Attached files
| file | filename |
|---|---|
| 8-K - CURRENT REPORT - STERIS CORP | d8k.htm |
| EX-99.1 - PRESS RELEASE ISSUED BY STERIS CORPORATION DATED APRIL 20, 2010 - STERIS CORP | dex991.htm |
Exhibit 99.2
UNITED STATES DISTRICT COURT
FOR THE NORTHERN DISTRICT OF OHIO
| UNITED STATES OF AMERICA, | ) | |||
| ) | ||||
| Plaintiff, |
) | |||
| ) | ||||
| v. |
) | |||
| ) | Civil Action No. | |||
| STERIS CORPORATION, a corporation; | ) | |||
| and WALTER M. ROSEBROUGH, JR., | ) | |||
| and PETER A. BURKE, individuals, | ) | |||
| ) | ||||
| Defendants. |
) |
CONSENT DECREE OF PERMANENT INJUNCTION
Plaintiff, the United States of America, by its undersigned attorneys, having filed a Complaint for Permanent Injunction (“Complaint”) against STERIS Corporation, located in Mentor, Ohio (“STERIS”); and Walter M. Rosebrough, Jr., President and Chief Executive Officer of STERIS (who assumed the President and CEO positions in October 2007), and Peter A. Burke, Senior Vice President and Chief Technology Officer of STERIS, individuals (collectively, “Defendants”), and Defendants having appeared and consented to entry of this Consent Decree of Permanent Injunction (“Decree”) without contest and before any testimony has been taken, and without admitting or denying the allegations in the Complaint, and the United States of America, having consented to this Decree;
IT IS HEREBY ORDERED, ADJUDGED, AND DECREED as follows:
1. This Court has jurisdiction over the subject matter of this action and has personal jurisdiction over all parties to this action under 21 U.S.C. § 332(a) and 28 U.S.C. § 1345.
2. Venue is proper in this District under 28 U.S.C. §§ 1391(b)-(c).
3. The Complaint states a cause of action against Defendants under the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. §§ 301-397 (“the Act”).
4. Defendants and each of their directors, officers, agents, representatives, employees, attorneys, successors, and assigns, and any and all persons in active concert or participation with any of them who have received actual notice of this Decree by personal service or otherwise, are permanently restrained and enjoined under 21 U.S.C. § 332(a) from directly or indirectly doing or causing to be done any act that:
A. violates 21 U.S.C. § 331(a), by introducing or causing to be introduced, or delivering or causing to be delivered for introduction, into interstate commerce any liquid chemical sterilizing device and/or any liquid chemical disinfecting device and any components or accessories thereto (“LCS Devices”) including, but not limited to, the STERIS SYSTEM 1, STERIS 20 sterilant concentrate, indicators, and/or trays/quick connects (“SSI”), articles of device within the meaning of 21 U.S.C. § 321(h), when any such device is adulterated within the meaning of 21 U.S.C. § 351(f)(1)(B) and/or misbranded within the meaning of 21 U.S.C. § 352(o); and
B. violates 21 U.S.C. § 331(k), by causing any LCS Device to become adulterated within the meaning of 21 U.S.C. § 351(f)(1)(B), while such article is held for sale after shipment of one or more of its components in interstate commerce.
5. Paragraph 4 shall not apply to the following:
A. Defendants may continue to support its customers’ use of the SS1 in strict accordance with the specific terms set forth in the Transition Plan Overview (attached hereto as Exhibit A and incorporated by reference herein), which requires, among other things: that
2
customers complete a Certificate of Medical Necessity form to obtain components, parts, accessories, and service so that they may continue to use the SS1 while making the transition to a legally marketed alternative product; that Defendants provide a Transition Guide document to customers; and that Defendants implement STERIS’s customer rebate program in accord with the Transition Plan Overview. A copy of the Certificate of Medical Necessity form and the STERIS Transition Guidance referenced in the Transition Plan Overview are attached hereto as Exhibits B and C, and incorporated by reference herein. The Transition Plan described in this paragraph and Exhibits A—C shall terminate no later than August 2, 2011. Defendants shall provide a report as described in the Transition Plan Overview. Defendants shall, at STERIS’s expense and under FDA supervision, destroy all SS1 devices, components, parts, and/or accessories that are in STERIS’s possession, custody, and control within the United States no later than September 30, 2011, or such an earlier or later time as FDA may require or allow in writing, following a written request by Defendants explaining the need for an extension. Within ten (10) calendar days after entry of this Decree, STERIS shall submit to FDA a written statement detailing its proposed timing and method of destruction, and STERIS shall not commence such destruction until it receives written authorization from FDA; FDA shall respond to STERIS’s written statement within ten (10) calendar days of receipt. The immediately preceding two sentences shall not apply to any unused SSI devices, components, parts, and accessories that may be lawfully distributed in interstate commerce provided, however, that STERIS first submits a written request to FDA on or before September 1, 2011, describing in detail such products and the legal basis for such distribution, and that FDA provides STERIS with a written authorization for such distribution. FDA’s written determination regarding the
3
distribution of such products shall be deemed final and reviewed under the standard set forth in paragraph 14;
B. Defendants may export any SS1 devices until the termination of the Transition Plan, provided: (1) STERIS identifies all SS1 products to be exported with a specific code, number, or identifier along with the serial and lot numbers that readily identifies the products as for export only; (2) STERIS establishes controls and documentation for SS1 devices to be exported to assist with the monitoring and tracking of the exported products and to prevent their reimportation into the United States; (3) STERIS provides FDA with an action plan it intends to implement in the event that STERIS becomes aware of a customer or supplier that has attempted to import, or has reimported, into the United States an SS1 that was intended for export only; and (4) the requirements of 21 U.S.C. § 381(e)(1)(A)-(C) or 21 U.S.C. § 382 have been satisfied with respect to any such device or component; and
C. Defendants may at their own risk manufacture, process, pack, label, and hold, but not distribute, quantities of LCS Devices in anticipation of FDA’s approval of a premarket approval application filed under 21 U.S.C. § 360e or clearance of a premarket notification filed under 21 U.S.C. § 360(k) (also referred to as 510(k) submission). Defendants understand and agree that any devices manufactured under this paragraph may have to be destroyed in the event FDA does not approve or clear such application or notification.
6. Representatives of FDA shall be permitted, without prior notice and as and when FDA deems necessary, to make inspections of Defendants’ places of business and take any other measures it deems necessary to monitor and ensure continuing compliance with the terms of this Decree. During such inspections, FDA representatives shall be permitted ready access to
4
Defendants’ places of business including, but not limited to, all buildings, equipment, finished and unfinished materials and products, containers, labeling, and other promotional material therein; to take photographs and make video recordings; to take samples of Defendants’ finished and unfinished materials and products, containers, labeling, and other promotional material; and to examine and copy all records relating to the SS1 Transition Plan and manufacture, processing, packing, labeling, holding, and distribution of any and all of Defendants’ devices, including components thereof, in order to ensure continuing compliance with the terms of this Decree, the Act, and its implementing regulations. The inspections shall be permitted upon presentation of a copy of this Decree and appropriate credentials. The inspection authority granted by this Decree is separate from, and in addition to, the authority to make inspections under the Act, 21 U.S.C. §374.
7. Defendant STERIS shall reimburse FDA for the costs of all FDA inspections, investigations, supervision, analyses, examinations, and reviews that FDA deems necessary to evaluate Defendants’ compliance with this Decree. The costs of such inspections shall be borne by Defendant STERIS at the standard rates in effect at the time the activities are accomplished. As of the date of entry of this Decree, these rates are: $87.57 per hour or fraction thereof per representative for inspection and investigative work; $104.96 per hour or fraction thereof per representative for laboratory and analytical work; $0.50 per mile for travel expenses by automobile; the government rate or the equivalent for travel by air or other means; and the published government per diem rate for subsistence expenses where necessary. In the event that the standard rates applicable to FDA supervision of court-ordered compliance are modified, these rates shall be increased or decreased without further order of the Court.
5
8. Within ten (10) business days of the date of entry of this Decree, Defendants shall provide a copy of the Decree, by personal service or certified mail (restricted delivery, return receipt requested), to each and all of the following “Associated Persons”: (i) directors, officers, agents, representatives, attorneys, successors, and assigns of STERIS, and any and all persons or entities in active concert or participation with any of them with respect to the manufacture, distribution, and promotion of, and implementation of the Transition Plan, for the SS1 Devices. In the event that Defendants become associated, at any time after the entry of this Decree, with new Associated Persons, Defendants shall within fifteen (15) calendar days of such association, (a) provide a copy of this Decree to such person(s) by personal service or certified mail (restricted delivery, return receipt requested) and (b) shall furnish FDA with an affidavit of compliance (signed by a person with personal knowledge of the facts) identifying the names, addresses, and positions of all new Associated Persons that received a copy of the Decree. Within twenty (20) calendar days of the date of entry of this Decree, Defendants shall provide a copy of this Decree to all of Defendants’ employees involved in the manufacture, processing, packing, storage, or distribution of LCS Devices at Defendant STERIS’s facilities and shall post a copy of this Decree in the employee common areas at all facilities where such employees are located. Defendants shall ensure that the Decree remains posted in the employee common areas for no less than twelve (12) months. Within thirty (30) calendar days of the date of entry of this Decree, Defendants shall provide to FDA an affidavit (signed by a person with personal knowledge of the facts) stating the fact and manner of their compliance with this paragraph, identifying the names, addresses, and positions of all persons who received a copy of this Decree pursuant to this paragraph.
6
9. Defendant STERIS shall notify FDA, in writing at least fifteen (15) calendar days before any change in ownership, character, or name of any of their businesses, including incorporation, reorganization, bankruptcy, assignment, sale resulting in the emergence of a successor business or corporation, the creation or dissolution of subsidiaries, franchisees, affiliates, or “doing business as” entities, or any other change in the structure or identity of Defendant STERIS (or any of any of its parents or subsidiaries), or the sale or assignment of any business assets, such as buildings, equipment, or inventory, that may affect obligations arising out of this Decree. Defendants shall provide a copy of this Decree to any prospective successor or assignee at least thirty (30) calendar days prior to any sale or assignment. Defendants shall furnish FDA with an affidavit of compliance with this paragraph no later than ten (10) business days prior to such assignment or change in ownership.
10. If, at any time after entry of this Decree, FDA determines, based on the results of an inspection, the analysis of a sample, a report or data prepared or submitted by Defendants, or any other information, that Defendants have failed to comply with any provision of this Decree, have violated the Act or its implementing regulations with respect to the LCS Device(s), or that additional corrective actions are necessary to achieve compliance with this Decree, the Act, or its implementing regulations with respect to the LCS Device(s), FDA may, as and when it deems necessary, order Defendants in writing to take appropriate corrective actions, including, but not limited to, the following:
A. Cease all manufacturing, processing, packing, repacking, labeling, holding, and/or distributing any or all device(s);
7
B. Recall, at Defendant STERIS’s expense, any device that is adulterated, misbranded, or otherwise in violation of this Decree, the Act, or its implementing regulations;
C. Revise, modify, or expand any report(s) or plan(s) prepared pursuant to this Decree;
D. Submit additional reports or information to FDA;
E. Submit any application or any supplement to an existing device application to FDA;
F. Issue a safety alert; and/or
G. Take any other corrective actions as FDA, in its discretion, deems necessary to bring Defendants into compliance with this Decree, the Act, or its implementing regulations.
11. The following process and procedures shall apply when FDA issues an order under paragraph 10:
A. Unless a different time frame is specified by FDA in its order, within ten (10) business days after receiving such order, Defendants shall notify FDA in writing either that: (1) Defendants are undertaking or have undertaken corrective action, in which event Defendants shall also describe the specific action taken or proposed to be taken and the proposed schedule for completing the action; or (2) Defendants do not agree with FDA’s order. If Defendants notify FDA that they do not agree with FDA’s order, Defendants shall explain in writing the basis for their disagreement; in so doing, Defendants also may propose specific alternative actions and specific time frames for achieving FDA’s objectives.
B. If Defendants notify FDA that they do not agree with FDA’s order, FDA will review Defendants’ notification and thereafter, in writing, affirm, modify, or withdraw its order,
8
as the Agency deems appropriate. If FDA affirms or modifies its order, it shall explain the basis for its decision in writing. The written notice of affirmation or modification shall constitute final agency action.
C. If FDA affirms or modifies its order, Defendants shall, upon receipt of FDA’s order, immediately implement the order (as modified, if applicable), and if they so choose, bring the matter before this Court on an expedited basis. Defendants shall continue to diligently implement FDA’s order, unless the Court sets aside, stays, reverses, vacates, or modifies FDA’s order. The Court’s decision under this paragraph shall be made in accordance with the terms set forth in paragraph 14.
D. The process and procedures set forth in paragraphs 11(A)-(C) shall not apply to any order issued pursuant to paragraph 10 if such order references this paragraph and states that, in FDA’s judgment, the order must be implemented immediately. In such case, Defendants shall, upon receipt of such order, immediately and fully comply with the terms of that order. Should Defendants seek to challenge any such order, the order shall be deemed to be final agency action and the Defendants shall begin compliance with the order while they petition this Court for relief.
12. Any cessation of operations or other action described in paragraphs 10-11 shall continue until Defendants receive written notification from FDA that Defendants appear to be in compliance with this Decree, the Act, and its implementing regulations, or an order from the Court overruling FDA’s order. The costs of FDA supervision, inspections, investigations, analyses, examinations, reviews, sampling, testing, travel time, and subsistence expenses to implement the remedies set forth in this paragraph and paragraphs 10-11, shall be borne by
9
Defendant STERIS at the rates specified in paragraph 7 of this Decree, unless the Court overrules FDA’s order.
13. If Defendants fail to comply with any of the provisions of this Decree, including any time frame imposed by this Decree, then, on motion of the United States in this proceeding, Defendant STERIS shall pay to the United States of America: fifteen thousand dollars ($15,000.00) in liquidated damages for each day such violation continues, and an additional sum of fifteen thousand dollars ($15,000.00) in liquidated damages for each violation of the Act, its implementing regulations, and/or this Decree relating to LCS Devices. Defendants understand and agree that the liquidated damages specified in this paragraph are not punitive in nature and their imposition does not in any way limit the ability of the United States to seek, or the power of the Court to impose, additional criminal or civil penalties or remedies based on conduct that may also be the basis for payment of liquidated damages pursuant to this paragraph. Liability under this paragraph shall not exceed, in any one calendar year, ten million dollars ($10,000,000).
14. Defendants shall abide by the decisions of FDA, and FDA’s decisions shall be final. All decisions conferred upon FDA in this Decree shall be vested in FDA’s discretion and, if contested, shall be reviewed by this Court under the review standard set forth in 5 U.S.C. § 706(2)(A) and shall be based exclusively on the written record before FDA at the time of the decision. No discovery shall be taken by either party.
15. All notifications, correspondence, and communications required to be sent to FDA by the terms of this Decree shall be marked “Consent Decree Correspondence” and shall be addressed to the District Director, FDA Cincinnati District Office, 6751 Steger Drive, Cincinnati, Ohio 45237.
10
16. If Defendants have continuously complied with the terms of this Decree, the Act, and all applicable laws and regulations with respect to the LCS Devices for a period of five (5) years after entry of this Decree, Defendants may petition this Court for relief from this Decree. If, at the time of the petition, in FDA’s judgment Defendants have met the foregoing criterion, Plaintiff will not oppose such petition.
17. The obligations under this Decree of each individual named herein shall apply only to the extent of his authorities, responsibilities, and/or conduct at STERIS. If, and for so long as, an individual Defendant or an employee of STERIS ceases to be employed by or act on behalf of STERIS, then that Defendant or employee shall not be subject to the terms of this Decree except as to such individual’s act(s) or failure(s) to act under this Decree prior to the time such individual ceased to be employed by or to act on behalf of STERIS.
18. Should Plaintiff bring, and prevail in, a contempt action to enforce the terms of this Decree, Defendants shall, in addition to other remedies, reimburse Plaintiff for its attorneys’ fees and costs, travel expenses incurred by attorneys and witnesses, expert witness fees, investigational and analytical expenses, and court costs relating to such contempt proceedings.
19. Defendants may petition FDA in writing to extend any deadline provided herein, and FDA may grant such extension without seeking leave of Court. However, any such petitions shall not become effective or stay the imposition of any payments under this Decree unless granted by FDA in writing.
20. This Court retains jurisdiction over this action and the parties thereto for the purpose of enforcing and modifying this Decree and for the purpose of granting such additional relief as may be necessary or appropriate.
11
SO ORDERED, this day of , 2010.
|
| ||||
| UNITED STATES DISTRICT JUDGE | ||||
| Entry consented to: | ||||
| For Defendants | For Plaintiff | |||
| /s/ Walter M. Rosebrough, Jr. |
||||
| WALTER M. ROSEBROUGH, JR. | ||||
| Individually and on behalf | ||||
| Of STERIS Corporation As its Chief Executive Officer and President |
||||
| /s/ Peter A. Burke |
||||
| PETER A. BURKE Individually |
EUGENE M. THIROLF DIRECTOR | |||
| /s/ Mark A. Heller |
/s/ Allan Gordus | |||
| MARK A. HELLER GOODWIN PROCTER LLP 901 New York Avenue, N.W. Washington, D.C. 20001
Counsel for Defendant STERIS |
ALLAN GORDUS Trial Attorney Office of Consumer Litigation U.S. Department of Justice P.O. Box 386 Washington, D.C. 20044 (202)307-1862
Of Counsel: | |||
| /s/ Stephen G. Sozio |
||||
| STEPHEN G. SOZIO JONES DAY 901 Lakeside Avenue Cleveland, OH 44114-1190
Counsel for Defendant STERIS |
DAVID S. CADE Acting General Counsel Department of Health and Human Services
RALPH S. TYLER Chief Counsel | |||
12
| Food and Drug Division | ||||
| /s/ Stuart M. Gerson |
ERIC M. BLUMBERG | |||
| STUART M. GERSON EPSTEIN BECKER & GREEN, P.C. 1227 25th Street, N.W. Suite 1200 Washington, D.C. 20036
Counsel for Individual Defendant Walter M. Rosebrough, Jr. |
Deputy Chief Counsel for Litigation
MARC L. CADEN Associate Chief Counsel Food and Drug Division U.S. Dept. of Health and Human Services Office of General Counsel 5600 Fishers Lane, GCF-1 Rockville, MD 20857 | |||
| /s/ Thomas O. Henteleff |
||||
| THOMAS O. HENTELEFF KLEINFELD KAPLAN & BECKER, LLP 1140 Nineteenth Street, N.W. Suite 900 Washington, D.C. 20036
Counsel for Individual Defendant Peter A. Burke |
||||
13
ATTACHMENT A
STERIS SYSTEM 1 Transition Plan Overview
This transition plan for SYSTEM 1 Customers in the U.S. was developed by STERIS with review and input from the United States Food and Drug Administration (FDA). STERIS believes that the transition plan will help to facilitate a smooth transition from SYSTEM 1 to a legally-marketed and acceptable alternative for sterilizing or high-level disinfecting temperature-sensitive devices.
Certificate of Medical Necessity (CN)
STERIS will continue to support its current Customers’ use of SYSTEM 1, sterilant, quick connects, service parts, filters, sterility assurance products and other accessories/consumables (“consumables”) during the transition period. In order for a Customer to receive this continued support, a Customer must submit a Certificate of Medical Necessity (CN) to STERIS that describes and justifies its immediate and continued medical necessity for STERIS to fully support its use of the STERIS SYSTEM 1. The CN shall be signed by one of the following: the Hospital or Health Care facility President or COO, head of the Institutional Review Board, CEO, and/or Chief Medical Officer. Healthcare facilities and distributors will have until June 18, 2010, to complete and return the CN to STERIS. After June 19, 2010, STERIS will cease support of its SYSTEM 1 at any facility that has not submitted a complete CN until such time as STERIS receives a completed CN from the facility.
Product Availability
STERIS will continue to provide consumables and services that are medically-necessary, for the purpose of maintaining and/or restoring the SYSTEM 1 units now in use to currently-labeled operating parameters until August 2, 2011, or the Customer is able to transition to a legally-marketed and acceptable alternative to the SYSTEM 1, whichever first occurs. STERIS will not replace any SYSTEM 1 units that have become unserviceable. Nor will STERIS sell any new types of quick connects (QCs) to accommodate newer instruments. Customers should not attempt to use or alter existing QCs or other components to attempt to connect devices to the SYSTEM 1 in an effort to sterilize them. Only instruments that have been validated for use in the SYSTEM 1 can be reprocessed in the device, and only until a legally-marketed and acceptable alternative for reprocessing those instruments has been placed into service.
STERIS will monitor the total volume of S20 sterilant supplied to Customers against the Customer’s average monthly sterilant purchases during the period January 1, 2009, to December 31, 2009 (“Average Monthly Usage”).
If the customer exceeds in two consecutive months 110% of its Average Monthly Usage, STERIS will:
1
| 1. | Contact the Customer in order for the Customer to determine and document with a new CN, reasons for the increased purchase volume (such as increased patient load in the facility), or |
| 2. | Limit further shipment of sterilant to the Customer to the Average Monthly Usage if there is no clinically-related reason for the increase. |
| 3. | When the Customer is a distributor, further shipments to the distributor shall be limited to the distributor’s Average Monthly Usage, unless a new CN(s) from a healthcare facility(ies) receiving S20 sterilant from the distributor documents the clinical need for the increased distributor orders of S20. |
When the Customer documents its reasons for the increased purchase volume, STERIS will submit that documentation to FDA at the Agency’s request. Provided, however, for Customers that purchase two or fewer cases of sterilant a month, the Average Monthly Usage limitation described above shall not apply.
Transition Guide
STERIS has prepared a guidance document titled “STERIS SYSTEM 1 Sterile Processing System: Transition Guidance” to help healthcare providers implement a process to consider the most appropriate alternative technologies to replace the SYSTEM 1. This document will help healthcare providers:
| 1. | Assess their current situation |
| 2. | Identify suitable legally-marketed alternatives for the devices they reprocess in SYSTEM 1 |
| 3. | Review relevant factors related to alternate technologies |
| 4. | Consider appropriate alternative technologies/processes |
| 5. | Identify the leading providers of alternative technologies |
| 6. | Identify additional sources of help and guidance including industry organizations such as AAMI, AORN, APIC and SGNA. |
| 7. | Minimize disruption and enhance patient care and safety during the transition period. |
This document is not a sales document, nor is it intended to be all-inclusive or identify all factors to be considered. Instead it is a guide to assist Customers through the transition.
SYSTEM 1 Customer Rebate Program
This Transition Plan and the STERIS FDA approved disposition plan are intended to facilitate the return and disposition of SYSTEM 1 units currently in use. This program also affords buying power to healthcare providers to purchase any technology or product they deem appropriate for their particular needs in return for their SYSTEM 1 unit(s). The proposed program has 2 options:
2
Option 1:
STERIS will provide a rebate to the healthcare provider based on net book value (NBV) for the SYSTEM 1 processor unit (based on AHA fixed asset depreciation guidelines of 7 years or 84 months, calculated based on the remaining economic life in number of months from the original processor shipment month to the return of the processor to STERIS) with a minimum rebate of $1,000 for fully depreciated SYSTEM 1 units. The cash may be used by a healthcare provider for any purpose, including towards the purchase of any legally-marketed STERIS or competitor technology or product.
Option 2:
STERIS will provide consideration greater in value than the NBV described in Option 1 to the healthcare provider toward the purchase of FDA-cleared or approved STERIS products, or any other STERIS product that is legally available.
Additionally, STERIS will provide full credit for the return of all SYSTEM 1 unused parts and consumables in unbroken packaging that are not past their shelf-lives. In addition, STERIS will credit all maintenance contract holders for the unused portion of any SYSTEM 1 service contract, after the unit has been de-installed and returned to STERIS.
Compliance with the Transition Plan
STERIS will monitor the following and generate a monthly report as indicated below:
| • | CN submissions by Customers. STERIS will generate a report that tracks the status of Customer compliance with the CN requirement. After June 19, 2010, STERIS will cease support of its System 1 at any facility that has not submitted a complete CN until such time as STERIS receives a complete CN from the facility. |
| • | The number of cases of S20 supplied to each Customer compared to the monthly average of cases supplied to the Customer during the period January 1, 2009, to December 31, 2009. |
| • | The number of consumables, both type and quantity, that have been supplied to each Customer per month compared to the monthly average and type during the period January 1, 2009, to December 31, 2009. |
| • | STERIS will document the return of SYSTEM 1 units from Customers pursuant to this Plan and the STERIS FDA approved disposition plan. |
STERIS shall provide FDA these reports for the first three months of the Transition Plan and thereafter, upon FDA’s request, shall make all of these reports available to FDA within 5 business days of the request.
3
ATTACHMENT B
CERTIFICATE OF MEDICAL NECESSITY (CN): SYSTEM 1 AND ACCESSORIES
Purpose: STERIS is using this clinical Certificate of Medical Necessity (CN) to continue to distribute SYSTEM 1, sterilant, quick connects, service parts, filters, sterility assurance products, other accessories/consumables, and to service your equipment.
NOTE: STERIS will not distribute SYSTEM 1, and related sterilant, quick connects, service parts, filters, sterility assurance products, other accessories/consumables after August 2, 2011.
Required information
| Hospital/Facility Name: |
|
Account #: |
|
| Address: |
|
| City: |
|
State: |
|
Postal Code |
|
Number of System 1 units in my facility/system (all departments/locations):
If you are submitting this certificate for multiple healthcare facilities within your health system, please attach a list with the names, account numbers, and addresses of the respective facilities and the number of System 1 units in each of those facilities.
I certify that STERIS 20 Sterilant Concentrate, Ongoing Service Parts & labor, and/or SYSTEM 1 Consumables and Accessories are an immediate and continued medical necessity for our facility, for the following reason(s):
|
|
|
|
|
|
|
|
|
|
|
|
| Authorized Signature: |
|
(Chairman of the Institutional Review Board, CEO, Chief Medical Officer, President, Chief Operating Officer)
| Name: |
| |
| Please Print |
| Title: |
| |
| Date: |
|
Telephone No.: |
|
email Address: |
|
Please complete and return this form by June 18, 2010, to continue to receive
SYSTEM 1 products and services from STERIS Corporation:
© 2009 STERIS Corporation. All Rights Reserved. Publication ID: 4436.
Please return this form to:
| Attn: | Scan and e-mail to: systeml@steris.com | |
| System 1 Response Team | Or Fax to: 440-392-8983 | |
| STERIS Corporation 5960 Heisley Road Mentor, OH 44060 |
© 2009 STERIS Corporation. All Rights Reserved. Publication ID: 4436.
2
ATTACHMENT C

STERIS SYSTEM 1® Sterile Processing System:
Transition Guidance
Table of Contents
| PAGE 1 |
BACKGROUND | |
| PAGE 2 |
PURPOSE OF THIS DOCUMENT | |
| PAGE 3-5 |
DEVICE REPROCESSING TRANSITION PROCEDURE | |
| PAGE 6 |
CONSIDERATIONS FOR REPLACING SYSTEM 1 | |
| PAGE 7-8 |
ALTERNATIVE REPROCESSING TECHNOLOGIES | |
| PAGE 8 |
ALTERNATIVE REPROCESSING TECHNOLOGY VENDORS | |
| PAGE 9 |
ADDITIONAL SOURCES OF INFORMATION | |
| PAGE 9 |
STERIS SUPPORT DURING THE TRANSITION | |
| PAGE 10 |
SYSTEM 1 TRANSITION DECISION TREE | |
BACKGROUND
STERIS Corporation was notified on December 3, 2009 that the U.S Food and Drug Administration (FDA) had issued a notice to healthcare facility administrators regarding the regulatory status of the STERIS SYSTEM 1® Sterile Processing System (hereafter SYSTEM 1). The notice outlined actions to be taken by healthcare facilities using the SYSTEM 1 device. The FDA has stated that healthcare administrators should transition to acceptable alternatives for device reprocessing.
STERIS is providing this Transition Guidance document to help our Customers evaluate acceptable alternatives to SYSTEM 1. The document offers recommendations for working through the evaluation and transition process; a list of alternative reprocessing methodologies for critical and semi-critical devices; plus observations regarding the potential impact of these alternatives on Customers’ operations. We have included links to additional information resources such as professional organizations and other companies that offer alternative sterilization and decontamination technologies.
STERIS has established a dedicated hotline at 800-548-4873 to support Customers through this transition. We will also continue providing the latest information via our website at www.steris.com.
PAGE 1
PURPOSE OF THIS DOCUMENT
This document is intended to support healthcare facilities and their assigned working committees in making informed decisions about the transition to acceptable reprocessing alternatives. At a high level, the decision-making process should include the following considerations:
| • | Identify all current low temperature re-usable medical devices currently processed in the SYSTEM 1 sterilizer. |
| • | Obtain and review all original equipment manufacturer’s instructions for use for these devices. In particular, make sure you understand which sterilization/high level disinfection modalities are currently cleared by FDA to process these devices. |
| • | Identify all current modalities available in your healthcare facility and the devices that can be moved away from SYSTEM 1 to an acceptable alternative process. |
| • | Compile a list of re-usable medical devices you can process given the healthcare facility’s current capabilities. |
| • | Determine whether there is another healthcare facility in your system with the appropriate sterilization/high level disinfection modalities and the operational capacity to process these devices. |
| • | If yes, can temporary arrangements be made for transportation and processing? |
| • | If no, is there a 3rd party low temperature service available in the area? |
After considering the factors listed above, if no alternatives exist within your facility or system, the following steps will guide you through the decision-making process.
These are merely our suggestions to help you through the transition decision-making process. Of course, you should defer to your own healthcare facility/system’s policies and procedures.
PAGE 2
SYSTEM 1* Sterile Processing System: Transition Guidance
DEVICE REPROCESSING TRANSITION PROCEDURE
| I. | OBJECTIVE: |
To establish a clear, comprehensive and consistent procedure for transitioning to acceptable alternative device reprocessing methodologies other than SYSTEM 1 in perioperative, endoscopy and sterile processing settings.
| II. | SCOPE: |
All perioperative, endoscopy and sterile processing areas of your facility.
| III. | EXCEPTIONS: |
None
| IV. | DEFINITIONS: |
| A. | Qualified Personnel - Members of staff who have the requisite product, process and quality knowledge and training in the operation of equipment used during the verification process: typically, instrument technicians or instrument specialists. |
| B. | User Verification - Documented procedures, performed in the user environment, for obtaining, recording and interpreting results data establishing that pre-determined specifications have been met. |
| C. | Validation - Documented procedure for obtaining, recording, and interpreting the results required to establish that a process will consistently yield product complying with predetermined specifications. |
NOTE 1:
Validation covers three activities:
| i. | Installation qualification |
| ii. | Operational qualification |
| iii. | Performance qualification |
NOTE 2:
Validation is performed by the device manufacturer. For example STERIS Corporation validates the AMSCO V-PRO™ 1 Low Temperature Sterilization System.
| V. | POLICY: |
Capital equipment and medical device procurement are collaborative processes requiring clinical, legal, and financial acumen. Patient care, product standardization and value analysis goals drive the selection of functional and reliable products that are safe; cost-effective; environmentally friendly; consistent with the mission of quality care; and that avoid duplication or rapid obsolescence.
PAGE 3
SYSTEM 1* Sterile Processing System: Transition Guidance
DEVICE REPROCESSING TRANSITION PROCEDURE
(continued...)
| VI. | PROCEDURE: |
| A. | Establish a multidisciplinary committee with representation from everyone affected by the new technology/product. For example, for a product related to low-temperature sterilization, representation includes (but is not limited to) infection prevention and control; the OR; endoscopy; sterile processing; risk management; general counsel; and staff development/training/education. |
Rationale: The selection of new sterilization/high-level disinfection modalities and/or products can be complex, requiring a systematic, thoughtful process to help facilitate the evaluation, procurement and implementation process.
| B. | Collect and distribute to the committee information related to the product. Such data includes (but is not limited to) the following: |
| 1. | FDA clearance documentation |
| 2. | Relevant research articles published in peer-reviewed journals |
| 3. | Manufacturers’ literature and instructions for use |
| 4. | Expert opinions |
| 5. | Reports from peers who are using or have trialed the technology/product |
| C. | In addition to evaluating the product’s intended application, consider the following: |
| 1. | Impact on patient safety |
| 2. | Any legal implications associated with use of the technology/product |
| 3. | Cost/value analysis (return on investment) |
| 4. | Staff training and education |
| 5. | Ease-of-use of technology/product |
| 6. | Related safety issues |
| 7. | Compatibility of product with existing equipment |
| 8. | Environmental impact |
| 9. | Availability of ongoing support from vendor for such services as maintenance |
| 10. | Impact on standardization of product inventory |
| D. | If a product trial is indicated, the following guidelines apply: |
| 1. | Establish a timeline for the trial |
| 2. | Identify the personnel and departments that should trial the product |
| 3. | Establish the amount of product that should be evaluated |
| 4. | Develop evaluation tools through the multidisciplinary committee identified above |
| 5. | Determine and implement the education and demonstrations needed for the trial |
| 6. | Define the desired outcome |
| 7. | Analyze the data and compare actual outcomes with desired outcomes |
PAGE 4
SYSTEM 1* Sterile Processing System: Transition Guidance
DEVICE REPROCESSING TRANSITION PROCEDURE
(continued...)
| VII. | THIRD PARTY REFERENCES FOR HIGH-LEVEL DISINFECTION AND STERILIZATION: |
| A. | ANSI/AAMI ST79:2008. “Comprehensive Guide to Steam Sterilization and Sterility Assurance in Health Care Facilities.” Arlington, VA: AAMI. |
| B. | ANSI/AAMI ST58:2005. “Chemical Sterilization and High Level Disinfection in Health Care Facilities.” |
| C. | The Association of periOperative Registered Nurses (AORN), “Recommended Practices for Product Selection in Preoperative Practice Settings.” Denver, CO: AORN. (2009). |
| D. | Association for Professionals in Infection Control and Epidemiology (APIC) “Guideline for Disinfection and Sterilization in Health Care Facilities.” Washington, D.C. APIC (2008). |
| E. | Society of Gastroenterology Nurses and Associates, Inc (SGNA) “Guidelines for Use of High-Level Disinfectants and Sterilants for Reprocessing Flexible Gastrointestinal Endoscopes.” |
Chicago, IL: SGNA. (2009).
| F. | CDC “Guideline for Disinfection and Sterilization in Health Care Facilities, 2008”, available on the CDC website. |
| VIII. RESPONSIBILITY: |
OR, endoscopy and sterile processing educators should be responsible for assuring that proper training is conducted with all employees responsible for the processing and sterilization of instruments and supplies in accordance with this work instruction.
The healthcare facility should be responsible for developing and/or revising this Transition Guidance document in accordance with the infection control department; sterilization policy; current industry standards; regulatory requirements; and manufacturers’ recommendations.
Service and maintenance of sterilizers should be the responsibility of the clinical engineering department. The cleaning of the outside surface should be the responsibility of sterile processing.
PAGE 5
SYSTEM 1* Sterile Processing System: Transition Guidance
CONSIDERATIONS FOR REPLACING SYSTEM 1
In order to help Customers determine an appropriate alternative to SYSTEM 1, STERIS provides this summary of some of the available alternative technologies:
CRITICAL DEVICES
| • | Ethylene Oxide Sterilization |
| • | Steam Sterilization |
| • | Ozone Sterilization |
| • | Hydrogen Peroxide Gas or Plasma Sterilization |
SEMI-CRITICAL DEVICES
| • | Automated Endoscope Reprocessor: High-Level Disinfection |
| • | Basin Soak: High-Level Disinfection |
ALTERNATIVE REPROCESSING TECHNOLOGIES
There are several relevant factors to evaluate in the transition decision-making process. The following matrix provides a high level comparison of the impact of selecting a particular technology. These factors include (but are not limited to): ability to re-process devices that are currently labeled for SYSTEM 1; time constraints; device inventory; labor issues (including worker safety); and relative cost.
Once it has been determined which appropriate alternative technology/product will meet the requirements of the task, Customers will need to implement a training plan. This will ensure consistency in performing all of the necessary steps to achieve a sterile or disinfected device.
THE FOLLOWING TABLE INCLUDES OUR ASSESSMENT OF THE APPLICABILITY OF ALTERNATIVE MEANS OF STERILIZING DEVICES CURRENTLY PROCESSED IN SYSTEM 1. MOST ETO STERILIZERS HAVE NOT BEEN CLEARED FOR MARKETING WITH SPECIFIC LUMEN CLAIMS STATING THE LENGTH AND DIAMETER OF THE LUMEN DEVICES WHICH CAN BE EFFECTIVELY REPROCESSED IN AN ETO STERILIZER. IF THE INSTRUCTIONS FOR USE OF THE ETO STERILIZER DO NOT SPECIFY WHAT LUMEN DEVICE SPECIFICATIONS CAN BE EFFECTIVELY STERILIZED, THEN ETO SHOULD BE USED ONLY IF THE DEVICE MANUFACTURER CAN PROVIDE VALIDATED ETO REPROCESSING CYCLE PARAMETERS WHICH ARE COMPATIBLE WITH THE ETO STERILIZER AVAILABLE TO THE USER.
PAGE 6
SYSTEM 1* Sterile Processing System: Transition Guidance
ALTERNATIVE REPROCESSING TECHNOLOGIES
(continued...)
| ALTERNATIVE | ABILITY TO REPROCESS SYSTEM 1 LABELED |
CYCLE TIME | DEVICE INVENTORY |
ADDITIONAL STEPS* | WORKER SAFETY | RELATIVE COST | ||||||
| OZONE | Medium | Approximately a 4 hour cycle time | Potential to require greater inventory investment | Devices must be wrapped, transported for processing and returned to the point-of-use | This is a safe technology | Operating cost is low. Acquisition cost is high. The technology requires installation of a source of oxygen. Facility modifications will be required for installation | ||||||
| HYDROGEN PEROXIDE | Low | 30 - 60minutes | Potential to require greater inventory investment | Devices must be wrapped, transported for processing and returned to the point-of-use | This is a safe technology | Operating cost is moderate. Acquisition cost is high. Facility modifications may be required for installation | ||||||
| ETHYLENE OXIDE (EtO) | High** | Cycle times are 12 hours or greater. Ensure users have and understand the EtO manufacturer’s parameters | Greater investment in device inventory to compensate for lengthy cycle-time | Devices must be wrapped, transported for processing and returned to the point-of-use | Ethylene oxide is a known carcinogen. OSHA has extensive monitoring and documentation requirements for use of this technology. EPA also has requirements for the use of this technology | Operating cost is relatively low. Acquisition cost is high. Indirect costs for additional inventory and monitoring are high. Facility modifications will be required for installation | ||||||
|
STEAM STERILIZATION |
Low | Cycle times are 45 minutes to 1 hour. | This will need to be carefully assessed by the facility | Devices must be wrapped, cooled, transported and returned to the point-of-use | This is a safe technology | Operating cost is relatively low | ||||||
| HIGH LEVEL DISINFECTION: AER |
High for flexible endoscopes only | Times vary significantly by manufacturer and product | No impact | Devices must be cleaned and prepared according to manufacturer’s instructions for reprocessing | This is a safe technology | Operating cost is low. Acquisition cost is high. Facility modification may be required for installation | ||||||
| HIGH LEVEL DISINFECTION: BASIN SOAK | High | 10 - 20+ minutes | No impact | All aspects of this process are manual. Processing staff training, detailed written instructions and close supervision are advised to ensure consistent results | Product has to be appropriately ventilated. PPE must be employed to minimize risk of exposure to aldehyde based solutions | Operating cost is low | ||||||
| * | “For all sterilization and disinfection methods, flexible and rigid endoscopes must be pre-cleaned, inspected, leak tested and manually cleaned according to the recommendations of the manufacturer.” |
| ** | Dependant on using a validated cycle, see page 7. |
PAGE 7
SYSTEM 1* Sterile Processing System: Transition Guidance
ALTERNATIVE REPROCESSING TECHNOLOGY VENDORS
The sterilization and high-level disinfection tables below list industry leading companies that provide acceptable alternatives to SYSTEM 1*. It is the responsibility of each healthcare institution to validate all devices that are to be processed in each one of these alternative technologies, according to manufacturer’s instructions for use. FDA has recommended that the healthcare provider also verify that alternative technologies have FDA clearance.
STERILIZATION
| TYPE | APPLICATION (CONSULT DEVICE MANUFACTURER’S INDICATIONS FOR USE) |
AVAILABLE FROM WWW.STERIS.COM 1-800-548-4873 |
LEADING SUPPLIERS | WEBSITE | ||||
| EtO (ETHYLENE OXIDE) | Heat-sensitive, immersible devices | Yes | STERIS 3M |
steris.com 3m.com | ||||
| VHP (Vaporous Hydrogen Peroxide) | Heat-sensitive, immersible devices | Yes | STERIS STERRAD |
steris.com sterrad.com | ||||
| Ozone | Heat-sensitive, immersible devices | No | TSO3 | tso3.com | ||||
| Steam Sterilization | Devices not sensitive to heat | Yes | STERIS Getinge Belimed |
steris.com getinge.com belimedusa.com | ||||
HIGH LEVEL DISINFECTION
| TYPE | APPLICATION (CONSULT DEVICE MANUFACTURER’S INDICATIONS FOR USE) |
AVAILABLE FROM 1-800-548-4873 |
ALTERNATE SUPPLIERS | WEBSITE | ||||
| HLD CHEMISTRY | Semi-Critical Devices | |||||||
| ALDEHYDE CHEMISTRIES | ||||||||
| GLUTARALDEHYDE | Heat-sensitive, immersible devices | No | Medivators Metrex ASP |
minntech.com metrex.com aspjj.com | ||||
|
ORTHO- PHTHALALDEHYDE |
Heat-sensitive, immersible devices | No | Metrex ASP | metrex.com aspjj.com | ||||
| OXIDATIVE CHEMISTRIES | ||||||||
| HYDROGEN PEROXIDE | Heat-sensitive, immersible devices | Yes - Resert HLD High Level Disinfectant | N/A | steris.com | ||||
| PERACETIC ACID | Heat-sensitive, immersible devices | Yes - Reliance EPS as a validated system (see below) | Medivators | steris.com minntech.com | ||||
| AER | ||||||||
| MEDIVATORS | Heat-sensitive, immersible devices | No | Medivators | minntech.com | ||||
| CUSTOM ULTRASONICS | Heat-sensitive, immersible devices | No | Custom Ultrasonics | customultrasonics.com | ||||
| ASP | Heat-sensitive, immersible devices | No | ASP | aspjj.com | ||||
|
STERIS RELIANCE EPS |
Heat-sensitive, immersible devices | Yes - Peracetic Acid | N/A | steris.com | ||||
| * | “List of suppliers is not intended to be comprehensive. Per the December 10, 2009 FDA teleconference, additional information on available technologies will be forthcoming from FDA.” |
PAGE 8
SYSTEM 1* Sterile Processing System: Transition Guidance
ADDITIONAL SOURCES OF INFORMATION
STERILIZATION STANDARDS & GUIDELINES
| Organization |
Website | |
| AAMI - Association for the Advancement of Medical Instrumentation (ANSI/AAMI ST79:2006) |
www.aami.org | |
| AORN - Association of Operating Room Nurses |
www.aorn.org | |
| APIC - Association for Professionals in Infection Control |
www.apic.org | |
| SGNA - Society of Gastroenterology Nurses and Associates |
www.sgna.org | |
STERIS SUPPORT DURING THE TRANSITION
STERIS Corporation is taking several steps to provide support during your transition from SYSTEM 1 to an acceptable alternative reprocessing method:
| • | We have established a SYSTEM 1 Hotline to address healthcare provider questions. Please call 1-800-548-4873. |
| • | Your local STERIS representative can also answer your SYSTEM 1 inquiries. |
| • | We are publishing constant updates on our website at www.steris.com/SS1. |
PAGE 9
| SYSTEM 1 Transition Decision Tree | This flow design was developed to help facilitate a smooth transistion to an alternative Sterilization or High Level Disinfection process for your current reusable medical devices being sterilized with your SYSTEM 1 . Please use in accordance with the device manufacturer’s instructions for use to include all cleaning and sterilization parameters. | |||
|
| ||||
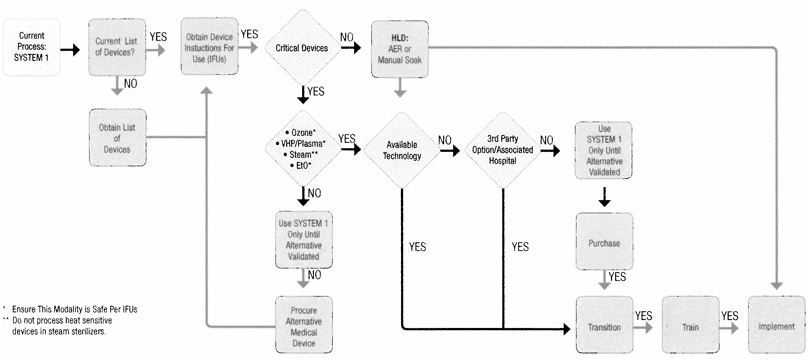
| • Current Process: SYSTEM 1 - Identify the total number of SYSTEM 1 units installed in your healthcare facility.
• Current List of Devices? - Do you have a complete list of reusable medical devices currently being processed through SYSTEM 1?
• Obtain List of Devices - Compile a list of all reusable medical devices currently being processed through SYSTEM 1 and obtain the necessary original equipment manufacturer’s instructions for use (IFUs) for these devices.
• Critical Devices or Hospital Policy - Is the reusable medical device a critical device or does hospital policy dictate this device be sterilized prior-to-use?
• EtO, Ozone, VHP/Plasma, Steam - Based on the original equipment manufacturer’s instructions for use is there an alternative modality of sterilization available? |
• Use SYSTEM 1 Only Until Alternative Validated - The originial equipment manufaturer for this device has specified SYSTEM 1 is the only sterilization method identified for sterilization.
• Procure Alternative Medical Device - Is there an alternative medical device available to support the medical procedure?
• HLD: AER or Manual Soak - If the device is identified as semi-critical and hospital policy will support High-level Disinfection, you may then implement the use of an AER or manual soaking method.
• Available Technology - Does the alternative sterilization modality currently exist In your healthcare facility?
• 3rd Party Option/Associated Hospital - Is there a 3rd party sterilization service in your area, or an associated healthcare facility currently using the alternative sterilization technology you seek? |
• Use SYSTEM 1 Only Until Alternative Validated - Continue using SYSTEM 1 until an alternative sterilization method has been validated.
• Purchase - Once the alternative sterilization technology has been Identified, pursue procurement options (once equipment specifications have been reviewed and can be satisfied given existing footprint).
• Transition - Install alternative sterilization technology.
• Train - Assure all staff and equipment operators are properly trained on the operation of said equipment, to include any accessories and new sterility assurance products supporting the alternative sterilization technology.
• Implement - Implement alternative sterilization technology.
|
|
© 2010 STERIS Corporation. All rights reserved. Document # M3399EN.2009-12, Rev. 3.31.10 Printed 04/2010 |
STERIS Corporation 5960 Heisley Road Mentor, OH 44060-1834 • USA 440-354-2600 • 800-548-4873 www.steris.com |