Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| x | Annual Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the fiscal year ended: December 31, 2009
or
| ¨ | Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
Commission file number: 000-51967
TRANSCEPT PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 33-0960223 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
1003 W. Cutting Blvd., Suite #110
Point Richmond, California 94804
(510) 215-3500
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive office)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of exchange on which registered | |
| Common Stock, par value $0.001 per share | NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer x | Smaller reporting company ¨ | |||
| (Do not check if a smaller reporting company) | ||||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the common stock of the registrant held by non-affiliates of the registrant on June 30, 2009, the last business day of the registrant’s second fiscal quarter was: $30,755,586.
As of March 26, 2010 there were 13,413,391 shares of the registrant’s common stock outstanding.
Documents incorporated by reference: Items 10, 11, 12, 13, and 14 of Part III incorporate information by reference from the Proxy Statement to be filed with the Commission within 120 days of the end of our fiscal year pursuant to General Instruction G(3) to Form 10-K.
Table of Contents
| Item No. | Page No. | |||||
| PART I |
||||||
| 1. | Business | 3 | ||||
| 1A. | Risk Factors | 30 | ||||
| 1B. | Unresolved Staff Comments | 49 | ||||
| 2. | Properties | 49 | ||||
| 3. | Legal Proceedings | 50 | ||||
| 4. | Reserved | 50 | ||||
| PART II |
||||||
| 5. | 51 | |||||
| 6. | 53 | |||||
| 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
54 | ||||
| 7A. | 68 | |||||
| 8. | 69 | |||||
| 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
99 | ||||
| 9A(T). | 99 | |||||
| 9B. | 100 | |||||
| PART III |
||||||
| 10. | 101 | |||||
| 11. | 101 | |||||
| 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
101 | ||||
| 13. | Certain Relationships and Related Transactions, and Director Independence |
102 | ||||
| 14. | 102 | |||||
| PART IV |
||||||
| 15. | 103 | |||||
| 103 | ||||||
| 107 | ||||||
Table of Contents
Special Note Regarding Forward-Looking Statements
This report contains forward-looking statements that are based upon current expectations within the meaning of the Private Securities Litigation Reform Act of 1995. Transcept Pharmaceuticals, Inc., or Transcept, intends that such statements be protected by the safe harbor created thereby. Forward-looking statements involve risks and uncertainties and actual Transcept results and the timing of events may differ significantly from those results discussed in the forward-looking statements. Examples of such forward-looking statements include, but are not limited to, statements about or relating to:
| • | expectations regarding plans to conduct a highway driving study and modify the Intermezzo® package presentation, and the sufficiency of such plans, to support the potential resubmission of a New Drug Application, or NDA, with the U.S. Food and Drug Administration, or FDA, for Intermezzo®; |
| • | expectations regarding our ability to resubmit the NDA for Intermezzo®, the expected timing of any such resubmission, and the expected timing of FDA review of any such resubmission; |
| • | the potential for Intermezzo® to be the first sleep aid approved specifically for use for middle of the night awakenings; |
| • | expected activities and responsibilities of us and Purdue Pharmaceutical Products L.P., or Purdue, under our United States License and Collaboration Agreement, or the Collaboration Agreement; |
| • | our potential receipt of revenue under the Collaboration Agreement, including milestone and royalty revenue; |
| • | the satisfaction of conditions under the Collaboration Agreement with Purdue required for continued commercialization, and the payment of potential milestone payments, royalties and fulfillment of other Purdue obligations under the Collaboration Agreement; |
| • | the potential benefits of, and markets for, Intermezzo® and other product candidates; |
| • | our plans for the manufacturing of Intermezzo®, including the manufacture of validation batches; |
| • | potential competitors and competitive products; |
| • | expectations with respect to our ability to carry out plans to promote Intermezzo® to psychiatrists in the United States through our co-promote option, if exercised, under the Collaboration Agreement, including development of a potential sales and marketing capability and the required size of our sales force; |
| • | guidance with respect to expected cash, cash equivalents and marketable securities, research and development expenses, and general and administrative expenses; |
| • | our ability to satisfy liquidity requirements for at least the next twelve months; |
| • | losses, costs, expenses, expenditures and cash flows, including the period of time over which we expect to recognize the revenue associated with the up-front payment under the Collaboration Agreement; |
| • | capital requirements and our needs for additional financing; |
| • | the ability and degree to which we may obtain and maintain market exclusivity from the FDA for Intermezzo® under Section 505(b)(2) of the Federal Food and Drug Cosmetic Act; |
| • | future payments under lease obligations and equipment financing lines; |
| • | our ability to obtain and maintain patent protection for Intermezzo® without violating the intellectual property rights of others; and |
| • | expected future sources of revenue and capital. |
1
Table of Contents
Transcept undertakes no obligation to, and expressly disclaims any obligation to, revise or update the forward-looking statements made herein or the risk factors whether as a result of new information, future events or otherwise. Forward-looking statements involve risks and uncertainties, which are more fully discussed in the “Risk Factors” section and elsewhere in this Annual Report, including, but not limited to, those risks and uncertainties relating to:
| • | whether additional data exists or can be generated from existing or new clinical studies to demonstrate sufficiently to the FDA that Intermezzo® would not present an unacceptable risk of residual effects, including residual effects that impair next day driving ability; |
| • | our ability to sufficiently demonstrate to the FDA that we can reduce inadvertent dosing errors of Intermezzo® in the middle of the night or that such dosing errors will not lead to unacceptable next day residual effects; |
| • | the potential for delays in or the inability to complete commercial partnership relationships, including additional marketing alliances for Intermezzo® outside the United States; |
| • | positive results in our clinical trials may not be sufficient to obtain FDA regulatory approval of Intermezzo® or to grant marketing exclusivity for Intermezzo® under Hatch-Waxman; |
| • | potential termination of the Collaboration Agreement by Purdue; |
| • | our satisfaction of conditions under the Collaboration Agreement with Purdue required for Purdue to carry out its obligations under such agreement; |
| • | difficulties or delays in building a sales organization in connection with any exercise of our co-promote option to psychiatrists under the Collaboration Agreement; |
| • | physician or patient reluctance to use Intermezzo®, if approved; |
| • | changing standards of care and the introduction of products by competitors, including generic products whose introduction could reduce our royalty rates under the Collaboration Agreement, or alternative therapies for the treatment of indications we target; |
| • | unexpected adverse side effects or inadequate therapeutic efficacy of our product candidates that could slow or prevent product approval or approval for particular indications; |
| • | inability to obtain additional financing, if necessary; |
| • | the uncertainty of protection for our intellectual property, through patents, trade secrets or otherwise; |
| • | potential infringement of the intellectual property rights or trade secrets of third parties; and |
| • | other difficulties or delays in development, testing, obtaining regulatory approval for, and undertaking production and marketing of our product candidates. |
Intermezzo®, Bimucoral®, and Transcept Pharmaceuticals, Inc.TM are registered and unregistered trademarks of ours in the United States and other jurisdictions. Other trademarks and trade names referred to in this Annual Report on Form 10-K are the property of their respective owners.
2
Table of Contents
PART I
| Item 1. | Business |
Merger of Novacea, Inc. and Transcept Pharmaceuticals, Inc.
Transcept Pharmaceuticals, Inc., or Transcept, was incorporated in Delaware in 2001 as Novacea, Inc., or Novacea. Novacea previously traded on the NASDAQ Global Market under the ticker symbol “NOVC.” On January 30, 2009, Novacea completed a business combination, or merger, with a privately held company, Transcept Pharmaceuticals, Inc., or TPI, pursuant to which TPI became a wholly owned subsidiary of Novacea and the corporate name of Novacea was changed to “Transcept Pharmaceuticals, Inc.” Prior to the merger, Novacea substantially ended its business of developing novel therapies for the treatment of cancer. Following the closing of the merger, the business conducted by TPI became the primary business of the combined entity and that business now operates through a wholly-owned subsidiary now known as Transcept Pharma, Inc. After the merger, former TPI stockholders, option holders and warrant holders as of January 30, 2009 owned approximately 61% of Transcept common stock on a fully-diluted basis. After the merger, the stockholders, option holders and warrant holders of Novacea prior to the merger owned approximately 39% of the Transcept common stock on a fully-diluted basis. Under generally accepted accounting principles in the United States, the merger is treated as a “reverse merger” under the purchase method of accounting. For accounting purposes, TPI is considered to have acquired Novacea.
Trading of Transcept Pharmaceuticals, Inc. securities on the NASDAQ Global Market under the ticker symbol “TSPT” commenced on February 2, 2009.
In this Annual Report, “Transcept,” “the Company,” “we,” “our” and “us” refers to the public company formerly known as Novacea and now known as Transcept Pharmaceuticals, Inc., and, as successor to the business of TPI, includes activities taking place with respect to the business of TPI prior to the merger of TPI and Novacea, as applicable.
Overview
Transcept Pharmaceuticals, Inc. is a specialty pharmaceutical company focused on the development and commercialization of proprietary products that address important therapeutic needs in neuroscience. Our most advanced product candidate, Intermezzo® (zolpidem tartrate sublingual tablet), is a sublingual low dose formulation of zolpidem that we are developing for use in the middle of the night at the time a patient awakens and has difficulty returning to sleep.
We submitted an NDA to the FDA on September 30, 2008. On October 28, 2009, we received a Complete Response Letter from the FDA regarding our NDA indicating that the NDA was not approved. The FDA stated in its Complete Response Letter that it believes we submitted substantial evidence of the effectiveness of Intermezzo® for its proposed indication. However, the FDA also noted that the intended use of Intermezzo® in the middle of the night represents a unique insomnia indication and dosing strategy for which safety has not been previously established and that we had not adequately demonstrated to the FDA that Intermezzo® can be reliably used safely.
Our proposed label for Intermezzo® indicates that Intermezzo® should only be taken when patients have at least four hours remaining in bed before being active again. In its Complete Response Letter, the FDA recognized that the Intermezzo® data we submitted did not indicate significant next day residual effects at four hours. However, the FDA requested additional data demonstrating that Intermezzo®, when taken as directed in the middle of the night, would not present an unacceptable risk of residual effects, with particular reference to next day driving ability. Based on communications with the FDA, we plan to conduct a pre-approval highway driving study to assess the effect of Intermezzo® on next morning driving ability beginning at approximately three hours and four hours after dosing Intermezzo® in the middle of the night.
3
Table of Contents
In the Complete Response Letter, the FDA also expressed two concerns regarding the possibility of patient dosing errors in the middle of the night that could lead to next day residual effects with particular reference to next day driving ability. Specifically, the FDA asked us to address methods to avoid inadvertent re-dosing in a single night and inadvertent dosing with less than four hours of bedtime remaining. To address these concerns, we plan to change our originally proposed Intermezzo® packaging from a multi-dose unit package to a bedside, single unit-dose package with revised patient instructions designed to reduce the possibility of inadvertent patient dosing errors.
In communications with the FDA, we discussed our proposed new packaging and how to assess its adequacy to address FDA concerns regarding the potential for inadvertent dosing errors. The FDA indicated that the revised packaging appeared to reduce the potential for inadvertently taking more than one dose in a single night, but expressed continuing concern about the risk of inadvertent dosing with less than four hours of time remaining in bed. The FDA agreed that our plan to conduct a highway driving study is a reasonable way to measure potential next day driving impairment from dosing Intermezzo® with four hours or less remaining in bed. The FDA and Transcept also discussed whether a pre-approval patient use study might help to define patient ability to properly follow instructions under actual conditions of use. We have no current plans to conduct a patient use study because of the challenges and limitations of such a study, and have submitted to the FDA our position in this regard. The FDA indicated that it would consider our position as part of the overall resubmission of the Intermezzo® NDA. We also plan to include data from on-going studies of patient comprehension of label instructions in our planned resubmission.
According to IMS Health, an independent market research firm, the number of prescriptions filled in the United States to treat insomnia grew from approximately 54 million in 2004 to approximately 79 million in 2009. Data from a major study from the Stanford Sleep Epidemiology Center published in 2008 indicate that middle of the night awakening is the most common form of insomnia in the United States and affects approximately one-third of the population at least three times each week. Data from a study published in Population Health Management in 2010, based on information from the United States National Health and Wellness Survey to evaluate the economic and humanistic burden of chronic insomnia characterized by nighttime awakenings, indicate that this condition was associated with a significant negative impact in health care utilization, health-related quality of life and work productivity. Despite the prevalence of middle of the night awakening, there is no sleep aid currently approved for use specifically in the middle of the night when patients awaken and have difficulty returning to sleep.
On July 31, 2009, we entered into the Collaboration Agreement with Purdue that provides Purdue with an exclusive license to commercialize Intermezzo® in the United States. We retained an option to co-promote Intermezzo® to psychiatrists in the United States. We also granted Purdue and an associated company the right to negotiate for the commercialization of Intermezzo® in Mexico and Canada, respectively, and we retained rights to commercialize Intermezzo® in the rest of the world. We plan to develop and market Intermezzo® through one or more development and marketing alliances in major markets outside the United States.
We have incurred net losses since inception as we have devoted substantially all of our resources to research and development, including contract manufacturing and clinical trials. As of December 31, 2009, we had cash, cash equivalents, and marketable securities of $88.9 million, including working capital of $74.3 million, and an accumulated deficit of $86.9 million.
Our ability to generate near term revenue is dependent upon the receipt of milestone and royalty payments under our Collaboration Agreement with Purdue, which are dependent upon the regulatory approval by the FDA of Intermezzo®. To achieve profitable operations, we must successfully develop and commercialize Intermezzo® or we may need to identify, develop and commercialize future product candidates. Even if approved, our products may not achieve market acceptance and will face competition from both generic and branded pharmaceutical products.
4
Table of Contents
We believe that Intermezzo® is positioned to be the first commercially available sleep aid specifically for use in the middle of the night when patients awaken and have difficulty returning to sleep. Intermezzo® has been uniquely designed for this indication and employs the following product features:
| • | Known active agent. The active pharmaceutical ingredient in Intermezzo® is zolpidem tartrate, cited by IMS Health as the most commonly prescribed agent for the treatment of insomnia in the United States, with over one billion zolpidem tablets prescribed in 2009 in the United States. Approved in 1992 as the active ingredient in Ambien®, a branded prescription sleep aid, zolpidem has a well established record of safety and efficacy. |
| • | Rapid bioavailability. We believe that rapid bioavailability, the delivery of the active pharmaceutical ingredient into systemic circulation, is a key product feature for a sleep aid intended to be used in the middle of night. Intermezzo® is formulated as a sublingual tablet, or a dosage form that dissolves under the tongue, using our proprietary technology to facilitate more rapid absorption as compared to swallowed zolpidem tablet formulations, such as Ambien®. |
| • | Low dose. We expect Intermezzo® to be commercially available in doses that are 65% and 72% lower than the comparable doses of Ambien® and Ambien CR®, a controlled release version of Ambien®, respectively. In Phase 3 clinical studies, patients who took this low dose returned to sleep rapidly and, about four hours after taking their medication in the middle of the night, showed no evidence of next day residual effects as compared to placebo. We believe that Intermezzo® 1.75 mg and 3.5 mg doses are the lowest doses of zolpidem that have been reported to induce sleep in a manner statistically superior to placebo. |
Our Business Strategy
Our goal is to become a leading developer and marketer of pharmaceutical products that fill important therapeutic needs in the field of neuroscience. Our efforts to achieve this goal are driven by the following key strategies:
| • | Obtain FDA approval for Intermezzo®. We are working to resolve the issues identified in the Complete Response Letter to our NDA. We plan to resubmit the Intermezzo® NDA in the late fourth quarter of 2010 after we generate and analyze additional data from our planned highway driving study. We expect that our resubmitted Intermezzo® NDA will result in a six-month review period at the FDA under the Prescription Drug User Fee Act, or PDUFA. |
| • | Maximize the market opportunity for Intermezzo® through marketing alliances. We granted Purdue an exclusive license to commercialize Intermezzo® in the United States. We also granted Purdue and an associated company the right to negotiate for the commercialization of Intermezzo® in Mexico and Canada, respectively. We retained rights to commercialize Intermezzo® in the rest of the world and have an active effort underway to enter into one or more development and marketing alliances with established pharmaceutical companies in major markets outside the United States. |
| • | Develop a specialty commercial organization focused on neuroscience. If Intermezzo® is approved in the United States, we plan to build a sales team focused on psychiatrists in the United States to co-promote Intermezzo®. Our collaboration agreement with Purdue gives us the option to co-promote Intermezzo® to psychiatrists in the United States as early as the first anniversary of commercial launch of Intermezzo®. For the 12 months ended December 31, 2009, IMS Health reports that approximately 5,700 psychiatrists wrote 66% of all insomnia prescriptions written by psychiatrists. Transcept believes it can begin to promote to this prescribing audience with a sales force of 50 to 60 representatives that could expand to 100 representatives depending on revenue results. |
| • | Develop a product pipeline to address unmet needs in the field of neuroscience. We completed two single blind exploratory clinical studies to examine the use of low doses of ondansetron for the treatment of obsessive-compulsive disorder, or OCD. We believe the exploratory results were encouraging and are evaluating product development strategies to pursue this opportunity. |
5
Table of Contents
| • | Identify and evaluate strategic product licensing opportunities. We are seeking additional development stage and marketed pharmaceutical product licensing opportunities in order to leverage the specialty marketing infrastructure that we plan to build in support of Intermezzo®. |
The Intermezzo® Opportunity
Overview of the insomnia market
According to IMS Health, an independent market research firm, the number of prescriptions filled in the United States to treat insomnia grew from approximately 54 million in 2004 to approximately 79 million in 2009.
Middle of the night awakening: the most common insomnia symptom
The 2003 National Sleep Foundation, or NSF, “Sleep in America” poll of the U.S. population between the ages of 55 and 84 described waking up during the night as the most prevalent insomnia symptom, affecting 33% of respondents. Based on the 2005 NSF poll data, we estimate that middle of the night awakening is 50% more common than difficulty going to sleep at bedtime among the general population. The 2009 NSF poll found that 46% of respondents described being “awake a lot during the night.”
Based on a study published in 2008 of nearly 9,000 individuals, the Stanford Sleep Epidemiology Research Center has estimated that about one-third of adults in the United States experience middle of the night awakenings at least three times each week. The study concluded that more than 90% of those subjects who reported middle of the night awakenings reported that this insomnia symptom persisted for at least six months. In the Stanford study, fewer than 25% of this middle of the night awakening group reported difficulty going to sleep at bedtime.
Data from a study published in Population Health Management in 2010, based on information from the United States National Health and Wellness Survey to evaluate the economic and humanistic burden of chronic insomnia characterized by nighttime awakenings, indicate that this condition was associated with a significant negative impact in health care utilization, health-related quality of life and work productivity.
The FDA has not previously approved any sleep aid specifically for use in the middle of the night when patients awaken and have difficulty returning to sleep. The most commonly prescribed sleep aids are formulated at doses that are sufficiently high to produce seven to eight hours of sleep. Such seven to eight hour products can be used at bedtime to prevent a middle of the night awakening. However, their prolonged duration of action makes them unsuitable for use in the middle of the night when an awakening occurs, as this may increase the risk of residual sedative effects the following morning.
Middle of the night awakenings typically do not occur every night, thus bedtime use of a high dose sleep aid to prevent an awakening requires that the patient either predict which night an awakening might occur, or take a seven to eight hour product every night. The result is that patients may use their sleep aid more often than necessary, and at a higher dose than necessary, as compared to a fast-acting, low dose sleep aid that would be used only on the nights and at the time when an awakening actually occurs.
Intermezzo®—potential to be the first sleep aid approved specifically for use in the middle of the night when patients awaken and have difficulty returning to sleep
We believe that Intermezzo®, if approved, will be the first sleep aid approved specifically for use in the middle of the night when patients awaken and have difficulty returning to sleep. In clinical trials, the unique Intermezzo® characteristics of rapid bioavailability and low dose enabled patients to return to sleep quickly and, about four hours after taking their medication in the middle of the night, showed no evidence of the residual effects that have been reported when seven to eight hour zolpidem products were taken in the middle of the night.
6
Table of Contents
Intermezzo® is a sublingual tablet utilizing a proprietary formulation intended to enhance the absorption of the active sleep medication, zolpidem. Zolpidem is the most frequently prescribed sleep aid in the United States, with, according to IMS Health, over one billion tablets prescribed in 2009 in the United States. We believe that Intermezzo® contains the lowest dose of zolpidem that has been reported to induce sleep in a manner statistically superior to placebo, and is expected to be available in doses that are 65% and 72% lower than the comparable doses of Ambien® and Ambien CR®, respectively.
Intermezzo®: Bimucoral® Technology
Intermezzo® differs from previous formulations of zolpidem through its combination of lower dose and sublingual route of administration, and is designed to be the first sleep aid approved specifically for use in the middle of the night when patients awaken and have difficulty returning to sleep. The Intermezzo® sublingual dosage form is formulated to rapidly deliver zolpidem to allow patients to return to sleep quickly. In order to permit patients to take Intermezzo® in the middle of the night and yet potentially awaken four hours later without hangover effects, Intermezzo® employs a significantly reduced zolpidem dose of 3.5 mg for adults under age 65 and 1.75 mg for those patients over age 65. We believe they are the lowest doses of zolpidem reported to be effective in inducing sleep in a manner that demonstrates statistical superiority to placebo.
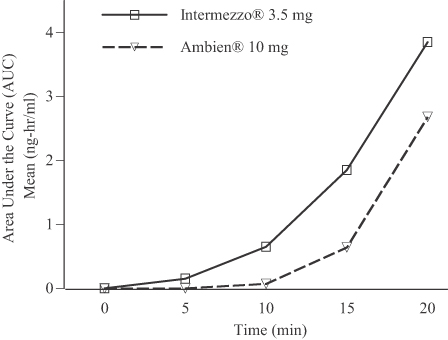
Intermezzo® utilizes Bimucoral® technology, a patented bicarbonate-carbonate binary buffer system, or a chemical combination that modifies the pH of saliva, to convert water-soluble zolpidem tartrate into its fat-soluble free-base form, which is more readily absorbed through the tissues of the mouth. We believe that this formulation facilitates rapid absorption, leading to measurable zolpidem blood levels within five minutes after administration of a 3.5 mg Intermezzo® tablet. Data from a comparative bioavailability study indicated that 10 to 20 minutes after dosing, zolpidem exposure, as delivered by Intermezzo®, was notably higher than that produced by a swallowed 10 mg zolpidem tablet. This occurred despite the fact that the 10 mg swallowed formulation contains nearly three times the Intermezzo® 3.5 mg dose. Data from this study demonstrating enhanced bioavailability, as demonstrated by the area under the curve, is illustrated below:
Bioavailability comparison study (n=33):
Intermezzo® 3.5 mg vs. Ambien® 10 mg (n=33)
7
Table of Contents
Intermezzo® Clinical Development Program
The Intermezzo® clinical development program that supported the filing of the NDA for Intermezzo® in September 2008 consisted of a total of 12 studies. Four studies were early stage bioavailability trials and utilized prototype formulations. These were completed prior to the submission of the investigational new drug application, or IND, in April 2005. Eight additional studies were conducted, including two Phase 3 clinical trials that were included in the Intermezzo® NDA submission.
The basis for clinical trial dose selection was initially provided by a pharmacokinetic and pharmacodynamic study, which demonstrated rapid bioavailability and also indicated that sedation reached peak levels within 20 minutes after dosing, as measured with the Digit Symbol Substitution Test, or DSST, a standard objective test of cognitive function to measure impairment. Despite this rapid effect, sedation levels returned to baseline within about three hours by most measures, suggesting that patients may be able to awaken without residual sedative effects four hours after taking a middle of the night dose of Intermezzo®.
The clinical safety and efficacy of Intermezzo® are supported by two Phase 3 clinical studies. The first Phase 3 trial was a double-blind crossover study conducted in sleep laboratories in 82 patients. This study analyzed both the objective and subjective effects of Intermezzo® on middle of the night awakenings. The second Phase 3 trial was a double-blind parallel group outpatient study in 294 patients which analyzed subjective outcomes when patients used Intermezzo® as needed at home at the time they awakened and had difficulty returning to sleep.
In both of these clinical trials, Intermezzo® met its primary clinical endpoint by enabling patients to return to sleep after a middle of the night awakening more rapidly than placebo. After going back to sleep, patients tended to remain asleep longer than those on placebo and awoke without evidence of residual effects as compared to placebo.
Pivotal Phase 3 sleep laboratory study
The Phase 3 sleep laboratory clinical trial was designed as an 82 patient randomized, double-blind, placebo controlled, three-way crossover study to evaluate the safety and efficacy of Intermezzo® 1.75 mg and 3.5 mg when taken for a scheduled middle of the night awakening in subjects with insomnia characterized by difficulty returning to sleep. The study was conducted in five U.S. clinical sites and each treatment period consisted of two consecutive nights of dosing followed by a 5 to 12 day washout period. The first period consisted of two baseline nights in the sleep laboratory, followed by randomized two night treatment periods using placebo and Intermezzo® 1.75 mg and 3.5 mg.
8
Table of Contents
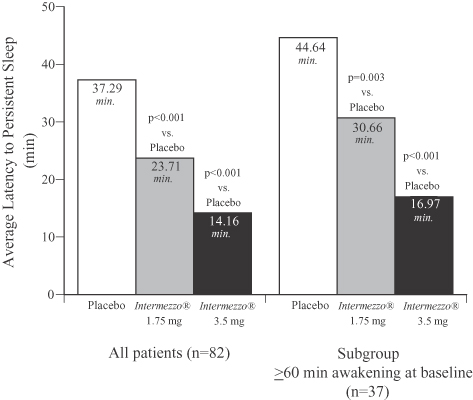
The figure below compares the time to sleep onset measured in the objective Phase 3 sleep laboratory study as produced by Intermezzo® 1.75 mg and 3.5 mg compared to placebo. The left hand bar graph compares sleep onset time in all patients in the study and demonstrates that 3.5 mg Intermezzo® returned patients to sleep in the middle of the night approximately 23 minutes faster than placebo. The right hand bar graph examines only those patients whose middle of the night awakenings were particularly prolonged, in that they experienced awakenings during the baseline observation period that lasted more than an hour. Despite the more prolonged middle of the night awakenings in this patient subset, the 3.5 mg Intermezzo® dose returned these patients to sleep approximately 28 minutes faster than placebo. All of these differences were statistically significant.
Phase 3 Sleep Laboratory Study (n=82)
Placebo vs. Intermezzo® 1.75 mg and 3.5 mg
Objective Latency to Persistent Sleep following a middle of the night awakening
9
Table of Contents
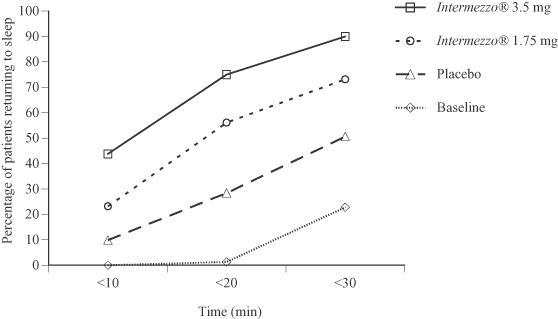
As the following figure shows, patients in the Phase 3 sleep laboratory study, when treated with either the 1.75 mg or 3.5 mg Intermezzo® dose, were more likely to fall asleep within 10 to 20 minutes than when these same patients received placebo. On the baseline nights, with one exception, no patients had returned to sleep within 20 minutes. However, on the subsequent treatment nights when patients were given Intermezzo® 3.5 mg, 75% of the same patients returned to sleep at or before the 20 minute time point.
Phase 3 Sleep Laboratory Study (n=82)
Baseline and placebo vs. Intermezzo® 1.75 mg and 3.5 mg:
Proportion of patients asleep vs. time following a middle of the night awakening
10
Table of Contents
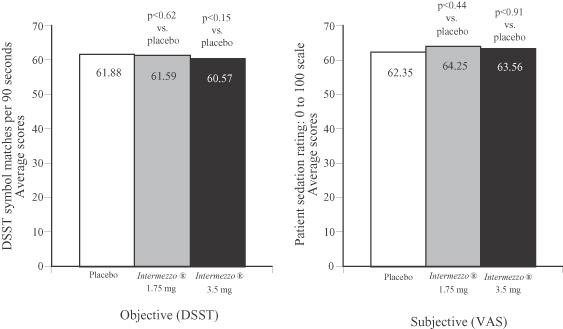
In the placebo-controlled sleep laboratory study, neither Intermezzo® dose produced residual hangover effects the morning after dosing. Residual hangover effects were measured objectively by the DSST and a subjective assessment of morning sleepiness and alertness utilizing a visual analog scale, or VAS. Results of the study are noted below.
Phase 3 Sleep Laboratory Study (n=82)
Residual effects of Intermezzo® 1.75 mg and 3.5 mg vs. placebo
DSST (objective) and VAS (subjective) scores
11
Table of Contents
Pivotal Phase 3 outpatient study
The Phase 3 outpatient clinical trial was designed as a 294-patient randomized, double-blind, placebo controlled study to evaluate the safety and efficacy of Intermezzo® 3.5 mg for use as-needed for the treatment of insomnia when a middle of the night awakening is followed by difficulty returning to sleep. The study was conducted in 25 U.S. clinical sites and the study duration included a two week baseline period, followed by a 28 day double-blind treatment period.
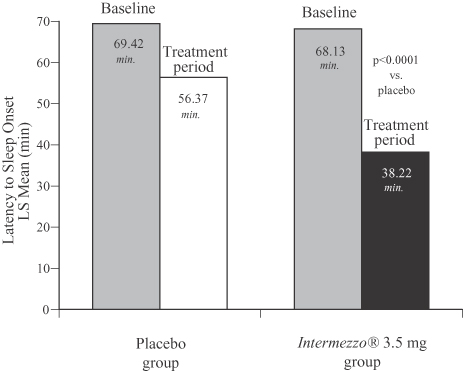
The Phase 3 outpatient study confirmed the positive results of the Phase 3 sleep laboratory study: Intermezzo® improved time to sleep onset after a middle of the night awakening by 18 minutes versus placebo, a difference that was statistically significant. The figure below compares the patient-reported time to sleep onset with Intermezzo® 3.5 mg as compared to that of placebo and at baseline.
Phase 3 Outpatient Study, Placebo vs. Intermezzo® 3.5 mg (n=294)
Latency to Sleep Onset (LSOMOTN) 4 week average
12
Table of Contents
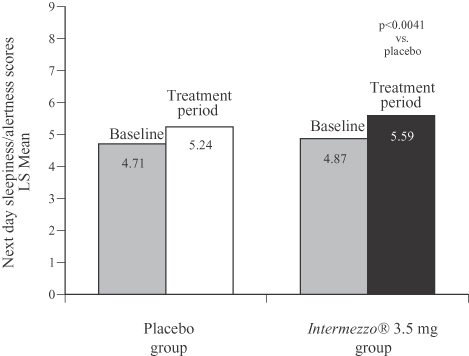
Each morning after awakening during the Phase 3 outpatient study patients reported their level of sleepiness on a nine-point scale. As the following graph shows, patients taking Intermezzo® 3.5 mg reported feeling less sleepy and more alert than patients taking placebo, a difference that was statistically significant.
Phase 3 Outpatient Study (n=294)
Next day sleepiness/alertness scores, 4-week average
(0 to 9 scale: 0 = sleepiness, 9 = awake and alert)
Intermezzo® Regulatory Review in the United States
Overview
On September 30, 2008, we submitted an NDA to the FDA to seek approval of Intermezzo® in the United States for use in the middle of the night at the time a patient awakens and has difficulty returning to sleep.
On October 28, 2009, we received a Complete Response Letter from the FDA regarding our NDA indicating that the NDA was not approved. The FDA stated in its Complete Response Letter that it believes we submitted substantial evidence of the effectiveness of Intermezzo® for its proposed indication. However, the FDA noted that the intended use of Intermezzo® in the middle of the night represents a unique insomnia indication and dosing strategy for which safety has not been previously established and that we had not adequately demonstrated to the FDA that Intermezzo® can be reliably used safely.
Our proposed label for Intermezzo® indicates that Intermezzo® should only be taken when patients have at least four hours remaining in bed before being active again. In its Complete Response Letter, the FDA recognized that the Intermezzo® data we submitted did not indicate significant next day residual effects at four hours, as measured by both the DSST and next day patient questionnaires. However, the FDA requested additional data demonstrating that Intermezzo®, when taken as directed in the middle of the night, would not present an unacceptable risk of residual effects, with particular reference to next day driving ability.
The FDA also expressed two concerns regarding the possibility of patient dosing errors in the middle of the night that could lead to next day residual effects with particular reference to next day driving ability. Specifically,
13
Table of Contents
the FDA asked us to address methods to avoid inadvertent dosing with less than four hours of bedtime remaining and inadvertent re-dosing in a single night.
On January 20, 2010, we met with the FDA to discuss the Complete Response Letter. In the briefing document submitted prior to the January 20, 2010 meeting, we proposed a new Intermezzo® bedside unit-dose package and patient instructions designed to reduce the possibility of inadvertent patient dosing errors. The FDA indicated in the meeting that the revised packaging appeared to reduce the potential for inadvertently taking more than one dose in a single night. However, the FDA expressed continuing concern that the revised packaging may not adequately address the risk of inadvertent dosing with less than four hours of time remaining in bed, with particular regard to the possibility of impaired driving.
On January 20, 2010, we also reviewed with the FDA the types of data that could support the evaluation of the proposed packaging and instructions, including data from pre-approval assessments of patient understanding of dosing instructions and a potential patient use study of the new Intermezzo® packaging.
On February 16, 2010, we proposed to the FDA to conduct a pre-approval highway driving study to assess the effect of Intermezzo® on driving ability beginning at approximately three hours and four hours post-dosing to further understand the safety of dosing Intermezzo® in the middle of the night. We also submitted additional supportive analyses of data from a 2005 Phase 1 Intermezzo® pharmacokinetic and pharmacodynamic study that measured cognitive effects through a battery of commonly recognized tests conducted at different time points up to five hours after dosing. As requested by the FDA, we also provided information on the challenges and limitations of pre-approval patient use studies, and submitted a plan to assess and optimize patient understanding of the new packaging and patient instructions.
On March 24, 2010, we had a teleconference with the FDA during which the FDA agreed that the proposal we submitted on February 16, 2010, to conduct a highway driving study, is a reasonable way to measure potential next day driving impairment as a result of dosing Intermezzo® in the middle of the night with four hours or less remaining in bed. During the teleconference, the FDA also indicated that it would consider, as part of the overall resubmission of the Intermezzo® NDA, our position on the challenges and limitations of a pre-approval patient use study. Such a study would seek to define patient ability to properly follow instructions under actual conditions of use. We have no current plans to conduct a patient use study because of the challenges and limitations of such a study. We submitted our position in this regard to the FDA on February 16, 2010 and currently plan to resubmit such position with the resubmission of the Intermezzo® NDA.
We plan to resubmit the Intermezzo® NDA in the late fourth quarter of 2010 after we generate and analyze additional data from our planned highway driving study. Because our planned resubmission will include additional clinical data, our resubmitted Intermezzo® NDA will result in a six-month review period at the FDA under the PDUFA.
Planned Highway Driving Study
We plan to conduct a pre-approval highway driving study to assess the effect of Intermezzo® on next morning driving ability beginning at approximately three hours and four hours after dosing Intermezzo® in the middle of the night. We believe this study will help to further characterize the safety profile of dosing Intermezzo® with four hours or less remaining in bed. The study will assess the potential residual effect of Intermezzo® on driving ability by tracking the deviation of subjects’ lateral position on the road over a one-hour driving period. The study is expected to assess the performance of 36 patients in a single center, double-blind, randomized, placebo-controlled crossover study design. We plan to conduct the study at the Maastricht University in the Netherlands, a leading center of research on the effects of drugs and alcohol on driving performance. Key comparisons to be analyzed in the study through their affect on subjects’ driving ability are:
| • | Intermezzo® 3.5mg verses placebo, 4 hours post-dose; |
| • | Intermezzo® 3.5mg verses placebo, 3 hours post-dose; and |
| • | Zopiclone 7.5mg verses placebo (positive control). |
14
Table of Contents
Phase 1 Safety Study
The Phase 1 study conducted in 2005 from which additional analyses were submitted to the FDA in February 2010, was conducted during the daytime in 24 normal healthy volunteers aged 21 to 44. Although not a measure of the presence or absence of potential driving impairment, the study did measure cognitive effects through a battery of commonly recognized objective and subjective measures of cognitive function. The tests included the Digit Symbol Substitution Test, or DSST, a standard objective test of cognitive function; a visual analog scale, or VAS, for subjective assessment of alertness; the Word Recall test, a generally accepted test of the effect of sedative hypnotics on memory; Choice Reaction Time, or CRT, a test that measures response time, lapses and errors, and is generally accepted as a test of performance on tasks that require sustained attention; and the Symbol Copying Test, or SCT, which is similar to DSST but without visual, search, memory or coding demands.
The Phase 1 study demonstrated that sedative activity was statistically different from placebo as early as 20 minutes in every test except SCT. SCT was not statistically different from placebo at any time-point. Scores returned to baseline at 2.5 hours or less as measured by DSST, VAS, Word Recall, CRT-response time and CRT-lapses. The CRT-error test scores were inconsistent. They were significantly different from placebo at 20 minutes and 3 hours after dosing, but not at hours 1, 1.5, 2.0, 2.5 and 4. Consistent with our proposed label for Intermezzo®, these results suggest that patients may be able to awaken without residual sedative effects four hours after taking a middle of the night dose of Intermezzo®.
Commercialization
Collaboration with Purdue
On July 31, 2009, we entered into the Collaboration Agreement with Purdue to commercialize Intermezzo® in the United States. Under the terms of our Collaboration Agreement:
| • | On August 4, 2009, Purdue paid us a $25.0 million non-refundable license fee; |
| • | We are obligated to seek FDA approval of Intermezzo® and to continue development of Intermezzo® at our expense until FDA approval; and |
| • | If Purdue elects not to terminate our collaboration after its review of an FDA approval of Intermezzo®: |
| • | Purdue is obligated to pay us up to $30.0 million, which amount is reduced by $2.0 million for each 30 day period that our receipt of an NDA approval for Intermezzo® is delayed beyond June 30, 2010; |
| • | We are obligated to transfer the Intermezzo® NDA to Purdue and Purdue is obligated to assume the expense associated with maintaining the NDA and further development of Intermezzo® in the United States, including any expense associated with post-approval studies; |
| • | Purdue is obligated to commercialize Intermezzo® in the United States at its expense; |
| • | Purdue is obligated to pay us tiered double-digit base royalties on net sales of Intermezzo® in the United States ranging up to the mid-twenty-percent level; |
| • | Purdue is obligated to pay us $10.0 million if either of two formulation patents are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book; and |
| • | Purdue is potentially obligated to pay us up to an additional $80.0 million upon meeting certain intellectual property milestones and upon the achievement of certain net sales targets for Intermezzo® in the United States. |
We retained an option to co-promote Intermezzo® to psychiatrists in the United States as early as the first anniversary of commercial launch of Intermezzo® and as late as 55 months after commercial launch. Upon entry into the market under the co-promotion option, we would receive an additional double-digit royalty from Purdue
15
Table of Contents
on sales generated by psychiatrists in the United States. This royalty is in addition to the base royalty on total United States net sales, and is set according to the time interval between initiation of sales of Intermezzo® in the United States and the time that we initiate our sales efforts in the market under the co-promote option.
Purdue has the right to terminate the collaboration agreement at any time upon 180 days notice and after review of any final FDA approved label for Intermezzo®. Our co-promote option may also be terminated by Purdue upon our acquisition by a third party or in the event of entry of generic competition to Intermezzo®. The royalty payments discussed above are subject to reduction in connection with, among other things, the entry of generic competition to Intermezzo®. The collaboration agreement expires on the later of 15 years from the date of first commercial sale in the United States or the expiration of patent claims related to Intermezzo®. The collaboration agreement is also subject to termination by Purdue in the event of FDA or governmental action that materially impairs Purdue’s ability to commercialize Intermezzo® or the occurrence of a serious event with respect to the safety of Intermezzo®. The collaboration agreement may also be terminated by us upon Purdue commencing an action that challenges the validity of Intermezzo® related patents. We also have the right to terminate the collaboration agreement immediately if Purdue is excluded from participation in federal healthcare programs. The collaboration agreement may also be terminated by either party in the event of a material breach or insolvency of the other party.
We granted Purdue and an associated company the right to negotiate for the commercialization of Intermezzo® in Mexico and Canada, respectively, and retained rights to commercialize Intermezzo® in the rest of the world.
Sales and Marketing
In our collaboration agreement with Purdue, we retained an option to co-promote Intermezzo® to psychiatrists in the United States. We can exercise this option to enter the market as early as the first anniversary of the commercial launch of Intermezzo® in the United States. Upon entry into the market under the co-promotion option, we would receive an additional double-digit royalty from Purdue on sales generated by psychiatrists in the United States.
Our co-promote option with Purdue provides us with the potential to develop our own United States specialty sales and marketing capabilities focused on the promotion of Intermezzo® to psychiatrists and other products that address unmet needs in the field of neuroscience. To achieve our goal of developing our own sales and marketing infrastructure, we must first obtain FDA approval of Intermezzo®, in-license another product opportunity or develop and obtain approval for another product. To achieve commercial success in marketing and selling Intermezzo® in the United States, we must work with our partner, Purdue, to effectively integrate our sales and marketing infrastructure and implement our sales and marketing efforts.
For the 12 months ended December 31, 2009, IMS Health reports that approximately 5,700 psychiatrists wrote 66% of all insomnia prescriptions written by psychiatrists. Transcept believes it can begin to promote to this prescribing audience with an initial sales force of 50 to 60 representatives that could expand to 100 representatives depending on revenue results.
Intermezzo® Commercialization Outside the United States
We have not yet applied for regulatory approval to sell Intermezzo® in any country other than the United States. We plan to market and sell our products that receive regulatory approval outside the United States through pharmaceutical companies that are established in their respective markets. We granted Purdue and an associated company the right to negotiate for the commercialization of Intermezzo® in Mexico and Canada, respectively. We retained rights to commercialize Intermezzo® and our other potential products in the rest of the world. We have an active effort underway to enter into one or more development and marketing alliances to develop and commercialize Intermezzo® with established pharmaceutical companies in major markets outside the United States.
16
Table of Contents
Exploratory Clinical Development Programs
We are seeking additional product opportunities that can be of importance in the field of neuroscience. In this regard, we are evaluating drug product concepts for the treatment of OCD in patients who do not respond to conventional therapeutics. Our strategy is to augment the therapeutic effects of fluoxetine and other similar selective serotonin reuptake inhibitors (SSRI) in OCD patients by promoting down-regulation of dopamine with ondansetron, currently marketed as Zofran® by GlaxoSmithKline, to provide more effective treatments to control OCD in patients who are resistant to conventional therapies. We have completed two single blind exploratory clinical studies to examine the use of a range of low doses of ondansetron in the treatment of this disorder. These studies have yielded initial results that we and our advisors believe to be encouraging and we are currently evaluating product development strategies to pursue this opportunity.
In-Licensing
We have an in-licensing effort underway to identify and secure licenses to patents and development rights relating to the use of existing drugs in the field of neuroscience, and to identify and secure the rights to one or more approved products that can be effectively sold by the specialty sales and marketing team that we plan to build.
Competition
If Intermezzo® receives FDA marketing approval, it will compete against well-established products currently used in the treatment of insomnia, both branded and generic. Potentially competitive products include branded formulations of zolpidem, such as Ambien® and Ambien CR® marketed by Sanofi-Aventis, generic formulations of zolpidem, Lunesta®, marketed by Dainippon-Sumitomo Pharma Co., Ltd., RozeremTM, marketed by Takeda Pharmaceuticals Company Limited, Sonata®, marketed by King Pharmaceuticals, Inc. and generic forms of this product, and a number of other pharmaceutical agents, including antidepressants and antipsychotics, that are prescribed off-label. None of the currently marketed sleep aids that have FDA approval are specifically approved for use in the middle of the night when patients awaken and have difficulty returning to sleep. However, many of these products can be used to prevent middle of the night awakenings by prophylactic use at bedtime.
The market for prescription sleep products has evolved significantly over the last 30 years. Until about 30 years ago, the market was dominated by barbiturate sedative-hypnotics such as Seconal® and Nembutal®. These were superseded by the benzodiazepine class of sedative-hypnotics including Dalmane®, RestorilTM and Halcion®. Zolpidem, which is a selective modulator of GABAA receptor and is a member of the non-benzodiazepine class of sleep aids, was introduced in the United States in 1993 under the Ambien® brand, and, according to IMS Health, rapidly achieved the dominant position in the prescription sleep aid market. The patent for Ambien® expired in April 2007, and shortly thereafter the FDA approved the generic manufacture of zolpidem by multiple pharmaceutical companies. The pricing of generically manufactured zolpidem is significantly lower than branded formulations of zolpidem and other non-generic sleep aids. Combined sales of generic zolpidem products accounted for approximately 45% of the U.S. prescription market for sleep aids. According to IMS Health, over one billion branded and generic zolpidem tablets were prescribed in the United States in 2009. An extended release version of zolpidem was launched successfully as Ambien CR® in 2005, and, according to IMS Health, held a 7.9% U.S. prescription market share in December 2009.
Other branded prescription sleep aids include Lunesta® (eszopiclone), marketed by Dainippon-Sumitomo Pharma Co., Ltd., which was approved in December 2004 by the FDA and launched in the first quarter of 2005, and Rozerem® (ramelteon), which is marketed by Takeda Pharmaceuticals Company Limited. According to IMS Health, in December 2009, Lunesta® held a 6.3% U.S. prescription market share and Rozerem® held a 0.8% U.S. prescription market share.
There exist a number of other agents that are used to treat insomnia. These include Sonata®, a short-acting sleep aid marketed by King Pharmaceuticals, Inc., which lost patent protection in June 2008. Although not
17
Table of Contents
approved or promoted for the treatment of middle of the night awakenings, some physicians prescribe Sonata® off-label for this purpose. EdluarTM, for which Orexo AB received marketing approval in March 2009, was launched in the U.S. market by Meda Pharmaceutals, Inc. in September 2009. EdluarTM employs the same 5mg and 10mg zolpidem doses as generic Ambien® and is designed to be used in the same manner at bedtime to produce seven to eight hours of sleep. There are also a number of other pharmaceutical agents including antidepressants and antipsychotics that are not approved for the treatment of insomnia but are frequently prescribed off-label owing to their ancillary sedative effects. For example, the antidepressant generic trazadone is widely prescribed off-label for the treatment of insomnia.
In addition to current products for the treatment of insomnia, a number of new prescription products may enter the insomnia market over the next several years. These may include the following:
| • | ZolpimistTM, an orally administered spray containing zolpidem, received marketing approval from the FDA in December 2008. ZolpimistTM employs the same 5mg and 10mg zolpidem doses as generic Ambien® and is designed to be used in the same manner at bedtime to produce seven to eight hours of sleep. In November 2009, the developers of Zolpimist™, NovaDel Pharma, Inc., announced it had licensed the U.S. and Canadian marketing rights for ZolpimistTM to ECR Pharmaceuticals Company, Inc., a wholly owned subsidiary of Hi-Tech Pharmacal Co., Inc. ECR Pharmaceuticals Company, Inc. stated in its press release announcing the agreement that it intends to launch ZolpimistTM in the first half of 2010. NovaDel Pharma, Inc. also recently announced that it commenced development of a low-dose version of Zolpimist™ for the treatment of middle-of-the-night awakenings with the intent to enter such product candidate into clinical trials. |
| • | Tasimelteon (VEC-162), a melatonin agonist being developed by Vanda Pharmaceuticals Inc., received an orphan designation from the FDA in January 2010 for treatment of non-24 hour sleep/wake disorder in blind individuals without light perception. |
| • | Silenor®, a low dose doxepin formulation intended for use at bedtime, for which Somaxon Pharmaceuticals, Inc. announced FDA approval in March 2010 for the treatment of both transient (short term) and chronic (long term) insomnia characterized by difficulty with sleep maintenance in both adults and elderly patients. In clinical trials, Silenor demonstrated maintenance of sleep into the 7th and 8th hours of the night, with no meaningful evidence of next day residual effects. In March 2010, Somaxon announced that it is focused on seeking a U.S. commercial partnership, building a U.S. commercial presence and preparing to launch Silenor® in the second half of 2010. |
| • | Indiplon, another agent that belongs to the GABAA receptor modulator class of compounds, and which has not been approved by the FDA, is being developed by Neurocrine Biosciences, Inc. for the treatment of sleep initiation insomnia and middle of the night dosing. The potential approval of indiplon pursuant to an NDA submitted by Neurocrine Biosciences, Inc. has been delayed and the regulatory future of this product is uncertain. |
| • | Almorexant, an orexin receptor agonist, is being co-developed by GlaxoSmithKline plc and Actelion Pharmaceuticals, Inc. for the treatment of insomnia. In December 2009, Actelion announced positive efficacy results from the first Phase 3 study of almorexant and that certain safety observations required further evaluation in another Phase 3 study. |
| • | SKP-1041, a controlled-release zaleplon formulation is being developed by Somnus Therapeutics Inc. targeting treatment of middle of the night awakenings with a formulation that is administered at bed time. |
| • | MK-4305, an orexin antagonist, is in Phase 3 trials and is being developed by Merck & Co., Inc. for the treatment of insomnia. |
| • | AZ-007, an inhaled version of zaleplon, is being developed by Alexza Pharmaceuticals, Inc. for the treatment of insomnia. Alexza Pharmaceuticals has commented publicly that they are evaluating AZ-007 for its suitability to treat middle of the night awakenings. |
18
Table of Contents
There are a variety of other drugs intended as sleep aids under earlier stages of development. With the exceptions of indiplon and a possible new formulation of ZolpimistTM, as noted above, we believe that all of these product candidates are intended to be taken at bedtime, and are not being developed for the as-needed treatment of middle of the night awakenings at the time they occur.
Manufacturing
We do not have or intend to develop internal clinical supply or commercial manufacturing capabilities for Intermezzo®, or other product candidates. We entered into an agreement with Patheon Inc., or Patheon, for the manufacture of Intermezzo® tablets. We have also entered into agreements with Plantex USA, Inc., or Plantex, as the sole source supplier of a special form of zolpidem tartrate, a specially manufactured form of the active pharmaceutical ingredient of Intermezzo®, and with SPI Pharma, Inc., or SPI, as a supplier of buffered soda and Pharmaburst®, a spray dried sugar, key excipients used in Intermezzo®. We have agreements with Anderson Packaging Inc., or Anderson, and Sharp Corporation, or Sharp, for packaging of Intermezzo® and have entered into agreements with Mikart, Inc., or Mikart, to qualify them as a backup commercial supplier of finished product and as a backup commercial supplier of a key Intermezzo® excipient. All of these supply and manufacturing agreements contain customary commercial terms for pharmaceutical companies regarding forecasting, payment, pricing, ordering, current good manufacturing practices, or cGMP, compliance and quality. All such agreements provide for us to pay for supplies within 30 days of being invoiced upon their shipment, and, except for the agreements with Mikart as described below, none of these agreements contain minimum purchase requirements. Other than the agreements with Sharp and Patheon, all agreements set forth four quarters of forecasting, with the first such quarter’s forecast being a binding firm order. The agreements with Sharp and Patheon contain similar forecasting provisions, except that the Sharp agreement sets forth a 12-month rolling forecast, with the first three months of such forecast being a binding firm order, and the Patheon agreement sets forth 18-month, non-binding forecasting, but with a requirement that firm orders be separately placed three months prior to expected delivery. A further description of the termination provisions and certain other terms is set forth below.
When we entered into the collaboration agreement with Purdue we amended our supply agreements with Patheon, Plantex, SPI, and Sharp effective upon notice to be provided to such manufacturers that the NDA for Intermezzo® has been transferred from us to Purdue. Once such notice has been delivered, these amendments allow Purdue to enter into direct supply agreements with such manufacturers for product supplied and sold in the United States. In connection with any termination of the Purdue collaboration agreement, the territory changes set forth in the amendments also terminate, and all supply arrangements for the U.S. territory return to Transcept.
Manufacturing Services Agreement with Patheon
In October 2006, we entered into the Manufacturing Services Agreement with Patheon. Under the agreement, we are required to obtain Intermezzo® tablets from Patheon, provided that we retain the ability to qualify a secondary supplier for a portion of its supply requirements from that secondary supplier. The initial term of the Manufacturing Services Agreement expires in December 2014, but is automatically renewed for three year periods, subject to 24 month prior notice of an election not to renew. The agreement may be terminated prior to the end of term by either party for breach or insolvency of the other party, and by us on 30 days’ notice in the event of regulatory prevention from, or six month notice for a determination by us to cease, commercialization of Intermezzo®, or upon 24 months’ prior notice for any business reason.
Supply Agreement with Plantex
In March 2006, we entered into the Supply Agreement with Plantex. Under the agreement, we are required to obtain specially manufactured zolpidem tartrate from Plantex, provided that we retain the ability to qualify a secondary supplier for a portion of its supply requirements from that secondary supplier. The initial term of the Supply Agreement expires on the earlier to occur of five years from the launch of Intermezzo® or ten years from
19
Table of Contents
the date of the agreement. The agreement may be terminated prior to the end of the term by either party for breach or insolvency of the other party, and may be terminated by Plantex upon 24 months’ prior notice if Plantex discontinues production of a special form of zolpidem tartrate.
Agreements with SPI
In June 2006, we entered into a Supply and License Agreement with SPI for the manufacture and supply of Pharmaburst®, a key excipient used in the manufacture of Intermezzo®. SPI is our sole supplier of Pharmaburst®. The term of the Supply and License Agreement expires in June 2016. This agreement may be terminated prior to the end of the term by either party for breach or insolvency of the other party.
In July, 2007, we entered into a Supply Agreement with SPI for the manufacture and supply of buffered soda, a key excipient used in the manufacture of Intermezzo®. Under the Supply Agreement, we obtained a license to patent rights for buffered soda, are required to obtain a large portion of our supply of buffered soda from SPI and are permitted to obtain up to a certain portion of alternative supply of buffered soda from a secondary supplier. A further description of the license under the Supply Agreement is in the section entitled “Business—Intellectual Property and Proprietary Technology.” The initial term of the Supply Agreement expires on the earlier to occur of the 10th anniversary of commercial sale of Intermezzo® or the 13th anniversary of the date of the agreement. This agreement may be terminated prior to the end of the term by either party for breach or insolvency of the other party, and may be terminated by SPI on 90 days’ notice if minimum annual purchase requirements are not met, or upon 12 months’ notice with each such termination not being effective until the third anniversary of certain qualifications of an alternative supplier. We have made minimum annual purchase requirements under the agreement through 2010.
Packaging and Supply Agreement with Anderson
In September 2006, we entered into a Packaging and Supply Agreement with Anderson. We plan to amend this agreement or enter into a packaging and supply agreement with a new supplier because we plan to change the packaging of Intermezzo® from a multi-dose unit package to a bedside, single unit-dose package. The initial term of the Packaging and Supply Agreement expires on the fifth anniversary of the execution of the agreement, and thereafter automatically renews for one year periods unless one year prior notice is given by either party of an intent not to renew. This agreement may be terminated prior to the end of the term by either party for breach or insolvency of the other party.
Packaging and Supply Agreement with Sharp
In June 2008, we entered into a Packaging and Supply Agreement with Sharp. We plan to amend this agreement or enter into a packaging and supply agreement with a new supplier because we plan to change the packaging of Intermezzo® from a multi-dose unit package to a bedside, single unit-dose package. The initial term of the Packaging and Supply Agreement expires in June 2018, and is renewable for three year terms upon our mutual agreement with Sharp prior to 180 days before the end of the then current term. This agreement may be terminated prior to the end of the term by either party for breach or insolvency of the other party.
Agreements with Mikart
In January 2008, we entered into a Supply and Sublicense Agreement with Mikart. Pursuant to the terms of the Supply and Sublicense Agreement, we granted to Mikart a non-exlusive sublicense in accordance with the terms of the Supply Agreement between us and SPI described above to allow Mikart to act as a back-up supplier of buffered soda. Such agreement requires us to purchase at least two batches of buffered soda (a total of approximately 420 kilograms) from Mikart within 24 months following the initial commercial sale of Intermezzo®, with the first such batch required to be purchased within 12 months of such date. The term of the Supply and Sublicense Agreement expires on the earlier to occur of the 10th anniversary of the first commercial sale of Intermezzo® or the 13th anniversary of the date of the agreement. This agreement may be terminated prior
20
Table of Contents
to the end of the term by either party for breach by the other party. In addition, we can terminate the agreement upon 45 days’ prior notice to Mikart, and payment to Mikart of a termination fee, at any time after the second anniversary of the first commercial sale of Intermezzo®.
In August 2008, we entered into a Manufacturing and Supply Agreement with Mikart for back-up supply of manufactured Intermezzo® tablets. Within the first 12 months after the FDA qualifies and approves Mikart as a supplier of Intermezzo® tablets, such agreement requires us to purchase at least three batches of tablets from Mikart, with any individual batch either containing 1.75 mg of zolpidem (in which case a batch means 500,000 tablets) or 3.5 mg of zolpidem (in which case a batch means 1,500,000 tablets). We and Mikart may also mutually agree on an alternate number of Intermezzo® tablets constituting a batch for purposes of such agreement. The term of the Manufacturing and Supply Agreement expires on the 10th anniversary of FDA qualification and approval of Mikart as a supplier of Intermezzo® tablets, but is automatically renewed for three year periods, subject to 18-month prior notice of an election not to renew. This agreement may be terminated prior to the end of the term by either party for breach by the other party, or by us if the FDA does not qualify Mikart as a supplier of Intermezzo® tablets.
Manufacturers and suppliers of our product candidates are subject to current cGMP requirements, U.S. Drug Enforcement Administration, or DEA, regulations and other rules and regulations prescribed by foreign regulatory authorities. We depend on third party suppliers and manufacturers for continued compliance with cGMP requirements and applicable foreign standards. We identified alternates for certain of the above-listed suppliers and plans to have such alternate suppliers qualified by the FDA and other regulatory authorities after potential approval of the Intermezzo® NDA.
Government Regulation
Prescription drug products are subject to extensive regulation by the FDA, including regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, distribution, import, export, advertising and promotion of such products under the Federal Food Drug and Cosmetic Act, or FFDCA, and its implementing regulations, and by comparable agencies and laws in foreign countries. Failure to comply with applicable FDA or other regulatory requirements may result in a variety of administrative or judicially imposed sanctions, including FDA refusal to approve pending applications, suspension or termination of clinical trials, Warning Letters, civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of a product from the market.
FDA approval is required before any new unapproved drug, including a new use or new dosage form of a previously approved drug, can be marketed in the United States. All applications for FDA approval must contain, among other things, information relating to safety and effectiveness, pharmaceutical formulation, stability, manufacturing, processing, packaging and labeling.
New Drug Approval
A new drug approval by the FDA generally involves, among other things:
| • | completion of extensive preclinical laboratory and animal testing in compliance with FDA good laboratory practice, or GLP, regulations; |
| • | submission to the FDA of an IND to conduct human clinical testing, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug product for each indication; |
| • | satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is produced to assess compliance with FDA cGMP, regulations; and |
| • | submission to and approval by the FDA of an NDA. |
21
Table of Contents
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates or any indications will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND automatically becomes effective 30 days after acceptance by the FDA, unless the FDA, within the 30 day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The submission of an IND may not result in FDA authorization to commence a clinical trial. Further, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice, or GCP, regulations, including regulations for obtaining informed consent by each patient.
For purposes of an NDA submission and approval, human clinical trials are typically conducted in the following four sequential phases, which may overlap:
| • | Phase 1: Studies are initially conducted in a limited population to test the product candidate for initial safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients. |
| • | Phase 2: Studies are generally conducted in a limited patient population to identify adverse effects and safety risks, to determine initial efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain additional information prior to beginning larger, more expensive and time consuming Phase 3 clinical trials. In some cases, a sponsor may decide to run what is referred to as a “Phase 2b” evaluation, which is a second, confirmatory Phase 2 trial that could, in limited situations, be accepted by the FDA and serve as one of the pivotal trials in the approval of a product candidate if the study is positive. |
| • | Phase 3: These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 trials are undertaken in larger patient populations in the target indication to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population, often at multiple, geographically-dispersed clinical trial sites. |
| • | Phase 4: In many cases, the FDA incorporates into the approval of an NDA the sponsor’s agreement to conduct additional clinical trials to further assess a drug’s safety and effectiveness after NDA approval. Such post approval trials are typically referred to as Phase 4 studies. |
Controlled clinical trials conducted for our drug candidates must be included in a clinical trials registry database that is available and accessible to the public through the internet. Failure to properly participate in the clinical trial database registry could result in significant civil monetary penalties.
The submission of an NDA is no guarantee that the FDA will find it complete and accept it for filing. The FDA reviews all NDAs submitted before it accepts them for filing. It may refuse to file the application and request additional information rather than accept the application for filing, in which case, the application must be resubmitted with the supplemental information. After the application is deemed filed by the FDA, agency staff of the FDA will review an NDA to determine, among other things, whether a product is safe and efficacious for its intended use.
22
Table of Contents
We submitted an NDA to the FDA on September 30, 2008. On October 28, 2009, we received a Complete Response Letter from the FDA regarding our NDA indicating that the NDA was not approved. We are working to resolve the issues identified in the Complete Response Letter. We plan to resubmit the Intermezzo® NDA after we generate and analyze any additional data from our planned highway driving study. The resubmission of an NDA after the receipt of a Complete Response Letter indicating that an NDA is not approved can be considered either a Class I resubmission in connection with which the goal of the FDA is to respond in two months after resubmission or a Class II resubmission in connection with which the goal of the FDA is to respond in six months after resubmission. A Class I resubmission is an application resubmitted after deficiencies in the final printed labeling, draft labeling, safety updates, stability updates, phase IV commitments, assay validation data, final release testing on the last 1-2 manufacturing lots (used to support approval), minor reanalysis of data previously submitted to the application or other minor clarifying information. A Class II resubmission is an application resubmitted after other deficiencies not under a Class I resubmission including items that require an advisory committee meeting. We believe that the planned resubmission of the Intermezzo® NDA will be considered a Class II submission because of the planned inclusion of the results of a new clinical study.
In 1992, under the Prescription Drug User Fee Act, or PDUFA, the FDA agreed to specific goals for improving the drug review time and created a two-tiered system of review times—Standard Review and Priority Review. Standard Review is applied to a drug that offers at most, only minor improvement over existing marketed therapies. The 2007 amendments to PDUFA set a goal that a Standard Review of an NDA be accomplished within a ten-month timeframe. A Priority Review designation is given to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. The goal of the FDA for completing a Priority Review is six months. The FDA strives to, and usually does, meet these review goals, but is not legally required to do so in every case. For example, the review of the Intermezzo® NDA was a Standard Review. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its NDA. As part of this review, the FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA may deny approval of an NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or additional pivotal Phase 3 clinical trials. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials may be subject to different interpretation, and the FDA may interpret data differently than we do.
Under new legislation in 2007 that granted significant new powers to the FDA, many of which are aimed at improving the safety of drug products before and after approval, the FDA may determine that a risk evaluation and mitigation strategy, or REMS, is necessary to ensure that the benefits of a new product outweigh its risks. If required, a REMS may include various elements, such as publication of a medication guide, patient package insert, a communication plan to educate healthcare providers of the drug’s risks, limitations on who may prescribe or dispense the drug, or other measures that the FDA deems necessary to assure the safe use of the drug.
Once the NDA is approved, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase 4 studies, and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are to be any material modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we will likely be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional and extensive preclinical studies and clinical trials.
23
Table of Contents
Section 505(b)(2) New Drug Applications
As an alternate path to FDA approval for modifications of products previously approved by the FDA, an applicant may submit an NDA under Section 505(b)(2) of the FFDCA. Section 505(b)(2) was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act. This statutory provision permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant, and for which the applicant has not obtained a right of reference from the owner of the data. The Hatch-Waxman Act permits the applicant to rely upon the FDA findings of safety and effectiveness of a drug that has obtained FDA approval based on preclinical or clinical studies conducted by others. In addition to relying on prior FDA findings of safety and effectiveness for a referenced drug product, the FDA may require companies to perform additional preclinical or clinical studies to support approval of the modification to the referenced product. We submitted the initial NDA for Intermezzo® under Section 505(b)(2). Our initial NDA relied on the extensive information that has been collected for immediate release zolpidem products, which contain the approved active drug agent that is incorporated in Intermezzo®. To the extent that a Section 505(b)(2) application relies on a prior FDA finding of safety and effectiveness of a previously approved product, the applicant is required to certify to the FDA concerning any patents listed for the referenced product in the FDA publication called “Approved Drug Products with Therapeutic Equivalence Evaluations,” otherwise known as the “Orange Book.” Specifically, the applicant must certify in the application that:
| • | there is no patent information listed for the reference drug; |
| • | the listed patent has expired for the reference drug; |
| • | the listed patent for the reference drug has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
| • | the listed patent for the reference drug is invalid, unenforceable, or will not be infringed by the manufacture, use or sale of the product for which the 505(b)(2) NDA is submitted. |
In the Intemezzo® NDA, we made appropriate certification based on the listed and unexpired patents, if any, for the referenced drug product. Currently, there are no unexpired patents for immediate release zolpidem products listed in the Orange Book.
In the event that one or more patents is listed in the Orange Book for the referenced product, including patents listed after we submitted the NDA for Intermezzo®, we may also be required to evaluate the applicability of these patents to Intermezzo® and submit additional patent certifications. A paragraph III certification, stating that a listed patent has not expired, but will expire on a particular date, may delay the approval of Intermezzo® until the expiration of the patent. A paragraph IV certification, stating that a listed patent is invalid, unenforceable, or not infringed by Intermezzo® may require us to notify the patent owner and the holder of the NDA for the referenced product of the existence of the Intermezzo® NDA, and may result in patent litigation against us and the entry of a 30-month stay of FDA ability to issue final approval to the Intermezzo® 505(b)(2) NDA.
If we obtain FDA approval for Intermezzo®, we could obtain three years of Hatch-Waxman marketing exclusivity for the product. Under this form of exclusivity, the FDA would be precluded from approving a marketing application for a duplicate of Intermezzo®, a product candidate that the FDA views as having the same conditions of approval as Intermezzo®, for example, the same indication, the same route of delivery and/or other conditions of use, or a 505(b)(2) NDA submitted to FDA with Intermezzo® as the reference drug, for a period of three years from the date of Intermezzo® approval, although the FDA may accept and commence review of such applications. This form of exclusivity may not prevent the FDA from approving an NDA that relies only on its own data to support the change or innovation. Further, if another company obtains approval for a product candidate for the same conditions of approval that we are studying for Intermezzo® before Intermezzo® were to receive approval, the Intermezzo® approval could be blocked until the other company’s three-year Hatch-Waxman marketing exclusivity expires.
24
Table of Contents
Manufacturing cGMP Requirements
We and our contract manufacturers are required to comply with applicable FDA manufacturing requirements contained in the FDA cGMP regulations. cGMP regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facilities for active pharmaceutical ingredients, or APIs, and finished drug products must meet cGMP requirements to the satisfaction of the FDA, and pass a pre-approval inspection before we can use them to manufacture our products. We and our third-party manufacturers are also subject to periodic inspections of facilities by the FDA and other authorities, including inspection of the procedures and operations used in the testing and manufacture of our products to assess continued compliance with applicable regulations.
The API used to manufacture some of our product candidates originates outside the United States. The FDA could increase its diligence with regard to foreign sourced materials and manufacturing processes which may result in increased costs of maintaining foreign manufacturing and could lengthen or delay the regulatory review process required to gain approval for our product candidates.
Failure to comply with statutory and regulatory requirements subjects a manufacturer to possible legal or regulatory action, including adverse publicity, Warning Letters, the seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations, and civil and criminal penalties. Adverse patient experiences with the product received by us must be reported to the FDA and could result in the imposition of market restriction through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
Other Regulatory Requirements
With respect to post-market product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards for direct-to-consumer advertising, industry-sponsored scientific and educational activities, and promotional activities involving the internet, as well as a prohibition on off-label promotion. The FDA has very broad enforcement authority under the FFDCA, and failure to abide by these regulations can result in penalties, including the issuance of a Warning Letter directing entities to correct deviations from FDA standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and state and federal civil and criminal investigations and prosecutions. Numerous other laws, not administered by the FDA, also apply to the promotion of pharmaceuticals, alleged violations of which may also result in state and federal civil and criminal investigation and prosecutions.
We are also subject to various laws and regulations regarding laboratory practices, the experimental use of animals, and the use and disposal of hazardous or potentially hazardous substances in connection with our activities. In each of these areas, as above, the FDA and other agencies have broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals, any one or more of which could have a material adverse effect on us.
DEA Regulation
Zolpidem, the active pharmaceutical ingredient in Intermezzo®, is classified as a schedule IV controlled substance by the DEA. As a result, manufacturing of zolpidem is subject to regulation by the DEA. Controlled substances are those drugs that appear on one of five schedules promulgated and administered by the DEA under the Controlled Substances Act, or CSA. The CSA governs, among other things, the distribution, record keeping, handling, security, and disposal of controlled substances. We, as well as our third-party suppliers who handle zolpidem, must be registered by the DEA in order to engage in these activities, and are subject to periodic and ongoing inspections by the DEA and similar state drug enforcement authorities to assess ongoing compliance
25
Table of Contents
with DEA regulations. Any failure by us or our third party suppliers to comply with these regulations could lead to a variety of sanctions, including the revocation, or a denial of renewal, of DEA registration, injunctions, or civil or criminal penalties and loss of supply.
Third-Party Reimbursement and Pricing Controls
In the United States and elsewhere, sales of pharmaceutical products depend in significant part on the availability of coverage and reimbursement to providers and the consumer from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. Our products may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow sales of our products on a competitive and profitable basis.
In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Medicare
Two principal payors in the United States are Medicaid and Medicare. We expect that in the United States many patients who are treated with Intermezzo® will be Medicare beneficiaries. The Centers for Medicare and Medicaid Services, or CMS, is the agency within the Department of Health and Human Services that administers both Medicare and Medicaid. CMS has the authority not to cover particular products or services if it determines that they are not “reasonable and necessary” for the treatment of Medicare beneficiaries. CMS may make a national coverage determination, or NCD, for a product, which establishes on a nationwide basis the indications that will be covered, and any restrictions or limitations. However, for most new drugs that are eligible for payment, CMS does not create an NCD, and we do not plan to request one. Nevertheless, CMS or a third party may request an NCD independently. If such request is made, we cannot assure investors that such NCD will contain favorable coverage terms.
If there is no NCD, the local Medicare contractors that are responsible for administering the program on a state or regional basis have the discretion to deny coverage and reimbursement for the drug or issue a local coverage determination, or LCD. These LCDs can include both coverage criteria for the drug and frequency limits for the administration of the drug. Overturning restrictive LCDs in the various regions can be a time-consuming and expensive process.
Effective January 1, 2006, Congress enacted a prescription drug benefit known as Medicare Part D. CMS contracts with numerous managed care plans and drug benefit plans to deliver the drug benefit. These plans develop formularies that determine which products are covered and at what co-pay level. If Medicare coverage for Intermezzo® is available, CMS will reimburse through Part D. While CMS evaluates Part D plans’ proposed formularies for potentially discriminatory practices, the plans have considerable discretion in establishing formularies, establishing tiered co-pay structures and placing prior authorization and other restrictions on the utilization of specific products. Moreover, Part D plan sponsors are permitted and encouraged to negotiate rebates with manufacturers. Revenue for Intermezzo® will be substantially affected by its formulary status on Part D plans and the rebates that Part D plan sponsors are able to negotiate.
Medicaid
Medicaid is a federal and state entitlement program that pays for medical assistance for certain individuals and families with low incomes and resources and who meet other eligibility requirements. Medicaid became law in 1965 and is jointly funded by the federal and state governments (including the District of Columbia and the territories) to assist states in furnishing medical assistance to eligible needy persons. Medicaid is the largest source of funding for medical and health-related services for the indigent population of the United States.
26
Table of Contents
We expect Intermezzo® to be eligible for reimbursement under Medicaid and, therefore, subject to rebates under the Medicaid Drug Rebate Program established by the Omnibus Budget Reconciliation Act of 1990. Under the Medicaid Drug Rebate Program, we would pay a rebate to each participating state agency for each unit of product reimbursed by Medicaid. The basic amount of the rebate for each product is the greater of 15.1% of the Average Manufacturer Price (AMP) of that product, or the difference between AMP and the best price available from us to any non-excluded customer. The rebate amount also includes an inflation adjustment if AMP increases faster than a specified inflation index. The rebate amount is calculated quarterly based on our reports of its current AMP and best price for each of its products to CMS. AMPs and best price may be recalculated after they are initially submitted based on the availability of additional data or because of additional analysis of prices that have been reported.
Several state Medicaid programs have implemented Preferred Drug Lists, or PDLs, and more states may adopt this practice. Products placed on a state Medicaid program’s PDL are not subject to restrictions on their utilization by Medicaid patients, such as the need to obtain authorization prior to prescribing. If Intermezzo® is not included on Medicaid PDLs, use of it in the Medicaid program may be adversely affected. In some states that have adopted PDLs, we may be required to provide substantial supplemental rebates to state Medicaid authorities in order for Intermezzo® to be included on the PDL.
Pharmaceutical manufacturers, as a condition of participation in the Medicaid Drug Rebate Programs, must enter into an agreement with the Secretary of the Department of Health and Human Services to participate in the 340B program, enacted by the Public Health Service, or PHS, Act. Under the 340B programs pharmaceutical manufacturers are required to extend discounts based on the Medicaid rebate to a variety of health care entities referred to as covered entities. These covered entities include health care providers that receive health services grants from the PHS, as well as certain hospitals that serve a disproportionate share of Medicare and Medicaid beneficiaries.
Section 603 of the Veteran’s Health Care Act of 1992 requires manufacturers of covered drugs to enter into a master agreement with the Secretary of the Department of Veteran Affairs, or VA, in order to have their drugs covered under Medicaid. The master agreement requires the manufacturer to make its products available for federal procurement by listing them on the Federal Supply Schedule. In addition, the master agreement requires the manufacturer to enter into a Pharmaceutical Pricing Agreement, or PPA, with the VA. Under the PPA, the manufacturer agrees to sell its drugs to the “Big Four” federal agencies—the VA, the Department of Defense, the PHS and the Coast Guard—at or below a Federal Ceiling Price, which is set at 76% of a calculation called the Non-Federal Average Manufacturer Price (non-FAMP), minus an additional discount.
Another source of reimbursement for drug products is state Pharmaceutical Assistance Programs, or SPAPs. Many of these programs were created by states to aid low-income elderly or persons with disabilities who do not qualify for Medicaid. We would pay rebates to some SPAPs and, if they are considered qualified programs by CMS, the prices we provided these entities would be excluded from our Medicaid best price.
Private Insurance Reimbursement
Commercial insurers usually offer pharmacy benefits. If private insurers decide to cover Intermezzo®, they will reimburse for the drug in a variety of ways, depending on the insurance plan’s policies, employer and benefit manager input and contracts with their physician network. Private insurers tend to adopt reimbursement methodologies for a product similar to those adopted by Medicare. Revenue for Intermezzo® may be materially and adversely affected if private payors make unfavorable reimbursement decisions or delay making favorable reimbursement decisions.
The continuing efforts of government and third-party payors to contain or reduce the costs of health care through various means may reduce potential revenues we may receive from sales of Intermezzo®, if approved. These payors’ efforts could decrease the price that we receive for products it may sell, including Intermezzo®. In
27
Table of Contents
addition, third-party insurance coverage may not be available to patients for our products at all, especially in light of the availability of low-cost generic zolpidem therapeutics, regardless of the fact that such products are not designed or approved to treat middle of the night awakenings. Third-party payors could also impose conditions that must be met by patients prior to providing coverage for use of our products. For instance, insurers may establish a prior authorization procedure or “step-edit” system that requires a patient to utilize a lower price alternative product prior to becoming eligible to purchase a higher price product that may be better targeted to the condition being treated. There can be no assurance that third-party payors will not similarly require a patient to first use generic zolpidem or other sleep aids prior to being eligible for insurance coverage of Intermezzo® use.
If government and third-party payors do not provide adequate coverage and reimbursement levels for our products, or if price controls or step-edit systems are enacted, our product royalties or revenues will suffer.
Intellectual Property and Proprietary Technology
Our success will depend in part on our ability to protect Intermezzo® and future products and product candidates by obtaining and maintaining a strong proprietary position both in the United States and in other countries. To develop and maintain our proprietary position, we will rely on patent protection, regulatory protection, trade secrets, know-how, continuing technological innovations and licensing opportunities.
The active ingredient and many of the inactive ingredients in Intermezzo® have been known and used for many years and, therefore, are no longer subject to patent protection. Accordingly, our patents and applications are directed to the particular formulations and methods of use of zolpidem, the active pharmaceutical ingredient in Intermezzo®. There can be no assurance that our issued patents that cover the formulation of Intermezzo® will prevent others from marketing formulations using the same active and inactive ingredients in similar but different formulations. Issued patents and currently pending patent applications that cover Intermezzo® have claims that are directed to both formulation and methods of use and are summarized below:
| • | Formulations of zolpidem. We have two issued U.S. patents, one pending U.S. patent application and 15 corresponding foreign patents or applications. Patents have been granted in China, New Zealand, Singapore, and South Africa. |
| • | Methods of use of zolpidem. We have seven pending U.S. patent applications and 13 foreign patents or applications. A patent has been granted in South Africa. |
| • | Buffered soda. We have one pending U.S. patent application, which is co-owned with SPI pursuant to the Supply Agreement between us and SPI, covering the compositions containing buffered soda and their method of use, which we refer to as Bimucoral® technology. Under the Supply Agreement, we have a royalty-free, fully paid-up exclusive license with respect to this patent application, with a right to grant sublicenses, for products incorporating both buffered soda and zolpidem. This license survives the termination of the Supply Agreement. |
In addition to the applications directed to Intermezzo®, we filed patent applications for various other formulations and methods of use of drugs including pilocarpine, ondansetron, and ondansetron in combination with atypical antipsychotic drugs. We currently have no plans to pursue development of products relating to the pilocarpine patent application, but are evaluating opportunities relating to ondansetron alone or in combination with atypical antipsychotic drugs. These applications are summarized below.
| • | One pending U.S. patent application relating to compositions and methods of use of pilocarpine |
| • | Three pending U.S. patent applications relating to compositions and methods of use of ondansetron and ondansetron in combination with atypical antipsychotics |
The patent positions of pharmaceutical companies are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued. Consequently, we do not know whether any of our patent applications will
28
Table of Contents
result in the issuance of patents or, if any of our issued patents will provide significant proprietary protection or will be circumvented or challenged and found to be unenforceable or invalid. In limited instances, patent applications in the United States and certain other jurisdictions are maintained in secrecy until patents issue, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain of the priority of inventions covered by pending patent applications. Moreover, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office to determine priority of invention or in opposition proceedings in a foreign patent office, any of which could result in substantial cost to us, even if the eventual outcome is favorable. There can be no assurance that a court of competent jurisdiction would hold the patents, if issued, valid. An adverse outcome could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties or require us to cease using such technology. To the extent we determine it to be prudent, we intend to bring litigation against third parties that we believe are infringing its patents.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose such technology or that we can meaningfully protect our trade secrets. However, we believe that the substantial costs and resources required to develop technological innovations will help us protect our products.
We require our employees, consultants and members of our scientific advisory board to execute confidentiality agreements upon the commencement of employment, consulting or collaborative relationships with us. These agreements provide that all confidential information developed or made known during the course of the relationship with us be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions resulting from work performed for us, utilizing the property or relating to our business and conceived or completed by the individual during employment shall be our exclusive property to the extent permitted by applicable law. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Employees
As of March 15, 2010, we had 29 employees, 6 of whom hold Ph.D., Pharm.D., or equivalent degrees. A total of 12 employees were engaged in research and development, 3 were in sales and marketing, and 14 were in administration and finance. None of our employees are represented by a labor union or subject to a collective bargaining agreement. We have not experienced any work stoppages and we consider our relations with our employees to be satisfactory.
Available Information
Availability of Reports. We are a reporting company under the Securities Exchange Act of 1934, as amended, or the 1934 Act, and file reports, proxy statements and other information with the Securities and Exchange Commission, or SEC. The public may read and copy any of our filings at the SEC’s Public Reference Room at 100 F Street N.E., Washington, D.C. 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. Because we make filings with the SEC electronically, you may access this information at the SEC’s Internet site: www.sec.gov. This site contains reports, proxies and information statements and other information regarding issuers that file electronically with the SEC.
Web Site Access. Our Internet web site address is www.transcept.com. We make available, free of charge at the “Investors” portion of our web site, annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the 1934 Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Reports of beneficial ownership filed pursuant to Section 16(a) of the 1934 Act are also available on our web site. Information in, or that can be accessed through, this web site is not part of this annual report on Form 10-K.
29
Table of Contents
| Item 1A. | Risk Factors |
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below and all information contained in this report before you decide to purchase our common stock. If any of the possible adverse events described below actually occurs, we may be unable to conduct our business as currently planned and our financial condition and operating results could be harmed. In addition, the trading price of our common stock could decline due to the occurrence of any of the events described below, and you may lose all or part of your investment.
In the following discussion, references to “Transcept,” the “Company,” “we,” “us” and “our” refer to the public company formerly known as Novacea, Inc. and now known as Transcept Pharmaceuticals, Inc., and (as successor to the business of the private company formerly known as Transcept Pharmaceuticals, Inc., or TPI, that is now the wholly owned subsidiary of Transcept following the merger of TPI and Novacea) also relates to activities taking place with respect to, and the financial information of, the business of TPI prior to the merger of TPI and Novacea.
We have had a brief operating history that may make it difficult for you to evaluate the potential success of our business and we have a history of incurring losses.
We were founded in January 2001 under our former name Novacea, Inc., and in January 2009 underwent a merger with TPI, a private company founded in 2002 whose business is currently conducted by us. Our operations to date have been limited to organizing and staffing, acquiring, developing and securing technology and undertaking preclinical studies and clinical trials. We have not yet demonstrated the ability to obtain regulatory approval and manufacture marketed products to the U.S. Food and Drug Administration, or FDA. We have also not demonstrated the ability to meet and adhere to other regulatory standards applicable to an FDA approved product, to conduct sales and marketing activities or to support commercialization efforts of a collaboration partner, such as our collaboration partner in the United States, Purdue Pharmaceutical Products L.P., or Purdue. Consequently, any predictions you make about our future success or viability may not be as accurate as they would be if we had a longer operating history.
Furthermore, our business is not profitable and has incurred losses in each year since the inception of TPI in 2002. Our net loss for the years ended December 31, 2009, 2008 and 2007 was $21.8 million, $20.0 million and $20.4 million, respectively. As of December 31, 2009, we had an accumulated deficit of $86.9 million. We expect to continue to incur losses for the foreseeable future unless Intermezzo® is approved by the FDA and we receive milestone and royalty revenue from our collaboration with Purdue that exceed our expenses. For the foreseeable future, we expect our accumulated deficit to increase as we continue our research, development, regulatory and pre-approval and pre-commercialization efforts with respect to Intermezzo® (both in support of our collaboration partner in the United States and potential collaboration partners worldwide) and with respect to other product candidates. If Intermezzo® or our other product candidates do not gain regulatory approval, are not commercialized or do not achieve market acceptance, we may not be able to generate any revenue. We cannot assure you that we will ever be profitable even if Intermezzo® or any other product candidate is commercialized or that we can sustain profitability, even if achieved. If we fail to achieve and maintain profitability, or if we are unable to fund our continuing losses, investors could lose all or part of their investment.
Our success depends substantially on our ability to obtain regulatory approval in the United States for our lead product candidate, Intermezzo®, and to overcome issues presented by the FDA in response to our New Drug Application.
Our success depends substantially on obtaining regulatory approval for our most advanced product candidate, Intermezzo®, for use as needed for the treatment of insomnia when a middle of the night awakening is followed by difficulty returning to sleep, or middle of the night awakening. Regulatory approval to market pharmaceutical products in the United States requires the completion of extensive non-clinical and clinical evaluations of a product candidate, referred to as clinical trials, to demonstrate substantial evidence of both safety
30
Table of Contents
and efficacy of the candidate, as well as development of manufacturing processes that demonstrate the ability to reliably and consistently produce the candidate under current Good Manufacturing Practice, or cGMP, regulations. Each of these elements requires pharmaceutical development companies to exercise certain judgments concerning applicable regulatory requirements and to predict what the regulatory authority will ultimately deem acceptable. There can be no assurance that the results of the clinical trials or manufacturing processes for Intermezzo® will satisfy the regulatory requirements for approval. A failure to meet these requirements would significantly delay or prevent approval of Intermezzo® and seriously harm our ability to generate revenue.
In September 2008, we submitted a New Drug Application, or NDA, to the FDA for Intermezzo®. On October 28, 2009, we received a Complete Response Letter from the FDA formally responding to our Intermezzo® NDA. The Complete Response Letter indicated that the FDA did not believe we adequately demonstrated that Intermezzo® could be reliably used safely. The NDA was therefore not approved.
In the Complete Response Letter, the FDA noted that we are seeking to gain approval of Intermezzo® in a unique insomnia indication for which safety has not previously been established, specifically, the as needed treatment for difficulty returning to sleep after a middle of the night awakening. The FDA’s Complete Response Letter noted that data presented in the Intermezzo® NDA indicated no significant residual effects four hours after dosing, as measured by both the Digit Symbol Substitution Test, a commonly used test to measure the impairment of patients taking sedative hypnotics, and next day patient questionnaires. However, the FDA requested additional data demonstrating that Intermezzo®, when taken as directed in the middle of the night, would not present an unacceptable risk of residual effects, with particular reference to a patient’s ability to drive the next morning.
The FDA also expressed two concerns in the Complete Response Letter regarding the possibility of patient dosing errors in the middle of the night that could lead to next day residual effects, with particular reference to next day driving ability. Specifically, the FDA asked us to address methods to avoid inadvertent dosing with less than four hours of bedtime remaining, and inadvertent re-dosing of Intermezzo® in a single night.
We plan to conduct a highway driving study to measure the potential for next day driving impairment from dosing Intermezzo® in the middle of the night with four hours or less remaining in bed. This study is designed to evaluate the effects of Intermezzo® when dosed in a manner that is consistent with the proposed label wherein dosing occurs with at least four hours remaining in bed, and when Intermezzo® is dosed in a manner that is inconsistent with the proposed label with at least three hours remaining in bed. We believe the general risks of study design and execution are heightened by the fact that, to our knowledge, this is the first study of residual effects on driving ability to be conducted in support of FDA approval of a new sleep agent. Therefore commonly accepted protocols, study endpoints and statistical evaluation methodologies have not been established. Thus, we cannot assure you that our planned study will generate results that will be sufficient to demonstrate the safety of Intermezzo®. Additional studies in support of the NDA, if later determined to be required, may also or alternatively include studies to assess other measurements of drug safety.
We also need to determine how best to demonstrate to the FDA that Intermezzo® would be used in a manner consistent with the proposed label by minimizing inadvertent dosing errors. We have discussed with the FDA our proposed change in packaging from a multi-dose unit package to a bedside, single unit-dose package with instructions designed to reduce inadvertent dosing errors. We have also discussed with the FDA how to assess the adequacy of the proposed new packaging to address FDA concerns regarding the potential for inadvertent dosing errors. The FDA expressed continuing concern about the risk of inadvertently dosing with less than four hours of time remaining in bed. If an evaluation of a new product presentation is required in support of FDA approval of Intermezzo®, there also can be no assurance that we will be able to effectively design or carry out such evaluation in a cost-effective manner, or at all, or that the FDA will find any data arising from such evaluation to be supportive of our efforts to gain approval for Intermezzo®. Accordingly, if the FDA believes that our new single unit-dose package with revised instructions does not adequately reduce inadvertent dosing errors, our planned resubmission of the Intermezzo® NDA could be denied and our business would be materially harmed.
31
Table of Contents
The FDA also discussed with us whether a pre-approval patient use study might help to define patient ability to properly follow instructions under actual conditions of use. We have no current plans to conduct a patient use study because of the challenges and limitations of such a study and have submitted to the FDA our position in this regard. Problems with pre-approval use studies include the inability to replicate actual in-use conditions and study subjects’ altering their behavior because they are under observation. The risks associated with designing and conducting such an evaluation are heightened by the fact that recognized standards for such evaluations have not been established. The FDA indicated that it would consider our position on the challenges and limitations of a pre-approval use study as part of the overall resubmission of the Intermezzo® NDA. The FDA may not agree with our proposal and may require us to conduct such a use study. If we are required to conduct a pre-approval patient use study, notification of such requirement may not be delivered to us until after review of our resubmitted NDA for Intermezzo®. If the FDA does not agree with our proposal to not conduct a use study, approval of a resubmitted Intermezzo® NDA could be denied and our business would be materially harmed.
Our proposed driving study has been designed, in part, to characterize the effect on driving of dosing Intermezzo® with at least three hours remaining in bed, which is in a manner that is inconsistent with the proposed label that requires patients dose with at least four hours remaining in bed. We believe the FDA will consider the adequacy of our new proposed packaging to minimize dosing errors in the context of the data from our proposed highway driving study that may reflect the presence or absence of impairment of driving ability from residual effects of Intermezzo®. However, even if our study shows the absence of residual effect on driving ability when Intermezzo® is dosed with three hours remaining in bed, the FDA may still request that we demonstrate the extent to which patients use Intermezzo® in the middle of the night in a manner inconsistent with the proposed label.
If our proposed new packaging is not acceptable to the FDA, we may need to develop a new presentation of the proposed product that may include new dosing instructions, packaging and/or dispensing methods for Intermezzo® designed to maximize the likelihood that Intermezzo® would be taken as directed. Any such new presentation could make Intermezzo® a less attractive commercial product and more costly to produce. There can be no assurance that we will be able to identify a new product presentation that will address the FDA’s concerns regarding inadvertent mis-dosing of Intermezzo® by patients to a degree sufficient to warrant FDA approval of Intermezzo®. We cannot assure you that any such new presentation, if identified or developed, will be cost-effective or easy to manufacture, and if the Intermezzo® NDA is approved after meeting FDA requirements, that such new presentation will not make Intermezzo® a less commercially attractive product.
Additionally, despite the FDA’s statement in the Complete Response Letter finding that that we presented substantial evidence of effectiveness of Intermezzo®, there can also be no assurance that the FDA will not come to a different interpretation of our previously submitted clinical trial data, including data from our two pivotal Phase 3 clinical trials that served as the basis for our Intermezzo® NDA, or otherwise alter its view and conclude that Intermezzo® is not sufficiently effective to warrant approval.
Because the FDA has not approved a pharmaceutical product specifically to treat middle of the night awakening, there can be no assurance that the FDA will approve this new indication within the insomnia category. While we expect to continue our efforts to obtain and to follow FDA guidance in order to obtain approval of Intermezzo®, the FDA may not agree that any new data or trial results we submit will be sufficient to support Intermezzo® approval or may reconsider its guidance, require more clinical trials or otherwise require additional data or studies to justify a new middle of the night awakening indication in the insomnia market.
In addition, we have limited experience in preparing, submitting and prosecuting regulatory filings, including NDAs and other applications necessary to gain regulatory approvals. Unless we receive regulatory approval from the FDA, Intermezzo® cannot be commercialized in the United States. Significant delay or the inability to commercialize Intermezzo® in the United States would significantly harm our business and financial prospects.
32
Table of Contents
Our clinical trials may fail to demonstrate adequately the safety and efficacy of our product candidates, which could prevent or delay regulatory approval and commercialization.
Before obtaining regulatory approvals for the commercial sale of our product candidates, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that the product candidate is both safe and effective for use in each target indication. The results obtained in completed clinical trials and non-clinical studies may not be predictive of results from ongoing or future trials. Our trial results may be negatively affected by factors that had not been fully anticipated prior to commencement of the trial. Such trials may fail to demonstrate efficacy in the treatment of the intended disorder or may fail to demonstrate that a product candidate is safe when used as directed or even when misused. The outcome of earlier trials may not be predictive of the outcome of later trials. For example, we previously conducted a Phase 1 pharmacokinetic and pharmacodynamic study of Intermezzo® that demonstrated rapid bioavailability and also indicated that sedation levels returned to baseline within about three hours by most measures, suggesting that patients may be able to awaken without residual sedative effects four hours after taking a middle of the night dose of Intermezzo®. These study results should not in any way be construed as predictive of the outcome of any future studies, including, without limitation, a repeat of the same or similar studies or the planned study to assess the effect of Intermezzo® on study subjects’ ability to drive or to be predictive of the sufficiency of current or future data that may be generated to support FDA approval of Intermezzo® for its intended indication. Actual results of any future studies may differ materially from past studies due to various risks and uncertainties, including, but not limited to,
| • | identical study designs evaluating identical endpoints may produce different study results; |
| • | different study designs intended to measure the same or similar endpoints may produce different results; |
| • | different studies in different or progressively larger patient populations could reveal more frequent, more severe or additional side effects that were not seen in earlier studies; and |
| • | the unpredictable nature of clinical trials generally. |
Although we seek to design our clinical trial protocols to address known factors that may negatively affect results, there can be no assurance that protocol designs will be adequate or that factors that we may or may not be aware of or anticipate will not have a negative effect on the results of our clinical trials. Once a study has commenced, we may voluntarily suspend or terminate the study if at any time we believe that there is an unacceptable safety risk to patients.
Further, side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities stopping further development of or denying approval of our product candidates. Based on results at any stage of clinical trials, we may decide to repeat or redesign a trial, modify our regulatory strategy or even discontinue development of one or more of our product candidates.
If our product candidates are not shown to be both safe and effective in clinical trials, the resulting delays in developing other compounds and conducting associated non-clinical testing and clinical trials, as well as the potential need for additional financing, would have a material adverse effect on our business, financial condition and results of operations.
Delays in the commencement or completion of clinical testing could result in increased costs to us and delay our ability to generate revenue.
We do not know whether future clinical trials will begin on time or be completed on schedule, if at all. The commencement and completion of clinical trials can be disrupted for a variety of reasons, including difficulties in:
| • | addressing issues raised by the FDA or other regulatory authorities regarding safety, design, scope and objectives of clinical studies; |
| • | recruiting and enrolling patients to participate in a clinical trial; |
33
Table of Contents
| • | obtaining regulatory approval to commence a clinical trial; |
| • | reaching agreement on acceptable terms with prospective clinical research organizations and trial sites; |
| • | manufacturing sufficient quantities of a product candidate; and |
| • | obtaining institutional review board approval to conduct a clinical trial at a prospective site. |
A clinical trial may also be suspended or terminated by us or the FDA or other regulatory authorities due to a number of factors, including:
| • | failure to conduct the clinical trial in accordance with regulatory requirements or in accordance with our clinical protocols; |
| • | inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| • | unforeseen safety issues; or |
| • | inadequate patient enrollment or lack of adequate funding to continue the clinical trial. |
The general risks of study design and execution applicable to our planned highway driving study are heightened by the fact that, to our knowledge, this is the first study of residual effects on driving ability to be conducted in support of FDA approval of a new sleep agent in that standards for evaluation of such a study have not been established.
In addition, changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols to reflect these changes, which could impact the cost, timing or successful completion of a clinical trial. If we experience delays in the commencement or completion of our clinical trials, the commercial prospects for our product candidates and our ability to generate product revenue will be harmed. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also lead to the denial of regulatory approval of a product candidate.
Our success depends on meeting the conditions for approval and market exclusivity under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or FFDCA.
We are seeking approval for Intermezzo® under Section 505(b)(2) of the FFDCA, enacted as part of the Drug Price Competition and Patent Restoration Act of 1984, otherwise known as the Hatch-Waxman Act, which permits applicants to rely in part on clinical and non-clinical data generated by third parties. Specifically, we are relying in part on third party data with respect to zolpidem, which is the active ingredient in Intermezzo® and the previously approved insomnia products Ambien® and Ambien CR®. There can be no assurance that the FDA will not require us to conduct additional non-clinical or clinical studies or otherwise obtain new supplementary data with respect to some or all of the data upon which we may rely prior to approving an Intermezzo® NDA.
Our NDA also relies on prior FDA findings of safety and effectiveness of previously-approved products, and we have made certifications in our NDA under Section 505(b)(2) requirements based on the listed patents in the FDA publication “Approved Drug Products with Therapeutics Equivalence Evaluations,” or the Orange Book, for certain of these referenced products. Currently, there are no unexpired patents for immediate release zolpidem products listed in the Orange Book. In the event that one or more patents is listed in the Orange Book for the referenced product after our submission of additional information in support of our NDA for Intermezzo®, we may also be required to evaluate the applicability of these patents to Intermezzo® and submit additional certifications. A paragraph III certification, stating that a listed patent has not expired, but will expire on a particular date, may delay the approval of Intermezzo® until the expiration of the patent. A paragraph IV certification, stating that a listed patent is invalid, unenforceable, or not infringed by Intermezzo® may require us to notify the patent owner and the holder of the NDA for the referenced product of the existence of the Intermezzo® NDA, and may result in patent litigation against us and the entry of a 30-month stay of FDA ability to issue final approval of the 505(b)(2) NDA for Intermezzo®.
34
Table of Contents
Our success also relies, in part, on obtaining Hatch-Waxman marketing exclusivity in connection with any approval of our NDA for Intermezzo®. Such exclusivity protection would preclude the FDA from approving a marketing application for a duplicate of Intermezzo®, a product candidate that the FDA views as having the same conditions of approval as Intermezzo® (for example, the same indication, the same route of delivery and/or other conditions of use), or a 505(b)(2) NDA submitted to the FDA with Intermezzo® as the reference product, for a period of three years from the date of Intermezzo® approval, although the FDA may accept and commence review of such applications. This form of exclusivity may not prevent FDA approval of an NDA that relies only on its own data to support the change or innovation. Similarly, if, prior to approval of the Intermezzo® NDA, another company obtains approval for a product candidate under, in the view of the FDA, the same conditions of approval that we are seeking for Intermezzo®, Intermezzo® could be blocked until the other company’s three-year Hatch-Waxman marketing exclusivity expires.
We are dependent upon the efforts of Purdue for commercializing Intermezzo® in the United States, and will be dependent on the efforts of other collaboration partners if we enter into additional strategic collaborations outside the United States.
The success of sales of Intermezzo® in the United States will be dependent on the ability of Purdue to successfully launch and commercialize Intermezzo®, if approved by the FDA, pursuant to the Collaboration Agreement we signed in July 2009. The terms of the Collaboration Agreement provide that Purdue has the ability to terminate such arrangement for any reason at any time upon 180 days notice and within 10 business days after review of documentation we receive from the FDA in connection with any approval of Intermezzo® in the United States. Thus, for example, even if the measures taken to address FDA concerns on the safety of Intermezzo® are successful to obtain FDA approval, Purdue may determine that such measures, or the outcome of any clinical trials from such measures, have made Intermezzo® a less attractive commercial product for Purdue and terminate our collaboration. If the Collaboration Agreement is terminated, our business and our ability to generate revenue from sales of Intermezzo® will be substantially harmed and we will be required to develop our own sales and marketing organization or enter into another strategic collaboration in order to commercialize Intermezzo® in the United States. Such efforts may not be successful and, even if successful, would require substantial time and resources to carry out.
The manner in which Purdue launches Intermezzo®, including the timing of launch and potential pricing, will have a significant impact on the ultimate success of Intermezzo® in the United States, and the success of the overall commercial arrangement with Purdue. If launch of commercial sales of Intermezzo® in the United States by Purdue is delayed or prevented, our revenue will suffer and our stock price will decline. Further, if launch and resulting sales of Intermezzo® are not deemed successful, our stock price will decline. The outcome of Purdue commercialization efforts could also have an effect on investors’ perception of potential sales of Intermezzo® outside of the United States, which could also cause a decline in our stock price and may make it more difficult to enter into strategic collaborations outside the United States.
The Collaboration Agreement provides for Purdue to be responsible for conducting any post-approval studies of Intermezzo®, both if such studies are required or requested in connection with FDA approval of Intermezzo®. The planning and execution of these studies will be primarily the responsibility of Purdue, and may not be carried out in accordance with our preferences, or could yield results that are detrimental to Purdue’s sales of Intermezzo® in the United States or detrimental to our efforts to develop or commercialize Intermezzo® outside the United States.
Our ability to receive any significant revenue from our product candidates covered by a strategic collaboration, such as our Collaboration Agreement with Purdue, will be dependent on the efforts of the collaboration partner and may result in lower levels of income than if we marketed or developed our product candidates entirely on our own. The collaboration partner may not fulfill its obligations or carry out marketing activities for our product candidates as diligently as we would like. We could also become involved in disputes with our partner, which could lead to delays in or termination of commercialization programs and
35
Table of Contents
time-consuming and expensive litigation or arbitration. If a collaboration partner terminates or breaches its agreement, or otherwise fails to complete its obligations in a timely manner, the chances of successfully developing or marketing our product candidates would be materially and adversely affected.
We are seeking to enter into additional strategic collaborations for the development and commercialization of Intermezzo® outside the United States. We may not be able to enter into additional collaborations on acceptable terms, if at all. Our establishment of Purdue as our commercial partner for Intermezzo® in the United States could also limit the potential collaboration options we have outside the United States or could render potential collaborators less inclined to enter into an agreement with us because of such relationship. Further, we have granted Purdue and an associated company an option to negotiate with us for a license to commercialize Intermezzo® in Mexico and Canada. While these options and subsequent negotiation periods continue, we are prevented from negotiating with and being able to enter into commercialization agreements with other potential strategic partners for development or commercialization of Intermezzo® in such countries.
If we choose to exercise our co-promotion option and are unable to establish a sales and marketing infrastructure in the United States, our potential revenues could be substantially harmed.
In order to commercialize Intermezzo® or any other product candidates successfully, we must enter into and maintain strategic collaborations to perform, and/or acquire or internally develop a sales, marketing and distribution infrastructure. We have entered into a strategic collaboration for commercialization of Intermezzo® in the United States with Purdue and may develop our own sales force and marketing infrastructure for Intermezzo® to co-promote Intermezzo® to psychiatrists in the United States. However, we have no experience in building a sales and marketing organization. If we exercise our co-promotion option and are unable to develop our own sales, marketing and distribution infrastructure to effectively commercialize Intermezzo® , our ability to generate additional revenue from potential sales of Intermezzo® to psychiatrists would be substantially harmed.
The development of sales, marketing and distribution infrastructure is difficult and time consuming, and requires substantial financial and other resources. Factors that may hinder our efforts to develop an internal sales, marketing and distribution infrastructure include:
| • | inability to recruit, retain and effectively manage adequate numbers of effective sales and marketing personnel; |
| • | the inability of sales personnel to obtain access to or convince adequate numbers of physicians to prescribe our products; |
| • | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
| • | unforeseen delays, costs and expenses associated with creating a sales and marketing organization. |
We may require substantial additional funding and may be unable to raise capital when needed.
We have no current source of product revenue. We have a limited operating history and have not yet commercialized any products. We had cash, cash equivalents and marketable securities of $88.9 million at December 31, 2009. We expect our negative cash flows from operations to continue for the foreseeable future as we determine and undertake activities to support the planned resubmission of the Intermezzo® NDA, pursue the regulatory approval and commercialization of Intermezzo® internationally and develop other product candidates. We do not know how long it will take to obtain regulatory approval of Intermezzo®, or if such approval is obtainable. We also expect negative cash flows beyond any potential regulatory approval and product launch of Intermezzo®. As a result, we will need to generate significant revenues to pay these costs and achieve profitability. We do not know whether or when we will become profitable because of the significant uncertainties with respect to our ability to gain regulatory approval of Intermezzo® and generate revenues from the sale of our products and from our existing and potential future collaborations.
36
Table of Contents
If the timing of potential product approval and launch is significantly delayed as a result of FDA or other regulatory approval delays, the Collaboration Agreement with Purdue is terminated or other factors arise, our cash, cash equivalents and marketable securities may prove insufficient to fund our operations through the commercial launch of Intermezzo®. Purdue is obligated to pay us up to $30.0 million if they elect to continue with our Collaboration Agreement after an FDA approval of Intermezzo®. This potential $30.0 million payment is reduced by $2.0 million for each 30-day period that our receipt of an NDA approval for Intermezzo® is delayed beyond June 30, 2010. Further, the development and potential regulatory approval of additional product candidates will likely require additional funding which may not be available at and as of the time needed on commercially reasonable terms, if at all.
We currently believe that our available cash, cash equivalents and marketable securities and interest income will be sufficient to fund our anticipated levels of operations for at least the next twelve months. However, our future capital requirements will depend on many factors, including:
| • | the success of our ability to obtain FDA approval of Intermezzo® and to develop and commercialize Intermezzo®; |
| • | the scope and results of our efforts to respond to the Complete Response Letter received by the FDA on the Intermezzo® NDA, including the cost, timing and results of any additional studies; |
| • | advancement of other product candidates into development; |
| • | potential acquisition or in-licensing of other products or technologies; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals; |
| • | the cost of development and manufacturing activities; |
| • | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims and other patent-related costs, including litigation costs and the results of such litigation; and |
| • | our ability to establish and maintain additional collaborative arrangements. |
Additional financing may not be available to us when we need it or it may not be available on favorable terms. If we are unable to obtain adequate financing on a timely basis, we may be required to significantly curtail one or more of our development, licensing or acquisition programs. We could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products which we would otherwise pursue on our own. If we raise additional funds by issuing equity securities, our then-existing stockholders will experience dilution and the terms of any new equity securities may have preferences over our common stock.
Intermezzo® and our other product candidates may not achieve market acceptance even if we obtain regulatory approvals.
Even if we receive regulatory approvals for the commercial sale of Intermezzo® or our other product candidates, the commercial success of these product candidates will depend upon, among other things, acceptance by physicians and patients. Market acceptance of, and demand for, any product that we develop and that are commercialized by us or our collaboration partner will depend on many factors, including:
| • | the ability to provide acceptable evidence of safety and efficacy of Intermezzo® or future products for their respective indications; |
| • | the ease of use of Intermezzo®; |
| • | the existence of generic or branded competition for Intermezzo®; |
| • | the ability to obtain adequate pricing and sufficient insurance coverage and reimbursement; |
| • | the availability, relative cost and relative efficacy and safety of alternative and competing treatments; |
37
Table of Contents
| • | the effectiveness of our or a collaboration partner’s sales, marketing and distribution strategies; and |
| • | the ability to produce commercial quantities sufficient to meet demand. |
If Intermezzo® or our other product candidates fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business.
We will face substantial competition from companies with established products.
We plan to seek approval of Intermezzo® for use as needed for the treatment of insomnia when a middle of the night awakening is followed by difficulty returning to sleep, an indication that we believe represents an opportunity in the broader insomnia therapeutic market. The insomnia market is large, deeply commercialized and characterized by intense competition among large, established pharmaceutical companies with well funded and staffed and experienced sales and marketing organizations and far greater name recognition than us or our collaboration partner.
If Intermezzo® receives marketing approval, it will compete in this large market against well-established branded products with significant advertising support, as well as with new market entrants and generic competitors selling zolpidem and other sleep aids at a fraction of the price at which we or our collaboration partner will most likely seek to sell Intermezzo®.
We believe that if Intermezzo® is approved on a timely basis, and with the label we have requested from the FDA, it will be the first sleep aid approved by the FDA specifically for use in the middle of the night when patients awaken and have difficulty returning to sleep. However, currently approved and marketed seven to eight hour therapeutics can also treat this condition when used to deliver a prophylactic dose of a sleep aid at the beginning of the night. The most directly competitive approved products in the United States are Ambien® and Ambien CR®, marketed by Sanofi-Aventis, and generic forms of zolpidem available from multiple manufacturers. Additionally, ZolpimistTM, an orally administered spray for which NovaDel Pharma, Inc. received marketing approval from the FDA in December 2008, and EdluarTM, a sublingual tablet for which Orexo AB received marketing approval from the FDA in March 2009, employ the same 5mg and 10mg zolpidem doses as generic Ambien® and are designed to be used in the same manner at bedtime to produce seven to eight hours of sleep. Zolpidem, in both its branded and generic forms, is the most widely-prescribed drug in the United States for treatment of insomnia.
Additionally, Lunesta® (eszopiclone), marketed by Dainippon-Sumitomo Pharma Co. Ltd. and Rozerem® (ramelteon), marketed by Takeda Pharmaceuticals Company Limited, can similarly treat middle of the night awakenings by providing a prophylactic dose at bed-time in order to avoid a middle of the night awakening, and short duration products such as Sonata®, which utilizes the active ingredient zaleplon and marketed by King Pharmaceuticals, Inc., have been used off-label for the as-needed treatment of middle of the night awakenings. In March 2010, Somaxon Pharmaceuticals, Inc. announced FDA approval Silenor®, a low dose doxepin formulation intended for use at bedtime, for the treatment of both transient (short term) and chronic (long term) insomnia characterized by difficulty with sleep maintenance in both adults and elderly patients. In clinical trials, Silenor® demonstrated maintenance of sleep into the 7th and 8th hours of the night, with no meaningful evidence of next day residual effects. In March 2010, Somaxon announced that it is focused on seeking a U.S. commercial partnership, building a U.S. commercial presence and preparing to launch Silenor® in the second half of 2010. In January 2010 Vanda Pharmaceuticals Inc. received an orphan indication from the FDA for VEC-162 (tasimelteon), a melatonin agonist similar to ramelteon, for treatment of non-24 hour sleep/wake disorder in blind individuals without light perception. Vanda may seek additional broader insomnia indications for this product. Other drugs, such as the antidepressant generic trazadone, are also widely prescribed off-label for the treatment of insomnia.
38
Table of Contents
Other companies may develop products to compete with Intermezzo®.
We are aware of several products currently in development which are seeking indication statements from the FDA for the treatment of middle of the night awakenings. Neurocrine Biosciences, Inc. received an approvable letter, pending additional clinical and pre-clinical studies, from the FDA for its product candidate, indiplon, proposed to be used for sleep initiation and middle of the night dosing. NovaDel Pharma, Inc. has announced that it commenced development of a low-dose version of Zolpimist™ for the treatment of middle-of-the-night awakenings with the intent to enter such product candidate into clinical trials, and Somnus Therapeutics Inc. has indicated that it is similarly targeting treatment of middle of the night awakenings with development of its controlled-release zaleplon formulation, SKP-1041. Additionally, Alexza Pharmaceuticals, Inc. has commented publicly that they are evaluating AZ-007, immediate release Staccato zaleplon, for its ability to treat middle of the night awakenings.
There are many other companies working to develop new products and other therapies to treat insomnia, including but not limited to Eli Lilly and Company, Merck and Co., Inc., and GlaxoSmithKline plc in conjunction with Actelion Pharmaceuticals Ltd. Several of these products are in late stage clinical trials.
Furthermore, new developments, including the development of other drug technologies and methods of treating conditions, occur in the biopharmaceutical industry at a rapid pace. Any of these developments may negatively affect the commercial prospects of Intermezzo®.
Many potential competitors, either alone or together with their partners, have substantially greater financial resources, research and development programs, clinical trial and regulatory experience, expertise in prosecution of intellectual property rights, and manufacturing, distribution and sales and marketing capabilities than us or our collaboration partner. As a result of these factors, these competitors may:
| • | develop product candidates and market products that are less expensive, safer, more effective or easier to use than our current product candidates and contemplated future products; |
| • | commercialize competing products before Intermezzo® or other product candidates can be launched; |
| • | initiate or withstand substantial price competition more successfully than we can; |
| • | have greater success in recruiting skilled scientific workers and experienced sales and marketing personnel from the limited pool of available talent; |
| • | more effectively negotiate third-party licenses and strategic collaborations; and |
| • | take advantage of acquisition or other opportunities more readily than us or our collaboration partner. |
Governmental and third-party payors may impose restrictions or reimbursement or pricing controls that could limit product revenue.
The continuing efforts of government and third-party payors to contain or reduce the costs of health care through various means may reduce potential revenue we may receive from sales of Intermezzo®, if approved. In particular, third-party insurance coverage may not be available to patients for Intermezzo® or our other products, especially in light of the availability of low-cost generic zolpidem therapeutics, regardless of the fact that such products are not specifically designed or indicated to specifically treat middle of the night awakening. Government and third-party payors could also impose price controls and other conditions that must be met by patients prior to providing coverage for use of our products. For example, insurers may establish a “step-edit” system that requires a patient to utilize a lower price alternative product prior to becoming eligible for reimbursement of a higher price product. If government and third-party payors do not provide adequate coverage and reimbursement levels for our products, or if price controls, prior authorization or step-edit systems are enacted, our product revenue will suffer.
39
Table of Contents
Negative publicity and documented side effects concerning products used to treat patients in the insomnia market may harm commercialization of Intermezzo® or our other product candidates.
Products containing zolpidem, the active ingredient in Intermezzo®, are widely marketed. Zolpidem use has been linked to negative effects, such as sleepwalking and amnesia, and has the potential to cause physical or psychological dependence. Furthermore, zolpidem is classified as a Schedule IV substance under the Comprehensive Drug Abuse and Prevention Control Act of 1970, and is subject to certain packaging, prescription and purchase volume limitations. There can be no assurance that additional negative publicity or increased governmental controls on the use of zolpidem or other compounds used in products for the insomnia market would not inhibit or prevent commercialization of Intermezzo® or our other product candidates. Furthermore, negative publicity concerning zolpidem and other hypnotic pharmaceuticals could cause the FDA to make approval of new products for the insomnia market more difficult, by requiring additional or different non-clinical or clinical studies or taking other actions, out of safety or other concerns, or could lead to reduced consumer usage of sleep aids, including zolpidem products and Intermezzo®.
Even if our product candidates receive regulatory approval, they will be subject to ongoing regulatory requirements and may face regulatory or enforcement action.
Any product candidate for which we receive regulatory approval, together with related third-party manufacturing facilities and processes, post-approval clinical data, and advertising and promotional activities for the product, will be subject to significant review, oversight and ongoing and changing regulation by the FDA and other regulatory agencies. Failure to comply with regulatory requirements may subject us to administrative and judicially-imposed sanctions. These may include warning letters, adverse publicity, civil and criminal penalties, injunctions, product seizures or detention, product recalls, total or partial suspension of production, and refusal to approve pending product marketing applications.
Even if we receive regulatory approval to market a particular product candidate, the approval could be conditioned on our conducting additional costly post-approval studies or could limit the indicated uses included in our labeling. Moreover, the product may later cause adverse effects that limit or prevent its widespread use, force us to withdraw it from the market or impede or delay the ability to obtain regulatory approvals in additional countries.
The FDA has also requested that all manufacturers of sedative-hypnotic pharmaceutical products modify their product labeling to include strong language concerning potential risks. These risks include severe allergic reactions and complex sleep-related behaviors, which may include sleep-driving. The FDA also recommended that pharmaceutical manufacturers conduct clinical studies to investigate the frequency with which sleep-driving and other complex behaviors occur in association with individual drug products. We have not conducted such studies, and it is unclear how and to what extent, if any, these requests and recommendations will affect Intermezzo® or our other product candidates.
If manufacturers supplying our product candidates fail to produce in the volumes and quality that are required on a timely basis, or to comply with stringent regulations applicable to pharmaceutical manufacturers, there may be delays in the development and commercialization of, or an inability to meet demand for, our products, if any, and we may lose potential revenue.
We do not manufacture Intermezzo®, and we do not plan to develop the capacity to do so. We have a primary manufacturing and supply agreement with Patheon, Inc. to manufacture a commercial supply of Intermezzo®. We also have agreements with Mikart, Inc. to qualify it as a backup commercial supplier of finished product, as well as a backup commercial manufacturer of a key excipient used in the manufacture of Intermezzo®. We currently have arrangements to use Anderson Packaging, Inc. as a primary packager of Intermezzo® and Sharp Corporation to supply sample packaging. We plan to amend our agreements with Anderson and Sharp or enter into packaging and supply agreements with new suppliers if we determine to change the packaging of Intermezzo®. We rely upon SPI Pharma, Inc. as a supplier for certain key excipients contained within Intermezzo®, for one of such excipients, Pharmaburst®, as the sole source, and upon Plantex USA, Inc. as
40
Table of Contents
our sole source for a special form of zolpidem tartrate. These agreements have set terms of duration, some of which automatically renew for successive one or three year periods. The first to expire among these agreements, the Packaging and Supply Agreement with Anderson Packaging, Inc., has a term that ends in September of 2011, although such agreement automatically renews for one year periods thereafter. Purdue is similarly dependent on these manufacturers for the commercial supply of Intermezzo® and has entered into direct agreements with certain of such manufacturers in connection with entry into the Collaboration Agreement that would take effect soon after an FDA approval of Intermezzo® if Purdue elects to continue with our collaboration. Any of the risks that we face with respect to these manufacturers are therefore similarly applicable to Purdue, and the realization of these risks by Purdue would have a significant impact on Purdue commercialization efforts and our ability to generate revenue under the Collaboration Agreement.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Third-party manufacturers and key suppliers may not perform as agreed, may terminate their agreements, or may experience manufacturing difficulties due to resource constraints or as a result of labor disputes, unstable political environments at foreign facilities or financial difficulties. For example, a supplier with a manufacturing facility in Israel may face geopolitical risk that could prevent it from providing supplies from such facility. Additionally, third-party manufacturers and key suppliers may become subject to claims of infringement of intellectual property rights of others, which could cause them to incur substantial expenses, and, if such claims were successful, could cause them to incur substantial damages or cease production of our products or product components. For example, SPI, the sole supplier of Pharmaburst®, a key excipient used in Intermezzo®, is the defendant in a lawsuit alleging patent infringement that may involve Pharmaburst®. In addition, several of our suppliers have only one facility qualified to supply key components of Intermezzo®, and transferring such supply to an alternate site could take substantial time and resources. Any interruption of supply from such facilities could materially impair our ability to manufacture and generate revenue from Intermezzo®. These manufacturers and suppliers may also choose, or be required, to seek licenses from the claimant, which may not be available on acceptable terms or at all. If these manufacturers or key suppliers were to encounter any of these difficulties, or otherwise fail to comply with their contractual obligations, ability to launch Intermezzo® or any other product candidate, if approved, would be jeopardized. Even if we were able to launch a product, these difficulties could cause increases in the prices we or our collaborators pay for supply of such product and its components which could substantially hinder or prevent commercialization efforts.
In addition, all manufacturers and suppliers of pharmaceutical products must comply with current Good Manufacturing Practice, or cGMP, requirements enforced by the FDA through its facilities inspection program. The FDA is likely to conduct inspections of third-party manufacturer and key supplier facilities as part of its review of any of our NDAs. If third-party manufacturers and key suppliers are not in compliance with cGMP requirements, it may result in a delay of approval, particularly if these sites are supplying single source ingredients required for the manufacture of Intermezzo®. These cGMP requirements include quality control, quality assurance and the maintenance of records and documentation. Furthermore, regulatory qualifications of manufacturing facilities are applied on the basis of the specific facility being used to produce supplies. As a result, if one of these manufacturers shifts production from one facility to another, the new facility must go through a complete regulatory qualification process and be approved by regulatory authorities prior to being used for commercial supply. Manufacturers may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any quantities supplied is compromised due to a third-party manufacturer or key supplier failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for our product candidates and, even if such approval is obtained, any resulting products may not be successfully commercialized.
41
Table of Contents
There are no alternate manufacturers qualified at this time with respect to the commercial supply of Intermezzo®, nor are there alternate manufacturers identified or qualified with respect to the commercial supply of several of the key ingredients and packaging materials used in Intermezzo® . If manufacturers are required to be changed, prior approval by the FDA and comparable foreign regulators will be required. In addition, we or Purdue would likely have to incur significant costs and expend significant efforts to educate the new manufacturer with respect to, or to help the new manufacturer independently develop, the processes necessary for production. Manufacturing and supply switching costs in the pharmaceutical industry can be very high, and switching manufacturers or key suppliers can frequently take 12 to 18 months to complete, although in certain circumstances such a switch may be significantly delayed or prevented by regulatory and other factors.
Any of these factors could cause the delay or suspension of regulatory submissions, required regulatory approvals or commercialization of Intermezzo® or any other product candidate that we develop, entail higher costs or result in an inability to effectively commercialize our products, if any are approved. Furthermore, if manufacturers fail to deliver the required commercial quantities of raw materials, including the active pharmaceutical ingredient, key excipients or finished product on a timely basis and at commercially reasonable prices, we or our strategic partners would be unable to meet demand for our products and we would lose potential revenue.
We rely on third parties to conduct our non-clinical and clinical trials. If these third parties do not perform as contractually required or otherwise expected, we may not be able to obtain regulatory approval for our product candidates.
We do not currently conduct non-clinical and clinical trials on our own, and instead rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories, to assist us with our non-clinical and clinical trials. We, and our third parties, are also required to comply with regulations and standards, commonly referred to as Good Clinical Practice, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. If these third parties do not successfully carry out their duties with regard to Intermezzo® development or fail to successfully carry out their duties to us as they relate to meeting future regulatory obligations or expected deadlines, if the third parties need to be replaced, or if the quality or accuracy of the data these third parties obtained during the development of Intermezzo® or in the future is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our non-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for our product candidates, including Intermezzo®.
We may face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit such candidate’s commercialization.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, health care providers, pharmaceutical companies or others selling our products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. We are also obligated under certain circumstances to indemnify suppliers and others with whom we have contractual relationships for product liability claims such entities might incur with respect to our products and product candidates. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for our products; |
| • | impairment of our business reputation; |
| • | withdrawal of clinical trial participants; |
| • | costs of related litigation; |
| • | substantial monetary awards to patients or other claimants; |
42
Table of Contents
| • | loss of revenue; and |
| • | the inability to commercialize our product candidates. |
Although we currently have product liability insurance coverage for our clinical trials with limits that we believe are customary and adequate to provide us with coverage for foreseeable risks associated with our development efforts, this insurance coverage may not reimburse us or may be insufficient to reimburse us for the actual expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for Intermezzo®, but we may be unable to obtain such product liability insurance on commercially reasonable terms.
We depend on key personnel and if we are not able to retain them our business will suffer.
We are highly dependent on the principal members of our management and scientific staff, including but not limited to Glenn A. Oclassen, our President and Chief Executive Officer, and Nikhilesh N. Singh, Ph.D., our Senior Vice President and Chief Scientific Officer. The competition for skilled personnel among biopharmaceutical companies in the San Francisco Bay Area is intense and the employment services of our scientific, management and other executive officers may be terminated at-will. If we lose one or more of these key employees, our ability to implement and execute our business strategy successfully could be seriously harmed. Replacing key employees may be difficult and may take an extended period of time because of the limited number of individuals in the biopharmaceutical industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. We do not carry key man life insurance on any of our key personnel other than Dr. Singh.
If we do not raise additional capital, we may be forced to delay, reduce or eliminate our development programs and commercialization efforts.
Our future capital requirements will depend on, and could increase significantly as a result of, many factors, including:
| • | the costs and timing of regulatory approval; |
| • | the need to conduct additional clinical trials; |
| • | the costs of establishing or contracting for sales and marketing capabilities, including potential costs of being required to engage in contracting for replacements for such capabilities if an existing arrangement is terminated; |
| • | the rate of progress to begin and conduct and cost of our clinical trials and other development activities; |
| • | the extent to which we acquire or in-license new products, technologies or businesses; |
| • | the effect of competing technological and market developments; and |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights. |
We may need to seek additional funding through strategic collaborations or through public or private sales of our equity securities. In addition, we may obtain equipment leases and may pursue opportunities to obtain debt financing in the future. There can be no assurance, however, that strategic collaborations, additional equity or debt financing will be available on reasonable terms, if at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our then existing or planned development, commercialization or expansion activities.
43
Table of Contents
Raising additional funds by issuing securities or through licensing arrangements may cause dilution to existing stockholders, restrict our operations or require us to relinquish proprietary rights.
To the extent that we raise additional capital by issuing equity securities, our existing stockholders’ ownership will be diluted. Any debt financing we enter into may involve covenants that restrict our operations. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of our assets, as well as prohibitions on our ability to create liens, pay dividends, redeem our stock or make investments. In addition, if we raise additional funds through licensing arrangements, it may be necessary to relinquish potentially valuable rights to potential products or proprietary technologies, or grant licenses on terms that are not favorable to us.
The commercial success, if any, of Intermezzo® depends, in part, on the rights we are seeking through certain patent applications.
The potential commercial success of Intermezzo® depends in part on two issued patents from the U.S. Patent and Trademark Office, or USPTO, covering the formulation of Intermezzo® and patents that may issue covering methods of use of zolpidem. In addition, we have pending certain foreign equivalent patent applications with respect to formulations and manufacture of zolpidem for use in treatment of middle of the night awakening, as well as applications covering combinations and methods of use of ondansetron in conjunction with atypical antipsychotic drugs. There can be no assurance that our pending patent applications and applications we may file in the future, or those applications we may license from third parties, will result in patents being issued in a timely manner, or at all. Even if patents issue, the claims in such patents may not issue in a form that will be advantageous to us, may not encompass Intermezzo® or our other product candidates and their unique features, and may not provide us with proprietary protection or competitive advantages. For instance, competitors may be able to engineer around our formulation patent applications with alternate formulations that deliver therapeutic effects similar to potential products covered by our zolpidem formulation patent applications. Other drug companies may also be able to develop generic versions of our products if we are unable to maintain our proprietary rights. For example, generic drug makers may attempt to introduce generic low dose zolpidem products similar to our products immediately after the expiration of Hatch-Waxman marketing exclusivity and prior to the expiration of patents that may be issued relating to Intermezzo® . Furthermore, among other limitations, the method of use patent applications that have been filed to encompass Intermezzo® are limited in scope to certain uses of zolpidem, so potential competitors could develop similar products using active pharmaceutical ingredients other than zolpidem. Any patents that have been allowed, we have obtained or do obtain may be challenged by re-examination, opposition, or other administrative proceeding, or may be challenged in litigation, and such challenges could result in a determination that the patent is invalid.
The active, and many of the inactive, ingredients in Intermezzo®, including generically manufactured zolpidem, have been known and used for many years and, therefore, are no longer subject to patent protection. Accordingly, certain of our pending patent applications are directed to the particular formulations of these ingredients in our products, and their use. Although we believe our formulations and their use are patentable and provide a competitive advantage, even if such patents are issued, such patents may not prevent others from marketing formulations using the same active and inactive ingredients in similar but different formulations. Moreover, if our patents were successfully challenged and ruled to be invalid, we would be exposed to a greater risk of direct competition.
Failure to obtain effective patent protection for Intermezzo® and our other product candidates would allow for products to be marketed by competitors that would undermine sales, marketing and collaboration efforts for our product candidates, and reduce or eliminate our revenue. In addition, both the patent application process and the process of managing patent disputes can be time consuming and expensive.
44
Table of Contents
If we are unable to maintain and enforce our proprietary rights, we may not be able to compete effectively or operate profitably.
Our commercial success will depend, in part, on obtaining and maintaining patent protection, trade secret protection and regulatory protection of our proprietary technology and information as well as successfully defending against third-party challenges to our proprietary technology and information. We will be able to protect our proprietary technology and information from use by third parties only to the extent that we have valid and enforceable patents, trade secrets or regulatory protection to cover them and we have exclusive rights to utilize them.
Our commercial success will continue to depend in part on the patent rights we own, the patent rights we have licensed, the patent rights of our suppliers and the patent rights we plan to obtain related to future products we may market. Our success also depends on our and our licensors’ and suppliers’ ability to maintain these patent rights against third-party challenges to their validity, scope or enforceability. Further, we do not fully control the patent prosecution of the patents and patent applications we have licensed. There is a risk that licensors to us will not devote the same resources or attention to the prosecution of the licensed patent applications as we would if we controlled the prosecution of the patent applications, and the resulting patent protection, if any, may not be as strong or comprehensive as if we had prosecuted the applications ourselves. The patent positions of biopharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the United States. The patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States or other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. For example:
| • | we or our licensors might not have been the first to make the inventions covered by pending patent applications and issued patents; |
| • | we or our licensors might not have been the first to file patent applications for these inventions; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | it is possible that none of our pending patent applications or any pending patent applications of our licensors will result in issued patents; |
| • | our patents, if issued, and the issued patents of our licensors may not provide a basis for commercially viable products, or may not provide us with any competitive advantages, or may be challenged and invalidated by third parties; |
| • | we may not develop additional proprietary technologies or product candidates that are patentable; or |
| • | the patents of others may have an adverse effect on our business. |
We also rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. While we seek to protect confidential information, in part, by confidentiality agreements with our employees, consultants, contractors, or scientific and other advisors, they may unintentionally or willfully disclose our information to competitors. If we were to enforce a claim that a third party had illegally obtained and was using our trade secrets, it would be expensive and time consuming, and the outcome would be unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets.
If we are not able to defend the patent or trade secret protection position of our technologies and product candidates, then we will not be able to exclude competitors from developing or marketing competing products, and we may not generate enough revenue from product sales, if any, to justify the cost of development of our product candidates and to achieve or maintain profitability.
45
Table of Contents
If we are sued for infringing intellectual property rights of other parties, such litigation will be costly and time consuming, and an unfavorable outcome would have a significant adverse effect on our business.
Although we believe that we would have valid defenses to allegations that our current product candidates, production methods and other activities infringe the valid and enforceable intellectual property rights of any third parties of which we are aware, we cannot be certain that a third party will not challenge our position in the future. Other parties may own patent rights that might be infringed by our products or other activities, or other parties may claim that their patent rights are infringed by excipients manufactured by others and contained in our products. For example, SPI, the sole supplier of Pharmaburst®, a key excipient used in Intermezzo®, is the defendant in a lawsuit that alleges that certain of SPI’s products infringe one or more patents of a third party. Although not specifically identified in the original complaint, subsequent press releases have indicated that Pharmaburst® products allegedly infringe such patents. SPI has informed us that the third party’s patent rights are not infringed by Pharmaburst® and that they intend to defend their rights vigorously. However, because Pharmaburst® is a key excipient in Intermezzo®, the interruption of supply of Pharmaburst® through an injunction or through voluntary cessation of production could materially harm our ability to supply Intermezzo®. There has been, and we believe that there will continue to be, significant litigation and demands for licenses in the life sciences industry regarding patent and other intellectual property rights. Competitors or other patent holders may assert that our products and the methods we employ are covered by their patents. These parties could bring claims against us that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages or possibly prevent us from commercializing our product candidates. Further, if a patent infringement suit were brought against us, we could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
As a result of patent infringement claims, or in order to avoid potential claims, we may choose to seek, or be required to seek, a license from the third party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we were able to obtain a license, the rights may be non-exclusive, which would give competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. This could harm our business significantly.
These risks of intellectual property infringement are similarly faced by our suppliers and collaborators, which could hinder or prevent them from manufacturing or commercializing our products.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
In the event a competitor infringes upon one of our patents or other intellectual property rights, litigation to enforce our intellectual property rights or to defend our patents against challenge, even if successful, could be expensive and time consuming and could require significant time and attention from management. We may not have sufficient resources to enforce our intellectual property rights or to defend our patents against challenges from others.
The pharmaceutical industry is characterized by extensive litigation and administrative proceedings over patent and other intellectual property rights. We could therefore become subject to litigation that could be costly, result in the diversion of management’s time and efforts, and require us to pay damages. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Our competitors may assert that they own U.S. or foreign patents containing claims that cover our products, components of our products, or the methods we employ in making or using our products. In addition, we may become a party to an interference proceeding declared by the USPTO to determine the priority of inventions. Because patent applications can take many years to issue, and in many instances, at least 18 months to publish, there may be applications now pending of which we are unaware, which may later result in issued patents that
46
Table of Contents
contain claims that cover our products. There could also be existing patents, of which we are unaware, that contain claims that cover one or more components of our products. As the number of participants in our industry increases, the possibility of patent infringement claims against us also increases.
Any interference proceeding, litigation, or other assertion of claims against us may cause us to incur substantial costs, could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. If the relevant patents were upheld as valid and enforceable and we were found to infringe, we could be required to pay substantial damages and/or royalties and could be prevented from selling our products unless we could obtain a license or were able to redesign our products to avoid infringement. Any such license may not be available on reasonable terms, if at all. If we fail to obtain any required licenses or make any necessary changes to our products or technologies, we may be unable to make, use, sell, or otherwise commercialize one or more of our products. In addition, if we are found to willfully infringe, we could be required to pay treble damages, among other penalties.
If we fail to comply with our obligations in the agreements under which we license rights to products or technology from third parties, we could lose license rights that are important to our business.
We are a party to a number of agreements that include technology licenses that are important to our business and expect to enter into additional licenses in the future. For example, we hold licenses from SPI Pharmaceuticals, Inc. relating to key excipients used in the manufacture of Intermezzo®. If we fail to comply with these agreements, the licensor may have the right to terminate the license, in which event we and our collaboration partners would not be able to market products covered by the license, including Intermezzo®.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of former employers.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we ourselves have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our or a collaboration partner’s ability to develop or commercialize certain potential products, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If our agreements with employees, consultants, advisors and corporate partners fail to protect our intellectual property, proprietary information or trade secrets, it could have a significant adverse effect on us.
We have taken steps to protect our intellectual property and proprietary technology by entering into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, advisors and corporate partners. However, such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements, and we may not be able to prevent such unauthorized disclosure. Monitoring unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate. Furthermore, the laws of some foreign countries may not protect our intellectual property rights to the same extent as do the laws of the United States.
47
Table of Contents
Our stock price is expected to be volatile.
The market price of our common stock could be subject to significant fluctuations. Market prices for securities of early-stage pharmaceutical, biotechnology and other life sciences companies have historically been particularly volatile. Some of the factors that may cause the market price of our common stock to fluctuate include:
| • | our ability to obtain regulatory approvals for Intermezzo® or other product candidates, and delays or failures to obtain such approvals; |
| • | the termination of key commercial partner agreements, such as our Collaboration Agreement with Purdue; |
| • | failure of any product candidates, if approved, to achieve commercial success; |
| • | issues in manufacturing approved products, if any, or product candidates; |
| • | the results of current and any future clinical trials of our product candidates; |
| • | the entry into, or termination of, key agreements, including additional commercial partner agreements; |
| • | the initiation of, material developments in, or conclusion of litigation to enforce or defend our intellectual property rights or defend against the intellectual property rights of others; |
| • | announcements by commercial partners or competitors of new commercial products, clinical progress or the lack thereof, significant contracts, commercial relationships or capital commitments; |
| • | adverse publicity relating to the insomnia market, including with respect to other products and potential products in such market; |
| • | the introduction of technological innovations or new therapies that compete with our potential products; |
| • | the loss of key employees; |
| • | changes in estimates or recommendations by securities analysts, if any, who cover our common stock; |
| • | future sales of our common stock; |
| • | general and industry-specific economic conditions that may affect our research and development expenditures; |
| • | changes in the structure of health care payment systems; and |
| • | period-to-period fluctuations in financial results. |
Moreover, the stock markets in general have experienced substantial volatility that has often been unrelated to the operating performance of individual companies. These broad market fluctuations may also adversely affect the trading price of our common stock.
In the past, following periods of volatility in the market price of a company’s securities, stockholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm our profitability and reputation.
Anti-takeover provisions in our Collaboration Agreement with Purdue, in our charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by stockholders to replace or remove management.
Provisions in our Collaboration Agreement with Purdue, certificate of incorporation and bylaws may delay or prevent an acquisition or a change in management. These provisions include an agreement with Purdue that prevents Purdue from acquiring above a certain percentage of our stock and engaging in certain other activities
48
Table of Contents
that may lead to an acquisition of our company. Such provisions in our charter documents include a classified board of directors, a prohibition on actions by written consent of stockholders and the ability of the board of directors to issue preferred stock without stockholder approval. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively will provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by stockholders to replace or remove the then-current management by making it more difficult for stockholders to replace members of the board of directors, which is responsible for appointing the members of management.
We have never paid dividends on our capital stock, and do not anticipate that we will pay any cash dividends in the foreseeable future.
We have not paid cash dividends on any of our classes of capital stock to date, and our current expectation is that we will retain our future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be the sole source of gain, if any, as a result of holding shares of our common stock, for the foreseeable future.
Future sales of shares by existing stockholders could cause our stock price to decline.
The market price of our common stock could decline as a result of sales by our existing stockholders in the market, or the perception that these sales could occur. These sales might also make it more difficult for us to issue and sell equity securities at a time and price that we deem appropriate. The lock-up agreements delivered by our executive officers and directors and certain other stockholders in connection with the merger of Novacea and TPI expired in July 2009. Subject to applicable securities law restrictions and other agreements between the company and certain of such stockholders, these shares are now freely tradable.
The highly concentrated ownership of our common stock may prevent stockholders from influencing significant corporate decisions and may result in conflicts of interest that could cause our stock price to decline.
Our executive officers, directors and their affiliates beneficially own or control approximately 55% of the outstanding shares of our common stock as of December 31, 2009. Accordingly, these executive officers, directors and their affiliates, acting as a group, will have substantial influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transactions. These stockholders may also delay or prevent a change of control of us, even if such a change of control would benefit the other stockholders. The significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
Our operational headquarters is located in Point Richmond, California, where we lease approximately 26,900 square feet of space under two separate leases, the first of which for approximately 14,600 square feet of space expires in May 2013, and the second of which for approximately 12,300 square feet of space also expires in May 2013, with an option to renew for an additional five years. Of the 26,900 square feet of space in Point Richmond, California, approximately 3,000 square feet is product development laboratory space and the remainder is general office space.
49
Table of Contents
Transcept also leases approximately 25,288 square feet of general office space in South San Francisco, California, under a lease that expires in October 2012. All of such space was subleased to a third party in May and June 2009.
Transcept believes that its current facilities are suitable and adequate for its current needs.
| Item 3. | Legal Proceedings |
From time to time, we may be involved in litigation relating to claims arising out of our operations. Transcept is not currently involved in any material legal proceedings.
SPI Pharmaceuticals, Inc., the sole supplier of Pharmaburst®, a key excipient used in Intermezzo®, is the defendant in a lawsuit brought by Roquette Frères, or Roquette, in the Federal District Court of Delaware on August 31, 2006 that alleges that certain of SPI’s products infringe one or more claims of a Roquette patent and seeks monetary damages and injunctive relief. Transcept has not been named in, and is not a party to, the lawsuit. Although not specifically identified in the original complaint, press releases have indicated that Pharmaburst® products are among the accused products. SPI has informed us that Roquette’s patent rights are not infringed by Pharmaburst® and that they intend to defend their rights vigorously. Because Pharmaburst® is a key excipient in Intermezzo®, the interruption of supply of Pharmaburst® through an injunction could materially harm our ability to supply Intermezzo® and our business. SPI has agreed to indemnify us against certain damages related to any such infringement suit under the terms of our agreement with SPI.
| Item 4. | Reserved |
50
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock is currently traded on the NASDAQ Global Market, under the symbol “TSPT.” Prior to February 2, 2009, our common stock was traded under the symbol “NOVC.” On January 30, 2009, in connection with the merger of Novacea and TPI, we completed a reverse stock split pursuant to which each five shares of our common stock was converted into one share of our common stock. The share-related information presented in this Form 10-K has been adjusted to reflect the reverse stock split.
The following table sets forth the range of high and low sales prices of our common stock for the quarterly periods indicated, as reported by the NASDAQ Global Market (adjusted for the 1-for-5 reverse stock split which occurred on January 30, 2009).
| Sales Price | ||||||
| High | Low | |||||
| Year ended December 31, 2008 |
||||||
| First quarter |
$ | 16.40 | $ | 11.00 | ||
| Second quarter |
$ | 17.00 | $ | 11.05 | ||
| Third quarter |
$ | 13.50 | $ | 7.20 | ||
| Fourth quarter |
$ | 8.70 | $ | 4.46 | ||
| Year ended December 31, 2009 |
||||||
| First quarter |
$ | 7.50 | $ | 2.56 | ||
| Second quarter |
$ | 6.36 | $ | 2.70 | ||
| Third quarter |
$ | 14.35 | $ | 4.54 | ||
| Fourth quarter |
$ | 15.40 | $ | 4.45 | ||
On January 30, 2009, Novacea completed a business combination with TPI. Novacea securities listed on the NASDAQ Global Market, trading under the ticker symbol “NOVC,” were suspended for trading as of the close of business on Friday, January 30, 2009 and trading of Transcept securities on the NASDAQ Global Market under the ticker symbol “TSPT” commenced on Monday, February 2, 2009.
The closing price of Transcept common shares as reported by the NASDAQ Global Market on March 26, 2010 was $7.8491 per share. As of March 26, 2010 there were approximately 84 holders of record of our common stock.
Dividend Policy
No dividends have been declared or paid on Transcept common stock. Transcept does not anticipate that it will pay any cash dividends on its common stock in the foreseeable future.
Issuer Purchases of Equity Securities
There were no repurchases of our common stock during the fourth quarter of fiscal 2009.
Securities Authorized For Issuance Under Equity Compensation Plans
Information relating to compensation plans under which equity securities are authorized for issuance is set forth under Item 12—“Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” of this Annual Report on Form 10-K.
51
Table of Contents
Performance Graph
The following graph compares:
| • | the performance of an investment in our common stock over the period of May 10, 2006 through December 31, 2009, beginning with an investment at the closing market price on May 10, 2006, the end of the first day our common stock traded on the NASDAQ Global Market following our initial public offering, and thereafter, based on the closing price of our common stock on the NASDAQ Global Market; with |
| • | an investment in the NASDAQ Composite Index and an investment in the NASDAQ Biotech Index, in each case, beginning with an investment at the closing price on May 10, 2006 and thereafter, based on the closing price of the index. |
The graph assumes $100 was invested on the starting date at the price indicated above and that all dividends were reinvested on the date of payment without payment of any commissions. We have not declared or paid any dividends on our common stock. The performance of our common stock shown in the graph below represents past performance and should not be considered an indication of future performance.
Comparison of Cumulative Total Return
For the Period from May 10, 2006 (trading commencement) through December 31, 2009
Among Transcept Pharmaceuticals, Inc., the NASDAQ Composite Index and the NASDAQ Biotechnology Index
52
Table of Contents
| Item 6. | Selected Financial Data |
The following selected financial data has been derived from our audited financial statements. The information below is not necessarily indicative of the results of future operations and should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and Item 1A, “Risk Factors,” of this Form 10-K, and the financial statements and related notes thereto included in Item 8 of this Form 10-K, in order to fully understand factors that may affect the comparability of the information presented below. All per share amounts reflect the conversion of TPI common stock to our common stock on January 30, 2009 at the rate of 0.14134 shares of common stock, after giving effect to the 1-for-5 reverse stock split, for each share of TPI common stock outstanding on January 30, 2009.
| For the year ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Statements of operations data |
||||||||||||||||||||
| License fee revenue |
$ | 5,208 | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
9,005 | 10,381 | 15,885 | 10,161 | 3,269 | |||||||||||||||
| General and administrative |
16,050 | 7,924 | 5,300 | 3,923 | 1,703 | |||||||||||||||
| Merger related transaction costs |
2,224 | 1,967 | — | — | — | |||||||||||||||
| Total operating expenses |
27,279 | 20,272 | 21,185 | 14,084 | 4,972 | |||||||||||||||
| Loss from operations |
(22,071 | ) | (20,272 | ) | (21,185 | ) | (14,084 | ) | (4,972 | ) | ||||||||||
| Interest and other income, net |
271 | 313 | 801 | 458 | 98 | |||||||||||||||
| Interest expense related to bridge loan warrants |
— | — | — | — | (535 | ) | ||||||||||||||
| Net loss |
$ | (21,800 | ) | $ | (19,959 | ) | $ | (20,384 | ) | $ | (13,626 | ) | $ | (5,409 | ) | |||||
| Deemed dividend—Series C preferred stockholders |
— | — | — | — | (458 | ) | ||||||||||||||
| Loss attributable to common stockholders |
$ | (21,800 | ) | $ | (19,959 | ) | $ | (20,384 | ) | $ | (13,626 | ) | $ | (5,867 | ) | |||||
| Basic and diluted net loss per share attributable to common stockholders |
$ | (1.79 | ) | $ | (49.77 | ) | $ | (68.86 | ) | $ | (60.83 | ) | $ | (33.15 | ) | |||||
| Weighted average common shares outstanding |
12,166 | 401 | 296 | 224 | 177 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Selected Balance Sheet Data |
||||||||||||||||||||
| Cash, cash equivalents, marketable securities and restricted cash |
$ | 89,102 | $ | 11,883 | $ | 35,434 | $ | 16,520 | $ | 20,910 | ||||||||||
| Total assets |
95,218 | 13,781 | 37,769 | 18,234 | 21,515 | |||||||||||||||
| Working capital |
74,293 | 6,875 | 29,612 | 12,627 | 20,561 | |||||||||||||||
| Convertible preferred stock |
— | 71,037 | 71,037 | 31,202 | 31,202 | |||||||||||||||
| Common stock and additional paid-in capital |
157,943 | 1,504 | 751 | 176 | 87 | |||||||||||||||
| Accumulated deficit |
(86,911 | ) | (65,111 | ) | (45,152 | ) | (24,768 | ) | (11,141 | ) | ||||||||||
| Total stockholders’ equity (net capital deficiency) |
71,071 | (63,581 | ) | (44,316 | ) | (24,589 | ) | (11,062 | ) | |||||||||||
53
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations contains certain statements that are not strictly historical and are “forward-looking” statements within the meaning of the Private Securities Litigation Reform Act of 1995 and involve a high degree of risk and uncertainty. Actual results may differ materially from those projected in the forward-looking statements due to other risks and uncertainties. All forward-looking statements included in this section are based on information available to us as of the date hereof, and we assume no obligation to update any such forward-looking statement, except as required by law.
Overview
We are a specialty pharmaceutical company focused on the development and commercialization of proprietary products that address important therapeutic needs in neuroscience. Our most advanced product candidate, Intermezzo® (zolpidem tartrate sublingual tablet), is a sublingual low dose formulation of zolpidem that is being developed for use in the middle of the night at the time a patient awakens and has difficulty returning to sleep.
We submitted a new drug application, or NDA, for Intermezzo® to the U.S. Food and Drug Administration, or FDA, on September 30, 2008. On October 28, 2009, we received a Complete Response Letter from the FDA regarding our NDA indicating that it was not approved.
In the Complete Response Letter, the FDA stated that it believes we had submitted substantial evidence of effectiveness for the use of Intermezzo® for its proposed indication. Our proposed label for Intermezzo® indicates that Intermezzo® should only be taken when patients have at least four hours remaining in bed before being active again. The FDA further recognized that the Intermezzo® data we submitted did not indicate significant next day residual effects at four hours after use. However, the FDA indicated that the intended use of Intermezzo® in the middle of the night represents a unique insomnia indication and dosing strategy for which safety has not been previously established and that we did not adequately demonstrate that Intermezzo® could reliably be used safely. The FDA requested additional data demonstrating that Intermezzo®, when taken as directed in the middle of the night, would not present an unacceptable risk of residual effects, with particular reference to next day driving ability. The FDA also expressed two concerns regarding the possibility of patient dosing errors in the middle of the night that could lead to next day residual effects with particular reference to next day driving ability. Specifically, the FDA has asked us to address methods to avoid inadvertent dosing with less than four hours of bedtime remaining, and inadvertent re-dosing in a single night.
On January 20, 2010, we met with the FDA to discuss the Complete Response Letter. Before the meeting, we proposed a plan to resubmit the Intermezzo® NDA. Our plan included new Intermezzo® bedside, single unit-dose packaging and patient instructions designed to reduce the possibility of inadvertent patient dosing errors. The FDA indicated in the meeting that the revised packaging appeared to reduce the potential for inadvertently taking more than one dose in a single night, but expressed continuing concern about the risk of dosing with less than four hours of time remaining in bed, with particular regard to the possibility of impaired driving. To further characterize the safety of dosing Intermezzo® in the middle of the night, we subsequently proposed to conduct a pre-approval highway driving study to assess the effect of Intermezzo® on driving ability beginning at approximately three hours and four hours post-dosing. The FDA and Transcept also discussed on January 20, 2010, whether a pre-approval patient use study might help to define patient ability to properly follow instructions under actual conditions of use.
On a March 24, 2010 teleconference, the FDA agreed that our plan to conduct a highway driving study is a reasonable way to measure potential next day driving impairment from dosing Intermezzo® with four hours or less remaining in bed.
Based on communications with the FDA, we plan to conduct a pre-approval highway driving study to assess the effect of Intermezzo® on next morning driving ability beginning at approximately three hours and four hours
54
Table of Contents
after dosing Intermezzo® in the middle of the night. We have no current plans to conduct a patient use study because of the challenges and limitations of such a study, and have submitted to the FDA our position in this regard. The FDA indicated that it would consider our position on patient use studies as part of the overall resubmission of the Intermezzo® NDA. We also plan to include data from on-going studies of patient comprehension of label instructions in our planned resubmission.
We plan to resubmit the Intermezzo® NDA in the late fourth quarter of 2010 after we generate and analyze additional data from our planned highway driving study. Because our planned resubmission will include additional clinical data, our resubmitted Intermezzo® NDA will result in a six-month review period at the FDA under the Prescription Drug User Fee Act, or PDUFA.
On July 31, 2009, we entered into the United States License and Collaboration Agreement, or the Collaboration Agreement, with Purdue Pharmaceutical Products, L.P., or Purdue, that provides for an exclusive license to Purdue to commercialize Intermezzo® in the United States and pursuant to which:
| • | On August 4, 2009, Purdue paid us a $25.0 million non-refundable license fee; |
| • | We are obligated to seek FDA approval of Intermezzo® and to continue development of Intermezzo® at our expense until FDA approval; and |
| • | If Purdue elects not to terminate our collaboration after its review of an FDA approval of Intermezzo®: |
| • | Purdue is obligated to pay us up to $30.0 million, which amount is reduced by $2.0 million for each 30 day period that our receipt of an NDA approval for Intermezzo® is delayed beyond June 30, 2010; |
| • | We are obligated to transfer the Intermezzo® NDA to Purdue and Purdue is obligated to assume the expense associated with maintaining the NDA and further development of Intermezzo® in the United States, including any expense associated with post-approval studies; |
| • | Purdue is obligated to commercialize Intermezzo® in the United States at its expense; |
| • | Purdue is obligated to pay us tiered double-digit base royalties on net sales of Intermezzo® in the United States ranging up to the mid-twenty-percent level; |
| • | Purdue is obligated to pay us $10.0 million if either of two formulation patents are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book; and |
| • | Purdue is potentially obligated to pay us up to an additional $80.0 million upon meeting certain intellectual property milestones and upon the achievement of certain net sales targets for Intermezzo® in the United States. |
We retained an option to co-promote Intermezzo® to psychiatrists in the United States as early as the first anniversary of commercial launch of Intermezzo® and upon entry into the market under the co-promotion option, we would receive an additional double-digit royalty from Purdue on sales generated by psychiatrists in the United States.
We also granted Purdue and an associated company the right to negotiate for the commercialization of Intermezzo® in Mexico and Canada, respectively, and retained rights to commercialize Intermezzo® in the rest of the world.
We have an active effort underway to enter into one or more development and marketing alliances to develop and commercialize Intermezzo® with established pharmaceutical companies in major markets outside the United States.
We have incurred net losses since inception as we have devoted substantially all of our resources to research and development, including contract manufacturing and clinical trials. As of December 31, 2009, we had cash,
55
Table of Contents
cash equivalents, and marketable securities of $88.9 million, working capital of $74.3 million and an accumulated deficit of $86.9 million.
Our ability to generate near term revenue is dependent upon the receipt of milestone and royalty payments under our Collaboration Agreement with Purdue, which are dependent upon the regulatory approval by the FDA of our lead product candidate, Intermezzo®. To achieve profitable operations, Intermezzo® must be successfully developed and commercialized and we may need to identify, develop and commercialize future product candidates. Even if approved, our products may not achieve market acceptance and will face competition from both generic and branded pharmaceutical products.
Merger with Novacea
Prior to January 30, 2009 we were known as Novacea, Inc. On January 30, 2009, we completed a business combination, referred to as the merger, with Transcept Pharmaceuticals, Inc., or TPI. For accounting purposes, TPI was deemed to be the acquiring entity in the merger. In connection with the merger, we changed our name to Transcept Pharmaceuticals, Inc. and effected a 1-for-5 reverse stock split of our common stock. All share and per share disclosures have been retroactively adjusted to reflect the exchange of shares in the merger, and the 1-for-5 reverse split of our common stock on January 30, 2009. All references to “Transcept,” “we,” “us,” “our” or the “Company” mean Transcept Pharmaceuticals, Inc., the publicly-traded combined company resulting from the merger and, as successor to the business of TPI, includes activities taking place with respect to the business of TPI prior to the merger of TPI and Novacea, as applicable.
The following analysis reflects the historical financial results of TPI prior to the merger and that of the combined company following the merger, and does not include the historical financial results of Novacea prior to the completion of the merger.
Financial Operations Overview
Revenue
We recognize revenue from the $25.0 million non-refundable license fee received pursuant to our Collaboration Agreement with Purdue ratably over an estimated 24-month period starting August 1, 2009 and ending on July 31, 2011. This represents the estimated period for which we have significant participatory obligations under the Collaboration Agreement. The revenue recognized in connection with the license fee during 2009 was $5.2 million.
Research and Development Expenses
Research and development expenses have represented approximately 33%, 51% and 75% of total operating expenses for the years ended December 31, 2009, 2008 and 2007, respectively. Research and development costs are expensed as incurred. Research and development expenses consist of expenses incurred in identifying, researching, developing and testing product candidates. These expenses primarily consist of the following:
| • | Salaries, benefits, travel and related expense of personnel associated with research and development activities; |
| • | Fees paid to professional service providers for services related to the conduct and analysis of clinical trials; |
| • | Contract manufacturing costs for formulations used in clinical trials, as well as scale up activities in anticipation of a potential launch of Intermezzo®; |
| • | Laboratory supplies and materials; |
| • | Depreciation of equipment; and |
| • | Allocated costs of facilities and infrastructure. |
56
Table of Contents
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related expenses for personnel in executive, marketing, finance and accounting, information technology and human resource functions. Other costs include facility costs not otherwise included in research and development expenses and professional fees for legal and accounting services.
Following the merger, we increased our general and administrative expenses for activities associated with operating as a publicly-traded company, including professional fees for consulting, legal and accounting as well as business licenses and fees. These increases include the hiring of additional personnel. As we continued working toward commercialization of Intermezzo® in the United States, we increased spending on our sales and marketing infrastructure, including increased headcount and marketing expenses necessary to prepare for the commercialization of Intermezzo® . Following our entry into the Collaboration Agreement with Purdue, the majority of our sales and marketing activities have been transitioned to Purdue.
Interest Income
We receive interest income from cash, cash equivalents, restricted cash and marketable securities held with certain financial institutions.
Interest Expense
We incurred interest expense on the outstanding balance from our $10.0 million venture debt facility agreement, which was repaid in full during the first quarter of 2009. We also incur interest expense on a $0.3 million loan for tenant improvements, payable to the landlord of our corporate facility in Point Richmond, California.
Other Income (Expense), Net
Other income (expense), net relates to the change in fair value of preferred stock warrants, gains or losses on sales of marketable securities and other miscellaneous items. In connection with the merger, the outstanding preferred stock warrants became outstanding common stock warrants and therefore are no longer treated as a liability requiring remeasurement to fair market value at each balance sheet date.
Results of Operations
Comparison of the Years Ended December 31, 2009 and 2008
The following table summarizes results of operations with respect to the items set forth below for the years ended December 31, 2009 and 2008, in thousands, together with the percentage change in those items.
| Year ended December 31, | |||||||||||||||
| 2009 | 2008 | $ Change |
% Change |
||||||||||||
| Revenue |
$ | 5,208 | $ | — | $ | 5,208 | — | ||||||||
| Research and development expenses |
9,005 | 10,381 | (1,376 | ) | (13 | %) | |||||||||
| General and administrative expenses |
16,050 | 7,924 | 8,126 | 103 | % | ||||||||||
| Merger related costs |
2,224 | 1,967 | 257 | 13 | % | ||||||||||
| Interest income |
282 | 742 | (460 | ) | (62 | %) | |||||||||
| Interest expense |
(179 | ) | (766 | ) | 587 | 77 | % | ||||||||
| Other income (expense), net |
168 | 337 | (169 | ) | (50 | %) | |||||||||
57
Table of Contents
Revenue
Revenue for 2009 relates to recognition of a portion of the $25.0 million non-refundable license fee we received from Purdue in connection with our entry into the Collaboration Agreement. We plan to recognize revenue over an estimated 24-month period starting in August 2009 through July 2011 as we have continuing participatory obligations under the agreement for the commercialization of Intermezzo®. Therefore, we have deferred the license fee and will recognize $1.04 million per month from the date we received the payment in August 2009 through July 2011.
Research and Development Expenses
Research and development expenses decreased 13% to $9.00 million for the year ended December 31, 2009 from $10.38 million for the comparable period in 2008. The decrease of $1.38 million is primarily attributable to Intermezzo® development costs as a result of the following:
| • | a decrease in clinical trial costs of $0.83 million due primarily to the Phase 3 outpatient trial being largely completed by year end 2007, with trial closeout activities occurring during the first half of 2008 and no clinical trials being conducted in 2009; and |
| • | a decrease of $0.66 million in registration and submission third party costs related to the filing of our NDA in September 2008; partially offset by |
| • | an increase in manufacturing costs of $0.24 million attributable to, in 2009, the manufacture of validation batches as well as tooling costs related to our proposed Intermezzo® packaging design and, in 2008, formulation development and packaging development as well as the purchase of raw materials, which was largely completed by September 2008 when we filed our NDA. |
The remaining $0.13 million reduction in research and development expenses is due to a reduction of staffing levels in our clinical development department during the latter half of 2008 resulting in reduced salary and related benefits during 2009 as compared to 2008. The decrease was partially offset by severance expenses incurred in connection with the restructuring announced in August 2009 and increased consulting expenses in 2009 related to the development of Intermezzo® in the European market.
General and Administrative Expenses
General and administrative expenses increased 103% to $16.05 million for the year ended December 31, 2009 from $7.92 million for the comparable period in 2008. The approximate $8.13 million increase consists of the following:
| • | Personnel costs and related expenses increased by $2.44 million due to an increase in headcount to operate as a publicly-traded company and to prepare for the potential commercialization of Intermezzo®. Headcount increases were in the marketing, finance, executive and operations functions. In addition, severance expenses related to the restructuring as noted above and higher non-cash stock-based compensation expense also contributed to increased general and administrative expenses in 2009; |
| • | Professional fees, including consulting, legal and accounting fees, increased by $2.86 million primarily attributable to costs associated with operating as a publicly-traded company, negotiating our Collaboration Agreement with Purdue and pursuing our patent applications; |
| • | Market research and other third party expenses increased by $0.83 million to support the development of the Intermezzo® commercialization plan; and |
| • | Other general and administrative expenses increased by $2.0 million due to increased insurance for operating as a publicly-traded company, costs associated with additional office space and increased travel to prepare for potential Intermezzo® commericalization. |
58
Table of Contents
Merger related transaction costs
Merger related transaction costs consisted primarily of $2.0 million in financial and advisory fees and $0.2 million in legal fees incurred in connection with the close of the merger transaction in January 2009. Merger related transaction costs in 2008 consist primarily of $1.97 million in professional fees incurred in connection with the merger.
Interest Income
Interest income decreased 62% to $282,000 for the year ended December 31, 2009 from $742,000 for the comparable period in 2008. The decrease of approximately $460,000 for the year ended December 31, 2009 is primarily attributable to changing the mix of investments toward lower risk, lower yield instruments due to the downturn in the U.S. and world economy during the second half of 2008. In addition, investments held during 2008 were, in the aggregate, purchased at a discount to face value whereas investments held during 2009 were primarily acquired at a premium. Amortization of bond premiums is recorded as a reduction of interest income.
Interest Expense
Interest expense decreased 77% to $179,000 for the year ended December 31, 2009 from $766,000 for the comparable period in 2008. The $587,000 decrease for the year ended December 31, 2009 was primarily attributable to lower average outstanding debt during the 2009 period as compared to the same period in the prior year due to the repayment in full of our debt under a Loan and Security Agreement with Hercules Technology Growth Capital during the first quarter 2009.
Other Income (Expense), Net
Other income (expense), net decreased 50% to $168,000 for the year ended December 31, 2009 from $337,000 for the comparable period in 2008. In 2008 through January 2009, the fair market value of the preferred stock warrants declined significantly, resulting in a reduction of the warrant liability and corresponding increase in other income. Other expense in 2009 included $143,000 of Delaware Franchise tax as compared to $15,000 during 2008. This increase in 2009 other expense was partially offset by $111,000 of realized gains on the sales of marketable securities
Comparison of the Years Ended December 31, 2008 and 2007
The following table summarizes results of operations with respect to the items set forth below for the years ended December 31, 2008 and 2007, in thousands, together with the percentage change in those items.
| Year ended December 31, | |||||||||||||||
| $ | % | ||||||||||||||
| 2008 | 2007 | Change | Change | ||||||||||||
| Research and development expenses |
$ | 10,381 | $ | 15,885 | $ | (5,504 | ) | (35 | %) | ||||||
| General and administrative expenses |
7,924 | 5,300 | 2,624 | 50 | % | ||||||||||
| Merger related transaction costs |
1,967 | — | 1,967 | — | |||||||||||
| Interest income |
742 | 2,029 | (1,287 | ) | (63 | %) | |||||||||
| Interest expense |
(766 | ) | (1,195 | ) | 429 | 36 | % | ||||||||
| Other income (expense), net |
337 | (33 | ) | 370 | 1121 | % | |||||||||
Research and Development Expenses
Research and development expenses decreased 35% to $10.38 million for the year ended December 31, 2008 from $15.88 million for the comparable period in 2007. The $5.50 million decrease is primarily attributable to Intermezzo® development costs as a result of the following:
| • | a decrease in clinical trial costs of $4.84 million due primarily to the Phase 3 outpatient trial being largely completed by year end 2007, with trial closeout activities occurring during the first half of 2008; |
59
Table of Contents
| • | a decrease in manufacturing costs of $1.06 million as formulation development and production of clinical batches had been completed in 2007; partially offset by |
| • | an increase of $0.72 million in registration and submission third party costs related to the filing of our NDA in September 2008. |
The remaining $0.32 million reduction in research and development expenses is related to the change in expenditures for earlier stage programs.
General and Administrative Expenses
General and administrative expenses increased 50% to $7.92 million in 2008 from $5.30 million in 2007. The approximate $2.62 million increase consists of the following:
| • | Personnel costs and related expenses increased by $1.2 million due to an increase in headcount in the executive, finance, accounting, marketing and operations functions to support the ramp-up of clinical development and NDA activities, as well as in preparation to operate as a publicly-traded company; |
| • | Market research and other third party marketing expenses increased by $0.91 million to support the development of the Intermezzo® commercialization plan; and |
| • | Other general and administrative expenses increased by $0.51 million due to increased professional fees incurred in support and anticipation of our merger with Novacea, Inc. |
Merger related transaction costs
Merger related transaction costs in 2008 consist primarily of $1.97 million in professional fees incurred in connection with our merger with Novacea, Inc.
Interest Income
Interest income decreased 63% to $0.72 million in 2008 from $2.03 million for the comparable period in 2007. The decrease of approximately $1.31 million is primarily attributable to lower average cash and investments balances due to cash used in operations during the 2008 period as compared to the same period in 2007.
Interest Expense
Interest expense decreased 36% to $0.77 million in 2008 from $1.20 million in 2007. The $0.43 million decrease was primarily attributable to lower average outstanding debt during 2008 compared to 2007.
Other Income (Expense), Net
Other income (expense), net increased to a gain of $337,000 in 2008 from a loss of $33,000 in 2007. In 2008, the fair market value of the preferred stock warrants declined significantly as compared to 2007, resulting in a reduction of the warrant liability and corresponding increase in other income.
Liquidity and Capital Resources
At December 31, 2009, we had cash, cash equivalents and marketable securities of $88.9 million.
From our inception through the completion of the merger, we financed our operations primarily through private placements of preferred stock, debt financing, the Collaboration Agreement and interest income. Through December 31, 2008, we received net proceeds of $71.0 million from the sale of preferred stock. In January 2009, through the merger, we acquired an additional $80.9 million in cash, cash equivalents and marketable securities. On August 4, 2009, we received a $25.0 million non-refundable license fee from Purdue in connection with our entry into the Collaboration Agreement.
60
Table of Contents
In February 2006, we entered into a $10.0 million venture debt facility agreement with Hercules Technology Growth Capital, Inc., or Hercules, and drew down $4.0 million in May 2006 and $6.0 million in December 2006, against which interest accrued at rates of 10.69% and 10.94%, respectively. Outstanding principal, accrued interest, and unpaid interest under the loan and security agreement became due and payable on certain change of control transactions. In conjunction with the merger of Novacea and TPI and pursuant to an agreement with Hercules, on February 3, 2009 we repaid in full all amounts outstanding related to this loan.
The following table summarizes our cash provided by (used in) operating, investing and financing activities (in thousands):
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Net cash provided by (used in) operating activities |
$ | 830 | $ | (20,130 | ) | $ | (18,822 | ) | ||||
| Net cash provided by (used in) investing activities |
14,652 | 22,438 | (19,492 | ) | ||||||||
| Net cash (used in) provided by financing activities |
(2,883 | ) | (3,572 | ) | 37,045 | |||||||
Net Cash Provided by (Used in) Operating Activities
Net cash provided by operating activities was $0.8 million for the year ended December 31, 2009. Net cash used in operating activities was $20.1 million and $18.8 million for the years ended December 31, 2008 and 2007, respectively. Net cash provided by operations during 2009 was primarily due to the receipt of the $25.0 million non-refundable license fee we received from Purdue in connection with our entry into the Collaboration Agreement partially offset by our net loss adjusted for noncash items such as depreciation, amortization, stock-based compensation charges and noncash interest expense, as well as net changes in working capital. Net cash used in operating activities for 2008 and 2007 consisted primarily of our net loss adjusted for noncash items such as depreciation, amortization, stock-based compensation charges and noncash interest expense, as well as net changes in working capital.
Net Cash Provided by (Used in) Investing Activities
Net cash provided by investing activities was $14.7 million for the year ended December 31, 2009 and $22.4 million for the year ended December 31, 2008. $80.9 million of net cash provided by investing activities during 2009 relates to the cash, cash equivalents and marketable securities that came from our merger with Novacea, Inc. This was partially offset by $65.9 million used in investing activities during 2009 due to purchases of marketable securities, net of sales and maturities. $22.7 million provided by investing activities in 2008 was due to maturities of marketable securities, net of purchases. $18.6 million used in investing activities during 2007 was due to purchases of marketable securities, net of maturities. Uses of cash in investing activities in all periods also included net purchases of property and equipment.
Net Cash (Used in) Provided by Financing Activities
Net cash used in financing activities for the years ended December 31, 2009 and 2008 was $2.9 million and $3.6 million, respectively. While both periods consisted primarily of debt repayment, the outstanding debt with Hercules Technology Growth Capital was fully repaid during the first quarter of 2009. Net cash provided by financing activities for the year ended December 31, 2007 of $37.0 million consisted primarily of net proceeds of $39.8 million from the Series D preferred stock issuance, partially offset by $3.0 million of debt repayment.
Capital Resources
Based on currently available information, we expect that our cash, cash equivalents, and marketable securities balances will be sufficient to satisfy our liquidity requirements for at least the next twelve months. If, at any time, our prospects for the commercialization of Intermezzo® diminish, we may decide to reduce operating
61
Table of Contents
expenses by limiting research and development efforts with respect to other potential product candidates. Alternatively, we may decide to raise funds through public or private financings, collaboration relationships or other arrangements. There can be no assurance that funding, if needed, will be available on attractive terms, or at all. Furthermore, any additional equity financing may be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants. Similarly, financing obtained through future collaborations may require us to forego certain commercialization and other rights to our drug candidates. Our failure to raise capital as and when needed could have a negative impact on our financial condition and our ability to pursue our business strategy.
Merger Related Uses of Cash
Shortly after the January 30, 2009 close of the merger with Novacea, we repaid in full our outstanding credit obligations to Hercules Technology Growth Capital in the approximate amount of $2.8 million including interest and prepayment penalty and also paid merger-related financial advisory fees of approximately $2.0 million.
Upon completion of the merger on January 30, 2009, we became liable to pay approximately $2.2 million in payments due to Novacea employees upon a change in control, all of which has been paid as of September 30, 2009. None of these payments required on-going services of the employees subsequent to the change in control.
Potential Impact of Global Market and Economic Conditions on Our Liquidity
In the United States and around the world, recent market and economic conditions have been unprecedented and challenging, with tighter credit conditions and slower growth through 2008. During 2008 and into 2009, continued concerns about the systemic impact of the availability and cost of credit, energy costs, geopolitical issues, the U.S. mortgage market, a declining real estate market in the U.S. and added concerns fueled by the federal government interventions in the U.S. financial and credit markets have contributed to instability in both U.S. and international capital and credit markets and diminished expectations for the U.S. and global economy. These conditions, combined with volatile oil prices, declining business and consumer confidence and increased unemployment have contributed to volatility of unprecedented levels and an economic slowdown.
As a result of these market conditions, the cost and availability of capital and credit has been and may continue to be adversely affected by illiquid credit markets and wider credit spreads. If volatile and adverse market conditions continue, they may limit our ability to timely borrow or access the capital and credit markets to meet liquidity needs, resulting in an adverse effect on our financial condition and results of operations. In addition, the bio-pharmaceutical industry has fluctuated significantly in the past and has experienced significant downturns in connection with, or in anticipation of, deterioration in general economic conditions, and we cannot accurately predict the severity or duration of any downturn.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in any off-balance sheet financing activities, including the use of structured finance, special purpose entities or variable interest entities.
Contingencies
There are no legal proceedings or other matters as of December 31, 2009 that are expected to have a material adverse effect on our financial position, results of operations or cash flows.
62
Table of Contents
Contractual Obligations and Commitments
Our contractual obligations and commitments as of December 31, 2009 include potential purchase commitments and future minimum lease payments under operating leases, as shown in the following table:
Total Contractual Obligations
(in thousands)
| Contractual Obligations |
Payments due by period | ||||||||||||||
| Total | Less than one year |
1 to 3 years |
3 to 5 years |
More than 5 years | |||||||||||
| Operating leases(1) |
$ | 3,777 | $ | 1,209 | $ | 2,568 | $ | — | $ | — | |||||
| Purchase commitments(2) |
350 | 350 | — | — | — | ||||||||||
| Loan payable(3) |
195 | 57 | 138 | — | — | ||||||||||
| Total contractual obligations |
$ | 4,322 | $ | 1,616 | $ | 2,706 | $ | — | $ | — | |||||
| (1) | Includes obligations under an operating lease for current corporate facilities of Transcept, as well as obligations under an operating lease for the former Novacea corporate facilities. In February 2006, we signed an operating lease for our corporate offices that include approximately 11,600 square feet of office and laboratory space in Point Richmond, California. The lease term is for seven years, commencing on June 1, 2006. In June 2007, we amended this operating lease to add approximately 3,000 square feet of additional office space. The lease term of this amendment coincides with the original lease agreement, with a separate commencement date of September 12, 2007. Both of these leases provide for periodic rent increases based upon previously negotiated or consumer price indexed adjustments. |
On February 20, 2009, we signed an operating lease for 12,257 square feet of general office space in Point Richmond, California. The lease term commenced in March 2009 and terminates on May 31, 2013, with an option to renew for an additional five years. Under the terms of the lease, we have the option of accelerating the termination date of the lease to May 31, 2011.
In June 2007, Novacea entered into an operating lease for its corporate facilities, located in South San Francisco, California. The Novacea lease for the corporate facilities is non-cancelable and has a five year term. The lease provides for periodic rent increases based upon previously negotiated or consumer price indexed adjustments. On March 25, 2009, we entered into a sublease agreement dated as of March 24, 2009 for 18,368 square feet of the 25,288 square feet located at our South San Francisco facilities. The term of the sublease commenced on June 1, 2009 and ends on October 31, 2012. On June 16, 2009, we entered into a sublease agreement dated for reference purposes as of June 11, 2009 for the remaining 6,920 square feet of the South San Francisco facility. The term of the sublease commenced on July 1, 2009 and ends on October 31, 2012. The above obligations do not include partially offsetting sublease income of approximately $1.4 million.
| (2) | Pursuant to the terms of our agreement with Plantex USA Inc., under the purchase order dated August 8, 2008, we are obligated to purchase $350,000 worth of zolpidem tartrate during 2010. |
| (3) | Loan payable represents a loan from the landlord of our corporate offices in Point Richmond, California for tenant improvements. |
Recently Adopted Accounting Standards
Effective January 1, 2009, we adopted Accounting Standards Codification, or ASC, Topic 820, Fair Value Measurements and Disclosures, or ASC Topic 820 (formerly Statement of Financial Accounting Standard, or SFAS, No. 157, Fair Value Measurements), for nonfinancial assets and liabilities. The adoption of ASC Topic 820 for nonfinancial assets and liabilities did not have a material effect on our results of operations and financial condition.
63
Table of Contents
Effective January 1, 2009, we adopted ASC Topic 805, Business Combinations, or ASC Topic 805 (formerly SFAS No. 141R, Business Combinations). ASC Topic 805 amends previous guidance, and retains the purchase method of accounting for acquisitions, but requires a number of changes, including changes in the way assets and liabilities are recognized in purchase accounting. It also changes the recognition of assets acquired and liabilities assumed arising from contingencies, requires the capitalization of in-process research and development at fair value, and requires the expensing of acquisition-related costs as incurred. It also provides disclosure requirements to enable users of the financial statements to evaluate the nature and financial effects of the business combination. We have recorded our acquisition of Novacea in accordance with the provision of ASC Topic 805. See Note 2, “Merger Agreement” for further details.
In June 2009, the Financial Accounting Standards Board, or FASB, issued ASC Topic 105, Generally Accepted Accounting Principles, which establishes the ASC as the sole source of authoritative generally accepted accounting principles. Pursuant to the provisions of ASC Topic 105, we have updated references to generally accepted accounting principles in our financial statements issued for the period ended December 31, 2009. The adoption of ASC Topic 105 did not impact our financial position or results of operations.
In September 2009, the FASB Emerging Issues Task Force reached a consensus on ASC Update 2009-13, or Topic 605-25, Multiple-Deliverable Revenue Arrangements, or ASC Update 2009-13. ASC Update 2009-13 applies to multiple-deliverable revenue arrangements that are currently within the scope of ASC Topic 605-25. ASC Update 2009-13 provides principles and application guidance on whether multiple deliverables exist and how the arrangement should be separated and the consideration allocated. ASC Update 2009-13 requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables, if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. The update eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. ASC Update 2009-13 should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. As a result, ASC Update 2009-13 will be effective for us no later than the first quarter of fiscal 2011. The Company is currently evaluating the impact of adopting ASC Update 2009-13 on its financial position and results of operations.
Critical Accounting Policies
This discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these consolidated financial statements requires management to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as revenue and expenses during the reporting periods. We evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believed were reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Therefore, actual results could differ materially from those estimates under different assumptions or conditions.
Significant accounting policies are described in Note 1 to the financial statements included in Part I, Item 1 of this annual report on Form 10-K. Some of these accounting policies require us to make difficult and subjective judgments, often as a result of the need to make estimates on matters that are inherently uncertain. We believe that the following critical accounting policies reflect the more significant estimates and assumptions used in the preparation of our financial statements.
64
Table of Contents
Revenue Recognition
We apply the revenue recognition criteria outlined in Staff Accounting Bulletin No. 104, Revenue Recognition in Financial Statements, and ASC Topic 605, Revenue Recognition, 25 Multiple-Element Arrangements.
Revenue arrangements with multiple components are divided into separate units of accounting if certain criteria are met, including whether the delivered component has stand-alone value to the customer, and whether there is objective and reliable evidence of the fair value of the undelivered items. Consideration received is allocated among the separate units of accounting based on their respective fair values. Applicable revenue recognition criteria are then applied to each of the units.
Revenue is recognized when the four basic criteria of revenue recognition are met:
| • | persuasive evidence of an arrangement exists; |
| • | transfer of technology has been completed or services have been rendered; |
| • | the fee is fixed or determinable; and |
| • | collectability is reasonably assured. |
For each source of revenue, we apply the above revenue recognition criteria in the following manner:
| • | Up-front license payments are assessed to determine whether or not the licensee is able to obtain any stand-alone value from the license. Where this is not the case, we do not consider the license deliverable to be a separate unit of accounting, and the revenue is deferred with revenue recognition for the license fee being assessed in conjunction with the other deliverables that constitute the combined unit of accounting. When the period of deferral cannot be specifically identified from the agreement, management estimates the period based upon provisions contained within the agreement and other relevant facts. We periodically review the estimated involvement period, which could impact the deferral period and, therefore, the timing and the amount of revenue recognized. It is possible that future adjustments will be made if actual conditions differ from our current plan and involvement assumptions. |
| • | Payments received that are related to substantive, performance-based “at-risk” milestones are recognized as revenue upon achievement of the milestone or event specified in the underlying contracts, which represents the culmination of the earnings process. Amounts received in advance, if any, are recorded as deferred revenue until the milestone is reached. |
| • | Royalty revenue from sales of our licensed products, if and when approved for marketing by the appropriate regulatory agency, will be recognized as earned in accordance with the contract terms when royalties from licensees can be reasonably estimated and collectability is reasonably assured. |
Clinical Trials
We accrue and expense costs for clinical trial activities performed by third parties, including clinical research organizations and clinical investigators, based upon estimates made of the work completed as of the reporting date, in accordance with agreements established with contract research organizations and clinical trial sites and the agreed upon fee to be paid for the services. We determine these estimates through discussion with internal personnel and outside service providers as to the progress or stage of completion of the trials or services. If the actual timing of performance of services or the level of effort varies from these estimates, the accrual will be adjusted accordingly. Costs of setting up clinical trial sites for participation in the trials are expensed as the activities are performed. Clinical trial site costs related to patient enrollment are accrued as patients are entered into the trial and reduced by any initial payment made to the clinical trial site when the first patient is enrolled. We adjust the estimates as actual costs become known. Through December 31, 2009, differences between actual
65
Table of Contents
and estimated activity levels for any particular study have not been material. However, if management does not receive complete and accurate information from vendors or underestimates activity levels associated with a study at a given point in time, we would have to record additional and potentially significant research and development expenses in future periods.
Stock-Based Compensation
We recognize stock based compensation in accordance with ASC Topic 718, Compensation—Stock Compensation, or ASC Topic 718 (formerly Statement of Financial Accounting Standards, or SFAS, No. 123(R), Share-Based Payment). ASC Topic 718 requires an entity to measure the cost of employee services received in exchange for an award of equity instruments based on the fair value of the award on the date of grant and to recognize the cost over the period during which the employee is required to provide service in exchange for the award. Additionally, we are required to include an estimate of the number of awards that will be forfeited in calculating compensation costs, which are recognized over the requisite service period of the awards on a straight-line basis.
Measurement and recognition of share-based compensation under ASC Topic 718 involve significant estimates and subjective inputs. The grant date fair value of stock option awards is determined using an option valuation model, such as the Black-Scholes model that we used, and the amount of expense recognized during the period is affected by many complex and subjective assumptions. These assumptions include estimates of our future volatility, employee exercise behavior, the expected term of the stock options, and the number of options expected to ultimately vest. Until the merger with Novacea, our stock did not have a readily available market. Consequently, expected future volatility is derived from the historical volatilities of several unrelated public companies within the specialty pharmaceutical industry. When making the selection of our industry peer companies to be used in the volatility calculation, consideration is given to the stage of development, size and financial leverage of potential comparable companies. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant for zero coupon U.S. Treasury notes with maturities approximately equal to each grant’s expected life. The assumed dividend yield was based on our expectations of not paying dividends in the foreseeable future. Given our limited history to accurately estimate the expected lives for the various employee groups, we used the “simplified” method as provided by Staff Accounting Bulletin No. 107, Share Based Payment. The “simplified” method is calculated as the average of the time-to-vesting and the contractual life of the options. Share-based compensation recorded in our Statements of Operations is based on awards expected to ultimately vest and has been reduced for estimated forfeitures. Estimated forfeitures may differ from actual forfeiture rates which would affect the amount of expense recognized during the period. Share-based compensation is adjusted to reflect the value of options which ultimately vest as such amounts become known in future periods.
If in the future, our management determines that another method is more reasonable, or if another method for calculating these input assumptions is prescribed by authoritative guidance, and, therefore, should be used to estimate volatility or expected life, the fair value calculated for our stock options could change significantly. Higher volatility and longer expected lives result in an increase to stock-based compensation expense determined at the date of grant. Stock-based compensation expense affects both our research and development expense and general and administrative expense.
There is inherent uncertainty in these estimates and if we had made different assumptions than those described above, the amount of stock-based compensation expense, net loss and net loss per share amounts could have been significantly different.
No related tax benefits of share-based compensation costs have been recognized since our inception.
66
Table of Contents
Fair Value Measurements.
On January 1, 2008, we adopted ASC Topic 820, Fair Value Measurements and Disclosures (formerly SFAS No. 157) as it applies to our financial assets and financial liabilities. ASC Topic 820 defines fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. Fair value is defined as the estimated exit price received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date rather than on an entry price which represents the purchase price of an asset or liability. ASC Topic 820 establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
| • | Level 1—Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| • | Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| • | Level 3—Unobservable inputs (i.e. inputs that reflect the reporting entity’s own assumptions about the assumptions that market participants would use in estimating the fair value of an asset or liability) are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
A financial instrument’s categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Where quoted prices are available in an active market, securities are classified as Level 1 of the valuation hierarchy. Level 1 securities include highly liquid money market funds. If quoted market prices are not available for the specific security, then we estimate fair value by using pricing models, quoted prices of securities with similar characteristics or discounted cash flows. Level 2 instruments include commercial paper, U.S. corporate debt, and U.S. government sponsored enterprise issues. In certain cases where there is limited activity or less transparency around inputs to valuation, securities are classified as Level 3 within the valuation hierarchy. Level 3 liabilities that are measured at fair value on a recurring basis consist of convertible preferred stock warrant liabilities. The fair values of the outstanding preferred stock warrants are measured using the Black-Scholes option-pricing model. Inputs used to determine fair market value include estimated market value of the underlying convertible preferred stock at the valuation measurement date, the remaining contractual term of the warrant, risk-free interest rates and expected dividends on and expected volatility of the price of the underlying convertible preferred stock.
During the year ended December 31, 2009, there were no significant changes to the valuation models used for purposes of determining the fair value of Level 2 assets or Level 3 liabilities. Concurrent with our merger with Novacea, Inc., the warrants to purchase preferred stock were converted into warrants to purchase common stock with the same exercise prices and expiration dates, and the aggregate fair value of these warrants was reclassified from a liability to additional paid-in capital, a component of stockholders’ equity (net capital deficiency).
Warrants to Purchase Convertible Preferred Stock
Effective July 1, 2005, we adopted the provisions of ASC Topic 480, Distinguishing Liabilities from Equity, or ASC Topic 480 (formerly FASB Staff Position No. 150-5, Issuer’s Accounting Under FASB Statement No. 150 for Freestanding Warrants and Other Similar Instruments with Characteristics of Both Liabilities and Equity, an interpretation of SFAS No. 150, Accounting for Certain Financial Instruments with Characteristics of Both Liabilities and Equity). Under ASC Topic 480, freestanding warrants to purchase shares of convertible preferred stock were classified as liabilities on the balance sheets at fair value because the warrants may conditionally obligate us to transfer assets at some point in the future. The warrants were subject to remeasurement at each balance sheet date, and any change in fair value was recognized as a component of other
67
Table of Contents
income (expense), net in the statements of operations. We estimated the fair value of these warrants at the respective balance sheet dates using the Black-Scholes option-pricing model as described in the stock-based compensation section above, based on the estimated market value of the underlying convertible preferred stock at the valuation measurement date, the remaining contractual term of the warrant, risk-free interest rates and expected dividends on and expected volatility of the price of the underlying convertible preferred stock. The assumptions used in the Black-Scholes option-pricing model, especially the market value of the underlying convertible preferred stock and the expected volatility, are highly judgmental.
We continued to record adjustments to the fair value of the warrants at each balance sheet date until the closing of the merger transaction on January 30, 2009, when they became warrants to purchase shares of common stock, wherein the warrants were no longer subject to ASC Topic 480. As of January 30, 2009, the current aggregate fair value of these warrants of $400,000 was reclassified from a liability to additional paid-in capital, a component of stockholders’ equity (net capital deficiency). We no longer record any related periodic fair value adjustments. Upon the closing of the merger transaction, the preferred stock warrants were converted into common stock warrants with the same exercise prices and expiration dates.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Our exposure to market risk is confined to cash, cash equivalents and marketable securities which have contractual maturities of eighteen months or less, bear interest rates at fixed rates and are denominated in, and pay interest in, U.S. dollars. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs, and fiduciary control of cash and investments. We also seek to achieve income from investments consistent with our investment policy. Investments are classified as available-for-sale. We do not use derivative financial instruments in our investment portfolio. To achieve our goals, we invest excess cash in securities with different maturities to match projected cash needs and limit concentration of credit risk by diversifying investments among a variety of high credit-quality issuers, including U.S. government agencies, commercial paper, corporate bonds and money market funds. The portfolio includes marketable securities with active secondary or resale markets to ensure portfolio liquidity, and we regularly review our portfolio against our policy. Our policy was further amended during 2009 to limit investments to U.S. Treasury debt or Securities and Exchange Commission, or SEC, registered money market funds effective September 30, 2009. A decline in short-term interest rates over time would reduce our interest income from our short-term investments. A hypothetical 100 basis point increase in interest rates would result in an approximate $248,000 decrease in the fair value of our marketable securities at December 31, 2009.
68
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
| Page | ||
| 70 | ||
| 71 | ||
| 72 | ||
| 73 | ||
| 74 | ||
| 75 |
69
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Transcept Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Transcept Pharmaceuticals, Inc. as of December 31, 2009 and 2008 and the related consolidated statements of operations, convertible preferred stock and stockholders’ equity (net capital deficiency), and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Transcept Pharmaceuticals, Inc. at December 31, 2009 and 2008, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
Palo Alto, California
March 30, 2010
70
Table of Contents
Transcept Pharmaceuticals, Inc.
Consolidated Balance Sheets
(in thousands, except for share and per share amounts)
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 17,031 | $ | 4,432 | ||||
| Marketable securities |
71,871 | 7,251 | ||||||
| Prepaid and other current assets |
1,276 | 382 | ||||||
| Restricted cash |
200 | 200 | ||||||
| Total current assets |
90,378 | 12,265 | ||||||
| Property and equipment, net |
1,052 | 1,450 | ||||||
| Goodwill |
2,962 | — | ||||||
| Other assets |
826 | 66 | ||||||
| Total assets |
$ | 95,218 | $ | 13,781 | ||||
| Liabilities, convertible preferred stock and stockholders’ equity (net capital deficiency) |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 728 | $ | 575 | ||||
| Accrued liabilities |
2,383 | 1,468 | ||||||
| Deferred revenue, short-term portion |
12,500 | — | ||||||
| Lease liability, short-term portion |
429 | — | ||||||
| Loan payable, short-term portion |
45 | 3,347 | ||||||
| Total current liabilities |
16,085 | 5,390 | ||||||
| Deferred revenue, long-term portion |
7,292 | — | ||||||
| Warrant liability |
— | 600 | ||||||
| Deposit for stock purchase |
41 | 88 | ||||||
| Deferred rent |
72 | 77 | ||||||
| Lease liability, long-term portion |
519 | — | ||||||
| Loan payable, long-term portion |
125 | 170 | ||||||
| Other liabilities |
13 | — | ||||||
| Total liabilities |
24,147 | 6,325 | ||||||
| Commitments and contingencies |
||||||||
| Convertible preferred stock: $0.001 par value; 7,593,091 shares authorized; |
— | 71,037 | ||||||
| Stockholders’ equity (net capital deficiency): |
||||||||
| Preferred stock: 5,000,000 shares authorized; 0 shares issued and outstanding |
— | — | ||||||
| Common stock: $0.001 par value; 100,000,000 shares authorized; 13,384,247 and 454,676 shares issued and outstanding at December 31, 2009 and 2008, respectively |
13 | — | ||||||
| Additional paid-in capital |
157,930 | 1,504 | ||||||
| Accumulated deficit |
(86,911 | ) | (65,111 | ) | ||||
| Accumulated other comprehensive income |
39 | 26 | ||||||
| Total stockholders’ equity (net capital deficiency) |
71,071 | (63,581 | ) | |||||
| Total liabilities, convertible preferred stock and stockholders’ equity (net capital deficiency) |
$ | 95,218 | $ | 13,781 | ||||
See accompanying notes.
71
Table of Contents
Transcept Pharmaceuticals, Inc.
Consolidated Statements of Operations
(in thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Revenue: |
||||||||||||
| License fee revenue |
$ | 5,208 | $ | — | $ | — | ||||||
| Operating expenses: |
||||||||||||
| Research and development |
9,005 | 10,381 | 15,885 | |||||||||
| General and administrative |
16,050 | 7,924 | 5,300 | |||||||||
| Merger related transaction costs |
2,224 | 1,967 | — | |||||||||
| Total operating expenses |
27,279 | 20,272 | 21,185 | |||||||||
| Loss from operations |
(22,071 | ) | (20,272 | ) | (21,185 | ) | ||||||
| Interest income |
282 | 742 | 2,029 | |||||||||
| Interest expense |
(179 | ) | (766 | ) | (1,195 | ) | ||||||
| Other income (expense), net |
168 | 337 | (33 | ) | ||||||||
| Net loss |
$ | (21,800 | ) | $ | (19,959 | ) | $ | (20,384 | ) | |||
| Basic and diluted net loss per share |
$ | (1.79 | ) | $ | (49.77 | ) | $ | (68.86 | ) | |||
| Weighted average shares outstanding |
12,166 | 401 | 296 | |||||||||
See accompanying notes.
72
Table of Contents
Transcept Pharmaceuticals, Inc.
Consolidated Statement of Convertible Preferred Stock and Stockholders’ Equity (Net Capital Deficiency)
(in thousands, except per share amounts)
| Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Accumulated Deficit |
Accumulated Other Comprehensive Income (Loss) |
Total Stockholders’ Equity (Net Capital Deficiency) |
||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||
| Balance at December 31, 2006 |
4,024 | $ | 31,202 | 245 | $ | — | $ | 177 | $ | (24,768 | ) | $ | 2 | $ | (24,589 | ) | |||||||||||||
| Issuance of Series D convertible preferred stock to investors for cash at $12.03 per share, net of issuance costs of $165 in February 2007 |
3,326 | 39,835 | — | — | — | — | — | — | |||||||||||||||||||||
| Exercise of options to purchase common stock |
— | — | 12 | — | 10 | — | — | 10 | |||||||||||||||||||||
| Stock-based compensation related to: |
|||||||||||||||||||||||||||||
| Employee stock option grants |
— | — | — | — | 470 | — | — | 470 | |||||||||||||||||||||
| Nonemployee stock option grants |
— | — | — | — | 13 | — | — | 13 | |||||||||||||||||||||
| Vested restricted stock |
— | — | 99 | — | 82 | — | — | 82 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (20,384 | ) | — | (20,384 | ) | |||||||||||||||||||
| Unrealized gain on marketable securities |
— | — | — | — | — | — | 83 | 83 | |||||||||||||||||||||
| Total comprehensive loss |
(20,301 | ) | |||||||||||||||||||||||||||
| Balance at December 31, 2007 |
7,350 | 71,037 | 356 | — | 752 | (45,152 | ) | 85 | (44,315 | ) | |||||||||||||||||||
| Exercise of options to purchase common stock |
— | — | 64 | — | 67 | — | — | 67 | |||||||||||||||||||||
| Stock-based compensation related to: |
|||||||||||||||||||||||||||||
| Employee stock option grants |
— | — | — | — | 613 | — | — | 613 | |||||||||||||||||||||
| Nonemployee stock option grants |
— | — | — | — | 24 | — | — | 24 | |||||||||||||||||||||
| Vested restricted stock |
— | — | 35 | — | 48 | — | — | 48 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (19,959 | ) | — | (19,959 | ) | |||||||||||||||||||
| Unrealized loss on marketable securities |
— | — | — | — | — | — | (59 | ) | (59 | ) | |||||||||||||||||||
| Total comprehensive loss |
(20,018 | ) | |||||||||||||||||||||||||||
| Balance at December 31, 2008 |
7,350 | 71,037 | 455 | — | 1,504 | (65,111 | ) | 26 | (63,581 | ) | |||||||||||||||||||
| Exercise of options to purchase common stock |
— | — | 293 | 1 | 384 | — | — | 385 | |||||||||||||||||||||
| Employee stock purchase under |
|||||||||||||||||||||||||||||
| Employee stock purchase plan |
— | — | 22 | — | 85 | — | — | 85 | |||||||||||||||||||||
| Stock-based compensation related to: |
|||||||||||||||||||||||||||||
| Employee stock option grants |
— | — | — | — | 1,057 | — | — | 1,057 | |||||||||||||||||||||
| Nonemployee stock option grants |
— | — | — | — | 121 | — | — | 121 | |||||||||||||||||||||
| Employee stock purchase plan |
— | — | — | — | 62 | — | — | 62 | |||||||||||||||||||||
| Stock option modifications |
— | — | — | — | 127 | — | — | 127 | |||||||||||||||||||||
| Vested restricted stock |
— | — | 33 | — | 47 | — | — | 47 | |||||||||||||||||||||
| Conversion of preferred shares to common stock |
(7,350 | ) | (71,037 | ) | 7,350 | 7 | 71,030 | — | — | 71,037 | |||||||||||||||||||
| Reclassification of warrant liability |
— | — | — | — | 400 | — | — | 400 | |||||||||||||||||||||
| Effect of the Merger (Note 2) |
— | — | 5,231 | 5 | 83,113 | — | — | 83,118 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (21,800 | ) | — | (21,800 | ) | |||||||||||||||||||
| Unrealized gain on marketable securities |
— | — | — | — | — | — | 13 | 13 | |||||||||||||||||||||
| Total comprehensive loss |
(21,787 | ) | |||||||||||||||||||||||||||
| Balance at December 31, 2009 |
— | $ | — | 13,384 | $ | 13 | $ | 157,930 | $ | (86,911 | ) | $ | 39 | $ | 71,071 | ||||||||||||||
See accompanying notes.
73
Table of Contents
Transcept Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Operating activities |
||||||||||||
| Net Loss |
$ | (21,800 | ) | $ | (19,959 | ) | $ | (20,384 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
550 | 505 | 379 | |||||||||
| Stock-based compensation |
1,367 | 637 | 483 | |||||||||
| Amortization of loan costs |
28 | 37 | 37 | |||||||||
| Amortization of discount (warrants) on debt |
47 | 151 | 216 | |||||||||
| Amortization of lease liability |
(281 | ) | — | — | ||||||||
| Remeasurement of preferred stock warrants |
(200 | ) | (368 | ) | 13 | |||||||
| Loss on disposals of fixed assets |
157 | 4 | — | |||||||||
| (Gain) loss on sale of marketable securities |
(111 | ) | 16 | — | ||||||||
| Amortization (accretion) of premium/discount on available for sale securities |
1,395 | (486 | ) | (1,250 | ) | |||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Prepaid and other current assets |
455 | 150 | (70 | ) | ||||||||
| Other assets |
(18 | ) | — | (6 | ) | |||||||
| Accounts payable |
154 | (242 | ) | 27 | ||||||||
| Accrued liabilities |
(700 | ) | (578 | ) | 1,720 | |||||||
| Deferred revenue |
19,792 | — | — | |||||||||
| Deferred rent |
(5 | ) | 3 | 13 | ||||||||
| Net cash provided by (used) in operating activities |
830 | (20,130 | ) | (18,822 | ) | |||||||
| Investing activities |
||||||||||||
| Purchases of property and equipment, net |
(318 | ) | (259 | ) | (643 | ) | ||||||
| Purchases of marketable securities |
(128,324 | ) | (22,857 | ) | (73,203 | ) | ||||||
| Maturities and sales of marketable securities |
95,307 | 45,554 | 54,554 | |||||||||
| Cash and cash equivalents received from the Merger |
47,987 | — | — | |||||||||
| Transfer to restricted cash |
— | — | (200 | ) | ||||||||
| Net cash provided by (used in) investing activities |
14,652 | 22,438 | (19,492 | ) | ||||||||
| Financing Activities |
||||||||||||
| Payments on long-term debt |
(3,353 | ) | (3,640 | ) | (3,008 | ) | ||||||
| Proceeds from issuances of convertible preferred stock, net |
— | — | 39,835 | |||||||||
| Proceeds from issuance of common stock, net |
470 | 68 | 218 | |||||||||
| Net cash (used in) provided by financing activities |
(2,883 | ) | (3,572 | ) | 37,045 | |||||||
| Net increase (decrease) in cash and cash equivalents |
12,599 | (1,264 | ) | (1,269 | ) | |||||||
| Cash and cash equivalents at beginning of period |
4,432 | 5,696 | 6,965 | |||||||||
| Cash and cash equivalents at end of period |
$ | 17,031 | $ | 4,432 | $ | 5,696 | ||||||
| Supplemental disclosure of cash flow information |
||||||||||||
| Cash paid during the year for interest |
$ | 136 | $ | 611 | $ | 921 | ||||||
| Supplemental disclosure of noncash investing and financing activities |
||||||||||||
| Purchase of leasehold improvements in exchange for loan payable |
$ | — | $ | — | $ | 258 | ||||||
| Acquisition of leasehold improvements paid by landlord |
$ | — | $ | — | $ | 60 | ||||||
See accompanying notes.
74
Table of Contents
Transcept Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
1. Organization and Summary of Significant Accounting Policies
Transcept Pharmaceuticals, Inc. (“Transcept” or the “Company”) is a specialty pharmaceutical company focused on the development and commercialization of proprietary products that address important therapeutic needs in neuroscience. The most advanced Transcept product candidate is Intermezzo® (zolpidem tartrate sublingual tablet), for which a New Drug Application (“NDA”) was submitted to the U.S. Food and Drug Administration (“FDA”) in September 2008 seeking approval as a prescription sleep aid for use in the middle of the night at the time a patient awakens and has difficulty returning to sleep. In October 2009, Transcept received a Complete Response Letter from the FDA on the Intermezzo® NDA and is working to respond to issues raised in the letter. On July 31, 2009, Transcept and Purdue Pharmaceutical Products, L.P. (“Purdue”) entered into the United States License and Collaboration Agreement (the “Collaboration Agreement”) in connection with the development and commercialization of Intermezzo® in the United States. As a result of the revenue recognized pursuant to the Collaboration Agreement and the late development stage of Intermezzo®, Transcept is no longer considered a Development Stage Company as of the third quarter 2009. Transcept operates in one business segment.
Prior to January 30, 2009, the name of the Company was Novacea, Inc. (“Novacea”). On August 29, 2008, Novacea, Pivot Acquisition, Inc., a Delaware corporation and a wholly-owned subsidiary of Novacea, and Transcept Pharmaceuticals, Inc., a private Delaware corporation (“TPI”), entered into an Agreement and Plan of Merger and Reorganization, which was amended on December 23, 2008 (collectively referred to as the “Merger Agreement”). On January 30, 2009, Novacea completed its business combination with TPI in accordance with the terms of the Merger Agreement, pursuant to which TPI became a wholly-owned subsidiary of Novacea (referred to as the “Merger”). Also on January 30, 2009, in connection with the Merger, Novacea effected a 1-for-5 reverse stock split of its common stock, and the name of Novacea was changed to “Transcept Pharmaceuticals, Inc.” The Merger, reverse stock split and the name change of Novacea were approved by the stockholders of Novacea at a special meeting of Novacea stockholders held on January 27, 2009. These financial statements reflect the historical results of TPI prior to the Merger and that of the combined company following the Merger, and do not include the historical results of Novacea prior to the completion of the Merger. All share and per share disclosures have been retroactively adjusted to reflect the exchange of shares in the Merger, and the 1-for-5 reverse stock split of the common stock on January 30, 2009.
Under the terms of the Merger Agreement, Novacea issued shares of common stock to the TPI stockholders at the rate of 0.14134 shares of common stock, after giving effect to the 1-for-5 reverse stock split, for each share of TPI common stock outstanding on January 30, 2009. Additionally, each share of common stock underlying TPI options and warrants as of January 30, 2009 was converted to 0.14134 shares of Transcept common stock. After consummation of the Merger, former TPI stockholders, option holders and warrant holders as of January 30, 2009 owned approximately 61% of Transcept common stock on a fully-diluted basis. The stockholders, option holders and warrant holders of Novacea prior to the Merger owned approximately 39% of the Transcept common stock on a fully-diluted basis following the Merger. Under generally accepted accounting principles in the United States, the Merger is treated as a “reverse merger” under the purchase method of accounting. For accounting purposes, TPI is considered to have acquired Novacea.
As part of the consummation of the Merger, Novacea securities listed on the NASDAQ Global Market, trading under the ticker symbol “NOVC,” were suspended for trading as of the close of business on January 30, 2009 and trading of Transcept securities on the NASDAQ Global Market under the ticker symbol “TSPT” commenced on February 2, 2009.
75
Table of Contents
Need to Raise Additional Capital
As of December 31, 2009, the Company had cash, cash equivalents and marketable securities of $88.9 million, working capital of $74.3 million, and an accumulated deficit of $86.9 million. Management expects to continue to incur additional losses in the foreseeable future as the Company continues its research and development activities and prepares for the potential commercialization of Intermezzo®. Management believes that cash, cash equivalents and marketable securities balances on hand at December 31, 2009 will be sufficient to fund planned expenditures for at least the next twelve months. Management recognizes the potential need to raise additional funds in the future. There can be no assurance that the Company will be successful in consummating any such financing transaction, or if the Company does consummate such a transaction, that the terms and conditions of such transaction will be favorable. Any failure to obtain additional funding may have a material negative effect on the Company and will likely result in a substantial reduction in the scope of the Company’s operations.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Management bases its estimates on historical experience and on assumptions believed to be reasonable under the circumstances. Actual results could differ materially from those estimates. Management makes estimates when preparing the financial statements including those relating to revenue recognition, clinical trials, stock-based compensation, restructuring and warrant liability valuation.
Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements include the results of operations of Transcept Pharmaceuticals, Inc. and its wholly-owned subsidiary, Transcept Pharma, Inc. All significant intercompany accounts and transactions have been eliminated in the consolidation.
Cash and Cash Equivalents
The Company invests its excess cash in bank deposits, money market accounts, and other marketable securities. The Company considers all highly liquid investments purchased with a maturity of three months or less from the date of purchase to be cash equivalents. Cash equivalents are carried at fair value. The Company invests in money market securities in a U.S. bank and is exposed to credit risk in the event of default by the financial institution to the extent of amounts recorded on the balance sheet.
Restricted cash represents a Certificate of Deposit (“CD”) which functions as security for the Company credit cards with the domestic financial institution that issued the credit cards. The CD will remain as security concurrent with the continuation of the Company credit card program.
Marketable Securities
All marketable securities have been classified as “available-for-sale” and are carried at fair value as determined based upon quoted market prices. Management determines the appropriate classification of its investments in debt securities at the time of purchase and reevaluates such designation as of each balance sheet date. Management views its investment portfolio as available for use in current operations and, accordingly, has reflected all such investments as current assets although the stated maturity of individual investments may be one year or more beyond the balance sheet date. Unrealized gains and losses are included in other comprehensive net income (loss) and reported as a separate component of stockholders’ equity (net capital deficiency). Realized gains and losses and declines in value judged to be other-than-temporary, if any, on available-for-sale securities are included in other income (expense), net. The cost of securities sold is based on the specific identification
76
Table of Contents
method. Interest on marketable securities is included in interest income. The net carrying value of debt securities classified as available-for-sale is adjusted for amortization of premiums and accretion of discounts to maturity, over the estimated life of the security. Such amortization is computed under the effective interest method and included in interest income.
Property and Equipment
Property and equipment is stated at cost and depreciated using the straight-line method over the estimated useful lives of the assets, ranging from two to five years. Leasehold improvements are amortized over the shorter of their estimated useful lives or the related lease term.
Long-Lived Assets
Long-lived assets include property and equipment. The carrying value of long-lived assets is reviewed for impairment whenever events or changes in circumstances indicate that the asset may not be recoverable. An impairment loss is recognized when the total of estimated future cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount or appraised value, as appropriate. Through December 31, 2009, there have been no such impairments.
Goodwill
Goodwill represents purchase consideration in excess of fair values assigned to the underlying net assets of acquired businesses. Goodwill is not amortized, but tested for impairment annually at September 30th, and at any time when events suggest impairment may have occurred. The Company’s goodwill impairment test is performed by comparing the fair value of the reporting unit to the carrying value of the reporting unit. The Company has one reporting unit to which the goodwill is assigned and tested for impairment. In the event the carrying value of a reporting unit exceeds its fair value, an impairment loss would be recognized to the extent the carrying amount of the reporting unit’s goodwill exceeds its implied fair value. Goodwill as of December 31, 2009 was approximately $3.0 million, and was recorded in connection with the Company’s merger with Novacea (see Note 2). Through December 31, 2009, there have been no impairments.
Revenue Recognition
The Company applies the revenue recognition criteria outlined in Staff Accounting Bulletin No. 104, Revenue Recognition in Financial Statements, and Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 605 Revenue Recognition, Sub-topic 25 Multiple-Element Arrangements.
Revenue arrangements with multiple components are divided into separate units of accounting if certain criteria are met, including whether the delivered component has stand-alone value to the customer, and whether there is objective and reliable evidence of the fair value of the undelivered items. Consideration received is allocated among the separate units of accounting based on their respective fair values. Applicable revenue recognition criteria are then applied to each of the units.
Revenue is recognized when the four basic criteria of revenue recognition are met: (1) persuasive evidence of an arrangement exists; (2) transfer of technology has been completed or services have been rendered; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured.
For each source of revenue, the Company applies the above revenue recognition criteria in the following manner:
| • | Up-front license payments are assessed to determine whether or not the licensee is able to obtain any stand-alone value from the license. Where this is not the case, the Company does not consider the license deliverable to be a separate unit of accounting, and the revenue is deferred with revenue |
77
Table of Contents
| recognition for the license fee being assessed in conjunction with the other deliverables that constitute the combined unit of accounting. When the period of deferral cannot be specifically identified from the agreement, management estimates the period based upon provisions contained within the agreement and other relevant facts. The Company periodically reviews the estimated involvement period, which could impact the deferral period and, therefore, the timing and the amount of revenue recognized. It is possible that future adjustments will be made if actual conditions differ from the Company’s current plan and involvement assumptions. |
| • | Payments received that are related to substantive, performance-based “at-risk” milestones are recognized as revenue upon achievement of the milestone or event specified in the underlying contracts, which represents the culmination of the earnings process. Amounts received in advance, if any, are recorded as deferred revenue until the milestone is reached. |
| • | Royalty revenue from sales of the Company’s licensed products, if and when approved for marketing by the appropriate regulatory agency, will be recognized as earned in accordance with the contract terms when royalties from licensees can be reasonably estimated and collectability is reasonably assured. |
Research and Development Costs
Research and development costs are expensed as incurred. Research and development costs consist of salaries and benefits, travel and related expenses, lab supplies and facility costs, as well as fees paid to other entities that conduct certain research and development activities on behalf of the Company.
Comprehensive Net Loss
The Company reports comprehensive net loss in accordance with FASB ASC Topic 220 Comprehensive Income (“ASC Topic 220”). Among other things, ASC Topic 220 requires unrealized gains or losses on the Company’s available-for-sale marketable securities to be included in other comprehensive loss and be reported as a separate component of stockholders’ equity (net capital deficiency).
Income Taxes
The Company utilizes the liability method of accounting for income taxes as required by FASB ASC Topic 740 Income Taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax reporting bases of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Uncertain tax positions are evaluated in accordance with this topic and if appropriate, the amount of unrecognized tax benefits are recorded within deferred tax assets. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized. Currently, there is no provision for income taxes as the Company has incurred operating losses to date.
Clinical Trials
The Company accrues and expenses costs for clinical trial activities performed by third parties, including clinical research organizations and clinical investigators, based upon estimates made of the work completed as of the reporting date, in accordance with agreements established with contract research organizations and clinical trial sites and the agreed upon fee to be paid for the services. The Company determines these estimates through discussion with internal personnel and outside service providers as to the progress or stage of completion of the trials or services. Costs of setting up clinical trial sites for participation in the trials are expensed immediately as research and development expenses. Clinical trial site costs related to patient enrollment are accrued as patients are entered into the trial and reduced by any initial payment made to the clinical trial site when the first patient is enrolled.
78
Table of Contents
Advertising
The Company expenses non-direct response advertising when the advertising expense takes place. Advertising expense was $125,000 during 2009. There was no comparable expense during 2008 or 2007.
Stock-Based Compensation
The Company records stock-based compensation in accordance with ASC Topic 718 Compensation—Stock Compensation (“ASC Topic 718”) (formerly Statement of Financial Accounting Standards (“SFAS”) No. 123(R), Share-Based Payment). ASC Topic 718 requires an entity to measure the cost of employee services received in exchange for an award of equity instruments based on the fair value of the award on the date of grant and to recognize the cost over the period during which the employee is required to provide service in exchange for the award. Additionally, the Company is required to include an estimate of the number of awards that will be forfeited in calculating compensation costs, which are recognized over the requisite service period of the awards on a straight-line basis.
During the years ended December 31, 2009, 2008 and 2007, the Company recorded employee share-based compensation costs of $1,246,000, $613,000 and $470,000, respectively, in accordance with the provisions of ASC Topic 718. No related tax benefits of share-based compensation costs have been recognized since the Company’s inception.
The Company accounts for equity instruments issued to nonemployees in accordance with the provisions of ASC Topic 505, subtopic 50, Equity-Based Payments to Non-Employees (formerly Emerging Issues Task Force No. 96-18, Accounting for Equity Instruments That are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling Goods or Services), using a fair-value approach. The equity instruments, consisting of stock options and warrants granted to lenders and consultants, are valued using the Black-Scholes valuation model. The measurement of stock-based compensation is subject to periodic adjustments as the underlying equity instruments vest and is recognized as an expense over the term of the related financing or the period over which services are received.
Warrants to Purchase Convertible Preferred Stock
Effective July 1, 2005, the Company adopted the provisions of ASC Topic 480 Distinguishing Liabilities from Equity (“ASC Topic 480”) (formerly FASB Staff Position No. 150-5, Issuer’s Accounting Under FASB Statement No. 150 for Freestanding Warrants and Other Similar Instruments with Characteristics of Both Liabilities and Equity, an interpretation of SFAS No. 150, Accounting for Certain Financial Instruments with Characteristics of Both Liabilities and Equity). Under ASC Topic 480, freestanding warrants to purchase shares of convertible preferred stock were classified as liabilities on the balance sheets at fair value because the warrants may conditionally obligate the Company to transfer assets at some point in the future. The warrants were subject to remeasurement at each balance sheet date, and any change in fair value was recognized as a component of other income (expense), net in the statements of operations.
The Company recorded $200,000, $368,000 and ($13,000) in other income (expense), net relating to changes in fair value of all preferred stock warrants during years ended December 31, 2009, 2008 and 2007, respectively. The Company continued to record adjustments to the fair value of the warrants until the closing of the merger transaction on January 30, 2009, when they became warrants to purchase shares of common stock, at which point the warrants were no longer subject to ASC Topic 480. As of January 30, 2009, $400,000, the then-current aggregate fair value of these warrants, was reclassified from a liability to additional paid-in capital, a component of stockholders’ equity (net capital deficiency).
79
Table of Contents
Concentration of Credit Risk
Financial instruments that are potentially subject to concentration of credit risk consist primarily of cash, cash equivalents, and marketable securities. The Company’s investment policy restricts investments to high-quality investments and limits the amounts invested with any one issuer other than U.S. Treasury debt obligations, U.S. agency debt obligations, or Securities and Exchange Commission (“SEC”) registered money market funds. This policy was further amended during 2009 to limit investments to U.S. Treasury debt or SEC registered money market funds effective September 30, 2009. The goals of the investment policy are as follows: preservation of capital, fulfillment of liquidity needs, and fiduciary control of cash and investments. The Company’s exposure to market risk is limited primarily to interest income sensitivity, which is affected by changes in the general level of United States interest rates, particularly because the majority of the Company’s investments are in short-term debt securities.
Recently Adopted Accounting Standards
Effective January 1, 2009, the Company adopted ASC Topic 820 Fair Value Measurements and Disclosures (“ASC Topic 820”) (formerly SFAS No. 157, Fair Value Measurements) for nonfinancial assets and liabilities. The adoption of ASC Topic 820 for nonfinancial assets and liabilities did not have a material effect on the Company’s results of operations and financial condition.
Effective January 1, 2009, the Company adopted ASC Topic 805 Business Combinations (“ASC Topic 805”) (formerly SFAS No. 141R, Business Combinations). ASC Topic 805 amends previous guidance, and retains the purchase method of accounting for acquisitions, but requires a number of changes, including changes in the way assets and liabilities are recognized in purchase accounting. It also changes the recognition of assets acquired and liabilities assumed arising from contingencies, requires the capitalization of in-process research and development at fair value, and requires the expensing of acquisition-related costs as incurred. It also provides disclosure requirements to enable users of the financial statements to evaluate the nature and financial effects of the business combination. The Company has recorded its acquisition of Novacea in accordance with the provision of ASC Topic 805. See Note 2, “Merger Agreement” for further details.
In June 2009, the FASB issued FASB ASC Topic 105 Generally Accepted Accounting Principles, which establishes the FASB ASC as the sole source of authoritative generally accepted accounting principles. Pursuant to the provisions of FASB ASC Topic 105, the Company has updated references to generally accepted accounting principles in its financial statements issued for the period ended December 31, 2009. The adoption of FASB ASC Topic 105 did not impact the Company’s financial position or results of operations.
In September 2009, the FASB Emerging Issues Task Force reached a consensus on ASC Update 2009-13 (“Topic 605-25”) Multiple-Deliverable Revenue Arrangements, or (“ASC Update 2009-13”). ASC Update 2009-13 applies to multiple-deliverable revenue arrangements that are currently within the scope of ASC Topic 605-25. ASC Update 2009-13 provides principles and application guidance on whether multiple deliverables exist and how the arrangement should be separated and the consideration allocated. ASC Update 2009-13 requires an entity to allocate revenue in an arrangement using estimated selling prices of deliverables, if a vendor does not have vendor-specific objective evidence or third-party evidence of selling price. The update eliminates the use of the residual method and requires an entity to allocate revenue using the relative selling price method and also significantly expands the disclosure requirements for multiple-deliverable revenue arrangements. ASC Update 2009-13 should be applied on a prospective basis for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, with earlier application permitted. As a result, ASC Update 2009-13 will be effective for the Company no later than the first quarter of fiscal 2011. The Company is currently evaluating the impact of adopting this ASC Update 2009-13 on its financial position and results of operations.
80
Table of Contents
2. Merger Agreement
As described in Note 1, the Company completed the Merger on January 30, 2009. Pursuant to the Merger, stockholders of TPI exchanged their shares of TPI stock for a total of 7,882,622 shares of Transcept common stock and a total of 156,007 warrants to purchase Transcept common stock. Immediately following the Merger, approximately 61% of the fully-diluted shares of Transcept common stock were owned by former stockholders of TPI. For accounting purposes, TPI was deemed to be the acquiring company, and the Merger was accounted for as a reverse acquisition.
The purchase consideration was approximately $83.1 million. The purchase consideration was determined based on the fair value of the net assets exchanged.
Transcept and Novacea completed the Merger principally to utilize the cash resources held by Novacea to continue the development of the late-stage product candidate held by TPI.
Allocation of total purchase consideration:
Under the purchase method of accounting, the total purchase consideration was allocated to the net tangible and identifiable intangible assets acquired and liabilities assumed based on their fair values as of January 30, 2009. The excess of the purchase price over the fair value of assets acquired and liabilities assumed was allocated to goodwill. The allocation of the purchase price is as follows (in thousands):
| Allocation of Purchase Price |
||||
| Cash, cash equivalents and marketable securities |
$ | 80,861 | ||
| Other assets |
3,794 | |||
| Goodwill |
2,962 | |||
| Existing assumed liabilities |
(1,466 | ) | ||
| Assumed lease liability |
(856 | ) | ||
| Assumed severance, retention and other merger related obligations |
(2,177 | ) | ||
| Total |
$ | 83,118 | ||
Goodwill is derived from the value obtained from the additional resources of the combined company. None of the goodwill will be deductible for tax purposes as the merger was structured as a stock purchase transaction.
Assumed severance, retention and other merger related obligations:
Upon completion of the Merger on January 30, 2009, the Company became liable to pay approximately $2.2 million in payments due to Novacea employees upon a change in control, all of which has been paid as of December 31, 2009. None of these payments required on-going services of the employees subsequent to the change in control.
81
Table of Contents
Pro forma information:
The following unaudited pro forma information presents a summary of the Company’s consolidated results of operations as if the Merger had taken place as of January 1, 2008 (in thousands, except per share information):
| Year Ended December 31, | |||||||
| 2009 | 2008 | ||||||
| Revenue(1) |
$ | 5,208 | $ | 60,621 | |||
| Net income (loss) |
$ | (22,572 | ) | $ | 14,863 | ||
| Pro forma net income (loss) per share: |
|||||||
| Basic |
$ | (1.71 | ) | $ | 1.15 | ||
| Diluted |
$ | (1.71 | ) | $ | 1.11 | ||
| Shares used in computing net income (loss) per share: |
|||||||
| Basic |
13,162 | 12,928 | |||||
| Diluted |
13,162 | 13,383 | |||||
| (1) | Revenue for 2009 related to the Collaboration Agreement between the Company and Purdue (see Note 11). Revenue for 2008 related to the collaboration agreement between Schering Corporation (“Schering”) and Novacea (the “Schering Collaboration Agreement”). Upon termination of the Schering Collaboration Agreement on April 4, 2008, Novacea recognized as revenue during the second quarter of 2008 the previously deferred revenue balance of $52.4 million related to the non-refundable upfront payments. The deferred revenue balance related to the non-refundable upfront payments from Schering was zero as of December 31, 2008. |
The unaudited pro forma results of operations are not necessarily indicative of what would have occurred had the Merger been completed at the beginning of the respective periods or of the results that may occur in the future.
3. Results of Operations
Net Loss Per Share
Basic net loss per share is computed by dividing net loss by the weighted average number of vested shares outstanding during the period. Diluted net loss per share is computed by giving effect to all potential dilutive common securities, including options and common stock subject to repurchase.
The following table presents the calculation of basic and diluted net loss per share (in thousands, except per share amounts):
| 2009 | 2008 | 2007 | ||||||||||
| Numerator: |
||||||||||||
| Loss attributable to common stockholders |
$ | (21,800 | ) | $ | (19,959 | ) | $ | (20,384 | ) | |||
| Denominator: |
||||||||||||
| Weighted average common shares outstanding |
12,205 | 475 | 345 | |||||||||
| Less: Weighted average common shares subject to repurchase |
(39 | ) | (74 | ) | (49 | ) | ||||||
| Denominator for basic and diluted net loss per share |
12,166 | 401 | 296 | |||||||||
| Basic and diluted net loss per share |
$ | (1.79 | ) | $ | (49.77 | ) | $ | (68.86 | ) | |||
82
Table of Contents
The following outstanding shares subject to options and warrants to purchase common stock, common stock subject to repurchase, convertible preferred stock and shares subject to warrants to purchase convertible preferred stock were antidilutive due to a net loss in the periods presented and, therefore, were excluded from dilutive securities computation as of the periods indicated below (in thousands):
| December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Shares subject to options to purchase common stock |
1,718 | 1,066 | 1,062 | |||
| Shares subject to warrants to purchase common stock |
156 | — | 3 | |||
| Common stock subject to repurchase |
22 | 56 | 91 | |||
| Convertible preferred stock (as-converted basis) |
— | 7,350 | 7,350 | |||
| Shares subject to warrants to purchase convertible preferred stock |
— | 156 | 156 | |||
| Total |
1,896 | 8,628 | 8,662 | |||
4. Cash, Cash Equivalents and Marketable Securities
The following is a summary of the fair value of cash, cash equivalents and available-for-sale securities (in thousands):
| December 31, 2009 | ||||||||||||
| Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value | |||||||||
| Cash |
$ | 803 | $ | — | $ | — | $ | 803 | ||||
| Certificates of deposit |
200 | — | — | 200 | ||||||||
| Money market funds |
16,228 | — | — | 16,228 | ||||||||
| U.S. Treasury securities |
71,832 | 39 | — | 71,871 | ||||||||
| $ | 89,063 | $ | 39 | $ | — | $ | 89,102 | |||||
| December 31, 2008 | ||||||||||||
| Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value | |||||||||
| Cash |
$ | 401 | $ | — | $ | — | $ | 401 | ||||
| Corporate notes |
998 | 2 | — | 1,000 | ||||||||
| Certificates of deposit |
200 | — | — | 200 | ||||||||
| Government sponsored enterprise issues |
5,727 | 24 | 5,751 | |||||||||
| Money market funds |
4,031 | — | — | 4,031 | ||||||||
| U.S. Treasury securities |
500 | — | — | 500 | ||||||||
| $ | 11,857 | $ | 26 | $ | — | $ | 11,883 | |||||
During 2009, proceeds from the sales of available-for-sale marketable securities totaled $47,282,000 with realized gains of $111,000. In 2008, proceeds from the sales of available-for-sale marketable securities totaled $3,805,000 with realized losses of $16,000. The amortized cost and estimated fair value of available-for-sale marketable securities at December 31, 2009 and 2008 were as follows (in thousands):
| 2009 | 2008 | |||||||||||
| Cost | Fair value | Cost | Fair value | |||||||||
| Cash equivalents |
$ | 17,031 | $ | 17,031 | $ | 4,432 | $ | 4,432 | ||||
| Marketable securities |
71,832 | 71,871 | 7,225 | 7,251 | ||||||||
| Restricted cash |
200 | 200 | 200 | 200 | ||||||||
| $ | 89,063 | $ | 89,102 | $ | 11,857 | $ | 11,883 | |||||
83
Table of Contents
All of the Company’s investments had contractual maturities of one year or less at December 31, 2009.
5. Fair Value
On January 1, 2008, the Company adopted ASC Topic 820 Fair Value Measurements and Disclosures (formerly SFAS No. 157) as it applies to its financial assets and financial liabilities. ASC Topic 820 defines fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. Fair value is defined as the estimated exit price received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date rather than on an entry price which represents the purchase price of an asset or liability. ASC Topic 820 establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
| • | Level 1—Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| • | Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| • | Level 3—Unobservable inputs (i.e. inputs that reflect the reporting entity’s own assumptions about the assumptions that market participants would use in estimating the fair value of an asset or liability) are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
A financial instrument’s categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Where quoted prices are available in an active market, securities are classified as Level 1 of the valuation hierarchy. Level 1 securities include highly liquid money market funds. If quoted market prices are not available for the specific security, then the Company estimates fair value by using pricing models, quoted prices of securities with similar characteristics or discounted cash flows. Level 2 instruments include commercial paper, U.S. corporate debt, and U.S. government sponsored enterprise issues. Level 3 liabilities that are measured at fair value on a recurring basis consist of convertible preferred stock warrant liabilities. The fair values of the outstanding preferred stock warrants are measured using the Black-Scholes option-pricing model. Inputs used to determine fair market value include estimated market value of the underlying convertible preferred stock at the valuation measurement date, the remaining contractual term of the warrant, risk-free interest rates and expected dividends on and expected volatility of the price of the underlying convertible preferred stock.
In accordance with ASC Topic 820, the following table represents the Company’s fair value hierarchy for its financial assets (cash equivalents and marketable securities) and liabilities (convertible preferred stock warrant liabilities) measured at fair value on a recurring basis as of December 31, 2009 (in thousands):
| December 31, 2009 |
Fair Value Measurements at Reporting Date Using | |||||||||||
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) | ||||||||||
| Assets |
||||||||||||
| Certificates of deposit |
$ | 200 | $ | 200 | $ | — | $ | — | ||||
| Money market funds |
16,228 | 16,228 | — | — | ||||||||
| U.S. Treasury securities |
71,871 | — | 71,871 | — | ||||||||
| $ | 88,299 | $ | 16,428 | $ | 71,871 | $ | — | |||||
| Liabilities |
||||||||||||
| Convertible preferred stock warrant liabilities |
$ | — | $ | — | $ | — | $ | — | ||||
84
Table of Contents
The following table represents the Company’s fair value hierarchy for its financial assets (cash equivalents and marketable securities) and liabilities (convertible preferred stock warrant liabilities) measured at fair value on a recurring basis as of December 31, 2008 (in thousands):
| December 31, 2008 |
Fair Value Measurements at Reporting Date Using | |||||||||||
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) | ||||||||||
| Assets |
||||||||||||
| Certificates of deposit |
$ | 200 | $ | 200 | $ | — | $ | — | ||||
| Money market funds |
4,031 | 4,031 | — | — | ||||||||
| U.S. corporate debt |
1,000 | — | 1,000 | — | ||||||||
| U.S. government sponsored enterprise issues |
5,751 | — | 5,751 | — | ||||||||
| U.S. Treasury securities |
500 | — | 500 | — | ||||||||
| $ | 11,482 | $ | 4,231 | $ | 7,251 | $ | — | |||||
| Liabilities |
||||||||||||
| Convertible preferred stock warrant liabilities |
$ | 600 | $ | — | $ | — | $ | 600 | ||||
The following table sets forth the changes in the fair value of the Company’s Level 3 financial liabilities (convertible preferred stock warrant liabilities), which were measured at fair value on a recurring basis for the period ended December 31, 2009 (in thousands):
| Fair value as of December 31, 2008 |
$ | 600 | ||
| Change in fair value |
(200 | ) | ||
| Transfer to equity upon conversion of preferred |
(400 | ) | ||
| Fair value as of December 31, 2009 |
$ | — | ||
The decrease in fair value of the convertible preferred stock warrant liabilities of $200,000 was recognized in other income (expense), net in the statements of operations. Concurrent with the Merger as described in Note 1 above, the warrants to purchase preferred stock were converted into warrants to purchase common stock with the same exercise prices and expiration dates, and the aggregate fair value of these warrants was reclassified from a liability to additional paid-in capital, a component of stockholders’ equity (net capital deficiency).
During the years ended December 31, 2009 and 2008, there were no significant changes to the valuation models used for purposes of determining the fair value of Level 2 assets and Level 3 liabilities. No other assets and liabilities were carried at fair value as of December 31, 2009.
6. Property and Equipment, Net
Property and equipment consisted of the following (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Computer equipment and software |
$ | 617 | $ | 585 | ||||
| Furniture and fixtures |
559 | 557 | ||||||
| Research equipment |
779 | 778 | ||||||
| Leasehold improvements |
624 | 624 | ||||||
| Construction in progress |
— | 6 | ||||||
| 2,579 | 2,550 | |||||||
| Less accumulated depreciation and amortization |
(1,527 | ) | (1,100 | ) | ||||
| Property and equipment, net |
$ | 1,052 | $ | 1,450 | ||||
85
Table of Contents
The Company recorded depreciation and amortization expense of $550,000, $505,000 and $379,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
7. Loans and Security Agreement
In February 2006, the Company entered into a Loan and Security Agreement (the “Agreement”) with Hercules Technology Growth Capital (“Hercules”). Under the terms of the Agreement, the Company was initially entitled to draw up to $4.0 million. This amount was raised to $10.0 million upon reaching certain development milestones, which were achieved in October 2006. Interest under the loan was fixed at prime plus 2.69% at the date of the initial draw. The Company drew down an advance of $4.0 million on May 31, 2006, and drew down the remaining available advance of $6.0 million on December 28, 2006, against which interest accrued at rates of 10.69% and 10.94%, respectively. The draw required interest only repayment for the period from initial borrowing to December 31, 2006. Principal and interest repayment commenced in January 2007, to be continued for 33 months. Under the terms of the Agreement, the Loan was secured by a perfected first priority security interest in all of the Company’s tangible and intangible assets owned or subsequently acquired, except for intellectual property. On February 3, 2009, the Company repaid the remaining outstanding principal and interest under this loan, in the amount of $2,763,000, which included a 2% prepayment charge. The prepayment charge of $54,000 was included in interest expense during the first quarter of 2009.
In connection with the Agreement, the Company was required to pay approximately $123,000 in facility and other fees. These fees were capitalized in other assets and were being amortized to interest expense over the term of the loan. In conjunction with the full repayment of the loan in February 2009, as noted above, the remaining facility and other fees of $28,000 were amortized to interest expense during the first quarter of 2009. Amortization expense was $28,000, $37,000 and $37,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
In addition, in connection with the Agreement in May and December 2006, the Company issued warrants to purchase 61,452 shares of Series C convertible preferred stock at an exercise price of $8.136 per share. These warrants were valued using the Black-Scholes valuation model, and the resulting estimated fair value of the warrants at the date of issuance was $440,000, which was recorded as a debt discount to the credit facility in 2006. The discount was amortized to interest expense over the repayment period. In conjunction with the full repayment of the loan in February 2009, as noted above, the remaining debt discount of $47,000 was charged to interest expense during the first quarter of 2009. Interest expense was $179,000, $707,000 and $1,152,000 for the years ended December 31, 2009, 2008 and 2007, respectively, of which $47,000, $151,000 and $216,000 related to amortization of the debt discount.
The Agreement did not have financial covenants. See Note 10 for discussion of the assumptions and methodology used to estimate the fair value of the warrant.
As discussed in Note 8, the Company leased additional office space in 2007. Under the terms of the agreement, the landlord agreed to finance approximately $258,000 of the costs for tenant improvements made to the space. The loan is being repaid at an interest rate of 8.25% over the period from September 2007 through May 2013.
86
Table of Contents
At December 31, 2009, future minimum principal payments related to long-term debt were as follows (in thousands):
| Year ending December 31, |
||||
| 2010 |
$ | 57 | ||
| 2011 |
57 | |||
| 2012 |
57 | |||
| 2013 |
24 | |||
| 2014 |
— | |||
| Thereafter |
— | |||
| 195 | ||||
| Less: Interest |
(25 | ) | ||
| Minimum principal payments |
170 | |||
| Less: Current minimum principal payments |
(45 | ) | ||
| Long-term minimum principal payments |
$ | 125 | ||
8. Commitments and Contingencies
Leases
In February 2006, the Company signed an operating lease for its corporate offices that included approximately 11,600 square feet of office and laboratory space in Point Richmond, California. The lease term is for seven years, commencing on June 1, 2006. In June 2007, the Company amended this operating lease to add approximately 3,000 square feet of additional office space. The lease term of this amendment coincides with the original lease agreement, with a separate commencement date of September 12, 2007. As part of this amendment, the landlord agreed to contribute $60,000 toward the costs of tenant improvements for the additional space. This landlord contribution is being amortized on a straight-line basis over the term of the lease as a reduction to rent expense.
On February 20, 2009, the Company signed an operating lease for 12,257 square feet of general office space in Point Richmond, California. The lease term commenced in March 2009 and terminates on May 31, 2013, with an option to renew for an additional five years. Under the terms of the lease, the Company has the option of accelerating the termination date of the lease to May 31, 2011. Total base rent payable by the Company from commencement of the lease through the end of the first term of the lease is approximately $0.8 million. In conjunction with the restructuring described in Note 12 below, the Company vacated this property in August 2009 and has recorded a charge to rent expense of $309,000 related to the fair value of the remaining lease payments reduced by estimated sublease income. This liability is being amortized using the effective interest method over the remaining life of the lease.
In June 2007, Novacea entered into an operating lease for 25,288 square feet for corporate facilities located in South San Francisco, California. The lease for the facilities is non-cancelable and has a five-year term with a total obligation of $3.6 million. The lease provides for periodic rent increases based upon previously negotiated or consumer price indexed adjustments, or in the case of an extension, market adjusted rates. As of December 31, 2009, the Company maintained a Certificate of Deposit acting as a security deposit of $770,000 required under conditions of the lease, which was recorded as a noncurrent asset on the Company’s balance sheet. On March 25, 2009, the Company entered into a sublease agreement dated as of March 24, 2009 for 18,368 square feet of the 25,288 square feet located in South San Francisco. The term of the sublease commenced on June 1, 2009 and ends on October 31, 2012. Total base rent payable by the sublessee through the end of the term of the sublease is approximately $1.1 million. In connection with this sublease, on April 6, 2009 the Company received an irrevocable standby letter of credit in the amount of $100,000, expiring May 31, 2010, as a security deposit. On June 16, 2009, the Company entered into a sublease agreement dated for reference purposes as of June 11, 2009 for the remaining 6,920 square feet of the South San Francisco facility. The term of the sublease commenced on July 1, 2009 and ends on October 31, 2012. Total base rent payable by the sublessee through the end of the term of the sublease is approximately $0.4 million.
87
Table of Contents
Future minimum payments under these leases as of December 31, 2009 are as follows (in thousands):
| Year ending December 31, |
|||
| 2010 |
$ | 1,209 | |
| 2011 |
1,254 | ||
| 2012 |
1,094 | ||
| 2013 |
220 | ||
| 2014 |
— | ||
| Thereafter |
— | ||
| Total |
$ | 3,777 | |
In addition, as noted above, The Company has subleased certain of these facilities under operating leases to third parties. The future minimum sublease payments due from lessees under those arrangements are $433,000 in 2010, $493,000 in 2011 and $422,000 in 2012.
Rent expense for the years ended December 31, 2009, 2008 and 2007 was $986,000, $278,000 and $232,000, respectively. Sublease income for the year ended December 31, 2009 was $123,000. There was no sublease income for the years ended December 31, 2008 or 2007.
Purchase Commitments
Pursuant to the terms of an agreement with Plantex USA Inc., the Company is obligated to purchase $350,000 worth of zolpidem tartrate during 2010.
Indemnity Agreements
The Company indemnifies its officers and directors for certain events or occurrences, subject to certain limits. The Company believes the fair value of these indemnification agreements is minimal. Accordingly, the Company has not recognized any liabilities relating to these agreements as of December 31, 2009.
Legal Proceedings
From time to time, we may be involved in litigation relating to claims arising out of our operations. Transcept is not currently involved in any material legal proceedings.
SPI Pharmaceuticals, Inc., the sole supplier of Pharmaburst®, a key excipient used in Intermezzo®, is the defendant in a lawsuit brought by Roquette Frères, or Roquette, in the Federal District Court of Delaware on August 31, 2006 that alleges that certain of SPI’s products infringe one or more claims of a Roquette patent and seeks monetary damages and injunctive relief. Transcept has not been named in, and is not a party to, the lawsuit. Although not specifically identified in the original complaint, press releases have indicated that Pharmaburst® products are among the accused products. SPI has informed us that Roquette’s patent rights are not infringed by Pharmaburst® and that they intend to defend their rights vigorously. Because Pharmaburst® is a key excipient in Intermezzo®, the interruption of supply of Pharmaburst® through an injunction could materially harm our ability to supply Intermezzo® and our business. SPI has agreed to indemnify us against certain damages related to any such infringement suit under the terms of our agreement with SPI.
88
Table of Contents
9. Accrued Liabilities
Accrued liabilities consist of the following (in thousands):
| December 31, | ||||||
| 2009 | 2008 | |||||
| Accrued payroll and related |
$ | 407 | $ | 449 | ||
| Accrued vacation pay |
157 | 133 | ||||
| Accrued professional fees |
478 | 290 | ||||
| Accrued merger related costs |
— | 221 | ||||
| Accrued tooling charges |
812 | — | ||||
| Accrued franchise taxes—Delaware |
117 | — | ||||
| Other accrued liabilities |
412 | 375 | ||||
| $ | 2,383 | $ | 1,468 | |||
10. Warrant liability
In conjunction with the sale of the 2005 subordinated convertible promissory notes, the Company issued warrants for the purchase of 94,554 shares of Series C convertible preferred stock at $8.136 per share in October 2005. These warrants have a contractual life of seven years. The fair value of these warrants was determined to be $535,000 at the date of issuance using the Black-Scholes Merton option valuation model with the following assumptions: a risk-free interest rate of 4.40%; no dividend yield; expected volatility of 70%; and an expected life of seven years.
In addition, the Company issued warrants to purchase 24,581 and 36,872 shares of Series C convertible preferred stock at an exercise price of $8.136 per share in May and December 2006, respectively. The aggregate fair value of these warrants was determined to be $440,000 at the dates of issuance using the Black-Scholes Merton option valuation model with the following assumptions: risk-free interest rates from 4.7% – 5.1%; no dividend yield; expected volatilities from 61% – 62%; and remaining contractual lives of 9.3 – 9.9 years.
Pursuant to ASC Topic 480, all preferred stock warrants were recorded as liabilities at the time of issuance and remeasured to current fair value at each reporting date.
In conjunction with the closing of the merger transaction on January 30, 2009, all outstanding warrants became warrants to purchase shares of common stock, at which point the warrants were no longer subject to ASC Topic 480. As of January 30, 2009, $400,000, the then-current aggregate fair value of these warrants, was reclassified from a liability to additional paid-in capital, a component of stockholders’ equity (net capital deficiency).
At January 30, 2009 and December 31, 2008 and 2007, the fair value of all outstanding Series C convertible preferred stock warrants was remeasured based on the then-current reassessed fair value of the Company’s convertible preferred stock and the assumptions in the following table:
| Period Ended January 30, 2009 |
Year Ended December 31, | |||||
| 2008 | 2007 | |||||
| Risk free interest rate |
1.32 - 2.27% | 1.87 - 2.25% | 3.70 - 3.85% | |||
| Remaining contractual terms |
3.8 - 7.2 years | 3.8 - 7.3 years | 4.8 - 8.3 years | |||
| Volatility |
79 - 92% | 79 - 92% | 61% | |||
The aggregate fair value of all outstanding Series C warrants was determined to be $600,000 and $968,000 as of December 31, 2008 and 2007, respectively. Net changes in fair value of $200,000, $368,000 and ($13,000) have been included in other income (expense), net for the years ended December 31, 2009, 2008 and 2007, respectively.
89
Table of Contents
11. Collaboration Agreement
On July 31, 2009, the Company signed the United States License and Collaboration Agreement (“Collaboration Agreement”) with Purdue Pharmaceutical Products L.P. (“Purdue”) that provides an exclusive license to Purdue to commercialize Intermezzo® in the United States and pursuant to which:
| • | On August 4, 2009, Purdue paid the Company a $25.0 million non-refundable license fee; |
| • | The Company is obligated to seek FDA approval of Intermezzo® and to continue development of Intermezzo® at its expense until FDA approval; and |
| • | If Purdue elects not to terminate the collaboration after its review of an FDA approval of Intermezzo®: |
| • | Purdue is obligated to pay the Company up to $30.0 million, which amount is reduced by $2.0 million for each 30-day period that the Company’s receipt of an NDA approval for Intermezzo® is delayed beyond June 30, 2010; |
| • | The Company is obligated to transfer the Intermezzo® NDA to Purdue and Purdue is obligated to assume the expense associated with maintaining the NDA and further development of Intermezzo® in the United States, including any expense associated with post-approval studies; |
| • | Purdue is obligated to commercialize Intermezzo® in the United States at its expense; |
| • | Purdue is obligated to pay the Company tiered double-digit base royalties on net sales of Intermezzo® in the United States ranging up to the mid-twenty-percent level; |
| • | Purdue is obligated to pay the Company $10.0 million if either of two formulation patents are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book; and |
| • | Purdue is potentially obligated to pay the Company up to an additional $80.0 million upon meeting certain intellectual property milestones and upon the achievement of certain net sales targets for Intermezzo® in the United States. |
The Company retained an option to co-promote Intermezzo® to psychiatrists in the United States as early as the first anniversary of commercial launch of Intermezzo® and upon entry into the market under the co-promotion option, the Company would receive an additional double-digit royalty from Purdue on sales generated by psychiatrists in the United States.
The Company also granted Purdue and an associated company the right to negotiate for the commercialization of Intermezzo® in Mexico and Canada, respectively, and retained rights to commercialize Intermezzo® in the rest of the world.
The Company’s co-promote option may be terminated by Purdue upon acquisition of the Company or in the event of entry of generic competition to Intermezzo®. The royalty payments discussed above are subject to reduction in connection with, among other things, the entry of generic competition to Intermezzo®.
The Company is recognizing revenue from the $25.0 million non-refundable license fee ratably over an estimated 24-month period starting August 1, 2009 and ending on July 31, 2011. This period represents the estimated period for which it has significant participatory obligations under the Collaboration Agreement. The revenue recognized in connection with the license fee in 2009 was $5.2 million.
12. Restructuring
On August 17, 2009, the Company implemented a reduction of approximately 30% of the Company’s workforce. Affected employees were notified on August 17, 2009. The reduction plan carried out a realignment of the Company’s workforce and operations after a reassessment of the Company’s development activities and
90
Table of Contents
corporate objectives in connection with the Company’s recent entry into the Collaboration Agreement described in Note 11 above. The Company substantially completed the reduction plan at December 31, 2009. Employees subject to the workforce reduction plan were eligible for one-time severance benefits that included severance pay of approximately $511,000 in total and one year accelerated vesting on outstanding options upon signing a separation and release agreement with the Company. Further, the affected employees were given the choice to extend the exercise period of their options to one year following termination. Total expense related to the modification of these stock option awards is expected to be approximately $79,000.
Additionally, the Company recorded a charge of approximately $309,000 related to the fair value of the remaining lease payments on 12,257 square feet of general office space in Point Richmond, California. This liability will be amortized over the remaining life of the lease, which expires on May 31, 2013.
The Company records restructuring activities in accordance with ASC topic 420 Exit or Disposal Cost Obligations (formerly SFAS 146, Accounting for Costs Associated with Exit or Disposal Activities.). The following tables summarize the charges recorded during the year ended December 31, 2009 related to the restructuring plan by type of activity (no such charges were recorded in the comparable prior year periods) (in thousands).
| Year ended December 31, 2009 |
Severance benefits |
Stock option modification |
Contract termination costs |
Total | ||||||||
| Research and development |
$ | 205 | $ | 24 | $ | — | $ | 229 | ||||
| General and administrative |
272 | 41 | 309 | 622 | ||||||||
| $ | 477 | $ | 65 | $ | 309 | $ | 851 | |||||
Since the inception of its restructuring plan through December 31, 2009, the Company has incurred approximately $477,000 of the estimated $511,000 of severance benefits expected to be incurred (in thousands).
| Severance benefits |
||||
| Restructuring liability as of January 1, 2009 |
$ | — | ||
| Expense |
477 | |||
| Payments |
(361 | ) | ||
| Restructuring liability as of December 31, 2009 |
$ | 116 | ||
The severance benefit liability is included in accrued liabilities while the contract termination costs are included in the lease liability in the balance sheet.
13. Convertible Preferred Stock
In connection with the Company’s merger with Novacea on January 30, 2009, all shares of convertible preferred stock were converted to common stock.
14. Stockholders’ Equity
Capital Stock
The authorized capital stock of the Company consists of 100,000,000 shares of common stock, par value $0.001 per share and 5,000,000 shares of preferred stock, par value $0.001 per share. There are no shares of preferred stock issued or outstanding and the Company has no present plans to issue any shares of preferred stock.
91
Table of Contents
Stock Options
Various employees, directors and consultants have been granted options to purchase common shares under equity incentive plans adopted in 2001, 2002 and 2006 (the “2001 Plan”, the “2002 Plan” and the “2006 Plan”). The 2001 Plan provided for the granting of incentive and non-statutory stock options to employees, officers, directors, and non-employees of the Company. The 2002 Plan provided for the granting of incentive and non-statutory stock options to employees, officers, directors, and consultants of the Company. Incentive stock options under all of these plans may be granted with exercise prices of not less than estimated fair value, and non-statutory stock options may be granted with an exercise price of not less than 85% of the estimated fair value of the common stock on the date of grant. Stock options granted to a stockholder owning more than 10% of voting stock of the Company must have an exercise price of not less than 110% of the estimated fair value of the common stock on the date of grant. The Company estimated the fair value of common stock until the Company became publicly traded. Stock options are generally granted with terms of up to ten years and vest over a period of four years. At December 31, 2009, there were no shares available for future grant under either the 2001 or the 2002 Plans.
The 2006 Plan became effective upon the completion of the Company’s initial public offering in 2006, and will terminate on the earlier of (i) ten years after its approval by the Company’s stockholders or (ii) when the Company’s compensation committee, with the approval of the Company’s board of directors, terminates the 2006 Plan. The 2006 Plan provides for the granting of incentive stock options, non-statutory stock options, restricted stock, performance share awards, performance stock units, dividend equivalents, restricted stock units, stock payments, deferred stock, performance-based awards and stock appreciation rights. The employee stock options generally vest over four years, are exercisable over a period not to exceed the contractual term of ten years from the date the stock options are issued and are granted at prices equal to the fair value of the Company’s common stock on the grant date. Stock option and restricted stock unit exercises are settled with newly issued common stock from the 2006 Plan’s previously authorized and available pool of shares. A total of 500,000 shares of common stock was originally authorized for issuance pursuant to the 2006 Plan, plus the number of shares of the Company’s common stock available for issuance under the 2001 Plan that are not subject to outstanding options, as of the effective date of the 2006 Plan (including shares that are subject to stock options outstanding under the 2001 Plan that expire, are cancelled or otherwise terminate unexercised, or shares that otherwise would have reverted to the share reserve of the 2001 Plan following the effective date of the 2006 Plan). In addition, the number of shares of common stock reserved for issuance under the 2006 Plan increases automatically on the first day of each fiscal year, beginning in 2007, by a number of shares equal to the least of: (i) 4.5% of shares of the Company’s common stock outstanding on a fully diluted basis on such date; (ii) 400,000 shares; or (iii) a smaller number determined by the Company’s board of directors. This provision resulted in an additional 400,000, 258,344 and 264,187 of the Company’s common stock becoming available for issuance on January 1, 2010, 2009 and 2008, respectively under the 2006 Plan.
At December 31, 2009, stock options to purchase 1,096,265 shares of common stock were vested and exercisable and 337,481 shares remain available for future grant under the 2006 Plan.
92
Table of Contents
The following table summarizes the Company’s stock option activity and related information through December 31, 2009:
| Options Outstanding | |||||||||
| Number of Shares Available for Grant |
Number of Shares |
Weighted-Average Exercise Price Per Share | |||||||
| Balance at December 31, 2006 |
152,572 | 609,992 | $ | 0.870 | |||||
| Options authorized |
607,762 | — | |||||||
| Options granted |
(650,060 | ) | 650,060 | $ | 1.797 | ||||
| Options exercised |
— | (176,164 | ) | $ | 1.238 | ||||
| Options forfeited |
21,853 | (21,853 | ) | $ | 1.160 | ||||
| Balance at December 31, 2007 |
132,127 | 1,062,035 | $ | 1.373 | |||||
| Options authorized |
28,267 | — | |||||||
| Options granted |
(173,477 | ) | 173,477 | $ | 5.526 | ||||
| Options exercised |
— | (63,441 | ) | $ | 1.061 | ||||
| Options forfeited |
106,151 | (106,151 | ) | $ | 1.274 | ||||
| Balance at December 31, 2008 |
93,068 | 1,065,920 | $ | 2.080 | |||||
| Options authorized |
258,344 | — | |||||||
| Options granted |
(553,016 | ) | 553,016 | $ | 4.009 | ||||
| Options exercised |
— | (292,760 | ) | $ | 1.313 | ||||
| Options forfeited |
125,571 | (125,571 | ) | $ | 9.613 | ||||
| 2002 Plan shares expired |
(150,576 | ) | — | ||||||
| Acquired in the merger |
564,090 | 517,653 | $ | 19.627 | |||||
| Balance at December 31, 2009 |
337,481 | 1,718,258 | $ | 7.494 | |||||
The total intrinsic value of options exercised during the years ended December 31, 2009 and 2008 was $2,420,000 and $281,000, respectively. The amount of cash received from exercise of stock options during the years ended December 31, 2009 and 2008 was $385,000 and $67,000, respectively.
Additional information related to the status of options at December 31, 2009 is as follows:
| Shares | Weighted- Average Exercise Price Per Share |
Weighted- Average Remaining Contractual Life (Years) |
Aggregate Intrinsic Value (in thousands) | |||||||
| Outstanding |
1,718,258 | $ | 7.494 | 6.46 | $ | 4,917 | ||||
| Vested and exercisable |
1,096,265 | $ | 9.706 | 5.20 | $ | 2,872 | ||||
The intrinsic value of options is the estimated fair value of the stock less the per share exercise price of the option multiplied by the number of shares.
As of December 31, 2009 and 2008, there were 22,078 and 55,855 restricted common shares outstanding subject to repurchase rights held by the Company. In accordance with ASC Topic 718, Compensation—Stock Compensation, the Company has shown $41,000 and $88,000 received as a liability in the balance sheet as of December 31, 2009 and 2008, respectively, and has not shown these shares as outstanding as of December 31, 2009 or 2008. These shares are subject to repurchase upon termination of the stockholders’ services to the Company and are subject to repurchase at the original issuance price. The Company’s right to repurchase these shares lapses at a rate of 2.08% per month.
93
Table of Contents
The following tables summarize information about stock options outstanding as of December 31, 2009:
| Range of Exercise Prices |
Number Outstanding |
Number Exercisable |
Weighted-Average Remaining contractual Life (Years) | |||
| $ 0.7075–$ 0.8844 |
212,557 | 211,966 | 5.76 | |||
| $1.7688 |
367,492 | 237,431 | 7.09 | |||
| $2.1225–$4.0328 |
239,988 | 51,218 | 8.78 | |||
| $4.1400 |
304,748 | 73,952 | 9.03 | |||
| $ 4.5000–$14.0000 |
253,660 | 181,885 | 6.12 | |||
| $14.0500–$15.1500 |
176,000 | 176,000 | 1.22 | |||
| $26.2500–$30.8500 |
94,233 | 94,233 | 4.70 | |||
| $31.0500–$32.5000 |
69,580 | 69,580 | 2.82 | |||
| 1,718,258 | 1,096,265 | 6.46 |
Stock Compensation Plans
The Company has recorded compensation expense for employee stock-based awards of approximately $1,057,000, $613,000 and $470,000 during 2009, 2008 and 2007, respectively.
The following table shows the weighted-average assumptions used to compute the share-based compensation costs for the stock options granted during the years ended December 31, 2009, 2008 and 2007 using the Black-Scholes option pricing model:
| Year Ended December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Risk free interest rate |
1.79 - 2.90% | 2.80 -3.49% | 4.57% | |||
| Expected life of the options |
5.27 - 6.08 years | 6.08 years | 6.02 - 6.08 years | |||
| Dividend yield |
None | None | None | |||
| Volatility |
82.82 - 90.25% | 70.29 - 77.59% | 61.17 - 61.38% | |||
The risk-free interest rate assumption was based on the United States Treasury’s rates for U.S. Treasury zero-coupon bonds with maturities similar to those of the expected term of the award being valued. The assumed dividend yield was based on the Company’s expectation of not paying dividends in the foreseeable future. The weighted-average expected life of the options was calculated using the simplified method as prescribed by the Securities and Exchange Commission (“SEC”) Staff Accounting Bulletin No. 107 and No. 110 (“SAB No. 107 and 110”). This decision was based on the lack of relevant historical data due to the Company’s limited historical experience. In addition, due to the Company’s limited historical data, the estimated volatility also reflects the application of SAB No. 107 and 110, incorporating the historical volatility of comparable companies whose share prices are publicly available.
The weighted-average grant-date fair value of stock options granted to employees during the years ended December 31, 2009, 2008 and 2007 was $2.894, $4.140 and $1.797 per share, respectively. As of December 31, 2009, there is approximately $1,888,000 of total unrecognized compensation cost related to the unvested share-based compensation arrangements granted under the Company’s equity incentive plan. The remaining unrecognized compensation cost will be recognized over a weighted-average period of 2.47 years.
As discussed in Note 1, the Company accounts for stock options granted to persons other than employees or directors at fair value using the Black-Scholes Merton option-pricing model in accordance with ASC Topic 505, subtopic 50 Equity-Based Payments to Non-Employees (formerly EITF Issue No. 96-18, Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services). Stock options granted to such persons and stock options that are modified and continue to vest when an
94
Table of Contents
employee has a change in employment status are subject to periodic revaluation over their vesting terms. The Company recognizes the resulting stock-based compensation expense during the service period over which the nonemployee provides services to the Company. In connection with the issuance of options to purchase shares of common stock to nonemployees, the Company recorded total stock-based compensation within stockholders’ equity totaling approximately $121,000, $24,000 and $13,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
During 2009, the Company granted 30,000 options to purchase shares of common stock to one nonemployee that vest over four years, with an exercise price of $2.96 per share. During 2008, the Company granted 3,516 options to purchase shares of common stock to four nonemployees that vest over four years, with an exercise price of $4.033 per share. During 2007, the Company granted 2,120 options to purchase shares of common stock to one nonemployee that vest over four years, with an exercise price of $1.769 per share. The following table shows the weighted-average assumptions used to compute the share-based compensation costs for stock options granted to nonemployees during the years ended December 31, 2009, 2008 and 2007 using the Black-Scholes Merton option pricing model:
| Year Ended December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Risk free interest rate |
2.28 - 3.85% | 1.89 - 3.98% | 4.71 - 4.72% | |||
| Expected life of the options |
6.25 - 9.92 years | 7.20 - 9.93 years | 8.21 - 9.26 years | |||
| Dividend yield |
None | None | None | |||
| Volatility |
80.47 - 88.36% | 73.97 - 85.11% | 65.40 - 66.29% | |||
Modification of Employee Stock-Based Awards
During the year ended December 31, 2009, the Company modified stock options of twelve of its employees in conjunction with their termination, primarily associated with those employees affected by the reduction-in-force described in Note 12. The modifications included accelerated vesting on certain options and extension of the exercise period after termination on certain of the options. These modifications resulted in additional compensation expense of $127,000 recorded in the year ended December 31, 2009. The Company accounted for the modifications of stock option awards in accordance with ASC Topic 718.
Employee Stock Purchase Plan
On June 3, 2009, at the annual meeting of stockholders, the stockholders of the Company approved the 2009 Employee Stock Purchase Plan (“ESPP”). The number of shares available for issuance over the term of the ESPP is limited to 500,000 shares. The ESPP is designed to allow eligible employees of the Company to purchase shares of common stock through periodic payroll deductions. The price of common stock purchased under the ESPP is equal to 85% of the lower of the fair market value of the common stock on the commencement date of each offering period or the specified purchase date. The first offering period began July 16, 2009 and ended on November 30, 2009 and resulted in the purchase of 22,060 of the Company’s common stock, leaving 477,940 shares available for issuance.
The weighted average assumptions used to value purchases under the ESPP plan during 2009 were as follows: risk free interest rate – 0.15% to 0.24%; expected life – 0.38 to 0.50 years; dividend yield – 0; and volatility – 75.48% to 122.57%. The risk-free interest rate assumption was based on the United States Treasury’s rates for U.S. Treasury zero-coupon bonds with maturities similar to those of the expected term of the award being valued. The assumed dividend yield was based on the Company’s expectation of not paying dividends in the foreseeable future. The weighted-average expected life is based on the duration of time in the purchasing period. In addition, due to the Company’s limited historical data, the estimated volatility also reflects the application of SAB No. 107 and 110, incorporating the historical volatility of comparable companies whose share prices are publicly available. The weighted average grant date fair value per share was $2.235. The Company has recorded compensation expense for employee stock-based purchase plan awards of approximately $62,000 during 2009.
95
Table of Contents
Reserved Shares
At December 31, 2009, the Company has reserved shares of common stock for future issuance as follows:
| Number of Shares | ||
| Employee stock purchase plan |
477,940 | |
| Stock option plans: |
||
| Subject to outstanding options |
1,718,258 | |
| Available for future grants |
337,481 | |
| Warrants |
156,007 | |
| Total |
2,689,686 | |
15. Income taxes
There is no provision for income taxes because the Company has incurred operating losses since inception. Income tax expense (benefit) differed from the amounts computed by applying the U.S. federal income tax rate of 35% to pretax losses from operations as a result of the following (in thousands):
| For the year ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Computed tax benefit at federal statutory rate |
$ | (7,630 | ) | $ | (6,979 | ) | $ | (7,135 | ) | |||
| State tax benefit, net of effect on Federal income taxes |
(1,253 | ) | (1,145 | ) | (1,166 | ) | ||||||
| State tax credits, net of Federal benefit |
(154 | ) | (161 | ) | (148 | ) | ||||||
| Federal tax credits |
(298 | ) | (301 | ) | (217 | ) | ||||||
| Permanent differences: |
||||||||||||
| Nondeductible stock option expense |
231 | 188 | 165 | |||||||||
| Merger related costs |
1,467 | — | — | |||||||||
| State tax effect from permanent differences |
272 | — | — | |||||||||
| Other |
(70 | ) | 26 | 31 | ||||||||
| Change in valuation allowance |
6,188 | 8,318 | 8,346 | |||||||||
| IRS section 382 NOL limitation |
1,196 | — | — | |||||||||
| Other, net |
51 | 54 | 124 | |||||||||
| Total tax expense |
$ | — | $ | — | $ | — | ||||||
Deferred income taxes reflect the net tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets are as follows (in thousands):
| December 31, | ||||||
| 2009 | 2008 | |||||
| Deferred tax assets: |
||||||
| Net operating loss carryforwards |
$ | 27,220 | $ | 24,652 | ||
| Depreciation |
46 | 19 | ||||
| Research and development credits |
1,467 | 1,347 | ||||
| Capitalized research and development expense |
3,764 | — | ||||
| Other |
800 | 1,091 | ||||
| 33,297 | 27,109 | |||||
| Valuation allowance |
33,297 | 27,109 | ||||
| Total deferred tax assets |
— | — | ||||
| Deferred tax liabilities: |
||||||
| Depreciation |
— | — | ||||
| Net deferred tax assets |
$ | — | $ | — | ||
96
Table of Contents
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $6,188,000 during 2009 and $8,318,000 during 2008.
As of December 31, 2009, the Company had federal net operating loss carryforwards of approximately $68,920,000, which expire in the years 2022 to 2029 if not utilized. The Company had net operating loss carryforwards for state income tax purposes of $53,918,000 which expire in the years 2012 to 2029 if not utilized.
The Company has carryforwards from the federal Credit for Increasing Research Expenditures of approximately $797,000, which expire in years 2023 through 2029. The Company also has state credit carryforwards of approximately $1,032,000 that carry forward indefinitely.
As a result of certain realization requirements of ASC Topic 718, the table of deferred tax assets and liabilities shown above does not include certain deferred tax assets at December 31, 2009 that arose directly from tax deductions related to equity compensation in excess of compensation recognized for financial reporting purposes. Equity will be increased by approximately $64,000 if and when such deferred tax assets are ultimately realized.
Utilization of the net operating loss and tax credit carryforwards may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986, as amended, and similar state provisions. The annual limitation may result in the expiration of net operating losses and credits before utilization.
The Company adopted ASC Topic 740, subtopic 10-50-15, Unrecognized Tax Benefit Related Disclosures (formerly FASB Interpretation 48, Accounting for Uncertainty in Income Taxes) on January 1, 2007.
The following table summarizes the activity related to our unrecognized tax benefits (in thousands):
| 2009 | 2008 | |||||||
| Balance as of January 1, |
$ | 1,312 | $ | 1,335 | ||||
| Decreases related to prior year tax positions |
(1,312 | ) | (38 | ) | ||||
| Increases related to current year tax positions |
— | 15 | ||||||
| Balance as of December 31, |
$ | — | $ | 1,312 | ||||
The beginning balance of unrecognized tax benefits from Novacea was related to Research and Development credits. As a result of the merger, those credits were limited. Therefore, the balance related to prior year tax positions was reduced to zero. There are no accrued interest or penalties associated with any unrecognized tax benefits.
The Company files U.S. and state income tax returns with varying statutes of limitations. The tax years from inception in 2002 forward remain open to examination due to the carryover of unused net operating losses and tax credits.
97
Table of Contents
16. Supplemental Financial Information
Quarterly Results of Operations (Unaudited)
The following table presents the unaudited statements of operations data for each of the eight quarters in the period ended December 31, 2009. The information has been presented on the same basis as the audited financial statements and all necessary adjustments, consisting only of normal recurring adjustments, have been included in the amounts below to present fairly the unaudited quarterly results when read in conjunction with the audited financial statements and related notes. The operating results for any quarter should not be relied upon as necessarily indicative of results for any future period.
Unaudited Quarterly Results of Operations
(in thousands, except per share amounts)
| Three months ended | Total for year 2009 |
|||||||||||||||||||
| March 31, 2009 |
June 30, 2009 |
September 30, 2009 |
December 31, 2009 |
|||||||||||||||||
| Revenue: |
||||||||||||||||||||
| License fee revenue |
$ | — | $ | — | $ | 2,083 | $ | 3,125 | $ | 5,208 | ||||||||||
| Total revenue |
— | — | 2,083 | 3,125 | 5,208 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
2,222 | 2,250 | 2,136 | 2,397 | 9,005 | |||||||||||||||
| General and administrative |
4,214 | 5,019 | 3,839 | 2,978 | 16,050 | |||||||||||||||
| Merger related transaction costs |
2,224 | — | — | — | 2,224 | |||||||||||||||
| Total operating expenses |
8,660 | 7,269 | 5,975 | 5,375 | 27,279 | |||||||||||||||
| Loss from operations |
(8,660 | ) | (7,269 | ) | (3,892 | ) | (2,250 | ) | (22,071 | ) | ||||||||||
| Interest income |
88 | 97 | 55 | 42 | 282 | |||||||||||||||
| Interest expense |
(166 | ) | (5 | ) | (4 | ) | (4 | ) | (179 | ) | ||||||||||
| Other income (expense), net |
200 | 64 | 36 | (132 | ) | 168 | ||||||||||||||
| Net loss |
$ | (8,538 | ) | $ | (7,113 | ) | $ | (3,805 | ) | $ | (2,344 | ) | $ | (21,800 | ) | |||||
| Basic and diluted net loss per share |
$ | (0.95 | ) | $ | (0.54 | ) | $ | (0.29 | ) | $ | (0.18 | ) | $ | (1.79 | ) | |||||
| Weighted average common shares outstanding |
9,003 | 13,070 | 13,175 | 13,357 | 12,166 | |||||||||||||||
| Three months ended | Total for year 2008 |
|||||||||||||||||||
| March 31, 2008 |
June 30, 2008 |
September 30, 2008 |
December 31, 2008 |
|||||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
$ | 3,611 | $ | 2,781 | $ | 2,453 | $ | 1,536 | $ | 10,381 | ||||||||||
| General and administrative |
1,512 | 1,712 | 2,141 | 2,559 | 7,924 | |||||||||||||||
| Merger related transaction costs |
— | — | — | 1,967 | 1,967 | |||||||||||||||
| Total operating expenses |
5,123 | 4,493 | 4,594 | 6,062 | 20,272 | |||||||||||||||
| Loss from operations |
(5,123 | ) | (4,493 | ) | (4,594 | ) | (6,062 | ) | (20,272 | ) | ||||||||||
| Interest income |
336 | 212 | 134 | 60 | 742 | |||||||||||||||
| Interest expense |
(237 | ) | (205 | ) | (177 | ) | (147 | ) | (766 | ) | ||||||||||
| Other income (expense), net |
(48 | ) | (22 | ) | 377 | 30 | 337 | |||||||||||||
| Net loss |
$ | (5,072 | ) | $ | (4,508 | ) | $ | (4,260 | ) | $ | (6,119 | ) | $ | (19,959 | ) | |||||
| Basic and diluted net loss per share |
$ | (14.09 | ) | $ | (12.09 | ) | $ | (10.07 | ) | $ | (13.66 | ) | $ | (49.77 | ) | |||||
| Weighted average common shares outstanding |
360 | 373 | 423 | 448 | 401 | |||||||||||||||
98
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Not applicable.
| Item 9A(T). | Controls and Procedures |
| (a) | Evaluation of disclosure controls and procedures |
Our management evaluated, with the participation and under the supervision of our Chief Executive Officer and our Chief Financial Officer, the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded, that our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission, or SEC, rules and forms, and that such information is accumulated and communicated to management as appropriate to allow timely decisions regarding required disclosures.
| (b) | Internal control over financial reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. We maintain a system of internal control that is designed to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Management assessed our internal control over financial reporting as of December 31, 2009, the end of our last fiscal year. Management based its assessment on criteria established in “Internal Control—Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management’s assessment included evaluation of such elements as the design and operating effectiveness of key financial reporting controls, process documentation, accounting policies and our overall control environment.
Based on our assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2009 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with generally accepted accounting principles.
There have not been any changes in our internal controls over financial reporting (as such item is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during our fiscal quarter ended December 31, 2009 that have materially affected, or are reasonably likely to materially affect, our internal controls over financial reporting.
This annual report does not include an attestation report of the company’s independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the company’s independent registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit the company to provide only management’s report in this annual report.
The information contained under this caption “Internal control over financial reporting” shall not be deemed to be filed with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that we specifically incorporate it by reference into such filing.
| (c) | Limitations on the Effectiveness of Controls |
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the controls are met. Because of the inherent limitations in all control systems, no
99
Table of Contents
evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected. Accordingly, our controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our control system are met.
| Item 9B. | Other Information |
None.
100
Table of Contents
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
Except as set forth below, the information required by this Item 10 is incorporated herein by reference to our Proxy Statement to be filed with the Commission within 120 days of the end of our fiscal year pursuant to General Instruction G(3) to Form 10-K.
Section 16(a) Beneficial Ownership Reporting Compliance
The information regarding our Section 16 beneficial ownership reporting compliance is incorporated by reference from our definitive Proxy Statement described above, where it appears under the heading “Section 16(a) Beneficial Ownership Reporting Compliance.”
Code of Business Conduct and Ethics
Our board of directors has adopted a code of business conduct and ethics. The code of business conduct applies to all of our employees, officers and directors. The full texts of our codes of business conduct and ethics are posted on our website at http://www.transcept.com under the Investor Relations section. We intend to disclose future amendments to our codes of business conduct and ethics, or certain waivers of such provisions, at the same location on our website identified above and also in public filings. The inclusion of our website address in this report does not include or incorporate by reference the information on our website into this report.
| Item 11. | Executive Compensation |
The information required by this Item 11 is incorporated by reference to our Proxy Statement to be filed with the Commission within 120 days of the end of our fiscal year pursuant to General Instruction G(3) to Form 10-K.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Except as set forth below, the information required by this Item 12 is incorporated by reference to our Proxy Statement to be filed with the Commission within 120 days of the end of our fiscal year pursuant to General Instruction G(3) to Form 10-K.
The following table provides certain information with respect to all of our equity compensation plans in effect as of December 31, 2009.
| Plan Category |
Number of Securities to be Issued Upon Exercise of Outstanding Options and Warrants |
Weighted Average Exercise Price of Outstanding Options and Warrants |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans(1) |
|||||||
| Equity compensation plans approved by stockholders |
1,718,258 | (2) | $ | 7.49 | (3) | 815,421 | (4) | |||
| Equity compensation plans not approved by stockholders |
156,007 | $ | 8.14 | — | ||||||
| Total |
1,874,265 | $ | 7.54 | 815,421 | ||||||
| (1) | The number of authorized shares under the 2006 Equity Incentive Plan, or the 2006 Plan, automatically increases on January 1 of each year by a number of shares equal to the lesser of (i) 400,000 shares, (ii) 4.5% of the outstanding shares on the last day of the immediately preceding fiscal year, or (iii) an amount determined by the Board of Directors. |
101
Table of Contents
| (2) | Includes 1,718,258 shares relating to outstanding options. |
| (3) | Represents the weighted average exercise price of outstanding options. |
| (4) | Includes 477,940 available under the 2009 Employee Stock Purchase Plan and 337,481 available under the 2006 Plan. |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this Item 13 is incorporated by reference to our Proxy Statement to be filed with the Commission within 120 days of the end of our fiscal year pursuant to General Instruction G(3) to Form 10-K.
| Item 14. | Principal Accountant Fees and Services |
The information required by this Item 14 is incorporated by reference to our Proxy Statement to be filed with the Commission within 120 days of the end of our fiscal year pursuant to General Instruction G(3) to Form 10-K.
102
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
(a)(1) Financial Statements
See Index to Financial Statements under Item 8 on page 69.
(a)(2) Financial Statement Schedules
Financial statement schedules are omitted because they are not applicable or are not required or the information required to be set forth therein is included in the Financial Statements or notes thereto.
(a)(3) Exhibits
The exhibits listed in the Exhibit Index below are filed or incorporated by reference as part of this report.
| Exhibit No. |
Description of Exhibit | |
| 2.1(1) | Agreement and Plan of Merger and Reorganization, dated as of August 29, 2008, by and among Novacea Inc. (“Novacea”), Pivot Acquisition, Inc. and Transcept Pharmaceuticals, Inc., a privately held corporation that is now named Transcept Pharma, Inc. (“TPI”) and is a wholly-owned subsidiary of Transcept Pharmaceuticals, Inc., a publicly-traded corporation formerly known as Novacea. | |
| 2.2(1) | Amendment to Agreement and Plan of Merger and Reorganization, dated as of December 23, 2008, by and among Novacea, Pivot Acquisition, Inc. and TPI. | |
| 3.1(2) | Amended and Restated Certificate of Incorporation of Transcept Pharmaceuticals, Inc. | |
| 3.2(2) | Bylaws of Transcept Pharmaceuticals, Inc., as amended. | |
| 4.1(3) | Specimen Common Stock certificate of Transcept Pharmaceuticals, Inc. | |
| 4.2(3) | Form of Preferred Stock Purchase Warrant issued to certain TPI investors as of March 21, 2005. | |
| 4.3(3) | Preferred Stock Purchase Warrant issued by TPI to Hercules Technology Growth Capital, Inc., dated as of April 13, 2006. | |
| 10.1(4) | Novacea 2001 Stock Option Plan and forms of agreements relating thereto. | |
| 10.2(5) | Novacea 2006 Equity Incentive Plan, as amended, and forms of agreements relating thereto. | |
| 10.3(3) | TPI Amended and Restated 2002 Stock Option Plan and forms of agreements relating thereto. | |
| 10.4(12) | Transcept Pharmaceuticals, Inc. 2009 Employee Stock Purchase Plan. | |
| 10.5(3) | Loan and Security Agreement, by and between Transcept Pharmaceuticals, Inc. and Hercules Technology Growth Capital, Inc. dated as of April 13, 2006. | |
| 10.6(3) | Secured Promissory Note issued to Hercules Technology Growth Capital, Inc., dated as of May 31, 2006. | |
| 10.7(6) | Office Lease, by and between Kashiwa Fudosan America, Inc. and Novacea, dated as of May 15, 2007. | |
| 10.8(8) | Sublease dated as of March 24, 2009 by and between Transcept Pharmaceuticals, Inc. and BiPar Sciences, Inc. | |
103
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.9(11) | Sublease dated for reference purposes as of June 11, 2009 by and between Transcept Pharmaceuticals, Inc. and Bay Area Bioscience Association. | |
| 10.10(3) | Lease, by and between TPI and Point Richmond R&D Associates, L.P., dated as of February 22, 2006. | |
| 10.11(3) | First Amendment to Lease, by and between TPI and Point Richmond R&D Associates, L.P., dated as of June 27, 2007. | |
| 10.12(7) | Second Amendment to Lease, by and between Transcept Pharmaceuticals, Inc. and Point Richmond R&D Associates, L.P., dated as of February 9, 2009. | |
| 10.13(7) | Lease, by and between Transcept and Point Richmond R&D Associates II, LLC, dated as of February 9, 2009. | |
| 10.14(3)† | Supply Agreement, by and between TPI and Plantex USA, Inc., dated as of March 31, 2006. | |
| 10.15(3)† | Letter Agreement, by and between TPI and Plantex USA, Inc., dated as of August 6, 2008. | |
| 10.16(13)† | First Amendment Plantex Supply Agreement dated as of July 31, 2009. | |
| 10.17(3)† | Packaging and Supply Agreement, by and between TPI and Anderson Packaging, Inc., dated as of September 14, 2006. | |
| 10.18(3)† | Amendment to Packaging and Supply Agreement, by and between TPI and Anderson Packaging, Inc., dated as of September 14, 2006. | |
| 10.19(3)† | Manufacturing Services Agreement, by and between TPI and Patheon Pharmaceuticals, Inc., dated as of October 6, 2006. | |
| 10.20(3)† | Amendment #1 to Manufacturing Services Agreement, by and between TPI and Patheon Pharmaceuticals, Inc., dated as of January 1, 2008. | |
| 10.21(13) | Amendment #2 to Manufacturing Services Agreement, by and between TPI and Patheon Pharmaceuticals, Inc., dated July 29, 2009. | |
| 10.22(3)† | Supply and Sublicense Agreement, by and between TPI and Mikart, Inc., dated as of January 22, 2008. | |
| 10.23(3)† | Manufacturing and Supply Agreement, by and between TPI and Mikart, Inc., dated as of August 21, 2008. | |
| 10.24(3)† | Packaging and Supply Agreement, by and between TPI and Sharp Corporation, dated as of June 16, 2008. | |
| 10.25(3)† | Supply and License Agreement, by and between TPI and SPI Pharma, Inc., dated as of June 27, 2006. | |
| 10.26(3)† | Amendment #1 to Supply and License Agreement, by and between TPI and SPI Pharma, Inc., dated as of March 14, 2008. | |
| 10.27(3)† | Supply Agreement, by and among TPI and SPI Pharma, Inc., dated as of July 23, 2007. | |
| 10.28(13) | Amendment #1 to Supply Agreement by and between Pivot Acquisition, Inc. and SPI Pharma, Inc. dated July 30, 2009. | |
| 10.29(13)† | Amendment #2 to Supply Agreement by and between Pivot Acquisition, Inc. and SPI Pharma, Inc dated July 30, 2009. | |
| 10.30(3) | Offer Letter dated April 15, 2008, by and between TPI and Terrence Moore, including Side Letter dated August 20, 2008 and Side Letter dated December 23, 2008. | |
104
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.31(10) | Amended and Restated Director Compensation Policy. | |
| 10.32(7) | Lease, by and between Transcept and Point Richmond R&D Associates II, LLC, dated as of February 20, 2009. | |
| 10.33(7) | Offer Letter dated March 4, 2009, by and between Transcept Pharmaceuticals, Inc. and Joseph Kennedy. | |
| 10.34(7) | Change of Control and Severance Benefits Agreement, by and between Transcept Pharmaceuticals, Inc. and Joseph Kennedy dated March 4, 2009. | |
| 10.35(9) | Form of Indemnification Agreement for officers and non-institutional investor affiliated directors. | |
| 10.36(9) | Form of Indemnification Agreement for institutional investor affiliated directors. | |
| 10.37(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Nipun Davar, Ph.D. dated April 30, 2009. | |
| 10.38(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Dennie Dyer dated April 30, 2009. | |
| 10.39(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Terrence Moore dated April 30, 2009. | |
| 10.40(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Glenn A. Oclassen dated April 30, 2009. | |
| 10.41(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Sharon Sakai, Ph.D. dated April 30, 2009. | |
| 10.42(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Nikhilesh Singh, Ph.D. dated April 30, 2009. | |
| 10.43(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Thomas P. Soloway dated April 30, 2009. | |
| 10.44(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Marilyn E. Wortzman dated April 30, 2009. | |
| 10.45(13)† | United States License and Collaboration Agreement by and between Transcept Pharmaceuticals, Inc. and Purdue Pharmaceutical Products L.P. dated July 31, 2009. | |
| 10.46(13)† | Letter agreement by and between Transcept Pharmaceuticals, Inc. and Purdue Pharmaceutical Products L.P. dated July 31, 2009. | |
| 10.47(13)† | Letter agreement by and between Transcept Pharmaceuticals, Inc. and LP Clover Limited dated July 31, 2009. | |
| 21.1 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 31.1 | Certification of the Company’s Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of the Company’s Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification of the Company’s Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
105
Table of Contents
| (1) | Incorporated by reference from the Registration Statement on Form S-4, Securities and Exchange Commission file number 333-153844, as declared effective on December 29, 2008. |
| (2) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2009. |
| (3) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2009. |
| (4) | Incorporated by reference from the Registration Statement on Form S-1, Securities and Exchange Commission file number 333-131741, filed on February 10, 2006. |
| (5) | Incorporated by reference from the Annual Report on Form 10-K filed with the Securities and Exchange Commission on April 2, 2007. |
| (6) | Incorporated by reference from the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 17, 2008. |
| (7) | Incorporated by reference from the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2009. |
| (8) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on March 31, 2009. |
| (9) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on April 9, 2009. |
| (10) | Incorporated by reference from the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 15, 2009. |
| (11) | Incorporated by reference from the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 14, 2009. |
| (12) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on June 9, 2009. |
| (13) | Incorporated by reference from the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 16, 2009. |
| † | Confidential treatment has been granted as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission. |
(b) Exhibits
See Exhibits listed under Item 15(a)(3) above.
(c) Financial Statement Schedules
Financial statement schedules are omitted because they are not applicable or are not required or the information required to be set forth therein is included in the Financial Statements or notes thereto.
106
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Point Richmond, State of California, on the 30th day of March, 2010.
| Transcept Pharmaceuticals, Inc. | ||
| By: |
/s/ GLENN A. OCLASSEN | |
| Glenn A. Oclassen President and Chief Executive Officer | ||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Glenn A. Oclassen and Thomas P. Soloway his true and lawful attorney-in-fact and agent, with full power of substitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this annual report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated opposite his/her name.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ GLENN A. OCLASSEN Glenn A. Oclassen |
President, Chief Executive Officer, and Director (Principal Executive Officer) |
March 30, 2010 | ||
| /s/ THOMAS P. SOLOWAY Thomas P. Soloway |
Senior Vice President, Operations and Chief Financial Officer (Principal Financial Officer) |
March 30, 2010 | ||
| /s/ MARILYN E. WORTZMAN Marilyn E. Wortzman |
Vice President, Finance (Principal Accounting Officer) |
March 30, 2010 | ||
| /s/ CHRISTOPHER B. EHRLICH Christopher B. Ehrlich |
Director | March 30, 2010 | ||
| /s/ THOMAS D. KILEY Thomas D. Kiley |
Director | March 30, 2010 | ||
| /s/ KATHLEEN D. LAPORTE Kathleen D. LaPorte |
Director | March 30, 2010 | ||
107
Table of Contents
| Signature |
Title |
Date | ||
| /s/ JAKE R. NUNN Jake R. Nunn |
Director | March 30, 2010 | ||
| /s/ G. KIRK RAAB G. Kirk Raab |
Chairman of the Board of Directors | March 30, 2010 | ||
| /s/ FREDERICK J. RUEGSEGGER Frederick J. Ruegsegger |
Director | March 30, 2010 | ||
| /s/ CAMILLE D. SAMUELS Camille D. Samuels |
Director | March 30, 2010 | ||
| /s/ DANIEL K. TURNER III Daniel K. Turner III |
Director | March 30, 2010 | ||
| /s/ JOHN P. WALKER John P. Walker |
Director | March 30, 2010 | ||
108
Table of Contents
Exhibit Index
| Exhibit No. |
Description of Exhibit | |
| 2.1(1) | Agreement and Plan of Merger and Reorganization, dated as of August 29, 2008, by and among Novacea, Inc. (“Novacea”), Pivot Acquisition, Inc. and Transcept Pharmaceuticals, Inc., a privately held corporation that is now named Transcept Pharma, Inc.(“TPI”) and is a wholly-owned subsidiary of Transcept Pharmaceuticals, Inc., a publicly-traded corporation formerly known as Novacea. | |
| 2.2(1) | Amendment to Agreement and Plan of Merger and Reorganization, dated as of December 23, 2008, by and among Novacea, Pivot Acquisition, Inc. and TPI. | |
| 3.1(2) | Amended and Restated Certificate of Incorporation of Transcept Pharmaceuticals, Inc. | |
| 3.2(2) | Bylaws of Transcept Pharmaceuticals, Inc., as amended. | |
| 4.1(3) | Specimen Common Stock certificate of Transcept Pharmaceuticals, Inc. | |
| 4.2(3) | Form of Preferred Stock Purchase Warrant issued to certain TPI investors as of March 21, 2005. | |
| 4.3(3) | Preferred Stock Purchase Warrant issued by TPI to Hercules Technology Growth Capital, Inc., dated as of April 13, 2006. | |
| 10.1(4) | Novacea 2001 Stock Option Plan and forms of agreements relating thereto. | |
| 10.2(5) | Novacea 2006 Equity Incentive Plan, as amended, and forms of agreements relating thereto. | |
| 10.3(3) | TPI Amended and Restated 2002 Stock Option Plan and forms of agreements relating thereto. | |
| 10.4(12) | Transcept Pharmaceuticals, Inc. 2009 Employee Stock Purchase Plan. | |
| 10.5(3) | Loan and Security Agreement, by and between Transcept Pharmaceuticals, Inc. and Hercules Technology Growth Capital, Inc. dated as of April 13, 2006. | |
| 10.6(3) | Secured Promissory Note issued to Hercules Technology Growth Capital, Inc., dated as of May 31, 2006. | |
| 10.7(6) | Office Lease, by and between Kashiwa Fudosan America, Inc. and Novacea, dated as of May 15, 2007. | |
| 10.8(8) | Sublease dated as of March 24, 2009 by and between Transcept Pharmaceuticals, Inc. and BiPar Sciences, Inc. | |
| 10.9(11) | Sublease dated for reference purposes as of June 11, 2009 by and between Transcept Pharmaceuticals, Inc. and Bay Area Bioscience Association. | |
| 10.10(3) | Lease, by and between TPI and Point Richmond R&D Associates, L.P., dated as of February 22, 2006. | |
| 10.11(3) | First Amendment to Lease, by and between TPI and Point Richmond R&D Associates, L.P., dated as of June 27, 2007. | |
| 10.12(7) | Second Amendment to Lease, by and between Transcept Pharmaceuticals, Inc. and Point Richmond R&D Associates, L.P., dated as of February 9, 2009. | |
| 10.13(7) | Lease, by and between Transcept and Point Richmond R&D Associates II, LLC, dated as of February 9, 2009. | |
| 10.14(3)† | Supply Agreement, by and between TPI and Plantex USA, Inc., dated as of March 31, 2006. | |
| 10.15(3)† | Letter Agreement, by and between TPI and Plantex USA, Inc., dated as of August 6, 2008. | |
1
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.16(13)† | First Amendment Plantex Supply Agreement dated as of July 31, 2009. | |
| 10.17(3)† | Packaging and Supply Agreement, by and between TPI and Anderson Packaging, Inc., dated as of September 14, 2006. | |
| 10.18(3)† | Amendment to Packaging and Supply Agreement, by and between TPI and Anderson Packaging, Inc., dated as of September 14, 2006. | |
| 10.19(3)† | Manufacturing Services Agreement, by and between TPI and Patheon Pharmaceuticals, Inc., dated as of October 6, 2006. | |
| 10.20(3)† | Amendment #1 to Manufacturing Services Agreement, by and between TPI and Patheon Pharmaceuticals, Inc., dated as of January 1, 2008. | |
| 10.21(13) | Amendment #2 to Manufacturing Services Agreement, by and between TPI and Patheon Pharmaceuticals, Inc., dated July 29, 2009. | |
| 10.22(3)† | Supply and Sublicense Agreement, by and between TPI and Mikart, Inc., dated as of January 22, 2008. | |
| 10.23(3)† | Manufacturing and Supply Agreement, by and between TPI and Mikart, Inc., dated as of August 21, 2008. | |
| 10.24(3)† | Packaging and Supply Agreement, by and between TPI and Sharp Corporation, dated as of June 16, 2008. | |
| 10.25(3)† | Supply and License Agreement, by and between TPI and SPI Pharma, Inc., dated as of June 27, 2006. | |
| 10.26(3)† | Amendment #1 to Supply and License Agreement, by and between TPI and SPI Pharma, Inc., dated as of March 14, 2008. | |
| 10.27(3)† | Supply Agreement, by and among TPI and SPI Pharma, Inc., dated as of July 23, 2007. | |
| 10.28(13) | Amendment #1 to Supply Agreement by and between Pivot Acquisition, Inc. and SPI Pharma, Inc. dated July 30, 2009. | |
| 10.29(13)† | Amendment #2 to Supply Agreement by and between Pivot Acquisition, Inc. and SPI Pharma, Inc dated July 30, 2009. | |
| 10.30(3) | Offer Letter dated April 15, 2008, by and between TPI and Terrence Moore, including Side Letter dated August 20, 2008 and Side Letter dated December 23, 2008. | |
| 10.31(10) | Amended and Restated Director Compensation Policy. | |
| 10.32(7) | Lease, by and between Transcept and Point Richmond R&D Associates II, LLC, dated as of February 20, 2009. | |
| 10.33(7) | Offer Letter dated March 4, 2009, by and between Transcept Pharmaceuticals, Inc. and Joseph Kennedy. | |
| 10.34(7) | Change of Control and Severance Benefits Agreement, by and between Transcept Pharmaceuticals, Inc. and Joseph Kennedy dated March 4, 2009. | |
| 10.35(9) | Form of Indemnification Agreement for officers and non-institutional investor affiliated directors. | |
| 10.36(9) | Form of Indemnification Agreement for institutional investor affiliated directors. | |
| 10.37(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Nipun Davar, Ph.D. dated April 30, 2009. | |
2
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.38(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Dennie Dyer dated April 30, 2009. | |
| 10.39(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Terrence Moore dated April 30, 2009. | |
| 10.40(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Glenn A. Oclassen dated April 30, 2009. | |
| 10.41(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Sharon Sakai, Ph.D. dated April 30, 2009. | |
| 10.42(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Nikhilesh Singh, Ph.D. dated April 30, 2009. | |
| 10.43(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Thomas P. Soloway dated April 30, 2009. | |
| 10.44(11) | Change of Control and Severance Benefits Agreement by and between Transcept Pharmaceuticals, Inc. and Marilyn E. Wortzman dated April 30, 2009. | |
| 10.45(13)† | United States License and Collaboration Agreement by and between Transcept Pharmaceuticals, Inc. and Purdue Pharmaceutical Products L.P. dated July 31, 2009. | |
| 10.46(13)† | Letter agreement by and between Transcept Pharmaceuticals, Inc. and Purdue Pharmaceutical Products L.P. dated July 31, 2009. | |
| 10.47(13)† | Letter agreement by and between Transcept Pharmaceuticals, Inc. and LP Clover Limited dated July 31, 2009. | |
| 21.1 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 31.1 | Certification of the Company’s Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of the Company’s Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification of the Company’s Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| (1) | Incorporated by reference from the Registration Statement on Form S-4, Securities and Exchange Commission file number 333-153844, as declared effective on December 29, 2008. |
| (2) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2009. |
| (3) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2009. |
| (4) | Incorporated by reference from the Registration Statement on Form S-1, Securities and Exchange Commission file number 333-131741, filed on February 10, 2006. |
| (5) | Incorporated by reference from the Annual Report on Form 10-K filed with the Securities and Exchange Commission on April 2, 2007. |
| (6) | Incorporated by reference from the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 17, 2008. |
| (7) | Incorporated by reference from the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2009. |
| (8) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on March 31, 2009. |
3
Table of Contents
| (9) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on April 9, 2009. |
| (10) | Incorporated by reference from the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 15, 2009. |
| (11) | Incorporated by reference from the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 14, 2009. |
| (12) | Incorporated by reference from the Current Report on Form 8-K filed with the Securities and Exchange Commission on June 9, 2009. |
| (13) | Incorporated by reference from the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 16, 2009. |
| † | Confidential treatment has been granted as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission. |
4