Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Lantheus Holdings, Inc. | ex321ceocfocertification.htm |
| EX-31.2 - EXHIBIT 31.2 - Lantheus Holdings, Inc. | ex312cfocertification.htm |
| EX-31.1 - EXHIBIT 31.1 - Lantheus Holdings, Inc. | ex311ceocertification.htm |
| EX-23.1 - EXHIBIT 23.1 - Lantheus Holdings, Inc. | ex231consent.htm |
| EX-21.1 - EXHIBIT 21.1 - Lantheus Holdings, Inc. | ex211subsidiaries.htm |
| EX-10.34 - EXHIBIT 10.34 - Lantheus Holdings, Inc. | ex1034priceandvolumeagreem.htm |
| EX-4.2 - EXHIBIT 4.2 - Lantheus Holdings, Inc. | ex42descriptionofregistran.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
_______________________________________________________________
FORM 10-K
_______________________________________________________________
(Mark One)
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2019
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 001-36569
_______________________________________________________________
LANTHEUS HOLDINGS, INC. |
(Exact name of registrant as specified in its charter) |
_______________________________________________________________
Delaware | 35-2318913 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
331 Treble Cove Road, North Billerica, MA | 01862 | |
(Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (978) 671-8001
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered |
Common Stock, $0.01 par value per share | LNTH | NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
_______________________________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ☐
Indicate by check mark whether the registrant has submitted every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes þ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | þ | Accelerated filer | ☐ | |||
Non-accelerated filer | ☐ | Smaller reporting company | ☐ | |||
Emerging growth company | ☐ | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined by Rule 12b-2 of the Act) Yes ☐ No þ
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant on June 30, 2019 was approximately $1,090.9 million based on the last reported sale price of the registrant’s common stock on the NASDAQ Global Market on June 28, 2019 of $28.30 per share.
As of February 19, 2020 the registrant had 39,252,651 shares of common stock, $0.01 par value, issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Listed hereunder are the documents, portions of which are incorporated by reference, and the parts of this Form 10-K into which such portions are incorporated:
The Registrant’s Definitive Proxy Statement for use in connection with the Annual Meeting of Stockholders to be held on April 23, 2020, portions of which are incorporated by reference into Parts II and III of this Form 10-K. The 2020 Proxy Statement will be filed with the Securities and Exchange Commission no later than 120 days after the close of our year ended December 31, 2019.
LANTHEUS HOLDINGS, INC.
ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
Page | ||
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Unless the context requires otherwise, references to “Lantheus,” “the Company,” “our company,” “we,” “us” and “our” refer to Lantheus Holdings, Inc. and, as the context requires, its direct and indirect subsidiaries, references to “Lantheus Holdings” refer to Lantheus Holdings, Inc. and references to “LMI” refer to Lantheus Medical Imaging, Inc., our wholly-owned subsidiary.
Some of the statements contained in this Annual Report on Form 10-K are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These forward-looking statements, including, in particular, statements about our plans, strategies, prospects and industry estimates are subject to risks and uncertainties. These statements identify prospective information and include words such as “anticipates,” “intends,” “plans,” “seeks,” “believes,” “estimates,” “expects,” “should,” “could,” “predicts,” “hopes” and similar expressions. Examples of forward-looking statements include statements we make relating to our outlook and expectations including, without limitation, in connection with: (i) continued market expansion and penetration for our commercial products, particularly DEFINITY in the face of segment competition and potential generic competition as a result of patent and regulatory exclusivity expirations; (ii) the global Molybdenum-99 (“Mo-99”) supply; (iii) our products manufactured at Jubilant HollisterStier (“JHS”); (iv) our efforts in new product development; and (v) our proposed acquisition (the “Progenics Transaction”) of Progenics Pharmaceuticals, Inc. (“Progenics”). Forward-looking statements are based on our current expectations and assumptions regarding our business, the economy and other future conditions. Because forward-looking statements relate to the future, such statements are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict. Our actual results may differ materially from those contemplated by the forward-looking statements. These statements are neither statements of historical fact nor guarantees or assurances of future performance. The matters referred to in the forward-looking statements contained in this Annual Report on Form 10-K may not in fact occur. We caution you, therefore, against relying on any of these forward-looking statements. Important factors that could cause actual results to differ materially from those in the forward-looking statements include regional, national or global political, economic, business, competitive, market and regulatory conditions and the following:
• | Our ability to continue to grow the appropriate use of DEFINITY in suboptimal echocardiograms in the face of segment competition from other echocardiography contrast agents, including Optison from GE Healthcare Limited (“GE Healthcare”) and Lumason from Bracco Diagnostics Inc. (“Bracco”), and potential generic competition as a result of patent and regulatory exclusivity expirations; |
• | The instability of the global Mo-99 supply, including (i) periodic outages at the NTP Radioisotopes (“NTP”) processing facility in South Africa in 2017, 2018 and 2019 and (ii) a current on-going outage at the Australian Nuclear Science and Technology Organisation’s (“ANSTO”) new Mo-99 processing facility in Australia, in each case resulting in our inability to fill some or all of the demand for our TechneLite generators on certain manufacturing days during the outage periods; |
• | Our dependence upon third parties for the manufacture and supply of a substantial portion of our products, raw materials and components, including DEFINITY at JHS; |
• | The extensive costs, time and uncertainty associated with new product development, including further product development relying on external development partners or developing internally; |
• | Our ability to identify and acquire or in-license additional products, businesses or technologies to drive our future growth; |
• | Our ability to protect our intellectual property and the risk of claims that we have infringed on the intellectual property of others; |
• | Risks associated with the technology transfer programs to secure production of our products at additional contract manufacturer sites, including a modified formulation of DEFINITY at Samsung BioLogics (“SBL”) in South Korea; |
• | Risks associated with our investment in, and construction of, additional specialized manufacturing capabilities at our North Billerica, Massachusetts facility, including our ability to bring the new capabilities online by 2021; |
• | Our dependence on key customers for our medical imaging products, and our ability to maintain and profitably renew our contracts with those key customers, including GE Healthcare, Cardinal Health (“Cardinal”), United Pharmacy Partners (“UPPI”), Jubilant Radiopharma formerly known as Triad Isotopes, Inc. (“Jubilant Radiopharma”) and PharmaLogic Holdings Corp (“PharmaLogic”); |
• | Risks associated with our lead agent in development, flurpiridaz F 18, which in 2017 we out-licensed to GE Healthcare, including: |
• | The ability to successfully complete the Phase 3 development program; |
• | The ability to obtain Food and Drug Administration (“FDA”) approval; and |
• | The ability to gain post-approval market acceptance and adequate reimbursement; |
1
• | Risks associated with our development agent, LMI 1195, for patient populations that would benefit from molecular imaging of the norepinephrine pathway, including designing and timely completing two Phase 3 clinical trials for the diagnosis and management of neuroendocrine tumors in pediatric and adult populations, respectively; |
• | Risks associated with the manufacturing and distribution of our products and the regulatory requirements related thereto; |
• | The dependence of certain of our customers upon third-party healthcare payors and the uncertainty of third-party coverage and reimbursement rates; |
• | The existence and market success of competitor products; |
• | Uncertainties regarding the impact of U.S. and state healthcare reform measures and proposals on our business, including measures and proposals related to reimbursement for our current and potential future products, controls over drug pricing, drug pricing transparency and generic drug competition; |
• | Our being subject to extensive government regulation and oversight, our ability to comply with those regulations and the costs of compliance; |
• | Potential liability associated with our marketing and sales practices; |
• | The occurrence of any serious or unanticipated side effects with our products; |
• | Our exposure to potential product liability claims and environmental, health and safety liability; |
• | Our ability to introduce new products and adapt to an evolving technology and medical practice landscape; |
• | Risks associated with prevailing economic or political conditions and events and financial, business and other factors beyond our control; |
• | Risks associated with our international operations, including potential global disruptions in air transport due to COVID-19 (coronavirus), which could adversely affect our international supply chains for radioisotopes and other critical materials as well as international distribution channels for our commercial products; |
• | Our ability to adequately qualify, operate, maintain and protect our facilities, equipment and technology infrastructure; |
• | Our ability to hire or retain skilled employees and key personnel; |
• | Our ability to utilize, or limitations in our ability to utilize, net operating loss carryforwards to reduce our future tax liability; |
• | Risks related to our outstanding indebtedness and our ability to satisfy those obligations; |
• | Costs and other risks associated with the Sarbanes-Oxley Act and the Dodd-Frank Act, including in connection with becoming a large accelerated filer as of December 31, 2019; |
• | Risks related to the ownership of our common stock; |
• | Risks related to the Progenics Transaction, including: |
• | We or Progenics may be unable to obtain stockholder approval as required; |
▪ | Conditions to the closing of the Progenics Transaction may not be satisfied; |
▪ | The Progenics Transaction may involve unexpected costs, liabilities or delays; |
▪ | The ability of our or Progenics’ business to retain and hire key personnel and maintain relationships with customers, suppliers and others with whom we or Progenics do business, or on our or Progenics’ operating results and business generally; |
▪ | Our or Progenics’ respective businesses may suffer as a result of uncertainty surrounding the Progenics Transaction and disruption of management’s attention due to the Progenics Transaction; |
▪ | The occurrence of any event, change or other circumstances that could give rise to the termination of our agreement with Progenics; |
▪ | Unanticipated risks to our integration plan including in connection with timing, talent, and the potential need for additional resources; |
▪ | New or previously unidentified manufacturing, regulatory, or research and development issues in the Progenics business; |
▪ | Risks that the anticipated benefits of the Progenics Transaction or other commercial opportunities may otherwise not be fully realized or may take longer to realize than expected; |
2
▪ | Risks that contractual contingent value rights (“CVRs”) we will issue as part of the Progenics Transaction may result in substantial future payments and could divert the attention of our management; |
▪ | Risks that in connection with the Progenics Transaction, the exercise of appraisal rights by dissenting stockholders could increase the aggregate amount we have to pay for Progenics; |
• | We or Progenics may be adversely affected by other economic, business, and/or competitive factors; |
▪ | The impact of legislative, regulatory, competitive and technological changes; |
▪ | Other risks to the consummation of the Progenics Transaction, including the risk that the Progenics Transaction will not be consummated within the expected time period or at all; and |
• | Other factors that are described in Part I, Item 1A. “Risk Factors” of this Annual Report on Form 10-K. |
Factors that could cause or contribute to such differences include, but are not limited to, those that are discussed in other documents we file with the Securities and Exchange Commission (“SEC”). Any forward-looking statement made by us in this Annual Report on Form 10-K speaks only as of the date on which it is made. Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them. We undertake no obligation to publicly update any forward-looking statement, whether as a result of new information, future developments or otherwise, except as may be required by law.
Trademarks
We own or have the rights to various trademarks, service marks and trade names, including, among others, the following: DEFINITY®, TechneLite®, Cardiolite®, Neurolite®, Vialmix®, Quadramet®, Luminity® and Lantheus Medical Imaging® referred to in this Annual Report on Form 10-K. Solely for convenience, we refer to trademarks and service marks in this Annual Report on Form 10-K without the TM, SM and ® symbols. Those references are not intended to indicate, in any way, that we will not assert, to the fullest extent permitted under applicable law, our rights to our trademarks and service marks. Each trademark, trade name or service mark of any other company appearing in this Annual Report on Form 10-K, such as Lumason®, OptisonTM, SonoVue®, Progenics®, Cerevast®, CarThera® and SonoCloud® are, to our knowledge, owned by that other company.
3
PART I
Item 1. Business
Overview
We are a global leader in the development, manufacture and commercialization of innovative diagnostic medical imaging agents and products that assist clinicians in the diagnosis and treatment of cardiovascular and other diseases. Clinicians use our imaging agents and products across a range of imaging modalities, including echocardiography and nuclear imaging. We believe that the resulting improved diagnostic information enables healthcare providers to better detect and characterize, or rule out, disease, potentially achieving improved patient outcomes, reducing patient risk and limiting overall costs for payers and the entire healthcare system.
Our commercial products are used by cardiologists, nuclear physicians, radiologists, internal medicine physicians, technologists and sonographers working in a variety of clinical settings. We sell our products to radiopharmacies, integrated delivery networks, hospitals, clinics and group practices.
We sell our products globally and currently operate our business in two reportable segments, which are further described below:
• | U.S. Segment produces and markets our medical imaging agents and products throughout the U.S. In the U.S., we primarily sell our products to radiopharmacies, integrated delivery networks, hospitals, clinics and group practices. |
• | International Segment operations consist of production and distribution activities in Puerto Rico and some direct distribution activities in Canada. Additionally, within our International Segment, we have established and maintain third-party distribution relationships under which our products are marketed and sold in Europe, Canada, Australia, Asia-Pacific and Latin America. |
Our Product Portfolio
Our current portfolio of ten commercial products is diversified across a range of imaging modalities. Our current products include an ultrasound contrast agent and medical radiopharmaceuticals (including Technetium generators).
• | Ultrasound contrast agents are compounds that are used in diagnostic procedures, such as cardiac ultrasounds or echocardiograms, to improve the clarity of the diagnostic image. |
• | Medical radiopharmaceuticals are radioactive pharmaceuticals used by clinicians to perform nuclear imaging procedures. |
• | In certain circumstances, a radioactive element, or radioisotope, is attached to a chemical compound to form the radiopharmaceutical. This act of attaching the radioisotope to the chemical compound is called radiolabeling, or labeling. |
• | In other circumstances, a radioisotope can be used as a radiopharmaceutical without attaching any additional chemical compound. |
• | Radioisotopes are most commonly manufactured in a nuclear research reactor, where a target is bombarded with subatomic particles, or in a cyclotron, which is a type of particle accelerator that also creates radioisotopes. |
• | Two common forms of nuclear imaging procedures are single-photon emission computed tomography (“SPECT”) which measures gamma rays emitted by a SPECT radiopharmaceutical, and positron emission tomography (“PET”) which measures positrons emitted by a PET radiopharmaceutical. |
As an example of the procedures in which our products may be used, in the diagnosis of cardiovascular disease, a typical diagnostic progression could include an electrocardiogram, followed by an echocardiogram (possibly using our agent DEFINITY) which delineates cardiac structure and function, and then a nuclear myocardial perfusion imaging (“MPI”) study using either SPECT or PET imaging (possibly using our Technetium generator and our Cardiolite SPECT-based MPI agent). An MPI study assesses blood flow distribution to the heart. MPI is also used for diagnosing the presence of coronary artery disease.
Progenics Transaction
On October 1, 2019, we entered into an Agreement and Plan of Merger (the “Initial Merger Agreement”) to acquire Progenics Pharmaceuticals, Inc. (NASDAQ: PGNX) in an all-stock transaction. Progenics is an oncology company developing innovative medicines and artificial intelligence to find, fight and follow cancer. Under the terms of the Initial Merger Agreement, we agreed to acquire all of the issued and outstanding shares of Progenics common stock at a fixed exchange ratio. Progenics stockholders would have received 0.2502 shares of our common stock for each share of Progenics common stock, representing an approximately 35% aggregate ownership stake in the combined company. The transaction contemplated by the Initial Merger Agreement was
4
unanimously approved by the Boards of Directors of both companies and was subject to the terms and conditions set forth in the Initial Merger Agreement, including, among other things, the affirmative vote of a majority of the outstanding shares of common stock of Progenics and a majority of votes cast by the holders of the common stock of the Company.
On February 20, 2020, we entered into an Amended and Restated Agreement and Plan of Merger (the “Amended Merger Agreement”) with Progenics, which amends and restates the Initial Merger Agreement. Under the terms of the Amended Merger Agreement, we will acquire all of the issued and outstanding shares of Progenics common stock at a fixed exchange ratio whereby Progenics stockholders will receive, for each share of Progenics stock held at the time of the closing of the merger, 0.31 of a share of our common stock, increased from 0.2502 under the Initial Merger Agreement, together with a non-tradeable CVR tied to the financial performance of PyLTM (18F-DCFPyL), Progenics’ prostate-specific membrane antigen targeted imaging agent designed to visualize prostate cancer currently in late stage clinical development (“PyL”). Each CVR will entitle its holder to receive a pro rata share of aggregate cash payments equal to 40% of U.S. net sales generated by PyL in 2022 and 2023 in excess of $100 million and $150 million, respectively. In no event will our aggregate payments under the CVRs exceed 19.9% of the total consideration we pay in the transaction. As a result of the increase in the exchange ratio, following the completion of the merger, former Progenics stockholders’ aggregate ownership stake will increase to approximately 40% of the combined company from approximately 35% under the Initial Merger Agreement. Progenics’ stockholders will also now be entitled to appraisal rights as provided under Delaware law. The transaction contemplated by the Amended Merger Agreement was unanimously approved by the Boards of Directors of both companies and requires, among other things, the affirmative vote of a majority of the outstanding shares of common stock of Progenics and a majority of votes cast by the holders of the common stock of the Company.
In addition, pursuant to the Amended Merger Agreement, the holder of each in-the-money option to purchase shares of Progenics common stock under any equity based compensation plan of Progenics (“Progenics Stock Option”) will be entitled to receive in exchange for each such in-the-money option (i) an option to purchase Lantheus Common Stock (each, a “Lantheus Stock Option”) converted based on the 0.31 exchange ratio, and (ii) a vested or unvested CVR depending on whether the underlying option is vested. Holders of out-of-the-money Progenics Stock Options will receive Lantheus Stock Options converted on an exchange ratio adjusted based on actual trading prices of common stock of Progenics and Lantheus Holdings prior to the effective time of the merger.
The Amended Merger Agreement also provides that on closing our board of directors will appoint Dr. Gerard Ber and Mr. Heinz Mausli, who are currently members of the board of directors of Progenics, to serve on our board of directors. In addition, our board of directors, subject to complying with applicable fiduciary duties, will use commercially reasonable efforts to cause Dr. Ber and Mr. Mausli to be nominated for reelection following the closing through 2023. Our board of directors will be reduced in size from ten to nine members at our annual meeting of stockholders on April 23, 2020 (or sooner if the transaction closes before then) and will be further reduced in size from nine to eight members prior to the date of our 2021 annual meeting of stockholders.
Except as described above, the material terms of the Amended Merger Agreement are substantially the same as the terms of the Initial Merger Agreement.
The transaction is currently expected to close in the second quarter of 2020. Upon completion of the acquisition, which the parties intend to report as tax-deferred to Progenics’ stockholders with respect to the stock component of the merger consideration for U.S. federal income tax purposes, the combined company will continue to be headquartered in North Billerica, Massachusetts and will trade on the NASDAQ under the ticker symbol LNTH. See our Current Reports on Form 8-K dated October 1, 2019 and February 20, 2020 for further information regarding the Initial Merger Agreement, the Amended Merger Agreement and the proposed Progenics acquisition.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our proposed acquisition of Progenics.
5
DEFINITY and the Expansion of Our Ultrasound Microbubble Franchise
DEFINITY is the leading ultrasound contrast imaging agent based on revenue and usage in the U.S., and is indicated for use in patients with suboptimal echocardiograms. Numerous patient conditions can decrease the quality of images of the left ventricle, the primary pumping chamber of the heart. The term DEFINITY includes its activated and non-activated forms.
DEFINITY is a clear, colorless, sterile liquid, which, upon activation in a Vialmix apparatus, a medical device specifically designed for DEFINITY, becomes a homogenous, opaque, milky white injectable suspension of perflutren-containing lipid microspheres. After activation and intravenous injection, DEFINITY opacifies the left ventricular chamber and improves the delineation of the left ventricular endocardial border, or innermost layer of tissue that lines the chamber of the left ventricle. Better visualization of the left ventricle allows clinicians to make more informed decisions about disease status.
DEFINITY offers flexible dosing and administration through an IV bolus or diluted bolus injection or continuous IV infusion. We believe DEFINITY’s synthetic lipid-cased coating gives the agent a distinct competitive advantage, because it provides a strong ultrasound signal and is the only perflutren-based echo contrast agent made without albumin.
There were approximately 35.1 million echocardiograms performed in the U.S. in 2019 according to a third party source. Assuming 20% of echocardiograms produce suboptimal images, as stated in the clinical literature, we estimate that approximately 7.0 million echocardiograms in 2019 produced suboptimal images. The use of DEFINITY during echocardiography allows physicians to significantly improve their assessment of the function of the left ventricle.
Since its launch in 2001, DEFINITY has been used in imaging procedures in more than 13.8 million studies throughout the world. We estimate that DEFINITY had over 80% share of the U.S. segment for contrast agents in echocardiography procedures as of December 2019. DEFINITY currently competes with Optison, a GE Healthcare product, Lumason, a Bracco product (known as SonoVue outside the U.S.) as well as echocardiography without contrast and non-echocardiography imaging modalities. DEFINITY, Optison and Lumason all carry an FDA-required boxed warning, which has been modified over time, to notify physicians and patients about potentially serious safety concerns or risks posed by the products. See Part I, Item 1A. “Risk Factors-Ultrasound contrast agents may cause side effects which could limit our ability to sell DEFINITY.”
As we continue to pursue expanding our microbubble franchise, our activities include:
• | Patents - We continue to actively pursue additional patents in connection with DEFINITY, both in the U.S. and internationally. In the U.S., we have an Orange Book-listed method of use patent expiring in March 2037 and additional manufacturing patents that are not Orange Book-listed expiring in 2021, 2023 and 2037. Outside of the U.S., while our DEFINITY patent protection and regulatory exclusivity have generally expired, we are currently prosecuting additional patent applications to try to obtain similar method of use and manufacturing patent protection as granted in the U.S. |
Hatch-Waxman Act - Even though our longest duration Orange Book-listed DEFINITY patent extends until March 2037, because our Orange Book-listed composition of matter patent expired in June 2019, we may face generic DEFINITY challengers in the near to intermediate term. Under the Hatch-Waxman Act, the FDA can approve Abbreviated New Drug Applications (“ANDAs”) for generic versions of drugs if the ANDA applicant demonstrates, among other things, that (i) its generic candidate is the same as the innovator product by establishing bioequivalence and providing relevant chemistry, manufacturing and product data, and (ii) the marketing of that generic candidate does not infringe an Orange Book-listed patent. With respect to any Orange Book-listed patent covering the innovator product, the ANDA applicant must give a notice to the innovator (a “Notice”) that the ANDA applicant certifies that its generic candidate will not infringe the innovator’s Orange Book-listed patent or that the Orange Book-listed patent is invalid. The innovator can then challenge the ANDA applicant in court within 45 days of receiving that Notice, and FDA approval to commercialize the generic candidate will be stayed (that is, delayed) for up to 30 months (measured from the date on which a Notice is received) while the patent dispute between the innovator and the ANDA applicant is resolved in court. The 30 month stay could potentially expire sooner if the courts determine that no infringement had occurred or that the challenged Orange Book-listed patent is invalid or if the parties otherwise settle their dispute.
As of the date of filing of this Annual Report on Form 10-K, we have not received any Notice from an ANDA applicant. If we were to (i) receive any such Notice in the future, (ii) bring a patent infringement suit against the ANDA applicant within 45 days of receiving that Notice, and (iii) successfully obtain the full 30 month stay, then the ANDA applicant would be precluded from commercializing a generic version of DEFINITY prior to the expiration of that 30 month stay period and, potentially, thereafter, depending on how the patent dispute is resolved. Solely by way of example and not based on any knowledge we currently have, if we received a Notice from an ANDA applicant in March 2020 and the full 30 month stay was obtained, then the ANDA applicant would be precluded from commercialization until at least September 2022. If we received a Notice some number of months in the future and the full 30 month stay was obtained, the commercialization date would roll forward in the future by the same calculation.
6
• | Modified Formulation - We are developing at SBL a modified formulation of DEFINITY. We believe this modified formulation will provide an enhanced product profile enabling storage as well as shipment at room temperature (DEFINITY’s current formulation requires refrigerated storage), will give clinicians additional choice, and will allow for greater utility of this formulation in broader clinical settings. We have a composition of matter patent on the modified formulation which runs through December 2035. If the modified formulation is approved by the FDA, then this patent would be eligible to be listed in the Orange Book. We currently believe that, if approved by the FDA, the modified formulation could become commercially available in early 2021, although that timing cannot be assured. Given its physical characteristics, the modified formulation may also be better suited for inclusion in kits requiring microbubbles for other indications and applications (including in kits developed by third parties of the type described in the next paragraph). |
• | New Clinical Applications - As we continue to look for other opportunities to expand our microbubble franchise, we are evaluating new indications and clinical applications beyond echocardiography and contrast imaging generally. For example, we recently announced a strategic development and commercial collaboration with Cerevast Medical, Inc. (“Cerevast”) in which our microbubble will be used in connection with Cerevast’s ocular ultrasound device to target improving blood flow in occluded retinal veins in the eye. Retinal vein occlusion is one of the most common causes of vision loss worldwide. We also recently announced a strategic commercial supply agreement with CarThera for the use of our microbubbles in combination with SonoCloud, a proprietary implantable device in development for the treatment of recurrent glioblastoma. Glioblastoma is a lethal and devasting from of brain cancer with median survival of 15 months after diagnosis. |
• | In-House Manufacturing - We are currently building specialized in-house manufacturing capabilities at our North Billerica, Massachusetts facility for DEFINITY and, potentially, other sterile vial products. We believe the investment in these efforts will allow us to better control DEFINITY manufacturing and inventory, reduce our costs in a potentially more price competitive environment, and provide us with supply chain redundancy. We currently expect to be in a position to use this in-house manufacturing capability by early 2021, although that timing cannot be assured. |
As part of our microbubble franchise strategy, we also conducted two Phase 3, open-label, multicenter studies to evaluate left ventricular ejection fraction (“LVEF”) measurement accuracy and reproducibility of DEFINITY contrast-enhanced and unenhanced echocardiography as compared to non-contrast cardiac magnetic resonance imaging (“CMRI”) used as the truth standard. The first of the two trials, BENEFIT 1, enrolled 145 subjects. After reviewing the study results from BENEFIT 1, we concluded there was no statistically significant improvement in the accuracy of LVEF values for contrast-enhanced echocardiography versus unenhanced echocardiography as compared to CMRI. In addition, analyses of the secondary endpoints revealed no improvement in inter-reader variability between the contrast-enhanced and unenhanced echocardiograms for LVEF assessments. A post-hoc analysis, however, did show statistically significant improvements in left ventricular diastolic, systolic and stroke volume measurements with contrast-enhanced versus unenhanced echocardiography when compared to CMRI. We will continue to analyze the BENEFIT 1 data, and when the data from BENEFIT 2 are available, we will compile the data sets to analyze the full results of the trials.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with DEFINITY and see Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations-Comparison of the Periods Ended December 31, 2019 and 2018-Revenues” for further information on total revenue contributed by DEFINITY in each of our last three fiscal years.
TechneLite
TechneLite is a self-contained system or generator of Technetium (“Tc-99m”), a radioactive isotope with a six hour half-life, used by radiopharmacies to prepare various nuclear imaging agents. Tc-99m results from the radioactive decay of Mo-99, itself a radioisotope with a 66-hour half-life produced in nuclear research reactors around the world from enriched uranium. The TechneLite generator is a little larger than a coffee can in size, and the self-contained system houses a vertical glass column at its core that contains Mo-99. During our manufacturing process, Mo-99 is added to the column within the generator where it is adsorbed onto alumina powder. The column is sterilized, enclosed in a lead shield and further sealed in a cylindrical plastic container, which is then immediately shipped to our radiopharmacy customers. Because of the short half-lives of Mo-99 and Tc-99m, radiopharmacies typically purchase TechneLite generators on a weekly basis pursuant to standing orders.
The Tc-99m produced by our TechneLite generator is the medical radioisotope that can be attached to a number of imaging agents, including our own Cardiolite products and Neurolite, during the radiolabeling process. To radiolabel a Tc-99m-based radiopharmaceutical, a vial of sterile saline and a vacuum vial are each affixed to the top of a TechneLite generator. The sterile saline is pulled through the generator where it attracts Tc-99m resulting from the radioactive decay of Mo-99 within the generator column. The Tc-99m-containing radioactive saline is then pulled into the vacuum vial and subsequently combined by a radiopharmacist with the applicable imaging agent, and individual patient-specific radiolabeled imaging agent doses are then prepared. When administered, the imaging agent binds to specific tissues or organs for a period of time, enabling the Tc-99m to illuminate the functional health of the imaged tissues or organs in a diagnostic image. Our ability to produce and market TechneLite is highly dependent on our supply of Mo-99. See “Raw Materials and Supply Relationships—Molybdenum-99” below.
7
TechneLite is produced in 13 sizes (based on amount of radioactivity) and is currently marketed primarily in North America and Latin America, largely to radiopharmacies that prepare unit doses of radiopharmaceutical imaging agents and ship these preparations directly to hospitals for administration to patients. In the U.S., we have supply contracts with the significant radiopharmacy groups, including GE Healthcare, Cardinal, UPPI, Jubilant Radiopharma and PharmaLogic. We also supply generators on a purchase order basis to other customers. We estimate that TechneLite had approximately one third of the U.S. generator market as of December 31, 2019, competing primarily with Tc-99m-based generators produced by Curium. In Puerto Rico, we also supply TechneLite to our wholly-owned radiopharmacy to prepare radiopharmaceutical imaging agent unit doses.
In Canada, we have a supply agreement (the “Isologic Supply Agreement”) with Isologic Innovative Radiopharmaceuticals Ltd. (“Isologic”). Under the Isologic Supply Agreement, we supply Isologic with certain of our products on commercial terms, including certain product purchase commitments by Isologic. The agreement expires in January 2021 and may be terminated upon the occurrence of specified events, including a material breach by the other party, bankruptcy by either party or certain force majeure events. In Australia, we have a supply agreement (the “GMS Supply Agreement”) with Global Medical Solutions (“GMS”). Under the GMS Supply Agreement, we supply GMS with certain of our products on commercial terms, including certain minimum product purchase commitments by GMS. The agreement expires in August 2020 and may be terminated in whole or in part on a product-by-product basis upon the occurrence of specified events, including a material breach by the other party, bankruptcy by either party or certain force majeure events.
The Mo-99 used in our TechneLite generators can be produced using targets made of either highly-enriched uranium (“HEU”) or low-enriched uranium (“LEU”). LEU consists of uranium that contains less than 20% of the uranium-235 isotope. HEU is often considered weapons grade material, with 20% or more of uranium-235. The American Medical Isotopes Production Act of 2012 encourages the domestic production of LEU Mo-99 and provides for the eventual prohibition of the export of HEU from the U.S. Although Medicare generally does not provide separate payment to hospitals for the use of diagnostic radiopharmaceuticals administered in an outpatient setting, since 2013, the Centers for Medicare and Medicaid Services (“CMS”), the federal agency responsible for administering the Medicare program, has provided an add-on payment (of $10) under the hospital outpatient prospective payment system for every Tc-99m diagnostic dose produced from non-HEU sourced Mo-99, to cover the marginal cost for radioisotopes produced from non-HEU sources. Our LEU TechneLite generator satisfies the reimbursement requirements under the applicable CMS rules.
TechneLite has patent protection in the U.S. and various foreign countries on certain component technology currently expiring in 2029. In addition, given the significant know-how and trade secrets associated with the methods of manufacturing and assembling the TechneLite generator, we believe we have a substantial amount of valuable and defensible proprietary intellectual property associated with the product. We believe that our substantial capital investments in our highly automated TechneLite production line and our extensive experience in complying with the stringent regulatory requirements for the handling of nuclear materials create significant and sustainable competitive advantages for us in generator manufacturing and distribution.
See Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations-Comparison of the Periods Ended December 31, 2019 and 2018-Revenues” for further information on total revenue contributed by TechneLite in each of our last three fiscal years.
Other Commercial Products
In addition to the products listed above, our portfolio of commercial products also includes important imaging agents in specific segments, which provide a stable base of recurring revenue. Most of these products have a favorable industry position as a result of our substantial infrastructure investment, specialized workforce, technical know-how and supplier and customer relationships.
• | Xenon Xe 133 Gas (“Xenon”) is a radiopharmaceutical gas that is inhaled and used to assess pulmonary function and also to image cerebral blood flow. Our Xenon is manufactured by a third party as a bi-product of Mo-99 production and is processed and finished by us. We are currently the leading provider of Xenon in the U.S. |
• | Neurolite is an injectable, Tc-99m-labeled imaging agent used with SPECT technology to identify the area within the brain where blood flow has been blocked or reduced due to stroke. We launched Neurolite in 1995. |
• | Cardiolite, also known by its generic name sestamibi, is an injectable, Tc-99m-labeled imaging agent used in MPI procedures to assess blood flow to the muscle of the heart using SPECT. Cardiolite was approved by the FDA in 1990 and its market exclusivity expired in July 2008. Included in Cardiolite revenues are branded Cardiolite and generic sestamibi revenues. |
• | Thallium TI 201 is an injectable radiopharmaceutical imaging agent used in MPI studies to detect cardiovascular disease. We have marketed Thallium since 1977 and manufacture the agent using cyclotron technology. |
• | FDG is an injectable, fluorine-18-radiolabeled imaging agent used with PET technology to identify and characterize tumors in patients undergoing oncologic diagnostic procedures. We manufacture and distribute FDG from our Puerto Rico radiopharmacy. |
8
• | Gallium (Ga 67) is an injectable radiopharmaceutical imaging agent used to detect certain infections and cancerous tumors, especially lymphoma. We manufacture Gallium using cyclotron technology. |
• | Quadramet, currently our only therapeutic product, is an injectable radiopharmaceutical used to treat severe bone pain associated with osteoblastic metastatic bone lesions. We serve as the direct manufacturer and supplier of Quadramet in the U.S. |
• | Cobalt (Co 57) is a non-pharmaceutical radiochemical used in the manufacture of sources for the calibration and maintenance of SPECT imaging cameras. |
Distribution, Marketing and Sales
The following table sets forth certain key market information for each of our commercial pharmaceutical products:
Product | Approved Markets |
DEFINITY | Australia, Canada, European Union, European Economic Area, India, Israel, Mexico, New Zealand, Singapore, South Korea, Taiwan, United States |
TechneLite | Australia, Brazil, Canada, China, Colombia, Costa Rica, New Zealand, Panama, South Korea, Taiwan, United States |
Xenon | Canada, United States |
Cardiolite | Australia, Canada, Costa Rica, Hong Kong, Israel, Japan, New Zealand, Panama, Philippines, South Korea, Taiwan, Thailand, United States |
Neurolite | Australia, Austria, Belgium, Canada, Costa Rica, Denmark, France, Germany, Hong Kong, Italy, Japan, Luxembourg, New Zealand, Philippines, Slovenia, South Korea, Spain, Taiwan, Thailand, United States |
Thallium Tl 201 | Australia, Canada, Colombia, New Zealand, Pakistan, Panama, South Korea, Taiwan, United States |
Gallium Ga 67 | Australia, Canada, Colombia, Costa Rica, New Zealand, Pakistan, Panama, South Korea, Taiwan, United States |
FDG | United States |
Quadramet | United States |
In the U.S. and Canada, we have a sales team of approximately 80 employees that call on healthcare providers in the echocardiography space, as well as radiopharmacy chains, integrated delivery networks and group purchasing organizations.
Our radiopharmaceutical products are sold in the U.S. through a subset of our sales team, primarily to radiopharmacies. We sell a majority of our radiopharmaceutical products in the U.S. to five radiopharmacy groups—namely GE Healthcare, Cardinal, UPPI, Jubilant Radiopharma and PharmaLogic. Our contractual distribution and other arrangements with these radiopharmacy groups are as follows:
• | GE Healthcare maintains approximately 31 radiopharmacies in the U.S. that purchase our TechneLite generators. We estimate that GE Healthcare distributed approximately 9% of the aggregate U.S. SPECT doses sold in the first half of 2019. We currently have an agreement with GE Healthcare for the distribution of TechneLite, Xenon and other products. The agreement provides that GE Healthcare will purchase a minimum percentage of TechneLite generators as well as certain other products from us. Our agreement, which expires on December 31, 2020, may be terminated by either party upon the occurrence of specified events including a material breach by either party, bankruptcy by either party, certain irresolvable regulatory changes or economic circumstances, or force majeure events. |
• | Cardinal maintains approximately 131 radiopharmacies that are typically located in large, densely populated urban areas in the U.S. We estimate that Cardinal’s radiopharmacies distributed approximately 44% of the aggregate U.S. SPECT doses sold in the first half of 2019 (the latest information currently available to us). Our written supply agreement with Cardinal relating to TechneLite, Xenon, Neurolite and other products expires on December 31, 2020. The agreement specifies pricing levels and requirements to purchase minimum percentages of certain products during certain periods. The agreement may be terminated upon the occurrence of specified events, including a material breach by the other party and certain force majeure events. |
9
• | UPPI is a cooperative purchasing group (roughly analogous to a group purchasing organization) of approximately 60 independently owned or smaller chain radiopharmacies located in the U.S. UPPI’s radiopharmacies are typically broadly dispersed geographically, with some urban presence and a substantial number of radiopharmacies located in suburban and rural areas of the country. We estimate that these independent radiopharmacies, together with approximately 9 unaffiliated, independent radiopharmacies, distributed approximately 19% of the aggregate U.S. SPECT doses sold in the first half of 2019. We currently have an agreement with UPPI for the distribution of TechneLite, Xenon and certain other products to radiopharmacies or families of radiopharmacies within the UPPI cooperative purchasing group. The agreement contains specified pricing levels based upon specified purchase amounts for UPPI. We are entitled to terminate the UPPI agreement upon 60 days written notice. The UPPI agreement expires on December 31, 2020. |
• | Jubilant Radiopharma maintains approximately 56 radiopharmacies in the U.S. that purchase a range of our products. We estimate that Jubilant Radiopharma distributed approximately 14% of the aggregate U.S. SPECT doses sold in the first half of 2019. We currently have an agreement with Jubilant Radiopharma for the distribution of TechneLite, Xenon, Neurolite and other products. The agreement specifies pricing levels and volume and percentage purchase requirements. The agreement will expire on December 31, 2020 and may be terminated upon the occurrence of specified events, including a material breach by the other party. |
• | PharmaLogic maintains approximately 23 radiopharmacies in the U.S. that purchase a range of our products. We estimate that PharmaLogic distributed approximately 4% of the aggregate U.S. SPECT doses sold in the first half of 2019. Our written supply agreement with PharmaLogic relating to TechneLite, Xenon, Cardiolite and other products expires on December 31, 2020. The agreement specifies pricing levels and requirements to purchase minimum percentages of certain products during certain periods. The agreement may be terminated upon the occurrence of specified events, including a material breach by the other party and certain force majeure events. |
In addition to the distribution arrangements for our radiopharmaceutical products described above, we also sell certain of our radiopharmaceutical products to independent radiopharmacies and directly to hospitals and clinics that maintain in-house radiopharmaceutical capabilities and operations. In the latter case, this represents a small percentage of overall sales because the majority of hospitals and clinics do not maintain these in-house capabilities.
In Puerto Rico, we own and operate one of the two radiopharmacies on the island, where we sell our own products as well as products of third parties to end-users. In Canada, we operate some direct distribution activities.
In Europe, Canada, Australia, Asia-Pacific and Latin America, we utilize third party distributor relationships to market, sell and distribute our products, either on a country-by-country basis or on a multi-country regional basis.
In March 2012, we entered into a development and distribution arrangement for DEFINITY in China, Hong Kong and Macau with Double-Crane Pharmaceutical Company (“Double-Crane”). With Double-Crane’s support, we are currently pursuing the Chinese regulatory approval required to commercialize DEFINITY. In July 2013, we submitted a clinical trial application to the Chinese Food and Drug Administration (“CFDA”) seeking an Import Drug License. After a very extensive waiting period caused by a large number of drugs seeking CFDA regulatory approval, in February 2016, the CFDA approved our clinical trial application. Double-Crane has conducted on our behalf three confirmatory clinical trials in pursuit of cardiac, liver and kidney imaging indications, as well as one small pharmacokinetic study. Double Crane is preparing an application to the CFDA for an Import Drug License for the cardiac indication. Double Crane is also analyzing the clinical results relating to the liver and kidney indications and will also prepare a CFDA application for those indications.
Seasonality
Our business has modest seasonality as patients may seek to schedule non-urgent diagnostic imaging procedures less frequently during the summer vacation months and over the year-end holidays.
Customers
No customer accounted for greater than 10% of revenues for the year ended December 31, 2019.
Backlog
Our backlog consists of orders for which a delivery schedule within the next twelve months has been specified. Orders included in backlog may be canceled or rescheduled by customers at any time with the exception of TechneLite orders. For TechneLite, customers must provide us with four weeks advanced notice to cancel an order. We do not believe that our backlog at any particular time is meaningful because it has historically been immaterial relative to our consolidated revenues and is not necessarily indicative of future revenues for any given period.
10
Competition
We believe that our key product characteristics, such as proven efficacy, reliability and safety, coupled with our core competencies, such as our efficient manufacturing processes, our established distribution network, our experienced field sales organization and our customer service focus, are important factors that distinguish us from our competitors.
The market for diagnostic medical imaging agents is highly competitive and continually evolving. Our principal competitors in existing diagnostic modalities include large, global companies that are more diversified than we are and that have substantial financial, manufacturing, sales and marketing, distribution and other resources. These competitors currently include Curium, GE Healthcare, Bracco and Jubilant Life Sciences, an affiliate of JHS and Jubilant Radiopharma, as well as other competitors, including NorthStar Medical Radioisotopes. We cannot anticipate their competitive actions in the same or competing diagnostic modalities, such as significant price reductions on products that are comparable to our own, development of new products that are more cost-effective or have superior performance than our current products or the introduction of generic versions after our proprietary products lose their current patent protection. In addition, distributors of our products could attempt to shift end-users to competing diagnostic modalities and products, or bundle the sale of a portfolio of products to the detriment of our specific products. Our current or future products could be rendered obsolete or uneconomical as a result of these activities.
Further, the radiopharmaceutical industry continues to evolve strategically, with several market participants either recently sold or for sale. In addition, the supply-demand dynamics of the industry are complex because of large market positions of some participants, legacy businesses, government subsidies (in particular, relating to the manufacture of radioisotopes), and group purchasing arrangements. We cannot predict what impact new owners and new operators may have on the strategic decision-making of our competitors, customers and suppliers.
Raw Materials and Supply Relationships
We rely on certain raw materials and supplies to produce our products. Due to the specialized nature of our products and the limited, and sometimes intermittent, supply of raw materials available in the market, we have established relationships with several key suppliers. For the year ended December 31, 2019, our largest suppliers of raw materials and supplies were Institute for Radioelements (“IRE”), ANSTO and NTP, which, in the aggregate, accounted for approximately 26% of our total purchases.
Molybdenum-99
Our TechneLite, Cardiolite and Neurolite products all rely on Mo-99, the radioisotope which is produced by bombarding uranium with neutrons in research reactors. With a 66-hour half-life, Mo-99 decays into, among other things, Tc-99m, another radioisotope with a half-life of six hours. Tc-99m is the isotope that is attached to radiopharmaceuticals, including our own Cardiolite and Neurolite, during the labeling process and is the most common radioisotope used for medical diagnostic imaging purposes.
We currently purchase finished Mo-99 from three of the four main processing sites in the world, namely IRE in Belgium, NTP in South Africa and ANSTO in Australia. These processing sites provide us Mo-99 from five of the six main Mo-99-producing reactors in the world, namely BR2 in Belgium, LVR-15 in the Czech Republic, HFR in The Netherlands, SAFARI in South Africa and OPAL in Australia.
Our agreement with IRE (the “IRE Agreement”) contains minimum percentage volume requirements and unit pricing. The IRE Agreement also requires IRE to provide certain favorable allocations of Mo-99 during periods of supply shortage or failure. The IRE Agreement also provides for an increased supply of Mo-99 derived from LEU targets upon IRE’s completion of its ongoing conversion program to modify its facilities and processes in accordance with Belgian nuclear security commitments. The IRE Agreement allows for termination upon the occurrence of certain events, including failure by IRE to provide our required amount of Mo-99, material breach of any provision by either party, bankruptcy by either party or force majeure events. The IRE Agreement expires on December 31, 2020, and is renewable by LMI on a year-to-year basis thereafter.
Our agreement with NTP (the “NTP Agreement”), with NTP acting for itself and on behalf of its subcontractor ANSTO, specifies LMI’s percentage purchase requirements and unit pricing, and provides for the supply of Mo-99 derived from LEU targets from NTP and ANSTO. ANSTO’s new Mo-99 processing facility, could eventually increase ANSTO’s production capacity from approximately 2,000 curies per week to 3,500 curies per week with additional committed financial and operational resources. At full ramp-up capacity, ANSTO’s new facility could provide incremental supply to our globally diversified Mo-99 supply chain and therefore mitigate some risk among our Mo-99 suppliers, although we can give no assurances to that effect and a prolonged disruption of service from one of our three Mo-99 processing sites or one of their main Mo-99-producing reactors could have a substantial negative effect on our business, results of operations, financial condition and cash flows.
Despite our globally diverse Mo-99 suppliers, we still face challenges in our Mo-99 supply chain. The NTP processing facility had periodic outages in 2017, 2018 and 2019. When NTP was not producing, we relied on Mo-99 supply from both IRE and ANSTO
11
to limit the impact of the NTP outages. In the second quarter of 2019, ANSTO experienced facility issues in its existing Mo-99 processing facility which resulted in a decrease in Mo-99 available to us. In addition, as ANSTO transitioned from its existing Mo-99 processing facility to its new Mo-99 processing facility in the second quarter of 2019, ANSTO experienced start-up and transition challenges, which also resulted in a decrease in Mo-99 available to us. Further, starting in late June 2019 and through the date of this filing, ANSTO’s new Mo-99 processing facility has experienced unscheduled production outages, and we are now relying on IRE and NTP to limit the impact of those ANSTO outages. Because of these various supply chain constraints, depending on reactor and processor schedules and operations, we have not been able to fill some or all of the demand for our TechneLite generators on certain manufacturing days.
We are also pursuing additional sources of Mo-99 from potential new producers around the world to further augment our current supply. In November 2014, we entered into a strategic arrangement with SHINE for the future supply of Mo-99. Under the terms of the supply agreement, SHINE will provide Mo-99 produced using its proprietary LEU-solution technology for use in our TechneLite generators once SHINE’s facility becomes operational and receives all necessary regulatory approvals, which SHINE now estimates will occur in 2022. However, we cannot assure you that SHINE or any other possible additional sources of Mo-99 will result in commercial quantities of Mo-99 for our business, or that these new suppliers together with our current suppliers will be able to deliver a sufficient quantity of Mo-99 to meet our needs.
Xenon
Xenon is a by-product of the Mo-99 production process. Under a strategic agreement we entered into in 2015, we receive from IRE bulk unprocessed Xenon, which we process and finish for our customers at our North Billerica, Massachusetts manufacturing facility. That contract runs through June 30, 2022, and is subject to further extension. Until we can qualify an additional source of bulk unprocessed Xenon, we will rely on IRE as a sole source provider.
Other Materials
We have additional supply arrangements for active pharmaceutical ingredients, excipients, packaging materials and other materials and components, none of which are exclusive, but a number of which are sole source, and all of which we currently believe are either in good standing or replaceable without any material disruption to our business.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our raw materials and supply arrangements.
Manufacturing
We maintain manufacturing operations at our North Billerica, Massachusetts facility. We manufacture TechneLite on a highly automated production line, Thallium and Gallium and certain radiochemicals using our cyclotron technology, and we process and finish Xenon and Quadramet using our hot cell infrastructure. We also maintain manufacturing operations at our San Juan, Puerto Rico radiopharmacy and PET manufacturing facility where we manufacture FDG using cyclotron technology. We manufacture, finish and distribute our radiopharmaceutical products on a just-in-time basis, and supply our customers with these products either by next day delivery services or by either ground or air custom logistics. We believe that our substantial capital investments in our highly automated generator production line, our cyclotrons and our extensive experience in complying with the stringent regulatory requirements for the handling of nuclear materials and operations in a highly regulated environment create significant and sustainable competitive advantages for us.
In addition to our in-house manufacturing capabilities, a substantial portion of our products are manufactured by third party contract manufacturing organizations, and in certain instances, we rely on them for sole source manufacturing. To ensure the quality of the products that are manufactured by third parties, the key raw materials used in those products are first sent to our North Billerica, Massachusetts facility, where we test them prior to the third party manufacturing of the final product. After the final products are manufactured, they are sent back to us for final quality control testing and then we ship them to our customers. We have expertise in the design, development and validation of complex manufacturing systems and processes, and our strong execution and quality control culture supports the just-in-time manufacturing model at our North Billerica, Massachusetts facility.
We are also in the final stages of an extensive, multi-year effort to add specialized manufacturing capabilities at our North Billerica, Massachusetts facility. This project is part of a larger corporate growth strategy to create a competitive advantage in specialized manufacturing. This project should not only deliver efficiencies and supply chain redundancy for our current portfolio but should also afford us increased flexibility as we consider external opportunities. We currently expect to be in a position to use this in-house manufacturing capability by early 2021. However, we can give no assurance that we will be successful in these efforts or that we will be able to successfully manufacture any additional commercial products at our North Billerica, Massachusetts facility.
12
Manufacturing and Supply Arrangements
We currently have the following technology transfer and manufacturing and supply agreements in place for some of our major products:
• | DEFINITY—In February 2012, we entered into a Manufacturing and Supply Agreement with JHS, for the manufacture of DEFINITY. Under the agreement, JHS manufactured DEFINITY for us for an initial term of five years. In September 2016, we extended the agreement through January 2022. The agreement contains automatic renewals for additional one-year periods thereafter. The agreement allows for termination upon the occurrence of certain events such as a material breach or default by either party, or bankruptcy by either party. The agreement also requires us to place orders for a minimum percentage of our requirements for DEFINITY with JHS. Based on our current projections, we believe that we will have sufficient supply of DEFINITY from JHS to meet expected demand. |
On May 3, 2016, we entered into a Manufacturing and Supply Agreement with SBL to perform technology transfer and process development services to manufacture and supply a modified formulation of DEFINITY. There are no minimum purchase requirements under this agreement, which has an initial term of five years from the date of first commercial sale and is renewable at our option for an additional five years. This agreement allows for termination upon the occurrence of certain events, including material breach or bankruptcy of either party. We cannot give any assurances as to when those technology transfer activities will be completed and when we will begin to receive supply of a modified formulation of DEFINITY from SBL.
• | Cardiolite—In May 2012, we entered into a Manufacturing and Supply Agreement with JHS for the manufacture of Cardiolite products. In the third quarter of 2016, we completed the technology transfer process and received FDA approval to manufacture Cardiolite at JHS. Under the agreement, JHS has agreed to manufacture products for an initial term of five years from the effective date. On November 9, 2017, we extended the term until December 31, 2020, and the agreement can be further extended for three additional one-year periods thereafter so long as the parties, using good faith, reasonable efforts, agree to new pricing for the upcoming additional term. The agreement allows for termination upon the occurrence of specified events, including material breach or bankruptcy by either party. The agreement requires us to place orders for 100% of our requirements for Cardiolite products with JHS during such term. Based on our current projections, we believe that we will have sufficient supply of Cardiolite products from JHS to meet expected demand. |
• | Neurolite—In May 2012, we entered into a Manufacturing and Supply Agreement with JHS for the manufacture of Neurolite, and in January 2015, the FDA granted approval to manufacture Neurolite at JHS. Under the agreement, JHS agreed to manufacture Neurolite for an initial term of five years from the effective date. On November 9, 2017, we extended the term of the agreement until December 31, 2020, and the agreement can be further extended for three additional one-year periods thereafter so long as the parties, using good faith, reasonable efforts, agree to new pricing for the upcoming additional term. The agreement allows for termination upon the occurrence of specified events, including material breach or bankruptcy by either party. The agreement also requires us to place orders for 100% of our requirements for Neurolite during such term. Based on our current projections, we believe that we will have sufficient supply of Neurolite from JHS to meet expected demand. |
Although we are pursuing additional third party manufacturing relationships to establish and secure additional long-term or alternative suppliers as described above, we are uncertain of the timing as to when these arrangements could provide meaningful quantities of our products.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our manufacturing relationships.
Clinical Development
For the years ended December 31, 2019, 2018 and 2017, we invested $20.0 million, $17.1 million and $18.1 million in research and development (“R&D”), respectively. Our R&D team includes our Medical Affairs and Medical Information functions, which educate physicians on the scientific aspects of our commercial products and the approved indications. In addition to the DEFINITY clinical trials in China described above, we currently have three active clinical development programs.
Flurpiridaz F 18—PET Myocardial Perfusion
We have developed flurpiridaz F 18, an internally discovered small molecule radiolabeled with fluorine-18, as an imaging agent used in PET MPI to assess blood flow to the heart.
13
Today, most MPI procedures use SPECT technology. Although SPECT imaging used in conjunction with a radiopharmaceutical imaging agent, such as Cardiolite, is most commonly used for MPI studies, PET imaging has gained considerable support in the field of cardiovascular imaging as it offers many advantages to SPECT imaging, including: higher image quality, increased diagnostic certainty, more accurate risk stratification and reduced patient radiation exposure. PET imaging has demonstrated broad utility for diagnosis, prognosis, disease staging and therapeutic response. When used in combination with an appropriate radiopharmaceutical imaging agent, PET imaging can provide important insights into physiologic and metabolic processes in the body and be useful in evaluating a variety of conditions including heart disease, neurological disease and cancer. In addition, PET MPI imaging could be particularly useful in difficult-to-image patients, including women and obese patients. The use of PET technology in MPI tests represents a broad emerging application for a technology more commonly associated with oncology and neurology. We anticipate that the adoption of PET technology in MPI tests will increase significantly in the future.
Flurpiridaz F 18 Clinical Overview and Phase 3 Program
We submitted an Investigational New Drug Application (“IND”) for flurpiridaz F 18 to the FDA in August 2006. Our clinical program to date has consisted of three Phase 1 studies, a Phase 2 clinical trial, conducted from 2007 to 2010, involving 176 subjects who received PET MPI performed with flurpiridaz F 18 and completed the trial, and a Phase 3 clinical trial (“301 Trial”) conducted from 2011 to 2013.
The 301 Trial was an open-label, multicenter, international study with 755 subjects with known or suspected coronary artery disease (“CAD”) and scheduled for coronary angiography and SPECT imaging who completed the trial and were included in the efficacy analysis. Subjects underwent flurpiridaz F 18 PET MPI and SPECT MPI studies with coronary angiography used as the truth standard for each. The study then compared MPI imaging using flurpiridaz F 18 versus SPECT imaging with primary endpoints of superiority for sensitivity (identifying disease) and non-inferiority for specificity (ruling out disease).
In the fourth quarter of 2013, we announced preliminary results from the 301 Trial, and in May 2015, after a re-read of the 301 Trial results, we announced the complete results from the 301 Trial. Flurpiridaz F 18 appeared to be well-tolerated from a safety perspective, and PET MPI with flurpiridaz F 18 consistently showed a balanced performance in sensitivity and specificity, when compared to coronary angiography, while SPECT imaging results were skewed with low sensitivity and high specificity when compared to coronary angiography. When results were compared to one another, flurpiridaz F 18 imaging substantially outperformed SPECT imaging in sensitivity but did not meet the non-inferiority endpoint in specificity, implying a substantial and unexpected under-diagnosis of CAD with SPECT imaging in the trial.
In subgroup analyses, the risk-benefit profile of flurpiridaz F 18 appeared to be favorable in women, obese patients, patients with multi-vessel disease and diabetics. A significantly higher percentage of images were rated as either excellent or good with flurpiridaz F 18 imaging as compared to SPECT imaging, leading to a greater diagnostic certainty of interpretation. Importantly, radiation exposure associated with flurpiridaz F 18 imaging was reduced to approximately 50% of SPECT imaging. In addition, no drug-related serious adverse events were observed.
GE Healthcare Collaboration
In April 2017, we announced that we entered into a definitive, exclusive Collaboration and License Agreement (the “License Agreement”) with GE Healthcare for the continued Phase 3 development and worldwide commercialization of flurpiridaz F 18. Under the License Agreement, GE Healthcare will complete the worldwide development of flurpiridaz F 18, pursue worldwide regulatory approvals and, if successful, lead a worldwide launch and commercialization of the agent, with us collaborating on both development and commercialization through a joint steering committee.
The second Phase 3 clinical trial is underway, as a prospective, open-label, international, multi-center trial of flurpiridaz F 18 for PET MPI in patients referred for invasive coronary angiography because of suspected CAD. The trial will enroll up to 650 participants, with a target completion date in the second half of 2020, although that timing cannot be assured. The primary outcome measure for the trial is the diagnostic efficacy of flurpiridaz F 18 PET MPI in the detection of significant CAD, with secondary outcome measures of diagnostic efficacy of flurpiridaz F 18 PET MPI compared with SPECT MPI in the detection of CAD in all patients. Secondary analysis will be performed in patients of special clinical interest, such as female, obese and diabetic patients, where current SPECT MPI technologies have shown certain limitations in the diagnostic performance.
LMI 1195 (flubrobenguan F18)
We have developed LMI 1195, an internally discovered small molecule imaging agent for the norepinephrine pathway. We originally pursued developing LMI 1195 for the diagnostic assessment of ischemic heart failure patients at risk of sudden cardiac death who may benefit from an implantable cardioverter defibrillator (ICD). However, after multiple interactions with the FDA, we have concluded that the cardiac clinical development program for this indication would be longer and more expensive than we had
14
initially envisioned. As a result, we have paused in pursuing this particular indication but may consider other possible cardiac indications in the future.
We are now designing two Phase 3 clinical trials for the use of LMI 1195 for the diagnosis and management of neuroendocrine tumors in pediatric and adult populations, respectively. The FDA has granted an Orphan Drug designation for the use of LMI 1195 in the management indication. We have also received notice of eligibility for a rare pediatric disease priority review voucher for a subsequent human drug application so long as LMI 1195 is approved by the FDA for its rare pediatric disease indication prior to September 30, 2022.
DEFINITY - LVEF
As part of our microbubble franchise strategy, we conducted two Phase 3, open-label, multicenter studies to evaluate LVEF measurement accuracy and reproducibility of DEFINITY contrast-enhanced and unenhanced echocardiography as compared to non-contrast CMRI used as the truth standard. The first of the two trials, BENEFIT 1, enrolled 145 subjects. After reviewing the study results from BENEFIT 1, we concluded there was no statistically significant improvement in the accuracy of LVEF values for contrast-enhanced echocardiography versus unenhanced echocardiography as compared to CMRI. In addition, analyses of the secondary endpoints revealed no improvement in inter-reader variability between the contrast-enhanced and unenhanced echocardiograms for LVEF assessments. A post-hoc analysis, however, did show statistically significant improvements in left ventricular diastolic, systolic and stroke volume measurements with contrast-enhanced versus unenhanced echocardiography when compared to CMRI. We will continue to analyze the BENEFIT 1 data, and when the data from BENEFIT 2 are available, we will compile the data sets to analyze the full results of the trials.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our clinical development programs.
Strategic Activities
We continue to evaluate a number of different opportunities to acquire or in-license additional products, businesses and technologies to drive our future growth. We are particularly interested in expanding our presence in oncology, in radiotherapeutics as well as diagnostics. In addition to the Progenics Transaction described above, we recently entered into a strategic collaboration and license agreement with NanoMab Technology Limited, a privately-held biopharmaceutical company focusing on the development of next generation radiopharmaceuticals for cancer precision medicine. We believe this collaboration will provide the first broadly-available imaging biomarker research tool to pharmaceutical companies and academic centers conducting research and development on PD-L1 immuno-oncology treatments, including combination therapies. We can give no assurance as to when or if this collaboration will be successful or accretive to earnings.
In addition, as described above, we continue to expand our microbubble franchise. We recently announced a strategic development and commercial collaboration with Cerevast in which our microbubble will be used in connection with Cerevast’s ocular ultrasound device to target improving blood flow in occluded retinal veins in the eye. Retinal vein occlusion is one of the most common causes of vision loss worldwide. We also recently announced a strategic commercial supply agreement with CarThera for the use of our microbubbles in combination with SonoCloud, a proprietary implantable device in development for the treatment of recurrent glioblastoma, Glioblastoma is a lethal and devasting from of brain cancer with median survival of 15 months after diagnosis.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our strategic activities.
Intellectual Property
Patents, trademarks and other intellectual property rights, both in the U.S. and foreign countries, are very important to our business. We also rely on trade secrets, manufacturing know-how, technological innovations, licensing agreements and confidentiality agreements to maintain and improve our competitive position. We review third party proprietary rights, including patents and patent applications, as available, in an effort to develop an effective intellectual property strategy, avoid infringement of third party proprietary rights, identify licensing opportunities and monitor the intellectual property owned by others. Our ability to enforce and protect our intellectual property rights may be limited in certain countries outside the U.S., which could make it easier for competitors to capture market position in those countries by utilizing technologies that are similar to those developed or licensed by us. Competitors also may harm our sales by designing products that mirror the capabilities of our products or technology without infringing our intellectual property rights. If we do not obtain sufficient protection for our intellectual property, or if we are unable to effectively enforce our intellectual property rights, our competitiveness could be impaired, which would limit our growth and future revenue.
15
Trademarks, Service Marks and Trade Names
We own various trademarks, service marks and trade names, including, among others, DEFINITY, TechneLite, Cardiolite, Neurolite, Vialmix, Quadramet, Luminity and Lantheus Medical Imaging. We have registered these trademarks, as well as others, in the U.S. and/or numerous foreign jurisdictions.
Patents
We actively seek to protect the proprietary technology that we consider important to our business, including chemical species, compositions and formulations, their methods of use and processes for their manufacture, as new intellectual property is developed. In addition to seeking patent protection in the U.S., we file patent applications in numerous foreign countries in order to further protect the inventions that we consider important to the development of our international business. We also rely upon trade secrets and contracts to protect our proprietary information. As of December 31, 2019, our patent portfolio included a total of 43 issued U.S. patents, 284 issued foreign patents, 22 pending U.S. patent applications and 160 pending foreign applications. These patents and patent applications include claims covering the composition of matter and methods of use for all of our preclinical and clinical stage agents.
We have patent protection on certain of our commercial products and on all of our clinical development candidates. We typically seek patent protection in major markets around the world, including, among others, the U.S., Canada, Western Europe, Asia, and Latin America.
DEFINITY - We continue to actively pursue additional patents in connection with DEFINITY, both in the U.S. and internationally. In the U.S., we have an Orange Book-listed method of use patent expiring in March 2037 and additional manufacturing patents that are not Orange Book-listed expiring in 2021, 2023 and 2037. Outside of the U.S., while our DEFINITY patent protection and regulatory exclusivity have generally expired, we are currently prosecuting additional patents to try to obtain similar method of use patent protection as granted in the U.S.
Even though our longest duration Orange Book-listed DEFINITY patent extends until March 2037, because our Orange Book-listed composition of matter patent expired in June 2019, we may face generic DEFINITY challengers in the near to intermediate term. Under the Hatch-Waxman Act, the FDA can approve Abbreviated New Drug Applications (“ANDAs”) for generic versions of drugs if the ANDA applicant demonstrates, among other things, that (i) its generic candidate is the same as the innovator product by establishing bioequivalence and providing relevant chemistry, manufacturing and product data, and (ii) the marketing of that generic candidate does not infringe an Orange Book-listed patent. With respect to any Orange Book-listed patent covering the innovator product, the ANDA applicant must give a notice to the innovator (a “Notice”) that the ANDA applicant certifies that its generic candidate will not infringe the innovator’s Orange Book-listed patent or that the Orange Book-listed patent is invalid. The innovator can then challenge the ANDA applicant in court within 45 days of receiving that Notice, and FDA approval to commercialize the generic candidate will be stayed (that is, delayed) for up to 30 months (measured from the date on which a Notice is received) while the patent dispute between the innovator and the ANDA applicant is resolved in court. The 30 month stay could potentially expire sooner if the courts determine that no infringement had occurred or that the challenged Orange Book-listed patent is invalid or if the parties otherwise settle their dispute.
As of the date of filing of this Annual Report on Form 10-K, we have not received any Notice from an ANDA applicant. If we were to (i) receive any such Notice in the future, (ii) bring a patent infringement suit against the ANDA applicant within 45 days of receiving such Notice, and (iii) successfully obtain the full 30 month stay, then the ANDA applicant would be precluded from commercializing a generic candidate prior to the expiration of such 30 month stay period and potentially thereafter depending on how a patent dispute is resolved. Solely by way of example and not based on any knowledge we currently have, if we received a Notice from an ANDA applicant in March 2020 and the full 30 month stay was obtained, then the ANDA applicant would be precluded from commercialization until at least September 2022. If we received a Notice some number of months in the future and the full 30 month stay was obtained, the commercialization date would roll forward in the future by the same calculation.
TechneLite - We currently have patent protection in the U.S. and various foreign countries on certain component technology expiring in 2029. In addition, given the significant know-how and trade secrets associated with the methods of manufacturing and assembling the TechneLite generator, we believe we have a substantial amount of valuable and defensible proprietary intellectual property associated with the product.
Other Nuclear Products - Neither Cardiolite nor Neurolite is covered any longer by patent protection in either the U.S. or the rest of the world. Xenon, Thallium and Gallium have no patent protection; however, we have patent protection in the U.S. that expires in October 2035 for an improved container for Xenon, and are pursuing similar patent protection outside the U.S.
16
Clinical Development Candidates - We have patents and patent applications in numerous jurisdictions covering composition, use, formulation and manufacturing of flurpiridaz F 18, including in the U.S. a composition patent expiring in 2026, a method of use patent expiring in 2028 and a method of manufacturing patent expiring in 2031, in the absence of any regulatory extension, and various patent applications, one of which, if granted, will expire in 2033 in the absence of any patent term adjustment or regulatory extensions. We also have patents and patent applications in numerous jurisdictions covering composition, use, and manufacture of LMI 1195, including in the U.S. a composition patent expiring in 2030, a method of use patent expiring in 2027, and manufacturing-related patents expiring in 2031 and 2032, in the absence of any regulatory extension, and patent applications which, if granted, will expire in 2027 and in 2031 in the absence of any patent term adjustment or regulatory extensions.
In addition to patents, we rely, where necessary, upon unpatented trade secrets and know-how, proprietary information and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our collaborators, employees, consultants and other third parties and invention assignment agreements with our employees. These confidentiality agreements may not prevent unauthorized disclosure of trade secrets and other proprietary information, and we cannot provide assurances that an employee or an outside party will not make an unauthorized disclosure of our trade secrets, other technical know-how or proprietary information. We may not have adequate monitoring abilities to discover, or adequate remedies for, any unauthorized disclosure. This might happen intentionally or inadvertently. It is possible that a competitor will make use of such information, and that our competitive position will be compromised, in spite of any legal action we might take against persons making such unauthorized disclosures. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
In addition, we license a limited number of third party technologies and other intellectual property rights that are incorporated into some elements of our drug discovery and development efforts. These licenses are not material to our business, and the technologies can be obtained from multiple sources. We are currently party to separate royalty-free, non-exclusive, cross-licenses with each of Bracco, GE Healthcare and Imcor Pharmaceutical Company. These cross-licenses give us freedom to operate in connection with contrast enhanced ultrasound imaging technology.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our intellectual property.
Regulatory Matters
Food and Drug Laws
The development, manufacture and commercialization of our agents and products are subject to comprehensive governmental regulation both within and outside the U.S. A number of factors substantially increase the time, difficulty and costs incurred in obtaining and maintaining the approval to market newly developed and existing products. These factors include governmental regulation, such as detailed inspection of and controls over research and laboratory procedures, clinical investigations, manufacturing, marketing, sampling, distribution, import and export, record keeping and storage and disposal practices, together with various post-marketing requirements. Governmental regulatory actions can result in the seizure or recall of products, suspension or revocation of the authority necessary for their production and sale as well as other civil or criminal sanctions.
Our activities related to the development, manufacture, packaging or repackaging of our pharmaceutical and medical device products subject us to a wide variety of laws and regulations. We are required to register for permits and/or licenses with, seek approvals from and comply with operating and security standards of, the FDA, the U.S. Nuclear Regulatory Commission (“NRC”), the U.S. Department of Health and Human Services (“HHS”), Health Canada, the European Medicines Agency (“EMA”), the U.K. Medicines and Healthcare Products Regulatory Agency (“MHRA”), the CFDA and various state and provincial boards of pharmacy, state and provincial controlled substance agencies, state and provincial health departments and/or comparable state and provincial agencies, as well as foreign agencies, and certain accrediting bodies depending upon the type of operations and location of product distribution, manufacturing and sale.
The FDA and various state regulatory authorities regulate the research, testing, manufacture, safety, labeling, storage, recordkeeping, premarket approval, marketing, advertising and promotion, import and export and sales and distribution of pharmaceutical products in the U.S. Prior to marketing a pharmaceutical product, we must first receive FDA approval. In the U.S., the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and implementing regulations. The process of obtaining regulatory approvals and compliance with appropriate federal, state, local, and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Currently, the process required by the FDA before a drug product may be marketed in the U.S. generally involves the following:
• | Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices regulations; |
17
• | Submission to the FDA of an IND which must become effective before human clinical studies may begin, including review and approval by any individual review board (“IRB”), serving any of the institutions participating in the clinical studies; |
• | Performance of adequate and well-controlled human clinical studies according to Good Clinical Practices and other requirements, to establish the safety and efficacy of the proposed drug product for its intended use; |
• | Submission to the FDA of a new drug application, or NDA, for a new drug; |
• | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug product is produced to assess compliance with current Good Manufacturing Practices (“cGMPs”) regulations; and |
• | FDA review and approval of the NDA. |
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain that any approvals for our agents in development will be granted on a timely basis, if at all. Once a pharmaceutical agent is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity, formulation, and stability, as well as animal studies to assess its potential safety and efficacy. This testing culminates in the submission of the IND to the FDA.
Once the IND becomes effective, including review and approval by any IRB, serving any of the institutions participating in the clinical trial, the clinical trial program may begin. Each new clinical trial protocol must be submitted to the FDA before the study may begin. Human clinical studies are typically conducted in three sequential phases that may overlap or be combined:
• | Phase 1. The agent is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the agent may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients with those diseases. |
• | Phase 2. Involves studies in a limited patient population to identify possible adverse effects and safety risks, to evaluate preliminarily the efficacy of the agent for specific targeted diseases and to determine dosage tolerance and optimal dosage and schedule. |
• | Phase 3. Clinical studies are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to collect sufficient safety and efficacy data to support the NDA for FDA approval. |
Clinical trial sponsors may request a Special Protocol Assessment (“SPA”) from the FDA. The FDA’s SPA process creates a written agreement between the sponsoring company and the FDA regarding the clinical trial design and other clinical trial issues that can be used to support approval of an agent. The SPA is intended to provide assurance that, if the agreed-upon clinical trial protocols are followed and the trial endpoints are achieved, then the data may serve as the primary basis for an efficacy claim in support of an NDA. However, the SPA agreement is not a guarantee of an approval of an agent or any permissible claims about the agent. In particular, the SPA is not binding on the FDA if public health concerns become evident that are unrecognized at the time that the SPA agreement is entered into, other new scientific concerns regarding product safety or efficacy arise, or if the clinical trial sponsor fails to comply with the agreed upon clinical trial protocols.
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. Submissions must also be made to inform the FDA of certain changes to the clinical trial protocol. Federal law also requires the sponsor to register the trials on public databases when they are initiated, and to disclose the results of the trials on public databases upon completion. Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the clinical trial sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, any IRB serving any of the institutions participating in the clinical trial can suspend or terminate approval of a clinical study at a relevant institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the agent has been associated with unexpected serious harm to patients. Failure to register a clinical trial or disclose study results within the required time periods could result in penalties, including civil monetary penalties.
Concurrent with clinical studies, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the agent and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the agent does not undergo unacceptable deterioration over its shelf life.
18
The results of product development, preclinical studies and clinical studies, along with descriptions of the manufacturing process, analytical tests conducted on the drug product, proposed labeling, and other relevant information, are submitted to the FDA as part of an NDA for a new drug, requesting approval to market the agent. The submission of an NDA is subject to the payment of a substantial user fee. A waiver of that fee may be obtained under certain limited circumstances. The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied. The FDA has substantial discretion in the product approval process, and it is impossible to predict whether and when the FDA will grant marketing approval. The FDA may on occasion require the sponsor of an NDA to conduct additional clinical studies or to provide other scientific or technical information about the product, and these additional requirements may lead to unanticipated delay or expense. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical studies are not always conclusive, and the FDA may interpret data differently than we interpret the same data.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical studies designed to further assess a drug product’s safety and effectiveness after NDA approval. The FDA also may impose a Risk Evaluation and Mitigation Strategy (“REMS”) to ensure that the benefits of a product outweigh its risks. A REMS could add training requirements for healthcare professionals, safety communications efforts and limits on channels of distribution, among other things. The sponsor would be required to evaluate and monitor the various REMS activities and adjust them if need be. Whether a REMS would be imposed on any of our products and any resulting financial impact is uncertain at this time.
Any drug products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements, and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on drug products that are placed on the market. Drugs may be promoted only for the approved indications and consistent with the provisions of the approved label and promotional claims must be appropriately balanced with important safety information and otherwise be adequately substantiated. Further, manufacturers of drugs must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented, and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Drug product manufacturers and other entities involved in the manufacturing and distribution of approved drugs products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain other agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug product. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release. In addition, manufacturers of commercial PET products, including radiopharmacies, hospitals and academic medical centers, are required to submit either an NDA or ANDA in order to produce PET drugs for clinical use, or produce the drugs under an IND.
The FDA also regulates the preclinical and clinical testing, design, manufacture, safety, efficacy, labeling, storage, record keeping, sales and distribution, post-market adverse event reporting, import/export and advertising and promotion of any medical devices that we distribute pursuant to the FDCA and FDA’s implementing regulations. The Federal Trade Commission shares jurisdiction with the FDA over the promotion and advertising of certain medical devices. The FDA can also impose restrictions on the sale, distribution or use of medical devices at the time of their clearance or approval, or subsequent to marketing. Currently, medical devices comprise only a small portion of our revenues.
The FDA may withdraw marketing authorization for a pharmaceutical or medical device product if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, civil monetary penalties, warning letters, holds on clinical studies, product recalls or seizures, product detention or refusal to permit the import or export of pharmaceuticals or medical device products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions, or civil or criminal penalties.
Because our operations include the manufacture and distribution of medical radioisotopes and other medical products, we are subject to regulation by the NRC and the departments of health of each state in which we operate and the applicable state boards of pharmacy. In addition, the FDA is also involved in the regulation of cyclotron facilities where PET products are produced in compliance with cGMP requirements and U.S. Pharmacopeia requirements for PET drug compounding.
19
Drug laws also are in effect in many of the non-U.S. markets in which we conduct business. These laws range from comprehensive drug approval requirements to requests for product data or certifications. In addition, inspection of and controls over manufacturing, as well as monitoring of adverse events, are components of most of these regulatory systems. Most of our business is subject to varying degrees of governmental regulation in the countries in which we operate, and the general trend is toward increasingly stringent regulation. The exercise of broad regulatory powers by the FDA continues to result in increases in the amount of testing and documentation required for approval or clearance of new drugs and devices, all of which add to the expense of product introduction. Similar trends also are evident in major non-U.S. markets, including Canada, the European Union, Australia and Japan.
To assess and facilitate compliance with applicable FDA, NRC and other state, federal and foreign regulatory requirements, we regularly review our quality systems to assess their effectiveness and identify areas for improvement. As part of our quality review, we perform assessments of our suppliers of the raw materials that are incorporated into products and conduct quality management reviews designed to inform management of key issues that may affect the quality of our products. From time to time, we may determine that products we manufactured or marketed do not meet our specifications, published standards, such as those issued by the International Standards Organization, or regulatory requirements. When a quality or regulatory issue is identified, we investigate the issue and take appropriate corrective action, such as withdrawal of the product from the market, correction of the product at the customer location, notice to the customer of revised labeling and other actions.
Hatch-Waxman Act
The Hatch-Waxman Act added two pathways for FDA drug approval. First, the Hatch-Waxman Act permits the FDA to approve ANDAs for generic versions of drugs if the ANDA applicant demonstrates, among other things, that its product is bioequivalent to the innovator product and provides relevant chemistry, manufacturing and product data. See “Item 1. Business - Patents.” Second, the Hatch-Waxman Act created what is known as a Section 505(b) (2) NDA, which requires the same information as a full NDA (known as a Section 505(b) (1) NDA), including full reports of clinical and preclinical studies but allows some of the information from the reports required for marketing approval to come from studies which the applicant does not own or have a legal right of reference. A Section 505(b) (2) NDA permits a manufacturer to obtain marketing approval for a drug without needing to conduct or obtain a right of reference for all of the required studies. The Hatch-Waxman Act also provides for: (1) restoration of a portion of a product’s patent term that was lost during clinical development and application review by the FDA; and (2) statutory protection, known as exclusivity, against the FDA’s acceptance or approval of certain competitor applications.
Patent term extension can compensate for time lost during product development and the regulatory review process by returning up to five years of patent life for a patent that covers a new product or its use. This period is generally one-half the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Patent term extensions, however, are subject to a maximum extension of five years, and the patent term extension cannot extend the remaining term of a patent beyond a total of 14 years. The application for patent term extension is subject to approval by the U.S. Patent and Trademark Office in conjunction with the FDA.
The Hatch-Waxman Act also provides for a period of statutory protection for new drugs that receive NDA approval from the FDA. If the FDA approves a Section 505(b) (1) NDA for a new drug that is a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active moiety, then the Hatch-Waxman Act prohibits the submission or approval of an ANDA or a Section 505(b) (2) NDA for a period of five years from the date of approval of the NDA, except that the FDA may accept an application for review after four years under certain circumstances. The Hatch-Waxman Act will not prevent the filing or approval of a full NDA, as opposed to an ANDA or Section 505(b) (2) NDA, for any drug, but the competitor would be required to conduct its own clinical trials, and any use of the drug for which marketing approval is sought could not violate another NDA holder’s patent claims. The Hatch-Waxman Act provides for a three-year period of exclusivity for an NDA for a new drug containing an active moiety that was previously approved by the FDA, but also includes new clinical data (other than bioavailability and bioequivalence studies) to support an innovation over the previously approved drug and those studies were conducted or sponsored by the applicant and were essential to approval of the application. This three-year exclusivity period does not prohibit the FDA from accepting an application from a third party for a drug with that same innovation, but it does prohibit the FDA from approving that application for the three-year period. The three-year exclusivity does not prohibit the FDA, with limited exceptions, from approving generic drugs containing the same active ingredient but without the new innovation.
20
Healthcare Reform and Other Laws Affecting Payment
We operate in a highly-regulated industry. The U.S. and state governments continue to propose and pass legislation that may affect the availability and cost of healthcare. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Healthcare Reform Act, substantially changes the way in which healthcare is financed by both governmental and private insurers and has a significant impact on the pharmaceutical industry. The Healthcare Reform Act contains a number of provisions that affect coverage, reimbursement and/or delivery of drug products and the medical imaging procedures in which our drug products are used. Key provisions that currently affect our business include the following:
• | increasing the presumed utilization rate for imaging equipment costing $1 million or more in the physician office and free-standing imaging facility setting which reduces the Medicare per procedure medical imaging reimbursement; which rate was further increased by subsequent legislation effective January 1, 2014; |
• | increasing drug rebates paid to state Medicaid programs under the Medicaid Drug Rebate Program for brand name prescription drugs and extending those rebates to Medicaid managed care organizations; |
• | imposing a non-deductible annual fee on pharmaceutical manufacturers or importers who sell brand name prescription drugs to specified federal government programs; and |
• | amending the federal self-referral laws to require referring physicians ordering certain diagnostic imaging services to inform patients under certain circumstances that the patients may obtain the services from other local and unaffiliated suppliers (which may affect the setting in which a patient obtains services). |
The Healthcare Reform Act also amended the federal self-referral laws, requiring referring physicians to inform patients under certain circumstances that the patients may obtain services, including MRI, computed tomography (“CT”), PET and certain other diagnostic imaging services, from a provider other than that physician, another physician in his or her group practice, or another individual under direct supervision of the physician or another physician in the group practice. The referring physician must provide each patient with a written list of other suppliers who furnish those services in the area in which the patient resides. These new requirements could have the effect of shifting where certain diagnostic medical imaging procedures are performed.
The Healthcare Reform Act has been subject to political and judicial challenges. For example, tax reform legislation was enacted at the end of 2017 that effectively eliminated the “individual mandate” to maintain health insurance coverage by eliminating the tax penalty for individuals who do not maintain sufficient health insurance coverage beginning in 2019. In December 2018, a federal district court judge, in a challenge brought by a number of state attorneys general, found the Healthcare Reform Act unconstitutional in its entirety because once Congress repealed the “individual mandate” provision, there was no longer a basis to rely on Congressional taxing authority to support enactment of the law. In December 2019, a federal appeals court agreed that the individual mandate provision was unconstitutional, but remanded the case back to the district court to assess more carefully whether any provisions of the Healthcare Reform Act were severable and could survive. Pending action by the district court and resolution of any appeals, which could take some time, the Healthcare Reform Act is still operational in all respects.
Recently, there has been considerable public and government scrutiny of pharmaceutical pricing and proposals to address the perceived high cost of pharmaceuticals. For example, in May 2018, President Trump and the Secretary of the Department of Health and Human Services released a “blueprint” to lower prescription drug prices and out-of-pocket costs. Certain proposals in the blueprint, and related drug pricing measures proposed since the blueprint, could cause significant operational and reimbursement changes for the pharmaceutical industry. As another example, in October 2018, CMS solicited public comments on potential changes to payment for certain Medicare Part B drugs, including reducing the Medicare payment amount for selected Medicare Part B drugs to more closely align with international drug prices. As another example, legislation passed in 2019 revised how certain prices reported by manufacturers under the Medicaid Drug Rebate Program are calculated, a revision that the Congressional Budget Office has estimated will save the federal government approximately $3 billion in the next ten years. Efforts by government officials or legislators to implement measures to regulate prices or payment for pharmaceutical products could limit our flexibility in establishing prices for our products or otherwise adversely affect our business if implemented. Changes could occur at the federal level or state level and may be adopted by statute, rule, or sub-regulatory policies. Recent state legislative efforts seek to address drug costs and generally have focused on increasing transparency around drug costs or limiting drug prices. Some of those efforts have been subject to legal challenge.
General legislative cost control measures may also affect reimbursement for our products or services provided with our products. The Budget Control Act, as amended by the Bipartisan Budget Act of 2019, resulted in the imposition of 2% reductions in Medicare (but not Medicaid) payments to providers beginning in 2013 and will remain in effect through 2029 unless additional Congressional action is taken. Any significant spending reductions affecting Medicare, Medicaid or other publicly funded or subsidized health programs that may be implemented and/or any significant taxes or fees that may be imposed on us could have an adverse impact on our business results of operations, financial condition and cash flows.
21
Healthcare Fraud and Abuse Laws
We are subject to various federal, state and local laws targeting fraud and abuse in the healthcare industry, including anti-kickback and false claims laws. Violations of fraud and abuse laws may be punishable by criminal or civil sanctions, including fines and civil monetary penalties, and/or exclusion from federal health care programs (including Medicare and Medicaid). Federal and state authorities are paying increased attention to enforcement of these laws within the pharmaceutical industry, and private individuals have been active in alleging violations of the laws and bringing suits on behalf of the government under the federal False Claims Act (“FCA”). Violations of international fraud and abuse laws could result in similar penalties, including exclusion from participation in health programs outside the U.S. If we were subject to allegations concerning, or were convicted of violating, these laws, our business could be harmed.
The federal Anti-Kickback Statute generally prohibits, among other things, a pharmaceutical manufacturer from directly or indirectly soliciting, offering, receiving, or paying any remuneration in cash or in kind where one purpose is either to induce the referral of an individual for, or the purchase or prescription of a particular drug that is payable by a federal health care program, including Medicare or Medicaid. The Healthcare Reform Act clarifies the intent requirements of the federal Anti-Kickback Statute, providing that a person or entity does not need to have actual knowledge of the statute or a specific intent to violate the statute. Violations of the federal Anti-Kickback Statute can result in exclusion from Medicare, Medicaid or other governmental programs as well as civil and criminal fines and penalties of up to $102,522 per violation and three times the amount of the unlawful remuneration. In addition, the Healthcare Reform Act revised the FCA to provide that a claim arising from a violation of the Federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. The majority of states also have anti-kickback, false claims, and similar fraud and abuse laws and although the specific provisions of these laws vary, their scope is generally broad, and there may not be regulations, guidance or court decisions that apply the laws to particular industry practices. There is therefore a possibility that our practices might be challenged under the anti-kickback statutes or similar laws.
Federal and state false claims laws generally prohibit anyone from knowingly and willfully, among other activities, presenting, or causing to be presented for payment to third party payors (including Medicare and Medicaid) claims for drugs or services that are false or fraudulent (which may include claims for services not provided as claimed or claims for medically unnecessary services). As discussed, a claim arising from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. False or fraudulent claims for purposes of the FCA carry fines and civil penalties for violations ranging from $11,181 to $22,363 for each false claim, plus up to three times the amount of damages sustained by the federal government and, most critically, may provide the basis for exclusion from federally funded healthcare programs. There is also a criminal FCA statute by which individuals or entities that submit false claims can face criminal penalties. In addition, under the federal Civil Monetary Penalty Law, the Department of Health and Human Services Office of Inspector General has the authority to exclude from participation in federal health care programs or to impose civil penalties against any person who, among other things, knowingly presents, or causes to be presented, certain false or otherwise improper claims. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws.
Laws and regulations have also been enacted by the federal government and various states to regulate the sales and marketing practices of pharmaceutical manufacturers. The laws and regulations generally limit financial interactions between manufacturers and health care providers; require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government; and/or require disclosure to the government and/or public of financial interactions (so-called “sunshine laws”). The Healthcare Reform Act requires manufacturers to submit information to the FDA on the identity and quantity of drug samples requested and distributed by a manufacturer during each year. Many of these laws and regulations contain ambiguous requirements or require administrative guidance for implementation. Given the lack of clarity in laws and their implementation, our activities could be subject to the penalty provisions of the pertinent federal and state laws and regulations.
Data Privacy and Security
We are subject to data protection laws and regulations that address privacy and data security. The legislative and regulatory landscape for data protection continues to evolve, and in recent years there has been an increasing focus on privacy and data security issues. In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws and federal and state consumer protection laws govern the collection, use, disclosure and protection of health-related and other personal information. Failure to comply with data protection laws and regulations could result in government enforcement actions, which could include civil or criminal penalties, private litigation and/or adverse publicity and could negatively affect our operating results and business. In addition, we may obtain health information from third parties (e.g., healthcare providers who prescribe our products) that are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act (collectively, “HIPAA”). While we believe that we are neither a “covered entity” nor “business associate” subject directly to regulation under HIPAA, HIPAA’s criminal provisions can apply to entities other than “covered entities” or “business associates” in certain
22
circumstances. Accordingly, we could be subject to criminal penalties if we knowingly obtain or disclose individually identifiable health information from a HIPAA-covered entity in a manner that is not authorized or permitted.
The collection and use of personal health data in the European Union is governed by the provisions of the General Data Protection Regulation, or GDPR, which came into effect in May 2018. This regulation imposes several requirements relating to the consent of the individuals to whom the personal data relates, the information provided to the individuals, notification of data processing obligations to the competent national data protection authorities and the security and confidentiality of the personal data. The GDPR also imposes strict rules on the transfer of personal data out of the European Union to the United States. Failure to comply with the requirements of the GDPR and the related national data protection laws of the European Union Member States may result in significant fines and other administrative penalties.
In the United States, several state legislatures are considering enacting new data privacy legislation. One example of such legislation that has already been passed is the California Consumer Privacy Act (“CCPA”), which took effect on January 1, 2020 and imposed many requirements on businesses that process the personal information of California residents. Many of the CCPA’s requirements are similar to those found in the GDPR, including requiring businesses to provide notice to data subjects regarding the information collected about them and how such information is used and shared, and providing data subjects the right to request access to such personal information and, in certain cases, request the erasure of such personal information. The CCPA also affords California residents the right to opt-out of “sales” of their personal information. The CCPA contains significant penalties for companies that violate its requirements. It also provides California residents a private right of action, including the ability to seek statutory damages, in the event of a breach involving their personal information. Compliance with the CCPA is a rigorous and time-intensive process that may increase the cost of doing business or require companies to change their business practices to ensure full compliance.
Antitrust and Competition Laws
The federal government and most states have enacted antitrust laws that prohibit specific types of anti-competitive conduct, including price fixing, wage fixing, concerted refusals to deal, price discrimination and tying arrangements, as well as monopolization and acquisitions of competitors that have, or may have, a substantial adverse effect on competition. Violations of federal or state antitrust laws can result in various sanctions, including criminal and civil penalties. We believe we are in compliance with such federal and state laws, but courts or regulatory authorities may reach a determination in the future that could adversely affect our business, results of operations, financial condition and cash flows. In addition, we are subject to similar antitrust and anti-competition laws in foreign countries. We believe we are in compliance with such laws, however, any violation could create a substantial liability for us and also cause a loss of reputation in both foreign and domestic markets.
Laws Relating to Foreign Trade
We are subject to various federal and foreign laws that govern our international business practices with respect to payments to government officials. Those laws include the Foreign Corrupt Practices Act (“FCPA”) which prohibits U.S. companies and their representatives from paying, offering to pay, promising, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate for the purpose of obtaining or retaining business or to otherwise obtain favorable treatment or influence a person working in an official capacity. In many countries, the healthcare professionals we regularly interact with may meet the FCPA’s definition of a foreign government official. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect their transactions and to devise and maintain an adequate system of internal accounting controls.
Those laws also include the U.K. Bribery Act (“Bribery Act”) which proscribes giving and receiving bribes in the public and private sectors, bribing a foreign public official, and failing to have adequate procedures to prevent employees and other agents from giving bribes. U.S. companies that conduct business in the United Kingdom generally will be subject to the Bribery Act. Penalties under the Bribery Act include potentially unlimited fines for companies and criminal sanctions for corporate officers under certain circumstances.
Our policies mandate compliance with these anti-bribery laws. Our operations reach many parts of the world that have experienced governmental corruption to some degree, and in certain circumstances strict compliance with anti-bribery laws may conflict with local customs and practices. Despite our training and compliance programs, our internal control policies and procedures may not always protect us from reckless or criminal acts committed by our employees or agents.
We are also subject to trade control regulations and trade sanctions laws that restrict the movement of certain goods, currency, products, materials, services and technology to, and certain operations in, various countries or with certain persons. Our ability to transfer people and products among certain countries may be subjected to these laws and regulations.
23
Health and Safety Laws
We are also subject to various federal, state and local laws, regulations and recommendations, both in the U.S. and abroad, relating to safe working conditions, laboratory and manufacturing practices and the use, transportation and disposal of hazardous or potentially hazardous substances.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks related to regulatory matters.
Environmental Matters
We are subject to various federal, state and local laws and regulations relating to the protection of the environment, human health and safety in the U.S. and in other jurisdictions in which we operate. Our operations, like those of other medical product companies, involve the transport, use, handling, storage, exposure to and disposal of materials and wastes regulated under environmental laws, including hazardous and radioactive materials and wastes. If we violate these laws and regulations, we could be fined, criminally charged or otherwise sanctioned by regulators. We believe that our operations currently comply in all material respects with applicable environmental laws and regulations.
Certain environmental laws and regulations assess liability on current or previous owners or operators of real property for the cost of investigation, removal or remediation of hazardous materials or wastes at those formerly owned or operated properties or at third-party properties at which they have disposed of hazardous materials or wastes. In addition to cleanup actions brought by governmental authorities, private parties could bring personal injury, property damage or other claims due to the presence of, or exposure to, hazardous materials or wastes. We currently are not party to any claims or any obligations to investigate or remediate any material contamination at any of our facilities.
We are required to maintain a number of environmental permits and nuclear licenses for our North Billerica, Massachusetts facility, which is our primary manufacturing, packaging and distribution facility. In particular, we must maintain a nuclear byproducts materials license issued by the Commonwealth of Massachusetts. This license requires that we provide financial assurance demonstrating our ability to cover the cost of decommissioning and decontaminating (“D&D”) the Billerica site at the end of its use as a nuclear facility. In addition, we have a radioactive production facility in San Juan, Puerto Rico, where we must also maintain a number of environmental permits and nuclear licenses. As of December 31, 2019, we currently estimate the D&D cost of both sites to be approximately $26.9 million. As of December 31, 2019 and 2018, we have a liability recorded associated with the fair value of the asset retirement obligations of $12.9 million and $11.6 million, respectively. We currently provide this financial assurance in the form of surety bonds. We generally contract with third parties for the disposal of wastes generated by our operations. Prior to disposal, we store any low level radioactive waste at our facilities until the materials are below regulatory limits, as allowed by our licenses and permits.
Environmental laws and regulations are complex, change frequently and have become more stringent over time. While we have budgeted for future capital and operating expenditures to maintain compliance with these laws and regulations, we cannot assure you that our costs of complying with current or future environmental protection, health and safety laws and regulations will not exceed our estimates or adversely affect our results of operations and financial condition. Further, we cannot assure you that we will not be subject to additional environmental claims for personal injury or cleanup in the future based on our past, present or future business activities. While it is not feasible to predict the future costs of ongoing environmental compliance, it is possible that there will be a need for future provisions for environmental costs that, in management’s opinion, are not likely to have a material effect on our financial condition, but could be material to the results of operations in any one accounting period.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with environmental matters.
Employees
As of December 31, 2019, we had 508 employees, of which 464 were located in the U.S. and 44 were located internationally. None of our employees are represented by a collective bargaining agreement, and we believe that our relationship with our employees is good.
Corporate History
Founded in 1956 as New England Nuclear Corporation, our medical imaging diagnostic business was purchased by E.I. du Pont de Nemours and Company (“DuPont”) in 1981. Bristol Myers Squibb (“BMS”) subsequently acquired our diagnostic medical imaging business as part of its acquisition of DuPont Pharmaceuticals in 2001. In January 2008, Avista Capital Partners, L.P., Avista Capital Partners (Offshore), L.P. and ACP-Lantern Co-Invest, LLC (collectively “Avista”) formed Lantheus Holdings and acquired our medical imaging business from BMS. On June 30, 2015, we completed an initial public offering (“IPO”) of our common stock. Our common stock is traded on the NASDAQ Global Market under the symbol “LNTH”.
24
Available Information
Our global Internet site is www.lantheus.com. We routinely make available important information, including copies of our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, as soon as reasonably practicable after those reports are electronically filed with, or furnished to, the SEC, free of charge on our website at www.investor.lantheus.com. We recognize our website as a key channel of distribution to reach public investors and as a means of disclosing material non-public information to comply with our disclosure obligations under SEC Regulation FD. Information contained on our website shall not be deemed incorporated into, or to be part of this Annual Report on Form 10-K, and any website references are not intended to be made through active hyperlinks.
Our reports filed with, or furnished to, the SEC are also available on the SEC’s website at www.sec.gov, and for Annual Reports on Form 10-K and Quarterly Reports on Form 10-Q, in an XBRL (Extensible Business Reporting Language) format. XBRL is an electronic coding language used to create interactive financial statement data over the Internet. The information on our website is neither part of nor incorporated by reference in this Annual Report on Form 10-K.
Item 1A. Risk Factors
You should carefully consider the following risks. These risks could materially affect our business, results of operations or financial condition, cause the trading price of our outstanding common stock to decline materially or cause our actual results to differ materially from those expected or those expressed in any forward-looking statements made by us or on our behalf. See “Cautionary Note Regarding Forward-Looking Statements” and the risks of our businesses described elsewhere in this Annual Report on Form 10‑K.
Risks Related to Our Current Products and Revenues
The growth of our business is substantially dependent on our ability to continue to grow the appropriate use of DEFINITY in suboptimal echocardiograms in the face of increased segment competition from other existing echocardiography agents and potential generic competitors as a result of future patent and regulatory exclusivity expirations.
The growth of our business is substantially dependent on our ability to continue to grow the appropriate use of DEFINITY in suboptimal echocardiograms. There were approximately 35.1 million echocardiograms in 2019 according to a third-party source. Assuming 20% of echocardiograms produce suboptimal images, as stated in the clinical literature, we estimate that approximately 7.0 million echocardiograms in 2019 produced suboptimal images. We estimate that DEFINITY held over 80% of the U.S. market for contrast agents in echocardiography procedures as of December 31, 2019. DEFINITY currently competes with Optison, a GE Healthcare product, Lumason, a Bracco product (known as SonoVue outside the U.S.), as well as echocardiography without contrast and other non-echocardiography agents.
We launched DEFINITY in 2001, and we continue to actively pursue patents in connection with DEFINITY, both in the U.S. and internationally. In the U.S., we have an Orange Book-listed method of use patent expiring in March 2037 and additional manufacturing patents that are not Orange Book-listed expiring in 2021, 2023 and 2037. Outside of the U.S., while our DEFINITY patent protection and regulatory exclusivity have generally expired, we are currently prosecuting additional patents to try to obtain similar method of use and manufacturing patent protection as granted in the U.S. We were also recently granted a composition of matter patent on the modified formulation of DEFINITY which runs through December 2035. If the modified formulation is approved by the FDA, then this patent would be eligible to be listed in the Orange Book.
Because our Orange Book-listed composition of matter patent expired in June 2019, we may face generic DEFINITY challengers in the near to intermediate term. Under the Hatch-Waxman Act, the FDA can approve ANDAs for generic versions of drugs before the expiration of an Orange Book-listed patent covering the innovator product if the ANDA applicant demonstrates, among other things, that (i) its generic candidate is the same as the innovator product by establishing bioequivalence and providing relevant chemistry, manufacturing and product data, and (ii) the marketing of that generic candidate does not infringe an Orange Book-listed patent or the Orange Book-listed patent is invalid. With respect to any Orange Book-listed patent covering the innovator product that expires after the ANDA applicant intends to begin commercialization, the ANDA applicant must certify that its generic candidate will not infringe the innovator’s Orange Book-listed patents or that the Orange Book-listed patents are invalid. The ANDA applicant must also give Notice to the innovator, which would then enable the innovator to challenge the ANDA applicant in court within 45 days of receiving such Notice. If the innovator challenges the ANDA applicant in court in a timely manner, then FDA approval to commercialize the generic candidate will be stayed (that is, delayed) for up to 30 months while the dispute between the innovator and the ANDA applicant is resolved in court. The 30 month stay can be shortened if the patent infringement suit is resolved in the ANDA applicant’s favor before the 30 month stay expires, and this may involve a successful challenge of the patent’s validity in U.S. Patent and Trademark Office, or USPTO, proceedings and appeals process.
25
As of the date of filing of this Annual Report on Form 10-K, we have not received any such Notice from any ANDA applicant but can give no assurance that we will not receive a Notice in the future. If we were to receive any such Notice in the future, we would review the Notice, evaluate the strength of any potential patent infringement claims, and be prepared to challenge the ANDA applicant in a timely fashion, which would thereby trigger the stay of up to 30 months. We can give no assurance that we would have grounds to file a patent infringement suit, that we would obtain the full 30 month stay, that we would be successful on the merits asserting that a generic candidate infringes our Orange Book-listed patent, or that we would be successful defending the validity of our Orange Book-listed patent in court or in a USPTO adversarial proceeding.
As part of our microbubble franchise strategy, (i) we are developing a modified formulation of DEFINITY, (ii) we look for other opportunities to expand our microbubble franchise, including new applications beyond echocardiography and contrast imaging generally such as our strategic arrangements with Cerevast and CarThera, and (iii) we continue to build specialized in-house manufacturing capabilities at our North Billerica facility for DEFINITY and, potentially, other products. However, we can give no assurance that our microbubble franchise strategy will be successful or that new manufacturing capabilities, a new indication, a modified formulation or new applications will grow our microbubble franchise.
We have on-going development and technology transfer activities for our modified formulation with SBL located in South Korea but can give no assurances as to when or if those development and technology transfer activities will be completed and when we will begin to receive a supply of our modified formulation from SBL. In addition, potential global disruptions in air transport due to COVID-19 (coronavirus) could adversely affect our ability to receive a supply of our modified formulation from SBL, which, depending upon the magnitude and duration of such disruptions, could delay the commercial launch of our modified formulation.
If we are not able to continue to (i) grow DEFINITY sales, which depend on one or more of the growth of echocardiograms, the growth in the appropriate use of contrast in suboptimal echocardiograms, and our ability to sustain and grow our leading position in the U.S. echocardiography contrast market, or (ii) be successful with our microbubble franchise strategy, we may not be able to continue to grow the revenue and cash flow of our business, which could have a negative effect on our business, results of operations and financial condition.
The global supply of Mo-99 is fragile and not stable. Our dependence on a limited number of third party suppliers for Mo-99 could prevent us from delivering some of our products to our customers in the required quantities, within the required timeframe, or at all, which could result in order cancellations and decreased revenues.
A critical ingredient of TechneLite is Mo-99. We currently purchase finished Mo-99 from three of the four main processing sites in the world, namely IRE in Belgium, NTP in South Africa and ANSTO in Australia. These processing sites provide us Mo-99 from five of the six main Mo-99-producing reactors in the world, namely BR2 in Belgium, LVR-15 in the Czech Republic, HFR in The Netherlands, SAFARI in South Africa and OPAL in Australia.
The NTP processing facility had periodic outages in 2017, 2018 and 2019. When NTP was not producing, we relied on Mo-99 supply from both IRE and ANSTO to limit the impact of the NTP outages. In the second quarter of 2019, ANSTO experienced facility issues in its existing Mo-99 processing facility which resulted in a decrease in Mo-99 available to us. In addition, as ANSTO transitioned from its existing Mo-99 processing facility to its new Mo-99 processing facility in the second quarter of 2019, ANSTO experienced start-up and transition challenges, which also resulted in a decrease in Mo-99 available to us. Further, starting in late June 2019 and through the date of this filing, ANSTO’s new Mo-99 processing facility has experienced unscheduled production outages, and we are now relying on IRE and NTP to limit the impact of those ANSTO outages. Because of these various supply chain constraints, depending on reactor and processor schedules and operations, we have not been able to fill some or all of the demand for our TechneLite generators on certain manufacturing days, consequently decreasing revenue and cash flow from this product line during the outage periods as compared to prior periods.
ANSTO’s new Mo-99 processing facility, could eventually increase ANSTO’s production capacity from approximately 2,000 curies per week to 3,500 curies per week with additional committed financial and operational resources. At full ramp-up capacity, ANSTO’s new facility could provide incremental supply to our globally diversified Mo-99 supply chain and therefore mitigate some risk among our Mo-99 suppliers, although we can give no assurances to that effect, and a prolonged disruption of service from one of our three Mo-99 processing sites or one of their main Mo-99-producing reactors could have a substantial negative effect on our business, results of operations, financial condition and cash flows.
26
We are also pursuing additional sources of Mo-99 from potential new producers around the world to further augment our current supply. In November 2014, we entered into a strategic arrangement with SHINE for the future supply of Mo-99. Under the terms of the supply agreement, SHINE will provide Mo-99 produced using its proprietary LEU-solution technology for use in our TechneLite generators once SHINE’s facility becomes operational and receives all necessary regulatory approvals, which SHINE now estimates will occur in 2022. However, we cannot assure you that SHINE or any other possible additional sources of Mo-99 will result in commercial quantities of Mo-99 for our business, or that these new suppliers together with our current suppliers will be able to deliver a sufficient quantity of Mo-99 to meet our needs.
U.S., Canadian and international governments have encouraged the development of a number of alternative Mo-99 production projects with existing reactors and technologies as well as new technologies. However, we cannot say when, or if, the Mo-99 produced from these projects will become available. As a result, there is a limited amount of Mo-99 available which could limit the quantity of TechneLite that we could manufacture, sell and distribute, resulting in a further substantial negative effect on our business, results of operations, financial condition and cash flows.
Most of the global suppliers of Mo-99 rely on Framatone-CERCA in France to fabricate uranium targets and in some cases fuel for research reactors from which Mo-99 is produced. Absent a new supplier, a supply disruption relating to uranium targets or fuel could have a substantial negative effect on our business, results of operations, financial condition and cash flows.
In addition, because we source our radioisotopes almost exclusively from international suppliers, potential global disruptions in air transport due to COVID-19 (coronavirus) could adversely affect our international supply chain for radioisotopes which, depending upon the magnitude and duration of such disruptions, could have a substantial negative effect on our business, results of operations, financial condition and cash flows.
The instability of the global supply of Mo-99, including supply shortages, has resulted in increases in the cost of Mo-99, which has negatively affected our margins, and more restrictive agreements with suppliers, which could further increase our costs.
With the general instability in the global supply of Mo-99, we have faced substantial increases in the cost of Mo-99 in comparison to historical costs. We expect these cost increases to continue in the future as the Mo-99 suppliers move closer to a full cost recovery business model. The Organization of Economic Cooperation and Development (“OECD”) defines full cost recovery as the identification of all of the costs of production and recovering these costs from the market. While we are generally able to pass Mo-99 cost increases on to our customers in our customer contracts, if we are not able to do so in the future, our margins may decline further with respect to our TechneLite generators, which could have a material adverse effect on our business, results of operations, financial condition and cash flows.
Our dependence upon third parties for the manufacture and supply of a substantial portion of our products could prevent us from delivering our products to our customers in the required quantities, within the required timeframes, or at all, which could result in order cancellations and decreased revenues.
We obtain a substantial portion of our products from third party manufacturers and suppliers. We rely on JHS as our sole source manufacturer of DEFINITY, Neurolite, Cardiolite and evacuation vials. We currently have additional on-going technology transfer activities for a modified formulation of DEFINITY with SBL. We currently believe that if approved by the FDA, the modified formulation could become commercially available in early 2021, although that timing cannot be assured. Currently, our DEFINITY, Neurolite, Cardiolite, evacuation vial and saline product supplies are approved for manufacture by a single manufacturer.
Based on our current estimates, we believe that we will have sufficient supply of DEFINITY, Neurolite, Cardiolite and evacuation vials from JHS, and sufficient supply of saline from our sole manufacturer, to meet expected demand. However, we can give no assurances that JHS or our other manufacturing partner will be able to manufacture and distribute our products in a high quality and timely manner and in sufficient quantities to allow us to avoid product stock-outs and shortfalls. Currently, regulatory authorities in certain countries have not yet approved JHS as a manufacturer of certain of our products. Accordingly, until those regulatory approvals have been obtained, our business, results of operations, financial condition and cash flows will continue to be adversely affected.
Xenon is captured as a by-product of the Mo-99 production process. We receive bulk unprocessed Xenon from IRE resulting from HEU Mo-99 production, which we process and finish for our customers. We do not yet receive Xenon resulting from LEU Mo-99 production at IRE and can give no assurances as to the timing of the availability of LEU Xenon. We believe we will have a sufficient supply of HEU and LEU Xenon to meet our customers’ needs. However, until IRE converts to LEU Xenon production or we can qualify an additional source of bulk unprocessed Xenon, we will rely on IRE as a sole source provider of HEU Xenon.
27
In addition to the products described above, for reasons of quality assurance or cost-effectiveness, we purchase certain components and raw materials from sole suppliers (including, for example, the lead casing for our TechneLite generators and the lipid blend material used in the processing of DEFINITY). Because we do not control the actual production of many of the products we sell and many of the raw materials and components that make up the products we sell, we may be subject to delays caused by interruption in production based on events and conditions outside of our control. At our North Billerica, Massachusetts facility, we manufacture TechneLite on a highly automated production line, as well as Thallium and Gallium using our older cyclotron technology and Xenon and Quadramet using our hot cell infrastructure. As with all manufacturing facilities, equipment and infrastructure age and become subject to increasing maintenance and repair. If we or one of our manufacturing partners experiences an event, including a labor dispute, natural disaster, fire, power outage, machinery breakdown, security problem, failure to meet regulatory requirements, product quality issue, technology transfer issue or other issue, we may be unable to manufacture the relevant products at previous levels or on the forecasted schedule, if at all. Due to the stringent regulations and requirements of the governing regulatory authorities regarding the manufacture of our products, we may not be able to quickly restart manufacturing at a third party or our own facility or establish additional or replacement sources for certain products, components or materials.
In addition to our existing manufacturing relationships, we are also pursuing new manufacturing relationships to establish and secure additional or alternative suppliers for our commercial products. We currently have additional on-going technology transfer activities for a modified formulation of DEFINITY with SBL. We are also in the final stages of an extensive, multi-year effort to add specialized manufacturing capabilities at our North Billerica, Massachusetts facility. This project is part of a larger corporate growth strategy to create a competitive advantage in specialized manufacturing. This project should not only deliver efficiencies and supply chain redundancy for our current portfolio but also should afford us increased flexibility as we consider external opportunities. However, we cannot assure you that these activities or any of our additional supply activities will be successful or that we will be able to avoid or mitigate interim supply shortages before new sources of product are fully functional and qualified. In addition, we cannot assure you that our existing manufacturers or suppliers or any new manufacturers or suppliers can adequately maintain either their financial health, technical capabilities or regulatory compliance to allow continued production and supply. A reduction or interruption in manufacturing, or an inability to secure alternative sources of raw materials or components, could eventually have a material adverse effect on our business, results of operations, financial condition and cash flows.
Our just-in-time manufacturing of radiopharmaceutical products relies on the timely receipt of radioactive raw materials and the timely shipment of finished goods, and any disruption of our supply or distribution networks could have a negative effect on our business.
Because a number of our radiopharmaceutical products, including our TechneLite generators, rely on radioisotopes with limited half-lives, we must manufacture, finish and distribute these products on a just-in-time basis, because the underlying radioisotope is in a constant state of radio decay. For example, if we receive Mo-99 in the morning of a manufacturing day for TechneLite generators, then we will generally ship finished generators to customers by the end of that same business day. Shipment of generators may be by next day delivery services or by either ground or air custom logistics. Any delay in us receiving radioisotopes from suppliers or being able to have finished products delivered to customers because of weather or other unforeseen transportation issues could have a negative effect on our business, results of operations, financial condition and cash flows.
Challenges with product quality or product performance, including defects, caused by us or our suppliers could result in a decrease in customers and revenues, unexpected expenses and loss of market share.
The manufacture of our products is highly exacting and complex and must meet stringent quality requirements, due in part to strict regulatory requirements, including the FDA’s cGMPs. Problems may be identified or arise during manufacturing quality review, packaging or shipment for a variety of reasons including equipment malfunction, failure to follow specific protocols and procedures, defective raw materials and environmental factors. Additionally, manufacturing flaws, component failures, design defects, off-label uses or inadequate disclosure of product-related information could result in an unsafe condition or the injury or death of a patient. Those events could lead to a recall of, or issuance of a safety alert relating to, our products. We also may undertake voluntarily to recall products or temporarily shut down production lines based on internal safety and quality monitoring and testing data.
Quality, regulatory and recall challenges could cause us to incur significant costs, including costs to replace products, lost revenue, damage to customer relationships, time and expense spent investigating the cause and costs of any possible settlements or judgments related thereto and potentially cause similar losses with respect to other products. These challenges could also divert the attention of our management and employees from operational, commercial or other business efforts. If we deliver products with defects, or if there is a perception that our products or the processes related to our products contain errors or defects, we could incur additional recall and product liability costs, and our credibility and the market acceptance and sales of our products could be materially adversely affected. Due to the strong name recognition of our brands, an adverse event involving one of our products could result in reduced market acceptance and demand for all products within that brand, and could harm our reputation and our ability to market our products in the future. In some circumstances, adverse events arising from or associated with the design, manufacture or marketing of our products could result in the suspension or delay of regulatory reviews of our applications for new product approvals. These challenges could have a material adverse effect on our business, results of operations, financial condition and cash flows.
28
In the U.S., we are heavily dependent on a few large customers and group purchasing organization arrangements to generate a majority of our revenues for our nuclear medical imaging products and our other products. Outside of the U.S., we rely primarily on distributors to generate a substantial portion of our revenue.
In the U.S., we have historically relied on a limited number of radiopharmacy customers, primarily GE Healthcare, Cardinal, UPPI, Jubilant Radiopharma and PharmaLogic, to distribute our current largest volume nuclear imaging products. Among the existing radiopharmacies in the U.S., continued consolidations, divestitures and reorganizations may have a negative effect on our business, results of operations, financial condition and cash flows. We generally have distribution arrangements with our major radiopharmacy customers pursuant to multi-year contracts, each of which is subject to renewal. If these contracts are terminated prior to expiration of their term, or are not renewed, or are renewed on terms that are less favorable to us, then such an event could have a material adverse effect on our business, results of operations, financial condition and cash flows.
For all of our medical imaging products, we continue to experience significant pricing pressures from our competitors, large customers and group purchasing organizations, and any significant, additional pricing pressures could lead to a reduction in revenue which could have a material adverse effect on our business, results of operations, financial condition and cash flows.
Outside of the U.S., Canada and Puerto Rico, we have no sales force and, consequently, rely on third-party distributors, either on a country-by-country basis or on a multi-country, regional basis, to market, sell and distribute our products. In Canada, we maintain our own direct sales force to sell DEFINITY. We formerly owned or operated radiopharmacies and we now sell radiopharmaceutical products under the Isologic Supply Agreement. In Australia, we also formerly owned or operated radiopharmacies, and we now sell DEFINITY and radiopharmaceutical products under the GMS Supply Agreement. In certain circumstances, distributors may also sell competing products to our own or products for competing diagnostic modalities and may have incentives to shift sales towards those competing products. As a result, we cannot assure you that our international distributors will increase or maintain current levels of unit sales or that we will be able to increase or maintain our current unit pricing, which, in turn, could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We face significant competition in our business and may not be able to compete effectively.
The market for diagnostic medical imaging agents is highly competitive and continually evolving. Our principal competitors in existing diagnostic modalities include large, global companies with substantial financial, manufacturing, sales and marketing and logistics resources that are more diversified than ours, such as GE Healthcare, Bracco, Curium and Jubilant Life Sciences, as well as other competitors, including NorthStar Medical Radioisotopes. We cannot anticipate their actions in the same or competing diagnostic modalities, such as significant price reductions on products that are comparable to our own, development or introduction of new products that are more cost-effective or have superior performance than our current products, the introduction of generic versions when our proprietary products lose their patent protection or the new entry into a generic market in which we are already a participant. In addition, distributors of our products could attempt to shift end-users to competing diagnostic modalities and products. Our current or future products could be rendered obsolete or uneconomical as a result of these activities. Our failure to compete effectively could cause us to lose market share to our competitors and have a material adverse effect on our business, results of operations, financial condition and cash flows.
Further, the radiopharmaceutical industry continues to evolve strategically, with several market participants either recently sold or for sale. In addition, the supply-demand dynamics of the industry are complex because of large market positions of some participants, legacy businesses, government subsidies (in particular, relating to the manufacture of radioisotopes), and group purchasing arrangements. We cannot predict what impact new owners and new operators may have on the strategic decision-making of our competitors, customers and suppliers, and such decision-making could have a material adverse effect on our business, results of operations, financial condition and cash flows.
29
Risks Related to Reimbursement and Regulation
Certain of our customers are highly dependent on payments from third party payors, including government sponsored programs, particularly Medicare, in the U.S. and other countries in which we operate, and reductions in third party coverage and reimbursement rates for our products (or services provided with our products) could adversely affect our business and results of operations.
A substantial portion of our revenue depends, in part, on the extent to which the costs of our products purchased by our customers (or services provided with our products) are reimbursed by third party payors, including Medicare, Medicaid, other U.S. government sponsored programs, non-U.S. governmental payors and private payors. These third party payors exercise significant control over patient access and increasingly use their enhanced bargaining power to secure discounted rates and impose other requirements that may reduce demand for our products. Our potential customers’ ability to obtain adequate reimbursement for products and services from these third party payors affects the selection of products they purchase and the prices they are willing to pay. For example, certain radiopharmaceuticals, when used for non-invasive imaging of the perfusion of the heart for the diagnosis and management of patients with known or suspected coronary artery disease, are currently subject to a Medicare National Coverage Determination (“NCD”). The NCD permits the coverage of such radiopharmaceuticals only when certain criteria are met. Our PET pipeline product flurpiridaz F 18, if approved, may become subject to this NCD, and may not be covered at all. If Medicare and other third party payors do not provide adequate reimbursement for the costs of our products (or services provided using our products), deny the coverage of the products (or those services), or reduce current levels of reimbursement, healthcare professionals may not prescribe our products and providers and suppliers may not purchase our products. In addition, demand for new products may be limited unless we obtain favorable reimbursement policies (including coverage, coding and payment) from governmental and private third party payors at the time of the product’s introduction, which will depend, in part, on our ability to demonstrate that a new agent has a positive impact on clinical outcomes. Third party payors continually review their coverage policies for existing and new products and procedures and can deny coverage for procedures that include the use of our products or revise payment policies such that payments do not adequately cover the cost of our products. Even if third party payors make coverage and reimbursement available, that reimbursement may not be adequate or these payors’ reimbursement policies may have an adverse effect on our business, results of operations, financial condition and cash flows.
Over the past several years, Medicare has implemented numerous changes to payment policies for imaging procedures in both the hospital setting and non-hospital settings (which include physician offices and freestanding imaging facilities). Some of these changes have had a negative impact on utilization of imaging services. Examples of these changes include:
• | Limiting payments for imaging services in physician offices and free-standing imaging facility settings based upon rates paid to hospital outpatient departments; |
• | Reducing payments for certain imaging procedures when performed together with other imaging procedures in the same family of procedures on the same patient on the same day in the physician office and free-standing imaging facility setting; |
• | Making significant revisions to the methodology for determining the practice expense component of the Medicare payment applicable to the physician office and free-standing imaging facility setting which results in a reduction in payment; |
• | Revising payment policies and reducing payment amounts for imaging procedures performed in the hospital outpatient setting; and |
• | Reducing prospective payment levels for applicable diagnosis-related groups in the hospital inpatient setting. |
In the physician office and free-standing imaging facility setting, services provided using our products are reimbursed under the Medicare physician fee schedule. Since 2015, payments under the Medicare physician fee schedule have been subject to specific annual updates: a 0.5% update through 2018; a 0.25% update in 2019; no updates from 2020 to 2025; and, beginning in 2026, differential updates based on whether the physician participates in advanced alternative payment models (with 0.75% updates for qualifying participants and 0.25% updates for non-qualifying participants) (which may be subject to budget neutrality adjustments). Since 2019, fee schedule payments have been adjusted for certain physicians based on their performance under a consolidated measurement system (that measures performance with respect to quality, resource utilization, meaningful use of certified electronic health records technology, and clinical practice improvement activities). From 2019 through payment year 2024, physicians may be eligible for a bonus based on the use of certain alternative payment models designated as “advanced” by CMS. The ongoing and future impact of these changes cannot be determined at this time.
30
We believe that Medicare changes to payment policies for imaging procedures applicable to non-hospital settings will continue to result in certain physician practices ceasing to provide these services and a further shifting of where certain medical imaging procedures are performed, from the physician office and free-standing imaging facility settings to the hospital outpatient setting. Changes applicable to Medicare payment in the hospital outpatient setting could also influence the decisions by hospital outpatient physicians to perform procedures that involve our products. Within the hospital outpatient setting, CMS payment policy is such that the use of many of our products are not separately payable by Medicare, although certain new drug products are eligible for separate (incremental) payment for the first three years after approval. Since 2013, although Medicare generally does not provide separate payment to hospitals for the use of diagnostic radiopharmaceuticals administered in an outpatient setting, CMS has had a policy to make a nominal additional payment ($10) to hospitals that utilize products with non-HEU, meaning the product is 95% derived from non-HEU sources. This payment policy continues in 2020. Although some of our TechneLite generators are manufactured using non-HEU, not all of our TechneLite generators currently meet CMS’s definition of non-HEU, and therefore this payment is not available for doses produced by the latter category of TechneLite generators used by our customers. Changes to the Medicare hospital outpatient prospective payment system payment rates, including reductions implemented for certain hospital outpatient sites, could influence the decisions by hospital outpatient physicians to perform procedures that involve our products.
We also believe that all these changes and their resulting pressures may incrementally reduce the overall number of diagnostic medical imaging procedures performed. These changes overall could slow the acceptance and introduction of next-generation imaging equipment into the marketplace, which, in turn, could adversely impact the future market adoption of certain of our imaging agents already in the market or currently in development. We expect that there will continue to be proposals to reduce or limit Medicare and Medicaid payment for diagnostic services.
We also expect increased regulation and oversight of advanced diagnostic testing in which our products are used. Beginning January 1, 2020, a professional who is ordering advanced diagnostic imaging services (which include MRI, CT, nuclear medicine (including PET) and other advanced diagnostic imaging services that the Secretary of HHS may specify) must consult a qualified clinical decision support mechanism, as identified by HHS, to determine whether the ordered service adheres to specified appropriate use criteria (“AUC”). Reimbursement penalties will apply in 2021 if this requirement is not met (and documented on the claim). To the extent that these types of changes have the effect of reducing the aggregate number of diagnostic medical imaging procedures performed in the U.S., our business, results of operations, financial condition and cash flows would be adversely affected.
Reforms to the U.S. healthcare system may adversely affect our business.
A significant portion of our patient volume is derived from U.S. government healthcare programs, principally Medicare, which are highly regulated and subject to frequent and substantial changes. The Healthcare Reform Act substantially changed the way healthcare is financed by both governmental and private insurers. The law contains a number of provisions that affect coverage and reimbursement of drug products and medical imaging procedures in which our drug products are used and/or that could potentially reduce the aggregate number of diagnostic medical imaging procedures performed in the U.S. Subsequently, the Medicare Access and CHIP Reauthorization Act of 2015 significantly revised the methodology for updating the Medicare physician fee schedule. And more recently, Congress enacted legislation in 2017 that eliminated the Healthcare Reform Act’s “individual mandate” beginning in 2019 Congress continues to consider other healthcare reform legislation. There is no assurance that the Healthcare Reform Act, as currently enacted or as amended in the future, will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative, judicial or administrative changes relating to healthcare reform will affect our business.
In addition, other legislative changes have been proposed and adopted since the Healthcare Reform Act was enacted. The Budget Control Act of 2011 and subsequent Congressional actions includes provisions to reduce the federal deficit. These provisions have resulted in the imposition of 2% reductions in Medicare payments to providers, which went into effect on April 1, 2013 and will remain in effect through 2029. Any significant spending reductions affecting Medicare, Medicaid or other publicly funded or subsidized health programs that may be implemented and/or any significant taxes or fees that may be imposed on us, as part of any broader deficit reduction effort or legislative replacement to the Budget Control Act, could have an adverse impact on our business, results of operations, financial condition and cash flows.
Further, changes in payor mix and reimbursement by private third party payors may also affect our business. Rates paid by some private third party payors are based, in part, on established physician, clinic and hospital charges and are generally higher than Medicare payment rates. Reductions in the amount of reimbursement paid for diagnostic medical imaging procedures and changes in the mix of our patients between non-governmental payors and government sponsored healthcare programs and among different types of non-government payor sources, could have a material adverse effect on our business, results of operations, financial condition and cash flows.
The full impact on our business of healthcare reforms and other new laws, or changes in existing laws, is uncertain. Nor is it clear whether additional legislative changes will be adopted or how those changes would affect our industry in general or our ability to successfully commercialize our products or develop new products.
31
Our business and industry are subject to complex and costly regulations. If government regulations are interpreted or enforced in a manner adverse to us or our business, we may be subject to enforcement actions, penalties, exclusion and other material limitations on our operations.
Both before and after the approval of our products and agents in development, we, our products, development agents, operations, facilities, suppliers, distributors, contract manufacturers, contract research organizations and contract testing laboratories are subject to extensive and, in certain circumstances, expanding regulation by federal, state and local government agencies in the U.S. as well as non-U.S. and transnational laws and regulations, with regulations differing from country to country, including, among other things, anti-trust and competition laws and regulations and the recently enacted General Data Protection Regulation (GDPR) in the European Union (the “EU”). In the U.S., the FDA regulates, among other things, the pre-clinical testing, clinical trials, manufacturing, safety, efficacy, potency, labeling, storage, record keeping, quality systems, advertising, promotion, sale, distribution, and import and export of drug products. We are required to register our business for permits and/or licenses with, and comply with the stringent requirements of the FDA, the NRC, the HHS, Health Canada, the EMA, the MHRA, the CFDA, state and provincial boards of pharmacy, state and provincial health departments and other federal, state and provincial agencies.
Under U.S. law, for example, we are required to report certain adverse events and production problems, if any, to the FDA. We also have similar adverse event and production reporting obligations outside of the U.S., including to the EMA and MHRA. Additionally, we must comply with requirements concerning advertising and promotion for our products, including the prohibition on the promotion of our products for indications that have not been approved by the FDA or a so-called “off-label use” or promotion that is inconsistent with the approved labeling. If the FDA determines that our promotional materials constitute unlawful promotion, it could request that we modify our promotional materials or subject us to regulatory or enforcement actions. Also, quality control and manufacturing procedures at our own facility and at third party suppliers must conform to cGMP regulations and other applicable law after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMPs and other applicable law, and, from time to time, makes those cGMPs more stringent. Accordingly, we and others with whom we work must expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production and quality control. If in the future issues arise at a third party manufacturer, the FDA could take regulatory action which could limit or suspend the ability of that third party to manufacture our products or have any additional products approved at the relevant facility for manufacture until the issues are resolved and remediated. Such a limitation or suspension could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We are also subject to laws and regulations that govern financial and other arrangements between pharmaceutical manufacturers and healthcare providers, including federal and state anti-kickback statutes, federal and state false claims laws and regulations and other fraud and abuse laws and regulations.
We must offer discounted pricing or rebates on purchases of pharmaceutical products under various federal and state healthcare programs, such as the Medicaid drug rebate program, the “federal ceiling price” drug pricing program, the 340B drug pricing program and the Medicare Part D Program. We must also report specific prices to government agencies under healthcare programs, such as the Medicaid drug rebate program and Medicare Part B. As a specific example, in 2010, we entered into a Medicaid Drug Rebate Agreement with the federal government for some but not all of our products, and in 2016 entered into a separate Medicaid Drug Rebate Agreement for the balance of our products. These agreements require us to report certain price information to the federal government. Determination of the rebate amount that we pay to state Medicaid programs for our products, of prices charged to government and certain private payors for our products, or of amounts paid for our products under government healthcare programs, depends upon information reported by us to the government. If we provide customers or government officials with inaccurate information about the products’ pricing or eligibility for coverage, or the products fail to satisfy coverage requirements, we could be terminated from the rebate program, be excluded from participation in government healthcare programs, or be subject to potential liability under the False Claims Act or other laws and regulations.
Failure to comply with other requirements and restrictions placed upon us or our third party manufacturers or suppliers by laws and regulations can result in fines, civil and criminal penalties, exclusion from federal healthcare programs and debarment. Possible consequences of those actions could include:
• | Substantial modifications to our business practices and operations; |
• | Significantly reduced demand for our products (if products become ineligible for reimbursement under federal and state healthcare programs); |
• | A total or partial shutdown of production in one or more of the facilities where our products are produced while the alleged violation is being remediated; |
• | Delays in or the inability to obtain future pre-market clearances or approvals; and |
• | Withdrawals or suspensions of our current products from the market. |
32
Regulations are subject to change as a result of legislative, administrative or judicial action, which may also increase our costs or reduce sales. Violation of any of these regulatory schemes, individually or collectively, could disrupt our business and have a material adverse effect on our business, results of operations, financial condition and cash flows.
Our marketing and sales practices may contain risks that could result in significant liability, require us to change our business practices and restrict our operations in the future.
We are subject to numerous domestic (federal, state and local) and foreign laws addressing fraud and abuse in the healthcare industry, including the FCA and federal Anti-Kickback Statute, self-referral laws, the FCPA, the Bribery Act, FDA promotional restrictions, the federal disclosure (sunshine) law and state marketing and disclosure (sunshine) laws. Violations of these laws are punishable by criminal or civil sanctions, including substantial fines, imprisonment and exclusion from participation in healthcare programs such as Medicare and Medicaid as well as health programs outside the U.S., and even settlement of alleged violations can result in the imposition of corporate integrity agreements that could subject us to additional compliance and reporting requirements and impact our business practices. These laws and regulations are complex and subject to changing interpretation and application, which could restrict our sales or marketing practices. Even minor and inadvertent irregularities could potentially give rise to a charge that the law has been violated. Although we believe we maintain an appropriate compliance program, we cannot be certain that the program will adequately detect or prevent violations and/or the relevant regulatory authorities may disagree with our interpretation. Additionally, if there is a change in law, regulation or administrative or judicial interpretations, we may have to change one or more of our business practices to be in compliance with these laws. Required changes could be costly and time consuming.
If our operations are found to be in violation of these laws or any other government regulations that apply to us, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, imprisonment, the curtailment or restructuring of our operations, or exclusion from state and federal healthcare programs including Medicare and Medicaid, any of which could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We no longer qualify as an “emerging growth company” as of December 31, 2019, and as a result, we will have to comply with increased disclosure and compliance requirements.
Based on the market value of our common stock held by non-affiliates which exceeded $700 million as of the last business day of June 2019, we no longer qualify as an “emerging growth company” but will instead be deemed to be a “large accelerated filer” as of December 31, 2019.
As a large accelerated filer, we will be subject to certain disclosure and compliance requirements that apply to other public companies but that did not previously apply to us due to our status as an emerging growth company. These requirements include, but are not limited to:
• | The requirement that our independent registered public accounting firm attest to the effectiveness of our internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act of 2002; |
• | Compliance with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor's report providing additional information about the audit and the financial statements; |
• | The requirement that we provide full and more detailed disclosures regarding executive compensation; and |
• | The requirement that we hold a non-binding advisory vote on executive compensation and obtain stockholder approval of any golden parachute payments not previously approved. |
We expect that the loss of emerging growth company status and compliance with the additional requirements of being a large accelerated filer will increase our legal, accounting and financial compliance costs and costs associated with investor relations activities, and cause management and other personnel to divert attention from operational and other business matters to devote substantial time to public company reporting requirements. In addition, if we are not able to comply with changing requirements in a timely manner, the market price of our stock could decline and we could be subject to sanctions or investigations by the stock exchange on which our common stock is listed, the SEC or other regulatory authorities, which would require additional financial and management resources.
As of the end of fiscal 2019, we are required to implement additional procedures and practices related to internal control over financial reporting, and we may identify deficiencies that we may not be able to remediate in time to meet the necessary deadline.
Pursuant to Section 404 of the Sarbanes-Oxley Act, our management is required to report upon the effectiveness of our internal control over financial reporting. Since we are deemed a large accelerated filer, our independent registered public accounting firm is required to attest to the effectiveness of our internal controls on an annual basis beginning with our Annual Report on Form 10-K for the year ended December 31, 2019. The rules governing the standards that must be met for our management and independent
33
registered public accounting firm to assess our internal controls are complex and require significant documentation, testing and possible remediation of our existing controls and the incurrence of significant additional expenditures. In connection with our evaluation of our internal controls, we may need to upgrade systems, including information technology; implement additional financial and management controls, reporting systems, and procedures; and hire additional accounting and finance staff.
Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. In addition, any testing by us or our independent registered public accounting firm conducted in connection with Section 404 of the Sarbanes-Oxley Act may reveal deficiencies in our internal controls that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Internal control deficiencies could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock. Internal control deficiencies could also result in a restatement of our financial results in the future. We could become subject to stockholder or other third party litigation, as well as investigations by the SEC or other regulatory authorities, which could require additional financial and management resources and could result in fines, trading suspensions, payment of damages or other remedies. Further, any delay in compliance with the auditor attestation provisions of Section 404 could subject us to a variety of administrative sanctions, including ineligibility for short-form resale registration, action by the SEC and the suspension or delisting of our common stock, which could reduce the trading price of our common stock and could harm our business.
Risks Related to Safety
Ultrasound contrast agents may cause side effects which could limit our ability to sell DEFINITY.
DEFINITY is an ultrasound contrast agent based on perflutren lipid microspheres. In 2007, the FDA received reports of deaths and serious cardiopulmonary reactions following the administration of ultrasound micro-bubble contrast agents used in echocardiography. Four of the 11 reported deaths were caused by cardiac arrest occurring either during or within 30 minutes following the administration of the contrast agent; most of the serious but non-fatal reactions also occurred in this time frame. As a result, in October 2007, the FDA requested that we and GE Healthcare, which distributes Optison, a competitor to DEFINITY, add a boxed warning to these products emphasizing the risk for serious cardiopulmonary reactions and that the use of these products was contraindicated in certain patients. In a strong reaction by the cardiology community to the FDA’s new position, a letter was sent to the FDA, signed by 161 doctors, stating that the benefit of these ultrasound contrast agents outweighed the risks and urging that the boxed warning be removed. In May 2008, the FDA substantially modified the boxed warning. On May 2, 2011, the FDA held an advisory committee meeting to consider the status of ultrasound micro-bubble contrast agents and the boxed warning. In October 2011, we received FDA approval of further modifications to the DEFINITY label, including: further relaxing the boxed warning; eliminating the sentence in the Indication and Use section “The safety and efficacy of DEFINITY with exercise stress or pharmacologic stress testing have not been established” (previously added in October 2007 in connection with the imposition of the box warning); and including summary data from the post-approval CaRES (Contrast echocardiography Registry for Safety Surveillance) safety registry and the post-approval pulmonary hypertension study. Further, in January 2017, the FDA approved an additional modification to the DEFINITY label, removing the contraindication statement related to use in patients with a known or suspected cardiac shunt. Bracco’s ultrasound contrast agent, Lumason, has substantially similar safety labeling as DEFINITY and Optison. If additional safety issues arise (not only with DEFINITY but also potentially with Optison and Lumason), this may result in unfavorable changes in labeling or result in restrictions on the approval of our product, including removal of the product from the market. Lingering safety concerns about DEFINITY among some healthcare providers or future unanticipated side effects or safety concerns associated with DEFINITY could limit expanded use of DEFINITY and have a material adverse effect on the unit sales of this product and our financial condition and results of operations.
A heightened public or regulatory focus on the radiation risks of diagnostic imaging could have an adverse effect on our business.
We believe that there has been heightened public and regulatory focus on radiation exposure, including the concern that repeated doses of radiation used in diagnostic imaging procedures pose the potential risk of long-term cell damage, cancer and other diseases. For example, starting in January 2012, CMS required the accreditation of facilities providing the technical component of advanced imaging services, including CT, MRI, PET and nuclear medicine, in non-hospital freestanding settings. In August 2011, The Joint Commission (an independent, not-for-profit organization that accredits and certifies more than 20,500 healthcare organizations and programs in the U.S.) issued an alert on the radiation risks of diagnostic imaging and recommended specific actions for providing “the right test and the right dose through effective processes, safe technology and a culture of safety.” The Joint Commission has revised accreditation standards for diagnostic imaging in recent years, including standards related to dose optimization.
34
Heightened regulatory focus on risks caused by the radiation exposure received by diagnostic imaging patients could lead to increased regulation of radiopharmaceutical manufacturers or healthcare providers who perform procedures that use our imaging agents, which could make the procedures more costly, reduce the number of providers who perform procedures and/or decrease the demand for our products. In addition, heightened public focus on or fear of radiation exposure could lead to decreased demand for our products by patients or by healthcare providers who order the procedures in which our agents are used. Although we believe that our diagnostic imaging agents when properly used do not expose patients and healthcare providers to unsafe levels of radiation, any of the foregoing risks could have an adverse effect on our business, results of operations, financial condition and cash flows.
In the ordinary course of business, we may be subject to product liability claims and lawsuits, including potential class actions, alleging that our products have resulted or could result in an unsafe condition or injury.
Any product liability claim brought against us, with or without merit, could be time consuming and costly to defend and could result in an increase of our insurance premiums. Although we have not had any such claims to date, claims that could be brought against us might not be covered by our insurance policies. Furthermore, although we currently have product liability insurance coverage with policy limits that we believe are customary for pharmaceutical companies in the diagnostic medical imaging industry and adequate to provide us with insurance coverage for foreseeable risks, even where the claim is covered by our insurance, our insurance coverage might be inadequate and we would have to pay the amount of any settlement or judgment that is in excess of our policy limits. We may not be able to obtain insurance on terms acceptable to us or at all, since insurance varies in cost and can be difficult to obtain. Our failure to maintain adequate insurance coverage or successfully defend against product liability claims could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We use hazardous materials in our business and must comply with environmental laws and regulations, which can be expensive.
Our operations use hazardous materials and produce hazardous wastes, including radioactive, chemical and, in certain circumstances, biological materials and wastes. We are subject to a variety of federal, state and local laws and regulations as well as non-U.S. laws and regulations relating to the transport, use, handling, storage, exposure to and disposal of these materials and wastes. Environmental laws and regulations are complex, change frequently and have become more stringent over time. We are required to obtain, maintain and renew various environmental permits and nuclear licenses. Although we believe that our safety procedures for transporting, using, handling, storing and disposing of, and limiting exposure to, these materials and wastes comply in all material respects with the standards prescribed by applicable laws and regulations, the risk of accidental contamination or injury cannot be eliminated. We place a high priority on these safety procedures and seek to limit any inherent risks. We generally contract with third parties for the disposal of wastes generated by our operations. Prior to disposal, we store any low level radioactive waste at our facilities to decay until the materials are no longer considered radioactive. Although we believe we have complied in all material respects with all applicable environmental, health and safety laws and regulations, we cannot assure you that we have been or will be in compliance with all such laws at all times. If we violate these laws, we could be fined, criminally charged or otherwise sanctioned by regulators. We may be required to incur further costs to comply with current or future environmental and safety laws and regulations. In addition, in the event of accidental contamination or injury from these materials, we could be held liable for any damages that result and any such liability could exceed our resources.
We lease a small portion of our North Billerica, Massachusetts facility to PerkinElmer for the manufacturing, finishing and packaging of certain radioisotopes, including Strontium-90, which has physical characteristics that make it more challenging to work with and dispose of than our own commercial radioisotopes, including a much longer half-life. We are fully indemnified by PerkinElmer under our lease for any property damage or personal injury resulting from their activities in our facility. If any release or excursion of radioactive materials took place from their leased space that resulted in property damage or personal injury, the indemnification obligations were not honored, and we were forced to cover any related remediation, clean-up or other expenses, depending on the magnitude, the cost of such remediation, clean-up or other expenses could have a material adverse effect on our business, results of operations, financial condition and cash flows.
While we have budgeted for current and future capital and operating expenditures to maintain compliance with these laws and regulations, we cannot assure you that our costs of complying with current or future environmental, health and safety laws and regulations will not exceed our estimates or adversely affect our results of operations and financial condition. Further, we cannot assure you that we will not be subject to additional environmental claims for personal injury, investigation or cleanup in the future based on our past, present or future business activities.
Risks Related to Our Business
35
Our business depends on our ability to successfully introduce new products and adapt to a changing technology and medical practice landscape.
The healthcare industry is characterized by continuous technological development resulting in changing customer preferences and requirements. The success of new product development depends on many factors, including our ability to fund development of new agents or new indications for existing agents, anticipate and satisfy customer needs, obtain regulatory approval on a timely basis based on performance of our agents in development versus their clinical study comparators, develop and manufacture products in a cost-effective and timely manner, maintain advantageous positions with respect to intellectual property and differentiate our products from our competitors. To compete successfully in the marketplace, we must make substantial investments in new product development, whether internally or externally through licensing or acquisitions. Our failure to introduce new and innovative products in a timely manner would have an adverse effect on our business, results of operations, financial condition and cash flows.
Even if we are able to develop, manufacture and obtain regulatory approvals for our new products, the success of these products would depend upon market acceptance and adequate reimbursement. Levels of market acceptance for our new products could be affected by a number of factors, including:
• | The availability of alternative products from our competitors; |
• | The breadth of indications in which alternative products from our competitors can be marketed; |
• | The price of our products relative to those of our competitors; |
• | The timing of our market entry; |
• | Our ability to market and distribute our products effectively; |
• | Market acceptance of our products; and |
• | Our ability to obtain adequate reimbursement. |
The field of diagnostic medical imaging is dynamic, with new products, including hardware, software and agents, continually being developed and existing products continually being refined. Our own diagnostic imaging agents compete not only with other similarly administered imaging agents but also with imaging agents employed in different and often competing diagnostic modalities, and in the case of DEFINITY, echocardiography procedures without contrast. New hardware, software or agents in a given diagnostic modality may be developed that provide benefits superior to the then-dominant hardware, software and agents in that modality, resulting in commercial displacement of the agents. Similarly, changing perceptions about comparative efficacy and safety including, among other things, comparative radiation exposure, as well as changing availability of supply may favor one agent over another or one modality over another. In addition, new or revised appropriate use criteria developed by professional societies, to assist physicians and other health care providers in making appropriate imaging decisions for specific clinical conditions, can and have reduced the frequency of and demand for certain imaging modalities and imaging agents. To the extent there is technological obsolescence in any of our products that we manufacture, resulting in lower unit sales or decreased unit sales prices, we will have increased unit overhead allocable to the remaining market share, which could have a material adverse effect on our business, results of operations, financial condition and cash flows.
The process of developing new drugs and obtaining regulatory approval is complex, time-consuming and costly, and the outcome is not certain.
We currently have three active clinical development programs in the U.S. - flurpiridaz F 18, LMI 1195 and DEFINITY for LVEF. To obtain regulatory approval for these agents in the indications being pursued, we must conduct extensive human tests, which are referred to as clinical trials, as well as meet other rigorous regulatory requirements, as further described in Part I, Item 1. “Business—Regulatory Matters.” Satisfaction of all regulatory requirements typically takes many years and requires the expenditure of substantial resources. A number of other factors may cause significant delays in the completion of our clinical trials, including unexpected delays in the initiation of clinical sites, slower than projected enrollment, competition with ongoing clinical trials and scheduling conflicts with participating clinicians, regulatory requirements, limits on manufacturing capacity and failure of an agent to meet required standards for administration to humans. In addition, it may take longer than we project to achieve study endpoints and complete data analysis for a trial or we may decide to slow down the enrollment in a trial in order to conserve financial resources.
36
Our agents in development are also subject to the risks of failure inherent in drug development and testing. The results of preliminary studies do not necessarily predict clinical success, and larger and later stage clinical trials may not produce the same results as earlier stage trials. Sometimes, agents that have shown promising results in early clinical trials have subsequently suffered significant setbacks in later clinical trials. Agents in later stage clinical trials may fail to show desired safety and efficacy traits, despite having progressed through initial clinical testing. In addition, the data collected from clinical trials of our agents in development may not be sufficient to support regulatory approval, or regulators could interpret the data differently and less favorably than we do. Further, the design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. Clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts. Regulatory authorities may require us or our partners to conduct additional clinical testing, in which case we would have to expend additional time and resources. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in regulatory policy that occur prior to or during regulatory review. The failure to provide clinical and preclinical data that are adequate to demonstrate to the satisfaction of the regulatory authorities that our agents in development are safe and effective for their proposed use will delay or preclude approval and will prevent us from marketing those products.
We are not permitted to market our agents in development in the U.S. or other countries until we have received requisite regulatory approvals. For example, securing FDA approval for a new drug requires the submission of an NDA to the FDA for our agents in development. The NDA must include extensive nonclinical and clinical data and supporting information to establish the agent’s safety and effectiveness for each indication. The NDA must also include significant information regarding the chemistry, manufacturing and controls for the product. The FDA review process can take many years to complete, and approval is never guaranteed. If a product is approved, the FDA may limit the indications for which the product may be marketed, require extensive warnings on the product labeling, impose restricted distribution programs, require expedited reporting of certain adverse events, or require costly ongoing requirements for post-marketing clinical studies and surveillance or other risk management measures to monitor the safety or efficacy of the agent. Markets outside of the U.S. also have requirements for approval of agents with which we must comply prior to marketing. Obtaining regulatory approval for marketing of an agent in one country does not ensure we will be able to obtain regulatory approval in other countries, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries. Also, any regulatory approval of any of our products or agents in development, once obtained, may be withdrawn. Approvals might not be granted on a timely basis, if at all.
In our flurpiridaz F 18 Phase 3 program, in May 2015, we announced complete results from the 301 trial. Although flurpiridaz F 18 appeared to be well-tolerated from a safety perspective and outperformed SPECT in a highly statistically significant manner in the co-primary endpoint of sensitivity and in the secondary endpoints of image quality and diagnostic certainty, the agent did not meet its other co-primary endpoint of non-inferiority for identifying subjects without disease. In April 2017, we entered into the License Agreement with GE Healthcare for the continued Phase 3 development and worldwide commercialization of flurpiridaz F 18. Under the License Agreement, GE Healthcare will, among other things, complete the worldwide development of flurpiridaz F 18 by conducting a second Phase 3 trial and pursue worldwide regulatory approvals. We cannot assure any particular outcome from GE Healthcare’s continued Phase 3 development of the agent or from regulatory review of either our or their Phase 3 study of the agent, that any of the data generated in either our or their sponsored Phase 3 study will be sufficient to support an NDA approval, that GE Healthcare will only have to conduct the one additional Phase 3 clinical study prior to filing an NDA, or that flurpiridaz F 18 will ever be approved as a PET MPI imaging agent by the FDA. Similarly, we can give no assurance that we will be successful in our clinical development program for LMI 1195 in the diagnosis and management of neuroendocrine tumors in pediatric and adult populations. For our DEFINITY for LVEF study, we did not achieve our primary or secondary endpoints in the first of two Phase 3 studies. Any failure or significant delay in completing clinical trials for our product candidates or in receiving regulatory approval for the sale of our product candidates may harm our business and delay or prevent us from being able to generate additional future revenue from product sales.
Even if our agents in development proceed successfully through clinical trials and receive regulatory approval, there is no guarantee that an approved product can be manufactured in commercial quantities at a reasonable cost or that such a product will be successfully marketed or distributed. The burden associated with the marketing and distribution of products like ours is substantial. For example, rather than being manufactured at our own facilities, both flurpiridaz F 18 and LMI 1195 would require the creation of a complex, field-based network involving PET cyclotrons located at radiopharmacies where the agent would need to be manufactured and distributed rapidly to end-users, given the agent’s 110-minute half-life. In addition, in the case of both flurpiridaz F 18 and LMI 1195, obtaining adequate reimbursement is critical, including not only coverage from Medicare, Medicaid, other government payors as well as private payors but also appropriate payment levels which adequately cover the substantially higher manufacturing and distribution costs associated with a PET agent in comparison to a Tc-99m-based agent. We can give no assurance even if either flurpiridaz F 18 or LMI 1195 obtains regulatory approval that a network of PET cyclotrons can be established or that adequate reimbursement can be secured to allow the approved agent or agents to become commercially successful.
37
Our future growth may depend on our ability to identify and acquire or in-license additional products, businesses or technologies, and if we do not successfully do so, or otherwise fail to integrate any new products, lines of business or technologies into our operations, we may have limited growth opportunities and it could result in significant impairment charges or other adverse financial consequences.
We are continuing to seek to acquire or in-license products, businesses or technologies that we believe are a strategic fit with our business strategy. Future acquisitions or in-licenses, however, may entail numerous operational and financial risks, including:
• | A reduction of our current financial resources; |
• | Incurrence of substantial debt or dilutive issuances of securities to pay for acquisitions; |
• | Difficulty or inability to secure financing to fund development activities for those acquired or in-licensed technologies; |
• | Higher than expected acquisition and integration costs; |
• | Disruption of our business, customer base and diversion of our management’s time and attention to develop acquired products or technologies; and |
• | Exposure to unknown liabilities. |
We may not have sufficient resources to identify and execute the acquisition or in-licensing of third party products, businesses and technologies and integrate them into our current infrastructure. In particular, we may compete with larger pharmaceutical companies and other competitors in our efforts to establish new collaborations and in-licensing opportunities. These competitors likely will have access to greater financial resources than we do and may have greater expertise in identifying and evaluating new opportunities. Furthermore, there may be overlap between our products or customers and the companies which we acquire that may create conflicts in relationships or other commitments detrimental to the integrated businesses. Additionally, the time between our expenditures to acquire or in-license new products, technologies or businesses and the subsequent generation of revenues from those acquired products, technologies or businesses (or the timing of revenue recognition related to licensing agreements and/or strategic collaborations) could cause fluctuations in our financial performance from period to period. Finally, if we devote resources to potential acquisitions or in-licensing opportunities that are never completed, or if we fail to realize the anticipated benefits of those efforts, we could incur significant impairment charges or other adverse financial consequences.
If we are unable to protect our intellectual property, our competitors could develop and market products with features similar to our products, and demand for our products may decline.
Our commercial success will depend in part on obtaining and maintaining patent and trade secret protection of our commercial products and technologies and agents in development as well as successfully enforcing and defending these patents and trade secrets against third parties and their challenges, both in the U.S. and in foreign countries. We will only be able to protect our intellectual property from unauthorized use by third parties to the extent that we maintain the secrecy of our trade secrets and can enforce our valid patents and trademarks.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. In addition, changes in either the patent laws or in interpretations of patent laws in the U.S. or other countries may diminish the value of our intellectual property and we may not receive the same degree of protection in every jurisdiction. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third party patents.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
• | We might not have been the first to make the inventions covered by each of our pending patent applications and issued patents, and we could lose our patent rights as a result; |
• | We might not have been the first to file patent applications for these inventions or our patent applications may not have been timely filed, and we could lose our patent rights as a result; |
• | Others may independently develop similar or alternative technologies or duplicate any of our technologies; |
• | It is possible that none of our pending patent applications will result in any further issued patents; |
• | Our issued patents may not provide a basis for commercially viable drugs, may not provide us with any protection from unauthorized use of our intellectual property by third parties, and may not provide us with any competitive advantages; |
• | Our patent applications or patents may be subject to interferences, oppositions, post-grant review, ex-parte re-examinations, inter partes review or similar administrative proceedings; |
38
• | While we generally apply for patents in those countries where we intend to make, have made, use or sell patented products, we may not be able to accurately predict all of the countries where patent protection will ultimately be desirable and may be precluded from doing so at a later date; |
• | We may choose not to seek patent protection in certain countries where the actual cost outweighs the perceived benefit at a certain time; |
• | Patents issued in foreign jurisdictions may have different scopes of coverage than our U.S. patents and so our products may not receive the same degree of protection in foreign countries as they would in the U.S.; |
• | We may not develop additional proprietary technologies that are patentable; or |
• | The patents of others may have an adverse effect on our business. |
Moreover, the issuance of a patent is not conclusive as to its validity or enforceability. A third party may challenge the validity or enforceability of a patent even after its issuance by the USPTO or the applicable foreign patent office. It is also uncertain how much protection, if any, will be afforded by our patents if we attempt to enforce them and they are challenged in court or in other proceedings, which may be brought in U.S. or non-U.S. jurisdictions to challenge the validity of a patent.
The initiation, defense and prosecution of intellectual property suits (including Hatch-Waxman related litigation), interferences, oppositions and related legal and administrative proceedings are costly, time consuming to pursue and result in a diversion of resources, including a significant amount of management time. The outcome of these proceedings is uncertain and could significantly harm our business. If we are not able to enforce and defend the patents of our technologies and products, then we will not be able to exclude competitors from marketing products that directly compete with our products, which could have a material and adverse effect on our business, results of operations, financial condition and cash flows.
For DEFINITY, our fastest growing and highest margin commercial product in 2019, we continue to actively pursue patents in both the U.S. and internationally. In the U.S., we now have an Orange Book-listed method of use patent expiring in March 2037 and additional manufacturing patents that are not Orange Book-listed expiring in 2021, 2023 and 2037. Outside of the U.S., while our DEFINITY patent protection and regulatory exclusivity have generally expired, we are currently prosecuting additional patents to try to obtain similar method of use and manufacturing patent protection as granted in the U.S. We were also recently granted a composition of matter patent on the modified formulation of DEFINITY which runs through December 2035. If the modified formulation is approved by the FDA, then this patent would be eligible to be listed in the Orange Book.
We will also rely on trade secrets and other know-how and proprietary information to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We use reasonable efforts to protect our trade secrets, but our employees, consultants, contractors, outside scientific partners and other advisors may unintentionally or willfully disclose our confidential information to competitors or other third parties. Enforcing a claim that a third party improperly obtained and is using our trade secrets is expensive, time consuming and resource intensive, and the outcome is unpredictable. In addition, courts outside the U.S. are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. We rely on confidentiality agreements with our collaborators, employees, consultants and other third parties and invention assignment agreements with our employees to protect our trade secrets and other know-how and proprietary information concerning our business. These confidentiality agreements may not prevent unauthorized disclosure of trade secrets and other know-how and proprietary information, and there can be no guarantee that an employee or an outside party will not make an unauthorized disclosure of our trade secrets, other technical know-how or proprietary information, or that we can detect such an unauthorized disclosure. We may not have adequate remedies for any unauthorized disclosure. This might happen intentionally or inadvertently. It is possible that a competitor will make use of that information, and that our competitive position will be compromised, in spite of any legal action we might take against persons making those unauthorized disclosures, which could have a material and adverse effect on our business, results of operations, financial condition and cash flows.
We rely on our trademarks, trade names and brand names to distinguish our products from the products of our competitors, and have registered or applied to register many of these trademarks, including, among others, DEFINITY, Cardiolite, TechneLite, Neurolite, Quadramet, Luminity and Lantheus Medical Imaging. We cannot assure you that any pending trademark applications will be approved. Third parties may also oppose our trademark applications, or otherwise challenge our use of the trademarks. If our trademarks are successfully challenged, we could be forced to re-brand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. Further, we cannot assure you that competitors will not infringe our trademarks, or that we will have adequate resources to enforce our trademarks.
39
We may be subject to claims that we have infringed, misappropriated or otherwise violated the patent or other intellectual property rights of a third party. The outcome of any of these claims is uncertain and any unfavorable result could adversely affect our business, financial condition and results of operations.
We may be subject to claims by third parties that we have infringed, misappropriated or otherwise violated their intellectual property rights. While we believe that the products that we currently manufacture using our proprietary technology do not infringe upon or otherwise violate proprietary rights of other parties or that meritorious defenses would exist with respect to any assertions to the contrary, we cannot assure you that we would not be found to infringe on or otherwise violate the proprietary rights of others.
We may be subject to litigation over infringement claims regarding the products we manufacture or distribute. This type of litigation can be costly and time consuming and could divert management’s attention and resources, generate significant expenses, damage payments (potentially including treble damages) or restrictions or prohibitions on our use of our technology, which could adversely affect our business, results of operations, financial condition and cash flows. In addition, if we are found to be infringing on proprietary rights of others, we may be required to develop non-infringing technology, obtain a license (which may not be available on reasonable terms, or at all), make substantial one-time or ongoing royalty payments, or cease making, using and/or selling the infringing products, any of which could have a material adverse effect on our business, results of operations, financial condition and cash flows.
We may be adversely affected by prevailing economic conditions and financial, business and other factors beyond our control.
Our ability to attract and retain customers, invest in and grow our business and meet our financial obligations depends on our operating and financial performance, which, in turn, is subject to numerous factors, including the prevailing economic conditions and financial, business and other factors beyond our control, such as the rate of unemployment, the number of uninsured persons in the U.S. and inflationary pressures. We cannot anticipate all the ways in which the current or future economic climate and financial market conditions could adversely impact our business. We are exposed to risks associated with reduced profitability and the potential financial instability of our customers, many of which may be adversely affected by volatile conditions in the financial markets. For example, unemployment and underemployment, and the resultant loss of insurance, may decrease the demand for healthcare services and pharmaceuticals. If fewer patients are seeking medical care because they do not have insurance coverage, our customers may experience reductions in revenues, profitability and/or cash flow that could lead them to modify, delay or cancel orders for our products. If customers are not successful in generating sufficient revenue or are precluded from securing financing, they may not be able to pay, or may delay payment of, accounts receivable that are owed to us. This, in turn, could adversely affect our financial condition and liquidity. To the extent prevailing economic conditions result in fewer procedures being performed, our business, results of operations, financial condition and cash flows could be adversely affected.
Our business is subject to international economic, political and other risks that could negatively affect our results of operations or financial position.
For the year ended December 31, 2019, we derived approximately 12% of our revenues from outside the fifty United States. Accordingly, our business is subject to risks associated with doing business internationally, including:
• | Less stable political and economic environments and changes in a specific country’s or region’s political or economic conditions; |
• | Changes in trade policies, regulatory requirements and other barriers, including, for example, U.S. trade sanctions against Iran and those countries and entities doing business with Iran, which could adversely impact international isotope production and, indirectly, our global supply chain; |
• | Potential global disruptions in air transport due to COVID-19 (coronavirus), which could adversely affect our international supply chains for radioisotopes and our modified formulation of DEFINITY as well as international distribution channels for our commercial products; |
• | Entering into, renewing or enforcing commercial agreements with international governments or provincial authorities or entities directly or indirectly owned or controlled by such governments or authorities, such as our Belgian, Australian and South African isotope suppliers, IRE, ANSTO and NTP, and our Chinese development and commercialization partner, Double-Crane Pharmaceutical Company; |
• | International customers which are agencies or institutions owned or controlled by foreign governments; |
• | Local business practices which may be in conflict with the U.S. Foreign Corrupt Practices Act and U.K. Bribery Act; |
• | Currency fluctuations; |
• | Unfavorable labor regulations; |
40
• | Greater difficulties in relying on non-U.S. courts to enforce either local or U.S. laws, particularly with respect to intellectual property; |
• | Greater potential for intellectual property piracy; |
• | Greater difficulties in managing and staffing non-U.S. operations; |
• | The need to ensure compliance with the numerous in-country and international regulatory and legal requirements applicable to our business in each of these jurisdictions and to maintain an effective compliance program to ensure compliance with these requirements, including in connection with the recently enacted GDPR in the EU; |
• | Changes in public attitudes about the perceived safety of nuclear facilities; |
• | Civil unrest or other catastrophic events; and |
• | Longer payment cycles of non-U.S. customers and difficulty collecting receivables in non-U.S. jurisdictions. |
These factors are beyond our control. The realization of any of these or other risks associated with operating outside the fifty United States could have a material adverse effect on our business, results of operations, financial condition and cash flows. As our international exposure increases and as we execute our strategy of international expansion, these risks may intensify.
We face currency and other risks associated with international sales.
We generate revenue from export sales, as well as from operations conducted outside the fifty United States. Operations outside the U.S. expose us to risks including fluctuations in currency values, trade restrictions, tariff and trade regulations, U.S. export controls, U.S. and non‑U.S. tax laws, shipping delays and economic and political instability. For example, violations of U.S. export controls, including those administered by the U.S. Treasury Department’s Office of Foreign Assets Control, could result in fines, other civil or criminal penalties and the suspension or loss of export privileges which could have a material adverse effect on our business, results of operations, financial conditions and cash flows.
Many of our customer relationships outside of the U.S. are, either directly or indirectly, with governmental entities, and we could be adversely affected by violations of the FCPA and similar worldwide anti-bribery laws outside the U.S.
The FCPA, the Bribery Act and similar worldwide anti-bribery laws in non-U.S. jurisdictions generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business.
The FCPA prohibits us from providing anything of value to foreign officials for the purposes of obtaining or retaining business or securing any improper business advantage. It also requires us to keep books and records that accurately and fairly reflect our transactions. Because of the predominance of government-sponsored healthcare systems around the world, many of our customer relationships outside of the U.S. are, either directly or indirectly, with governmental entities and are therefore subject to the FCPA and similar anti-bribery laws in non-U.S. jurisdictions. In addition, the provisions of the Bribery Act extend beyond bribery of foreign public officials and are more onerous than the FCPA in a number of other respects, including jurisdiction, non-exemption of facilitation payments and penalties.
Our policies mandate compliance with these anti-bribery laws. We operate in many parts of the world that have experienced governmental corruption to some degree, and in certain circumstances strict compliance with anti-bribery laws may conflict with local customs and practices. Despite our training and compliance programs, our internal control policies and procedures may not always protect us from reckless or criminal acts committed by our employees or agents. Violations of these laws, or allegations of those violations, could disrupt our business and result in a material adverse effect on our results of operations, financial condition and cash flows.
41
Our business depends on the continued effectiveness and availability of our information technology infrastructure, and failures of this infrastructure could harm our operations.
To remain competitive in our industry, we must employ information technologies to support manufacturing processes, quality processes, distribution, R&D and regulatory applications and that capture, manage and analyze large streams of data in compliance with applicable regulatory requirements. We rely extensively on technology, some of which is managed by third-party service providers, to allow the concurrent conduct of work sharing around the world. As with all information technology, our equipment and infrastructure age and become subject to increasing maintenance and repair and our systems generally are vulnerable to potential damage or interruptions from fires, natural disasters, power outages, blackouts, machinery breakdown, telecommunications failures and other unexpected events, as well as to break-ins, sabotage, increasingly sophisticated intentional acts of vandalism or cybersecurity threats which, due to the nature of such attacks, may remain undetected for a period of time. As these threats continue to evolve, we may be required to expend additional resources to enhance our information security measures or to investigate and remediate any information security vulnerabilities. Given the extensive reliance of our business on technology, any substantial disruption or resulting loss of data that is not avoided or corrected by our backup measures could harm our business, reputation, operations and financial condition.
A disruption in our computer networks, including those related to cybersecurity, could adversely affect our operations or financial position.
We rely on our computer networks and systems, some of which are managed by third parties, to manage and store electronic information (including sensitive data such as confidential business information and personally identifiable data relating to employees), and to manage or support a variety of critical business processes and activities. We may face threats to our networks from unauthorized access, security breaches and other system disruptions. Despite our security measures, our infrastructure may be vulnerable to external or internal attacks. Any such security breach may compromise information stored on our networks and may result in significant data losses or theft of sensitive or proprietary information. A cybersecurity breach could hurt our reputation by adversely affecting the perception of customers and potential customers of the security of their orders and personal information, as well as the perception of our manufacturing partners of the security of their proprietary information. In addition, a cybersecurity attack could result in other negative consequences, including disruption of our internal operations, increased cybersecurity protection costs, lost revenue, regulatory actions or litigation. Any disruption of internal operations could also have a material adverse impact on our results of operations, financial condition and cash flows. To date, we have not experienced any material cybersecurity attacks.
We may be limited in our ability to utilize, or may not be able to utilize, net operating loss carryforwards to reduce our future tax liability.
As of December 31, 2019, we had federal income tax loss carryforwards of approximately $174.0 million, which will begin to expire in 2032 and will completely expire in 2037. We may be limited in our ability to use these tax loss carryforwards to reduce our future U.S. federal income tax liabilities if we were to experience another “ownership change” as specified in Section 382 of the Internal Revenue Code including if we were to issue a certain amount of equity securities, certain of our stockholders were to sell shares of our common stock, or we were to enter into certain strategic transactions.
We may not be able to hire or retain the number of qualified personnel, particularly scientific, medical and sales personnel, required for our business, which would harm the development and sales of our products and limit our ability to grow.
Competition in our industry for highly skilled scientific, healthcare and sales personnel is intense. Although we have not had any material difficulty in the past in hiring or retaining qualified personnel, if we are unable to retain our existing personnel, or attract and train additional qualified personnel, either because of competition in our industry for these personnel or because of insufficient financial resources, then our growth may be limited and it could have a material adverse effect on our business.
If we lose the services of our key personnel, our business could be adversely affected.
Our success is substantially dependent upon the performance, contributions and expertise of our chief executive officer, executive leadership and senior management team. Mary Anne Heino, our Chief Executive Officer and President, and other members of our executive leadership and senior management team play a significant role in generating new business and retaining existing customers. We have an employment agreement with Ms. Heino and a limited number of other individuals on our executive leadership team, although we cannot prevent them from terminating their employment with us. We do not maintain key person life insurance policies on any of our executive officers. While we have experienced both voluntary and involuntary turnover on our executive leadership team, to date we have been able to attract new, qualified individuals to lead our company and key functional areas. Our inability to retain our existing executive leadership and senior management team, maintain an appropriate internal succession program or attract and retain additional qualified personnel could have a material adverse effect on our business.
Risks Related to Our Capital Structure
42
We have a substantial amount of indebtedness which may limit our financial and operating activities and may adversely affect our ability to incur additional debt to fund future needs.
As of December 31, 2019, we had approximately $195.0 million of total principal indebtedness remaining under our five‑year secured term loan facility, which matures on June 30, 2024 (the “2019 Term Facility” and the loans thereunder, the “2019 Term Loans”) and availability of $200.0 million under our five-year revolving credit facility (the “2019 Revolving Facility” and, together with the 2019 Term Facility, the “2019 Facility”). Our substantial indebtedness and any future indebtedness we incur could:
• | Require us to dedicate a substantial portion of cash flow from operations to the payment of interest on and principal of our indebtedness, thereby reducing the funds available for other purposes; |
• | Make it more difficult for us to satisfy and comply with our obligations with respect to our outstanding indebtedness, namely the payment of interest and principal; |
• | Make it more difficult to refinance the outstanding indebtedness; |
• | Subject us to increased sensitivity to interest rate increases; |
• | Make us more vulnerable to economic downturns, adverse industry or company conditions or catastrophic external events; |
• | Limit our ability to withstand competitive pressures; |
• | Reduce our flexibility in planning for or responding to changing business, industry and economic conditions; and |
• | Place us at a competitive disadvantage to competitors that have relatively less debt than we have. |
In addition, our substantial level of indebtedness could limit our ability to obtain additional financing on acceptable terms, or at all, for working capital, capital expenditures and general corporate purposes. Our liquidity needs could vary significantly and may be affected by general economic conditions, industry trends, performance and many other factors not within our control.
We may not be able to generate sufficient cash flow to meet our debt service obligations.
Our ability to generate sufficient cash flow from operations to make scheduled payments on our debt obligations will depend on our future financial performance, which will be affected by a range of economic, competitive and business factors, many of which are outside of our control. If we do not generate sufficient cash flow from operations to satisfy our debt obligations, including interest and principal payments, our credit ratings could be downgraded, and we may have to undertake alternative financing plans, such as refinancing or restructuring our debt, selling assets, entering into additional corporate collaborations or licensing arrangements for one or more of our products or agents in development, reducing or delaying capital investments or seeking to raise additional capital. We cannot assure you that any refinancing would be possible, that any assets could be sold, licensed or partnered, or, if sold, licensed or partnered, of the timing of the transactions and the amount of proceeds realized from those transactions, that additional financing could be obtained on acceptable terms, if at all, or that additional financing would be permitted under the terms of our various debt instruments then in effect. Furthermore, our ability to refinance would depend upon the condition of the financial and credit markets. Our inability to generate sufficient cash flow to satisfy our debt obligations, or to refinance our obligations on commercially reasonable terms or on a timely basis, would have an adverse effect on our business, results of operations and financial condition.
Despite our substantial indebtedness, we may incur more debt, which could exacerbate the risks described above.
We and our subsidiaries may be able to incur substantial additional indebtedness in the future subject to the limitations contained in the agreements governing our debt, including the 2019 Facility. Although these agreements restrict us and our restricted subsidiaries from incurring additional indebtedness, these restrictions are subject to important exceptions and qualifications. For example, we are generally permitted to incur certain indebtedness, including indebtedness arising in the ordinary course of business, indebtedness among restricted subsidiaries and us and indebtedness relating to hedging obligations. If we or our subsidiaries incur additional debt, the risks that we and they now face as a result of our leverage could intensify. In addition, 2019 Facility will not prevent us from incurring obligations that do not constitute indebtedness under the agreements.
Our 2019 Facility contains restrictions that will limit our flexibility in operating our business.
Our 2019 Facility contains various covenants that limit our ability to engage in specified types of transactions. These covenants limit our and our restricted subsidiaries’ ability to, among other things:
• | Maintain net leverage above certain specified levels; |
• | Maintain interest coverage below certain specified levels; |
• | Incur additional debt; |
• | Pay dividends or make other distributions; |
43
• | Redeem stock; |
• | Issue stock of subsidiaries; |
• | Make certain investments; |
• | Create liens; |
• | Enter into transactions with affiliates; and |
• | Merge, consolidate or transfer all or substantially all of our assets. |
A breach of any of these covenants could result in a default under the 2019 Facility. We may also be unable to take advantage of business opportunities that arise because of the limitations imposed on us by the restrictive covenants under our indebtedness.
U.S. credit markets may impact our ability to obtain financing or increase the cost of future financing, including interest rate fluctuations based on macroeconomic conditions that are beyond our control.
During periods of volatility and disruption in the U.S., European, or global credit markets, obtaining additional or replacement financing may be more difficult and the cost of issuing new debt or replacing our 2019 Facility could be higher than under our current 2019 Facility. Higher cost of new debt may limit our ability to have cash on hand for working capital, capital expenditures and acquisitions on terms that are acceptable to us. Additionally, our 2019 Facility has variable interest rates. By its nature, a variable interest rate will move up or down based on changes in the economy and other factors, all of which are beyond our control. If interest rates increase, our interest expense could increase, affecting earnings and reducing cash flows available for working capital, capital expenditures and acquisitions.
Our stock price could fluctuate significantly, which could cause the value of your investment to decline, and you may not be able to resell your shares at or above your purchase price.
Securities markets worldwide have experienced, and may continue to experience, significant price and volume fluctuations. This market volatility, as well as general economic, market or political conditions, could reduce the market price of our common stock regardless of our operating performance. The trading price of our common stock is likely to be volatile and subject to wide price fluctuations in response to various factors, including:
• | Market conditions in the broader stock market; |
• | Actual or anticipated fluctuations in our quarterly financial and operating results; |
• | Issuance of new or changed securities analysts’ reports or recommendations; |
• | Investor perceptions of us and the medical technology and pharmaceutical industries; |
• | Sales, or anticipated sales, of large blocks of our stock; |
• | Acquisitions or introductions of new products or services by us or our competitors; |
• | Positive or negative results from our clinical development programs; |
• | Additions or departures of key personnel; |
• | Regulatory or political developments; |
• | Loss of intellectual property protections; |
• | Litigation and governmental investigations; and |
• | Changing economic conditions. |
These and other factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management from our business, which could significantly harm our profitability and reputation.
44
If securities or industry analysts do not publish research or reports about our business, if they adversely change their recommendations regarding our stock or if our results of operations do not meet their expectations, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline. Moreover, if one or more of the analysts who cover us downgrade our stock, or if our results of operations do not meet their expectations, our stock price could also decline.
We do not anticipate paying any cash dividends for the foreseeable future.
We currently intend to retain our future earnings, if any, for the foreseeable future, to repay indebtedness and to fund the development and growth of our business. We do not intend to pay any dividends to holders of our common stock and the agreements governing our senior secured credit facilities limit our ability to pay dividends. As a result, capital appreciation in the price of our common stock, if any, will be your only source of gain on an investment in our common stock.
Risks Related to the Progenics Transaction
The Progenics Transaction may not occur, and if it does, it may not be accretive and may cause dilution to our earnings per share, which may negatively affect the market price of our common stock.
Although we currently anticipate that the Progenics Transaction will occur and will be accretive to adjusted earnings per share by 2022 and GAAP-reported earnings per share by 2023, these expectations are based on assumptions about our and Progenics’ business and preliminary forecasts, which may change materially. Certain other expenses to be paid in connection with the Progenics Transaction may cause dilution to our earnings per share or decrease or delay the expected accretive effect of the Progenics Transaction and could cause a decrease in the market price of our common stock. In addition, the Progenics Transaction may not occur or we could encounter additional transaction-related costs or other factors such as the failure to realize all of the benefits anticipated in the Progenics Transaction, including synergies, cost savings, innovation and operational efficiencies and revenue growth from the combination. All of these factors could cause dilution to our earnings per share or decrease or delay the expected accretive effect of the Progenics Transaction and cause a decrease in the market price of our common stock.
The Progenics Transaction is subject to conditions, some or all of which may not be satisfied, or completed on a timely basis, if at all. Failure to complete the Progenics Transaction could have material adverse effects on our business.
The completion of the Progenics Transaction is subject to a number of conditions, including, among others, the approval of the Merger Agreement by a majority of votes cast by the holders of the common stock of the Company and a majority of the outstanding shares of Progenics common stock, the absence of any law or order prohibiting the consummation of the Progenics Transaction or the issuance of the shares of our common stock as deal consideration, the effectiveness of a registration statement covering the issuance of shares of our common stock to the stockholders of Progenics, the absence of a material adverse effect on us or Progenics, and other conditions customary for a transaction of this type, which make the completion of the Progenics Transaction and timing thereof uncertain. In addition, the Merger Agreement contains certain termination rights for both us and Progenics, including, among other things (i) if the Progenics Transaction is not consummated on or before the “outside date” of July 1, 2020, (ii) if the required approval of our stockholders or the Progenics stockholders is not obtained, (iii) if the other party willfully breaches its non-solicitation obligations in the Merger Agreement, (v) if the other party materially breaches its representations, warranties or covenants and fails to cure such breach, (vi) if any law or order prohibiting the Progenics Transaction or the issuance of the shares of our common stock forming part of the merger consideration has become final and non-appealable, or (vii) if the board of directors of the other party fails to include such party’s recommendation in favor of the Progenics Transaction in the joint proxy statement/prospectus or changes its recommendation in connection with the Progenics Transaction. If the Progenics Transaction is not completed, our ongoing business may be materially adversely affected and, without realizing any of the benefits that we could have realized had the Progenics Transaction been completed, we will be subject to a number of risks, including the following:
• | The market price of our common stock could decline; |
• | We could owe substantial termination fees to Progenics under certain circumstances; |
• | Time and resources committed by our management to matters relating to the Progenics Transaction could otherwise have been devoted to pursuing other beneficial opportunities; |
• | We may experience negative reactions from the financial markets or from our customers, suppliers or employees; and |
• | We will be required to pay our costs relating to the Progenics Transaction, such as legal, accounting, certain financial advisory, consulting and printing fees, whether or not the Progenics Transaction is completed. |
45
Upon termination of the Merger Agreement, we will be required to pay to Progenics a termination fee of $18.34 million if: (i) we willfully breach our nonsolicitation obligations in the Merger Agreement; (ii) our Board changes its recommendation in support of the merger as a result of a superior proposal or intervening event; or (iii) our stockholders do not approve the issuance of common stock in connection with the merger (if at such time Progenics has the right to terminate the Merger Agreement because we willfully breached our nonsolicitation obligations in the Merger Agreement or our board changed its recommendation in support of the merger as a result of a superior proposal or intervening event). In addition, we will be required to pay to Progenics the termination fee if we receive an acquisition proposal, the Merger Agreement is later terminated under certain circumstances and within twelve months after termination we enter into an agreement with respect to (or consummate) an acquisition proposal for 50% or more of our stock or assets.
In addition, if the Progenics Transaction is not completed, we could be subject to litigation related to any failure to complete the Progenics Transaction or related to any enforcement proceeding commenced against us to perform our obligations under the Merger Agreement. If any such risk materializes, it could adversely impact our ongoing business. Similarly, delays in the completion of the Progenics Transaction could, among other things, result in additional transaction costs, loss of revenue or other negative effects associated with uncertainty about completion of the Progenics Transaction and cause us not to realize some or all of the benefits that we expect to achieve if the Progenics Transaction is successfully completed within its expected timeframe. We cannot assure you that the conditions to the closing of the Progenics Transaction will be satisfied or waived or that the Progenics Transaction will be consummated.
We and Progenics are each subject to business uncertainties and contractual restrictions while the Progenics Transaction is pending, which could adversely affect the business and operations of us or the combined company.
In connection with the pendency of the Progenics Transaction, it is possible that some customers, suppliers and other persons with whom we or Progenics has a business relationship may delay or defer certain business decisions or might decide to seek to terminate, change or renegotiate their relationships with us or Progenics, as the case may be, as a result of the Progenics Transaction, which could negatively affect our current or the combined company’s future revenues, earnings and cash flows, as well as the market price of our common stock, regardless of whether the Progenics Transaction is completed. Under the terms of the Merger Agreement, we and Progenics are each subject to certain restrictions on the conduct of our businesses prior to completing the Progenics Transaction, which could adversely affect each party’s ability to execute certain of its business strategies. Such limitations could adversely affect each party’s business and operations prior to the completion of the Progenics Transaction. Each of the risks described above may be exacerbated by delays or other adverse developments with respect to the completion of the Progenics Transaction.
Uncertainties associated with the Progenics Transaction may cause a loss of management personnel and other key employees, and we and Progenics may have difficulty attracting and motivating management personnel and other key employees, which could adversely affect the future business and operations of the combined company.
We and Progenics are each dependent on the experience and industry knowledge of our respective management personnel and other key employees to execute our business plans. The combined company’s success after the completion of the Progenics Transaction will depend in part upon the ability of each of us and Progenics to attract, motivate and retain key management personnel and other key employees. Prior to completion of the Progenics Transaction, current and prospective employees of each of us and Progenics may experience uncertainty about their roles within the combined company following the completion of the Progenics Transaction, which may have an adverse effect on the ability of each of us and Progenics to attract, motivate or retain management personnel and other key employees. In addition, no assurance can be given that the combined company will be able to attract, motivate or retain management personnel and other key employees of each of us and Progenics to the same extent that we and Progenics have previously been able to attract or retain their own employees.
The Progenics Transaction is subject to the expiration or termination of applicable waiting periods and the receipt of approvals, consents or clearances from regulatory authorities that may impose conditions that could have an adverse effect on us or the combined company or, if not obtained, could prevent completion of the Progenics Transaction.
Before the Progenics Transaction may be completed, any approvals, consents or clearances required in connection with the Progenics Transaction must have been obtained, in each case, under applicable law. Consummation of the Progenics Transaction is conditioned upon, among other things, the expiration or termination of the waiting period (and any extensions thereof) applicable to the Progenics Transaction under the HSR Act, which has been obtained by grant of early termination of the HSR Act waiting period on October 25, 2019. Notwithstanding the grant of early termination, at any time before or after the Progenics Transaction is consummated, the Antitrust Division of the United States Department of Justice, the Federal Trade Commission or U.S. state attorneys general could take action under the antitrust laws in opposition to the Progenics Transaction, including seeking to enjoin completion of the Progenics Transaction, condition completion of the Progenics Transaction upon the divestiture of assets, or impose restrictions on post-merger operations. Any such requirements or restrictions may prevent or delay completion of the Progenics Transaction or may reduce the anticipated benefits of the Progenics Transaction.
46
The Merger Agreement limits our ability to pursue alternatives to the merger and may discourage other companies from trying to acquire us.
The Merger Agreement contains a “no solicitation” covenant that restricts our ability to solicit, initiate, seek or support, or knowingly encourage or facilitate, any inquiries or proposals with respect to certain acquisition proposal relating to the Company; engage or participate in negotiations with respect to any acquisition proposal; provide a third party confidential information with respect to, or have or participate in any discussions with, any person relating to any acquisition proposals; or enter into any acquisition agreement with respect to certain unsolicited proposals relating to an acquisition proposal. In the event we receive an unsolicited acquisition proposal, we must promptly communicate the receipt of such proposal and provide copies of material communications and information, including the terms and conditions of such proposal, to the other party. If, in response to such proposals and subject to certain conditions, we intend to effect a change in our board of directors’ recommendation to stockholders, we must provide Progenics an opportunity to offer to modify the terms of the Merger Agreement in response to such competing acquisition proposal before our board may withdraw or qualify its respective recommendation. The Merger Agreement further provides that in the event of a termination of the Merger Agreement under certain specified circumstances, including a termination by Progenics following a change in recommendation by our board or a willful and material breach of the no-solicitation provision applicable to us, we may be required to pay Progenics a termination fee equal to $18,340,000.
These provisions could discourage a potential third-party acquirer that might have an interest in acquiring all or a significant portion of the Company from considering or proposing that acquisition, even if it were prepared to pay consideration with a higher per share cash or total value than the total value proposed to be paid in the merger. These provisions might also result in a potential third-party acquirer proposing to pay a lower price in an acquisition proposal than it might otherwise have proposed to pay because of the added expense of the termination fee and other fees and expenses that may become payable in certain circumstances.
Current stockholders will have a reduced ownership and voting interest in the Company after the Progenics Transaction and will exercise less influence over the management of the combined company.
Upon completion of the Progenics Transaction, we expect to issue approximately 26.9 million shares of our common stock to Progenics stockholders. As a result, it is expected that, immediately after completion of the Progenics Transaction, former Progenics stockholders will own approximately 40% of our outstanding shares of common stock. In addition, shares of our common stock may be issued from time to time following the Progenics Transaction to holders of Progenics equity awards on the terms set forth in the Merger Agreement. Consequently, our current stockholders in the aggregate will have less influence over the management and policies of the Company than they currently have.
We and Progenics may be targets of securities class action and derivative lawsuits that could result in substantial costs and may delay or prevent the Progenics Transaction from being completed.
Securities class action lawsuits and derivative lawsuits are often brought against public companies that have entered into merger agreements. As of the date of filing of this report, six securities class action lawsuits have been filed against Progenics and its board of directors alleging inadequate disclosure by Progenics under the Registration Statement on Form S-4 related to the Progenics Transaction (which registration statement will be revised to describe, among other things, the revised terms in the Amended Merger Agreement). Two of those lawsuits against Progenics also name us or one of our affiliates as defendants, although we do not believe that those lawsuits have merit or will result in any material monetary damages payable by us. Even if lawsuits are without merit, defending against any legal claims can result in substantial costs and divert management time and resources. An adverse judgment in a securities class action lawsuit or derivative lawsuit alleging significant monetary loss by the plaintiffs could result in monetary damages for us and Progenics, which could have a negative impact on our and Progenics’ respective liquidity and financial condition. Additionally, if a plaintiff is successful in obtaining an injunction prohibiting completion of the Progenics Transaction, then that injunction may delay or prevent the Progenics Transaction from being completed, or from being completed within the expected timeframe, which may adversely affect our business, financial position and results of operation.
Completion of the Progenics Transaction may trigger change in control or other provisions in certain agreements to which Progenics or its subsidiaries are a party, which may have an adverse impact on the combined company’s business and results of operations.
The completion of the Progenics Transaction may trigger change in control and other provisions in certain agreements to which Progenics or its subsidiaries are a party. If we and Progenics are unable to negotiate waivers of those provisions, the counterparties may exercise their rights and remedies under the agreements, potentially terminating the agreements or seeking monetary damages. Even if we and Progenics are able to negotiate waivers, the counterparties may require a fee for such waivers or seek to renegotiate the agreements on terms less favorable to Progenics or the combined company. Any of the foregoing or similar developments may have an adverse impact on the combined company’s business and results of operations.
Progenics stockholders have appraisal rights under Delaware law.
47
Under Delaware law, Progenics stockholders who do not vote in favor of adoption of the Amended Merger Agreement and who otherwise properly perfect their rights will be entitled to “appraisal rights” in connection with the Progenics Transaction, which generally entitle stockholders to receive, in lieu of the merger consideration, a cash payment of an amount determined by the Delaware Court of Chancery to be equal to the fair value of their Progenics common stock as of the effective time of the merger. The appraised value would be determined by the Court of Chancery and could be less than, the same as, or more than the merger consideration. Under Delaware law, stockholders are generally entitled to statutory interest on an appraisal award at a rate equal to 5% above the Federal Reserve discount rate compounded quarterly from the closing date of the merger until the award is actually paid. Stockholders who have properly demanded appraisal rights must file a petition for appraisal with the Court of Chancery within 120 days after the effective date of the merger. Should a material number of Progenics stockholders exercise appraisal rights and should the Court determine that the fair value of such shares of Progenics common stock is materially greater than the merger consideration, we will be required to pay significantly more than anticipated in connection with the Progenics Transaction, which could adversely affect the liquidity and financial condition of the combined company.
The combined company may be unable to successfully integrate the Progenics business into our business and realize the anticipated benefits of the Progenics Transaction.
The success of the Progenics Transaction will depend, in part, on the combined company’s ability to successfully combine the business of Progenics with our business, which currently operate as independent public companies, and realize the anticipated benefits, including synergies, cost savings, innovation and operational efficiencies and revenue growth from the combination. If the combined company is unable to achieve these objectives within the anticipated time frame, or at all, the anticipated benefits may not be realized fully or at all, or may take longer to realize than expected and the value of its common stock may be harmed. Additionally, as a result of the Progenics Transaction, rating agencies may take negative actions against the combined company's credit ratings, which may increase the combined company’s financing costs.
The Progenics Transaction involves the integration of Progenics’ business into our existing business, which is expected to be a complex, costly and time-consuming process. We and Progenics have not previously completed a transaction comparable in size or scope to the Progenics Transaction. The integration may result in material challenges, including, without limitation:
• | The diversion of management’s attention from ongoing business concerns and performance shortfalls at one or both of the companies as a result of the devotion of management’s attention to the Progenics Transaction; |
• | Managing a larger combined company; |
• | Maintaining employee morale and attracting, motivating and retaining management personnel and other key employees; |
• | Unanticipated risks to our integration plan including in connection with timing, talent, and the potential need for additional resources; |
• | New or previously unidentified manufacturing, regulatory, or research and development issues in the Progenics business; |
• | Retaining existing business and operational relationships and attracting new business and operational relationships; |
• | Integrating corporate and administrative infrastructures in geographically separate organizations and eliminating duplicative expenses; |
• | Unanticipated issues in integrating information technology, communications and other systems; |
• | Unanticipated changes in federal or state laws or regulations; and |
• | Unforeseen expenses or delays associated with the Progenics Transaction. |
Many of these factors will be outside of the combined company’s control and any one of them could result in delays, increased costs, decreases in the amount of expected revenues and diversion of management’s time and energy, which could materially affect the combined company’s financial position, results of operations and cash flows. We and Progenics have operated, and until completion of the Progenics Transaction will continue to operate, independently. We and Progenics are currently permitted to conduct only limited planning for the integration of the two companies following the Progenics Transaction and have not yet determined the exact nature of how the businesses and operations of the two companies will be combined after the combination. The actual integration of Progenics with our business may result in additional or unforeseen expenses, and the anticipated benefits of the integration plan may not be realized. These integration matters could have an adverse effect on (i) each of us and Progenics during this transition period and (ii) the combined company for an undetermined period after completion of the Progenics Transaction. In addition, any actual cost savings of the Progenics Transaction could be less than anticipated.
The future results of the combined company may be adversely impacted if the combined company does not effectively manage its expanded operations following the completion of the Progenics Transaction.
48
Following the completion of the Progenics Transaction, the size of the combined company’s business will be significantly larger than the current size of our business. The combined company’s ability to successfully manage this expanded business will depend, in part, upon management’s ability to design and implement strategic initiatives that address not only the integration of two independent stand-alone companies, but also the increased scale and scope of the combined business with its associated increased costs and complexity. The combined company may not be successful or may not realize the expected operating efficiencies, cost savings and other benefits currently anticipated from the Progenics Transaction.
The CVRs we will issue as part of the Progenics Transaction may result in substantial future payments and could divert the attention of our management.
As part of the consideration for the Progenics Transaction, we will issue CVRs to the stockholders of Progenics and holders of in-the-money Progenics equity awards entitling them to future cash payments of 40% of PyL net sales over $100 million in 2022 and over $150 million in 2023. These payments could be substantial and could adversely impact our liquidity. In addition, we are obligated to exercise a level of effort, expertise and resources consistent with those normally used in a medical diagnostics business similar to our size and resources with respect to developing, seeking regulatory approval for and commercializing a product of similar market potential at a similar stage in its development or product life to PyL. We are also required to produce net sales statements for PyL that may be reviewed and challenged by CVR holders, with any disagreement to be resolved by an independent accountant. These requirements could divert management time and resources and result in additional costs.
The financial analyses and forecasts considered by Lantheus Holdings and Progenics and their respective financial advisors may not be realized, which may adversely affect the market price of Lantheus Holdings common stock following the completion of the merger.
In performing their financial analyses and rendering their opinions related to the merger, each of the respective financial advisors to Lantheus Holdings and Progenics relied on, among other things, internal stand-alone financial analyses and forecasts as separately provided by Lantheus Holdings and Progenics. These analyses and forecasts were prepared by, or as directed by, the management of Lantheus Holdings or the management of Progenics, as applicable. None of these analyses or forecasts were prepared with a view towards public disclosure or compliance with the published guidelines of the SEC, the U.S. Generally Accepted Accounting Principles. These projections are inherently based on various estimates and assumptions that are subject to the judgment of those preparing them. These projections are also subject to significant economic, competitive, industry and other uncertainties and contingencies, all of which are difficult or impossible to predict and many of which are beyond the control of Lantheus Holdings and Progenics. There can be no assurance that Lantheus Holdings’ or Progenics’ financial condition or results of operations will be consistent with those set forth in such analyses and forecasts, which could have an adverse impact on the market price of Lantheus Holdings common stock or the financial position of the combined company following the merger.
The combined company is expected to incur substantial expenses related to the completion of the Progenics Transaction and the integration of the Progenics business with our business.
The combined company is expected to incur substantial expenses in connection with the completion of the Progenics Transaction, including seeking approval from our stockholders, and the integration of the Progenics business with our business. There are a large number of processes, policies, procedures, operations, technologies and systems that must be integrated, including purchasing, accounting and finance, sales, payroll, pricing, revenue management, marketing and benefits. The substantial majority of these costs will be non-recurring expenses related to the Progenics Transaction, facilities and systems consolidation costs. The combined company may incur additional costs to maintain employee morale and to attract, motivate or retain management personnel or key employees. We will also incur transaction fees and costs related to formulating integration plans for the combined business, and the execution of these plans may lead to additional unanticipated costs. Additionally, as a result of the Progenics Transaction, rating agencies may take negative actions with regard to the combined company’s credit ratings, which may increase the combined company’s financing costs. These incremental transaction and acquisition-related costs may exceed the savings the combined company expects to achieve from the elimination of duplicative costs and the realization of other efficiencies related to the integration of the businesses, particularly in the near term and in the event there are material unanticipated costs.
Item 1B. Unresolved Staff Comments
None.
49
Item 2. Properties
The following table summarizes information regarding our significant leased and owned properties, as of December 31, 2019:
Location | Purpose | Segment | Square Footage | Ownership | Lease Term End | ||||||
U.S. | |||||||||||
North Billerica, Massachusetts | Corporate Headquarters, Manufacturing, Laboratory, Mixed Use and Other Office Space | U.S. Segment | 431,000 | Owned | N/A | ||||||
Canada | |||||||||||
Quebec | Mixed Use and Office Space | International Segment | 1,106 | Leased | April 2020 | ||||||
Quebec | Distribution Center and Office Space | International Segment | 1,433 | Leased | May 2022 | ||||||
Puerto Rico | |||||||||||
San Juan | Manufacturing, Laboratory, Mixed Use and Office Space | International Segment | 9,550 | Leased | October 2024 | ||||||
We believe all of these facilities are well-maintained and suitable for the office, radiopharmacy, manufacturing or warehouse operations conducted in them and provide adequate capacity for current and foreseeable future needs.
Item 3. Legal Proceedings
From time to time, we are a party to various legal proceedings arising in the ordinary course of business. In addition, we have in the past been, and may in the future be, subject to investigations by governmental and regulatory authorities which expose us to greater risks associated with litigation, regulatory or other proceedings, as a result of which we could be required to pay significant fines or penalties. The costs and outcome of litigation, regulatory or other proceedings cannot be predicted with certainty, and some lawsuits, claims, actions or proceedings may be disposed of unfavorably to us. In addition, intellectual property disputes often have a risk of injunctive relief which, if imposed against us, could materially and adversely affect our financial condition or results of operations.
In October 2019, we were awarded a total of approximately $3.5 million, consisting of damages, pre-judgment interest, and certain arbitration fees, compensation and expenses in our arbitration with Pharmalucence in connection with a Manufacturing and Supply Agreement dated November 12, 2013, under which Pharmalucence agreed to manufacture and supply DEFINITY for us. The commercial arrangement contemplated by that agreement was repeatedly delayed and ultimately never successfully realized. After extended settlement discussions between Sun Pharma, the ultimate parent of Pharmalucence, and us, which did not lead to a mutually acceptable outcome, on November 10, 2017, we filed an arbitration demand (and later an amended arbitration demand) with the American Arbitration Association against Pharmalucence, alleging breach of contract, breach of the covenant of good faith and fair dealing, tortious misrepresentation and violation of the Massachusetts Consumer Protection Law, also known as Chapter 93A. In November 2019, we received proceeds of approximately $3.5 million, which we recorded in other expense (income) in the consolidated statement of operations.
As of December 31, 2019, except as disclosed above we had no material ongoing litigation in which we were a party. In addition, we had no material ongoing regulatory or other proceeding and no knowledge of any investigations by governmental or regulatory authorities in which we are a target, in either case that we believe could have a material and adverse effect on our current business.
Item 4. Mine Safety Disclosures
Not applicable
50
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
The Company’s common stock began trading on the NASDAQ Global Market under the symbol “LNTH” on June 25, 2015. Prior to that time, there was no established public trading market for our common stock.
Holders of Record
On February 19, 2020, there were approximately 8 stockholders of record of our common stock. This number does not include stockholders for whom shares are held in “nominee” or “street” name.
Performance Graph
The performance graph set forth below shall not be deemed “soliciting material” or to be “filed” with the SEC. This graph will not be deemed “incorporated by reference” into any filing under the Securities Act or the Exchange Act, whether such filing occurs before or after the date hereof, except to the extent that the Company explicitly incorporates it by reference into in such filing.
The following graph provides a comparison of the cumulative total shareholder return on our common shares with that of the cumulative total shareholder return on the (i) Russell 2000 Index and (ii) the NASDAQ US Small Cap Index, commencing on June 25, 2015 and ending December 31, 2019. The graph assumes a hypothetical $100 investment in our common stock and in each of the comparative indices on June 25, 2015. Our historic share price performance is not necessarily indicative of future share price performance.
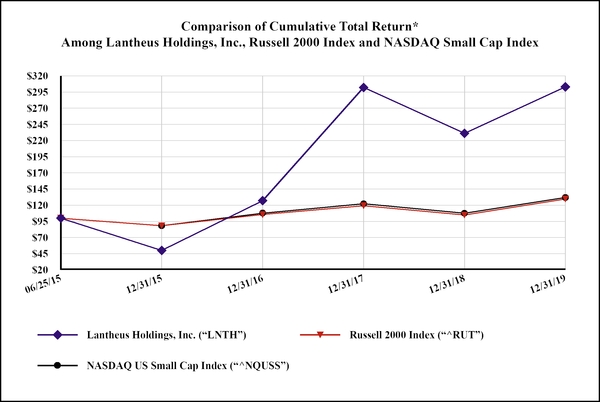
________________________________
* Assumes hypothetical investment of $100 in our common stock and each of the indices on June 25, 2015, the date of our IPO, including reinvestment of dividends.
51
Performance Graph Data
The following table sets forth the cumulative total shareholder return on the hypothetical $100 investment in the Company’s common stock and each of the comparative indices on June 25, 2015:
Date | Lantheus Holdings, Inc. (“LNTH”) | Russell 2000 Index (“^RUT”) | NASDAQ US Small Cap Index (“^NQUSS”) | |||||||||
06/25/15 | $ | 100.00 | $ | 100.00 | $ | 100.00 | ||||||
12/31/15 | $ | 49.93 | $ | 88.26 | $ | 88.24 | ||||||
12/31/16 | $ | 127.03 | $ | 105.45 | $ | 107.67 | ||||||
12/31/17 | $ | 302.07 | $ | 119.31 | $ | 122.28 | ||||||
12/31/18 | $ | 231.17 | $ | 104.79 | $ | 107.62 | ||||||
12/31/19 | $ | 302.95 | $ | 129.64 | $ | 131.70 | ||||||
Issuer Purchase of Equity Securities
None.
Dividend Policy
We did not declare or pay any dividends and we do not currently intend to pay dividends in the foreseeable future. We currently expect to retain future earnings, if any, for the foreseeable future, to finance the growth and development of our business and to repay indebtedness. Our ability to pay dividends is restricted by our financing arrangements. See Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources—External Sources of Liquidity” for further information.
Recent Sales of Unregistered Securities
None.
Repurchases
The following table presents information with respect to purchases of common stock we made during the quarter ended December 31, 2019. The Company does not currently have a share repurchase program in effect. The 2015 Equity Incentive Plan, adopted by the Company on June 24, 2015, as amended on April 26, 2016 and as further amended on April 27, 2017 and April 24, 2019 (the “2015 Plan”), provides for the withholding of shares to satisfy minimum statutory tax withholding obligations. It does not specify a maximum number of shares that can be withheld for this purpose. The shares of common stock withheld to satisfy minimum tax withholding obligations may be deemed to be “issuer purchases” of shares that are required to be disclosed pursuant to this Item 5. These shares are then sold in compliance with Rule 10b5-1 into the market to allow the Company to satisfy the tax withholding requirements in cash.
Period | Total Number of Shares Purchased | Average Price Paid per Share | Total Number of Shares Purchased as Part of Publicly Announced Programs | Approximate Dollar Value of Shares that May Yet Be Purchased Under the Program | |||||||
October 2019 ** | 735 | $ | 18.83 | * | * | ||||||
November 2019 ** | 1,149 | $ | 21.29 | * | * | ||||||
December 2019 ** | 243 | $ | 20.60 | * | * | ||||||
Total | 2,127 | * | |||||||||
________________________________
* These amounts are not applicable as the Company does not have a share repurchase program in effect.
** Reflects shares withheld to satisfy minimum statutory tax withholding amounts due from employees related to the receipt of stock which resulted from the exercise for vesting of equity awards.
Securities Authorized for Issuance under Equity Compensations Plans
The information required with respect to this item is incorporated herein by reference to our Definitive Proxy Statement for our 2020 Annual Meeting of Stockholders to be filed with the SEC no later than 120 days after the close of our year ended December 31, 2019.
52
Item 6. Selected Financial Data
Basis of Financial Information
The consolidated financial statements have been prepared in U.S. Dollars, in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). The consolidated financial statements include the accounts of Lantheus Holdings, Inc. (“Holdings”) and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
Selected Financial Data
In the table below, we provide you with our selected consolidated financial data for the periods presented. We have prepared this information using our audited consolidated financial statements for the years ended December 31, 2019, 2018, 2017, 2016 and 2015.
The following selected consolidated financial information should be read in conjunction with our consolidated financial statements, the related notes and Part II, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K. The results indicated below and elsewhere in this Annual Report on Form 10-K are not necessarily indicative of results to be expected for any future period.
Year Ended December 31, | |||||||||||||||||||
2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
Statement of Operations | (in thousands, except per share data) | ||||||||||||||||||
Revenues | $ | 347,337 | $ | 343,374 | $ | 331,378 | $ | 301,853 | $ | 293,461 | |||||||||
Cost of goods sold | 172,526 | 168,489 | 169,243 | 164,073 | 157,939 | ||||||||||||||
Sales and marketing | 41,888 | 43,159 | 42,315 | 36,542 | 34,740 | ||||||||||||||
General and administrative | 61,244 | 50,167 | 49,842 | 38,832 | 43,894 | ||||||||||||||
Research and development | 20,018 | 17,071 | 18,125 | 12,203 | 14,358 | ||||||||||||||
Gain on sales of assets | — | — | — | 6,385 | — | ||||||||||||||
Operating income | 51,661 | 64,488 | 51,853 | 56,588 | 42,530 | ||||||||||||||
Interest expense | 13,617 | 17,405 | 18,410 | 26,618 | 38,715 | ||||||||||||||
Debt retirement costs | — | — | — | 1,896 | — | ||||||||||||||
Loss on extinguishment of debt | 3,196 | — | 2,442 | — | 15,528 | ||||||||||||||
Other expense (income) | 6,221 | (2,465 | ) | (8,638 | ) | (220 | ) | 65 | |||||||||||
Income (loss) before income taxes | 28,627 | 49,548 | 39,639 | 28,294 | (11,778 | ) | |||||||||||||
Income tax (benefit) expense(a) | (3,040 | ) | 9,030 | (83,746 | ) | 1,532 | 2,968 | ||||||||||||
Net income (loss) | $ | 31,667 | $ | 40,518 | $ | 123,385 | $ | 26,762 | $ | (14,746 | ) | ||||||||
Net income (loss) per common share: | |||||||||||||||||||
Basic | $ | 0.81 | $ | 1.06 | $ | 3.31 | $ | 0.84 | $ | (0.60 | ) | ||||||||
Diluted | $ | 0.79 | $ | 1.03 | $ | 3.17 | $ | 0.82 | $ | (0.60 | ) | ||||||||
Weighted-average common shares: | |||||||||||||||||||
Basic | 38,988 | 38,233 | 37,276 | 32,044 | 24,440 | ||||||||||||||
Diluted | 40,113 | 39,501 | 38,892 | 32,656 | 24,440 | ||||||||||||||
_______________________________
(a) The 2017 amount reflects the release of our valuation allowance of $141.1 million against its deferred tax assets offset by a provision of $45.1
million for remeasuring the Company’s deferred tax assets for the change in tax rates enacted under the Tax Cuts and Jobs Act of 2017.
December 31, | |||||||||||||||||||
2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
Balance Sheet Data | (in thousands) | ||||||||||||||||||
Cash and cash equivalents | $ | 92,919 | $ | 113,401 | $ | 76,290 | $ | 51,178 | $ | 28,596 | |||||||||
Total assets | $ | 405,919 | $ | 439,831 | $ | 383,858 | $ | 255,898 | $ | 242,379 | |||||||||
Long-term debt, net | $ | 183,927 | $ | 263,709 | $ | 265,393 | $ | 274,460 | $ | 349,858 | |||||||||
Total liabilities | $ | 291,318 | $ | 368,829 | $ | 360,567 | $ | 362,414 | $ | 427,668 | |||||||||
Total stockholders’ equity (deficit) | $ | 114,601 | $ | 71,002 | $ | 23,291 | $ | (106,516 | ) | $ | (185,289 | ) | |||||||
53
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read together with Item 6, “Selected Financial Data” and the consolidated financial statements and the related notes included in Item 8 of this Annual Report on Form 10-K. This discussion contains forward-looking statements related to future events and our future financial performance that are based on current expectations and subject to risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those set forth in Part I—Item 1A. “Risk Factors” and “Cautionary Note Regarding Forward Looking Statements.” included in this Annual Report on Form 10-K.
Overview
Our Business
We are a global leader in the development, manufacture and commercialization of innovative diagnostic medical imaging agents and products that assist clinicians in the diagnosis and treatment of cardiovascular and other diseases. Clinicians use our imaging agents and products across a range of imaging modalities, including echocardiography and nuclear imaging. We believe that the resulting improved diagnostic information enables healthcare providers to better detect and characterize, or rule out, disease, potentially achieving improved patient outcomes, reducing patient risk and limiting overall costs for payers and the entire healthcare system.
Our commercial products are used by cardiologists, nuclear physicians, radiologists, internal medicine physicians, technologists and sonographers working in a variety of clinical settings. We sell our products to radiopharmacies, integrated delivery networks, hospitals, clinics and group practices.
We sell our products globally and operate our business in two reportable segments, which are further described below:
• | U.S. Segment produces and markets our medical imaging agents and products throughout the U.S. In the U.S., we primarily sell our products to radiopharmacies, integrated delivery networks, hospitals, clinics and group practices. |
• | International Segment operations consist of production and distribution activities in Puerto Rico and some direct distribution activities in Canada. Additionally, within our International Segment, we have established and maintain third-party distribution relationships under which our products are marketed and sold in Europe, Canada, Australia, Asia-Pacific and Latin America. |
Our Product Portfolio
Our product portfolio includes an ultrasound contrast agent, nuclear imaging products and a radiotherapeutic product. Our principal products include the following:
• | DEFINITY is a microbubble contrast agent used in ultrasound exams of the heart, also known as echocardiography exams. DEFINITY contains perflutren-containing lipid microspheres and is indicated in the U.S. for use in patients with suboptimal echocardiograms to assist in imaging the left ventricular chamber and left endocardial border of the heart in ultrasound procedures. |
• | TechneLite is a Tc-99m generator that provides the essential nuclear material used by radiopharmacies to radiolabel Cardiolite, Neurolite and other Tc-99m-based radiopharmaceuticals used in nuclear medicine procedures. TechneLite uses Mo-99 as its active ingredient. |
Sales of our microbubble contrast agent, DEFINITY, are made in the U.S. and Canada through a DEFINITY direct sales team. In the U.S., our nuclear imaging products, including TechneLite, Xenon, Neurolite and Cardiolite, are primarily distributed through commercial radiopharmacies, the majority of which are controlled by or associated with GE Healthcare, Cardinal, UPPI, Jubilant Radiopharma and PharmaLogic. A small portion of our nuclear imaging product sales in the U.S. are made through our direct sales force to hospitals and clinics that maintain their own in-house radiopharmaceutical preparation capabilities. We own one radiopharmacy in Puerto Rico, where we sell our own products as well as products of third parties to end-users.
We also maintain our own direct sales force in Canada for certain of our products. In Europe, Australia, Asia-Pacific and Latin America, we generally rely on third-party distributors to market, sell and distribute our nuclear imaging and contrast agent products, either on a country-by-country basis or on a multi-country regional basis.
Progenics Transaction
On October 1, 2019, we entered into the Initial Merger Agreement to acquire Progenics in an all-stock transaction. Under the terms of the Initial Merger Agreement, we agreed to acquire all of the issued and outstanding shares of Progenics common stock at a fixed exchange ratio. Progenics stockholders would have received 0.2502 shares of our common stock for each share of Progenics common stock, representing an approximately 35% aggregate ownership stake in the combined company. The transaction
54
contemplated by the Initial Merger Agreement was unanimously approved by the Boards of Directors of both companies and was subject to the terms and conditions set forth in the Initial Merger Agreement, including, among other things, the affirmative vote of a majority of the outstanding shares of common stock of Progenics and a majority of votes cast by the holders of the common stock of the Company.
On February 20, 2020, we entered into the Amended Merger Agreement with Progenics, which amends and restates the Initial Merger Agreement. Under the terms of the Amended Merger Agreement, Lantheus will acquire all of the issued and outstanding shares of Progenics common stock at a fixed exchange ratio whereby Progenics stockholders will receive, for each share of Progenics stock held at the time of the closing of the merger, 0.31 of a share of our common stock, increased from 0.2502 under the Initial Merger Agreement, together with a non-tradeable CVR tied to the financial performance of PyL, such that each CVR will entitle its holder to receive a pro rata share of aggregate cash payments equal to 40% of U.S. net sales generated by PyL in 2022 and 2023 in excess of $100 million and $150 million, respectively. In no event will our aggregate payments under the CVRs exceed 19.9% of the total consideration we pay in the transaction. As a result of the increase in the exchange ratio, following the completion of the merger, former Progenics stockholders’ aggregate ownership stake will increase to approximately 40% of the combined company from approximately 35% under the Initial Merger Agreement. Progenics’ stockholders will also now be entitled to appraisal rights as provided under Delaware law. The transaction contemplated by the Amended Merger Agreement was unanimously approved by the Boards of Directors of both companies and requires, among other things, the affirmative vote of a majority of the outstanding shares of common stock of Progenics and a majority of votes cast by the holders of the common stock of the Company.
In addition, pursuant to the Amended Merger Agreement, the holder of each in-the-money Progenics Stock Option will be entitled to receive in exchange for each such in-the-money option (i) a Lantheus Stock Option converted based on the 0.31exchange ratio and (ii) a vested or unvested CVR depending on whether the underlying option is vested. Holders of out-of-the-money Progenics Stock Options will receive Lantheus Stock Options converted on an exchange ratio adjusted based on actual trading prices of common stock of Progenics and Lantheus Holdings prior to the effective time of the merger.
The Amended Merger Agreement also provides that on closing our board of directors will appoint Dr. Gerard Ber and Mr. Heinz Mausli, who are currently members of the board of directors of Progenics, to serve on our board of directors. In addition, our board of directors, subject to complying with applicable fiduciary duties, will use commercially reasonable efforts to cause Dr. Ber and Mr. Mausli to be nominated for reelection following the closing through 2023. Our board of directors will be reduced in size from ten to nine members at our annual meeting of stockholders on April 23, 2020 (or sooner if the transaction closes before then) and will be further reduced in size from nine to eight members prior to the date of our 2021 annual meeting of stockholders.
Except as described above, the material terms of the Amended Merger Agreement are substantially the same as the terms of the Initial Merger Agreement.
The transaction is currently expected to close in the second quarter of 2020. Upon completion of the acquisition, which the parties intend to report as tax-deferred to Progenics’ stockholders with respect to the stock component of the merger consideration for U.S. federal income tax purposes, the combined company will continue to be headquartered in North Billerica, Massachusetts and will trade on the NASDAQ under the ticker symbol LNTH.
See Part I, Item 1A. “Risk Factors” for information regarding certain risks associated with our proposed acquisition of Progenics.
Key Factors Affecting Our Results
Our business and financial performance have been, and continue to be, affected by the following:
Anticipated Continued Growth of DEFINITY and Expansion of Our Ultrasound Microbubble Franchise
We believe the market opportunity for our ultrasound microbubble contrast agent, DEFINITY, continues to be significant. DEFINITY is our fastest growing and highest margin commercial product. We anticipate DEFINITY sales will continue to grow and that DEFINITY will constitute a greater share of our overall product mix in 2020 as compared to prior years. As we continue to educate the physician and healthcare provider community about the benefits and risks of DEFINITY, we believe we will be able to continue to grow the appropriate use of DEFINITY in suboptimal echocardiograms. In a U.S. market with three echocardiography contrast agents approved by the FDA, we estimate that DEFINITY had over 80% of the market as of December 31, 2019.
As we continue to pursue expanding our microbubble franchise, our activities include:
• | Patents - We continue to actively pursue additional patents in connection with DEFINITY, both in the U.S. and internationally. In the U.S., we have an Orange Book-listed method of use patent expiring in March 2037 and additional manufacturing patents that are not Orange Book-listed expiring in 2021, 2023 and 2037. Outside of the U.S., while our |
55
DEFINITY patent protection and regulatory exclusivity have generally expired, we are currently prosecuting additional patents to try to obtain similar method of use and manufacturing patent protection as granted in the U.S.
Hatch-Waxman Act - Even though our longest duration Orange Book-listed DEFINITY patent extends until March 2037, because our Orange Book-listed composition of matter patent expired in June 2019, we may face generic DEFINITY challengers in the near to intermediate term. Under the Hatch-Waxman Act, the FDA can approve ANDAs for generic versions of drugs if the ANDA applicant demonstrates, among other things, that (i) its generic candidate is the same as the innovator product by establishing bioequivalence and providing relevant chemistry, manufacturing and product data, and (ii) the marketing of that generic candidate does not infringe an Orange Book-listed patent. With respect to any Orange Book-listed patent covering the innovator product, the ANDA applicant must give Notice to the innovator that the ANDA applicant certifies that its generic candidate will not infringe the innovator’s Orange Book-listed patent or that the Orange Book-listed patent is invalid. The innovator can then challenge the ANDA applicant in court within 45 days of receiving that Notice, and FDA approval to commercialize the generic candidate will be stayed (that is, delayed) for up to 30 months (measured from the date on which a Notice is received) while the patent dispute between the innovator and the ANDA applicant is resolved in court. The 30 month stay could potentially expire sooner if the courts determine that no infringement had occurred or that the challenged Orange Book-listed patent is invalid or if the parties otherwise settle their dispute.
As of the date of filing of this Annual Report on Form 10-K, we have not received any Notice from an ANDA applicant. If we were to (i) receive any such Notice in the future, (ii) bring a patent infringement suit against the ANDA applicant within 45 days of receiving that Notice, and (iii) successfully obtain the full 30 month stay, then the ANDA applicant would be precluded from commercializing a generic version of DEFINITY prior to the expiration of that 30 month stay period and, potentially, thereafter, depending on how the patent dispute is resolved. Solely by way of example and not based on any knowledge we currently have, if we received a Notice from an ANDA applicant in March 2020 and the full 30 month stay was obtained, then the ANDA applicant would be precluded from commercialization until at least September 2022. If we received a Notice some number of months in the future and the full 30 month stay was obtained, the commercialization date would roll forward in the future by the same calculation.
• | Modified Formulation - We are developing at SBL a modified formulation of DEFINITY. We believe this modified formulation will provide an enhanced product profile enabling storage as well as shipment at room temperature (DEFINITY’s current formulation requires refrigerated storage), will give clinicians additional choice, and will allow for greater utility of this formulation in broader clinical settings. We were recently granted a composition of matter patent on the modified formulation which runs through December 2035. If the modified formulation is approved by the FDA, then this patent would be eligible to be listed in the Orange Book. We currently believe that, if approved by the FDA, the modified formulation could become commercially available in early 2021, although that timing cannot be assured. Given its physical characteristics, the modified formulation may also be better suited for inclusion in kits requiring microbubbles for other indications and applications (including in kits developed by third parties of the type described in the next paragraph). |
• | New Clinical Applications - As we continue to look for other opportunities to expand our microbubble franchise, we are evaluating new indications and clinical applications beyond echocardiography and contrast imaging generally. For example, we recently announced a strategic development and commercial collaboration with Cerevast in which our microbubble will be used in connection with Cerevast’s ocular ultrasound device to target improving blood flow in occluded retinal veins in the eye. Retinal vein occlusion is one of the most common causes of vision loss worldwide. We also recently announced a strategic commercial supply agreement with CarThera for the use of our microbubbles in combination with SonoCloud, a proprietary implantable device in development for the treatment of recurrent glioblastoma. Glioblastoma is a lethal and devastating form of brain cancer with median survival of 15 months after diagnosis. |
• | In-House Manufacturing - We are currently building specialized in-house manufacturing capabilities at our North Billerica, Massachusetts facility for DEFINITY and, potentially, other sterile vial products. We believe the investment in these efforts will allow us to better control DEFINITY manufacturing and inventory, reduce our costs in a potentially more price competitive environment, and provide us with supply chain redundancy. We currently expect to be in a position to use this in-house manufacturing capability by early 2021, although that timing cannot be assured. |
Global Mo-99 Supply
We currently have Mo-99 supply agreements with IRE, running through December 31, 2022, and renewable by us on a year-to-year basis thereafter, and with NTP and ANSTO, running through December 31, 2021. We also have a Xenon supply agreement with IRE which runs through June 30, 2022, and which is subject to further extension.
Although we have a globally diverse Mo-99 supply with IRE in Belgium, NTP in South Africa and ANSTO in Australia, we still face challenges in our Mo-99 supply chain. The NTP processing facility has had periodic outages in 2017, 2018 and 2019. When NTP was not producing, we relied on Mo-99 supply from both IRE and ANSTO to limit the impact of the NTP outages. In the second quarter of 2019, ANSTO experienced facility issues in its existing Mo-99 processing facility which resulted in a decrease in Mo-99 available to us. In addition, as ANSTO transitioned from its existing Mo-99 processing facility to its new Mo-99 processing facility in
56
the second quarter of 2019, ANSTO experienced start-up and transition challenges, which also resulted in a decrease in Mo-99 available to us. Further, starting in late June 2019 and through the date of this filing, ANSTO’s new Mo-99 processing facility has experienced unscheduled production outages, and we are now relying on IRE and NTP to limit the impact of those ANSTO outages. Because of these various supply chain constraints, depending on reactor and processor schedules and operations, we have not been able to fill some or all of the demand for our TechneLite generators on certain manufacturing days.
ANSTO’s new Mo-99 processing facility, could eventually increase ANSTO’s Mo-99 production capacity from approximately 2,000 curies per week to 3,500 curies per week with additional committed financial and operational resources. At full ramp-up capacity, ANSTO’s new facility could provide incremental supply to our globally diversified Mo-99 supply chain and therefore mitigate some risk among our Mo-99 suppliers, although we can give no assurances to that effect. In addition, we also have a strategic arrangement with SHINE, a Wisconsin-based company, for the future supply of Mo-99. Under the terms of that agreement, SHINE will provide us Mo-99 once SHINE’s facility becomes operational and receives all necessary approvals, which SHINE now estimates will occur in 2022.
Inventory Supply
We obtain a substantial portion of our imaging agents from third-party suppliers, JHS is currently our sole source manufacturer of DEFINITY, Neurolite, Cardiolite and evacuation vials, the latter being an ancillary component for our TechneLite generators. We are currently seeking approval from certain foreign regulatory authorities for JHS to manufacture certain of our products. Until we receive these approvals, we will face continued limitations on where we can sell those products outside of the U.S.
In addition to JHS, we are also currently working to secure additional alternative suppliers for our key products as part of our ongoing supply chain diversification strategy. We have ongoing development and technology transfer activities for a modified formulation of DEFINITY with SBL, which is located in South Korea. We currently believe that if approved by the FDA, the modified formulation could be commercially available in early 2021, although that timing cannot be assured. We are also building in-house specialized manufacturing capabilities at our North Billerica, Massachusetts facility, as part of a larger strategy to create a competitive advantage in specialized manufacturing, which will also allow us to optimize our costs and reduce our supply chain risk. We can give no assurance as to when or if we will be successful in these efforts or that we will be able to successfully manufacture any additional commercial products at our North Billerica, Massachusetts facility.
Radiopharmaceuticals are decaying radioisotopes with half-lives ranging from a few hours to several days. These products cannot be kept in inventory because of their limited shelf lives and are subject to just-in-time manufacturing, processing and distribution, which takes place at our North Billerica, Massachusetts facility.
Research and Development Expenses
To remain a leader in the marketplace, we have historically made substantial investments in new product development. As a result, the positive contributions of those internally funded research and development programs have been a key factor in our historical results and success. On April 25, 2017, we announced entering into a definitive, exclusive Collaboration and License Agreement with GE Healthcare for the continued Phase 3 development and worldwide commercialization of flurpiridaz F 18. For LMI 1195, our PET-based molecular imaging agent for the norepinephrine pathway, we are currently designing two Phase 3 clinical trials for the use of LMI 1195 for the diagnosis and management of neuroendocrine tumors in pediatric and adult populations, respectively. The FDA has granted an Orphan Drug designation for the use of LMI 1195 in the management indication. We have also received notice of eligibility for a rare pediatric disease priority review voucher for a subsequent human drug application so long as LMI 1195 is approved by the FDA for its rare pediatric disease indication prior to September 30, 2022. Our investments in these additional clinical activities will increase our operating expenses and impact our results of operations and cash flow, and we can give no assurances as to whether or when LMI 1195 would be approved.
As part of our microbubble franchise strategy, we also conducted two Phase 3, open-label, multicenter studies to evaluate LVEF measurement accuracy and reproducibility of DEFINITY contrast-enhanced and unenhanced echocardiography as compared to non-contrast CMRI used as the truth standard. The first of the two trials, BENEFIT 1, enrolled 145 subjects. After reviewing the study results from BENEFIT 1, we concluded there was no statistically significant improvement in the accuracy of LVEF values for contrast-enhanced echocardiography versus unenhanced echocardiography as compared to CMRI. In addition, analyses of the secondary endpoints revealed no improvement in inter-reader variability between the contrast-enhanced and unenhanced echocardiograms for LVEF assessments. A post-hoc analysis, however, did show statistically significant improvements in left ventricular diastolic, systolic and stroke volume measurements with contrast-enhanced versus unenhanced echocardiography when compared to CMRI. We will continue to analyze the BENEFIT 1 data, and when the data from BENEFIT 2 are available, we will compile the data sets to analyze the full results of the trials.
New Initiatives
57
We continue to evaluate a number of different opportunities to acquire or in-license additional products, businesses and technologies to drive our future growth. We are particularly interested in expanding our presence in oncology, in radiotherapeutics as well as diagnostics. In addition to the Progenics Transaction described above, we recently entered into a strategic collaboration and license agreement with NanoMab Technology Limited, a privately-held biopharmaceutical company focusing on the development of next generation radiopharmaceuticals for cancer precision medicine. We believe this collaboration will provide the first broadly-available imaging biomarker research tool to pharmaceutical companies and academic centers conducting research and development on PD-L1 immuno-oncology treatments, including combination therapies. We can give no assurance as to when or if this collaboration will be successful or accretive to earnings.
In addition, as described above, we continue to expand our microbubble franchise. We recently announced a strategic development and commercial collaboration with Cerevast in which our microbubble will be used in connection with Cerevast’s ocular ultrasound device to target improving blood flow in occluded retinal veins in the eye. We also recently announced a strategic commercial supply agreement with CarThera for the use of our microbubbles in combination with SonoCloud, a proprietary implantable device in development for the treatment of recurrent glioblastoma.
Results of Operations
The following is a summary of our consolidated results of operations:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
Revenues | $ | 347,337 | $ | 343,374 | $ | 331,378 | |||||
Cost of goods sold | 172,526 | 168,489 | 169,243 | ||||||||
Gross profit | 174,811 | 174,885 | 162,135 | ||||||||
Operating expenses | |||||||||||
Sales and marketing | 41,888 | 43,159 | 42,315 | ||||||||
General and administrative | 61,244 | 50,167 | 49,842 | ||||||||
Research and development | 20,018 | 17,071 | 18,125 | ||||||||
Total operating expenses | 123,150 | 110,397 | 110,282 | ||||||||
Operating income | 51,661 | 64,488 | 51,853 | ||||||||
Interest expense | 13,617 | 17,405 | 18,410 | ||||||||
Loss on extinguishment of debt | 3,196 | — | 2,442 | ||||||||
Other expense (income) | 6,221 | (2,465 | ) | (8,638 | ) | ||||||
Income before income taxes | 28,627 | 49,548 | 39,639 | ||||||||
Income tax (benefit) expense | (3,040 | ) | 9,030 | (83,746 | ) | ||||||
Net income | $ | 31,667 | $ | 40,518 | $ | 123,385 | |||||
58
Comparison of the Periods Ended December 31, 2019 and 2018
Revenues
Segment revenues are summarized by product as follows:
Year Ended December 31, | 2019 vs. 2018 | 2018 vs. 2017 | |||||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | Change $ | Change % | Change $ | Change % | ||||||||||||||||||
U.S. | |||||||||||||||||||||||||
DEFINITY | $ | 211,777 | $ | 178,440 | $ | 153,581 | $ | 33,337 | 18.7 | % | $ | 24,859 | 16.2 | % | |||||||||||
TechneLite | 72,534 | 74,042 | 90,489 | (1,508 | ) | (2.0 | )% | (16,447 | ) | (18.2 | )% | ||||||||||||||
Other nuclear | 36,231 | 48,935 | 54,822 | (12,704 | ) | (26.0 | )% | (5,887 | ) | (10.7 | )% | ||||||||||||||
Rebates and allowances | (16,553 | ) | (12,837 | ) | (8,890 | ) | (3,716 | ) | 28.9 | % | (3,947 | ) | 44.4 | % | |||||||||||
Total U.S. Revenues | 303,989 | 288,580 | 290,002 | 15,409 | 5.3 | % | (1,422 | ) | (0.5 | )% | |||||||||||||||
International | |||||||||||||||||||||||||
DEFINITY | 5,731 | 4,633 | 3,687 | 1,098 | 23.7 | % | 946 | 25.7 | % | ||||||||||||||||
TechneLite | 14,058 | 24,816 | 14,155 | (10,758 | ) | (43.4 | )% | 10,661 | 75.3 | % | |||||||||||||||
Other nuclear | 23,574 | 25,349 | 23,558 | (1,775 | ) | (7.0 | )% | 1,791 | 7.6 | % | |||||||||||||||
Rebates and allowances | (15 | ) | (4 | ) | (24 | ) | (11 | ) | 275.0 | % | 20 | (83.3 | )% | ||||||||||||
Total International Revenues | 43,348 | 54,794 | 41,376 | (11,446 | ) | (20.9 | )% | 13,418 | 32.4 | % | |||||||||||||||
Worldwide | |||||||||||||||||||||||||
DEFINITY | 217,508 | 183,073 | 157,268 | 34,435 | 18.8 | % | 25,805 | 16.4 | % | ||||||||||||||||
TechneLite | 86,592 | 98,858 | 104,644 | (12,266 | ) | (12.4 | )% | (5,786 | ) | (5.5 | )% | ||||||||||||||
Other nuclear | 59,805 | 74,284 | 78,380 | (14,479 | ) | (19.5 | )% | (4,096 | ) | (5.2 | )% | ||||||||||||||
Rebates and allowances | (16,568 | ) | (12,841 | ) | (8,914 | ) | (3,727 | ) | 29.0 | % | (3,927 | ) | 44.1 | % | |||||||||||
Total Revenues | $ | 347,337 | $ | 343,374 | $ | 331,378 | $ | 3,963 | 1.2 | % | $ | 11,996 | 3.6 | % | |||||||||||
2019 vs. 2018
The increase in U.S. segment revenues during the year ended December 31, 2019, as compared to the prior year is primarily due to a $33.3 million increase in DEFINITY revenue as a result of higher unit volume. This increase was offset, in part, by decreases primarily associated with lower Xenon and other nuclear product volume, an increase in rebate and allowance provisions and lower TechneLite revenue driven by temporary supplier disruptions.
The decrease in International segment revenues during the year ended December 31, 2019, as compared to the prior year is primarily due to a decrease of $10.8 million in TechneLite revenue primarily driven by opportunistic incremental demand in the prior year period and temporary supplier disruptions in the current period, lower volumes of other nuclear products and a negative exchange rate impact of approximately $0.4 million, offset in part, by higher DEFINITY revenue driven by increased volume.
Rebates and Allowances
Estimates for rebates and allowances represent our estimated obligations under contractual arrangements with third parties. Rebate accruals and allowances are recorded in the same period the related revenue is recognized, resulting in a reduction to revenue and the establishment of a liability which is included in accrued expenses. These rebates and allowances result from performance-based offers that are primarily based on attaining contractually specified sales volumes and growth, Medicaid rebate programs for our products, administrative fees of group purchasing organizations and certain distributor related commissions. The calculation of the accrual for these rebates and allowances is based on an estimate of the third-party’s buying patterns and the resulting applicable contractual rebate to be earned over a contractual period.
An analysis of the amount of, and change in, reserves is summarized as follows:
(in thousands) | Rebates and Allowances | ||
Balance, January 1, 2019 | $ | 4,654 | |
Provision related to current period revenues | 16,729 | ||
Adjustments relating to prior period revenues | (161 | ) | |
Payments or credits made during the period | (14,237 | ) | |
Balance, December 31, 2019 | $ | 6,985 | |
59
Gross Profit
Gross profit is summarized by segment as follows:
Year Ended December 31, | 2019 vs. 2018 | 2018 vs. 2017 | |||||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | Change $ | Change % | Change $ | Change % | ||||||||||||||||||
U.S. | $ | 164,051 | $ | 161,760 | $ | 154,671 | $ | 2,291 | 1.4 | % | $ | 7,089 | 4.6 | % | |||||||||||
International | 10,760 | 13,125 | 7,464 | (2,365 | ) | (18.0 | )% | 5,661 | 75.8 | % | |||||||||||||||
Total Gross profit | $ | 174,811 | $ | 174,885 | $ | 162,135 | $ | (74 | ) | — | % | $ | 12,750 | 7.9 | % | ||||||||||
2019 vs. 2018
The increase in U.S. segment gross profit for the year ended December 31, 2019, as compared to the prior year is primarily attributable to higher DEFINITY unit volume. This was offset by lower TechneLite, Xenon and other nuclear product unit volume, as well as an increase in rebate and allowance provisions.
The decrease in International segment gross profit for the year ended December 31, 2019, as compared to the prior year is primarily attributable to lower volume of TechneLite and other nuclear products, offset in part, by higher DEFINITY gross profit driven by increased volume.
Sales and Marketing
Sales and marketing expenses consist primarily of salaries and other related costs for personnel in field sales, marketing and customer service functions. Other costs in sales and marketing expenses include the development and printing of advertising and promotional material, professional services, market research and sales meetings.
Sales and marketing expense is summarized by segment as follows:
Year Ended December 31, | 2019 vs. 2018 | 2018 vs. 2017 | |||||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | Change $ | Change % | Change $ | Change % | ||||||||||||||||||
U.S. | $ | 39,672 | $ | 40,579 | $ | 39,471 | $ | (907 | ) | (2.2 | )% | $ | 1,108 | 2.8 | % | ||||||||||
International | 2,216 | 2,580 | 2,844 | (364 | ) | (14.1 | )% | (264 | ) | (9.3 | )% | ||||||||||||||
Total Sales and marketing | $ | 41,888 | $ | 43,159 | $ | 42,315 | $ | (1,271 | ) | (2.9 | )% | $ | 844 | 2.0 | % | ||||||||||
2019 vs. 2018
The decrease in the U.S. segment sales and marketing expenses for the year ended December 31, 2019, as compared to the prior year period is primarily due to lower market research activities and employee-related costs.
The decrease in the International segment sales and marketing expenses for the for the year ended December 31, 2019, as compared to the prior year period is primarily due to lower employee-related costs.
General and Administrative
General and administrative expenses consist of salaries and other related costs for personnel in executive, finance, legal, information technology and human resource functions. Other costs included in general and administrative expenses are professional fees for information technology services, external legal fees, consulting and accounting services as well as bad debt expense, certain facility and insurance costs, including director and officer liability insurance.
General and administrative expense is summarized by segment as follows:
Year Ended December 31, | 2019 vs. 2018 | 2018 vs. 2017 | |||||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | Change $ | Change % | Change $ | Change % | ||||||||||||||||||
U.S. | $ | 60,752 | $ | 49,149 | $ | 49,269 | $ | 11,603 | 23.6 | % | $ | (120 | ) | (0.2 | )% | ||||||||||
International | 492 | 1,018 | 573 | (526 | ) | (51.7 | )% | 445 | 77.7 | % | |||||||||||||||
Total General and administrative | $ | 61,244 | $ | 50,167 | $ | 49,842 | $ | 11,077 | 22.1 | % | $ | 325 | 0.7 | % | |||||||||||
60
2019 vs. 2018
The increase in U.S. segment general and administrative expenses for the year ended December 31, 2019, as compared to the prior year is driven primarily by an increase in acquisition-related costs associated with the pending acquisition of Progenics and higher employee-related costs. This increase was offset, in part, by lower campus consolidation and information technology costs as a result of prior year efficiency projects.
The International segment general and administrative expenses decreased for the year ended December 31, 2019, as compared to the prior year, driven primarily by an insurance benefit received in the current period.
Research and Development
Research and development expenses relate primarily to the development of new products to add to our portfolio and costs related to our medical affairs, medical information and regulatory functions. We do not allocate research and development expenses incurred in the U.S. to our International segment.
Research and development expense is summarized by segment as follows:
Year Ended December 31, | 2019 vs. 2018 | 2018 vs. 2017 | |||||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | Change $ | Change % | Change $ | Change % | ||||||||||||||||||
U.S. | $ | 19,352 | $ | 15,705 | $ | 16,692 | $ | 3,647 | 23.2 | % | $ | (987 | ) | (5.9 | )% | ||||||||||
International | 666 | 1,366 | 1,433 | (700 | ) | (51.2 | )% | (67 | ) | (4.7 | )% | ||||||||||||||
Total Research and development | $ | 20,018 | $ | 17,071 | $ | 18,125 | $ | 2,947 | 17.3 | % | $ | (1,054 | ) | (5.8 | )% | ||||||||||
2019 vs. 2018
The increase in U.S. segment research and development expenses for the year ended December 31, 2019, as compared to the prior year is primarily attributable to clinical research expenses related to DEFINITY studies, a one-time payment relating to a collaboration and license agreement entered into in Q2 2019, and higher employee-related costs.
The decrease in the International segment research and development expenses for the year ended December 31, 2019, as compared to the prior year period is driven by a European Phase 4 study for one of our products in the prior year.
Interest Expense
Interest expense for the year ended December 31, 2019 decreased $3.8 million as compared to the prior year period due to the refinancing of our existing indebtedness.
Loss on Extinguishment of Debt
During the year ended December 31, 2019, we incurred a $3.2 million loss on extinguishment of debt in connection with the refinancing of our existing indebtedness.
Other Expense (Income)
Other expense (income) changed by $8.7 million for the year ended December 31, 2019 as compared to the prior year, due to the reduction of indemnified receivables related to the release of our uncertain tax positions. The offset was recorded in income tax (benefit) expense. Refer to Note 5, Income Taxes. This expense, in part, was offset by the impact of proceeds received related to an arbitration award and an increase in interest income.
Income Tax (Benefit) Expense
Income tax (benefit) expense is summarized as follows:
Year Ended December 31, | 2019 vs. 2018 | 2018 vs. 2017 | |||||||||||||||||||||||
(in thousands) | 2019 | 2018 | 2017 | Change $ | Change % | Change $ | Change % | ||||||||||||||||||
Income tax (benefit) expense | $ | (3,040 | ) | $ | 9,030 | $ | (83,746 | ) | $ | (12,070 | ) | (133.7 | )% | $ | 92,776 | (110.8 | )% | ||||||||
61
The income tax benefit for the year ended December 31, 2019 was primarily due to the benefits arising from the release of uncertain tax positions and stock compensation deductions, netted with the tax effect on income generated in the period and the accrual of interest associated with uncertain tax positions. In accordance with the Company’s accounting policy, the change in the tax liability, penalties and interest associated with these uncertain tax positions (net of any offsetting federal or state benefit) is recognized within income tax (benefit) expense. Contemporaneously, changes in the tax indemnification receivable are recognized within other expense (income) in the consolidated statement of operations. Accordingly, as these reserves change, adjustments are included in income tax (benefit) expense with an offsetting adjustment included in other expense (income). Assuming that the receivable from BMS continues to be considered recoverable by the Company, there will be no effect on net income and no net cash outflows related to these liabilities. Refer to Note 5, Income Taxes.
The income tax expense for the year ended December 31, 2018 was primarily due to the income generated in the period and the accrual of interest associated with uncertain tax positions, offset by the release of the valuation allowance against our Canada deferred tax assets and tax benefits arising from stock compensation deductions.
We regularly assess our ability to realize our deferred tax assets. Assessing the realizability of deferred tax assets requires significant management judgment. In determining whether our deferred tax assets are more-likely-than-not realizable, we evaluate all available positive and negative evidence, and weigh the objective evidence and expected impact. We released the full valuation allowance recorded against our Canada deferred tax assets during the year ended December 31, 2018. We continue to record a valuation allowance against certain of our foreign net deferred tax assets.
Our effective tax rate for each reporting period is presented as follows:
Year Ended December 31, | |||||
2019 | 2018 | 2017 | |||
Effective tax rate | (10.6)% | 18.2% | (211.3)% | ||
Our effective tax rate in fiscal 2019 differs from the U.S. statutory rate of 21% principally due to the release of uncertain tax positions and stock compensation deductions, offset by the impact of U.S. state taxes and the accrual of interest on uncertain tax positions.
The decrease in the effective income tax rate for the year ended December 31, 2019 as compared to the prior year period is primarily due to the release of uncertain tax positions in the current period.
Comparison of the Periods Ended December 31, 2018 and 2017
For a comparison of our results of operations for the fiscal years ended December 31, 2018 and December 31, 2017, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our Annual Report on Form 10-K for the fiscal year ended December 31, 2018, filed with the SEC on February 20, 2019.
Liquidity and Capital Resources
Cash Flows
The following table provides information regarding our cash flows:
Year Ended December 31, | ||||||||||||
(in thousands) | 2019 | 2018 | 2017 | |||||||||
Net cash provided by operating activities | $ | 80,384 | $ | 61,193 | $ | 54,777 | ||||||
Net cash used in investing activities | $ | (22,061 | ) | $ | (19,132 | ) | $ | (16,309 | ) | |||
Net cash used in financing activities | $ | (78,881 | ) | $ | (4,668 | ) | $ | (13,450 | ) | |||
For a discussion of our liquidity and capital resources related to our cash flow activities for the fiscal year ended December 31, 2017, see “Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our annual report on Form 10-K for the fiscal year ended December 31, 2018, filed with the SEC on February 20, 2019.
62
Net Cash Provided by Operating Activities
Net cash provided by operating activities of $80.4 million in the year ended December 31, 2019 was driven primarily by net income of $31.7 million plus $13.4 million of depreciation, amortization and accretion expense, changes in long-term income tax payable and other long-term liabilities of $13.2 million, stock-based compensation expense of $12.5 million, changes in long-term income tax receivable of $10.6 million, changes in deferred taxes of $9.7 million and debt extinguishment expense of $3.2 million. These net sources of cash were further increased by a net increase of $9.0 million related to movements in our working capital accounts during the period. The overall increases in cash from our working capital accounts were primarily driven by accrued expenses and the timing of purchases.
Net cash provided by operating activities of $61.2 million in the year ended December 31, 2018 was driven primarily by net income of $40.5 million plus $13.9 million of depreciation, amortization and accretion expense, $8.7 million of stock-based compensation expense and changes in deferred taxes of $5.8 million. These net sources of cash were offset by a net decrease of $14.0 million related to movements in our working capital accounts during the period. The overall decreases in cash from our working capital accounts were primarily driven by the strategic inventory build during the period to mitigate sole supplier risk as well as higher accounts receivable due to increased sales.
Net Cash Used in Investing Activities
Net cash used in investing activities during the year ended December 31, 2019 reflected $22.1 million in capital expenditures.
Net cash used in investing activities during the year ended December 31, 2018 reflected $20.1 million in capital expenditures offset by the cash proceeds of $1.0 million received from the sale of land.
Net Cash Used in Financing Activities
Net cash used in financing activities during the year ended December 31, 2019 is primarily attributable to the net cash outflow of approximately $73 million in connection with the refinancing of our previous 2017 Facility, payments on long-term debt of $5.0 million related to the 2019 Term Facility and payments for minimum statutory tax withholding related to net share settlement of equity awards of $2.5 million. Starting in 2019, we require certain senior executives to cover tax liabilities resulting from the vesting of their equity awards pursuant to sell-to-cover transactions under 10b5-1 plans.
Net cash used in financing activities during the year ended December 31, 2018 reflected payments for minimum statutory tax withholding related to net share settlement of equity awards of $3.4 million, payments on long-term debt of $2.9 million, offset by proceeds of $1.2 million from the exercise of stock options.
External Sources of Liquidity
In June 2019, we refinanced our 2017 $275 million five-year term loan facility with the 2019 Term Facility. In addition, we replaced our $75 million revolving facility with the 2019 Revolving Facility. The terms of the 2019 Facility are set forth in the Credit Agreement, dated as of June 27, 2019, by and among us, the lenders from time to time party thereto and Wells Fargo Bank, N.A., as administrative agent and collateral agent. We have the right to request an increase to the 2019 Term Facility or request the establishment of one or more new incremental term loan facilities, in an aggregate principal amount of up to $100 million, plus additional amounts, in certain circumstances.
We are permitted to voluntarily prepay the 2019 Term Loans, in whole or in part, without premium or penalty. The 2019 Term Facility requires us to make mandatory prepayments of the outstanding 2019 Term Loans in certain circumstances. The 2019 Term Facility amortizes at 5.00% per year through September 30, 2022 and 7.5% thereafter, until its June 27, 2024 maturity date.
Under the terms of the 2019 Revolving Facility, the lenders thereunder agreed to extend credit to us from time to time until June 27, 2024 consisting of revolving loans in an aggregate principal amount not to exceed $200 million at any time outstanding. The 2019 Revolving Facility includes a $20 million sub-facility for the issuance of Letters of Credit. The 2019 Revolving Facility includes a $10 million sub-facility for Swingline Loans. The Letters of Credit, Swingline Loans and the borrowings under the 2019 Revolving Facility are expected to be used for working capital and other general corporate purposes.
Please refer to Note 11, Long-term debt, net and other borrowings, for further details on the 2019 Facility.
Our ability to fund our future capital needs will be affected by our ability to continue to generate cash from operations and may be affected by our ability to access the capital markets, money markets or other sources of funding, as well as the capacity and terms of our financing arrangements.
63
We may from time to time repurchase or otherwise retire our debt and take other steps to reduce our debt or otherwise improve our balance sheet. These actions may include prepayments of our term loans or other retirements or refinancing of outstanding debt, privately negotiated transactions or otherwise. The amount of debt that may be retired, if any, could be material and would be decided at the sole discretion of our Board of Directors and will depend on market conditions, our cash position and other considerations.
Funding Requirements
Our future capital requirements will depend on many factors, including:
• | The costs of acquiring or in-licensing, developing, obtaining regulatory approval for, and commercializing, new products, businesses or technologies, together with the costs of pursuing opportunities that are not eventually consummated; |
• | The pricing environment and the level of product sales of our currently marketed products, particularly DEFINITY and any additional products that we may market in the future; |
• | Revenue mix shifts and associated volume and selling price changes that could result from contractual status changes with key customers and additional competition; |
• | Our investment in the further clinical development and commercialization of existing products and development candidates; |
• | The costs of investing in our facilities, equipment and technology infrastructure; |
• | The costs and timing of establishing manufacturing and supply arrangements for commercial supplies of our products and raw materials and components; |
• | Our ability to have product manufactured and released from JHS and other manufacturing sites in a timely manner in the future; |
• | The costs of further commercialization of our existing products, particularly in international markets, including product marketing, sales and distribution and whether we obtain local partners to help share such commercialization costs; |
• | The extent to which we choose to establish collaboration, co-promotion, distribution or other similar arrangements for our marketed products; |
• | The legal costs relating to maintaining, expanding and enforcing our intellectual property portfolio, pursuing insurance or other claims and defending against product liability, regulatory compliance or other claims; and |
• | The cost of interest on any additional borrowings which we may incur under our financing arrangements. |
Until we successfully become dual sourced for our principal products, we are vulnerable to future supply shortages. Disruption in our financial performance could also occur if we experience significant adverse changes in product or customer mix, broad economic downturns, adverse industry or company conditions or catastrophic external events, including natural disasters and political or military conflict. If we experience one or more of these events in the future, we may be required to implement expense reductions, such as a delay or elimination of discretionary spending in all functional areas, as well as scaling back select operating and strategic initiatives.
If our capital resources become insufficient to meet our future capital requirements, we would need to finance our cash needs through public or private equity offerings, debt financings, assets securitizations, sale-leasebacks or other financing or strategic alternatives, to the extent such transactions are permissible under the covenants of our Credit Agreement. Additional equity or debt financing, or other transactions, may not be available on acceptable terms, if at all. If any of these transactions require an amendment or waiver under the covenants in our Credit Agreement, which could result in additional expenses associated with obtaining the amendment or waiver, we will seek to obtain such a waiver to remain in compliance with those covenants. However, we cannot be assured that such an amendment or waiver would be granted, or that additional capital will be available on acceptable terms, if at all.
At December 31, 2019, our only current committed external source of funds is our borrowing availability under our 2019 Revolving Facility. We had $92.9 million of cash and cash equivalents at December 31, 2019. Our 2019 Facility contains a number of affirmative, negative, reporting and financial covenants, in each case subject to certain exceptions and materiality thresholds. Incremental borrowings under the 2019 Revolving Facility may affect our ability to comply with the covenants in the 2019 Facility, including the financial covenants restricting consolidated net leverage and interest coverage. Accordingly, we may be limited in utilizing the full amount of our 2019 Revolving Facility as a source of liquidity.
In addition, in connection with the Progenics Transaction, which we expect to close in the second quarter of 2020, although the merger is structured as a stock-for-stock exchange, we will incur legal, accounting, financial advisory, consulting and printing fees, and transition, integration and other costs which we intend to fund from our available cash and the available cash of Progenics. The
64
CVRs we will issue in the Progenics Transaction will entitle holders thereof to future cash payments of 40% of PyL net sales over $100 million in 2022 and $150 million in 2023, which, if payable, we currently intend to fund from our then-available cash.
Based on our current operating plans, we believe that our existing cash and cash equivalents, results of operations and availability under our 2019 Revolving Facility will be sufficient to continue to fund our liquidity requirements for the foreseeable future.
Contractual Obligations
Contractual obligations represent future cash commitments and liabilities under agreements with third parties and exclude contingent contractual liabilities for which we cannot reasonably predict future payment, including contingencies related to potential future development, financing, certain suppliers, contingent royalty payments and/or scientific, regulatory, or commercial milestone payments under development agreements. The following table summarizes our contractual obligations as of December 31, 2019:
Payments Due by Period | |||||||||||||||||||
(in thousands) | Total | Less than 1 Year | 1 - 3 Years | 3 -5 Years | More than 5 Years | ||||||||||||||
Debt obligations (principal) | $ | 195,000 | $ | 10,000 | $ | 21,250 | $ | 163,750 | $ | — | |||||||||
Interest on debt obligations(a) | 27,334 | 6,872 | 12,622 | 7,840 | — | ||||||||||||||
Operating lease obligations(b) | 1,130 | 238 | 476 | 416 | — | ||||||||||||||
Purchase obligations(c) | 10,330 | 4,132 | 6,198 | — | — | ||||||||||||||
Finance lease obligations | 354 | 135 | 219 | — | — | ||||||||||||||
Other long-term liabilities(d) | — | — | — | — | — | ||||||||||||||
Asset retirement obligations(e) | — | — | — | — | — | ||||||||||||||
Total contractual obligations | $ | 234,148 | $ | 21,377 | $ | 40,765 | $ | 172,006 | $ | — | |||||||||
________________________________
(a) | Amounts relate to the estimated interest under our 2019 Term Facility based on interest rates in effect as of December 31, 2019. |
(b) | Operating leases include minimum payments under leases for our facilities. |
(c) | Excludes purchase orders for inventory in the normal course of business. |
(d) | Our other long-term liabilities in the consolidated balance sheet include unrecognized tax benefits and related interest and penalties. As of December 31, 2019, we had unrecognized tax benefits of $27.0 million, which included interest and penalties, classified as noncurrent liabilities. At this time, we are unable to make a reasonably reliable estimate of the timing of payments in individual years in connection with these tax liabilities; therefore, such amounts are not included in the above contractual obligation table. |
(e) | We have excluded asset retirement obligations from the table above due to the uncertainty of the timing of the future cash outflows related to the decommissioning of our radioactive operations. As of December 31, 2019, the liability, which was approximately $12.9 million, was measured at the present value of the obligation expected to be incurred of approximately $26.9 million. |
Off-Balance Sheet Arrangements
We are required to provide the U.S. Nuclear Regulatory Commission and Massachusetts Department of Public Health financial assurance demonstrating our ability to fund the decommissioning of our North Billerica, Massachusetts production facility upon closure, though we do not intend to close the facility. We have provided this financial assurance in the form of a $28.2 million surety bond.
Since inception, we have not engaged in any other off-balance sheet arrangements, including structured finance, special purpose entities or variable interest entities.
Effects of Inflation
We do not believe that inflation has had a significant impact on our revenues or results of operations since inception. We expect our cost of product sales and other operating expenses will change in the future in line with periodic inflationary changes in price levels. Because we intend to retain and continue to use our property and equipment, we believe that the incremental inflation related to the replacement costs of those items will not materially affect our operations. However, the rate of inflation affects our expenses, such as those for employee compensation and contract services, which could increase our level of expenses and the rate at which we use our resources. While we generally believe that we will be able to offset the effect of price-level changes by adjusting our product prices and implementing operating efficiencies, any material unfavorable changes in price levels could have a material adverse effect on our financial condition, results of operations and cash flows.
65
Recent Accounting Standards
Refer to Note 2, “Summary of Significant Accounting Policies,” in the accompanying consolidated financial statements located under Item 8 of this Annual Report on Form 10-K for information regarding recently issued accounting standards that may have a significant impact on our business.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these consolidated financial statements require us to make estimates and judgments that affect our reported assets and liabilities, revenues and expenses, and other financial information. Actual results may differ materially from these estimates under different assumptions and conditions. In addition, our reported financial condition and results of operations could vary due to a change in the application of a particular accounting standard.
We believe the following represent our critical accounting policies and estimates used in the preparation of our financial statements.
Revenue from Contracts with Customers
We adopted ASC 606 on January 1, 2018 using the modified retrospective method for all contracts not completed as of the date of adoption. The reported results for 2019 and 2018 reflect the application of ASC 606 guidance while the reported results for 2017 were prepared under the guidance of ASC 605, Revenue Recognition (“ASC 605”). For our accounting policy for revenue recognition under ASC 605, refer to Item 8 of the Annual Report on Form 10-K for the year ended December 31, 2017. The adoption of ASC 606 did not have a material impact on our consolidated balance sheet, results of operations, equity or cash flows as of the adoption date or for the periods presented.
Revenue is measured based on a consideration specified in a contract with a customer, and excludes any sales incentives and amounts collected on behalf of third parties. We recognize revenue when we satisfy our performance obligations by transferring control over products or services to our customers. The amount of revenue we recognize reflects the consideration to which we expect to be entitled to receive in exchange for these goods or services. To achieve this core principle, we apply the following five steps: (1) identify the contracts with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) we satisfy performance obligations.
We derive our revenues through arrangements with customers for product sales as well as licensing and royalty arrangements. We sell our products principally to hospitals and clinics, radiopharmacies, and distributors and we consider customer purchase orders, which in some cases are governed by master sales or group purchasing organization agreements, to be contracts with our customers. In addition to these arrangements, we also enter into licensing agreements under which we license certain rights to third parties. The terms of these arrangements typically include payment to us of one or more of the following: non-refundable, up-front license fees; development, regulatory and commercial milestone payments; and royalties on net sales of licensed products. We analyze various factors requiring management judgment when applying the five-step model to our contracts with customers.
Our product revenues are recorded at the net sales price (transaction price), which represents our sales price less estimates related to reserves which are established for items such as discounts, returns, rebates and allowances that may be provided for in certain contracts with our customers. Judgment is used in determining and updating our reserves on an on-going basis, and where appropriate, these estimates take into consideration a range of possible outcomes which are probability-weighted for relevant factors such as our historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the terms of the contract. Actual amounts of consideration ultimately received may differ from the Company’s estimates.
For our licensing and royalty arrangements, we use judgment in determining the number of performance obligations in a license agreement by assessing whether the license is distinct or should be combined with another performance obligation as well as the nature of the license. As part of the accounting for these arrangements, we develop assumptions that require judgment to determine the stand-alone selling price for each performance obligation identified in a contract. These key assumptions may include market conditions, reimbursement rates for personnel costs, development timelines and probabilities of regulatory success.
66
Income Taxes
We account for income taxes using an asset and liability approach. Income tax (benefit) expense represents income taxes paid or payable for the current year plus the change in deferred taxes during the year. Deferred taxes result from differences between the financial and tax bases of our assets and liabilities. Deferred tax assets and liabilities are measured using the currently enacted tax rates that apply to taxable income in effect for the years in which those tax attributes are expected to be recovered or paid, and are adjusted for changes in tax rates and tax laws when such changes are enacted.
On December 22, 2017, the United States enacted the Tax Cuts and Jobs Act of 2017 (the “Act”). The Act is significant and has wide-ranging effects. The primary material impact to the Company was on our net U.S. deferred tax assets, which were reduced as a result of the reduction in U.S. corporate tax rates from 35% to 21% for years beginning on or after January 1, 2018. We recorded tax expense of $45.1 million during the year ended December 31, 2017, to reflect the impact of the Act on our net deferred tax assets carrying value. We have reviewed the guidance issued by the U.S. Treasury concerning the repatriation transition tax. The repatriation transition tax impacted U.S. entities with accumulated yet unrepatriated or 'untaxed' foreign earnings. As of December 31, 2017, we had no accumulated unrepatriated foreign earnings, and therefore were not affected by the new provisions of the Act concerning the repatriation transition tax.
We regularly assess our ability to realize our deferred tax assets, and that assessment requires significant management judgment. In determining whether our deferred tax assets are more-likely-than-not realizable, we evaluate all available positive and negative evidence, and weigh that evidence based on its objective verifiability and expected impact.
During the fourth quarter of 2017, we determined, based on our consideration of the weight of positive and negative evidence, that there was sufficient positive evidence that our U.S. federal and state deferred tax assets were more-likely-than-not realizable as of December 31, 2017. Our conclusion was primarily driven by the achievement of a sustained level of profitability, the expectation of sustained future profitability, and mitigating factors related to external supplier and customer risk sufficient to outweigh the available negative evidence. Accordingly, we released the valuation allowance previously recorded against our U.S. net deferred tax assets resulting in an income tax benefit of $141.1 million. We have continued to assess the level of the valuation allowance required and if the weight of negative evidence exists in future periods to again support the recording of a partial or full valuation allowance against our U.S. deferred tax assets, that would likely have a material negative impact on our results of operations in that future period.
During the fourth quarter of 2018, we further determined that there was sufficient positive evidence that our Canada deferred tax assets were more-likely-than-not realizable as of December 31, 2018. Our conclusion was primarily driven by the achievement of a sustained level of profitability and the expectation of sustained future profitability. Accordingly, we released the valuation allowance previously recorded against our Canada net deferred tax assets resulting in an income tax benefit of $4.0 million. We continue to maintain a valuation allowance of $1.2 million on foreign net deferred tax assets generated where there is still an insufficient history of cumulative profitability in the relevant jurisdiction.
We account for uncertain tax positions using a recognition threshold and measurement analysis method for determining the financial statement impact of uncertain tax positions taken or expected to be taken in a tax return. Differences between tax positions taken in a tax return and amounts recognized in the financial statements are recorded as adjustments to income taxes payable or receivable, or adjustments to deferred taxes, or both. We record the related interest and penalties to income tax (benefit) expense.
We have a tax indemnification agreement with BMS related to certain uncertain tax positions that arose prior to the acquisition of the business from BMS. The uncertain tax positions are recognized as long-term liabilities, and a tax indemnification receivable is recognized within other long-term assets. Changes in the tax indemnification receivable are recognized within other expense (income) in the consolidated statements of operations, and changes in the liabilities are recorded within the tax provision. Accordingly, as these reserves change, adjustments are included in income tax (benefit) expense with an offsetting adjustment included in other expense (income). Assuming that we continue to consider the receivable from BMS to be fully recoverable, there is no net effect on net income related to these liabilities and no net cash outflows.
During the fourth quarter of 2019, we reassessed the indemnified uncertain tax positions and obtained, with the assistance of third-party tax experts, additional technical insights with respect to the indemnified uncertain tax positions. On the basis of new information, we changed our estimate with respect to some of the indemnified uncertain tax positions. For the year ended December 31, 2019, we released $17.1 million of the liability for uncertain tax positions, including $12.7 million of accrued interest and penalties, recorded to income tax (benefit) expense, offset by a reduction in deferred tax assets of $3.3 million and a $13.8 million reduction of the indemnification receivable recorded in other expense (income).
The calculation of our uncertain tax positions involves certain estimates, assumptions and the application of complex tax regulations in multiple jurisdictions worldwide. Any material change in our estimates or assumptions, or the tax regulations, may have a material impact on our results of operations.
67
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
We are exposed to market risk from changes in interest rates and foreign currency exchange rates. Beginning in 2019, we may from time to time use derivative financial instruments or other financial instruments to hedge these economic exposures related to foreign currencies. We do not hold or issue financial instruments for trading purposes.
Interest Rate Risk
Under our 2019 Facility, we have substantial variable rate debt. Fluctuations in interest rates may affect our business, financial condition, results of operations and cash flows. As of December 31, 2019, we had $195.0 million outstanding principal under our 2019 Term Facility with variable interest rates.
Furthermore, we are subject to interest rate risk in connection with our 2019 Revolving Facility, which is variable rate indebtedness. Interest rate changes could increase the amount of our interest payments and thus negatively impact our future earnings and cash flows. As of December 31, 2019, there was availability of $200.0 million on the 2019 Revolving Facility. Any increase in the interest rate under the 2019 Revolving Facility may have a negative impact on our future earnings to the extent we have outstanding borrowings under the 2019 Revolving Facility. The effect of a 100 basis points adverse change in market interest rates on our 2019 Term Facility, in excess of applicable minimum floors, on our interest expense would be approximately $2.4 million.
Historically, we have not used derivative financial instruments or other financial instruments to hedge such economic exposures.
Foreign Currency Risk
We face exposure to movements in foreign currency exchange rates whenever we, or any of our subsidiaries, enter into transactions with third parties that are denominated in currencies other than ours, or that subsidiary’s, functional currency. Intercompany transactions between entities that use different functional currencies also expose us to foreign currency risk.
During the years ended December 31, 2019, 2018 and 2017, the net impact of foreign currency changes on transactions was a gain of less than $0.1 million, a loss of $0.6 million and a gain of $0.3 million, respectively. In 2019, we have entered into foreign currency forward contracts primarily to reduce the effects of fluctuating foreign currency exchange rates. We may enter into additional foreign currency forward contracts when deemed appropriate. We do not enter into foreign currency forward contracts for speculative or trading purposes.
The Canadian dollar presents the primary currency risk on our earnings. At December 31, 2019, a hypothetical 10% change in value of the U.S. dollar relative to the Canadian dollar would not have materially affected our financial instruments.
68
Item 8. Financial Statements and Supplementary Data
LANTHEUS HOLDINGS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Page | |
69
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of
Lantheus Holdings, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Lantheus Holdings, Inc. and subsidiaries (the "Company") as of December 31, 2019 and 2018, the related consolidated statements of operations, comprehensive income, stockholders’ equity (deficit), and cash flows, for each of the three years in the period ended December 31, 2019, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 25, 2020, expressed an unqualified opinion on the Company's internal control over financial reporting.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the US federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current-period audit of the financial statements that was communicated or required to be communicated to the audit committee and that (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Asset Retirement Obligations - Refer to Notes 2 and 8 to the financial statements
Critical Audit Matter Description
The Company records asset retirement obligations associated with relevant federal, state, local, and foreign environmental laws and regulations that may require the Company to remove or mitigate the effects of the disposal or release of chemical substances in jurisdictions where the Company does business or maintains properties. The Company establishes accruals when those costs are legally obligated and can be reasonably estimated. The asset retirement obligations are estimated, which may include the assistance of third-party environmental specialists, and are based on currently available information, regulatory requirements, remediation strategies, historical experience, the relative shares of the total remediation costs, a relevant discount rate, and the time periods of when estimated costs can be reasonably predicted. The asset retirement obligations balance was $12.9 million as of December 31, 2019.
We identified asset retirement obligations related to the decommissioning of certain facilities as a critical audit matter because the expected costs involve significant estimation by management in order to comply with relevant regulatory requirements. This required a high degree of auditor judgement and an increased extent of effort, including the need to involve environmental specialists.
70
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to the asset retirement obligations included the following, among others:
• | We tested the effectiveness of controls related to the determination of asset retirement obligations, including management’s controls over the review of the expected decommissioning costs used in the determination of the asset retirement obligations. |
• | We evaluated the methods and assumptions used by management to estimate the expected decommissioning costs used in the determination of the asset retirement obligations. |
• | We made inquiries of management regarding the relevant regulatory requirements. |
• | With the assistance of auditor specialists who have expertise in environmental matters and specialized skills and training, we: |
– | Evaluated the relevant professional experience of management’s third-party environmental specialist. |
– | Evaluated the completeness of the expected decommissioning costs by conducting a search of new or revised relevant regulatory requirements that would impact the Company’s cost estimate. |
– | Evaluated the accuracy of management’s methods and assumptions to estimate the cost to remove, mitigate, or remediate the effects of the disposal or release of chemical substances through comparison of the expected decommissioning costs with the relevant regulatory requirements. |
/s/ Deloitte & Touche LLP |
Boston, Massachusetts |
February 25, 2020 |
We have served as the Company’s auditor since 2007.
71
Lantheus Holdings, Inc.
Consolidated Balance Sheets
(in thousands, except par value)
December 31, | |||||||
2019 | 2018 | ||||||
Assets | |||||||
Current assets | |||||||
Cash and cash equivalents | $ | 92,919 | $ | 113,401 | |||
Accounts receivable, net | 43,529 | 43,753 | |||||
Inventory | 29,180 | 33,019 | |||||
Other current assets | 7,283 | 5,242 | |||||
Total current assets | 172,911 | 195,415 | |||||
Property, plant and equipment, net | 116,497 | 107,888 | |||||
Intangibles, net | 7,336 | 9,133 | |||||
Goodwill | 15,714 | 15,714 | |||||
Deferred tax assets, net | 71,834 | 81,449 | |||||
Other long-term assets | 21,627 | 30,232 | |||||
Total assets | $ | 405,919 | $ | 439,831 | |||
Liabilities and stockholders’ equity | |||||||
Current liabilities | |||||||
Current portion of long-term debt and other borrowings | $ | 10,143 | $ | 2,750 | |||
Accounts payable | 18,608 | 17,955 | |||||
Accrued expenses and other liabilities | 37,360 | 32,050 | |||||
Total current liabilities | 66,111 | 52,755 | |||||
Asset retirement obligations | 12,883 | 11,572 | |||||
Long-term debt, net and other borrowings | 183,927 | 263,709 | |||||
Other long-term liabilities | 28,397 | 40,793 | |||||
Total liabilities | 291,318 | 368,829 | |||||
Commitments and contingencies (see Note 15) | |||||||
Stockholders’ equity | |||||||
Preferred stock ($0.01 par value, 25,000 shares authorized; no shares issued and outstanding) | — | — | |||||
Common stock ($0.01 par value, 250,000 shares authorized; 39,251 and 38,466 shares issued and outstanding, respectively) | 393 | 385 | |||||
Additional paid-in capital | 251,641 | 239,865 | |||||
Accumulated deficit | (136,473 | ) | (168,140 | ) | |||
Accumulated other comprehensive loss | (960 | ) | (1,108 | ) | |||
Total stockholders’ equity | 114,601 | 71,002 | |||||
Total liabilities and stockholders’ equity | $ | 405,919 | $ | 439,831 | |||
The accompanying notes are an integral part of these consolidated financial statements.
72
Lantheus Holdings, Inc.
Consolidated Statements of Operations
(in thousands, except per share data)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Revenues | $ | 347,337 | $ | 343,374 | $ | 331,378 | |||||
Cost of goods sold | 172,526 | 168,489 | 169,243 | ||||||||
Gross profit | 174,811 | 174,885 | 162,135 | ||||||||
Operating expenses | |||||||||||
Sales and marketing | 41,888 | 43,159 | 42,315 | ||||||||
General and administrative | 61,244 | 50,167 | 49,842 | ||||||||
Research and development | 20,018 | 17,071 | 18,125 | ||||||||
Total operating expenses | 123,150 | 110,397 | 110,282 | ||||||||
Operating income | 51,661 | 64,488 | 51,853 | ||||||||
Interest expense | 13,617 | 17,405 | 18,410 | ||||||||
Loss on extinguishment of debt | 3,196 | — | 2,442 | ||||||||
Other expense (income) | 6,221 | (2,465 | ) | (8,638 | ) | ||||||
Income before income taxes | 28,627 | 49,548 | 39,639 | ||||||||
Income tax (benefit) expense | (3,040 | ) | 9,030 | (83,746 | ) | ||||||
Net income | $ | 31,667 | $ | 40,518 | $ | 123,385 | |||||
Net income per common share: | |||||||||||
Basic | $ | 0.81 | $ | 1.06 | $ | 3.31 | |||||
Diluted | $ | 0.79 | $ | 1.03 | $ | 3.17 | |||||
Weighted-average common shares outstanding: | |||||||||||
Basic | 38,988 | 38,233 | 37,276 | ||||||||
Diluted | 40,113 | 39,501 | 38,892 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
73
Lantheus Holdings, Inc.
Consolidated Statements of Comprehensive Income
(in thousands)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Net income | $ | 31,667 | $ | 40,518 | $ | 123,385 | |||||
Other comprehensive income (loss): | |||||||||||
Foreign currency translation | 148 | (74 | ) | (87 | ) | ||||||
Total other comprehensive income (loss) | 148 | (74 | ) | (87 | ) | ||||||
Comprehensive income | $ | 31,815 | $ | 40,444 | $ | 123,298 | |||||
The accompanying notes are an integral part of these consolidated financial statements.
74
Lantheus Holdings, Inc.
Consolidated Statements of Changes in Stockholders’ Equity (Deficit)
(in thousands)
Common Stock | Additional Paid-In Capital | Accumulated Deficit | Accumulated Other Comprehensive Loss | Total Stockholders’ Equity (Deficit) | |||||||||||||||||||
Shares | Amount | ||||||||||||||||||||||
Balance, January 1, 2017 | 36,756 | $ | 367 | $ | 226,462 | $ | (332,398 | ) | $ | (947 | ) | $ | (106,516 | ) | |||||||||
Net income | — | — | — | 123,385 | — | 123,385 | |||||||||||||||||
Other comprehensive loss | — | — | — | — | (87 | ) | (87 | ) | |||||||||||||||
Stock option exercises and employee stock plan purchases | 478 | 5 | 3,429 | — | — | 3,434 | |||||||||||||||||
Vesting of restricted stock awards | 744 | 8 | (8 | ) | — | — | — | ||||||||||||||||
Shares withheld to cover taxes | (214 | ) | (2 | ) | (2,851 | ) | — | — | (2,853 | ) | |||||||||||||
Stock-based compensation | — | — | 5,928 | — | — | 5,928 | |||||||||||||||||
Balance, December 31, 2017 | 37,765 | 378 | 232,960 | (209,013 | ) | (1,034 | ) | 23,291 | |||||||||||||||
Net income | — | — | — | 40,518 | — | 40,518 | |||||||||||||||||
Forfeiture of dividend equivalent right | — | — | — | 355 | — | 355 | |||||||||||||||||
Other comprehensive loss | — | — | — | — | (74 | ) | (74 | ) | |||||||||||||||
Stock option exercises and employee stock plan purchases | 223 | 2 | 1,578 | — | — | 1,580 | |||||||||||||||||
Vesting of restricted stock awards | 672 | 7 | (7 | ) | — | — | — | ||||||||||||||||
Shares withheld to cover taxes | (194 | ) | (2 | ) | (3,384 | ) | — | — | (3,386 | ) | |||||||||||||
Stock-based compensation | — | — | 8,718 | — | — | 8,718 | |||||||||||||||||
Balance, December 31, 2018 | 38,466 | 385 | 239,865 | (168,140 | ) | (1,108 | ) | 71,002 | |||||||||||||||
Net income | — | — | — | 31,667 | — | 31,667 | |||||||||||||||||
Other comprehensive income | — | — | — | — | 148 | 148 | |||||||||||||||||
Stock option exercises and employee stock plan purchases | 95 | 1 | 1,745 | — | — | 1,746 | |||||||||||||||||
Vesting of restricted stock awards and units | 796 | 8 | (8 | ) | — | — | — | ||||||||||||||||
Shares withheld to cover taxes | (106 | ) | (1 | ) | (2,453 | ) | — | — | (2,454 | ) | |||||||||||||
Stock-based compensation | — | — | 12,492 | — | — | 12,492 | |||||||||||||||||
Balance, December 31, 2019 | 39,251 | $ | 393 | $ | 251,641 | $ | (136,473 | ) | $ | (960 | ) | $ | 114,601 | ||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
75
Lantheus Holdings, Inc.
Consolidated Statements of Cash Flows
(in thousands)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Operating activities | |||||||||||
Net income | $ | 31,667 | $ | 40,518 | $ | 123,385 | |||||
Adjustments to reconcile net income to net cash flows from operating activities: | |||||||||||
Depreciation, amortization and accretion | 13,379 | 13,929 | 19,231 | ||||||||
Amortization of debt related costs | 978 | 1,279 | 1,361 | ||||||||
Provision for bad debt | 146 | 321 | 136 | ||||||||
Provision for excess and obsolete inventory | 1,851 | 2,875 | 1,215 | ||||||||
Stock-based compensation | 12,492 | 8,718 | 5,928 | ||||||||
Loss on impairment of land | — | — | 912 | ||||||||
Loss on extinguishment of debt | 3,196 | — | 2,442 | ||||||||
Deferred taxes | 9,725 | 5,762 | (86,946 | ) | |||||||
Long-term income tax receivable | 10,635 | (2,855 | ) | (8,413 | ) | ||||||
Long-term income tax payable and other long-term liabilities | (13,156 | ) | 3,219 | 2,793 | |||||||
Other | 422 | 1,399 | 1,049 | ||||||||
Increases (decreases) in cash from operating assets and liabilities: | |||||||||||
Accounts receivable | 156 | (3,985 | ) | (3,407 | ) | ||||||
Inventory | 1,994 | (8,690 | ) | (9,620 | ) | ||||||
Other current assets | (2,411 | ) | (661 | ) | (388 | ) | |||||
Accounts payable | 3,233 | (2,886 | ) | 604 | |||||||
Accrued expenses and other liabilities | 6,077 | 2,250 | 4,495 | ||||||||
Net cash provided by operating activities | 80,384 | 61,193 | 54,777 | ||||||||
Investing activities | |||||||||||
Capital expenditures | (22,061 | ) | (20,132 | ) | (17,543 | ) | |||||
Proceeds from sale of assets | — | 1,000 | 1,234 | ||||||||
Net cash used in investing activities | (22,061 | ) | (19,132 | ) | (16,309 | ) | |||||
Financing activities | |||||||||||
Proceeds from issuance of common stock | 573 | 428 | 187 | ||||||||
Payments for public offering costs | — | — | (74 | ) | |||||||
Proceeds from issuance of long-term debt | 199,461 | — | 274,313 | ||||||||
Payments on long-term debt and other borrowings | (275,376 | ) | (2,862 | ) | (286,694 | ) | |||||
Deferred financing costs | (2,258 | ) | — | (1,576 | ) | ||||||
Proceeds from stock option exercises | 1,173 | 1,152 | 3,247 | ||||||||
Payments for minimum statutory tax withholding related to net share settlement of equity awards | (2,454 | ) | (3,386 | ) | (2,853 | ) | |||||
Net cash used in financing activities | (78,881 | ) | (4,668 | ) | (13,450 | ) | |||||
Effect of foreign exchange rates on cash and cash equivalents | 76 | (282 | ) | 94 | |||||||
Net (decrease) increase in cash and cash equivalents | (20,482 | ) | 37,111 | 25,112 | |||||||
Cash and cash equivalents, beginning of year | 113,401 | 76,290 | 51,178 | ||||||||
Cash and cash equivalents, end of year | $ | 92,919 | $ | 113,401 | $ | 76,290 | |||||
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Supplemental disclosure of cash flow information | |||||||||||
Cash paid during the period for: | |||||||||||
Interest | $ | 12,253 | $ | 15,869 | $ | 16,653 | |||||
Income taxes, net of refunds of $2, $35 and $17, respectively | $ | 274 | $ | 90 | $ | 106 | |||||
Schedule of non-cash investing and financing activities | |||||||||||
Additions of property, plant and equipment included in liabilities | $ | 4,175 | $ | 7,395 | $ | 2,738 | |||||
The accompanying notes are an integral part of these consolidated financial statements.
76
Lantheus Holdings, Inc.
Notes to Consolidated Financial Statements
1. Description of Business
Lantheus Holdings, Inc., a Delaware corporation, is the parent company of Lantheus Medical Imaging, Inc. (“LMI”), also a Delaware corporation.
The Company develops, manufactures and commercializes innovative diagnostic medical imaging agents and other products that assist clinicians in the diagnosis and treatment of cardiovascular and other diseases.
The Company’s commercial products are used by cardiologists, nuclear physicians, radiologists, internal medicine physicians, technologists and sonographers working in a variety of clinical settings. The Company sells its products to radiopharmacies, integrated delivery networks, hospitals, clinics and group practices.
The Company sells its products globally and has operations in the U.S., Puerto Rico and Canada and third-party distribution relationships in Europe, Canada, Australia, Asia-Pacific and Latin America.
The Company’s product portfolio includes an ultrasound contrast agent, nuclear imaging products and a radiotherapeutic product. The Company’s principal products include the following:
• | DEFINITY is a microbubble contrast agent used in ultrasound exams of the heart, also known as echocardiography exams. DEFINITY contains perflutren-containing lipid microspheres and is indicated in the U.S. for use in patients with suboptimal echocardiograms to assist in imaging the left ventricular chamber and left endocardial border of the heart in ultrasound procedures. |
• | TechneLite is a Tc-99m generator that provides the essential nuclear material used by radiopharmacies to radiolabel Cardiolite, Neurolite and other Tc-99m-based radiopharmaceuticals used in nuclear medicine procedures. TechneLite uses Mo-99 as its active ingredient. |
Sales of the Company’s microbubble contrast agent, DEFINITY, are made in the U.S. and Canada through a DEFINITY direct sales team. In the U.S., the Company’s nuclear imaging products, including TechneLite, Xenon, Neurolite and Cardiolite, are primarily distributed through commercial radiopharmacies, the majority of which are controlled by or associated with GE Healthcare, Cardinal, UPPI, Jubilant Radiopharma and PharmaLogic. A small portion of the Company’s nuclear imaging product sales in the U.S. are made through the Company’s direct sales force to hospitals and clinics that maintain their own in-house radiopharmaceutical preparation capabilities. The Company owns one radiopharmacy in Puerto Rico where they sell their own products as well as products of third parties to end-users.
The Company also maintains its own direct sales force in Canada for certain of its products. In Europe, Australia, Asia-Pacific and Latin America, the Company generally relies on third-party distributors to market, sell and distribute its nuclear imaging and contrast agent products, either on a country-by-country basis or on a multi-country regional basis.
Progenics Transaction
On October 1, 2019, the Company entered into an Agreement and Plan of Merger (the “Initial Merger Agreement”) to acquire Progenics Pharmaceuticals, Inc. (NASDAQ: PGNX) (“Progenics”) in an all-stock transaction. Progenics is an oncology company developing innovative medicines and artificial intelligence to find, fight and follow cancer. Under the terms of the Initial Merger Agreement, the Company agreed to acquire all of the issued and outstanding shares of Progenics common stock at a fixed exchange ratio. Progenics stockholders would have received 0.2502 shares of the Company’s common stock for each share of Progenics common stock, representing an approximately 35% aggregate ownership stake in the combined company. The transaction contemplated by the Initial Merger Agreement was unanimously approved by the Boards of Directors of both companies and was subject to the terms and conditions set forth in the Initial Merger Agreement, including, among other things, the affirmative vote of a majority of the outstanding shares of common stock of Progenics and a majority of votes cast by the holders of the common stock of the Company. The Initial Merger Agreement further provides that in the event of a termination of the Initial Merger Agreement under certain circumstances, one party may be required to pay the other party a termination fee equal to $18.3 million. In the event of a termination of the Merger Agreement as a result of Progenics stockholders failing to adopt the Initial Merger Agreement, Progenics may be required to reimburse reasonable and documented out-of-pocket expenses incurred by the Company in connection with the Merger Agreement not to exceed $5.3 million.
On February 20, 2020, the Company entered into an Amended and Restated Agreement and Plan of Merger (the “Amended Merger Agreement”) with Progenics that, among other things, increases the exchange ratio and provides for the issuance of CVRs in connection with the transaction. See Note 20, “Subsequent Events” for further details on the Amended Merger Agreement.
77
2. Summary of Significant Accounting Policies
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements have been prepared in accordance with U.S. GAAP. The consolidated financial statements include the accounts of the Company and its direct and indirect wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make certain estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the consolidated financial statements, and the reported amounts of revenues and expenses during the reporting period. The more significant estimates reflected in the Company’s consolidated financial statements include, but are not limited to, certain judgments regarding revenue recognition, goodwill, tangible and intangible asset valuation, inventory valuation, asset retirement obligations, income tax liabilities and related indemnification receivable, deferred tax assets and liabilities and accrued expenses. Actual results could materially differ from those estimates or assumptions.
Revenue Recognition
The Company recognizes revenue when it transfers control of promised goods or services to its customers in an amount that reflects the consideration to which the Company expects to be entitled to in exchange for those goods and services. See Note 3, “Revenue from Contracts with Customers” for further discussion on revenues.
Accounts Receivable, net
Accounts receivable consist of amounts billed and currently due from customers. The Company maintains an allowance for doubtful accounts for estimated losses. In determining the allowance, consideration includes the probability of recoverability based on past experience and general economic factors. Certain accounts receivable may be fully reserved when the Company becomes aware of any specific collection issues.
Income Taxes
The Company accounts for income taxes using an asset and liability approach. Income tax (benefit) expense represents income taxes paid or payable for the current year plus the change in deferred taxes during the year. Deferred taxes result from differences between the financial and tax bases of the Company’s assets and liabilities. Deferred tax assets and liabilities are measured using the currently enacted tax rates that apply to taxable income in effect for the years in which those tax attributes are expected to be recovered or paid, and are adjusted for changes in tax rates and tax laws when such changes are enacted.
The Company recognizes deferred tax assets to the extent that the Company believes that these assets are more-likely-than-not to be realized. Valuation allowances are recorded to reduce deferred tax assets when it is more-likely-than-not that the future tax benefit will not be realized. The assessment of whether or not a valuation allowance is required involves weighing both positive and negative evidence, including both historical and prospective information, with greater weight given to evidence that is objectively verifiable. A history of recent losses is negative evidence that is difficult to overcome with positive evidence. In evaluating prospective information there are four sources of taxable income: reversals of taxable temporary differences, items that can be carried back to prior tax years (such as net operating losses), pre-tax income, and prudent and feasible tax planning strategies. Adjustments to the deferred tax valuation allowances are made in the period when those assessments are made.
The Company accounts for uncertain tax positions using a two-step recognition threshold and measurement analysis method to determine the financial statement impact of uncertain tax positions taken or expected to be taken in a tax return. Differences between tax positions taken in a tax return and amounts recognized in the financial statements are recorded as adjustments to other long-term assets and liabilities, or adjustments to deferred taxes, or both. The Company records the related interest and penalties to income tax (benefit) expense.
Net Income per Common Share
Basic earnings per common share is computed by dividing net income by the weighted-average number of shares of common stock outstanding during the period. Diluted earnings per common share is computed by dividing net income by the weighted-average number of shares of common stock outstanding during the period, plus the potential dilutive effect of other securities if those securities were converted or exercised. During periods in which the Company incurs net losses, both basic and diluted loss per common share is calculated by dividing the net loss by the weighted-average shares of common stock outstanding and potentially dilutive securities are excluded from the calculation because their effect would be antidilutive.
78
Cash and Cash Equivalents
Cash and cash equivalents include savings deposits, certificates of deposit and money market funds that have original maturities of three months or less when purchased.
Concentration of Risks and Limited Suppliers
Financial instruments which potentially subject the Company to concentrations of credit risk consist principally of trade accounts receivable. The Company periodically reviews its accounts receivable for collectability and provides for an allowance for doubtful accounts to the extent that amounts are not expected to be collected. The Company sells primarily to large national distributors, which in turn, resell the Company’s products.
As of December 31, 2019, no customer accounted for greater than 10% of accounts receivable, net. One customer accounted for approximately 11% of accounts receivable, net for the year ended December 31, 2018. No customer accounted for greater than 10% of revenues for the years ended December 31, 2019 and December 31, 2018. Three customers accounted for approximately 12%, 10% and 10% of revenues for the year ended December 31, 2017.
The Company relies on certain materials used in its development and manufacturing processes, some of which are procured from only one or a few sources. The failure of one of these suppliers to deliver on schedule could delay or interrupt the manufacturing or commercialization process and would adversely affect the Company’s operating results. In addition, a disruption in the commercial supply of, or a significant increase in the cost of one of the Company’s materials from these sources could have a material adverse effect on the Company’s business, financial position and results of operations.
The Company has Mo-99 supply agreements with IRE of Belgium, running through December 31, 2022, and renewable by the Company on a year-to-year basis thereafter, and with ANSTO and NTP, running through December 31, 2021. The Company also has a Xenon supply agreement with IRE which runs through June 30, 2022, and which is subject to further extension. The Company currently relies on IRE as the sole supplier of bulk-unprocessed Xenon which the Company processes and finishes for its customers. The Company currently relies on JHS as its sole source manufacturer of DEFINITY, Neurolite, Cardiolite and evacuation vials for TechneLite.
The following table sets forth revenues for each of the Company’s products representing 10% or more of revenues:
Year Ended December 31, | |||||
2019 | 2018 | 2017 | |||
DEFINITY | 62.6% | 53.3% | 47.5% | ||
TechneLite | 24.9% | 28.8% | 31.6% | ||
Inventory
Inventory includes material, direct labor and related manufacturing overhead and is stated at the lower of cost and net realizable value on a first-in, first-out basis. The Company records inventory when the Company takes title to the product.
The Company assesses the recoverability of inventory to determine whether adjustments for excess and obsolete inventory are required. Inventory that is in excess of future requirements is written down to its estimated net realizable value based on product shelf life, forecasted demand and other factors.
Inventory costs associated with product that has not yet received regulatory approval are capitalized if the Company believes there is probable future commercial use of the product and future economic benefits of the asset. If future commercial use of the product is not probable, then inventory costs associated with such product are expensed as incurred. As of December 31, 2019 and 2018, the Company had no capitalized inventories associated with product that did not have regulatory approval, respectively.
79
Property, Plant and Equipment, net
Property, plant & equipment are stated at cost. Replacements of major units of property are capitalized, and replaced properties are retired. Replacements of minor components of property and repair and maintenance costs are charged to expense as incurred. Certain costs to obtain or develop computer software are capitalized and amortized over the estimated useful life of the software. Depreciation and amortization is computed on a straight-line basis over the estimated useful lives of the related assets. The estimated useful lives of the major classes of depreciable assets are as follows:
Class | Range of Estimated Useful Lives | |
Buildings | 10 - 50 years | |
Land improvements | 15 - 40 years | |
Machinery and equipment | 3 - 15 years | |
Furniture and fixtures | 15 years | |
Leasehold improvements | Lesser of lease term or 15 years | |
Computer software | 3 - 5 years | |
Upon retirement or other disposal of property, plant & equipment, the cost and related amount of accumulated depreciation are removed from the asset and accumulated depreciation accounts, respectively. The difference, if any, between the net asset value and the proceeds is included in operating income.
Included within machinery, equipment and fixtures are spare parts. Spare parts include replacement parts relating to plant & equipment and are either recognized as an expense when consumed or reclassified and capitalized as part of the related asset and depreciated over the remaining useful life of the related asset.
Goodwill
Goodwill is not amortized but is instead tested for impairment at least annually and whenever events or circumstances indicate that it is more likely-than-not that they may be impaired. The Company has elected to perform the annual test for goodwill impairment as of October 31 of each year. All goodwill has been allocated to the U.S. reporting unit.
In performing the Company’s annual assessment, the Company is permitted to first perform a qualitative test and if necessary, perform a quantitative test. If the Company is required to perform the quantitative impairment test of goodwill, the Company compares the fair value of a reporting unit to its carrying value. If the reporting unit’s carrying value exceeds its fair value, the Company would record an impairment loss to the extent that the carrying value of goodwill exceeds its implied fair value. The Company estimates the fair value of its reporting unit using discounted cash flow or other valuation models, such as comparative transactions and market multiples. The Company did not recognize any goodwill impairment charges during the years ended December 31, 2019, 2018 or 2017.
Intangible and Long-Lived Assets
The Company tests intangible and long-lived assets for recoverability whenever events or changes in circumstances suggest that the carrying value of an asset or group of assets may not be recoverable. The Company measures the recoverability of assets to be held and used by comparing the carrying amount of the asset to future undiscounted net cash flows expected to be generated by the asset. If those assets are considered to be impaired, the impairment equals the amount by which the carrying amount of the assets exceeds the fair value of the assets. Any impairments are recorded as permanent reductions in the carrying amount of the assets. Long-lived assets, other than goodwill and other intangible assets that are held for sale are recorded at the lower of the carrying value or the fair market value less the estimated cost to sell.
Intangible assets, consisting of patents, trademarks and customer relationships related to the Company’s products are amortized in a method equivalent to the estimated utilization of the economic benefit of the asset.
Contingencies
In the normal course of business, the Company is subject to loss contingencies, such as legal proceedings and claims arising out of its business, that cover a wide range of matters, including, among others, product and environmental liability. The Company records accruals for those loss contingencies when it is probable that a liability will be incurred and the amount of loss can be reasonably estimated. The Company does not recognize gain contingencies until realized.
80
Fair Values of Financial Instruments
The estimated fair values of the Company’s financial instruments, including its cash and cash equivalents, accounts receivable, accounts payable and accrued expenses approximate the carrying values of these instruments due to their short term nature. The estimated fair value of the Company’s long term debt approximates its carrying values as the applicable interest rates are subject to change with market interest rates.
Advertising and Promotion Costs
Advertising and promotion costs are expensed as incurred. During the years ended December 31, 2019, 2018 and 2017, the Company incurred $3.8 million, $4.0 million and $4.4 million, respectively in advertising and promotion costs, which are included in sales and marketing in the consolidated statements of operations.
Research and Development
Research and development costs are expensed as incurred and relate primarily to the development of new products to add to the Company’s portfolio and costs related to its medical affairs and medical information functions. Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and recognized as an expense as the goods are delivered or the related services are performed.
Foreign Currency
The consolidated statements of operations of the Company’s foreign subsidiaries are translated into U.S. Dollars using weighted-average exchange rates. The net assets of the Company’s foreign subsidiaries are translated into U.S. Dollars using the end of period exchange rates. The impact from translating the net assets of these subsidiaries at changing rates are recorded in the foreign currency translation adjustment account, which is included in accumulated other comprehensive loss in the consolidated balance sheets.
Remeasurement of the Company’s foreign currency denominated transactions are included in net income. Transaction gains and losses are reported as a component of other expense (income) in the consolidated statements of operations.
Stock-Based Compensation
The Company’s stock-based compensation cost is measured at the grant date of the stock-based award based on the fair value of the award and is recognized as expense over the requisite service period, which generally represents the vesting period, and includes an estimate of the awards that will be forfeited. The Company estimates the fair value of each stock-based award on its measurement date using either the current market price of the stock, the Black-Scholes option valuation model or the Monte Carlo Simulation valuation model, whichever is most appropriate. The Black-Scholes and Monte Carlo Simulation valuation models incorporate assumptions such as stock price volatility, the expected life of options or awards, a risk-free interest rate and dividend yield.
Expense for performance restricted stock awards is recognized based upon the fair value of the awards on the date of grant and the number of shares expected to vest based on the terms of the underlying award agreement and the requisite service period(s).
Other Expense (Income)
Other expense (income) consisted of the following:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
Foreign currency (gains) losses | $ | (33 | ) | $ | 557 | $ | (253 | ) | |||
Tax indemnification expense (income), net | 10,635 | (2,855 | ) | (8,367 | ) | ||||||
Interest income | (686 | ) | (167 | ) | (18 | ) | |||||
Arbitration award | (3,453 | ) | — | — | |||||||
Other income | (242 | ) | — | — | |||||||
Total other expense (income) | $ | 6,221 | $ | (2,465 | ) | $ | (8,638 | ) | |||
Comprehensive Income (Loss)
Comprehensive income (loss) consists of net income and other gains and losses affecting stockholders’ equity that, under U.S. GAAP, are excluded from net income. For the Company, other comprehensive income (loss) consists of foreign currency translation gains and losses. The accumulated other comprehensive loss balance consists entirely of foreign currency translation gains and losses.
81
Asset Retirement Obligations
The Company’s compliance with federal, state, local and foreign environmental laws and regulations may require it to remove or mitigate the effects of the disposal or release of chemical substances in jurisdictions where it does business or maintains properties. The Company establishes accruals when those costs are legally obligated and can be reasonably estimated. Accrual amounts are estimated, which may include the assistance of third-party environmental specialists, and are based on currently available information, regulatory requirements, remediation strategies, historical experience, the relative shares of the total remediation costs, a relevant discount rate, and the time periods of when estimated costs can be reasonably predicted. Changes in these assumptions could impact the Company’s future reported results.
The Company has production facilities which manufacture and process radioactive materials at its North Billerica, Massachusetts and San Juan, Puerto Rico sites. The Company considers its legal obligation to remediate its facilities upon a decommissioning of its radioactive-related operations as an asset retirement obligation. The fair value of a liability for asset retirement obligations is recognized in the period in which the liability is incurred. The liability is measured at the present value of the obligation expected to be incurred and is adjusted in subsequent periods as accretion expense is recorded. The corresponding asset retirement costs are capitalized as part of the carrying values of the related long-lived assets and depreciated over the assets’ useful lives.
The Company has identified conditional asset retirement obligations related to the future removal and disposal of asbestos contained in certain of the buildings located on the Company’s North Billerica, Massachusetts campus. The Company believes the asbestos is appropriately contained and it is compliant with all applicable environmental regulations. If these properties undergo major renovations or are demolished, certain environmental regulations are in place, which specify the manner in which asbestos must be handled and disposed. The Company is required to record the fair value of these conditional liabilities if they can be reasonably estimated. As of December 31, 2019 and 2018, sufficient information was not available to estimate a liability for such conditional asset retirement obligations as the obligations to remove the asbestos from these properties have indeterminable settlement dates. As such, no liability for conditional asset retirement obligations has been recorded in the accompanying consolidated balance sheets as of December 31, 2019 and 2018.
Self-Insurance Reserves
The Company’s consolidated balance sheets at both December 31, 2019 and 2018 include $0.6 million of accrued liabilities associated with employee medical costs that are retained by the Company. The Company estimates the required liability of those claims on an undiscounted basis based upon various assumptions which include, but are not limited to, the Company’s historical loss experience and projected loss development factors. The required liability is also subject to adjustment in the future based upon changes in claims experience, including changes in the number of incidents (frequency) and change in the ultimate cost per incident (severity). The Company also maintains a separate cash account to fund these medical claims and must maintain a minimum balance as determined by the plan administrator. The balance of this restricted cash account was approximately $0.2 million and $0.1 million at December 31, 2019 and 2018, respectively, and is included in other current assets.
82
Recent Accounting Pronouncements
Standard | Description | Effective Date for Company | Effect on the Consolidated Financial Statements |
Recently Issued Accounting Standards Not Yet Adopted | |||
ASU 2016-13, “Financial Instruments-Credit Losses (Topic 326)” | This ASU will require financial instruments measured at amortized cost and accounts receivable to be presented at the net amount expected to be collected. The new model requires an entity to estimate credit losses based on historical information, current information and reasonable and supportable forecasts that affect the collectability of the reported amount. ASU 2016-13 is effective for annual reporting periods beginning after December 15, 2019. | January 1, 2020 | The Company has completed its assessment on the impact of the standard and concluded that upon adoption of this standard there will not be a material impact to its consolidated financial statements. |
Standard | Description | Effective Date for Company | Effect on the Consolidated Financial Statements |
Accounting Standards Adopted During the Year Ended December 31, 2019 | |||
ASU 2016-02, “Leases (Topic 842)” | This ASU supersedes existing guidance on accounting for leases in “Leases (Topic 840)” and generally requires all leases to be recognized on the balance sheet. In July 2018, an amendment was made that allows companies the option of using the effective date of the new standard as the initial application date (at the beginning of the period in which it is adopted, rather than at the beginning of the earliest comparative period). | January 1, 2019 | See Note 13, "Leases" for the required disclosures related to the impact of adopting this standard. The adoption of this standard resulted in the recording of an additional lease asset and lease liability of approximately $1.1 million as of January 1, 2019. |
3. Revenue from Contracts with Customers
Adoption of ASC Topic 606, “Revenue from Contracts with Customers” (“ASC 606”)
The Company adopted ASC 606 on January 1, 2018 using the modified retrospective method for all contracts not completed as of the date of adoption. The reported results for 2019 and 2018 reflect the application of ASC 606 guidance while the reported results for 2017 were prepared under the guidance of ASC 605. The adoption of ASC 606 did not have a material impact on the Company’s consolidated balance sheet, results of operations, equity or cash flows as of the adoption date or for the periods presented.
Revenue Recognition
Revenue is recognized when a customer obtains control of promised goods or services. The amount of revenue recognized reflects the consideration to which the Company expects to be entitled to receive in exchange for these goods or services. To achieve this core principle, the Company applies the following five steps: (1) identify the contracts with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the Company satisfies a performance obligation.
Disaggregation of Revenue
83
The following table summarizes revenue by revenue source and reportable segment as follows:
Year Ended December 31, | ||||||||
Major Products/Service Lines by Segment (in thousands) | 2019 | 2018 | ||||||
U.S. | ||||||||
Product revenue, net(1) | $ | 303,989 | $ | 288,580 | ||||
Total U.S. revenues | 303,989 | 288,580 | ||||||
International | ||||||||
Product revenue, net(1) | 41,287 | 52,556 | ||||||
License and royalty revenues | 2,061 | 2,238 | ||||||
Total International revenues | 43,348 | 54,794 | ||||||
Total revenues | $ | 347,337 | $ | 343,374 | ||||
______________________________
(1) | The Company’s principal products include DEFINITY and TechneLite and are categorized within product revenue, net. The Company applies the same revenue recognition policies and judgments for all of its principal products. |
Product Revenue, Net
The Company sells its products principally to hospitals and clinics, radiopharmacies and distributors. The Company considers customer purchase orders, which in some cases are governed by master sales or group purchasing organization agreements, to be the contracts with a customer.
For each contract, the Company considers the promise to transfer products, each of which is distinct, to be the identified performance obligations. In determining the transaction price, the Company evaluates whether the price is subject to refund or adjustment to determine the net consideration to which the Company expects to be entitled.
The Company typically invoices customers upon satisfaction of identified performance obligations. As the Company’s standard payment terms are 30 to 60 days from invoicing, the Company has elected to use the significant financing component practical expedient.
The Company allocates the transaction price to each distinct product based on their relative standalone selling price. The product price as specified on the purchase order is considered the standalone selling price as it is an observable input which depicts the price as if sold to a similar customer in similar circumstances.
Revenue is recognized when control of the product is transferred to the customer (i.e., when the Company’s performance obligation is satisfied), which typically occurs upon delivery to the customer. Further, in determining whether control has transferred, the Company considers if there is a present right to payment and legal title, along with risks and rewards of ownership having transferred to the customer.
Frequently, the Company receives orders for products to be delivered over multiple dates that may extend across several reporting periods. The Company invoices for each delivery upon shipment and recognizes revenues for each distinct product delivered, assuming transfer of control has occurred.
The Company generally does not separately charge customers for shipping and handling costs, but any shipping and handling costs charged to customers are included in product revenue, net. Taxes collected from customers relating to product sales and remitted to governmental authorities are excluded from revenues.
Variable Consideration
Revenues from product sales are recorded at the net sales price (transaction price), which includes estimates of variable consideration for which reserves are established for discounts, returns, rebates and allowances that are offered within contracts between the Company and its customers. These reserves are based on the amounts earned or to be claimed on the related sales and are classified as a current liability. Where appropriate, these estimates take into consideration a range of possible outcomes which are probability-weighted for relevant factors such as the Company’s historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. Overall, these reserves reflect the Company’s best estimates of the amount of consideration to which it is entitled based on the
84
terms of the contract. The amount of variable consideration which is included in the transaction price may be constrained, and is included in the net sales price only to the extent that it is probable that a significant reversal in the amount of the cumulative revenue recognized will not occur in a future period. Actual amounts of consideration ultimately received may differ from the Company’s estimates. If actual results in the future vary from the Company’s estimates, the Company adjusts these estimates, which would affect product revenue and earnings in the period such variances become known.
Rebates and Allowances: The Company provides certain customers with rebates and allowances that are explicitly stated in the Company’s contracts and are recorded as a reduction of revenue in the period the related product revenue is recognized. The Company establishes a liability for such amounts, which is included in accrued expenses in the accompanying consolidated balance sheets. These rebates and allowances result from performance-based offers that are primarily based on attaining contractually specified sales volumes and administrative fees the Company is required to pay to group purchasing organizations. The Company estimates the amount of rebates and allowances that are explicitly stated in the Company’s contracts based on a combination of actual purchases and an estimate of the customer’s buying patterns.
Product Returns: The Company generally offers customers a limited right of return due to non-conforming product. The Company estimates the amount of its product sales that may be returned by its customers and records this estimate as a reduction of revenue in the period the related product revenue is recognized. The Company currently estimates product return liabilities using its historical product return information and considers other factors that it believes could significantly impact its expected returns, including product recalls. Reserves for product returns are not significant to the Company due to the nature of its products including radiopharmaceutical products with limited half-lives.
License and Royalty Revenues
The Company has entered into licensing agreements, under which it licenses certain rights to third parties. The terms of these arrangements typically include payment to the Company of one or more of the following: non-refundable, up-front license fees; development, regulatory and commercial milestone payments; and royalties on net sales of licensed products. The Company also has distribution licenses which are treated as combined performance obligations with the delivery of its products and are classified as product revenue, net.
In determining the appropriate amount of revenue to be recognized as it fulfills its obligations under each of its agreements, the Company performs the five-step approach stated earlier. The Company uses judgment in determining the number of performance obligations in a license agreement by assessing whether the license is distinct or should be combined with another performance obligation, as well as the nature of the license. As part of the accounting for these arrangements, the Company must develop assumptions that require judgment to determine the stand-alone selling price for each performance obligation identified in the contract. The Company uses key assumptions to determine the stand-alone selling price, which may include market conditions, reimbursement rates for personnel costs, development timelines and probabilities of regulatory success.
Licenses of intellectual property: If the license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenues from non-refundable, up-front fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are bundled with other promises, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, up-front fees. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition.
Milestone Payments: At the inception of each arrangement that includes development milestone payments, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the control of the Company or the licensee, such as regulatory approvals, are not considered probable of being achieved until those approvals are received. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which the Company recognizes revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, the Company re-evaluates the probability of achievement of such development milestones and any related constraint, and if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect license and royalty revenues and earnings in the period of adjustment. At December 31, 2019, the Company is constraining variable consideration related to milestone payments requiring regulatory approvals.
85
Royalty Revenues: For arrangements that include sales-based royalties, including milestone payments based on the level of sales, and the license is deemed to be the predominant item to which the royalties relate, the Company recognizes revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied).
Contract Costs
The Company recognizes an asset for incremental costs of obtaining a contract with a customer if it expects to recover those costs. The Company’s sales incentive compensation plans qualify for capitalization since these plans are directly related to sales achieved during a period of time. However, the Company has elected the practical expedient to expense the costs as they are incurred, within sales and marketing expenses, since the amortization period is less than one year.
The Company recognized certain revenues as follows:
Year Ended December 31, | |||||||
(in thousands) | 2019 | 2018 | |||||
Amounts included in the contract liability at the beginning of the period | $ | 33 | $ | 33 | |||
Performance obligations satisfied (or partially satisfied) in previous periods | $ | — | $ | — | |||
The Company’s performance obligations are typically part of contracts that have an original expected duration of one year or less. As such, the Company is not disclosing the aggregate amount of the transaction price allocated to performance obligations that are unsatisfied (or partially satisfied) as of the end of the reporting period.
4. Fair Value of Financial Instruments
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. In order to increase consistency and comparability of fair value measurements, financial instruments are categorized based on a hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
• | Level 1 — Inputs are unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. |
• | Level 2 — Inputs include quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (i.e., interest rates, yield curves, etc.) and inputs that are derived principally from or corroborated by observable market data by correlation or other means (market corroborated inputs). |
• | Level 3 — Unobservable inputs that reflect a Company’s estimates about the assumptions that market participants would use in pricing the asset or liability. The Company develops these inputs based on the best information available, including its own data. |
The Company’s financial assets measured at fair value on a recurring basis consist of money market funds. The Company invests excess cash from its operating cash accounts in overnight investments and reflects these amounts in cash and cash equivalents in the consolidated balance sheets at fair value using quoted prices in active markets for identical assets.
86
The tables below present information about the Company’s assets and liabilities measured at fair value on a recurring basis:
December 31, 2019 | |||||||||||||||
(in thousands) | Total Fair Value | Level 1 | Level 2 | Level 3 | |||||||||||
Money market | $ | 39,530 | $ | 39,530 | $ | — | $ | — | |||||||
Total | $ | 39,530 | $ | 39,530 | $ | — | $ | — | |||||||
December 31, 2018 | |||||||||||||||
(in thousands) | Total Fair Value | Level 1 | Level 2 | Level 3 | |||||||||||
Money market | $ | 61,391 | $ | 61,391 | $ | — | $ | — | |||||||
Total | $ | 61,391 | $ | 61,391 | $ | — | $ | — | |||||||
5. Income Taxes
The components of income before income taxes is summarized as follows:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
U.S. | $ | 25,432 | $ | 46,945 | $ | 39,559 | |||||
International | 3,195 | 2,603 | 80 | ||||||||
Income before income taxes | $ | 28,627 | $ | 49,548 | $ | 39,639 | |||||
The income tax (benefit) expense is summarized as follows:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
Current | |||||||||||
Federal | $ | 287 | $ | (21 | ) | $ | (58 | ) | |||
State | (13,166 | ) | 3,424 | 3,242 | |||||||
International | 114 | (135 | ) | 16 | |||||||
(12,765 | ) | 3,268 | 3,200 | ||||||||
Deferred | |||||||||||
Federal | 8,712 | 7,821 | (71,742 | ) | |||||||
State | 790 | 1,411 | (15,220 | ) | |||||||
International | 223 | (3,470 | ) | 16 | |||||||
9,725 | 5,762 | (86,946 | ) | ||||||||
Income tax (benefit) expense | $ | (3,040 | ) | $ | 9,030 | $ | (83,746 | ) | |||
87
The reconciliation of income taxes at the U.S. federal statutory rate to the actual income taxes is as follows:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
U.S. statutory rate | $ | 6,012 | $ | 10,405 | $ | 13,873 | |||||
Permanent items | 3,737 | 505 | (1,916 | ) | |||||||
Uncertain tax positions | (13,156 | ) | 3,227 | 3,128 | |||||||
Other tax credits | (1,685 | ) | (742 | ) | (175 | ) | |||||
State and local taxes | 1,914 | 2,125 | 1,252 | ||||||||
Impact of rate change on deferred taxes | — | — | 45,129 | ||||||||
True-up of prior year tax | — | — | 7 | ||||||||
Foreign tax rate differential | (238 | ) | 30 | 97 | |||||||
Valuation allowance | (22 | ) | (4,073 | ) | (141,094 | ) | |||||
Benefit of windfall related to stock compensation | (2,768 | ) | (1,760 | ) | (2,723 | ) | |||||
Increase in indemnification deferred tax asset | 2,531 | (731 | ) | (1,055 | ) | ||||||
Other | 635 | 44 | (269 | ) | |||||||
Income tax (benefit) expense | $ | (3,040 | ) | $ | 9,030 | $ | (83,746 | ) | |||
The components of deferred income tax assets (liabilities) are as follows:
December 31, | |||||||
(in thousands) | 2019 | 2018 | |||||
Deferred Tax Assets | |||||||
Federal benefit of state tax liabilities | $ | 5,278 | $ | 7,809 | |||
Reserves, accruals and other | 15,026 | 11,005 | |||||
Inventory obsolescence | 550 | 428 | |||||
Capitalized research and development | 5,086 | 7,491 | |||||
Amortization of intangibles other than goodwill | 1,569 | 2,809 | |||||
Net operating loss carryforwards | 47,095 | 55,938 | |||||
Depreciation | 56 | — | |||||
Deferred tax assets | 74,660 | 85,480 | |||||
Deferred Tax Liabilities | |||||||
Reserves, accruals and other | (881 | ) | (1,078 | ) | |||
Customer relationships | (707 | ) | (986 | ) | |||
Depreciation | — | (727 | ) | ||||
Deferred tax liability | (1,588 | ) | (2,791 | ) | |||
Less: valuation allowance | (1,238 | ) | (1,240 | ) | |||
$ | 71,834 | $ | 81,449 | ||||
Recorded in the accompanying consolidated balance sheets as: | |||||||
Noncurrent deferred tax assets, net | $ | 71,834 | $ | 81,449 | |||
On December 22, 2017, the United States enacted the Tax Cuts and Jobs Act of 2017 (the “Act”). The Act is significant and has wide-ranging effects.
The Company has completed its study of the ramifications of the Act, and has confirmed the primary material impact of the Act to be the remeasurement of the Company’s deferred tax assets, which was recorded in fiscal 2017 as a result of the reduction in U.S. corporate tax rates from 35% to 21%. As of December 31, 2017, the Company determined it had no accumulated unrepatriated foreign earnings, and therefore recorded no liability for the repatriation transition tax.
The Company has also completed its evaluation of and accounting for all other relevant changes resulting from the Act, and has determined that through December 31, 2018, these changes do not materially impact the Company's effective tax rate.
88
The Company regularly assesses its ability to realize its deferred tax assets. Assessing the realizability of deferred tax assets requires significant management judgment. In determining whether its deferred tax assets are more-likely-than-not realizable, the Company evaluated all available positive and negative evidence, and weighed the objective evidence and expected impact. During the fourth quarter of fiscal year 2018, the Company's Canada subsidiary entered an accumulated three year period of profitability, removing a strong item of negative evidence previously supporting the recording of a full valuation allowance. Management has determined that the weight of the relevant positive evidence outweigh the negative evidence, and released the valuation allowance against its Canada subsidiary's net deferred tax assets, resulting in an income tax benefit of $4.0 million in fiscal 2018. The Company continues to record a valuation allowance of $1.2 million against the net deferred tax assets of its U.K. subsidiary.
During the fourth quarter of 2017, the Company determined based on its consideration of the weight of positive and negative evidence that there was sufficient positive evidence that its U.S. federal and state deferred tax assets were more-likely-than-not realizable. The Company’s conclusion was primarily driven by the achievement of a sustained level of U.S. profitability, the expectation of sustained future profitability, and mitigating factors related to external supplier and customer risk sufficient to outweigh the available negative evidence. Accordingly, the Company released the valuation allowance previously recorded against its U.S. net deferred tax assets, resulting in a fiscal 2017 income tax benefit of $141.1 million.
The Company will continue to assess the level of the valuation allowance required. If the weight of negative evidence exists in future periods to again support the recording of a partial or full valuation allowance against the Company’s deferred tax assets, there would likely be a material negative impact on the Company’s results of operations in that future period.
A summary of the changes in the Company’s valuation allowance is summarized below:
(in thousands) | Amount | ||
Balance, January 1, 2018 | $ | 5,368 | |
Charged to income tax (benefit) expense | (103 | ) | |
Foreign currency | (56 | ) | |
Release valuation allowance | (3,969 | ) | |
Balance, December 31, 2018 | 1,240 | ||
Charged to income tax (benefit) expense | (22 | ) | |
Foreign currency | 20 | ||
Release valuation allowance | — | ||
Balance, December 31, 2019 | $ | 1,238 | |
The Company’s U.S. federal income tax returns are subject to examination for three years. The state and foreign income tax returns are subject to examination for periods varying from three to four years depending on the specific jurisdictions’ statutes of limitation.
At December 31, 2019, the Company has U.S. federal net operating loss carryovers of approximately $174.0 million, which will expire between 2032 and 2037, and U.S. federal research credits of $1.4 million which will begin to expire in 2037. The Company has state research credit carryforwards of $3.0 million, which will expire between 2024 and 2033. The Company has state investment tax credit carryforwards of $2.1 million, of which $0.7 million have no expiration date, and the remainder of which will begin to expire in 2020 and fully expire in 2022.
89
A reconciliation of the Company’s changes in uncertain tax positions for 2019 and 2018 is as follows:
(in thousands) | Amount | ||
Balance of uncertain tax positions as of January 1, 2018 | $ | 9,866 | |
Additions related to current year tax positions | — | ||
Reductions related to prior year tax positions | (4 | ) | |
Settlements | — | ||
Lapse of statute of limitations | (74 | ) | |
Balance of uncertain tax positions as of December 31, 2018 | 9,788 | ||
Additions related to current year tax positions | — | ||
Reductions related to prior year tax positions | (4,496 | ) | |
Settlements | — | ||
Lapse of statute of limitations | — | ||
Balance of uncertain tax positions as of December 31, 2019 | $ | 5,292 | |
In connection with the Company’s acquisition of the medical imaging business from Bristol-Myers Squibb (“BMS”) in 2008, the Company recorded a liability for uncertain tax positions related to the acquired business and simultaneously entered into a tax indemnification agreement with BMS under which BMS agreed to indemnify the Company for any payments made to settle those uncertain tax positions with the taxing authorities. Accordingly, a long-term receivable is recorded to account for the expected value to the Company of future indemnification payments, net of actual tax benefits received, to be paid on behalf of the Company by BMS. The tax indemnification receivable is recorded within other long-term assets.
In accordance with the Company’s accounting policy, the change in the tax liability, penalties and interest associated with these uncertain tax positions (net of any offsetting federal or state benefit) is recognized within income tax (benefit) expense. Contemporaneously, changes in the tax indemnification receivable are recognized within other expense (income) in the consolidated statement of operations. Accordingly, as these reserves change, adjustments are included in income tax (benefit) expense with an offsetting adjustment included in other expense (income). Assuming that the receivable from BMS continues to be considered recoverable by the Company, there will be no effect on net income and no net cash outflows related to these liabilities.
For the year ended December 31, 2019, the Company released $17.1 million of liabilities for uncertain tax positions, including interest and penalties of $12.7 million. This included a release of a liability of $1.9 million, including interest and penalties of $1.4 million, arising from a settlement during the year. The remaining release of $15.2 million of liability was due to a change in estimate with respect to the Company’s indemnified uncertain tax positions. In late 2019 the Company reassessed its indemnified uncertain tax positions and obtained, with the assistance of third-party tax experts, additional technical insights with respect to the indemnified uncertain tax positions. On the basis of the new information obtained, the Company changed its estimate with respect to certain of its indemnified uncertain tax positions and consequently released $15.2 million of related reserves.
The combined release of $17.1 million was recorded to income tax (benefit) expense and offset by a reduction in deferred tax assets of $3.3 million and a $13.8 million reduction of the indemnification receivable recorded to other expense (income). The amount due from BMS as of December 31, 2019, was also increased by $3.2 million, due to the accrual of interest on the liability with respect to the remaining uncertain tax positions. Similarly, the amount due from BMS increased by $3.3 million in 2018, due to the accrual of interest on the existing liability for uncertain tax positions. In 2017, the amount due from BMS increased by $8.4 million, primarily due to the decrease in U.S. corporate tax rates effective January 1, 2018. As noted above, there is no effect on net income or net cash flows in any period due to the indemnification agreement in place.
As of December 31, 2019 and 2018, total liabilities for uncertain tax positions including interest and penalties were $27.0 million and $40.2 million, respectively, consisting of uncertain tax positions of $5.3 million and $9.8 million, interest accruals of $20.7 million and $28.2 million, and penalty accruals of $1.0 million and $2.2 million, respectively. As of December 31, 2019 and 2018, all of these liabilities were included in other long-term liabilities. Included in the 2019, 2018 and 2017 tax provisions are a benefit of $13.2 million and expense of $3.2 million and $3.1 million, respectively, relating to accrual of interest, net of benefits for reversals of uncertain tax positions, recognized upon settlements, effective settlements or lapses of relevant statutes of limitation.
The total long-term asset related to the indemnification was $18.9 million and $29.5 million at December 31, 2019 and 2018, respectively. Included in other expense (income) for the years ended December 31, 2019, 2018 and 2017, is tax indemnification expense (income), net of $10.6 million, $(2.9) million and $(8.4) million, respectively. For the year ended December 31, 2017, $6.5 million of the tax indemnification income is related to the impact of the U.S. federal tax rate reduction, and the remainder arises from increases in the indemnified liabilities.
90
6. Inventory
Inventory consisted of the following:
December 31, | |||||||
(in thousands) | 2019 | 2018 | |||||
Raw materials | $ | 11,417 | $ | 11,100 | |||
Work in process | 9,450 | 4,261 | |||||
Finished goods | 8,313 | 17,658 | |||||
Total inventory | $ | 29,180 | $ | 33,019 | |||
7. Property, Plant and Equipment, Net
Property, plant and equipment, net, consisted of the following:
December 31, | |||||||
(in thousands) | 2019 | 2018 | |||||
Land | $ | 13,450 | $ | 13,450 | |||
Buildings | 75,654 | 64,444 | |||||
Machinery, equipment and fixtures | 87,763 | 69,298 | |||||
Computer software | 20,739 | 19,266 | |||||
Construction in progress | 10,546 | 24,169 | |||||
208,152 | 190,627 | ||||||
Less: accumulated depreciation and amortization | (91,655 | ) | (82,739 | ) | |||
Total property, plant and equipment, net | $ | 116,497 | $ | 107,888 | |||
Depreciation and amortization expense related to property, plant & equipment, net, was $10.3 million, $10.1 million and $14.8 million for the years ended December 31, 2019, 2018 and 2017, respectively.
8. Asset Retirement Obligations
The Company considers its legal obligation to remediate its facilities upon a decommissioning of its radioactive-related operations as an asset retirement obligation. The Company has production facilities which manufacture and process radioactive materials at its North Billerica, Massachusetts and San Juan, Puerto Rico sites. As of December 31, 2019, the liability is measured at the present value of the obligation expected to be incurred, of approximately $26.9 million.
The following table provides a summary of the changes in the Company’s asset retirement obligations:
(in thousands) | Amount | ||
Balance, January 1, 2019 | $ | 11,572 | |
Revisions in estimated cash flows | 20 | ||
Accretion expense | 1,291 | ||
Balance, December 31, 2019 | $ | 12,883 | |
The Company is required to provide the U.S. Nuclear Regulatory Commission and Massachusetts Department of Public Health financial assurance demonstrating the Company’s ability to fund the decommissioning of its North Billerica, Massachusetts production facility upon closure, although the Company does not intend to close the facility. The Company has provided this financial assurance in the form of a $28.2 million surety bond.
91
9. Intangibles, Net
Intangibles, net, consisted of the following:
December 31, 2019 | |||||||||||||
(in thousands) | Amortization Method | Cost | Accumulated Amortization | Net | |||||||||
Trademarks | Straight-Line | $ | 13,540 | $ | (10,407 | ) | $ | 3,133 | |||||
Customer relationships | Accelerated | 99,019 | (94,816 | ) | 4,203 | ||||||||
Total | $ | 112,559 | $ | (105,223 | ) | $ | 7,336 | ||||||
December 31, 2018 | |||||||||||||
(in thousands) | Amortization Method | Cost | Accumulated Amortization | Net | |||||||||
Trademarks | Straight-Line | $ | 13,540 | $ | (9,856 | ) | $ | 3,684 | |||||
Customer relationships | Accelerated | 98,912 | (93,463 | ) | 5,449 | ||||||||
Patents | Straight-Line | 6,570 | (6,570 | ) | — | ||||||||
Total | $ | 119,022 | $ | (109,889 | ) | $ | 9,133 | ||||||
The Company recorded amortization expense for its intangible assets of $1.8 million, $2.6 million and $3.3 million for the years ended December 31, 2019, 2018 and 2017, respectively.
The below table summarizes the estimated aggregate amortization expense expected to be recognized on the above intangible assets:
(in thousands) | Amount | ||
2020 | $ | 1,568 | |
2021 | 1,311 | ||
2022 | 1,174 | ||
2023 | 579 | ||
2024 | 496 | ||
2025 and thereafter | 2,208 | ||
Total | $ | 7,336 | |
10. Accrued Expenses and Other Liabilities
Accrued expenses are comprised of the following:
December 31, | |||||||
(in thousands) | 2019 | 2018 | |||||
Compensation and benefits | $ | 15,100 | $ | 15,962 | |||
Freight, distribution and operations | 6,260 | 7,721 | |||||
Accrued rebates, discounts and chargebacks | 6,985 | 4,654 | |||||
Accrued professional fees | 6,917 | 1,673 | |||||
Other | 2,098 | 2,040 | |||||
Total accrued expenses and other liabilities | $ | 37,360 | $ | 32,050 | |||
11. Long-Term Debt, Net, and Other Borrowings
In June 2019, the Company refinanced its previous $275 million five-year term loan agreement (the “2017 Term Facility”) with a new five-year $200 million term loan facility (the “2019 Term Facility” and the loans thereunder, the “2019 Term Loans”). In addition, the Company replaced its previous $75 million five-year revolving credit facility (the “2017 Revolving Facility”) with a new $200 million five-year revolving credit facility (the “2019 Revolving Facility” and, together with the 2019 Term Facility, the “2019 Facility”). The terms of the 2019 Facility are set forth in the Credit Agreement, dated as of June 27, 2019 (the “2019 Credit Agreement”), by and among Holdings, the Company, the lenders from time to time party thereto and Wells Fargo Bank, N.A., as
92
administrative agent and collateral agent. The Company has the right to request an increase to the 2019 Term Facility or request the establishment of one or more new incremental term loan facilities, in an aggregate principal amount of up to $100 million, plus additional amounts, in certain circumstances.
The net proceeds of the 2019 Term Facility, together with approximately $73 million of cash on hand, were used to refinance in full the aggregate remaining principal amount of the loans outstanding under the 2017 Term Facility and pay related interest, transaction fees and expenses. No amounts were outstanding under the 2017 Revolving Facility at that time. The Company accounted for the refinancing of the 2017 Term Facility as a debt extinguishment and the 2017 Revolving Facility as a debt modification by evaluating the refinancing on a creditor by creditor basis. The Company recorded a loss on extinguishment of debt of $3.2 million related to the write-off of unamortized debt issuance costs and debt discounts. In addition, the Company incurred and capitalized $2.8 million of new debt issuance costs and debt discounts related to the refinancing.
2019 Term Facility
The 2019 Term Loans under the 2019 Term Facility bear interest, with pricing based from time to time at the Company’s election at (i) LIBOR plus a spread ranging from 1.25% to 2.25% as determined by the Company’s total net leverage ratio (as defined in the 2019 Credit Agreement) or (ii) the Base Rate (as defined in the 2019 Credit Agreement) plus a spread ranging from 0.25% to 1.25% as determined by the Company’s total net leverage ratio. The use of the LIBOR is expected to be phased out by the end of 2021. The 2019 Credit Agreement allows for a replacement interest rate in the event the LIBOR is phased out. At December 31, 2019, the Company’s interest rate under the 2019 Term Facility was 3.55%.
The Company is permitted to voluntarily prepay the 2019 Term Loans, in whole or in part, without premium or penalty. The 2019 Term Facility requires the Company to make mandatory prepayments of the outstanding 2019 Term Loans in certain circumstances. The 2019 Term Loans mature in June 2024.
As of December 31, 2019, the Company’s maturities of principal obligations under its long-term debt and other borrowings are as follows:
(in thousands) | Amount | ||
2020 | $ | 10,000 | |
2021 | 10,000 | ||
2022 | 11,250 | ||
2023 | 15,000 | ||
2024 | 148,750 | ||
Total principal outstanding | 195,000 | ||
Unamortized debt discount | (485 | ) | |
Unamortized debt issuance costs | (774 | ) | |
Finance lease liabilities | 329 | ||
Total | 194,070 | ||
Less: current portion | (10,143 | ) | |
Total long-term debt | $ | 183,927 | |
2019 Revolving Facility
Under the terms of the 2019 Revolving Facility, the lenders thereunder agreed to extend credit to the Company from time to time until June 27, 2024 consisting of revolving loans (the “Revolving Loans” and, together with the 2019 Term Loans, the “Loans”) in an aggregate principal amount not to exceed $200 million (the “Revolving Commitment”) at any time outstanding. The 2019 Revolving Facility includes a $20 million sub-facility for the issuance of letters of credit (the “Letters of Credit”). The 2019 Revolving Facility includes a $10 million sub-facility for swingline loans (the “Swingline Loans”). The Letters of Credit, Swingline Loans and the borrowings under the 2019 Revolving Facility are expected to be used for working capital and other general corporate purposes.
The Revolving Loans under the 2019 Revolving Facility bear interest, with pricing based from time to time at the Company’s election at (i) LIBOR plus a spread ranging from 1.25% to 2.25% as determined by the Company’s total net leverage ratio or (ii) the Base Rate plus a spread ranging from 0.25% to 1.25% as determined by the Company’s total net leverage ratio. The 2019 Revolving Facility also includes a commitment fee, which ranges from 0.15% to 0.30% as determined by the Company’s total net leverage ratio.
The Company is permitted to voluntarily prepay the Revolving Loans, in whole or in part, or reduce or terminate the Revolving Commitment, in each case, without premium or penalty. On any business day on which the total amount of outstanding Revolving
93
Loans and Letters of Credit exceeds the total Revolving Commitment, the Company must prepay the Revolving Loans in an amount equal to such excess. As of December 31, 2019, there were no outstanding borrowings under the 2019 Revolving Facility.
2019 Facility Covenants
The 2019 Facility contains a number of affirmative, negative, reporting and financial covenants, in each case subject to certain exceptions and materiality thresholds. The 2019 Facility requires the Company to be in quarterly compliance, measured on a trailing four quarter basis, with two financial covenants. The minimum interest coverage ratio, commencing with the fiscal quarter ending September 30, 2019, must be at least 3.00 to 1.00. The maximum total net leverage ratio permitted by the financial covenant is displayed in the table below:
2019 Facility Financial Covenant | |
Period | Consolidated Leverage Ratio |
Q1 2020 to Q2 2020 | 4.00 to 1.00 |
Q3 2020 to Q2 2021 | 3.75 to 1.00 |
Thereafter | 3.50 to 1.00 |
The Company may elect to increase the maximum total net leverage ratio by 0.50 to 1.00 (subject to a maximum of 4.25 to 1.00) up to two separate times during the term of the 2019 Facility in connection with any Material Acquisition (as defined in the Credit Agreement).
The 2019 Facility contains usual and customary restrictions on the ability of the Company and its subsidiaries to: (i) incur additional indebtedness (ii) create liens; (iii) consolidate, merge, sell or otherwise dispose of all or substantially all of its assets; (iv) sell certain assets; (v) pay dividends on, repurchase or make distributions in respect of capital stock or make other restricted payments; (vi) make certain investments; (vii) repay subordinated indebtedness prior to stated maturity; and (viii) enter into certain transactions with its affiliates.
Upon an event of default, the administrative agent under the Credit Agreement will have the right to declare the Loans and other obligations outstanding immediately due and payable and all commitments immediately terminated or reduced.
The 2019 Facility is guaranteed by Holdings and Lantheus MI Real Estate, LLC, and obligations under the 2019 Facility are generally secured by first priority liens over substantially all of the assets of each of LMI, Holdings and Lantheus MI Real Estate, LLC (subject to customary exclusions set forth in the transaction documents) owned as of June 27, 2019 or thereafter acquired.
12. Stock-Based Compensation
Equity Incentive Plans
As of December 31, 2019, the Company’s approved equity incentive plans included the 2015 Equity Incentive Plan (“2015 Plan”), the 2013 Equity Incentive Plan (“2013 Plan”), and the 2008 Equity Incentive Plan (“2008 Plan”). These plans are administered by the Board of Directors and permit the granting of stock options, stock appreciation rights, restricted stock, restricted stock units and dividend equivalent rights (“DERs”) to employees, officers, directors and consultants of the Company.
The Company has certain stock option and restricted stock awards outstanding under each of its equity incentive plans but, upon adoption of the 2015 Plan, no longer grants new equity awards under its 2008 and 2013 Plans. The Company adopted its 2015 Plan in June 2015 and subsequently amended the plan in April 2016, 2017 and 2019 which increased the common stock reserved for issuance under the plan to an aggregate 6,580,277 shares.
Stock-based compensation expense recognized in the consolidated statements of operations is summarized below:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
Cost of goods sold | $ | 2,091 | $ | 1,140 | $ | 1,692 | |||||
Sales and marketing | 1,953 | 1,244 | 640 | ||||||||
General and administrative | 6,990 | 4,990 | 2,964 | ||||||||
Research and development | 1,458 | 1,344 | 632 | ||||||||
Total stock-based compensation expense | $ | 12,492 | $ | 8,718 | $ | 5,928 | |||||
94
Stock Options
Stock option awards under the 2015 Plan are granted with an exercise price equal to the fair value of the Company’s common stock at the date of grant. All option awards have a ten-year contractual term.
A summary of option activity for 2019 is presented below:
Total Stock Options | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | |||||||||
Balance at January 1, 2019 | 357,075 | $ | 17.50 | |||||||||
Options granted | — | $ | — | |||||||||
Options exercised | (67,558 | ) | $ | 17.37 | ||||||||
Options cancelled and expired | (17,293 | ) | $ | 18.66 | ||||||||
Outstanding at December 31, 2019 | 272,224 | $ | 17.44 | 3.7 | 1,096,000 | |||||||
Exercisable at December 31, 2019 | 272,224 | $ | 17.44 | 3.7 | 1,096,000 | |||||||
During the years ended December 31, 2019, 2018 and 2017, 67,558, 192,550 and 465,232 options were exercised having aggregate intrinsic values of $0.6 million, $2.4 million and $5.1 million, respectively.
Restricted Stock
A summary of restricted stock awards and restricted stock units activity for 2019 is presented below:
Shares | Weighted- Average Grant Date Fair Value Per Share | |||||
Nonvested balance at January 1, 2019 | 1,508,539 | $ | 9.51 | |||
Granted | 409,821 | $ | 23.33 | |||
Vested | (795,503 | ) | $ | 8.52 | ||
Forfeited | (91,085 | ) | $ | 15.84 | ||
Nonvested balance at December 31, 2019 | 1,031,772 | $ | 15.20 | |||
As of December 31, 2019, there was $9.9 million of unrecognized compensation expense related to outstanding restricted stock, which is expected to be recognized over a weighted-average period of 2.0 years.
The weighted average grant-date fair value for restricted stock granted during the fiscal years ended December 31, 2019, 2018 and 2017 was $23.33, $15.46 and $12.94 per share, respectively. The total fair value of restricted stock vested in fiscal years 2019, 2018 and 2017 was $6.8 million, $4.3 million and $2.9 million, respectively.
Performance Restricted Stock Awards
Performance awards vest based on the requisite service period subject to the achievement of specific financial performance targets. The Company monitors the probability of achieving the performance targets on a quarterly basis and may adjust periodic stock compensation expense accordingly. The performance targets include the achievement of internal performance targets only.
95
A summary of performance restricted stock award activity for 2019 is presented below:
Shares | Weighted- Average Grant Date Fair Value Per Share | |||||
Nonvested balance at January 1, 2019 | 241,880 | $ | 16.71 | |||
Granted | — | $ | — | |||
Vested | — | $ | — | |||
Forfeited | (15,870 | ) | $ | 18.10 | ||
Nonvested balance at December 31, 2019 | 226,010 | $ | 16.62 | |||
As of December 31, 2019, there was $0.4 million of unrecognized compensation expense related to outstanding performance restricted stock which is expected to be recognized over a weighted-average period of 0.2 years.
The weighted average grant-date fair value for performance restricted stock granted during the fiscal year ended December 31, 2017 was $16.69 per share.
Total Stockholder Return Restricted Stock Awards (“TSR Awards”)
During the years ended December 31, 2019 and 2018, the Company granted total stockholder return (“TSR”) Awards that include a three-year market condition where the performance measurement period is three years. Vesting of the TSR Awards is based on the Company’s level of attainment of specified TSR targets relative to the percentage appreciation of a specified index of companies for the respective three-year period and is also subject to the continued employment of the grantees. The number of shares that are earned over the performance period ranges from 0% to 200% of the initial award. The fair value of these awards are based on a Monte Carlo Simulation valuation model with the following assumptions:
Year Ended December 31, | ||||||
2019 | 2018 | |||||
Expected volatility | 71.7 | % | 84.3 | % | ||
Risk-free interest rate | 2.4 | % | 2.4 | % | ||
Expected life (in years) | 2.9 | 2.8 | ||||
Expected dividend yield | — | — | ||||
A summary of TSR Award activity for 2019 is presented below:
Shares | Weighted- Average Grant Date Fair Value Per Share | |||||
Nonvested balance at January 1, 2019 | 179,913 | $ | 22.76 | |||
Granted | 152,869 | $ | 39.92 | |||
Vested | — | $ | — | |||
Forfeited | (26,552 | ) | $ | 31.64 | ||
Nonvested balance at December 31, 2019 | 306,230 | $ | 30.56 | |||
As of December 31, 2019, there was $5.8 million of unrecognized compensation expense related to outstanding performance restricted stock which is expected to be recognized over a weighted-average period of 1.9 years.
The weighted average grant-date fair value for TSR Awards granted during the fiscal years ended December 31, 2019 and 2018 was $39.92 and $22.76 per share, respectively.
96
Employee Stock Purchase Plan
In April 2017, the Company’s stockholders approved the 2017 Employee Stock Purchase Plan (“2017 ESPP”), which authorized the issuance of up to 250,000 shares of common stock thereunder. Under the terms of the 2017 ESPP, eligible U.S. employees can elect to acquire shares of the Company’s common stock through periodic payroll deductions during a series of six month offering periods, which will generally begin in March and September of each year. Purchases under the 2017 ESPP are effected on the last business day of each offering period at a 15% discount to the closing price on that day. The 2017 ESPP was implemented, subject to stockholder approval, on March 10, 2017, and the first purchases thereunder were made on September 13, 2017.
13. Leases
Adoption of ASC Topic 842, “Leases”
The Company adopted ASC 842 on January 1, 2019, using the prospective approach which provides a method for recording existing leases at adoption using the effective date of the standard as its initial application date. ASC 842 generally requires all leases to be recognized on the balance sheet. In addition, the Company elected the relief package of practical expedients permitted under the transition guidance within the new standard, which, among other things, allowed the Company not to reassess whether any expired or existing contracts are or contain leases, the lease classification for any expired or existing leases and initial direct costs for any existing leases. The reported results for 2019 reflect the application of ASC 842 guidance while the reported results for 2018 were prepared under the guidance of ASC 840, Leases. The adoption of ASC 842 resulted in the recording of an additional lease asset and lease liability of approximately $1.1 million as of January 1, 2019. ASC 842 did not materially impact the Company’s consolidated results of operations, equity or cash flows as of the adoption date or for the periods presented.
Leases
The Company determines if an arrangement is a lease at inception. The Company has operating and finance leases for vehicles, corporate offices and certain equipment.
Operating lease right-of-use (“ROU”) assets and operating lease liabilities are recognized based on the present value of the future minimum lease payments over the lease term at commencement date. Lease agreements with lease and non-lease components are accounted for separately. As the Company’s leases do not provide an implicit rate, the Company used the incremental borrowing rate based on the information available at commencement date in determining the present value of future payments. The operating lease ROU asset also includes any lease payments made and excludes lease incentives and initial direct costs incurred. The lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. Lease expense for minimum lease payments is recognized on a straight-line basis over the lease term.
Leases with an initial term of 12 months or less are not recorded on the balance sheet as the Company has elected to apply the short-term lease exemption. The Company recognizes lease expense for these leases on a straight-line basis over the lease term.
Operating and finance lease assets and liabilities are as follows:
(in thousands) | Classification | December 31, 2019 | ||
Assets | ||||
Operating | Other long-term assets | $ | 935 | |
Finance | Property, plant and equipment, net | 348 | ||
Total leased assets | $ | 1,283 | ||
Liabilities | ||||
Current | ||||
Operating | Accrued expenses and other liabilities | $ | 193 | |
Finance | Current portion of long-term debt and other borrowings | 143 | ||
Noncurrent | ||||
Operating | Other long-term liabilities | 812 | ||
Finance | Long-term debt, net and other borrowings | 186 | ||
Total leased liabilities | $ | 1,334 | ||
The components of lease expense were as follows:
97
(in thousands) | Year Ended December 31, 2019 | ||
Operating lease expense | $ | 223 | |
Finance lease expense | |||
Amortization of ROU assets | 167 | ||
Interest on lease liabilities | 11 | ||
Short-term lease expense | 91 | ||
Total lease expense | $ | 492 | |
Other information related to leases were as follows:
December 31, 2019 | |
Weighted-average remaining lease term (Years): | |
Operating leases | 4.8 |
Finance leases | 2.5 |
Weighted-average discount rate: | |
Operating leases | 5.1% |
Finance leases | 5.4% |
(in thousands) | Year Ended December 31, 2019 |
Cash paid for amounts included in the measurement of lease liabilities: | |
Operating cash flows from operating leases | 230 |
Operating cash flows from finance leases | 11 |
Financing cash flows from finance leases | 190 |
ROU assets obtained in exchange for lease obligations: | |
Operating leases | — |
Finance leases | 379 |
Future minimum lease payments under non-cancellable leases as of December 31, 2019 were as follows:
(in thousands) | Operating Leases | Finance Leases | |||||
2020 | $ | 238 | $ | 135 | |||
2021 | 238 | 136 | |||||
2022 | 238 | 83 | |||||
2023 | 238 | — | |||||
2024 | 178 | — | |||||
Total future minimum lease payments | 1,130 | 354 | |||||
Less: interest | 125 | 25 | |||||
Total | $ | 1,005 | $ | 329 | |||
98
14. Net Income Per Common Share
A summary of net income per common share is presented below:
Year Ended December 31, | |||||||||||
(in thousands, except per share amounts) | 2019 | 2018 | 2017 | ||||||||
Net income | $ | 31,667 | $ | 40,518 | $ | 123,385 | |||||
Basic weighted-average common shares outstanding | 38,988 | 38,233 | 37,276 | ||||||||
Effect of dilutive stock options | 75 | 61 | 288 | ||||||||
Effect of dilutive restricted stock | 1,050 | 1,207 | 1,328 | ||||||||
Diluted weighted-average common shares outstanding | 40,113 | 39,501 | 38,892 | ||||||||
Basic income per common share | $ | 0.81 | $ | 1.06 | $ | 3.31 | |||||
Diluted income per common share | $ | 0.79 | $ | 1.03 | $ | 3.17 | |||||
Antidilutive securities excluded from diluted net income per common share | 50 | 424 | 604 | ||||||||
15. Commitments and Contingencies
Purchase Commitments
The Company has entered into purchasing arrangements in which minimum quantities of goods or services have been committed to be purchased on an annual basis.
As of December 31, 2019, future payments required under purchase commitments are as follows:
(in thousands) | Amount | ||
2020 | $ | 4,132 | |
2021 | 4,132 | ||
2022 | 2,066 | ||
Total | $ | 10,330 | |
The Company has entered into agreements which contain certain percentage volume purchase requirements. The Company has excluded these future purchase commitments from the table above since there are no minimum purchase commitments or payments under these agreements.
Legal Proceedings
From time to time, the Company is a party to various legal proceedings arising in the ordinary course of business. In addition, the Company has in the past been, and may in the future be, subject to investigations by governmental and regulatory authorities, which expose it to greater risks associated with litigation, regulatory or other proceedings, as a result of which the Company could be required to pay significant fines or penalties. The costs and outcome of litigation, regulatory or other proceedings cannot be predicted with certainty, and some lawsuits, claims, actions or proceedings may be disposed of unfavorably to the Company and could have a material adverse effect on the Company’s results of operations or financial condition. In addition, intellectual property disputes often have a risk of injunctive relief which, if imposed against the Company, could materially and adversely affect its financial condition or results of operations.
In October 2019, the Company was awarded a total of approximately $3.5 million, consisting of damages, pre-judgment interest, and certain arbitration fees, compensation and expenses in its arbitration with Pharmalucence in connection with a Manufacturing and Supply Agreement dated November 12, 2013, under which Pharmalucence agreed to manufacture and supply DEFINITY for the Company. The commercial arrangement contemplated by that agreement was repeatedly delayed and ultimately never successfully realized. After extended settlement discussions between Sun Pharma, the ultimate parent of Pharmalucence, and the Company, which did not lead to a mutually acceptable outcome, on November 10, 2017, the Company filed an arbitration demand (and later an amended arbitration demand) with the American Arbitration Association against Pharmalucence, alleging breach of contract, breach of the covenant of good faith and fair dealing, tortious misrepresentation and violation of the Massachusetts Consumer Protection Law,
99
also known as Chapter 93A. In November 2019, the Company received proceeds of $3.5 million, which is recorded in other expense (income) in the consolidated statement of operations.
As of December 31, 2019, except as disclosed above the Company had no material ongoing litigation in which the Company was a party. In addition, the Company had no material ongoing regulatory or other proceedings and no knowledge of any investigations by government or regulatory authorities in which the Company is a target, in either case that the Company believes could have a material and adverse effect on its current business.
16. 401(k) Plan
The Company maintains a qualified 401(k) plan (the “401(k) Plan”) for its U.S. employees. The 401(k) Plan covers U.S. employees who meet certain eligibility requirements. Under the terms of the 401(k) Plan, the employees may elect to make tax-deferred contributions through payroll deductions within statutory and plan limits, and the Company may elect to make non-elective discretionary contributions. The Company may also make optional contributions to the 401(k) Plan for any plan year at its discretion.
Expense recognized by the Company for matching contributions made to the 401(k) Plan was $2.1 million, $1.8 million and $1.8 million for the years ended December 31, 2019, 2018 and 2017, respectively.
17. Segment Information
The Company reports two operating segments, U.S. and International, based on geographic customer base. The results of these operating segments are regularly reviewed by the Company’s chief operating decision maker, the President and Chief Executive Officer. The Company’s segments derive revenues through the manufacture, marketing, selling and distribution of medical imaging products, focused primarily on cardiovascular diagnostic imaging. All goodwill has been allocated to the U.S. operating segment. The Company does not identify or allocate assets to its segments.
Selected information regarding the Company’s segments are provided as follows:
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
Revenue by product from external customers | |||||||||||
U.S. | |||||||||||
DEFINITY | $ | 211,777 | $ | 178,440 | $ | 153,581 | |||||
TechneLite | 72,534 | 74,042 | 90,489 | ||||||||
Other nuclear | 36,231 | 48,935 | 54,822 | ||||||||
Rebates and allowances | (16,553 | ) | (12,837 | ) | (8,890 | ) | |||||
Total U.S. Revenues | 303,989 | 288,580 | 290,002 | ||||||||
International | |||||||||||
DEFINITY | 5,731 | 4,633 | 3,687 | ||||||||
TechneLite | 14,058 | 24,816 | 14,155 | ||||||||
Other nuclear | 23,574 | 25,349 | 23,558 | ||||||||
Rebates and allowances | (15 | ) | (4 | ) | (24 | ) | |||||
Total International Revenues | 43,348 | 54,794 | 41,376 | ||||||||
Worldwide | |||||||||||
DEFINITY | 217,508 | 183,073 | 157,268 | ||||||||
TechneLite | 86,592 | 98,858 | 104,644 | ||||||||
Other nuclear | 59,805 | 74,284 | 78,380 | ||||||||
Rebates and allowances | (16,568 | ) | (12,841 | ) | (8,914 | ) | |||||
Total Revenues | $ | 347,337 | $ | 343,374 | $ | 331,378 | |||||
100
Year Ended December 31, | |||||||||||
(in thousands) | 2019 | 2018 | 2017 | ||||||||
Geographical revenues | |||||||||||
U.S. | $ | 303,989 | $ | 288,580 | $ | 290,002 | |||||
International | 43,348 | 54,794 | 41,376 | ||||||||
Total revenues | $ | 347,337 | $ | 343,374 | $ | 331,378 | |||||
Operating income | |||||||||||
U.S. | $ | 44,275 | $ | 56,327 | $ | 49,239 | |||||
International | 7,386 | 8,161 | 2,614 | ||||||||
Operating income | 51,661 | 64,488 | 51,853 | ||||||||
Interest expense | 13,617 | 17,405 | 18,410 | ||||||||
Loss on extinguishment of debt | 3,196 | — | 2,442 | ||||||||
Other expense (income) | 6,221 | (2,465 | ) | (8,638 | ) | ||||||
Income before income taxes | $ | 28,627 | $ | 49,548 | $ | 39,639 | |||||
Depreciation and amortization | |||||||||||
U.S. | $ | 11,673 | $ | 12,278 | $ | 17,672 | |||||
International | 414 | 491 | 517 | ||||||||
Total depreciation and amortization | $ | 12,087 | $ | 12,769 | $ | 18,189 | |||||
December 31, | |||||||
(in thousands) | 2019 | 2018 | |||||
Long-lived assets | |||||||
U.S. | $ | 115,560 | $ | 106,755 | |||
International | 937 | 1,133 | |||||
Total long-lived assets | $ | 116,497 | $ | 107,888 | |||
18. Valuation and Qualifying Accounts
(in thousands) | Balance at Beginning of Year | Charged to Income | Deductions from Reserves(1) | Other Adjustments | Balance at End of Year | |||||||||||||||
Allowance for doubtful accounts | ||||||||||||||||||||
Year ended December 31, 2019 | $ | 1,119 | $ | 146 | $ | (323 | ) | $ | — | $ | 942 | |||||||||
Year ended December 31, 2018 | $ | 977 | $ | 321 | $ | (179 | ) | $ | — | $ | 1,119 | |||||||||
Year ended December 31, 2017 | $ | 969 | $ | 136 | $ | (128 | ) | $ | — | $ | 977 | |||||||||
Rebates and allowances | ||||||||||||||||||||
Year ended December 31, 2019 | $ | 4,654 | $ | 16,729 | $ | (14,237 | ) | $ | (161 | ) | $ | 6,985 | ||||||||
Year ended December 31, 2018 | $ | 2,860 | $ | 13,202 | $ | (11,047 | ) | $ | (361 | ) | $ | 4,654 | ||||||||
Year ended December 31, 2017 | $ | 2,297 | $ | 9,568 | $ | (8,351 | ) | $ | (654 | ) | $ | 2,860 | ||||||||
________________________________
(1) | Amounts charged to deductions from allowance for doubtful accounts represent the write-off of uncollectible balances and represent payments for rebates and allowances. |
101
19. Quarterly Consolidated Financial Data (Unaudited)
Summarized quarterly consolidated financial data is presented below:
Quarterly Periods During the Year Ended December 31, 2019 | |||||||||||||||
Q1 | Q2 | Q3 | Q4 | ||||||||||||
(in thousands, except per share data) | |||||||||||||||
Revenues | $ | 86,510 | $ | 85,705 | $ | 85,776 | $ | 89,346 | |||||||
Gross profit | $ | 44,084 | $ | 44,573 | $ | 41,589 | $ | 44,565 | |||||||
Net income | $ | 9,949 | $ | 6,412 | $ | 4,856 | $ | 10,450 | |||||||
Basic income per weighted-average share(a) | $ | 0.26 | $ | 0.16 | $ | 0.12 | $ | 0.27 | |||||||
Diluted income per weighted-average share(a) | $ | 0.25 | $ | 0.16 | $ | 0.12 | $ | 0.26 | |||||||
Quarterly Periods During the Year Ended December 31, 2018 | |||||||||||||||
Q1 | Q2 | Q3 | Q4 | ||||||||||||
(in thousands, except per share data) | |||||||||||||||
Revenues | $ | 82,630 | $ | 85,573 | $ | 88,900 | $ | 86,271 | |||||||
Gross profit | $ | 42,309 | $ | 43,846 | $ | 44,885 | $ | 43,845 | |||||||
Net income | $ | 8,211 | $ | 9,745 | $ | 9,269 | $ | 13,293 | |||||||
Basic income per weighted-average share(a) | $ | 0.22 | $ | 0.25 | $ | 0.24 | $ | 0.35 | |||||||
Diluted income per weighted-average share(a) | $ | 0.21 | $ | 0.25 | $ | 0.24 | $ | 0.34 | |||||||
________________________________
(a) | Quarterly and annual computations are prepared independently. Accordingly, the sum of each quarter may not necessarily total the fiscal year period amounts noted elsewhere within this Annual Report on Form 10-K. |
102
20. Subsequent Events
On February 20, 2020, the Company entered into the Amended Merger Agreement with Progenics, which amends and restates the Initial Merger Agreement. Under the terms of the Amended Merger Agreement, the Company will acquire all of the issued and outstanding shares of Progenics common stock at a fixed exchange ratio whereby Progenics stockholders will receive, for each share of Progenics stock held at the time of the closing of the merger, 0.31 of a share of the Company’s common stock, increased from 0.2502 under the Initial Merger Agreement, together with a non-tradeable contingent value right (a “CVR”) tied to the financial performance of PyLTM (18F-DCFPyL), Progenics’ prostate-specific membrane antigen targeted imaging agent designed to visualize prostate cancer currently in late stage clinical development (“PyL”). Each CVR will entitle its holder to receive a pro rata share of aggregate cash payments equal to 40% of U.S. net sales generated by PyL in 2022 and 2023 in excess of $100 million and $150 million, respectively. In no event will the Company’s aggregate payments under the CVRs exceed 19.9% of the total consideration the Company pays in the transaction. As a result of the increase in the exchange ratio, following the completion of the merger, former Progenics stockholders’ aggregate ownership stake will increase to approximately 40% of the combined company from approximately 35% under the Initial Merger Agreement. Progenics’ stockholders will also now be entitled to appraisal rights as provided under Delaware law. The transaction contemplated by the Amended Merger Agreement was unanimously approved by the Boards of Directors of both companies and requires, among other things, the affirmative vote of a majority of the outstanding shares of common stock of Progenics and a majority of votes cast by the holders of the common stock of the Company.
In addition, pursuant to the Amended Merger Agreement, the holder of each in-the-money option to purchase shares of Progenics common stock under any equity based compensation plan of Progenics (“Progenics Stock Option”) will be entitled to receive in exchange for each such in-the-money option (i) an option to purchase Lantheus Common Stock (each, a “Lantheus Stock Option”) converted based on the 0.31 exchange ratio and (ii) a vested or unvested CVR depending on whether the underlying option is vested. Holders of out-of-the-money Progenics Stock Options will receive Lantheus Stock Options converted on an exchange ratio adjusted based on actual trading prices of common stock of Progenics and Lantheus Holdings prior to the effective time of the merger.
The Amended Merger Agreement also provides that on closing the Company’s board of directors will appoint Dr. Gerard Ber and Mr. Heinz Mausli, who are currently members of the board of directors of Progenics, to serve on the Company’s board of directors. In addition, the Company’s board of directors, subject to complying with applicable fiduciary duties, will use commercially reasonable efforts to cause Dr. Ber and Mr. Mausli to be nominated for reelection following the closing through 2023. The Company’s board of directors will be reduced in size from ten to nine members at the Company’s annual meeting of stockholders on April 23, 2020 (or sooner if the transaction closes before then) and will be further reduced in size from nine to eight members prior to the date of the Company’s 2021 annual meeting of stockholders.
Except as described above, the material terms of the Amended Merger Agreement are substantially the same as the terms of the Initial Merger Agreement.
The transaction is currently expected to close in the second quarter of 2020. Upon completion of the acquisition, which the parties intend to report as tax-deferred to Progenics’ stockholders with respect to the stock component of the merger consideration for U.S. federal income tax purposes, the combined company will continue to be headquartered in North Billerica, Massachusetts and will trade on the NASDAQ under the ticker symbol LNTH.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
The Company’s management, with the participation of the Company’s Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), its principal executive officer and principal financial officer, respectively, has evaluated the effectiveness of the Company’s disclosure controls and procedures as defined in Rule 13a-15(e) and 15d-15(e) of the Exchange Act. Based on that evaluation, the Company’s CEO and CFO concluded that the Company’s disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) were effective as of the period covered by this report.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management, with the participation of our CEO and CFO, is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control system is designed to provide reasonable assurance to our management and Board of Directors regarding the preparation and fair presentation of published financial statements.
103
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2019. In making its assessment of internal control over financial reporting, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control—Integrated Framework (2013). Based on this assessment, management concluded that, as of December 31, 2019, our internal control over financial reporting was effective.
Deloitte & Touche LLP, an independent registered public accounting firm that audited our financial statements for the fiscal year ended December 31, 2019, included in this report, has issued an attestation report on the effectiveness of our internal control over financial reporting. This report is set forth below:
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Lantheus Holdings, Inc.
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of Lantheus Holdings, Inc. and subsidiaries (the “Company”) as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated financial statements as of and for the year ended December 31, 2019, of the Company and our report dated February 25, 2020, expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
February 25, 2020
104
Changes in Internal Controls Over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2019 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
None.
105
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Pursuant to Section 406 of the Sarbanes-Oxley Act of 2002, we have adopted a code of conduct and ethics (our “Code of Conduct”) for all of our employees, including our CEO, CFO and other senior financial officers, or persons performing similar functions, and each of the non-employee directors on our Board of Directors. Our Code of Conduct is currently available on our website, www.lantheus.com. The information on our web site is not part of, and is not incorporated into, this Annual Report on Form 10-K. We intend to provide any required disclosure of any amendment to or waiver from such code that applies to our CEO, CFO and other senior financial officers, or persons performing similar functions, in a Current Report on Form 8-K filed with the SEC.
The additional information required with respect to this item will be incorporated herein by reference to our Definitive Proxy Statement for our 2020 Annual Meeting of Stockholders or an amendment of this report to be filed with the SEC no later than 120 days after the close of our year ended December 31, 2019.
Item 11. Executive Compensation
The information required with respect to this item will be incorporated herein by reference to our Definitive Proxy Statement for our 2020 Annual Meeting of Stockholders or an amendment of this report to be filed with the SEC no later than 120 days after the close of our year ended December 31, 2019.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required with respect to this item will be incorporated herein by reference to our Definitive Proxy Statement for our 2020 Annual Meeting of Stockholders or an amendment of this report to be filed with the SEC no later than 120 days after the close of our year ended December 31, 2019.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required with respect to this item will be incorporated herein by reference to our Definitive Proxy Statement for our 2020 Annual Meeting of Stockholders or an amendment of this report to be filed with the SEC no later than 120 days after the close of our year ended December 31, 2019.
Item 14. Principal Accountant Fees and Services
The information required with respect to this item will be incorporated herein by reference to our Definitive Proxy Statement for our 2020 Annual Meeting of Stockholders or an amendment of this report to be filed with the SEC no later than 120 days after the close of our year ended December 31, 2019.
106
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a)(1) Financial Statements
The following consolidated financial statements of Lantheus Holdings, Inc. are filed as part of this Annual Report on Form 10-K under Part II, Item 8. Financial Statements and Supplementary Data:
Page | |
(a)(2) Schedules
All schedules are omitted because they are not applicable, not required, or because the required information is included in the consolidated financial statements or notes thereto.
(a)(3) Exhibits
EXHIBIT INDEX
Incorporated by Reference | ||||||||||
Exhibit Number | Description of Exhibits | Form | File Number | Exhibit | Filing Date | |||||
2.1 | 8-K | 001-36569 | 10.1 | October 2, 2019 | ||||||
3.1 | 8-K | 001-36569 | 3.1 | April 27, 2018 | ||||||
3.2 | 8-K | 001-36569 | 3.2 | April 27, 2018 | ||||||
4.1 | 8-K | 001-36569 | 4.1 | June 30, 2015 | ||||||
4.2* | ||||||||||
10.1† | S-4 | 333-169785 | 10.9 | December 23, 2010 | ||||||
10.2† | S-4 | 333-169785 | 10.10 | December 1, 2010 | ||||||
10.3† | 10-Q | 333-169785 | 10.1 | May 13, 2011 | ||||||
10.4+ | S-4 | 333-169785 | 10.18 | October 6, 2010 | ||||||
10.5+ | S-4 | 333-169785 | 10.19 | October 6, 2010 | ||||||
10.6+ | S-4 | 333-169785 | 10.20 | October 6, 2010 | ||||||
10.7+ | S-4 | 333-169785 | 10.21 | October 6, 2010 | ||||||
10.9† | 10-Q | 333-169785 | 10.2 | May 15, 2012 | ||||||
10.10† | 10-Q | 333-169785 | 10.1 | August 14, 2012 | ||||||
10.11† | 10-Q | 001-36569 | 10.53 | May 2, 2018 | ||||||
10.12+ | 8-K | 333-169785 | 10.1 | May 6, 2013 | ||||||
10.13+ | 8-K | 333-169785 | 10.2 | May 6, 2013 | ||||||
10.14+ | 8-K | 333-169785 | 10.3 | May 6, 2013 | ||||||
10.15+ | S-1 | 333-196998 | 10.37 | June 24, 2015 | ||||||
107
Incorporated by Reference | ||||||||||
Exhibit Number | Description of Exhibits | Form | File Number | Exhibit | Filing Date | |||||
10.16+ | S-1 | 333-196998 | 10.38 | June 24, 2015 | ||||||
10.17+ | S-1 | 333-196998 | 10.39 | June 24, 2015 | ||||||
10.18+ | S-1 | 333-196998 | 10.40 | June 24, 2015 | ||||||
10.19+ | S-1 | 333-196998 | 10.41 | June 24, 2015 | ||||||
10.20+ | 8-K | 001-36569 | 10.1 | April 28, 2016 | ||||||
10.21† | 10-Q | 001-36569 | 10.2 | November 1, 2016 | ||||||
10.22+ | 8-K | 001-36569 | 10.1 | April 28, 2017 | ||||||
10.23+ | 8-K | 001-36569 | 10.2 | April 28, 2017 | ||||||
10.24† | 10-Q | 001-36569 | 10.1 | August 1, 2017 | ||||||
10.25† | 10-K | 001-36569 | 10.65 | February 7, 2018 | ||||||
10.26*+ | 10-K | 001-36569 | 10.68 | February 20, 2019 | ||||||
10.27*+ | 10-K | 001-36569 | 10.69 | February 20, 2019 | ||||||
10.28*+ | 10-K | 001-36569 | 10.70 | February 20, 2019 | ||||||
10.29*+ | 10-K | 001-36569 | 10.71 | February 20, 2019 | ||||||
10.30+ | 10-Q | 001-36569 | 10.1 | April 30, 2019 | ||||||
10.31+ | 10-Q | 001-36569 | 10.1 | July 25, 2019 | ||||||
10.32+ | 10-Q | 001-36569 | 10.2 | July 25, 2019 | ||||||
10.33 | 10-Q | 001-36569 | 10.3 | July 25, 2019 | ||||||
10.34*†† | ||||||||||
21.1* | ||||||||||
23.1* | ||||||||||
24.1* | ||||||||||
31.1* | ||||||||||
31.2* | ||||||||||
32.1** | ||||||||||
101.INS* | XBRL Instance Document | |||||||||
101.SCH* | XBRL Taxonomy Extension Schema | |||||||||
101.CAL* | XBRL Taxonomy Extension Calculation | |||||||||
101.DEF* | XBRL Taxonomy Extension Definition | |||||||||
101.LAB* | XBRL Taxonomy Extension Labels | |||||||||
101.PRE* | XBRL Taxonomy Extension Presentation | |||||||||
________________________________
* | Filed herewith. |
** | Furnished herewith. |
†† | Portions of this exhibit have been omitted for confidential treatment pursuant to Item 601(b)(10)(iv) of Regulation S-K. |
+ | Indicates management contract or compensatory plan or arrangement. |
† | Confidential treatment requested as to certain portions, which portions have been filed separately with the Securities and Exchange Commission |
108
Item 16. Form 10-K Summary
None.
109
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
LANTHEUS HOLDINGS, INC. | |
By: | /S/ MARY ANNE HEINO |
Name: | Mary Anne Heino |
Title: | President and Chief Executive Officer |
Date: | February 25, 2020 |
We, the undersigned directors and officers of Lantheus Holdings, Inc., hereby severally constitute and appoint Mary Anne Heino, Robert J. Marshall, Jr. and Michael P. Duffy, and each of them individually, with full powers of substitution and resubstitution, our true and lawful attorneys, with full powers to them and each of them to sign for us, in our names and in the capacities indicated below, any and all amendments to this Annual Report on Form 10-K filed with the SEC, granting unto said attorneys-in-fact and agents, each acting alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that any such attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
/S/ MARY ANNE HEINO | Chief Executive Officer, President and Director (Principal Executive Officer) | February 25, 2020 | ||
Mary Anne Heino | ||||
/S/ ROBERT J. MARSHALL, JR. | Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) | February 25, 2020 | ||
Robert J. Marshall, Jr. | ||||
/S/ BRIAN MARKISON | Chairman of the Board of Directors | February 25, 2020 | ||
Brian Markison | ||||
/S/ JAMES C. CLEMMER | Director | February 25, 2020 | ||
James C. Clemmer | ||||
/S/ SAMUEL R. LENO | Director | February 25, 2020 | ||
Samuel R. Leno | ||||
/S/ JULIE H. MCHUGH | Director | February 25, 2020 | ||
Julie H. McHugh | ||||
/S/ GARY J. PRUDEN | Director | February 25, 2020 | ||
Gary J. Pruden | ||||
/S/ KENNETH J. PUCEL | Director | February 25, 2020 | ||
Kenneth J. Pucel | ||||
/S/ DR. FREDERICK A. ROBERTSON | Director | February 25, 2020 | ||
Dr. Frederick A. Robertson | ||||
/S/ DR. DERACE L. SCHAFFER | Director | February 25, 2020 | ||
Dr. Derace L. Schaffer | ||||
/S/ DR. JAMES H. THRALL | Director | February 25, 2020 | ||
Dr. James H. Thrall | ||||
110