Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - BIOVIE INC. | bivi-20180630_10kex32z2.htm |
| EX-32.1 - EXHIBIT 32.1 - BIOVIE INC. | bivi-20180630_10kex32z1.htm |
| EX-31.2 - EXHIBIT 31.2 - BIOVIE INC. | bivi-20180630_10kex31z2.htm |
| EX-31.1 - EXHIBIT 31.1 - BIOVIE INC. | bivi-20180630_10kex31z1.htm |
UNITED
STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| ☒ | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
FOR THE FISCAL YEAR ENDED JUNE 30, 2018
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ____________to _____________
Commission File Number: 333-190635
BIOVIE INC.
(Exact name of registrant as specified in its charter)
| Nevada | 46-2510769 | |
| (State or other jurisdiction of | (I.R.S. Empl. Ident. No.) | |
| incorporation or organization) |
| 100 Cummings Center, Suite 247-C | ||
| Beverly, MA 01915 | ||
| (Address of principal executive offices, Zip Code) | ||
| (312)-283-5793 | ||
| (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(g) of the Act:
| $.0001 par value common stock | Over the Counter Bulletin Board |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the past 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer," "smaller reporting company" and "emerging growth company" in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer | ☐ | Accelerated Filer | ☐ |
| Non-Accelerated Filer | ☐ | Smaller reporting company | ☒ |
| (Do not check if a smaller reporting company) | Emerging growth company | ☐ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ☐ No ☒
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act
Yes ☐ No ☒
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
The Aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed fourth fiscal quarter, June 30, 2018 was $5,319,173.
There were 315,053,673 shares of the Registrant’s $0.0001 par value common stock outstanding as of September 28, 2018.
BIOVIE INC.
PART I
| Item 1. | Description of Business | 1 |
| Item 1A. | Risk Factors | 8 |
| Item 1B. | Unresolved Staff Comments | 19 |
| Item 2. | Description of Property | 19 |
| Item 3. | Legal Proceedings | 19 |
| Item 4. | Mine Safety Disclosure | 19 |
PART II
| Item 5. | Market for Common Equity and Related Stockholder Matters | 19 |
| Item 6. | Selected Financial Data | 20 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 20 |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 25 |
| Item 8. | Financial Statements and Supplementary Data | 26 |
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 27 |
| Item 9A(T). | Controls and Procedures | 27 |
| Item 9B. | Other information | 27 |
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance | 28 |
| Item 11. | Executive Compensation | 31 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 32 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 34 |
| Item 14. | Principal Accountant Fees and Services | 34 |
PART IV
| Item 15. | Exhibits and Financial Statement Schedules | 35 |
| Signatures | 35 | |
| Certifications |
BIOVIE INC.
This Annual Report on Form 10-K and the documents incorporated herein by reference contain forward-looking statements that have been made pursuant to the provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements are based on current expectations, estimates and projections about BioVie Inc.’s industry, management beliefs, and assumptions made by management. Words such as “anticipates,” “expects,” “intends,” “plans,” “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements. These statements are not guarantees of future performance and are subject to certain risks, uncertainties and assumptions that are difficult to predict; therefore, actual results and outcomes may differ materially from what is expressed or forecasted in any such forward-looking statements.
PART I
| ITEM 1. | DESCRIPTION OF BUSINESS |
Introduction
BioVie Inc. (the “Company”) is a clinical-stage company pursuing the discovery, development, and commercialization of innovative drug therapies. The Company is currently focused on developing and commercializing BIV201, a novel approach to the treatment of ascites due to chronic liver cirrhosis. In March 2017, the Company received notification from the FDA that it could initiate a Phase 2a US clinical trial. In April 2017, the Company signed a Cooperative Research and Development Agreement (CRADA) with the McGuire Research Institute Inc. in Richmond, VA, and began dosing patients with BIV201 in September 2017. As of June 2018, three patients had been treated with BIV201 therapy in this ongoing Phase 2a clinical trial.
BIV201 has the potential to improve the health of thousands of patients suffering from life-threatening complications of liver cirrhosis due to hepatitis, NASH, and alcoholism. It has FDA Fast-Track status and Orphan Drug designation for the most common of these complications, ascites, which represents a significant unmet medical need. The FDA has never approved any drug specifically for treating ascites.
The BIV201 development program began at LAT Pharma LLC. On April 11, 2016, the Company acquired LAT Pharma LLC and the rights to its BIV201 development program. The Company currently owns all development and marketing rights to its drug candidate. The Company and PharmaIN, Corp. (“PharmaIN”), LAT Pharma’s former partner focused on the development of new modified drug candidates in the same therapeutic field but not including BIV201, have agreed to pay royalties equal to less than 1% of future net sales of each company's ascites drug development programs, or if such program is licensed to a third party, less than 5% of each company's net license revenues. The Company’s relationship with PharmaIN could advance into a collaboration or be terminated. The Company has an issued US Patent covering the use of BIV201 for the treatment of ascites patients in the outpatient setting using ambulatory pump infusion, and has filed patent applications for its drug candidate in Japan, and Europe, and China.
The Company’s activities are subject to significant risks and uncertainties including failure to secure additional funding to properly execute the Company’s business plan.
About Ascites and Liver Cirrhosis
About 600,000 Americans and millions worldwide suffer from liver cirrhosis. Cirrhosis is the 12th leading cause of death due to disease in the US, killing more than 30,000 people each year. The condition results primarily from hepatitis, alcoholism, and fatty liver disease linked to obesity. Ascites is a common complication of advanced liver cirrhosis, involving kidney dysfunction and the accumulation of large amounts of fluid in the abdominal cavity.
The Need for an Ascites Therapy
With no medications approved by the FDA specifically for treating ascites, an estimated 40% of patients die within two years of diagnosis. Certain drugs approved for other uses such as diuretics may provide initial relief, but patients may fail to respond to treatment as ascites worsens. This represents a critical unmet medical need. US treatment costs for liver cirrhosis, including ascites and other complications, are estimated at more than $4 billion annually.
-1-
The Ascites Development Pathway
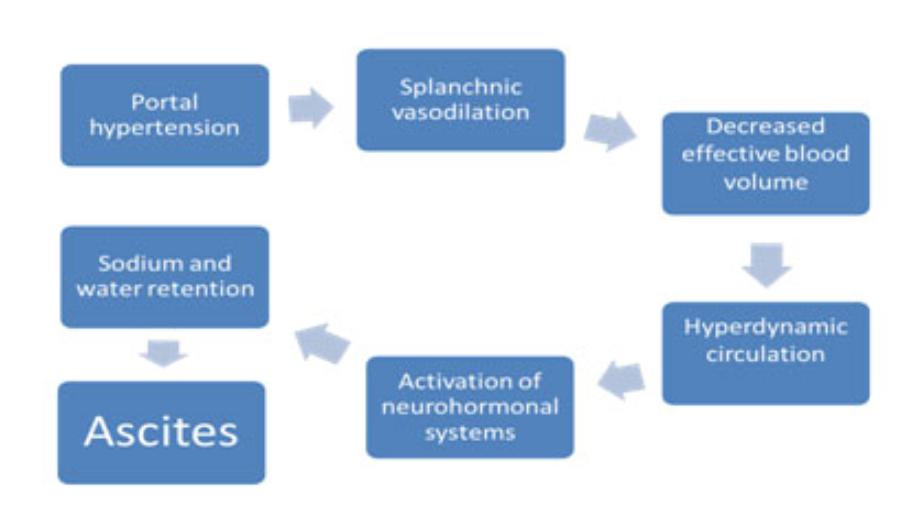
Most experts agree that ascites develops through a sequence of events illustrated by the above diagram. High blood pressure in the vein that supplies blood to the liver, called “portal hypertension,” occurs as increasing liver damage (fibrosis) impedes blood flow through the liver. This causes vasodilation and blood pooling in the central or “splanchnic” region of the body and low blood volume in the arteries. The decrease in effective blood volume activates a signaling pathway (“neurohormonal systems”) which tells the kidneys to retain large amounts of salt and water in an effort to increase blood volume. Ultimately the retention of excess sodium and water leads to the formation of ascites as these substances “weep” from the liver and lymph system and collect in the patient’s abdomen.
The BIV201 Mechanism of Action
BIV201 is being developed by BioVie with the goal of alleviating the portal hypertension and correcting splanchnic vasodilation, thereby increasing effective blood volume and reducing the signals to the kidneys to retain excess salt and water. If successful, BIV201 could halt the cycle of accelerating fluid generation in ascites patients and reduce the need for the frequent and painful paracentesis procedures many of these patients currently require.
Future Possible BIV201 Indications
Based on investigative studies around the world of the active agent in BIV201, terlipressin, our new drug candidate has potential future applications in other life-threatening conditions due to liver cirrhosis, such as those listed below. Securing marketing approvals for any of these new uses will require well-controlled clinical trials to satisfy the FDA and/or other countries’ regulatory requirements, none of which have commenced at this time. The Company may be unable to, or chose not to, pursue the development BIV201 for these indications.
| · | Bleeding Esophageal Varices (BEV): The bursting of blood vessels lining the esophagus due to high blood pressure (“portal hypertension”) in the vein which supplies blood to the liver resulting as a result of advanced liver cirrhosis. This situation requires emergency treatment to avoid blood loss and death. |
| · | Hepatorenal syndrome (HRS): As their disease progresses liver cirrhosis patients’ kidneys may begin to fail, and this deadly condition may set in. It often occurs once a patient no longer responds to (off-label) drugs used to control ascites. The second stage is called “type 1 HRS” and requires hospitalization as multiple organ failure and death may occur. |
-2-
Joint Venture and Possible Access to Early-Stage Compounds
The Company has an Agreement with PharmaIN providing certain limited rights and information on their program to develop novel modified terlipressin compounds. Although at an early stage, these compounds hold the promise of simpler and potentially safer dosing for patients outside the hospital. If this program makes significant advances, BioVie may contact PharmaIN to explore a licensing opportunity.
Efflux Pump Antibiotics Program
Prior to the Merger of Lat Pharma LLC and NanoAntibiotics Inc. in April 2016, the Company was exclusively developing novel nanotechnology anti-infective drugs to combat multi-drug resistant bacteria. We are at an early stage of discovery and development of broad spectrum antibiotics for gram-negative and gram-positive bacterial infections. Developing this technology in-house is resource-intensive with respect to time, personnel and capital necessary for scientific discovery. For further development of our nanoantibiotic technology we will need to find and license additional nanotechnology to complete our planned products. Presently this program is inactive as we are focusing our efforts on BIV201.
Intellectual Property
BioVie relies on a combination of trade secrecy and patent strategy to protect our confidential information and seek market exclusivity for our products. In May 2017 the Company announced issuance of a US patent covering the use of BIV201 in the treatment of ascites due to liver cirrhosis with administration via ambulatory pump. In July 2017 the Company announced filing an application for similar patent coverage in Japan, and subsequently filed for patent protection in Europe and China. BioVie has secured Orphan Drug designation for BIV201 in the treatment of ascites from the US Food and Drug Administration (FDA). The Company has applied for two additional Orphan Drug designations which could be granted in late 2018 or early 2019.
Research and Development
For the year ended June 30, 2018, the Company spent $370,853 in research and development activities.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing. Any pharmaceutical candidate that we develop must be approved by the FDA before it may be legally marketed in the United States and by the appropriate foreign regulatory agency before it may be legally marketed in foreign countries.
United States Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act, or FDCA, and implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. Biologics are subject to regulation by the FDA under the FDCA, the Public Health Service Act, or the PHSA, and related regulations, and other federal, state and local statutes and regulations. Biological products include, among other things, viruses, therapeutic serums, vaccines and most protein products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
-3-
The process required by the FDA before a drug or biological product may be marketed in the United States generally involves the following:
• Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other applicable regulations;
• Submission to the FDA of an Investigational New Drug Application, or an IND, which must become effective before human clinical trials may begin;
• Performance of adequate and well-controlled human clinical trials according to the FDA's current good clinical practices, or GCPs, to establish the safety and efficacy of the proposed drug or biologic for its intended use;
•Submission to the FDA of a New Drug Application, or an NDA, for a new drug product, or a Biologics License Application, or a BLA, for a new biological product;
•Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the drug or biologic is to be produced to assess compliance with the FDA's current good manufacturing practice standards, or cGMP, to assure that the facilities, methods and controls are adequate to preserve the drug's or biologic's identity, strength, quality and purity;
• Potential FDA audit of the nonclinical and clinical trial sites that generated the data in support of the NDA or BLA; and
• FDA review and approval of the NDA or BLA.
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources. There can be no certainty that approvals will be granted.
Clinical trials involve the administration of the drug or biological candidate to healthy volunteers or patients having the disease being studied under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor's control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted in accordance with the FDA's good clinical practices requirements. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until it is completed.
Human clinical trials prior to approval are typically conducted in three sequential Phases that may overlap or be combined:
• Phase 1. The drug or biologic is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients having the specific disease.
• Phase 2. The drug or biologic is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule for patients having the specific disease.
• Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials, which usually involve more subjects than earlier trials, are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. Generally, at least two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA or BLA.
-4-
Post-approval studies, or Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and may be required by the FDA as part of the approval process.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA by the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements or if the drug or biologic has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and develop additional information about the chemistry and physical characteristics of the drug or biologic as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug or biological candidate and, among other things, must include methods for testing the identity, strength, quality and purity of the final drug or biologic. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug or biological candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug or biologic, proposed labeling and other relevant information are submitted to the FDA as part of an NDA or BLA requesting approval to market the product. The submission of an NDA or BLA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances.
The FDA reviews all NDAs and BLAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA or BLA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA or BLA.
After the NDA or BLA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product's identity, strength, quality and purity. The FDA reviews a BLA to determine, among other things, whether the product is safe, pure and potent and the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product's continued safety, purity and potency. In addition to its own review, the FDA may refer applications for novel drug or biological products or drug or biological products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the approval process, the FDA also will determine whether a risk evaluation and mitigation strategy, or REMS, is necessary to assure the safe use of the drug or biologic. If the FDA concludes that a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS; the FDA will not approve the NDA or BLA without a REMS, if required.
Before approving an NDA or BLA, the FDA will inspect the facilities at which the product is to be manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with cGMP. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable it will outline the deficiencies in the submission and often will request additional testing or information.
-5-
The NDA or BLA review and approval process is lengthy and difficult and the FDA may refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA will issue a "complete response" letter if the agency decides not to approve the NDA or BLA. The complete response letter usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical trials designed to further assess a product's safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA or BLA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has Orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors, however, may receive approval of different products for the indication for which the Orphan product has exclusivity or obtain approval for the same product but for a different indication for which the Orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same drug or biological product as defined by the FDA or if our drug or biological candidate is determined to be contained within the competitor's product for the same indication or disease. If a drug or biological product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity. Orphan drug status in the European Union has similar but not identical benefits in the European Union.
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drug and biological products that meet certain criteria. Specifically, new drug and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA or BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA or BLA, the FDA agrees to accept sections of the NDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA or BLA.
-6-
Any product submitted to the FDA for marketing approval, including those submitted to a Fast Track program, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared with marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA generally requires that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies to establish safety and efficacy for the approved indication. Failure to conduct such studies or conducting such studies that do not establish the required safety and efficacy may result in revocation of the original approval. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch or subsequent marketing of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Post-Approval Requirements
Any drug or biological products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information on an annual basis or as required more frequently for specific events, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, prohibitions against promoting drugs and biologics for uses or in patient populations that are not described in the drug's or biologic's approved labeling (known as "off-label use"), rules for conducting industry-sponsored scientific and educational activities, and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including the immediate discontinuation of noncomplying materials, adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available drugs and biologics for off-label uses, manufacturers may not market or promote such off-label uses.
We will need to rely, on third parties for the production of our product candidates. Manufacturers of our product candidates are required to comply with applicable FDA manufacturing requirements contained in the FDA's cGMP regulations. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of comprehensive records and documentation. Drug and biologic manufacturers and other entities involved in the manufacture and distribution of approved drugs and biologics are also required to register their establishments and list any products made there with the FDA and comply with related requirements in certain states, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in serious and extensive restrictions on a product, manufacturer, or holder of an approved NDA or BLA, including suspension of a product until the FDA is assured that quality standards can be met, continuing oversight of manufacturing by the FDA under a "consent decree," which frequently includes the imposition of costs and continuing inspections over a period of many years, and possible withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could otherwise restrict the distribution or use of the product.
-7-
Employees
Our business is managed by our officers. Our Chief Operating Officer, Jonathan Adams, began devoting full-time efforts to the Company on July 1st, 2017. Our Corporate Secretary, Julie Anderson, devote part time efforts to the Company’s activities. There are no additional employees. The Company relies on a team of highly experienced scientific, medical, and regulatory consultants to conduct its drug development activities.
| ITEM 1A. | RISK FACTORS |
THE SECURITIES BEING OFFERED INVOLVE A HIGH DEGREE OF RISK AND, THEREFORE, SHOULD BE CONSIDERED EXTREMELY SPECULATIVE. THEY SHOULD NOT BE PURCHASED BY PERSONS WHO CANNOT AFFORD THE POSSIBILITY OF THE LOSS OF THE ENTIRE INVESTMENT. PROSPECTIVE INVESTORS SHOULD READ THE ENTIRE PROSPECTUS, INCLUDING ALL EXHIBITS, AND CAREFULLY CONSIDER, AMONG OTHER FACTORS THE FOLLOWING RISK FACTORS.
Risks Relating to Our Business and Industry
We are a development stage company with a limited operating history, making it difficult for you to evaluate our business and your investment.
BioVie Inc. was incorporated on April 10, 2013. We are a development stage biopharmaceutical company with a potential therapy that we have not evaluated in clinical trials, and our operations are subject to all of the risks inherent in the establishment of a new business enterprise, including but not limited to the absence of an operating history, the lack of commercialized products, insufficient capital, expected substantial and continual losses for the foreseeable future, limited experience in dealing with regulatory issues, the lack of manufacturing experience and limited marketing experience, possible reliance on third parties for the development and commercialization of our proposed products, a competitive environment characterized by numerous, well-established and well capitalized competitors and reliance on key personnel.
Since inception, we have not established any revenues or operations that shall provide financial stability in the long term, and there can be no assurance that the Company will realize its plans on its projected timetable in order to reach sustainable or profitable operations.
Investors are subject to all the risks incident to the creation and development of a new business and each Investor should be prepared to withstand a complete loss of his, her or its investment. Furthermore, the accompanying financial statements have been prepared assuming that the Company will continue as a going concern. The Company has not emerged from the development stage, and may be unable to raise further equity. These factors raise substantial doubt about its ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Because we are subject to these risks, you may have a difficult time evaluating our business and your investment in our Company. Our ability to become profitable depends primarily on our ability to develop drugs, to obtain approval for such drugs, and if approved, to successfully commercialize our drugs, our R&D efforts, including the timing and cost of clinical trials; and our ability to enter into favorable alliances with third-parties who can provide substantial capabilities in clinical development, regulatory affairs, sales, marketing and distribution.
Even if we successfully develop and market our drug candidates, we may not generate sufficient or sustainable revenue to achieve or sustain profitability, which could cause us to cease operations and cause you to lose all of your investment
-8-
If the Company fails to maintain an effective system of internal controls, it may not be able to accurately report its financial results or detect fraud. Consequently, investors could lose confidence in the Company’s financial reporting and this may decrease the trading price of its stock.
The Company must maintain effective internal controls to provide reliable financial reports and detect fraud. The Company has concluded that its disclosure controls and procedures internal controls, as well as internal controls over financial reporting, are ineffective. Failure to implement changes to the Company’s internal controls or any others that it identifies as necessary to establish an effective system of internal controls could harm its operating results and cause investors to lose confidence in the Company’s reported financial information. Any such loss of confidence would have a negative effect on the trading price of the Company’s stock.
We have no products approved for commercial sale, have never generated any revenues and may never achieve revenues or profitability, which could cause us to cease operations.
We have no products approved for commercial sale and, to date, we have not generated any revenues. Our ability to generate revenue depends heavily on (a) successful development program and thereafter demonstration in human clinical trials that BIV201, our drug candidate, is safe and effective; (b) our ability to seek and obtain regulatory approvals, including, without limitation, with respect to the indications we are seeking; (c) successful commercialization of our product candidates; and (d) market acceptance of our products. There are no assurances that we will achieve any of the forgoing objectives. Furthermore, our drug candidate is in the development stage, and we have not evaluated it in human clinical trials. If we do not successfully develop and commercialize our drug candidate we will not achieve revenues or profitability in the foreseeable future, if at all. If we are unable to generate revenues or achieve profitability, we may be unable to continue our operations.
We will need to raise substantial additional capital in the future to fund our operations and we may be unable to raise such funds when needed and on acceptable terms, which could have a materially adverse effect on our business.
Developing biopharmaceutical products, including conducting pre-clinical studies and clinical trials and establishing manufacturing capabilities, requires substantial funding. As of June 30, 2018, we had cash and cash equivalents totaling $45,800. Additional financing will be required to fund the research and development of our product candidates. We have not generated any product revenues, and do not expect to generate any revenues until, and only if, we develop, and receive approval to sell our drug candidates from the FDA and other regulatory authorities for our product candidates.
We may not have the resources to complete the development and commercialization of any of our proposed drug candidate. We will require additional financing to further the clinical development of our drug candidate. In the event that we cannot obtain the required financing, we will be unable to complete the development necessary to file an investigational new drug application with the FDA for BIV201, our drug candidate. This will delay research and development programs, preclinical studies and clinical trials, material characterization studies, regulatory processes, the establishment of our own laboratory or a search for third party marketing partners to market our products for us, which could have a materially adverse effect on our business.
The amount of capital we may need will depend on many factors, including the progress, timing and scope of our research and development programs, the progress, timing and scope of our preclinical studies and clinical trials, the time and cost necessary to obtain regulatory approvals, the time and cost necessary to establish our own marketing capabilities or to seek marketing partners, the time and cost necessary to respond to technological and market developments, changes made or new developments in our existing collaborative, licensing and other commercial relationships, and new collaborative, licensing and other commercial relationships that we may establish.
Until we can generate a sufficient amount of product revenue, if ever, we expect to finance future cash needs, through public or private equity offerings, debt financings, or corporate collaboration and licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. In addition, we could be forced to discontinue product development and reduce or forego attractive business opportunities. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional significant dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates, or grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
-9-
Our fixed expenses, such as rent and other contractual commitments, will likely increase in the future, as we may enter into leases for new facilities and capital equipment; enter into additional licenses and collaborative agreements. Therefore, if we fail to raise substantial additional capital to fund these expenses, we could be forced to cease operations, which could cause you to lose all of your investment.
We have limited experience in drug development and may not be able to successfully develop any drugs, which would cause us to cease operations.
The Company has never successfully developed a new drug and brought it to market. Our management and clinical teams have experience in drug development but they may not be able to successfully develop any drugs. Our ability to achieve revenues and profitability in our business will depend on, among other things, our ability to develop products internally or to obtain rights to them from others on favorable terms; complete laboratory testing and human studies; obtain and maintain necessary intellectual property rights to our products; successfully complete regulatory review to obtain requisite governmental agency approvals; enter into arrangements with third parties to manufacture our products on our behalf; and enter into arrangements with third parties to provide sales and marketing functions. If we are unable to achieve these objectives we will be forced to cease operations and you will lose all of your investment.
Development of pharmaceutical products is a time-consuming process, subject to a number of factors, many of which are outside of our control. Consequently, if we are unsuccessful or fail to timely develop new drugs, we could be forced to discontinue our operations.
Our lead drug candidate, BIV201, has been cleared by the US Food and Drug Administration (FDA) to begin a mid-stage (Phase 2a) clinical trial. Further development and extensive testing will be required to determine its technical feasibility and commercial viability. Our success will depend on our ability to achieve scientific and technological advances and to translate such advances into reliable, commercially competitive drugs on a timely basis. Drugs that we may develop are not likely to be commercially available for a few years, if ever. The proposed development schedules for our drug candidate may be affected by a variety of factors, including technological difficulties, proprietary technology of others, and changes in government regulation, many of which will not be within our control. Any delay in the development, introduction or marketing of our drug candidates could result either in such drugs being marketed at a time when their cost and performance characteristics would not be competitive in the marketplace or in the shortening of their commercial lives. In light of the long-term nature of our projects and other risk factors described elsewhere in this document, we may not be able to successfully complete the development or marketing of any drugs which could cause us to cease operations.
We may fail to successfully develop and commercialize our drug candidate(s) if it is found to be unsafe or ineffective in clinical trials; does not receive necessary approval from the FDA or foreign regulatory agencies; fails to conform to a changing standard of care for the disease it seeks to treat; or is less effective or more expensive than current or alternative treatment methods.
Drug development failure can occur at any stage of clinical trials and as a result of many factors and there can be no assurance that we or our collaborators will reach our anticipated clinical targets. Even if we or our collaborators complete our clinical trials, we do not know what the long-term effects of exposure to our drug candidate will be. Furthermore, our drug candidate may be used in combination with other treatments and there can be no assurance that such use will not lead to unique safety issues. Failure to complete clinical trials or to prove that our drug candidate is safe and effective would have a material adverse effect on our ability to generate revenue and could require us to reduce the scope of or discontinue our operations, which could cause you to lose all of your investment.
-10-
We have no manufacturing experience, and the failure to comply with all applicable manufacturing regulations and requirements could have a materially adverse effect on our business.
The Company has never manufactured products in the highly regulated environment of pharmaceutical manufacturing, and our team has limited experience in the manufacture of drug therapies. There are numerous regulations and requirements that must be maintained to obtain licensure and permitting required prior to the commencement of manufacturing, as well as additional requirements to continue manufacturing pharmaceutical products. We do not own or lease facilities currently that could be used to manufacture any products that might be developed by the Company, nor do we have the resources at this time to acquire or lease suitable facilities. If we fail to comply with regulations, to obtain the necessary licenses and knowhow or to obtain the requisite financing in order to comply with all applicable regulations and to own or lease the required facilities in order to manufacture our products, we could be forced to cease operations, which would cause you to lose all of your investment.
We do not currently have the sales and marketing personnel necessary to sell products, and the failure to hire and retain such staff could have a materially adverse effect on our business.
We are an early stage development Company with limited resources. Even if we had products available for sale, which we currently do not, we have not secured sales and marketing staff at this early stage of operations to sell products. We cannot generate sales without sales or marketing staff and must rely on officers to provide any sales or marketing services until such personnel are secured, if ever. If we fail to hire and retain the requisite expertise in order to market and sell our products or fail to raise sufficient capital in order to afford to pay such sales or marketing staff, then we could be forced to cease operations and you could lose all of your investment.
Even if we were to successfully develop approvable drugs, we will not be able to sell these drugs if we or our third-party manufacturers fail to comply with manufacturing regulations, which could have a materially adverse effect on our business.
If we were to successfully develop approvable drugs, before we can begin selling these drugs, we must obtain regulatory approval of our manufacturing facility and process or the manufacturing facility and process of the third party or parties with whom we may outsource our manufacturing activities. In addition, the manufacture of our products must comply with the FDA's current Good Manufacturing Practices regulations, commonly known as GMP regulations. The GMP regulations govern quality control and documentation policies and procedures. Our manufacturing facilities, if any in the future, and the manufacturing facilities of our third-party manufacturers will be continually subject to inspection by the FDA and other state, local and foreign regulatory authorities, before and after product approval. We cannot guarantee that we, or any potential third-party manufacturer of our products, will be able to comply with the GMP regulations or other applicable manufacturing regulations. The failure to comply with all necessary regulations would have a materially adverse effect on our business and could force us to cease operations and you could lose all of your investment.
We must comply with significant and complex government regulations, compliance with which may delay or prevent the commercialization of our drug candidate, which could have a materially adverse effect on our business.
The R&D, manufacture and marketing of drug candidates are subject to regulation, primarily by the FDA in the United States and by comparable authorities in other countries. These national agencies and other federal, state, local and foreign entities regulate, among other things, R&D activities (including testing in animals and in humans) and the testing, manufacturing, handling, labeling, storage, record keeping, approval, advertising and promotion of the product that we are developing. Noncompliance with applicable requirements can result in various adverse consequences, including approval delays or refusals to approve drug licenses or other applications, suspension or termination of clinical investigations, revocation of approvals previously granted, fines, criminal prosecution, recalls or seizures of products, injunctions against shipping drugs and total or partial suspension of production and/or refusal to allow a company to enter into governmental supply contracts.
The process of obtaining FDA approval has historically been costly and time consuming. Current FDA requirements for a new human drug or biological product to be marketed in the United States include: (a) the successful conclusion of pre-clinical laboratory and animal tests, if appropriate, to gain preliminary information on the product's safety; (b) filing with the FDA of an IND application to conduct human clinical trials for drugs or biologics; (c) the successful completion of adequate and well-controlled human clinical investigations to establish the safety and efficacy of the product for its recommended use; and (d) filing by a company and acceptance and approval by the FDA of a New Drug Application (NDA) for a drug product or a biological license application (BLA) for a biological product to allow commercial distribution of the drug or biologic. A delay in one or more of the procedural steps outlined above could be harmful to us in terms of getting our drug candidates through clinical testing and to market, which could have a materially adverse effect on our business.
-11-
The FDA reviews the results of the clinical trials and may order the temporary or permanent discontinuation of clinical trials at any time if it believes the drug candidate exposes clinical subjects to an unacceptable health risk. Investigational drugs used in clinical studies must be produced in compliance with current good manufacturing practice (GMP) rules pursuant to FDA regulations.
Sales outside the United States of products that we develop will also be subject to regulatory requirements governing human clinical trials and marketing for drugs and biological products and devices. The requirements vary widely from country to country, but typically the registration and approval process takes several years and requires significant resources.
If we experience delays or discontinuations of our clinical trials by the FDA or comparable authorities in other countries, or if we fail to obtain registration or other approvals of our products or devices then we could be forced to cease our operations and you will lose all of your investment.
Even if we are successful in developing BIV201, our drug candidate, we have limited experience in conducting or supervising clinical trials that must be performed to obtain data to submit in concert with applications for approval by the FDA. The regulatory process to obtain approval for drugs for commercial sale involves numerous steps. Drugs are subjected to clinical trials that allow development of case studies to examine safety, efficacy, and other issues to ensure that sale of drugs meets the requirements set forth by various governmental agencies, including the FDA. In the event that our protocols do not meet standards set forth by the FDA, or that our data is not sufficient to allow such trials to validate our drugs in the face of such examination, we might not be able to meet the requirements that allow our drugs to be approved for sale which could have a materially adverse effect on our business.
We can provide no assurance that our drug candidates will obtain regulatory approval or that the results of clinical studies will be favorable.
The business plan we have developed for the next twelve months is to complete the work necessary to commence the Phase 2 clinical development program for our lead new drug candidate BIV201 and to pursue other key milestones such as additional US Orphan Drug designations. Due to our financial constraints, we may not have the resources necessary to complete our application. If the results of our planned initial Phase 2a clinical trial are satisfactory to the FDA, we will aim to proceed to a larger Phase 2b clinical trials in the US. There is no guarantee the FDA will approve a Phase 2b trial, and even if they do our financial constraints may prevent us from undertaking clinical trials.
The testing, marketing and manufacturing of any product for use in the United States will require approval from the FDA. We cannot predict with any certainty the amount of time necessary to obtain such FDA approval and whether any such approval will ultimately be granted. Preclinical and clinical trials may reveal that one or more products are ineffective or unsafe, in which event further development of such products could be seriously delayed or terminated. Moreover, obtaining approval for certain products may require testing on human subjects of substances whose effects on humans are not fully understood or documented. Delays in obtaining FDA or any other necessary regulatory approvals of any proposed drug and failure to receive such approvals would have an adverse effect on the drug's potential commercial success and on our business, prospects, financial condition and results of operations. In addition, it is possible that a proposed drug may be found to be ineffective or unsafe due to conditions or facts that arise after development has been completed and regulatory approvals have been obtained. In this event, we may be required to withdraw such proposed drug from the market. To the extent that our success will depend on any regulatory approvals from government authorities outside of the United States that perform roles similar to that of the FDA, uncertainties similar to those stated above will also exist and should it result in our drug candidates failing to receive regulatory approval you could lose all of your investment.
-12-
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information and disclosure of our trade secrets or proprietary information could compromise any competitive advantage that we have, which could have a materially adverse effect on our business.
We depend heavily upon confidentiality agreements with our officers, employees, consultants and subcontractors to maintain the proprietary nature of our technology. These measures may not afford us complete or even sufficient protection, and may not afford an adequate remedy in the event of an unauthorized disclosure of confidential information. In addition, others may independently develop technology similar to ours, otherwise avoiding the confidentiality agreements, or produce patents that would materially and adversely affect our business, prospects, financial condition and results of operations in which event and you could lose all of your investment.
We may be unable to obtain or protect intellectual property rights relating to our products, and we may be liable for infringing upon the intellectual property rights of others, which could have a materially adverse effect on our business.
Our ability to compete effectively will depend on our ability to maintain the proprietary nature of our technologies. In 2017 the US Patent and Trademark Office issued a patent covering the Company’s lead drug candidate BIV201 for use in ascites patients administered by an ambulatory pump. There can be no assurance that any future patent applications we have filed will ultimately result in the issuance of a patent with respect to the technology owned by us or licensed to us. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the United States Patent and Trademark Office use to grant patents are not always applied predictably or uniformly and can change. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents. Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others. Further, we rely on a combination of trade secrets, know-how, technology and nondisclosure, and other contractual agreements and technical measures to protect our rights in the technology. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business and financial condition could be materially adversely affected.
We do not believe that BIV201, the drug candidate we are currently developing, infringes upon the rights of any third parties nor are they infringed upon by third parties. However, there can be no assurance that our technology will not be found in the future to infringe upon the rights of others or be infringed upon by others. In such a case, others may assert infringement claims against us, and should we be found to infringe upon their patents, or otherwise impermissibly utilize their intellectual property, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties' patent rights. In addition to any damages we might have to pay, we may be required to obtain licenses from the holders of this intellectual property, enter into royalty agreements, or redesign our drug candidates so as not to utilize this intellectual property, each of which may prove to be uneconomical or otherwise impossible. Conversely, we may not always be able to successfully pursue our claims against others that infringe upon our technology. Thus, the proprietary nature of our technology or technology licensed by us may not provide adequate protection against competitors.
Moreover, the cost to us of any litigation or other proceeding relating to our patents and other intellectual property rights, even if resolved in our favor, could be substantial, and the litigation would divert our management's efforts. Uncertainties resulting from the initiation and continuation of any litigation could limit our ability to continue our operations and you could lose all of your investment.
We depend upon our management and their loss or unavailability could put us at a competitive disadvantage which could have a material adverse effect on our business.
We currently depend upon the efforts and abilities of our management team of Jonathan Adams, our Chief Operating Officer, and Julie Anderson, our Company Secretary. Mr. Adams serves the Company full-time and Ms. Anderson serves the Company part-time. The loss or unavailability of the services of either of these individuals for any significant period of time could have a material adverse effect on our business, prospects, financial condition and results of operations which may cause you to lose all of your investment. We have not obtained, do not own, nor are we the beneficiary of key-person life insurance.
-13-
We may not be able to attract and retain highly skilled personnel, which could have a materially adverse effect on our business.
Our ability to attract and retain highly skilled personnel is critical to our operations and expansion. We face competition for these types of personnel from other pharmaceutical companies and more established organizations, many of which have significantly larger operations and greater financial, technical, human and other resources than us. We may not be successful in attracting and retaining qualified personnel on a timely basis, on competitive terms, or at all. If we are not successful in attracting and retaining these personnel, our business, prospects, financial condition and results of operations will be materially and adversely affected.
The biotechnology and biopharmaceutical industries are characterized by rapid technological developments and a high degree of competition. We may be unable to compete with enterprises equipped with more substantial resources than us, which could cause us to curtail or cease operations.
The biotechnology and biopharmaceutical industries are characterized by rapid technological developments and a high degree of competition based primarily on scientific and technological factors. These factors include the availability of patent and other protection for technology and products, the ability to commercialize technological developments and the ability to obtain government approval for testing, manufacturing and marketing.
We compete with biopharmaceutical firms in the United States, Europe and elsewhere, as well as a growing number of large pharmaceutical companies that are applying biotechnology to their operations. Many biopharmaceutical companies have focused their development efforts in the human therapeutics area. Many major pharmaceutical companies have developed or acquired internal biotechnology capabilities or made commercial arrangements with other biopharmaceutical companies. These companies, as well as academic institutions, government agencies and private research organizations, also compete with us in recruiting and retaining highly qualified scientific personnel and consultants. Our ability to compete successfully with other companies in the pharmaceutical field will also depend to a considerable degree on the continuing availability of capital to us.
Although there are not currently any therapies approved by the FDA specifically for the treatment of ascites due to liver cirrhosis, the Company still faces significant competitive and market risk. Other companies, such as Mallinckrodt Inc., are developing therapies for severe complications of advanced liver cirrhosis, which may in future be developed for the treatment of ascites, and these therapies could compete indirectly or directly with our drug candidate. There may be other competitive development programs of which we are unaware. Even if our drug candidate is ultimately approved by the FDA, there is no guarantee that once it is on the market doctors will adopt it in favor of current ascites treatment procedures such as diuretics and paracentesis. These competitive and market risks could have a material adverse effect on our business, prospects, financial condition and results of operations which may cause you to lose all of your investment.
Our competition will be determined in part by the potential indications for which drugs are developed and ultimately approved by regulatory authorities. Additionally, the timing of the market introduction of some of our potential drug candidate or of competitors' products may be an important competitive factor. Accordingly, the relative speed with which we can develop drugs, complete pre-clinical testing, clinical trials, approval processes and supply commercial quantities to market are important competitive factors. We expect that competition among drugs approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price and patent protection.
The successful development of biopharmaceuticals is highly uncertain. A variety of factors including, pre-clinical study results or regulatory approvals, could cause us to abandon the development of our drug candidates.
Successful development of biopharmaceuticals is highly uncertain and is dependent on numerous factors, many of which are beyond our control.
Products that appear promising in the early phases of development may fail to reach the market for several reasons. Pre-clinical study results may show the product to be less effective than desired (e.g., the study failed to meet its primary objectives) or to have harmful or problematic side effects. Products may fail to receive the necessary regulatory approvals or may be delayed in receiving such approvals. Among other things, such delays may be caused by slow enrollment in clinical studies, length of time to achieve study endpoints, additional time requirements for data analysis or a IND and later NDA, preparation, discussions with the FDA, an FDA request for additional pre-clinical or clinical data or unexpected safety or manufacturing issues; manufacturing costs, pricing or reimbursement issues, or other factors that make the product not economical. Proprietary rights of others and their competing products and technologies may also prevent the product from being commercialized.
-14-
Success in pre-clinical and early clinical studies does not ensure that large-scale clinical studies will be successful. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. The length of time necessary to complete clinical studies and to submit an application for marketing approval for a final decision by a regulatory authority varies significantly from one product to the next, and may be difficult to predict. There can be no assurance that any of our products will develop successfully, and the failure to develop our products will have a materially adverse effect on our business and will cause you to lose all of your investment.
There may be conflicts of interest among our officers, directors and stockholders.
Certain of our executive officers and directors and their affiliates are engaged in other activities and have interests in other entities on their own behalf or on behalf of other persons. Neither we nor any of our shareholders will have any rights in these ventures or their income or profits. In particular, our executive officers or directors or their affiliates may have an economic interest in or other business relationship with partner companies that invest in us or are engaged in competing drug development. Our executive officers or directors may have conflicting fiduciary duties to us and third parties. The terms of transactions with third parties may not be subject to arm's length negotiations and therefore may be on terms less favorable to us than those that could be procured through arm's length negotiations. Although the Company is not aware of any conflict that has arisen to date, we do not have any policy in place to deal with such should such a conflict arise.
We may enter into employment agreements with our executive officers and compensation payable thereunder may not be based on arms-length negotiations.
The Company’s current executive officers also serve as directors of the Company, and the Company does not have an independent compensation committee to determine compensation and to approve employment agreements. Therefore, compensation which may be paid by the Company to its management may not be determined based on arms-length negotiations. The Company may grant stock options and other equity incentives to its executive officers and directors that are consistent with the nature of the pharmaceutical industry. There can be no assurance made that the consideration which may be payable to management will reflect the true market value of services provided to the Company.
RISKS RELATING TO OUR COMMON STOCK
There is a risk of dilution of your percentage ownership of Common Stock in the Company.
The Company has the right to raise additional capital or incur borrowings from third parties to finance its business. The Company may also implement public or private mergers, business combinations, business acquisitions and similar transactions pursuant to which it would issue substantial additional capital stock to outside parties, causing substantial dilution in the ownership of the Company by its existing stockholders. Our Board of Directors has the authority, without the consent of any of the stockholders, to cause the Company to issue more shares of Common Stock and/or preferred stock at such price and on such terms and conditions as are determined by the Board in its sole discretion. The issuance of additional shares of capital stock by the Company will dilute your ownership percentage in the Company and could impair our ability to raise capital in the future through the sale of equity securities.
Certain stockholders who are also officers and directors of the Company may have significant control over our management.
The directors and executive officers of the Company currently own an aggregate 440,181,137 shares, which currently constitutes 83.6% of the Common Stock of the Company. As a result, directors and executive officers may have a significant influence on the affairs and management of the Company, as well as on all matters requiring member approval, including electing and removing members of the Company’s Board of Directors, causing the Company to engage in transactions with affiliated entities, causing or restricting the sale or merger of the Company, and certain other matters. Such concentration of ownership and control could have the effect of delaying, deferring or preventing a change in control of the Company even when such a change of control would be in the best interests of the Company’s stockholders.
-15-
There is very little liquidity in our Common Stock and we may not be successful at obtaining a quotation on a recognized quotation service. In such event it may be difficult for you to sell your shares.
The OTC Bulletin Board and similar quotation services are often characterized by low trading volumes, and price volatility, which may make it difficult for an investor to sell our Common Stock on acceptable terms. If trades in our Common Stock are not quoted on a quotation facility, it may be very difficult for an investor to find a buyer for their shares in our Company.
Our Common Stock is subject to the “penny stock” rules of the SEC and the trading market in our securities is limited, which makes transactions in our stock cumbersome and may reduce the value of an investment in our stock.
Under U.S. federal securities legislation, our Common Stock will constitute “penny stock”. Penny stock is any equity security that has a market price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require that a broker or dealer approve a potential investor’s account for transactions in penny stocks, and the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. In order to approve an investor’s account for transactions in penny stocks, the broker or dealer must obtain financial information and investment experience objectives of the person, and make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prepared by the Securities and Exchange Commission relating to the penny stock market, which, in highlight form sets forth the basis on which the broker or dealer made the suitability determination. Brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock. Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
We may, in the future, issue additional common stock, which would reduce investors’ percent of ownership and may dilute our share value.
As of June 30, 2018, our Articles of Incorporation authorize the issuance of 300,000,000 shares of Common Stock. As of June 30, 2018, the Company had 98,503,199 shares of Common Stock outstanding. Accordingly, we may issue up to an additional 201,496,801 shares of Common Stock. The future issuance of Common Stock may result in substantial dilution in the percentage of our Common Stock held by our then existing shareholders. We may value any Common Stock in the future on an arbitrary basis. The issuance of Common Stock for future services or acquisitions or other corporate actions may have the effect of diluting the value of the shares held by our investors, might have an adverse effect on any trading market for our Common Stock and could impair our ability to raise capital in the future through the sale of equity securities.
We have a large number of restricted shares outstanding, a portion of which may be sold under Rule 144 which may reduce the market price of our shares.
Of the 98,503,199 shares of Common Stock issued as of June 30, 2018 and outstanding, and assuming no Warrants are exercised, 86,603,765 shares are held by non-affiliates and 11,899,434 are owned by affiliates of the Company, consisting of our officers and directors. The majority of our Common Stock, including all of the Affiliates’ securities are deemed “restricted securities” within the meaning of Rule 144 as promulgated under the Securities Act.
It is anticipated that all of the “restricted securities” will be eligible for resale under Rule 144. In general, under Rule 144, subject to the satisfaction of certain other conditions, a person, who is not an affiliate (and who has not been an affiliate for a period of at least three months immediately preceding the sale) and who has beneficially owned restricted shares of our common stock for at least six months is permitted to sell such shares without restriction, provided that there is sufficient public information about us as contemplated by Rule 144. An affiliate who has beneficially owned restricted shares of our common stock for a period of at least one year may sell a number of shares equal to one percent of our issued and outstanding common stock approximately every three months.
-16-
The respective holding periods for the shares issued to affiliates and non-affiliates holding restricted securities commenced and were issued between May 17, 2013 and June 30, 2013. The possibility that substantial amounts of our Common Stock may be sold under Rule 144 into the public market may adversely affect prevailing market prices for the Common Stock and could impair our ability to raise capital in the future through the sale of equity securities.
The lack of public company experience of our management team could adversely impact our ability to comply with the reporting requirements of U.S. securities laws, which could have a materially adverse effect on our business.
Our officers have limited public company experience, which could impair our ability to comply with legal and regulatory requirements such as those imposed by Sarbanes-Oxley Act of 2002. Our officers and directors have never been responsible for managing a publicly traded company. Such responsibilities include complying with federal securities laws and making required disclosures on a timely basis. Any such deficiencies, weaknesses or lack of compliance could have a materially adverse effect on our ability to comply with the reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), which is necessary to maintain our public company status. If we were to fail to fulfill those obligations, our ability to continue as a U.S. public company would be in jeopardy in which event you could lose your entire investment in our Company.
The Company is considered a smaller reporting company and is exempt from certain disclosure requirements, which could make our stock less attractive to potential investors.
Rule 12b-2 of the Exchange Act defines a “smaller reporting company” as an issuer that is not an investment company, an asset-backed issuer, or a majority-owned subsidiary of a parent that is not a smaller reporting company and that:
| · | Had a public float of less than $250 million as of the last business day of its most recently completed fiscal quarter, computed by multiplying the aggregate number of worldwide number of shares of its voting and non-voting common equity held by non-affiliates by the price at which the common equity was last sold, or the average of the bid and asked prices of common equity, in the principle market for the common equity; or |
| · | In the case of an initial registration statement under the Securities Act or the Exchange Act for shares of its common equity, had a public float of less than $250 million as of a date within 30 days of the date of the filing of the registration statement, computed by multiplying the aggregate worldwide number of such shares held by non-affiliates before the registration plus, in the case of a Securities Act registration statement, the number of such shares included in the registration statement by the estimated public offering price of the shares; or |
| · | In the case of an issuer who had annual revenue of less than $100 million during the most recently completed fiscal year for which audit financial statements are available, had a public float as calculated under paragraph (1) or (2) of this definition that was either zero or less than $700 million. |
As a “smaller reporting company” (in addition to and without regard to our status as an “emerging growth company”) we are not required and may not include a Compensation Discussion and Analysis (“CD&A”) section in our proxy statements; we provide only 3 years of business development information; provide fewer years of selected data; and have other “scaled” disclosure requirements that are less comprehensive than issuers that are not “smaller reporting companies” which could make our stock less attractive to potential investors, which could make it more difficult for you to sell your shares.
The Company is considered an “emerging growth company” and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our Common Stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart our Business Startups Act of 2012, and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies, including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved.
-17-
We will remain an emerging growth company until the earlier of (i) the last day of the fiscal year (A) following the fifth anniversary of our first sale of common equity securities pursuant to an effective registration statement, (B) in which we have total annual gross revenue of at least $1.0 billion, or (C) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, and (ii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We cannot predict if investors will find our Common Stock less attractive because we will rely on these exemptions. If some investors find our Common Stock less attractive as a result, there may be a less active trading market for our Common Stock and our stock price may be more volatile when trading occurs.
We are subject to the periodic reporting requirements of the Exchange Act, which require us to incur audit fees and legal fees in connection with the preparation of such reports. These additional costs will negatively affect our ability to earn a profit.
We are required to file periodic reports with the Securities and Exchange Commission pursuant to the Exchange Act and the rules and regulations thereunder. In order to comply with such requirements, our independent registered auditors have to review our financial statements on a quarterly basis and audit our financial statements on an annual basis. Moreover, our legal counsel have to review and assist in the preparation of such reports. Factors such as the number and type of transactions that we engage in and the complexity of our reports cannot accurately be determined at this time and may have a major negative effect on the cost and amount of time to be spent by our auditors and attorneys. However, the incurrence of such costs are an expense to our operations and thus have a negative effect on our ability to meet our overhead requirements and earn a profit.
However, for as long as we remain an “emerging growth company” we intend to take advantage of certain exemptions from various reporting requirements until we are no longer an “emerging growth company.”
We also qualify as a smaller reporting company, and so long as we remain a smaller reporting company, we benefit from the same exemptions and exclusions as an emerging growth company. In the event that we cease to be an emerging growth company as a result of a lapse of the five-year period, but continue to be a smaller reporting company, we would continue to be subject to the exemptions available to emerging growth companies until such time as we were no longer a smaller reporting company.
After, and if ever, we are no longer an “emerging growth company,” we expect to incur significant additional expenses and devote substantial management effort toward ensuring compliance with those requirements applicable to companies that are not “emerging growth companies,” including Section 404 of the Sarbanes-Oxley Act.
The JOBS Act allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies, which means that our financial statements may not be comparable to companies that comply with public company effective dates, which could make our Common Stock less attractive to investors.
Since we have elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act, this election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates.
Because we do not intend to pay any cash dividends on our common stock, our stockholders will not be able to receive a return on their shares unless they sell them.
We intend to retain any future earnings to finance the development and expansion of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. Unless we pay dividends, our stockholders will not be able to receive a return on their shares unless they sell them. There is no assurance that stockholders will be able to sell shares when desired.
-18-
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | DESCRIPTION OF PROPERTY |
On January 1, 2014, the company executed a lease agreement with Cummings Properties for the company’s office of 270 square feet at 100 Cummings Center, Suite 247-C, Beverly, MA 01915. The lease is for a term of five years from January 1, 2014 to December 30, 2018 and requires monthly payments of $379. The Company has notified Cummings Properties that it shall cancel the lease effective December 30, 2018 and relocate its headquarters.
| ITEM 3. | LEGAL PROCEEDINGS |
To our knowledge, neither the Company nor any of our officers or directors is a party to any material legal proceeding or litigation and such persons know of no material legal proceeding or contemplated or threatened litigation. There are no judgments against us or our officers or directors. None of our officers or directors has been convicted of a felony or misdemeanor relating to securities or performance in corporate office.
| ITEM 4. | MINE SAFETY DISCLOSURE |
None
PART II
| ITEM 5. | MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS |
The Company’s Common Stock trades on the over-the-counter market on the National Association of Securities Dealers, Inc. OTC Bulletin Board System (“OTCQB”) under the symbol “BIVI.” The following table sets forth the range of high and low closing bid quotations of the Common Stock as reported by the OTCQB for each fiscal quarter for the year ended June 30, 2017 and 2018. High and low bid quotations reflect inter-dealer prices without adjustment for retail mark-ups, markdowns or commissions and may not necessarily represent actual transactions.
| Bid Prices | ||
| Low | High | |
| Quarter ended June 30, 2018 | $ 0.01 | $ 0.08 |
| Quarter ended March 31, 2018 | $ 0.02 | $ 0.19 |
| Quarter ended December 31, 2017 | $ 0.14 | $ 0.27 |
| Quarter ended September 30, 2017 | $ 0.16 | $ 0.35 |
| Quarter ended June 30, 2017 | $ 0.21 | $ 0.45 |
| Quarter ended March 31, 2017 | $ 0.16 | $ 0.40 |
| Quarter ended December 31, 2016 | $ 0.16 | $ 0.45 |
| Quarter ended September 30, 2016 | $ 0.21 | $ 0.40 |
On June 30, 2018, the closing bid price of the Company’s Common Stock as reported by the OTC was $0.05.
DIVIDENDS
We have not paid any cash dividends on our common or preferred stock and do not anticipate paying any such cash dividends in the foreseeable future. Earnings, if any, will be retained to finance future growth. We may issue shares of our common stock and preferred stock in private or public offerings to obtain financing, capital or to acquire other businesses that can improve our performance and growth. Issuance and or sales of substantial amounts of common stock could adversely affect prevailing market prices in our common stock.
Common Stock
During the year ended June 30, 2018, there was no modification of any instruments defining the rights of holders of the Company’s common stock and no limitation or qualification of the rights evidenced by the Company’s common stock as a result of the issuance of any other class of securities or the modification thereof.
-19-
| ITEM 6. | SELECTED FINANCIAL DATA |
Not Required
| ITEM 7. |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
This report contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, and Section 27A of the Securities Act of 1933. Any statements contained in this report that are not statements of historical fact may be forward-looking statements. When we use the words “intends,” “estimates,” “predicts,” “potential,” “continues,” “anticipates,” “plans,” “expects,” “believes,” “should,” “could,” “may,” “will” or the negative of these terms or other comparable terminology, we are identifying forward-looking statements. Forward-looking statements involve risks and uncertainties, which may cause our actual results, performance or achievements to be materially different from those expressed or implied by forward-looking statements. These factors include our; research and development activities, distributor channel; compliance with regulatory impositions; and our capital needs. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements.
Except as may be required by applicable law, we do not undertake or intend to update or revise our forward-looking statements, and we assume no obligation to update any forward-looking statements contained in this report as a result of new information or future events or developments. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in such forward-looking statements. You should carefully review and consider the various disclosures we make in this report and our other reports filed with the Securities and Exchange Commission that attempt to advise interested parties of the risks, uncertainties and other factors that may affect our business.
All statements other than statements of historical fact are statements that could be deemed forward-looking statements. The Company assumes no obligation and does not intend to update these forward-looking statements, except as required by law. When used in this report, the terms “BioVie”, “Company”, “we”, “our”, and “us” refer to BioVie Inc.
The following discussion of the Company’s financial condition and the results of operations should be read in conjunction with the Financial Statements and Notes thereto appearing elsewhere in this document.
The Private Securities Litigation Reform Act of 1995 provides a safe harbor for forward-looking statements. In order to comply with the terms of the safe harbor, the Company notes that in addition to the description of historical facts contained herein, this report contains certain forward-looking statements that involve risks and uncertainties as detailed herein and from time to time in the Company’s other filings with the Securities and Exchange Commission and elsewhere. Such statements are based on management’s current expectations and are subject to a number of factors and uncertainties, which could cause actual results to differ materially from those, described in the forward-looking statements. These factors include, among others: (a) the Company’s fluctuations in sales and operating results; (b) risks associated with international operations; (c) regulatory, competitive and contractual risks; (d) product development risks; (e) the ability to achieve strategic initiatives, including but not limited to the ability to achieve sales growth across the business segments through a combination of enhanced sales force, new products, and customer service; and (f) pending litigation.
Management’s Discussion
We are a clinical stage biotechnology company engaged in the discovery, development and commercialization of therapies targeting life-threatening complications of liver cirrhosis. Our initial disease target is ascites, a serious medical condition affecting about 100,000 Americans and many times more worldwide. Our therapeutic drug candidate BIV201 is based on a drug that is approved in about 40 countries to treat related complications of liver cirrhosis (part of the same disease pathway as ascites), but not yet available in the US. The active agent in BIV201, terlipressin, is a potent vasoconstrictor which is in use for various medical conditions around the world. The goal is for BIV201 to interrupt the ascites disease pathway, thereby halting the cycle of accelerating fluid generation in ascites patients.
-20-
BioVie accomplished the following key milestones during the twelve months ended June 30th, 2018:
| - | In July 2017 the Company filed for BIV201 patent protection in Japan. |
| - | In September 2017, the first patient was enrolled and treated with BIV201 in our US Phase 2a clinical trial. |
| - | In September 2017, the Company engaged Mr. R. Richard Wieland II as our part-time chief financial officer. |
| - | In October 2017, the Company recruited Mr. Michael Sherman, a former investment banker and securities lawyer to join our Board of Directors. |
| - | In November 2017, the Company recruited Ms. Mina Sooch, the former chief executive officer for Gemphire Therapeutics, Inc. (NASDAQ: GEMP) to join our Board of Directors. |
| - | In December 2017, the Company received FDA Fast Track Designation for BIV201 for the treatment of refractory ascites. |
| - | In January 2018, an independent Data Safety Monitoring Board reviewed the clinical data for the first 2 patients treated with BIV201 and recommended continuing the trial. |
| - | In May 2018, the Company signed a Promissory Note with Acuitas Group Holdings, LLC in the amount of $250,000 as the first step in a multi-million dollar equity investment which closed in July 2018. |
| - | As of June 2018, 3 refractory ascites patients had been treated with BIV201 in our ongoing open-label 6-patient Phase 2a clinical trial. |
Comparison of the Year Ended June 30, 2018 to the Year Ended June 30, 2017
Research and Development
Research and development expenses were $370,853 for the fiscal year ended June 30, 2018, a decrease of $95,501, compared to $466,354 for the fiscal year ended June 30, 2017. The research and development expenses were primarily due to the expenses incurred for clinical development activities.
Selling, General and Administrative
Selling, general and administrative expenses were $129,270 for the fiscal year ended June 30, 2018, an increase of $60,148, compared to $69,122 for the fiscal year ended June 30, 2017. The increase in selling, general and administrative expenses was primarily due to travel and conference expenses associated with financing activities.
Professional Fees
Professional fees were $1,331,142 for the fiscal year ended June 30, 2018, an increase of $827,773 compared to $503,369 for the fiscal year ended June 30, 2017. The increase in professional fees related to a large expense for financial and strategic advisory services paid in BioVie common stock.
Payroll Expenses
Payroll expenses were $311,525 for the fiscal year ended June 30, 2018, an increase of $26,133 compared to $285,392 for the fiscal year ended June 30, 2017. Payroll expenses were related to accrued salary for the Chief Operating Officer, Jonathan Adams. The payroll expenses for fiscal year ended June 30, 2018 included a $30,547 adjustment made for fiscal year ended June 30, 2017. The adjustment was due to a correction made to the valuation of Stock Options issued to the Chief Operating Officer.
We have incurred $2,372,166 of operating expenses for the year ended June 30, 2018. We are now engaged in organizational activities and sourcing compounds and materials. We anticipate incurring other costs associated with equipment purchases and general and administrative expenses, including employee salaries and benefits, legal expenses, and other costs associated with an early stage, publicly-traded company.
The amounts that we actually spend for any specific purpose may vary significantly, and will depend on a number of factors including, but not limited to, the pace of progress of our research and development, market conditions, and our ability to qualify vendors. In addition, we may use a portion of any net proceeds to acquire complementary compounds; however, we do not have plans for any acquisitions at this time. We will have significant discretion in the use of any net proceeds. Investors will be relying on the judgment of our management regarding the application of the proceeds of any sale of our Common Stock.
-21-
Requirement for Additional Capital
The Company incurred $370,853 in research and development activities for fiscal year ended June 30, 2018. The Company received an equity investment from Acuitas Group Holding, LLC (“Acuitas”) and other purchasers which generated $3.2 million in cash proceeds. This funding is expected to be sufficient to cover our operational costs for the next twelve (12) months.
The Acuitas investment agreement also stipulated that if the clinical development of BIV201 continues, Acuitas may invest an additional $3 million to fund operations in year two, less any federal or FDA grant funding received by the Company.
We currently have not secured funding that will be required to pay for operating expenses incurred after the 2-year period expires.
The Company had approximately $2,255,000 of cash on hand at August 31, 2018.
The Company has limited experience with pharmaceutical drug development. As such these budget estimates may not be accurate. In addition, the actual work to be performed can only be broadly projected, as is normal with any scientific work. As further work is performed, additional work may become necessary or change in plans or workload may occur. Such changes may have an adverse impact on our estimated budget. Such changes may also have an adverse impact on our projected timeline of drug development.
Capital Resources and Liquidity
As of August 31, 2018, we had approximately $2,255,000 of cash on hand in our corporate bank account. The Company is considered to be a development stage company and will continue in the development stage until generating revenues from the sales of its products or services.
We cannot assure you that our drug candidate will be developed, work, or receive regulatory approval; that we will ever earn revenues sufficient to support our operations or that we will ever be profitable. Furthermore, since we have no committed source of financing, we cannot assure you that we will be able to raise money as and when we need it to continue our operations. If we cannot raise funds as and when we need them, we may be required to severely curtail, or even to cease, our operations.
If we are unable to raise additional funds in the future, we will need to do one or more of the following:
| ● | delay, scale-back or eliminate some or all of our research and product development programs; |
| ● | provide licenses to third parties to develop and commercialize products or technologies that we would otherwise seek to develop and commercialize ourselves; |
| ● | seek strategic alliances or business combinations; |
| ● | attempt to sell our company; |
| ● | cease operations; or |
| ● | declare bankruptcy. |
On July 3, 2018, the Company raised $3.2 million in equity investment proceeds. We believe that our existing cash and cash equivalents, including the amounts received after the end of the fiscal year, will be sufficient to meet our operating and capital requirements for at least 12 months from the date of this report.
-22-
Our management intends to attempt to secure additional required funding primarily through additional equity or debt financings. We may also seek to secure required funding through sales or out-licensing of intellectual property assets, seeking partnerships with other pharmaceutical companies or third parties to co-develop and fund research and development efforts, or similar transactions. However, there can be no assurance that we will be able to obtain required funding. If we are unsuccessful in securing funding from any of these sources, we will defer, reduce or eliminate certain planned expenditures in our research protocols. If we do not have sufficient funds to continue operations, we could be required to seek bankruptcy protection or other alternatives that could result in our stockholders losing some or all of their investment in us.
Emerging Growth Company
We are an “emerging growth company” under the federal securities laws and will be subject to reduced public company reporting requirements. In addition, Section 107 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are choosing to take advantage of the extended transition period for complying with new or revised accounting standards.
Off-Balance Sheet Arrangements
The Company has no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect or change on the Company’s financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors. The term “off-balance sheet arrangement” generally means any transaction, agreement or other contractual arrangement to which an entity unconsolidated with the Company is a party, under which the Company has (i) any obligation arising under a guarantee contract, derivative instrument or variable interest; or (ii) a retained or contingent interest in assets transferred to such entity or similar arrangement that serves as credit, liquidity or market risk support for such assets.
Critical Accounting Policies and Estimates
Basis of Presentation
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash
Cash is maintained at financial institutions and, at times, balances may exceed federally insured limits. The Company has never experienced any losses related to these balances. All of the Company’s cash balances were fully insured at June 30, 2018.
Financial Instruments
The Company’s financial instruments include cash, accounts payable, related party loans and a demand promissory note. The carrying amounts of cash and accounts payable approximate their fair value, due to the short-term nature of these items.
-23-
Long-Term Notes Payable
The Company’s long-term notes payable include accrued payroll to officers and accrued payments to third party consultants.
Research and Development
Research and development costs are charged to operations when incurred and are included in operating expenses.
Income Taxes
Deferred income tax assets and liabilities arise from temporary differences associated with differences between the financial statements and tax basis of assets and liabilities, as measured by the enacted tax rates, which are expected to be in effect when these differences reverse. Deferred tax assets and liabilities are classified as current or non-current, depending on the classification of the assets or liabilities to which they relate. Deferred tax assets and liabilities not related to an asset or liability are classified as current or non-current depending on the periods in which the temporary differences are expected to reverse.
The Company follows the provisions of FASB ASC 740-10 “Uncertainty in Income Taxes” (ASC 740-10), January 1, 2007. The Company has not recognized a liability as a result of the implementation of ASC 740-10. A reconciliation of the beginning and ending amount of unrecognized tax benefits has not been provided since there are no unrecognized benefits at June 30, 2018 and since the date of adoption. The Company has not recognized interest expense or penalties as a result of the implementation of ASC 740-10. If there were an unrecognized tax benefit, the Company would recognize interest accrued related to unrecognized tax benefits in interest expense and penalties in operating expenses.
Earnings (Loss) per Share
Basic earnings per share are computed by dividing net income by the weighted average number of shares of common stock outstanding during the year. Diluted earnings per common share are computed by dividing net income by the weighted average number of shares of common stock outstanding and dilutive options outstanding during the year. For the years ended June 30, 2017 and 2018 all outstanding options have been excluded from the calculation of the diluted net loss per share since their effect was anti-dilutive.
Stock-based Compensation
The Company has accounted for stock-based compensation under the provisions of FASB ASC 718 – “Stock Compensation” which requires the use of the fair-value based method to determine compensation for all arrangements under which employees and others receive shares of stock or equity instruments (stock options and common stock purchase warrants). For employee awards, the fair value of each stock option award is estimated on the date of grant using the Black-Scholes valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. For non-employees, the fair value of each stock option award is estimated on the measurement date using the Black-Scholes valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. For non-employees, the Company utilizes the graded vesting attribution method under which the entity treats each separately vesting portion (tranche) as a separate award and recognizes compensation cost for each tranche over its separate vesting schedule. Expected volatilities are based on historical volatility of peer companies and other factors estimated over the expected term of the stock options. For employee awards, the expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for the period of the expected term.
Goodwill
Goodwill is recorded when the purchase price paid for an acquisition exceeds the fair value of net identified tangible and intangible assets acquired. The Company performs an annual impairment test of goodwill and further periodic tests to the extent indicators of impairment develop between annual impairment tests. The Company’s impairment review process compares the fair value of the reporting unit to its carrying value, including the goodwill related to the reporting unit. To determine the fair value of the reporting unit, the Company may use various approaches including an asset or cost approach, market approach or income approach or any combination thereof. These approaches may require the Company to make certain estimates and assumptions including future cash flows, revenue and expenses. These estimates and assumptions are reviewed each time the Company tests goodwill for impairment and are typically developed as part of the Company’s routine business planning and forecasting process. While the Company believes its estimates and assumptions are reasonable, variations from those estimates could produce materially different results.
-24-
Impairment of Long-Lived Assets
Long-lived assets, including intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset.
If the carrying amount of an asset exceeds its undiscounted estimated future cash flows, an impairment review is performed. An impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. Generally, fair value is determined using valuation techniques such as expected discounted cash flows or appraisals, as appropriate. Assets to be disposed of would be separately presented in the balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated or amortized. The assets and liabilities of a disposed group classified as held for sale would be presented separately in the appropriate asset and liability sections of the balance sheet.
New Accounting Pronouncements
For a description of recent accounting standards, including the expected dates of adoption and estimated effects, if any, on our financial statements, see “Note 3: Significant Accounting Polices: Recent Accounting Standards” in Part II, Item 8 of this Form 10-K.
| ITEM 7A. | QUANTATITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Not Applicable.
-25-
| ITEM 8. | FINANCIAL STATEMENTS |
BioVie, Inc.
Financial Statements
Contents
| Report of Independent Registered Public Accounting Firm | F-1 | |||
| Financial Statements: | ||||
| Balance Sheets | F-2 | |||
| Statements of Operations | F-3 | |||
| Statements of Changes in Stockholders’ Equity | F-4 | |||
| Statements of Cash Flows | F-5 | |||
| Notes to Financial Statements | F-6 | |||
-26-

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of BioVie, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of BioVie, Inc. (the Company) as of June 30, 2018 and 2017, and the related statements of operations, stockholders’ equity, and cash flows for each of the years then ended, and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2018 and 2017, and the results of its operations and its cash flows for each of the two years ended June 30, 2018 and 2017, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
D. Brooks and Associates CPA’s, P.A.
We have served as the Company’s auditor since 2017.
Palm Beach Gardens, Florida
October 4, 2018
F-1
BioVie Inc.
Balance Sheets
| June 30, | June 30, | |||||||||
| 2018 | 2017 | |||||||||
| ASSETS | ||||||||||
| CURRENT ASSETS: | ||||||||||
| Cash | $ | 45,800 | $ | 5,140 | ||||||
| Total Current Assets | 45,800 | 5,140 | ||||||||
| OTHER ASSETS: | ||||||||||
| Intangible Assets (Net of Amortization) | 1,783,980 | 2,013,357 | ||||||||
| Goodwill | 345,711 | 345,711 | ||||||||
| Total Other Assets | 2,129,691 | 2,359,068 | ||||||||
| TOTAL ASSETS | $ | 2,175,491 | $ | 2,364,209 | ||||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||||
| CURRENT LIABILITIES: | ||||||||||
| Accounts Payable and accrued expenses | $ | 884,207 | $ | 470,973 | ||||||
| Related Party Loan | — | 35,000 | ||||||||
| Accrued Payroll | 354,167 | 125,000 | ||||||||
| Total Current Liabilities | 1,238,374 | 630,973 | ||||||||
| LONG-TERM LIABILITIES: | ||||||||||
| Demand Promissory Note | 250,000 | — | ||||||||
| Notes Payable, Related Parties | 575,918 | 575,918 | ||||||||
| Total Long-Term Liabilities | 825,918 | 575,918 | ||||||||
| TOTAL LIABILITIES | 2,064,292 | 1,206,891 | ||||||||
| STOCKHOLDERS' EQUITY | ||||||||||
| Preferred stock; $0.001 par value; 10,000,000 shares authorized; 0 shares issued and outstanding | — | — | ||||||||
| Common stock, $0.0001 par value; 300,000,000 shares authorized; 98,503,199 and 91,925,000 shares issued and outstanding, respectively | 9,850 | 9,192 | ||||||||
| Additional paid in capital | 4,870,475 | 3,483,134 | ||||||||
| Accumulated deficit | (4,769,126 | ) | (2,335,009 | ) | ||||||
| Total Stockholders' Equity | 111,199 | 1,157,317 | ||||||||
| TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY | $ | 2,175,491 | $ | 2,364,209 | ||||||
The accompanying notes are an integral part of the financial statements.
F-2
BioVie Inc.
Statements of Operations
| For the Twelve Months | For the Twelve Months | |||||||
| Ended | Ended | |||||||
| June 30, | June 30, | |||||||
| 2018 | 2017 | |||||||
| REVENUE | $ | — | $ | — | ||||
| OPERATING EXPENSES: | ||||||||
| Amortization | 229,377 | 229,377 | ||||||
| Research and development expenses | 370,853 | 466,354 | ||||||
| Payroll expenses | 311,525 | 285,392 | ||||||
| Professional fees | 1,331,142 | 503,369 | ||||||
| Selling, general and administrative expenses | 129,270 | 69,122 | ||||||
| TOTAL OPERATING EXPENSES | 2,372,166 | 1,553,614 | ||||||
| LOSS FROM OPERATIONS | (2,372,166 | ) | (1,553,614 | ) | ||||
| OTHER EXPENSE (INCOME): | ||||||||
| Other Income | — | (222,928 | ) | |||||
| Interest expense | 40,960 | — | ||||||
| Interest income | (4 | ) | (14 | ) | ||||
| TOTAL OTHER EXPENSE (INCOME), NET | 40,956 | (222,942 | ) | |||||
| NET LOSS | $ | (2,413,122 | ) | $ | (1,330,672 | ) | ||
| Deemed dividend | (20,995 | ) | — | |||||
| NET LOSS ATTRIBUTABLE TO COMPANY STOCKHOLDERS | (2,434,117 | ) | — | |||||
| NET LOSS PER COMMON SHARE, BASIC AND DILUTED | $ | (0.03 | ) | $ | (0.01 | ) | ||
| WEIGHTED AVERAGE NUMBER OF | ||||||||
| COMMON SHARES OUTSTANDING, BASIC AND DILUTED | 95,758,079 | 89,391,302 | ||||||
The accompanying notes are an integral part of the financial statements.
F-3
BioVie Inc.
Statement of Changes in Stockholders’ Equity
For the Years Ended June 30, 2018 and 2017
| Common | Common | Additional | Total | |||||||||||||||||
| Stock | Stock | Paid in | Accumulated | Stockholders' | ||||||||||||||||
| Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||
| Balance, June 30, 2016 | 87,160,001 | $ | 8,716 | $ | 2,911,560 | $ | (1,004,337 | ) | $ | 1,915,939 | ||||||||||
| Issuance of shares and warrants for cash | 4,764,999 | 477 | 479,523 | — | 479,999 | |||||||||||||||
| Options vested | — | — | 92,051 | — | 92,051 | |||||||||||||||
| Net loss | — | — | — | (1,330,672 | ) | (1,330,672 | ) | |||||||||||||
| Balance, June 30, 2017 | 91,925,000 | 9,193 | 3,483,134 | (2,335,009 | ) | 1,157,317 | ||||||||||||||
| Issuance of shares and warrants for cash | 1,729,699 | 172 | 444,827 | — | 444,999 | |||||||||||||||
| Issuance of shares for services | 4,748,500 | 475 | 642,375 | — | 642,850 | |||||||||||||||
| Options vested | — | — | 238,165 | — | 238,165 | |||||||||||||||
| Exercise of options for cash | 100,000 | 10 | 1,990 | — | 2,000 | |||||||||||||||
| Issuance of warrants for services | — | — | 12,469 | — | 12,469 | |||||||||||||||
| Issuance of warrants with debt | — | — | 26,519 | — | 26,519 | |||||||||||||||
| Deemed dividends for ratchet adjustment to warrants | — | — | 20,995 | (20,995 | ) | — | ||||||||||||||
| Net loss | — | — | — | (2,413,122 | ) | (2,413,122 | ) | |||||||||||||
| Balance, June 30, 2018 | 98,503,199 | $ | 9,850 | $ | 4,870,474 | $ | (4,769,126 | ) | $ | 111,197 | ||||||||||
The accompanying notes are an integral part of the financial statements.
F-4
BioVie Inc.
Statements of Cash Flows
| For the Twelve Months | For the Twelve Months | |||||||
| Ended | Ended | |||||||
| Ended June 30, | Ended June 30, | |||||||
| 2018 | 2017 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (2,413,122 | ) | $ | (1,330,672 | ) | ||
| Adjustments to reconcile net loss to net cash to cash used in operating activities: | ||||||||
| Amortization of intangible assets | 229,377 | 229,377 | ||||||
| Amortization of debt discount | 26,519 | — | ||||||
| Share based compensation expense | 893,484 | 92,052 | ||||||
| Changes in operating assets and liabilities | ||||||||
| Decrease in prepaid expenses | — | 6,982 | ||||||
| Increase in: | ||||||||
| Accounts payable | 413,234 | 350,674 | ||||||
| Accrued payroll | 229,167 | 27,972 | ||||||
| Net cash used in operating activities | (621,341 | ) | (623,615 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Repayment of loan | (35,000 | ) | — | |||||
| Proceeds from related party | — | 25,000 | ||||||
| Proceeds from demand promissory note | 250,000 | — | ||||||
| Proceeds from issuance of common stock and warrants | 344,999 | 479,999 | ||||||
| Proceeds from exercise of options | 2,000 | — | ||||||
| Proceeds from issuance of warrants | 100,000 | — | ||||||
| Net cash provided by financing activities | 661,999 | 504,999 | ||||||
| Net Increase (decrease) in cash | 40,658 | (118,616 | ) | |||||
| Cash, beginning of period | 5,140 | 123,757 | ||||||
| Cash, end of period | $ | 45,800 | $ | 5,140 | ||||
| SUPPLEMENTAL CASH FLOW INFORMATION: | ||||||||
| Non-cash Investing and Financing Activities: | ||||||||
| Issuance of warrants with debt | $ | 26,519 | — | |||||
The accompanying notes are an integral part of the financial statements.
F-5
BioVie Inc.
Notes to Financial Statements
For the Years Ended June 30, 2018 and 2017
| 1. | Background Information |
BioVie Inc. (the “Company”) is a clinical-stage company pursuing the discovery, development, and commercialization of innovative drug therapies. The Company is currently focused on developing and commercializing BIV201, a novel approach to the treatment of ascites due to chronic liver cirrhosis. In March 2017, the Company received notification from the US Food and Drug Administration (FDA) that it could initiate a Phase 2a US clinical trial. In April the Company signed a Cooperative Research and Development Agreement (CRADA) with the McGuire Research Institute/VA in Richmond, VA, and began dosing patients with BIV201 in September 2017. As of June 2018, three patients had been treated with BIV201 therapy in this ongoing Phase 2a clinical trial.
BIV201 has the potential to improve the health of thousands of patients suffering from life-threatening complications of liver cirrhosis due to hepatitis, nonalcoholic steatohepatitis (NASH), and alcoholism. It has FDA Fast-Track status and Orphan Drug designation for the most common of these complications, ascites, which represents a significant unmet medical need. The FDA has never approved any drug specifically for treating ascites.
The BIV201 development program began at LAT Pharma LLC. On April 11, 2016, the Company acquired LAT Pharma LLC and the rights to its BIV201 development program. The Company currently owns all development and marketing rights to its drug candidate. The Company and PharmaIN, Corp. (“PharmaIN”), LAT Pharma’s former partner focused on the development of new modified drug candidates in the same therapeutic field but not including BIV201, agreed to pay royalties equal to less than 1% of future net sales of each company's ascites drug development programs, or if such program is licensed to a third party, less than 5% of each company's net license revenues. The Company’s relationship with PharmaIN could advance into a collaboration or be terminated. The Company has an issued US Patent covering the use of BIV201 for the treatment of ascites patients in the outpatient setting using ambulatory pump infusion, and has filed patent applications for its drug candidate in Japan, and Europe, and China.
The Company’s activities are subject to significant risks and uncertainties including failure to secure additional funding to properly execute the Company’s business plan.
| 2. | Liquidity |
We believe that our existing cash and cash equivalents, including the amounts received after the end of the fiscal year, will be sufficient to meet our operating and capital requirements for at least 12 months from the date of this report.
On July 3, 2018, the Company, entered into a Securities Purchase Agreement (the “Purchase Agreement”) with Acuitas Group Holdings, LLC (“Acuitas”) and certain other purchasers identified in the Purchase Agreement (together with Acuitas, the “Purchasers”) pursuant to which (i) the Purchasers agreed to purchase an aggregate of 2,133,332 shares of the Company’s newly created Series A Convertible Preferred Stock (the “Preferred Stock”) at a price per share of $1.50 per share of Preferred Stock (the “Initial Sale”) and (ii) the Company will issue associated warrants (the “Warrants”) to purchase 213,333,200 shares of the Company’s Class A Common Stock (the “Common Stock”), each subject to the terms and conditions set forth in the Purchase Agreement, for an aggregate consideration of $3.2 million.
F-6
| 3. | Significant Accounting Policies |
Basis of Presentation
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash
Cash is maintained at financial institutions and, at times, balances may exceed federally insured limits. The Company has never experienced any losses related to these balances. All of the Company’s cash balances were fully insured at June 30, 2018.
Financial Instruments
The Company’s financial instruments include cash, accounts payable, related party loans and a demand promissory note. The carrying amounts of cash and accounts payable approximate their fair value, due to the short-term nature of these items.
Long-Term Notes Payable
The Company’s long-term notes payable include accrued payroll to officers and accrued payments to third party consultants.
Research and Development
Research and development costs are charged to operations when incurred and are included in operating expenses. The Company expensed $370,853 and $466,354 for research and development for the years ended June 30, 2018 and 2017, respectively.
Income Taxes
Deferred income tax assets and liabilities arise from temporary differences associated with differences between the financial statements and tax basis of assets and liabilities, as measured by the enacted tax rates, which are expected to be in effect when these differences reverse. Deferred tax assets and liabilities are classified as current or non-current, depending on the classification of the assets or liabilities to which they relate. Deferred tax assets and liabilities not related to an asset or liability are classified as current or non-current depending on the periods in which the temporary differences are expected to reverse.
The Company follows the provisions of FASB ASC 740-10 “Uncertainty in Income Taxes” (ASC 740-10), January 1, 2007. The Company has not recognized a liability as a result of the implementation of ASC 740-10. A reconciliation of the beginning and ending amount of unrecognized tax benefits has not been provided since there are no unrecognized benefits at June 30, 2018 and since the date of adoption. The Company has not recognized interest expense or penalties as a result of the implementation of ASC 740-10. If there were an unrecognized tax benefit, the Company would recognize interest accrued related to unrecognized tax benefits in interest expense and penalties in operating expenses. The Company’s tax returns for the years ended June 30, 2015, 2016, 2017 and 2018 remain open to examination by taxing authorities.
F-7
Earnings (Loss) per Share
Basic earnings per share are computed by dividing net income by the weighted average number of shares of common stock outstanding during the year. Diluted earnings per common share are computed by dividing net income by the weighted average number of shares of common stock outstanding and dilutive options outstanding during the year. For the years ended June 30, 2017 and 2018 all outstanding options have been excluded from the calculation of the diluted net loss per share since their effect was anti-dilutive.
The following potentially dilutive securities were excluded from the computation of diluted loss per share for the years ended June 30, 2017 and 2018:
| 2017 | 2018 | ||
| Number of Shares (Thousands) | Number of Shares (Thousands) | ||
| Stock Options | 4,000 | 5,150 | |
| Warrants | 6,174 | 4,774 | |
| Total | 10,174 | 9,924 |
Stock-based Compensation
The Company has accounted for stock-based compensation under the provisions of FASB ASC 718 – “Stock Compensation” which requires the use of the fair-value based method to determine compensation for all arrangements under which employees and others receive shares of stock or equity instruments (stock options and common stock purchase warrants). For employee awards, the fair value of each stock option award is estimated on the date of grant using the Black-Scholes valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. For non-employees, the fair value of each stock option award is estimated on the measurement date using the Black-Scholes valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. For non-employees, the Company utilizes the graded vesting attribution method under which the entity treats each separately vesting portion (tranche) as a separate award and recognizes compensation cost for each tranche over its separate vesting schedule. Expected volatilities are based on historical volatility of peer companies and other factors estimated over the expected term of the stock options. For employee awards, the expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for the period of the expected term.
Goodwill
Goodwill is recorded when the purchase price paid for an acquisition exceeds the fair value of net identified tangible and intangible assets acquired. The Company performs an annual impairment test of goodwill and further periodic tests to the extent indicators of impairment develop between annual impairment tests. The Company’s impairment review process compares the fair value of the reporting unit to its carrying value, including the goodwill related to the reporting unit. To determine the fair value of the reporting unit, the Company may use various approaches including an asset or cost approach, market approach or income approach or any combination thereof. These approaches may require the Company to make certain estimates and assumptions including future cash flows, revenue and expenses. These estimates and assumptions are reviewed each time the Company tests goodwill for impairment and are typically developed as part of the Company’s routine business planning and forecasting process. While the Company believes its estimates and assumptions are reasonable, variations from those estimates could produce materially different results. The Company did not recognize any goodwill impairments for the years ended June 30th, 2017 and June 30th, 2018.
F-8
Impairment of Long-Lived Assets
Long-lived assets, including intangible assets, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset.
If the carrying amount of an asset exceeds its undiscounted estimated future cash flows, an impairment review is performed. An impairment charge is recognized in the amount by which the carrying amount of the asset exceeds the fair value of the asset. Generally, fair value is determined using valuation techniques such as expected discounted cash flows or appraisals, as appropriate. Assets to be disposed of would be separately presented in the balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated or amortized. The assets and liabilities of a disposed group classified as held for sale would be presented separately in the appropriate asset and liability sections of the balance sheet.
Recent accounting pronouncements
The Company has reviewed recent accounting pronouncements issued by the FASB (including its EITF), the AICPA, and the SEC and did not or are not believed by management to have a material impact on the Company’s financial statements.
4. Intangible Assets
The company’s intangible assets consist of intellectual property acquired from LAT Pharma, Inc., and are amortized over their estimated useful lives as indicated below. The following is a summary of the intangible assets as of June 30, 2018 and 2017.
| June 30, 2018 | June 30, 2017 | |||||||
| Intangible Assets subject to Amortization | $ | 2,293,770 | $ | 2,293,770 | ||||
| Accumulated Amortization | 509,790 | 280,413 | ||||||
| Intangible Assets (Net of Amortization) | $ | 1,783,980 | $ | 2,013,357 | ||||
Future expected Amortization of intangible assets is as follows:
| Year Ending June 30, | |
| 2019 | $ 229,377 |
| 2020 | 229,377 |
| 2021 | 229,377 |
| 2022 | 229,377 |
| 2023 | 229,377 |
| Thereafter | 637,095 |
| $ 1,783,980 |
F-9
5. Demand Note
On May 21, 2018, the Company received $250,000 in exchange for a promissory note from Acuitas Group Holding LLC. The promissory note carries 10% interest per year and has a maturity date of 10 business days of demand by Payee. The promissory note also has a provision that in the event of a superseding equity financing transaction, the Payor will receive 50% warrant coverage on same terms as if the superseding transactions occurs. On July 3, 2018, the Company entered into an equity financing transaction which resulted in Acuitas Group Holding LLC., receiving 833,333 warrants that expire on July 3rd 2024 with an exercise price of 1.8 cents per share. The Company valued the warrants at $29,666 using the Black Scholes Model and the following assumptions were used: volatility – 169%; Term – 6 years; Risk Free Rate – 2.96%; dividend rate – 0.00%. Based on their relative fair value, the Company allocated $26,519 of the proceeds to the warrants, which was recorded as additional interest expense for the year ended June 30, 2018. As the promissory note was converted into common stock subsequent to June 30, 2018, the balance as of June 30, 2018 is classified as long term on the accompanying balance sheet.
6. Related Party Transactions
Notes Payable
LAT Pharma was given a zero-interest bearing loan by the Company’s CEO, Jonathan Adams in the amount of $5,000 in August 2015 and $5,000 in November 2015. The total of $10,000 was outstanding when the Company merged with LAT Pharma. On June 16th, 2017, the Company was given an additional $25,000 zero-interest bearing loan by Jonathan Adams. During the quarter ended December 31, 2017, the Company repaid the $35,000 outstanding balance of the loan. During the quarter ended March 31, 2018, the Company was given an additional $25,000 zero-interest bearing loan by Jonathan Adams. During the quarter ended June 30, 2018, the Company repaid the $25,000 outstanding balance of the loan. The outstanding balance of the loan was $35,000 as of June 30, 2017 and zero as of June 30, 2018.
On March 23, 2017, Barrett Ehrlich agreed to defer the payment of his consulting fee debt of $173,333 until December 31, 2019, through the issuance of a Promissory note. The promissory note does not carry any interest charge as long as the amount is paid in full before December 31, 2019. The consulting fee debt has thereby been reclassified from a current liability to a long-term liability on the balance sheet. Any portion of the balance due under the note that remains unpaid after December 31, 2019 will accrue interest at a rate of 5% per annum until paid in full.
On March 23, 2017, Elliot Ehrlich agreed to forgive 50% of his salary debt of $444,056. The adjusted salary debt is $222,028.13. Elliot Ehrlich also agreed to defer the payment of his salary debt of $222,028 until December 31, 2019, through the issuance of a Promissory note. The promissory note does not carry any interest charge as long as the amount is paid in full before December 31, 2019. The salary debt has thereby been reclassified from a current liability to a long-term liability on the balance sheet and the salary debt forgiven has been reflected on the income statement as other income. Any portion of the balance due under the note that remains unpaid after December 31, 2019 will accrue interest at a rate of 5% per annum until paid in full.
On March 23, 2017, Jonathan Adams agreed to defer the payment of his salary debt of $180,555 until December 31, 2019, through the issuance of a Promissory note. The promissory note does not carry any interest charge as long as the amount is paid in full before December 31, 2019. The salary debt has thereby been reclassified from a current liability to a long-term liability on the balance sheet. Any portion of the balance due under the note that remains unpaid after December 31, 2019 will accrue interest at a rate of 5% per annum until paid in full.
The outstanding balance of the long-term note payable was $575,917 as of June 30, 2017 and $575,917 as of June 30, 2018.
F-10
Sale of Common Stock and Warrants
In January 2018, the Company sold an aggregate of 333,333 shares of common stock and warrants to purchase 333,333 shares of common stock to a member of its board of directors for aggregate gross proceeds of $50,000. The purchase price for the common stock and warrants was $0.15 per share. The warrants are exercisable at an exercise price of $0.15 at any time from date of issuance until 7 years from the date of issuance.
Common Stock issued for Services
In January 2018, the Company issued 1,400,000 shares of common stock as compensation for the Board of Directors. The shares were valued at $0.15 per share which was the trading price on date of issuance, and the value of the compensation was $210,000.
7. Commitments and Contingencies
Office Lease
On January 1, 2014, the Company executed a lease agreement with Cummings Properties for the Company’s office of 270 square feet at 100 Cummings Center, Suite 247-C, Beverly, MA 01915. The lease is for a term of five years from January 1, 2014 to December 30, 2018 and requires monthly payments of $379. The Company notified the lessor that it will terminate the lease on December 30, 2018.
Employment Agreements
On April 11, 2016, the Company entered into employment agreement with CEO Jonathan Adams. The Company’s agreement provides for a three-year term with minimum annual base salary of $250,000 per year.
Challenge to US Patent
On April 30, 2018, the Company received notice that Mallinckrodt Pharmaceuticals Ireland Limited had petitioned the US Patent and Trademark Office (USPTO) to institute an Inter Partes Review of BioVie’s US Patent No. 9,655,945 titled “Treatment of Ascites” (the ‘945 patent).
Inter Partes Review is a trial proceeding conducted with the USPTO Patent Trial and Appeal Board (PTAB) to review the patentability of one or more claims of a patent. Such review is limited to grounds of novelty and obviousness on the basis of prior art consisting of patents and printed publications. Although a petition for Inter Partes Review has been filed, grant of the petition by the PTAB is required for the proceeding to be instituted.
On August 15, 2018, BioVie submitted a Preliminary Response to the PTAB providing a rationale as to why, in the Company’s opinion, Mallinckrodt’s request to institute the IPR should not be granted. If he IPR is allowed to proceed, BioVie will seek to defend the ‘945 patent and/or pursue a favorable settlement. As of June 30, 2018, no adjustments or accruals are reflected as the Company is unable to determine a likely outcome at this time.
Royalty Agreements
Pursuant to the Agreement and Plan of Merger entered into on April 11, 2016 between LAT Pharma LLC and NanoAntibiotics, Inc., BioVie is obligated to pay a low single digit royalty on net sales of BIV201 (continuous infusion terlipressin) to be shared among LAT Pharma Members, PharmaIn Corporation; and The Barrett Edge, Inc.
Pursuant to the Technology Transfer Agreement entered into on July 25, 2016 between BioVie and the University of Padova (Italy), BioVie is obligated to pay a low single digit royalty on net sales of all terlipressin products covered by US patent no. 9,655,645 and any future foreign issuances capped at significantly less than $500,000 per year.
The Company and PharmaIN, Corporation, LAT Pharma’s former partner focused on the development of new modified drug candidates in the same therapeutic field but not including BIV201, agreed to pay royalties equal to less than 1% of future net sales of each company's ascites drug development programs, or if such program is licensed to a
third party, less than 5% of each company's net license revenues. The Company’s relationship with PharmaIN could advance into a collaboration or be terminated. The Company has an issued US Patent covering the use of BIV201 for the treatment of ascites patients in the outpatient setting using ambulatory pump infusion, and has filed patent applications for its drug candidate in Japan, and Europe, and China.
F-11
8. Stockholders’ Equity
Stock Options
During the year ended June 30, 2017 and 2018, the Company issued stock options to consultants and board of directors for services provided to the Company.
The following is a summary of stock option activity for the years ended June 30, 2017 and 2018.
| Shares (Thousands) | Weighted-Average Exercise Price | Weighted Remaining Average Contractual Term | |||
| Options | |||||
| Outstanding at July 1, 2016 | 3,000 | $ 0.06 | 6.5 | ||
| Granted | 1,000 | $ 0.24 | 4.0 | ||
| Outstanding at June 30, 2017 | 4,000 | $ 0.10 | 5.9 | ||
| Granted | 1,250 | $ 0.15 | 5.0 | ||
| Options Exercised | (100) | $ 0.02 | - | ||
| Outstanding at June 30, 2018 | 5,150 | $ 0.12 | 5.8 | ||
| Exercisable at June 30, 2018 | 4,150 | $ 0.13 | 4.8 |
The following is a summary of stock options outstanding and exercisable by exercise price as of June 30, 2018.
| Weighted Average Contract Life | |||
| Exercise Price | Outstanding | Exercisable | |
| $ 0.06 | 3,100,000 | 6.5 | 2,100,000 |
| $ 0.10 | 500,000 | 4.8 | 500,000 |
| $ 0.20 | 200,000 | 4.5 | 200,000 |
| $ 0.21 | 550,000 | 4.1 | 550,000 |
| $ 0.22 | 100,000 | 4.0 | 100,000 |
| $ 0.23 | 200,000 | 4.4 | 200,000 |
| $ 0.25 | 500,000 | 3.6 | 500,000 |
| Total | 5,150,000 | 4,150,000 |
F-12
The fair value of options granted during the year ended June 30, 2018 was estimated using the Black Scholes Method and the following assumptions: volatility - 124.7% to 143.46%; Term – 5 years; Risk Free Rate – 2.45% to 2.61%; dividend rate – 0.00%. The fair value of options granted during the year ended June 30, 2017 was estimated using the Black Scholes Method and the following assumptions: volatility – 136.8% to 143.1%; Term – 4 years; Risk Free Rate – 0.53% to 1.4%; dividend rate – 0.00%.
The compensation expense for the year ended June 30, 2018 includes $30,978 related to the stock options described above and an adjustment for year ended June 30, 2017 of $30,547. The adjustment was due to a correction made to the valuation of Stock Options issued to the Chief Operating Officer. The legal and professional expenses for the year ended June 30, 2018 includes $176,641 related to the stock options described above. The Company expects to recognize $11,486 of future expenses related to the vesting of stock options through April 11, 2019.
The compensation expense for the year ended June 30, 2017 includes $35,392 related to the stock options described above. The legal and professional expenses for the year ended June 30, 2017 includes $56,660 related to the stock options described above.
Extension of Maturity
In November 2017, the Company extended the maturity date of stock options to acquire 800,000 shares at exercise prices ranging from $0.21 to $0.25 issued to the board of directors between November 2016 and December 2016 by 3 years. The Company recorded an incremental expense of $79,491 based on the increase in fair value of the options.
Offerings of Common Stock and Warrants
Issuance of Shares for Cash
In September 2016, the Company sold and issued an aggregate of 49,999 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $5,000. The purchase price for the common stock was $0.10 per share.
In October 2016, the Company sold and issued an aggregate of 225,000 shares of common stock and warrants to purchase 112,500 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $45,000. The purchase price for the common stock and warrants was $0.20 per share. The warrants are exercisable at an exercise price of $0.50 at any time from date of issuance until 5 years from the date of issuance.
In November 2016, the Company sold and issued an aggregate of 250,000 shares of common stock and warrants to purchase 125,000 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $50,000. The purchase price for the common stock and warrants was $0.20 per share. The warrants are exercisable at an exercise price of $0.50 at any time from date of issuance until 5 years from the date of issuance.
In December 2016, the Company sold and issued an aggregate of 100,000 shares of common stock and warrants to purchase 50,000 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $20,000. The purchase price for the common stock and warrants was $0.20 per share. The warrants are exercisable at an exercise price of $0.50 at any time from date of issuance until 5 years from the date of issuance.
In January 2017, the Company, entered into a common stock purchase agreement (the “Purchase Agreement”) with Aspire Capital Fund, LLC, an Illinois limited liability company (“Aspire Capital”) which provides that, on the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $12.0 million of shares of the Company’s common stock over the 30-month term of the Purchase Agreement (“Aspire Equity Line”). On execution of the Purchase Agreement, the Company agreed to sell to Aspire Capital 1,000,000 shares of common stock and warrants to purchase 500,000 shares of common stock for proceeds of $200,000. The Warrant Shares will each have a five-year term and will be exercisable at $0.50 per share. Concurrently with entering into the Purchase Agreement, the Company also entered into a registration rights agreement with Aspire Capital (the “Registration Rights Agreement”), in which the Company agreed to file one or more registration statements, as permissible and necessary to register under the Securities Act of 1933, as amended (the “Securities Act”), registering the sale of the shares of the Company’s common stock that have been and may be issued to Aspire Capital under the Purchase Agreement.
F-13
Under the Purchase agreement, after the Securities and Exchange Commission (the “SEC”) has declared effective the registration statement referred to above, on any trading day selected by the Company, the Company has the right, in its sole discretion, to present Aspire Capital with a purchase notice (each, a “Purchase Notice”), directing Aspire Capital (as principal) to purchase up to 100,000 shares of the Company’s common stock per business day, up to $12.0 million of the Company’s common stock in the aggregate at a per share price (the “Purchase Price”) equal to the lesser of:
- the lowest sale price of the Company’s common stock on the purchase date; or
- the arithmetic average of the three (3) lowest closing sale prices for the Company’s common stock during the twelve (12) consecutive trading days ending on the trading day immediately preceding the purchase date.
In addition, on any date on which the Company submits a Purchase Notice to Aspire Capital in an amount equal to 100,000 shares and the closing sale price of our stock is equal to or greater than $0.30 per share, the Company also has the right, in its sole discretion, to present Aspire Capital with a volume-weighted average price purchase notice (each, a “VWAP Purchase Notice”) directing Aspire Capital to purchase an amount of stock equal to up to 30% of the aggregate shares of the Company’s common stock traded on its principal market on the next trading day (the “VWAP Purchase Date”), subject to a maximum number of shares the Company may determine. The purchase price per share pursuant to such VWAP Purchase Notice is generally 95% of the volume-weighted average price for the Company’s common stock traded on its principal market on the VWAP Purchase Date.
The Purchase Price will be adjusted for any reorganization, recapitalization, non-cash dividend, stock split, or other similar transaction occurring during the period(s) used to compute the Purchase Price. The Company may deliver multiple Purchase Notices and VWAP Purchase Notices to Aspire Capital from time to time during the term of the Purchase Agreement, so long as the most recent purchase has been completed.
The Purchase Agreement provides that the Company and Aspire Capital shall not affect any sales under the Purchase Agreement on any purchase date where the closing sale price of the Company’s common stock is less than $0.10. There are no trading volume requirements or restrictions under the Purchase Agreement, and the Company will control the timing and amount of sales of the Company’s common stock to Aspire Capital. Aspire Capital has no right to require any sales by the Company but is obligated to make purchases from the Company as directed by the Company in accordance with the Purchase Agreement. There are no limitations on use of proceeds, financial or business covenants, restrictions on future funding, rights of first refusal, participation rights, penalties or liquidated damages in the Purchase Agreement. In consideration for entering into the Purchase Agreement, concurrently with the execution of the Purchase Agreement, the Company issued to Aspire Capital 2,400,000 shares of the Company’s common stock (the “Commitment Shares”). The Purchase Agreement may be terminated by the Company at any time, at its discretion, without any cost to the Company. Aspire Capital has agreed that neither it nor any of its agents, representatives and affiliates shall engage in any direct or indirect short-selling or hedging of the Company’s common stock during any time prior to the termination of the Purchase Agreement. Any proceeds that the Company receives under the Purchase Agreement are expected to be used for working capital and general corporate purposes.
F-14
In March 2017, the Company sold and issued an aggregate of 500,000 shares of common stock and warrants to purchase 250,000 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $100,000. The purchase price for the common stock and warrants was $0.20 per share. The warrants are exercisable at an exercise price of $0.50 at any time from date of issuance until 5 years from the date of issuance.
In May 2017, the Company sold and issued an aggregate of 240,000 shares of common stock and warrants to purchase 120,000 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $60,000. The purchase price for the common stock and warrants was $0.25 per share. The warrants are exercisable at an exercise price of $0.60 at any time from date of issuance until 5 years from the date of issuance. In August 2017, the Company issued an aggregate of 32,727 shares of common stock and 16,364 warrants to compensate these investors who purchased common stock at a $0.25 share price in a Series C offering prior to a reduction in the offering price to $0.22 per share.
In July 2017 and August 2017, the Company sold and issued an aggregate of 886,364 shares of common stock and warrants to purchase 443,182 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $195,000. The purchase price for the common stock and warrants was $0.22 per share. The warrants are exercisable at an exercise price of $0.60 at any time from date of issuance until 5 years from the date of issuance.
Between July 2017 and September 2017, the Company sold an aggregate of 250,000 shares of common stock in transactions under the Aspire Equity Line for aggregate gross proceeds of $50,000. The average purchase price for the common stock was $0.20 per share.
In October 2017, the Company sold and issued an aggregate of 159,091 shares of common stock and warrants to purchase 79,545 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $35,000. The purchase price for the common stock and warrants was $0.22 per share. The warrants are exercisable at an exercise price of $0.60 at any time from date of issuance until 5 years from the date of issuance.
In November 2017, the Company also sold and issued an aggregate of 68,182 shares of common stock and warrants to purchase 34,091 shares of common stock in a private placement transaction for aggregate gross proceeds of approximately $15,000. The purchase price for the common stock and warrants was $0.22 per share. The warrants are exercisable at an exercise price of $0.60 at any time from date of issuance until 5 years from the date of issuance.
In January 2018, the Company sold an aggregate of 333,333 shares of common stock and warrants to purchase 333,333 shares of common stock to a member of its board of directors for aggregate gross proceeds of $50,000. The purchase price for the common stock and warrants was $0.15 per share. The warrants are exercisable at an exercise price of $0.15 at any time from date of issuance until 7 years from the date of issuance.
In June 2018, 100,000 shares of stock options were exercised for $2,000.
Issuance of Shares for Services
In August 2017, the Company issued 1,500,000 shares of common stock to Aspire Capital in exchange for services. The shares were valued at $0.22 per share which was the trading price on date of issuance, and the value of the services were $330,000.
In November 2017, the Company issued 150,000 shares of common in exchange for services. The shares were valued at $0.23 per share which was the trading price on date of issuance, and the value of the services were $34,500.
In January 2018, The Company issued 30,000 shares of common stock in exchange for services. The shares were valued at $0.13 per share which was the trading price on date of issuance, and the value of the services were $3,900.
F-15
In January 2018, the Company issued 1,400,000 shares of common stock as compensation for the Board of Directors. The shares were valued at $0.15 per share which was the trading price on date of issuance, and the value of the compensation was $210,000.
In February 2018, the Company issued 600,000 shares of common stock in exchange for services. The shares were valued at $0.0475 per share which was the trading price on date of issuance, and the value of the services were $28,500.
In April 2018, the Company issued 300,000 shares of common in exchange for services. The shares were valued at $0.045 per share, and the value of the services were $13,500. In April 2018, the Company issued 150,000 shares of common in exchange for services. The shares were valued at $0.024 per share which was the trading price on date of issuance, and the value of the services were $3,600.
In May 2018, the Company issued 250,000 shares of common in exchange for services. The shares were valued at $0.018 per share which was the trading price on date of issuance, and the value of the services were $4,500.
In May 2018, the Company issued 68,500 shares of common in exchange for services. The shares were valued at $0.10 per share which was the trading price on date of issuance, and the value of the services were $6,850.
In June 2018, the Company issued 300,000 shares of common in exchange for services. The shares were valued at $0.025 per share which was the trading price on date of issuance, and the value of the services were $7,500.
Issuance of Warrants for Cash
In December 2017, the Company issued warrants to purchase 2,500,000 shares of common stock in a private placement transaction for aggregate gross proceeds of $100,000. The purchase price for the warrants were $0.04 per warrant. The warrants are exercisable at an exercise price of $0.20 at any time from date of issuance until 7 years from the date of issuance.
Issuance of Warrants for Services
In January 2018, the Company issued warrants to purchase 105,000 shares of common stock in exchange for services. The warrants are exercisable at an exercise price of $0.15 any time from the date of issuance until 7 years from the date of issuance. The warrants were valued at $9,444. The fair value of the warrants granted was estimated using the Black Scholes Method and the following assumptions: volatility – 166.7%; Term – 7 years; Risk Free Rate – 2.48%; dividend rate – 0.00%
In February 2018, the Company issued warrants to purchase 105,000 shares of common stock in a termination agreement. The warrants are exercisable at an exercise price of $0.15 any time from the date of issuance until 7 years from the date of issuance. The warrants were valued at $3,025. The fair value of the warrants granted was estimated using the Black Scholes Method and the following assumptions: volatility – 166.7%; Term – 7 years; Risk Free Rate – 2.81%; dividend rate – 0.00%
Warrant Price Adjustment
In December 2017, the Company issued warrants to purchase 2,500,000 shares of common stock in a private placement transaction for aggregate gross proceeds of $100,000. The warrants were exercisable at an exercise price of $0.20 at any time from date of issuance until 7 years from the date of issuance. The warrants have a down round feature that reduces the exercise price if the Company sells stock for a lower price. In January 2018, the Company sold shares at $0.15, which therefore triggered the reduction in the strike price. The Company calculated the difference in fair value of the warrants between the stated exercise price and the reduced exercise price and recorded $20,995 as a deemed dividend. The fair value of the warrants granted was estimated using the Black Scholes Method and the following assumptions: volatility – 164.7%; Term – 7 years; Risk Free Rate – 2.47%; dividend rate – 0.00%.
F-16
The following table summarizes the warrants that have been issued:
| Weighted Average | Weighted Average | |||||||||||||
| Number of Shares | Exercise Price | Remaining Life (Years) | ||||||||||||
| Outstanding at June 30, 2016 | 5,000,000 | $ | 0.50 | — | ||||||||||
| Granted | 1,173,864 | $ | 0.51 | 3.5 | ||||||||||
| Outstanding at June 30, 2017 | 6,173,864 | $ | 0.50 | 0.7 | ||||||||||
| Granted | 3,600,151 | $ | 0.22 | 6.1 | ||||||||||
| Expired | (5,000,000 | ) | $ | 0.50 | — | |||||||||
| Outstanding at June 30, 2018 | 4,774,015 | $ | 0.29 | 5.5 | ||||||||||
The following table summarizes the warrants by price:
| Weighted Average | Weighted Average | |||||||||
| Exercise Price | Number of Shares | Remaining Life (Years) | ||||||||
| $ | 0.15 | 3,043,333 | 6.4 | |||||||
| $ | 0.50 | 1,037,501 | 3.5 | |||||||
| $ | 0.60 | 693,181 | 4.1 | |||||||
| 4,774,015 | 5.5 | |||||||||
| 9. | Income Taxes |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. At June 30, 2018, the Company has a Net Operating Loss (“NOL”) carryforward of approximately $1,800,000. The NOL expires during the years 2032 to 2037. Realization of any portion of the $832,186 of net deferred tax assets at June 30, 2018 is not considered more likely than not by management; accordingly, a valuation allowance has been established for the full amount. The valuation allowance as of June 30, 2018 was $832,186. The change in the valuation allowance during the year ended June 30, 2018 amounted to $357,231. The Company does not have any uncertain tax positions or events leading to uncertainty in a tax position. The Company’s 2015, 2016 and 2017 Corporate Income Tax Returns are subject to Internal Revenue Service examination.
On December 22, 2017, the Tax Cuts and Jobs Act (the “Act”) was signed into law. The Act decreases the U.S. corporate federal income tax rate from a maximum of 35% to a flat 21% effective January 1, 2018. The Act also includes a number of other provisions including, among others, the elimination of net operating loss carrybacks and limitations on the use of future losses, the repeal of the Alternative Minimum Tax regime and the repeal of the domestic production activities deduction. These provisions are not expected to have a material effect on the Corporation.
F-17
Given the significant complexity of the Act and anticipated additional implementation guidance from the Internal Revenue Service, further implications of the Act may be identified in future periods.
Significant components of the Company’s deferred tax assets are as follows:
| June 30, 2018 | June 30, 2017 | |||||||
| Deferred tax assets: | ||||||||
| Tax loss carryforward | $ | 555,064 | $ | 446,071 | ||||
| Intangible assets | 19,277 | 3,735 | ||||||
| Stock based compensation | 257,845 | 25,149 | ||||||
| Valuation Allowance | (832,186 | ) | (474,955 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
Since management of the Company believes that it is more likely than not that the net deferred tax assets will not provide future benefit, the Company has established a 100 percent valuation allowance on the net deferred tax assets as of June 30, 2018.
Reconciliation of the differences between income tax benefit computed at the federal and state statutory tax rates and the provision for income tax benefit for the years ended June 30, 2018 and 2017 is as follows:
| 2018 | 2017 | |||||||
| Income tax expense (benefit) at federal statutory rate | 34 | % | 34 | % | ||||
| State taxes, net of federal benefit | 5 | % | 5 | % | ||||
| Change in valuation allowance | -39 | % | -39 | % | ||||
| — | — | |||||||
10. Subsequent Events
On July 3, 2018, BioVie, Inc., the Company, entered into a Securities Purchase Agreement (the “Purchase Agreement”) with Acuitas Group Holdings, LLC (“Acuitas”) and certain other purchasers identified in the Purchase Agreement (together with Acuitas, the “Purchasers”) pursuant to which (i) the Purchasers agreed to purchase an aggregate of 2,133,332 shares of the Company’s newly created Series A Convertible Preferred Stock (the “Preferred Stock”) at a price per share of $1.50 per share of Preferred Stock (the “Initial Sale”) and (ii) the Company will issue associated warrants (the “Warrants”) to purchase 213,333,200 shares of the Company’s Class A Common Stock (the “Common Stock”), each subject to the terms and conditions set forth in the Purchase Agreement, for an aggregate consideration of $3.2 million. Acuitas also received an additional 833,333 Warrants in connection with the payoff of a note issued by the Company in favor of Acuitas. The Initial Sale and issuance of the Warrants occurred on July 3, 2018. In addition, Acuitas has the option to purchase up to an additional 200,000,000 shares of Common Stock at a price per share of $0.015, and associated warrants on the same terms as the Warrants, within two weeks following the one year anniversary of the closing of the Initial Sale (the “Subsequent Sale”) in the event that the Company has not obtained $3,000,000 of funding through various non-dilutive grants prior to the one year anniversary of the closing of the Initial Sale.
F-18
Each share of Preferred Stock automatically converted into 100 shares of Common Stock upon the filing with the Secretary of State of the State of Nevada of a Certificate of Amendment to the Company’s Articles of Incorporation (the “Amendment”) on August 13, 2018 that increased the number of authorized shares of Common Stock to 800,000,000. The Amendment was approved by the written consent of the holders of more than a majority of the Company’s issued and outstanding Common Stock on July 3, 2018, and was filed with the Secretary of State of the State of Nevada 20 calendar days following the distribution of the Company’s Definitive Information to be filed with the Securities and Exchange Commission.
See the heading “Series A Convertible Preferred Stock” below for additional information related to the Preferred Stock. The purchase price of the Preferred Stock in the Initial Sale, the exercise price of the Warrants, and the Common Stock in the Subsequent Sale is subject to adjustment. First, in the event that the volume weighted average price of the Common Stock during the five-trading day period following July 3, 2018 is less than $0.015 per share, the price per share of Common Stock, the associated conversion ratio of the Preferred Stock, and the exercise price of the Warrants shall be retroactively adjusted to reflect such lower price. Second, in the event that Mallinckrodt Pharmaceuticals Ireland Limited prevails in any proceeding which results in the useful life of the Company’s current intellectual property rights being reduced by more than 75 percent, then the price per share of Common Stock, the associated conversion ratio of the Preferred Stock, and the exercise price of the Warrants shall be retroactively adjusted to 50 percent of the then-effective price per share of Common Stock under the Purchase Agreement (for example, if the then-effective price per share of Common Stock is $0.015, then following such event, the price per share will be $0.0075). In each case, the Company may be required to issue additional shares of Common Stock, but in no event will the Company be required to pay cash, to reflect such lower price per share.
The Purchase Agreement contained customary representations and warranties. In connection with the disclosure schedule associated with the representations and warranties, the Company also disclosed customary information, including the following: (i) the existence of the Mallinckrodt Pharmaceuticals Ireland Limited petition before the US Patent Trial and Appeal Board, (ii) the current capitalization of the Company, (iii) the Company’s obligation to pay a royalty on the net sales of BIV201 (continuous infusion terlipressin) in the amount of five percent to be allocated four percent to LAT Pharma LLC members, 0.4 percent to PharmaIn Corporation and 0.6 percent to The Barrett Edge, Inc. pursuant to the Agreement and Plan of Merger, dated April 11, 2016, by and between LAT Pharma LLC and the Company, (iv) the Company’s obligation to pay a royalty of five percent on net sales of all terlipressin products covered by specified patents up to a maximum of $200,000 per year pursuant to the Technology Transfer Agreement, dated July 25, 2016, by and between the Company and the University of Padova (Italy), and (v) certain recent issuances of Common Stock by the Company.
Pursuant to the Purchase Agreement, Terren Peizer, the Chairman of Acuitas, was appointed as a member of the Company’s Board of Directors (the “Board”) and as the Chief Executive Officer of the Company, effective July 3, 2018. The issuance of the Preferred Stock, the Warrants and the underlying common stock under the Purchase Agreement is exempt from registration under the Securities Act of 1933, as amended (the “Securities Act”), pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(a)(2) of the Securities Act.
On July 9, 2018, Elliot Ehrlich entered into an Accord and Debt Satisfaction Agreement with the Company in which he agreed to release the Company from all liabilities including the original contract to defer payment of his accrued salary dated March 23, 2017 totaling the amount of $222,028.13 and received the settled sum of $22,203 and 222,028 common shares in satisfaction. This settlement reduced the Company’s debt by $222,028.13.
On July 9, 2018, Jonathan Adams entered into an Accord and Debt Satisfaction Agreement with the Company in which he agreed to release the Company from all liabilities including the original contract to defer payment of his accrued salary dated March 23, 2017 and subsequent unpaid salary, totaling the amount of $513,889, and received the settled sum of $25,694 in satisfaction. This settlement reduced the Company’s debt by $513,889.
F-19
On July 19, 2018, Geis-Hides Consulting LLC entered into an Accord and Debt Satisfaction Agreement with the Company in which the consulting firm agreed to release the Company from all liabilities arising from the Original Contract and Debt Repayment Plan dated December 15, 2013 totaling $130,000 and received the settled sum of $65,000 and 260,000 common shares in satisfaction. This settlement reduced the Company’s debt by $130,000.
As a result of the conversion of the Series A Preferred Stock in July 2018, the exercise of warrants to purchase 2,500,000 shares of common stock was reduced from $0.15 per share to $0.015 per share. On August 4, 2018, the Company issued 2,241,913 shares of common stock pursuant to a cash less exercise of warrants to purchase 2,500,000 shares at an exercise price of $0.015 per share.
On August 8, 2018, Barrett Ehrlich on behalf of The Barrett Edge Inc. (“Barrett”) entered into an Accord and Debt Satisfaction Agreement with the Company in which Barrett agreed to release the Company from all liabilities including the original contract to defer payment of Barrett’s accrued salary dated March 23, 2017, loan to the Company for $14,000, and subsequent unpaid consulting fees, totaling $493,333, and received the settled sum of $131,333 and 493,333 common shares in satisfaction. This settlement reduced the Company’s debt by $507,333.
F-20
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A(T). | CONTROLS AND PROCEDURES |
Disclosure Controls and Procedures
The Company’s Chief Operating Officer has evaluated the effectiveness of the Company’s disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the fiscal period ending June 30, 2018 covered by this Annual Report on Form 10-K. Based upon such evaluation, the Chief Operating Officer has concluded that, as of the end of such period, the Company’s disclosure controls and procedures were not effective as required under Rules 13a – 15(e) and 15d – 15(e) under the Exchange Act. This conclusion by the Company Operating Officer does not relate to reporting periods after June 30, 2018.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rue 13a-15(f) and 15d – 15(f) of the Exchange Act) of the Company. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
The Company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies and procedures may deteriorate.
Management, under the supervision of the Company’s Chief Operating Officer, conducted an evaluation of the effectiveness of internal control over financial reporting based on the framework in Internal control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, management concluded that the Company’s internal control over financial reporting was not effective as of June 30, 2018 under the criteria set forth in the Internal Control – Integrated Framework.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. Management has determined that material weaknesses exist due to a lack of formalized controls and procedures as well as a lack of segregation of duties, as well as the absence of an independent audit committee chair, resulting from the Company’s limited resources.
-27-
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the fourth fiscal quarter ended June 30, 2018 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Part III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Set forth below is certain information concerning the directors and executive officers of the Company as of June 30, 2018.
| Name | Age | Position | ||||
| Terren Peizer | 59 | Chairman & Chief Executive Officer | ||||
| Jonathan Adams | 55 | President & Chief Operating Officer | ||||
| Cuong Do | 53 | Independent Director | ||||
| Jim Lang | 55 | Independent Director | ||||
| Julie Anderson | 62 | Corporate Secretary and Independent Director | ||||
| Hari Kumar | 63 | Independent Director | ||||
| Michael Sherman | 59 | Independent Director | ||||
| Mina Sooch | 50 | Independent Director | ||||
According to our Bylaws, the directors shall be elected at the annual meeting of the stockholders and each director shall be elected to serve until his successor shall be elected and shall qualify. A director need not be a stockholder. Directors shall not receive any stated salary for their services as directors or as members of committees, but by resolution of the Board a fixed fee and expenses of attendance may be allowed for attendance at each meeting. The Bylaws shall not be construed to preclude any director from serving the Company in any other capacity as an officer, agent or otherwise, and receiving compensation therefor.
There are no familial relationships among any of our Directors or officers. Jim Lang currently also serves as a Director at OptimizeRX, a US reporting Company that is listed on the Nasdaq stock exchange. None of our other Directors or officers is or has been a Director or has held any form of directorship in any other U.S. reporting companies except as mentioned above. None of our Directors or officers has been affiliated with any Company that has filed for bankruptcy within the last five years. The Company is not aware of any proceedings to which any of the Company’s Officers or Directors, or any associate of any such officer or Director, is a party that are adverse to the Company. We are also not aware of any material interest of any of our officers or directors that is adverse to our own interests.
Information
Mr. Terren Peizer, Chairman of the Board of Directors and Chief Executive Officer, is an entrepreneur, investor, and financier with a particular interest in healthcare, having founded and successfully commercialized several healthcare companies. Mr. Peizer is the founder of Catasys, Inc., a leader in behavioral and mental health management services, having served as the Company’s Chairman of the Board of Directors and CEO since the Company’s inception in 2003. Mr. Peizer also is the Founder, Chairman and CEO and majority shareholder of NeurMedix, Inc., a biotechnology
Company with a focus on inflammatory, neurological and neuro-degenerative diseases. Mr. Peizer is Chairman of Acuitas Group Holdings, LLC, his personal holding Company that owns his portfolio Company interests. Through Acuitas, he owns Crede Capital Group, LLC, an industry leader in investing in micro and small capitalization public equities, having invested over $1.2 billion directly into portfolio companies. Previously he was Chairman of Cray, Inc., the leading supercomputing Company, and held senior executive positions at various publicly-traded growth companies and with the investment banking firms Goldman Sachs, First Boston, and Drexel Burnham Lambert. He received his B.S.E. in finance from The Wharton School of Finance and Commerce.
-28-
Mr. Jonathan Adams has served as the Company’s Chief Executive Officer and Chief Financial Officer since it acquired LAT Pharma LLC on April 11, 2016 until July 2018. In July 2018, he began serving as the Company’s President and Chief Operating Officer. He founded LAT Pharma LLC and served as its Chief Executive Officer prior to its acquisition. Mr. Adams has over 26 years of biopharmaceutical industry experience, including corporate finance, Company acquisitions and licensing deals, marketing and sales support. At Searle Pharmaceuticals he was a member of the global launch team for Celebrex, and he has worked on launching numerous new drugs and medical devices. Mr. Adams earned a BS at Cornell University and an MBA at the Tuck School at Dartmouth.
Mr. Cuong Do is currently President, Global Strategy Group, at Samsung. Mr. Do helps to set the strategic direction for Samsung Group’s diverse business portfolio. He was previously the Chief Strategy Officer for Merck, a leading US pharmaceuticals Company, Tyco Electronics, and Lenovo. Mr. Do is a former senior partner at McKinsey & Company, where he spent 17 years and helped build the healthcare, high tech and corporate finance practices. He holds a BA from Dartmouth College, and an MBA from the Tuck School of Business at Dartmouth.
Mr. Jim Lang is currently CEO of Water Street Capital’s and JLL Partner’s Global Life Sciences Services Platform. He formerly served as the CEO of Decision Resources Group (DRG), which he transformed into a leading healthcare data and analytics firm. Prior to that, Jim was CEO of IHS Cambridge Energy Research Associates (IHS CERA), a recognized leader in energy industry subscription information products, and formerly the President of Strategic Decisions Group (SDG), a leading global strategy consultancy. Mr. Lang holds a BS summa cum laude in electrical and computer engineering from the University of New Hampshire and an MBA with Distinction from the Tuck School of Business. Jim Lang currently also serves as a Director at OptimizeRX, a Nasdaq listed Company.
Ms. Julie Anderson has decades of pharmaceutical industry marketing and new drug commercialization experience. She most recently served Catheter Connections, Inc. as its Vice President of Marketing until the Company was sold. Previously she was Senior Director of Marketing for Durata Therapeutics, Inc. contributing to Company growth which led to the Company being acquired by Actavis (now Allergan) in 2014 in a deal valued at about $675 million. Previously she worked for Sanofi-Synthelabo, Inc., Bayer Pharmaceuticals, and G.D. Searle. She originally trained as a nurse and earned a Masters of Management at Northwestern University.
Hari Kumar, PhD held positions of increasing responsibility at Roche Pharma culminating in serving as Global Business Development Director, and in 2007 assumed the role of Chief Business Officer for Amira Pharmaceuticals. He led the sale of Amira to Bristol-Myers Squibb in 2011 for $475 million. He then served as Chief Executive Officer (CEO) for Panmira Pharmaceuticals LLC, which is developing anti-inflammatory compounds, and in 2013 became CEO for Adheron Therapeutics, which Roche Pharma acquired in 2015 for $580 million. Dr. Kumar earned a PhD in immunology in 1984.
Mina Sooch, is a successful entrepreneur, executive, and venture capitalist in the life sciences sector. From 2014 to 2017, she served as President, CEO, and Board member of Gemphire Therapeutics, advancing its drug candidate through multiple clinical trials, raising nearly $60 million in funding, and taking the Company public. Prior to Gemphire, she co-founded and served as CEO of ProNAi, an oncology Company, where she raised over $70 million from venture capital investors. Prior to her CEO roles, she spent over a decade in life sciences venture capital as a Founder of Apjohn Ventures with several portfolio companies developing treatments for kidney and liver diseases. Mina received an MBA from Harvard Business School and holds a BS from Wayne State University.
Michael Sherman JD recently retired from his position as a Managing Director at Barclays Plc, where he had worked since 2008. Previously he was a Managing Director at Lehman Brothers, Inc. He has worked in investment banking for 30 years. Mr. Sherman has significant experience in healthcare finance, most recently assisting on a $450 million convertible transaction for Neurocrine Biosciences. He has worked on successful financial transactions for Teva Pharmaceutical Industries, Amgen Inc., Cubist Pharmaceuticals, Merck & Co., and Cardinal Health, among other companies. After graduating from the University of Pennsylvania, Michael Sherman received his JD, cum laude, from the Harvard Law School.
-29-
Qualifications
Terrent Peizer’s qualifications to serve on our Board of Directors are primarily based on his experience as an entrepreneur, investor, and financier with a particular interest in healthcare, having founded and successfully commercialized several healthcare companies. Mr. Peizer is the founder of Catasys, Inc., a leader in behavioral and mental health management services, having served as the Company’s Chairman of the Board of Directors and CEO since the Company’s inception in 2003. Mr. Peizer also is the Founder, Chairman and CEO and majority shareholder of NeurMedix, Inc., a biotechnology Company with a focus on inflammatory, neurological and neuro-degenerative diseases. Mr. Peizer is Chairman of Acuitas Group Holdings, LLC, his personal holding Company that owns his portfolio Company interests. Through Acuitas, he owns Crede Capital Group, LLC, an industry leader in investing in micro and small capitalization public equities, having invested over $1.2 billion directly into portfolio companies.
Jonathan Adams’s qualifications to serve on our Board of Directors are primarily based on his founding of LAT Pharma LLC and his over 26 years of biopharmaceutical industry experience. As Chief Executive of LAT Pharma LLC, Mr. Adams worked to develop CIPT Technology and secured Orphan Drug Designation for a BIV201 analogue (this new drug candidate is no longer in development). Mr. Adams’s biopharmaceutical experience includes work in corporate finance, Company acquisitions and licensing deals, marketing and sales support.
Cuong Do’s qualifications to serve on our Board of Directors are primarily based on his decades of experience as an executive in the pharma, biotech, and other high technology industries. He was previously the Chief Strategy Officer for Merck, a leading US pharmaceuticals Company, Tyco Electronics, and Lenovo. Mr. Do is a former senior partner at McKinsey & Company, where he spent 17 years and helped build the healthcare, high tech and corporate finance practices.
Jim Lang’s qualifications to serve on our Board of Directors are primarily based on his decades of experience as a strategy consultant, broad industry expertise, and senior-level management experience running several healthcare and information technology companies. This includes his experience as CEO of Decision Resources Group, CEO of IHS Cambridge Energy Research Associates (IHS CERA), and President of Strategic Decisions Group (SDG), a leading global strategy consultancy.
Julie Anderson’s qualifications to serve on our Board of Directors are primarily based on her decades of successful pharmaceutical marketing and new drug commercialization expertise. For Searle she led the global launch of the multi-billion-dollar drug Celebrex, and more recently for Durata Therapeutics she led the marketing efforts which resulted in a sale of the Company for about $675 million. Originally trained as a critical care nurse, Julie treated patients at risk of death due to complications caused by chronic liver cirrhosis, and deeply understands the unmet medical need targeted by BioVie.
Hari Kumar’s qualifications to serve on our Board of Directors are primarily based on his decades of biopharma industry experience including serving as the chief executive officer at multiple companies, extensive technical and business knowledge, and outstanding track record for delivering value to investors. He led the sale of Amira to Bristol-Myers Squibb in 2011 for $475 million, and as CEO for Adheron Therapeutics, he led the sale of this Company to Roche Pharma for $580 million in 2015.
Mina Sooch’s qualifications to serve on our Board of Directors are primarily based on her decades of biopharma industry experience including as a successful entrepreneur, executive, and venture capitalist in the life sciences sector. She has served as President, CEO, and a Board member for multiple biopharma companies. Prior to her CEO roles, she spent over a decade in life sciences venture capital with several portfolio companies developing treatments for kidney and liver diseases.
-30-
Michael Sherman’s qualifications to serve on our Board of Directors are primarily based on his decades of finance industry experience including as a Managing Director at Barclays Plc and as a Managing Director at Lehman Brothers, Inc. He has worked in investment banking for 30 years. Mr. Sherman has significant experience in healthcare finance including having worked on successful financial transactions for several pharmaceutical and healthcare focused companies.
AUDIT COMMITTEE
We do not have an audit committee or an audit committee financial expert. Our corporate financial affairs are simple at this stage of development and each financial transaction can be viewed by any officer or Director at will.
CODE OF ETHICS
We have adopted a code of ethics meeting the requirements of Section 406 of the Sarbanes-Oxley Act of 2002. We believe our code of ethics is reasonably designed to deter wrongdoing and promote honest and ethical conduct; provide full, fair, accurate, timely and understandable disclosure in public reports; comply with applicable laws; ensure prompt internal reporting of violations; and provide accountability for adherence to the provisions of the code of ethic. Our code of ethics is filed as an exhibit to this Form 10-K.
| ITEM 11. | EXECUTIVE COMPENSATION |
We have not paid any compensation to any of our executive officers, however, we did accrue the Chief Executive Officer’s salary per the employment agreements effective July 1, 2013 and subsequently April 11, 2016.
Summary Compensation Table
| Annual Compensation | Long Term Compensation | |||||||||||||||||||||||||||||||||||
| Name and Principal Position | Year (1) | Salary | Bonus | Stock Awards | Option Awards | Non-Equity Incentive Plan Compensation | Nonqualified Deferred Compensation Earnings | All Other Compensation | Total | |||||||||||||||||||||||||||
| Jonathan Adams | ||||||||||||||||||||||||||||||||||||
| Chief Executive Officer and Chief Financial Officer, Treasurer and Corporate Secretary | 2016 | $ | 250,000 | $ | — | $ | — | $ | 69,659 | $ | — | $ | — | $ | — | $ | 319,659 | |||||||||||||||||||
| 2017 | $ | 250,000 | $ | 65,939 | $ | 315,939 | ||||||||||||||||||||||||||||||
| 2018 | $ | 250,000 | $ | — | $ | — | $ | 30,978 | $ | 280,978 | ||||||||||||||||||||||||||
| Elliot Ehrlich | ||||||||||||||||||||||||||||||||||||
| Chief Executive Officer and Chief Financial Officer, Treasurer and Corporate Secretary | 2014 | $ | 150,000 | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | 150,000 | |||||||||||||||||||
| 2015 | $ | 150,000 | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | 150,000 | ||||||||||||||||||||
_____________________________________
(1) We were incorporated on April 10, 2013.
-31-
Employment Agreement
On April 11, 2016, the Company entered into an employment agreement with the Company’s Chief Executive Officer paying $250,000 in annual salary. The agreement was effective beginning April 11, 2016 and expires on April 10, 2019.
Option/SAR Grants
In connection with the employment agreement signed with the Chief Financial Officer on April 11, 2016, Jonathan Adams received options to acquire 3 million shares exercisable at $0.06 per share, the closing price on that date. These Options Group A shall become vested and exercisable (i) as to 1 million shares on April 11, 2017, (ii) as to 1 million shares on April 11, 2018, and (iii) as to 1 million shares on April 11, 2019.
Between 11/16/2016 and 5/19/2017, the Company issued options to acquire 1 million shares exercisable at an average price of $0.24 per share to consultants and board of directors for services provided to the Company.
Long-Term Incentive Plans and Awards
Other than the options granted to the Chief Executive Officer as described above, the Company does not have any long-term incentive plans that provide compensation intended to serve as incentive for performance. Since prior to this grant, no individual grants or agreements regarding future payouts under non-stock price-based plans had been made to any executive officer or any Director or any employee or consultant since our inception, no future payouts under non-stock price-based plans or agreements had been granted or entered into or exercised by our officer or Director or employees or consultants.
Compensation of Directors
There are no arrangements pursuant to which our Directors are or will be compensated in the future for any services provided to the Company, except that each Director shall receive stock options and common share grants as remuneration for their service in lieu of cash compensation. For fiscal year 2018, each Director received 100,000 stock options on the one-year anniversary of his or her service to the Company with an exercise price equal to the closing stock price on the day of the option grant. The total value of the options granted to Directors for FY 2018 was $50,482 based on the Black-Scholes option value method. Each Director also receives a stock grant of 200,000 common shares for every year of service. On January 2, 2018, the seven (7) Directors received a combined grant of 1.4 million common shares with a face value of $210,000 based on the closing stock price of $0.15 on the grant date.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Long-Term Incentive Plans and Awards
Other than the options granted to the Chief Executive Officer on April 11, 2016 as described previously, the Company does not have any long-term incentive plans that provide compensation intended to serve as incentive for performance. Since prior to this grant, no individual grants or agreements regarding future payouts under non-stock price-based plans had been made to any executive officer or any Director or any employee or consultant since our inception, no future payouts under non-stock price-based plans or agreements had been granted or entered into or exercised by our officer or Director or employees or consultants.
-32-
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information concerning the ownership of the Common Stock by (a) each person who, to the best of our knowledge, beneficially owned on that date more than 5% of our outstanding Common Stock, (b) each of our Directors and executive officers and (c) all current Directors and executive officers as a group.
| Name and Address of Beneficial Owner | Number of Common Shares of Beneficial Ownership (1) | Percentage of Beneficial Ownership | ||||||
| Terren Peizer | 400,833,333 | 80.30 | % | |||||
| Jonathan Adams | 8,136,248 | 8.10 | % | |||||
| Julie Anderson | 368,500 | * | ||||||
| Cuong Do | 20,537,888 | 17.70 | % | |||||
| James Lang | 5,328,788 | 5.20 | % | |||||
| Hari Kumar | 427,272 | — | ||||||
| Michael Sherman | 3,935,472 | 3.90 | % | |||||
| Mina Sooch | 613,636 | * | ||||||
| All Directors and executive officers as a group (eight persons): | 440,181,137 | 83.60 | % | |||||
| Other 5% or Greater Beneficial Owners: | ||||||||
| Acuitas Group Holdings, LLC | ||||||||
| 1601 Wilsire Boulevard | ||||||||
| Suite 1100 | ||||||||
| Los Angeles, CA 90025 | 400,833,333 | 80.3 | % | |||||
| Elliot Ehrlich | ||||||||
| 9511 Collins Ave # 807 Surfside, FL 33154 | 7,662,500 | 7.8 | % | |||||
| Leo and Helene Ehrlich | ||||||||
| 7846 Tennyson Ct, Baton Raton, FL 33433 | 8,500,000 | 8.6 | % | |||||
| Rebecca Guttman | ||||||||
| 655 Ibsen St., Woodmere, NY 11598 | 8,500,000 | 8.6 | % | |||||
| RGN Brothers Trust | ||||||||
| 2715 Avenue L, Brooklyn, NY 11210 | 8,500,000 | 8.6 | % | |||||
_________________________________
(1) Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission and generally includes voting or investment power with respect to securities. In accordance with SEC rules, shares of Common Stock issuable upon the exercise of options or warrants which are currently exercisable or which become exercisable within 60 days following the date of the information in this table are deemed to be beneficially owned by, and outstanding with respect to, the holder of such option or warrant, however none of the persons listed hereinabove has the right to acquire beneficial ownership in any other shares of the Company. Subject to community property laws where applicable, to our knowledge, each person listed is believed to have sole voting and investment power with respect to all shares of Common Stock owned by such person.
-33-
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
None
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The following table shows what the auditor billed for the audit and other services for the years ended June 30, 2018 and 2017.
Year Ended June 30, 2018 | Year Ended June 30, 2017 | |||||||
| Audit Fees | $ | 22,000 | $ | 12,000 | ||||
| Audit-Related Fees | — | — | ||||||
| Tax Fees | — | — | ||||||
| All Other Fees | — | — | ||||||
| Total | $ | 22,000 | $ | 12,000 | ||||
Audit Fees—This category includes the audit of the Company’s annual financial statements, review of financial statements included in the Company’s Form 10-Q Quarterly Reports and services that are normally provided by the independent auditors in connection with engagements for those years.
Audit-Related Fees —N/A
Tax Fees—N/A
Overview —The Company’s Board reviews, and in its sole discretion pre-approves, our independent auditors’ annual engagement letter including proposed fees and all audit and non-audit services provided by the independent auditors. Accordingly, all services described under “Audit Fees,” “Audit-Related Fees,” and “Tax Fees” were pre-approved by our Company’s Board. The Board may not engage the independent auditors to perform the non-audit services proscribed by law or regulation.
-34-
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES | |
|
Regulation Number |
Exhibit | |
| 14.1 | Code of Ethics | |
| 31.1 | Rule 13a-14(a) Certification | |
| 31.2 | Rule 13a-14(a) Certification | |
| 32.1 | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 32.2 | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 101.INS | XBRL Instance Document | |
| 101.SCH | XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | XBRL Taxonomy Calculation Linkbase Document. | |
| 101.LAB | XBRL Taxonomy Label Linkbase Document | |
| 101.PRE | XBRL Taxonomy Presentation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document. |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
BIOVIE Inc.
| Signature | Titles | Date | ||
| /s/ Terren Peizer | ||||
| Terren Peizer | Chairman and Chief Executive Officer (Principal Executive Officer) | October 5, 2018 | ||
| /s/ Jonathan Adams | October 5, 2018 | |||
| Jonathan Adams | Chief Operating Officer (Principal Financial Officer) | |||
-35-