Attached files
| file | filename |
|---|---|
| 8-K/A - FORM 8-K/A - Molecular Templates, Inc. | d266142d8ka.htm |
| EX-99.2 - EX-99.2 - Molecular Templates, Inc. | d266142dex992.htm |
| EX-10.3 - EX-10.3 - Molecular Templates, Inc. | d266142dex103.htm |
| EX-99.3 - EX-99.3 - Molecular Templates, Inc. | d266142dex993.htm |
| EX-99.5 - EX-99.5 - Molecular Templates, Inc. | d266142dex995.htm |
| EX-99.4 - EX-99.4 - Molecular Templates, Inc. | d266142dex994.htm |
| EX-23.1 - EX-23.1 - Molecular Templates, Inc. | d266142dex231.htm |
Exhibit 99.1
The following is an excerpt of portions of the prospectus contained in the Form S-4 registration statement (File No. 333-217993) as declared effective by the Securities and Exchange Commission on June 30, 2017. Such information is as of June 30, 2017 (unless an earlier date is indicated).
MOLECULAR BUSINESS
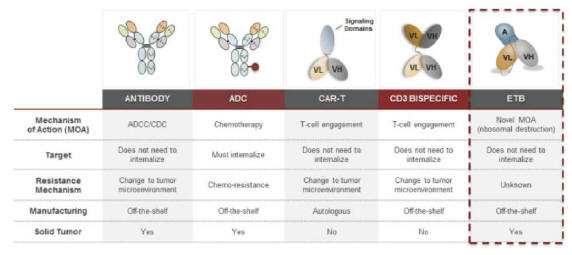
Molecular is a clinical-stage oncology company focused on the discovery and development of differentiated, targeted, biologic therapeutics for cancer. Molecular believes its proprietary biologic drug platforms, which it refers to as engineered toxin bodies, or ETBs, provide a differentiated mechanism of action that solves problems associated with currently available cancer therapeutics. ETBs use a genetically engineered version of the Shiga-like Toxin A subunit, or SLTA, a ribosome inactivating bacterial protein. In its wild-type form, SLT is thought to induce its own entry into a cell when proximal to the cell surface membrane, self-route to the cytosol, and enzymatically and irreversibly shut down protein synthesis via ribosome inactivation. SLTA is normally coupled to its cognate Shiga-like Toxin B subunit, or SLTB, to target the CD77 cell surface marker, a non-internalizing glycosphingolipid. In Molecular’s scaffold, a genetically engineered SLTA subunit with no cognate SLTB component is genetically fused to antibody domains or fragments specific to a cancer target, resulting in a biologic therapeutic that can identify the particular target cell and specifically kill the target cell. The antibody domains may be substituted with other antibody domains having different specificities to allow for the rapid development of new drugs to selected targets in cancer.
ETBs combine the specificity of an antibody with SLTA’s potent mechanism of cell destruction. In Molecular’s second and third-generation ETBs, Molecular has modified the SLTA further to reduce immunogenicity and deliver additional payloads into a target cell, respectively. Immunogenicity is the ability of a particular substance, such as an antigen or epitope, to provoke an immune response. ETBs have relatively predictable pharmacokinetic, or PK, and absorption, distribution, metabolism and excretion, or ADME, profiles and can be rapidly screened for desired activity in robust cell-based and animal-model assays. Because SLTA can induce internalization against non- and poorly-internalizing receptors, the universe of targets for ETBs may be substantially larger than that seen with antibody-drug conjugates, or ADCs, which may not be effective if the target is not able to internalize them.
ETBs have a differentiated mechanism of cell-kill in cancer therapeutics (the inhibition of protein synthesis via ribosome destruction), and Molecular has preclinical and clinical data demonstrating the utility of these molecules in chemotherapy-refractory cancers. ETBs have shown good safety data in multiple animal models as well as in Molecular’s clinical study. Molecular believes the target specificity of ETBs, their ability to self-internalize, their potent and differentiated mechanism of cell kill and their safety profile provide opportunities for the clinical development of these agents to address multiple cancer types.
Molecular’s approach to drug development in oncology involves the selection of lead compounds to validated targets in cancer and continuous improvements to Molecular’s ETB platform. Molecular has developed ETBs for various targets, including CD20, CD38, HER2, PD-L1, and CD45. CD20 is central to B cell malignancies and is clinically validated as a target for the treatment of lymphomas and autoimmune disease. CD38 has been validated as a meaningful clinical target in the treatment of multiple myeloma. PD-L1 is central to the immune checkpoint pathways and is a target expressed in a variety of solid tumor cancers. CD45 is expressed on most lymphocytes and has been studied as a potential key target for lymphodeletion strategies related to stem cell transplant therapy. Molecular’s lead compound, MT-3724, is a first generation ETB that recognizes CD20, a B cell marker. The dose escalation portion of its first Phase I clinical trial has been completed for MT-3724. Molecular anticipates advancing one or more additional ETBs into clinical trials in 2018.
Molecular has built up multiple core competencies around the creation and development of ETBs. Molecular developed the ETB technology in-house and continues to make iterative improvements in the scaffold and identify new uses of the technology. Molecular also developed the process for manufacturing ETBs under GMP standards and continues to make improvements to its manufacturing processes. Molecular has conducted multiple GMP manufacturing runs with its lead compound and believes this process is robust and could support commercial production with economics similar to those seen with antibodies.
Challenges in Oncology
Existing mechanisms of action, the specific biochemical interaction through which a drug substance produces its pharmacological effect, are subject to numerous limitations in oncology. The clinical benefit of a given drug is a function of the biological properties of the drug, the target with which the drug interacts and the tumor indication being treated, but the relative contribution of each of these factors is difficult to separate. To date, identifying the most appropriate cancer targets, applying the most effective mechanisms of action and selecting the appropriate disease indications and most responsive patient populations for a particular drug have presented significant challenges, including the following:
The limited number of addressable cancer targets given current available mechanisms of action; for example, targets appropriate for antibody-drug conjugate, or ADC, approaches are relegated to those extracellular targets that already readily and efficiently self-internalize;
| • | The level of cell surface expression of these addressable cancer targets and the shedding of the target by the tumor as a means of resistance to therapy may impact a drug’s effectiveness; |
| • | ADC approaches generally use small molecule payloads which damage DNA, or disrupt or prevent microtubule assembly, and can be subject to the same mechanisms of resistance as in general chemotherapy; |
| • | Established single-agent therapies are only effective in a minority of cancer patients; |
| • | Current approaches to target prioritization are not comprehensively systematic and do not leverage a complete understanding of a drug’s effect on a given tumor type to best identify high value targets in certain patient populations; |
| • | In vitro epitope selection on a given target may not be predictive of clinical optimization; and |
| • | Predictive biomarkers, the value and use of which are relatively new, are not uniformly used to proactively select responsive patient populations and/or preferred indications, which can drive longer development timelines with higher associated costs. |
Molecular’s Differentiated Approach
Molecular was founded on the principle that differentiated mechanisms of action are crucial for improving outcomes in oncology. Molecular has created a new scaffold with a differentiated mechanism of action, coupled with a predictable PK and ADME profile. Molecular’s ETB scaffold permits rapid screening ability for lead identification and easily scalable production, which Molecular believes offers an opportunity to provide meaningful clinical benefits in oncology with more efficient capital expenditures than current treatments. Molecular believes the differentiated biological activity innate to the ETB scaffold, particularly the ability to induce internalization and employ a differentiated mechanism of cell kill, may allow for differentiated clinical benefit in patients with relapsed or refractory disease as well as potential combination with standard of care therapies in earlier stage disease.
Molecular likens the extensive de-immunization work it has conducted on SLTA to the chimerization of monoclonal antibodies. Monoclonal antibody chimerization is a process for reducing immunogenicity when an antibody from one species is introduced into a different species. Chimerization allows for the wide-spread use of antibodies as human therapeutics across multiple disease settings. Molecular believes that the de-immunization of SLTA may allow for ETB use across multiple indications in oncology, including solid tumors.
Molecular has seen in both preclinical models and in its Phase I study that the differentiated mechanism of action employed by its ETBs can be effective in chemo-resistant tumor cells. Molecular believes this creates the potential for a rapid characterization of efficacy in carefully designed clinical trials in relapsed and refractory settings, particularly when targeting tumor markers that persist after treatment with multiple lines of therapy and whose targeting has been shown to provide a survival benefit. Molecular also has seen preclinically that its ETBs
2
can have additive or synergistic activity in combination with a number of small molecule agents including chemotherapeutics, immunomodulatory agents and tyrosine kinase inhibitors. Molecular believes that the ability of ETBs to be additive or synergistic to a variety of current treatments may allow for combination therapy in earlier lines of disease.
Molecular believes its efforts have allowed and will continue to allow Molecular to develop ETBs against well-validated targets and new targets, enabling a phenotypically based clinical trial design that may result in shorter development timelines with lower associated costs. More specifically:
| • | Molecular’s research and design platform allows it to rapidly select lead ETBs from a comprehensive screen. Molecular’s ETB platform utilizes a suite of integrated technologies to screen ETB libraries for lead identification. Molecular performs initial preclinical screens on ETBs with lead selection around potency, affinity and expression. These screens typically take six to eight weeks. Critical components of Molecular’s approach include: |
| • | The proprietary optimization of the genetic fusion between the immunoglobulin-targeting domain and Molecular’s proprietary SLTA scaffold; |
| • | The proprietary de-immunizing modifications made to the SLTA scaffold, which reduce both adaptive and innate immune responses to ETBs; |
| • | Rapid screening for potency, affinity and specificity against target expressing versus non-expressing cells; and |
| • | Early evaluation of protein expression and stability of potential lead ETB candidates. |
| • | Molecular’s ability to create lead ETBs to well-validated targets reduces the risk of target-mediated side effects and increases the likelihood of obtaining meaningful clinical benefit. Molecular has deployed its technology against targets in oncology that are central to disease progression and that are known to persist after a given modality has failed. Molecular believes these targets reduce the risk of clinical failure from either unacceptable target-mediated adverse events or from a failure to impact disease outcome because of loss of the target. For example, Molecular’s lead compound, MT-3724, targets the B-cell surface marker CD20. CD20 appears central to B-cell malignancies, and the FDA has approved multiple antibody therapies targeting CD20. Destruction of CD20-expressing cells has not been found to cause significant damage to the patient, known as severe toxicity. CD20 cell surface expression persists in the majority of patients who have progressed after treatment with a CD20 monoclonal antibody. Because of its centrality to disease progression, lack of associated toxicities and persistence after treatment failure, Molecular chose targeting of CD20 for Molecular’s lead ETB program. Molecular used a similar rationale in the selection of Molecular’s current pipeline, including ETBs targeting CD38 and HER2, which are targets central to disease outcome that persist after a given modality has failed. |
| • | Molecular’s ETB platform allows Molecular to rapidly identify ETBs to targets and select patients in the Phase I clinical trials that phenotypically match that ETB program. Molecular can screen a library of single chain variable fragments, or scFvs, expressed in Molecular’s ETB scaffold to a given target in six to eight weeks. The pharmacokinetic and ADME profile of these compounds are similar and relatively predictive in humans based on animal models. Once the lead is selected and IND-enabling studies are completed, Molecular can enrich a Phase I trial with only patients expressing the target of the ETB. In these Phase I trials, Molecular can get a faster read on safety as well as efficacy than is possible in many drug development programs. Molecular’s current Phase I trial with MT-3724 established the PK, ADME, dose-limiting toxicity, or DLT, recommended Phase II dose and monotherapy efficacy after just 21 patients were treated. |
3
Molecular’s goal is to bring the right ETBs to the right patients to provide long-lasting benefits that ultimately improve patients’ lives. To achieve its goal, Molecular is:
| • | Implementing development strategies that capitalize on the differentiated pharmacological features of Molecular’s ETB technology and the validated nature of the targets it has chosen. Molecular believes the target specificity of its ETBs, their ability to self-internalize, their potent and differentiated mechanism of cell kill and their safety profiles will provide opportunities for the clinical development of these agents to address multiple cancer types. For example, Molecular is aggressively developing its lead product MT-3724 as a single agent therapy for relapsed and refractory diffuse large B-cell lymphoma, or DLBCL, patients and in combination with approved therapies in earlier stages of high-risk DLBCL. The targeting of CD20 with antibody therapeutics is known to confer clinical benefit in these settings. MT-3724’s differentiated mechanism of action, safety and pharmacological profiles targeting CD20 may provide an advantage over other modalities. Given the unique mechanism of direct cell-kill, via ribosome inactivation, Molecular believes there is the potential for combination or sequential drug strategies that may be unique to its ETB drug candidates. Further, although the safety data for MT-3724 is still preliminary, Molecular believes the different PK and ADME profiles of its ETBs may allow them to be more appropriate therapies for certain patient populations, particularly those who are unable to tolerate intensive chemotherapy as primary or conditioning therapy. Molecular believes all of these attributes will enable Molecular to pursue development strategies not feasible with other therapeutic approaches. |
| • | Efficiently building a broad pipeline of ETB therapeutics targeting defined patient populations through the use of Molecular’s research and design platform. Molecular believes its research and design platform is an efficient and productive discovery and development engine that can identify new targets across multiple cell types with the aim of creating a portfolio of novel, targeted ETBs. By selecting tumor targets best suited to ETB biology, Molecular can prioritize indications, including potential niche indications and/or niche subsets of indications. Molecular believes this will enable Molecular to build a clinical population of patients who may be more likely to respond to its therapies, allowing Molecular to potentially shorten development timelines and lower associated costs. |
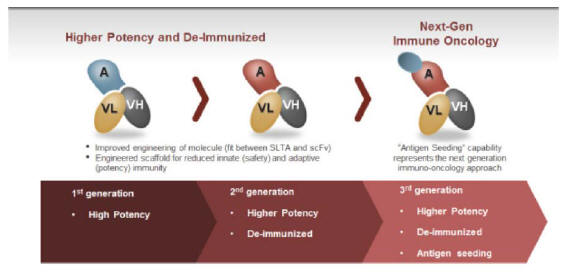
| • | Maximizing the value of Molecular’s early pipeline through the continual improvement of Molecular’s technology. Since the founding of the company, Molecular has made substantial progress in improving its ETB technology. Molecular’s lead compound, MT-3724, is a first-generation ETB utilizing a minimally altered SLTA and a first-generation fusion between the SLTA and the antibody domains. Molecular’s second-generation ETBs utilize a proprietary SLTA that has been heavily modified to dramatically reduce innate and adaptive immunogenicity. In addition, the second-generation compounds utilize a new approach for the genetic fusion of the SLTA and antibody domain that enhances the potency of Molecular’s ETBs. Molecular has now created a third generation of ETBs that retain the properties of the second generation but add the ability to deliver foreign class I antigens into target cells for expression in complex with MHC class I molecules on the target cell’s surface. Molecular has shown preclinically that certain foreign antigens can be functionally recognized by human T-cells and believes this represents a differentiated approach to immuno-oncology. |
| • | Building a fully integrated discovery-to-commercial oncology company focused on compounds with unique and differentiated biology. Molecular believes that differentiated mechanisms of action are crucial for improving outcomes in oncology. Molecular has created a robust translational platform that Molecular believes allows it to create a sustainable, novel pipeline of ETBs with differentiated mechanisms of tumor destruction, relatively predictable PK and ADME, and scalable and economical manufacturing. If MT-3724, MT-4019, MT-5111 or any future product candidates Molecular may develop are approved, Molecular will consider commercializing them itself in select markets. |
Molecular’s Engineered Toxin Body (ETB) Platform Technology
Although chemotherapy remains the cornerstone of treatment for most cancers, the advent of new and targeted classes of therapies has dramatically changed outcomes in the treatment of disease. The advent of monoclonal antibodies, signal transduction inhibitors and, most recently, immune-oncologics have provided substantial clinical benefit in both the relapsed and refractory setting and in combination in earlier lines of therapy. Molecular believes that ETBs can represent a new class of targeted agents with differentiated biology that are well-positioned to potentially improve outcomes in cancer patients.
4
ETBs appear to induce the internalization of non- or poorly-internalizing targets, have a differentiated mechanism of action (enzymatic and irreversible ribosome inactivation), have relatively predictable PK and ADME profiles and can be readily manufactured to GMP standards. Molecular’s research and design platform allows for the rapid (six to eight weeks) in vitro selection of a lead ETB to a given target based on affinity and specificity, potency and expression. Lead selection is confirmed through the use of animal models to verify PK, ADME and potency. Molecular’s first generation ETB is represented by MT-3724. Molecular’s first generation ETBs possess potent direct cell killing effects via a differentiated mechanism of action, can force receptor internalization, but do not use a de-immunized scaffold. Because MT-3724 is being developed for treating B-cell malignancies, where patients are typically immuno-compromised, Molecular did not believe de-immunization was critical in most patients; this hypothesis has been supported by clinical data in DLBCL patients.
Molecular’s second-generation ETBs have higher potency than its first-generation ETBs and possess a de-immunized scaffold that elicits significantly reduced innate and adaptive immunogenic responses as demonstrated in Molecular’s preclinical and animal studies (presented at the 2017 American Association for Cancer Research, or AACR, Annual Meeting). With Molecular’s third-generation ETBs, Molecular has now begun to explore a unique approach to immuno-oncology, which is referred to herein as “Antigen Seeding.” Molecular is currently building out animal models to further validate and screen ETB candidates support this approach.
Molecular believes that its proprietary ETB technology platform represents a differentiated approach in oncology. ETBs possess the targeting specificity of antibody-based therapeutic approaches but deliver highly potent payloads that disrupt protein synthesis, a fundamental function of a cancer cell, in a manner not subject to traditional chemotherapy resistance mechanisms or target internalization limitations, as with ADCs. Molecular also is seeking to exploit the ETB’s ability to force internalization against receptors that do not normally internalize to expand the universe of potential targets subject to pharmaceutical treatments. MT-3724 highlights this capability and approach. MT-3724 targets CD20, which is a canonical non-internalizing receptor not susceptible to traditional chemo-based ADC approaches. Also as described earlier, ETBs are easily manufactured to GMP standards and do not require intensive logistical and manufacturing infrastructure to support their manufacture and distribution.
Novel mechanisms of action are needed in oncology treatment, and Molecular believes that differentiated mechanisms of action that are innate to the ETB platform technology puts its ETBs into a distinct class of biologic therapies that may offer unique benefits over existing treatment modalities.
5
ETB Product Pipeline
Molecular is developing a pipeline of ETBs that Molecular believes will provide a meaningful and long-lasting benefit to cancer patients. Molecular plans to develop each of these as single agents and/or in combination with other therapies, as applicable. The following table depicts Molecular’s current pipeline:

MT-3724—ETB Targeting CD20
Overview
CD20 is expressed on 90% of B-cell non-Hodgkin’s lymphoma, or NHL, cells and is a non-internalizing receptor. Rituxan (rituximab), an antibody to CD20, is approved for treatment of NHL in both the front and second-line settings. However, Rituxan has limited direct cell-kill effects against CD20-expressing cells. Instead, it works through indirect methods of recruiting immune responses to CD20-expressing cells through antibody dependent cell-mediated cytotoxicity, or ADCC, and/or complement dependent cytotoxicity, or CDC. Rituxan’s indirect cell-kill mechanism’s reliance on a favorable tumor microenvironment for immune stimulation is problematic because it allows multiple points where resistance can emerge. Therefore, direct cell-kill approaches that target CD20-expressing lymphomas are attractive. Two such agents are currently approved, the radioisotope-conjugated antibodies Bexxar, developed by GlaxoSmithKline, and Zevalin, developed by IDEC Pharmaceuticals (now part of Biogen), both of which use ionizing radiation to induce direct cell-kill without internalization being necessary.
6
These radioisotope conjugated antibodies are more effective than naked anti-CD20 antibody approaches such as Rituxan and HuMax-CD20 in the relapsed or refractory indolent NHL setting because they are far less dependent on the physiology of the tumor. However, despite their favorable efficacy profile, Bexxar and Zevalin are considered commercial disappointments and have not been widely adopted by oncologists primarily due to the constraints associated with the administration of nuclear medicines. Radioimmunotherapies are difficult to administer, with few institutions licensed for nuclear medicine. Because of these factors, the combined use of Bexxar and Zevalin accounted for only 4% of all administered second-line therapies for indolent NHL patients worldwide (seven major markets) despite superior clinical data in this setting. Molecular believes this provides a significant opportunity for a CD20-targeting therapy, such as MT-3724, that directly kills cells without the use of radioisotopes, preferably through a mechanism of action of cell kill that is also not subject to cross-resistance with chemotherapy or antibody approaches.
MT-3724 is a first-generation ETB specific to the B-cell marker CD20 protein. Molecular developed MT-3724 to provide a non-radioactive means of direct cell-kill targeted to CD20 for the treatment of NHL. The differentiated mechanism of action of MT-3724 involves binding to the surface protein CD20, forcing internalization into the target cell, retrograde transport to the cytosol and subsequent enzymatic and permanent ribosome-inactivation. Molecular is currently conducting a Phase I study of MT-3724 in patients with relapsed/refractory NHL.
Preclinical Overview
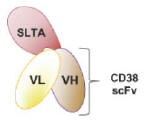
MT-3724 is a fusion protein which is comprised of the variable regions of the heavy (VH) and light chains (VL) of an anti-CD20 antibody connected with a short linker peptide (Figure 1) that make up a single-chain variable fragment, or scFv. This binding domain is genetically fused to a proprietarily engineered form of SLTA. Because MT-3724 lacks the fragment crystallizable, or Fc, portion of an intact antibody, MT-3724 does not rely on host antibody-dependent cellular toxicity, or ADCC, complement-dependent cytotoxicity, or CDC, or complement-mediated lysis to induce cell death. Naked antibody therapies rely on the induction of ADCC/CDC as the primary mechanisms of indirect cell-kill. Thus, Molecular believes MT-3724 may avoid the mechanisms of lymphoma cell resistance identified with the currently available anti-CD20 antibodies.
The three key biological properties of MT-3724 that reflect the differentiated biology of ETBs include:
| • | forced internalization against CD20, a receptor that does not normally internalize; |
| • | self-routing through the cell to the cytosol; and |
| • | irreversible and enzymatic inactivation of target cell ribosomes. |
Figure 1. MT-3724 drug product

Molecular conducted a study to evaluate the binding affinity and selectivity of MT-3724 to CD20+ cells, which demonstrated that MT-3724 bound to CD20+ expressing cell lines with specificity. MT-3724 gains entry into target cells through CD20-dependent binding. The binding of MT-3724 to CD20 is a critical step in cellular cytotoxicity induced by MT-3724.
7
In Vivo Results
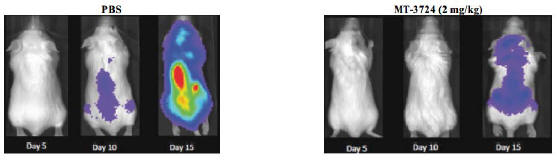
MT-3724 has demonstrated potent and specific activity against a wide panel of CD20 expressing cancer cell lines, including Rituxan refractory patient samples. In addition to in vitro activity, Molecular has evaluated MT-3724 in a series of preclinical efficacy models that show its potent activity in destroying CD20 expressing human tumors. MT-3724 was generally well tolerated in these animal models. In one model, tumor responses were measured on Days 5, 10, 15 and 20 by bioluminescent imaging of Raji-luc tumors. Treatment with MT-3724 was tolerated and resulted in a statistically significant survival advantage in this model as shown in Figure 2:
Figure 2. Disseminated Raji-Luc Imaging
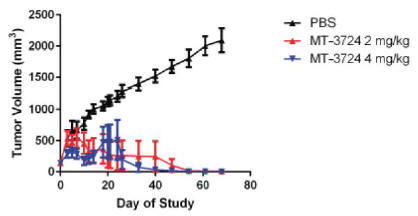
Molecular performed a study to determine the therapeutic potential of MT-3724 to inhibit the growth of CD20-expressing human lymphoma cells in a subcutaneous implant model in athymic nude mice. Molecular observed a significant anti-tumor response in MT-3724 treated mice. Specifically, Molecular concluded that administration of MT-3724 at both 2 mg/kg/dose and 4 mg/kg/dose demonstrated cytotoxic activity against human lymphoma cells in this xenograft tumor model, as shown in Figure 3. Treatment with MT-3724 was generally well tolerated in the animals.
Figure 3. Subcutaneous Raji Xenograft Tumor Volumes
Clinical Overview
MT-3724 is being developed for the treatment of patients with relapsed or refractory NHL who have relapsed following response to one or more anti-CD20 antibody therapies with or without other front-line therapies and for whom higher priority approved therapies (biologic, chemotherapeutic or stem cell transplantation) are not an
8
option. The primary objectives of the multicenter Phase I clinical trial of MT-3724 were to establish the maximum tolerated dose, or MTD, and an appropriate dose for Phase II clinical trials. The secondary objectives of the Phase I clinical trial were to assess the safety, tolerability and pharmacokinetic profile of MT-3724 after intravenous dosing as well as to assess any biological and clinical activity. This Phase I clinical trial was not designed to show statistical significance of the study endpoints.
Molecular initially filed an IND application with the FDA on July 31, 2014, and Molecular received the notification from the FDA that it could proceed with the Phase I trial on August 29, 2014. The Phase I trial was a multi-center, open-label, multiple-dose Phase I/Ib, dose-escalation study of MT-3724 in subjects with relapsed, refractory B-cell NHL or chronic lymphocytic leukemia, or CLL. A total of 21 patients were treated with MT-3724 with doses ranging from 5 to 100 mcg/kg dose. Patients were dosed on a 3 times per week schedule over two weeks (6 doses) with a two-week hiatus for the first cycle as mandated by the FDA. Subsequent cycles were dosed over two weeks with a one-week hiatus. Originally, up to five cycles of treatment were allowed per protocol. This was subsequently amended to allow for extended dosing beyond five cycles.
Twenty-one patients were treated with escalating doses of MT-3724 starting at the 5 mcg/kg dose level. Nearly all patients experienced at least one adverse event, with peripheral edema, diarrhea, myalgia, cough, fatigue, constipation, nausea, anemia, stomatitis, pyrexia, dizziness, headache, insomnia, dyspnea, being the more commonly reported adverse events. During the study, there were no treatment-related deaths.
The first two patients treated in the 100 mcg/kg/dose cohort developed signs and symptoms of a systemic inflammatory response (a constellation of adverse events including a grade 2 decrease in serum albumin levels, which together were consistent with capillary leak syndrome) in the first cycle of treatment. Upon thorough evaluation of each case, the Data Monitoring Committee, or DMC, deemed the capillary leak syndrome the DLT and determined that the 100 mcg/kg/dose had exceeded the MTD and the cohort was closed to further enrollment. The symptoms related to the DLT were non-life threatening and resolved upon cessation of dosing MT-3724. Six patients were dosed at a reduced dose level of 75 mcg/kg cohort with no DLTs reported. The recommended Phase II dose will be 75 mcg/kg.
To date, 31 SAEs have been reported. Most these events were attributed to exacerbation of a pre-existing condition or disease progression. Both subjects in the 100 mcg/kg/dose cohort were withdrawn in cycle 1 for SAEs which the investigator and DMC assessed as DLTs and determined that the MTD had been exceeded.
Molecular has observed promising signals of efficacy for MT-3724 in treating non-Hodgkin’s lymphoma. Patients in the Phase I trial were of older age (median = 67) and heavily pre-treated with a median number of prior therapies of four. Those patients with £ four prior therapies (n=5) were generally chemo-intolerant patients who could not sustain multiple lines of chemo-based regiments. The majority of patients were of the DLBCL subtype (n=16). Of the 14 DLBCL patients who received at least one cycle of MT-3724, eight patients entered the trial with low levels of serum anti-CD20 antibody while six patients had high levels of anti-CD20 antibody. As reported in Molecular’s presentation to the 2016 American Society of Hematology Annual Meeting, or the 2016 ASH Meeting, patients with high anti-CD20 antibody did not respond to MT-3724, presumably due to target inaccessibility. In the eight DLBCL patients with low CD20 antibody, the observed objective response rate, or ORR was 25% (2/8) including a partial response, or PR, and a complete metabolic response, or CMR. Molecular observed clinical responses starting at the lowest dose level of 5 mcg/kg as shown in Figure 4. The patient who achieved a CMR was eligible for and received an allogeneic stem cell transplant, or SCT. Three patients had stable disease, or SD, with tumor reductions of 19% (10 mcg/kg), 48% (75 mcg/kg), and 49% (100 mcg/kg), respectively. The patient at 100 mcg/kg with 49% tumor reduction had received only a single dose of MT-3724 at the time of measurement. The remaining three patients had progressive disease, or PD. Notably, three of the eight DLBCL patients received fewer than two cycles of MT-3724 due to early withdrawal from the study (including the two patients at the DLT dose of 100 mcg/kg). Lower levels of anti-drug antibodies, or ADAs, were observed among DLBCL patients and did not appear to neutralize the efficacy of MT-3724 in patients.
9
Figure 4. PET images for DLBCL patient in the 5 mcg/kg dose cohort
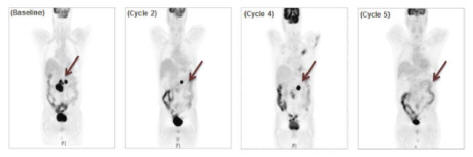
Based on the clinical effect observed among DLBCL patients, Molecular has opened up an expansion study as part of the Phase I trial to further explore the potential of MT-3724 in DLBCL. Molecular expects to enroll up to nine additional DLBCL patients at the 75 mcg/kg dose level. Molecular expects to start reporting on this expansion study in early 2018. Furthermore, Molecular is planning to develop MT-3724 in combination with chemotherapy-based standards of care for high-risk, treatment-naïve DLBCL patients. A patient is considered to be treatment naive if they have never undergone treatment for DLBCL. Although treatment with the CD20 antibody Rituxan in combination with chemotherapy remains the standard of care treatment with curative intent for all treatment-naïve DLBCL patients, the presence of certain prognostic markers is associated with relapse and rapid disease progression. Molecular plans to combine MT-3724 with the standard of care for these high-risk patients to potentially improve overall survival and cure rates in this disease. Molecular plans on initiating its Phase II trials in treatment-naïve DLBCL patients in the second half of 2017.
Recent Presentations
MT-3724 AACR presentation: In April 2017, Molecular presented preclinical data for Molecular’s MT-3724 lead compound at the AACR annual conference. MT-3724 is a first-generation compound and, as such, is not de-immunized. Nevertheless, to date, Molecular has not seen a high level of neutralizing antibodies in patients treated with MT-3724, likely because of the nature of their disease (B-cell malignancy) and their prior therapies (B-cell depleting agents). The MT-3724 presentation at AACR demonstrated the reduction in anti-drug antibodies, or ADAs, seen when MT-3724 was co-administered with sirolimus in both murine and non-human primate, or NHP, models. These data may be useful in guiding clinical development of MT-3724 if Molecular does begin to see significant levels of ADAs to patients treated with MT-3724. Additionally, researchers at MD Anderson Cancer Center presented preclinical data on MT-3724 potency against mantle cell lymphoma samples. Researchers demonstrated a substantial survival advantage in a xenograft model using a patient-derived mantle cell lymphoma.
MT-4019—ETB Targeting CD38
Overview
CD38 is a single-chain type II transmembrane glycoprotein that is expressed by a variety of hematologic cells in an activation- and differentiation-dependent manner. Its cellular functions are involved in the regulation of cell proliferation and survival. CD38 is expressed at high rates on patient myeloma samples, making it an important marker and potential target in the development of targeted biologics.
Daratumumab (Genmab/Johnson and Johnson) received FDA approval in 2015. Daratumumab is a monoclonal antibody that binds CD38 on multiple myeloma cells and induces cell death indirectly. A careful analysis of patients treated with daratumumab in the Phase II pivotal trial for approval in fourth-line myeloma patients reveals that CD38 expression persists after patients have progressed on daratumumab and that the myeloma cells of patients who relapsed after daratumumab treatment showed an increase in cell surface receptors (CD55 and CD59) that inhibit daratumumab’s ability to recruit an immune response to the myeloma cells (Nijhof et al., 2016). Persistence of a surface marker central to disease strongly suggests that a different modality targeting that surface marker and that is not cross-resistant to antibody therapy may provide substantial clinical benefit in myeloma.
10
Despite cell specific expression, an antibody-drug conjugate, or ADC, approach to CD38 has not been developed, likely because CD38 does not efficiently internalize, thereby limiting the amount of drug that could be delivered to myeloma cell. Because SLTA can force its own internalization and enzymatically inhibit ribosome function, Molecular theorized that the lack of internalization seen with CD38 might not prevent the engineering of a potent and specific ETB targeted to CD38.
MT-4019 is Molecular’s most advanced second-generation ETB and specifically targets CD38. The compound was evaluated in many of the same preclinical assays as daratumumab. Daratumumab (trade name Darzalex®) is an anti-cancer drug originally developed by Genmab. Based on published daratumumab xenograft data, MT-4019 appears to have more potent direct cell-kill activity and more rapid and pronounced activity when tested in the identical xenograft model. However, the mechanism of action of MT-4019 is wholly different than daratumumab, and Molecular believes that MT-4019 may be active in CD38+ myeloma patients that have failed treatment with an anti-CD38 antibody.
The proposed development plan for MT-4019 is modeled on that of daratumumab. After a robust response rate in its Phase I trial, daratumumab was granted Breakthrough Therapy Designation, and its expanded Phase II trial (N=106) was considered sufficient for registration. If similar efficacy is seen with MT-4019, Molecular believes its CPRIT grant funding dedicated to this program may be sufficient to advance MT-4019 through a pivotal trial.
Preclinical Data with MT-4019
MT-4019 Structure
MT-4019 utilizes Molecular’s second-generation scaffold in which the fusion of the scFv to the SLTA has been optimized and in which the SLTA portion of the ETB has been de-immunized. MT-4019 has high affinity for the CD38 receptor and potent and specific cell-kill activity against CD38-expressing cells.
Figure 5. MT-4019 Drug Product
De- immunized SLTA scaffold
The host immune response to bacterial proteins used in the treatment of solid tumors has historically prevented prolonged dosing and limited the utility of immunotoxins as a class of molecules. There has been much greater success with immunotoxins in hematological malignancies, as patients tend to be immunosuppressed due both to the nature of their disease and the drugs used in treatment (Kreitman et al., 2006). Multiple myeloma patients show a decreased immune response to bacterial proteins (Jacobson, et al., 1986), and Molecular has further reduced the likelihood of neutralizing antibodies by using its proprietary de-immunized SLTA, as shown in Molecular’s MT-4019 presentation at the 2017 AACR Annual Meeting.
MT-4019 Binding Specificity
MT-4019 showed high-affinity binding to recombinant CD38 protein and to the CD38+ myeloma H929 cell line. MT-4019 shows no binding to a non-specific protein.
11
MT-4019 In Vitro Activity
MT-4019 shows extremely potent and specific cell-kill activity against cells that express CD38. MT-4019 was tested for cell-kill activity on H929 and HDLM-2 cells, two commonly used cell lines that are CD38+ and CD38-, respectively. The IC50 (the concentration at which 50% of cells are killed) for MT-4019 was calculated as 16 picomolar (pM) against H929 cells, but Molecular did not observe any measurable cell-kill with MT-4019 against CD38-HDLM-2 cells. A full summary of cell kill is presented in Table 1. There is a relationship between cell-kill potency and level of CD38 expression.
Table 1. Summary of Cell-Kill Activity for MT-4019
| Cell Line |
Type |
CD38 Expression Level |
CD50 ( I) | |||
| H929 |
Multiple myeloma | +++ | 16 pM | |||
| Daudi |
B-lymphoblast | +++ | 58 pM | |||
| ST486 |
B-lymphoblast | +++ | 41 pM | |||
| MOLP-8 |
Multiple myeloma | ++ | 228 pM | |||
| BC3 |
B-lymphocyte | ++ | 180 pM | |||
| IM-9 |
Multiple myeloma | — | >>100 nM | |||
| HDLM-2 |
B-lymphoblast | — | >> 100 nM | |||
| Ll236 |
B-lymphoblast | — | >> 100 nM |
| (1) | pM = picomolar; nM = nanomolar |
The potency of MT-4019 compares favorably with that reported for daratumumab, but direct comparisons are difficult as daratumumab requires the addition of effector cells for cytotoxicity. In an assay measuring the potency of CDC-mediated cell-kill of daratumumab against Daudi cells, the CD50 was reported to be approximately 800pM compared to 58pM with MT-4019 (de Weers et al., 2011). What is likely to be more important than the improved potency seen with MT-4019, though, is its wholly distinct mechanism of action. In patients who have progressed after CD38 treatment but still retain CD38 expression, the direct mechanism of cell kill seen with MT-4019 may be relevant.
MT-4019 In Vivo Activity: MTD Study
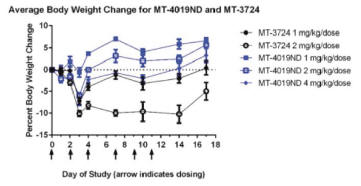
MT-4019 and MT-3724 were tested in CB17 SCID mice to determine the maximal tolerated dose, or MTD, of the drug. Mice were dosed via IP injection with either MT-3724 at 1 or 2mg/kg or MT-4019 at 1, 2, or 4 mg/kg. Dosing was three times weekly for two weeks, and cageside observations and body weight measurements were conducted. The doses of MT-4019 were selected based on experience with MT-3724.
The MTD for MT-4019 was not identified within the dose range tested. No deaths were observed during dosing or the recovery period. Average body weight loss appeared dose-dependent with the highest loss for MT-4019 occurring in the 4 mg/kg arm, but even in this arm mean body weight loss was still no more than 5% of baseline (Figure 6). By comparison, at the 2 mg/kg dose for MT-3724, mean body weight loss was 10%.
12
Figure 6. Murine Safety Study
MT-4019 In Vivo Activity
Molecular replicated the Daudi cell xenograft model used with daratumumab (de Weers, et al.) with MT-4019 to confirm in vivo activity. Molecular implanted luciferase-expressing Daudi cells (2.5 X 106 Daudi cells as in the daratumumab study) in SCID mice and administered varying doses of MT-4019. Due to the smaller size of the younger age mice used in the MT-4019 study (5-6 weeks), compared to the daratumumab study (8-10 weeks), the tumor burden per mass was larger for mice in the MT-4019 study. There was variability in tumor enlargement between the daratumumab and MT-4019 models. As measured by the integrated light intensity, tumors were significantly larger at peak in the MT-4019 model than in the daratumumab model (1.5X1011 photons per second in control animals for MT-4019 vs. up to 1.0X107 integrated light intensity in control animals for daratumumab). Because of the much shorter half-life of MT-4019, six administrations were given over two weeks as opposed to one administration of daratumumab.
By Day 40, a statistically significant difference was seen between mice treated with the vehicle control and mice treated with MT-4019 (Figure 7A). Tumor imaging clearly shows the difference between the treated and untreated mice by day 22 (Figure 7B).
Figure 7. MT-4019 Daudi-luc Disseminated Xenograft
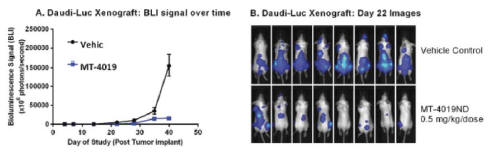
Figure 7. SCID mice were injected intraveneously with 2.5 X 106 Daudi cells expressing luciferase. After 1hr, the first dose of MT-4019 or Vehicle was administered intraperitoneally. In total, six doses of MT-4019 were administered over two weeks on a Monday-Wednesday-Friday schedule. Total BLI was measured. Representative imaging for mice treated with Vehicle or 0.5 mg/Kg of MT-4019 at Day 22 are shown in Figure 7B.
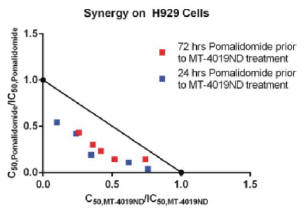
MT-4019 Combination Activity
MT-4019 was combined in vitro with pomalidomide, an immunomodulatory imide drug, or IMiD, and approved standard of care for refractory multiple myeloma. H929 cells were pre-treated for either 24 or 72 hours with pomalidomide and then treated with MT-4019. An isobologram was calculated to determine whether there was a synergistic effect between the two agents. Strong synergy was demonstrated (Figure 8) which is likely due to both
13
the differences in mechanism of action between the agents as well as the target for MT-4019 as pomalidomide has been shown to increase the expression of CD38 (Boxhammer, et al., 2015). The differences in mechanism of cell-kill and the effect of pomalidomide on CD38 expression may make the combination of these agents worth exploring in the clinic.
Figure 8. Combination Study with Pomalidomide
Clinical and Regulatory Plan
Molecular has begun to pursue GMP manufacturing for MT-4019. Molecular has substantial expertise with the GMP manufacture of ETBs based on its successful production of MT-3724. Molecular has a non-GMP facility in-house and has conducted seven GMP campaigns with MT-3724. From its experience with MT-3724,
Molecular believes it can transfer expression of MT-4019 and complete manufacturing for GLP toxicity studies within six months. Based on expression and process improvements, MT-4019 is expected to have similar or better yields than MT-3724.
Molecular expects to initiate IND-enabling studies to fully characterize MT-4019 based on toxicology and pharmacology in 2017. Molecular expect to initiate a Phase I clinical trial for MT-4019 in 2018. The Phase I trial will be conducted as a single-arm, open-label, multi-center, dose escalation study in patients with CD38+ relapsed/refractory multiple myeloma. Molecular was awarded a $15.2 million grant from CPRIT for the development of MT-4019. Molecular expects this grant to cover the cost of IND-enabling studies and the Phase I and Phase II trials for MT-4019.
Recent Presentations
MT-4019 AACR presentation. Molecular presented data on MT-4019, Molecular’s second-generation ETB targeting CD38, at the AACR Meeting in April 2017. The CD38 receptor has been shown to persist in patients after they stop responding to daratumumab, which is a monoclonal antibody that works with a person’s immune system. Monoclonal antibodies attach themselves to multiple myeloma cells and directly kill them and/or signal to the immune system to destroy them. CD38 is a poorly internalizing receptor, rendering it unsuitable for targeting with standard antibody-drug conjugates, or ADCs. Unlike chemotherapy, ADCs are intended to target and kill only the cancer cells and spare healthy cells. ADCs are complex molecules composed of an antibody linked to a biologically active cytotoxic (anticancer) payload or drug. Molecular believes CD38 is an excellent target for Molecular’s ETB technology. After a robust screening process, Molecular identified MT-4019 as Molecular’s lead ETB to CD38. MT-4019 utilizes Molecular’s second-generation ETB scaffold and, in the AACR presentation, Molecular demonstrated potent cell-kill activity against CD38-expressing tumor cells with 50% inhibitory concentrations (IC50) achieved at picomolar concentrations of the drug. MT-4019 also demonstrated reduced innate and adaptive immunity in murine and NHP models. Molecular believes this level of decreased immunogenicity has not been previously reported. Molecular anticipates moving MT-4019 into clinical trials in 2018.
14
Next Generation ETB Targets
Molecular has launched additional programs against the key targets HER2 and PD-L1. Molecular selected HER2 as a target because of its validated role in breast cancer. Targeting HER2 with different modalities (antibody, small molecule and ADC) has shown clinical benefit, and the target is known to persist after a given modality has failed. The clinical benefit seen with Kadcyla (an ADC to HER2) strongly suggests that a direct cell-kill approach to HER2 can be well tolerated in patients. Molecular believes that attacking HER2-expressing tumor cells with a differentiated mechanism of destruction may provide meaningful clinical benefits, even in patients whose disease has progressed on other HER2-targeted modalities. Molecular’s lead HER2 ETB, MT-5111, has shown potent picomolar activity in Kadcyla insensitive HER2+ cell lines and has shown additive or synergistic benefit in vitro with Kadcyla in HER2+ cell lines.
In the case of Molecular’s ETB program targeting the PD-L1 receptor, Molecular has focused on targeting PD-L1 with a direct cell-kill approach rather than using it to induce an immune response. PD-L1 is a focal point for immuno-oncology checkpoint antibodies; its expression on tumors is known to downregulate CD8 T-cell activity against tumor cells. Molecular believes that targeting PD-L1 in this manner may overcome resistance to checkpoint inhibitors dependent on T-cell infiltration and changes in the tumor microenvironment.
ETB Research & Development Partnerships
Takeda Pharmaceuticals
In October 2016, Molecular entered into a collaboration and option agreement with Takeda to discover and develop CD38-targeting ETBs, which includes MT-4019 for evaluation by Takeda. Under the terms of the agreement, Molecular is responsible for providing to Takeda (i) new ETBs generated using Takeda’s proprietary fully human antibodies targeting CD38 and (ii) MT-4019 for in vitro and in vivo pharmacological and anti-tumor efficacy evaluations. Molecular granted Takeda an exclusive option to negotiate and obtain an exclusive worldwide license to develop and commercialize any ETB that may result from this collaboration, including MT-4019. Molecular is entitled to receive up to $2.0 million in technology access fees and cost reimbursement associated with Molecular’s performance and completion of Molecular’s obligations under the agreement. To date, Molecular has received $1.0 million under this agreement.
In June 2017, Molecular entered into a Multi-Target Collaboration and License Agreement with Takeda in which Molecular will collaborate with Takeda to identify, generate and evaluate engineered toxin bodies, or ETBs, against certain targets designated by Takeda. Takeda will designate certain targets of interest as the focus of the research. Takeda will provide to Molecular targeting moieties against the designated targets. Molecular will create and characterize ETBs against those targets and provide them to Takeda for further evaluation. Each party grants to the other nonexclusive rights in its intellectual property for purposes of the conduct of the research, and Molecular agrees to work exclusively with Takeda with respect to the designated targets.
Under the agreement, Takeda has an option to acquire an exclusive license under Molecular’s intellectual property to develop, manufacture, commercialize and otherwise exploit ETBs against the designated targets. Upon exercise of the option, Takeda is obliged to use commercially reasonable efforts to develop and obtain regulatory approval of any licensed ETBs in major market countries, and thereafter to commercialize licensed ETBs in those countries. Molecular is obligated to manufacture ETBs to support research and clinical development through Phase I clinical trials, provided that Takeda can assume manufacturing responsibility at any time.
Molecular will receive an upfront fee and may receive net milestone payments of $25.0 million in aggregate through the exercise of the option to license ETBs. Post option exercise, Molecular is entitled to receive up to approximately $545.0 million in additional milestone payments through preclinical and clinical development and commercialization. Molecular is also entitled to tiered royalty payments of a mid-single to low-double digit percentage of net sales of any licensed ETBs, subject to certain reductions.
The agreement will expire on the expiration of the option period for the designated targets if Takeda does not exercise its options, or, following exercise of the option, on the later of the expiration of patent rights claiming the licensed ETB or ten years from first commercial sale of a the licensed ETB. The agreement may be sooner terminated by Takeda for convenience or upon a Molecular change of control, or by either party for an uncured material breach of the agreement.
15
Other Research & Development Collaborations
Henry M. Jackson Foundation
In July 2014, Molecular entered into a non-exclusive license agreement for certain biological materials for use in conjunction with the development of Molecular’s lead clinical stage ETB MT-3724. Under the terms of the agreement, Molecular is required to pay the Henry M. Jackson Foundation aggregate payments totaling $110,000 with respect to this license.
Manufacturing
Molecular relies on third-party contract manufacturing organizations, known as CMOs, to manufacture and supply Molecular with GMP drug substance and drug product materials to support Molecular’s clinical trials and anticipate doing so for the foreseeable future. The manufacturing processes for MT-3724, MT-4019 and other preclinical ETB candidates have been developed by Molecular’s manufacturing staff. Once a process is developed and defined for an ETB, it is transferred to CMOs to scale-up and optimize for manufacturing that conforms to current GMP (“cGMP”) standards.
Molecular has established well-defined, cost efficient manufacturing under GMP, including bioanalytical, quality control and quality assurance, logistics, distribution and supply chain management. After manufacturing, Molecular’s ETB candidates are tested and released by Molecular’s analytical and quality systems staff in conjunction with some select contract research organizations, or CROs. The quality control organization performs a series of release assays designed to ensure that the product meets all applicable specifications. Molecular’s quality assurance staff also reviews manufacturing and quality control records prior to batch release in an effort to assure conformance with cGMP as mandated by the FDA and foreign regulatory agencies.
Molecular’s manufacturing staff is trained and routinely evaluated for conformance to rigorous manufacturing procedures and quality standards. This oversight is intended to ensure compliance with FDA and foreign regulations and to provide consistent ETB output. Molecular’s quality control and quality assurance staff is similarly trained and evaluated as part of Molecular’s effort to ensure consistency in the testing and release of the product, as well as consistency in materials, equipment and facilities.
For the purposes of internal research and support for Molecular’s ongoing collaborations, Molecular has small scale manufacturing capabilities that are sufficient to manufacture drug materials for preclinical research.
Intellectual Property Portfolio
Molecular seeks to protect proprietary rights to its platform technologies through a combination of patents and patent applications, trade secrets and know-how. Molecular’s platform technologies include ETBs directed to specific molecular targets, in which a Shiga toxin A subunit construct is linked to an immunoglobulin domains directed to the target, and their uses for treating cancer, killing cancer cells and selectively delivering payload molecules into a target cell. Molecular’s platform technologies also include various ETB scaffolds regardless of target, and the Shiga toxin components of ETBs, including improved Shiga toxin A subunit constructs having disruptions of B-cell epitopes and/or T-cell epitopes for reduced immunogenicity when used in ETB scaffolds.
To cover its proprietary technologies and its current pipeline of proprietary ETB products and related methods, such as methods of use, Molecular has filed patent applications representing 11 international patent families, together covering 72 pending regional and national applications worldwide, including 12 pending U.S. patent applications and 60 foreign patent applications currently pending in the regional European Patent Office and nine other jurisdictions outside of the U.S. (Australia, Canada, China, Hong Kong, Israel, India, Japan, South Korea and Mexico). Molecular also has provisional rights pending in seven U.S. provisional patent applications.
16
Molecular’s patent families covering ETBs and modified ETB scaffolds for the targeted killing of cancer cells or for the selective delivery of molecules into a target cell include 10 internationally filed patent families. Patent rights in these patent families, if granted, will expire without extension in 2034-2036. Molecular also has a patent family directed to the screening of large ETB libraries, in which patent rights, if granted, will expire without extension in 2035. With respect to its ETB pipeline, Molecular’s lead compound which targets CD20, MT-3724, and pharmaceutical compositions and uses of MT-3724, are covered by two international patent families. Patent rights in these patent families, if granted, will expire without extension in 2034 and 2036. Molecular’s current pipeline also includes ETBs which target CD38 (MT-4019) and HER2 (MT-5111), covered by at least one international patent family from which patent rights, if granted, will expire without extension in 2036.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and biological products, such as MT-3724, MT-4019, and any future product candidates. Generally, before a new drug or biologic can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. drug development
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations and biologics under the FDCA, the Public Health Service Act, or PHSA, and their implementing regulations. Both drugs and biologics also are subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or post-market may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls or market withdrawals, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on Molecular. MT-3724, MT-4019 and any ETB product candidates must be approved by the FDA through either a NDA or Biologics Licensing Application, BLA, process before they may be legally marketed in the United States. The process generally involves the following:
| • | Completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with GLP requirements; |
| • | Submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| • | Approval by an independent institutional review board, or IRB, or ethics committee at each clinical trial site before a trial may be initiated at that site; |
| • | Performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, good clinical practice requirements, or GCP, and other clinical trial-related requirements to establish the safety and efficacy of the investigational product for each proposed indication; |
| • | Submission to the FDA of an NDA or BLA; |
17
| • | A determination by the FDA within 60 days of its receipt of an NDA or BLA that the NDA or BLA is sufficiently complete to permit a substantial review, in which case the NDA or BLA is filed; |
| • | Satisfactory completion of a FDA pre-approval inspection of the manufacturing facility or facilities where the drug or biologic will be produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug or biologic’s identity, strength, quality and purity; |
| • | Potential FDA audit of the preclinical and/or clinical trial sites that generated the data in support of the NDA or BLA; and |
| • | FDA review and approval of the NDA or BLA, including consideration of the views of an FDA advisory committee, if one was involved, prior to any commercial marketing or sale of the drug or biologic in the United States. |
The preclinical testing, clinical trials and the approval process requires substantial time, effort and financial resources, and Molecular cannot be certain that any approvals for MT-3724, MT-4019 and any future product candidates will be granted on a timely basis, or at all. The data required to support an NDA or BLA are generated in two distinct developmental stages: preclinical and clinical. The preclinical developmental stage generally involves laboratory evaluations of drug chemistry, formulation and stability, as well as studies to evaluate toxicity in animals, which support subsequent clinical testing. The sponsor must submit the results of the preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational new drug to humans, and must become effective before human clinical trials may begin.
The clinical stage of development involves the administration of the investigational product to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical trial must be reviewed and approved by an IRB at each institution at which the clinical trial will be conducted to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative, and must monitor the clinical trial until completed. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries.
A sponsor who wishes to conduct a clinical trial outside of the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of an NDA or BLA. The FDA will accept a well-designed and well-conducted foreign clinical trial not conducted under an IND if the trial was conducted in accordance with GCP requirements, and the FDA is able to validate the data through an onsite inspection if deemed necessary.
Preclinical Studies and IND
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as in vitro and animal studies to assess the potential for adverse events and in some cases to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical trials, among other things, to the FDA as part of an IND. Some long-term preclinical testing, such as animal tests of effects on reporduction
18
and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time, the FDA raises concerns or questions and places the IND on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical trials
Clinical trials generally are conducted in three sequential phases, known as Phase I, Phase II and Phase III, which may overlap.
| • | Phase I clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug. |
| • | Phase II clinical trials involve studies in disease-affected patients to determine the dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified and a preliminary evaluation of efficacy is conducted. |
| • | Phase III clinical trials generally involve a large number of patients at multiple sites and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. |
Post-approval trials, sometimes referred to as Phase IV clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase IV clinical trials as a condition of approval of an NDA or BLA.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the drug, findings from animal or in vitro testing that suggest a significant risk for human subjects and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure.
Phase I, Phase II and Phase III clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug or biologic has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether a trial may move forward at designated check points based on access to certain data from the trial. Concurrent with clinical trials, companies may perform additional animal studies and develop additional information about the chemistry and physical characteristics of the drug or biologic as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that MT-3724, MT-4019 and any future product candidates do not undergo unacceptable deterioration over their shelf life.
19
NDA/BLA and FDA Review Process
Following completion of the clinical trials, data are analyzed to assess whether the investigational product is safe and effective for the proposed indicated use or uses. The results of preclinical studies and clinical trials are then submitted to the FDA as part of an NDA or BLA, along with proposed labeling, chemistry and manufacturing information to ensure product quality and other relevant data. In short, the NDA or BLA is a request for approval to market the drug or biologic for one or more specified indications and must contain proof of safety and efficacy for a drug or safety, purity and potency for a biologic. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of FDA. FDA approval of an NDA or BLA must be obtained before a drug or biologic may be marketed in the United States.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each NDA or BLA must be accompanied by a user fee. FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fee schedule, for fiscal year 2017, the user fee for an application requiring clinical data, such as an NDA or BLA, is $2,038,100. PDUFA also imposes an annual product fee for human drugs and biologics (approximately $97,750) and an annual establishment fee (approximately $0.51 million) on facilities used to manufacture prescription drugs and biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs or BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA reviews all submitted NDAs and BLAs before it accepts them for filing, or it may refuse to file the application and request additional information. The FDA must make a decision on accepting an NDA or BLA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA or BLA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has 10 months, from the filing date, in which to complete its initial review of a new molecular-entity (NME) or nonNME NDA or original BLA and respond to the applicant, and six months from the filing date of a NME NDA or original BLA designated for priority review. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs or BLAs, and the review process is often extended by FDA requests for additional information or clarification.
Before approving an NDA or BLA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMP requirements. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA also may audit data from clinical trials to ensure compliance with GCP requirements. Additionally, the FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions, if any. The FDA is not bound by recommendations of an advisory committee, but it considers such recommendations when making decisions on approval. The FDA likely will reanalyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. After the FDA evaluates an NDA or BLA, it will issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application will not be approved in its present form. A Complete Response Letter usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The Complete Response Letter may require additional clinical data, including additional pivotal Phase III clinical trial(s) and/or other significant and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than Molecular interpret the same data.
20
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years from the date of such approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety or providing a major contribution to patient care or in instances of drug supply issues. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication.
Expedited Development and Review Programs
The FDA has a fast track program that is intended to expedite or facilitate the process for reviewing new drugs and biologics that meet certain criteria. Specifically, new drugs and biologics are eligible for fast track designation if they are intended to treat a serious or life threatening condition and preclinical or clinical data demonstrate the potential to address unmet medical needs for the condition. Fast track designation applies to both the product and the specific indication for which it is being studied. The sponsor can request the FDA to designate the product for fast track status any time before receiving NDA or BLA approval, but ideally no later than the pre-NDA or pre-BLA meeting.
Any product submitted to the FDA for marketing, including under a fast track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it treats a serious or life-threatening condition and, if approved, would provide a significant improvement in safety and effectiveness compared to available therapies.
The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biologic designated for priority review in an effort to facilitate the review. A product may also be eligible for accelerated approval, if it treats a serious or life-threatening condition and generally provides a meaningful advantage over available therapies. In addition, it must demonstrate an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, or IMM, that is reasonably likely to predict an effect on IMM or other clinical benefit. As a condition of approval, the FDA may require that a sponsor of a drug or biologic receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. If the FDA concludes that a drug or biologic shown to be effective can be safely used only if distribution or use is restricted, it will require such post-marketing restrictions, as it deems necessary to assure safe use of the product.
Additionally, a drug or biologic may be eligible for designation as a breakthrough therapy if the product is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over currently approved therapies on one or more clinically significant endpoints. The benefits of breakthrough therapy designation include the same benefits as fast track designation, plus intensive guidance from the FDA to ensure an efficient drug development program. Fast track designation, priority review, accelerated approval and breakthrough therapy designation do not change the standards for approval, but may expedite the development or approval process.
21
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, an NDA or BLA or supplement to a NDA or BLA must contain data to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. The Food and Drug Administration Safety and Innovation Act, or FDASIA, amended the FDCA to require that a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within 60 days of an end-of-Phase II meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase III or Phase II/III study. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials and/or other clinical development programs.
Post-marketing Requirements
Following approval of a new product, the manufacturer and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and record-keeping activities, reporting of adverse experiences, and complying with promotion and advertising requirements, which include restrictions on promoting approved drugs for unapproved uses or patient populations (known as “off-label use”). Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such uses. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Further, if there are any modifications to the drug or biologic, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA/BLA or NDA/BLA supplement, which may require additional data from preclinical studies or clinical trials.
The FDA may also place other conditions on approvals including the requirement for REMS to assure the safe use of the product. If the FDA concludes a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS. The FDA will not approve the NDA or BLA without an approved REMS, if required. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing. FDA regulations require that products be manufactured in specific approved facilities and in accordance with cGMPs. Molecular rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of Molecular’s products in accordance with cGMPs. These manufacturers must comply with cGMPs that require, among other things, quality control and quality assurance, the maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved drugs or biologics are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP requirements and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. The discovery of violative conditions, including failure to conform to cGMPs, could result in enforcement actions, and the discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA or BLA, including recall.
Companion Diagnostics and Complementary Diagnostics
Molecular believes that the success of Molecular’s product candidates may depend, in part, on the development and commercialization of either a companion diagnostic or complementary diagnostic. Companion diagnostics and complementary diagnostics can identify patients who are most likely to benefit from a particular therapeutic product; identify patients likely to be at increased risk for serious side effects as a result of treatment
22
with a particular therapeutic product; or monitor response to treatment with a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness. Companion diagnostics and complementary diagnostics are regulated as medical devices by the FDA. The level of risk combined with available controls to mitigate risk determines whether a companion diagnostic device requires Premarket Approval Application, or PMA, approval or is cleared through the 510(k) premarket notification process. For a novel therapeutic product for which a companion diagnostic device is essential for the safe and effective use of the product, the companion diagnostic device should be developed and approved or 510(k)-cleared contemporaneously with the therapeutic. The use of the companion diagnostic device will be stipulated in the labeling of the therapeutic product.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval may also be subject to regulation by other regulatory authorities in the United States in addition to the FDA, Depending on the nature of the product, those authorities may include the CMS, other divisions of the Department of Health and Human Services, the Department of Justice, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, OSHA, the Environmental Protection Agency and state and local governments.
For example, in the United States, sales and marketing must comply with state and federal fraud and abuse laws. These laws include the federal Anti-Kickback Statute, which makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce or reward referrals, including the purchase, recommendation, order or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties and exclusion from participation in federal healthcare programs. In addition, the ACA, among other things, amends the intent requirement of the federal Anti-Kickback Statute and criminal healthcare fraud statutes created by HIPAA. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. Moreover, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in the ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities also are potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with any of these laws or regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, requests for recall, seizure of products, total or partial suspension of production, denial or withdrawal of product approvals or refusal to allow a firm to enter into supply contracts, including government contracts. Any action against Molecular for violation of these laws, even if Molecular successfully defend against it, could cause Molecular to incur significant legal expenses and divert Molecular’s management’s attention from the operation of Molecular’s business. Prohibitions or restrictions on sales or withdrawal of future products marketed by Molecular could materially affect Molecular’s business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact Molecular’s business in the future by requiring, for example: (i) changes to Molecular’s manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of Molecular’s products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of Molecular’s business.
23
U.S. Patent-term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of MT-3724, MT-4019 and any future product candidates, some of Molecular’s U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit restoration of the patent term of up to five years as compensation for patent term lost during product development and FDA regulatory review process. Patent-term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent-term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA or BLA plus the time between the submission date of an NDA or BLA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, Molecular may apply for restoration of patent term for Molecular’s currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA or BLA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of a NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for a NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
An abbreviated approval pathway for biological products shown to be similar to, or interchangeable with, an FDA-licensed reference biological product was created by the Biologics Price Competition and Innovation Act of 2009, or BPCIA, as part of the ACA. This amendment to the PHSA, in part, attempts to minimize duplicative testing. Biosimilarity, which requires that the biological product be highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there be no clinically meaningful differences between the product and the reference product in terms of safety, purity and potency, can be shown through analytical studies, animal studies and a clinical trial or trials. Interchangeability requires that a biological product be biosimilar to the reference product and that the product can be expected to produce the same clinical results as the reference product in any given patient and, for products administered multiple times to an individual, that the product and the reference product may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product without such alternation or switch. Complexities associated with the larger, and often more complex, structure of biological products as compared to small molecule drugs, as well as the processes by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
24
A reference biological product is granted 12 years of data exclusivity from the time of first licensure of the product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. Date of first licensure does not include the date of licensure of (and a new period of exclusivity is not available for) a biological product if the licensure is for a supplement for the biological product or for a subsequent application by the same sponsor or manufacturer of the biological product (or licensor, predecessor in interest, or other related entity) for a change (not including a modification to the structure of the biological product) that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength, or for a modification to the structure of the biological product that does not result in a change in safety, purity, or potency. Whether a subsequent application, if approved, warrants exclusivity as the “first licensure” of a biological product is determined on a case-by-case basis with data submitted by the sponsor.
Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing regulatory exclusivity periods. This six-month exclusivity, which attaches to both the twelve-year and four-year exclusivity periods for reference biologics, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial. Furthermore, a biological product seeking licensure as biosimilar to or interchangeable with a reference product indicated for a rare disease or condition and granted seven years of orphan drug exclusivity may not be licensed by the FDA for the protected orphan indication until after the expiration of the seven-year orphan drug exclusivity period or the 12-year reference product exclusivity, whichever is later.
European Union Drug Development
In the European Union, Molecular’s future products also may be subject to extensive regulatory requirements. As in the United States, drugs, which are referred to as medicinal products can be marketed only if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of preclinical and clinical research in the European Union are subject to significant regulatory controls. Although the EU Clinical Trials Directive 2001/20/EC has sought to harmonize the EU clinical trials regulatory framework, setting out common rules for the control and authorization of clinical trials in the EU, the EU Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the member state regimes. Under the current regime, before a clinical trial can be initiated, a clinical trial application must be approved in each of the EU countries where the trial is to be conducted by two distinct bodies: the National Competent Authority, or NCA, and one or more Ethics Committees, or ECs. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the Member State where they occurred. The EU clinical trials legislation currently is undergoing a transition process mainly aimed at harmonizing and streamlining clinical-trial authorization, simplifying adverse-event reporting procedures, improving the supervision of clinical trials and increasing their transparency.
European Union Drug Review and Approval
In the European Economic Area, or EEA, which is comprised of the 27 Member States of the European Union (including Norway and excluding Croatia), Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations.
| • | The Community MA is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced-therapy medicines such as gene-therapy, somatic cell-therapy or tissue-engineered medicines and medicinal products containing a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and other immune dysfunctions and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU. |
25
| • | National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State, or RMS. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the Member States (i.e., in the RMS and the Member States Concerned). |
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
European Union New Chemical Entity Exclusivity
In the European Union, new chemical entities, sometimes referred to as new active substances, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. The data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application for eight years, after which generic marketing authorization can be submitted, and the innovator’s data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of 11 years if, during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are determined to bring a significant clinical benefit in comparison with currently approved therapies.
European Union Orphan Designation and Exclusivity
In the European Union, the EMA’s Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union community (or where it is unlikely that the development of the medicine would generate sufficient return to justify the investment) and for which no satisfactory method of diagnosis, prevention or treatment has been authorized (or, if a method exists, the product would be a significant benefit to those affected).
In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
26
Rest of the World Regulation
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. Additionally, the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If Molecular fail to comply with applicable foreign regulatory requirements, Molecular may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Reimbursement
Sales of Molecular’s products will depend, in part, on the extent to which Molecular’s products will be covered by third-party payors, such as government health programs, commercial insurance and managed care organizations. In the United States no uniform policy of coverage and reimbursement for drug or biological products exists. Accordingly, decisions regarding the extent of coverage and amount of reimbursement to be provided for any of Molecular’s products will be made on a payor-by-payor basis. As a result, the coverage determination process is often a time-consuming and costly process that will require Molecular to provide scientific and clinical support for the use of Molecular’s products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained.
The United States government, state legislatures and foreign governments have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price-controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. For example, the ACA contains provisions that may reduce the profitability of drug products through increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Adoption of general controls and measures, coupled with the tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceutical drugs. The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. The ACA made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate on most branded prescription drugs from 15.1% of average manufacturer price, or AMP, to 23.1% of AMP and adding a new rebate calculation for “line extensions” (i.e., new formulations, such as extended release formulations) of solid oral dosage forms of branded products, as well as potentially impacting their rebate liability by modifying the statutory definition of AMP. The ACA also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and by enlarging the population potentially eligible for Medicaid drug benefits. Congress and President Trump have expressed their intention to repeal or repeal and replace the ACA. If that is done, many if not all of the provisions of the ACA may no longer apply to prescription drugs.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. While all Medicare drug plans must give at least a standard level of coverage set by Medicare, Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for products for which Molecular receive marketing approval. However, any negotiated prices for Molecular’s products covered by a Part D prescription drug plan likely will be lower than the prices Molecular might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
27
For a drug product to receive federal reimbursement under the Medicaid or Medicare Part B programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the AMP and Medicaid rebate amounts reported by the manufacturer. As of 2010, the ACA expanded the types of entities eligible to receive discounted 340B pricing, although, under the current state of the law, with the exception of children’s hospitals, these newly eligible entities will not be eligible to receive discounted 340B pricing on orphan drugs. In addition, as 340B drug pricing is determined based on AMP and Medicaid rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discount to increase.
As noted above, the marketability of any products for which Molecular receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. An increasing emphasis on cost containment measures in the United States has increased and Molecular expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which Molecular receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
In addition, in most foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing and reimbursement vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of Molecular’s products. Historically, products launched in the European Union do not follow price structures of the United States and generally prices tend to be significantly lower.
Competition
Molecular competes directly with companies that focus on oncology as well as companies dedicating their resources to novel forms of cancer therapies. Molecular also faces competition from academic research institutions, governmental agencies and various other public and private research institutions. With the proliferation of new drugs and therapies into oncology, Molecular expects to face increasingly intense competition as new technologies become available. Any ETB candidates that Molecular successfully develops and commercializes will compete with existing therapies and new therapies that may become available in the future.
Many of Molecular’s competitors have significantly greater financial, manufacturing, marketing, drug development, technical and human resources than Molecular does. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of Molecular’s competitors. Smaller or early-stage companies also may prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with Molecular in recruiting and retaining top qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, Molecular’s programs.
The key competitive factors affecting the success of all of Molecular’s ETB candidates, if approved, are likely to be their efficacy, safety, dosing convenience, price, the effectiveness of companion diagnostics in guiding the use of related therapeutics, the level of generic competition and the availability of reimbursement from government and other third-party payors.
28
Molecular’s commercial opportunity could be reduced or eliminated if its competitors develop and commercialize products that are safer, more effective, less expensive, more convenient or easier to administer, or have fewer or less severe effects than any products that Molecular may develop. Molecular’s competitors also may obtain FDA, EMA or other regulatory approval for their products more rapidly than Molecular may obtain approval for its products, which could result in Molecular’s competitors establishing a strong market position before Molecular is able to enter the market. Even if Molecular’s ETB candidates achieve marketing approval, they may be priced at a significant premium over competitive products if any have been approved by then.
In addition to currently marketed therapies, there are also a number of products in late-stage clinical development directed to the same biological targets as Molecular’s programs, including antibodies, antibody drug conjugates and bi-specific antibodies.
| • | Approved antibody-based products targeting CD20 include rituximab (Genentech/Roche), ofatumumab (Novartis), obinutuzumab (Genentech/Roche) and ibritumomab tiuxetan (Spectrum Pharmaceuticals). |
| • | Antibody-based products, including bi-specific antibodies, targeting CD20 in development include veltuzumab (Immunomedics), ocaratuzumab (Mentrik Biotech), REGN1979 (Regeneron Pharmaceuticals), RG7828 (Genentech/Roche), XmAb13676 (Novartis/Xencor) and CD3-CD20 Duobody (Genmab). |
| • | The approved antibody-based product targeting CD38 is daratumumab (Janssen/Genmab). |
| • | Antibody-based products, including bi-specific antibodies, targeting CD38 in development include MOR02 (Morphosys), isatuximab (Sanofi) and XmAb13551 (Amgen/Xencor) |
| • | Approved antibody-based products, including antibody drug conjugates, targeting HER2 include trastuzumab, pertuzumab and trastuzumab emtansine (all from Genentech/Roche) |
| • | Antibody-based products, including bi-specific antibodies, targeting HER2 in development include margetuximab (Macrogenics), MEDI4276 (AstraZeneca), MM-111 (Merrimack Pharmaceuticals), FS102 (Bristol-Myers Squibb/F-star) and MCLA-128 (Merus). |
| • | Approved antibody-based products targeting PD-L1 include atezolizumab (Genentech/Roche) and avelumab (Merck KGaA/Pfizer) |
| • | Antibody-based products targeting PD-L1 in development include durvalumab (AstraZeneca), LY3300054 (Lilly) and BMS-936559 (Bristol-Myers Squibb) |
Employees
As of May 5, 2017, Molecular had 24 full-time employees. 9 of Molecular’s employees have Ph.D., PharmD or M.D. degrees, and 16 of Molecular’s employees are engaged in research and development activities. None of Molecular’s employees are subject to a collective bargaining agreement. Molecular believes that Molecular has good relations with Molecular’s employees.
Corporate Information
Molecular (formerly D5 Pharma, Inc., a Delaware corporation) was incorporated in Delaware on February 19, 2009. Its principal executive offices are at 9301 Amberglen Boulevard, Suite 100, Austin, Texas 78729, and its telephone number is (512) 869 1555.
29
Properties
Molecular currently subleases Molecular’s research and development and corporate offices in Austin, TX, occupying approximately 18,000 square feet. This sublease expires at the end of May 2017. In October 2016, Molecular entered into a five-year lease agreement for the Austin, TX space that will be effective June 2017 through May 2022 and includes an option to renew for one additional five-year period at Molecular’s discretion. In January 2017, Molecular entered into an amendment of the lease to add an additional 3,000 square feet, consisting mostly of research and development space. During March 2017, Molecular entered into a second amendment to the Austin, TX lease that allows for adding an additional 11,000 square feet. This amendment becomes effective automatically if and when Molecular receives at least $30.0 million in additional aggregate debt or equity funding, but excluding any CPRIT grant funds, on or before June 23, 2017. Molecular also has the right to waive this financing requirement at Molecular’s discretion. Upon the amendment becoming effective, the term of Molecular’s lease for the Austin, TX lease will extend to 72 months and expire May 2023.
Molecular also leases office facilities occupying approximately 4,600 square feet in Jersey City, NJ under a lease expiring in September 2019.
Molecular believes substantially all of Molecular’s property and equipment is in good condition and that Molecular has sufficient capacity to meet its current operational needs.
Legal Proceedings
Molecular is not a party to any material legal proceedings.
30