Attached files
| file | filename |
|---|---|
| EX-99.1 - EX-99.1 - Spero Therapeutics, Inc. | d698178dex991.htm |
| 8-K - 8-K - Spero Therapeutics, Inc. | d698178d8k.htm |
Exhibit 99.2
Additional information regarding interim data from the Company’s Phase 1 dose-selection clinical trial of SPR994
Based on our pre-investigational new drug application meeting with the U.S. Food and Drug Administration, or FDA, we initiated a Phase 1 pharmacokinetics and safety clinical trial of SPR994 in Australia in October 2017. We have reported positive interim data from the trial, as described below, and expect to report top-line data from the multiple-ascending dose, or MAD, portion of the trial in the third quarter of 2018. Following completion of this trial, we intend to request a pre-Phase 3 meeting with the FDA to confirm that no additional clinical trials or nonclinical studies are required prior to initiating a Phase 3 clinical trial. Subject to feedback from the FDA, and using know-how we have licensed from Meiji Seika Pharma Co. Ltd., or Meiji, we hope to obtain agreement on the clinical trial protocol in late 2018 and expect to submit an investigational new drug application and initiate the pivotal Phase 3 clinical trial of SPR994 for the treatment of complicated urinary tract infections, or cUTI, around year-end 2018. We believe that the data from this Phase 3 trial will form the basis for the clinical trial data package that we may submit to the FDA in support of an NDA.
The FDA has designated SPR994 as a Qualified Infectious Disease Product, or QIDP, for the treatment of cUTI, community-acquired bacterial pneumonia, or CABP, and diabetic foot infections, or DFI, under the Generating Antibiotics Incentives Now Act, which enables priority review for regulatory approval by the FDA. The QIDP designation for SPR994, however, does not guarantee a faster development process or ensure FDA approval. Further, if SPR994 is successfully developed and approved for the treatment of cUTI, CABP or DFI, the FDA’s QIDP designation for SPR994 should extend any non-patent exclusivity period awarded to SPR994 in the United States for five years, such as a five-year New Chemical Entity data exclusivity granted under The Drug Price Competition and Patent Term Restoration Act of 1984.
Our ongoing Phase 1 clinical trial of SPR994 is assessing the safety, tolerability and pharmacokinetics of orally administered SPR994, including the impact of utilizing immediate and sustained release formulations to optimize the pharmacokinetic profile of the drug. In addition, the impact of administration in the fed and fasted state has been evaluated. We expect to use data from the trial to refine a pharmacokinetic/pharmacodynamics model to establish an in vitro/in vivo relationship to support dose and schedule administration for our planned pivotal Phase 3 clinical trial based on drug concentration and inter-patient variability.
To date, the trial has enrolled 115 healthy adult volunteers into 14 single-ascending dose, or SAD, cohorts and one MAD cohort evaluating an immediate-release and various extended-release formulations of SPR994 at single oral doses ranging from 100 mg to 900 mg in the SAD cohorts and repeated doses of 300 mg orally three times daily for 14 days in the MAD cohort. Interim results from the Phase 1 clinical trial have demonstrated that, to date:
| • | Oral administration of SPR994 has been well-tolerated at all doses tested. There has been a linear increase in plasma concentration following oral administration of doses ranging from 100 mg to 900 mg of SPR994. |
| • | Administration of SPR994 following a high fat meal has not substantially altered the plasma exposure as compared with administration to volunteers in a fasted state. We believe these data indicate that SPR994 can be administered without regard to meals. |
| • | Administration of SPR994 in an immediate-release formulation produced plasma exposure (AUC) comparable to that observed with extended-release formulations, supporting our decision to utilize an immediate-release formulation of SPR994 in our planned pivotal Phase 3 clinical trial of SPR994. |
| • | The mean plasma free drug concentrations versus time observed following administration of SPR994 300 mg (immediate-release formulation) to healthy adult volunteers (fasted and fed) are presented in the figure below. The mean plasma concentrations of tebipenem remain above the MIC90 for the relevant bacterial pathogens for >50% of an 8-hour dosing interval (three-times daily administration). This exposure is predicted to be effective as a treatment for cUTI based on preclinical pharmacodynamic models. |
Tebipenem Pharmacokinetic Profile Following Administration of 300 mg of SPR994 (IR)
to Healthy Volunteers in the Fasted or Fed State (Mean +/- SD)
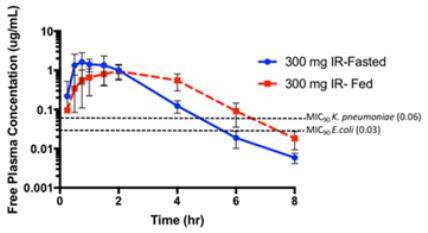
| • | Furthermore, the tebipenem plasma exposure (AUC 0-24) following the administration of SPR994 300 mg orally three times daily was comparable to the exposure level which reduced infections in the Phase 2 UTI trials conducted by Meiji. |
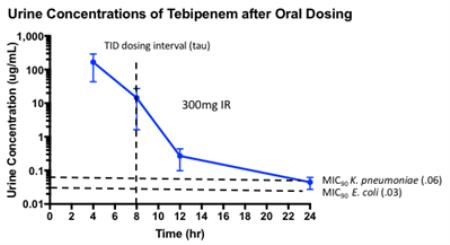
| • | Renal elimination of tebipenem has resulted in urine concentrations that are approximately 100-fold higher than the maximum plasma free drug concentrations following the administration of 300 mg of SPR994 and in excess of the MICs of the most prevalent urinary pathogens for greater than the 8-hour dosing interval, as presented in the figure below. We believe this provides an added margin of exposure for the treatment of cUTI with SPR994 300 mg administered orally three times per day. |
| • | The tebipenem plasma free drug exposure predicted following the administration of SPR994 300 mg orally three times daily (expressed as a function of the dosing interval, tau) was comparable to that observed following the administration of ertapenem (1 g) administered intravenously every 24 hours. Since both ertapenem and tebipenem exhibit time and concentration-dependent antibacterial activity, we believe that the similar plasma free drug exposures over time mitigate the risk of comparing the efficacy of oral tebipenem with IV-administered ertapenem in our planned pivotal Phase 3 clinical trial of SPR994. |
| • | The most common adverse event in the Phase 1 study has been diarrhea, occurring in 9/107 (8.4%) of Phase 1 subjects dosed to date with SPR994 or placebo (3:1 randomization; data remain blinded). These events have been mild in severity and self-limited and the frequency is consistent with prior experience with Orapenem® (9.5%) and other carbapenem antibiotics, including ertapenem (9.2-10.3% of patients in clinical trials, Invanz® USPI). |
| • | 2/107 (1.9%) subjects dosed to date with SPR994 or placebo (3:1 randomization; data remain blinded) have experienced a clinically significant increase in transaminase levels to 4-8 times the upper limit of normal (ULN). There have been no Hy’s law cases or other evidence of drug induced liver injury. Alanine transaminase (ALT) elevations in these patients returned toward normal after dosing in one subject (SAD) and despite continued dosing in the other subject (MAD). As these events were considered to be monitorable and reversible, no change in the Phase 1 dose escalation for SPR994 was made by the Safety Monitoring Group. Transaminase elevation is a known class effect of carbapenem antibiotics, including Orapenem, reported as <1% in clinical trials and the post-marketing surveillance study, and the data received to date for SPR994 suggest that its effect on transaminase levels is comparable to that observed in other carbapenems in independent studies. In third-party clinical studies that did not involve a comparison of such drugs with SPR994, transaminase elevations were reported for ertapenem (8%), ceftaroline (2%) and aztreonam (10-38%) (as reported in the LiverTox database maintained by the National Institute of Diabetes and Digestive and Kidney Diseases), as well as for doripenem (4.0%) and ceftazidime-avibactam (4.6%) (as reported in the FDA summary basis for approval of ceftazidime-avibactam (Avycaz®)). |
The MAD portion of the Phase 1 clinical trial is designed to assess the safety, tolerability and pharmacokinetics of SPR994 administered orally for 14 days to healthy volunteers (8 subjects per cohort). The initial cohort MAD received 300 mg (immediate-release formulation) orally three times per day. This dose was chosen because, in clinical trials to date, 300 mg of SPR994 was well tolerated as a single dose, the plasma drug exposure observed with such dose was consistent with that predicted for microbial efficacy based on preclinical infection models, and such dose was previously demonstrated to reduce infections in a Phase 2 UTI trial utilizing Orapenem tablets. Dosing of this cohort has been completed with tolerability consistent with Orapenem clinical trials and the post-marketing surveillance data including a 3,540 patient Japanese post-marketing study.