Attached files
| file | filename |
|---|---|
| EX-31.1 - EX-31.1 - Sarepta Therapeutics, Inc. | a09-35851_1ex31d1.htm |
| EX-23.1 - EX-23.1 - Sarepta Therapeutics, Inc. | a09-35851_1ex23d1.htm |
| EX-32.1 - EX-32.1 - Sarepta Therapeutics, Inc. | a09-35851_1ex32d1.htm |
| EX-31.2 - EX-31.2 - Sarepta Therapeutics, Inc. | a09-35851_1ex31d2.htm |
| EX-21.1 - EX-21.1 - Sarepta Therapeutics, Inc. | a09-35851_1ex21d1.htm |
| EX-10.78 - EX-10.78 - Sarepta Therapeutics, Inc. | a09-35851_1ex10d78.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2009
o TRANSITION REPORT PURSUANT TO SECTION 13 OF 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission File Number: 001-14895
AVI BioPharma, Inc.
(Exact name of registrant as specified in its charter)
|
Oregon |
|
93-0797222 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
|
3450 Monte Villa Parkway, Suite 101, Bothell, Washington |
|
98021 |
|
(Address of principal executive offices) |
|
(Zip Code) |
Registrant’s telephone number, including area code: (425) 354 5038
Securities registered under Section 12(b) of the Act: None
Securities registered under Section 12(g) of the Act:
Common Stock with $.0001 par value
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer o |
|
Accelerated filer x |
|
Non-accelerated filer o |
|
Smaller Reporting Company o |
|
(Do not check if a smaller reporting company) |
|
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of the voting stock held by non-affiliates of the Registrant as of June 30, 2009 was approximately $128,954,972. This determination of affiliate status is not necessarily a conclusive determination for other purposes. The number of outstanding shares of the Registrant’s Common Stock as of the close of business on March 15, 2010 was 110,395,587.
Documents Incorporated by Reference
The issuer has incorporated into Part III of this annual report on Form 10-K, by reference, portions of its definitive Proxy Statement for its 2010 annual meeting.
FORM 10-K INDEX
General Overview
AVI BioPharma is a biopharmaceutical company specializing in the discovery and development of novel, RNA-based drugs targeting a range of diseases (in this report, “we,” “our,” “us,” “AVI,” and “Company” refers to AVI BioPharma, Inc.).
We are focused on the discovery and development of RNA-based drugs utilizing antisense technologies employing proprietary chemistries developed and optimized by AVI. These drugs can be designed to target disease mechanisms through distinct mechanisms of action. Unlike other RNA-based approaches, AVI’s antisense technology has been used to directly target both messenger RNA (mRNA) and precursor messenger RNA (pre-mRNA) to either down-regulate (inhibit) or up-regulate (promote) targeted genes or proteins. We believe that these broad capabilities give the Company a highly competitive RNA-based technology platform and a strong intellectual property position, both of which are the result of advances across several areas of science, including over 20 years of research and development work in chemistry and biology. Our patent estate includes 191 patents (foreign and domestic) issued to or licensed by us and 190 pending patent applications (domestic and foreign).
AVI is leveraging its discovery and development capabilities to build a pipeline of RNA-based drug candidates to develop in collaboration with larger pharmaceutical and biotechnology partners. Current applications of AVI’s RNA technology platform include genetic diseases (Duchenne muscular dystrophy), infectious diseases (including Ebola, Marburg and H1N1 Influenza viruses), and other early discovery targets. Several of our antiviral programs, including Ebola, Marburg, Junín and H1N1, have been or are currently funded by the U.S. government (see U.S. Department of Defense Agreements below). Many of our other programs have received funding from non-government sources.
Business Strategy
We believe that our RNA-based technology is applicable to the development of potential pharmaceutical products in many therapeutic areas and we intend to use data-driven decision making to exploit our core technology appropriately.
Our strategy is to:
focus on near-term opportunities in the genetic disease, immunological and infectious diseases areas;
manage drug discovery, pre-clinical and clinical development in-house;
utilize biodefense funding to advance our chemistry and its application to our antiviral programs;
bring products through early phase clinical development;
enter into collaborative development agreements with strategic partners for specific molecular targets or selected disease indications or commercialization and development of stockpiling inventories.
AVI Chemistries
AVI’s core chemistry is based on phosphordiamidate — linked morpholino oligomers or “PMOs”. PMOs are synthetic molecules based on a fundamental redesign of the natural nucleic acid structure of DNA and RNA. PMOs bind to complementary sequences of RNA by standard nucleic acid base—pairing. Structurally, the difference between PMOs on the one hand and DNA and RNA on the other hand is that while PMOs have standard nucleic acid bases, those bases are bound to morpholine rings instead of deoxyribose (in DNA) or ribose (in RNA) rings, and they are linked through phosphorodiamidate groups instead of phosphates. Replacement of anionic phosphates with the uncharged phosphorodiamidate groups eliminates ionization in the usual physiological pH range, so PMOs in organisms or cells are uncharged molecules. The entire backbone of a PMO is made from these modified subunits; they act in a drug-like manner as steric blockers and do not activate biologic mechanisms — such as RNAse H — for their mode of action.
New synthetic analogs of this original chemistry differentiate AVI’s PMO-based drug candidates from earlier-generation RNA antisense compounds and many current antisense drug candidates. We believe that these key differences provide pharmaceutical properties that are preferable for advanced antisense technology to achieve broader drug characteristics and greater potential clinical utility than the other antisense compounds.
AVI has advanced its original PMO chemistry through the addition of two new series of PMO analogues. The first is a peptide conjugated phosphorodiamidate morpholino oligomer, or PPMO, where cellular uptake of the active PMOs, as well as their potency and specificity of tissue targeting, may be significantly enhanced by the conjugation of chemical moieties to a PMO, which broadly can be considered as arginine-rich cell-penetrating peptides (CPPs).
The second analogue series includes the addition of positive charges to certain monomers in the PMO backbone. We believe that this new series, the PMOplus™, may be effective in overcoming the viral mutations that make certain RNA viruses drug-resistant. We continue to advance additional discoveries to further optimize our core proprietary chemistry as well as to develop further novel analogues that we believe could provide benefit in the key characteristics for drug action, such as potency, bioavailability, therapeutic index and tissue selectivity.
As a part of our new corporate research and development, headquartered in Bothell, Washington, we established a dedicated research chemistry group to improve certain drug-like properties of our PMO chemistry.
Translation Suppressing Oligomers (TSOs)
Translation Suppressing Oligomers (TSOs) are PMO-based antisense compounds that interfere with gene expression — and other RNA-dependent cellular processes — by binding to their specific target sequence in RNA. The primary application of TSOs is to stop or suppress the translation of a specific protein through this binding process, thus inducing a desired therapeutic effect. TSOs demonstrate tight and selective RNA binding and act by a direct steric-blocking mechanism instead of by RNAse H-mediated or RISC-mediated RNA degradation.
Splice Switching Oligomers (SSOs)
Splice Switching Oligomers (SSOs) are PMO-based compounds that can direct alternative splicing by forcing the cellular splicing machinery towards desired splicing outcomes, such as promoting the expression of a mRNA for desired protein. Sometimes these desired splicing pathways are entirely novel, i.e. they are not normally seen in the human body, and could produce important therapeutic outcomes. SSOs exploit pre-mRNA splicing to control gene function and are intended to produce a therapeutic benefit in which a protein is inhibited, increased in its expression level, changed in the overall profile of protein isoforms or results in the expression of a unique novel protein. We believe this powerful mechanism may provide significant discrimination when used for intervention in disease-causing processes.
We believe that the field of directed alternative splicing is positioned at the crucial interface of genomes, regulatory networks and evolution, and represents a rapidly emerging mechanism for gene regulation. The genetic information stored in human DNA is dispersed in short DNA stretches, called exons that code for fragments of the protein and are separated by long non-coding pieces of DNA called introns. During processing of precursor or pre-mRNA, which is copied from the DNA template, introns are removed and exons spliced together to create the mature mRNA. In mRNA, the exons are brought together, the genetic information is made contiguous and a functional protein can be translated. In alternative splicing, different pre-mRNA exons are combined, thus, creating multiple messenger RNAs and, hence, multiple proteins, all from the same gene. We believe our SSOs can be used to manipulate splicing in a way that is distinct from conventional antisense or siRNA or RNAi-based approaches.
By targeting elements in precursor RNA that are essential for splicing, SSO compounds force the cellular machinery to skip over targeted exons, creating an altered mRNA template. In a disease situation, SSOs can be designed to prevent formation of harmful proteins and/or help to restore beneficial proteins. When the exon contains a disease-causing mutation, for example, forced skipping of the harmful exon produces an altered protein which may have its function restored, partially restored or neutralized. This approach may be used to overcome the devastating consequences of certain disease-causing mutations, which are known as genetic diseases.
The Human Genome Project revealed that humans have far fewer genes than would have been predicted from the number of unique proteins that are expressed in the human proteome. Latest estimates indicate that approximately 90% of human genes are alternatively spliced. Thus, for the majority of genes, alternative splicing produces multiple proteins that can have slightly or profoundly different functions. Some pairs of splice variants code for proteins that have exactly opposite effects. Alternative splicing pathways are affected in many different diseases such that pathological protein isoforms are overproduced and the physiological isoforms are decreased. AVI’s PMO-based SSO technology enables manipulation of splicing to restore production of desired proteins, and, therefore, represents a novel therapeutic platform, with significant therapeutic potential in some previously untreatable diseases.
Therapeutic applications of SSOs include:
· Inhibition of mRNA production through a kinetically favored process
· Functional repair of RNA mutations
· Expression of novel proteins
· Alteration of protein compartmentalization
· The ability to flip an on/off control switch on specific gene targets
· Alteration of the profile of protein isoforms
The Company believes that the field of directed alternative splicing represents an exciting opportunity and has emerged as a ubiquitous and dynamic mechanism for gene regulation. Supported by a growing stream of new insights and discoveries derived from the fields of genomics, bioinformatics and molecular biology, we believe that this area promises to be a rich source of therapeutic applications.
This annual report includes our trademarks and registered trademarks, including NeuGene®, Avicine®, Resten-NG®, Resten-CP™, Oncomyc-NG™, and PMOplus™. Each other trademark, trade name or service mark appearing in this annual report belongs to its holder.
Development Programs
Our RNA-based drug programs are being clinically evaluated for the treatment of Duchenne Muscular Ddystrophy and have demonstrated promising antiviral activity in Ebola, Marburg and H1N1 virus diseases and may prove applicable to other viral targets such as HCV, Junín and Dengue viruses. We currently have products at various stages of development including those summarized below.
|
Program |
|
Mechanism |
|
Chemistry |
|
Status |
|
Developer / |
|
AVI-4658 Duchenne muscular dystrophy DMD — Exon 51 |
|
SSO |
|
PMO |
|
Phase 1 intramuscular (IM) study complete. Phase 1/2 —IV study ongoing. Orphan status granted in U.S. and EU. |
|
Proprietary |
|
AVI-5038 Duchenne muscular dystrophy DMD — Exon 50 |
|
SSO |
|
PPMO |
|
Preclinical Development. Orphan status granted in U.S. and EU. |
|
Proprietary |
|
AVI — 6002 Ebola virus |
|
TSO |
|
PMOplus™ |
|
Open IND |
|
Proprietary/U.S. Government |
|
AVI — 6003 Marburg virus |
|
TSO |
|
PMOplus™ |
|
Open IND |
|
Proprietary/U.S. Government |
|
AVI — 7367 H1N1 virus |
|
TSO |
|
PMOplus™ |
|
Discovery |
|
Proprietary/U.S. Government |
Duchenne Muscular Dystrophy (DMD) programs. We are developing a series of drugs for the potential treatment of DMD. We believe that a series of drugs skipping different exons could treat the various genotypically distinct forms of DMD. Patients with DMD have a mutation in the DNA that codes for the production dystrophin, a critical muscle protein localized beneath the sarcolemmal membrane of muscle cells. The absence of dystrophin in muscle cells leads to cell damage, an abnormally permeable cell membrane and ultimately causes muscle cell death and fibrotic replacement. We currently have two programs underway with drugs that skip exon 51 (AVI-4658) and exon 50 (AVI-5038) and are identifying lead candidates for exons 44, 45 and 53.
AVI-4658. A phase 1 human clinical trial in patients with Duchenne Muscular Dystrophy (DMD) was completed by the MDEX consortium in the United Kingdom. We announced the successful outcome of that trial in January 2009. Our SSO drug, AVI-4658, targets the most frequent mutations resulting in DMD, forces the genetic machinery to skip over an adjacent contiguous piece (one or more exons) of RNA, thus restoring the ability of the cell to express a new truncated, but functional, dystrophin protein. We believe that this may restore, prevent or slow deterioration of muscle function. This was the first clinical application of our SSO technology and entailed administration of the drug directly into an affected muscle in DMD patients. We have an ongoing phase 1b/2 systemic clinical trial with this product at the Institute of Child Health in London, UK and the University of Newcastle, UK. AVI-4658 has been granted Orphan Status in the U.S. and the EU. A set of Good Laboratory Practice (GLP) compliant preclinical studies has recently been completed and will be submitted to the FDA to support release from the full clinical hold imposed on the IND for AVI-4658. No U. S. patients have yet been treated with AVI-4658.
AVI-5038. We are conducting Good Laboratory Practice (GLP) compliant preclinical evaluations with our SSO drug, AVI-5038, which is designed to skip exon 50 and potentially overcome the mutational block and restore dystrophin expression. This drug utilizes our PPMO chemistry with the aim to enhance potency, tissue selectivity and bioavailability when compared to first generation PMO drug candidates.
AVI-5126 Prevention of Restenosis. AVI partnered with Global Therapeutics, a Cook Medical Company, to evaluate AVI’s cardiovascular restenosis drug candidate for use on a chromium cobalt drug-eluting stent (DES) for the treatment of cardiovascular restenosis. In November 2009, we announced that we believe Cook discontinued development of our drug candidate, AVI-5126, on its cobalt-chromium stent because of an unexpectedly high rate of restenosis.
Infectious Disease Programs. Our infectious disease programs are currently focusing on single-stranded RNA viruses using our proprietary TSO technology with our PMOplus™ chemistry backbone to target the often fatal diseases such as Ebola and Marburg Hemorrhagic Fevers, and H1N1 viruses, as well as other items included on the Department of Homeland Security’s list of bioterrorism agents, including Dengue and Junín viruses, anthrax and ricin.
AVI-6002 Ebola virus program, Ebola virus causes a highly lethal disease with no effective current therapy. We have demonstrated significant survival in mice, guinea pigs and monkeys when they are treated with AVI-6002 post infection with Ebola virus. In November 2008, we filed an Investigational New Drug application (IND) with the United States Food and Drug Administration (FDA). In December 2008, we received approval to move into the initial clinical study. We plan to pursue development and approval of AVI-6002 under the “Animal Rule.” The Animal Rule states that in selected circumstances, when it is unethical or infeasible to conduct human efficacy studies, the FDA may grant marketing approval based on adequate and well-controlled animal studies when the results of those studies establish that the drug or biological product is reasonably likely to produce clinical benefit in humans. Demonstration of the product’s safety in humans is still necessary. We have responded to a U.S. government request for proposal to develop AVI-6002 through to NDA. Future development of AVI-6002 is dependent on continued U.S. government funding.
AVI-6003 Marburg virus program, Marburg virus is a highly lethal virus with no effective current therapy. We have demonstrated significant survival in mice, guinea pigs and monkeys when they are treated with AVI-6003 post-infection with Marburg virus. In November 2008, we filed an Investigational New Drug application (IND) with the FDA. In December 2008, we received approval to move into the initial clinical study. We plan to pursue development and approval of AVI-6003 under the Animal Rule. Our development of AVI-6003 is currently funded by the U.S. government. Future development of AVI-6003 is dependent on continued U.S. government funding.
AVI-7367 H1N1 virus program, Pandemic H1N1 virus, also known as H1N1 or swine origin influenza virus, is often treated with the standard of care drug oseltamivir, also known as Tamiflu. AVI’s lead RNA-based candidate drug showed a statistically significantly greater reduction in viral titer and clinical scores in infected ferrets than was seen with the scrambled sequence control, the saline control or the positive control using oseltamivir.
Strategic Alliances and Other Material Agreements
We believe that our RNA-based technology could be broadly applicable for the potential development of pharmaceutical products in many therapeutic areas. To exploit our core technology as fully as possible, our strategy is to enter into strategic research and development alliances with larger pharmaceutical and biotechnology companies for specific AVI indentified molecular targets or selected disease indications, where these entities would partner in the research and commercial development of such targets and diseases. We also plan to pursue opportunities to access intellectual property rights through license agreements or other arrangements that complement our portfolio of patents and patent applications.
We currently have strategic alliances with the following companies and institutions
Isis — Ercole Agreement
In May of 2003, Ercole Biotech and Isis Pharmaceuticals entered into a collaboration and license agreement related to RNA splicing. This agreement established a cross-license between the parties granting each party certain exclusive and nonexclusive rights under a selected set of the other parties’ patents and patent applications for the research, development, and commercialization of antisense therapeutics using RNA splicing. The agreement also established that certain gene targets become exclusive to each party under and during the term of the agreement.
Subject to the satisfaction of certain milestones triggering the obligation to make any such payments, AVI may be obligated to make milestone payments of up to $23,450,000 in the aggregate for each product developed under a licensed patent under this agreement.
As of December 31, 2009, AVI has not made, and is not under any current obligation to make, any such milestone payments, as the conditions triggering any such milestone payment obligations have not been satisfied. The range of percentage royalty payments required to be made by AVI under the terms of this agreement is in the single digits.
Subject to the satisfaction of certain milestones triggering the obligation to make any such payments, Isis may be obligated to make milestone payments of up to $21,050,000 in the aggregate for each product developed under a licensed patent under this agreement. As of December 31, 2009, Isis has not made, and is not under any current obligation to make, any such milestone payments, as the conditions triggering any such milestone payment obligations have not been satisfied. The range of percentage royalty payments required to be made by Isis under the terms of this agreement is in the single digits.
This agreement will terminate on the expiration date of the last to expire patent licensed in the agreement, which expiration date is in March, 2020. Research collaboration activity defined in the agreement expired in 2006. This agreement became an obligation of the Company as a part of the acquisition of Ercole Biotech.
Chiron Agreement
In January 2006, we entered into an agreement with Chiron Corporation that granted us a nonexclusive license to Chiron’s patents and patent applications for research, development, and commercialization of antisense therapeutics against hepatitis C virus (HCV). Chiron scientists were the first to clone HCV and Chiron has been granted more than 100 HCV-related patents.
The license agreement with Chiron further strengthened our patent position on our HCV antisense product candidates, which are already covered by issued U.S. patent claims. In conjunction with the license agreement, AVI issued Chiron shares of AVI common stock as an initial license fee payment.
Subject to the satisfaction of certain milestones triggering the obligation to make any such payments, AVI may be obligated to make milestone payments of up to $5 million in the aggregate under this agreement. As of December 31, 2009, AVI has not made, and is not under any current obligation to make, any such milestone payments, as the conditions triggering any such milestone payment obligations have not been satisfied. The range of percentage royalty payments required to be made by AVI under the terms of this agreement is in the single digits. Chiron is not obligated to make any milestone payments or royalty payments under the agreement. This agreement will terminate as of the later of (i) the 20th anniversary of the effective date of the agreement, or (ii) the expiration date of the last to expire patent among certain patents issued to Chiron, which expiration date is in 2016.
Cook Group Agreement
In March 2006, we entered into agreements with Cook Group Incorporated (“Cook”) for the development and commercialization of products for vascular diseases. Cook is the world’s largest privately-held manufacturer of medical devices and is a leading designer, manufacturer and global distributor of minimally invasive medical device technologies for diagnostic and therapeutic procedures. Pursuant to our agreements, Cook licensed AVI-5126 for down-regulating c-Myc expression in the field of cardiovascular disease. Cook has taken over the clinical development of device-related programs for cardiovascular restenosis, including our AVI-5126 drug-eluting stent (DES) program, Resten-MP™ microparticle delivery program, and a program for catheter delivery of Resten-NG®.
Based on the agreements, we expect Cook to fully fund the development, clinical and regulatory costs of licensed programs in the U.S. and Europe leading to commercialization. The Company is not obligated to make any milestone payments under the agreements with Cook. Subject to the satisfaction of a commercialization milestone relating to net sales of products developed under the agreement, Cook is obligated to make a one-time milestone payment of $10 million under the license and development agreement. As of December 31, 2009, Cook has not made, and is not under any current obligation to make, any such milestone payment, as the condition triggering such milestone payment obligation has not been satisfied. The license and development agreement also provides for payment to AVI of a double-digit percentage royalty on net sales by Cook. Cook has the right to terminate the agreements upon 90 days’ written notice to AVI. AVI has the right to terminate the agreements upon 60 days’ written notice to Cook if, following an assignment of Cook’s rights under the agreements in connection with a merger or sale of assets, the assignee terminates its development efforts under the license and development agreement. In the absence of any such termination by Cook or AVI, the agreement terminates by its own terms with the expiration of the last to expire patent among certain patents. AVI does not expect to be entitled to or receive any milestone payment from Cook under this agreement. In November 2009, we announced that we believe Cook discontinued development of our drug candidate, AVI-5126, on its cobalt-chromium stent because of an unexpectedly high rate of restenosis.
Ercole Agreement
In December 2006, AVI and Ercole entered into a collaboration and license agreement for purposes of identifying and developing drugs that direct the splicing of precursor messenger RNA (pre-mRNA) to treat a variety of genetic and acquired diseases. Under the collaboration and license agreement, each party selected gene targets for their research and development efforts. Subject to the satisfaction of certain development-related milestones, Ercole was obligated to pay milestone payments to AVI of up to $2.2 million in the aggregate with respect to each therapeutic candidate resulting from Ercole’s work on the gene targets selected by Ercole. AVI had a reciprocal obligation to Ercole with respect to each therapeutic candidate resulting from AVI’s work on the gene targets selected by AVI.
Subject to the satisfaction of certain commercialization milestones relating to net sales of drugs successfully developed under the agreement, AVI was also obligated under the collaboration and license agreement to pay Ercole $20 million for each drug resulting from AVI’s work on the gene targets selected by AVI, up to a maximum aggregate amount of $100 million in such payments. The parties also had reciprocal obligations to pay a single-digit percentage royalty to one another on net sales of drugs developed from the gene targets they selected. The collaboration and license agreement provided that it would terminate as of the later of (i) the expiration date of the last to expire patent among certain patents, which expiration date is in 2020, or (ii) if all such patents were found to be invalid or unenforceable, 10 years. The obligations of the parties to one another under this agreement were terminated by operation of law when AVI acquired Ercole in March 2008. At the time the acquisition was completed, no milestone payments had been made by one party to the other, and no royalties had been paid under the agreement.
In May 2007, the parties entered into a second collaboration and license agreement for purposes of expanding the collaboration between the parties to include the discovery and development of drugs to treat muscular dystrophy and beta thalassemia. The parties agreed to share certain research and development costs under this agreement. The second collaboration and license agreement provided that it would terminate as of the later of (i) the expiration date of the last to expire patent among certain patents, which expiration date is in 2020, or (ii) if all such patents were found to be invalid or unenforceable, 10 years. The obligations of the parties to one another under this agreement were likewise terminated by operation of law when AVI acquired Ercole in March 2008.
Eleos Agreement
In January 2007, we announced that we had entered into a cross-license agreement with Eleos Inc. (“Eleos”) for the development of antisense drugs targeting p53, a well-studied human protein that controls cellular response to genetic damage. Under the terms of the agreement, AVI granted Eleos an exclusive license to AVI’s NeuGene® third-generation antisense chemistry to treat cancer with p53-related drugs. In return, Eleos granted an exclusive license to its patents to AVI for treatment of most viral diseases with drugs that target p53. The companies are sharing rights in other medical fields where targeting p53 may be therapeutically useful. Subject to the satisfaction of certain development and commercialization milestones, Eleos may be obligated to make milestone payments of up to $19.5 million in the aggregate with respect to drugs resulting from Eleos’ use of AVI intellectual property licensed to Eleos under the agreement. AVI has a reciprocal obligation to Eleos with respect to drugs resulting from AVI’s use of Eleos’ intellectual property licensed to AVI under the agreement. As of December 31, 2009, neither Eleos nor AVI has made, and neither Eleos nor AVI is under any current obligation to make, any such milestone payments, as the conditions triggering any such milestone payment obligations have not been satisfied. Percentage royalty payments required to be made by Eleos to AVI under the terms of this agreement range from single digits to low teens on net sales of drugs resulting from Eleos’ use of AVI’s intellectual property licensed to Eleos under the agreement. AVI is required to pay to Eleos a low-teens percentage royalty on net sales of drugs resulting from AVI’s use of Eleos’ intellectual property. For the fiscal years ending December 31, 2009, 2008 and 2007, AVI recognized $125,000, $125,000, and $125,000, respectively, in revenue from this agreement. This agreement will terminate as of the later of (i) the expiration date of the last to expire patent among certain patents licensed under the agreement having claims covering a product using AVI or Eleos intellectual property licensed under the agreement, which expiration date is in 2024, or (ii) 10 years from the date of the first commercial sale of a product using AVI or Eleos intellectual property licensed under the agreement.
Charley’s Fund Agreement
In October 2007, AVI and Charley’s Fund, Inc., a nonprofit organization that funds drug development and discovery initiatives specific to Duchenne muscular dystrophy (DMD), announced that AVI had been awarded a $2.45 million research grant from Charley’s Fund for the purposes of supporting a new product development program using proprietary exon skipping technologies developed by AVI to overcome the effects of certain genetic errors in the dystrophin gene. The parties entered into a sponsored research agreement in October 2007. The parties subsequently entered into an amendment of the sponsored research agreement in May 2009.
At the time of the execution of the amendment by the parties, of the $2.45 million to be paid, $2 million had already been paid to AVI by Charley’s Fund, of which $1.35 million had been spent by AVI under the terms of the agreement. The May 2009 amendment allocated the remaining $650,000 already received by AVI, but not yet spent by AVI, toward a revised list of research and development tasks to be performed by AVI. Under the terms of the May 2009 amendment, subject to the satisfaction of certain milestones, Charley’s Fund agreed that it would pay up to an additional $3 million to AVI in milestone payments over and above the $2 million it had already paid to AVI at the time of the execution of the amendment. As of December 31, 2009, Charley’s Fund has made an aggregate of $3.4 million in milestone payments to AVI, an amount which includes the $2 million amount paid to AVI prior to the execution of the amendment to the sponsored research agreement. Revenue associated with this research and development arrangement is recognized based on proportional performance applied to non-fundable payments received. AVI recognized $0, $22,500 and $37,500, respectively, in revenue from Charley’s Fund for the years ended December 31, 2009, 2008 and 2007.
Under the terms of the Sponsored Research Agreement, as amended, if AVI and any of its strategic partner(s) elect to discontinue the development and commercialization of any product containing any molecular candidate arising or derived from the research sponsored by Charley’s Fund other than for safety or efficacy, AVI has granted to Charley’s Fund an exclusive, royalty-bearing, fully-paid, worldwide license, with right of sublicense, to any such product. Depending on the timing of the obtainment of a license by Charley’s Fund to any such product, percentage royalty payments on net sales required to be made by Charley’s Fund to AVI under the terms of the sponsored research agreement, as amended, range from single digits to low teens. Under the terms of the sponsored research agreement, as amended, if the parties are able to successfully commercialize any molecular candidate arising or derived from the research sponsored by Charley’s Fund either through sales of products or through licensing or partnership arrangements with a third party that include rights for such third party to sell, distribute, promote or market such products or the underlying intellectual property, then AVI is obligated to repay the research funds paid to AVI by Charley’s Fund, up to an amount equal to the total amount of funds provided by Charley’s Fund to AVI. In connection with this repayment obligation, AVI agreed that it would pay a single-digit percentage royalty on net sales of products containing any molecular candidate arising or derived from the research sponsored by Charley’s Fund (up to an amount equal to the total amount of funds provided by Charley’s Fund to AVI). This agreement will terminate by its own terms at the completion of the research being sponsored by Charley’s Fund. The Company technology upon which the agreement is based is covered by certain patents, the last of which expires following the termination of the agreement.
U.S. Department of Defense Agreements
The Company currently has several contracts with the U.S. Department of Defense and its agencies funding its programs, including the Company’s clinical stage programs for the Ebola, Marburg, and Junín and Swine Flu viruses. The continued funding of these programs from the U.S. government is critical to the ongoing development of these programs. Future funding of these programs is subject to availability of budgeted funds from the U.S. Department of Defense. As of December 31, 2009, the Company had received an aggregate of $61.7 million in contract awards from the U.S. government, and an aggregate of $46.3 million in milestones payments had been made by the U.S. government to AVI under such awards. AVI is not required to make any milestone payments or royalty payments to the U.S. government under these contracts. Unless terminated earlier by the U.S. government, these agreements terminate upon completion of the research funded by the award. The Company technology upon which the agreements are based is covered by certain patents, all of which expire after the termination of the agreements.
Manufacturing
We believe we have developed proprietary manufacturing techniques that could allow synthesis and purification of our products to support up to Phase 2 clinical development. We are in the process of establishing relationships with multiple third parties that have established Good Manufacturing Practices (“GMP”) that will facilitate production of our products at a greater scale. We believe that we will be successful at utilizing outsourced GMP manufacturing facilities to provide sufficient manufacturing capacity to continue to meet our clinical trial requirements for the foreseeable future and allow us to produce products incorporating our technology.
Marketing Strategy
Initially, we plan to market products for which we obtain regulatory approval through marketing arrangements or other licensing arrangements with other pharmaceutical or biotechnology companies. Implementation of this strategy will depend on many factors, including the market potential of any products we develop, and our financial resources. We do not expect to establish a direct sales capability for therapeutic compounds for at least the next several years, if at all. To market products that could serve a large, geographically diverse patient population, we expect to enter into licensing, distribution, or partnering agreements with pharmaceutical companies that have, established sales organizations. We believe that the timing of our entry into marketing arrangements or other licensing arrangements with pharmaceutical companies will depend on many factors, including successful product development and regulatory approval within the regulatory framework established by the Federal Food, Drug and Cosmetics Act, as amended, and regulations promulgated thereunder and, to the extent our products are distributed outside of the United States, within the regulatory framework established in other countries. Although the implementation of initial aspects of our marketing strategy may be undertaken before this process is completed, the development and approval process typically takes at least three to five years after the filing of an IND application to complete and, thus, our marketing strategy may not be implemented for several years. See “Drug Approval Process and Other Governmental Regulation.”
Patents and Proprietary Rights
We have developed or acquired a comprehensive body of intellectual property rights. The proprietary nature of, and protection for, our product candidates, processes and know-how are important to our business. We plan to prosecute and aggressively defend our patents and proprietary technology. Our policy is to patent the technology, inventions, and improvements that we believe are important to the development of our business. We also depend upon trade secrets, know-how, and continuing technological innovation to develop and maintain our competitive position.
A patent estate including 191 patents (domestic and foreign) issued or licensed to us, and 190 pending patent applications (domestic and foreign) has been developed for purposes of protecting our technologies. We intend to protect our proprietary technology with additional filings as appropriate. Some of our patents on core technologies expired in 2008, including that for our basic PMO chemistry. Based on patented improvements and additional support to such core patents, however, we believe our patent protection for those products and other products.
We have licensed certain technology to supplement and support certain of our core technologies. We have certain obligations and minimum royalties under those agreements, which costs are not deemed material to our business.
Drug Approval Process and Other Government Regulation
The process for obtaining marketing approval of a drug in the United States is highly regulated. In the U.S. the drug discovery, testing and regulatory process to successfully develop a single product from research through market approval is estimated by the Pharmaceutical Research and Manufacturers Association to cost approximately $1.2 to $1.3 billion and take between 10 and 15 years. (Pharmaceutical Research and Manufacturers Association, Profile 2009 report)
Drug Discovery
Drug discovery is the process of discovering or designing compounds with potential therapeutic benefit. The compounds are tested in cells or animal models of against a disease target, often a protein or gene, and modified to make them more effective. From this process lead drug candidates with desired drug properties can be identified.
Preclinical Development
Using a lead candidate from the discovery process, the preclinical development stage employs a set of extensive laboratory and animal studies to establish biological activity and to gain insight into whether it might be safe and effective in humans.
Investigational New Drug Application
When a company intends to initiate human clinical trials it submits an Investigation New Drug (IND) Application to the FDA containing all the preclinical data it generated along with a clinical trial plan. The Food and Drug Administration “FDA” may prevent or delay the start of human clinical trials if not satisfied with the IND and request clarification or further data.
Phase 1I Clinical Trials
After an IND becomes effective, Phase 1 human clinical trials in the U.S. may begin. These trials usually involve small groups of healthy volunteers and are intended to provide an initial assessment of safety and evaluate how the compound affects the body.
Phase 2 Clinical Trials
Phase 2 clinical trials are generally conducted with volunteer patients with the targeted disease. The purpose of these tests is to evaluate the safety and effectiveness of the drug on the volunteer patients, to determine an optimal dose at which the test drug is deemed safe and effective.
Phase 3 Clinical Trials
Phase 3 clinical trials are normally larger scale trials to further study efficacy and to observe and report any reactions in a larger group of patients each of whom received the optimal. It is customary to agree with the FDA on the data that needs to be generated from these studies before they are started.
New Drug Application
After successful completion of clinical trials a New Drug Application (NDA) is submitted to the FDA. The NDA is a comprehensive set of documents, including all information obtained from each clinical trial, as well as all data pertaining to the manufacturing and testing of the product, as well as the results from all preclinical toxicology testing. Based upon the NDA, the FDA will either approve or deny permission to market the compound as a drug.
Marketing Approval
If the FDA approves the NDA, the drug becomes available for physicians to prescribe. Periodic reports must be submitted to the FDA, including descriptions of any adverse reactions reported. The FDA may request additional studies (Phase 4) to answer certain specific questions or concerns.
Phase 4 Clinical Trials and Post Marketing Studies
In addition to studies requested by the FDA after approval, these trials and studies are conducted to explore new indications. The purpose of these trials and studies and related publications may be to broaden the application and use of the drug and its acceptance in the medical community. Sometimes an NDA is approved with the sponsoring company being required as a condition of approval to perform specific additional work, which should be provided to the FDA in due course to maintain the product’s approval.
Many other countries and jurisdictions have similar drug development and regulatory review processes.
Competition
The biotechnology and pharmaceutical industries are highly competitive and subject to potentially rapid significant change. It can be expected that competing technologies will emerge and present a competitive challenge to us. We may also experience significant competition for drug candidates we are developing, or that we may decide to develop, because other companies may achieve significant competitive advantages over us. We may also compete against existing therapies that have an established history of safe and effective use.
We face competition from organizations pursuing the same or similar technologies that we are pursuing, as well as from organizations that are developing drugs that would compete with drug candidates we are developing. We may not be able to compete successfully against these other organizations, which include larger biotechnology and pharmaceutical companies that have substantial drug development capabilities and experience as well as greater financial, scientific and marketing resources than we do.
We believe that other biotechnology and pharmaceutical companies share a focus on RNA-based drug discovery and development. Competitors with respect to our RNA-based technologies include Alnylam Pharmaceuticals, Isis Pharmaceuticals, and Santaris.
Competitors with respect to our Duchenne muscular dystrophy (DMD) program include Prosensa and GlaxoSmithKline (“GSK), and BioMarin Pharmaceuticals. A European based clinical trial evaluating the systemic administration of the Prosensa/GSK lead DMD drug candidate started several months before the start of our similar clinical trial. The Prosensa/GSK drug candidate may, or may not, prove to be safer and more efficacious than our product candidate and it could gain marketing approval before our product candidate.
Research and Development
We expensed $24.4, $27.3 and $31.1 million on research and development activities during the years ended December 31, 2009, 2008 and 2007, respectively. Research and development (R&D) expenses include related salaries, contractor fees, materials, utilities and allocations of corporate costs. R&D expenses consist of independent R&D costs and costs associated with collaborative development arrangements. In addition, the Company funded R&D at other companies and research institutions under agreements. Research and development costs are expensed as incurred.
Employees
As of December 31, 2009, we had 63 employees. Of these employees, 23 hold advanced degrees. 45 are engaged directly in research and development activities, and 18 are in administration. None of our employees are covered by collective bargaining agreements and we consider relations with our employees to be good.
Where You Can Find Additional Information
We are a reporting company and file annual, quarterly and current reports, proxy statements and other information with the SEC. For further information with respect to us, you may read and copy our reports, proxy statements and other information, at the SEC’s public reference room at Room 1580, 100 F Street, NE, Washington, D.C. 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the public reference rooms. Our SEC filings are also available at the SEC’s web site at “http://www.sec.gov.”
Copies of our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, our proxy statement and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”) as well as our corporate governance guidelines, outline of directorship qualifications, code of business conduct and ethics and the charters of our audit committee, compensation committee, and nominating and corporate governance committees are all available on our website (www.avibio.com) or by sending a request for a paper copy to: AVI BioPharma, Inc., 3450 Monte Villa Parkway, Suite 101, Bothell WA 98021, attn. Investor Relations.
Risks Affecting Future Operating Results
The following factors should be considered in evaluating our business and prospects for the future. If risks described below actually occur, our operating results and financial condition would likely suffer and the trading price of our common stock may fall, causing a loss of some or all of an investment in our common stock. In addition, there may be additional risks not known to us or understood by us which may adversely affect our financial condition, results of operations, and the price of our stock.
We will need additional funds to conduct our planned research and development efforts. If we fail to continue to attract significant capital, we may be unable to continue to successfully develop our products.
Since we began operations, we have obtained operating funds primarily by selling shares of our common stock. Based on our current plans, we believe that current cash balances will be sufficient to meet our operating needs for the next twelve months. Furthermore, the actual amount of funds that we will need will be determined by many factors, some of which are beyond our control. These factors include the success of our research and development efforts, the status of our pre-clinical and clinical testing, costs relating to securing regulatory approvals and the costs and timing of obtaining new patent rights, regulatory changes, competition and technological developments in the market. An unforeseen change in these factors might increase our need for additional capital. We may need funds sooner than currently anticipated.
If necessary, potential sources of additional funding could include strategic relationships, public or private sales of shares of our stock, debt, or other arrangements. We may not be able to obtain additional funding when we need it on terms that will be acceptable to us or at all. If we raise funds by selling additional shares of our common stock or securities convertible into our common stock, the ownership interest of our existing shareholders will be diluted. If we were unable to obtain financing when needed, our business and future prospects would be materially adversely affected.
Our products are in an early stage of research and development and may not be determined to be safe or effective.
We are in the early stages of clinical development with respect to our RNA-based pharmaceutical products. We have devoted almost all of our resources to research and development of our product candidates, protecting our proprietary rights and establishing strategic alliances. Our potential products are in the pre-clinical or clinical stages of research and development and will require significant further research, development, clinical testing and regulatory approvals. We have no products available for sale and we do not expect to have any products available for sale for several years. Our products could be found to be ineffective or toxic, or could fail to receive necessary regulatory approvals. We have not received any significant revenues from the sale of products and we may not successfully develop marketable products that will produce sales and, given adequate margins, make us profitable. Third parties may develop superior or equivalent, but less expensive, products.
We rely on U.S. government contracts to support several important R&D programs.
We rely on U.S. government contracts and awards to fund several of our development programs, including those for the Ebola, Marburg, Junín and H1N1 viruses. The termination of one or more of these contracts, whether due to lack of funding, for convenience, or otherwise, or the occurrence of delays or product failures in connection with one or more of these contracts, could negatively impact our financial condition. Furthermore, we can give no assurance that we would be able to procure new U.S. government contracts to offset the revenues lost as a result of any termination of our contracts.
The funding of U.S. government programs is subject to Congressional appropriations. Congress generally appropriates funds on a fiscal year basis even though a program may extend over several fiscal years. Consequently, programs are often only partially funded initially and additional funds are committed only as Congress makes further appropriations. In the event that appropriations for one of our programs become unavailable, or are reduced or delayed our contracts may be terminated or adjusted by the government, which could have a negative impact on our future sales under such a contract or subcontract. From time to time, when a formal appropriation bill has not been signed into law before the end of the U.S. government’s fiscal year, Congress may pass a continuing resolution that authorizes agencies of the U.S. government to continue to operate, generally at the same funding levels from the prior year, but does not authorize new spending initiatives, during a certain period. During such a period (or until the regular appropriation bills are passed), delays can occur in government procurement due to lack of funding and such delays can affect our operations during the period of delay.
In addition, U.S. government contracts generally also permit the government to terminate the contract, in whole or in part, without prior notice, at the government’s convenience or for default based on performance. If one of our contracts is terminated for convenience, we would generally be entitled to payments for our allowable costs and would receive some allowance for profit on the work performed. If one of our contracts is terminated for default, we would generally be entitled to payments for our work that has been accepted by the government. A termination arising out of our default could expose us to liability and have a negative impact on our ability to obtain future contracts.
If we fail to receive or experience delays in receiving necessary regulatory approvals, we will be unable to develop and commercialize our product in a timely manner.
All of our products are subject to extensive regulation by the United States FDA, and by comparable agencies in other countries. The FDA and these agencies require new pharmaceutical products to undergo lengthy and detailed preclinical and clinical testing procedures and other costly and time-consuming compliance procedures. We do not know when, or if, we will be able to submit our products for regulatory review. Even if we submit a new drug application, there may be delays in obtaining regulatory approvals, if we are able to obtain them at all. Sales of our products outside the United States will also be subject to regulatory requirements governing clinical trials and product approval. These requirements vary from country to country and could delay introduction of our products in those countries. We cannot guarantee that any of our products will receive marketing approval from the FDA or comparable foreign agencies. We expect to develop the therapeutic product candidates to treat Ebola Virus and Marburg Virus under defined regulatory pathways using the Animal Rule mechanism. This mechanism has become available only relatively recently and has been infrequently used. This process has yet to be well tested and may present challenges for gaining final regulatory approval for these product candidates.
If we lose key personnel or are unable to attract and retain additional, highly skilled personnel required for our activities, our business will suffer.
The loss of key employees could significantly delay the achievement of our goals. Competition for qualified personnel in our industry is intense, and our success will depend on our ability to attract and retain highly skilled personnel. To date, we have been successful in attracting and retaining key personnel. We now have added emphasis on product development in our business plan. In addition, we are building a new chemistry-led research capability in Bothell, Washington and have outsourced our large scale manufacturing capability in Corvallis, Oregon. This short term transformation of our skill base has placed additional emphasis on our ability to attract and retain skilled personnel.
Asserting, defending and maintaining our intellectual property rights could be challenging and costly, and our failure to do so could harm our ability to compete and impair the outcome of our operations. The pharmaceutical, biotechnology and academic environments are highly competitive and competing intellectual property could limit our ability to protect our products.
Our success will depend in significant part on our existing patents and licenses (191 patents (domestic and foreign) issued or licensed to us and 190 (domestic and foreign) pending patent applications) and our ability to obtain additional patents in the future. We license patents from other parties for certain complementary technologies.
Some of our patents on core technologies expired in 2008, including for our general PMO chemistry. Based on patented improvements and inventive additions to such core patents, however, we believe the patent protection for our products in development extends beyond 2020.
We cannot be certain that pending patent applications will result in patents being issued in the United States or foreign countries. In addition, the patents that have been or will be issued may not afford meaningful protection for our technology and products. Competitors may develop products similar to ours that do not conflict with our patents. Pharmaceutical research and development is highly competitive; others may file patents first. We are aware of a patent that was issued that may provide the basis for the patent holder to assert that our drug AVI-4658 infringes on such patent. We intend to vigorously defend against any such claim if one should be asserted and believe that we may be able to invalidate some or all of the claims covered by this patent. In any case, we believe that we have freedom to operate and are moving forward with our ongoing clinical trials and drug development efforts for this drug candidate.
Others may challenge our patents and, as a result, our patents could be narrowed or invalidated. The patent position of pharmaceutical and biotechnology firms, as well as academia, is generally highly uncertain, involves complex legal and factual questions, and has recently been the subject of much litigation. No consistent policy has emerged from the United States Patent and Trademark Office (USPTO) or the courts regarding the breadth of claims allowed or the degree of protection afforded under biotechnology patents. In addition, there is a substantial backlog of pharmaceutical and biotechnology patent applications at the USPTO and the approval or rejection of patents may take several years.
Our success will also depend partly on our ability to operate without infringing upon the proprietary rights of others as well as our ability to prevent others from infringing on our proprietary rights. We may be required at times to take legal action to protect our proprietary rights and, despite our best efforts, we may be sued for infringing on the patent rights of others. We have not received any communications or other indications from owners of related patents or others that such persons believe our products or technology may infringe on their patents. Patent litigation is costly and, even if we prevail, the cost of such litigation could adversely affect our financial condition. If we do not prevail, in addition to any damages we might have to pay, we could be required to stop the infringing activity or obtain a license. Any required license may not be available to us on acceptable terms, or at all. If we fail to obtain a license, our business might be materially adversely affected.
To help protect our proprietary rights in unpatented trade secrets, we require our employees, consultants and advisors to execute confidentiality agreements. However, such agreements may not provide us with adequate protection if confidential information is used or disclosed improperly. In addition, in some situations these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants or advisors have prior employment or consulting relationships. Further, others may independently develop substantially equivalent proprietary information and techniques, or otherwise gain access to our trade secrets.
We depend on our partners and contractors for critical functions. Therefore, if our collaborations or strategic relationships are unsuccessful, our business could be harmed.
Our strategic relationships are important to our success. The discovery, development and marketing of many of our key therapeutic products are or will be dependent in large part on the efforts of our strategic partners. Our strategic partners may be unsuccessful in their attempt to develop our potential products due to circumstances that are beyond our control. The transactions contemplated by our agreements with strategic partners, including the equity purchases and cash payments, are subject to numerous risks and conditions. The occurrence of any of these events could severely harm our business.
We plan to enter into relationships with pharmaceutical or biotechnology companies to conduct late stage clinical trials and to market our products. We also plan to use contract manufacturing for late stage clinical and commercial quantities of our products. We may be unable to enter into partnerships or other relationships at all or on favorable terms, which could impede our ability to bring our products to market. Any such partnerships, if entered into at all, may be on less than favorable terms and may not result in the successful development or marketing of our products. If we are unsuccessful in establishing advantageous clinical testing, manufacturing and marketing relationships, we are not likely to generate significant revenues and become profitable.
To fully realize the potential of our products, including development, production and marketing, we may need to establish other strategic relationships.
We may get unexpected positive or negative results or outcomes during any stage of product development.
Clinical studies at all phases of clinical investigation, must be granted permission to proceed by the regulatory authorities and Institutional Review Boards (IRBs) or Ethics Committees (ECs) before they may start and these agencies review progress and safety information during the course of any study. Clinical studies are experiments designed to test a theory or hypothesis, and by their very nature, the result is unknown at the time the study is started, therefore, unexpected results or outcomes may occur that may provide positive or negative new information. Examples of unexpected results or outcomes that may occur include results that are either better than or not as good as predicted during hypothesis generation, including for example a result where the product performs better than expected and as a result of the clinical benefit a longer time is taken to reach a predefined clinical endpoint that is based on worsening disease factors, or the product may not demonstrate the predicted level of effectiveness in a specific study, or an unexpected serious adverse event may. These kinds of results or outcomes may result in the company voluntarily discussing the clinical study design, results and outcomes, and future development plans with the regulatory agencies, IRBs or ECs; any such discussions, changes in study designs or changes to the overall development plans may either accelerate or delay development of the product. Similarly, unexpected results or outcomes may occur in nonclinical animal studies or in manufacturing and quality control. This might also lead to changes in the developmental plan which could shorten or extend the time to a final study result.
We have incurred net losses since our inception and we may not achieve or sustain profitability.
We incurred a net loss of $25.2 million and $24.0 million for the years ended December 31, 2009 and 2008, respectively. As of December 31, 2009, our accumulated deficit was $275.5 million. Our losses have resulted principally from expenses incurred in research and development of our technology and products and from general and administrative expenses that we have incurred while building our business infrastructure. We expect to continue to incur significant operating losses in the future as we continue our research and development efforts and seek to obtain regulatory approval of our products. Our ability to achieve profitability depends on our ability to raise additional capital, complete development of our products, obtain regulatory approvals and market our products. It is uncertain when, if ever, we will become profitable.
Our ability to be successful against our competitors cannot be assured.
The biopharmaceutical industry is highly competitive, with a number of well-established firms performing leading-edge research for the development of new products to treat a wide range of diseases. These companies generate patents for their intellectual property rights that could preclude other companies from using similar technologies in their product development. Moreover, companies that are focused on the treatment of similar diseases are in effect competing for the same finite number of potential patients. Even if we are able to develop new products for market, there can be no assurance that we will be able to compete effectively or profitably against our competitors.
We may be subject to clinical trial claims and our insurance may not be adequate to cover damages.
We believe we carry adequate insurance for our current product development activities. In the future, commercial sale and use of our products might expose us to the risk of clinical trial claims. Although we intend to obtain product liability insurance coverage, product liability insurance may not continue to be available to us on acceptable terms and our coverage may not be sufficient to cover all claims against us. A product liability claim, even one without merit or for which we have substantial coverage, could result in significant legal defense costs, thereby increasing our expenses, lowering our earnings and, depending on revenues, potentially resulting in additional losses.
We use hazardous substances in our research activities.
We use organic and inorganic solvents and reagents in our research and development efforts that are customarily used in pharmaceutical research and development. Some of these chemicals may be classified as hazardous substances, are flammable and, if exposed to human skin, can cause anything from irritation to severe burns. We receive, store, use and dispose of such chemicals in compliance with all applicable laws with containment storage facilities and contained handling and disposal safeguards and procedures. We are routinely inspected by federal, state and local governmental and public safety agencies regarding our storage, use and disposal of such chemicals, including the federal Occupational, Safety and Health Agency (“OSHA”), the Oregon Department of Environmental Quality (“DEQ”) and the Washington Department of Ecology (‘DOE”) and local fire departments, without any material noncompliance issues in such inspections to date. Based on our limited use of such chemicals, the nature of such chemicals and the safeguards undertaken by the Company for storage, use and disposal, we believe we do not have any material exposure for toxic tort liability. Further, the cost of such compliance is not a material cost in our operating budget. While we do not have toxic tort liability insurance at this time, we believe our other insurance coverage is adequate to cover most liabilities that may arise from our use of such substances. If we are wrong in any of our beliefs, we could incur a liability in certain circumstances that would be material to our finances and the value of an investment in our securities.
Risks Related to Share Ownership
Our right to issue preferred stock, and our classified Board of Directors and Oregon Anti-Takeover laws may delay a takeover attempt and prevent or frustrate any attempt to replace or remove the then current management and Board of Director’s.
Our authorized capital consists of 200 million shares of common stock and 20 million shares of preferred stock. Our Board of Directors, without any further vote by the shareholders, has the authority to issue preferred shares and to determine the price, preferences, rights and restrictions, including voting and dividend rights, of these shares. The rights of holders of any preferred shares that our Board of Directors may issue in the future may affect the rights of the holders of shares of common stock. For example, our Board of Directors may allow the issuance of preferred shares with more voting rights, preferential dividend payments or more favorable rights upon dissolution than the shares of common stock or special rights to elect directors.
In addition, we have a “classified” Board of Directors, which means that approximately one-half of our directors are eligible for election each year. Therefore, if shareholders wish to change the composition of our Board of Directors, it could take at least two years to remove a majority of the existing directors or to change all directors. Having a classified Board of Directors may, in some cases, delay mergers, tender offers or other possible transactions that may be favored by some or a majority of our shareholders and may delay or frustrate action by shareholders to change the then current Board of Directors and management.
The Oregon Control Share Act and Business Combination Act may limit parties that acquire a significant amount of voting shares from exercising control over us for specific periods of time. These acts may lengthen the period for a proxy contest or for a person to vote their shares to elect the majority of our Board and change management.
Our stock price is volatile and may fluctuate due to factors beyond our control.
Historically, the market price of our stock has been highly volatile. The following types of announcements could have a significant impact on the price of our common stock: positive or negative results of testing and clinical trials by ourselves, strategic partners, or competitors; delays in entering into corporate partnerships; technological innovations or commercial product introductions by ourselves or competitors; changes in government regulations; developments concerning proprietary rights, including patents and litigation matters; public concern relating to the commercial value or safety of any of our products; financing or other corporate transactions; or general stock market conditions.
The significant number of our shares of common stock eligible for future sale may cause the price of our common stock to fall.
We have outstanding 110,495,587 shares of common stock as of December 31, 2009 and all are eligible for sale under Rule 144 or are otherwise freely tradable. In addition:
· Our employees and others hold options to buy a total of 8,932,811 shares of common stock, of which 5,119,227 options were exercisable at December 31, 2009. The options outstanding have exercise prices between $0.60 and $7.35 per share. The shares of common stock to be issued upon exercise of these options have been registered, and, therefore, may be freely sold when issued.
· There are outstanding warrants to buy 32,332,996 shares of common stock as of December 31, 2009 with exercise prices ranging from $.0003 to $35.63 per share. Outstanding warrants to buy 30,203,466 shares of common stock are issuable upon exercise of outstanding warrants for common stock registered for resale and may be freely sold when issued, subject to the limitations imposed by applicable securities laws.
Warrants to purchase an aggregate of 2,129,530 shares of common stock are not registered for resale. These warrants include warrants to purchase an aggregate of shares of common stock were issued to Isis Pharmaceuticals, Inc. (“ISIS”) in exchange for warrants to purchase shares of Ercole capital stock previously issued by Ercole to ISIS prior to the Company’s acquisition of Ercole. Warrants to purchase an aggregate of 1,683,545 shares of common stock issued in 2000 and prior were issued as a part of a technology licensing agreement and to a former employee.
· We may issue options to purchase up to an additional 681,955 shares of common stock as of December 31, 2009 under our stock option plans, which also will be fully saleable when issued except to the extent limited under Rule 144 for resales by our officers and directors.
Sales of substantial amounts of shares into the public market could lower the market price of our common stock.
Our common stock is listed on The NASDAQ Global Market and we may not be able to maintain that listing, which may make it more difficult for investors to sell shares of our common stock.
Our common stock is listed on The NASDAQ Global Market. The NASDAQ Global Market has several quantitative and qualitative requirements with which companies must comply in order to maintain this listing, including a $1.00 minimum bid price per share and $50 million minimum value of listed securities. If a listed company fails to meet the $1.00 minimum bid price per share requirement for 30 consecutive days, it will receive a notice from NASDAQ mandating that the company achieve compliance with the minimum bid price per share listing requirement within 180 calendar days. Our stock price is currently above $1.00; however, our stock price was priced at $0.99 as recently as May 11, 2009. There can be no assurance that we will be able to maintain compliance with the minimum bid price per share requirement in the future.
In addition to the foregoing, if we are not listed on The NASDAQ Stock Market and/or if our public float remains below $75 million, we may be limited in our ability to file new shelf registration statements on SEC Form S-3 and/or to fully use one or more registration statements on SEC Form S-3. We have relied significantly on shelf registration statements on SEC Form S-3 for most of our financings in recent years, so any such limitations might have a material adverse effect on our ability to raise any future capital we might need.
We do not expect to pay dividends in the foreseeable future.
We have never paid dividends on our shares of common stock and do not intend to pay dividends in the foreseeable future. Therefore, you should only invest in our common stock with the expectation of realizing a return through capital appreciation on your investment. You should not invest in our common stock if you are seeking dividend income.
Item 1B. Unresolved Staff Comments.
None.
We occupy 53,000 square feet of leased laboratory and office space at 4575 SW Research Way, Suite 200, Corvallis, Oregon 97333. This lease expires in December 2020. In March 2007, we purchased an additional facility, totaling 34,000 square feet, in Corvallis, Oregon which was acquired with the intention of providing the Company with future expansion space for the manufacture of potential products and components. In September we engaged a commercial real estate agent and have listed this property for sale.
In July 2009, we added a second location and leased 19,000 square feet of laboratory and office space at 3450 Monte Villa Parkway, Suite 101, Bothell, Washington. We believe that our facilities are suitable and adequate for our present operational requirements for the foreseeable future.
As of March 6, 2010, there were no material, pending legal proceedings to which we are a party. From time to time, we become involved in ordinary, routine or regulatory legal proceedings incidental to our business.
Item 4. Submission of Matters to a Vote of Security Holders.
No matters were submitted to a vote of our shareholders during the quarter ended December 31, 2009.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our Common Stock is quoted on the Nasdaq Global Market (“Nasdaq”) under the symbol “AVII.” The following table sets forth the high and low closing sales prices as reported by Nasdaq for each quarterly period in the two most recent years:
|
|
|
High |
|
Low |
|
||
|
2008 |
|
|
|
|
|
||
|
Quarter 1 |
|
$ |
1.84 |
|
$ |
1.07 |
|
|
Quarter 2 |
|
1.88 |
|
1.12 |
|
||
|
Quarter 3 |
|
1.26 |
|
0.96 |
|
||
|
Quarter 4 |
|
1.11 |
|
0.48 |
|
||
|
|
|
|
|
|
|
||
|
2009 |
|
|
|
|
|
||
|
Quarter 1 |
|
$ |
1.41 |
|
$ |
0.55 |
|
|
Quarter 2 |
|
1.87 |
|
0.67 |
|
||
|
Quarter 3 |
|
2.47 |
|
1.25 |
|
||
|
Quarter 4 |
|
1.92 |
|
1.39 |
|
||
Holders
As of March 1, 2010, we had 602 shareholders of record.
Dividends
There were no cash dividends declared or paid in years 2009 or 2008. We do not anticipate declaring such dividends in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plans
Equity Compensation Plan Information
|
Plan category |
|
Number of securities to |
|
Weighted-average |
|
Number of securities |
|
|
|
Equity compensation plans approved by security holders |
|
8,932,811 |
|
$ |
2.79 |
|
681,955 |
|
|
|
|
|
|
|
|
|
|
|
|
Equity compensation plans not approved by security holders |
|
-0- |
|
— |
|
-0- |
|
|
|
Total |
|
8,932,811 |
|
$ |
2.79 |
|
681,955 |
|
(1) The number of securities remaining available for future issuance under equity compensation plans includes shares from the Company’s 2002 Equity Incentive Plan (the “2002 Plan”). The number of shares reserved for issuance is increased by an automatic annual share increase pursuant to which the number of shares available for issuance under the 2002 Plan automatically increases on the first trading day of each year (the “First Trading Day”), beginning with the 2003 fiscal year and continuing through the fiscal year 2011, by an amount equal to two percent (2%) of the total number of shares outstanding on the last trading day of the immediately preceding fiscal year; such increases being subject to the limitation in the next sentence. The 2002 Plan provides that, following any such adjustment, the number of then outstanding options under the Company’s stock option plans and stock purchase plans, together with options in the reserve then available for future grants under the Company’s stock option plans, will not exceed twenty percent (20%) of the then outstanding voting shares of capital stock of the Company, and all the actually outstanding stock options under the Company’s stock option plans, together with all shares in the reserve then available for future grants under the Company’s stock option and stock purchase plans. This automatic share increase feature is designed to assure that a sufficient reserve of Common Stock remains available for the duration of the 2002 Plan to attract and retain the services of key individuals essential to the Company’s long-term growth and success. This feature is also designed to eliminate the uncertainty inherent in seeking an individual increase to the reserve each year as to what number of shares will be available in the reserve for option grants. Creating a certain rate of growth under the 2002 Plan assists the Company as it makes strategic personnel decisions in an effort to expand its growth, as the Company will know the approximate number of shares that will become available for issuance under the 2002 Plan. At the same time, the Company has attempted to minimize the dilutive effect that the issuance of Common Stock upon the exercise of options can have on stockholders’ percentage of ownership in the Company by adopting only a 2% growth rate for the 2002 Plan. This rate, while it provides room for growth in the 2002 Plan, is a rate which the Company believes it can reasonably sustain, minimizing the risk to stockholders that the option reserve grows faster than the Company itself. The twenty percent (20%) limitation discussed above further protects shareholders by capping the size of the 2002 Plan in relation to the Company’s other securities.
Performance Graph
The following graph compares the performance of the Company’s Common Stock for the periods indicated with the performance of the NASDAQ Composite Index and the Amex Biotech Index. This graph assumes an investment of $100 on December 31, 2004 in each of the Company’s common stock, the NASDAQ Composite Index and the Amex Biotech Index, and assumes reinvestment of dividends, if any. The stock price performance shown on the graph below is not necessarily indicative of future stock price performance.
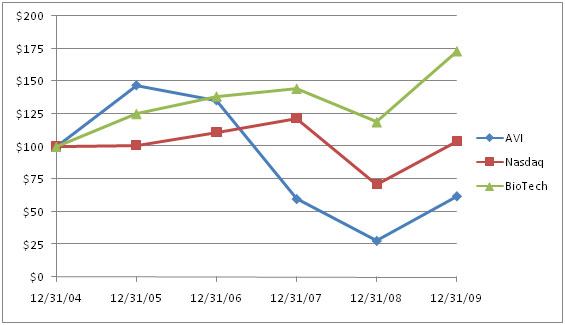
|
|
|
AVII |
|
NASDAQ |
|
Amex |
|
|||
|
|
|
|
|
|
|
|
|
|||
|
End of Fiscal 2004 |
|
$ |
100.00 |
|
$ |
100.00 |
|
$ |
100.00 |
|
|
End of Fiscal 2005 |
|
$ |
146.81 |
|
$ |
101.24 |
|
$ |
125.11 |
|
|
End of Fiscal 2006 |
|
$ |
135.32 |
|
$ |
110.88 |
|
$ |
138.59 |
|
|
End of Fiscal 2007 |
|
$ |
60.00 |
|
$ |
121.76 |
|
$ |
144.51 |
|
|
End of Fiscal 2008 |
|
$ |
28.09 |
|
$ |
71.19 |
|
$ |
118.91 |
|
|
End of Fiscal 2009 |
|
$ |
62.13 |
|
$ |
104.17 |
|
$ |
173.11 |
|
Recent Sales of Unregistered Securities.
None.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
None.
Item 6. Selected Financial Data.
The following selected financial data is derived from our audited financial statements and should be read in conjunction with, and is qualified in its entirety by, Item 7. “Management’s Discussion and Analysis or Plan of Operation” and Item 8. “Financial Statements.”
|
|
|
YEAR ENDED DECEMBER 31, |
|
|||||||||||||
|
(in thousands) |
|
2009 |
|
2008 |
|
2007 |
|
2006 |
|
2005 |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Operations data: |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Revenues |
|
$ |
17,585 |
|
$ |
21,258 |
|
$ |
10,985 |
|
$ |
115 |
|
$ |
4,784 |
|
|
Research and development |
|
24,396 |
|
27,331 |
|
31,058 |
|
25,346 |
|
17,118 |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
General and administrative |
|
8,696 |
|
11,469 |
|
13,035 |
|
7,753 |
|
5,182 |
|
|||||
|
Acquired in-process research and development |
|
— |
|
9,916 |
|
— |
|
— |
|
— |
|
|||||
|
Operating Loss |
|
(15,507 |
) |
(27,458 |
) |
(33,108 |
) |
(32,984 |
) |
(17,516 |
) |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Interest (expense) income, and other net |
|
(454 |
) |
344 |
|
984 |
|
1,910 |
|
840 |
|
|||||
|
Decrease (increase) on warrant valuation |
|
(9,198 |
) |
3,161 |
|
4,956 |
|
2,386 |
|
(1,530 |
) |
|||||
|
Net loss |
|
(25,159 |
) |
(23,953 |
) |
(27,168 |
) |
(28,688 |
) |
(18,206 |
) |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Net loss per share - basic and diluted |
|
$ |
(0.27 |
) |
$ |
(0.34 |
) |
$ |
(0.50 |
) |
$ |
(0.54 |
) |
$ |
(0.41 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Balance sheet data: |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Cash and investments |
|
$ |
48,446 |
|
$ |
11,474 |
|
$ |
25,074 |
|
$ |
33,152 |
|
$ |
47,051 |
|
|
Working capital |
|
17,803 |
|
9,756 |
|
18,959 |
|
25,596 |
|
38,327 |
|
|||||
|
Total assets |
|
60,027 |
|
25,536 |
|
38,638 |
|
40,863 |
|
56,408 |
|
|||||
|
Shareholders’ equity |
|
23,630 |
|
15,732 |
|
26,382 |
|
32,519 |
|
46,082 |
|
|||||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operation
Forward-Looking Information
This report contains forward-looking statements regarding our plans, expectations, estimates and beliefs. Our actual results could differ materially from those discussed in, or implied by, these forward-looking statements. Forward-looking statements are identified by words such as “believe,” “anticipate,” “expect,” “intend,” “plan,” “will,” “may,” and other similar expressions. In addition, any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements. We have based these forward-looking statements largely on our expectations. Forward-looking statements in this report include, but are not necessarily limited to, those relating to:
· our intention to introduce new products,
· receipt of any required FDA or other regulatory approval for our products,
· our expectations about the markets for our products,
· acceptance of our products, when introduced, in the marketplace,
· our expectations about availability of government funding for certain projects,
· our future capital needs,
· results of our research and development efforts, and
· success of our patent applications.
Forward-looking statements are subject to risks and uncertainties, certain of which are beyond our control. Actual results could differ materially from those anticipated as a result of the factors described in the “Risk Factors” and detailed herein and in our other Securities and Exchange Commission filings, including among others:
· the effect of regulation by the FDA and other governmental agencies,
· delays in obtaining, or our inability to obtain, approval by the FDA or other regulatory authorities for our products,
· research and development efforts, including delays in developing, or the failure to develop, our products,
· uncertainty of government funding for certain projects,
· the development of competing or more effective products by other parties,
· the results of pre-clinical and clinical testing and our ability to conduct these tests,
· uncertainty of market acceptance of our products,
· problems that we may face in manufacturing, marketing, and distributing our products,
· our inability to raise additional capital when needed,
· delays in the issuance of, or the failure to obtain, patents for certain of our products and technologies, and
· problems with important suppliers and business partners.
Because of these risks and uncertainties, the forward-looking events and circumstances discussed in this report or incorporated by reference might not occur. Factors that cause actual results or conditions to differ from those anticipated by these and other forward-looking statements include those more fully described in the “Risk Factors” section and elsewhere in this report.
Overview
From our inception in 1980, we have devoted our resources primarily to fund our research and development efforts. We have been unprofitable since inception and we have had no material revenues from the sale of products or from other sources other than from government grants and research contracts; we do not expect material revenues for the foreseeable future. We expect to continue to incur losses for the foreseeable future as we continue our research and development efforts and enter into additional collaborative efforts. As of December 31, 2009, our accumulated deficit was $275.5 million.
Results of Operations
Summary of Results for Fiscal Years 2009, 2008, and 2007
|
(In thousands, except percentages and per share amounts) |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
Revenue |
|
$ |
17,585 |
|
$ |
21,258 |
|
$ |
10,985 |
|
|
Operating loss |
|
$ |
(15,507 |
) |
$ |
(27,458 |
) |
$ |
(33,108 |
) |
|
Net loss |
|
$ |
(25,159 |
) |
$ |
(23,953 |
) |
$ |
(27,168 |
) |
|
Diluted earnings per share |
|
$ |
(0.27 |
) |
$ |
(0.34 |
) |
$ |
(0.50 |
) |
Year Ended December 31, 2009 Compared with Year Ended December 31, 2008.
Revenues decreased to $17.6 million in 2009 compared to $21.3 million in 2008. The decrease in research contracts revenues was the result of the decline in revenues from government research contracts, due to lower levels of project activities related to the DTRA Ebola, Marburg and Junín contract.
Operating loss decreased to $15.5 million in 2009 compared to $27.5 million in 2008. In 2008, the operating loss included a charge of $9.9 million for acquired in-process research and development associated with the acquisition of Ercole Biotech, Inc. The operating loss in 2009 also decreased compared to 2008 as a result of lower research and development and general and administrative expenses.
Research and development expenses decreased to $24.4 million in 2009 compared to $27.3 million 2008. Lower research and development expenses in 2009 compared to 2008 result primarily from less spending on government research projects.
General and administrative expenses decreased to $8.7 million in 2009 compared to $11.5 million in 2008. The decrease in general and administrative expenses was due primarily to non-cash costs recognized in 2008 primarily due to stock issued to Ercole executives related to the prior year acquisition, 2008 severance and stock compensation expenses related to the resignation of former executive officers, and relocation costs for new executive officers.
Interest income and other expense, net declined $0.8 million in 2009 as compared to 2008 primarily due to declines in market rates of interest on the Company’s interest-earning investments and the write off of valuations for patents, property and equipment.
The increase on warrant liability valuation of $9.2 million in 2009 compared to the decrease on warrant liability valuation of $3.2 million, in 2008, is a non-cash expense and is the result of the new warrants issued in 2009 and the increase in the Company’s stock price. The decrease or increase on the warrant liability valuation fluctuates as the market price of the Company’s stock fluctuates.
The net loss increased to $25.2 million in 2009 from $24.0 million in 2008. The net loss increased primarily due to the increase in the warrant valuation, a non-cash expense that resulted from the fluctuation in the Company’s stock price.
Year Ended December 31, 2008 Compared with Year Ended December 31, 2007.
Revenues, from license fees, grants and research contracts, increased to $21.3 million 2008 from $11.0 million in 2007. The increase in revenues for 2008 was primarily due to the increase from government funding for work performed on viral disease research projects.
Operating loss decreased to $27.5 million in 2008 compared to $33.1 million in 2007. In 2008, the operating loss included a one-time charge of $9.9 million for acquired in-process research and development associated with the acquisition of Ercole Biotech, Inc. The operating loss in 2008 also decreased compared to 2007 as a result of higher revenue and lower research and development expenses and lower general and administrative expenses.
Research and development expenses decreased to $27.3 million in 2008 compared to $31.1 million 2007. Lower research and development expenses in 2008 compared to 2007 results primarily from lower spending on manufacturing compounds for use in clinical trials.
General and administrative expenses decreased to $11.5 million in 2008 compared to $13.0 million in 2007. The decrease in general and administrative expenses was due primarily to decreases in compensation costs, legal and investor relation expenses.
Net interest income declined in 2008 as compared to 2007 primarily due to declines in the average balances in cash and cash equivalents, combined with lower market rates of interest on the Company’s interest-earning investments.
The decrease on the warrant liability valuation of $3.2 million in 2008 compared to the decrease on warrant liability valuation of $5.0 million in 2007 is a non-cash expense and is the result of the decrease in the Company’s stock price. The decrease or increase on the warrant liability valuation fluctuates as the market price of the Company’s stock fluctuates.
Research and Development Expenses
Our research and development costs allocated by focus area for the year ended December 31, 2009 were as follows:
(in thousands)
|
As of December 31, |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
(in thousands) |
|
|
|
|
|
|
|
|||
|
Focus areas: |
|
|
|
|
|
|
|
|||
|
Government funded projects |
|
$ |
12,978 |
|
$ |
17,581 |
|
$ |
8,033 |
|
|
Other internal projects |
|
11,418 |
|
9,750 |
|
23,025 |
|
|||
|
|
|
|
|
|
|
|
|
|||
|
Total research and development expense |
|
$ |
24,396 |
|
$ |
27,331 |
|
$ |
31,058 |
|
Direct research and development costs associated with our programs include clinical trial site costs, clinical manufacturing costs, costs incurred for consultants and other outside services, such as data management and statistical analysis support, and materials and supplies used in support of the clinical programs, as well as other direct research. Indirect costs of our clinical program include wages, payroll taxes and other employee-related expenses including rent, restructuring, stock based compensation, utilities and other facilities-related maintenance. Costs attributable to our discovery research programs represent our efforts to develop and expand our product pipeline. The amount and timing of future research and development expense will depend on the Company’s ability to obtain U.S. government awards to fund the advanced development of its antiviral therapeutic candidates. Without future government awards, the Company would likely drastically reduce its spending in these areas. Future research and development costs may also increase if our internal projects, such as Duchene Muscular Dystrophy, enter later stage clinical development.
While we believe our programs are promising, we do not know whether any commercially successful products will result from our research and development efforts. Thus, we believe that the nature, timing, and estimated costs of the efforts necessary to complete the projects and the anticipated completion dates, are not estimable due to many factors, including the following:
· Delivery strategies and potency enhancements of the Company’s compounds are still being developed and explored;
· Variability among different disease categories result in successful delivery strategies or potency enhancements not necessarily being applicable across different disease categories;
· Costs of clinical trials, like costs of all forms of medical care, are rapidly changing;
· Variability among different disease categories in terms of such factors as dosages, duration of treatment, method of administration, and others exist;
· Rules surrounding filings and conduct of clinical trials are changing;
· Confidentiality surrounding commercialization is heightening; and
· Clinical endpoints are in a constant state of flux.
Liquidity and Capital Resources
Since its inception in 1980 through December 31, 2009, the Company has incurred losses of approximately $275.5 million, substantially all of which resulted from expenditures related to research and development, general and administrative charges and acquired in-process research and development resulting from two acquisitions. The Company has not generated any material revenue from product sales to date, and there can be no assurance that revenues from product sales will be achieved. Moreover, even if the Company does achieve revenues from product sales, the Company expects to incur operating losses over the next several years.
Our cash, cash equivalents and short-term securities were $48.4 million at December 31, 2009, compared with $11.5 million at December 31, 2008, respectively. The increase was due primarily to net proceeds of $47.8 million from the sale of common stock and issuance of stock warrants from two separate equity financing transactions that closed in January and August of 2009. The cash from financing activities was partially offset by cash used in operations of $8.8 million, costs of $2.0 million related to acquisitions of patents and fixed assets and debt repayments of $0.1 million.
On January 30, 2009, the Company closed a registered equity financing for net proceeds of $15.5 million with several institutional investors. The Company sold 14,224,202 shares of common stock at $1.16 per share, and also issued warrants for the purchase of 14,224,202 common shares at $1.16 per share. The warrants had fair value at the date of issue of $8.2 million. These warrants are exercisable starting July 30, 2009 and expire on July 30, 2014. In connection with the equity financing, the placement agent received a warrant for the purchase of an additional 426,726 common shares at $1.45 per share. This warrant is exercisable starting January 30, 2009 and expires on January 30, 2014. All of these warrants have been classified as liabilities as they require the issuance of registered shares. These warrants are non-cash liabilities; The Company does not expect to expend any cash to settle these liabilities.
On August 25, 2009, the Company closed a registered equity financing for net proceeds of $32.3 million with several institutional investors. The Company sold 24,295,775 shares of common stock at $1.42 per share, and also issued warrants for the purchase of 9,718,310 common shares at $1.78 per share. The warrants had fair value at the date of issue of $9.0 million. These warrants are exercisable starting February 25, 2010 and expire on August 25, 2014. All of these warrants have been classified as liabilities as, as they require the issuance of registered shares. These warrants are non-cash liabilities; The Company does not expect to expend any cash to settle these liabilities.
The Company believes it has sufficient cash to fund operations at least through the following twelve months, exclusive of future receipts from billings on existing government contracts. For 2010, the Company expects expenditures for operations, net of government funding, including collaborative efforts and research and development activities to be approximately $23 to $27 million. The Company believes it will continue to receive funding from government and other sources to pursue the development of its product candidates, and has assumed certain revenues from these awards in providing this guidance. Should the Company not continue to receive funding from its current contracts or receive additional funding, or should the timing be delayed, it may have a significant negative impact on the Company’s guidance.
Because of the cost (up to $1.318 billion) and timeframe (of 10 to 15 years) as published by Wiley InterScience, ã2007 John Wiley and Sons, LTD, generally associated with developing a potential drug or pharmaceutical product to the point of approval by the FDA or other regulatory agencies for human use, our general business strategy is to develop our products up through human clinical trials and then look for third parties to fund further development of the product and to market the product through strategic partnerships, license agreements or other relationships. We also look for collaborative and other efforts to utilize other technology to increase the potential variety and reduce the cost of identifying products. We believe that this strategy could reduce the potential costs we would otherwise incur in developing a product and increase the likelihood of getting a product to market. Our expected costs under our various contracts and for various drug development products can be estimated for the next year or two, but not thereafter with much certainty due to the uncertainty of clinical trial results, research results and the timing of securing one or more partners to develop and market a potential drug.
Because of the various factors noted above and the expectation that, until we establish revenue sources, we will license or jointly develop our prospective products to or with strategic partners, we review, at least annually, each research program and clinical trial based on results and progress during the prior year and estimate our needs for that program or trial for the coming year, making adjustments based on the progress of the program during the year.
The Company currently has a total of $61.7 million of contracted development studies. As of December 31, 2009, $48.4 million has been billed to the government. The Company has $13.3 in development contracts remaining that have not yet been completed and have not been billed. The Company expects to complete the remaining contract activity and receive the contracted revenue in 2010 and early 2011.
Summary of Material Revenue Contracts
In December 2006, the Company announced the execution of a two-year $28 million research contract with the Defense Threat Reduction Agency (DTRA), an agency of the United States Department of Defense (DoD). The contract is directed toward funding the Company’s development of antisense therapeutics to treat the effects of Ebola, Marburg and Junín hemorrhagic viruses, which are seen by DoD as potential biological warfare and bioterrorism agents. In May 2009, the Company received an amendment from DTRA to extend the contract performance period to November 29, 2009 and a cost modification of an additional $5.9 million, increasing the total contract amount to $33.9 million. In September 2009, the Company received a second amendment from DTRA to extend the contract performance period to February 28, 2011 and a cost modification of an additional $11.5 million, increasing the total contract amount to $45.4 million.
During the twelve month period ended December 31, 2009, 2008 and 2007, the Company recognized $10.4 million, $16.8 million and $8.0 million, respectively, in research contract revenue from this contract. To date, the Company has recognized revenues of $35.2 million from this contract. Funding of the remainder of the contract is anticipated in 2010 and 2011.
In January 2006, the Company announced that the final version of the 2006 defense appropriations act had been approved, which included an allocation of $11.0 million to fund the Company’s ongoing defense-related programs. Net of government administrative costs, it is anticipated that the Company will receive up to $9.8 million under this allocation. The Company’s technology is expected to be used to continue developing RNA based drugs agents against Ebola and Marburg viruses. The Company has received signed contracts for all of these projects. The Company expects that funding under these signed contracts will be completed over the next 12 months. During the twelve month period ended December 31, 2009, 2008 and 2007, the Company recognized $2.3 million, $4.2 million and $2.7 million, respectively, in research contract revenue from this contract. To date, the Company has recognized revenues of $9.2 million on these contracts. Funding of the remainder of these contracts is anticipated in 2010.
In May 2009, the Company entered into a contract with DTRA to develop H1N1 drugs. Under this contract, DTRA will pay up to $4.1 million to the Company for the work to be performed by the Company. The work will involve the application of analogs based on the Company’s proprietary PMO chemistry and the Company plans to conduct preclinical development of at least one drug candidate and demonstrate it is effective by testing it in virus-infected animals. During the twelve month period ended December 31, 2009, the Company recognized $1.7 million in revenue under this contract.
In September 2009, the Company and Charley’s Fund, Inc. (“Charley’s Fund”), a nonprofit organization that funds drug discovery and development initiatives specific to DMD, entered into the First Amendment to an existing Sponsored Research Agreement (the “Amendment”). The Amendment pertains to certain provisions of the Sponsored Research Agreement by and between the Company and Charley’s Fund entered into effective October 12, 2007 (the “Agreement”). Under the terms of the Amendment, the Company was awarded up to an additional $3 million in sponsored research funds, for a total of $5 million from Charley’s Fund to support a new product development program using proprietary exon skipping technologies developed by the Company to overcome the effects of certain genetic errors in the dystrophin gene. In December 2009, the Company received $1.4 million that was recorded as deferred revenue. Revenue associated with this research and development arrangement is recognized under the proportional performance method, using the payment received method. The Company did not recognize any revenue under this contract in 2009.
We do not expect any material revenues in 2010 from our business activities except for revenues from U.S. government contracts and other agreements. We expect that our cash requirements for the next twelve months will be satisfied by existing cash resources and these revenues. To fund our operations beyond the next twelve months, we may need to raise additional capital. We will continue to look for opportunities to finance our ongoing activities and operations through accessing corporate partners or the public equity markets, as we currently have no credit facility and do not intend to seek one.
The likelihood of the long-term success of the Company must be considered in light of the expenses, difficulties and delays frequently encountered in the development and commercialization of new pharmaceutical products, competitive factors in the marketplace as well as the complex regulatory environment in which the Company operates. There can be no assurance that the Company will ever achieve significant revenues or profitable operations.
Off-Balance Sheet Arrangements
The Company’s off-balance sheet arrangements are limited to operating leases and rents on certain facilities and equipment and license agreements for which it is obligated to pay the licensors a minimum annual royalty. These off-balance sheet arrangements are expensed as incurred. In 2009, these expenses totaled $1,467,000 for operating leases and $75,000 for royalty payments.
Contractual Payment Obligations
A summary of our contractual commitments and obligations as of December 31, 2009 is as follows:
|
|
|
Payments Due By Period |
|
|||||||||||||
|
Contractual Obligations (in |
|
Total |
|
2010 |
|
2011 and 2012 |
|
2013 and 2014 |
|
2015 and |
|
|||||
|
Operating leases |
|
$ |
19,682 |
|
$ |
2,073 |
|
$ |
4,405 |
|
$ |
4,069 |
|
$ |
9,135 |
|
|
Royalty payments |
|
1,110 |
|
100 |
|
160 |
|
135 |
|
715 |
|
|||||
|
|
|
$ |
20,792 |
|
$ |
2,173 |
|
$ |
4,565 |
|
$ |
4,204 |
|
$ |
9,850 |
|
Our future expenditures and capital requirements depend on numerous factors, most of which are difficult to project beyond the short term. These requirements include the progress of our research and development programs and our pre-clinical and clinical trials, the time and costs involved in obtaining regulatory approvals, the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, competing technological and market developments, our ability to establish collaborative arrangements and the terms of any such arrangements, and the costs associated with commercialization of our products. Our cash requirements are expected to increase as we advance our research projects. There can be no assurance, however, that we will ever be able to generate product revenues or achieve or sustain profitability.
New Accounting Pronouncements
See Note 2 of Notes to Financial Statements with this report on Form 10-K included under Part III, Item 15.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to stock-based compensation, valuation of investments, long-lived assets, and revenue recognition. We base our estimates on historical experience and on various other assumptions. Actual results may differ from these estimates under different assumptions or conditions. We believe the following critical accounting policies and the related judgments and estimates affect the preparation of our financial statements.
Revenue Recognition
Revenue is recorded from research contracts and grants as the services are performed and payment is reasonably assured. Upfront, nonrefundable fees and other fees associated with license and development arrangements are recognized as revenue ratably over the performance period. Revenue associated with research and development arrangements is recognized under the proportional performance method, using the payment received method. To date, revenue from research and development arrangements has not been material.
Long-Lived Asset Impairment
Long-lived assets held and used by us and intangible assets with determinable lives are reviewed for impairment whenever events or circumstances indicate that the carrying amount of assets may not be recoverable in accordance with generally accepted accounting principles. We evaluate recoverability of assets to be held and used by comparing the carrying amount of an asset to future net undiscounted cash flows to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amount of the assets exceeds the fair value of the assets. Such reviews assess the fair value of the assets based upon estimates of future cash flows that the assets are expected to generate.
Stock-based Compensation Expense
Stock-based compensation costs are generally based on the fair value calculated from the Black-Scholes option-pricing model on the date of grant for stock options and on the date of enrollment for the Plan. The fair value of stock grants are amortized as compensation expense on a straight-line basis over the vesting period of the grants. Compensation expense recognized is shown in the operating activities section of the statements of cash flows. Stock options granted to employees are service-based and typically vest over three years.
The fair market values of stock options granted were measured on the date of grant using the Black-Scholes option-pricing model, with weighted average assumptions for the risk-free interest rate, expected dividend yield, expected lives, and expected volatility. The Company is required to estimate potential forfeiture of stock grants and adjust compensation cost recorded accordingly. The estimate of forfeitures will be adjusted over the requisite service period to the extent that actual forfeitures differ, or are expected to differ, from such estimates. Changes in estimated forfeitures will be recognized through a cumulative catch-up in the period of change and will also impact the amount of stock compensation expense to be recognized in future periods.
The assumptions used in calculating the fair value of stock-based compensation expense represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and the Company uses different assumptions, its stock-based compensation expense could be materially different in the future. See Note 3 to Notes to Financial Statements for a further discussion of stock-based compensation.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
The primary objective of our cash investment activities is to preserve principal and avoid significant market risk. As of December 31, 2009, we held more than 99% of our cash in a sweep money market account, with the remainder held in non-interest bearing checking accounts or short-term certificates of deposit. We have no holdings of derivative financial or commodity instruments. We do not anticipate making significant changes in how we hold our cash. Accordingly, we believe our credit risk is immaterial.
Item 8. Financial Statements and Supplementary Data.
The information required by this Item 8 begins on page F-1 in Item 15 of Part III of this report on Form 10-K and is incorporated into this item by reference.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Disclosure Controls and Procedures
We carried out an evaluation as of the end of period covered by this report, under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of our disclosure controls and procedures pursuant to paragraph (b) of Rule 13a-15 and 15d-15 under the Exchange Act. Based on that review, the Chief Executive Officer and the Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that information required to be disclosed by the Company in the reports it files or submits under the Exchange Act (1) is recorded, processed, summarized, and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and (2) is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
The Company does not expect that its disclosure controls and procedures will prevent all error and all fraud. A control procedure, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control procedure are met. Because of the inherent limitations in all control procedures, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The Company considered these limitations during the development of it disclosure controls and procedures, and will continually reevaluate them to ensure they provide reasonable assurance that such controls and procedures are effective.
Internal Control over Financial Reporting
Management’s Annual Report on Internal Control over Financial Reporting
The management of AVI BioPharma, Inc. (the Company or AVI) is responsible for establishing and maintaining adequate internal control over financial reporting. The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
· Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company;
· Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and
· Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2009. In making this assessment, the Company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in “Internal Control-Integrated Framework.” Based on management’s assessment and those criteria, we believe that, as of December 31, 2009, the Company’s internal control over financial reporting is effective.
Changes in Internal Control over Financial Reporting
There have not been any changes in the Company’s internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the Company’s fourth fiscal quarter that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
AVI BioPharma, Inc:
We have audited AVI BioPharma, Inc.’s (a development stage enterprise) internal control over financial reporting as of December 31, 2009, based on criteria established in lnternal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). AVI BioPharma, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Managements‘ Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, AVI BioPharma, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of AVI BioPharma, Inc. (a development stage enterprise) as of December 31, 2009 and 2008, and the related statements of operations, shareholders’ equity and comprehensive income (loss), and cash flows for each of the years in the three-year period ended December 31, 2009 and the information included in the cumulative from inception presentations for the period January 1,2002 to December 31,2009 (not separately presented herein). These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. The financial statements of AVI BioPharma, Inc. for the period July 22, 1980 (inception) to December 31, 2001 were audited by other auditors who have ceased operations. Those auditors expressed an unqualified opinion on those financial statements in their report dated February 21, 2002. Our report dated March 16, 2010 expressed an unqualified opinion on those financial statements.
|
/s/ KPMG LLP |
|
|
|
|
|
Portland, Oregon |
|
|
March 16, 2010 |
|
None.
Item 10. Directors, Executive Officers and Corporate Governance.
Information regarding our directors and executive officers required by this item is included in our definitive proxy statement for our 2010 annual meeting of shareholders to be filed with the Commission not later than 120 days after the end of the fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 11. Executive Compensation.
The information required by this item is included in our definitive proxy statement for our 2010 annual meeting of shareholders to be filed with the Commission not later than 120 days after the end of the fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item is included in our definitive proxy statement for our 2010 annual meeting of shareholders to be filed with the Commission not later than 120 days after the end of the fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item is included in our definitive proxy statement for our 2010 annual meeting of shareholders to be filed with the Commission not later than 120 days after the end of the fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services.
The information required by this item is included in our definitive proxy statement for our 2010 annual meeting of shareholders to be filed with the Commission not later than 120 days after the end of the fiscal year covered by this Annual Report and is incorporated herein by reference.
Item 15. Exhibits, Financial Statement Schedules.
(a) The following documents are filed as part of this Report:
(1) Financial Statements
The following financial statements of the Company and the Report of KPMG LLP, Independent Auditors, are included in Part IV of this Report on the pages indicated:
|
Report of KPMG LLP, Independent Registered Public Accounting Firm |
|
F-1 |
|
|
F-2 |
|
|
|
F-3 |
|
|
|
F-4 |
|
|
Statements of Shareholders’ Equity and Comprehensive Income (Loss) |
|
F-5 |
|
|
F-6 |
|
|
|
F-7 |
(2) Financial Statement Schedules
All schedules are omitted because they are not applicable or the required information is shown in the financial statements or the notes thereto.
(3) Exhibits
The following exhibits are filed herewith and this list is intended to constitute the exhibit index:
|
Exhibit No. |
|
Description |
|
1.1 |
|
Underwriting Agreement dated November 14, 2005. (15) |
|
1.2 |
|
Placement Agency Agreement between AVI BioPharma, Inc. and Citigroup Global Markets Inc., Oppenheimer & Co. Inc., and Maxim Group, LLC, dated December 12, 2007. (22) |
|
1.3 |
|
Engagement Letter dated January 28, 2009 between AVI BioPharma, Inc. and Rodman & Renshaw, LLC. (41) |
|
2.1 |
|
Agreement and Plan of Merger dated March 12, 2008 by and among AVI BioPharma, Inc., EB Acquisition Corp., Ercole Biotech, Inc. and the Stockholder Representative. (35) |
|
3.1 |
|
Third Restated Articles of Incorporation of AntiVirals Inc. (1) |
|
3.2 |
|
First Restated Bylaws of AVI BioPharma, Inc. (28) |
|
3.3 |
|
First Amendment to Third Restated Articles of Incorporation. (4) |
|
3.4 |
|
Amendment to Article 2 of the Company Third Restated Articles of Incorporation. (11) |
|
4.1 |
|
Form of Specimen Certificate for Common Stock. (1) |
|
4.2 |
|
Warrant to purchase 485,290 shares of the Company’s common stock dated November 14, 2005. (16) |
|
4.3 |
|
Form of Warrant to Purchase Common Stock, issued in connection with the Placement Agency Agreement dated December 12, 2007. (23) |
|
4.4 |
|
Form of Common Stock Purchase Warrant. (42) |
|
4.5 |
|
Form of Common Stock Purchase Warrant. (51) |
|
10.1† |
|
1992 Stock Incentive Plan (as amended through May 11, 2000). (1) |
|
10.2† |
|
Employment Agreement with Denis R. Burger, Ph.D. dated November 4, 1996. (1) |
|
10.3† |
|
Employment Agreement with Alan P. Timmins dated November 4, 1996. (1) |
|
10.4† |
|
Employment Agreement with Dwight Weller, Ph.D. dated November 4, 1996. (1) |
|
10.5 |
|
Technology Transfer Agreement between Anti-Gene Development Group and AntiVirals Inc., dated February 9, 1992. (1) |
|
10.6 |
|
Amendment to Technology Transfer Agreement between Anti-Gene Development Group and AntiVirals Inc. dated January 20, 1996. (1) |
|
10.7 |
|
License and Option Agreement between Anti-Gene Development Group and AntiVirals Inc., dated February 9, 1993. (1) |
|
10.8 |
|
Commercial Lease between Research Way Investments, Landlord, and AntiVirals Inc., Tenant, dated June 15, 1992. (1) |
|
10.9 |
|
Lease between Benjamin Franklin Plaza, Inc., Landlord, and AntiVirals Inc., Tenant, dated June 17, 1992. (1) |
|
10.10 |
|
First Amendment to Lease between Benjamin Franklin Plaza, Inc., Landlord, and AntiVirals Inc., Tenant, dated July 24, 1995. (1) |
|
10.11† |
|
Employment Agreement with Patrick L. Iversen, Ph.D. dated July 14, 1997. (2) |
|
10.12† |
|
ImmunoTherapy Corporation 1997 Stock Option Plan. (3) |
|
10.13 |
|
License Agreement between ImmunoTherapy Corporation and Ohio State University, dated March 12, 1996. (3) |
|
10.14 |
|
License Agreement between ImmunoTherapy Corporation and Ohio State University, dated December 26, 1996. (3) |
|
10.15 |
|
Amendment to License Agreement between ImmunoTherapy Corporation and Ohio State University, dated September 23, 1997. (3) |
|
10.16 |
|
Purchase Agreement, dated December 15, 1999, by and between AVI BioPharma, Inc. and certain Investors. (5) |
|
10.17 |
|
Registration Rights Agreement, dated December 15, 1999, by and between AVI BioPharma, Inc. and certain Investors. (5) |
|
10.18 |
|
Purchase Agreement, dated December 16, 1999, by and between AVI BioPharma, Inc. and certain Investors. (5) |
|
10.19 |
|
Registration Rights Agreement, dated December 16, 1999, by and between AVI BioPharma, Inc. and certain Investors. (5) |
|
10.20 |
|
Subscription Agreement, dated December 1, 1999, by and between SuperGen, Inc. and AVI BioPharma, Inc. (5) |
|
10.21 |
|
2000 Amendment to Technology Transfer Agreement between Anti-Gene Development Group and AVI BioPharma, Inc. (6) |
|
10.22 |
|
United States of America Sales, Distribution, and Development Agreement, dated April 4, 2000, between SuperGen, Inc. and AVI BioPharma, Inc. (7) |
|
10.23 |
|
Common Stock and Warrant Purchase Agreement, dated April 4, 2000, between SuperGen, Inc. and AVI BioPharma, Inc. (7) |
|
10.24 |
|
Registration Rights Agreement, dated April 14, 2000, between SuperGen, Inc. and AVI BioPharma, Inc. (7) |
|
10.25† |
|
2000 Employee Share Purchase Plan. (8) |
|
10.26† |
|
Employment Agreement with Mark M. Webber dated May 11, 2000. (9) |
|
10.27 |
|
Lease Agreement with Spieker Partners, LP dated May 8, 2001. (9) |
|
10.28* |
|
Investment Agreement dated May 22, 2001 between the Company and Medtronic Asset Management, Inc. (9) |
|
10.29 |
|
Warrant dated June 20, 2001 issued to Medtronic Asset Management, Inc. (9) |
|
10.30 |
|
Registration Rights Agreement dated June 20, 2001 between the Company and Medtronic Asset Management, Inc. (9) |
|
10.31* |
|
License and Development Agreement dated June 20, 2001 between the Company and Medtronic, Inc. (9) |
|
10.32* |
|
Supply Agreement dated June 20, 2001 between the Company and Medtronic, Inc. (9) |
|
10.33 |
|
Securities Purchase Agreement dated March 25, 2002 between the Company and certain purchasers (“2002 SPA”). (10) |
|
10.34 |
|
Form of Warrant issued by the Company to certain purchasers under the 2002 SPA. (10) |
|
10.35 |
|
Registration Rights Agreement dated March 25, 2002 between the Company and certain purchasers. (10) |
|
10.36† |
|
2002 Equity Incentive Plan. (11) |
|
10.37 |
|
Securities Purchase Agreement dated January 19, 2005 between the Company and certain purchasers (“2005 SPA”). (12) |
|
10.38 |
|
Form of Purchase Warrant issued by the Company to certain purchasers under the 2005 SPA. (12) |
|
10.39† |
|
Amendment to employment agreement of Denis R. Burger, Ph.D. (14) |
|
10.40† |
|
Amendment to employment agreement of Alan P. Timmins. (14) |
|
10.41† |
|
Amendment to employment agreement of Patrick L. Iversen, Ph.D. (14) |
|
10.42† |
|
Amendment to employment agreement of Dwight D. Weller, Ph.D. (14) |
|
10.43† |
|
Amendment to employment agreement of Peter D. O’Hanley, M.D., Ph.D. (14) |
|
10.44† |
|
Amendment to employment agreement of Mark M. Webber. (14) |
|
10.45 |
|
Securities Purchase Agreement dated November 14, 2005 between the Company and certain purchasers. (16) |
|
10.46* |
|
Supply Agreement, dated March 10, 2006, by and between Cook Group Incorporated and AVI BioPharma, Inc. (17) |
|
10.47* |
|
License and Development Agreement, dated March 10, 2006, by and between Cook Group Incorporated and AVI BioPharma, Inc. (17) |
|
10.48* |
|
Investment Agreement, dated March 10, 2006, by and between Cook Group Incorporated and AVI BioPharma, Inc. (17) |
|
10.49* |
|
License Agreement dated January 26, 2006 by and between with Chiron Corporation and AVI BioPharma, Inc. (18) |
|
10.50 |
|
Stock Purchase Agreement dated January 26, 2006 by and between with Chiron Corporation and AVI BioPharma, Inc. (18) |
|
10.51 |
|
Second Lease Extension and Modification Agreement dated January 24, 2006 by and between Research Way Investments and AVI BioPharma, Inc. (19) |
|
10.52* |
|
Collaboration and License Agreement, dated December 19, 2006, by and between Ercole Biotech, Inc. and AVI BioPharma, Inc. (20) |
|
10.53 |
|
Series A-2 Preferred Stock and Common Stock Purchase Agreement, dated December 19, 2006, by and between Ercole Biotech, Inc. and AVI BioPharma, Inc. (21) |
|
10.54* |
|
Cross License Agreement dated January 8, 2007 by and between Eleos, Inc. and AVI BioPharma, Inc. (24) |
|
10.55 |
|
Separation and Release Agreement dated March 27, 2007 by and between Denis R. Burger, Ph.D. and AVI BioPharma, Inc. (25) |
|
10.56* |
|
Second License and Collaboration Agreement dated May 1, 2007 by and between Ercole Biotech. Inc. and AVI BioPharma, Inc. (26) |
|
10.57 |
|
Real Property Purchase Agreement, dated April 19, 2007, by and between WKL Investments Airport, LLC and AVI BioPharma, Inc. (27) |
|
10.58* |
|
Sponsored Research Agreement between AVI BioPharma, Inc. and Charley’s Fund, Inc., effective October 12, 2007. (29) |
|
10.59 |
|
Shareholder’s Trust Agreement between and among AVI BioPharma, Inc., AVI Shareholder Advocacy Trust, The Shareholder Advocate LLC, and Richard Macary, dated October 29, 2007. (30) |
|
10.60† |
|
Amended and Restated Employment Agreement between Alan P. Timmins and AVI BioPharma, Inc., dated October 26, 2007. (31) |
|
10.61 |
|
Professional Services Agreement between James B. Hicks Ph.D., LLC and AVI BioPharma, Inc., dated October 26, 2007. (32) |
|
10.62 |
|
Letter Agreement executed by George Haywood, dated October 29, 2007. (33) |
|
10.63† |
|
Employment Agreement dated February 8, 2008 by and between AVI BioPharma, Inc. and Leslie Hudson, Ph.D. (34) |
|
10.64 |
|
Ercole Biotech, Inc. Convertible Promissory Note dated March 12, 2008. (36) |
|
10.65† |
|
Employment Agreement dated April 10, 2008 by and between AVI BioPharma, Inc. and Dr. Ryszard Kole. (37) |
|
10.66*† |
|
Employment Agreement dated July 24, 2008 by and between AVI BioPharma, Inc. and J. David Boyle II. (38) |
|
10.67*† |
|
Amendment No. 1 to Employment Agreement dated August 1, 2008 by and between AVI BioPharma, Inc. and J. David Boyle II. (39) |
|
10.68† |
|
Severance and Release Agreement effective October 27, 2008 by and between AVI BioPharma, Inc. and Peter O’Hanley. (40) |
|
10.69† |
|
Employment Agreement dated January 26, 2009 between AVI BioPharma, Inc. and Stephen Bevan Shrewsbury, M.D. (43) |
|
10.70 |
|
Securities Purchase Agreement dated January 29, 2009 between AVI BioPharma, Inc. and the Purchasers. (44) |
|
10.71† |
|
Letter Agreement Regarding Board of Director Representation between AVI BioPharma, Inc. and Eastbourne Capital Management, LLC. (45) |
|
10.72 |
|
Agreement between AVI BioPharma, Inc. and the U.S. Defense Threat Reduction Agency dated May 5, 2009. (46) |
|
10.73*† |
|
Employment Agreement dated May 19, 2009 between AVI BioPharma, Inc. and Paul Medeiros. (47) |
|
10.74 |
|
Agreement between AVI BioPharma, Inc. and the U.S. Defense Threat Reduction Agency dated May 28, 2009. (48) |
|
10.75* |
|
First Amendment to Sponsored Research Agreement between AVI BioPharma, Inc. and Charley’s Fund, Inc. dated June 2, 2009. (49) |
|
10.76 |
|
Lease dated July 24, 2009 by and between BMR-3450 Monte Villa Parkway, LLC and AVI BioPharma, Inc. (50) |
|
10.77 |
|
Amendment of Contract between AVI BioPharma, Inc. and the U.S. Defense Threat Reduction Agency (contract no HDTRA 1-07-C0010), effective September 30, 2009. (52) |
|
10.78* |
|
Collaboration and License Agreement between Isis Pharmaceuticals and Ercole Biotech, Inc. dated May 16, 2003 |
|
14.1 |
|
Code of Business Conduct and Ethics. (13) |
|
21.1 |
|
Subsidiaries of the Registrant. |
|
23.1 |
|
Consent of Independent Registered Public Accounting Firm. |
|
31.1 |
|
Certification of the Company’s Chief Executive Officer, Leslie Hudson, Ph.D., pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
31.2 |
|
Certification of the Company’s Chief Financial Officer, J. David Boyle II, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
32.1 |
|
Certification of CEO and CFO Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
(1) |
Incorporated by reference to Exhibits to Registrant’s Registration Statement on Form SB-2, as amended and filed with the Securities and Exchange Commission on May 29, 1997 (Commission Registration No. 333-20513). |
|
|
|
|
(2) |
Incorporated by reference to Exhibits to Registrant’s Annual Report on Form 10-KSB for the fiscal year ended December 31, 1997, and filed with the Securities and Exchange Commission on March 30, 1998. |
|
|
|
|
(3) |
Incorporated by reference to Exhibits to Registrant’s Registration Statement on Form S-4, as amended, and filed with the Securities and Exchange Commission on August 7, 1998 (Commission Registration No. 333-60849). |
|
|
|
|
(4) |
Incorporated by reference to Exhibits to Registrant’s current report on Form 8-K, as filed with the Securities and Exchange Commission on September 30, 1998 (Commission Registration No. 000-22613). |
|
|
|
|
(5) |
Incorporated by reference to Exhibits to Registrant’s Registration Statement on Form S-3, as amended, and filed with the Securities and Exchange Commission on December 21, 1999 (Commission Registration No. 333-93135). |
|
|
|
|
(6) |
Incorporated by reference to Exhibits to Registrant’s Registration Statement on Form S-1 and filed with the Securities and Exchange Commission on June 16, 2000 (Commission Registration No. 333-39542). |
|
|
|
|
(7) |
Incorporated by reference to Exhibits to Registrant’s Registrations Statement on Form S-3, and filed with the Securities and Exchange Commission on September 15, 2000 (Commission Registration No. 333-45888). |
|
|
|
|
(8) |
Incorporated by reference to Appendix A to Registrant’s Definitive Proxy Statement on Form 14-A, as amended, filed with the Securities and Exchange Commission on April 12, 2000. |
|
|
|
|
(9) |
Incorporated by reference to Exhibits to Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2001, and filed with the Securities and Exchange Commission on August 14, 2001, as amended on April 23, 2002. |
|
|
|
|
(10) |
Incorporated by reference to Exhibits to Registrant’s current report on Form 8-K, as filed with the Securities and Exchange Commission on April 2, 2002. |
|
|
|
|
(11) |
Incorporated by reference to appendixes to Registrant’s Definitive Proxy Statement on Schedule 14-A, as filed with the Securities and Exchange Commission on April 11, 2002. |
|
|
|
|
(12) |
Incorporated by reference to registrants current report on Form 8-K, as filed with the Securities and Exchange Commission on January 20, 2005. |
|
|
|
|
(13) |
Incorporated by reference to Exhibits to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2003, and filed with the Securities and Exchange Commission on March 15, 2004. |
|
(14) |
Incorporated by reference to Registrant’s current report on Form 8-K, as filed with the Securities and Exchange Commission on February 28, 2005. |
|
|
|
|
(15) |
Incorporated by reference to Registrant’s current report on Form 8-K, as filed with the Securities and Exchange Commission on November 21, 2005. |
|
|
|
|
(16) |
Incorporated by reference to Exhibits to Registrant’s Annual Report on Form 10-K for the fiscal year ended December 31, 2005, and filed with the Securities and Exchange Commission on March 16, 2006. |
|
|
|
|
(17) |
Incorporated by reference to Exhibits to Registrant’s Registrations Statement on Form S-3, and filed with the Securities and Exchange Commission on April 11, 2006 (Commission Registration No. 333-133211). |
|
|
|
|
(18) |
Incorporated by reference to Exhibits to Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2006, and filed with the Securities and Exchange Commission on May 10, 2006. |
|
|
|
|
(19) |
Incorporated by reference to Exhibits to Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2006, and filed with the Securities and Exchange Commission on August 9, 2006. |
|
|
|
|
(20) |
Incorporated by reference to Exhibit 10.56 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2006, filed with the Securities and Exchange Commission on March 16, 2007. |
|
|
|
|
(21) |
Incorporated by reference to Exhibit 10.57 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2006, filed with the Securities and Exchange Commission on March 16, 2007. |
|
|
|
|
(22) |
Incorporated by reference to Exhibit 1.01 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on December 13, 2007. |
|
|
|
|
(23) |
Incorporated by reference to Exhibit 4.5 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on December 13, 2007. |
|
|
|
|
(24) |
Incorporated by reference to Exhibit 10.58 to the Registrant’s Form 10-Q for the quarterly period ended March 31, 2007, filed with the Securities and Exchange Commission on May 10, 2007. |
|
|
|
|
(25) |
Incorporated by reference to Exhibit 10.59 to the Registrant’s Form 10-Q for the quarterly period ended March 31, 2007, filed with the Securities and Exchange Commission on May 10, 2007. |
|
|
|
|
(26) |
Incorporated by reference to Exhibit 10.60 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007. |
|
|
|
|
(27) |
Incorporated by reference to Exhibit 10.61 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007. |
|
|
|
|
(28) |
Incorporated by reference to Exhibit 3.5 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on February 7, 2008. |
|
|
|
|
(29) |
Incorporated by reference to Exhibit 10.58 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2007, filed with the Securities and Exchange Commission on March 17, 2008. |
|
|
|
|
(30) |
Incorporated by reference to Exhibit 10.59 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2007, filed with the Securities and Exchange Commission on March 17, 2008. |
|
|
|
|
(31) |
Incorporated by reference to Exhibit 10.60 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2007, filed with the Securities and Exchange Commission on March 17, 2008. |
|
|
|
|
(32) |
Incorporated by reference to Exhibit 10.61 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2007, filed with the Securities and Exchange Commission on March 17, 2008. |
|
|
|
|
(33) |
Incorporated by reference to Exhibit 10.62 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2007, filed with the Securities and Exchange Commission on March 17, 2008. |
|
|
|
|
(34) |
Incorporated by reference to Exhibit 10.63 to the Registrant’s Form 10-Q for the quarterly period ended March 31, 2008, filed with the Securities and Exchange Commission on May 12, 2008. |
|
|
|
|
(35) |
Incorporated by reference to Exhibit 2.1 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on March 13, 2008. |
|
(36) |
Incorporated by reference to Exhibit 10.62 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on March 13, 2008. |
|
|
|
|
(37) |
Incorporated by reference to Exhibit 10.64 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2008, filed with the Securities and Exchange Commission on August 11, 2008. |
|
|
|
|
(38) |
Incorporated by reference to Exhibit 10.65 to the Registrant’s Form 10-Q for the quarterly period ended September 30, 2008, filed with the Securities and Exchange Commission on November 10, 2008. |
|
|
|
|
(39) |
Incorporated by reference to Exhibit 10.66 to the Registrant’s Form 10-Q for the quarterly period ended September 30, 2008, filed with the Securities and Exchange Commission on November 10, 2008. |
|
|
|
|
(40) |
Incorporated by reference to Exhibit 10.68 to the Registrant’s Form 10-K for the fiscal year ended December 31, 2008, filed with the Securities and Exchange Commission on March 10, 2009. |
|
|
|
|
(41) |
Incorporated by reference to Exhibit 1.3 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on January 30, 2009. |
|
|
|
|
(42) |
Incorporated by reference to Exhibit 4.4 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on January 30, 2009. |
|
|
|
|
(43) |
Incorporated by reference to Exhibit 10.71 to the Registrant’s Form 10-Q for the quarterly period ended March 31, 2009, filed with the Securities and Exchange Commission on May 11, 2009. |
|
|
|
|
(44) |
Incorporated by reference to Exhibit 10.67 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on January 30, 2009. |
|
|
|
|
(45) |
Incorporated by reference to Exhibit 10.68 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on January 30, 2009. |
|
|
|
|
(46) |
Incorporated by reference to Exhibit 10.72 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2009, filed with the Securities and Exchange Commission on August 10, 2009. |
|
|
|
|
(47) |
Incorporated by reference to Exhibit 10.73 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2009, filed with the Securities and Exchange Commission on August 10, 2009. |
|
|
|
|
(48) |
Incorporated by reference to Exhibit 10.74 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2009, filed with the Securities and Exchange Commission on August 10, 2009. |
|
|
|
|
(49) |
Incorporated by reference to Exhibit 10.75 to the Registrant’s Form 10-Q for the quarterly period ended June 30, 2009, filed with the Securities and Exchange Commission on August 10, 2009. |
|
|
|
|
(50) |
Incorporated by reference to Exhibit 10.76 to the Registrant’s Form 10-Q for the quarterly period ended September 30, 2009, filed with the Securities and Exchange Commission on November 9, 2009. |
|
|
|
|
(51) |
Incorporated by reference to Exhibit 4.1 to the Registrant’s Form 8-K filed with the Securities and Exchange Commission on August 24, 2009. |
|
|
|
|
(52) |
Incorporated by reference to Exhibit 10.77 to the Registrant’s Form 10-Q for the quarterly period ended September 30, 2009, filed with the Securities and Exchange Commission on November 9, 2009. |
(b) Exhibits.
The exhibits listed under Item 15(a)(3) hereof are filed as part of this Form 10-K other than Exhibit 32.1, which shall be deemed furnished.
(c) Financial Statement Schedules.
All schedules are omitted because they are not applicable or the required information is shown in the financial statements or the notes thereto.
* A Confidential Treatment Request for certain information in this document has been filed with the Securities and Exchange Commission. The information for which treatment has been sought has been deleted from such exhibit and the deleted text replaced by an asterisk (*).
† Indicates management contract or compensatory plan, contract or arrangement.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Dated: |
March 16, 2010 |
AVI BIOPHARMA, INC. |
|
|
|
|
|
|
|
|
|
By: |
/s/ Leslie Hudson, Ph.D. |
|
|
|
Leslie Hudson, Ph.D. |
|
|
|
|
President and Chief Executive Officer |
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in their capacities indicated on March 16, 2010:
|
Signature |
|
Title |
|
|
|
|
|
/s/ LESLIE HUDSON, Ph.D. |
|
President and Chief Executive Officer; Director |
|
Leslie Hudson, Ph.D. |
|
(Principal Executive Officer) |
|
|
|
|
|
/s/ J. DAVID BOYLE II |
|
Chief Financial Officer |
|
J. David Boyle II |
|
(Principal Financial and Accounting Officer) |
|
|
|
|
|
/s/ MICHAEL D. CASEY |
|
Chairman of the Board |
|
Michael D. Casey |
|
|
|
|
|
|
|
/s/ M. KATHLEEN BEHRENS Ph.D. |
|
Director |
|
M. Kathleen Behrens, Ph.D. |
|
|
|
|
|
|
|
/s/ K. MICHAEL FORREST |
|
Director |
|
K. Michael Forrest |
|
|
|
|
|
|
|
/s/ WILLIAM A. GOOLSBEE |
|
Director |
|
William A. Goolsbee |
|
|
|
|
|
|
|
/s/ CHRISTOPHER S. HENNEY, Ph.D., D.Sc. |
|
Director |
|
Christopher S. Henney, Ph.D., D.Sc. |
|
|
|
|
|
|
|
/s/ JOHN C. HODGMAN |
|
Director |
|
John C. Hodgman |
|
|
|
|
|
|
|
/s/ GIL PRICE, M.D. |
|
Director |
|
Gil Price, M.D. |
|
|
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
AVI BioPharma, Inc:
We have audited the accompanying balance sheets of AVI BioPharma, Inc. (a development stage enterprise) as of December 31, 2009 and 2008, and the related statements of operations, shareholders’ equity and comprehensive income (loss), and cash flows for each of the years in the three-year period ended December 31, 2009 and the information included in the cumulative from inception presentations for the period January 1, 2002 to December 31, 2009 (not separately presented herein). These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. The financial statements of AVI BioPharma, Inc. for the period July 22, 1980 (inception) to December 31, 2001 were audited by other auditors who have ceased operations. Those auditors expressed an unqualified opinion on those financial statements in their report dated February 21, 2002.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of AVI BioPharma, Inc. (a development stage enterprise) as of December 31, 2009 and 2008, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2009 and the information included in the cumulative from inception presentations for the period January I, 2002 to December 31, 2009 (not separately presented herein), in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of AVI BioPharma, Inc.’s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report, dated March 16, 2010 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
|
/s/ KPMG LLP |
|
|
|
|
|
Portland, Oregon |
|
|
March 16, 2010 |
|
THIS REPORT IS A CONFORMED COPY OF THE REPORT PREVIOUSLY ISSUED BY ARTHUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY THAT FIRM.
Report of Independent Public Accountants
To the Board of Directors and Shareholders of
AVI BioPharma, Inc.
We have audited the accompanying balance sheet of AVI BioPharma, Inc. (an Oregon corporation in the development stage) as of December 31, 2001, and the related statements of operations, shareholders’ equity and cash flows for each of the two years in the period ended December 31, 2001 and for the period from inception (July 22, 1980) to December 31, 2001. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of AVI BioPharma, Inc. as of December 31, 2001, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2001 and for the period from inception (July 22, 1980) to December 31, 2001, in conformity with accounting principles generally accepted in the United States.
|
/s/ Arthur Andersen LLP |
|
|
|
|
|
Portland, Oregon |
|
|
February 21, 2002 |
|
AVI BioPharma, Inc.
(A Development Stage Company)
|
|
|
December 31, |
|
December 31, |
|
||
|
|
|
2009 |
|
2008 |
|
||
|
Assets |
|
|
|
|
|
||
|
Current Assets: |
|
|
|
|
|
||
|
Cash and cash equivalents |
|
$ |
48,275 |
|
$ |
11,192 |
|
|
Short-term securities—available-for-sale |
|
171 |
|
282 |
|
||
|
Accounts receivable |
|
2,085 |
|
4,971 |
|
||
|
Other current assets |
|
779 |
|
599 |
|
||
|
Total Current Assets |
|
51,310 |
|
17,044 |
|
||
|
|
|
|
|
|
|
||
|
Property held for sale |
|
2,372 |
|
_ |
|
||
|
Property and Equipment, net of accumulated depreciation and amortization of $14,026 and $12,919 |
|
2,466 |
|
5,189 |
|
||
|
Patent Costs, net of accumulated amortization of $1,762 and $1,927 |
|
3,759 |
|
3,268 |
|
||
|
Other assets |
|
120 |
|
35 |
|
||
|
Total Assets |
|
$ |
60,027 |
|
$ |
25,536 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
Liabilities and Shareholders’ Equity |
|
|
|
|
|
||
|
Current Liabilities: |
|
|
|
|
|
||
|
Accounts payable |
|
$ |
1,381 |
|
$ |
2,014 |
|
|
Accrued employee compensation |
|
922 |
|
1,306 |
|
||
|
Long-term debt, current portion |
|
77 |
|
74 |
|
||
|
Warrant valuation |
|
27,609 |
|
1,254 |
|
||
|
Deferred revenue |
|
3,428 |
|
2,190 |
|
||
|
Other liabilities |
|
90 |
|
450 |
|
||
|
Total Current Liabilities |
|
33,507 |
|
7,288 |
|
||
|
|
|
|
|
|
|
||
|
Commitments and Contingencies |
|
— |
|
— |
|
||
|
|
|
|
|
|
|
||
|
Long-term debt, non-current portion |
|
1,924 |
|
2,001 |
|
||
|
Other long-term liabilities |
|
966 |
|
515 |
|
||
|
|
|
|
|
|
|
||
|
Shareholders’ Equity: |
|
|
|
|
|
||
|
Preferred stock, $.0001 par value, 20,000,000 shares authorized; none issued and outstanding |
|
— |
|
— |
|
||
|
Common stock, $.0001 par value, 200,000,000 shares authorized; 110,495,587 and 71,101,738 issued and outstanding |
|
11 |
|
7 |
|
||
|
Additional paid-in capital |
|
299,088 |
|
266,035 |
|
||
|
Deficit accumulated during the development stage |
|
(275,469 |
) |
(250,310 |
) |
||
|
Total Shareholders’ Equity |
|
23,630 |
|
15,732 |
|
||
|
Total Liabilities and Shareholders’ Equity |
|
$ |
60,027 |
|
$ |
25,536 |
|
See accompanying notes to financial statements.
AVI BioPharma, Inc.
(A Development Stage Company)
|
|
|
|
|
|
|
|
|
July 22, 1980 |
|
||||
|
|
|
Year ended December 31, |
|
(Inception) through |
|
||||||||
|
(in thousands) |
|
2009 |
|
2008 |
|
2007 |
|
December 31, 2009 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Revenues from license fees, grants and research contracts |
|
$ |
17,585 |
|
$ |
21,258 |
|
$ |
10,985 |
|
$ |
59,809 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
||||
|
Research and development |
|
24,396 |
|
27,331 |
|
31,058 |
|
230,432 |
|
||||
|
General and administrative |
|
8,696 |
|
11,469 |
|
13,035 |
|
74,020 |
|
||||
|
Acquired in-process research and development |
|
— |
|
9,916 |
|
— |
|
29,461 |
|
||||
|
Operating loss |
|
(15,507 |
) |
(27,458 |
) |
(33,108 |
) |
(274,104 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Other non-operating (loss) income: |
|
|
|
|
|
|
|
|
|
||||
|
Interest (expense) income and other, net |
|
(454 |
) |
344 |
|
984 |
|
8,323 |
|
||||
|
(Increase) decrease on warrant valuation |
|
(9,198 |
) |
3,161 |
|
4,956 |
|
3,450 |
|
||||
|
Realized gain on sale of short-term securities— available-for-sale |
|
— |
|
— |
|
— |
|
3,863 |
|
||||
|
Write-down of short-term securities— available-for-sale |
|
— |
|
— |
|
— |
|
(17,001 |
) |
||||
|
|
|
(9,652 |
) |
3,505 |
|
5,940 |
|
(1,365 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net loss |
|
$ |
(25,159 |
) |
$ |
(23,953 |
) |
$ |
(27,168 |
) |
$ |
(275,469 |
) |
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net loss per share - basic and diluted |
|
$ |
(0.27 |
) |
$ |
(0.34 |
) |
$ |
(0.50 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Weighted average number of common shares outstanding for computing basic and diluted loss per share |
|
93,090 |
|
69,491 |
|
53,942 |
|
|
|
||||
See accompanying notes to financial statements.
AVI BioPharma, Inc.
(A Development Stage Company)
Statements of Shareholders’ Equity and Comprehensive Income (Loss)
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
Deficit |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
Other |
|
Accumulated |
|
|
|
||||
|
|
|
|
|
|
|
|
|
Additional |
|
Comprehensive |
|
During the |
|
Total |
|
||||
|
|
|
Partnership |
|
Common Stock |
|
Paid-In |
|
Income |
|
Development |
|
Shareholders’ |
|
||||||
|
(in thousands) |
|
Units |
|
Shares |
|
Amount |
|
Capital |
|
(Loss) |
|
Stage |
|
Equity |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
BALANCE AT JULY 22, 1980 (Inception) |
|
— |
|
— |
|
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
Issuance of partnership units, warrants and common stock |
|
3,615 |
|
8,273 |
|
1 |
|
33,733 |
|
— |
|
— |
|
33,734 |
|
||||
|
Compensation expense related to issuance of warrants for common stock and partnership units |
|
— |
|
— |
|
— |
|
537 |
|
— |
|
— |
|
537 |
|
||||
|
Exercise of warrants for partnership units and common stock |
|
42 |
|
2,236 |
|
— |
|
4,152 |
|
— |
|
— |
|
4,152 |
|
||||
|
Exercise of options for common stock |
|
— |
|
990 |
|
— |
|
4,005 |
|
— |
|
— |
|
4,005 |
|
||||
|
Issuance of common stock for ESPP |
|
— |
|
252 |
|
— |
|
810 |
|
— |
|
— |
|
810 |
|
||||
|
Issuance of common stock and warrants for cash and securities, net of offering costs |
|
— |
|
37,185 |
|
4 |
|
162,348 |
|
— |
|
— |
|
162,352 |
|
||||
|
Issuance of common stock and warrants for the acquisition of ImmunoTherapy Corporation |
|
— |
|
2,132 |
|
— |
|
17,167 |
|
— |
|
— |
|
17,167 |
|
||||
|
Issuance of common stock and warrants for services |
|
— |
|
536 |
|
— |
|
2,469 |
|
— |
|
— |
|
2,469 |
|
||||
|
Compensation expense related to issuance of options for common stock |
|
— |
|
— |
|
— |
|
6,842 |
|
— |
|
— |
|
6,842 |
|
||||
|
Conversion of debt into common stock and partnership units |
|
9 |
|
10 |
|
— |
|
88 |
|
— |
|
— |
|
88 |
|
||||
|
Issuance of common stock in exchange for partnership units |
|
(1,810 |
) |
1,633 |
|
— |
|
(0 |
) |
— |
|
— |
|
— |
|
||||
|
Withdrawal of partnership net assets upon conveyance of technology |
|
(1,856 |
) |
— |
|
— |
|
(177 |
) |
— |
|
— |
|
(177 |
) |
||||
|
Common stock subject to rescission, net |
|
— |
|
(64 |
) |
— |
|
(289 |
) |
— |
|
— |
|
(289 |
) |
||||
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Write-down of short-term securities— available-for-sale |
|
— |
|
— |
|
— |
|
— |
|
17,001 |
|
— |
|
17,001 |
|
||||
|
Realized gain on sale of short-term securities— available-for-sale |
|
— |
|
— |
|
— |
|
— |
|
(3,766 |
) |
— |
|
(3,766 |
) |
||||
|
Unrealized loss on short-term securities— available-for-sale |
|
— |
|
— |
|
— |
|
— |
|
(13,217 |
) |
— |
|
(13,217 |
) |
||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(199,189 |
) |
(199,189 |
) |
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
(199,171 |
) |
||||
|
BALANCE AT DECEMBER 31, 2006 |
|
— |
|
53,183 |
|
5 |
|
$ |
231,685 |
|
$ |
18 |
|
$ |
(199,189 |
) |
$ |
32,519 |
|
|
Exercise of warrants for common stock |
|
— |
|
12 |
|
— |
|
29 |
|
— |
|
— |
|
29 |
|
||||
|
Exercise of options for common stock |
|
— |
|
39 |
|
— |
|
90 |
|
— |
|
— |
|
90 |
|
||||
|
Issuance of common stock for ESPP |
|
— |
|
518 |
|
— |
|
1,450 |
|
— |
|
— |
|
1,450 |
|
||||
|
Issuance of common stock to vendors |
|
— |
|
|
|
|
|
|
|
— |
|
— |
|
— |
|
||||
|
Compensation expense related to issuance of options for common stock |
|
— |
|
— |
|
— |
|
313 |
|
— |
|
— |
|
313 |
|
||||
|
Issuance of common stock for cash and securities, net of offering costs |
|
— |
|
10,697 |
|
1 |
|
14,447 |
|
— |
|
— |
|
14,448 |
|
||||
|
Stock-based compensation |
|
— |
|
— |
|
— |
|
4,719 |
|
— |
|
— |
|
4,719 |
|
||||
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Unrealized gain on short-term securities— available-for-sale, net |
|
— |
|
— |
|
— |
|
— |
|
(18 |
) |
— |
|
(18 |
) |
||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(27,168 |
) |
(27,168 |
) |
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
(27,186 |
) |
||||
|
BALANCE AT DECEMBER 31, 2007 |
|
— |
|
64,449 |
|
6 |
|
$ |
252,733 |
|
$ |
— |
|
$ |
(226,357 |
) |
$ |
26,382 |
|
|
Exercise of options for common stock |
|
— |
|
7 |
|
— |
|
9 |
|
— |
|
— |
|
9 |
|
||||
|
Issuance of common stock for ESPP |
|
— |
|
84 |
|
— |
|
72 |
|
— |
|
— |
|
72 |
|
||||
|
Issuance of common stock and warrants to vendors |
|
— |
|
324 |
|
— |
|
828 |
|
— |
|
— |
|
828 |
|
||||
|
Compensation expense to non-employees on issuance of options and warrants to purchase common stock |
|
— |
|
— |
|
— |
|
180 |
|
— |
|
— |
|
180 |
|
||||
|
Compensation expense on issuance of restricted stock |
|
— |
|
100 |
|
— |
|
166 |
|
— |
|
— |
|
166 |
|
||||
|
Stock-based compensation |
|
— |
|
326 |
|
— |
|
3,656 |
|
— |
|
— |
|
3,656 |
|
||||
|
Issuance of common stock for acquisition of Ercole |
|
— |
|
5,812 |
|
1 |
|
8,391 |
|
— |
|
— |
|
8,392 |
|
||||
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Unrealized gain on short-term securities— available-for-sale, net |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(23,953 |
) |
(23,953 |
) |
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
(23,953 |
) |
||||
|
BALANCE AT DECEMBER 31, 2008 |
|
— |
|
71,102 |
|
7 |
|
$ |
266,035 |
|
$ |
— |
|
$ |
(250,310 |
) |
$ |
15,732 |
|
|
Exercise of options for common stock |
|
— |
|
62 |
|
— |
|
76 |
|
— |
|
— |
|
76 |
|
||||
|
Issuance of common stock for ESPP |
|
— |
|
124 |
|
— |
|
85 |
|
— |
|
— |
|
85 |
|
||||
|
Issuance of common stock for cash and securities, net of offering costs |
|
— |
|
38,520 |
|
4 |
|
30,518 |
|
— |
|
— |
|
30,522 |
|
||||
|
Compensation expense on issuance of restricted stock |
|
— |
|
427 |
|
— |
|
203 |
|
— |
|
— |
|
203 |
|
||||
|
Stock-based compensation |
|
— |
|
261 |
|
— |
|
2,171 |
|
— |
|
— |
|
2,171 |
|
||||
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Unrealized gain on short-term securities— available-for-sale, net |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
||||
|
Net loss |
|
— |
|
— |
|
— |
|
— |
|
— |
|
(25,159 |
) |
(25,159 |
) |
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
(25,159 |
) |
||||
|
BALANCE AT DECEMBER 31, 2009 |
|
— |
|
110,496 |
|
11 |
|
$ |
299,088 |
|
$ |
— |
|
$ |
(275,469 |
) |
$ |
23,630 |
|
See accompanying notes to financial statements.
AVI BioPharma, Inc.
(A Development Stage Company)
|
|
|
|
|
|
|
|
|
For the Period |
|
||||
|
|
|
|
|
|
|
|
|
July 22, 1980 |
|
||||
|
|
|
Year ended December 31, |
|
(Inception) through |
|
||||||||
|
(in thousands) |
|
2009 |
|
2008 |
|
2007 |
|
December 31, 2009 |
|
||||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
||||
|
Net loss |
|
$ |
(25,159 |
) |
$ |
(23,953 |
) |
$ |
(27,168 |
) |
$ |
(275,469 |
) |
|
Adjustments to reconcile net loss to net cash flows used in operating activities: |
|
|
|
|
|
|
|
|
|
||||
|
Depreciation and amortization |
|
1,379 |
|
1,469 |
|
2,014 |
|
17,682 |
|
||||
|
Loss on disposal of assets |
|
347 |
|
584 |
|
59 |
|
1,305 |
|
||||
|
Realized gain on sale of short-term securities— available-for-sale |
|
— |
|
— |
|
— |
|
(3,863 |
) |
||||
|
Write-down of short-term securities—available-for-sale |
|
— |
|
— |
|
— |
|
17,001 |
|
||||
|
Impairment charge on property held for sale |
|
128 |
|
800 |
|
|
|
928 |
|
||||
|
Stock-based compensation |
|
2,374 |
|
4,830 |
|
5,732 |
|
22,697 |
|
||||
|
Conversion of interest accrued to common stock |
|
— |
|
— |
|
— |
|
8 |
|
||||
|
Acquired in-process research and development |
|
— |
|
9,916 |
|
— |
|
29,461 |
|
||||
|
Increase (decrease) on warrant valuation |
|
9,198 |
|
(3,161 |
) |
(4,956 |
) |
(3,450 |
) |
||||
|
(Increase) decrease in: |
|
|
|
|
|
|
|
|
|
||||
|
Accounts receivable and other assets |
|
2,621 |
|
(1,850 |
) |
(2,849 |
) |
(2,900 |
) |
||||
|
Net increase in accounts payable, accrued employee compensation, and other liabilities |
|
312 |
|
(975 |
) |
2,491 |
|
5,274 |
|
||||
|
Net cash used in operating activities |
|
(8,800 |
) |
(12,340 |
) |
(24,677 |
) |
(191,326 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
||||
|
Purchase of property and equipment |
|
(931 |
) |
(369 |
) |
(1,270 |
) |
(17,869 |
) |
||||
|
Patent costs |
|
(1,063 |
) |
(848 |
) |
(857 |
) |
(7,243 |
) |
||||
|
Purchase of marketable securities |
|
— |
|
(11 |
) |
(110 |
) |
(112,986 |
) |
||||
|
Sale of marketable securities |
|
111 |
|
— |
|
12,813 |
|
117,724 |
|
||||
|
Acquisition costs |
|
— |
|
(11 |
) |
— |
|
(2,389 |
) |
||||
|
Net cash (used in) provided by investing activities |
|
(1,883 |
) |
(1,239 |
) |
10,576 |
|
(22,763 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
||||
|
Proceeds from sale of common stock, warrants and partnership units, net of offering costs, and exercise of options and warrants |
|
47,840 |
|
81 |
|
18,745 |
|
262,937 |
|
||||
|
Repayments of long-term debt |
|
(74 |
) |
(113 |
) |
— |
|
(187 |
) |
||||
|
Buyback of common stock pursuant to rescission offering |
|
— |
|
— |
|
— |
|
(289 |
) |
||||
|
Withdrawal of partnership net assets |
|
— |
|
— |
|
— |
|
(177 |
) |
||||
|
Issuance of convertible debt |
|
— |
|
— |
|
— |
|
80 |
|
||||
|
Net cash (used in) provided by financing activities |
|
47,766 |
|
(32 |
) |
18,745 |
|
262,364 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Increase (decrease) in cash and cash equivalents |
|
37,083 |
|
(13,611 |
) |
4,644 |
|
48,275 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Cash and cash equivalents: |
|
|
|
|
|
|
|
|
|
||||
|
Beginning of period |
|
11,192 |
|
24,803 |
|
20,159 |
|
— |
|
||||
|
End of period |
|
$ |
48,275 |
|
$ |
11,192 |
|
$ |
24,803 |
|
$ |
48,275 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: |
|
|
|
|
|
|
|
|
|
||||
|
Cash paid during the year for interest |
|
$ |
97 |
|
$ |
104 |
|
$ |
104 |
|
$ |
305 |
|
|
SUPPLEMENTAL SCHEDULE OF NONCASH INVESTING ACTIVITIES AND FINANCING ACTIVITIES: |
|
|
|
|
|
|
|
|
|
||||
|
Short-term securities—available-for-sale received in connection with the private offering |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
17,897 |
|
|
Change in unrealized gain (loss) on short-term securities—available-for-sale |
|
$ |
— |
|
$ |
— |
|
$ |
(18 |
) |
$ |
— |
|
|
Issuance of common stock and warrants in satisfaction of liabilities |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
545 |
|
|
Issuance of common stock for building purchase |
|
$ |
— |
|
$ |
— |
|
$ |
750 |
|
$ |
750 |
|
|
Assumption of long-term debt for building purchase |
|
$ |
— |
|
$ |
— |
|
$ |
2,200 |
|
$ |
2,200 |
|
|
Issuance of common stock for Ercole assets |
|
$ |
— |
|
$ |
8,075 |
|
$ |
— |
|
$ |
8,075 |
|
|
Assumption of liabilities for Ercole assets |
|
$ |
— |
|
$ |
2,124 |
|
$ |
— |
|
$ |
2,124 |
|
See accompanying notes to financial statements.
AVI BioPharma, Inc.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
1. ORGANIZATION AND NATURE OF BUSINESS:
AVI BioPharma, Inc. (the “Company” or “AVI”) was incorporated in the State of Oregon on July 22, 1980. The mission of the Company is to develop and commercialize improved therapeutic products based upon antisense and cancer immunotherapy technology.
Through May 1993, the financial statements included the combined accounts of the Company and ANTI-GENE DEVELOPMENT GROUP, a limited partnership (AGDG or the Partnership) founded in 1981 and registered in the State of Oregon. Substantially all income generated and proceeds from the Partnership unit sales through that date have been paid to the Company under the terms of research and development contracts entered into by the Partnership and the Company. Significant transactions between the Company and the Partnership through that date have been eliminated.
In March 1993, the Company offered to all partners in the Partnership the opportunity to exchange their partnership units or warrants to purchase partnership units (unit warrants) for common stock or warrants to purchase common stock. Under the terms of the offer, which was completed May 1, 1993, each partner could elect to exchange each unit held or unit warrant held for 1,100 shares of common stock or warrants to purchase 1,100 shares of common stock of the Company, respectively. Total shares and warrants to purchase shares issued in the exchange offer were 1,632,950 and 381,700, respectively.
Effective May 19, 1993, the Company and the Partnership entered into a Technology Transfer Agreement wherein the Partnership conveyed all intellectual property then within its control to the Company. As part of the conveyance, the Company tendered to the Partnership for liquidation all partnership units received pursuant to the exchange offer and received a 49.37 percent undivided interest in the intellectual property. The Company then purchased the remaining undivided interest in the intellectual property for rights to payments of 4.05 percent of gross revenues in excess of $200 million, from sales of products, which would, in the absence of the Technology Transfer Agreement, infringe a valid claim under any patent transferred to the Company. The Company also granted to the Partnership a royalty-bearing license to make, use and sell small quantities of product derived from the intellectual property for research purposes only.
In March 2000, the Company and AGDG amended the Technology Transfer Agreement to give to AGDG and Gene Tools LLC, related organizations, exclusive, non royalty-bearing rights to in vitro diagnostic applications of the intellectual property. In consideration for this amendment, Gene Tools paid the Company $1 million and reduced the royalty that the Company would pay to AGDG under the Technology Transfer Agreement on future sales of therapeutic products from 4.05% to 3.00%.
The remaining net assets of the Partnership, $177,000 of cash, were no longer combined with those of the Company in May 1993. Under the terms of the Technology Transfer Agreement, the Partnership ceased active sales of partnership units and income generating activities and no longer will enter into research and development contracts with the Company. The Partnership currently exists primarily for the purpose of collecting potential future payments from the Company as called for in the Technology Transfer Agreement.
Acquisition of Ercole
On March 20, 2008, the Company acquired all of the stock of Ercole Biotechnology, Inc. (“Ercole”) in exchange for 5,811,721 shares of AVI common stock. The transaction included the assumption of approximately $1.8 million in liabilities of Ercole. The AVI common stock was valued at approximately $8.4 million. AVI also issued warrants to purchase AVI stock to settle certain outstanding warrants held in Ercole, which were valued at $437,000. These warrants are classified as equity. The acquisition was aimed at consolidating AVI’s position in directed alternative RNA splicing therapeutics. Ercole and the Company had been collaborating since 2006 to develop drug candidates, including AVI-4658, currently in clinical testing in the United Kingdom for the treatment of Duchenne muscular dystrophy. Ercole has other ongoing discovery research programs.
The total estimated purchase price of $10.2 million has been allocated as follows:
|
Accounts Receivable |
|
$ |
76,000 |
|
|
Prepaid Expenses |
|
$ |
7,000 |
|
|
Fixed Assets |
|
$ |
10,000 |
|
|
Patents |
|
$ |
190,000 |
|
|
Acquired In-Process Research and Development |
|
$ |
9,916,000 |
|
The pending patents acquired as part of the Ercole acquisition have an expected expiration date of 2026. Acquired in-process research and development consists of other discovery research programs in areas including beta thalassemia and soluble tumor necrosis factor receptor. As these programs were in development at the time of acquisition, there were significant risks associated with completing these projects, and there were no alternative future uses for these projects, the associated value has been considered acquired in-process research and development.
Ercole has been a development stage company since inception and does not have a product for sale. The Company has retained a limited number of Ercole employees and plans on incorporating in-process technology of Ercole into the Company’s processes. The acquisition of Ercole did not meet the definition of a business and it is therefore being accounted for as an asset acquisition.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES:
Basis of Accounting
The financial statements have been prepared in accordance with U.S. generally accepted accounting principles as outlined in the FASB Accounting Standards CodificationTM.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant items subject to such estimates and assumptions include the valuation of investments and liability classified warrants, long-lived asset impairment, and revenue recognition.
Reclassifications
Certain prior year amounts have been reclassified to conform to current year presentation. These changes did not have a significant impact on Company’s net loss, assets, liabilities, shareholders’ equity or cash flows.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less from the date of purchase to be cash equivalents.
Short-Term Securities—Available-For-Sale
Short-term securities include certificates of deposit, commercial paper and other highly liquid investments with original maturities in excess of 90 days at the time of purchase and less than one year from the balance sheet date. The Company classifies its investment securities as available-for-sale and, accordingly, such investment securities are stated on the balance sheet at their fair market value with unrealized gains (losses) recorded as a separate component of shareholders’ equity and comprehensive income (loss). There were no unrealized gains or losses on the Company’s investments in marketable securities on its balance sheets as of December 31, 2009 and 2008.
Accounts Receivable
Accounts receivable are stated at invoiced amount and do not bear interest as they are due within twelve months. Because a majority of accounts receivable are from the U.S. government and historically no amounts have been written off, an allowance for doubtful accounts receivable is not considered necessary. Amounts included in accounts receivable are as follows:
|
As of December 31, |
|
2009 |
|
2008 |
|
||
|
(in thousands) |
|
|
|
|
|
||
|
Research contract |
|
$ |
2,085 |
|
$ |
4,971 |
|
|
|
|
|
|
|
|
||
|
Accounts receivable |
|
$ |
2,085 |
|
$ |
4,971 |
|
Property and Equipment
Property and equipment is stated at cost and depreciated over the estimated useful lives of the assets, generally five years, using the straight-line method. Leasehold improvements are amortized over the shorter of the lease term or the estimated useful life of the asset, generally five years, using the straight-line method. Expenditures for repairs and maintenance are expensed as incurred. Expenditures that increase the useful life or value are capitalized.
Amounts included in property held for sale:
|
As of December 31, |
|
2009 |
|
2008 |
|
||
|
(in thousands) |
|
|
|
|
|
||
|
Property held for sale |
|
$ |
2,372 |
|
$ |
— |
|
The Company has listed for sale an industrial property located in Corvallis Oregon for a sales price of $2.5 million. Selling and closing expenses are estimated to be $0.1 million. The Company has decided to outsource its large scale manufacturing activities and has listed this property for sale with a commercial real estate agent.
Amounts included in property and equipment are as follows:
|
As of December 31, |
|
2009 |
|
2008 |
|
||
|
(in thousands) |
|
|
|
|
|
||
|
Building |
|
$ |
— |
|
$ |
2,500 |
|
|
Lab equipment |
|
5,933 |
|
5,676 |
|
||
|
Office equipment |
|
970 |
|
741 |
|
||
|
Leasehold improvements |
|
9,589 |
|
9,191 |
|
||
|
|
|
16,492 |
|
18,108 |
|
||
|
Less accumulated depreciation |
|
(14,026 |
) |
(12,919 |
) |
||
|
Property and equipment, net |
|
$ |
2,466 |
|
$ |
5,189 |
|
Depreciation expense was $1,154,000, $1,212,000 and $1,718,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
Patent Costs
Patent costs consist primarily of legal and filing fees incurred to file patents on proprietary technology developed by the Company. Patent costs are amortized on a straight-line basis over the shorter of the estimated economic lives or the legal lives of the patents, generally 20 years. Patent amortization was $225,000, $257,000 and $296,000 for the years ended December 31, 2009, 2008 and 2007, respectively. The Company also expensed the remaining net book value of previously capitalized patents that were later abandoned of $347,000, $580,000 and $0, for 2009, 2008 and 2007 respectively. The Company expects to incur amortization expense of approximately $146,000 per year over the following five fiscal years.
Revenue Recognition
The Company records revenue from research contracts and grants as the services are performed and payment is reasonably assured. In 2009, 2008 and 2007, the Company recognized $17,585,000, $21,258,000 and $10,985,000, respectively, in research contracts revenues from government funding for work performed on viral disease projects and other grants and contracts. Revenue associated with research and development arrangements is recognized under the proportional performance method, using the payment received method. To date, revenue from research and development arrangements has not been material.
Research and Development
Research and development (R&D) expenses include related salaries, contractor fees, materials, utilities and allocations of corporate costs. R&D expenses also consist of independent R&D costs and costs associated with collaborative development arrangements. In addition, the Company funds R&D at other companies and research institutions under agreements. Research and development costs are expensed as incurred.
Other Current Assets
Amounts included in other current assets are as follows:
|
As of December 31, |
|
2009 |
|
2008 |
|
||
|
(in thousands) |
|
|
|
|
|
||
|
Prepaid expenses |
|
$ |
337 |
|
$ |
316 |
|
|
Prepaid rents |
|
158 |
|
— |
|
||
|
Restricted cash |
|
284 |
|
283 |
|
||
|
|
|
|
|
|
|
||
|
Other current assets |
|
$ |
779 |
|
$ |
599 |
|
Starting in April 2006, the Company was required to pledge $150,000 as collateral for company credit cards issued to certain employees. Starting in April 2007, the Company was required to pledge $125,000 as collateral for payments on long-term debt. The Company classifies these amounts as restricted cash. As of December 31, 2009, restricted cash including accrued interest was $284,000. The remaining components of other current assets include normally occurring prepaid expenses and rents.
Stock-based Compensation
The Company issues stock-based compensation to certain employees, officers and directors. These principles require companies to account for stock options using the fair value method, which results in the recognition of compensation expense over the vesting period of the options. See Note 3.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered and settled. A valuation allowance is recorded to reduce the net deferred tax asset to zero because it is more likely than not that the deferred tax asset will not be realized. The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained upon an examination.
Fair Value of Financial Instruments
The Company measures at fair value certain financial assets and liabilities. Generally accepted accounting principles specify a hierarchy of valuation techniques based on whether the inputs to those valuation techniques are observable or unobservable. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect the Company’s market assumptions. These two types of inputs have created the following fair-value hierarchy:
Level 1—Quoted prices for identical instruments in active markets;
Level 2—Quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, and model-derived valuations in which all significant inputs and significant value drivers are observable in active markets; and
Level 3—Valuations derived from valuation techniques in which one or more significant value drivers are unobservable.
The Company’s assets measured at fair value on a recurring basis consisted of the following as of December 31, 2009:
|
|
|
Fair Value Measurement as of December 31, 2009 |
|
||||||||||
|
(in thousands) |
|
Total |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
||||
|
Short-term securities-available- for-sale and restricted cash |
|
$ |
48,730 |
|
$ |
48,275 |
|
$ |
455 |
|
$ |
— |
|
|
Total |
|
$ |
48,730 |
|
$ |
48,275 |
|
$ |
455 |
|
$ |
— |
|
The Company’s liabilities measured at fair value on a recurring basis consisted of the following as of the date indicated:
|
|
|
Fair Value Measurement as of December 31, 2009 |
|
||||||||||
|
(in thousands) |
|
Total |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
||||
|
Warrants |
|
$ |
27,609 |
|
$ |
— |
|
$ |
— |
|
$ |
27,609 |
|
|
Total |
|
$ |
27,609 |
|
$ |
— |
|
$ |
— |
|
$ |
27,609 |
|
The Company’s assets measured at fair value on a recurring basis consisted of the following as of December 31, 2008:
|
|
|
Fair Value Measurement as of December 31, 2008 |
|
||||||||||
|
(in thousands) |
|
Total |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
||||
|
Short-term securities-available- for-sale and restricted cash |
|
$ |
11,757 |
|
$ |
11,192 |
|
$ |
565 |
|
$ |
— |
|
|
Total |
|
$ |
11,757 |
|
$ |
11,192 |
|
$ |
565 |
|
$ |
— |
|
The Company’s liabilities measured at fair value on a recurring basis consisted of the following as of the date indicated:
|
|
|
Fair Value Measurement as of December 31, 2008 |
|
||||||||||
|
(in thousands) |
|
Total |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
||||
|
Warrants |
|
$ |
1,254 |
|
$ |
— |
|
$ |
— |
|
$ |
1,254 |
|
|
Total |
|
$ |
1,254 |
|
$ |
— |
|
$ |
— |
|
$ |
1,254 |
|
A reconciliation of the change in value of the Company’s warrants for the year ended December 31, 2009 is as follows:
|
|
|
Fair Value Measurements |
|
|
|
(in thousands) |
|
(Level 3) |
|
|
|
|
|
|
|
|
|
Balance at January 1, 2009 |
|
$ |
1,254 |
|
|
Change in value of warrants |
|
9,198 |
|
|
|
Issuances |
|
17,157 |
|
|
|
Balance at December 31, 2009 |
|
$ |
27,609 |
|
A reconciliation of the change in value of the Company’s warrants for the year ended December 31, 2008 is as follows:
|
|
|
Fair Value Measurements |
|
|
|
(in thousands) |
|
(Level 3) |
|
|
|
|
|
|
|
|
|
Balance at January 1, 2008 |
|
$ |
4,415 |
|
|
Change in value of warrants |
|
(3,161 |
) |
|
|
|
|
|
|
|
|
Balance at December 31, 2008 |
|
$ |
1,254 |
|
The carrying amounts reported in the balance sheets for cash and cash equivalents, accounts receivable, accounts payable, and other current monetary assets and liabilities approximate fair value because of the immediate or short-term maturity of these financial instruments.
Warrants
Certain of the Company’s warrants issued in connection with financing arrangements are classified as liabilities in accordance with the generally accepted accounting pronouncements, whereby, the fair market value of these warrants is recorded on the balance sheet at issuance and marked to market at each financial reporting period. The change in the fair value of the warrants is recorded in the Statement of Operations as a non-cash gain (loss) and is estimated using the Black-Scholes option-pricing model with the following assumptions:
|
Year Ended December 31, |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
Risk-free interest rate |
|
0.2%-2.69 |
% |
0.3%-3.0 |
% |
3.1%-3.5 |
% |
|||
|
Expected dividend yield |
|
0 |
% |
0 |
% |
0 |
% |
|||
|
Expected lives |
|
0.4-4.7 years |
|
0.2-4.2 years |
|
0.9-5.0 years |
|
|||
|
Expected volatility |
|
86.0%-102.1 |
% |
63.6%-104.8 |
% |
58.2%-80.7 |
% |
|||
|
Warrants classified as liabilities |
|
30,203,466 |
|
7,994,229 |
|
9,607,866 |
|
|||
|
Warrants classified as equity |
|
2,129,530 |
|
2,129,530 |
|
4,248,545 |
|
|||
|
Market value of stock at beginning of year |
|
$ |
0.66 |
|
$ |
1.41 |
|
$ |
3.18 |
|
|
Market value of stock at end of year |
|
$ |
1.46 |
|
$ |
0.66 |
|
$ |
1.41 |
|
The risk-free interest rate is estimated using an average of treasury bill interest rates. The expected dividend yield is zero as the Company has not paid any dividends to date and does not expect to pay dividends in the future. The expected lives are based on the remaining contractual lives of the related warrants. The expected volatility is estimated using historical calculated volatility and considers factors such as future events or circumstances that could impact volatility.
For warrants classified as permanent equity, the fair value of the warrants is recorded as additional paid-in capital and no further adjustments are made.
Comprehensive Income (Loss)
Comprehensive income (loss) includes charges or credits to equity that did not result from transactions with shareholders. The Company’s only component of “other comprehensive income (loss)” is unrealized gain (loss) on cash equivalents and short-term securities—available-for-sale.
Rent Expense
The Company’s operating lease agreements for its Corvallis, Oregon facility and its Bothell, Washington facility provide for scheduled annual rent increases throughout the lease’s term. In accordance with generally accepted accounting principles the Company recognizes the effects of the scheduled rent increases on a straight-line basis over the full term of the leases, which expires in 2020 and 2014.
During the years ended December 31, 2009, 2008 and 2007, the Company recognized $230,000, $133,000 and $155,000, respectively, in additional rent expense from the amortization of future scheduled rent increases.
Commitments and Contingencies.
In the normal course of business, the Company may be named as a party to various legal claims, actions and complaints, including matters involving employment, intellectual property, effects from the use of drugs utilizing our technology, or others. It is impossible to predict with certainty whether any resulting liability would have a material adverse effect on the Company’s financial position, results of operations or cash flows.
Financial Instruments.
The carrying amounts reported in the balance sheets for cash and cash equivalents, accounts receivable, accounts payable, and other current monetary assets and liabilities approximate fair value because of the immediate or short-term maturity of these financial instruments.
License Arrangements.
License arrangements may consist of non-refundable upfront license fees, data transfer fees, research reimbursement payments, exclusive licensed rights to patented or patent pending compounds, technology access fees, various performance or sales milestones and future product royalty payments. Some of these arrangements are multiple element arrangements.
The Company defers recognition of non-refundable upfront fees if it has continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of Company performance under the other elements of the arrangement. In addition, if the Company has continuing involvement through research and development services that are required because its know-how and expertise related to the technology is proprietary to the Company, or can only be performed by the Company, then such up-front fees are deferred and recognized over the period of continuing involvement.
Payments related to substantive, performance-based milestones in a research and development arrangement are recognized as revenue upon the achievement of the milestones as specified in the underlying agreements when they represent the culmination of the earnings process.
Long-Lived Asset Impairment
Long-lived assets held and used by us and intangible assets with determinable lives are reviewed for impairment whenever events or circumstances indicate that the carrying amount of assets may not be recoverable in accordance with generally accepted accounting pronouncements. We evaluate recoverability of assets to be held and used by comparing the carrying amount of an asset to future net undiscounted cash flows to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amount of the assets exceeds the fair value of the assets. Such reviews assess the fair value of the assets based upon estimates of future cash flows that the assets are expected to generate.
At December 31, 2008, the Company determined that the ongoing decline in the real estate market had adversely impacted the fair value of a building purchased by the Company for $3.3 million in 2007. Based on an independent third-party appraisal, the Company estimated that the current fair value of the building had declined to approximately $2.5 million. Accordingly, an impairment charge of $800,000 was recorded for the year ended December 31, 2008. The Company completed a second third party appraisal in November of 2009, based on this revised estimate the Company believes the property to have a current fair value, net of costs to sell of $2.4 million. This property was listed for sale in November of 2009. Selling and closing expenses are estimated to be $0.1 million. The Company has decided to outsource its large scale manufacturing activities and has listed this property for sale with a commercial real estate agent.
In addition, at December 31, 2009, the Company conducted an evaluation of the status of its patents each quarter during 2009. Pursuant to these evaluations, the Company has recorded a write-off of $347,000, in 2009 for previously capitalized costs related to patents that have expired or were abandoned.
Government Research Contract Revenue.
The Company recognizes revenues from federal research contracts during the period in which the related expenditures are incurred. The Company receives reimbursement of costs incurred, overhead and, in some cases, a fixed fee. The Company presents these revenues and related expenses at gross in the consolidated financial statements in accordance with the generally accepted accounting pronouncements.
Recent Accounting Pronouncements
Recently adopted accounting guidance:
During the first fiscal quarter of 2009, the Financial Accounting Standards Board issued Staff Positions ASC 820 10 65-65-4, “Determining Fair Value When the Volume and Level of Activity for the Asset or Liability has Significantly Decreased and the Identifying Transactions That Are Not Orderly”, ASC 320 10 65-65-1, “Recognition and Presentation of Other-Than-Temporary Impairments”, and ASC 825 10 65 65-1, “Interim Disclosures about Fair Value of Financial Instruments”. These Staff Positions were issued to clarify the application of ASC 820 10 65-65-4, “Fair Value Measurements” in the current economic environment, modify the recognition of other-than-temporary impairments of debt securities, and require companies to disclose the fair value of financial instruments in interim periods. The Staff Positions are effective for interim and annual periods ending after September 15, 2009, with early adoption permitted for periods ending after March 15, 2009, if all three Staff Positions or both the fair-value measurement and other-than-temporary impairment Staff Positions are adopted simultaneously. The Company has adopted the Staff Positions in the third quarter of fiscal 2009, and there was no material impact on the Company’s Financial Statements or related disclosures.
In April 2009, the FASB issued FASB Staff Position ASC 320 10 65-65-1, “Recognition and Presentation of Other-Than-Temporary Impairments”, which requires the Company to disclose information for interim and annual periods that enables users of its financial statements to understand the types of available-for-sale and held-to-maturity debt and equity securities held, including information about investments in an unrealized loss position for which an other-than-temporary impairment has or has not been recognized. The provisions of this pronouncement were adopted in the second quarter of 2009. There was no material impact on the Company’s financial statements.
In April 2009, the FASB issued FSP ASC 825 10 65 65-1, “Interim Disclosures about Fair Value of Financial Instruments” , which requires publicly traded companies to include disclosures about the fair value of its financial instruments whenever it issues summarized financial information for interim reporting periods. The provisions of these pronouncements were adopted in the second quarter of 2009. There was no material impact on the Company’s financial statements.
Recent accounting guidance not yet adopted:
In January 2010, the FASB issued guidance to amend the disclosure requirements related to recurring and nonrecurring fair value measurements. The guidance requires new disclosures on the transfers of assets and liabilities between Level 1 (quoted prices in active market for identical assets or liabilities) and Level 2 (significant other observable inputs) of the fair value measurement hierarchy, including the reasons and the timing of the transfers. The guidance will become effective for us with the reporting period beginning January 1, 2010, except for the disclosure on the roll forward activities for Level 3 fair value measurements, which will become effective for us with the reporting period beginning July 1, 2011. Other than requiring additional disclosures, adoption of this new guidance will not have a material impact on our financial statements.
3. STOCK-BASED COMPENSATION:
Stock-based compensation costs are generally based on the fair value calculated from the Black-Scholes option-pricing model on the date of grant for stock options and on the date of enrollment for the Plan. The fair value of stock grants is amortized as compensation expense on a straight-line basis over the vesting period of the grants. Stock options granted to employees are service-based and typically vest over three years.
The fair market values of stock options granted during 2009, 2008 and 2007 were measured on the date of grant using the Black-Scholes option-pricing model, with the following weighted average assumptions:
|
Year Ended December 31, |
|
2009 |
|
2008 |
|
2007 |
|
|
Risk-free interest rate |
|
1.2%-1.8 |
% |
1.1%-3.4 |
% |
4.4%-5.1 |
% |
|
Expected dividend yield |
|
0 |
% |
0 |
% |
0 |
% |
|
Expected lives |
|
3.6-9.1 Years |
|
3.6-9.1 Years |
|
3.7-9.1 Years |
|
|
Expected volatility |
|
92.0%-94.4 |
% |
81.0%-90.7 |
% |
84.1%-90.6 |
% |
The risk-free interest rate is estimated using an average of treasury bill interest rates. The expected dividend yield is zero as the Company has not paid any dividends to date and does not expect to pay dividends in the future. The expected lives are estimated using expected and historical exercise behavior. The expected volatility is estimated using historical calculated volatility.
The Company is required to estimate potential forfeiture of stock grants and adjust compensation cost recorded accordingly. The estimate of forfeitures is adjusted over the requisite service period to the extent that actual forfeitures differ, or are expected to differ, from such estimates. Changes in estimated forfeitures are recognized through a cumulative catch-up in the period of change and impact the amount of stock compensation expense to be recognized in future periods.
A summary of the Company’s stock option activity with respect to the years ended December 31, 2009, 2008 and 2007 is presented in the following table:
|
|
|
2009 |
|
2008 |
|
2007 |
|
|||||||||
|
For the Year Ended |
|
Shares |
|
Weighted |
|
Shares |
|
Weighted Price |
|
Shares |
|
Weighted |
|
|||
|
Options outstanding at beginning of year |
|
7,540,873 |
|
$ |
3.34 |
|
6,304,453 |
|
$ |
4.60 |
|
5,571,470 |
|
$ |
5.12 |
|
|
Granted |
|
2,727,000 |
|
1.10 |
|
2,743,607 |
|
1.27 |
|
1,263,548 |
|
2.80 |
|
|||
|
Exercised |
|
(62,711 |
) |
1.68 |
|
(6,761 |
) |
1.31 |
|
(11,639 |
) |
2.49 |
|
|||
|
Canceled |
|
(1,272,351 |
) |
2.72 |
|
(1,500,426 |
) |
4.82 |
|
(518,926 |
) |
5.88 |
|
|||
|
Options outstanding at end of year |
|
8,932,811 |
|
2.79 |
|
7,540,873 |
|
3.34 |
|
6,304,453 |
|
4.60 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
Exercisable at end of year |
|
5,119,227 |
|
$ |
3.94 |
|
4,779,603 |
|
$ |
4.18 |
|
4,497,526 |
|
$ |
4.76 |
|
|
Vested at December 31, 2009 and expected to vest |
|
8,856,539 |
|
$ |
2.80 |
|
|
|
|
|
|
|
|
|
||
The following table summarizes information about stock options outstanding at December 31, 2009:
|
|
|
Outstanding Options |
|
Exercisable Options |
|
||||||||
|
Range of Exercise Prices |
|
Number of Shares |
|
Weighted Average Exercise |
|
Weighted Average |
|
Number of |
|
Weighted Price |
|
||
|
$0.60-$1.09 |
|
2,013,950 |
|
$ |
0.96 |
|
8.54 |
|
200,617 |
|
$ |
1.06 |
|
|
$1.10-$1.39 |
|
1,895,430 |
|
$ |
1.24 |
|
8.64 |
|
447,181 |
|
$ |
1.28 |
|
|
$1.42-$2.53 |
|
1,969,357 |
|
$ |
2.18 |
|
6.29 |
|
1,569,524 |
|
$ |
2.30 |
|
|
$2.55-$5.75 |
|
2,106,291 |
|
$ |
4.49 |
|
3.08 |
|
1,954,122 |
|
$ |
4.60 |
|
|
$5.88-$7.35 |
|
947,783 |
|
$ |
7.21 |
|
4.26 |
|
947,783 |
|
$ |
7.21 |
|
|
Total |
|
8,932,811 |
|
$ |
2.79 |
|
6.33 |
|
5,119,227 |
|
$ |
3.94 |
|
The weighted average fair value per share of stock-based payments granted to employees during 2009, 2008 and 2007 was $1.09, $1.04 and $2.27, respectively. During 2009, 2008 and 2007, the total intrinsic value of stock options exercised was $105,301, $1,831 and $4,937, and the total fair value of stock options that vested was $1,740,000, $3,040,000 and $3,661,000, respectively.
As of December 31, 2009, there was $2,278,000 of total unrecognized compensation cost related to nonvested share-based compensation arrangements granted under the Plan. These costs are expected to be recognized over a weighted-average period of 2.3 years.
During the year ended December 31, 2009, $76,000 was received for the exercise of stock options. The Company is obligated to issue shares from the 2002 Equity Incentive Plan upon the exercise of stock options. The Company does not currently expect to repurchase shares from any source to satisfy its obligations under the Plan. The Company may issue options to purchase up to an additional 681,995 shares of Common Stock at December 31, 2009 under stock option plans.
The following are the stock-based compensation costs recognized in the Company’s statements of operations:
(in thousands)
|
|
|
Year Ended |
|
Year Ended |
|
Year Ended |
|
|||
|
Research and development |
|
$ |
1,192 |
|
$ |
1,689 |
|
$ |
1,878 |
|
|
General and administrative |
|
1,182 |
|
3,141 |
|
3,854 |
|
|||
|
Total |
|
$ |
2,374 |
|
$ |
4,830 |
|
$ |
5,732 |
|
On March 27, 2007, in connection with his resignation, the Company entered into a Separation and Release Agreement with AVI’s former Chairman and Chief Executive Officer. Pursuant to this agreement, he may exercise his previously granted options until the earlier of the termination date specified in the respective stock option grant agreements or March 28, 2010. This modification of these stock options in the first quarter of 2007 increased compensation costs by $1,057,000.
In the first quarter of 2008, the Company granted 333,000 shares of restricted stock to its new Chief Executive Officer. These shares vest over a period of four years. The Company recognized compensation expense related to these shares of for the years ended December 31, 2009 and 2008, of $63,000 and $166,000.
In the third quarter of 2008, the Company’s President and Chief Operating Officer resigned. In accordance with his existing employment agreement, he may exercise his previously granted options until the earlier of the termination date specified in the respective stock option grant agreements or March 18, 2010. This acceleration of the vesting of these stock options resulted in additional compensation costs of $382,000 for the year ended December 31, 2008. As of December 31, 2009, these options were outstanding.
In the second quarter of 2009, the Company granted a total of 25,000 shares of restricted stock to members of its Board of Directors. These shares vest over a period of one year. During year ended December 31, 2009, the Company recognized compensation expense related to these shares of $58,000.
Also in the second quarter of 2009, the Company granted 100,000 shares of restricted stock to its Vice President of Business Development. These shares vest upon the achievement of certain performance milestones. During the year ended December 31, 2009, the Company did not recognize any compensation expense related to these shares since the performance milestones was not achieved and these shares were canceled.
In the first quarter of 2009, the Company granted 60,000 shares of restricted stock to its Chief Medical Officer. These shares vest over a period of 181 days. During the year ended December 31, 2009 the Company recognized compensation expense related to these shares of $82,000.
The Company records the fair value of stock options granted to non-employees in exchange for services in accordance with generally accepted accounting principles. The fair value of the options granted is expensed when the measurement date is known. The performance for services was satisfied on the grant date for stock options granted to non-employees.
The total fair value of the options granted to non-employees during the years ended December 31, 2009, 2008 and 2007 was $141,000, $180,000 and $313,000 respectively, which was expensed to general and administration.
4. NET LOSS PER SHARE:
Basic EPS is calculated using the weighted average number of common shares outstanding for the period and diluted EPS is computed using the weighted average number of common shares and dilutive common equivalent shares outstanding. Given that the Company is in a loss position, there is no difference between basic EPS and diluted EPS since the common stock equivalents would be antidilutive.
|
Year Ended December 31, |
|
2009 |
|
2008 |
|
2007 |
|
|||
|
(in thousands) |
|
|
|
|
|
|
|
|||
|
Net loss |
|
$ |
(25,159 |
) |
$ |
(23,953 |
) |
$ |
(27,168 |
) |
|
Weighted average number of shares of common stock and common stock equivalents outstanding: |
|
|
|
|
|
|
|
|||
|
Weighted average number of common shares outstanding for computing basic earnings per share |
|
93,090 |
|
69,491 |
|
53,942 |
|
|||
|
Dilutive effect of warrants and stock options after application of the treasury stock method |
|
* |
|
* |
|
* |
|
|||
|
Weighted average number of common shares outstanding for computing diluted earnings per share |
|
93,090 |
|
69,491 |
|
53,942 |
|
|||
|
Net loss per share - basic and diluted |
|
$ |
(0.27 |
) |
$ |
(0. 34) |
|
$ |
(0. 50) |
|
* Warrants and stock options to purchase 41,266,000, 17,665,000 and 20,161,000 shares of common stock as of December 31, 2009, 2008 and 2007, respectively, were excluded from the earnings per share calculation as their effect would have been antidilutive.
5. LIQUIDITY:
Since its inception in 1980 through December 31, 2009, the Company has incurred losses of approximately $275.5 million, substantially all of which resulted from expenditures related to research and development, general and administrative charges and acquired in-process research and development resulting from two acquisitions. The Company has not generated any material revenue from product sales to date, and there can be no assurance that revenues from product sales will be achieved. Moreover, even if the Company does achieve revenues from product sales, the Company expects to incur operating losses over the next several years.
The Company believes it has sufficient cash to fund operations at least through the following twelve months, exclusive of future receipts from billings on existing government contracts. For 2010, the Company expects expenditures for operations, net of government funding, including collaborative efforts and research and development activities to be approximately $23 to $27 million. The Company believes it will continue to receive funding from government and other sources to pursue the development of its product candidates, and has assumed certain revenues from these awards in providing this guidance. Should the Company not continue to receive funding from its current contracts or receive additional funding, or should the timing be delayed, it may have a significant negative impact on the Company’s guidance.
Our cash, cash equivalents and short-term securities were $48.4 million at December 31, 2009, compared with $11.5 million at December 31, 2008, respectively. The increase of $36.9 million was due primarily to net proceeds of $47.8 million from the sale of common stock and issuance of stock warrants from two separate equity financing transactions that closed in January and August of 2009. The cash from financing activities was partially offset by cash used in operations of $8.8 million, costs of $2.0 million related to acquisitions of patents and fixed assets and debt repayments of $0.1 million.
In January 2009, we raised net proceeds of $15.5 million in financing through the sale of 14,224,202 shares of common stock pursuant to a registered direct offering to a select group of institutional investors. The investors also received warrants to purchase 14,224,202 shares of the Company’s common stock at an exercise price of $1.16 per share. These warrants are exercisable starting July 30, 2009 and expire on July 30, 2014. In addition, the placement agent used for the equity financing received a warrant for the purchase of an additional 426,726 common shares at $1.45 per share. This warrant is exercisable starting January 30, 2009 and expires on January 30, 2014. All of these warrants have been classified as liabilities as discussed in Note 7, as they require the issuance of registered shares. These warrants are non-cash liabilities; the Company is not required to expend any cash to settle these liabilities.
On August 25, 2009, the Company closed a registered equity financing for net proceeds of $32.3 million with several institutional investors. The Company sold 24,295,775 shares of common stock at $1.42 per share, and also issued warrants for the purchase of 9,718,310 common shares at an exercise price of $1.78 per share. These warrants are exercisable starting February 25, 2010 and expire on August 25, 2014. All of these warrants have been classified as liabilities as discussed in Note 7, as they require the issuance of registered shares. These warrants are non-cash liabilities; the Company is not required to expend any cash to settle these liabilities.
The Company currently has a total of $61.7 million of contracted development studies. As of December 31, 2009, $48.4 million has been billed to the government. The Company has $13.3 in development contracts remaining that have not yet been completed and have not been billed. The Company expects to complete the remaining contract activity and receive the contracted revenue in 2010 and early 2011.
In December 2006, the Company announced the execution of a two-year $28 million research contract with the Defense Threat Reduction Agency (DTRA), an agency of the United States Department of Defense (DoD). The contract is directed toward funding the Company’s development of antisense therapeutics to treat the effects of Ebola, Marburg and Junín hemorrhagic viruses, which are seen by DoD as potential biological warfare and bioterrorism agents. In May 2009, the Company received an amendment from DTRA to extend the contract performance period to November 29, 2009 and a cost modification of an additional $5.9 million, increasing the total contract amount to $33.9 million. In September 2009, the Company received a second amendment from DTRA to extend the contract performance period to February 28, 2011 and a cost modification of an additional $11.5 million, increasing the total contract amount to $45.4 million. During the twelve month period ended December 31, 2009, 2008 and 2007, the Company recognized $10.4 million, $16.8 million and $8.0 million, respectively, in research contract revenue from this contract. To date, the Company has recognized revenues of $35.2 million from this contract. Funding of the remainder of the contract is anticipated in 2010 and 2011.
In January 2006, the Company announced that the final version of the 2006 defense appropriations act had been approved, which included an allocation of $11.0 million to fund the Company’s ongoing defense-related programs. Net of government administrative costs, it is anticipated that the Company will receive up to $9.8 million under this allocation. The Company’s technology is expected to be used to continue developing RNA based drugs against Ebola and Marburg viruses. The Company has received signed contracts for all of these projects. The Company expects that funding under these signed contracts will be completed over the next 12 months. During the twelve month period ended December 31, 2009, 2008 and 2007, the Company recognized $2.3 million, $4.2 million and $2.7 million, respectively, in research contract revenue from this contract. To date, the Company has recognized revenues of $9.2 million on these contracts. Funding of the remainder of these contracts is anticipated in 2010.
In May 2009, the Company entered into a contract with DTRA to develop H1N1 drugs. Under this contract, DTRA will pay up to $4.1 million to the Company for the work to be performed by the Company. The work will involve the application of analogs based on the Company’s proprietary PMO and PMOplus antisense chemistry and the Company plans to conduct preclinical development of at least one drug candidate and demonstrate it is effective by testing it in virus infected animals. During the twelve month period ended December 31, 2009, the Company recognized $1.7 million in revenue under this contract.
In July 2009, the Company entered into a lease agreement with BMR-3450 Monte Villa Parkway LLC relating to the lease of 19,108 square feet of laboratory and office space in Bothell, Washington. The Company began occupying this space in August 2009, and has moved its headquarters and R&D functions to this new location. The term of the lease is approximately 63 months, although the Company has a one-time option to terminate the lease after 3 years’ time upon payment of a termination fee. The Company will commence paying base rent of approximately $43,000 per month after approximately 3 months. The amount of base rent is subject to an annual increase of 3%.
The likelihood of the long-term success of the Company must be considered in light of the expenses, difficulties and delays frequently encountered in the development and commercialization of new pharmaceutical products, competitive factors in the marketplace as well as the complex regulatory environment in which the Company operates. There can be no assurance that the Company will ever achieve significant revenues or profitable operations.
6. LONG-TERM DEBT
The Company has two loans outstanding which are collateralized by a parcel of real property purchased in April 2007 in Corvallis, Oregon. These loans bear interest at 4.75% and mature in February 2027. At December 31, 2009, these loans had unpaid principal balances of $1,275,000 and $726,000, for a total indebtedness of $2,001,000. The Company incurred interest expense on these loans of $97,000, $104,000 and $104,000, respectively, for the years ended December 31, 2009, 2008 and 2007.
The following table sets forth the expected future principal payments on these loans:
(in thousands)
|
Year ending December 31, |
|
|
|
|
|
2010 |
|
$ |
77 |
|
|
2011 |
|
81 |
|
|
|
2012 |
|
85 |
|
|
|
2013 |
|
90 |
|
|
|
2014 |
|
92 |
|
|
|
Thereafter |
|
1,576 |
|
|
|
Total scheduled loan principal payments |
|
$ |
2,001 |
|
7. SHAREHOLDERS’ EQUITY AND WARRANT LIABILITY:
In December 2007, the Company closed a private equity financing for net proceeds of $14,448,250 with several institutional investors. The Company sold 10,696,616 shares of common stock at $1.90 per share. These investors also received warrants for the purchase of 5,348,308 common shares at $2.45 per share. These warrants are exercisable starting June 19, 2008 and expire on December 18, 2012.
On January 30, 2009, the Company closed a registered equity financing for net proceeds of $15.5 million with several institutional investors. The Company sold 14,224,202 shares of common stock at $1.16 per share, and also issued warrants for the purchase of 14,224,202 common shares at $1.16 per share and a fair value at the date of issue of $8.2 million. These warrants are exercisable starting July 30, 2009 and expire on July 30, 2014. In connection with the equity financing, the placement agent received a warrant for the purchase of an additional 426,726 common shares at $1.45 per share. This warrant is exercisable starting January 30, 2009 and expires on January 30, 2014. All of these warrants have been classified as liabilities as they require the issuance of registered shares. These warrants are non-cash liabilities; The Company does not expect to expend any cash to settle these liabilities.
On August 25, 2009, the Company closed a registered equity financing for net proceeds of $32.3 million with several institutional investors. The Company sold 24,295,775 shares of common stock at $1.42 per share, and also issued warrants for the purchase of 9,718,310 common shares at $1.78 per share and a fair value at the date of issue of $9.0 million. These warrants are exercisable starting February 25, 2010 and expire on August 25, 2014. All of these warrants have been classified as liabilities as, as they require the issuance of registered shares. These warrants are non-cash liabilities; The Company does not expect to expend any cash to settle these liabilities.
The Company has two stock option plans, the 2002 Equity Incentive Plan and the 1997 Stock Option Plan (the Plans). The 2002 Plan provides for the issuance of incentive stock options to employees and nonqualified stock options, stock appreciation rights and bonus rights to employees, directors of the Company and consultants. The 1997 Plan provides for the assumption of the ImmunoTherapy Options under the Merger Agreement. The Company has reserved 11,828,111 shares of common stock for issuance under the Plans. Options issued under the Plans generally vest ratably over three years and expire five to ten years from the date of grant. At December 31, 2009, 8,932,811 options were outstanding at a weighted-average exercise price of $2.79 under equity compensations plans approved by security holders. At December 31, 2009, 681,955 options were available for issuance under equity compensation plans approved by security holders. See Note 3—“Stock-Based Compensation” for a summary of the status of the Company’s stock option plans and changes for the years ended December 31, 2009, 2008 and 2007.
The Company has also issued warrants for the purchase of common stock in conjunction with financing and compensation arrangements. A summary of the status and activity with respect to the Company’s warrants is presented in the following table:
|
|
|
2009 |
|
2008 |
|
2007 |
|
|||||||||
|
For the Year Ended |
|
Shares |
|
Weighted |
|
Shares |
|
Weighted |
|
Shares |
|
Weighted |
|
|||
|
Warrants outstanding at beginning of year |
|
10,123,759 |
|
$ |
8.54 |
|
13,856,411 |
|
$ |
8.12 |
|
8,508,103 |
|
$ |
11.68 |
|
|
Granted |
|
24,369,238 |
|
1.41 |
|
445,985 |
|
1.77 |
|
5,348,308 |
|
2.45 |
|
|||
|
Exercised |
|
— |
|
— |
|
— |
|
— |
|
— |
|
— |
|
|||
|
Expired |
|
(2,160,001 |
) |
5.00 |
|
(4,178,637 |
) |
6.42 |
|
— |
|
— |
|
|||
|
Warrants outstanding at end of year |
|
32,332,996 |
|
3.40 |
|
10,123,759 |
|
8.54 |
|
13,856,411 |
|
8.12 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
Exercisable at end of year |
|
20,948,808 |
|
$ |
1.60 |
|
8,457,881 |
|
$ |
3.21 |
|
6,842,225 |
|
$ |
5.85 |
|
The following table summarizes information about warrants outstanding at December 31, 2009:
|
Exercise |
|
Outstanding Warrants at |
|
Weighted Average |
|
Exercisable Warrants |
|
|
|
$ |
0.0003 |
|
16,667 |
|
No expiration date |
|
16,667 |
|
|
0.1679 |
|
238,228 |
|
2.87 |
|
238,228 |
|
|
|
1.14 |
|
1,000 |
|
No expiration date |
|
1,000 |
|
|
|
1.16 |
|
14,224,202 |
|
4.58 |
|
14,224,202 |
|
|
|
1.45 |
|
426,726 |
|
4.08 |
|
426,726 |
|
|
|
1.78 |
|
9,718,310 |
|
4.67 |
|
0 |
|
|
|
2.45 |
|
5,348,308 |
|
2.97 |
|
5,348,308 |
|
|
|
3.61 |
|
207,757 |
|
0.37 |
|
207,757 |
|
|
|
5.00 |
|
485,920 |
|
0.37 |
|
485,920 |
|
|
|
35.63 |
|
1,665,878 |
|
0.25 |
|
— |
|
|
|
|
|
32,332,996 |
|
|
|
20,948,808 |
|
|
The warrants issued in 2009 and 2007 do not require net cash settlement. However, because the warrants require settlement in registered shares, the Company has recorded the warrants as liabilities on the accompanying balance sheet. There is no effect on cash flows from these warrants, as the mark-to-market adjustment is reflected as a non-cash charge within the Company’s Statements of Operations. There were 30,203,466, 9,607,866, and 4,259,558 outstanding warrants classified as liabilities at December 31, 2009, 2008, and 2007, respectively.
8. SIGNIFICANT AGREEMENTS:
On January 27, 2007, the Company announced that it had entered into a definitive License Agreement with Chiron Corporation (“Chiron”) granting the Company a nonexclusive license to Chiron’s patents and patent applications for the research, development, and commercialization of antisense therapeutics against hepatitis C virus, in exchange for the payment of certain milestone and royalty payments to Chiron. In lieu of the first milestone payment due under the License Agreement, the Company and Chiron also entered into a separate agreement under which the Company issued to Chiron 89,012 shares of the Company’s common stock with a market value of $500,000 and which was expensed to research and development. There may be future payments made to Chiron by the Company based on milestones in the License Agreement.
On March 13, 2007, the Company announced that it had entered into agreements with Cook Group Inc. (“Cook”) for Cook’s development and commercialization of products for vascular and cardiovascular diseases. In November 2009, we announced that we believe Cook discontinued development of our drug candidate, AVI-5126, on its cobalt-chromium stent because of an unexpectedly high rate of restenosis.
Effective January 1, 2006, the Company extended the lease on its facility located at 4575 SW Research Way, Suite 200, Corvallis, OR 97333. This lease now expires on December 31, 2020. As of December 31, 2005, the Company had an accrued rent payable of $615,163 related to back rent payments. During the first half of 2006 the Company issued 31,154 shares of the Company’s common stock with a market value of $175,000, paid cash and sold fixed assets to Research Way Investments to pay off the accrued rent payable related to back rent payments.
In January 2006, the Company issued 30,000 shares of the Company’s common stock with a market value of $200,000 to the Oregon State University Foundation to secure access to certain university research facilities, which was expensed to research and development.
On January 8, 2007, the Company announced that it had entered into a cross-license agreement with Eleos Inc. for the development of antisense drugs targeting p53, a well-studied human protein that controls cellular response to genetic damage. Under the terms of the agreement, the Company granted Eleos Inc. an exclusive license to the Company’s NEUGENE® third-generation antisense chemistry to treat cancer with p53-related drugs. In return, Eleos Inc. granted an exclusive license to its patents to the Company for treatment of most viral diseases with drugs that target p53. The companies are sharing rights in other medical fields where targeting p53 may be therapeutically useful. Each company will make milestone payments and royalty payments to the other on development and sales of products that utilize technology licensed under the agreement. In addition, Eleos Inc. made an upfront payment of $500,000 to the Company. The Company recognized $125,000 in license fees for each of the years ended December 31, 2009, 2008 and 2007; the remaining $125,000 has been classified as deferred revenue.
On March 27, 2007, in connection with the resignation of AVI’s former Chairman and Chief Executive Officer, the Company entered into a Separation and Release Agreement, pursuant to which the former Chairman and CEO is entitled to receive his base compensation for 18 months ($562,500 in the aggregate) and medical insurance for the same 18 month period and may exercise his previously granted options until the earlier of the termination date of the respective stock option grant agreements or March 28, 2010. The Company recognized $1,619,872 in total compensation expense to general and administrative in 2007, including $562,500 in cash compensation and $1,057,372 in stock-based compensation.
On April 19, 2007, the Company entered into a real property purchase agreement with WKL Investments Airport, LLC (“WKL”) to purchase a parcel of real property, including improvements situated on the land and intangibles related to the land, for $3,300,000. The Company paid the purchase price as follows: $350,000 in cash, assumption of two loans secured by the property in the amount of $2,200,000, and issuance of 270,758 shares of AVI common stock (at $2.77 per share or $750,000 in the aggregate).
On October 15, 2007, the Company and Charley’s Fund, Inc. announced that the Company had been awarded a $2.45 million research grant from Charley’s Fund, a nonprofit organization that funds drug development and discovery initiatives specific to Duchenne muscular dystrophy (DMD). This award will support a new product development program using proprietary exon skipping technologies developed by the Company to overcome the effects of certain genetic errors in the dystrophin gene. The award will allow AVI to accelerate its development of new therapeutics for DMD. Through December 31, 2009, the Company had received $2.0 million from Charley’s Fund, and recorded the advances as Deferred Revenue, to be recognized upon the attainment of certain milestones as specified in the agreement. In September 2009, the Company amended the agreement with Charley’s Fund. The Amendment pertains to certain provisions of the Sponsored Research Agreement by and between the Company and Charley’s Fund entered into effective October 12, 2007 (the “Agreement”). Under the terms of the Amendment, the Company was awarded up to an additional $3 million in sponsored research funds, for a total of $5 million from Charley’s Fund to support a new product development program using proprietary exon skipping technologies developed by the Company to overcome the effects of certain genetic errors in the dystrophin gene. Revenue associated with this research and development arrangement is recognized under the proportional performance method, using the payment received method. For the years ended December 31, 2009, 2008 and 2007, the Company recognized $ 0, $23,000 and $38,000, respectively, in revenues from Charley’s Fund.
On September 18, 2008, the Company’s President and Chief Operating Officer resigned. In accordance with his employment agreement, he is entitled to receive severance payments totaling $630,000. Of this amount, one-third ($210,000) was paid on the effective date of his termination, and the remaining $420,000 was paid in monthly installments of $35,000 over the following 12 months. The Company recognized compensation expense of $630,000 in 2008 pursuant to his resignation, of which $280,000 was classified as a deferred liability as of December 31, 2008. In 2009 the Company recognized $315,000 of compensation expense. In addition, in accordance with his employment agreement, he may exercise his previously granted stock options until the earlier of the termination date specified in the respective stock option grant agreements or March 18, 2010. This acceleration of the vesting of these stock options resulted in additional compensation costs of $382,419 for the year ended December 31, 2008.
In July 2009, the Company entered into a lease agreement with BMR-3450 Monte Villa Parkway LLC relating to the lease of 19,108 square feet of laboratory and office space in Bothell, Washington. The Company began occupying this space in August 2009, and has moved its headquarters and R&D functions to this new location. The term of the lease is approximately 63 months, although the Company has a one-time option to terminate the lease after 3 years’ time upon payment of a termination fee. The Company will commence paying base rent of approximately $43,000 per month after approximately 3 months. The amount of base rent is subject to an annual increase of 3%.
9. INCOME TAXES:
As of December 31, 2009 the Company has federal and state net operating loss carryforwards of approximately $211,108,000 and $225,611,000, respectively, available to reduce future taxable income, which expire 2009 through 2028. Of these amounts, approximately $2,007,000 and $2,046,000, respectively, relate to federal and state net operating losses assumed as part of the Ercole acquisition. Utilization of the Ercole net operating losses is limited to approximately $425,000 per year. In addition, the Internal Revenue Code rules under Section 382 and related state laws could limit the future use of the remaining net operating losses based on ownership changes and the value of the Company’s stock. Approximately $3,930,000 of the Company’s carryforwards were generated as a result of deductions related to exercises of stock options. When utilized, this portion of the Company’s carryforwards, as tax affected, will be accounted for as a direct increase to contributed capital rather than as a reduction of the year’s provision for income taxes. The principal differences between net operating loss carryforwards for tax purposes and the accumulated deficit result from depreciation, amortization, investment write-downs, treatment of research and development costs, limitations on the length of time that net operating losses may be carried forward, and differences in the recognition of stock-based compensation.
The Company had net deferred tax assets of $110,539,000 and $102,881,000 at December 31, 2009 and 2008, respectively, primarily from net operating loss carryforwards and research and development credit carryforwards. A valuation allowance was recorded to reduce the net deferred tax asset to zero because it is more likely than not that the deferred tax asset will not be realized. The net change in the valuation allowance for deferred tax assets was an increase of approximately $7,658,000 and $8,250,000 for the years ended December 31, 2009 and 2008, respectively, mainly due to the increase in the net operating loss carryforwards, research and development tax credits, and a decrease in the asset related to short term securities due to the expiration of the capital loss carryforward period as of December 31, 2009.
Deferred tax assets assumed as part of the Ercole acquisition total approximately $1,407,000 and primarily relate to accrual to cash adjustment, net operating losses, and research & development credits. A valuation allowance was recorded to reduce the net deferred tax assets to zero because it is more likely than not that the deferred tax asset will not be realized. When such deferred tax assets are utilized or at such time when the valuation allowance is lifted, this portion of the Company’s deferred tax assets, as tax affected, will be accounted for as a direct increase to equity rather than as a reduction of that year’s provision for income taxes.
An analysis of the deferred tax assets (liabilities) is as follows:
|
December 31, |
|
2009 |
|
2008 |
|
||
|
(in thousands) |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
Net operating loss carryforwards |
|
$ |
83,057 |
|
$ |
75,509 |
|
|
Difference in depreciation and amortization |
|
2,544 |
|
2,276 |
|
||
|
Capital loss carryforward |
|
8 |
|
8 |
|
||
|
Research and development tax credits |
|
18,436 |
|
20,404 |
|
||
|
stock compensation |
|
4,197 |
|
3,326 |
|
||
|
Stock options for consulting services |
|
1,012 |
|
957 |
|
||
|
Deferred Rent |
|
378 |
|
244 |
|
||
|
Deferred Revenue |
|
805 |
|
— |
|
||
|
Other |
|
102 |
|
157 |
|
||
|
|
|
110,539 |
|
102,881 |
|
||
|
|
|
|
|
|
|
||
|
Valuation allowance |
|
(110,539 |
) |
(102,881 |
) |
||
|
|
|
|
|
|
|
||
|
|
|
$ |
— |
|
$ |
— |
|
The Company’s policy is to recognize interest and/or penalties related to income tax matters in income tax expense. The Company had no accrual for interest or penalties on its balance sheet at December 31, 2009 and at December 31, 2008, and has not recognized interest and/or penalties in the statement of operations for the years ended December 31, 2009, 2008 or 2007.
10. COMMITMENTS AND CONTINGENCIES:
Lease Obligations
The Company leases office and laboratory facilities under various noncancelable operating leases through December 2020. Rent expense under these leases was $1,467,000, $1,429,000 and $1,388,000 for the years ended December 31, 2009, 2008 and 2007, respectively, and $12,837,000 for the period from July 22, 1980 through December 31, 2009.
At December 31, 2009, the aggregate non-cancelable future minimum payments under these leases are as follows:
(in thousands)
|
Year ending December 31, |
|
|
|
|
|
2010 |
|
$ |
2,073 |
|
|
2011 |
|
2,175 |
|
|
|
2012 |
|
2,230 |
|
|
|
2013 |
|
2,036 |
|
|
|
2014 |
|
2,033 |
|
|
|
Thereafter |
|
9,135 |
|
|
|
Total minimum lease payments |
|
$ |
19,682 |
|
Royalty Obligations
The Company has license agreements for which it is obligated to pay the licensors a minimum annual royalty. Royalty payments under these agreements were $75,000, $75,000 and $125,000 for the years ended December 31, 2009, 2008 and 2007, respectively, and $1,259,000 for the period from July 22, 1980 through December 31, 2009.
At December 31, 2009, the aggregate future minimum royalty payments under these agreements are as follows:
( in thousands)
|
Year ending December 31, |
|
|
|
|
|
2010 |
|
$ |
100 |
|
|
2011 |
|
80 |
|
|
|
2012 |
|
80 |
|
|
|
2013 |
|
80 |
|
|
|
2014 |
|
55 |
|
|
|
Thereafter |
|
715 |
|
|
|
Total minimum royalty payments |
|
$ |
1,110 |
|
Litigation
In the ordinary course of business, the Company defends its patents as deemed necessary. There are no material asserted claims as of 12/31/09.
11. FINANCIAL INFORMATION BY QUARTER (UNAUDITED):
|
2009 for quarter ended (in thousands) |
|
December 31 |
|
September 30 |
|
June 30 |
|
March 31 |
|
||||
|
Revenues from grants and research contracts |
|
$ |
5,141 |
|
$ |
6,349 |
|
$ |
2,945 |
|
$ |
3,150 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
||||
|
Research and development |
|
6,624 |
|
7,473 |
|
5,804 |
|
4,495 |
|
||||
|
General and administrative |
|
2,470 |
|
1,800 |
|
2,206 |
|
2,220 |
|
||||
|
Operating loss |
|
(3,953 |
) |
(2,924 |
) |
(5,065 |
) |
(3,565 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Other income (loss): |
|
|
|
|
|
|
|
|
|
||||
|
Interest income, net |
|
(312 |
) |
(127 |
) |
(31 |
) |
16 |
|
||||
|
Increase (decrease) on warrant valuation |
|
7,791 |
|
(5,039 |
) |
(14,572 |
) |
2,622 |
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net income (loss) |
|
$ |
3,526 |
|
$ |
(8,090 |
) |
$ |
(19,668 |
) |
$ |
(927 |
) |
|
Net income (loss) per share — basic |
|
$ |
0.03 |
|
$ |
(0.08 |
) |
$ |
(0.23 |
) |
$ |
(0.01 |
) |
|
Net income (loss) per share — diluted |
|
$ |
0.03 |
|
$ |
(0.08 |
) |
$ |
(0.23 |
) |
$ |
(0.01 |
) |
|
Shares used in per share calculations — basic |
|
110,266 |
|
95,261 |
|
85,664 |
|
80,759 |
|
||||
|
Shares used in per share calculations — diluted |
|
125,647 |
|
95,261 |
|
85,664 |
|
80,759 |
|
||||
|
2008 for quarter ended (in thousands) |
|
December 31 |
|
September 30 |
|
June 30 |
|
March 31 |
|
||||
|
Revenues from grants and research contracts |
|
$ |
5,479 |
|
$ |
5,171 |
|
$ |
4,983 |
|
$ |
5,625 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
||||
|
Research and development |
|
5,070 |
|
7,680 |
|
7,678 |
|
6,903 |
|
||||
|
General and administrative |
|
3,303 |
|
3,429 |
|
2,184 |
|
2,553 |
|
||||
|
Acquired in process research and development |
|
— |
|
— |
|
— |
|
9,916 |
|
||||
|
Operating loss |
|
(2,894 |
) |
(5,938 |
) |
(4,879 |
) |
(13,747 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Other income (loss): |
|
|
|
|
|
|
|
|
|
||||
|
Interest income, net |
|
36 |
|
60 |
|
81 |
|
167 |
|
||||
|
Increase (decrease) on warrant valuation |
|
1,718 |
|
(169 |
) |
3,047 |
|
(1,435 |
) |
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net income (loss) |
|
$ |
(1,140 |
) |
$ |
(6,047 |
) |
$ |
(1,751 |
) |
$ |
(15,015 |
) |
|
Net income (loss) per share — basic |
|
$ |
(0.01 |
) |
$ |
(0.08 |
) |
$ |
(0.02 |
) |
$ |
(0.23 |
) |
|
Net income (loss) per share — diluted |
|
$ |
(0.01 |
) |
$ |
(0.08 |
) |
$ |
(0.02 |
) |
$ |
(0.23 |
) |
|
Shares used in per share calculations — basic |
|
71,074 |
|
71,151 |
|
70,986 |
|
65,189 |
|
||||
|
Shares used in per share calculations — diluted |
|
71,074 |
|
71,151 |
|
70,986 |
|
65,189 |
|
||||
12. SUBSEQUENT EVENTS:
During the first quarter of 2010, the independent directors of the Company’s Board of Directors completed a voluntary review regarding prior disclosures made by the Company concerning the status of the clinical development of a drug-eluting stent using AVI-5126, licensed to Global Therapeutics, a Cook Medical Company (the “Cook Trial”). The independent directors initiated the review following questions from a shareholder with respect to the Cook Trial. The independent directors asked the Company’s regular outside general counsel to identify one of his partners with expertise in commercial, business and securities issues, who had not previously preformed any work on behalf of the Company, to interview employees, review documents and emails and prepare a report to the independent directors. The Company’s outside general counsel was supportive in these activities. Following the delivery of counsels’ report to the independent directors, the independent directors concluded that no further disclosures were required with respect to the Cook Trial. Following that conclusion of but prior to communicating it to the shareholder, on March 15, 2009 the Company received a letter from the shareholder’s counsel making similar demands. Based on the review undertaken prior to receipt of this letter, the Company believes that this matter will not have a material impact on the Company's statements or operations.