Attached files
| file | filename |
|---|---|
| EX-32 - EX-32 - CytoDyn Inc. | d840102dex32.htm |
| EX-31.2 - EX-31.2 - CytoDyn Inc. | d840102dex312.htm |
| EX-31.1 - EX-31.1 - CytoDyn Inc. | d840102dex311.htm |
| EX-24 - EX-24 - CytoDyn Inc. | d840102dex24.htm |
| EX-23 - EX-23 - CytoDyn Inc. | d840102dex23.htm |
| EX-4.12 - EX-4.12 - CytoDyn Inc. | d840102dex412.htm |
| EX-4.11 - EX-4.11 - CytoDyn Inc. | d840102dex411.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended May 31, 2018
or
| ☐ | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 000-49908

CYTODYN INC.
(Exact name of registrant as specified in its charter)
| Delaware | 75-3056237 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 1111 Main Street, Suite 660 Vancouver, Washington |
98660 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s Telephone Number, including area code: (360) 980-8524
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act:
Title of class
Common Stock, par value $0.001 per share
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by checkmark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by checkmark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☒ | |||
| Non-accelerated filer | ☐ (Do not check if a smaller reporting company) | Smaller reporting company | ☐ | |||
| Emerging growth company | ☐ | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
State the aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked price of such common equity, as of the last business day of the registrant’s most recently completed second fiscal quarter: $89,898,278 as of November 30, 2017.
Indicate the number of shares outstanding of each of the registrant’s classes of common stock, as of the latest practicable date. As of June 30, 2018, the registrant had 218,692,779 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
| Document |
Parts Into Which Incorporated | |
| Portions of the Proxy Statement for the 2018 Annual Meeting of Stockholders |
Part III |
CYTODYN INC.
FORM 10-K FOR THE YEAR ENDED MAY 31, 2018
Table of Contents
1
FORWARD-LOOKING STATEMENTS
This annual report contains certain forward-looking statements that involve risks, uncertainties and assumptions that are difficult to predict, including statements regarding the proposed transaction with ProstaGene (as defined herein), the likelihood of closing the proposed transaction with ProstaGene, our clinical focus and our current and proposed trials. Words and expressions reflecting optimism, satisfaction or disappointment with current prospects, as well as words such as “believes,” “hopes,” “intends,” “estimates,” “expects,” “projects,” “plans,” “anticipates” and variations thereof, or the use of future tense, identify forward-looking statements, but their absence does not mean that a statement is not forward-looking. Our forward-looking statements are not guarantees of performance and actual results could differ materially from those contained in or expressed by such statements. In evaluating all such statements we urge you to specifically consider various risk factors identified in this prospectus, including the matters set forth under the heading “Risk Factors,” any of which could cause actual results to differ materially from those indicated by our forward-looking statements.
Our forward-looking statements reflect our current views with respect to future events and are based on currently available financial, economic, scientific, and competitive data and information on current business plans. You should not place undue reliance on our forward-looking statements, which are subject to risks and uncertainties relating to, among other things: (i) the sufficiency of our cash position and our ongoing ability to raise additional capital to fund our operations, (ii) our ability to complete our Phase2b/3 pivotal combination therapy trial for PRO 140 (CD02) and to meet the requirements of the U.S. Food and Drug Administration (“FDA”) with respect to safety and efficacy to support the filing of a Biologics License Application, (iii) our ability to meet our debt obligations, if any, (iv) our ability to identify patients to enroll in our clinical trials in a timely fashion, (v) our ability to achieve approval of a marketable product, (vi) design, implementation and conduct of clinical trials, (vii) the results of our clinical trials, including the possibility of unfavorable clinical trial results, (viii) the market for, and marketability of, any product that is approved, (ix) the existence or development of vaccines, drugs, or other treatments for infection with the Human Immunodeficiency Virus (“HIV”) that are viewed by medical professionals or patients as superior to our products, (x) regulatory initiatives, compliance with governmental regulations and the regulatory approval process, (xi) general economic and business conditions, (xii) changes in foreign, political, and social conditions, (xiii) the specific risk factors discussed under the heading “Risk Factors” below, and (xiv) various other matters, many of which are beyond our control. Should one or more of these risks or uncertainties develop, or should underlying assumptions prove to be incorrect, actual results may vary materially and adversely from those anticipated, believed, estimated, or otherwise indicated by our forward-looking statements.
We intend that all forward-looking statements made in this annual report on Form 10-K will be subject to the safe harbor protection of the federal securities laws pursuant to Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), to the extent applicable. Except as required by law, we do not undertake any responsibility to update these forward-looking statements to take into account events or circumstances that occur after the date of this prospectus. Additionally, we do not undertake any responsibility to update you on the occurrence of any unanticipated events which may cause actual results to differ from those expressed or implied by these forward-looking statements.
| Item 1. | Business. |
Overview/Corporate History
CytoDyn Inc. was originally incorporated under the laws of Colorado on May 2, 2002 under the name RexRay Corporation (our previous name). Effective August 27, 2015, we completed a reincorporation from Colorado to Delaware. Our principal business office is 1111 Main Street, Suite 660, Vancouver, Washington 98660. Our website can be found at www.cytodyn.com. We will make available on our website, free of charge, the proxy statements and reports on Forms 8-K, 10-K, and 10-Q that we file with the United States Securities and Exchange Commission (“SEC”) as soon as reasonably practicable, after such material is electronically filed with or furnished to, the SEC. The SEC also maintains a website that contains our SEC filings. The address of the site is www.sec.gov. Further, a copy of this Annual Report on Form 10-K is located at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330.We do not intend to incorporate any contents from our website into this annual report. Unless the context otherwise requires, references in this prospectus to “CytoDyn,” the “Company,” “we,” “our,” or “us” are to CytoDyn Inc. and its subsidiaries.
We are a clinical-stage biotechnology company focused on the clinical development and potential commercialization of humanized monoclonal antibodies to treat HIV infection. Our lead product candidate, PRO 140, belongs to a class of HIV therapies known as entry inhibitors that block HIV from entering into and infecting certain cells. We believe that monoclonal antibodies are a new emerging class of therapeutics for the treatment of HIV to address unmet medical needs in the area of HIV and other immunologic indications, such as Graft versus Host Disease (“GvHD”) and certain types of cancer. As we progress in evaluating PRO 140 in different pathways of human disease and inflammation, we are encouraged by the opportunity to build a broad pipeline of indications.
2
The preclinical and clinical development of PRO 140 was led by Progenics Pharmaceuticals, Inc. (“Progenics”) through 2011. We acquired the asset from Progenics in October 2012, as described in “PRO 140 Acquisition and Licensing Arrangements” below, and have recently advanced PRO 140 through achieving primary endpoint in a Phase 2b/3 pivotal trial for HIV combination therapy and are progressing forward with the preparation and filing of a biologics license application.
PRO 140
We believe the PRO 140 antibody shows promise as a powerful anti-viral agent with the advantage of fewer side effects, lower toxicity and less frequent dosing requirements, as compared to daily drug therapies currently in use for the treatment of HIV. The PRO 140 antibody belongs to a class of HIV therapies known as entry inhibitors that block HIV from entering into and infecting certain cells. PRO 140 blocks HIV from entering a cell by binding to a molecule called the C-C chemokine receptor type 5 (“CCR5”), a normal cell surface receptor protein to which certain strains of HIV, referred to as “R5” strains, attach as part of HIV’s entry into a cell.
PRO 140 does not affect the normal function of the CCR5 co-receptor for HIV. Instead, PRO 140 binds to a precise site on CCR5 that R5 strains of HIV use to enter the cell and, in doing so, inhibits the ability of these strains of HIV to infect the cell without affecting the cell’s normal function. The R5 strains of HIV currently represent approximately 67% of all HIV infections in the U.S. As a result, we believe PRO 140 represents a distinct class of CCR5 inhibitors with advantageous virological and immunological properties and may provide a unique tool to treat HIV infected patients.
We believe PRO 140 is uniquely positioned to address a growing HIV market, as an alternative, or in addition to current therapies, which are failing primarily due to compliance, which causes drug resistance. There are several factors giving rise to compliance issues, such as, toxicity and side effects, coupled with the need for a strict regimen of daily dosing. In seven clinical trials previously conducted, PRO 140 was generally well tolerated, and no drug-related serious adverse events (“SAE’s”), or dose-proportional adverse events (“AE’s”), were reported. In addition, there were no dose-limiting toxicities or patterns of drug-related toxicities observed during these trials. The results of these studies established that PRO 140’s antiviral activity was potent, rapid, prolonged, dose-dependent, and statistically significant following a single dose. Because PRO 140’s mechanism of action (for a monoclonal antibody use in HIV) is a relatively new therapeutic approach, it provides a very useful method of suppressing the virus in treatment-experienced patients who have failed a prior HIV regimen and need new treatment options. PRO 140, as a single agent therapy, has also demonstrated that it could replace highly active antiretroviral therapy (“HAART”) altogether for a subpopulation of R5 patients who have suppressed viral load with HAART, but are seeking an alternative treatment that allows the patient an improved quality of life, with the advantages of fewer side effects, lower toxicity and less frequent dosing requirements.
To date, PRO 140 has been tested and administered to patients either intravenously or as a subcutaneous injection. We believe that, if PRO 140 is approved for use as an injectable by the FDA, it may nonetheless be an attractive and marketable therapeutic option for patients, particularly in the following scenarios:
| • | Patients desiring a break from existing treatment regimens, whether due to side-effects or for any personal reasons; |
| • | Patients with difficulty adhering to daily drug regimens; |
| • | Patients who poorly tolerate existing therapies; |
| • | Patients with compromised organ function, such as hepatitis C (“HCV”) co-infection; |
| • | Patients with complex concomitant medical requirements; and |
| • | Patients who choose not to start their HAART regimen immediately after being infected with HIV. |
We believe PRO 140 has demonstrated potent (as compared to existing treatments) antiretroviral activity and an encouraging safety profile in prior clinical testing, that PRO 140 has the potential to be the first long-acting (weekly or every other week), self-administered HIV therapy, and that PRO 140 inhibits CCR5-tropic HIV while preserving CCR5’s natural function. We believe PRO 140 represents a distinct class of CCR5 inhibitors with unique virological and immunological properties and may provide another distinct tool to treat HIV-infected patients.
Our ongoing HIV-related clinical trials, described in greater detail below, have been designed to demonstrate the proof of concept that PRO 140 monotherapy can continue to suppress the viral load in certain HIV-infected, treatment-experienced patients who had suppressed viral load on HAART, but would like an alternative treatment that provides a higher quality of life with one dose a week through a self-injection. Once the viral load is undetectable, weekly administration of PRO 140 can help maintain the suppressed viral load in a subpopulation of R5 patients over an extended period of time (currently shown to be approaching four years). Based on the preliminary results of such studies, we believe that a PRO 140 treatment option could also address the unmet medical need for therapy options for certain HIV-infected patients with uncontrolled viral load, despite conventional HAART treatments.
3
To facilitate our self-funded and sponsored clinical research plans and trials, we engaged Amarex Clinical Research, LLC (“Amarex”), as our principal contract research organization (“CRO”), to provide comprehensive clinical trial management services.
Proposed ProstaGene Transaction
On July 12, 2018, we announced a strategic expansion of our clinical focus to include the evaluation of PRO 140 in certain cancer and immunological indications where CCR5 antagonism has shown initial promise. In connection with this expansion, we signed a non-binding letter of intent regarding a proposed acquisition of intellectual property and other assets of ProstaGene LLC (“ProstaGene”), a privately held company focused on prostate cancer diagnostics and therapeutics aimed at blocking cancer metastasis by blocking CCR5. At the same time, we remain committed to advancing our clinical programs with PRO 140 in HIV and GvHD, and is continuing with our previously announced plans to submit a Biologics License Application (“BLA”) to the U.S. Food and Drug Administration (the “FDA”) for PRO 140 as a combination therapy for HIV.
As part of the proposed transaction with ProstaGene, Richard G. Pestell, M.D., Ph.D., M.B.A., F.A.C.P., F.R.A.C.P., the Chief Executive Officer of ProstaGene and President of the Pennsylvania Cancer and Regenerative Medicine Research Center, will join as our Chief Medical Officer. It is also expected that Dr. Pestell will join our Board of Directors at the closing of the transaction. The transaction is subject to completion of due diligence review, customary definitive documentation, deal structure and requisite corporate and regulatory approvals. The final terms of the transaction will be available upon the execution of definitive documentation.
Current Clinical Trials
PRO 140 is currently being studied in four clinical trials:
| • | Our first ongoing clinical trial is an extension study of our Phase 2b treatment substitution trial, which was initially completed in January 2015. There are a total of six patients in this extension study and each has surpassed three and one-half years of suppressed viral load with PRO 140 as a single agent therapy. |
| • | Our second ongoing clinical trial is a pivotal Phase 2b/3 trial for PRO 140 as a combination therapy to existing HAART drug regimens. This trial was a double blind placebo controlled trial. In late February 2018, the Company reported that it had enrolled 52 patients and the trial’s primary endpoint was achieved with a p-value of 0.0032. The primary endpoint for efficacy was defined as 0.5log of viral load drop after one week of therapy with PRO 140 in combination with the patient’s failing HAART regimen. Patients for the safety analysis in a Biologics License Application will be provided by all of our HIV trials, providing that those patients have been on a PRO 140 therapy for 24 weeks, with the same or higher dose as the combination therapy trial. |
| • | The third ongoing trial is an investigative Phase 2b/3 trial including 300 patients to assess the treatment strategy of using PRO 140 subcutaneously as a long-acting single-agent maintenance therapy. This is a 48-week trial in patients with suppressed viral load with CCR5-tropic HIV-1 infection. The primary endpoint is the number of patients who can maintain suppressed viral load under a PRO 140 monotherapy replacing their HAART regimen for 48 weeks. The secondary endpoint is the number of weeks a patient is off of their ART regimen. The Company is currently exploring a higher dose arm (525 mg), which is a 50% increase from the original dosage (350 mg), as well as a 700mg dose. The Company believes that the higher dose will result in a higher response rate, which is supported by preliminary data. |
| • | Our fourth ongoing trial of PRO 140 is a Phase 2 study for GvHD and is the first non-HIV, immunologic indication for PRO 140. This trial is designed to evaluate the feasibility of the use of PRO 140 as an add-on therapy to standard GvHD prophylaxis treatment for prevention of acute GvHD in adult patients with acute myeloid leukemia (“AML”) or myelodysplastic syndrome (“MDS”) undergoing allogeneic hematopoietic stem cell transplantation (“HST”). On October 5, 2017, we announced that the FDA had granted orphan drug designation to PRO 140 for the prevention of GvHD. In March 2018, we announced the completion of a planned interim analysis of trial data on the first 10 patients enrolled, which resulted in certain amendments to the trial protocol described in greater detail below. |
We will require a significant amount of additional capital to complete the foregoing clinical trials for HIV and to prepare and file our BLA submission. See “Liquidity and Capital Resources” under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations” below.
4
Phase 2b Extension Study for HIV, as Monotherapy
Currently, there are a total of six patients in this ongoing extension study and each has surpassed three and one-half years of suppressed viral load with PRO 140 as a single agent therapy. These patients are continuing in extension studies of this monotherapy trial by self-administering a weekly injection of PRO 140.
Phase 2b/3 Pivotal Trial for HIV, as Combination Therapy
This is a pivotal 25-week trial for PRO 140 as a combination therapy to existing HAART drug regimens. This trial was a double blind, placebo controlled trial. In late February 2018, the Company reported that it had enrolled 52 patients and the trial’s primary endpoint was achieved with a p-value of 0.0032. The primary endpoint for efficacy was defined as 0.5log reduction in viral load after one week of therapy with PRO 140 in combination with the patient’s failing HAART regimen. Following the achievement of primary endpoint, the trial is continuing to enroll under an open label for safety analysis. Nearly all patients have successfully completed this trial and most of the patients have transitioned into a FDA-cleared rollover study, in order to provide continued access to PRO 140 therapy, at the request of their treating physician.
Rollover Study for HIV, as Combination Therapy
This study is designed for patients who successfully complete the Phase 2b/3 Combination Therapy trial and for whom the treating physicians request a continuation of PRO 140 therapy in order to maintain suppressed viral load.
Phase 2b/3 Investigative Trial for HIV, as Long-term Monotherapy
This is a trial of 300 patients that assesses using PRO 140 subcutaneously as a long-acting single-agent maintenance therapy for 48 weeks in patients with suppressed viral load with CCR5-tropic HIV-1 infection. The primary endpoint is the proportion of participants with a suppressed viral load to those who experienced virologic failure. The secondary endpoint is length of time to virologic failure. We are currently exploring a high-dose arm with a 50% increase in dosage (525mg), as well as a 700mg dose, within the trial’s protocol in order to evaluate an increased response rate of all patients. Patients who are completing this trial are transitioning to a roll-over protocol, as requested by the treating physicians to enable the patients to have continued access to PRO 140 as a single-agent, self-injection maintenance therapy. The first patients were enrolled in December 2016 and enrollment is now continuing beyond 300 patients in order to evaluate the response rate of two higher dose arms.
Phase 2 Trial for Graft-versus-Host Disease
This Phase 2 multi-center 100-day study with 60 patients is designed to evaluate the feasibility of the use of PRO 140 as an add-on therapy to standard GvHD prophylaxis treatment for prevention of acute GvHD in adult patients with acute myeloid leukemia (“AML”) or myelodysplastic syndrome (“MDS”) undergoing allogeneic hematopoietic stem cell transplantation (“HST”). Enrollment of the first patient was announced in May of 2017. On October 5, 2017, the Company announced that the FDA had granted orphan drug designation to PRO 140 for the prevention of GvHD.
In March 2018, we announced that the Independent Data Monitoring Committee (“IDMC”) for PRO 140 Phase 2 trial in GvHD had completed a planned interim analysis of trial data on the first 10 patients enrolled. Following this review of data from the first 10 patients in the Phase 2 trial, we filed amendments to the protocol with the FDA. The amendments included switching the pre-treatment conditioning regimen from aggressive myeloablative (“MA”) conditioning to a reduced intensity conditioning (“RIC”), and switching from a blinded one-for-one randomized placebo-controlled design to an open-label design under which all enrollees receive PRO 140. The amendments also provide for a 50% increase in the dose of PRO 140 (to 525 mg) to more closely mimic preclinical dosing. The next review of data by the IDMC will occur following enrollment of 10 patients under the amended protocol after each patient has been dosed for 30 days.
Immunological Applications for PRO 140
We are continuing to explore opportunities for clinical applications for PRO 140 involving the CCR5 receptor, other than HIV-related treatments, such as inflammatory conditions, autoimmune diseases and cancer.
The target of PRO 140 is the important immunologic receptor CCR5. The CCR5 receptor is more than the door for HIV to enter T-cells; it is also a crucial component in inflammatory responses. This opens the potential for multiple pipeline opportunities for PRO 140.
5
The CCR5 receptor is a protein located on the surface of white blood cells that serves as a receptor for chemical attractants called chemokines. Chemokines are the key orchestrators of leukocyte trafficking by attracting immune cells to the sites of inflammation.
At the site of an inflammatory reaction, chemokines are released. These chemokines are specific for CCR5 and cause the migration of T-cells to these sites promoting further inflammation. The mechanism of action of PRO 140 has the potential to block the movement of T-cells to inflammatory sites, which could be instrumental in diminishing or eliminating inflammatory responses. Some disease processes that could benefit from CCR5 blockade include new reactions to cancer, transplantation rejection, autoimmunity and chronic inflammation such as rheumatoid arthritis and psoriasis.
Due to our mechanism of action, we believe PRO 140 has significant advantages in terms of safety and reduced side effects over other CCR5 antagonists. Prior studies have demonstrated that PRO 140 does not cause direct activation of T-cells. We have already reported encouraging human safety data for our clinical trials with PRO 140 in HIV-infected patients.
We have initiated our first clinical trial with PRO 140 in an immunological indication – a Phase 2 clinical trial with PRO 140 for GvHD in patients with AML or MDS who are undergoing bone marrow stem cell transplantation. GvHD represents an unmet medical need, with patients who contract GvHD during stem cell transplant having a significantly decreased 1-year survival rate with relapsed GvHD as the leading cause of death. Our pre-clinical study in GvHD has been published in the peer-reviewed journal Biology of Blood and Marrow Transplantation.
We are also expanding the clinical focus with PRO 140 to include the evaluation in certain cancer and immunological indications where CCR5 antagonism has shown initial promise.
Other Product Candidates
Except as otherwise disclosed above, until the clinical trials for PRO 140 have advanced further, we do not plan to devote any resources towards the development, research, testing, approval or commercialization of other product candidates.
PRO 140 Acquisition and Licensing Arrangements
We originally acquired PRO 140, as well as certain other related assets, including the existing inventory of PRO 140 bulk drug substance, intellectual property, and FDA regulatory filings, pursuant to an Asset Purchase Agreement, dated as of July 25, 2012 (the “Progenics Purchase Agreement”), between CytoDyn and Progenics Pharmaceuticals, Inc. (“Progenics”). Pursuant to the Progenics Purchase Agreement, we are required to pay Progenics a remaining milestone payment and royalties as follows: (i) $5,000,000 at the time of the first U,S. new drug application approval by the FDA or other non-U.S. approval for the sale of PRO 140; and (ii) royalty payments of up to 5% on net sales during the period beginning on the date of the first commercial sale of PRO 140 until the later of (a) the expiration of the last to expire patent included in the acquired assets, and (b) 10 years, in each case determined on a country-by country basis. To the extent that such remaining milestone payment and royalties are not timely made, under the terms of the Progenics Purchase Agreement, Progenics has certain repurchase rights relating to the assets sold to us thereunder.
Payments to Progenics are in addition to payments due under a Development and License Agreement, dated April 30, 1999 (the “PDL License”), between Protein Design Labs (now AbbVie Inc.) and Progenics, which was assigned to us in the Progenics Purchase Agreement, pursuant to which we have an exclusive worldwide license to develop, make, have made, import, use, sell, offer to sell or have sold products that incorporate the humanized form of the PRO 140 antibody developed under the agreement. Pursuant to the PDL License, we are required to pay AbbVie Inc. remaining milestone payments and royalties as follows: (i) $500,000 upon filing a Biologic License Application with the FDA or non-U.S. equivalent regulatory body; (ii) $500,000 upon FDA approval or approval by another non-U.S. equivalent regulatory body; and (iii) royalties of up to 7.5% of net sales for the longer of 10 years and the date of expiration of the last to expire licensed patent. Additionally, the PDL License provides for an annual maintenance fee of $150,000 until royalties paid exceed that amount. To the extent that such remaining milestone payments and royalties are not timely made, under the terms of the PDL License, AbbVie Inc. has certain termination rights relating to our license of PRO 140 thereunder.
Effective July 29, 2015, we entered into a License Agreement (the “Lonza Agreement”) with Lonza Sales AG (“Lonza”) covering Lonza’s “system know-how” technology with respect to our use of proprietary cell lines to manufacture new PRO 140 material. The Lonza Agreement provides for an annual license fee and future royalty payments, both of which varies based on whether Lonza, or we or our strategic partner manufactures PRO 140. We currently use an independent party as a contract manufacturer for PRO 140. Therefore, if this arrangement continues, an annual license fee of £300,000 (approximately US$400,000 given current exchange rate) would continue to apply, as well as a royalty, up to, 2% of the net selling price upon commercialization of PRO 140, excluding value added taxes and similar amounts.
6
Patents, Proprietary Technology and Data Exclusivity
Protection of our intellectual property rights is important to our business. We may file patent applications in the U.S., Canada, China, and Japan, European countries that are party to the European Patent Convention and other countries on a selective basis in order to protect inventions we consider to be important to the development of our business.
Generally, patents issued in the U.S. are effective for either (i) 20 years from the earliest asserted filing date, if the application was filed on or after June 8, 1995, or (ii) the longer of 17 years from the date of issue or 20 years from the earliest asserted filing date, if the application was filed prior to that date. A U.S. patent, to be selected by us upon receipt of FDA regulatory approval, may be subject to up to a five-year patent term extension in certain instances. While the duration of foreign patents varies in accordance with the provisions of applicable local law, most countries provide for a patent term of 20 years measured from the application filing date and some may also allow for patent term extension to compensate for regulatory approval delay. We pursue opportunities for seeking new meaningful patent protection on an ongoing basis. We currently anticipate that, absent patent term extension, patent protection relating to the PRO 140 antibody itself will start to expire in 2023, certain methods of using PRO 140 will start to expire in 2026, and certain formulations comprising PRO 140 will start to expire in 2031.
Patents do not enable us to preclude competitors from commercializing drugs in direct competition with our products that are not covered by granted and enforceable patent claims. Consequently, patents may not provide us with any meaningful competitive advantage. See related risk factors under the heading “Risk Factors” below. We may also rely on data exclusivity, trade secrets and proprietary know-how to develop and attempt to achieve a competitive position with our product candidates. We generally require our employees, consultants and partners who have access to our proprietary information to sign confidentiality agreements in an effort to protect our intellectual property.
Separate from and in addition to the patent rights noted above, we expect that PRO 140 will be subject to at least a 12-year data exclusivity period measured from the first date of FDA licensure, during which period no other applications referencing PRO 140 will be approved by FDA. Further, no other applications referencing PRO 140 will be accepted by FDA for a 4-year period measured from the first date of FDA licensure. Accordingly, this period of data exclusivity is expected to provide at least a 12-year term of protection against competing products shown to be biosimilar or interchangeable with PRO 140. Similar data exclusivity or data protection periods of up to about five years or more are provided in at least Australia, Canada, Europe, Japan, and New Zealand.
We note that data exclusivity is not an extension of patent rights, and it does not prevent the introduction of generic versions of the innovative drug during the data exclusivity period, as long as the marketing approval of the generic version does not use or rely upon the innovator’s test data. Patents and data exclusivity are different concepts, protect different subject matter, arise from different efforts, and have different legal effects over different time periods.
Information with respect to our current patent portfolio as of May 31, 2018, is set forth below.
| Number of Patents | Expiration Dates(1) |
Number of Patent Applications |
||||||||||||||||||
| Product Candidates |
U.S. | International | U.S. | International | ||||||||||||||||
| PRO 140(2) |
9 | 33 | 2018-2032 | 11 | 27 | |||||||||||||||
| (1) | Patent term extensions and pending patent applications may extend periods of patent protection. |
| (2) | PRO 140 patents and applications relate to HIV-1, GvHD, and cancer treatments. |
Research, development and commercialization of a biopharmaceutical product often require choosing between alternative development and optimization routes at various stages in the development process. Preferred routes depend upon current—and may be affected by subsequent—discoveries and test results, availability of financial resources, and other factors, and cannot be identified with certainty. There are numerous third-party patents in fields in which we work, and we may need to obtain licenses under patents of others in order to pursue a preferred development route of one or more of our product candidates. The need to obtain a license would decrease the ultimate value and profitability of an affected product. If we cannot negotiate such a license, we might have to pursue a less desirable development route or terminate the program altogether. See “Risk Factors” below.
7
Government Regulation
Regulation of Health Care Industry
The health care industry is highly regulated, and state and federal health care laws and regulations are applicable to certain aspects of our business. For example, there are federal and state health care laws and regulations that apply to the operation of clinical laboratories, the business relationships between health care providers and suppliers, the privacy and security of health information and the conduct of clinical research.
Regulation of Products
The design, testing, manufacture, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion of our products is regulated by numerous third parties, including the FDA, foreign governments, independent standards auditors and our customers.
In the United States, biological products have long been subject to regulation by various federal and state agencies, primarily as to product safety, efficacy, manufacturing, advertising, labeling, import, export and safety reporting. The exercise of broad regulatory powers by the FDA through its Center for Devices and Radiological Health and its Center for Biological Evaluation and Research continues to result in increases in the amounts of testing and documentation for FDA clearance of current and new biologic products. The FDA can ban certain biological products; detain or seize adulterated or misbranded biological products; order repair, replacement or refund of these products; and require notification of health professionals and others with regard to biological products that present unreasonable risks of substantial harm to the public health. The FDA may also enjoin and restrain certain violations of the Federal Food, Drug and Cosmetic Act, as amended, or the Public Health Service Act pertaining to certain biological products or initiate action for criminal prosecution of such violations.
The lengthy process of seeking drug approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Failure to comply with applicable regulations can result in refusal by the FDA to approve product license applications. The FDA also has the authority to revoke previously granted product approvals.
Pharmaceutical products such as PRO 140 may not be commercially marketed without prior approval from the FDA and comparable agencies in foreign countries. In the United States, the process for obtaining FDA approval for products like PRO 140 typically includes pre-clinical studies, the filing of an Investigational New Drug application (“IND”), human clinical trials and filing and approval of either a New Drug Application (“NDA”), for chemical pharmaceutical products, or a Biologics License Application (“BLA”) for biological pharmaceutical products, such as PRO 140. The results of pre-clinical testing, which include laboratory evaluation of product chemistry and formulation, animal studies to assess the potential safety and efficacy of the product and its formulations, details concerning the drug manufacturing process and its controls, and a proposed clinical trial protocol and other information must be submitted to the FDA as part of an IND that must be reviewed and become effective before clinical testing can begin. The study protocol and informed consent information for patients in clinical trials must also be submitted to an independent institutional review board (“IRB”), for approval. Once a sponsor submits an IND, the sponsor must wait 30 calendar days before initiating any clinical trials, during which time the FDA has an opportunity to review the IND and raise concerns or questions relating to the proposed clinical trials outlined in the IND. If the FDA has comments or questions, they must be resolved to the satisfaction of the FDA before clinical trials can begin. In addition, the FDA, an IRB or we may, at any time, impose a clinical hold on ongoing clinical trials. If the FDA imposes a clinical hold, clinical trials cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA. Our non-clinical and clinical studies must conform to the FDA’s Good Laboratory Practice (“GLP”), and Good Clinical Practice (“GCP”), requirements, which are designed to ensure the quality and integrity of submitted data and protect the rights and well-being of study patients. Information for certain clinical trials also must be publicly disclosed within certain time limits on the clinical trial registry and results databank maintained by the National Institutes of Health (“NIH”).
The results of the pre-clinical and clinical testing, chemistry, manufacturing and control information and proposed labeling are then submitted to the FDA in the form of either an NDA or BLA for review and potential approval to commence commercial sales. In responding to an NDA or BLA, the FDA may grant marketing approval, request additional information in a complete response letter, or deny the approval if it determines that the NDA or BLA does not provide an adequate basis for approval. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of an NDA or BLA and may require additional testing. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter, which authorizes commercial marketing of the product with specific prescribing information for specific indications, and sometimes with specified post-marketing commitments. Any approval required from the FDA might not be obtained on a timely basis, if at all.
8
Among the conditions for an NDA or BLA approval is the requirement that the manufacturing operations conform on an ongoing basis with current Good Manufacturing Practices (“cGMPs”). In complying with cGMPs, we must expend time, money and effort in the areas of training, production and quality control within our own organization and at our contract manufacturing laboratories. A successful inspection of the manufacturing facility by the FDA is a prerequisite for final approval of a biological product like PRO140. Following approval of the NDA or BLA, we and our third-party manufacturers remain subject to periodic inspections by the FDA. We also face similar inspections coordinated by the European Medicines Agency by inspectors from particular European Union (“EU”) member countries that conduct inspections on behalf of the EU and from other foreign regulatory authorities. Any determination by the FDA or other regulatory authorities of manufacturing or other deficiencies could materially adversely affect our business.
Regulatory requirements and approval processes in EU countries are similar in principle to those in the United States and can be at least as costly and uncertain. The EU has established a unified centralized filing and approval system administered by the Committee for Medicinal Products for Human Use designed to reduce the administrative burden of processing applications for pharmaceutical products derived from new technologies. In addition to obtaining regulatory approval of products, it is generally necessary to obtain regulatory approval of the facility in which the product will be manufactured.
We use and plan to continue to use third-party manufacturers to produce our products in clinical and commercial quantities. Future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product, including new safety risks, or the failure to comply with requirements may result in restrictions on a product, manufacturer or holder of an approved NDA or BLA, including withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay further marketing. Newly discovered or developed safety or efficacy data may require changes to an approved product’s approved labeling, including the addition of new warnings and contraindications, the imposition of additional mandatory post-market studies or clinical trials, or the imposition of or revisions to a risk evaluation mitigation strategies (“REMS “) program, including distribution and/or use restrictions.
Once a BLA is approved, a product will be subject to certain post-approval requirements, including requirements for adverse event reporting and submission of periodic reports to the FDA, recordkeeping, product sampling and distribution, and, as discussed above, may be subject to mandatory post-market study and REMS requirements. In addition, the FDA strictly regulates the promotional claims that may be made about prescription drug products and biologics. In particular, a drug or biologic may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. The FDA also requires substantiation of any claims of superiority of one product over another, including the requirement that such claims be proven by adequate and well-controlled head-to-head clinical trials. The FDA also requires all promotional materials that discuss the use or effectiveness of a prescription drug or biologic to disclose in a balanced manner the risks and safety profile of the product.
Regulation of Laboratory Operations
Clinical laboratories that perform laboratory testing (except for research purposes only) on human patients are subject to regulation under Clinical Laboratory Improvement Amendments (“CLIA”). CLIA regulates clinical laboratories by requiring that the laboratory be certified by the federal government, licensed by the state and comply with various operational, personnel and quality requirements intended to ensure that clinical laboratory test results are accurate, reliable and timely. State law and regulations also apply to the operation of clinical laboratories.
State Governments
Most states in which we operate have regulations that parallel federal regulations. Most states conduct periodic unannounced inspections and require licensing under such state’s procedures. Our research and development activities and the manufacture and marketing of our products are and will be subject to rigorous regulations relating to product safety and efficacy by numerous governmental authorities in the United States and other countries.
Other Laws and Regulations
We are subject to various laws and regulations relating to safe working conditions, clinical, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research. The extent of government regulation applying to our business that might result from any legislative or administrative action cannot be accurately predicted.
9
Environmental
We are subject to a variety of federal, state and local environmental protection measures. We believe that our operations comply in all material respects with applicable environmental laws and regulations. Our compliance with these regulations did not have during the past year and is not expected to have a material effect upon our capital expenditures, cash flows, earnings or competitive position.
Registrational Clinical Trials Process
Described below is the traditional registrational drug development track. Our current business strategy is to focus primarily on our PRO 140 Phase 2b/3 pivotal and investigative trials, to manage our Phase 2 trial for GvHD and to continue to explore other immunologic indications for PRO 140, as described above.
Phase 1
Phase 1 includes the initial introduction of an investigational new drug or biologic into humans. These studies are closely monitored and may be conducted in patients, but are usually conducted in a small number of healthy volunteer patients. These studies are designed to determine the metabolic and pharmacologic actions of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. During Phase 1, sufficient information about the investigational product’s pharmacokinetics and pharmacological effects are obtained to permit the design of well-controlled, scientifically valid, Phase 2 studies. Phase 1 studies of PRO 140 have been conducted and completed by or on behalf of Progenics by certain principal investigators prior to our acquisition of PRO 140.
Phase 2
Phase 2 includes the early controlled clinical studies conducted to obtain some preliminary data on the effectiveness of the drug for a particular indication or indications in patients with the disease or condition. This phase of testing also helps determine the common short-term side effects and risks associated with the drug. Phase 2 studies are typically well-controlled, closely monitored, and conducted in a relatively small number of patients, often involving several hundred people. In some cases, depending upon the need for a new drug, a particular drug candidate may be licensed for sale in interstate commerce after a “pivotal” Phase 2 trial.
Phase 2 is often broken into Phase 2a, which can be used to refer to “pilot trials,” or more limited trials evaluating exposure response in patients, and Phase 2b trials that are designed to evaluate dosing efficacy and ranges.
Phase 3
Phase 3 studies are expanded controlled clinical studies. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained in Phase 2, and are intended to gather the additional information about effectiveness and safety that is needed to evaluate the overall benefit/risk relationship of the drug. Phase 3 studies also provide an adequate basis for extrapolating the results to the general population and transmitting that information in the physician labeling. Phase 3 studies usually involve significantly larger groups of patients, and considerable additional expense. We were required to pay significant fees to third parties upon the first patient dosing in a Phase 3 trial of PRO 140, and we may be required to make additional fee payments to third parties upon the completion of additional milestones. See the discussion under the subheading “PRO 140 Acquisition and Licensing Arrangements” above.
Competition
The pharmaceutical and biotechnology industries are characterized by rapidly evolving technology and intense competition. Our development efforts may compete with more established biotechnology companies that have significantly greater financial and managerial resources than we do.
Advancing PRO 140 to commercialization is our highest priority. PRO 140 blocks a cell receptor called CCR5, which is the entry point for most strains of HIV virus. Pfizer’s Maraviroc (Selzentry®) is the only currently approved CCR5 blocking agent. Maraviroc like all other HIV approved drugs must be taken daily and are believed to have side effects and toxicity. For these reasons, we believe that our lead product, PRO 140, a monoclonal antibody, may prove to be useful in patients that cannot tolerate existing HIV therapies or desire a respite from those therapies. Nonetheless, manufacturers of current therapies, such as Pfizer, Gilead Sciences, Merck, Bristol-Myers Squibb and ViiV Healthcare, are very large, multi-national corporations with significant resources. We expect that these companies will compete fiercely to defend and expand their market share.
10
To construct a HAART regimen, three drugs from two classes of drugs are typically needed. Currently there are only five different classes of drugs from which four are primarily used to construct a HAART regimen. Each of these four classes of drugs has many drugs available in its respective class, except the entry inhibitor (“EI”) class, which has only two drugs available. The only two drugs in the EI class approved by the FDA are Maraviroc, a small molecule drug (which is taken orally once or twice a day) and Ibalizumab (which is an IV infusion administered once every two weeks). If approved, we believe that PRO 140 will be only the second approved drug outside of the main four classes of drugs approved for HIV since 2007.
The only other monoclonal antibody that recently received FDA approval is TMB-335, also referred to as Ibalizumab, which was developed by TaiMed Biologics. Ibalizumab targets the CD4 receptor on T-cells which is one of the two co-receptors required for HIV entry into T-cells.
Our potential competitors include entities that develop and produce therapeutic agents. These include numerous public and private academic and research organizations and pharmaceutical and biotechnology companies pursuing production of, among other things, biologics from cell cultures, genetically engineered drugs and natural and chemically synthesized drugs. Our competitors may succeed in developing potential drugs or processes that are more effective or less costly than any that may be developed by us or that gain regulatory approval prior to our potential drug candidates. Worldwide, there are many antiviral drugs for treating HIV. In seeking to manufacture, distribute and market the potential drugs we hope to have approved, we face competition from established pharmaceutical companies. Many of these potential competitors have substantially greater capital resources, management expertise, research and development capabilities, manufacturing and marketing resources and experience than we do.
We also expect that the number of our competitors and potential competitors will increase as more potential drugs receive commercial marketing approvals from the FDA or analogous foreign regulatory agencies. Any of these competitors may be more successful than us in manufacturing, marketing and distributing HIV treatments.
Manufacturing
We do not own or operate manufacturing facilities for the production of PRO 140. We expect to depend on third-party manufacturing organizations and suppliers for all of our clinical trial quantities of PRO 140, in addition to previously manufactured supplies of PRO 140. We continue to explore alternative manufacturing sources, in order to ensure that we have access to sufficient manufacturing capacity in order to meet potential demand for PRO 140 in a cost-efficient manner.
We have engaged a contract manufacturing organization (“CMO”) to initiate the scale-up to commercial batch quantities of product, and develop the necessary controls and specifications to manufacture product on a consistent and reproducible manner. We have also contracted with a suitable CMO to fill, label, and package product into the final commercial package for commercial use. In order to commercialize product, this scaled-up material will need to be validated under best practices, and demonstrated to meet approved specifications on an ongoing basis. GMP material will be produced as needed to support clinical trials for all therapeutic indications and until commercial product is approved by the FDA. We will rely on CMO’s for all of our developmental and commercial needs.
Research and Development Costs
Our research and development expenses totaled approximately $38.2 million, $20.2 million and $13.7 million for the fiscal years ended May 31, 2018, May 31, 2017 and May 31, 2016, respectively. We expect our research and development expenses to continue to increase in future periods as the activity within our clinical trials expands and our biologics manufacturing processes and related regulatory compliance activities increase.
Employees and Consultants
We currently have six full-time employees, as well as several independent consultants assisting us with our clinical trials of PRO 140 and manufacturing activities. There can be no assurances, however, that we will be able to identify or hire and retain additional employees or consultants on acceptable terms in the future.
| Item 1A. | Risk Factors. |
The risks enumerated below are not the only risks we face, and the listed risk factors are not intended to be an all-inclusive discussion of all of the potential risks relating to our business. Any of the risk factors described below could significantly and adversely affect our business, prospects, financial condition and results of operations. Additional risks and uncertainties not currently known or that are currently considered to be immaterial may also materially and adversely affect our business.
11
Risks Related to Our Business
We are a biotechnology company and have a history of significant operating losses; we expect to continue to incur operating losses, and we may never achieve or maintain profitability.
We have not generated any revenue from product sales, licensing, or other potential sales to date. Since our inception, we have incurred operating losses in each year due to costs incurred in connection with research and development activities and general and administrative expenses associated with our operations. Our current drug candidate is in the later stages of clinical trials, and we expect to continue with significant additional clinical trial activities and the ongoing preparation of a BLA for an anticipated filing in 2019, before we can seek the regulatory approvals necessary to begin commercial sales. During the fiscal years ended May 31, 2018 and 2017, we incurred net losses of approximately $50.1 million and $25.8 million, respectively, and at May 31, 2018, we had an accumulated deficit of approximately $173.1 million and a stockholders’ deficit of $13.2 million. We expect to incur losses for the foreseeable future as we continue development of, and seek regulatory approvals for, our drug candidate and commercialize any approved product usages. If our current drug candidate fails to gain regulatory approval, or if it or other candidates we own do not achieve approval and market acceptance, we will not be able to generate any revenue, or explore other opportunities to enhance stockholder value, such as through a sale. If we fail to generate revenue and eventually become and remain profitable, or if we are unable to fund our continuing losses, our shareholders could lose all or part of their investments.
Any failure to attract and retain skilled directors, executives, employees and consultants could impair our drug development and commercialization activities.
Our business depends on the skills, performance, and dedication of our directors, executive officers and key scientific and technical advisors. All of our current scientific advisors are independent contractors and are either self-employed or employed by other organizations. As a result, they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, which may affect their ability to provide services to us in a timely manner. We may need to recruit additional directors, executive management employees, and advisers, particularly scientific and technical personnel, which will require additional financial resources. In addition, there is currently intense competition for skilled directors, executives and employees with relevant scientific and technical expertise, and this competition is likely to continue. If we are unable to attract and retain persons with sufficient scientific, technical and managerial experience, we may be forced to limit or delay our product development activities or may experience difficulties in successfully conducting our business, which would adversely affect our operations and financial condition.
We expect to rely on third party manufacturers and will be dependent on their quality and effectiveness.
Our primary product candidate and potential drug candidates require precise, high-quality manufacturing. The failure to achieve and maintain high manufacturing standards, including failure to detect or control unexpected events or unanticipated manufacturing errors or the frequent occurrence of such errors, could result in patient injury or death, discontinuance or delay of ongoing or planned clinical trials, delays or failures in product testing or delivery, cost overruns, product recalls or withdrawals and other problems that could seriously hurt our business. Contract manufacturers of biopharmaceutical drugs can encounter difficulties involving manufacturing processes, facilities, operations, production yields, quality control, compliance and shortages of qualified personnel. These manufacturers are subject to stringent regulatory requirements, including the FDA’s current good-manufacturing-practices (cGMP) regulations and similar foreign laws and standards. If our contract manufacturers fail to maintain ongoing compliance at any time, we may be unable to obtain regulatory approval for our products. In addition, the production of our product candidates could be interrupted, resulting in delays or discontinuance of our clinical trials, disruption in our release of commercial supplies, or other factors that could cause increases in costs and loss of potential revenues.
We do not have internal research and development personnel, making us dependent on consulting relationships and strategic alliances with industry partners.
We currently have no research and development staff or coordinators. We rely and intend to continue to rely on third parties for many of these functions. We engaged Amarex, a full service clinical research organization, to manage our clinical trials. As a result, we will be dependent on consultants and strategic partners in our development and commercialization activities, and it may be administratively challenging to monitor and coordinate these relationships. If we do not appropriately manage our relationships with third parties, we may not be able to successfully manage development, testing, and preparation of our BLA filing for our PRO 140 drug candidate or other products or commercialize any products that are approved, which would have a material and adverse effect on our business, financial condition and stock price.
12
We will need to outsource and rely on third parties for the clinical development and manufacture, sales and marketing of product candidates, and our future success will be dependent on the timeliness and effectiveness of the efforts of these third parties.
We are dependent on third parties for important aspects of our product development strategy. We do not have the required financial and human resources to carry out independently the pre-clinical and clinical development for our product candidate, and do not have the capability or resources to manufacture, market or sell our current product candidate. As a result, we contract with and rely on third parties for important functions, including testing, storing, and manufacturing our products and managing and conducting clinical trials from which we may obtain a benefit. We have recently entered into several agreements with third parties for such services. If problems develop in our relationships with third parties, or if such parties fail to perform as expected, it could lead to delays or lack of progress, significant cost increases, changes in our strategies, and even failure of our product initiatives.
We will need substantial additional funding to complete our Phase 2b and Phase 2b/3 clinical trials for PRO 140 for HIV-related treatments, to continue our Phase 2 clinical trial for GvHD, to fund development of PRO 140 for other indications, such as cancer and immunologic indications, and to operate our business, and such funding may not be available or, if it is available, such financing is likely to substantially dilute our existing stockholders.
The discovery, development, and commercialization of new treatments, such as our PRO 140 product candidate, entail significant costs. We expect the total estimated expenses for our pivotal Phase 2b/3 combination therapy trial may range from approximately $12 million to $13 million, including the open label portion, and the total estimated expenses for our Phase 2b/3 monotherapy trial may range from $22 million to $25 million. Our total estimated expenses for the Phase 2 GvHD trial are approximately $3.5 million to $4 million. In addition, to the extent further development and clinical trials of PRO 140 for other indications, such as cancer, and of other products continue to appear promising and we elect to fund its development and commercialization, we will need to raise substantial additional capital, or enter into strategic partnerships, to enable us to:
| • | fund clinical trials and seek regulatory approvals; |
| • | access manufacturing and commercialization capabilities; |
| • | pay required license fees, milestone payments, and maintenance fees to Progenics, Lonza and AbbVie Inc.; |
| • | develop, test, and, if approved, market our product candidate; |
| • | acquire or license additional internal systems and other infrastructure; |
| • | hire and support additional management and scientific personnel; and |
| • | explore additional indications for PRO 140, such as in the area of immunology. |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never achieve, we expect to finance our cash needs primarily through public or private equity offerings, debt financings or through strategic alliances. We cannot be certain that additional funding will be available on acceptable terms or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of, or eliminate one or more of our clinical trials, collaborative development programs or future commercialization initiatives. In addition, any additional funding that we do obtain will dilute the ownership held by our existing security holders. The amount of this dilution may be substantially increased if the trading price of our common stock is lower at the time of any financing. Regardless, the economic dilution to stockholders will be significant if our stock price does not increase significantly, or if the effective price of any sale is below the price paid by a particular shareholder. Any debt financing could involve substantial restrictions on activities and creditors could seek a pledge of some or all of our assets. We have not identified potential sources for the additional financing that we will require, and we do not have commitments from any third parties to provide any future financing. If we fail to obtain additional funding as needed, we may be forced to cease or scale back operations, and our results, financial condition and stock price would be adversely affected.
The amount of financing we require will depend on a number of factors, many of which are beyond our control. Our results of operations, financial condition and stock price are likely to be adversely affected if our funding requirements increase or are otherwise greater than we expect.
Our future funding requirements will depend on many factors, including, but not limited to:
| • | the costs of our two Phase 2b/3 clinical trials for PRO 140 for HIV-related treatments, our Phase 2 clinical trial for GvHD and other clinical trials and development activities conducted by us directly, and our ability to successfully conclude the studies and achieve favorable results; |
| • | our ability to attract strategic partners to pay for or share costs related to our product development efforts; |
13
| • | the costs and timing of seeking and obtaining regulatory approvals and making related milestone payments due to Progenics, Lonza and AbbVie Inc.; |
| • | the costs of filing, prosecuting, maintaining and enforcing patents and other intellectual property rights and defending against potential claims of infringement; |
| • | decisions to hire additional scientific or administrative personnel or consultants; |
| • | our ability to manage administrative and other costs of our operations; and |
| • | the presence or absence of adverse developments in our research program. |
If any of these factors cause our funding needs to be greater than expected, our operations, financial condition, ability to continue operations and stock price may be adversely affected.
Our future cash requirements may differ significantly from our current estimates.
Our cash requirements may differ significantly from our estimates from time to time, depending on a number of factors, including:
| • | the costs and results of our two Phase 2b/3 clinical trials for HIV-related treatments, our Phase 2 clinical trial for GvHD, and other clinical trials we are undertaking or may in the future pursue with PRO 140; |
| • | the time and costs involved in our Chemistry, Manufacturing, and Controls (“CMC”) activities; |
| • | the time and costs involved in our BLA preparation activities; |
| • | the time and costs involved in obtaining regulatory approvals; |
| • | whether our outstanding convertible notes are converted into equity or we receive additional cash upon the exercise of our outstanding common stock warrants; |
| • | whether we receive additional cash upon the exercise of our outstanding options and warrants for common stock; |
| • | whether we are able to obtain funding under future licensing agreements, strategic partnerships, or other collaborative relationships, if any; |
| • | the costs of compliance with laws, regulations, or judicial decisions applicable to us; and |
| • | the costs of general and administrative infrastructure required to manage our business and protect corporate assets and stockholder interests. |
If we fail to raise additional funds on a timely basis we will need to scale back our business plans, which would adversely affect our business, financial condition, and stock price, and we may even be forced to discontinue our operations and liquidate our assets.
Clinical trials are expensive, time-consuming and subject to delay.
Clinical trials are subject to rigorous regulatory requirements and are expensive and time-consuming to design, implement and manage. The length of time and number of trial sites and patients required for clinical trials vary substantially based on the type, complexity, novelty, intended use and any safety concerns relating to a drug candidate. We estimate that it may take as much as up to two years to complete the necessary clinical trials, obtain regulatory approval from the FDA or other non-U.S. regulatory agency, and begin to commercialize PRO 140, even if trials are successful, of which there can be no assurance. Clinical trials for our other drug candidates may take significantly longer to complete, if they are pursued at all.
The commencement and completion of clinical trials could be delayed or prevented by many factors, including, but not limited to:
| • | periodic amendments to clinical trial protocols to address certain variables which arise during the course of a trial must be negotiated with and approved by the FDA; |
| • | slower than expected rates of patient recruitment and enrollment which has occurred in connection with certain of our trials, including as a result of competition with other clinical trials for patients, limited numbers of patients that meet the enrollment criteria, or the introduction of alternative therapies or drugs by others; |
| • | our ability to obtain regulatory or other approvals to commence and conduct clinical trials in the manner we or our partners consider appropriate for timely development; |
14
| • | our ability to identify and reach agreement on acceptable terms with prospective clinical trial sites and entities involved in the conduct of our clinical trials; |
| • | unforeseen issues with our relationship with our contract clinical management services provider; |
| • | delays in paying third-party vendors of biopharmaceutical services; |
| • | lack of effectiveness of our drug candidates during clinical trials; or |
| • | unforeseen safety issues. |
The results of previous clinical trials may not be predictive of future results, and the results of our current and planned clinical trials may not satisfy the requirements of the FDA or non-U.S. regulatory authorities
We must successfully complete clinical trials for PRO 140 before we can apply for marketing approval. Although test results have been positive thus far, the process of obtaining approval of a drug product for use in humans is extremely lengthy and time-consuming, and numerous factors may prevent our successful development of PRO 140, including negative results in ongoing and future clinical trials, and inability to obtain sufficient additional funding to continue to pursue development. Our clinical trials may be unsuccessful, which would materially harm our business. Even if our ongoing clinical trials are successful, we may be required to conduct additional clinical trials and other testing to establish PRO 140’s safety and efficacy before a BLA can be filed with the FDA for marketing approval of PRO 140.
The results from the prior clinical trials of PRO 140 may not necessarily be predictive of the results of future clinical trials or preclinical studies. Even if we are able to complete our planned clinical trials of PRO 140 according to our current development timeline, the results from our prior clinical trials of PRO 140 may not be replicated in these future trials. Clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in prior clinical trials nonetheless have failed to obtain FDA approval. If we fail to produce positive results in our clinical trials of PRO 140, the development timeline and regulatory approval and commercialization prospects for PRO 140 and our business and financial prospects, would be adversely affected.
Further, PRO 140 may not be approved even if it achieves its primary endpoint in our ongoing Phase 3 clinical trials. The FDA or non-U.S. regulatory authorities may disagree with our trial design and our interpretation of data from preclinical studies and clinical trials. In addition, any of these regulatory authorities may change its requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a pivotal clinical trial that, if successful, would potentially form the basis for an application for approval by the FDA or another regulatory authority. The FDA may require us to procure the development of a companion diagnostic test to help identify patients who may be more likely to respond to PRO 140 for certain uses. Furthermore, any of these regulatory authorities may also approve PRO 140 for fewer or more limited indications than we request or may grant approval contingent on the performance of costly post-marketing clinical trials.
The FDA and non-U.S. regulatory authorities retain broad discretion in evaluating the results of our clinical trials and in determining whether the results demonstrate that PRO 140 is safe and effective. If prior to approval, we are required to conduct additional preclinical trials, clinical studies or other types of testing of PRO 140, including after the completion of our current and planned later phase clinical trials, we will need substantial additional funds, and there is no assurance that the results of any such additional clinical trials will be sufficient for approval.
Clinical trials may fail to demonstrate the desired safety and efficacy of our product candidates, which could prevent or significantly delay completion of clinical development and regulatory approval.
Prior to receiving approval to commercialize PRO 140 or any other product candidates, we must adequately demonstrate to the FDA and any foreign regulatory authorities in jurisdictions in which we seek approval that it or any other product candidate is sufficiently safe and effective with substantial evidence from well-controlled clinical trials. In clinical trials, we will need to demonstrate efficacy for the treatment of specific indications and monitor safety throughout the clinical development process and following approval. If clinical work by us or others leads to undesirable adverse effects in patients, it could delay or prevent us from furthering the regulatory approval process or cause us to cease clinical trials with respect to any drug candidate. If our current or future preclinical studies or clinical trials are unsuccessful, our business will be significantly harmed and our stock price would be negatively affected.
Our product candidates are subject to the risks of failure inherent in drug-related product development. Preclinical studies may not yield results that adequately support our regulatory applications. Even if these applications are filed with respect to our product candidates, the results of preclinical studies do not necessarily predict the results of clinical trials. In addition, even if we believe the data collected from clinical trials of our product candidates are promising, these data may not be sufficient to support approval by the FDA or foreign regulatory authorities. If regulatory authorities do not approve our products, or if we fail to maintain regulatory compliance, we would be unable to commercialize our products, and our business, results of operations and financial condition would be harmed.
15
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on the rate at which we can recruit patients to participate in testing our product candidates. If patients are unwilling to participate in our trials because of negative publicity from adverse events in the biotechnology industries, public perception of vaccine safety issues or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting studies and obtaining regulatory approval of potential products may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of the clinical trials altogether.
We may not be able to identify, recruit and enroll a sufficient number of patients, or those with the required enrollment criteria, to complete our clinical trials in a timely manner. Patient enrollment is affected by several factors, including:
| • | severity of the disease under investigation; |
| • | design of the trial protocol; |
| • | size of the patient population; |
| • | eligibility criteria for the trial in question; |
| • | perceived risks and benefits of the product candidate being tested; |
| • | proximity and availability of clinical trial sites for prospective patients; |
| • | availability of competing vaccines and/or therapies and related clinical trials; |
| • | efforts to facilitate timely enrollment in clinical trials; |
| • | patient referral practices of physicians; and |
| • | ability to monitor patients adequately during and after treatment. |
We may not be able to initiate or continue clinical trials if we cannot enroll a sufficient number of eligible patients to participate in the clinical trials required by regulatory agencies.
Even if we enroll a sufficient number of eligible patients to initiate our clinical trials, we may be unable to maintain participation of these patients throughout the course of the clinical trial as required by the clinical trial protocol, in which event we may be unable to use the research results from those patients. If we have difficulty enrolling, and maintaining the enrollment of a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an adverse effect on our business.
PRO 140 may cause undesirable side effects or have other properties that delay or prevent its regulatory approval or limit their commercial potential.
Undesirable side effects caused by our product candidates or even competing products in development that utilize a common mechanism of action could cause regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities and potential product liability claims. While PRO 140 was generally well tolerated and no drug-related serious adverse events or dose-proportional adverse events were reported, our understanding of the relationship between adverse events reported in future clinical trials of other product candidates may change as we gather more information, and unexpected adverse events may be observed. Routine review and analysis of post-marketing safety surveillance and clinical trials will provide additional information, for example, potential evidence of rare, population-specific or long-term adverse reactions, and may adversely affect the commercialization of the product, and even lead to the suspension or withdrawal of product marketing authorization.
If we or others identify undesirable side effects caused by PRO 140 either before or after receipt of marketing approval, a number of potentially significant negative consequences could result, including:
| • | our clinical trials may be put on hold; |
| • | we may be unable to obtain regulatory approval for our product candidates; |
| • | regulatory authorities may withdraw approvals of our products; |
| • | regulatory authorities may require additional warnings on the label; |
| • | a medication guide outlining the risks of such side effects for distribution to patients may be required; |
| • | we could be sued and held liable for harm caused to patients; and |
16
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining marketing approvals for and market acceptance of our product candidate and could have a material adverse effect on our business and financial results.
We may not be able to identify, negotiate and maintain the strategic alliances necessary to develop and commercialize our products and technologies, and we will be dependent on our corporate partners if we do.
We may seek to enter into a strategic alliance with a pharmaceutical company for the further development and approval of one or more of our product candidates. Strategic alliances potentially provide us with additional funds, expertise, access, and other resources in exchange for exclusive or non-exclusive licenses or other rights to the technologies and products that we are currently developing or may explore in the future. We cannot give any assurance that we will be able to enter into strategic relationships with a pharmaceutical company or others in the near future or at all, or maintain our current relationships. In addition, we cannot assure you that any agreements we do reach will achieve our goals or be on terms that prove to be economically beneficial to us. When we do enter into strategic or contractual relationships, we become dependent on the successful performance of our partners or counterparties. If they fail to perform as expected, such failure could adversely affect our financial condition, lead to increases in our capital needs, or hinder or delay our development efforts.
Although PRO 140 has been designated for fast track approval by the FDA, our ability to obtain accelerated approval may be lost.
The FDA designated PRO 140 for fast track consideration in 2006. The letter ascribing this designation stated that, if the clinical development program pursued for PRO 140 did not continue to meet the criteria for fast track designation, the IND application would not be reviewed under the fast track program. There is no assurance that the FDA will ultimately consider PRO 140 for approval on an accelerated basis. Failure to maintain eligibility for fast track review will likely result in requirements for longer or additional clinical trials and a slower approval process, resulting in additional costs and, therefore, additional capital, which will likely result in further delay in the potential realization of revenues from commercialization of PRO 140.
Although we have applied with the FDA for breakthrough therapy designation for PRO 140, for certain HIV-related treatments, such a designation may not lead to a faster development or regulatory review or approval process, and it may not increase the likelihood that PRO 140 will receive marketing approval in the United States.
We applied with the FDA for breakthrough therapy designation for PRO 140, for certain HIV-related treatments. The FDA, in its comments to us, requested additional trial data to support our request for such designation. We currently plan to submit additional data to the FDA as it becomes available to us from our pivotal Phase 2b/3 combination trial. A breakthrough therapy is defined as a product that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and for which preliminary clinical evidence indicates substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For drugs and biologics that have been designated as breakthrough therapies, interaction and communication between the FDA and the applicant can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Products designated as breakthrough therapies by the FDA may, in some cases, also be eligible for accelerated approval.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe PRO 140 meets the criteria for designation as a breakthrough therapy, the FDA may disagree. In any event, the receipt of a breakthrough therapy designation for PRO 140 may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA procedures and, in any event, does not assure ultimate approval by the FDA. In addition, even if PRO 140 does qualify as a breakthrough therapy, the FDA may later decide that PRO 140 no longer meet the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. The foregoing considerations could result in additional costs and/or delay in the potential realization of revenues from commercialization of PRO 140.
If we are not able to obtain any required regulatory approvals for PRO 140, we will not be able to commercialize our primary product candidate, which would materially and adversely affect our business, financial condition and stock price.
Our efforts to commercialize PRO 140 are dependent on obtaining FDA or other non-U.S. regulatory agency approval of its use in HIV-infected patients. Even if we complete our clinical trials, it does not assure FDA approval. We have never commercialized any product candidates and do not have any compounds other than PRO 140 in current clinical testing. We cannot be certain that PRO 140 will prove to be sufficiently effective and safe to meet applicable regulatory standards for any indication. If we fail to win approval for PRO 140 as a treatment for HIV, GvHD or any other indication, it would have a material and adverse effect on our business, financial condition and stock price, and would threaten our ability to continue to operate our business.
17
Even if we obtain marketing approval for PRO 140, we must successfully commercialize it. We currently have no sales and marketing organization. If we are unable to secure a sales and marketing partner or establish satisfactory sales and marketing capabilities, we may not successfully commercialize it.
Approval of PRO 140 is no guarantee of commercial success. The sale and marketing of drug products is a complicated and multifaceted process, and many approved drugs are not commercially successful.
At present, we have no sales or marketing personnel. In order to commercialize products that are approved for commercial sales, we must either collaborate with third parties that have such commercial infrastructure or develop our own sales and marketing infrastructure. If we are not successful in entering into appropriate collaboration arrangements, or recruiting sales and marketing personnel or in building a sales and marketing infrastructure, we will have difficulty successfully commercializing PRO 140, which would adversely affect our business, operating results and financial condition.
If approved for marketing, the commercial success of PRO 140 will depend upon its acceptance by customers and other stakeholders, including physicians, patients and health care payors. The degree of market acceptance of PRO 140 will depend on a number of factors, including:
| • | demonstration of clinical safety and efficacy; |
| • | relative convenience and ease of administration; |
| • | the prevalence and severity of any adverse effects; |
| • | the willingness of physicians to prescribe PRO 140 and of the target patient population to try new therapies; |
| • | safety, tolerability and efficacy of PRO 140 compared to competing products; |
| • | the introduction of any new products that may in the future become available to treat indications for which PRO 140 may be approved; |
| • | pricing and cost-effectiveness; |
| • | the inclusion or omission of PRO 140 in applicable treatment guidelines; |
| • | the effectiveness of our or any future collaborators’ sales and marketing strategies; |
| • | limitations or warnings contained in FDA approved labeling; |
| • | our ability to obtain and maintain sufficient third party coverage or reimbursement from government health care programs, including Medicare and Medicaid, private health insurers and other third party payors; and |
| • | the willingness of patients to pay out-of-pocket in the absence of third party coverage or reimbursement. |
Even if we obtain marketing approval for PRO 140 we will be subject to ongoing regulatory obligations and oversight.
Even if we obtain marketing approval for PRO 140, we will be subject to ongoing obligations and continued regulatory review, which will result in significant risks and significant additional expenses. Additionally, PRO 140 could be subject to labeling and other restrictions and withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements, or if we experience unanticipated problems with PRO 140.
Even if we obtain FDA approval of PRO 140 for an indication, the FDA may still impose significant restrictions on its indicated uses or marketing or the conditions of approval, or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials, and post-market surveillance to monitor safety and efficacy. PRO 140 will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market information. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current cGMPs, which are requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
18
Although the FDA has granted orphan drug designation for PRO 140 for the prevention of GvHD, we may not be able to obtain or maintain orphan drug exclusivity for PRO 140.
We have received orphan drug designation by the FDA for PRO 140 in connection with our Phase 2 trial for GvHD. We may not be able to obtain or maintain orphan drug exclusivity for PRO 140. Generally, a product with orphan drug designation only becomes entitled to orphan drug exclusivity if it receives the first marketing approval for the indication for which it has such designation, in which case the FDA will be precluded from approving another marketing application for the same drug for that indication for the applicable exclusivity period. The applicable exclusivity period is seven years in the United States. Orphan drug exclusivity may be lost if the FDA determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition.
Even with orphan drug exclusivity for PRO 140, such exclusivity may not effectively protect the product from competition, because FDA has taken the position that, under certain circumstances, another drug with the same active moiety can be approved for the same condition. Specifically, the FDA’s regulations provide that it can approve another drug with the same active moiety for the same condition, if the FDA concludes that the later drug is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care.
Our competitors may develop drugs that are more effective, safer and less expensive than ours.
We are engaged in the HIV treatment sector of the biopharmaceutical industry, which is intensely competitive. There are current treatments that are quite effective at controlling the effects of HIV, and we expect that new developments by other companies and academic institutions in the areas of HIV treatment will continue. If approved for marketing by the FDA, depending on the approved clinical indication, our product candidates may be competing with existing and future antiviral treatments for HIV.
Our competitors may:
| • | develop drug candidates and market drugs that increase the levels of safety or efficacy that our product candidates will need to show in order to obtain regulatory approval; |
| • | develop drug candidates and market drugs that are less expensive or more effective than ours; |
| • | commercialize competing drugs before we or our partners can launch any products we are working to develop; |
| • | hold or obtain proprietary rights that could prevent us from commercializing our products; or |
| • | introduce therapies or market drugs that render our potential product candidates obsolete. |
We expect to compete against large pharmaceutical and biotechnology companies and smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. These competitors, in nearly all cases, operate research and development programs that have substantially greater financial resources than we do. Our competitors also have significantly greater experience in:
| • | developing drug and other product candidates; |
| • | undertaking preclinical testing and clinical trials; |
| • | building relationships with key customers and opinion-leading physicians; |
| • | obtaining and maintaining FDA and other regulatory approvals; |
| • | formulating and manufacturing drugs; |
| • | launching, marketing and selling drugs; and |
| • | providing management oversight for all of the above-listed operational functions. |
If we fail to achieve superiority over other existing or newly developed treatments, we may be unable to obtain regulatory approval. If our competitors market drugs that are less expensive, safer or more effective than our potential product candidates, or that gain or maintain greater market acceptance, we may not be able to compete effectively.
We may not be able to successfully manufacture our product candidates in sufficient quantities for late-stage clinical development, and scale-up manufacturing processes for commercial production, which would delay or prevent us from developing our product candidates and commercializing approved products, if any.
In order to conduct larger-scale or late-stage clinical trials, we need to maintain sufficient product inventory. A failure to manufacture a product candidate in a timely manner or unexpected failure of product in inventory due to unacceptable test results may lead to significant delays in clinical development. For commercialization of any resulting product, if that candidate is approved for
19
sale, we will need to manufacture it in larger quantities while preserving its quality. Our contract manufacturing organization may not be able to successfully increase the manufacturing capacity for any of our product candidates in a timely or cost-effective manner, or at all. In addition, quality issues may arise during development, scale-up and validation of commercial manufacturing processes. If we are unable to successfully develop robust, commercial-scale processes to manufacture our product candidates in sufficient quality and quantity, the regulatory approval or commercial launch of such product candidates may be delayed, which could significantly harm our business.
We may be subject to potential product liability and other claims that could materially affect our business and financial condition.
The development and sale of medical products exposes us to the risk of significant damages from product liability and other claims, and the use of our product candidates in clinical trials may result in adverse effects. We cannot predict all the possible harms or adverse effects that may result. We maintain a modest amount of product liability insurance to provide some protections from claims. Nonetheless, we may not have sufficient resources to pay for any liabilities resulting from a personal injury or other claim, even if it is partially covered by insurance. In addition to the possibility of direct claims, we may be required to indemnify third parties against damages and other liabilities arising out of our development, commercialization and other business activities, which would increase our liability exposure. If third parties that have agreed to indemnify us fail to do so, we may be held responsible for those damages and other liabilities as well.
Legislative, regulatory, or medical cost reimbursement changes may adversely affect our business.
New laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, that relate to the health care system in the U.S. and in other jurisdictions may change the nature of and regulatory requirements relating to drug discovery, clinical testing and regulatory approvals, limit or eliminate payments for medical procedures and treatments, or subject the pricing of pharmaceuticals to government control. Outside the U.S., and particularly in the EU, the pricing of prescription pharmaceuticals is subject to governmental control. In addition, third-party payers in the U.S. are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drug products. Consequently, significant uncertainty exists as to the reimbursement status of newly approved health care products. Significant changes in the health care system in the U.S. or elsewhere, including changes resulting from adverse trends in third-party reimbursement programs, could have a material adverse effect on our projected future operating results and our ability to raise capital, commercialize products, and remain in business.
If we are unable to effectively maintain a system of internal control over financial reporting, we may not be able to accurately or timely report our financial results and our stock price could be adversely affected.
Section 404 of the Sarbanes-Oxley Act of 2002 and related regulations require us to evaluate the effectiveness of our internal control over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal control over financial reporting in our Annual Report on Form 10-K for that fiscal year. Management determined that as of May 31, 2018, our disclosure controls and procedures and internal control over financial reporting were effective. Prior to the fiscal year ended May 31, 2017, our disclosure controls and procedures and internal control over financial reporting were not effective, due to material weaknesses in our internal control over financial reporting related to inadequate segregation of duties over authorization, review and recording of transactions, as well as the financial reporting of such transactions. Any failure to maintain our controls or operation of these controls, could harm our operations, decrease the reliability of our financial reporting, and cause us to fail to meet our financial reporting obligations, which could adversely affect our business and reduce our stock price.
Our success depends substantially upon our ability to obtain and maintain intellectual property protection relating to our product candidates and research technologies.
Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering pharmaceutical inventions and the claim scope of patents, our ability to enforce our existing patents and to obtain and enforce patents that may issue from any pending or future patent applications is uncertain and involves complex legal, scientific and factual questions. To date, no consistent policy has emerged regarding the breadth of claims allowed in biotechnology and pharmaceutical patents. Thus, we cannot be sure that any patents will issue from any pending or future patent applications owned by or licensed to us. Even if patents do issue, we cannot be sure that the claims of these patents will be held valid or enforceable by a court of law, will provide us with any significant protection against competing products, or will afford us a commercial advantage over competitive products. If one or more products resulting from our product candidates is approved for sale by the FDA and we do not have adequate intellectual property protection for those products, competitors could duplicate them for approval and sale in the United States without repeating the extensive testing required of us or our partners to obtain FDA approval.
20
Certain agreements and related license agreements require us to make significant milestone, royalty, and other payments, which will require additional financing and, in the event we do commercialize our PRO 140 product, decrease the revenues we may ultimately receive on sales. To the extent that such milestone, royalty and other payments are not timely made, the counterparties to such agreements in certain cases have repurchase and termination rights thereunder with respect to PRO 140.
Under the Progenics Purchase Agreement, the PDL License and the Lonza Agreement, we must pay to Progenics, AbbVie Inc. and Lonza significant milestone payments, license fees for “system know-how” technology and royalties. In order to make the various milestone and license payments that are required, we will need to raise additional funds. In addition, our royalty obligations will reduce the economic benefits to us of any future sales if we do receive regulatory approval and seek to commercialize PRO 140. To the extent that such milestone payments and royalties are not timely made, under each their respective agreements, Progenics has certain repurchase rights relating to the assets sold to us, and AbbVie Inc. has certain termination rights relating to our license of PRO 140 under the PDL License. For more information, see “Business—PRO 140 Acquisition and Licenses,” as well as the Progenics Purchase Agreement, the PDL License and the Lonza Agreement, each of which are filed, respectively, as Exhibits 2.1, 10.13 and 10.19 to this Form 10-K.
Known third party patent rights could delay or otherwise adversely affect our planned development and sale of PRO 140. We have identified but not exhaustively analyzed other patents that could relate to our proposed products.
We are aware of patent rights held by a third party that may cover certain compositions within our PRO 140 candidate. The patent holder has the right to prevent others from making, using, or selling a drug that incorporates the patented compositions, while the patent remains in force. While we believe that the third party’s patent rights will not affect our planned development, regulatory clearance, and eventual marketing, commercial production, and sale of PRO 140, there can be no assurance that this will be the case. The relevant patent expires before we expect to commercially introduce PRO 140. In addition, the Hatch-Waxman exemption to U.S. patent law permits all uses of compounds in clinical trials and for other purposes reasonably related to obtaining FDA clearance of drugs that will be sold only after patent expiration, so our use of PRO 140 in those FDA-related activities does not infringe the patent holder’s rights. However, were the patent holder to assert its rights against us before expiration of the patent for activities unrelated to FDA clearance, the development and ultimate sale of a PRO 140 product could be significantly delayed, and we could incur the expense of defending a patent infringement suit and potential liability for damages for periods prior to the patent’s expiration.
In connection with our acquisition of rights to PRO 140, our patent counsel conducted a freedom-to-operate search that identified other patents that could relate to our proposed PRO 140 candidate. Sufficient research and analysis was conducted to enable us to reach the conclusion that PRO 140 likely does not infringe those patent rights. However, we did not have an exhaustive analysis conducted as to the identified patent rights, because doing so would have been more costly than appeared to be justified. If any of the holders of the identified patents were to assert patent rights against us, the development and sale of PRO 140 could be delayed, we could be required to spend time and money defending patent litigation, and we could incur liability for infringement or be enjoined from producing our products if the patent holders prevailed in an infringement suit.
If we are sued for infringing on third-party intellectual property rights, it will be costly and time-consuming, and an unfavorable outcome would have a significant adverse effect on our business.
Our ability to commercialize our product candidates depends on our ability to use, manufacture and sell those products without infringing the patents or other proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the monoclonal antibody therapeutic area in which we are developing product candidates and seeking new potential product candidates. There may be existing patents, unknown to us, on which our activities with our product candidates could infringe.
If a third party claims that our actions infringe on its patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including, but not limited to:
| • | infringement and other intellectual property claims that, even if meritless, can be costly and time-consuming, delay the regulatory approval process and divert management’s attention from our core business operations; |
| • | substantial damages for infringement, if a court determines that our products or technologies infringe a third party’s patent or other proprietary rights; |
| • | a court prohibiting us from selling or licensing our products or technologies unless the holder licenses the patent or other proprietary rights to us, which it is not required to do; and |
| • | even if a license is available from a holder, we may have to pay substantial royalties or grant cross-licenses to our patents or other proprietary rights. |
21
If any of these events occur, it could significantly harm our operations and financial condition and negatively affect our stock price.
We may undertake infringement or other legal proceedings against third parties, causing us to spend substantial resources on litigation and exposing our own intellectual property portfolio to challenge.
We may come to believe that third parties are infringing on our patents or other proprietary rights. To prevent infringement or unauthorized use, we may need to file infringement and/or misappropriation suits, which are very expensive and time-consuming and would distract management’s attention. Also, in an infringement or misappropriation proceeding a court may decide that one or more of our patents is invalid, unenforceable, or both, in which case third parties may be able to use our technology without paying license fees or royalties. Even if the validity of our patents is upheld, a court may refuse to stop the other party from using the technology at issue on the ground that the other party’s activities are not covered by our patents.
We may become involved in disputes with our present or future contract partners over intellectual property ownership or other matters, which would have a significant effect on our business.
Inventions discovered in the course of performance of contracts with third parties may become jointly owned by our strategic partners and us, in some cases, and the exclusive property of one of us, in other cases. Under some circumstances, it may be difficult to determine who owns a particular invention or whether it is jointly owned, and disputes could arise regarding ownership or use of those inventions. Other disputes may also arise relating to the performance or alleged breach of our agreements with third parties. Any disputes could be costly and time-consuming, and an unfavorable outcome could have a significant adverse effect on our business.
We are subject to the oversight of the SEC and other regulatory agencies. Investigations by those agencies could divert management’s focus and could have a material adverse effect on our reputation and financial condition.
We are subject to the regulation and oversight of the SEC and state regulatory agencies, in addition to the FDA. As a result, we may face legal or administrative proceedings by these agencies. We are unable to predict the effect of any investigations on our business, financial condition or reputation. In addition, publicity surrounding any investigation, even if ultimately resolved in our favor, could have a material adverse effect on our business.
Our information technology systems could fail to perform adequately or we may fail to adequately protect such information technology systems against data corruption, cyber-based attacks, or network security breaches.
We rely on information technology networks and systems, including the Internet, to process, transmit, and store electronic information. In particular, we depend on our information technology infrastructure to effectively manage our business data, accounting, and other business processes and electronic communications between our personnel and corporate partners. If we do not allocate and effectively manage the resources necessary to build and sustain an appropriate technology infrastructure, our business, and financial condition therefore could be materially adversely affected. In addition, security breaches or system failures of this infrastructure can create system disruptions, shutdowns, or unauthorized disclosure of confidential information. If we are unable to prevent such breaches or failures, our operations could be disrupted, or we may suffer financial damage or loss because of lost or misappropriated information.
If the FDA or comparable foreign regulatory authorities approve generic or biosimilar versions of any product candidate that receive marketing approval, or if any product approval we obtain does not provide us with the exclusivity periods we hope to achieve, sales of our product could be adversely affected.
As part of the ongoing efforts of governmental authorities to lower health care costs by facilitating generic competition to pharmaceutical products, the BPCIA enacted as part of the Health Care Reform Law, created a new abbreviated regulatory approval pathway in the United States for biological products that are found to be “biosimilar” to or “interchangeable” with a biological “reference product” previously licensed under a BLA. This abbreviated approval pathway is intended to permit a biosimilar to come to market more quickly and less expensively by relying to some extent on the data generated by the reference product’s sponsor and the FDA’s previous review and approval of the reference product. Under the BPCIA, a biosimilar sponsor’s ability to seek or obtain approval through the abbreviated pathway is limited by periods of exclusivity granted by the FDA to the holder of the reference product’s BLA, and no biosimilar application may be accepted by the FDA for review until four years after the date the reference product was first licensed by the FDA, and no biosimilar application, once accepted, may receive final approval until 12 years after the reference product was first licensed by the FDA.
22
Once approved, biosimilars likely would compete with, and in some circumstances may be deemed under applicable laws to be “interchangeable with,” the previously approved reference product. The extent to which a biosimilar, once approved, will be substituted for any one of our product candidates, if approved, in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. Although there is uncertainty regarding the impact of this new program, it seems likely that if any of our product candidates are approved by the FDA, there is risk that the approval of a biosimilar competitor to one of our products could have an adverse impact on our business. In particular, a biosimilar could be significantly less costly to bring to market and priced significantly lower than our product, if approved by the FDA.
We may also be subject to competition from biosimilar products in Europe. To date a number of biosimilar products have been authorized by the EMA. As in the United States the regulatory approval pathway for biosimilar products in Europe is abbreviated. A biosimilar sponsor must however still provide all of the preclinical and clinical data required to demonstrate the similarity of their product with the reference product. The level of data required is assessed on a case by case basis but it will be less than that required for an original biological product. The pathway is more complex than the abridged procedure that may be followed to obtain authorization of a generic version of a non-biological product but it would still allow the biosimilar product to be brought to market more quickly and less expensively than our original product. That said, in Europe applications for marketing authorizations in relation to biosimilar products are subject to the same data and market exclusivity as apply to generic non-biologic products so no biosimilar product could be approved or placed on the market during the periods such exclusivity applies to our product. Marketing authorization of a biosimilar product in Europe does not guarantee that the biosimilar product may be substituted for the reference product. Interchangeability of a biosimilar product with the reference product is not assessed by the EMA but this determination is left to each of the member states. We cannot know at this stage the extent to which any biosimilar product would be interchangeable with our reference product, and this may vary between member states.
Our auditors have issued a going concern opinion, and we will not be able to achieve our objectives and will have to cease operations if we cannot adequately fund our operations.
Our auditors issued an opinion, which includes a going concern exception, in connection with the audit of our annual financial statements for the fiscal year ended May 31, 2018. A going concern exception to an audit opinion means that there is substantial doubt that we can continue as an ongoing business for the next 12 months. If we are unable to continue as a going concern, we might have to liquidate our assets and the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements. In addition, the inclusion of an explanatory paragraph regarding substantial doubt about our ability to continue as a going concern and our lack of cash resources may materially adversely affect our share price and our ability to raise new capital or to enter into critical contractual relations with third parties. There is no assurance that we will be able to adequately fund our operations in the future.
Risks Relating to Our Proposed Transaction with ProstaGene
Failure to complete and to successfully integrate our proposed transaction with ProstaGene could negatively affect our stock price and future business and financial results.
We may not be able to complete and successfully integrate our proposed transaction with ProstaGene. Without realizing any of the benefits of the transaction, we will be subject to a number of risks, including our ability to continue to raise the capital necessary to fund our business and potential litigation related to the failure to complete the transaction. Achieving the benefits of the proposed transaction with ProstaGene will depend in part on the successful integration of ProstaGene, its intellectual property and the expertise of Dr. Pestell with our existing operations in a timely and efficient manner. This process may be difficult and unpredictable. If any of these risks materializes, we may not realize the expected benefits of the transaction, which could adversely affect our stock price, future business and financial results.
Integrating ProstaGene may divert management’s attention away from our operations.
The successful completion and integration of the proposed transaction with ProstaGene may place a significant burden on our management and internal resources. The diversion of management’s attention and any difficulties encountered in the transition and integration process could result in delays in the companies’ clinical trial programs and could otherwise harm our business, financial condition and operating results.
23
Risks Relating to Our Common Stock
The significant number of shares of common stock issuable upon the exercise of outstanding common stock options and warrants could adversely affect the trading price of our common stock.
If our existing stockholders sell, or indicate an intent to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline significantly. In addition, as of May 31, 2018, we have 10,751,691 shares subject to outstanding options under our stock option plans 4,858,870 shares reserved for future issuance under our equity compensation plan and 121,633,578 shares issuable upon exercise of outstanding warrants. If our existing stockholders sell substantial amounts of our common stock in the public market, or if the public perceives that such sales could occur, this could have an adverse impact on the market price of our common stock, even if there is no relationship between such sales and the performance of our business.
The price of our common stock has been and could remain volatile, and the market price of our common stock may decrease.
The market price of our common stock has historically experienced and may continue to experience significant volatility. From June 1, 2016 through May 31, 2018, the market price of our common stock has fluctuated from a high of $1.49 per share in the quarter ended August 31, 2016, to a low of $0.45 per share in quarter ending May 31, 2018. The volatile nature of our common share price may cause investment losses for our stockholders. In addition, the market price of stock in small capitalization biotech companies is often driven by investor sentiment, expectation and perception, all of which may be independent of fundamental, objective and intrinsic valuation metrics or traditional financial performance metrics, thereby exacerbating volatility. In addition, our common stock is quoted on the OTCQB of the OTC Markets marketplace, which may increase price quotation volatility and could limit liquidity, all of which may adversely affect the market price of our shares.
If we implement a reverse stock split, there can be no assurances that the price per share of our common stock will increase proportionately with the reverse stock split, or at all.
Our stockholders have currently authorized our Board of Directors to implement a reverse stock split at a ratio of any whole number between one-for-two and one-for-fifteen, as determined by our Board of Directors, with no reduction in the total number of authorized shares of our common stock, at any time before August 24, 2018, if and as determined by our Board of Directors. We may in the future seek approval from our stockholders to effect a reverse stock split at another time.
Reducing the number of outstanding shares of our common stock through a reverse stock split is intended, absent other factors, to increase the per share market price of our common stock, including in preparation for a potential uplisting to a national securities exchange. However, other factors, such as our financial results, market conditions and the market perception of our business, may adversely affect the market price of our common stock. As a result, there can be no assurance that a reverse stock split, if completed, will result in making our common stock more attractive to a broader range of institutional and other investors, that the per share market price of our common stock will increase following a reverse stock split or that the per share market price of our common stock will not decrease in the future. Additionally, we cannot assure shareholders that the per share market price per share of our common stock after a reverse stock split, if completed, will increase in proportion to the reduction in the number of shares of our common stock outstanding before the reverse stock split. Accordingly, the total market capitalization of our common stock after a reverse stock split may be lower than the total market capitalization before the reverse stock split.
If the beneficial ownership of our stock becomes highly concentrated, it may prevent you and other stockholders from influencing significant corporate decisions.
Our significant stockholders may exercise substantial influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets, or any other significant corporate transactions. These stockholders may also vote against a change of control, even if such a change of control would benefit our other stockholders. See “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” below.
Our common stock is classified as “penny stock” and trading of our shares may be restricted by the SEC’s penny stock regulations.
Rules 15g-1 through 15g-9 promulgated under the Securities Exchange Act of 1934 (the “Exchange Act”) impose sales practice and disclosure requirements on certain brokers-dealers who engage in transactions involving a “penny stock.” The SEC has adopted regulations which generally define “penny stock” to be any equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exceptions. Our common stock is covered by the penny stock rules, which impose additional sales practice requirements on broker-dealers who sell to persons other than established customers and “accredited investors.” The penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document in a form prepared by the SEC which provides information about penny stocks and the nature and level of risks in the penny stock market. The broker-dealer also must provide the customer with current bid and offer
24
quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer’s account. In addition, the penny stock rules require that, prior to a transaction in a penny stock that is not otherwise exempt, the broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for stock that is subject to these penny stock rules. Consequently, these penny stock rules may affect the ability of broker-dealers to trade our securities. We believe that the penny stock rules may discourage investor interest in and limit the marketability of our common stock.
Future sales of our securities could adversely affect the market price of our common stock and our future capital-raising activities could involve the issuance of equity securities, which would dilute your investment and could result in a decline in the trading price of our common stock.
We may sell securities in the public or private equity markets if and when conditions are favorable, or at prices per share below the current market price of our common stock, even if we do not have an immediate need for additional capital at that time. Sales of substantial amounts of our common stock, or the perception that such sales could occur, could adversely affect the prevailing market price of our shares and our ability to raise capital. We may issue additional shares of common stock in future financing transactions or as incentive compensation for our executive management and other key personnel, consultants and advisors. Issuing any equity securities would be dilutive to the equity interests represented by our then-outstanding shares of common stock. Moreover, sales of substantial amounts of shares in the public market, or the perception that such sales could occur, may adversely affect the prevailing market price of our common stock and make it more difficult for us to raise additional capital.
Purchasers in future offerings may experience immediate and substantial dilution.
The current trading price of the common stock is higher than the current net tangible book value per share of our common stock. Therefore, if you purchase shares of common stock in future offerings, if any, you may incur immediate and substantial dilution in the pro forma net tangible book value per share of common stock from the price per share that you pay for the common stock. In addition, you will experience dilution when we issue additional shares of common stock that we are permitted or required to issue under outstanding options and warrants and under our equity incentive plan or other compensation plans.
Our certificate of incorporation allows for our Board of Directors to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock. Currently, our Board of Directors has the authority to designate and issue up to 4,600,000 shares of our preferred stock without further stockholder approval. As a result, our Board of Directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our common stock. In addition, our Board of Directors could authorize the issuance of a series of preferred stock that has greater voting power than our common stock or that is convertible into our common stock, which could decrease the relative voting power of our common stock or result in dilution to our existing stockholders.
We do not expect any cash dividends to be paid on our shares in the foreseeable future.
We have never declared or paid a cash dividend and we do not anticipate declaring or paying dividends for the foreseeable future. We expect to use future financing proceeds and earnings, if any, to fund operating expenses. Consequently, stockholders’ only opportunity to achieve a return on their investment is if the price of our stock appreciates and they sell their shares at a profit. We cannot assure stockholders of a positive return on their investment when they sell their shares or that stockholders will not lose the entire amount of their investment.
Anti-takeover provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our Board of Directors and management.
Certain provisions of our amended and restated certificate of incorporation and bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for shares of common stock. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our Board of Directors. These provisions also could limit the price that investors might be willing to pay in the future for our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. Among other things, these provisions:
25
| • | allow us to designate and issue shares of preferred stock, without stockholder approval, that could adversely affect the rights, preferences and privileges of the holders of our common stock and could make it more difficult or less economically beneficial to acquire or seek to acquire us. |
| • | provide that special meetings of stockholders may be called only by the Board of Directors acting pursuant to a resolution approved by the affirmative majority of the entire Board of Directors. |
| • | provide that stockholders may, at a special stockholders meeting called for the purpose of removing directors, remove the entire Board of Directors or any lesser number, but only with cause, by a majority vote of the shares entitled to vote at an election of directors. |
| • | do not include a provision for cumulative voting in the election of directors. Under cumulative voting, a minority stockholder holding a sufficient number of shares may be able to ensure the election of one or more directors. The absence of cumulative voting may have the effect of limiting the ability of minority stockholders to effect changes in our Board of Directors. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
| Item 1B. | Unresolved Staff Comments. |
None.
| Item 2. | Properties. |
Our principal office location is 1111 Main Street, Suite 660, Vancouver, Washington 98660. We lease 1,812 square feet in a commercial office building pursuant to a lease that expires on April 30, 2021 at a current cost of $3,404 per month, plus modest annual increases.
| Item 3. | Legal Proceedings. |
On April 17, 2018, the staff of the Division of Enforcement (the “SEC Staff”) of the SEC issued a “Wells Notice” indicating that the SEC Staff is considering recommending that the SEC institute proceedings against us alleging that we violated certain provisions of the federal securities laws pertaining to the disclosure of material weaknesses in our Internal Controls over Financial Reporting (“ICFR”) during the period from 2008 to 2016. A Wells Notice is neither a formal allegation of wrongdoing nor a finding that any violations of law have occurred. Rather, it provides us with an opportunity to respond to issues raised by the Commission and offer our perspective prior to any SEC decision to institute proceedings. Subsequently, we entered into negotiations with the SEC Staff during which we reached an agreement in principle to settle this matter. As part of the proposed Offer of Settlement, we would (i) neither admit nor deny any wrongdoing, (ii) pay a monetary penalty of $35,000, and (iii) agree to cease-and-desist from committing or causing any violations or future violations of those provisions of the federal securities laws. During the course of these negotiations, we took steps to improve our ICFR and we have remediated the material weaknesses in ICFR and determined that, as we previously disclosed, our ICFR was effective as of May 31, 2017. As a result of our remediation efforts, the SEC Staff lowered the monetary penalty in the proposed Offer of Settlement to $35,000 from $50,000. Final resolution of this matter is subject to mutual agreement on the language of the Offer of Settlement, as well as final approval by the SEC. Accordingly, there can be no assurance that our efforts to resolve this matter will be successful or that the settlement terms will be as anticipated.
From time to time, we are involved in claims and suits that arise in the ordinary course of our business. Management currently believes that the resolution of any such claims against us, if any, will not have a material adverse effect on our business, financial condition or results of operations.
| Item 4. | Mine Safety Disclosures. |
Not applicable.
26
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market Information
Our common stock is presently quoted on the OTCQB of the OTC Markets marketplace under the trading symbol CYDY. Historically, trading in our stock has been very limited and the trades that have occurred cannot be characterized as amounting to an established public trading market. As a result, the trading prices of our common stock may not reflect the price that would result if our stock was actively traded.
The following are high and low bid prices quoted on the OTCQB during the periods indicated. The quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions:
| High | Low | |||||||
| Fiscal Year Ended May 31, 2018: |
||||||||
| First quarter ended August 31, 2017 |
$ | 0.79 | $ | 0.56 | ||||
| Second quarter ended November 30, 2017 |
$ | 0.70 | $ | 0.56 | ||||
| Third quarter ended February 28, 2018 |
$ | 0.84 | $ | 0.52 | ||||
| Fourth quarter ended May 31, 2018 |
$ | 0.79 | $ | 0.45 | ||||
| Fiscal Year Ended May 31, 2017: |
||||||||
| First quarter ended August 31, 2016 |
$ | 1.49 | $ | 0.96 | ||||
| Second quarter ended November 30, 2016 |
$ | 1.08 | $ | 0.61 | ||||
| Third quarter ended February 28, 2017 |
$ | 0.84 | $ | 0.66 | ||||
| Fourth quarter ended May 31, 2017 |
$ | 0.73 | $ | 0.46 | ||||
27
The stock performance graph has been prepared assuming that $100 was invested on June 1, 2013 in our common stock and the indexes shown. The stock price performance reflected in the graph may not be indicative of future price performance.
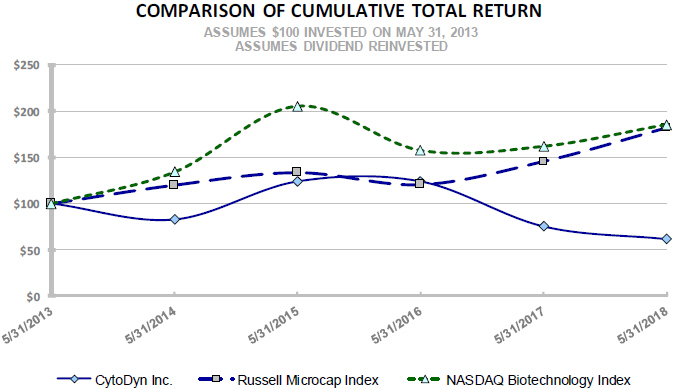
Holders
The number of record holders of our common stock on June 30, 2018, was approximately 670.
Dividends
Holders of our common stock are entitled to receive dividends as may be declared from time to time by our Board. While we have no restrictions on our ability to pay dividends, we have not paid any cash dividends since inception on our common stock and do not anticipate paying any in the foreseeable future. Our current policy is to retain earnings, if any, for use in our operations.
Holders of 92,100 shares of Series B Preferred Stock are entitled to receive, in preference to the common stock, annual cumulative dividends equal to $0.25 per share per annum from the date of issuance, which shall accrue, whether or not declared. At the time shares of Series B Preferred Stock are converted into common shares, accrued and unpaid dividends will be paid in cash or with common shares. In the event we elect to pay dividends with common shares, the shares issued will be valued at $0.50 per share. As of June 30, 2018, if we declared a dividend and elected pay such dividend in the form of common stock, approximately 391,000 shares of common stock would be issued in the form of dividend.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
During the year ended May 31, 2018 the Company purchased 159,011 shares of $0.001 par value treasury stock.
| Item 6. | Selected Consolidated Financial Data. |
The following selected financial data should be read in conjunction with our consolidated financial statements and the accompanying notes incorporated into Item 7 of Part II., “Management’s Discussion and Analysis of Financial Condition and Result of Operations.” Historical results are not necessarily indicative of future results.
28
| Year Ended May 31, | ||||||||||||||||||||
| 2018 | 2017 | 2016 | 2015 | 2014 | ||||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| General and administrative |
$ | 7,340,605 | $ | 6,758,606 | $ | 7,082,475 | $ | 4,079,579 | $ | 3,778,831 | ||||||||||
| Research and development |
38,222,580 | 20,205,743 | 13,731,426 | 15,156,365 | 3,981,468 | |||||||||||||||
| Amortization and depreciation |
356,128 | 366,385 | 361,610 | 360,582 | 352,429 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
45,919,313 | 27,330,734 | 21,175,511 | 19,596,526 | 8,112,728 | |||||||||||||||
| Operating loss |
(45,919,313 | ) | (27,330,734 | ) | (21,175,511 | ) | (19,596,526 | ) | (8,112,728 | ) | ||||||||||
| Other income (expense): |
||||||||||||||||||||
| Interest income |
3,620 | 15,167 | 7,918 | 2,199 | 7,767 | |||||||||||||||
| Gain on settlement of accounts payable |
— | — | 72,898 | — | 183,944 | |||||||||||||||
| Loss on extinguishment of convertible notes |
— | — | (584,177 | ) | — | — | ||||||||||||||
| Change in fair value of derivative liability |
1,690,935 | 2,164,533 | 646,505 | (838,643 | ) | — | ||||||||||||||
| Interest expense: |
||||||||||||||||||||
| Amortization of discount on convertible notes |
(1,666,017 | ) | — | (1,791,967 | ) | (2,145,010 | ) | (3,807,320 | ) | |||||||||||
| Amortization of discounts on related party convertible notes |
— | — | (94,344 | ) | (523,614 | ) | — | |||||||||||||
| Amortization of debt issuance costs |
(435,609 | ) | — | (604,625 | ) | (103,598 | ) | (120,000 | ) | |||||||||||
| Interest related to derivative liability |
— | (540,330 | ) | — | — | — | ||||||||||||||
| Interest expense associated with warrant extension |
(826,252 | ) | (72,437 | ) | (866,713 | ) | (555,887 | ) | — | |||||||||||
| Inducement interest related to warrant tender offer |
(393,685 | ) | — | — | — | — | ||||||||||||||
| Inducement interest related to convertible notes |
(2,352,045 | ) | — | (1,194,887 | ) | (970,367 | ) | — | ||||||||||||
| Interest on convertible notes payable |
(251,315 | ) | — | (118,709 | ) | (356,624 | ) | (583,076 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total interest expense |
(5,924,923 | ) | (612,767 | ) | (4,671,245 | ) | (4,655,100 | ) | (4,510,396 | ) | ||||||||||
| Loss before income taxes |
(50,149,681 | ) | (25,763,801 | ) | (25,703,612 | ) | (25,088,070 | ) | (12,431,413 | ) | ||||||||||
| Provision for taxes on income |
— | — | — | — | — | |||||||||||||||
| Net loss |
$ | (50,149,681 | ) | $ | (25,763,801 | ) | $ | (25,703,612 | ) | $ | (25,088,070 | ) | $ | (12,431,413 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted (loss) per share |
$ | (0.29 | ) | $ | (0.19 | ) | $ | (0.27 | ) | $ | (0.43 | ) | $ | (0.27 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted weighted average common shares outstanding |
174,885,422 | 138,004,461 | 95,437,594 | 58,375,637 | 46,900,643 | |||||||||||||||
| Selected balance sheet data: |
||||||||||||||||||||
| Cash |
$ | 1,231,445 | $ | 1,775,583 | $ | 9,641,776 | $ | 1,050,060 | $ | 4,886,122 | ||||||||||
| Current assets |
3,320,627 | 6,120,938 | 11,494,342 | 2,037,809 | 5,443,235 | |||||||||||||||
| Current liabilities |
16,733,237 | 6,144,005 | 3,580,681 | 10,725,252 | 2,166,355 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Working capital (deficit) surplus |
(13,412,610 | ) | (23,067 | ) | 7,913,661 | (8,687,443 | ) | 3,276,880 | ||||||||||||
| Total assets |
4,898,998 | 8,055,438 | 13,786,131 | 4,679,261 | 8,427,271 | |||||||||||||||
| Notes payable, net |
— | 1,058,611 | — | 4,272,076 | 2,338,684 | |||||||||||||||
| Derivative liability |
1,323,732 | 3,014,667 | — | 2,008,907 | — | |||||||||||||||
| Stockholders’ (deficit) equity |
(13,157,971 | ) | (1,103,234 | ) | 10,205,450 | (10,692,516 | ) | 3,922,232 | ||||||||||||
29
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with the other sections of this Annual Report, including our consolidated financial statements and related notes set forth in Item 8. This discussion and analysis contains forward-looking statements, including information about possible or assumed results of our financial condition, operations, plans, objectives and performance that involve risks, uncertainties and assumptions. The actual results may differ materially from those anticipated and set forth in such forward-looking statements.
Business Highlights
Over the past three fiscal years ending May 31, 2018, 2017 and 2016, we commenced several initiatives to advance our lead product candidate, PRO 140. The following is a brief summary of key accomplishments:
| • | Raised approximately $80 million, net of offering costs, in capital through equity offerings; |
| • | Raised approximately $6.0 million in capital through short-term private convertible debt offerings; |
| • | Received approximately $4.0 million, net of offering costs, in proceeds from the exercise of warrants; |
| • | Induced the conversion of approximately $5.8 million in aggregate principal amount of convertible promissory notes into common stock resulting in the conversion of nearly all previously outstanding notes; |
| • | Successfully concluded the Phase 2b clinical trial for a PRO 140 monotherapy study (CD01); |
| • | Published results from the Phase 2b monotherapy trial (CD01) in the peer reviewed journal HIV Clinical Trials; |
| • | Initiated an extension study of the Phase 2b monotherapy trial (CD01); |
| • | Initiated two Phase 2b/3 clinical trials for patients with HIV, one of which is a pivotal trial; |
| • | Successfully achieved the primary endpoint for our Phase 2b/3 pivotal combination therapy trial; |
| • | Initiated a Phase 2 trial for GvHD, the first non-HIV or immunologic clinical indication for PRO 140; |
| • | Received FDA grant of orphan drug designation to PRO 140 for the prevention of GvHD; |
| • | Published results from our preclinical study in GvHD in the peer-reviewed journal Biology of Blood and Marrow Transplantation; |
| • | Obtained FDA clearance for a compassionate use protocol to accommodate one patient and obtained FDA clearance for a roll over protocol for other patients who are successfully completing the Phase 2b/3 combination therapy trial (CD02) and wish to continue with PRO 140 as part of their treatment; |
| • | Further advanced manufacturing of new cGMP PRO 140 antibody material with the production of three cGMP batches. These batches are considered pivotal stability batches and the basis for product commercial stability and shelf life, essential for the BLA submission and ultimate FDA approval; |
| • | Organized and implemented a comprehensive team to commence preparation of our biologics license application; |
| • | Formation of a Scientific Advisory Board to advise on development of PRO 140 in certain immunologic disorders; and |
| • | Expanded the exploration of certain cancer and immunologic indications for PRO 140. |
Proposed ProstaGene Transaction
On July 12, 2018, we announced a strategic expansion of our clinical focus to include the evaluation of PRO 140 in certain cancer and immunological indications where CCR5 antagonism has shown initial promise. In connection with this expansion, we signed a non-binding letter of intent regarding a proposed acquisition of intellectual property and other assets of ProstaGene LLC (“ProstaGene”), a privately held company focused on prostate cancer diagnostics and therapeutics aimed at blocking cancer metastasis by blocking CCR5. At the same time, we remain committed to advancing our clinical programs with PRO 140 in HIV and GvHD, and is continuing with our previously announced plans to submit a Biologics License Application to the U.S. FDA for PRO 140 as a combination therapy for HIV. As part of the proposed transaction with ProstaGene, Richard G. Pestell, M.D., Ph.D., M.B.A., F.A.C.P., F.R.A.C.P., the Chief Executive Officer of ProstaGene and President of the Pennsylvania Cancer and Regenerative Medicine Research Center, will join as our Chief Medical Officer. It is also expected that Dr. Pestell will join our Board of Directors at the closing of the transaction. The transaction is subject to completion of due diligence review, customary definitive documentation, deal structure and requisite corporate and regulatory approvals. The final terms of the transaction will be available upon the execution of definitive documentation.
30
Results of Operations
Clinical Trials Update
Phase 2b Extension Study for HIV, as Monotherapy (CD01). Currently, there are a total of six patients in this ongoing extension study and each has surpassed three and one-half years of suppressed viral load with PRO 140 as a single agent therapy.
Phase 2b/3 Pivotal Trial for HIV, as Combination Therapy (CD02). In late February 2018, the Company reported that it had enrolled 52 patients and the trial’s primary endpoint was achieved with a p-value of 0.0032. Following the achievement of primary endpoint, the trial is continuing to enroll under an open label for safety analysis. This is a pivotal 25-week double blind, placebo controlled trial for PRO 140 as a combination therapy to existing HAART drug regimens. Nearly all patients have completed this trial and most of the patients have transitioned to a FDA-cleared rollover study, as requested by the treating physicians to enable the patients to have continued access to PRO 140. Management projects that the total estimated costs for this trial, including the open label portion, may range from $12 million to $13 million.
Rollover Study for HIV as Combination Therapy. This study is designed for patients who successfully complete the pivotal Phase 2b/3 Combination Therapy trial (CD02) and for whom the treating physicians request a continuation of PRO 140 therapy in order to maintain suppressed viral load. If this study enrolls 50 patients from the Phase 2b/3 trial and all patients remain in the rollover study for one year, management estimates the cost of this study to be approximately $5 million to $6 million.
Phase 2b/3 Investigative Trial for HIV, as Long-term Monotherapy (CD03). This is a trial of 300 patients that assesses using PRO 140 subcutaneously as a long-acting single-agent maintenance therapy for 48 weeks in patients with suppressed viral load with CCR5-tropic HIV-1 infection. The primary endpoint is the proportion of participants with a suppressed viral load to those who experienced virologic failure. The secondary endpoint is length of time to virologic failure. Enrollment of the first several patients was announced in December 2016. We are currently evaluating a higher dose arm (525 mg), with a 50% increase from the original dosage (350 mg), as well as a 700mg dose. We believe that a higher dose will result in a higher response rate, which is supported by preliminary data. We expect to increase the number of sites in order to accelerate enrollment following the completion of enrollment of the pivotal combination therapy trial and the availability of additional capital. The estimates for the total cost of this trial currently range from $22 million to $25 million, but such estimates will be updated upon the determination of the increased number of sites, the rate of patient enrollment and the overall duration of the trial, all of which could cause the total trial costs to vary. We expect enrollment to be completed in 2018, subject to the foregoing variables. Patients who are completing this trial are transitioning to a FDA-cleared rollover study, as requested by the treating physicians to enable the patients to have continued access to PRO 140 as a single-agent maintenance therapy.
Phase 2 Trial for Graft-versus-Host Disease. This Phase 2, randomized, double-blind, placebo-controlled, multi-center 100-day study with 60 patients is designed to evaluate the feasibility of the use of PRO 140 as an add-on therapy to standard GvHD prophylaxis treatment for prevention of acute GvHD in adult patients with AML or MDS undergoing allogeneic hematopoietic stem cell transplantation. Enrollment of the first patient was announced in May of 2017. On October 5, 2017, the Company announced that the FDA had granted orphan drug designation to PRO 140 for the prevention of GvHD.
In March 2018, we announced that the IDMC for our PRO 140 Phase 2 trial in GvHD had completed a planned interim analysis of trial data on the first 10 patients enrolled. Following this review of data from the first 10 patients in the Phase 2 trial, we filed amendments to the protocol with the FDA. The amendments included switching the pre-treatment conditioning regimen from aggressive MA conditioning to an RIC, and switching from a blinded one-for-one randomized placebo-controlled design to an open-label design under which all enrollees receive PRO 140. The amendments also provide for a 50% increase in the dose of PRO 140 (to 525 mg) to more closely mimic preclinical dosing. The next review of data by the IDMC will occur following enrollment of 10 patients under the amended protocol after each patient has been dosed for 30 days. Management estimates the cost of this trial to be approximately $3.5 million to $4 million.
We will require a significant amount of additional capital to complete the foregoing clinical trials for HIV and prepare its BLA submission. See “Liquidity and Capital Resources” below.
Results of operations for the year ended May 31, 2018, 2017 and 2016, are as follows:
For the years ended May 31, 2018, May 31, 2017 and 2016, we had no activities that produced revenues from operations. The following schedule sets forth the percentage of total expenses as a percent of net loss for the years ended May 31, 2018, 2017 and 2016.
31
| Percentage of Total Net Loss | ||||||||||||||||||||||||
| Years Ended May 31, | ||||||||||||||||||||||||
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| General and administrative |
$ | 7,340,605 | (0.15 | )% | $ | 6,758,606 | (0.26 | )% | $ | 7,082,475 | (0.28 | )% | ||||||||||||
| Research and development |
38,222,580 | (0.76 | ) | 20,205,743 | (0.78 | ) | 13,731,426 | (0.53 | ) | |||||||||||||||
| Amortization and depreciation |
356,128 | (0.01 | ) | 366,385 | (0.01 | ) | 361,610 | (0.01 | ) | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total operating expenses |
45,919,313 | (0.92 | ) | 27,330,734 | (1.06 | ) | 21,175,511 | (0.82 | ) | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Operating loss |
(45,919,313 | ) | (0.92 | ) | (27,330,734 | ) | (1.06 | ) | (21,175,511 | ) | (0.82 | ) | ||||||||||||
| Other income (expense): |
||||||||||||||||||||||||
| Interest income |
3,620 | 0.00 | 15,167 | 0.00 | 7,918 | 0.00 | ||||||||||||||||||
| Change in fair value of derivative liability |
1,690,935 | 0.03 | 2,164,533 | 0.08 | 646,505 | 0.03 | ||||||||||||||||||
| Gain on settlement of accounts payable |
— | — | — | — | 72,898 | 0.00 | ||||||||||||||||||
| Loss on extinguishment of convertible notes |
— | — | — | — | (584,177 | ) | (0.02 | ) | ||||||||||||||||
| Interest expense: |
— | — | — | — | — | |||||||||||||||||||
| Amortization of discount on convertible notes |
(1,666,017 | ) | (0.03 | ) | — | — | (1,791,967 | ) | (0.07 | ) | ||||||||||||||
| Amortization of discounts on related party convertible notes |
— | — | — | — | (94,344 | ) | (0.00 | ) | ||||||||||||||||
| Amortization of debt issuance costs |
(435,609 | ) | (0.01 | ) | — | — | (604,625 | ) | (0.02 | ) | ||||||||||||||
| Interest related to derivative liability |
— | — | (540,330 | ) | (0.02 | ) | — | — | ||||||||||||||||
| Inducement interest related to warrant tender offer |
(393,685 | ) | (0.01 | ) | — | — | — | — | ||||||||||||||||
| Inducement interest related to warrant extension |
(826,252 | ) | (0.02 | ) | (72,437 | ) | (0.00 | ) | (866,713 | ) | (0.03 | ) | ||||||||||||
| Inducement interest related to convertible notes |
(2,352,045 | ) | (0.05 | ) | — | — | (1,194,887 | ) | (0.05 | ) | ||||||||||||||
| Interest on convertible notes payable |
(251,315 | ) | (0.01 | ) | — | — | (118,709 | ) | (0.00 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total interest expense |
(5,924,923 | ) | (0.12 | ) | (612,767 | ) | (0.02 | ) | (4,671,245 | ) | (0.18 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Loss before income taxes |
(50,149,681 | ) | (1.00 | ) | (25,763,801 | ) | (1.00 | ) | (25,703,612 | ) | (1.00 | ) | ||||||||||||
| Provision for taxes on income |
— | — | — | |||||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Net loss |
$ | (50,149,681 | ) | (1.00 | )% | $ | (25,763,801 | ) | (1.00 | )% | $ | (25,703,612 | ) | (1.00 | )% | |||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Basic and diluted loss per share |
$ | (0.29 | ) | $ | (0.19 | ) | $ | (0.27 | ) | |||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Basic and diluted weighted average common shares outstanding |
174,885,422 | 138,004,461 | 95,437,594 | |||||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
Results of operations for the years ended May 31, 2018 and 2017
For the years ended May 31, 2018 and 2017, we had a net loss of approximately $50.1 million and $25.8 million, respectively. The increase in net loss of approximately $24.4 million for fiscal 2018 over 2017 was primarily attributable to increased R&D expenses of approximately $18.0 million, an increase in interest expense of approximately $5.3 million and a modest increase in general and administrative expense of approximately $0.6 million, combined with a reduction in the non-cash benefit in fair value of derivative liability of approximately $0.5 million. The loss per share for the fiscal year ended May 31, 2018 was $(0.29) compared to $(0.19) for the prior fiscal year.
Total operating expenses for the years ended May 31, 2018 and 2017 were approximately as follows:
| 2018 | 2017 | |||||||
| General and administrative: |
||||||||
| Salaries and other compensation |
$ | 2,454,000 | $ | 2,332,000 | ||||
| Stock-based compensation |
1,291,000 | 1,205,000 | ||||||
| Other |
3,596,000 | 3,222,000 | ||||||
|
|
|
|
|
|||||
| Total general and administrative |
7,341,000 | 6,759,000 | ||||||
| Research and development |
38,223,000 | 20,206,000 | ||||||
| Amortization and depreciation |
356,000 | 366,000 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
$ | 45,920,000 | $ | 27,331,000 | ||||
|
|
|
|
|
|||||
32
For the fiscal year ended May 31, 2018 and May 31, 2017, operating expenses totaled approximately $45.9 million and $27.3 million, respectively, consisting primarily of research and development (“R&D”) expenses of $38.2 million, general and administrative expenses of approximately $7.3 million and amortization and depreciation of approximately $0.4 million. The increase in operating expenses over the comparable 2017 period was attributable to an increase in R&D expenses of approximately $18.0 million owing to higher clinical trial and manufacturing-related expenses and a modest increase in general and administrative expenses of approximately $0.6 million primarily related to an increase in consulting services and employee-related expenses.
General and administrative expenses, totaled approximately $7.3 million and $6.8 million, respectively, for fiscal 2018 and 2017. General and administrative expenses were comprised of salaries and benefits, non-cash stock-based compensation expense, professional fees, insurance and various other expenses. The increase in general and administrative expenses of approximately $0.6 million, or 8.6%, for the fiscal year ended May 31, 2018 over the comparable 2017 period was primarily due to increased consulting services and employee-related expenses.
R&D expenses, which totaled approximately $38.2 million for the fiscal year ended May 31, 2018, increased approximately $18.0 million, or 89.2%, over the same 2017 period. This increase was attributable to higher clinical trial expenses, combined with an expansion of our CMC activities in connection with the preparation of a BLA. We expect R&D expenses to maintain at this level, as the two ongoing Phase 2b/3 trials with PRO 140 for HIV therapy continue, along with their related rollover studies, combined with the Phase 2 GvHD trial, and the expenses to continue activities related to manufacturing cGMP PRO 140 material for the BLA and for future use.
We record research and development expenses where directly identifiable, and approximated the following, for the years ended May 31, 2018 and 2017:
| Year Ended May 31, | ||||||||
| 2018 | 2017 | |||||||
| Research and development: |
||||||||
| Clinical |
$ | 22,543,000 | $ | 9,846,000 | ||||
| Non-Clinical |
887,000 | 691,000 | ||||||
| CMC |
14,240,000 | 8,998,000 | ||||||
| Licenses and patent fees |
553,000 | 671,000 | ||||||
|
|
|
|
|
|||||
| Total research and development |
$ | 38,223,000 | $ | 20,206,000 | ||||
|
|
|
|
|
|||||
For the fiscal year ended May 31, 2018, we recognized an unrealized non-cash benefit from the decrease in derivative liability of approximately $1.7 million, as compared to an approximate non-cash benefit of $2.2 million in the comparable 2017 period. The warrants that contain a contingent cash settlement provision, which gives rise to a derivative liability, originated in September 2016. For each reporting period, we determine the fair value of the derivative liability and record a corresponding non-cash benefit or non-cash charge, as a consequence of a decrease or increase, respectively, in the calculated derivative liability.
Interest expense for the fiscal year ended May 31, 2018 of approximately $5.9 million increased approximately $5.3 million over the 2017 fiscal year due primarily to non-cash interest of approximately $3.6 million related to inducement interest on (i) convertible notes; (ii) the expiration date extension of certain warrants and (iii) the warrant tender offer that was completed in March 2018, coupled with cash interest expense of approximately $0.3 on a convertible note and an increase in amortization of debt discount and issuance costs of approximately $2.1 million, offset by a reduction in interest related to derivative liability of approximately $0.5 million.
The future trends of all expenses will be driven, in large part, by the future outcomes of clinical trials and the correlative effect on research and development expenses, as well as general and administrative expenses, in addition to the manufacturing of new commercial PRO 140, along with the increasing activities to prepare and file a BLA. We require a significant amount of additional capital, and our ability to continue to fund operations will continue to depend on our ability to raise such capital. See in particular, “Liquidity and Capital Resources” below See, in particular, Item 1A “Risk Factors” above.
Results of operation for the years ended May 31, 2017 and 2016
For the years ended May 31, 2017 and 2016, we had a net loss of approximately $25.8 million and $25.7 million, respectively. The increase in net loss of approximately $0.1 million for fiscal 2017 over 2016 was primarily attributable to an increase in research and development, offset by the non-cash benefit from a change in derivative liability and substantially lower interest expense. The loss per shares for the fiscal year ended May 31, 2017 was $(0.19) compared to $(0.27) for the prior fiscal year.
33
The operating expenses for the years ended May 31, 2017 and 2016 and approximated as follows:
| 2017 | 2016 | |||||||
| General and administrative: |
||||||||
| Salaries and other compensation |
$ | 2,332,000 | $ | 1,341,000 | ||||
| Stock-based compensation |
1,205,000 | 2,353,000 | ||||||
| Other |
3,222,000 | 3,388,000 | ||||||
|
|
|
|
|
|||||
| Total general and administrative |
6,759,000 | 7,082,000 | ||||||
| Research and development |
20,206,000 | 13,731,000 | ||||||
| Amortization and depreciation |
366,000 | 362,000 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
$ | 27,331,000 | $ | 21,175,000 | ||||
|
|
|
|
|
|||||
The increase in fiscal 2017 total operating expenses of approximately $6.2 million, or 29%, over fiscal 2016 was primarily related to the increased research and development, offset in part by moderately lower general and administrative expenses.
Salaries and other compensation increased approximately $1.0 million in fiscal year 2017 over fiscal year 2016 due to additional employees and related expenses.
Stock-based compensation (a non-cash expense) decreased approximately $1.1 million, or 49%, in fiscal 2017, from fiscal 2016 due to the granting of long-term incentive stock options covering a fewer number of shares for management.
Other operating expenses of approximately $3.2 million for fiscal 2017 decreased approximately $166,000, or 5%, from fiscal 2016 as a result of increased insurance costs and travel, offset in part by reductions in professional fee expenses.
R&D expenses of approximately $20.2 million for fiscal 2017 increased approximately $6.5 million over fiscal 2016 principally due to additional clinical trial and manufacturing expenses and the acceleration of non-clinical related expenses required for the BLA submission. The fiscal 2017 R&D expenditures were primarily devoted to: (1) advancing clinical trials of PRO 140 for the extension study of the Phase 2b monotherapy trial, one pivotal Phase 2b/3 combination therapy trial, one investigative Phase 2b/3 monotherapy trial, initiation of a Phase 2 GvHD trial, (2) increased CMC activities to address regulatory compliance requirements of a future BLA filing and to advance the preparations for manufacturing new PRO 140 and (3) preparation of the non-clinical section necessary to complete the BLA filing with the FDA.
We expect R&D expenses to continue to increase in future periods, as the activity within our clinical trials expands and the biologics manufacturing processes and related regulatory compliance activities increase, all of which support our objectives to advance the preparation for an anticipated BLA filing.
We record research and development expenses where directly identifiable, approximately as follows for the years ended May 31, 2017 and 2016:
| Year Ended May 31, | ||||||||
| 2017 | 2016 | |||||||
| Research and development: |
||||||||
| Clinical |
$ | 9,846,000 | $ | 6,468,000 | ||||
| Non-Clinical |
691,000 | 21,000 | ||||||
| CMC |
8,998,000 | 6,214,000 | ||||||
| Licenses and patent fees |
671,000 | 1,028,000 | ||||||
|
|
|
|
|
|||||
| Total research and development |
$ | 20,206,000 | $ | 13,731,000 | ||||
|
|
|
|
|
|||||
For the fiscal years ended May 31, 2017 and May 31, 2016, we recognized non-cash benefits associated with the fair value of a derivative liability of approximately $2.2 million and $0.6 million, respectively. For each reporting period, we determine the fair value of the derivative liability and record a corresponding non-cash benefit or a non-cash charge.
The derivative liability for fiscal 2017 arose from our issuance of warrants to investors and the placement agent in connection with the September 2016 registered direct equity offering. The warrants contain a provision for net cash settlement in the event that there is a fundamental transaction (contractually defined as a merger, sale of substantially all assets, tender offer or share exchange). If a fundamental transaction occurs in which the consideration issued consists principally of cash or stock in a successor entity, then the warrantholder has the option to receive cash, equal to the fair value of the remaining unexercised portion of the warrant. Due to this contingent cash settlement provision, the investor and placement agent warrants require liability classification as derivatives in accordance with ASC 480 and ASC 815 and are recorded at fair value.
34
The derivative liability for fiscal 2016 related to a provision for potential adjustment of the conversion rate of a note, commonly known as an anti-dilution or “down round” provision. The note was converted into common stock in June 2015.
Interest expense for fiscal 2017 totaled approximately $0.6 million, all of which was non-cash, and was primarily due to the derivative liability arising from the issuance of certain warrants. Interest expense for fiscal 2017 declined approximately $4.1 million from fiscal 2016 owing to the non-comparable inducement interest expense incurred in fiscal 2016 and the conversion or repayment of all previously outstanding debt.
Fluctuations in Quarterly Operating Results
We have historically experienced significant fluctuations in our quarterly operating results and we expect such fluctuations to continue in the future. Our operating results may fluctuate due to a number of factors, such as the timing of product manufacturing, patient enrollment or completion rates in various trials, coupled with comments from FDA. As a non-revenue generating company, we are regularly conducting offerings to raise capital, which can create various forms of amortization of issuance costs or non-cash inducement interest expense. In addition, the aforementioned derivative liability is tied to a fundamental transaction and stock price, which can vary substantially from quarter to quarter, creating a non-cash charge or benefit.
Liquidity and Capital Resources
Our Company’s cash position at May 31, 2018 decreased approximately $0.5 million to approximately $1.2 million, as compared to a balance of approximately $1.8 million, as of May 31, 2017. The net decrease in cash from a year ago was attributable to net cash used in operating activities of approximately $29.9 million, offset in part by net cash provided by financing activities of approximately $29.4 million.
As of May 31, 2018, we had negative working capital of approximately $13.4 million compared to negative working capital of approximately $23,000 at May 31, 2017, an increase in negative working capital of approximately $13.4 million attributable primarily to cash used in operations.
Due to our current liquidity condition, our CMO has suspended certain preparations for future commercialization activities which are integral for the timely completion of a BLA filing. Relations with the CMO remain accommodative and resumption of certain CMC activities will be contingent on a material improvement in our liquidity. Certain other CMO activities related to BLA preparations remain on schedule at present, and provided liquidity improves, we are confident that existing BLA schedules will be maintained. Several qualification batches that may be commercially suitable remain on schedule for year-end 2018, but may be delayed into early 2019, unless liquidity improves in the near future.
Cash Flows
Net cash used in operating activities totaled approximately $29.9 million during the fiscal year end 2018, which reflects an increase of approximately $3.2 million of net cash used in operating activities over the approximate $26.7 million in fiscal 2017. The increase in net cash used in operating activities was due to an increase in net loss of approximately $24.4 million, which was mitigated in part by the effect of a comparative net change in working capital components totaling approximately $15.6 million, coupled with an increase in non-cash interest expense components of approximately $5.1 million, and an approximate $0.5 million decrease in the non-cash benefit from the change in fair value of the derivative liability.
There were no investing activities during the fiscal year ended May 31, 2018.
Net cash provided by financing activities of approximately $29.4 million for the year ended May 31, 2018 increased approximately $10.5 million over $18.9 million of net cash provided by financing activities during fiscal year ended May 31, 2017. The increase in net cash provided from financing activities was attributable to an increase in net proceeds from the sale of common stock and warrants of approximately $4.4 million, coupled with an increase of approximately $2.8 million in proceeds from the exercise of warrants in the 2018 fiscal year and an increase in proceeds of approximately $3.7 million in fiscal 2018 from the sale of convertible notes, reduced by payments to retire debt of approximately $0.3 million during the fiscal year ended May 31, 2018.
Capital Requirements
We have not generated revenue to date, and will not generate product revenue in the foreseeable future. We expect that we will continue to incur operating losses as expenses continue to increase as it proceeds with clinical trials with respect to PRO 140 and continues to advance it through the product development and regulatory process. The future trends of all expenses will be driven, in large part, by the future outcomes of the clinical trials and their correlative effect on general and administrative expenses, in addition to the manufacturing of new commercial PRO 140, along with the increasing activities to prepare and file a BLA. We will require a significant amount of additional capital in the future for our clinical trials and to advance our manufacturing activities of PRO 140 necessary for completion of our BLA filing.
35
In connection with this undertaking, we have entered into an arrangement with a third party CMO to provide process transfer, validation and manufacturing services for PRO 140. Management believes the CMO will best serve our strategic objectives for the anticipated BLA filing and, if approved, the long-term commercial manufacturing capabilities for PRO 140. Management will continue to assess manufacturing capacity requirements as new market information becomes available. In the event that we terminate the agreement with our CMO, we may incur certain financial penalties which would become payable to the CMO. Conditioned on the timing of termination, the financial penalties may range up to an approximate high of $3.2 million. These CMO undertakings are anticipated to require approximately $15 million of additional capital over the next several fiscal quarters, including the estimated costs to fill, label, and package product into the final commercial package for commercial sale.
We have entered into project work orders for each of our clinical trials with our CRO and related laboratory vendors. Under the terms of these agreements, we have prepaid certain execution fees for direct services costs. In connection with our clinical trials, we have entered into separate project work orders for each trial with our CRO. In the event that we terminate any trial, we may incur certain financial penalties which would become payable to the CRO. Conditioned upon the form of termination of any one trial, the financial penalties may range from an approximate low of $0.1 million to an approximate high of $1.1 million. In the remote circumstance that we terminate all clinical trials, the collective financial penalties may range from an approximate low of $0.8 million to an approximate high of $2.4 million.
Under the Progenics Purchase Agreement, we are required to pay Progenics the following ongoing milestone payments and royalties: (i) $5.0 million at the time of the first U.S. new drug application approval by the FDA or other non-U.S. approval for the sale of PRO 140; and (ii) royalty payments of up to five percent (5%) on net sales during the period beginning on the date of the first commercial sale of PRO 140 until the later of (a) the expiration of the last to expire patent included in the acquired assets, and (b) 10 years, in each case determined on a country-by country basis. In addition, under a Development and License Agreement, dated April 30, 1999 (the “PDL License”), between Protein Design Labs (now AbbVie Inc.) and Progenics, which was previously assigned to us, we are required to pay AbbVie Inc. additional milestone payments and royalties as follows: (i) $0.5 million upon filing a Biologic License Application with the FDA or non-U.S. equivalent regulatory body; (ii) $0.5 million upon FDA approval or approval by another non-U.S. equivalent regulatory body; and (iii) royalties of up to 7.5% of net sales for the longer of 10 years and the date of expiration of the last to expire licensed patent. Additionally, the PDL License provides for an annual maintenance fee of $150,000 until royalties paid exceed that amount.
As of the date of this filing, it is management’s conclusion that the probability of achieving the subsequent future clinical development and regulatory milestones is not reasonably determinable, thus the future milestone payments payable to Progenics and its sub-licensors are deemed contingent consideration and, therefore, are not currently accruable.
Subsequent to fiscal year end, on June 26, 2018, we entered into a securities purchase agreement pursuant to which we issued a convertible promissory note in the initial principal amount of $5.7 million. In addition to a conversion feature into which principal can be converted into common stock at $0.55 per share, the investor may redeem any portion of the note, at any time after six months from the issue date, subject to a maximum monthly amount of $350,000.
Going Concern
As reported in the accompanying financial statements, during the year ended May 31, 2018, May 31, 2017 and May 31, 2016, we incurred net losses of approximately $50.1 million, $25.8 million and $25.7 million respectively. We have no activities that produced revenue in the periods presented and has sustained operating losses since inception.
We currently require and will continue to require a significant amount of additional capital to fund operations, pay our accounts payables, and our ability to continue as a going concern is dependent upon our ability to raise such additional capital, commercialize our product and achieve profitability. If we are not able to raise such additional capital on a timely basis or on favorable terms, we may need to scale back our operations or slow down or cease certain clinical trials or CMO activities, which could materially delay the timeframe to BLA submission. In extreme cases, we could be forced to file for bankruptcy protection, discontinue our operations or liquidate our assets.
Since inception, we have financed our activities principally from the sale of public and private equity securities and proceeds from convertible notes payable and related party notes payable. We intend to finance our future operating activities and our working capital needs largely from the sale of equity and debt securities, combined with additional funding from other traditional financing sources. As of the date of this filing, we have approximately 75 million shares of common stock authorized and available for issuance under our certificate of incorporation, as amended, and approximately $166 million available for future registered offerings of securities under our universal shelf registration statement on Form S-3, which was declared effective on March 9, 2018 (assuming the full exercise of outstanding warrants, at the currently applicable exercise prices, that were previously issued in registered transactions thereunder).
36
The sale of equity and convertible debt securities to raise additional capital may result in dilution to stockholders and those securities may have rights senior to those of common shares. If we raise additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these activities or other debt could contain covenants that would restrict our operations. Any other third-party funding arrangements could require us to relinquish valuable rights. We may require additional capital beyond currently anticipated needs. Additional capital, if available, may not be available on reasonable or non-dilutive terms. Please refer to the matters discussed under the heading “Risk Factors” above.
The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. We have incurred losses for all periods presented and have a substantial accumulated deficit. As of May 31, 2018, these factors, among several others, raise substantial doubt about our ability to continue as a going concern.
The consolidated financial statements do not include any adjustments relating to the recoverability and classification of liabilities that might be necessary should we be unable to continue as a going concern. Our continuation as a going concern is dependent upon our ability to obtain additional operating capital, complete development of our product candidates, obtain FDA approval, outsource manufacturing of our products, and ultimately to attain profitability. We intend to seek additional funding through equity or debt offerings, licensing agreements or strategic alliances to implement our business plan. There are no assurances, however, that we will be successful in these endeavors.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Contractual Obligations
| As of May 31, 2018 Payments Due by Period |
||||||||||||||||
| Total | Less than 1 year | 1-3 years | 3-5 years | |||||||||||||
| Operating leases |
$ | 136,268 | $ | 50,864 | $ | 85,404 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
We believe that the following critical policies affect our more significant judgments and estimates used in preparation of our consolidated financial statements.
We follow the provisions of FASB ASC 815-Derivatives and Hedging (“ASC 815”), FASB ASC 480-Distinguishing liabilities from equity (“ASC 480”), ASC 470- Debt and debt with conversion and other options (“ASC 470”). We have issued instruments that meet the criteria of derivative liabilities. Derivative financial instruments consist of financial instruments that contain a notional amount and one or more underlying variable (e.g., contingent cash settlement provision), require no initial net investment and permit net settlement. Derivative financial instruments may be free-standing or embedded in other financial instruments. We have induced conversion of certain instruments with bifurcated conversion options. We have followed the general extinguishment model to record certain conversion and the extinguishment of derivative liabilities. We utilized a Binomial Lattice Model to value the conversion options, which utilizes assumptions that market participants would likely consider in negotiating the transfer of the convertible options, including early conversions. The assumptions in the model are subject to estimates and judgement.
We use the Black-Scholes option pricing model to estimate the fair value of stock-based awards on the date of grant utilizing certain assumptions that require judgments and estimates. These assumptions include estimates for volatility, expected term and risk-free interest rates in determining the fair value of the stock-based awards.
We periodically issue stock options and warrants to consultants for various services. Costs for these transactions are measured at the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more readily measurable. This determination requires judgment in terms of the consideration being measured.
37
We have historically issued convertible promissory notes with detachable warrants to purchase common stock. The conversion options are fixed, but may be beneficial to the note holders at the respective commitment dates. The valuation of the beneficial conversion feature of the notes and of the warrants gives rise to the recognition of a debt discount, which requires the use of certain assumptions inherent in the Black-Scholes option pricing model, including various judgments and estimates.
As discussed in Notes 8 and 9 to the consolidated financial statements, we have significant license and contingent milestone and royalty liabilities. We must estimate the likelihood of paying these contingent liabilities periodically based on the progress of our clinical trials.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk. |
Our exposure to market risk is limited to changes in the market price of its common stock and to a lesser extent foreign currency exchange risk.
Common Stock Price Risk
We do not use derivative instruments to hedge risks relating to our ongoing business operations or for speculative purposes. However, as described in greater detail in Note 5 (Derivative Liability) to the accompanying financial statements, we are required to account for certain outstanding series of warrants as derivative instruments.
All derivative instruments are required to be recorded on the balance sheet at their fair values. Each quarter, management determines the fair value of the warrants accounted for as derivative instruments using a binomial lattice valuation model. The key inputs in determining fair value of such derivative liabilities include our stock price and stock price volatility, and the then applicable risk free interest rate. Changes in these inputs affect the valuation of such derivatives and result in non-cash gain or loss each quarterly period. For example, a 10% increase or decrease in stock price would increase or decrease the value of the warrant derivative liability by approximately $0.2 million, resulting in a non-cash loss (for an increase) or gain (for a decrease) of the same amount. Similarly, a 10% increase or decrease in stock price volatility would increase or decrease the value of the warrant derivative liability by approximately $0.1 million, resulting in a non-cash loss (for an increase) or gain (for a decrease) of the same amount. Finally, a 10% increase or decrease in the risk free interest rate would not have a material effect on the value of the warrant derivative liability. Management’s discretion is required to estimate certain other factors, as described in Note 5 to the accompanying financial statements, which also contribute to the fair value estimates of such derivative liability.
During the fiscal year ended May 31, 2018, we recorded a non-cash benefit, or unrealized non-cash gain, from a decrease in the fair value of the derivative liability associated with certain warrants of approximately $1.7 million, due primarily to decreases in our common stock price and calculated stock price volatility.
Foreign Currency Exchange Risk
We may face certain exposure to fluctuation in foreign currency exchange rates, due primarily to a license agreement with a third-party licensor under which we are required to pay annual license fees and/or royalties denominated in British pounds sterling. For more information about this license agreement, see Note 8 (License Agreements) to the accompanying financial statements. Nevertheless, fluctuations in foreign exchange rates have not previously had, nor does management believe that they will have, any material impact on our earnings, cash flows or other financial results.
| Item 8. | Financial Statements and Supplementary Data. |
CYTODYN INC.
38
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
CytoDyn Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of CytoDyn Inc. (the Company) as of May 31, 2018 and 2017 and the related consolidated statements of operations, changes in stockholders’ (deficit) equity, and cash flows for each of the years in the three-year period ended May 31, 2018, and the related notes (collectively referred to as the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of May 31, 2018 and 2017, and the results of its operations and its cash flows for each of the years in the three-year period ended May 31, 2018, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of May 31, 2018, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated July 27, 2018, expressed an unqualified opinion.
The Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company incurred a net loss of $50,149,681 for the year ended May 31, 2018 and has an accumulated deficit of $173,139,396 through May 31, 2018, which raises substantial doubt about its ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ Warren Averett, LLC |
We have served as the Company’s auditor since 2007.
Birmingham, Alabama
July 27, 2018
39
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of CytoDyn Inc.
Opinion on Internal Control over Financial Reporting
We have audited CytoDyn Inc.’s (the Company’s) internal control over financial reporting as of May 31, 2018, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of May 31, 2018, based on criteria established in Internal Control—Integrated Framework (2013) issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the balance sheets and the related statements of operations, changes in stockholders’ (deficit) equity, and cash flows of the Company, and our report dated July 27, 2018, expressed an unqualified opinion.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Warren Averett, LLC
Birmingham, Alabama
July 27, 2018
40
CytoDyn Inc.
| May 31, 2018 | May 31, 2017 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash |
$ | 1,231,445 | $ | 1,775,583 | ||||
| Prepaid expenses |
227,173 | 207,314 | ||||||
| Prepaid service fees |
1,862,009 | 4,138,041 | ||||||
|
|
|
|
|
|||||
| Total current assets |
3,320,627 | 6,120,938 | ||||||
| Furniture and equipment, net |
11,228 | 17,281 | ||||||
| Intangibles, net |
1,567,143 | 1,917,219 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 4,898,998 | $ | 8,055,438 | ||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ (Deficit) Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 15,841,859 | $ | 4,281,204 | ||||
| Accrued compensation |
699,795 | 637,190 | ||||||
| Accrued license fees |
133,600 | 167,000 | ||||||
| Accrued liabilities |
57,983 | — | ||||||
| Convertible notes payable, net |
— | 1,058,611 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
16,733,237 | 6,144,005 | ||||||
|
|
|
|
|
|||||
| Long-term liabilities: |
||||||||
| Derivative liability |
1,323,732 | 3,014,667 | ||||||
|
|
|
|
|
|||||
| Total long-term liabilities |
1,323,732 | 3,014,667 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
18,056,969 | 9,158,672 | ||||||
| Commitments and Contingencies |
— | — | ||||||
| Stockholders’ (Deficit) Equity |
||||||||
| Series B convertible preferred stock, $0.001 par value; 400,000 shares authorized, 92,100 shares issued and outstanding at May 31, 2018 and |
||||||||
| May 31, 2017, respectively |
92 | 92 | ||||||
| Common stock, $0.001 par value; 375,000,000 and 350,000,000 shares authorized, 216,881,790 and 149,468,244 issued and 216,722,779 and 149,468,244 outstanding at May 31, 2018 and May 31, 2017, respectively |
216,881 | 149,468 | ||||||
| Additional paid-in capital |
159,764,611 | 121,736,921 | ||||||
| Accumulated (deficit) |
(173,139,396 | ) | (122,989,715 | ) | ||||
| Less: treasury stock, at par (159,011 shares at $0.001) |
(159 | ) | — | |||||
|
|
|
|
|
|||||
| Total stockholders’ (deficit) |
(13,157,971 | ) | (1,103,234 | ) | ||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ (deficit) equity |
$ | 4,898,998 | $ | 8,055,438 | ||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
41
CytoDyn Inc.
Consolidated Statements of Operations
| Years ended May 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Operating expenses: |
||||||||||||
| General and administrative |
$ | 7,340,605 | $ | 6,758,606 | $ | 7,082,475 | ||||||
| Research and development |
38,222,580 | 20,205,743 | 13,731,426 | |||||||||
| Amortization and depreciation |
356,128 | 366,385 | 361,610 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
45,919,313 | 27,330,734 | 21,175,511 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating loss |
(45,919,313 | ) | (27,330,734 | ) | (21,175,511 | ) | ||||||
| Other income (expense): |
||||||||||||
| Interest income |
3,620 | 15,167 | 7,918 | |||||||||
| Change in fair value of derivative liability |
1,690,935 | 2,164,533 | 646,505 | |||||||||
| Gain on settlement of accounts payable |
— | — | 72,898 | |||||||||
| Loss on extinguishment of convertible notes |
— | — | (584,177 | ) | ||||||||
| Interest expense: |
||||||||||||
| Amortization of discount on convertible notes |
(1,666,017 | ) | — | (1,791,967 | ) | |||||||
| Amortization of discounts on related party convertible notes |
— | — | (94,344 | ) | ||||||||
| Amortization of debt issuance costs |
(435,609 | ) | — | (604,625 | ) | |||||||
| Interest related to derivative liability |
— | (540,330 | ) | — | ||||||||
| Inducement interest related to warrant tender offer |
(393,685 | ) | ||||||||||
| Inducement interest related to warrant extension |
(826,252 | ) | (72,437 | ) | (866,713 | ) | ||||||
| Inducement interest related to convertible notes |
(2,352,045 | ) | — | (1,194,887 | ) | |||||||
| Interest on convertible notes payable |
(251,315 | ) | — | (118,709 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Total interest expense |
(5,924,923 | ) | (612,767 | ) | (4,671,245 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes |
(50,149,681 | ) | (25,763,801 | ) | (25,703,612 | ) | ||||||
| Provision for taxes on income |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (50,149,681 | ) | $ | (25,763,801 | ) | $ | (25,703,612 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted loss per share |
$ | (0.29 | ) | $ | (0.19 | ) | $ | (0.27 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted weighted average common shares outstanding |
174,885,422 | 138,004,461 | 95,437,594 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
42
CytoDyn Inc.
Consolidated Statements of Changes in Stockholders’ (Deficit) Equity
| Preferred Stock | Common Stock | |||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||
| Balance May 31, 2015 |
95,100 | $ | 95 | 63,644,348 | $ | 63,644 | ||||||||||
| Warrant issued related to conversion inducement |
— | — | — | — | ||||||||||||
| Modification of warrants to related conversion inducement |
— | — | — | — | ||||||||||||
| Debt discount due to reduction in note conversion price from ($0.75 to $0.675/share) |
— | — | — | — | ||||||||||||
| Conversion of convertible debt & accrued, unpaid interest to common stock ($0.675/share) |
— | — | 4,095,008 | 4,095 | ||||||||||||
| Conversion of convertible debt and extinguishment of derivative liability |
— | — | 5,274,656 | 5,275 | ||||||||||||
| Conversion of convertible debt & accrued, unpaid interest to common stock ($0.75/share) |
— | — | 792,201 | 792 | ||||||||||||
| Exercise of common stock warrants (cashless) |
— | — | 74,490 | 74 | ||||||||||||
| Exercise of common stock warrants ($0.50/share) |
— | — | 192,307 | 192 | ||||||||||||
| Exercise of common stock warrants ($0.75/share) |
— | — | 603,286 | 604 | ||||||||||||
| Stock-based compensation |
— | — | — | — | ||||||||||||
| Proceeds from private equity offering ($0.75/share) |
— | — | 44,357,838 | 44,358 | ||||||||||||
| Proceeds from private equity offering ($1.00/share) |
— | — | 4,301,500 | 4,302 | ||||||||||||
| Deferred offering costs |
— | — | — | — | ||||||||||||
| Net (loss) for the year ended May 31, 2016 |
— | — | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Balance May 31, 2016 |
95,100 | $ | 95 | 123,335,634 | $ | 123,336 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Interest expense related to warrant extension |
— | — | — | 36 | ||||||||||||
| Stock-based compensation |
— | — | — | — | ||||||||||||
| Legal fees in connection with registered offerings |
— | — | — | — | ||||||||||||
| Proceeds from private equity offering ($1.00/share) |
— | — | 729,500 | 730 | ||||||||||||
| Proceeds from registered direct offering ($0.75/share) |
24,538,994 | 24,539 | ||||||||||||||
| Offering costs related to equity offering |
— | — | — | — | ||||||||||||
| Debt discount related to convertible notes payable |
— | — | — | — | ||||||||||||
| Conversion of Series B Preferred ($0.50/share) |
(3,000 | ) | (3 | ) | 40,602 | 3 | ||||||||||
| Proceeds from warrant exercise ($0.50/share) |
— | — | 730,765 | 730 | ||||||||||||
| Proceeds from warrant exercise ($0.75/share) |
— | — | 43,332 | 44 | ||||||||||||
| Cashless exercise of warrants |
— | — | 49,417 | 50 | ||||||||||||
| Net (loss) for the year ended May 31, 2017 |
— | — | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Balance May 31, 2017 |
92,100 | $ | 92 | 149,468,244 | $ | 149,468 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Stock-based compensation |
— | — | — | — | ||||||||||||
| Stock issued for board compensation |
— | — | — | — | ||||||||||||
| Stock issued for bonuses and tendered for income tax |
— | — | 310,526 | 311 | ||||||||||||
| Proceeds from private equity offering ($0.50/share) |
— | — | 35,286,904 | 35,286 | ||||||||||||
| Offering costs related to private equity offering |
— | — | — | — | ||||||||||||
| Proceeds from registered direct offering ($0.50/share) |
— | — | 25,493,853 | 25,494 | ||||||||||||
| Offering costs related to registered direct offering |
— | — | — | — | ||||||||||||
| Legal fees in connection with equity offerings |
— | — | — | — | ||||||||||||
| Proceeds from warrant exercise ($0.50/share) |
— | — | 6,322,263 | 6,322 | ||||||||||||
| Offering costs related to warrant tender offer |
— | — | — | — | ||||||||||||
| Debt discount related to convertible notes payable |
— | — | — | — | ||||||||||||
| Interest expense related to warrant extension |
— | — | — | — | ||||||||||||
| Interest expense realted to warrant tender offer |
— | — | — | — | ||||||||||||
| Interest expense related to conversion of notes payable |
— | — | — | — | ||||||||||||
| Net (loss) for the year ended May 31, 2018 |
— | — | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Balance May 31, 2018 |
92,100 | $ | 92 | 216,881,790 | $ | 216,881 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
43
CytoDyn Inc.
Consolidated Statements of Changes in Stockholders’ (Deficit) Equity
| Treasury Stock | Additional | Accumulated | ||||||||||||||||||
| Shares | Amount | Paid-In Capital | Deficit | Total | ||||||||||||||||
| Balance May 31, 2015 |
— | $ | — | $ | 60,766,047 | $ | (71,522,302 | ) | $ | (10,692,516 | ) | |||||||||
| Warrant issued related to conversion inducement |
— | — | 757,871 | — | 757,871 | |||||||||||||||
| Modification of warrants to related conversion inducement |
— | — | 866,713 | — | 866,713 | |||||||||||||||
| Debt discount due to reduction in note conversion price from |
||||||||||||||||||||
| ($0.75 to $0.675/share) |
— | — | 329,524 | — | 329,524 | |||||||||||||||
| Conversion of convertible debt & accrued, unpaid interest to common stock ($0.675/share) |
— | — | 2,760,059 | — | 2,764,154 | |||||||||||||||
| Conversion of convertible debt and extinguishment of derivative liability |
— | — | 4,727,239 | — | 4,732,514 | |||||||||||||||
| Conversion of convertible debt & accrued, unpaid interest to common stock ($0.75/share) |
— | — | 593,360 | — | 594,152 | |||||||||||||||
| Exercise of common stock warrants (cashless) |
— | — | (74 | ) | — | |||||||||||||||
| Exercise of common stock warrants ($0.50/share) |
— | — | 95,962 | — | 96,154 | |||||||||||||||
| Exercise of common stock warrants ($0.75/share) |
— | — | 451,861 | — | 452,465 | |||||||||||||||
| Stock-based compensation |
— | — | 2,353,194 | — | 2,353,194 | |||||||||||||||
| Proceeds from private equity offering ($0.75/share) |
— | — | 33,224,106 | — | 33,268,464 | |||||||||||||||
| Proceeds from private equity offering ($1.00/share) |
— | — | 4,297,198 | — | 4,301,500 | |||||||||||||||
| Deferred offering costs |
— | — | (3,915,127 | ) | — | (3,915,127 | ) | |||||||||||||
| Net (loss) for the year ended May 31, 2016 |
— | — | — | (25,703,612 | ) | (25,703,612 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance May 31, 2016 |
— | $ | — | $ | 107,307,933 | $ | (97,225,914 | ) | $ | 10,205,450 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Interest expense related to warrant extension |
— | — | 72,398 | — | 72,434 | |||||||||||||||
| Stock-based compensation |
— | — | 1,204,791 | — | 1,204,791 | |||||||||||||||
| Legal fees in connection with registered offerings |
— | — | (280,883 | ) | — | (280,883 | ) | |||||||||||||
| Proceeds from private equity offering ($1.00/share) |
— | — | 728,770 | — | 729,500 | |||||||||||||||
| Proceeds from registered direct offering ($0.75/share) |
— | — | 14,019,713 | 14,044,252 | ||||||||||||||||
| Offering costs related to equity offering |
— | — | (1,804,249 | ) | — | (1,804,249 | ) | |||||||||||||
| Debt discount related to convertible notes payable |
— | — | 91,389 | 91,389 | ||||||||||||||||
| Conversion of Series B Preferred ($0.50/share) |
— | — | — | — | — | |||||||||||||||
| Proceeds from warrant exercise ($0.50/share) |
— | — | 364,653 | — | 365,383 | |||||||||||||||
| Proceeds from warrant exercise ($0.75/share) |
— | — | 32,456 | — | 32,500 | |||||||||||||||
| Cashless exercise of warrants |
— | — | (50 | ) | — | — | ||||||||||||||
| Net (loss) for the year ended May 31, 2017 |
— | — | — | (25,763,801 | ) | (25,763,801 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance May 31, 2017 |
— | $ | — | $ | 121,736,921 | $ | (122,989,715 | ) | $ | (1,103,234 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Stock-based compensation |
— | — | 1,290,777 | — | 1,290,777 | |||||||||||||||
| Stock issued for board compensation |
— | — | 260,190 | — | 260,190 | |||||||||||||||
| Stock issued for bonuses and tendered for income tax |
159,011 | (159 | ) | 104,394 | — | 104,546 | ||||||||||||||
| Proceeds from private equity offering ($0.50/share) |
— | — | 17,608,165 | — | 17,643,451 | |||||||||||||||
| Offering costs related to private equity offering |
— | — | (1,717,597 | ) | — | (1,717,597 | ) | |||||||||||||
| Proceeds from registered direct offering ($0.50/share) |
— | — | 13,585,925 | — | 13,611,419 | |||||||||||||||
| Offering costs related to registered direct offering |
— | — | (857,149 | ) | — | (857,149 | ) | |||||||||||||
| Legal fees in connection with equity offerings |
— | — | (533,436 | ) | — | (533,436 | ) | |||||||||||||
| Proceeds from warrant exercise ($0.50/share) |
— | — | 3,154,810 | — | 3,161,132 | |||||||||||||||
| Offering costs related to warrant tender offer |
— | — | (85,382 | ) | — | (85,382 | ) | |||||||||||||
| Debt discount related to convertible notes payable |
— | — | 1,645,011 | — | 1,645,011 | |||||||||||||||
| Interest expense related to warrant extension |
— | — | 826,252 | — | 826,252 | |||||||||||||||
| Interest expense realted to warrant tender offer |
— | — | 393,685 | — | 393,685 | |||||||||||||||
| Interest expense related to conversion of notes payable |
— | — | 2,352,045 | — | 2,352,045 | |||||||||||||||
| Net (loss) for the year ended May 31, 2018 |
— | — | — | (50,149,681 | ) | (50,149,681 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance May 31, 2018 |
159,011 | $ | (159 | ) | $ | 159,764,611 | $ | (173,139,396 | ) | $ | (13,157,971 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
See accompanying notes to consolidated financial statements
44
CytoDyn Inc.
Consolidated Statements of Cash Flows
| Years Ended May 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (50,149,681 | ) | $ | (25,763,801 | ) | $ | (25,703,612 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Amortization and depreciation |
356,128 | 366,385 | 361,610 | |||||||||
| Amortization of debt issuance costs |
435,609 | — | 604,625 | |||||||||
| Amortization of discount on convertible notes |
1,666,017 | — | 1,791,967 | |||||||||
| Amortization of discounts on related party notes |
— | — | 94,344 | |||||||||
| Gain on extinguishment of accounts payable |
— | — | 72,898 | |||||||||
| Loss on extinguishment of convertible notes |
— | — | 584,177 | |||||||||
| Inducement interest expense |
— | 72,437 | 1,954,108 | |||||||||
| Interest expense associated with warrant extension |
826,252 | — | — | |||||||||
| Interest expense association with warrant tender offer |
393,685 | — | — | |||||||||
| Interest expense associated with conversion of notes |
2,352,045 | — | — | |||||||||
| Interest expense associated with derivative liability |
— | 540,330 | — | |||||||||
| Change in fair value of derivative liability |
(1,690,935 | ) | (2,164,533 | ) | (646,505 | ) | ||||||
| Stock-based compensation |
1,290,777 | 1,204,791 | 2,353,194 | |||||||||
| Changes in current assets and liabilities: |
||||||||||||
| Decrease (increase) in prepaid expenses |
2,256,173 | (2,492,789 | ) | (864,817 | ) | |||||||
| Increase (decrease) in accounts payable and accrued expenses |
12,365,959 | 1,504,712 | (5,412,640 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(29,897,971 | ) | (26,732,468 | ) | (24,810,651 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Furniture and equipment purchases |
— | (11,114 | ) | (11,949 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
— | (11,114 | ) | (11,949 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from sale of common stock and warrants |
25,224,212 | 19,133,755 | 37,569,964 | |||||||||
| Proceeds from warrant exercises |
3,161,131 | 397,883 | 548,619 | |||||||||
| Proceeds from convertible notes payable |
4,888,500 | 1,150,000 | — | |||||||||
| Payment of offering costs |
(3,558,789 | ) | (1,804,249 | ) | (3,915,127 | ) | ||||||
| Payment of payroll taxes related to tender of common stock for income tax witholding |
(102,064 | ) | — | — | ||||||||
| Repayment of principal and interest on convertible note |
(259,157 | ) | — | (789,140 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
29,353,833 | 18,877,389 | 33,414,316 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net change in cash |
(544,138 | ) | (7,866,193 | ) | 8,591,716 | |||||||
| Cash, beginning of period |
1,775,583 | 9,641,776 | 1,050,060 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash, end of period |
$ | 1,231,445 | $ | 1,775,583 | $ | 9,641,776 | ||||||
|
|
|
|
|
|
|
|||||||
45
CytoDyn Inc.
Consolidated Statements of Cash Flows
| Years Ended May 31, | ||||||||||||
| 2018 | 2017 | 2016 | ||||||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid during the period for interest |
$ | 9,157 | $ | — | $ | 26,890 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash investing and financing transactions: |
||||||||||||
| Debt discount associated with convertible notes payable |
$ | 1,574,628 | $ | 91,389 | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Financing costs associated with investor warrants |
$ | — | $ | 819,200 | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Common stock issued for accrued bonus compensation |
$ | 214,263 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Common stock issued for board compensation |
$ | 260,190 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Common stock issued upon conversion of convertible debt |
$ | 5,788,500 | $ | — | $ | 7,947,342 | ||||||
|
|
|
|
|
|
|
|||||||
| Common stock issued for accrued interest payable |
$ | 242,158 | $ | — | $ | 143,479 | ||||||
|
|
|
|
|
|
|
|||||||
| Financing costs associated with placement agent warrants |
$ | 70,383 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Accounts payable extinguished through settlements |
$ | — | $ | — | $ | 72,898 | ||||||
|
|
|
|
|
|
|
|||||||
| Derivative liability associated with warrants |
$ | — | $ | 5,179,200 | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes to consolidated financial statements.
46
CYTODYN INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
AS OF MAY 31, 2018
Note 1 – Organization
CytoDyn Inc. (the “Company”) was originally incorporated under the laws of Colorado on May 2, 2002 under the name RexRay Corporation (its previous name) and, effective August 27, 2015, reincorporated under the laws of Delaware. We are a clinical-stage biotechnology company focused on the clinical development and potential commercialization of humanized monoclonal antibodies to treat Human Immunodeficiency Virus (“HIV”) infection. Our lead product candidate, PRO 140, belongs to a class of HIV therapies known as entry inhibitors. These therapies block HIV from entering into and infecting certain cells.
The Company has developed a class of therapeutic monoclonal antibodies to address unmet medical needs in the areas of HIV and GvHD. In addition, we are expanding the clinical focus with PRO 140 to include the evaluation in certain cancer and immunological indications where CCR antagonism has shown initial promise.
Note 2 – Summary of Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements include the accounts of CytoDyn Inc. and its wholly owned subsidiaries, AGTI and CVM, both of which are dormant entities. All intercompany transactions and balances are eliminated in consolidation.
Reclassifications
Certain prior year amounts shown in the accompanying consolidated financial statements have been reclassified to conform to the 2018 presentation. These reclassifications did not have any effect on total current assets, total assets, total current liabilities, total liabilities, total stockholders’ (deficit) equity, net loss or earnings per share.
Going Concern
The consolidated accompanying financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. As shown in the accompanying consolidated financial statements, the Company had losses for all periods presented. The Company incurred a net loss of $50,149,681, $25,763,801 and $25,703,612 for the years ended May 31, 2018, May 31, 2017 and May 31, 2016, respectively, and has an accumulated deficit of $173,139,396 as of May 31, 2018. These factors, among several others, raise substantial doubt about the Company’s ability to continue as a going concern.
The consolidated financial statements do not include any adjustments relating to the recoverability of assets and classification of liabilities that might be necessary should the Company be unable to continue as a going concern. The Company’s continuation as a going concern is dependent upon its ability to obtain additional operating capital, complete development of its product candidates, obtain U.S. FDA approval, outsource manufacturing of the product candidates, and ultimately achieve initial revenues and attain profitability. The Company is currently engaging in significant research and development activities related to these product candidates, and expects to incur significant research and development expenses in the future primarily related to its clinical trials. These research and development activities are subject to significant risks and uncertainties. The Company intends to finance its future development activities and its working capital needs largely from the sale of equity and debt securities, combined with additional funding from other traditional sources. There can be no assurance, however, that the Company will be successful in these endeavors.
Use of Estimates
The preparation of the consolidated financial statements in accordance with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash
Cash is maintained at federally insured financial institutions and, at times, balances may exceed federally insured limits. The Company has never experienced any losses related to these balances. Balances in excess of federally insured limits at May 31, 2018 and May 31, 2017 approximated $1.1 million and $1.5 million, respectively.
Identified Intangible Assets
The Company follows the provisions of FASB ASC Topic 350 Intangibles-Goodwill and Other, which establishes accounting standards for the impairment of long-lived assets such as intangible assets subject to amortization. The Company reviews long-lived assets to be held and used for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets
47
may not be recoverable. If the sum of the undiscounted expected future cash flows over the remaining useful life of a long-lived asset group is less than its carrying value, the asset is considered impaired. Impairment losses are measured as the amount by which the carrying amount of the asset group exceeds the fair value of the asset. There were no impairment charges for the years ended May 31, 2018, May 31, 2017 and May 31, 2016. The value of the Company’s patents would be significantly impaired by any adverse developments as they relate to the clinical trials pursuant to the patents acquired as discussed in Notes 7 and 9.
Research and Development
Research and development costs are expensed as incurred. Clinical trial costs incurred through third parties are expensed as the contracted work is performed. Where contingent milestone payments are due to third parties under research and development collaboration arrangements or other contractual agreements, the milestone payment obligations are expensed when the milestone conditions are probable and the amount of payment is reasonably estimable.
Pre-launch Inventory
The Company may scale-up and make commercial quantities of its product candidate prior to the date it anticipates that such product will receive final FDA approval. The scale-up and commercial production of pre-launch inventories involves the risk that such products may not be approved for commercial use by the FDA on a timely basis, or ever. This risk notwithstanding, the Company may scale-up and build pre-launch inventories of product that have not yet received final governmental approval when the Company believes that such action is appropriate in relation to the commercial value of the product launch opportunity. The determination to capitalize is made once the Company (or its third party development partners) has filed a BLA, that has been acknowledged by the FDA as containing sufficient information to allow the FDA to conduct its review in an efficient and timely manner and management is reasonably certain that all regulatory and legal hurdles will be cleared. This determination is based on the particular facts and circumstances relating to the expected FDA approval of the drug product being considered. As of May 31, 2018 and May 31, 2017, the Company did not have pre-launch inventory that qualified for capitalization pursuant to U.S. GAAP ASC 330 Inventory.
Fair Value of Financial Instruments
At May 31, 2018 and May 31, 2017, the carrying value of the Company’s cash, accounts payable and accrued liabilities approximate their fair value due to the short-term maturity of the instruments. The Company carries derivative financial instruments at fair value as required by U.S. GAAP.
Derivative financial instruments consist of financial instruments that contain a notional amount and one or more underlying variables (e.g., interest rate, security price, variable conversion rate or other variables), require no initial net investment and permit net settlement. Derivative financial instruments may be free-standing or embedded in other financial instruments. The Company follows the provisions of ASC 815, as their instruments are recorded as a derivative liability, at fair value, and ASC 480, as it relates to warrant liability, with changes in fair value reflected in income.
Fair Value Hierarchy
The three levels of inputs that may be used to measure fair value are as follows:
Level 1. Quoted prices in active markets for identical assets or liabilities.
Level 2. Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities, quoted prices in markets with insufficient volume or infrequent transactions (less active markets), or model-derived valuations in which all significant inputs are observable or can be derived principally from or corroborated with observable market data for substantially the full term of the assets or liabilities. Level 2 inputs also include non-binding market consensus prices that can be corroborated with observable market data, as well as quoted prices that were adjusted for security-specific restrictions.
Level 3. Unobservable inputs to the valuation methodology are significant to the measurement of the fair value of assets or liabilities. These Level 3 inputs also include non-binding market consensus prices or non-binding broker quotes that the Company was unable to corroborate with observable market data.
48
Liability measured at fair value on a recurring basis by level within the fair value hierarchy as of May 31, 2018 and May 31, 2017 is as follows:
| Fair Value Measurement at | Fair Value Measurement at | |||||||||||||||
| May 31, 2018 (1) | May 31, 2017 (1) | |||||||||||||||
| Using | Using | |||||||||||||||
| Level 3 | Total | Level 3 | Total | |||||||||||||
| Liability: |
||||||||||||||||
| Derivative liability |
$ | 1,323,732 | $ | 1,323,732 | $ | 3,014,667 | $ | 3,014,667 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total liability |
$ | 1,323,732 | $ | 1,323,732 | $ | 3,014,667 | $ | 3,014,667 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| (1) | The Company did not have any assets or liabilities measured at fair value using Level 1 or 2 of the fair value hierarchy as of May 31, 2018 and May 31, 2017. |
A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurements. These instruments are not quoted on an active market, so the Company uses a Binomial Lattice Model to estimate the value of the derivative liability. A Binomial Lattice Model was used because management believes it reflects all the assumptions that market participants would likely consider in negotiating the transfer of the warrant. The Company’s derivative liability is classified within Level 3 of the fair value hierarchy because certain unobservable inputs were used in the valuation model.
The following is a reconciliation of the beginning and ending balances for liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) during the year ended May 31, 2018 and May 31, 2017:
| Balance at May 31, 2016 |
$ | — | ||
| Investor warrants issued with registered direct equity offering |
4,360,000 | |||
| Placement agent warrants issued with registered direct equity offering |
819,200 | |||
| Fair value adjustments |
(2,164,533 | ) | ||
|
|
|
|||
| Balance at May 31, 2017 |
3,014,667 | |||
|
|
|
|||
| Fair value adjustments |
(1,690,935 | ) | ||
|
|
|
|||
| Balance at May 31, 2018 |
$ | 1,323,732 | ||
|
|
|
Stock-Based Compensation
U.S. GAAP requires companies to measure the cost of employee services received in exchange for the award of equity instruments based on the fair value of the award at the date of grant. The expense is to be recognized over the period during which an employee is required to provide services in exchange for the award (requisite service period) or when designated milestones have been achieved.
The Company accounts for stock-based awards established by the fair market value of the instrument using the Black-Scholes option pricing model utilizing certain weighted average assumptions including stock price volatility, expected term and risk-free interest rates, as of the grant date. The risk-free interest rate assumption is based upon observed interest rates appropriate for the expected term of the stock-based award. The expected volatility is based on the historical volatility of the Company’s common stock on monthly intervals. The computation of the expected option term is based on the “simplified method,” as the Company issuances are considered “plain vanilla” options. For stock-based awards with defined vesting, the Company recognizes compensation expense over the requisite service period or when designated milestones have been achieved. The Company estimates forfeitures at the time of grant and revised, if necessary, in subsequent periods, if actual forfeitures differ from those estimates. Based on limited historical experience of forfeitures, the Company estimated future unvested forfeitures at 0% for all periods presented.
Common Stock
On March 18, 2016, at a special meeting of stockholders, a proposal was approved to increase the total number of authorized shares of common stock of the Company from 200,000,000 to 250,000,000. Subsequently, on August 24, 2016, at the Annual Meeting of Stockholders, a proposal was approved to increase the total number of authorized shares of common stock from 250,000,000 to 350,000,000. On August 24, 2017, at the 2017 Annual Meeting of Stockholders, a proposal was approved to increase the total number of authorized shares of common stock from 350,000,000 to 375,000,000. On June 7, 2018, at a special meeting of stockholders, a proposal was approved to increase the total number of authorized shares of common stock of the Company from 375,000,000 to 450,000,000.
49
On November 1, 2017, the Company held a special meeting of stockholders, at which the stockholders approved a proposal to effect a reverse stock split at a ratio of any whole number between one-for-two and one-for-fifteen, as determined by the board of directors, and a simultaneous reduction in the total number of authorized shares of common stock to 200,000,000 at any time before August 24, 2018, if and as determined by the board of directors.
Preferred Stock
The Company’s Board of Directors is authorized to issue up to 5,000,000 shares of preferred stock without stockholder approval. As of May 31, 2018, the Company has authorized the issuance of 400,000 shares of Series B convertible preferred stock, of which 92,100 shares were outstanding. The remaining preferred shares authorized have no specified rights.
Treasury Stock
Treasury stock purchases are accounted for under the par value method, whereby the cost of the acquired stock is recorded at par value. During the year ended May 31, 2018, the Company purchased 159,011 shares of $0.001 par value treasury stock for shares tendered in satisfaction of income tax withholding, in connection with incentive compensation paid to certain officers in the form of common stock.
Debt Discount
During the years ended May 31, 2018, May 31, 2017 and May 31, 2016, the Company incurred approximately $ 1.5 million, $92,000 and $-0-, respectively, of debt discount related to the issuance of short-term convertible promissory notes issued with detachable warrants, as described in Note 4. The discount was amortized over the life of the convertible promissory notes and the Company recognized approximately $1.6 million, $-0- and $1.9 million, of related amortization expense for the years ended May 31, 2018, May 31, 2017 and May 31, 2016, respectively.
Debt Issuance Costs
During the year ended May 31, 2018, the Company incurred direct costs associated with the issuance of short-term convertible promissory notes, as described in Note 4, and recorded approximately $0.4 million of debt issuance costs. The Company recognized approximately $0.4 million, -0- and $0.6 million of related amortization expense for the year ended May 31, 2018, May 31, 2017 and May 31, 2016, respectively.
Offering Costs
During the years ended May 31, 2018, May 31, 2017 and May 31, 2016, the Company incurred approximately $3.5 million, $1.8 million and $3.9 million respectively, in direct incremental costs associated with the sale of equity securities. The offering costs were recorded as a component of equity upon receipt of the proceeds, as fully described in Notes 10 and 11.
Stock for Services
The Company periodically issues warrants to consultants for various services. The Black-Scholes option pricing model, as described more fully above, is utilized to measure the fair value of the equity instruments on the date of issuance. The Company recognizes the compensation expense associated with the equity instruments over the requisite service or vesting period.
Loss per Common Share
Basic loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted loss per share would include the weighted average common shares outstanding and potentially dilutive common share equivalents. Because of the net losses for all periods presented, the basic and diluted weighted average shares outstanding are the same since including the additional shares would have an anti-dilutive effect on the loss per share. For this reason, common stock options and warrants to purchase 132,385,269; 77,859,626 and 63,307,150 shares of common stock were not included in the computation of basic and diluted weighted average common shares outstanding for the years ended May 31, 2018, May 31, 2017 and May 31, 2016, respectively. As of May 31, 2018 and May 31, 2017 shares of Series B convertible preferred stock in the aggregate of 92,100 shares can potentially convert into 921,000 shares of common stock.
Income Taxes
Deferred taxes are provided on the asset and liability method, whereby deferred tax assets are recognized for deductible temporary differences and operating loss and tax credit carry forwards and deferred tax liabilities are recognized for taxable temporary differences. Temporary differences are the differences between the reported amounts of assets and liabilities and their tax bases. Future tax benefits for net operating loss carry forwards are recognized to the extent that realization of these benefits is considered more likely than not. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized.
The Company follows the provisions of FASB ASC 740-10 Uncertainty in Income Taxes (“ASC 740-10”). A reconciliation of the beginning and ending amount of unrecognized tax benefits has not been provided since there are no unrecognized benefits for all periods presented. The Company has not recognized interest expense or penalties as a result of the implementation of ASC 740-10. If there were an unrecognized tax benefit, the Company would recognize interest accrued related to unrecognized tax benefit in interest expense and penalties in operating expenses.
50
The Tax Cuts and Jobs Act (the “Act”) was enacted on December 22, 2017. The Act reduces the U.S. federal corporate tax rate from 35% to 21% effective as of January 1, 2018. In accordance with Section 15 of the Internal Revenue Code, we utilized a blended rate of 28.62% for our fiscal 2018 tax year, by applying a prorated percentage of the number of days prior to and subsequent to the January 1, 2018 effective date. For the fiscal year ended May 31, 2018 and May 31, 2017, we recorded provisional charges for the re-measurement of the deferred tax assets and reduced our deferred taxes before the valuation allowance by $17,497,051 to our income tax expense. The net tax expense for the year ended May 31, 2018, is zero, due to the reduction in the deferred tax valuation allowance. The Company has a full valuation allowance as of May 31, 2018, as management does not consider it more than likely than not that the benefits from the deferred taxes will be realized.
Note 3 – Recent Accounting Pronouncements
Recent accounting pronouncements, other than below, issued by the Financial Accounting Standards Board(“ FASB”) (including its EITF), the AICPA and the SEC did not or are not believed by management to have a material effect on the Company’s present or future financial statements.
In July 2017, the FASB issued Accounting Standards Update (“ASU”) No. 2017-11, Earnings Per Share (Topic 260), Distinguishing Liabilities from Equity (Topic 480), Derivatives and Hedging (Topic 815). The amendments in Part I of this Update change the classification analysis of certain equity-linked financial instruments (or embedded features) with down round features. When determining whether certain financial instruments should be classified as liabilities or equity instruments, a down round feature no longer precludes equity classification when assessing whether the instrument is indexed to an entity’s own stock. The amendments also clarify existing disclosure requirements for equity-classified instruments. As a result, a freestanding equity-linked financial instrument (or embedded conversion option) no longer would be accounted for as a derivative liability at fair value as a result of the existence of a down round feature. For freestanding equity classified financial instruments, the amendments require entities that present earnings per share (EPS) in accordance with Topic 260 to recognize the effect of the down round feature when it is triggered. That effect is treated as a dividend and as a reduction of income available to common shareholders in basic EPS. Convertible instruments with embedded conversion options that have down round features are now subject to the specialized guidance for contingent beneficial conversion features (in Subtopic 470-20, Debt—Debt with Conversion and Other Options), including related EPS guidance (in Topic 260). The amendments in Part II of this Update recharacterize the indefinite deferral of certain provisions of Topic 480 that now are presented as pending content in the Codification, to a scope exception. Those amendments do not have an accounting effect. For public business entities, the amendments in Part I of this Update are effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018. Early adoption is permitted for all entities, including adoption in an interim period. If an entity early adopts the amendments in an interim period, any adjustments should be reflected as of the beginning of the fiscal year that includes that interim period. Management is currently assessing the impact the adoption of ASU 2017-11 will have on the Company’s Consolidated Financial Statements.
In May 2017, the FASB issued ASU 2017-09, Compensation-Stock Compensation (Topic 718), Scope of Modification Accounting. The amendments in this Update provide guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. The amendments in this Update are effective for all entities for annual periods, and interim periods within those annual periods, beginning after December 15, 2017. Early adoption is permitted, including adoption in any interim period, for public business entities for reporting periods for which financial statements have not yet been issued. Management is currently assessing the impact the adoption of ASU 2017-09 will have on the Company’s Consolidated Financial Statements.
Note 4 – Convertible Instruments
Series B Convertible Preferred Stock
During fiscal 2010, the Company issued 400,000 shares of Series B, $0.001 par value Convertible Preferred Stock (“Series B”) at $5.00 per share for cash proceeds totaling $2,009,000, of which 92,100 shares remain outstanding at May 31, 2018. Each share of the Series B is convertible into ten shares of the Company’s $0.001 par value common stock, including any accrued dividends, with an effective fixed conversion price of $0.50 per share. The holders of the Series B can only convert their shares to common shares provided the Company has sufficient authorized common shares at the time of conversion. Accordingly, the conversion option was contingent upon the Company increasing its authorized common shares, which occurred in April 2010, when the Company’s stockholders approved an increase in the authorized shares of common stock to 100,000,000. At the commitment date, which occurred upon such stockholder approval, the conversion option related to the Series B was beneficial. The intrinsic value of the conversion option at the commitment date resulted in a constructive dividend to the Series B holders of approximately $6,000,000. The constructive dividend increased and decreased additional paid-in capital by identical amounts. The Series B has liquidation preferences over the common shares at $5.00 per share plus any accrued dividends. Dividends are payable to the Series B holders when declared by the board of directors at the rate of $0.25 per share per annum. Such dividends are cumulative and accrue whether or not declared and whether or not there are any profits, surplus or other funds or assets of the Company legally available. The Series B holders have no voting rights. As of May 31, 2018 the undeclared, accrued dividends were approximately $199,000 or 387,000 shares of common stock.
51
AVCP Convertible Notes
During the year ended May 31, 2015, the Company issued a two-year term unsecured convertible promissory note (the “AVCP Two-Year Note”) in the aggregate principal amount of $2,000,000 to Alpha Venture Capital Partners, L.P. (“AVCP”), an affiliate of one of the Company’s directors as described under Note 15 below. As described in greater detail below, along with the AVCP Bridge Note, the AVCP Two-Year Note has subsequently been converted in a transaction occurring during the year ended May 31, 2016. The AVCP Two-year Note bore simple interest at the annual rate of 5%, payable quarterly. The principal balance of the AVCP Two-Year Note was due and payable in full on September 26, 2016. The principal amount of the AVCP Two-Year Note plus unpaid accrued interest was convertible at the election of the holder into shares of the Company’s common stock at any time prior to maturity at an initial conversion price of $1.00 per share. The conversion price was subject to adjustment on the same terms, and contained similar consent rights to the issuance of additional indebtedness, as the AVCP Bridge Note below. During the year ended May 31, 2015, the Company issued a three-month unsecured convertible promissory note (the “AVCP Bridge Note” and together with the AVCP Two-Year Note, the “AVCP Convertible Notes”) in the aggregate principal amount of $1,500,000 to AVCP. The AVCP Bridge Note, along with the AVCP Two-Year Note, were subsequently converted in a transaction occurring during the year ended May 31, 2016. The principal amount of the AVCP Bridge Note plus unpaid accrued interest was convertible into shares of the Company’s common stock prior to maturity at an initial conversion price of $1.00 per share. The AVCP Bridge Note bore simple interest of 1.2% per month, payable at maturity on May 5, 2015, and monthly thereafter, upon the Company’s election to exercise a one-time option to extend the maturity by an additional three months, which the Company exercised on April 1, 2015 (extending the maturity date to August 5, 2015). The conversion price was subject to (i) adjustment for stock splits and similar corporate events and (ii) reduction to a price per share that is 10% below the lowest sale price that is below $.9444 per share, for shares of common stock sold or deemed sold in future securities offerings, including sales to AVCP and its designees subject to certain exempt transactions. Without AVCP’s prior written consent, the Company was not permitted to incur additional indebtedness for borrowed money, other than up to an additional $6.0 million in convertible promissory notes that may be issued to AVCP or related parties, unless such indebtedness was subordinated in right of payment to the Company’s obligations under the AVCP Bridge Note and any additional notes issued to AVCP or related parties.
As a result of the private placement of approximately $4.0 million in convertible notes during the fourth quarter of fiscal year ended May 31, 2015, as described below, the conversion price of the AVCP Convertible Notes was reduced to $0.675 per share of common stock, which was 90% of the weighted-average price of the deemed issued shares of $0.75 related to the approximately $4 million offering of 2015 Short Term Convertible Notes described below. The decrease in the conversion price caused the number of shares of common stock issuable upon conversion of the AVCP Convertible Notes to increase from 3,500,000 to 5,185,185 shares of common stock. The Company accounted for the AVCP Convertible Notes and related warrants, fully described below, as a financing transaction, wherein proceeds were allocated to the financial instruments issued. Prior to making the accounting allocation, the AVCP Convertible Notes and warrants were evaluated for proper classification under ASC 480 and ASC 815. The debt discounts associated with the notes were amortized over the term of the notes and the Company recognized approximately $94,000 in non-cash amortization expense for the period ended May 31, 2016. ASC 815 generally requires embedded terms and features that have characteristics of derivatives to be evaluated for bifurcation and separate accounting in instances where their economic risks and characteristics are not clearly and closely related to the risks of the host contract. The embedded derivative features consisted of the conversion price being subject to (i) adjustment for stock splits and similar corporate events and (ii) reduction to a conversion price per share that is 10% below the lowest sale price that is below $0.9444 per share for common stock sold or deemed sold in future securities offerings, subject to certain exempt transactions. The note conversion round down (or anti-dilution) provision terms were not consistent with the definition for financial instruments indexed to the Company’s stock. As such, the conversion option and conversion reset price protection in the AVCP Convertible Notes required bifurcation as a derivative liability. In connection with the original issuance of the two AVCP Convertible Notes, the Company issued warrants to AVCP covering 250,000 and 75,000 shares of the Company’s common stock exercisable at a price of $0.50 per share on September 26, 2014 and February 6, 2015, respectively. The warrants are currently exercisable in full, include a cashless exercise feature, and will expire on December 31, 2019 and February 29, 2020, respectively. The aforementioned warrants have a term of five years from inception and an exercise price of $0.50 per share and meet the conditions for equity classification per ASC 815. The fair value of the warrants was determined using a Black-Scholes option model using the following assumptions:
| Warrants issued on | Warrants issued on | |||||||
| September 26, 2014 | February 6, 2015 | |||||||
| Risk free interest rate |
1.82 | % | 1.48 | % | ||||
| Expected life |
5 years | 5 years | ||||||
| Expected volatility |
136 | % | 119 | % | ||||
| Dividend yield |
0.00 | % | 0.00 | % | ||||
52
Based on the previous conclusions, the Company allocated the cash proceeds first to the derivative liability at its fair value and then to the warrants at their relative fair value, with the residual allocated to the host AVCP Convertible Notes as presented below. On June 23, 2015, the Company, Alpha Venture Capital Management, LLC and AVCP entered into a Debt Conversion and Termination Agreement pursuant to which (i) AVCP agreed to convert the $3,535,627 in aggregate indebtedness as of June 23, 2015 under the AVCP Convertible Notes in exchange for 5,237,966 shares of the Company’s common stock; (ii) subject to the conversion of the two AVCP Convertible Notes, the Company agreed to issue AVCP an additional five-year warrant covering 1,000,000 shares of common stock at an exercise price of $0.675 per share and (iii) subject to the AVCP’s receipt of the common shares and warrant, the parties agreed to (a) terminate the subscription agreements; and (b) release and discharge each other party from all claims and obligations arising under the two AVCP Convertible Notes and subscription agreements. As a result of the debt conversion, the Company recognized a loss on extinguishment of the AVCP Convertible Notes of $584,177, a non-cash gain on the change in the fair value of the derivative liability of approximately $647,000 and non-cash inducement interest expense of approximately $758,000 arising from the aforementioned warrant.
| Year Ended May 31, 2016 | ||||||||||||||||||||
| May 31, 2015 | Debt Discount | Fair Value | Conversion | May 31, 2016 | ||||||||||||||||
| AVCP Convertible notes payable |
$ | 2,637,618 | $ | 94,344 | $ | — | $ | (2,731,962 | ) | $ | — | |||||||||
| Compound embedded derivative |
2,008,907 | — | (646,505 | ) | (1,362,402 | ) | — | |||||||||||||
| Warrants (equity allocation) |
215,732 | — | — | — | — | |||||||||||||||
| Accrued interest on notes payable |
— | — | — | (35,627 | ) | — | ||||||||||||||
| Fair Value of Common Stock Issued |
— | — | — | 4,714,168 | — | |||||||||||||||
| Loss on Conversion |
— | — | — | (584,177 | ) | — | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| $ | 4,862,257 | $ | 94,344 | $ | (646,505 | ) | $ | — | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
2015 Short-Term Convertible Notes
During the year ended May 31, 2015, the Company issued approximately $4.0 million of six-month unsecured convertible promissory notes (the “2015 Notes”) and related warrants to investors for cash. Each 2015 Note was originally convertible, at the election of the holder, at any time into common shares at a $0.75 per share. The 2015 Notes bore interest of 7% per annum, payable in cash upon maturity. In connection with the issuance of the 2015 Notes, the Company also issued warrants with a five-year term to purchase a total of 1,061,586 shares of common stock at an exercise price of $0.75. The Company determined the fair value of the warrants using the Black-Scholes option pricing model utilizing certain weighted-average assumptions, such as expected stock price volatility, term of the warrants, risk-free interest rate and expected dividend yield at the commitment date.
The Company utilized the following weighted-average assumptions to value the above investor warrants:
| 2015 | ||
| Expected dividend yield |
0% | |
| Stock price volatility |
88.79% | |
| Expected term |
5 year | |
| Risk-free interest rate |
1.46%-1.58% | |
| Grant-date fair value |
$0.52-$0.76 |
Additionally, at the commitment date, the Company determined that the conversion feature related to the 2015 Notes was beneficial to the investors. As a result, the Company determined the intrinsic value of the beneficial conversion feature utilizing the fair value of the underlying common stock at the commitment date and the effective conversion price after discounting the 215 Notes for the fair value of the warrants. The fair value of the warrants and the intrinsic value of the conversion feature were recorded as a debt discounts to the 2015 Notes, and a corresponding increase to additional paid-in capital. The debt discounts were amortized over the life of the 2015 Notes. The Company recognized approximately $ 1,784,000 as interest expense related to the amortization of the debt during the year ended May 31, 2016. There were no 2015 Notes outstanding at May 31, 2016. The unamortized discounts were fully amortized upon conversion of the 2015 Notes before maturity. During the year ended May 31, 2016, the Company tendered an offer to settle the balances of the 2015 Notes. The Company offered to exchange the 2015 Notes for (i) the issuance of restricted shares of common stock, for the settlement of the balance of the 2015 Notes, principal and accrued but unpaid interest as of September 21, 2015, which was the commitment date, at a conversion price of $0.675 per share, and (ii) the amendment of the related warrants to reduce the exercise price to $0.675 per share. The offer represented a 10.0% discount to $0.75, which was the current conversion price of the 2015 Notes and current exercise price of the related warrants. On September 21, 2015, the offering period and withdrawal rights
53
for the exchange offer expired, and the Company completed the exchange offer for approximately $2.7 million in aggregate original principal amount of 2015 Notes. Following the consummation of the exchange offer described above, an aggregate principal amount of $525,000 and accrued but unpaid interest of $17,830 converted into 723,773 shares of common stock. The principal and interest for 2015 Notes that were not exchanged in the exchange offer, or that are not otherwise converted pursuant to their terms, became due and payable between October 30, 2015 and November 15, 2015, six months from their issuance. The Company repaid the remaining aggregate principal and interest of approximately $789,000 on the 2015 Notes on their respective maturity dates. Related to the tender offer conversions, the Company recognized approximately $330,000 in non-cash interest expense and approximately $108,000 commission expense to assist the Company in conversion of the debt at the commitment date.
Activity related to the 2015 Notes for fiscal year ended May 31, 2016 was as follows:
| May 31, 2016 | ||||
| Face amount of Notes |
$ | 3,981,050 | ||
|
|
|
|||
| Unamortized discount |
— | |||
| Tender offer conversions |
(2,693,800 | ) | ||
| Conversions |
(525,000 | ) | ||
| Payments upon maturity |
(762,250 | ) | ||
|
|
|
|||
| Total carrying value of Notes |
$ | — | ||
|
|
|
|||
2017 Short-Term Convertible Notes
During the year ended May 31, 2017, the Company issued $1.15 million of unsecured convertible promissory notes (the “2017 Notes”), with a maturity date of January 31, 2018, and related warrants to investors for cash. The principal amount of the 2017 Notes, including any accrued but unpaid interest thereon, was convertible at the election of the holder at any time into shares of common shares at any time prior to maturity at a conversion price of $0.75 per share. The 2017 Notes bore simple interest at the annual rate of 7%. Principal and accrued interest, to the extent not previously paid or converted, was due and payable on the maturity date.
On June 14, 2017, the Company’s Board of Directors approved a modification in the warrant terms issued in connection with the 2017 Notes. The warrant coverage was increased from 25% to 50% and the exercise price of the warrant was reduced from $1.35 to $1.00 per share. On June 19, 2017, in connection with the new terms, the Company issued an incremental 383,333 warrant shares to these previous 2017 Note holders.
During the year May 31, 2018, the Company issued approximately $4.89 million in aggregate principal of additional 2017 Notes and related warrants, as described above. At the commitment dates, the Company determined that the conversion feature related to these 2017 Notes to be beneficial to the investors. As a result, the Company determined the intrinsic value of the beneficial conversion feature utilizing the fair value of the underlying common stock on the commitment dates and the effective conversion price after discounting the 2017 Notes for the fair value of the related warrants.
In connection with the sale of the 2017 Notes for the years ended May 31, 2018 and May 31, 2017, detachable common stock warrants to purchase a total of 4,025,656 common shares, with an exercise price of $1.00 per share and a five-year term were issued to the 2017 Note holders. The Company determined the fair value of the warrants at issuance using the Black-Scholes option pricing model utilizing certain weighted average assumptions, such as expected stock price volatility, expected term of the warrants, risk-free interest rates and expected dividend yield at the grant date.
| 2018 | 2017 | |||
| Expected dividend yield |
0% | 0% | ||
| Stock price volatility |
69.8% | 69% | ||
| Expected term |
5 year | 5 year | ||
| Risk-free interest rate |
1.77-1.93% | 1.75% | ||
| Grant-date fair value |
$0.30-$0.39 | $0.24 |
54
The fair value of the warrants, coupled with the beneficial conversion features, were recorded as a debt discount to the 2017 Notes and a corresponding increase to additional paid-in capital was amortized over the term of the 2017 Notes. The Company incurred debt discount of approximately $1.5 million and $92,000 during the years ended May 31, 2018 and May 31, 2017, respectively, related to the beneficial conversion feature and detachable warrants issued with the 2017 Notes. Accordingly, the Company recognized approximately $1.6 million and $-0- of non-cash debt discount during the year ended May 31, 2018 and May 31, 2017, respectively. In connection with the 2017 Notes, the Company incurred direct issuance costs of approximately $0.4 million during the year ended May 31, 2018. The issuance costs were amortized over the term of the 2017 Notes and accordingly, the Company recognized approximately $0.4 million of debt issuance costs during the year ended May 31, 2018.
On January 31, 2018, in connection with a registered direct equity offering, as fully described in Note 11, the 2017 Notes in an aggregate principal amount of $5,788,500, plus accrued unpaid interest of approximately $243,000 were sold for 12,062,728 shares of common stock. The 2017 Note investors also received warrants to purchase 7,718,010 shares of common stock. The securities were sold at a combined purchase price of $0.50 per share of common stock and related warrants, for aggregate gross proceeds to the Company of approximately $6.0 million. The Company repaid one 2017 Note, including accrued interest in the aggregate of approximately $259,000.
Activity related to the 2017 Notes was as follows:
| For the year ended May 31, | ||||||||
| 2018 | 2017 | |||||||
| Face amount of Notes |
$ | 6,038,500 | $ | 1,150,000 | ||||
| Unamortized discount |
— | (92,000 | ) | |||||
| Registered direct equity offering |
(5,788,500 | ) | — | |||||
| Note repayment |
(250,000 | ) | — | |||||
|
|
|
|
|
|||||
| Carrying value of Notes, net |
$ | — | $ | 1,058,000 | ||||
|
|
|
|
|
|||||
Note 5—Derivative Liability
The investor warrants issued with the September 2016 registered direct equity offering, and the placement agent warrants issued in conjunction with the offering, as fully described in Note 11, contain a provision for net cash settlement in the event that there is a fundamental transaction (contractually defined as a merger, sale of substantially all assets, tender offer or share exchange). If a fundamental transaction occurs in which the consideration issued consists principally of cash or stock in a successor entity, then the warrantholder has the option to receive cash, equal to the fair value of the remaining unexercised portion of the warrant. Due to this contingent cash settlement provision, the investor and placement agent warrants require liability classification as derivatives in accordance with ASC 480 and ASC 815 and are recorded at fair value.
The following tables summarize the fair value of the warrant derivative liability and related common shares as of inception date September 15, 2016, May 31, 2017 and May 31, 2018:
| Shares Indexed | Derivative Liability | |||||||
| Inception to date September 15, 2016 |
7,333,334 | $ | 5,179,200 | |||||
| Balance May 31, 2017 |
7,733,334 | 3,014,667 | ||||||
| Balance May 31, 2018 |
7,733,334 | $ | 1,323,732 | |||||
Changes in the fair value of the derivative liability are reported as “Change in fair value of derivative liability” in the Consolidated Statements of Operations. During the years ended May 31, 2018 and May 31, 2017, the Company recognized a net, non-cash gain of approximately $1.7 million and $2.2 million, respectively, due to the changes in the fair value of the liability associated with such classified warrants. In connection with the derivative liability, the Company recognized a non-cash interest expense of approximately $540,000 during the year ended May 31, 2017.
ASC 820 provides requirements for disclosure of liabilities that are measured at fair value on a recurring basis in periods subsequent to the initial recognition. Fair values for the warrants were determined using a Binomial Lattice Model.
55
The Company estimated the fair value of the warrant derivative liability as of inception, May 31, 2017 and May 31, 2018, using the following assumptions:
| September 15, 2016 |
May 31, 2017 |
May 31, 2018 |
||||||||||
| Fair value of underlying stock |
$ | 0.78 | $ | 0.60 | $ | 0.49 | ||||||
| Risk free rate |
1.20 | % | 1.71 | % | 2.63 | % | ||||||
| Expected term (in years) |
5.00 | 4.29 | 3.30 | |||||||||
| Stock price volatility |
106 | % | 94 | % | 64 | % | ||||||
| Expected dividend yield |
— | — | — | |||||||||
| Probability of Fundamental Transaction |
50 | % | 50 | % | 50 | % | ||||||
| Probability of holder requesting cash payment |
50 | % | 50 | % | 50 | % | ||||||
Due to the fundamental transaction provisions, which could provide for early redemption of the warrants, the model also considered subjective assumptions related to the fundamental transaction provision. The fair value of the warrants will be significantly influenced by the fair value of the Company’s stock price, stock price volatility, changes in interest and managements assumptions related to the fundamental transaction provision.
Note 6 – Stock Options and Warrants
The Company has one active stock-based equity plan at May 31, 2018, the CytoDyn Inc. 2012 Equity Incentive Plan (the “2012 Plan”) and one stock-based equity plan that is no longer active, but under which certain prior awards remain outstanding, the CytoDyn Inc. 2004 Stock Incentive Plan (the “2004 Plan” and, together with the 2012 Plan, the “Incentive Plans”). The 2012 Plan was approved by stockholders at the Company’s 2012 annual meeting to replace the 2004 Plan. The 2012 Plan was amended by stockholder approval in February 2015 to increase the number of shares available for issuance from 3,000,000 to 5,000,000 shares of common stock and in March 2016 to increase the number of shares available for issuance from 5,000,000 to 7,000,000 shares of common stock. At the annual meeting of stockholders held on August 24, 2017, the stockholders approved an amendment to the 2012 Plan to increase the number of shares available for issuance from 7,000,000 to 15,000,000 shares of common stock. As of May 31, 2018, the Company had 4,858,870 shares available for future stock-based grants under the 2012 Plan, as amended.
Stock Options
During the year ended May 31, 2018, the Company granted annual stock option awards to directors to purchase a total of 450,000 shares of common stock with an exercise price of $0.57 per share. These option awards vest quarterly over one year and have a ten-year term. The grant date fair value related to these options was $0.36 per share.
During the year ended May 31, 2018, the Company granted stock option awards to directors to purchase a total of 836,055 shares of common stock with an exercise price of $0.56 per share. The option awards were issued in lieu of accrued and unpaid cash board compensation for the previous quarters ended May 31, 2017, August 31, 2017, November 30, 2017 and February 28, 2018. The options awards fully vest upon grant, have a ten-year term and a grant date fair value of $0.31 per share.
During the year ended May 31, 2018, the Company granted to its Chief Science Officer a stock option award covering 600,000 shares of common stock with an exercise price of $0.57 per share. This option award vests annually over three years, has a ten-year term and a grant date fair value of $0.35 per share.
During the year ended May 31, 2018, the Company granted, to executive management and employees, stock options covering an aggregate of 800,000 shares of common stock, with exercise prices of $0.57 per share. The option awards vest annually over three years, have a ten-year term and grant date fair values of $0.35 per share.
During the year ended May 31, 2018, the Company issued replacement stock options, to executive management and directors, covering an aggregate of 1,050,000 shares of common stock. The replacement options retained the original exercise price of $0.80 per share and have a five-year term, to reflect the corrected term of approximately ten years from the original grant date. These options have a grant date fair value of $0.42 per share. In connection with the modification, the Company recognized non-cash stock based compensation of approximately $321,000.
During the year ended May 31, 2017, the Company’s Compensation Committee of the Board of Directors granted a time-based option covering 550,000 shares of common stock and a milestone-based option covering 450,000 shares of common stock to the Executive Chairman. The time-based option has an exercise price of $0.76 and a ten-year term. The option vests in equal monthly installments over the next two years and has a grant date fair value of $0.64 per share. The grant of the milestone-based option is conditioned on stockholder approval of the increase in the number of shares authorized for issuance under the 2012 Plan, as discussed above. The milestone-based option will not be exercisable unless and until approval of the share increase, for the 2012 Plan, as discussed above, is obtained from the stockholders. At that time the vesting will be contingent upon the achievement of certain strategic milestones specified in the option agreement.
56
During the year ended May 31, 2017, the Company granted annual stock option awards to directors to purchase a total of 300,000 shares of common stock with an exercise price of $1.09 per share. These option awards vest quarterly over one year and have a ten-year term. The grant date fair value related to these options was $0.78 per share. An additional stock option covering 100,000 shares of common stock was granted to a director. The option has an exercise price of $0.68 and vests 25% immediately with the remainder ratably over one year, has a ten-year term and grant date fair value of $0.53 per share. In April 2017, an option award was granted to the Company’s newly appointed director, subject to stockholder approval of the increase in the number of shares authorized for issuance under the 2012 Plan, in a pro-rata amount covering 7,123 shares of common stock with an exercise price of $0.61 per share. The option vested May 31, 2017 and has a ten-year term and grant date fair value of $0.36 per share.
During the year ended May 31, 2017, the Company granted options covering an aggregate of 1,050,000 shares of common stock to executive management and certain employees with exercise prices of $1.09 and $1.10 per share. The options vest annually over three years, have a ten-year term and grant date fair values of $0.75 and $0.76 per share, respectively.
Warrants
During the year ended May 31, 2018, the Company granted, to a consultant, a warrant covering an aggregate of 200,000 shares of common stock, with an exercise price of $0.64 per share. The warrant vests 25% upon grant date, 25% on December 31, 2017 and 50% upon achieving certain future milestones. The warrant has a five-year term and a grant date fair value of $0.26 per share.
During the year ended May 31, 2018, the Company granted to a consultant a warrant covering an aggregate of 100,000 shares of common stock, with an exercise price of $0.75 per share. The warrant vests immediately, has a five-year term and a grant date fair value of $0.29 per share.
During the year ended May 31, 2018, the Company granted to a consultant a warrant covering an aggregate of 50,000 shares of common stock, with an exercise price of $0.76 per share. The warrant vests immediately, has a five-year term and a grant date fair value of $0.26 per share.
During the year ended May 31, 2018, in connection with a private equity offering, as fully described in Note 10, the Company issued common stock warrants covering a total of 35,286,904 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $0.75 per share. In connection with this offering, the Company also issued common stock warrants covering 2,813,491 shares of common stock to the placement agent. The placement agent warrants have a five-year term and an exercise price of $0.55, and include a cashless exercise provision.
During the year ended May 31, 2018, the Company determined to extend the expiration dates of certain warrants from May 31, 2017 to June 30, 2017 covering 3,295,000 shares of common stock. The warrants were originally issued in connection with 2013 convertible promissory notes and had an exercise price of $1.00 per share. The extension to June 30, 2017 was contingent upon immediate exercise of the warrants at a reduced exercise price of $0.50 per share. The Company received proceeds of approximately $1.6 million and, pursuant to U.S. GAAP, the Company recognized non-cash inducement interest expense of approximately $0.8 million, which represented the incremental increase in the fair value of the extended warrants.
The Company determined the fair value of the warrant extension using the Black-Scholes option pricing model utilizing certain weighted-average assumptions, such as expected stock price volatility, term of the warrants, risk-free rate and expected dividend yield at date of exercise.
| 2017 | ||
| Expected dividend yield |
0% | |
| Stock price volatility |
61.48% | |
| Expected term |
1 month | |
| Risk-free interest rate |
0.84% | |
| Grant-date fair value |
$0.25 |
On January 23, 2018, in connection with a registered direct equity offering, as fully described in Note 11, the Company issued warrants covering 3,071,014 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $0.75 per share. In connection with this offering, the Company also issued warrants covering 245,681 shares of common stock to the placement agent. The placement agent warrants have a five-year term and an exercise price of $0.55 per share, and include a cashless exercise provision.
57
On January 31, 2018, in connection with a registered direct equity offering, as fully described in Note 11, the Company issued warrants covering 7,718,010 shares of common stock to investors. The warrants have a five-year term and an exercise price of $0.75 per share.
On May 22, 2018, in connection with a registered direct equity offering, as fully described in Note 11, the Company issued warrants covering 4,640,000 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $0.75 per share. In connection with this offering, the Company also issued warrants covering 315,200 shares of common stock to the placement agent. The placement agent warrants have a five-year term and an exercise price of $0.55 per share, and include a cashless exercise provision.
On May 31, 2017, in connection with the sale of the 2017 Notes, as fully described in Note 4, the Company issued common stock warrants covering 383,333 shares of common stock to note holders. The warrants have a five-year term and an exercise price of $1.35 per share.
On June 14, 2017, the Company’s Board of Directors approved a modification in the warrant terms issued in connection with the 2017 Notes, as fully described in Note 4. The warrant coverage ratio was increased from 25% to 50% and the per share exercise price of the warrant was reduced to $1.00 from $1.35. On June 19, 2017, in connection with new terms, the Company issued incremental warrants covering 383,333 shares to certain 2017 Note holders whose investment was completed on May 31, 2017.
During the year ended May 31, 2018, in connection with the issuance of the 2017 Notes, as fully described in Note 4, and more fully described in Note 11 below, the Company issued common stock warrants, covering 3,258,990 shares of common stock to additional 2017 Note holders. The warrants have a five-year term and an exercise price of $1.00 per share. In connection with the 2017 Notes, the Company issued warrants covering 350,766 to the placement agent. The placement agent warrants have a five-year term and an exercise price of $0.825, and include a cashless exercise provision.
In connection with the January 31, 2018, registered direct offering, as fully described below in Note 11, the exercise price of all detachable warrants issued with the 2017 Notes described in Note 4, was reduced further to $0.75 per share. As a result of this modification, the Company recognized non-cash inducement interest expense of approximately $2.4 million.
On September 8, 2017, in connection with a registered direct equity offering, as fully described in Note 11, the Company issued common stock warrants covering 1,668,163 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $1.00 per share. In connection with this offering, the Company also issued common stock warrants covering 213,573 shares of common stock to the placement agent. The placement agent warrants have a five-year term and an exercise price of $0.825 per share, and include a cashless provision. In connection with the Make-Whole Offering, fully described in Note 10, the exercise price of the investor and placement agent warrants were reduced to $0.75 and $0.715 per share, respectively.
On October 11, 2017, in connection with a registered direct equity offering, as fully described in Note 11, the Company issued common stock warrants covering 940,380 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $0.75 per share. In connection with this offering, the Company also issued common stock warrants covering 150,461 shares of common stock to the placement agent. The placement agent warrants have a five-year term and an exercise price of $0.715 per share, and include a cashless exercise provision.
On November 24, 2017, the Company filed an “Offer to Amend and Exercise” (the “Offer”) certain warrants covering an aggregate of 51,090,113 shares of common stock, at a potentially reduced exercise price of $0.50 per share. The original exercise price on these certain warrants ranged from $0.50 to $1.35 per share and have expiration dates beginning October 2018 continuing through October 2022. The Offer was originally scheduled to expire December 22, 2017, but was subsequently extended three times to March 23, 2018. The Offer was subject to the completion of an election to participate and exercise by the holder, certain representations and warranties by the holder and remittance of exercise proceeds to the Company. Upon expiration of the Offer, warrants to purchase up to 3,027,263 shares of common stock were accepted for gross cash proceeds to the Company of approximately $1.5 million. Solicitation fees of approximately $73,000 were paid to the solicitating agent in the Offer. Pursuant to U.S. GAAP, the Company recognized non-cash inducement expense of approximately $0.4 million, due to the reduction in warrant exercise price, related to this Offering.
On November 30, 2017, in connection with a registered direct equity offering dated September 8, 2017, as fully described in Note 11, the Company issued incremental common stock warrants covering 251,504 shares of common stock to investors. The investor warrants have a five-year term from initial investment date, September 8, 2017, and an exercise price of $0.75 per share. In connection with this offering, the Company also issued common stock warrants covering 26,702 shares of common stock to the placement agent.
58
The placement agent warrants have a five-year term from September 8, 2017, and an exercise price of $0.715 per share, and include a cashless exercise provision.
In connection with a private equity offering completed in June 2016, as fully described in Note 10, the Company issued common stock warrants covering 182,375 shares of common stock to investors. The warrants have a five-year term and an exercise price of $1.35 per share.
During the year ended May 31, 2017, in connection with the December, January and February registered direct equity offerings, as fully described in Note 11, the Company issued common stock warrants covering 5,602,821 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $1.00 per share. In connection with these offerings, the Company also issued common stock warrants covering 576,451 shares of common stock to the placement agent. The placement agent warrants have a five-year term, an exercise price of $0.825 per share, and include a cashless exercise provision.
In January 2017, the Company determined to extend the expiration dates of certain warrants (“2013 Warrants”) to May 31, 2017, covering an aggregate of 6,310,667 shares of common stock. The 2013 Warrants were originally issued in connection with the sale of 2013 convertible promissory notes. The 2013 Warrants currently have an exercise price of $1.00 per share, and all but two warrants were exercisable through October 2016. One 2013 Warrant, for the purchase of 186,667 shares of common stock, was exercisable through December 2016 and one 2013 Warrant, for the purchase of 160,000 shares of common stock, was exercisable until January 15, 2017. The extension to May 31, 2017 was contingent upon the execution of a release of claims by each of the warrantholders, the delivery of the “form of exercise,” and the receipt of the exercise proceeds to the Company. Pursuant to U.S. GAAP the Company recognized non-cash interest expense of approximately $72,000, which represented the incremental increase in fair value of the extended warrants.
During the fiscal year ended May 31, 2016, the board of directors approved a one-year extension of expiration dates of the 2013 Warrants, further extended, as mentioned above. The 2013 Warrants, which had an original term of two years, covering approximately 6.3 million shares of common stock, with an exercise price of $1.00 per share. The first extension of expiration dates ranged from October 2015 through January 2016 and the second extension deferred the expiration dates to October 2016 through January 2017. The extensions were effective upon the receipt of certain executed documentation from the warrantholders. Pursuant to U.S. GAAP, the Company recognized non-cash interest expense in fiscal year ended May 31, 2016 of approximately $867,000, in connection with these extensions, which represented the incremental increase in the fair value of the modified warrants
During the year ended May 31, 2017, holders of warrants covering 774,097 shares of common stock exercised the right to purchase such shares at either $0.50 or $0.75 per share and the Company received proceeds of approximately $398,000. Additionally, warrants covering 138,864 shares with an exercise price of $0.75 per share were exercised pursuant to a cashless exercise provision.
In connection with a registered direct equity offering completed in September 2016, as fully described in Note 11, the Company issued common stock warrants covering 6,666,667 shares of common stock to investors. The investor warrants have a five-year term and an exercise price of $1.00 per share. In connection with this offering, the Company also issued common stock warrants covering 1,066,667 shares of common stock to the placement agent. The placement agent warrants have a five-year term, an exercise price of $0.825 per share, and include a cashless exercise provision.
Compensation expense related to stock options and warrants for the fiscal years ended May 31, 2018, May 31, 2017 and May 31, 2016 was approximately $1.3 million, $1.2 million and $2.4 million, respectively. The grant date fair value of options and warrants vested during the fiscal years ended May 31, 2018, May 31, 2017 and May 31, 2016, was approximately $1.4 million, $0.9 million and $1.7 million, respectively. As of May 31, 2018, there was approximately $0.6 million of unrecognized compensation expense related to share-based payments for unvested options, with is expected to be recognized over a weighted-average period of approximately 1.13 years.
59
The following table represents stock option and warrant activity for the years ended May 31, 2018, May 31, 2017 and May 31, 2016:
| Weighted | ||||||||||||||||
| Average | ||||||||||||||||
| Weighted | Remaining | |||||||||||||||
| Number of | Average | Contractual Life | Aggregate | |||||||||||||
| Shares | Exercise Price | in Years | Intrinsic Value | |||||||||||||
| Options and warrants outstanding—May 31, 2015 |
31,008,915 | $ | 0.88 | 2.94 | $ | 5,538,335 | ||||||||||
| Granted |
33,838,536 | 0.80 | — | — | ||||||||||||
| Exercised |
(1,050,301 | ) | 0.70 | — | — | |||||||||||
| Forfeited/expired/cancelled |
(490,000 | ) | 2.01 | — | — | |||||||||||
|
|
|
|||||||||||||||
| Options and warrants outstanding—May 31, 2016 |
63,307,150 | $ | 0.83 | 3.20 | $ | 9,863,492 | ||||||||||
|
|
|
|||||||||||||||
| Granted |
16,935,437 | 0.99 | — | — | ||||||||||||
| Exercised |
(912,961 | ) | 0.55 | — | — | |||||||||||
| Forfeited/expired/cancelled |
(1,470,000 | ) | 1.56 | — | — | |||||||||||
|
|
|
|||||||||||||||
| Options and warrants outstanding—May 31, 2017 |
77,859,626 | $ | 0.86 | 3.40 | $ | 40,250 | ||||||||||
|
|
|
|||||||||||||||
| Granted |
65,420,227 | 0.75 | — | — | ||||||||||||
| Exercised |
(6,322,263 | ) | 0.50 | — | — | |||||||||||
| Forfeited/expired/cancelled |
(4,572,321 | ) | 0.94 | — | — | |||||||||||
|
|
|
|||||||||||||||
| Options and warrants outstanding—May 31, 2018 |
132,385,269 | 0.80 | 3.78 | 3,673 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding exercisable—May 31, 2018 |
128,872,852 | $ | 0.80 | 3.66 | $ | 3,673 | ||||||||||
|
|
|
|||||||||||||||
Note 7 – Acquisition of patents
As discussed in Note 9 below, the Company consummated an asset purchase on October 16, 2012, and paid $3,500,000 for certain assets, including intellectual property, certain related licenses and sublicenses, FDA filings and various forms of the PRO 140 drug substance. The Company followed the guidance in Financial Accounting Standards Topic 805 to determine if the Company acquired a business. Based on the prescribed accounting, the Company acquired assets and not a business. As of May 31, 2018, the Company has recorded and is amortizing $3,500,000 of intangible assets in the form of patents. The Company estimates the acquired patents have an estimated life of ten years. Subsequent to the acquisition date, the Company has continued to expand, amend and file new patents central to its current clinical trial strategies, which, in turn, have extended the protection period for certain methods of using PRO 140 and formulations comprising PRO 140 out through at least 2031 and 2038, respectively, in various countries.
The following presents intangible assets activity:
| May 31, 2018 | May 31, 2017 | |||||||
| Gross carrying amounts |
$ | 3,500,000 | $ | 3,500,000 | ||||
| Accumulated amortization |
(1,968,846 | ) | (1,618,770 | ) | ||||
|
|
|
|
|
|||||
| Total amortizable intangible assets, net |
1,531,154 | 1,881,230 | ||||||
| Patents currently not amortized |
35,989 | 35,989 | ||||||
|
|
|
|
|
|||||
| Carrying value of intangibles, net |
$ | 1,567,143 | $ | 1,917,219 | ||||
|
|
|
|
|
|||||
Amortization expense related to intangible patents was approximately $350,000 for each of the fiscal years ended May 31, 2018, May 31, 2017 and May 31, 2016. The estimated aggregate future amortization expense related to the Company’s intangible assets with finite lives is estimated to be approximately $350,000 per year over the next four to five years.
Note 8 – License Agreements
The Company has a license agreement with a third-party licensor covering the licensor’s “system know-how” technology with respect to the Company’s use of proprietary cell lines to manufacture new PRO 140 material. The Company accrues an annual license fee of £300,000 (approximately US$400,000 utilizing current exchange rates), which is payable annually in December, except for the December 2017 payment, which was extended to March 15, 2018. Future annual license fees and royalty rate will vary depending on whether the Company manufactures PRO 140, utilizes the third-party licensor as a contract manufacturer, or utilizes an independent party as a contract manufacturer. The licensor does not charge an annual license fee when it serves as the manufacturer. In addition, the Company will incur royalties of up to 2% of net sales when the Company commences their first commercial sale, which will continue as long as the license agreement is maintained.
60
Note 9 – Commitments and Contingencies
Under the Progenics Purchase Agreement, the Company acquired rights to the HIV viral-entry inhibitor drug candidate PRO 140, a humanized anti-CCR5 monoclonal antibody, as well as certain other related assets, including the existing inventory of bulk PRO 140 drug product, intellectual property, certain related licenses and sublicenses, and FDA regulatory filings. In connection with purchase, the Company has two remaining milestone payments, (i) $0.5 million upon filing a BLA with the FDA or non-U.S. equivalent regulatory body and (ii) $5.0 million, which will become due at the time of the first U.S. new drug application approval by the FDA or other non-U.S. approval for the sale of PRO 140. In addition, the Company will incur royalty payments of up to 5% on net sales during the period beginning on the date of the first commercial sale of PRO 140 until the later of (a) the expiration of the last to expire patent included in the acquired assets, and (b) 10 years, in each case determined on a country-by country basis.
During the year ended May 31, 2016 the Company paid a milestone obligation of $1.5 million owed to Progenics as a result of the first dosing in a U.S. Phase 3 trial. To the extent that the remaining milestone payment and royalties are not timely made, under the terms of the Progenics Purchase Agreement, Progenics has certain repurchase rights relating to the assets sold to the Company thereunder. As of the date of this filing, it is management’s conclusion that the probability of achieving the subsequent future scientific research milestone is not reasonably determinable, thus the future milestone payments payable to Progenics and its sub-licensors are deemed contingent consideration and, therefore, are not currently accruable.
Payments to the third-party licensor and to Progenics are in addition to payments due under a Development and License Agreement, dated April 30, 1999 (the “PDL License”), between Protein Design Labs (now AbbVie Inc.) (“PDL”) and Progenics, which was assigned to the Company in the Progenics Purchase Agreement, pursuant to which the Company has an exclusive worldwide license to develop, make, have made, import, use, sell, offer to sell or have sold products that incorporate the humanized form of the PRO 140 antibody developed by PDL under the agreement the Company has paid various milestone obligations, with one remaining milestone payment of $0.5 million, which will become due upon FDA approval or approval by another non-U.S. equivalent regulatory body. In addition, the Company will incur royalties of up to 7.5% of net sales for the longer of 10 years and the date of expiration of the last to expire licensed patent. Additionally, the PDL License provides for an annual maintenance fee of $150,000 or until annual royalties paid exceed that amount. To the extent the remaining milestone payment and royalties are not timely made, under the terms of the PDL License, AbbVie Inc. has certain termination rights relating to the Company’s license of PRO 140 thereunder. As of the date of this filing, it is management’s conclusion that the probability of achieving the subsequent future scientific research milestones is not reasonably determinable, thus the future milestone payments payable to PDL, Progenics and its sub-licensors are deemed contingent consideration and, therefore, are not currently accruable.
The Company has entered into project work orders, as amended, for each of its CRO and related laboratory vendors. Under the terms of these agreements, the Company incurs execution fees for direct services costs, which are recorded as a current asset. In the event the Company were to terminate any trial, it may incur certain financial penalties which would become payable to the CRO. Conditioned upon the form of termination of any one trial, the financial penalties may range from an approximate low of $0.1 million to an approximate high of $1.1 million. In the remote circumstance that the Company would terminate all clinical trials, the collective financial penalties may range from an approximate low of $0.8 million to an approximate high of $2.4 million.
During the year ended May 31, 2017, the Company entered into agreements with contract manufacturing companies. Under the terms of the agreements, the Company incurred approximately $2.1 million of execution fees for process validation and manufacturing activities, of which the remaining $0.7 million is reflected as a current asset, as of May 31, 2018. In the event the Company were to terminate any of the agreements, it may incur certain financial penalties which would become payable to the manufacturers. Conditioned on the timing of termination, the financial penalties may range up to an approximate high of $3.2 million.
From time to time, the Company is involved in routine litigation that arises in the ordinary course of business. There are no pending significant legal proceedings to which the Company is a party for which management believes the ultimate outcome would have a material adverse effect on the Company’s financial position.
Note 10—Private Securities Offerings
During the year ended May 31, 2016, the Company conducted private equity offerings (the “Equity Offerings”), in which accredited investors purchased unregistered common stock at $0.75 and $1.00 per share with warrant coverage of 50% and 25%, respectively, based on the number of shares of common stock purchased. Pursuant to the Equity Offerings, the Company sold a total of 48,659,338 shares of common stock, $0.001 par value, for aggregate gross proceeds of approximately $37.6 million and issued five-year warrants covering 23,254,230 shares of common stock. In conjunction with the Equity Offerings, the Company paid an aggregate cash fee of approximately $3.9 million to the placement agent and issued warrants covering an aggregate of 4,960,314 shares of common stock to the placement agent as additional compensation. The placement agent warrants had aggregate Black-Scholes valuations of approximately $2.7 million at issuance.
61
During the year ended May 31, 2017, the Company conducted a private equity offering, in which accredited investors purchased unregistered common stock at $1.00 per share with warrant coverage ratio of 25%, based on the number of shares of common stock purchased. Pursuant to the offering, the Company sold a total of 729,500 shares of common stock, $0.001 par value, for aggregate gross proceeds of $729,500 and issued to the investors five-year warrants covering 182,375 shares of common stock with an exercise price of $1.35 per share.
In connection with the September 2017 Offering, as fully described below in Note 11, on November 30, 2017, the Company completed an offer and sale (the “Make-Whole Offering”) of an aggregate of 503,015 shares of Common Stock (the “Make-Whole Shares”) and warrants to purchase up to 251,504 shares of common stock (the “Make-Whole Warrants” and, collectively with the Make-Whole Shares, the “Make-Whole Securities”). The Make-Whole Securities issued were unregistered.
The Make-Whole Securities were offered pursuant to a form of Waiver and Subscription Agreement (the “Waiver and Subscription Agreement”). The Make-Whole Securities represent the difference in the numbers of shares of Common Stock and warrants that would have been sold to investors in the September 2017 Offering had the reduced purchase price of $0.65 per share of Common Stock and related Warrants in the October 2017 Offering, registered direct offering (as compared to $0.75 in the September 2017 Offering) and the reduced warrant exercise price of $0.75 in the October 2017 Offering (as compared to $1.00 in the September 2017 Offering) applied to the September 2017 Offering as well. The Make-Whole Securities were offered as consideration for the release of potential claims by participating investors. In connection with these arrangements, the exercise prices of any warrants previously sold in the September 2017 Offering to participating investors has also been reduced to $0.75 from $1.00. In addition, warrants previously issued to the placement agent (or its designees) in respect of participating investors have also been proportionately adjusted to reflect a reduced exercise price of $0.715 (as compared to $0.825 in the September 2017 Offering) and 26,702 additional shares.
During the year ended May 31, 2018, the Company conducted a private equity offering, in which accredited investors purchased unregistered common stock at $0.50 per share with warrant coverage ratio of 100%, based on the number of shares of common stock purchased. Pursuant to the offering, the Company sold a total of 35,286,904 shares of common stock for aggregate gross proceeds of $17.6 million and issued warrants covering an aggregate of 35,286,904 shares of common stock with a five-year term and an exercise price of $0.75 per share. In connection with the offering, the placement agent received a warrant covering 2,813,491 shares of common stock, with a five-year term, an exercise price of $0.55 per share, and include a cashless exercise provision.
In connection with the November 24, 2017, Offer to amend and exercise certain eligible warrants at a reduced exercise price of $0.50 per share of common stock, as fully described in Note 6 above, on March 23, 2018, the Company issued 2,470,585 shares of common stock to warrant holders who participated in the Offer, in exchange for their eligible warrants, in a private securities offering.
Note 11—Registered Direct Equity Offerings
In September 2016, the Company entered into securities purchase agreements with certain institutional investors for the sale of 13,333,334 shares of common stock at a purchase price of $0.75 per share in a registered direct equity offering (the “Registered Offering”), pursuant to a registration statement on Form S-3. The investors in this Registered Offering also received warrants to purchase 6,666,667 shares of common stock with a five-year term and an exercise price of $1.00 per share. The Company received net proceeds from the offering of approximately $9.0 million after placement fees of 8% of the gross proceeds and various expenses. In addition, the placement agent received warrants covering 1,066,667 shares (or 8% of total shares sold to investors) with a five-year term and an exercise price of $0.825 per share, and included a cashless exercise provision.
A summary of the cash proceeds of the offering is as follows:
| Gross proceeds from sale of common stock |
$ | 10,000,000 | ||
| Placement agent fees and expenses |
1,010,000 | |||
|
|
|
|||
| Total net proceeds |
$ | 8,990,000 | ||
|
|
|
As fully described in Note 5 above, the investor warrants and the placement agent warrants issued in connection with the Registered Offering are required to be accounted for in accordance with ASC 480 and ASC 815.
A summary of the ASC 480 allocation of the proceeds of the offering is as follows:
| Allocated to common stock and additional paid in capital |
$ | 6,334,417 | ||
| Allocated to warrant liabilities |
2,655,583 | |||
|
|
|
|||
| Total net proceeds |
$ | 8,990,000 | ||
|
|
|
62
Closing costs included 1,066,667 warrants valued at $819,200 for placement agent fees. Based upon the estimated fair value of the stock and warrants in the units, the Company allocated $241,986 to financing expense and $577,214 as stock issuance costs.
On December 12, 2016, the Company entered into securities purchase agreements with certain investors for the sale of 4,000,000 shares of common stock at a purchase price of $0.75 per share in a registered direct offering (the “December Offering”), pursuant to a registration statement on Form S-3. The investors in the December Offering also received warrants to purchase 2,000,000 shares of common stock with an exercise price of $1.00 per share and a five-year term. The Company received net proceeds from the December Offering of $3.0 million.
On January 31, 2017, the Company entered into subscription agreements with certain investors for the sale of 1,534,999 shares of common stock at a purchase price of $0.75 per share in a registered direct offering (the “January Offering”), pursuant to a registration statement on Form S-3. The investors in the January Offering also received warrants to purchase 767,498 shares of common stock with a five-year term and an exercise price of $1.00 per share. The Company received net proceeds from the January Offering of approximately $1.0 million after placement fees of 9% of the gross proceeds and various expenses. In addition, the placement agent received warrants covering 122,799 shares (or 8% of total shares sold to investors) with a five-year term and an exercise price of $0.825 per share, including a cashless exercise provision).
On February 28, 2017, the Company entered into subscription agreements with certain investors for the sale of 5,670,661 shares of common stock at a purchase price of $0.75 per share in a registered direct offering (the “February Offering”), pursuant to a registration statement on Form S-3. The investors in the February Offering also received warrants to purchase 2,835,323 shares of common stock with a five-year term and an exercise price of $1.00 per share term. The Company received net proceeds from the February Offering of approximately $3.8 million after placement fees of 9% of the gross proceeds and various expenses. In addition, the placement agent received warrants covering 453,652 shares (or 8% of total shares sold to investors) with a five-year term and an exercise price of $0.825 per share, including a cashless exercise provision.
On September 8, 2017, the Company entered into subscription agreements with certain investors for the sale of 3,336,331 shares of common stock at a purchase price of $0.75 per shares in a registered direct offering (the “September 2017 Offering), pursuant to a registration statement on Form S-3. The investors in this September 2017 Offering also received warrants to purchase 1,668,163 shares of common stock with a five-year term and an exercise price of $1.00 per share. The Company received net proceeds from the September 2017 Offering of approximately $2.3 million after placement fees of 9% of the gross proceeds and various expenses. In addition, the placement agent received warrants covering 213,573 shares (or 8% of total shares sold to investors) with a five-year term and an exercise price of $0.825 per share, including a cashless exercise provision. As fully described in Note 10 above, the Company completed the Make-Whole Offering, in which incremental shares of common stock and warrants were issued. Simultaneously, the exercise price of the investor and placement agent warrants related to the September 2017 Offering were reduced to $0.75 and $0.715 per share, respectively.
On October 11, 2017, the Company entered into subscription agreements with certain investors for the sale of 1,880,765 shares of common stock at a purchase price of $0.65 per shares in a registered direct offering (the “October 2017 Offering), pursuant to a registration statement on Form S-3. The investors in this October 2017 Offering also received warrants to purchase 940,380 shares of common stock with a five-year term and an exercise price of $0.75 per share. The Company received net proceeds from the October 2017 Offering of approximately $1.1 million. In addition, the placement agent received warrants covering 150,461 shares (or 8% of total shares sold to investors) with a five-year term and an exercise price of $0.715 per share, including a cashless exercise provision.
On January 23, 2018, the Company entered into subscription agreements with certain investors for the sale of 3,071,014 shares of common stock at a purchase price of $0.50 per shares in a registered direct offering (the “January 23 Offering”), pursuant to a registration statement on Form S-3. The investors in the January 23 Offering also received warrants to purchase 3,071,014 shares of common stock with an exercise price of $0.75 per share and a five-year term. The Company received net proceeds from the January 23 Offering of approximately $1.4 million. In addition, the placement agent received warrants covering 245,681shares of common stock (or 8% of total shares sold to investors) with a five-year term and an exercise price of $0.55 per share, including a cashless exercise provision.
On January 31, 2018, the Company entered into subscription agreements with certain investors who owned the 2017 Notes, more fully described in Note 4, for the sale by the Company of 12,062,728 shares of common stock in a registered direct offering (the “January 31 Offering”). The investors in the January 31 Offering also received warrants to purchase 7,718,010 shares of common stock. The securities were sold at a combined purchase price of $0.50 per share of common stock and related warrants, for aggregate gross proceeds to the Company of approximately $6.0 million. The 2017 Notes matured on January 31, 2018, upon which date the Company became obligated to pay the principal amount of approximately $6.0 million on the 2017 Notes, plus accrued but unpaid interest of approximately $0.3 million, for aggregate payment obligations at maturity of approximately $6.3 million. The common stock and warrants were issued in full satisfaction of approximately $6.0 million of such payment obligations, with one holder of an aggregate principal amount and accrued unpaid interest of approximately $0.3 million electing to be repaid in cash instead of participating in the January 31, 2018 Offering. As a result, all of the proceeds from the Offering were used to satisfy the Company’s payment obligations
63
pursuant to the 2017 Notes. The warrants will be exercisable for a period of five years commencing on their issuance date, at an exercise price of $0.75 per share of common stock, subject to certain ownership limitations and adjustments as provided under the terms of the warrants. The number of shares of common stock underlying the warrant issued to each investor was calculated as the difference between (x) the number of shares of common stock issued to each investor in the January 31, 2018 Offering in respect of the payment obligations relating solely to principal amounts on the 2017 Notes and (y) the number of shares of common stock underlying certain warrants originally issued to such investor in the original 2017 Note offering. The effect was to bring each investor from 50% warrant coverage in the original offering of Notes, assuming conversion of the principal amount thereof at an original conversion price of $0.75 per share, to 100% warrant coverage after the January 31, 2018 Offering, assuming reinvestment of the principal amount on the 2017 Notes at $0.50 per share. The warrants in the January 31, 2018 Offering, had an original exercise price of $1.00 per share, therefore, due to the reduction of exercise price to $0.75 per share, and the reduction in the conversion rate of the 2017 notes form $0.75 to $0.50 per share, the Company recognized a non-cash inducement interest expense of approximately $2.4 million due to these modifications. In connection with this offering, the Company paid a commission of $164,425 to the placement agent.
In connection with the November 24, 2017 Offer to amend and exercise certain eligible warrants at a reduced exercise price of $0.50 per share of common stock, as fully described in Note 6 above, on March 23, 2018, the Company issued 556,678 shares of common stock to warrant holders who participated in the Offer, in exchange for their eligible warrants, pursuant to an effective registration statement.
On May 22, 2018, the Company entered into subscription agreements with certain investors for the sale of 4,640,000 shares of common stock at a purchase price of $0.50 per shares in a registered direct offering (the “May 22 Offering”), pursuant to a registration statement on Form S-3. The investors in the May 22 Offering also received warrants to purchase 4,640,000 shares of common stock with a five-year term and an exercise price of $0.75 per share. The Company received net proceeds from the May 22 Offering of approximately $2.1 million. In addition, the placement agent received warrants covering 315,200 shares of common stock (or 8% of total shares sold to investors, for which they introduced to the May 22 Offering) with a five-year term and an exercise price of $0.55 per share, including a cashless exercise provision.
Note 12 – Employee Benefit Plan
The Company has an employee savings plan (the “Plan”) pursuant to Section 401(k) of the Internal Revenue Code (the “Code”), covering all of its employees. The Company makes a qualified non-elective contribution of 3%, which consequently vests immediately. In addition, participants in the Plan may contribute a percentage of their compensation, but not in excess of the maximum allowed under the Code. During the year ended May 31, 2018, May 31, 2017 and May 31, 2016, the Company incurred an expense of approximately $61,000, $40,300 and $22,000, respectively, for qualified non-elective contributions.
Note 13 – Income Taxes
Deferred taxes are recorded for all existing temporary differences in the Company’s assets and liabilities for income tax and financial reporting purposes. Due to the valuation allowance for deferred tax assets, as noted below, there was no net deferred tax benefit or expense for the periods ended May 31, 2018, May 31, 2017 and May 31, 2016.
Reconciliation of the federal statutory blended income tax rate of 28.6% for the year ended May 31, 2018 and the federal statutory rate of 34% for the years ended May 31, 2017 and May 31, 2016, to the effective income tax rate is as follows for all periods presented:
| 2018 | 2017 | 2016 | ||||||||||
| Income tax provision at statutory rate: |
28.6 | % | 34.0 | % | 34.0 | % | ||||||
| State income taxes net |
— | — | — | |||||||||
| Rate Change |
(34.8 | ) | — | — | ||||||||
| Loss on debt conversion |
— | — | (0.8 | ) | ||||||||
| Derivative gain/loss |
1.00 | 2.80 | 0.9 | |||||||||
| Non-deductible debt issuance costs |
(0.2 | ) | — | — | ||||||||
| Non-deductible interest on conversion |
(0.1 | ) | — | — | ||||||||
| Inducement charge |
(2.0 | ) | (1.00 | ) | (1.0 | ) | ||||||
| Other |
(1.1 | ) | — | — | ||||||||
| Miscellaneous |
(0.1 | ) | (0.10 | ) | (0.5 | ) | ||||||
| Current year credits generated |
4.4 | — | — | |||||||||
| Credit carry forward generated (recorded currently) |
4.1 | — | — | |||||||||
| Valuation allowance |
0.3 | (35.7 | ) | (32.9 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| 0.0 | % | 0.0 | % | 0.0 | % | |||||||
|
|
|
|
|
|
|
|||||||
64
Net deferred tax assets and liabilities are comprised of the following as of May 31, 2018 and 2017:
| 2018 | 2017 | |||||||
| Deferred tax asset (liability) non-current: |
||||||||
| Net Operating Loss |
$ | 29,230,279 | $ | 32,530,436 | ||||
| Credits |
4,260,470 | — | ||||||
| ASC 718 Expense on NQO’s |
2,916,585 | 4,284,246 | ||||||
| Charitable Contribution—Carry forward |
— | 25,500 | ||||||
| Accrued Expenses |
117,880 | 216,645 | ||||||
| Fixed Assets |
174 | 1,300 | ||||||
| Amortization |
139,875 | 186,772 | ||||||
| Capitalized Debt Issuance Costs |
— | 157,992 | ||||||
| Debt Discount |
— | (31,072 | ) | |||||
| Valuation allowance |
(36,665,263 | ) | (37,371,817 | ) | ||||
|
|
|
|
|
|||||
| $ | — | $ | — | |||||
|
|
|
|
|
|||||
| Noncurrent asset |
$ | 36,665,263 | $ | 37,371,817 | ||||
| Valuation Allowance |
(36,665,263 | ) | (37,371,817 | ) | ||||
|
|
|
|
|
|||||
| $ | — | $ | — | |||||
|
|
|
|
|
|||||
The income tax benefit for the period presented is offset by a valuation allowance established against deferred tax assets arising from operating losses and other temporary differences, the realization of which could not be considered more likely than not. In future periods, tax benefits and related tax deferred assets will be recognized when management considers realization of such amounts to be more likely than not.
At May 31, 2018, May 31, 2017 and May 31, 2016 the Company had available net operating loss carry forwards of approximately $139.2 million, $95.6 million and $69.1 million, respectively, which expire beginning in 2023.
The Company’s income tax returns remain subject to examination by all tax jurisdictions for tax years May 31, 2015 through 2017.
Note 14 – Quarterly Financial Information (Unaudited)
| Quarter Ended | ||||||||||||||||
| August 31, | November 30, | February 28, | May 31, | |||||||||||||
| Year ended May 31, 2018: |
||||||||||||||||
| Operating expenses |
$ | (9,807,001 | ) | $ | (10,775,051 | ) | $ | (14,140,507 | ) | $ | (11,196,754 | ) | ||||
| Other income (expense) |
(361,879 | ) | 830,022 | (740,404 | ) | 1,966,816 | ||||||||||
| Interest expense |
(1,459,393 | ) | (1,002,656 | ) | (3,069,189 | ) | (393,685 | ) | ||||||||
| Net (loss) |
(11,628,273 | ) | (10,947,685 | ) | (17,950,100 | ) | (9,623,623 | ) | ||||||||
| Basic and diluted loss per share |
$ | (0.08 | ) | $ | (0.07 | ) | $ | (0.10 | ) | $ | (0.04 | ) | ||||
| Year ended May 31, 2017: |
||||||||||||||||
| Operating expenses |
$ | (5,358,120 | ) | $ | (6,156,117 | ) | $ | (8,016,917 | ) | $ | (7,799,580 | ) | ||||
| Other income (expense) |
3,735 | 1,229,114 | (23,078 | ) | 969,929 | |||||||||||
| Interest expense |
— | (540,330 | ) | (72,437 | ) | — | ||||||||||
| Net (loss) |
(5,354,385 | ) | (5,467,333 | ) | (8,112,432 | ) | (6,829,651 | ) | ||||||||
| Basic and diluted loss per share |
$ | (0.04 | ) | $ | (0.04 | ) | $ | (0.06 | ) | $ | (0.05 | ) | ||||
| Year ended May 31, 2016: |
||||||||||||||||
| Operating expenses |
$ | (6,657,481 | ) | $ | (2,893,779 | ) | $ | (5,049,037 | ) | $ | (6,575,214 | ) | ||||
| Other income (expense) |
62,686 | 211 | 2,202 | 78,045 | ||||||||||||
| Interest expense |
(2,300,220 | ) | (2,371,025 | ) | — | — | ||||||||||
| Net (loss) |
(8,895,015 | ) | (5,264,593 | ) | (5,046,835 | ) | (6,497,169 | ) | ||||||||
| Basic and diluted loss per share |
$ | (0.12 | ) | $ | (0.06 | ) | $ | (0.05 | ) | $ | (0.04 | ) | ||||
65
Note 15 – Related Party Transactions
On September 26, 2014, the Company entered into a $2.0 million convertible promissory note with AVCP, as more fully described in Note 4 above. In October of 2014, Mr. Carl C. Dockery, the principal of AVCP, was appointed a director of the Company. On February 6, 2015, the Company entered into a second convertible promissory note in the aggregate principal amount of $1.5 million, as more fully described in Note 4 above. On June 23, 2015, these notes and accrued but unpaid interest were converted into shares of common stock. In connection with the Debt Conversion and Termination Agreement dated June 23, 2015, the Company issued to AVCP a warrant covering 1,000,000 shares of common stock, as more fully described in Note 4.
On May 31, 2017, Anthony D. Caracciolo, Executive Chairman of the Company, participated in the private placement of 2017 Notes, as fully described in Note 4 above. Mr. Caracciolo purchased a promissory note, bearing interest of 7%, for $1,000,000 and received a warrant covering 333,333 shares of common stock at an exercise price of $1.35. The exercise price was subsequently reduced to $1.00 per share, as further described in Note 4 above. The terms and conditions of Mr. Caracciolo’s investment was identical to those offered to all other investors in the offering and his investment was approved by the Audit Committee of the Board of Directors.
On July 26, 2017, Jordan G. Naydenov, a director with the Company, participated in the private placement of 2017 Notes, as fully described in Note 4 above. Mr. Naydenov purchased a promissory note, bearing interest of 7%, for $100,000 in aggregate principal and received a warrant covering 66,666 shares of common stock at an exercise price of $1.00. The terms and conditions of Mr. Naydenov’s investment were identical to those offered to all other investors in the offering and his investment was approved by the Audit Committee of the Board of Directors.
On July 28, 2017, AVCP, participated in the private placement of 2017 Notes, as fully described in Note 4 above. Carl C. Dockery, the principal of AVCP, is a director of the Company. AVCP purchased a promissory note, bearing interest of 7%, for $50,000 in aggregate principal and received a warrant covering 33,333 shares of common stock at an exercise price of $1.00. The terms and conditions of the AVCP investment were identical to those offered to all other investors in the offering and his investment was approved by the Audit Committee of the Board of Directors.
On November 8, 2017, in connection with a private equity offering, a limited liability company in which Anthony D. Caracciolo, Executive Chairman of the Company, holds a partial ownership interest, purchased $100,000 of common stock and warrants on terms identical to those applicable to the other investors in the private equity offering.
On January 31, 2018 each of Mr. Caracciolo, Mr. Naydenov and AVCP participated with other investors in the offering of common stock and warrants in satisfaction of the payment obligations relating to the 2017 Notes, as fully described in Note 11 above.
The Audit Committee of the Board of Directors, comprised of independent directors, reviews and approves all related party transactions. The above terms and amounts are not necessarily indicative of the terms and amounts that would have been incurred had comparable transactions been entered into with independent parties.
Note 16 – Subsequent Events
On June 7, 2018, at a special meeting of stockholders, a proposal was approved to increase the total number of authorized shares of common stock of the Company from 375,000,000 to 450,000,000.
On June 8, 2018, the Company issued to directors, in connection with their annual compensation, stock options covering 600,000 shares of common stock. The stock options have an exercise price of $0.49, a ten-year term and vest quarterly over one year. These awards reflect an increase in the annual non-employee director stock option award from 75,000 to 100,000 shares per year effective for fiscal year 2019, which begins June 1, 2018. Additionally, in conjunction with annual incentive compensation, the Company issued stock options covering 1,825,000 shares of common stock to management and employees. The stock options have an exercise price of $0.49, a ten-year term and 875,000 shares vest annually over three years and 950,000 shares vest ratably over twenty four months.
On June 15, 2018, the Company entered into subscription agreements with certain investors for the sale of 1,970,000 shares of common stock at a purchase price of $0.50 per shares in a registered direct offering, pursuant to a registration statement on Form S-3. The investors in the offering also received warrants to purchase 1,970,000 shares of common stock with an exercise price of $0.75 per share and a five-year term. The Company received net proceeds from the offering of approximately $0.9 million. In addition, the placement agent received warrants covering 133,600 shares of common stock (or 8% of total shares sold to investors) with a per share exercise price of $0.55, a five-year term and include a cashless exercise provision.
On June 26, 2018, the Company entered into a securities purchase agreement, pursuant to which the Company issued a convertible promissory note (the “2018 Note”) with a two-year term to an institutional accredited investor in initial principal amount of $5.7 million. The investor gave consideration of $5.0 million, reflecting original issue discount of $0.6 million and expenses payable by the Company of $0.1 million.
66
The 2018 Note is the general unsecured obligation of the Company and ranks pari passu with all other creditors of the Company. Interest accrues on the outstanding balance of the 2018 Note at 10% per annum. Upon the occurrence of an event of default, interest accrues at the lesser of 22% per annum or the maximum rate permitted by applicable law. The 2018 Note contains customary default provisions, including provisions for potential acceleration. The investor may convert all or any part the outstanding balance of the 2018 Note into shares of common stock, par value $0.001 per share, of the Company at an initial conversion price of $0.55 per share, at any time after six months from the issue date upon five trading days’ notice, subject to certain adjustments and ownership limitations specified in the 2018 Note. The investor may redeem any portion of the 2018 Note, at any time after six months from the issue date upon five trading days’ notice, subject to a maximum monthly redemption amount of $350,000. The Company may prepay the outstanding balance of the 2018 Note, in part or in full, at a 15% premium to par value, at any time upon five trading days’ notice.
On July 12, 2018, the Company announced certain leadership changes in connection with the strategic expansion and entry into certain cancer and immunologic indications. In connection with such leadership changes and effective July 11, 2018 Denis R. Burger, Ph.D. and A. Bruce Montgomery, M.D. resigned as members of the Company’s Board of Directors and Dr. Burger has also resigned as Chief Science Officer of the Company, which is not an executive officer position.
In connection with the resignations of Dr. Burger and Dr. Montgomery, on July 10, 2018, the Company’s Board of Directors approved a motion to accelerate all outstanding unvested stock options held by Dr. Burger and Dr. Montgomery, to vest immediately upon the effectiveness of their resignations and to retain the stock options’ exercise period through their respective expiration date. Stock options covering 500,000 shares held by Dr. Burger and stock options covering 100,000 shares held by Dr. Montgomery were subject to acceleration. The terms of the stock options remained otherwise unchanged.
Effective July 11, 2018, Anthony D. Caracciolo resigned as Executive Chairman of the Company, which is an executive officer position. Mr. Caracciolo will continue to serve as a member and the non-executive Chairman of the Company’s Board of Directors.
67
| Item 9. | Changes In and Disagreements with Accountants on Accounting and Financial Disclosure. |
None.
| Item 9A. | Controls and Procedures. |
Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and our Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, we have evaluated the effectiveness of our disclosure controls and procedures as of May 31, 2018. Based on that evaluation, our Chief Executive Officer and our Chief Financial Officer concluded that our disclosure controls and procedures were effective as of May 31, 2018.
Internal Control Over Financial Reporting.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed by, or under the supervision of, our Chief Executive Officer and our Chief Financial Officer and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes policies and procedures that (i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect our transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures of the Company’s assets are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of May 31, 2018. This evaluation was based on the framework established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “2013 COSO Framework”). Based upon that evaluation, our management concluded that our internal control over financial reporting was effective as of May 31, 2018.
Attestation Report of Independent Registered Public Accounting Firm
The effectiveness of our internal control over financial reporting has been audited by Warren Averett, LLC, an independent registered public accounting firm, as stated in their report, which appears herein.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting that have materially affected, or are reasonably likely to materially affect our internal control over financial reporting during the quarter ended May 31, 2018.
| Item 9B. | Other Information. |
None.
68
| Item 10. | Directors, Executive Officers and Corporate Governance. |
The information required by Item 10 relating to our directors, executive officers and corporate governance is incorporated herein by reference to our definitive proxy statement for the 2018 Annual Meeting of Stockholders, to be filed with the SEC within 120 days of the end of the Company’s fiscal year, May 31, 2018 (the “2018 Proxy Statement”).
We have adopted a Code of Ethics for our Senior Executive Officers (the Chief Executive Officer, Chief Financial Officer, Treasurer, and Secretary), as well as an Insider Trading Policy for the Company. Copies are available on our website at www.cytodyn.com.
| Item 11. | Executive Compensation. |
The information required by Item 11 relating to executive compensation is incorporated herein by reference to our 2018 Proxy Statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
The information required by Item 12 relating to security ownership of certain beneficial owners and management and related stockholders matters is incorporated herein by reference to our 2018 Proxy Statement.
| Item 13. | Certain Relationships and Related Transactions and Director Independence. |
The information required by Item 13 relating to certain relationships and related transactions and director independence is incorporated herein by reference to our 2018 Proxy Statement.
| Item 14. | Principal Accountant Fees and Services. |
The information required by Item 14 relating to principal accountant fees and services is incorporated herein by reference to our 2018 Proxy Statement.
| Item 15. | Exhibits and Financial Statement Schedules. |
The following are filed as part of this Annual Report on Form 10-K:
Consolidated Financial Statements
The Consolidated Financial Statements for the years ended May 31, 2018 and 2017 are included under Item 8 of this report.
Exhibits
Exhibits are listed in the Exhibit Index which appears immediately following the signature page of this report.
| Item 16. | Form 10-K Summary. |
None.
69
EXHIBIT INDEX
| Exhibit Number |
Description | |
| Certifications | ||
| 31.1 | Certification of Chief Executive Officer under Rule 13a-14(a). | |
| 31.2 | Certification of Chief Financial Officer under Rule 13a-14(a). | |
| 32** | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350. | |
| XBRL | ||
| 101.INS | XBRL Instance Document. | |
| 101.SCH | XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document. | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document. | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document. | |
| * | Management contract or compensatory plan or arrangement. |
| ** | Furnished herewith. |
Note: All exhibits incorporated by reference to filings other than registration statements are incorporated by reference to filings that have SEC File No. 000-49908.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: July 27 , 2018 |
CYTODYN INC. | |||||
| (Registrant) | ||||||
| By: | /s/ Nader Z. Pourhassan | |||||
| Nader Z. Pourhassan, Ph. D. | ||||||
| President and Chief Executive Officer | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated on July 2018.
Principal Executive Officer and Director:
| /s/ Nader Z. Pourhassan |
| Nader Z. Pourhassan, Ph. D. President and Chief Executive Officer, Director |
Principal Financial and Accounting Officer:
| /s/ Michael D. Mulholland |
| Michael D. Mulholland Chief Financial Officer, Treasurer and Corporate Secretary |
Remaining Directors:
| *
|
| Anthony D. Caracciolo |
| *
|
| Carl C. Dockery |
| *
|
| Gregory A. Gould |
| *
|
| Jordan G. Naydenov |
| *
|
| Scott A. Kelly, M.D. |
| * By |
/s/ Michael D. Mulholland | |
| Michael D. Mulholland | ||
| Attorney-In-Fact |