Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
___________________________________________
FORM 10-K
___________________________________________
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2019
OR
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to | |
Commission File Number: 000-19756
___________________________________________

PDL BioPharma, Inc.
(Exact name of registrant as specified in its charter)
___________________________________________
Delaware | 94-3023969 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
932 Southwood Boulevard
Incline Village, Nevada 89451
(Address of principal executive offices)
Registrant’s telephone number, including area code
(775) 832-8500
___________________________________________
Securities registered pursuant to Section 12(b) of the Act:
Title of Class | Trading Symbol | Name of Exchange on which Registered |
Common Stock, par value $0.01 per share | PDLI | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
___________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
Large accelerated filer ¨ | Accelerated filer ý | |||
Non-accelerated filer ¨ | Smaller reporting company ¨ | |||
Emerging growth company ¨ | ||||
If an emerging growth company, indicated by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act ¨ | ||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No ý
The aggregate market value of shares of common stock held by non-affiliates of the registrant, based on the closing sale price of a share of common stock on June 28, 2019 (the last business day of the registrant’s most recently completed second fiscal quarter), as reported on the Nasdaq Global Select Market, was $354,547,520.
As of February 28, 2020, the registrant had outstanding 123,591,824 shares of common stock.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s proxy statement to be delivered to stockholders with respect to the registrant’s 2020 Annual Meeting of Stockholders to be filed by the registrant with the U.S. Securities and Exchange Commission are incorporated by reference into Part III of this Annual Report on Form 10-K. The registrant intends to file its proxy statement within 120 days after its fiscal year end.
PDL BIOPHARMA, INC.
2019 Form 10-K Annual Report
Table of Contents
PART I | |||
Item 1 | |||
Item 1A | |||
Item 1B | |||
Item 2 | |||
Item 3 | |||
Item 4 | |||
PART II | |||
Item 5 | |||
Item 6 | |||
Item 7 | |||
Item 7A | |||
Item 8 | |||
Item 9 | |||
Item 9A | |||
Item 9B | |||
PART III | |||
Item 10 | |||
Item 11 | |||
Item 12 | |||
Item 13 | |||
Item 14 | |||
PART IV | |||
Item 15 | |||
Item 16 | |||
PART I
Forward-looking Statements
This Annual Report contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). All statements other than statements of historical facts are “forward-looking statements” for purposes of these provisions, including any projections of earnings, revenues or other financial items, any statements of the plans and objectives of management for future operations, including any statements concerning the timing, implementation or success of our monetization strategy/plan of complete liquidation, any statements regarding future economic conditions or performance, and any statement of assumptions underlying any of the foregoing. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. In some cases, forward-looking statements can be identified by the use of terminology such as “may,” “will,” “intends,” “plans,” “believes,” “targets,” “anticipates,” “expects,” “estimates,” “predicts,” “potential,” “continue” or “opportunity,” or the negative thereof or other comparable terminology. The forward-looking statements in this Annual Report are only predictions. Although we believe that the expectations presented in the forward-looking statements contained herein are reasonable at the time of filing, there can be no assurance that such expectations or any of the forward-looking statements will prove to be correct. These forward-looking statements, including with regards to our future financial condition and results of operations, are subject to inherent risks and uncertainties, including but not limited to the risk factors set forth below, and for the reasons described elsewhere in this Annual Report. All forward-looking statements and reasons why results may differ included in this Annual Report are made as of the date hereof. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
We own or have rights to certain trademarks, trade names, copyrights and other intellectual property used in our business, including PDL BioPharma, Inc. and the PDL logo, each of which is considered a registered trademark. All other company names, product names, trade names and trademarks included in this Annual Report are trademarks, registered trademarks or trade names of their respective owners.
3
ITEM 1. BUSINESS
Overview
In this report all references to “PDL,” “we,” “us,” “our” or the “Company” mean collectively PDL BioPharma, Inc. and its subsidiaries, except where it is made clear that the term means only PDL BioPharma, Inc.
Throughout our history, our mission has been to improve the lives of patients by aiding in the successful development of innovative therapeutics and healthcare technologies. PDL BioPharma was founded in 1986 as Protein Design Labs, Inc. when it pioneered the humanization of monoclonal antibodies, enabling the discovery of a new generation of targeted treatments that have had a profound impact on patients living with different cancers as well as a variety of other debilitating diseases. In 2006, we changed our name to PDL BioPharma, Inc.
In September 2019, we engaged financial and legal advisors and initiated a review of our strategy. In December 2019, we disclosed that we planned to halt the execution of our growth strategy, cease making additional strategic transactions and investments and instead pursue a formal process to unlock the value of our portfolio by monetizing our assets and ultimately distributing net proceeds to stockholders. Over the subsequent months, our board of directors and management analyzed, together with outside financial and legal advisors, how to best capture value pursuant to our monetization strategy and best return the significant intrinsic value of the high-quality assets in our portfolio to our stockholders. In February 2020, our board of directors determined to seek stockholder approval to dissolve the company pursuant to a plan of complete liquidation under Delaware law at our next annual meeting of the stockholders. In the event that the board of directors concludes that the whole company sale process is unlikely to maximize the value that can be returned to the stockholders from our monetization process, the company would, if approved by the stockholders, file a Certificate of Dissolution in Delaware and proceed to wind-down and dissolve the company in accordance with Delaware law. Pursuant to our monetization strategy, we are exploring a variety of potential transactions, including a whole company sale, divestiture of assets, spin-offs of operating entities, merger opportunities or a combination thereof. In addition, we have analyzed, and continue to analyze, the optimal mechanisms for returning value to stockholders in a tax-efficient manner, including via share repurchases, cash dividends and other distributions of assets. We have not set a definitive timeline and intend to pursue monetization in a disciplined and cost-effective manner seeking to maximize returns to stockholders. We recognize, however, that accelerating the timeline, while continuing to seek to optimize asset value, could increase returns to stockholders due to reduced general and administrative expenses as well as potentially provide faster returns to stockholders. While, as noted above, we cannot provide a definitive timeline for the monetization and wind-down process, we are targeting the end of 2020 for completing the monetization of our key assets.
In conjunction with our intent to seek stockholder approval for complete dissolution of the company, a proxy statement will be presented to the stockholders that identifies in detail the rationale for the board of director’s decision to seek stockholder approval for dissolution and further presents the risk factors associated with such dissolution. We will continue to be receptive to offers to purchase the entire company throughout the monetization process, with all or less than all of our current assets, should such an offer be made. However, if we conclude that a whole company sale is unlikely or that the value from a whole company sale will not maximize the returns we can provide to our stockholders, we expect that the proposed wind-down will ultimately conclude with dissolution in accordance with Delaware law.
To assist us in our monetization strategy, we have retained Bank of America Securities to advise us in a process for a sale of the Company. We have also retained SVB Leerink to advise us generally regarding the monetization strategy. In the event that we conclude that a whole company sale will not maximize value, and that a sale of the assets of the Company, separately or in combination, will provide more significant stockholder value, we have retained Torreya Partners to advise us in our monetization of our Noden asset and shares of Evofem Biosciences, Inc. (“Evofem”), SVB Leerink to advise us in our monetization of LENSAR, Inc. (“LENSAR”), and Bank of America Securities to advise us in a sale of our royalty assets.
Historically, we generated a substantial portion of our revenues through the license agreements related to patents covering the humanization of antibodies, which we refer to as the Queen et al. patents. In 2012, and in anticipation of declining revenues from the Queen et al. patents, we began providing alternative sources of capital through royalty monetizations and debt facilities, and, in 2016, we began acquiring commercial-stage products and launching specialized companies dedicated to the commercialization of these products. Prior to 2016, we had a history of paying quarterly dividends to stockholders. The dividend payments were first reduced and then eliminated in 2016 due to the declining revenues from the Queen et al. patents and a change in strategy. Beginning in March 2017, we began repurchasing shares of our common stock in lieu of paying cash dividends.
Based on the nature of our investments entered into between 2012 through 2016 and further discussed below, our operations were structured in three segments designated as Medical Devices, Pharmaceutical and Income Generating Assets.
4
In early 2019, and as a further evolution of our strategy, we began to enter into strategic transactions involving innovative late clinical-stage or early commercial-stage therapeutics with attractive revenue growth potential with the intention to provide a significant return for our stockholders.
Consistent with this strategy, on April 10, 2019, we entered into a securities purchase agreement with Evofem, pursuant to which we invested $60.0 million in a private placement of securities. The transaction was structured in two tranches. The first tranche comprised $30.0 million, which was funded on April 11, 2019. We had the right to invest an additional $30.0 million in a second tranche, which we did on June 10, 2019, alongside two existing Evofem stockholders, who each invested an additional $10.0 million. These investments were expected to provide funding for Evofem's pre-commercial activities for Amphora®, its investigational, non-hormonal, on-demand prescription contraceptive gel for women. We believe this investment provided us the ability to take a significant position in a promising company at a critical stage of development where we could provide meaningful contributions through our capital and expertise.
As a result of this investment in Evofem we established a fourth segment, “Strategic Positions.”
Our Medical Devices segment consists of revenue from the sale and lease of the LENSAR® Laser System, which may include equipment, Patient Interface Devices (“PIDs”), procedure licenses, training, installation, warranty and maintenance agreements.
Our Strategic Positions segment consists of an investment in Evofem (NASDAQ: EVFM). Our investment includes shares of common stock and warrants to purchase additional shares of common stock. Evofem is a pre-commercial company and, as such, is not yet engaged in revenue-generating activities.
Our Pharmaceutical segment consists of revenue derived from branded prescription medicine products sold under the name Tekturna® and Tekturna HCT® in the United States, Rasilez® and Rasilez HCT® in the rest of the world and revenue generated from the sale of an authorized generic form of Tekturna in the United States (collectively, the “Noden Products”).
Our Income Generating Assets segment consists of revenue derived from (i) notes and other long-term receivables, (ii) royalty rights and hybrid notes/royalty receivables, (iii) equity investments and (iv) royalties from issued patents in the United States and elsewhere covering the humanization of antibodies, which we refer to as the Queen et al. patents.
Financial information about our segments, including our revenues and net (loss) income for the years ended December 31, 2019, 2018 and 2017, and select long-lived assets as of December 31, 2019 and 2018, is included in our Consolidated Financial Statements and accompanying notes in Item 8.
Medical Devices
LENSAR
LENSAR is a medical device company focused on delivering next generation femtosecond cataract laser technology used in refractive cataract surgical procedures. LENSAR’s femtosecond laser uses advanced imaging and laser technology to customize planning and treatments, allowing faster visual recovery and improved outcomes, as compared to conventional cataract surgery, a more manual procedure combined with ultrasound, referred to as phacoemulsification. LENSAR has developed the LENSAR® Laser System, which is the only femtosecond cataract laser built specifically for refractive cataract surgery.
Cataract surgery is the highest volume surgical procedure performed worldwide with 30 million surgeries projected in 2020, the majority of which use conventional phacoemulsification techniques. LENSAR is currently focusing its research and development efforts on an advanced integrated workstation combining an enhanced LENSAR® Laser System and a phacoemulsification device in a single, compact workstation, designed to fit directly in the surgical theater. LENSAR’s recent acquisitions of certain intellectual property uniquely position LENSAR to develop a system that can perform all cataract surgeries in a single platform.
The LENSAR® Laser System offers cataract surgeons automation and customization for their astigmatism treatment planning and other essential steps of the refractive cataract surgery procedure with the highest levels of precision, accuracy, and efficiency. These features assist surgeons in managing their astigmatism treatment plans for optimal overall visual outcomes.
The LENSAR® Laser System has been cleared by the U.S. Food and Drug Administration (“FDA”) for anterior capsulotomy, lens fragmentation, corneal and arcuate incisions. The LENSAR Laser with Augmented Reality™ provides an accurate 3-D model of the relevant anatomical features of each patient’s anterior segment, allowing precise laser delivery and enhanced surgical
5
confidence in performing accurate corneal incisions, precise size, shape and location of free-floating capsulotomies, and efficient lens fragmentation for all grades of cataracts. The LENSAR® Laser System - fs 3D (LLS-fs 3D) with Streamline™ includes the integration with multiple pre-operative diagnostic devices, utilizing automated Iris Registration with automatic cyclorotation adjustment. IntelliAxis-C™ (corneal) and IntelliAxis-L™ (lens capsule) markers provide the surgeon tools for simple and precise alignment without errors associated with manually transposing the preoperative data, and marking the eye for incisions and implantation of Toric IOLs as well as treatment planning tools for precision guided laser treatments. The corneal incision-only mode, expanded remote diagnostics capabilities, additional pre-programmable preferences, thoughtful ergonomics, and up to 20 seconds faster laser treatment times with Streamline allow for seamless integration and maximum surgical efficiency with patient comfort.
Intellectual Property
LENSAR has over 85 granted patents in the United States and rest of the world and over 60 pending patent applications in the United States and rest of the world. LENSAR acquired a number of patents in 2019 to support the development and eventual commercialization of its second generation laser system which will combine a femtosecond cataract laser system with a phacoemulsification system in a single machine.
Manufacturing
Through our LENSAR subsidiary, we currently manufacture our LENSAR® Laser System at a facility in Orlando, Florida.
LENSAR purchases both custom and off-the-shelf components from a small number of suppliers and subjects them to stringent quality specifications and processes. Some of the components necessary for the assembly of the LENSAR® Laser System are currently provided by sole-sourced suppliers (the only recognized supply source available to us) or single-sourced suppliers (the only approved supply source for us among other sources). LENSAR purchases the majority of its components and major assemblies through purchase orders with limited long-term supply agreements and generally does not maintain large volumes of finished goods.
LENSAR has entered into various supply agreements for the manufacture and supply of certain components. The supply agreements commit LENSAR to a minimum purchase obligation of approximately $10.4 million over the next twenty-four months of which $9.6 million is due in the next 12 months. LENSAR expects to meet these requirements.
Sales and Distribution
LENSAR markets and sells the LENSAR® Laser System to ophthalmic ambulatory surgical centers, specialty ophthalmic hospitals and multi-specialty hospitals in the United States through a direct sales force. Outside of the United States, LENSAR typically sells the LENSAR® Laser System through distributors. A distributor in Asia and a distributor in Europe represent 25% and 11%, respectively, of the net sales in our Medical Devices segment for the year ended December 31, 2019.
Competition
The LENSAR® Laser System is a femtosecond cataract laser for refractive cataract surgery. We estimate that the market penetration of femtosecond cataract laser surgery is approximately 10.7% of total procedures in the United States and approximately 2.8% of the total cataract surgeries performed globally. Femtosecond cataract laser procedures are forecast to grow approximately 5.0% annually through 2024.
Employees
As of December 31, 2019, we had 75 full-time employees at LENSAR, who manage its business and operations.
Strategic Positions
Evofem
We invested $60.0 million in Evofem in the second quarter of 2019, representing approximately a 28% ownership interest in the company as of December 31, 2019. The transaction was structured in two tranches. The first tranche comprised $30.0 million, which was funded on April 11, 2019. We invested an additional $30.0 million in a second tranche on June 10, 2019, alongside two existing Evofem shareholders, who each invested an additional $10.0 million. These investments were expected to provide
6
funding for Evofem's pre-commercial activities for Amphora®, its investigational, non-hormonal, on-demand prescription contraceptive gel for women. We believe this investment provided us the ability to take a significant position in a promising company at a critical stage of development where we could provide meaningful contributions through our capital and expertise.
Evofem is a clinical-stage biopharmaceutical company committed to developing and commercializing innovative products to address unmet needs in women's sexual and reproductive health. Evofem is leveraging its proprietary Multipurpose Vaginal pH Regulator (MVP-R™) platform to develop Amphora (L-lactic acid, citric acid and potassium bitartrate) for hormone-free birth control. In 2015, Evofem submitted a new New Drug Application (“NDA”) for prevention of pregnancy to the FDA. In April 2016, the FDA issued a Complete Response Letter (“CRL”) with respect to the Amphora NDA, citing certain clinical deficiencies. In the fourth quarter of 2019, Evofem resubmitted the Amphora NDA, which included results from a subsequent Phase 3 trial. In December 2019, the FDA acknowledged receipt of the NDA and assigned a six-month review period and a Prescription Drug User Fee Act (“PDUFA”) goal date of May 25, 2020.
Amphora is also being studied for the prevention of chlamydia and gonorrhea. In December 2019, Evofem announced positive top-line results from AMPREVENCE, a Phase 2b clinical trial evaluating the efficacy and safety of Amphora for the prevention of urogenital chlamydia and gonorrhea in women. Further analysis is ongoing and final results are subject to change based on a comprehensive review by the company and the FDA.
Pharmaceutical
Noden
On July 1, 2016, our subsidiary, Noden Pharma DAC, entered into an asset purchase agreement (“Noden Purchase Agreement”) whereby it purchased from Novartis Pharma AG (“Novartis”) the exclusive worldwide rights to manufacture, market and sell the Noden Products and certain related assets and assumed certain related liabilities (the “Noden Transaction”). Noden Pharma DAC and Noden Pharma USA, Inc., together, and including their respective subsidiaries represent deployed capital of $191.2 million.
Tekturna (or Rasilez outside of the United States) contains aliskiren, a direct renin inhibitor, for the treatment of hypertension. While indicated as a first line treatment, it is more commonly used as a third line treatment in those patients who are intolerant of angiotensin-receptor blockers (“ARBs”) or angiotensin converting enzyme inhibitors (“ACEIs”). Studies indicate that approximately 12% of hypertension patients are ARB/ACEI intolerant. Tekturna and Rasilez are not indicated for use with ARBs and ACEIs in patients with diabetes or renal impairment and are contraindicated for use by pregnant women.
Tekturna HCT is a combination of aliskiren and hydrochlorothiazide, a diuretic, for the treatment of hypertension in patients not adequately controlled by monotherapy and as an initial therapy in patients likely to need multiple drugs to achieve their blood pressure goals. It is not indicated for use with ACEIs and ARBs in patient with diabetes or renal impairment, or for use in patients with known anuria or hypersensitivity to sulfonamide derived drugs and is contraindicated for use by pregnant women.
The Noden Purchase Agreement provides for various transition periods for development and commercialization activities relating to the Noden Products. Initially, Novartis distributed the Noden Products on behalf of Noden worldwide and Noden received a profit transfer on such sales. Generally, the profit transfer to Noden was defined as gross revenues less product cost and a low single digit percentage fee to Novartis. The profit transfer terminated upon the transfer of the marketing authorization from Novartis to Noden in each country. In the United States, the duration of the profit transfer ran from July 1, 2016 through October 4, 2016. Outside the United States, the profit transfer ended in the first quarter of 2018. Prior to the transfer of the marketing authorization, revenue was presented on a “net” basis; after the transfer of the marketing authorization, revenue is presented on a “gross” basis, meaning product costs are reported separately and there is no fee to Novartis. Except for the sales outside of the United States preceding the final profit transfer that occurred in the first quarter of 2018, revenues of the Noden Products for the periods herein are presented on a gross basis.
Intellectual Property
The Noden Products are protected by multiple patents worldwide, which specifically cover the composition of matter, the pharmaceutical formulations and methods of production. In the United States, the FDA Orange Book for Tekturna lists U.S. Patent No. 8,617,595 (the “’595 Patent”), which covers certain compositions comprising aliskiren, together with other formulation components, and will expire on February 19, 2026.
The FDA Orange Book for Tekturna HCT lists U.S. patent Nos. 8,618,172, which expires on July 13, 2028 and 9,023,893, which expires March 3, 2022, which patents cover certain compositions comprising aliskiren and hydrochlorothiazide, together with other formulation components. In Europe, European patent No. 678 503B (the “’503B Patent”) expired in 2015. However,
7
numerous Supplementary Protection Certificates (“SPCs”) have been granted which are based on the ‘503B Patent and which provide for extended protection. These SPCs generally expire in April of 2020. European Patent Publication Number 2 305 232, which covers certain pharmaceutical compositions comprising aliskiren and HCT, will expire in December 2021.
On June 12, 2017, our subsidiary Noden Pharma DAC (“Noden DAC”) filed a complaint against Anchen Pharmaceuticals, Inc. (“Anchen”) and Par Pharmaceutical (“Par”) for infringement of the ‘595 Patent based on Anchen’s submission of an Abbreviated New Drug Application (“ANDA”) seeking authorization from the FDA to market a generic version of aliskiren hemifumarate tablets, 150 mg and 300 mg, in the United States. Noden DAC’s suit triggered a 30-month stay of FDA approval of that application under the Hatch Waxman Act. Par filed a counterclaim seeking a declaratory judgment with respect to their proposed generic version of aliskiren hemifumarate hydrochlorothiazide tablets (150 mg eq. base/12.5 mg HCT, 150 mg eq. base/25 mg HCT, 300 mg eq. base/12.5 mg HCT, and 300 mg eq. base/25 mg HCT), described in a separate ANDA submitted by Par to the FDA, of noninfringement of U.S. Patent No. 8,618,172 (the “’172 Patent”), also owned by Noden DAC. This case was filed in the United States District Court for the District of Delaware. In March 2018, each of the parties to the proceeding filed a joint stipulation of dismissal of the defendants’ counterclaim seeking a declaratory judgment of non-infringement of the ‘172 Patent. In the stipulation, Anchen and Par agreed that they will not seek, or otherwise join or assist in, any post-grant review, including inter partes review, of the ‘172 Patent or U.S. Patent No. 9,023,893 (the “’893 Patent”). The defendants further stipulated that they will not seek marketing approval of Par’s ANDA or submit any other ANDA seeking approval to market aliskiren hemifumarate hydrochlorothiazide prior to the expiration of the ‘172 Patent in July of 2028. Both the ‘172 Patent and the ‘893 Patent are listed in the Orange Book for Tekturna HCT.
On June 8, 2018, Noden and Anchen entered into a settlement agreement (the “Settlement Agreement”). Under the Settlement Agreement, the parties agreed to file a stipulation of dismissal with the court to facilitate dismissal of the litigation in its entirety, with prejudice. In the Settlement Agreement, Noden granted Anchen a non-exclusive, royalty free, fully paid up and non-transferable license to manufacture and commercialize in the United States a generic version of aliskiren which is described in Anchen’s ANDA, and Anchen agreed not to commercialize its generic version of aliskiren prior to March 1, 2019. The license grant excludes certain formulations covered by the ‘595 Patent which closely relate to the commercial formulation of Tekturna marketed by Noden. The Settlement Agreement includes a release by each party for liabilities associated with the litigation and an acknowledgment from Anchen that the ‘595 Patent claims are valid and enforceable.
As a result of the Settlement Agreement and the imminent launch of a generic version of aliskiren by Par Pharmaceuticals in the United States, management evaluated the ongoing value of the Noden DAC asset group and concluded that the Noden DAC acquired product rights and customer relationship long-lived assets, with a carrying amount of $192.5 million, were no longer recoverable and wrote them down to their estimated fair value of $40.1 million, resulting in an impairment charge of $152.3 million in the second quarter of 2018.
On March 4, 2019, we announced the U.S commercial launch of an authorized generic form of Tekturna, aliskiren hemifumarate 150 mg and 300 mg tablets with the same drug formulation as Tekturna. The authorized generic launch was carried out by Prasco, LLC d/b/a Prasco Laboratories. On March 22, 2019, the FDA approved Anchen’s generic form of aliskiren.
As of December 31, 2019, given our monetization strategy and updated forecasts for Noden, we revised our estimates of future cash flows and as a result of this analysis, determined that the sum of undiscounted cash flows was not greater than the carrying value of the assets. As a result, we concluded that the Noden DAC acquired product rights and customer relationship long-lived assets, with a carrying amount of $32.6 million, were no longer recoverable and wrote them down to their estimated fair value of $10.1 million, resulting in an impairment charge of $22.5 million.
Manufacturing
Noden DAC and Novartis entered into a supply agreement pursuant to which Novartis will manufacture and supply to Noden DAC a bulk tableted form of the Noden Products, and for the supply of active pharmaceutical ingredient (“API”). In May 2019, Noden DAC and Novartis entered into an amended supply agreement pursuant to which Novartis will supply to Noden DAC a bulk tableted form of the Noden Products through 2020 and API through June 2021. For additional details regarding the amended supply agreement, see Note 15, Commitments and Contingencies, to the Consolidated Financial Statements included in Item 8. Under the terms of the amended supply agreement, Noden DAC is committed to purchase certain quantities of bulk product and API that would amount to approximately $61.7 million through June 2021, of which $39.8 million is committed over the next twelve months, which are guaranteed by us. To date, Novartis has met our manufacturing requirements and we expect that it is capable of providing sufficient quantities of the Noden Products to meet anticipated demands. We have contracted with an additional third-party located outside of the United States for the manufacture of Noden Products once the agreement with Novartis expires.
8
Sales and Distribution
In anticipation of the commercialization of Par’s generic version of aliskiren and in order to optimize profitability, in the third quarter of 2018, Noden discontinued its direct sales force and transitioned to a non-personal promotion strategy. All investments in non-personal promotion were in turn discontinued shortly after the launch of the authorized generic form of Tekturna by Prasco Laboratories.
We entered into an arrangement with a third-party logistics provider to distribute the Noden Products within the United States on our behalf. The Noden Products are sold directly to wholesalers from a distribution center owned by the third-party logistics provider.
As of December 31, 2019, the Noden Products were distributed in 13 countries outside of the United States. During 2018, we ceased distribution of the Noden Products in several European countries where they were not profitable or had extremely low gross margins.
Prasco, LLC, PHOENIX Pharma-Einkauf GmbH, and the pharmaceutical industry’s largest U.S. wholesale distributors, Amerisource Bergen Corporation, McKesson Corporation and Cardinal Health, Inc., accounted for 23%, 15%, 8%, 8% and 7%, respectively, of our total net pharmaceutical product sales for the year ended December 31, 2019, and 0%, 11%, 15%, 17% and 13%, respectively, of our total net pharmaceutical product sales for the year ended December 31, 2018.
Competition
The pharmaceutical industry is characterized by rapid innovation and intense competition which is applicable to the therapeutic area our Noden Products are approved. The Noden Products are direct renin inhibitors approved for the treatment of hypertension. They compete against a number of classes of treatments including changes in diet, exercise, thiazide diuretics, ACEIs, ARBs, calcium channel blockers, cardioselective beta blockers, alpha blockers, direct vasodilators and centrally acting agents. With the exception of diet and exercise, there are numerous drugs within each of the classes enumerated above, most of which have generic versions that are less expensive than Tekturna and Tekturna HCT. Physicians may also treat hypertension patients by combining one or more of the enumerated classes of treatments. Diet, thiazide diuretics, ACEIs, ARBs and calcium channel blockers are most commonly used as first line treatments for hypertension and dominate the market, in part, because of the availability of low cost generics in each category. Renin inhibitors, such as Tekturna and Tekturna HCT which are the only approved direct renin inhibitors, and beta blockers are used thereafter followed by direct vasodilators, central acting agents and alpha blockers. Tekturna and Tekturna HCT are generally perceived as alternatives for patients who do not respond to, or are intolerant of, the first line therapies. In 2019, we launched an authorized generic form of Tekturna, aliskiren hemifumarate 150 mg and 300 mg tablets with the same drug formulation as Tekturna. There is currently one other generic form of aliskiren hemifumarate available on the market in the United States.
Employees
As of December 31, 2019, we had 14 full-time employees at Noden, who manage its business and operations.
Income Generating Assets
We have pursued income generating assets when such assets can be acquired on terms that we believe allow us to increase return to our stockholders. The income generating assets typically consist of (i) notes and other long-term receivables, (ii) royalty rights and hybrid notes/royalty receivables, (iii) equity investments and (iv) royalties from the Queen et. al patents. While we currently maintain a portfolio of income generating assets, our intention is to no longer pursue these transactions while we focus on our monetization strategy.
9
Investment | Investment Type | Segment | Deployed Capital 3 (in millions) | |||||
Assertio Therapeutics, Inc. (“Assertio”) 1 | Royalty | Income Generating Assets | $ | 260.5 | ||||
The Regents of the University of Michigan (“U-M”) | Royalty | Income Generating Assets | $ | 65.6 | ||||
AcelRx Pharmaceuticals, Inc. (“AcelRx”) | Royalty | Income Generating Assets | $ | 65.0 | ||||
Viscogliosi Brothers, LLC (“VB”) | Royalty | Income Generating Assets | $ | 15.5 | ||||
KYBELLA® | Royalty | Income Generating Assets | $ | 9.5 | ||||
CareView Communications, Inc. (“CareView”) | Debt | Income Generating Assets | $ | 20.0 | ||||
Wellstat Diagnostics, LLC (“Wellstat Diagnostics”) 2 | Royalty/debt hybrid | Income Generating Assets | $ | 44.0 | ||||
_______________
1 | Formerly Depomed, Inc. |
2 | Also known as Defined Diagnostic, LLC. The Wellstat Diagnostics investment also includes our note receivable with Hyperion Catalysis International, Inc. (“Hyperion”). |
3 | Excludes transaction costs. |
Notes and Other Long-Term Receivables
We have entered into credit agreements with borrowers across the healthcare industry, under which we made available cash loans to be used by the borrower. Obligations under these credit agreements are typically secured by a pledge of substantially all the assets of the borrower and any of its subsidiaries. As of December 31, 2019, we had two notes receivable transactions outstanding, CareView and Wellstat Diagnostics, which are summarized below:
CareView
Technology
CareView is a provider of products and on-demand application services for the healthcare industry by specializing in bedside video monitoring, archiving and patient care documentation systems and patient entertainment services.
Deal Summary
In June 2015, we entered into a credit agreement with CareView, whereby we made available to CareView up to $40.0 million in loans comprised of two tranches of $20.0 million each, subject to CareView’s attainment of specified milestones and under which we have a security interest in substantially all of CareView’s assets. In October 2015, we and CareView entered into an amendment of the credit agreement to modify certain definitions related to the first and second tranche milestones and we funded the first tranche of $20.0 million, net of fees, based on CareView’s attainment of the first milestone, as amended. The second $20.0 million tranche was not funded due to CareView’s failure to meet the funding milestone and we have no further funding obligation at this time. The outstanding borrowing under the credit agreement initially bore interest at the rate of 13.5% per annum payable quarterly in arrears. Principal repayment was to commence on the ninth quarterly interest payment date and continue in equal installments until final maturity of the loan in October 2020.
In February 2018, we entered into a modification agreement with CareView (the “February 2018 Modification Agreement”) whereby we agreed, effective as of December 28, 2017, to modify the credit agreement before remedies could otherwise have become available to us under the credit agreement in relation to certain obligations of CareView that would potentially not be met, including the requirement to make principal payments. Under the February 2018 Modification Agreement, we agreed that (i) a lower liquidity covenant would be applicable and (ii) principal repayment would be delayed for a period of up to December 31, 2018. In exchange for agreeing to these modifications, among other things, the exercise price of our warrants to purchase 4.4 million shares of common stock of CareView was reduced and, subject to the occurrence of certain events, CareView agreed to grant us additional equity interests. In each of September 2018, December 2018, May 2019, September 2019 and December 2019, we entered into amendments to the February 2018 Modification Agreement with CareView whereby we agreed to deferrals of principal repayments and interest payments. In the May 2019 amendment we also increased the interest rate to 15.5% and removed the liquidity covenant under the credit agreement. In January 2020 we agreed to a further amendment of the February 2018 Modification
10
Agreement that deferred principal repayment and interest payments until April 30, 2020, which was conditioned upon CareView raising additional financing from third parties.
Wellstat Diagnostics
Technology
Wellstat Diagnostics is a private company formerly dedicated to the development, manufacture, sale and distribution of small point of care diagnostic systems.
Deal Summary
In March 2012, we executed a $7.5 million two-year senior secured note receivable with the holders of the equity interests in Wellstat Diagnostics. In August 2012, we and Wellstat Diagnostics amended the note receivable, providing a senior secured note receivable of $10.0 million, bearing interest at 12% per annum, to replace the original $7.5 million note receivable. This $10.0 million note receivable was repaid on November 2, 2012, using the proceeds of the $40.0 million credit facility we entered into on the same date.
In November 2012, we entered into a $40.0 million credit agreement with Wellstat Diagnostics pursuant to which we were to accrue quarterly interest payments at the rate of 5% per annum. In January 2013, Wellstat Diagnostics defaulted on the credit agreement, and as a result both parties agreed to enter into a forbearance agreement whereby we agreed to provide additional funding. In August 2013, we entered into an amended and restated credit agreement with terms substantially the same as those of the original credit agreement. However, pursuant to the amended and restated credit agreement the principal amount was reset to approximately $44.1 million.
We, Wellstat Diagnostics, and Samuel J. Wohlstadter, Nadine H. Wohlstadter, Duck Farm, Inc., Hebron Valley Farms, Inc., HVF, Inc., Hyperion Catalysis EU Limited, Hyperion, NHW, LLC, Wellstat AVT Investment, LLC, Wellstat Biocatalysis, LLC, Wellstat Biologics Corporation, Wellstat Diagnostics, Wellstat Immunotherapeutics, LLC, Wellstat Management Company, LLC, Wellstat Ophthalmics Corporation, Wellstat Therapeutics Corporation, Wellstat Therapeutics EU Limited, Wellstat Vaccines, LLC and SJW Properties, Inc., the guarantors of Wellstat Diagnostics’ obligations to us (collectively, the “Wellstat Diagnostics Guarantors”) were involved in a series of legal actions. A further discussion of the Wellstat litigation is included in Note 25, Legal Proceedings, to the Consolidated Financial Statements included in Item 8.
The Wellstat Diagnostics investment also includes our note receivable with Hyperion. A further discussion is included in Note 7, Notes and Other Lon-term Receivables.
Royalty Rights - At Fair Value
We have entered into various royalty purchase agreements with counterparties, whereby the counterparty conveys to us the right to receive royalties that are typically payable on sales revenue generated by the sale, distribution or other use of the counterparties’ products.
We record the royalty rights at fair value using discounted cash flows related to the expected future cash flows to be received. We use significant judgment in determining our valuation inputs, including estimates as to the probability and timing of future sales of the licensed product. A third-party expert is generally engaged to assist us with the development of our estimate of the expected future cash flows. The estimated fair value of the asset is subject to variation should those cash flows vary significantly from our estimates. At each reporting period, an evaluation is performed to assess those estimates, discount rates utilized and general market conditions affecting fair market value.
11
At December 31, 2019, we had a total of five royalty rights transactions outstanding, which are summarized below in chronological order:
Assertio
Deal Summary
In October 2013, we entered into a Royalty Purchase and Sale Agreement (the “Assertio Royalty Agreement”) with Assertio, whereby we acquired the rights to receive royalties and milestones payable on sales of five Type 2 diabetes products licensed by Assertio in exchange for a $240.5 million cash payment.
In August 2018, we entered into an amendment to the Assertio Royalty Agreement pursuant to which we purchased Assertio’s remaining interests in royalty and milestone payments payable on sales of Type 2 diabetes products licensed by Assertio for $20.0 million.
The Assertio Royalty Agreement terminates on the third anniversary following the date upon which the later of the following occurs: (a) October 25, 2021, or (b) at such time as no royalty payments remain payable under any license agreement and each of the license agreements has expired by its terms.
Technology
The rights acquired include Assertio’s royalty and milestone payments accruing from and after October 1, 2013: (a) from Santarus, Inc., which was subsequently acquired by Salix Pharmaceuticals, Inc., which itself was acquired by Valeant Pharmaceuticals International, Inc. (“Valeant”), which, in July 2018 changed its name to Bausch Health Companies Inc. (“Bausch Health”) with respect to sales of Glumetza® (metformin HCL extended-release tablets) in the United States; (b) from Merck & Co., Inc. with respect to sales of Janumet XR® (sitagliptin and metformin HCL extended-release); (c) from Janssen Pharmaceuticals N.V. with respect to potential future development milestones and sales of its approved fixed-dose combination of Invokana® (canagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor) and extended-release metformin tablets, marketed as Invokamet XR®; (d) from Boehringer Ingelheim GmbH (“Boehringer Ingelheim”) and Eli Lilly and Company (“Eli Lilly”) with respect to potential future development milestones and sales of the investigational fixed-dose combinations of drugs and extended-release metformin subject to Assertio’s license agreement with Boehringer Ingelheim including its approved products, Jentadueto XR® and Synjardy XR®; and (e) from Bausch Health for sales of extended-release metformin tablets in Korea and Canada, respectively.
On May 31, 2016, Boehringer Ingelheim and Eli Lilly announced that the FDA approved Jentadueto XR (a fixed dose combination of Linagliptin, a dipeptidyl peptidase-4 inhibitor and extended-release metformin tablets) for the treatment of type 2 diabetes in adults, which will be marketed by both companies. This approval triggered the payment of a milestone to us of $6.0 million. On September 21, 2016, Janssen Pharmaceuticals announced that the FDA approved Invokamet XR for the treatment of type 2 diabetes in adults. This approval triggered the payment of a milestone to us of $5.0 million. On December 13, 2016, Boehringer Ingelheim and Eli Lilly announced that the FDA approved Synjardy® XR (a fixed dose combination of Empagliflozin, a SGLT2 inhibitor, and extended-release metformin tablets) for the treatment of type 2 diabetes in adults, which will be marketed by both companies. This approval triggered the payment of a milestone to us of $6.0 million. In 2017, we started to receive royalties on the net sales of these three newly approved products.
In February 2013, a generic equivalent to Glumetza was approved by the FDA and in August 2016, two additional generic equivalents to Glumetza were approved by the FDA. In February 2016, Lupin Pharmaceuticals, Inc., in August 2017, Teva Pharmaceutical Industries Ltd., and in July 2018, Sun Pharmaceutical, Inc. each announced a launch of a generic equivalent approved product. Multiple generic versions of extended release metformin hydrochloride are currently approved by the FDA.
In May 2017, we received notification that a subsidiary of Valeant had launched an authorized generic equivalent product in February 2017, and we received royalties on such authorized generic equivalent product under the same terms as the branded Glumetza product, retroactive to February 2017. We continue to monitor whether the generic competition further affects sales of Glumetza and thus royalties on such sales paid to us, and the impact of the launched authorized generic equivalent.
12
Viscogliosi Brothers
Deal Summary
In June 2014, we entered into a Royalty Purchase and Sale Agreement (the “VB Royalty Agreement”) with VB, whereby VB conveyed to us the right to receive royalties on net sales of a spinal implant that has received pre-market approval from the FDA held by VB and commercialized by Paradigm Spine, LLC (“Paradigm Spine”), in exchange for a $15.5 million cash payment, less fees. Paradigm Spine was acquired in March 2019 by RTI Surgical Holdings, Inc.
The royalty rights acquired include royalties accruing from and after April 1, 2014. Under the terms of the VB Royalty Agreement, we receive all royalty payments due to VB pursuant to certain technology transfer agreements between VB and Paradigm Spine until we have received payments equal to 2.3 times the cash payment it made to VB, after which all rights to receive royalties will be returned to VB. VB’s ability to repurchase the royalty right for a specified amount expired on June 26, 2018.
Technology
The coflex® Interlaminar technology is an Interlaminar Stabilization® device indicated for use in one or two level lumbar stenosis from L1-L5 in skeletally mature patients with at least moderate impairment in function.
University of Michigan
Deal Summary
In November 2014, we acquired a portion of all royalty payments of the Regents of the University of Michigan’s (“U-M”) worldwide royalty interest in Cerdelga® (eliglustat) for $65.6 million pursuant to the Royalty Purchase and Sale Agreement with U-M (the “U-M Royalty Agreement”). Under the terms of the U-M Royalty Agreement, we receive 75% of all royalty payments due under U-M’s license agreement with Genzyme Corporation, a Sanofi company (“Genzyme”), until expiration of the licensed patents, excluding any patent term extension.
Technology
Cerdelga, an oral therapy for adult patients with Gaucher disease type 1, was developed by Genzyme. Cerdelga was approved in the United States in August 2014, in the European Union (“EU”) in January 2015 and in Japan in March 2015. In addition, marketing applications for Cerdelga are under review by other regulatory authorities. While marketing applications have been approved in the United States, the EU and Japan, national pricing and reimbursement decisions are delayed in some countries.
AcelRx
Deal Summary
In September 2015, we entered into a royalty interest assignment agreement (the “AcelRx Royalty Agreement”) with ARPI LLC, a wholly-owned subsidiary of AcelRx Pharmaceuticals, Inc., whereby we acquired the rights to receive a portion of the royalties and certain milestone payments on sales of Zalviso® (sufentanil sublingual tablet system) in the EU, Switzerland and Australia by AcelRx’s commercial partner, Grünenthal, in exchange for a $65.0 million cash payment. Under the terms of the AcelRx Royalty Agreement, we receive 75% of all royalty payments and 80% of the first four commercial milestone payments, due under AcelRx’s license agreement with Grüenthal until the earlier to occur of (i) receipt by us of payments equal to three times the cash payments made to AcelRx and (ii) the expiration of the licensed patents. Zalviso received marketing approval by the European Commission in September 2015. Grünenthal launched Zalviso in the second quarter of 2016 and we started to receive royalties in the third quarter of 2016.
Due to the slower than expected adoption of the product since its initial launch relative to our estimates and the increased variance noted between our forecast model and actual results in the three months ended June 30, 2019, we utilized a third-party expert in the second quarter of 2019 to reassess the market and expectations for the Zalviso product. Key findings from the third-party study included: the post-surgical PCA (Patient-Controlled Analgesia) market being smaller than previously forecasted; the higher price of the product relative to alternative therapies, the product not being used as
13
a replacement for systemic opioids and the design of the delivery device, which is pre-filled for up to three days of treatment, which limited its use in procedures with anticipated shorter recovery times. Based on this analysis and the impact to the projected sales-based royalties and milestones, we wrote down the fair value of the royalty asset by $60.0 million in the second quarter of 2019.
Technology
Zalviso is a combination drug and device product which, using a patient controlled dispenser, delivers a sub-lingual formulation of sufentanil, an opioid with a high therapeutic index.
KYBELLA
Deal Summary
In July 2016, we entered into a royalty purchase and sales agreement with an individual, whereby we acquired that individual’s rights to receive certain royalties on sales of KYBELLA® by Allergan plc in exchange for a $9.5 million cash payment and up to $1.0 million in future milestone payments based upon product sales targets. We started to receive royalty payments during the third quarter of 2016.
Technology
KYBELLA is an FDA approved injectable treatment for adults with moderate-to-severe fat below the chin, known as submental fat. KYBELLA contains deoxycholic acid which destroys fat cells and allows for a safer and less invasive alternative to surgical procedures.
Royalties from Queen et al. patents
We have been issued patents in the United States and elsewhere, covering the humanization of antibodies, which we refer to as our Queen et al. patents. Our Queen et al. patents, for which final patent expiry was in December 2014, covered, among other things, humanized antibodies, methods for humanizing antibodies, polynucleotide encoding in humanized antibodies and methods of producing humanized antibodies.
We previously entered into licensing agreements under our Queen et al. patents with numerous entities that are independently developing or have developed humanized antibodies. Under our licensing agreements, we are entitled to receive a flat-rate royalty based upon our licensees’ net sales of covered antibodies, although the royalties under these agreements have substantially ended.
Solanezumab is a Lilly-licensed humanized monoclonal antibody being tested in a study of older individuals who may be at risk of memory loss and cognitive decline due to Alzheimer’s disease. Lilly has characterized the study as an assessment of whether an anti-amyloid investigational drug in older individuals who do not yet show symptoms of Alzheimer's disease cognitive impairment or dementia can slow memory loss and cognitive decline. The study will also test whether solanezumab treatment can delay the progression of Alzheimer’s disease related brain injury on imaging and other biomarkers. If solanezumab is approved and commercialized pursuant to this clinical trial or another, we would be entitled to receive a royalty based on a "know-how" license for technology provided in the design of this antibody. The 2% royalty on net sales is payable for 12.5 years after the product's first commercial sale. The above described study is currently in Phase 3 testing with results expected in July of 2022.
For the years ended December 31, 2019, 2018 and 2017, royalties from Queen et al. patents accounted for less than 1%, 2%, and 11% of our total revenues, respectively.
Competition
The underlying products associated with our income generating assets compete with existing products and are vulnerable to new branded or generic entrants in the marketplace.
Governmental Regulation
The research and development, manufacturing and marketing of pharmaceutical and medical device products are subject to regulation by numerous governmental authorities in the United States and other countries. We and our borrowers and royalty-agreement counterparties, depending on specific activities performed, are subject to these regulations. In the United States,
14
pharmaceuticals and medical devices are subject to regulation by both federal and various state authorities, including the FDA. The Federal Food, Drug and Cosmetic Act (“FFDCA”) governs the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of pharmaceutical and medical device products, and with respect to biologics, compliance with the Public Health Service Act is also required. There are also comparable laws and regulations that apply at the state level and in other countries as well. For both currently marketed and products in development, failure to comply with applicable regulatory requirements can, among other things, result in delays, the suspension of regulatory approvals, as well as possible civil and criminal sanctions.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
• | completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations; |
• | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
• | approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated; |
• | performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP, requirements to establish the safety and efficacy of the proposed drug product for each indication; |
• | submission to the FDA of a new drug application, or NDA; |
• | satisfactory completion of an FDA advisory committee review, if applicable; |
• | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practice (“cGMP”), requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
• | FDA review and approval of the NDA. |
Preclinical Studies
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
• | Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
• | Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
• | Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. |
15
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to the FDA because the FDA has 60 days to make a “filing” decision.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements. After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or preclinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit
16
further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval. Moreover, after approval of an NDA, a company may decide to launch an “authorized generic” version of the drug, which is an approved brand name drug that is marketed without the brand name on its label. Other than the fact that it does not have the brand name on its label, it is the exact same drug product as the branded product. While a separate NDA is not required for marketing an authorized generic, the FDA requires that the NDA holder notify the FDA if it markets an authorized generic. The NDA holder may market both the authorized generic and the brand-name product at the same time.
Special FDA Expedited Review and Approval Programs
The FDA has various programs, including, but not limited to, fast track designation, accelerated approval, priority review, and breakthrough therapy designation, which are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life-threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
To be eligible for a fast track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA will determine that a product will fill an unmet medical need if it will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. The FDA may review sections of the NDA for a fast track product on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA. The FDA may give a priority review designation to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. Under the new PDUFA agreement, these six and ten-month review periods are measured from the “filing” date rather than the receipt date for NDAs for new molecular entities, which typically adds 60 days to the timeline for review and decision from the date of submission. Most products that are eligible for fast track designation are also likely to be considered appropriate to receive a priority review.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures.
Moreover, under the provisions of the Food and Drug Administration Safety and Innovation Act, or FDASIA, passed in July 2012, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Post-Approval Regulation
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved
17
product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a Risk Evaluation and Mitigation Strategies (“REMS”) program. Other potential consequences include, among other things:
• | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
• | fines, warning letters or holds on post-approval clinical trials; |
• | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; |
• | product seizure or detention, or refusal to permit the import or export of products; or |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
Medical Devices Regulation in the United States
Under the FFDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to ensure safety and effectiveness. Class I devices are those for which safety and effectiveness can be assured by adherence to the FDA’s general controls for medical devices, which include compliance with the applicable portions of the FDA’s Quality System Regulation, or QSR, facility registration and product listing, reporting of adverse medical events, and appropriate, truthful and non-misleading labeling, advertising, and promotional materials. Some Class I devices also require premarket clearance by the FDA through the 510(k) premarket notification process described below. Class II devices are subject to the FDA’s general controls, and any other special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification procedure, unless exempt. A Class III product is a product which has a new intended use or uses advanced technology that is not substantially equivalent to that of a legally marketed device. The safety and effectiveness of Class III devices cannot be assured solely by the General Controls and the other requirements described above. These devices almost always require formal clinical studies to demonstrate safety and effectiveness. Our current medical device products are classified as Class II medical devices.
When a 510(k) is required, the manufacturer must submit to the FDA a premarket notification submission demonstrating that the device is “substantially equivalent” to either: a device that was legally marketed prior to May 28, 1976, the date upon which the Medical Device Amendments of 1976 were enacted, and for which the FDA has not yet called for the submission of pre-market approval applications (“PMAs”), or is a device that has been reclassified from Class III to either Class II or I.
If the FDA agrees that the device is substantially equivalent to a predicate device, it will grant clearance to commercially market the device in the U.S. The FDA’s 510(k) clearance process usually takes from three to twelve months from the date the application
18
is submitted and filed with the FDA, but may take significantly longer and clearance is never assured. Although many 510(k) pre-market notifications are cleared without clinical data, in some cases, the FDA requires significant clinical data to support substantial equivalence. In reviewing a pre-market notification, the FDA may request additional information, including clinical data, which may significantly prolong the review process. If the FDA determines that the device, or its intended use, is not “substantially equivalent,” the FDA may deny the request for clearance. After a device receives 510(k) clearance, any subsequent modification of the device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new 510(k) clearance or could require pre-market approval. The FDA requires each manufacturer to make this determination initially, but the FDA may review any such decision and may disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination, the FDA may require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or pre-market approval is obtained. We have modified aspects of some of our devices since receiving regulatory clearance and we have made the determination that new 510(k) clearances or pre-market approvals were not required.
In addition, over the last several years, the FDA has proposed reforms to its 510(k) clearance process, and such proposals could include increased requirements for clinical data and a longer review period, or could make it more difficult for manufacturers to utilize the 510(k) clearance process for their products. For example, in November 2018, FDA officials announced forthcoming steps that the FDA intends to take to modernize the premarket notification pathway under Section 510(k) of the FFDCA. Among other things, the FDA announced that it planned to develop proposals to drive manufacturers utilizing the 510(k) pathway toward the use of newer predicates. These proposals included plans to potentially sunset certain older devices that were used as predicates under the 510(k) clearance pathway, and to potentially publish a list of devices that have been cleared on the basis of demonstrated substantial equivalence to predicate devices that are more than 10 years old. In May 2019, the FDA solicited public feedback on these proposals. These proposals have not yet been finalized or adopted, and the FDA may work with Congress to implement such proposals through legislation.
More recently, in September 2019, the FDA finalized guidance describing an optional “safety and performance based” premarket review pathway for manufacturers of “certain, well-understood device types” to demonstrate substantial equivalence under the 510(k) clearance pathway by showing that such device meets objective safety and performance criteria established by the FDA, thereby obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA intends to develop and maintain a list of device types appropriate for the “safety and performance based” pathway and will continue to develop product-specific guidance documents that identify the performance criteria for each such device type, as well as the testing methods recommended in the guidance documents, where feasible.
Although unlikely for the types of medical devices marketed by us, the FDA may classify the device, or the particular use of the device, into Class III, and the device sponsor must then fulfill more rigorous PMA requirements. A PMA application, which is intended to demonstrate that a device is safe and effective, must be supported by extensive data, including extensive technical and manufacturing data and data from preclinical studies and human clinical trials. After a PMA application is submitted and filed, the FDA begins an in-depth review of the submitted information, which typically takes between one and three years, but may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also, during the review period, an advisory panel of experts from outside the FDA will usually be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of the manufacturing facility to ensure compliance with the QSR, which impose elaborate design development, testing, control, documentation and other quality assurance procedures in the design and manufacturing process. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution and collection of long-term follow-up data from patients in the clinical study that supported approval. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including the loss or withdrawal of the approval. New PMA applications or PMA supplements are required for significant modifications to the manufacturing process, labeling of the product and design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an original PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA, and may not require as extensive clinical data or the convening of an advisory panel.
A clinical trial is typically required to support a PMA application and is sometimes required for a 510(k) pre-market notification. Clinical trials generally require submission of an application for an Investigational Device Exemption, or IDE, to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the investigational protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Clinical trials for a significant risk device may begin once the IDE application is approved by the FDA as well as the appropriate institutional review boards at the clinical trial sites, and the informed consent of the patients
19
participating in the clinical trial is obtained. After a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to an unacceptable health risk. Any trials we conduct must be conducted in accordance with FDA regulations as well as other federal regulations and state laws concerning human subject protection and privacy.
In addition, after a device is placed on the market, numerous FDA and other regulatory requirements continue to apply. These include establishment registration and device listing with the FDA; compliance with medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and compliance with corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FFDCA that may present a risk to health. The FDA and the Federal Trade Commission also regulate the advertising and promotion of our products to ensure that the claims we make are consistent with our regulatory clearances, that there is scientific data to substantiate the claims and that our advertising is neither false nor misleading. In general, we may not promote or advertise our products for uses not within the scope of our intended use statement in our clearances or make unsupported safety and effectiveness claims. Many regulatory jurisdictions outside of the United States have similar regulations to which we are subject.
Foreign Regulation of Drugs and Medical Devices
In order for us to market our products in countries outside the United States, we must obtain regulatory approvals and comply with extensive product and quality system regulations in other countries. These regulations, including the requirements for approvals or clearance and the time required for regulatory review, vary from country to country. Some countries have regulatory review processes which are substantially longer than U.S. processes. Failure to obtain regulatory authorizations or approvals in a timely manner and to meet all local requirements including language and specific safety standards in any foreign country in which we plan to market our products could prevent us from marketing products in such countries or subject us to sanctions and fines.
Commercialization of medical devices in Europe is regulated by the EU. The EU presently requires that all medical products bear the Conformité Européenne (“CE”) mark, for compliance with the Medical Device Directive (93/42/EEC) as amended. The CE mark is an international symbol of adherence to certain essential principles of safety and performance mandated in applicable European medical device directives, which once affixed, enables a product to be sold in member countries of the EU and those affiliated countries which accept the CE mark. The CE mark is also recognized in many countries outside of the EU, such as Australia, and can assist in the clearance process. In order to affix the CE mark on products, a recognized European Notified Body must certify a manufacturer’s quality system and design dossier for compliance with international and European requirements. To maintain authorization to apply the CE mark, we are subject to annual surveillance audits and periodic re-certification audits. In September 2013, the European Commission adopted a recommendation indicating that all Notified Bodies, including Presafe, an accredited certification body, should carry out unannounced audits, at least once every third year, of the manufacturers whose medical devices they have certified. These unannounced audits can also extend to the manufacturer’s critical suppliers or sub-contractors (those that supply a critical input or perform a critical function for the manufacturer).
Federal, State and Foreign Fraud and Abuse and Physician Payment Transparency Laws
We are also subject to federal and state healthcare laws and regulations pertaining to fraud and abuse, physician payment transparency, privacy, and security laws and regulations. These laws include, without limitation: foreign, federal, and state anti-kickback and false claims laws, as well as transparency laws regarding payments or other items of value provided to healthcare providers. The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind to induce or in return for purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value, including stock, stock options and the compensation derived through ownership interests.
Recognizing that the federal Anti-Kickback Statute is broad and may prohibit many innocuous or beneficial arrangements within the healthcare industry, the Department of Health and Human Services issued regulations in July 1991, which the Department has referred to as “safe harbors.” These safe harbor regulations set forth certain provisions which, if met in form and substance, will assure pharmaceutical, biotechnology and medical device manufacturers, healthcare providers and other parties that they will not be prosecuted under the federal Anti-Kickback Statute. Additional safe harbor provisions providing similar protections have been published intermittently since 1991. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve
20
remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal Anti-Kickback Statute has been violated. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Moreover, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act (described below).
Violations of the federal Anti-Kickback Statute may result in civil monetary penalties, plus up to three times the remuneration involved. Civil penalties for such conduct can further be assessed under the federal False Claims Act. Violations can also result in criminal penalties, including criminal fines and imprisonment. Similarly, violations can result in exclusion from participation in government healthcare programs, including Medicare and Medicaid. Liability under the federal Anti-Kickback Statute may also arise because of the intentions or actions of the parties with whom we do business. Conduct and business arrangements that do not fully satisfy one of these safe harbor provisions may result in increased scrutiny by government enforcement authorities. The majority of states also have anti-kickback laws which establish similar prohibitions and, in some cases may apply more broadly to items or services covered by any third-party payor, including commercial insurers and self-pay patients.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. The federal civil False Claims Act also applies to false submissions that cause the government to be paid less than the amount to which it is entitled, such as a rebate. Intent to deceive is not required to establish liability under the civil federal civil False Claims Act.
In addition, private parties may initiate “qui tam” whistleblower lawsuits against any person or entity under the federal civil False Claims Act in the name of the government and share in the proceeds of the lawsuit. Penalties for federal civil False Claim Act violations include fines for each false claim, plus up to three times the amount of damages sustained by the federal government and, most critically, may provide the basis for exclusion from the federally funded healthcare program. On May 20, 2009, the Fraud Enforcement Recovery Act of 2009, or FERA, was enacted, which modifies and clarifies certain provisions of the federal civil False Claims Act. In part, the FERA amends the federal civil False Claims Act such that penalties may now apply to any person, including an organization that does not contract directly with the government, who knowingly makes, uses or causes to be made or used, a false record or statement material to a false or fraudulent claim paid in part by the federal government. The government may further prosecute conduct constituting a false claim under the federal criminal False Claims Act. The criminal False Claims Act prohibits the making or presenting of a claim to the government knowing such claim to be false, fictitious or fraudulent and, unlike the federal civil False Claims Act, requires proof of intent to submit a false claim. When an entity is determined to have violated the federal civil False Claims Act, the government may impose civil fines and penalties, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs.
The federal Civil Monetary Penalty Act of 1981 imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent, or offering or transferring remuneration to a federal healthcare beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier.
The Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors; knowingly and willfully embezzling or stealing from a healthcare benefit program; willfully obstructing a criminal investigation of a healthcare offense; and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Many foreign countries have similar laws relating to healthcare fraud and abuse. Foreign laws and regulations may vary greatly from country to country. For example, the advertising and promotion of our products is subject to EU Directives concerning misleading and comparative advertising and unfair commercial practices, as well as other EEA Member State legislation governing the advertising and promotion of medical devices. These laws may limit or restrict the advertising and promotion of our
21
products to the general public and may impose limitations on our promotional activities with healthcare professionals. Also, many U.S. states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs.
Additionally, there has been a recent trend of increased foreign, federal, and state regulation of payments and transfers of value provided to healthcare professionals or entities. The federal Physician Payments Sunshine Act imposes annual reporting requirements on certain drug, biologics, medical supplies and device manufacturers for which payment is available under Medicare, Medicaid or Children’s Health Insurance Program for payments and other transfers of value provided by them, directly or indirectly, to physicians (including physician family members) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. A manufacturer’s failure to submit timely, accurately and completely the required information for all payments, transfers of value or ownership or investment interest may result in civil monetary penalties. Manufacturers must submit reports by the 90th day of each calendar year. Certain foreign countries and U.S. states also mandate implementation of commercial compliance programs, impose restrictions on device manufacturer marketing practices and require tracking and reporting of gifts, compensation and other remuneration to healthcare professionals and entities.
Coverage and reimbursement
In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products or the products for which we receive royalty revenue unless coverage is provided, and reimbursement is adequate to cover a significant portion of the cost. Sales of any products therefore depend, in part, on the availability of coverage and adequate reimbursement from third-party payors. Third-party payors include government authorities, managed care plans, private health insurers and other organizations.
The process for determining whether a third-party payor will provide coverage for a pharmaceutical or device product typically is separate from the process for setting the price of such product or for establishing the reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication. A decision by a third-party payor not to cover our products could reduce physician utilization of our products and have a material adverse effect on our sales, results of operations and financial condition. Moreover, a third-party payor’s decision to provide coverage for a pharmaceutical or device product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. Additionally, coverage and reimbursement for products can differ significantly from payor to payor. One third-party payor’s decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service, or will provide coverage at an adequate reimbursement rate.
For our medical device business, the reimbursement to the facility from third-party payors is intended to cover the overall cost of treatment, including the cost of our devices used during the procedure as well as the overhead cost associated with the facility where the procedure is performed. We do not directly bill any third-party payors; instead, we receive payment from the hospital or other facility that uses our devices. Failure by physicians, hospitals, and other users of our devices to obtain sufficient coverage and reimbursement from healthcare payors for procedures in which our devices are used, or adverse changes in government and private third-party payors’ policies could have a material adverse effect on our business, financial condition, results of operations and future growth prospects.
In addition, there are periodic changes to reimbursement. Third-party payors regularly update reimbursement amounts and also from time to time revise the methodologies used to determine reimbursement amounts. This includes annual updates to payments to physicians, hospitals and other facilities for procedures during which our devices are used. Because the cost of our devices generally is recovered by the healthcare provider as part of the payment for performing a procedure and not separately reimbursed, these updates could directly impact the demand for our devices. An example of such payment updates is the Medicare program’s updates to hospital and physician payments, which are done on an annual basis using a prescribed statutory formula. In the past, with respect to reimbursement for physician services under the Medicare Physician Fee Schedule, when the application of the formula resulted in lower payment, Congress has passed interim legislation to prevent the reductions.
The containment of healthcare costs is a priority of federal, state and foreign governments, and the prices of pharmaceutical or device products have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost-effectiveness of pharmaceutical products, medical devices and medical services, in addition to questioning safety and efficacy. If these third-party payors do not consider our
22
products to be cost-effective compared to other available therapies, they may not cover our products or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. Current and future legislative proposals to further reform healthcare or reduce healthcare costs may limit coverage of or lower reimbursement for the procedures associated with the use of our products. The cost containment measures that payors and providers are instituting, and the effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our products.
The implementation of the Affordable Care Act, (the “ACA”), in the United States, for example, has changed healthcare financing and delivery by both governmental and private insurers substantially, and affected medical device manufacturers significantly. The ACA imposed, among other things, a 2.3% federal excise tax, with limited exceptions, on any entity that manufactures or imports Class I, II and III medical devices offered for sale in the United States that began on January 1, 2013. Through a series of legislative amendments, the tax was suspended for 2016 through 2019. The device excise tax was repealed on December 20, 2019. The ACA also provided incentives to programs that increase the federal government’s comparative effectiveness research, and implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models. Additionally, the ACA has expanded eligibility criteria for Medicaid programs and created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. We do not yet know the full impact that the ACA will have on our business.
There have been judicial and Congressional challenges to certain aspects of the ACA, and we expect additional challenges and amendments in the future. Moreover, the Trump Administration and the U.S. Congress may take further action regarding the ACA, including, but not limited to, repeal or replacement.
Moreover, other legislative changes have been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011, among other things, included reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2025 unless additional Congressional action is taken. Additionally, the American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
We expect additional state and federal healthcare reform measures to be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
In addition, changes in existing regulations could have a material adverse effect on us or our licensees, borrowers or royalty-agreement counterparties. For a discussion of the risks associated with government regulations, see Item 1A, “Risk Factors.”
Manufacturing
Our manufacturing processes are required to comply with the FDA’s cGMP requirements, which for medical devices, are contained in its QSR and associated regulations and guidance. The QSR covers, among other things, the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation, and servicing of all medical devices intended for human use. The QSR also requires maintenance of extensive records which demonstrate compliance with FDA regulation, the manufacturer’s own procedures, specifications, and testing as well as distribution and post-market experience. Compliance with the QSR is necessary to receive FDA clearance or approval to market new products and is necessary for a manufacturer to be able to continue to market cleared or approved product offerings in the United States. A company’s facilities, records, and manufacturing processes are subject to periodic scheduled or unscheduled inspections by the FDA, which may issue reports known as Forms FDA 483 or Notices of Inspectional Observations which list instances where the FDA inspector believes the manufacturer has failed to comply with applicable regulations and/or procedures. If the observations are sufficiently serious or the manufacturer fails to respond appropriately, the FDA may issue Warning Letters, or Untitled Letters, which are notices of potential enforcement actions against the manufacturer. If a Warning Letter or Untitled Letter is not addressed to the satisfaction of the FDA, or if the FDA becomes aware of any other serious issue with a manufacturer’s products
23
or facilities, it could result in fines, injunctions, civil penalties, delays, suspension or withdrawal of clearances, seizures or recalls of products, operating restrictions, total shutdown of production facilities, prohibition on export or import and criminal prosecution. Such actions may have further indirect consequences for the manufacturer outside of the United States, and may adversely affect the reputation of the manufacturer and the product. In the United States, routine FDA inspections usually occur every two years, and may occur more often for cause.
To a greater or lesser extent, most other countries require some form of quality system and regulatory compliance, which may include periodic inspections, inspections by third-party auditors, and specialized documentation. Failure to meet all the requirements of these countries could jeopardize our ability to import, market, support, and receive reimbursement for the use of our products in these countries. In addition to the above, we may seek to conduct clinical studies or trials in the United States or other countries on products that have not yet been cleared or approved for a particular indication. Products manufactured outside the United States by or for us are subject to U.S. Customs and FDA inspection upon entry into the United States. We must demonstrate compliance of such products to U.S. regulations and carefully document the eventual distribution or re-exportation of such products. Failure to comply with all applicable regulations could prevent us from having access to products or components critical to the manufacture of finished products and lead to shortages and delays.
Employees
As of December 31, 2019, we had 20 full-time employees managing our intellectual property, acquisitions, operations and other corporate activities, including providing management oversight, accounting, legal and tax support and administrative assistance to our subsidiaries, as well as performing certain essential functions of a public company. In addition, we had 89 full-time employees at our operating subsidiaries, Noden and LENSAR, who manage the subsidiaries’ businesses and operations. Geographically, 96 employees were based in the United States and 13 employees were located internationally. None of our employees are covered by a collective bargaining agreement, and we consider our relationship with our employees to be good.
About PDL
We were incorporated under the laws of the state of Delaware in 1986 under the name Protein Design Labs, Inc. In 2006, we changed our name to PDL BioPharma, Inc. Our business previously included a biotechnology operation that was focused on the discovery and development of novel antibodies. We spun-off the operation to our stockholders as Facet Biotech Corporation (“Facet”) in December 2008. Our principal executive offices are located at 932 Southwood Boulevard, Incline Village, Nevada, 89451, (775) 832-8500, and our website address is www.pdl.com. The information in or accessible through our website is not incorporated into, and is not considered part of, this filing.
Available Information
We file electronically with the U.S. Securities and Exchange Commission (the “SEC”) our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that website is www.sec.gov.
We make available free of charge on or through our website at www.pdl.com our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and proxy statements, as well as amendments to these reports and statements, as soon as practicable after we have electronically filed such material with, or furnished them to, the SEC. You may also obtain copies of these filings free of charge by calling us at (775) 832-8500. Also, our Audit Committee Charter, Compensation Committee Charter, Nominating and Governance Committee Charter, Litigation Committee Charter, Corporate Governance Guidelines and Code of Business Conduct, as well as amendments thereto, are also available free of charge on our website or by calling the number listed above. The information in or accessible through the SEC and our website is not incorporated into, and is not considered part of, this filing.
We operate our business as four segments as defined by U.S. generally accepted accounting principles. Our financial results for the years ended December 31, 2019 and 2018 are discussed in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this annual report. For management’s discussion covering the fiscal year ended December 31, 2017, please refer to Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Form 10-K for the fiscal year ended December 31, 2018. Our financial results for the years ended December 31, 2019, 2018 and 2017 are discussed in “Item 8. Financial Statements and Supplementary Data” of this Annual Report.
24
ITEM 1A. RISK FACTORS
You should carefully consider and evaluate all of the information included and incorporated by reference in this Annual Report, including the risk factors listed below. Any of these risks, as well as other risks and uncertainties, could materially and adversely affect our business, results of operations and financial condition, which in turn could materially and adversely affect the trading price of shares of our common stock. Additional risks not currently known or currently material to us may also harm our business.
We are exploring and evaluating potential transactions pursuant to our monetization strategy and plan of complete liquidation and there can be no assurance that we will be successful in identifying or completing any potential transaction or otherwise providing value to our stockholders or successfully implementing the strategy, that any such potential transactions will yield significant value for stockholders or that the process will not have an adverse impact on our business.
In September 2019, in an effort to enhance stockholder value, we commenced a review of strategic alternatives, including a possible sale or liquidation of our company. In December 2019, we announced that we had completed the strategic review process that we had initiated in September 2019 and that, as a result, we had decided to halt the execution of its growth strategy, cease additional strategic investments and pursue a formal process that is intended to unlock value by monetizing the Company’s assets and returning any available net proceeds to stockholders. We further announced in December 2019 that we would explore a variety of potential transactions in connection with such monetization strategy, including a sale of our company, divestiture of our assets or businesses, a spin-off transaction, a merger or a combination thereof. In February of 2020, the board of directors approved, consistent with our monetization strategy, a plan of complete liquidation and passed a resolution to seek stockholder approval to dissolve the Company under Delaware law at its next annual stockholder meeting. However, there can be no assurance that the exploration of one or more potential monetization transactions will result in the identification or consummation of any transaction, the period of time it will take to effect the strategy, or that we will be successful in implementing the strategy.
The success of our strategy will depend on our ability to identify and complete one or more transactions that will capture value for our assets, and on numerous other factors, many of which are beyond our control. Such factors include market conditions, industry trends, the interest of third parties in our business and assets and the availability of financing to potential buyers. Our stock price or the value of net proceeds we are able to generate from the monetization process may be adversely affected if the process is delayed, does not result in one or more successful transactions or if we are not able to execute the strategy. Even if one or more transactions are completed, there can be no assurance that they will have a positive effect on stockholder value. Our board of directors may determine to modify, amend or terminate the strategy at any time. If our board of directors were to so determine, there could be a material adverse effect on our business, financial condition and results of operations, and we would need to continue to operate our business and seek to grow it and create stockholder value.
In addition, our financial results and operations may be adversely affected by our monetization strategy and by the uncertainty regarding its outcome. Management and our board of directors have been and will continue to focus on our monetization strategy as well as on the continued operations of our business until such time, if any, that the monetization strategy has been successfully executed or abandoned. Additionally, we have directed capital resources to the strategy that otherwise could have been used in our business operations, and we expect to continue to do so until the process is completed or a determination is made that we will no longer pursue the strategy. We expect to incur substantial expenses associated with identifying and evaluating potential transactions, including those related to employee retention payments, equity compensation, severance pay, directors and officers insurance, taxes, and legal, accounting and financial advisory fees. The process of exploring potential transactions is expected to be time consuming and disruptive to our current business operations and, if we are unable to effectively manage the process, our business, financial condition and results of operations would be adversely affected.
Furthermore, speculation regarding any developments related to the review of strategic alternatives and perceived uncertainties related to the implementation of the monetization strategy could cause our stock price to fluctuate significantly.
We cannot assure you that we will be able to successfully implement our monetization strategy or that any transaction we may enter into pursuant to the strategy would yield significant value for our stockholders. We also cannot assure you that any potential transaction or other strategic alternative, if identified, evaluated and consummated, will provide greater value to our stockholders than that reflected in the current stock price.
Our efforts to enhance stockholder value through our monetization strategy may not be successful.
We cannot assure you that our efforts to enhance stockholder value through the conduct of our monetization strategy will succeed. There will be risks associated with any potential divestiture transaction, including whether we will attract potential acquirers for
25
the Company, its assets or its businesses, and whether offers made by such potential acquirors, if any, will be at valuations that we deem reasonable. Moreover, we are not able to predict how long it will take to implement our monetization strategy. The timing and terms of any transaction will depend on a variety of factors, many of which are beyond our control. A delay in, or failure to complete, any such transaction could have a material effect on our stock price and the amount of any potential distributions to our stockholders.
If we were to pursue a plan of dissolution, there can be no assurance as to the amount, if any, of cash or other property that could be distributed to our stockholders or the timing of any such future distribution.
Assuming successful implementation of our monetization strategy and plan of complete liquidation, and an inability to sell the Company as a whole, together with all or less than all of its assets, we expect to pursue a plan of dissolution as the most effective mechanism for wind-down of the Company and resolution of outstanding claims. If such a plan of dissolution were to be approved by our stockholders, once implemented through the filing of a Certificate of Dissolution and in accordance with Delaware law, known liabilities would be paid or provided for, reserves would be established for contingent known and unknown liabilities and any remaining assets would be monetized with net proceeds ultimately distributed to stockholders. The period for claimants to file claims against the Company following the filing of a Certificate of Dissolution is set at three years by Delaware statute. However, to the extent the Company is subject to pending litigation, the Company would potentially continue its existence through the claims resolution process beyond the three-year period, with attendant expenses, until such litigation is resolved. After the filing of a Certificate of Dissolution, it is possible that remaining assets not sold during the pre-dissolution period would be monetized and net proceeds ultimately distributed, subject to the claims resolution process. We expect that we would have limited or no new revenue generation sources or activities and that we would not engage in further business activities following the adoption of a dissolution plan and the filing of a Certificate of Dissolution except for winding up our business, selling or disposing of any of our remaining saleable assets, satisfying and providing for our liabilities and claims, and distributing net proceeds to stockholders. The amount and timing of any distributions to stockholders would be determined by our board of directors (or the trustee of a liquidating trust if our assets and liabilities are transferred to a liquidating trust pursuant to a plan of liquidation and dissolution), in its sole discretion, and would depend, in part, on our ability to settle or otherwise resolve and provide for all of our remaining liabilities and contingencies and convert any remaining assets into cash. In addition, after the filing of a Certificate of Dissolution, the Company expects to follow a process which will involve appearances before the Delaware courts to obtain the court’s confirmation that stockholder distributions are in compliance with Delaware law, including whether the Company has set aside sufficient funds as a reserve for claims, and these proceedings may delay or limit such post-dissolution distributions. If we pursue liquidation and dissolution, uncertainties as to the amount of our liabilities and the disposition value, if any, of our remaining assets make it impractical to predict the net value which might ultimately be distributable to our stockholders. No assurance can be given that available cash and any amounts received on any sale of assets will be adequate to provide for our obligations, liabilities, expenses and claims and to make cash distributions to stockholders. We also cannot assure you that the value of any distribution in liquidation would equal the price or prices at which our common stock has recently traded or may trade in the future.
We cannot predict the timing, amount or mechanics of any potential distributions to our stockholders.
Many unknown variables will affect the amount, timing and mechanics of any potential distributions to stockholders. Factors that could have a material effect on the amount of any potential future distributions include, but are not limited to, decreases in the purchase price that third parties are willing to pay for our assets, a failure to sell our assets, the amount of assets and corporate wind-down related operating and other expenses, the Company’s tax treatment, any required reserves to address potential liabilities, including retained and contingent liabilities (including, but not limited to those arising from any sales of the Company’s assets), and/or other unforeseen events. These and other factors, such as the procedures established under Delaware law for the dissolution of a corporation, could also delay the timing of any potential distributions.
A delay in the sales of our assets is likely to decrease the funds available for distribution to stockholders.
Potential liabilities and expenses from operations (including, but not limited to operating costs such as salaries, directors’ fees, directors’ and officers’ insurance, federal and state income taxes, payroll and local taxes, legal, investment banking, consulting and accounting fees and miscellaneous office expenses) will continue to be incurred by us as we seek to sell our assets and wind-down our operations. In the event that any sales of our assets are delayed, we may incur additional liabilities and expenses from operations, that will reduce the net funds ultimately available for distribution to our stockholders.
26
Focus on our monetization strategy could materially and adversely affect our existing operating businesses.
Until such time, if any, as we are able to successfully implement our monetization strategy, we are subject to operational risks related to our existing business. As a result of our monetization strategy, our management’s focus and attention on such efforts may be diverted, which could cause disruption of our ongoing business or inconsistencies in standards and controls that could negatively affect our ability to maintain third-party relationships. Moreover, we do not anticipate raising additional capital, which could result in a shortfall in our cash resources that would limit our ability to operate our business profitably, and could otherwise have a material adverse effect on our business, results of operations, financial condition and prospects. Management will be required to devote sufficient attention to both the monetization strategy and continued development of our businesses until the strategy is fully executed.
Our revenues from our Pharmaceutical segment consist entirely of sales of the Noden Products, and our revenues from our Medical Devices segment consist entirely of sales and leasing of the LENSAR Laser System. The success of Noden is dependent upon the success of the prescription pharmaceutical products sold under the brand names Tekturna, Tekturna HCT, Rasilez and Rasilez HCT, and there can be no assurance that in the future we will be able to continue to successfully attain and maintain significant market acceptance of our products among physicians, patients, third-party payors and others in the health care community. Failure to do so could adversely affect the value we receive from any sale of such products or businesses as part of our monetization strategy.
Also, we have experienced generic product competition for our products, which may increase in the future and reduce our market share. In March 2019, under an agreement with Prasco Laboratories, and in anticipation of the launch of third party generic aliskiren products by Par Pharmaceuticals, we launched in the United States an authorized generic form of Tekturna. There can be no assurance that we will be able to maintain meaningful revenues from sales of this authorized generic product, and there can be no assurance that we will be able to maintain market acceptance of this authorized generic product.
In addition, our acquisition of LENSAR resulted in establishing our Medical Devices segment. There can be no assurance that we will be able to continue to successfully develop this segment on a commercial scale. Any failures or perceived difficulties in developing these assets on a commercial sale in the future could have an adverse impact on our business, and could limit the value we may realize with respect to LENSAR.
Our strategic investment in the common stock of Evofem is our sole asset in our Strategic Positions segment. The value of our Evofem common stock is dependent upon the success of the product candidates under development by Evofem. Evofem is a pre-commercial company and, as such, is not yet engaged in revenue-generating activities. As a minority stockholder of Evofem, we have no control or oversight over the management of the Evofem business, and the value of our Evofem common stock will depend on the Evofem management team’s ability to develop, raise capital and successfully market and sell Amphora, the failure of which would have a material adverse effect on our investment and the value thereof. There can be no assurance as to the value we are able to achieve regarding monetization of our investment in Evofem.
We are substantially dependent on our key employees to facilitate the consummation of our monetization strategy and to continue to operate our business until the strategy is fully executed, and it may be difficult to retain such employees.
In December 2019, we implemented a strategic process to monetize our existing assets and return net proceeds to our stockholders. In order to successfully operate our business prior to finalization of our monetization process, we must retain certain of our key personnel. Certain of our employees have a significant amount of know-how and experience in our company, and the loss of one or more of them could have a material and adverse effect on our operations or ability to implement our monetization strategy. In an effort to retain key personnel for our monetization strategy, we implemented a wind-down retention plan that provides certain benefits to employees in consideration for their continued employment with the company. Despite our efforts to retain these employees, one or more may terminate their employment with us on short notice. The loss of the services of any of these employees could potentially harm our ability to implement our monetization strategy and evaluate and pursue strategic transactions, continue to operate our business during the execution of the strategy and fulfill our reporting obligations as a public company. If we are unsuccessful in retaining qualified personnel, our ability to execute our monetization strategy may be adversely affected.
Distributions to our stockholders may be treated as a dividend to the extent of our current and accumulated earnings and profits, rather than a distribution in exchange for our stock.
27
Generally, a distribution by us to our stockholders would constitute dividends for U.S. federal income tax purposes to the extent of our current and accumulated earnings and profits, as determined under U.S. federal income tax principles. However, distributions by us to our stockholders in “complete liquidation” would be treated as payment in exchange for our stock. The term “complete liquidation” is not defined in the Internal Revenue Code. While we expect our monetization strategy to be implemented in a manner so as to cause distributions to our stockholders to qualify as one or more distributions in “complete liquidation” for federal income tax purposes, there can be no assurance that efforts to do so will be successful or that the Internal Revenue Service will not take a contrary position. To the extent that any distributions do not qualify as distributions in “complete liquidation”, they will be treated for U.S. federal income tax purposes as dividends to our stockholders to the extent of our current and accumulated earnings and profits.
Our results of operations and/or our monetization strategy could be materially negatively affected by market fluctuations, business or economic disruptions or public health risks.
Our results of operations and/or our monetization strategy could be materially negatively affected by economic conditions generally, both in the United States and elsewhere around the world. Concerns over inflation, energy costs, geopolitical issues, public health emergencies, the availability and cost of credit, and the U.S. financial markets have in the past contributed to, and may continue in the future to contribute to, increased volatility and diminished expectations for the economy and the markets. For example, in December 2019, a novel strain of coronavirus surfaced in Wuhan, China. The coronavirus has impacted the global economy, including limiting business activities in China and South Korea, and may impact our operations including, among other things, sales to international customers (including those in China and South Korea) and the potential interruption of LENSAR’s supply chain. Further, entities in which we have invested, including Evofem, may be negatively impacted by the coronavirus, which could decrease the value of our investment and impact our ability to liquidate our investment on favorable terms, or at all. The extent to which public health issues or pandemics, including the coronavirus, will impact our results of operations and the results of operations of the entities in which we have invested will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of the coronavirus and the actions to contain the coronavirus or treat its impact, among others. In addition, domestic and international equity and debt markets have experienced and may continue to experience heightened volatility and turmoil based on domestic and international economic conditions and concerns. In the event these economic conditions and concerns continue or worsen and the markets continue to remain volatile, our results of operations could be adversely affected by those factors in many ways and our stock price or the value of our assets may decline. In addition, we maintain significant amounts of cash and cash equivalents at one or more financial institutions that are not federally insured. If economic instability continues, we cannot provide assurance that we will not experience losses on these investments. The occurrence of any of the foregoing events could have a material adverse effect on our business, results of operations and/or our ability to return value to our stockholders, including through our monetization strategy.
Our ability to successfully complete our monetization strategy could be materially negatively affected by public health risks such as the recent outbreak of the coronavirus.
We are exploring and evaluating potential transactions in furtherance of our monetization strategy, the success of which may be impacted by the growing spread of the coronavirus globally. In order to successfully monetize our assets, we must identify and complete one or more transactions with third parties. The business and assets and the availability of potential buyers of our company or certain of our assets may be significantly impacted by public health issues or pandemics, including the coronavirus. The uncertain severity and impact of the coronavirus could result in reduced demand for our assets by third parties globally as well as potentially affect our own ability to operate.
Even if we are able to identify potential transactions in furtherance of our monetization strategy, such buyers may be operationally constrained or unable to locate financing on attractive terms or at all, which risk may be heightened due to the uncertainty of the coronavirus and its impact. We are subject to increased risk that the growing spread of the coronavirus will affect the geographies, both in the near term and in the future, of any third parties we identify as possible counterparties to any monetization transaction. If financing is unavailable to potential buyers of our company or assets, or if potential buyers are unwilling to engage in various transactions due to the uncertainty in the market, our ability to complete such acquisition would be significantly impaired.
Any negative impact on such third parties due to any of the foregoing events could cause costly delays and have a material adverse effect on our ability to return value to our stockholders, including our ability to realize full value from a sale or other disposition of our assets as part of our monetization strategy. In addition, if members of our management team were to be affected by COVID-19, this could significantly delay or impair our ability to execute our monetization strategy.
Our ability to realize value from our investments in Evofem and LENSAR may depend on whether they can successfully develop, gain approval for and commercialize products. Failure of Evofem to successfully develop, gain approval or commercialize Amphora for prevention of pregnancy would likely cause its business to fail, which would diminish or
28
eliminate the value of our investment in Evofem. Failure of LENSAR to successfully continue its development and commercialization of a next generation cataract laser surgery system could negatively affect our ability to realize full value from a sale or other disposition of the business as part of our asset monetization strategy.
The value of our investment in Evofem and LENSAR may be dependent on their ability to successfully develop their respective products.
Evofem resubmitted an NDA for Amphora for the prevention of pregnancy in 2019; however, there is no assurance that the FDA will approve Amphora for this indication or for any other indication. Evofem has not received regulatory approval for any product. Even though Evofem was able to successfully complete its clinical trial for Amphora for prevention of pregnancy, it may be unable to obtain regulatory approval for Amphora for prevention of pregnancy. The state of development of Amphora, including the FDA review and approval process, at the time we determine optimal to dispose of our investment position in Evofem will have a significant impact in the potential value realized and is entirely out of our control. The commercial success of Amphora will also depend in significant measure upon Evofem’s ability to obtain marketing approval from the FDA or other regulatory authorities including an indication and labeling of sufficient scope to be commercially meaningful. Failure to achieve marketing approval from the FDA or other regulatory authorities of a commercially meaningful indication and labeling may substantially limit Evofem’s ability to market and promote Amphora and our ability to monetize or otherwise dispose of our investment in Evofem. In addition, to obtain marketing approval of Amphora on schedule, manufacturing facilities operated by third parties with which Evofem has contracted for the purpose of the supply of Amphora will need to pass a regulatory inspection. Failure of the FDA to approve manufacture of Amphora at such third party facilities may delay approval, and consequently affect the value of our investment in Evofem. Evofem will also likely incur significant costs associated with launching and Amphora, including the development of a successful commercial team and strategy. The failure of Evofem to successfully develop, gain marketing approval and commercialize Amphora would have a material adverse impact on our investment in Evofem and could limit any upside, or result in a loss, on our investment, or limit our ability to generate significant value for stockholders in connection with the potential disposition of our investment in furtherance of our monetization strategy.
LENSAR is developing and intends to commercialize a next generation cataract laser surgery system. The value we are able to obtain from our investment in LENSAR may depend on the value investors and/or potential acquirors perceive in the next generation system, which may not be in condition for approval or commercialization prior to our monetization of LENSAR. The development process for new devices in the eye care industry and more general healthcare industry can sometimes be expensive, prolonged and entail considerable uncertainty. Because of the complexities and uncertainties associated with ophthalmic research and development in particular, and healthcare related research and development in general, products LENSAR is currently developing, or that may be developed in the future, are subject to the risk that LENSAR may not complete the development process or obtain the regulatory approvals required to market such products successfully. These complexities and risks could significantly reduce the value we are able to achieve during the monetization process from our investment in LENSAR.
We may be unable to monetize our investment in Evofem due to the illiquidity of our shares, and our position in Evofem may be subject to dilution and fluctuations in value, each of which could have a material and adverse impact on our financial condition, results of operations and/or ability to monetize or otherwise dispose of our position.
As of December 31, 2019, we held approximately 28% of the shares of common stock of Evofem. Our shares of Evofem were acquired in a private placement and, absent registration, are deemed to be restricted stock. A such, it may be difficult to sell our Evofem shares in connection with our monetization strategy, and any such sale may be delayed due to the lead time required to register such shares with the Securities and Exchange Commission.
Furthermore, it is expected that Evofem will seek to raise significant additional capital to finance its operations in the future. Raising additional capital may cause substantial dilution in our investment and such financing activities could limit our ability to generate a meaningful return or sell our investment as we attempt to monetize or otherwise dispose of our position.
We account for our investment in Evofem using the fair value method. Because a mark to market valuation will occur at the end of each quarterly reporting period, changes in fair value will vary based upon the volatility of the stock price, and such changes in fair value could have a material and adverse impact on our financial condition and results of operations.
If we do not meet the requirements for continued listing on The Nasdaq Stock Market, our common stock could be delisted which would adversely affect the ability of our stockholders to sell shares of our common stock.
Our common stock is currently listed on The Nasdaq Stock Market. The Nasdaq Stock Market imposes continued listing requirements that companies must meet to remain listed on that market. These requirements include, among other things,
29
minimum levels of assets and revenues. The Nasdaq Stock Market also considers factors like the number of employees of a company and a company’s ongoing revenue generating operations and plans. If we do not meet the requirements for continued listing on The Nasdaq Stock Market, whether as a result of our monetization and distribution of net proceeds to stockholders or otherwise, our common stock could be delisted. Further, upon dissolution, we anticipate that our stock would then be delisted from Nasdaq and our stock transfer books closed, after which time it would not be possible for stockholders to publicly trade our stock. At that point, the proportionate interests of all of our stockholders will be fixed on the basis of their respective stock holdings at the close of business on the final record date, and, after the final record date, in general any distributions made by us will be made solely to the stockholders of record at the close of business on the final record date. The delisting of our common stock would have an adverse effect on the liquidity of our common stock, such as the ability of our stockholders to sell their shares of our common stock, and its trading price. If our common stock ceases to be traded or quoted on any of the NYSE, the NASDAQ Global Select Market or the NASDAQ Global Market, a “fundamental change” under each of our convertible notes indentures would occur, which entitles the holders of the convertible notes issued under such indentures to require us to repurchase the convertible notes of such holders. Delisting could also have other negative results, including, but not limited to, the potential loss of confidence by employees, the loss of institutional investor interest and fewer opportunities with counterparties to potential monetization transactions.
If we remain an independent company, we may need to obtain funds from additional financings or other sources for our business activities. If we do not receive these funds, we may need to reduce, delay or eliminate some of our expenditures.
If we are not successful in implementing, or the board of directors decides to terminate, the monetization strategy, and we were to therefore remain an independent company, we may need to raise additional capital to accomplish our business plan over the next several years through debt or equity financing, joint ventures, license agreements, sale of assets, as well as various other financing arrangements. If we obtain additional funds by issuing equity securities, dilution to stockholders may occur. There can be no assurance as to the availability or terms upon which such financing and capital might be available.
Through our investments in Noden and Evofem, and our acquisition of LENSAR, we have a significant investment in the commercialization of products worldwide, and our returns from these assets are subject to a number of risks associated with international operations that could materially and adversely affect our business, results of operations and cash flows and/or our ability to monetize or otherwise dispose of such assets.
As a result of our Noden and LENSAR operating businesses, and our strategic investment in Evofem, we are directly and indirectly subject to a number of risks related to the sale of products worldwide, including:
• | international regulatory requirements for drug and medical device marketing and pricing in foreign countries; |
• | varied standards of care in various countries that could complicate the commercial success of products; |
• | varied drug and medical device import and export rules; |
• | varying standards for the protection of intellectual property rights which may result in reduced or compromised exclusivity in certain countries; |
• | unexpected changes in tariffs, trade barriers and regulatory requirements; |
• | varied reimbursement systems and different competitive drugs indicated to treat the indications for which Noden Products are being commercialized and medical devices indicated to treat the indications for which LENSAR products are being commercialized; |
• | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
• | widespread outbreak of health epidemics that could impact international sales and operations; |
• | compliance with tax, employment, immigration and labor laws applicable to foreign operations; |
• | compliance with the U.S. Foreign Corrupt Practices Act (“FCPA”), the UK Bribery Act, and other anti-corruption and anti-bribery laws; |
• | foreign taxes and duties; |
• | foreign currency fluctuations and other obligations incident to doing business in another country; |
• | workforce uncertainty in countries where labor unrest is more common than in the United States; |
• | reliance on management, contract services organizations and other third parties that may be less experienced with manufacturing and commercialization than the party from whom the Noden Products were acquired; |
• | potential liability resulting from product liability laws or the activities of foreign distributors; and |
• | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters. |
30
In addition, our international operations could be affected by capital and exchange controls, expropriation and other restrictive government actions as well as by political unrest, unstable governments and legal systems and inter-governmental disputes. Any of these circumstances could materially and adversely affect our business, results of operations and cash flows, as well as adversely affect our ability to monetize or dispose of such assets and/or reduce the proceeds we realize in such a monetization or disposition.
The terms of our convertible notes indentures could negatively affect our ability to execute the monetization strategy.
In addition to the requirement under our convertible notes indentures for us to repurchase the convertible notes upon the election of the holders of such convertible notes upon a “fundamental change,” each of the convertible notes indentures includes a merger covenant that requires any successor company that purchases “substantially all” of our property and assets to assume the obligations under such convertible notes indenture. There is no precise established definition of the phrase “substantially all” under New York law, the law which governs each of our convertible notes indentures, and whether a transaction or series of transactions constitutes “substantially all” of our property and assets depends on the consideration of both quantitative and qualitative factors. Consequently, depending on the transactions pursued by us in connection with the monetization strategy and the sequencing and timing of these transactions, our monetization strategy could result in a transfer of “substantially all” of our properties and assets under each of our convertible notes indentures, which would require the transferee under such monetization strategy to assume the obligations under such convertible notes indenture and could thus negatively affect our ability to execute the monetization strategy. In order to minimize the impact of the merger covenants in our convertible notes indentures on our ability to execute the monetization strategy, we could either conduct a tender offer for our outstanding convertible notes or could solicit consents from the holders of our outstanding convertible notes to waive the requirements of such merger covenants, or could pursue a combination of tender offers and consent solicitations, but we cannot assure you that any such tender offers or consent solicitations would become effective or that they would be agreed upon on commercially acceptable terms.
We have in the past and are currently involved in, and expect that in the future we will from time to time be involved in, litigation, either as a defendant or a plaintiff, which could have a negative impact on our business, results of operations and cash flows and/or our monetization strategy.
Monitoring and defending against or prosecuting legal actions is time-consuming for our management, will require litigation related expenses, and may detract from our ability to fully focus our internal resources on our operations and monetization strategy. Moreover, we may be subject to additional litigation as we pursue potential transactions in furtherance of our monetization strategy, and expect to be subject to increased risk of litigation following the completion of the divestiture of all or a portion of our assets or businesses, a spin-off transaction, a merger, or any combination thereof. In addition, the stock markets have experienced significant price and volume fluctuations that have affected the market prices for the common stock of companies. These broad market fluctuations as well a broad range of other factors, including the realization of any of the risks described in these “Risk Factors,” may cause the market price of our common stock to decline. In the past, securities class action litigation has often been brought against a company involved in significant corporate transaction or following a decline in the market price of its securities. Legal fees and costs incurred in connection with litigation may be significant. Depending on the nature of the lawsuit, a decision adverse to our interests could result in the payment of substantial damages and could have a material adverse effect on our cash flow, results of operations, financial position and ability to return value to stockholders, or impact our rights in an adverse way. In addition, if we elect to file a certificate of dissolution, we may be subject to litigation with potential or unknown claimants, which litigation will have an effect on the timing and ability of the Company to make distributions to stockholders, and will be accompanied by litigation related costs and operations.
We rely on third-party manufacturers to manufacture our pharmaceutical products, and these third parties may not perform adequately.
We do not have any operating manufacturing facilities for Noden Products, and do not expect to independently manufacture our products under the Pharmaceutical segment. We currently rely on Novartis for a specified period of time to manufacture and package the Noden Products.
Risks arising from reliance on third-party manufacturers include:
• | reduced control and additional burdens of oversight as a result of using third-party manufacturers for all aspects of manufacturing activities, including regulatory compliance and quality control and assurance; |
• | termination or non-renewal of manufacturing and supply agreements with third parties in a manner or at a time that may negatively impact commercialization activities; and |
• | disruption in the operations of third-party manufacturers or suppliers unrelated to our products, including the bankruptcy of the manufacturer or supplier or a catastrophic event affecting the third manufacturers or suppliers. |
31
In addition, difficulties or delays in product manufacturing and reliance on third-party manufacturing could adversely affect performance of Noden and the Noden Products by virtue of regulatory actions, shut-downs, approval delays, withdrawals, recalls, penalties, supply disruptions or shortages or force majeure events, reputational harm, product liability, unanticipated costs or otherwise. Examples of such difficulties or delays include, but are not limited to, the possibility that the supply of incoming materials may be delayed or become unavailable or be subject to increased costs and that the quality of incoming materials may be substandard and not detected; the possibility that third-party manufacturers may fail to maintain appropriate quality standards throughout the internal and external supply network and/or comply with cGMPs and other applicable regulations such as tracking and tracing of products in the supply chain to enhance patient safety; risks to supply chain continuity as a result of natural or man-made disasters at a supplier or vendor; or failure to maintain the integrity of the supply chains against intentional and criminal acts such as economic adulteration, product diversion, product theft, and counterfeit goods. Any of these events could adversely affect our ability to successfully commercialize our products and/or our ability to sell our products as part of our monetization strategy or realize expected value in such a sale.
Our products are subject to the risks and uncertainties of branded pharmaceutical and medical device products.
If our products become subject to problems such as changes in prescription growth rates, product utilization, product liability litigation, unexpected side effects, regulatory proceedings, manufacturing issues, publicity affecting doctor or patient confidence, pressure from existing competitive products, changes in labeling, loss of patent protection (when applicable), or, if a new, more effective treatment should be introduced, the adverse impact on our revenues could be significant. The occurrence of any of the foregoing problems, or additional problems that may arise in the future, could materially and adversely affect our business, results of operations and financial condition, and/or our ability to successfully monetize the underlying assets pursuant to our monetization strategy.
For as long as we own our Noden business, we will continue to depend upon a limited number of wholesalers for a significant portion of our revenues from the Noden Products, and the loss of, or significant reduction in sales to, any one of these wholesalers could materially and adversely affect our business, results of operations and financial condition and/or our ability to sell Noden or its assets, as well as materially reduce the amount of value we could realize in such a sale.
We sell the Noden Products primarily to wholesalers. Wholesalers sell the Noden Products to hospitals and pharmacies. We do not promote the Noden Products to wholesalers, and they do not set or determine demand for Noden Products. Our ability to successfully commercialize Noden Products will depend, in part, on the extent to which we are able to provide adequate distribution of the Noden Products to patients. Although we have contracted with a number of wholesalers, they are expected generally to carry a very limited inventory and may be reluctant to be part of our distribution network in the future if demand for the product does not increase.
The use of pharmaceutical wholesalers involves certain risks, including, but not limited to, risks that these pharmaceutical wholesalers will not provide us accurate or timely information regarding their inventories, demand from wholesaler customers buying the Noden Products or complaints about the Noden Products, that these wholesalers will reduce their efforts or discontinue to sell or support or otherwise not effectively sell or support the Noden Products, or not devote the resources necessary to sell the Noden Products in the volumes and within the time frames that we expect.
Further, it is possible that these wholesalers could decide to change their policies or fees, or both, at some time in the future. This could result in their refusal to carry smaller volume products such as Noden Products, or lower margins or the need to find alternative methods of distributing the Noden Products. Although we believe we can find alternative channels to distribute the Noden Products on relatively short notice, our revenue during that period of time may suffer and we may incur additional costs to replace any such wholesaler. The loss of any large wholesaler as part of our distribution network, a significant reduction in sales we make to wholesalers, or any failure to pay for the Noden Products we have shipped to them could materially and adversely affect our business, results of operations and financial condition and/or our ability to sell Noden, as well as materially reduce the amount of value we could realize in such a sale.
Certain of our income generating assets are secured by collateral, and we may be, or may become, under-secured by the collateral, or such collateral may not have a value equal to our investment in the event of a default by our counterparties, or may lose value, each of which could negatively affect our ability recuperate our capital expenditures in such income generating assets.
Among our current income generating assets, the Wellstat Diagnostics loan and the CareView loan have exposed us to credit risk due to default or potential default by the counterparty. To mitigate this risk at the initiation of the loans, we obtained security
32
interests as collateral in the assets of such counterparty. Our credit risk in respect of such counterparty may be exacerbated when the collateral held by us cannot be realized upon or is liquidated at prices not sufficient to recover the full amount we are due pursuant to the terms of the particular credit agreement. This could occur in circumstances where the original collateral was not sufficient to cover a complete loss (e.g., our interests were only partially secured) or may result from the deterioration in value of the collateral, so that, in either such case, we are unable to recover our full capital outlay and any anticipated return. Obligations under these credit agreements are secured by a pledge of substantially all the assets of the borrower and any of its subsidiaries and, in the instance of Wellstat Diagnostics, by assets held by its affiliates. Although these loans are secured, we cannot guarantee that we will be able to collect all or part of the amounts owed to us. If we are unable to collect any amount, or amounts collected are not equal to our investment in the event of default by our counterparties, the value of our assets may decrease, which could materially and adversely affect our business, results of operations and financial condition, and/or our ability to successfully monetize the underlying assets pursuant to our monetization strategy. Additionally, we may face difficulty in collection efforts with respect to the Wellstat Diagnostics and CareView loans. Such difficulties with the CareView loan could lead to litigation or other legal procedures which may or may not be successful, and which will require significant financial and management resources to address. We have been engaged in multiple legal proceedings with Wellstat Diagnostics and its affiliates related to their credit agreement default, which is described in more detail in Note 25, Legal Proceedings, of this Annual Report. Any such losses resulting therefrom could materially and adversely affect our business, results of operations and financial condition.
We are exposed to the credit risk of some of our customers, which could result in material losses.
In our Medical Devices segment, customers may finance through leasing the acquisition of certain devices, and we believe there has been an increase in demand for customer financing through leasing in recent years. We may experience loss from a customer’s failure to make payments according to the contractual lease terms. Our exposure to the credit risks relating to our lease financing arrangements may increase if our customers are adversely affected by changes in healthcare laws, coverage and reimbursement, economic pressures or uncertainty, or other customer-specific factors. The factors affecting our customers’ ability to make timely payments according to the contractual lease terms are out of our control, and as a result, exposes us to additional risks that may materially and adversely affect our business and results of operations. The occurrence of any such factors affecting our customers may cause delays in payments or, in some cases, defaults on payment obligations, which could result in material losses.
Although we have programs in place that are designed to monitor and mitigate the associated risk, there can be no assurance that such programs will be effective in reducing credit risks relating to these lease financing arrangements. If the level of credit losses we experience in the future exceed our expectations, such losses could have a material adverse effect on our financial condition or results of operations or adversely affect our ability to sell such assets as part of our monetization strategy.
We, our licensees, borrowers and royalty-agreement counterparties and the companies in which we invest may be unable to maintain regulatory approvals for currently licensed products, or to obtain regulatory approvals or favorable pricing for new products, and we or they may voluntarily remove currently licensed products from marketing and commercial distribution. Any of such events, whether due to safety issues or other factors, could reduce our revenues or return on investments, and could limit our ability to generate expected returns from the monetization of such assets.
We and our licensees, borrowers and royalty-agreement counterparties and companies in which we have invested, are subject to stringent regulation with respect to product safety and efficacy by various international, federal, state and local authorities. Of particular significance are the FDA requirements covering research and development, testing, manufacturing, quality control, labeling and promotion of drugs and medical devices for human use in the United States. As a result of these requirements, the length of time, the level of expenditures and the laboratory and clinical information required for approval of a biologic license application or new drug application and approval or clearance of a medical device are substantial and can require a number of years. In addition, even if our products, our licensees’, borrowers’ and royalty-agreement counterparties’ products or products of companies in which we invest receive regulatory approval, we and they will remain subject to ongoing FDA and other international regulations including, but not limited to, obligations to conduct additional clinical trials or other testing, changes to the product label, new or revised regulatory requirements for manufacturing practices, written advisements to physicians and/or a product recall or withdrawal. We, our licensees, borrowers and royalty-agreement counterparties and the companies in which we invest may not maintain necessary regulatory approvals for our or their existing licensed products or we or our licensees may not obtain necessary regulatory approvals on a timely basis, if at all, for any of our products, or the licensed products our licensees are developing or manufacturing. Moreover, the current political environment in the United States is focused on potential reductions in pricing for pharmaceutical and other healthcare products, which may negatively impact any existing or new products from which our revenues would be derived. We are unable to control the pricing strategies used by our licensees, borrowers and royalty-agreement counterparties and may have limited influence over the pricing strategies used by companies in which we invest, and if they fail to use appropriate pricing strategies, or receive negative reactions to their pricing strategies, it could negatively impact our revenues or return on investment. We may also select pricing strategies for our own products and medical
33
devices that are less competitive than those of our competitors, or we may fail to obtain acceptable prices or an adequate level of reimbursement for products or medical devices from third-party payors or governmental agencies, which could negatively impact our revenues or return on investment and could limit our ability to generate expected returns from the monetization of such assets.
In addition, communications from government officials regarding pricing for pharmaceutical and other health care products could have a negative impact on our stock price or the value of the assets we are seeking to monetize, even if such communications do not ultimately impact our products, our licensees’, borrowers’ and royalty-agreement counterparties’ products or the products of companies in which we invest. The occurrence of adverse events reported by any licensee, borrower or royalty-agreement counterparty or company in which we invest may result in the revocation of regulatory approvals or decreased sales of the applicable product due to a change in physicians’ willingness to prescribe, or patients’ willingness to use the applicable product. We, our licensees, borrowers and royalty-agreement counterparties and the companies in which we invest could also choose to voluntarily remove licensed products from marketing and commercial distribution. For example, in November 2011, the FDA removed the indication for breast cancer from Avastin’s label. In 2005, Tysabri was temporarily suspended and then returned to the market. In any of these cases, our revenues or return on investment could be materially and adversely affected. Moreover, any value we may receive upon the potential sale of our company or other potential transaction in furtherance of our monetization strategy could be materially and adversely affected.
In addition, the current regulatory framework could change, or additional regulations could arise at any stage during our licensees’ or invested company’s product development or marketing which may affect their ability to obtain or maintain approval of their respective products. Delays in our licensees or the companies in which we have invested receiving regulatory approval for their respective products or their failure to maintain existing regulatory approvals could have a material adverse effect on our business or our ability to successfully implement our monetization strategy.
Many of our pharmaceutical products, investments, medical devices and income generating assets are in companies or assets that have limited commercialized revenue-generating products or are dependent on the actions of unrelated third parties, which may negatively impact the value we are able to realize as part of our monetization strategy.
Our ability to realize full value for certain of our assets (including in connection with executing a successful monetization strategy) depends on the progress of such assets towards commercialization, and the ability to further the development in a competitive and highly regulated market. Our or our counterparties or their licensees inability to do so would negatively affect our investment returns and could limit our ability to generate expected returns from the monetization of such assets.
In addition, in connection with our income generating assets and our investments in our Strategic Positions segment, we are dependent, to a large extent, on third parties to enforce certain rights for our benefit. For example, when we acquired certain royalty rights from Assertio (formerly Depomed), which, as the licensor of certain patents, retains various rights, including the contractual right to audit its licensees and to ensure those licensees are complying with the terms of the underlying license agreements. Assertio also retained full responsibility to protect and maintain the intellectual property rights underlying the licenses. While we have contractual rights to require Assertio to take action regarding many of these rights, because Assertio’s economic interest in the license agreements is limited, it may not enforce or protect those rights as it otherwise would have had it retained the full economic interest in the payments under the license agreements. Moreover, in respect of the royalty stream relating to the Glumetza diabetes medication that we acquired from Assertio, which is the royalty right producing the highest revenues from our Assertio acquired royalties, a single generic manufacturer was approved by the FDA in February 2013 and in August 2016, two additional generic manufacturers were approved by the FDA to enter the US as provided for in settlement agreements between Assertio and these generic manufacturers. In February 2016, Lupin Pharmaceuticals, Inc., and in August 2017, Teva Pharmaceutical Industries Ltd., and in July 2018, Sun Pharmaceutical, Inc., each launched a generic equivalent approved product. We were aware of these settlement agreements, considered them in the cost of the acquiring this asset and expected the entry of these generic products to reduce our Glumetza revenues. We are further aware of additional approved generic extended release metformin products, which could negatively affect Glumetza revenues.
We and our companies’ licensees, borrowers and royalty-agreement counterparties face significant market pressures with respect to our and their products, and the amount of revenues from our or their pharmaceutical products or medical devices, or from our income generating assets that we receive are subject to various competitive and market factors that may be outside of our control.
We and our companies, licensees, borrowers and royalty-agreement counterparties face competition from other pharmaceutical, biotechnology, device and diagnostic companies. The introduction of new competitive products may result in lost market share for us or our licensees, borrowers and royalty-agreement counterparties, reduced use of our or their products or devices, lower prices and/or reduced sales, any of which could reduce our royalty revenues, or the revenues on which we rely to produce the returns on
34
our acquisitions, and have a material adverse effect on our results of operations. Any such reduction to revenues could further limit any realized value we may obtain in connection with the evaluation of potential transactions to monetize such assets.
The amount of any royalties or revenues, and the subsequent returns on our investments that we receive from our pharmaceutical products, medical devices and/or income generating assets will depend on many factors, including the following:
• | the timing and availability of generic product or devices competition for our products or devices, and our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices; |
• | potential challenges or design-arounds to product, use or manufacturing related patents which provide exclusivity for products and assets before their expiration by generic pharmaceutical manufacturers; |
• | the size of the market for our products or devices, and our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices; |
• | the extent and effectiveness of the sales and marketing and distribution support for our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices and commercial infrastructure with respect to our products or devices; |
• | the existence of novel or superior products or devices to our products or devices, or our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices; |
• | the availability of reduced pricing and discounts applicable to our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices; |
• | stocking and inventory management practices related to our products or our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices; |
• | limitations on indications for which our products or devices or our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices can be marketed; the competitive landscape for approved products or devices and developing therapies that compete with our products or devices or our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices; |
• | the ability of patients to be able to afford our products or devices, or our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices or obtain healthcare coverage that covers those products or devices; |
• | acceptance of, and ongoing satisfaction with, our products or devices and our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices by the care providers, patients receiving therapy and third-party payors; or |
• | the unfavorable outcome of any potential litigation relating to our products or devices and our licensees’, borrowers’ and royalty-agreement counterparties’ products or devices. |
For example, in mid-2019, Bausch Health announced potential price decreases on Glumetza, a royalty-bearing product under our Assertio Royalty Agreement. These price decreases could negatively affect revenues and thus our royalties. Due to the uncertainties caused by changes in pricing by third parties that are outside our control, including as a result of generic competition, we may not be able to accurately estimate the impact on royalties on such sales paid to us for Glumetza or any other product.
Additionally, Noden’s ’111 Patent, which was previously extended by virtue of pediatric testing requirements, expired in January 2019. While Noden has additional patent coverage related to drug formulation and manufacturing technology which relate to our commercialization of Tekturna in the United States which has not yet expired, the expiration of the composition of matter patents related to our Tekturna products will allow entry of competitors which have been able to design around the remaining formulation patents. For example, in 2018, Noden settled a paragraph IV challenge with Anchen Pharmaceuticals, Inc. which allowed entry into the market of a generic aliskiren product in March 2019, which generic product was subsequently made commercially available through Par Pharmaceuticals, Inc. and competes with Tekturna and our authorized generic product. While we are unaware of the entry of any additional third-party generic product at the present time, we cannot preclude the possibility that another third-party generic version of aliskiren will be available at some time in the future. In March 2019, under an agreement with Prasco Laboratories, we launched in the United States an authorized generic form of Tekturna which currently competes with the Par Pharmaceuticals generic aliskiren product. The increase in competition and additional generic competition may have a material and adverse effect on our ability to realize significant value for stockholders on the disposition or sale of the Noden Products in furtherance of our monetization strategy.
We, our licensees and the companies in which we have invested must continue to protect our and their intellectual property rights for us to protect the value of our assets and/or execute a successful monetization strategy.
Our success in protecting the value of our assets (including in connection with executing a successful monetization strategy) is dependent in significant part on our ability and the ability of third parties in control of the assets in which we’ve invested to
35
protect the scope, validity and enforceability of our and their intellectual property, including the patents, SPCs and license agreements, all of which support our revenues. The scope, validity, enforceability and effective term of patents and SPCs can be highly uncertain and often involve complex legal and factual questions and proceedings. In addition, the legal principles applicable to patents in any given jurisdiction may be altered through changing court precedent and legislative action, and such changes may affect the scope, strength and enforceability of our patent rights or the nature of proceedings which may be brought related to the relevant patent rights. A finding in a proceeding related to patent rights which support our revenues which narrows the scope or which affects the validity or enforceability of some or all of our patent rights could have a material impact on our ability to continue to collect royalty payments from our investments or collect revenue from our sales of our pharmaceutical products and medical devices. If the scope, validity and enforceability of our and their intellectual property were to be negatively impacted prior to monetizing any such assets, our expected return on such assets could be materially and adversely affected.
Our reliance on sole and single source suppliers could harm our ability to meet demand for our products or devices in a timely manner or within budget.
Some of the components necessary for the assembly of our Medical Devices segment are currently provided to us by sole-sourced suppliers or single-sourced suppliers. We generally purchase components through purchase orders rather than long-term supply agreements and generally do not maintain large volumes of inventory. While alternative suppliers exist and could be identified for sole-sourced components, the disruption or termination of the supply of components could cause a significant increase in the costs of these components, which could affect our operating results. A disruption or termination in the supply of components could also result in our inability to meet demand for our products, which could harm our ability to generate revenues, lead to customer dissatisfaction and damage our reputation. Furthermore, if we are required to change the manufacturer of a key component of our products, we may be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could delay our ability to manufacture our products in a timely manner or within budget, which may have a material adverse impact on our business, financial condition, results of operations, or cash flows, as well as our ability to successfully monetize or otherwise dispose of such products and/or related businesses.
Recently enacted and future legislation is expected to increase the difficulty and costs to maintain revenues from our products, and in particular may negatively impact the pricing of our products and/or the ability to realize value as part of our monetization strategy.
In the United States and some foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could, among other things, affect our ability to profitably sell our products.
For example, in the United States in March 2010, the Patient Protection and Affordable Care Act (“ACA”) was enacted to increase access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and the health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The law has continued the downward pressure on pharmaceutical pricing, especially under the Medicare program, and increased the industry’s regulatory burdens and operating costs. Among the provisions of the ACA of importance are the following:
• | manufacturers and importers of certain branded prescription drugs with annual sales of more than $5 million made to or covered by specified federal healthcare programs are required to pay an annual, non-tax deductible fee based on each company’s market share of all such sales in the prior year; |
• | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program for brand and generic drugs; |
• | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
• | extension of manufacturers’ Medicaid rebate liability to individuals enrolled in Medicaid managed care organizations; |
• | expansion of eligibility criteria for Medicaid programs in certain states; |
• | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries under their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Subsequent legislative amendments have increased the point-of-sale discounted to 70%, effective 2019; |
• | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program, commonly referred to as the “340B Program”; |
36
• | a new requirement to annually report drug samples that manufacturers and distributors provide to physicians, also known as the “Physicians Payment Sunshine Act”; and |
• | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
The potential financial impact of the ACA over the next few years will depend on a number of factors including policies reflected in implementing regulations and guidance and changes in sales volumes for products affected by the new system of rebates, discounts and fees. The taxes imposed by the ACA and the expansion in the government’s role in the U.S. healthcare industry may result in decreased profits to us, lower reimbursement by payors for our products, and/or reduced medical procedure volumes, all of which may have a material adverse effect on our business, financial condition and results of operations. The Trump Administration and the U.S. Congress may take further action regarding the ACA, including, but not limited to, repeal or replacement. For example, the Tax Cuts and Jobs Act of 2017 (the “2017 Tax Act”) was enacted, which, among other things, removes penalties for not complying with the individual mandate to carry health insurance. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the 2017 Tax Act, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the United States Court of Appeals for the Fifth Circuit affirmed the portion of the district court’s ruling declaring the individual mandate unconstitutional and remanded for the district court to conduct analysis in the first instance on which provisions of the statute are severable from it and thus remain intact. On March 2, 2020, the Supreme Court of the United States granted certiorari to hear the case. While the Trump Administration and the Centers for Medicare & Medicaid Services have both stated that the ruling will have no immediate effect, it is unclear how this decision, subsequent appeals, including the aforementioned appeal, and other efforts to repeal and replace the ACA will impact the ACA. There may be additional challenges and amendments to the ACA in the future, including continued litigation and legislation, and all or a portion of the ACA and related subsequent legislation may be modified, repealed or otherwise invalidated through judicial challenge. We cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us and will continue to evaluate the effect that the ACA and its possible repeal and replacement has on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. These changes included automatic aggregate reductions to Medicare payments to providers of 2% per fiscal year as part of the federal budget sequestration under the Budget Control Act of 2011, which went into effect in April 2013 and, due to subsequent legislative amendments, will remain in effect through 2025 unless additional action is taken by Congress. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers and increased the statute of limitations period in which the government may recover overpayments to providers from three to five years. In addition, recently there has been heightened governmental scrutiny over the manner in which drug manufacturers set prices for their commercial products and regulatory action aimed at reducing the cost of prescription drugs. For example, in December 2019, the U.S. Department of Health and Human Services and the FDA issued a proposed rule and draft guidance concerning two new pathways for importing lower-cost drugs into the United States. The proposed rule, if finalized, would allow certain prescription drugs to be imported from Canada. The draft guidance describes procedures for drug manufacturers to facilitate the importation of FDA-approved drugs and biologics manufactured abroad and originally intended for sale in a foreign country into the United States. Additionally, President Trump’s administration has proposed to establish an international pricing index that would tie domestic prices for certain drugs and biologics to the prices in other countries with more aggressive drug price regulation. The implementation of cost containment measures or other healthcare reforms may limit us from being able to generate revenue, attain profitability, or commercializing our products, which could have a material adverse effect on business and results of operations.
In any event, we expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for pharmaceutical products, which could result in reduced demand for our products or our counterparties’ products or additional pricing pressures on our products or our counterparties’ products, as well as negatively affect our ability to successfully execute our monetization strategy.
Changes in third-party coverage and reimbursement may affect sales of our products, product sales from which we receive royalty revenues and the products our borrowers sell to generate revenues and the growth of managed care organizations (“MCOs”), is expected to increase pricing pressures on our products in the United States.
Sales of our products, product sales from which we receive royalties and the products our borrowers sell to generate revenues will depend significantly on the extent to which reimbursement for the cost of such products and related treatments will be available to physicians and patients from various levels of United States and international government health authorities, private health insurers and other organizations. Third-party payers and government health administration authorities increasingly attempt to limit and/or regulate the coverage and reimbursement of medical products and services, including branded prescription drugs. Changes
37
in government legislation or regulation, such as the ACA, and changes in formulary or compendia listing or changes in private third-party payers’ policies toward reimbursement for such products may reduce reimbursement of the cost of such products to physicians, pharmacies and distributors. Decreases in third-party reimbursement could reduce usage of such products and sales to collaborators, which may have a material adverse effect on our revenues derived from our products, those from which we receive royalties from the business of our borrowers. In addition, macroeconomic factors may affect the ability of patients to pay or co-pay for costs or otherwise pay for our products or the products from which we, our royalty counterparties and borrowers generate revenues by, for example, decreasing the number of patients covered by insurance policies or increasing costs associated with such policies. All of these factors could negatively impact our ability to sell or otherwise dispose of our assets or reduce the value we realize as we seek to execute our monetization strategy.
In the United States in particular, the influence of MCOs has increased in recent years due to the growing number of patients receiving coverage through MCOs. The growth of MCOs has increased pressure on drug prices as well as revenues for pharmaceutical companies. One objective of MCOs is to contain and, where possible, reduce healthcare expenditures. MCOs typically use formularies as a means to negotiate prices with pharmaceutical providers; physician protocols requiring prior authorization for a branded product if a generic product is available or requiring the patient to first fail on one or more generic products before permitting access to a branded medicine; volume purchasing; and long-term contracts. In addition, by placing branded medicines on higher-tier status in their formularies or non-preferred tier status, MCOs transfer a portion of the cost of those medicines to the patient (through and increase in co-payment requirements), resulting in significant out-of-pocket expenses for the patient. This financial disincentive is a means by which MCOs manage drug costs and influence patients to use medicines preferred by the MCOs.
Exclusion of a product from a formulary or other MCO-implemented restrictions can significantly impact drug usage in the MCO patient population. Consequently, pharmaceutical companies compete to gain access to formularies for their products. Unique product features, such as greater efficacy, better patient ease of use, or fewer side effects, are generally beneficial to achieving access to formularies. Larger pharmaceutical companies have the ability to bundle available products and discounts in an effort to place and maintain products on formulary. We will be responsible for meeting the requirements of MCO’s in the United States and ensuring the competitive use of our products in a highly uncertain and changing environment. There can be no assurance that we will be able to maintain the level of use of our products, and their inability to do so could have a material adverse impact on the value we receive from any sale of our products or related businesses as part of our asset monetization strategy.
Generic products may increase pricing pressures on our products.
Although we believe that our products benefit from issued and/or pending patents as well as proprietary manufacturing technology, one competitive challenge that our branded pharmaceuticals products face is or will be from generic pharmaceutical manufacturers. Upon the expiration or loss of patent protection for a product, especially a small molecule product, the major portion of revenues for that product may be dramatically reduced in a very short period of time. Several such competitors make a regular practice of challenging product patents before their expiration. Also, manufacturers of generic pharmaceutical products may file or have already filed an abbreviated NDA (“ANDA”) with the FDA seeking to market generic forms of our products prior to the expiration of relevant patents owned by Noden. In June 2018, Noden Pharma DAC entered into a settlement agreement with Anchen pursuant to which Anchen, the sole ANDA filer for Tekturna of which the Company is aware, was granted a license from Noden to commercialize its generic form of aliskiren starting March 1, 2019. Under their license, Anchen may commercialize their formulation of aliskiren, but is not permitted to commercialize a generic version of aliskiren which closely relates to the formulation of Tekturna. As a result of a settlement with Anchen, Par Pharmaceuticals has commercialized a generic form of aliskiren. Further patent litigation and other challenges to Noden’s patents would be costly and unpredictable, would require extensive management time and resources, and may ultimately deprive us of market exclusivity for our products in a given geographical territory. The FDA ANDA approval process exempts generics from costly and time-consuming clinical trials to demonstrate their safety and efficacy, as long as they can scientifically demonstrate that their product performs in the same manner as the innovator drug, thus allowing generic manufacturers to rely on the safety and efficacy data of the innovator’s product. Generic competitors do not generally need to conduct clinical trials and can market a competing version of a product after the expiration or loss of patent or regulatory exclusivity and often charge significantly lower prices. In addition, as noted above, MCOs that focus primarily on the immediate cost of medicines often favor generics over branded drugs. Many governments also encourage the use of generics as alternatives to brand-name drugs in their healthcare programs. Additionally, certain foreign governments have indicated that compulsory licenses to patents may be granted in the case of national emergencies or in other circumstances, which could diminish or eliminate sales and profits from those regions, negatively affect our results of operations and cash flows, lead to an impairment charge of our long-lived assets or result in a material decline of our revenue. In March 2019, under an agreement with Prasco Laboratories, we launched in the United States an authorized generic form of Tekturna which competes with the Par Pharmaceuticals generic aliskiren product. Pricing pressure could reduce the value of the product, and consequently of Noden, as we seek to sell it as part of our monetization strategy.
38
Our ability to generate revenue from an authorized generic may be limited in the future.
In March 2019, we launched in the United States aliskiren hemifumarate tablets, 150 mg and 300 mg, an authorized generic of Tekturna, through Prasco Laboratories under an agreement with Noden Pharma DAC. Our ability to generate revenue related to our authorized generic may be limited for a number of reasons, including, without limitation, the entry into the market of additional generic products, the ratings for any existing or potential additional generic products, and the effect of increased generic competition on pricing. All of these factors could materially and adversely affect Noden’s business and the value we are able to realize as we seek to sell Noden as part of our asset monetization strategy.
Our products may develop undesirable side effects or have other properties impacting safety or efficacy.
Undesirable side effects caused by our products or similar products sold or developed by other companies, could reveal a high and unacceptable severity and prevalence of side effects or adverse events resulting in a number of potentially significant negative consequences, including:
• | regulatory authorities may withdraw approvals of such product; |
• | regulatory authorities may require additional warnings on the label; |
• | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
• | we could be sued and held liable for harm caused to patients; and |
• | our reputation may suffer. |
Any of these events could result in a material decline of our revenue negatively affecting the value of our assets and the proceeds we realize from a sale of such assets as part of our monetization strategy.
We may have significant product liability exposure and our insurance may not cover all potential claims.
We are exposed to product liability and other claims in the event that our technologies or products are alleged to have caused harm. Any potential product liability claims could exceed the amount of our insurance coverage or may be excluded from coverage under the terms of our policies. Our insurance may not be renewed at a cost and level of coverage comparable to that then in effect. Any of these events could materially and adversely affect our business, results of operations and financial condition, as well as the ability to successfully execute our monetization strategy.
Our third-party contractors as well as our own employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could result in significant liability for us and harm our reputation.
We are exposed to the risk of fraud or other misconduct in connection with international business operations and our reliance on third-party contractors to manage and conduct those activities with respect to our products. These risks include potential failures to:
• | comply with FDA regulations or similar regulations of comparable foreign regulatory authorities; |
• | provide accurate information to the FDA or comparable foreign regulatory authorities; |
• | comply with manufacturing standards applicable to our products; |
• | comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities; |
• | comply with the FCPA, the UK Bribery Act, and other anti-bribery laws; |
• | report financial information or data and our business affairs accurately; or |
• | disclose unauthorized activities to us. |
Our investment in Noden, an Irish entity, subjects us to both United States and international tax laws with respect to the structure and operations of our business and the business conducted by Noden, which are subject to continued scrutiny and change by governments and may result in additional liabilities that may affect our results of operations or reduce the proceeds we are able to realize as we seek to sell Noden or its assets as part of our monetization strategy.
Noden is incorporated in Ireland and maintains the performance of certain functions and ownership of certain assets in a more tax-efficient jurisdiction than the United States. Taxing authorities, such as the United States Internal Revenue Service (“IRS”),
39
actively audit and otherwise challenge these types of arrangements, and have regularly done so in the pharmaceutical industry. We remain subject to reviews and audits by the IRS and other taxing authorities from time to time, and the IRS or other taxing authority may challenge our structure and intra-company arrangements through an audit or lawsuit. Responding to or defending against those and other challenges from taxing authorities could be expensive and, in any event, would consume time and other resources, and divert management’s time and focus, as well as restrict or delay our ability to sell Noden. We generally cannot predict whether taxing authorities will conduct an audit or file a lawsuit challenging our current structure, the cost involved in responding to any inquiry or audit or lawsuit, or the outcome. If we are unsuccessful, we may be required to consolidate income and pay greater taxes as well as interest, fines or penalties, and may be obligated to pay increased taxes in the future, any of which could have a material adverse effect on our results of operations or the proceeds we are able to realize from a sale of Noden.
The regulatory clearance and approval processes of the FDA are lengthy, time-consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory clearance or approval for any new product candidates or modifications to existing products, our business or monetization strategy could be harmed.
The time required to obtain approval or clearance of a drug or device, respectively, by the FDA is unpredictable but typically takes many years following the commencement of clinical trials, if required, and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval or clearance policies, regulations, or the type and amount of clinical data necessary to gain marketing authorization may change during the course of a product candidate’s development and may vary among jurisdictions. We are not permitted to market any new product candidates in the United States until we receive regulatory approval of an NDA for any new drug product candidate or clearance of a 510(k) premarket notification (or approval of a PMA application) for any new medical device from the FDA, unless the device is exempt from such requirements.
Prior to obtaining approval to commercialize a drug product candidate in the United States or abroad, we or our collaborators must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA, that such product candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if we believe the preclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. The FDA may also require us to conduct additional preclinical studies or clinical trials for our product candidates either prior to or post-approval, or it may object to elements of our clinical development program.
In the United States, before we can market a new medical device, or a new use of, new claim for or significant modification to an existing product, we must first receive either clearance under Section 510(k) of the FFDCA or approval of a PMA application from the FDA, unless an exemption applies. In the 510(k) clearance process, before a device may be marketed, the FDA must determine that a proposed device is “substantially equivalent” to a legally-marketed “predicate” device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (pre-amendments device), a device that was originally on the U.S. market pursuant to an approved PMA and later down-classified, or a 510(k)-exempt device. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence. In the PMA process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices.
Modifications to products that are approved through a PMA application generally require FDA approval. Similarly, certain modifications made to products cleared through a 510(k) may require a new 510(k) clearance. Both the PMA approval and the 510(k) clearance process can be expensive, lengthy and uncertain. The FDA’s 510(k) clearance process usually takes from three to 12 months, but can last longer. The process of obtaining a PMA is much costlier and more uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is filed with the FDA. In addition, a PMA generally requires the performance of one or more clinical trials. Despite the time, effort and cost, we cannot assure you that any particular device will be approved or cleared by the FDA. Any delay or failure to obtain necessary regulatory approvals could harm our business or monetization strategy.
In the United States, we have obtained 510(k) premarket clearance from the FDA to market the LENSAR device. An element of our strategy is to continue to add new features and seek new indications. We expect that any such modifications may require new 510(k) clearance; however, future modifications may be subject to the substantially more costly, time-consuming and uncertain PMA process. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to
40
existing products than we had expected, product introductions or modifications could be delayed or canceled, which could cause our sales to decline.
The FDA can delay, limit or deny clearance or approval of our product candidates or require us to conduct additional preclinical or clinical testing or abandon a program for many reasons, including:
• | the FDA’s disagreement with the design or implementation of our clinical trials; |
• | negative or ambiguous results from our clinical trials; |
• | results that may not meet the level of statistical significance required by the FDA for approval or clearance; |
• | serious and unexpected drug-related adverse events experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates; |
• | our inability to demonstrate to the satisfaction of the FDA that our product candidates are safe and effective for the proposed indication or, in the case of our medical devices, are substantially equivalent to our proposed predicate device; |
• | the FDA’s disagreement with the interpretation of data from preclinical studies or clinical trials; |
• | our inability to demonstrate that the clinical and other benefits of our product candidates outweigh any safety or other perceived risks; |
• | the FDA’s requirement for additional preclinical studies or clinical trials; |
• | the FDA’s disagreement regarding the formulation, labeling or the specifications of our product candidates; |
• | the FDA’s agency’s failure to approve the manufacturing processes or facilities of third-party manufacturers with which we contract; or |
• | the potential for approval policies or regulations of the FDA to significantly change in a manner rendering our clinical data insufficient for approval. |
Of the large number of products in development, only a small percentage successfully complete the FDA marketing authorization process and become commercialized. The lengthy process as well as the unpredictability of outcomes from future clinical trials may result in our failing to obtain regulatory authorization to market our product candidates.
Even if we eventually complete clinical testing and receive approval of an NDA, 510(k), or similar foreign marketing application for our product candidates, the FDA may grant approval contingent on the performance of costly additional clinical trials, including Phase 4 clinical trials, or in the case of our drugs, the implementation of a Risk Evaluation and Mitigation Strategy (“REMS”), which may be required to ensure safe use of the drug after approval. The FDA also may authorize a product candidate for a more limited indication or patient population than we originally requested, and the FDA may not authorize us to market the product with the labeling that we believe is necessary or desirable for the successful commercialization of a product candidate. Any delay in obtaining, or inability to obtain, applicable regulatory authorization would delay or prevent commercialization of that product candidate. In addition, our drugs may become subject to class-wide REMS that implicate all manufactures of a particular class of drugs, which could significantly impact our ability to commercialize our drugs and could reduce their market potential.
The safety and efficacy of our medical device products is not yet supported by long-term clinical data, which could limit sales, and our products might therefore prove to be less safe or effective than initially thought.
Our medical device products have received premarket clearance under Section 510(k) of the FFDCA. In the 510(k) clearance process, before a device may be marketed the FDA must determine that a proposed device is “substantially equivalent” to a legally-marketed “predicate” device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (pre-amendments device), a device that was originally on the U.S. market pursuant to an approved PMA application and later down-classified, or a 510(k)-exempt device. This process is typically shorter and generally requires the submission of less supporting documentation than the FDA’s PMA application process and does not always require long-term clinical studies.
In the European Economic Area (“EEA”) manufacturers of medical devices are required by the Medical Devices Directive to collect post-marketing clinical data in relation to their CE marked medical devices. Post-market surveillance includes the conduct of post-market clinical follow-up studies permitting manufacturers to gather information concerning quality, safety or performance of medical devices after they have been placed on the market in the EEA. All information collected as part of the post-market surveillance process must be reviewed, investigated and analyzed on a regular basis in order to determine whether trending conclusions can be made concerning the safety or performance of the medical device and decisions must be taken in relation to the continued marketing of medical devices currently on the market. If development of these products is continued by
41
us, we would expect to incur ongoing costs to comply with these post-market clinical obligations in EEA markets for so long as we continue to market and sell products in those markets.
Given the foregoing regulatory environment in which we operate, we lack the breadth of published long-term clinical data supporting the safety and efficacy of our medical devices and the benefits they offer that might have been generated in connection with other approval processes. For these reasons, the market may be slow to adopt our products, we may not have comparative data that our competitors have or are generating, and we may be subject to greater regulatory and product liability risks.
In addition, while our LENSAR® Laser Systems were first cleared in 2010 in the United States and in 2013 in EEA, we have limited complication or patient success rate data with respect to uses of our products. In addition, if future studies and experience indicate that our products cause unexpected or serious complications or other unforeseen negative effects, we could be subject to mandatory product recalls or suspension or withdrawal of clearance in the United States or the EEA, and our reputation with physicians, patients and healthcare providers may suffer.
The misuse or off-label use of our products may harm our reputation in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.
Our products have been approved or cleared by the FDA for specific indications. Our marketing does not promote our products for uses outside of these cleared or approved indications for use, known as “off-label uses.” We cannot, however, prevent a physician from using or prescribing our products off-label, when in the physician’s independent professional medical judgment he or she deems it appropriate. There may be increased risk of injury to patients if physicians prescribe or use our products off-label. Furthermore, the use of our products for indications other than those cleared or approved by the FDA or any foreign regulatory body may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients.
If the FDA or any foreign regulatory body determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance or imposition of a warning letter or an untitled letter, which is used for violators that do not necessitate a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action under other regulatory authority, such as false claims laws, if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs and the curtailment of our operations.
Moreover, if our products do not comply with regulatory requirements, including with respect to labeling and promotion, or are misused or used with improper technique, we may become subject to costly litigation. Product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance. In addition, any of the events described above could harm our business and/or negatively affect our ability to monetize such assets or the value we may receive from such a monetization.
Even though we have received regulatory approval for our drug product candidates and clearance of a premarket notification for our devices, we are subject to ongoing regulatory obligations and continued regulatory review, which results in significant additional expense, and we may be subject to penalties, if we fail to comply with regulatory requirements or experience unanticipated problems with our product candidates.
Any regulatory approvals or clearances that we receive may be subject to limitations on the indicated uses for which the product may be marketed or the conditions of approval, or contain requirements for potentially costly post-market testing and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a REMS as a condition of approval of our product candidates, which could include requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
In addition, once the FDA or a comparable foreign regulatory authority authorizes a product for marketing, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping are subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs and GCP requirements for any clinical trials that we conduct post-approval. For example, we are subject to the medical device reporting requirements for our medical device products, which require us to report to the FDA when we receive or become aware of
42
information that reasonably suggests that one or more of our medical devices may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of the product. If we fail to comply with our reporting obligations, the FDA could take enforcement action against us. We are subject to similar post-market reporting requirements with respect to our drug products.
Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• | restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
• | fines, warning letters or holds on clinical trials; |
• | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of approvals; |
• | product seizure or detention, or refusal to permit the import or export of our product candidates; and |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could impact our business. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may fail to obtain any marketing approvals, lose any marketing approval that we have obtained and we may not achieve or sustain profitability.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. For example, certain policies of the Trump administration may impact our business and industry. Namely, the Trump administration has taken several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. If these executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
We are subject to extensive healthcare laws, regulation and enforcement, and our failure to comply with those laws could have a material adverse effect on our results of operations and financial condition.
We are subject to potential risk of civil and criminal enforcement by the federal government and the states and foreign governments due to the acquisitions of Noden and LENSAR. Our business practices and relationships with providers are subject to scrutiny under these laws. We are also be subject to privacy and security regulation related to patient, customer, employee and other third-party information by both the federal government and the states and foreign jurisdictions in which we conduct our business. The laws, regulations and codes that may affect us in the United States include:
• | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. The U.S. government has interpreted this law broadly to apply to the marketing and sales activities of manufacturers. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Violations of the federal Anti-Kickback Statute may result in civil monetary penalties plus up to three times the remuneration involved. Civil penalties for such conduct can further be assessed under the federal False Claims Act. Violations can also result in criminal penalties and imprisonment of up to 10 years. Similarly, violations can result in exclusion from participation in government healthcare programs, including Medicare and Medicaid; |
43
• | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent. Private individuals can bring False Claims Act ‘‘qui tam’’ actions, on behalf of the government and such individuals, commonly known as ‘‘whistleblowers,’’ may share in amounts paid by the entity to the government in fines or settlement. When an entity is determined to have violated the federal civil False Claims Act, the government may impose civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs; |
• | the federal Civil Monetary Penalties Law, which prohibits, among other things, offering or transferring remuneration to a federal healthcare beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier; |
• | The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
• | HIPAA, as amended by the Health Information for Economic and Clinical Health Act of 2009 (“HITECH”), and its implementing regulations, which imposes certain requirements on certain covered healthcare providers, health plans and healthcare clearinghouses as well as their business associates that perform services for them that involve individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information, without appropriate authorization, including mandatory contractual terms as well as directly applicable privacy and security standards and requirements. Failure to comply with the HIPAA privacy and security standards can result in civil monetary penalties for each violation and, in certain circumstances, criminal penalties with fines for each violation and/or imprisonment. State attorneys general can also bring a civil action to enjoin a HIPAA violation or to obtain statutory damages on behalf of residents of his or her state; |
• | the federal physician sunshine requirements under the ACA, which require manufacturers of drugs, devices, biologics, and medical supplies to report annually to the Centers for Medicare and Medicaid Services (“CMS”), information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members. Applicable manufacturers are required to submit annual reports to CMS. Failure to submit required information may result in civil monetary penalties for all payments, transfers of value or ownership or investment interests that are not timely, accurately, and completely reported in an annual submission, and may result in liability under other federal laws or regulations; |
• | guidelines promulgated by the Office of Inspector General of the U.S. Department of Health and Human Services related to pharmaceutical and medical device company regulatory compliance programs and the PhRMA Code on Interactions with Healthcare Professionals and the AdvaMed Code of Ethics, as amended; |
• | foreign and state law equivalents of each of the above federal laws, such as the FCPA, anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; |
• | state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; |
• | state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and |
• | state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts. |
These laws and regulations, among other things, constrain our business, marketing and other promotional activities by limiting the kinds of financial arrangements, including sales programs, we may have with hospitals, physicians or other potential purchasers of our products. Due to the breadth of these laws, the narrowness of statutory exceptions and regulatory safe harbors available, and the range of interpretations to which they are subject, it is possible that some of our current or future practices might be challenged under one or more of these laws.
To enforce compliance with the healthcare regulatory laws, certain enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Additionally, as a result of these investigations, healthcare providers and entities may have to agree to additional compliance and reporting requirements as part of a consent decree or corporate integrity
44
agreement. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity and be costly to respond to.
We do not have experience in establishing the compliance programs necessary to comply with this complex and evolving regulatory environment and our reliance on Noden and LENSAR to operate and address these requirements appropriately increases the risks that we may be found to violate the applicable laws and regulations. If we are found to be in violation of any of such laws or any other governmental regulations, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, the exclusion from participation in federal and state healthcare programs, imprisonment, contractual damages, reputational harm, disgorgement and the curtailment or restructuring of our operations, any of which could materially and adversely affect interests in our products, including having a material adverse effect on our financial results or our ability to monetize such products or related businesses as part of our monetization strategy.
An impairment charge with respect to intangible assets could have a material impact on our results of operations and/or negatively affect our monetization strategy.
We periodically evaluate our intangible assets to determine whether all or a portion of their carrying values may be impaired, in which case a charge to earnings may be necessary. The occurrence of certain events, changes in business strategy, government regulations or economic or market conditions may cause us to remeasure the fair value of certain assets and liabilities. Our judgments regarding the existence of impairment indicators are based on, among other things, legal factors, market conditions, and operational performance. If an event or events occur that would cause us to revise our estimates and assumptions used in analyzing the value of our intangible assets, such revision could result in an impairment charge that could have a material impact on our results of operations in the period in which the impairment occurs. For example, at December 31, 2019, we recorded an impairment charge of $22.5 million for the Noden intangible assets related to our monetization strategy and updated forecasts for Noden. As a result of this impairment charge, which was based on the estimated fair value of the assets, the remaining carrying value of these intangible assets were determined to be $10.1 million. For additional information on the impairment charge, see Note 10, Intangible Assets.
We may use a certain amount of cash from time to time in order to repurchase or satisfy obligations relating to our convertible notes. The maturity or conversion of any of our convertible notes could materially and adversely affect our business, results of operations and financial condition.
We are required to repay the full principal amount of approximately $19.2 million in principal amount outstanding under the 2.75% Convertible Senior Notes due December 1, 2021 (the “December 2021 Notes”) and approximately $11.5 million in principal amount outstanding plus an additional accreted amount of $1.5 million under the 2.75% Convertible Senior Notes due December 1, 2024 (the “December 2024 Notes”) if not previously converted or repurchased.
Our ability to make scheduled payments of the principal of, to pay interest on, to pay any cash due upon conversion of, or to refinance, our indebtedness, depends on our future performance, which is subject to economic, financial, competitive and other
factors beyond our control. Our business may not generate cash flow from operations in the future sufficient to service our debt and make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
Holders of the December 2021 Notes may convert their notes at their option under the following conditions at any time prior to the close of business on the business day immediately preceding June 1, 2021: (i) during any fiscal quarter (and only during such fiscal quarter) commencing after the fiscal quarter ending June 30, 2017, if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive), in the period of 30 consecutive trading days, ending on, and including, the last trading day of the immediately preceding fiscal quarter, exceeds 130% of the conversion price for the notes on each applicable trading day; (ii) during the five business day period immediately after any five consecutive trading-day period (the measurement period), in which the trading price per $1,000 principal amount of the December 2021 Notes for each trading day of that measurement period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate for the notes for each such trading day; or (iii) upon the occurrence of specified corporate events. Holders of the December 2024 Notes may convert their notes at their option under the following conditions at any time prior to the close of business on the business day immediately preceding June 1, 2024: (i) during any fiscal quarter (and only during such fiscal quarter) if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive), in the period of 30 consecutive trading days, ending on, and including, the last trading day of the immediately preceding fiscal quarter, exceeds 130% of the
45
conversion price for the notes on each applicable trading day; (ii) during the five business day period immediately after any five consecutive trading-day period (the measurement period), in which the trading price per $1,000 principal amount of the December 2024 Notes for each trading day of that measurement period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate for the notes for each such trading day; or (iii) upon the occurrence of specified corporate events.
The December 2021 Notes and the December 2024 Notes may be settled by paying or delivering, as applicable, cash, shares of our common stock or a combination of cash and shares of our common stock, at our election. During the year ended December 31, 2019 we repurchased approximately $44.8 million in principal amount of our December 2021 Notes and approximately $74.6 million in principal amount of our December 2024 Notes. In furtherance of our monetization strategy, we expect to use a certain amount of cash from time to time in order to repurchase or satisfy obligations relating to our convertible notes which could adversely affect the amount or timing of any distribution to our stockholders.
The conversion or any future exchanges of any of the December 2021 Notes or the December 2024 Notes into shares of our common stock would have a dilutive effect that could cause our stock price to go down.
Until June 1, 2021, the December 2021 Notes are convertible into shares of our common stock only if specified conditions are met and thereafter convertible at any time, at the option of the holder. Until June 1, 2024, the December 2024 Notes are convertible into shares of our common stock only if specified conditions are met and thereafter convertible at any time, at the option of the holder. We have reserved shares of our authorized common stock for issuance upon conversion of the December 2021 Notes and the December 2024 Notes. Upon conversion, the principal amount of our convertible notes may be settled in, at our option, cash, common stock or a combination of cash and common stock. If any or all of these convertible notes are converted into shares of our common stock, our existing stockholders will experience immediate dilution of voting rights and our common stock price may decline. Furthermore, the perception that such dilution could occur may cause the market price of our common stock to decline. If our stock price is negatively impacted, any value we may receive upon the potential sale of our company or other potential transaction in furtherance of our monetization strategy could be materially and adversely affected.
We entered into capped call transactions in connection with the issuance of our December 2021 Notes and our December 2024 Notes that may affect the value of our common stock and any desired dilution mitigation will be limited to the extent that our stock price rises above the cap price of the applicable capped call transactions.
In connection with the issuance of our December 2021 Notes and our December 2024 Notes, we entered into capped call transactions, with hedge counterparties, which we expect to reduce the potential dilution upon conversion of the December 2021 Notes and the December 2024 Notes in the event that the market price per share of our common stock, as measured under the terms of the applicable capped call transaction, at the time of exercise is greater than the strike price of the applicable capped call transaction, which corresponds to the initial conversion price of the applicable notes and is subject to certain adjustments similar to those contained in the applicable notes. If, however, the market price per share of our common stock, as measured under the terms of the applicable capped call transaction, exceeds the cap price of the applicable capped call transaction, there would nevertheless be dilution to the extent that such market price exceeds the cap price of the applicable capped call transaction.
In connection with hedging the capped call transactions, the hedge counterparties or their affiliates:
• | expect to purchase our common stock in the open market and/or enter into various derivatives and/or enter into various derivative transactions with respect to our common stock; and |
• | may enter into or unwind various derivatives and/or purchase or sell our common stock in secondary market transactions. |
These activities could have the effect of increasing or preventing a decline in the price of our common stock concurrently with or following the pricing of the applicable notes and could have the effect of decreasing the price of our common stock during the period immediately prior to a conversion of the applicable notes.
The hedge counterparties or their affiliates are likely to modify their hedge positions in relation to the capped call transactions from time to time prior to conversion or maturity of the applicable notes by purchasing and selling our common stock, other of our securities, or other instruments they may wish to use in connection with such hedging.
In addition, we intend to exercise options we hold under the capped call transaction whenever the notes are converted. In order to unwind its hedge positions with respect to those exercised options, the counterparties or affiliates thereof expect to sell our common stock in secondary market transactions or unwind various derivative transactions with respect to our common stock during the period immediately prior to conversion of the applicable notes. We have also agreed to indemnify the hedge
46
counterparties and affiliates thereof for losses incurred in connection with a potential unwinding of their hedge positions under certain circumstances.
The effect, if any, of any of these transactions and activities on the market price of our common stock will depend in part on market conditions and cannot be ascertained at this time, but any of these activities could adversely affect the value of our common stock. For further information regarding the mechanics of our capped call transactions, refer to our discussion in Note 13, Convertible Senior Notes, to the Consolidated Financial Statements.
We have implemented a corporate structure taking into consideration our limited operations and potentially applicable tax impact on our royalty and other income, and any changes in applicable tax laws and regulations or enforcement positions of tax authorities may negatively impact our financial condition and operating results.
We have established our corporate structure to be closely aligned with the financial nature of our business. There can be no assurance that the applicable tax laws and regulations will continue in effect or that the taxing authorities in any or all of the applicable jurisdictions will not challenge one or more aspects or characterizations of our corporate structure and the treatment of transactions or agreements within our corporate structure, or determine that the manner in which we operate our business is not consistent with our corporate structure. For example, recently enacted U.S. tax legislation may result in an increased tax liability as a result of our current corporate structure. We may also have disputes with one or more state tax authorities regarding whether we are subject to that state’s tax and, if we are subject to such state’s tax, what proportion of our revenues is subject to taxation in such state. For example, we are currently subject to an audit by the California Franchise Tax Board and, while we may disagree with their conclusions regarding such issues, the proceedings may extend over a long period of time and we may ultimately be required to pay taxes either in a settlement or upon a final decision of an agency or court. In addition, an inability to resolve our audit with the Franchise Tax Board in a satisfactory manner without extensive legal proceedings may result in an extended period of operation prior to wind-down or dissolution. Any unfavorable changes in laws and regulations or positions by tax authorities could harm our financial position, results of operations and cash flows.
We may have exposure to additional tax liabilities.
We are subject to taxes in the United States and other jurisdictions. Tax rates in these jurisdictions may be subject to significant change due to economic and/or political conditions. A number of other factors may also impact our future effective tax rate including:
• | the jurisdictions in which profits are determined to be earned and taxed; |
• | the resolution of issues arising from tax audits with various tax authorities; |
• | changes in valuation of our deferred tax assets and liabilities; |
• | increases in expenses not deductible for tax purposes, including write-offs of acquired intangibles and impairment of goodwill in connection with acquisitions; |
• | changes in availability of tax credits, tax holidays, and tax deductions; |
• | changes in share-based compensation; and |
• | changes in tax laws or the interpretation of such tax laws and changes in generally accepted accounting principles. |
On December 22, 2017, the U.S. federal government enacted the 2017 Tax Act. The 2017 Tax Act significantly changed the existing U.S. corporate income tax laws by, among other things, lowering the corporate tax rate (from a top rate of 35% to a flat rate of 21%), implementing elements of a territorial tax system, and imposing a one-time deemed repatriation transition tax on cumulative undistributed foreign earnings, for which we have not previously paid U.S. taxes. The Company recognized in its Consolidated Financial Statements for the year ended December 31, 2017 estimated tax impacts related to the revaluation of deferred tax assets and liabilities. The ultimate impact did not differ materially from these provisional amounts after additional analysis, changes in interpretations and assumptions the Company made and additional regulatory guidance that was issued. The accounting was completed when the Company’s 2017 U.S. corporate income tax return was filed in 2018. We have made a policy election with respect to our treatment of potential GILTI to account for taxes on GILTI as a current-period expense as incurred.
In addition, certain activities conducted by our foreign subsidiaries may give rise to United States corporate income tax, even if there are no distributions to the United States. These taxes would be imposed on us when our subsidiaries that are controlled foreign corporations generate income that is subject to Subpart F of the U.S. Internal Revenue Code (“Subpart F”) or Global intangible low-taxed income (“GILTI”). Passive income, such as rents, royalties, interest and dividends, is among the types of income subject to taxation under Subpart F. Any income taxable under Subpart F or GILTI is taxable in the United States at federal corporate income tax rates of 21%. Subpart F income that is taxable to us, even if it is not distributed to us, may also include income from intercompany transactions between our U.S. and non-U.S. subsidiaries, or where our non-U.S. subsidiaries
47
make an “investment in U.S. property,” within the meaning of Subpart F, such as holding the stock in, or making a loan to, a U.S. corporation.
While we may mitigate this increase in our effective tax rate through claiming a foreign tax credit against our U.S. federal income taxes or potentially have foreign or U.S. taxes reduced under applicable income tax treaties, we are subject to various limitations on claiming foreign tax credits and we may lack treaty protections in certain jurisdictions that will potentially limit any reduction of the increased effective tax rate. A higher effective tax rate may also result to the extent that losses are incurred in non-U.S. subsidiaries that do not reduce our U.S. taxable income.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited.
At December 31, 2019, we had federal, state and foreign net operating loss carryforwards of $108.6 million, $63.9 million and $125.6 million, respectively, and federal and state tax credit carryforwards of $2.2 million and $19.3 million, respectively. There may be limitations on our ability to use our net operating loss carryforwards or other tax assets. For example, under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income may be limited. We or our subsidiaries may have experienced, or may in the future experience, “ownership changes” as a result of shifts in stock ownership. Tax attributes acquired from LENSAR may be subject to separate return limitations that may limit the corporation’s ability to use the acquired net operating losses and credits. Any limitations on our ability to use our net operating loss carryforwards and other tax assets could materially and adversely impact our financial condition and results of operations. Furthermore, under the 2017 Tax Act, although the treatment of tax losses generated in taxable years ending before December 31, 2017 has generally not changed, tax losses generated in taxable years beginning after December 31, 2017 may only be utilized to offset 80% of taxable income annually. This change may require us to pay additional federal income taxes in future years.
We depend on our licensees and royalty-agreement counterparties for the determination of royalty payments. While we have rights to audit our licensees and royalty-agreement counterparties, the independent auditors may have difficulty determining the correct royalty calculation, we may not be able to detect errors and payment calculations may call for retroactive adjustments. We may have to exercise legal remedies to resolve any disputes resulting from the audit or otherwise related to non-performance by a licensee or royalty counterparty.
The royalty payments we receive are determined by our licensees based on their reported sales. Each licensee’s calculation of the royalty payments is subject to and dependent upon the adequacy and accuracy of its sales and accounting functions, and errors may occur from time to time in the calculations made by a licensee. Our license and royalty agreements provide us the right to audit the calculations and sales data for the associated royalty payments; however, our right to conduct such audits may be limited in terms of the covered periods, and such audits may occur many months following our recognition of the royalty revenue, may require us to adjust our royalty revenues in later periods and may require incurring additional expenses on our part. Further, our licensees and royalty-agreement counterparties may be uncooperative or have insufficient records, which may complicate and delay the audit process.
Although we regularly exercise our royalty audit rights, and reference publicly available information in the assessment of the paid royalties, we rely in the first instance on our licensees and royalty-agreement counterparties to accurately report sales and calculate and pay applicable royalties and, upon exercise of such royalty audit rights, we rely on licensees’ and royalty-agreement counterparties’ cooperation in performing such audits. In the absence of such cooperation, we may be forced to exercise legal remedies to enforce our agreements.
We may experience increases and decreases in our revenues due to fluctuations in foreign currency exchange rates and we may be unsuccessful in our attempts to mitigate this risk.
Our operating results are subject to volatility due to fluctuations in foreign currency exchange rates. Our primary exposure to fluctuations in foreign currency exchange rates relates to revenue and operating expenses denominated in currencies other than the U.S. dollar. Fluctuations in foreign currency rates, particularly the Euro, relative to the U.S. dollar can significantly affect our revenues and operating results. While foreign currency conversion terms vary by license agreement, generally most agreements require that royalties first be calculated in the currency of sale and then converted into U.S. dollars using the average daily exchange rates for that currency for a specified period at the end of the calendar quarter. For example, when the U.S. dollar weakens in relation to other currencies, the converted amount is greater than it would have been had the U.S. dollar exchange rates remained unchanged. Our revenues may fluctuate due to changes in foreign currency exchange rates and is subject to foreign currency exchange risk.
48
To compensate for Euro currency fluctuations, we may hedge Euro currency exposures with Euro forward and option contracts, to offset the risks associated with these Euro currency exposures. We may suspend the use of these contracts from time to time or we may be unsuccessful in our attempt to hedge our Euro currency risk. We will continue to experience foreign currency related fluctuations in our royalty revenues in certain instances when we do not enter into foreign currency exchange contracts or where it is not possible or cost effective to hedge our foreign currency related exposures. Currency related fluctuations in our royalty revenues will vary based on the currency exchange rates associated with these exposures and changes in those rates, whether we have entered into foreign currency exchange contracts to offset these exposures and other factors. All of these factors could materially impact our results of operations, financial position and cash flows, the timing of which is variable and generally outside of our control.
Legislative or regulatory reforms in the United States or the EU may make it more difficult and costly for us to obtain regulatory clearances or approvals for our medical device products or to manufacture, market or distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulation of medical devices. In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions, which may prevent or delay approval or clearance of our future products under development or impact our ability to modify our currently cleared products on a timely basis. Over the last several years, the FDA has proposed reforms to its 510(k) clearance process, and such proposals could include increased requirements for clinical data and a longer review period, or could make it more difficult for manufacturers to utilize the 510(k) clearance process for their products. For example, in November 2018, FDA officials announced forthcoming steps that the FDA intends to take to modernize the premarket notification pathway under Section 510(k) of the FFDCA. Among other things, the FDA announced that it planned to develop proposals to drive manufacturers utilizing the 510(k) pathway toward the use of newer predicates. These proposals included plans to potentially sunset certain older devices that were used as predicates under the 510(k) clearance pathway, and to potentially publish a list of devices that have been cleared on the basis of demonstrated substantial equivalence to predicate devices that are more than 10 years old. In May 2019, the FDA solicited public feedback on these proposals. These proposals have not yet been finalized or adopted, and the FDA may work with Congress to implement such proposals through legislation. Accordingly, it is unclear the extent to which any proposals, if adopted, could impose additional regulatory requirements on us that could delay our ability to obtain new 510(k) clearances, increase the costs of compliance, or restrict our ability to maintain our current clearances, or otherwise create competition that may negatively affect our business or monetization strategy.
More recently, in September 2019, the FDA finalized guidance describing an optional “safety and performance based” premarket review pathway for manufacturers of “certain, well-understood device types” to demonstrate substantial equivalence under the 510(k) clearance pathway by showing that such device meets objective safety and performance criteria established by the FDA, thereby obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA intends to develop and maintain a list of device types appropriate for the “safety and performance based” pathway and will continue to develop product-specific guidance documents that identify the performance criteria for each such device type, as well as the testing methods recommended in the guidance documents, where feasible. The FDA may establish performance criteria for classes of devices for which we or our competitors seek or currently have received clearance, and it is unclear the extent to which such performance standards, if established, could impact our ability to obtain new 510(k) clearances or otherwise create competition that may negatively affect our business or monetization strategy.
In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new statutes, regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of any future products or make it more difficult to obtain clearance or approval for, manufacture, market or distribute our products. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require: additional testing prior to obtaining clearance or approval; changes to manufacturing methods; recall, replacement or discontinuance of our products; or additional record keeping.
On April 5, 2017, the European Parliament passed the Medical Devices Regulation (Regulation 2017/745), which repeals and replaces the EU Medical Devices Directive and the Active Implantable Medical Devices Directive. Unlike directives, which must be implemented into the national laws of the EEA member states, the regulations would be directly applicable, i.e., without the need for adoption of EEA member state laws implementing them, in all EEA member states and are intended to eliminate current differences in the regulation of medical devices among EEA member States. The Medical Devices Regulation, among other
49
things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices and ensure a high level of safety and health while supporting innovation.
The Medical Devices Regulation will, however, only become applicable three years after publication (in 2020). Once applicable, the new regulations will among other things:
• | strengthen the rules on placing devices on the market and reinforce surveillance once they are available; |
• | establish explicit provisions on manufacturers’ responsibilities for the follow‑up of the quality, performance and safety of devices placed on the market; |
• | improve the traceability of medical devices throughout the supply chain to the end‑user or patient through a unique identification number; |
• | set up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; |
• | strengthened rules for the assessment of certain high‑risk devices, such as implants, which may have to undergo an additional check by experts before they are placed on the market. |
These modifications may have an effect on the way we conduct our business in the EEA.
Our agreements with Facet may not reflect terms that would have resulted from arm’s-length negotiations between unaffiliated third parties.
The agreements associated with the spin-off of Facet in December 2008 (the “Spin-Off”), including the Separation and Distribution Agreement, Tax Sharing and Indemnification Agreement and Cross License Agreement, were negotiated in the context of the Spin-Off while Facet was still part of us and, accordingly, may not reflect more favorable terms that may have resulted from arm’s-length negotiations between unaffiliated third parties.
We may have obligations for which we may not be able to collect under our indemnification rights from Facet.
Under the terms of the Separation and Distribution agreement with Facet, we and Facet agreed to indemnify the other from and after the Spin-Off with respect to certain indebtedness, liabilities and obligations that were retained by our respective companies. These indemnification obligations could be significant. The ability to satisfy these indemnities, if called upon to do so, will depend upon our future financial strength. We cannot assure you that, if Facet has to indemnify us for any substantial obligations, Facet will have the ability to satisfy those obligations. If Facet does not have the ability to satisfy those obligations, we may be required to satisfy those obligations instead. For example, in connection with the Spin-Off, we entered into amendments to the leases for the facilities in Redwood City, California, which formerly served as our corporate headquarters, under which Facet was added as a co-tenant under the leases and a Co-Tenancy Agreement under which Facet agreed to indemnify us for all matters related to the leases attributable to the period after the Spin-Off date. Should Facet default under its lease obligations, we would be held liable by the landlord as a co-tenant and, thus, we have in substance guaranteed the payments under the lease agreements for the Redwood City facilities, the disposition of which could have a material adverse effect on the amount or timing of any distribution to our stockholders. As of December 31, 2019, the total lease payments for the duration of the guarantee, which runs through December 2021, are approximately $22.6 million. We would also be responsible for lease-related payments including utilities, property taxes and common area maintenance that may be as much as the actual lease payments. In April 2010, Abbott Laboratories acquired Facet and renamed the company Abbott Biotherapeutics Corp., and in January 2013, Abbott Biotherapeutics Corp. was renamed AbbVie Biotherapeutics, Inc. and spun off from Abbott as a subsidiary of AbbVie Inc. We do not know how Abbott’s acquisition of Facet will impact our ability to collect under our indemnification rights or whether Facet’s ability to satisfy its obligations will change. In addition, we have limited information rights under the Co-Tenancy Agreement. As a result, we are unable to determine definitively whether Facet continues to occupy the space and whether it has subleased the space to another party or the basis upon which our potential co-tenant obligation may be triggered. See “Item 2-Properties.”
As we continue to operate our business and as we implement our monetization strategy, our mix of assets and sources of income may require that we register with the SEC as an “investment company” in accordance with the Investment Company Act of 1940.
We are not registered and have no intention to register as an “investment company” under the Investment Company Act of 1940 (the “40 Act”). As a result, we are not and do not expect to become subject to regulation under the 40 Act, including its reporting and corporate governance requirements and restrictions on leverage and affiliate transactions.
50
Generally, to avoid being regulated as an “investment company” under the 40 Act an issuer must:
• | not be engaged or hold itself out as being engaged primarily in the business of investing, reinvesting or trading in securities and not own or propose to acquire “investment securities” with a value of more than 40% of the value of its total assets (exclusive of U.S. government securities and cash items) on an unconsolidated basis; or |
• | be able to rely on an exception from the definition of “investment company” under the ’40 Act or an exemptive rule. |
“Investment securities” are any securities other than U.S. government securities and securities issued by a majority-owned subsidiary that is not itself either an “investment company” or a private investment company, meaning a company that is excluded from the definition of “investment company” by Section 3(c)(1) or Section 3(c)(7) of the 40 Act.
We have in the past and may in the future rely on one or more exceptions to the definition of “investment company” under the 40 Act, including the exception under Section 3(c)(5) of the 40 Act. To rely on Section 3(c)(5), as interpreted by the staff of the SEC, we would be required to have at least 55% of our total assets in certain qualifying assets. In a no-action letter issued to Royalty Pharma on August 13, 2010, the SEC staff stated that certain royalty interests of the type we own can be treated as qualifying assets.
Ensuring that we do not fall within the definition of “investment company” under the 40 Act may limit our ability to make certain investments (including divesting certain assets), or require us to take or forego certain actions, that could materially and adversely affect our financial condition and results of operation. In addition, if the SEC, its staff or the courts changes their interpretation of certain provisions of the 40 Act, including Section 3(c)(5), we may need to take additional steps in order to avoid becoming subject to regulation under the 40 Act, which could materially and adversely affect our financial condition and results of operation.
If we were required to register as an “investment company,” the obligations imposed on us by the 40 Act would likely require substantial changes in the way we do business and would result in significant additional regulatory and administrative burdens and costs. In order to remain outside the scope of regulation under the 40 Act, we may need to take various actions which we might otherwise not pursue. These actions may include restructuring our company and modifying our mixture of assets and income, including divesting certain desirable assets immediately, and could have a material and adverse effect on us.
Failure in our information technology and storage systems could significantly disrupt the operation of our business.
Our ability to execute our business plan depends, in part, on the continued and uninterrupted performance of our information technology (“IT”) systems. IT systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers may be vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our IT systems, sustained or repeated system failures that interrupt our ability to generate and maintain data could adversely affect our ability to operate our business.
Changes to financial accounting standards may affect our reported results of operations.
A change in accounting standards or practices can have a significant effect on our reported results and may even affect our reporting of transactions completed before the change is effective. New accounting pronouncements and varying interpretations of accounting pronouncements have occurred and may occur in the future. Changes to existing standards or the reevaluation of current practices may adversely affect our reported financial results or the way we conduct our business.
We use estimates, make judgments, and apply certain methods in measuring the progress of our business in determining our financial results and in applying our accounting policies. As these estimates, judgments, and methods change, our assessment of the progress of our business and our results of operations could vary.
The methods, estimates, and judgments we use in applying our accounting policies have a significant impact on our results of operations. Such methods, estimates, and judgments are, by their nature, subject to substantial risks, uncertainties, and assumptions, and factors may arise over time may lead us to change our methods, estimates, and judgments. Changes in any of our assumptions may adversely affect our reported financial results.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
51
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. We are required, under Section 404 of the Sarbanes-Oxley Act (“Section 404”), to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment must include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting.
Our compliance with Section 404 requires that we incur substantial accounting expense and expend significant management efforts. Our acquired businesses may have limited experience complying with Section 404 and if in the future we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. Furthermore, we cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by Nasdaq, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Medical Devices Segment
LENSAR leases an office and manufacturing facility of approximately 33,900 square feet in Orlando, Florida, which serves as the office managing all medical device operations. The lease expires in July 2021.
Pharmaceutical Segment
Noden Pharma DAC leases approximately 3,100 square feet of office space in Dublin, Ireland, which serves as the office managing all pharmaceutical operations. The lease expires in September 2025. Noden Pharma DAC has the option to terminate the lease in September 2021.
Income Generating Assets Segment
We lease approximately 5,900 square feet of office space in Incline Village, Nevada, which serves as our corporate headquarters. The lease expires in May 2022.
In July 2006, we entered into two leases and a sublease for facilities in Redwood City, California, which formerly served as our corporate headquarters and cover approximately 450,000 square feet of office space. Under the amendments to the leases entered into in connection with the Spin-Off, Facet was added as a co-tenant under the leases. As a co-tenant, Facet is bound by all of the terms and conditions of the leases. We and Facet are jointly and severally liable for all obligations under the leases, including the payment of rental obligations. The guarantee runs through December 2021. We also entered into a Co-Tenancy Agreement with Facet in connection with the Spin-Off and the lease amendments under which we assigned to Facet all rights under the leases, including, but not limited to, the right to amend the leases, extend the lease terms or terminate the leases, and Facet assumed all of our obligations under the leases. Under the Co-Tenancy Agreement, we also relinquished any right or option to regain possession, use or occupancy of these facilities. Facet agreed to indemnify us for all matters associated with the leases attributable to the period after the Spin-Off date and we agreed to indemnify Facet for all matters associated with the leases attributable to the period before the Spin-Off date. In addition, in connection with the Spin-Off, we assigned the sublease to Facet. In April 2010, Abbott Laboratories acquired Facet and later renamed the entity AbbVie Biotherapeutics, Inc. (“AbbVie”). To date, AbbVie has satisfied all obligations under the Redwood City leases.
52
We believe that our existing facilities are adequate to meet our business requirements for the reasonably foreseeable future and that additional space will be available on commercially reasonable terms, if required.
ITEM 3. LEGAL PROCEEDINGS
The information set forth in Note 25, Legal Proceedings, to the Consolidated Financial Statements included in Item 8, “Financial Statements and Supplementary Data” of this Annual Report is incorporated by reference herein.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
53
PART II
ITEM 5. MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
As of February 28, 2020, we had approximately 120 common stockholders of record. Most of our outstanding shares of common stock are held of record by one stockholder, Cede & Co., as nominee for the Depository Trust Company. Many brokers, banks and other institutions hold shares of common stock as nominees for beneficial owners that deposit these shares of common stock in participant accounts at the Depository Trust Company. The actual number of beneficial owners of our stock is likely significantly greater than the number of stockholders of record; however, we are unable to reasonably estimate the total number of beneficial owners.
Equity Compensation Plan Information
See Part III, Item 12, “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” for information regarding securities authorized for issuance under equity compensation plans.
Recent Sales of Unregistered Securities
On September 12, 2019, we entered into separate, privately negotiated agreements with a limited number of holders of our 2.75% Convertible Senior Notes due 2021 (the “December 2021 Notes”) to exchange an aggregate of approximately $86.1 million principal amount of the 2021 Notes for (i) an aggregate of approximately $86.1 million original principal amount of new 2.75% Convertible Senior Notes due 2024 (the “December 2024 Notes” or the “Exchanged Notes”) and (ii) an aggregate of $6.0 million in cash (such transactions, collectively, the “September Exchange”).
The 2024 Notes were issued in private placements exempt from registration in reliance on Section 4(a)(2) of the Securities Act of 1933, as amended (the “Securities Act”). We did not receive any cash proceeds from the issuance of the 2024 Notes. In connection with the September Exchange, we entered into a capped call transaction with Royal Bank of Canada (“RBC”). The capped call transaction covers, subject to customary anti-dilution adjustments, the number of shares of our common stock that will initially underlie the Exchange Notes. The capped call transaction is intended to reduce the dilutive impact of the conversion feature of the Exchange Notes on the Company’s outstanding shares of common stock and/or offset any cash payments the Company will be required to make in excess of the original principal amount upon any conversion of the Exchange Notes, with such offset subject to a cap. In connection with the September Exchange, we also entered into an unwind agreement with RBC in order to partially unwind the previous capped call transaction entered into by the Company related to the 2021 Notes that were exchanged.
On December 12, 2019, we entered into separate, privately negotiated exchange agreements pursuant to which we repurchased $119.3 million in aggregate principal amount of our December 2021 and December 2024 convertible notes for (i) $97.9 million excluding accrued and unpaid interest and (ii) 13.4 million shares of our common stock in exchange for $44.8 million in aggregate principal amount of our outstanding December 2021 Notes and $74.6 million in aggregate principal amount of our outstanding December 2024 Notes (the “December Exchange”). The issuance of shares of common stock was exempt from registration in reliance on Section 3(a)(9) of the Securities Act of 1933, as amended.
Issuer purchases of Equity Securities
On December 9, 2019, we announced that our board of directors authorized the repurchase of issued and outstanding shares of our common stock and convertible notes up to an aggregate value of $200.0 million pursuant to a repurchase program. Repurchases under the new repurchase program will be made from time to time in the open market or in privately negotiated transactions and funded from our working capital. The amount and timing of repurchases of shares of our common stock or convertible notes will depend upon the price and availability of shares, general market conditions and the availability of cash. Repurchases may also be made under a trading plan under Rule 10b5-1, which would permit shares or convertible notes to be repurchased when we might otherwise be precluded from doing so because of self-imposed trading blackout periods or other regulatory restrictions. All shares of common stock repurchased under the share repurchase program are expected to be retired and restored to authorized but unissued shares of common stock. All convertible notes repurchased under the program will be retired. This repurchase program may be suspended or discontinued at any time without notice. On December 16, 2019, we announced that our board of directors approved a $75.0 million increase to the previous $200.0 million repurchase program to acquire outstanding common stock and convertible notes.
54
In connection with the December Exchange, we unwound a pro rata portion of the capped call transactions we entered into with the issuance of the December 2021 Notes and the December 2024 Notes. We received cash proceeds from RBC of $6.7 million as a result of the partial unwinding of the capped call agreements. Pursuant to the partial unwinding of the capped call agreements, we entered into an agreement with RBC to purchase 3.2 million shares of our common stock previously acquired by RBC to hedge the capped calls. We acquired the common stock at its closing price on December 12, 2019.
The following table contains information relating to the repurchases of our common stock made by us in the three months ended December 31, 2019 (in thousands, except per share amounts):
Fiscal Period | Total Number of Shares Repurchased | Average Price Paid Per Share | Total Number of Shares Purchased As Part of a Publicly Announced Program | Approximate Dollar Amount of Shares That May Yet be Purchased Under the Program | |||||||||||||
October 1, 2019 | to | October 31, 2019 | — | $ | — | — | $ | — | |||||||||
November 1, 2019 | to | November 30, 2019 | — | $ | — | — | $ | — | |||||||||
December 1, 2019 | to | December 31, 2019 | 3,209 | (1) | $ | 3.43 | 3,209 | $ | 120,204 | (2) | |||||||
Total during three months ended December 31, 2019 | 3,209 | $ | 3.43 | 3,209 | $ | 120,204 | |||||||||||
________________
(1) Purchases in December 2019 were made pursuant to capped call options the Company entered into in connection with the issuance of the December 2021 Notes and the December 2024 Notes. For additional information on the capped call transactions, see Note 13, Convertible Notes.
(2) The approximate dollar amount of shares that may yet be purchased under the share repurchase program was reduced by the cash and PDL common stock issued as consideration to repurchase the convertible notes in December 2019.
55
Comparison of Stockholder Returns
The line graph below compares the cumulative total stockholder return on our common stock between December 31, 2014, and December 31, 2019, with the cumulative total return of (i) the Nasdaq Biotechnology Index and (ii) the Nasdaq Composite Index over the same period. This graph assumes that $100.00 was invested on December 31, 2014, in our common stock at the closing sales price for our common stock on that date and at the closing sales price for each index on that date and that all dividends were reinvested. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns and are not intended to be a forecast.
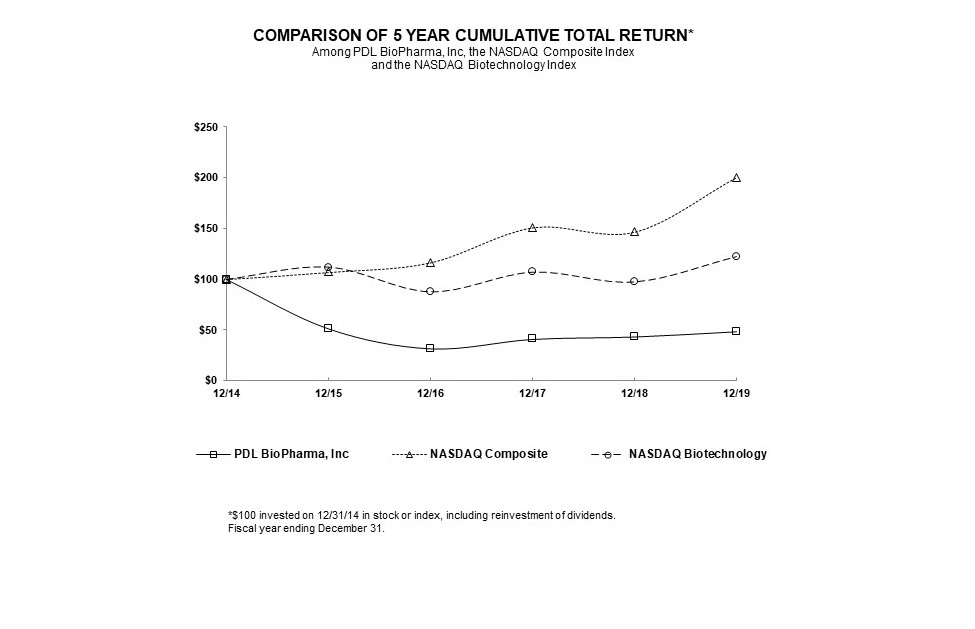
12/31/2014 | 12/31/2015 | 12/31/2016 | 12/31/2017 | 12/31/2018 | 12/31/2019 | ||||||||||||||||||
PDL BioPharma, Inc. | $ | 100.00 | $ | 50.20 | $ | 30.98 | $ | 40.04 | $ | 42.38 | $ | 48.55 | |||||||||||
Nasdaq Composite Index | $ | 100.00 | $ | 140.56 | $ | 112.25 | $ | 133.67 | $ | 121.24 | $ | 200.49 | |||||||||||
Nasdaq Biotechnology Index | $ | 100.00 | $ | 122.81 | $ | 133.19 | $ | 172.11 | $ | 165.84 | $ | 121.92 | |||||||||||
The information in this section shall not be deemed to be “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that we specifically incorporate it by reference in such filing.
56
ITEM 6. SELECTED CONSOLIDATED FINANCIAL DATA
The following selected consolidated financial information has been derived from our Consolidated Financial Statements. The information below is not necessarily indicative of the results of future operations and should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” Item 1A, “Risk Factors” and the Consolidated Financial Statements and related notes thereto included in Item 8, “Financial Statements and Supplementary Data” in order to fully understand factors that may affect the comparability of the information presented below.
Consolidated Statements of Operations Data
For the Years Ended December 31, | ||||||||||||||||||||
(in thousands, except per share data) | 2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
Revenues: | ||||||||||||||||||||
Product revenue, net | $ | 85,835 | $ | 105,448 | $ | 84,123 | $ | 31,669 | $ | — | ||||||||||
Royalty rights - change in fair value | (31,042 | ) | 85,256 | 162,327 | 16,196 | 68,367 | ||||||||||||||
Royalties from Queen et al. patents | 9 | 4,536 | 36,415 | 166,158 | 485,156 | |||||||||||||||
Interest revenue | — | 2,337 | 17,744 | 30,404 | 36,202 | |||||||||||||||
License and other | (45 | ) | 533 | 19,451 | (126 | ) | 723 | |||||||||||||
Total revenues | 54,757 | 198,110 | 320,060 | 244,301 | 590,448 | |||||||||||||||
Operating expenses: | ||||||||||||||||||||
Cost of product revenue (excluding intangible asset amortization and impairment) | 53,619 | 48,460 | 30,537 | 4,065 | — | |||||||||||||||
Amortization of intangible assets | 6,306 | 15,831 | 24,689 | 12,028 | — | |||||||||||||||
General and administrative expenses | 45,598 | 45,420 | 45,641 | 39,790 | 36,090 | |||||||||||||||
Sales and marketing | 8,482 | 17,139 | 17,683 | 538 | — | |||||||||||||||
Research and development | 7,308 | 2,955 | 7,381 | 3,820 | — | |||||||||||||||
Impairment of intangible assets | 22,490 | 152,330 | — | — | — | |||||||||||||||
Asset impairment loss | 10,768 | 8,200 | — | 3,735 | — | |||||||||||||||
Acquisition-related costs | — | — | — | 3,564 | — | |||||||||||||||
Loss on extinguishment of notes receivable | — | — | — | 51,075 | 3,979 | |||||||||||||||
Change in fair value of anniversary payment and contingent consideration | — | (41,631 | ) | 349 | (3,716 | ) | — | |||||||||||||
Total operating expenses | 154,571 | 248,704 | 126,280 | 114,899 | 40,069 | |||||||||||||||
Operating (loss) income | (99,814 | ) | (50,594 | ) | 193,780 | 129,402 | 550,379 | |||||||||||||
Non-operating income (expense), net: | ||||||||||||||||||||
Equity affiliate - change in fair value | 36,402 | — | — | — | — | |||||||||||||||
Gain on bargain purchase | — | — | 9,309 | — | — | |||||||||||||||
Other non-operating expense, net | (10,328 | ) | (5,328 | ) | (18,562 | ) | (20,032 | ) | (20,241 | ) | ||||||||||
Non-operating income (expense), net | 26,074 | (5,328 | ) | (9,253 | ) | (20,032 | ) | (20,241 | ) | |||||||||||
(Loss) income before income taxes | (73,740 | ) | (55,922 | ) | 184,527 | 109,370 | 530,138 | |||||||||||||
Income tax (benefit) expense | (3,049 | ) | 12,937 | 73,826 | 45,711 | 197,343 | ||||||||||||||
Net (loss) income | (70,691 | ) | (68,859 | ) | 110,701 | 63,659 | 332,795 | |||||||||||||
Less: Net (loss) income attributable to noncontrolling interests | (280 | ) | — | (47 | ) | 53 | — | |||||||||||||
Net (loss) income attributable to PDL’s stockholders | $ | (70,411 | ) | $ | (68,859 | ) | $ | 110,748 | $ | 63,606 | $ | 332,795 | ||||||||
Net (loss) income per basic share | $ | (0.59 | ) | $ | (0.47 | ) | $ | 0.71 | $ | 0.39 | $ | 2.04 | ||||||||
Net (loss) income per diluted share | $ | (0.59 | ) | $ | (0.47 | ) | $ | 0.71 | $ | 0.39 | $ | 2.03 | ||||||||
Cash dividends declared and paid | $ | — | $ | — | $ | — | $ | 0.10 | $ | 0.60 | ||||||||||
57
Consolidated Balance Sheet Data
December 31, | ||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
Cash, cash equivalents, short-term investments and restricted investments | $ | 193,451 | $ | 394,590 | $ | 532,114 | $ | 242,141 | $ | 220,352 | ||||||||||
Working capital | $ | 268,202 | $ | 464,747 | $ | 447,334 | $ | 267,716 | $ | 245,969 | ||||||||||
Total assets | $ | 716,119 | $ | 963,736 | $ | 1,243,123 | $ | 1,215,387 | $ | 1,012,205 | ||||||||||
Long-term obligations, less current portion | $ | 77,148 | $ | 181,487 | $ | 204,124 | $ | 329,649 | $ | 279,512 | ||||||||||
Retained earnings | $ | 670,832 | $ | 828,547 | $ | 945,614 | $ | 857,116 | $ | 810,036 | ||||||||||
Total stockholders’ equity | $ | 593,278 | $ | 729,779 | $ | 845,890 | $ | 755,423 | $ | 695,952 | ||||||||||
58
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with “Selected Consolidated Financial Data” and the Consolidated Financial Statements and related Notes included elsewhere in this Report.
Overview
Throughout our history, our mission has been to improve the lives of patients by aiding in the successful development of innovative therapeutics and healthcare technologies. PDL BioPharma was founded in 1986 as Protein Design Labs, Inc. when it pioneered the humanization of monoclonal antibodies, enabling the discovery of a new generation of targeted treatments that have had a profound impact on patients living with different cancers as well as a variety of other debilitating diseases. In 2006, we changed our name to PDL BioPharma, Inc.
In September 2019, we engaged financial and legal advisors and initiated a review of our strategy. This review was completed in December 2019. At such time, we disclosed that we planned to halt the execution of our growth strategy, cease making additional strategic transactions and investments and pursue a formal process to unlock the value of our portfolio by monetizing our assets and ultimately distributing net proceeds to stockholders. Over the subsequent months, our board of directors and management analyzed, together with our outside financial and legal advisors, how to best capture value pursuant to our monetization strategy and best return the significant intrinsic value of the high-quality assets in our portfolio to the stockholders. In February of 2020, the board of directors approved a plan of complete liquidation of our assets and passed a resolution to seek stockholder approval to dissolve the Company under Delaware law at its next annual meeting of the stockholders. In the event that the board of directors concludes that the whole company sale process is unlikely to maximize the value that can be returned to the stockholders from our monetization process, we would, if approved by the stockholders, file a Certificate of Dissolution in Delaware and proceed to wind-down and dissolve the Company in accordance with Delaware law. Pursuant to its monetization strategy, we are exploring a variety of potential transactions, including a whole company sale, divestiture of assets, spin-offs of operating entities, merger opportunities or a combination thereof. In addition, we have analyzed, and continue to analyze, the optimal mechanisms for returning value to stockholders in a tax-efficient manner, including via share repurchases, cash dividends and other distributions of assets. We have not set a definitive timeline and intend to pursue monetization in a disciplined and cost-effective manner to maximize returns to stockholders. We recognize, however, that accelerating the timeline, while continuing to optimize asset value, could increase returns to stockholders due to reduced general and administrative expenses as well as provide faster returns to stockholders. While, as noted above, we cannot provide a definitive timeline for the monetization and wind-down process, we are targeting the end of 2020 for completing the monetization of our key assets, at which time we may be in a position to file a certificate of dissolution under Delaware law.
In conjunction with our intent to seek stockholder approval for complete dissolution of the Company, a proxy statement will be presented to the stockholders that identifies in detail the rationale for our decision to seek stockholder approval for dissolution and which further presents the risk factors associated with such dissolution. While we ultimately expect that wind-down will conclude with dissolution in accordance with Delaware law, we will continue to be receptive to offers to purchase the entire company throughout the monetization process, with all or less than all of our current assets, should such an offer be made. However, if we conclude that a whole company sale is unlikely or that the value from a whole company sale will not maximize the returns we can provide to our stockholders, we expect that wind-down will ultimately conclude with dissolution in accordance with Delaware law.
Historically, we generated a substantial portion of our revenues through the license agreements related to patents covering the humanization of antibodies, which we refer to as the Queen et al. patents. In 2012, we began providing alternative sources of capital through royalty monetizations and debt facilities, and, in 2016, we began acquiring commercial-stage products and launching specialized companies dedicated to the commercialization of these products. In 2019, we entered into a securities purchase agreement with Evofem, pursuant to which we invested $60.0 million in a private placement of securities. These investments are expected to provide funding for Evofem's pre-commercial activities for Amphora, its investigational, non-hormonal, on-demand prescription contraceptive gel for women. To date, we have consummated eighteen transactions, the following ten of which are active and outstanding:
59
Investment | Investment Type | Segment | Deployed Capital 4 (in millions) | |||||
LENSAR, Inc. (“LENSAR”) | Converted equity and loan | Medical Devices | $ | 47.0 | ||||
Evofem | Equity | Strategic Positions | $ | 60.0 | ||||
Noden 1 | Equity and loan | Pharmaceutical | $ | 191.2 | ||||
CareView communications, Inc. (“CareView”) | Debt | Income Generating Assets | $ | 20.0 | ||||
Wellstat Diagnostics, LLC (“Wellstat Diagnostics”) 2 | Royalty/debt hybrid | Income Generating Assets | $ | 44.0 | ||||
Assertio Therapeutics, Inc. (“Assertio”) 3 | Royalty | Income Generating Assets | $ | 260.5 | ||||
The Regents of the University of Michigan (“U-M”) | Royalty | Income Generating Assets | $ | 65.6 | ||||
AcelRx Pharmaceuticals, Inc. (“AcelRx”) | Royalty | Income Generating Assets | $ | 65.0 | ||||
Viscogliosi Brothers, LLC (“VB”) | Royalty | Income Generating Assets | $ | 15.5 | ||||
KYBELLA | Royalty | Income Generating Assets | $ | 9.5 | ||||
_______________
1 | Noden Pharma DAC and Noden Pharma USA, Inc. (together, and including their respective subsidiaries, “Noden”) |
2 | Also known as Defined Diagnostic, LLC. The Wellstat Diagnostics investment also includes our note receivable with Hyperion Catalysis International, Inc. (“Hyperion”). |
3 | Formerly Depomed, Inc. |
4 | Excludes transaction costs. |
In connection with our investment in Evofem in April 2019, we established our Strategic Positions operating segment and began operating in four operating segments designated as Medical Devices, Strategic Positions, Pharmaceutical and Income Generating Assets. The creation of the Strategic Positions’ segment had no impact on our prior segment reporting structure.
Our Medical Devices segment consists of revenue derived from the sale and lease of the LENSAR® Laser System, which may include equipment, PIDs, procedure licenses, training, installation, warranty and maintenance agreements. Our Strategic Positions segment consists of an investment in Evofem. Our Evofem investment includes shares of common stock and warrants to purchase additional shares of common stock. Evofem is a pre-commercial company and, as such, is not yet engaged in revenue-generating activities. Our Pharmaceutical segment consists of revenue derived from branded prescription medicine products sold under the name Tekturna and Tekturna HCT in the United States, and Rasilez and Rasilez HCT in the rest of the world and revenue generated from the sale of an authorized generic of Tekturna in the United States (collectively, the “Noden Products”). Our Income Generating Assets segment consists of revenue derived from (i) notes and other long-term receivables, (ii) royalty rights and hybrid notes/royalty receivables, (iii) equity investments and (iv) royalties from issued patents in the United States and elsewhere covering the humanization of antibodies, which we refer to as the Queen et al. patents.
Prospectively, we do not expect to make any additional acquisitions in our four operating segments as we pursue our monetization strategy. We do, however, expect to continue supporting the development of LENSAR’s next generation equipment by providing necessary funding.
Critical Accounting Policies and Significant Estimates
The preparation of financial statements and related disclosures in conformity with U.S. Generally Accepted Accounting Principles (“GAAP”) and the discussion and analysis of our financial condition and operating results require our management to make judgments, assumptions and estimates that affect the amounts reported in its Consolidated Financial Statements and accompanying notes. Note 2, Summary of Significant Accounting Policies, to the Consolidated Financial Statements included in Item 8 describes the significant accounting policies and methods used in the preparation of our Consolidated Financial Statements. Management bases its estimates on historical experience and on various other assumptions it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates and such differences may be material.
While our significant accounting policies are more fully described in the notes to our Consolidated Financial Statements appearing elsewhere in this Annual Report, management believes that the following accounting policies related to notes receivable and other long-term receivables, inventory, intangible assets, convertible notes, product revenue, royalty rights - at fair value, income taxes, and business combination are critical because they are both important to the portrayal of our financial
60
condition and operating results, and they require management to make judgments and estimates about inherently uncertain matters.
Notes Receivable and Other Long-Term Receivables
We account for our notes receivable at amortized cost, net of unamortized origination fees, if any, and adjusted for any impairment losses. Interest is accreted or accrued to “Interest revenue” using the effective interest method. When and if supplemental payments are received from certain of these notes and other long-term receivables, an adjustment to the estimated effective interest rate is affected prospectively.
We evaluate the collectability of both interest and principal for each note receivable or loan to determine whether it is impaired. A note receivable or loan is considered to be impaired when, based on current information and events, we determine it is probable that it will be unable to collect amounts due according to the existing contractual terms. When a note receivable or loan is considered to be impaired, the amount of loss is calculated by comparing the carrying value of the financial asset to the value determined by discounting the expected future cash flows at the loan’s effective interest rate or to the estimated fair value of the underlying collateral, less costs to sell, if the loan is collateralized and we expect repayment to be provided solely by the collateral. Impairment assessments require significant judgments and are based on significant assumptions related to the borrower’s credit risk, financial performance, expected sales, and estimated fair value of the collateral.
We record interest on an accrual basis and recognize it as earned in accordance with the contractual terms of the applicable credit agreement, to the extent that the underlying note receivable or loan is not impaired and such amounts are expected to be collected. When a note receivable or loan becomes past due, or if management otherwise does not expect that principal, interest, and other obligations due will be collected in full, we will generally place the note receivable or loan on an impaired status and cease recognizing interest income on that note receivable or loan until all principal and interest due has been paid or until such time that we believe the borrower has demonstrated the ability to repay its current and future contractual obligations. Any uncollected interest related to prior periods is reversed from income in the period that collection of the interest receivable is determined to be doubtful. However, we may make exceptions to this policy if the investment has sufficient collateral value and is in the process of collection.
As of December 31, 2019, we had three notes receivable investments which we determined to be impaired with an aggregate carrying value and fair value of approximately $52.1 million and $57.3 million, respectively, compared to three note receivable investments which we determined to be impaired as of December 31, 2018 with an aggregate carrying value and fair value of approximately $62.8 million and $70.0 million, respectively. We did not recognize any losses on extinguishment of notes receivable during the years ended December 31, 2019 and 2018. During the year ended December 31, 2017, we recognized a loss on extinguishment of notes of $51.1 million. During the years ended December 31, 2019 and 2018, we recorded impairment losses of $10.8 million and $8.2 million, respectively, related to the CareView note receivable. There were no impairment losses on notes receivable for the year ended December 31, 2017. For the year ended December 31, 2019, we did not recognize any interest income for note receivable investments as all such note receivable investments were on an impaired status and no cash interest payments were received. For the years ended December 31, 2018 and 2017, we recognized $2.3 million and $3.1 million, respectively, of interest revenue for the CareView note receivable investment as result of cash interest payments made during the respective fiscal years.
Inventory
Inventory, which consists of raw materials, work-in-process and finished goods, is stated at the lower of cost or net realizable value. We determine cost using the first-in, first-out method. Inventory levels are analyzed periodically and written down to their net realizable value if they have become obsolete, have a cost basis in excess of its expected net realizable value or are in excess of expected requirements. We analyze current and future product demand relative to the remaining product shelf life to identify potential excess inventory. We build demand forecasts by considering factors such as, but not limited to, overall market potential, market share, market acceptance and patient usage. The Company classifies inventory as current on the Consolidated Balance Sheets when the Company expects inventory to be consumed for commercial use within the next twelve months. During the years ended December 31, 2019 and 2018 we recognized reductions in the inventory reserve of $0.3 million and $1.2 million, respectively. During the year ended December 31, 2017, we recognized an inventory write-down of approximately $2.0 million.
Intangible Assets
Intangible assets with finite useful lives consist primarily of acquired product rights and acquired technology and are amortized on a straight-line basis over their estimated useful lives (seven to 20 years). The estimated useful lives associated with finite-lived
61
intangible assets are consistent with the estimated lives of the associated products and may be modified when circumstances warrant. Such assets are reviewed for impairment when events or circumstances indicate that the carrying value of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of an asset and its eventual disposition are less than its carrying amount. The amount of any impairment loss is measured as the difference between the carrying amount and the fair value of the impaired asset.
In June 2018, a settlement agreement with Anchen was reached that granted Anchen a nonexclusive royalty-free license to manufacture and commercialize a generic version of aliskiren in the United States. In return, Anchen agreed not to commercialize its generic version of aliskiren prior to March 1, 2019. As a result of this settlement agreement, we performed an impairment assessment of our Noden asset group and concluded that the sum of undiscounted cash flows was not greater than the carrying value of the assets. Therefore, we performed a discounted cash flow analysis to estimate the fair value of the asset group in accordance with Accounting Standard Codification (“ASC”) 360, Impairment or Disposal of Long-lived Assets, resulting in an impairment charge of $152.3 million in the second quarter of 2018.
On March 4, 2019, we announced the U.S. commercial launch of an authorized generic form of Tekturna, with the same drug formulation as Tekturna. Future events, such as FDA approval of a third-party generic version of aliskiren or publicly announced plans of a launch of a generic version of aliskiren, may be further indicators of impairment which may require us to perform additional impairment testing. On March 22, 2019, the FDA approved Anchen’s generic form of aliskiren.
In December 2019, given our monetization strategy and updated forecasts for Noden, we revised our estimates of Noden’s future cash flows and as a result of this analysis, determined that the sum of undiscounted cash flows was not greater than the carrying value of the assets. Therefore, we performed a discounted cash flow analysis to estimate the fair value of the asset group in accordance with ASC 360, resulting in an impairment charge of $22.5 million in the fourth quarter of 2019.
Future events, such as FDA approval of another third-party generic form of aliskiren or publicly announced plans of a launch of another generic form of aliskiren, may be further indicators of impairment which may require us to perform additional impairment testing.
Convertible Notes
We perform an assessment of all embedded features of a debt instrument to determine if (i) such features should be bifurcated and separately accounted for, and (ii) if bifurcation requirements are met, whether such features should be classified and accounted for as equity or debt instruments. If the embedded feature meets the requirements to be bifurcated and accounted for as a liability, the fair value of the embedded feature is measured initially, included as a liability on the Consolidated Balance Sheets, and re-measured to fair value at each reporting period. Any changes in fair value are recorded in the Consolidated Statement of Operations. We monitor, on an ongoing basis, whether events or circumstances could give rise to a change in our classification of embedded features.
We issued the December 2021 Notes with an option to settle conversions by paying or delivering, as applicable, cash, shares of our common stock or a combination of cash and shares of our common stock, at our election. In accordance with accounting guidance for convertible debt instruments that may be settled in cash or other assets on conversion, we separated the principal balance between the fair value of the liability component and the common stock conversion feature using a market interest rate for a similar nonconvertible instrument at the date of issuance.
The fair value of the liability component of the December 2021 Notes was estimated at $109.1 million at issuance. Therefore, the difference between the face value of the December 2021 Notes at issuance and the estimated fair value of the liability component was being amortized to interest expense over the term of the December 2021 Notes using the effective interest method.
On September 17, 2019, we exchanged $86.1 million aggregate principal of December 2021 Notes for an identical aggregate original principal amount of December 2024 Notes, plus a cash payment of $70.00 for each $1,000 principal amount exchanged (the “September Exchange Transaction”). We pay interest at 2.75% on the December 2024 Notes semiannually in arrears on June 1 and December 1 of each year, beginning December 1, 2019. The original principal of the December 2024 Notes will accrete at a rate of 2.375% per year (“Accretion Interest”) commencing September 17, 2019 through the maturity of the December 2024 Notes. The accreted principal amount of the December 2024 Notes is payable in cash upon maturity. We issued the December 2024 Notes with an option to settle conversions by paying or delivering, as applicable, cash, shares of our common stock or a combination of cash and shares of our common stock, at our election. In accordance with accounting guidance for convertible debt instruments that may be settled in cash or other assets on conversion, we separated the principal balance between the fair
62
value of the liability component and the common stock conversion feature using a market interest rate for a similar nonconvertible instrument at the date of issuance.
The September Exchange Transaction qualified as a debt extinguishment and we recognized a loss on exchange of the convertible notes of $3.9 million, which is included in Non-operating income (expense), net in the Consolidated Statement of Operations for the year ended December 31, 2019.
In accordance with the accounting guidance for an extinguishment of convertible debt instruments with a cash conversion feature, we were required to allocate the fair value of the consideration transferred between the liability component and the equity component. To calculate the fair value of the debt immediately prior to derecognition, the carrying value was recalculated in a manner that reflected the estimated market interest rate for a similar nonconvertible instrument at the date of issuance. Using an assumed borrowing rate of 7.05%, we calculated the fair value of the debt representing the amount allocated to the liability component of the December 2024 Notes with the remainder of the consideration allocated to the equity conversion feature, to reflect the reacquisition of the embedded conversion option. The conversion feature together with the fees allocated to the debt are accounted for as a debt discount. As a result of the September Exchange Transaction, we recorded a total debt discount of $9.4 million, which included the common stock conversion feature of $8.1 million and the debt issuance fees of $1.3 million, charged $5.5 million to Additional paid-in capital ($13.5 million charge to Additional paid-in capital representing the reduction to the 2021 equity component, partially offset by the $8.1 million allocated to equity for the 2024 notes) and recorded $1.2 million to deferred tax liability. The net amount charged to Additional paid-in capital represents the difference between the consideration paid for the September Exchange Transaction and the fair value of the convertible debt prior to the extinguishment.
In connection with the September Exchange Transaction, we entered into a capped call transaction with a counterparty on similar terms and conditions as the capped call transaction entered into between the two parties when the December 2021 Notes were issued. The aggregate cost of the capped call transaction was $4.5 million. We evaluated the capped call transaction under authoritative accounting guidance and determined that it should be accounted for as a separate transaction and classified as a net reduction to Additional paid-in capital within stockholders’ equity with no recurring fair value measurement recorded. Also with the September Exchange Transaction, we and the counterparty unwound a portion of the capped call entered into when the December 2021 Notes were issued as they were no longer scheduled to mature in 2021. The $0.9 million proceeds from the unwind of the capped call, which reflected the value of the options outstanding at the time of the September Exchange Transaction and the average share price of our common stock, were included as an increase to Additional paid-in capital within stockholders’ equity.
On December 12, 2019, we initiated the repurchase of $119.3 million in aggregate principal amount of our December 2021 and December 2024 convertible notes for $97.9 million in cash and 13.4 million shares of our common stock in privately negotiated transactions (the “December Exchange Transaction”). The closing of the December Exchange Transaction occurred on December 17, 2019. We determined that the repurchase of the principal amount should be accounted for as a partial extinguishment of the December 2021 Notes and December 2024 Notes and a loss on extinguishment of $4.5 million was recorded at closing of the transaction. The loss on extinguishment included the de-recognition of a proportional share of the deferred issuance costs of $1.5 million. In connection with the December Exchange Transaction, we unwound a corresponding portion of the capped call related to the convertible notes and repurchased 3.2 million shares of our common stock from the capped call counterparty. We paid the capped call counterparty $4.3 million representing $11.0 million for the common stock repurchased, net of $6.7 million owed to us for unwinding the capped call, which reflected the value of the options outstanding at the time of the December Exchange Transaction. The common stock repurchased was reflected as a decrease to Retained earnings within stockholders’ equity. The proceeds from the capped call were included as an increase to Additional paid-in capital within stockholders’ equity. In furtherance of our monetization strategy, we expect to continue to repurchase or satisfy obligations relating to our convertible notes.
The estimated fair value of the liability components at the date of issuance for the December 2021 Notes and December 2024 Notes were determined using valuation models and are complex and subject to judgment. Significant assumptions within the valuation models included an implied credit spread, the expected volatility and dividend yield of our common stock and the risk-free interest rate for notes with a similar term.
Product Revenue
General
In accordance with ASC 606, revenue is recognized from the sale of products and services when a customer obtains control of such promised products and services. The amount of revenue recognized reflects the consideration to which we expect to be
63
entitled to receive in exchange for these products and services. A five-step model is utilized to achieve the core principle and includes the following steps: (1) identify the customer contract; (2) identify the contract’s performance obligations; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations; and (5) recognize revenue when the performance obligations are satisfied.
The following is a description of principal activities - separated by reportable segments - from which we generate our revenue.
Medical Devices
We principally generate revenue in our Medical Devices segment from the sale and lease of the LENSAR® Laser System, which may include equipment, PIDs, procedure licenses, and training, installation, warranty and maintenance agreements.
For bundled packages, we account for individual products and services separately if they are distinct - i.e. if a product or service is separately identifiable from other promises in the bundled package and if the customer can benefit from it on its own or with other resources that are readily available to the customer. The LENSAR® Laser system, training and installation services are one performance obligation. All other elements are separate performance obligations. PIDs, procedure licenses, warranty and maintenance services are also sold on a stand-alone basis.
As we both sell and lease the LENSAR® Laser System, the consideration (including any discounts) is first allocated between lease and non-lease components and then allocated between the separate products and services based on their stand-alone selling prices. The stand-alone selling prices for the PIDs and procedure licenses are determined based on the prices at which we separately sell the PIDs and procedure licenses. The LENSAR® Laser System and warranty stand-alone selling prices are determined using the expected cost plus a margin approach.
For LENSAR® Laser System sales, we recognize revenue in product revenue when a customer takes possession of the system. This usually occurs after the customer signs a contract, LENSAR installs the system, and LENSAR performs the requisite training for use of the system. For LENSAR® Laser System leases, we recognized revenue in Product revenue over the length of the lease in accordance with ASC Topic 840, Leases through December 31, 2018 and recognizes Product revenue in accordance with ASC Topic 842, Leases, after January 1, 2019.
The LENSAR® Laser System requires both a consumable, a PID, and a procedure license to perform each procedure. We recognize revenue for PIDs in product revenue when the customer takes possession of the PID. PIDs are sold by the case. We recognize revenue for procedure licenses in product revenue when a customer purchases a procedure license from the web portal. Typically, consideration for PIDs and procedure licenses is considered fixed consideration except for certain customer agreements that provide for tiered volume discount pricing which is considered variable consideration.
We offer an extended warranty that provides additional services beyond the standard warranty. We recognize revenue from the sale of extended warranties in product revenue over the warranty period. Customers have the option of renewing the warranty period, which is considered a new and separate contract.
Pharmaceutical
We principally generate revenue in our Pharmaceutical segment from products sold to wholesalers and distributors. Customer orders are generally fulfilled within a few days of receipt resulting in minimal order backlog. Contractual performance obligations are usually limited to transfer of the product to the customer. The transfer occurs either upon shipment or upon receipt of the product in certain countries outside the United States after considering when the customer obtains control of the product. In addition, for some non-U.S. countries, we sell product on a consignment basis where control is not transferred until the customer resells the product to an end user. At these points, customers are able to direct the use of and obtain substantially all of the remaining benefits of the product.
Sales to customers are initially invoiced at contractual list prices. Payment terms are typically 30 to 90 days based on customary practice in each country. Revenue is reduced from the list price at the time of recognition for expected chargebacks, discounts, rebates, sales allowances and product returns, which are referred to as gross-to-net adjustments. These reductions are attributed to various commercial agreements, managed healthcare organizations and government programs such as Medicare, Medicaid, and the 340B Drug Pricing Program containing various pricing implications such as mandatory discounts, pricing protection below wholesaler list price and other discounts when Medicare Part D beneficiaries are in the coverage gap. These various reductions in the transaction price have been estimated using either a most likely amount, in the case of prompt pay discounts, or expected value method for all other variable consideration and have been reflected as liabilities and are settled through cash payments,
64
typically within time periods ranging from a few months to one year. Significant judgment is required in estimating gross-to-net adjustments considering legal interpretations of applicable laws and regulations, historical experience, payer channel mix, current contract prices under applicable programs, unbilled claims, processing time lags and inventory levels in the distribution channel.
Reserves for chargebacks, discounts, rebates, sales allowances and product returns are included within current liabilities in our Consolidated Balance Sheets.
For the period from July 1, 2016 through October 4, 2016, all of the Noden Products were distributed by Novartis under the terms of the Noden Purchase Agreement while transfer of the marketing authorization rights were pending. We presented revenue under the Novartis transition arrangement on a “net” basis and established a reserve for retroactive adjustment to the profit split with Novartis during this time. Beginning on October 5, 2016, Noden Pharma USA, Inc. distributed the Noden Products in the United States and we began presenting revenue for all sales in the United States on a “gross” basis and established a reserve for allowances.
Beginning on September 1, 2017, Noden Pharma DAC began distributing the Noden Products to select countries outside the United States and we began presenting revenue for Noden Products sold by Novartis outside of the United States on a “gross” basis as the marketing authorization rights were transferred. Noden Pharma DAC completed the marketing authorization transfers for all territories in the third quarter of 2018.
Income Generating Assets
Royalty Rights - At Fair Value
We account for our investments in royalty rights at fair value with changes in fair value presented in earnings. The fair value of the investments in royalty rights is determined by using a discounted cash flow analysis related to the expected future cash flows to be received. For each arrangement, we are entitled to royalty payments based on revenue generated by the net sales of the product.
These assets are classified as Level 3 assets within the fair value hierarchy, as our valuation estimates utilize significant unobservable inputs, including estimates. Critical estimates may include probability and timing of future sales of the related products, product demand and market growth assumptions, inventory target levels, product approval and pricing assumptions. Factors that could cause a change in estimates of future cash flows include a change in estimated market size, market share of the products on which we receive royalties, a change in pricing strategy or reimbursement coverage, a delay in obtaining regulatory approval, changes to forecast volume and pricing as a result of generic competition, a change in dosage of the product, and a change in the number of treatments.
The changes in the estimated fair value from investments in royalty rights along with cash receipts in each reporting period are presented together on our Consolidated Statements of Operations as a component of revenue under the caption, “Royalty rights - change in fair value.”
Realized gains and losses on Royalty Rights are recognized as they are earned and when collection is reasonably assured. Royalty Rights revenue is recognized over the respective contractual arrangement period. Transaction-related fees and costs are expensed as incurred.
In the second quarter of 2019, due to the slower than expected adoption of Zalviso (the AcelRx Royalty Agreement product) since its initial launch relative to our estimates and the increased variance noted between our forecast model and actual results in the three months ended June 30, 2019, we utilized a third-party expert to reassess the market and expectations for the product. Based on this analysis and the impact to the projected sales-based royalties and milestones, we wrote down the fair value of the royalty asset by $60.0 million.
In the fourth quarter of 2019, management re-evaluated, with assistance of a third-party expert, the market share data, the gross-to-net revenue adjustment assumptions, expected ex-U.S. launch dates and demand data for our Assertio royalty asset and wrote down the fair value of the asset by $46.3 million.
Income Taxes
The provision for income taxes is determined using the asset and liability approach. Tax laws require items to be included in tax filings at different times than the items are reflected in the financial statements. A current liability is recognized for the estimated
65
taxes payable for the current year. Deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. Deferred taxes are adjusted for enacted changes in tax rates and tax laws. Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized.
We recognize tax benefits from uncertain tax positions only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the Consolidated Financial Statements from such positions are then measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. We adjust the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions. Any interest and penalties on uncertain tax positions are included within the tax provision.
The Tax Cuts and Jobs Act of 2017 (the “2017 Tax Act”) significantly changed the existing U.S. corporate income tax laws by, among other things, lowering the corporate tax rate (from a top rate of 35% to a flat rate of 21%), implementing elements of a territorial tax system, and imposing a one-time deemed repatriation transition tax on cumulative undistributed foreign earnings, for which we have not previously paid U.S. taxes.
Business Combination
We apply ASC 805, Business combinations, pursuant to which the cost of an acquisition is measured as the aggregate of the fair values at the date of exchange of the assets given, liabilities incurred, and equity instruments issued. The costs directly attributable to the acquisition are expensed as incurred. Identifiable assets, liabilities and contingent liabilities acquired or assumed are measured separately at their fair value as of the acquisition date, irrespective of the extent of any noncontrolling interests. The excess of the (i) the total of cost of acquisition, fair value of the noncontrolling interests and acquisition date fair value of any previously held equity interest in the acquiree over (ii) the fair value of the identifiable net assets of the acquiree is recorded as goodwill. If the cost of acquisition is less than the fair value of the net assets of the subsidiary acquired, the difference is recognized directly in the Consolidated Statements of Operations.
The determination and allocation of fair values to the identifiable assets acquired and liabilities assumed is based on various assumptions and valuation methodologies requiring considerable management judgment. The most significant variables in these valuations are discount rates, terminal values, the number of years on which to base the cash flow projections, as well as the assumptions and estimates used to determine the cash inflows and outflows. Management determines discount rates to be used based on the risk inherent in the related activity’s current business model and industry comparisons. Terminal values are based on the expected life of products and forecasted life cycle and forecasted cash flows over that period. Although management believes that the assumptions applied in the determination are reasonable based on information available at the date of acquisition, actual results may differ from the forecasted amounts and the difference could be material.
Recently Issued Accounting Standards
See Part II, Item 8, “Financial Statements and Supplementary Data”, Note 2, Summary of Significant Accounting Policies, to the Consolidated Financial Statements for a discussion of recently adopted accounting pronouncements and recently issued accounting pronouncements not yet adopted as of December 31, 2019.
Recent Developments
Repurchase Program
From January 1, 2020 to March 10, 2020, the Company repurchased approximately 3.8 million shares of its common stock at a weighted-average price of $3.42 per share for a total of $12.9 million and repurchased $3.2 million in aggregate principal amount of December 2021 Convertible Notes and $10.5 million in aggregate principal amount of December 2024 Convertible Notes.
Amendment to CareView Modification Agreement
As further discussed in Note 7, Notes and Other Long-Term Receivables, to the Consolidated Financial Statement, in January 2020 we entered into an additional amendment with CareView whereby we agreed that the principal and interest payments would be further deferred until April 30, 2020, which was conditioned upon CareView raising additional financing from third parties.
Plan of Liquidation
66
On February 7, 2020, our board of directors approved a plan of complete liquidation which triggered the change in control clause in the Amended and Restated 2005 Equity Incentive Plan, accelerating the vesting of a significant portion of our outstanding equity awards.
Summary of 2019 and 2018 Financial Results
This section provides an analysis of our financial results for the fiscal year ended December 31, 2019 compared to the fiscal year ended December 31, 2018. For the discussion covering the fiscal year ended December 31, 2017, please refer to Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Form 10-K for the fiscal year ended December 31, 2018.
• | Our net loss for the years ended December 31, 2019 and 2018 was $70.4 million and $68.9 million, respectively; |
• | At December 31, 2019, we had cash and cash equivalents of $193.5 million as compared with $394.6 million at December 31, 2018; |
• | At December 31, 2019, we had $716.1 million in total assets as compared with $963.7 million at December 31, 2018; and |
• | At December 31, 2019, we had $122.8 million in total liabilities as compared with $234.0 million at December 31, 2018. |
Revenues
A summary of our revenues for the years ended December 31, 2019 and 2018, is presented below:
(Dollars in thousands) | 2019 | 2018 | Change from Prior Year % | ||||||||
Revenues: | |||||||||||
Product revenue, net | $ | 85,835 | $ | 105,448 | (19 | )% | |||||
Royalty rights - change in fair value | (31,042 | ) | 85,256 | (136 | )% | ||||||
Royalties from Queen et al. patents | 9 | 4,536 | (100 | )% | |||||||
Interest revenue | — | 2,337 | N/M | ||||||||
License and other | (45 | ) | 533 | (108 | )% | ||||||
Total revenues | $ | 54,757 | $ | 198,110 | (72 | )% | |||||
___________________
N/M Not meaningful
Our total revenues decreased by 72%, or $143.4 million, for the year ended December 31, 2019, when compared to the year ended December 31, 2018. The decrease was primarily due to:
• | decreased royalty asset revenues due in part to a decrease in fair value of the AcelRx and Assertio royalty assets, |
• | a decrease in product revenue from the sale of the Noden Products in our Pharmaceutical segment, |
• | decreased interest revenue related to the CareView note receivable asset, |
• | lower license and other revenue, partially offset by |
• | an increase in product revenue from sales of the LENSAR Laser System in our Medical Devices segment. |
Revenue from our Medical Devices segment for the year ended December 31, 2019 was $30.7 million, an increase of 25% compared to the year ended December 31, 2018. The increase is attributable to higher net revenues in both North America and the rest of the world, with the majority of the increase outside of North America. Revenue from LENSAR product consists of revenue from the sale and lease of the LENSAR® Laser System, which may include equipment, PIDs, procedure licenses, training, installation, warranty and maintenance agreements.
Revenue from our Pharmaceutical segment for the year ended December 31, 2019 was $55.1 million, a decrease of 32% when compared to the same period in 2018. The decrease in revenue from our Pharmaceutical segment reflects lower net revenues in the United States and the rest of the world. The decrease in revenue from our Pharmaceutical segment in the United States for the year ended December 31, 2019 reflects the introduction of our authorized generic form of Tekturna and a third-party generic form of aliskiren during the year ended December 31, 2019. The decrease in revenue for the rest of the world is due to lower sales
67
volume of Rasilez in certain territories. All revenues from our Pharmaceutical segment were derived from sales of the Noden Products.
The following table provides a summary of activity with respect to our sales allowances and accruals for the year ended December 31, 2019:
(in thousands) | Discount and Distribution Fees | Government Rebates and Chargebacks | Assistance and Other Discounts | Product Return | Total | |||||||||||||||
Balance as of December 31, 2018 | $ | 3,094 | $ | 8,901 | $ | 3,457 | $ | 4,681 | $ | 20,133 | ||||||||||
Allowances for current period sales | 5,090 | 12,104 | 5,003 | 1,720 | 23,917 | |||||||||||||||
Allowances for prior period sales | 50 | 1,848 | 142 | 46 | 2,086 | |||||||||||||||
Credits/payments for current period sales | (3,813 | ) | (8,843 | ) | (4,186 | ) | (276 | ) | (17,118 | ) | ||||||||||
Credits/payments for prior period sales | (3,076 | ) | (10,393 | ) | (3,411 | ) | (2,295 | ) | (19,175 | ) | ||||||||||
Balance as of December 31, 2019 | $ | 1,345 | $ | 3,617 | $ | 1,005 | $ | 3,876 | $ | 9,843 | ||||||||||
We record revenue from our Pharmaceutical segment net of estimated product returns, pricing discounts, including rebates offered pursuant to mandatory federal and state government programs, chargebacks, prompt pay discounts, distribution fees and co-pay assistance for product sales each period.
Revenue from our Income Generating Assets segment for the year ended December 31, 2019 was $(31.1) million, a decrease of 134%, or $123.7 million, when compared to the same period in 2018. The decrease was primarily due to:
• | a decrease in the estimated fair value of the AcelRx and Assertio royalty assets in 2019, |
• | decreasing royalties from the Queen et al. patents as the patents have expired, |
• | the absence of interest revenue recognized from our CareView note receivable in 2019, and |
• | lower license and other revenue. |
The adjustment to the fair value of the AcelRx royalty asset in the second quarter of 2019 was due to the slower than expected adoption of Zalviso since its initial launch relative to our estimates and the increased variance noted between our forecast model and actual results in the second quarter of 2019. We engaged a third-party expert in the second quarter of 2019 to reassess the market and expectations for the product. Key findings from the third-party study included: the post-surgical PCA (Patient-Controlled Analgesia) market being smaller than previously forecasted; the higher price of the product relative to alternative therapies, the product not being used as a replacement for systemic opioids and the design of the delivery device, which is pre-filled for up to three days of treatment, which limited its use for procedures with anticipated shorter recovery times.
The adjustment to the fair value of the Assertio royalty asset in the fourth quarter of 2019 was due to a decrease in the sales forecast for the Assertio products. We engaged a third-party expert in the fourth quarter of 2019 to reassess the market and expectations for the royalty asset. Key findings from the third-party study included: an anticipated decrease in the Glumetza net sales forecast due to an accelerated shift in the channel mix resulting in a substantial decline in net selling prices, particularly in the fourth quarter of 2019 and beyond, as previously announced by Bausch Health and the delayed launch dates of the extended release products in the Assertio royalty asset portfolio outside of the United States.
The following tables provide a summary of activity with respect to our royalty rights - change in fair value for the years ended December 31, 2019 and 2018:
Year Ended December 31, 2019 | ||||||||||||
Change in | ||||||||||||
(in thousands) | Cash Royalties | Fair Value | Total | |||||||||
Assertio | $ | 72,225 | $ | (45,699 | ) | $ | 26,526 | |||||
VB | 966 | (518 | ) | 448 | ||||||||
U-M | 5,664 | (5,197 | ) | 467 | ||||||||
AcelRx | 307 | (57,428 | ) | (57,121 | ) | |||||||
KYBELLA | 110 | (1,472 | ) | (1,362 | ) | |||||||
$ | 79,272 | $ | (110,314 | ) | $ | (31,042 | ) | |||||
68
Year Ended December 31, 2018 | ||||||||||||
Change in | ||||||||||||
(in thousands) | Cash Royalties | Fair Value | Total | |||||||||
Assertio | $ | 71,502 | $ | 12,333 | $ | 83,835 | ||||||
VB | 1,062 | (272 | ) | 790 | ||||||||
U-M | 4,631 | (1,174 | ) | 3,457 | ||||||||
AcelRx | 249 | (2,514 | ) | (2,265 | ) | |||||||
Avinger | 366 | (396 | ) | (30 | ) | |||||||
KYBELLA | 159 | (690 | ) | (531 | ) | |||||||
$ | 77,969 | $ | 7,287 | $ | 85,256 | |||||||
The following table summarizes the percentage of our total revenues earned, which individually accounted for 10% or more of our total revenues for the years ended December 31, 2019 and 2018:
Year Ended December 31, | ||||||||
Source | Product Name | 2019 | 2018 | |||||
AcelRx | Zalviso | (104 | )% | (1 | )% | |||
Assertio | Glumetza, Janumet XR1, Jentadueto XR, Invokamet XR and Synjardy XR | 48 | % | 42 | % | |||
LENSAR | LENSAR Laser System | 56 | % | 12 | % | |||
Noden | Tekturna, Tekturna HCT, Rasilez and Rasilez HCT | 101 | % | 41 | % | |||
_______________
1 | Royalties received through the third quarter of 2018. |
Foreign currency exchange rates also impact our reported revenues from royalty assets and product sales. Our revenues may fluctuate due to changes in foreign currency exchange rates and are subject to foreign currency exchange risk. While foreign currency conversion terms vary by license agreement, generally most agreements require that royalties first be calculated in the currency of sale and then converted into U.S. dollars using the average daily exchange rates for that currency for a specified period at the end of the calendar quarter. Accordingly, when the U.S. dollar weakens against other currencies, the converted amount is greater than it would have been had the U.S. dollar not weakened. In addition, our Noden Product sales in markets outside the United States are typically denominated in foreign currencies and can cause fluctuations in our reported revenue from period to period. The impact of changes in foreign currency exchange rates to our reported revenue was insignificant for the years ended December 31, 2019 and 2018.
69
Operating Expenses
A summary of our operating expenses for the years ended December 31, 2019 and 2018 is presented below:
(Dollars in thousands) | 2019 | 2018 | Change from Prior Year % | ||||||||
Costs of product revenue (excluding intangible amortization and impairment) | $ | 53,619 | $ | 48,460 | 11 | % | |||||
Amortization of intangible assets | 6,306 | 15,831 | (60 | )% | |||||||
General and administrative | 45,598 | 45,420 | 0 | % | |||||||
Sales and marketing | 8,482 | 17,139 | (51 | )% | |||||||
Research and development | 7,308 | 2,955 | 147 | % | |||||||
Impairment of intangible assets | 22,490 | 152,330 | (85 | )% | |||||||
Asset impairment loss | 10,768 | 8,200 | 31 | % | |||||||
Change in fair value of contingent consideration | — | (41,631 | ) | N/M | |||||||
Total operating expenses | $ | 154,571 | $ | 248,704 | (38 | )% | |||||
Percentage of total revenues | 282 | % | 126 | % | |||||||
___________________
N/M Not meaningful
Total operating expenses decreased by 38%, or $94.1 million for the year ended December 31, 2019, when compared to the year ended December 31, 2018. The decrease was primarily a result of:
• | a $22.5 million impairment of the Noden intangible asset in the current year compared to a $152.3 million impairment in 2018, |
• | lower amortization expense for the Noden intangible assets in 2019 resulting from the impairment recorded in 2018 due to the increased probability of a third-party generic form of aliskiren being launched in the United States, |
• | lower sales and marketing expenses reflecting the cost savings from the change in our marketing strategy to a non-personal promotion strategy for the Noden Products in anticipation of a launch of a third-party generic form of aliskiren. This non-personal promotion strategy was subsequently discontinued upon the launch of our authorized generic form of Tekturna in the first quarter of 2019, partially offset by |
• | the favorable adjustment to the Noden acquisition related contingent consideration, which was first reduced in the second quarter of 2018 prompted by the increased probability of a third-party generic form of aliskiren being launched in the United States and subsequently eliminated in the fourth quarter of 2018 when the launch was imminent, |
• | an increase in research and development expenses in our Medical Devices segment primarily due to the exclusive licensing of intellectual property from a third party for $3.5 million in cash for use in developing its next generation technology, |
• | higher cost of product revenue, due to increased sales in our Medical Devices segment and costs associated with the amended Novartis supply agreement in our Pharmaceutical segment, |
• | a small increase in our general and administrative expenses, as detailed below, and |
• | a $10.8 million impairment loss on the CareView note receivable recorded in 2019 compared to the $8.2 million impairment loss on the CareView note receivable recorded in 2018. |
70
General and administrative expenses for the years ended December 31, 2019 and 2018 by segment are summarized in the tables below:
Year Ended December 31, 2019 | ||||||||||||||||
(in thousands) | Pharmaceutical | Medical Devices | Income Generating Assets | Total | ||||||||||||
Compensation | $ | 1,858 | $ | 4,109 | $ | 16,656 | $ | 22,623 | ||||||||
Salaries and Wages (including taxes) | 1,306 | 1,883 | 6,277 | 9,466 | ||||||||||||
Bonuses (including accruals) | 344 | 1,260 | 3,643 | 5,247 | ||||||||||||
Equity | 208 | 966 | 6,736 | 7,910 | ||||||||||||
Asset management | — | — | 2,741 | 2,741 | ||||||||||||
Business development | — | — | 1,282 | 1,282 | ||||||||||||
Accounting and tax services | 1,115 | 759 | 4,400 | 6,274 | ||||||||||||
Other professional services | 654 | 403 | 2,262 | 3,319 | ||||||||||||
Other | 2,645 | 1,713 | 5,001 | 9,359 | ||||||||||||
Total general and administrative | $ | 6,272 | $ | 6,984 | $ | 32,342 | $ | 45,598 | ||||||||
_______________
No general and administrative expenses were attributable to the Strategic Positions segment for the years ended December 31, 2019 or 2018.
Year Ended December 31, 2018 | ||||||||||||||||
(in thousands) | Pharmaceutical | Medical Devices | Income Generating Assets | Total | ||||||||||||
Compensation | $ | 1,971 | $ | 3,627 | $ | 10,204 | $ | 15,802 | ||||||||
Salaries and Wages (including taxes) | 1,506 | 1,871 | 6,193 | 9,570 | ||||||||||||
Bonuses (including accruals) | 325 | 991 | (203 | ) | 1,113 | |||||||||||
Equity | 140 | 765 | 4,214 | 5,119 | ||||||||||||
Asset management | — | — | 5,386 | 5,386 | ||||||||||||
Business development | 203 | — | 1,168 | 1,371 | ||||||||||||
Accounting and tax services | 1,528 | 39 | 4,288 | 5,855 | ||||||||||||
Other professional services | 3,891 | 825 | 1,921 | 6,637 | ||||||||||||
Other | 3,781 | 1,399 | 5,189 | 10,369 | ||||||||||||
Total general and administrative | $ | 11,374 | $ | 5,890 | $ | 28,156 | $ | 45,420 | ||||||||
Non-operating Income (Expense), Net
A summary of our non-operating expense, net, for the years ended December 31, 2019 and 2018, is presented below:
(Dollars in thousands) | 2019 | 2018 | Change from Prior Year % | ||||||||
Interest and other income, net | $ | 6,030 | $ | 6,065 | (1 | )% | |||||
Interest expense | (11,404 | ) | (12,157 | ) | (6 | )% | |||||
Equity affiliate - change in fair value | 36,402 | — | N/M | ||||||||
Gain on sale of intangible assets | 3,476 | — | N/M | ||||||||
Gain on investments | — | 764 | N/M | ||||||||
Loss on exchange and extinguishment of convertible notes | (8,430 | ) | — | N/M | |||||||
Total non-operating income (expense), net | $ | 26,074 | $ | (5,328 | ) | (589 | )% | ||||
___________________
N/M Not meaningful
71
For the year ended December 31, 2019, compared to December 31, 2018
Total non-operating income (expense), net, changed from expense of $5.3 million for the year ended December 31, 2018 to income of $26.1 million for the year ended December 31, 2019, primarily due to:
• | an increase to the fair value of our investment in common stock and warrants of Evofem subsequent to our acquisition earlier in 2019, |
• | the decrease in interest expense, and |
• | the gain recognized on the sale of our Direct Flow Medical, Inc. (“Direct Flow Medical”) intangible assets, partially offset by |
• | the losses on the exchange and extinguishment of a portion of our December 2021 Notes and December 2024 Notes, |
• | the absence of a realized gain on investments in 2019, and |
• | lower interest and other income. |
Income Taxes
Income tax (benefit) expense for the years ended December 31, 2019 and 2018 was $(3.0) million and $12.9 million, respectively, which resulted primarily from applying the federal statutory income tax rate to (loss) income before income taxes. The tax rate of 4.1% in 2019 differs from the statutory tax rate of 21% primarily as a result of increases in our valuation allowance. The tax rate of (23.1)% in 2018 differs from the statutory tax rate of 21%, primarily as a result of the foreign rate differential on income or loss at our foreign subsidiaries and increases in our valuation allowance.
During 2019, the amount of our unrecognized tax benefits increased by $3.4 million. The future impact of the unrecognized tax benefits of $84.2 million, if recognized, is comprised of $27.9 million, which would affect the effective tax rate, and $56.3 million, which would result in adjustments to deferred tax assets and our valuation allowance.
Estimated interest and penalties associated with unrecognized tax benefits increased our income tax expense in the Consolidated Statements of Operations by $1.6 million during the year ended December 31, 2019 and $1.0 million during the year ended December 31, 2018. In general, our income tax returns are subject to examination by U.S. federal, state and local tax authorities for tax years 2000 forward. Interest and penalties associated with unrecognized tax benefits accrued on the balance sheet were $9.7 million and $8.0 million as of December 31, 2019 and 2018, respectively. We are currently under income tax examination by the State of California for tax years 2009 through 2015 and by the Internal Revenue Service for the tax year 2016. The timing of the resolution of income tax examinations is highly uncertain, and the amounts ultimately paid, if any, upon resolution of the issues raised by the taxing authorities may differ materially from the amounts accrued for each year. We do not anticipate any material change to the amount of our unrecognized tax benefit over the next 12 months.
Net (Loss) Income per Share
Net (loss) income per share for the years ended December 31, 2019 and 2018, is presented below:
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
Net (loss) per basic share | $ | (0.59 | ) | $ | (0.47 | ) | |
Net (loss) per diluted share | $ | (0.59 | ) | $ | (0.47 | ) | |
Liquidity and Capital Resources
We have previously financed our operations primarily through royalty and other license-related revenues, public and private placements of debt and equity securities, interest income on invested capital and cash generated from pharmaceutical and medical device product sales. We plan to continue to finance our operations in the near term primarily through royalty and other license-related revenues and cash generated from product sales.
In September 2019, we engaged financial and legal advisors and initiated a review of our strategy. In December 2019, we disclosed that we planned to halt the execution of our growth strategy, cease making additional strategic transactions and investments and pursue a formal process to unlock the value of our portfolio by monetizing our assets and ultimately returning net proceeds to our stockholders. Over the subsequent months, our board of directors and management analyzed, together with its
72
outside financial and legal advisors, how to best capture value pursuant to its monetization strategy and best return the significant intrinsic value of the high-quality assets in its portfolio to the stockholders. In February of 2020, the board of directors approved a plan of complete liquidation of our assets and passed a resolution to seek stockholder approval to dissolve the Company under Delaware law at its next annual meeting of the stockholders. In the event that the Board concludes that the whole company sale process is unlikely to maximize the value that can be returned to the stockholders from our monetization process, we would, if approved by the stockholders, file a certificate of dissolution in Delaware and proceed to wind-down and dissolve the Company in accordance with Delaware law. Pursuant to its monetization strategy, we are exploring a variety of potential transactions, including a whole company sale, divestiture of assets, spin-offs of operating entities, merger opportunities or a combination thereof. In addition, we have analyzed, and continue to analyze, the optimal mechanisms for returning value to stockholders in a tax-efficient manner, including via share repurchases, cash dividends and other distributions of assets. We have not set a definitive timeline and intend to pursue monetization in a disciplined and cost-effective manner to maximize returns to stockholders. We recognize, however, that accelerating the timeline, while continuing to optimize asset value, could increase returns to stockholders due to reduced general and administrative expenses as well as provide faster returns to stockholders. While we cannot provide a definitive timeline for the monetization and wind-down process, we are targeting the end of 2020 for completing the monetization of its key assets.
As a result of this monetization strategy, we expect to generate additional cash from the sale of one or more of the assets in our portfolio with the intention of managing the successful wind down of our business and distributing the remaining net proceeds to our stockholders.
Our future capital requirements are difficult to forecast and will depend upon many factors, including the type of distributions we make, the amount of net cash proceeds we receive, after transaction costs, and the time it takes to monetize our assets. Our future capital requirements will also depend on the amount of common stock and convertible notes we repurchase under our repurchase program, both of which we expect to pursue as part of our monetization strategy.
The general cash needs of our Medical Devices, Strategic Positions, Pharmaceutical and Income Generating Assets segments can vary significantly.
• | In our Medical Devices segment, the primary factor determining cash needs is the funding of operations, which we expect to continue to expand as the business grows, and enhancing our product offerings through the research and development of our next generation device which will integrate a femtosecond laser and a phacoemulsification system in a single, compact workstation. |
• | In our Pharmaceutical segment, cash needs tend to be driven primarily by material purchases. |
• | The cash needs of our Income Generating Assets segment tend to be driven by legal and professional service fees required for operating a publicly traded company, as well as the funding of potential repurchases of our common stock and convertible notes. |
• | The current cash needs for our Strategic Positions segment are insignificant. |
On December 9, 2019, we announced that our board of directors authorized the repurchase of issued and outstanding shares of our common stock and convertible notes up to an aggregate value of $200.0 million pursuant to a share repurchase program. On December 16, 2019, we announced that our board of directors approved a $75.0 million increase to this repurchase program. Repurchases under the new repurchase program will be made from time to time in the open market or in privately negotiated transactions and funded from our working capital. The amount and timing of such repurchases will depend upon the price and availability of shares or convertible notes, general market conditions and the availability of cash. Common stock and convertible note repurchases may also be made under a trading plan under Rule 10b5-1, which would permit shares and convertible notes to be repurchased when we might otherwise be precluded from doing so because of self-imposed trading blackout periods or other regulatory restrictions. All shares of common stock repurchased under our repurchase program are expected to be retired and restored to authorized but unissued shares of common stock. All convertible notes repurchased under the program will be retired. As of December 31, 2019, we had repurchased $44.8 million in aggregate principal amount of December 2021 Notes and $74.5 million in aggregate principal amount of December 2024 Notes under the board authorized program for aggregate consideration consisting of a cash payment of $97.9 million and the issuance of 13.4 million shares of our common stock. Pursuant to the convertible note repurchase transactions and the unwinding of a proportional amount of the capped call transaction entered into for the notes, we also repurchased 3.2 million shares of our common stock under this program directly from our capped call counterparty. This repurchase program may be suspended at any time without notice.
Our debt service obligations consists of interest payments and repayment of our December 2021 Notes and December 2024 Notes. We have and may continue to repurchase the remaining outstanding convertible notes, which could adversely affect the amount or timing of any distributions to our stockholders. We expect to finance such repurchases with cash on hand.
73
We had cash and cash equivalents in the aggregate of $193.5 million and $394.6 million at December 31, 2019 and 2018, respectively, representing a decrease of $201.1 million. The decrease was primarily attributable to the repurchase of the December 2021 Notes and December 2024 Notes for $97.9 million, the repurchase of stock of $86.9 million, the investment in Evofem of $60.0 million and cash used in operating activities of $32.4 million, partially offset by cash received from royalties of $79.3 million.
We believe that cash on hand and cash generated from future revenues and from asset sales, net of operating expenses, debt service and income taxes, will be sufficient to fund our operations until all net proceeds are distributed to our stockholders. Our continued success is dependent on our ability to execute on our planned strategy to monetize our assets, in order to return capital to our stockholders and service our remaining debt.
Off-Balance Sheet Arrangements
As of December 31, 2019, we did not have any off-balance sheet arrangements, as defined under SEC Regulation S-K Item 303(a)(4)(ii).
Contractual Obligations
The following table summarizes our contractual obligations and commercial commitments as of December 31, 2019:
Payments Due by Period | ||||||||||||||||||||
(in thousands) | Less than 1 year | 1-3 years | 3-5 years | Thereafter | Total | |||||||||||||||
Operating leases 1 | $ | 958 | $ | 776 | $ | — | $ | — | $ | 1,734 | ||||||||||
Convertible notes 2 | 843 | 20,329 | 13,636 | — | 34,808 | |||||||||||||||
Inventory 3 | 49,419 | 22,639 | — | — | 72,058 | |||||||||||||||
Total contractual obligations | $ | 51,220 | $ | 43,744 | $ | 13,636 | $ | — | $ | 108,600 | ||||||||||
_____________________________
1 Amounts represent the lease for our headquarters in Incline Village, Nevada, the lease for the Noden Pharma DAC office in Dublin, Ireland, the lease for the LENSAR office and manufacturing facility in Orlando, Florida and operating leases for office equipment.
2 Amounts represent principal and cash interest payments due on the December 2021 Notes and the December 2024 Notes and accretion interest on the December 2024 Notes.
3 Consist of minimum purchase obligation under the Novartis supply agreement for API and bulk tablets and inventory components for LENSAR, as discussed in “Purchase Obligations” below.
In addition to amounts in the table above, we are contractually obligated to pay $1.0 million to a third party upon achievement of product sales milestones for KYBELLA, which we do not believe are probable. As such, this contingent payment is not recorded on our Consolidated Balance Sheets. See “Kybella Royalty Agreement” below for further discussion.
Our liability for uncertain tax positions was $37.6 million as of December 31, 2019, all of which has been excluded from the table above due to the uncertainty in the timing of the settlement of these positions.
Purchase Obligations
Noden DAC and Novartis entered into a supply agreement pursuant to which Novartis will manufacture and supply to Noden DAC a bulk tableted form of the Noden Products and API. In May 2019, Noden DAC and Novartis entered into an amended supply agreement pursuant to which Novartis will supply to Noden DAC a bulk tableted form of the Noden Products through 2020 and API through June 2021. The supply agreement may be terminated by either party for material breach that remains uncured for a specified time period. Under the terms of the amended supply agreement, Noden DAC is committed to purchase certain quantities of bulk product and API that would amount to approximately $61.7 million through June 2021, of which $39.8 million is committed over the next twelve months, which are guaranteed by us. While the supply agreement provides that the parties will agree to reasonable accommodations with respect to changes in firm orders, we expect that Noden DAC will meet the requirements of the supply agreement, unless otherwise negotiated.
74
LENSAR entered into various supply agreements for the manufacture and supply of certain components. The supply agreements commit LENSAR to a minimum purchase obligation of approximately $10.4 million over the next twenty-four months of which $9.6 million is due in the next 12 months, a portion of which are guaranteed by us. LENSAR expects to meet these requirements.
Kybella Royalty Agreement
On July 8, 2016, we entered into a royalty purchase and sales agreement with an individual, whereby we acquired that individual’s rights to receive certain royalties on sales of KYBELLA by Allergan plc, in exchange for a $9.5 million cash payment and up to a $1.0 million future milestone payment based upon product sales targets.
Guarantees
Redwood City Lease Guarantee
In connection with the spin-off of Facet in December 2008, we entered into amendments to the leases for our former facilities in Redwood City, California, under which Facet was added as a co-tenant and a Co-Tenancy Agreement, under which Facet agreed to indemnify us for all matters related to the leases attributable to the period after the spin-off date. In April 2010, Abbott Laboratories acquired Facet and later renamed the entity AbbVie Biotherapeutics, Inc. (“AbbVie”). If AbbVie were to default under its lease obligations, we could be held liable by the landlord as a co-tenant, and, thus, we have in substance guaranteed the payments under the lease agreements for the Redwood City facilities. As of December 31, 2019, the total lease payments for the duration of the guarantee, which runs through December 2021, are approximately $22.6 million. For additional information regarding our lease guarantee, see Note 15, Commitments and Contingencies.
Escrow Receivable
On September 21, 2017, we entered into an agreement (the “kaléo Note Sale Agreement”) with MAM-Kangaroo Lender, LLC, a Delaware limited liability company (the “kaléo Purchaser”), pursuant to which we sold our entire interest in the notes issued by Accel 300, LLC (“Accel 300”) pursuant to that certain Indenture, dated as of April 1, 2014, by and between Accel 300 and U.S. Bank National Association, as the current trustee of the notes described therein (the “kaléo Note”).
Pursuant to the kaléo Note Sale Agreement, the kaléo Purchaser paid to us an amount equal to 100% of the then outstanding principal, a premium of 1% of such amount and accrued interest under the kaléo Notes, for an aggregate cash purchase price of $141.7 million.
The aggregate purchase price of $1.4 million was deposited into an escrow account as a potential payment against certain contingencies for 18 months. The escrow period ended on March 20, 2019 and the escrow agent released the entire $1.4 million to us.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Sensitive Financial Instruments
Our exposure to market risk for changes in interest rates relates primarily to our excess cash investments and our convertible notes.
Our excess cash investments consist of Rule 2a-7 money market funds and had a fair value of approximately $131.3 million at December 31, 2019 and $226.7 million at December 31, 2018. Due to the short duration of these investments, with a maximum weighted average maturity of 60 days or less, if market interest rates were to increase or decrease by 1%, there would be no material impact on the fair value of our portfolio.
The aggregate fair value of our convertible notes was estimated to be $33.9 million at December 31, 2019, and $151.4 million at December 31, 2018, based on available pricing information. At December 31, 2019 our convertible notes consisted of the December 2021 Notes, and the December 2024 Notes, both of which have a fixed interest rate of 2.75%, and also for the December 2024 Notes, a principal accretion rate of 2.375% per year. As of December 31, 2018, our convertible notes consisted of the December 2021 Notes. Changes in interest rates do not affect interest expense on fixed rate debt. While changes in interest rates do not impact the amount of interest we pay, these obligations are subject to interest rate risk because changes in interest rates would affect the fair values of fixed rate debt.
75
The following table presents information about our material debt obligation that is sensitive to changes in interest rates. The table presents principal amounts and related interest rates by year of expected maturity for our debt obligations or the earliest year in which the holders may put the debt to us. The convertible notes may be converted to our common stock prior to the maturity date under certain conditions.
(in thousands) | 2020 | 2021 | 2022 | 2023 | 2024 | Total | Fair Value | ||||||||||||||||||||||
Convertible notes | |||||||||||||||||||||||||||||
Fixed Rate | $ | — | $ | 19,170 | $ | — | $ | — | $ | 11,500 | $ | 30,670 | $ | 33,931 | (1) | ||||||||||||||
Average Interest Rate | 3.64 | % | 3.69 | % | 5.13 | % | 5.13 | % | 5.13 | % | |||||||||||||||||||
_________________________
(1) | The fair value of the remaining payments under our December 2021 Notes and December 2024 Notes was estimated based on the trading value of these notes at December 31, 2019. |
Equity Price Risk
Our investments in equity securities expose us to equity price risk. Equity price risk results from fluctuations in quoted market prices for equity securities and instruments that derive their value from such securities. The fair value of our investments that are subject to equity price risk as of December 31, 2019 was approximately $96.4 million. The impact of a 10% decrease in the quoted market price related to these investments would have been approximately a $10.0 million decrease to pre-tax income. Due to equity securities being measured at fair value with net unrealized gains and losses from changes in the fair value recognized in earnings, fluctuations in quoted market prices for equity securities could have a material effect on our results of operations and our financial position.
Foreign Currency Sensitive Financial Instruments
Our international operations are affected by fluctuations in the value of the U.S. dollar as compared to foreign currencies, predominantly the euro. Increases and decreases in our international product sales from movements in foreign currency exchange rates are offset partially by the corresponding increases or decreases in our international operating expenses. Our revenues, expenses and cash flows may fluctuate due to changes in foreign currency exchange rates and are subject to foreign currency exchange risk. While foreign currency conversion terms vary by agreement, generally most agreements require that royalties first be calculated in the currency of sale and then converted into U.S. dollars using the average daily exchange rates for that currency for a specified period at the end of the calendar quarter. Similarly, sales outside of the United States of our Noden Products as well as a portion of the cash balances and expenses of our Irish subsidiary are denominated in currencies other than the U.S. dollar. Accordingly, when the U.S. dollar weakens against other currencies, the converted amount is greater than it would have been had the U.S. dollar not weakened and when the U.S. dollar strengthens against other currencies, the converted amount is less.
76
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index to Consolidated Financial Statements
Item | Page |
77
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of PDL BioPharma, Inc.
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated balance sheets of PDL BioPharma, Inc. and its subsidiaries (the “Company”) as of December 31, 2019 and 2018, and the related consolidated statements of operations, of comprehensive (loss) income, of stockholders’ equity and of cash flows for each of the three years in the period ended December 31, 2019, including the related notes (collectively referred to as the “consolidated financial statements”). We also have audited the Company's internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the COSO.
Basis for Opinions
The Company's management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on the Company’s consolidated financial statements and on the Company's internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Subsequent Event
As discussed in Note 26 to the consolidated financial statements, on February 7, 2020, the Company’s board of directors approved a plan of liquidation.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide
78
reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP | |||
San Francisco, California
March 11, 2020
We have served as the Company’s auditor since 2014.
79
PDL BIOPHARMA, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except per share data)
December 31, | |||||||
2019 | 2018 | ||||||
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 193,451 | $ | 394,590 | |||
Accounts receivable, net | 13,552 | 21,648 | |||||
Notes receivable | 52,583 | 63,042 | |||||
Inventory | 39,773 | 18,942 | |||||
Prepaid and other current assets | 14,536 | 18,995 | |||||
Total current assets | 313,895 | 517,217 | |||||
Property and equipment, net | 5,520 | 7,387 | |||||
Royalty rights - at fair value | 266,196 | 376,510 | |||||
Investment in equity affiliate | 82,267 | — | |||||
Notes and other receivables, long-term | 827 | 771 | |||||
Long-term deferred tax assets | — | 1,539 | |||||
Intangible assets, net | 23,298 | 51,319 | |||||
Other assets | 24,116 | 8,993 | |||||
Total assets | $ | 716,119 | $ | 963,736 | |||
Liabilities and Stockholders’ Equity | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 17,370 | $ | 13,142 | |||
Accrued liabilities | 28,306 | 39,312 | |||||
Accrued income taxes | 17 | 16 | |||||
Total current liabilities | 45,693 | 52,470 | |||||
Convertible notes payable | 27,250 | 124,644 | |||||
Other long-term liabilities | 49,898 | 56,843 | |||||
Total liabilities | 122,841 | 233,957 | |||||
Commitments and contingencies (Note 15) | |||||||
Stockholders’ equity: | |||||||
Preferred stock, par value $0.01 per share, 10,000 shares authorized; no shares issued and outstanding | — | — | |||||
Common stock, par value $0.01 per share, 350,000 shares authorized; 124,303 and 136,513 shares issued and outstanding at December 31, 2019 and 2018, respectively | 1,243 | 1,365 | |||||
Additional paid-in capital | (78,875 | ) | (98,030 | ) | |||
Treasury stock, at cost (zero and 750 shares held) | — | (2,103 | ) | ||||
Retained earnings | 670,832 | 828,547 | |||||
Total PDL’s stockholders’ equity | 593,200 | 729,779 | |||||
Noncontrolling interests | 78 | — | |||||
Total stockholders’ equity | 593,278 | 729,779 | |||||
Total liabilities and stockholders’ equity | $ | 716,119 | $ | 963,736 | |||
See accompanying notes.
80
PDL BIOPHARMA, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Revenues: | |||||||||||
Product revenue, net | $ | 85,835 | $ | 105,448 | $ | 84,123 | |||||
Royalty rights - change in fair value | (31,042 | ) | 85,256 | 162,327 | |||||||
Royalties from Queen et al. patents | 9 | 4,536 | 36,415 | ||||||||
Interest revenue | — | 2,337 | 17,744 | ||||||||
License and other | (45 | ) | 533 | 19,451 | |||||||
Total revenues | 54,757 | 198,110 | 320,060 | ||||||||
Operating expenses | |||||||||||
Cost of product revenue (excluding intangible asset amortization and impairment) | 53,619 | 48,460 | 30,537 | ||||||||
Amortization of intangible assets | 6,306 | 15,831 | 24,689 | ||||||||
General and administrative | 45,598 | 45,420 | 45,641 | ||||||||
Sales and marketing | 8,482 | 17,139 | 17,683 | ||||||||
Research and development | 7,308 | 2,955 | 7,381 | ||||||||
Impairment of intangible assets | 22,490 | 152,330 | — | ||||||||
Asset impairment loss | 10,768 | 8,200 | — | ||||||||
Change in fair value of anniversary payment and contingent consideration | — | (41,631 | ) | 349 | |||||||
Total operating expenses | 154,571 | 248,704 | 126,280 | ||||||||
Operating (loss) income | (99,814 | ) | (50,594 | ) | 193,780 | ||||||
Non-operating income (expense), net | |||||||||||
Interest and other income, net | 6,030 | 6,065 | 1,659 | ||||||||
Interest expense | (11,404 | ) | (12,157 | ) | (20,221 | ) | |||||
Equity affiliate - change in fair value | 36,402 | — | — | ||||||||
Gain on sale of intangible assets | 3,476 | — | — | ||||||||
Gain on bargain purchase | — | — | 9,309 | ||||||||
Gain on investments | — | 764 | — | ||||||||
Loss on exchange and extinguishment of convertible notes | (8,430 | ) | — | — | |||||||
Total non-operating income (expense), net | 26,074 | (5,328 | ) | (9,253 | ) | ||||||
(Loss) income before income taxes | (73,740 | ) | (55,922 | ) | 184,527 | ||||||
Income tax (benefit) expense | (3,049 | ) | 12,937 | 73,826 | |||||||
Net (loss) income | (70,691 | ) | (68,859 | ) | 110,701 | ||||||
Less: Net (loss) income attributable to noncontrolling interests | (280 | ) | — | (47 | ) | ||||||
Net (loss) income attributable to PDL’s stockholders | $ | (70,411 | ) | $ | (68,859 | ) | $ | 110,748 | |||
Net (loss) income per share | |||||||||||
Basic | $ | (0.59 | ) | $ | (0.47 | ) | $ | 0.71 | |||
Diluted | $ | (0.59 | ) | $ | (0.47 | ) | $ | 0.71 | |||
Weighted-average shares outstanding | |||||||||||
Basic | 118,631 | 145,669 | 155,394 | ||||||||
Diluted | 118,631 | 145,669 | 156,257 | ||||||||
See accompanying notes.
81
PDL BIOPHARMA, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE (LOSS) INCOME
(In thousands)
Year Ended December 31, | ||||||||||||
2019 | 2018 | 2017 | ||||||||||
Net (loss) income | $ | (70,691 | ) | $ | (68,859 | ) | $ | 110,701 | ||||
Other comprehensive (loss) income, net of tax | ||||||||||||
Change in unrealized gains on investments in available-for-sale securities: | ||||||||||||
Change in fair value of investments in available-for-sale securities, net of tax | — | (578 | ) | 1,181 | ||||||||
Adjustment for net (gains) losses realized and included in net (loss) income, net of tax | — | (603 | ) | — | ||||||||
Total change in unrealized gains (losses) on investments in available-for-sale securities, net of tax(a) | — | (1,181 | ) | 1,181 | ||||||||
Comprehensive (loss) income | (70,691 | ) | (70,040 | ) | 111,882 | |||||||
Less: Comprehensive (loss) income attributable to noncontrolling interests | (280 | ) | — | (47 | ) | |||||||
Comprehensive (loss) income attributable to PDL’s stockholders | $ | (70,411 | ) | $ | (70,040 | ) | $ | 111,929 | ||||
___________________________________
(a) Net of tax of ($314) and $314 for the years ended December 31, 2018 and 2017, respectively.
See accompanying notes.
82
PDL BIOPHARMA, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share amounts)
PDL’s Stockholders Equity | ||||||||||||||||||||||||||||||
Common Stock | Treasury Stock | Additional Paid-In Capital | Retained Earnings | Accumulated Other Comprehensive Income (Loss) | Non-controlling Interest | Total Stockholders’ Equity | ||||||||||||||||||||||||
Shares | Amount | |||||||||||||||||||||||||||||
Balance at December 31, 2016 | 165,538,447 | $ | 1,655 | $ | — | $ | (107,628 | ) | $ | 857,116 | $ | — | $ | 4,280 | $ | 755,423 | ||||||||||||||
Issuance of common stock, net of forfeitures | 1,582,698 | 16 | — | (16 | ) | — | — | — | — | |||||||||||||||||||||
Stock-based compensation expense | — | — | — | 3,138 | — | — | — | 3,138 | ||||||||||||||||||||||
Repurchase and retirement of common stock | (13,346,389 | ) | (133 | ) | — | — | (29,867 | ) | — | — | (30,000 | ) | ||||||||||||||||||
Acquisition of Noden common stock | — | — | — | 2,063 | — | — | (4,233 | ) | (2,170 | ) | ||||||||||||||||||||
Cumulative effect from change in accounting principles | — | — | — | — | 7,617 | — | — | 7,617 | ||||||||||||||||||||||
Comprehensive income: | ||||||||||||||||||||||||||||||
Net income | — | — | — | — | 110,748 | — | (47 | ) | 110,701 | |||||||||||||||||||||
Change in unrealized gains and losses on investments in available-for-sale securities, net of tax | — | — | — | — | — | 1,181 | — | 1,181 | ||||||||||||||||||||||
Total comprehensive income | — | — | — | — | — | — | — | 111,882 | ||||||||||||||||||||||
Balance at December 31, 2017 | 153,774,756 | 1,538 | — | (102,443 | ) | 945,614 | 1,181 | — | 845,890 | |||||||||||||||||||||
Issuance of common stock, net of forfeitures | (601,668 | ) | (6 | ) | — | 6 | 58 | — | — | 58 | ||||||||||||||||||||
Stock-based compensation expense | — | — | — | 4,407 | — | — | — | 4,407 | ||||||||||||||||||||||
Repurchase and retirement of common stock | (16,660,566 | ) | (167 | ) | (2,103 | ) | — | (48,266 | ) | — | — | (50,536 | ) | |||||||||||||||||
Comprehensive loss: | ||||||||||||||||||||||||||||||
Net loss | — | — | — | — | (68,859 | ) | — | — | (68,859 | ) | ||||||||||||||||||||
Change in unrealized gains and losses on investments in available-for-sale securities, net of tax | — | — | — | — | — | (1,181 | ) | — | (1,181 | ) | ||||||||||||||||||||
Total comprehensive loss | — | — | — | — | — | — | — | (70,040 | ) | |||||||||||||||||||||
Balance at December 31, 2018 | 136,512,522 | 1,365 | (2,103 | ) | (98,030 | ) | 828,547 | — | — | 729,779 | ||||||||||||||||||||
Issuance of common stock, net of forfeitures | 729,191 | 7 | — | (7 | ) | 8 | — | — | 8 | |||||||||||||||||||||
Stock-based compensation expense | — | — | — | 6,907 | — | — | — | 6,907 | ||||||||||||||||||||||
Repurchase and retirement of common stock | (26,321,293 | ) | (263 | ) | 2,103 | — | (87,312 | ) | — | — | (85,472 | ) | ||||||||||||||||||
Transfer of subsidiary shares to non-controlling interest | — | — | — | 426 | — | — | 358 | 784 | ||||||||||||||||||||||
Exchange of convertible notes | — | — | — | (36,963 | ) | — | — | — | (36,963 | ) | ||||||||||||||||||||
Issuance of common stock in connection with repurchase of convertible notes | 13,382,196 | 134 | — | 45,767 | — | — | — | 45,901 | ||||||||||||||||||||||
Capped call transactions | — | — | — | 3,025 | — | — | — | 3,025 | ||||||||||||||||||||||
Comprehensive loss: | ||||||||||||||||||||||||||||||
Net loss | — | — | — | — | (70,411 | ) | — | (280 | ) | (70,691 | ) | |||||||||||||||||||
Total comprehensive loss | — | — | — | — | — | — | — | (70,691 | ) | |||||||||||||||||||||
Balance at December 31, 2019 | 124,302,616 | $ | 1,243 | $ | — | $ | (78,875 | ) | $ | 670,832 | $ | — | $ | 78 | $ | 593,278 | ||||||||||||||
See accompanying notes.
83
PDL BIOPHARMA, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Cash flows from operating activities | |||||||||||
Net (loss) income | $ | (70,691 | ) | $ | (68,859 | ) | $ | 110,701 | |||
Adjustments to reconcile net (loss) income to net cash (used in) provided by operating activities: | |||||||||||
Amortization of convertible notes conversion options and debt issuance costs | 7,237 | 7,609 | 11,038 | ||||||||
Accreted interest on convertible note principal | 79 | — | — | ||||||||
Amortization of intangible assets | 6,306 | 15,831 | 24,689 | ||||||||
Amortization of right-of-use assets | 886 | — | — | ||||||||
Impairment of intangible asset | 22,490 | 152,330 | — | ||||||||
Asset impairment loss | 10,768 | 8,200 | — | ||||||||
Change in fair value of royalty rights - at fair value | 31,042 | (85,256 | ) | (162,327 | ) | ||||||
Change in fair value of equity affiliate | (31,641 | ) | — | — | |||||||
Change in fair value of derivative assets | (4,715 | ) | (33 | ) | 49 | ||||||
Change in fair value of anniversary payment and contingent consideration | — | (41,631 | ) | 349 | |||||||
Other amortization, depreciation and accretion of embedded derivative | 2,901 | 3,696 | 2,366 | ||||||||
Loss on exchange and extinguishment of convertible notes | 8,430 | — | — | ||||||||
Gain on sale of intangible assets | (3,476 | ) | — | — | |||||||
Gain on sale of available-for-sale securities | — | (764 | ) | (108 | ) | ||||||
Loss on disposal of property and equipment | 1,200 | 66 | — | ||||||||
Escrow receivable | — | — | (1,400 | ) | |||||||
Bargain purchase gain | — | — | (9,309 | ) | |||||||
Stock-based compensation expense | 7,119 | 4,758 | 3,138 | ||||||||
Deferred income taxes | (10,617 | ) | 13,846 | 39,172 | |||||||
Changes in assets and liabilities: | |||||||||||
Accounts receivable | 8,195 | 9,349 | 11,008 | ||||||||
Prepaid and other current assets | 4,464 | (5,025 | ) | (9,100 | ) | ||||||
Accrued interest on notes receivable | — | — | 1,475 | ||||||||
Inventory | (21,923 | ) | (9,508 | ) | 892 | ||||||
Other assets | (156 | ) | (2,120 | ) | (1,400 | ) | |||||
Accounts payable | 4,191 | (6,642 | ) | 10,840 | |||||||
Accrued liabilities | (9,341 | ) | (7,449 | ) | 13,120 | ||||||
Accrued income taxes | 1 | (1,361 | ) | (3,346 | ) | ||||||
Other long-term liabilities | 4,808 | (462 | ) | (1,223 | ) | ||||||
Net cash (used in) provided by operating activities | (32,443 | ) | (13,425 | ) | 40,624 | ||||||
Cash flows from investing activities | |||||||||||
Purchases of investments | — | — | (23,213 | ) | |||||||
Investment in equity affiliate | (60,000 | ) | — | — | |||||||
Maturities of investments-other | — | — | 75,000 | ||||||||
Payment of contingent consideration | — | (858 | ) | — | |||||||
Proceeds from sales of available-for-sale securities | — | 4,116 | 39,956 | ||||||||
Purchase of royalty rights - at fair value | — | (20,000 | ) | — | |||||||
Proceeds from royalty rights - at fair value | 79,272 | 77,969 | 107,253 | ||||||||
Purchase of intangible assets | (1,700 | ) | — | — | |||||||
Sale of royalty rights - at fair value | — | — | 108,169 | ||||||||
Proceeds from the sale of intangible assets | 5,000 | — | — | ||||||||
Repayment of notes receivable | — | — | 144,829 | ||||||||
Proceeds from sales of assets held for sale | — | — | 8,190 | ||||||||
Purchase of property and equipment | (763 | ) | (4,523 | ) | (1,297 | ) | |||||
Net cash provided by investing activities | 21,809 | 56,704 | 458,887 | ||||||||
Cash flows from financing activities | |||||||||||
Repurchase of convertible notes | (97,889 | ) | — | — | |||||||
Repayment of convertible notes | — | (126,447 | ) | — | |||||||
Payment to exchange convertible notes | (7,451 | ) | — | — | |||||||
Capped call transactions | 3,025 | — | — | ||||||||
Payment of anniversary payment | — | — | (87,007 | ) | |||||||
Payment of contingent consideration | (1,071 | ) | — | — | |||||||
Cash paid for purchase of noncontrolling interest | — | — | (2,170 | ) | |||||||
Repurchase of Company common stock | (86,898 | ) | (49,109 | ) | (30,000 | ) | |||||
Cash dividends paid | (9 | ) | (48 | ) | (222 | ) | |||||
Net settlement of stock-based compensation awards | (212 | ) | (351 | ) | — | ||||||
Net cash used in financing activities | (190,505 | ) | (175,955 | ) | (119,399 | ) | |||||
Net (decrease) increase in cash and cash equivalents | (201,139 | ) | (132,676 | ) | 380,112 | ||||||
Cash and cash equivalents at beginning of the year | 394,590 | 527,266 | 147,154 | ||||||||
Cash and cash equivalents at end the year | $ | 193,451 | $ | 394,590 | $ | 527,266 | |||||
See accompanying notes
84
PDL BIOPHARMA, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS, continued
(In thousands)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Supplemental cash flow information | |||||||||||
Cash (refunded) paid for income taxes | $ | (2,689 | ) | $ | 3,805 | $ | 43,366 | ||||
Cash paid for interest | $ | 4,265 | $ | 6,654 | $ | 9,286 | |||||
Supplemental schedule of non-cash investing and financing activities | |||||||||||
Convertible notes due December 2021 exchanged for convertible notes due December 2024 | $ | 86,053 | $ | — | $ | — | |||||
Common stock used to settle convertible notes payable | $ | 45,901 | $ | — | $ | — | |||||
Assets held for sale reclassified from other assets to intangible assets | $ | — | $ | 1,811 | $ | — | |||||
Asset held for sale reclassified from notes receivable to other assets | $ | — | $ | — | $ | 10,000 | |||||
Extinguishment of notes receivable | $ | — | $ | — | $ | 43,909 | |||||
See accompanying notes
85
PDL BIOPHARMA, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2019
1. Organization and Business
Throughout our history, the Company’s mission has been to improve the lives of patients by aiding in the successful development of innovative therapeutics and healthcare technologies. PDL BioPharma was founded in 1986 as Protein Design Labs, Inc. when it pioneered the humanization of monoclonal antibodies, enabling the discovery of a new generation of targeted treatments that have had a profound impact on patients living with different cancers as well as a variety of other debilitating diseases. In 2006, the Company changed its name to PDL BioPharma, Inc.
In December 2019, the Company announced that it had completed a strategic review process and decided to halt the execution of its growth strategy, cease additional strategic investments and pursue a formal process to unlock value by monetizing our assets and returning net proceeds to stockholders (the “monetization strategy”). The Company further announced in December 2019 that it would explore a variety of potential transactions in connection with the monetization strategy, including a sale of the Company, divestiture of the Company’s assets or businesses, a spin-off transaction, a merger or a combination thereof.
Historically, the Company generated a substantial portion of its revenues through the license agreements related to patents covering the humanization of antibodies, which it refers to as the Queen et al. patents. In 2012, the Company began providing alternative sources of capital through royalty monetizations and debt facilities, and, in 2016, the Company began acquiring commercial-stage products and launching specialized companies dedicated to the commercialization of these products. In 2019, and as a further evolution of the Company’s strategy, it began to enter into strategic transactions involving innovative late clinical-stage or early commercial-stage therapeutics. Consistent with this strategy, on April 10, 2019, the Company entered into a securities purchase agreement with Evofem Biosciences, Inc. (“Evofem”), pursuant to which it invested $60.0 million in a private placement of securities structured in two tranches. To date, the Company has consummated eighteen transactions, ten of which are active and outstanding. Pursuant to the Company’s monetization strategy, the Company does not expect to enter into any additional similar transactions.
Based on the composition of its existing investment portfolio, the Company currently operates in four segments designated as Medical Devices, Strategic Positions, Pharmaceutical and Income Generating Assets. With the investment in Evofem in the second quarter of 2019, the Company added the Strategic Positions segment. This did not have any impact on its prior segment reporting structure.
Our Medical Devices segment consists of revenue derived from the sale and lease of the LENSAR® Laser System made by the Company’s majority-owned subsidiary, LENSAR, Inc. (“LENSAR”), which may include equipment, Patient Interface Devices (“PIDs”), procedure licenses, training, installation, warranty and maintenance agreements.
Our Strategic Positions segment consists of an investment in Evofem. Evofem is a publicly-traded (NASDAQ: EVFM) clinical-stage biopharmaceutical company committed to developing and commercializing innovative products to address unmet needs in women's sexual and reproductive health. Evofem is leveraging its proprietary Multipurpose Vaginal pH Regulator (MVP-R™) platform to develop Amphora® (L-lactic acid, citric acid and potassium bitartrate) for hormone-free birth control. Our investments are expected to provide funding for Evofem's pre-commercial activities for Amphora and include shares of common stock and warrants to purchase additional shares of common stock. Evofem is a pre-commercial company and, as such, is not yet engaged in revenue-generating activities.
Our Pharmaceutical segment consists of revenue derived from branded prescription medicine products sold under the name Tekturna® and Tekturna HCT® in the United States and Rasilez® and Rasilez HCT® in the rest of the world and revenue generated from the sale of an authorized generic form of Tekturna in the United States (collectively, the “Noden Products”). The branded prescription Noden Products were acquired from Novartis AG, Novartis Pharma AG and Speedel Holding AG (collectively, “Novartis”) in July 2016 (the “Noden Transaction”) by the Company’s wholly-owned subsidiary, Noden Pharma DAC (“Noden DAC”). The Company, through its wholly-owned subsidiary, Noden Pharma USA Inc. (“Noden USA”) launched its authorized generic form of Tekturna in the United States in March 2019.
Our Income Generating Assets segment consists of revenue derived from (i) notes and other long-term receivables, (ii) royalty rights and hybrid notes/royalty receivables, (iii) equity investments and (iv) royalties from the Queen et al. patents.
86
In September 2019, the Company engaged financial advisors and initiated a review of its strategy. This review was completed in December 2019. At such time, it was decided to halt the execution of the Company’s growth strategy, cease making additional strategic transactions and investments and pursue a formal process to unlock the value of its portfolio by monetizing its assets and ultimately distributing net proceeds to stockholders.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying Consolidated Financial Statements of PDL Biopharma, Inc. and its subsidiaries (collectively, the “Company” or “PDL”) have been prepared in accordance with Generally Accepted Accounting Principles (United States) (“GAAP”).
Principles of Consolidation
The Consolidated Financial Statements include the accounts of the Company and its wholly-owned and majority-owned subsidiaries. All significant intercompany balances and transactions have been eliminated upon consolidation.
A subsidiary is an entity in which the Company, directly or indirectly, controls more than one half of the voting power; has the power to appoint or remove the majority of the members of the board of directors; to cast a majority of votes at the meeting of the board of directors or to govern the financial and operating policies of the investee under a statute or agreement among the stockholders or equity holders.
The Company applies the guidance codified in ASC 810, Consolidations, which requires certain variable interest entities to be consolidated by the primary beneficiary of the entity in which it has a controlling financial interest. The Company identifies an entity as a variable interest entity if either: (1) the entity does not have sufficient equity investment at risk to permit the entity to finance its activities without additional subordinated financial support, or (2) the entity’s equity investors lack the essential characteristics of a controlling financial interest. The Company performs ongoing qualitative assessments of its variable interest entities to determine whether the Company has a controlling financial interest in any variable interest entity and therefore is the primary beneficiary, and if it has the power to direct activities that impact the activities of the entity.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the Consolidated Financial Statements and accompanying Notes to the Consolidated Financial Statements. The accounting estimates that require management’s most significant, difficult and subjective judgments include the valuation of royalty rights - at fair value, product revenue recognition and allowance for customer rebates and allowances, the valuation of notes receivable and inventory, the assessment of recoverability of intangible assets and their estimated useful lives, the valuation and recognition of stock-based compensation, the recognition and measurement of current and deferred income tax assets and liabilities, and the valuation of warrants to acquire shares of common stock. Actual results could differ from those estimates.
Segment Reporting
Under ASC 280, Segment Reporting, operating segments are defined as components of an enterprise about which separate financial information is available that is regularly evaluated by the entity’s chief operating decision maker, in deciding how to allocate resources and in assessing performance. The Company has evaluated its operating segments in accordance with ASC 280 as of December 31, 2019, and has identified four reportable segments: Medical Devices, Strategic Positions, Pharmaceutical and Income Generating Assets.
Cash Equivalents
The Company considers all highly liquid investments with initial maturities of three months or less at the date of purchase to be cash equivalents. The Company places its cash and cash equivalents with high credit quality financial institutions and, by policy, limits the amount of credit exposure in any one financial instrument.
87
Accounts Receivable
As of December 31, 2019, the Company concluded that an allowance for doubtful accounts was not required. As of December 31, 2018, the Company had $78,000 in its allowance for doubtful accounts. The Company provides an allowance for doubtful accounts based on experience and specifically identified risks. Accounts receivable are carried at fair value and charged off against the allowance for doubtful accounts when the Company determines that recovery is unlikely and the Company ceases collection efforts.
Investments
As of December 31, 2019 and 2018, the Company’s investments were comprised of an investment in a publicly traded company and a privately-held company.
The Company’s investment in Evofem qualifies for equity method accounting given its percentage ownership in Evofem and the ability to exercise significant influence. The Company elected the fair value method to account for its investment in Evofem as it believes it better reflects economic reality, the financial reporting of the investment and the current value of the asset. Changes in fair value of the Evofem equity investment are presented in Non-operating income (expense), net on the Consolidated Statement of Operations.
The Company’s equity security investment in Alphaeon Corporation (“Alphaeon”) qualifies to be measured at fair value, although it has been determined that the fair value of the investment is not readily determinable as Alphaeon’s shares are not publicly traded. The Company evaluates the fair value of this investment by performing a qualitative assessment each reporting period. If the results of this qualitative assessment indicate that the fair value is less than the carrying value, the investment is written down to its fair value. There have been no such write downs since the Company acquired these shares. This investment is included in other long-term assets. For additional information on the Alphaeon investment, see Note 9, Notes and Other Long-Term Receivables.
Fair Value Measurements
The fair value of the Company’s financial instruments are estimates of the amounts that would be received if the Company were to sell an asset or the Company paid to transfer a liability in an orderly transaction between market participants at the measurement date or exit price. The assets and liabilities are categorized and disclosed in one of the following three categories:
Level 1 – based on quoted market prices in active markets for identical assets and liabilities;
Level 2 – based on observable inputs other than quoted prices in active markets for identical assets and liabilities, quoted prices for identical or similar assets or liabilities in inactive markets, or other inputs that are or can be corroborated by observable market data for substantially the full term of the assets or liabilities, and
Level 3 – based on unobservable inputs using management’s best estimate and assumptions when inputs are unavailable.
Notes Receivable and Other Long-Term Receivables
The Company accounts for its notes receivable at amortized cost, net of unamortized origination fees, if any, and adjusted for any impairment losses. Interest is accreted or accrued to “Interest revenue” using the effective interest method. When and if supplemental payments are received from certain of these notes and other long-term receivables, an adjustment to the estimated effective interest rate is affected prospectively.
The Company evaluates the collectability of both interest and principal for each note receivable and loan to determine whether it is impaired. A note receivable or loan is considered to be impaired when, based on current information and events, the Company determines it is probable that it will be unable to collect amounts due according to the existing contractual terms. When a note receivable or loan is considered to be impaired, the amount of loss is calculated by comparing the carrying value of the financial asset to the value determined by discounting the expected future cash flows at the loan’s effective interest rate or to the estimated fair value of the underlying collateral, less costs to sell, if the loan is collateralized and the Company expects repayment to be provided solely by the collateral. Impairment assessments require significant judgments and are based on significant assumptions related to the borrower’s credit risk, financial performance, expected sales, and estimated fair value of the collateral.
88
The Company records interest on an accrual basis and recognizes it as earned in accordance with the contractual terms of the credit agreement, to the extent that such amounts are expected to be collected. When a note receivable or loan becomes past due, or if management otherwise does not expect that principal, interest, and other obligations due will be collected in full, the Company will generally place the note receivable or loan on an impaired status and cease recognizing interest income on that note receivable or loan on an accrual basis until all principal and interest due has been paid or until such time that the Company believes the borrower has demonstrated the ability to repay its current and future contractual obligations. Any uncollected interest related to prior periods is reversed from income in the period that collection of the interest receivable is determined to be doubtful. However, the Company may make exceptions to this policy if the investment has sufficient collateral value and is in the process of collection. Any interest payments received for notes receivable or loans on an impaired status are recognized as interest income on a cash basis.
For the year ended December 31, 2019, the Company did not recognize any interest revenue for the CareView Communications, Inc. (“CareView”) note receivable while on impaired status. For the years ended December 31, 2018 and 2017, the Company recognized $2.3 million and $3.1 million, respectively, of interest revenue for the CareView note receivable as a result of cash interest payments made during these years.
As of December 31, 2019, the Company had three notes receivable investments which were determined to be impaired with a cumulative investment cost and fair value of approximately $52.1 million and $57.3 million, respectively. The same three note receivable investments were determined to be impaired as of December 31, 2018 with a cumulative investment cost and fair value of approximately $62.8 million and $70.0 million, respectively as of this date. During the years ended December 31, 2019, 2018, and 2017, the Company did not recognize any losses on extinguishment of notes receivable.
During the years ended December 31, 2019 and 2018, the Company recorded an impairment loss of $10.8 million and $8.2 million, respectively, related to the CareView note receivable. There were no impairment losses on notes receivable for the year ended December 31, 2017. For additional information about the impairment loss recorded on the CareView note receivable, see Note 7, Notes and Other Long-Term Receivables.
Inventory
Inventory, which consists of raw materials, work-in-process and finished goods, is stated at the lower of cost or net realizable value. The Company determines cost using the first-in, first-out method. Inventory levels are analyzed periodically and written down to their net realizable value if they have become obsolete, have a cost basis in excess of its expected net realizable value or are in excess of expected requirements. The Company analyzes current and future product demand relative to the remaining product shelf life to identify potential excess inventory. The Company builds demand forecasts by considering factors such as, but not limited to, overall market potential, market share, market acceptance and patient usage. The Company classifies inventory as current on the Consolidated Balance Sheets when the Company expects inventory to be consumed for commercial use within the next twelve months.
Intangible Assets
Intangible assets with finite useful lives consist primarily of acquired product rights and acquired technology and are amortized on a straight-line basis over their estimated useful lives, over seven years to 20 years. The estimated useful lives associated with finite-lived intangible assets are consistent with the estimated lives of the associated products and may be modified when circumstances warrant. Such assets are reviewed for impairment when events or circumstances indicate that the carrying value of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of an asset and its eventual disposition are less than its carrying amount. The amount of any impairment is measured as the difference between the carrying amount and the fair value of the impaired asset.
89
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the following estimated useful lives:
Leasehold improvements | Lesser of useful life or term of lease | |
Manufacturing equipment | 3-5 years | |
Computer and office equipment | 3 years | |
Transportation equipment | 3 years | |
Furniture and fixtures | 7 years | |
Equipment under lease | Greater of lease term or 5-10 years | |
Convertible Notes
The Company has previously issued convertible notes with settlement features that allow the Company to settle the notes by paying or delivering, as applicable, cash, shares of the Company’s common stock or a combination of cash and shares of our common stock, at the Company’s election. In accordance with accounting guidance for convertible debt instruments that may be settled in cash or other assets on conversion, the Company separated the principal balance between the fair value of the liability component and the common stock conversion feature using a market interest rate for a similar nonconvertible instrument at the date of issuance.
Financing Costs Related to Long-term Debt
Costs associated with obtaining long-term debt are deferred and amortized over the term of the related debt using the effective interest method. Such costs are presented as reductions from the carrying amount of the long-term debt liability, consistent with debt discounts, on the Company’s Consolidated Balance Sheets.
Revenue Recognition
The reported results for 2019 and 2018 reflect the application of ASC 606, Revenue from Contracts with Customers (“ASC 606”), while the reported results for 2017 were prepared under the guidance of ASC 605, which is also referred to herein as “legacy GAAP” or the “previous guidance”.
Policy Elections and Practical Expedients Taken
Upon the Company’s adoption of ASC 606, it elected the following practical expedients:
Taxes assessed by a governmental authority that are both imposed on and concurrent with a specific revenue-producing transaction, that are collected by the Company from a customer, are excluded from revenue.
Shipping and handling costs associated with outbound freight after control over a product has transferred to a customer are accounted for as a fulfillment cost and are included in cost of product revenue.
Sales commissions and other incremental costs of obtaining contracts are expensed as incurred as the amortization periods are less than one year.
General
In accordance with ASC 606, revenue is recognized from the sale of products when a customer obtains control of promised products and services. The amount of revenue recognized reflects the consideration to which the Company expects to be entitled to receive in exchange for these products and services. A five-step model is utilized to achieve the core principle and includes the following steps: (1) identify the customer contract; (2) identify the contract’s performance obligations; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations; and (5) recognize revenue when the performance obligations are satisfied.
90
The following is a description of principal activities - separated by reportable segments - from which the Company generates its revenue. For more detailed information about reportable segments, see Note 20, Segment Information.
Pharmaceutical
The Company’s Pharmaceutical segment consists of revenue derived from sales of the Noden Products.
The agreement between Novartis and Noden DAC provided for various transition periods for development and commercialization activities relating to the Noden Products. For the period from July 1, 2016 through October 4, 2016, all of the Noden Products were distributed by Novartis under the terms of the Noden Purchase Agreement while transfer of the marketing authorization rights were pending. During this time, the Company presented revenue under the Novartis transition arrangement on a “net” basis and established a reserve for retroactive adjustment to the profit transfer with Novartis. As of the third quarter of 2018, Noden Pharma DAC completed the marketing authorization transfers for all territories.
In the United States, the duration of the profit transfer ran from July 1, 2016 through October 4, 2016. Beginning on October 5, 2016, Noden Pharma USA, Inc. distributed the Noden Products in the United States. At such time, the Company presented revenue for all sales in the United States on a “gross” basis, meaning product costs were reported separately and there was no fee to Novartis, and established a reserve for discounts and allowances further described below.
Initially, Novartis distributed the Noden Products on behalf of Noden DAC worldwide and Noden DAC received a profit transfer on such sales. Generally, the profit transfer to Noden DAC was defined as gross revenues less product cost and a low single-digit percentage fee to Novartis. The profit transfer terminated upon the transfer of the marketing authorization from Novartis to Noden DAC in each country. For the period from October 5, 2016 to August 31, 2017, Novartis continued to distribute the Noden Products outside of the United States. Beginning on September 1, 2017, Noden Pharma DAC began distributing the Noden Products to select countries outside the United States. Outside the United States, the profit transfer ended in the first quarter of 2018.
Except for the sales in certain countries outside of the United States preceding the final profit transfer that occurred in the first quarter of 2018, revenues of the Noden Products for the periods herein are presented on a gross basis.
Noden USA launched an authorized generic of Tekturna in the United States in March 2019.
The Pharmaceutical segment principally generates revenue from products sold to wholesalers and distributors. Customer orders are generally fulfilled within a few days of receipt resulting in minimal order backlog. Contractual performance obligations are usually limited to transfer of the product to the customer. The transfer occurs either upon shipment or upon receipt of the product in certain countries outside the United States after considering when the customer obtains control of the product. In addition, in some countries outside of the United States, the Company sells product on a consignment basis where control is not transferred until the customer resells the product to an end user. At these points, customers are able to direct the use of and obtain substantially all of the remaining benefits of the product.
Sales to customers are initially invoiced at contractual list prices. Payment terms are typically 30 to 90 days based on customary practice in each country. Revenue is reduced from the list price at the time of recognition for expected chargebacks, discounts, rebates, sales allowances and product returns, which are collectively referred to as gross-to-net adjustments. These reductions are attributed to various commercial agreements, managed healthcare organizations and government programs such as Medicare, Medicaid, and the 340B Drug Pricing Program containing various pricing implications such as mandatory discounts, pricing protection below wholesaler list price and other discounts when Medicare Part D beneficiaries are in the coverage gap. These various reductions in the transaction price have been estimated using either a most likely amount, in the case of prompt pay discounts, or expected value method for all other variable consideration and have been reflected as liabilities and are settled through cash payments, typically within time periods ranging from a few months to one year. Significant judgment is required in estimating gross-to-net adjustments considering legal interpretations of applicable laws and regulations, historical experience, payer channel mix, current contract prices under applicable programs, unbilled claims, processing time lags and inventory levels in the distribution channel. A description of gross-to-net adjustments are described below.
Customer Credits: The Company’s customers are offered various forms of consideration, including allowances, service fees and prompt payment discounts. The Company expects customers will earn prompt payment discounts and, therefore, the Company deducts the full amount of these discounts from total product sales when revenues are recognized. Service fees are also deducted from total product sales as they are earned.
91
Rebates and Discounts: Allowances for rebates include mandated discounts under the Medicaid Drug Rebate Program in the United States and mandated discounts in the European Union (“EU”) in markets where government-sponsored healthcare systems are the primary payers for healthcare. Rebates are amounts owed after the final dispensing of the product to a benefit plan participant and are based upon contractual agreements or legal requirements with public sector benefit providers. The accrual for rebates is based on negotiated discount rates and expected utilization as well as historical data. Estimates for expected utilization of rebates are based on data received from the customers. Rebates are generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters’ unpaid rebates. If actual future rebates vary from estimates, the Company may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Chargebacks: Chargebacks are discounts that occur when certain contracted customers, which currently consist primarily of group purchasing organizations, Public Health Service institutions, non-profit clinics, and Federal government entities purchasing via the Federal Supply Schedule, purchase directly from the Company’s wholesalers. Contracted customers generally purchase the product at a discounted price. The wholesalers, in turn, charges back to the Company the difference between the price initially paid by the wholesalers and the discounted price paid by the contracted customers. In addition to actual chargebacks received, the Company maintains an accrual for chargebacks based on the estimated contractual discounts on products sold for which the chargeback has not been billed. If actual future chargebacks vary from these estimates, the Company may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Medicare Part D Coverage Gap: Medicare Part D prescription drug benefit mandates manufacturers to fund 70% in 2019 and 50% in 2018 and 2017 of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Estimates for the expected Medicare Part D coverage gap are based on historical invoices received and in part from data received from the Company’s customers. Funding of the coverage gap is generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters. If actual future funding varies from estimates, the Company may need to adjust prior period accruals, which would affect revenue in the period of adjustment.
Co-payment Assistance: Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. The Company accrues a liability for co-payment assistance based on actual program participation and estimates of program redemption using data provided by third-party administrators.
Returns: Returns are generally estimated and recorded based on historical sales and returns information. Products that exhibit unusual sales or return patterns due to dating, competition or other marketing matters are specifically investigated and analyzed as part of the accounting for sales returns accruals.
Reserves for chargebacks, discounts, rebates, sales allowances and product returns are included within current liabilities in the Company’s Consolidated Balance Sheets.
For licenses that are bundled with other promises, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, up-front license fees. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition.
Medical Devices
The Medical Devices segment principally generates revenue from the sale and lease of the LENSAR® Laser System, which may include equipment, PIDs or consumables, procedure licenses, training, installation, warranty and maintenance agreements.
For bundled packages, the Company accounts for individual products and services separately if they are distinct - i.e. if a product or service is separately identifiable from other items in the bundled package and if the customer can benefit from it on its own or with other resources that are readily available to the customer. The LENSAR® Laser System, standard warranty, training and installation services are one performance obligation. All other elements are separate performance obligations. PIDs, procedure licenses, warranty and maintenance services are also sold on a stand-alone basis.
As the Company both sells and leases the LENSAR® Laser System, the consideration (including any discounts) is first allocated between lease and non-lease components and then allocated between the separate products and services based on their stand-alone selling prices. The stand-alone selling prices for the PIDs and procedure licenses are determined based on the prices at which the
92
Company separately sells the PIDs and procedure licenses. The LENSAR® Laser System and warranty stand-alone selling prices are determined using the expected cost plus a margin approach.
For LENSAR® Laser System sales, the Company recognizes Product revenue when a customer takes possession of the system. This usually occurs after the customer signs a contract, LENSAR installs the system, and LENSAR performs the requisite training for use of the system. For LENSAR® Laser System leases, the Company recognized Product revenue over the length of the lease in accordance with ASC Topic 840, through December 31, 2018 and recognizes Product revenue in accordance with ASC Topic 842, Leases, after January 1, 2019. For additional information regarding accounting for leases, see Note 8, Leases.
The LENSAR® Laser System requires both a consumable and a procedure license to perform each procedure. The Company recognizes Product revenue for PIDs when the customer takes possession of the PID. PIDs are sold by the case. The Company recognizes Product revenue for procedure licenses when a customer purchases a procedure license from the web portal. Typically, consideration for PIDs and procedure licenses is considered fixed consideration except for certain customer agreements that provide for tiered volume discount pricing, which is considered variable consideration.
The Company offers an extended warranty that provides additional services beyond the standard warranty. The Company recognizes Product revenue from the sale of extended warranties over the warranty period. Customers have the option of renewing the warranty period, which is considered a new and separate contract.
Income Generating Assets
For licenses of intellectual property, if the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenues from non-refundable, up-front fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license.
In January 2018, DFM, LLC, a wholly-owned subsidiary of the Company, granted an exclusive license related to certain Direct Flow Medical, Inc. assets in exchange for $0.5 million in cash and up to $2.0 million in royalty payments. The $0.5 million payment was accounted for in accordance with ASC 606 under which the full cash payment was recognized as revenue in the first quarter of 2018 as DFM, LLC had fulfilled its performance obligation under the agreement. In September 2019, the remaining assets of DFM, LLC were sold for $5.0 million.
Queen et al. Royalty Revenues
Under the Company’s license agreements related to the Queen et al. patents, the Company receives royalty payments based upon its licensees’ net sales of covered products. Royalties qualify for the sales-and-usage exemption under ASC 606 as (i) royalties are based strictly on the sales-and-usage by the licensee; and (ii) a license of intellectual property is the sole or predominant item to which such royalties relate. Based on this exemption, these royalties are earned under the terms of a license agreement in the period the products are sold by the Company's partner and the Company has a present right to payment. Generally, under these agreements, the Company receives royalty reports from its licensees approximately one quarter in arrears; that is, generally in the second month of the quarter after the licensee has sold the royalty-bearing product. The Company recognizes royalty revenues when it can reliably estimate such amounts and collectability is reasonably assured. Under this accounting policy, the royalty revenues the Company reports are not based upon estimates, and such royalty revenues are typically reported in the same period in which the Company receives payment from its licensees.
Although the last of the Queen et al. patents expired in December 2014, the Company has received royalties beyond expiration based on the terms of its licenses and its legal settlement. Under the terms of the legal settlement between Genentech, Inc. (“Genentech”) and the Company, the first quarter of 2016 was the last period for which Genentech paid royalties to the Company for Avastin®, Herceptin®, Xolair®, Perjeta® and Kadcyla®. Other products from the Queen et al. patent licenses, such as Tysabri®, entitle the Company to royalties following the expiration of its patents with respect to sales of licensed product manufactured prior to patent expiry in jurisdictions providing patent protection licenses. In November 2017, the Company was notified by Biogen, Inc. that product supply for Tysabri® that was manufactured prior to patent expiry, and for which the Company would receive royalties on, had been extinguished in the United States and was rapidly being reduced in other countries. As a result, royalties from product sales of Tysabri were substantially lower in 2018 and 2019 and no additional royalties are expected.
93
Royalty Rights - At Fair Value
The Company accounts for its investments in royalty rights at fair value with changes in fair value presented in earnings. The fair value of the investments in royalty rights is determined by using a discounted cash flow analysis related to the expected future cash flows to be received. These assets are classified as Level 3 assets within the fair value hierarchy, as the Company’s valuation estimates utilize significant unobservable inputs, including estimates as to the probability and timing of future sales of the related products. Transaction-related fees and costs are expensed as incurred.
The changes in the estimated fair value from investments in royalty rights along with cash receipts in each reporting period are presented together on the Company’s Consolidated Statements of Operations as a component of revenue under the caption, “Royalty rights - change in fair value.”
Realized gains and losses on royalty rights are recognized as they are earned and when collection is reasonably assured. Royalty Rights revenue is recognized over the respective contractual arrangement period. Critical estimates may include product demand and market growth assumptions, inventory target levels, product approval, pricing assumptions and the impact of competition from other branded or generic products. Factors that could cause a change in estimates of future cash flows include a change in estimated market size, a change in pricing strategy or reimbursement coverage, a delay in obtaining regulatory approval, a change in dosage of the product a change in the number of treatments and the entrants of new competitors or generic products. For each arrangement, the Company is entitled to royalty payments based on revenue generated by the net sales of the product.
Research and Development
The Company expenses research and development costs as incurred. Research and development expenses consist primarily of engineering, product development, clinical studies to develop and support the Company’s products, regulatory expenses, and other costs associated with products and technologies that are in development. Research and development expenses include employee compensation, including stock-based compensation, supplies, consulting, prototyping, testing, materials, travel expenses, and depreciation.
Foreign Currency Translation
The Company uses the U.S. dollar predominately as the functional currency of its foreign subsidiaries. For foreign subsidiaries where the U.S. dollar is the functional currency, gains and losses from remeasurement of foreign currency balances into U.S. dollars are included in the Consolidated Statements of Operations. The aggregate net (losses) gains resulting from foreign currency transactions and remeasurement of foreign currency balances into U.S. dollars that were included in the Consolidated Statements of Operations amounted to a loss of $0.5 million and $0.7 million for the years ended December 31, 2019 and 2018, respectively and a $0.1 million gain for the year ended December 31, 2017.
Comprehensive (Loss) Income
Comprehensive (loss) income comprises net (loss) income adjusted for other comprehensive (loss) income, using the specific identification method, which includes the changes in unrealized gains and losses on cash flow hedges and changes in unrealized gains and losses on the Company’s investments in available-for-sale securities, all net of tax, which are excluded from the Company’s net (loss) income.
Income Taxes
The provision for income taxes is determined using the asset and liability approach. Tax laws require items to be included in tax filings at different times than the items are reflected in the Consolidated Financial Statements. A current liability is recognized for the estimated taxes payable for the current year. Deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. Deferred taxes are adjusted for enacted changes in tax rates and tax laws. Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized.
The Company recognizes tax benefits from uncertain tax positions only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities, based on the technical merits of the position. The tax benefits recognized in the consolidated financial statements from such positions are then measured based on the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement. The Company adjusts the level of the liability to reflect any
94
subsequent changes in the relevant facts surrounding the uncertain positions. Any interest and penalties on uncertain tax positions are included within the tax provision.
The Tax Cuts and Job Act of 2017 (the “2017 Tax Act”) significantly changed the existing U.S. corporate income tax laws by, among other things, lowering the corporate tax rate (from a top rate of 35% to a flat rate of 21%), implementing elements of a territorial tax system, and imposing a one-time deemed repatriation transition tax on cumulative undistributed foreign earnings, for which the Company has not previously paid U.S. taxes. The Company recognized the estimated tax impact related to the revaluation of deferred tax assets and liabilities in its Consolidated Financial Statements for the year ended December 31, 2017. The ultimate impact did not differ materially from these provisional amounts after additional analysis, changes in interpretations and assumptions the Company made and additional regulatory guidance that was issued. The accounting was completed when the Company’s 2017 U.S. corporate income tax return was filed in 2018. The Company has made a policy election with respect to its treatment of potential global intangible low-taxed income (“GILTI”) to account for taxes on GILTI as a current-period expense as incurred.
Business Combination
The Company applies ASC 805, Business combinations (“ASC 805”), pursuant to which the cost of an acquisition is measured as the aggregate of the fair values at the date of exchange of the assets given, liabilities incurred, and equity instruments issued. The costs directly attributable to the acquisition are expensed as incurred. Identifiable assets, liabilities and contingent liabilities acquired or assumed are measured separately at their fair value as of the acquisition date, irrespective of the extent of any noncontrolling interests. The excess of the (i) the total of cost of acquisition, fair value of the noncontrolling interests and acquisition date fair value of any previously held equity interest in the acquiree over (ii) the fair value of the identifiable net assets of the acquiree is recorded as goodwill. If the cost of an acquisition is less than the fair value of the net assets of the subsidiary acquired, the difference is recognized directly in the Consolidated Statements of Operations as a bargain purchase gain.
Leases
General
In February 2016, the FASB issued ASU No. 2016-02, Leases, that supersedes ASC 840, Leases. Subsequently, the FASB issued several updates to ASU No. 2016-02, codified in ASC Topic 842 (“ASC 842”). The Company adopted ASC 842, Leases, on January 1, 2019 using the modified retrospective method for all leases not substantially completed as of the date of adoption. The reported results for the year ended December 31, 2019 reflect the application of ASC 842 guidance while the reported results for the years ended December 31, 2018 and 2017 were prepared under the guidance of ASC 840, which is also referred to herein as “legacy GAAP” or the “previous guidance”. The cumulative impact of the adoption of ASC 842 was not material, therefore, the Company did not record any adjustments to retained earnings. As a result of adopting ASC 842, the Company recorded operating lease right-of-use (“ROU”) assets of $2.1 million and operating lease liabilities of $2.1 million, primarily related to corporate office leases, based on the present value of the future lease payments on the date of adoption. Changes to lessor accounting focused on conforming with certain changes made to lessee accounting and the recently adopted revenue recognition guidance. The adoption of ASC 842 did not materially change how the Company accounts for lessor arrangements.
The Company determines if an arrangement is a lease or contains an embedded lease at inception if it contains the right to control the use of an identified asset under a leasing arrangement with an initial term greater than 12 months. The Company determines whether a contract conveys the right to control the use of an identified asset for a period of time if the contract contains both the right to obtain substantially all of the economic benefits from the use of the identified asset and the right to direct the use of the identified asset. The Company has lease arrangements with lease and non-lease components, which are accounted for separately.
Policy Elections and Practical Expedients Taken
For leases that commenced before the effective date of ASC 842, the Company elected the practical expedients to not reassess the following: (i) whether any expired or existing contracts contain leases; (ii) the lease classification for any expired or existing leases; and (iii) initial direct costs for any existing leases.
The Company adopted a policy of expensing short-term leases, defined as 12 months or less, as incurred.
The Company has a policy to exclude from the consideration in a lessor contract all taxes assessed by a governmental authority that are both imposed on and concurrent with a specific lease revenue-producing transaction and collected by the Company from a lessee.
95
Lessee arrangements
Lessee operating leases are included in Other assets, Accrued liabilities, and Other long-term liabilities in the Company’s Consolidated Balance Sheet. The Company does not have lessee financing leases.
Operating lease ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent its obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. The Company uses the implicit rate when readily determinable at lease inception. As most of the Company’s leases do not provide an implicit rate, the Company uses its incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments. The Company’s remaining lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. Lease expense for lease payments is recognized on a straight-line basis as operating expense in the Consolidated Statements of Operations over the lease term.
For lease arrangements with lease and non-lease components where the Company is the lessee, the Company separately accounts for lease and non-lease components, which consists primarily of taxes and common area maintenance costs. Non-lease components are expensed as incurred.
Lessor arrangements
The Company leases medical device equipment to customers in both operating lease and sales-type lease arrangements generated from its Medical Devices segment.
For sales-type leases, the Company derecognizes the carrying amount of the underlying asset and capitalizes the net investment in the lease, which consists of the total minimum lease payments receivable from the lessee, at lease inception. The Company does not estimate an unguaranteed residual value of the equipment at lease termination because the equipment transfers to the lessee upon completion of the lease. Selling profit or loss is recognized at lease inception. Initial direct costs are recognized as an expense, unless there is no selling profit or loss. If there is no selling profit or loss, initial direct costs are deferred and recognized over the lease term. The Company recognizes interest income from the lease receivable over the lease term in Interest and other income, net in the Consolidated Statements of Operations.
For operating leases, rental income is recognized on a straight-line basis over the lease term. The cost of customer-leased equipment is recorded within Property and equipment, net in the accompanying Consolidated Balance Sheets and depreciated over the equipment’s estimated useful life. Depreciation expense associated with the leased equipment under operating lease arrangements is reflected in Cost of product revenue in the accompanying Consolidated Statements of Operations. Some of the Company’s operating leases include a purchase option for the customer to purchase the leased asset at the end of the lease arrangement. The Company manages its risk on its investment in the equipment through pricing and the term of the leases. Lessees do not provide residual value guarantees on leased equipment. Equipment returned to the Company may be leased or sold to other customers. Initial direct costs are deferred and recognized over the lease term.
Leases are generally not cancellable until after an initial term and may or may not require the customer to purchase a minimum number of procedures and consumables throughout the contract term.
For lease arrangements with lease and non-lease components where the Company is the lessor, the Company allocates the contract’s transaction price to the lease and non-lease components on a relative standalone selling price basis using the Company’s best estimate of the standalone selling price of each distinct product or service in the contract. Allocation of the transaction price is determined at the inception of the lease arrangement. The Company’s leases primarily consist of leases with fixed lease payments. For those leases with variable lease payments, the variable lease payment is typically based upon use of the leased equipment or the purchase of procedure licenses and consumables used with the leased equipment. Non-lease components are accounted for under ASC 606. For additional information regarding ASC 606, see Note 19, Revenue from Contracts with Customers.
Adopted Accounting Pronouncements
Intangibles-Goodwill and Other
In January 2017, the FASB issued ASU 2017-04, Intangibles-Goodwill and Other: Simplifying the Test for Goodwill Impairment, to simplify the subsequent measurement of goodwill by eliminating step two from the goodwill impairment test. Under the amendments, an entity will recognize an impairment charge for the amount by which the carrying value exceeds the fair value.
96
The amendments are effective for fiscal years and interim periods within those years beginning after December 15, 2019 on a prospective basis and early adoption is permitted. Effective January 1, 2019, the Company adopted the requirements of ASU No. 2017-04. The adoption did not have an effect on the Consolidated Financial Statements on the adoption date and no adjustment to prior year Consolidated Financial Statements was required.
Recently Issued Accounting Pronouncements
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments - Credit Losses: Measurement of Credit Losses on Financial Instruments. The new guidance amends the impairment model to utilize an expected loss methodology in place of the currently used incurred loss methodology, which will result in more timely recognition of losses. ASU No. 2016-13 has an effective date of the fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. The Company does not expect this guidance to have a significant impact on its financial statements and related disclosures.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement. The new guidance modifies disclosure requirements related to fair value measurement. The amendments in ASU No. 2018-13 are effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2019. Implementation on a prospective or retrospective basis varies by specific disclosure requirement. Early adoption is permitted. The standard also allows for early adoption of any removed or modified disclosures upon issuance of ASU No. 2018-13 while delaying adoption of the additional disclosures until their effective date. The Company does not expect this guidance to have a significant impact on its financial statements and related disclosures.
In August 2018, the FASB issued ASU No. 2018-15, Intangibles-Goodwill and Other-Internal-Use Software. The new guidance reduces complexity for the accounting for costs of implementing a cloud computing service arrangement and aligns the requirements for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software (and hosting arrangements that include an internal use software license). For public companies, the amendments in ASU No. 2018-15 are effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2019, with early adoption permitted. Implementation should be applied either retrospectively or prospectively to all implementation costs incurred after the date of adoption. The Company does not expect this guidance to have a significant impact on its financial statements and related disclosures.
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes: Simplifying the Accounting for Income Taxes. This guidance removes certain exceptions related to the approach for intraperiod tax allocation, the methodology for calculating income taxes in an interim period, and the recognition of deferred tax liabilities for outside basis differences. This guidance also clarifies and simplifies other areas of ASC 740. This ASU will be effective for public companies for fiscal years, and interim periods within those fiscal years beginning after December 15, 2020. Early adoption is permitted. Certain amendments in this update must be applied on a prospective basis, certain amendments must be applied on a retrospective basis, and certain amendments must be applied on a modified retrospective basis through a cumulative-effect adjustment to retained earnings/(deficit) in the period of adoption. The Company is currently evaluating the impact this ASU will have on the Company’s financial statements and related disclosures as well as the timing of adoption.
3. Investment in Evofem Biosciences, Inc.
On April 10, 2019, the Company entered into a securities purchase agreement with Evofem and two other purchasers, pursuant to which the Company purchased $60.0 million of Evofem securities in a private placement. The transaction was structured in two tranches.
The first tranche closed on April 11, 2019, pursuant to which the Company invested $30.0 million to purchase 6,666,667 shares of Evofem common stock at $4.50 per share and was also issued warrants to purchase up to 1,666,667 shares of Evofem common stock. The warrants are exercisable beginning six months after the issuance date for a period of seven years from the issuance date at an exercise price of $6.38 per share.
The second tranche closed on June 10, 2019, pursuant to which the Company invested an additional $30.0 million to purchase an additional 6,666,667 shares of Evofem common stock at $4.50 per share and was also issued warrants to purchase up to an additional 1,666,667 shares of Evofem common stock with the same terms as the warrants issued in the first tranche. Following the closing of the second tranche, the Company has a right to appoint one member to Evofem’s board of directors and has a limited right to have one board observer participate in Evofem board meetings. In December 2019, the Company’s representatives
97
resigned from these positions. Since that time, the Company has elected not to appoint a director or board observer to the Evofem board of directors but retains the right to do so.
The Company has registration rights on customary terms for all Evofem shares issued under the securities purchase agreement, including the shares underlying the warrants.
As of December 31, 2019, the Company owned approximately 28% of Evofem’s common stock. The Company’s investment in Evofem qualifies for equity method accounting given its percentage ownership in Evofem and the ability to exercise significant influence. The Company elected the fair value method to account for its investment in Evofem as it believes it better reflects economic reality, the financial reporting of the investment and the current value of the asset. Changes in fair value of the Evofem equity investment are presented in Non-operating income (expense), net on the Consolidated Statement of Operations. Because the mark to market valuation will occur at the end of each quarterly reporting period, changes in fair value will vary based upon the volatility of the stock price. The Evofem equity investment is presented on the Consolidated Balance Sheet as an Investment in equity affiliate and reflects the fair value of the equity investment at the end of the reporting period.
For the year ended December 31, 2019, the Company had an unrealized gain of $36.4 million on its investment in Evofem, of which $31.6 million was related to Evofem common stock and $4.8 million was related to Evofem warrants.
The latest Evofem financial statements can be found on their corporate website at www.evofem.com or filed with the SEC at www.sec.gov.
4. Cash and Cash Equivalents
As of December 31, 2019 and 2018, the Company had invested its excess cash balances primarily in cash and money market funds. The fair values of cash equivalents approximate their carrying values due to the short-term nature of such financial instruments.
The following table summarizes the Company’s cash and cash equivalents by significant investment category reported as cash and cash equivalents as of December 31, 2019 and 2018:
Reported as: | ||||||||||||
(in thousands) | Amortized Cost | Estimated Fair Value | Cash and Cash Equivalents | |||||||||
December 31, 2019 | ||||||||||||
Cash | $ | 62,187 | $ | 62,187 | $ | 62,187 | ||||||
Money market funds | 131,264 | 131,264 | 131,264 | |||||||||
Total | $ | 193,451 | $ | 193,451 | $ | 193,451 | ||||||
December 31, 2018 | ||||||||||||
Cash | $ | 167,871 | $ | 167,871 | $ | 167,871 | ||||||
Money market funds | 226,719 | 226,719 | 226,719 | |||||||||
Total | $ | 394,590 | $ | 394,590 | $ | 394,590 | ||||||
The Company recognized approximately $0.8 million and $0.1 million, respectively, of gains on sales of available-for-sale securities in the years ended December 31, 2018 and 2017, respectively. As of December 31, 2019 and 2018 the Company did not have any available-for-sale securities.
98
5. Inventories
Inventories consisted of the following:
December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Raw materials | $ | 24,727 | $ | 6,214 | ||||
Work in process | 3,700 | 549 | ||||||
Finished goods | 11,346 | 12,179 | ||||||
Total inventories | $ | 39,773 | $ | 18,942 | ||||
As of December 31, 2019 and 2018, the Company deferred approximately $0.1 million and $0.5 million, respectively, of costs associated with inventory transfers made under the Company’s third-party logistic provider service arrangement. These costs have been recorded as Prepaid and other current assets on the Company’s Consolidated Balance Sheets as of December 31, 2019 and 2018. The Company will recognize the cost of product sold as inventory is transferred from its third-party logistics provider to the Company’s customers.
During the years ended December 31, 2019 and 2018, the Company recognized reductions in the inventory reserve of $0.3 million and $1.2 million. During the year ended December 31, 2017, the Company recognized an inventory write-down of $2.0 million, related to the Noden Products that the Company determined it would not be able to sell prior to their expiration.
6. Fair Value Measurements
Assets/Liabilities Measured and Recorded at Fair Value on a Recurring Basis
The following table presents the fair value of the Company’s financial instruments measured at fair value on a recurring basis by level within the valuation hierarchy, as discussed in Note 2, Summary of Significant Accounting Policies:
December 31, 2019 | December 31, 2018 | |||||||||||||||||||||||||||||||
(in thousands) | Level 1 | Level 2 | Level 3 | Total | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||||||||||
Financial assets: | ||||||||||||||||||||||||||||||||
Money market funds | $ | 131,264 | $ | — | $ | — | $ | 131,264 | $ | 226,719 | $ | — | $ | — | $ | 226,719 | ||||||||||||||||
Corporate securities 1 | 82,267 | — | — | 82,267 | — | — | — | — | ||||||||||||||||||||||||
Warrants 2 | — | 14,152 | — | 14,152 | — | 62 | — | 62 | ||||||||||||||||||||||||
Royalty rights - at fair value | — | — | 266,196 | 266,196 | — | — | 376,510 | 376,510 | ||||||||||||||||||||||||
Total | $ | 213,531 | $ | 14,152 | $ | 266,196 | $ | 493,879 | $ | 226,719 | $ | 62 | $ | 376,510 | $ | 603,291 | ||||||||||||||||
Financial liabilities: | ||||||||||||||||||||||||||||||||
Contingent consideration, current 3 | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | 1,071 | $ | 1,071 | ||||||||||||||||
Total | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | 1,071 | $ | 1,071 | ||||||||||||||||
___________________
1 Corporate securities are classified as “Investment in equity affiliate” on the December 31, 2019 Consolidated Balance Sheet.
2 Warrants are included in “Other assets” on the December 31, 2019 and 2018 Consolidated Balance Sheets.
3 Contingent consideration, current is included in “Accrued liabilities” on the December 31, 2018 Consolidated Balance Sheet.
There have been no transfers between levels during the periods presented in the table above. The Company recognizes transfers between levels on the date of the event or change in circumstances that caused the transfer.
Money Market Funds - The fair values of cash equivalents approximate their carrying values due to the short-term nature of such financial instruments.
99
Corporate Securities - Corporate securities consists of common stock shares of Evofem, a clinical-stage biopharmaceutical company listed on Nasdaq. For additional information on the Evofem investment, see Note 3, Investment in Evofem.
Warrants - Warrants consist of rights to purchase shares of common stock in Evofem and CareView, see Note 3, Investment in Evofem, and Note 7, Notes and Other Long-Term Receivables. The fair value of the warrants is estimated using recently quoted market prices of the underlying equity security and the Black-Scholes option pricing model.
Royalty Rights - At Fair Value
Assertio (Depomed) Royalty Agreement
On October 18, 2013, the Company entered into the Royalty Purchase and Sale Agreement (the “Assertio Royalty Agreement”) with Assertio Therapeutics, Inc. (formerly known as Depomed, Inc.), and Depo DR Sub, LLC (together, “Assertio”), whereby the Company acquired the rights to receive royalties and milestones payable on sales of five Type 2 diabetes products licensed by Assertio in exchange for a $240.5 million cash payment. Total consideration was $241.3 million, which was comprised of the $240.5 million cash payment to Assertio and $0.8 million in transaction costs.
The rights acquired include Assertio’s royalty and milestone payments accruing from and after October 1, 2013: (a) from Santarus, Inc., which was subsequently acquired by Salix Pharmaceuticals, Inc., which itself was acquired by Valeant Pharmaceuticals International, Inc. (“Valeant”), which, in July 2018, changed its name to Bausch Health Companies Inc. (“Bausch Health”) with respect to sales of Glumetza (metformin HCL extended-release tablets) in the United States; (b) from Merck & Co., Inc. with respect to sales of Janumet® XR (sitagliptin and metformin HCL extended-release tablets); (c) from Janssen Pharmaceutica N.V. with respect to potential future development milestones and sales of its approved fixed-dose combination of Invokana® (canagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor) and extended-release metformin tablets, marketed as Invokamet XR®; (d) from Boehringer Ingelheim and Eli Lilly and Company with respect to potential future development milestones and sales of the investigational fixed-dose combinations of drugs and extended-release metformin subject to Assertio’s license agreement with Boehringer Ingelheim, including its approved products, Jentadueto XR® and Synjardy XR®; and (e) from Bausch Health for sales of extended-release metformin tablets in Korea and Canada, respectively.
In February 2013, a generic equivalent to Glumetza was approved by the U.S. Food and Drug Administration (“FDA”) and in August 2016, two additional generic equivalents to Glumetza were approved by the FDA. In February 2016, Lupin Pharmaceuticals, Inc., in August 2017, Teva Pharmaceutical Industries Ltd., and in July 2018, Sun Pharmaceutical, Inc. (“Sun”) each launched a generic equivalent approved product. In May 2017, the Company received notification that a subsidiary of Valeant had launched an authorized generic equivalent product in February 2017, and the Company received royalties on such authorized generic equivalent product under the same terms as the branded Glumetza product, retroactive to February 2017. The Company continues to monitor whether the generic competition further affects sales of Glumetza and thus royalties on such sales paid to the Company, and the impact of the launched authorized generic equivalent. Due to the uncertainty around Bausch Health’s marketing and pricing strategy, as well as Sun’s recently launched generic product and limited historical demand data after generic market entrance, the Company may need to further evaluate future cash flows in the event of more rapid reduction or increase in market share of Glumetza and its authorized generic equivalent product and/or a further erosion in net pricing.
The Company determined that its royalty purchase interest in Depo DR Sub, LLC represented a variable interest in a variable interest entity. However, the Company did not have the power to direct the activities of Depo DR Sub, LLC that most significantly impact Depo DR Sub, LLC’s economic performance and was not the primary beneficiary of Depo DR Sub, LLC; therefore, Depo DR Sub, LLC was not subject to consolidation by the Company.
On August 2, 2018, PDL Investment Holding, LLC (“PDLIH”), a wholly-owned subsidiary of the Company and assignee from the Company under the Assertio Royalty Agreement, entered into an amendment to the Assertio Royalty Agreement with Assertio. Pursuant to the amendment, PDLIH purchased all of Assertio’s remaining interests in royalty and milestone payments payable on sales of Type 2 diabetes products licensed by Assertio for $20.0 million. Prior to the amendment, the Assertio Royalty Agreement provided that the Company would have received all royalty and milestone payments due under license agreements between Assertio and its licensees until the Company received payments equal to two times the cash payment it made to Assertio, or approximately $481.0 million, after which all net payments received by Assertio would have been shared equally between the Company and Assertio. Following the amendment, the Assertio Royalty Agreement provides that the Company will receive all royalty and milestone payments due under the license agreements between Assertio and its licensees. The Company has elected to continue to follow the fair value option and carry the financial asset at fair value.
100
The Assertio Royalty Agreement terminates on the third anniversary following the date upon which the later of the following occurs: (a) October 25, 2021, or (b) at such time as no royalty payments remain payable under any license agreement and each of the license agreements has expired by its terms.
As of December 31, 2018, in conjunction with the amendment described above, the Company was provided the power to direct the activities of Depo DR Sub, LLC and is the primary beneficiary of Depo DR Sub, LLC; therefore, Depo DR Sub, LLC is subject to consolidation by the Company. As of December 31, 2019 and 2018, Depo DR Sub, LLC did not have any assets or liabilities of value for consolidation with the Company.
The financial asset acquired represents a single unit of accounting. This financial asset is classified as a Level 3 asset within the fair value hierarchy, as the Company’s valuation utilized significant unobservable inputs, including estimates as to the probability and timing of future commercialization for products not yet approved by regulatory agencies outside of the United States. The estimated fair value of the financial asset acquired was determined by using a discounted cash flow analysis related to the expected future cash flows to be generated by each licensed product. The discounted cash flows are based upon expected royalties from sales of licensed products over approximately a nine-year period. The estimated fair value of the asset is subject to variation should those cash flows vary significantly from the Company’s estimates. The Company periodically assesses the expected future cash flows and to the extent such payments are greater or less than its initial estimates, or the timing of such payments is materially different than the original estimates, the Company will adjust the estimated fair value of the asset. A third-party expert is engaged to assist management with the development of its estimate of the expected future cash flows, when deemed necessary. Should the expected royalties increase or decrease by 2.5%, the fair value of the asset could increase or decrease by $5.5 million, respectively. Significant judgment is required in selecting appropriate discount rates. The discount rates utilized range from 10% to 24%. At December 31, 2019, an evaluation was performed to assess those rates and general market conditions potentially affecting the fair market value of the financial asset. Should these discount rates increase or decrease by 2.5%, the fair value of the asset could decrease by $17.5 million or increase by $20.5 million, respectively.
In February 2016, at the Company’s request and pursuant to the Assertio Royalty Agreement, Assertio exercised its audit right with respect to Glumetza royalties. The independent auditor engaged to perform the royalty audit completed it in July 2017, and based upon the results of the audit, Assertio, on behalf of the Company, filed a lawsuit on September 7, 2017, against Valeant and one of its subsidiaries, claiming damages for unpaid royalties, fees and interest. Valeant (now Bausch Health), Assertio and the Company entered into a settlement agreement on October 27, 2017 whereby the parties agreed to dismiss the litigation, with prejudice, and Valeant agreed to pay to Assertio $13.0 million. The full amount of the settlement payment was transferred to the Company under the terms of the Assertio Royalty Agreement in November 2017. In October 2018, PDL submitted notice of its intent to exercise its audit right under the Assertio Royalty Agreement with respect to the period beginning January 1, 2016 and ending December 31, 2018. No material adjustments were identified in connection with this audit.
As of December 31, 2019, the Company’s discounted cash flow analysis reflects its expectations as to the amount and timing of future cash flows up to the valuation date for the above described royalty streams.
On May 31, 2016, the Company obtained a notification indicating that the FDA approved Jentadueto XR for use in patients with Type 2 diabetes. In June 2016, the Company received a $6.0 million FDA approval milestone pursuant to the terms of the Assertio Royalty Agreement. The product approval was earlier than initially expected. Based on the FDA approval and anticipated timing of the product launch, the Company adjusted the timing of future cash flows and discount rate used in the discounted cash flow model at June 30, 2016. At year-end 2017, management re-evaluated, with assistance of a third-party expert, the cash flow assumptions for Jentadueto XR and revised the discounted cash flow model. As of December 31, 2019, the Company’s discounted cash flow analysis reflects its expectations as to the amount and timing of future cash flows up to the valuation date.
On September 21, 2016, the Company obtained a notification indicating that the FDA approved Invokamet XR for use in patients with Type 2 diabetes. The product approval triggered a $5.0 million approval milestone payment to the Company pursuant to the terms of the Assertio Royalty Agreement. Based on the FDA approval and timing of the product launch, the Company adjusted the timing of future cash flows and discount rate used in the discounted cash flow model at December 31, 2017.
On December 13, 2016, the Company obtained a notification indicating that the FDA approved Synjardy XR for use in patients with Type 2 diabetes. The product approval triggered a $6.0 million approval milestone payment to the Company pursuant to the terms of the Assertio Royalty Agreement. Based on the FDA approval and the April 2017 launch of Synjardy XR by Boehringer Ingelheim, the Company adjusted the timing of future cash flows and discount rate used in the discounted cash flow model at December 31, 2017.
101
In the fourth quarter of 2019, management re-evaluated, with assistance of a third-party expert, the market share data, the gross-to-net revenue adjustment assumptions and Glumetza demand data and re-evaluated the assumptions, including the expected ex-U.S. launch dates, underlying the fair values of the non-Glumetza Type 2 extended release diabetes products comprising the Assertio royalty asset portfolio. These data and assumptions are based on available but limited information. Key findings from the third-party study included: an anticipated decrease in the Glumetza net sales forecast due to an accelerated shift in the channel mix resulting in a substantial decline in net selling prices, particularly in the fourth quarter of 2019 and beyond, as previously announced by Bausch Health, and the delayed launch dates of the extended release products in the Assertio royalty asset portfolio outside of the United States. As a result of this analysis, the Company wrote down the fair value of the Assertio asset by $46.3 million.
As of December 31, 2019, the fair value of the asset acquired as reported in the Company’s Consolidated Balance Sheet was $218.7 million and the maximum loss exposure was $218.7 million.
Viscogliosi Brothers Royalty Agreement
On June 26, 2014, the Company entered into a Royalty Purchase and Sale Agreement (the “VB Royalty Agreement”) with Viscogliosi Brothers, LLC (“VB”), whereby VB conveyed to the Company the right to receive royalties payable on sales of a spinal implant that has received pre-market approval from the FDA held by VB and commercialized by Paradigm Spine, LLC (“Paradigm Spine”), in exchange for a $15.5 million cash payment, less fees. Paradigm Spine was acquired in March 2019 by RTI Surgical Holdings, Inc.
The royalty rights acquired include royalties accruing from and after April 1, 2014. Under the terms of the VB Royalty Agreement, the Company receives all royalty payments due to VB pursuant to certain technology transfer agreements between VB and Paradigm Spine until the Company has received payments equal to 2.3 times the cash payment made to VB, after which all rights to receive royalties will be returned to VB. VB’s ability to repurchase the royalty right for a specified amount expired on June 26, 2018.
The estimated fair value of the royalty rights at December 31, 2019, was determined by using a discounted cash flow analysis related to the expected future cash flows to be received. This asset is classified as a Level 3 asset, as the Company’s valuation utilized significant unobservable inputs, including estimates as to the probability and timing of future sales of the licensed product. The discounted cash flow was based upon expected royalties from sales of licensed product over approximately a ten-year period. The estimated fair value of the asset is subject to variation should those cash flows vary significantly from the Company’s estimates. The Company periodically assesses the expected future cash flows and to the extent such payments are greater or less than its initial estimates, or the timing of such payments is materially different than the original estimates, the Company will adjust the estimated fair value of the asset. A third-party expert is engaged to assist management with the development of its estimate of the expected future cash flows, when deemed necessary. Should the expected royalties increase or decrease by 2.5%, the fair value of the asset could increase or decrease by $0.3 million, respectively. Significant judgment is required in selecting the appropriate discount rate. The discount rate utilized was 15.0%. Should this discount rate increase or decrease by 2.5%, the fair value of this asset could decrease by $1.3 million or increase by $1.6 million, respectively.
As of December 31, 2019, the Company’s discounted cash flow analysis reflects its expectations as to the amount and timing of future cash flows up to the valuation date.
As of December 31, 2019, the fair value of the asset acquired as reported in the Company’s Consolidated Balance Sheet was $13.6 million and the maximum loss exposure was $13.6 million.
University of Michigan Royalty Agreement
On November 6, 2014, the Company acquired a portion of all royalty payments of the U-M worldwide royalty interest in Cerdelga® (eliglustat) for $65.6 million pursuant to the Royalty Purchase and Sale Agreement with U-M (the “U-M Royalty Agreement”). Under the terms of the U-M Royalty Agreement, the Company receives 75% of all royalty payments due under the U-M license agreement with Genzyme Corporation, a Sanofi company (“Genzyme”), until expiration of the licensed patents, excluding any patent term extension. Cerdelga, an oral therapy for adult patients with Gaucher disease type 1, was developed by Genzyme. Cerdelga was approved in the United States in August 2014, in the EU in January 2015, and in Japan in March 2015. In addition, marketing applications for Cerdelga are under review by other regulatory authorities. While marketing applications have been approved in the United States, the EU and Japan, national pricing and reimbursement decisions are delayed in some countries.
102
The estimated fair value of the royalty right at December 31, 2019, was determined by using a discounted cash flow analysis related to the expected future cash flows to be received. This asset is classified as a Level 3 asset, as the Company’s valuation utilized significant unobservable inputs, including estimates as to the probability and timing of future sales of the licensed product. The discounted cash flow was based upon expected royalties from sales of licensed product over approximately a three-year period. Based on the results of the Company’s analysis, which considered input from a third-party expert and the variance between the Company’s forecast model and actual results, the Company wrote down the fair value of the royalty asset by $3.1 million in the third quarter ended September 30, 2019. The estimated fair value of the asset is subject to variation should those cash flows vary significantly from the Company’s estimates. An evaluation of those estimates, discount rate utilized and general market conditions affecting fair market value is performed in each reporting period. A third-party expert is engaged to assist management with the development of its estimate of the expected future cash flows, when deemed necessary. Should the expected royalties increase or decrease by 2.5%, the fair value of the asset could increase or decrease by $0.5 million, respectively.
Significant judgment is required in selecting the appropriate discount rate. The discount rate utilized was approximately 12.8%. Should this discount rate increase or decrease by 2.5%, the fair value of this asset could decrease or increase by $0.6 million. As of December 31, 2019, the Company’s discounted cash flow analysis reflects its expectations as to the amount and timing of future cash flows.
As of December 31, 2019, the fair value of the asset acquired as reported in the Company’s Consolidated Balance Sheet was $20.4 million and the maximum loss exposure was $20.4 million.
ARIAD Royalty Agreement
On July 28, 2015, the Company entered into the revenue interest assignment agreement (the “ARIAD Royalty Agreement”) with ARIAD, whereby the Company acquired the rights to receive royalties from ARIAD’s net revenues generated by the sale, distribution or other use of Iclusig® (ponatinib), a cancer medicine for the treatment of adult patients with chronic myeloid leukemia, in exchange for up to $200.0 million in cash payments. The purchase price of $100.0 million was payable in two tranches of $50.0 million each, with the first tranche having been funded on July 28, 2015 and the second tranche having been funded on July 28, 2016. Upon the occurrence of certain events, including a change of control of ARIAD, the Company had the right to require ARIAD to repurchase the royalty rights for a specified amount. The Company elected the fair value option to account for the hybrid instrument in its entirety. Any embedded derivative shall not be separated from the host contract. The asset acquired pursuant to the ARIAD Royalty Agreement represents a single unit of accounting.
In February 2017, Takeda Pharmaceutical Company Limited (“Takeda”) acquired ARIAD and the Company exercised its put option on the same day, which resulted in an obligation by Takeda to pay the Company a 1.2x multiple of the $100.0 million funded by the Company under the ARIAD Royalty Agreement, less royalty payments already received by the Company.
On March 30, 2017, Takeda fulfilled its obligations under the put option and paid the Company the repurchase price of $108.2 million for the royalty rights under the ARIAD Royalty Agreement.
AcelRx Royalty Agreement
On September 18, 2015, the Company entered into a royalty interest assignment agreement (the “AcelRx Royalty Agreement”) with ARPI LLC, a wholly-owned subsidiary of AcelRx Pharmaceuticals, Inc. (“AcelRx”), whereby the Company acquired the rights to receive a portion of the royalties and certain milestone payments on sales of Zalviso® (sufentanil sublingual tablet system) in the EU, Switzerland and Australia by AcelRx’s commercial partner, Grünenthal, in exchange for a $65.0 million cash payment. Under the terms of the AcelRx Royalty Agreement, the Company receives 75% of all royalty payments and 80% of the first four commercial milestone payments due under AcelRx’s license agreement with Grünenthal until the earlier to occur of (i) receipt by the Company of payments equal to three times the cash payments made to AcelRx and (ii) the expiration of the licensed patents. Zalviso received marketing approval by the European Commission in September 2015. Grünenthal launched Zalviso in the second quarter of 2016 and the Company started to receive royalties in the third quarter of 2016.
As of December 31, 2019 and 2018, the Company determined that its royalty rights under the AcelRx Royalty Agreement represented a variable interest in a variable interest entity. However, the Company does not have the power to direct the activities of ARPI LLC that most significantly impact ARPI LLC’s economic performance and is not the primary beneficiary of ARPI LLC; therefore, ARPI LLC is not subject to consolidation by the Company.
Due to the slower than expected adoption of the product since its initial launch relative to the Company’s estimates and the increased variance noted between the Company’s forecast model and actual results in the three months ended June 30, 2019, the Company utilized a third-party expert in the second quarter of 2019 to reassess the market and expectations for the Zalviso
103
product. Key findings from the third-party study included: the post-surgical PCA (Patient-Controlled Analgesia) market being smaller than previously forecasted; the higher price of the product relative to alternative therapies, the product not being used as a replacement for systemic opioids and the design of the delivery device, which is pre-filled for up to three days of treatment, which limited its use for procedures with anticipated shorter recovery times. Based on this analysis and the impact to the projected sales-based royalties and milestones, the Company wrote down the fair value of the royalty asset by $60.0 million in the second quarter of 2019.
The estimated fair value of the royalty right at December 31, 2019, was determined by using a discounted cash flow analysis related to the expected future cash flows to be received. This asset is classified as a Level 3 asset, as the Company’s valuation utilized significant unobservable inputs, including estimates as to the probability and timing of future sales of the licensed product. The discounted cash flow was based upon expected royalties from sales of licensed product over approximately a thirteen-year period. The estimated fair value of the asset is subject to variation should those cash flows vary significantly from the Company’s estimates. An evaluation of those estimates, discount rate utilized and general market conditions affecting fair market valuation is performed for each reporting period. A third-party expert is engaged to assist management with the development of its estimate of the expected future cash flows, when deemed necessary. Should the expected royalties increase or decrease by 2.5%, the fair value of the asset could increase or decrease by $0.3 million, respectively. Significant judgment is required in selecting the appropriate discount rate. The discount rate utilized was approximately 13.4%. Should this discount rate increase or decrease by 2.5%, the fair value of this asset could decrease by $1.2 million or increase by $1.4 million, respectively. As of December 31, 2019, the Company’s discounted cash flow analysis reflects its expectations as to the amount and timing of future cash flows up to the valuation date.
As of December 31, 2019, the fair value of the asset acquired as reported in the Company’s Consolidated Balance Sheet was $13.0 million and the maximum loss exposure was $13.0 million.
Kybella Royalty Agreement
On July 8, 2016, the Company entered into a royalty purchase and sales agreement with an individual, whereby the Company acquired that individual’s rights to receive certain royalties on sales of KYBELLA® by Allergan plc in exchange for a $9.5 million cash payment and up to $1.0 million in future milestone payments based upon product sales targets. The Company started to receive royalty payments during the third quarter of 2016.
The estimated fair value of the royalty right at December 31, 2019, was determined by using a discounted cash flow analysis related to the expected future cash flows to be received. This asset is classified as a Level 3 asset, as the Company’s valuation utilized significant unobservable inputs, including estimates as to the probability and timing of future sales of the licensed product. The discounted cash flow was based upon expected royalties from sales of a licensed product over approximately a six-year period. The estimated fair value of the asset is subject to variation should those cash flows vary significantly from the Company’s estimates. An evaluation of those estimates, discount rate utilized and general market conditions affecting fair market value is performed in each reporting period. A third-party expert is engaged to assist management with the development of its estimate of the expected future cash flows, when deemed necessary. Should the expected royalties increase or decrease by 2.5%, the fair value of the asset could increase or decrease by less than $0.1 million, respectively. Significant judgment is required in selecting the appropriate discount rate. The discount rate utilized was approximately 14.4%. Should this discount rate increase or decrease by 2.5%, the fair value of this asset could decrease or increase by less than $0.1 million, respectively.
As of December 31, 2019, the Company’s discounted cash flow analysis reflects its expectations as to the amount and timing of future cash flows up to the valuation date.
As of December 31, 2019, the fair value of the asset acquired as reported in the Company’s Consolidated Balance Sheet was $0.6 million and the maximum loss exposure was $0.6 million.
104
The following tables summarize the changes in Level 3 Royalty Right Assets and the gains and losses included in earnings for the year ended December 31, 2019:
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) - Royalty Rights Assets | ||||||||||
(in thousands) | Royalty Rights - At Fair Value | |||||||||
Fair value as of December 31, 2018 | $ | 376,510 | ||||||||
Total net change in fair value for the period | ||||||||||
Change in fair value of royalty rights - at fair value | $ | (31,042 | ) | |||||||
Proceeds from royalty rights - at fair value | $ | (79,272 | ) | |||||||
Total net change in fair value for the period | (110,314 | ) | ||||||||
Fair value as of December 31, 2019 | $ | 266,196 | ||||||||
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) - Royalty Rights Assets | ||||||||||||
Fair Value as of | Royalty Rights - | Fair Value as of | ||||||||||
(in thousands) | December 31, 2018 | Change in Fair Value | December 31, 2019 | |||||||||
Assertio | $ | 264,371 | $ | (45,699 | ) | $ | 218,672 | |||||
VB | 14,108 | (518 | ) | 13,590 | ||||||||
U-M | 25,595 | (5,197 | ) | 20,398 | ||||||||
AcelRx | 70,380 | (57,428 | ) | 12,952 | ||||||||
KYBELLA | 2,056 | (1,472 | ) | 584 | ||||||||
$ | 376,510 | $ | (110,314 | ) | $ | 266,196 | ||||||
The following table summarizes the changes in Level 3 liabilities and the gains and losses included in earnings for the year ended December 31, 2019:
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) - Liabilities | |||||||
(in thousands) | Contingent Consideration | ||||||
Fair value as of December 31, 2018 | $ | (1,071 | ) | ||||
Settlement of financial instrument 1 | 1,071 | ||||||
Fair value as of December 31, 2019 | $ | — | |||||
___________________
1 Represents the final conversion consideration and earn out liability for the LENSAR acquisition of assets from Precision Eye Services (“PES”).
105
Gains and losses from changes in Level 3 assets included in earnings for each period are presented in “Royalty rights - change in fair value” and gains and losses from changes in Level 3 liabilities included in earnings for each period are presented in “Change in fair value of anniversary payment and contingent consideration” as follows:
Year Ended December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Total change in fair value for the period included in earnings for royalty right assets held at the end of the reporting period | $ | (31,042 | ) | $ | 85,256 | |||
Total change in fair value for the period included in earnings for liabilities held at the end of the reporting period | $ | — | $ | 41,631 | ||||
Assets/Liabilities Measured and Recorded at Fair Value on a Nonrecurring Basis
The Company remeasures the fair value of certain assets and liabilities upon the occurrence of certain events. Such assets consist of long-lived assets, including property and equipment and intangible assets and the shares of Alphaeon Class A common stock, received in connection with the loans made to LENSAR by the Company prior to its acquisition of LENSAR.
During the year ended December 31, 2019, the Company recorded an impairment charge of $22.5 million for the Noden intangible assets given the Company’s monetization strategy and updated forecasts for Noden. As a result of this impairment charge, which was based on the estimated fair value of the assets, the remaining carrying value of these intangible assets was determined to be $10.1 million. During the three months ended June 30, 2018, the Company recorded an impairment charge of $152.3 million for the Noden intangible assets related to the increased probability of a generic form of aliskiren being launched in the United States. As a result of this impairment charge, which was based on the estimated fair value of the assets, the remaining carrying value of these intangible assets was determined to be $40.1 million. These intangible asset fair value calculations included level 3 inputs. For additional information on the Noden intangible asset, see Note 10, Intangible Assets.
The Company’s carrying value of the 1.7 million shares of Alphaeon common stock as of both December 31, 2019 and December 31, 2018 is $6.6 million based on an estimated per share value of $3.84, which was established by a valuation performed when the shares were acquired. The value of the Company’s investment in Alphaeon is not readily determinable as Alphaeon’s shares are not publicly traded. The Company evaluates the fair value of this investment by performing a qualitative assessment each reporting period. If the results of this qualitative assessment indicate that the fair value is less than the carrying value, the investment is written down to its fair value. There have been no such write downs since the Company acquired these shares. This investment is included in Other long-term assets. For additional information on the Alphaeon investment, see Note 7, Notes and Other Long-Term Receivables.
Assets/Liabilities Not Subject to Fair Value Recognition
The following tables present the fair value of assets and liabilities not subject to fair value recognition by level within the valuation hierarchy:
December 31, 2019 | December 31, 2018 | |||||||||||||||||||||||
(in thousands) | Carrying Value | Fair Value Level 2 | Fair Value Level 3 | Carrying Value | Fair Value Level 2 | Fair Value Level 3 | ||||||||||||||||||
Assets: | ||||||||||||||||||||||||
Wellstat Diagnostics note receivable | $ | 50,191 | $ | — | $ | 55,389 | $ | 50,191 | $ | — | $ | 57,322 | ||||||||||||
Hyperion note receivable | 1,200 | — | 1,200 | 1,200 | — | 1,200 | ||||||||||||||||||
CareView note receivable | 690 | — | 690 | 11,458 | — | 11,458 | ||||||||||||||||||
Total | $ | 52,081 | $ | — | $ | 57,279 | $ | 62,849 | $ | — | $ | 69,980 | ||||||||||||
Liabilities: | ||||||||||||||||||||||||
December 2021 Notes | $ | 16,950 | $ | 20,978 | $ | — | $ | 124,644 | $ | 151,356 | $ | — | ||||||||||||
December 2024 Notes | 10,300 | 12,953 | — | — | — | — | ||||||||||||||||||
Total | $ | 27,250 | $ | 33,931 | $ | — | $ | 124,644 | $ | 151,356 | $ | — | ||||||||||||
106
During the years ended December 31, 2019 and 2018 the Company recorded impairment losses of $10.8 million and $8.2 million, respectively, for the note receivable with CareView. There were no impairment losses on notes receivable in the year ended December 31, 2017.
As of December 31, 2019 the estimated fair value of the CareView note receivable was determined using a liquidation analysis. A liquidation analysis considers the asset side of the balance sheet and adjusts the value in accordance with the relative risk associated with the asset and the probable liquidation value. The asset recovery rates varied by asset. At December 31, 2018, the estimated fair value of the CareView note receivable was determined using discounted cash flow models, using a discount rate of 30%, incorporating expected principal and interest payments and also considered the recoverability of the note receivable balance utilizing third-party revenue multiples for small cap healthcare technology companies. As of December 31, 2019 and 2018, the estimated fair value of the Wellstat Diagnostics and Hyperion Catalysis International, Inc. (“Hyperion”) notes receivable were determined by using an asset approach and discounted cash flow model related to the underlying collateral and adjusted to consider estimated costs to sell the assets.
The Company determined its notes receivable assets are Level 3 assets as the Company’s valuations utilized significant unobservable inputs, including estimates of future revenues, discount rates, expectations about settlement, terminal values, required yield and the value of underlying collateral. The Company engages third-party valuation experts when deemed necessary to assist in evaluating its investments and the related inputs needed to estimate the fair value of certain investments.
The CareView note receivable is secured by substantially all assets of, and equity interests in, CareView. The Wellstat Diagnostics note receivable is secured by substantially all assets of Wellstat Diagnostics and is supported by a guaranty from the Wellstat Diagnostics Guarantors (as defined in Note 7, Notes and Other Long-Term Receivables).
On December 31, 2019, the carrying value of one of the Company’s notes receivable assets differed from its estimated fair value. This is the result of inputs used in estimating the fair value of the collateral, including appraisals, projected cash flows of collateral assets and discount rates used when performing a discounted cash flow analysis.
The fair values of the Company’s convertible senior notes were determined using quoted market pricing.
The following table represents significant unobservable inputs used in determining the estimated fair value of the Wellstat Diagnostics note receivable investment:
Asset | Valuation Technique | Unobservable Input | December 31, 2019 | December 31, 2018 | ||||
Wellstat Diagnostics | ||||||||
Wellstat Diagnostics Guarantors intellectual property | Income Approach | |||||||
Discount rate | 12% | 12% | ||||||
Undiscounted royalty amount | $21 million | $21 million | ||||||
Settlement Amount | Income Approach | |||||||
Discount rate | 15% | 15% | ||||||
Undiscounted settlement amount | $28 million | $34 million | ||||||
Real Estate Property | Market Approach | |||||||
Estimated annual appreciation | —% | 4% | ||||||
Estimated realtor fee | 6% | 6% | ||||||
Undiscounted market value | $16 million | $16 million | ||||||
7. Notes and Other Long-Term Receivables
Notes and other long-term receivables included the following significant agreements:
Wellstat Diagnostics Note Receivable and Credit Agreement and Related Litigation
On November 2, 2012, the Company and Wellstat Diagnostics entered into a $40.0 million credit agreement pursuant to which the Company was to accrue quarterly interest payments at the rate of 5% per annum (payable in cash or in kind). In addition, the Company was to receive quarterly royalty payments based on a low double-digit royalty rate of Wellstat Diagnostics’ net
107
revenues, generated by the sale, distribution or other use of Wellstat Diagnostics’ products, if any, commencing upon the commercialization of its products. A portion of the proceeds of the $40.0 million credit agreement were used to repay certain notes receivable which Wellstat Diagnostics entered into in March 2012.
In January 2013, the Company was informed that, as of December 31, 2012, Wellstat Diagnostics had used funds contrary to the terms of the credit agreement and breached Sections 2.1.2 and 7 of the credit agreement. The Company sent Wellstat Diagnostics a notice of default on January 22, 2013, and accelerated the amounts owed under the credit agreement. In connection with the notice of default, the Company exercised one of its available remedies and transferred approximately $8.1 million of available cash from a bank account of Wellstat Diagnostics to the Company and applied the funds to amounts due under the credit agreement. On February 28, 2013, the parties entered into a forbearance agreement whereby the Company agreed to refrain from exercising additional remedies for 120 days. During such forbearance period, the Company provided approximately $1.3 million to Wellstat Diagnostics to fund ongoing operations of the business. During the year ended December 31, 2013, approximately $8.7 million was advanced pursuant to the forbearance agreement.
On August 15, 2013, the Company entered into an amended and restated credit agreement with Wellstat Diagnostics. The Company determined that the new agreement should be accounted for as a modification of the existing agreement.
Except as otherwise described herein, the material terms of the amended and restated credit agreement are substantially the same as those of the original credit agreement, including quarterly interest payments at the rate of 5% per annum (payable in cash or in kind). In addition, the Company was to continue to receive quarterly royalty payments based on a low double-digit royalty rate of Wellstat Diagnostics’ net revenues. However, pursuant to the amended and restated credit agreement: (i) the principal amount was reset to approximately $44.1 million, which was comprised of approximately $33.7 million original loan principal and interest, $1.3 million term loan principal and interest and $9.1 million forbearance principal and interest; (ii) the specified internal rates of return increased; (iii) the default interest rate was increased; (iv) Wellstat Diagnostics’ obligation to provide certain financial information increased in frequency to monthly; (v) internal financial controls were strengthened by requiring Wellstat Diagnostics to maintain an independent, third-party financial professional with control over fund disbursements; (vi) the Company waived the existing events of default; and (vii) the owners and affiliates of Wellstat Diagnostics were required to contribute additional capital to Wellstat Diagnostics upon the sale of an affiliate entity. The amended and restated credit agreement had an ultimate maturity date of December 31, 2021 (but has subsequently been accelerated as described below).
In June 2014, the Company received information from Wellstat Diagnostics showing that it was generally unable to pay its debts as they became due, constituting an event of default under the amended and restated credit agreement.
On August 5, 2014, the Company delivered a notice of default to Wellstat Diagnostics, which accelerated all obligations under the amended and restated credit agreement and demanded immediate payment in full in an amount equal to approximately $53.9 million, (which amount, in accordance with the terms of the amended and restated credit agreement, included an amount that, together with interest and royalty payments already made to the Company, would generate a specified internal rate of return to the Company), plus accruing fees, costs and interest, and demanded that Wellstat Diagnostics protect and preserve all collateral securing its obligations.
On August 7, 2014, the Company delivered a notice to each of the guarantors of Wellstat Diagnostics’ obligations to the Company (collectively, the “Wellstat Diagnostics Guarantors”) under the credit agreement, which included a demand that the guarantors remit payment to the Company in the amount of the outstanding obligations. The guarantors include certain affiliates and related companies of Wellstat Diagnostics, including Wellstat Therapeutics and Wellstat Diagnostics’ stockholders.
On September 24, 2014, the Company filed an ex-parte petition for appointment of receiver with the Circuit Court of Montgomery County, Maryland, which was granted on the same day. Wellstat Diagnostics remained in operation during the period of the receivership with incremental additional funding from the Company. On May 24, 2017, Wellstat Diagnostics transferred substantially all of its assets to the Company pursuant to a credit bid. The credit bid reduced the outstanding balance of the loan by an immaterial amount.
On September 4, 2015, the Company filed in the Supreme Court of New York a motion for summary judgment in lieu of complaint which requested that the court enter judgment against certain of the Wellstat Diagnostics Guarantors for the total amount due on the Wellstat Diagnostics debt, plus all costs and expenses including lawyers’ fees incurred by the Company in enforcement of the related guarantees. On September 23, 2015, the Company filed in the same court an ex parte application for a temporary restraining order and order of attachment of the Wellstat Diagnostics Guarantor defendants’ assets. Although the court denied the Company’s request for a temporary restraining order at a hearing on September 24, 2015, it ordered that assets of the Wellstat Diagnostics Guarantor defendants should be held in status quo ante and only used in the normal course of business.
108
On July 29, 2016, the Supreme Court of New York granted the Company’s motion for summary judgment and held that the Wellstat Diagnostics Guarantor defendants are liable for all “Obligations” owed by Wellstat Diagnostics to the Company.
After appeal by the Wellstat Diagnostics Guarantor defendants on February 14, 2017, the Appellate Division of the Supreme Court of New York reversed on procedural grounds a portion of the Memorandum of Decision granting the Company summary judgment in lieu of complaint, but affirmed the portion of the Memorandum of Decision denying the Wellstat Diagnostics Guarantor defendants’ motion for summary judgment in which they sought a determination that the guarantees had been released. As a result, the litigation has been remanded to the Supreme Court of New York to proceed on the Company’s claims as a plenary action. On June 21, 2017, the Supreme Court of New York ordered the Company to file a Complaint, which was filed by the Company on July 20, 2017. The Wellstat Diagnostics Guarantors filed their answer on August 9, 2017, including counterclaims against the Company alleging breach of contract, breach of fiduciary duty, and tortious interference with prospective economic advantage.
On October 14, 2016, the Company sent a notice of default and reference to foreclosure proceedings to certain of the Wellstat Diagnostics Guarantors which are not defendants in the New York action, but which are owners of real estate assets over which a deed of trust in favor of the Company securing the guarantee of the loan to Wellstat Diagnostics had been executed. On March 2, 2017, the Company sent a second notice to foreclose on the real estate assets, and noticed the sale for March 29, 2017. The sale was taken off the calendar by the trustee under the deed of trust and has not been re-scheduled yet. On March 6, 2017, the Company sent a letter to the Wellstat Diagnostics Guarantors seeking information in preparation for a UCC Article 9 sale of some or all of the intellectual property-related collateral of the Wellstat Diagnostics Guarantors. The Wellstat Diagnostics Guarantors did not respond to the Company’s letter, but on March 17, 2017, filed an order to show cause with the Supreme Court of New York to enjoin the Company’s sale of the real estate or enforcing its security interests in the Wellstat Diagnostics Guarantors’ intellectual property during the pendency of any action involving the guarantees at issue. On February 6, 2018, the Supreme Court of New York issued an order from the bench which enjoins the Wellstat Diagnostics Guarantors from selling, encumbering, removing, transferring or altering the collateral pending the outcome of the proceedings before it. The Supreme Court of New York also issued an order precluding the Company from foreclosing on certain of the Wellstat Diagnostics Guarantors’ collateral pending the outcome of the proceedings before it. In September of 2018, discovery in the New York action was completed. Summary judgment motions were filed by Wellstat Diagnostics and the Company in 2018 and a hearing was held on May 22, 2019. On September 11, 2019, the Supreme Court of New York granted the Company’s summary judgment motion, the court holding that the guarantees executed by the Wellstat Diagnostics Guarantors are valid and enforceable, and that the Wellstat Diagnostics Guarantors are liable for the amount owed under the loan agreement. The court ordered a damages inquest before a special referee to calculate the amount owed under the loan agreement between Wellstat Diagnostics and the Company. On September 12, 2019, the Wellstat Diagnostics Guarantors filed a notice of appeal in relation to the court’s decision. On September 17, 2019, the Wellstat Diagnostics Guarantors requested a stay of the enforcement of the New York Supreme Court’s decision pending their appeal of the decision, which was denied on November 21, 2019. A damages hearing was scheduled to begin before a judicial hearing officer on December 17, 2019. At the request of the judicial hearing officer, the parties agreed to mediate their dispute prior to the commencement of the damages hearing. As a result, no decision has been made by the hearing officer with respect to the amount of damages owed to the Company.
In an unrelated litigation, Wellstat Therapeutics filed a lawsuit against BTG International, Inc. for breach of contract (the “BTG Litigation”). In September 2017, the Delaware Chancery Court found in favor of Wellstat Therapeutics and awarded a judgment of $55.8 million in damages, plus interest. In October 2017, the Company filed a motion with the Supreme Court of New York requesting a pre-judgement attachment of the award. In June 2018, the Delaware Supreme Court largely affirmed the September 2017 decision of the Delaware Chancery Court, including the $55.8 million awarded in judgment. In August of 2018, in a letter to the Company’s counsel, Wellstat Diagnostics Guarantors’ counsel confirmed that the Wellstat Diagnostics Guarantors are preserving the BTG Litigation judgment award proceeds consistent with the New York Court’s prior directions.
On October 22, 2015, certain of the Wellstat Diagnostics Guarantors filed a separate complaint against the Company in the Supreme Court of New York seeking a declaratory judgment that certain contractual arrangements entered into between the parties subsequent to Wellstat Diagnostics’ default, and which relate to a split of proceeds in the event that the Wellstat Diagnostics Guarantors voluntarily monetize any assets that are the Company’s collateral, is of no force or effect. This case has been joined for all purposes, including discovery and trial, and consolidated with the pending case filed by the Company. The Wellstat Diagnostic Guarantors filed a summary judgment motion with regard to this case, which was also heard by the court at the hearing on May 22, 2019. The court, in its September 11, 2019 decision, denied in its entirety the Wellstat Diagnostics Guarantors’ motion for summary judgment.
109
Effective April 1, 2014, and as a result of the event of default, the Company determined the loan to be impaired and it ceased to accrue interest revenue. At that time and as of December 31, 2019, it has been determined that an allowance on the carrying value of the note was not necessary, as the Company believes the value of the collateral securing Wellstat Diagnostics’ obligations exceeds the carrying value of the asset and is sufficient to enable the Company to recover the current carrying value of $50.2 million. The Company continues to closely monitor the timing and expected recovery of amounts due, including litigation and other matters related to Wellstat Diagnostics Guarantors’ assets. There can be no assurance that an allowance on the carrying value of the notes receivable investment will not be necessary in a future period depending on future developments.
Hyperion Agreement
On January 27, 2012, the Company and Hyperion (which is also a Wellstat Diagnostics Guarantor) entered into an agreement whereby Hyperion sold to the Company the royalty streams accruing from January 1, 2012 through December 31, 2013 due from Showa Denko K.K. (“SDK”) related to a certain patent license agreement between Hyperion and SDK dated December 31, 2008. In exchange for the lump sum payment to Hyperion of $2.3 million, in addition to any royalties from SDK, the Company was to receive two equal payments of $1.2 million on March 5, 2013 and March 5, 2014. The first payment of $1.2 million was paid on March 5, 2013, but the second payment that was due on March 5, 2014 has not been made by Hyperion. Effective as of such date and as a result of the event of default, the Company ceased to accrue interest revenue. As of December 31, 2019, the estimated fair value of the collateral was determined to be in excess of the carrying value. There can be no assurance of realizing value from such collateral in the event of the Company’s foreclosure on the collateral.
Avinger Credit and Royalty Agreement
On April 18, 2013, the Company entered into a credit agreement with Avinger, Inc. (the “Avinger Credit and Royalty Agreement”). Under the terms of the Avinger Credit and Royalty Agreement, the Company received a low, single-digit royalty on Avinger’s net revenues until April 2018. Commencing in October 2015, after Avinger repaid $21.4 million pursuant to its note payable to the Company prior to its maturity date, the royalty on Avinger’s net revenues was reduced by 50%, subject to certain minimum payments from the prepayment date until April 18, 2018. The Company accounted for the royalty rights in accordance with the fair value option. As of April 18, 2018, there were no further obligations owed to the Company.
LENSAR Credit Agreement
On October 1, 2013, the Company entered into a credit agreement with LENSAR, pursuant to which the Company made available to LENSAR up to $60.0 million to be used by LENSAR in connection with the commercialization of its currently marketed LENSAR™ Laser System. Of the $60.0 million available to LENSAR, an initial $40.0 million, net of fees, was funded by the Company at the close of the transaction. The remaining $20.0 million was never funded. Outstanding borrowings under the loans bore interest at the rate of 15.5% per annum, payable quarterly in arrears.
On May 12, 2015, the Company entered into a forbearance agreement with LENSAR, pursuant to which the Company agreed to refrain from exercising certain remedies available to it resulting from the failure of LENSAR to comply with a liquidity covenant and make interest payments due under the credit agreement. Under the forbearance agreement, the Company agreed to provide LENSAR with up to an aggregate of $8.5 million in weekly increments through the period ended September 30, 2015 plus employee retention amounts of approximately $0.5 million in the form of additional loans, subject to LENSAR meeting certain milestones related to LENSAR obtaining additional capital to fund or to sell the business and repay outstanding amounts under the credit agreement. In exchange for the forbearance, LENSAR agreed to additional reporting covenants, the engagement of a chief restructuring officer and an increase on the interest rate to 18.5%, applicable to all outstanding amounts under the credit agreement.
On September 30, 2015, the Company agreed to extend the forbearance agreement until October 9, 2015 and provide for up to an additional $0.8 million in funding while LENSAR negotiated a potential sale of its assets. On October 9, 2015, the forbearance agreement expired, but the Company agreed to fund LENSAR’s operations while LENSAR continued to negotiate a potential sale of its assets.
On November 15, 2015, LENSAR, LLC (“LENSAR/Alphaeon”), a wholly-owned subsidiary of Alphaeon, and LENSAR entered into the Asset Purchase Agreement whereby LENSAR/Alphaeon agreed to acquire certain assets of LENSAR and assumed certain liabilities of LENSAR. The acquisition was consummated on December 15, 2015.
In connection with the closing of the acquisition, LENSAR/Alphaeon entered into an amended and restated credit agreement with the Company, assuming $42.0 million in loans as part of the borrowings under the Company’s prior credit agreement with
110
LENSAR. In addition, Alphaeon issued 1.7 million shares of its Class A common stock to the Company which were valued at $6.6 million at the time the shares were received. For additional information on this investment in Alphaeon, see Note 6, Fair Value Measurements.
In December 2016, LENSAR, re-acquired the assets from LENSAR/Alphaeon and the Company entered into a second amended and restated credit agreement with LENSAR whereby LENSAR assumed all obligations under the amended and restated credit agreement with LENSAR/Alphaeon. Also in December, LENSAR filed for a voluntary petition under Chapter 11 of the U.S. Bankruptcy Code (“Chapter 11 case”) with the support of the Company. In January 2017, the Company agreed to provide debtor-in-possession financing of up to $2.8 million in new advances to LENSAR so that it could continue to operate its business during the Chapter 11 case. LENSAR filed a Chapter 11 plan of reorganization with the Company’s support under which LENSAR would issue all of its equity interests to the Company in exchange for the cancellation of the Company’s claims as a secured creditor in the Chapter 11 case, other than with respect to the debtor-in-possession financing, and would thereby become an operating wholly-owned subsidiary of the Company. On April 26, 2017, the bankruptcy court approved the plan of reorganization.
Pursuant to the plan of reorganization, LENSAR emerged from bankruptcy on May 11, 2017 as a wholly-owned subsidiary of the Company, and the Company started to consolidate LENSAR’s financial statements under the voting interest model beginning May 11, 2017.
For additional information on LENSAR please refer to Note 10, Intangible Assets, Note 20, Segment Information and Note 24, Business Combinations.
Direct Flow Medical Credit Agreement
On November 5, 2013, the Company entered into a credit agreement with Direct Flow Medical, Inc. (“Direct Flow Medical”) under which the Company agreed to provide up to $50.0 million to Direct Flow Medical. Of the $50.0 million available to Direct Flow Medical, the first tranche of $35.0 million, net of fees, was funded by the Company at the close of the transaction.
On November 10, 2014, the Company and Direct Flow Medical agreed to an amendment to the credit agreement to permit Direct Flow Medical to borrow an additional $15.0 million (in a second tranche) upon receipt by Direct Flow Medical of a specified minimum amount of proceeds from an equity offering prior to December 31, 2014. In exchange, the parties amended the credit agreement to provide for additional fees associated with certain liquidity events, such as a change of control or the consummation of an initial public offering, and granted the Company certain board of director observation rights. On November 19, 2014, upon Direct Flow Medical satisfying the amended tranche two milestone, the Company funded the $15.0 million second tranche to Direct Flow Medical, net of fees.
Outstanding borrowings under tranche one bore interest at the rate of 15.5% per annum, payable quarterly in arrears, until the occurrence of the second tranche. Upon occurrence of the borrowing of this second tranche, the interest rate applicable to all loans under the credit agreement was decreased to 13.5% per annum, payable quarterly in arrears.
Under the terms of the credit agreement, Direct Flow Medical’s obligation to repay loan principal commenced on the twelfth interest payment date, September 30, 2016. The principal amount outstanding at commencement of repayment was required to be repaid in equal installments until final maturity of the loans. The loans were scheduled to mature on November 5, 2018. The obligations under the credit agreement were secured by a pledge of substantially all of the assets of Direct Flow Medical and any of its subsidiaries.
On December 21, 2015, Direct Flow Medical and the Company entered into a waiver to the credit agreement in anticipation of Direct Flow Medical being unable to comply with the liquidity covenant and make interest payments due under the credit agreement, which was subsequently extended on January 14, 2016, and further delayed the timing of the interest payments through the period ending September 30, 2016 while Direct Flow Medical sought additional financing to operate its business.
On January 28, 2016, the Company funded an additional $5.0 million to Direct Flow Medical in the form of a short-term secured promissory note.
On February 26, 2016, the Company and Direct Flow Medical entered into the fourth amendment to the credit agreement that, among other things, (i) converted the $5.0 million short-term secured promissory note into a loan under the credit agreement with substantially the same interest and payment terms as the existing loans, (ii) added a conversion feature whereby the $5.0 million loan would convert into equity of Direct Flow Medical upon the occurrence of certain events and (iii) provided for a second $5.0 million convertible loan tranche commitment, to be funded at the option of the Company. The commitment for the second tranche
111
was not funded and has since expired. In addition, (i) the Company agreed to waive the liquidity covenant and delay the timing of the unpaid interest payments until September 30, 2016 and (ii) Direct Flow Medical agreed to issue to the Company a specified amount of warrants to purchase shares of convertible preferred stock on the first day of each month for the duration of the waiver period at an exercise price of $0.01 per share.
On July 15, 2016, the Company and Direct Flow Medical entered into the fifth amendment and limited waiver to the credit agreement. The Company funded an additional $1.5 million to Direct Flow Medical in the form of a note with substantially the same interest and payment terms as the existing loans and a conversion feature whereby the $1.5 million loan would convert into equity of Direct Flow Medical upon the occurrence of certain events. In addition, Direct Flow Medical agreed to issue to the Company warrants to purchase shares of convertible preferred stock at an exercise price of $0.01 per share.
On September 12, 2016, the Company and Direct Flow Medical entered into the sixth amendment and limited waiver to the credit agreement under which the Company funded an additional $1.5 million to Direct Flow Medical in the form of a note with substantially the same interest and payment terms as the existing loans. In addition, Direct Flow Medical agreed to issue to the Company a specified amount of warrants to purchase shares of convertible preferred stock at an exercise price of $0.01 per share.
On September 30, 2016, the Company and Direct Flow Medical entered into a waiver to the credit agreement where the parties agreed, among other things, to (i) delay payment on all overdue interest payments until October 31, 2016, (ii) waive the initial principal repayment until October 31, 2016 and (iii) continue to waive the liquidity requirements until October 31, 2016. Further, Direct Flow Medical agreed to issue to the Company a specified amount of warrants to purchase shares of convertible preferred stock at an exercise price of $0.01 per share.
On October 31, 2016, the Company agreed to extend the waivers described above until November 30, 2016 and on November 14, 2016, the Company advanced an additional $1.0 million loan while Direct Flow Medical continued to seek additional financing.
On November 16, 2016, Direct Flow Medical advised the Company that its potential financing source had modified its proposal from an equity investment to a loan with a substantially smaller amount and under less favorable terms. Direct Flow Medical shut down its operations in December 2016 and in January 2017 made an assignment for the benefit of creditors. The Company then initiated foreclosure proceedings, resulting in the Company obtaining ownership of most of the Direct Flow Medical assets through the Company’s wholly-owned subsidiary, DFM, LLC. The assets were held for sale and carried at the lower of carrying amount or fair value, less estimated selling costs, which was primarily based on supporting data from market participant sources, and valid offers from third parties.
At December 31, 2016, the Company completed an impairment analysis and concluded that the situation qualified as a troubled debt restructuring and recognized an impairment loss of $51.1 million.
In January 2017, the Company started to actively market the asset held for sale. On January 23, 2017, the Company and DFM, LLC entered into an Intellectual Property Assignment Agreement with Hong Kong Haisco Pharmaceutical Co., Limited (“Haisco”), a Chinese pharmaceutical company, whereby Haisco acquired former Direct Flow Medical clinical, regulatory and commercial information and intellectual property rights exclusively in China for $7.0 million. The Company, through DFM, LLC, also sold Haisco certain manufacturing equipment for $450,000 and collected $692,000 on outstanding Direct Flow Medical accounts receivable during the year ended December 31, 2017.
On January 6, 2018, DFM, LLC, a wholly-owned subsidiary of the Company, and HaisThera Advisors Co., Limited (“HaisThera”) entered into a license agreement whereby DFM, LLC granted HaisThera an exclusive license to develop, manufacture and commercialize percutaneously implanting stentless aortic valves in the EU. The consideration for the license agreement was $500,000 upfront and up to $2.0 million in royalty payments. In August 2019, the remaining assets of DFM, LLC were sold for $5.0 million.
kaléo Note Purchase Agreement
On April 1, 2014, the Company entered into a note purchase agreement with Accel 300, LLC (“Accel 300”), a wholly-owned subsidiary of kaléo, Inc. (“kaléo”), pursuant to which the Company acquired $150.0 million of secured notes due 2029 (the “kaléo Note”). The kaléo Note was issued pursuant to an indenture between Accel 300 and U.S. Bank, National Association, as trustee, and was secured by 20% of net sales of its first approved product, Auvi-Q® (epinephrine auto-injection, USP) (known as Allerject® in Canada) and 10% of net sales of kaléo’s second proprietary auto-injector based product, EVZIO (naloxone hydrochloride injection ) (the “kaléo Revenue Interests”), and a pledge of kaléo’s equity ownership in Accel 300.
112
On September 21, 2017, the Company entered into an agreement (the “kaléo Note Sale Agreement”) with MAM-Kangaroo Lender, LLC, a Delaware limited liability company (the “kaléo Purchaser”), pursuant to which the Company sold its entire interest in the kaléo Note.
Pursuant to the kaléo Note Sale Agreement, the kaléo Purchaser paid to the Company an amount equal to all of the then outstanding principal, a premium of 1% of such amount and accrued interest under the kaléo Note, for an aggregate cash purchase price of $141.7 million, subject to an 18-month escrow holdback of $1.4 million against certain potential contingencies. The escrow period ended on March 20, 2019 and the escrow agent released the entire $1.4 million to the Company. For a further discussion on this topic, see Note 15, Commitments and Contingencies.
CareView Credit Agreement
On June 26, 2015, the Company entered into a credit agreement with CareView, under which the Company made available to CareView up to $40.0 million in loans comprised of two tranches of $20.0 million each, subject to CareView’s attainment of specified milestones relating to the placement of CareView Systems. On October 7, 2015, the Company and CareView entered into an amendment of the credit agreement to modify certain definitions related to the first and second tranche milestones and the Company funded the first tranche of $20.0 million, net of fees, based on CareView’s attainment of the first milestone, as amended. The second $20.0 million tranche was not funded due to CareView’s failure to achieve the related funding milestones and there is no additional funding obligation due from the Company. Outstanding borrowings under the credit agreement bear interest at the rate of 13.5% per annum and are payable quarterly in arrears.
As part of the original credit agreement, the Company received a warrant to purchase approximately 4.4 million shares of common stock of CareView at an exercise price of $0.45 per share. The Company has accounted for the warrant as derivative asset with an offsetting credit as debt discount. At each reporting period the warrant is marked to market for changes in fair value.
In connection with the October 2015 amendment of the credit agreement, the Company and CareView also agreed to amend the warrant to purchase common stock agreement by reducing the warrant’s exercise price from $0.45 to $0.40 per share.
In February 2018, the Company entered into a modification agreement with CareView (the “February 2018 Modification Agreement”) whereby the Company agreed, effective December 28, 2017, to modify the credit agreement before remedies could otherwise have become available to the Company under the credit agreement in relation to certain obligations of CareView that would potentially not be met, including the requirement to make principal payments. Under the February 2018 Modification Agreement, the Company agreed that (i) a lower liquidity covenant would be applicable and (ii) principal repayment would be delayed until December 31, 2018. In exchange for agreeing to these modifications, among other things, the exercise price of the Company’s warrants to purchase 4.4 million shares of common stock of CareView was repriced from $0.40 to $0.03 per share and, subject to the occurrence of certain events, CareView agreed to grant the Company additional equity interests. As a result of the February 2018 Modification Agreement, the Company determined the loan to be impaired and it ceased to accrue interest revenue effective October 1, 2017.
In September 2018, the Company entered into an amendment to the February 2018 Modification Agreement with CareView whereby the Company agreed, effective as of September 28, 2018, that a lower liquidity covenant would be applicable. In December 2018, the Company further modified the loan by agreeing that (i) a lower liquidity covenant would be applicable, (ii) the first principal payment would be deferred until January 31, 2019, and (iii) the scheduled interest payment due December 31, 2018 would be deferred until January 31, 2019. In December 2018, and in consideration of the further modification to the credit agreement, the Company completed an impairment analysis and determined that the note was impaired and recorded an impairment loss of $8.2 million. The principal repayment and interest payment were subsequently deferred until May 15, 2019 under additional amendments. In May 2019, and in consideration of additional capital raised by CareView, the Company further modified the loan by agreeing that (i) the first principal and interest payments would be deferred until September 30, 2019 (ii) the remaining liquidity covenant would be removed, and (iii) the interest rate would be increased to 15.5%. Pursuant to further amendments to the February 2018 Modification Agreement in September 2019 and December 2019, the Company agreed to defer principal and interest payments until December 31, 2019.
In December 2019, and in consideration of the further modification to the credit agreement and February 2018 Modification Agreement, the Company updated its impairment analysis and determined that an additional impairment was necessary and recorded an impairment loss of $10.8 million. At December 31, 2019, the Company estimated the fair value of the warrant to be less than $0.1 million. For additional information please refer to Note 6, Fair Value Measurements.
113
In January 2020 the Company agreed to a further amendment of the February 2018 Modification Agreement that deferred principal repayment and interest payments until April 30, 2020, which was conditioned upon CareView raising additional financing from third parties.
8. Leases
Lessee arrangements
The Company has operating leases for corporate offices and certain equipment. The Company’s operating leases have remaining lease terms ranging from one to seven years, some of which include options to extend the leases for up to five years, and some of which include options to terminate the leases within two years.
The components of lease expense are as follows:
Year Ended December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Operating lease cost | $ | 919 | $ | 1,188 | ||||
Short-term lease cost | 81 | 50 | ||||||
Total lease cost | $ | 1,000 | $ | 1,238 | ||||
Supplemental cash flow information related to leases is as follows:
Year Ended December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Cash paid for amounts included in the measurement of lease liabilities: | ||||||||
Operating cash flows from operating leases | $ | 921 | $ | 1,188 | ||||
Right-of-use-assets obtained in exchange for lease obligations: | ||||||||
Operating leases | $ | 2,534 | N/A | |||||
_______________
N/A Not applicable
The following table presents the lease balances within the Consolidated Balance Sheet, weighted-average remaining lease term, and weighted-average discount rates related to the Company’s operating leases (in thousands):
Operating Leases | Classification | December 31, 2019 | ||||
Operating lease ROU assets | Other assets | $ | 1,638 | |||
Operating lease liabilities, current | Accrued liabilities | $ | 918 | |||
Operating lease liabilities, long-term | Other long-term liabilities | 754 | ||||
Total operating lease liabilities | Total operating lease liabilities | $ | 1,672 | |||
Weighted-average remaining lease term | 1.9 years | |||||
Weighted-average discount rate | 6.4 | % | ||||
114
Maturities of operating lease liabilities as of December 31, 2019 are as follows (in thousands):
Fiscal Year | Amount | |||
2020 | $ | 958 | ||
2021 | 686 | |||
2022 | 90 | |||
2023 | — | |||
2024 | — | |||
Thereafter | — | |||
Total operating lease payments | 1,734 | |||
Less: imputed interest | 62 | |||
Total operating lease liabilities | $ | 1,672 | ||
Future minimum operating lease payments as of December 31, 2018 were as follows (in thousands):
Fiscal Year | Amount | |||
2019 | $ | 1,140 | ||
2020 | 1,003 | |||
2021 | 559 | |||
2022 | — | |||
2023 | — | |||
Thereafter | — | |||
Total | $ | 2,702 | ||
As of December 31, 2019, the Company had no additional significant operating or finance leases that had not yet commenced.
Lessor arrangements
The Company has operating and sales-type leases for medical device equipment generated from its medical devices segment. The Company’s leases have remaining lease terms of less than one year to five years, some of which include options to extend the leases on a month-to-month basis if the customer does not notify the Company of the intention to return the equipment at the end of the lease term. The Company typically does not offer options to terminate the leases before the end of the lease term.
The components of lease income are as follows:
Year Ended December 31, | ||||||||||
(in thousands) | Classification | 2019 | 2018 | |||||||
Sales-type lease selling price | Product revenue, net | $ | 542 | $ | 746 | |||||
Cost of underlying asset | (109 | ) | (344 | ) | ||||||
Operating profit | $ | 433 | $ | 402 | ||||||
Interest income on the lease receivable | Interest and other income, net | $ | 53 | $ | 51 | |||||
Initial direct costs incurred | Operating expense | $ | (35 | ) | $ | (41 | ) | |||
Operating lease income | Product revenue, net | $ | 5,180 | $ | 7,264 | |||||
115
Net investment in sales-type leases are as follows:
(in thousands) | Classification | December 31, 2019 | December 31, 2018 | |||||||
Lease payment receivable, current | Accounts receivable, net and Notes receivable, current | $ | 502 | $ | 472 | |||||
Lease payment receivable, long-term | Notes receivable, long-term and Other assets | 827 | 753 | |||||||
Total lease payment receivable | $ | 1,329 | $ | 1,225 | ||||||
Equipment under lease is stated at cost less accumulated depreciation and is classified as Property and equipment, net on the Consolidated Balance Sheets. Depreciation is computed using the straight-line method over an estimated useful life of the greater of the lease term or five years to ten years. Equipment under lease is as follows:
(in thousands) | December 31, 2019 | December 31, 2018 | ||||||
Equipment under lease | $ | 6,652 | $ | 6,529 | ||||
Less accumulated depreciation | (5,231 | ) | (3,665 | ) | ||||
Equipment under lease, net | $ | 1,421 | $ | 2,864 | ||||
Depreciation expense on equipment under lease amounted to $2.1 million, $2.7 million and $1.6 million for the years ended December 31, 2019, 2018 and 2017, respectively.
Maturities of sales-type lease receivables as of December 31, 2019 are as follows (in thousands):
Fiscal Year | Amount | |||
2020 | $ | 538 | ||
2021 | 396 | |||
2022 | 350 | |||
2023 | 126 | |||
2024 | — | |||
Thereafter | — | |||
Total undiscounted cash flows | 1,410 | |||
Present value of lease payments (recognized as lease receivables) | 1,329 | |||
Difference between undiscounted and discounted cash flows | $ | 81 | ||
Maturities of operating lease receivables as of December 31, 2019 are as follows (in thousands):
Fiscal Year | Amount | |||
2020 | $ | 2,287 | ||
2021 | 1,218 | |||
2022 | 426 | |||
2023 | 81 | |||
2024 | — | |||
Thereafter | — | |||
Total undiscounted cash flows | $ | 4,012 | ||
116
9. Property and Equipment
The following table provides details of the property and equipment, net:
December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Leasehold improvements | $ | 350 | $ | 322 | ||||
Manufacturing equipment | 1,749 | 1,669 | ||||||
Computer and office equipment | 9,991 | 9,451 | ||||||
Furniture and fixtures | 175 | 162 | ||||||
Equipment under lease | 6,652 | 6,529 | ||||||
Transportation equipment | 67 | 67 | ||||||
Total | 18,984 | 18,200 | ||||||
Less accumulated depreciation | (17,058 | ) | (14,203 | ) | ||||
Construction in progress | 3,594 | 3,390 | ||||||
Property and equipment, net | $ | 5,520 | $ | 7,387 | ||||
Depreciation expense on property and equipment amounted to $2.9 million, $2.7 million and $2.3 million for the years ended December 31, 2019, 2018 and 2017, respectively.
10. Intangible Assets
Noden
On June 8, 2018, Noden DAC entered into a Settlement Agreement (the “Settlement Agreement”) with Anchen Pharmaceuticals, Inc. and its affiliates (“Anchen”) to resolve the patent litigation relating to infringement of U.S. Patent No. 8,617,595 (the “‘595 Patent”) based on their submission of an Abbreviated New Drug Application (“ANDA”) seeking authorization from the FDA to market a generic version of aliskiren, the active ingredient in the Tekturna and Tekturna HCT drug. Under the Settlement Agreement, Anchen, the sole ANDA filer of which the Company is aware, agreed to not commercialize its generic version of aliskiren prior to March 1, 2019. Per the Settlement Agreement, Anchen may commercialize their formulation of aliskiren, but is not permitted to commercialize a copy of Tekturna.
Accordingly, management evaluated the ongoing value of the Noden DAC asset group based upon the probability of Anchen’s market entry of a generic version of aliskiren in the United States and the associated cash flows and conducted a test for impairment. Due to the increased probability of a generic version of aliskiren being launched in the United States, the Company revised its estimates of future cash flows and as a result of this analysis, determined that the sum of undiscounted cash flows was not greater than the carrying value of the assets. Therefore, the Company performed a discounted cash flow analysis to estimate the fair value of the asset group in accordance with ASC 360, Impairment or Disposal of Long-lived Assets. The cash flows used in this analysis are those expected to be generated by market participants, discounted to reflect an appropriate amount of risk, which was determined to be 21%. The Company concluded that the Noden DAC acquired product rights and customer relationship long-lived assets, with a carrying amount of $192.5 million, were no longer recoverable and wrote them down to their estimated fair value of $40.1 million, resulting in an impairment charge of $152.3 million in the second quarter of 2018. This write-down is included in Impairment of intangible assets in the Consolidated Statement of Operations and the Consolidated Statement of Cash Flows for the year ended December 31, 2018.
At December 31, 2019, due to the Company’s monetization strategy and updated forecasts for Noden, the Company revised its estimates of future cash flows and as a result of this analysis, determined that the sum of undiscounted cash flows was not greater than the carrying value of the assets. Therefore, the Company performed a discounted cash flow analysis to estimate the fair value of the asset group in accordance with ASC 360. The cash flows used in this analysis are those expected to be generated by market participants, discounted to reflect an appropriate amount of risk, which was determined to be 19%. The Company concluded that the Noden DAC acquired product rights and customer relationship long-lived assets, with a carrying amount of $32.6 million, were no longer recoverable and wrote them down to their estimated fair value of $10.1 million, resulting in an impairment charge of $22.5 million in the fourth quarter of 2019. This write-down is included in Impairment of intangible assets in the Consolidated Statement of Operations and the Consolidated Statement of Cash Flows for the year ended December 31, 2019.
117
During the fourth quarter of 2019, while performing its impairment analysis on its Noden intangible assets, the Company identified an error in the 2018 impairment charge recorded on its Noden intangible assets, which resulted in a $10.5 million overstatement of the 2018 impairment charge. As of December 31, 2018, the net carrying value of the intangible asset was understated by $9.8 million with a corresponding overstatement of net loss for the year ended December 31, 2018. This prior year impairment expense error was corrected as an out of period adjustment in 2019 in connection with the further impairment of the intangible asset to $10.1 million. The Company determined that these errors were not material to its current and previously issued financial statements.
Based on an analysis of Accounting Standards Codification (“ASC”) 250, Accounting Changes and Error Corrections (“ASC 250”), Staff Accounting Bulletin 99, Materiality (“SAB 99”) and Staff Accounting Bulletin 108, Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements (“SAB 108”), the Company determined that these errors were immaterial to the previously issued annual and interim financial statements. The amount of the intangible assets and accumulated amortization have been corrected as of December 31, 2019.
LENSAR
In April 2019, LENSAR acquired certain intellectual property from a third-party for $2.0 million in cash and obligations to pay a $0.3 million milestone payment and royalties upon the completion of certain events, which were met prior to December 31, 2019.
In September 2019, LENSAR exclusively licensed certain intellectual property from a third-party for $3.5 million in cash for use in research and development activities. The amount was immediately expensed and is included in Research and development expense in the Consolidated Statement of Operations for the year ended December 31, 2019.
The components of intangible assets as of December 31, 2019 and 2018 were as follows:
December 31, 2019 | December 31, 2018 | |||||||||||||||||||||||
(in thousands) | Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | ||||||||||||||||||
Finite-lived intangible assets: | ||||||||||||||||||||||||
Acquired products rights 1 | $ | 9,108 | $ | — | $ | 9,108 | $ | 36,143 | $ | (2,258 | ) | $ | 33,885 | |||||||||||
Customer relationships 1, 2, 4 | 5,049 | (884 | ) | 4,165 | 8,028 | (782 | ) | 7,246 | ||||||||||||||||
Acquired technology 2, 3, 5 | 11,500 | (1,741 | ) | 9,759 | 11,011 | (1,203 | ) | 9,808 | ||||||||||||||||
Acquired trademarks 2 | 570 | (304 | ) | 266 | 570 | (190 | ) | 380 | ||||||||||||||||
$ | 26,227 | $ | (2,929 | ) | $ | 23,298 | $ | 55,752 | $ | (4,433 | ) | $ | 51,319 | |||||||||||
_______________
1 The Company acquired certain intangible assets as part of the Noden transaction which were subsequently impaired. The amount remaining at December 31, 2019 will be amortized on a straight-line basis over a weighted-average period of seven years.
2 The Company acquired certain intangible assets as part of its acquisition of LENSAR in May 2017. They are being amortized on a straight-line basis over a weighted-average period of 15 years. The intangible assets for customer relationships are being amortized using a double-declining method of amortization as such method better represents the economic benefits to be obtained. For a further discussion of the LENSAR transaction, see Note 24, Business Combinations.
3 The Company acquired certain intangible assets as part of the foreclosure on certain of Direct Flow Medical assets. In August 2019, the Company sold the DFM, LLC intangible assets for $5.0 million in cash and a single-digit percentage of any net final award received as part of the acquirer’s monetization process using the intangible assets. Prior to the sale, these intangible assets were being amortized on a straight-line basis over a weighted-average period of 10 years.
4 LENSAR acquired certain intangible assets for customer relationships from PES, which are being amortized using a double-declining method over a period of 20 years.
5 LENSAR acquired certain intangible assets from a third-party, which are being amortized on a straight-line basis over a period of 15 years.
Amortization expense for the years ended December 31, 2019, 2018 and 2017 was $6.3 million, $15.8 million and $24.7 million, respectively.
118
Based on the intangible assets recorded at December 31, 2019, and assuming no subsequent additions to or impairment of the underlying assets, the remaining amortization expense is expected to be as follows (in thousands):
Fiscal Year | Amount | |||
2020 | $ | 2,753 | ||
2021 | 2,721 | |||
2022 | 2,616 | |||
2023 | 2,553 | |||
2024 | 2,530 | |||
Thereafter | 10,125 | |||
Total remaining estimated amortization expense | $ | 23,298 | ||
11. Asset Acquisition
On January 8, 2018, LENSAR entered into an Asset Purchase Agreement with PES to purchase assets used in PES’ laser-assisted cataract surgery business. The assets purchased include equipment, inventory and PES’ customer contracts. No workforce was transferred as part of the transaction.
The Company assessed the acquisition of PES assets under ASC 805. Under ASC 805, the Company determined that the acquired assets did not constitute a business and that the transaction would be accounted for as an asset acquisition.
The following table summarizes the fair values of the identifiable assets acquired and liabilities assumed at the acquisition date:
(in thousands) | Amount | |||
Equipment and inventory | $ | 848 | ||
Fixed assets | 67 | |||
Intangible assets (customer relationships) | 1,845 | |||
Total identifiable assets | $ | 2,760 | ||
Consideration paid at closing, cash | $ | 1,200 | ||
Conversion consideration | 920 | |||
Contingent consideration | 640 | |||
Total fair value of consideration | $ | 2,760 | ||
12. Accrued Liabilities
Accrued liabilities consist of the following:
December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Accrued rebates, chargebacks and other revenue reserves | $ | 9,843 | $ | 20,133 | ||||
Deferred revenue | 3,907 | 8,811 | ||||||
Compensation | 7,354 | 4,468 | ||||||
Interest | 70 | 344 | ||||||
Legal | 931 | 623 | ||||||
Other | 6,201 | 4,933 | ||||||
Total | $ | 28,306 | $ | 39,312 | ||||
119
The following table provides a summary of activity with respect to the Company’s sales allowances and accruals for the year ended December 31, 2019:
(in thousands) | Discount and Distribution Fees | Government Rebates and Chargebacks | Assistance and Other Discounts | Product Returns | Total | |||||||||||||||
Balance as of December 31, 2018 | $ | 3,094 | $ | 8,901 | $ | 3,457 | $ | 4,681 | $ | 20,133 | ||||||||||
Allowances for current period sales | 5,090 | 12,104 | 5,003 | 1,720 | 23,917 | |||||||||||||||
Allowances for prior period sales | 50 | 1,848 | 142 | 46 | 2,086 | |||||||||||||||
Credits/payments for current period sales | (3,813 | ) | (8,843 | ) | (4,186 | ) | (276 | ) | (17,118 | ) | ||||||||||
Credits/payments for prior period sales | (3,076 | ) | (10,393 | ) | (3,411 | ) | (2,295 | ) | (19,175 | ) | ||||||||||
Balance as of December 31, 2019 | $ | 1,345 | $ | 3,617 | $ | 1,005 | $ | 3,876 | $ | 9,843 | ||||||||||
13. Convertible Senior Notes
February 2018 Notes
On February 12, 2014, the Company issued $300.0 million in aggregate principal amount, at par, of the 4.0% Convertible Senior Notes due February 1, 2018 (the “February 2018 Notes”) Notes in an underwritten public offering, for net proceeds of $290.2 million. The February 2018 Notes were due February 1, 2018, and the Company paid interest at 4.0% on the February 2018 Notes semiannually in arrears on February 1 and August 1 of each year, beginning August 1, 2014. A portion of the proceeds from the February 2018 Notes, net of amounts used for purchased call option transactions and provided by the warrant transactions described below, were used to redeem $131.7 million of the Company’s 2.975% Convertible Senior Notes due February 17, 2016.
In accordance with the accounting guidance for convertible debt instruments that may be settled in cash or other assets on conversion, the Company was required to separately account for the liability component of the instrument in a manner that reflected the market interest rate for a similar nonconvertible instrument at the date of issuance. As a result, the Company separated the principal balance of the February 2018 Notes between the fair value of the debt component and the fair value of the common stock conversion feature. Using an assumed borrowing rate of 7.0%, which represented the estimated market interest rate for a similar nonconvertible instrument available to the Company on the date of issuance, the Company recorded a total debt discount of $29.7 million, allocated $19.3 million to additional paid-in capital and allocated $10.4 million to deferred tax liability. The discount was being amortized to interest expense over the term of the February 2018 Notes and increased interest expense during the term of the February 2018 Notes from the 4.0% cash coupon interest rate to an effective interest rate of 6.9%.
In connection with the issuance of the February 2018 Notes, the Company entered into purchased call option transactions with two hedge counterparties. The Company paid an aggregate amount of $31.0 million for the purchased call options with terms substantially similar to the embedded conversion options in the February 2018 Notes. The purchased call options covered, subject to anti-dilution and certain other customary adjustments substantially similar to those in the February 2018 Notes, approximately 13.8 million shares of the Company’s common stock. Outstanding purchased call options expired on February 1, 2018.
In addition, the Company sold to the hedge counterparties warrants exercisable, on a cashless basis, for the sale of rights to receive shares of common stock underlying the February 2018 Notes at a strike price of $10.3610 per share, which represented a premium of approximately 30% over the last reported sale price of the Company’s common stock of $7.97 on February 6, 2014. The Company received an aggregate amount of $11.4 million for the sale from the two counterparties.
The purchased call options and warrants were considered indexed to the Company stock, required net-share settlement and met all criteria for equity classification at inception and in subsequent periods. The purchased call options cost of $31.0 million, less deferred taxes of $10.8 million, and the $11.4 million received for the warrants, were recorded as adjustments to additional paid-in capital.
On November 20, 2015, the Company’s agent initiated the repurchase of $53.6 million in aggregate principal amount of its February 2018 Notes for $43.7 million in cash in four open market transactions. The closing of these transactions occurred on November 30, 2015. It was determined that the repurchase of the principal amount should be accounted for as a partial extinguishment of the February 2018 Notes. As a result, a gain on extinguishment of $6.5 million was recorded at closing of the transaction. The $6.5 million gain on extinguishment included the de-recognition of a proportional share of the original issuance discount of $3.1 million, outstanding deferred issuance costs of $0.9 million and agent fees of $0.1 million. In connection with this repurchase of the February 2018 Notes, the Company unwound a corresponding portion of the purchased call options related
120
to the notes. As a result of this unwinding, the Company received $0.3 million in cash. The payments received have been recorded as an increase to additional paid-in-capital. In addition, the Company unwound a corresponding portion of the warrants issued in connection with the notes for $0.2 million in cash, payable by the Company. The payments have been recorded as a decrease to additional paid-in-capital.
On November 22, 2016, the Company repurchased $120.0 million in aggregate principal amount of its February 2018 Notes for approximately $121.5 million in cash (including $1.5 million of accrued interest) in open market transactions. It was determined that the repurchase of the principal amount be accounted for as an extinguishment. The extinguishment included the de-recognition of a proportional share of the original issuance discount of $4.3 million and outstanding deferred issuance costs of $1.3 million. In connection with the repurchase of the February 2018 Notes, the Company unwound a corresponding portion of the purchased call options. The transaction did not result in any cash payments between the parties. In addition, the Company and the counterparties agreed to unwind a corresponding portion of the warrants, which also did not result in any cash payments between the parties.
On February 1, 2018, upon maturity of the February 2018 Notes, the Company repaid a total cash amount of $129.0 million to the custodian, The Bank of New York Mellon Trust Company, N.A., which was comprised of $126.4 million in principal amount and $2.6 million in accrued interest, to retire the February 2018 Notes.
Interest expense for the February 2018 Notes on the Company’s Consolidated Statements of Operations was as follows:
Year Ended December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
Contractual coupon interest | $ | — | $ | 422 | $ | 5,058 | ||||||
Amortization of debt issuance costs | — | 88 | 1,022 | |||||||||
Amortization of debt discount | — | 293 | 3,449 | |||||||||
Total | $ | — | $ | 803 | $ | 9,529 | ||||||
December 2021 Notes
On November 22, 2016, the Company issued $150.0 million in aggregate principal amount, at par, of 2.75% Convertible Senior Notes due December 1, 2021 (the “December 2021 Notes”) in an underwritten public offering, for net proceeds of $145.7 million. The December 2021 Notes are due December 1, 2021, and the Company pays interest at 2.75% on the December 2021 Notes semiannually in arrears on June 1 and December 1 of each year, beginning June 1, 2017. A portion of the proceeds from the December 2021 Notes, net of amounts used for the capped call transaction described below, was used to extinguish $120.0 million of the February 2018 Notes.
In September 2019, the Company entered into privately negotiated exchange agreements with certain holders of approximately $86.1 million aggregate principal amount of outstanding December 2021 Notes. The Company exchanged $86.1 million aggregate principal of December 2021 Notes for an identical principal amount of 2.75% Convertible Senior Notes due December 1, 2024 (the “December 2024 Notes”), plus a cash payment of $70.00 for each $1,000 principal amount tendered (“September Exchange Transaction”). See “December 2024 Notes” below. The terms of the remaining December 2021 Notes remained unchanged.
The September Exchange Transaction qualified as a debt extinguishment and the Company recognized a loss on exchange of the convertible notes of $3.9 million, which is included in Non-operating income (expense), net in the Consolidated Statement of Operations for the year ended December 31, 2019.
Upon the occurrence of a fundamental change, as defined in the indenture entered into in connection with the December 2021 Notes (the “December 2021 Notes Indenture”), holders have the option to require the Company to repurchase their December 2021 Notes at a purchase price equal to 100% of the principal, plus accrued interest.
The December 2021 Notes are convertible under any of the following circumstances at any time prior to the close of business on the business day immediately preceding June 1, 2021 (or at any time beginning on June 1, 2021 until the close of business on the second scheduled trading day immediately preceding the stated maturity):
• | During any fiscal quarter (and only during such fiscal quarter) commencing after the fiscal quarter ended June 30, 2017, if the last reported sale price of Company common stock for at least 20 trading days (whether or not consecutive), in the |
121
period of 30 consecutive trading days, ending on, and including, the last trading day of the immediately preceding fiscal quarter, exceeds 130% of the conversion price for the notes on each applicable trading day;
• | During the five business-day period immediately after any five consecutive trading-day period, which the Company refers to as the measurement period, in which the trading price per $1,000 principal amount of notes for each trading day of that measurement period was less than 98% of the product of the last reported sale price of Company common stock and the conversion rate for the notes for each such trading day; or |
• | Upon the occurrence of specified corporate events as described in the December 2021 Notes Indenture. |
The initial conversion rate for the December 2021 Notes is 262.2951 shares of the Company’s common stock per $1,000 principal amount of December 2021 Notes, which is equivalent to an initial conversion price of approximately $3.81 per share of common stock, subject to adjustments upon the occurrence of certain specified events as set forth in the December 2021 Notes Indenture.
In accordance with the accounting guidance for convertible debt instruments that may be settled in cash or other assets on conversion, the Company was required to separately account for the liability component of the instrument in a manner that reflects the market interest rate for a similar nonconvertible instrument at the date of issuance. As a result, the Company separated the principal balance of the December 2021 Notes between the fair value of the debt component with the remainder of the consideration being allocated to the equity component. Using an assumed borrowing rate of 9.5%, which represented the estimated market interest rate for a similar nonconvertible instrument available to the Company on the date of issuance, the Company recorded a debt discount of $4.3 million, allocated $23.8 million to Additional paid-in capital for the conversion feature and allocated $12.8 million to deferred tax liability. The debt discount, including the conversion feature and issuance costs allocated to debt, which remained after amortization and the effect of the September Exchange Transaction, is being amortized to interest expense over the term of the December 2021 Notes and increases interest expense during the term of the December 2021 Notes from the 2.75% cash coupon interest rate to an effective interest rate of 9.7%. As of December 31, 2019, the remaining discount amortization period is 1.9 years.
On December 17, 2019, the Company repurchased $44.8 million in aggregate principal amount of its December 2021 Notes for $39.9 million in cash and 3.5 million shares of its common stock in privately negotiated transactions (the “December Exchange Transaction”). It was determined that the repurchase of the principal amount should be accounted for as a partial extinguishment of the December 2021 Notes. As a result, a loss on extinguishment of $2.5 million was recorded at closing of the transaction. The loss on extinguishment included the de-recognition of a proportional share of the original issuance discount of $0.3 million and outstanding deferred issuance costs of less than $0.1 million.
The carrying value and unamortized discount of the December 2021 Notes were as follows:
(in thousands) | December 31, 2019 | December 31, 2018 | ||||||
Principal amount of the December 2021 Notes | $ | 19,170 | $ | 150,000 | ||||
Unamortized discount of liability component | (2,220 | ) | (25,356 | ) | ||||
Net carrying value of the December 2021 Notes | $ | 16,950 | $ | 124,644 | ||||
Interest expense for the December 2021 Notes included in the Company’s Consolidated Statements of Operations was as follows:
Year Ended December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Contractual coupon interest | $ | 3,390 | $ | 4,125 | ||||
Amortization of debt issuance costs | 64 | 76 | ||||||
Amortization of debt discount | 459 | 542 | ||||||
Amortization of conversion feature | 5,973 | 6,611 | ||||||
Total | $ | 9,886 | $ | 11,354 | ||||
As of December 31, 2019 and 2018, the December 2021 Notes are not convertible.
Capped Call Transaction
In connection with the offering of the December 2021 Notes, the Company entered into a privately-negotiated capped call transaction with an affiliate of the underwriter of such issuance. The aggregate cost of the capped call transaction was $14.4
122
million. The capped call transaction is generally expected to reduce the potential dilution upon conversion of the December 2021 Notes and/or partially offset any cash payments the Company is required to make in excess of the principal amount of converted December 2021 Notes in the event that the market price per share of the Company’s common stock, as measured under the terms of the capped call transaction, is greater than the strike price of the capped call transaction. This initially corresponds to the approximate $3.81 per share conversion price of the December 2021 Notes and is subject to anti-dilution adjustments substantially similar to those applicable to the conversion rate of the December 2021 Notes. The cap price of the capped call transaction was initially $4.88 per share and is subject to certain adjustments under the terms of the capped call transaction. The Company will not be required to make any cash payments to the option counterparty upon the exercise of the options that are a part of the capped call transaction, but the Company will be entitled to receive from it an aggregate amount of cash and/or number of shares of the Company’s common stock, based on the settlement method election chosen for the related convertible senior notes, with a value equal to the amount by which the market price per share of the Company’s common stock, as measured under the terms of the capped call transaction, is greater than the strike price of the capped call transaction during the relevant valuation period under the capped call transaction, with such number of shares of the Company’s common stock and/or amount of cash subject to the cap price.
The Company evaluated the capped call transaction under authoritative accounting guidance and determined that it should be accounted for as a separate transaction and classified as a net reduction to additional paid-in capital within stockholders’ equity with no recurring fair value measurement recorded.
In connection with the September Exchange Transaction, the Company unwound a portion of the capped call entered into when the December 2021 Notes were issued, as they were no longer scheduled to mature in 2021. This generated proceeds, of which $0.9 million, was paid to the Company. The $0.9 million proceeds from the unwind of the capped call, which reflected the value of the options outstanding at the time of the September Exchange Transaction and the average share price of the Company’s common stock were included as an increase to Additional paid-in capital within stockholders’ equity.
In connection with the December Exchange Transaction, the Company unwound a corresponding portion of the capped call related to the notes and repurchased 1.6 million shares of its common stock from the counterparty. The Company paid the capped call counterparty $3.1 million, representing $5.6 million for the common stock repurchased from the counterparty, net of $2.5 million owed from the counterparty to the Company for unwinding the capped call. The common stock repurchased was reflected as a decrease to Retained earnings within stockholders’ equity. The proceeds from the capped call were included as an increase to Additional paid-in capital within stockholders’ equity.
December 2024 Notes
On September 17, 2019, in connection with the September Exchange Transaction, the Company exchanged $86.1 million aggregate principal of December 2021 Notes for an identical aggregate original principal amount of December 2024 Notes, plus a cash payment of $70.00 for each $1,000 principal amount exchanged, totaling approximately $6.0 million. The December 2024 Notes are due December 1, 2024, and the Company pays interest at 2.75% on the December 2024 Notes semiannually in arrears on June 1 and December 1 of each year, beginning December 1, 2019. The original principal of the December 2024 Notes will accrete at a rate of 2.375% per year (“Accretion Interest”) commencing September 17, 2019 through the maturity of the December 2024 Notes. The accreted principal amount of the December 2024 Notes is payable in cash upon maturity and is included in Other long-term liabilities.
Upon the occurrence of a fundamental change, as defined in the indenture entered into in connection with the December 2024 Notes (the “December 2024 Notes Indenture”), holders have the option to require the Company to repurchase their December 2024 Notes at a purchase price equal to 100% of the accreted principal amount of such December 2024 Notes, plus accrued interest on the original principal amount thereon.
The December 2024 Notes are convertible under any of the following circumstances at any time prior to the close of business on the business day immediately preceding June 1, 2024 (or at any time beginning on June 1, 2024 until the close of business on the second scheduled trading day immediately preceding the stated maturity):
• | During any fiscal quarter (and only during such fiscal quarter) commencing after the fiscal quarter ended December 31, 2019, if the last reported sale price of Company common stock for at least 20 trading days (whether or not consecutive), in the period of 30 consecutive trading days, ending on, and including, the last trading day of the immediately preceding fiscal quarter, exceeds 130% of the conversion price for the notes on each applicable trading day; |
• | During the five business-day period immediately after any five consecutive trading-day period, which the Company refers to as the measurement period, in which the trading price per $1,000 original principal amount of notes for each |
123
trading day of that measurement period was less than 98% of the product of the last reported sale price of Company common stock and the conversion rate for the notes for each such trading day;
• | Upon the occurrence of specified corporate events or upon a redemption of the notes, in each case as described in the December 2024 Notes Indenture; or |
• | On or after June 1, 2024, at the option of the holder prior to the second scheduled trading day preceding December 1, 2024. |
In accordance with the terms of the December 2024 Notes Indenture, the Company has the right, but not the obligation, to redeem all or any portion of the December 2024 Notes that is equal to $1,000 original principal amount or an integral multiple of $1,000 prior to their scheduled maturity on a redemption date beginning on or after December 1, 2021 and on or before the 60th scheduled trading day before December 1, 2024, for a cash purchase price equal to the redemption price, but only if the last reported sale price of Company common stock exceeds 128% of the conversion price for the December 2024 Notes on (i) each of at least 20 trading Days (whether or not consecutive) during the 30 consecutive trading days ending on, and including, the trading day immediately before the redemption notice date for such redemption; and (ii) the trading day immediately before such redemption notice date. The redemption price for the December 2024 Notes called for redemption is equal to the then accreted principal amount of such December 2024 Notes plus accrued but unpaid interest on the original principal amount thereon. The calling of any December 2024 Notes for redemption will constitute a make-whole fundamental change with respect to such notes, entitling the holders who convert such December 2024 Notes called for redemption prior to the applicable redemption date to receive an increase in the applicable conversion rate, as described in the December 2024 Notes Indenture.
The initial conversion rate for the December 2024 Notes is 262.2951 shares of the Company’s common stock per $1,000 original principal amount of December 2024 Notes, which is equivalent to an initial conversion price of approximately $3.81 per share of common stock, subject to adjustments upon the occurrence of certain specified events as set forth in the December 2024 Notes Indenture.
In accordance with the accounting guidance for an extinguishment of convertible debt instruments with a cash conversion feature, the Company was required to allocate the fair value of the consideration transferred between the liability component and the equity component. To calculate the fair value of the debt immediately prior to derecognition, the carrying value was recalculated in a manner that reflected the estimated market interest rate for a similar nonconvertible instrument at the date of issuance. Using an assumed borrowing rate of 7.05% the Company calculated the fair value of the debt representing the amount allocated to the liability component of the December 2024 Notes with the remainder of the consideration allocated to the equity conversion feature, to reflect the reacquisition of the embedded conversion option. The conversion feature together with the fees allocated to the debt are accounted for as a debt discount. As a result of the September Exchange Transaction, the Company recorded a total debt discount of $9.4 million, which included the cash conversion feature of $8.1 million and the debt issuance fees of $1.3 million, charged $5.5 million to Additional paid-in capital ($13.5 million charge to Additional paid-in capital representing the reduction to the 2021 equity component, partially offset by the $8.1 million allocated to equity for the 2024 notes) and recorded $1.2 million to deferred tax liability. The net amount charged to Additional paid-in capital represents the difference between the consideration paid for the September Exchange Transaction and the fair value of the convertible debt prior to the extinguishment.
The Accretion Interest and debt discount, including the conversion feature and issuance costs allocated to debt, are being amortized to interest expense over the term of the December 2024 Notes which increases interest expense during the term of the December 2024 Notes from the 2.75% cash coupon interest rate to an effective interest rate of 7.5%. As of December 31, 2019, the remaining discount amortization period is 4.9 years.
On December 17, 2019, in connection with the December Exchange Transaction, the Company repurchased $74.6 million in aggregate principal amount of its December 2024 Notes for $58.0 million in cash and 9.9 million shares of its common stock in privately negotiated transactions. It was determined that the repurchase of the principal amount should be accounted for as a partial extinguishment of the December 2024 Notes. As a result, a loss on extinguishment of $2.1 million was recorded at closing of the transaction. The loss on extinguishment included the de-recognition of a proportional share of the deferred issuance costs of $1.1 million.
The carrying value, accretion and unamortized discount of the December 2024 Notes were as follows:
(in thousands) | December 31, 2019 | |||
Principal amount of the December 2024 Notes | $ | 11,500 | ||
Unamortized discount of liability component | (1,200 | ) | ||
Net carrying value of the December 2024 Notes | $ | 10,300 | ||
124
Interest expense for the December 2024 Notes included in the Company’s Consolidated Statement of Operations was as follows:
Year Ended | ||||
(in thousands) | December 31, 2019 | |||
Contractual coupon interest | $ | 598 | ||
Accretion Interest on outstanding principal | 517 | |||
Amortization of debt issuance costs | 53 | |||
Amortization of conversion feature | 350 | |||
Total | $ | 1,518 | ||
Capped Call Transaction
In connection with the issuance of the December 2024 Notes in the September Exchange Transaction, the Company entered into a privately-negotiated capped call transaction with an affiliate of the underwriter of such issuance. The aggregate cost of the capped call transaction was $4.5 million. The capped call transaction is generally expected to reduce the potential dilution upon conversion of the December 2024 Notes and/or partially offset any cash payments the Company is required to make in excess of the principal amount of converted December 2024 Notes in the event that the market price per share of the Company’s common stock, as measured under the terms of the capped call transaction, is greater than the strike price of the capped call transaction. This initially corresponds to the approximate $3.81 per share conversion price of the December 2024 Notes and is subject to anti-dilution adjustments substantially similar to those applicable to the conversion rate of the December 2024 Notes. The cap price of the capped call transaction was initially $4.88 per share and is subject to certain adjustments under the terms of the capped call transaction. The Company will not be required to make any cash payments to the option counterparty upon the exercise of the options that are a part of the capped call transaction, but the Company will be entitled to receive from it an aggregate amount of cash and/or number of shares of the Company’s common stock, based on the settlement method election chosen for the related convertible senior notes, with a value equal to the amount by which the market price per share of the Company’s common stock, as measured under the terms of the capped call transaction, is greater than the strike price of the capped call transaction during the relevant valuation period under the capped call transaction, with such number of shares of the Company’s common stock and/or amount of cash subject to the cap price.
The Company evaluated the capped call transaction under authoritative accounting guidance and determined that it should be accounted for as a separate transaction and classified as a net reduction to additional paid-in capital within stockholders’ equity with no recurring fair value measurement recorded.
In connection with the December Exchange Transaction, the Company unwound a corresponding portion of the capped call related to the notes and repurchased 1.6 million shares of its common stock from the counterparty. The Company paid the capped call counterparty $1.2 million, representing $5.4 million for the common stock repurchased from the counterparty, net of $4.2 million owed from the counterparty to the Company for unwinding the capped call. The common stock repurchased was reflected as a decrease to Retained earnings within stockholders’ equity. The proceeds from the capped call were included as an increase to Additional paid-in capital within stockholders’ equity.
The Company evaluated the capped call transaction under authoritative accounting guidance and determined that it should be accounted for as separate transaction from the debt as it was entered into with a separate counterparty and does not relate to the same risk. The $4.5 million premium for the capped call was classified as a reduction to Additional paid-in capital within stockholders’ equity and will not be subject to recurring fair value measurement.
125
As of December 31, 2019, the future minimum principal payments under the December 2021 and December 2024 Notes were:
(in thousands) | December 2021 Notes | December 2024 Notes | Total | |||||||||
2020 | $ | — | $ | — | $ | — | ||||||
2021 | 19,170 | — | 19,170 | |||||||||
2022 | — | — | — | |||||||||
2023 | — | — | — | |||||||||
2024 | — | 11,500 | 11,500 | |||||||||
Thereafter | — | — | — | |||||||||
Total | $ | 19,170 | $ | 11,500 | $ | 30,670 | ||||||
14. Other Long-Term Liabilities
Other long-term liabilities consist of the following:
December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Uncertain tax positions | $ | 37,574 | $ | 31,706 | ||||
Deferred tax liability | 484 | 13,847 | ||||||
Accrued lease liability | 10,700 | 10,700 | ||||||
Long-term incentive | — | 125 | ||||||
Other | 1,140 | 465 | ||||||
Total | $ | 49,898 | $ | 56,843 | ||||
15. Commitments and Contingencies
Lease Guarantee
In connection with the spin-off by the Company of Facet Biotech Corporation (“Facet”), the Company entered into amendments to the leases for the Company’s former facilities in Redwood City, California, under which Facet was added as a co-tenant, and a Co-Tenancy Agreement, under which Facet agreed to indemnify the Company for all matters related to the leases attributable to the period after the spin-off date. In April 2010, Abbott Laboratories acquired Facet and later renamed the entity AbbVie Biotherapeutics, Inc. (“AbbVie”). If AbbVie were to default under its lease obligations, the Company could be held liable by the landlord as a co-tenant and, thus, the Company has in substance guaranteed the payments under the lease agreements for the Redwood City facilities. As of December 31, 2019, the total lease payments for the duration of the guarantee, which runs through December 2021, are approximately $22.6 million.
The Company prepared a discounted, probability weighted cash flow analysis to calculate the estimated fair value of the lease guarantee as of the spin-off. The Company was required to make assumptions regarding the probability of Facet’s default on the lease payment, the likelihood of a sublease being executed and the times at which these events could occur. These assumptions are based on information that the Company received from real estate brokers and the then-current economic conditions, as well as expectations of future economic conditions. The fair value of this lease guarantee was charged to Additional paid-in capital upon the spin-off and any future adjustments to the carrying value of the obligation will also be recorded in Additional paid-in capital.
The Company has recorded a liability of $10.7 million on its Consolidated Balance Sheets as of December 31, 2019 and 2018, related to this guarantee. In future periods, the Company may adjust this liability for any changes in the ultimate outcome of this matter that are both probable and estimable.
Irrevocable Letters of Credit
On June 30, 2016, the Company purchased a $75.0 million certificate of deposit, which is designated as cash collateral for the $75.0 million letter of credit issued on July 1, 2016 with respect to the first anniversary payment under the Noden Purchase Agreement. In addition, the Company provided an irrevocable and unconditional guarantee to Novartis, to pay up to $14.0 million
126
of the remaining amount of the first anniversary payment not covered by the letter of credit. The Company concluded that both guarantees were contingent obligations and should be accounted for in accordance with ASC 450, Contingencies. Further, it was concluded that both guarantees did not meet the conditions to be accrued at June 30, 2016 and December 31, 2016. On July 3, 2017, the first anniversary payment of $89.0 million was paid pursuant to the Noden Purchase Agreement and the $14.0 million guarantee expired. On July 31, 2017, the $75.0 million certificate of deposit matured, and on August 1, 2017, the letter of credit terminated.
Purchase Obligations
Noden DAC and Novartis entered into a supply agreement pursuant to which Novartis will manufacture and supply to Noden DAC a bulk tableted form of the Noden Products and active pharmaceutical ingredient (“API”). In May 2019, Noden DAC and Novartis entered into an amended supply agreement pursuant to which Novartis will supply to Noden DAC a bulk tableted form of the Noden Products through 2020 and API through June 2021. The supply agreement may be terminated by either party for material breach that remains uncured for a specified time period. Under the terms of the amended supply agreement, Noden DAC is committed to purchase certain quantities of bulk product and API that would amount to approximately $61.7 million through June 2021, of which $39.8 million is committed over the next twelve months, which are guaranteed by the Company. While the supply agreement provides that the parties will agree to reasonable accommodations with respect to changes in firm orders, the Company expects that Noden DAC will meet the requirements of the supply agreement, unless otherwise negotiated.
LENSAR entered into various supply agreements for the manufacture and supply of certain components. The supply agreements commit LENSAR to a minimum purchase obligation of approximately $10.4 million over the next twenty-four months of which $9.6 million is due in the next 12 months, a portion of which are guaranteed by the Company. LENSAR expects to meet these requirements.
Escrow Receivable
On April 1, 2014, the Company entered into a note purchase agreement with Accel 300, a wholly-owned subsidiary of kaléo, Inc. (“kaléo”), pursuant to which the Company acquired $150.0 million of secured notes due 2029 (the “kaléo Note”). The kaléo Note was issued pursuant to an indenture between Accel 300 and U.S. Bank, National Association, as trustee, and was secured by 20% of net sales of its first approved product, Auvi-Q® (epinephrine auto-injection, USP) (known as Allerject® in Canada) and 10% of net sales of kaléo’s second proprietary auto-injector based product, EVZIO (naloxone hydrochloride injection) (the “kaléo Revenue Interests”), and a pledge of kaléo’s equity ownership in Accel 300.
On September 21, 2017, the Company entered into an agreement (the “kaléo Note Sale Agreement”) with MAMKangaroo Lender, LLC, a Delaware limited liability company (the kaléo Purchaser”), pursuant to which the Company sold its entire interest in the kaléo Note for an aggregate cash purchase price of $141.7 million.
Pursuant to the terms of the kaléo Note Sale Agreement, $1.4 million of the aggregate purchase price was deposited into an escrow account as a potential payment against certain contingencies. The escrow period ended on March 20, 2019 and the escrow agent released the entire $1.4 million to the Company.
16. Stockholders’ Equity
Stock Repurchase Program
On March 1, 2017, the Company announced that its board of directors authorized the repurchase through March 2018 of issued and outstanding shares of the Company’s common stock having an aggregate value of up to $30.0 million pursuant to a share repurchase program. The repurchases under the share repurchase program were made from time to time in the open market or in privately negotiated transactions and were funded from the Company’s working capital. All shares of common stock repurchased under the Company’s share repurchase program were retired and restored to authorized but unissued shares of common stock at June 30, 2017. The Company repurchased 13.3 million shares of its common stock under the share repurchase program during the fiscal year ended December 31, 2017 for an aggregate purchase price of $30.0 million, or an average cost of $2.25 per share, including trading commissions.
On September 25, 2017, the Company announced that its board of directors authorized the repurchase of issued and outstanding shares of the Company’s common stock having an aggregate value of up to $25.0 million pursuant to a share repurchase program. The repurchases under the share repurchase program were made from time to time in the open market or in privately negotiated transactions and were funded from the Company’s working capital. All shares of common stock repurchased under this share repurchase program were retired and restored to authorized but unissued shares of common stock. The Company repurchased 8.7
127
million shares of its common stock under the share repurchase program during the fiscal year ended December 31, 2018, for an aggregate purchase price of $25.0 million, or an average cost of $2.86 per share, including trading commissions.
On September 24, 2018, the Company announced that its board of directors authorized the repurchase of issued and outstanding shares of the Company’s common stock having an aggregate value of up to $100.0 million pursuant to a share repurchase program. Repurchases under this share repurchase program were made from time to time in the open market or in privately negotiated transactions and funded from the Company’s working capital. All shares of common stock repurchased under this repurchase program were retired and restored to authorized but unissued shares of common stock at July 31, 2019. The Company repurchased 31.0 million shares of its common stock under this share repurchase program for an aggregate purchase price of $100.0 million, or an average cost of $3.22 per share, including trading commissions.
On December 9, 2019, the Company announced that its board of directors authorized the repurchase of issued and outstanding shares of the Company's common stock and convertible notes up to an aggregate value of $200 million. On December 16, 2019, the Company announced that its board of directors approved a $75 million increase to the aforementioned $200 million repurchase program to acquire outstanding PDL common stock and convertible notes. Repurchases under the new repurchase program will be made from time to time in the open market or in privately negotiated transactions and funded from the Company’s working capital. The amount and timing of such repurchases will depend upon the price and availability of shares or convertible notes, general market conditions and the availability of cash. Repurchases may also be made under a trading plan under Rule 10b5-1, which would permit shares or convertible notes to be repurchased when the Company might otherwise be precluded from doing so because of self-imposed trading blackout periods or other regulatory restrictions. All shares of common stock repurchased under the Company’s new share repurchase program are expected to be retired and restored to authorized but unissued shares of common stock. All convertible notes repurchased under the program will be retired. As of December 31, 2019, the Company had repurchased $44.8 million in aggregate principal amount of 2021 Convertible Notes and $74.6 million in aggregate principal amount of 2024 Convertible Notes for consideration consisting of a cash payment of $97.9 million and the issuance of 13.4 million shares of the Company’s common stock. As of December 31, 2019, the Company had not repurchased any shares of common stock under this program. This repurchase program may be suspended at any time without notice.
17. Accumulated Other Comprehensive Income
Comprehensive income is comprised of net (loss) income and other comprehensive (loss) income. The Company includes unrealized net gains (losses) on investments held in its available-for-sale securities and unrealized gains (losses) on its cash flow hedges in other comprehensive (loss) income, and presents the amounts net of tax. The Company’s other comprehensive (loss) income is included in the Company’s Consolidated Statements of Comprehensive (Loss) Income.
The balance of “accumulated other comprehensive (loss) income,” net of tax, was as follows:
(in thousands) | Unrealized gains (losses) on available-for- sale securities | Total Accumulated Other Comprehensive Income | ||||||
Balance at December 31, 2016 | $ | — | $ | — | ||||
Activity for the year ended December 31, 2017 | 1,181 | 1,181 | ||||||
Ending Balance at December 31, 2017 | 1,181 | 1,181 | ||||||
Activity for the year ended December 31, 2018 | (1,181 | ) | (1,181 | ) | ||||
Ending Balance at December 31, 2018 | — | — | ||||||
Activity for the year ended December 31, 2019 | — | — | ||||||
Ending Balance at December 31, 2019 | $ | — | $ | — | ||||
18. Stock-Based Compensation
The Company grants restricted stock awards and stock options pursuant to a stockholder approved stock-based incentive plan.
128
The following table summarizes the Company’s stock option and restricted stock award compensation expense during the years ended December 31, 2019, 2018 and 2017:
Year Ended December 31, | ||||||||||||
Stock-based Compensation | 2019 | 2018 | 2017 | |||||||||
(in thousands) | ||||||||||||
Employees and directors | $ | 6,907 | $ | 4,758 | $ | 3,138 | ||||||
The fair value of each stock option grant is estimated on the date of grant using the Black-Scholes option-pricing model. Expected volatility is based on the historical volatility of our common stock over the estimated expected life of the options. The expected term represents the period of time the options are expected to be outstanding. The expected term is based on the “simplified method” as defined by the SEC Staff Accounting Bulletin No. 110 (Topic 14.D.2). The Company uses the “simplified method” due to the lack of sufficient historical exercise data to provide a reasonable basis upon which to otherwise estimate the expected life of the options. The risk-free rate is based on yields on U.S. Treasury securities with a maturity similar to the estimated expected term of the options. The fair value of restricted stock awards is based on the closing price of the Company’s common stock on the grant date.
The fair value of our stock options was estimated assuming no expected dividends and the following weighted-average assumptions:
Year Ended December 31, | ||||||||||||
2019 | 2018 | 2017 | ||||||||||
Range of expected term (in years) | 3.5 | - | 6.1 | 3.5 | - | 6.0 | 3.7 | |||||
Range of risk-free interest rate | 1.5% | - | 3.0% | 2.7% | - | 3.0% | 2.0% | |||||
Expected volatility | 40% | 40% | 44% | |||||||||
Stock-Based Incentive Plans
2005 Equity Incentive Plan
The Company currently has one active stock-based incentive plan under which it may grant stock-based awards to the Company’s employees, directors and non-employees.
Under the Company’s Amended and Restated 2005 Equity Incentive Plan effective June 8, 2018 (the “2005 Equity Incentive Plan”), the Company is authorized to issue a variety of incentive awards, including stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, performance share and performance unit awards, deferred compensation awards and other stock-based or cash-based awards. As of December 31, 2019, awards granted under the 2005 Equity Incentive Plan consisted of stock options and restricted stock awards. There were no other grants of any other award types under the 2005 Equity Incentive Plan.
In June 2018, the Company’s stockholders approved an amendment and restatement of the 2005 Equity Incentive Plan that increased the number of shares available for grant by 15,000,000 to 26,200,000. The number of shares of common stock authorized for issuance, shares of common stock issued upon exercise of options or grant of restricted stock awards, shares of common stock subject to outstanding awards and shares available for grant under this plan as of December 31, 2019, are as follows:
Title of Plan | Total Shares of Common Stock Authorized | Total Shares of Common Stock Issued | Total Shares of Common Stock Available for Grant | ||||||
2005 Equity Incentive Plan | 26,200,000 | 15,889,993 | 10,310,007 | ||||||
129
Stock Options
The following table summarizes the option activity under the 2005 Equity Incentive Plan for the year ended December 31, 2019:
Options | Weighted-Average Exercise Price | Weighted-Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | ||||||||||
(in thousands) | (in thousands) | ||||||||||||
Outstanding at beginning of year | 6,908 | $ | 2.76 | 9.1 | $ | 1,099 | |||||||
Granted | 5,796 | $ | 3.58 | ||||||||||
Forfeited | (1,052 | ) | $ | 3.27 | |||||||||
Outstanding at end of year | 11,652 | $ | 3.12 | 8.5 | $ | 3,473 | |||||||
Exercisable at end of year | 3,154 | $ | 2.77 | 8.1 | $ | 1,526 | |||||||
Options to purchase common stock generally vest over a 3 or 4-year period and are generally granted for a term of 10 years.
The weighted-average grant-date fair value of options granted during the year ended December 31, 2019 was $1.49. The total fair value of options vested during the year ended December 31, 2019 was approximately $5.6 million. Total unrecognized compensation expense of $7.8 million related to options will be recognized over a weighted-average period of 1.6 years.
Restricted Stock Awards
Restricted stock has the same rights as other issued and outstanding shares of the Company’s common stock, including, in some cases, the right to accrue dividends, which are held in escrow until the award vests. The compensation expense related to these awards is determined using the fair market value of the Company’s common stock on the date of the grant, and the compensation expense is recognized ratably over the vesting period. Under the Company’s restricted stock plans, restricted stock awards typically vest over one to five years and compensation expense associated with these awards is recognized on a straight-line basis over the vesting period. In addition to service requirements, vesting of restricted stock awards may be subject to the achievement of specified performance goals set by the Compensation Committee. If the performance goals are not met, no compensation expense is recognized and any previously recognized compensation expense is reversed.
The following table summarizes the restricted stock award activity under the 2005 Equity Incentive Plan for the year ended December 31, 2019:
2019 | ||||||
Number of shares | Weighted-average grant-date fair value per share | |||||
(in thousands) | ||||||
Unvested at beginning of year | 723 | $ | 2.79 | |||
Awards granted | 917 | $ | 3.62 | |||
Awards vested | (519 | ) | $ | 2.79 | ||
Withheld related to net settlement | (64 | ) | $ | 2.78 | ||
Forfeited | (124 | ) | $ | 3.18 | ||
Unvested at end of year | 933 | $ | 3.56 | |||
The total fair value of restricted stock awards vested during the years ended December 31, 2019, 2018 and 2017 was approximately $1.4 million, $2.1 million and $2.8 million, respectively.
The weighted-average grant date fair value for restricted stock awards granted under the 2005 Equity Incentive Plan for the years end December 31, 2019, 2018 and 2017 was $3.62, $2.61 and $2.15, respectively.
130
At December 31, 2019, there was approximately $1.6 million of total unrecognized compensation expense related to restricted stock awards granted under the 2005 Equity Incentive Plan, which is expected to be recognized over a weighted-average period of 1.2 years.
Inducement Award Agreements
On September 12, 2017, the Company granted 961,000 shares of common stock in the form of a non-statutory inducement stock option grant pursuant to a non-statutory inducement stock option agreement and granted 240,200 shares of our common stock in the form of an inducement restricted stock grant pursuant to an inducement restricted stock agreement. These inducement awards were not granted under the 2005 Equity Incentive Plan.
Inducement Stock Option Activity
As of December 31, 2019, all stock option awarded under the non-statutory inducement stock option agreement were outstanding and 373,719 shares were exercisable. The total fair value of options vested during the year ended December 31, 2019 was approximately $0.5 million. Total unrecognized compensation expense of $0.2 million related to these options will be recognized over a weighted-average period of 1.8 years.
Inducement Restricted Stock
As of December 31, 2019, 80,067 shares of restricted stock awarded under the non-statutory inducement restricted stock agreement were outstanding and unvested. The total fair value of the restricted stock awards vested during the year ended December 31, 2019 was approximately $0.3 million.
Compensation expense associated with unvested restricted stock awards is recognized on a straight-line basis over the vesting period. At December 31, 2019, there was approximately $0.1 million of total unrecognized compensation expense related to restricted stock awards granted under the non-statutory inducement restricted stock agreement, which is expected to be recognized over a weighted-average period of 1.0 year.
19. Revenue from Contracts with Customers
Disaggregation of Revenue
The Company disaggregates its revenue from contracts with customers by segment and geographic location as the Company believes it best depicts how the nature, amount, timing and uncertainty of its revenue and cash flows are affected by economic factors. In the following table, revenue is disaggregated by segment and primary geographical market for the years ended December 31, 2019 and 2018:
Year Ended December 31, 2019 | Year Ended December 31, 2018 | ||||||||||||||
(in thousands) | Medical Devices | Pharmaceutical | Medical Devices | Pharmaceutical | |||||||||||
Primary geographical markets: | |||||||||||||||
North America | $ | 10,155 | $ | 26,034 | $ | 7,425 | $ | 41,900 | |||||||
Europe | 3,438 | 22,816 | 2,451 | 25,259 | |||||||||||
Asia | 11,536 | 6,243 | 7,136 | 13,637 | |||||||||||
Other | 433 | — | 377 | — | |||||||||||
Total revenue from contracts with customers 1 | $ | 25,562 | $ | 55,093 | $ | 17,389 | $ | 80,796 | |||||||
_______________
1 The table above does not include lease revenue from the Company’s Medical Devices segment of $5.2 million and $7.3 million for the years ended December 31, 2019 and 2018, respectively. For additional information, see Note 8, Leases.
131
Contract Balances
The following table provides information about receivables, contract assets and contract liabilities from contracts with customers:
(in thousands) | December 31, 2019 | December 31, 2018 | ||||||
Receivables, current and non-current, net | $ | 10,777 | $ | 20,655 | ||||
Contract assets | $ | 3,512 | $ | 2,595 | ||||
Contract liabilities | $ | 4,024 | $ | 8,938 | ||||
Receivables, Net—Receivables, net, include amounts billed and due from customers. The amounts due are stated at their net estimated realizable value and are classified as current or noncurrent based on the timing of when the Company expects to receive payment. The Company maintains an allowance for doubtful accounts to provide for the estimated amount of receivables that will not be collected. The allowance is based upon an assessment of customer creditworthiness, historical payment experience, the age of outstanding receivables and collateral to the extent applicable.
Contract Assets—The Company’s contract assets represent revenue recognized for performance obligations completed before an unconditional right to payment exists, and therefore invoicing or associated reporting from the customer regarding the computation of the net product sales has not yet occurred. The Company classifies contract assets in Prepaid and other current assets in the Company’s Consolidated Balance Sheets based on the timing of when it expects to receive payment.
(in thousands) | Medical Devices | Pharmaceutical | Total | |||||||||
Contract assets at December 31, 2018 | $ | — | $ | 2,595 | $ | 2,595 | ||||||
Contract assets recognized | — | 12,259 | 12,259 | |||||||||
Payments received | — | (11,342 | ) | (11,342 | ) | |||||||
Contract assets at December 31, 2019 | $ | — | $ | 3,512 | $ | 3,512 | ||||||
Contract Liabilities—The Company’s contract liabilities consist of deferred revenue for products sold to customers for which the performance obligation has not been completed by the Company. The Company classifies deferred revenue as current or noncurrent based on the timing of when it expects to recognize revenue. The noncurrent portion of deferred revenue is included in Other long-term liabilities in the Company’s Consolidated Balance Sheets.
(in thousands) | Medical Devices | Pharmaceutical | Total | |||||||||
Contract liabilities at December 31, 2018 | $ | 1,167 | $ | 7,771 | $ | 8,938 | ||||||
Additions | 916 | 2,123 | 3,039 | |||||||||
Amounts recognized in revenue | (1,008 | ) | (6,945 | ) | (7,953 | ) | ||||||
Contract liabilities at December 31, 2019 | $ | 1,075 | $ | 2,949 | $ | 4,024 | ||||||
Transaction Price Allocated to Future Performance Obligations
The following table includes estimated revenue expected to be recognized in the future related to performance obligations that are unsatisfied (or partially unsatisfied) at the end of the reporting period.
Twelve months ended | ||||||||||||
(in thousands) | December 31, 2020 | Thereafter | Total | |||||||||
Pharmaceutical product sales | $ | 2,326 | $ | — | $ | 2,326 | ||||||
Medical device sales | $ | 5,473 | $ | 6,280 | $ | 11,753 | ||||||
The Company does not disclose the value of unsatisfied performance obligations for (i) contracts with original expected lengths of one year or less or (ii) contracts for which the Company recognizes revenue at the amount to which it has the right to invoice for the products delivered or services performed.
132
20. Segment Information
In connection with its investment in Evofem in the second quarter of 2019, the Company added a fourth reportable segment, “Strategic Positions.” This had no impact on the Company’s prior segment reporting structure.
Information regarding the Company’s segments for the year ended December 31, 2019 and 2018 is as follows:
Revenues by segment | Year Ended December 31, | |||||||
(in thousands) | 2019 | 2018 | ||||||
Medical Devices | $ | 30,742 | $ | 24,652 | ||||
Strategic Positions | — | — | ||||||
Pharmaceutical | 55,093 | 80,796 | ||||||
Income Generating Assets | (31,078 | ) | 92,662 | |||||
Total revenues | $ | 54,757 | $ | 198,110 | ||||
(Loss) income by segment | Year Ended December 31, | |||||||
(in thousands) | 2019 | 2018 | ||||||
Medical Devices | $ | (5,230 | ) | $ | (5,086 | ) | ||
Strategic Positions | 28,758 | — | ||||||
Pharmaceutical | (19,048 | ) | (98,368 | ) | ||||
Income Generating Assets | (74,891 | ) | 34,595 | |||||
Total net (loss) income | $ | (70,411 | ) | $ | (68,859 | ) | ||
Long-lived assets by segment | Year Ended December 31, | |||||||
(in thousands) | 2019 | 2018 | ||||||
Medical Devices | $ | 2,435 | $ | 3,545 | ||||
Strategic Positions | — | — | ||||||
Pharmaceutical | 2,960 | 3,682 | ||||||
Income Generating Assets | 125 | 160 | ||||||
Total long-lived assets | $ | 5,520 | $ | 7,387 | ||||
The operations for the Medical Devices segment are primarily located in the United States and the operations for the Pharmaceutical segment are primarily located in Italy, Ireland and the United States.
21. Concentration of Credit Risk
Product Line Concentration
The percentage of total revenue recognized, which individually accounted for 10% or more of the Company’s total revenues in one or more of the periods presented below, was as follows:
Year Ended December 31, | |||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||
AcelRx | (104 | )% | (1 | )% | 2 | % | |||
Assertio | 48 | % | 42 | % | 52 | % | |||
Biogen | — | % | 2 | % | 11 | % | |||
LENSAR | 56 | % | 12 | % | 5 | % | |||
Noden | 101 | % | 41 | % | 22 | % | |||
133
Total revenues by geographic area are based on the country of domicile of the counterparty to the agreement are as follows:
Year Ended December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
United States | $ | 10,143 | $ | 148,622 | $ | 291,448 | ||||||
Europe | 26,254 | 27,709 | 16,144 | |||||||||
Rest of world | 18,360 | 21,779 | 12,468 | |||||||||
Total revenues | $ | 54,757 | $ | 198,110 | $ | 320,060 | ||||||
The following tables presents total receivables which individually account for 10% or more of the Company’s total receivables balance:
December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Assertio | $ | 3,176 | $ | — | ||||
Cardinal Health | $ | 1,071 | $ | 2,732 | ||||
McKesson | $ | 1,311 | $ | 2,352 | ||||
AmerisourceBergen | $ | 823 | $ | 4,330 | ||||
22. Income Taxes
For financial reporting purposes, (loss) income before income taxes includes the following components:
Years Ended December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
United States | $ | (54,861 | ) | $ | 48,844 | $ | 195,865 | |||||
Foreign | (18,879 | ) | (104,766 | ) | (11,338 | ) | ||||||
Total | $ | (73,740 | ) | $ | (55,922 | ) | $ | 184,527 | ||||
The provision for income taxes for the years ended December 31, 2019, 2018 and 2017 consisted of the following:
Year Ended December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
Current income tax expense (benefit) | ||||||||||||
Federal | $ | 3,750 | $ | 2,169 | $ | 31,338 | ||||||
State | 2,141 | 1,029 | 2,843 | |||||||||
Foreign | 1,677 | (4,107 | ) | 529 | ||||||||
Total current | 7,568 | (909 | ) | 34,710 | ||||||||
Deferred income tax (benefit) expense | ||||||||||||
Federal | (11,062 | ) | 11,497 | 36,911 | ||||||||
State | 445 | 1,313 | 2,591 | |||||||||
Foreign | — | 1,036 | (386 | ) | ||||||||
Total deferred | (10,617 | ) | 13,846 | 39,116 | ||||||||
Total provision | $ | (3,049 | ) | $ | 12,937 | $ | 73,826 | |||||
134
A reconciliation of the income tax provision computed using the U.S. statutory federal income tax rate compared to the income tax provision for income included in the Consolidated Statements of Operations is as follows:
Year Ended December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
Tax at U.S. statutory rate on (loss) income before income taxes | $ | (15,485 | ) | $ | (11,744 | ) | $ | 64,589 | ||||
Change in valuation allowance | 5,570 | 11,226 | 1,807 | |||||||||
State taxes | 2,149 | 1,376 | 1,496 | |||||||||
Change in uncertain tax positions | 1,513 | 809 | 681 | |||||||||
Foreign income | — | 1,048 | 3,231 | |||||||||
Foreign rate differential | 1,603 | 8,936 | 1,356 | |||||||||
Change in tax rate reform | — | — | 716 | |||||||||
True-ups | 249 | 939 | — | |||||||||
Other | 1,352 | 347 | (50 | ) | ||||||||
Total | $ | (3,049 | ) | $ | 12,937 | $ | 73,826 | |||||
Deferred tax assets and liabilities are determined based on the differences between financial reporting and income tax bases of assets and liabilities, as well as net operating loss carryforwards and are measured using the enacted tax rates and laws in effect when the differences are expected to reverse. The significant components of the Company’s net deferred tax assets and liabilities are as follows:
December 31, | ||||||||
(in thousands) | 2019 | 2018 | ||||||
Deferred tax assets: | ||||||||
Net operating loss carryforwards | $ | 19,789 | $ | 11,713 | ||||
Research and other tax credits | 1,448 | 1,580 | ||||||
Intangible assets | — | 2,203 | ||||||
Stock-based compensation | 1,781 | 1,130 | ||||||
Accruals | 1,969 | 2,362 | ||||||
Debt modifications | 7,189 | 4,661 | ||||||
Capital loss carryforward | 1,213 | 1,866 | ||||||
Other | 9,609 | 6,642 | ||||||
Total deferred tax assets | 42,998 | 32,157 | ||||||
Valuation allowance | (22,143 | ) | (13,271 | ) | ||||
Total deferred tax assets, net of valuation allowance | 20,855 | 18,886 | ||||||
Deferred tax liabilities: | ||||||||
Debt modifications | (308 | ) | (2,981 | ) | ||||
Intangible assets | (20,720 | ) | (28,214 | ) | ||||
Other | (311 | ) | — | |||||
Total deferred tax liabilities | (21,339 | ) | (31,195 | ) | ||||
Net deferred tax liabilities | $ | (484 | ) | $ | (12,309 | ) | ||
As of December 31, 2019 and 2018, the Company had federal net operating loss carryforwards of $108.6 million and $101.7 million, respectively. As of December 31, 2019 and 2018, the Company also had state net operating loss carryforwards of $63.9 million and $70.8 million, respectively, excluding $215.5 million of California net operating losses available to offset assessments, if any, resulting from the current audit by the California Franchise Tax Board. The federal and state net operating loss carryforwards will begin expiring in the year 2023, if not utilized. As of December 31, 2019 and 2018, the Company had $2.2 million of federal tax credits that will begin expiring in the year 2025, if not utilized. As of December 31, 2019 and 2018, the Company had $19.3 million of state tax credit carryforwards that do not expire. As of December 31, 2019 and 2018, the Company had $125.6 million and $73.0 million, respectively, of net operating loss carryforwards in Ireland that do not expire.
135
Utilization of the federal and state net operating loss and tax credit carryforwards may be subject to a substantial annual limitation due to the “change in ownership” provisions of the Internal Revenue Code of 1986. The annual limitation may result in the expiration of net operating losses and credits before utilization. Of the Company’s $108.6 million of federal net operating loss carryforwards as of December 31, 2019, $28.7 million are subject to an annual limitation of $1.8 million for each of the years ending December 31, 2019 to 2022, and $1.3 million for the year ending December 31, 2023. As of December 31, 2019, the Company estimates that at least $22.0 million of federal net operating loss carryforwards and none of the state net operating losses will expire unutilized. Tax attributes acquired from LENSAR may be subject to separate return limitations that may limit the corporation’s ability to use the acquired net operating losses and credits. Furthermore, under the 2017 Tax Act, although the treatment of tax losses generated in taxable years ending before December 31, 2017 has not changed, tax losses generated in taxable years beginning after December 31, 2017 may only be utilized to offset 80% of taxable income annually. This change may require the Company to pay additional federal income taxes in future years if additional losses are generated post 2017.
As of December 31, 2019, the Company determined that it was more likely than not that certain deferred tax assets would not be realized in the near future and had a $22.1 million valuation allowance against deferred tax assets. The net change in total valuation allowance for each of the years ending December 31, 2019 and 2018, was an increase of $8.9 million and $11.2 million, respectively. $1.2 million of the valuation allowance at December 31, 2019, is related to capital losses that have limited carryforward utilization. The Company does not have an expectation of future capital gains against which such losses could be utilized and as such determined that it was more likely than not that such deferred tax assets would not be realized. $14.7 million of the valuation allowance at December 31, 2019, is related to Ireland deferred tax assets that the Company determined it was more likely than not would not be realized. $6.2 million of the valuation allowance at December 31, 2019 is related to federal and state deferred tax assets that the Company determined it was more likely than not would be realized.
The cumulative amount of earnings of our foreign subsidiaries are expected to be permanently invested in the foreign subsidiaries. Deferred taxes have not been provided on the excess of book basis over tax basis, or the excess tax basis over book basis in the shares of our foreign subsidiaries because these basis differences are not expected to reverse in the foreseeable future and are essentially permanent in duration. Our intention is to reinvest the earnings of the foreign subsidiaries indefinitely.
The Tax Cuts and Job Act of 2017 significantly changed the existing U.S. corporate income tax laws by, among other things, lowering the corporate tax rate (from a top rate of 35% to a flat rate of 21%), implementing elements of a territorial tax system, and imposing a one-time deemed repatriation transition tax on cumulative undistributed foreign earnings, for which the Company has not previously paid U.S. taxes.
A reconciliation of the Company’s unrecognized tax benefits, excluding accrued interest and penalties, for 2019, 2018 and 2017 is as follows:
December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
Balance at the beginning of the year | $ | 80,783 | $ | 79,179 | $ | 59,429 | ||||||
Increases related to tax positions from prior fiscal years | 3,927 | 1,604 | 783 | |||||||||
Increases related to tax positions taken during current fiscal year | — | — | 18,967 | |||||||||
Decreases related to tax positions from prior fiscal years | (497 | ) | — | — | ||||||||
Balance at the end of the year | $ | 84,213 | $ | 80,783 | $ | 79,179 | ||||||
The future impact of the unrecognized tax benefit of $84.2 million, if recognized, is as follows: $27.9 million would affect the effective tax rate and $56.3 million would result in adjustments to deferred tax assets and valuation allowances. The Company periodically evaluates its exposures associated with our tax filing positions. The Company is currently under audit by the California Franchise Tax Board and the Internal Revenue Service. The timing of the audit resolution and the amount to be ultimately paid (if any) is uncertain. The outcome of these audits could result in the payment of tax amounts that differ from the amounts the Company has reserved for uncertain tax positions for the periods under audit resulting in incremental expense or a reversal of the Company’s reserves in a future period. At this time, the Company does not anticipate a material change in the unrecognized tax benefits related to the California or Internal Revenue Service audits that would affect the effective tax rate, deferred tax assets or valuation allowances over the next 12 months.
Estimated interest and penalties associated with unrecognized tax benefits increased our income tax expense in the Consolidated Statements of Operations by $1.6 million, during the year ended December 31, 2019 and $1.0 million during each of the years ended December 31, 2018 and 2017, respectively. In general, our income tax returns are subject to examination by U.S. federal, state and local tax authorities for tax years 2000 forward. Interest and penalties associated with unrecognized tax benefits accrued
136
on the balance sheet were $9.7 million and $8.0 million as of December 31, 2019 and 2018, respectively. The Company is currently under income tax examination by the State of California for the tax years 2009 through 2015 and by the Internal Revenue Service for the tax year 2016.
23. Net (Loss) Income per Share
Net (Loss) Income per Basic and Diluted Share | Year Ended December 31, | ||||||||||
(in thousands, except per share amounts) | 2019 | 2018 | 2017 | ||||||||
Numerator | |||||||||||
(Loss) income attributable to the PDL’s stockholders used to compute net (loss) income per basic and diluted share | $ | (70,411 | ) | $ | (68,859 | ) | $ | 110,748 | |||
Denominator | |||||||||||
Total weighted-average shares used to compute net (loss) income attributable to PDL's stockholders, per basic share | 118,631 | 145,669 | 155,394 | ||||||||
Restricted stock outstanding | — | — | 863 | ||||||||
Shares used to compute net (loss) income attributable to PDL’s stockholders, per diluted share | 118,631 | 145,669 | 156,257 | ||||||||
Net (loss) income attributable to PDL’s stockholders, per share - basic | $ | (0.59 | ) | $ | (0.47 | ) | $ | 0.71 | |||
Net (loss) income attributable to PDL’s stockholders, per share - diluted | $ | (0.59 | ) | $ | (0.47 | ) | $ | 0.71 | |||
The Company computes net (loss) income per diluted share using the sum of the weighted-average number of common and common equivalent shares outstanding common equivalent shares used in the computation of net (loss) income per diluted share include shares that may be issued pursuant to outstanding stock options and restricted stock awards in each case, on a weighted-average basis for the period they were outstanding, including, if applicable, the underlying shares using the treasury stock method.
The February 2018 Notes that were repaid on February 1, 2018, the December 2021 Notes and the December 2024 Notes allow, or previously allowed, for the settlement entirely or partially in cash, and are accounted for under the treasury stock method. Under the treasury stock method, the shares issuable upon conversion of the notes are not included in the calculation of diluted earnings per share except to the extent that the conversion value of the notes exceeds their principal amount. The effect of which, for diluted earnings per share purposes, is that only the number of shares of common stock that would be necessary to settle such excess, if the Company elected to settle such excess in shares, are included in the computation.
December 2021 Notes and December 2024 Notes Capped Call Potential Dilution
In November 2016, the Company issued $150.0 million in aggregate principal of the December 2021 Notes. In September 2019, the Company entered into the September Exchange Transaction through which it exchanged a portion of the December 2021 Notes for the December 2024 Notes. Both the December 2021 Notes and the December 2024 Notes provide in certain situations for the conversion of the outstanding principal amount into shares of the Company’s common stock at a predefined conversion rate. In conjunction with the issuance of the December 2021 Notes and the issuance of the December 2024 Notes pursuant to the September Exchange Transaction, the Company entered into capped call transactions, with a hedge counterparty. The capped call transactions are expected generally to reduce the potential dilution, and/or offset, to an extent, the cash payments the Company may choose to make in excess of the principal amount, upon conversion of the December 2021 Notes or the December 2024 Notes. The Company has excluded the capped call transaction from the net (loss) income per diluted share computation as such securities would have an anti-dilutive effect and those securities should be considered separately rather than in the aggregate in determining whether their effect on net (loss) income per diluted share would be dilutive or anti-dilutive. For additional information regarding the conversion rates and the capped call transactions related to the Company’s December 2021 Notes and December 2024 Notes; see Note 13, Convertible Senior Notes.
Anti-Dilutive Effect of Restricted Stock Awards and Stock Options
For the years ended December 31, 2019, 2018 and 2017, the Company excluded approximately 1,013,000, 1,139,000, and 1,830,000 shares, respectively, underlying restricted stock awards, calculated on a weighted-average basis, from it’s net (loss) income per diluted share calculations because their effect was anti-dilutive.
137
For the years ended December 31, 2019, 2018 and 2017, the Company excluded approximately 11,192,000, 3,892,000 and 502,000 shares underlying outstanding stock options, respectively, calculated on a weighted-average basis, from the Company’s net (loss) income per diluted share calculations because their effect was anti-dilutive.
24. Business Combinations
LENSAR TRANSACTION
Description of the LENSAR Transaction
In December 2016, LENSAR filed the Chapter 11 case with the support of the Company, as its largest senior secured creditor under a credit agreement, as amended, that the Company and LENSAR had entered into in 2013. For more information regarding the credit agreement between the Company and LENSAR, please see Note 7, Notes and Other Long-Term Receivables. In January 2017, the Company agreed to provide debtor-in-possession financing of up to $2.8 million in new advances to LENSAR so that it could continue to operate its business during the remainder of the Chapter 11 case. As part of the Chapter 11 case, LENSAR filed a Chapter 11 plan of reorganization, with the Company’s support, under which LENSAR would issue 100% of its equity securities to the Company in exchange for the cancellation of the Company’s claims as a secured creditor in the Chapter 11 case. Following consummation of the Chapter 11 plan of reorganization, LENSAR would become an operating subsidiary of the Company and the Company provided LENSAR a new, senior-secured, first-priority term loan facility (the “Exit Facility”).
On April 26, 2017, the bankruptcy court approved the plan of reorganization. On May 11, 2017, LENSAR and the Company consummated the plan of reorganization and LENSAR emerged from bankruptcy. Pursuant to the plan of reorganization, the Company obtained control of 100% of the outstanding voting shares of LENSAR. All assets of the LENSAR bankruptcy estate re-vested in reorganized LENSAR free and clear of all liens, claims or charges. Upon consummation of the plan of reorganization, all debt owed to the Company was eliminated other than the Exit Facility. Liabilities to other creditors, including general unsecured creditors, were satisfied through the plan of reorganization.
The Company concluded that the LENSAR transaction should be accounted for by applying the acquisition method in accordance with ASC 805 that did not involve a transfer of consideration (“combinations by contract”).
Fair Value of Consideration Transferred
Contemporaneously with the cancellation of the Company’s notes receivable with a carrying value of $43.9 million, the Company acquired 100% equity interests in LENSAR, at fair value, for $31.7 million resulting in a loss on extinguishment of notes receivable of $10.6 million. The fair value of the equity interest in LENSAR was determined primarily using the “income method,” which starts with a forecast of all expected future cash flows of the acquired business. The acquisition resulted in a gain on bargain purchase because the fair value of assets acquired and liabilities assumed exceeded the total of the fair value of the equity interest in LENSAR by approximately $9.3 million, net of loss on extinguishment of notes receivables, which was recorded in the Consolidated Statement of Operations for the year ended December 31, 2017.
138
Assets Acquired and Liabilities Assumed
The following table summarizes the fair values of the identifiable intangible assets acquired and liabilities assumed at the acquisition date:
(in thousands) | Amount | |||
Cash | $ | 1,983 | ||
Tangible assets | 18,647 | |||
Intangible assets 1 | 11,970 | |||
Net deferred tax assets | 25,723 | |||
Total identifiable assets | 58,323 | |||
Current liabilities | (6,673 | ) | ||
Total liabilities assumed | (6,673 | ) | ||
Net loss on derecognition of notes receivables | (10,615 | ) | ||
Gain on bargain purchase, net of loss on extinguishment of notes receivable | (9,309 | ) | ||
Total fair value of consideration | $ | 31,726 | ||
______________
1 As of the effective date of the transaction, identifiable intangible assets are required to be measured at fair value. The fair value measurement is based on significant inputs that are unobservable in the market and thus represents a Level 3 measurement. The Company used an income approach to estimate the preliminary fair value of the intangibles which includes technology, trademarks and customer relationships. The assumptions used to estimate the cash flows of the business included a discount rate of 16%, estimated gross margins ranging from 37-72%, income tax rate of 35%, and operating expenses consisting of direct costs based on the anticipated level of revenues. The intangible assets have a weighted-average useful life of approximately 15 years. The intangible assets for acquired technology and trademarks are being amortized over their estimated useful lives using the straight-line method of amortization. The intangible assets for customer relationship are being amortized using a double-declining method of amortization as such method better represents the economic benefits to be obtained.
25. Legal Proceedings
PDL BioPharma, Inc. v Merck Sharp & Dohme, Corp.
On January 22, 2016, the Company filed a complaint against Merck Sharp & Dohme, Corp (“Merck”) for patent infringement in the United States District Court for the District of New Jersey. In the complaint, the Company alleged that manufacture and sales of certain of Merck’s Keytruda product infringed one or more claims of the Company’s U.S. Patent No. 5,693,761 (the “761 Patent”). The Company requested judgment that Merck infringed the 761 Patent, an award of damages due to the infringement, a finding that such infringement was willful and deliberate and trebling of damages therefore, and a declaration that the case is exceptional and warrants an award of attorney’s fees and costs.
On April 21, 2017, the Company entered into a settlement agreement with Merck to resolve the patent infringement lawsuit between the parties pending in the U.S. District Court for the District of New Jersey related to Merck’s Keytruda humanized antibody product. Under the terms of the agreement, Merck paid the Company a one time, lump-sum payment of $19.5 million, and the Company granted Merck a fully paid-up, royalty free, non-exclusive license to certain of the Company’s rights to issued patents in the United States and elsewhere, covering the humanization of antibodies (the “Queen et al. patent”) for use in connection with Keytruda as well as a covenant not to sue Merck for any royalties regarding Keytruda. In addition, the parties agreed to dismiss all claims in the relevant legal proceedings.
Wellstat Litigation
On September 4, 2015, the Company filed in the Supreme Court of New York a motion for summary judgment in lieu of complaint which requested that the court enter judgment against Wellstat Diagnostics Guarantors for the total amount due on the Wellstat Diagnostics debt, plus all costs and expenses including lawyers’ fees incurred by the Company in enforcement of the related guarantees. On July 29, 2016, the court issued its Memorandum of Decision granting the Company’s motion for summary judgment and denying the Wellstat Diagnostics Guarantors’ cross-motion for summary judgment seeking a determination that
139
they were no longer liable under the guarantees. The Supreme Court of New York held that the Wellstat Diagnostics Guarantors are liable for all “Obligations” owed by Wellstat Diagnostics to the Company. It did not set a specific dollar amount due, but ordered that a judicial hearing officer or special referee be designated to determine the amount of the Obligations owing, and awarded the Company its attorneys’ fees and costs in an amount to be determined. On July 29, 2016, the Wellstat Diagnostics Guarantors filed a notice of appeal from the Memorandum of Decision to the Appellate Division of the Supreme Court of New York. On February 14, 2017, the Appellate Division reversed the summary judgment decision of the Supreme Court in the Company’s favor, but affirmed the denial of the Wellstat Diagnostics Guarantors’ cross-motion for summary judgment. The Appellate Division determined that the action was inappropriate for summary judgment pursuant to New York Civil Practice Law & Rules section 3213 on procedural grounds, but specifically made no determination regarding whether the Company was entitled to a judgment on the merits. Pursuant to this decision, the action was remanded to the Supreme Court for further proceedings on the merits. The proceeding has been conducted as a plenary proceeding, with both parties having the opportunity to take discovery and file dispositive motions in accordance with New York civil procedure. On September 11, 2019, the Supreme Court of New York granted the Company’s summary judgment motion, the court holding that the guarantees executed by the Wellstat Diagnostics Guarantors are valid and enforceable, and that the Wellstat Diagnostics Guarantors are liable for the amount owed under the loan agreement. The court ordered a damages hearing before a special referee to calculate the amount owed under the loan agreement between Wellstat Diagnostics and the Company. On September 12, 2019, the Wellstat Diagnostics Guarantors filed a notice of appeal of the Supreme Court of New York’s decision on summary judgment. On September 17, 2019, the Wellstat Diagnostics Guarantors requested a stay of the enforcement of the New York Supreme Court’s decision pending their appeal of the decision, which was denied on November 21, 2019. A damages hearing was scheduled to begin before a judicial hearing officer on December 17, 2019. At the request of the judicial hearing officer, the parties agreed to mediate their dispute prior to the commencement of the damages hearing. As a result, no decision has been made by the hearing officer with respect to the amount of damages owed to the Company.
Glumetza Class Action Antitrust Litigation
On September 18, 2019, the City of Providence filed a civil antitrust suit on behalf of a putative class of payors in the Northern District of California against Bausch Health Companies, Inc., Salix Pharmaceuticals, Inc., Santarus, Inc., Assertio Therapeutics, Inc., Lupin Pharmaceuticals, Inc. and the Company, inter alia, alleging that a patent settlement agreement between Assertio and Lupin unlawfully restrained competition in an alleged market for Glumetza and its AB-rated generic equivalents sold in the United States. The plaintiffs claim that the settlement agreement violated the federal Sherman Act and various state antitrust laws. The Company was a named defendant by certain End Payor Plaintiffs (EPPs) due to its purchase from Assertio in 2013 of a royalty asset based on sales of Glumetza. On January 21, 2020, the EPPs voluntarily dismissed their claims against the Company, without prejudice. The Company has agreed to toll the running of statute of limitations for a limited period of time and to respond to certain discovery requests, subject to reasonable objections.
Noden Pharma DAC v Anchen Pharmaceuticals, Inc. et al
On June 12, 2017, Noden Pharma DAC filed a complaint against Anchen and Par Pharmaceutical (“Par”) for infringement of U.S. Patent No. 8,617,595 based on their submission of an ANDA seeking authorization from the FDA to market a generic version of Tekturna® aliskiren hemifumarate tablets, 150 mg and 300 mg, in the United States. Noden Pharma DAC’s suit triggered a 30-month stay of FDA approval of that application under the Hatch Waxman Act. Par filed a counterclaim seeking a declaratory judgment that their proposed generic version of Tekturna HCT® aliskiren hemifumarate hydrochlorothiazide tablets (150 mg eq. base/12.5 mg HCT, 150 mg eq. base/25 mg HCT, 300 mg eq. base/12.5 mg HCT, and 300 mg eq. base/25 mg HCT), described in a separate ANDA submitted by Par to FDA, alleging noninfringement of U.S. Patent No. 8,618,172 (“the ‘172 Patent”), also owned by Noden Pharma DAC. This case was litigated in the United States District Court for the District of Delaware. In March of 2018, the Parties filed a joint stipulation of dismissal of the defendants’ counterclaim seeking a declaratory judgment of non-infringement of the ‘172 Patent. In the stipulation, Anchen and Par agreed that they will not seek, or otherwise join or assist in, any post-grant review, including inter partes review, of the ‘172 patent or U.S. Patent No. 9,023,893. The defendants further stipulated that they will not seek marketing approval of Par’s ANDA or submit any other ANDA seeking approval to market aliskiren hemifumarate hydrochlorothiazide prior to the expiration of the ‘172 Patent in July of 2028. Both the ‘172 Patent and the ‘893 Patent are listed in the Orange Book for Tekturna HCT. On June 8, 2018, Noden and Anchen entered into the Settlement Agreement. Under the Settlement Agreement, the parties agreed to file a stipulation of dismissal with the court to facilitate dismissal of the litigation in its entirety, with prejudice. In the Settlement Agreement, Noden granted Anchen a non-exclusive, royalty free, fully paid up and non-transferable license to manufacture and commercialize in the United States a generic version of aliskiren which is described in Anchen’s ANDA, and Anchen agreed not to commercialize its generic version of aliskiren prior to March 1, 2019. The license grant excludes certain formulations covered by the ‘595 Patent which closely relate to the commercial formulation of Tekturna marketed by Noden. The Settlement Agreement includes a release by each party for liabilities associated with the litigation and an acknowledgment from Anchen that the ‘595 Patent claims are valid and enforceable.
140
Depomed, Inc. vs. Valeant Pharmaceuticals, Inc.
On October 27, 2017, Valeant, Depomed and the Company entered into a settlement agreement (“Depomed Settlement Agreement”) to resolve all matters addressed in the lawsuit. Under the terms of the Depomed Settlement Agreement, the litigation was dismissed, with prejudice, and Valeant paid to Depomed a one-time, lump-sum payment of $13.0 million. In addition, Depomed and the Company released Valeant and its subsidiary from any and all claims against them as a result of the audit, Valeant’s obligation to pay additional royalties under the commercialization agreement and/or the litigation; and Valeant released Depomed and the Company against any and all claims against them as a result of the audit and/or the litigation. The settlement payment was transferred to the Company under the terms of the Depomed Royalty Agreement in November of 2017.
Other Legal Proceedings
From time to time, the Company is involved in lawsuits, arbitrations, claims, investigations and proceedings, consisting of intellectual property, commercial, employment and other matters, which arise in the ordinary course of business. The Company makes provisions for liabilities when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Such provisions are reviewed at least quarterly and adjusted to reflect the impact of settlement negotiations, judicial and administrative rulings, advice of legal counsel, and other information and events pertaining to a particular case. Litigation is inherently unpredictable. If any unfavorable ruling were to occur in any specific period, there exists the possibility of a material adverse impact on the results of the Company’s operations of that period and on its cash flows and liquidity.
26. Subsequent Events
Repurchase Program
From January 1, 2020 to March 10, 2020, the Company repurchased approximately 3.8 million shares of its common stock at a weighted-average price of $3.42 per share for a total of $12.9 million and repurchased $3.2 million in aggregate principal amount of December 2021 Convertible Notes and $10.5 million in aggregate principal amount of December 2024 Convertible Notes.
Amendment to CareView Modification Agreement
As further discussed in Note 7, Notes and Other Long-Term Receivables, to the Consolidated Financial Statements, in January 2020 we entered into an amendment of the February 2018 Modification Agreement with CareView that deferred principal repayment and interest payments until April 30, 2020, conditioned upon CareView raising additional financing from third parties.
Plan of Liquidation
In December 2019, the Company announced that it had completed a strategic review process and decided to halt the execution of its growth strategy, cease additional strategic transactions and investments and pursue a formal process to unlock value by monetizing its assets and returning net proceeds to stockholders. Over the subsequent months, the Company’s board of directors and management analyzed, together with outside financial and legal advisors, how to best capture value pursuant to its monetization strategy and return the significant intrinsic value of the high-quality assets in its portfolio to its stockholders. On February 7, 2020, the Company’s board of directors approved a plan of complete liquidation for the Company’s assets and a resolution to seek stockholder approval to dissolve the Company at its next annual meeting of the stockholders.
Pursuant to the board’s decisions of February 7, 2020, noted above, the change in control clause in the Amended 2005 Equity Incentive Plan was triggered, accelerating the vesting of a significant portion of the Company’s outstanding equity awards resulting in incremental stock-based compensation expense of $16.3 million to be recorded in the first quarter of 2020.
141
27. Quarterly Financial Data (Unaudited)
Three Months Ended | ||||||||||||||||
(in thousands, except per share data) | December 31, 2019 | September 30, 2019 | June 30, 2019 | March 31, 2019 | ||||||||||||
Total revenues | $ | (5,795 | ) | $ | 44,165 | $ | (22,526 | ) | $ | 38,913 | ||||||
Net (loss) income attributable to PDL’s stockholders | $ | (54,888 | ) | $ | (17,784 | ) | $ | (4,419 | ) | $ | 6,680 | |||||
Net (loss) income per basic share | $ | (0.48 | ) | $ | (0.16 | ) | $ | (0.04 | ) | $ | 0.05 | |||||
Net (loss) income per diluted share | $ | (0.48 | ) | $ | (0.16 | ) | $ | (0.04 | ) | $ | 0.05 | |||||
Three Months Ended | ||||||||||||||||
(in thousands, except per share data) | December 31, 2018 | September 30, 2018 | June 30, 2018 | March 31, 2018 | ||||||||||||
Total revenues | $ | 45,119 | $ | 67,898 | $ | 46,575 | $ | 38,518 | ||||||||
Net income (loss) attributable to PDL’s stockholders | $ | 16,279 | $ | 25,556 | $ | (112,296 | ) | $ | 1,602 | |||||||
Net income (loss) per basic share | $ | 0.12 | $ | 0.18 | $ | (0.76 | ) | $ | 0.01 | |||||||
Net income (loss) per diluted share | $ | 0.11 | $ | 0.18 | $ | (0.76 | ) | $ | 0.01 | |||||||
142
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
The Company’s management has evaluated, with the participation of the chief executive officer and the chief financial officer, the effectiveness of the Company’s disclosure controls and procedures (as defined in Rule 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934 (Exchange Act)) as of the end of the period covered by this report. Based on this evaluation, management concluded that the Company’s disclosure controls and procedures were effective as of December 31, 2019.
Management’s Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in the Exchange Act Rules 13a-15(f) and 15d-15(f). The Company’s management, including the chief executive officer and chief financial officer, conducted an evaluation of the effectiveness of the Company’s internal control over financial reporting based on the Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on the results of this evaluation, the Company’s management concluded that internal control over financial reporting was effective as of December 31, 2019.
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2019, has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in its report included herein.
Changes in Internal Control over Financial Reporting
During the quarter ended December 31, 2019, there were no changes in the Company’s internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
Not applicable.
143
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K and is incorporated herein by reference to our definitive Proxy Statement for our 2019 Annual Meeting of Stockholders (the “Proxy Statement”), which we intend to file pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended, within 120 days after December 31, 2019.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item 10 will be contained in the Proxy Statement and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item 11 will be contained in the Proxy Statement and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item 12 will be contained in the Proxy Statement and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item 13 will be contained in the Proxy Statement and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this Item 14 will be contained in the Proxy Statement and is incorporated herein by reference.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) | The following documents are filed as part of this Annual Report on Form 10-K: |
(1) | Financial Statements - See Index to Consolidated Financial Statements at Item 8 of this Annual Report on Form 10-K. |
(2) | Financial Statement Schedules |
The financial statement schedules are omitted because the information is not applicable, not required under the instructions, or the information requested is set forth in our Consolidated Financial Statements or related notes thereto.
(3) | Exhibits required by Item 601 of Regulation S-K |
The information required by this Section (a)(3) of Item 15 is set forth on the exhibit index that precedes the Signatures page of this Annual Report on Form 10-K.
ITEM 16. FORM 10-K SUMMARY
None.
144
EXHIBIT INDEX
Exhibit Number | Exhibit Title |
2.1 | |
2.2 | |
3.1 | Restated Certificate of Incorporation effective March 23, 1993 (incorporated by reference to Exhibit 3.1 to Annual Report on Form 10-K filed March 31, 1993) |
3.2 | |
3.3 | |
3.4 | |
3.5 | |
3.6 | |
4.1 | |
4.2 | |
4.3 | |
4.4 | |
4.5 | |
4.6 | |
4.7 | |
4.8# | |
10.1* | |
10.2* | |
10.3* | |
145
10.4 | |
10.5 | |
10.6 | |
10.7 | |
10.8 | |
10.9 | |
10.10 | |
10.11* | |
10.12 | |
10.13 | |
10.14 | |
10.15* | |
10.16* | |
10.17 | |
10.18 | |
10.19* | |
10.20 | |
10.21 | |
10.22 | |
10.23 | |
10.24 | |
146
10.25 | |
10.26 | |
10.27* | |
10.28 | |
10.29 | |
10.30 | |
10.31 | |
10.32 | |
10.33* | |
10.34* | |
10.35 | |
10.36* | |
10.37* | |
10.38 | |
10.39* | |
10.40 | |
10.41 | |
10.42 | |
10.43 | |
10.44 | |
10.45* | |
147
10.46* | |
10.47 | |
10.48* | |
10.49* | |
10.50* | |
10.51* | |
10.52 | |
10.53* | |
10.54* | |
10.55* | |
10.56* | |
10.57 | |
10.58* | |
10.59* | |
10.60 | |
10.61* | |
10.62* | |
10.63† | |
10.64* | |
10.65 | |
10.66# | |
10.67#* | |
148
10.68#* | |
10.69#* | |
10.70#* | |
10.71#* | |
10.72#* | |
10.73#* | |
10.74#* | |
10.75#* | |
10.76#* | |
21.1# | |
23.1# | |
31.1# | |
31.2# | |
32.1#+ | |
101.INS | XBRL Instance Document |
101.SCH | XBRL Taxonomy Extension Schema |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase |
101.DEF | XBRL Taxonomy Extension Definition Linkbase |
101.LAB | XBRL Taxonomy Extension Label Linkbase |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase |
____________________________________
# | Filed herewith. |
* | Management contract or compensatory plan or arrangement. |
† | Certain information in this exhibit has been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request under 17 C.F.R. Sections 200.80(b)(4) and 24b-2. |
+ | The certifications attached as Exhibit 32.1 accompany this Annual Report on Form 10-K pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, and shall not be deemed “filed” by the Registrant for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. |
149
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
PDL BIOPHARMA, INC. | |||
By: | /S/ DOMINIQUE MONNET | ||
(Dominique Monnet) | |||
President and Chief Executive Officer | |||
Date: | March 11, 2020 | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date |
/S/ DOMINIQUE MONNET | President and Chief Executive Officer (Principal Executive Officer) | March 11, 2020 |
(Dominique Monnet) | ||
/S/ EDWARD A. IMBROGNO | Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | March 11, 2020 |
(Edward A. Imbrogno) | ||
ALAN BAZAAR | Director | March 11, 2020 |
(Alan Bazaar) | ||
/S/ DAVID GRYSKA | Director | March 11, 2020 |
(David Gryska) | ||
/S/ NATASHA A. HERNDAY | Director | March 11, 2020 |
(Natasha A. Hernday) | ||
/S/ JOHN P. MCLAUGHLIN | Director | March 11, 2020 |
(John P. McLaughlin) | ||
/S/ ELIZABETH O’FARRELL | Director | March 11, 2020 |
(Elizabeth O’Farrell) | ||
/S/ PAUL W. SANDMAN | Director | March 11, 2020 |
(Paul W. Sandman) | ||
/S/ SHLOMO YANAI | Director | March 11, 2020 |
(Shlomo Yanai) | ||
150