Attached files
| file | filename |
|---|---|
| EX-32.1 - CASI Pharmaceuticals, Inc. | v178789_ex32-1.htm |
| EX-31.1 - CASI Pharmaceuticals, Inc. | v178789_ex31-1.htm |
| EX-31.2 - CASI Pharmaceuticals, Inc. | v178789_ex31-2.htm |
| EX-32.2 - CASI Pharmaceuticals, Inc. | v178789_ex32-2.htm |
| EX-23.1 - CASI Pharmaceuticals, Inc. | v178789_ex23-1.htm |
FORM
10-K
SECURITIES
AND EXCHANGE COMMISSION
WASHINGTON,
D. C., 20549
ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15 (d) OF
THE
SECURITIES EXCHANGE ACT OF 1934
For the
fiscal year ended December 31, 2009
Commission
file number 0-20713
ENTREMED,
INC.
|
Delaware
|
58-1959440
|
|
|
(State of Incorporation)
|
(I.R.S. Employer Identification No.)
|
|
|
9640 Medical Center Drive, Rockville, MD
|
20850
|
|
|
(Address of principal executive offices)
|
(Zip Code)
|
Registrant's
telephone number, including area code: (240)
864-2600
Securities
registered pursuant to Section 12(b) of the Act:
|
Common Stock, $0.01 par value
|
The NASDAQ Stock Market LLC
|
|
|
(Title of each class)
|
(Name of each exchange on which registered)
|
Securities
registered pursuant to Section 12(g) of the Act: NONE
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes ¨ No
x
Indicate
by check mark if the registrant is not required to file reports pursuant to
Section 13 or 15 (d) of the Act. Yes ¨ No x
Indicate
by check mark whether the registrant (1) has filed all reports required to be
filed by Section 13 or 15 (d) of the Securities Exchange Act of 1934 during the
preceding 12 months (or for such shorter period that the registrant was required
to file such reports), and (2) has been subject to such filing requirements for
the past 90 days. Yes x No ¨
Indicate
by check mark whether the registrant has submitted electronically and posted on
its corporate Web site, if any, every Interactive Data File required to be
submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding
12 months (or for such shorter period that the registrant was required to submit
and post such files). Yes ¨ No ¨
Indicate
by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K is not contained herein, and will not be contained, to the best
of registrant's knowledge, in definitive proxy or information statements
incorporated by reference in Part III of this form 10-K or any amendment to this
Form 10-K x
Indicate
by check mark whether the registrant is a large accelerated filer, an
accelerated filer, a non-accelerated filer, or a smaller reporting
company. See the definitions of “large accelerated filer,”
“accelerated filer” and “smaller reporting company” in Rule 12b-2 of the
Exchange Act. (Check one):
|
Large
accelerated filer ¨
|
Accelerated
filer ¨
|
|
Non-accelerated
filer x
|
Smaller
reporting company ¨
|
Indicate
by check mark whether the registrant is a shell company (as defined in Rule
12b-2 of the Act). Yes ¨ No x
As of
June 30, 2009, the aggregate market value of the shares of common stock held by
non-affiliates was approximately $38,148,385.
As of
March 15, 2010, 95,694,736 shares of the Company’s common stock were
outstanding.
Documents
Incorporated By Reference
The
registrant intends to file a definitive proxy statement pursuant to
Regulation 14A within 120 days of the end of the fiscal year ended
December 31, 2009. The proxy statement is incorporated herein by reference
into the following parts of the Form 10K:
Part III,
Item 10, Directors, Executive Officers and Corporate
Governance;
Part III,
Item 11, Executive Compensation;
Part III,
Item 12, Security Ownership of Certain Beneficial Owners and Management and
Related Stockholder
Matters;
Part III,
Item 13, Certain Relationships and Related Transactions, and Director
Independence; and
Part III,
Item 14, Principal Accounting Fees and Services.
ENTREMED,
INC.
FORM
10-K - FISCAL YEAR ENDED DECEMBER 31, 2009
Contents
and Cross Reference Sheet
|
Form 10-K
Part No.
|
Form 10-K
Item No.
|
Description
|
Form 10-K
Page No.
|
|||
|
I
|
1
|
Business
|
3
|
|||
|
|
||||||
|
1A
|
Risk
Factors
|
10
|
||||
|
1B
|
Unresolved
Staff Comments
|
19
|
||||
|
2
|
Properties
|
20
|
||||
|
3
|
Legal
Proceedings
|
20
|
||||
|
4
|
Reserved
|
20
|
||||
|
II
|
5
|
Market
for Registrant's Common Equity, Related Stockholder Matters And Issuer
Purchases of Equity Securities
|
20
|
|||
|
6
|
Selected
Financial Data
|
23
|
||||
|
7
|
Management's
Discussion and Analysis of Financial Condition and Results of
Operations
|
24
|
||||
|
7A
|
Quantitative
and Qualitative Disclosures About Market Risk
|
33
|
||||
|
8
|
Financial
Statements and Supplementary Data
|
33
|
||||
|
9
|
Changes
in and Disagreements with Accountants On Accounting and Financial
Disclosure
|
33
|
||||
|
9A
|
Controls
and Procedures
|
34
|
||||
|
9B
|
Other
Information
|
36
|
||||
|
III
|
10
|
Directors,
Executive Officers and Corporate Governance
|
36
|
|||
|
11
|
Executive
Compensation
|
36
|
||||
|
12
|
Security
Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters
|
36
|
||||
|
|
||||||
|
13
|
Certain
Relationships and Related Transactions, and Director
Independence
|
37
|
||||
|
14
|
Principal
Accounting Fees and Services
|
37
|
||||
|
IV
|
15
|
Exhibits
and Financial Statement Schedules
|
37
|
|||
|
Signatures
|
42
|
|||||
|
Audited
Consolidated Financial Statements
|
F-1
|
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This
report contains certain forward-looking statements within the meaning of Section
27A of the Securities Exchange Act of 1933, as amended and Section 21E of the
Securities Exchange Act of 1934, as amended. Forward-looking
statements also may be included in other statements that we make. All
statements that are not descriptions of historical facts are forward-looking
statements. These statements can generally be identified by the use
of forward-looking terminology such as “believes,” “expects,” “intends,” “may,”
“will,” “should,” or “anticipates” or similar terminology. These
forward-looking statements include, among others, statements regarding the
timing of our clinical trials, our cash position and future expenses, and our
future revenues.
Our
forward-looking statements are based on information available to us today, and
we will not update these statements.
1
Actual
results could differ materially from those currently anticipated due to a number
of factors, including the risk that we may be unable to continue as a
going concern as a result of our inability to raise sufficient capital for our
operational needs; the possibility that we may be delisted from trading on the
Nasdaq Capital Market; the volatility of our common stock; risks relating to the
need for additional capital and the uncertainty of securing additional funding
on favorable terms; the failure to consummate a transaction to monetize our
Thalomid® royalty
stream for any reason, including our inability to obtain the required
third-party consents; declines in actual sales of Thalomid®
resulting in reduced royalty payments; risks associated with our product
candidates; the early-stage products under development; results in preclinical
models are not necessarily indicative of clinical results; uncertainties
relating to preclinical and clinical trials, including delays to the
commencement of such trials; success in the clinical development of any
products; dependence on third parties; and risks relating to the
commercialization, if any, of our proposed products (such as
marketing, safety, regulatory, patent, product liability, supply, competition
and other risks). Additional information about the factors and risks
that could affect our business, financial condition and results of operations,
are contained in Section 1A, “Risk Factors,” of this Annual Report on Form 10-K
and our other filings with the U.S. Securities and Exchange Commission (SEC),
which are available at www.sec.gov.
2
PART
I
ITEM
1. BUSINESS.
ENMD-2076
EntreMed, Inc. (“EntreMed” or “the
Company”) (Nasdaq: ENMD) is a clinical-stage pharmaceutical company focused on
developing ENMD-2076, an Aurora A and angiogenic kinase inhibitor for the
treatment of cancer. ENMD-2076 is currently in Phase 1 studies in
advanced cancers, multiple myeloma and leukemia. We anticipate the
initiation of a Phase 2 study in ovarian cancer during the second quarter of
2010.
ENMD-2076
is a novel orally-active, Aurora A/angiogenic kinase inhibitor with potent
activity against Aurora A and multiple tyrosine kinases linked to cancer and
inflammatory diseases. ENMD-2076 is relatively selective for the
Aurora A isoform in comparison to Aurora B. Aurora kinases are key
regulators of the process of mitosis, or cell division, and are often
over-expressed in human cancers. ENMD-2076 exerts its effects through multiple
mechanisms of action, including antiproliferative activity and the inhibition of
angiogenesis. ENMD-2076 has demonstrated significant, dose-dependent preclinical
activity as a single agent, including tumor regression, in multiple xenograft
models (e.g. breast, colon, leukemia), as well as activity towards ex
vivo-treated human leukemia patient cells.
ENMD-2076
has received orphan drug designation from the United States Food and Drug
Administration (the “FDA”) for the treatment of ovarian cancer, multiple myeloma
and acute myeloid leukemia (“AML”). In the United States, the
Orphan Drug Act is intended to encourage companies to develop therapies for the
treatment of diseases that affect fewer than 200,000 people in this
country. Orphan drug designation provides us with seven years of
market exclusivity that begins once ENMD-2076 receives FDA marketing
approval. It also provides certain financial incentives that can help
support the development of ENMD-2076.
The table
below illustrates the current status of our product pipeline.
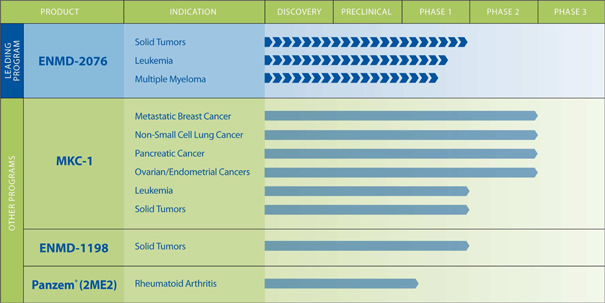
3
OTHER
DRUG CANDIDATES
ENMD-2076
is the only program currently under active clinical evaluation by the
Company. The selection of ENMD-2076 as our lead product allows us to
direct the majority of our resources to accelerate its clinical
development. However, we also own the intellectual property of our
other early-stage therapeutic candidates. We do not intend to
initiate additional new studies for programs other than ENMD-2076 unless
additional significant financing becomes available to us. Our other
therapeutic candidates include MKC-1, an oral cell-cycle inhibitor with activity
against the mTOR pathway that has completed multiple Phase 2 clinical trials for
cancer, and ENMD-1198, a novel antimitotic agent that has completed a Phase 1
study in advanced cancers. We also have an approved Investigational
New Drug Application (“IND”) for the use of Panzem® in
rheumatoid arthritis (“RA”) treatment. All of our candidates are
multi-mechanism in that they target disease cells and the blood vessels that
nourish them, which we believe can be developed to be safe and convenient, and
provide the potential for improved patient outcomes.
MKC-1 for
Oncology. MKC-1 is an orally-active, small molecule, cell
cycle inhibitor with in
vitro and in
vivo efficacy against a broad range of human solid tumor cell lines,
including multi-drug resistant cell lines. MKC-1 acts through multiple
mechanisms of action, arresting cellular mitosis and inducing cell death
(apoptosis) by binding to a number of different cellular proteins
including tubulin and members of the importin β family. MKC-1 also
inhibits activation of the oncogenic kinase Akt and the mTOR pathway through a
mechanism that is a subject of investigation by EntreMed
scientists.
MKC-1 has
demonstrated broad antitumor effects in multiple preclinical models, including
paclitaxel-resistant models, and was evaluated in several Phase 1 and 2 clinical
studies involving nearly 270 patients prior to the licensing of the drug from
Hoffman La Roche. These studies have provided extensive pharmacokinetic and
safety data. Since acquired by EntreMed, MKC-1 has shown single agent antitumor
activity in breast cancer patients and in combination with Alimta® in NSCLC
patients. MKC-1 has completed multiple Phase 2 clinical trials for
cancer and has a robust data set.
ENMD-1198 for
Oncology. ENMD-1198 is a potent, orally-active, antimitotic
agent that induces cell cycle arrest and apoptosis in tumor cells. ENMD-1198 has
shown pronounced antitumor activity as well as substantial synergistic effects
in combination with standard of care chemotherapeutic agents in numerous
preclinical models.
ENMD-1198
does not exhibit sensitivity to multi-drug resistance mechanisms in preclinical
studies. ENMD-1198 also has shown preclinical activity towards
taxane and vinca alkaloid resistant tumor cells. Additionally, ENMD-1198
decreases the activity of three oncogenic proteins (HIF-1α, NF-κB and STAT3)
that are known to promote tumor growth and
progression. ENMD-1198 has been evaluated in a Phase 1
clinical trial for safety, tolerability, pharmacokinetics, and clinical benefit
in advanced cancer patients.
Panzem® (2ME2) for Rheumatoid
Arthritis. Panzem® is an
orally active compound that has antiproliferative, antiangiogenic and
anti-inflammatory properties. The inhibition of angiogenesis is an important
approach to the treatment of both cancer and rheumatoid arthritis. Panzem® has
potential as a single agent in rheumatoid arthritis based on its antiangiogenic,
anti-inflammatory, and anti-osteoclastic (bone resorption)
properties.
An IND to
develop Panzem®
(2-methoxyestradiol, 2ME2) in rheumatoid arthritis (RA) was accepted based on
its safety profile demonstrated throughout the oncology development effort, and
preclinical research results showing that 2ME2 has disease modifying or “DMARD”
activity in a variety of animal models of RA. DMARDs are drugs that
have the ability to slow down disease progression in rheumatoid arthritis and
other autoimmune diseases, in contrast to non-steroidal anti-inflammatory drugs
which only treat the immune reaction resulting from tissue damage. We
have generated substantial preclinical data demonstrating the positive effects
of 2ME2 treatment on inflammation and disease progression in standard animal
models of RA. Radiographic and immunohistochemical staining results from these
preclinical studies have shown consistent therapeutic effects on the hallmarks
of the disease, including the inhibition of the highly angiogenic pannus,
infiltrating cells, cartilage lesions and bone resorption.
4
PANZEM® NCD
(2ME2) for Oncology
In
2008, we discontinued clinical development of 2ME2 (Panzem® NCD) for
oncology. Substantial clinical trial and manufacturing/process
development costs would be required to narrow the oncology indications for
larger registration-track randomized studies. These expenditures
would require the commitment of a disproportionate amount of resources and limit
clinical development efforts on other assets.
PRECLINICAL
Our focus
is on clinical development. Accordingly, we are not devoting any significant
resources to preclinical activities.
2010
OUTLOOK
In 2010,
we will continue to focus on three principal objectives:
|
|
·
|
to
initiate Phase 2 for ENMD-2076 in the second quarter and to continue to
concentrate our resources on ENMD-2076 in order to accelerate clinical
objectives so that we can provide a more direct path forward to product
registration and ultimately to the
market;
|
|
|
·
|
to
conserve our cash by deferring new program initiatives and reducing
expenses; and
|
|
|
·
|
to
be opportunistic in raising capital and in seeking collaborations for our
principal assets.
|
More specifically, in order to further
advance our commercial objectives, we may seek strategic alliances,
co-development partnerships and other collaborations with other companies to
develop our drug candidates. Our focus will continue to be clinical
development and, as such, will not devote any significant resources to
preclinical activities.
OPERATING
LOSSES
To date,
we have been engaged exclusively in research and development
activities. As a result, we have incurred operating losses through
December 31, 2009 and expect to continue to incur operating losses for the
foreseeable future before commercialization of any products. We spent $7,902,000 on
research and development in 2009, which includes costs associated with ENMD-2076
of $5,036,000, with MKC-1 of $449,000, with ENMD-1198 of $177,000 and with
Panzem® oncology
of $126,000, as compared to $20,069,000 in 2008 and $23,739,000 in
2007. To accomplish our business goals, we, or prospective
development partners, will be required to conduct substantial development
activities for all proposed products that we intend to pursue to
commercialization. We intend to continue to pursue strategic relationships to
provide resources for the further development of our product candidates. There
can be no assurance, however, that these discussions will result in
relationships or additional funding. In addition, we may continue to
seek capital through the public or private sale of securities. There can be no
assurance that we will be successful in seeking such additional
capital.
MANAGEMENT
EntreMed's
management team has aligned the Company's business strategy with its core
scientific strengths, while maintaining prudent resource management, fiscal
responsibility and accountability. The team has redirected EntreMed's
financial resources and R&D strategy to focus primarily on the development
of our Aurora A/angiogenic kinase inhibitor, ENMD-2076.
The
current senior management team includes: Carolyn F. Sidor, M.D., Vice President
& Chief Medical Officer; Mark R. Bray, Ph.D., Vice President, Research;
Cynthia W. Hu, JD, Chief Operating Officer, General Counsel & Secretary; and
Kathy R. Wehmeir-Davis, Principal Accounting Officer. This senior
management team reports directly to the Executive Committee of the Board
comprised of three directors: Michael M. Tarnow, Dwight L. Bush and Jennie
Hunter-Cevera, Ph.D. Mr. Tarnow serves as our Executive Chairman and
Mr. Bush serves as our Vice Chairman.
5
SCIENTIFIC
FOUNDATION
We
developed our drug candidates based on comprehensive research into the
relationship between malignancy and angiogenesis (the growth of new blood
vessels). This research led to a focus on drug candidates that act on the
cellular pathways that affect biological processes important in multiple
diseases, specifically angiogenesis and cell cycle regulation through the
inhibition of key kinases. Our drug candidate, ENMD-2076, has
potential applications in oncology and other diseases that are dependent on the
regulation of these processes.
Kinase
Inhibition. Kinases are enzymes that are primary regulators of
many essential processes in living cells. There are approximately 500 different
kinases encoded in the human genome, and these proteins act together in
intricate communication networks and pathways to control virtually every aspect
of cellular function. The reliance of the cell on kinases to regulate function
can be disastrous when kinase signaling becomes aberrant. Many human diseases
have been linked to these enzymes including all forms of cancer, arthritis,
inflammation, diabetes, and cardiovascular disease. The inhibition of kinases as
a targeted therapeutic approach has now been validated by several drugs that
have advanced successfully through clinical trials to the marketplace. The
integral role kinases play in angiogenesis and cell cycle regulation has led
EntreMed to develop inhibitors to key kinases involved in these
processes.
Cell Cycle
Regulation. Precise regulation of the cell cycle is essential
for healthy cell functions including the replication, growth, and
differentiation. One specific aspect of cell cycle regulation is the programmed
control of cell death (apoptosis). In certain diseases, such as cancer, the
balance between cell proliferation and cell death is altered, resulting in
inappropriate cell growth. Our compounds impact biochemical pathways in cells
that result in their death via apoptosis. We believe that the selective
induction of apoptosis through drugs that induce cell cycle arrest can either
stabilize or cause the regression of cancer, inflammation and other disease
processes characterized by inappropriate cell growth.
Angiogenesis. Angiogenesis
is a multi-step process whereby new blood vessels are formed. This tightly
regulated process involves the migration, proliferation and differentiation of
endothelial cells. In normal physiology, angiogenesis is a necessary component
of the menstrual cycle and wound healing, where the process is regulated through
appropriate shifts in the balance of pro-angiogenic and antiangiogenic signals.
This tight regulation of angiogenesis in normal physiology is absent or aberrant
in multiple disease settings that are characterized by persistent, inappropriate
blood vessel development. Inappropriate angiogenesis occurs in more
than 80 diseases, particularly in various cancers where the growth of new blood
vessels is necessary to sustain tumor growth.
BUSINESS
DEVELOPMENT AND COMMERCIALIZATION STRATEGY
Oncology
is our principal clinical and commercial focus. Based on the
compound’s strong preclinical antitumor activity, favorable safety profile and
bioavailability, we believe that ENMD-2076 has significant therapeutic potential
in a broad range of tumor types and continue to focus our resources on ENMD-2076
as our priority program. We believe that ENMD-2076 represents a potential Phase
2 partnering opportunity. As a result, our strategy is to pursue the
development of ENMD-2076 for oncology, obtain additional clinical data while
being selective and opportunistic in exploring strategic alliances for this and
other compounds in our pipeline. We may pursue co-development
partners for our other pipeline product candidates to help accelerate their
development and strengthen the development program with complementary
expertise. We can also provide our co-development partners with
substantial know-how relating to small molecules that inhibit angiogenesis and
inflammation, as well as regulate cell cycle pathways.
RELATIONSHIPS RELATING TO CLINICAL
PROGRAMS
Contract
Manufacturing. The manufacturing efforts for the production of
our clinical trial materials are performed by contract manufacturing
organizations. Established relationships, coupled with supply agreements, have
secured the necessary resources to ensure adequate supply of clinical materials
to support our clinical development program. We believe that our
current strategy of outsourcing manufacturing is cost-effective and allows for
the flexibility we require.
6
Sponsored Research
Agreements. To complement our in-house research and
development efforts, we have entered into sponsored research agreements with
outside scientists to conduct specific projects. Under these
agreements, we have secured the rights to intellectual property and to develop
under exclusive license any discoveries resulting from these collaborations. The
funds we provide in accordance with these agreements partially support the
scientists’ laboratory, research personnel and research supplies.
Clinical Trial
Centers. As of March 12, 2010, we are conducting clinical
trials for ENMD-2076 at the following institutions:
|
|
- Dana-Farber
Cancer Institute, Boston, MA
|
|
|
- Massachusetts
General Hospital, Boston, MA
|
|
|
- Indiana
University Cancer Center, Indianapolis,
IN
|
|
|
- Princess
Margaret Hospital, Toronto, Ontario
|
|
|
- University
of Colorado Cancer Center, Aurora,
CO
|
PATENTS,
LICENSES AND PROPRIETARY RIGHTS
Our
success will depend in part on our ability to obtain patent protection for our
products, both in the United States and abroad. The patent position of
biotechnology and pharmaceutical companies, in general, is highly uncertain and
involves complex legal and factual questions.
All of
our programs are backed by strong intellectual property rights. Each
product candidate is covered by issued or pending composition, method and use
patents. With respect to our leading program, ENMD-2076, we directly
own patent applications for the lead compound, ENMD-2076, and also for a library
of over 600 analogs. Ownership includes 1 issued US patent, 3 pending
US applications, 2 issued foreign patents and 43 pending foreign
applications. Our issued patents for ENMD-2076 expire September 9,
2026, and in the case of the issued US patent, March 5, 2027. Pending
patent applications for ENMD-2076 provide coverage through
2026. Patent applications pending for the over 600 analogs provide
coverage ranging from 2025 to 2028.
With
respect to our entire patent estate for all of our product candidates, we
directly own 40 U.S. issued patents and patent applications and 48 foreign
issued patents and patent applications; and have exclusively in-licensed 9 U.S.
issued patents and 135 foreign issued patents and patent
applications. We review and assess our portfolio on a regular basis
to ensure protection and to align our patent strategy with our overall business
strategy.
We have
registered the trademarks ENTREMED, MIIKANA, and PANZEM in
the U.S. Patent and Trademark Office and have applications pending for
registration of the trademarks in selected foreign countries.
GOVERNMENT
REGULATION
Our
development, manufacture, and potential sale of therapeutics in the United
States are subject to extensive regulations.
In the
United States, the FDA regulates our product candidates currently being
developed as drugs or biologics. New drugs are subject to regulation
under the Federal Food, Drug, and Cosmetic Act (FFDCA), and biological products,
in addition to being subject to certain provisions of that Act, are regulated
under the Public Health Service Act (PHSA). We believe that the FDA
is likely to regulate the products currently being developed by us or our
collaborators as new drugs. Both the FFDCA and PHSA and corresponding
regulations govern, among other things, the testing, manufacturing, safety,
efficacy, labeling, storage, recordkeeping, advertising and other promotion of
biologics or new drugs, as the case may be. FDA clearances or
approvals must be obtained before clinical testing, and before manufacturing and
marketing of biologics or drugs.
7
Preparing
drug candidates for regulatory approval has historically been a costly and
time-consuming process. Generally, in order to gain FDA permission to
test a new agent, a developer first must conduct preclinical studies in the
laboratory and in animal model systems to gain preliminary information on an
agent's effectiveness and to identify any safety problems. The
results of these studies are submitted as a part of an IND application for a
drug or biologic, which the FDA must review before human clinical trials of an
investigational drug can begin. In addition to the known safety and
effectiveness data on the drug or biologic, the IND must include a detailed
description of the clinical investigations proposed to be
undertaken. Based on the current FDA organizational structure,
ENMD-2076 is regulated as a new drug by the FDA’s Center for Drug Evaluation and
Research (CDER). Generally, as new chemical entities like our small
molecules are discovered, formal IND-directed toxicology studies are required
prior to initiating human testing. Clinical testing may begin 30 days
after submission of an IND to the FDA unless FDA objects to the initiation of
the study or has outstanding questions to discuss with the IND
sponsor.
In order
to commercialize any drug or biological products, we or our collaborators must
sponsor and file an IND and conduct clinical studies to demonstrate the safety
and effectiveness necessary to obtain FDA approval of such
products. For studies conducted under INDs sponsored by us or our
collaborators, we or our collaborators will be required to select qualified
investigators (usually physicians within medical institutions) to supervise the
administration of the products, test or otherwise assess patient results, and
collect and maintain patient data; monitor the investigations to ensure that
they are conducted in accordance with applicable requirements, including the
requirements set forth in the general investigational plan and
protocols contained in the IND; and comply with applicable reporting and
recordkeeping requirements.
Clinical
trials of drugs or biologics are normally done in three phases, although the
phases may overlap. Phase 1 trials for drug candidates to be used to treat
cancer patients are concerned primarily with the safety and preliminary
effectiveness of the drug, involve a small group ranging from 15 - 40 subjects,
and may take from six months to over one year to complete. Phase 2
trials normally involve 30 - 200 patients and are designed primarily to
demonstrate effectiveness in treating or diagnosing the disease or condition for
which the drug is intended, although short-term side effects and risks in people
whose health is impaired may also be examined. Phase 3 trials are
expanded clinical trials with larger numbers of patients which are intended to
evaluate the overall benefit-risk relationship of the drug and to gather
additional information for proper dosage and labeling of the
drug. Phase 3 clinical trials generally take two to five years to
complete, but may take longer. The FDA receives reports on the
progress of each phase of clinical testing, as well as reports of unexpected
adverse experiences occurring during the trial. FDA may require the
modification, suspension, or termination of clinical trials, if it concludes
that an unwarranted risk is presented to patients, or, in Phase 2 and 3, if it
concludes that the study protocols are deficient in design to meet their stated
objectives.
If
clinical trials of a new drug candidate are completed successfully, the sponsor
of the product may seek FDA marketing approval. If the product is
classified as a new drug, an applicant must file a New Drug Application (NDA)
with the FDA and receive approval before commercial marketing of the
drug. The NDA must include detailed information about the product and
its manufacture and the results of product development, preclinical studies and
clinical trials.
The
testing and approval processes require substantial time and effort and there can
be no assurance that any approval will be obtained on a timely basis, if at
all. Although it is the policy of the FDA to complete the review of
the initial submission of NDAs within six to twelve months, the entire FDA
review process may take several years. Notwithstanding the submission
of relevant data, the FDA may ultimately decide that the NDA does not satisfy
its regulatory criteria and deny the approval. Further, the FDA may
require additional clinical studies before making a decision on approval. In
addition, the FDA may condition marketing approval on the conduct of specific
post-marketing studies to further evaluate safety and
effectiveness. Even if FDA regulatory clearances are obtained, a
marketed product is subject to continuing regulatory requirements and review
relating to Good Manufacturing Practices, adverse event reporting, promotion and
advertising, and other matters. Discovery of previously unknown
problems or failure to comply with the applicable regulatory requirements may
result in restrictions on the marketing of a product or withdrawal of the
product from the market, as well as possible civil or criminal
sanctions.
8
COMPETITION
Competition
in the pharmaceutical, biotechnology and biopharmaceutical industries is intense
and based significantly on scientific and technological factors, the
availability of patent and other protection for technology and products, the
ability and length of time required to obtain governmental approval for testing,
manufacturing and marketing and the ability to commercialize products in a
timely fashion. Moreover, the biopharmaceutical industry is
characterized by rapidly evolving technology that could result in the
technological obsolescence of any products that we develop.
We
compete with many specialized biopharmaceutical firms, as well as a growing
number of large pharmaceutical companies that are applying biotechnology to
their operations. Many biopharmaceutical companies have focused their
development efforts in the human therapeutics area, including oncology and
inflammation, and many major pharmaceutical companies have developed or acquired
internal biotechnology capabilities or made commercial arrangements with other
biopharmaceutical companies. These companies, as well as academic institutions,
governmental agencies and private research organizations, also compete with us
in recruiting and retaining highly qualified scientific personnel and
consultants.
Our
competition will be determined in part by the potential indications for which
our product candidates may be developed and ultimately approved by regulatory
authorities. We may rely on third parties to commercialize our products, and
accordingly, the success of these products will depend in significant part on
these third parties' efforts and ability to compete in these markets. The
success of any collaboration will depend in part upon our collaborative
partners' own competitive, marketing and strategic considerations, including the
relative advantages of alternative products being developed and marketed by our
collaborative partners and our competitors.
Many of
our existing or potential competitors have substantially greater financial,
technical and human resources than we do and may be better equipped to develop,
manufacture and market products. In addition, many of these competitors have
extensive experience in preclinical testing and human clinical trials and in
obtaining regulatory approvals. The existence of competitive products, including
products or treatments of which we are not aware, or products or treatments that
may be developed in the future, may adversely affect the marketability of
products that we may develop.
EMPLOYEES
In
December 2008, we selected ENMD-2076 as our leading program and accordingly
realigned our human resources to directly support and accelerate the clinical
development of ENMD-2076. Our work force currently consists of 13
full-time employees and 1 part-time employee dedicated to the core areas of our
operations. Certain of our activities, such as manufacturing and
clinical trial operations, are outsourced at the present time. We may
hire additional personnel, in addition to utilizing part-time or temporary
consultants, on an as-needed basis. None of our employees are
represented by a labor union, and we believe our relations with our employees
are satisfactory.
CORPORATE
HEADQUARTERS
We were
incorporated under Delaware law in 1991. Our principal executive
offices are located at 9640 Medical Center Drive, Rockville, Maryland 20850, and
our telephone number is (240) 864-2600. We also lease office space in
Durham, North Carolina where our clinical and regulatory operations are based
and lease laboratory space in Toronto, Ontario where we conduct correlative
research to support our development of ENMD-2076.
AVAILABLE
INFORMATION
Through our website at
www.entremed.com, we make available, free
of charge, our filings with the Securities and Exchange Commission (“SEC”),
including our annual proxy statements, annual reports on Form 10-K, quarterly
reports on Form 10-Q and current reports on Form 8-K, and all amendments
thereto, as soon as reasonably practicable after such reports are filed
with or furnished to the Securities and Exchange Commission. Our
filings are also available through the Securities and Exchange Commission via
their website, http://www.sec.gov. You
may also read and copy any materials we file with the SEC at the SEC’s Public
Reference Room at 100 F Street, N.E., Washington, D.C. 20549. You may
obtain information on the operation of the Public Reference Room by calling the
SEC at 1-800-SEC-0330. The information contained on our website is
not incorporated by reference in this annual report on Form 10-K and should not
be considered a part of this report.
9
|
ITEM 1A.
|
RISK
FACTORS.
|
We
Have an Immediate Need for Capital and Will Need to Raise Additional Capital in
the Future to Continue our Business, and our Independent Registered Public
Accounting Firm Has Expressed Substantial Doubt as to our Ability to Continue as
a Going Concern
We estimate that our current capital
resources will not be sufficient to fund our operations significantly beyond
September 2010 without an additional curtailment of operating expenditures or
new equity or debt financing. In addition, our financial statements have been
prepared on the assumption that we will continue as a going concern, which
contemplates the realization of assets and liquidation of liabilities in the
normal course of business. However, our independent registered public accounting
firm’s report on our financial statements as of and for the fiscal year ended
December 31, 2009, includes an explanatory paragraph that states that our
recurring losses from operations raise substantial doubt about our ability to
continue as a going concern.
Our ability to continue as a going
concern is dependent on our success at raising additional capital sufficient to
meet our obligations on a timely basis and to ultimately attain profitability.
In the event we are unable to successfully raise additional capital, it is
unlikely that we will have sufficient cash flows and liquidity to finance our
business operations as currently contemplated. Accordingly, in the event new
financing is not obtained, we will likely reduce general and administrative
expenses and delay clinical development activity until we are able to obtain
sufficient financing to do so. These factors could significantly limit our
ability to continue as a going concern.
We
Have a History of Losses and Anticipate Future Losses
To date,
we have been engaged primarily in research and development activities. Although
we receive limited revenues on royalties from sales of Thalomid® and in
the past have received license fees and research and development funding from a
former collaborator and limited revenues from certain research grants, we have
not derived significant revenues from operations.
We have experienced losses in each year
since inception. Through December 31, 2009, we had an accumulated
deficit of approximately $366 million. We expect that it will
be very difficult to raise capital to continue our operations and our
independent registered public accounting firm has issued an opinion with an
explanatory paragraph to the effect that there is substantial doubt about our
ability to continue as a going concern. Although we have been
successfully funded to date by attracting investors in our equity securities and
through royalty payments, there is no assurance that our capital-raising efforts
will be able to attract the capital needed to sustain our operations beyond that
date or that such royalty payments will continue to provide revenues at
historical levels. If we are unable to obtain additional funding for
operations, we may not be able to continue operations as proposed, requiring us
to modify our business plan, curtail various aspects of our operations or cease
operations. In such event, investors may lose a portion or all of their
investment.
Losses
have continued since December 31, 2009. We will also be required to
conduct substantial research and development and clinical testing activities for
ENMD-2076. We expect that these activities will result in operating losses for
the foreseeable future before we commercialize any products, if
ever. In addition, to the extent we rely on others to develop and
commercialize our products, our ability to achieve profitability will depend
upon the success of these other parties. To support our research and
development of certain product candidates, we may seek and rely on cooperative
agreements from governmental and other organizations as a source of support. If
a cooperative agreement were to be reduced to any substantial extent, it may
impair our ability to continue our research and development
efforts. Even if we do achieve profitability, we may be unable to
sustain or increase it.
10
Our
Common Stock May be Delisted From The NASDAQ Capital Market, Which Could
Negatively Impact the Price of Our Common Stock and Our Ability to Access the
Capital Markets
NASDAQ Minimum $1.00 Closing Bid
Price
On
April 4, 2008, we received a letter from The NASDAQ Stock Market LLC
(“NASDAQ”) advising that for the previous 30 consecutive business days, the bid
price of the Company’s common stock had closed below the minimum $1.00 per share
requirement for continued inclusion on The NASDAQ Global Market. The
letter also advised us that failure to comply with this minimum bid price
requirement, or any other listing standard applicable to issuers listed on The
NASDAQ Global Market, by October 1, 2008, would result in our common stock
being ineligible for quotation on The NASDAQ Global Market. Our stock price has
not closed above $1.00 for ten consecutive trading days since the date of the
receipt of the letter from NASDAQ.
On
September 22, 2008, we submitted an application to transfer the trading of
our common stock to The NASDAQ Capital Market. On October 1, 2008, we
received a letter from The NASDAQ Listing Qualifications Department stating that
our application had been approved and that our common stock would commence
trading on The NASDAQ Capital Market on October 3, 2008. The NASDAQ Capital
Market operates in substantially the same manner as The NASDAQ Global Market.
Our trading symbol remains as “ENMD” and the trading of our stock was unaffected
by the transfer.
NASDAQ
suspended the enforcement of the minimum bid price rule, because of the current
extraordinary market conditions, until July 31, 2009. Upon
reinstatement of the rule on August 3, 2009, under NASDAQ rules, we had
until January 15, 2010, to regain compliance with the minimum bid price
standard. We did not regain compliance with the minimum bid price
standard of the NASDAQ rules by January 15, 2010.
On
January 19, 2010, we received a Staff Determination letter from NASDAQ stating
that we were not in compliance with the continued listing rules and that our
Common Stock would be delisted unless we requested an appeal of such
determination. On January 20, 2010, we filed an appeal of the
Staff's determination to a NASDAQ Hearings Panel (the “Panel”), pursuant to the
procedures set forth in the NASDAQ Marketplace Rules. The hearing request
stayed the delisting of the Company’s securities pending the Panel's
decision. On February 25, 2010, we met with the Panel providing them
a plan of action, with the intention of returning to compliance with NASDAQ’s
requirements. On March 23, 2010, we received a decision letter from
the Panel granting our request to extend the compliance date for an additional
180 days or until July 16, 2010. As a result of this decision by the
Panel, our stock will continue to trade on The Nasdaq Capital Market until such
date, by which time our minimum bid price must have exceeded $1.00 for ten
consecutive trading days.
In the
event that our common stock continues to trade under $1.00, we will consider all
alternatives in order to maintain the public trading status of our stock,
including electing to implement a reverse stock split with the approval of our
stockholders. We
have not made a final determination as to whether to affect a reverse split, as
we continue to have an opportunity to meet the $1.00 minimum closing bid price
during the 180-day extension period. An alternative to not having to
meet both the NASDAQ $1.00 minimum closing bid price and the $2.5 million
stockholders’ equity is to apply to have our common stock traded on the
Over-The-Counter Bulletin Board (the “OTCBB”), an electronic quotation system
that displays stock quotes by market makers. If such course of action
is taken, there can be no assurance that our common stock would be timely
admitted for trading on that market. This alternative may result in a
less liquid market available for existing and potential shareholders to buy and
sell shares of our common stock and could further depress the price of our
stock.
The
delisting of our common stock from a national exchange could significantly
affect the ability of investors to trade our securities and could negatively
affect the value and liquidity of our common stock. Delisting could also have
other negative results, including the potential loss of confidence by employees,
the loss of institutional investor interest and fewer business development
opportunities. In addition, the delisting of our common stock could materially
adversely affect our ability to raise capital on terms acceptable to us or at
all.
11
NASDAQ
Minimum $2.5 Million Stockholders’ Equity
Additionally,
we must continually maintain (1) stockholders’ equity of at least $2.5 million
or (2) a minimum of $35 million in market value of our listed securities for ten
consecutive trading days to be in compliance with the continued listing
standards for The NASDAQ Capital Market. At December 31, 2009, our
consolidated stockholders’ deficit was approximately $1.9 million and the market
value of our listed securities was $70.9 million. There can be no
assurance that we will be able to meet either of these listing standards in the
future. If we do not meet one of these NASDAQ listing requirements,
we will be in a deficiency period and will submit a plan of compliance with
NASDAQ. After the deficiency period, if we are unable to successfully
appeal to the NASDAQ Panel for an extension of time to regain compliance, our
common stock could be delisted from The NASDAQ Capital Market.
The
Current Capital and Credit Market Conditions May Adversely Affect the Company’s
Access to Capital, Cost of Capital, and Ability to Execute its Business Plan as
Scheduled
Access to
capital markets is critical to our ability to operate. Traditionally,
biopharmaceutical companies (such as we) have funded their research and
development expenditures through raising capital in the equity
markets. Declines and uncertainties in these markets over the past
two years have severely restricted raising new capital and have affected our
ability to continue to expand or fund existing research and development efforts.
We require significant capital for research and development for our product
candidates and clinical trials. In recent years, the general economic and
capital market conditions in the United States have deteriorated significantly
and have adversely affected our access to capital and increased the cost of
capital, and there is no certainty that a recovery in the capital and credit
markets, enabling us to raise capital in an amount to sufficiently fund our
short-term and long-term plans, will occur in 2010. If these economic
conditions continue or become worse, the Company’s future cost of equity or debt
capital and access to the capital markets could be adversely affected. In
addition, an inability by the Company to access the capital markets on favorable
terms because of our low stock price, or upon our delisting from the NASDAQ
Stock Market if we fail to satisfy a listing requirement, could affect our
ability to execute our business plan as scheduled. Moreover, we rely and intend
to rely on third parties, including our clinical research organizations, third
party manufacturers, and certain other important vendors and consultants. As a
result of the current volatile and unpredictable global economic situation,
there may be a disruption or delay in the performance of our third-party
contractors and suppliers. If such third parties are unable to adequately
satisfy their contractual commitments to us in a timely manner, our business
could be adversely affected.
We
are Uncertain Whether Additional Funding Will Be Available For Our Future
Capital Needs and Commitments, and If We Cannot Raise Additional Funding, or
Access the Credit Markets, We May Be Unable to Complete Development of Our
Product Candidates
We will
require substantial funds in addition to our existing working capital to develop
our product candidates and otherwise to meet our business objectives. We have
never generated sufficient revenue during any period since our inception to
cover our expenses and have spent, and expect to continue to spend, substantial
funds to continue our research and development and clinical programs. Any one of
the following factors, among others, could cause us to require additional funds
or otherwise cause our cash requirements in the future to increase
materially:
|
|
·
|
results of research and
development activities;
|
|
|
·
|
progress of our preclinical
studies or clinical trials;
|
|
|
·
|
results of clinical
trials;
|
|
|
·
|
changes in or terminations of our
relationships with strategic
partners;
|
|
|
·
|
changes in the focus, direction,
or costs of our research and development
programs;
|
|
|
·
|
competitive and technological
advances;
|
|
|
·
|
establishment of marketing and
sales capabilities;
|
|
|
·
|
manufacturing;
|
|
|
·
|
the regulatory approval process;
or
|
|
|
·
|
product
launch.
|
12
At
December 31, 2009, we had cash, cash equivalents and short-term investments of
approximately $6,366,253. During the first quarter of 2010, we
raised an additional $4.6 million. We currently have no commitments
or arrangements for any financing. We may continue to seek additional
capital through public or private financing or collaborative agreements. Our
operations require significant amounts of cash. We may be required to seek
additional capital, whether from sales of equity or debt or additional
borrowings, for the future growth and development of our business. We can give
no assurance as to the availability of such additional capital or, if available,
whether it would be on terms acceptable to us. In addition, we may continue to
seek capital through the public or private sale of securities, if market
conditions are favorable for doing so. If we are successful in raising
additional funds through the issuance of equity securities, stockholders will
likely experience substantial dilution, or the equity securities may have
rights, preferences, or privileges senior to those of the holders of our common
stock. If we raise funds through the issuance of debt securities, those
securities would have rights, preferences, and privileges senior to those of our
common stock. The current credit environment has negatively affected the
economy, and we have considered how it might affect our business. Events
affecting credit market liquidity could increase borrowing costs or limit
availability of funds, and due to the continued adverse trends in the credit
market, it may not be possible to refinance our existing credit facility to take
advantage of lower interest rates. Moreover, the covenants of our term loan
agreement contain provisions that may restrict the debt we may incur in the
future. If we are not successful in obtaining sufficient capital because we are
unable to access the capital markets at financially economical interest rates,
it could reduce our research and development efforts and may materially
adversely affect our future growth, results of operations and financial results,
and we may be required to curtail significantly, or eliminate at least
temporarily, one or more of our drug development programs.
We Rely Exclusively on the Royalty
Payments Based upon Thalomid ® Sales by a Third Party to Produce our
Revenues
We
entered into a licensing agreement in 2001 regarding royalty payments for
Thalomid®, and in
2004, certain provisions of that agreement were satisfied, which then entitled
the Company to share in royalty payments received by Royalty Pharma Finance
Trust on annual Thalomid® sales
above a certain threshold. Based on the licensing agreement royalty formula,
annual royalty sharing commences with Thalomid® annual
sales of approximately $225 million. During the year ended
December 31, 2009, royalty payments from Thalomid® sales by
Celgene Corporation accounted for substantially all of our total
revenues. As Thalomid® is
distributed and sold by Celgene and/or its affiliates, we are reliant on a third
party for our revenues. Our royalty payment in the third quarter of
2009 experienced a decline as compared to the same quarter in 2008, and our
total revenues earned in 2009 were lower than 2008. A wide
variety of events could cause Thalomid® sales to
decline, resulting in a material reduction in our revenues. For
example, if a competing drug gains greater market share or wider acceptance, or
if regulatory approvals for certain uses of Thalomid® are
withdrawn, such events could adversely affect our royalty revenue. In
the event that Celgene determines to cease selling Thalomid® or
target its sales efforts to other proprietary drugs, or unexpected adverse
effects are reported by patients or doctors in connection with the use of
Thalomid®, patient
and physician confidence in Thalomid® as a
treatment could be adversely affected. The inability of one of these third
parties to perform these functions, or the failure of any of these parties to
perform successfully, could cause our revenues to suffer. Additionally, if a
competitor to Celgene successfully introduces a generic pharmaceutical product
equivalent to Thalomid® at a
relatively lower price and bypasses Celgene’s S.T.E.P.S.®
proprietary distribution program, such action could have the effect of reducing
the market share and profitability of Thalomid®, thus
potentially causing a material adverse effect on our revenues and cash flow.
Because we are dependent on sales of Thalomid®, any
reduction in Thalomid® sales
for any reason, including, but not limited to, the reasons described, would
cause our results of operations to suffer.
13
The
Market Price of Our Common Stock May Be Highly Volatile or May Decline
Regardless of Our Operating Performance
Our
common stock price has fluctuated from year-to-year and quarter-to-quarter and
will likely continue to be volatile. Our stock has not traded at the
minimum closing bid price of $1.00 for a period of ten or more days in the past
twelve months. The valuations of many biotechnology companies without
consistent product revenues and earnings are extraordinarily high based on
conventional valuation standards, such as price to earnings and price to sales
ratios. These trading prices and valuations may not be sustained. In the future,
our operating results in a particular period may not meet the expectations of
any securities analysts whose attention we may attract, or those of our
investors, which may result in a decline in the market price of our common
stock. Any negative change in the public’s perception of the prospects of
biotechnology companies could depress our stock price regardless of our results
of operations. These factors may materially and adversely affect the market
price of our common stock.
Our
Existing Term Loan Contains Affirmative and Negative Covenants That May Restrict
our Business and Financing Activities
We entered into a $20 million loan
agreement with General Electric Capital Corporation, as agent for the lenders
party thereto, on September 12, 2007. The loan agreement is
secured by a pledge of all of our assets other than intellectual property,
including the shares of the outstanding capital stock, or other equity
interests, of each of our subsidiaries, and contains a variety of operational
covenants, including limitations on our ability to incur liens or additional
debt, make dispositions, pay dividends, redeem our stock, make certain
investments and engage in certain merger, consolidation or asset sale
transactions and transactions with affiliates, among other restrictions. Any
future debt financing we enter into may involve similar or more onerous
covenants that restrict our operations. Our borrowings under the loan agreement
or any future debt financing we do will need to be repaid, which creates
additional financial risk for our company, particularly if our business, or
prevailing financial market conditions, are not conducive to paying-off or
refinancing our outstanding debt obligations. Furthermore, our failure to comply
with the covenants in the loan agreement could result in an event of default
that, if not cured or waived, could result in the acceleration of all or a
substantial portion of our debt, which could have a material adverse effect on
our cash position, business, prospects, financial condition and results of
operations. Our final scheduled payment under the loan agreement is
in January 2011.
Our
Secured Lender and Preferred Stockholder Would Have Priority in Distributions
Over our Common Stockholders Following a Liquidation Event Affecting the
Company. As a Result, in the Event of a Liquidation Event, our Common
Stockholders Would Receive Distributions Only After Priority Distributions Are
Paid
In the
event of a Liquidation Event (as such term is defined in our Certificate of
Designation of Series A Convertible Preferred Stock), our senior lender would be
repaid first out of the proceeds received. Celgene, the sole holder of our
outstanding Series A Convertible Preferred Stock (“Series A
Preferred”), then would be paid an amount equal to the Series A Preferred
liquidation preference of $10.00 per share of Series A Preferred, plus all
accrued and unpaid dividends on such shares of Series A Preferred, which
totals approximately $7 million as of December 31,
2009. Additionally, unless waived, the holder of the Series A
Preferred would be entitled to receive the Series A liquidation preference
plus all accrued and unpaid dividends prior to any distributions to our common
stockholders upon the occurrence of certain other liquidation
events. As a result, in the event of a Liquidation Event, our common
stockholders’ ability to realize value for their shares would be subject to the
payment of such priority distributions.
Development
of Our Products is at an Early Stage and is Uncertain
Our
proposed products and research programs are in the early stage of clinical
development and require significant, time-consuming and costly research and
development, testing and regulatory clearances. In developing our products, we
are subject to risks of failure that are inherent in the development of these
products and therapeutic procedures. For example, it is possible that any or all
of our proposed products will be ineffective or toxic, or otherwise will fail to
receive necessary FDA clearances. There is a risk that the proposed products
will be uneconomical to manufacture or market or will not achieve market
acceptance. There is also a risk that third parties may hold proprietary rights
that preclude us from marketing our proposed products or that others will market
a superior or equivalent product. Further, our research and development
activities might never result in commercially viable products.
14
A number
of companies in the pharmaceutical and biotechnology industries have suffered
significant setbacks in advanced clinical trials even after promising results in
earlier trials.
In
particular, given the current conditions in the financial markets, an
unfavorable outcome in our Phase 2 trials for ENMD 2076, or a significant delay
in initiating such trials, may require us to delay, reduce the scope of, or
eliminate this program and could have a material adverse effect on our company
and the value of our common stock.
Once a
clinical trial has begun, it may be delayed, suspended or terminated due to a
number of factors, including:
|
|
·
|
ongoing
discussions with regulatory authorities regarding the scope or design of
our clinical trials or requests by them for supplemental information with
respect to our clinical trial
results;
|
|
|
·
|
failure
to conduct clinical trials in accordance with regulatory
requirements;
|
|
|
·
|
lower
than anticipated retention rate of patients in clinical
trials;
|
|
|
·
|
serious
adverse events or side effects experienced by participants;
and
|
|
|
·
|
insufficient
supply or deficient quality of product candidates or other materials
necessary for the conduct of our clinical
trials.
|
Many of
these factors may also ultimately lead to denial of regulatory approval of a
current or potential product candidate. If we experience delays,
suspensions or terminations in a clinical trial, the commercial prospects for
the related product candidate will be harmed, and our ability to generate
product revenues will be delayed.
Our
product candidates are at the clinical stage of development. Although several of
our product candidates have demonstrated some promising results in early
clinical (human) trials and preclinical (animal) studies, they may not
prove to be effective in humans. For example, testing on animals may occur under
different conditions than testing in humans and therefore the results of animal
studies may not accurately predict human experience. Likewise, early clinical
studies may not be predictive of eventual safety or effectiveness results in
larger-scale pivotal clinical trials.
There are
many regulatory steps that must be taken before any of these product candidates
will be eligible for FDA approval and subsequent sale, including the completion
of preclinical and clinical trials. We do not expect that these product
candidates will be commercially available for several years, if
ever.
Technological
Developments By Competitors May Render Our Products Obsolete
If
competitors were to develop superior technologies, our technologies could be
rendered noncompetitive or obsolete, resulting in a material adverse effect to
our business. Developments in the biotechnology and pharmaceutical industries
are expected to continue at a rapid pace. Success depends upon achieving and
maintaining a competitive position in the development of products and
technologies. Competition from other biotechnology and pharmaceutical companies
can be intense. Many competitors have substantially greater research and
development capabilities, marketing, financial and managerial resources and
experience in the industry. Even if a competitor creates a technology that is
not superior, we may not be able to compete with such technology.
We
Must Show the Safety and Efficacy of Our Product Candidates Through Clinical
Trials, the Results of Which are Uncertain
Before
obtaining regulatory approvals for the commercial sale of our products, we must
demonstrate, through preclinical studies (animal testing) and clinical trials
(human testing), that our proposed products are safe and effective for use in
each target indication. Testing of our product candidates will be required, and
failure can occur at any stage of testing. Clinical trials may not demonstrate
sufficient safety and efficacy to obtain the required regulatory approvals or
result in marketable products. The failure to adequately demonstrate the safety
and efficacy of a product under development could delay or prevent regulatory
approval of the potential product.
15
Clinical
trials for the product candidates we are developing may be delayed by many
factors, including that potential patients for testing are limited in number.
The failure of any clinical trials to meet applicable regulatory standards could
cause such trials to be delayed or terminated, which could further delay the
commercialization of any of our product candidates. Newly emerging safety risks
observed in animal or human studies also can result in delays of ongoing or
proposed clinical trials. Any such delays will increase our product development
costs. If such delays are significant, they could negatively affect our
financial results and the commercial prospects for our products.
The
Independent Clinical Investigators and Contract Research Organizations That We
Rely Upon to Assist in the Conduct of Our Clinical Trials May Not Be Diligent,
Careful or Timely, and May Make Mistakes, in the Conduct of Our
Trials
We depend
on independent clinical investigators and contract research organizations, or
CROs, to assist in the conduct of our clinical trials under their agreements
with us. The investigators are not our employees, and we cannot control the
amount or timing of resources that they devote to our programs. If independent
investigators fail to devote sufficient time and resources to our drug
development programs, or if their performance is substandard, it will delay the
approval of our FDA applications and our introduction of new drugs. The CROs we
contract with to assist with the execution of our clinical trials play a
significant role in the conduct of the trials and the subsequent collection and
analysis of data. Failure of the CROs to meet their obligations could adversely
affect clinical development of our products.
The
Success of Our Business Depends Upon the Members of Our Senior Management Team,
Our Scientific Staff and Our Ability to Continue to Attract and Retain Qualified
Scientific, Technical and Business Personnel
We are
dependent on the principal members of our reconstituted senior management team
and scientific staff for our business success. The loss of any of these people
could impede the achievement of our development and business objectives. We do
not carry key man life insurance on the lives of any of our key personnel. There
is intense competition for human resources, including management, in the
scientific fields in which we operate and there can be no assurance that we will
be able to attract and retain qualified personnel necessary for the successful
development of ENMD-2076, and any expansion into areas and activities requiring
additional expertise. In addition, there can be no assurance that such personnel
or resources will be available when needed. In addition, we rely on a
significant number of consultants to assist us in formulating our clinical
strategy and other business activities. All of our consultants may have
commitments to, or advisory or consulting agreements with, other entities that
may limit their availability to us.
We
May Need New Collaborative Partners to Further Develop and Commercialize
Products, and if We Enter Into Such Arrangements, We May Give Up Control Over
the Development and Approval Process and Decrease our Potential
Revenue
We plan
to develop and commercialize our product candidates both with and without
corporate alliances and partners. Nonetheless, we intend to explore
opportunities for new corporate alliances and partners to help us develop,
commercialize and market our product candidates. We expect to grant to our
partners certain rights to commercialize any products developed under these
agreements, and we may rely on our partners to conduct research and development
efforts and clinical trials on, obtain regulatory approvals for, and manufacture
and market any products licensed to them. Each individual partner will seek to
control the amount and timing of resources devoted to these activities
generally. We anticipate obtaining revenues from our strategic partners under
such relationships in the form of research and development payments and payments
upon achievement of certain milestones. Since we generally expect to obtain a
royalty for sales or a percentage of profits of products licensed to third
parties, our revenues may be less than if we retained all commercialization
rights and marketed products directly. In addition, there is a risk that our
corporate partners will pursue alternative technologies or develop competitive
products as a means for developing treatments for the diseases targeted by our
programs.
16
We may
not be successful in establishing any collaborative arrangements. Even if we do
establish such collaborations, we may not successfully commercialize any
products under or derive any revenues from these arrangements. Our strategy also
involves entering into multiple, concurrent strategic alliances to pursue
commercialization of our core technologies. There is a risk that we will be
unable to manage simultaneous programs successfully. With respect to existing
and potential future strategic alliances and collaborative arrangements, we will
depend on the expertise and dedication of sufficient resources by these outside
parties to develop, manufacture, or market products. If a strategic alliance or
collaborative partner fails to develop or commercialize a product to which it
has rights, we may not recognize any revenues on that particular
product.
We
Have No Current Manufacturing or Marketing Capacity and Rely on Only One
Supplier For Some of Our Products
We do not
expect to manufacture or market products in the near term, but we may try to do
so in certain cases. We do not currently have the capacity to manufacture or
market products and we have limited experience in these activities. The
manufacturing processes for all of the small molecules we are developing have
not yet been tested at commercial levels, and it may not be possible to
manufacture these materials in a cost-effective manner. If we elect
to perform these functions, we will be required to either develop these
capacities, or contract with others to perform some or all of these tasks. We
may be dependent to a significant extent on corporate partners, licensees, or
other entities for manufacturing and marketing of products. If we engage
directly in manufacturing or marketing, we will require substantial additional
funds and personnel and will be required to comply with extensive regulations.
We may be unable to develop or contract for these capacities when required to do
so in connection with our business.
We depend
on our third-party manufacturers to perform their obligations effectively and on
a timely basis. These third parties may not meet their obligations and any such
non-performance may delay clinical development or submission of products for
regulatory approval, or otherwise impair our competitive position. Any
significant problem experienced by one of our suppliers could result in a delay
or interruption in the supply of materials to us until such supplier resolves
the problem or an alternative source of supply is located. Any delay or
interruption would likely lead to a delay or interruption of manufacturing
operations, which could negatively affect our operations. Although we have
identified alternative suppliers for our product candidates, we have not entered
into contractual or other arrangements with them. If we needed to use an
alternate supplier for any product, we would experience delays while we
negotiated an agreement with them for the manufacture of such product. In
addition, we may be unable to negotiate manufacturing terms with a new supplier
as favorable as the terms we have with our current suppliers.
Problems
with any manufacturing processes could result in product defects, which could
require us to delay shipment of products or recall products previously shipped.
In addition, any prolonged interruption in the operations of the manufacturing
facilities of one of our sole-source suppliers could result in the cancellation
of shipments. A number of factors could cause interruptions, including equipment
malfunctions or failures, or damage to a facility due to natural disasters or
otherwise. Because our manufacturing processes are or are expected to be highly
complex and subject to a lengthy FDA approval process, alternative qualified
production capacity may not be available on a timely basis or at all.
Difficulties or delays in our manufacturing could increase our costs and damage
our reputation.
The
manufacture of pharmaceutical products can be an expensive, time consuming, and
complex process. Manufacturers often encounter difficulties in scaling-up
production of new products, including quality control and assurance and
shortages of personnel. Delays in formulation and scale-up to commercial
quantities could result in additional expense and delays in our clinical trials,
regulatory submissions, and commercialization.
17
Failure
of Manufacturing Facilities Producing Our Product Candidates to Maintain
Regulatory Approval Could Delay or Otherwise Hinder Our Ability to Market Our
Product Candidates
Any
manufacturer of our product candidates will be subject to applicable Good
Manufacturing Practices (GMP) prescribed by the FDA or other rules and
regulations prescribed by foreign regulatory authorities. We and any of our
collaborators may be unable to enter into or maintain relationships either
domestically or abroad with manufacturers whose facilities and procedures comply
or will continue to comply with GMP and who are able to produce our small
molecules in accordance with applicable regulatory standards. Failure by a
manufacturer of our products to comply with GMP could result in significant time
delays or our inability to obtain marketing approval or, should we have market
approval, for such approval to continue. Changes in our manufacturers could
require new product testing and facility compliance inspections. In the United
States, failure to comply with GMP or other applicable legal requirements can
lead to federal seizure of violated products, injunctive actions brought by the
federal government, inability to export product, and potential criminal and
civil liability on the part of a company and its officers and
employees.
We
Depend on Patents and Other Proprietary Rights, Some of Which are
Uncertain
Our
success will depend in part on our ability to obtain patents for our products,
both in the United States and abroad. The patent position of biotechnology and
pharmaceutical companies in general is highly uncertain and involves complex
legal and factual questions. Risks that relate to patenting our products include
the following:
|
|
·
|
our failure to obtain additional
patents;
|
|
|
·
|
challenge, invalidation, or
circumvention of patents already issued to
us;
|
|
|
·
|
failure of the rights granted
under our patents to provide sufficient
protection;
|
|
|
·
|
independent development of
similar products by third parties;
or
|
|
|
·
|
ability of third parties to
design around patents issued to our collaborators or
us.
|
Our
potential products may conflict with composition, method, and use of patents
that have been or may be granted to competitors, universities or others. As the
biotechnology industry expands and more patents are issued, the risk increases
that our potential products may give rise to claims that may infringe the
patents of others. Such other persons could bring legal actions against us
claiming damages and seeking to enjoin clinical testing, manufacturing and
marketing of the affected products. Any such litigation could result in
substantial cost to us and diversion of effort by our management and technical
personnel. If any of these actions are successful, in addition to any potential
liability for damages, we could be required to obtain a license in order to
continue to manufacture or market the affected products. We may not prevail in
any action and any license required under any needed patent might not be made
available on acceptable terms, if at all.
We are a
party to license agreements that require us to make milestone payments upon
attainment of certain regulatory milestones. Failure to meet such milestones
could result in the loss of certain rights to compounds covered under such
license agreements.
We also
rely on trade secret protection for our confidential and proprietary
information. However, trade secrets are difficult to protect and others may
independently develop substantially equivalent proprietary information and
techniques and gain access to our trade secrets and disclose our technology. We
may be unable to meaningfully protect our rights to unpatented trade secrets. We
require our employees to complete confidentiality training that specifically
addresses trade secrets. All employees, consultants, and advisors are required
to execute a confidentiality agreement when beginning an employment or a
consulting relationship with us. The agreements generally provide that all trade
secrets and inventions conceived by the individual and all confidential
information developed or made known to the individual during the term of the
relationship automatically become our exclusive property. Employees and
consultants must keep such information confidential and may not disclose such
information to third parties except in specified circumstances. However, these
agreements may not provide meaningful protection for our proprietary information
in the event of unauthorized use or disclosure of such
information.
18
To the
extent that consultants, key employees, or other third parties apply
technological information independently developed by them or by others to our
proposed projects, disputes may arise as to the proprietary rights to such
information. Any such disputes may not be resolved in our favor. Certain of our
consultants are employed by or have consulting agreements with other companies
and any inventions discovered by them generally will not become our
property.
Our
Potential Products Are Subject to Government Regulatory Requirements and an
Extensive Approval Process
Our
research, development, preclinical and clinical trials, manufacturing, and
marketing of most of our product candidates are subject to an extensive
regulatory approval process by the FDA and other regulatory agencies in the
United States and abroad. The process of obtaining FDA and other required
regulatory approvals for drug and biologic products, including required
preclinical and clinical testing, is time consuming and expensive. Even after
spending time and money, we may not receive regulatory approvals for clinical
testing or for the manufacturing or marketing of any products. Our collaborators
or we may encounter significant delays or costs in the effort to secure
necessary approvals or licenses. Even if we obtain regulatory clearance for a
product, that product will be subject to continuing review. Later discovery of
previously unknown defects or failure to comply with the applicable regulatory
requirements may result in restrictions on a product’s marketing or withdrawal
of the product from the market, as well as possible civil or criminal
penalties.
Potential
Products May Subject Us to Product Liability for Which Insurance May Not Be
Available
The use
of our potential products in clinical trials and the marketing of any
pharmaceutical products may expose us to product liability claims. We have
obtained a level of liability insurance coverage that we believe is adequate in
scope and coverage for our current stage of development. However, our present
insurance coverage may not be adequate to protect us from liabilities we might
incur. In addition, our existing coverage will not be adequate as we further
develop products and, in the future, adequate insurance coverage and
indemnification by collaborative partners may not be available in sufficient
amounts or at a reasonable cost. If a product liability claim or series of
claims are brought against us for uninsured liabilities, or in excess of our
insurance coverage, the payment of such liabilities could have a negative effect
on our business and financial condition.
We
Acquired Miikana in 2006 in a Strategic Transaction and May Engage in Other
Strategic Transactions, Which Could Negatively Affect Our Business and
Earnings
In
January 2006, we acquired Miikana Therapeutics, Inc., a clinical-stage
biopharmaceutical company. In 2010, we may consider strategic
and other corporate transactions as opportunities present
themselves. There are risks associated with such activities. These
risks include, among others, incorrectly assessing the quality of a prospective
strategic partner, encountering greater than anticipated costs in integration,
being unable to profitably deploy assets acquired in the transaction, such as
drug candidates, possible dilution to our stockholders, and the loss of key
employees due to changes in management. Further, strategic transactions may
place additional constraints on our resources by diverting the attention of our
management from our business operations. Our newly constituted senior management
team does not have substantial experience with acquisitions. To the extent we
issue securities in connection with additional transactions, these transactions
and related issuances may have a dilutive effect on earnings per share and our
ownership. Our earnings, financial condition, and prospects after an acquisition
depend in part on our ability to successfully integrate the operations of the
acquired business or technologies. We may be unable to integrate operations
successfully or to achieve expected cost savings. Any cost savings which are
realized may be offset by losses in revenues or other charges to
earnings.
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS.
|
None.
19
|
ITEM 2.
|
PROPERTIES.
|
As of
December 31, 2009, EntreMed leased approximately 8,554 square feet of space in
Rockville, Maryland where its headquarters are located. In addition,
as of December 31, 2009, the Company leased office space in Durham, North
Carolina where our clinical and regulatory operations are based and laboratory
space in Toronto, Ontario where our research facility is currently
based. On March 4, 2010, we amended the Rockville lease to reduce the
rented space from 8,554 square feet to 4,367 square feet and accordingly reduced
the rent on a proportional basis.
|
ITEM 3.
|
LEGAL
PROCEEDINGS.
|
EntreMed
is subject in the normal course of business to various legal proceedings in
which claims for monetary or other damages may be
asserted. Management does not believe such legal proceedings, except
as otherwise disclosed herein, are material.
|
ITEM 4.
|
RESERVED.
|
Reserved.
PART
II
|
ITEM 5.
|
MARKET
FOR REGISTRANT'S COMMON EQUITY RELATED STOCKHOLDER MATTERS AND ISSUER
PURCHASES OF EQUITY SECURITIES.
|
Market
for Common Equity
On
April 4, 2008, we received a letter from The NASDAQ Stock Market LLC
(“NASDAQ”) advising that for the previous 30 consecutive business days, the bid
price of the Company’s common stock had closed below the minimum $1.00 per share
requirement for continued inclusion on The NASDAQ Global Market. The
letter also advised us that failure to comply with this minimum bid price
requirement, or any other listing standard applicable to issuers listed on The
NASDAQ Global Market, by October 1, 2008, would result in our common stock
being ineligible for quotation on The NASDAQ Global Market.
On
September 22, 2008, we submitted an application to transfer the trading of
our common stock to The NASDAQ Capital Market and our common stock commenced
trading on The NASDAQ Capital Market on October 3, 2008. Our trading symbol
remained as “ENMD” and the trading of our stock was unaffected by the
transfer.
NASDAQ
suspended the enforcement of the minimum bid price rule, because of the current
extraordinary market conditions, until July 31, 2009. Upon
reinstatement of the rule on August 3, 2009, under NASDAQ rules, we had
until January 15, 2010, to regain compliance with the minimum bid price
standard. We did not regain compliance with the minimum bid price
standard of the NASDAQ rules by January 15, 2010.
On
January 19, 2010, we received a Staff Determination letter from NASDAQ stating
that we were not in compliance with the continued listing rules and that our
Common Stock would be delisted unless we requested an appeal of such
determination. On January 20, 2010, we filed an appeal of the
Staff's determination to a NASDAQ Hearings Panel (the “Panel”), pursuant to the
procedures set forth in the NASDAQ Marketplace Rules. The hearing request
stayed the delisting of the Company’s securities pending the Panel's
decision. On February 25, 2010, we met with the Panel providing them
a plan of action, with the intention of returning to compliance with NASDAQ’s
requirements. On March 23, 2010, we received a decision letter from
the Panel granting our request to extend the compliance date for an additional
180 days or until July 16, 2010.
20
The
following table sets forth the high and low closing price for our common stock
by quarter, as reported by the Nasdaq Stock Market, for the periods
indicated:
|
HIGH
|
LOW
|
|||||||
|
2008:
|
||||||||
|
First
Quarter
|
$ | 1.25 | $ | 0.57 | ||||
|
Second
Quarter
|
0.90 | 0.55 | ||||||
|
Third
Quarter
|
0.62 | 0.33 | ||||||
|
Fourth
Quarter
|
0.45 | 0.16 | ||||||
|
2009:
|
||||||||
|
First
Quarter
|
$ | 0.49 | $ | 0.16 | ||||
|
Second
Quarter
|
0.81 | 0.39 | ||||||
|
Third
Quarter
|
0.62 | 0.37 | ||||||
|
Fourth
Quarter
|
1.38 | 0.46 | ||||||
|
2010:
|
||||||||
|
First
Quarter (through March 25, 2010)
|
$ | 1.08 | $ | 0.65 | ||||
On March
25, 2010, the closing price of our common stock, as reported by the Nasdaq
Capital Market, was $0.71 per share. As of March 25, 2010 there were
approximately 829 holders of record of our common stock.
Dividend
Policy
Since our
initial public offering in 1996, we have not paid cash dividends on our common
stock. We currently anticipate that any earnings will be retained for the
continued development of our business and we do not anticipate paying any cash
dividends on our common stock in the foreseeable future.
Our Series A Convertible Preferred
Stock (“Series A Preferred”) is held exclusively by Celgene
Corporation. The Series A Preferred accrues dividends on each share
of Series A Preferred at 6% per year of the original issue price of the Series A
Preferred Stock, which is $5.00 per share. In the event of any
liquidation, dissolution or winding up the Company, or the bankruptcy of the
Company, all accrued and unpaid dividends will be added to the liquidation
preference of such shares of Series A Preferred. As of December 31,
2009, accrued Series A Preferred dividends totaled $7,035,000.
21
STOCK
PRICE PERFORMANCE PRESENTATION
The
following chart compares the cumulative total stockholder return on the
Company’s Shares with the cumulative total stockholder return of the NASDAQ
Stock Market – U. S. Index, and the NASDAQ Pharmaceuticals Index.
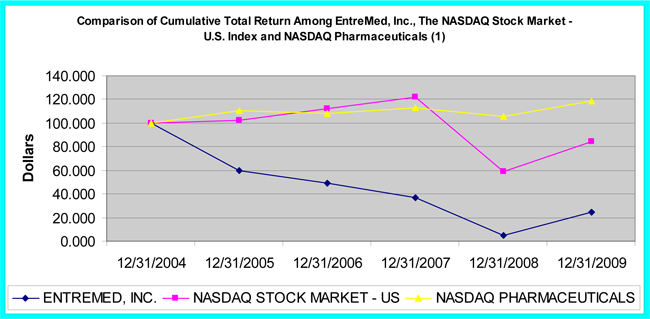
|
12/31/2004
|
12/31/2005
|
12/31/2006
|
12/31/2007
|
12/31/2008
|
12/31/2009
|
|||||||||||||||||||
|
ENTREMED,
INC.
|
100.000 | 59.877 | 48.765 | 37.037 | 4.938 | 24.691 | ||||||||||||||||||
|
NASDAQ
STOCK MARKET - US
|
100.000 | 102.135 | 112.187 | 121.681 | 58.639 | 84.282 | ||||||||||||||||||
|
NASDAQ
PHARMACEUTICALS
|
100.000 | 110.123 | 107.793 | 113.364 | 105.476 | 118.522 | ||||||||||||||||||
|
(1)
|
Assumes
$100 invested on December 31, 2004 and assumes dividends are
reinvested. Measurement points begin with the date of the
assumed investment and include the last day of each of the subsequent 5
years through and including December 31, 2009. The material in
this chart is not soliciting material, is not deemed filed with the SEC
and is not incorporated by reference in any filing of the Company under
the Securities Act of 1933, as amended, (the “1933 Act”) or the 1934 Act,
whether made before or after the date of this proxy statement and
irrespective of any general incorporation language in such
filing.
|
22
|
ITEM 6.
|
SELECTED
FINANCIAL DATA.
|
The
following selected consolidated financial data set forth below has been derived
from our audited consolidated financial statements. The data should be read in
conjunction with the consolidated financial statements and related notes,
Management's Discussion and Analysis of Financial Condition and Results of
Operations in Item 7 and other financial information included elsewhere in this
annual report on Form 10-K.
|
Year Ended December 31,
|
||||||||||||||||||||
|
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
|
STATEMENTS
OF OPERATIONS DATA:
|
||||||||||||||||||||
|
Revenues
|
||||||||||||||||||||
|
License
fees
|
$ | - | $ | - | $ | - | $ | - | $ | 590,992 | ||||||||||
|
Royalty
revenues
|
4,989,725 | 7,472,061 | 7,393,463 | 6,881,799 | 5,310,439 | |||||||||||||||
|
Other
|
294,333 | 5,158 | 2,188 | 12,559 | 16,624 | |||||||||||||||
|
Total
revenues
|
5,284,058 | 7,477,219 | 7,395,651 | 6,894,358 | 5,918,055 | |||||||||||||||
|
Expenses:
|
||||||||||||||||||||
|
Research
and development
|
7,901,522 | 20,069,229 | 23,739,392 | 21,671,117 | 17,325,048 | |||||||||||||||
|
General
and administrative
|
4,104,287 | 7,764,532 | 7,386,570 | 7,393,722 | 5,920,455 | |||||||||||||||
|
Acquired
In-Process R&D
|
- | 2,000,000 | - | 29,481,894 | - | |||||||||||||||
|
Interest
expense
|
1,493,400 | 2,216,163 | 793,393 | 156,787 | - | |||||||||||||||
|
Fixed
asset impairment loss
|
- | 171,576 | - | - | - | |||||||||||||||
|
Other
expense
|
70,929 | - | - | - | - | |||||||||||||||
|
Investment
income
|
(69,702 | ) | (882,253 | ) | (2,112,583 | ) | (1,867,204 | ) | (1,010,771 | ) | ||||||||||
|
Gain
on sale of asset
|
- | - | - | (52,901 | ) | (3,420 | ) | |||||||||||||
|
Net
loss
|
$ | (8,216,378 | ) | $ | (23,862,028 | ) | $ | (22,411,121 | ) | $ | (49,889,057 | ) | $ | (16,313,257 | ) | |||||
|
Dividends
on Series A convertible preferred stock
|
( 1,005,000 | ) | ( 1,005,000 | ) | ( 1,005,000 | ) | ( 1,005,000 | ) | ( 1,005,000 | ) | ||||||||||
|
Net
loss attributable to common shareholders
|
$ | ( 9,221,378 | ) | $ | ( 24,867,028 | ) | $ | ( 23,416,121 | ) | $ | ( 50,894,057 | ) | $ | ( 17,318,257 | ) | |||||
|
Net
loss per share
|
$ | (0.11 | ) | $ | (0.29 | ) | $ | (0.28 | ) | $ | (0.71 | ) | $ | (0.36 | ) | |||||
|
Weighted
average number of shares outstanding
|
87,757,765 | 86,479,768 | 84,166,552 | 71,873,734 | 48,176,914 | |||||||||||||||
|
As of December 31,
|
||||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
||||||||||||||||
|
BALANCE
SHEET DATA:
|
||||||||||||||||||||
|
Cash
and cash equivalents and short-term investments
|
$ | 6,366,253 | $ | 24,291,173 | $ | 47,748,191 | $ | 50,570,097 | $ | 30,082,388 | ||||||||||
|
Working
capital
|
(1,395,158 | ) | 14,782,482 | 42,929,602 | 46,268,554 | 28,510,176 | ||||||||||||||
|
Total
assets
|
10,067,028 | 28,923,395 | 53,014,908 | 55,719,534 | 36,431,885 | |||||||||||||||
|
Accumulated
deficit
|
(366,126,413 | ) | (357,910,035 | ) | (334,048,007 | ) | (311,636,886 | ) | (261,747,829 | ) | ||||||||||
|
Total
stockholders' (deficit) equity
|
(1,925,980 | ) | 5,922,166 | 26,896,978 | 46,963,219 | 29,369,190 | ||||||||||||||
23
|
ITEM 7.
|
MANAGEMENT'S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS.
|
The
following discussion should be read in conjunction with the Consolidated
Financial Statements and Notes thereto appearing elsewhere in this report. See
also "Risk Factors" in Item 1A of this Annual Report.
OVERVIEW
We are a
clinical-stage pharmaceutical company focused on developing ENMD-2076, an Aurora
A and angiogenic kinase inhibitor for the treatment of
cancer. ENMD-2076 is currently in Phase 1 studies in advanced
cancers, multiple myeloma and leukemia, and we expect the initiation of a Phase
2 study in ovarian cancer in the second quarter of 2010. ENMD-2076 is
a novel orally-active, Aurora A/angiogenic kinase inhibitor with potent activity
against Aurora A and multiple tyrosine kinases linked to cancer and inflammatory
diseases. ENMD-2076 is relatively selective for the Aurora A isoform
in comparison to Aurora B. Aurora kinases are key regulators of the
process of mitosis, or cell division, and are often over-expressed in human
cancers. ENMD-2076 exerts its effects through multiple mechanisms of action,
including antiproliferative activity and the inhibition of angiogenesis.
ENMD-2076 has demonstrated significant, dose-dependent preclinical activity as a
single agent, including tumor regression, in multiple xenograft models (e.g.
breast, colon, leukemia), as well as activity towards ex vivo-treated human
leukemia patient cells.
ENMD-2076
has received orphan drug designation for the treatment of ovarian cancer,
multiple myeloma and acute myeloid leukemia (“AML”).
ENMD-2076
is our only program currently under active clinical evaluation. This
prioritization allows us to direct the majority of our resources to the clinical
development of ENMD-2076. However, we also own intellectual property
for our other therapeutic candidates. So that we can continue to
accelerate the development of ENMD-2076, we do not intend to initiate additional
new studies for these programs unless additional significant financing becomes
available to us. Our other therapeutic candidates include MKC-1, an
oral cell-cycle inhibitor with activity against the mTOR pathway that has
completed multiple Phase 2 clinical trials for cancer, and ENMD-1198, a novel
antimitotic agent that has completed a Phase 1 study in advanced
cancers. We also have an approved Investigational New Drug
Application (IND) for the use of Panzem® in
rheumatoid arthritis (RA) treatment. All of our candidates are
multi-mechanism drugs that target disease cells and the blood vessels that
nourish them, which we believe can be developed to be safe and convenient, and
provide the potential for improved patient outcomes.
We have
incurred substantial operating losses since our inception due in large part to
expenditures for our research and development activities. At December 31, 2009,
we had an accumulated deficit of $366 million. We expect to continue to
incur expenses, resulting in operating losses, for the foreseeable future due
to, among other factors, our continuing clinical trials, planned future clinical
trials, and other anticipated research and development activities. Based on
current plans, we expect our current available cash and cash equivalents to meet
our cash requirements into the fourth quarter of fiscal 2010. Therefore, there
exists substantial doubt about our ability to continue as a going concern. We
will require significant additional funding prior to January 1, 2011 to
fund operations until such time, if ever, we become profitable. We intend to
augment our cash and cash equivalent balances as of December 31, 2009 by
pursuing other forms of capital infusion, including strategic alliances or
collaborative development opportunities with organizations that have
capabilities and/or products that are complementary to our capabilities and
products in order to continue the development of our potential product candidate
that we intend to pursue to commercialization. However, there can be no
assurance that adequate additional financing under such arrangements will be
available to us on terms that we deem acceptable, if at all.
On
January 11, 2010, we sold 3,125,000 shares of our common stock, par value $0.01
per share, to an institutional investor for an aggregate purchase price of
$2,500,000 or $0.80 per share. On February 3, 2010, we sold 3,846,154
shares of our common stock to the same institutional investor for an aggregate
purchase price of $2,500,000, or $0.65 per share. Our net proceeds
from these two offerings were approximately $4.6 million. Additional
funds raised by issuing equity securities, may result in dilution to existing
shareholders. If we fail to obtain additional capital when needed, we
may be required to delay, scale back, or eliminate our clinical
program.
24
CRITICAL
ACCOUNTING POLICIES AND THE USE OF ESTIMATES
The
preparation of our financial statements in conformity with accounting principles
generally accepted in the U.S. requires management to make estimates and
assumptions that affect the amounts reported in our consolidated financial
statements and accompanying notes. Actual results could differ materially from
those estimates. Our critical accounting policies, including the
items in our financial statements requiring significant estimates and judgments,
are as follows:
|
|
-
|
Going
Concern - A fundamental principle of the preparation
of financial statements in accordance with GAAP is the assumption that an
entity will continue in existence as a going concern, which contemplates
continuity of operations and the realization of assets and settlement of
liabilities occurring in the ordinary course of business. This
principle is applicable to all entities except for entities in liquidation
or entities for which liquidation appears imminent. In
accordance with this requirement, our policy is to prepare our
consolidated financial statements on a going concern basis unless we
intend to liquidate or have no other alternative but to
liquidate. As a result of our operational losses and the
potential that we may be unable to meet our cash requirements for the next
twelve months, there is substantial doubt about our ability to continue as
a going concern. While we have prepared our consolidated
financial statements on a going concern basis, if we do not receive
additional funding, our ability to continue as a going concern may be
impacted. Our consolidated financial statements included in
this Annual Report on Form 10-K do not reflect any adjustments that might
specifically result from the outcome of this
uncertainty.
|
|
|
-
|
Revenue
Recognition - We recognize revenue in accordance with the provisions of
authoritative guidance issued, whereby revenue is not recognized until it
is realized or realizable and earned. Revenue is recognized
when all of the following criteria are met: persuasive evidence
of an arrangement exists, delivery has occurred or services have been
rendered, the price to the buyer is fixed and determinable and
collectibility is reasonably
assured.
|
|
|
-
|
Royalty
Revenue – Royalties from licenses are based on third-party sales and
recorded as earned in accordance with contract terms, when third-party
results are reliably measured and collectibility is reasonably assured.
The majority of our 2009 revenues were from royalties on the sale of
Thalomid®,
which we began to recognize in the third quarter. In 2004,
certain provisions of a purchase agreement dated June 14, 2001 by and
between Bioventure Investments kft (“Bioventure”) and the Company were
satisfied and, as a result, beginning in 2005 we became entitled to share
in the royalty payments received by Royalty Pharma Finance Trust,
successor to Bioventure, on annual Thalomid®
sales above a certain threshold. Based on the licensing
agreement royalty formula, annual royalty sharing commences with
Thalomid®
annual sales of approximately $225
million.
|
|
|
-
|
The
Company is also eligible to receive royalties from Oxford Biomedica, PLC
based on a portion of the net sales of products developed for the
treatment of ophthalmic (eye) diseases based in part on
Endostatin. We received our first royalty in the amount of
$386,000 under this agreement in 2009 and have accrued at December 31,
2009 a portion of this payment payable to Children’s Medical Center
Corporation under the Company’s original Endostatin license agreement with
Children’s.
|
|
|
-
|
In
the future, royalty payments, if any, will be recorded as revenue when
received and/or when collectibility is reasonably
assured.
|
|
|
-
|
Research
and Development - Research and development expenses consist primarily of
compensation and other expenses related to research and development
personnel, research collaborations, costs associated with preclinical
testing and clinical trials of our product candidates, including the costs
of manufacturing drug substance and drug product, regulatory maintenance
costs, and facilities expenses. Research and development costs
are expensed as incurred.
|
25
|
|
-
|
Expenses
for Clinical Trials – Expenses for clinical trials are incurred from
planning through patient enrollment to reporting of the
data. We estimate expenses incurred for clinical trials that
are in process based on patient enrollment and based on clinical data
collection and management. Costs that are associated with
patient enrollment are recognized as each patient in the clinical trial
completes the enrollment process. Estimated clinical trial
costs related to enrollment can vary based on numerous factors, including
expected number of patients in trials, the number of patients that do not
complete participation in a trial, and when a patient drops out of a
trial. Costs that are based on clinical data collection and
management are recognized in the reporting period in which services are
provided. In the event of early termination of a clinical
trial, we would accrue an amount based on estimates of the remaining
non-cancelable obligations associated with winding down the clinical
trial.
|
|
|
-
|
Stock-Based
Compensation – All share-based payment transactions are recognized in the
financial statements at their fair values. Using the
straight-line expense attribution method over the requisite service
period, which is generally the option vesting term of three years,
share-based compensation expense recognized in the years ended December
31, 2009, 2008 and 2007 totaled $320,000, $1,090,000 and $1,455,000,
respectively.
|
The
determination of fair value of stock-based payment awards on the date of grant
using the Black-Scholes model is affected by our stock price, as well as the
input of other subjective assumptions. These assumptions include, but
are not limited to, the expected forfeiture rate and expected term of stock
options and our expected stock price volatility over the term of the
awards. Changes in the assumptions can materially affect the fair
value estimates.
Any
future changes to our share-based compensation strategy or programs would likely
affect the amount of compensation expense recognized.
RESULTS
OF OPERATIONS
Years
Ended December 31, 2009, 2008 and 2007.
Revenues. Revenues decreased
29% in 2009 to $5,284,000 from $7,477,000 in 2008 after increasing slightly in
2008 from $7,396,000 in 2007. The three years presented reflect royalty
revenues. The decrease in 2009 revenues results from decreased
royalty revenue earned on sales of Thalomid®. Beginning
in 2005, we became entitled to share in the royalty payments received by Royalty
Pharma Finance Trust on annual Thalomid® sales
above approximately $225 million. Thalomid® sales in
2009, 2008 and 2007 surpassed the annual revenue targets in the third quarter of
each fiscal year and we recorded royalty revenues of $4,990,000, $7,472,000 and
$7,393,000, respectively.
Research and Development
Expenses. Our 2009 research and development expenses totaled
$7,902,000, a 61% decrease from costs incurred in 2008. The 2009 amount reflects
direct project costs for ENMD-2076 of $5,036,000, $449,000 for MKC-1, $177,000
for ENMD-1198 and $126,000 for Panzem®
oncology. In 2008, our research and development expenses totaled
$20,069,000, reflecting direct project costs for ENMD-2076 of $3,707,000,
$3,716,000 for MKC-1, $3,298,000 for ENMD-1198 and $3,343,000 for Panzem®
oncology. Research and development expenses totaled $23,739,000 in
2007, which included direct project costs of $3,958,000 for ENMD-2076,
$3,241,000 for MKC-1, $2,200,000 for ENMD-1198 and $7,673,000 for Panzem®
oncology. The significant decrease in 2009 research and development
spending as compared to costs incurred in 2008 and in 2007, results primarily
from our decision to focus our resources on the clinical development of
ENMD-2076. Our 2008 and 2007 costs reflect clinical and manufacturing
activities in our discontinued programs.
At December 31, 2009, accumulated
direct project expenses for Panzem® oncology
were $54,244,000; direct ENMD-1198 project expenses totaled $13,143,000; and,
since acquired, accumulated direct project expenses for ENMD-2076 totaled
$14,158,000 and for MKC-1, accumulated project expenses totaled
$10,406,000. Our research and development expenses also include
non-cash stock-based compensation totaling $116,000, $233,000 and $353,000,
respectively, for 2009, 2008 and 2007. The decreases in stock-based
compensation expense are related to lower fair values on fewer stock options
granted in 2009 and 2008. The balance of our research and development
expenditures includes facility costs and other departmental overhead, and
expenditures related to the non-clinical support of our
programs.
26
We expect our research and development
expenses in 2010 to increase as compared to expenses in 2009, as we expand
ENMD-2076 into Phase 2 and continue to focus on the clinical development of
ENMD-2076 and correlative research. We will continue to conduct research
on ENMD-2076 in order to comply with stipulations made by the FDA, as well as to
increase understanding of the mechanism of action and toxicity parameters of
ENMD-2076 and its metabolites. Completion of clinical development may take
several years or more, but the length of time generally varies substantially
according to the type, complexity, novelty and intended use of a product
candidate.
We estimate that clinical trials of the
type we generally conduct are typically completed over the following
timelines:
|
CLINICAL PHASE
|
ESTIMATED
COMPLETION
PERIOD
|
|
|
Phase
I
|
1-2 Years
|
|
|
Phase
II
|
2-3 Years
|
|
|
Phase
III
|
2-4 Years
|
The duration and the cost of clinical
trials may vary significantly over the life of a project as a result of
differences arising during the clinical trial protocol, including, among others,
the following:
|
|
-
|
the
number of patients that ultimately participate in the
trial;
|
|
|
-
|
the
duration of patient follow-up that seems appropriate in view of the
results;
|
|
|
-
|
the
number of clinical sites included in the trials;
and
|
|
|
-
|
the
length of time required to enroll suitable patient
subjects.
|
We test our potential product
candidates in numerous preclinical studies to identify indications for which
they may be product candidates. We may conduct multiple clinical
trials to cover a variety of indications for each product
candidate. As we obtain results from trials, we may elect to
discontinue clinical trials for certain product candidates or for certain
indications in order to focus our resources on more promising product candidates
or indications.
Our proprietary drug candidates have
also not yet achieved FDA regulatory approval, which is required before we can
market them as therapeutic products. In order to proceed to subsequent clinical
trial stages and to ultimately achieve regulatory approval, the FDA must
conclude that our clinical data establish safety and
efficacy. Historically, the results from preclinical testing and
early clinical trials have often not been predictive of results obtained in
later clinical trials. A number of new drugs and biologics have shown promising
results in clinical trials, but subsequently failed to establish sufficient
safety and efficacy data to obtain necessary regulatory approvals.
Our business strategy now includes
entering into collaborative arrangements with third parties to complete the
development and commercialization of our products. In the event that
third parties take over the clinical trial process for one of our product
candidates, the estimated completion date would largely be under the control of
that third party rather than us. We cannot forecast with any degree
of certainty which proprietary products or indications, if any, will be subject
to future collaborative arrangements, in whole or in part, and how such
arrangements would affect our capital requirements.
27
As a result of the uncertainties
discussed above, among others, we are unable to estimate the duration and
completion costs of our research and development projects. Our
inability to complete our research and development projects in a timely manner
or our failure to enter into collaborative agreements, when appropriate, could
significantly increase our capital requirements and could adversely impact our
liquidity. These uncertainties could force us to seek additional,
external sources of financing from time to time in order to continue with our
business strategy. There can be no assurance that we will be able to
successfully access external sources of financing in the future. Our
inability to raise additional capital, or to do so on terms reasonably
acceptable to us, would jeopardize the future success of our
business.
Research and development expenses
consist primarily of compensation and other expenses related to research and
development personnel, research collaborations, costs associated with internal
and contract preclinical testing and clinical trials of our product candidates,
including the costs of manufacturing drug substance and drug product, regulatory
maintenance costs, and facilities expenses. Overall research and
development expenses decreased to $7,902,000 in 2009 from $20,069,000 in 2008
and from $23,739,000 in 2007.
The fluctuations in research and
development expenses were specifically impacted by the following:
|
|
-
|
Outside
Services – We utilize outsourcing to conduct our product development
activities. Larger-scale small molecule synthesis, in vivo testing and
data analysis are examples of the services that we
outsource. We spent $430,000 in 2009, $1,170,000 in 2008 and
$2,871,000 in 2007 on these activities. The decrease in 2009 as
compared to 2008 primarily reflects the absence in 2009 of analytical
testing and preclinical support services in 2ME2 (2-methoxyestradiol) and
ENMD-1198 studies. The decrease in 2008 as compared to 2007
reflects the absence in 2008 of certain IND-directed expenses associated
with the IND submissions for Panzem®
NCD for the treatment of rheumatoid arthritis and for ENMD-2076 in
oncology during 2007.
|
|
|
-
|
Clinical
Trial Costs – Clinical trial costs decreased to $2,518,000 in 2009, from
$5,194,000 in 2008, which increased from $4,282,000 in
2007. The 2009 decrease relates to the Company’s restructuring
and the reprioritization of our clinical programs, focusing on the
clinical development of ENMD-2076. Our increase in 2008
clinical expenses resulted primarily from the initiation of clinical
trials for ENMD-2076 and also the expanded MKC-1 clinical
program. Costs of such trials include the clinical site fees,
monitoring costs and data management
costs.
|
|
|
-
|
Contract
Manufacturing Costs – The costs of manufacturing the material used in
clinical trials for our product candidates is reflected in contract
manufacturing. These costs include bulk manufacturing, encapsulation and
fill and finish services, and product release costs. Contract
manufacturing costs decreased in 2009 to $965,000 from $3,418,000 in 2008,
and from $5,681,000 in 2007. The 2009 decrease reflects the
absence of contract manufacturing activities for ENMD-1198 and Panzem®
NCD. The most significant component of the 2008 decrease was our
acquisition of bulk API (Active Pharmaceutical Ingredient) to support the
Panzem®
NCD trials in 2007. The absence of Panzem®
NCD API costs in 2008 was partially offset by an increase in contract
manufacturing activities for ENMD-1198. The 2007 contract
manufacturing expenses reflect the costs of supplying finished drug
product to support new and ongoing trials for three clinical drug
candidates and also the cost of securing clinical material to support
Phase 1 trials for ENMD-2076, which started in
2008.
|
|
|
-
|
Personnel
Costs — Personnel costs decreased to $2,320,000 in 2009 from $5,751,000 in
2008, which increased from $5,301,000 in 2007. The 2009
decreased expenses result from our corporate restructuring at the end of
2008, at which time, we eliminated certain management positions and
significantly reduced our research and development staff and
operations. The increase in 2008 is attributed primarily to
severance costs related to the December 2008 reduction in force, offset by
the decision not to accrue bonuses in
2008.
|
|
|
-
|
Also
reflected in our 2009 research and development expenses are patent costs
of $487,000, facility and related expenses of $606,000, laboratory
supplies and animal costs of $33,000, consulting fees of $301,000 and
travel expenses of $83,000. In 2008, these expenses totaled
$767,000, $1,483,000, $985,000, $451,000 and $193,000, respectively, and
in 2007, these expenses totaled $1,251,000, $1,500,000, $1,111,000,
$627,000 and $243,000, respectively. The reduction in expenses
in 2009 resulted from the decision to focus our resources on the
development of ENMD-2076 and the elimination of costs associated with our
other product candidates.
|
28
General and Administrative
Expenses. General and administrative expenses include
compensation and other expenses related to finance, business development and
administrative personnel, professional services and facilities.
General
and administrative expenses decreased to approximately $4,175,000 in 2009 from
$7,765,000 in 2008, which increased from $7,387,000 in 2007. The
decrease in 2009 is primarily related to our corporate restructuring, which
resulted in reductions in our headcount and the elimination of three executive
officer positions in December 2008. Such executive officer positions
have not been opened to date. The increase in 2008 is primarily
related to accrued severance costs associated with the termination of those
corporate officers.
In-process R&D. In
January 2006, we acquired Miikana Therapeutics, a private biotechnology
company. Pursuant to the Merger Agreement, we acquired all of the
outstanding capital stock of Miikana Therapeutics, Inc. in exchange for 9.96
million shares of common stock and the assumption of certain
obligations. In 2008, EntreMed initiated a Phase 1 clinical trial
with its Aurora A and angiogenic kinase inhibitor, ENMD-2076, in patients with
solid tumors. Dosing of the first patient with ENMD-2076 triggered a
purchase price adjustment milestone of $2 million, which we opted to pay in
stock. In June 2008, 2,564,104 shares of common stock were issued as
consideration for the satisfaction of the milestone payment. The
additional payment of $2 million was recorded to expense as in-process research
and development since the research and development project related to the Aurora
Kinase Program had not reached technical feasibility and has no future
alternative use. Under the terms of the merger agreement, the former
Miikana stockholders may earn up to an additional $16 million of potential
payments upon the satisfaction of additional clinical and regulatory
milestones. If ENMD-2076 advances to Phase 2, dosing of the first
patient in a Phase 2 study will trigger a milestone payment of $3 million, which
we expect to occur in the second quarter of 2010 and may be paid in cash or
stock at our sole discretion.
Interest
Expense. Interest expense for the year ended December 31, 2009
decreased to approximately $1,493,000 (including $133,000 of non-cash interest)
from $2,216,000 (including $199,000 of non-cash interest). Interest
expense increased in 2008 from $793,000 in 2007. The increase in 2008
results from a financing transaction with General Electric Capital Corporation
(GECC) in September 2007 and the related interest expense. The 2007
interest expense primarily related to debt with Venture Lending & Leasing
IV, LLC, which was paid in full in September 2007.
Investment
Income. Investment income decreased in 2009 to $70,000 from
$882,000 in 2008 and $2,113,000 in 2007 as a result of lower yields on lower
invested balances in interest-bearing cash accounts and
investments.
Dividends on Series A Convertible
Preferred Stock. The Consolidated Statements of Operations for
the years ended December 31, 2009, 2008 and 2007 reflect accrued, but unpaid,
dividends of $1,005,000 relating to Series A Convertible Preferred Stock held by
Celgene pursuant to a Securities Purchase Agreement dated December 31,
2002. The holders of Series A Preferred Stock will accumulate
dividends at a rate of 6% and will participate in dividends declared and paid on
the common stock, if any. All accumulated dividends must be paid
before any dividends may be declared or paid on the Common Stock. We
have no plans to pay any dividends in the foreseeable future.
LIQUIDITY
AND CAPITAL RESOURCES
To date, we have been engaged primarily
in research and development activities. As a result, we have incurred and expect
to continue to incur operating losses in 2010 and the foreseeable future before
we commercialize any products. Based on our current plans, we expect
our current available cash and cash equivalents to meet our cash requirements
into September of fiscal 2010.
Our
independent registered public accounting firm, Ernst & Young LLP, has
concluded that substantial doubt exists about our ability to continue as a going
concern, and has included an explanatory paragraph to describe this uncertainty
in its audit report on our consolidated financial statements for the year ended
December 31, 2009 included in this Annual Report on
Form 10-K. We did not include any adjustments to the
consolidated financial statements included in this Annual Report on Form 10-K to
reflect the possible future effects that may result from the uncertainty of our
ability to continue as a going concern because we believe that we will continue
to be a financially sound and viable business, continuing to conduct clinical
trials for, and further development of, ENMD-2076 and provide value to our
shareholders.
29
We will
require significant additional funding prior to January 1, 2011 to fund
operations until such time, if ever, we become profitable. We intend
to augment our cash and cash equivalent balances as of December 31, 2009 by
pursuing other forms of capital infusion, including strategic alliances or
collaborative development opportunities with organizations that have
capabilities and/or products that are complementary to our capabilities and
products in order to continue the development of our potential product
candidates that we intend to pursue to commercialization.
We intend
to explore one or more of the following alternatives to raise additional capital
so that the Company can continue as a going concern through 2010:
|
|
·
|
selling
additional equity securities;
|
|
|
·
|
out-licensing
technologies or product candidates to one or more corporate
partners;
|
|
|
·
|
monetizing
our Thalomid®
royalty payment stream
|
|
|
·
|
completing
an outright sale of non-priority assets;
and/or
|
|
|
·
|
engaging
in one or more strategic
transactions.
|
We also
will consider additional actions to reduce our expenses.
There can
be no assurance that adequate additional financing under such arrangements will
be available to us on terms that we deem acceptable, if at all. If
additional funds are raised by issuing equity securities, dilution to existing
shareholders may result. If we fail to obtain additional capital when
needed, we may be required to delay or scale back our Phase 2 plans for
ENMD-2076.
At
December 31, 2009, we had cash and short-term investments of $6,366,253, with a
working capital deficit of ($1,395,158).
In
January 2006, we acquired Miikana Therapeutics, a private biotechnology company,
in exchange for 9.96 million shares of our common stock and the assumption of
certain obligations of Miikana. We acquired certain drug candidates
in connection with the acquisition, including the lead molecule in the Aurora
Kinase Program, ENMD-2076, which advanced into clinical development in
2008. ENMD-2076 is a kinase inhibitor with activity towards Aurora A
and multiple other kinases linked to promoting cancer. Dosing of the
first patient in ENMD-2076 trials triggered a milestone payment of $2 million to
the former Miikana stockholders payable in stock or cash, at the Company’s
discretion. In June 2008, 2,564,105 shares of common stock were
issued to the former Miikana stockholders as consideration for the satisfaction
of the milestone payment. Under the terms of the merger agreement,
dosing of the first patient in a Phase 2 trial for ENMD-2076 will trigger an
additional milestone payment of $3 million, payable in stock or cash, at the
Company’s sole discretion. We expect this milestone to occur during
the second quarter of 2010. Beyond this milestone, the former Miikana
stockholders may earn up to an additional $13 million of potential payments upon
the satisfaction of additional clinical and regulatory
milestones. Through the Miikana acquisition, we also acquired rights
to MKC-1, a Phase 2 clinical candidate licensed from Roche by Miikana in April
2005. Under the terms of the agreement, Roche may be entitled to
receive future payments upon successful attainment of certain clinical,
regulatory and commercialization milestones; however, ENMD-2076 is the only
program currently under active clinical evaluation by the Company, we do not
expect to trigger any of these milestone payments during fiscal
2010.
30
FINANCING
ACTIVITIES
In September 2007, we entered into a
$20 million term loan agreement with General Electric Capital Corporation
(“GECC”), as agent, Merrill Lynch Capital and Oxford Finance Corporation
(collectively with GECC, the “Lenders”). The loan accrues
interest in arrears at a fixed annual interest rate of 10.47% until fully
repaid. The loan is to be repaid by the Company to GECC, for the
ratable benefit of the Lenders, as nine consecutive monthly payments of interest
only, each in the amount of $174,500, which commenced on November 1, 2007, and
thirty consecutive monthly payments of principal and interest, each in the
amount of $760,606, which commenced on August 1, 2008. In connection
with the transaction, we issued warrants with an exercise price of $2.00 per
share to the Lenders providing rights to purchase their respective pro
rata share of 250,000
shares of common stock of the Company. The unpaid balance of the loan
at December 31, 2009 is approximately $9,311,000.
The Loan
Agreement contains customary affirmative and negative operating and financial
covenants. We were in compliance with such covenants as of December
31, 2009.
On August
6, 2009, we filed a Form S-3 registration statement with the SEC utilizing a
“shelf” registration process. On October 9, 2009, the Form S-3
registration statement was declared effective by the SEC. Pursuant to
this shelf registration statement, we may sell debt or equity securities in one
or more offerings up to a total public offering price of $30.0
million. We believe that this shelf registration statement currently
provides us additional flexibility with regard to potential financings that we
may undertake when market conditions permit or our financial condition may
require. To date, we have completed two offerings under the shelf
registration statement.
On
January 11, 2010, we sold 3,125,000 shares of our common stock to an
institutional investor for an aggregate purchase price of $2,500,000 or $0.80
per share.
On
February 3, 2010, we sold 3,846,154 shares of our common stock to the same
institutional investor for an aggregate purchase price of $2,500,000, or $0.65
per share.
To
accomplish our business plans, we will be required to continue to conduct
substantial development activities for ENMD-2076. Under our current operating
plans for 2010, we expect to have ENMD-2076, our leading program, under clinical
investigation and we expect our 2010 results of operations to reflect a net loss
of approximately $8,000,000, including non-cash charges of approximately
$1,400,000. These projections are subject to judgment and estimation
and could significantly change.
We expect
that the majority of our 2010 revenues will continue to be from royalties on the
sales of Thalomid®. Thalomid® is sold
by a third-party and is subject to competition from other products and generic
drugs, and we have no control over such party’s sales efforts or the resources
devoted to Thalomid®
sales. In 2009 and 2008, we received royalty-sharing revenue in the
amount of $4,990,000 and $7,472,000, respectively. There can be no
assurance on the future trends of Thalomid® sales
and the amount of revenue we will receive and record in 2010. In
addition, under our licensing agreement with Oxford Biomedica, PLC and Oxford
Biomedica (UK) Limited Oxford, we are entitled to receive payments upon the
achievement of certain milestones with respect to the development of gene
therapies for ophthalmic (eye) diseases. In connection with the
announced collaboration between Oxford Biomedica and Sanofi Aventis which
included certain compounds that are governed by our licensing agreement, we
received $368,000 in 2009, representing a share of the upfront payment received
by Oxford Biomedica. Accordingly, a sub-royalty payment of $74,000 to
be remitted to Children’s Medical Center Corporation has been accrued at
December 31, 2009. We do not control the drug development efforts of
Oxford and have no information or control over when or whether any milestones
will be reached that would result in additional payments to us.
INFLATION
AND INTEREST RATE CHANGES
Management
does not believe that our working capital needs are sensitive to inflation and
changes in interest rates.
31
CONTRACTUAL
OBLIGATIONS
The table
below sets forth our contractual obligations at December 31, 2009.
|
CONTRACTUAL OBLIGATIONS
|
PAYMENTS DUE BY PERIOD
|
|||||||||||||||||||
|
Total
|
Less than 1
year
|
1-3 years
|
3 - 5 years
|
More than
5 years
|
||||||||||||||||
|
Operating
Leases Obligations
|
$ | 74,000 | $ | 74,000 | $ | — | $ | — | $ | — | ||||||||||
|
Loan
Payable, including interest
|
9,311,000 | 8,555,000 | 756,000 | |||||||||||||||||
|
Obligations
under Licensing and Miikana Merger Agreements (1)
|
— | — | — | — | — | |||||||||||||||
|
Purchase
Obligations
|
||||||||||||||||||||
|
Clinical
Trial Contracts
|
2,698,000 | 1,350,000 | 1,348,000 | — | — | |||||||||||||||
|
Contract
Manufacturing
|
505,000 | 253,000 | 252,000 | — | — | |||||||||||||||
|
Total Contractual Cash
Obligations
|
$ | 12,588,000 | $ | 10,232,000 | $ | 2,356,000 | $ | — | $ | — | ||||||||||
|
(1)
|
In
the event that we reach certain development milestones for Panzem®
and MKC-1, such as initiation of Phase 3 trials and multiple regulatory
approvals (US, Europe and Japan), we could be obligated to make future
milestone payments of up to $35.75 million under the related license
agreements. Of this amount, up to $10 million could become
payable while these product candidates are in clinical
development. We would also be obligated to make
annual-sales-based-royalty payments if we successfully commercialize
either compound. Our other development programs are in Phase 1
or earlier stages of development. Under the terms of the
Miikana merger agreement we could be obligated to make additional payments
to Miikana’s selling shareholders of $16 million upon the attainment of
certain clinical milestones for two acquired preclinical
programs. In addition, under the terms of our license agreement
with Celgene, we could make future development and commercialization
milestone payments, including payments for approvals in the U.S. and other
countries, totaling $25.25 million. Of the milestones, $5.25 million would
be payable if a product candidate successfully moves through clinical
trials. We would also be required to pay
annual-sales-based-royalties under this agreement. We cannot
forecast with any degree of certainty whether any of our product
candidates will reach additional developmental milestones. We
therefore have excluded the milestone amounts and any royalty payments
from the above table.
|
OFF-BALANCE-SHEET
ARRANGEMENTS
We had no
material off-balance sheet arrangements during fiscal year 2009.
NEW
ACCOUNTING PRONOUNCEMENTS
In
July 2009, the FASB Accounting Standards Codification (“ASC” or
“Codification”) became the authoritative source of accounting principles
generally accepted in the United States (“GAAP”) recognized by the FASB. All
existing FASB accounting standards and guidance were superseded by the ASC.
Instead of issuing new accounting standards in the form of statements, FASB
staff positions and Emerging Issues Task Force abstracts, the FASB now issues
Accounting Standards Updates that update the Codification. Rules and
interpretive releases of the Securities and Exchange Commission (“SEC”) under
authority of federal securities laws continue to be additional sources of
authoritative GAAP for SEC registrants. This Codification is
effective for financial statements issued for interim and annual periods ending
after September 15, 2009. There has been no change to our consolidated
financial statements due to the implementation of the Codification other than
changes in reference to various authoritative accounting pronouncements in the
consolidated financial statements.
32
On June
30, 2009, we adopted authoritative guidance which requires a publicly-traded
company to include disclosures about the fair value of its financial instruments
whenever it issues summarized financial information for interim reporting
periods. Such disclosures include the fair value of all financial instruments,
for which it is practicable to estimate that value, whether recognized or not
recognized in the statement of financial position; the related carrying amount
of these financial instruments; and the methods and significant assumptions used
to estimate the fair value. Other than the required disclosures (see the Debt
note), the adoption of this guidance had no impact on our consolidated financial
statements.
In May 2009, the FASB issued
authoritative guidance regarding subsequent events which is intended to
establish general standards of accounting for, and disclosure of, events that
occur after the balance sheet date but before financial statements are issued or
are available to be issued. Specifically, the guidance sets forth the
period after the balance sheet date during which management of a reporting
entity should evaluate events or transactions that may occur for potential
recognition or disclosure in the financial statements, the circumstances under
which an entity should recognize events or transactions occurring after the
balance sheet date in its financial statements, and the disclosures that an
entity should make about events or transactions that occurred after the balance
sheet date. The adoption of this pronouncement, which is effective for financial
statements issued for interim and annual financial periods ending after June 15,
2009, did not have a material impact on our financial position and results of
operations.
In April 2009, the FASB issued
authoritative accounting guidance on how to determine the fair value of assets
and liabilities in the current economic environment, which reemphasizes that the
objective of a fair value measurement remains an exit price. If we were to
conclude that there has been a significant decrease in the volume and level of
activity of the asset or liability in relation to normal market activities,
quoted market values may not be representative of fair value and we may conclude
that a change in valuation technique or the use of multiple valuation techniques
may be appropriate. Additional guidance issued in April 2009 modifies the
requirements for recognizing other-than-temporarily impaired debt securities and
revises the existing impairment model for such securities by modifying the
current intent and ability indicator in determining whether a debt security is
other-than-temporarily impaired. The FASB also issued guidance to enhance the
disclosure of financial instruments for both interim and annual periods. The
adoption of this guidance had no material effect on our consolidated results of
operations, financial position or liquidity.
In
June 2008, the FASB issued authoritative guidance which mandates a two-step
process for evaluating whether an equity-linked financial instrument or embedded
feature is indexed to the entity’s own stock. The adoption of this guidance as
of January 1, 2009 had no material effect on our consolidated results of
operations, financial position or liquidity.
|
ITEM 7A.
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET
RISK.
|
The
primary objective of our investment activities is to preserve our capital until
it is required to fund operations while at the same time maximizing the income
we receive from our investments without incurring investment market volatility
risk. Our investment income is sensitive to the general level of U.S. interest
rates. In this regard, changes in the U.S. interest rates affect the interest
earned on our cash and cash equivalents. Due to the short-term nature of our
cash and cash equivalent holdings, a 10% movement in market interest rates would
not materially impact on the total fair market value of our portfolio as of
December 31, 2009.
|
ITEM 8.
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA.
|
The
response to this item is submitted in a separate section of this report. See
Index to Consolidated Financial Statements on Page F-1.
|
ITEM 9.
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE.
|
None.
33
|
ITEM 9A.
|
CONTROLS
AND PROCEDURES.
|
Disclosure
Controls and Procedures
As of the
end of the period covered by this Annual Report, we carried out an evaluation,
under the supervision and with the participation of our management, including
our Executive Chairman and Principal Accounting Officer (its principal executive
officer and principal financial officer), of the effectiveness of the design and
operation of our disclosure controls and procedures (as defined in the
Securities Exchange Act of 1934 Rules 13a-15(e) and
15d-15(e)). Our Executive Chairman and Principal Accounting Officer
have concluded that our disclosure controls and procedures are effective to
ensure that information required to be disclosed by us in the reports that we
file or submit under the Exchange Act is recorded, processed, summarized and
reported within the time periods specified in the rules and forms of the
Securities and Exchange Commission and that such information is accumulated and
communicated to our management (including our Executive Chairman and Principal
Accounting Officer) to allow timely decisions regarding required
disclosures. Based on such evaluation, our Executive Chairman and
Principal Accounting Officer have concluded these disclosure controls are
effective as of December 31, 2009.
Changes
in Internal Control Over Financial Reporting
There
have not been any changes in our internal control over financial reporting
during the fiscal quarter ended December 31, 2009 that have materially affected,
or are reasonably likely to materially affect, our internal control over
financial reporting.
Management's
Report on Internal Control Over Financial Reporting
Our
management is responsible for establishing and maintaining adequate internal
control over financial reporting, as such term is defined in Securities Exchange
Act Rules 13a-15(f) and 15d-15(f). Our internal control over
financial reporting is designed to provide reasonable assurance to our
management and board of directors regarding the reliability of financial
reporting and the preparation and fair presentation of financial statements for
external purposes in accordance with generally accepted accounting principles.
Any internal control over financial reporting, no matter how well designed, has
inherent limitations. As a result of these inherent limitations,
internal control over financial reporting may not prevent or detect
misstatements. Therefore, even those internal controls determined to
be effective can provide only reasonable assurance with respect to reliability
of financial reporting and the preparation of financial statements for external
purposes in accordance with generally accepted accounting
principles.
Under the
supervision and with the participation of our management, including our Chief
Operating Officer and Principal Accounting Officer, we conducted an assessment
of the effectiveness of our internal control over financial reporting using the
criteria set forth by the Committee of Sponsoring Organizations of the Treadway
Commission (COSO) in Internal
Control — Integrated Framework. Based on our assessment,
we concluded that our internal control over financial reporting was effective as
of December 31, 2009.
Ernst & Young LLP, the
independent registered public accounting firm that has audited our consolidated
financial statements included herein, has issued an attestation report on the
effectiveness of our internal control over financial reporting as of December
31, 2009, a copy of which is included in this Annual Report on Form
10-K.
34
Report
of Independent Registered Public Accounting Firm
The
Board of Directors and Stockholders of EntreMed, Inc.
We have audited EntreMed, Inc.’s
internal control over financial reporting as of December 31, 2009, based on
criteria established in Internal Control—Integrated Framework issued by the
Committee of Sponsoring Organizations of the Treadway Commission (the COSO
criteria). EntreMed, Inc.’s management is responsible for maintaining effective
internal control over financial reporting, and for its assessment of the
effectiveness of internal control over financial reporting included in the
accompanying “Management’s Report on Internal Control over Financial Reporting”.
Our responsibility is to express an opinion on the company’s internal control
over financial reporting based on our audit.
We conducted our audit in accordance
with the standards of the Public Company Accounting Oversight Board (United
States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control over financial
reporting was maintained in all material respects. Our audit included obtaining
an understanding of internal control over financial reporting, assessing the
risk that a material weakness exists, testing and evaluating the design and
operating effectiveness of internal control based on the assessed risk, and
performing such other procedures as we considered necessary in the
circumstances. We believe that our audit provides a reasonable basis for our
opinion.
A company’s internal control over
financial reporting is a process designed to provide reasonable assurance
regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted
accounting principles. A company’s internal control over financial reporting
includes those policies and procedures that (1) pertain to the maintenance of
records that, in reasonable detail, accurately and fairly reflect the
transactions and dispositions of the assets of the company; (2) provide
reasonable assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally accepted
accounting principles, and that receipts and expenditures of the company are
being made only in accordance with authorizations of management and directors of
the company; and (3) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use or disposition of the company’s
assets that could have a material effect on the financial
statements.
Because of its inherent limitations,
internal control over financial reporting may not prevent or detect
misstatements. Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become inadequate because of
changes in conditions, or that the degree of compliance with the policies or
procedures may deteriorate.
In our opinion, EntreMed, Inc.
maintained, in all material respects, effective internal control over financial
reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance
with the standards of the Public Company Accounting Oversight Board (United
States), the consolidated balance sheets of EntreMed, Inc. as of December 31,
2009 and 2008, and the related consolidated statements of operations,
stockholders’ equity, and cash flows for each of the three years in the period
ended December 31, 2009 of EntreMed, Inc. and our report dated March 29, 2010
expressed an unqualified opinion thereon that included an explanatory paragraph
regarding EntreMed, Inc.’s ability to continue as a going concern.
/s/ Ernst
& Young LLP
McLean,
Virginia
March 29,
2010
35
|
ITEM 9B.
|
OTHER
INFORMATION
|
None.
PART
III
|
ITEM 10.
|
DIRECTORS,
EXECUTIVE OFFICERS AND CORPORATE
GOVERNANCE
|
Information required by this item will
be contained in our Definitive Proxy Statement for our 2010 Annual Meeting of
Stockholders, to be filed pursuant to Regulation 14A with the Securities and
Exchange Commission within 120 days of December 31, 2009. Such
information is incorporated herein by reference.
We have adopted a Code of Ethics, as
defined in applicable SEC and NASD rules, that applies to directors, officers
and employees, including our principal executive officer and principal
accounting officer. The Code of Ethics is available on the Company’s
website at www.entremed.com.
|
ITEM 11.
|
EXECUTIVE
COMPENSATION
|
Information
required by this item will be contained in our Definitive Proxy Statement for
our 2010 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A
with the Securities and Exchange Commission within 120 days of December 31,
2009. Such information is incorporated herein by
reference.
|
ITEM 12.
|
SECURITY OWNERSHIP OF CERTAIN
BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER
MATTERS
|
Information required by this item will
be contained in our Definitive Proxy Statement for our 2010 Annual Meeting of
Stockholders, to be filed pursuant to Regulation 14A with the Securities and
Exchange Commission within 120 days of December 31, 2009. Such
information is incorporated herein by reference.
Options
under Employee Benefit Plans
The following table discloses certain
information about the options issued and available for issuance under all
outstanding Company option plans, as of December 31, 2009.
|
(a)
|
(b)
|
(c)
|
||||||||||
|
Plan category
|
Number of securities to
be issued upon exercise
of outstanding options,
warrants and rights
|
Weighted-average
exercise price of
outstanding options,
warrants and rights
|
Number of securities
remaining available for
future issuance under
equity compensation
plans [excluding
securities reflected in
column (a)]
|
|||||||||
|
Equity
compensation plans approved by security holders
|
6,984,606 | $ | 5.50 | 3,135,043 | ||||||||
|
Equity
compensation plans not approved by security holders
|
0 | $ | 0.00 | 0 | ||||||||
|
Total
|
6,984,606 | $ | 5.50 | 3,135,043 | ||||||||
Warrants issued under the unauthorized
plans represent compensation for consulting services rendered by the
holders.
36
|
ITEM 13.
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
|
Information
required by this item will be contained in our Definitive Proxy Statement for
our 2010 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A
with the Securities and Exchange Commission within 120 days of December 31,
2009. Such information is incorporated herein by
reference.
|
ITEM 14.
|
PRINCIPAL
ACCOUNTING FEES AND SERVICES.
|
Information
required by this item will be contained in our Definitive Proxy Statement for
our 2010 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A
with the Securities and Exchange Commission within 120 days of December 31,
2009. Such information is incorporated herein by
reference.
PART
IV
|
ITEM 15.
|
EXHIBITS,
FINANCIAL STATEMENT SCHEDULES.
|
(a) 1.
FINANCIAL STATEMENTS - See index to Consolidated Financial
Statements.
2.
Schedules
All
financial statement schedules are omitted because they are not applicable, not
required under the instructions or all the information required is set forth in
the financial statements or notes thereto.
3.
Exhibits
|
2.1(1)
|
Agreement
and Plan of Merger, dated as of December 22, 2005 among EntreMed, Inc.,
E.M.K. Sub, Inc., Miikana Therapeutics, Inc., and Andrew
Schwab
|
|
|
3.1(2)
|
Amended
and Restated Certificate of Incorporation of EntreMed,
Inc.
|
|
|
3.2(3)
|
By-laws
of EntreMed, Inc.
|
|
|
4.1(4)
|
Certificate
of Designations of the Series A Convertible Preferred
Stock
|
|
|
4.2(5)
|
Warrant
to Purchase Common Stock, dated January 13, 2003, issued by EntreMed, Inc.
in favor of Celgene Corporation
|
|
|
4.3(6)
|
Warrant
to Purchase Common Stock, dated September 12, 2007, issued by EntreMed,
Inc. in favor of General Electric Capital Corporation
|
|
|
4.4(6)
|
Warrant
to Purchase Common Stock, dated September 12, 2007, issued by EntreMed,
Inc. in favor of Merrill Lynch Capital
|
|
|
4.5(6)
|
Warrant
to Purchase Common Stock, dated September 12, 2007, issued by EntreMed,
Inc. in favor of Oxford Finance Corporation
|
|
|
10.1(7)
|
1992
Stock Incentive Plan*
|
|
|
10.2(7)
|
Amended
and Restated 1996 Stock Option Plan*
|
|
|
10.3(7)
|
|
Form
of Stock Option Agreement, under Amended and Restated 1996 Stock Option
Plan*
|
37
|
10.4(8)
|
License
Agreement between Children's Hospital Medical Center Corporation and
EntreMed, Inc. signed December 20, 1996 regarding Estrogenic Compounds as
Anti-Mitotic Agents
|
|
|
10.5(9)
|
Amendment
to the 1996 Stock Option Plan*
|
|
|
10.6(10)
|
License
Agreement between Celgene Corporation and EntreMed, Inc. signed December
9, 1998 regarding thalidomide intellectual property
|
|
|
10.7(10)
|
Lease
Agreement between EntreMed, Inc. and Red Gate III Limited Partnership,
dated June 10, 1998
|
|
|
10.8(11)
|
1999
Long-Term Incentive Plan*
|
|
|
10.9(12)
|
EntreMed,
Inc. 2001 Long-Term Incentive Plan*
|
|
|
10.10.1(13)
|
Purchase
Agreement between Bioventure Investments kft and EntreMed, Inc., dated
June 15, 2001+
|
|
|
10.10.2(13)
|
Amendment
1 to Purchase Agreement between Bioventure Investments kft and EntreMed,
Inc., date July 13, 2001
|
|
|
10.10.3(13)
|
Amendment
2 to Purchase Agreement between Bioventure Investments kft and EntreMed,
Inc., dated July 30, 2001
|
|
|
10.10.4(13)
|
Amendment
3 to Purchase Agreement between Bioventure Investments kft and EntreMed,
Inc., dated August 3, 2001
|
|
|
10.11(14)
|
Board
Service Agreement, dated February 5, 2003, between Michael M. Tarnow and
EntreMed, Inc. *
|
|
|
10.12(15)
|
Securities
Purchase Agreement by and among EntreMed, Inc., and Celgene Corporation,
dated as of December 31, 2002
|
|
|
10.13(15)
|
Investor
and Registration Rights Agreement by and between EntreMed, Inc. and
Celgene Corporation, dated as of December 31, 2002
|
|
|
10.14(16)
|
Employment
Agreement between EntreMed and Carolyn F. Sidor, M.D. effective December
1, 2004, as amended*
|
|
|
10.15(17)
|
EntreMed,
Inc. 2001 Long Term Incentive Plan Non-Qualified Stock Option Grant
Agreement (Director)*
|
|
|
10.16(17)
|
EntreMed,
Inc. 2001 Long Term Incentive Plan Non-Qualified Stock Option Grant
Agreement (Non-Director Employee)*
|
|
|
10.17(17)
|
Form
of Change in Control Agreement*
|
|
|
10.18(17)
|
Amendment
to Employment Agreement by and between the Company and Carolyn F. Sidor,
effective April 16, 2007*
|
|
|
10.19(17)
|
Amendment
to Employment Agreement by and between the Company and Cynthia Wong Hu,
effective April 16, 2007*
|
|
|
10.20(18)
|
Form
of Restricted Stock Award under EntreMed, Inc. 2001 Long Term Incentive
Plan*
|
|
|
10.21(19)
|
|
License
Agreement between EntreMed and Celgene Corporation signed March 24, 2005
regarding the development and commercialization of Celgene’s small
molecule tubulin inhibitor compounds for the treatment of
cancer+
|
38
|
10.22(20)
|
Employment
Agreement by and between EntreMed and Cynthia Wong, dated as of June 1,
2006, as amended*
|
|
|
10.23(21)
|
License
Agreement, dated January 9, 2006, by and between Elan Pharma International
Limited and EntreMed, Inc. +
|
|
|
10.24(21)
|
Research,
Development and Commercialization Agreement, dated as of April 20, 2005,
by and between Hoffman-La Roche Inc. and F. Hoffman La Roche Ltd.
(together, “Roche”), and Miikana Therapeutics Inc.+
|
|
|
10.25(22)
|
Loan
and Security Agreement dated September 12, 2007 among General Electric
Capital Corporation, Oxford Finance Corporation, Merrill Lynch Capital, as
the lenders and EntreMed, Inc. and Miikana Therapeutics, Inc. as the loan
parties
|
|
|
10.26(22)
|
Promissory
Note dated September 12, 2007 to General Electric Capital
Corporation
|
|
|
10.27(22)
|
Promissory
Note dated September 12, 2007 to Merrill Lynch Capital
|
|
|
10.28(22)
|
Promissory
Note dated September 12, 2007 to Oxford Finance
Corporation
|
|
|
10.29(23)
|
Amendment
to Lease between EntreMed, Inc. and Red Gate III Limited Partnership,
dated January 27, 2009
|
|
|
10.30(23)
|
Employment
Agreement by and between the Company and Kathy Wehmeir-Davis, dated as
January 1, 2009*
|
|
|
10.31(23)
|
Employment
Agreement by and between the Company and Mark R. Bray, dated as January 1,
2009*
|
|
|
23.1
|
Consent
of Independent Registered Public Accounting Firm
|
|
|
31.1
|
Rule
13a-14(a) Certification of President and CEO
|
|
|
31.2
|
Rule
13a-14(a) Certification of Chief Financial Officer
|
|
|
32.1
|
Rule
13a-14(b) Certification by President and CEO
|
|
|
32.2
|
Rule
13a-14(b) Certification by Chief Financial Officer
|
|
|
*
|
Compensatory
Plan, Contract or Arrangement.
|
|
|
+
|
Certain
portions of this exhibit have been omitted based upon a request for
confidential treatment. The omitted portions have been filed with the
Commission pursuant to our application for confidential
treatment.
|
|
|
(1)
|
Incorporated
by reference from our Form 8-K previously filed with the Securities and
Exchange Commission on December 29, 2005.
|
|
|
(2)
|
Incorporated
by reference from our Form 10-Q for the quarter ended June 30, 2006
previously filed with the Securities and Exchange
Commission.
|
|
|
(3)
|
|
Incorporated
by reference from our Form 8-K previously filed with the Securities and
Exchange Commission on December 6,
2007
|
39
|
(4)
|
Incorporated
by reference to Exhibit 99.4 of our Form 8-K dated December 31, 2002
previously filed with the Securities and Exchange Commission on January
15, 2003.
|
|
|
(5)
|
Incorporated
by reference to Exhibit 99.5 of our Form 8-K dated December 31, 2002
previously filed with the Securities and Exchange Commission on January
15, 2003.
|
|
|
(6)
|
Incorporated
by reference from our Form 10-Q for the quarter ended September 30, 2007
previously filed with the Securities nd Exchange
Commission.
|
|
|
(7)
|
Incorporated
by reference from our Registration Statement on Form S-1 declared
effective by the Securities and Exchange Commission on June 11,
1996.
|
|
|
(8)
|
Incorporated
by reference from our Form 10-K for the year ended December 31, 1996
previously filed with the Securities and Exchange
Commission.
|
|
|
(9)
|
Incorporated
by reference from our Form 10-K for the year ended December 31, 1997
previously filed with the Securities and Exchange
Commission.
|
|
|
(10)
|
Incorporated
by reference from our Form 10-K for the year ended December 31, 1998
previously filed with the Securities and Exchange
Commission.
|
|
|
(11)
|
Incorporated
by reference from our Form 10-Q for the quarter ended June 30, 1999
previously filed with the Securities and Exchange
Commission.
|
|
|
(12)
|
Incorporated
by reference from Exhibit A to our Definitive Proxy Statement filed with
the Securities and Exchange Commission on May 12, 2006.
|
|
|
(13)
|
Incorporated
by reference from our Form 10-Q for the quarter ended June 30, 2001
previously filed with the Securities and Exchange
Commission.
|
|
|
(14)
|
Incorporated
by reference from our Form 10-K/A for the year ended December 31, 2002
previously filed with the Securities and Exchange
Commission.
|
|
|
(15)
|
Incorporated
by reference from our Form 8-K dated December 31, 2002 previously filed
with the Securities and Exchange Commission on January 15,
2003.
|
|
|
(16)
|
Incorporated
by reference from our Form 8-K filed with the Securities and Exchange
Commission on December 6, 2004, as amended on Form 8-K filed on April 17,
2007 with the Securities and Exchange Commission.
|
|
|
(17)
|
Incorporated
by reference from our Form 8-K previously filed with the Securities and
Exchange Commission on April 17, 2007.
|
|
|
(18)
|
Incorporated
by reference from our Form 8-K previously filed with the Securities and
Exchange Commission on March 11, 2005.
|
|
|
(19)
|
Incorporated
by reference from our Form 10-Q for the quarter ended March 31, 2005
previously filed with the Securities and Exchange
Commission.
|
|
|
(20)
|
Incorporated
by reference from our Form 8-K previously filed with the Securities and
Exchange Commission on June 6, 2006.
|
|
|
(21)
|
|
Incorporated
by reference from our Form 10-Q for the quarter ended March 31, 2006
previously filed with the Securities and Exchange
Commission.
|
40
|
(22)
|
Incorporated
by reference from our Form 10-Q for the quarter ended September 30, 2007
previously filed with the Securities and Exchange
Commission.
|
|
|
(23)
|
|
Incorporated
by reference from our Form 10-K for the fiscal year ended December 31,
2008, previously filed with the Securities and Exchange
Commission.
|
41
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of
1934, the registrant has duly caused this report to be signed on its behalf by
the undersigned, thereunto duly authorized.
March 29,
2010
|
ENTREMED,
INC.
|
|
|
By:
|
/s/Michael M. Tarnow
|
|
Michael
M. Tarnow
|
|
|
Executive
Chairman
|
|
Pursuant
to the requirements of the Securities Act of 1934, this report has been signed
below by the following persons in the capacities and on the dates
indicated.
|
SIGNATURE
|
TITLE
|
DATE
|
||
|
/s/Michael
M. Tarnow
|
Executive
Chairman
|
March
29, 2010
|
||
|
Michael
M. Tarnow
|
||||
|
/s/
Kathy Wehmeir-Davis
|
Principal
Accounting Officer
|
March
29, 2010
|
||
|
Kathy
Wehmeir-Davis
|
||||
|
/s/Donald
S. Brooks
|
Director
|
March
25, 2010
|
||
|
Donald
S. Brooks
|
||||
|
/s/Dwight
L. Bush
|
Director
|
March
25, 2010
|
||
|
Dwight
L. Bush
|
||||
|
/s/Jennie
C. Hunter-Cevera
|
Director
|
March
29, 2010
|
||
|
Jennie
C. Hunter-Cevera
|
||||
|
/s/Mark
C. M. Randall
|
Director
|
March
29, 2010
|
||
|
Mark
C. M. Randall
|
||||
|
/s/Peter
S. Knight
|
Director
|
March
29, 2010
|
||
|
Peter
S. Knight
|
42
The
following consolidated financial statements of EntreMed, Inc. are included in
Item 8:
|
Report
of Independent Registered Public Accounting Firm
|
F-2
|
|
Consolidated
Balance Sheets as of December 31, 2009 and 2008
|
F-3
|
|
Consolidated
Statements of Operations for the years ended December 31, 2009, 2008 and
2007
|
F-4
|
|
Consolidated
Statements of Stockholders' Equity for the years ended December 31, 2009,
2008 and 2007
|
F-5
|
|
Consolidated
Statements of Cash Flows for the years ended December 31, 2009, 2008 and
2007
|
F-6
|
|
Notes
to Consolidated Financial Statements
|
F-7
|
F-1
Report
of Independent Registered Public Accounting Firm
The Board
of Directors and Stockholders of EntreMed, Inc.:
We have audited the accompanying
consolidated balance sheets of EntreMed, Inc. as of December 31, 2009 and 2008,
and the related consolidated statements of operations, stockholders’ equity, and
cash flows for each of the three years in the period ended December, 31 2009.
These financial statements are the responsibility of the Company’s management.
Our responsibility is to express an opinion on these financial statements based
on our audits.
We conducted our audits in accordance
with the standards of the Public Company Accounting Oversight Board (United
States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are free of material
misstatement. An audit includes examining, on a test basis, evidence supporting
the amounts and disclosures in the financial statements. An audit also includes
assessing the accounting principles used and significant estimates made by
management, as well as evaluating the overall financial statement presentation.
We believe that our audits provide a reasonable basis for our
opinion.
In our opinion, the financial
statements referred to above present fairly, in all material respects, the
consolidated financial position of EntreMed, Inc. at December 31, 2009 and 2008,
and the consolidated results of its operations and its cash flows for each of
the three years in the period ended December 31, 2009, in conformity with U.S.
generally accepted accounting principles.
The accompanying consolidated financial
statements have been prepared assuming that EntreMed, Inc. will continue as a
going concern. As more fully described in Note 1, the Company has incurred
recurring operating losses and negative cash flows from operations. These
conditions raise substantial doubt about the Company’s ability to continue as a
going concern. Management’s plans in regard to these matters are also described
in Note 1. The 2009 consolidated financial statements do not include any
adjustments to reflect the possible future effects on the recoverability and
classification of assets or the amounts and classification of liabilities that
may result from the outcome of this uncertainty.
We also have audited, in accordance
with the standards of the Public Company Accounting Oversight Board (United
States), EntreMed, Inc.’s internal control over financial reporting as of
December 31, 2009, based on criteria established in Internal Control –
Integrated Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission and our report dated March 29, 2010 expressed an unqualified
opinion thereon.
/s/Ernst
& Young LLP
McLean,
Virginia
March 29,
2010
F-2
EntreMed,
Inc.
Consolidated
Balance Sheets
|
DECEMBER 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
ASSETS
|
||||||||
|
Current
assets:
|
||||||||
|
Cash
and cash equivalents
|
$ | 6,312,182 | $ | 16,743,129 | ||||
|
Short-term
investments
|
54,071 | 7,548,044 | ||||||
|
Accounts
receivable, net of allowance for doubtful accounts of $72,145 at December
31, 2009 and $54,145 at December 31, 2008
|
3,286,858 | 3,880,108 | ||||||
|
Prepaid
expenses and other
|
220,925 | 420,940 | ||||||
|
Total
current assets
|
9,874,036 | 28,592,221 | ||||||
|
Property
and equipment, net
|
171,498 | 263,715 | ||||||
|
Other
assets
|
21,494 | 67,459 | ||||||
|
Total
assets
|
$ | 10,067,028 | $ | 28,923,395 | ||||
|
LIABILITIES
AND STOCKHOLDERS' (DEFICIT) EQUITY
|
||||||||
|
Current
liabilities:
|
||||||||
|
Accounts
payable
|
$ | 1,968,607 | $ | 3,496,198 | ||||
|
Accrued
liabilities
|
745,183 | 2,584,327 | ||||||
|
Current
portion of loan payable
|
8,555,404 | 7,708,451 | ||||||
|
Deferred
rent
|
- | 20,763 | ||||||
|
Total
current liabilities
|
11,269,194 | 13,809,739 | ||||||
|
Loan
payable, less current portion
|
723,814 | 9,191,490 | ||||||
|
Stockholders'
(deficit) equity:
|
||||||||
|
Convertible
preferred stock, $1.00 par value; 5,000,000 shares authorized and
3,350,000 shares issued and outstanding at December 31, 2009 and 2008
(liquidation value - $33,500,000 at December 31, 2009 and
2008)
|
3,350,000 | 3,350,000 | ||||||
|
Common
stock, $.01 par value: 170,000,000 shares authorized at December 31, 2009
and 2008; 88,671,445 and 88,489,278 shares issued and outstanding at
December 31, 2009 and 2008, respectively
|
886,715 | 884,893 | ||||||
|
Additional
paid-in capital
|
367,997,962 | 367,689,943 | ||||||
|
Treasury
stock, at cost: 874,999 shares held
at
|
||||||||
|
December
31, 2009 and 2008
|
(8,034,244 | ) | (8,034,244 | ) | ||||
|
Accumulated
other comprehensive income
|
- | (58,391 | ) | |||||
|
Accumulated
deficit
|
(366,126,413 | ) | (357,910,035 | ) | ||||
|
Total
stockholders' (deficit) equity
|
(1,925,980 | ) | 5,922,166 | |||||
|
Total
liabilities and stockholders' (deficit) equity
|
$ | 10,067,028 | $ | 28,923,395 | ||||
See
accompanying notes.
F-3
EntreMed,
Inc.
Consolidated
Statements of Operations
|
YEAR
ENDED DECEMBER 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Revenues:
|
||||||||||||
|
Royalties
|
4,989,725 | 7,472,061 | 7,393,463 | |||||||||
|
Other
|
294,333 | 5,158 | 2,188 | |||||||||
| 5,284,058 | 7,477,219 | 7,395,651 | ||||||||||
|
Costs
and expenses:
|
||||||||||||
|
Research
and development
|
7,901,522 | 20,069,229 | 23,739,392 | |||||||||
|
General
and administrative
|
4,104,287 | 7,764,532 | 7,386,570 | |||||||||
|
Acquired
In-Process R&D
|
- | 2,000,000 | - | |||||||||
| 12,005,809 | 29,833,761 | 31,125,962 | ||||||||||
|
Investment
income
|
69,702 | 882,253 | 2,112,583 | |||||||||
|
Interest
expense
|
(1,493,400 | ) | (2,216,163 | ) | (793,393 | ) | ||||||
|
Fixed
asset impairment loss
|
- | (171,576 | ) | - | ||||||||
|
Other
expense
|
(70,929 | ) | - | - | ||||||||
|
Net
loss
|
(8,216,378 | ) | (23,862,028 | ) | (22,411,121 | ) | ||||||
|
Dividends
on Series A convertible preferred stock
|
(1,005,000 | ) | (1,005,000 | ) | (1,005,000 | ) | ||||||
|
Net
loss attributable to common shareholders
|
$ | (9,221,378 | ) | $ | (24,867,028 | ) | $ | (23,416,121 | ) | |||
|
Net
loss per share (basic and diluted)
|
$ | (0.11 | ) | $ | (0.29 | ) | $ | (0.28 | ) | |||
|
Weighted
average number of shares outstanding (basic and diluted)
|
87,757,765 | 86,479,768 | 84,166,552 | |||||||||
See
accompanying notes.
F-4
ENTREMED,
INC.
CONSOLIDATED
STATEMENT OF STOCKHOLDERS' EQUITY
YEARS
ENDED DECEMBER 31, 2009, 2008, AND 2007
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
Deferred
|
Other
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Treasury
|
Paid-in
|
Stock
|
Comprehensive
|
Accumulated
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Stock
|
Capital
|
Compensation
|
Income
(Loss)
|
Deficit
|
Total
|
|||||||||||||||||||||||||||||||
|
Balance
at December 31, 2006
|
3,350,000 | $ | 3,350,000 | 83,964,586 | $ | 848,400 | $ | (8,034,244 | ) | $ | 362,318,737 | $ | - | $ | 117,212 | $ | (311,636,886 | ) | $ | 46,963,219 | ||||||||||||||||||||
|
Issuance
of common stock for options exercised
|
- | - | 75,000 | 750 | - | 81,000 | - | - | - | 81,750 | ||||||||||||||||||||||||||||||
|
Issuance
of common stock for warrants exercised
|
- | - | 675,000 | 6,750 | - | 666,774 | - | - | - | 673,523 | ||||||||||||||||||||||||||||||
|
Fair
value of warrants issued pursuant to debt settlement
agreements
|
- | - | - | - | - | 190,000 | - | - | - | 190,000 | ||||||||||||||||||||||||||||||
|
Restricted
stock grants, net of issuance cost
|
- | - | 123,407 | 1,225 | - | 187,729 | - | - | - | 188,954 | ||||||||||||||||||||||||||||||
|
Stock-based
compensation expense, net of forfeitures
|
- | - | - | - | - | 1,260,910 | - | - | - | 1,260,910 | ||||||||||||||||||||||||||||||
|
Comprehensive
loss:
|
||||||||||||||||||||||||||||||||||||||||
|
Net
loss
|
- | - | - | - | - | - | - | - | (22,411,121 | ) | (22,411,121 | ) | ||||||||||||||||||||||||||||
|
Unrealized
loss on investments
|
- | - | - | - | - | - | - | (50,258 | ) | - | (50,258 | ) | ||||||||||||||||||||||||||||
|
Comprehensive
loss
|
- | - | - | - | - | - | - | - | - | (22,461,379 | ) | |||||||||||||||||||||||||||||
|
Balance
at December 31, 2007
|
3,350,000 | $ | 3,350,000 | 84,837,993 | $ | 857,125 | $ | (8,034,244 | ) | $ | 364,705,150 | $ | - | $ | 66,954 | $ | (334,048,007 | ) | $ | 26,896,977 | ||||||||||||||||||||
|
Issuance
of common stock for milestone payment, net of stock issuance
costs
|
- | - | 2,564,104 | 25,641 | - | 1,897,403 | 1,923,044 | |||||||||||||||||||||||||||||||||
|
Restricted
stock grants
|
- | - | 212,182 | 2,127 | - | 190,929 | - | - | - | 193,056 | ||||||||||||||||||||||||||||||
|
Stock-based
compensation expense, net of forfeitures
|
- | - | - | - | - | 896,461 | - | - | - | 896,461 | ||||||||||||||||||||||||||||||
|
Comprehensive
loss:
|
||||||||||||||||||||||||||||||||||||||||
|
Net
loss
|
- | - | - | - | - | - | - | - | (23,862,028 | ) | (23,862,028 | ) | ||||||||||||||||||||||||||||
|
Unrealized
loss on investments
|
- | - | - | - | - | - | - | (125,345 | ) | - | (125,345 | ) | ||||||||||||||||||||||||||||
|
Comprehensive
loss
|
- | - | - | - | - | - | - | - | - | (23,987,373 | ) | |||||||||||||||||||||||||||||
|
Balance
at December 31, 2008
|
3,350,000 | $ | 3,350,000 | 87,614,279 | $ | 884,893 | $ | (8,034,244 | ) | $ | 367,689,943 | $ | - | $ | (58,391 | ) | $ | (357,910,035 | ) | $ | 5,922,166 | |||||||||||||||||||
|
Issuance
of common stock for options exercised (less options used for pymt of
taxes)
|
- | - | 67,802 | 678 | - | (10,993 | ) | - | - | - | (10,315 | ) | ||||||||||||||||||||||||||||
|
Restricted
stock grants
|
- | - | 114,365 | 1,144 | - | 72,050 | - | - | - | 73,194 | ||||||||||||||||||||||||||||||
|
Stock-based
compensation expense, net of forfeitures
|
- | - | - | - | - | 246,962 | - | - | - | 246,962 | ||||||||||||||||||||||||||||||
|
Comprehensive
loss:
|
||||||||||||||||||||||||||||||||||||||||
|
Net
loss
|
- | - | - | - | - | - | - | 58,391 | (8,216,378 | ) | (8,157,987 | ) | ||||||||||||||||||||||||||||
|
Unrealized
loss on investments
|
- | - | - | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Comprehensive
loss
|
- | - | - | - | - | - | - | - | - | (8,157,986 | ) | |||||||||||||||||||||||||||||
|
Balance
at December 31, 2009
|
3,350,000 | $ | 3,350,000 | 87,796,446 | $ | 886,715 | $ | (8,034,244 | ) | $ | 367,997,962 | $ | - | $ | - | $ | (366,126,413 | ) | $ | (1,925,980 | ) | |||||||||||||||||||
See
accompanying notes.
F-5
EntreMed,
Inc.
Consolidated
Statements of Cash Flows
|
YEAR
ENDED DECEMBER 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
CASH
FLOWS FROM OPERATING ACTIVITIES
|
||||||||||||
|
Net
loss
|
$ | (8,216,378 | ) | $ | (23,862,028 | ) | $ | (22,411,121 | ) | |||
|
Adjustments
to reconcile net loss to net cash used in operating
|
||||||||||||
|
activities:
|
||||||||||||
|
Depreciation
and amortization
|
95,106 | 319,230 | 333,827 | |||||||||
|
Write-off
of in-process R&D
|
- | 2,000,000 | - | |||||||||
|
Fixed
asset impairment loss
|
- | 171,576 | - | |||||||||
|
Investment
impairment loss
|
70,929 | - | - | |||||||||
|
Stock-based
compensation expense
|
320,156 | 1,089,517 | 1,454,864 | |||||||||
|
Amortization
of deferred stock-based compensation
|
- | - | - | |||||||||
|
Amortization
of discount on short-term investments
|
(21,270 | ) | (573,647 | ) | (1,112,976 | ) | ||||||
|
Non-cash
interest
|
133,082 | 198,543 | 62,428 | |||||||||
|
Changes
in operating assets and liabilities:
|
||||||||||||
|
Accounts
receivable
|
593,250 | 21,446 | (56,554 | ) | ||||||||
|
Interest
receivable
|
37,329 | 106,789 | (70,296 | ) | ||||||||
|
Prepaid
expenses and other
|
162,688 | 80,944 | (86,085 | ) | ||||||||
|
Accounts
payable
|
(1,527,591 | ) | (1,054,694 | ) | (1,395,960 | ) | ||||||
|
Accrued
liabilities
|
(1,839,146 | ) | 908,512 | (152,352 | ) | |||||||
|
Deferred
rent
|
(20,763 | ) | (119,595 | ) | (89,848 | ) | ||||||
|
Net
cash used in operating activities
|
(10,212,608 | ) | (20,713,407 | ) | (23,524,073 | ) | ||||||
|
CASH
FLOWS FROM INVESTING ACTIVITIES
|
||||||||||||
|
Purchases
of short term investments
|
(7,992,859 | ) | (51,556,886 | ) | (43,999,618 | ) | ||||||
|
Maturities
of short term investments
|
15,500,000 | 62,525,000 | 56,664,000 | |||||||||
|
Unrealized
gain on short term investments
|
(4,436 | ) | 4,436 | - | ||||||||
|
Purchases
of furniture and equipment
|
(2,889 | ) | (134,065 | ) | (106,721 | ) | ||||||
|
Net
cash provided by (used in) investing activities
|
7,499,816 | 10,838,485 | (12,557,661 | ) | ||||||||
|
CASH
FLOWS FROM FINANCING ACTIVITIES
|
||||||||||||
|
Repayment
of loan
|
(7,707,840 | ) | (2,980,893 | ) | (751,093 | ) | ||||||
|
Proceeds
from issuance of loan and warrants, net of discount
|
- | - | 19,900,000 | |||||||||
|
Debt
issuance costs
|
- | - | (153,012 | ) | ||||||||
|
Stock
issuance costs
|
(10,315 | ) | (76,955 | ) | - | |||||||
|
Net
proceeds from sale of common stock
|
- | - | 750,275 | |||||||||
|
Net
cash (used in) provided by financing activities
|
(7,718,155 | ) | (3,057,848 | ) | 19,746,170 | |||||||
|
Net
(decrease) increase in cash and cash equivalents
|
(10,430,947 | ) | (12,932,770 | ) | 8,779,758 | |||||||
|
Cash
and cash equivalents at beginning of year
|
16,743,129 | 29,675,899 | 20,896,141 | |||||||||
|
Cash
and cash equivalents at end of year
|
$ | 6,312,182 | $ | 16,743,129 | $ | 29,675,899 | ||||||
|
Supplemental
disclosure of cash flow information:
|
||||||||||||
|
Cash
paid during the year for interest
|
$ | 1,360,319 | $ | 2,017,619 | $ | 554,660 | ||||||
|
Non-cash
investing activity:
|
||||||||||||
|
Stock
issued in connection with milestone payment related to the acquisition of
Miikana
|
$ | - | $ | 2,000,000 | $ | - | ||||||
F-6
EntreMed,
Inc.
Notes to
Consolidated Financial Statements
December
31, 2009
1. DESCRIPTION
OF BUSINESS AND BASIS OF PRESENTATION
EntreMed, Inc. (“EntreMed” or “the
Company”) (Nasdaq: ENMD) is a clinical-stage pharmaceutical company focused on
developing ENMD-2076, an Aurora A and angiogenic kinase inhibitor for the
treatment of cancer. ENMD-2076 is currently in Phase 1 studies in
advanced cancers, multiple myeloma and leukemia. The Company anticipates
the initiation of a Phase 2 study in ovarian cancer in the second quarter
2010.
ENMD-2076
is a novel orally-active, Aurora A/angiogenic kinase inhibitor with potent
activity against Aurora A and multiple tyrosine kinases linked to cancer and
inflammatory diseases. ENMD-2076 is relatively selective for the
Aurora A isoform in comparison to Aurora B. Aurora kinases are key
regulators of the process of mitosis, or cell division, and are often
over-expressed in human cancers. ENMD-2076 exerts its effects through multiple
mechanisms of action, including antiproliferative activity and the inhibition of
angiogenesis. ENMD-2076 has demonstrated significant, dose-dependent preclinical
activity as a single agent, including tumor regression, in multiple xenograft
models (e.g. breast, colon, leukemia), as well as activity towards ex
vivo-treated human leukemia patient cells.
ENMD-2076
has received orphan drug designation for the treatment of ovarian cancer,
multiple myeloma and AML.
ENMD-2076
is the only program currently under active clinical evaluation by the
Company. The selection of ENMD-2076 as its lead drug candidate allows
the Company to direct the majority of its resources to its clinical
development. However, the Company also owns intellectual property
around its other drug candidates. So that EntreMed can continue to
accelerate the development of ENMD-2076, the Company does not intend to initiate
additional new studies for these programs unless additional significant
financing becomes available. EntreMed’s other drug candidates include
MKC-1, an oral cell-cycle inhibitor with activity against the mTOR pathway that
has completed multiple Phase 2 clinical trials for cancer, and ENMD-1198, a
novel antimitotic agent that has completed a Phase 1 study in advanced
cancers. The Company also has an approved Investigational New Drug
Application (IND) for the use of Panzem® in
rheumatoid arthritis (RA) treatment. All of EntreMed’s candidates are
multi-mechanism in that they target disease cells and the blood vessels that
nourish them. The Company believes they can be developed to be safe
and convenient, and provide the potential for improved patient
outcomes.
LIQUIDITY
RISKS AND MANAGEMENT’S PLANS
The accompanying financial statements
have been prepared on a going-concern basis, which contemplates the realization
of assets and satisfaction of liabilities in the normal course of
business. As of December 31 2009, the Company has operating and
liquidity concerns. Since inception, the Company has incurred
significant losses from operations and has incurred an accumulated deficit of
$366 million. The Company expects to continue to incur expenses,
resulting in operating losses, for the foreseeable future due to, among other
factors, its continuing clinical trials, planned future clinical trials, and
other anticipated research and development activities. Based on
current plans, the Company expects its current available cash and cash
equivalents to meet its cash requirements into the fourth quarter of fiscal
2010.
The Company’s ability to continue as a
going concern is dependent on its success at raising additional capital
sufficient to meet its obligations on a timely basis, and to ultimately attain
profitability. Management is actively engaged in raising capital
through a planned common stock financing, and is confident of its ability to
raise the necessary funds for the Company’s growth and development
activities. However, there is no assurance that the Company will
raise capital sufficient to enable the Company to continue its operations for
the next 12 months. If additional funds are raised by issuing equity
securities, dilution to existing shareholders may result. There can
be no assurance that adequate additional financing will be available to the
Company on terms that it deems acceptable, if at all.
F-7
In the event the Company is unable to
successfully raise additional capital, it is unlikely that the Company will have
sufficient cash flows and liquidity to finance its business operations as
currently contemplated. Accordingly, in the event new financing is
not obtained, the Company will likely reduce general and administrative expenses
and delay, scale back, or interrupt the clinical development of ENMD-2076 until
it is able to obtain sufficient financing to do so.
These factors could significantly limit
the Company’s ability to continue as a going concern. The financial
statements do not include any adjustments relating to recoverability and
classification of recorded asset amounts or the amounts and classification of
liabilities that might be necessary should the Company be unable to continue in
existence.
The
accompanying consolidated financial statements include the accounts of the
Company’s controlled subsidiary, Miikana Therapeutics, Inc.
(Miikana). All inter-company balances and transactions have been
eliminated in consolidation. The Company refers to EntreMed and its
consolidated subsidiary.
Material
subsequent events have been considered for disclosure and recognition through
the filing date of these consolidated financial statements.
2. SUMMARY
OF SIGNIFICANT ACCOUNTING POLICIES
SEGMENT
INFORMATION
The
Company currently operates in one business segment, which is the development of
therapeutics primarily for the treatment of cancer. The Company is
managed and operated as one business. EntreMed’s senior management
team reports to the Executive Committee of the Board of Directors and is
responsible for aligning the Company’s business strategy with its core
scientific strengths, while maintaining prudent resource management, fiscal
responsibility and accountability. The team has redirected EntreMed’s financial
resources and R&D strategy to focus on the development of its
Aurora/angiogenic kinase inhibitor, ENMD-2076.
The
Company does not operate separate lines of business with respect to its products
or product candidates. Accordingly, the Company does not have separately
reportable segments as defined by authoritative guidance issued by the Financial
Accounting Standards Board (FASB).
RESEARCH
AND DEVELOPMENT
Research
and development expenses consist primarily of compensation and other expenses
related to research and development personnel, research collaborations, costs
associated with pre-clinical testing and clinical trials of our product
candidates, including the costs of manufacturing drug substance and drug
product, regulatory maintenance costs, and facilities expenses. Research and
development costs are expensed as incurred, including costs incurred in filing,
defending and maintaining patents.
PROPERTY
AND EQUIPMENT
Furniture
and equipment and leasehold improvements are stated at cost and are depreciated
over their estimated useful lives of 3 to 10 years. Depreciation is determined
on a straight-line basis. Depreciation expense was $95,106, $319,230
and $333,827 in 2009, 2008 and 2007, respectively.
F-8
Property
and equipment consists of the following:
|
DECEMBER
31,
|
||||||||
|
2009
|
2008
|
|||||||
|
Furniture
and equipment
|
$ | 4,990,578 | $ | 4,987,689 | ||||
|
Leasehold
improvements
|
1,325,031 | 1,325,031 | ||||||
| 6,315,609 | 6,312,720 | |||||||
|
Less: accumulated
depreciation
|
(6,144,111 | ) | (6,049,005 | ) | ||||
| $ | 171,498 | $ | 263,715 | |||||
IMPAIRMENT
OF LONG-LIVED ASSETS
In accordance with authoritative
guidance issued by FASB, the Company periodically evaluates the value reflected
in its balance sheet of long-lived assets, such as equipment, when events and
circumstances indicate that the carrying amount of an asset may not be
recovered. Such events and circumstances include the use of the asset
in current research and development projects, any potential alternative uses of
the asset in other research and development projects in the short to medium term
and restructuring plans entered into by the Company. Determination of
recoverability is based on an estimate of undiscounted future cash flows
resulting from the use of the asset and its eventual disposition. In
the event that such cash flows are not expected to be sufficient to recover the
carrying amount of the assets, the assets are written down to their estimated
fair values. At December 31, 2008, the Company recorded a fixed asset
impairment charge of approximately $172,000 in connection with its restructuring
plan discussed later in Note 2. No impairment charges were recorded
in 2009.
CASH
AND CASH EQUIVALENTS
Cash and
cash equivalents include cash and highly liquid investments with original
maturities of less than 90 days. Substantially all of the Company's cash
equivalents are held in short-term money market accounts of banks and brokerage
houses.
SHORT-TERM
INVESTMENTS
Short-term
investments at December 31, 2009 consist of equity securities. The
Company has classified these investments as available for sale. Such
securities are carried at fair market value. The cost of securities sold is
calculated using the specific identification method. Unrealized gains
and losses on these securities, if any, are reported as accumulated other
comprehensive income (loss), which is a separate component of stockholders’
equity. Net unrealized losses of $12,538 and $125,345 were recorded in 2009 and
2008, respectively. Realized gains and losses and declines in value
judged to be other than temporary on securities available for sale, if any, are
included in operations. There were realized losses of approximately $71,000 for
the year ended December 31, 2009. There were no realized gains or
losses for the year ended December 31, 2008. Short-term investments
are principally uninsured and subject to normal credit risk.
The
following is a summary of available-for-sale securities at December 31,
2009:
|
Available-for-Sale Securities
|
||||||||||||||||
|
Amortized
|
Gross
Unrealized
|
Gross
Realized
|
Estimated Fair
Value (Net
Carrying
|
|||||||||||||
|
Cost
|
Gains
|
Losses
|
Amount)
|
|||||||||||||
|
Commercial
Paper
|
$ | - | $ | - | $ | - | $ | - | ||||||||
|
Government-sponsored
Enterprise Obligations
|
- | - | - | - | ||||||||||||
|
Equity
Securities
|
125,000 | - | (70,929 | ) | 54,071 | |||||||||||
|
Total
|
$ | 125,000 | $ | - | $ | (70,929 | ) | $ | 54,071 | |||||||
F-9
The
following is a summary of available-for-sale securities at December 31,
2008:
|
Available-for-Sale Securities
|
||||||||||||||||
|
Amortized
|
Gross
Unrealized
|
Gross
Unrealized
|
Estimated Fair
Value (Net
Carrying
|
|||||||||||||
|
Cost
|
Gains
|
Losses
|
Amount)
|
|||||||||||||
|
Commercial Paper
|
$ | 3,965,832 | $ | 33,558 | $ | - | $ | 3,999,390 | ||||||||
|
Government-sponsored Enterprise
Obligations
|
3,515,196 | 4,491 | - | 3,519,687 | ||||||||||||
|
Equity
Securities
|
125,000 | - | (96,033 | ) | 28,967 | |||||||||||
|
Total
|
$ | 7,606,028 | $ | 38,049 | $ | (96,033 | ) | $ | 7,548,044 | |||||||
ACCOUNTS
RECEIVABLE
Accounts receivable are stated net of
allowances for doubtful accounts. Allowances for doubtful accounts are
determined on a specific item basis. Management reviews the credit
worthiness of individual customers and past payment history to determine the
allowance for doubtful accounts. There is an allowance for doubtful
accounts of $72,145 and $54,145 at December 31, 2009 and 2008,
respectively.
As of December 31, 2009 and 2008, one
individual customer represented 100% and 99%, respectively, of the total
accounts receivable.
EXPENSES
FOR CLINICAL TRIALS
Expenses
for clinical trials are incurred from planning through patient enrollment to
reporting of the data. The Company estimates expenses incurred for
clinical trials that are in process based on patient enrollment and based on
clinical data collection and management. Costs that are associated
with patient enrollment are recognized as each patient in the clinical trial
completes the enrollment process. Estimated clinical trial costs
related to enrollment can vary based on numerous factors, including expected
number of patients in trials, the number of patients that do not complete
participation in a trial, and when a patient drops out of a
trial. Costs that are based on clinical data collection and
management are recognized in the reporting period in which services are
provided. In the event of early termination of a clinical trial, the
Company would accrue an amount based on estimates of the remaining
non-cancelable obligations associated with winding down the clinical
trial. As of December 31, 2009 and 2008, clinical trial accruals were
$1,425,725 and $1,600,975, respectively, and are included in Accounts Payable in
the accompanying consolidated balance sheets.
INCOME
TAXES
Income
tax expense is accounted for in accordance with authoritative guidance issued by
FASB. Income tax expense has been provided using the liability
method. Deferred tax assets and liabilities are determined based on the
difference between the financial statement and tax bases of assets and
liabilities as measured by the enacted tax rates that will be in effect when
these differences reverse. The Company provides a valuation allowance against
net deferred tax assets if, based upon the available evidence, it is not more
likely than not that the deferred tax assets will be
realized.
Effective
January 1, 2007, the Company adopted a new interpretation of an existing
accounting standard that specifies how tax benefits for uncertain tax positions
are to be recognized, measured and derecognized in financial statements;
requires certain disclosures of uncertain tax matters; specifies how reserves
for uncertain tax positions should be classified on the balance sheet; and
provides transition and interim-period guidance, among other
provisions.
F-10
At the
date of adoption of the authoritative guidance, EntreMed had an unrecognized tax
benefit of $2,751,000 related to net R&D tax credit
carryforwards. The Company had a full valuation allowance on the net
deferred tax asset recognized in the consolidated financial
statements. As a result, the adoption of the authoritative guidance
effective January 1, 2007, had no effect on the Company’s financial
position.
The
Company’s policy is to recognize interest and penalties related to the
underpayment of income taxes as a component of income tax expense. To date,
there have been no interest or penalties charged to the Company in relation to
the underpayment of income taxes.
REVENUE
RECOGNITION
Revenue is not recognized until it is
realized or realizable and earned. Revenue is recognized when all of
the following criteria are met: persuasive evidence of an arrangement
exists, delivery has occurred or services have been rendered, the price to the
buyer is fixed and determinable and collectibility is reasonably
assured.
Royalty
Revenue - Royalties from licenses are based on third-party sales and recorded as
earned in accordance with contract terms, when third-party results are reliably
measured and collectibility is reasonably assured. The majority of the Company’s
2009, 2008 and 2007 revenues were from royalties on the sale of Thalomid®, which
the Company began to recognize in the third quarter of each year. In
2004, certain provisions of a purchase agreement dated June 14, 2001 by and
between Bioventure Investments kft (“Bioventure”) and the Company were
satisfied, and, as a result, in 2005 the Company became entitled to share in the
royalty payments received by Royalty Pharma Finance Trust, successor to
Bioventure, on annual Thalomid® sales
above a certain threshold. Based on the licensing agreement royalty
formula, annual royalty sharing commences with Thalomid® annual
sales of approximately $225 million. The Company may also be eligible
to receive royalty payments under a February 2004 agreement with Children’s
Medical Center Corporation (“CMCC”) and Alchemgen Therapeutics. Under
the agreement, Alchemgen received rights to market endostatin and angiostatin in
Asia. In April 2008, the Company was advised that Alchemgen
Therapeutics ceased operations, therefore eliminating our ability to receive any
royalties from Alchemgen under the agreement. Through a sublicense to
OxFord Biomedica PLC, the Company is eligible to receive royalties from it based
on a portion of the net sales of products based on Endostatin and angiostatin
developed for the treatment of ophthalmic (eye) diseases. The Company
received its first royaltiy payment under this agreement in 2009, and has
accrued a sub-royalty payment in the amount of $74,000 at December 31, 2009 as a
payable to CMCC.
NET
LOSS PER SHARE
Net loss
per share (basic and diluted) was computed by dividing net loss attributable to
common shareholders by the weighted average number of shares of common stock
outstanding. Preferred Series A common stock equivalents totaling 16,750,000 for
2009, 2008 and 2007 were anti-dilutive and, therefore, were not included in the
computation of weighted average shares used in computing diluted loss per
share.
COMPREHENSIVE
LOSS
The
Company presents comprehensive loss and its components as part of the
consolidated financial statements. Comprehensive loss is comprised of net loss
and unrealized gain and loss on investments as follows:
|
Years ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Net
loss
|
$ | (8,157,987 | ) | $ | (23,862,028 | ) | $ | (22,411,121 | ) | |||
|
Other
comprehensive loss
|
- | (125,345 | ) | (50,258 | ) | |||||||
|
Comprehensive
loss
|
$ | (8,157,987 | ) | $ | (23,987,373 | ) | $ | (22,461,379 | ) | |||
F-11
FAIR
VALUE MEASUREMENT
The Company adopted a new accounting
standard, effective January 1, 2008, which defines fair value as the price that
would be received to sell an asset or paid to transfer a liability (“the exit
price”) in an orderly transaction between market participants at the measurement
date. The standard outlines a valuation framework and creates a fair
value hierarchy in order to increase the consistency and comparability of fair
value measurements and the related disclosures. In determining fair
value, EntreMed primarily uses prices and other relevant information generated
by market transactions involving identical or comparable assets (“market
approach”). The Company has determined that the fair value
measurements are in accordance with the guidance; therefore, the implementation
of the new accounting standard did not have any impact on its consolidated
financial condition or results of operations. The implementation of
the new accounting standard resulted in expanded disclosures about securities
measured at fair value, as discussed below.
The guidance established a three-level
hierarchy for fair value measurements that distinguishes between market
participant assumptions developed based on market data obtained from sources
independent of the reporting entity (“observable inputs”) and the reporting
entity’s own assumptions about market participant assumptions developed based on
the best information available in the circumstances (“unobservable
inputs”). EntreMed currently does not have non-financial assets and
non-financial liabilities that are required to be measured at fair value on a
recurring basis. The Company’s financial assets and liabilities are
measured using inputs from the three levels of the fair value hierarchy, defined
as follows:
Level 1 – Inputs are unadjusted quoted
prices in active markets for identical assets or liabilities that we have the
ability to access at the measurement date.
Level 2 – Inputs are quoted prices for
similar assets and liabilities in active markets, quoted prices for identical or
similar assets or liabilities in markets that are not active, inputs other than
quoted pries that are observable for the asset or liability (i.e., interest
rates, yield curves, etc.), and inputs that are derived principally from or
corroborated by observable market data by correlation or other means (market
corroborated inputs).
Level 3 – Unobservable inputs that
reflect our own assumptions, based on the best information available, including
our own data.
In accordance with the fair value
hierarchy described above, the following table shows the fair value of the
Company’s financial assets and liabilities that are required to be measured at
fair value as of December 31, 2009 and 2008:
|
Fair Value Measurements at December 31, 2009
|
||||||||||||||||
|
Total Carrying
Value at
|
Quoted prices in
active markets
|
Significant other
observable inputs
|
Significant
unobservable
inputs
|
|||||||||||||
|
December 31, 2009
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Cash
equivalents
|
$ | 4,105,868 | $ | 4,105,868 | $ | — | $ | — | ||||||||
|
Available
for sale securities*
|
54,051 | 54,051 | — | — | ||||||||||||
* Realized
losses related to available for sale securities are included in operations, as
disclosed in Footnote 2.
F-12
|
Fair Value Measurements at December 31, 2008
|
||||||||||||||||
|
Total Carrying
Value at
|
Quoted prices in
active markets
|
Significant other
observable inputs
|
Significant
unobservable
inputs
|
|||||||||||||
|
December 31, 2008
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Cash
equivalents
|
$ | 16,743,129 | $ | 14,491,454 | $ | 2,251,675 | $ | — | ||||||||
|
Available
for sale securities*
|
7,548,044 | 28,967 | 7,519,077 | — | ||||||||||||
* Unrealized
gains and losses related to available for sale securities are reported as
accumulated other comprehensive income (loss), as disclosed in Footnote
2.
The Company’s Level 1 assets include
money market instruments and equity securities with quoted prices in active
markets. Level 2 assets include government-sponsored enterprise
securities, commercial paper and mortgage-backed securities. The
Level 2 securities are valued using matrix pricing, which is an acceptable,
practical expedient for inputs, under the new accounting standard.
SHARE-BASED
COMPENSATION
The
Company has adopted incentive and nonqualified stock option plans for executive,
scientific and administrative personnel of the Company as well as outside
directors and consultants, of which 3,135,043 shares remain available for grant
under the Company’s 2001 Long-Term Incentive Plan as of December 31,
2009. There are 6,984,606 shares issuable under options previously
granted under the plans and currently outstanding, with exercise prices ranging
from $0.16 to $58.38. Options granted under the plans vest over
periods varying from immediately to three years, are not transferable and
generally expire ten years from the date of grant.
The
Company records compensation expense associated with stock options and other
equity-based compensation in accordance with provisions of authoritative
guidance. Compensation costs are recognized based on a straight-line
method over the requisite service period, which is generally the option vesting
term of three years.
NEW
ACCOUNTING PRONOUNCEMENTS
In July 2009, the FASB Accounting
Standards Codification (“ASC” or “Codification”) became the authoritative source
of accounting principles generally accepted in the United States (“GAAP”)
recognized by the FASB. All existing FASB accounting standards and guidance were
superseded by the ASC. Instead of issuing new accounting standards in the form
of statements, FASB staff positions and Emerging Issues Task Force abstracts,
the FASB now issues Accounting Standards Updates that update the Codification.
Rules and interpretive releases of the Securities and Exchange Commission
(“SEC”) under authority of federal securities laws continue to be additional
sources of authoritative GAAP for SEC registrants. This Codification
is effective for financial statements issued for interim and annual periods
ending after September 15, 2009. There has been no change to the Company’s
consolidated financial statements due to the implementation of the Codification
other than changes in reference to various authoritative accounting
pronouncements in the consolidated financial statements.
On June
30, 2009, the Company adopted authoritative guidance which requires a publicly
traded company to include disclosures about the fair value of its financial
instruments whenever it issues summarized financial information for interim
reporting periods. Such disclosures include the fair value of all financial
instruments, for which it is practicable to estimate that value, whether
recognized or not recognized in the statement of financial position; the related
carrying amount of these financial instruments; and the methods and significant
assumptions used to estimate the fair value. Other than the required disclosures
(see the Debt note), the adoption of this guidance had no impact on the
Company’s consolidated financial statements.
F-13
In May 2009, the FASB issued
authoritative guidance regarding subsequent events which is intended to
establish general standards of accounting for, and disclosure of, events that
occur after the balance sheet date but before financial statements are issued or
are available to be issued. Specifically, the guidance sets forth the
period after the balance sheet date during which management of a reporting
entity should evaluate events or transactions that may occur for potential
recognition or disclosure in the financial statements, the circumstances under
which an entity should recognize events or transactions occurring after the
balance sheet date in its financial statements, and the disclosures that an
entity should make about events or transactions that occurred after the balance
sheet date. The adoption of this pronouncement, which is effective for financial
statements issued for interim and annual financial periods ending after June 15,
2009, did not have a material impact on the Company’s financial position and
results of operations. See Footnote 1 for the Company’s disclosure regarding its
evaluation of subsequent events.
In April 2009, the FASB issued
authoritative accounting guidance on how to determine the fair value of assets
and liabilities in the current economic environment, which reemphasizes that the
objective of a fair value measurement remains an exit price. If the Company were
to conclude that there has been a significant decrease in the volume and level
of activity of the asset or liability in relation to normal market activities,
quoted market values may not be representative of fair value and the Company may
conclude that a change in valuation technique or the use of multiple valuation
techniques may be appropriate. Additional guidance issued in April 2009
modifies the requirements for recognizing other-than-temporarily impaired debt
securities and revises the existing impairment model for such securities by
modifying the current intent and ability indicator in determining whether a debt
security is other-than-temporarily impaired. The FASB also issued guidance to
enhance the disclosure of financial instruments for both interim and annual
periods. The adoption of this guidance had no material effect on the Company’s
consolidated results of operations, financial position or
liquidity.
In
June 2008, the FASB issued authoritative guidance which mandates a two-step
process for evaluating whether an equity-linked financial instrument or embedded
feature is indexed to the entity’s own stock. The adoption of this guidance as
of January 1, 2009 had no material effect on the Company’s consolidated
results of operations, financial position or liquidity.
EntreMed has implemented all new
accounting pronouncements that are in effect and that may impact the Company’s
consolidated financial statements and the Company does not believe that there
are any other new accounting pronouncements that have been issued that might
have a material impact on its consolidated financial statements.
FAIR
VALUE OF FINANCIAL INSTRUMENTS AND CONCENTRATIONS OF RISK
Financial instruments that potentially
subject the Company to concentrations of credit risk consist principally of cash
and cash equivalents, short-term investments, accounts receivable and note
receivable. The Company maintains its cash and cash equivalents in
bank deposit accounts, which, at times, may exceed federally insured amounts.
The Company believes it is not exposed to any significant credit risk on cash
and cash equivalents or short-term investments. The carrying amount
of current assets and liabilities approximates their fair values due to their
short-term maturities. The carrying value and estimated fair value of
debt, before discount, were $9,311,000 and approximately $9,437,000,
respectively, at December 31, 2009. The fair value was estimated
based on the quoted market price.
USE
OF ESTIMATES
The
preparation of consolidated financial statements in conformity with accounting
principles generally accepted in the United States requires management to make
estimates and assumptions that affect the amounts reported in the financial
statements and accompanying notes. Actual results may differ from those
estimates, and such differences may be material to the consolidated financial
statements. In December 2009, the Company received $1.7 million
in royalty revenue from the sales of Thalomid® during
the third quarter of 2009, compared to estimated royalty revenue of $3.3
million. This resulted in a change in accounting estimate which has
been accounted for prospectively.
F-14
3. RESTRUCTURING
On December 15, 2008, the Company
announced its plan to focus and accelerate the Company’s clinical objectives for
ENMD-2076 as its leading program and effectively become a clinically-focused
operation, thereby lowering operating costs and preserving capital. In
connection with that plan, the Company reduced its workforce to align with the
new business structure. The workforce reductions resulted in the elimination of
approximately sixty percent of the Company’s total positions across all areas of
business and were substantially completed by December 31,
2008. Accordingly, the Company incurred charges for severance and
related benefits totaling $1,748,634 in 2008, all of which is included in
accrued liabilities at December 31, 2008. Additionally, the Company
incurred charges of $35,139 in 2009 related to the December 2008 reduction in
force.
On December 7, 2009, the Company
announced its plan to further reduce its workforce, which resulted in the
elimination of five full-time positions effective December 31, 2009 and one
full-time position effective February 28, 2010. The Company
incurred charges for severance and related benefits totaling $228,754 in the
fourth quarter of 2009, which is included in accrued liabilities at December 31,
2009. The Company has accounted for this restructuring as proscribed
by authoritative guidance.
A summary of changes in the accrued
liability during the twelve months ended December 31, 2009, is as
follows:
|
R&D
|
G&A
|
|||||||||||
|
Expenses
|
Expenses
|
|||||||||||
|
Balance
at December 31, 2008
|
$ | 1,748,634 | ||||||||||
|
Termination
benefits incurred
|
$ | 225,416 | $ | 38,477 | 263,893 | |||||||
|
Cash
payments
|
(757,440 | ) | (1,026,333 | ) | (1,783,773 | ) | ||||||
|
Balance
at December 31, 2009
|
$ | 228,754 | ||||||||||
Costs incurred in 2009 related to the
2008 and 2009 reductions in force are $264,000, of which, approximately $39,000
have been expensed as general and administrative costs and $225,000 have been
expensed as research and development costs. The Company does not
expect to incur additional costs in connection with these
terminations.
4. IN-PROCESS
R&D
In January 2006, the Company acquired
Miikana Therapeutics, a private biotechnology company. Pursuant to
the Merger Agreement, the Company acquired all of the outstanding capital stock
of Miikana Therapeutics, Inc. in exchange for 9.96 million shares of common
stock and the assumption of certain obligations. In 2008, EntreMed
initiated a Phase 1 clinical trial with its Aurora A and angiogenic kinase
inhibitor, ENMD-2076, in patients with solid tumors. A dosing of the
first patient with ENMD-2076 triggered a purchase price adjustment milestone of
$2 million, which the Company opted to pay in stock. In June 2008,
2,564,104 shares of common stock were issued as consideration for the
satisfaction of the milestone payment. The additional payment of $2
million was recorded to expense as in-process research and development since the
research and development project related to the Aurora Kinase Program had not
reached technical feasibility and has no future alternative use.
5. LOAN
PAYABLE
On
September 12, 2007, EntreMed, Inc. and Miikana Therapeutics, Inc., its wholly
owned subsidiary, entered into a Loan and Security Agreement (“Loan Agreement”)
with General Electric Capital Corporation (“GECC”), as agent, Merrill Lynch
Capital and Oxford Finance Corporation (collectively, “the
Lenders”). The Loan Agreement provides for (i) a term loan (“Term
Loan”) issued by the Lenders to the Company in the aggregate amount of
$20,000,000 and (ii) the issuance and sale to the Lenders of stock purchase
warrants evidencing the Lenders’ right to acquire their respective pro rata share of 250,000
shares of common stock of the Company (“Warrants”).
The Term
Loan will accrue interest in arrears at a fixed annual interest rate of 10.47%
until the Term Loan is fully repaid. The Term Loan will be repaid by
the Company to GECC, for the ratable benefit of the Lenders, as: (i) nine
consecutive monthly payments of interest only, each in the amount of $174,500,
commencing on November 1, 2007 and (ii) thirty consecutive monthly payments of
principal and interest, each in the amount of $760,606, commencing on August 1,
2008. The Term Loan expires on the earlier of (i) January 1, 2011 or
(ii) the date the Term Loan otherwise becomes due and payable under the Loan
Agreement, whether by acceleration of the obligations under the Term Loan or
otherwise.
F-15
The
Company will have the right to voluntarily prepay the Term Loan, in full or in
part, upon five business days’ written notice to GECC. Under certain
circumstances, the prepayment of the aggregate amount outstanding under the Term
Loan triggers a prepayment penalty equal to: (i) 3% on such prepayment amount,
if such prepayment is made on or before the one year anniversary of the closing
date, (ii) 2% on such prepayment amount, if such prepayment is made after the
one year anniversary of the closing date but on or before the two year
anniversary of the closing date, and (iii) 1% on such prepayment amount, if such
prepayment is made after the two year anniversary of the closing date but on or
before the Term Loan maturity date. The Loan Agreement contains
customary events of default that permits GECC to accelerate the Company’s
outstanding obligations if an event of default occurs and it is not cured within
the applicable grace periods. The Loan Agreement also provides for
automatic acceleration upon bankruptcy and other insolvency events.
The Term
Loan will be used for general corporate purposes and is secured by the personal
property owned by the Company, except for any intellectual property owned by the
Company. Notwithstanding the foregoing, the collateral for the Term
Loan includes (i) all cash, royalty fees and other proceeds that consist of
rights of payment or proceeds from the sale, licensing or other disposition of
all or any part of, or rights in, the intellectual property and the Thalidomide
Royalty Agreement and (ii) the Company’s rights under the Thalidomide Royalty
Agreement.
The Loan
Agreement contains customary affirmative and negative covenants. The
Company was in compliance with such covenants as of December 31,
2009.
As of
December 31, 2009, principal payments due are as follows:
|
Less
than one year
|
$ | 8,555,404 | ||
|
One
to two years
|
755,863 | |||
|
Total
|
$ | 9,311,267 |
The
Warrants are exercisable by the Lenders until September 12, 2012 at an exercise
price of $2.00 per share. The fair value of the Warrants issued was
$190,000, calculated using a Black-Scholes value of $.76 with an expected and
contractual life of 5 years, an assumed volatility of 98%, and a risk-free
interest rate of 4.11%. The value of the Warrants, and an upfront
underwriting fee of $100,000 paid to one of the Lenders, are recorded as a
discount on the loan and are amortized as interest expense over the life of the
loan. The Company also incurred certain debt issuance costs that were
deferred and are included in other assets in the Company’s balance sheet as of
December 31, 2009. Amortization of these fees and the discount
results in an effective interest rate of 11.40%. Non-cash interest
expense related to the amortization of debt issuance costs and debt discount was
$133,081, $198,543 and $62,428 for the years ended December 31, 2009, 2008 and
2007, respectively.
6. LICENSE
AGREEMENTS
Pursuant
to a purchase agreement dated June 14, 2001 by and between Bioventure
Investments kft ("Bioventure") and the Company, as amended July 13, 2001, July
30, 2001 and August 3, 2001 (the "Purchase Agreement"), Bioventure purchased all
of the Company’s right, title and interest to the net royalty payments payable
by Celgene Corporation ("Celgene") to the Company under the agreement dated as
of December 9, 1998 by and between the Company and Celgene (the "Celgene
Sublicense").
A
provision of the Bioventure purchase agreement provided the potential for an
adjustment in the purchase price if cumulative sales of Thalomid® exceeded
$800 million by December 31, 2004. Based on Thalomid® sales
reported publicly by Celgene, the Company concluded that cumulative
Thalomid® sales
had reached this milestone by December 31, 2004, thus triggering a royalty
sharing provision. Beginning the year after cumulative sales reach
$800 million, EntreMed is entitled to share in the royalty payments received by
Royalty Pharma Finance Trust, successor to Bioventure, on annual Thalomid® sales
above a certain threshold. In 2009, 2008 and 2007 Thalomid® sales
surpassed the royalty-sharing point and the Company recognized royalty revenues
of $4,990,000, $7,472,000 and $7,393,000, respectively. There can be
no assurance that the Company will receive additional material royalties under
the royalty sharing provision in the future.
F-16
In
March 2005, the Company entered into an exclusive worldwide license
agreement with Celgene Corporation for the development and commercialization of
Celgene’s small molecule tubulin inhibitor compounds for the treatment of
cancer. Under the terms of the agreement, Celgene received an upfront licensing
fee and may receive additional payments upon successful completion of certain
clinical, regulatory and sales milestones. No such milestones have been reached
through December 31, 2009. Our preliminary work on the program was
completed in 2008 and we do not expect to devote any significant resources to
this program in 2010.
In
January 2006, the Company entered into a License Agreement with Elan
Corporation, plc (“Elan”) in which the Company has been granted rights to
utilize Elan’s proprietary NanoCrystal Technology in connection with the
development of the oncology product candidate, Panzem® NCD.
Under the terms of the License Agreement, Elan is eligible to receive payments
upon the achievement of certain clinical, manufacturing, and regulatory
milestones and to receive royalty payments based on sales of Panzem®
NCD. Additionally, under the agreement and the corresponding Services
Agreement, Elan has the right to manufacture EntreMed’s Panzem®
NCD. Milestones related to the initiation of Phase 2 clinical trials
for Panzem® NCD have
been paid and there are no additional milestones achieved as of December 31,
2009. We have discontinued clinical development of Panzem® NCD for
oncology.
7. RELATED
PARTY TRANSACTIONS
There
were no expenses from related parties in 2009, 2008 and 2007 and no payables as
of December 31, 2009.
8. INCOME
TAXES
The
Company has net operating loss carryforwards for income tax purposes of
approximately $333,486,000 at December 31, 2009 ($332,862,000 at December 31,
2008 and $309,997,000 at December 31, 2007) that expire in years 2010 through
2029. The Company also has research and development tax credit carryforwards of
approximately $8,868,000 as of December 31, 2009 that expire in years 2010
through 2028. These net operating loss carryforwards include approximately
$20,000,000, related to exercises of stock options for which the income tax
benefit, if realized, would increase additional paid-in capital. The utilization
of the net operating loss and research and development carryforwards may be
limited in future years due to changes in ownership of the Company pursuant to
Internal Revenue Code Section 382. For financial reporting purposes, a valuation
allowance has been recognized to reduce the net deferred tax assets to zero due
to uncertainties with respect to the Company's ability to generate taxable
income in the future sufficient to realize the benefit of deferred income tax
assets.
Deferred
income taxes reflect the net effect of temporary differences between the
carrying amounts of assets and liabilities for financial reporting purposes and
the amounts used for income tax purposes. Significant components of
the Company's deferred income tax assets and liabilities as of December 31, 2009
and 2008 are as follows:
|
|
DECEMBER 31,
|
|||||||
|
2009
|
2008
|
|||||||
|
Deferred
income tax assets (liabilities):
|
||||||||
|
Net
operating loss carryforwards
|
$ | 131,478,000 | $ | 131,232,000 | ||||
|
Research
and development credit carryforward
|
8,868,000 | 9,179,000 | ||||||
|
Equity
investment
|
72,000 | 72,000 | ||||||
|
Other
|
3,624,000 | 3,869,000 | ||||||
|
Depreciation
|
325,000 | 293,000 | ||||||
|
Valuation
allowance for deferred income tax assets
|
(144,367,000 | ) | ( 144,645,000 | ) | ||||
|
Net
deferred income tax assets
|
$ | - | $ | - | ||||
F-17
A reconciliation of the provision for
income taxes to the federal statutory rate is as follows:
|
2009
|
2008
|
2007
|
||||||||||
|
Tax
benefit at statutory rate
|
$ | ( 2,794,000 | ) | $ | ( 8,113,000 | ) | $ | (7,620,000 | ) | |||
|
State
taxes
|
( 445,000 | ) | ( 1,293,000 | ) | (1,282,000 | ) | ||||||
|
Net
R&D credit adjustment
|
412,000 | 114,000 | 2,125,000 | |||||||||
|
Attribute
expiration and other
|
3,062,000 | (520,000 | ) | 703,000 | ||||||||
|
Permanent
M-1s
|
6,000 | 10,000 | (13,000 | ) | ||||||||
|
Change
in valuation allowance
|
(241,000 | ) | 8,884,000 | 9,351,000 | ||||||||
|
Change
in estimated effective rate
|
- | 918,000 | (3,264,000 | ) | ||||||||
| $ | - | $ | - | $ | - | |||||||
The
Company had $3,063,000 of unrecognized tax benefits as of January 1, 2009
related to net R&D tax credit carryforwards. EntreMed had a full valuation
allowance on the net deferred tax asset recognized in the consolidated financial
statements. As a result, the adoption of the accounting guidance
effective January 1, 2007 had no effect on the Company’s retained earnings as of
such date, or on net operating losses available to offset future taxable
income. For the period ended December 31, 2009, there was an
additional unrecognized tax benefit of $94,000 related to R&D tax credits,
and a reduction in unrecognized tax benefits of $198,000 related to R&D
credit carryforwards expiring in 2009. The Company has a full
valuation allowance at January 1, 2009 and at December 31, 2009 against the full
amount of its net deferred tax assets and therefore, there was no impact on the
Company’s financial position.
A reconciliation of the beginning and
ending amount of gross unrecognized tax benefits is as follows:
|
2009
|
2008
|
2007
|
||||||||||
|
Unrecognized
tax benefits balance at January 1
|
$ | 3,063,000 | $ | 2,959,000 | $ | 2,751,000 | ||||||
|
Additions
for Tax Positions of Prior Periods
|
94,000 | 114,000 | 208,000 | |||||||||
|
Reductions
for Tax Positions of Prior Periods
|
(198,000 | ) | (10,000 | ) | - | |||||||
|
Additions
for Tax Positions of Current Period
|
- | - | - | |||||||||
|
Reductions
for Tax Positions of Current Period Settlements
|
- - | - - | - - | |||||||||
|
Lapse
of statute of limitations
|
- | - | - | |||||||||
|
Unrecognized
tax benefits balance at December 31
|
$ | 2,959,000 | $ | 3,063,000 | $ | 2,959,000 | ||||||
The Company recognizes interest and
penalties related to uncertain tax positions as a component of income tax
expense. As of January 1, 2009 and December 31, 2009, the Company had
no accrued interest or penalties related to uncertain tax positions,
respectively.
The tax returns for all years in the
Company’s major tax jurisdictions are not settled as of December 31,
2009. Due to the existence of tax attribute carryforwards (which are
currently offset by a full valuation allowance), the Company treats all years’
tax positions as unsettled due to the taxing authorities’ ability to modify
these attributes.
The Company believes that the total
unrecognized tax benefit, if recognized, would impact the effective rate,
however, such reversal may be offset by a corresponding adjustment to the
valuation allowance.
9. STOCKHOLDERS'
EQUITY
In
2002, the Company
issued 3,350,000 shares of Series A Preferred Stock to Celgene. The
Series A Preferred Stock is convertible, at the option of Celgene, at any time,
into common stock at an initial per common share conversion price of $1.00 (1
share of preferred converts into 5 shares of common). The value of
the common stock at the date the Series A Preferred Stock was issued was
$0.86. The conversion price is subject to change for certain dilutive
events, as defined. The Company may cause the Series A Preferred
Stock to convert automatically provided all of the following conditions are
met:
F-18
|
|
(i)
|
As
of the conversion date, the common stock is traded and was traded during
the 60 trading days preceding the conversion date, on a national
securities exchange;
|
|
|
(ii)
|
The
average per share closing price of the common stock is greater than $5.00
over a 60-trading day period ending on the conversion date,
and
|
|
|
(iii)
|
A
registration statement with respect to resale of the common stock issuable
in the conversion to the holders of the Series A Preferred Stock has been
filed with the SEC, such registration statement is effective and the
Company has agreed to maintain the effectiveness of the registration
statement for at least 180 consecutive days beginning with the conversion
date.
|
The
Series A Preferred Stock accrues and accumulates dividends at a rate of 6% and
will participate in dividends declared and paid on the common stock, if
any. At December 31, 2009, cumulative unpaid preferred stock
dividends totaled $7,035,000 or $2.10 per share. All unpaid preferred
stock dividends must be paid before any dividends may be declared or paid on the
Common Stock, and will be added to the liquidation preference of the Series A
Preferred Stock payable upon the liquidation, dissolution or winding up of the
Company. The liquidation preference is equal to the greater
of:
|
|
(i)
|
Two
times the original per share purchase price plus accrued and unpaid
dividends or
|
|
|
(ii)
|
The
amount per share that would be payable to a holder of shares of the Series
A Preferred Stock had all of the shares been converted to common stock
immediately prior to a liquidation
event.
|
The
liquidation preference of the Series A Preferred Stock on a converted basis at
December 31, 2009 totaled approximately $33,500,000, excluding cumulative unpaid
preferred stock dividends as discussed above. This value is
calculated based on the contractual liquidation preference articulated in the
Series A Preferred Stock agreement. There can be no assurance what
impact the conversion of the Series A Preferred to common stock would have on
the trading value of the Company’s common stock.
Holders
of the Series A Preferred Stock generally vote together with the holders of
common stock, with each share of Series A Preferred Stock representing the
number of votes equal to that number of shares of common stock into which it is
then convertible.
In
December 2004, the Company completed a private placement of 5,490,198 shares of
its common stock and warrants to purchase a total of 1,098,040 shares of common
stock at an exercise price of $3.67, resulting in gross proceeds, prior to the
deduction of fees and commissions of approximately $14.0 million (net proceeds
of $13.3 million).
In March
2005, the Company issued 7,000,000 shares of its common stock pursuant to the
exercise of a warrant held by Celgene Corporation. The warrant,
exercisable at $1.50 per share was issued to Celgene as part of the 2002
transaction and resulted in gross proceeds, prior to the deduction of fees and
commissions of $10.5 million (net proceeds of $9.9 million).
In
January 2006, the Company acquired Miikana Therapeutics, a private biotechnology
company. Pursuant to the merger Agreement, the Company acquired all
of the outstanding capital stock of Miikana Thereapeutics, Inc. in exchange for
9.96 million shares of common stock and the assumption of certain
obligations.
In
February 2006, the Company completed a private placement of 12,972,966 shares of
its common stock and warrants to purchase a total of 6,486,484 shares of common
stock at an exercise price of $2.50, resulting in gross proceeds, prior to the
deduction of fees and commissions, of approximately $30 million (net proceeds of
approximately $27.9 million). The fair value of warrants issued was
$11,156,752, calculated using a Black-Scholes value of $1.72 with an expected
and contractual life of 5 years with no dividend yield. Volatility was assumed
to be 103.84%, and the risk free interest rate was 4.52%.
In
December 2006, the Company completed a registered direct offering of 10,727,500
shares of its common stock resulting in gross proceeds, prior to the deduction
of fees and commissions, of approximately $17 million (net proceeds of
approximately $15.9 million).
F-19
In
September 2007, as discussed in Note 3, the Company issued
250,000 warrants with an exercise price of $2.00 per share to the Lenders of the
Term Loan.
In
December 2007, the Company issued 675,000 shares of its common stock pursuant to
the exercise of certain warrants, resulting in net proceeds of approximately
$674,000.
In June
2008, the Company issued 2,564,104 shares of common stock as consideration for
the satisfaction of a purchase price adjustment milestone payment of $2,000,000
triggered by the dosing of the first patient in the ENMD-2076 clinical
trials. ENMD-2076 is the lead molecule in the Aurora Kinase Program,
which advanced into clinical development in 2008.
10. SHARE-BASED
COMPENSATION
The
Company has adopted incentive and nonqualified stock option plans for executive,
scientific and administrative personnel of the Company as well as outside
directors and consultants, of which 3,135,043 shares remain available for grant
under the Company’s 2001 Long-term Incentive Plan as of December 31,
2009. There are 6,984,606 shares issuable under options
previously granted under the plans and currently outstanding, with exercise
prices ranging from $0.16 to $58.38. Options granted under the plans
vest over periods varying from immediately to three years, are not transferable
and generally expire ten years from the date of grant.
The
Company recorded non-cash compensation charges of $73,000, $193,000 and $194,000
in 2009, 2008 and 2007, respectively, related to the issuance of restricted
stock to members of its Board of Directors. In 2008, each
non-employee director received an annual retainer fee of $25,000 that was
payable in restricted stock. Additionally, one Board member elected
to receive restricted stock in lieu of a cash retainer totaling
$20,000. No restricted stock was issued in 2009.
The Company’s net loss for the years
ended December 31, 2009, 2008 and 2007 includes $320,156, $1,089,517 and
$1,454,864, respectively, of compensation expense related to the Company’s
share-based compensation awards, including the charges noted above related to
restricted stock issued to members of its Board of Directors. The
compensation expense related to the Company’s share-based compensation
arrangements is recorded as components of general and administrative expense and
research and development expense, as follows:
|
2009
|
2008
|
2007
|
||||||||||
|
Research
and development
|
$ | 115,635 | $ | 233,201 | $ | 352,999 | ||||||
|
General
and administrative
|
204,521 | 856,316 | 1,101,865 | |||||||||
|
Share-based
compensation expense
|
$ | 320,156 | $ | 1,089,517 | $ | 454,864 | ||||||
|
Net
share-based compensation expense, per common share:
|
||||||||||||
|
Basic
and diluted
|
$ | 0.004 | $ | 0.013 | $ | 0.017 | ||||||
Stock Options. The
Company uses the Black-Scholes-Merton valuation model to estimate the fair value
of stock options granted to employees. Option valuation models, including
Black-Scholes-Merton, require the input of highly subjective assumptions, and
changes in the assumptions used can materially affect the grant date fair value
of an award. These assumptions include the risk free rate of
interest, expected dividend yield, expected volatility, and the expected life of
the award.
Expected
Volatility—Volatility is a measure of the amount by which a financial variable
such as a share price has fluctuated (historical volatility) or is expected to
fluctuate (expected volatility) during a period. The Company uses the historical
volatility based on the weekly price observations of its common stock during the
period immediately preceding the share-based award grant that is equal in length
to the award’s expected term (up to a maximum of five years). EntreMed believes
that historical volatility within the last five years represents the best
estimate of future long term volatility.
Risk-Free
Interest Rate—This is the average interest rate consistent with the yield
available on a U.S. Treasury note (with a term equal to the expected term of the
underlying grants) at the date the option was granted.
F-20
Expected
Term of Options—This is the period of time that the options granted are expected
to remain outstanding. EntreMed adopted a simplified method for estimating the
expected term of share-based awards granted during the year ended December 31,
2007.
Expected
Dividend Yield—EntreMed has never declared or paid dividends on its common stock
and does not anticipate paying any dividends in the foreseeable future. As such,
the dividend yield percentage is assumed to be zero.
Forfeiture
Rate—This is the estimated percentage of options granted that are expected to be
forfeited or cancelled on an annual basis before becoming fully vested. The
Company estimates the forfeiture rate based on historical forfeiture experience
for similar levels of employees to whom options were granted.
Following
are the weighted-average assumptions used in valuing the stock options granted
to employees during the years ended December 31, 2009, 2008 and
2007:
|
Years ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Expected
Volatility
|
78.02 | % | 72.84 | % | 94.46 | % | ||||||
|
Risk
free interest rate
|
2.08 | % | 4.69 | % | 4.50 | % | ||||||
|
Expected
term of option
|
5
years
|
5
years
|
5
years
|
|||||||||
|
Forfeiture
rate
|
*5.00 | % | **5.00 | % | 5.00 | % | ||||||
|
Expected
dividend yield
|
- | - | - | |||||||||
* - Throughout
2009, forfeitures were estimated at 5%; the actual forfeiture rate for 2009 was
8.26%.
**- Throughout
2008, forfeitures were estimated at 5%; the actual forfeiture rate for 2008 was
88.26% due to a 59% reduction in force effective 12/31/2008.
The
weighted average fair value of stock options granted was $0.10, $0.46 and $1.03
in 2009, 2008 and 2007, respectively.
Share-based
compensation expense recognized in the Consolidated Statement of Operations is
based on awards ultimately expected to vest, net of estimated
forfeitures. The authoritative guidance requires forfeitures to be
estimated at the time of grant and revised, if necessary, in subsequent periods
if actual forfeitures differ from those estimates.
A summary
of the Company's stock option plans and of changes in options outstanding under
the plans during the years ended December 31, is as follows:
|
Number of Options
|
Weighted Average
Exercise Price
|
Weighted Average
Remaining
Contractual Term
|
Aggregate Intrinsic
Value
|
|||||||||||||
|
In
years
|
||||||||||||||||
|
Outstanding
at December 31, 2006
|
8,111,086 | $ | 7.87 | |||||||||||||
|
Exercised
|
(75,000 | ) | $ | 1.09 | ||||||||||||
|
Granted
|
1,327,732 | $ | 1.39 | |||||||||||||
|
Expired
|
(628,784 | ) | $ | 10.18 | ||||||||||||
|
Forfeited
|
(63,726 | ) | $ | 1.73 | ||||||||||||
|
Outstanding
at December 31, 2007
|
8,671,308 | $ | 6.81 | |||||||||||||
|
Exercised
|
- | $ | - | |||||||||||||
|
Granted
|
626,664 | $ | 0.74 | |||||||||||||
|
Expired
|
(405,474 | ) | $ | 9.65 | ||||||||||||
|
Forfeited
|
(546,570 | ) | $ | 0.98 | ||||||||||||
|
Outstanding
at December 31, 2008
|
8,345,928 | $ | 6.60 | |||||||||||||
|
Exercised
|
86,250 | $ | 0.16 | |||||||||||||
|
Granted
|
1,313,500 | $ | 0.16 | |||||||||||||
|
Expired
|
(2,480,032 | ) | $ | 6.77 | ||||||||||||
|
Forfeited
|
(108,540 | ) | $ | 0.44 | ||||||||||||
|
Outstanding
at December 31, 2009
|
6,984,606 | $ | 5.50 | 4.37 | $ | 763,658 | ||||||||||
|
Vested
and expected to vest at December 31, 2009
|
6,947,536 | $ | 5.53 | 4.10 | $ | 744,434 | ||||||||||
|
Exercisable
at December 31, 2009
|
6,243,210 | $ | 6.12 | 3.82 | $ | 377,483 | ||||||||||
F-21
The
aggregate intrinsic value is calculated as the difference between (i) the
closing price of the common stock at December 31, 2009 and (ii) the weighted
average exercise price of the underlying awards, multiplied by the number of
options that had an exercise price less than the closing price on the last
trading day of 2009. The aggregate intrinsic value of options
exercised that had an exercise price less than the closing price on the last
trading day of the year, was $55,200 and $8,250 for the years ended December 31,
2009 and 2007, respectively. There were no options exercised in the
year ended December 31, 2008.
Cash
received from option exercises under all share-based payment arrangements for
the years ended December 31, 2009 and 2007 was $13,800 and $81,750,
respectively. Due to the availability of net operating loss
carryforwards and research tax credits, tax deductions for option exercises were
not recognized in the years ended December 31, 2009 and 2007. There
were no options exercised in the year ended December 31, 2008.
The
following summarizes information about stock options granted to employees and
directors outstanding at December 31, 2009:
|
Options Outstanding
|
Options Exercisable
|
|||||||||||||||||||
|
Range of
Exercise Prices
|
Number
Outstanding
at 12/31/09
|
Weighted
Average
Remaining
Contractual
Life in Years
|
Weighted
Average
Exercise
Price
|
Number
Exercisable
at 12/31/09
|
Weighted
Average
Exercise
Price
|
|||||||||||||||
|
$
|
0.00
- $1.50
|
2,653,823 |
6.3
|
$ | 0.68 | 1,912,731 | $ | 0.85 | ||||||||||||
|
$
|
1.51
- $3.00
|
1,537,320 |
5.3
|
$ | 1.90 | 1,537,016 | $ | 1.90 | ||||||||||||
|
$
|
3.01
- $4.50
|
569,976 |
4.2
|
$ | 3.63 | 569,976 | $ | 3.63 | ||||||||||||
|
$
|
4.51
- $6.00
|
211,280 |
2.8
|
$ | 5.00 | 211,280 | $ | 5.00 | ||||||||||||
|
$
|
6.01
- $10.00
|
841,817 |
1.8
|
$ | 8.71 | 841,817 | $ | 8.71 | ||||||||||||
|
$
|
10.01
- $15.00
|
370,402 |
1.6
|
$ | 12.30 | 370,402 | $ | 12.30 | ||||||||||||
|
$
|
15.01
- $25.00
|
377,575 |
1.4
|
$ | 17.12 | 377,575 | $ | 17.12 | ||||||||||||
|
$
|
25.01
- $35.00
|
400,290 |
0.3
|
$ | 27.62 | 400,290 | $ | 27.62 | ||||||||||||
|
$
|
35.01
- $50.00
|
3,611 |
0.6
|
$ | 44.82 | 3,611 | $ | 44.82 | ||||||||||||
|
$
|
50.01
- $65.00
|
18,512 |
0.3
|
$ | 53.20 | 18,512 | $ | 53.20 | ||||||||||||
| 6,984,606 |
4.4
|
$ | 5.50 | 6,243,210 | $ | 6.12 | ||||||||||||||
As of
December 31, 2009, there was approximately $88,000 of total unrecognized
compensation cost related to nonvested employee stock options. That cost is
expected to be recognized over a weighted-average period of 1.4
years.
Warrants. Warrants
granted generally expire after 5 years from the date of grant. Stock warrant
activity to non-employees is as follows:
|
Number of Shares
|
Weighted Average
Exercise Price
|
|||||||
|
Outstanding
at December 31, 2006
|
10,347,599 | $ | 2.96 | |||||
|
Granted
|
250,000 | $ | 1.13 | |||||
|
Exercised
|
(675,000 | ) | 1.00 | |||||
|
Expired
|
(123,336 | ) | $ | 11.61 | ||||
|
Outstanding
at December 31, 2007
|
9,799,263 | $ | 2.94 | |||||
|
Granted
|
- | $ | - | |||||
|
Exercised
|
- | $ | - | |||||
|
Expired
|
(1,938,626 | ) | $ | 4.07 | ||||
|
Outstanding
at December 31, 2008
|
7,860,637 | $ | 2.69 | |||||
|
Granted
|
- | $ | - | |||||
|
Exercised
|
- | $ | - | |||||
|
Expired
|
(1,123,153 | ) | $ | 3.96 | ||||
|
Outstanding
at December 31, 2009
|
6,737,484 | $ | 2.48 | |||||
|
Exercisable
at December 31, 2009
|
6,737,484 | $ | 2.48 | |||||
F-22
11.
COMMITMENTS AND CONTINGENCIES
Commitments
In
January 2006, the Company acquired Miikana Therapeutics, a private biotechnology
company. Pursuant to the Merger Agreement, the Company acquired all
of the outstanding capital stock of Miikana Therapeutics, Inc. in exchange for
9.96 million shares of common stock and the assumption of certain
obligations. In 2008, EntreMed initiated a Phase 1 clinical trial
with its Aurora A and angiogenic kinase inhibitor, ENMD-2076, in patients with
solid tumors. A dosing of the first patient with ENMD-2076 triggered
a purchase price adjustment milestone of $2 million, which the Company opted to
pay in stock. Under the terms of the merger agreement, the former
Miikana stockholders may earn up to an additional $16 million of potential
payments upon the satisfaction of additional clinical and regulatory
milestones. If ENMD-2076 successfully completes Phase 1 clinical
trials and advances to Phase 2, the dosing of the first patient will trigger an
additional purchase price adjustment milestone of $3 million, which could occur
and be paid (in cash or stock at our sole discretion) in 2010. Through the acquisition,
the Company acquired rights to MKC-1, a Phase 2 clinical candidate licensed from
Hoffman-LaRoche, Inc. (“Roche”) by Miikana in April 2005. Under the
terms of the agreement, Roche may be entitled to receive future payments upon
successful completion of Phase 3 developmental milestones. The
Company does not anticipate reaching any of these milestones in
2010. Roche is also eligible to receive royalties on sales and
certain one-time payments based on attainment of annual sales milestones. The
Company is also obligated to make certain “success fee” payments to ProPharma
based on successful completion of developmental milestones under the Roche
license agreement.
In
January 2006, the Company entered into a License Agreement with Elan
Corporation, plc (“Elan”) in which the Company has been granted rights to
utilize Elan’s proprietary NanoCrystal Technology in connection with the
development of the oncology product candidate, Panzem® NCD.
Under the terms of the License Agreement, Elan is eligible to receive payments
upon the achievement of certain clinical, manufacturing, and regulatory
milestones and to receive royalty payments based on sales of Panzem®
NCD. Additionally, under the agreement and the corresponding Services
Agreement, Elan has the right to manufacture EntreMed’s Panzem®
NCD. Milestones related to the initiation of Phase 2 clinical trials
for Panzem® NCD have
been paid and there are no additional milestones achieved as of December 31,
2009. The Company has discontinued clinical development of Panzem® NCD for
oncology.
In
March 2005, the Company entered into an exclusive worldwide license
agreement with Celgene Corporation for the development and commercialization of
Celgene’s small molecule tubulin inhibitor compounds for the treatment of
cancer. Under the terms of the agreement, Celgene received an upfront licensing
fee of $1,000,000 and may receive additional payments up to approximately $25.25
million based upon the attainment of certain milestones. No such
milestones have been reached through December 31, 2009. Our
preliminary work on the program was completed in 2008 and we did not devote
resources to this program in 2009.
The
Company entered into two license agreements with Children's Hospital, Boston for
the exclusive, worldwide, royalty-bearing licenses to make, use and sell
Endostatin and 2-methoxyestradiol (“2ME2”), both inhibitors of angiogenesis. In
February 2004, the Company transferred rights to Endostatin in an agreement with
Children’s Medical Center Corporation (“CMCC”) and Alchemgen Therapeutics. In
April 2008, the Company was advised that Alchemgen Therapeutics ceased
operations, therefore eliminating the ability to receive any royalties from
Alchemgen under the agreement. The Company is eligible to receive
royalties from Oxford Biomedica, PLC based on a portion of the net sales of
products developed for the treatment of ophthalmic (eye)
diseases. The Company received its first royalty payment in the
amount of $368,000 from Oxford Biomedica, PLC under this agreement in 2009, and
has accrued a sub-royalty payment of $74,000 at December 31, 2009 payable to
CMCC. In consideration for retaining the 2ME2 rights, the Company
must pay a royalty on any sublicensing fees, as defined in the agreement, to
Children's Hospital, Boston. The agreement obligates the Company to pay up to
$1,000,000 "upon the attainment of certain milestones." As of December 31, 2009,
the Company has paid $500,000 under this agreement for the milestones that have
been achieved to date. The Company has discontinued research and
development of 2ME2.
F-23
Pursuant
to the commitments detailed above, in aggregate, the Company could potentially
pay up to $75.5 million if each licensed technology is fully developed and
approved for commercial use in all of the major territories of the
world. In this event, the Company would also be obligated to pay
annual sales-based royalties under the license agreements. However,
the Company cannot forecast with any degree of certainty whether any of the
other product candidates will reach additional developmental
milestones. As such, the timing of any future payments, if any,
cannot be determined. All of the milestone payments under these
agreements are contingent upon successful development and ultimate advancement
to commercialization, thus, there can be no assurances that all or any of the
triggering events will occur. The Company has discontinued clinical
development of 2ME2 (Panzem® NCD) for
oncology.
As of
December 31, 2009, the Company also has purchase obligation commitments, in the
normal course of business, for clinical trial contracts totaling $2,698,000 and
for contract manufacturing totaling $505,000.
The
Company leases its primary corporate facilities under a lease agreement that
continues through February 2010. As a result of the Company’s prioritization of
ENMD-2076 as the leading program and subsequent realignment of personnel
consistent with the strategy, in January 2009, the Company amended its lease in
order to reduce our occupied space to 8,554 square feet which provided
significant savings in rent expense to the Company in 2009. Rent
expense is recognized under the straight-line method. Additionally,
the Company leases office equipment under operating leases.
The
future minimum payments under its facilities and equipment leases as of December
31, 2009 have been significantly reduced, reflecting the impact of the Company’s
restructuring. The future minimum payments are as
follows:
|
2010
|
74,316 | |||
|
Thereafter
|
- | |||
|
Total
minimum payments
|
$ | 74,316 |
Rental
expense for the years ended December 31, 2009, 2008 and 2007 was $507,000,
$980,000, and $955,000, respectively.
Contingencies
EntreMed
is subject in the normal course of business to various legal proceedings in
which claims for monetary or other damages may be
asserted. Management does not believe such legal proceedings, unless
otherwise disclosed herein, are material.
12. EMPLOYEE
RETIREMENT PLAN
The
Company sponsors the EntreMed, Inc. 401(k) and Trust. The plan covers
substantially all employees and enables participants to contribute a portion of
salary and wages on a tax-deferred basis. Contributions to the plan by the
Company are discretionary. Contributions by the Company totaled approximately
$57,000, $117,000 and $103,000 in 2009, 2008 and 2007,
respectively.
13. SUBSEQUENT
EVENTS
Effective
March 1, 2010, the Company amended its lease for its principal executive offices
in Rockville, MD in order to reduce the occupied space to 4,367 square feet
further providing significant savings in rent expense to the Company in
2010. As a result of the amendment, the Company reduced its monthly
rent for the Rockville facility by 50%.
On
February 3, 2010, the Company consummated the issuance and sale of 3,846,154
shares of its common stock, par value $0.01 per share, to an institutional
investor for an aggregate purchase price of $2,500,000, or $0.65 per
share. The offering was made pursuant to a stock purchase agreement
dated as of February 3, 2010 between the Company and the
investor.
F-24
On
January 19, 2010, the Company received a NASDAQ Staff Determination letter
indicating that the Company failed to comply with NASDAQ Listing Rule 5450(a)(1)
for continued listing on The NASDAQ Capital Market. Rule 5450(a)(1)
requires that a listed company maintain a minimum closing bid price of at least
$1.00 per share. The Company had initially been notified by NASDAQ of
such minimum bid price deficiency on April 4, 2008. As a result of
suspensions by NASDAQ of enforcement of the bid price due to extraordinary
market conditions, the Company had until January 15, 2010 to regain
compliance. As the Company has not regained compliance with such
NASDAQ rule, the NASDAQ Staff has determined to delist its securities
from the NASDAQ Capital Market on January 28, 2010, unless the Company requests
an appeal of the determination. On January 20, 2010, the Company
filed an appeal of the Staff's determination to a NASDAQ Hearings Panel (the
“Panel”),
pursuant to the procedures set forth in the NASDAQ Marketplace Rules. The
hearing request stayed the delisting of the Company’s securities pending the
Panel's decision. On February 25, 2010, the Company met with the
Panel providing them a plan of action, with the intention of returning to
compliance with NASDAQ’s requirements. At its discretion, the Panel
has the authority to grant the Company up to an additional 180 days from the
date of the Staff Determination letter to execute its plan of
compliance. On March 23, 2010, the Company received a decision
letter from the Panel granting its request to extend the compliance date for an
additional 180 days or until July16, 2010.
On January 11, 2010, the Company
consummated the issuance and sale of 3,125,000 shares of its common stock, par
value $0.01 per share, to an institutional investor for an aggregate purchase
price of $2,500,000 or $0.80 per share. The offering was made
pursuant to a stock purchase agreement dated as of January 8, 2010 between the
Company and the investor.
F-25
14. QUARTERLY
FINANCIAL INFORMATION (UNAUDITED)
Summarized
unaudited quarterly financial information for the years ended December 31, 2009
and 2008 is as follows:
|
|
QUARTER ENDED
|
|||||||||||||||
|
MARCH 31,
|
JUNE 30,
|
SEPTEMBER 30,
|
DECEMBER 31,
|
|||||||||||||
|
2009
|
||||||||||||||||
|
Revenues
|
$ | - | $ | - | $ | 3,668,333 | $ | 1,615,725 | ||||||||
|
Research
and development costs
|
1,953,460 | 1,659,474 | 2,326,969 | 1,961,619 | ||||||||||||
|
General
and administrative expenses
|
1,159,721 | 1,010,733 | 911,816 | 1,022,017 | ||||||||||||
| 3,113,181 | 2,670,207 | 3,238,785 | 2,983,636 | |||||||||||||
|
Investment
income
|
50,187 | 18,744 | - | 771 | ||||||||||||
|
Interest
expense
|
(453,761 | ) | (399,730 | ) | (344,634 | ) | (295,275 | ) | ||||||||
|
Other
expense
|
- | - | - | (70,929 | ) | |||||||||||
|
Net
income (loss)
|
(3,516,755 | ) | (3,051,193 | ) | 84,914 | (1,733,344 | ) | |||||||||
|
Dividends
on Series A convertible preferred stock
|
(251,250 | ) | (251,250 | ) | (251,250 | ) | (251,250 | ) | ||||||||
|
Net
loss attributable to common Shareholders
|
(3,768,005 | ) | (3,302,443 | ) | (166,336 | ) | (1,984,594 | ) | ||||||||
|
Net
loss per share (basic and diluted)
|
$ | (0.04 | ) | $ | (0.04 | ) | $ | (0.00 | ) | $ | (0.03 | ) | ||||
|
2008
|
||||||||||||||||
|
Revenues
|
$ | - | $ | - | $ | 3,501,307 | $ | 3,975,912 | ||||||||
|
Research
and development costs
|
6,187,203 | 5,484,857 | 4,957,067 | 3,440,102 | ||||||||||||
|
General
and administrative expenses
|
1,982,994 | 1,739,691 | 1,551,900 | 2,489,947 | ||||||||||||
|
In-process
R&D
|
- | 2,000,000 | - | - | ||||||||||||
| 8,170,197 | 9,224,548 | 6,508,967 | 5,930,049 | |||||||||||||
|
Fixed
asset impairment loss
|
- | - | - | (171,576 | ) | |||||||||||
|
Investment
income
|
398,787 | 229,451 | 168,444 | 85,571 | ||||||||||||
|
Interest
expense
|
(575,046 | ) | (575,046 | ) | (558,661 | ) | (507,410 | ) | ||||||||
|
Net
loss
|
(8,346,456 | ) | (9,570,143 | ) | (3,397,877 | ) | (2,547,552 | ) | ||||||||
|
Dividends
on Series A convertible
|
||||||||||||||||
|
preferred
stock
|
(251,250 | ) | (251,250 | ) | (251,250 | ) | (251,250 | ) | ||||||||
|
Net
loss attributable to common
|
||||||||||||||||
|
Shareholders
|
(8,597,706 | ) | (9,821,393 | ) | (3,649,127 | ) | (2,798,802 | ) | ||||||||
|
Net
loss per share (basic and diluted)
|
$ | (0.10 | ) | $ | ( 0.11 | ) | $ | (0.04 | ) | $ | (0.04 | ) | ||||
F-26