Attached files
| file | filename |
|---|---|
| EX-31.4 - EXHIBIT 31.4 - 9 METERS BIOPHARMA, INC. | tv496133_ex31-4.htm |
| EX-31.3 - EXHIBIT 31.3 - 9 METERS BIOPHARMA, INC. | tv496133_ex31-3.htm |
| EX-10.2 - EXHIBIT 10.2 - 9 METERS BIOPHARMA, INC. | tv496133_ex10-2.htm |
| EX-10.1 - EXHIBIT 10.1 - 9 METERS BIOPHARMA, INC. | tv496133_ex10-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K/A
(Amendment No. 1)
(Mark One)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ________________ to ________________
Commission file number 001-37797
INNOVATE BIOPHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 27-3948465 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) |
8480 Honeycutt Road, Suite 120
Raleigh, North Carolina 27615
(Address of principal executive offices, including zip code)
(919) 275-1933
(Registrant’s telephone number, including area code)
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
| Title of each class | Name of each exchange on which registered |
| Common Stock $0.0001 Par Value | The Nasdaq Stock Market LLC |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT: None
Indicate by check mark if the registrant is a well-known seasoned issuer as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the issuer is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if the disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or an amendment to this 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |
| Non-accelerated filer | ¨ | Smaller reporting company | þ | |
| (Do not check if smaller reporting company) | Emerging growth company | þ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. þ
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate value of common stock held by non-affiliates of the registrant as of June 30, 2017, the last business day of the registrant’s most recently completed second fiscal quarter, was $3,878,345 (based on the last reported closing sale price on the Nasdaq Capital Market on that date of $4.90 per share).
As of March 12, 2018, the registrant had 25,691,680 shares of common stock, par value $0.0001 per share, issued and outstanding.
TABLE OF CONTENTS
Form 10-K/A
(Amendment No. 1)
| EXPLANATORY NOTE | ||
| PART I | ||
| ITEM 1. | BUSINESS | 3 |
| PART IV | ||
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES | 36 |
| SIGNATURES | 41 | |
| 2 |
Innovate Biopharmaceuticals, Inc. (the “Company”) is filing this Amendment No. 1 on Form 10-K/A (this “Amendment”) to amend the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2017, as filed with the Securities and Exchange Commission (the “SEC”) on March 14, 2018 (the “Form 10-K”). This Amendment is being filed solely to (i) update “Item 1. Business” to reflect comments received from the SEC Staff on the Company’s Form 8-K filed with the SEC on February 2, 2018, as amended and previously reflected therein, (ii) re-file Exhibits 10.1 and 10.2 to the Form 10-K (the “Exhibits”) and (iii) amend Part IV, “Item 15. Exhibits, Financial Statement Schedules” of the Form 10-K, including revising the Exhibit Index. The Exhibits, as re-filed on this Amendment, restore certain provisions that had previously been redacted in accordance with the Company’s application for confidential treatment in response to comments received from the SEC Staff. In addition, as required by Rule 12b-15 under the Securities Exchange Act of 1934, as amended, new certifications by the Company’s principal executive officer and principal financial officer are filed as exhibits to this Amendment.
No attempt has been made in this Amendment to modify or update the other disclosures presented in the Form 10-K. This Amendment does not reflect events occurring after the filing of the Form 10-K (i.e., those events occurring after March 14, 2018) or modify or update disclosures that may be affected by subsequent events. Such subsequent matters are addressed in subsequent reports filed with the SEC. Accordingly, this Amendment should be read in conjunction with the Form 10-K and the Company’s other filings with the SEC.
Merger of Monster Digital, Inc. and Innovate Biopharmaceuticals Inc.
On January 29, 2018, Monster Digital, Inc. (“Monster”) and privately held Innovate Biopharmaceuticals Inc. (“Private Innovate”) completed a reverse recapitalization in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated July 3, 2017, as amended (the “Merger Agreement”), by and among Monster, Monster Merger Sub, Inc. (“Merger Sub”) and Private Innovate, which changed its name in connection with the transaction to IB Pharmaceuticals Inc. (“IB Pharmaceuticals”). Pursuant to the Merger Agreement, Merger Sub merged with and into IB Pharmaceuticals with IB Pharmaceuticals surviving as the wholly owned subsidiary of Monster (the “Merger”). Immediately following the Merger, Monster changed its name to Innovate Biopharmaceuticals, Inc. (“Innovate”). In connection with the closing of the Merger, Innovate’s common stock began trading on the Nasdaq Capital Market under the ticker symbol “INNT” on February 1, 2018. Prior to the Merger, Monster was incorporated in Delaware in November 2010 under the name “Monster Digital, Inc.”
Except as otherwise noted or where the context otherwise requires, as used in this report, the words “we,” “us,” “our,” the “Company” and “Innovate” refer to Innovate Biopharmaceuticals, Inc. as of and following the closing of the Merger on January 29, 2018 and, where applicable, the business of Private Innovate prior to the Merger. All references to “Monster” refer to Monster Digital, Inc. prior to the closing of the Merger.
Overview
Prior to the Merger, Monster’s primary business focus was the design, development and marketing of premium products under the “Monster Digital” brand for use in high-performance consumer electronics, mobile products and computing applications.
After the Merger, we are a clinical-stage biopharmaceutical company developing novel medicines for autoimmune and inflammatory diseases with unmet needs. Our pipeline includes drug candidates for celiac disease, nonalcoholic steatohepatitis (NASH), Crohn’s disease (CD) and ulcerative colitis (UC). Our lead program, INN-202 (larazotide acetate or larazotide) is entering Phase 3 registration trials, targeted for the second half of 2018, and has the potential to be the first-to-market therapeutic for celiac disease, an unmet medical need, which affects an estimated 1% of the North American population or approximately 3 million individuals. Celiac patients have no treatment alternative other than a strict lifelong adherence to a gluten-free diet, which is difficult to maintain and can be deficient in key nutrients. Additionally, current FDA labeling standards allow up to 20 parts per million (ppm) of gluten in “gluten-free” labelled foods, which are sufficient to cause celiac symptoms in many patients, including abdominal pain, abdominal cramping, bloating, gas, headaches, ataxia, ’‘brain fog’’ and fatigue. Long-term ramifications of celiac disease include enteropathy associated T-cell lymphoma (EATL), osteoporosis and anemia.
| 3 |
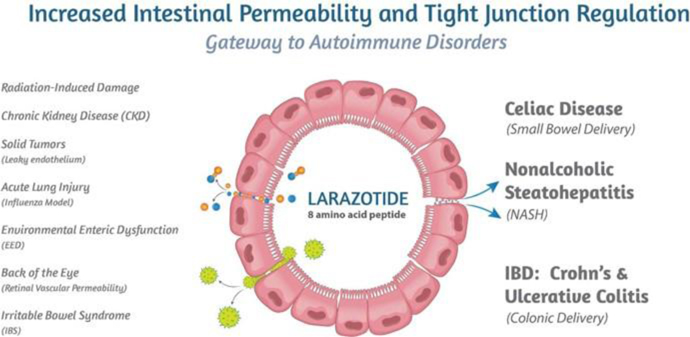
Figure 1: Larazotide’s mechanism of action is applicable to multiple diseases.
Larazotide is an 8-amino acid peptide orally administered in a capsule which has been tested in more than 500 celiac patients with statistically significant improvement in celiac symptoms. The FDA has granted larazotide Fast Track Designation for celiac disease. Larazotide’s safety profile was similar to placebo primarily because larazotide is not systemically absorbed into the blood circulation. Additionally, larazotide’s mechanism of action (MoA) as a tight junction regulator is a new approach to treating autoimmune diseases, such as celiac disease. Pre-clinical studies have shown larazotide causes a reduction in permeability across the intestinal epithelial barrier, making it the only drug candidate known to us which is in clinical trials with this MoA. Increased intestinal permeability underlies several diseases in addition to celiac disease, including NASH, Crohn’s disease, ulcerative colitis and irritable bowel syndrome (IBS), among others (Figure 1). We are engaging in multiple research collaborations to expand larazotide’s clinical indications with a shorter time to proof-of-concept.
With the release of the Phase 2b trial data in 342 celiac patients at the 2014 Digestive Disease Week (DDW) conference, larazotide became the first and the only drug for the treatment of celiac disease (published data), which met its primary efficacy endpoint with statistical significance. The Phase 2b data showed statistically significant (p=0.022) reduction in abdominal and non-GI (headache) symptoms as measured by the CeD PRO. After a successful End-of-Phase 2 meeting with the FDA, which confirmed the regulatory path forward, we expect to launch the Phase 3 registration program later this year with topline data expected by 2019.
Larazotide is being investigated as an adjunct to a gluten-free diet for celiac patients who continue to experience symptoms despite adhering to a gluten-free diet. Due to the difficulty of maintaining a gluten-free diet due to lack of easy access to and the higher cost of gluten-free foods, contamination from gluten as well as social pressures, it is estimated that more than half the celiac population experiences multiple, potentially debilitating, symptoms per month. A study from the UK indicates that more than 70% of patients diagnosed with celiac disease consume gluten either intentionally or inadvertently (Hall et al. 2013).
| 4 |
Another indication for which larazotide is currently being developed is NASH. NASH is an unmet need disease affecting approximately 5%-6% of the US adult population. There are currently several drugs in development; however, to our knowledge, none have larazotide’s MoA. We are developing a proprietary formulation of larazotide, INN-217, for efficient delivery to the intestine. INN-217 has the potential to reduce the transport of lipopolysaccharide (LPS), which is produced by gram negative bacteria in the gut, from the intestinal lumen to the liver via the portal circulation. Several studies have shown the link between NASH and increased levels of LPS, which translocates across an inflamed, “leaky” epithelial barrier to the liver thus directly damaging liver cells via an inflammatory cascade. INN-217 can potentially decrease the flux of LPS across the leaky epithelial barrier, which is known to play an important role in the pathogenesis of NASH. Since none of the NASH drugs in development currently target intestinal permeability, INN-217 has the potential to affect NASH alone and work synergistically with late stage NASH drugs in development, which are primarily focused on metabolic targets such as farnesoid X receptor (FXR) and acetyl-CoA carboxylase (ACC).

Figure 2: LPS (Lipopolysaccharide) is a toxin produced by intestinal bacteria and can translocate via the leaky epithelial barrier to the liver and damage liver cells. Thus LPS has been implicated in the pathogenesis of NASH.
| 5 |
INN-108, is in development for the treatment of mild-to-moderate UC. INN-108 is expected to be delivered orally using an azo-bonded pro-drug approach linking mesalamine or 5-ASA (5-amino salicylic acid) to 4-APAA (approved as Actarit in Japan in 1994 for the treatment of rheumatoid arthritis). After having completed a successful Phase 1 trial at currently approved doses of mesalamine, INN-108 is expected to enter a proof-of-concept Phase 2 trial. The azo-bond protects INN-108 (Figure 2) from the low pH in the stomach, allowing it to transit to the colon where the UC lesions are primarily located. In the colon, the azo bond is broken enzymatically by azoreductases, leading to the separation of mesalamine and 4-APAA which has a synergistic anti-inflammatory effect. Although the majority of patients present with mild-to-moderate UC, the focus of drug development has been in moderate-to-severe UC with little innovation or drug development for mild-to-moderate UC. The mainstay of treatment for mild-to-moderate UC continues to be various oral reformulations of mesalamine such as Shire’s Lialda (approved 2007) and Pentasa (approved 1993), Allergan’s Asacol HD (approved 2008) and Valeant/Salix’s Apriso (approved 2008).
We also own the global rights to INN-329, a proprietary formulation of secretin, a peptide hormone which is used to improve visualization in magnetic resonance cholangiopancreatography (MRCP) procedures. Secretin is a 27-amino acid long hormone which rapidly stimulates release of pancreatic secretions, thus improving visualization of the pancreatic ducts during imaging procedures.
| 6 |
Our Strategy
Our goal is to become a leading biopharmaceutical company by developing novel therapeutics that have the potential to transform current treatment paradigms for patients and to address unmet medical needs. We are currently pursuing the development of drugs for autoimmune and inflammatory diseases that target established biological pathways. The critical components of our strategy are as follows:
| • | Advance INN-202 (larazotide) for celiac disease into Phase 3 clinical trials. Our highest clinical priority is to initiate the Phase 3 trials for larazotide for the treatment of celiac disease. We had a successful End-of-Phase 2 meeting with the FDA in 2017. With the guidance and agreement reached with the FDA, we plan to initiate our Phase 3 trials during the second half of 2018. |
| • | Accelerate development of INN-217 (larazotide) for NASH. Increased intestinal permeability leads to LPS translocation to the liver and is one of the key recognized pathogenic factors in NASH. Larazotide’s mechanism of action to decrease intestinal permeability could thus have a therapeutic effect in NASH. We plan to develop larazotide alone and in combination with select NASH therapies in clinical trials with the potential for synergistic therapeutic benefit. |
| • | Further the study INN-108 for Ulcerative colitis . We are currently developing plans to initiate the proof of concept Phase 2 trials for INN-108 for the treatment of UC. We plan to initially develop INN-108 for mild-to-moderate UC in adults. |
| • | Further the study of INN-289 (larazotide) for Crohn’s disease. The mechanism of action of larazotide to decrease intestinal permeability can have a therapeutic effect in inflammatory bowel disease (IBD). In an IL-10 knockout animal model, larazotide showed promising data which can position it for a proof-of-concept study using a proprietary formulation of larazotide, INN-289, alone and in combinations with select approved immunological therapies. |
| • | Seek partnerships to commercialize late stage pipeline drugs. With large addressable markets, such as celiac disease, we plan to seek out partners with established presences and histories of successful commercialization. |
| • | Leverage and protect our existing intellectual property portfolio and secure patents for additional indications. We intend to continue to expand our intellectual property protection strategy, grounded in securing composition of matter patents and method of use patents for newer indications. We plan to develop newer formulations for the product candidates for other indications and improved performance of existing indications. |
| • | In-license additional intellectual property and pipeline drugs to expand our presence in the treatment of autoimmune and inflammatory diseases . In addition to broadening our current pipeline through indication expansion, we plan to explore expansion of our product pipeline through in-licensing, strategic partnerships and product acquisitions, as we did in 2016 by in-licensing of larazotide from Alba Therapeutics Corporation. We expect that future pipeline expansion decisions will be based on the unmet medical needs in autoimmune and inflammatory disease areas including, but not limited to, celiac disease and ulcerative colitis, the commercial opportunity, and the ability to rapidly develop and commercialize a product candidate. |
| • | Leverage the expertise of our management team and network of scientific advisors and key opinion leaders . We are led by a strong management team with deep experience in drug development, collaborations, operations, and corporate finance. Our team has been involved in a broad spectrum of R&D activities leading to successful outcomes, including FDA approvals and drug launches. We will continue to leverage the collective experience and talent of our management team, network of leading scientific experts, and key opinion leaders to strategize and implement our development and eventually our commercialization strategy. |
| • | Out-license our non-core assets/indications and establish research collaborations . From time to time, we review our internal research priorities and therapeutic focus areas and may decide to out-license non-core assets/indications that arise from current and future available data. We may seek research collaborations that leverage the capabilities of our core assets to monetize and expand upon the breadth of opportunities that may be accessible through our drug candidates. |
| • | Outsource capital intensive operations . We plan to continue to outsource capital intensive operations, including most clinical development and all manufacturing operations of our product candidates, to facilitate the rapid development of our pipeline by using high quality specialist vendors and consultants in a capital efficient manner. |
| 7 |
Our Drug Product Pipeline
Our current pipeline is focused on clinical stage assets with large markets and unmet medical needs. We continue to leverage additional proof-of-concept work for larazotide to expand into additional indications, including NASH, Crohn’s disease and ulcerative colitis. The following table summarizes key information about our pipeline of drug product candidates to date (Table 1):
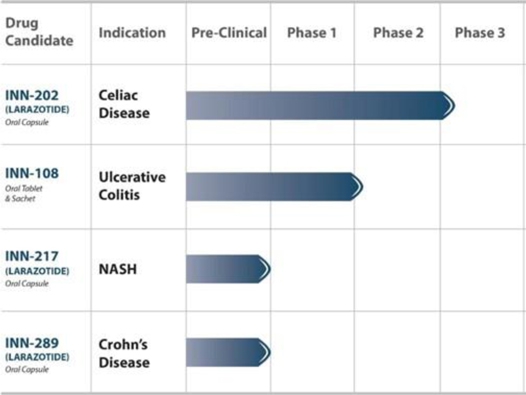
Table 1: Our key pipeline products are clinical stage and address large markets with chronically dosed therapies.
INN-202 (Larazotide) for Celiac Disease
Larazotide is being developed for the treatment of celiac disease and has successfully completed a Phase 2b trial showing statistically significant reduction in abdominal and non-GI (headache) symptoms. We are planning to launch the Phase 3 trials in the second half of 2018.
Larazotide is an orally administered, locally acting, non-systemic, synthetic 8-amino acid (Figure 3), tight junction regulator being investigated as an adjunct to a gluten-free diet in celiac disease patients who still experience persistent GI symptoms despite being on a gluten-free diet. Because of larazotide’s lack of absorption into the blood circulation, we believe that fewer complications, if any, are likely to develop for individuals taking chronically dosed lifetime medication.
The larazotide drug product is an enteric coated drug product formulated as enteric coated multiparticulate beads filled into hard gelatin capsules for oral delivery. The enteric coating is designed to allow the bead particles to bypass the stomach and release larazotide upon entry into the small intestine (duodenum). A mixed bead formulation is used to allow partial release of larazotide upon entry into the duodenum and to release the remaining larazotide approximately 30 minutes later. In clinical trials, larazotide has been dosed 15 minutes before meals allowing time for its effect in the small bowel before exposure to gluten.
| 8 |
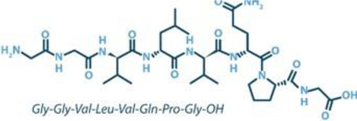
Figure 3: Larazotide acetate is an 8-amino acid peptide in an oral capsule using a proprietary formulation
Larazotide’s Mechanism of Action
In research studies supportive of the mechanism of action, larazotide has been shown to stimulate recovery of mucosal barrier function via the regulation of tight junctions both in vitro and in vivo , including in a celiac disease mouse model (Gopalakrishnan, 2012). In doing so, it is proposed that larazotide reduces the symptoms associated with celiac disease.
In several autoimmune diseases, this increased intestinal permeability or paracellular leakage allows increased exposure to a triggering antigen and a consequent inflammatory response, the characteristics of which are determined by the particular disease and the genetic makeup of the individual. A new paradigm for autoimmune diseases is that there are three contributing factors to the development of disease:
| 1. | A genetically susceptible immune system that allows the host to react abnormally to an environmental antigen; |
| 2. | An environmental antigen that triggers the disease process; and |
| 3. | The ability of the environmental antigen to interact with the immune system. |
Larazotide regulates tight junction opening triggered by both gluten and inflammatory cytokines, thus reducing uptake of gluten. Larazotide also disrupts the intestinal permeability-inflammation loop, and has been shown to reduce symptoms associated with celiac disease.
Larazotide’s Dose Response
Previously published in vitro work using Caco-2 cells has shown a linear dose response for larazotide in reducing permeability of the epithelial barrier by tightening the tight junctions (Gopalakrishnan, 2012). In several clinical trials, larazotide has exhibited clinical benefit by reducing celiac symptoms at lower doses while inhibition of this activity occurs at the higher doses. To better understand this observation, Dr. Anthony Blikslager from North Carolina State University evaluated the pharmacology of larazotide at the luminal surface of the small intestine in an ex vivo porcine model. A section of the porcine intestine was ligated, placed in an Ussing chamber and changes in permeability were measured by electrical resistance. Multiple experiments demonstrated that following an ischemic insult causing increased intestinal permeability, full length larazotide is capable of restoring intestinal wall integrity to that of the non-ischemic control. Subsequently, it was discovered that a specific aminopeptidase located within the brush borders of the intestinal epithelium cleaves larazotide into two fragments which lack either one or both N-terminus glycine (G) residues ( GG VLVQPG). Both cleaved fragments, GVLVQPG and VLVQPG, do not decrease intestinal permeability. Moreover, when these two fragments are administered in combination with the active full-length larazotide, they inhibit larazotide’s activity to restore intestinal wall integrity or reduce permeability. These data demonstrate that higher doses of larazotide lead to local buildup of breakdown fragments, which then compete with and block activity of larazotide after threshold concentration is reached. The in vitro experiments using Caco-2 monolayers did not show the same pharmacology and dose response because they lack the brush border and therefore lack the aminopeptidase which cleaves larazotide. These data also provide an explanation for the clinical observations of an optimal lower dose of larazotide, which avoids the reservoir of competing inactive fragments generated at high doses of larazotide.
| 9 |
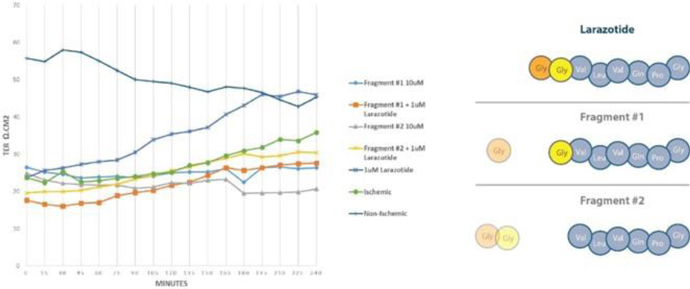
Figure 4: An aminopeptidase in the brush border cleaves larazotide into two fragments, #1 and #2, which then act as inhibitors of larazotide

Figure 5: Illustrative effect of gluten ingestion, breakdown to gliadin which can cross a “leaky” epithelial barrier in the small bowel thus activating the intestinal-inflammatory loop and leading to symptoms and villous atrophy.
The Intestinal Barrier, Tight Junctions, and Intestinal Permeability
The intestine is one of the largest interfaces between a person and his or her environment, and an intact intestinal barrier is essential in maintaining overall health. An important function of the intestinal barrier is to regulate the trafficking of macromolecules between the environment and the host. Together with gut-associated lymphoid tissue and the neuroendocrine network, the intestinal epithelial barrier controls the equilibrium between tolerance and immunity to non self-antigens. When the finely tuned trafficking of macromolecules is dysregulated, both intestinal and extra-intestinal autoimmune disorders can occur in genetically susceptible individuals (Figure 5).
| 10 |
Transcellular fluxes (through the cell membrane) allow nutrients and small molecules to enter the cell from the luminal side of the intestine and exit on the serosal side (internal milieu). Paracellular fluxes (between cells) in contrast are limited by size and charge constraints imposed by the tight junctions between epithelial cells. The paracellular pathway is the key regulator of intestinal permeability to larger more complex macromolecules that may be immunogenically significant.
Intestinal epithelial cells adhere to each other through junction complexes. The tight junction, also referred to as zonula occludens, represents the major barrier to diffusion within the paracellular space between intestinal cells. Multiple proteins that make up the tight junction have been identified including occludin, claudin family members, and junctional adhesion protein (JAM). These interact with cytosolic proteins (ZO-1, ZO-2, and ZO-3) that function as adaptors between the tight junction proteins and actin and myosin contractile elements within the cell. Acting together, they open and close the paracellular junctions between cells. It is now apparent that tight junctions are dynamic structures that are involved in developmental, physiological, and pathological processes.
The role of tight junction dysfunction in the pathogenesis of autoimmune diseases is under active investigation. Many autoimmune populations have increased intestinal permeability, and it is believed that this may play a fundamental role in the development of autoimmunity. In susceptible populations, the opening of tight junctions between intestinal epithelial cells may lead to exposure to oral antigens via paracellular transport and a consequent autoimmune response. A wide range of gastrointestinal and systemic inflammatory diseases are associated with abnormal intestinal permeability including celiac disease, type 1 diabetes, inflammatory bowel diseases (Crohn’s disease and UC), and ankylosing spondylitis.
Summary of Key Clinical Trials using Larazotide in Celiac Disease
Larazotide has been administered to humans in seven clinical trials. These include three Phase 1 trials: (two trials in healthy subjects and a Phase 1b proof of concept (POC) trial in subjects with celiac disease), two Phase 2 gluten challenge studies in subjects with controlled celiac disease, and additionally two Phase 2 trials in subjects with active celiac disease (Table 2). After the Phase 1 studies, larazotide was tested to explore which endpoint would be suitable for celiac disease. After looking at permeability changes in the gut, which turned out to be highly variable in a large trial setting, and then mucosal healing, which likely requires a longer-term study, symptom reduction showed the most consistent and reliable reduction both in a gluten challenge and a ’‘real-life’’ trial. Importantly, after exposure in more than 800 subjects, the safety profile of larazotide remained similar to placebo due to its lack of absorption into the bloodstream, which we believe is an important advantage for a chronically dosed drug.
The initial Investigational New Drug Application (IND) for the treatment of celiac disease was filed with the FDA by Alba Therapeutics Corporation (Alba) on 12 August 2005 for the use of larazotide acetate (INN-202). The IND was transferred from Alba to Innovate effective March 8, 2016. Over the course of the seven clinical studies, 5 patients experienced a serious adverse event, of which 2 received placebo and 3 received larazotide. These events included inflammation of the gallbladder, gall stones, depression, allergic reaction to penicillin, and appendicitis. We do not believe that any of these events were considered related to treatment with study medication.
| Trial | Study Date | Clinical Trial | No. of Subjects | |||
| -001 | 2005 | Phase 1: Single Escalating Doses in Healthy Volunteers | 24 | |||
| -002 | 2005-06 | Phase 1b: Multiple Dose POC in Celiac Patients – Gluten Challenge | 21 | |||
| -003 | 2006 | Phase 1: Multiple Escalating Dose in Volunteers | 24 | |||
| -004 | 2006-07 | Phase 2a: Multiple Dose POC in Celiac Patients Gluten Challenge 2 weeks | 86 | |||
| -006 | 2008 | Phase 2b: Dose Ranging, in Celiac Patients Gluten Challenge, 6 weeks | 184 | |||
| -011 | 2008-09 | Phase 2b: POC and Dose Ranging in Active Celiac Patients | 105 | |||
| -06B | 2008 | Phase 2b: Similar to -006, in Celiac Patients | 42 | |||
| -012 | 2011-13 | Phase 2b: Multiple dose in Celiac patients with Symptoms on a Gluten-Free Diet | 342 |
Table 2: Significant drug exposure in more than 800 subjects in multiple clinical trials consistently showed a safety profile similar to placebo, which we believe is an important advantage for chronic lifetime administration.
| 11 |
Clinical Trial (’006) Results Revealed Key Insight into Symptom Reduction as a Primary Endpoint
A Phase 2b study with a gluten challenge (CLIN1001-006) was conducted in 184 subjects with well-controlled celiac disease on a gluten-free diet. Subjects were randomized to one of four treatment groups, (placebo, 1 mg, 4 mg, or 8 mg larazotide) and asked to take treatment 15 minutes prior to each meal (TID). Nine hundred (900) mg of gluten was taken with each meal. Subjects remained on their gluten-free diet throughout the duration of the trial. The trial results revealed key insights into how to move the program forward by focusing on reduction of symptoms. The 1-mg dose prevented the development of gluten-induced symptoms as measured by GSRS (a patient-reported outcome (PRO) devised and validated by AstraZeneca), and all drug treatment groups had lower anti-transglutaminase antibody levels than the placebo group. Results of pre-specified secondary endpoints suggest that larazotide reduced antigen exposure as manifested by reduced production of anti-tissue transglutaminase (tTG) levels and immune reactivity towards gluten and gluten-related gastrointestinal symptoms in subjects with celiac disease undergoing a gluten challenge.

Figure 6: The overall trial designs for Phase 2b and Phase 3 are similar with a screening period followed by 12 weeks of randomization to larazotide vs. placebo.
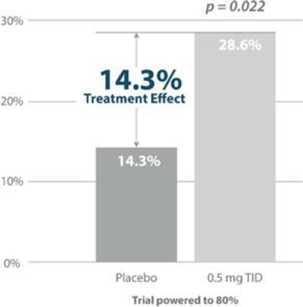
Figure 7: Responder Rate Analysis: Larazotide is the only drug in development for celiac disease to meet its primary endpoint with statistical significance as measured by the copyrighted CeD PRO (celiac disease patient reported outcome), an FDA-agreed upon primary endpoint for Phase 3 (shown above). Source: Gastroenterology 2015; 148:1311–1319; p. 1315
Clinical Trial (’012) Met the Primary Endpoint with Statistical Significance (CeD-GSRS/CeD PRO)
The purpose of the ’012 study was to assess the efficacy (reduction and relief of signs and symptoms of celiac disease) of 3 different doses of larazotide (0.5 mg, 1 mg, and 2 mg TID) versus placebo for the treatment of celiac disease in adults as an adjunct to a gluten-free diet. Larazotide or placebo which was administered TID, 15 minutes prior to each meal. After a screening period, subjects were asked to continue following their current gluten-free diet into a placebo-run in phase for 4 weeks after which they were randomized to drug versus placebo. Subjects maintained an electronic diary capturing: daily symptoms celiac disease patient reported outcome (CeD-PRO), weekly symptoms (CeD-GSRS), bowel movements (BSFS), and a self-reported daily general well-being assessment (Figure 6).
| 12 |
The primary endpoint of average on-treatment CeD GSRS score throughout the treatment period was met at the 0.5 mg TID dose. In addition, a number of pre-specified secondary and exploratory endpoints, such as symptomatic days and symptom-free days, collectively demonstrated that a dose of 0.5 mg TID was superior to placebo and higher doses of larazotide. No difference was observed between the two higher dose levels (1 and 2 mg TID) or placebo, suggesting a narrow dose range around the 0.5mg dose which seems to correlate with pre-clinical data.
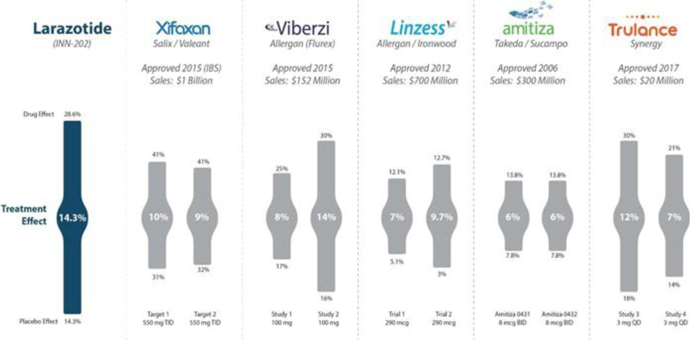
Figure 8: Treatment effect of larazotide from the Phase 2b trial (’012) compared to approved IBS/CIC drugs with varying treatment effects mostly in the mid to high single digit range. Source: Gastroenterology 2015; 148:1311–1319; p. 1315 and FDA Drug Labels
The CeD PRO, a copyrighted PRO created specifically for celiac disease and wholly owned by us, showed a statically significant (p=0.022) treatment effect of 14.3% (drug responder rate minus placebo responder rate). Although to our knowledge there are no celiac drugs approved as a comparator, the treatment effect was greater than several other GI dugs approved for IBS and chronic idiopathic constipation (CIC) which use a similar clinical trial design (Figure 8).
| 13 |
Path Forward to Phase 3 Trials
After a successful End-of-Phase 2 meeting with the FDA, agreements were reached on the key aspects of the Phase 3 trials. The FDA agreed on using the previously validated CeD PRO as the primary endpoint with two doses of larazotide which bracket the range of efficacy in previous trials. Two Phase 3 trials with a size of about 500 patients each would allow for more than a 90% power to replicate the Phase 2b trial results. Most other criteria, such as inclusion, exclusion and site selection/coordination, are expected to remain similar to the ’012 Phase 2b trial.
About Celiac Disease
Celiac disease is a genetic autoimmune disease triggered by the ingestion of gluten-containing foods such as wheat, barley, and rye. Individuals with celiac disease have increased intestinal permeability, commonly referred to as a ’‘leaky’’ gut. This allows macromolecules that normally remain on the luminal side of the intestine to pass through to the serosal side through tight junctions via paracellular diffusion (Figure 9). In the case of celiac disease, this permeability may allow gluten break-down products, the triggering antigens of celiac disease, to reach gut-associated lymphoid tissue(GALT), initiating an inflammatory response. Celiac disease is characterized by chronic inflammation of the small intestinal mucosa that may result in diverse symptoms, malabsorption, atrophy of intestinal villi, and a variety of clinical manifestations.

Figure 9: The epithelial barrier separates the intestinal content from the immune system (lamina propria) and the vasculature.
| 14 |
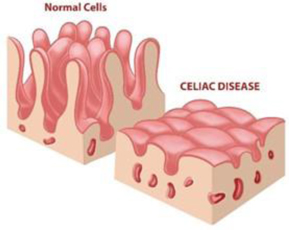
Figure 10: Intestinal villi atrophy in celiac patients, a characteristic finding upon biopsy of the small intestine.
Large Population — Unmet Need (no drug approved); Serious Long-Term Consequences
Celiac disease affects an estimated 1% of the Western population (Dubé, 2005). Currently, there are no therapeutics available to treat celiac disease, and the current management of celiac disease is a life-long adherence to a gluten-free diet. Changes in dietary habits are difficult to maintain, and foods labeled as gluten-free may still contain small amounts of gluten (up to 20 ppm per FDA labeling standards). Dietary compliance is imperfect in a large fraction of patients (Rostom, 2006) and difficult to adhere to on an ongoing basis (Green, 2007). In a survey conducted in the United Kingdom non-adherence to the gluten-free diet was found to be as high as 70% (Hall, 2013).
There are serious long-term consequences to exposure to gluten in patients with celiac disease, including the risk of developing osteoporosis, stomach, esophageal, or colon cancers, and T-cell lymphoma (Green 2003, Green 2007). The continuous GI symptoms often result in significant morbidity with a substantial reduction in quality of life. In addition, not all patients respond to a gluten-free diet. Patients with known celiac disease may continue to have or re-develop symptoms despite being on a gluten-free diet (Rostom 2006). This suggests a need for a therapeutic agent for the treatment of celiac disease (Green, 2007; Hall, 2013).
Celiac disease represents a model of an autoimmune disorder in which the following elements are known:
| 1. | The triggering environmental factor is glutenin or gliadin, the proline, glutamine and glycine rich glycoprotein fractions of gluten; |
| 2. | There is a close genetic association with HLA haplotypes DQ2 and/or DQ8; and |
| 3. | A highly specific humoral autoimmune response occurs. |
Genetics of Celiac Disease
The high incidence of celiac disease in first degree relatives of celiac patients (10 − 15%) and high concordance rate in monozygotic twins (80%) suggest a strong genetic component. Gliadin deamidation by tissue transglutaminase (tTG) enhances the recognition of gliadin peptides by human leukocyte antigen (HLA) DQ2 and DQ8 T cells in genetically predisposed subjects, which in turn may initiate the cascade of autoimmune reactions responsible for mucosal destruction. This interaction implies that gliadin and/or its breakdown peptides in some way cross the intestinal epithelial barrier and reach the lamina propria of the intestinal mucosa where they are recognized by antigen-presenting cells. The enhanced paracellular permeability of individuals with celiac disease would allow passage of macromolecules through the paracellular spaces with resulting autoimmune inflammation. There is a strong genetic predisposition to celiac disease, with major risk associated with HLA DQ2 (approximately 95% of celiac disease patients) and HLA-DQ8 (approximately 5% of celiac disease patients). The prevalence of celiac disease in the U.S. is estimated to be approximately 1%; however approximately 30% of the general U.S. population is HLA DQ2 positive (Figure 11), indicating that additional factors are involved in the development of celiac disease.
| 15 |
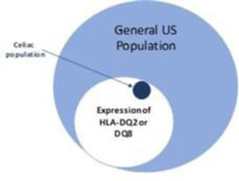
Figure 11: Distribution of HLA-DQ2/DQ8 in the general US population and in celiac disease. Source: J. Clin. Invest. 2007 Jan 2;117(1):41.
In celiac disease, an inflammatory reaction occurs in the intestine that is characterized by infiltration of immune cells in the lamina propria and epithelial compartments with chronic inflammatory cells and progressive architectural changes to the mucosa. Both adaptive and innate branches of the immune system are involved. The adaptive response is mediated by gluten-reactive CD4+ T cells in the lamina propria that recognize gluten-derived peptides when presented by the HLA class II molecules DQ2 or DQ8. The CD4+ T cells then produce pro-inflammatory cytokines such as interferon gamma. This results in an inflammatory cascade with the release of cytokines, anti-tTG antibodies, T cells, and other tissue-damaging mediators leading to villous injury and crypt hyperplasia in the intestine. Anti-human tissue transglutaminase (anti-tTG) antibodies are also produced, which form the basis of serological diagnosis of celiac disease.
Anti-tTG Antibodies: Highly Sensitive and Specific Blood-based ELISA Diagnostic Test
The current approach for diagnosis of celiac disease is to use anti-tissue transglutaminase-2 (tTG-2) antibody tests as an initial screen with definitive diagnosis from biopsy of the small intestine mucosa. The diagnosis of celiac disease is confirmed by demonstration of characteristic histologic changes in the small intestinal mucosa, which are scored based on criteria initially put forth by Marsh and later modified. In 2012, the European Society of Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) Guidelines allowed symptomatic children with serum anti-tTG antibody levels ≥10 times upper limit of normal to avoid duodenal biopsies after positive human leukocyte (HLA) test and serum anti-endomysial antibodies.
The need for multiple clinical and laboratory findings to diagnose celiac disease makes monitoring disease progression difficult. International guidelines give standardized definitions and criteria for the diagnosis of celiac disease, however there are not clear standards for follow-up and monitoring of treatment. This is particularly true for celiac patients diagnosed as adults, who respond differently and less completely to a gluten-free diet than do celiac patients diagnosed as children. It is not clear who should perform follow-up of patients with celiac disease and at what frequency but the American College of Gastroenterology suggests that an annual follow-up seems reasonable. Recommendations for monitoring disease progression include assessing symptoms and dietary compliance, and repeating serology tests. Markers of celiac disease progression and improvement that are both validated and provide a timely assessment of disease activity are lacking.
Role of Tissue Transglutaminase in Celiac Disease
Anti-tTG-2 antibodies are produced in the small-intestinal mucosa (Picarelli et al. 1996), where they can bind tTG-2 present in the basement membrane and around blood vessels and form deposits characteristic of the disease. tTG-2 has been implicated in a variety of human disorders including several neurodegenerative conditions and cancer. Transglutaminases (TGs) were first discovered in the 1950s and are a family of enzymes which catalyze Ca2+-dependent post-translational modification of proteins. Of the seven isoforms discovered so far all share the same basic four-domain tertiary structure, with minor variations, although their catalytic mechanism is conserved, resembling that of the cysteine proteases. tTGs cause transamidation, esterification, and hydrolysis, all of which lead to post-translational modifications in the target proteins. Characteristically, tTG’s mediate selective protein cross-linking by forming covalent isopeptide linkages between two target proteins. The resulting cross-linked products in many cases have high molecular masses and are unusually resistant to proteolytic degradation and mechanical strain. As in the case of the gliadin fragments in celiac disease, they are able to pass thru the leaky paracellular pathway from the lumen to the lamina propria , where the immune cells reside and are then activated.
| 16 |
Gliadin fragments, in addition to being rich in proline, also have high glutamine content, which makes them suitable substrates for tTG-2, which targets glutamine residues. For augmented DQ2/8 binding, the conversion of glutamine residues to glutamic acid is catalyzed by tTG-2 as a deamidation reaction. After deamidation, the gliadin peptides become highly negatively charged in key anchor positions, thereby increasing their affinity to the HLA molecules. CD4+ T cells recognize the deamidated gliadin peptides bound to the HLA DQ2 or DQ8 molecules by their T cell receptors, thus activating intestinal inflammation leading to villous atrophy.
Gluten and Food Labeling
Gluten is a complex molecule contained in several grains such as wheat, rye and barley. Gluten can be subdivided into two major protein subgroups according to its solubility in alcohol and aqueous solutions. These subclasses consist of gliadins, soluble in 40 − 70% ethanol and glutenins which are large, polymeric molecules insoluble in both alcohol and aqueous solutions. The gliadins and glutenins can be further subdivided into groups according to their molecular weight. Glutenins can be subdivided into low and high molecular weight proteins, while the gliadin protein family contains α-, β-, γ- and ω- types. Both glutenins and gliadins are characterized by a high amount of prolines (20%) and glutamines (40%) that protect them from complete degradation in the gastrointestinal tract and make them difficult to digest. Currently 31 nine-amino acid peptide sequences in the prolamins of wheat and related species have been defined as being celiac toxic or celiac ’‘epitopes.’’ These epitopes are located in the repetitive domains of the prolamins, which are proline and glutamine-rich, and the high levels of proline make the peptide resistant to proteolysis. In addition, the prolamin-reactive T cells also recognize these epitopes to a greater extent when specific glutamine residues in their sequences have been deamidated to glutamic acid by tTG-2. The immunodominant sequence after wheat challenge corresponds to a well-characterized 33 residue peptide from α-gliadin, ’’33-mer,’’ that is resistant to gastrointestinal digestion (with pepsin and trypsin) and was initially identified as the major celiac toxic peptide in the gliadins.
The FDA finalized a standard definition of ’‘gluten-free’’ in August 2013. As of August 5, 2014, all manufacturers of FDA-regulated packaged food making a gluten-free claim must comply with the guidelines outlined by the FDA ( www.fda.gov/gluten-freelabeling ). A ’‘gluten free’’ claim still allows up to 20 ppm of gluten which leads to more than 100mg/day up to 500 mg/day of gluten exposure. Due to presence of gluten in foods, beer/liquor, cosmetics and household products, exposure is virtually impossible to completely avoid, and with cross-contamination, celiac patients cannot avoid exposure to gluten therefore, making symptoms more frequent than expected.
| CNS | Endocrine | Oncology/Heme | Skin | Other | ||||
| Headaches | Type 1 Diabetes |
Enteropathy associated T-cell lymphoma (EATL) |
Dermatitis herpetiformis | Rheumatoid arthritis (RA) | ||||
| Gluten ataxia |
Autoimmune Thyroid |
anemia | Alopecia areata |
Reduced bone Density | ||||
| Peripheral neuropathies | Addison’s disease | Vitiligo | Sjogren’s syndrome |
Table 3: Diseases associated with celiac disease
Non-GI Manifestations of Celiac Disease and Co-Morbidities
Headache, Gluten Ataxia: Nervous System Manifestation of Celiac Disease. The association between celiac disease and neurologic disorders has been supported by numerous studies over the past 40 years. While peripheral neuropathy and ataxia have been the most frequently reported neurologic extra-intestinal manifestations of celiac disease a growing body of literature has established headache as a common presentation of celiac disease as well. The exact prevalence of headache among patients ranges from about 30% to 6% (Lebwohl, 2016).
Dermatitis herpetiformis: Skin Manifestation of Celiac Disease. Dermatitis herpetiformis (DH) is an inflammatory cutaneous disease characterized by intensely pruritic polymorphic lesions with a chronic-relapsing course, first described by Duhring in 1884. DH’s only treatment is a strict lifelong gluten-free diet, for achieving and maintaining a permanent control. It appears in around 25% patients with celiac disease, at any age of life, mainly in adults and is a very characteristic clinical presenting symptom.
| 17 |
INN-217: Non-alcoholic steatohepatitis (NASH) and The Microbiome
NASH is a growing epidemic affecting approximately 5 – 6% of the general population. An additional 10% to 20% of the general population who ingest little (< 70 g/week for females and <140 g/week for males) to no alcohol are characterized with fat accumulation in the liver, without inflammation or damage, a condition called nonalcoholic fatty liver disease (NAFLD). The progression of fatty liver to NAFLD to NASH to cirrhosis is a serious condition which has no approved FDA treatment. Evidence supporting a role for the gut-liver axis in the pathogenesis of NAFLD/NASH has been accumulating over the past 20 years. LPS or endotoxin translocation is thought to be a primary cause of downstream signaling in the liver causing inflammation and damage. NASH is associated with increased gut permeability caused by disruption of intercellular tight junctions in the intestine allowing LPS from bacteria to pass into the portal circulation to the liver directly damaging hepatocytes. LPS constitutes the outer leaflet of the outer membrane of most gram-negative bacteria. LPS is comprised of a hydrophilic polysaccharide and a hydrophobic component known as lipid A which is responsible for the major bioactivity of endotoxin. When released and translocated into the bloodstream from the gut, LPS can cause a variety of cytokine activity and inflammation in the host.
The disrupted barrier along with an altered microbiome in the gut contribute to NASH as recently demonstrated by a group from Emory University, Rahman et. al. , in Gastroenterology (2016). Knockout mice missing the junctional adhesion molecule A (JAM-A) ( F11r-/- ), which have a defect in the intestinal epithelial barrier thus making it “leaky,” develop more severe steatohepatitis. JAM-A is a component of the tight junction complex that regulates intestinal epithelial paracellular permeability. F11r-/- mice therefore have leaky tight junctions that allow for translocation of gut bacteria to peripheral organs. By restoring the leaky tight junctions, larazotide could potentially have a beneficial therapeutic effect by blocking translocation of bacterial toxins via the paracellular pathway and may also help normalize the dysbiotic microbiome found in NASH.
According to a marketing and research firm, GlobalData, the market for NASH therapeutics is expected to grow significantly. GlobalData estimates that the market in the United States, France, Germany, Italy, Spain, the United Kingdom, and Japan for such therapeutics will be approximately $25.3 billion by 2026. We expect that this market could be addressed by larazotide, although we are unable to estimate what portion until the effect in this population has been studied in clinical trials to understand the specific patient-type in NASH that may derive benefit.

Figure 12: Growing NASH population up to 5%-6% of adults in the US alone.
INN-289: Crohn’s Disease: Chronic Disease with need to oral therapeutics
Innovate is working on a proprietary formulation of larazotide for Crohn’s disease, INN-289. Animal data has shown the effect of larazotide on disease attenuation in an IL-10 knockout mouse model (Arrieta, 2009), which has been well established and used for several drug development programs. Larazotide was placed in the drinking water of the mice at a low dose (0.1 mg/ml) or high dose (1.0 mg/ml) during the period from 4 to 17 weeks of age. Results were compared to wild type mice, IL-10 knockout mice with no treatment, and IL-10 knockout mice treated with probiotics. Intestinal and colonic permeability was significantly reduced in the high dose larazotide treatment group, but not in the untreated IL-10 knockout group. Larazotide treatment caused a reduction in all tissue markers of colonic inflammation (IFNγ and TNFα) and in histological inflammation.
| 18 |
Other Indications using Larazotide’s Mechanism of Action
Larazotide for Environmental Enteric Dysfunction (EED): Positive in vitro Data;
Environmental enteric dysfunction (EED) is a rare pediatric tropical disease in the U.S. and Europe, however, more than 165 million children in developing countries in Africa and Asia suffer from it. As per section 524 of the Federal Food, Drug, and Cosmetic Act (FD&C) Act, EED would likely fall under ’‘Current List of Tropical Disease’’ number ’S,’ thus making a drug approved for EED in the U.S. potentially eligible for a Priority Review Voucher.
The histological presentation of EED is very similar to celiac disease with villous atrophy and chronic inflammation of the small bowel and the pathogenesis of EED is linked to increased intestinal permeability. We have tested larazotide against some of the pathogens commonly found in EED (unpublished) and found positive in vitro results which will need to be confirmed in animal models before starting a clinical trial in EED.
INN-108: Mild-to-Moderate Ulcerative Colitis
INN-108 is in development for mild-to-moderate UC and is expected to enter a proof-of-concept Phase 2 trial in the second half of 2018 after a successful Phase 1 trial at currently approved doses of mesalamine. UC is an IBD that affects more than 1.25 million people in the major markets of the United States, France, Germany, Italy, Spain, the United Kingdom, and Japan and is characterized by inflammation and ulcers in the colon and rectum. UC is a chronic disease that can be debilitating and sometimes lead to life-threatening complications. While poorly understood, a multitude of environmental factors and genetic vulnerabilities are thought to lead to the dysregulation of the immune response via a defective epithelial barrier. Although the majority of patients present with mild-to-moderate UC which can progress to severe UC, the focus of drug development has been in moderate-to-severe UC with little innovation or drug development for mild-to-moderate UC. The mainstay of treatment for mild-to-moderate UC remain various oral reformulations of mesalamine or 5-ASA (5-amino salicylic acid) such as Shire’s Lialda (approved 2007) and Pentasa (approved 1993), Allergan’s Asacol HD (approved 2008) and Valeant/Salix’s Apriso (approved 2008).
The initial IND was filed with the FDA by Nobex Corporation on May 15, 2003 for the use of APAZA (INN-108) for the treatment of ulcerative colitis. The IND was then transferred from Seachaid Corporation to Innovate effective March 19, 2014. Two Phase 1 studies in healthy subjects and patients with ulcerative colitis were conducted by Nobex with INN-108. No serious adverse events were reported during either study.
INN-108 uses an azo-bonded pro-drug approach linking mesalamine to 4-APAA. Mitsubishi Pharma developed 4-APAA as Actarit in Japan which was approved in 1994 for rheumatoid arthritis. IBD drugs were all originally approved for RA, from the oldest 5-ASA, sulfasalazine, to the latest biologics, Humira and Enbrel. 4-APAA has more than two decades of safety data as a standalone drug and has an MoA which is differentiated from mesalamine though the ultimate effect for both is anti-inflammatory (Figure 13). Taken orally as a tablet, the azo-bond protects INN-108 from the low pH in the stomach, thus allowing it to transit to the colon where the UC lesions are located. In the colon, the azo bond is broken enzymatically leading to the release of mesalamine and 4-APAA which have a synergistic anti-inflammatory effect. With the addition of 4-APAA, which is not approved in the U.S. or EU, to the already approved mesalamine, the synergistic effect could lead to superior clinical efficacy over the currently approved oral mesalamines.
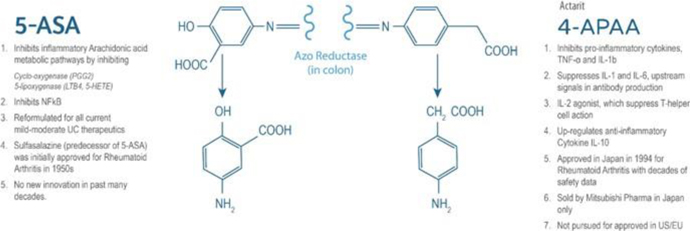
Figure 13: 4-APAA is covalently bonded to 5-ASA via a high energy azo-bond which is only enzymatically cleaved in the colon. The anti-inflammatory effect of each of 5-ASA and 4-APAA via different pathways which could lead to a potential synergistic anti-inflammatory effect as seen in animal studies.
| 19 |
INN-108: UC Animal Model Data Shows Synergy between 4-APAA and Mesalamine
The effects of chronic treatment with INN-108 on Clostridium diffıcile toxin A — induced colitis of the colon is shown in Figure 14. Orally administered INN-108 was significantly more potent than sulfasalazine or 4-APAA alone (McVey, 2005).
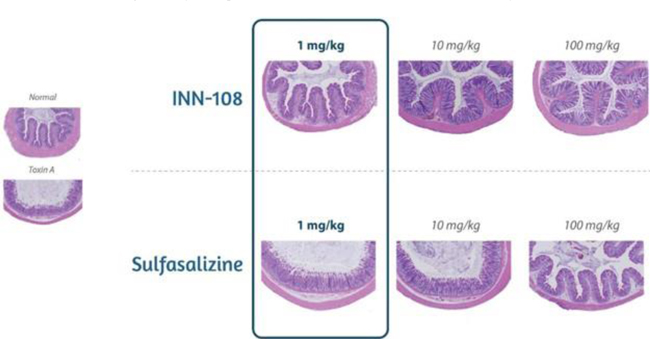
Figure 14: A rat UC model using toxin A induced-colitis as the insult leads to sloughing of the colonic epithelium with increasing doses. Using sulfasalazine vs. INN-108 to protect against the toxin A injury showed INN-108 was significantly more potent that sulfasalazine. Source: McVey DC et al. Digestive Diseases and Sciences. 2005 Mar 1;50(3):565-73.
INN-108 Clinical Development Pathway
After completing two Phase 1 studies, the first of which was conducted between June 2003 and July 2003, and the second of which was conducted between and February 2004 and June 2004, a profile was established with dosing of mesalamine and 4-APAA at 2 grams each for a total of 4 grams three times a day.
The typical dose of the various approved mesalamine formulations range from 1.5g to 2.4g per day, thus INN-108’s mesalamine content is within the established approved dose range. The addition of 4-APAA is thought to improve the efficacy above mesalamine, which would allow INN-108 to be used either after or instead of current mesalamines. In a Phase 2 trial, we plan to compare INN-108 to mesalamine seeking to demonstrate a greater clinical effect than mesalamine alone.
Ulcerative Colitis: Lack of Innovation in New Drug Development for Past Several Decades
Conventional therapies broadly inhibit mechanisms involved in the inflammatory process and are commonly used to effectively treat patients experiencing a mild-to-moderate form of the disease. For mild-to-moderate UC, oral mesalamine has an established efficacy and safety profile. However, gastroenterologists cite the need for new therapies for mild-to-moderate UC.
Patients who do not respond to mesalamine are typically eventually transitioned to biologics. The primary targets for biologics have been to control the immune response and inflammatory cascade, by inhibiting or downregulating molecules such as TNF-α, NF-κB, IL-1β and IFN1-γ. We believe INN-108 bridges the gap between mesalamine and biologics by its mechanism of action of both inhibiting the inflammatory process and down-regulating the cytokines.
About Ulcerative Colitis
UC is a chronic intermittent relapsing inflammatory disorder of the large intestine and rectum. While poorly understood, a multitude of environmental factors and genetic vulnerabilities are thought to lead to the dysregulation of the immune response via a defective epithelial barrier. As a result, chronic inflammation and ulceration of the colon occurs. UC is specific to the colon and affects only the mucosal lining of the colon. Common symptoms of UC include diarrhea, bloody stools, and abdominal pain. The majority of patients are intermittent in their disease course, in that they experience a relapse among periods of remission. However, some patients experience only a single episode of the disease prior to maintaining remission whereas other patients are chronically symptomatic and may require a proctocolectomy to treat their condition.
| 20 |
History of Drug Development in Mild-to-Moderate Ulcerative Colitis
The original compound used in UC was sulfasalazine (Azulfidine), a conjugate of 5-ASA linked to sulfapyridine by an azo bond, which is split into the two molecules by bacterial azoreductases in the colon. The 5-ASA component or mesalamine is the active therapeutic moiety of sulfasalazine, with sulfapyridine thought to have little if any therapeutic effect. Sulfapyridine, however, is the cause of most of the significant adverse side effects of sulfasalazine.
This led to the development of other 5-ASA preparations utilizing azo chemistry to deliver high concentrations of mesalamine or 5-ASA to the colon by preventing early absorption of the drug in the small intestine. Such preparations include olsalazine (Dipentum), consisting of two molecules of 5-ASA bonded together by an azo bond, and balsalazide (Colazal), consisting of 5-ASA azo bonded to an inert carrier (4-aminobenzoyl-β-alanine). The efficacy of these newer oral forms of 5-ASA is comparable to that of sulfasalazine, but they are better tolerated. However, some side effects persist which prevent wider use. In each of these preparations, the only active moiety is mesalamine or 5-ASA, an anti-inflammatory agent.
INN-329
INN-329 is a proprietary formulation of secretin, a peptide hormone which is used to improve visualization in a magnetic resonance cholangiopancreatography (MRCP) procedures. Secretin is a 27-amino acid long hormone which rapidly stimulates release of pancreatic secretions, thus improving visualization of the pancreatic ducts during imaging procedures. Secretin has also been tested in a variety of central nervous system conditions such as autism, though currently approved only for pancreatic function testing and imaging with endoscopic retrograde cholangiopancreatography (ERCP).. We acquired the assets of secretin from Repligen Corporation in December 2014.
The initial IND and was filed with the FDA by Repligen on July 29, 2005 for MRCP. The IND was transferred from Repligen to Innovate in January 2015. The New Drug Application (NDA) for MRCP was filed with the FDA on December 21, 2011 and was transferred to Innovate in January 2015.
MRCP has been used for more than 20 years as a non-invasive tool for imaging pancreatic ducts. With the addition of secretin pancreatic secretions are increased leading to significantly improved visualization of the pancreatic ducts for detection of abnormalities, including pancreatic cancer. The gold standard for pancreatic duct imaging had been ERCP, an expensive and invasive procedure with complications such as pancreatitis (3 − 5%), bleeding (1 − 2%), perforation (1%), infection (1 − 2%) and death (1/250). More than a half-million ERCP procedures are performed annually in the U.S. and as the role of ERCP diminishes for screening, it will further the need for approval of secretin for S-MRCP. We expect to repeat a Phase 3 trial with a partner, if and when secured, as per previous discussion with the FDA to look at improvement in visualization of the pancreatic duct via MRCP with and without secretin.
Our Intellectual Property
We strive to protect the proprietary technology that we believe is important to our business, including our product candidates and our processes. We seek patent protection in the United States and internationally for our product candidates, their methods of use, and processes of manufacture and any other technology to which we have rights, as appropriate. Additionally, we have licensed the rights to intellectual property related to certain of our product candidates, including patents and patent applications that cover the products or their methods of use or processes of manufacture. The terms of the licenses are described below under the heading “Licensing Agreements.” The patent families related to the intellectual property covered by the licenses include 29 U.S. patents and 107 foreign patents with expiration dates ranging from 2018 to 2035. We also rely on trade secrets that may be important to the development of our business.
Our success will in part depend on the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to our business, the validity and enforceability of our patents, the continued confidentiality of our trade secrets, and our ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may own or license in the future, nor can we be sure that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology and products. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors—Risks Related to Our Intellectual Property.”
| 21 |
CeD PRO: Copyrighted Primary Endpoint for Celiac Disease Tested in a Successful Clinical Trial
The patient reported outcome (PRO) primary end point for celiac disease (CeD PRO) was developed based on FDA guidance and is copyrighted in the United States effective October 13, 2011. The copyright registration is in effect for 95 years from the year of first publication or 120 years from the year of creation, whichever expires first. If the drug is approved by the FDA and is the first drug to be approved for celiac disease, Innovate believes that the PRO will become the standard for assessing efficacy in celiac disease. Competitor companies seeking to use a PRO to establish efficacy in this indication would either need to develop their own PRO or would be required to license the CeD PRO from Innovate, thus providing an additional barrier to competitor entry into the marketplace.
Strategic Collaborations and License Agreements
We have entered into collaboration agreements with several academic institutions and other contract research organizations to investigate pre-clinical studies for the use of our product candidates in potential other indications or to further broaden our understanding of the current indications.
Licensing Agreements
License with Alba Therapeutics Corporation
In February 2016, we entered into a license agreement (the “Alba License”) with Alba Therapeutics Corporation (“Alba”) to obtain an exclusive worldwide license to certain intellectual property relating to larazotide and related compounds.
Our initial area of focus for this asset relates to the treatment of celiac disease. We now refer to this program as INN-202. The license agreement gives us the rights to (i) patent families owned by University of Maryland, Baltimore (UMB) and licensed to Alba, (ii) certain patent families owned by Alba, and (iii) one patent family that is jointly owned. In connection with the Alba License, we also entered into a sublicense agreement with Alba under which Alba sublicensed the UMB patents to us (the “Alba Sublicense”).
As consideration for the Alba License, we agreed to pay (i) a one-time, non-refundable fee of $0.4 million at the time of execution and (ii) set payments totaling up to $151.5 million upon the achievement of certain milestones in connection with the development of the product, which milestones include the dosing of the first patient in the Phase 3 clinical trial, acceptance and approval of the New Drug Application, the first commercial sale, and the achievement of certain net sales targets. The last milestone payment is due upon the achievement of annual net sales of INN-202 in excess of $1.5 billion. Upon the first commercial sale of INN-202, the license becomes perpetual and irrevocable. The term of the Alba Sublicense, for which we paid a one-time, non-refundable fee of $0.1 million, extends until the earlier of (i) the termination of the Alba License, (ii) the termination of the underlying license agreement, or (iii) an assignment of the underlying license agreement to us. After we make the first milestone payment after the dosing of the first patient in the Phase 3 clinical trial and are able to demonstrate sufficient financial resources to complete the trial, we have the exclusive option to purchase the assets covered by the license.
The patents covering the composition-of-matter for the larazotide peptide expire in 2018 (2019 outside the United States). The Alba Therapeutics patent estate nevertheless provides product exclusivity for INN-202 in the U.S. until June 4, 2031, not including patent term extensions that may apply upon product approval.
Significant patents in the INN-202 patent estate include issued patents in the U.S. for methods of treating celiac disease with larazotide (US Patents 8,034,776 and 9,279,807), of which the last to expire has a term to July 16, 2030.
Other significant patents include the larazotide formulation patent family, which has three issued U.S. patents as well as 39 filings outside the U.S. (31 issued). The significant patents in the INN-202 patent estate formulation patent family includes patents covering the composition-of-matter (US Patent 9,265,811) and corresponding methods of treatment (US Patents 8,168,594 and 9,241,969) for the larazotide formulation, with the last to expire patent having an expiration in the U.S. of June 4, 2031.
| 22 |
License with Seachaid Pharmaceuticals, Inc.
In April 2013, we entered into a license agreement (the “Seachaid License”) with Seachaid Pharmaceuticals, Inc. (“Seachaid”) to further develop and commercialize the licensed product, the compound known as APAZA. This program is now referred to as INN-108 by us.
The license agreement gives us the exclusive rights to (i) commercialize products covered by the patents owned or controlled by Seachaid related to the composition, formulation or use of any APAZA compound in the territory that includes the U.S., Canada, Japan, and most countries in Europe, and (ii) use, research, develop, export and make products worldwide for the purposes of such commercialization.
As consideration for the Seachaid License, we agreed to pay a one-time, non-refundable fee of $0.2 million at the earlier of the time we meet certain financing levels or 18 months following the execution of the agreement and set payments totaling up to $6.0 million upon the achievement of certain milestones in connection with the development of the product, filing of the New Drug Application, the first commercial sale, and payments ranging from $1.0 million to $2.5 million based on the achievement of certain net sales targets. There are future royalty payments in the single digits based on achieving sales targets, and we are required to pay Seachaid a portion of any sublicense revenue. The royalty payments continue for each licensed product and in each applicable country until the earlier of (i) the date of expiration of the last valid claim for such products to expire or (ii) the date that one or more generic equivalents if such product makes up 50 percent or more of sales in the applicable country. The term of the Seachaid License extends on a product-by-product and country-by-country basis until the expiration of the royalty period for the applicable product in the applicable country.
The INN-108 patent estate includes issued patents for:
(i.) immunoregulatory compounds and derivatives and methods of treating diseases therewith, of which the last to expire has a term to December 17, 2021 (in the U.S.) and August 28, 2021 (in Europe);
(ii.) methods and compositions employing 4-aminophenylacetic acid, of which the last to expire has a term to to August 29, 2021 (in the U.S.);
(iii.) 5-ASA derivatives having anti-inflammatory and antibiotic activity, of which the last to expire has a term to August 29, 2021 (in the U.S.) and August 28, 2021 (in Europe); and
(iv.) synthesis of azo bonded immunoregulatory compounds, of which the last to expire has a term to May 31, 2028 (in the U.S.) and July 7, 2025 (in Europe).
The corresponding European patent application for (ii.) methods and compositions employing 4-aminophenylacetic acid is still pending, but if issued would provide a term to March 22, 2025 in the countries where the application is validated.
The INN-108 patent estate includes also provisional patent applications for pharmaceutical compositions, delivery compositions, methods of prophylaxis and methods of treatment. These patent applications have not yet been issued, and so it is impossible to know the expiration date of any intellectual property that might result from these applications.
| 23 |
Asset Purchase Agreement
In December 2014, we entered into an Asset Purchase Agreement (the “Asset Purchase Agreement”) with Repligen Corporation (“Repligen”) to acquire Repligen’s RG-1068 program for the development of secretin for the pancreatic imaging market and MRCP procedures. We now refer to this program as INN-329. As consideration for the Asset Purchase Agreement, we agreed to make a non-refundable cash payment on the date of the agreement and future royalty payments consisting of a percentage between five and fifteen of annual net sales, with such royalty payment percentage increasing as annual net sales increase. The royalty payments are made on a product-by-product and country-by-country basis and the obligation to make the payments expires with respect to each country upon the later of (i) the expiration of regulatory exclusivity for the product in that country or (ii) ten years after the first commercial sale in that country. The royalty amount is subject to reduction in certain situations, such as the entry of generic competition in the market.
Manufacturing and Supply
We contract with third parties for the manufacturing of all of our product candidates, including INN-108, INN-202 and INN-329, for pre-clinical and clinical studies and intend to continue to do so in the future. We do not own or operate any manufacturing facilities, and we have no plans to build any owned clinical or commercial scale manufacturing capabilities. We believe that the use of contract manufacturing organizations (CMOs) eliminates the need to directly invest in manufacturing facilities, equipment and additional staff. Although we rely on contract manufacturers, our personnel or consultants have extensive manufacturing experience overseeing CMOs.
As we further develop our molecules, we expect to consider secondary or back-up manufacturers for both active pharmaceutical ingredient and drug product manufacturing. To date, our third-party manufacturers have met the manufacturing requirements for our product candidates in a timely manner. We expect third-party manufacturers to be capable of providing sufficient quantities of our product candidates to meet anticipated full-scale commercial demands but we have not assessed these capabilities beyond the supply of clinical materials to date. We currently engage CMOs on a ’‘fee for services’’ basis based on our current development plans. We plan to identify CMOs and enter into longer term contracts or commitments as we move our product candidates into Phase 3 clinical trials.
We believe alternate sources of manufacturing will be available to satisfy our clinical and future commercial requirements; however we cannot guarantee that identifying and establishing alternative relationships with such sources will be successful, cost effective, or completed on a timely basis without significant delay in the development or commercialization of our product candidates. All of the vendors we use are required to conduct their operations under current Good Manufacturing Practices, or cGMP, a regulatory standard for the manufacture of pharmaceuticals.
Commercialization
We own or control exclusive rights to all three of our product candidates in the markets of the United States, France, Germany, Italy, Spain, the United Kingdom, and Japan. We plan to pursue regulatory approvals for our products in the United States and the European Union, and may independently commercialize these products in the United States. In doing so, we may engage strategic partners to assist with the sales and promotion of our products.
Our anticipated commercialization strategy in the United States would target key prescribing physicians, including specialists such as gastroenterologists, as well as provide patients with support programs to ensure product access. Outside of the United States, we plan to seek partners to commercialize our products via out-licensing agreements or other similar commercial arrangements.
| 24 |
Competition
The pharmaceutical industry is highly competitive and characterized by intense and rapidly changing competition to develop new technologies and proprietary products. Our potential competitors include both major and specialty pharmaceutical companies worldwide. Our success will be based in part on our ability to identify, develop and manage a portfolio of product candidates that are safer and more effective than competing products.
The competitive landscape in celiac disease is currently limited, which we believe is due to lack of significant past R&D investments and lack of recognition and education around the disease. To our knowledge, there are no late stage competitors entering Phase 3 clinical trials or any who have successfully completed Phase 2 studies to date. However, in recent years large pharmaceutical companies have begun to expand their focus areas to autoimmune diseases such as celiac disease, and given the unmet medical needs in these areas, we anticipate increasing competition. A few early stage programs are active, with time to enter Phase 1 clinical trials still several years away, including Roche/Genetech’s RG7625 (cathepsin S inhibitor), Takeda/PvP’s KumaMax (gluten degrading enzyme), Celimmune/Amgen’s AMG-714 (an IL-15 MAb) and Dr. Falk Pharma/Zeria’s ZED-1227 (a tTG-2 inhibitor). ImmunogenX’s IMGX003 (two gluten degrading enzymes) failed to meet its primary endpoint in a Phase 2b trial in 2015.
| Product | Status | Mechanism | Company | Route | Product Type | |||||
| AMG 714 | Phase 2 |
Anti-IL-15 MAb |
Celimmune/ Amgen |
Subcutaneous; 2x/month |
MAb (humanized) | |||||
| ZED-1227 | Phase 1b | TGase-2 inhibitor |
Zedira GmbH/ Dr Falk Pharma |
Oral |
Small molecule (peptidomimetic) | |||||
| Nexvax2 | Phase 1 | Tolerizing vaccine | ImmusanT | Intradermal | 3 gliadin epitopes (peptides) | |||||
| KumaMax | Pre-clinical | Enzymatic degradation of gluten |
Takeda/PvP Biologics |
Oral | Recombinant enzyme |
Table 4: Current celiac drugs in development are still in pre-clinical to early Phase 2 proof-of-concept stage. No drugs have completed a successful Phase 2b efficacy trial other than larazotide.
Ulcerative colitis drug development has historically been primarily focused on the moderate-to-severe UC population with little investment and research and development in mild-to-moderate UC, which is the majority of the patient populations. Current treatments for mild-to-moderate UC include the mesalamine reformulations that are pictured in Figure 15 below and described above under the heading “History of Drug Development in Mild to Moderate Ulcerative Colitis,” as well as Lialda, Pentasa, Asacol HD and Apriso, Valeant/Salix’s Uceris (oral MMX-formulated budesonide; a corticosteroid) and 5-mercaptopurine (severe side effects). Eventually, half of the mild-to-moderate UC patients progress from mesalamine to the more expensive biologics, which creates a significant potential market opportunity for any drug that is more effective than mesalamine and less expensive than the biologics.
| 25 |

Figure 15: Other than various reformulations of mesalamine which have been used for the past several decades, no new drugs have been approved for mild-to-moderate UC
Government Regulations
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of drugs, such as those we are developing. Along with third-party contractors, we will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
Government Regulation of Drugs
The process required by the FDA before drug product candidates may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices, or GLP, regulation; |
| • | submission to the FDA of an Investigational New Drug application, or IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| • | approval by an independent Institutional Review Board, or IRB, or ethics committee for each clinical site before a clinical trial can begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed product candidate for its intended purpose; |
| • | preparation of and submission to the FDA of a New Drug Application, or NDA, after completion of all required clinical trials; |
| 26 |
| • | a determination by the FDA within 60 days of its receipt of a NDA to file the application for review; |
| • | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with current Good Manufacturing Practices, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the product’s continued safety, purity and potency, and of selected clinical investigational sites to assess compliance with current Good Clinical Practices, or cGCPs; and |
| • | FDA review and approval of the NDA to permit commercial marketing of the product for particular indications for use in the United States, which must be updated annually and when significant changes are made. |
The testing and approval processes require substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. Prior to beginning the first clinical trial with a product candidate, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent Institutional Review Board, or IRB, for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site, and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
For purposes of NDA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| • | Phase 1. The drug product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, the initial human testing is often conducted in patients. |
| • | Phase 2. The drug product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| • | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling. |
| • | Phase 4. In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be required as a condition to approval of the NDA. |
| 27 |
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the drug characteristics of the product candidate, and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
NDA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a NDA requesting approval to market the product for one or more indications. The NDA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a NDA requires payment of a substantial User Fee to FDA, and the sponsor of an approved NDA is also subject to annual product and establishment user fees. These fees are typically increased annually. A waiver of user fees may be obtained under certain limited circumstances.
Within 60 days following submission of the application, the FDA reviews an NDA to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any NDA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the NDA must be resubmitted with the additional information. Once a NDA has been filed, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process may be significantly extended by FDA requests for additional information or clarification. The FDA reviews a NDA to determine, among other things, whether a product is safe and effective for the indication being pursued, and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety and effectiveness. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a NDA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a NDA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and we may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. After the FDA evaluates a NDA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may request additional information or clarification. The FDA may delay or refuse approval of a NDA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the NDA with a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
| 28 |
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs that meet certain criteria. Specifically, new drug products are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a fast track product, the FDA may consider sections of the NDA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A fast track designated product candidate may also qualify for priority review, under which the FDA sets the target date for FDA action on the NDA at six months after the FDA accepts the application for filing. Priority review is granted when there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the accelerated approval program, the FDA may approve an NDA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Products subject to accelerated approval must have associated marketing materials submitted for pre-approval by the FDA’s Office of Prescription Drug Promotion during the pre-approval review period. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the product’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit. In addition, the Food and Drug Administration Safety and Innovation Act, or FDASIA, which was enacted and signed into law in 2012, established breakthrough therapy designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Sponsors may request the FDA to designate a breakthrough therapy at the time of or any time after the submission of an IND, but ideally before an end-of-Phase 2 meeting with FDA. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the therapy; providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and considering alternative clinical trial designs when scientifically appropriate, which may result in smaller or more efficient clinical trials that require less time to complete and may minimize the number of patients exposed to a potentially less efficacious treatment. Breakthrough designation also allows the sponsor to file sections of the NDA for review on a rolling basis. We may seek designation as a breakthrough therapy for some or all of our product candidates.
Fast Track designation, priority review and breakthrough therapy designation do not change the standards for approval but may expedite the development or approval process.
Orphan Drug Status
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drug candidates intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that costs of research and development of the drug for the indication can be recovered by sales of the drug in the United States. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Although there may be some increased communication opportunities, orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a drug candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same drug for the same indication for seven years, except in very limited circumstances, such as if the second applicant demonstrates the clinical superiority of its product or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
| 29 |
Orphan drug exclusivity could block the approval of our drug candidates for seven years if a competitor obtains approval of the same product as defined by the FDA or if our drug candidate is determined to be contained within the competitor’s product for the same indication or disease.
As in the United States, designation as an orphan drug for the treatment of a specific indication in the European Union, must be made before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including up to 10 years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product.
The FDA and foreign regulators expect holders of exclusivity for orphan drugs to assure the availability of sufficient quantities of their orphan drugs to meet the needs of patients. Failure to do so could result in the withdrawal of marketing exclusivity for the orphan drug.
Post-Approval Requirements
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, distribution, and advertising and promotion of the product. After approval, most changes to the approved product labeling, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMP, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. We cannot be certain that we or our present or future suppliers will be able to comply with the cGMP regulations and other FDA regulatory requirements. If our present or future suppliers are not able to comply with these requirements, the FDA may, among other things, halt their clinical trials, require them to recall a product from distribution, or withdraw approval of the NDA.
Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing.
The FDA may withdraw approval of an NDA if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market, or product recalls; |
| • | fines, warning letters, or holds on post-approval clinical studies; |
| • | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; or |
| 30 |
| • | injunctions or the imposition of civil or criminal penalties. |
The FDA closely regulates the marketing, labeling, advertising and promotion of drugs and biologics. A company can make only those claims relating to safety and efficacy that are consistent with the FDA approved label and with FDA regulations governing marketing of prescription products. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.
Other Healthcare Laws and Compliance Requirements
Our sales, promotion, medical education, clinical research and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services and state and local governments. Our promotional and scientific/educational programs and interactions with healthcare professionals must comply with the federal Anti-Kickback Statute, the civil False Claims Act, physician payment transparency laws, privacy laws, security laws, anti-bribery and corruption laws, and additional federal and state laws similar to the foregoing.
The federal Anti-Kickback Statute prohibits, among other things, the knowing and willing, direct or indirect offer, receipt, solicitation or payment of remuneration in exchange for or to induce the referral of patients, including the purchase, order or lease of any good, facility, item or service that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, formulary managers, and beneficiaries on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to increased scrutiny and review if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal Anti-Kickback Statute has been violated. The government has enforced the federal Anti-Kickback Statute to reach large settlements with healthcare companies based on sham research or consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. Many states have similar laws that apply to their state health care programs as well as private payers.
Federal false claims and false statement laws, including the federal civil False Claims Act, or FCA, impose liability on persons and/or entities that, among other things, knowingly present or cause to be presented claims that are false or fraudulent or not provided as claimed for payment or approval by a federal health care program. The FCA has been used to prosecute persons or entities that “cause” the submission of claims for payment that are inaccurate or fraudulent, by, for example, providing inaccurate billing or coding information to customers, promoting a product off-label, submitting claims for services not provided as claimed, or submitting claims for services that were provided but not medically necessary. Actions under the FCA may be brought by the Attorney General or as a qui tam action by a private individual, or whistleblower, in the name of the government. Violations of the FCA can result in significant monetary penalties and treble damages. The federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other illegal sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, certain companies that were found to be in violation of the FCA have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or corporate integrity agreements, restricting the manner in which they conduct their business.
| 31 |
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers; knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services; and willfully obstructing a criminal investigation of a healthcare offense. Like the federal Anti-Kickback Statute, the Affordable Care Act amended the intent standard for certain healthcare fraud statutes under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Given the significant size of actual and potential settlements, it is expected that the federal government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws. Many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payer, in addition to items and services reimbursed under Medicaid and other state programs. To the extent that our products, once commercialized, are sold in a foreign country, we may be subject to similar foreign laws.
There has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposed new reporting requirements on certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, for payments or other transfers of value made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Covered manufacturers are required to collect and report detailed payment data and submit legal attestation to the accuracy of such data to the government each year. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Additionally, entities that do not comply with mandatory reporting requirements may be subject to a corporate integrity agreement. Certain states also mandate implementation of commercial compliance programs, impose restrictions on covered manufacturers’ marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare professionals.
We may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations impose specified requirements on certain health care providers, plans and clearinghouses (collectively, “covered entities”) and their “business associates,” relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain states have their own laws that govern the privacy and security of health information in certain circumstances, many of which differ from each other and/or HIPAA in significant ways and may not have the same effect, thus complicating compliance efforts.
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to them, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, disgorgement, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs, imprisonment, contractual damages, reputational harm, and diminished profits and future earnings, any of which could adversely affect our ability to operate our business and our financial results.
In addition to the foregoing health care laws, we are also subject to the U.S. Foreign Corrupt Practices Act, or FCPA, and similar worldwide anti-bribery laws, which generally prohibit companies and their intermediaries from making improper payments to government officials or private-sector recipients for the purpose of obtaining or retaining business. We have plans to adopt an anti-corruption policy, which will become effective upon the completion of this transaction, and expect to prepare and implement procedures to ensure compliance with such policy. The anti-corruption policy mandates compliance with the FCPA and similar anti-bribery laws applicable to our business throughout the world. However, we cannot assure you that such a policy or procedures implemented to enforce such a policy will protect us from intentional, reckless or negligent acts committed by our employees, distributors, partners, collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on our business, results of operations and reputation.
| 32 |
Coverage and Reimbursement
Sales of pharmaceutical products depend significantly on the extent to which coverage and adequate reimbursement are provided by third-party payers. Third-party payers include state and federal government health care programs, managed care providers, private health insurers and other organizations. Although we currently believe that third-party payers will provide coverage and reimbursement for our product candidates, if approved, we cannot be certain of this. Third-party payers are increasingly challenging the price, examining the cost-effectiveness, and reducing reimbursement for medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. The U.S. government, state legislatures and foreign governments have continued implementing cost containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. We may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of our products. The product candidates that we develop may not be considered cost-effective and thus may not be covered or sufficiently reimbursed. It is time consuming and expensive for us to seek coverage and reimbursement from third-party payers, as each payer will make its own determination as to whether to cover a product and at what level of reimbursement. Thus, one payer’s decision to provide coverage and adequate reimbursement for a product does not assure that another payer will provide coverage or that the reimbursement levels will be adequate. Moreover, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Reimbursement may not be available or sufficient to allow them to sell our products on a competitive and profitable basis.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could materially affect our ability to sell our products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
By way of example, in 2010 the Affordable Care Act was signed into law, intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the Affordable Care Act of importance to our potential drug candidates are:
| • | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively; |
| • | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
| 33 |
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include, among others, the Budget Control Act of 2011, which mandates aggregate reductions to Medicare payments to providers of up to 2% per fiscal year effective in 2013, and, due to subsequent legislative amendments, will remain in effect through 2024, unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for our product candidates, if approved, and, accordingly, our financial operations.
We expect that healthcare reform measures that may be adopted in the future, including the possible repeal and replacement of the Affordable Care Act which the Trump administration has stated is a priority, are unpredictable, and the potential impact on our operations and financial position are uncertain, but may result in more rigorous coverage criteria and lower reimbursement, and place additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our drugs.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products to the extent we choose to develop or sell any products outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement, and privacy, can vary greatly from country to country.
Research and Development Expenses
Private Innovate had research and development expenses of $4.0 million and $1.9 million for the years ended December 31, 2017 and December 31, 2016, respectively.
Employees
Following completion of the Merger, we now have five full-time employees. We also engage consultants to provide services to us, including clinical development, manufacturing support, regulatory support, business development, and general business operational support.
Corporate Information
Private Innovate was incorporated under the laws of North Carolina under the name “GI Therapeutics, Inc.” in 2012 and changed its name to “Innovate Biopharmaceuticals Inc.” when it converted to a Delaware corporation in 2014. In January 2018, Merger Sub merged with and into Private Innovate with Private Innovate surviving as a wholly owned subsidiary of the Company, and the Company changed its name to Innovate Biopharmaceuticals, Inc. Our principal executive offices are located at 8480 Honeycutt Road, Suite 120, Raleigh, NC 27615 and our telephone number is (919) 275-1933. Our corporate website address is http://www.innovatebiopharma.com . Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to reports filed pursuant to Sections 13(a) and 15(d) of the Exchange Act, will be made available free of charge on our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the U.S. Securities and Exchange Commission, or the SEC. The contents of our website are not incorporated into this Annual Report on Form 10-K and our reference to the URL for our website is intended to be an inactive textual reference only.
This Annual Report on Form 10-K contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this Annual Report on Form 10-K, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other company.
| 34 |
We are an “emerging growth company” as defined in the JOBS Act, and therefore we may take advantage of certain exemptions from various public company reporting requirements. As an “emerging growth company:”
| • | we will present no more than two years of audited financial statements and no more than two years of related management’s discussion and analysis of financial condition and results of operations; | |
| • | we will avail ourselves of the exemption from the requirement to obtain an attestation and report from our auditors on the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act; | |
| • | we will provide less extensive disclosure about our executive compensation arrangements; and | |
| • | we will not require stockholder non-binding advisory votes on executive compensation or golden parachute arrangements. |
However, we have
chosen to irrevocably opt out of the extended transition periods available under the JOBS Act for complying with new or revised
accounting standards. We will remain an “emerging growth company” for up to five years, although we will cease to be
an “emerging growth company” upon the earliest of (1) December 31, 2021, (2) the last day of the first fiscal year
in which our annual gross revenues are $1.07 billion or more, (3) the date on which we have, during the previous rolling three-year
period, issued more than $1 billion in non-convertible debt securities, and (4) the date on which we are deemed to be a “large
accelerated filer” as defined in the Exchange Act.
| 35 |
Item 15. Exhibits, Financial Statement Schedules.
| (a)(1) | Financial Statements |
The financial statements required by this item were previously submitted in a separate section beginning on page F-1 of the Form 10-K filed on March 14, 2018.
| (a)(2) | Financial Statement Schedules |
Financial statement schedules have been omitted because they are either not required, not applicable, or the information is otherwise included.
| (a)(3) | Exhibits |
| 36 |
EXHIBIT INDEX
| 37 |
| 38 |
| 39 |
| + | Pursuant to Regulation S-K Item 601(b)(2), certain schedules (or similar attachments) to this exhibit have not been filed herewith. A list of omitted schedules (or similar attachments) is included in the agreement. The Company agrees to furnish supplementally a copy of any such schedule (or similar attachment) to the Securities and Exchange Commission upon request; provided, however, that the Company may request confidential treatment of omitted items. | |
| † | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment pursuant to Rule 24b-2 under the Securities Exchange Act of 1934. | |
| # | Indicates management contract or compensatory plan or arrangement. |
| * | Indicates that the exhibit was previously filed or furnished on or with the Company’s Annual Report on Form 10-K on March 14, 2018, as applicable. |
| 40 |
Pursuant to the requirements of the Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Amendment No. 1 to its Annual Report on Form 10-K/A to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: June 29, 2018 | Innovate Biopharmaceuticals, Inc. | |
| By | /s/ Christopher Prior, Ph.D. | |
| Name: Christopher Prior, Ph.D. | ||
| Title: Chief Executive Officer | ||
| 41 |