Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 001-33498
BIONOVO, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 20-5526892 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 5858 Horton Street, Suite 400 Emeryville, California 94608 |
(510) 601-2000 | |
| (Address of principal executive offices, including zip code) |
(Telephone Number) | |
Indicate by check mark whether the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act). (Check one):
| Large accelerated filer ¨ |
Accelerated filer ¨ | Non-accelerated filer þ | Smaller reporting company ¨ | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act of 1934). Yes ¨ No þ
The aggregate market value of the voting stock held by non-affiliates of the Registrant based upon the last trade price of the common stock reported on The NASDAQ Capital Market on June 30, 2009 was approximately $32.7 million.*
As of March 11, 2010, 107,518,690 shares of the registrant’s common stock, par value $0.0001, were outstanding.
Table of Contents
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s Proxy Statement for the 2010 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K. Such Proxy Statement will be filed within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
| * | Excludes 19,249,188 shares of Common Stock held by directors, officers and stockholders whose beneficial ownership exceeds 5% of the Registrant’s Common Stock outstanding. The number of shares owned by stockholders whose beneficial ownership exceeds 5% was determined based upon information supplied by such persons and upon Schedules 13D and 13G, if any, filed with the SEC. Exclusion of shares held by any person should not be construed to indicate that such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the Registrant, that such person is controlled by or under common control with the Registrant, or that such persons are affiliates for any other purpose. |
Table of Contents
| PART I | ||||
| Item 1. |
Business | 1 | ||
| Item 1A. |
Risk Factors | 15 | ||
| Item 1B. |
Unresolved Staff Comments | 29 | ||
| Item 2. |
Properties | 29 | ||
| Item 3. |
Legal Proceedings | 29 | ||
| Item 4. |
Reserved | 29 | ||
| PART II | ||||
| Item 5. |
30 | |||
| Item 6. |
32 | |||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
33 | ||
| Item 7A. |
43 | |||
| Item 8. |
44 | |||
| Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
70 | ||
| Item 9A. |
70 | |||
| Item 9B. |
70 | |||
| PART III | ||||
| Item 10. |
71 | |||
| Item 11. |
71 | |||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Shareholder Matters |
71 | ||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
71 | ||
| Item 14. |
71 | |||
| PART IV | ||||
| Item 15. |
72 | |||
| 72 | ||||
| 72 | ||||
| 75 | ||||
i
Table of Contents
PART I
Forward Looking Statements
Statements made in this document other than statements of historical fact, including statements about us and the future of our respective clinical trials, research programs, product pipelines, current and potential corporate partnerships, licenses and intellectual property, the adequacy of capital reserves and anticipated operating results and cash expenditures, are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. When used in this Annual Report the words “anticipate,” “objective,” “may,” “might,” “should,” “could,” “can,” “intend,” “expect,” “believe,” “estimate,” “predict,” “potential,” “plan” or the negative of these and similar expressions identify forward-looking statements. These statements reflect our current views with respect to uncertain future events and are based on imprecise estimates and assumptions and subject to risk and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. While we believe our plans, intentions and expectations reflected in those forward-looking statements are reasonable, these plans, intentions or expectations may not be achieved. Our actual results, performance or achievements could differ materially from those contemplated, expressed or implied by the forward-looking statements contained in this Annual Report for a variety of reasons, including those set forth in Item 1A “Risk Factors” and elsewhere in this Annual Report.
All forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the risk factors and other cautionary statements set forth in this Annual Report. Other than as required by applicable securities laws, we are under no obligation, and we do not intend, to update any forward-looking statement, whether as result of new information, future events or otherwise.
The following should be read in conjunction with our consolidated financial statements located elsewhere in this Annual Report on Form 10-K for the year ended December 31, 2009 and other documents filed by us from time to time with the Securities and Exchange Commission.
| Item 1. | Business |
Overview
We are a clinical stage drug discovery and development company focusing on women’s health and cancer, two large markets with significant unmet needs. Building on our understanding of the biology of menopause and cancer, we design new drugs derived from botanical sources which have novel mechanisms of action. Based on the results of our clinical trials and preclinical studies to date, we believe that we have discovered new classes of drug candidates with the potential to be leaders in their markets.
Our lead drug candidate, Menerba (formerly MF-101), represents a new class of receptor sub-type selective estrogen receptor modulator (SERM), for the treatment of vasomotor symptoms of menopause, or “hot flashes.” We have designed Menerba to selectively modulate estrogen receptor beta (ERß) and to provide a safe and effective alternative to existing FDA approved therapies that pose a significant risk to women for developing breast cancer, stroke, cardiovascular disease, blood clots and other serious diseases. In preclinical studies, Menerba does not lead to tumor formation in either breast or uterine tissues and it does not increase the risk for clotting. This activity, if confirmed in clinical testing, would differentiate Menerba from some existing therapies and other therapies in clinical development. We announced results of our completed, multicenter, randomized, double-blinded, placebo-controlled Phase 1 trial, involving 217 women, which showed the higher of two Menerba doses tested was well tolerated and resulted in a statistically significant reduction of hot flashes after 12 weeks of treatment. In addition, treatment with the higher of the two doses of Menerba lead to a statistically significant reduction in nighttime awakenings when compared to placebo, which represents superior efficacy to existing non-hormonal therapies. We are seeking FDA approval to conduct further clinical testing at multiple clinical sites in the U.S. We believe that Menerba’s novel mechanism of action could lead to a more favorable safety profile than currently approved hormone therapies (HT) and other therapies under development and testing.
1
Table of Contents
We are also developing Bezielle, an oral anticancer agent for advanced breast cancer. Unlike most other anticancer drugs and drug candidates, which try to control cancer through genomic and proteomic signaling pathways, Bezielle is designed to take advantage of a unique metabolism of cancer cells, with a well-characterized mechanism of action which leads to very selective tumor cell DNA damage and the death of cancer cells, without lasting harm to normal cells. This is accomplished by Bezielle’s secondary inhibition of glycolysis, a metabolic pathway on which cancer cells rely for energy production. Glycolysis inhibition leads to energy collapse in the cells and results in death of cancer cells. Since normal cells do not rely for the most part on glycolysis for energy production they are not killed by Bezielle. Based on our clinical and pre-clinical studies we believe that Bezielle may have a preferential effect on hormone-independent cancers, a subset with few treatment options. To date, 48 women with metastatic breast cancer have been treated with Bezielle in two separate Phase 1 clinical trials. As predicted by the mechanism of action, Bezielle had very limited toxicities with an extremely favorable tolerability profile. Moreover, Bezielle showed encouraging clinical activity among a cohort of women with metastatic breast cancer who had been heavily pretreated with other regimens. A Phase 2 trial has been approved by the FDA and the institutional review boards (“IRBs”) at several prestigious breast cancer clinical sites in the U.S. Bionovo is awaiting funding to commence this open-label, non-randomized trial in 80 women diagnosed with metastatic breast cancer who have failed no more than two prior chemotherapy regimens. We believe that Bezielle’s novel mechanism of action and favorable tolerability profile could lead to preferential use over existing drugs in the treatment of advanced breast cancer, and potentially in the broader, adjuvant care of breast cancer. We also plan to evaluate Bezielle for the treatment of other forms of cancer, including pancreatic cancer and adjuvant use in breast cancer.
We have a diverse pipeline of additional preclinical drug candidates in both women’s health and cancer. We are prepared to proceed with an investigational new drug, or IND, application and a Phase 1/2 trial for our second SERM drug candidate, Seala, for the treatment of postmenopausal vulvar and vaginal atrophy, or “vaginal dryness.” Further, we have identified or begun preclinical work on other drug candidates for a variety of indications within women’s health and cancer.
We have internally discovered and developed all of our drug candidates using our proprietary biological and chemical techniques. Our drug development process targets herbs and other botanical sources, used in Traditional Chinese Medicine, and believed to produce biologically active compounds. We apply our clinical knowledge, experience with natural compounds and knowledge of proper scientific screening tools to isolate and purify botanical compounds and extracts for pharmaceutical development. There are approximately 13,000 species used historically in Chinese medicine. To date, only few were exploited for pharmaceutical development. There are numerous examples of drugs that were derived from plants such as paclitaxel, digitalis and aspirin, and a few from Chinese medicine such as ephedrine and camptothecin. In June 2004, the FDA released a document to provide drug developers with guidance on the approval for botanical drugs. This guidance states that applicants may submit reduced documentation of nonclinical (preclinical) safety to support an IND application for initial clinical studies of botanicals. The first botanical extract drug developed pursuant to these guidelines was approved by the FDA in October 2006. To date, all of our drug candidates are derived from botanical extracts and are being developed in accordance with this FDA guidance. In addition, we have identified the active chemical components underpinning the mechanism of action for our novel drugs, and in some cases, we have developed synthetic methods of production.
We expect to continue to incur significant operating losses over at least the next several years, and do not expect to generate profits until and unless our drug candidates have been approved and are being marketed with commercial partners.
2
Table of Contents
Formation of the Company
The Company’s history began in February 2002 as a private company operating in California. Now Bionovo, Inc. is incorporated in the state of Delaware and was made public through a reverse merger transaction between Bionovo Biopharmaceuticals, Inc.1 and Lighten Up Enterprises International, Inc.2 on April 6, 2005. Immediately following the reverse merger, the Company operated as Lighten Up Enterprises International, Inc. until June 29, 2005 when the Company assumed the name, Bionovo, Inc.
Reverse Merger
On April 6, 2005, Lighten Up Enterprises International, Inc. acquired all the outstanding shares of Bionovo Biopharmaceuticals, Inc. in exchange for 42,112,448 restricted shares of its common stock in a reverse triangular merger. The acquisition was accounted for as a reverse merger in which Bionovo Biopharmaceuticals, Inc. was deemed to be the accounting acquirer. Accordingly, the historical financial information presented herein is that of Bionovo Biopharmaceuticals, Inc. (as adjusted for any difference in the par value of each entity’s stock with an offset to capital in excess of par value). Financial information subsequent to the reverse merger is that of the merged entity, Bionovo, Inc.
Development Stage Company
We have not generated any significant revenue since inception. Accordingly, the accompanying financial statements have been prepared using the accounting formats prescribed by the development stage entities Topic of the FASB Accounting Standards Codification. Although the Company has recognized some nominal amount of revenue, we still believe that we are devoting, substantially, all our efforts toward developing the business and, therefore, still qualify as a Development Stage Enterprise (DSE).
We are a DSE and are primarily engaged in the development of pharmaceuticals, derived from botanical sources, to treat cancer and women’s health. The initial focus of our research and development efforts will be the generation of products for the treatment of breast, and other cancers and to alleviate the symptoms of menopause. The production and marketing of our products and our ongoing research and development activities are and will continue to be subject to extensive regulation by numerous governmental authorities in the United States. Prior to marketing in the United States, any drug we develop must undergo rigorous preclinical and clinical testing and an extensive regulatory approval process implemented by the Food and Drug Administration (FDA) under the Food, Drug and Cosmetic Act. We have limited experience in conducting and managing the preclinical and clinical testing necessary to obtain regulatory approval. There can be no assurance that we will not encounter problems in clinical trials that will cause us or the FDA to delay or suspend clinical trials.
Our success will depend in part on our ability to obtain patents and product license rights, maintain trade secrets, and operate without infringing on the proprietary rights of others, both in the United States and other countries. There can be no assurance that patents issued to us will not be challenged, invalidated, or circumvented, or that the rights granted thereunder will provide proprietary protection or competitive advantages to us.
| 1 | Bionovo Biopharmaceuticals, Inc. was originally formed as “Bionovo, Inc.” and began operations in the state of California in February 2002. In March of 2004, it reincorporated in the state of Delaware and in June of 2005 changed its name from Bionovo, Inc. to Bionovo Pharmaceuticals, Inc. |
| 2 | Lighten Up Enterprises International, Inc. was incorporated in Nevada in January of 1998. |
3
Table of Contents
Business Strategy
Our goal is to achieve a position of sustainable leadership in the biopharmaceutical industry. Our strategy consists of the following key elements:
Integrate Scientific Discoveries with Natural Substances Used in Traditional Chinese Medicine
For more than 2,000 years, Chinese people have used Traditional Chinese Medicine (TCM), for the prevention and treatment of disease. Many significant drugs and therapies have been derived from botanical sources, such as aspirin and paclitaxel, and some from botanicals used in TCM, such as ephedrine. Advances in science and technology and analytical methodology for natural products can be harnessed for the discovery and development of new drugs from the botanicals used in TCM. We intend to continue to integrate cutting edge scientific discoveries and modern medicine with our expertise in natural substances used in TCM to discover and screen novel formulations derived from botanicals.
Focus on Cancer and Women’s Health
We have intentionally directed our focus on initial medical applications with urgent needs and very large potential markets. With this strategy, even a small market penetration should result in relatively substantial revenue streams. Under this strategy, we have initially directed our attention to the design of drugs to target cancer and women’s health.
According to the American Cancer Society (ACS) 2005 Cancer Facts and Figures, cancer is a leading cause of death in the U.S., yet there remain unmet needs, and current treatments remain ineffective and inadequate for some populations. We believe our addressable market for advanced breast cancer is approximately 500,000—600,000 patients per year. In the current health care regulatory environment, the FDA regulatory requirements for approval of cancer drugs has been modified because of the urgent need for safe and effective treatments of this disease.
There are approximately 27 million women suffering from menopausal symptoms such as hot flashes and vaginal dryness. To date, pharmaceutical interventions offered for women suffering from menopausal symptoms are often deemed unsatisfactory, or they have significant undesirable side effects. Relying in part on what we believe to be our novel system for the assessment of selective estrogen receptors ((a) and (ß)) modulators, or SERMs, their downstream co-regulatory proteins and their transcriptional outcome as well as cytotoxic agents, we intend to continue to target this significant market opportunity for drugs targeting cancer and women’s health.
Develop Our Existing Product Portfolio
We have one drug, Menerba, designed to alleviate the symptoms of menopause, which has completed a Phase 2 clinical trial. We have completed two Phase 1 clinical trials of a second drug, Bezielle, an oral anticancer agent for metastatic breast cancer and we have FDA approval for a Phase 2 clinical trial to further evaluate Bezielle for metastatic breast cancer. We are evaluating the potential of Bezielle in other indications, including the treatment of pancreatic cancer. We also are in the process of preparing a Phase 1 trial for another drug, Seala, an intra-vaginal drug (VG101) for the treatment of postmenopausal vulvar and vaginal atrophy (“vaginal dryness”). We also continued the development of two additional anticancer agents, BN107 and BN108. These drug candidates have unique mechanisms of action which may have a potential safety and efficacy advantage over existing drugs. We intend to further develop these drugs both by expanding our internal resources and by collaborating with leading governmental and educational institutions as well as other companies.
Foster Academic and Industry Collaborations
We have developed research and development relationships with various faculty members at the University of California at San Francisco (UCSF), the University of California at Berkeley (UCB), the University of
4
Table of Contents
California at Davis (UCD), University of Southern California (USC), University of Texas MD Anderson Cancer Center at Houston, Case Western Reserve University, the University of Colorado Health Sciences College (UCHSC), Houston University, Baylor University, and the Methodist Hospital Research Institute in Houston, Texas. We also have an active Scientific Advisory Board (SAB) and Medical Advisory Board (MAB). These collaborations provide access to leading intellectual and physical resources, and we believe will augment funding for academic development and accelerate technology transfer of promising innovations. We intend to continue our collaborations in order to leverage the intellectual resources of other major research centers by seeking additional academic collaborations.
We will also seek strategic scientific collaborations with other biotechnology and pharmaceutical companies in order to expand and accelerate the process to product development. We believe this will enhance our research and development capabilities as well as provide potential sales channels for our products. We plan to target specific biotech and pharmaceutical companies in need of our proprietary technology or potential products in the endeavor to reach licensing and development agreements.
Diversify Application of Drug Candidates for Extended Indications
Many of our initial products under development are not specific for women’s health, although the initial clinical trials will focus on this application. Anticancer therapeutics, for example, will apply to a wide range of oncological applications. Similarly, hormonally-active drugs could potentially be used to treat or prevent some forms of cancer or osteoporosis. Accordingly, we intend to pursue alternative applications for our drug candidates when deemed appropriate, to increase our chances of commercial success.
Scientific Discovery Platforms
Screening Philosophy
A useful strategy for the discovery of biologically active compounds from plants utilizes information about the traditional medicinal use of these botanical agents. An advantage to this strategy over random screening is that the extensive clinical tradition and literature may allow for some rationalization with respect to the biological potential for their reputed use. Since most organisms living today evolved under similar adaptation pressures, it is plausible that plants can interact with mammalian organic processes, along similar lines as nutrition from food, and therefore, can be utilized to regulate pathological conditions as they do normal physiological functioning.
As an example, experimental antineoplastic (inhibiting or preventing the growth or development of malignant cells) agents derived from botanicals have been under study in China since the mid-fifties. Agents discovered through this effort include: campothecin (“CPT”) and hydroxycampothecin (“OPT”) from CAMPOTHECA ACUMINATA Decne, Harringtonine and homoheringtonine from several species of CEPHALOTAXUS, Indirubin from BAPHICACANTHES CUSIA Bremek and INDIGOFERA TINCTORIA L. and Kanglaite from COIX LACHRYMA-JOBI L.
Our strategic advantage for drug development is our extensive clinical knowledge and experience with natural compounds coupled with definitive knowledge of the proper scientific tools for screening. Instead of creating massive screens, we select the most likely candidate compounds and test them for efficacy and toxicity with state of the art screening models such as estrogen receptor regulation or induction of cancer cell death. To date, the success rate from our screens has been approximately 25% in the cytotoxic drug discovery and approximately 40% in our estrogen drug discovery efforts.
Our clinical knowledge also accelerates the preclinical testing due to the longstanding anecdotal knowledge regarding the toxicities of these agents. The shorter time to clinic provides an opportunity to exercise major savings and prolong the exploitation of the patent.
5
Table of Contents
This approach provides a new rational paradigm for drug discovery, whereby the prior knowledge of the indication coupled with the molecular mechanism prevents much of the guess work experienced by other linear approaches where the relationship between the molecular target and the indication is not always known.
Scientific Discovery Platform I: Anticancer Drugs
A number of Chinese medicinal herbs have traditionally been used to prevent and treat cancer. These herbal preparations are purported to have many biological effects including direct anti-proliferative effects on cancer cells, anti-mutagenic activity, and other signaling pathways such as nuclear receptor regulation resulting in prevention of growth or death.
Aqueous and organic extracts from 71 Chinese medicinal herbs historically used for the treatment of illness were evaluated for antiproliferative activity on five breast cancer cell lines. Approximately 26% (19/71) of the extracts demonstrated greater than 50% growth inhibition on 80% of the cancer cell lines tested while five more herbs showed the same activity on fewer cell lines. These results, as well as our data from dose-response curves, DNA fragmentation and flow cytometric analyses, indicate that many of the herbs have significant growth inhibitory effects on various cancer cells in vitro. Furthermore, in vivo tests of some of the extracts in a mouse xenograph model show a significant inhibition of tumor formation with oral administration, with no toxicity or compromise to the mice activity including fluid and food intake.
Scientific Discovery Platform II: Hormone Replacement Therapy and Selective Estrogen Receptor Modulators
Over 300 plants synthesize compounds that interact with estrogen receptors (ER), known as phytoestrogens. Elucidating how phytoestrogens regulate ER transcriptional and cell proliferation pathways could have a profound impact on women. For example, phytoestrogens may prevent some cancers that are common in postmenopausal women. In fact, the lowest rates of breast, endometrial and colon cancers are observed in countries that have a high consumption of phytoestrogens in their diet.
Our goal is to identify compounds effective at preventing or treating breast cancer as well as potential compounds for desired hormone activity. We have tested 71 herbs that are used in TCM for their ability to regulate transcriptional activity in the presence of ERa or ERß. Over forty five percent (46.4%) of the herbs showed selective activity on the two ERs. In these studies, we identified the herbs that selectively regulate ERa or ERß and result in different transcriptional activity as well as in tissue selectivity.
These studies have the potential to identify natural selective SERMs, such as the drug tamoxifen, which may be used, with FDA approval, to prevent and treat breast cancer. In addition, we anticipate these studies will provide leads that do not increase the risk of breast cancer. Other indications for estrogenic compounds or SERMs are osteoporosis, obesity, autoimmune disorders, cardiovascular disease prevention, arthritis and menopausal symptomatic management (such as hot flashes, insomnia, vaginal dryness and decreased libido).
Pharmacology
After assessing the functional activity of whole herb extracts in our established assay systems, we aim to isolate anticancer and estrogenic compounds from herbal extracts to identify their chemical structure and to evaluate their pharmacodynamic and pharmacokinetic properties. The following studies are or will be conducted for all extracts in order to comply with FDA regulatory demands:
| • | Activity Guided Isolation. These studies will be done in order to discover the active components. Once determined, the chemical structure can then be used as a marker for quantification in our manufacturing process, or as a basis for developing a synthetic equivalent. |
| • | Evaluation of the Pharmacokinetic and Metabolism of Isolated Compounds. Since the drugs are designed to have greater selectivity and less toxicity, these studies will allow us to further determine their dynamic range, application and safety. |
6
Table of Contents
| • | Botanical Drug Consistency Measures. We have developed methods for simultaneous intra batch and inter batch consistency measures using state of the art analytical technology. |
| • | Biological Measures. Specific biochemical assays will be employed to measure biological specificity and effect as well as manufacturing consistency similar to other biological drugs. |
We will also repeat all quantifiable biological measures, pertaining to the drug, available through our proposed drug platforms. This will ensure qualitative efficacy control of proposed drugs. By identifying compounds both biologically as well as pharmacologically, we believe we will be able to overcome any FDA hurdles regarding drug manufacturing consistency.
Clinical Trials Design
Many companies with good science suffer from a lack of sufficient clinical knowledge, poor clinical trials design expertise or limited access to reputable clinical facilities to conduct their early trials. Since our approach to drug design relies heavily on clinical experience and expertise, in our respective fields, we emphasize sound clinical trial design as one of our strengths.
All of our drug trials follow traditional methods for assessment as well as auxiliary clinical and objective measures in order to strengthen our primary and secondary claims.
Scientific Consultants
We use consultants to provide us with expert advice and consultation on our scientific programs and strategies. They also serve as contacts for us throughout the broader scientific community. We have consulting agreements with a number of academic scientists and clinicians, who collectively serve as our SAB and MAB. These individuals serve as key consultants with respect to our product development programs and strategies. They possess expertise in numerous scientific fields, including chemistry, pharmacology, cancer, estrogen receptor biology and clinical drug testing.
We retain each consultant according to the terms of a consulting agreement. Under such agreements, we pay them a consulting fee. In addition, some consultants hold options to purchase our common stock, subject to the vesting requirements contained in the consulting agreements. Our consultants are employed by institutions other than ours, and therefore may have commitments to, or consulting or advisory agreements with other entities or academic institutions that may limit their availability to us.
Research and Development
Our pre-clinical research is conducted at our main office in Emeryville, with some research work also conducted at UCSF, UCB, UCD, Baylor University, University of Houston and UCHSC. During the fiscal years ended December 31, 2009, 2008 and 2007, we incurred research and development expenses of $12.5 million, $11.4 million and $9.9 million, respectively. We further expect that research and development expenses will increase as and when we continue development of our drugs.
Intellectual Property and Patent Protection
Patent protection is important to our business. The patent position of companies in the pharmaceutical field generally is highly uncertain, involves complex legal and factual questions, and has recently been the subject of much litigation. Therefore, we cannot be certain that any patent applications relating to our products or processes will result in patents being issued, or that the resulting patents, if any, will provide protection against competitors who successfully challenge our patents, obtain patents that may have an adverse effect on our ability to conduct business, or are able to circumvent our patent position. It is possible that other parties have conducted or are conducting research and could make discoveries of compounds or processes that would precede any of our
7
Table of Contents
discoveries. Finally, there can be no assurance that others will not independently develop similar pharmaceutical products which will compete against ours, or cause our drug product candidates and compounds to become obsolete. At present, we have 74 patent applications pending in the United States Patent and Trademark Office, as well as in the Patent Cooperation Treaty, the European Patent Office, Japan, and other markets. One patent (US 7482029 B1) for Menerba, entitled “Composition for Treatment of Menopause”, was issued on March 29, 2006 and expires in August of 2026.
Our competitive position is also dependent upon unpatented trade secrets. We have a policy of requiring our employees, consultants and advisors to execute proprietary information and invention assignment agreements upon commencement of employment or consulting relationships with us. These agreements will provide that all confidential information developed or made known to the individual during the course of their relationship with us must be kept confidential, except in specified circumstances. However, we cannot assure you that these agreements will provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure of confidential information. Further, invention assignment agreements executed by consultants and advisors may conflict with, or be subject to, the rights of third parties with whom such individuals have employment or consulting relationships. In addition, we cannot assure you that others will not independently develop equivalent proprietary information and techniques or otherwise gain access to our trade secrets, that such trade secrets will not be disclosed, or that we can effectively protect our rights to unpatented trade secrets.
We may be required to obtain licenses to patents or proprietary rights of others. We cannot assure you that any licenses required under any such patents or proprietary rights would be made available on terms acceptable to us or at all. If we do not obtain such licenses, we could encounter delays in product market introductions while we attempt to design around such patents, or could find that the development, manufacture, or sale of products requiring such licenses could be foreclosed. Litigation may be necessary to defend against or assert claims of infringement to enforce patents issued to us or exclusively licensed to us, to protect trade secrets or know-how possessed by us, or to determine the scope and validity of the proprietary rights of others. In addition, we may become involved in oppositions in foreign jurisdictions or interference proceedings declared by the United States Patent and Trademark Office to determine the priority of inventions with respect to our patent applications or those of our licensors. Litigation, opposition, or interference proceedings could result in substantial costs to and diversion of effort by, and may have a material adverse impact on, us. In addition, we cannot assure you that our efforts will be successful.
Government Regulation and Environmental Issues
Regulation by governmental authorities in the United States and foreign countries is a significant factor in the development, manufacture, and expected marketing of our drug product candidates and in our ongoing research and development activities. The nature and extent to which such regulation will apply to us will vary depending on the nature of any products developed. We anticipate that all of our drug product candidates will require regulatory approval by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical and clinical testing and other approval procedures of the FDA and similar regulatory authorities in foreign countries. Various federal statutes and regulations also govern or influence testing, manufacturing, safety, labeling, storage, and record-keeping related to such products and their marketing. The process of obtaining these approvals and the subsequent compliance with the appropriate federal statutes and regulations requires substantial time and financial resources. Any failure by us or our collaborators to obtain, or any delay in obtaining, regulatory approval could adversely affect the marketing of any products developed by us, our ability to receive product revenues, and our liquidity and capital resources.
The development, manufacturing, marketing, and distribution of drug products are extensively regulated by the FDA in the U.S. and similar regulatory agencies in other countries.
8
Table of Contents
The steps ordinarily required before a new drug may be marketed in the U.S., which are similar to steps required in most other countries, include:
| • | adequate and well-controlled clinical trials to establish the safety and efficacy of the drug; |
| • | the submission of a new drug application to the FDA; and |
| • | FDA review and approval of the new drug application (NDA) or biologics license application (or BLA). |
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal studies. The results of preclinical testing are submitted to the FDA as part of an investigational new drug (IND) application. A 30-day waiting period after the filing of each IND application is required prior to commencement of clinical testing in humans. At any time during the 30-day period or at any time thereafter, the FDA may halt proposed or ongoing clinical trials until the FDA authorizes trials under specified terms. The IND application process may be extremely costly and substantially delay development of our drug product candidates. Moreover, positive results of preclinical tests will not necessarily indicate positive results in subsequent clinical trials. The FDA may require additional animal testing after an initial IND is approved and prior to Phase 3 trials. These additional studies are customary for drugs intended for use by healthy populations. Our menopausal drug, Menerba, may be subjected to such studies, which may delay or damage our ability to complete trials and obtain a marketing license.
Clinical trials to support new drug applications are typically conducted in three sequential phases, although the phases may overlap. During Phase 1, clinical trials are conducted with a small number of subjects to assess metabolism, pharmacokinetics, pharmacological actions, various dosage levels, and safety, including potential side effects associated with increasing doses. Phase 2 usually involves studies in a limited patient population to:
| • | assess the efficacy of the drug in specific, targeted indications; |
| • | assess dosage tolerance and optimal dosage; |
| • | identify possible adverse effects and safety risks; and |
| • | correlate drug product potency to efficacy via pharmacological or biological studies. |
If a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to further demonstrate clinical efficacy and to further test for safety within an expanded patient population at geographically dispersed clinical trial sites.
After successful completion of the required clinical trials, an NDA is generally submitted. The NDA contains scientific and clinical data, which is intended to demonstrate that the drug is safe and effective for its proposed indication(s). The FDA may request additional information before accepting the NDA for filing, in which case the NDA must be resubmitted with the additional information. Once the submission has been accepted for filing, the FDA reviews the NDA and responds to the applicant. FDA requests for additional information or clarification often significantly extend the review process. The FDA may refer the NDA to an appropriate advisory committee for review, evaluation and recommendation as to whether the NDA should be approved, although the FDA is not bound by the recommendation of an advisory committee.
If the FDA evaluations of the application and the manufacturing facilities are favorable, the FDA may issue an approval letter or an “approvable” letter. An approvable letter will usually contain a number of conditions that must be met in order to secure final approval of the NDA and authorization of commercial marketing of the drug for certain indications. The FDA may also refuse to approve the NDA or issue a “not approvable” letter outlining the deficiencies in the submission and often requiring additional testing or information.
The FDA’s Modernization Act codified the FDA’s policy of granting “fast track” approval of cancer therapies and other therapies intended to treat severe or life threatening diseases and having potential to address
9
Table of Contents
unmet medical needs. Previously, the FDA approved cancer therapies primarily based on patient survival rates or data on improved quality of life. The FDA considered evidence of partial tumor shrinkage, while often part of the data relied on for approval, insufficient by itself to warrant approval of a cancer therapy, except in limited situations. Under the FDA’s revised policy, which became effective in 1998, the FDA has broadened authority to consider evidence of partial tumor shrinkage or other clinical outcomes for approval, such as progression-free survival. This revised policy is intended to facilitate the study of cancer therapies and shorten the total time for marketing approvals. We intend to take advantage of this policy; however, it is too early to tell what effect, if any, these provisions may have on the approval of our oncology drug product candidates.
Sales outside the United States of products we develop will also be subject to regulatory requirements governing human clinical trials and marketing for drugs. The requirements vary widely from country to country, but typically the registration and approval process takes several years and requires significant resources. In most cases, if the FDA has not approved a product for sale in the United States, the product may be exported for sale outside of the United States, only if it has been approved in any one of the following: the European Union, Canada, Australia, New Zealand, Japan, Israel, Switzerland and/or South Africa. There are specific FDA regulations that govern this process.
We are also subject to various federal, state and local laws, regulations and recommendations relating to safe working conditions, laboratory and manufacturing practices, and the use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research work. We cannot accurately predict the extent of government regulation that might result from future legislation or administrative action.
Manufacturing
We currently have no clinical or commercial manufacturing capabilities. To date, we have engaged domestic and foreign manufacturers experienced in FDA Good Manufacturing Practices (or GMP) for drug production, for the supply of our product candidates and other compounds solely for our pre-clinical research and development activities. We have not entered into formal written agreements with these manufacturers, and submit purchase orders on an as needed basis. We believe that numerous alternative manufacturers exist that would be capable of fulfilling our current product supply needs in the event we were unable to obtain product from our current manufacturer. We also intend to establish a demonstration or pilot plant capable of demonstrating our manufacturing processes and capable of use for producing certain pre-clinical and early clinical supplies.
In order to successfully commercialize our drug product candidates, we, or third parties with whom we contract, must be able to manufacture products in commercial quantities in compliance with the FDA’s current GMP (or cGMP) at acceptable costs and in a timely manner. As we do not own a cGMP manufacturing facility, we expect to contract with third parties to provide us with cGMP production capacity when appropriate.
Marketing and Sales
We currently have no sales activities, and no employees engaged in selling any of our products, as we have not received FDA approval to do so. Marketing activities are related to web-site design, and attendance at industry tradeshows and conferences, where we promote the results of our research activities.
Competition
Competition in the pharmaceutical and biotechnology industries is intense. Our competitors include pharmaceuticals companies and biotechnology companies, as well as universities and public and private research institutions. In addition, companies that are active in different but related fields represent substantial competition for us. Many of our competitors have significantly greater capital resources, larger research and development
10
Table of Contents
staffs and facilities and greater experience in drug development, regulation, manufacturing and marketing than we do. These organizations also compete with us to recruit qualified personnel, attract partners for joint ventures or other collaborations, and license technologies that are competitive with ours. To compete successfully in this industry we must identify novel and unique drugs or methods of treatment and then complete the development of those drugs as treatments in advance of our competitors.
The drugs that we are attempting to develop will have to compete with existing therapies. In addition, a large number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. Other companies have products or drug candidates in various stages of pre-clinical or clinical development to treat diseases for which we are also seeking to discover and develop drug candidates. Some of these potential competing drugs are further advanced in development than our drug candidates and may be commercialized earlier.
In particular, there are numerous companies attempting to discover and develop drugs to treat cancer. Many of them are targeting pathways similar to those targeted by us. However, we believe few of the companies are attempting to develop drugs derived from natural products and fewer companies are trying to discover drugs from botanical extracts. Moreover, we are not aware of many companies attempting to discover and develop more selective estrogen receptor modulators.
Our lead product candidate for metastatic breast cancer, Bezielle, may be used for patients with either hormone receptor positive or negative tumors, and is designed for use in both premenopausal and postmenopausal patients and patients who are both HER2 positive and negative. Accordingly, it can be expected to compete with most forms of current therapies for metastatic breast cancer, including hormonal therapy, chemotherapy or biologic therapy. Leading hormonal agents include Nolvadex and Arimidex by AstraZeneca and leading chemotherapy agents include Gemzar by Eli Lilly, Taxotere by Aventis and Xeloda by Roche. The leading biologic agents include Avastin and Herceptic by Genentech and Tykerb by GlaxoSmithKline.
We believe that Bezielle’s novel mechanism of action and favorable tolerability profile could lead to preferential use over existing drugs in the treatment of advanced breast cancer, and potentially in the broader, adjuvant care of breast cancer. While further clinical testing has yet to be completed to demonstrate this, certain of the above drug therapies may still have advantages relative to Bezielle. These advantages may include: selective patient population, lower pricing and greater efficacy.
Our lead product candidate for the treatment of menopausal symptoms, Menerba, would be expected to compete with postmenopausal hormone replacement therapy which has been the primary treatment of menopausal symptoms such as hot flashes. Leading hormonal agents include Premarin and Prempro by Wyeth Pharmaceuticals, Cenestin, Estradiol (generic) and Medroxy-Progesterone Acetate by Barr Pharmaceuticals, and Ogen, Provera and Estring by Pfizer. In addition, Menerba may be expected to compete with newer generation anti-depressants used off-label to treat hot flash frequency, such as venlafaxine by Wyeth, fluoxetine by Eli Lilly and paroxetine by Glaxo Smith Kline. The makers of these would compete directly with us relative to Menerba.
We believe that Menerba’s novel mechanism of action could lead to a more favorable safety profile than currently approved hormone therapies (HT) and other therapies under development and testing for postmenopausal vasomotor symptoms. While further clinical testing has yet to be completed to demonstrate this, certain hormone therapies may still have advantages relative to Menerba. These advantages may include lower pricing and greater efficacy.
We believe we possess the competitive advantage of using unique technologies with extracts that are considered to be safe and tolerable in humans. We also believe that finding lead drugs that are orally tolerable and potentially safe from the start of the discovery process provides an advantage in the pharmaceutical industry.
11
Table of Contents
Company Information
We were incorporated in Delaware in 2005. Our principal executive offices are located at 5858 Horton Street, Suite 400, Emeryville, California 94608, and our main telephone number is (510) 601-2000. Investors can obtain access to this annual report on Form 10-K, our quarterly reports on Form 10-Q, our current reports on Form 8-K and all amendments to these reports, free of charge, on our website at http://www.bionovo.com as soon as reasonably practicable after such filings are electronically filed with the Securities and Exchange Commission (“SEC”).
We have adopted a code of ethics, which is part of our Code of Business Conduct and Ethics that applies to all of our employees, including our principal executive officers. This code of ethics is posted on our website.
Employees
As of February 28, 2010, we had 58 employees, none of which are represented by a collective bargaining agreement. We believe that our employee relations are good.
Executive Officers and Directors
Our directors and executive officers and their ages as of February 28, 2010 are as follows:
| Name |
Age | Position | ||
| Isaac Cohen, O.M.D.,L.Ac. |
47 | Chairman, Chief Executive Officer, | ||
| Mary Tagliaferri, M.D., L.Ac |
44 | President, Chief Medical Officer, Chief Regulatory Officer and Director | ||
| Thomas C. Chesterman |
50 | Senior Vice President and Chief Financial Officer | ||
| John Baxter, M.D. (1)(2) |
69 | Director | ||
| George Butler, Ph.D (2) |
62 | Director | ||
| Louis Drapeau, (1) |
66 | Director | ||
| David Naveh, Ph.D. (1)(2) |
58 | Director | ||
| Michael D. Vanderhoof |
50 | Director |
| (1) | Member of the Audit Committee. |
| (2) | Member of the Compensation and Nominations Committee. |
Isaac Cohen, O.M.D., L.Ac., 47, is a co-founder of Bionovo Pharmaceuticals, Inc. (“Bionovo Pharmaceuticals”), and has served as its Chairman, President, Chief Executive Officer, and Chief Scientific Officer and as a director since February 2002. He became Bionovo, Inc.’s Chairman, Chief Executive Officer and Chief Scientific Officer and a Director in April 2005. Mr. Cohen has been a Guest Scientist at the University of California, San Francisco (UCSF) Cancer Research Center and UCSF Center for Reproductive Endocrinology since 1996. Mr. Cohen was in private practice at The American Acupuncture Center, located in Berkeley, California from 1989-2005.
Mary Tagliaferri, M.D., L.Ac., 44, is a co-founder of Bionovo Pharmaceuticals, and has served as its Chief Regulatory Officer, Chief Medical Officer, Secretary and Treasurer and as a director since February 2002. She became Vice President, Chief Medical Officer, Chief Regulatory Officer, Secretary and Treasurer of Bionovo, Inc. in April 2005, and a director effective in May 2005. She became President of Bionovo, Inc. in addition to continuing her functions as the Company’s Chief Medical Officer, Secretary, Treasurer and Director in May 2007. Dr. Tagliaferri was conducting translational research at the University of California, San Francisco from 1996 to 2002.
12
Table of Contents
Thomas C. Chesterman, M.B.A., 50, has served as our Senior Vice President, Chief Financial Officer and Assistant Secretary since July 2007. From January 2002 to June 2007, Mr. Chesterman was Sr. Vice President and Chief Financial Officer at Aradigm, Corporation, a drug development company. From March 1996 to December 2001, Mr. Chesterman was Vice President and Chief Financial Officer at Bio-Rad Laboratories, Inc., a life-science research products and clinical diagnostics company. From 1993 to 1996, Mr. Chesterman was Vice President of Strategy and Chief Financial Officer of Europolitan AB, a telecommunications company.
John D. Baxter, M.D., 69, has been a Director since April 14, 2008. Dr. Baxter is currently Senior Member and Co-Director of the Center for Diabetes Research at The Methodist Hospital Research Institute, and Head of Endocrinology at the University of Houston. He was associated with the University of California, San Francisco (or “UCSF”) from 1970-2008 where he was a Professor of Medicine from 1979-2008, Chief of the Endocrinology, Parnassus Campus from 1980 to 1997, and Director of UCSF’s Metabolic Research Unit from 1981 to 1999. Dr. Baxter was President of The Endocrine Society from June 2002 to June 2003. He was a founder and served as a director of California Biotechnology, Inc., a drug development company (now Scios, Inc., a division of Johnson & Johnson) from 1982 until 1992 and was a Founder and Director of Karo Bio A.B., a Swedish biotechnology company and SciClone Pharmaceuticals, a drug development company. Dr. Baxter has also been elected to the National Academy of Sciences and the Institute of Medicine of the National Academy of Sciences, and he received the Outstanding Investigator Award from the Howard Hughes Medical Institute. Dr. Baxter is an independent and unaffiliated director, and serves on our Audit Committee, and on our Compensation and Nominations Committee.
George Butler, Ph.D., 62, has been a Director since March 11, 2008. Currently, Dr. Butler serves as the Chairman and President of SingEval (Singapore) Pte. Ltd., a drug development company based in Singapore and the US. Dr. Butler was formerly the vice president, Customer Relationships, Global Development of AstraZeneca, plc, a global pharmaceutical company. Prior to this, he was vice president, head of regulatory affairs. Prior to his time at AstraZeneca, Dr. Butler was vice president and head of regulatory affairs with Novartis AG. Dr. Butler has over 30 years of healthcare research and business experience, primarily in a development environment in multiple biopharmaceutical companies and has also been active for many years in advancing Asian-Western development/regulatory single programs. Dr. Butler is an independent and unaffiliated director, and serves on our Compensation and Nominations Committee.
Louis Drapeau, CPA, MBA, 66, has been a Director since March 14, 2008. Currently, Mr. Drapeau serves as the Chief Executive Officer of InSite Vision since November 2008 and as its Chief Financial Officer since October 1, 2007. Prior to InSite Vision, he served as Chief Financial Officer, Senior Vice President, Finance, at Nektar Therapeutics, a biopharmaceutical company headquartered in San Carlos, California from January 2006 to August 2007. Prior to Nektar, Mr. Drapeau served as Acting Chief Executive Officer from August 2004 to May 2005 and as Senior Vice President and Chief Financial Officer from August 2002 to August 2005 for BioMarin Pharmaceutical Inc, a pharmaceutical company. Previously, Mr. Drapeau spent more than 30 years at Arthur Andersen. Mr. Drapeau also serves on the boards of Intermune, Inc., a pharmaceutical company and Bio-Rad Laboratories, a life science and clinical diagnosis company. Mr. Drapeau is an independent and unaffiliated director, and serves as the chair of our Audit Committee. Mr. Drapeau meets the qualifications as a “Financial Expert”, according to the definition in Item 407 (d)(5)(ii) on Regulation S-K.
David Naveh, Ph.D., MBA, 58, has been a Director of Bionovo Pharmaceuticals since August 2003. He became a director of the Company in May 2005. In 2007, Dr. Naveh retired from Bayer Corporation, a pharmaceutical company where he had worked since 1992, and served as Chief Technological Officer of Bayer Biological Products, Worldwide. Dr. Naveh is an independent and unaffiliated director, serves on our Audit Committee, and serves as the chair of our Compensation and Nominations Committee.
Michael Vanderhoof, 50, has been a Director since June 2005. Mr. Vanderhoof is Chairman of Cambria Asset Management, LLC, which owns Cambria Capital, LLC, a NASD registered broker dealer with offices in Los Angeles and Salt Lake City. Mr. Vanderhoof is also a co-manager of Cambria Investment Fund LP.
13
Table of Contents
Mr. Vanderhoof has over 20 years of experience in the capital markets. Mr. Vanderhoof is also a director of Auxilio, Inc., a healthcare document management services company. Mr. Vanderhoof is an outside director, though he would be considered an affiliated party due to his relationship with Cambria Capital, LLC, with whom the Company has done business in the past year.
Scientific Advisory Board
We have assembled a scientific advisory board that includes several prominent scientific and product development advisors who provide expertise, but are employed elsewhere on a full-time basis, in the areas of women’s health including menopause, breast cancer, cell biology, immunology, hormonal and metabolic disorders, biostatistics and pharmaceutical development. As of March 11, 2010, the board consisted of the following individuals:
| Name |
Affiliation |
Area of Expertise | ||
| John D. Baxter, M.D. |
The Methodist Hospital Research Institute | Endocrinology | ||
| Len Bjeldanes, Ph.D. |
University of California, Berkeley | Molecular Toxicology/Bioactive Compound Isolation and Identification | ||
| Paul Pui-Hay But, Ph.D. |
Food and Drug Authentication Laboratory Ltd., Hong Kong | Botanical Authentication and Chinese Medicine Quality Control | ||
| Michael J. Campbell, Ph.D. |
University of California, San Francisco | Cell Biology/Immunology | ||
| Uwe Christians M.D., Ph.D. |
University of Colorado | Pharmacology | ||
| Isaac Cohen, O.M.D., L.Ac. |
Bionovo, Inc. | Herbology, Pharmacology, Cell Biology and Pharmaceutical development | ||
| Gary L. Firestone, Ph.D. |
University of California, Berkeley | Molecular and Cell Biology | ||
| Richard Gless, Ph.D. |
Arete Therapeutics | Chemical Research Management | ||
| Jan Ake Gustafsson, M.D., Ph.D. |
Houston University | Nuclear Receptors and Cell Signaling | ||
| Craig Henderson, M.D. |
University of California, San Francisco | Breast Cancer | ||
| Willa A. Hsueh, M.D. |
The Methodist Hospital Research Institute | Nuclear Receptor Regulation | ||
| Dale Leitman, M.D., Ph.D. |
University of California, Berkeley | Estrogen Receptor Biology | ||
| Bert W. O’Malley, M.D. |
Baylor College of Medicine | Molecular and Cell Biology, Nuclear Receptor Regulation | ||
| Moshe Rosenberg, D.Sc. |
University of California, Davis | Microencapsulation Properties of Proteins, Lipids and Carbohydrates | ||
| Terry Speed, Ph.D. |
Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia | Bioinformatics | ||
14
Table of Contents
Medical Advisory Board
We have assembled a medical advisory board that includes several prominent clinical and scientific advisors who provide expertise, but are employed elsewhere on a full-time basis, in the areas of women’s health, cancer treatment, and related areas. These individuals provide critical guidance in our planning and execution of clinical trials for our drug candidates. As of March 11, 2010, the board consisted of the following individuals:
| Name |
Affiliation |
Area of Expertise | ||
| Mary Cushman, M.D. |
University of Vermont | Hematology, Epidemiology | ||
| Marco Gambacciani, M.D. |
Santa Chiara University Hospital | Menopause | ||
| Steven Goldstein, M.D. |
New York University | Menopause, Uterine Safety | ||
| Deborah Grady, M.D. |
University of California, San Francisco | Menopause | ||
| Mary Tagliaferri, M.D., L.Ac. |
Bionovo, Inc. | Menopause, Breast Cancer | ||
| Debasish Tripathy, M.D. |
University of Southern California | Breast Cancer | ||
| Wulf H. Utian, M.D. |
Rapid Medical Research, Inc. | Gynecological Endocrinology | ||
| Ethan Weiss, M.D. |
University of California, San Francisco | Cardiology | ||
| Janet Wittes, Ph.D. |
Statistics Collaborative, Inc. | Clinical Biostatistics | ||
| Item 1A. | Risk Factors |
Stockholders should carefully consider the following risk factors, together with the other information included and incorporated by reference in this Annual Report on Form 10-K. We operate in a rapidly changing environment that involves a number of risks, some of which are beyond our control. This discussion highlights some of the risks which may affect future operating results. These are the risks and uncertainties we believe are most important for you to consider. Additional risks and uncertainties not presently known to us, which we currently deem immaterial or which are similar to those faced by other companies in our industry or business in general, may also impair our business operations. If any of the following risks or uncertainties actually occurs, our business, financial condition and operating results would likely suffer.
Risks Relating to Our Business and Industry
We have a history of net losses, which we expect to continue for the foreseeable future and, as a result, we are unable to predict the extent of any future losses or when, if ever, we will become profitable or if we will be able to continue as a going concern.
We are a development stage company which commenced operations in 2002, and we are subject to all of the risks associated with having a limited operating history and pursuing the development of new products. Our cash flows may be insufficient to meet expenses relating to our operations and the development of our business, and may be insufficient to allow us to develop our products. We currently conduct research and development to develop anticancer drugs and women’s health products. We do not know whether we will be successful in the development of such products. As of December 31, 2009, we have incurred $55.8 million in cumulative net loss from our inception in 2002, and we expect losses to continue for the foreseeable future. Our net loss for fiscal year ended December 31, 2009 was $16.4 million and for the fiscal year ended December 31, 2008 was $16.7 million. Our net loss for the fiscal year ended December 31, 2007 was $12.9 million, and for the fiscal year ended December 31, 2006 was $5.6 million. To date, we have mainly recognized revenues from a technology license, research services and a National Institute of Health (NIH) grant drawdown. On October 15, 2007, we terminated the technology license agreement. We do not anticipate generating significant revenues from sales of
15
Table of Contents
our products, if approved, for at least several years, if at all. All of our product candidates are in development and none have been approved for commercial sale. We expect to increase our operating expenses over the next several years as we expand clinical trials for our product candidates currently in clinical development, including Menerba and Bezielle, advance our other anticancer and women’s health product candidates into clinical trials, expand our research and development activities, and seek regulatory approvals and eventually engage in commercialization activities in anticipation of potential FDA approval of our product candidates. Because of the numerous risks and uncertainties associated with developing our product candidates and their potential for commercialization, we are unable to predict the extent of any future losses or when we will become profitable, if at all. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. If we are unable to achieve and sustain profitability, the market value of our common stock will likely decline and we may not be able to continue as a going concern.
We have a limited operating history and are considered a development stage company.
We are considered a development stage company for accounting purposes because we have not generated any material revenues to date. Accordingly, we have limited relevant operating history upon which an evaluation of our performance and prospects can be made. We are subject to all of the business risks associated with a new enterprise, including risks of unforeseen capital requirements, failure of market acceptance, failure to establish business relationships and competitive disadvantages as against larger and more established companies.
Our product development and commercialization involves a number of uncertainties, and we may never generate sufficient revenues from the sale of potential products to become profitable.
We have generated no significant revenues to date. To generate revenue and to achieve profitability, we must successfully develop, clinically test, market and sell our potential products. Even if we generate revenue and successfully achieve profitability, we cannot predict the level of that profitability or whether it will be sustainable. We expect that our operating results will fluctuate from period to period as a result of differences in when we incur expenses and receive revenues from sales of our potential products, collaborative arrangements and other sources. Some of these fluctuations may be significant.
All of our products in development will require extensive additional development, including preclinical testing and human studies, as well as regulatory approvals, before we can market them. We cannot predict if or when any of the products we are developing or those being co-developed with us will be approved for marketing. There are many reasons that we or our collaborative partners may fail in our efforts to develop our potential products, including the possibility that:
| • | preclinical testing or human studies may show that our potential products are ineffective or cause harmful side effects; |
| • | the products may fail to receive necessary regulatory approvals from the FDA or foreign authorities in a timely manner, or at all; |
| • | the products, if approved, may not be produced in commercial quantities or at reasonable costs; |
| • | the potential products, once approved, may not achieve commercial acceptance; |
| • | regulatory or governmental authorities may apply restrictions to our potential products, which could adversely affect their commercial success; or |
| • | the proprietary rights of other parties may prevent us or our partners from marketing our potential products. |
16
Table of Contents
Our drug development programs will require substantial additional future funding which could hurt our operational and financial condition.
Our drug development programs require substantial additional capital to successfully complete them, arising from costs to:
| • | conduct research, preclinical testing and human studies; |
| • | establish pilot scale and commercial scale manufacturing processes and facilities; and |
| • | establish and develop quality control, regulatory and administrative capabilities to support these programs. |
Our future operating and capital needs will depend on many factors, including the following:
| • | the pace of scientific progress in our research and development programs and the magnitude of these programs; |
| • | the scope and results of preclinical testing and human studies; |
| • | the time and costs involved in obtaining regulatory approvals; |
| • | the time and costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims; |
| • | competing technological and market developments; |
| • | our ability to establish additional collaborations; |
| • | changes in our existing collaborations; |
| • | the cost of manufacturing scale-up; and |
| • | the effectiveness of our commercialization activities. |
We base our outlook regarding the need for funds on many uncertain variables. Such uncertainties include regulatory approvals, the timing of events outside our direct control such as negotiations with potential strategic partners and other factors. Any of these uncertain events can significantly change our cash requirements as they determine such one-time events as the receipt of major milestones and other payments.
If additional funds are required to support our operations and we are unable to obtain them on favorable terms, we may be required to cease or reduce further development or commercialization of our potential products, to sell some or all of our technology or assets or to merge with another entity.
Our potential products face significant regulatory hurdles prior to marketing which could delay or prevent sales. These regulatory hurdles include both clinical development requirements and manufacturing and quality control (CMC) requirements.
Before we obtain the approvals necessary to sell any of our potential products, we must show through preclinical studies and human testing that each potential product is safe and effective. We have a number of products moving toward or currently in clinical trials. Failure to show any potential product’s safety and effectiveness would delay or prevent regulatory approval of the product and could adversely affect our business. The clinical trials process is complex and uncertain. The results of preclinical studies and initial clinical trials may not necessarily predict the results from later large-scale clinical trials. In addition, clinical trials may not demonstrate a potential product’s safety and effectiveness to the satisfaction of the regulatory authorities. A number of companies have suffered significant setbacks in advanced clinical trials or in seeking regulatory approvals, despite promising results in earlier trials.
17
Table of Contents
The rate at which we complete our clinical trials depends on many factors, including our ability to obtain adequate supplies of the potential products to be tested and patient enrollment. Patient enrollment is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites and the eligibility criteria for the trial. Delays in patient enrollment may result in increased costs and longer development times. In addition, future collaborative partners may have rights to control product development and clinical programs for products developed under the collaborations. As a result, these collaborators may conduct these programs more slowly or in a different manner than we had expected. Even if clinical trials are completed, we or our collaborative partners still may not apply for FDA approval in a timely manner or the FDA still may not grant approval.
In addition, the process development and manufacturing of potential products is subject to extensive government regulation, including by the FDA, DEA and state and other territorial authorities. The FDA administers processes to assure that marketed products are safe, effective, and consistently uniform, of high quality and marketed only for approved indications. These quality assurance processes are complex and difficult for a new company to implement. Failure to comply with applicable regulatory requirements can result in sanctions up to the suspension of regulatory approval as well as civil and criminal sanctions. A number of companies have suffered significant setbacks in clinical development and in seeking regulatory approvals, despite their best efforts in the development of quality systems. In our case these requirements are more complex, since the published guidance document from the FDA (the Botanical Guidance) is relatively new, and there are limited precedent botanical drug candidates which have been submitted to the FDA for final approval.
Failure to obtain regulatory approvals in foreign jurisdictions would prevent us from marketing our products internationally.
We intend to have our product candidates marketed outside the United States. In order to market products in the European Union, Asia and many other non-U.S. jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. We may be unable to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. The approval procedure varies among countries and can involve additional and costly clinical testing and data review. The time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory agencies in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory agencies in other foreign countries or by the FDA. The failure to obtain these approvals could harm our business and result in decreased revenues.
We expect to rely heavily on collaborative relationships and termination of any of these programs could reduce the financial resources available to us, including research funding and milestone payments.
Our strategy for developing and commercializing many of our potential products, including products aimed at larger markets, includes entering into collaborations with corporate partners, licensors, licensees and others. These collaborations will provide us with funding and research and development resources for potential products. These agreements also will give our collaborative partners significant discretion when deciding whether or not to pursue any development program. Our collaborations may not be successful.
In addition, our collaborators may develop drugs, either alone or with others, which compete with the types of drugs they currently are developing with us. This would result in less support and increased competition for our programs. If any of our collaborative partners breach or terminate their agreements with us or otherwise fail to conduct their collaborative activities successfully, our product development under these agreements will be delayed or terminated.
We may have disputes in the future with our collaborators, including disputes concerning who owns the rights to any technology developed. These and other possible disagreements between us and our collaborators
18
Table of Contents
could delay our ability and the ability of our collaborators to achieve milestones or our receipt of other payments. In addition, any disagreements could delay, interrupt or terminate the collaborative research, development and commercialization of certain potential products, or could result in litigation or arbitration. The occurrence of any of these problems could be time-consuming and expensive and could adversely affect our business.
There is a substantial risk of product liability claims in our business. If we are unable to obtain sufficient insurance, a product liability claim against us could adversely affect our business.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face even greater risks upon any commercialization by us of our product candidates. We have product liability insurance covering our clinical trials at a level appropriate for the industry, which we currently believe is adequate to cover any product liability exposure we may have. Clinical trial and product liability insurance is volatile and may become increasingly expensive. As a result, we may be unable to obtain sufficient insurance or increase our existing coverage at a reasonable cost to protect us against losses that could have a material adverse effect on our business. An individual may bring a product liability claim against us if one of our products or product candidates causes, or is claimed to have caused, an injury or is found to be unsuitable for consumer use. Any product liability claim brought against us, with or without merit, could result in:
| • | liabilities that substantially exceed our product liability insurance, which we would then be required to pay from other sources, if available; |
| • | an increase of our product liability insurance rates or the inability to maintain insurance coverage in the future on acceptable terms, or at all; |
| • | withdrawal of clinical trial volunteers or patients; |
| • | damage to our reputation and the reputation of our products, resulting in lower sales; |
| • | regulatory investigations that could require costly recalls or product modifications; |
| • | litigation costs; and |
| • | the diversion of management’s attention from managing our business. |
Claims relating to any improper handling, storage or disposal of biological and hazardous materials by us could be time-consuming and costly.
Our research and development activities in our Aurora, Colorado and Emeryville, California facilities involve the controlled storage, use and disposal of hazardous materials. We are subject to government regulations relating to the use, manufacture, storage, handling and disposal of hazardous materials and waste products. Although we believe that our safety procedures for handling and disposing of these hazardous materials comply with the standards prescribed by applicable laws and regulations, the risk of accidental contamination or injury from hazardous materials cannot be completely eliminated. In the event of an accident, we could be held liable for any damages that result or we could be penalized with fines, and any liability could exceed the limits of or fall outside our insurance coverage. We may not be able to maintain insurance on acceptable terms, or at all. Further, we could be required to incur significant costs to comply with current or future environmental laws and regulations.
We do not have any manufacturing experience, nor do we currently have any manufacturing facilities. We currently rely upon third-party manufacturers to manufacture all clinical quantities of our product candidates. We depend on these third-party manufacturers to perform their obligations in a timely manner and in accordance with applicable governmental regulations. Our third-party manufacturers may encounter difficulties with meeting our requirements, including problems involving:
| • | inconsistent production yields; |
| • | poor quality control and assurance or inadequate process controls; and |
19
Table of Contents
| • | lack of compliance with regulations set forth by the FDA or other foreign regulatory agencies. |
These contract manufacturers may not be able to manufacture our product candidates at a cost or in quantities necessary to make them commercially viable. We also have no control over whether third-party manufacturers breach their agreements with us or whether they may terminate or decline to renew agreements with us. To date, our third party manufacturers have met our manufacturing requirements, but we cannot assure you that they will continue to do so. Furthermore, changes in the manufacturing process or procedure, including a change in the location where the drug is manufactured or a change of a third-party manufacturer, may require prior FDA review and approval in accordance with the FDA’s current Good Manufacturing Practices, or cGMPs. There are comparable foreign requirements. This review may be costly and time-consuming and could delay or prevent the launch of a product. The FDA or similar foreign regulatory agencies at any time may also implement new standards, or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of products. If we or our contract manufacturers are unable to comply, we or they may be subject to regulatory action, civil actions or penalties.
If we cannot rely on third-party manufacturers, we will be required to incur significant costs and devote significant efforts to establish our own manufacturing facilities and capabilities. If we are unable to enter into agreements with additional manufacturers on commercially reasonable terms, or if there is poor manufacturing performance on the part of our third party manufacturers, we may not be able to complete development of, or market, our product candidates.
If we lose the services of our co-founders who serve as directors and officers of our company, our operations could be disrupted and our business could be harmed.
Our business plan relies significantly on the continued services of our co-founders, Isaac Cohen and Mary Tagliaferri. If we were to lose the services of one or both of them, our ability to continue to execute our business plan could be materially impaired. Neither Mr. Cohen nor Dr. Tagliaferri has indicated they intend to leave our company, and we are not aware of any facts or circumstances that suggest either of them might leave us.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results. As a result, current and potential investors could lose confidence in our financial reporting, which could harm our business and have an adverse effect on our stock price.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, we are required to annually furnish a report by our management on our internal control over financial reporting. Such report must contain, among other matters, an assessment by our principal executive officer and our principal financial officer on the effectiveness of our internal control over financial reporting, including a statement as to whether or not our internal control over financial reporting is effective as of the end of our fiscal year. This assessment must include disclosure of any material weakness in our internal control over financial reporting identified by management. Performing the system and process documentation and evaluation needed to comply with Section 404 is both costly and challenging. We have in the past discovered, and may in the future discover, areas of internal controls that need improvement. For example, during the second quarter of 2009, we discovered that we did not timely file our 2009 proxy statement within the required timeline set by the SEC. We determined that we had a deficiency in our internal control over financial reporting as of June 30, 2009, but it was not a material weakness. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
In the future if any material weaknesses are identified in our internal control over financial reporting, neither our management nor our independent registered public accounting firm will be able to assert that our internal control over financial reporting or our disclosure controls and procedures are effective, and we could be required to further implement expensive and time-consuming remedial measures. We cannot be certain that any measures
20
Table of Contents
we take will ensure that we implement and maintain adequate internal control over financial reporting and that we will remediate any material weaknesses. As a result of recent reductions in our workforce and other personnel departures, we have experienced turnover in our personnel responsible for performing activities related to our internal control over financial reporting. We have used third-party contractors to maintain effective internal control over financial reporting since 2007. However, if we fail to maintain effective internal control over financial reporting or disclosure controls and procedures we could lose investor confidence in the accuracy and completeness of our financial reports, which could have a material adverse effect on our stock price.
The current global economic environment poses severe challenges to our business strategy, which relies on access to capital from the markets and our collaborators, and creates other financial risks for us.
The global economy, including credit markets and the financial services industry, has been experiencing a period of substantial turmoil and uncertainty. These conditions have generally made equity and debt financing more difficult to obtain, and may negatively impact our ability to complete financing transactions. The duration and severity of these conditions is uncertain, as is the extent to which they may adversely affect our business and the business of current and prospective collaborators and vendors. If the global economy does not improve or worsens, we may be unable to secure additional funding to sustain our operations or to find suitable partners to advance our internal programs, even if we receive positive results from our research and development or business development efforts.
We maintain a portfolio of investments in marketable debt securities which are recorded at fair value. Although we have established investment guidelines relative to diversification and maturity with the objectives of maintaining safety of principal and liquidity, credit rating agencies may reduce the credit quality of our individual holdings which could adversely affect their value. Lower credit quality and other market events, such as changes in interest rates and further deterioration in the credit markets, may have an adverse effect on the fair value of our investment holdings and cash position.
Our operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Our corporate headquarters are located at a single business park in Emeryville, California. Important documents and records, including copies of our regulatory documents and other records for our product candidates, are located at our facilities and we depend on our facilities for the continued operation of our business. Natural disasters and other catastrophic events, such as wildfires and other fires, earthquakes and extended power interruptions, which have not severely impacted Emeryville businesses in the past, and terrorist attacks, drought or flood, could significantly disrupt our operations and result in additional, unplanned expense. As a small company, we have limited capability to establish and maintain a comprehensive disaster recovery program. Any significant natural disaster or catastrophic event could delay our development and commercialization efforts. Even though we believe we carry commercially reasonable insurance, we might suffer losses that exceed the coverage available under these insurance policies. In addition, we are not insured against terrorist attacks or earthquakes.
We may be negatively impacted by the effects of climate change.
With respect to the impacts of climate change, the Company has no means of assessing or determining that climate change is a known trend likely to come to fruition with a material impact on the Company. If climate change were to have a impact on the Company’s results of operations, the physical impacts of climate change could have an adverse effect on the sourcing of our raw materials. The company may be required to pay more for the raw materials and/or move to alternative suppliers in new geographical areas.
21
Table of Contents
Risks Relating to Our Products, Technology and Know-how
Our products, if and when any of them are approved, could be subject to restrictions or withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements, or if such products exhibit unacceptable problems.
Any product for which we obtain marketing approval, together with the manufacturing processes, and advertising and promotional activities for such product, will be subject to continued regulation by the FDA and other regulatory agencies. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Any adverse effects observed after the approval and marketing of a product candidate could result in the withdrawal of any approved products from the marketplace. Absence of long-term safety data may also limit the approved uses of our products, if any, to short-term use. Later discovery of previously unknown problems with our products or their manufacture, or failure to comply with regulatory requirements, may result in one or more of the following:
| • | restrictions on such products or manufacturing processes; |
| • | withdrawal of the products from the market; |
| • | voluntary or mandatory recalls; |
| • | fines; |
| • | suspension of regulatory approvals; |
| • | product seizures; or |
| • | injunctions or the imposition of civil or criminal penalties. |
If we are slow to adapt, or unable to adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements or policies, we may lose marketing approval for them when and if any of them are approved, resulting in decreased revenue from milestones, product sales or royalties.
If our competitors develop and market products that are more effective than our existing product candidates or any products that we may develop, or if they obtain marketing approval for their products before we do, our commercial opportunity will be reduced or eliminated.
The pharmaceutical and biotechnology industry is highly competitive. Pharmaceutical and biotechnology companies are under increasing pressure to develop new products, particularly in view of lengthy product development and regulatory timelines, expiration of patent protection and recent setbacks experienced by several products previously approved for marketing. We compete with many companies that are developing therapies for the treatment of cancer and the symptoms of menopause. Several companies are developing products with technologies that are similar to ours. We also face competition in the field of cancer treatment and therapies for the symptoms of menopause from academic institutions and governmental agencies. Many of our competitors may have greater financial and human resources or more experience in research and development than we have, and they may have established sales, marketing and distribution capabilities. If we or our collaborators receive regulatory approvals for our product candidates, some of our products will compete with well-established, FDA-approved therapies that have generated substantial sales over a number of years. In addition, we will face competition based on many different factors, including the following:
| • | the safety and effectiveness of our products; |
| • | the timing and scope of regulatory approvals for these products; |
| • | the availability and cost of manufacturing, marketing and sales capabilities; |
22
Table of Contents
| • | the effectiveness of our or our collaborators marketing and sales capabilities; |
| • | the price of our products; |
| • | the availability and amount of third-party reimbursement; and |
| • | the strength of our patent position. |
We also anticipate that we will face increased competition in the future as new companies enter our target markets and scientific developments surrounding cancer therapies and drugs for menopause continue to accelerate. Competitors may develop more effective or more affordable products, or may achieve patent protection or commercialize products before us or our collaborators. In addition, the health care industry is characterized by rapid technological change. New product introductions, technological advancements, or changes in the standard of care for our target diseases could make some or all of our products obsolete.
While we are developing Bezielle to minimize many of the adverse side effects associated with the above breast cancer treatments, further clinical testing has yet to be completed, and certain drug therapies may have advantages relative to Bezielle, which may include selective patient population, greater efficacy and lower cost.
Our lead product candidate for the treatment of menopausal symptoms, Menerba, would be expected to compete with postmenopausal hormone replacement therapy which has been the primary treatment of menopausal symptoms such as hot flashes. Leading hormonal agents include Premarin and Prempro by Wyeth Pharmaceuticals, Cenestin, Estradiol (generic) and Medroxy-Progesterone Acetate by Barr Pharmaceuticals, and Ogen, Provera and Estring by Pfizer. In addition, Menerba may be expected to compete with newer generation anti-depressants used to treat hot flash frequency, such as desvenlafaxine by Wyeth, fluoxetine by Eli Lilly and paroxetine by Glaxo Smith Kline. The makers of these hormonal agents would compete directly with us relative to Menerba.
While we are developing Menerba to minimize many of the risks associated with long-term use of HT indicated in recent studies and further clinical testing has yet to be completed, certain hormone therapies may have advantages relative to Menerba. These advantages may include greater efficacy and lower cost.
We will face uncertainty in any commercialization of our product candidates relating to coverage, pricing and reimbursement due to health care reform and heightened scrutiny from third-party payers, which may make it difficult or impossible to sell our product candidates on commercially reasonable terms.
Sales of prescription drugs depend significantly on access to the formularies, or lists of approved prescription drugs, of third-party payers such as government and private insurance plans, as well as the availability of reimbursement to the consumer from these third party payers. These third party payers frequently require drug companies to provide predetermined discounts from list prices, and they are increasingly challenging the prices charged for medical products and services. Our potential products may not be considered cost-effective, may not be added to formularies and reimbursement to the consumer may not be available or sufficient to allow us to sell our potential products on a competitive basis.
In addition, the efforts of governments and third-party payers to contain or reduce the cost of health care will continue to affect the business and financial condition of drug companies such as us. A number of legislative and regulatory proposals to change the health care system have been discussed in recent years, including price caps and controls for pharmaceuticals. These proposals could reduce or cap the prices for our potential products or reduce government reimbursement rates for such products. In addition, an increasing emphasis on managed care in the United States has and will continue to increase pressure on drug pricing. We cannot predict whether legislative or regulatory proposals will be adopted or what effect those proposals or managed care efforts may have on our business. The announcement or adoption of such proposals or efforts could adversely affect our business.
23
Table of Contents
Failure to secure patents and other proprietary rights or challenges to those patents and rights may significantly hurt our business.
Our success will depend on our ability to obtain and maintain patents and proprietary rights for our potential products and to avoid infringing the proprietary rights of others, both in the United States and in foreign countries. Patents may not be issued from any of these applications currently on file, or, if issued, may not provide sufficient protection.
At present, we have 74 patent applications pending in the United States Patent and Trademark Office, as well as in the Patent Cooperation Treaty, the European Patent Office, Japan, and other markets. One patent (US 7482029 B1) for Menerba, entitled “Composition for Treatment of Menopause”, was issued on March 29, 2006. Our patent position, like that of many pharmaceutical companies, is uncertain and involves complex legal and technical questions for which important legal principles are unresolved. We may not develop or obtain rights to products or processes that are patentable. Even if we do obtain patents, they may not adequately protect the technology we own or have licensed. In addition, others may challenge, seek to invalidate, infringe, or circumvent any patents we own or license, and rights we receive under those patents may not provide competitive advantages to us.
Several drug companies and research and academic institutions have developed technologies, filed patent applications, or received patents for technologies that may be related to our business. In addition, we may not be aware of all patents or patent applications that may impact our ability to make, use, or sell any of our potential products. For example, US patent applications may be kept confidential while pending in the Patent and Trademark Office and patent applications filed in foreign countries are often first published six months or more after filing. Any conflicts resulting from the patent rights of others could limit our ability to obtain meaningful patent protection. If other companies obtain patents with conflicting claims, we may be required to obtain licenses to those patents or to develop or obtain alternative technology. We may not be able to obtain any such licenses on acceptable terms, or at all. Any failure to obtain such licenses could delay or prevent us from pursuing the development or commercialization of our potential products.
We may also need to initiate litigation, which could be time-consuming and expensive, to enforce our proprietary rights or to determine the scope and validity of others’ rights. If any of our competitors have filed patent applications in the United States which claim technology we also have invented, the Patent and Trademark Office may require us to participate in expensive interference proceedings to determine who has the right to a patent for the technology.
If third parties successfully assert that we have infringed their patents and proprietary rights or challenge the validity of our patents and proprietary rights, we may become involved in intellectual property disputes and litigation that would be costly, time consuming, and delay or prevent the development of our product candidates.
The manufacture, use, or sale of our potential products may infringe the patent rights of others. Any litigation to determine the scope and validity of such third party patent rights would be time consuming and expensive. If we are found to infringe on the patent or intellectual property rights of others, we may be required to pay damages, stop the infringing activity, or obtain licenses covering the patents or other intellectual property in order to use, manufacture, or sell our products. Any required license may not be available to us on acceptable terms or at all. If we succeed in obtaining these licenses, payments under these licenses would reduce any earnings from our products. In addition, some licenses may be non-exclusive and, accordingly, our competitors may have access to the same technology as that which is licensed to us. If we fail to obtain a required license or are unable to alter the design of our product candidates to make the licenses unnecessary, we may be unable to commercialize one or more of them, which could significantly affect our ability to achieve, sustain, or grow our commercial business.
24
Table of Contents
If we are unable to protect our trade secrets, we may be unable to protect our interests in proprietary know-how that is not patentable or for which we have elected not to seek patent protection.
In an effort to protect our unpatented proprietary technology, processes, and know-how, we require our employees, consultants, collaborators, and advisors to execute confidentiality agreements. These agreements, however, may not provide us with adequate protection against improper use or disclosure of confidential information. These agreements may be breached, and we may not become aware of, or have adequate remedies in the event of, any such breach. In addition, in some situations, these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants, collaborators, or advisors have previous employment or consulting relationships. Also, others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets.
Our product candidates may have difficulties with market acceptance even after FDA approval.
Even if we receive regulatory approvals for the commercial sale of our product candidates, the commercial success of these products will depend on, among other things, their acceptance by physicians, patients, third-party payers and other members of the medical community as a therapeutic and cost-effective alternative to competing products and treatments. If our product candidates fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business. Market acceptance of, and demand for, any product candidate that we may develop and commercialize will depend on many factors, including the following:
| • | our ability to provide acceptable evidence of safety and efficacy; |
| • | the prevalence and severity of side effects or other reactions; |
| • | the convenience and ease of use; |
| • | availability, relative cost and relative efficacy of alternative and competing products and treatments; |
| • | the effectiveness of our marketing and distribution strategy; |
| • | the publicity concerning our products or competing products and treatments; and |
| • | our ability to obtain third-party insurance coverage and adequate payment levels. |
If our product candidates do not become widely accepted by physicians, patients, third-party payers and other members of the medical community, we may be unable to achieve profitability.
Risks Related to Our Common Stock
We will need additional capital to conduct our operations and develop our products, and our ability to obtain the necessary funding is uncertain.
We will require substantial capital resources in order to conduct our operations and develop our therapeutic products, and we cannot assure you that our existing capital resources and the exercise of outstanding warrants and interest income will be sufficient to fund our current and planned operations. The timing and degree of any future capital requirements will depend on many factors.
We do not have any committed sources of capital. Additional financing through grants, strategic collaborations, public or private equity financings or other financing sources may not be available on acceptable terms, or at all. The receptivity of the public and private equity markets to proposed financings is substantially affected by the general economic, market and political climate and by other factors which are unpredictable and over which we have no control. Additional equity financings, if we obtain them, could result in significant dilution to shareholders. Further, in the event that additional funds are obtained through arrangements with collaborative partners, these arrangements may require us to relinquish rights to some of our technologies, product candidates, or products that we would otherwise seek to develop and commercialize ourselves.
25
Table of Contents
To conserve funds, we may pursue less expensive but higher-risk development paths or suspend all development activity. Historically, we have limited our product development activities to the minimum we felt was sufficient to support our development and commercialization goals, in particular, with respect to Menerba. While we successfully completed Phase 1 clinical trials of Menerba, without extensive product development experience, we may lack the information necessary and the resources to complete product development and may be unable to manufacture successfully at commercial-scale. If we are unable to complete our product development, we may be unable to effectively commercialize our products, if approved.
If we are unable to raise sufficient additional capital, we may seek to merge with or be acquired by another company and that transaction may adversely affect our business and the value of our securities.
If we are unable to raise sufficient additional capital, we may seek to merge with another company with a stronger cash position, complementary work force, or product candidate portfolio or for other reasons. We believe the market price for our common stock may not accurately reflect the value of our business. While we will continue to seek to maximize the value of our business to our stockholders, the most attractive option for doing so may require us to consummate a transaction involving an exchange of our common stock with that of another company. There are numerous risks associated with merging or being acquired. These risks include, among others, incorrectly assessing the quality of a prospective acquirer or merger-partner, encountering greater than anticipated costs in integrating businesses, facing resistance from employees and being unable to profitably deploy the assets of the new entity. The operations, financial condition, and prospects of the post-transaction entity depend in part on our and our acquirer/merger-partner’s ability to successfully integrate the operations related to our product candidates, business and technologies. We may be unable to integrate operations successfully or to achieve expected cost savings and any cost savings which are realized may be offset by losses in revenues or other charges to operations. As a result, our stockholders may not realize the full value of their investment.
If our securities are removed from NASDAQ listing, you may have more difficulty purchasing and selling our common stock.
Our securities are currently listed and traded on The NASDAQ Capital Market. To maintain our listing, we must meet NASDAQ’s continued listing standards, including minimum stockholders’ equity, number of stockholders, and bid price. As previously disclosed in our filings with the SEC, on September 15, 2009, we received a letter from The NASDAQ Stock Market stating that we were not in compliance with NASDAQ listing rules because we failed to maintain a minimum bid price of $1.00 per share. NASDAQ has granted us a period of 180 calendar days, or until March 16, 2010, to regain compliance. If, at anytime before March 16, 2010, the bid price of our common stock closes at $1.00 per share or more for a minimum of 10 consecutive business days, NASDAQ has stated that it will provide us with written notification that compliance with the listing rule has been regained.
In the event we do not regain compliance with the listing rule prior to the expiration of the compliance period, NASDAQ has indicated that we will be eligible for an additional grace period of 180 days if we meet NASDAQ Capital Market initial listing standards, with the exception of the bid price requirement. We believe we are in compliance with the initial listing standards, excluding the bid price requirement.
We intend to use our best efforts to regain compliance with NASDAQ’s minimum bid requirement and expect that our shares will continue to be listed on The NASDAQ Capital Market during this process under the symbol “BNVI.”
There can be no assurance that we will comply with the continued listing standards in the future. If we are delisted from The NASDAQ Capital Market, we may not be able to secure listing on other exchanges or quotation systems. As a result, we believe that investors will have significantly less liquidity and more difficulty purchasing and selling shares of our common stock, which could have a material adverse effect on the market price of our common stock.
26
Table of Contents
If our common stock were delisted from The NASDAQ Capital Market and determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock in the secondary market.
If our common stock were removed from listing with The NASDAQ Capital Market, it may be subject to the so-called “penny stock” rules. The SEC has adopted regulations that define a “penny stock” to be any equity security that has a market price per share of less than $5.00, subject to certain exceptions, such as any securities listed on a national securities exchange. For any transaction involving a “penny stock,” unless exempt, the rules impose additional sales practice requirements on broker-dealers, subject to certain exceptions. If our common stock were delisted and determined to be a “penny stock,” a broker-dealer may find it more difficult to trade our common stock and an investor may find it more difficult to acquire or dispose of our common stock on the secondary market. Investors in penny stocks should be prepared for the possibility that they may lose their whole investment.
Volatility of our stock price could adversely affect stockholders.
The market price of our common stock has been volatile, and fluctuates widely in price in response to various factors which are beyond our control. The price of our common stock is not necessarily indicative of our operating performance or long term business prospects. In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of our common stock. During the period from January 1, 2005 to December 31, 2009, the lowest and highest reported trading prices of our common stock on The NASDAQ Capital Market, or for periods before May 29, 2007, as reported on the OTC Bulletin Board, were $0.11 and $6.80. Factors such as the following could cause the market price of our common stock to fluctuate substantially:
| • | the results of research or development testing of our or our competitors’ products; |
| • | technological innovations related to diseases we are studying; |
| • | new commercial products introduced by our competitors; |
| • | government regulation of our industry; |
| • | receipt of regulatory approvals by our competitors; |
| • | our failure to receive regulatory approvals for products under development; |
| • | developments concerning proprietary rights; or |
| • | litigation or public concern about the safety of our products. |
The stock market in general has recently experienced extreme price and volume fluctuations. In particular, market prices of securities of drug development companies have experienced fluctuations that often have been unrelated or disproportionate to the operating results of these companies. Continued market fluctuations could result in extreme volatility in the price of our common stock, which could cause a decline in the value of our common stock. Price volatility might be worse if the trading volume of our common stock is low.
Our principal stockholders have significant voting power and may take actions that may not be in the best interest of other stockholders.
Our officers, directors, and principal stockholders control approximately 19% of our currently outstanding common stock. This concentration of ownership may not be in the best interests of all our stockholders. If these stockholders act together, they may be able to exert significant control over our management and affairs requiring stockholder approval, including approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control and might adversely affect the market price of our common stock.
27
Table of Contents
There may be sales of our stock by our executive officers and directors, and these sales could adversely affect our stock price.
Sales of our stock by our executive officers and directors, or the perception that such sales may occur, could adversely affect the market price of our stock. None of our executive officers have adopted trading plans under SEC Rule 10b5-1 to dispose of a portion of their stock. Any of our executive officers or directors may adopt such trading plans in the future.
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable.
Provisions of our certificate of incorporation, bylaws, and provisions of applicable Delaware law may discourage, delay, or prevent a merger or other change in control that a stockholder may consider favorable.
Pursuant to our certificate of incorporation, our board of directors may issue additional shares of common or preferred stock. Any additional issuance of common stock could have the effect of impeding or discouraging the acquisition of control of us by means of a merger, tender offer, proxy contest or otherwise, including a transaction in which our stockholders would receive a premium over the market price for their shares, and thereby protects the continuity of our management. Specifically, if in the due exercise of fiduciary obligations, the board of directors were to determine that a takeover proposal was not in our best interest, shares could be issued by our board of directors without stockholder approval in one or more transactions that might prevent or render more difficult or costly the completion of the takeover by any of the following:
| • | diluting the voting or other rights of the proposed acquirer or insurgent stockholder group; |
| • | putting a substantial voting bloc in institutional or other hands that might undertake to support the incumbent board of directors; or |
| • | effecting an acquisition that might complicate or preclude the takeover. |
Our certificate of incorporation also allows our board of directors to fix the number of directors in the by-laws. Cumulative voting in the election of directors is specifically denied in our certificate of incorporation. The effect of these provisions may be to delay or prevent a tender offer or takeover attempt that a stockholder may determine to be in his, her or its best interest, including attempts that might result in a premium over the market price for the shares held by the stockholders.
We also are subject to Section 203 of the Delaware General Corporation Law. In general, these provisions prohibit a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder, unless the transaction in which the person became an interested stockholder is approved in a manner presented in Section 203 of the Delaware General Corporation Law. Generally, a “business combination” is defined to include mergers, asset sales, and other transactions resulting in financial benefit to a stockholder. In general, an “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years, did own, 15% or more of a corporation’s voting stock. This statute could prohibit or delay mergers or other takeover or change in control attempts and, accordingly, may discourage attempts to acquire us.
We do not anticipate paying cash dividends for the foreseeable future, and therefore investors should not buy our stock if they wish to receive cash dividends.
We have never declared or paid any cash dividends or distributions on our capital stock. We currently intend to retain our future earnings to support operations and to finance expansion and therefore we do not anticipate paying any cash dividends on our common stock in the foreseeable future.
28
Table of Contents
A significant number of our shares are eligible for sale and their sale or potential sale may depress the market price of our common stock.
Sales of a significant number of shares of our common stock in the public market could harm the market price of our common stock. As of December 31, 2009, we have two effective registration statements registering the resale of up to 57,025,186 shares of our outstanding common stock and shares of common stock issuable upon exercise of warrants, which represents a significant majority of our currently outstanding shares of common stock. As additional shares of our common stock become available for resale in the public market pursuant to that registration statement, and otherwise, the supply of our common stock will increase, which could decrease its price. Some or all of such shares of common stock may be offered from time to time in the open market and these sales may have a depressive effect on the market for our shares of common stock.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
The Company leases approximately 32,000 square feet of laboratory and office facilities located at 5858 Horton Street, Suite 400, Emeryville, CA 94608, under a long-term, non-cancelable operating lease agreement that expires in December of 2018. The lease provides for increases in future minimum annual rental payments and the agreement requires the Company to pay executory costs (real estate taxes, insurance, and repairs). In November of 2009, the Company signed a one-year lease for additional laboratory space in Emeryville, California.
The Company also maintains laboratory facilities in Aurora, Colorado under a lease that expired in November of 2009. The Company is currently evaluating lease renewal options.
| Item 3. | Legal Proceedings |
On October 16, 2007, a former officer of the Company filed a complaint against Bionovo, Inc. and two of its officers, alleging breach of contract resulting from the rescission of the former officer’s employment agreement with the Company in September 2007. This complaint was filed with the Superior Court of the State of California in and for the County of Alameda. As a part of his compensation, the former officer was to receive 600,000 shares of common stock, over a two-year period from the date of grant, subject to various limitations and vesting. In his complaint, he alleges that the Company failed to accelerate the vesting of 300,000 shares to him, thereby breaching an agreement.
In September of 2008 the Company was notified of an order by the Superior Court, dated August 29, 2008, granting in part a motion to compel binding arbitration for a subset of the claims. The arbitration was held on May 11 and 12, 2009 and on June 10, 2009 the arbitration decision was issued denying all claims alleged by the former officer against the Company and its officers.
In February of 2010, the Company reached a settlement agreement for the residual claims that were not subject to the arbitration. The settlement amount was included as an expense accrual in the Company’s financial statements for the year ended December 31, 2009; however it did not have a material impact on the Company’s financial position, results of operations or cash flows.
| Item 4. | Reserved |
29
Table of Contents
PART II
| Item 5. | Market For Registrant’s Common Equity, Related Stockholder Matters And Issuer Purchases Of Equity Securities |
Our common stock trades on The NASDAQ Capital Market under the symbol “BNVI”. The following table sets forth the high and low closing sale prices of our common stock for the periods indicated.
| High | Low | |||
| 2008 |
||||
| First Quarter |
2.15 | 1.12 | ||
| Second Quarter |
1.55 | 0.76 | ||
| Third Quarter |
1.49 | 0.70 | ||
| Fourth Quarter |
0.93 | 0.17 | ||
| 2009 |
||||
| First Quarter |
0.40 | 0.18 | ||
| Second Quarter |
0.95 | 0.22 | ||
| Third Quarter |
1.15 | 0.36 | ||
| Fourth Quarter |
0.79 | 0.34 | ||
Holders
On February 28, 2010, there were 160 stockholders of record of our common stock and the last reported sales price on The NASDAQ Capital Market for our common stock was $0.58. The market for our common stock is highly volatile.
Dividends
We did not repurchase any shares of our equity securities during the year ended December 31, 2009. We have never declared or paid any cash dividends on our common stock and we do not currently intend to pay any cash dividends on our common stock for the foreseeable future.
The information required by this item regarding equity compensation plans is incorporated by reference to the information in Item 11 of this Annual Report.
30
Table of Contents
Stockholder Return Comparison
The following graph shows the total shareholder return of an investment of $100 in cash on December 31, 2004 for (i) our common stock, (ii) the Nasdaq composite Market Index (the “NASDAQ Index”) and (iii) The NASDAQ Pharmaceutical Index (the “NASDAQ-Pharmaceutical”). The NASDAQ Index tracks the aggregate price performance of equity securities traded on the NASDAQ. The NASDAQ-Pharmaceutical tracks the aggregate price performance of equity securities of pharmaceutical companies traded on The NASDAQ Index.
PERFORMANCE MEASUREMENT COMPARISON *
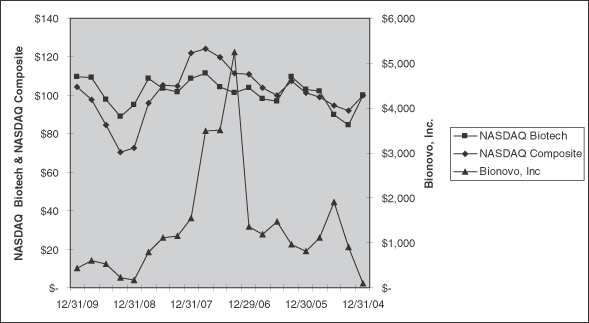
| * | This section is not “soliciting material,” is not deemed “filed” with the SEC, and is not to be incorporated by reference into any filing of the Company under the 1933 Act or 1934 Act, whether made before or after the date hereof and irrespective of any general incorporation language contained in such filing. |
31
Table of Contents
| Item 6. | Selected Financial Data |
The following selected financial data should be read in conjunction with the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and notes thereto included in this Report on Form 10-K.
| For the Years Ended December 31, | Accumulated from February 1, 2002 (Date of inception) to December 31, 2009 |
|||||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||||||
| Statements of Operations Data: |
||||||||||||||||||||||||
| Revenues |
$ | 288 | $ | 233 | $ | 582 | $ | 15 | $ | 15 | $ | 1,180 | ||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| Research and development |
12,499 | 11,416 | 9,938 | 4,021 | 1,535 | 39,706 | ||||||||||||||||||
| General and administrative |
4,053 | 6,097 | 4,284 | 1,799 | 1,056 | 17,602 | ||||||||||||||||||
| Merger cost |
— | — | — | — | 1,964 | 1,964 | ||||||||||||||||||
| Total operating expenses |
16,552 | 17,513 | 14,222 | 5,820 | 4,555 | 59,272 | ||||||||||||||||||
| Loss from operations |
(16,264 | ) | (17,280 | ) | (13,640 | ) | (5,805 | ) | (4,540 | ) | (58,092 | ) | ||||||||||||
| Change in fair value of warrant liability |
— | — | — | — | 831 | 831 | ||||||||||||||||||
| Interest income |
84 | 730 | 850 | 261 | 148 | 2,074 | ||||||||||||||||||
| Interest expense |
(95 | ) | (129 | ) | (87 | ) | (47 | ) | (74 | ) | (460 | ) | ||||||||||||
| Other expense |
(88 | ) | (17 | ) | (21 | ) | (24 | ) | (2 | ) | (153 | ) | ||||||||||||
| Loss before income tax |
(16,363 | ) | (16,696 | ) | (12,898 | ) | (5,615 | ) | (3,637 | ) | (55,800 | ) | ||||||||||||
| Income tax provision |
(1 | ) | (4 | ) | (3 | ) | (3 | ) | (1 | ) | (14 | ) | ||||||||||||
| Net loss |
$ | (16,364 | ) | $ | (16,700 | ) | $ | (12,901 | ) | $ | (5,618 | ) | $ | (3,638 | ) | $ | (55,814 | ) | ||||||
| Basic and diluted net loss per common share |
$ | (0.20 | ) | $ | (0.22 | ) | $ | (0.20 | ) | $ | (0.11 | ) | $ | (0.09 | ) | $ | (1.18 | ) | ||||||
| Shares used in computing basic and diluted net loss per common share |
83,623 | 76,353 | 65,763 | 49,923 | 40,063 | 47,345 | ||||||||||||||||||
| For the Years Ended December 31, | Accumulated from February 1, 2002 (Date of inception) to December 31, 2009 |
|||||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||||||
| Cash, cash equivalents and short term investments |
$ | 15,934 | $ | 13,562 | $ | 33,296 | $ | 3,055 | $ | 6,448 | $ | 15,934 | ||||||||||||
| Working capital |
14,524 | 12,166 | 31,271 | 2,153 | 5,954 | 14,524 | ||||||||||||||||||
| Total assets |
24,149 | 22,504 | 38,165 | 4,972 | 7,094 | 24,149 | ||||||||||||||||||
| Long-term obligations |
217 | 545 | 526 | 301 | 240 | 217 | ||||||||||||||||||
| Accumulated deficit |
(55,814 | ) | (39,450 | ) | (22,750 | ) | (9,849 | ) | (4,231 | ) | (55,814 | ) | ||||||||||||
| Total shareholders’ equity (deficit) |
21,896 | 19,632 | 34,922 | 3,496 | 6,201 | 21,896 | ||||||||||||||||||
32
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following should be read in conjunction with our consolidated financial statements located elsewhere in this Annual Report on Form 10-K for the year ended December 31, 2009 and other documents filed by us from time to time with the Securities and Exchange Commission. Statements made in this Item other than statements of historical fact, including statements about us and our subsidiaries and our respective clinical trials, research programs, product pipeline, current and potential corporate partnerships, licenses and intellectual property, the adequacy of capital reserves and anticipated operating results and cash expenditures, are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. As such, they are subject to a number of uncertainties that could cause actual results to differ materially from the statements made, including risks associated with the success of research and product development programs, the issuance and validity of patents, the development and protection of proprietary technologies, the ability to raise capital, operating expense levels and the ability to establish and retain corporate partnerships. Reference is made to discussions about risks associated with product development programs, intellectual property and other risks which may affect us under Item 1A, “Risk Factors” above. We do not undertake any obligation to update forward-looking statements.
Overview
Our company’s history began in February 2002 as a private company operating in California. Now Bionovo, Inc. is incorporated in the state of Delaware and was made public through a reverse merger transaction between Bionovo Biopharmaceuticals, Inc.1 and Lighten Up Enterprises International, Inc.2 on April 6, 2005. Immediately following the reverse merger, the Company operated as Lighten Up Enterprises International, Inc. until June 29, 2005 when the Company assumed the name, Bionovo, Inc.
On April 6, 2005, Lighten Up Enterprises International, Inc. acquired all the outstanding shares of Bionovo Biopharmaceuticals, Inc. in exchange for 42,112,448 restricted shares of its common stock in a reverse triangular merger. The acquisition was accounted for as a reverse merger in which Bionovo Biopharmaceuticals, Inc. was deemed to be the accounting acquirer. Accordingly, the historical financial information presented herein is that of Bionovo Biopharmaceuticals, Inc. (as adjusted for any difference in the par value of each entity’s stock with an offset to capital in excess of par value). Financial information subsequent to the reverse merger is that of the merged entity, Bionovo, Inc.
Business
We are a clinical stage drug discovery and development company focusing on women’s health and cancer, two large markets with significant unmet needs. Building on our understanding of the biology of menopause and cancer, we design new drugs derived from botanical sources which have novel mechanisms of action. Based on the results of our clinical trials and preclinical studies to date, we believe that we have discovered new classes of drug candidates with the potential to be leaders in their markets.
Our lead drug candidate, Menerba (formerly MF-101), represents a new class of receptor sub-type selective estrogen receptor modulator (SERM), for the treatment of vasomotor symptoms of menopause, or “hot flashes.” We have designed Menerba to selectively modulate estrogen receptor beta (ERß) and to provide a safe and effective alternative to existing FDA approved therapies that pose a significant risk to women for developing breast cancer, stroke, cardiovascular disease, blood clots and other serious diseases. In preclinical studies, Menerba does not lead to tumor formation in either breast or uterine tissues and it does not increase the risk for clotting. This activity, if confirmed in clinical testing, would differentiate Menerba from some existing therapies and other therapies in clinical development. We announced results in 2007 of our completed, multicenter,
| 1 | Bionovo Biopharmaceuticals, Inc. was originally formed as “Bionovo, Inc.” and began operations in the state of California in February 2002. In March of 2004, it reincorporated in the state of Delaware and in June of 2005 changed its name from Bionovo, Inc. to Bionovo Pharmaceuticals, Inc. |
| 2 | Lighten Up Enterprises International, Inc. was incorporated in Nevada in January of 1998. |
33
Table of Contents
randomized, double-blinded, placebo-controlled Phase 1 trial, involving 217 women, which showed the higher of two Menerba doses tested was well tolerated and resulted in a statistically significant reduction of hot flashes after 12 weeks of treatment. In addition, treatment with the higher dose of Menerba lead to a statistically significant reduction in nighttime awakenings when compared to placebo, which represents superior efficacy to existing non-hormonal therapies. We are seeking FDA approval to conduct further clinical testing at multiple clinical sites in the U.S. We believe that Menerba’s novel mechanism of action could lead to a more favorable safety profile than currently approved hormone therapies (HT) and other therapies under development and testing.
We are also developing Bezielle (previously referred to as “BZL101”), an oral anticancer agent for advanced breast cancer. Unlike most other anticancer drugs and drug candidates, which try to control cancer through genomic and proteomic signaling pathways, Bezielle is designed to take advantage of the unique metabolism of cancer cells. Bezielle inhibits glycolysis, a metabolic pathway on which cancer cells rely. Glycolysis inhibition leads to DNA damage and death of cancer cells without harm to normal cells. We believe that Bezielle may have a preferential effect on hormone-independent cancers, a subset with few treatment options. To date, 48 women with metastatic breast cancer have been treated with Bezielle in two separate Phase 1 clinical trials. As predicted by the mechanism of action, Bezielle had very limited toxicities with an extremely favorable tolerability profile. Moreover, Bezielle showed encouraging clinical activity among a cohort of women with metastatic breast cancer who had been heavily pretreated with other regimens. A Phase 2 trial has been approved by the FDA and the IRBs at several prestigious breast cancer clinical sites in the U.S. Bionovo is awaiting funding to commence this open-label, non-randomized trial in 80 women diagnosed with metastatic breast cancer who have failed no more than two prior chemotherapy regimens. We plan to evaluate Bezielle for the treatment of other forms of cancer, including pancreatic cancer and adjuvant use in breast cancer.
We have a diverse pipeline of additional preclinical drug candidates in both women’s health and cancer, including our second women’s health drug candidate, Seala (formerly referred to as “VG101”), for the treatment of postmenopausal vulvar and vaginal atrophy, or “vaginal dryness”.
We have identified or begun preclinical work on other drug candidates for a variety of indications within women’s health and cancer. We have internally discovered and developed all of our drug candidates using our proprietary biological and chemical techniques.
Our drug development process targets herbs and other botanical sources, used in Traditional Chinese Medicine, believed to produce biologically active compounds. We apply our clinical knowledge, experience with natural compounds and knowledge of proper scientific screening tools to isolate and purify botanical compounds and extracts for pharmaceutical development. In June 2004, the FDA released a document to provide drug developers with guidance on the approval for botanical drugs. This guidance states that applicants may submit reduced documentation of nonclinical (preclinical) safety to support an IND application for initial clinical studies of botanicals. The first botanical extract drug developed pursuant to these guidelines was approved by the FDA in October 2006. To date, all of our drug candidates are derived from botanical extracts and are being developed in accordance with this FDA guidance. In addition, we have identified the active chemical components underpinning the mechanism of action for our novel drugs, and in some cases, we have developed synthetic methods of production.
We expect to continue to incur significant operating losses for the foreseeable future, and do not expect to generate profits until and unless our drug candidates have been approved and are being marketed with commercial partners. As of December 31, 2009, we had an accumulated deficit of $55.8 million. Historically we have funded our operations primarily through private placements and public offerings of our capital stock, equipment lease financings, license fees and interest earned on investments.
We will need to raise and are pursuing additional funds through grants, strategic collaborations, public or private equity or debt financing, or other funding sources. This funding may not be available on acceptable terms, or at all, and may dilutive to shareholder interests.
34
Table of Contents
Development Stage Company
We have not generated any significant revenue since inception. The accompanying financial statements have, therefore, been prepared using the accounting formats prescribed for a development stage enterprise (DSE). Although the Company has recognized some nominal amount of revenue, the Company still believes it is devoting, substantially, all its efforts on developing the business and, therefore, still qualifies as a DSE.
The Company is primarily engaged in the development of pharmaceuticals, derived from botanical sources, to treat cancer and women’s health. The initial focus of the Company’s research and development efforts will be the generation of products for the treatment of breast and other cancers, and to alleviate the symptoms of menopause. The production and marketing of the Company’s products and its ongoing research and development activities are and will continue to be subject to extensive regulation by numerous governmental authorities in the United States. Prior to marketing in the United States, any drug developed by the Company must undergo rigorous preclinical and clinical testing and an extensive regulatory approval process implemented by the Food and Drug Administration (FDA) under the Food, Drug and Cosmetic Act. The Company has limited experience in conducting and managing the preclinical and clinical testing necessary to obtain regulatory approval. There can be no assurance that the Company will not encounter problems in clinical trials that will cause the Company or the FDA to delay or suspend clinical trials.
The Company’s success will depend in part on its ability to obtain patents and product license rights, maintain trade secrets, and operate without infringing on the proprietary rights of others, both in the United States and other countries. There can be no assurance that patents issued to the Company will not be challenged, invalidated, or circumvented, or that the rights granted thereunder will provide proprietary protection or competitive advantage resulting in the Company’s transition from Development Stage Enterprise reporting.
Critical Accounting Policies and Estimates
The accompanying discussion and analysis of our financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires management to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses, and related disclosures. On an ongoing basis, we evaluate these estimates, including those related to clinical trial accruals, income taxes (including the valuation allowance for deferred tax assets), restructuring costs and share-based compensation. Estimates are based on historical experience, information received from third parties and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could therefore differ materially from those estimates under different assumptions or conditions.
We consider the following accounting policies related to revenue recognition, clinical trial accruals, share-based compensation, income tax and research and development expenses to be the most critical accounting policies, because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
Our revenues are derived from collaborative research and development arrangements, technology licenses, and government grants.
Revenue is recognized when the four basic criteria of revenue recognition are met: (i) a contractual agreement exists; (ii) transfer of technology has been completed or services have been rendered; (iii) the fee is fixed or determinable, and (iv) collectability is reasonably assured.
35
Table of Contents
Revenue from technology licenses or other payments under collaborative agreements where we have a continuing obligation to perform is recognized as revenue over the expected period of the continuing performance obligation.
Revenue on government contracts is recognized on a qualified cost reimbursement basis.
Accruals
As part of the process of preparing financial statements, we are required to estimate accrued expenses. This process involves communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. We periodically confirm the accuracy of our estimates with selected service providers and make adjustments, if necessary. Examples of estimated accrued expenses include:
| • | fees paid to contract research organizations in connection with preclinical and toxicology studies and clinical trials; |
| • | fees paid to investigative sites in connection with clinical trials; |
| • | fees paid to contract manufacturers in connection with the production of clinical trial materials; and |
| • | professional service fees. |
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
Share-Based Compensation
Share-based compensation to outside consultants is recorded at fair market value in general and administrative expense. We record expenses relating to stock options granted to employees based on the fair value at the time of grant. The fair value of stock options and warrants is calculated using the Black-Scholes pricing method on the date of grant. This option valuation model requires input of highly subjective assumptions. Because employee stock options have characteristics significantly different from those of traded options, and because changes in the subjective input assumptions can materially affect the fair value estimate, in our management’s opinion, the existing model does not necessarily provide a reliable single measure of fair value of our employee stock options.
In order to determine the fair value of our stock for periods prior to the date of our reverse merger transaction, we estimated the fair value per share by reviewing values of other development stage biopharmaceutical organizations, comparing products in development, status of clinical trials, and capital received from government and private organizations. Once a total value was determined, we then factored the number of shares outstanding, or possibly outstanding, resulting in an estimated value per share. Once we completed our reverse merger transaction on April 6, 2005, the trading price of our common stock was used.
36
Table of Contents
For periods prior to our reverse merger transaction, we chose not to obtain contemporaneous valuations of our stock by any unrelated valuation specialist after realizing the cost of services would be substantial and that the benefit derived would not be substantially different from our estimate as we had used a multi-tiered approach to estimate the value of our stock.
Income Tax
We file income tax returns in the U.S. federal jurisdiction and the California state jurisdiction. To date, we have not been audited by the Internal Revenue Service or any state income tax jurisdiction. Our policy is to recognize interest and penalties related to the underpayment of income taxes as a component of income tax expense. As of December 31, 2009, there have been no interest or penalties charged to us in relation to the underpayment of income taxes.
We generated net losses since inception through the year ended December 31, 2009 and accordingly did not record a provision for federal income taxes. Deferred tax assets of $21.8 million as of December 31, 2009 were primarily comprised of federal and state tax net operating loss, or NOL, carryforwards. Due to uncertainties surrounding our ability to continue to generate future taxable income to realize these tax assets, a full valuation allowance has been established to offset our deferred tax assets. Additionally, the future utilization of our NOL carry-forwards to offset future taxable income may be subject to an annual limitation as a result of ownership changes that may have occurred previously or that could occur in the future. We have not yet determined whether such an ownership change has occurred. If necessary, the deferred tax assets will be reduced by any carryforwards that expire prior to utilization as a result of such limitations, with a corresponding reduction of the valuation allowance.
Research and Development Activities
Research and development costs are charged to expense when incurred. The major components of research and development costs include clinical manufacturing costs, pre-clinical and clinical trial expenses, consulting and other third-party costs, salaries and employee benefits, share-based compensation expense, supplies and materials, and facilities costs.
Our cost accruals for clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with numerous clinical trial sites and clinical research organizations. In the normal course of business we contract with third parties to perform various clinical trial activities in the on-going development of potential products. The financial terms of these agreements are subject to negotiation and variation from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the achievement of certain events, the successful enrollment of patients, and the completion of portions of the clinical trial or similar conditions. The objective of our accrual policy is to match the recording of expenses in our financial statements to the actual services received and efforts expended. As such, expense accruals related to clinical trials are recognized based on our estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract. We monitor service provider activities to the extent possible; however, if we underestimate activity levels associated with various studies at a given point in time, we could record significant research and development expenses in future periods.
During the years ended December 31, 2009, 2008 and 2007, we incurred research and development expenses of $12.5 million, $11.4 million and $9.9 million, respectively. We expect that research and development expenses will continue to increase as we continue our development of Menerba, Bezielle and other drug candidates.
Most of our product development programs are at an early stage. Accordingly, the successful development of our product candidates is highly uncertain and may not result in approved products. Completion dates and completion costs can vary significantly for each product candidate and are difficult to predict. Product candidates that may appear promising at early stages of development may not reach the market for a number of reasons.
37
Table of Contents
Product candidates may be found ineffective or cause harmful side effects during clinical trials, may take longer to progress through clinical trials than anticipated, may fail to receive necessary regulatory approvals and may prove impracticable to manufacture in commercial quantities at a reasonable cost and with acceptable quality. The lengthy process of seeking FDA approvals requires the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining regulatory approvals could adversely affect our product development efforts. Because of these risks and uncertainties, we cannot predict when or whether we will successfully complete the development of our product candidates or the ultimate product development cost or whether we will obtain any approval required by the FDA on a timely basis, if at all.
Results of Operations
Results of Operations for the Years Ended December 31, 2009, 2008, and 2007
Revenues
| Years Ended December 31, | % Increase (Decrease) | ||||||||||||||
| 2009 | 2008 | 2007 | 2008 to 2009 |
2007 to 2008 |
|||||||||||
| (in thousands) | |||||||||||||||
| NIH grant |
$ | 233 | $ | 233 | $ | 479 | 0 | % | -51 | % | |||||
| Licensing |
— | — | 103 | — | -100 | % | |||||||||
| Service revenue |
55 | — | — | * | * | ||||||||||
| Total |
$ | 288 | $ | 233 | $ | 582 | 24 | % | -60 | % | |||||
Revenues over the fiscal years 2009, 2008 and 2007 consist primarily of licensing fees and proceeds from a US government grant. Total revenue in 2009 is greater than 2008 due to limited research services provided to one customer. We expect to continue performing the research services into 2010; however, the total revenue recognized is not expected to be significant to our operations. While the National Institute of Health (NIH) grant revenue was consistent in 2009 and 2008, the grant is scheduled to expire in 2010 and we do not expect revenues from this grant to continue.
Revenues were $0.2 million in 2008, compared to $0.6 million in 2007. The $0.4 million decrease in 2008 from 2007 was due to a decrease in proceeds from the NIH grant activity and the termination of our licensing and technology transfer agreement with a Taiwanese company in 2007, which resulted in the recognition of $0.1 million in revenue in 2007 from the remaining deferred revenue balance.
Research and Development Expenses
| Years Ended December 31, | % Increase (Decrease) | ||||||||||||||
| 2009 | 2008 | 2007 | 2008 to 2009 |
2007 to 2008 |
|||||||||||
| (in thousands) | |||||||||||||||
| Research and development expenses |
$ | 12,499 | $ | 11,416 | $ | 9,938 | 9 | % | 15 | % | |||||
Research and development (R&D) expenses reflect costs for the development of drugs and include salaries, contractor and consultant fees and other support costs, including employee share-based compensation expense. R&D expenses were $12.5 million in 2009 compared with $11.4 million in 2008. The increase of $1.1 million is primarily due to increases in R&D related depreciation, facilities and information technology charges combined with an increase in purchases of lab supplies and raw materials to support the Menerba manufacturing process development, partially offset by decreases in our clinical trial expenses.
R&D expenses were $11.4 million in 2008 compared to $9.9 million in 2007. The increase of $1.5 million in 2008 from 2007 is due primarily to clinical trials expenses related to the development of our lead drug candidates.
38
Table of Contents
General and Administrative Expenses
| Years Ended December 31, | % Increase (Decrease) | ||||||||||||||
| 2009 | 2008 | 2007 | 2008 to 2009 |
2007 to 2008 |
|||||||||||
| (in thousands) | |||||||||||||||
| General and administrative expense |
$ | 4,053 | $ | 6,097 | $ | 4,284 | -34 | % | 42 | % | |||||
General and administrative (G&A) expense includes personnel costs for finance, administration, information systems, and public relations related to our drug development and clinical trials, participation in conventions and tradeshows, website related expenses, facilities expenses, professional fees, legal expenses, and other administrative costs. We do not currently have any dedicated sales support or personnel. The $2.0 million decrease in G&A expenses in 2009 compared to 2008 was primarily due to decreases in G&A related depreciation, facilities and information technology expenses caused by a decrease in the proportion of office to laboratory space as compared with our old location and the implementation of cost reduction initiatives to preserve our cash position.
The $1.8 million increase in G&A expenses in 2008 as compared to 2007 was primarily due to activities in support of our business including an increase in facility rental, legal expenses and an increase in payroll expenses related to an increase in G&A management personnel.
Interest Income, Interest Expense and Other Expense
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||
| 2009 | 2008 | 2007 | 2008 to 2009 |
2007 to 2008 |
||||||||||||||
| (in thousands) | ||||||||||||||||||
| Interest income |
$ | 84 | $ | 730 | $ | 850 | -88 | % | -14 | % | ||||||||
| Interest expense |
(95 | ) | (129 | ) | (87 | ) | -26 | % | 48 | % | ||||||||
| Other expense, net |
(88 | ) | (17 | ) | (21 | ) | 418 | % | -19 | % | ||||||||
| Total |
$ | (99 | ) | $ | 584 | $ | 742 | -117 | % | -21 | % | |||||||
Interest income is derived from cash balances and short term investments. Interest income was $0.1 million in 2009 compared to $0.7 million in 2008 and $0.9 million in 2007. The decreases in both years reflect lower average cash balances. We expect interest income to increase in 2010 due to our increase in cash balance from our financing transaction in October 2009.
Interest expense includes interest expense from capital lease agreements for our laboratory equipment and two notes payable to finance $0.2 million of leasehold improvements made in 2009.
The increase in other expense from 2008 to 2009 is due to approximately $91,000 in loss on fixed asset disposal primarily associated with the move to our new offices.
Liquidity and Capital Resources
Since our inception, we have incurred losses, and we have relied primarily on leasing and public and private financing to fund our operations.
As of December 31, 2009 we had cash, cash equivalents and short term investments of $15.9 million compared to $13.6 million at December 31, 2008. This increase in cash, cash equivalents, and short term investments for the year ended December 31, 2009 is primarily due to $17.4 million in net proceeds from our financing transaction in October 2009 partially offset by net cash used in operating activities of $13.3 million for the twelve months ended December 31, 2009, $0.9 million in expenditures for capital assets and patent costs, and $0.7 million for payments on our capital lease agreements and notes payable.
39
Table of Contents
Net cash used by investing activities was $3.9 million for the year ended December 31, 2009 compared to net cash used in investing activities of $9.1 million for the year ended December 31, 2008. The decrease of $5.2 million was due to $2.6 million decrease in capital expenditures combined with $2.5 million decrease in cash used in short-term investment activities. The decrease in capital expenditures in 2009 was primarily due to leasehold improvement expenditures in 2008 in preparation for the move to our new headquarters (completed in January 2009). The decrease in net cash used in short-term investing activities was due to lower cash and short-term investment balances throughout 2009 as compared with 2008.
Net cash provided by financing activities in 2009 was $16.8 million compared with net cash used by financing activities of $0.9 million in 2008 due to our public offering completed on October 7, 2009. We did not have any equity financing transactions in 2008.
Registered Public Offering
On October 7, 2009, we completed a registered public offering and issued 30,933,140 shares of our common stock and 29,324,570 warrants. In addition, we granted the placement agent 1,546,657 warrants, each warrant entitling the holder to purchase one share of common stock at $0.97 per share at any time for a period commencing six months after the date of the closing, and continuing for five years following the closing date of the offering. The warrants may also be executed on a cashless basis, in accordance with the terms of the warrant. The offering resulted in gross proceeds of $19.1 million and we received net proceeds of $17.4 million after related issuance costs.
We will need to obtain substantial amounts of cash to achieve our objectives of internally developing drugs, which take many years and potentially significantly more amounts of cash to develop. If we decide to market and commercialize any other drug candidate independently or with a partner, we may need to invest heavily in associated manufacturing, marketing and commercialization costs. Such costs will be substantial and some will need to be incurred prior to receiving marketing approval. We do not currently have adequate internal liquidity to meet these objectives in the long term. To do so, we will need to continue our partnering activities and look to other external sources of liquidity, including the public and private financial markets and strategic partners.
As of December 31, 2009, we had an accumulated deficit of $55.8 million, working capital of $14.5 million, and shareholders’ equity of $21.9 million.
We expect that we will need to raise substantial additional capital to fully pursue our business plan before the end of 2010. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our development programs. We are currently pursuing a variety of funding options, including government grants, partnering/co-investment, venture debt and equity offerings.
The length of time that our current cash and cash equivalents, short-term investments and any available borrowings will sustain our operations will be based on, among other things, our prioritization decisions regarding funding for our programs, our progress in our clinical and earlier-stage programs, the time and costs related to current and future clinical trials and regulatory decisions, our research, development, manufacturing and commercialization costs (including personnel costs), the progress in our collaborations, costs associated with intellectual property, our capital expenditures, and costs associated with securing any future licensing opportunities. Based on the proceeds of our recent public offering, funding we expect to be available to us, and our ability to prioritize spending, we believe we have sufficient cash to meet our operating needs for at least the next 12 months.
Notes Payable
In connection with the move to our new headquarters, we financed leasehold improvements under two notes payable with the lessor of the building. The first note of $100,000 bears interest at 9.5% and is payable in
40
Table of Contents
monthly payments of interest and principal amounting to approximately $1,000 beginning May 1, 2009 and continuing over the remaining lease term with a due date of December 31, 2018. The second note of $104,000 bears interest at 9.5% and is payable in 24 monthly payments of interest and principal amounting to approximately $5,000 beginning August 1, 2009. The future minimum payments as of December 31, 2009 are as follows (in thousands):
| Principal | Interest | |||||
| 2010 |
59 | 15 | ||||
| 2011 |
40 | 9 | ||||
| 2012 |
9 | 7 | ||||
| 2013 |
9 | 6 | ||||
| 2014 |
10 | 6 | ||||
| Thereafter |
53 | 11 | ||||
| Total minimum payments |
$ | 180 | $ | 54 | ||
Commitments and Contingencies
Our contractual obligations and future minimum lease payments that are non-cancelable at December 31, 2009 are disclosed in the following table (in thousands).
| Total | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 and beyond | |||||||||||||||
| Operating lease obligations |
$ | 18,460 | $ | 1,850 | $ | 1,868 | $ | 1,924 | $ | 1,982 | $ | 2,041 | $ | 8,795 | |||||||
| Notes payable |
234 | 74 | 49 | 16 | 15 | 16 | 64 | ||||||||||||||
| Capital lease obligations |
602 | 501 | 94 | 7 | — | — | — | ||||||||||||||
| Total contractual commitments |
$ | 19,296 | $ | 2,425 | $ | 2,011 | $ | 1,947 | $ | 1,997 | $ | 2,057 | $ | 8,859 | |||||||
Recent Accounting Pronouncements
In June of 2009, the FASB issued guidance now codified as FASB ASC Topic 105, “Generally Accepted Accounting Principles,” as the single source of authoritative nongovernmental U.S. GAAP. FASB ASC Topic 105 does not change current U.S. GAAP, but is intended to simplify user access to all authoritative U.S. GAAP by providing all authoritative literature related to a particular topic in one place. All existing accounting standard documents will be superseded and all other accounting literature not included in the FASB Codification will be considered non-authoritative. These provisions of FASB ASC Topic 105 are effective for interim and annual periods ending after September 15, 2009 and, accordingly, are effective for the Company for the current fiscal reporting period. The adoption of this pronouncement did not have an impact on our financial condition or results of operations, but will impact our financial reporting process by eliminating all references to pre-codification standards. On its effective date, the Codification superseded all then-existing non-SEC accounting and reporting standards, and all other non-grandfathered non-SEC accounting literature not included in the Codification became non-authoritative.
In April of 2009, the FASB issued guidance in the Fair Value Measurements and Disclosures Topic of the Codification on determining fair value when the volume and level of activity for an asset or liability have significantly decreased and identifying transactions that are not orderly. The guidance emphasizes that even if there has been a significant decrease in the volume and level of activity, the objective of a fair value measurement remains the same. Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction (that is, not a forced liquidation or distressed sale) between market participants. The guidance provides a number of factors to consider when evaluating whether there has been a significant decrease in the volume and level of activity for an asset or liability in relation to normal market
41
Table of Contents
activity. In addition, when transactions or quoted prices are not considered orderly, adjustments to those prices based on the weight of available information may be needed to determine the appropriate fair value. The guidance is effective for interim or annual reporting periods ending after June 15, 2009, and shall be applied prospectively. We adopted this guidance effective for the quarter ending June 30, 2009 and there is no impact of the adoption on our financial statements as of December 31, 2009.
In April of 2009, FASB issued guidance in the Financial Instruments Topic of the Codification on interim disclosures about fair value of financial instruments. The guidance requires disclosures about the fair value of financial instruments for both interim reporting periods, as well as annual reporting periods. The guidance is effective for all interim and annual reporting periods ending after June 15, 2009 and shall be applied prospectively. The adoption of this guidance had no impact on our financial statements as of December 31, 2009.
In May of 2009, the FASB issued SFAS No. 165, Subsequent Events (ASC Topic 855). This guidance establishes general standards of accounting for and disclosures of events that occur after the balance sheet date but before financial statements are issued or are available to be issued. This guidance was effective for us in the second quarter of 2009 and its adoption did not have a material impact on our consolidated financial statements. On February 24, 2010, the FASB issued ASU No. 2010-09 amending Topic 855 to no longer require entities to disclose the date through which management evaluated subsequent events in the financial statements. The update was effective upon release and the adoption of the update did not have a material impact on our financial statements as of December 31, 2009.
In October of 2009, the FASB issued the following ASU No. 2009-13, Revenue Recognition (ASC Topic 605)—Multiple-Deliverable Revenue Arrangements, a consensus of the FASB Emerging Issues Task Force. This guidance modifies the fair value requirements of ASC subtopic 605-25 Revenue Recognition-Multiple Element Arrangements by allowing the use of the “best estimate of selling price” for determining the selling price of a deliverable. A vendor is now required to use its best estimate of the selling price when vendor specific objective evidence or third-party evidence of the selling price cannot be determined. In addition, the residual method of allocating arrangement consideration is no longer permitted. This guidance is effective in 2011. We are currently evaluating the impact of adopting this update on its consolidated financial statements.
In January of 2010, the FASB issued an amendment to the accounting standards related to the disclosures about an entity’s use of fair value measurements. Among these amendments, entities will be required to provide enhanced disclosures about transfers into and out of the Level 1 (fair value determined based on quoted prices in active markets for identical assets and liabilities) and Level 2 (fair value determined based on significant other observable inputs) classifications, provide separate disclosures about purchases, sales, issuances and settlements relating to the tabular reconciliation of beginning and ending balances of the Level 3 (fair value determined based on significant unobservable inputs) classification and provide greater disaggregation for each class of assets and liabilities that use fair value measurements. Except for the detailed Level 3 roll-forward disclosures, the new standard is effective for interim and annual reporting periods beginning after December 31, 2009. The requirement to provide detailed disclosures about the purchases, sales, issuances and settlements in the roll-forward activity for Level 3 fair value measurements is effective for interim and annual reporting periods beginning after December 31, 2010. We do not expect that the adoption of this new standard will have a material impact to our consolidated financial statements.
Off-Balance Sheet Financings and Liabilities
Other than contractual obligations incurred in the normal course of business, we do not have any off-balance sheet financing arrangements or liabilities, guarantee contracts, retained or contingent interests in transferred assets or any obligation arising out of a material variable interest in an unconsolidated entity.
Subsequent Events
On January 12, 2010, we granted approximately 657,000 options to employees in lieu of cash bonuses based on fiscal year 2009 performance. The options were fully vested upon grant date and approved by the Company’s
42
Table of Contents
Board of Directors in December of 2009. Accordingly, we have included an accrual of the related share-based compensation expense in the financial statements as of December 31, 2009 of approximately $0.2 million, which is the estimated fair value as of the reporting date.
In February of 2010, we reached a settlement agreement with a former officer of the Company who filed a complaint against Bionovo, Inc. and two of its officers, alleging breach of contract resulting from the rescission of the former officer’s employment agreement with the Company in September 2007. The settlement amount is included as an expense accrual in our financial statements for the year ended December 31, 2009; however, the amount did not have a material impact on our financial position, results of operations or cash flows.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
In the normal course of business, our financial position is routinely subject to a variety of risks, including market risk associated with interest rate movement. We regularly assess these risks and have established policies and business practices intended to protect against these and other exposures. As a result, we do not anticipate material potential losses in these areas.
As of December 31, 2009, we had cash, cash equivalents, and short-term investments of $15.9 million, consisting of cash, cash equivalents and highly liquid short-term investments. Our short-term investments will likely decline by an immaterial amount if market interest rates increase and, therefore, we believe our exposure to interest rate changes is immaterial. Declines of interest rates over time will however, reduce our interest income from short-term investments.
43
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Bionovo, Inc.
We have audited the accompanying consolidated balance sheets of Bionovo, Inc., a development stage company, as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’ equity and cash flows for the years ended December 31, 2009, 2008, 2007 and the period from inception (February 1, 2002) to December 31, 2009. Bionovo, Inc.’s management is responsible for these consolidated financial statements. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of Bionovo, Inc. as of December 31, 2009 and 2008 and the consolidated results of its operations and its consolidated cash flows for each of the years in the three-year period ended December 31, 2009, 2008, 2007 and the period from inception (February 1, 2002) to December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
| /s/ PMB Helin Donovan, LLP |
| PMB Helin Donovan, LLP |
| San Francisco, California |
March 15, 2010
44
Table of Contents
(A Development Stage Company)
Consolidated Balance Sheets
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (in thousands) | ||||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 2,799 | $ | 3,270 | ||||
| Short-term investments |
13,135 | 10,292 | ||||||
| Receivables |
251 | 126 | ||||||
| Prepaid expenses |
214 | 334 | ||||||
| Other current assets |
161 | 471 | ||||||
| Total current assets |
16,560 | 14,493 | ||||||
| Property and equipment, net |
6,197 | 6,938 | ||||||
| Patent pending, net |
822 | 575 | ||||||
| Other assets |
570 | 498 | ||||||
| Total assets |
$ | 24,149 | $ | 22,504 | ||||
| LIABILITIES AND SHAREHOLDERS’ EQUITY | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 311 | $ | 521 | ||||
| Accrued compensation and benefits |
367 | 456 | ||||||
| Current portion of lease obligations |
476 | 682 | ||||||
| Current portion of notes payable |
59 | — | ||||||
| Other current liabilities |
823 | 668 | ||||||
| Total current liabilities |
2,036 | 2,327 | ||||||
| Non-current portion of lease obligations |
96 | 545 | ||||||
| Non-current portion of notes payable |
121 | — | ||||||
| Total liabilities |
2,253 | 2,872 | ||||||
| Commitments and contingencies |
||||||||
| Shareholders’ equity: |
||||||||
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized; none issued and outstanding |
— | — | ||||||
| Common stock $0.0001 par value, 190,000,000 shares authorized, 107,518,690 and 76,363,101 shares outstanding at December 31, 2009 and 2008, respectively |
11 | 8 | ||||||
| Additional paid-in capital |
77,704 | 59,050 | ||||||
| Accumulated other comprehensive (loss) income |
(5 | ) | 24 | |||||
| Accumulated deficit |
(55,814 | ) | (39,450 | ) | ||||
| Total shareholders’ equity |
21,896 | 19,632 | ||||||
| Total liabilities and shareholders’ equity |
$ | 24,149 | $ | 22,504 | ||||
See accompanying notes to these consolidated financial statements
45
Table of Contents
(A Development Stage Company)
Consolidated Statements of Operations
| For the Years Ended December 31, | Accumulated from February 1, 2002 (Date of inception) to December 31, 2009 |
|||||||||||||||
| 2009 | 2008 | 2007 | ||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenues |
$ | 288 | $ | 233 | $ | 582 | $ | 1,180 | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
12,499 | 11,416 | 9,938 | 39,706 | ||||||||||||
| General and administrative |
4,053 | 6,097 | 4,284 | 17,602 | ||||||||||||
| Merger cost |
— | — | — | 1,964 | ||||||||||||
| Total operating expenses |
16,552 | 17,513 | 14,222 | 59,272 | ||||||||||||
| Loss from operations |
(16,264 | ) | (17,280 | ) | (13,640 | ) | (58,092 | ) | ||||||||
| Change in fair value of warrant liability |
— | — | — | 831 | ||||||||||||
| Interest income |
84 | 730 | 850 | 2,074 | ||||||||||||
| Interest expense |
(95 | ) | (129 | ) | (87 | ) | (460 | ) | ||||||||
| Other expense |
(88 | ) | (17 | ) | (21 | ) | (153 | ) | ||||||||
| Loss before income tax |
(16,363 | ) | (16,696 | ) | (12,898 | ) | (55,800 | ) | ||||||||
| Income tax provision |
(1 | ) | (4 | ) | (3 | ) | (14 | ) | ||||||||
| Net loss |
$ | (16,364 | ) | $ | (16,700 | ) | $ | (12,901 | ) | $ | (55,814 | ) | ||||
| Basic and diluted net loss per common share |
$ | (0.20 | ) | $ | (0.22 | ) | $ | (0.20 | ) | $ | (1.18 | ) | ||||
| Shares used in computing basic and diluted net loss per share |
83,623 | 76,353 | 65,763 | 47,345 | ||||||||||||
See accompanying notes to these consolidated financial statements
46
Table of Contents
(A Development Stage Company)
Consolidated Statements of Cash Flow
| For the Years Ended December 31, | Accumulated from February 1, 2002 (Date of inception) to December 31, 2009 |
|||||||||||||||
| 2009 | 2008 | 2007 | ||||||||||||||
| Cash flows from operating activities: |
||||||||||||||||
| Net loss |
$ | (16,364 | ) | $ | (16,700 | ) | $ | (12,901 | ) | $ | (55,814 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||||||
| Depreciation and amortization |
1,578 | 1,076 | 738 | 3,680 | ||||||||||||
| Stock-based compensation expense |
1,186 | 1,371 | 1,280 | 4,467 | ||||||||||||
| Amortization and accretion of investments |
79 | 67 | (5 | ) | 141 | |||||||||||
| Non-cash compensation for warrants issued |
— | — | — | 1,964 | ||||||||||||
| Amortization of note discount |
— | — | — | 139 | ||||||||||||
| Amortization of deferred stock compensation |
— | — | — | 16 | ||||||||||||
| Change in fair value of warrrant liability |
— | — | — | (831 | ) | |||||||||||
| Loss on disposal of property and equipment |
91 | 2 | 7 | 113 | ||||||||||||
| Noncash adjustment to available-for-sale securities |
(1 | ) | — | — | (1 | ) | ||||||||||
| Changes in assets and liabilities: |
||||||||||||||||
| Receivables |
(125 | ) | 160 | (284 | ) | (251 | ) | |||||||||
| Prepaid expenses and other current assets |
430 | (400 | ) | (222 | ) | (375 | ) | |||||||||
| Deposits and other non-current assets |
(75 | ) | (467 | ) | 5 | (537 | ) | |||||||||
| Accounts payable |
(210 | ) | 221 | (247 | ) | 311 | ||||||||||
| Accrued compensation and benefits |
(89 | ) | 8 | 348 | 367 | |||||||||||
| Other accrued liabilities |
155 | (593 | ) | 1,130 | 824 | |||||||||||
| Deferred revenue |
— | — | (103 | ) | — | |||||||||||
| Net cash used in operating activities |
(13,345 | ) | (15,255 | ) | (10,254 | ) | (45,787 | ) | ||||||||
| Cash flows from investing activities: |
||||||||||||||||
| Capital expenditures |
(629 | ) | (3,211 | ) | (1,744 | ) | (6,596 | ) | ||||||||
| Acquisition of intangible assets |
(299 | ) | (362 | ) | (242 | ) | (966 | ) | ||||||||
| Proceeds from sales and maturities of investments |
10,235 | 10,520 | 493 | 27,494 | ||||||||||||
| Purchases of available-for-sale securities |
(13,185 | ) | (16,036 | ) | (4,823 | ) | (40,772 | ) | ||||||||
| Net cash used in investing activities |
(3,878 | ) | (9,089 | ) | (6,316 | ) | (20,840 | ) | ||||||||
| Cash flows from financing activities: |
||||||||||||||||
| Proceeds from issuance of common stock and warrants, net |
17,351 | — | 38,180 | 67,085 | ||||||||||||
| Payments under capital lease obligation |
(695 | ) | (876 | ) | (571 | ) | (2,498 | ) | ||||||||
| Payments under notes payable |
(24 | ) | — | — | (24 | ) | ||||||||||
| Proceeds from exercise of warrants and options |
120 | 18 | 4,862 | 5,000 | ||||||||||||
| Payments of convertible notes payable |
— | — | — | (50 | ) | |||||||||||
| Payments for financing costs for convertible notes |
— | — | — | (87 | ) | |||||||||||
| Net cash provided by (used in) financing activities |
16,752 | (858 | ) | 42,471 | 69,426 | |||||||||||
| Net (decrease) increase in cash and cash equivalents |
(471 | ) | (25,202 | ) | 25,901 | 2,799 | ||||||||||
| Cash and cash equivalents at the beginning of the period |
3,270 | 28,472 | 2,571 | — | ||||||||||||
| Cash and cash equivalents at the end of the period |
$ | 2,799 | $ | 3,270 | $ | 28,472 | $ | 2,799 | ||||||||
See accompanying notes to these consolidated financial statements
47
Table of Contents
(A Development Stage Company)
Statements of Shareholders’ Equity
| Common Stock | Additional Paid in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit Development Stage |
Total Sharesholders’ Equity |
||||||||||||||||
| Number of Shares |
Par | |||||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Balance at inception (February 1, 2002)—Adjusted to reflect effect of stock split on June 17, 2004 and March 4, 2004, and reverse merger on April 6, 2005 |
20,400 | $ | 2.0 | $ | (2 | ) | $ | — | $ | — | $ | — | ||||||||
| Balances at December 31, 2002 |
20,400 | 2.0 | (2 | ) | — | — | — | |||||||||||||
| Comprehensive loss: |
||||||||||||||||||||
| Net loss |
— | — | — | — | (56 | ) | (56 | ) | ||||||||||||
| Balances at December 31, 2003 |
20,400 | 2.0 | (2 | ) | — | (56 | ) | (56 | ) | |||||||||||
| Noncash compensation expense for options issued |
— | — | 30 | — | 30 | |||||||||||||||
| Net loss |
— | — | — | — | (538 | ) | (538 | ) | ||||||||||||
| Balances at December 31, 2004 |
20,400 | 2.0 | 28 | — | (594 | ) | (564.0 | ) | ||||||||||||
| Issuance of shares for reverse merger |
4,000 | 0.4 | — | — | — | 0.4 | ||||||||||||||
| Issuance of common stock for funds received by private placement net of financing cost |
20,461 | 2.1 | 9,930 | — | — | 9,932.1 | ||||||||||||||
| Issuance of common stock for conversion on notes payable |
1,251 | 0.1 | 462 | — | — | 462.1 | ||||||||||||||
| Amortization of deferred share-based compensation |
— | — | 16 | — | — | 16.0 | ||||||||||||||
| Net loss |
— | — | — | — | (3,637 | ) | (3,637.0 | ) | ||||||||||||
| Balances at December 31, 2005 |
46,112 | 4.6 | 10,436 | — | (4,231 | ) | 6,209.6 | |||||||||||||
| Issuance of common stock for exercise of warrants |
5,025 | 0.5 | 2,304 | — | — | 2,304.5 | ||||||||||||||
| Issuance of common stock for services |
200 | — | 165 | — | — | 165.0 | ||||||||||||||
| Amortization of deferred share-based compensation |
— | — | — | — | — | — | ||||||||||||||
| Share-based compensation |
— | — | 435 | — | — | 435.0 | ||||||||||||||
| Net loss |
— | — | — | — | (5,618 | ) | (5,618.0 | ) | ||||||||||||
| Balances at December 31, 2006 |
51,337 | $ | 5.1 | $ | 13,340 | $ | — | $ | (9,849 | ) | $ | 3,496 | ||||||||
| Comprehensive loss: |
||||||||||||||||||||
| Net loss |
— | (12,901 | ) | (12,901 | ) | |||||||||||||||
| Change in net unrealized gain on available for sale investments |
4 | — | 4 | |||||||||||||||||
| Total comprehensive loss |
(12,897 | ) | ||||||||||||||||||
| Issuance of common stock from private placement, net of financing cost |
10,521 | 1.1 | 14,505 | — | — | 14,506.1 | ||||||||||||||
| Issuance of common stock from public financing, net of financing cost |
10,000 | 1.0 | 23,674 | — | — | 23,675 | ||||||||||||||
| Issuance of common stock upon exercise of warrants |
4,108 | 0.4 | 4,537 | — | — | 4,537.4 | ||||||||||||||
| Issuance of common stock upon exercise of stock options |
377 | — | 325 | — | — | 325 | ||||||||||||||
| Share-based compensation related to issuance of stock option grants |
— | — | 1,280 | — | — | 1,280 | ||||||||||||||
| Balances at December 31, 2007 |
76,343 | 7.6 | 57,661 | 4 | (22,750 | ) | 34,923 | |||||||||||||
48
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Statements of Shareholders’ Equity—(Continued)
| Common Stock | Additional Paid in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit Development Stage |
Total Sharesholders’ Equity |
||||||||||||||||
| Number of Shares |
Par | |||||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Balances at December 31, 2007 |
76,343 | 7.6 | 57,661 | 4 | (22,750 | ) | 34,923 | |||||||||||||
| Comprehensive loss: |
||||||||||||||||||||
| Net loss |
— | (16,700 | ) | (16,700 | ) | |||||||||||||||
| Change in net unrealized gain on available for sale investments |
20 | — | 20 | |||||||||||||||||
| Total comprehensive loss |
— | — | — | — | — | (16,680 | ) | |||||||||||||
| Issuance of common stock upon exercise of stock options |
20 | — | 18 | — | — | 18 | ||||||||||||||
| Share-based compensation related to issuance of stock option grants |
— | — | 1,371 | — | — | 1,371 | ||||||||||||||
| Balances at December 31, 2008 |
76,363 | 7.6 | 59,050 | 24 | (39,450 | ) | 19,632 | |||||||||||||
| Comprehensive loss: |
||||||||||||||||||||
| Net loss |
— | (16,364 | ) | (16,364 | ) | |||||||||||||||
| Change in net unrealized gain on available for sale investments |
(29 | ) | — | (29 | ) | |||||||||||||||
| Total comprehensive loss |
— | — | — | — | — | (16,393 | ) | |||||||||||||
| Issuance of common stock upon exercise of warrants |
222 | — | 120 | — | — | 120 | ||||||||||||||
| Issuance of common stock from public financing, net of financing cost |
30,934 | 3.1 | 17,348 | — | — | 17,351 | ||||||||||||||
| Share-based compensation related to issuance of stock option grants |
— | — | 1,186 | — | — | 1,186 | ||||||||||||||
| Balances at December 31, 2009 |
107,519 | $ | 10.7 | $ | 77,704 | $ | (5 | ) | $ | (55,814 | ) | $ | 21,896 | |||||||
See accompanying notes to these consolidated financial statements
49
Table of Contents
(A Development Stage Company)
Notes to Consolidated Financial Statements
1. BUSINESS AND ORGANIZATION
Formation of the Company
The Company’s history began in February 2002 as a private company operating in California. Now Bionovo, Inc. is incorporated in the state of Delaware and was made public through a reverse merger transaction between Bionovo Biopharmaceuticals, Inc.1 and Lighten Up Enterprises International, Inc.2 on April 6, 2005. Immediately following the reverse merger, the Company operated as Lighten Up Enterprises International, Inc. until June 29, 2005 when the Company assumed the name, Bionovo, Inc.
Reverse Merger
On April 6, 2005, Lighten Up Enterprises International, Inc. acquired all the outstanding shares of Bionovo Biopharmaceuticals, Inc. in exchange for 42,112,448 restricted shares of its common stock in a reverse triangular merger. The acquisition was accounted for as a reverse merger in which Bionovo Biopharmaceuticals, Inc. was deemed to be the accounting acquirer. Accordingly, the historical financial information presented herein is that of Bionovo Biopharmaceuticals, Inc. (as adjusted for any difference in the par value of each entity’s stock with an offset to capital in excess of par value). Financial information subsequent to the reverse merger is that of the merged entity, Bionovo, Inc.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Development Stage Company
The Company has not generated any significant revenue since inception. Accordingly, the accompanying financial statements have been prepared using the accounting formats prescribed by the Development Stage Entity Topic of the Financial Accounting Standards Board (FASB) Codification for a development stage enterprise (DSE). Although the Company has recognized some nominal amount of revenue, the Company still believes it is devoting, substantially, all its efforts on developing the business and, therefore, still qualifies as a DSE.
Liquidity
The Company has sustained recurring losses and negative cash flows from operations. At December 31, 2009, the Company had an accumulated deficit of $55.8 million, working capital of $14.5 million and shareholders’ equity of $21.9 million. Over the past years, the Company’s growth has been funded through a combination of private equity, debt, lease financing and public offering. As of December 31, 2009, the Company had $2.8 million in cash and cash equivalents and $13.1 million in short-term securities, for a total of $15.9 million.
In October of 2009, the Company obtained additional financings of $17.4 million, net of financing cost, through the sale of its common stock and warrants. The Company believes that, as a result of this, it currently has sufficient cash and financing commitments to meet its funding requirements over the next twelve months.
However, the Company has experienced and continues to experience operating losses and negative cash flows from operations, as well as an ongoing requirement for substantial additional capital investment. The Company expects that it will need to raise substantial additional capital to fully pursue its business plan before
| 1 | Bionovo Biopharmaceuticals, Inc. was originally formed as “Bionovo, Inc.” and began operations in the state of California in February 2002. In March of 2004, it reincorporated in the state of Delaware and in June of 2005 changed its name from Bionovo, Inc. to Bionovo Pharmaceuticals, Inc. |
| 2 | Lighten Up Enterprises International, Inc. was incorporated in Nevada in January of 1998. |
50
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
the end of 2010. If adequate funds are not available, the Company may be required to delay, reduce the scope of, or eliminate one or more of its development programs. The Company is currently pursuing a variety of funding options, including government grants, partnering/co-investment, venture debt and equity offerings. There can be no assurance as to the availability or terms upon which such financing and capital might be available. If the Company is not successful in its efforts to raise additional funds by the first half of 2011, we may not be able to continue as a going concern.
Use of Estimates and Reclassifications
The preparation of consolidated financial statements, in conformity with accounting principles generally accepted in the United States of America, requires management to make estimates and assumptions that affect the amounts in the financial statements and accompanying notes. Actual results could differ from those estimates.
Management makes estimates that affect certain accounts including deferred income tax assets, estimated useful lives of property and equipment, accrued expenses, fair value of equity instruments and reserves for any other commitments or contingencies. Any adjustments applied to estimates are recognized in the year in which such adjustments are determined.
Certain reclassifications of prior period amounts have been made to our consolidated financial statements to conform to the current period presentation.
Cash, Cash Equivalents and Short-Term Investments
As part of our cash management program, the Company maintains a portfolio of marketable investment securities. The securities are investment grade and generally mature within one year and may include tax exempt securities, and certificates of deposit. Marketable securities that are bought and held principally for the purpose of selling them in the near term are classified as trading securities and are reported at fair value, with unrealized gains and losses recognized in earnings. The fair value of substantially all securities is determined by quoted market prices. The estimated fair value of securities for which there are no quoted market prices is based on similar types of securities that are traded in the market. The Company maintains its cash in bank accounts, which at times may exceed federally insured limits. The Company has not experienced any losses on such accounts. The Company considers all highly liquid investments purchased with an original maturity of three months or less at the time of purchase to be cash equivalents.
Property and Equipment
Property and equipment are recorded at cost. Expenditures for major additions and improvements are capitalized, and minor replacements, maintenance, and repairs are charged to expense as incurred. When property and equipment are retired or otherwise disposed of, the cost and accumulated depreciation are removed from the accounts and any resulting gain or loss is included in the results of operations for the respective period. Depreciation is provided over the estimated useful lives of the related assets using the straight-line method for financial statement purposes. The Company uses other depreciation methods (generally accelerated) for tax purposes where appropriate. The estimated useful lives for significant property and equipment categories are as follows:
| Office and laboratory equipment |
3 to 5 years | |
| Computer equipment and software |
3 years | |
| Leasehold improvements |
Term of lease agreement |
51
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
The following is a summary of property and equipment, at cost less accumulated depreciation, at December 31, 2009 and 2008 (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Office and lab equipment |
$ | 5,967 | $ | 5,290 | ||||
| Leasehold improvements |
3,359 | 3,440 | ||||||
| Computer equipment and software |
266 | 249 | ||||||
| 9,592 | 8,979 | |||||||
| Less: accumulated depreciation |
(3,395 | ) | (2,041 | ) | ||||
| Property and equipment, net |
$ | 6,197 | $ | 6,938 | ||||
Leasehold improvements include $0.2 million in gross assets acquired under two notes payable with the lessor of the Company’s headquarters (see Note 5, “Notes Payable” for further details). Office and lab equipment include gross assets acquired under capital leases of approximately $1.6 million and $3.0 million as of December 31, 2009 and 2008, respectively. Amortization of assets under capital leases is included in depreciation expense. For the years ended December 31, 2009, 2008 and 2007, the Company recorded depreciation expense of approximately $1.5 million, $1.0 million and $0.7 million, respectively.
Assets Held under Capital Leases
Assets held under capital leases are recorded at the lower of the net present value of the minimum lease payments or the fair value of the leased asset at the inception of the lease. Amortization expense is computed using the straight-line method over the shorter of the estimated useful lives of the assets or the period of the related lease.
Intangible assets—Patent Costs
Intangible assets consist of patent licensing costs incurred to date. The Company is amortizing the patent cost incurred to date, over a 15 year period. If the patents are not awarded, the costs related to those patents will be expensed in the period that determination is made. The Company has capitalized $0.8 million in patent licensing costs as of December 31, 2009. Amortization expense charged to operations was approximately $55,000 for 2009, $33,000 for 2008 and $18,000 for 2007.
Revenue Recognition
Revenue is generated from collaborative research and development arrangements, technology licenses, and government grants. Revenue is recognized when the four basic criteria of revenue recognition are met: (i) a contractual agreement exists; (ii) transfer of technology (intellectual property) has been completed or services have been rendered; (iii) the fee is fixed or determinable, and (iv) collectibility is reasonably assured.
Revenue from technology licenses or other payments under collaborative agreements where the Company has a continuing obligation to perform is recognized as revenue over the expected period of the continuing performance obligation. Revenue on government contracts is recognized on a qualified cost reimbursement basis.
In fiscal year 2009, the Company began performing limited research services for one customer and for the year ended December 31, 2009 has recognized approximately $55,000 in related revenue. Prior to 2009, only revenue from technology licenses and grant proceeds had been received.
52
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
Research and Development
Research and development expenses include internal and external costs. Internal costs include salaries and employment related expenses and allocated facility costs. External expenses consist of costs associated with outsourced clinical research organization activities, sponsored research studies, product registration, and investigator sponsored trials. All research and development costs are charged to expense as incurred.
Basic and Diluted Loss per Share
In accordance with Earnings per Share Topic of the FASB Codification, the basic loss per share is computed by dividing the loss attributable to common stockholders by the weighted average number of common shares outstanding during the period. Basic net loss per share excludes the dilutive effect of stock options or warrants and convertible notes. Diluted net earnings (loss) per common share is determined using the weighted-average number of common shares outstanding during the period, adjusted for the dilutive effect of common stock equivalents, consisting of shares that might be issued upon exercise of common stock options and warrants. In periods where losses are reported, the weighted-average number of common shares outstanding excludes common stock equivalents, because their inclusion would be anti-dilutive.
The potential shares, which are excluded from the determination of basic and diluted net loss per share as their effect is anti-dilutive, are as follows (in thousands):
| December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Options to purchase common stock |
5,110 | 5,215 | 4,750 | |||
| Options to purchase common stock—outside plan |
103 | 103 | 103 | |||
| Warrants to purchase common stock |
43,245 | 10,466 | 10,467 | |||
| Potential equivalent shares excluded |
48,458 | 15,784 | 15,320 | |||
Comprehensive Loss
Comprehensive loss includes net loss and other comprehensive income, which for us is primarily comprised of unrealized holding gains or losses on our available-for-sale securities that are excluded from the statement of operations in computing net loss and reported separately in shareholders’ equity (deficit). Comprehensive loss and its components are as follows (in thousands):
| December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Net loss |
$ | (16,364 | ) | $ | (16,700 | ) | $ | (12,901 | ) | |||
| Changes in unrealized gain (loss) on securities available-for-sale |
(29 | ) | 20 | 4 | ||||||||
| Comprehensive loss |
$ | (16,393 | ) | $ | (16,680 | ) | $ | (12,897 | ) | |||
53
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
Other Current Liabilities
Other current liabilities include the following as of December 31, 2009 and 2008 (in thousands):
| December 31, | ||||||
| 2009 | 2008 | |||||
| Accrued clinical trial expenses |
$ | 12 | $ | 73 | ||
| Accrued consulting fees |
29 | 29 | ||||
| Accrued legal fees |
191 | 41 | ||||
| Accrued accounting fees |
114 | 85 | ||||
| Deferred rent |
286 | — | ||||
| Other current liabilities |
191 | 440 | ||||
| Total other current liabilities |
$ | 823 | $ | 668 | ||
Income Taxes
The Company accounts for its income taxes based on the Income Taxes Topic of the FASB ASC which requires the establishment of a deferred tax asset or liability for the recognition of future deductible or taxable amounts and operating loss and tax credit carryforwards. Deferred tax expense or benefit is recognized as a result of timing differences between the recognition of assets and liabilities for book and tax purposes during the year.
Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Deferred tax assets are recognized for deductible temporary differences and operating loss, and tax credit carryforwards. A valuation allowance is established, when necessary, to reduce that deferred tax asset if it is “more likely than not” that the related tax benefits will not be realized.
Share-based Compensation
The Company records share-based compensation in accordance with the provisions of the Share-based Compensation Topic of the FASB Codification. The guidance requires the use of option-pricing models that require the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. The fair value of each option grant is estimated on the date of grant using the Black-Scholes option valuation model, and the resulting charge is expensed using the straight-line attribution method over the vesting period. Both the expected stock price volatility and the weighted-average expected life assumptions were determined using historical data. Stock compensation expense recognized during the period is based on the value of share-based awards that were expected to vest during the period adjusted for estimated forfeitures.
The estimated fair value of grants of stock options and warrants to non-employees of the Company is charged to expense, if applicable, in the financial statements. These options vest in the same manner as the employee options granted under each of the option plans as described above.
Recent Accounting Pronouncements
In June of 2009, the FASB issued guidance now codified as FASB ASC Topic 105, “Generally Accepted Accounting Principles,” as the single source of authoritative nongovernmental U.S. GAAP. FASB ASC Topic
54
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
105 does not change current U.S. GAAP, but is intended to simplify user access to all authoritative U.S. GAAP by providing all authoritative literature related to a particular topic in one place. All existing accounting standard documents will be superseded and all other accounting literature not included in the FASB Codification will be considered non-authoritative. These provisions of FASB ASC Topic 105 are effective for interim and annual periods ending after September 15, 2009 and, accordingly, are effective for the Company for the current fiscal reporting period. The adoption of this pronouncement did not have an impact on the Company’s financial condition or results of operations, but will impact the financial reporting process by eliminating all references to pre-codification standards. On its effective date, the Codification superseded all then-existing non-SEC accounting and reporting standards, and all other non-grandfathered non-SEC accounting literature not included in the Codification became non-authoritative.
In April of 2009, the FASB issued guidance in the Fair Value Measurements and Disclosures Topic of the Codification on determining fair value when the volume and level of activity for an asset or liability have significantly decreased and identifying transactions that are not orderly. The guidance emphasizes that even if there has been a significant decrease in the volume and level of activity, the objective of a fair value measurement remains the same. Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction (that is, not a forced liquidation or distressed sale) between market participants. The guidance provides a number of factors to consider when evaluating whether there has been a significant decrease in the volume and level of activity for an asset or liability in relation to normal market activity. In addition, when transactions or quoted prices are not considered orderly, adjustments to those prices based on the weight of available information may be needed to determine the appropriate fair value. The guidance is effective for interim or annual reporting periods ending after June 15, 2009, and shall be applied prospectively. The Company adopted this guidance effective for the quarter ending June 30, 2009. There was no impact of the adoption on the Company’s financial statements for the year ended December 31, 2009.
In April of 2009, FASB issued guidance in the Financial Instruments Topic of the Codification on interim disclosures about fair value of financial instruments. The guidance requires disclosures about the fair value of financial instruments for both interim reporting periods, as well as annual reporting periods. The guidance is effective for all interim and annual reporting periods ending after June 15, 2009 and shall be applied prospectively. The adoption of this guidance had no impact on the Company’s financial statements as of December 31, 2009.
In May of 2009, the FASB issued SFAS No. 165, Subsequent Events (ASC Topic 855). This guidance establishes general standards of accounting for and disclosures of events that occur after the balance sheet date but before financial statements are issued or are available to be issued. This guidance was effective for the Company in the second quarter of 2009 and its adoption did not have a material impact on the Company’s condensed consolidated financial statements. On February 24, 2010, the FASB issued ASU No. 2010-09 amending Topic 855 to no longer require entities to disclose the date through which management evaluated subsequent events in the financial statements. The update was effective upon release and the adoption of the update did not have a material impact on our financial statements as of December 31, 2009. (See Note 11, “Subsequent Events”).
In October of 2009, the FASB issued the following ASU No. 2009-13, Revenue Recognition (ASC Topic 605)—Multiple-Deliverable Revenue Arrangements, a consensus of the FASB Emerging Issues Task Force. This guidance modifies the fair value requirements of ASC subtopic 605-25 Revenue Recognition-Multiple Element Arrangements by allowing the use of the “best estimate of selling price” for determining the selling price of a deliverable. A vendor is now required to use its best estimate of the selling price when vendor specific objective
55
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
evidence or third-party evidence of the selling price cannot be determined. In addition, the residual method of allocating arrangement consideration is no longer permitted. This guidance is effective in 2011. The Company is currently evaluating the impact of adopting this update on its condensed consolidated financial statements.
In January of 2010, the FASB issued an amendment to the accounting standards related to the disclosures about an entity’s use of fair value measurements. Among these amendments, entities will be required to provide enhanced disclosures about transfers into and out of the Level 1 (fair value determined based on quoted prices in active markets for identical assets and liabilities) and Level 2 (fair value determined based on significant other observable inputs) classifications, provide separate disclosures about purchases, sales, issuances and settlements relating to the tabular reconciliation of beginning and ending balances of the Level 3 (fair value determined based on significant unobservable inputs) classification and provide greater disaggregation for each class of assets and liabilities that use fair value measurements. Except for the detailed Level 3 roll-forward disclosures, the new standard is effective for the Company for interim and annual reporting periods beginning after December 31, 2009. The requirement to provide detailed disclosures about the purchases, sales, issuances and settlements in the roll-forward activity for Level 3 fair value measurements is effective for the Company for interim and annual reporting periods beginning after December 31, 2010. The Company does not expect that the adoption of this new standard will have a material impact to its consolidated financial statements.
3. CASH, CASH EQUIVALENTS AND INVESTMENTS
As part of our cash management program, the Company maintains a portfolio of marketable investment securities. The securities are investment grade and generally mature within one year and may include tax exempt securities and certificates of deposit. The fair value of substantially all securities is determined by quoted market prices. The estimated fair value of securities for which there are no quoted market prices is based on similar types of securities that are traded in the market. The Company maintains its cash in bank accounts, which at times may exceed federally insured limits. The Company has not experienced any losses on such accounts. The Company considers all highly liquid investments purchased with an original maturity of three months or less at the time of purchase to be cash equivalents.
The following is a summary of cash, cash equivalents, and available-for-sale securities at December 31, 2009 and December 31, 2008 (in thousands):
| December 31, 2009 | |||||||||||||
| Cost Basis |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value | ||||||||||
| Cash and cash equivalents: |
|||||||||||||
| Cash |
$ | 124 | $ | — | $ | — | $ | 124 | |||||
| Money market funds |
1,073 | — | — | 1,073 | |||||||||
| Corporate notes |
1,602 | — | — | 1,602 | |||||||||
| Total cash and cash equivalents |
$ | 2,799 | $ | — | $ | — | $ | 2,799 | |||||
| Available-for-sale investments: |
|||||||||||||
| US govt. agency obligations |
$ | 7,812 | $ | — | $ | (5 | ) | $ | 7,807 | ||||
| Corporate notes |
5,328 | — | — | 5,328 | |||||||||
| Total available-for-sale investments |
$ | 13,140 | $ | — | $ | (5 | ) | $ | 13,135 | ||||
56
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
| December 31, 2008 | ||||||||||||
| Cost Basis |
Unrealized Gains |
Unrealized Losses |
Estimated Fair Value | |||||||||
| Cash and cash equivalents: |
||||||||||||
| Cash |
$ | 378 | $ | — | $ | — | $ | 378 | ||||
| Money market funds |
2,433 | — | — | 2,433 | ||||||||
| US term deposits |
458 | 1 | — | 459 | ||||||||
| Total cash and cash equivalents |
$ | 3,269 | $ | 1 | $ | — | $ | 3,270 | ||||
| Available-for-sale investments: |
||||||||||||
| US govt. agency obligations |
$ | 6,500 | $ | 20 | $ | — | $ | 6,520 | ||||
| Corporate notes |
3,769 | 3 | — | 3,772 | ||||||||
| Total available-for-sale investments |
$ | 10,269 | $ | 23 | $ | — | $ | 10,292 | ||||
As of December 31, 2009 and 2008, unrealized losses of approximately $5,000 and unrealized gains of approximately $24,000 were included in accumulated other comprehensive income in the accompanying consolidated balance sheets, respectively.
Fair Value Hierarchy
U.S. GAAP establishes a fair value hierarchy which has three levels based on the reliability of the inputs used to determine the fair value. These levels include: Level 1, defined as inputs such as unadjusted quoted prices in active markets for identical assets or liabilities; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs for use when little or no market data exists, therefore requiring an entity to develop its own assumptions.
The following table represents the Company’s fair value hierarchy for its financial assets (cash equivalents and short-term investments) measured at fair value on a recurring basis as of December 31, 2009 (in thousands):
| Fair Value Measurements at Reporting Date Using | |||||||||
| Total | (Level 1) | (Level 2) | |||||||
| December 31, 2009 |
|||||||||
| Money market funds |
$ | 1,073 | $ | 1,073 | $ | — | |||
| US government agencies |
7,807 | — | 7,807 | ||||||
| US corporate debt |
6,930 | 6,930 | |||||||
| Total |
$ | 15,810 | $ | 1,073 | $ | 14,737 | |||
| Total | (Level 1) | (Level 2) | |||||||
| December 31, 2008 |
|||||||||
| Money market funds |
$ | 2,433 | $ | 2,433 | $ | — | |||
| US term deposits |
459 | — | 459 | ||||||
| US government agencies |
6,520 | — | 6,520 | ||||||
| US corporate debt |
3,772 | — | 3,772 | ||||||
| Total |
$ | 13,184 | $ | 2,433 | $ | 10,751 | |||
57
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
4. SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION
The following table shows supplemental cash flows for the years ended December 31, 2009, 2008 and 2007 and inception to date from February 1, 2002 through December 31, 2009 (in thousands).
| For the Years Ended December 31, | Accumulated from February 1, 2002 (Date of inception) to December 31, 2009 | |||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| Cash paid during the year for: |
||||||||||||
| Interest paid |
$ | 89 | $ | 128 | $ | 92 | $ | 444 | ||||
| Income taxes paid |
1 | 4 | 3 | 13 | ||||||||
| Supplemental disclosure of non-cash investing and financing: |
||||||||||||
| Non-cash warrant expense for warrants issued |
$ | — | $ | — | $ | — | $ | 1,964 | ||||
| Adjustment in warrant liability |
— | — | — | 7,030 | ||||||||
| Conversion of notes payable to common stock |
— | — | — | 450 | ||||||||
| Assets acquired under capital lease |
40 | 870 | 1,211 | 3,071 | ||||||||
| Assets acquired under notes payable |
204 | — | — | 204 | ||||||||
| Issuance of common stock for services |
— | — | — | 165 | ||||||||
| Conversion of accrued interest payable |
— | — | — | 12 | ||||||||
| Issuance of common stock with reverse merger |
— | — | — | 4 | ||||||||
5. NOTES PAYABLE
In January of 2009, the Company relocated to its new headquarters. Related to the relocation, the Company financed leasehold improvements under a two notes payable with the lessor of the building. The first note of $100,000 bears interest at 9.5% and is payable in monthly payments of interest and principal amounting to approximately $1,000 beginning May 1, 2009 and continuing over the remaining lease term with a due date of December 31, 2018. The second note of $104,000 bears interest at 9.5% and is payable in 24 monthly payments of interest and principal amounting to approximately $5,000 beginning August 1, 2009. The future minimum payments as of December 31, 2009 are as follows (in thousands):
| Principal | Interest | |||||
| 2010 |
59 | 15 | ||||
| 2011 |
40 | 9 | ||||
| 2012 |
9 | 7 | ||||
| 2013 |
9 | 6 | ||||
| 2014 |
10 | 6 | ||||
| Thereafter |
53 | 11 | ||||
| Total minimum payments |
$ | 180 | $ | 54 | ||
6. SHAREHOLDERS’ EQUITY AND SHARE-BASED COMPENSATION
Common Stock
In April and May of 2005 the Company completed private placements selling 20,461,000 shares of common stock to accredited investors at a price of $0.50 per share. The Company received gross proceeds of $10,230,500.
58
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
As part of the closing of the private placements we issued five-year warrants to purchase a total of 2,557,625 shares of common stock at an exercise price of $0.75 per share and 2,557,625 shares of common stock at an exercise price of $1.00 per share.
In connection with the closing of the private placements, on April 6, 2005, the convertible notes resulting from the September 30, 2004 bridge financing of $500,000 were converted into common stock at $0.36 per share or paid. The aggregate principal amount of the convertible secured notes of $450,000 was converted into a total of 1,251,448 shares of common stock. The remaining $50,000 principal was repaid.
On March 17, 2006, the Company issued a call notice to all holders of the common stock purchase warrants ($0.75 exercise price) issued in the April 6, 2005 and May 5, 2005 private placements. Section 3(c) of the warrants permit the Company to call for cancellation all or a part of the unexercised portion of the warrants upon the occurrence of certain events. The events required are (i) an effective registration statement registering the resale of the shares of common stock underlying the warrants, (ii) the closing bid price of the common stock for each of the ten (10) consecutive trading days equals or exceeds $0.9375 and (iii) the average daily trading volume of the common stock for such ten (10) days period equals or exceeds 100,000 shares. As of March 16, 2006, each of the foregoing events had occurred. The holders of these warrants had until April 10, 2006 to exercise all or a portion of the unexercised portion of such warrants, and any such warrants not so exercised will automatically be canceled on that date. Pursuant to the call notice, warrant holders purchased 2,671,062 shares, warrants exercisable at $0.75 per share represented 2,495,125 shares, while warrants exercisable at $1.00 per share represented 175,937 shares. Warrants representing 62,500 shares exercisable at $0.75 per share, were cancelled, as the warrant holders failed to exercise pursuant to the call notice.
Unrelated to the call notice, during the year ending December 31, 2006, certain warrant holders exercised common stock purchase warrants representing 2,353,714 shares, for which the Company received $257,776.
On January 19, 2007, the Company completed a private placement to accredited investors of approximately 10,521,000 shares of our common stock, at a purchase price of $1.50 per share, which is equal to a 4.5% discount from the average closing market price of its common stock over the twenty day period ending on the pricing date of January 12, 2007, for gross proceeds of $15,781,500. As part of the private placement, the investors were issued five-year warrants to purchase up to an aggregate of 3,682,350 shares of our common stock, at an initial exercise price of $2.25. The warrants are callable by the Company when the trailing 10-day average of the closing market share price of our common stock equals or exceeds $2.75.
On March 9, 2007, the Company issued a call notice to all holders of the common stock purchase warrants ($1.00 exercise price) issued in the April 6, 2005 and May 5, 2005 private placements. Section 3(c) of the warrants permit the Company to call for cancellation all or a part of the unexercised portion of the warrants upon the occurrence of certain events. The events required are (i) an effective registration statement registering the resale of the shares of common stock underlying the warrants, (ii) the closing bid price of the common stock for each of the ten (10) consecutive trading days equals or exceeds $1.25 and (iii) the average daily trading volume of the common stock for such ten (10) days period equals or exceeds 100,000 shares. As of March 8, 2007, each of the foregoing events had occurred. The holders of these warrants had until March 31, 2007 to exercise all or a portion of the unexercised portion of such warrants, and any such warrants not so exercised will automatically be canceled on that date. As of March 31, 2007, all of the $1.00 warrants from the April 6, 2005 and May 5, 2005 private placements had been exercised pursuant to this call notice, or exercised prior to the call notice.
On October 31, 2007, the Company received $24.0 million, net of underwriting discount, from the closing of its public offering of 10.0 million shares of common stock at a price to the public of $2.50 per share and 5.3 million 5-year warrants to purchase shares of common stock at $3.50 per share at a price of $0.10 per warrant.
59
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
On October 7, 2009, the Company completed a registered public offering and issued 30,933,140 shares of its common stock and 29,324,570 warrants at a price of $6.20 per unit including 10 shares of common stock and 10 warrants to purchase common stock at $0.85 per share and $6.00 per unit of 10 shares of common stock exclusive of warrants. In addition, the Company granted the placement agent 1,546,657 warrants, each warrant entitling the holder to purchase one share of common stock at $0.97 per share at any time for a period commencing six months after the date of the closing, and continuing for five years following the closing date of the offering. The warrants may also be executed on a cashless basis, in accordance with the terms of the warrant.
Stock Option Plan
On April 6, 2005, in connection with the completion of the reverse merger, the board of directors assumed and adopted the Stock Incentive Plan, as amended, (the “Plan”) of Bionovo Biopharmaceuticals and 3,496,788 shares of common stock for issuance under the Plan. In May 2006, shareholders approved an increase of 3,000,000 additional shares for the Plan. In April 2008, the Board adopted Amendment No. 5 to the Plan, approved by stockholders in June 2008, to increase the number of shares covered by, and reserved for issuance under, the Plan from 6,496,788 shares to 9,496,788 shares. Such share reserve consists of the number of shares that remain available for issuance under the Plan and shares subject to outstanding options.
Under the Plan, incentive options to purchase the Company’s common stock may be granted to employees at prices not lower than fair market value at the date of grant as determined by the Board of Directors. Non-statutory options (options that do not qualify as incentive options) may be granted to employees and consultants at prices no lower than 85% of fair market value at the date of grant as determined by the Board of Directors. In addition, incentive or non-statutory options may be granted to persons owning more than 10% of the voting power of all classes of stock at prices no lower than 110% of the fair market value at the date of grant as determined by options (no longer than ten years from the date of grant, five years in certain instances). Options granted generally have a five-year life and typically vest over four years.
As of December 31, 2009, we had reserved 4.0 million shares of common stock for future issuance under the Plan.
Share-based Compensation Expense
To estimate the value of an award, the Company uses the Black-Scholes option pricing model. This model requires inputs such as expected life, expected volatility and risk-free interest rate. The forfeiture rate also impacts the amount of aggregate compensation expense. These inputs are subjective and generally require significant analysis and judgment to develop. While estimates of expected life, volatility and forfeiture rate are derived primarily from the Company’s historical data, the risk-free rate is based on the yield available on U.S. Treasury zero-coupon issues. For the years ended December 31, 2009, 2008 and 2007, the weighted-average assumptions used were:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | |||
| Expected volatility |
127 | % | 93 | % | 83 | % | |||
| Risk-free interest rate |
1.42 | % | 2.44 | % | 3.45 | % | |||
| Expected life |
3.75 years | 3.11 years | 4.75 years | ||||||
60
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
The following table summarizes share-based compensation expense related to employee and non-employee stock options for the years ended December 31, 2009, 2008 and 2007 (in thousands):
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Research and development |
$ | 686 | $ | 512 | $ | 743 | |||
| General and administrative |
500 | 859 | 537 | ||||||
| Total share-based compensation expense |
$ | 1,186 | $ | 1,371 | $ | 1,280 | |||
Total share-based compensation expense for the year ended December 31, 2009 includes an accrual of $0.2 million for approximately 657,000 options granted to employees on January 12, 2010. The options were fully vested upon grant date and were granted to employees in lieu of cash bonuses based on fiscal year 2009 performance.
There was no capitalized share-based compensation cost as of December 31, 2009 and there were no recognized tax benefits during the years ended December 31, 2009, 2008 and 2007.
Share option activity for the year ended December 31, 2009 was as follows:
| Options (in thousands) |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (Years) |
Aggregate Intrinsic Value (in thousands) | ||||||||
| Options outstanding at December 31, 2008 |
5,215 | $ | 1.89 | ||||||||
| Granted |
524 | 0.48 | |||||||||
| Exercised |
— | — | |||||||||
| Forfeited |
(515 | ) | 2.34 | ||||||||
| Expired or canceled |
(114 | ) | 3.25 | ||||||||
| Options outstanding at December 31, 2009 |
5,110 | $ | 1.67 | 3.11 | $ | 195 | |||||
| Options exercisable at December 31, 2009 |
3,514 | $ | 1.52 | 3.08 | $ | 152 | |||||
| Options exercisable and options expected to vest at December 31, 2009 |
4,949 | $ | 1.67 | 2.95 | $ | 183 | |||||
The weighted-average grant date fair value of options granted for each of the last three years was as follows:
| Grant Year |
Options Granted (in thousands) |
Weighted Average Grant Date Fair Value | |||
| 2007 |
2,673 | $ | 1.97 | ||
| 2008 |
814 | 0.71 | |||
| 2009 |
524 | 0.38 | |||
The intrinsic value of options exercised for the years December 31, 2008 and 2007 was approximately $2,000 and $474,000, respectively. There were no stock option exercises for the year ended December 31, 2009. As of December 31, 2009, there was $1.3 million of total future compensation cost related to unvested stock options to be recognized over a weighted-average period of 1.8 years.
61
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
Unvested share activity for the year ended December 31, 2009 is summarized below:
| Unvested Number of Options (in thousands) |
Weighted Average Grant Date Fair Value | |||||
| Unvested balance at December 31, 2008 |
2,304 | $ | 1.62 | |||
| Granted |
524 | 0.38 | ||||
| Vested |
(717 | ) | 1.48 | |||
| Forfeited |
(515 | ) | 1.73 | |||
| Unvested balance at December 31, 2009 |
1,596 | $ | 1.25 | |||
The following table summarizes information about stock options outstanding as of December 31, 2009:
| Options Outstanding | Options Exercisable | |||||||||||
| Range of Exercise Price |
Number Outstanding (in thousands) |
Weighted Average Remaining Contractual Life (in Years) |
Weighted Average Exercise Price |
Number Exercisable (in thousands) |
Weighted Average Exercise Price | |||||||
| $0.01 – $1.00 |
2,426 | 3.7 | $ | 0.61 | 1,947 | $ | 0.62 | |||||
| $1.01 – $2.00 |
1,056 | 2.6 | 1.46 | 650 | 1.49 | |||||||
| $2.01 – $3.00 |
1,148 | 2.6 | 2.85 | 630 | 2.85 | |||||||
| $3.01 – $4.00 |
50 | 2.1 | 3.94 | 50 | 3.94 | |||||||
| $4.01 – $5.00 |
300 | 2.7 | 4.40 | 150 | 4.40 | |||||||
| $5.01 – $6.00 |
130 | 2.3 | 5.75 | 87 | 5.75 | |||||||
| 5,110 | 3.1 | 1.67 | 3,514 | 1.52 | ||||||||
Shares Reserved for Future Issuance
The Company has reserved shares of common stock for future issuance as follows (in thousands):
| December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Options to purchase common stock—Plan |
5,110 | 5,215 | 4,750 | |||
| Options to purchase common stock—Outside plan |
103 | 103 | 103 | |||
| Shares available for option grants |
3,991 | 3,886 | 1,370 | |||
| Warrants to purchase common stock |
43,245 | 10,466 | 10,467 | |||
| Total |
52,449 | 19,670 | 16,690 | |||
7. WARRANTS
September 2004 Bridge Financing
In September 2004, in connection with Bionovo Biopharmaceuticals’ $500,000 principal amount of 6% convertible secured notes bridge financing completed on September 30, 2004, Bionovo Biopharmaceuticals issued to investors in the bridge financing warrants, or bridge warrants, exercisable for 556,123 shares of Bionovo Biopharmaceuticals common stock at an exercise price of $0.53 per share. The bridge warrants were
62
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
exercisable until the earlier of September 30, 2011 and the fifth anniversary of Bionovo Biopharmaceuticals’ merger with a company required to file reports pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”). Upon the closing of our reverse merger transaction, the bridge warrants were amended to become bridge warrants to purchase shares of our common stock upon the same terms and conditions as the bridge warrants issued by Bionovo Biopharmaceuticals. The bridge warrants expire on April 6, 2010.
In connection with the closing of the bridge financing, Bionovo Biopharmaceuticals issued five-year warrants to Duncan Capital, LLC as partial compensation for acting as placement agent in the transaction. Upon the closing of our reverse merger transaction, these placement agent warrants were amended to become warrants to purchase shares of our common stock upon the same terms and conditions as the placement agent warrants issued in the bridge financing by Bionovo Biopharmaceuticals. The bridge placement agent warrants are exercisable in whole or in part, at an exercise price of $0.35 per share, before September 30, 2009 for up to 132,421 shares of common stock. These warrants contain provisions that protect the holders thereof against dilution by adjustment of the purchase price in certain events, such as stock splits or reverse stock splits, stock dividends, recapitalizations or similar events. The holders of these warrants will not possess any rights as stockholders unless and until they exercise their warrants. The bridge placement agent warrants do not confer upon holders any voting or any other rights as stockholders.
As of December 31, 2009, all bridge warrants and related placement agent warrants had been exercised.
April 2005 Private Placement
In April 2005, as part of the closing of Bionovo Biopharmaceuticals’ April 6, 2005 private placement, Bionovo Biopharmaceuticals issued five-year warrants to purchase a total of 2,023,875 shares of Bionovo Biopharmaceuticals common stock at an exercise price of $0.75 per share and 2,023,875 shares of Bionovo Biopharmaceuticals common stock at an exercise price of $1.00 per share. The warrants were exercisable in whole or in part until April 6, 2010. Upon the closing of our reverse merger transaction, the April 2005 private placement warrants were amended to become warrants to purchase shares of our common stock upon the same terms and conditions as the April 2005 private placement warrants issued by Bionovo Biopharmaceuticals. As a result of the call noticed issued in March of 2007 (discussed in Note 6, “Shareholders’ Equity and Share-based Compensation”), all warrants issued through the April 2005 private placement have been exercised or cancelled as of December 31, 2009.
In connection with the closing of the April 2005 private placement, Bionovo Biopharmaceuticals issued five-year warrants to Duncan Capital, LLC as partial compensation for acting as placement agent in the transaction. Upon the closing of our reverse merger transaction, these placement agent warrants were amended to become warrants to purchase shares of our common stock upon the same terms and conditions as the placement agent warrants issued in the April 2005 private placement by Bionovo Biopharmaceuticals. The April 2005 placement agent warrants are exercisable in whole or in part, at an exercise price of $0.50 per share, before April 6, 2010 for up to 1,709,100 shares of common stock. These warrants contain provisions that protect the holders thereof against dilution by adjustment of the purchase price in certain events, such as stock splits or reverse stock splits, stock dividends, recapitalizations or similar events. The holders of these warrants will not possess any rights as stockholders unless and until they exercise their warrants. The April 2005 placement agent warrants do not confer upon holders any voting or any other rights as stockholders. As of December 31, 2009, 926,040 placement agent warrants remain outstanding.
63
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
April 2005 Reverse Merger
In connection with the closing of our reverse merger transaction on April 6, 2005, we issued five-year warrants to Duncan Capital, LLC as partial compensation for its advisory services relating to the merger. These reverse merger warrants are exercisable in whole or in part, at an exercise price of $0.01 per share, before April 6, 2010 for up to 1,979,630 shares of common stock. These warrants contain provisions that protect the holders thereof against dilution by adjustment of the purchase price in certain events, such as stock splits or reverse stock splits, stock dividends, recapitalizations or similar events. The holders of these warrants will not possess any rights as stockholders unless and until they exercise their warrants. The reverse merger warrants do not confer upon holders any voting or any other rights as stockholders. As of December 31, 2009, all warrants associated with the Company’s reverse merger had been exercised.
May 2005 Private Placement
In May 2005, as part of the closing of our May 5, 2005 private placement, we issued five-year warrants to purchase a total of 533,750 shares of common stock at an exercise price of $0.75 per share and 533,750 shares of common stock at an exercise price of $1.00 per share. The warrants are exercisable in whole or in part until May 5, 2010. As a result of the call noticed issued in March of 2007 (discussed in Note 6, “Shareholders’ Equity and Share-based Compensation”), all warrants issued through the May 2005 private placement have been exercised or cancelled as of December 31, 2009.
In connection with the closing of our May 2005 private placement, we issued five-year warrants to Duncan Capital, LLC as partial compensation for acting as placement agent in the transaction. These May 2005 placement agent warrants are exercisable in whole or in part, at an exercise price of $0.50 per share, before May 5, 2010 for up to 427,000 shares of common stock. These warrants contain provisions that protect the holders thereof against dilution by adjustment of the purchase price in certain events, such as stock splits or reverse stock splits, stock dividends, recapitalizations or similar events. The holders of these warrants will not possess any rights as stockholders unless and until they exercise their warrants. The May 2005 placement agent warrants do not confer upon holders any voting or any other rights as stockholders. As of December 31, 2009, 402,000 placement agent warrants remain outstanding.
January 2007 Private Placement
In connection with the closing of our January 2007 private placement, the investors were issued five-year warrants to purchase up to an aggregate of 3,682,350 shares of our common stock, at an initial exercise price of $2.25. The warrants are callable by the Company when the trailing 10-day average of the closing market share price of our common stock equals or exceeds $2.75. As of December 31, 2009, 2,988,184 warrants remain outstanding.
In connection with the closing of our January 2007 private placement, we issued five-year warrants to the brokers as partial compensation for acting as placement agent in the transaction. These January 2007 private placement agent warrants are exercisable in whole or in part, at an exercise price of $1.50 per share, before January 19, 2012 for up to 736,470 shares of common stock. These warrants contain provisions that protect the holders thereof against dilution by adjustment of the purchase price in certain events, such as stock splits or reverse stock splits, stock dividends, recapitalizations or similar events. The holders of these warrants will not possess any rights as stockholders unless and until they exercise their warrants. The January 2007 placement agent warrants do not confer upon holders any voting or any other rights as stockholders. As of December 31, 2009, 604,185 placement agent warrants remain outstanding.
64
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
October 2007 Public Offering
In connection with the closing of our October 31, 2007 public offering of common shares, the investors were issued five-year warrants to purchase up to an aggregate of 5,323,775 shares of our common stock, at an initial exercise price of $3.50 per share and a market price of $0.10 per warrant. The warrants contain a subsequent equity sales provision that provides for repricing of the warrants if subsequent warrants or options are sold at with an exercise price of less than $3.50 per share. The exercise price would be reduced to the greater of (a) the price of the new warrants or (b) $2.50 per share and the total warrants outstanding would be increased such that the total aggregate exercise price shall be equal to the aggregate exercise price prior to the adjustment. The closing of our October 7, 2009 public offering triggered the subsequent equity sales provision of the investor warrants issued on October 31, 2007. As such, the 5,323,775 warrants outstanding at an exercise price of $3.50 were cancelled and 7,453,285 shares were issued with an exercise price of $2.50. As of December 31, 2009, all 7,453,285 warrants remain outstanding.
October 2009 Public Offering
In connection with the closing of our October 7, 2009 public offering of common shares, the investors were issued five-year warrants to purchase up to an aggregate 29,324,570 shares of our common stock at an initial exercise price of $0.85 per share. In addition, the Company granted the placement agent 1,546,657 warrants, each warrant entitling the holder to purchase one share of common stock at $0.97 per share at any time for a period commencing six months after the date of the closing, and continuing for five years following the closing date of the offering. The warrants may also be executed on a cashless basis, in accordance with the terms of the warrant. As of December 31, 2009, all October 2009 public offering warrants and related placement agent warrants remain outstanding.
The following warrants are each exercisable into one share of common stock:
| Number of Shares (in thousands) |
Weighted Average Exercise Price |
Aggregate Price (in thousands) |
||||||||
| Balance at December 31, 2008 |
10,467 | $ | 2.58 | $ | 27,047 | |||||
| Warrants granted |
38,324 | 1.18 | 45,059 | |||||||
| Warrants exercised |
(222 | ) | 0.54 | (120 | ) | |||||
| Warrants cancelled or forfeited |
(5,324 | ) | 3.50 | (18,633 | ) | |||||
| Unvested balance at December 31, 2009 |
43,245 | 1.23 | $ | 53,353 | ||||||
Warrants to purchase 222,449 shares of common stock were exercised during the year ended December 31, 2009. As of December 31, 2009, there were 43.2 million warrants to purchase Bionovo shares outstanding with a weighted average price of $1.23 and an aggregate price of $53.4 million.
The following table summarizes information about all warrants outstanding as of December 31, 2009:
| Issue Date |
Expiration Date | Number Outstanding (in thousands) |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (years) | ||||
| April 2005 |
April 2010 | 926 | 0.57 | 0.26 | ||||
| May 2005 |
May 2010 | 402 | 0.77 | 0.34 | ||||
| January 2007 |
January 2012 | 3,593 | 2.13 | 2.05 | ||||
| October 2007 |
October 2012 | 7,453 | 2.50 | 2.84 | ||||
| October 2009 |
October 2014 | 30,871 | 0.86 | 4.77 | ||||
| 43,245 | ||||||||
65
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
The estimated fair value of the warrants was calculated using the Black-Scholes valuation model. The following assumptions were used: (i) no expected dividends, (ii) risk free interest rates of 2.2%–4.0%, (iii) expected volatility of 90%–187%, and (iv) expected life in the stated life of the warrant. The fair value of the warrants ranged from $0.10 to $0.89. The fair value of warrants granted is included in additional paid-in capital along with the proceeds from issuance of common stock.
Employee Benefit Plan
The Company has a 401(k) Plan that covers substantially all of its employees. Under the 401(k) Plan, eligible employees may contribute up to 15 percent of their eligible compensation, subject to certain Internal Revenue Service restrictions. The Company does not match employee contributions in the 401(k) Plan.
8. LEASES, COMMITMENTS AND CONTINGENCIES
Leases
The Company leases certain office and laboratory equipment under agreements that are classified as capital leases. The cost of equipment under capital leases is included in the Balance Sheets as property and equipment was $1.6 million at December 31, 2009 and $3.0 million at December 31, 2008. Accumulated amortization of leased equipment at December 31, 2009 was $0.7 million and as of December 31, 2008 was $1.0 million. Amortization of assets under capital leases is included in depreciation expense. The Company’s leases with General Electric Capital Corporation (GE) required security deposits of 25% of the purchase price. As of December 31, 2009 and 2008, security deposits of $0.2 million and $0.3 million, respectively, were included in the Balance Sheets as other assets.
The Company leases its laboratory and office facilities for various terms under long-term, non-cancelable operating lease agreements. The leases expire at various dates through 2019. The leases provide for increases in future minimum annual rental payments and the agreements generally require the Company to pay executory costs (real estate taxes, insurance, and repairs), which are approximately $41,000 per month. In addition, the Company pays monthly payments of approximately $6,000 toward $0.2 million in notes payable to the lessor for leasehold improvements associated with the Company’s relocation to new office and laboratory facilities. In November of 2009, the Company signed a new lease for additional laboratory space in Emeryville, California, with fixed monthly rent of $3,300. This lease expires in November of 2010.
Operating lease expense for the years ended December 31, 2009, 2008 and 2007 totaled $2.1 million, $0.7 million and $0.6 million, respectively.
66
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
Future minimum lease payments under non-cancelable capital and operating leases are as follows (in thousands):
| Capital Leases | Operating Leases | ||||||
| 2010 |
$ | 501 | $ | 1,850 | |||
| 2011 |
94 | 1,868 | |||||
| 2012 |
7 | 1,924 | |||||
| 2013 |
— | 1,982 | |||||
| 2014 |
— | 2,041 | |||||
| Thereafter |
— | 8,795 | |||||
| Total minimum lease payments |
$ | 602 | $ | 18,460 | |||
| Less: Amount representing interest |
(30 | ) | |||||
| Present value of minimum lease payments |
$ | 572 | |||||
| Less: Current portion |
(476 | ) | |||||
| Obligations under capital lease, net of current portion |
$ | 96 | |||||
9. INCOME TAX
The provision for income taxes on the statements of operations consists of $800 and $3,700 for the years ended December 31, 2009 and 2008, respectively.
Deferred tax assets (liabilities) are comprised of the following at December 31, 2009 and 2008 (in thousands):
| Year Ended December 31, | ||||||||
| 2009 | 2008 | |||||||
| Net operating loss carryforward |
$ | 20,569 | $ | 14,326 | ||||
| Fixed assets |
(269 | ) | (290 | ) | ||||
| Stock compensation |
1,505 | 1,055 | ||||||
| Loss on fixed asset disposal and other |
(23 | ) | (11 | ) | ||||
| 21,782 | 15,080 | |||||||
| Less valuation allowance |
(21,782 | ) | (15,080 | ) | ||||
| Net deferred tax assets |
$ | — | $ | — | ||||
Deferred taxes arise from temporary differences in the recognition of certain expenses for tax and financial reporting purposes. At December 31, 2009 and 2008, management determined that realization of these benefits is not assured and has provided a valuation allowance for the entire amount of such benefits. At December 31, 2009, net operating loss carryforwards were approximately $51.7 million for federal tax purposes that expire at various dates from 2023 through 2029 and $51.6 million for state tax purposes that expire in 2014 through 2019.
Utilization of net operating loss carryforwards may be subject to substantial annual limitations due to the “change in ownership” provisions of the Internal Revenue Code of 1986, as amended, and similar state regulations. The annual limitation may result in the expiration of substantial net operating loss carryforwards before utilization.
67
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
The provision for income taxes differs from the amount computed by applying the U.S. federal statutory tax rate (34% in 2009 and 2008) to income taxes as follows (in thousands):
| Year Ended December 31, | ||||||||
| 2009 | 2008 | |||||||
| Tax benefit computed at 34% |
$ | (5,563 | ) | $ | (5,677 | ) | ||
| Change in valuation allowance |
6,702 | 6,609 | ||||||
| Change in carryovers and tax attributes |
(1,138 | ) | (928 | ) | ||||
| Tax provision |
$ | 1 | $ | 4 | ||||
Management believes that, based on a number of factors, it is not determinable that it is more likely than not that the deferred tax assets will be realized.
10. QUARTERLY RESULTS OF OPERATIONS (UNAUDITED)
Following is a summary of the quarterly results of operations for the years ended December 31, 2009 and 2008 (in thousands, except per share data):
| March 31, 2009 |
June 30, 2009 |
September 30, 2009 |
December 31, 2009 |
|||||||||||||
| Revenues |
$ | — | $ | 7 | $ | 155 | $ | 126 | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
3,601 | 2,954 | 2,938 | 3,006 | ||||||||||||
| General and administrative |
1,009 | 1,175 | 926 | 943 | ||||||||||||
| Total operating expenses |
4,610 | 4,129 | 3,864 | 3,949 | ||||||||||||
| Loss from operations |
(4,610 | ) | (4,122 | ) | (3,709 | ) | (3,823 | ) | ||||||||
| Interest income |
54 | 16 | 5 | 9 | ||||||||||||
| Interest expense |
(33 | ) | (22 | ) | (22 | ) | (18 | ) | ||||||||
| Other expense |
(77 | ) | (6 | ) | — | (5 | ) | |||||||||
| Loss before income taxes |
(4,666 | ) | (4,134 | ) | (3,726 | ) | (3,837 | ) | ||||||||
| Provision for income taxes |
(2 | ) | — | — | 1 | |||||||||||
| Net loss |
$ | (4,668 | ) | $ | (4,134 | ) | $ | (3,726 | ) | $ | (3,836 | ) | ||||
| Basic and diluted net loss per common share |
$ | (0.06 | ) | $ | (0.05 | ) | $ | (0.05 | ) | $ | (0.04 | ) | ||||
| Shares used in computing basic and diluted net loss per common share |
76,363 | 76,363 | 76,368 | 105,501 | ||||||||||||
68
Table of Contents
Bionovo, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements—(Continued)
| March 31, 2008 |
June 30, 2008 |
September 30, 2008 |
December 31, 2008 |
|||||||||||||
| Revenues |
$ | — | $ | — | $ | — | $ | 233 | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
2,387 | 2,553 | 3,942 | 2,534 | ||||||||||||
| General and administrative |
1,822 | 1,808 | 1,222 | 1,245 | ||||||||||||
| Total operating expenses |
4,209 | 4,361 | 5,164 | 3,779 | ||||||||||||
| Loss from operations |
(4,209 | ) | (4,361 | ) | (5,164 | ) | (3,546 | ) | ||||||||
| Interest income |
306 | 190 | 143 | 91 | ||||||||||||
| Interest expense |
(27 | ) | (36 | ) | (36 | ) | (30 | ) | ||||||||
| Other expense |
(16 | ) | — | (1 | ) | — | ||||||||||
| Loss before income taxes |
(3,946 | ) | (4,207 | ) | (5,058 | ) | (3,485 | ) | ||||||||
| Provision for income taxes |
(3 | ) | — | — | (1 | ) | ||||||||||
| Net loss |
$ | (3,949 | ) | $ | (4,207 | ) | $ | (5,058 | ) | $ | (3,486 | ) | ||||
| Basic and diluted net loss per common share |
$ | (0.05 | ) | $ | (0.06 | ) | $ | (0.07 | ) | $ | (0.05 | ) | ||||
| Shares used in computing basic and diluted net loss per common share |
76,343 | 76,344 | 76,363 | 76,363 | ||||||||||||
11. SUBSEQUENT EVENTS
2009 Performance Bonus Options
On January 12, 2010, the Company granted approximately 657,000 options to employees in lieu of cash bonuses based on fiscal year 2009 performance. The options were fully vested upon grant date and approved by the Company’s Board of Directors in December of 2009. Accordingly, the Company has included an accrual of the related share-based compensation expense in the financial statements as of December 31, 2009 of approximately $0.2 million.
Litigation Settlement
In February of 2010, the Company reached a settlement agreement with a former officer of the Company who filed a complaint against Bionovo, Inc. and two of its officers, alleging breach of contract resulting from the rescission of the former officer’s employment agreement with the Company in September 2007. The settlement amount is included as an expense accrual in the Company’s financial statements for the year ended December 31, 2009; however the amount did not have a material impact on the Company’s financial position, results of operations or cash flows.
69
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of disclosure controls and procedures
We have performed an evaluation under the supervision and with the participation of our management, including our Chief Executive Officer (CEO) and Chief Financial Officer (CFO), of the effectiveness of our disclosure controls and procedures, as defined in Rule 13a-15(e) under the Exchange Act. Based on that evaluation, our management, including our CEO and CFO, concluded that our disclosure controls and procedures were effective as of December 31, 2009 to provide reasonable assurance that information required to be disclosed by us in the reports filed or submitted by us under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
Management’s Annual Report on Internal Control Over Financial reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Under the supervision and with the participation of the Company’s management, including the chief executive officer and principal financial officer, we evaluated the effectiveness of our internal control over financial reporting as of December 31, 2009. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control-Integrated Framework. The Company’s management has concluded that, as of December 31, 2009, our internal control over financial reporting is effective based on these criteria.
Inherent Limitations on Effectiveness of Controls
We believe that a controls system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected. Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives, and our CEO and our CFO have concluded that these controls and procedures are effective at the “reasonable assurance” level.
Changes in internal controls
There were no significant changes in our internal controls over financial reporting during the period covered by this report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting
| Item 9B. | Other Information |
None.
70
Table of Contents
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
(a) The information required by this Item concerning our directors and executive officers will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before April 30, 2010 and is incorporated herein by reference.
(b) The information required by this Item concerning our executive officers is set forth in the section entitled “Executive Officers” in Part I of this Form 10-K and is incorporated by reference into this section.
We have adopted a code of ethics that applies to all of our employees, including our principal executive officer, our principal financial officer and our principal accounting officer. This code of ethics, which is part of our Code of Business Conduct and Ethics that applies to all of our employees, is posted on our website and can be accessed from the following link: http://www.bionovo.com/home.php?menu=investors&submenu=corporateinvestors/governance
| Item 11. | Executive Compensation |
The information required by this Item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before April 30, 2010 and is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before April 30, 2010 and is incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions and Director Independence |
The information required by this Item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before April 30, 2010 and is incorporated herein by reference.
| Item 14. | Principal Accountant Fees and Services |
The information required by this Item will be set forth in the Company’s proxy statement to be filed with the Securities and Exchange Commission on or before April 30, 2010 and is incorporated herein by reference.
71
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statements Schedules |
(a) The following documents are included as part of this Annual Report on Form 10-K.
(1) Index to Financial Statements.
Included in Part II of this Report:
| Page in Form 10-K | ||
| 44 | ||
| 45 | ||
| 46 | ||
| 47 | ||
| 48 | ||
| 50 |
(2) Financial Statement Schedules.
All financial statement schedules are omitted because they are not applicable or not required or because the required information is included in the financial statements or notes thereto.
(3) Exhibits.
| Exhibit No. |
Description | |
| 2.1(1) | Agreement of Merger and Plan of Reorganization, dated as of April 6, 2005, among Lighten Up Enterprises International, Inc., LTUP Acquisition Corp. and Bionovo, Inc. | |
| 2.2(2) | Agreement and Plan of Merger, dated as of June 28, 2005, between Lighten Up Enterprises International, Inc. and Bionovo, Inc. | |
| 3.1(4) | Amended and Restated Certificate of Incorporation of Bionovo, Inc. | |
| 3.2(12) | Amended and Restated By-Laws of Bionovo, Inc. | |
| 4.1(5) | Form of Common Stock Purchase Warrant used in Private Placement. | |
| 4.2(9) | Form of Common Stock Certificate. | |
| 4.3(3) | Form of Bridge Warrant. | |
| 4.4(6) | Form of Private Placement Warrant | |
| 4.5(2) | Registration Rights Agreement, dated September 30, 2004 | |
| 4.6(8) | First Amendment to Registration Rights Agreement | |
| 4.7(8) | Amendment to Registration Rights Agreement | |
| 4.8(13) | Form of Common Stock Purchase Warrant. | |
| 4.9(14) | Common Stock Purchase Warrant | |
72
Table of Contents
| Exhibit No. |
Description | |
| 4.10(14) | Registration Rights Agreement | |
| 4.11(19) | Form of Common Stock Purchase Warrant (2009 Unit Offering) | |
| 4.12(5) | Form of Private Placement Registration Rights Agreement | |
| 10.1(5) | Form of Private Placement Subscription Agreement | |
| 10.4(5) | Form of Private Placement Acknowledgement and Amendment | |
| 10.5(10) | Stock Incentive Plan, as amended. | |
| 10.5(18) | Amended and Restated Executive Employment Agreement effective January 1, 2009, between Bionovo, Inc. and Isaac Cohen. | |
| 10.6(11) | Assignment and Assumption Agreement, dated April 6, 2005, among the Registrant, Bionovo, Inc. and Isaac Cohen regarding Employment Agreement | |
| 10.7(18) | Amended and Restated Executive Employment Agreement effective January 1, 2009, between Bionovo, Inc. and Mary Tagliaferri | |
| 10.8(11) | Assignment and Assumption Agreement, dated April 6, 2005, among the Registrant, Bionovo, Inc. and Mary Tagliaferri regarding Employment Agreement. | |
| 10.9(7) | Licensing and Technology Transfer Agreement, dated as of November 6, 2003, with United Biotech Corporation (certain terms of this agreement have been omitted and are subject to confidential treatment granted by the SEC) | |
| 10.10(3) | Landlord Consent to Sublease, dated as of December 30, 2003, with Extensity, Inc. and CA- Emeryville Properties Limited Partnership | |
| 10.11(3) | Landlord Consent to Addendum to Sublease, dated July 16, 2004, with CA-Emeryville Properties Limited Partnership and Geac Enterprise Solutions, Inc.. | |
| 10.12(3) | Office Lease, dated as of July 6, 2005, with Emery Station Joint Venture, LLC. | |
| 10.13(3) | Merrill Lynch Bank USA Irrevocable Letter of Credit | |
| 10.14(13) | Underwriting Agreement, dated October 31, 2007, between Bionovo, Inc. and BMO Capital Markets Corp. | |
| 10.15(14) | Subscription Agreement | |
| 10.16(15) | Offer Letter, dated May 29, 2007, from Bionovo, Inc. to Thomas C. Chesterman | |
| 10.17(16) | Amendment Two to Space Lease between Bionovo, Inc. and Fitzsimons Redevelopment Authority | |
| 10.27(19) | Letter Agreement, dated September 10, 2009, with Dawson James Securities, Inc. | |
| 10.28(19) | First Amendment to Letter Agreement, dated September 23, 2009, with Dawson James Securities, Inc. | |
| 10.29(19) | Second Amendment to Letter Agreement, dated September 28, 2009, with Dawson James Securities, Inc. | |
| 10.30(19) | Third Amendment to Letter Agreement, dated September 29, 2009, with Dawson James Securities, Inc. | |
| 16.1(17) | Letter regarding change in certifying accountant | |
| 23.1* | Consent of PMB Helin Donovan, LLP | |
| 31.1* | Certification of Principal Executive Officer | |
| 31.2* | Certification of Principal Financing Officer | |
| 32.1* | Certification of the Chief Executive Officer and the Chief Financial Officer | |
73
Table of Contents
| * | Filed herewith. |
| (1) | Incorporated by reference to the Company’s Current Report on Form 8-K filed with the SEC on April 8, 2005. |
| (2) | Incorporated by reference to the Company’s Definitive Proxy Statement on Schedule 14C filed with the SEC on June 3, 2005. |
| (3) | Incorporated by reference to the Company’s Registration Statement on Form SB-2, as amended, initially filed with the SEC on July 5, 2005. |
| (4) | Incorporated by reference to the Company’s Quarterly report on Form 10-Q for the period ended September 30, 2008, filed with the SEC on November 4, 2008. |
| (5) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on May 11, 2005. |
| (6) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on January 22, 2005. |
| (7) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on October 19, 2005. |
| (8) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on March 10, 2006. |
| (9) | Incorporated by reference from Exhibit 4 to the Registrant’s Form 10SB 12G filed with the SEC on November 7, 2002. |
| (10) | Incorporated by reference to the Registrant’s Schedule 14A filed with the SEC on April 17, 2006 |
| (11) | Incorporated by reference to the Registrant’s Form 8-K/A, Amendment No. 1, filed with the SEC on June 3, 2005. |
| (12) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on January 7, 2008. |
| (13) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on November 6, 2007. |
| (14) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on January 22, 2007. |
| (15) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on July 13, 2007. |
| (16) | Incorporated by reference to the Company’s Quarterly report on Form 10-QSB for the period ended March 31, 2007, filed with the SEC on May 8, 2007. |
| (17) | Incorporated by reference to the Company’s Current Report on Form 8K filed with the SEC on February 1, 2007. |
| (18) | Incorporated by reference to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2008, filed with the SEC on March 13, 2009. |
| (19) | Incorporated by reference to the Company’s Registration Statement on Form S-1, as amended, initially filed with the SEC on September 10, 2009. |
(b) Index to exhibits
See exhibits listed under Item 15(a) (3).
(c) Financial Statement Schedules.
All financial statement schedules are omitted because they are not applicable or not required or because the required information is included in the financial statements or notes thereto.
74
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized on the 15th day of March 2010.
BIONOVO, INC.
| By: |
/s/ ISAAC COHEN | |
| Isaac Cohen | ||
| Chairman and Chief Executive Officer |
KNOWN ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints, jointly and severally, Isaac Cohen and Thomas C. Chesterman, and each one of them, attorneys-in-fact for the undersigned, each with power of substitution, for the undersigned in any and all capacities, to sign any and all amendments to this Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or their substitutes, may do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated opposite his name.
Pursuant to the requirements of the Securities and Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ ISAAC COHEN Isaac Cohen |
Chairman, Chief Executive Officer and Director (Principal Executive Officer) | March 15, 2010 | ||
| /s/ THOMAS C. CHESTERMAN Thomas C. Chesterman |
Sr. VP and Chief Financial Officer (Principal Financial and Accounting Officer) | March 15, 2010 | ||
| /s/ MARY TAGLIAFERRI Mary Tagliaferri |
President, Chief Medical Officer and Director | March 15, 2010 | ||
| /s/ JOHN BAXTER John Baxter |
Director | March 15, 2010 | ||
| /s/ GEORGE BUTLER George Butler |
Director | March 15, 2010 | ||
| /s/ LOUIS DRAPEAU Louis Drapeau |
Director | March 15, 2010 | ||
| /s/ DAVID NAVEH David Naveh |
Director | March 15, 2010 | ||
| /s/ MICHAEL D. VANDERHOOF Michael D. Vanderhoof |
Director | March 15, 2010 | ||
75