportionately negatively impacted by the Sarbanes-Oxley Act of 2002 and related regulations which have increased our costs in the past and have required additional management resources.
The Sarbanes-Oxley Act of 2002 and rules adopted by the SEC and the Nasdaq have resulted in significant costs to us. In particular, our efforts to comply with Section 404 of the Sarbanes-Oxley Act of 2002 and related regulations regarding the required assessment of our internal control over financial reporting, and our
29
Table of Contents
independent registered public accounting firm’s audit of internal control over financial reporting, have required commitments of significant management time. We expect these commitments to continue. Additionally, these laws and regulations could make it more difficult for us to attract and retain qualified members for our Board of Directors, particularly independent directors, or qualified executive officers.
Our internal control over financial reporting (as defined in Rules 13a-15 of the Securities Exchange Act) is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our consolidated financial statements for external purposes in accordance with U.S. generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect all deficiencies or weaknesses in our financial reporting. While our management has concluded that there were no material weaknesses in our internal control over financial reporting as of December 31, 2010, our procedures are subject to the risk that our controls may become inadequate because of changes in conditions or as a result of a deterioration in compliance with such procedures. No assurance is given that our procedures and processes for detecting weaknesses in our internal control over financial reporting will be effective.
| Item 1B. | Unresolved Staff Comments |
We have received no written comments from the staff of the SEC regarding our periodic or current reports that (1) we believe are material, (2) were issued not less than 180 days before the end of our 2010 fiscal year, and (3) remain unresolved.
| Item 2. | Properties |
We maintain our corporate offices in Lexington, Massachusetts, in a 162,000 square foot facility under a lease agreement that terminates in August 2013. We have an option to renew this lease for two additional ten-year periods. We have sublet a portion of this facility.
In addition, we leased approximately 40,000 square feet of laboratory, office, and manufacturing space in Framingham, Massachusetts under a lease agreement that terminated in September 2010. We had sublet this entire facility.
We also lease approximately 5,400 square feet in an office building in New York, New York. Our New York lease terminates in April 2012.
We believe substantially all of our property and equipment is in good condition and that we have sufficient capacity to meet our current operational needs. We do not anticipate experiencing significant difficulty in retaining occupancy of any of our manufacturing or office facilities and will do so through lease renewals prior to expiration or through replacing them with equivalent facilities.
| Item 3. | Legal Proceedings |
Agenus, our Chairman and CEO, Garo H. Armen, Ph.D., and two investment banking firms that served as underwriters in our initial public offering were named as defendants in a federal civil class action lawsuit in the United States District Court for the Southern District of New York. Substantially similar actions were filed concerning the initial public offerings for more than 300 different issuers, and the cases were coordinated for pre-trial purposes as In re Initial Public Offering Securities Litigation, 21 MC 92. The suit alleges that the brokerage arms of the investment banking firms charged secret excessive commissions to certain of their customers in return for allocations of our stock in the offering. The suit also alleges that shares of our stock were allocated to certain of the investment banking firms’ customers based upon agreements by such customers to purchase additional shares of our stock in the secondary market. The parties have reached a global settlement of the litigation. Under the settlement, the insurers will pay the full amount of settlement share allocated to the defendants, and the defendants will bear no financial liability. Agenus and the other defendants will receive complete dismissals from the case. In October 2009, the Court entered an order granting final approval of the
30
Table of Contents
settlement, and subsequently judgment was entered. Various objectors have filed appeals. If for any reason the settlement does not become effective, we believe we have meritorious defenses to the claims and intend to defend the action vigorously. We are unable to predict the likelihood of an unfavorable outcome or estimate our potential liability, if any.
We may currently be a party, or may become a party, to other legal proceedings, claims and investigations that arise in the ordinary course of business such as, but not limited to, patent, employment, commercial and environmental matters, as well. While we currently believe that the ultimate outcome of any of these proceedings will not have a material adverse effect on our financial position, results of operations, or liquidity, litigation is subject to inherent uncertainty. Furthermore, litigation consumes both cash and management attention.
| Item 4. | (Removed and Reserved) |
Executive Officers of the Registrant
Set forth below is certain information regarding our current and certain former executive officers, including their age, as of March 1, 2011:
| Name |
Age | Title | ||||
| Garo H. Armen, Ph.D. |
58 | Chairman of the Board and Chief Executive Officer | ||||
| Shalini Sharp |
36 | Vice President and Chief Financial Officer | ||||
| Christine M. Klaskin |
45 | Vice President, Finance and Principal Accounting Officer | ||||
| Karen H. Valentine |
39 | Vice President and General Counsel | ||||
| Kerry A. Wentworth |
38 | Vice President, Clinical, Regulatory & Quality | ||||
Garo H. Armen, PhD—Dr. Armen is Chairman and Chief Executive Officer of Agenus Inc., the biotechnology company he co-founded with Pramod Srivastava in 1994. From mid-2002 through 2004, he was Chairman of the Board of Directors for the biopharmaceutical company Elan Corporation, plc. Dr. Armen is also the founder and President of the Children of Armenia Fund, a charitable organization established in 2000 that is dedicated to the positive development of the children and youth of Armenia.
Shalini Sharp—Ms. Sharp is Chief Financial Officer of Agenus Inc. Prior to joining Agenus Inc. in 2003, Ms. Sharp was director of strategic planning at Elan Corporation, plc., where she served as chief of staff to the chairman of the board during the restructuring process and drove to completion a number of strategic corporate and financial transactions. Ms. Sharp was previously a management consultant at McKinsey & Company, specializing in pharmaceuticals and medical devices. Ms. Sharp received her BA and MBA from Harvard University.
Christine M. Klaskin—Christine M. Klaskin is Vice President, Finance and Principal Accounting Officer. Since joining Agenus Inc. in 1996 as finance manager, Ms. Klaskin has held various positions within the finance department and has been involved in all equity and debt offerings of the Company including its IPO. Prior to joining Agenus, Ms. Klaskin was employed by Arthur Andersen as an audit manager. Ms. Klaskin received her Bachelor of Accountancy from The George Washington University.
Karen H. Valentine—Karen Higgins Valentine is Vice President and General Counsel and also serves as Secretary and Chief Compliance Officer of the Company. Prior to joining Agenus Inc. in 2004, Ms. Valentine was an associate in the biotechnology practice of Palmer & Dodge LLP (now Edwards, Angell, Palmer & Dodge LLP). While at the law firm, she provided corporate law services to a broad range of both public and private corporations, and developed an expertise in the areas of licensing and strategic collaborations. Ms. Valentine graduated cum laude with a bachelor’s degree in neuroscience from Colgate University, and received her law degree, magna cum laude, from Boston University School of Law.
31
Table of Contents
Kerry A. Wentworth—Kerry Wentworth is Vice President, Clinical, Regulatory & Quality. Before joining Agenus Inc. in 2005, Ms. Wentworth served as senior director of regulatory affairs at Genelabs Technologies, where she was responsible for the business’ regulatory and quality functions. There she focused on the late-stage clinical development and subsequent US and European commercial application filings for the company’s lead product Prestara™. Prior to Genelabs, Ms. Wentworth held various positions in regulatory affairs at Shaman Pharmaceuticals and at Genzyme Corporation. Ms. Wentworth received a BS in pre-veterinary medicine from the University of New Hampshire.
32
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock is currently listed on The Nasdaq Capital Market under the symbol “AGEN.”
The following table sets forth, for the periods indicated, the high and low sale prices per share of our common stock.
| High | Low | |||||||
| 2009 |
||||||||
| First Quarter |
$ | 0.60 | $ | 0.19 | ||||
| Second Quarter |
3.34 | 0.43 | ||||||
| Third Quarter |
3.11 | 1.46 | ||||||
| Fourth Quarter |
2.24 | 0.63 | ||||||
| 2010 |
||||||||
| First Quarter |
1.20 | 0.60 | ||||||
| Second Quarter |
1.72 | 0.70 | ||||||
| Third Quarter |
1.12 | 0.73 | ||||||
| Fourth Quarter |
1.12 | 0.87 | ||||||
As of March 1, 2011, there were approximately 1,900 holders of record and approximately 25,000 beneficial holders of our common stock.
We have never paid cash dividends on our common stock, and we do not anticipate paying any cash dividends in the foreseeable future. We currently intend to retain future earnings, if any, for the future operation and expansion of our business. Any future payment of dividends on our common stock will be at the discretion of our Board of Directors and will depend upon, among other things, our earnings, financial condition, capital requirements, level of indebtedness, and other factors that our Board of Directors deems relevant.
Stock Performance
The following graph shows the cumulative total stockholder return on our common stock over the period from December 31, 2005 to December 31, 2010, as compared with that of the Nasdaq Stock Market (U.S. Companies) Index and the Nasdaq Biotechnology Index, based on an initial investment of $100 in each on December 31, 2005. Total stockholder return is measured by dividing share price change plus dividends, if any, for each period by the share price at the beginning of the respective period, and assumes reinvestment of dividends.
This stock performance graph shall not be deemed “filed” with the SEC or subject to Section 18 of the Securities Exchange Act, nor shall it be deemed incorporated by reference in any of our filings under the Securities Act of 1933, as amended (the “Securities Act”).
33
Table of Contents
COMPARISON OF CUMULATIVE TOTAL RETURN OF AGENUS INC.,
NASDAQ STOCK MARKET (U.S. COMPANIES) INDEX
AND NASDAQ BIOTECHNOLOGY INDEX
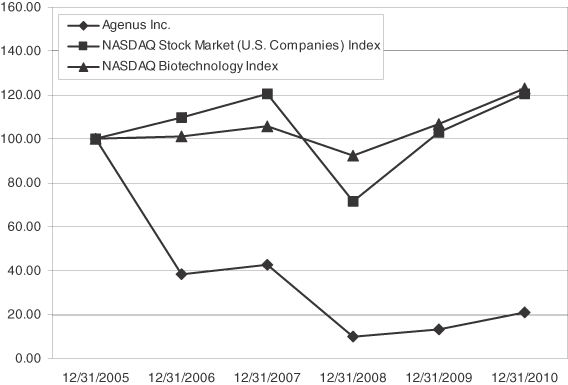
| 12/31/2005 | 12/31/2006 | 12/31/2007 | 12/31/2008 | 12/31/2009 | 12/31/2010 | |||||||||||||||||||
| Agenus Inc. |
100.00 | 38.45 | 42.86 | 10.08 | 13.45 | 21.22 | ||||||||||||||||||
| NASDAQ Stock Market (U.S. Companies) Index |
100.00 | 109.52 | 120.27 | 71.51 | 102.89 | 120.29 | ||||||||||||||||||
| NASDAQ Biotechnology Index |
100.00 | 101.02 | 105.65 | 92.31 | 106.74 | 122.76 | ||||||||||||||||||
Recent Sales of Unregistered Securities
The below listed payments in 2008 relate to compensation to a third-party consultant, Raifarm Limited or its affiliates (collectively, “Raifarm”), for services rendered in relation to the registration and commercialization activities in Russia for Oncophage pursuant to a Master Services Agreement between us and Raifarm, as amended from time to time. The below listed payments in 2010 relate to compensation to a third-party consultant, Hamilton Communications (“Hamilton”), for services rendered in connection with our rebranding effort pursuant to a Services Agreement between us and Hamilton, as amended. The offer, issuance and delivery of the below listed shares of common stock in the manner contemplated by the applicable agreements, did not require registration under Section 5 of the Securities Act because the transactions were exempted transactions under Section 4(2) of the Securities Act. This determination was based upon and assuming the accuracy of representations and warranties we obtained from Raifarm and Hamilton and compliance by Raifarm and Hamilton with the offering and transfer procedures and restrictions described in the applicable agreements and related documents.
| Date Issued |
Title of Each Class of Security |
Amount
of Securities Amount Issued |
Nature of Transaction Nature of Transaction | |||||
| Various dates, February – July, 2008 |
Common Stock, par value $0.01 | 346,509 | Shares issued for services rendered | |||||
| September 16, 2010 |
Common Stock, par value $0.01 | 111,111 | Shares issued for services rendered | |||||
| November 30, 2010 |
Common Stock, par value $0.01 | 54,945 | Shares issued for services rendered | |||||
34
Table of Contents
Information concerning our equity compensation plans is set forth in our Definitive Proxy Statement with respect to our 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission no later than 120 days after the end of the fiscal year under the heading “Equity Plans,” which is incorporated herein by reference.
| Item 6. | Selected Financial Data |
We have derived the consolidated balance sheet data set forth below as of December 31, 2010 and 2009, and the consolidated statement of operations data for each of the years in the three-year period ended December 31, 2010, from our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
You should read the selected consolidated financial data in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” our consolidated financial statements, and the notes to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
Given our history of incurring operating losses, management believes that it is more likely than not that any deferred tax assets will not be realized through future earnings. Therefore, no income tax benefit has been recognized in the consolidated statements of operations because of the loss before income taxes, and the need to recognize a valuation allowance on the portion of our deferred tax assets, which will not be offset by the reversal of deferred tax liabilities (see Note (1) below).
Changes in cash, cash equivalents, and short-term investments, total current assets, total assets, and stockholders’ deficit in the periods presented below include the effects of the receipt of net proceeds from our debt offerings, equity offerings, the exercise of stock options and warrants, and employee stock purchases that totaled approximately $11.6 million, $18.7 million, $46.9 million, $4.6 million, and $25.4 million in the years ended December 31, 2010, 2009, 2008, 2007, and 2006, respectively.
35
Table of Contents
| For the Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (In thousands, except per share data) | ||||||||||||||||||||
| Consolidated Statement of Operations Data: |
||||||||||||||||||||
| Revenue |
$ | 3,360 | $ | 3,334 | $ | 2,651 | $ | 5,552 | $ | 692 | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Cost of goods sold |
(123 | ) | — | — | — | — | ||||||||||||||
| Research and development |
(12,878 | ) | (16,903 | ) | (20,663 | ) | (21,789 | ) | (28,643 | ) | ||||||||||
| General and administrative |
(12,112 | ) | (14,110 | ) | (19,832 | ) | (17,041 | ) | (21,288 | ) | ||||||||||
| Restructuring costs |
— | — | — | — | (1,374 | ) | ||||||||||||||
| Loss from operations |
(21,753 | ) | (27,679 | ) | (37,844 | ) | (33,278 | ) | (50,613 | ) | ||||||||||
| Non-operating income |
4,680 | 2,568 | 12,356 | 1 | 141 | |||||||||||||||
| Interest expense, net |
(4,834 | ) | (5,207 | ) | (5,313 | ) | (4,658 | ) | (2,287 | ) | ||||||||||
| Net loss (1) |
(21,907 | ) | (30,318 | ) | (30,801 | ) | (37,935 | ) | (52,759 | ) | ||||||||||
| Dividends on series A convertible preferred stock |
(790 | ) | (790 | ) | (790 | ) | (790 | ) | (790 | ) | ||||||||||
| Net loss attributable to common stockholders |
$ | (22,697 | ) | $ | (31,108 | ) | $ | (31,591 | ) | $ | (38,725 | ) | $ | (53,549 | ) | |||||
| Net loss attributable to common stockholders per common share, basic and diluted |
$ | (0.23 | ) | $ | (0.39 | ) | $ | (0.50 | ) | $ | (0.83 | ) | $ | (1.17 | ) | |||||
| Weighted average number of shares outstanding, basic and diluted |
96,650 | 79,017 | 63,249 | 46,512 | 45,809 | |||||||||||||||
| December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents, and short-term investments |
$ | 19,782 | $ | 30,065 | $ | 34,463 | $ | 18,679 | $ | 40,095 | ||||||||||
| Total current assets |
20,854 | 31,533 | 35,486 | 20,782 | 42,298 | |||||||||||||||
| Total assets |
30,907 | 45,874 | 56,822 | 44,351 | 72,726 | |||||||||||||||
| Total current liabilities |
5,416 | 5,355 | 6,997 | 8,383 | 9,078 | |||||||||||||||
| Long-term debt, less current portion |
34,050 | 49,494 | 64,126 | 71,524 | 68,276 | |||||||||||||||
| Stockholders’ deficit |
(14,707 | ) | (16,975 | ) | (20,330 | ) | (41,370 | ) | (10,563 | ) | ||||||||||
| (1) | Given our history of incurring operating losses, no income tax benefit has been recognized in our consolidated statements of operations because of the loss before income taxes, and the need to recognize a valuation allowance on the portion of our deferred tax assets which will not be offset by the reversal of deferred tax liabilities. |
36
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
Overview
Our current research and/or development activities are focused on developing technologies and product candidates to treat cancers and infectious diseases. Since our inception in March 1994, our activities have primarily been associated with the development of our heat shock protein technology, primarily our lead autologous cancer immunotherapies (formerly referred to as Oncophage), the Prophage Series of cancer vaccines (vitespen; HSPPC-96). The first product derived from the Prophage Series of vaccines (R-100, still referred to in Russia and Europe as Oncophage), represents the only approved treatment for adjuvant or non-metastatic renal cell carcinoma (RCC; kidney cancer) patients at intermediate risk for disease recurrence. In a registry following patients from a large randomized Phase 3 trial in non-metastatic RCC, patients at intermediate risk of recurrence who were in the treatment arm demonstrated an approximately 46 percent lower risk of death compared with those in the control arm (n = 362; P < 0.05; hazard ratio = 0.54). Phase 2 trials testing the Prophage Series candidates G-100 and G-200 are underway in both newly diagnosed and recurrent glioma, respectively, where promising data has been generated to date. Additional trials are planned in metastatic RCC (R-200) and metastatic melanoma (M-200) in combination with potentially synergistic therapies, as well as in pediatric neurological tumors (NP-150). Our business activities have included product research and development, intellectual property prosecution, manufacturing, regulatory and clinical affairs, corporate finance and development, market development, and support of our collaborations.
We have incurred significant losses since our inception. As of December 31, 2010, we had an accumulated deficit of $584.4 million. Since our inception, we have financed our operations primarily through the sale of equity and convertible notes, interest income earned on cash, cash equivalents, and short-term investment balances, and debt provided through secured lines of credit. We believe that, based on our current plans and activities, our working capital resources at December 31, 2010, anticipated revenues, and the estimated proceeds from our license, supply, and collaborative agreements will be sufficient to satisfy our liquidity requirements through 2011. We expect to attempt to raise additional funds in advance of depleting our current funds. We may attempt to raise additional funds by: (1) licensing technologies or products to one or more collaborative partners, (2) renegotiating third party agreements, (3) completing an outright sale of assets, (4) securing additional debt financing, and/or (5) selling additional equity securities. Satisfying long-term liquidity needs may require the successful commercialization of (1) our product, Oncophage and/or one or more partnering arrangements for Oncophage, (2) vaccines containing QS-21 under development by our licensees, and/or (3) potentially other product candidates, each of which will require additional capital.
In April 2008, the Russian Ministry of Public Health issued a registration certificate for the use of Oncophage for the adjuvant treatment of kidney cancer patients at intermediate risk for disease recurrence and, in September 2008, the Food & Drug Administration (“FDA”) granted the necessary permission to allow for the export of Oncophage from the United States for patient administration in Russia. The Russian registration was our first product approval from a regulatory authority, and the first approval of a patient-specific therapeutic cancer vaccine in a major market.
In October 2008, we announced the submission of a marketing authorization application (“MAA”) to the European Medicines Agency (“EMA”) requesting conditional authorization of Oncophage in earlier-stage, localized kidney cancer. On November 20, 2009 we announced that the Committee for Medicinal Products for Human Use (“CHMP”) of the EMA formally adopted a negative opinion on this MAA. Subsequently we withdrew our application and we have been evaluating a variety of options to potentially bring Oncophage to patients and physicians globally. Although we are no longer in active discussions with a potential partner for the European market, we continue to actively seek partnership discussions for multiple products generated from our Prophage Series.
37
Table of Contents
Guidance received from past interaction with the FDA indicated that an additional Phase 3 clinical study must be conducted to demonstrate the efficacy and safety of Oncophage. Because the primary evidence of efficacy comes from a subgroup analysis of the pre-specified primary and secondary endpoints and was not demonstrated in the intent-to-treat population, our Phase 3 renal cell carcinoma trial is likely not sufficient as sole support for product approval based on existing standards in the United States and potentially in other territories.
On March 3, 2011, we were notified by the Listing Qualifications Staff of Nasdaq (the “Staff”) indicating that we are not in compliance with the Bid Nasdaq Marketplace Rule 5550(a)(2) (the “Bid Price Requirement”) because the bid price for our common stock had closed below the minimum $1.00 per share requirement for 30 consecutive business days. In accordance with Nasdaq Marketplace Rule 5810(c)(3)(A), we have been provided 180 calendar days, or until August 30, 2011, to regain compliance with the Bid Price Requirement. If compliance is not demonstrated within the applicable compliance period, the Staff will notify us that our securities will be delisted from the Nasdaq Capital Market. However, we may appeal the Staff’s determination to delist our securities to a Hearings Panel. During any appeal process, shares of our common stock would continue to trade on the Nasdaq Capital Market. There can be no assurance that we will meet the requirements for continued listing on the Nasdaq Capital Market or whether any appeal would be granted by the Hearings Panel.
Historical Results of Operations
Year Ended December 31, 2010 Compared to the Year Ended December 31, 2009
Revenue: We generated revenue of $3.4 million and $3.3 million during the years ended December 31, 2010 and 2009, respectively. Revenue includes revenue earned on shipments of QS-21 to our QS-21 licensees, license fees, royalties earned, and in 2010, grants earned and Oncophage sales. In the years ended December 31, 2010 and 2009, we recorded $1.5 million each period from the amortization of deferred revenue related to our QS-21 partnered programs.
Research and Development: Research and development expenses include the costs associated with our internal research and development activities, including compensation and benefits, occupancy costs, clinical manufacturing costs, costs of consultants, and administrative costs. Research and development expense decreased 24% to $12.9 million for the year ended December 31, 2010 from $16.9 million for the year ended December 31, 2009. The decrease included declines of $1.7 million for personnel related expenses and $367,000 for facility related costs primarily due to cost containment efforts, and $1.8 million for various outside services primarily related to the status of our efforts in Russia and other territories.
General and Administrative: General and administrative expenses consist primarily of personnel costs, facility expenses, and professional fees. General and administrative expenses decreased 14% to $12.1 million for the year ended December 31, 2010 from $14.1 million for the year ended December 31, 2009. This decrease is largely attributable to declines of $1.5 million for various outside services primarily relating to the status of our efforts in Russia and other territories, and $145,000 in employee and director noncash share-based compensation expense.
Non-operating Income: Non-operating income of $4.7 million for the year ended December 31, 2010 consists of a net gain of $2.8 million on the extinguishment of a portion of our 2005 Notes and the change in the fair value of our derivative liability since December 31, 2009 of $1.9 million.
Interest Expense: Interest expense decreased to $4.9 million for the year ended December 31, 2010 from $5.3 million for the year ended December 31, 2009. This decrease is related to the repurchase of a portion of our 2005 Notes. Interest on our 2006 Notes is payable semi-annually on December 30 and June 30 in cash or, at our option, in additional notes or a combination thereof. During the years ended December 31, 2010 and 2009, interest expense included $2.6 million and $2.4 million, respectively, paid in the form of additional 2006 Notes.
38
Table of Contents
Interest Income: Interest income decreased 73% to $38,000 for the year ended December 31, 2010 from $137,000 for the year ended December 31, 2009. This decrease is primarily attributable to a decrease in our average cash, cash equivalents and short-term investments balance coupled with declining interest rates earned on our cash, cash equivalents, and short-term investments. Our average interest rate earned decreased from 0.49% for the year ended December 31, 2009 to 0.15% for the year ended December 31, 2010.
Year Ended December 31, 2009 Compared to the Year Ended December 31, 2008
Revenue: We generated revenue of $3.3 million and $2.7 million during the years ended December 31, 2009 and 2008, respectively. Revenue includes revenue earned on shipments of QS-21 to our QS-21 licensees, license fees, and royalties earned. In the years ended December 31, 2009 and 2008, we recorded $1.5 million each period from the amortization of deferred revenue related to our QS-21 partnered programs.
Research and Development: Research and development expenses include the costs associated with our internal research and development activities, including compensation and benefits, occupancy costs, clinical manufacturing costs, costs of consultants, and administrative costs. Research and development expense decreased 18% to $16.9 million for the year ended December 31, 2009 from $20.7 million for the year ended December 31, 2008. The decrease included declines of $1.5 million for personnel related expenses and $241,000 for facility related costs primarily due to cost containment efforts, and $1.5 million for various outside services primarily related to the status of our efforts in Russia and other territories.
General and Administrative: General and administrative expenses consist primarily of personnel costs, facility expenses, and professional fees. General and administrative expenses decreased 29% to $14.1 million for the year ended December 31, 2009 from $19.8 million for the year ended December 31, 2008. This decrease is largely attributable to declines of $2.3 million for various outside services primarily relating to the status of our efforts in Russia and other territories, $1.5 million in personnel related expenses due to cost containment efforts, $1.0 million in employee and director noncash share-based compensation expense and a $332,000 decrease in our foreign currency exchange loss.
Non-operating Income: Non-operating income of $2.6 million for the year ended December 31, 2009 consists primarily of a gain on the extinguishment of a portion of our 2005 Notes.
Interest Expense: Interest expense decreased to $5.3 million for the year ended December 31, 2009 from $6.3 million for the year ended December 31, 2008. This decrease is related to the repurchase of a portion of our 2005 Notes during the fourth quarter of 2008 and the second quarter of 2009. Interest on our 2006 Notes is payable semi-annually on December 30 and June 30 in cash or, at our option, in additional notes or a combination thereof. During the years ended December 31, 2009 and 2008, interest expense included $2.4 million and $2.2 million, respectively, paid in the form of additional 2006 Notes.
Interest Income: Interest income decreased 86% to $137,000 for the year ended December 31, 2009 from $966,000 for the year ended December 31, 2008. This decrease is primarily attributable to a decrease in our average cash, cash equivalents and short-term investments balance coupled with declining interest rates earned on our cash, cash equivalents, and short-term investments. Our average interest rate earned decreased from 2.4% for the year ended December 31, 2008 to 0.49% for the year ended December 31, 2009.
39
Table of Contents
Research and Development Programs
Prior to 2002, we did not track costs on a per project basis, and therefore have estimated the allocation of our total research and development costs to our largest research and development programs for that time period. During 2010, these research and development programs consisted largely of our Prophage Series vaccines and QS-21, as indicated in the following table (in thousands).
| Research and Development Program |
Product | Year Ended December 31, | Prior
to 2008 |
Total | ||||||||||||||||||||
| 2010 | 2009 | 2008 | ||||||||||||||||||||||
| Heat shock proteins for cancer |
|
Prophage Series Vaccines |
|
$ | 10,960 | $ | 15,309 | $ | 17,156 | $ | 238,426 | $ | 281,851 | |||||||||||
| Heat shock proteins for infectious diseases |
HerpV | 644 | 262 | 1,377 | 16,071 | 18,354 | ||||||||||||||||||
| Vaccine adjuvant * |
QS-21 | 1,185 | 1,071 | 648 | 9,500 | 12,404 | ||||||||||||||||||
| Other research and development programs |
89 | 261 | 1,482 | 31,695 | 33,527 | |||||||||||||||||||
| Total research and development expenses |
$ | 12,878 | $ | 16,903 | $ | 20,663 | $ | 295,692 | $ | 346,136 | ||||||||||||||
| * | Prior to 2000, costs were incurred by Aquila Biopharmaceuticals, Inc., a company we acquired in November 2000. |
Research and development program costs include compensation and other direct costs plus an allocation of indirect costs, based on certain assumptions and our review of the status of each program. Our Prophage Series vaccines are in various stages of development as described below. Significant additional expenditures will be required if we start new trials, encounter delays in our programs, apply for regulatory approvals, continue development of our technologies, expand our operations, and/or bring our product candidates to market. The eventual total cost of each clinical trial is dependent on a number of factors such as trial design, length of the trial, number of clinical sites, and number of patients. The process of obtaining and maintaining regulatory approvals for new therapeutic products is lengthy, expensive, and uncertain. Because the further development of our Prophage Series vaccines is subject to evaluation and uncertainty, and because HerpV is an early-stage clinical development candidate and generally on hold due to cost-containment efforts, we are unable to reliably estimate the cost of completing our research and development programs, the timing of bringing such programs to various markets, and, therefore, when, if ever, material cash inflows are likely to commence. Programs involving QS-21 depend on our collaborative partners or licensees successfully completing clinical trials, successfully manufacturing QS-21 to meet demand, obtaining regulatory approvals and successfully commercializing product candidates containing QS-21.
Product Development Portfolio
Prophage Series of Cancer Vaccines
We started enrolling patients in our first clinical trial studying a Prophage Series vaccine at Memorial Sloan-Kettering Cancer Center in New York, New York in November 1997. To date, we have treated nearly 850 cancer patients in our clinical trials. Because Prophage Series vaccines are novel therapeutic vaccines that are patient-specific, meaning derived from the patient’s own tumor, they are experiencing a long development process and high development costs, either of which could delay or prevent our commercialization efforts. For additional information regarding regulatory risks and uncertainties, please read the risks identified under Part I-Item 1A. “Risk Factors” of this Annual Report on Form 10-K.
We believe that the collective results from our clinical trials thus far show that the Prophage Series vaccines have a favorable safety profile. We also believe that available results from clinical trials suggest that treatment with Prophage Series vaccines can generate immunological and anti-tumor responses.
40
Table of Contents
We initiated a Phase 3, multicenter, international trial for non-metastatic RCC into which the first patient was randomized in February 2001. As announced on March 24, 2006, the trial did not reach statistical significance in its primary endpoint of recurrence-free survival in the total patient population, though a positive trend was observed. During the protocol design process in 1999 and 2000, key opinion leaders were consulted, and the non-metastatic RCC patient population designated for enrollment in the trial was thought to be a relatively uniform group. In 2006, the Eastern Cooperative Oncology Group (“ECOG”) initiated a trial in adjuvant RCC with sorafenib and sunitinib that stratified their patient population into intermediate-risk, high-risk, and very high-risk recurrence categories. Using these ECOG defined criteria, analysis of the intermediate risk patients (362 of the 604 eligible patients) in the trial showed a statistically significant difference in recurrence-free survival in favor of the Oncophage arm. In part because the intermediate-risk category was not prospectively delineated prior to the trial’s initiation, the FDA has indicated that, by itself, part I of our Phase 3 clinical trial in renal cell carcinoma is not sufficient to support a biologics license application (“BLA”) filing.
We opened a subsequent protocol that continued to follow patients from this trial in the format of a registry in order to collect overall survival information, as well as investigator reports of disease recurrence. The registry, which is expected to provide additional data on the effectiveness of the vaccine, followed patients until March 2010, an additional three years from closure of the initial trial, providing more than five years of data collection following the enrollment of the last patient in the trial. Final analysis of this data is in process. At the 2009 American Society of Clinical Oncology (“ASCO”) annual meeting, we announced results of an interim analysis from the ongoing global patient survival registry, which showed that patients with kidney cancer at intermediate risk of disease recurrence demonstrated an approximately 46 percent lower risk of death in the treatment arm compared with the control arm (n = 362; P < 0.05; hazard ratio = 0.54).
In addition to the patient registry, patient enrollment has commenced into a small study in non-metastatic RCC to assess immune response in the intermediate-risk patient population. The results of this study, continued data collection through the survival registry, and ongoing analysis are uncertain, and may not positively affect the acceptability of the overall results of the trial and, even if clinically meaningful, may not meet the requirements of the FDA or other regulatory authorities for submission and approval of a marketing application or similar applications for product approval outside the United States.
In April 2008, the Russian Ministry of Public Health issued a registration certificate for the use of Oncophage for the treatment of kidney cancer patients at intermediate risk for disease recurrence. Because, among other things, we have limited resources and minimal sales and marketing experience, commercialization of Oncophage has been slow, and only modest sales of Oncophage in Russia have occurred during the year ended December 31, 2010. The Russian registration was our first product approval from a regulatory authority, and the first approval of a patient-specific therapeutic cancer vaccine in a major market. Since this approval we have been focusing our efforts in Russia on securing one or more distribution and/or partnering arrangements and related commercialization activities. The amount of any future revenue generated from the sale of Oncophage in Russia will depend on our ability to successfully execute on these efforts and identify and obtain adequate reimbursement, as well as on decisions of physicians and patients, among other factors. Furthermore, we may experience significant delays in the receipt of payment for Oncophage, or an inability to collect payments at all.
In October 2008, we announced the submission of a MAA to the EMA requesting conditional authorization of Oncophage in earlier-stage, localized kidney cancer. On November 20, 2009, we announced that the CHMP of the EMA formally adopted a negative opinion on this MAA. Subsequently we withdrew our application and have been evaluating a variety of options to potentially bring Oncophage to patients and physicians globally. Although we are no longer in active discussions with a potential partner for the European market, we continue to actively seek partnership discussions for multiple products generated from our portfolio of Prophage Series vaccines.
A Phase 2 clinical trial with Prophage Series G-200 vaccine in recurrent, high-grade glioma is currently ongoing. This study is being led by the Brain Tumor Research Center at the University of California, San Francisco (“UCSF”), with grants from the American Brain Tumor Association and the National Cancer Institute
41
Table of Contents
Special Programs of Research Excellence. Phase 1 results, presented at the Society for Neuro-Oncology Annual Meeting Conference in November 2008, showed that vaccination following brain cancer surgery increased overall median survival to approximately 10.5 months, with four patients surviving beyond 12 months and one patient surviving almost 2.5 years. The study also showed that all 12 treated patients demonstrated a significant immune response after vaccination with G-200 ( P < 0.001) and that patients with minimal residual disease at time of first vaccination (n = 7) were more likely to survive beyond nine months compared with patients with significant residual disease.
The study, which is designed to enroll approximately 50 patients, has expanded to include New York-Presbyterian Hospital/Columbia University Medical Center and University Hospitals/Case Western Reserve. Interim data was presented at the Society for Neuro-Oncology meeting in October 2009, which showed a median survival of 10.1 months in the first 20 patients treated with G-200, and that to date six patients (30 percent) had survived at or beyond 12 months. This early data shows an improvement in overall survival over the previous long-standing historical median survival of 6.5 months, and is also slightly favorable to the recently reported median survival of 9.2 months with Avastin® (bevacizumab) in patients with recurrent high-grade glioma. In May 2010, data presented at the International Conference on Brain Tumor Research and Therapy suggested that vaccination with this candidate may improve overall survival in patients with recurrent high-grade glioma. An overall median survival of 44 weeks after tumor resection was observed. Approximately 70% of the evaluable patients survived beyond 36 weeks, and 41% survived up to or longer than one year. Additional data from this trial will be reported by mid-2011. UCSF also initiated an additional Phase 2 clinical trial in newly diagnosed glioma testing Prophage Series G-100 vaccine in combination with Temodar® (temozolomide). This trial is currently enrolling, with a target of 50 patients. Based on promising trends to date, this trial is being expanded to include up to 10 clinical sites.
QS-21
QS-21 Stimulon® adjuvant is an adjuvant, or a substance added to a vaccine or other immunotherapy, that is intended to enhance immune response. The key licensees of QS-21 are GSK and JANSSEN Alzheimer Immunotherapy. There are 14 vaccines containing QS-21 in clinical development, including four in Phase 3 testing for malaria, melanoma, non-small cell lung cancer and shingles. The first products containing QS-21 are expected to be launched in the 2013-2014 timeframe. The pipeline of product candidates containing QS-21 is extraordinarily diverse, encompassing prophylactic as well as therapeutic vaccines for infectious diseases, multiple cancer types, and Alzheimer’s disease. The Company does not incur clinical development costs for these products and is generally reimbursed for any related expenses by its licensees.
In return for rights to use QS-21, our licensees have generally agreed to pay us license fees, manufacturing payments, milestone payments, and royalties on product sales for a minimum of 10 years after commercial launch. In addition to our corporate licensing arrangements, we have developed a number of academic collaborations to test new vaccine concepts and products containing QS-21. From time to time our collaborators or licensees initiate and/or cease programs containing QS-21. For example, an undisclosed infectious disease Phase 3 program was recently discontinued by one of our collaborators.
On January 16, 2009, we entered into an Amended and Restated Manufacturing Technology Transfer and Supply Agreement, under which GSK has the right to manufacture all of its requirements of commercial grade QS-21. GSK is obligated to supply us (or our affiliates, licensees, or customers) certain quantities of commercial grade QS-21 for a stated period of time. We understand that QS-21 is a key component included in several of GSK’s proprietary adjuvant systems and that a number of GSK’s vaccine candidates currently under development are formulated using adjuvant systems containing QS-21. GSK has initiated Phase 3 studies evaluating its investigational MAGE-A3 Antigen-Specific Cancer Immunotherapeutic containing QS-21 in non-small cell lung cancer and melanoma. GSK has also initiated a Phase 3 clinical trial in malaria and a Phase 3 clinical trial in shingles. Revenues recognized with respect to this agreement were $1.3 million for each of the years ended December 31, 2010 and 2009.
42
Table of Contents
Elan Pharmaceuticals, Inc. and/or its affiliates (“Elan”) had a commercial license for the use of QS-21 in the research and commercialization of products. Effective September 14, 2009, we entered into an Amended and Restated License Agreement (“Amended License Agreement”) with Elan. On September 17, 2009, the Amended License Agreement was assigned to JANSSEN Alzheimer Immunotherapy. Under the terms of the Amended License Agreement assigned to JANSSEN Alzheimer Immunotherapy, they will have the right to develop, make, have made, use, sell, offer for sale, import, and have sold, the Alzheimer’s disease vaccine that contains QS-21 (“Licensed Product”). In addition, pursuant to the terms of the Amended License Agreement, JANSSEN Alzheimer Immunotherapy has the right to manufacture all of its requirements of QS-21 for use in the Licensed Product and we have no further supply obligations. Under the terms of the Amended License Agreement, we are entitled to receive future milestone payments and product royalties in the event of the successful development of the Licensed Product. In 2007, Elan initiated a Phase 2 study of their vaccine. Revenues recognized with respect to this agreement were $160,000 in the year ended December 31, 2010.
Liquidity and Capital Resources
We have incurred annual operating losses since inception, and we had an accumulated deficit of $584.4 million as of December 31, 2010. We expect to incur significant losses over the next several years as we continue our clinical trials, apply for regulatory approvals, prepare for commercialization, and continue development of our technologies. Since our inception, we have financed our operations primarily through the sale of equity and convertible notes, interest income earned on cash, cash equivalents, and short-term investment balances, and debt provided through secured lines of credit. From our inception through December 31, 2010, we have raised aggregate net proceeds of $506.3 million through the sale of common and preferred stock, the exercise of stock options and warrants, proceeds from our employee stock purchase plan, and the issuance of convertible notes, and borrowed $20.5 million under two credit facilities. During February 2010, we entered into an At the Market Sales Agreement with McNicoll, Lewis & Vlak LLC and Wm Smith & Co (the “Sales Agents”) under which we may sell an aggregate of up to 20 million shares of our common stock from time to time through the Sales Agents. To date we have issued approximately 7.0 million shares of our common stock in at the market offerings through the Sales Agents and raised net proceeds of approximately $8.8 million after deducting offering costs of approximately $331,000. As of December 31, 2010, we had debt outstanding of $34.9 million in principal, including $34.7 million in principal of our 2006 Notes maturing August 31, 2014 and $100,000 in principal of our 2005 Notes maturing February 20, 2025. The 2005 Notes are subject to redemption at the option of the holders or us beginning February 1, 2012.
Our cash, cash equivalents, and short-term investments at December 31, 2010 were $19.8 million, a decrease of $10.3 million from December 31, 2009. Based on our current plans and activities, we anticipate that our net cash burn (defined as cash used in operating activities plus capital expenditures and dividend payments) will be in the $16-$18 million range for the year ending December 31, 2011. In addition, we hope to generate royalties from our QS-21 product in the 2013-2014 timeframe.
We believe that, based on our current plans and activities, our working capital resources at December 31, 2010, anticipated revenues, and the estimated proceeds from our license, supply, and collaborative agreements will be sufficient to satisfy our liquidity requirements into 2012. We closely monitor our cash needs. We continue to monitor the likelihood of success of our key initiatives and are prepared to discontinue funding of such activities if they do not prove to be commercially feasible. In addition, we will continue to adjust other spending as needed in order to preserve liquidity. We expect to attempt to raise additional funds in advance of depleting our current funds. In order to fund our operations through 2011 and beyond, we will need to contain costs and raise additional funds. We may attempt to raise additional funds by: (1) licensing technologies or products to one or more collaborative partners, (2) renegotiating third party agreements, (3) completing an outright sale of selected assets, (4) securing additional debt financing, and/or (5) selling additional equity securities. Our ability to successfully enter into any such arrangements is uncertain, and if funds are not available, or not available on terms acceptable to us, we may be required to revise our planned clinical trials, other development activities, capital expenditures, and/or the scale of our operations. As noted above, we expect to attempt to raise additional funds in advance of depleting our current funds; however, we may not be able to
43
Table of Contents
raise funds or raise amounts sufficient to meet the long-term needs of the business. Satisfying long-term liquidity needs may require the successful commercialization of Oncophage and/or one or more partnering arrangements for our other Prophage Series vaccines, successful commercialization of vaccines containing QS-21 under development by our licensees, and potentially successful commercialization of other product candidates, each of which will require additional capital, as discussed above. Please see the “Forward-Looking Statements” section and the risks highlighted under Part I-Item 1A. “Risk Factors” of this Annual Report on Form 10-K.
Our future cash requirements include, but are not limited to, efforts to make Oncophage available in Russia and other jurisdictions we are currently exploring, as well as supporting our clinical trial and regulatory efforts and continuing our other research and development programs. Since inception, we have entered into various agreements with institutions and clinical research organizations to conduct and monitor our clinical studies. Under these agreements, subject to the enrollment of patients and performance by the applicable institution of certain services, we have estimated our payments to be $47.2 million over the term of the studies. Through December 31, 2010, we have expensed $46.5 million as research and development expenses and $46.3 million has been paid related to these clinical studies. The timing of expense recognition and future payments related to these agreements is subject to the enrollment of patients and performance by the applicable institution of certain services.
We have also entered into sponsored research agreements related to our product candidates that required payments of $6.5 million, all of which has been paid as of December 31, 2010. We plan to enter into additional sponsored research agreements, and we anticipate significant additional expenditures will be required to advance our clinical trials, apply for regulatory approvals, continue development of our technologies, and bring our product candidates to market. Part of our strategy is to develop and commercialize some of our product candidates by continuing our existing collaborative arrangements with academic and collaborative partners and licensees and by entering into new collaborations. As a result of our collaborative agreements, we will not completely control the efforts to attempt to bring those product candidates to market. We have various agreements, for example, with collaborative partners and/or licensees, which allow the use of our QS-21 adjuvant in numerous vaccines. These agreements grant exclusive worldwide rights in some fields of use and co-exclusive or non-exclusive rights in others. These agreements generally provide us with rights to manufacture and supply QS-21 to the collaborative partner or licensee and also call for royalties to be paid to us on future sales of licensed vaccines that include QS-21, which may or may not be achieved. Significant investment in manufacturing capacity could be required if we were to retain our manufacturing and supply rights.
Net cash used in operating activities for the year ended December 31, 2010 and 2009 was $14.8 million and $24.2 million, respectively. We continue to support and develop our QS-21 partnering collaborations, with the goal of generating royalties from this product in the 2013-2014 timeframe. Our future ability to generate cash from operations will depend on achieving regulatory approval of our product candidates, and market acceptance of Oncophage and our product candidates, achieving benchmarks as defined in existing collaborative agreements, and our ability to enter into new collaborations. Please see the “Forward-Looking Statements” section and the risks highlighted under Part I-Item 1A. “Risk Factors” of this Annual Report on Form 10-K.
The table below summarizes our contractual obligations as of December 31, 2010 (in thousands).
| Payments Due by Period | ||||||||||||||||||||
| Total | Less than 1 Year |
1 – 3 Years | 3 – 5 Years | More than 5 Years |
||||||||||||||||
| Long-term debt (1) |
$ | 46,542 | $ | 207 | $ | 103 | $ | 46,232 | $ | — | ||||||||||
| Operating leases |
5,771 | 2,224 | 3,547 | — | — | |||||||||||||||
| Total |
$ | 52,313 | $ | 2,431 | $ | 3,650 | $ | 46,232 | $ | — | ||||||||||
| (1) | Assumes the 2006 Notes are not converted and are paid in 2014. In certain circumstances, the 2006 Notes could be converted before then. Also includes fixed interest payments, some of which may be paid in kind, and assumes that the 2005 Notes are not converted and are paid on February 1, 2012. In certain |
44
Table of Contents
| circumstances, the 2005 Notes could be converted before then. In addition, the holders of the 2005 Notes can require us to purchase debt from them at certain dates between 2012 and 2020. If the 2005 Notes are not converted and we are not required to purchase the debt, the 2005 Notes mature on February 1, 2025. If the 2005 Notes were outstanding until maturity, there would be additional interest payments of $68,000 for the period 2012 through 2025. |
Effective July 19, 2002, we sublet part of our Framingham facility to GTC Biotherapeutics, Inc. (“GTC”) and we leased related leasehold improvements and equipment under agreements that were to expire on December 31, 2006. GTC exercised its option to extend this lease until September 2010, the date our original lease expired. Under the terms of our original lease, we were obligated to pay our landlord approximately 7% of our rental income. Effective March 17, 2004, we sublet an additional part of our Framingham facility to PP Manufacturing, whose lease also expired in September 2010. Since September 30, 2010, we are no longer a party to any lease or subleasing arrangements for this facility. Effective July 30, 2010, we sublet part of our Lexington facility to Cubist Pharmaceuticals, Inc. whose lease expires in July 2012. Our Lexington facility and New York office leases expire August 2013 and April 2012, respectively.
We are currently involved in certain legal proceedings as detailed in Item 3 above and Note 16 of the notes to our consolidated financial statements. While we currently believe that the ultimate outcome of any of these proceedings will not have a material adverse effect on our financial position, results of operations, or liquidity, litigation is subject to inherent uncertainty. Furthermore, litigation consumes both cash and management attention.
Inflation
We believe that inflation has not had a material adverse effect on our business, results of operations, or financial condition to date.
Related Parties
In March 1995, we entered into a consulting agreement with Dr. Pramod Srivastava, our scientific founder and a former member of our Board of Directors, and upon its expiration in March 2006, we entered into a new consulting agreement, effective March 28, 2006, with Dr. Srivastava. The agreement has an initial term ending March 31, 2011. In exchange for the timely performance of services, as defined in the agreement, Dr. Srivastava is entitled to receive compensation to be established by the Compensation Committee of the Agenus Board of Directors. For the twelve-month period ending March 31, 2011, Dr. Srivastava will receive $50,000. Dr. Srivastava is also eligible to receive an annual bonus and stock options at the discretion of the Compensation Committee of our Board of Directors. During the year ended December 31, 2009, we paid Dr. Srivastava an additional $50,000 for his work related to our MAA submitted to the EMEA.
On January 9, 2008, we entered into a private placement agreement (the “January 2008 private placement”) that included (i) 8,708,717 shares of common stock, (ii) warrants to acquire up to 8,708,717 shares of common stock at $3.00 per share, and (iii) unit warrants, which, if exercisable due to a triggering event as that term is defined in the applicable warrant, permit a holder to acquire up to 8,708,717 shares of common stock at $3.00 per share and warrants to acquire up to an additional 8,708,717 shares of common stock at $3.00 per share. In conjunction with this private placement, we sold 542,050 shares of common stock to Garo H. Armen, Ph.D., our Chairman and Chief Executive Officer, and 1,166,667 shares of common stock to Armen Partners LP. Garo H. Armen is the general partner of Armen Partners LP and owns a controlling interest therein. In addition to the common stock acquired by Garo H. Armen and Armen Partners LP, each acquired an equal number of both warrants and unit warrants. The unit warrants expired January 9, 2010.
Critical Accounting Policies and Estimates
The SEC defines “critical accounting policies” as those that require the application of management’s most difficult, subjective, or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain and may change in subsequent periods.
45
Table of Contents
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. We base those estimates on historical experience and on various assumptions that are believed to be reasonable under the circumstances. Actual results could differ from those estimates.
The following listing is not intended to be a comprehensive list of all of our accounting policies. Our significant accounting policies are described in Note 2 of the notes to our consolidated financial statements. In many cases, the accounting treatment of a particular transaction is dictated by U.S. generally accepted accounting principles, with no need for our judgment in its application. There are also areas in which our judgment in selecting an available alternative would not produce a materially different result. We have identified the following as our critical accounting policies.
Share-Based Compensation
In accordance with the fair value recognition provisions of ASC 718, Compensation—Stock Compensation, we recognize share-based compensation expense net of an estimated forfeiture rate and only recognize compensation expense for those share-based awards expected to vest. Compensation expense is recognized on a straight-line basis over the requisite service period of the award.
Stock options granted to certain non-employees have been accounted for based on the fair value method of accounting in accordance with ASC 505-50, Equity- Equity-Based Payments to Non-Employees. As a result, the noncash charge to operations for non-employee options with vesting or other performance criteria is affected each reporting period, until the non-employee options vest, by changes in the fair value of our common stock. Under the provisions of ASC 505-50, the change in fair value of vested options issued to non-employees is reflected in the statement of operations each reporting period, until the options are exercised or expire.
Determining the appropriate fair value model and calculating the fair value of share-based awards requires the use of highly subjective assumptions, including the expected life of the share-based awards and stock price volatility. The assumptions used in calculating the fair value of share-based awards represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and we use different assumptions, our share-based compensation expense could be materially different in the future. In addition, if our actual forfeiture rate is materially different from our estimate, the share-based compensation expense could be significantly different from what we have recorded in the current period. See Note 10 of the notes to our consolidated financial statements for a further discussion on share-based compensation.
Fair Value Accounting—Derivative Liability
As a result of the adoption of certain guidance within ASC 815-40, Derivatives and Hedging- Contracts In Entity’s Own Equity, as of January 1, 2009, the conversion feature embedded in our 2006 Notes is treated as a derivative and recorded at its fair value, with period to period changes in the fair value recorded as a gain or loss in our consolidated statement of operations.
We measure fair value based on a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the inputs that market participants would use in pricing the asset or liability and are developed based on the best information available in the circumstances. Our derivative liability is valued based on significant unobservable inputs.
46
Table of Contents
Revenue Recognition
Revenue for services under research and development contracts are recognized as the services are performed, or as clinical trial materials are provided. Non-refundable milestone payments that represent the completion of a separate earnings process are recognized as revenue when earned. License fees and royalties are recognized as they are earned. Revenue recognized from collaborative agreements is based upon the provisions of Accounting Standards Codification (“ASC”) 605-25, Revenue Recognition—Multiple Element Arrangements.
Recent Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board (“FASB”) revised authoritative guidance on multiple-deliverable revenue arrangements providing a greater ability to separate and allocate arrangement consideration in a multiple element revenue arrangement by requiring the use of estimated selling prices to allocate arrangement consideration if neither vendor-specific objective evidence nor third party evidence of selling price is available, thereby eliminating the use of the residual method of allocation. The revised guidance also requires expanded qualitative and quantitative disclosures surrounding multiple deliverable revenue arrangements. This guidance is effective for fiscal years beginning after June 15, 2010 and may be applied retrospectively or prospectively for new or materially modified arrangements. Early adoption is permitted. We will evaluate the impact of this standard on future revenue arrangements that we may enter into.
In April 2010, the FASB codified the consensus reached in Emerging Issues Task Force Issue No. 08-09, “Milestone Method of Revenue Recognition” issuing Accounting Standard Update (“ASU”) No. 2010-17 Milestone Method of Revenue Recognition, to limit the scope of this ASU to research or development arrangements and require that guidance in this ASU be met for an entity to apply the milestone method (which allows entities to record the milestone payment in its entirety in the period achieved). However, the FASB clarified that, even if the requirements in this ASU are met, entities would not be precluded from making an accounting policy election to apply another appropriate accounting policy that results in the deferral of some portion of the arrangement consideration. The ASU was effective for periods beginning on or after June 15, 2010. Early application was permitted. Entities can apply this guidance prospectively to milestones achieved after adoption. However, retrospective application to all prior periods was also permitted. We will evaluate the impact of this standard on future revenue arrangements that we may enter into.
In January 2010, the FASB issued ASU No. 2010-06, “Fair Value Measurements and Disclosures (Topic 820)—Improving Disclosures about Fair Value Measurements” (“ASU 2010-06”). ASU 2010-06 requires new disclosures regarding significant transfers in and out of Levels 1 and 2 fair value measurements, as well as information about activity in Level 3 fair value measurements, including presenting information about purchases, sales, issuances and settlements on a gross versus a net basis in the Level 3 activity roll forward. In addition, ASU 2010-06 also clarifies existing disclosures regarding input and valuation techniques, as well as the level of disaggregation for each class of assets and liabilities. ASU No. 2010-06 was effective for interim and annual periods beginning after December 15, 2009, except for the disclosures pertaining to purchases, sales, issuances and settlements in the roll forward of Level 3 activity; those disclosures are effective for interim and annual periods beginning after December 15, 2010. The adoption of ASU 2010-06 had no current impact and is expected to have no subsequent impact on our consolidated financial position, results of operations or cash flows. Required disclosure requirements of ASU 2010-06 have been included in Note 15 to our consolidated financial statements.
In December 2010, the FASB issued additional guidance on when to perform Step 2 of the goodwill impairment test for reporting units with zero or negative carrying amounts. The criteria for evaluating Step 1 of the goodwill impairment test and proceeding to Step 2 was amended for reporting units with zero or negative carrying amounts and requires performing Step 2 if qualitative factors indicate that it is more likely than not that a goodwill impairment exists. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2010. Upon adoption of the amended guidance, any impairment will be recorded as an adjustment to beginning retained earnings. We are currently evaluating the impact of adoption on our consolidated financial statements.
47
Table of Contents
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
In the normal course of business, we are exposed to fluctuations in interest rates as we seek debt financing and invest excess cash. We are also exposed to foreign currency exchange rate fluctuation risk related to our transactions denominated in foreign currencies. We do not currently employ specific strategies, such as the use of derivative instruments or hedging, to manage these exposures. Our currency exposures vary, but are primarily concentrated in the Euro. During the year ended December 31, 2010, there has been no material change with respect to our interest rate and foreign currency exposures or our approach toward those exposures. However, we are exploring possible commercialization of Oncophage outside of the U.S., which could result in increased foreign currency exposure.
The information below summarizes our market risks associated with debt obligations as of December 31, 2010. Fair value included herein has been estimated taking into consideration the nature and terms of each instrument and the prevailing economic and market conditions at December 31, 2010. The table presents principal payments by year of maturity based on the terms of the debt (in thousands).
| Estimated Fair Value (2) |
Outstanding Principal Amount December 31, 2010 |
Year of Maturity | ||||||||||||||||||
| 2011 | 2012 | 2014 | ||||||||||||||||||
| Long-term debt (1) |
$ | 30,829 | $ | 34,916 | $ | 146 | $ | 100 | $ | 34,670 | ||||||||||
| (1) | Fixed interest rates range from 5.25% to 8%. The above table is based on the assumptions that future interest on the 2006 Notes is paid in cash and that these notes are not converted at maturity August 31, 2014. In certain circumstances, the 2006 Notes could be converted before then. In addition, the table is based on the assumption that the 2005 Notes are redeemed on February 1, 2012. In certain circumstances, the 2005 Notes could be converted on or before February 1, 2012. The note holders of our 2005 Notes can require us to redeem debt at certain dates between 2012 and 2020. If the 2005 Notes are not converted and we are not required to purchase the notes, they mature on February 1, 2025. |
| (2) | The estimated fair value of our long-term debt was derived by evaluating the nature and terms of each note and considering the prevailing economic and market conditions at the balance sheet date. In addition, the fair value of our 2005 Notes was estimated based on the most recent market transactions. |
We had cash and cash equivalents at December 31, 2010 of $19.8 million, which are exposed to the impact of interest and foreign currency exchange rate changes, and our interest income fluctuates as interest rates change. Due to the short-term nature of our investments in money market funds, our carrying value approximates the fair value of these investments at December 31, 2010, however, we are subject to investment risk.
We invest our cash, cash equivalents, and short-term investments in accordance with our Investment Policy. The primary objectives of our Investment Policy are to preserve principal, maintain proper liquidity to meet operating needs, and maximize yields. We review our Investment Policy annually and amend it as deemed necessary. Currently, the Investment Policy prohibits investing in any structured investment vehicles and asset-backed commercial paper. Although our investments are subject to credit risk, our Investment Policy specifies credit quality standards for our investments and limits the amount of credit exposure from any single issue, issuer, or type of investment. Our investments are also subject to interest rate risk and will decrease in value if market interest rates increase. However, due to the conservative nature of our investments and relatively short duration, interest rate risk is mitigated. We do not invest in derivative financial instruments. Accordingly, we do not believe that there is currently any material market risk exposure with respect to derivative or other financial instruments that would require disclosure under this item.
48
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
49
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Agenus Inc.:
We have audited the accompanying consolidated balance sheets of Agenus Inc. and subsidiaries as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity (deficit) and comprehensive loss, and cash flows for each of the years in the three-year period ended December 31, 2010. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Agenus Inc. and subsidiaries as of December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Agenus Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated March 16, 2011, expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
As discussed in Note 14 to the consolidated financial statements, in 2009 the Company retrospectively changed its method of accounting for certain convertible debt instruments that may be settled in cash upon conversion due to the adoption of new accounting requirements issued by the FASB. In addition, as discussed in Note 14 to the consolidated financial statements, the Company changed its method of evaluating when adjustment features within contracts are considered to be equity–indexed due to the adoption of new accounting requirements issued by the FASB, as of January 1, 2009.
/s/ KPMG LLP
Boston, Massachusetts
March 16, 2011
50
Table of Contents
CONSOLIDATED BALANCE SHEETS
| December 31, 2010 | December 31, 2009 | |||||||
| ASSETS | ||||||||
| Cash and cash equivalents |
$ | 19,781,976 | $ | 20,066,817 | ||||
| Short-term investments |
— | 9,998,294 | ||||||
| Inventories |
26,432 | 324,035 | ||||||
| Accounts receivable |
35,000 | — | ||||||
| Prepaid expenses |
704,744 | 751,960 | ||||||
| Other current assets |
306,008 | 391,723 | ||||||
| Total current assets |
20,854,160 | 31,532,829 | ||||||
| Plant and equipment, net of accumulated amortization and depreciation of $24,993,225 and $28,612,631 at December 31, 2010 and 2009, respectively |
6,194,465 | 8,891,124 | ||||||
| Goodwill |
2,572,203 | 2,572,203 | ||||||
| Core and developed technology, net of accumulated amortization of $10,443,247 and $9,753,106 at December 31, 2010 and 2009, respectively |
— | 1,319,523 | ||||||
| Debt issuance costs, net of accumulated amortization of $1,270,492 and $1,139,807 at December 31, 2010 and 2009, respectively |
29,841 | 293,575 | ||||||
| Other long-term assets |
1,255,990 | 1,264,833 | ||||||
| Total assets |
$ | 30,906,659 | $ | 45,874,087 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Current portion, long-term debt |
$ | 146,061 | $ | 146,061 | ||||
| Current portion, deferred revenue |
1,540,385 | 1,501,902 | ||||||
| Accounts payable |
698,554 | 895,338 | ||||||
| Accrued liabilities |
2,684,609 | 2,597,056 | ||||||
| Other current liabilities |
346,314 | 214,591 | ||||||
| Total current liabilities |
5,415,923 | 5,354,948 | ||||||
| Convertible notes |
34,050,033 | 49,494,119 | ||||||
| Deferred revenue |
3,612,156 | 2,976,538 | ||||||
| Derivative liability |
755,000 | 2,665,156 | ||||||
| Other long-term liabilities |
1,780,759 | 2,358,293 | ||||||
| Commitments and contingencies (Notes 13 and 16) |
||||||||
| STOCKHOLDERS’ DEFICIT |
||||||||
| Preferred stock, par value $0.01 per share; 25,000,000 shares authorized: |
||||||||
| Series A convertible preferred stock; 31,620 shares designated, issued, and outstanding at December 31, 2010 and 2009; liquidation value of $31,817,625 at December 31, 2010 |
316 | 316 | ||||||
| Series B2 convertible preferred stock; 3,105 shares designated, issued, and outstanding at December 31, 2010 and 2009 |
31 | 31 | ||||||
| Common stock, par value $0.01 per share; 250,000,000 shares authorized; 111,885,759 and 90,015,425 shares issued at December 31, 2010 and 2009, respectively |
1,118,858 | 900,154 | ||||||
| Additional paid-in capital |
568,916,796 | 544,961,442 | ||||||
| Treasury stock, at cost; 260,944 shares of common stock at December 31, 2010 and 2009 |
(324,792 | ) | (324,792 | ) | ||||
| Accumulated deficit |
(584,418,421 | ) | (562,512,118 | ) | ||||
| Total stockholders’ deficit |
(14,707,212 | ) | (16,974,967 | ) | ||||
| Total liabilities and stockholders’ deficit |
$ | 30,906,659 | $ | 45,874,087 | ||||
See accompanying notes to consolidated financial statements.
51
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
For the Years Ended December 31, 2010, 2009, and 2008
| 2010 | 2009 | 2008 | ||||||||||
| Revenue: |
||||||||||||
| Product revenue |
$ | 52,500 | $ | — | $ | — | ||||||
| Grant revenue |
424,720 | — | — | |||||||||
| Research and development revenue |
2,882,391 | 3,334,444 | 2,651,081 | |||||||||
| Total revenues |
3,359,611 | 3,334,444 | 2,651,081 | |||||||||
| Operating expenses: |
||||||||||||
| Cost of goods sold |
(122,946 | ) | — | — | ||||||||
| Research and development |
(12,877,695 | ) | (16,902,537 | ) | (20,662,987 | ) | ||||||
| General and administrative |
(12,111,507 | ) | (14,110,514 | ) | (19,831,858 | ) | ||||||
| Operating loss |
(21,752,537 | ) | (27,678,607 | ) | (37,843,764 | ) | ||||||
| Other income (expense): |
||||||||||||
| Non-operating income |
4,680,120 | 2,568,545 | 12,355,677 | |||||||||
| Interest expense |
(4,871,446 | ) | (5,344,713 | ) | (6,278,492 | ) | ||||||
| Interest income |
37,560 | 137,482 | 965,843 | |||||||||
| Net loss |
(21,906,303 | ) | (30,317,293 | ) | (30,800,736 | ) | ||||||
| Dividends on series A convertible preferred stock |
(790,500 | ) | (790,500 | ) | (790,500 | ) | ||||||
| Net loss attributable to common stockholders |
$ | (22,696,803 | ) | $ | (31,107,793 | ) | $ | (31,591,236 | ) | |||
| Per common share data, basic and diluted: |
||||||||||||
| Net loss attributable to common stockholders |
$ | (0.23 | ) | $ | (0.39 | ) | $ | (0.50 | ) | |||
| Weighted average number of common shares outstanding, basic and diluted |
96,650,120 | 79,017,143 | 63,249,458 | |||||||||
See accompanying notes to consolidated financial statements.
52
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT) AND COMPREHENSIVE LOSS
For the Years Ended December 31, 2010, 2009, and 2008
| Series
A Convertible Preferred Stock |
Series
B1 Convertible Preferred Stock |
Series
B2 Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Treasury Stock | Accumulated Deficit |
Total | |||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Amount | |||||||||||||||||||||||||||||||||||||||||||
| Balance at January 1, 2008 |
31,620 | 316 | 10,000 | 100 | 5,250 | 53 | 47,557,007 | 475,570 | 459,539,406 | 5,953 | (12,168 | ) | (501,372,841 | ) | (41,369,564 | ) | ||||||||||||||||||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | — | — | — | (30,800,736 | ) | (30,800,736 | ) | |||||||||||||||||||||||||||||||||||||
| Share-based compensation |
— | — | — | — | — | — | — | — | 5,265,530 | — | — | — | 5,265,530 | |||||||||||||||||||||||||||||||||||||||
| Shares issued in private placement |
— | — | — | — | — | — | 15,708,717 | 157,087 | 45,382,134 | — | — | — | 45,539,221 | |||||||||||||||||||||||||||||||||||||||
| Shares sold at the market |
— | — | — | — | — | — | 271,762 | 2,718 | 801,238 | — | — | — | 803,956 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 28,469 | 285 | 46,277 | — | — | — | 46,562 | |||||||||||||||||||||||||||||||||||||||
| Employee share purchases |
— | — | — | — | — | — | 171,113 | 1,711 | 285,219 | — | — | — | 286,930 | |||||||||||||||||||||||||||||||||||||||
| Conversion of series B1 convertible preferred stock |
— | — | (10,000 | ) | (100 | ) | — | — | 1,585,197 | 15,852 | (15,752 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Shares issued under Directors’ Deferred Compensation Plan |
— | — | — | — | — | — | 61,938 | 619 | 228,381 | — | — | — | 229,000 | |||||||||||||||||||||||||||||||||||||||
| Shares issued to a consultant |
— | — | — | — | — | — | 346,509 | 3,465 | 814,161 | — | — | — | 817,626 | |||||||||||||||||||||||||||||||||||||||
| Reclassification of liability classified option grants |
— | — | — | — | — | — | — | — | (100,771 | ) | — | — | — | (100,771 | ) | |||||||||||||||||||||||||||||||||||||
| Vesting of nonvested shares |
— | — | — | — | — | — | 766,990 | 7,670 | (7,670 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Treasury stock received for vested share tax payments |
— | — | — | — | — | — | — | — | — | 137,078 | (257,681 | ) | — | (257,681 | ) | |||||||||||||||||||||||||||||||||||||
| Dividends on series A convertible preferred stock ($25 per share) |
— | — | — | — | — | — | — | — | (790,500 | ) | — | — | — | (790,500 | ) | |||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2008 |
31,620 | 316 | — | — | 5,250 | 53 | 66,497,702 | 664,977 | 511,447,653 | 143,031 | (269,849 | ) | (532,173,577 | ) | (20,330,427 | ) | ||||||||||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
53
Table of Contents
AGENUS INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT) AND COMPREHENSIVE LOSS—(Continued)
For the Years Ended December 31, 2010, 2009, and 2008
| Series
A Convertible Preferred Stock |
Series
B1 Convertible Preferred Stock |
Series
B2 Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Treasury Stock | Accumulated Deficit |
Total | |||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Amount | |||||||||||||||||||||||||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | — | — | — | (30,317,293 | ) | (30,317,293 | ) | |||||||||||||||||||||||||||||||||||||
| Adoption of EITF 07-5 |
— | — | — | — | — | — | — | — | (1,352,317 | ) | — | — | (21,248 | ) | (1,373,565 | ) | ||||||||||||||||||||||||||||||||||||
| Share-based compensation |
— | — | — | — | — | — | — | — | 3,115,642 | — | — | — | 3,115,642 | |||||||||||||||||||||||||||||||||||||||
| Shares issued in private placements |
— | — | — | — | — | — | 9,385,965 | 93,860 | 18,478,795 | — | — | — | 18,572,655 | |||||||||||||||||||||||||||||||||||||||
| Conversion of series B2 preferred shares |
— | — | — | — | (2,145 | ) | (22 | ) | 5,929,212 | 59,292 | (59,270 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Shares issued to repurchase convertible senior notes |
— | — | — | — | — | — | 5,597,362 | 55,974 | 14,078,215 | — | — | — | 14,134,189 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 79,276 | 792 | 140,520 | — | — | — | 141,312 | |||||||||||||||||||||||||||||||||||||||
| Employee share purchases |
— | — | — | — | — | — | 41,300 | 413 | 16,520 | — | — | — | 16,933 | |||||||||||||||||||||||||||||||||||||||
| Shares issued under Directors’ Deferred Compensation Plan |
— | — | — | — | — | — | 15,376 | 154 | 21,346 | — | — | — | 21,500 | |||||||||||||||||||||||||||||||||||||||
| Shares issued to CEO in lieu of cash compensation |
— | — | — | — | — | — | 130,143 | 1,302 | 108,698 | — | — | — | 110,000 | |||||||||||||||||||||||||||||||||||||||
| Reclassification of liability classified option grants |
— | — | — | — | — | — | — | — | (220,470 | ) | — | — | — | (220,470 | ) | |||||||||||||||||||||||||||||||||||||
| Vesting of nonvested shares |
— | — | — | — | — | — | 2,339,089 | 23,390 | (23,390 | ) | — | — | ||||||||||||||||||||||||||||||||||||||||
| Treasury stock received for vested share tax payments |
— | — | — | — | — | — | — | — | — | 117,913 | (54,943 | ) | — | (54,943 | ) | |||||||||||||||||||||||||||||||||||||
| Dividends on series A convertible preferred stock ($25 per share) |
— | — | — | — | — | — | — | — | (790,500 | ) | — | — | — | (790,500 | ) | |||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2009 |
31,620 | $ | 316 | — | $ | — | 3,105 | $ | 31 | 90,015,425 | $ | 900,154 | $ | 544,961,442 | 260,944 | $ | (324,792 | ) | $ | (562,512,118 | ) | $ | (16,974,967 | ) | ||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
54
Table of Contents
AGENUS INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT) AND COMPREHENSIVE LOSS—(Continued)
For the Years Ended December 31, 2010, 2009, and 2008
| Series
A Convertible Preferred Stock |
Series
B1 Convertible Preferred Stock |
Series
B2 Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Treasury Stock | Accumulated Deficit |
Total | |||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Amount | |||||||||||||||||||||||||||||||||||||||||||
| Net loss and comprehensive loss |
— | — | — | — | — | — | — | — | — | — | — | (21,906,303 | ) | (21,906,303 | ) | |||||||||||||||||||||||||||||||||||||
| Share-based compensation |
— | — | — | — | — | — | — | — | 2,813,304 | — | — | — | 2,813,304 | |||||||||||||||||||||||||||||||||||||||
| Shares issued in private placements |
— | — | — | — | — | — | 3,199,451 | 31,995 | 2,847,511 | — | — | — | 2,879,506 | |||||||||||||||||||||||||||||||||||||||
| Shares sold at the market |
— | — | — | — | — | — | 6,820,070 | 68,201 | 8,577,529 | — | — | — | 8,645,730 | |||||||||||||||||||||||||||||||||||||||
| Shares issued to repurchase convertible senior notes |
— | — | — | — | — | — | 9,855,266 | 98,553 | 10,263,367 | — | — | — | 10,361,920 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 959 | 10 | 709 | — | — | — | 719 | |||||||||||||||||||||||||||||||||||||||
| Employee share purchases |
— | — | — | — | — | — | 89,725 | 897 | 47,706 | — | — | — | 48,603 | |||||||||||||||||||||||||||||||||||||||
| Shares issued to consultants for services |
— | — | — | — | — | — | 166,056 | 1,660 | 148,340 | — | — | — | 150,000 | |||||||||||||||||||||||||||||||||||||||
| Shares issued to CEO in lieu of cash compensation |
— | — | — | — | — | — | 152,905 | 1,528 | 130,472 | — | — | — | 132,000 | |||||||||||||||||||||||||||||||||||||||
| Reclassification of liability classified option grants |
— | — | — | — | — | — | — | — | (67,224 | ) | — | — | — | (67,224 | ) | |||||||||||||||||||||||||||||||||||||
| Vesting of nonvested shares |
— | — | — | — | — | — | 1,585,902 | 15,860 | (15,860 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Dividends on series A convertible preferred stock ($25 per share) |
— | — | — | — | — | — | — | — | (790,500 | ) | — | — | — | (790,500 | ) | |||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2010 |
31,620 | $ | 316 | — | $ | — | 3,105 | $ | 31 | 111,885,759 | $ | 1,118,858 | $ | 568,916,796 | 260,944 | $ | (324,792 | ) | $ | (584,418,421 | ) | $ | (14,707,212 | ) | ||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
55
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the Years Ended December 31, 2010, 2009, and 2008
| 2010 | 2009 | 2008 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (21,906,303 | ) | $ | (30,317,293 | ) | $ | (30,800,736 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
3,437,767 | 4,108,538 | 4,634,186 | |||||||||
| Share-based compensation |
3,151,537 | 3,130,804 | 5,581,731 | |||||||||
| Noncash interest expense |
4,053,272 | 4,014,840 | 3,474,115 | |||||||||
| Loss on monetization of receivable |
— | 317,512 | — | |||||||||
| Gain on extinguishment of debt |
(2,761,426 | ) | (2,653,387 | ) | (7,734,042 | ) | ||||||
| Asset impairment |
629,382 | — | — | |||||||||
| Gain on sale of patent applications |
— | — | (4,619,325 | ) | ||||||||
| Change in fair value of derivative liability |
(1,910,156 | ) | (47,707 | ) | — | |||||||
| Loss on disposal of assets |
161,188 | 51,584 | 17,053 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(35,000 | ) | — | 318,707 | ||||||||
| Inventories |
297,603 | (97,659 | ) | 284,496 | ||||||||
| Prepaid expenses |
47,216 | (141,498 | ) | 226,613 | ||||||||
| Accounts payable |
(198,116 | ) | 296,094 | (133,944 | ) | |||||||
| Deferred revenue |
674,101 | (440,404 | ) | 467,309 | ||||||||
| Accrued liabilities and other current liabilities |
(246,879 | ) | (2,120,876 | ) | (690,733 | ) | ||||||
| Other operating assets and liabilities |
(152,221 | ) | (293,559 | ) | 63,395 | |||||||
| Net cash used in operating activities |
(14,758,035 | ) | (24,193,011 | ) | (28,911,175 | ) | ||||||
| Cash flows from investing activities: |
||||||||||||
| Proceeds from maturities of available-for-sale securities |
40,000,000 | 30,000,000 | 24,117,910 | |||||||||
| Purchases of available-for-sale securities |
(29,989,763 | ) | (29,986,794 | ) | (29,911,527 | ) | ||||||
| Proceeds from sale of equipment |
50,299 | 53,550 | — | |||||||||
| Purchases of plant and equipment |
(130,437 | ) | (243,868 | ) | (206,010 | ) | ||||||
| Sale of patent applications |
— | — | 2,000,000 | |||||||||
| Collection of receivable from sale of patent applications |
— | 2,337,475 | — | |||||||||
| Net cash provided by (used in) investing activities |
9,930,099 | 2,160,363 | (3,999,627 | ) | ||||||||
| Cash flows from financing activities: |
||||||||||||
| Net proceeds from sales of equity |
11,525,236 | 18,572,655 | 46,545,177 | |||||||||
| Proceeds from exercise of stock options |
719 | 141,312 | 46,562 | |||||||||
| Proceeds from employee stock purchases |
48,603 | 16,933 | 286,930 | |||||||||
| Treasury stock received to satisfy minimum tax withholding requirements |
— | (54,943 | ) | (257,681 | ) | |||||||
| Payments of series A convertible preferred stock dividends |
(790,500 | ) | (790,500 | ) | (790,500 | ) | ||||||
| Payments of long-term debt |
(6,240,963 | ) | (255,000 | ) | (2,930,000 | ) | ||||||
| Net cash provided by financing activities |
4,543,095 | 17,630,457 | 42,900,488 | |||||||||
| Net (decrease) increase in cash and cash equivalents |
(284,841 | ) | (4,402,191 | ) | 9,989,686 | |||||||
| Cash and cash equivalents, beginning of year |
20,066,817 | 24,469,008 | 14,479,322 | |||||||||
| Cash and cash equivalents, end of year |
$ | 19,781,976 | $ | 20,066,817 | $ | 24,469,008 | ||||||
| Supplemental cash flow information: |
||||||||||||
| Cash paid for interest |
$ | 1,122,473 | $ | 1,573,906 | $ | 2,802,858 | ||||||
| Non-cash investing and financing activities: |
||||||||||||
| Issuance of senior secured convertible notes as payment in-kind for interest |
$ | 2,615,667 | $ | 2,418,332 | $ | 2,235,883 | ||||||
| Issuance of note receivable for assignment of certain patent applications |
— | — | 2,619,325 | |||||||||
| Issuance of common stock, $0.01 par value, as payment of long-term debt including accrued and unpaid interest |
10,361,920 | 14,134,189 | — | |||||||||
See accompanying notes to consolidated financial statements.
56
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(1) Description of Business
Agenus Inc., formerly Antigenics Inc., (including its subsidiaries, also referred to as “Agenus,” the “Company,” “we,” “us,” and “our”) is a biotechnology company developing and commercializing technologies to treat cancers and infectious diseases, primarily based on immunological approaches. Our most advanced product, Oncophage® (vitespen), is a patient-specific therapeutic cancer vaccine registered for use in Russia. As resources allow, we explore potential opportunities to seek product approval in other jurisdictions. Our Prophage Series of cancer vaccines has been tested in Phase 3 clinical trials for the treatment of renal cell carcinoma, the most common type of kidney cancer, as Oncophage, and for metastatic melanoma, and it has also been tested in Phase 1 and Phase 2 clinical trials in a range of indications. It is currently in Phase 2 clinical trials in glioma, a type of brain cancer, and adjuvant renal cell carcinoma, validating immune response. Our product candidate portfolio includes (1) QS-21 Stimulon ® adjuvant, or QS-21, which is used in numerous vaccines under development in trials, some as advanced as Phase 3, for a variety of diseases, including human immunodeficiency virus, cancer, Alzheimer’s disease, malaria, and tuberculosis, and (2) HerpV, a therapeutic vaccine program tested in a Phase 1 clinical trial for the treatment of genital herpes. Further clinical development of HerpV will be pursued if a development partnership can be successfully established. Our business activities have included product research and development, intellectual property prosecution, manufacturing, regulatory and clinical affairs, corporate finance and development activities, market development, and support of our collaborations.
Our product candidates require clinical trials and approvals from regulatory agencies, as well as acceptance in the marketplace. Part of our strategy is to develop and commercialize some of our product candidates by continuing our existing arrangements with academic and corporate collaborators and licensees and by entering into new collaborations.
We have incurred significant losses since our inception. As of December 31, 2010, we had an accumulated deficit of $584.4 million. Since our inception, we have financed our operations primarily through the sale of equity and convertible notes, interest income earned on cash, cash equivalents, and short-term investment balances, and debt provided through secured lines of credit. We believe that, based on our current plans and activities, our working capital resources as of December 31, 2010, anticipated revenues, and the estimated proceeds from our license, supply, and collaborative agreements will be sufficient to satisfy our liquidity requirements into 2012. We continue to monitor the likelihood of success of our key initiatives and are prepared to discontinue funding of such activities if they do not prove to be feasible.
Research and development program costs include compensation and other direct costs plus an allocation of indirect costs, based on certain assumptions, and our review of the status of each program. Our product candidates are in various stages of development and significant additional expenditures will be required if we start new trials, encounter delays in our programs, apply for regulatory approvals, continue development of our technologies, expand our operations, and/or bring our product candidates to market. The eventual total cost of each clinical trial is dependent on a number of factors such as trial design, length of the trial, number of clinical sites, and number of patients. The process of obtaining and maintaining regulatory approvals for new therapeutic products is lengthy, expensive, and uncertain. Because the development of our Prophage Series vaccines is subject to further evaluation and uncertainty, and because HerpV is in early-stage clinical development and requires a partner for further development, we are unable to reliably estimate the cost of completing research and development programs, the timing of bringing such programs to various markets, and, therefore, are unable to determine when, if ever, material cash inflows from operating activities are likely to commence. We will continue to adjust other spending as needed in order to preserve liquidity.
As of December 31, 2010, we had debt outstanding of $34.9 million in principal, including $34.7 million in principal of our 8% senior secured convertible notes due August 2014 (the “2006 Notes”) and $100,000 in
57
Table of Contents
principal of our 5.25% convertible senior notes due February 2025 (the “2005 Notes”). The 2005 Notes are subject to redemption at the option of the holders or us beginning February 1, 2012. We expect to attempt to raise additional funds in advance of depleting our current funds. We may attempt to raise funds by: (1) licensing technologies or products to one or more collaborative partners, (2) renegotiating third party agreements, (3) completing an outright sale of assets, (4) securing additional debt financing, and/or (5) selling additional equity securities. Satisfying long-term liquidity needs may require the successful commercialization and/or partnering arrangements for our products and product candidates including Oncophage, HerpV and vaccines containing QS-21 under development by our licensees and will require additional capital. If we incur operating losses for longer than we expect and/or we are unable to raise additional capital, we may become insolvent and be unable to continue our operations.
(2) Summary of Significant Accounting Policies
(a) Basis of Presentation and Principles of Consolidation
The consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles and include the accounts of Agenus and our wholly-owned subsidiaries. All significant intercompany transactions and accounts have been eliminated in consolidation. Certain prior period amounts have been retrospectively adjusted in order to conform to the current period’s presentation.
(b) Segment Information
We are managed and operated as one business. The entire business is managed by a single executive operating committee that reports to the chief executive officer. We do not operate separate lines of business with respect to any of our product candidates. Accordingly, we do not prepare discrete financial information with respect to separate product areas or by location and do not have separately reportable segments as defined by ASC 280, Segment Reporting.
(c) Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. We base those estimates on historical experience and on various assumptions that are believed to be reasonable under the circumstances. Actual results could differ from those estimates.
(d) Cash and Cash Equivalents
We consider all highly liquid investments purchased with maturities at acquisition of three months or less to be cash equivalents. As of December 31, 2010 and 2009, cash equivalents consist primarily of money market funds.
(e) Investments
We classify investments in marketable securities at the time of purchase. At December 31, 2009, all marketable securities are classified as available-for-sale and as such, the investments are recorded at fair value. Gains and losses on the sale of marketable securities are recognized in operations based on the specific identification method. At December 31, 2010, our investments consisted of institutional money market funds and at December 31, 2009, U.S. treasury bills.
58
Table of Contents
(f) Concentrations of Credit Risk
Financial instruments that potentially subject us to concentrations of credit risk are primarily cash equivalents, investments, and accounts receivable. We invest our cash, cash equivalents and investments in accordance with our Investment Policy, which specifies high credit quality standards and limits the amount of credit exposure from any single issue, issuer, or type of investment. We carry balances in excess of federally insured levels, however, we have not experienced any losses to date from this practice. Credit risk on accounts receivable is minimized by the financial position of the entities with which we do business. Credit losses from our customers have been immaterial.
(g) Inventories
Inventories are stated at the lower of cost or market. Cost has been determined using standard costs that approximate the first-in, first-out method.
(h) Plant and Equipment
Plant and equipment, including software developed for internal use, are carried at cost. Depreciation is computed using the straight-line method over the estimated useful lives of the assets. Amortization of leasehold improvements is computed over the shorter of the lease term or estimated useful life of the asset. Additions and improvements are capitalized, while repairs and maintenance are charged to expense as incurred. Amortization and depreciation of plant and equipment was $2.6 million, $2.8 million, and $3.3 million for the years ended December 31, 2010, 2009 and 2008, respectively.
(i) Fair Value of Financial Instruments
The estimated fair values of all of our financial instruments, excluding debt, approximate their carrying amounts in the consolidated balance sheets. As of December 31, 2010, the fair value of our 2005 Notes was estimated based on the most recent market transactions. The fair value of our 2006 Notes exclusive of the conversion option is based on a present value methodology. The outstanding principal amount of debt, including the current portion, is $34.9 million and $52.2 million at December 31, 2010 and 2009, respectively.
(j) Revenue Recognition
Revenue for services under research and development contracts are recognized as the services are performed, or as clinical trial materials are provided. Non-refundable milestone payments that represent the completion of a separate earnings process are recognized as revenue when earned. License fees and royalties are recognized as they are earned. Revenue recognized from collaborative agreements is based upon the provisions of ASC 605-25, Revenue Recognition – Multiple-Element Arrangements. Product revenue is recognized as product is shipped. For the years ended December 31, 2010, 2009, and 2008, 39%, 51%, and 68%, respectively, of our revenue was earned from one research partner. In addition, 31%, 32% and 27% of our revenue for the years ended December 31, 2010, 2009 and 2008 was earned from one of our licensees.
(k) Foreign Currency Transactions
Gains and losses from our euro based currency accounts and foreign currency transactions, such as those resulting from the translation and settlement of receivables and payables denominated in foreign currencies, are included in the consolidated statements of operations. We do not currently use derivative financial instruments to manage the risks associated with foreign currency fluctuations. We recorded foreign currency losses of $45,000, $32,000, and $378,000, for the years ended December 31, 2010, 2009, and 2008, respectively. Such losses are included as a component of operating expenses.
(l) Research and Development
Research and development expenses include the costs associated with our internal research and development activities, including salaries and benefits, share-based compensation, occupancy costs, clinical manufacturing
59
Table of Contents
costs, related administrative costs, and research and development conducted for us by outside advisors, such as sponsored university-based research partners and clinical study partners. We account for our clinical study costs by estimating the total cost to treat a patient in each clinical trial and recognizing this cost based on estimates of when the patient receives treatment, beginning when the patient enrolls in the trial. Research and development expenses also include the cost of clinical trial materials shipped to our research partners. Research and development costs are expensed as incurred.
(m) Share-Based Compensation
We account for share-based compensation in accordance with the provisions of ASC 718, Compensation—Stock Compensation and ASC 505-50, Equity-Based Payments to Non-Employees. Share-based compensation expense is recognized based on the estimated grant date fair value, and is recognized net of an estimated forfeiture rate such that we recognize compensation cost for those shares expected to vest. Compensation cost is recognized on a straight-line basis over the requisite service period of the award. See Note 10 for a further discussion on share-based compensation.
(n) Income Taxes
Income taxes are accounted for under the asset and liability method with deferred tax assets and liabilities recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis and net operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which such items are expected to be reversed or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the consolidated statement of operations in the period that includes the enactment date. Deferred tax assets are recorded when they more likely than not are expected to be realized.
(o) Net Loss Per Share
Basic income and loss per common share is calculated by dividing the net loss attributable to common stockholders by the weighted average number of common shares outstanding (including common shares issuable under our Directors’ Deferred Compensation Plan). Diluted income per common share is calculated by dividing net income attributable to common stockholders by the weighted average number of common shares outstanding (including common shares issuable under our Directors’ Deferred Compensation Plan) plus the dilutive effect of outstanding instruments such as warrants, stock options, nonvested shares, convertible preferred stock, and convertible notes. Because we have reported a net loss attributable to common stockholders for all annual periods presented, diluted loss per common share is the same as basic loss per common share, as the effect of utilizing the fully diluted share count would have reduced the net loss per common share. Therefore, shares underlying the warrants outstanding or issuable to acquire 19,856,302 shares, the outstanding stock options to acquire 7,272,850 shares, the 513,449 nonvested shares, the 2,000,000 common shares underlying the 31,620 outstanding shares of series A convertible preferred stock, and the impact of conversion of our 2005 Notes and our 2006 Notes are not included in the calculation of diluted net loss per common share.
(p) Goodwill and Acquired Intangible Assets
Goodwill represents the excess of cost over the fair value of net assets of businesses acquired. Goodwill is not amortized, but instead tested for impairment at least annually. Intangible assets with estimable useful lives are amortized over their respective estimated useful lives to their estimated residual values, and reviewed for impairment as deemed necessary.
Annually we assess whether there is an indication that goodwill is impaired, or more frequently if events and circumstances indicate that the asset might be impaired during the year. We perform our annual impairment test as of October 31 of each year. We consider ourselves a single reporting unit for purposes of the impairment test. We determine our fair value using the quoted market price of our common stock, adjusted for certain
60
Table of Contents
factors, and compare it to our net book value at the date of our evaluation. To the extent our net book value exceeds the fair value, there is an indication that the reporting unit goodwill may be impaired and a second step of the impairment test is performed to determine the amount of the impairment to be recognized, if any.
The costs of core and developed technology are presented at their estimated fair value as of their acquisition date. These costs were being amortized on a straight-line basis over their estimated useful lives of 10 years.
(q) Accounting for Asset Retirement Obligations
We record the fair value of an asset retirement obligation as a liability in the period in which we incur a legal obligation associated with the retirement of tangible long-lived assets that result from the acquisition, construction, development, and/or normal use of the assets. A legal obligation is a liability that a party is required to settle as a result of an existing or enacted law, statute, ordinance, or contract. We are also required to record a corresponding asset that is depreciated over the life of the asset. Subsequent to the initial measurement of the asset retirement obligation, the obligation will be adjusted at the end of each period to reflect the passage of time (accretion) and changes in the estimated future cash flows underlying the obligation. Changes in the liability due to accretion are charged to the consolidated statement of operations, whereas changes due to the timing or amount of cash flows are an adjustment to the carrying amount of the related asset. Our asset retirement obligations primarily relate to the expiration of our facility leases and anticipated costs to be incurred based on our lease terms.
(r) Long-lived Assets
Recoverability of assets to be held and used, other than goodwill and intangible assets not being amortized, is measured by a comparison of the carrying amount of an asset to the undiscounted future net cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future undiscounted cash flows, an impairment charge is recognized for the amount by which the carrying amount of the asset exceeds the fair value of the asset. Authoritative guidance requires companies to separately report discontinued operations and extends that reporting to a component of an entity that either has been disposed of (by sale, abandonment, or in a distribution to owners) or is classified as held for sale. Assets to be disposed of are reported at the lower of the carrying amount or fair value less costs to sell.
(s) Recent Accounting Pronouncements
In October 2009, the FASB revised authoritative guidance on multiple-deliverable revenue arrangements providing a greater ability to separate and allocate arrangement consideration in a multiple-deliverable revenue arrangement by requiring the use of estimated selling prices to allocate arrangement consideration if neither vendor-specific objective evidence nor third party evidence of selling price is available, thereby eliminating the use of the residual method of allocation. The revised guidance also requires expanded qualitative and quantitative disclosures surrounding multiple-deliverable revenue arrangements. This guidance is effective for fiscal years beginning after June 15, 2010 and may be applied retrospectively or prospectively for new or materially modified arrangements. Early adoption is permitted. We will evaluate the impact of this standard on future revenue arrangements that we may enter into.
In April 2010, the FASB codified the consensus reached in Emerging Issues Task Force Issue No. 08-09, “Milestone Method of Revenue Recognition” by issuing Accounting Standard Update (“ASU”) No. 2010-17 Milestone Method of Revenue Recognition, to limit the scope of this ASU to research or development arrangements and require that guidance in this ASU be met for an entity to apply the milestone method (which allows entities to record the milestone payment in its entirety in the period achieved). However, the FASB clarified that, even if the requirements in this ASU are met, entities would not be precluded from making an accounting policy election to apply another appropriate accounting policy that results in the deferral of some portion of the arrangement consideration. The ASU was effective for periods beginning on or after June 15, 2010. Early application was permitted. Entities can apply this guidance prospectively to milestones achieved
61
Table of Contents
after adoption. However, retrospective application to all prior periods was also permitted. We will evaluate the impact of this standard on future revenue arrangements that we may enter into.
In January 2010, the FASB issued ASU No. 2010-06, “Fair Value Measurements and Disclosures (Topic 820)—Improving Disclosures about Fair Value Measurements” (“ASU 2010-06”). ASU 2010-06 requires new disclosures regarding significant transfers in and out of Levels 1 and 2 fair value measurements, as well as information about activity in Level 3 fair value measurements, including presenting information about purchases, sales, issuances and settlements on a gross versus a net basis in the Level 3 activity roll forward. In addition, ASU 2010-06 also clarifies existing disclosures regarding input and valuation techniques, as well as the level of disaggregation for each class of assets and liabilities. ASU No. 2010-06 was effective for interim and annual periods beginning after December 15, 2009, except for the disclosures pertaining to purchases, sales, issuances and settlements in the roll forward of Level 3 activity; those disclosures are effective for interim and annual periods beginning after December 15, 2010. The adoption of ASU 2010-06 had no current impact and is expected to have no subsequent impact on our consolidated financial position, results of operations or cash flows. Required disclosure requirements of ASU 2010-06 have been included in Note 15.
In December 2010, the FASB issued additional guidance on when to perform Step 2 of the goodwill impairment test for reporting units with zero or negative carrying amounts. The criteria for evaluating Step 1 of the goodwill impairment test and proceeding to Step 2 was amended for reporting units with zero or negative carrying amounts and requires performing Step 2 if qualitative factors indicate that it is more likely than not that a goodwill impairment exists. This guidance is effective for fiscal years, and interim periods within those years, beginning after December 15, 2010. Upon adoption of the amended guidance, any impairment will be recorded as an adjustment to beginning retained earnings. We are currently evaluating the impact of adoption on our consolidated financial statements.
(3) Inventories
The components of inventories are as follows as of December 31, 2010 and 2009 (in thousands).
| 2010 | 2009 | |||||||
| Work in process |
$ | — | $ | 242 | ||||
| Finished goods |
26 | 82 | ||||||
| $ | 26 | $ | 324 | |||||
(4) Investments
Cash Equivalents and Short-term Investments
Cash equivalents and short-term investments consisted of the following as of December 31, 2010 and 2009 (in thousands).
| 2010 | 2009 | |||||||||||||||
| Cost | Estimated Fair Value |
Cost | Estimated Fair Value |
|||||||||||||
| Institutional money market funds |
$ | 19,782 | $ | 19,782 | $ | 19,468 | $ | 19,468 | ||||||||
| U.S. treasury bills |
— | — | 9,998 | 9,998 | ||||||||||||
| $ | 19,782 | $ | 19,782 | $ | 29,466 | $ | 29,466 | |||||||||
Proceeds from maturities of available-for-sale securities amounted to $ 40.0 million, $30.0 million, and $24.1 million, for the years ended December 31, 2010, 2009, and 2008, respectively. No available-for-sale
62
Table of Contents
securities were sold before their maturity in 2010, 2009, or 2008. Gross realized gains and gross realized losses included in net loss as a result of those maturities were immaterial for each of the years in the three-year period ended December 31, 2010. As a result of the short-term nature of our investments, there were no unrealized holding gains or losses as of December 31, 2010, 2009 and 2008.
Of the investments listed above, $19.8 million and $19.5 million have been classified as cash equivalents on our consolidated balance sheet as of December 31, 2010 and 2009, respectively. Approximately $10.0 million were classified as short-term investments as of December 31, 2009.
(5) Plant and Equipment
Plant and equipment as of December 31, 2010 and 2009 consists of the following (in thousands).
| 2010 | 2009 | Estimated Depreciable Lives |
||||||||||
| Furniture, fixtures, and other |
$ | 1,649 | $ | 1,648 | 3 to 10 years | |||||||
| Laboratory and manufacturing equipment |
5,546 | 6,817 | 4 to 10 years | |||||||||
| Leasehold improvements |
18,218 | 22,778 | 2 to 12 years | |||||||||
| Software and computer equipment |
5,774 | 6,070 | 3 years | |||||||||
| Construction in progress |
— | 191 | ||||||||||
| 31,187 | 37,504 | |||||||||||
| Less accumulated depreciation and amortization |
(24,993 | ) | (28,613 | ) | ||||||||
| $ | 6,194 | $ | 8,891 | |||||||||
During the year ended December 31, 2010, plant and equipment with a net book value of approximately $155,000 was retired from service and disposed.
(6) Other Intangible Assets
The following table presents certain information on our intangible assets as of December 31, 2010 and 2009 (in thousands).
| Weighted Average Amortization Period |
As of December 31, 2010 | As of December 31, 2009 | ||||||||||||||||||||||||||||||
| Gross Carrying Amount |
Impairment Charge |
Accumulated Amortization |
Net Carrying Amount |
Gross Carrying Amount |
Accumulated Amortization |
Net Carrying Amount |
||||||||||||||||||||||||||
| Amortizing intangible assets: |
||||||||||||||||||||||||||||||||
| Core and developed technology |
10 years | $ | 11,073 | $ | 630 | $ | 10,443 | $ | — | $ | 11,073 | $ | 9,753 | $ | 1,320 | |||||||||||||||||
Our intangible assets were being amortized over their estimated useful lives of 10 years, with no estimated residual values. Amortization expense related to core and developed technology was $690,000, $1.1 million, and $1.1 million, in 2010, 2009 and 2008 respectively. As further development of Aroplatin, a liposomal chemotherapeutic tested in a Phase 1 clinical trial for the treatment of solid malignancies and B-cell lymphomas, was discontinued, we determined that an impairment had occurred and accordingly recorded a loss of approximately $630,000 during the year ended December 31, 2010, representing the net carrying value of the intangible asset related to liposomal technology at the time development was discontinued. This impairment charge is included in research and development expenses.
63
Table of Contents
(7) Income Taxes
We are subject to taxation in the U.S. and various state, local, and foreign jurisdictions. We remain subject to examination by U.S. Federal, state, local, and foreign tax authorities for tax years 2007 through 2010. With a few exceptions, we are no longer subject to U.S. Federal, state, local, and foreign examinations by tax authorities for the tax year 2006 and prior. However, net operating losses from the tax year 2006 and prior would be subject to examination if and when used in a future tax return to offset taxable income. Our policy is to recognize income tax related penalties and interest, if any, in our provision for income taxes and, to the extent applicable, in the corresponding income tax assets and liabilities, including any amounts for uncertain tax positions.
As of December 31, 2010, we have available net operating loss carryforwards of $481.8 million and $120.7 million for Federal and state income tax purposes, respectively, which are available to offset future Federal and state taxable income, if any, and expire between 2011 and 2030. Our ability to use these net operating losses is limited by change of control provisions under Internal Revenue Code Section 382 and may expire unused. In addition, we have $7.9 million and $6.4 million of Federal and state research and development credits, respectively, available to offset future taxable income. These Federal and state research and development credits expire between 2012 and 2030 and 2015 and 2025, respectively. The potential impacts of such provisions are among the items considered and reflected in management’s assessment of our valuation allowance requirements.
The tax effect of temporary differences and net operating loss and tax credit carryforwards that give rise to significant portions of the deferred tax assets and deferred tax liabilities as of December 31, 2010 and 2009 are presented below (in thousands).
| 2010 | 2009 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 170,171 | $ | 167,263 | ||||
| Research and development tax credits |
12,122 | 12,929 | ||||||
| Other |
13,042 | 13,618 | ||||||
| Total deferred tax assets |
195,335 | 193,810 | ||||||
| Less: valuation allowance |
(195,052 | ) | (192,292 | ) | ||||
| Net deferred tax assets |
283 | 1,518 | ||||||
| Deferred tax liabilities |
(283 | ) | (1,518 | ) | ||||
| Net deferred tax |
$ | — | $ | — | ||||
In assessing the realizablility of deferred tax assets, we consider whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which the net operating loss and tax credit carryforwards can be utilized or the temporary differences become deductible. We consider projected future taxable income and tax planning strategies in making this assessment. In order to fully realize the deferred tax asset, we will need to generate future taxable income sufficient to utilize net operating losses prior to their expiration. Based upon our history of not generating taxable income due to our business activities focused on product development, we believe that it is more likely than not that deferred tax assets will not be realized through future earnings. Accordingly, a valuation allowance has been established for deferred tax assets which will not be offset by the reversal of deferred tax liabilities. The valuation allowance on the deferred tax assets increased by $2.8 million during the year ended December 31, 2010 and decreased $3.4 million during the year ended December 31, 2009. The net operating loss includes amounts pertaining to tax deductions relating to stock exercises for which any subsequently recognized tax benefit will be recorded as an increase to additional paid-in capital.
64
Table of Contents
Income tax benefit was nil for each of the years ended December 31, 2010, 2009, and 2008, and differed from the amounts computed by applying the U.S. Federal income tax rate of 34% to loss before income taxes as a result of the following (in thousands).
| 2010 | 2009 | 2008 | ||||||||||
| Computed “expected” Federal tax benefit |
$ | (7,451 | ) | $ | (10,308 | ) | $ | (10,472 | ) | |||
| (Increase) reduction in income taxes benefit resulting from: |
||||||||||||
| Change in valuation allowance |
2,760 | (3,415 | ) | 5,311 | ||||||||
| Increase due to uncertain tax positions |
67 | 241 | 4,615 | |||||||||
| State and local income benefit, net of Federal income tax benefit |
(534 | ) | (1,498 | ) | (1,799 | ) | ||||||
| Net operating loss expirations |
4,363 | 14,759 | — | |||||||||
| Other, net |
795 | 221 | 2,345 | |||||||||
| $ | — | $ | — | $ | — | |||||||
As of December 31, 2010 and 2009, our gross unrecognized tax benefits totaled $5.4 and $5.3 million, respectively. These unrecognized tax benefits would all impact the effective tax rate if recognized. There are no positions which we anticipate could change within the next twelve months.
A reconciliation of the beginning and ending amount of gross unrecognized tax benefits is as follows (in thousands):
| Balance, December 31, 2009 |
$ | 5,349 | ||
| Increase related to current year positions |
62 | |||
| Increase related to previously recognized positions |
18 | |||
| Balance, December 31, 2010 |
$ | 5,429 | ||
(8) Accrued Liabilities
Accrued liabilities consist of the following as of December 31, 2010 and 2009 (in thousands)
| 2010 | 2009 | |||||||
| Payroll |
$ | 1,086 | $ | 155 | ||||
| Professional fees |
888 | 915 | ||||||
| Clinical contractors |
89 | 295 | ||||||
| Accrued interest |
2 | 437 | ||||||
| Other |
620 | 795 | ||||||
| $ | 2,685 | $ | 2,597 | |||||
(9) Equity
Our authorized capital stock consists of 250,000,000 shares of $0.01 par value per share of common stock and 25,000,000 shares of preferred stock, $0.01 par value per share. Our Board of Directors is authorized to issue the preferred stock and to set the voting, conversion, and other rights.
In a private placement in September 2003, we sold 31,620 shares of our series A convertible preferred stock, par value $0.01 per share, for net proceeds of $31.6 million. Under the terms and conditions of the Certificate of Designation creating the series A convertible preferred stock, this stock is convertible by the holder at any time into our common stock, is non-voting, carries a 2.5% annual dividend yield, has an initial conversion price of $15.81 per common share, subject to adjustment, and is redeemable by us at its face amount ($31.6 million) on or
65
Table of Contents
after September 24, 2013. The Certificate of Designation does not contemplate a sinking fund. The series A convertible preferred stock ranks senior to our common stock. In a liquidation, dissolution, or winding up of the Company, the series A convertible preferred stock’s liquidation preference must be fully satisfied before any distribution could be made to the holders of the common stock. Other than in such a liquidation, no terms of the series A convertible preferred stock affect our ability to declare or pay dividends on our common stock as long as the series A convertible preferred stock’s dividends are accruing. The liquidation value of this series A convertible preferred stock is equal to $1,000 per share outstanding plus any accrued unpaid dividends. Accrued and unpaid dividends of the series A convertible preferred stock aggregated $197,625 or $6.25 per share, at December 31, 2010.
On September 10, 2007, we issued 1,623,377 shares of our common stock at a price of $3.08 per share to a single institutional investor. In conjunction with this transaction, we also issued to the investor 10,000 shares of our new series B1 convertible preferred stock and 5,250 shares of our new series B2 convertible preferred stock. Shares of the series B1 convertible preferred stock permitted the investor, within one year of the anniversary of closing, to purchase up to an additional $10.0 million of common shares at a purchase price equal to the lesser of $3.08 per share or a price calculated based on the then-prevailing price of our common stock minus $0.30 per share. Gross proceeds of $5.0 million were received as a result of this transaction. Net proceeds, after deducting the placement agent fees and offering expenses paid by us, were $4.7 million. The class B convertible preferred stock has been recorded as an equity classified instrument in accordance with the applicable authoritative guidance. In April 2008, we issued 1,585,197 shares of our common stock upon conversion of 10,000 shares of our series B1 convertible preferred stock via a cashless conversion. These shares were issued pursuant to an effective shelf registration statement. Shares of the series B2 convertible preferred stock permit the investor to purchase common shares for consideration of up to 35 percent of the total dollar amount previously invested pursuant to the agreement with the investor, including conversions of the series B1 convertible preferred stock, at a purchase price equal to the lesser of $4.16 per common share or a price calculated based on the then-prevailing price of our common stock, and such right expires seven years from the date of issuance. In April 2009, we issued 5,929,212 shares of our common stock upon conversion of 2,145 shares of our series B2 convertible preferred stock via cashless conversions. Upon completion of the conversions, 3,105 shares of our series B2 convertible preferred stock are still outstanding although no further shares can be converted into shares of common stock as the maximum number of shares (as defined in the agreement) have been issued. The total number of shares of common stock issued or issuable to the holder of the class B convertible preferred stock cannot exceed 19.9% of our outstanding common stock. No dividends are paid on the class B convertible preferred stock and there are no liquidation preferences.
On January 9, 2008, we entered into a private placement agreement (the “January 2008 private placement”) pursuant to which we sold 8,708,717 shares of common stock. Investors also received (i) 10-year warrants to purchase, at an exercise price of $3.00 per share, up to 8,708,717 shares of common stock and (ii) unit warrants to purchase, at an exercise price of $3.00 per unit, contingent upon a triggering event as defined in the January 2008 private placement documents, (a) up to 8,708,717 shares of common stock and (b) additional 10-year warrants to purchase, at an exercise price of $3.00 per share, up to 8,708,717 additional shares of common stock. We raised net proceeds in the January 2008 private placement of $25.8 million, after deducting offering costs of $296,000.
In accordance with the terms of the January 2008 private placement, the 10-year warrants became exercisable for a period of 9.5 years as of July 9, 2008. Our private placement in April 2008 qualified as a triggering event, and therefore the unit warrants became exercisable for a period of eighteen months as of July 9, 2008. The unit warrants expired unexercised in January 2010.
In February 2008, we filed a registration statement covering the resale of the 8,708,717 shares of common stock issued and the 8,708,717 shares issuable upon the exercise of the 10-year warrants issued in the January 2008 private placement. The Securities and Exchange Commission (the “SEC”) declared the resale registration statement effective on February 14, 2008.
66
Table of Contents
On April 8, 2008, we entered into a private placement agreement (the “April 2008 private placement”) under which we sold (i) 7,000,000 shares of common stock and (ii) five-year warrants to acquire up to 7,000,000 shares of common stock at an exercise price of $3.75 per share, for $3.00 for each share and warrant sold. The warrants became exercisable for a period of 4.5 years as of October 10, 2008. We raised net proceeds in the April 2008 private placement of $19.7 million, after deducting offering costs of $1.3 million.
In April 2008, we filed a registration statement covering the resale of the 7,000,000 shares of common stock issued and the 7,000,000 shares issuable upon the exercise of the related warrants issued in the April 2008 private placement. The SEC declared the resale registration statement effective on May 7, 2008.
On July 30, 2009, we entered into a private placement agreement under which we issued and sold (i) 5,000,000 shares of our common stock, (ii) six-month warrants to purchase up to 2,500,000 additional shares of common stock at an exercise price of $2.00 per share, and (iii) four-year warrants to purchase up to 2,173,900 additional shares of common stock at an exercise price of $2.30 per share, for $2.00 for each share sold generating gross proceeds of $10.0 million. The six-month warrants expired unexercised in January 2010. Subsequently, we filed, and the SEC declared effective, a registration statement covering the resale of the 5,000,000 shares of common stock issued and the 4,673,900 shares issuable upon the exercise of the related warrants issued in this private placement.
On August 3, 2009, we entered into a private placement agreement under which we issued and sold (i) 4,385,965 shares of our common stock, (ii) six-month warrants to purchase up to 2,192,982 additional shares of common stock at an exercise price of $2.31 per share, and (iii) four-year warrants to purchase up to 1,973,685 additional shares of common stock at an exercise price of $2.50 per share, for $2.28 for each share sold generating gross proceeds of $10.0 million. The warrants were not exercisable for the first six months following the closing, which occurred on August 4, 2009. The six-month warrants expired unexercised in July 2010. Subsequently, we filed, and the SEC declared effective, a registration statement covering the resale of the 4,385,965 shares of our common stock issued and the 4,166,667 shares issuable upon the exercise of the related warrants issued in this private placement. In connection with the two private placements during 2009, we raised net proceeds of $18.6 million, after deducting offering costs of $1.4 million.
As part of all private placement agreements, we agreed to register the shares of common stock and the shares of common stock underlying the warrants (with the exception of the unit warrants from the January 2008 private placement) issued to the investors with the SEC within contractually specified time periods. As noted above, we filed registration statements covering all required shares. We have also agreed to use our best efforts to keep the registration statements continuously effective. If we are unable to keep the registration statements continuously effective in accordance with the terms of the private placements, we are subject to liquidated damages of up to a maximum of 10% of the aggregate purchase price paid by the original investors, or up to $3.8 million as of December 31, 2010.
In April 2008, we issued and sold a total of 271,762 shares of our common stock through our placement agent, Wm Smith & Co., and raised net proceeds of $804,000, after deducting offering costs of $38,000, in at the market transactions. Proceeds from the offering were used for general corporate purposes. During the year ended December 31, 2010, we issued approximately 6.8 million shares of our common stock under an At the Market Sales Agreement through our sales agents, McNicoll, Lewis & Vlak LLC and Wm Smith & Co. and raised net proceeds of approximately $8.6 million after deducting offering costs of approximately $325,000. These offerings were made under effective shelf registration statements. Approximately 13 million shares remain available for sale under this agreement.
On December 13, 2010, we entered into subscription agreements under which we issued and sold 3,199,451 shares of our common stock for the aggregate purchase price of $2,879,506. Additionally, within 90 calendar days of the date of the subscription agreements, the investors have the right and option to purchase up to an additional 639,890 shares of our common stock for the aggregate purchase price of up to $575,901. The offering and sale of these common shares were made under an effective shelf registration statement.
67
Table of Contents
During the years ended December 31, 2009, and 2008, certain employees, in lieu of paying withholding taxes on the vesting of nonvested stock awarded under our 1999 Equity Incentive Plan, as amended (the “1999 EIP”), authorized the withholding of an aggregate of 117,913, and 137,078 shares, respectively, of common stock to satisfy the minimum tax withholding requirements related to such vesting. We recorded these shares as treasury stock using the cost method at the market price of the common stock on the vesting dates.
(10) Share-based Compensation Plans
Our 1999 EIP authorized awards of incentive stock options within the meaning of Section 422 of the Internal Revenue Code (the “Code”), non-qualified stock options, nonvested (restricted) stock, and unrestricted stock for up to 12,000,000 shares of common stock (subject to adjustment for stock splits and similar capital changes and exclusive of options exchanged at the consummation of mergers) to employees and, in the case of non-qualified stock options, nonvested (restricted) stock, and unrestricted stock, to consultants and directors as defined in the 1999 EIP. The plan terminated on November 15, 2009. On March 12, 2009, our Board of Directors adopted, and on June 10, 2009, our stockholders approved, our 2009 Equity Incentive Plan (the “2009 EIP”). The 2009 EIP provides for the grant of incentive stock options intended to qualify under Section 422 of the Code, nonstatutory stock options, restricted stock, unrestricted stock and other equity-based awards, such as stock appreciation rights, phantom stock awards, and restricted stock units, which we refer collectively as Awards, for up to 13,000,000 shares of our common stock (subject to adjustment in the event of stock splits and other similar events). The Board of Directors appointed the Compensation Committee to administer the 1999 EIP and the 2009 EIP.
Under the 1999 Employee Stock Purchase Plan, as amended (the “1999 ESPP”), eligible employees purchased shares of common stock at a discount from fair value. There were 450,000 shares of common stock reserved for issuance under the 1999 ESPP. The 1999 ESPP, which terminated on November 15, 2009, was intended to qualify as an employee stock purchase plan within the meaning of Section 423 of the Code. On March 12, 2009, our Board of Directors adopted, and on June 10, 2009, our stockholders approved, the 2009 Employee Stock Purchase Plan (the “2009 ESPP”) to provide eligible employees the opportunity to acquire our common stock in a program also designed to comply with Section 423 of the Code. There are 500,000 shares of common stock reserved for issuance under the 2009 ESPP subject to adjustment as defined in the plan. Rights to purchase common stock under the 2009 ESPP are granted at the discretion of the Compensation Committee, which determines the frequency and duration of individual offerings under the plan and the dates when stock may be purchased. Eligible employees participate voluntarily and may withdraw from any offering at any time before the stock is purchased. Participation terminates automatically upon termination of employment. The purchase price per share of common stock in an offering is 85% of the lesser of its fair value at the beginning of the offering period or on the applicable exercise date and may be paid through payroll deductions, periodic lump sum payments, the delivery of our common stock, or a combination thereof. Unless otherwise permitted by the Board of Directors, no participant may acquire more than 20,000 shares of stock in any offering period. No participant is allowed to purchase shares under the 2009 ESPP if such employee would own or would be deemed to own stock possessing 5% or more of the total combined voting power or value of the Company. No offerings will be made under the 2009 ESPP after June 10, 2019.
Our Director’s Deferred Compensation Plan, as amended, permits each outside director to defer all, or a portion of, their cash compensation until their service as a director ends or until a specified date into a cash account or a stock account. There are 450,000 shares of our common stock reserved for issuance under this plan. As of December 31, 2010, 92,946 shares have been issued. Amounts deferred to a cash account will earn interest at the rate paid on one-year Treasury bills with interest added to the account annually. Amounts deferred to a stock account will be converted on a quarterly basis into a number of units representing shares of our common stock equal to the amount of compensation which the participant has elected to defer to the stock account divided by the applicable price for our common stock. The applicable price for our common stock has been defined as the average of the closing price of our common stock for all trading days during the calendar quarter preceding the conversion date as reported by The Nasdaq Capital Market. Pursuant to this plan, a total of 426,789 units, each
68
Table of Contents
representing a share of our common stock at a weighted average common stock price of $1.86, have been credited to participants’ stock accounts as of December 31, 2010. The compensation charges for this plan were immaterial for all periods presented.
We use the Black-Scholes option pricing model to value options granted to employees, and non-employees as well as options granted to members of our Board of Directors. All stock option grants have 10-year terms and generally vest ratably over a four-year period. The non-cash charge to operations for the non-employee options with vesting or other performance criteria is affected each reporting period, until the non-employee options vest, by changes in the fair value of our common stock.
The fair value of each option granted during the periods was estimated on the date of grant using the following weighted average assumptions:
| 2010 | 2009 | 2008 | ||||||||||
| Expected volatility |
104 | % | 94 | % | 71 | % | ||||||
| Expected term in years |
6 | 6 | 5 | |||||||||
| Risk-free interest rate |
2.1 | % | 2.7 | % | 2.8 | % | ||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
Expected volatility is based exclusively on historical volatility data of our common stock. The expected term of stock options granted is based on historical data and other factors and represents the period of time that stock options are expected to be outstanding prior to exercise. The risk-free interest rate is based on U.S. Treasury strips with maturities that match the expected term on the date of grant.
A summary of option activity for 2010 is presented below:
| Options | Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2009 |
6,148,621 | $ | 2.93 | |||||||||||||
| Granted |
2,021,700 | 0.80 | ||||||||||||||
| Exercised |
(958 | ) | 0.75 | |||||||||||||
| Forfeited |
(281,989 | ) | 1.60 | |||||||||||||
| Expired |
(614,524 | ) | 4.69 | |||||||||||||
| Outstanding at December 31, 2010 |
7,272,850 | $ | 2.24 | 7.1 | $ | 456,372 | ||||||||||
| Vested or expected to vest at December 31, 2010 |
7,028,559 | $ | 2.28 | 7.1 | $ | 408,652 | ||||||||||
| Exercisable at December 31, 2010 |
4,686,716 | $ | 2.76 | 6.6 | $ | 130,898 | ||||||||||
The weighted average grant-date fair values of options granted during the years ended December 31, 2010, 2009, and 2008, was $0.61, $ 1.21, and $1.03, respectively.
The aggregate intrinsic value in the table above represents the difference between our closing stock price on the last trading day of fiscal 2010 and the exercise price, multiplied by the number of in-the-money options that would have been received by the option holders had all option holders exercised their options on December 31, 2010 (the intrinsic value is considered to be zero if the exercise price is greater than the closing stock price). This amount changes based on the fair market value of our stock. The total intrinsic value of options exercised during the years ended December 31, 2010, 2009, and 2008, determined on the dates of exercise, was $0 and $54,000 and $21,000, respectively.
69
Table of Contents
During 2010, 2009, and 2008, all options were granted with exercise prices equal to the market value of the underlying shares of common stock on the grant date.
As of December 31, 2010, $1.3 million of total unrecognized compensation cost related to stock options granted to employees and directors is expected to be recognized over a weighted average period of 1.6 years.
As of December 31, 2010, unrecognized expense for options granted to outside advisors for which performance (vesting) has not yet been completed but the exercise price of the option is known is $64,000. Such amount is subject to change each reporting period based upon changes in the fair value of our common stock, expected volatility, and the risk-free interest rate, until the outside advisor completes his or her performance under the option agreement.
Certain employees and consultants have been granted nonvested stock. The fair value of nonvested stock is calculated based on the closing sale price of our common stock on the date of issuance.
A summary of nonvested stock activity for 2010 is presented below:
| Nonvested Shares |
Weighted Average Grant Date Fair Value |
|||||||
| Outstanding at December 31, 2009 |
200,029 | $ | 1.13 | |||||
| Granted |
1,949,844 | 0.81 | ||||||
| Vested |
(1,589,249 | ) | 0.88 | |||||
| Forfeited |
(47,175 | ) | 1.08 | |||||
| Outstanding at December 31, 2010 |
513,449 | 0.77 | ||||||
As of December 31, 2010, there was $295,000 of unrecognized share-based compensation expense related to these nonvested shares. This cost is expected to be recognized over a weighted average period of 1.8 years. The total intrinsic value of shares vested during the years ended December 31, 2010, 2009, and 2008, was $1.6 million, $1.5 million, and $1.3 million, respectively.
Cash received from option exercises and purchases under our 1999 ESPP and our 2009 ESPP (collectively the “ESPPs”) for the years ended December 31, 2010, 2009, and 2008, was $49,000, $158,000, and $333,000, respectively. We issue new shares upon option exercises, purchases under our ESPPs, vesting of nonvested stock, and under the Director’s Deferred Compensation Plan. During the years ended December 31, 2010, 2009, and 2008, 89,725 shares, 41,300 shares, and 171,113 shares, were issued under the ESPPs, respectively. During the year ended December 31, 2010, 1,585,902 shares were issued as a result of the vesting of nonvested stock. During the year ended December 31, 2009, 2,221,176 shares, net of 117,913 shares withheld to cover personal income tax withholding, were issued as a result of the vesting of nonvested stock. During the year ended December 31, 2008, 629,912 shares, net of 137,078 shares withheld to cover personal income tax withholding, were issued as a result of the vesting of nonvested stock. The shares withheld were recorded as treasury stock using the cost method, at weighted average prices of $ 0.47 per share and $1.88 per share during the years ended December 31, 2009, and 2008, respectively, based on the closing sale price of our common stock on the vesting dates, for a total of approximately $55,000, and $258,000, respectively.
For the years ended December 31, 2009, and 2008, 15,376 shares, and 61,938 shares, respectively, were issued under our Directors’ Deferred Compensation Plan. No shares were issued during the year ended December 31, 2010.
70
Table of Contents
The impact on our results of operations from share-based compensation for the years ended December 31, 2010, 2009, and 2008, was as follows (in thousands).
| 2010 | 2009 | 2008 | ||||||||||
| Research and development |
$ | 1,058 | $ | 864 | $ | 1,517 | ||||||
| General and administrative |
2,094 | 2,267 | 4,065 | |||||||||
| Total share-based compensation expense |
$ | 3,152 | $ | 3,131 | $ | 5,582 | ||||||
(11) License, Research, and Other Agreements
In November 1994, we entered into a Patent License Agreement with the Mount Sinai School of Medicine, or Mount Sinai (the “Mount Sinai Agreement”). Through the Mount Sinai Agreement, we obtained an exclusive worldwide license to patent rights relating to the heat shock protein technology that resulted from the research and development performed by Dr. Pramod Srivastava, our founding scientist and a former member of our Board of Directors. We agreed to pay Mount Sinai a royalty on the net sales of products covered by the licensed patent rights and also provided Mount Sinai with a 0.45% equity interest in the Company (approximately 62,000 shares valued at $90,000 at the time of issuance). The term of the Mount Sinai Agreement ends when the last of the licensed patents expires (2018) or becomes no longer valid. If we fail to pay royalties that are due under the agreement, Mount Sinai may issue written notice to us. If we continue to fail to pay royalties after 60 days from receipt of the written notice, Mount Sinai can terminate the agreement. The Mount Sinai Agreement requires us to use due diligence to make the products covered by the licensed patent rights commercially available, including a requirement for us to use best efforts to reach a number of developmental milestones which have been achieved. If we fail to comply with the due diligence provisions of the agreement, Mount Sinai could take actions to convert our exclusive license to a non-exclusive license after six months written notice. The Mount Sinai Agreement does not contain any milestone payment provisions.
During 1995, Dr. Srivastava moved his research to Fordham University (“Fordham”). We entered into a sponsored research and technology license agreement with Fordham (the “Fordham Agreement”) in March 1995 relating to the continued development of the heat shock protein technology and agreed to make payments to Fordham to sponsor Dr. Srivastava’s research. Through the Fordham Agreement, we obtained an exclusive, perpetual, worldwide license to all of the intellectual property, including all the patent rights which resulted from the research and development performed by Dr. Srivastava at Fordham. We also agreed to pay Fordham a royalty on the net sales of products covered by the Fordham Agreement through the last expiration date on the patents under the agreement (2018) or when the patents become no longer valid. The agreement does not contain any milestone payment provisions or any diligence provisions. Dr. Srivastava moved his research to the University of Connecticut Health Center (“UConn”) during 1997 and, accordingly, the parts of the agreement related to payments for sponsored research at Fordham terminated in mid-1997. During the term of the agreement, we paid $2.4 million to Fordham.
We entered into a license agreement with UConn in May 2001 (the “License Agreement”) that provides us with the exclusive, worldwide rights to technologies discovered and developed under the research agreement. The term of the License Agreement ends when the last of the licensed patents expires (2019) or becomes no longer valid. UConn may terminate the License Agreement: (1) if, after 30 days written notice for breach, we continue to fail to make any payments due under the License Agreement, or (2) we cease to carry on our business related to the patent rights or if we initiate or conduct actions in order to declare bankruptcy. We may terminate the License Agreement upon 90 days written notice. The License Agreement contains aggregate milestone payments of $1.2 million for each product we develop covered by the licensed patent rights. These milestone payments are contingent upon regulatory filings, regulatory approvals and commercial sales of products. We have also agreed to pay UConn a royalty on the net sales of products covered by the License Agreement as well as annual license maintenance fees beginning in May 2006. Royalties otherwise due on the net sales of products covered by the License Agreement may be credited against the annual license maintenance fee obligations. To date, we have paid $240,000 to UConn under the License Agreement. The License Agreement gives us complete
71
Table of Contents
discretion over the commercialization of products covered by the licensed patent rights, but also requires us to use commercially reasonable diligent efforts to introduce commercial products within and outside the United States. If we fail to meet these diligence requirements, UConn may be able to terminate the License Agreement.
In March 2003, we entered into an amendment agreement that amended certain provisions of the License Agreement. The amendment agreement granted us a license to additional patent rights. In consideration for execution of the amendment agreement, we agreed to pay UConn an upfront payment and to make future payments for licensed patents or patent applications. Through December 31, 2010, we have paid approximately $100,000 to UConn under the License Agreement, as amended.
We have entered into various agreements with institutions and contract research organizations to conduct clinical studies. Under these agreements, subject to the enrollment of patients and performance by the institution of certain services, we have estimated our payments to be $47.2 million over the term of the studies. For the years ended December 31, 2010, 2009, and 2008, $361,000, $170,000, and $123,000, respectively, have been expensed in the accompanying consolidated statements of operations related to these clinical studies. Through December 31, 2010, $46.3 million of this estimate has been paid. The timing of our expense recognition and future payments related to these agreements is dependent on the enrollment of patients and documentation received from the institutions.
We have various comprehensive agreements with collaborative partners that allow for the use of QS-21, an investigational adjuvant used in numerous vaccines under development for a variety of diseases including, but not limited to, hepatitis, HIV, influenza, cancer, Alzheimer’s disease, malaria, and tuberculosis. These agreements grant exclusive worldwide rights in some fields of use, and co-exclusive or non-exclusive rights in others. The agreements call for royalties to be paid to us by the collaborative partner on the future sales of licensed vaccines that include QS-21.
On July 6, 2006, we and GlaxoSmithKline Biologicals SA (“GSK”) entered into an expanded license agreement (the “GSK License Agreement”) and an expanded Manufacturing Technology Transfer and Supply Agreement (the “2006 GSK Supply Agreement”) for the use of QS-21. Under the terms of the agreements, we agreed to supply QS-21 to GSK through 2014. In addition, we agreed to transfer manufacturing technologies under the 2006 GSK Supply Agreement. In conjunction with the GSK License Agreement and the 2006 GSK Supply Agreement, we received a $3.0 million upfront non-refundable payment in July 2006. In February 2007, we received and recorded $2.0 million as revenue as a result of the achievement of a milestone related to the transfer of manufacturing technologies to GSK.
On July 20, 2007, we executed a letter (the “GSK Letter”) with GSK amending the 2006 GSK Supply Agreement to accelerate GSK’s commercial grade QS-21 manufacturing rights previously granted in July 2006. On January 16, 2009, we entered into an Amended and Restated Manufacturing Technology Transfer and Supply Agreement (the “Amended GSK Supply Agreement”) reflecting the provisions of the letter. Accordingly, from the effective date of the GSK Letter, GSK has the right to manufacture all of its requirements of commercial grade QS-21. In addition, the parties have amended their purchase and supply obligations with respect to pre-commercial grade QS-21. In accordance with the terms of the Amended GSK Supply Agreement, upon our election, GSK is obligated to supply us (or our affiliates, licensees, or customers) certain quantities of commercial grade QS-21 for a stated period of time.
As consideration for our entering into the GSK Letter, we received a $2.0 million upfront non-refundable payment from GSK in August 2007, in lieu of a milestone payment that would have otherwise been payable under the 2006 GSK Supply Agreement. In addition, GSK is obligated to make payments to us totaling $5.25 million through December 2012, of which $3.5 million has been received, for manufacturing profits that were anticipated to have otherwise been earned under the 2006 GSK Supply Agreement. Except as expressly provided in the Amended GSK Supply Agreement, all other financial obligations of GSK under the 2006 GSK Supply Agreement, including royalty payments, remain unchanged. The Amended GSK Supply Agreement does not
72
Table of Contents
affect the rights and obligations of the parties under the GSK License Agreement. We are entitled to receive royalties on the net sales for a period of at least 10 years after the first commercial sale of a resulting GSK product.
During the years ended December 31, 2010, 2009, and 2008, we recognized revenue of $1.3 million each year related to payments received under our license and supply agreements with GSK. Deferred revenue of $3.6 million related to our agreements with GSK is included in deferred revenue on our consolidated balance sheet as of December 31, 2010.
Effective September 14, 2009, we entered into an Amended and Restated License Agreement (“Amended License Agreement”) with Elan Corporation, plc and/or its affiliates (“Elan”) and Elan Pharmaceuticals, Inc. On September 17, 2009, the Amended License Agreement was assigned to JANSSEN Alzheimer’s Immunotherapy, a subsidiary of Johnson & Johnson. Under the terms of the Amended License Agreement assigned to JANSSEN Alzheimer Immunotherapy, they will have the right to develop, make, have made, use, sell, offer for sale, import, and have sold the Alzheimer’s disease vaccine that contains QS-21 (the “Licensed Product”). In addition, pursuant to the terms of the Amended License Agreement, JANSSEN Alzheimer Immunotherapy has the right to manufacture all of its requirements of QS-21 for use in the Licensed Product and we have no further supply obligations. To date, we have received $1.5 million in upfront and milestone payments under this agreement and are entitled to receive future payments contingent upon successful milestone achievements. In addition, we are entitled to receive royalties on a country-by-country basis on net sales of the Licensed Product for a period of at least 10 years after first commercial sale in that country. Deferred revenue of $1.3 million related to this Amended License Agreement is included in deferred revenue on our consolidated balance sheet as of December 31, 2010.
(12) Certain Related Party Transactions
In March 1995, we entered into a consulting agreement with Dr. Pramod Srivastava, our founding scientist and a former member of our Board of Directors, and upon its expiration in March 2006, we entered into a new consulting agreement, effective March 28, 2006, with Dr. Srivastava. The agreement with Dr. Srivastava has an initial term of five years and is automatically extended for successive terms of one year unless either party notifies the other at least 90 days prior to the expiration of the original or any extension term that the agreement is not to be extended. The agreement may be terminated without cause by us during its term, subject to the payment of compensation for twelve months at the then current rate provided for under the agreement. In exchange for the timely performance of services, as defined in the agreement, Dr. Srivastava is entitled to receive compensation to be established by the Compensation Committee of the Agenus Board of Directors. During the year ended December 31, 2009, we paid Dr. Srivastava an additional $50,000 for his work related to our marketing authorization application submitted to the European Medicines Agency.
On January 9, 2008, we entered into the January 2008 private placement that included (i) 8,708,717 shares of common stock, (ii) warrants to acquire up to 8,708,717 shares of common stock at $3.00 per share, and (iii) unit warrants, which, if exercisable due to a triggering event as that term is defined in the applicable warrant, permit a holder to acquire up to 8,708,717 shares of common stock at $3.00 per share and additional ten-year warrants to acquire up to an additional 8,708,717 shares of common stock at $3.00 per share. In conjunction with this private placement, we sold 542,050 shares of common stock to Garo H. Armen, Ph.D., our Chairman and Chief Executive Officer, and 1,166,667 shares of common stock to Armen Partners LP. Garo H. Armen is general partner of Armen Partners LP and owns a controlling interest therein. In addition to the common stock acquired by Garo H. Armen and Armen Partners LP, each acquired an equal number of both warrants and unit warrants. The unit warrants expired unexercised on January 9, 2010.
(13) Leases
We lease manufacturing, research and development, and office facilities under various long-term lease arrangements. Rent expense (before sublease income) was $2.6 million, $2.9 million, and $2.9 million, for the years ended December 31, 2010, 2009, and 2008, respectively.
73
Table of Contents
We lease a 162,000 square foot facility in Lexington, Massachusetts. We currently occupy 94,000 square feet of this facility. The future minimum rental payments under our leases of our New York City facility, which expires in 2012, and our Lexington headquarters, which expires in 2013, are as follows (in thousands).
| Year ending December 31, |
||||
| 2011 |
$ | 2,224 | ||
| 2012 |
2,141 | |||
| 2013 |
1,406 | |||
| Total |
$ | 5,771 | ||
In connection with the Lexington facility, we maintain a fully collateralized letter of credit of $1.0 million. No amounts have been drawn on the letter of credit as of December 31, 2010. In addition, for the office space in New York City, we were required to deposit $161,000 with the landlord as an interest-bearing security deposit pursuant to our obligations under the lease.
We sublet a portion of our Lexington and Framingham facilities and received rental payments of $1.1 million in 2010. For the years ended December 31, 2009, and 2008, we received sublease rental payments of $1.2 million in each period with respect to our subleased facilities. We are contractually entitled to receive rental payments of $530,000 and $309,000 in 2011 and 2012, respectively.
(14) Debt
As of December 31, 2010, we have $34.9 million in principal of debt outstanding: $34.7 million due in 2014 (2006 Notes), $100,000 due in 2025 (2005 Notes) and $146,000 currently due.
Convertible Notes—2006 Notes
On October 30, 2006 (the “Issuance Date”), we issued $25.0 million of the 2006 Notes to a group of accredited investors (“Investors”). These 2006 Notes bear interest at 8% (an effective rate of 8.10%) payable semi-annually on December 30 and June 30 in cash or, at our option, in additional notes or a combination thereof and had an original maturity date of August 30, 2011. During the years ended December 31, 2010, 2009 and 2008, we issued additional 2006 Notes in the amount of $2.6 million, $2.4 million and $2.2 million, respectively, as payment for interest due.
On November 11, 2008, we entered into an Amendment of Rights Agreement with the majority holder of our 2006 Notes. The Amendment of Rights Agreement amended the definition of an Event of Default under the 2006 Notes to exclude the redemption and repurchase of up to $15 million of our 2005 Notes and modified certain anti-dilutive rights of the holders of the 2006 Notes upon our issuance and sale of certain new securities up to the aggregate dollar amount expended by us for the repurchase of the 2005 Notes. On July 31 and August 3, 2009, the majority holder of our 2006 Notes agreed to waive certain anti-dilutive rights of the holders of the 2006 Notes upon our issuance and sale of certain new securities up to the aggregate dollar amount expended by us for the repurchase of the 2005 Notes during 2009. In connection with the waiver in August 2009, the fixed conversion price was adjusted from $3.50 to $3.00 per share. During 2010, the majority holder of our 2006 Notes agreed to again waive certain anti-dilutive rights of the holders of the 2006 Notes upon our issuance and sale of certain new securities up to the aggregate dollar amount expended by us for the repurchase of the 2005 Notes during 2010.
On February 23, 2011, we entered into a Ninth Amendment of Rights Agreement (the “Amendment”) to the 2006 Notes. The Amendment extends the maturity date of the 2006 Notes to August 31, 2014, and waives the rights of the note holders to convert the 2006 Notes into our common stock. The Amendment also removes substantially all restrictions on us incurring indebtedness subordinate to the 2006 Notes and substantially all
74
Table of Contents
restrictions to issue our common stock. We have also agreed to waive our right to prepay these notes in the event that our shares trade at a weighted average price over $7.00 for a 30-day period.
As of December 31, 2010, the 2006 Notes were convertible into our common stock at a fixed conversion price of $3.00 per share at the option of the Investors. If, prior to the original maturity date of these notes, we were to issue or sell, or in accordance with the terms of the 2006 Notes we were deemed to have issued or sold, any shares of our common stock (including the issuance or sale of shares of our common stock owned or held by or for our account, but excluding certain excluded securities) for a consideration per share of less than $3.00 (the “New Issuance Price”), then immediately after such issuance, the fixed conversion price then in effect was to be reduced to an amount equal to a 16.66% premium to the New Issuance Price. Effective with the Amendment this conversion provision is removed from the terms of the 2006 Notes. The 2006 Notes can be converted into an interest in one of our wholly-owned subsidiaries that holds the rights or patents to QS-21 and HerpV. If converted into an interest of this subsidiary, the ownership interest in the subsidiary is determined by multiplying the quotient of the conversion amount divided by $25.0 million, by 30%.
Prior to the Amendment, at any time after October 30, 2009, we were able to call the 2006 Notes and accrued interest at face value for cash if our shares had a minimum average trading price during the prior 30-day period of $7.00 or higher. This provision was removed with the Amendment. If the Investors elect at any time to convert the 2006 Notes into ownership of the subsidiary holding the rights or patents to QS-21 and HerpV, we have the right, within 60 days, to redeem the 2006 Notes, including accrued interest, at a redemption price providing a 30-percent internal rate of return to the Investors. The 2006 Notes are secured by our equity ownership in this subsidiary.
Upon the maturity of the 2006 Notes, we may elect to repay the outstanding balance in cash or in common stock, subject to certain limitations. If we elect to satisfy the outstanding balance with common stock at maturity, the number of shares issued will be determined by dividing the cash obligation by 90 percent of the weighted average price of the common shares for the 20 trading days preceding the maturity date of the 2006 Notes. This right is subject to our market capitalization exceeding $300 million at such time.
In no event will any Investor be obligated to accept equity that would result in an Investor owning in excess of 9.99% of the Company’s outstanding common stock at any given time in connection with any conversion, redemption, or repayment of the 2006 Notes. Prior to the Amendment, the note agreements included material restrictions on the Company’s incurrence of debt and liens while the 2006 Notes were outstanding, as well as other customary covenants. The Amendment removes substantially all restrictions on the Company incurring indebtedness subordinate to the 2006 Notes. The note agreements also include a change of control provision whereby the holders of the 2006 Notes could require us to redeem all or a portion of the then outstanding 2006 Notes at a price equal to 101% of the conversion amount being redeemed and a right of first refusal provision for the holders of the 2006 Notes on any sales of equity of the subsidiary holding the rights or patents to QS-21 and HerpV, to purchase up to 50% of such sales of equity on the same terms as the third-party purchaser.
Prior to the Amendment, if we at any time on or after the Issuance Date subdivided (by any stock split, stock dividend, recapitalization, or otherwise) one or more classes of our outstanding shares of common stock into a greater number of shares, the fixed conversion price in effect immediately prior to such subdivision would have been proportionately reduced. If we at any time on or after the Issuance Date combined (by combination, reverse stock split, or otherwise) one or more classes of our outstanding shares of common stock into a smaller number of shares, the fixed conversion price in effect immediately prior to such combination would have been proportionately increased. If any event occurred of the type contemplated above but not expressly provided for by such provisions (including, without limitation, the granting of stock appreciation rights, phantom stock rights, or other rights with equity features), then our Board of Directors would have made an appropriate adjustment in the fixed conversion price then in effect so as to protect the rights of the holders of the 2006 Notes; provided that no such adjustment would have increased the fixed conversion price then in effect as otherwise determined. The Amendment removes these provisions from the terms of the 2006 Notes.
75
Table of Contents
Convertible Notes—2005 Notes
On January 25, 2005, we issued $50.0 million of our 2005 Notes. Proceeds from the sale of the 2005 Notes were approximately $48.0 million net of issuance costs. Issuance costs are being amortized using the effective interest method over seven years, the expected life of the 2005 Notes based on the earliest date on which the holders can require redemption. During 2008, we repurchased $11.8 million in principal of these 2005 Notes for $2.9 million plus accrued interest of $178,000. We recorded a gain of $7.7 million in non-operating income, which is net of related debt issuance costs that were relieved. During 2009, we repurchased $18.2 million in principal of our 2005 Notes for $255,000 and approximately 5,482,000 shares of our common stock. In connection with these 2009 repurchases we recorded a net gain of $2.7 million in non-operating income, which is comprised of inducement expense of $9.8 million and a gain on extinguishment of debt of $12.5 million. During 2010, we repurchased $19.9 million in principal of the 2005 Notes for $6.2 million and approximately 9,643,000 shares of our common stock. In connection with these 2010 repurchases we recorded a net gain of $2.8 million in non-operating income, which is comprised of inducement expense of $8.9 million and a gain on extinguishment of debt of $11.7 million. At December 31, 2010, $100,000 of the 2005 Notes remains outstanding.
The 2005 Notes, which mature in 2025, bear interest payable semi-annually on February 1 and August 1 of each year, at a rate of 5.25% per annum (an effective rate of 5.94%) and are convertible into common stock at an initial conversion price of $10.76 per share. On or after February 1, 2012, we may redeem the 2005 Notes for cash, at a redemption price equal to 100% of the principal amount of the 2005 Notes, plus any accrued and unpaid interest. On each of February 1, 2012, February 1, 2015 and February 1, 2020, holders may require us to purchase their 2005 Notes for cash equal to 100% of the principal amount of the 2005 Notes, plus any accrued and unpaid interest. At December 31, 2010, $100,000 of the 2005 Notes remain outstanding.
Convertible Notes—Conversion Option
As of January 1, 2009, we adopted revised guidance that addressed certain matters applicable to convertible debt instruments and retrospectively applied this change in accounting to all prior periods presented for which we had applicable outstanding convertible debt, as required by this new guidance. Under this new method of accounting, the debt and equity components of our 2005 Notes and our 2006 Notes are bifurcated and accounted for separately based on the fair value and related interest rate of a non-convertible debt security with the same terms. The fair value of a non-convertible debt instrument at the original issuance dates of our 2005 Notes and our 2006 Notes was determined to be $42.6 million and $23.6 million, respectively. The equity (conversion options) components of our convertible debt securities have been included in additional paid-in capital on our consolidated balance sheet and, accordingly, the initial carrying value of the debt securities was reduced by $8.8 million. Our previously reported net loss for the years ended December 31, 2008 was increased by $2.1 million primarily due to recognizing the accretion of the reduced carrying values of our convertible debt securities to their face amount as additional non-cash interest expense.
Additionally, as a result of the adoption of revised guidance for evaluating when adjustment features within contracts are considered to be equity-indexed, as of January 1, 2009, the conversion feature embedded in our 2006 Notes is now treated as a derivative liability and recorded at its fair value, with period to period changes in the fair value recorded as a gain or loss in our consolidated statement of operations. Accordingly, upon adoption we recorded a reduction to additional paid-in capital of $1.4 million, an increase to debt discount of $1.3 million, a derivative liability of $2.7 million, and a charge to opening accumulated deficit of $21,000. As of December 31, 2010 and 2009, our debt discount balance was $720,000, and $2.5 million, respectively. During the year ended December 21, 2010, we recorded a gain of $1.9 million due to the change in the fair value of the derivative. For the year ended December 31, 2009, we recorded a charge to other income of $48,000 due to changes in the fair value of the derivative and noncash interest expense of $1.3 million due to the adoption of this revised guidance.
Other
At December 31, 2010, approximately $146,000 of debentures we assumed in our merger with Aquila Biopharmaceuticals are outstanding. These debentures carry interest at 7% and are callable by the holders. Accordingly they are classified as part of the current portion of long-term debt.
76
Table of Contents
(15) Fair Value Measurements
We measure fair value based on a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that market participants would use in pricing the asset or liability based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the inputs that market participants would use in pricing the asset or liability and are developed based on the best information available in the circumstances. The fair value hierarchy is broken down into three levels based on the source of inputs as follows:
Level 1—Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access;
Level 2—Valuations based on quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active and models for which all significant inputs are observable, either directly or indirectly; and
Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement.
The availability of observable inputs can vary among the various types of financial assets and liabilities. To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, for financial statement disclosure purposes, the level in the fair value hierarchy within which the fair value measurement is categorized is based on the lowest level input that is significant to the overall fair value measurement.
We measure our short-term investments and derivative liability at fair value. Our short-term investments are comprised of U.S. Treasury securities that are valued using quoted market prices with no valuation adjustments applied. Accordingly, these securities are categorized in Level 1. Our derivative liability is classified within Level 3 because it is valued using a modified Black-Scholes model due to the potential at December 31, 2010 for the note holders to convert into shares of either our common stock or an interest in on of our wholly-owned subsidiaries. Certain inputs into this model were valued using a combination of income and market approaches which are unobservable in the market and are significant.
The estimated fair values of all of our financial instruments, excluding long-term debt, approximate their carrying amounts in the consolidated balance sheets. The fair value of our long-term debt was derived by evaluating the nature and terms of each note and considering the prevailing economic and market conditions at the balance sheet date.
Liabilities measured at fair value are summarized below (in thousands):
| Description |
December 31, 2010 | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Unobservable Inputs (Level 3) |
|||||||||
| Liabilities: |
||||||||||||
| Derivative Liability |
$ | 755 | — | $ | 755 | |||||||
| Description |
December 31, 2009 | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Unobservable Inputs (Level 3) |
|||||||||
| Assets: |
||||||||||||
| Short-term investments |
$ | 9,998 | $ | 9,998 | — | |||||||
| Liabilities: |
||||||||||||
| Derivative Liability |
$ | 2,665 | — | $ | 2,665 | |||||||
77
Table of Contents
The following table presents our liabilities measured at fair value using significant unobservable inputs (Level 3), as of December 31, 2010 (amounts in thousands):
| Balance, December 31, 2009 |
$ | 2,665 | ||
| Decrease in fair value for the year ended December 31, 2010 |
(1,910 | ) | ||
| Balance, December 31, 2010 |
$ | 755 | ||
The decrease in fair value of the derivative liability is included in non-operating income in our consolidated statement of operations for the year ended December 31, 2010.
As of December 31, 2010, $100,000 in principal of the 2005 Notes are outstanding with an estimated fair value of $87,000 based on recent market transactions. As of December 31, 2010, $34.7 million in principal of the 2006 Notes are outstanding with a fair value of the debt portion exclusive of the conversion option estimated to be $30.8 million based on a present value methodology.
(16) Contingencies
Agenus, our Chairman and CEO, Garo H. Armen, Ph.D., and two investment banking firms that served as underwriters in our initial public offering were named as defendants in a federal civil class action lawsuit in the United States District Court for the Southern District of New York. Substantially similar actions were filed concerning the initial public offerings for more than 300 different issuers, and the cases were coordinated for pre-trial purposes as In re Initial Public Offering Securities Litigation, 21 MC 92. The suit alleges that the brokerage arms of the investment banking firms charged secret excessive commissions to certain of their customers in return for allocations of our stock in the offering. The suit also alleges that shares of our stock were allocated to certain of the investment banking firms’ customers based upon agreements by such customers to purchase additional shares of our stock in the secondary market. The parties have reached a global settlement of the litigation. Under the settlement, the insurers will pay the full amount of settlement share allocated to the defendants, and the defendants will bear no financial liability. Agenus and the other defendants will receive complete dismissals from the case. In October 2009, the Court entered an order granting final approval of the settlement, and subsequently judgment was entered. Various objectors have filed appeals. If for any reason the settlement does not become effective, we believe we have meritorious defenses to the claims and intend to defend the action vigorously. We are unable to predict the likelihood of an unfavorable outcome or estimate our potential liability, if any. No accrual has been recorded at December 31, 2010 for this action.
We may currently be, or may become a party, to other legal proceedings as well. While we currently believe that the ultimate outcome of any of these proceedings will not have a material adverse effect on our financial position, results of operations, or liquidity, litigation is subject to inherent uncertainty. Furthermore, litigation consumes both cash and management attention.
(17) 401(k) Plan
We sponsor a defined contribution 401(k) savings plan for all eligible employees, as defined. Participants may contribute up to 60% of their compensation, as defined in the savings plan, with a maximum contribution of $16,500 for individuals under 50 years old and $22,000 for individuals 50 years old and older in 2010. Each participant is fully vested in his or her contributions and related earnings and losses. The Company matched 50% of the participant’s contribution, subject to a maximum of 6% of compensation through February 2009. Such matching contributions vest over four years. In 2010 we made a discretionary contribution to the savings plan of approximately $42,000. For the years ended December 31, 2010, 2009, and 2008, we expensed $42,000, $37,000, and $163,000, respectively, for the Company’s contributions to the 401(k) plan.
78
Table of Contents
(18) Restructuring Costs
On February 2, 2009, we initiated a plan of restructuring that resulted in a reduction of our workforce by approximately 20%, or 19 positions. We engaged in this workforce reduction in order to reduce operating expenses in light of market conditions and to focus our resources on near-term commercial opportunities. This restructuring action resulted in total charges of approximately $177,000 in severance and outplacement expenses in the quarter ended March 31, 2009, with $42,000 included in research and development expenses and $135,000 included in general and administrative expenses in our consolidated statement of operations. The charge to operations was reduced by $10,000 during the quarter ended June 30, 2009 based on actual activities. All amounts were paid during 2009.
(19) Quarterly Financial Data (Unaudited)
| Quarter Ended, | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| (In thousands, except per share data) | ||||||||||||||||
| 2010 |
||||||||||||||||
| Revenue |
$ | 936 | $ | 806 | $ | 624 | $ | 994 | ||||||||
| Net loss |
(8,811 | ) | (4,972 | ) | (5,707 | ) | (2,416 | ) | ||||||||
| Net loss attributable to common stockholders |
(9,009 | ) | (5,170 | ) | (5,905 | ) | (2,613 | ) | ||||||||
| Per common share, basic and diluted: |
||||||||||||||||
| Net loss attributable to common stockholders |
$ | (0.10 | ) | $ | (0.05 | ) | $ | (0.06 | ) | $ | (0.03 | ) | ||||
| Quarter Ended, | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| (In thousands, except per share data) |
||||||||||||||||
| 2009 |
||||||||||||||||
| Revenue |
$ | 621 | $ | 1,270 | $ | 896 | $ | 547 | ||||||||
| Net (loss) income |
(9,476 | ) | (12,087 | ) | (10,612 | ) | 1,858 | |||||||||
| Net (loss) income attributable to common stockholders |
(9,674 | ) | (12,285 | ) | (10,810 | ) | 1,661 | |||||||||
| Per common share, basic and diluted: |
||||||||||||||||
| Net (loss) income attributable to common stockholders |
$ | (0.14 | ) | $ | (0.17 | ) | $ | (0.13 | ) | $ | 0.02 | |||||
Net (loss) income attributable to common stockholders per share is calculated independently for each of the quarters presented. Therefore, the sum of the quarterly net loss per share amounts will not necessarily equal the total for the full fiscal year.
79
Table of Contents
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
Not applicable.
| Item 9A. | Controls and Procedures |
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer concluded that our disclosure controls and procedures were functioning effectively as of the end of the period covered by this Annual Report on Form 10-K to provide reasonable assurance that the Company can meet its disclosure obligations.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Securities Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2010.
KPMG LLP, our independent registered public accounting firm, has issued their report, included herein, on the effectiveness of our internal control over financial reporting.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the fourth quarter of 2010 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
80
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Agenus Inc.:
We have audited Agenus Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Agenus Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Agenus Inc. and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Agenus Inc. and subsidiaries as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity (deficit) and comprehensive loss, and cash flows for each of the years in the three-year period ended December 31, 2010, and our report dated March 16, 2011 expressed an unqualified opinion on those consolidated financial statements.
/s/ KPMG LLP
Boston, Massachusetts
March 16, 2011
81
Table of Contents
| Item 9B. | Other Information |
None.
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
The response to this item is incorporated by reference from “Executive Officers of the Registrant” found in Part I of this Annual Report on Form 10-K, following Item 4 of this Annual Report on Form 10-K, and from sections entitled “Proposal 1 – Election of Directors,” “Our Corporate Governance,” and “Section 16(a) Beneficial Ownership Reporting Compliance” in our Proxy Statement relating to our 2011 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of our 2010 fiscal year (the “2011 Proxy Statement”).
| Item 11. | Executive Compensation |
The response to this item is incorporated by reference into this Annual Report on Form 10-K from sections entitled “Our Corporate Governance,” “Compensation Discussion and Analysis,” “Compensation Committee Report,” “Compensation of Executive Officers,” and “Director Compensation” in our 2011 Proxy Statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The response to this item is incorporated by reference into this Annual Report on Form 10-K from sections entitled “Equity Plans” and “Ownership of Our Common Stock” in our 2011 Proxy Statement.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The response to this item is incorporated by reference into this Annual Report on Form 10-K from the sections entitled “Our Corporate Governance” and “Certain Relationships and Related Transactions” in our 2011 Proxy Statement.
| Item 14. | Principal Accountant Fees and Services |
The response to this item is incorporated by reference into this Annual Report on Form 10-K from the section entitled “Proposal 3—Ratify the Appointment of KPMG LLP as our Independent Registered Public Accounting Firm for the Fiscal Year Ending December 31, 2011” in our 2011 Proxy Statement.
82
Table of Contents
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
(a) 1. Consolidated Financial Statements
The consolidated financial statements are listed under Item 8 of this Annual Report on Form 10-K.
2. Financial Statement Schedules
The financial statement schedules required under this Item and Item 8 are omitted because they are not applicable or the required information is shown in the consolidated financial statements or the footnotes thereto.
3. Exhibits
The exhibits are listed below under Part IV Item 15(b).
(b) Exhibits
Exhibit Index
| Exhibit No. |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation of Antigenics Inc. Filed as Exhibit 3.1 to our Current Report on Form 8-K (File No. 0-29089) filed on June 10, 2002 and incorporated herein by reference. | |
| 3.1.1 | Certificate of Amendment to the Amended and Restated Certificate of Incorporation of Antigenics Inc. Filed as Exhibit 3.1 to our Current Report on Form 8-K (File No. 0-29089) filed on June 11, 2007 and incorporated herein by reference. | |
| 3.1.2 | Certificate of Ownership and Merger changing the name of the corporation to Agenus Inc. Filed as Exhibit 3.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 6, 2011 and incorporated herein by reference. | |
| 3.2 | Fifth Amended and Restated By-laws of Agenus Inc. Filed as Exhibit 3.2 to our Current Report on Form 8-K (File No. 0-29089) filed on January 6, 2011 and incorporated herein by reference. | |
| 3.3 | Certificate of Designation, Preferences and Rights of the Series A Convertible Preferred Stock of Agenus Inc. filed with the Secretary of State of the State of Delaware on September 24, 2003. Filed as Exhibit 3.1 to our Current Report on Form 8-K (File No. 0-29089) filed on September 25, 2003 and incorporated herein by reference. | |
| 3.4 | Certificate of Designations, Preferences and Rights of the Class B Convertible Preferred Stock of Agenus Inc. Filed as Exhibit 3.1 to our Current Report on Form 8-K (File No. 0-29089) filed on September 5, 2007 and incorporated herein by reference. | |
| 4.1 | Form of Common Stock Certificate. Filed as Exhibit 4.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 6, 2011 and incorporated herein by reference. | |
| 4.2 | Indenture, dated January 25, 2005, between the Registrant and HSBC Bank USA, National Association. Filed as Exhibit 4.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 25, 2005 and incorporated herein by reference. | |
| 4.3 | Registration Rights Agreement, dated January 25, 2005, between the Registrant and the initial purchasers. Filed as Exhibit 4.2 to our Current Report on Form 8-K (File No. 0-29089) filed on January 25, 2005 and incorporated herein by reference. | |
| 4.4 | Form of Amended and Restated Note under the Securities Purchase Agreement dated as of October 30, 2006 (as amended), by and among Agenus Inc. Inc., a Delaware corporation and the investors listed on the Schedule of Buyers thereto. Filed herewith. | |
83
Table of Contents
| Exhibit No. |
Description | |
| 4.5 | Form of Amended and Restated PIK Note under the Securities Purchase Agreement dated as of October 30, 2006 (as amended), by and among Agenus Inc. Inc., a Delaware corporation and the investors listed on the Schedule of Buyers thereto. Filed herewith. | |
| 4.6 | Pledge of Security Agreement dated as of October 30, 2006 by and among Agenus Inc. Inc., a Delaware corporation and the investors listed on the Schedule of Buyers thereto. Filed as Exhibit 4.3 to our Current Report on Form 8-K (File No. 0-29089) filed on October 31, 2006 and incorporated herein by reference. | |
| 4.7 | Guaranty dated as of October 30, 2006 by and between Agenus Inc. Inc., a Massachusetts corporation and Ingalls & Snyder LLC, as Collateral Agent for the Buyers. Filed as Exhibit 4.4 to our Current Report on Form 8-K (File No. 0-29089) filed on October 31, 2006 and incorporated herein by reference. | |
| 4.8 | Guaranty dated as of October 30, 2006 by and between Aronex Pharmaceuticals, Inc. and Ingalls & Snyder LLC, as Collateral Agent for the Buyers. Filed as Exhibit 4.5 to our Current Report on Form 8-K (File No. 0-29089) filed on October 31, 2006 and incorporated herein by reference. | |
| 4.9 | Securities Purchase Agreement dated as of October 30, 2006 by and among Agenus Inc. Inc., a Delaware corporation and the investors listed on the Schedule of Buyers thereto. Filed as Exhibit 4.6 to our Current Report on Form 8-K (File No. 0-29089) filed on October 31, 2006 and incorporated herein by reference. | |
| 4.10 | Form of Warrant under the Securities Purchase Agreement dated January 9, 2008. Filed as Exhibit 4.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 11, 2008 and incorporated herein by reference. | |
| 4.11 | Purchase Agreement dated August 31, 2007 by and between Agenus Inc. and Fletcher International. Filed as Exhibit 99.1 to our Current Report on Form 8-K (File No. 0-29089) filed on September 5, 2007 and incorporated herein by reference. | |
| 4.12 | Securities Purchase Agreement dated April 8, 2008. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on April 10, 2008 and incorporated herein by reference. | |
| 4.13 | Form of Warrant to purchase common stock dated April 9, 2008. Filed as Exhibit 4.1 to our Current Report on Form 8-K (File No. 0-29089) filed on April 10, 2008 and incorporated herein by reference. | |
| 4.14 | Securities Purchase Agreement by and between Agenus Inc. and the investors identified on Schedule I attached to the agreement, dated January 9, 2008. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 11, 2008 and incorporated herein by reference. | |
| 4.15 | Form of 4 Year Warrant under the Securities Purchase Agreement dated July 30, 2009. Filed as Exhibit 4.2 to our Current Report on Form 8-K (File No. 0-29089) filed on August 3, 2009 and incorporated herein by reference. | |
| 4.16 | Form of 4 Year Warrant under the Securities Purchase Agreement dated August 3, 2009. Filed as Exhibit 4.2 to our Current Report on Form 8-K (File No. 0-29089) filed on August 5, 2009 and incorporated herein by reference. | |
| 4.17 | Ninth Amendment of Rights with respect to Events of Default and Issuance of Other Securities by and between Agenus Inc. and Ingalls & Snyder Value Partners L.P. dated February 23, 2011. Filed herewith. | |
84
Table of Contents
| Exhibit No. |
Description | |
| 10.1* | 1999 Equity Incentive Plan, as amended. Filed as Exhibit 10.1 to our Annual Report on Form 10-K (File No. 0-29089) for the year ended December 31, 2008 and incorporated herein by reference. | |
| 10.1.2* | Form of Non-Statutory Stock Option. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on December 15, 2004 and incorporated herein by reference. | |
| 10.1.3* | Form of 2007 Restricted Stock Award Agreement. Filed as Exhibit 10.1.5 to our Annual Report on Form 10-K (File No. 0-29089) for the year ended December 31, 2007 and incorporated herein by reference. | |
| 10.1.4* | Form of 2008 Restricted Stock Award Agreement. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on March 11, 2008 and incorporated herein by reference. | |
| 10.1.5* | Sixth Amendment to the Agenus Inc. 1999 Equity Incentive Plan. Filed as Appendix D to our Definitive Proxy Statement on Schedule 14A filed on April 27, 2009 and incorporated herein by reference. | |
| 10.2* | 1999 Employee Stock Purchase Plan, as amended. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on June 11, 2007 and incorporated herein by reference. | |
| 10.3 | Founding Scientist’s Agreement between Agenus Inc. and Pramod K. Srivastava, Ph.D. dated March 28, 1995. Filed as Exhibit 10.3 to our registration statement on Form S-1 (File No. 333-91747) and incorporated herein by reference. | |
| 10.3.1(1) | Amendment to Founding Scientist’s Agreement dated January 1, 2003. Filed as Exhibit 10.29 to our Annual Report on Form 10-K (File No. 0-29089) for the year ended December 31, 2002 and incorporated herein by reference. | |
| 10.4 | Form of Indemnification Agreement between Agenus Inc. and its directors and executive officers. These agreements are materially different only as to the signatories and the dates of execution. Filed as Exhibit 10.4 to our registration statement on Form S-1 (File No. 333-91747) and incorporated herein by reference. | |
| 10.4.1 | Current schedule indentifying the directors and executive officers who are party to an Indemnification Agreement, the form of which was filed as Exhibit 10.4 to our registration statement on Form S-1 (File No. 333-91747). Filed herewith. | |
| 10.5(1) | Patent License Agreement between Agenus Inc. and Mount Sinai School of Medicine dated November 1, 1994, as amended on June 5, 1995. Filed as Exhibit 10.8 to our registration statement on Form S-1 (File No. 333-91747) and incorporated herein by reference. | |
| 10.6(1) | Sponsored Research and Technology License Agreement between Agenus Inc. and Fordham University dated March 28, 1995, as amended on March 22, 1996. Filed as Exhibit 10.9 to our registration statement on Form S-1 (File No. 333-91747) and incorporated herein by reference. | |
| 10.7 | Lease of Premises at 3 Forbes Road, Lexington, Massachusetts dated as of December 6, 2002 from BHX, LLC, as Trustee of 3 Forbes Realty Trust, to Agenus Inc. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 8, 2003 and incorporated herein by reference. | |
| 10.7.1 | First Amendment of Lease dated as of August 15, 2003 from BHX, LLC, as trustee of 3 Forbes Road Realty, to Agenus Inc. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended March 31, 2004 and incorporated herein by reference. | |
85
Table of Contents
| Exhibit No. |
Description | |
| 10.7.2 | Second Amendment of Lease dated as of March 7, 2007 from BHX, LLC as trustee of 3 Forbes Road Realty, to Agenus Inc. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended March 31, 2007 and incorporated herein by reference. | |
| 10.7.3 | Third Amendment to Lease dated April 23, 2008 between TBCI, LLC, as successor to BHX, LLC, as Trustee of 3 Forbes Road Realty Trust, and Agenus Inc. Filed as Exhibit 10.2 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2008 and incorporated herein by reference. | |
| 10.7.4 | Fourth Amendment to Lease dated September 30, 2008 between TBCI, LLC, as successor to BHX, LLC, as Trustee of 3 Forbes Road Realty Trust, and Agenus Inc. Filed as Exhibit 10.2 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2008 and incorporated herein by reference. | |
| 10.8* | Agenus Inc. Directors’ Deferred Compensation Plan, as amended. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on June 11, 2007 and incorporated herein by reference. | |
| 10.8.1* | Third Amendment to Directors’ Deferred Compensation Plan. Filed as Appendix E to our Definitive Proxy Statement on Schedule 14A filed on April 27, 2009 and incorporated herein by reference. | |
| 10.8.2* | Fourth Amendment to Directors’ Deferred Compensation Plan. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on December 14, 2010 and incorporated herein by reference. | |
| 10.9(1) | License Agreement between the University of Connecticut Health Center and Agenus Inc. dated May 25, 2001, as amended on March 18, 2003. Filed as Exhibit 10.2 to the Amendment No. 1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended March 31, 2003 and incorporated herein by reference. | |
| 10.9.1(1) | Letter Agreement by and between Agenus Inc. and The University of Connecticut Health Center dated May 11, 2009. Filed as Exhibit 10.5 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2009 and incorporated herein by reference. | |
| 10.9.2(1) | Amendment Number Two to License Agreement by and between Agenus Inc. and The University of Connecticut Health Center dated June 5, 2009. Filed as Exhibit 10.6 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2009 and incorporated herein by reference. | |
| 10.10* | Employment Agreement dated February 20, 2007 between Agenus Inc. and Shalini Sharp. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on February 26, 2007 and incorporated herein by reference. | |
| 10.10.1* | First Amendment to Employment Agreement dated July 2, 2009 between Agenus Inc. and Shalini Sharp. Filed as Exhibit 10.2 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference. | |
| 10.10.2* | Second Amendment to Employment Agreement dated December 15, 2010 between Agenus Inc. and Shalini Sharp. Filed herewith. | |
| 10.11* | Employment Agreement dated February 20, 2007 between Agenus Inc. and Kerry Wentworth. Filed as Exhibit 10.2 to our Current Report on Form 8-K (File No. 0-29089) filed on February 26, 2007 and incorporated herein by reference. | |
86
Table of Contents
| Exhibit No. |
Description | |
| 10.11.1* | First Amendment to Employment Agreement dated July 2, 2009 between Agenus Inc. and Kerry Wentworth. Filed as Exhibit 10.4 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference. | |
| 10.11.2* | Second Amendment to Employment Agreement dated December 15, 2010 between Agenus Inc. and Kerry Wentworth. Filed herewith. | |
| 10.12* | Employment Agreement dated December 1, 2005 between Agenus Inc. and Garo Armen. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on December 7, 2005 and incorporated herein by reference. | |
| 10.12.1* | First Amendment to Employment Agreement dated July 2, 2009 between Agenus Inc. and Garo Armen. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference | |
| 10.12.2* | Second Amendment to Employment Agreement dated December 15, 2010 between Agenus Inc. and Garo Armen. Filed herewith. | |
| 10.13* | Amended and Restated Executive Change-in-Control Plan. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on November 3, 2010 and incorporated herein by reference. | |
| 10.14* | 2004 Executive Incentive Plan, as amended. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on January 27, 2011 and incorporated herein by reference. | |
| 10.15* | Consulting Agreement dated March 28, 2006 between Agenus Inc. and Pramod Srivastava. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on March 28, 2006 and incorporated herein by reference. | |
| 10.16(1) | License Agreement by and between Agenus Inc. and GlaxoSmithKline Biologicals SA dated July 6, 2006. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2006 and incorporated herein by reference. | |
| 10.17 | Standard Form of Loft Lease effective October 24, 2006 between 162 Fifth Avenue Associates LLC and Agenus Inc. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2006 and incorporated herein by reference. | |
| 10.18 | Form of the Johns Hopkins University Uniform Provisions for Board Service. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on September 15, 2006 and incorporated herein by reference. | |
| 10.19 | At Market Issuance Sales Agreement between Antigenics Inc. and McNicoll, Lewis & Vlak LLC and Wm Smith & Co., dated February 26, 2010. Filed as Exhibit 1.1 to our Current Report on Form 8-K (File No. 0-29089) filed on March 1, 2010 and incorporated herein by reference. | |
| 10.20* | Employment Agreement dated September 16, 2008 between Agenus Inc. and Karen Valentine. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on September 19, 2008 and incorporated herein by reference. | |
| 10.20.1* | First Amendment to Employment Agreement dated July 2, 2009 between Agenus Inc. and Karen Valentine. Filed as Exhibit 10.3 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference. | |
| 10.20.2* | Second Amendment to Employment Agreement dated December 15, 2010 between Agenus Inc. and Karen Valentine. Filed herewith. | |
87
Table of Contents
| Exhibit No. |
Description | |
| 10.21(1) | Amended and Restated License Agreement by and between Antigenics Inc., a Massachusetts corporation and wholly owned subsidiary of Agenus Inc., Elan Pharma International Limited, and Elan Pharmaceuticals, Inc. dated September 14, 2009. Filed as Exhibit 10.5 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference. | |
| 10.22 | Notice of Assignment of Amended and Restated License Agreement by and between Antigenics Inc., a Massachusetts corporation and wholly owned subsidiary of Agenus Inc., Elan Pharma International Limited, and Elan Pharmaceuticals, Inc. dated September 17, 2009. Filed as Exhibit 10.6 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference. | |
| 10.23(1) | Supply Agreement by and between Agenus Inc. and ISSI-Strategy LLC dated July 9, 2009. Filed as Exhibit 10.7 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended September 30, 2009 and incorporated herein by reference. | |
| 10.24(1) | Binding Letter of Intent by and between Agenus Inc. and ISSI-Strategy dated May 24, 2009. Filed as Exhibit 10.7 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2009 and incorporated herein by reference. | |
| 10.25 | Securities Purchase Agreement dated as of July 30, 2009 by and among Agenus Inc. and the investors listed on the Schedule of Buyers thereto. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on August 3, 2009 and incorporated herein by reference. | |
| 10.26 | Securities Purchase Agreement dated as of August 3, 2009 by and among Agenus Inc. and the investors listed on the Schedule of Buyers thereto. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on August 5, 2009 and incorporated herein by reference. | |
| 10.27(1) | Amended and Restated Manufacturing Technology Transfer and Supply Agreement by and between Agenus Inc. and GlaxoSmithKline Biologicals SA dated January 19, 2009. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended March 31, 2009 and incorporated herein by reference. | |
| 10.28* | Summary of oral agreement between Garo H. Armen, PhD and Agenus Inc. Agenus Inc. modifying the base salary of Dr. Armen. Filed as Exhibit 10.3 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended March 31, 2009 and incorporated herein by reference. | |
| 10.29 | Securities Exchange Agreement by and between Agenus Inc. and Tang Capital Partners, LP dated June 3, 2009. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2009 and incorporated herein by reference. | |
| 10.30 | Securities Exchange Agreement by and between Agenus Inc. and The Conus Fund L.P., The Conus Fund Offshore Master Fund Ltd., and The Conus Fund (QP) L.P. dated June 4, 2009. Filed as Exhibit 10.2 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2009 and incorporated herein by reference. | |
| 10.31 | Securities Exchange Agreement by and between Agenus Inc. and The Wolverine Convertible Arbitrage Fund Trading Limited dated June 4, 2009. Filed as Exhibit 10.3 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2009 and incorporated herein by reference. | |
| 10.32* | Agenus Inc. 2009 Equity Incentive Plan. Filed as Appendix A to our Definitive Proxy Statement on Schedule 14A filed on April 27, 2009 and incorporated herein by reference. | |
88
Table of Contents
| Exhibit No. |
Description | |
| 10.32.1* | Form of Restricted Stock Agreement for the Agenus Inc. Agenus Inc. 2009 Equity Incentive Plan. Filed as Exhibit 10.2 to our Current Report on Form 8-K (File No. 0-29089) filed on June 15, 2009 and incorporated herein by reference. | |
| 10.32.2* | Form of Stock Option Agreement for the Agenus Inc. 2009 Equity Incentive Plan. Filed as Exhibit 10.3 to our Current Report on Form 8-K (File No. 0-29089) filed on June 15, 2009 and incorporated herein by reference. | |
| 10.33* | Agenus Inc. 2009 Employee Stock Purchase Plan. Filed as Appendix B to our Definitive Proxy Statement on Schedule 14A filed on April 27, 2009 and incorporated herein by reference. | |
| 10.34 | Landlord, Sublessor and Sublessee Agreement dated August 4, 2010 between Agenus Inc., Cubist Pharmaceuticals, Inc., and TBCI, LLC, as Trustee of 3 Forbes Road Realty Trust. Filed as Exhibit 10.1 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2010 and incorporated herein by reference. | |
| 10.35 | Sublease Agreement by and between Agenus Inc. and Cubist Pharmaceuticals, Inc. dated July 30, 2010. Filed as Exhibit 10.2 to our Quarterly Report on Form 10-Q (File No. 0-29089) for the quarter ended June 30, 2010 and incorporated herein by reference. | |
| 10.36 | Form of Subscription Agreement. Filed as Exhibit 10.1 to our Current Report on Form 8-K (File No. 0-29089) filed on December 14, 2010 and incorporated herein by reference. | |
| 10.37 | Securities Exchange Agreement by and between Agenus Inc. and Invus Public Equities L.P. dated April 22, 2010. Filed herewith. | |
| 10.38 | Securities Exchange Agreement by and between Agenus Inc. and Bruce Fund Inc. dated November 18, 2010. Filed herewith. | |
| 10.39 | Securities Exchange Agreement by and between Agenus Inc. and Professional Life and Casualty dated November 18, 2010. Filed herewith. | |
| 10.40 | Securities Repurchase Agreement by and between Agenus Inc. and Ingalls & Snyder Value Partners L.P. dated December 7, 2010. Filed herewith. | |
| 10.41 | Securities Exchange Agreement by and between Agenus Inc. and Ingalls & Snyder Value Partners L.P. dated December 28, 2010. Filed herewith. | |
| 21 | Subsidiaries of Agenus Inc. Filed herewith. | |
| 23 | Consent of KPMG LLP, independent registered public accounting firm. Filed herewith. | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as amended. Filed herewith. | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as amended. Filed herewith. | |
| 32.1(2) | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. Submitted herewith. | |
| * | Indicates a management contract or compensatory plan. |
| (1) | Certain confidential material contained in the document has been omitted and filed separately with the Securities and Exchange Commission pursuant to Rule 406 of the Securities Act or Rule 24b-2 of the Securities Exchange Act. |
| (2) | This certification accompanies the Annual Report on Form 10-K and is not filed as part of it. |
89
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| AGENUS INC. | ||
| By: | /s/ GARO H. ARMEN, PH.D. | |
| Garo H. Armen, Ph.D. | ||
| Chief Executive Officer and | ||
| Chairman of the Board | ||
Dated: March 16, 2011
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities indicated as of March 16, 2011.
| Signature |
Title | |
| /S/ GARO H. ARMEN, PH.D. Garo H. Armen, Ph.D. |
Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) | |
| /S/ SHALINI SHARP Shalini Sharp |
Vice President and Chief Financial Officer (Principal Financial Officer) | |
| /S/ CHRISTINE M. KLASKIN Christine M. Klaskin |
Vice President, Finance (Principal Accounting Officer) | |
| /S/ BRIAN CORVESE Brian Corvese |
Director | |
| /S/ TOM DECHAENE Tom Dechaene |
Director | |
| /S/ JOHN HATSOPOULOS John Hatsopoulos |
Director | |
| /S/ WADIH JORDAN Wadih Jordan |
Director | |
| /S/ HYAM I. LEVITSKY, MD Hyam I. Levitsky, MD |
Director | |
| /S/ TIMOTHY ROTHWELL Timothy Rothwell |
Director | |
| /S/ TIMOTHY R. WRIGHT Timothy R. Wright |
Director | |
90