Attached files
| file | filename |
|---|---|
| EX-32.1 - Greenwich LifeSciences, Inc. | ex32-1.htm |
| EX-31.1 - Greenwich LifeSciences, Inc. | ex31-1.htm |
| EX-4.2 - Greenwich LifeSciences, Inc. | ex4-2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| [X] | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2020
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition Period from to
Commission File Number: 001-39555
GREENWICH LIFESCIENCES, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 20-5473709 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 3992 Bluebonnet Dr., Building 14, Stafford, Texas 77477 | 77477 | |
| (Address of principal executive offices) | (Zip Code) |
| (832) 819-3232 |
| (Registrant’s telephone number, including area code) |
| Securities registered pursuant to Section 12(b) of the Act: |
| Title of each class | Name of each exchange on which registered | |
| Common Stock, $0.001 par value | The NASDAQ Capital Market | |
| Securities registered pursuant to Section 12(g) of the Act: None | ||
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes [ ] No [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer [ ] | Accelerated filer [ ] | Non-accelerated filer [X] |
Smaller reporting company [X] | Emerging growth company [X] |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes [ ] No [X]
The Registrant was not a public company as of the last business day of its most recently completed second fiscal quarter and, therefore, cannot calculate the aggregate market value of its voting and non-voting common equity held by non-affiliates as of such date.
As of March 15, 2021, 12,846,897 shares of the registrant’s common stock, $0.001 par value per share, were issued and outstanding.
| 2 |
Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements which are made pursuant to the safe harbor provisions of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These statements may be identified by such forward-looking terminology as “may,” “should,” “expects,” “intends,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue” or the negative of these terms or other comparable terminology. Our forward-looking statements are based on a series of expectations, assumptions, estimates and projections about our company, are not guarantees of future results or performance and involve substantial risks and uncertainty. We may not actually achieve the plans, intentions or expectations disclosed in these forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in these forward-looking statements. Our business and our forward-looking statements involve substantial known and unknown risks and uncertainties, including the risks and uncertainties inherent in our statements regarding:
| ● | our projected financial position and estimated cash burn rate; |
| ● | our estimates regarding expenses, future revenues and capital requirements; |
| ● | our ability to continue as a going concern; |
| ● | our need to raise substantial additional capital to fund our operations; |
| ● | the success, cost and timing of our clinical trials; |
| ● | our dependence on third parties in the conduct of our clinical trial; |
| ● | our ability to obtain the necessary regulatory approvals to market and commercialize our product candidate; |
| ● | the ultimate impact of the current coronavirus pandemic, or any other health epidemic, on our business, our clinical trials, our research programs, healthcare systems or the global economy as a whole; |
| ● | the potential that results of preclinical and clinical trials indicate our current product candidate or any future product candidates we may seek to develop are unsafe or ineffective; |
| ● | the results of market research conducted by us or others; |
| ● | our ability to obtain and maintain intellectual property protection for our current product candidate; |
| ● | our ability to protect our intellectual property rights and the potential for us to incur substantial costs from lawsuits to enforce or protect our intellectual property rights; |
| ● | the possibility that a third party may claim we or our third-party licensors have infringed, misappropriated or otherwise violated their intellectual property rights and that we may incur substantial costs and be required to devote substantial time defending against claims against us; |
| ● | our reliance on third-party suppliers and manufacturers; |
| ● | the success of competing therapies and products that are or become available; |
| ● | our ability to expand our organization to accommodate potential growth and our ability to retain and attract key personnel; |
| ● | the potential for us to incur substantial costs resulting from product liability lawsuits against us and the potential for these product liability lawsuits to cause us to limit our commercialization of our product candidate; |
| ● | market acceptance of our product candidate, the size and growth of the potential markets for our current product candidate and any future product candidates we may seek to develop, and our ability to serve those markets; and |
| ● | the successful development of our commercialization capabilities, including sales and marketing capabilities. |
All of our forward-looking statements are as of the date of this Annual Report on Form 10-K only. In each case, actual results may differ materially from such forward-looking information. We can give no assurance that such expectations or forward-looking statements will prove to be correct. An occurrence of, or any material adverse change in, one or more of the risk factors or risks and uncertainties referred to in this Annual Report on Form 10-K or included in our other public disclosures or our other periodic reports or other documents or filings filed with or furnished to the U.S. Securities and Exchange Commission (the “SEC”) could materially and adversely affect our business, prospects, financial condition and results of operations. Except as required by law, we do not undertake or plan to update or revise any such forward-looking statements to reflect actual results, changes in plans, assumptions, estimates or projections or other circumstances affecting such forward-looking statements occurring after the date of this Annual Report on Form 10-K, even if such results, changes or circumstances make it clear that any forward-looking information will not be realized. Any public statements or disclosures by us following this Annual Report on Form 10-K that modify or impact any of the forward-looking statements contained in this Annual Report on Form 10-K will be deemed to modify or supersede such statements in this Annual Report on Form 10-K.
This Annual Report on Form 10-K may include market data and certain industry data and forecasts, which we may obtain from internal company surveys, market research, consultant surveys, publicly available information, reports of governmental agencies and industry publications, articles and surveys. Industry surveys, publications, consultant surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but the accuracy and completeness of such information is not guaranteed. While we believe that such studies and publications are reliable, we have not independently verified market and industry data from third-party sources.
| 3 |
Risk Factor Summary
Our business is subject to significant risks and uncertainties that make an investment in us speculative and risky. Below we summarize what we believe are the principal risk factors but these risks are not the only ones we face, and you should carefully review and consider the full discussion of our risk factors in the section titled “Risk Factors”, together with the other information in this Annual Report on Form 10-K. If any of the following risks actually occurs (or if any of those listed elsewhere in this Annual Report on Form 10-K occur), our business, reputation, financial condition, results of operations, revenue, and future prospects could be seriously harmed. Additional risks and uncertainties that we are unaware of, or that we currently believe are not material, may also become important factors that adversely affect our business.
Risks Relating to Our Financial Position and Capital Needs
We have incurred substantial losses since our inception and anticipate that we will continue to incur substantial and increasing losses for the foreseeable future.
We need significant additional financing to fund our operations and complete the development and, if approved, the commercialization of our product candidate. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our product candidate on unfavorable terms to us.
We currently have no source of revenues. We may never generate revenues or achieve profitability.
Risks Related to the Development and Regulatory Approval of Our Product Candidate
Clinical-stage biopharmaceutical companies with product candidates in clinical development face a wide range of challenging activities which may entail substantial risk.
We may find it difficult to enroll patients in our clinical trials given the limited number of patients who have the diseases for which our product candidate is being studied which could delay or prevent the start of clinical trials for our product candidate.
The results of preclinical studies or earlier clinical trials are not necessarily predictive of future results. Our existing product candidate in clinical trials, and any other product candidates that may advance into clinical trials, may not have favorable results in later clinical trials or receive regulatory approval.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome.
Our current and future product candidates, the methods used to deliver them or their dosage levels may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label or result in significant negative consequences following any regulatory approval.
Our product development program may not uncover all possible adverse events that patients who take our product candidate may experience. The number of subjects exposed to our product candidate and the average exposure time in the clinical development program may be inadequate to detect rare adverse events or chance findings that may only be detected once the product is administered to more patients and for greater periods of time.
Our future success is dependent on the regulatory approval of our product candidate.
| 4 |
Our current product candidate and future product candidates could fail to receive regulatory approval from the FDA.
Even if our current candidate receive regulatory approval, it may still face future development and regulatory difficulties.
Risks Related to Our Manufacturing
We have limited to no manufacturing, sales, marketing or distribution capability and must rely upon third parties for such.
We are subject to a multitude of manufacturing risks, any of which could substantially increase our costs and limit supply of our product candidate.
In the clinical trials using GP2, GM-CSF is also administered and its availability is dependent upon a third-party manufacturer, which may or may not reliably provide GM-CSF, thus jeopardizing the completion of the trials.
Risks Related to Our Dependence on Third Parties and Our License Agreements
We rely on third parties to conduct our preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, or if we lose any of our CROs or other key third-party vendors, we may not be able to obtain regulatory approval for or commercialize our current or future product candidates on a timely basis, if at all.
We are dependent on technologies we license, and if we lose the right to license such technologies or we fail to license new technologies in the future, our ability to develop new products would be harmed, and if we fail to meet our obligations under our license agreements, we may lose the ability to develop our product candidate.
Risks Related to Our Intellectual Property
We rely on an exclusive license granted to us by HJF with respect to GP2, and if HJF does not adequately defend such license, our business may be harmed.
Risks Related to Commercialization of Our Current Product Candidate and Future Product Candidates
Our commercial success depends upon attaining significant market acceptance of our current product candidate and future product candidates, if approved, among physicians, patients, healthcare payors and cancer treatment centers.
Even if we are able to commercialize our current product candidate or any future product candidates, the products may not receive coverage and adequate reimbursement from third-party payors in the U.S. and in other countries in which we seek to commercialize our products, which could harm our business.
Risks Related to Healthcare Compliance Regulations
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings. If we or they are unable to comply with these provisions, we may become subject to civil and criminal investigations and proceedings that could have a material adverse effect on our business, financial condition and prospects.
Risks Related to our Business Operations
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
| 5 |
Our business may be adversely affected by the ongoing coronavirus pandemic
Risks Related to Owning our Common Stock
The price of our common stock may fluctuate substantially.
Because certain of our stockholders control a significant number of shares of our common stock, they may have effective control over actions requiring stockholder approval.
Future sales and issuances of our common stock could result in additional dilution of the percentage ownership of our stockholders and could cause our share price to fall.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows.
BUSINESS
Overview
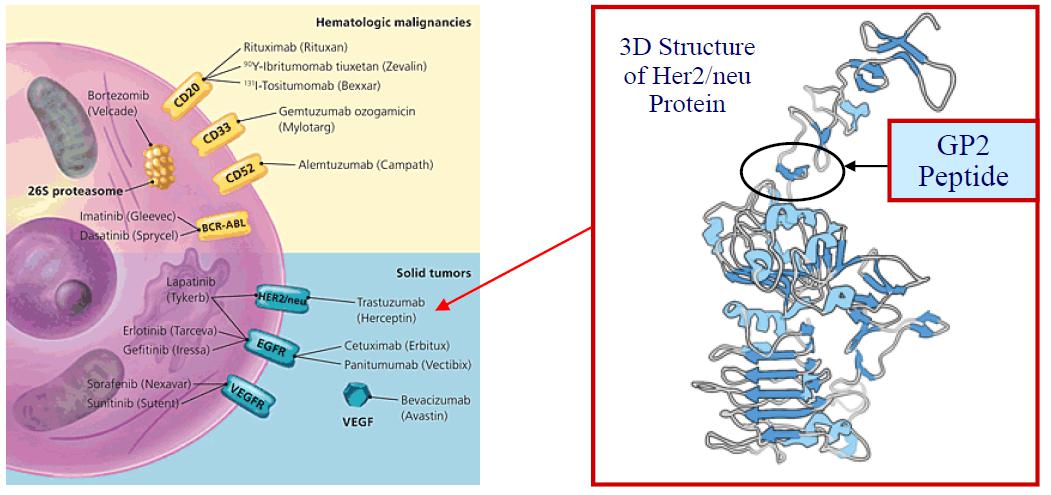
We are a biopharmaceutical company that is developing GP2, an immunotherapy designed to prevent the recurrence of breast cancer following surgery. GP2 is a 9 amino acid transmembrane peptide of the HER2/neu protein, a cell surface receptor protein that is expressed in a variety of common cancers, including expression in 75% of breast cancers at low (1+), intermediate (2+), and high (3+ or over-expressor) levels. In a completed Phase IIb clinical trial led by MD Anderson Cancer Center, no recurrences were observed in the HER2/neu 3+ adjuvant setting after median 5 years of follow-up, if the patient received the 6 primary intradermal injections over the first 6 months. We are planning to commence a Phase III clinical trial in 2021.
Our Product Candidate
GP2 is a HER2/neu transmembrane peptide that elicits a targeted immune response against HER2/neu-expressing cancers. Below is an image of a cell surface showing therapeutically relevant cell surface proteins in cancer. Breast cancers and other solid tumors with elevated expression of HER2/neu protein are highly aggressive with an increased disease recurrence and a worse prognosis.
GM-CSF Immunoadjuvant
Recombinant human granulocyte macrophage colony-stimulating factor or GM-CSF (sargramostim, Leukine®) has been shown to enhance monocyte as well as neutrophil cytotoxicity against melanoma tumor cells and to enhance activity-dependent cellular cytotoxicity of monocytes and neutrophils against targets coated with the anti-ganglioside antibodies. GP2 will be delivered in combination with GM-CSF to induce GP2 peptide specific immunity. GP2 treatment is administered via an intradermal injection by mixing GP2 peptide and GM-CSF at the time of administration.
GM-CSF is available in both liquid and lyophilized forms exclusively from one manufacturer, and we will continue to be dependent on such manufacturer for our supply of GM-CSF in combination with GP2 in our ongoing GP2 trials and upon potential commercialization of GP2. Although GM-CSF is currently approved for sale in the U.S. by the FDA and is available in other countries on a name patient basis through a specialized company that focuses on making products approved in the U.S. available globally, GM-CSF may be registered for sale in other countries by such manufacturer in the future.
Cancer Immunotherapy
Cancer immunotherapies seek to stimulate an individual’s own immune system to selectively attack cancer cells while not affecting normal cells or delivering certain immune system components in order to inhibit the spread of cancer. Cancer immunotherapy drugs are a new method of cancer treatment which are in addition to more established treatment options such as surgery, chemotherapy, targeted therapy, and radiation therapy. Therefore, cancer immunotherapy is an important and rapidly emerging field, which has led to new clinical research studies and garnered the attention of biotechnology and pharmaceutical companies, regulatory agencies, payors and hospital systems, cancer patients and their families, and the general public at large.
| 6 |
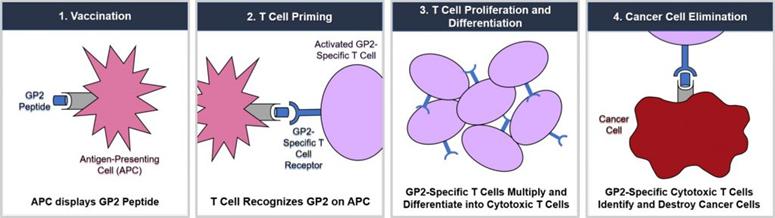
Cancer immunotherapy harnesses the body’s natural immune system response to fight and/or prevent tumor growth. An essential characteristic of the immune system, which is a network of tissues, cells, and signaling molecules that work to protect the body, is its ability to differentiate foreign threats, including cancerous growths, from normal cells. Despite the fact that tumor cells originate from normal cells, tumor cells can be recognized as foreign threats because of their ability to elicit the production of tumor antigens. These antigens may be released in the interstitial tissues, and eventually in the bloodstream or may remain on the surface of cognate cancer cells. The HER2/neu protein is one of the most widely expressed tumor antigens in multiple malignances.
Several cell types play an important role in the development and maintenance of immune responses against cancer. The most important cell types with regard to immune response are antigen-presenting cells (“APCs”) and lymphocytes. APCs include various subtypes, such as dendritic cells, monocytes and macrophages. Once a patient is exposed to a tumor antigen (either by the presence of cancer itself or through active immunization through a vaccine type immunotherapeutic), the tumor antigen gets recognized by the APC and becomes “processed” through digestion into smaller fragments within the APC. Subsequently, the APC “communicates” with a specific type of lymphocyte called a T-cell. Inactive T-cells search for tumor antigens by transiently binding to antigens presented by major histocompatibility complexes (“MHCs”) on the APCs. There is great variability in the expression of different subtypes of MHCs in the human population. The MHC system expresses human leukocyte antigens (“HLAs”) and these HLA subtypes determine the vigor and duration of any given T-cell response to a cancer among different patients.
As shown below, following GP2 immunotherapy, CD8+ cytotoxic T lymphocytes recognize and destroy HER2/neu-expressing cancer cells. GP2 is administered in combination with an FDA-approved immunoadjuvant GM-CSF, which stimulates the proliferation of antigen presenting cells. Preclinical studies have shown that T cells sensitized against the GP2 peptide demonstrate significant recognition of HER2/neu-expressing tumors. Both ovarian and breast cancer-specific CTLs recognize GP2, which is widely expressed in HER2/neu-expressing tumors and is capable of inducing tumor-specific CTL populations in vitro.
Breast Cancer Treatment Approach — Adjuvant & Neoadjuvant Treatments
As shown below, in the adjuvant setting, a HER2/neu 3+ patient typically receives Herceptin in the first year following breast cancer surgery, with the hope that their breast cancer will not recur, and with the odds of recurrence slowly decreasing over the first 5 years following surgery. Herceptin has been shown to reduce recurrence rates by approximately 50%, from 25% to 12%, in the adjuvant setting. In the neoadjuvant setting, a HER2/neu 3+ patient receives treatment before surgery and based on the results of a biopsy at surgery, will receive Herceptin or Kadcyla, a more potent form of Herceptin, following surgery. Kadcyla has been shown to reduce recurrence rates by 50%, from 22% to 11%, in the neoadjuvant setting. Accordingly, we believe that GP2 may be effective in safely addressing the 50% of recurring patients who do not respond to either Herceptin or Kadcyla.
GP2 is administered in combination with the immunoadjuvant GM-CSF in years 2-4, following the first year of treatment with Herceptin, in a series of 11 intradermal injections comprising 6 primary injections over 6 months (1 injection per month) followed by 5 booster injections every 6 months thereafter. Furthermore, we believe that recently approved drugs such as Perjeta and Nerlynx do not fully address this unmet need, even in their most efficacious subpopulations, and that in the initial GP2 indication, approximately 17,000 new patients may be eligible for GP2 treatment per year, which could save approximately 1,500 to 2,000 lives per year.
| 7 |
As only injection site reactions were observed (which speaks to the immunogenicity of GP2) and no SAEs were reported in the GP2 Phase IIb clinical trial, GP2 may be positioned as the final treatment for patients post-surgery. Furthermore, we believe that clinicians and patients are seeking a de-escalation and a return to normal life free of toxic treatments, especially if the chance of recurrence is reduced substantially. Lastly, we believe that GP2 may be the treatment that will synergistically overlap with or follow Herceptin, Kadcyla, or Enhertu (fam-trastuzumab deruxtecan-nxki, DS-8201) or any of the other Herceptin derivatives or antibody drug conjugates being developed.
We believe that U.S. academic centers will be moving higher risk, node positive patients into neoadjuvant treatment and will use Kadcyla if residual disease is observed at the time of surgery; however community centers and international markets may not move as quickly or at all, due to the high dual therapy costs and the lack of approval or reimbursement of Kadcyla in markets outside of the U.S. and Europe. GP2 will be pursued in both the adjuvant and neoadjuvant settings in HER2/neu 3+ patients in our planned Phase III trial.
GP2 Clinical Data & Planned Phase III Trial
In the Phase IIb and three Phase I clinical trials where 138 patients received GP2 immunotherapy, there were no SAEs reported in any of the trials, including for GP2 and GM-CSF combination treatments or any other GP2 combination treatments.
| Clinical Trial Description | Status | ||
| GP2 Phase IIb Clinical Trial | Trial Completed | ||
| ● | Prospective, Randomized, Single-Blinded, Multi-Center Phase II Trial of the HER2/neu Peptide GP2 + GM-CSF Vaccine versus GM-CSF Alone in HLA-A02+ Node-Positive and High-Risk Node-Negative Breast Cancer Patients to Prevent Recurrence | ||
| ● | 89 patients treated with GP2 + GM-CSF, 91 placebo patients treated with GM-CSF | ||
| GP2 Phase I Clinical Trial — Combination with AE37 | Trial Completed | ||
| ● | Phase I Safety Trial of the GP2 + GM-CSF Vaccine in Combination with the Helper Peptide AE37 + GM-CSF Vaccine | ||
| ● | 14 patients treated with GP2 + AE37 + GM-CSF | ||
| GP2 Phase I Clinical Trial — Combination with Trastuzumab | Trial Completed | ||
| ● | Phase Ib Trial of Combination Immunotherapy with HER2/neu Peptide GP2 + GM-CSF Vaccine and Trastuzumab in Breast Cancer Patients | ||
| ● | 17 patients treated with GP2 + GM-CSF + trastuzumab | ||
| First GP2 Phase I Clinical Trial | Trial Completed | ||
| ● | Phase Ib Trial of HER2/neu Peptide (GP2) Vaccine in Breast Cancer Patients | ||
| ● | 18 patients treated with GP2 + GM-CSF | ||
| 8 |
Phase I Clinical Trials
First GP2 Phase I Clinical Trial
As shown in the table above, the first GP2 Phase I clinical trial was conducted at Walter Reed Army Medical Center. The study was conducted in patients over the age of 18 years with a diagnosis of HER2/neu 1-3+, node negative breast cancer who had undergone primary surgical and medical therapies and who were without evidence of disease at the time of enrollment into the study. Patients were HLA typed and HLA-A02 patients were skin tested for recall antigens. HLA-A02 patients found to be immunologically intact received the vaccine. There were no grade 3-5 toxicities among the 18 patients receiving a total of 108 doses of GP2 + GM-CSF. Among all patients, the maximum local toxicity occurring during the entire series was grade 1 in 38.9% and grade 2 in 61.1% of the patients. The maximum systemic toxicity during the series was grade 0 in 5.6%, grade 1 in 61.1%, and grade 2 in 33.3% of the patients. The most common local reactions included erythema and induration (100% of patients), pruritis (25%), and inflammation (23%). The most common systemic reactions were grade 1 fatigue (40%) and grade 1 arthralgia/myalgia (15%). There were no recurrences and no deaths reported in study subjects. Additional data analysis included topics such as pre-existing immunity, dosing, and epitope spreading.
GP2 Phase I Clinical Trial — Combination with Trastuzumab
Preclinical research has previously demonstrated that a synergy may exist between trastuzumab and GP2 peptide-stimulated CTLs ex vivo. Pretreatment of breast cancer cells with trastuzumab followed by incubation with GP2 peptide-induced CTLs resulted in enhanced cytotoxicity in 3 tumor cell lines compared to treatment with trastuzumab or GP2-specific CTLs alone. These results suggest that concurrent GP2 vaccination during trastuzumab therapy may be a possible combination immunotherapy.
As shown in the table above, a Phase I trial evaluating the combination therapy of GP2 + GM-CSF administered simultaneously with trastuzumab was conducted. The combination therapy was found to be well tolerated when given concurrently in 17 clinically disease-free, HER2/neu over-expressing breast cancer patients.
GP2 Phase I Clinical Trial — Combination with AE37
As shown in the table above, a Phase I trial evaluating the combination therapy of GP2 + GM-CSF administered simultaneously with HER2/neu peptide AE37 in 14 clinically disease-free, HER2/neu breast cancer and ovarian cancer patients was conducted. While 28 patients enrolled, 14 patients completed the 6 vaccination series. Initial results suggest that combining GP2 and AE37 peptides is well tolerated at all tested dosing levels. Additionally, we believe the combination is capable of stimulating strong peptide-specific in vivo immune responses.
During the primary vaccination series, an AE37/GP2+GM-CSF dual peptide vaccine resulted in robust T-cell proliferation. However, significant immune responses became more variable at 6 and 12 months post vaccination suggesting the need for boosters in some individuals.
Phase II Clinical Trial
GP2 Phase IIb Clinical Trial Overview
In a prospective, randomized, single-blinded, placebo-controlled, multi-center (16 sites led by MD Anderson Cancer Center) Phase IIb clinical trial of HLA-A02 breast cancer patients, the combination of GP2-GMCSF-Herceptin treatment resulted in no recurrences in 46 HER2/neu 3+ over-expressor patients who were fully treated with GP2 versus 50 placebo patients who were treated with GMCSF-Herceptin and who recurred at a rate similar to historical recurrence rates for patients treated with Herceptin. After median 5 years of follow-up, there were 0% cancer recurrences in the HER2/neu 3+ patients treated with GP2-GMCSF-Herceptin, if the patient received the 6 primary intradermal injections over the first 6 months, versus an 11% cancer recurrence rate in the placebo arm treated with GMCSF-Herceptin (p = 0.0338). Thus, sequentially combining Herceptin in year 1 and GP2-GMCSF in years 2-4 may dramatically lower breast cancer recurrences in this patient population.
| 9 |
The design of the Phase IIb trial was as follows:
| ● | Prospective, randomized, single-blinded, placebo-controlled phase IIb clinical trial of GP2 + GM-CSF or GM-CSF alone in HER2/neu 1-3+, HLA-A02 patients. | |
| ● | High-risk breast cancer patients (Node Positive, High Risk Node Negative) who were disease-free and immunocompetent after having completed standard of care therapy. | |
| ● | The primary endpoint was to determine if GP2 + GM-CSF reduces breast cancer recurrence rates versus GM-CSF alone. A recurrence is defined as either a pathologically confirmed recurrence or a new radiographic finding during standard of care follow-up. |

The Phase IIb clinical trial closed in December 2018. The final median 5 year follow-up data from this Phase IIb clinical trial is currently being collected and analyzed.
| GP2
+ GM-CSF Treated Patients Recurrence Rate | GM-CSF
Placebo Patients Recurrence Rate | Hazard Ratio | Kaplan-Meier
Survival Analysis | |||||||||
| 0.0% | 11.0% | 0.00 | p = 0.0338 | |||||||||
Of the total 180 intent-to-treat patients enrolled, 168 patients completed the 6 primary intradermal injection series over the first 6 months. HER2/neu status was determined based on the expression levels of the HER2/neu protein in each patient using standard of care HER2/neu diagnostic technology. The trial was prospectively designed to analyze these fully treated patients by 2 distinct patient populations, namely HER2/neu 3+ (over expressors) and HER2/neu 1-2+ (low to intermediate expressors):
| ● | HER2/neu 3+ Over Expressors: In the 96 HER2/neu 3+, HLA-A02 patients, no recurrences were observed if the patient received the 6 primary intradermal injections over the first 6 months following the first year of Herceptin treatment. This is the target population for our planned Phase III trial. | |
| ● | HER2/neu 1-2+ Low to Intermediate Expressors: In the 72 HER2/neu 1-2+, HLA-A02 patients, no reduction in recurrence rates were observed, but Herceptin was not administered to these patients. Thus, we may pursue a future trial with GP2 in combination with Herceptin therapy. |
| 10 |
San Antonio Breast Cancer Symposium Poster Presentation of Median 5 Year Top-Line Data
The median 5 year top-line data described below was presented at the San Antonio Breast Cancer Symposium in a poster on December 9, 2020, entitled “Five year median follow-up data from a prospective, randomized, placebo-controlled, single-blinded, multicenter, phase IIb study evaluating the reduction of recurrences using HER2/neu peptide GP2 + GM-CSF vs. GM-CSF alone after adjuvant trastuzumab in HER2 positive women with operable breast cancer.”
The final analysis of the GP2 prospective, randomized, placebo-controlled, single-blinded, multicenter Phase IIb trial investigating GP2+GM-CSF administered in the adjuvant setting to node-positive and high-risk node-negative breast cancer patients with tumors expressing any degree of HER2 (immuno-histochemistry [IHC] 1-3+) (NCT00524277) is now complete with 5 year follow-up. The trial enrolled HLA-A02 patients randomized to receive GP2+GM-CSF versus GM-CSF alone. The trial’s primary objective was to determine if treatment with GP2, a HER2-derived peptide, reduces recurrence rates.
Each enrolled and consented subject was randomized and scheduled to receive a total of 6 GP2+GM-CSF (500 mcg GP2:125 mcg GM-CSF) or placebo (125 mcg GM-CSF alone) intradermal injections every 3-4 weeks as part of the Primary Immunization Series (“PIS”) for the first 6 months and 4 GP2+GM-CSF booster or placebo intradermal injections every 6 months thereafter. Boosters were introduced during the trial, thus some patients did not receive all 4 boosters.
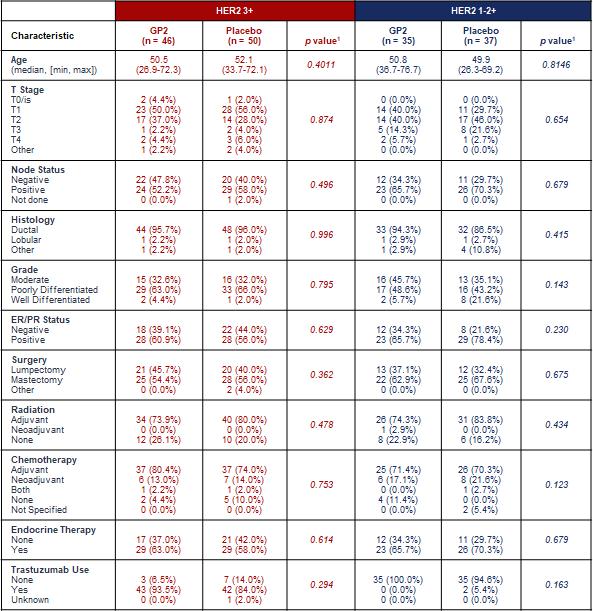
This 168 patient (Intent to Treat, “ITT”: n=180) basket trial across 16 clinical sites explored 96 HER2 3+ patients, who received a standard course of trastuzumab after surgery and subsequently completed the full PIS or placebo, starting the PIS at median 17.1 months after surgery, and 72 HER2 1-2+ patients, who did not receive trastuzumab after surgery and subsequently completed the full PIS or placebo, starting the PIS at median 10.8 months after surgery. Subject disease characteristics are described in Table 1.
Since GP2 is synergistic with trastuzumab, and the HER2 1-2+ patients did not receive trastuzumab, it was prespecified to compare recurrence rates ITT versus per protocol in these 2 distinct, independently reported populations, excluding those patients who did not complete the PIS. Figure 1 depicts evidence that disease free survival (“DFS”) is more likely in HER2 3+ GP2-treated subjects (p = 0.0338). Figure 2 provides DFS for the HER2 1-2+ group.
GP2 was shown to be well tolerated with no SAEs and elicited a potent immune response measured by local skin tests and immunological assays, which suggest peak immunity is reached at 6 months upon completion of the PIS.
Table 1: Clinicopathologic Characteristics by Treatment Group for HER2 3+ and HER2 1-2+ Subjects Who Completed the PIS(1)
(1) Continuous variables difference between treatment groups assessed by t-test. Categorical variables difference between treatment group distribution assessed by chi-square test.
| 11 |

As shown above, after 5 years of follow-up, the Kaplan-Meier estimated 5-year DFS rate in the 46 HER2 3+ patients treated with GP2+GM-CSF, if the patient completed the PIS, was 100% versus 89.4% (95% CI:76.2, 95.5%) in the 50 placebo patients treated with GM-CSF (p = 0.0338). As shown in Table 1, the treated versus placebo HER2 3+ patients were well-matched, where approximately 53% were stage T1, 41% were stages T2-T4, 55% were node positive, 58% were hormone receptor positive and received endocrine therapy, 77% received adjuvant radiation, 77% received adjuvant chemotherapy, and 89% received trastuzumab.
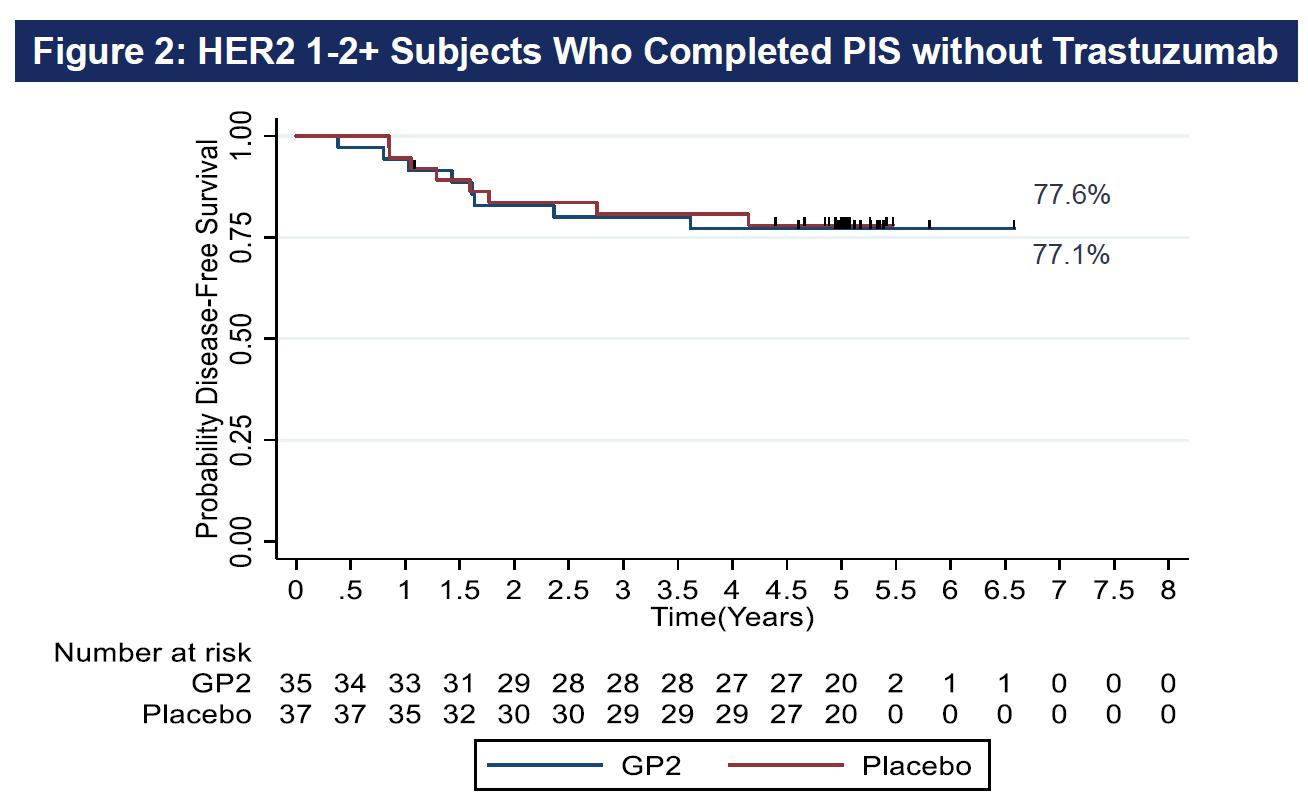
As shown above, after 5 years of follow-up, the Kaplan-Meier estimated 5-year DFS rate in the 35 HER2 1-2+ patients treated with GP2+GM-CSF, if the patient completed the PIS, was 77.1% (95% CI:59.5, 87.9%) versus 77.6% (95% CI:60.1, 88.2%) in the 37 placebo patients treated with GM-CSF (p = 0.9142).
Safety & Immune Response Data of GP2 Phase II Trial – Median 3 Year Data
A median 3 year interim analysis of the GP2 Phase II trial was published in 2016 and presented efficacy, safety, and immunological data, and a median 4 year interim analysis of the GP2 Phase II trial was published in April 2020. The safety and immunological data is shown below.
| 12 |
In both patient populations, GP2 was shown to be well tolerated, consisting of primarily injection site reactions which are caused by GM-CSF and can be mitigated by reducing the GM-CSF dose (and then the GP2 dose, if necessary). No SAEs were reported in the GP2 treated patients. Maximum local and systemic toxicities were primarily grade 1 and grade 2. Toxicities ranged from redness at injection site to flu-like symptoms and can be largely attributed to GM-CSF, and not to GP2.

Toxicity: The maximum local and systemic toxicity experienced by patients administered the GP2+GM-CSF vaccine were comparable to those experienced by patients receiving GM-CSF alone. For patients receiving GP2 + GM-CSF, maximum local toxicities experienced during the primary vaccination series were grade 1 (70%), grade 2 (28%), or grade 3 (1%). The most common toxicities included erythema, induration and pruritis; the grade 3 toxicity was induration. Maximum systemic toxicities were grade 0 (13%), grade 1 (71%), grade 2 (15%), or grade 3 (1%). The most common systemic toxicities included fatigue, headache, and myalgias. The grade 3 toxicity was a diffuse maculopapular rash. The toxicities were comparable for patients receiving GM-CSF only, with maximum local toxicities being grade 1 (75%) or grade 2 (25%); and maximum systemic toxicities being grade 0 (21%), grade 1 (60%), grade 2 (15%), or grade 3 (3%). The grade 3 systemic toxicities in this group included diffuse urticarial reactions, syncope and extremity pain.
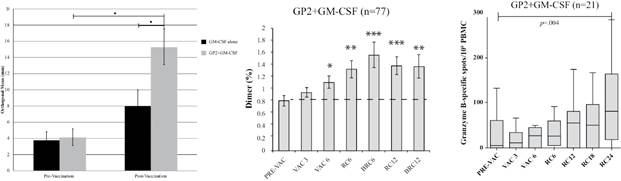
GP2 immunotherapy elicited a potent immune response in HLA-A02 patients after they received the 6 primary intradermal injections over the first 6 months. The immune response was measured by a local skin test and immunological assays. Further, booster injections given every 6 months thereafter prolonged the immune response, thereby providing longer term protection.
| ● | Immune response was observed peaking after 6 months compared to baseline, measured by Delayed Type Hypersensitivity ((“DTH”) skin test using GP2) and immunological assay. DTH response rate for treated patients is very high. Orthogonal mean baseline versus six months: 4.1±1.1mm versus 15.3± 2.2mm (± standard error). | |
| ● | Boosters were administered every 6 months to sustain immunity. |
| 13 |
Planned Phase III Trial
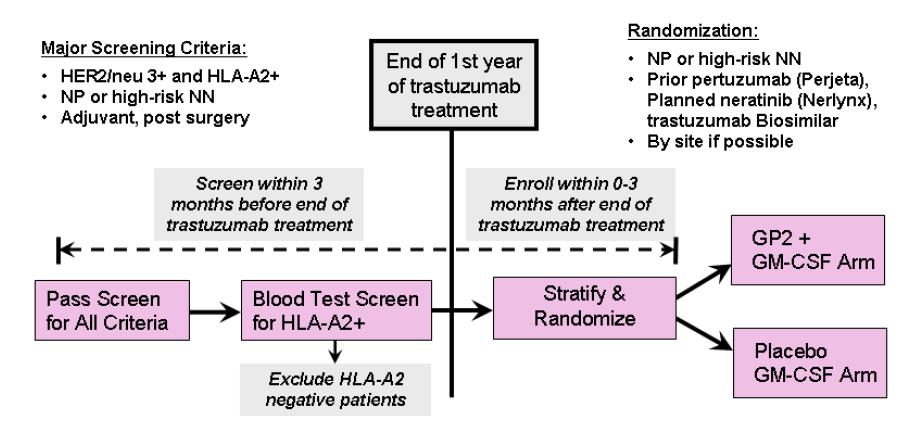
We are planning to launch a Phase III clinical trial in 2021, using a similar treatment regime as the Phase IIb clinical trial. The manufacturing plan and the Phase III trial protocol have been reviewed by the FDA, and final revisions to the Phase III trial protocol are under way, which may include an interim analysis/adaptive trial design that will result in the finalization of the size of the trial. The primary endpoint of the Phase III clinical trial will compare recurrence rate of GP2 + GM-CSF treated patients versus placebo patients at various time points using standard of care follow-up. We believe that it may require up to 2 years to fully enroll all patients for the trial, and that we may follow-up patients for up to a median 5 years following enrollment in such trial; however the addition of an interim analysis may reduce the time required to report clinical data and to file a BLA application. These design features of the Phase III clinical trial are currently being finalized by our clinical advisors.
An overview of the Phase III clinical trial design is shown below.
We have commenced GP2 manufacturing, and we are currently in the process of finalizing our engagement of CMOs and CROs for the Phase III clinical trial.
Large Initial & Expandable Breast Cancer Market
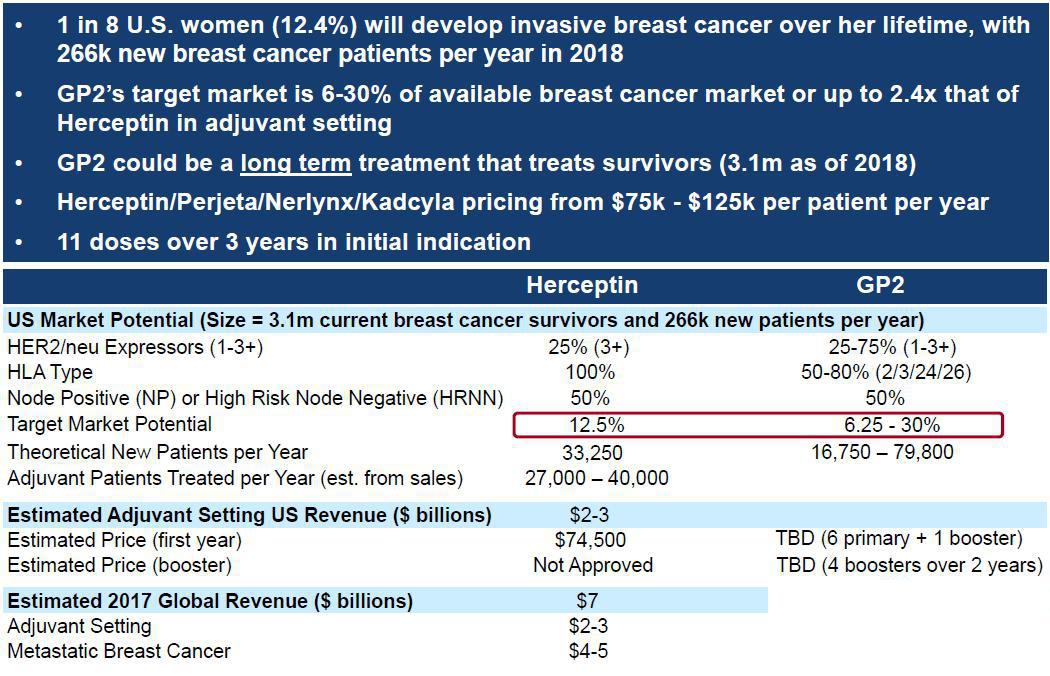
We believe that the potential market for the proposed initial and follow-on indications is large. HER2/neu 3+ breast cancer patients comprise approximately 25% of all breast cancer patients. Approximately 40% to 50% of the U.S. population contains the HLA-A02 allele, while node positive and high risk node negative patients comprise approximately 50% of the market. Therefore, we believe that the initial market for GP2 could be the combination of the three populations above which together comprises 6% of breast cancer patients. We believe that follow-on indications could include additional HLA types (an additional 30% of the U.S. population) and the low to intermediate expressors of HER2/neu 1-2+ patients (an additional 50% of all breast cancer patients) which would expand the GP2 market from our estimated initial 6% to 30% of breast cancer patients who undergo surgery. Thus the market for GP2, including follow-on indications, could be 2.4 times the current Herceptin adjuvant setting market, which constitutes approximately 12.5% of breast cancer patients.
| 14 |
We believe that the potential market for GP2 could be estimated as follows, with the long term multi-billion dollar annual revenue potential of GP2 based on 16,750 to 79,800 potential new patients treated per year and Herceptin’s 2018 annual per patient price of $74,500:
Competition
Cancer immunotherapy has become a significant growth area for the biopharmaceutical industry, attracting large pharmaceutical companies as well as small niche players. Generally, our principal competitors in the cancer immunotherapy market comprise both types of companies with currently approved products for various indications, such as manufacturers of approved bispecific antibodies, CAR-T cells, and checkpoint inhibitors, as well as companies currently engaged in cancer immunotherapy clinical development. The large and medium-size players who have successfully obtained approval for cancer immunotherapy products include Bristol-Myers Squib Company, Merck & Co., Inc., Genentech, Inc. (a subsidiary of Roche Holding AG), AstraZeneca PLC, Celgene Corporation, Johnson & Johnson, Amgen, Novartis, Juno Therapeutics, Inc. (a subsidiary of Celgene), Kite Pharma, Inc., a wholly-owned subsidiary of Gilead Sciences, Inc. and Pfizer, Inc./EMD Serono, Inc. Most of these companies, either alone or together with their collaborative partners, have substantially greater financial resources than we do.
Companies developing novel products with similar indications to those we are pursuing are expected to influence our ability to penetrate and maintain market share. For patients with early stage breast cancer, adjuvant therapy is often given to prevent recurrence and increase the chance of long-term disease free survival. Adjuvant therapy for breast cancer can include chemotherapy, hormonal therapy, radiation therapy, or combinations thereof. In addition, the HER2 targeted drug Herceptin (trastuzumab) alone or in combination with Perjeta (pertuzumab), both manufactured and marketed by Roche/Genentech, may be given to patients with tumors with high expression of HER2/neu.
| 15 |
There are a number of approved HER2/neu targeted therapies, some of which include the following: Genentech’s Herceptin, Perjeta and Kadcyla (TDM-1, ado-trastuzumab emtansine); Puma’s Nerlynx; Daichi Sanko’s Enhertu (DS-8201, fam-trastuzumab deruxtecan-nxki), and Seattle Genetics’ (Tukysa, tucatanib). In addition, the following biosimilars to trastuzumab have been approved: Biocon/Mylan’s (Ogivri — trastuzumab-dkst; Celltrion/Teva’s (Herzuma — trastuzumab-pkrb); Samsung/Biogen/Merck’s (Ontruzant — trastuzumab-dttb); Pfizer’s (Trazimera — trastuzumab-qyyp); and Allergan/Amgen’s (Kanjinti; trastuzumab-anns). Furthermore, the following immune checkpoint inhibitors have also been approved or are under review by the FDA to treat breast cancer patients: Merck’s Keytruda (pembrolizumab) and Genentech’s Tecentriq (atezolumab). Moreover we believe that drug candidates from Sellas (formerly Galena), Marker (formerly TapImmune), Epithany, Antigen Express (Generex subsidiary), and various companies pursuing neoantigen technologies are in clinical development and are being pursued for different sub-populations or are behind GP2 in clinic development.
We believe that GP2 will act synergistically with Herceptin, Perjeta, Nerlynx, and the newest entrants Kadcyla and Enhertu.

 |
 |
| 16 |
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do, and more experience in obtaining FDA and other regulatory approvals of treatments and in commercializing those treatments. Accordingly, our competitors may be more successful than us in obtaining approval for cancer immunotherapy products and achieving widespread market acceptance. Our competitors’ treatments may be more effectively marketed and sold than any products we may commercialize, thus causing limited market share before we can recover the expenses of developing and commercializing our cancer immunotherapy product candidate.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These activities may lead to consolidated efforts that allow for more rapid development of cancer immunotherapy product candidates.
These competitors also compete with us in the recruiting and retaining of qualified scientific and management personnel, the ability to work with specific clinical contract organizations due to conflict of interest, and the conduct of trials in the ability to recruit clinical trial sites and subjects for our clinical trials.
We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, price, and the availability of coverage and reimbursement from government and other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are viewed as safer, more convenient, or less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our current product candidate or any other future product candidate, which could result in our competitors establishing a strong market position before we are able to enter the market.
Manufacturing
We do not own or operate manufacturing facilities for the production of our product candidate nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently depend on third-party contract manufacturers for all of our required raw materials, active pharmaceutical ingredients (“APIs”), and finished product candidate for our clinical trials. We do not have any current contractual arrangements for the manufacture of commercial supplies of our product candidate.
For prior clinical trials, GP2 was formulated, filled, labeled, stored, tested, packaged, and distributed to clinical sites by the pharmacy at the Walter Reed Medical Center and the HJF. For future clinical trials, we anticipate that GP2 will be formulated, filled, labeled, stored, tested, packaged, and distributed to clinical sites in licensed cGMP manufacturing facilities as we evaluate and select primary and secondary facilities which may also serve as commercial facilities.
Exclusive License
The Henry M. Jackson Foundation out-licenses technology of the United States military and it conducts research and manages clinical trials. HJF managed the GP2 Phase IIb clinical which was led by MD Anderson Cancer Center, oversaw all regulatory filings with the FDA for all 4 GP2 clinical trials (including the three Phase I and the Phase IIb clinical trials), and possesses all patient and manufacturing data from such trials.
In April 2009, we entered into an exclusive license agreement, as amended, with HJF pursuant to which HJF granted us exclusive worldwide rights to several U.S. and foreign patents and patent applications covering methods of using GP2 as an immunotherapy that elicits a targeted immune response against HER2/neu-expressing cancers. In consideration for such licensed rights, we issued HJF 202,619 shares of our common stock. In addition, we are required to pay an annual maintenance fee and milestone payments of up to an aggregate of $5.7 million. We are also required to make 2.5-5% royalty payments based on the sales of GP2 and to reimburse HJF for patent expenses. To date we have not been required to make any milestone or royalty payments to HJF. The term of the exclusive license shall terminate at such time that the last licensed patent or patent application expires or is abandoned, unless terminated earlier pursuant to the terms of the exclusive license agreement. We may terminate the license by giving 90 days notice. HJF may terminate the license if we do not make required payments, if we default in our performance obligations, if we do not sufficiently develop and advance GP2 towards commercialization, and for various other reasons.
| 17 |
In connection with the exclusive license agreement with HJF, we were the financial and corporate sponsors of the GP2 Phase IIb clinical trial. HJF has provided us with all FDA correspondences and GP2 patient and manufacturing data for the history of the drug’s development for all 4 clinical trials, and we have incorporated this data into our corporate investigational new drug application (“IND”) with the FDA.
Intellectual Property Portfolio
Our commercial success depends in part on our ability to avoid infringing the proprietary rights of third parties, our ability to obtain and maintain proprietary protection for our technologies where applicable, and our ability to prevent others from infringing our proprietary rights. We intend to protect our proprietary technologies by, among other methods, evaluating relevant patents, establishing defensive positions, monitoring European Union oppositions and pending intellectual property rights, preparing litigation strategies in view of the U.S. legislative framework, and filing U.S. and international patent applications on technologies, inventions and improvements that are important to our business. Patents and other intellectual property rights are crucial to our success. We intend to protect our intellectual property rights through available means including filing and prosecuting patent applications in the U.S. and other countries, protecting trade secrets, and utilizing regulatory protections such as data exclusivity. In addition, we include restrictions regarding use and disclosure of our proprietary information in our contracts with third parties, and utilize customary confidentiality agreements with our employees, consultants, clinical investigators, and scientific advisors to protect our confidential information and know-how. Together with our licensors, we also rely on trade secrets to protect our combined technology especially where we do not believe patent protection is appropriate or obtainable. It is our policy to operate without knowingly infringing on, or misappropriating, the proprietary rights of others.
An international patent law treaty (“PCT”) provides a unified procedure for filing patent applications to protect inventions in each of its contracting states. Thus, a single PCT application can be converted into a national stage patent application in any of the more than 145 PCT contracting states, and is considered a simple, cost-effective means for seeking patent protection in numerous regions or countries. This nationalization (converting into an application in any of the contracting states) typically occurs 18 months after the PCT application filing date. We also rely on trade secrets, know-how, and continuing technological innovation to develop and maintain our proprietary position.
The term of individual patents depends upon the legal term of the patents in countries in which they are obtained. In most countries, including the U.S., the patent term is generally 20 years from the earliest date of filing a non-provisional patent application in the applicable country. In the U.S., a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in examining and granting a patent or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
HJF License
Pursuant to our exclusive license agreement with HJF, we were granted exclusive worldwide rights to several U.S. and foreign patents and patent applications covering methods of using GP2. The GP2 issued patents provide protection ranging from 2026 through 2032 in major markets such as the U.S., Europe, Japan, Australia, and Canada, with ongoing prosecution of pending patent applications in other markets. We plan to register GP2 as a biologic, which may be subject to 10-12 years market exclusivity in the U.S. upon receiving marketing approval.
The following summarizes the two patent families subject to our exclusive license agreement with HJF. We have licensed rights to issued patents and pending patent applications in certain countries with respect to the two patent families below and do not own or have rights to any other patents or patent applications for GP2 or any other products:
| ● | GP2 + GM-CSF Patent Family — A patent application has been filed and licensed describing methods and compositions for the induction of a cytotoxic T-cell response to the GP2 peptide with the effect of inducing and maintaining a protective or therapeutic immunity against breast cancer. Patent claims describe the use of the GP2 technology including dosing, formulation, identification of patients, and use in combination with GM-CSF. Patents issued in the U.S. will expire in 2032 and 2029 and international patents will expire in 2029. |
| 18 |
| ● | GP2 + Herceptin Patent Family — A patent application has been filed and licensed describing methods and compositions of GP2 peptide in combination with a HER2/neu targeting antibody such as Herceptin. U.S. and certain foreign patent claims describe the method and timing of administration. Patents issued in the U.S. will expire in 2028 and 2026 and international patents will expire in 2026. |
Corporate Strategy
We do not have a sales, marketing, or product distribution strategy for our GP2 immunotherapy or any future product candidates because GP2 is still in clinical development. Our future commercial strategy may include the use of strategic partners, distributors, a contract sales force, or the establishment of our own commercial and specialty sales force for the U.S. market, as well as similar strategies for regions and territories outside the U.S. We plan to further evaluate these options as we approach approval for the use of our product candidate for one or more indications.
The GP2 issued patents provide protection ranging from 2026 through 2032 in various markets, and we plan to register GP2 as a biologic, which may be subject to 10-12 years market exclusivity in the U.S. upon receiving marketing approval. During this period of exclusivity, we intend to advance GP2 into a Phase III clinical trial in the U.S. and pursue a European and global clinical trial strategy to support GP2 registration outside of the U.S. We are considering various options to fund the Phase III clinical trial including financing and/or strategic transactions. Our strategy during such time also includes building a commercialization team, pursuing additional funding after this offering, and pursuing strategic collaborations to support the future global marketing and sales of GP2. A long term global and regional licensing process has been initiated and will continue as the Phase III trial commences.
Pipeline Strategy — Including GP2 In Other HER2/neu-Expressing Cancers
We are developing follow-on indications for GP2 by designing and planning additional clinical trials to expand the breast cancer patient population and to pursue additional HER2/neu-expressing cancers. Pending the receipt of sufficient capital, the planned Phase III clinical trial can be supplemented with the following pipeline investments:
| ● | The efficacy of GP2-GMCSF-Herceptin can be explored in (1) other HLA patients in the same HER2/neu 3+ breast cancer patient population, (2) breast cancer patients who are low to intermediate expressors of HER2/neu (1-2+) and who comprise two-thirds of the triple negative market, or (3) other HER2/neu-expressing cancers including, but not limited to, ovarian, gastrointestinal, and colon cancers. | |
| ● | We may acquire a preclinical platform that can be quickly advanced into IND-enabling GMP manufacturing and GLP toxicology studies followed by initial human clinical trials. |
Government Regulations
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of biologics such as those we are developing. Along with third-party contractors, we will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our current product candidate or any future product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label.
The process required by the FDA before biologic product candidates may be marketed in the U.S. generally involves the following:
| ● | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices, or GLP, regulations; | |
| ● | submission to the FDA of an IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| 19 |
| ● | approval by an independent IRB or ethics committee at each clinical site before the trial is begun; | |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed biologic product candidate for its intended purpose; | |
| ● | preparation of and submission to the FDA of a BLA, after completion of all pivotal clinical trials; | |
| ● | satisfactory completion of an FDA Advisory Committee review, if applicable; | |
| ● | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; | |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with cGMP and to assure that the facilities, methods and controls are adequate to preserve the biological product’s continued safety, purity and potency, and of selected clinical investigations to assess compliance with GCP; and | |
| ● | FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the U.S., which must be updated annually when significant changes are made. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our current product candidate or any future product candidates will be granted on a timely basis, if at all. Prior to beginning the first clinical trial with a product candidate, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site and must monitor the clinical trial until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by a Data and Safety Monitoring Board, or DSMB, organized by the clinical trial sponsor, which provides authorization for whether or not a clinical trial may move forward at designated check points based on access to certain data from the clinical trial and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical trial results to public registries.
For purposes of BLA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| ● | Phase 1 — The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. |
| 20 |
| ● | Phase 2 — The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. | |
| ● | Phase 3 — The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. | |
| ● | Phase 4 — In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be made a condition to approval of the BLA. |
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product, or for biologics, the safety, purity and potency. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
BLA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a BLA requires payment of a substantial user fee to FDA, and the sponsor of an approved BLA is also subject to annual product and establishment user fees. These fees are typically increased annually. A waiver of user fees may be obtained under certain limited circumstances.
Once a BLA has been submitted, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and we may encounter difficulties or unanticipated costs in its efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our product. After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may request additional information or clarification. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
| 21 |
If regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our product under development.
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a product candidate with Fast Track designation, the FDA may consider sections of the BLA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A Fast Track designated product candidate may also qualify for priority review, under which the FDA sets the target date for FDA action on the BLA at six months after the FDA accepts the application for filing. Priority review is granted when there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the Accelerated Approval program, the FDA may approve a BLA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the biologic’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit.
In addition, a sponsor may seek FDA designation of its product candidate as a Breakthrough Therapy, if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application. Breakthrough designation also allows the sponsor to file sections of the BLA for review on a rolling basis.
Fast Track, Priority Review and Breakthrough Therapy designations do not change the standards for approval but may expedite the development or approval process.
Other Healthcare Laws and Compliance Requirements
Our sales, promotion, medical education and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the U.S. in addition to FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services, other divisions of the Department of Health and Human Services and state and local governments. Our promotional and scientific/educational programs must comply with the federal Anti-Kickback Statute, the Foreign Corrupt Practices Act, the False Claims Act, or FCA, the Veterans Health Care Act, physician payment transparency laws, privacy laws, security laws, and additional state laws similar to the foregoing.
| 22 |
The federal Anti-Kickback Statute prohibits, among other things, the offer, receipt, or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham research or consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. Many states have similar laws that apply to their state health care programs as well as private payors.
The FCA imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. Actions under the FCA may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the FCA can result in significant monetary penalties and treble damages. The federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multibillion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, companies have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or corporate integrity agreements, restricting the manner in which they conduct their business. The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created federal criminal statutes that prohibit, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposed new reporting requirements on drug manufacturers for payments or other transfers of value made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties. Certain states also mandate implementation of commercial compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare professionals.
We may also be subject to data privacy and security regulation by both the federal government and the states in which it conducts its business. HIPAA, as amended by HITECH, and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect.
| 23 |
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to it, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Also, the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to foreign officials for the purpose of obtaining or retaining business. We cannot assure you that our internal control policies and procedures will protect us from reckless or negligent acts committed by our employees, future distributors, partners, collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on our business, results of operations and reputation.
Coverage and Reimbursement
Sales of pharmaceutical products depend significantly on the availability of third-party coverage and reimbursement. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. Although we currently believe that third-party payors will provide coverage and reimbursement for our product candidate, if approved, these third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of our product candidate. Seeking coverage and reimbursement from third-party payors can be time consuming and expensive. Moreover, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Reimbursement may not be available or sufficient to allow us to sell our product on a competitive and profitable basis.
Foreign Regulation
In addition to regulations in the U.S., we are and will be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and commercial sales and distribution of our product, if approved.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-U.S. countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the U.S. have processes that require the submission of a clinical trial application much like an IND prior to the commencement of human clinical trials. In Europe, for example, a clinical trial application, or CTA, must be submitted to the competent national health authority and to independent ethics committees in each country in which a company plans to conduct clinical trials. Once the CTA is approved in accordance with a country’s requirements, clinical trials may proceed in that country.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country, even though there is already some degree of legal harmonization in the European Union member states resulting from the national implementation of underlying E.U. legislation. In all cases, the clinical trials are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of a new drug or medicinal product in the European Union, a sponsor must obtain approval of a marketing authorization application. The way in which a medicinal product can be approved in the European Union depends on the nature of the medicinal product.
The centralized procedure results in a single marketing authorization granted by the European Commission that is valid across the European Union, as well as in Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human drugs that are: (i) derived from biotechnology processes, such as genetic engineering, (ii) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions and viral diseases, (iii) officially designated as “orphan drugs” and (iv) advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines. The centralized procedure may, at the request of the applicant, also be used for human drugs which do not fall within the above mentioned categories if the human drug (a) contains a new active substance which was not authorized in the European Community; or (b) the applicant shows that the medicinal product constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization in the centralized procedure is in the interests of patients or animal health at the European Community level.
| 24 |
Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application by the EMA is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Committee for Medicinal Products for Human Use, or CHMP), with adoption of the actual marketing authorization by the European Commission thereafter. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest from the point of view of therapeutic innovation, defined by three cumulative criteria: the seriousness of the disease to be treated; the absence of an appropriate alternative therapeutic approach, and anticipation of exceptional high therapeutic benefit. In this circumstance, EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days and the opinion issued thereafter.
The mutual recognition procedure, or MRP, for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the European Union. The MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is applicable to the majority of conventional medicinal products, and is based on the principle of recognition of an already existing national marketing authorization by one or more member states.
The characteristic of the MRP is that the procedure builds on an already existing marketing authorization in a member state of the E.U. that is used as reference in order to obtain marketing authorizations in other E.U. member states. In the MRP, a marketing authorization for a drug already exists in one or more member states of the E.U. and subsequently marketing authorization applications are made in other European Union member states by referring to the initial marketing authorization. The member state in which the marketing authorization was first granted will then act as the reference member state. The member states where the marketing authorization is subsequently applied for act as concerned member states.
The MRP is based on the principle of the mutual recognition by European Union member states of their respective national marketing authorizations. Based on a marketing authorization in the reference member state, the applicant may apply for marketing authorizations in other member states. In such case, the reference member state shall update its existing assessment report about the drug in 90 days. After the assessment is completed, copies of the report are sent to all member states, together with the approved summary of product characteristics, labeling and package leaflet. The concerned member states then have 90 days to recognize the decision of the reference member state and the summary of product characteristics, labeling and package leaflet. National marketing authorizations shall be granted within 30 days after acknowledgement of the agreement.
Should any Member State refuse to recognize the marketing authorization by the reference member state, on the grounds of potential serious risk to public health, the issue will be referred to a coordination group. Within a timeframe of 60 days, member states shall, within the coordination group, make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the Commission, for the start of the decision-making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products or Veterinary Medicinal Products, as appropriate.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the other applicable regulatory requirements.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
| 25 |
Employees
As of March 15, 2021 we had 1 full-time employee and 2 part-time employees. We are not a party to any collective bargaining agreements. We believe that we maintain good relations with our employees.
An investment in our securities involves a high degree of risk. An investor should carefully consider the risks described below as well as other information contained in this Annual Report on Form 10-K and our other reports filed with the U.S. Securities and Exchange Commission (“SEC”). The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our securities could decline, and investors in our company may lose all or part of their investment.
Risks Relating to Our Financial Position and Capital Needs
We have incurred substantial losses since our inception and anticipate that we will continue to incur substantial and increasing losses for the foreseeable future.
We are a clinical stage biopharmaceutical company focused on the development of our novel cancer immunotherapy GP2, for breast cancer and potentially for a broad range of other HER2/neu-expressing cancers. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that a product candidate will fail to prove effective, gain regulatory approval or become commercially viable. We do not have any products approved by regulatory authorities and have not generated any revenues from collaboration and licensing agreements or product sales to date, and have incurred significant research, development and other expenses related to our ongoing operations and expect to continue to incur such expenses. As a result, we have not been profitable and have incurred significant operating losses since our inception. For the years ended December 31, 2020 and 2019, we reported a net loss of $1.9 million and $3.4 million, respectively. As of December 31, 2020, we had an accumulated deficit of $29.0 million.
We do not expect to generate revenues for many years, if at all. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses to increase as we continue to research, develop and seek regulatory approvals for our product candidate and any additional product candidates we may acquire, and potentially begin to commercialize product candidates that may achieve regulatory approval. We may also encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. Our expenses will further increase as we:
| ● | conduct clinical trials of our lead product candidate, GP2; | |
| ● | in-license or acquire the rights to, and pursue development of, other products, product candidates or technologies; | |
| ● | hire additional clinical, manufacturing, quality control, quality assurance and scientific personnel; | |
| ● | seek marketing approval for any product candidates that successfully complete clinical trials; | |
| ● | develop our outsourced manufacturing and commercial activities and establish sales, marketing and distribution capabilities, if we receive, or expect to receive, marketing approval for any product candidates; | |
| ● | maintain, expand and protect our intellectual property portfolio; and | |
| ● | add operational, financial and management information systems and personnel. |
| 26 |
We need significant additional financing to fund our operations and complete the development and, if approved, the commercialization of our product candidate. If we are unable to raise capital when needed, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts.
We expect our existing cash as of December 31, 2020 will enable us to fund our operating expenses through and capital expenditure requirements for at least twelve months from the date of this Annual Report on Form 10-K; however, our existing cash will not be sufficient to complete development and obtain regulatory approval for our product candidate, and we will need to raise significant additional capital to help us do so. In addition, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned.
We expect to expend substantial resources for the foreseeable future to continue the clinical development and manufacturing of our product candidate and the advancement and expansion of our preclinical research pipeline. These expenditures will include costs associated with research and development, potentially acquiring new product candidates or technologies, conducting preclinical studies and clinical trials and potentially obtaining regulatory approvals and manufacturing products, as well as marketing and selling products approved for sale, if any.
We believe that it may cost approximately $12 million to $15 million to complete an interim analysis of the safety and efficacy of our Phase III trial. Furthermore, the total cost to complete an interim analysis and file a BLA application for drug approval in the U.S. could exceed $16 million, and the total cost to complete our Phase III trial as planned could exceed $30 million; however, we believe that we have budget flexibility with respect to the design of the Phase III clinical trial. We believe that we may be able to alter the cost of our Phase III clinical trial by adjusting the enrollment rate, the number of patients, and/or the number of immunological assays. While our budget for such Phase III trial may be flexible, our ability to reduce or modify costs may be adversely effected by, among other things, unexpected or higher costs associated with the trial, time required to complete the trial and other factors that may be beyond our control. Our budgets and future capital requirements depend on many factors, including:
| ● | the scope, progress, results and costs of our ongoing and planned development programs for our product candidate, as well as any additional clinical trials we undertake to obtain data sufficient to seek marketing approval for our product candidate; | |
| ● | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidate if our clinical trials are successful; | |
| ● | the cost of commercialization activities for our product candidate, if our product candidate is approved for sale, including marketing, sales and distribution costs; | |
| ● | the cost of manufacturing our product candidate for clinical trials in preparation for regulatory approval, including the cost and timing of process development, manufacturing scale-up and validation activities; | |
| ● | our ability to establish and maintain strategic licensing or other arrangements and the financial terms of such agreements; | |
| ● | the costs to in-license future product candidates or technologies; | |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, expanding, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; | |
| ● | the costs in defending and resolving future derivative and securities class action litigation; | |
| ● | our operating expenses; and | |
| ● | the emergence of competing technologies or other adverse market developments. |
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. We have no committed source of additional capital. If adequate funds are not available to us on a timely basis, we may not be able to continue as a going concern or we may be required to delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for our product candidate or target indications, or delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize our product candidate.
We may consider strategic alternatives in order to maximize stockholder value, including financings, strategic alliances, acquisitions or the possible sale of the Company. We may not be able to identify or consummate any suitable strategic alternatives.
We may consider all strategic alternatives that may be available to us to maximize stockholder value, including financings, strategic alliances, acquisitions or the possible sale of the Company. We currently have no agreements or commitments to engage in any specific strategic transactions, and our exploration of various strategic alternatives may not result in any specific action or transaction. To the extent that this engagement results in a transaction, our business objectives may change depending upon the nature of the transaction. There can be no assurance that we will enter into any transaction as a result of the engagement. Furthermore, if we determine to engage in a strategic transaction, we cannot predict the impact that such strategic transaction might have on our operations or stock price. We also cannot predict the impact on our stock price if we fail to enter into a transaction.
| 27 |
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our product candidate on unfavorable terms to us.
We may seek additional capital through a variety of means, including through private and public equity offerings and debt financings, collaborations, strategic alliances and marketing, distribution or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, or through the issuance of shares under management or other types of contracts, or upon the exercise or conversion of outstanding derivative securities, the ownership interests of our stockholders will be diluted, and the terms of such financings may include liquidation or other preferences, anti-dilution rights, conversion and exercise price adjustments and other provisions that adversely affect the rights of our stockholders, including rights, preferences and privileges that are senior to those of our holders of common stock in the event of a liquidation. In addition, debt financing, if available, could include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, making capital expenditures, entering into licensing arrangements, or declaring dividends and may require us to grant security interests in our assets, including our intellectual property. If we raise additional funds through collaborations, strategic alliances, or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, product or product candidate or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may need to curtail or cease our operations.
We currently have no source of revenues. We may never generate revenues or achieve profitability.
Currently, we do not generate any revenues from product sales or otherwise. Even if we are able to successfully achieve regulatory approval for our product candidate, we do not know when we will generate revenues or become profitable, if at all. Our ability to generate revenues from product sales and achieve profitability will depend on our ability to successfully commercialize products, including our current product candidate, GP2, and other product candidates that we may develop, in-license or acquire in the future. Our ability to generate revenues and achieve profitability also depends on a number of additional factors, including our ability to:
| ● | successfully complete development activities, including the necessary clinical trials; | |
| ● | complete and submit either Biologics License Applications, or BLAs, or New Drug Applications, or NDAs, to the FDA and obtain U.S. regulatory approval for indications for which there is a commercial market; | |
| ● | complete and submit applications to foreign regulatory authorities; | |
| ● | obtain regulatory approval in territories with viable market sizes; | |
| ● | obtain coverage and adequate reimbursement from third parties, including government and private payors; |
| ● | set commercially viable prices for our product, if any; | |
| ● | establish and maintain supply and manufacturing relationships with reliable third parties and/or build our own manufacturing facility and ensure adequate, legally globally compliant manufacturing of bulk drug substances and drug products to maintain that supply; | |
| ● | develop distribution processes for our product candidate; | |
| ● | develop commercial quantities of our product candidate, once approved, at acceptable cost levels; obtain additional funding, if required to develop and commercialize our product candidate; | |
| ● | develop a commercial organization capable of sales, marketing and distribution for any products we intend to sell ourselves, in the markets in which we choose to commercialize on our own; | |
| ● | achieve market acceptance of our product; | |
| ● | attract, hire and retain qualified personnel; and | |
| ● | protect our rights in our intellectual property portfolio. |
Our revenues for any product candidate for which regulatory approval is obtained will be dependent, in part, upon the size of the markets in the territories for which it gains regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of our addressable disease patients is not as significant as our estimates, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenues from sales of such products, even if approved. In addition, we anticipate incurring significant costs associated with commercializing any approved product candidate. As a result, even if we generate revenues, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and may be forced to reduce our operations.
| 28 |
The Tax Cuts and Jobs Act could adversely affect our business and financial condition.
H.R. 1, “An Act to provide for reconciliation pursuant to title II and V of the concurrent resolution on the budget for fiscal year 2018,” informally entitled the Tax Cuts and Jobs Act (“Tax Act”) enacted on December 22, 2017, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a single rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted taxable income (except for certain small businesses), limitation of the deduction for net operating losses carried forward from taxable years beginning after December 31, 2017 to 80% of current year taxable income and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, elimination of U.S. tax on foreign earnings (subject to certain important exceptions), providing immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits (including reduction of tax credits under the Orphan Drug Act). Notwithstanding the reduction in the corporate income tax rate, the overall impact of the Tax Act is uncertain and our business and financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the Tax Act.
Our ability to use net operating losses to offset future taxable income may be subject to limitations.
As of December 31, 2020, we had federal net operating loss, or NOLs, carryforwards of approximately $5.6 million. Our NOLs generated in tax years ending on or prior to December 31, 2017 are only permitted to be carried forward for 20 years under applicable U.S. tax laws, and will begin to expire, if not utilized, beginning in 2027. These NOL carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under the Tax Act, federal NOLs incurred in tax years ending after December 31, 2017 may be carried forward indefinitely, but the deductibility of such federal NOLs is limited. It is uncertain if and to what extent various states will conform to the Tax Act, or whether any further regulatory changes may be adopted in the future that could minimize its applicability. In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, and certain corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in the ownership of its equity over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income may be limited.
Risks Related to the Development and Regulatory Approval of Our Product Candidate
Clinical-stage biopharmaceutical companies with product candidates in clinical development face a wide range of challenging activities which may entail substantial risk.
We are a clinical-stage biopharmaceutical company with a product candidate in clinical development. The success of our product candidate will depend on several factors, including the following:
| ● | designing, conducting and successfully completing preclinical development activities, including preclinical efficacy and IND-enabling studies, for our product candidate or product candidates we may, in the future, in-license or acquire; | |
| ● | designing, conducting and completing clinical trials for our product candidate with positive results; | |
| ● | receipt of regulatory approvals from applicable authorities; | |
| ● | obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidate; | |
| ● | making arrangements with third-party manufacturers, receiving regulatory approval of our manufacturing processes and our third-party manufacturers’ facilities from applicable regulatory authorities and ensuring adequate supply of drug product; | |
| ● | manufacturing our product candidate at an acceptable cost; | |
| ● | effectively launching commercial sales of our product candidate, if approved, whether alone or in collaboration with others; | |
| ● | achieving acceptance of our product candidate, if approved, by patients, the medical community and third-party payors; | |
| ● | effectively competing with other therapies; | |
| ● | if our product candidate is approved, obtaining and maintaining coverage and adequate reimbursement by third-party payors, including government payors, for our product candidate; | |
| ● | complying with all applicable regulatory requirements, including FDA current Good Clinical Practices (“GCP”), current Good Manufacturing Practices (“cGMP”), and standards, rules and regulations governing promotional and other marketing activities; | |
| ● | maintaining a continued acceptable safety profile of the product during development and following approval; and | |
| ● | maintaining and growing an organization of scientists and business people who can develop and commercialize our product and technology. |
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully develop and commercialize our product candidate, which could materially harm our business.
| 29 |
We may find it difficult to enroll patients in our clinical trials given the limited number of patients who have the diseases for which our product candidate is being studied which could delay or prevent the start of clinical trials for our product candidate.
Identifying and qualifying patients to participate in clinical trials of our product candidate is essential to our success. The timing of our clinical trials depends in part on the rate at which we can recruit patients to participate in clinical trials of our product candidate, and we may experience delays in our clinical trials if we encounter difficulties in enrollment. If we experience delays in our clinical trials, the timeline for obtaining regulatory approval of our product candidate will most likely be delayed.
Many factors may affect our ability to identify, enroll and maintain qualified patients, including the following:
| ● | eligibility criteria of our ongoing and planned clinical trials with specific characteristics appropriate for inclusion in our clinical trials; | |
| ● | design of the clinical trial; | |
| ● | size and nature of the patient population; | |
| ● | patients’ perceptions as to risks and benefits of the product candidate under study and the participation in a clinical trial generally in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating; | |
| ● | the availability and efficacy of competing therapies and clinical trials; | |
| ● | pendency of other trials underway in the same patient population; | |
| ● | willingness of physicians to participate in our planned clinical trials; | |
| ● | severity of the disease under investigation; | |
| ● | proximity of patients to clinical sites; | |
| ● | patients who do not complete the trials for personal reasons; and | |
| ● | issues with CROs and/or with other vendors that handle our clinical trials. |
We may not be able to initiate or continue to support clinical trials of our product candidate for one or more indications, or any future product candidates if we are unable to locate and enroll a sufficient number of eligible participants in these trials as required by the FDA or other regulatory authorities. Even if we are able to enroll a sufficient number of patients in our clinical trials, if the pace of enrollment is slower than we expect, the development costs for our product candidate may increase and the completion of our trials may be delayed or our trials could become too expensive to complete.
If we experience delays in the completion of, or termination of, any clinical trials of our product candidate, the commercial prospects of our product candidate could be harmed, and our ability to generate product revenue from any of our product candidate could be delayed or prevented. In addition, any delays in completing our clinical trials would likely increase our overall costs, impair product candidate development and jeopardize our ability to obtain regulatory approval relative to our current plans. Any of these occurrences may harm our business, financial condition, and prospects significantly.
The results of preclinical studies or earlier clinical trials are not necessarily predictive of future results. Our existing product candidate in clinical trials, and any other product candidates that may advance into clinical trials, may not have favorable results in later clinical trials or receive regulatory approval.
Success in preclinical studies and early clinical trials does not ensure that later clinical trials will generate adequate data to demonstrate the efficacy and safety of an investigational drug. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience than us, have suffered significant setbacks in clinical trials, even after seeing promising results in earlier preclinical studies or clinical trials.
Despite the results reported in earlier preclinical studies or clinical trials for our product candidate, we do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market our product candidate for a particular indication, in any particular jurisdiction. Efficacy data from prospectively designed trials may differ significantly from those obtained from retrospective subgroup analyses. If later-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for our product candidate may be adversely impacted. Even if we believe that we have adequate data to support an application for regulatory approval to market our current product candidate or any future product candidates, the FDA or other regulatory authorities may not agree and may require that we conduct additional clinical trials.
| 30 |
Clinical drug development involves a lengthy and expensive process with an uncertain outcome.
Clinical testing is expensive and can take many years to complete, with the outcome inherently uncertain. Failure can occur at any time during the clinical trial process. Before obtaining approval from regulatory authorities for the sale of our product candidate, we must conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidate in humans. Prior to initiating clinical trials, a sponsor must complete extensive preclinical testing of a product candidate, including, in most cases, preclinical efficacy experiments as well as IND-enabling toxicology studies. These experiments and studies may be time-consuming and expensive to complete. The necessary preclinical testing may not be completed successfully for a preclinical product candidate and a potentially promising product candidate may therefore never be tested in humans. Once it commences, clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products. We may experience numerous unforeseen events during drug development that could delay or prevent our ability to receive marketing approval or commercialize our product candidate. In particular, clinical trials of our product candidate may produce inconclusive or negative results. We have limited data regarding the safety, tolerability and efficacy of GP2 administered in combination with GM-CSF. Clinical trials also require the review and oversight of an institutional review board (“IRB”). An inability or delay in obtaining IRB approval could prevent or delay the initiation and completion of clinical trials, and the FDA may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB review and approval.
We may experience delays in our ongoing or future clinical trials, and we do not know whether planned clinical trials will begin or enroll subjects on time, will need to be redesigned or will be completed on schedule, if at all. There can be no assurance that the FDA will not put clinical trials of our product candidate on hold in the future. Clinical trials may be delayed, suspended or prematurely terminated for a variety of reasons, such as:
| ● | delay or failure in reaching agreement with the FDA or a comparable foreign regulatory authority on a clinical trial design that we are able to execute; | |
| ● | delay or failure in obtaining authorization to commence a trial or inability to comply with conditions imposed by a regulatory authority regarding the scope or design of a trial; | |
| ● | delay or failure in reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | delay or failure in obtaining IRB approval or the approval of other reviewing entities, including comparable foreign regulatory authorities, to conduct a clinical trial at each site; | |
| ● | withdrawal of clinical trial sites from our clinical trials or the ineligibility of a site to participate in our clinical trials; | |
| ● | delay or failure in recruiting and enrolling suitable subjects to participate in a trial; | |
| ● | delay or failure in subjects completing a trial or returning for post-treatment follow-up; | |
| ● | clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial; | |
| ● | inability to identify and maintain a sufficient number of trial sites, many of which may already be engaged in other clinical trial programs, including some that may be for the same indication; | |
| ● | failure of our third-party clinical trial managers, CROs, clinical trial sites, contracted laboratories or other third-party vendors to satisfy their contractual duties, meet expected deadlines or return trustworthy data; |
| ● | delay or failure in adding new trial sites; | |
| ● | interim results or data that are ambiguous or negative or are inconsistent with earlier results or data; | |
| ● | alteration of trial design necessitated by re-evaluation of design assumptions based upon observed data; | |
| ● | feedback from the FDA, the IRB or a comparable foreign regulatory authority, or results from earlier stage or concurrent preclinical studies and clinical trials, that might require modification to the protocol for a trial; | |
| ● | a decision by the FDA, the IRB, a comparable foreign regulatory authority, or us to suspend or terminate clinical trials at any
time for safety issues or for any other reason; | |
| ● | unacceptable risk-benefit profile, unforeseen safety issues or adverse side effects; | |
| ● | failure to demonstrate a benefit from using a product candidate; | |
| ● | difficulties in manufacturing or obtaining from third parties sufficient quantities of a product candidate to start or to use in clinical trials; | |
| ● | lack of adequate funding to continue a trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional studies or increased expenses associated with the services of our CROs and other third parties; or | |
| ● | changes in governmental regulations or administrative actions or lack of adequate funding to continue a clinical trial. |
| 31 |
If we experience delays in the completion or termination of any clinical trial of our product candidate, the approval and commercial prospects of our product candidate will be harmed, delaying our ability to generate product revenues from such product candidate and our costs will most likely increase. The required regulatory approvals may also be delayed, thereby jeopardizing our ability to commence product sales and generate revenues and the period of commercial exclusivity for our product may be decreased. Regulatory approval of our product candidate may be denied for the same reasons that caused the delay.
Risks associated with operating in foreign countries could materially adversely affect our product development.
We may conduct future studies in countries outside of the U.S. Consequently, we may be subject to risks related to operating in foreign countries. Risks associated with conducting operations in foreign countries include:
| ● | differing regulatory requirements for drug approvals and regulation of approved drugs in foreign countries; more stringent privacy requirements for data to be supplied to our operations in the U.S., e.g., General Data Protection Regulation in the European Union; | |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements; economic weakness, including inflation, or political instability in particular foreign economies and markets; compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; foreign taxes, including withholding of payroll taxes; | |
| ● | differing payor reimbursement regimes, governmental payors or patient self-pay systems and price controls; | |
| ● | foreign currency fluctuations, which could result in increased operating expenses or reduced revenues, and other obligations incident to doing business or operating in another country; | |
| ● | workforce uncertainty in countries where labor unrest is more common than in the U.S.; | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and | |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism. |
Our current and future product candidates, the methods used to deliver them or their dosage levels may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label or result in significant negative consequences following any regulatory approval.
Undesirable side effects caused by our current or future product candidates, their delivery methods or dosage levels could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval or termination of clinical trials by the FDA or other comparable foreign regulatory authorities; or an IRB, that approves and, monitors biomedical research to protect the rights and welfare of human subjects. As a result of safety or toxicity issues that we may experience in our clinical trials, or negative or inconclusive results from the clinical trials of others for drug candidates similar to our own, we may not receive approval to market our current product candidate or any product candidates we may pursue, which could prevent us from ever generating revenues or achieving profitability. Results of our trials could reveal an unacceptably high severity and incidence of side effects. In such an event, our trials could be suspended or terminated, and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our current or any future product candidates for any or all targeted indications. The drug-related side effects could also affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims. Any of these occurrences may have a material adverse effect on our business, results of operations, financial condition, cash flows and future prospects.
Additionally, if our product candidate receives regulatory approval, and we or others later identify undesirable side effects caused by such product, a number of potentially significant negative consequences could result, including that:
| ● | we may be forced to suspend marketing of such product; | |
| ● | regulatory authorities may withdraw their approvals of such product; | |
| ● | regulatory authorities may require additional warnings on the label that could diminish the usage or otherwise limit the commercial success of such product; | |
| ● | we may be required to conduct post-marketing studies; | |
| ● | we may be required to change the way the product is administered; | |
| ● | we could be sued and held liable for harm caused to subjects or patients; and | |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of our product candidate, if approved.
| 32 |
Our product development program may not uncover all possible adverse events that patients who take our product candidate may experience. The number of subjects exposed to our product candidate and the average exposure time in the clinical development program may be inadequate to detect rare adverse events or chance findings that may only be detected once the product is administered to more patients and for greater periods of time.
Clinical trials by their nature utilize a sample of the potential patient population. However, with a limited number of subjects and limited duration of exposure, we cannot be fully assured that rare and severe side effects of our product candidate will be uncovered. Such rare and severe side effects may only be uncovered with a significantly larger number of patients exposed to our product candidate. If such safety problems occur or are identified after our product candidate reaches the market, the FDA may require that we amend the labeling of the product or recall the product, or may even withdraw approval for the product.
Our future success is dependent on the regulatory approval of our product candidate.
Our business is dependent on our ability to obtain regulatory approval for our product candidate in a timely manner. We cannot commercialize our product candidate in the U.S. without first obtaining regulatory approval for the product from the FDA. Similarly, we cannot commercialize our product candidate outside of the U.S. without obtaining regulatory approval from comparable foreign regulatory authorities. Before obtaining regulatory approvals for the commercial sale of our product candidate for a target indication, we must demonstrate with substantial evidence gathered in preclinical studies and clinical trials, that the product candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate with respect to such product candidate.
The time required to obtain approval by the FDA and comparable foreign regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions.
Even if a product candidate were to successfully obtain approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, or may be subject to burdensome post-approval study or risk management requirements. Also, any regulatory approval of our current product candidate or any future product candidates we may pursue, once obtained, may be withdrawn.
Our current product candidate and future product candidates could fail to receive regulatory approval from the FDA.
We have not obtained regulatory approval for our product candidate and it is possible that our existing product candidate or any future product candidates will not obtain regulatory approval, for many reasons, including:
| ● | disagreement with the regulatory authorities regarding the scope, design or implementation of our clinical trials; | |
| ● | failure to demonstrate that a product candidate is safe and effective for our proposed indication; | |
| ● | failure of clinical trials to meet the level of statistical significance required for approval; | |
| ● | failure to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; | |
| ● | disagreement with our interpretation of data from preclinical studies or clinical trials; | |
| ● | the insufficiency of data collected from clinical trials of our product candidate to support the submission and filing of a BLA, NDA or other submission or to obtain regulatory approval; | |
| ● | failure to obtain approval of our manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies or our own manufacturing facility; or | |
| ● | changes in the approval policies or regulations that render our preclinical and clinical data insufficient for approval. |
The FDA or a comparable foreign regulatory authority may require more information, including additional preclinical or clinical data to support approval or additional studies, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. If we were to obtain approval, regulatory authorities may approve our current product candidate and any future product candidates we may pursue for fewer or more limited indications than we request (including failing to approve the most commercially promising indications), may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate.
If we are unable to obtain regulatory approval for our product candidate in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding to continue the development of that product or generate revenues attributable to that product candidate.
| 33 |
Failure to obtain regulatory approval in international jurisdictions would prevent our product candidate from being marketed abroad.
In addition to regulations in the U.S., to market and sell our product candidate in the European Union, United Kingdom, many Asian countries and other jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the U.S. does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. The regulatory approval process outside the U.S. generally includes all of the risks associated with obtaining FDA approval as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. We may not be able to obtain approvals from regulatory authorities outside the U.S. on a timely basis, if at all. Clinical trials accepted in one country may not be accepted by regulatory authorities in other countries. In addition, many countries outside the U.S. require that a product be approved for reimbursement before it can be approved for sale in that country. A product candidate that has been approved for sale in a particular country may not receive reimbursement approval in that country.
We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our product in any market. If we are unable to obtain approval of any of our current product candidate or any future product candidates we may pursue by regulatory authorities in the European Union, United Kingdom, Asia or elsewhere, the commercial prospects of that product candidate may be significantly diminished, our business prospects could decline and this could materially adversely affect our business, results of operations and financial condition.
Even if our current candidate receive regulatory approval, it may still face future development and regulatory difficulties.
Even if we obtain regulatory approval for our product candidate, that approval would be subject to ongoing requirements by the FDA and comparable foreign regulatory authorities governing the manufacture, quality control, further development, labeling, packaging, storage, distribution, adverse event reporting, safety surveillance, import, export, advertising, promotion, recordkeeping and reporting of safety and other post-marketing information. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance by us and/or our CMOs and CROs for any post-approval clinical trials that we may conduct. The safety profile of any product will continue to be closely monitored by the FDA and comparable foreign regulatory authorities after approval. If the FDA or comparable foreign regulatory authorities become aware of new safety information after approval of our product candidate, they may require labeling changes or establishment of a risk evaluation and mitigation strategy, impose significant restrictions on such product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance.
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP, GCP, and other regulations. If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. If we, our product candidate or the manufacturing facilities for our product candidate fail to comply with applicable regulatory requirements, a regulatory agency may:
| ● | issue warning letters or untitled letters; | |
| ● | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; | |
| ● | require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; | |
| ● | seek an injunction or impose civil or criminal penalties or monetary fines; | |
| ● | suspend or withdraw regulatory approval; | |
| ● | suspend any ongoing clinical trials; | |
| ● | refuse to approve pending applications or supplements to applications filed by us; | |
| ● | suspend or impose restrictions on operations, including costly new manufacturing requirements; or | |
| ● | seize or detain products, refuse to permit the import or export of products, or require us to initiate a product recall. |
The occurrence of any event or penalty described above may inhibit our ability to successfully commercialize our product and generate revenues.
Advertising and promotion of any product candidate that obtains approval in the U.S. is heavily scrutinized by the FDA, the Department of Justice, the Office of Inspector General of Health and Human Services, state attorneys general, members of Congress and the public. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. Additionally, advertising and promotion of any product candidate that obtains approval outside of the U.S. is heavily scrutinized by comparable foreign regulatory authorities. Violations, including actual or alleged promotion of our product for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil and criminal sanctions by the FDA, as well as prosecution under the federal False Claims Act. Any actual or alleged failure to comply with labeling and promotion requirements may have a negative impact on our business.
| 34 |
Risks Related to Our Manufacturing
We have limited to no manufacturing, sales, marketing or distribution capability and must rely upon third parties for such.
We currently have purchase orders with various third-party manufacturing facilities for production of our product candidate for research and development and testing purposes. We depend on these manufacturers to meet our deadlines, quality standards and specifications. Our reliance on third parties for the manufacture of our active pharmaceutical ingredient and drug product and, in the future, any approved products, creates a dependency that could severely disrupt our research and development, our clinical testing, and ultimately our sales and marketing efforts if the source of such supply proves to be unreliable or unavailable. If the contracted manufacturing source is unreliable or unavailable, we may not be able to manufacture clinical drug supplies of our product candidate, and our preclinical and clinical testing programs may not be able to move forward and our entire business plan could fail.
The active pharmaceutical ingredient for our product candidate is currently sourced from Polypeptide Laboratories located in San Diego, California. We believe this single source is currently capable of supplying all anticipated needs of our proposed clinical studies, as well as initial commercial introduction. We will be developing a source or sources for drug product manufacturing. If we are able to commercialize our product in the future, there is no assurance that our manufacturers will be able to meet commercialized scale production requirements in a timely manner or in accordance with applicable standards or cGMP. Once the nature and scope of additional indications and their commensurate drug product demands are established, we will seek secondary suppliers of both the active pharmaceutical ingredient and drug product for our product candidate, but we cannot assure that such secondary suppliers will be found on terms acceptable to us, or at all.
We are subject to a multitude of manufacturing risks, any of which could substantially increase our costs and limit supply of our product candidate.
We and our CMOs will need to conduct significant development work for our product candidate for each target indication for studies, trials and commercial launch readiness. Developing commercially viable manufacturing processes is a difficult, expensive and uncertain task, and there are risks associated with scaling to the level required for advanced clinical trials or commercialization, including cost overruns, potential problems with process scale-up, process reproducibility, stability issues, consistency and timely availability of reagents or raw materials. The manufacturing facilities in which our product candidate will be made could be adversely affected by earthquakes and other natural disasters, medical pandemics, equipment failures, labor shortages, power failures, and numerous other factors.
Additionally, the process of manufacturing our product candidate is complex, highly regulated and subject to several risks, including but not limited to:
| ● | product loss due to contamination, equipment failure or improper installation or operation of equipment, or vendor or operator error; | |
| ● | reduced production yields, product defects, and other supply disruptions due to deviations, even minor, from normal manufacturing and distribution processes; |
| ● | unexpected product defects; and | |
| ● | microbial, viral, or other contaminations in our product candidate or in the manufacturing facilities in which our product candidate is made, which may result in the closure of such manufacturing facilities for an extended period of time to allow for the investigation and remediation of the contamination. |
Any adverse developments affecting manufacturing operations for our product candidate may result in shipment delays, inventory shortages, lot failures, withdrawals or recalls or other interruptions in the supply of our drug substance and drug product, which could delay the development of our product candidate. We may also have to write off inventory, incur other charges and expenses for supply of drug product that fails to meet specifications, undertake costly remediation efforts, or seek more costly manufacturing alternatives. Inability to meet the demand for our product candidate could damage our reputation and the reputation of our product among physicians, healthcare payors, patients or the medical community, and cancer treatment centers, which could adversely affect our ability to operate our business and our results of operations.
In the clinical trials using GP2, GM-CSF is also administered and its availability is dependent upon a third-party manufacturer, which may or may not reliably provide GM-CSF, thus jeopardizing the completion of the trials.
GP2 is administered in combination with GM-CSF which is available in both liquid and lyophilized forms exclusively from one manufacturer. We will continue to be dependent on such manufacturer for our supply of GM-CSF in combination with GP2 in the ongoing GP2 trials and upon the potential commercialization of GP2. We have not entered into a supply agreement with the manufacturer for GM-CSF, and instead rely on purchase orders to meet our supply needs. Any temporary interruptions or discontinuation of the availability of GM-CSF could have a material adverse effect on our operations.
| 35 |
If any of our CMOs’ clinical manufacturing facilities are damaged or destroyed or production at such facilities is otherwise interrupted, our business and prospects would be negatively affected.
If our CMOs’ manufacturing facilities or the equipment in them is damaged or destroyed, we may not be able to quickly or inexpensively replace our manufacturing capacity or replace it at all. In the event of a temporary or protracted loss of this facility or equipment, we might not be able to transfer manufacturing to another CMO. Even if we could transfer manufacturing to another CMO, the shift would likely be expensive and time-consuming, particularly because the new facility would need to comply with the necessary regulatory requirements and we would need FDA approval before selling any products manufactured at that facility. Such an event could delay our clinical trials or reduce our product sales.
Although we do not currently maintain insurance coverage against damage to our property and to cover business interruption and research and development restoration expenses, any insurance coverage we obtain in the future may not reimburse us, or may not be sufficient to reimburse us, for any expenses or losses we may suffer. We may be unable to meet our requirements for our product candidate if there were a catastrophic event or failure of our current manufacturing facility or processes.
Risks Related to Our Dependence on Third Parties and Our License Agreements
We rely on third parties to conduct our preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, or if we lose any of our CROs or other key third-party vendors, we may not be able to obtain regulatory approval for or commercialize our current or future product candidates on a timely basis, if at all.
Our internal capacity for clinical trial execution and management is limited and therefore we rely heavily on third parties. We have relied upon and plan to continue to rely upon third-party CROs, vendors and contractors to monitor and manage data for our ongoing preclinical and clinical programs. For example, our collaborating investigators along with their clinical and clinical operations teams may manage the conduct of any future clinical trials for GP2 as well as perform the analysis, publication and presentation of data and results related to this program.
We plan to rely on CROs and other third-party vendors for all currently contemplated clinical studies. We rely on these parties for the execution of our preclinical studies and clinical trials, including the proper and timely conduct of our clinical trials, and we control only some aspects of their activities. Outsourcing these functions involves risk that third parties may not perform to our standards, may not produce results or data in a timely manner or may fail to perform at all.
While we may have agreements governing the commitments of our third-party vendor services, we will have limited influence over their actual performance. Nevertheless, we will be responsible for ensuring that each of our trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on the CROs will not relieve us of our regulatory responsibilities.
If our Company, or any of our partners or CROs, fail to comply with applicable regulations and good clinical practices, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our regulatory applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with applicable requirements. In addition, our clinical trials must be conducted with product produced under cGMP and other requirements. We are also required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, clinicaltrials.gov, within a specified timeframe. Failure to comply also would violate federal requirements in the U.S. and could result in other penalties, which would delay the regulatory approval process and result in adverse publicity.
Our CROs, third-party vendors and contractors are not and will not be our employees, and except for remedies available to us under our agreements with such CROs, third-party vendors and contractors, we cannot control whether or not they devote sufficient time and resources, including experienced staff, to our ongoing clinical, nonclinical and preclinical programs. They may also have relationships with other entities, some of which may be our competitors. If CROs, third-party vendors and contractors do not successfully carry out their contractual duties or obligations or meet expected deadlines or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our current or future product candidates. CRO, vendor or contractor errors could cause our results of operations and the commercial prospects for our current or future product candidates to be harmed, our costs to increase and our ability to generate revenues to be delayed.
In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. To the extent we are unable to identify and successfully manage the performance of third-party service providers in the future, our business may be adversely affected. Though, once engaged, we intend to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
We are dependent on technologies we license, and if we lose the right to license such technologies or we fail to license new technologies in the future, our ability to develop new products would be harmed, and if we fail to meet our obligations under our license agreements, we may lose the ability to develop our product candidate.
We currently are dependent on a license from HJF for technologies relating to our product candidate. The license imposes, and any future licenses we enter into are likely to impose, various development, funding, royalty, diligence, sublicensing, insurance and other obligations on us. If our license with respect to any of these technologies is terminated for any reason, the development of the products contemplated by the licenses would be delayed, or suspended altogether, while we seek to license similar technology or develop new non-infringing technology which could have a material adverse effect on our business.
| 36 |
We may not realize the benefits of our strategic alliances that we may form in the future.
We may form strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties that we believe will complement or augment our existing business. These relationships, or those like them, may require us to incur nonrecurring and other charges, increase our near- and long-term expenditures, issue securities that dilute our existing stockholders or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic alliances and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic alliance or other alternative arrangements for or current product candidate or any future product candidates and programs because our research and development pipeline may be insufficient, our current product candidate and future product candidates and programs may be deemed to be at too early a stage of development for collaborative effort and third parties may not view such product candidates and programs as having the requisite potential to demonstrate safety and efficacy. If we license products or acquire businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture. We cannot be certain that, following a strategic transaction or license, we will achieve the revenues or specific net income that justifies such transaction. Any delays in entering into new strategic alliances agreements related to our current product candidate or future product candidates could also delay the development and commercialization of such product candidates and reduce their competitiveness even if they reach the market.
Our business involves the use of hazardous materials and we and our third-party manufacturers and suppliers must comply with environmental, health and safety laws and regulations, which can be expensive and restrict how we do business.
Our third-party manufacturers’ and suppliers’ activities involve the controlled storage, use and disposal of hazardous materials. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials even after we sell or otherwise dispose of the products. In some cases, these hazardous materials and various wastes resulting from their use will be stored at our contractors or manufacturers’ facilities pending use and disposal. We cannot completely eliminate the risk of contamination, which could cause injury to our employees and others, environmental damage resulting in costly cleanup and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we expect that the safety procedures utilized by our third-party contractors and manufacturers for handling and disposing of these materials will generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this will be the case or eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources. We do not currently carry biological or hazardous waste insurance coverage and any future property and casualty, and general liability insurance policies may exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination.
We may not be able to establish or maintain the third-party relationships that are necessary to develop or potentially commercialize our product candidate.
We expect to depend on collaborators, partners, licensees, CROs and other third parties to formulate our product candidate, to manufacture our product candidate, and to conduct clinical trials for our product candidate. We cannot guarantee that we will be able to successfully negotiate agreements for or maintain relationships with collaborators, partners, licensees, clinical investigators, vendors and other third parties on favorable terms, if at all. Our ability to successfully negotiate such agreements will depend on, among other things, potential partners’ evaluation of the superiority of our technology over competing technologies and the quality of the preclinical and clinical data that we have generated, and the perceived risks specific to developing our product candidate. If we are unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidate. We cannot necessarily control the amount or timing of resources that our contract partners will devote to our product candidate, and we cannot guarantee that these parties will fulfill their obligations to us under these arrangements in a timely fashion. We may not be able to readily terminate any such agreements with contract partners even if such contract partners do not fulfill their obligations to us.
In addition, we may receive notices from third parties from time to time alleging that our technology or product candidate infringes upon the intellectual property rights of those third parties. Any assertion by third parties that our activities or product candidate infringes upon the intellectual property rights of third parties may adversely affect our ability to secure strategic partners or licensees for our technology or product candidate or our ability to secure or maintain manufacturers for our compounds.
Risks Related to Our Intellectual Property
We rely on an exclusive license granted to us by HJF with respect to GP2, and if HJF does not adequately defend such license, our business may be harmed.
We have been granted an exclusive license to GP2, our product candidate, from HJF. The GP2 patent rights were assigned to HJF by certain third parties including the Uniformed Services University of the Health Sciences. We rely on HJF to maintain the patents already issued with respect to GP2, to continue to pursue patent applications pending in certain countries with respect to GP2, and otherwise protect the intellectual property covered by our exclusive license agreement. We have limited control over the activities of HJF or over any other intellectual property that may be related to GP2. For example, we cannot be certain that activities by HJF have been or will be conducted in compliance with applicable laws and regulations and/or any agreements between HJF and the third party assignors. We have no control or input over whether, and in what manner, HJF may enforce or defend the patents against a third-party. HJF may enforce or defend the patent less vigorously than if we had enforced or defended the patents ourselves. Further, HJF may not necessarily seek enforcement in scenarios in which we would feel that enforcement was in our best interests. For example, HJF may not enforce the patents against a competitor of ours who is not a direct competitor of HJF. If our in-licensed intellectual property is found to be invalid or unenforceable, then HJF may not be able to enforce the patents against a competitor of ours. If we fail to meet our obligations under our exclusive license agreement with HJF, then HJF may terminate such agreement. Although we may choose to terminate our license agreement with HJF, doing so would allow a third party to seek and obtain an exclusive license to GP2. If a third party obtains an exclusive license to intellectual property with respect to GP2, then the third party may seek to enforce the intellectual property against us which may have a material adverse effect on our business.
| 37 |
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection. If our patent position does not adequately protect our product candidate, others could compete against us more directly, which would harm our business, possibly materially.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our current product candidate and future product candidates, the processes used to manufacture them and the methods for using them, as well as successfully defending these patents against third-party challenges. As of the date of this Annual Report on Form 10-K, we only have licensed rights from HJF to certain issued patents as well as patent applications which are currently pending in certain countries with respect to GP2. Our ability to stop third parties from making, using, selling, offering to sell or importing our product candidate is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities.
The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in pharmaceutical patents has emerged to date in the U.S. or in foreign jurisdictions outside of the U.S. Changes in either the patent laws or interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be enforced in the patents that may be issued from the applications we currently or may in the future own or license from third parties. Further, if any patents we obtain or license are deemed invalid and unenforceable, our ability to commercialize or license our technology could be adversely affected.
Others have filed, and in the future are likely to file, patent applications covering products and technologies that are similar, identical or competitive to ours or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed or in-licensed by us, or that we or our licensors will not be involved in interference, opposition, reexamination, review, reissue, post grant review or invalidity proceedings before U.S. or non-U.S. patent offices.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| ● | others may be able to make compounds that are similar to our product candidate, but that are not covered by the claims of our licensed patents; | |
| ● | HJF might not have been the first to make the inventions covered by its pending patent applications; |
| ● | we or HJF might not have been the first to file patent applications for these inventions; | |
| ● | HJF’s pending patent applications may not result in issued patents; | |
| ● | the claims of HJF’s issued patents or patent applications when issued may not cover our product or product candidate; | |
| ● | any patents that we obtain from licensing or otherwise may not provide us with any competitive advantages; | |
| ● | any granted patents that we rely upon may be held invalid or unenforceable as a result of legal challenges by third parties; and | |
| ● | the patents of others may have an adverse effect on our business. |
If we fail to comply with our obligations in the agreements under which we may license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose rights that are important to our business.
We may be required to enter into intellectual property license agreements that are important to our business. These license agreements may impose various diligence, milestone payment, royalty and other obligations on us. For example, we may enter into exclusive license agreements with various universities and research institutions, we may be required to use commercially reasonable efforts to engage in various development and commercialization activities with respect to licensed products, and may need to satisfy specified milestone and royalty payment obligations. If we fail to comply with any obligations under our agreements with any of these licensors, we may be subject to termination of the license agreement in whole or in part; increased financial obligations to our licensors or loss of exclusivity in a particular field or territory, in which case our ability to develop or commercialize products covered by the license agreement will be impaired.
In addition, disputes may arise regarding intellectual property subject to a license agreement, including:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; | |
| ● | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
| 38 |
| ● | our diligence obligations under the license agreement and what activities satisfy those obligations; | |
| ● | if a third-party expresses interest in an area under a license that we are not pursuing, under the terms of certain of our license agreements, we may be required to sublicense rights in that area to a third party, and that sublicense could harm our business; and | |
| ● | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us. |
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize our product candidate.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidate. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize our product candidate, which could harm our business significantly.
We may incur substantial costs as a result of litigation or other proceedings relating to patents and other intellectual property rights.
If we choose to commence a proceeding or litigation to prevent another party from infringing HJF’s patents, that party will have the right to ask the examiner or court to rule that such patents are invalid or should not be enforced against them. There is a risk that the examiner or court will decide that HJF’s patents are not valid and that HJF does not have the right to stop the other party from using the related inventions. There is also the risk that, even if the validity of such patents is upheld, the examiner or court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights to such patents. In addition, the U.S. Supreme Court has recently modified some tests used by the U.S. Patent and Trademark Office (the “USPTO”) in granting patents over the past 20 years, which may decrease the likelihood that we or HJF will be able to obtain patents and increase the likelihood of challenge to any patents we obtain or license. Any proceedings or litigation to enforce our intellectual property rights or defend ourselves against claims of infringement of third-party intellectual property rights could be costly and divert the attention of managerial and scientific personnel, regardless of whether such litigation is ultimately resolved in our favor. We may not have sufficient resources to bring these actions to a successful conclusion. Moreover, if we are unable to successfully defend against claims that we have infringed the intellectual property rights of others, we may be prevented from using certain intellectual property and may be liable for damages, which in turn could materially adversely affect our business, financial condition or results of operations.
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our product candidate.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. We cannot guarantee that our product candidate, or manufacture or use of our product candidate, will not infringe third-party patents. Furthermore, a third party may claim that we are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidate. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. Some of these third parties may be better capitalized and have more resources than us. There is a risk that a court would decide that we are infringing the third party’s patents and would order us to stop the activities covered by the patents. In that event, we may not have a viable way around the patent and may need to halt commercialization of our product candidate. In addition, there is a risk that a court will order us to pay the other party damages for having violated the other party’s patents. In addition, we may be obligated to indemnify our licensors and collaborators against certain intellectual property infringement claims brought by third parties, which could require us to expend additional resources. The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform.
If we are sued for patent infringement, we would need to demonstrate that our product candidate or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Proving invalidity is difficult. For example, in the U.S., proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and divert management’s time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, which may not be available, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid, we may incur substantial monetary damages, encounter significant delays in bringing our product candidate to market and be precluded from manufacturing or selling our product candidate.
We cannot be certain that others have not filed patent applications for technology covered by HJF’s pending applications, or that HJF the first to invent the technology, because:
| ● | some patent applications in the U.S. may be maintained in secrecy until the patents are issued; | |
| ● | patent applications in the U.S. are typically not published until 18 months after the priority date; and | |
| ● | publications in the scientific literature often lag behind actual discoveries. |
| 39 |
Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours. Any such patent application may have priority over HJF’s patent applications, which could require us to obtain rights to issued patents covering such technologies. If another party has filed U.S. patent applications on inventions similar to HJF that claims priority to any applications filed prior to the priority dates of HJF’s applications, HJF may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the U.S. It is possible that such efforts would be unsuccessful if, unbeknownst to HJF, the other party had independently arrived at the same or similar inventions prior to HFJ’s inventions, resulting in a loss of HFJ’s U.S. patent position with respect to such inventions which could in turn have a material adverse effect on our operations. Other countries have similar laws that permit secrecy of patent applications, and may be entitled to priority over our applications in such jurisdictions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than us or the third parties from whom we license intellectual property because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and product could be significantly diminished.
We also rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Furthermore, any license agreements we enter into in the future may require us to notify, and in some cases license back to the licensor, certain additional proprietary information or intellectual property that we developed using the rights licensed to us under these agreements. Any such licenses back to the licensor could allow our licensors to use that proprietary information or intellectual property in a manner that could harm our business. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its transparency initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed alleged trade secrets.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we could lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Our intellectual property may not be sufficient to protect our product candidate from competition, which may negatively affect our business as well as limit our partnership or acquisition appeal.
We may be subject to competition despite the existence of intellectual property we license or own. We can give no assurances that our intellectual property claims will be sufficient to prevent third parties from designing around patents we own or license and developing and commercializing competitive products. The existence of competitive products that avoid our intellectual property could materially adversely affect our operating results and financial condition. Furthermore, limitations, or perceived limitations, in our intellectual property may limit the interest of third parties to partner, collaborate or otherwise transact with us, if third parties perceive a higher than acceptable risk to commercialization of our product candidate or future product candidates.
We may elect to sue a third party, or otherwise make a claim, alleging infringement or other violation of patents, trademarks, trade dress, copyrights, trade secrets, domain names or other intellectual property rights that we either own or license from a third party. If we do not prevail in enforcing our intellectual property rights in this type of litigation, we may be subject to:
| ● | paying monetary damages related to the legal expenses of the third party; | |
| ● | facing additional competition that may have a significant adverse effect on our product pricing, market share, business operations, financial condition, and the commercial viability of our product; and | |
| ● | restructuring our company or delaying or terminating select business opportunities, including, but not limited to, research and development, clinical trial, and commercialization activities, due to a potential deterioration of our financial condition or market competitiveness. |
| 40 |
A third party may also challenge the validity, enforceability or scope of the intellectual property rights that we license or own; and, the result of these challenges may narrow the scope or claims of or invalidate patents that are integral to our product candidate in the future. There can be no assurance that we will be able to successfully defend patents we own or license in an action against third parties due to the unpredictability of litigation and the high costs associated with intellectual property litigation, amongst other factors.
Intellectual property rights and enforcement may be less extensive in jurisdictions outside of the U.S.; thus, we may not be able to protect our intellectual property and third parties may be able to market competitive products that may use some or all of our intellectual property.
Changes to patent law, including the Leahy-Smith America Invests Act, AIA or Leahy-Smith Act, of 2011 and the Patent Reform Act of 2009 and other future article of legislation, may substantially change the regulations and procedures surrounding patent applications, issuance of patents, and prosecution of patents. We can give no assurances that the patents of our licensor can be defended or will protect us against future intellectual property challenges, particularly as they pertain to changes in patent law and future patent law interpretations.
In addition, enforcing and maintaining our intellectual property protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by the USPTO, courts and foreign government patent agencies, and HJF’s patent protection could be reduced or eliminated for non-compliance with these requirements which may have a material adverse effect on our business.
Risks Related to Commercialization of Our Current Product Candidate and Future Product Candidates
Our commercial success depends upon attaining significant market acceptance of our current product candidate and future product candidates, if approved, among physicians, patients, healthcare payors and cancer treatment centers.
Even if we obtain regulatory approval for our current product candidate or any future product candidates, the products may not gain market acceptance among physicians, healthcare payors, patients or the medical community, including cancer treatment centers. Market acceptance of any product candidates for which we receive approval depends on a number of factors, including:
| ● | the efficacy and safety of such product candidates as demonstrated in clinical trials; | |
| ● | the clinical indications and patient populations for which the product candidate is approved; | |
| ● | acceptance by physicians, major cancer treatment centers and patients of the drug as a safe and effective treatment; | |
| ● | the adoption of novel immunotherapies by physicians, hospitals and third-party payors; | |
| ● | the potential and perceived advantages of product candidates over alternative treatments; | |
| ● | the safety of product candidates seen in a broader patient group, including our use outside the approved indications; | |
| ● | any restrictions on use together with other medications; | |
| ● | the prevalence and severity of any side effects; | |
| ● | product labeling or product insert requirements of the FDA or other regulatory authorities; | |
| ● | the timing of market introduction of our product as well as competitive products; |
| ● | the development of manufacturing and distribution processes for commercial scale manufacturing for our current product candidate and any future product candidates; | |
| ● | the cost of treatment in relation to alternative treatments; | |
| ● | the availability of coverage and adequate reimbursement from third-party payors and government authorities; | |
| ● | relative convenience and ease of administration; and | |
| ● | the effectiveness of our sales and marketing efforts and those of our collaborators. |
If our current product and any future product candidates are approved but fail to achieve market acceptance among physicians, patients, healthcare payors or cancer treatment centers, we will not be able to generate significant revenues, which would compromise our ability to become profitable.
| 41 |
Even if we are able to commercialize our current product candidate or any future product candidates, the products may not receive coverage and adequate reimbursement from third-party payors in the U.S. and in other countries in which we seek to commercialize our products, which could harm our business.
Our ability to commercialize any product successfully will depend, in part, on the extent to which coverage and adequate reimbursement for such product and related treatments will be available from third-party payors, including government health administration authorities, private health insurers and other organizations.
Third-party payors determine which medications they will cover and establish reimbursement levels. A primary trend in the healthcare industry is cost containment. Third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. Third-party payors may also seek additional clinical evidence, beyond the data required to obtain regulatory approval, demonstrating clinical benefit and value in specific patient populations before covering our product for those patients. We cannot be sure that coverage and adequate reimbursement will be available for any product that we commercialize and, if coverage is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain regulatory approval. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize any product candidate for which we obtain regulatory approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may only be temporary. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by third-party payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. No uniform policy for coverage and reimbursement exists in the U.S., and coverage and reimbursement can differ significantly from payor to payor. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies, but also have their own methods and approval process apart from Medicare determinations. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved product that we develop could have a material adverse effect on our operating results, ability to raise capital needed to commercialize our product and overall financial condition.
Healthcare legislative measures aimed at reducing healthcare costs may have a material adverse effect on our business and results of operations.
Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In both the U.S. and certain international jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our product profitably. In particular, in 2010, the Affordable Care Act (“ACA”) was enacted, which, among other things, subjected biologic products to potential competition by lower-cost biosimilars, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, and provided incentives to programs that increase the federal government’s comparative effectiveness research. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the current U.S. administration to repeal or repeal and replace certain aspects of the ACA. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, or the Texas District Court Judge, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as a part of the Tax Act, the remaining provisions of the ACA are invalid as well. While the Texas District Court Judge, as well as the Trump Administration and CMS, have stated that the ruling will have no immediate effect, it is unclear how this decision, subsequent appeals and other efforts to repeal and replace the ACA will impact the ACA. Until there is more certainty concerning the future of the ACA, it will be difficult to predict its full impact and influence on our business.
In addition, other legislative changes have been proposed and adopted in the U.S. since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in 2013, and will remain in effect through 2027 unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012 further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
| ● | the demand for our product candidate, if we obtain regulatory approval; | |
| ● | our ability to receive or set a price that we believe is fair for our product; | |
| ● | our ability to generate revenue and achieve or maintain profitability; |
| 42 |
| ● | the level of taxes that we are required to pay; and | |
| ● | the availability of capital. |
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, lower reimbursement and new payment methodologies. This could lower the price that we receive for any approved product. Any denial in coverage or reduction in reimbursement from Medicare or other government-funded programs may result in a similar denial or reduction in payments from private payors, which may prevent us from being able to generate sufficient revenue, attain profitability or commercialize our product candidate, if approved.
Price controls may be imposed in foreign markets, which may adversely affect our future profitability.
In some countries, particularly member states of the European Union, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of regulatory approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices.
In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidate to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our product is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
Risks Related to Healthcare Compliance Regulations
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings. If we or they are unable to comply with these provisions, we may become subject to civil and criminal investigations and proceedings that could have a material adverse effect on our business, financial condition and prospects.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we obtain regulatory approval. Our current and future arrangements with healthcare providers, healthcare entities, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we research, develop and will market, sell and distribute our product. As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include the following:
| ● | the federal healthcare Anti-Kickback Statute which prohibits, among other things, individuals and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid; | |
| ● | federal civil and criminal false claims laws, including the federal False Claims Act that can be enforced through civil whistleblower or qui tam actions, and civil monetary penalty laws, prohibit individuals or entities from knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment or approval that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also created federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”) which imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information on entities subject to the law, such as certain healthcare providers, health plans, and healthcare clearinghouses, known as covered entities, and their respective business associates that perform services for them that involve the creation, use, maintenance or disclosure of, individually identifiable health information; | |
| ● | the federal physician sunshine requirements under the ACA which requires certain manufacturers of drugs, devices, biologics and medical supplies, with certain exceptions, to report annually to HHS information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations; |
| 43 |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; some state laws which require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures or pricing information; and certain state and local laws which require the registration of pharmaceutical sales representatives; and |
| ● | state and foreign laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, integrity oversight and reporting obligations, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and integrity oversight and reporting obligations.
Product liability lawsuits against us could cause us to incur substantial liabilities and to limit commercialization of any products that we may develop.
We face an inherent risk of product liability exposure related to the testing of our current product candidate or future product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. Product liability claims may be brought against us by subjects enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our product. If we cannot successfully defend ourselves against claims that our product candidate or product caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for any product candidates or products that we may develop; | |
| ● | termination of clinical trial sites or entire clinical trial programs; | |
| ● | injury to our reputation and significant negative media attention; | |
| ● | withdrawal of clinical trial participants; | |
| ● | significant costs to defend the related litigation; |
| ● | substantial monetary awards to trial subjects or patients; | |
| ● | loss of revenue; | |
| ● | diversion of management and scientific resources from our business operations; and | |
| ● | the inability to commercialize any products that we may develop. |
| 44 |
Prior to engaging in future clinical trials, we intend to obtain product liability insurance coverage at a level that we believe is customary for similarly situated companies and adequate to provide us with insurance coverage for foreseeable risks; however, we may be unable to obtain such coverage at a reasonable cost, if at all. If we are able to obtain product liability insurance, we may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise and such insurance may not be adequate to cover all liabilities that we may incur. Furthermore, we intend to expand our insurance coverage for products to include the sale of commercial products if we obtain regulatory approval for our product candidate in development, but we may be unable to obtain commercially reasonable product liability insurance for any products that receive regulatory approval. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
Risks Related to our Business Operations
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
We face competition from numerous pharmaceutical and biotechnology enterprises, as well as from academic institutions, government agencies and private and public research institutions for our current product candidate. Our commercial opportunities will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than any products that we may develop. Competition could result in reduced sales and pricing pressure on our current product candidate, if approved, which in turn would reduce our ability to generate meaningful revenues and have a negative impact on our results of operations. In addition, significant delays in the development of our product candidate could allow our competitors to bring products to market before we do and impair our ability to commercialize our product candidate. The biotechnology industry, including the cancer immunotherapy market, is intensely competitive and involves a high degree of risk. We compete with other companies that have far greater experience and financial, research and technical resources than us. Potential competitors in the U.S. and worldwide are numerous and include pharmaceutical and biotechnology companies, educational institutions and research foundations, many of which have substantially greater capital resources, marketing experience, research and development staffs and facilities than ours. Some of our competitors may develop and commercialize products that compete directly with those incorporating our technology or may introduce products to market earlier than our product or on a more cost-effective basis. Our competitors compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our technology. We may face competition with respect to product efficacy and safety, ease of use and adaptability to various modes of administration, acceptance by physicians, the timing and scope of regulatory approvals, availability of resources, reimbursement coverage, price and patent position, including the potentially dominant patent positions of others. An inability to successfully complete our product development or commercializing our product candidate could result in our having limited prospects for establishing market share or generating revenue.
Many of our competitors or potential competitors have significantly greater established presence in the market, financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do, and as a result may have a competitive advantage over us. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or potentially advantageous to our business.
As a result of these factors, these competitors may obtain regulatory approval of their products before we are able to obtain patent protection or other intellectual property rights, which will limit our ability to develop or commercialize our current product candidate. Our competitors may also develop drugs that are safer, more effective, more widely used and cheaper than ours, and may also be more successful than us in manufacturing and marketing their products. These appreciable advantages could render our product candidate obsolete or noncompetitive before we can recover the expenses of development and commercialization.
Our business may be adversely affected by the ongoing coronavirus pandemic.
The outbreak of the novel coronavirus (COVID-19) has evolved into a global pandemic. The coronavirus has spread to many regions of the world. The extent to which the coronavirus impacts our business and operating results will depend on future developments that are highly uncertain and cannot be accurately predicted, including new information that may emerge concerning the coronavirus and the actions to contain the coronavirus or treat its impact, among others.
As a result of the continuing spread of the coronavirus, our business operations could be delayed or interrupted. For instance, our clinical trials may be affected by the pandemic. Site initiation, participant recruitment and enrollment, participant dosing, distribution of clinical trial materials, study monitoring and data analysis may be paused or delayed due to changes in hospital or university policies, federal, state or local regulations, prioritization of hospital resources toward pandemic efforts, or other reasons related to the pandemic. If the coronavirus continues to spread, some participants and clinical investigators may not be able to comply with clinical trial protocols. For example, quarantines or other travel limitations (whether voluntary or required) may impede participant movement, affect sponsor access to study sites, or interrupt healthcare services, and we may be unable to conduct our clinical trials. Further, if the spread of the coronavirus pandemic continues and our operations are adversely impacted, we risk a delay, default and/or nonperformance under existing agreements which may increase our costs. These cost increases may not be fully recoverable or adequately covered by insurance.
Infections and deaths related to the pandemic may disrupt the United States’ healthcare and healthcare regulatory systems. Such disruptions could divert healthcare resources away from, or materially delay FDA review and/or approval with respect to, our clinical trials. It is unknown how long these disruptions could continue, were they to occur. Any elongation or de-prioritization of our clinical trials or delay in regulatory review resulting from such disruptions could materially affect the development and study of our product candidates.
| 45 |
We currently utilize third parties to, among other things, manufacture raw materials. If any third-party parties in the supply chain for materials used in the production of our product candidates are adversely impacted by restrictions resulting from the coronavirus outbreak, our supply chain may be disrupted, limiting our ability to manufacture our product candidates for our clinical trials and research and development operations.
As a result of the shelter-in-place order and other mandated local travel restrictions, our employees conducting research and development or manufacturing activities may not be able to access their laboratory or manufacturing space which may result in our core activities being significantly limited or curtailed, possibly for an extended period of time.
The spread of the coronavirus, which has caused a broad impact globally, including restrictions on travel and quarantine policies put into place by businesses and governments, may have a material economic effect on our business. While the potential economic impact brought by and the duration of the pandemic may be difficult to assess or predict, it has already caused, and is likely to result in further, significant disruption of global financial markets, which may reduce our ability to access capital either at all or on favorable terms. In addition, a recession, depression or other sustained adverse market event resulting from the spread of the coronavirus could materially and adversely affect our business and the value of our common stock.
The ultimate impact of the current pandemic, or any other health epidemic, is highly uncertain and subject to change. We do not yet know the full extent of potential delays or impacts on our business, our clinical trials, our research programs, healthcare systems or the global economy as a whole. However, these effects could have a material impact on our operations, and we will continue to monitor the situation closely.
Significant disruptions of information technology systems, computer system failures or breaches of information security could adversely affect our business.
We rely to a large extent upon sophisticated information technology systems to operate our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information (including, but not limited to, personal information and intellectual property). The size and complexity of our information technology and information security systems, and those of our third-party vendors with whom we may contract, make such systems potentially vulnerable to service interruptions or to security breaches from inadvertent or intentional actions by our employees or vendors, or from malicious attacks by third parties. Such attacks are of ever-increasing levels of sophistication and are made by groups and individuals with a wide range of motives (including, but not limited to, industrial espionage and market manipulation) and expertise. While we intend to invest in the protection of data and information technology, there can be no assurance that our efforts will prevent service interruptions or security breaches.
Our internal computer systems, and those of our CROs, our CMOs, and other business vendors on which we may rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, fire, terrorism, war and telecommunication and electrical failures. We exercise little or no control over these third parties, which increases our vulnerability to problems with their systems. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. Any interruption or breach in our systems could adversely affect our business operations and/or result in the loss of critical or sensitive confidential information or intellectual property, and could result in financial, legal, business and reputational harm to us or allow third parties to gain material, inside information that they use to trade in our securities. For example, the loss of clinical trial data from completed or ongoing clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability, the further development of our current and future product candidates could be delayed and our business could be otherwise adversely affected.
We will need to grow the size of our organization in the future, and we may experience difficulties in managing this growth.
As of March 31, 2021, we had 1 full-time employee and 2 part-time employees. We will need to grow the size of our organization in order to support our continued development and potential commercialization of our product candidate. As our development and commercialization plans and strategies continue to develop, our need for additional managerial, operational, manufacturing, sales, marketing, financial and other resources may increase. Our management, personnel and systems currently in place may not be adequate to support this future growth. Future growth would impose significant added responsibilities on members of management, including:
| ● | managing our clinical trials effectively; | |
| ● | identifying, recruiting, maintaining, motivating and integrating additional employees; | |
| ● | managing our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors and other third parties; | |
| ● | improving our managerial, development, operational, information technology, and finance systems; and | |
| ● | expanding our facilities. |
| 46 |
If our operations expand, we will also need to manage additional relationships with various strategic partners, suppliers and other third parties. Our future financial performance and our ability to commercialize our product candidate and to compete effectively will depend, in part, on our ability to manage any future growth effectively, as well as our ability to develop a sales and marketing force when appropriate for our company. To that end, we must be able to manage our development efforts and preclinical studies and clinical trials effectively and hire, train and integrate additional management, research and development, manufacturing, administrative and sales and marketing personnel. The failure to accomplish any of these tasks could prevent us from successfully growing our company.
Our future success depends on our ability to retain our executive officers and to attract, retain and motivate qualified personnel.
We are highly dependent upon our personnel, including Snehal Patel, our Chief Executive Officer and member of our board of directors. The loss of Mr. Patel’s services could impede the achievement of our research, development and commercialization objectives. We have not obtained, do not own, nor are we the beneficiary of, key-person life insurance. Our future growth and success depend on our ability to recruit, retain, manage and motivate our employees. The loss of any member of our senior management team or the inability to hire or retain experienced management personnel could compromise our ability to execute our business plan and harm our operating results. Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the pharmaceutical field is intense and as a result, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Risks Related to Owning our Common Stock
The price of our common stock may fluctuate substantially.
You should consider an investment in our common stock to be risky, and you should invest in our common stock only if you can withstand a significant loss and wide fluctuations in the market value of your investment. Some factors that may cause the market price of our common stock to fluctuate, in addition to the other risks mentioned in this “Risk Factors” section and elsewhere in this Annual Report on Form 10-K, are:
| ● | sale of our common stock by our stockholders, executives and directors; | |
| ● | volatility and limitations in trading volumes of our shares of common stock; | |
| ● | our ability to obtain financings to conduct and complete research and development activities including, but not limited to, our clinical trials, and other business activities; | |
| ● | possible delays in the expected recognition of revenue due to lengthy and sometimes unpredictable sales timelines; | |
| ● | the timing and success of introductions of new products by us or our competitors or any other change in the competitive dynamics of our industry, including consolidation among competitors, customers or strategic partners; | |
| ● | network outages or security breaches; | |
| ● | our ability to attract new customers; | |
| ● | our ability to secure resources and the necessary personnel to conduct clinical trials on our desired schedule; | |
| ● | commencement, enrollment or results of our clinical trials for our product candidate or any future clinical trials we may conduct; |
| 47 |
| ● | changes in the development status of our product candidate; | |
| ● | any delays or adverse developments or perceived adverse developments with respect to the FDA’s review of our planned preclinical and clinical trials; | |
| ● | any delay in our submission for studies or product approvals or adverse regulatory decisions, including failure to receive regulatory approval for our product candidate; | |
| ● | unanticipated safety concerns related to the use of our product candidate; | |
| ● | failures to meet external expectations or management guidance; | |
| ● | changes in our capital structure or dividend policy, future issuances of securities, sales of large blocks of common stock by our stockholders; | |
| ● | our cash position; | |
| ● | announcements and events surrounding financing efforts, including debt and equity securities; | |
| ● | our inability to enter into new markets or develop new products; | |
| ● | reputational issues; | |
| ● | competition from existing technologies and products or new technologies and products that may emerge; | |
| ● | announcements of acquisitions, partnerships, collaborations, joint ventures, new products, capital commitments, or other events by us or our competitors; | |
| ● | changes in general economic, political and market conditions in or any of the regions in which we conduct our business; | |
| ● | changes in industry conditions or perceptions; |
| ● | changes in valuations of similar companies or groups of companies; | |
| ● | analyst research reports, recommendation and changes in recommendations, price targets, and withdrawals of coverage; | |
| ● | departures and additions of key personnel; | |
| ● | disputes and litigations related to intellectual properties, proprietary rights, and contractual obligations; | |
| ● | changes in applicable laws, rules, regulations, or accounting practices and other dynamics; and | |
| ● | other events or factors, many of which may be out of our control. |
In addition, if the market for stocks in our industry or industries related to our industry, or the stock market in general, experiences a loss of investor confidence, the trading price of our common stock could decline for reasons unrelated to our business, financial condition and results of operations. If any of the foregoing occurs, it could cause our stock price to fall and may expose us to lawsuits that, even if unsuccessful, could be costly to defend and a distraction to management.
Market and economic conditions may negatively impact our business, financial condition and share price.
Concerns over medical epidemics, energy costs, geopolitical issues, the U.S. mortgage market and a deteriorating real estate market, unstable global credit markets and financial conditions, and volatile oil prices have led to periods of significant economic instability, diminished liquidity and credit availability, declines in consumer confidence and discretionary spending, diminished expectations for the global economy and expectations of slower global economic growth, increased unemployment rates, and increased credit defaults in recent years. Our general business strategy may be adversely affected by any such economic downturns (including the downturn related to the current COVID-19 pandemic), volatile business environments and continued unstable or unpredictable economic and market conditions. If these conditions continue to deteriorate or do not improve, it may make any necessary debt or equity financing more difficult to complete, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance, and share price and could require us to delay or abandon development or commercialization plans.
If securities or industry analysts do not publish research or reports, or publish unfavorable research or reports about our business, our stock price and trading volume may decline.
The trading market for our common stock relies in part on the research and reports that industry or financial analysts publish about us, our business, our markets and our competitors. We do not control these analysts. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect the market price of our common stock. Furthermore, if one or more of the analysts who do cover us downgrade our stock or if those analysts issue other unfavorable commentary about us or our business, our stock price would likely decline. If one or more of these analysts cease coverage of us or fails to regularly publish reports on us, we could lose visibility in the market and interest in our stock could decrease, which in turn could cause our stock price or trading volume to decline and may also impair our ability to expand our business with existing customers and attract new customers.
| 48 |
Because certain of our stockholders control a significant number of shares of our common stock, they may have effective control over actions requiring stockholder approval.
As of March 15, 2020, our directors, executive officers and principal stockholders, and their respective affiliates, beneficially own approximately 69% of our outstanding shares of common stock. As a result, these stockholders, acting together, have the ability to control the outcome of matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these stockholders, acting together, have the ability to control the management and affairs of our company. Accordingly, this concentration of ownership might harm the market price of our common stock by:
| ● | delaying, deferring or preventing a change in corporate control; | |
| ● | impeding a merger, consolidation, takeover or other business combination involving us; or | |
| ● | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
Future sales and issuances of our common stock could result in additional dilution of the percentage ownership of our stockholders and could cause our share price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations, including increased marketing, hiring new personnel, commercializing our product, and continuing activities as an operating public company. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders.
We do not intend to pay cash dividends on our shares of common stock so any returns will be limited to the value of our shares.
We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will therefore be limited to the increase, if any, of our share price.
We are an “emerging growth company” and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. In addition, pursuant to Section 107 of the JOBS Act, as an “emerging growth company” we intend to take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of the completion of our initial public offering; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
We may be at risk of securities class action litigation.
We may be at risk of securities class action litigation. In the past, biotechnology and pharmaceutical companies have experienced significant stock price volatility, particularly when associated with binary events such as clinical trials and product approvals. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and results in a decline in the market price of our common stock.
| 49 |
Our common stock is currently listed on The Nasdaq Capital Market. If we are unable to maintain listing of our securities on Nasdaq or any stock exchange, our stock price could be adversely affected and the liquidity of our stock and our ability to obtain financing could be impaired and it may be more difficult for our stockholders to sell their securities.
Although our common stock is currently listed on The Nasdaq Capital Market, we may not be able to continue to meet the exchange’s minimum listing requirements or those of any other national exchange. If we are unable to maintain listing on Nasdaq or if a liquid market for our common stock does not develop or is sustained, our common stock may remain thinly traded.
The listing rules of Nasdaq require listing issuers to comply with certain standards in order to remain listed on its exchange. If, for any reason, we should fail to maintain compliance with these listing standards and Nasdaq should delist our securities from trading on its exchange and we are unable to obtain listing on another national securities exchange, a reduction in some or all of the following may occur, each of which could have a material adverse effect on our stockholders:
| ● | the liquidity of our common stock; | |
| ● | the market price of our common stock; | |
| ● | our ability to obtain financing for the continuation of our operations; | |
| ● | the number of institutional and general investors that will consider investing in our common stock; | |
| ● | the number of market makers in our common stock; | |
| ● | the availability of information concerning the trading prices and volume of our common stock; and | |
| ● | the number of broker-dealers willing to execute trades in shares of our common stock. |
Our second amended and restated certificate of incorporation (“Amended and Restated Certificate of Incorporation”) and our second amended and restated bylaws (the “Amended and Restated Bylaws”) and Delaware law may have anti-takeover effects that could discourage, delay or prevent a change in control, which may cause our stock price to decline.
Our Amended and Restated Certificate of Incorporation and our Amended and Restated Bylaws and Delaware law could make it more difficult for a third party to acquire us, even if closing such a transaction would be beneficial to our stockholders. We are authorized to issue up to 10 million shares of preferred stock. This preferred stock may be issued in one or more series, the terms of which may be determined at the time of issuance by our board of directors without further action by stockholders. The terms of any series of preferred stock may include voting rights (including the right to vote as a series on particular matters), preferences as to dividend, liquidation, conversion and redemption rights and sinking fund provisions. The issuance of any preferred stock could materially adversely affect the rights of the holders of our common stock, and therefore, reduce the value of our common stock. In particular, specific rights granted to future holders of preferred stock could be used to restrict our ability to merge with, or sell our assets to, a third party and thereby preserve control by the present management.
Provisions of our Amended and Restated Certificate of Incorporation and our Amended and Restated Bylaws and Delaware law also could have the effect of discouraging potential acquisition proposals or making a tender offer or delaying or preventing a change in control, including changes a stockholder might consider favorable. Such provisions may also prevent or frustrate attempts by our stockholders to replace or remove our management. In particular, the certificate of incorporation and bylaws and Delaware law, as applicable, among other things:
| ● | provide the board of directors with the ability to alter the Amended and Restated Bylaws without stockholder approval; | |
| ● | place limitations on the removal of directors; | |
| ● | establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings; and | |
| ● | provide that vacancies on the board of directors may be filled by a majority of directors in office, although less than a quorum. |
Financial reporting obligations of being a public company in the U.S. are expensive and time-consuming, and our management is required to devote substantial time to compliance matters.
As a publicly traded company we incur significant additional legal, accounting and other expenses. The obligations of being a public company in the U.S. require significant expenditures and place significant demands on our management and other personnel, including costs resulting from public company reporting obligations under the Exchange Act and the rules and regulations regarding corporate governance practices, including those under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, and the listing requirements of The Nasdaq Capital Market. These rules require the establishment and maintenance of effective disclosure and financial controls and procedures, internal control over financial reporting and changes in corporate governance practices, among many other complex rules that are often difficult to implement, monitor and maintain compliance with. Moreover, despite recent reforms made possible by the JOBS Act, the reporting requirements, rules, and regulations will make some activities more time-consuming and costly, particularly after we are no longer an “emerging growth company.” Our management and other personnel will need to devote a substantial amount of time to ensure that we comply with all of these requirements and to keep pace with new regulations, otherwise we may fall out of compliance and risk becoming subject to litigation or being delisted, among other potential problems.
| 50 |
Our Amended and Restated Bylaws provides that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for substantially all disputes between the Company and its stockholders, which could limit stockholders’ ability to obtain a favorable judicial forum for disputes with the Company or its directors, officers or employees.
Our Amended and Restated Bylaws provides that unless we consent in writing to the selection of an alternative forum, the State of Delaware is the sole and exclusive forum for: (i) any derivative action or proceeding brought on behalf of us, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of our Company to us or our stockholders, (iii) any action asserting a claim against us, our directors, officers or employees arising pursuant to any provision of the Delaware General Corporation Law (the “DGCL”) or our Amended and Restated Certificate of Incorporation or our Amended and Restated Bylaws, or (iv) any action asserting a claim against us, our directors, officers, employees or agents governed by the internal affairs doctrine, except for, as to each of (i) through (iv) above, any claim as to which the Court of Chancery determines that there is an indispensable party not subject to the jurisdiction of the Court of Chancery (and the indispensable party does not consent to the personal jurisdiction of the Court of Chancery within ten days following such determination), which is vested in the exclusive jurisdiction of a court or forum other than the Court of Chancery, or for which the Court of Chancery does not have subject matter jurisdiction. This exclusive forum provision would not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. To the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder.
Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. However, our Amended and Restated Bylaws contain a federal forum provision which provides that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America will be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock are deemed to have notice of and consented to this provision.
These choice of forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find our choice of forum provisions contained in either our Amended and Restated Bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our business, results of operations, and financial condition.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. We are required to furnish a report by management on, among other things, the effectiveness of internal control over financial reporting. This assessment will include disclosure of any material weaknesses identified by management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from an issuer’s independent registered public accounting firm on the effectiveness of its internal control over financial reporting. However, for as long as we remain an emerging growth company under the JOBS Act, we may take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 of the Sarbanes-Oxley Act may require that we incur substantial accounting expense and expend significant management efforts. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we may be unable to assert that our internal control over financial reporting is effective. In connection with management’s assessment of internal controls over financial reporting for the quarter ended September 30, 2020, we identified a material weakness due to inadequate segregation of duties within our accounting processes due to limited personnel and insufficient written policies and procedures for accounting, IT and financial reporting and record keeping. Although we are developing a plan to remediate the material weaknesses, we cannot assure you that we will be able to remediate such weaknesses or that there will not be new material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, we could lose investor confidence in the accuracy and completeness of our financial reports, the value of our common stock could decline, and we could be subject to sanctions or investigations by regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We sublease a facility to support our clinical trial operations and contract research and development.
We may be involved from time to time in ordinary litigation, negotiation, and settlement matters that will not have a material effect on our operations or finances. We are not currently party to any material legal proceedings, and we are not aware of any pending or threatened litigation against us.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 51 |
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market information
Our common stock has traded on The Nasdaq Capital Market under the symbol “GLSI” since September 25, 2020.
Number of Stockholders
As of March 15, 2021, we had approximately 19 stockholders of record of our common stock.
Dividend Policy
Historically, we have not paid any dividends to the holders of shares of our common stock and we do not expect to pay any such dividends in the foreseeable future as we expect to retain our future earnings for use in the operation and expansion of our business.
Securities Authorized for Issuance under Equity Compensation Plans
The following table summarizes information about our equity compensation plans as of December 31, 2020.
| Number of Shares of Common Stock to be Issued upon Exercise of Outstanding Options, Warrants and Rights | Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Options Remaining Available for Future Issuance Under Equity Compensation Plans (excluding securities reflected in column (a)) | ||||||||||
| (a) | (b) | (c) | ||||||||||
| Equity Compensation Plans Approved by Stockholders | — | — | 1,498,128 | |||||||||
| Equity Compensation Plans Not Approved by Stockholders | — | — | — | |||||||||
| Total | — | — | 1,498,128 | |||||||||
ITEM 6. SELECTED FINANCIAL DATA
Not applicable.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We are a biopharmaceutical company that is developing GP2, an immunotherapy designed to prevent the recurrence of breast cancer following surgery. GP2 is a 9 amino acid transmembrane peptide of the HER2/neu protein, a cell surface receptor protein that is expressed in a variety of common cancers, including expression in 75% of breast cancers at low (1+), intermediate (2+), and high (3+ or over-expressor) levels. In a completed Phase IIb clinical trial led by MD Anderson Cancer Center, no recurrences were observed in the HER2/neu 3+ adjuvant setting after median 5 years of follow-up, if the patient received the 6 primary intradermal injections over the first 6 months. We are planning to commence a Phase III clinical trial in 2021.
To date, we have not generated any revenue and we have incurred net losses. Our net losses were approximately $1.9 million and $3.4 million for the years ended December 31, 2020 and 2019, respectively.
Our net losses have resulted from costs incurred in developing the drug in our pipeline, planning and preparing for clinical trials and general and administrative activities associated with our operations. We expect to continue to incur significant expenses and corresponding increased operating losses for the foreseeable future as we continue to develop our pipeline. Our costs may further increase as we conduct clinical trials and seek regulatory approval for and prepare to commercialize our product candidate. We expect to incur significant expenses to continue to build the infrastructure necessary to support our expanded operations, clinical trials, commercialization, including manufacturing, marketing, sales and distribution functions. We will also experience increased costs associated with operating as a public company.
| 52 |
Basis of Presentation
The accompanying financial statements are presented in conformity with accounting principles generally accepted in the United States of America (“GAAP”) and pursuant to the rules and regulations of the SEC.
Results of Operations For the Years Ended December 31, 2020 and 2019
Research and Development Expenses
Research and development expenses decreased by $1,548,814, or 59%, to $1,057,606 for the year ended December 31, 2020 from $2,606,420 for the year ended December 31, 2019. The decrease was primarily the result of a decrease in compensation expenses.
General and Administrative Expenses
General and administrative expenses decreased by $12,699, or 2% to $806,188 for the year ended December 31, 2020 from $818,887 for the year ended December 31, 2019. The change was negligible.
Liquidity and Capital Resources
Since our inception in 2006, we have devoted most of our cash resources to research and development and general and administrative activities. We have not yet achieved commercialization of our product and have a cumulative net loss from our operations. We will continue to incur net losses for the foreseeable future. Our financial statements have been prepared assuming that we will continue as a going concern.
We will require additional capital to meet our long-term operating requirements. We expect to raise additional capital through the sale of equity and/or debt securities; however, there is no assurance that we will be successful at raising additional capital in the future. If our plans are not achieved and/or if significant unanticipated events occur, we may have to further modify our business plan, which may require us to raise additional capital. As of December 31, 2020 and December 31, 2019, our principal source of liquidity was our cash, which totaled $28,660,375 and $6,835, respectively, and additional loans and accrued unreimbursed expenses from related parties. Historically, our principal sources of cash have included proceeds from the sale of common stock and preferred stock and related party loans. Our principal uses of cash have included cash used in operations. We expect that the principal uses of cash in the future will be for continuing operations, funding of research and development, including our clinical trials, and general working capital requirements.
Cash Flow Activities for the Years Ended December 31, 2020 and 2019
We incurred net losses of $1,862,962 and $3,425,307 during the years ended December 31, 2020 and 2019, respectively, and the decrease was primarily due to an increase in compensation expense, advisory and audit expenses, license expenses, and the GMP manufacturing of GP2. Cash was $6,835 at December 31, 2019 and $28,660,375 at December 31, 2020 and increased due to the following reasons:
Operating Activities
Net cash used in operating activities was $1,152,962 for the year ended December 31, 2020 and $293,267 for the year ended December 31, 2019. The increase was primarily due to an increase in advisory and audit expenses and the GMP manufacturing of GP2.
Investing Activities
We did not use or generate cash from investing activities during the year ended December 31, 2020 and December 31, 2019.
Financing Activities
Net cash provided by financing activities was $29,806,502 during the year ended December 31, 2020, attributable to the completion of the Company’s initial public offering and a follow-on offering. Net cash provided by financing activities was $215,000 during the year ended December 31, 2019, attributable to related party loans.
| 53 |
Off-Balance Sheet Arrangements
As of December 31, 2020, we did not have any off-balance sheet arrangements as described by Item 303(a)(4) of Regulation S-K.
Critical Accounting Policies
Stock-Based Compensation
Compensation expense related to warrants and stock granted to employees and non-employees is measured at the grant date based on the estimated fair value of the award and is recognized on a straight-line basis over the requisite service period. Forfeitures are recognized as a reduction of stock-based compensation expense as they occur. Stock-based compensation expense for an award with a performance condition is recognized when the achievement of such performance condition is determined to be probable. If the outcome of such performance condition is not determined to be probable or is not met, no compensation expense is recognized and any previously recognized compensation expense is reversed.
Recent Accounting Pronouncements
We have evaluated the following recent accounting pronouncements through the date the financial statements were issued and filed with the SEC and believe that none of them will have a material effect on our financial statements:
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, “Leases: Topic 842” (“ASU 2016-02”), to supersede nearly all existing lease guidance under GAAP. The guidance would require lessees to recognize most leases on their balance sheets as lease liabilities with corresponding right-of-use assets. ASU 2016-02 is effective for the Company in the first quarter of its fiscal year ending December 31, 2019 using a modified retrospective approach with the option to elect certain practical expedients. The Company has no material leases, thus the adoption of ASU 2016-02 will have no material impact on the Company’s financial statements.
| 54 |
In May 2016, the FASB issued ASU 2016-12, Revenue from Contracts from Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients. The amendments in this update affect the guidance in ASU 2014-09. The core principle of the guidance in Topic 606 is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The amendments in ASU 2016-12 do not change the core principle of the guidance in Topic 606, but instead affect only the narrow aspects noted in Topic 606. Topic 606 became effective for the Company on December 1, 2018. The Company has no revenue, thus the adoption of ASU 2016-12 will have no material impact on the Company’s financial statements.
In June 2018, the FASB issued ASU 2018-07, “Compensation-Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting,” which modifies the accounting for share-based payment awards issued to nonemployees to largely align it with the accounting for share-based payment awards issued to employees. ASU 2018-07 is effective for us for annual periods beginning January 1, 2019. The Company evaluated ASU 2018-07 and determined that the adoption of this new accounting standard did not have a material impact on the Company’s financial statements.
JOBS Act
On April 5, 2012, the JOBS Act was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended (“Securities Act”) for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
We have chosen to take advantage of the extended transition periods available to emerging growth companies under the JOBS Act for complying with new or revised accounting standards until those standards would otherwise apply to private companies provided under the JOBS Act. As a result, our financial statements may not be comparable to those of companies that comply with public company effective dates for complying with new or revised accounting standards.
Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain of these exemptions, including, without limitation, (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (ii) complying with any requirement that may be adopted by the Public Company Accounting Oversight Board (“PCAOB”) regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an “emerging growth company” until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of the completion of our initial public offering; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
| 55 |
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
Our cash and cash equivalent primarily consists of deposits managed by commercial banks as of December 31, 2020. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of interest rates, particularly because our investments are in deposits as of December 31, 2020. Due to the short-term duration of our investment portfolio and the relatively low risk profile of our investments, a sudden change in interest rates would not have a material effect on the fair market value of our portfolio, nor our operating results or cash flows.
We do not believe our cash and cash equivalents have significant risk of default issues; however, we maintain significant amounts of cash and cash equivalents at one or more financial institutions that are in excess of federally insured limits. Given the current stability of financial institutions, we believe that we will not experience losses on these deposits.
Foreign Currency Risk
We face the foreign currency risk as a result of entering into transactions denominated in currencies other than U.S. dollars. Changes in foreign currency exchange rates can create foreign exchange gains or losses to us. We did not incur significant foreign currency gains or losses for the years ended December 31, 2020 and December 31, 2019.
Effects of Inflation
We do not believe that inflation and changing prices during the years ended December 31, 2020 and 2019 had a significant impact on our results of operations.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
All financial information required by this Item is attached hereto at the end of this report beginning on page F-1 and is hereby incorporated by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as defined in Rule 13a-15(e) and Rule 15d-15(e) under the Exchange Act that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure.
Our management, with the participation of our principal executive officer and principal accounting and financial officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of the end of the period covered by this Annual Report on Form 10-K. Based on such evaluation, our principal executive officer and principal accounting and financial officer has concluded that as of December 31, 2020, our disclosure controls and procedures were not effective as of such date as a result of material weaknesses in our internal control over financial reporting due to inadequate segregation of duties within account processes due to limited personnel and insufficient written policies and procedures for accounting, IT and financial reporting and record keeping. Under the direction of our principal executive officer and principal financial and accounting officer, we are developing a plan to remediate the material weaknesses.
Management’s Report on Internal Control Over Financial Reporting
Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting during the quarter ended December 31, 2020 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
| 56 |
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Executive Officers, Directors and Key Employees
The following table sets forth the name, age and position of each of our executive officers, key employees and directors as of March 15, 2021. All directors hold office until the next annual meeting of stockholders and the election and qualification of their successors. Officers serve at the discretion of the board.
| Name | Age | Position | ||
| Snehal Patel | 57 | Chief Executive Officer, Chief Financial Officer and Director | ||
| F. Joseph Daugherty | 70 | Chief Medical Officer and Director | ||
| Jaye Thompson | 55 | Vice President Clinical & Regulatory Affairs | ||
| David McWilliams | 77 | Chairman of the Board | ||
| Eric Rothe | 45 | Director | ||
| Kenneth Hallock | 72 | Director |
Biographies
The principal occupations for the past five years (and, in some instances, for prior years) of each of our directors and executive officers are as follows:
Snehal Patel. Snehal Patel has over 30 years of experience in executive management, corporate development, operations, and investment banking in the healthcare industry. Mr. Patel has served as our Chief Executive Officer since June 2016 and our Chief Financial Officer and a member of our board of directors since February 2010. In addition, since 2009, Mr. Patel has served as a consultant, manager, and advisor at various levels in multiple private start-up biotech companies helping to develop clinical and pre-clinical assets in cancer and other therapeutic areas. Prior to 2010, Mr. Patel served as a consultant to public and private companies focused on stem cell therapy, multiple sclerosis t-cell therapy, oncolytic viruses, and disposable biotech manufacturing equipment. In addition, Mr. Patel previously served as an investment banker at Sanders Morris Harris, Ferghana Partners, and JP Morgan Chase focusing on healthcare and biotech financing and strategic transactions. Mr. Patel also previously worked in operations and business development at Bayer Corporation and in design and operations consulting firms. Mr. Patel received a Bachelor of Science degree in chemical engineering and a Master of Science degree in biochemical engineering from the Massachusetts Institute of Technology and a Masters of Business Administration degree from the University of Chicago. We believe Mr. Patel is qualified to serve as a member of our board of directors because of his executive and management experience working with biotech companies.
F. Joseph Daugherty. F. Joseph Daugherty has over 35 years of experience in managing and overseeing biotechnology and biomedical projects. Dr. Daugherty has served as our Chief Medical Officer since September 2019 and a member of our board of directors since September 2019. In addition, since 2002, Dr. Daugherty has served as the Managing Partner of Phenolics, LLC and PharmaPrint, LLC which was spun off from Phenolics, LLC, both of which are nutraceutical companies. From 2002 until 2018, he served first as President, and since 2008 as Chief Executive Officer, Chief Medical Officer and the Chairman of the board of directors of Eleos Inc., a clinical stage private biotech company focused on anti-sense technology in cancer. Dr. Daugherty also served in various other capacities as a management consultant as well as an officer and director to over 20 public and private biomedical companies including Dupont. In addition, Dr. Daugherty was President of ConAgra’s biotech division. Dr. Daugherty received a Bachelor of Arts degree in biology from Washington University, a Doctor of Medicine degree from the University of Nebraska Medical Center and a Masters of Science in Industrial Administration from Carnegie-Mellon University (Tepper). We believe Dr. Daugherty is qualified to serve as a member of our board of directors because of his executive and management experience, including his experience working with biotech companies.
Jaye Thompson. Jaye Thompson has over 30 years of experience in pharmaceutical and device product development. Dr. Thompson has served as our Vice President Clinical & Regulatory Affairs since September 2019. Since December 2017, Dr. Thompson has served as a co-founder and Chief Operating Officer of Proxima Clinical Research, Inc., a clinical research service provider. Dr. Thompson previously served as Senior Vice President of Clinical and Regulatory Affairs of Repros Therapeutics, a reproductive health company, from March 2013 to May 2017 and as a member of the board of directors of Repros Therapeutics from November 2009 to March 2013. Dr. Thompson previously served as Senior Vice President of Clinical Development and Regulatory Affairs of Opexa Therapeutics, a multiple sclerosis cell therapy company, from September 2009 to March 2013. In addition, Dr. Thompson has served at clinical stage biotech companies, in various senior clinical and regulatory roles and at inVentiv Clinical Solutions, a clinical research service provider. Dr. Thompson was the president and founder of SYNERGOS, Inc., a clinical research service provider, which was founded in 1991, and acquired by inVentiv Health, as a wholly-owned subsidiary in 2006. Dr. Thompson has advised several of the region’s leading life science companies on strategic and regulatory planning as well as clinical product development. She has directed and managed statistical analysis, data management, report writing, and the conduct of clinical trials for a wide variety of indications. Dr. Thompson has been actively involved in over 200 clinical trials for drugs, biologics and devices, and has been associated with numerous FDA regulatory submissions. Dr. Thompson has often represented sponsor companies at FDA meetings and advisory committee meetings, and she was appointed to the Governor’s Texas Emerging Technology Fund Advisory Committee. Dr. Thompson received a BS in applied mathematics from Texas A&M University and an MS and a PhD in biostatistics from the University of Texas Health Science Center in Houston.
| 57 |
David McWilliams. David McWilliams has over 40 years of experience in building biopharmaceutical and healthcare companies. Mr. McWilliams has served as a member of our board of directors since February 2009. He previously served as the Chief Executive Officer from February 2010 to June 2016 and Chairman of the board of directors of the Company since February 2009. In addition, since 2008, Mr. McWilliams has served as a consultant and an advisor at various levels in multiple private start-up biotech companies to help develop clinical and pre-clinical assets in cancer and other therapeutic areas. Mr. McWilliams previously served as the Chief Executive Officer and a member of the board of directors of Opexa Therapeutics, Inc., a multiple sclerosis cell therapy company, from 2004 until 2008. Mr. McWilliams also previously served as the Chief Executive Officer, President and a member of the board of directors of Bacterial Barcodes, Inc., a bacteria and fungi diagnostic company, and the Chief Executive Officer and a member of the board of directors of Signase, Inc., a cancer therapeutics company. Mr. McWilliams has also served in various other capacities including Chief Executive Officer, President and a member of the board of directors of both Encysive Pharmaceuticals, Inc. and Repros Therapeutics Inc.; Chief Executive Officer and President of Kallestad Diagnostics (Erbamont); President of Harleco Diagnostics Division (EM Industries); General Manager and Program Manager of Abbott Laboratories; and Management Consultant at McKinsey & Company. In addition to the foregoing, Mr. McWilliams currently serves as the Chairman of the board of directors of BioHouston, an advocate of the life sciences industry in Houston. Mr. McWilliams received a Bachelor of Arts degree in chemistry from Washington and Jefferson College and a Master of Business Administration degree from the University of Chicago. We believe Mr. McWilliams is qualified to serve as a member of our board of directors because of his executive experience, management experience and experience working with biotech companies.
Eric Rothe. Eric Rothe is the founder of the Company and has over 12 years of industry and academic experience in gene-based therapies and vaccines, including six years of laboratory experience. Mr. Rothe previously served as President of the Company from October 2006 to February 2010, Chief Executive Officer of the Company from October 2007 to February 2010 and Chairman of the Company’s board of directors from October 2006 to February 2009. In addition, Mr. Rothe has served as a member of the Company’s board of directors since August 2006. Since August 2017, Mr. Rothe has served as the Global Product Line Leader at Baker Hughes, an energy technology company. Previously, from September 2014 until its acquisition by GE Oil & Gas’ acquisition of Baker Hughes in July 2017, Mr. Rothe served as Vice President of Mid-Continent and NE US Geomarket and Global Product Line Leader of GE Oil & Gas. From 2012 to 2014, Mr. Rothe served as the International Sales and Operations Director at National Oilwell Varco, one of the world’s largest oil field equipment providers. Before joining the oil & gas sector, Mr. Rothe was Director of the Clinical Cancer Genetics program at U.T. M.D. Anderson Cancer Center, Project Manager at Introgen, a developer of cancer products in advanced clinical trials, and provided consulting services for start-up/small biotechnology companies in Texas. Mr. Rothe received a Bachelor of Arts degree in molecular and cell biology from the University of California at Berkeley and a Master of Business Administration degree from Rice University. We believe Mr. Rothe is qualified to serve as a member of our board of directors because of his expertise in cancer immunology, GMP manufacturing, and clinical research, and his experience in various senior management positions in global commercial operations at large corporations.
Kenneth Hallock. Kenneth Hallock has over 40 years of experience in general management and new venture start-ups and is a major investor in our Company. Mr. Hallock has served as a member of our board of directors since September 2019. Mr. Hallock is currently a senior manager and partner in a private start-up equipment manufacturing company and has been in this role for over 10 years. Previously, Mr. Hallock worked in large industrial corporations such as NL Industries and Anderson Clayton, which were subsequently acquired. Mr. Hallock received a Bachelor of Engineering degree in chemical engineering from Princeton University and a Master of Business Administration degree from Harvard Business School. We believe Mr. Hallock is qualified to serve as a member of our board of directors because of his experience in various management positions for several Fortune 500 companies.
Family Relationships and Other Arrangements
There are no family relationships among our directors and executive officers. There are no arrangements or understandings between or among our executive officers and directors pursuant to which any director or executive officer was or is to be selected as a director or executive officer.
| 58 |
Board Leadership Structure and Role in Risk Oversight
We have historically separated the roles of Chairman of the Board (“Chairman”) and Chief Executive Officer. Although the separation of roles has been appropriate for us, in the view of the Board, the advisability of the separation of these roles depends upon the specific circumstances and dynamics of our leadership.
The Board, as a unified body and through committee participation, organizes the execution of its monitoring and oversight roles and does not expect its Chairman to organize those functions.
The Board has three standing committees-Audit, Compensation and Corporate Governance/Nominating. The membership of each of the committees of the Board is comprised of independent directors, with each of the committees having a chairman, each of whom is an independent director. Our non-management members of the Board meet in executive session at each regular Board meeting.
Risk is inherent with every business, and how well a business manages risk can ultimately determine its success. Management is responsible for the day-to-day management of the risks we face, while the Board, as a whole and through its committees, has responsibility for the oversight of risk management. In its risk oversight role, the Board is responsible for satisfying itself that the risk management processes designed and implemented by management are adequate and functioning as designed.
The Board believes that establishing the right “tone at the top” and that full and open communication between executive management and the Board are essential for effective risk management and oversight. Our CEO communicates frequently with members of the Board to discuss strategy and challenges facing our company. Senior management usually attends our regular quarterly Board meetings and is available to address any questions or concerns raised by the Board on risk management-related and any other matters. Each quarter, the Board receives presentations from senior management on matters involving our key areas of operations.
Committees of Our Board of Directors
Our Board directs the management of our business and affairs, as provided by Delaware law, and conducts its business through meetings of the Board and its standing committees. We have a standing audit committee and compensation committee. Our entire Board serves in place of a nominating and corporate governance committee. In addition, from time to time, special committees may be established under the direction of the Board when necessary to address specific issues.
Audit Committee
Our audit committee is responsible for, among other things:
| ● | approving and retaining the independent auditors to conduct the annual audit of our financial statements; | |
| ● | reviewing the proposed scope and results of the audit; | |
| ● | reviewing and pre-approving audit and non-audit fees and services; | |
| ● | reviewing accounting and financial controls with the independent auditors and our financial and accounting staff; | |
| ● | reviewing and approving transactions between us and our directors, officers and affiliates; | |
| ● | establishing procedures for complaints received by us regarding accounting matters; | |
| ● | overseeing internal audit functions, if any; and | |
| ● | preparing the report of the audit committee that the rules of the SEC require to be included in our annual meeting proxy statement. |
Our audit committee consists of David McWilliams, Eric Rothe and Kenneth Hallock, with David McWilliams serving as chair. Our board of directors has affirmatively determined that David McWilliams, Eric Rothe and Kenneth Hallock each meet the definition of “independent director” under the Nasdaq rules, and that they meet the independence standards under Rule 10A-3. Each member of our audit committee meets the financial literacy requirements of the Nasdaq rules. In addition, our board of directors has determined that David McWilliams qualifies as an “audit committee financial expert,” as such term is defined in Item 407(d)(5) of Regulation S-K. Our board of directors adopted a written charter for the audit committee, which is available on our principal corporate website at www.greenwichlifesciences.com.
| 59 |
Compensation Committee
Our compensation committee is responsible for, among other things:
| ● | reviewing and recommending the compensation arrangements for management, including the compensation for our president and chief executive officer; | |
| ● | establishing and reviewing general compensation policies with the objective to attract and retain superior talent, to reward individual performance and to achieve our financial goals; | |
| ● | administering our stock incentive plans; and | |
| ● | preparing the report of the compensation committee that the rules of the SEC require to be included in our annual meeting proxy statement. |
Our compensation committee consists of David McWilliams, Eric Rothe and Kenneth Hallock, with David McWilliams serving as chair. Our board has determined that David McWilliams, Eric Rothe and Kenneth Hallock are independent directors under Nasdaq rules. Our board of directors adopted a written charter for the compensation committee, which is available on our principal corporate website at www.greenwichlifesciences.com.
Nominating and Governance Committee
Although our entire board of directors serves in place of a nominating and corporate governance committee, our independent directors on the board are responsible for, among other things:
| ● | nominating members of the board of directors; | |
| ● | developing a set of corporate governance principles applicable to our company; and | |
| ● | overseeing the evaluation of our board of directors. |
Our entire board of directors serves in place of a nominating and corporate governance committee. Our board of directors adopted resolutions addressing, among other things, the nomination process.
Code of Business Conduct and Ethics
We have adopted a formal Code of Business Conduct and Ethics applicable to all Board members, officers and employees. Our Code of Business Conduct and Ethics can be found on our website (www.greenwichlifesciences.com). A copy of our Code of Business Conduct and Ethics may be obtained without charge upon written request to Secretary, Greenwich LifeSciences, Inc., 3992 Bluebonnet Dr., Building 14, Stafford, TX 77477. If we make any substantive amendments to our Code of Business Conduct and Ethics or grant any waiver from a provision of the Code of Business Conduct and Ethics to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website (www.greenwichlifesciences.com) and/or in our public filings with the SEC.
Hedging and Pledging Policies
As part of our Insider Trading Policy, all of our officers, all of our directors, certain of our employees and consultants and family members or others sharing a household with any of the foregoing are prohibited from engaging in short sales of our securities, any hedging or monetization transactions involving our securities and in transactions involving puts, calls or other derivative securities based on our securities. Our Insider Trading Policy further prohibits such persons from purchasing our securities on margin, borrowing against any account in which our securities are held or pledging our securities as collateral for a loan unless pre-cleared by our Insider Trading Compliance Officer. As of March 15, 2021, none of our directors or executive officers had pledged any shares of our common stock.
| 60 |
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation Table
The following table presents the compensation awarded to, earned by or paid to each of our named executive officers for the year ended December 31, 2020.
| Name and Principal Position | Year |
Salary ($) |
Bonus ($) |
Stock awards ($)(1) |
Option awards ($) |
Nonequity incentive plan compensation ($) |
Nonqualified deferred compensation earnings ($) |
All other compensation ($)(2) |
Total ($) |
|||||||||||||||||||||||||||
| Snehal Patel, Chief Executive Officer | 2020 | 114,966 | 392,516 | 491,589 | — | — | — | — | 999,071 | |||||||||||||||||||||||||||
| 2019 | — | — | 122,750 | — | — | — | 16,423 | 139,173 | ||||||||||||||||||||||||||||
| (1) | For 2020 fiscal year, Mr. Patel received 218,484 shares of our common stock for services rendered and as incentive for services to be rendered. Mr. Patel did not receive any options or warrants for the 2020 fiscal year. For 2019 fiscal year, Mr. Patel received 148,254 shares of our common stock for services rendered and as incentive for services to be rendered. Mr. Patel did not receive any options or warrants for the 2019 fiscal year. |
| (2) | For fiscal year 2019, Mr. Patel received (i) 4,494,383 shares of our common stock in exchange for related party payables for the periods from January 1, 2010 through September 30, 2019 and (ii) 1,656,607 shares of our common stock in exchange for warrants to purchase shares of our common stock. |
Outstanding Equity Awards at Fiscal Year-End
The following table provides information regarding awards held by each of our named executive officers that were outstanding as of December 31, 2020.
| Option Awards(1) | Stock Awards | |||||||||||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | Number of shares or units of stock that have not vested (#) | Market value of shares or units of stock that have not vested ($) | ||||||||||||||||||
| Snehal Patel | 382,326 | (1) | 860,234 | |||||||||||||||||||||
| (1) | We granted Mr. Patel shares of common stock on September 30, 2019 for compensation and incentives of which 93,633 vested immediately upon grant, 273,105 vested between October 1 2019 and December 31, 2020 over the 15 month period, and the balance, or 382,347 shares of common stock vest over 21 equal monthly installments commencing on January 1, 2021. |
Non-Employee Director Compensation
The following table presents the total compensation for each person who served as a non-employee member of our Board and received compensation for such service during the fiscal year ended December 31, 2020. Other than as set forth in the table and described more fully below, we did not pay any compensation, make any equity awards or non-equity awards to, or pay any other compensation to any of the non-employee members of our Board in 2020.
| Name |
Fees Earned or Paid in Cash ($) |
Stock Awards ($) | All Other Compensation ($)(4) |
Total ($) |
||||||||||||
| David McWilliams(1) | 21,087 | 21,087 | ||||||||||||||
| Eric Rothe(2) | 14,067 | 14,067 | ||||||||||||||
| Kenneth Hallock(3) | 14,067 | 14,067 | ||||||||||||||
| (1) | On September 30, 2019, we authorized the issuance of 28,090 shares of its common stock to Mr. McWilliams. The shares vest in 36 equal monthly installments with the first installment vesting on October 1, 2019. Of such shares, 9,372 shares of common stock vested during the fiscal year ended December 31, 2020. Mr. McWilliams did not receive any options or warrants during the 2020 fiscal year. |
| 61 |
| (2) | On September 30, 2019, we authorized the issuance of 18,727 shares of its common stock to Mr. Rothe. The shares vest in 36 equal monthly installments with the first installment vesting on October 1, 2019. Of such shares, 6,252 shares of common stock vested during the fiscal year ended December 31, 2020. Mr. Rothe did not receive any options or warrants during the 2020 fiscal year. |
| (3) | On September 30, 2019, we authorized the issuance of 18,727 shares of its common stock to Mr. Hallock. The shares vest in 36 equal monthly installments with the first installment vesting on October 1, 2019. Of such shares, 6,252 shares of common stock vested during the fiscal year ended December 31, 2020. Mr. Hallock did not receive any options or warrants during the 2020 fiscal year. |
Employment Agreements
Snehal Patel Employment Agreement
On September 29, 2020, we entered into an employment agreement (the “Employment Agreement”) with Snehal Patel, our Chief Executive Officer in connection with our initial public offering (the “IPO”). The term of the Employment Agreement will continue until December 31, 2021 and automatically renews for successive one year periods at the end of each term until either party delivers written notice of their intent not to renew at least 60 days prior to the expiration of the then effective term. Pursuant to the terms of the Employment Agreement, Mr. Patel shall, among other things, (i) receive a base salary of $450,000, subject to increase, (ii) shall be eligible to receive equity grants, (iii) shall be eligible to receive an annual bonus of up to 50% of his then base salary and (iv) shall be eligible to receive a strategic transaction bonus. In addition, Mr. Patel shall also be eligible to participate in all employee welfare and benefit plans and shall receive such other fringe benefits as we offer to our senior executives and directors.
In the event Mr. Patel’s employment is terminated by us for Cause (as defined in the Employment Agreement), as a result of Mr. Patel’s death or Disability (as defined in the Employment Agreement), voluntarily by Mr. Patel without Good Reason (as defined in the Employment Agreement), or upon expiration of the term, we shall pay Mr. Patel (i) a lump sum amount equal to (A) any unpaid base salary and equity grants then due plus (B) any bonus earned but not paid and (ii) any unpaid expenses (collectively, the “Patel Compensation”). In addition, if Mr. Patel’s employment is terminated for death, Disability or as a result of the expiration of the term of the Employment Agreement as a result of the non-renewal of such term by us, we shall pay Mr. Patel any pro-rated bonus for the target year in which the termination occurs. In the event Mr. Patel’s employment is terminated by us without Cause or by Mr. Patel for Good Reason, we shall pay Mr. Patel (i) the Patel Compensation, (ii) any pro-rated bonus for the target year in which the termination occurs and (iii) provided that Mr. Patel executes the Release (as defined in the Employment Agreement), (A) the Severance Payment (as defined in the Employment Agreement) and (B) COBRA premiums for twelve months from the date of termination. In the event of Mr. Patel’s termination (i) by us without Cause or by Mr. Patel for Good Reason within six months prior to the consummation of a Change of Control (as defined in the Employment Agreement) transaction, if, prior to or as of such termination, a Change of Control transaction was Pending (as defined in the Employment Agreement), at any time during such six month period, (ii) by Mr. Patel for Good Reason at any time within twelve months after the consummation of a Change of Control, or (iii) by us without Cause at any time within twelve months after the consummation of a Change of Control, Mr. Patel shall receive (A) the Patel Compensation, (B) any pro-rated bonus for the target year in which the termination occurs and (C) provided that Mr. Patel executes the Release, (a) a lump sum amount equal to twelve months of Mr. Patel’s then base salary and equity grants at the rate in effect as of the date of termination and (b) COBRA premiums for six months from the date of termination. Furthermore, all of the shares that are then unvested shall immediately vest and, all options, warrants and other convertible securities beneficially held by Mr. Patel shall become fully exercisable for (i) a period of six months following the date of termination only if at the time of such termination there is a Change of Control transaction Pending but in no event beyond expiration of the original term of the award or (ii) if clause (i) does not apply, then such period of time set forth in the agreement evidencing the security. The Employment Agreement also contains covenants restricting Mr. Patel from: (i) engaging in any activity competitive with our business during the term of the Employment Agreement and for a period of one year thereafter; and (ii) soliciting our customers, suppliers or employees during the term of the Employment Agreement and for a period of one year thereafter.
| 62 |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding the beneficial ownership of our common stock as of March 15, 2021 by:
| ● | each of our named executive officers; | |
| ● | each of our directors; | |
| ● | all of our current directors and executive officers as a group; and | |
| ● | each stockholder known by us to own beneficially more than 5% of our common stock. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Shares of common stock that may be acquired by an individual or group within 60 days of March 15, 2021, pursuant to the exercise of options or warrants, vesting of common stock or conversion of preferred stock or convertible debt, are deemed to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table. Percentage of ownership is based on 12,846,897 shares of common stock issued and outstanding as of March 15, 2021.
Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them, based on information provided to us by such stockholders. Unless otherwise indicated, the address for each director and executive officer listed is: c/o Greenwich LifeSciences, Inc., 3992 Bluebonnet Dr, Building 14, Stafford, TX 77477.
| 63 |
| Number of Shares | Percentage of Common Stock | |||||||
| Name of Beneficial Owner | Beneficially Owned | Beneficially Owned | ||||||
| Directors and Named Executive Officers | ||||||||
| Snehal Patel | 7,488,467 | (1) | 58.13 | % | ||||
| F. Joseph Daugherty | 57,886 | (2) | * | |||||
| David McWilliams | 607,631 | (3) | 4.73 | % | ||||
| Eric Rothe | 304,919 | (4) | 2.37 | % | ||||
| Kenneth Hallock | 388,697 | (5) | 3.03 | % | ||||
| All current named executive officers and directors as a group (5 persons) | 8,847,600 | 68.64 | % | |||||
| * | Represents beneficial ownership of less than 1%. |
| (1) | Consists of (i) 895,548 shares of common stock owned by Snehal Patel, (ii) 1,408,033 shares of common stock owned by Snehal Patel IRA, (iii) 2,405,670 shares of common stock owned by Patel Family Trust 1, (iv) 1,320,226 shares of common stock owned by Patel Family Trust 2, (v) 1,329,590 shares of common stock owned by Patel Family Trust 3, and (vi) 129,400 shares of common stock owned by Kinnary Patel IRA. Excludes 291,291 shares of common stock held by Snehal Patel which vest in 16 equal monthly installments. Snehal Patel and Kinnary Patel, the spouse of Snehal Patel, are the Trustees of the Patel Family Trust 1, Patel Family Trust 2 and Patel Family Trust 3. Snehal Patel is the Trustee of the Snehal Patel IRA. Kinnary Patel is the Trustee of the Kinnary Patel IRA. In such capacities, Snehal Patel is deemed to hold voting and dispositive power over the securities held by such entities. |
| (2) | Excludes 29,124 shares of common stock which vest in 16 equal monthly installments. |
| (3) | Excludes 12,470 shares of common stock which vest in 16 equal installments. |
| (4) | Excludes 8,307 shares of common stock which vest in 16 equal monthly installments. |
| (5) | Excludes 8,307 shares of common stock which vest in 16 equal monthly installments. Kenneth Hallock and Annette Hallock are the Trustees of the Hallock Trust and in such capacities share voting and dispositive power over the securities held by such entity. |
Section 16(A) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our officers and directors, and persons who own more than ten percent of a registered class of our equity securities, to file reports of ownership and changes in ownership with the SEC. Officers, directors and greater than ten percent stockholders are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file.
Based on a review of the copies of such forms received, we believe that during 2020, all filing requirements applicable to our officers, directors and greater than ten percent beneficial owners were complied with.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The following includes a summary of transactions since January 1, 2020 to which we have been a party, including transactions in which the amount involved in the transaction exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end for the last two completed fiscal years, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described elsewhere in this Annual Report on Form 10-K. We are not otherwise a party to a current related party transaction, and no transaction is currently proposed, in which the amount of the transaction exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end for the last two completed fiscal years and in which a related person had or will have a direct or indirect material interest.
On October 9, 2019, Eric Rothe, a director, loaned us $15,000 which is payable on demand, is not secured, and does not incur interest, all of which was repaid on November 20, 2020.
On May 30, 2018 and October 2, 2019, the Kenneth and Annette Hallock Revocable Trust loaned us $100,000 and $200,000, respectively, which is payable on demand, is not secured, and does not incur interest, of which $120,000 remained outstanding as of December 31, 2020 and was subsequently fully paid off as of March 15, 2021. Kenneth Hallock, a director, is one of the Trustees of the Hallock Trust.
Between November 2014 and August 2017, Snehal Patel, our Chief Executive Officer and director, loaned us an aggregate of $320,154, which is payable on demand, is not secured, and does not incur interest, of which $155,154 remained outstanding, as of December 31, 2020 and was subsequently fully paid off as of March 15, 2021. In addition, as of December 31, 2020, Snehal Patel is owed $59,367 for reimbursable expenses.
| 64 |
As of September 30, 2019, related party payables to our officers and directors since January 1, 2010 totaled $12 million. As of September 30, 2019, our officers and directors owned outstanding warrants to acquire 2,565,521 shares of our common stock. On September 30, 2019, the officers and directors exchanged all related party payables and outstanding warrants for an aggregate of 7,902,603 shares of our common stock, leaving us with no related party payables and no outstanding warrants on September 30, 2019.
On December 15, 2020, we announced we had entered into an option agreement with Westport Bio to in-license a pre-clinical coronavirus vaccine program that is currently at the stage of pre-clinical animal testing. The option is exercisable at our discretion. In exchange for the option, we have agreed to sponsor research with Westport Bio in an aggregate amount of up to $250,000, plus additional license and assignment fees. The founder of Westport Bio is our Chief Executive Officer and director, Snehal Patel
Related Person Transaction Policy
We adopted a related person transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related person transactions. For purposes of our policy only, a related person transaction is a transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we and any related person are, were or will be participants in which the amount involved exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end. Transactions involving compensation for services provided to us as an employee or director are not covered by this policy. A related person is any executive officer, director or beneficial owner of more than 5% of any class of our voting securities, including any of their immediate family members and any entity owned or controlled by such persons.
Under the policy, if a transaction has been identified as a related person transaction, including any transaction that was not a related person transaction when originally consummated or any transaction that was not initially identified as a related person transaction prior to consummation, our management must present information regarding the related person transaction to our audit committee, or, if audit committee approval would be inappropriate, to another independent body of our board of directors, for review, consideration and approval or ratification. The presentation must include a description of, among other things, the material facts, the interests, direct and indirect, of the related persons, the benefits to us of the transaction and whether the transaction is on terms that are comparable to the terms available to or from, as the case may be, an unrelated third party or to or from employees generally. Under the policy, we will collect information that we deem reasonably necessary from each director, executive officer and, to the extent feasible, significant stockholder to enable us to identify any existing or potential related-person transactions and to effectuate the terms of the policy. In addition, under our code of business conduct and ethics, our employees and directors have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest. In considering related person transactions, our audit committee, or other independent body of our board of directors, will take into account the relevant available facts and circumstances including, but not limited to:
| ● | the risks, costs and benefits to us; | |
| ● | the impact on a director’s independence in the event that the related person is a director, immediate family member of a director or an entity with which a director is affiliated; | |
| ● | the availability of other sources for comparable services or products; and | |
| ● | the terms available to or from, as the case may be, unrelated third parties or to or from employees generally. |
The policy requires that, in determining whether to approve, ratify or reject a related person transaction, our audit committee, or other independent body of our board of directors, must consider, in light of known circumstances, whether the transaction is in, or is not inconsistent with, our best interests and those of our stockholders, as our audit committee, or other independent body of our board of directors, determines in the good faith exercise of its discretion.
Director Independence
Our board of directors undertook a review of the independence of our directors and considered whether any director has a relationship with us that could compromise that director’s ability to exercise independent judgment in carrying out that director’s responsibilities. Our board of directors has affirmatively determined that David McWilliams, Eric Rothe and Kenneth Hallock are each an “independent director,” as defined under the Nasdaq rules.
| 65 |
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Audit Fees
The aggregate fees billed to us by MaloneBailey, LLP, our independent registered public accounting firm, for the indicated services for each of the last two fiscal years were as follows:
| 2020 | 2019 | |||||||
| Audit fees (1) | $ | 69,000 | $ | 15,000 | ||||
| (1) | Audit fees consist of fees for professional services performed by MaloneBailey for the audit and review of our financial statements, preparation and filing of our registration statements, including issuance of comfort letters. |
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Auditors
Consistent with SEC policies and guidelines regarding audit independence, the Audit Committee is responsible for the pre-approval of all audit and permissible non-audit services provided by our independent registered public accounting firm on a case-by-case basis. Our Audit Committee has established a policy regarding approval of all audit and permissible non-audit services provided by our principal accountants. Our Audit Committee pre-approves these services by category and service. Our Audit Committee has pre-approved all of the services provided by our independent registered public accounting firm.
| 66 |
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
Exhibit Number |
Description of Exhibit | |
| (a)(1) Financial Statements | ||
| The financial statements required by this item are submitted in a separate section beginning on page F-1 of this Annual Report on Form 10-K. |
| + | Indicates a management contract or compensatory plan or arrangement. |
None.
| 67 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| GREENWICH LIFESCIENCES, INC. | |
| /s/ Snehal Patel | |
| March 31, 2021 | Chief Executive Officer (Principal Executive Officer and Principal Accounting and Financial Officer) |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Snehal Patel as his or her attorney-in-fact, with full power of substitution and resubstitution, for him or her in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| SIGNATURE | TITLE | DATE | ||
| /s/ Snehal Patel | Chief Executive Officer and Director | March 31, 2021 | ||
| Snehal Patel | (Principal Executive Officer and Principal Accounting and Financial Officer) | |||
| /s/ F. Joseph Daugherty | Chief Medical Officer and Director | March 31, 2021 | ||
| F. Joseph Daugherty | ||||
| /s/ David McWilliams | Director | March 31, 2021 | ||
| David McWilliams | ||||
| /s/ Eric Rothe | Director | March 31, 2021 | ||
| Eric Rothe | ||||
| /s/ Kenneth Hallock | Director | March 31, 2021 | ||
| Kenneth Hallock |
| 68 |
GREENWICH LIFESCIENCES, INC.
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
Greenwich Lifesciences, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Greenwich LifeSciences, Inc. (the “Company”) as of December 31, 2020 and 2019, and the related statements of operations, stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ MaloneBailey, LLP
www.malonebailey.com
We have served as the Company’s auditor since 2019.
Houston, Texas
March 31, 2021
| F-2 |
BALANCE SHEETS
AS OF DECEMBER 31, 2020 AND 2019
| December 31, 2020 | December 31, 2019 | |||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash | $ | 28,660,375 | $ | 6,835 | ||||
| Total current assets | 28,660,375 | 6,835 | ||||||
| Acquired patents, net | 16,227 | 19,836 | ||||||
| Total assets | $ | 28,676,602 | $ | 26,671 | ||||
| Liabilities and stockholders’ deficit | ||||||||
| Current liabilities | ||||||||
| Accounts payable & accrued interest | $ | 710,971 | $ | 730,309 | ||||
| Unreimbursed expenses | 59,367 | 11,626 | ||||||
| Advance from related party/shareholder | 275,154 | 635,154 | ||||||
| Total current liabilities | 1,045,492 | 1,377,089 | ||||||
| Total liabilities | 1,045,492 | 1,377,089 | ||||||
| Stockholders’ equity (deficit) | ||||||||
| Common stock, $0.001 par value; 100,000,000 shares authorized; 12,703,541 and 8,458,048 shares issued and outstanding as of December 31, 2020 and 2019, respectively | 12,704 | 8,458 | ||||||
| Preferred stock, $0.001 par value; 10,000,000 shares authorized; | ||||||||
| Series A preferred stock: No shares as of December 31, 2020 and 1,520,937 shares issued and outstanding as of December 31, 2019 | — | 1,521 | ||||||
| Series B preferred stock: No shares as of December 31, 2020 and 129,267 shares issued and outstanding as of December 31, 2019 | — | 129 | ||||||
| Series C preferred stock: No shares as of December 31, 2020 and 66,575 shares issued and outstanding as of December 31, 2019 | — | 67 | ||||||
| Series D preferred stock: No shares as of December 31, 2020 and 263,586 shares issued and outstanding as December 31, 2019 | — | 264 | ||||||
| Additional paid-in capital | 56,695,359 | 25,853,134 | ||||||
| Accumulated deficit | (29,076,953 | ) | (27,213,991 | ) | ||||
| Total stockholders’ equity (deficit) | 27,631,110 | (1,350,418 | ) | |||||
| Total liabilities and stockholders’ equity (deficit) | $ | 28,676,602 | $ | 26,671 | ||||
See accompanied notes to financial statements.
| F-3 |
STATEMENTS OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2020 AND 2019
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Revenue | $ | — | $ | — | ||||
| Operating expenses | ||||||||
| Research and development | 1,057,606 | 2,606,420 | ||||||
| General and administrative | 806,188 | 818,887 | ||||||
| Total operating expenses | 1,863,794 | 3,425,307 | ||||||
| Loss from operations | (1,863,794 | ) | (3,425,307 | ) | ||||
| Interest income | 832 | |||||||
| Net loss | $ | (1,862,962 | ) | $ | (3,425,307 | ) | ||
| Per share information: | ||||||||
| Net loss per common share, basic and diluted | $ | (0.20 | ) | $ | (1.52 | ) | ||
| Weighted average common shares outstanding, basic and diluted | 9,499,155 | 2,257,979 | ||||||
See accompanied notes to financial statements.
| F-4 |
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
FOR THE YEARS ENDED DECEMBER 31, 2020 AND 2019
| Common Stock | Preferred Stock | Additional | Total Stockholders’ | |||||||||||||||||||||||||
| Shares | Par Amount | Shares | Par Amount | Paid-in Capital | Accumulated Deficit | Equity (Deficit) | ||||||||||||||||||||||
| Balances, December 31, 2018 | 202,996 | $ | 203 | 1,980,365 | $ | 1,981 | $ | 13,666,446 | $ | (23,788,684 | ) | $ | (10,120,054 | ) | ||||||||||||||
| Exchange of related party payables and warrants for common stock | 8,012,684 | 8,013 | — | — | 11,991,987 | — | 12,000,000 | |||||||||||||||||||||
| Stock-based compensation | 242,368 | 242 | — | — | 194,701 | — | 194,943 | |||||||||||||||||||||
| Net loss | (3,425,307 | ) | (3,425,307 | ) | ||||||||||||||||||||||||
| Balances, December 31, 2019 | 8,458,048 | 8,458 | 1,980,365 | 1,981 | 25,853,134 | (27,213,991 | ) | (1,350,418 | ) | |||||||||||||||||||
| Stock-based compensation | 301,854 | 302 | — | — | 677,686 | — | 677,988 | |||||||||||||||||||||
| Issuance of common stock in initial public offering, net of offering costs | 1,260,870 | 1,261 | — | — | 6,206,241 | — | 6,207,502 | |||||||||||||||||||||
| Additional preferred stock issued due to anti-dilution | — | — | 42,404 | 42 | (42 | ) | — | — | ||||||||||||||||||||
| Conversion of preferred to common stock | 2,022,769 | 2,023 | (2,022,769 | ) | (2,023 | ) | — | — | — | |||||||||||||||||||
| Issuance of common stock in follow-on offering, net of offering costs | 660,000 | 660 | — | — | 23,958,340 | — | 23,959,000 | |||||||||||||||||||||
| Net loss | (1,862,962 | ) | (1,862,962 | ) | ||||||||||||||||||||||||
| Balances, December 31, 2020 | 12,703,541 | $ | 12,704 | — | $ | — | $ | 56,695,359 | $ | (29,076,953 | ) | $ |
27,631,110 | |||||||||||||||
See accompanied notes to financial statements.
| F-5 |
STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2020 AND 2019
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (1,862,962 | ) | $ | (3,425,307 | ) | ||
| Adjustments required to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization | 3,609 | 3,607 | ||||||
| Stock-based compensation | 677,988 | 194,943 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts payable | (92,550 | ) | 393,402 | |||||
| Accrued interest | 73,212 | 59,353 | ||||||
| Unreimbursed expenses (accrued) | 47,741 | (19,263 | ) | |||||
| Related party payable | — | 2,500,000 | ||||||
| Net cash used in operating activities | (1,152,962 | ) | (293,267 | ) | ||||
| Investing activities: | ||||||||
| Financing activities: | ||||||||
| Net proceeds from initial public offering and follow-on offering of common stock | 30,166,502 | — | ||||||
| Repayment to related party/shareholder | (360,000 | ) | — | |||||
| Advance from related party/shareholder | — | 215,000 | ||||||
| Net cash provided by (used in) financing activities | 29,806,502 | 215,000 | ||||||
| Net increase (decrease) in cash | 28,653,540 | (78,267 | ||||||
| Cash, beginning of period | 6,835 | 85,102 | ||||||
| Cash, end of period | $ | 28,660,375 | $ | 6,835 | ||||
| Non-cash investing and financing activities: | ||||||||
| Common stock to settle related party payable | — | 12,000,000 | ||||||
| Conversion of preferred stock to common | 2,023 | — | ||||||
| Issuance of preferred stock due to antidilution | 42 | — | ||||||
See accompanied notes to financial statements.
| F-6 |
GREENWICH LIFESCIENCES, INC.
1. Organization and Description of the Business
Greenwich LifeSciences, Inc. (the “Company”) was incorporated in the state of Delaware in 2006 under the name Norwell, Inc. In March 2018, Norwell, Inc. changed its name to Greenwich LifeSciences, Inc. The Company is developing a breast cancer immunotherapy focused on preventing the recurrence of breast cancer following surgery.
2. Going Concern
On August 27, 2014, the Financial Accounting Standards Board issued Accounting Standards Update (“ASU”) 2014-05, Disclosure of Uncertainties about an Entity’s ability to Continue as a Going Concern (“ASU 2014-05”), which requires management to assess a company’s ability to continue as a going concern within one year from financial statement issuance and to provide related footnote disclosures in certain circumstances.
The accompanying financial statements and notes have been prepared assuming the Company will continue as a going concern. During the year ended December 31, 2019, the Company suffered from recurring losses from operations and negative cash flows from operations, resulting in a need for, among other things, capital resources. As of December 31, 2019, the Company had cash of $6,835 and disclosed that its ability to continue as a going concern was predicated on the Company’s ability to raise capital and to sustain adequate working capital to finance its operations. In September 2020, the Company completed its initial public offering and raised $7,250,002 in gross proceeds and $6,207,502 in net proceeds, after deducting underwriting discounts and commissions and other offering expenses. In December 2020, the Company completed a follow-on offering and raised $26,400,000 in gross proceeds and $23,959,000 in net proceeds, after deducting underwriting discounts and commissions and other offering expenses. The Company met and exceeded those predications thus mitigating any substantial doubt about the Company’s ability to continue as a going concern as defined by ASU 2014-05 and its ability to satisfy the estimated liquidity needs for the twelve months from the issuance of the financial statements.
As of December 31, 2020, the Company had cash of $28,660,375.
3. Significant Accounting Policies
Basis of Presentation
The accompanying financial statements are presented in conformity with accounting principles generally accepted in the United States of America (“GAAP”) and pursuant to the rules and regulations of US Securities and Exchange Commission (“SEC”).
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in its financial statements and accompanying notes. On an ongoing basis, management evaluates these estimates and judgments, which are based on historical and anticipated results and trends and on various other assumptions that management believes to be reasonable under the circumstances. By their nature, estimates are subject to an inherent degree of uncertainty and, as such, actual results may differ from management’s estimates.
Cash
Cash consists primarily of deposits with commercial banks and financial institutions.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment when events or changes in circumstances indicate the carrying value of the assets may not be recoverable. Recoverability is measured by comparison of the book values of the assets to future net undiscounted cash flows that the assets or the asset groups are expected to generate. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the book value of the assets exceed their fair value, which is measured based on the estimated discounted future net cash flows arising from the assets or asset groups. No impairment losses on long-lived assets have been recorded through December 31, 2020.
Stock-Based Compensation
Compensation expense related to warrants and stock granted to employees and non-employees is measured at the grant date based on the estimated fair value of the award and is recognized on a straight-line basis over the requisite service period. Forfeitures are recognized as a reduction of stock-based compensation expense as they occur. Stock-based compensation expense for an award with a performance condition is recognized when the achievement of such performance condition is determined to be probable. If the outcome of such performance condition is not determined to be probable or is not met, no compensation expense is recognized and any previously recognized compensation expense is reversed.
| F-7 |
GREENWICH LIFESCIENCES, INC.
NOTES TO FINANCIAL STATEMENTS
3. Significant Accounting Policies (cont.)
Research and Development Costs
Research and development expenses are charged to operations as incurred. Research and development expenses include, among other things, salaries, costs of outside collaborators and outside services, and supplies.
Income Taxes
The Company’s income tax returns are based on calculations and assumptions that are subject to examination by the Internal Revenue Service and other tax authorities. In addition, the calculation of tax liabilities involves dealing with uncertainties in the application of complex tax regulations.
Basic and Diluted Loss per Share
The Company computes loss per share in accordance with Accounting Standards Codification (“ASC”) 260 — Earnings per Share. ASC 260 requires presentation of both basic and diluted earnings per share (“EPS”) on the face of the statements of operations. Basic EPS is computed by dividing net loss available to common shareholders (numerator) by the weighted average number of common shares outstanding (denominator) during the period. Diluted EPS gives effect to all dilutive potential common shares outstanding during the period using the treasury stock method and convertible notes payable using the if-converted method. Diluted EPS excludes all dilutive potential shares if their effect is antidilutive. During periods of net loss, all common stock equivalents are excluded from the diluted EPS calculation because they are antidilutive.
As of December 31, 2020, the Company has common stock equivalents related to warrants outstanding to acquire 100,869 shares of the Company’s common stock. As of December 31, 2019, the Company had no warrants.
As of December 31, 2020, the Company has no common stock equivalents related to convertible preferred stock issued and outstanding. As of December 31, 2019, the Company had common stock equivalents related to 1,520,937 shares of the Company’s common stock issuable upon conversion of the Company’s Series A Preferred Stock, 129,267 shares of the Company’s common stock issuable upon conversion of the Company’s Series B Preferred Stock, 66,575 shares of the Company’s common stock issuable upon conversion of the Company’s Series C Preferred Stock, and 263,586 shares of the Company’s common stock issuable upon conversion of the Company’s Series D Preferred Stock issued and outstanding.
Recent Accounting Pronouncements
The Company has evaluated the following recent accounting pronouncements through the date the financial statements were issued and filed with the SEC and believes that none of them will have a material effect on the Company’s financial statements:
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-02, “Leases: Topic 842 (ASU 2016-02)”, to supersede nearly all existing lease guidance under GAAP. The guidance would require lessees to recognize most leases on their balance sheets as lease liabilities with corresponding right-of-use assets. ASU 2016-02 is effective for the Company in the first quarter of its fiscal year ending December 31, 2019 using a modified retrospective approach with the option to elect certain practical expedients. The Company has no material leases, thus the adoption of ASU 2016-02 will have no material impact on the Company’s financial statements.
In May 2016, the FASB issued ASU 2016-12, Revenue from Contracts from Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients. The amendments in this update affect the guidance in ASU 2014-09. The core principle of the guidance in Topic 606 is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The amendments in ASU 2016-12 do not change the core principle of the guidance in Topic 606, but instead affect only the narrow aspects noted in Topic 606. Topic 606 became effective for the Company on December 1, 2018. The Company has no revenue, thus the adoption of ASU 2016-12 will have no material impact on the Company’s financial statements.
| F-8 |
GREENWICH LIFESCIENCES, INC.
NOTES TO FINANCIAL STATEMENTS
3. Significant Accounting Policies (cont.)
In June 2018, the FASB issued ASU 2018-07, “Compensation-Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting,” which modifies the accounting for share-based payment awards issued to nonemployees to largely align it with the accounting for share-based payment awards issued to employees. ASU 2018-07 is effective for us for annual periods beginning January 1, 2019. The Company evaluated ASU 2018-07 and determined that the adoption of this new accounting standard did not have a material impact on the Company’s financial statements.
4. Related Party Transactions
Unreimbursed expenses have been accrued and incurred by management, which total $59,367 as of December 31, 2020 and $11,626 as of December 31, 2019. In October 2019, the Kenneth Hallock and Annette Hallock Revocable Trust loaned $200,000 to the Company and Eric Rothe, a director of the Company, loaned $15,000 to the Company, both of which are payable on demand, are not secured, and do not incur interest. Kenneth Hallock, a director of the Company, is one of the Trustees of the Hallock Trust. In 2018, the Kenneth Hallock and Annette Hallock Revocable Trust loaned $100,000 to the Company that is payable on demand, not secured, and does not incur interest. In total, Snehal Patel, Company’s Chief Executive Officer and director, Eric Rothe, and the Kenneth Hallock and Annette Hallock Revocable Trust have loaned capital to the Company that is payable on demand, is not secured, and does not incur interest, which in the aggregate totals $275,154 as of December 31, 2020 and $635,154 as of December 31, 2019. In 2020, an aggregate of $360,000 of the outstanding loan balance as of December 31, 2019 was paid off by the Company to the related parties.
| F-9 |
GREENWICH LIFESCIENCES, INC.
NOTES TO FINANCIAL STATEMENTS
4. Related Party Transactions (cont.)
Related party payables to the Company’s officers and directors since January 1, 2010 total $12.0 million as of September 30, 2019. Related party payables were decreased from $12.0 million to $0 and all of the Company’s 2,675,602 warrants were cancelled on September 30, 2019, as all related party payables and all warrants were exchanged for an aggregate of 8,012,684 shares of the Company’s common stock on September 30, 2019. There are no related party payables as of December 31, 2020 and December 31, 2019.
5. Income Taxes
Significant components of the Company’s deferred tax assets and liabilities were as follows:
| December 31, | ||||||||
| 2020 | 2019 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | 1,039,177 | 790,333 | ||||||
| Valuation allowance | (1,039,177 | ) | (790,333 | ) | ||||
| Total deferred tax assets | — | — | ||||||
The federal income tax rate used for 2020 and 2019 was 21%. At December 31, 2020, the Company had federal net operating loss (“NOL”) carryforwards of approximately $4.9 million that will expire in tax years up through 2037. The NOLs generated in tax years 2018 and forward will carry forward indefinitely, but the deductibility of such federal net operating losses is limited. The NOL and tax credit carryforwards may be further subject to the application of Section 382 of the Internal Revenue Code of 1986, as amended (the “Code”), as discussed further below. The Company has provided a valuation allowance to offset the deferred tax assets due to the uncertainty of realizing the benefits of the net deferred tax asset.
The Company’s issuances of common and preferred stock have likely resulted in ownership changes as defined by Section 382 of the Code; however, the Company has not conducted a Section 382 study to date. It is possible that a future analysis may result in the conclusion that a substantial portion, or perhaps substantially all of the Company’s NOL carryforwards and R&D tax credit carryforwards will expire due to the limitations of Sections 382 and 383 of the Code. As a result, the utilization of the carryforwards may be limited and a portion of the carryforwards may expire unused.
The Company is subject to U.S. federal tax examinations by tax authorities for the years 2010 to 2009 due to the fact that NOL carryforwards exist going back to 2010 that may be utilized on a current or future year tax return.
6. Commitments and Contingencies
License Obligation, Legal Expenses, and Manufacturing Agreements
The Company entered into an exclusive license agreement with The Henry M. Jackson Foundation (“HJF”) in April 2009, as amended, pursuant to which it acquired exclusive marketing rights to GP2, the Company’s product candidate. In consideration for such licensed rights, the Company issued HJF 202,619 shares of the Company’s common stock valued at $0.267 per share, which is amortized over 15 years at $3,607 per year. Pursuant to the exclusive license agreement, the Company is required to pay an annual maintenance fee, milestone payments and royalty payments based on sales of GP2 and to reimburse HJF for patent expenses related to GP2. The Company currently depends on third-party contract manufacturers for all required raw materials, active pharmaceutical ingredients, and finished product candidate for the Company’s clinical trials.
Accounts payable includes accrued patent and license obligations to HJF, including accrued interest, plus accrued expenses for manufacturing of GP2 for the upcoming Phase III clinical trial through purchase orders with Polypeptide Laboratories and Stratum Medical, and legal expenses with Sheppard Mullin, which total $710,971 as of December 31, 2020 and $730,309 as of December 31, 2019.
| F-10 |
GREENWICH LIFESCIENCES, INC.
NOTES TO FINANCIAL STATEMENTS
6. Commitments and Contingencies (cont.)
Legal Proceedings
From time to time, the Company may be involved in disputes, including litigation, relating to claims arising out of operations in the normal course of business. Any of these claims could subject the Company to costly legal expenses and, while management generally believes that there will be adequate insurance to cover different liabilities at such time the Company becomes a public company and commences clinical trials, the Company’s future insurance carriers may deny coverage or policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on the results of operations and financial position. Additionally, any such claims, whether or not successful, could damage the Company’s reputation and business. The Company is currently not a party to any legal proceedings, the adverse outcome of which, in management’s opinion, individually or in the aggregate, could have a material adverse effect on our results of operations or financial position.
7. Stockholders’ Equity
In 2019, an aggregate total of 8,255,052 shares of the Company’s common stock were issued to retire all related party payables, to cancel all warrants, and to compensate and incentivize management, directors, and consultants.
On September 30, 2019, the board of directors (the “Board”) and stockholders of the Company adopted the Greenwich LifeSciences, Inc. 2019 Equity Incentive Plan setting aside and reserving 1,498,128 shares of common stock without any issuance of common stock or options under the plan. In addition, on September 30, 2019, the Board authorized the Company to enter into a lock-up/leak-out agreement with its shareholders, the size of the Board was increased from three to five members, two new members were appointed to the Board, $12 million of related party payables and 2,675,602 warrants were exchanged for 8,012,684 shares of the Company’s common stock, and 155,433 shares of the Company’s common stock were issued upfront at no value in consideration for services and 908,242 shares of the Company’s common stock were authorized to be issued at $2,037,000 value based on various vesting schedules that start monthly vesting on October 1, 2019 and on the first day of each subsequent month.
As of December 31, 2020, 379,425 shares of the 908,242 shares of the common stock grant had vested at approximately $853,706 value and 528,817 shares remain unvested and unrecognized at approximately $1,189,838 value. In 2020, 301,854 shares of the common stock grant vested at approximately $677,988 value.
On December 30, 2019, the Company issued a consultant 9,364 shares of the Company’s common stock for services rendered at approximately $21,000 value.
Pursuant to ASU 2018-07, “Compensation-Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting,” the Company’s warrants were valued using the Black-Scholes option pricing model. Assumptions used in the valuation include the following: a) market value of stock on measurement date of $0.00; b) risk-free rate of 0.49%; c) volatility factor of 109%; d) dividend yield of 0.00%. Based on the valuation, the warrants had no value on the grant date of September 30, 2019.
In addition, the Company modified the exercise price of all 2,675,602 warrants to $0 on the modification date of September 30, 2019, and thus the Company exchanged the 2,675,602 warrants for 2,675,602 shares of the Company’s common stock at no value on the modification date. The warrants were valued using the Black-Scholes option pricing model. Assumptions used in the valuation include the following: a) market value of stock on measurement date of $0.00; b) risk-free rate of 0.49%; c) volatility factor of 109%; d) dividend yield of 0.00%. Based on the valuation, the modified warrants had no value on the modification date of September 30, 2019. Therefore, no incremental expense was recorded due to the modification.
On June 22, 2020, the Company filed an amendment to its Amended and Restated Certificate of Incorporation, as amended (the “Certificate of Incorporation”), to effectuate a 1-for-2.67 reverse stock split of the Company’s issued and outstanding common and preferred stock. No fractional shares were issued and any fractional shares resulting from the stock split were rounded up to the nearest whole share. All common and preferred stock share and per-share data and conversion or exercise price data for applicable common stock equivalents included in these financial statements have been retroactively adjusted to reflect the reverse stock split.
| F-11 |
GREENWICH LIFESCIENCES, INC.
NOTES TO FINANCIAL STATEMENTS
No new equity was raised in 2019.
Initial Public Offering (IPO)
On September 25, 2020, the Company completed its initial public offering (the “IPO”) pursuant to which it issued and sold 1,260,870 shares of its common stock at a public offering price of $5.75 per share for gross proceeds of $7,250,002 and net proceeds of $6,207,502, after deducting underwriting discounts and commissions and offering expenses borne by the Company, which totaled $1,042,500. In addition, the Company granted the underwriters a 45-day option to purchase up to 189,130 additional shares of common stock at the public offering price, less offering expenses, to cover over-allotments, if any.
On September 29, 2020, in connection with the completion of the IPO, the Company converted all of the outstanding shares of Series A Preferred Stock into an aggregate of 1,520,937 shares of common stock, all of the outstanding shares of Series B Preferred Stock into an aggregate of 129,267 shares of common stock, all of the outstanding shares of Series C Preferred Stock into an aggregate of 66,575 shares of common stock and all of the outstanding shares of Series D Preferred Stock into an aggregate of 305,990 shares of common stock upon the closing of the IPO, which included the issuance of an aggregate of 42,404 additional shares of common stock upon the issuance and conversion of an additional 42,404 shares of Series D Preferred Stock issuable in connection with the IPO as a result of the anti-dilution protection set forth in the Company’s Certificate of Incorporation; based upon the IPO price of $5.75 per share.
On September 29, 2020, in connection with the completion of the IPO, the Board and stockholders of the Company approved the Company’s Second Amended and Restated Bylaws and the filing of the Company’s Second Amended and Restated Certificate of Incorporation with the Delaware Secretary of State which authorizes the Company to issue 100,000,000 shares of common stock with a par value of $0.001 per share and 10,000,000 shares of preferred stock with a par value of $0.001 per share. In addition, on September 29, 2020, the Company entered into an employment agreement with Snehal Patel pursuant to which Mr. Patel will serve as the Company’s Chief Executive Officer as described in the Company Current Report on Form 8-K filed with the SEC on October 1, 2020.
Follow-On Offering
On December 22, 2020, the Company completed a follow-on offering pursuant to which it issued and sold 660,000 shares of its common stock at a public offering price of $40.00 per share for gross proceeds of $26,400,000 and net proceeds of $23,959,000, after deducting underwriting discounts and commissions and offering expenses borne by the Company, which totaled $2,441,000. In addition, the Company granted the underwriters a 45-day option to purchase up to 99,000 additional shares of common stock at the public offering price, less offering expenses, to cover over-allotments, if any.
| F-12 |
Warrants
Prior to the IPO, there were no outstanding warrants to purchase shares of common stock accounted for as equity or liabilities.
On September 25, 2020, in connection with the IPO, the underwriter, Aegis Capital Corp., was issued a warrant to purchase 100,870 shares of common stock, representing 8% of the number of shares sold in the IPO, excluding the over-allotment option. The warrants will be exercisable at any time and from time to time, in whole or in part, during a period commencing March 24, 2021 and expiring September 24, 2025. The warrants will be exercisable at a price equal to $7.1875 per share, which represents 125% of the public offering price per share of common stock sold in the IPO. In the event that a registration statement registering the common stock underlying the warrants is not effective, the warrants may be exercised on a cashless basis. If the warrants are exercised for cash within the first six months of the period in which they are exercisable, the exercise price will be equal to 97% of 125% of the public offering price or $6.9718 per share.
At December 31, 2020, outstanding warrants to purchase shares of common stock accounted for as equity or liabilities were as follows with an aggregate intrinsic value as of December 31, 2020 of $2,953,726 based on the December 31, 2020 closing share price of $36.47:
| Shares Underlying | ||||||||
| Outstanding | Exercise | Expiration | ||||||
| Warrants | Price(1) | Date(1) | ||||||
| 100,870 | $ | 7.1875 | September 24, 2025 | |||||
| 100,870 | ||||||||
| (1) | The warrants are exercisable at any time and from time to time, in whole or in part, during a period commencing March 24, 2021 and expiring September 24, 2025. The exercise price of the warrants is $7.1875 per share or $6.9718 per share if the warrants are exercised for cash within the first six months of the period in which they are exercisable. |
8. Subsequent Events
On January 29, 2021, the underwriter exercised its option to purchase 70,000 additional shares of common stock at the public offering price of $40.00 per share for gross proceeds of $2,800,000 and net proceeds of $2,548,000, after deducting underwriting discounts and commissions and offering expenses borne by the Company, which totaled $252,000.
Between January 1, 2021 and March 15, 2021, the Company paid off the remaining related party loans of $155,154 and $120,000 to Snehal Patel and the Kenneth Hallock and Annette Hallock Revocable Trust, respectively.
An aggregate of 73,356 shares of common stock were vested in January, February, and March 2021 in consideration for services rendered.
| F-13 |