Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
—————
FORM 10-K
——————
|
ý
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the fiscal year ended March 31, 2018
OR
|
☐
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from _________ to _________
Commission File Number 001-38355
NEMAURA MEDICAL INC.
(Exact name of registrant as specified in its charter)
|
Nevada
|
46-5027260
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
Advanced Technology Innovation Centre,
Loughborough University Science and Enterprise Parks
5 Oakwood Drive,
Loughborough, Leicestershire
LE11 3QF
United Kingdom
(Address of principal executive offices) (Zip Code)
Registrant's telephone number, including area code: + 44 1509 222912
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act:
|
Title of each class
|
|
Common Stock,$0.001 Par Value
|
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ý.
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ý.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ☐.
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ☐.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer", "smaller reporting company" and "emerging growth company" in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐
|
Accelerated filer ý
|
|
Non-accelerated filer ☐ (Do not check if a smaller reporting company)
|
Smaller reporting company ☐
|
|
Emerging growth company ý
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ý.
The aggregate market value of the registrant’s common stock held by non-affiliates computed based on the closing sales price of such stock on September 30, 2017 was $291,923,562.
The number of shares outstanding of the registrant's common stock, as of June 7, 2018 was 205,000,000.
NEMAURA MEDICAL INC.
INDEX TO ANNUAL REPORT ON FORM 10-K
|
|
Page
|
|
||||||
|
PART I
|
|
|
|
|||||
|
|
|
|
||||||
|
Item 1.
|
|
|
Business.
|
|
|
|
2
|
|
|
Item 1A.
|
|
|
Risk Factors.
|
|
|
|
14
|
|
|
Item 1B.
|
|
|
Unresolved Staff Comments.
|
|
|
|
23
|
|
|
Item 2.
|
|
|
Properties.
|
|
|
|
23
|
|
|
Item 3.
|
|
|
Legal Proceedings.
|
|
|
|
23
|
|
|
Item 4.
|
|
|
Mine Safety Disclosures
|
|
|
|
23
|
|
|
|
|
|
||||||
|
PART II
|
|
|
|
|||||
|
|
|
|
||||||
|
Item 5.
|
|
|
Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
|
|
|
|
23
|
|
|
Item 6.
|
|
|
Selected Financial Data.
|
|
|
|
23
|
|
|
Item 7.
|
|
|
Management's Discussion and Analysis of Financial Condition and Results of Operations.
|
|
|
|
23
|
|
|
Item 7A.
|
|
|
Quantitative and Qualitative Disclosures About Market Risk.
|
|
|
|
29
|
|
|
Item 8.
|
|
|
Financial Statements and Supplementary Data.
|
|
|
|
F-1
|
|
|
Item 9.
|
|
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
|
|
|
|
30
|
|
|
Item 9A.
|
|
|
Controls and Procedures.
|
|
|
|
30
|
|
|
Item 9B.
|
|
|
Other Information.
|
|
|
|
31 |
|
|
|
|
|
||||||
|
PART III
|
|
|
|
|||||
|
|
|
|
||||||
|
Item 10.
|
|
|
Directors, Executive Officers and Corporate Governance.
|
|
|
|
32
|
|
|
Item 11.
|
|
|
Executive Compensation.
|
|
|
|
35
|
|
|
Item 12.
|
|
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
|
|
|
|
36
|
|
|
Item 13.
|
|
|
Certain Relationships and Related Transactions, and Director Independence.
|
|
|
|
37
|
|
|
Item 14.
|
|
|
Principal Accountant Fees and Services.
|
|
|
|
37
|
|
|
|
|
|
||||||
|
PART IV
|
|
|
|
|||||
|
|
|
|
||||||
|
Item 15.
|
|
|
Exhibits, Financial Statement Schedules.
|
|
|
|
38
|
|
DISCLOSURE REGARDING FORWARD-LOOKING STATEMENTS
Certain statements contained in this Annual Report that are not historical facts constitute forward-looking statements, within the meaning of the Private Securities Litigation Reform Act of 1995, and are intended to be covered by the safe harbors created by that Act. Reliance should not be placed on forward-looking statements because they involve known and unknown risks, uncertainties, and other factors, which may cause actual results, performance, or achievements to differ materially from those expressed or implied. Any forward-looking statement speaks only as of the date made. We undertake no obligation to update any forward-looking statements to reflect events or circumstances after the date on which they are made.
The words "believe," "anticipate," "design," "estimate," "plan," "predict," "seek," "expect," "intend," "may," "could," "should," "potential," "likely," "projects," "continue," "will," and "would" and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These forward-looking statements are not guarantees of the future as there are a number of meaningful factors that could cause Nemaura Medical Inc.’s (“Nemaura Medical”) actual results to vary materially from those indicated by such forward-looking statements. These statements are based on certain assumptions made based on experience, expected future developments and other factors Nemaura Medical believes are appropriate in the circumstances. Factors which could cause actual results to differ from expectations, many of which are beyond Nemaura Medical’s control, include, but are not limited to, obtaining regulatory approval for our sugarBEAT device, conducting successful clinical trials, executing agreements required to successfully advance the Company's objectives; retaining the management and scientific team to advance the product; overcoming adverse changes in market conditions and the regulatory environment; obtaining and enforcing intellectual property rights; obtaining adequate financing in the future through product licensing, public or private equity or debt financing or otherwise; dealing with general business conditions and competition; and other factors referenced herein in “Risk Factors.” Except as required by law, we do not assume any obligation to update any forward-looking statement. We disclaim any intention or obligation to update or revise any forward-looking statement, whether as a result of new information, future events or otherwise.
1
PART I
ITEM 1. BUSINESS.
Business Overview
We are a medical technology company developing sugarBEAT®, a non-invasive, affordable and flexible continuous glucose monitoring system for adjunctive use by persons with diabetes. SugarBEAT consists of a disposable adhesive skin-patch connected to a rechargeable wireless transmitter and displays glucose readings at regular five minute intervals via a mobile app. SugarBEAT works by extracting glucose from the skin into a chamber in the patch that is in direct contact with an electrode-based sensor. The transmitter sends the raw data to a mobile app where it is processed by an algorithm and displayed as a glucose reading, with the ability to track and trend the data over days, weeks and months. While sugarBEAT requires once per day calibration by the patient using a blood sample obtained by a finger stick, we believe sugarBEAT will be adopted by non-insulin dependent persons with diabetes alongside insulin-injecting persons with diabetes who all perform multiple daily finger sticks to manage their disease. We have applied for an on-site CE Mark review and approval for sugarBEAT, and expect this to be completed by the end of 2018 to enable commercial launch in Europe. CE approval is the process to achieve a mandatory conformity marking for the sugarBEAT device to allow it to be legally sold in the European Union. It is a manufacturers' declaration that the product meets the requirements of the applicable European laws. We also plan to commence a 525 patient days clinical trial in the U.S. to support a Pre-Market Approval Application ("PMA") for FDA approval to market sugarBEAT in the U.S. The timing of such clinical trial has not yet been determined.
We previously developed a wristwatch-based version of sugarBEAT for which we obtained CE approval in February 2016. Since then we have developed sugarBEAT using the same underlying technology as the wristwatch. The European clinical trial programme for sugarBEAT evaluated 525 patient days across 75 Type 1 and Type 2 diabetic patients and was completed in December 2017. We are currently awaiting CE approval to allow sugarBEAT to be legally sold in the European Union. CE approval is disclosed by the use of the CE Mark, a manufacturers' declaration that the product meets the requirements of the applicable European laws. Although at this time we may not seek to commercially market and sell the wristwatch-based version of sugarBeat, we may continue to use it in clinics, or sell it as an introductory product in the event there are delays in securing CE approval for the skin-patch version.
We believe there are additional applications for sugarBEAT and the underlying BEAT technology platform, which may include:
| · |
a web-server accessible by physicians and diabetes professionals to track the condition remotely, thereby reducing healthcare costs and managing the condition more effectively;
|
·
| · |
a complete virtual doctor that monitors a person's vital signs and transmits results via the web; and
|
·
| · |
other patches using the BEAT technology platform to measure alternative analytes, including lactate, uric acid, lithium and drugs. This would be a step-change in the monitoring of conditions, particularly in the hospital setting. Lactate monitoring is currently used to determine the relative fitness of professional athletes.
|
Our Business Strategy
We intend to lead in the discovery, development and commercialization of innovative and targeted diagnostic medical devices that improve disease monitoring, management and overall patient care. Specifically, we intend to focus on the monitoring of molecules that can be drawn out through the skin non-invasively using our technology platform. In addition to glucose, such molecules may include lactic acid monitoring and the monitoring of prescription drugs and blood biomarkers that may help in the diagnosis, prevention or management of diseases, such as diabetes. We plan to take the following steps to implement our broad business strategy. Our key commercial strategies post-approval will first be implemented in Europe and then in parts of the Middle East and Asia, and then the U.S., as follows:
· Commercialise sugarBEAT in the United Kingdom and Republic of Ireland with Dallas Burston Pharma (Jersey) Limited, with whom we have an exclusive marketing rights agreement for these two countries.
We have also signed a full commercial agreement with Dallas Burston Ethitronix (Europe) Limited in May 2018 for all other European territories as part of an equal joint venture agreement. The joint venture intends to seek sub-license rights opportunities to one or more leading companies in the diabetes monitoring space, to leverage their network, infrastructure and resources.
Dallas Burston (Jersey) Limited was founded by Dr. Dallas Burston, MBBS, an entrepreneur who has founded and sold several companies specializing in marketing pharmaceuticals. For example, in 1999, he sold 49% of Ashbourne Pharmaceuticals to HSBC Private Equity for £32 million and Bartholomew-Rhodes to Galen Ltd. for £19.8 million. More recently, in 2015, he sold DB Ashbourne Limited, a provider of off-patent branded pharmaceuticals for the UK market, to Ethypharm. At the time of the sale, DB Ashbourne Limited was estimated to have revenue of approximately £90 million.
· Establish licensing or joint venture agreements with other parties to market sugarBEAT in other geographies. We are in detailed discussions and negotiations with several other parties worldwide for licensing or joint venture agreements for the sale of the sugarBEAT device
·
· Conduct a clinical trial in the U.S. to support a PMA for sugarBEAT. We will conduct further clinical trials to support a PMA for FDA approval to market sugarBEAT in the U.S, timings for which are yet to be confirmed.
· Expand the indications for which the sugarBEAT device may be used. We believe that the sugarBEAT device may offer significant benefits as compared to those found in the non-acute setting for the monitoring of other diseases. This includes monitoring of lactic acid for performance athletics, and the monitoring of drugs. We intend to complete initial proof of concept in laboratory settings followed by a clinical program.
2
· Expand our product pipeline through our proprietary platform technologies, acquisitions and strategic licensing arrangements. We intend to leverage our proprietary platform technologies to grow our portfolio of product candidates for the diagnosis of diabetes and other diseases. In addition, we intend to license our product and acquire products and technologies that are consistent with our research and development and business focus and strategies. This may include drug delivery products for the improved management of diabetes, for example improved insulin injector systems, and/or combination drug products for diabetes related drugs.
Product Development
Management has extensive experience in regulatory and clinical development of diagnostic medical devices. We intend to take advantage of this experience in the field of diagnostic medical devices in an attempt to increase the probability of product approval. The overall regulatory process for diagnostic medical devices for diabetes is currently similar to those governing other diagnostic devices. The timelines are shorter than where for example new drugs or completely invasive diagnostic devices are trialled in clinics. We believe that the non-invasive nature of sugarBEAT means the device can be tested and evaluated for its clinical output, in this case the accuracy and safety with which it can trend blood glucose levels, which is in the order of several hours and days to see the endpoint, as compared to several months and years for an invasive device. In addition, because the results are instantaneous, and the device is worn for 24 hours at any given time, the clinical trials do not initially require long term follow-up for primary endpoints, which ordinarily would otherwise take significant periods of time to evaluate. As we continue to raise funds for marketing the device in some European Union territories, we also intend to seek collaborations with future licensees and marketing partners to achieve our product development and meet our projected milestones.
The table below provides our current estimate of our timeline:
Product Development Timelines
|
Milestone
|
Target Start Date
|
Target Completion Date
|
|
Completion of clinical studies in Type 1 and Type 2 diabetic subjects to define final device claims and for submission for CE Mark approval with final device claims.
|
July 2017
|
Completed
|
|
Scale up of commercial sensor/patch manufacturing
(Scale up means we have started looking at larger scales - sufficient for product launch in the UK. It refers to the manufacturing process for sensors.)
|
January 2017
|
Completed
|
|
Scale up of device (transmitter) manufacturing
|
January 2017
|
Ongoing
|
|
CE Mark for body worn transmitter device
|
April 2018
|
December 2018
|
|
Commercial launch in the UK, followed by major territories in Europe
|
Q4 2018
|
Staggered launch
|
|
Commence clinical trial to support U.S. PMA submission
|
To be determined
|
To be determined
|
Market Opportunity for the Company's Products
According to the International Diabetes Federation Atlas (the "IDF"), there are approximately 425 million people in the world who had diabetes as of December 2017. The IDF is predicting that by 2035 this will rise to 592 million people. The number of people with Type 2 diabetes is increasing in every country and currently eighty percent (80%) of people with diabetes live in low- and middle-income countries. The greatest number of people with diabetes is between 40 and 59 years of age.
Statistics published by the IDF report that diabetes is a huge and growing problem, and the costs to society are high and escalating. In addition, Europe has the highest prevalence of children with Type 1 diabetes.
Statistical Data for Diabetes in Europe
|
2013
|
2035
|
|
|
Adult population
(20-79 years, millions)
|
659
|
669
|
|
Diabetes (20 – 79 years)
|
||
|
Regional prevalence (%)
|
8.5
|
10.3
|
|
Comparative prevalence (%)
|
6.8
|
7.1
|
|
Number of people with diabetes
(millions)
|
56.3
|
68.9
|
|
Impaired Glucose Tolerance (20 – 79 years)
|
||
|
Regional prevalence (%)
|
9.2
|
11.0
|
|
Comparative prevalence (%)
|
8.1
|
8.9
|
|
Number of people with IGT (millions)
|
60.6
|
73.7
|
|
Type 1 diabetes (0 – 14 years)
|
||
|
Number of children with Type 1
diabetes (thousands)
|
129.4
|
-
|
|
Number of newly diagnosed cases per year (thousands)
|
20.0
|
-
|
Each year approximately 600,000 people die from diabetes in Europe.
3
Deaths From Diabetes
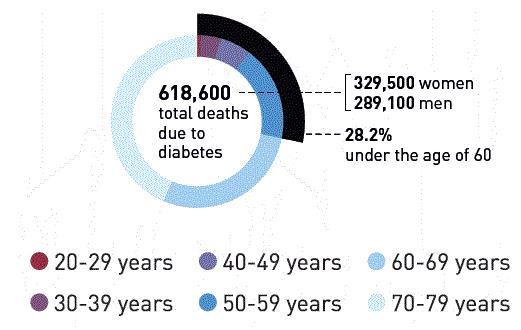
Europe has the highest incidence of children with Type 1 diabetes according to data supplied from IDF.org. The top five countries for the number of people afflicted with diabetes in Europe are listed in the table below.
Top 5 Countries In Europe For People Afflicted With Diabetes 20-79 Years (2013)
|
Countries/Territories
|
Millions
|
|
Russian Federation
|
10.9
|
|
Germany
|
7.6
|
|
Turkey
|
7.0
|
|
Spain
|
3.8
|
|
Italy
|
3.6
|
Type 1 diabetes, once known as juvenile diabetes or insulin-dependent diabetes, is a chronic condition in which the pancreas produces little or no insulin, a hormone needed to allow sugar (glucose) to enter cells to produce energy. The far more common Type 2 diabetes occurs when the body becomes resistant to the effects of insulin or doesn't make enough insulin.
Various factors may contribute to Type 1 diabetes including genetics and exposure to certain viruses. Although Type 1 diabetes typically appears during childhood or adolescence, it also can develop in adults.
Despite active research, Type 1 diabetes has no cure, although it can be managed. With proper treatment, people who have Type 1 diabetes can expect to live longer, healthier lives than they did in the past. Type 1 diabetes includes autoimmune Type 1 diabetes (Type 1a) which is characterized by having positive autoantibodies, as well as idiopathic Type 1 diabetes (Type 1b) where autoantibodies are negative and c-peptide is low. Patients with Type 1 diabetes (insulin dependent) require long term treatment with exogenous insulin and these patients perform self-monitoring of blood glucose (SMBG) to calculate the appropriate dose of insulin. SMBG is done by using blood samples obtained by finger sticks but frequent SMBG does not detect all the significant deviations in blood glucose, specifically in patients who have rapidly fluctuating glucose levels.
Type 2 diabetes, once known as adult-onset or noninsulin-dependent diabetes, is a chronic condition that affects the way your body metabolizes sugar (glucose), your body's main source of fuel. With Type 2 diabetes, your body either resists the effects of insulin, a hormone that regulates the movement of sugar into your cells, or doesn't produce enough insulin to maintain a normal glucose level. Untreated, Type 2 diabetes can be life-threatening.
More common in adults, Type 2 diabetes increasingly affects children as childhood obesity increases. There's no cure for Type 2 diabetes, but it can be managed by eating well, exercising and maintaining a healthy weight. If diet and exercise don't control the blood sugar, diabetes medications or insulin therapy may be required.
Each year, millions of patients undergo diabetes testing in the European Union and in the U.S. The main reason for this testing is to detect and evaluate diabetes in patients with symptoms of diabetes. These studies provide clinical benefit in the initial evaluation of patients with suspected but unproven diabetes, and in those patients in whom a diagnosis of diabetes has been established and information on prognosis or risk is required.
We believe that our market opportunity is a direct function of the number of persons tested, diagnosed and treated for either Type 1 or Type 2 diabetes. The IDF indicates that the total world market opportunity for a continuous glucose monitoring device is in the billions of dollars and is projected to grow annually through the year 2035.
We do not believe it is possible to estimate the number of diabetes patients that undergo finger pricks or other types of invasive glucose monitoring. However, we are unaware of any product currently on the market that may allow for non-invasive continuous glucose monitoring. We believe the sugarBEAT device may be readily adopted by the medical community for the assessment of a patient continuously.
We believe our non-invasive sugarBEAT device possesses many significant advantages and may represent an ideal device for the detection of discordances in an individual's blood sugar levels. If approved for commercialization, we believe the sugarBEAT device may represent a best in class non-invasive continuous glucose monitoring device to reach those afflicted with diabetes. While we cannot estimate the market share that our sugarBEAT device may capture, we believe that the sugarBEAT device will capture a significant share of the non-invasive continuous glucose monitoring market, in-particular the market that has been established by the Abbott Freestyle Libre device for glucose trending, as well as be adopted by non-insulin dependent diabetics who have not historically used continuous glucose monitoring devices due to their invasiveness.
Commercialization Plan
We intend to develop our products through the completion of European CE mark and FDA PMA approvals, to verify the claims that the device may be used as an adjunct to a finger-stick measurement, and/or a glucose trending device such as those claims made by the Abbott Freestyle Libre device. We will seek to partner with organizations that may facilitate the further development and distribution of our products at all stages of development. We also intend to seek strategic partners early in the research and development cycle for programs that may fall outside of our core competencies.
4
Competitive Landscape
We expect to compete with several medical device manufacturing companies including Dexcom, Abbott, and Senseonics. Our competitors may:
· develop and market products that are less expensive or more effective than our future product;
· commercialize competing products before we or our partners can launch any products developed by us;
· operate larger research and development programs or have substantially greater financial resources than we do;
· initiate or withstand substantial price competition more successfully than we can;
· have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent;
· more effectively negotiate third-party licenses and strategic relationships; and
· take advantage of acquisition or other opportunities more readily than we can.
We will compete for market share against large pharmaceutical and biotechnology companies, smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their partners, may develop new products that will compete with ours, and these competitors may, and in certain cases do, operate larger research and development programs or have substantially greater financial resources than we do.
We anticipate that we will have competition from specific companies. Although it is difficult to analyze our major competitors since currently there are no non-invasive diagnostic medical devices to continuously monitor blood glucose levels, we anticipate that specific companies may compete with us in the future.
Information relating to our competitors is listed in the table below.
|
FreeStyle Libre™(1)
|
Platinum G6®(2)
|
Platinum G5®(3)
|
Eversense™(4)
|
SugarBEAT®
|
|
|
Manufacturer
|
Abbott
|
Dexcom
|
Dexcom
|
Senseonics
|
Nemaura Medical
|
|
Technology
|
Inserted Sensor
|
Inserted Sensor
|
Inserted Sensor
|
Implanted Sensor
|
Non-invasive Sensor
|
|
Reliability (Overall MARD)
|
11.4%
|
9.8%
|
9.0%
|
11.4%
|
<14%*
|
|
Reliability (Clarke Error Grid A+B zone)
|
99%
|
Not available
|
97.0%
|
99.1%
|
>95.0%
|
|
Patients Studied
|
72
|
324
|
97
|
44
|
>75
|
|
Patient Days Studies
|
14
|
10
|
9
|
90
|
1 to 4
|
|
Warm-up Time
|
1 hour
|
2 hours
|
2 hours
|
NA
|
30-60 min
|
|
Daily Calibration
|
None
|
None
|
2x
|
2x
|
1x
|
|
Glucose Display Frequency
|
On manual activation of sensor
|
Every 5 min
|
Every 5 min
|
Every 5 min
|
Every 5 min
|
|
Patch/Senor Life
|
14 days
|
10 days
|
7 days
|
90 days
|
1 day
|
|
Regulatory Approvals
|
EU
|
US
|
Worldwide
|
EU
|
EU **
|
|
Basis for reimbursement
|
Finger stick
|
Not available
|
CGM
|
CGM
|
Finger stick
|
|
Daily Avg. Reimbursement Cost
|
$2.50 (Germany)
|
Not available
|
$9 (US)
|
Not available
|
$2.50***
|
|
Daily Retail Cost UK (exc. VAT)
|
£3.50 (Patch)
£50 (Reader)
|
Not available
|
£7.30 (Patch)
£475 (Hardware)
|
Not available
|
£2*** (Daily Patch)
£30*** (Transmitter)
|
Sources: (1) Diabetes Technology & Therapeutics, Timothy Bailey, MD, et al., Nov. 2015; (2) Dexcom’s press release, Mar. 2018; Dexcom G6 user’s guide (3) Dexcom’s press release, Aug. 2015; Dexcom G5 user’s guide; (4) SenseonicsHoldings’ 8-K, Dec. 2015. * based on summary data released in January 2018; ** CE Mark obtained on watch format, with CE Mark for patch format applied for and expected before the end of calendar year 2018. *** Estimated
Regulatory Requirements
Our device has been electrically safety tested, and all biocompatibility conformance also demonstrated, against the relevant European Medical Device Directives. When new materials are introduced, these undergo a biocompatibility risk assessment, and further testing where necessary. Batches of the device and patches were manufactured for human clinical studies that took place between November 2014 and December 2015. This was a functional watch device with a wire connection to a skin adhered sensor and electrode. Subsequent to studies conducted in India the device received a CE mark approval in February 2016. The device has since been upgraded to include wireless communication from a body worn/adhered transmitter and also to reduce the device size, and with an enhanced sensor system. Further clinical studies are ongoing to confirm the accuracy of the device and absence of skin irritation after which a further CE submission will be made to include new claims on the basis of which the body worn device will be launched in those territories that accept the CE mark approval. The watch device has been further miniaturised and in light of its somewhat obtrusive nature, it has been reserved for future clinical studies in critical care settings.
Prior to launching commercial sales of our product, we must complete key material points:
| · |
Completion of the technical dossier, documenting the entire design process including the industrial design, electronic design and software design for the final commercial product, incorporating the final aesthetics and materials for product launch.
|
| · |
Completion of human clinical studies in Type I and Type II diabetic patients against a defined clinical protocol, the outcome of which must support the claims for the device; additional ethics committee approvals and regulatory body approvals will be required if the device is to be tested in clinics other than those where ethics approval has already been obtained, or if clinical studies are planned in other countries, respectively.
|
| · |
CE approval in Europe and subsequent regulatory approvals in other territories with new claims; and
|
| · |
Prepare the body worn transmitter, and sensor-electrode system for manufacturing for commercial sales, i.e., in large volumes. The patches (containing the sensors) and the device have been manufactured in small batches sufficient for clinical studies and laboratory testing. The scale up of the processes will be undertaken to mass-produce the sensors and patches and the devices in a scale that allows large volume batches to be produced cost effectively. This is necessary to ensure that the manufacturing costs of our products are minimized in order to effectively meet market demands.
|
5
Intellectual Property
We believe that clear and extensive intellectual property relating to our technologies is central to long-term success and we intend to invest accordingly. This applies to both domestic and international patent coverage, and trade secrets, and trademarks.
The SugarBEAT technology is protected by our portfolio of intellectual property comprised of issued and pending patents and trade secrets covering a range of claims, including the methods and apparatus for measuring glucose extracted from human skin in a non-invasive manner, the formula for the cumulative measurement of an analyte, and the formulation and process for preparation of the enzyme solution used in the sensor.
On May 8, 2014, NDM Technologies Limited, a related company, assigned the UK patent application 1208950.4 and International (PCT) patent application PCT/GB2013/051322 entitled "Cumulative Measurement of an Analyte" to Dermal Diagnostics Limited (“DDL”) for a nominal consideration.
A further patent is planned for filing which allows the application of the sugarBEAT device and sensor to the skin in a single step, eliminating several manual steps.
Additionally, we retain substantial trade secrets relating to the sensor formulation, which have taken over five years to develop, and will prove very difficult to reverse engineer as it consist of formulation components in addition to processing methods in unique combinations that are unique to the final functional sensor. Patents will not be filed on this aspect of the technology to avoid any public dissemination of the know-how.
These patents and know-how cover aspects of the technology platform. Furthermore the trademark BEAT and sugarBEAT has been registered in all major territories globally. Accordingly, all intellectual property essential to the sugarBEAT product is owned by us, and not subject to royalty payments. We intend to take the lead in the preservation and/or prosecution of these patents and patent applications going forward as required. We intend to file additional patents as the development progresses, where deemed to be of value to protecting the technology platform and future modifications and improvements. Where patents cannot be secured, the intellectual property will be limited to know-how and trade secrets, and these will be diligently guarded.
Trade Secrets, Trademarks, and Patents Filed, Granted and Pending
|
IP: Patent (Core Claim), Know-how, Trademark
|
Expiration Date
|
Jurisdictions in which Granted/ Issued
|
Jurisdictions in which
Pending |
Ongoing Royalty or Milestone Payments
|
||
|
Patent: Cumulative Measurement of an Analyte*.
|
May 20, 2032
|
Australia, France, Germany, Italy, Poland, Spain, Netherlands, UK
|
Brazil, Canada, China, India, Japan, Qatar, United Arab Emirates, U.S.
|
None
|
||
|
Patent: Patches for Reverse Iontophoresis**
|
July 1, 2029
|
Australia, Germany, France, UK, Italy, Netherlands, Switzerland, China, Hong Kong, Japan.
|
None
|
None
|
||
|
Know-how: Sensor Formulation
|
N/A
|
Trade Secret
|
N/A
|
N/A
|
||
|
Patent: Patch Application to the skin: Device for applying the sugarBEAT to the skin in a single step, to eliminate multiple manual steps.
|
To be filed
|
N/A
|
N/A
|
N/A
|
||
|
Trademark: BEAT
|
Renewal due in 2026
|
UK, China, EU, India, Japan
|
Canada
|
N/A
|
||
|
Trademark: sugarBEAT
|
Renewal due in 2025
|
UK, Australia, Switzerland, China, Egypt, EU, Israel, India, Iran, Japan, North Korea, Morocco, Mexico, Norway, New Zealand, Russia, Singapore, Tunisia, Turkey, USA
|
Canada
|
N/A
|
* This patent provides a formula for calculating the amount of glucose extracted over a defined period of time by deducting the difference between two readings to allow rapid sensing without needing to deplete the analyte being measured.
** This patent provides a reverse iontophoresis patch with means for releasing a conductive medium onto the skin during use and means for transporting analyte to a separate location for analysis.
6
Clinical Trials
Our clinical testing is conducted by contract clinical research organizations in various centres around the world to cover a wide demographic – including Asia and Europe – and is managed by our in-house management team.
We have had 2 pre-submission meetings with the FDA in June 2016, whereby the regulatory approval route has been defined by the FDA as being PMA and a clinical roadmap clarified. As a result, a detailed clinical plan has been developed and approved internally and a clinical site in Europe has been selected and audited and approved for commencement of clinical studies using the body worn transmitter device version of the sugarBEAT.
In August 2017, we commenced a European three-stage 75 patient clinical study, consisting of 80% Type 1 and 20% Type 2 diabetics. The study was designed as a single centre open-label, single arm, within-subject comparison of sugarBEAT, with blood samples drawn from a venous catheter at corresponding time points, with glucose concentration measured using a laboratory blood glucose analyser, ARCHITECT C8000. The European clinical trial programme consisted of a total of 525 patient days, with each patient continuously wearing sugarBEAT for 14 hours on seven consecutive days in a combination of home and clinic settings. Three of the seven days were in-clinic where venous blood samples were taken at 15 minute intervals over a continuous 12 hour period. The clinical study was completed in December 2017. An interim analysis of the data has thus far indicated a precision of 1.07 and accuracy as determined by the Mean Absolute Relative Deviation (MARD) of less than 14%, with no serious or major adverse events. The precision and accuracy of sugarBEAT observed in the study was similar to other CE Mark approved continuous glucose monitors. The full clinical evaluation is expected to be reported in June 2018.
Research and development
We spent $993,833 and $1,034,605 in 2018 and 2017, respectively, on research and development. We anticipate that for the year ending March 2019, research and development expenditures will increase to further develop the device for commercial launch in the UK and Europe.
Development and clinical test costs in support of our current product, as well as costs to file patents and revise and update previous filings on our technologies, will continue to be substantial as we assess the next steps to advance the product.
Manufacturing
The manufacture and sale of CE certified medical devices are controlled and governed by guidelines stipulated in the International Organisation for Standardisation (ISO), more specifically ISO13485; sugarBEAT will be manufactured and marketed according to ISO13485 quality standards.
In preparation for our anticipated commercial launch of sugarBEAT in the UK during the fourth quarter of 2018 we initiated scale-up manufacturing of the various sugarBEAT components alongside facilities for final assembly and packaging. As part of this process, we expanded our manufacturing and assembly capabilities by occupying additional space within our existing headquarters site at Loughborough Science Park in the UK.
Manufacturers for our sensors are Parlex (a division of Johnson Electrics), Isle of White, UK; Polarseal Limited, Surrey, England for our patches; and Datalink Limited located in Loughborough, UK manufactures our electronics.
We expect to enter into the following types of agreements during 2018:
| - |
Manufacturing agreements for the sensor manufacture
|
| - |
Manufacturing agreements for the patch manufacture
|
| - |
Manufacturing agreements for the CGM watch device and transmitter device manufacture
|
Sales and Marketing
An Exclusive Marketing Rights agreement for the UK and Republic of Ireland was signed on March 31, 2014 with Dallas Burston Pharma, a Jersey (Channel Island) based company who has pharmaceutical product marketing operations in the UK and has demonstrated a very successful model for the marketing of prescription medical products directly to general practitioners. We received a non-refundable upfront payment of $1.67 million in return for providing the company with the exclusive right to sell the sugarBEAT device in the UK and Republic of Ireland, both direct to consumer and through prescriptions by general practitioners. Subsequently, on April 4, 2014, a Letter of Intent was entered into outlining the basic terms of the cost at which the patches and watch will be supplied and minimum order quantities in the first two (2) years. The key terms of the Exclusive Marketing Rights Agreement were concluded in a Commercial Agreement signed in August 2015.
In addition, a joint venture agreement was entered into with Dallas Burston Ethitronix (Europe) in May 2018, whereby we will share equally the costs and net profits of the sales of our sugarBEAT system in all territories in Europe, with the exception of the United Kingdom, which is the subject of a separate agreement with Dallas Burston Pharma (Jersey) Limited.
7
Regulatory matters
Government Regulation
Our business is subject to extensive federal, state, local and foreign laws and regulations, including those relating to the protection of the environment, health and safety. Some of the pertinent laws have not been definitively interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of subjective interpretations. In addition, these laws and their interpretations are subject to change, or new laws may be enacted.
Both federal and state governmental agencies continue to subject the healthcare industry to intense regulatory scrutiny, including heightened civil and criminal enforcement efforts. We believe that we have structured our business operations to comply with all applicable legal requirements. However, it is possible that governmental entities or other third parties could interpret these laws differently and assert otherwise. We discuss below the statutes and regulations that are most relevant to our business.
United Kingdom and Wales and the European Union regulations
Government authorities in the United Kingdom and Wales and the European Union as well as other foreign countries extensively regulate, among other things, the research, development, testing, manufacture, labelling, promotion, advertising, distribution, sampling, marketing and import and export of medical devices, including patches and other pharmaceutical products. Our body worn transmitter devices in the United Kingdom and Wales will be subject to strict regulation and require regulatory approval prior to commercial distribution. The process of obtaining governmental approvals and complying with ongoing regulatory requirements requires the expenditure of substantial time and financial resources. In addition, statutes, rules, regulations and policies may change and new legislation or regulations may be issued that could delay such approvals. If we fail to comply with applicable regulatory requirements at any time during the product development process, approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the authority's refusal to approve pending applications, withdrawals of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of our operations, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on us.
The European Commission on Public Health (the "ECPH") provides the regulation for the development and commercialization of new medical diagnostic devices. Any medical device placed on the European market must comply with the relevant legislation, notably with Directive 93/42/EEC, with the active implantable devices Directive 90/385/EEC or with the in vitro devices Directive 98/79/EC. We must first determine whether the device we intend to manufacture or import falls under any of these directives. All medical devices must fulfil the essential requirements set out in the above-mentioned directives. Where available, relevant standards may be used to demonstrate compliance with the essential requirements defined in the devices Directives.
Manufacturers also need to determine the appropriate conformity assessment route. For devices falling under Directive 93/42/EEC, other than custom-made devices and devices intended for clinical investigation, the conformity assessment route depends on the class of the device, to be determined in accordance with certain rules set forth in the directives. Once the applicable class or list has been determined, manufacturers need to follow the appropriate conformity assessment procedure. Subject to the type of the device, this may require manufacturers to have their quality systems and technical documentation reviewed by a Notified Body before they can place their products on the market. A Notified Body is a third party body that can carry out a conformity assessment recognized by the European Union. The Notified Body will need to assure itself that relevant requirements have been met before issuing relevant certification. Manufacturers can then place the CE marking on their products to demonstrate compliance with the requirements.
The CE approval is the process of achieving a mandatory conformity marking for the sugarBEAT device to allow it to be legally sold in the European Union. It is a manufacturers' declaration that the product meets the requirements of the applicable European laws. The process for the sugarBEAT device CE submission and approval will involve the following:
1. The device is classified depending on certain categories described by the European Directive with Class I products being low risk (e.g. band aid plasters), through Class III devices being the highest risk. The classes are Class I, IIa, IIb and III. Risk is based upon the potential harm to the patient should a problem arise with a product or its use. The sugarBEAT device is classified as a IIa device.
2. A 'technical file' containing all of the information required to demonstrate that the product meets the essential requirements of the European directive will be prepared. This includes information relating to performance and safety of the device such as product specifications, labelling, instructions for use, risk analysis and specific test information/clinical evidence relating to the product that support the claims being made for the product.
3. Clinical evidence included in the technical file is expected to demonstrate that the device is safe and meets defined performance requirements. This clinical evidence can be in the form of literature data where substantial published data exists that utilizes the same technique for glucose extraction and measurement (albeit in a different device format), or data from actual clinical studies performed using the sugarBEAT device. The first CE mark submission will be based on literature evaluation of 3rd party published clinical data available in the public domain. The final CE mark submission with final claims will be based on literature evaluation and actual clinical data from human clinical studies performed using the sugarBEAT device. The clinical data will be generated to show that the sugarBEAT device can trend blood glucose levels in a human subject by taking measurements up to 4 times per hour. The clinical trial data must demonstrate the sugarBEAT device blood glucose trend can be used to supplement normal finger prick measurements.
4. The technical file will be assessed by an independent inspector (the Notified Body), regulated by the competent authority, (Medicines and Healthcare products Regulatory Agency, MHRA in the United Kingdom). The Notified Body (an organization in the European Union that has been accredited by a member state to determine whether a medical device complies with the European medical device directives), will then notify The European Commission on Public Health (the "ECPH") of the approval and a certificate will be issued to the Company by the notified body and we will then be able to apply the CE mark to the device, and legally offer the product for sale in the European Economic Area (EEA).
5. The review of the technical file typically takes a matter of days although the lead time can be 90 days depending upon the notified body, and approval is usually attained within 3-6 months of submission and review.
6. Generating the information required to complete the technical file takes the most time and this information is collated throughout the product development cycle. Delays arise where the company has not consulted its Notified Body prior to technical file review and elements may require further detail before the Notified Body can confirm that the device meets the essential requirements. This could delay an approval process by several weeks or in more drastic cases by several months depending on the time taken to provide any additional information requested by the Notified Body. Nemaura has been in regular communication with the Notified Body throughout the development of the sugarBEAT device, and continues to do so for the forthcoming CE approval.
8
U.S. Food and Drug Administration regulation of medical devices.
The FDCA and FDA regulations establish a comprehensive system for the regulation of medical devices intended for human use. sugarBeat is a medical device that is subject to these, as well as other federal, state, local and foreign, laws and regulations. The FDA is responsible for enforcing the laws and regulations governing medical devices in the United States.
The FDA classifies medical devices into one of three classes (Class I, Class II, or Class III) depending on their level of risk and the types of controls that are necessary to ensure device safety and effectiveness. The class assignment is a factor in determining the type of premarketing submission or application, if any, that will be required before marketing in the United States. sugarBeat falls under Class III.
| ● |
Class I devices present a low risk and are not life-sustaining or life-supporting. The majority of Class I devices are subject only to "general controls" (e.g., prohibition against adulteration and misbranding, registration and listing, good manufacturing practices, labeling, and adverse event reporting. General controls are baseline requirements that apply to all classes of medical devices.)
|
| ● |
Class II devices present a moderate risk and are devices for which general controls alone are not sufficient to provide a reasonable assurance of safety and effectiveness. Devices in Class II are subject to both general controls and "special controls" (e.g., special labeling, compliance with performance standards, and post market surveillance. Unless exempted, Class II devices typically require FDA clearance before marketing, through the premarket notification (510(k)) process.)
|
| ● |
Class III devices present the highest risk. These devices generally are life-sustaining, life-supporting, or for a use that is of substantial importance in preventing impairment of human health, or present a potential unreasonable risk of illness or injury. Class III devices are devices for which general controls, by themselves, are insufficient and for which there is insufficient information to determine that application of special controls would provide a reasonable assurance of safety and effectiveness. Class III devices are subject to general controls and typically require FDA approval of a premarket approval ("PMA") application before marketing.
|
Unless it is exempt from premarket review requirements, a medical device must receive marketing authorization from the FDA prior to being commercially marketed, distributed or sold in the United States. The most common pathways for obtaining marketing authorization are 510(k) clearance and PMA. After preliminary discussions with the FDA in June 2016 as part of a pre-submission meeting it was determined that the pathway for sugarBeat would be a PMA approval.
Premarket approval pathway
The PMA approval process requires an independent demonstration of the safety and effectiveness of a device. PMA is the most stringent type of device marketing application required by the FDA. PMA approval is based on a determination by the FDA that the PMA contains sufficient valid scientific evidence to ensure that the device is safe and effective for its intended use(s). A PMA application generally includes extensive information about the device including the results of clinical testing conducted on the device and a detailed description of the manufacturing process.
After a PMA application is accepted for review, the FDA begins an in-depth review of the submitted information. FDA regulations provide 180 days to review the PMA and make a determination; however, in reality, the review time is normally longer (e.g., 1-3 years). During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the data supporting the application and provide recommendations to the FDA as to whether the data provide a reasonable assurance that the device is safe and effective for its intended use. In addition, the FDA generally will conduct a preapproval inspection of the manufacturing facility to ensure compliance with Quality System Regulation, which imposes comprehensive development, testing, control, documentation and other quality assurance requirements for the design and manufacturing of a medical device.
Based on its review, the FDA may (i) issue an order approving the PMA, (ii) issue a letter stating the PMA is "approvable" (e.g., minor additional information is needed), (iii) issue a letter stating the PMA is "not approvable," or (iv) issue an order denying PMA. A company may not market a device subject to PMA review until the FDA issues an order approving the PMA. As part of a PMA approval, the FDA may impose post-approval conditions intended to ensure the continued safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution, and requiring the collection of additional clinical data. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including withdrawal of the approval.
Most modifications to a PMA approved device, including changes to the design, labeling, or manufacturing process, require prior approval before being implemented. Prior approval is obtained through submission of a PMA supplement. The type of information required to support a PMA supplement and the FDA's time for review of a PMA supplement vary depending on the nature of the modification.
Clinical trials
Clinical trials of medical devices in the United States are governed by the FDA's Investigational Device Exemption ("IDE") regulation. This regulation places significant responsibility on the sponsor of the clinical study including, but not limited to, choosing qualified investigators, monitoring the trial, submitting required reports, maintaining required records, and assuring investigators obtain informed consent, comply with the study protocol, control the disposition of the investigational device, submit required reports, etc.
Clinical trials of significant risk devices (e.g., implants, devices used in supporting or sustaining human life, devices of substantial importance in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health) require FDA and Institutional Review Board ("IRB") approval prior to starting the trial. FDA approval is obtained through submission of an IDE application. Clinical trials of non-significant risk ("NSR"), devices (i.e., devices that do not meet the regulatory definition of a significant risk device) only require IRB approval before starting. The clinical trial sponsor is responsible for making the initial determination of whether a clinical study is significant risk or NSR; however, a reviewing IRB and/or FDA may review this decision and disagree with the determination.
9
An IDE application must be supported by appropriate data, such as performance data, animal and laboratory testing results, showing that it is safe to evaluate the device in humans and that the clinical study protocol is scientifically sound. There is no assurance that submission of an IDE will result in the ability to commence clinical trials. Additionally, after a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to an unacceptable health risk.
As noted above, the FDA may require a company to collect clinical data on a device in the post-market setting.
The collection of such data may be required as a condition of PMA approval. The FDA also has the authority to order, via a letter, a post-market surveillance study for certain devices at any time after they have been cleared or approved.
Pervasive and continuing FDA regulation
After a device is placed on the market, regardless of its classification or premarket pathway, numerous additional FDA requirements generally apply. These include, but are not limited to:
| ● |
Establishment registration and device listing requirements;
|
| ● |
Quality System Regulation ("QSR"), which governs the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labelling, storage, installation, and servicing of finished devices;
|
| ● |
Labelling requirements, which mandate the inclusion of certain content in device labels and labelling, and generally require the label and package of medical devices to include a unique device identifier ("UDI"), and which also prohibit the promotion of products for uncleared or unapproved, i.e., "off-label," uses;
|
| ● |
Medical Device Reporting ("MDR") regulation, which requires that manufacturers and importers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and
|
| ● |
Reports of Corrections and Removals regulation, which requires that manufacturers and importers report to the FDA recalls (i.e., corrections or removals) if undertaken to reduce a risk to health posed by the device or to remedy a violation of the Federal Food, Drug and Cosmetic Act that may present a risk to health; manufacturers and importers must keep records of recalls that they determine to be not reportable.
|
The FDA enforces these requirements by inspection and market surveillance. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include, but is not limited to, the following sanctions:
| ● |
Untitled letters or warning letters;
|
| ● |
Fines, injunctions and civil penalties;
|
| ● |
Recall or seizure of our products;
|
| ● |
Operating restrictions, partial suspension or total shutdown of production;
|
| ● |
Refusing our request for 510(k) clearance or premarket approval of new products;
|
| ● |
Withdrawing 510(k) clearance or premarket approvals that are already granted; and
|
| ● |
Criminal prosecution.
|
We would be subject to unannounced device inspections by the FDA, as well as other regulatory agencies overseeing the implementation of and compliance with applicable state public health regulations. These inspections may include our suppliers' facilities.
Other Regulation in the United Kingdom and Wales and the EU
Healthcare Reimbursement
Government and private sector initiatives to limit the growth of healthcare costs, including price regulation, competitive pricing, coverage and payment policies, and managed-care arrangements, are continuing in many countries where we do business, including the United Kingdom and Wales. These changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective medical products. Government programs, private healthcare insurance and managed-care plans have attempted to control costs by limiting the amount of reimbursement they will pay for particular procedures or treatments. This has created an increasing level of price sensitivity among customers for products. Some third-party payers must also approve coverage for new or innovative devices or therapies before they will reimburse healthcare providers who use the medical devices or therapies. Even though a new medical product may have been cleared for commercial distribution, we may find limited demand for the product until reimbursement approval has been obtained from governmental and private third-party payers.
Environmental Regulation
We are also subject to various environmental laws and regulations both within and outside the United Kingdom and Wales. Like many other medical device companies, our operations involve the use of substances, including hazardous wastes, which are regulated under environmental laws, primarily manufacturing and sterilization processes. We do not expect that compliance with environmental protection laws will have a material impact on our consolidated results of operations, financial position or cash flow. These laws and regulations are all subject to change, however, and we cannot predict what impact, if any, such changes might have on our business, financial condition or results of operations.
10
Foreign Regulation
Whether or not we obtain regulatory approval for a product, we must obtain approval from the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for EC approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement also vary greatly from country to country.
Under European Union regulatory systems, we may submit marketing authorization applications under a decentralized procedure. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval. This procedure is referred to as the mutual recognition procedure, or called the MRP.
In addition, regulatory approval of prices is required in most countries other than the United States. We face the risk that the prices which result from the regulatory approval process would be insufficient to generate an acceptable return to us or our collaborators.
EU General Data Protection Regulation
The EU General Data Protection Regulation (the “GDPR”) came into force in all EU Member States from 25 May 2018 and replaced previous EU data privacy laws. Although a number of basic existing principles will remain the same, the GDPR introduces new obligations on data controllers and rights for data subjects, including, among others:
• accountability and transparency requirements, which will require data controllers to demonstrate and record compliance with the GDPR and to provide more detailed information to data subjects regarding processing;
• enhanced data consent requirements, which includes “explicit” consent in relation to the processing of sensitive data;
• obligations to consider data privacy as any new products or services are developed and limit the amount of information collected, processed, stored and its accessibility;
• constraints on using data to profile data subjects;
• providing data subjects with personal data in a useable format on request and erasing personal data in certain circumstances; and
• reporting of breaches without undue delay (72 hours where feasible).
The GDPR also introduces new fines and penalties for a breach of requirements, including fines for serious breaches of up to the higher of 4% of annual worldwide turnover or €20m and fines of up to the higher of 2% of annual worldwide turnover or €10m (whichever is highest) for other specified infringements. The GDPR identifies a list of points to consider when imposing fines (including the nature, gravity and duration of the infringement).
11
The Company has assessed the implications of the GDPR on all personal data it holds and has implemented measures to ensure that personal data shall be:
| - |
Processed lawfully, fairly and in a transparent manner in relation to the data subject.
|
| - |
Collected for a specified, explicit and legitimate purpose and not further processed in a manner that is incompatible with those purposes.
|
| - |
Adequate, relevant and limited to what is necessary in relation to the purposes for which they are processed.
|
| - |
Kept in a form which permits identification of data subjects for no longer than is necessary for the purposes for which the personal data are processed.
|
| - |
Processed in a manner that ensures appropriate security of the personal data, including protection against unauthorised or unlawful processing and against accidental loss, destruction or damage, using appropriate technical or organisational measures.
|
| - |
Maintained accurately and up to date and that every reasonable step is taken to ensure that personal data that are inaccurate, having regard to the purposes for which they are processed, are erased or rectified without delay.
|
At the current stage of the Company’s development and, with being pre-revenue at this stage, the scope of data held, and consequently the impact of GDPR, is limited. Increased application of GDPR will be assessed and implemented prior to further Company developments that warrant additional GDPR measures. As the Company progresses with product commercialization, the extent to which GDPR will affect the Company will increase, which will require additional changes to the Company’s procedures and policies which could adversely impact operational and compliance costs. Further, there is a risk that the measures will not be implemented correctly or that individuals within the business will not be fully compliant with the new procedures. If there are breaches of these measures, the Company could face significant administrative and monetary sanctions as well as reputational damage which may have a material adverse effect on its operations, financial condition and prospects.
Corporate Information
Our principal executive offices are located at The Advanced Technology Centre, Oakwood Drive, Loughborough, Leicestershire, LE113QF, UK. Our website is located at www.nemauramedical.com and our telephone number is +44 1509 222912. Information found on, or accessible through, our website is not a part of, and is not incorporated into, this Annual Report, and you should not consider it part of the Annual Report.
Employees
We currently employ 8 personnel. We believe our relationships with our employees and contractors are good.
Corporate History and Restructuring
We are a holding corporation that owns one hundred percent (100%) of a diagnostic medical device company specializing in discovering, developing and commercializing specialty medical devices. We were organized on December 24, 2013 under the laws of the State of Nevada. We own one hundred percent (100%) of Region Green Limited, a British Virgin Islands corporation formed on December 12, 2013. Region Green Limited owns one hundred percent (100%) of the stock in Dermal Diagnostic (Holdings) Limited, an England and Wales corporation formed on December 11, 2013. Dermal Diagnostics (Holdings) Limited owns one hundred percent (100%) of the stock in Dermal Diagnostics Limited, an England and Wales corporation formed on January 20, 2009, and one hundred percent (100%) of the stock in Trial Clinic Limited, an England and Wales corporation formed on January 12, 2011.
In December 2013, we restructured the Company and re-domiciled as a domestic corporation in the United States. The corporate re-organization was accomplished to preserve the tax advantages under the laws of the England and Wales tax laws for the benefit of the shareholders of both Dermal Diagnostics Limited (“DDL”) and Trial Clinic Limited (“TCL”).
DDL is a diagnostic medical device company headquartered in Loughborough, Leicestershire, England. DDL was founded on January 20, 2009 to engage in the discovery, development and commercialization of diagnostic medical devices. The Company’s initial focus has been on the development of a novel continuous glucose monitoring (CGM) device.
On July 10, 2017 the Registrant’s Board of Directors amended the Company’s By-laws to provide that any action required or permitted by law, or by the Articles of Incorporation to be taken at any meeting of the stockholders may be taken without a meeting, without prior notice, and without a vote, if a written consent, setting forth the action so taken, shall be signed by the holders of outstanding stock having not less than the minimum number of votes that would be necessary to authorize or take such action at a meeting by less than unanimous written consent shall be given to those stockholders who have not consented in writing thereto.
12
On October 5, 2017, the Registrant entered into common stock exchange agreements (the “Agreements”) with each of its three largest shareholders, (i) Dewan F.H. Chowdhury, who is the Registrant’s Chief Executive Officer and serves as a Director on the Board of Directors of the Registrant (the “Board”), (ii) Bashir Timol, who serves as a Director on the Board, and (iii) Sufyan Ismail. Pursuant to the Agreements, the shareholders would exchange, in the aggregate, 137,324,000 shares of the Registrant’s common stock (the “Shares”) for 137,324 shares of Series A Convertible Preferred Stock (the “Transaction”). On October 10, 2017, the Registrant filed a Certificate of Designation with the Secretary of State of the State of Nevada to designate two hundred thousand shares of preferred stock as Series A Convertible Preferred Stock. Each share of Series A Convertible Preferred Stock is convertible into 1,000 shares of the Company’s common stock, automatically upon the occurrence of certain triggering events, as set forth in the Certificate of Designation, or voluntarily by the holder after February 1, 2019, if these triggering events have not occurred and the Board, in its sole and absolute discretion, could change the date of voluntary conversion. Each holder of issued and outstanding Series A Convertible Preferred Stock is entitled to a number of votes equal to the number of shares of common stock into which the Series A Convertible Preferred Stock is convertible. Holders of Series A Convertible Preferred Stock are entitled to vote on any and all matters presented to stockholders of the Company, except as provided by law. The Series A Convertible Preferred Stock has preference to the common stock as to dividends or distributions of assets upon liquidation or winding up of the Company. On November 6, 2017, the transaction was consummated and the Shares were cancelled. As a result, the Registrant has 67,676,000 shares of common stock issued and outstanding.
On February 7, 2018, the Board by unanimous written consent, approved to change the date on which the holders of Series A Convertible Preferred Stock can voluntarily convert their shares of Series A Convertible Preferred Stock from February 1, 2019 to February 7, 2018.
On February 7, 2018, the holders of the Series A Convertible Preferred Stock waived their rights to have any preferential rights to dividends or distributions of assets upon liquidation or winding up of the Company.
On June 5, 2018, the three holders of our Series A Convertible Preferred Stock (the “Series A Preferred”) each delivered notices of conversion to voluntarily convert their Series A Preferred, in the aggregate amount of 137,324 shares, into 137,324,000 shares of our common stock. The holders had the right to voluntarily convert each share of Series A Preferred into 1,000 shares of common stock of the Company. As a result of the conversion, we currently have 205,000,000 shares of common stock outstanding.
13
ITEM 1A. — RISK FACTORS
If any of the following risks actually occur, they could materially adversely affect our business, financial condition or operating results. In that case, the trading price of our common stock could decline.
Risks Related to Our Product Candidate and Operation
We are largely dependent on the success of our sole product candidate, the sugarBEAT device, and we may not be able to successfully commercialize this potential product.
We have incurred and will continue to incur significant costs relating to the development and marketing of our sole product candidate, the sugarBEAT device. We have not obtained approval to market this potential product in any jurisdiction and we may never be able to obtain approval or, if approvals are obtained, to commercialize this product successfully.
If we fail to successfully commercialize our product(s), we may be unable to generate sufficient revenue to sustain and grow our business, and our business, financial condition and results of operations will be adversely affected.
If we fail to obtain regulatory approval of the sugarBEAT device or any of our other future products, we will be unable to commercialize these potential products.
The development, testing, manufacturing and marketing of our product is subject to extensive regulation by governmental authorities in Great Britain and the European Union. In particular, the process of obtaining CE approval by a Notified Body, a third party that can carry out a conformity assessment recognized by the European Union, is costly and time consuming, and the time required for such approval is uncertain. Our product must undergo rigorous preclinical and clinical testing and an extensive regulatory approval process mandated for the CE. Such regulatory review includes the determination of manufacturing capability and product performance. As of the date of this filing we have not applied for CE approval for the latest version of our product.
Whilst we have received a CE approval on our current sugarBEAT device, which excludes any wireless communication capabilities, we can give no assurance that our future products will be approved by the European Union or Great Britain or any other governmental body. In addition, there can be no assurance that all necessary approvals will be granted for future products or that CE review or actions will not involve delays caused by requests for additional information or testing that could adversely affect the time to market for and sale of our product. Further failure to comply with applicable regulatory requirements can, among other things; result in the suspension of regulatory approval as well as possible civil and criminal sanctions.
Failure to enroll patients in our clinical trials may cause delays in developing the sugarBEAT device or any of our future products.
We may encounter delays in the development and commercialization, or fail to obtain marketing approval, of the sugarBEAT device or any other future products if we are unable to enroll enough patients to complete clinical trials. Our ability to enroll sufficient numbers of patients in our clinical trials depends on many factors, including the severity of illness of the population, the size of the patient population, the nature of the clinical protocol, the proximity of patients to clinical sites, and the eligibility criteria for the trial and competing clinical trials. Delays in planned patient enrolment may result in increased costs and harm our ability to complete our clinical trials and obtain regulatory approval.
Delays in clinical testing could result in increased costs to us and delay our ability to generate revenue.
Significant delays in clinical testing could materially adversely impact our product development costs. We do not know whether planned clinical trials will begin on time, will need to be restructured or will be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence and continue a study, delays in reaching agreement on acceptable clinical study terms with prospective sites, delays in obtaining institutional review board approval to conduct a study at a prospective site and delays in recruiting patients to participate in a study.
Significant delays in testing or regulatory approvals for any of our current or future products, including the sugarBEAT device, could prevent or cause delays in the commercialization of such product candidates, reduce potential revenues from the sale of such product candidates and cause our costs to increase.
Our clinical trials for any of our current or future products may produce negative or inconclusive results and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing for these products or cease our trials.
We will only receive regulatory approval to commercialize a product candidate if we can demonstrate to the satisfaction of the applicable regulatory agency that the product is safe and effective. We do not know whether our future clinical trials will demonstrate safety and efficacy sufficiently to result in marketable products. Because our clinical trials for the sugarBEAT device may produce negative or inconclusive results, we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing for this product or cease our clinical trials. If this occurs, we may not be able to obtain approval for this product or our anticipated time to market for this product may be substantially delayed and we may also experience significant additional development costs. We may also be required to undertake additional clinical testing if we change or expand the indications for our product.
If approved, the commercialization of our product, the sugarBEAT device, may not be profitable due to the need to develop sales, marketing and distribution capabilities, or make arrangements with a third party to perform these functions.
In order for the commercialization of our potential product to be profitable, our product must be cost-effective and economical to manufacture on a commercial scale. Subject to regulatory approval, we expect to incur significant sales, marketing, distribution, and to the extent we do not outsource manufacturing, manufacturing expenses in connection with the commercialization of the sugarBEAT device and our other potential products. We do not currently have a dedicated sales force or manufacturing capability, and we have no experience in the sales, marketing and distribution of medical diagnostic device products. In order to commercialize the sugarBEAT device or any of our other potential products that we may develop, we must develop sales, marketing and distribution capabilities or make arrangements with a third party to perform these functions. Developing a sales force is expensive and time-consuming, and we may not be able to develop this capacity. If we are unable to establish adequate sales, marketing and distribution capabilities, independently or with others, we may not be able to generate significant revenue and may not become profitable. Our future profitability will depend on many factors, including, but not limited to:
14
· the costs and timing of developing a commercial scale manufacturing facility or the costs of outsourcing the manufacturing of the sugarBEAT device;
· receipt of regulatory approval of the sugarBEAT device;
· the terms of any marketing restrictions or post-marketing commitments imposed as a condition of approval by regulatory authorities;
· the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
· costs of establishing sales, marketing and distribution capabilities;
· the effect of competing technological and market developments; and
· the terms and timing of any collaborative, licensing and other arrangements that we may establish.
Even if we receive regulatory approval for the sugarBEAT device or any other product candidates, we may never receive significant revenues from any of them. To the extent that we are not successful in commercializing our potential products, we will incur significant additional losses if we do not successfully commercialize our products.
Our proprietary rights may not adequately protect our intellectual property and product and if we cannot obtain adequate protection of our intellectual property and product, we may not be able to successfully market our product.
Our commercial success will depend in part on obtaining and maintaining intellectual property protection for our technologies and product. We will only be able to protect our technologies and product from unauthorized use by third parties to the extent that valid and enforceable patents cover them, or that other market exclusionary rights apply. While we have issued enforceable patents covering the sugarBEAT device, the patent positions of companies like ours can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in Great Britain and the European Union. The general patent environment outside the United States involves significant uncertainty. Accordingly, we cannot predict the breadth of claims that may be allowed or that the scope of these patent rights would provide a sufficient degree of future protection that would permit us to gain or keep our competitive advantage with respect to this product and technology. Additionally, companies like ours are dependent on creating a pipeline of products. We may not be able to develop additional proprietary technologies or products that produce commercially viable products or that are themselves patentable.
Our issued patents may be subject to challenge and possibly invalidated by third parties. Changes in either the patent laws or in the interpretations of patent laws in Great Britain or the European Union or other countries may diminish the market exclusionary ability of our intellectual property.
In addition, others may independently develop similar or alternative technologies that may be outside the scope of our intellectual property. Should third parties obtain patent rights to similar technology, this may have an adverse effect on our business.
To the extent that consultants or key employees apply technological information independently developed by them or by others to our product, disputes may arise as to the proprietary rights of the information, which may not be resolved in our favour. Consultants and key employees that work with our confidential and proprietary technologies are required to assign all intellectual property rights in their discoveries to us. However, these consultants or key employees may terminate their relationship with us, and we cannot preclude them indefinitely from dealing with our competitors. If our trade secrets become known to competitors with greater experience and financial resources, the competitors may copy or use our trade secrets and other proprietary information in the advancement of their products, methods or technologies. If we were to prosecute a claim that a third party had illegally obtained and was using our trade secrets, it would be expensive and time consuming and the outcome would be unpredictable. In addition, courts in Great Britain and the European Union are sometimes less willing to protect trade secrets than courts in the United States. Moreover, if our competitors independently develop equivalent knowledge, we would lack any contractual claim to this information, and our business could be harmed.
Our ability to commercialize our product will depend on our ability to sell such products without infringing the patent or proprietary rights of third parties. If we are sued for infringing intellectual property rights of third parties, such litigation will be costly and time consuming and an unfavorable outcome would have a significant adverse effect on our business.
Our ability to commercialize our product will depend on our ability to sell such products without infringing the patents or other proprietary rights of third parties. Third-party intellectual property in the field of diagnostic medical devices is complicated, and third-party intellectual property rights in this field are continuously evolving. We have not performed searches for third-party intellectual property rights that may raise freedom-to-operate issues, and we have not obtained legal opinions regarding commercialization of our product other than patent research prior to the filing of our patent applications, and search and examination reports from the respective patent examination offices.
In addition, because patent applications are published months after their filing, and because applications can take several years to issue, there may be currently pending third-party patent applications that are unknown to us, which may later result in issued patents. If a third-party claims that we infringe on its patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including:
· infringement claims that, with or without merit, can be costly and time consuming to litigate, can delay the regulatory approval process and can divert management’s attention from our core business strategy;
· substantial damages for past infringement which we may have to pay if a court determines that our products or technologies infringe upon a competitor’s patent or other proprietary rights;
· if a license is available from a holder, we may have to pay substantial royalties or grant cross licenses to our patents or other proprietary rights; and
· Re-designing our process so that it does not infringe the third-party intellectual property, which may not be possible, or which may require substantial time and expense including delays in bringing our own products to market.
Such actions could harm our competitive position and our ability to generate revenue and could result in increased costs.
15
Nemaura Medical Inc. is an Emerging Growth Company (EGC) as defined under the Jumpstart Our Business Startups (JOBS) Act.
An “emerging growth company” is an issuer whose initial public offering was or will be completed after Dec. 8, 2011, and had total annual gross revenues of less than $1 billion during its most recently completed fiscal year. An issuer’s EGC status terminates on the earliest of:
· The last day of the first fiscal year of the issuer during which it had total annual gross revenues of $1 billion or more;
· The last day of the fiscal year of the issuer following the fifth anniversary of the date of the issuer’s initial public offering;
· The date on which such issuer has issued more than $1 billion in non-convertible debt securities during the prior three-year period determined on a rolling basis; or
· The date on which the issuer is deemed to be a “large accelerated filer” under the Exchange Act, which means, among other things, that it has a public float in excess of $700 million.
Pursuant to the JOBS Act of 2012, as an emerging growth company the Company can elect to opt out of the extended transition period for any new or revised accounting standards that may be issued by the PCAOB or the SEC. The Company has elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public or private companies, the Company, as an emerging growth company, can adopt the standard for the private company. This may make comparison of the Company's financial statements with any other public company which is not either an emerging growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible as possible different or revised standards may be used.
The Company has elected to use the extended transition period for complying with new or revised financial accounting standards available under Section 102(b)(2)(B) of the Act. Among other things, this means that the Company's independent registered public accounting firm will not be required to provide an attestation report on the effectiveness of the Company's internal control over financial reporting so long as it qualifies as an emerging growth company, which may increase the risk that weaknesses or deficiencies in the internal control over financial reporting go undetected. Likewise, so long as it qualifies as an emerging growth company, the Company may elect not to provide certain information, including certain financial information and certain information regarding compensation of executive officers that would otherwise have been required to provide in filings with the SEC, which may make it more difficult for investors and securities analysts to evaluate the Company. As a result, investor confidence in the Company and the market price of its common stock may be adversely affected.
If our product, the sugarBEAT device, does not gain market acceptance among physicians, patients and the medical community, we will be unable to generate significant revenue, if any.
The sugarBEAT device that we developed may not achieve market acceptance among physicians, patients, third-party payers and others in the medical community. If we receive the regulatory approvals necessary for commercialization, the degree of market acceptance will depend upon a number of factors, including:
· limited indications of regulatory approvals;
· the establishment and demonstration in the medical community of the clinical efficacy and safety of our product and its potential advantages over existing diagnostic medical devices;
· the prevalence and severity of any side effects;
· our ability to offer our product at an acceptable price;
· the relative convenience and ease of use of our product;
· the strength of marketing and distribution support; and
· sufficient third-party coverage or reimbursement.
The market may not accept the sugarBEAT device based on any number of the above factors. If the sugarBEAT device is approved, there may be other therapies available which directly compete for the same target market. The market may choose to continue utilizing the existing products for any number of reasons, including familiarity with or pricing of these existing products. The failure of any of our product to gain market acceptance could impair our ability to generate revenue, which could have a material adverse effect on our future business.
We have no commercial manufacturing facility for our sugarBEAT device and no experience in manufacturing products for commercial purposes and the failure to find manufacturing partners or create a manufacturing facility ourselves could have an adverse impact on our ability to grow our business.
We have no commercial manufacturing facility for the sugarBEAT device and no experience in manufacturing commercial quantities of our product. As such, we are dependent on third parties to supply our product according to our specifications, in sufficient quantities, on time, in compliance with appropriate regulatory standards and at competitive prices. We cannot be sure that we will be able to obtain an adequate supply of our product candidates on acceptable terms, or at all.
Manufacturers supplying diagnostic medical devices must comply with regulations which require, among other things, compliance with evolving regulations under Medical Device Directives stipulated under ISO13485. The manufacturing of products at any facility will be subject to strict quality control, testing and record keeping requirements, and continuing obligations regarding the submission of safety reports and other post-market information. Both the sensor and patch manufacturing facilities for the sugarBEAT device are currently ISO13485 certified. We cannot guarantee that the facilities will continue to pass regulatory inspection, or that future changes to ISO13485 standards will not also affect the manufactures of the sensors and patches.
If we fail to attract and retain senior management, consultants, advisors and scientific and technical personnel, our product development and commercialization efforts could be impaired.
Our performance is substantially dependent on the performance of our senior management and key scientific and technical personnel, particularly Dr. Dewan Fazlul Hoque Chowdhury, President, Chairman and Chief Executive Officer. Although we have entered into an employment agreement with Dr. Chowdhury, there is no assurance that he will remain in our employ for the entire term of such employment agreement. The loss of the services of any member of our senior management or our scientific or technical staff may significantly delay or prevent the development of our product and other business objectives by diverting management’s attention to transition matters and identification of suitable replacements, if any, and could have a material adverse effect on our business, operating results and financial condition.
We also rely on consultants and advisors to assist us in formulating our research and development strategy. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, that may affect their ability to contribute to us.
In addition, we believe that we will need to recruit additional executive management and scientific and technical personnel. There is currently intense competition for skilled executives and employees with relevant scientific and technical expertise, and this competition is likely to continue. The inability to attract and retain sufficient scientific, technical and managerial personnel could limit or delay our product development efforts, which would adversely affect the development of our product and commercialization of our potential product and growth of our business.
16
We expect to expand our research, development, clinical research and marketing capabilities and, as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to have significant growth in expenditures, the number of our employees and the scope of our operations, in particular with respect to those potential products that we elect to commercialize independently or together with others. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to train qualified personnel. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plan or disrupt our operations.
We will need to raise additional funds in order to finance the anticipated commercialization of our product by incurring indebtedness, through collaboration and licensing arrangements, or by issuing securities which may cause dilution to existing stockholders, or require us to relinquish rights to our technologies and our product.
Developing our product, conducting clinical trials, establishing manufacturing facilities and developing marketing and distribution capabilities is expensive. We will need to finance future cash needs through additional public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. We cannot be certain that additional funding will be available to us on acceptable terms, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience dilution. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product or grant licenses on terms that are not favorable to us.
We have a limited operating history and you should not rely on our historical financial data as an indicator of our future financial performance.
We have a limited operating history in the medical device industry. You should consider our business and prospects in light of the risks and difficulties we face with our limited operating history and should not rely on our past results as an indication of our future performance. In particular, we may face challenges in planning our growth strategy and forecasting market demand accurately as a result of our limited historical data and limited experience in implementing and evaluating our business strategies. If we are unable to successfully address these risks, difficulties and challenges as a result of our limited operating history, our ability to implement our strategic initiatives could be adversely affected, which may in turn have a material adverse effect on our business, financial condition, results of operations and prospects.
We have a history of losses and may not achieve or maintain profitability.
We have incurred net losses every year since our inception in 2009 and have not generated revenue from the period of our inception from product sales or licenses to date. As of March 31, 2018, we had an accumulated deficit of approximately $9.0 million. We may expect to incur losses for the next several years and cannot be certain that we will ever achieve profitability. As a result, our business is subject to all of the risks inherent in the development of a new business enterprise, such as the risk that we may not obtain substantial additional capital needed to support the expenses of developing our technology and commercializing our potential products; develop a market for our potential products; successfully transition from a company with a research focus to a company capable of either manufacturing and selling potential products or profitably licensing our potential products to others; and/or attract and retain qualified management, technical and scientific staff.
We currently have not generated any revenue from product sales and may never become profitable.
To date, we have generated no revenue for product sales and we do not know when or if our product will generate revenue. Our ability to generate revenue depends on a number of factors, including our ability to successfully complete clinical trials for the sugarBEAT device and obtain regulatory approval to commercialize these potential products. Even then, we will need to establish and maintain sales, marketing, distribution and to the extent we do not outsource manufacturing, manufacturing capabilities. We plan to rely on one or more strategic collaborators to help generate revenues in markets outside of Great Britain however, we cannot be sure that our collaborators, if any, will be successful. Our ability to generate revenue will also be impacted by certain challenges, risks and uncertainties frequently encountered in the establishment of new technologies and products in emerging markets and evolving industries. These challenges include our ability to:
· execute our business model;
· create brand recognition;
· manage growth in our operations;
· create a customer base cost-effectively;
· retain customers;
· access additional capital when required; and
· attract and retain key personnel.
We cannot be certain that our business model will be successful or that it will successfully address these and other challenges, risks and uncertainties. If we are unable to generate significant revenue, we may not become profitable, and we may be unable to continue our operations. Even if we are able to commercialize the sugarBEAT device, we may not achieve profitability for at least several years, if at all, after generating material revenue.
17
Fluctuations in foreign exchange rates may adversely affect our financial condition and results of operations.
Our functional currency is the Great Britain Pound Sterling (“GBP”). The reporting currency is the United States dollar (US$). Income and expenditures are translated at the average exchange rates prevailing during the reporting period. Assets and liabilities are translated at the exchange rates as of balance sheet date. Stockholder’s equity is translated into United States dollars from GBP at historical exchange rates. Currency fluctuations and restrictions on currency exchange may adversely affect our business, including limiting our ability to convert GBP into foreign currencies and, if the GBP were to decline in value, reducing our revenue in U.S. dollar terms. To the extent the U.S. dollar strengthens against foreign currencies, the translation of these foreign currencies denominated transactions results in reduced revenue, operating expenses and net income for our international operations. Similarly, to the extent the U.S. dollar weakens against foreign currencies, the translation of these foreign currency denominated transactions results in increased revenue, operating expenses and net income for our international operations. We are also exposed to foreign exchange rate fluctuations as we convert the financial statements of our foreign subsidiaries into U.S. dollars in consolidation. If there is a change in foreign currency exchange rates, the conversion of the foreign subsidiaries’ financial statements into U.S. dollars will lead to a translation gain or loss which is recorded as a component of other comprehensive income (loss). We have not entered into agreements or purchased instruments to hedge our exchange rate risks. The availability and effectiveness of any hedging transaction may be limited and we may not be able to successfully hedge our exchange rate risks.
In addition, following the UK’s Brexit vote to leave the EU, there has been a weakening of GBP against many currencies. We expect to have to pay some of our service providers and vendors in USD and we will pay approximately 10% more at present than we would have done prior to the Brexit vote. The currency exchange rate continues to be very unstable and therefore the future impact or further weakening of GBP is not known at this time.
Risks Related to Our Industry
Our competitors may develop products that are less expensive, safer or more effective, which may diminish or eliminate the commercial success of any potential products that we may commercialize.
If our competitor’s market products that are less expensive, safer or more effective than our future products developed from our product candidates, or that reach the market before our products, we may not achieve commercial success. For example, if approved, the sugarBEAT device’s primary competition in the glucose monitoring device setting will be companies such as Dexcom, Abbott, and Senseonics who produce glucose monitoring devices. The market may choose to continue utilizing the existing products for any number of reasons, including familiarity with or pricing of these existing products. The failure of our product to compete with products marketed by our competitors would impair our ability to generate revenue, which would have a material adverse effect on our future business, financial condition and results of operations.
We expect to compete with several companies including Dexcom, Abbott, and Senseonics, and our competitors may:
· develop and market products that are less expensive or more effective than our future product;
· commercialize competing products before we can launch any products developed from our product candidate;
· operate larger research and development programs or have substantially greater financial resources than we do;
· initiate or withstand substantial price competition more successfully than we can;
· have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent;
· more effectively negotiate third-party licenses and strategic relationships; and
· take advantage of acquisition or other opportunities more readily than we can.
We expect to compete for market share against large medical diagnostic device manufacturing companies, smaller companies that are collaborating with larger companies, new companies, and other public and private research organizations.
In addition, our industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Our competitors may render our technologies obsolete by advances in existing technological approaches or the development of new or different approaches, potentially eliminating the advantages in our product discovery process that we believe we derive from our research approach and proprietary technologies.
The use of hazardous materials in our operations may subject us to environmental claims or liabilities.
Our research and development activities involve the use of hazardous chemical materials. Injury or contamination from these materials may occur and we could be held liable for any damages, which could exceed our available financial resources. This liability could materially adversely affect our business, financial condition and results of operations.
We are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. We may be required to incur significant costs to comply with environmental laws and regulations in the future that could materially adversely affect our business, financial condition and results of operations.
If we fail to comply with extensive regulations enforced by regulatory agencies with respect to diagnostic medical device products, the commercialization of our product could be prevented, delayed or halted.
Research, preclinical development, clinical trials, manufacturing and marketing of our product is subject to extensive regulation by various government authorities. We have not received marketing approval for the sugarBEAT device. The process of obtaining the required regulatory approvals is lengthy and expensive, and the time required for such approvals is uncertain. The approval process is affected by such factors as:
· the indication and claims of the diagnostic device;
· the quality of submission relating to the product;
· the product’s clinical efficacy and safety;
· the manufacturing facility compliance;
· the availability of alternative devices;
· the risks and benefits demonstrated in clinical trials; and
· the patent status and marketing exclusivity rights of certain innovative products.
18
Any regulatory approvals that we or our partners receive for our product may also be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies. The subsequent discovery of previously unknown problems with the product, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product and withdrawal of the product from the market.
Manufacturing, labelling, storage and distribution activities also are subject to strict regulation and licensing by government authorities. The manufacturing facilities for our product will be subject to periodic inspection by the regulatory authorities and from time to time, these agencies may send notice of deficiencies as a result of such inspections. Our failure or the failure of our manufacturing facilities, to continue to meet regulatory standards or to remedy any deficiencies could result in corrective action by the authorities, including the interruption or prevention of marketing, closure of our manufacturing facilities, and fines or penalties.
Regulatory authorities also will require post-marketing surveillance to monitor and report potential adverse effects of our product. If approved, any of our products’ subsequent failure to comply with applicable regulatory requirements could, among other things, result in warning letters, fines, suspension or revocation of regulatory approvals, product recalls or seizures, operating restrictions, injunctions and criminal prosecutions.
Government policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our product. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action. If we are not able to maintain regulatory compliance, we might not be permitted to market our product and our business could suffer.
In the future, we hope to distribute and sell our product outside of the United Kingdom and the European Union, which will subject us to further regulatory risk.
In addition to seeking approval from the United Kingdom and the European Union for the sugarBEAT device, we may seek regulatory approval from Saudi Arabia and the United Arab Emirates, Hong Kong, Australia, and the USA, to market the sugarBEAT device, however, there is no guarantee we will do so. We may in the future also seek approvals for additional countries. The regulatory review process varies from country to country, and approval by foreign government authorities is unpredictable, uncertain and generally expensive. The ability to market our product could be substantially limited due to delays in receipt of, or failure to receive, the necessary approvals or clearances. Marketing of our product in these countries, and in most other countries, is not permitted until we have obtained required approvals or exemptions in each individual country. Failure to obtain necessary regulatory approvals could impair our ability to generate revenue from international sources.
Market acceptance of our product will be limited if users are unable to obtain adequate reimbursement from third-party payers.
Government health administration authorities, private health insurers and other organizations generally provide reimbursement for products like our product and our commercial success will depend in part on these third-party payers agreeing to reimburse patients for the costs of our product. Even if we succeed in bringing our product to market, we cannot assure you that third-party payers will consider our product cost effective or provide reimbursement in whole or in part for its use.
Significant uncertainty exists as to the reimbursement status of newly approved health care products. Our product is intended to replace or alter existing therapies or procedures. These third-party payers may conclude that our product is less safe, effective or cost-effective than existing therapies or procedures. Therefore, third-party payers may not approve our product for reimbursement.
If third-party payers do not approve our product for reimbursement or fail to reimburse for them adequately, sales will suffer as some physicians or their patients will opt for a competing product that is approved for reimbursement or is adequately reimbursed. Even if third-party payers make reimbursement available, these payers’ reimbursement policies may adversely affect our ability and the ability of our potential collaborators to sell our product on a profitable basis.
The trend toward managed healthcare, the growth of organizations such as health maintenance organizations and legislative proposals to reform healthcare and government insurance programs could significantly influence the purchase of healthcare services and products, resulting in lower prices and reduced demand for our product which could adversely affect our business, financial condition and results of operations.
In addition, legislation and regulations affecting the pricing of our product may change in ways adverse to us before or after the regulatory agencies approve our product for marketing. While we cannot predict the likelihood of any of these legislative or regulatory proposals, if any government or regulatory agencies adopt these proposals, they could materially adversely affect our business, financial condition and results of operations.
Product liability claims may damage our reputation and, if insurance proves inadequate, the product liability claims may harm our business.
We may be exposed to the risk of product liability claims that is inherent in the diagnostic medical device. A product liability claim may damage our reputation by raising questions about our product’s safety and efficacy and could limit our ability to sell our product by preventing or interfering with commercialization of our product.
In addition, product liability insurance for our industry is generally expensive to the extent it is available at all. There can be no assurance that we will be able to obtain and maintain such insurance on acceptable terms or that we will be able to secure increased coverage if the commercialization of our product progresses, or that future claims against us will be covered by our product liability insurance. Moreover, there can be no assurance that any product liability coverage from any insurance policy and/or any rights of indemnification and contribution that we may have will offset any future claims. We currently do not maintain product liability insurance. A successful claim against us with respect to uninsured liabilities and not subject to any indemnification or contribution could have a material adverse effect on our business, financial condition and results of operations.
19
We could be negatively impacted by the application or enforcement of fraud and abuse laws, including anti-kickback laws and other anti-referral laws.
We are not aware of any current business practice which is in violation of any fraud and abuse law. However, continued vigilance to assure compliance with all potentially applicable laws will be a necessary expense associated with product development. For example, all product marketing efforts must be strictly scrutinized to assure that they are not associated with improper remunerations to referral sources in violation of any anti-kickback statutes. Remunerations may include potential future activities for our product, including discounts, rebates and bundled sales, which must be appropriately structured to take advantage of statutory and regulatory “safe harbors.” From time to time we may engage physicians in consulting activities. In addition, we may decide to sponsor continuing medical education activities for physicians or other medical personnel. We also may award or sponsor study grants to physicians from time to time. All relationships with physicians, including consulting arrangements, continuing medical education and study grants, must be similarly reviewed for compliance with any anti-kickback statute to assure that remuneration is not provided in return for referrals. Patient inducements may also be unlawful. Inaccurate reports of product pricing, or a failure to provide a product at an appropriate price to various governmental entities, could also serve as a basis for an enforcement action under various theories.
Claims which are “tainted” by virtue of kickbacks or a violation of self-referral rules may be alleged as false claims if other elements of a violation are established. Because our potential customers may seek payments from healthcare programs for our product, even during the clinical trial stages, we must assure that we take no actions which could result in the submission of false claims. For example, free product samples which are knowingly or with reckless disregard billed to healthcare programs could constitute false claims. If the practice was facilitated or fostered by us, we could be liable. Moreover, inadequate accounting for or a misuse of grant funds used for product research and development could be alleged as a violation of relevant statutes.
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations, and additional legal or regulatory change.
Risks Related to Our Common Stock
Our stock price may be volatile.
The stock market, particularly in recent years, has experienced significant volatility particularly with respect to pharmaceutical, biotechnology and other diagnostic medical device company stocks. The volatility of pharmaceutical, biotechnology and other diagnostic medical device company stocks often does not relate to the operating performance of the companies represented by the stock. Factors that could cause this volatility in the market price of our Common Stock include:
· results from and any delays in our clinical trials;
· failure or delays in entering our product into clinical trials;
· failure or discontinuation of any of our research programs;
· delays in establishing new strategic relationships;
· delays in the development or commercialization of our product;
· market conditions in the diagnostic medical device sectors and issuance of new or changed securities analysts’ reports or recommendations;
· actual and anticipated fluctuations in our financial and operating results;
· developments or disputes concerning our intellectual property or other proprietary rights;
· introduction of technological innovations or new commercial products by us or our competitors;
· issues in manufacturing our product;
· market acceptance of our product;
· third-party healthcare reimbursement policies;
· regulatory actions affecting us or our industry;
· litigation or public concern about the safety of our product; and
· additions or departures of key personnel.
These and other external factors may cause the market price and demand for our Common Stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of Common Stock and may otherwise negatively affect the liquidity of our Common Stock. In the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management.
We have not paid and may not pay any dividends on our Common Stock.
We have paid no dividends on our Common Stock to date and may not pay dividends to holders of our Common Stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, it is currently anticipated that any earnings will be retained to finance our future expansion and for the implementation of our business plan. As an investor, you should take note of the fact that a lack of a dividend can further affect the market value of our stock, and could significantly affect the value of any investment in our Company.
We are subject to the reporting requirements of federal securities laws. This can be expensive and may divert resources from other projects, and thus impairing our ability to grow.
We are subject to the information and reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and other federal securities laws, including compliance with the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”). The costs of preparing and filing annual and quarterly reports, proxy statements and other information with the SEC (including reporting of any Merger that may occur in the future) and furnishing audited reports to stockholders will cause our expenses to be higher than they would have been if we had remained privately held.
20
If we fail to establish and maintain an effective system of internal control, we may not be able to report our financial results accurately or to prevent fraud. Any inability to report and file our financial results accurately and timely could harm our reputation and adversely impact the trading price of our Common Stock.
We are subject to reporting obligations under the U.S. securities laws. The Securities and Exchange Commission, or the SEC, as required by Section 404 of the Sarbanes- Oxley Act of 2002, or the Sarbanes-Oxley Act, adopted rules requiring every public company to include a management report on such company’s internal control over financial reporting in its annual report, which contains management’s assessment of the effectiveness of the company’s internal control over financial reporting. Our management has concluded that our internal control over our financial reporting is not effective. Our reporting obligations as a public company will place a significant strain on our management, operational and financial resources and systems for the foreseeable future.
Prior to 2014, we were a private company with a short operating history and limited accounting personnel and other resources with which to address our internal control and procedures over financial reporting. We have identified material weaknesses, which include (i) our size has prevented us from being able to employ sufficient resources to enable us to have an adequate level of supervision and segregation of duties within our internal control system, (ii) a lack of adequate financial expertise related to the assessment of complex transactions and a lack of adequate resources to review out of the ordinary transactions and arrangements of the Company, (iii) limited policies and procedures over related party transactions. We will continue to implement measures to remedy these material weaknesses as well as other deficiencies. If we fail to timely achieve and maintain the adequacy of our internal controls, we may not be able to conclude that we have effective internal control over financial reporting. Moreover, effective internal control over financial reporting is necessary for us to produce reliable financial reports and is important to help prevent fraud. As a result, our failure to achieve and maintain effective internal control over financial reporting could result in the loss of investor confidence in the reliability of our financial statements, which in turn could harm our business and negatively impact the market price of our commons stock.
Effective internal control is necessary for us to provide reliable financial reports and prevent fraud. If we cannot provide reliable financial reports or prevent fraud, we may not be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed. As a result, our small size and any current internal control deficiencies may adversely affect our financial condition, results of operation and access to capital. We have not performed an in-depth analysis to determine if historical un-discovered failures of internal controls exist and may in the future discover areas of our internal control that need improvement.
We have disclosed a material weakness in our internal control over financial reporting which could have an adverse effect on our ability to report our financial condition, results of operations or cash flows accurately and on a timely basis.
We have disclosed a material weakness in our internal control over financial reporting due to (i) our size has prevented us from being able to employ sufficient resources to enable us to have an adequate level of supervision and segregation of duties within our internal control system, (ii) a lack of adequate financial expertise related to the assessment of complex transactions and a lack of adequate resources to review out of the ordinary transactions and arrangements of the Company, (iii) limited policies and procedures over related party transactions. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company's annual or interim financial statements will not be prevented or detected on a timely basis. We have determined that further improvements are required in our accounting processes and personnel before we can consider the material weakness remediated. Management's procedures and testing identified errors that, although not material to the consolidated financial statements, led management to conclude that control deficiencies exist related to the timely production and filing of financial information. As a result of these deficiencies, it is reasonably possible that internal controls over financial reporting may not have prevented or detected errors from occurring that could have been material, either individually or in the aggregate.
A material weakness in our internal control over financial reporting could adversely impact our ability to provide timely and accurate financial information. While considerable actions have been taken and are underway to improve our internal controls in response to the identified material weaknesses and further action steps to strengthen controls have been taken, additional work continues to address and remediate the identified material weakness. If we are unsuccessful in implementing or following our remediation plan, we may not be able to timely or accurately report our financial condition, results of operations or cash flows or maintain effective internal controls over financial reporting. If we are unable to report financial information timely and accurately or to maintain effective disclosure controls and procedures, we could be subject to, among other things, regulatory or enforcement actions by the SEC, which could adversely affect the valuation of our common stock and could adversely affect our business prospects.
Public company compliance may make it more difficult to attract and retain officers and directors.
The Sarbanes-Oxley Act and new rules subsequently implemented by the SEC have required changes in corporate governance practices of public companies. As a public company, we expect these new rules and regulations to increase our compliance costs in 2018 and beyond and to make certain activities more time consuming and costly. As a public company, we also expect that these new rules and regulations may make it more difficult and expensive for us to obtain director and officer liability insurance in the future and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors or as executive officers.
Our Common Stock will be deemed a “penny stock,” which makes it more difficult for our investors to sell their shares.
Our Common Stock will be subject to the “penny stock” rules adopted under Section 15(g) of the Exchange Act. The penny stock rules generally apply to companies whose common stock is not listed on The Nasdaq Stock Market or other national securities exchange and trades at less than $5.00 per share, other than companies that have had average revenue of at least $6,000,000 for the last three years or that have tangible net worth of at least $5,000,000 ($2,000,000 if the company has been operating for three or more years). These rules require, among other things, that brokers who trade penny stock to persons other than “established customers” complete certain documentation, make suitability inquiries of investors and provide investors with certain information concerning trading in the security, including a risk disclosure document and quote information under certain circumstances. Many brokers have decided not to trade penny stocks because of the requirements of the penny stock rules and, as a result, the number of broker-dealers willing to act as market makers in such securities is limited. If we remain subject to the penny stock rules for any significant period, it could have an adverse effect on the market, if any, for our securities. If our securities are subject to the penny stock rules, investors will find it more difficult to dispose of our securities.
Offers or availability for sale of a substantial number of shares of our Common Stock may cause the price of our Common Stock to decline.
If our stockholders sell substantial amounts of our Common Stock in the public market upon the expiration of any statutory holding period, under Rule 144, or issued upon the exercise of outstanding options or warrants, it could create a circumstance commonly referred to as an “overhang” and in anticipation of which the market price of our Common Stock could fall. The existence of an overhang, whether or not sales have occurred or are occurring, also could make more difficult our ability to raise additional financing through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate.
21
The interests of Dr. Chowdhury, or the controlling shareholders, may not always coincide with the interests of us and our other shareholders, and the controlling shareholders may exert significant control or substantial influence over us and may take actions that are not in, or may conflict with, public shareholders’ best interests.
The controlling shareholders will control the exercise of voting rights of over 50 % of the shares eligible to vote in any of our annual or special meeting. Therefore, these controlling shareholders will be able to exercise significant influence over all matters that require us to obtain shareholder approval, including the election of directors to our board and approval of significant corporate transactions that we may consider, such as a merger or other sale of our company or its assets. The controlling shareholders may cause us to take actions that are not in, or may conflict with, the interests of us or the public shareholders. In the case where the interests of the controlling shareholders conflict with those of our other shareholders, or if the controlling shareholders choose to cause us to pursue objectives that would conflict with the interests of our other shareholders, such other shareholders could be left in a disadvantageous position by such actions caused by the controlling shareholders and the price of our common stock could be adversely affected.
We are subject to the anti-takeover provisions of the Nevada Revised Statutes governing business combinations and control share acquisition.
Applicability of the Nevada business combination statute would discourage parties interested in taking control of our company if they cannot obtain the approval of our board of directors. These provisions could prohibit or delay a merger or other takeover or change in control attempt and, accordingly, may discourage attempts to acquire our company even though such a transaction may offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
The effect of the Nevada control share statute is that the acquiring person, and those acting in association with the acquiring person, will obtain only such voting rights in the control shares as are conferred by a resolution of the stockholders at an annual or special meeting of the stockholders. The Nevada control share law, if applicable, could have the effect of discouraging takeovers of our company based on our organizational structure.
We are subject to compliance with multiple tax jurisdictions.
As we transact out of both the UK and United States we must comply with tax filing requirements in both jurisdictions.
We may not manage to implement changes to our control environment within the timeframes required
We have identified changes that we need to make to our control environment in order to move to SOX compliance. Whilst we have an action plan in place, it may not be possible for us to implement all of the changes required by the required date.
22
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
ITEM 2. PROPERTIES.
Our offices are located at ATIC Building, 5 Oakwood Drive, Loughborough, Leicestershire, United Kingdom. The offices house our headquarters; offices; laboratory; and small in-house manufacturing facility. The monthly rent is $2,500. The lease is on flexible terms with annual renewal. We believe that we will be able to continue on a year to year lease for as long as necessary.
ITEM 3. LEGAL PROCEEDINGS.
We do not know of any material, active, pending or threatened proceeding against us or our subsidiaries, nor are we, or any subsidiary, involved as a plaintiff or defendant in any material proceeding or pending litigation.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market Information
Our common stock began quotation on the OTCBB under the symbol “NMRD” on November 4, 2014. On June 30, 2017, our common stock began quotation on the OTCQB.
On January 25, 2018, the Company’s common stock commenced trading on the NASDAQ Capital Market under its existing trading symbol, “NMRD”. For the periods indicated, the following table sets forth the high and low bid prices per share of common stock. The following quotations reflect the high and low bids for our shares of common stock based on inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions.
|
Fiscal Year 2017
|
High Bid
|
Low Bid
|
||||||
|
First Quarter
|
1.99
|
1.50
|
||||||
|
Second Quarter
|
1.95
|
1.90
|
||||||
|
Third Quarter
|
1.90
|
1.50
|
||||||
|
Fourth Quarter
|
2.50
|
1.40
|
||||||
|
Fiscal Year 2018
|
High Bid
|
Low Bid
|
||||||
|
First Quarter
|
9.00
|
2.00
|
||||||
|
Second Quarter
|
6.99
|
4.04
|
||||||
|
Third Quarter
|
6.49
|
4.20
|
||||||
|
Fourth Quarter*
|
6.80
|
4.50
|
||||||
|
Fiscal Year 2019
|
High Bid
|
Low Bid
|
||||||
|
First Quarter (through June 1, 2018)
|
4.95
|
2.76
|
||||||
* Our common stock commenced trading on the NASDAQ Capital Market.
As of June 4, 2018, we had approximately 92 holders on record of our common stock.
23
Dividends
Since incorporation, we have not paid any dividend on any class of equity securities. We anticipate that for the foreseeable future all earnings will be retained for use in our business and no cash dividends will be paid to stockholders. Any payment of cash dividends in the future on the Company’s common stock or preferred stock, will be dependent upon our financial condition, results of operations, current and anticipated cash requirements, plans for expansion, as well as other factors that the Board of Directors deems relevant.
Securities Authorized for Issuance Under Equity Compensation Plans
Currently, there is no equity compensation plan in place.
Performance Graph
The graph below charts the cumulative total return of our common stock listed on Nasdaq, the Nasdaq Composite index and the S&P 1500 Health Care Technology Index, for the fiscal years ended March 31, 2015, 2016, 2017 and 2018. Our stock began trading on Nasdaq on January 25, 2018.
Unregistered Sales of Securities
None.
Purchases of Equity Securities by the Registrant and Affiliated Purchasers
We have not repurchased any shares of our common stock during the fiscal year ended March 31, 2018.
24
ITEM 6. SELECTED FINANCIAL DATA.
Financial highlights
|
Year Ended March 31,
|
2018
|
2017
|
2016
|
2015
|
2014
|
|||||||||||||||
|
Net loss
|
$
|
(1,820,449
|
)
|
$
|
(1,551,266
|
)
|
$
|
(1,539,637
|
)
|
$
|
(1,319,840
|
)
|
$
|
(586,233
|
)
|
|||||
|
Diluted loss per share
|
$
|
(0.01
|
)
|
$
|
*
|
$
|
*
|
$
|
*
|
*
|
||||||||||
|
Cash, cash equivalents, and short-term investments
|
$
|
5,733,886
|
$
|
2,779,309
|
$
|
9,403,965
|
$
|
354,749
|
$
|
1,873,141
|
||||||||||
|
Total assets
|
$
|
6,255,402
|
$
|
7,401,906
|
$
|
9,732,783
|
$
|
913,108
|
$
|
2,049,774
|
||||||||||
|
Long-term obligations
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
(170,000
|
)
|
$
|
-
|
|||||||||
|
Deferred Revenue
|
$
|
1,333,128
|
$
|
1,183,035
|
$
|
1,396,005
|
$
|
1,538,300
|
$
|
1,667,200
|
||||||||||
|
Stockholders’ equity/(deficit)
|
$
|
4,110,965
|
$
|
5,366,500
|
$
|
7,678,765
|
$
|
(917,411
|
)
|
$
|
373,900
|
|||||||||
 |
||||||||||||||||||||
* less than $0.01
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The discussion and analysis below includes certain forward-looking statements that are subject to risks, uncertainties and other factors, as described in “Risk Factors” and elsewhere in this Annual Report on Form 10-K, that could cause our actual growth, results of operations, performance, financial position and business prospects and opportunities for this fiscal year and the periods that follow to differ materially from those expressed in, or implied by, those forward-looking statements.
Corporate Overview
Since inception we have devoted substantially all of our efforts establishing a new business and while operations have commenced we have generated no revenue from our limited operations. We are a holding company for a diagnostic medical device company and a clinical trial company specializing in discovering, developing and commercializing diagnostic medical devices with initial applications in the area of diabetes.
We are a holding corporation that owns one hundred percent (100%) of a diagnostic medical device company specializing in discovering, developing and commercializing specialty medical devices. We were organized on December 24, 2013 under the laws of the State of Nevada. We own one hundred percent (100%) of Region Green Limited, a British Virgin Islands corporation formed on December 12, 2013. Region Green Limited owns one hundred percent (100%) of the stock in Dermal Diagnostic (Holdings) Limited, an England and Wales corporation formed on December 11, 2013. Dermal Diagnostics (Holdings) Limited owns one hundred percent (100%) of the stock in DDL, an England and Wales corporation formed on January 20, 2009, and one hundred percent (100%) of the stock in Trial Clinic Limited, an England and Wales corporation formed on January 12, 2011.
In December 2013, we restructured the Company and re-domiciled as a domestic corporation in the United States. The corporate re-organization was accomplished to preserve the tax advantages under the laws of the England and Wales tax laws for the benefit of the shareholders of both DDL and TCL.
Affiliated Company Relationships
Pharma was incorporated in November 2005. Through October 2013, all technology development and related transactions were incurred by Pharma. As new technology platforms were invented and developed, additional companies were set up to contain these new technology platforms to aid in the process of raising further investments to progress the development of these subsequent technologies. However, due to the small size of the operations, low number of employees and laboratory and office space required, only one payroll was maintained initially. Invoices were posted in Pharma and recharges were made as required. Prior to the year ended March 31, 2016, recharges included a proportion of the overhead allocated based on management’s assessment. Management believes that the allocation methodologies are reasonable.
Dr. D. F Chowdhury and Mr. Bashir Timol are officers of Pharma. However, Pharma plans a management restructuring and a new management team is planned to be recruited in due course, aligned with commercial launch plans. The current management at DDL, including Dr. D. F. Chowdhury will allocate 15%-20% of their time to oversee the current operations at Pharma and the implementation of the new management team and to provide ongoing support in an advisory role. Pharma is a drug delivery company, which means that its activities are entirely related to the delivery of drugs to the body of a human or animal subject. DDL is a diagnostic company, which means it is entirely focused on extracting molecules from the human or animal subject and analyzing it to make a diagnosis or to monitor the level of a particular molecule such as glucose. These are two independent businesses engaged in different activities, therefore there is no conflict of interest between the two and management does not see any conflicts arising from the allocations of some of DDL management time to overseeing the operations of Pharma.
Payments made solely for work that Dr. Chowdhury performed/performs for Pharma in his capacity as Manager are not recharged to Nemaura Medical Inc. and are not included in our financial statements.
25
RESULTS OF OPERATIONS
Management’s plans and basis of presentation
The Company has experienced recurring losses and negative cash flows from operations. At March 31, 2018, the Company had approximate cash and fixed rate cash account balances of $5,734,000, working capital of $5,187,000, total stockholders’ equity of $4,111,000 and an accumulated deficit of $8,973,000. To date, the Company has in large part relied on equity financing to fund its operations. Additional funding has come from related party contributions. The Company expects to continue to incur losses from operations for the near-term and these losses could be significant as product development, regulatory activities, clinical trials and other commercial and product development related expenses are incurred.
Management’s strategic assessment includes the following potential options:
|
●
|
obtaining regulatory approval for the sugarBEAT device: CE mark review and approval in Europe is anticipated in 2018, and FDA clinical program and PMA submission timing is yet to be determined.
|
|
●
|
pursuing additional capital raising opportunities;
|
|
●
|
exploring licensing opportunities; and
|
|
●
|
undertaking manufacturing development and scale-up of the sugarBEAT device for commercialization.
|
Results of Operations
Year Ended March 31, 2018 Compared To The Year Ended March 31, 2017
Revenue
There was no revenue recognized in the years ended March 31, 2018 and March 31, 2017. In 2014, we received an upfront non-refundable cash payment of £1 million (approximately $1.40 million at March 31, 2018) in connection with an Exclusive Marketing Rights Agreement with an unrelated third party that provides the third party the exclusive right to market and promote the sugarBEAT device and related patch under its own brand in the United Kingdom and the Republic of Ireland. We have deferred this licensing revenue until we complete our continuing performance obligations, which include securing successful CE marking of the sugarBEAT patch, and we expect to record the revenue in income over an approximately 10 year term from the date CE marking approval is obtained. Although the revenue is deferred at March 31, 2018 and 2017, the cash payment became immediately available and was being used to fund our operations, including research and development costs associated with obtaining the CE marking approval.
Research and Development Expenses
Research and development expenses were $993,833 and $1,034,605 for the years ended March 31, 2018 and 2017, respectively. This amount related to clinical trials and improvements made to the sugarBEAT device, and expenditures included sub-contractor activities, and consultancy fees and wages. We expect research and development expenses to continue to be a significant cost in future periods as we continue our clinical studies of our sugarBEAT device and pursue strategic opportunities.
General and Administrative Expenses
General and administrative expenses were $915,132 and $516,661 for the years ended March 31, 2018 and 2017, respectively. These consisted primarily of legal, professional and audit fees plus wages and charitable contributions. General and administrative expenses will be expected to significantly increase as we commence product manufacture and commercialisation.
Other Comprehensive Income
For the years ended March 31, 2018 and 2017 other comprehensive income/(loss) was $564,914 and ($760,999), respectively, arising from foreign currency translation adjustments.
Year Ended March 31, 2017 Compared To The Year Ended March 31, 2016
Revenue
There was no revenue recognized in the years ended March 31, 2017 and March 31, 2016. In 2014, we received an upfront non-refundable cash payment of approximately $1.67 million in connection with an Exclusive Marketing Rights Agreement with an unrelated third party that provides the third party the exclusive right to market and promote the sugarBEAT device and related patch under its own brand in the United Kingdom and the Republic of Ireland. We have deferred this licensing revenue until we complete our continuing performance obligations, which include securing successful CE marking of the sugarBEAT patch, and we expect to record the revenue in income over an approximately 10 year term from the date CE marking approval is obtained. Although the revenue is deferred at March 31, 2017 and 2016, the cash payment became immediately available and was being used to fund our operations, including research and development costs associated with obtaining the CE marking approval
26
Research and Development Expenses
Research and development expenses were $1,034,605 and $1,028,224 for the years ended March 31, 2017 and 2016, respectively. This amount consisted primarily of expenditure on sub-contractor activities, consultancy fees and wages and demonstrated continuing expenditure for improvements made to the sugarBEAT device. We expect research and development expenses to continue to be a significant cost in future periods as we continue our clinical studies of our sugarBEAT device and pursue strategic opportunities.
General and Administrative Expenses
General and administrative expenses were $516,661 and $511,413 for the years ended March 31, 2017 and 2016, respectively. These consisted primarily of legal, professional and audit fees plus wages. General and administrative expenses will be expected to significantly increase as we commence product manufacture and commercialisation.
Other Comprehensive Income
For the years ended March 31, 2017 and 2016 other comprehensive (loss)/income was ($760,999) and $135,813 respectively, arising from foreign currency translation adjustments.
Liquidity and Capital Resources
We have experienced net losses and negative cash flows from operations since our inception. We have sustained cumulative losses of $8,973,082 through March 31, 2018. We have historically financed our operations through the issuances of equity, UK government grants and contributions of services from related entities.
At March 31, 2018, the Company had net working capital of $5,187,224 which included cash and short-term fixed rate cash account balances of $5,733,886. The Company reported a net loss of $1,820,449 for the year ended March 31, 2018.
While our current cash level (including fixed rate cash accounts) is sufficient for the completion of the clinical studies and the initial scale up of our manufacturing, our long term business plan is contingent upon our ability to raise additional funds. This may include a combination of debt, equity and licensing fees. If we are not successful in raising the funds needed in the specified timelines, the target dates for the achievement of the milestones will be extended.
We believe the cash position as of March 31, 2018 is adequate for our current level of operations through June 2019, and for the achievement of certain of our product development milestones. Our plan is to utilize the cash on hand to complete the following:
- Establish commercial manufacturing operations for commercial supply of the sugarBEAT device and patches.
- Complete clinical studies for CE approval of the body worn miniaturised device with Bluetooth connectivity.
In November 2015 we received proceeds of $10,000,000 in connection with the private placement of 5 million shares and warrants for up to 10 million shares of our common stock.
Operating activities
Net cash consumed by our operating activities for the year ended March 31, 2018 was $2,136,977 which reflected our net loss of $1,820,449, increased by an increase in accounts payable, an increase in prepayments, a decrease in liability due to related parties and an increase in accrued interest receivable of $452,535, and offset by an increase in accruals of $106,751.
Net cash consumed by our operating activities for the year ended March 31, 2017 was $1,192,828 which reflected our net loss of $1,551,266, and offset by a net increase in accounts payable, liability due to related parties and accrued expenses of $252,638, and by a decrease in prepayments and other receivables of $85,367.
Net cash consumed by our operating activities for the year ended March 31, 2016 was $1,209,365 which reflected our net loss of $1,539,637, a decrease in accounts payable and accrued expenses of $160,983 and offset by a decrease in prepayments and other receivables of $224,392 and decrease in prepayment to related party of $249,459.
Net cash generated by investing activities was $1,949,215 for the year ended March 31, 2018, which reflected the cash received from the maturity of a fixed rate savings account of $1,994,475 offset by expenditures made in developing intellectual property, primarily related to patent filings of $45,260.
Net cash used in investing activities was $6,306,089 for the year ended March 31, 2017, which reflected the expenditures made in developing intellectual property, primarily related to patent filings of $73,070, property and equipment of $6,519 and $6,226,500 invested in fixed rate savings account.
Net cash used in investing activities was $87,564 for the year ended March 31, 2016, which reflected the expenditures made in developing intellectual property, primarily related to patent filings of $78,197 and property and equipment of $9,367.
For the years ended March 31, 2018 and 2017, there were no financing activities.
Net cash provided by financing activities was $10,299,434 for the year ended March 31, 2016. Net cash provided by financing activities represents proceeds from the issuance of common stock for cash of $10,000,000 and costs paid for by a related party of $299,434.
27
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements, including unrecorded derivative instruments that have or are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Contractual Obligations
None
Critical Accounting Policies
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (GAAP) requires management to make estimates and assumptions about future events that affect the amounts reported in the financial statements and accompanying notes. Future events and their effects cannot be determined with absolute certainty. Therefore, the determination of estimates requires the exercise of judgment. Actual results inevitably will differ from those estimates, and such differences may be material to the financial statements. The most significant accounting estimates inherent in the preparation of our financial statements include estimates associated with research and development, income taxes and intangible assets.
The Company’s financial position, results of operations and cash flows are impacted by the accounting policies the Company has adopted. In order to get a full understanding of the Company’s financial statements, one must have a clear understanding of the accounting policies employed. A summary of the Company’s critical accounting policies follows:
Research and Development Expenses: The Company charges research development expenses to operations as incurred. Research and Development expenses primarily consist of salaries and related expenses for personnel and outside contractor and consulting services. Other research and development expenses include the costs of materials and supplies used in research and development, prototype manufacturing, clinical studies, related information technology and an allocation of facilities costs.
Income taxes: Income taxes are accounted for under the asset and liability method. Deferred income tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, and operating loss carry forwards. Deferred income tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which those temporary differences are expected to be recovered or settled. The effect on deferred income tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is provided to reduce the carrying amount of deferred income tax assets if it is considered more likely than not that some portion, or all, of the deferred income tax assets will not be realized.
The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. The Company has elected to classify interest and penalties related to unrecognized tax benefits as part of income tax expense in the consolidated statements of comprehensive income (loss).
Intangible Assets: Intangible assets primarily represent legal costs and filings associated with obtaining patents on the Company’s new discoveries. The Company amortizes these costs over the shorter of the legal life of the patent or its estimated economic life using the straightline method. The Company tests intangible assets with finite lives upon significant changes in the Company’s business environment and any resulting impairment charges are recorded at that time.
Revenue Recognition: Revenue is recognized when the four basic criteria of revenue recognition are met: (1) a contractual agreement exists; (2) transfer of rights has been completed; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured.
The Company may enter into product development and other agreements and with collaborative partners. The terms of the agreements may include non-refundable signing and licensing fees, milestone payments and royalties on any product sales derived from collaborations.
The Company recognizes up front license payments as revenue upon delivery of the license only if the license has stand-alone value to the customer. However, where further performance criteria must be met, revenue is deferred and recognized on a straight-line basis over the period the Company is expected to complete its performance obligations.
Royalty revenue will be recognized upon the sale of the related products provided the Company has no remaining performance obligations under the agreement.
28
|
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
Interest Rate Risk
The Company’s exposure to interest rate risk is minimal. We have no bank borrowings and, although we have placed funds on deposit to earn interest during the year, these are of fixed-term and fixed-rate and therefore offer little exposure to interest rate risk.
Foreign Exchange Risk
Our foreign currency exposure gives rise to market risk associated with exchange rate movements against the US dollar, our reporting currency. Currently, the majority of our expenses and cash and fixed rate deposits are denominated in Pounds Sterling, with the remaining portion denominated in US dollars. Fluctuations in exchange rates, primarily the US dollar against the Pound Sterling, will affect our financial position. At March 31, 2018, the Company held approximately USD 5.7 million in GBP-denominated bank and fixed rate cash accounts. Based on this balance, a 1% depreciation of the Pound against the US dollar would cause an approximate USD 57 thousand reduction in cash and fixed rate deposit account balances.
We have not utilized any hedging instruments in order to mitigate the foreign currency risk.
Inflation
Historically, with UK inflation rates having been low in recent years, inflation has not had a significant effect on our business in the UK, the location of the substantial part of our activities.
29
NEMAURA MEDICAL INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
MARCH 31, 2018 AND 2017
|
|
Page
|
|||
|
Reports of Independent Registered Public Accounting Firms
|
F-2
|
|||
|
Consolidated Balance Sheets as of March 31, 2018 and 2017
|
F-4
|
|||
|
Consolidated Statements of Comprehensive Loss for the years ended March 31, 2018, 2017 and 2016
|
F-5
|
|||
|
Consolidated Statements of Changes of Stockholders’ Equity for the years ended March 31, 2018, 2017 and 2016
|
F-6
|
|||
|
Consolidated Statement of Cash Flows for the years ended March 31, 2018, 2017 and 2016
|
F-7
|
|||
|
Notes to Consolidated Financial Statements
|
F-8-15
|
|||
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Stockholders and the Board of Directors of Nemaura Medical Inc.
Loughborough, United Kingdom
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Nemaura Medical Inc. (the "Company") as of March 31, 2018 and 2017, the related consolidated statements of comprehensive loss, changes in stockholders’ equity (deficit), and cash flows for each of the two years in the period ended March 31, 2018, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2018 and 2017, and the results of its operations and its cash flows for each of the two years in the period ended March 31, 2018, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting in accordance with the standards of the PCAOB. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion in accordance with the standards of the PCAOB.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Emphasis of Matter – Related Party Transactions
The Company has significant transactions and relationships with related parties that are described in Note 7 to the consolidated financial statements.
/s/ Crowe Horwath LLP
We have served as the Company's auditor since 2017.
Denver, Colorado
June 12, 2018
F-2
THIS REPORT IS A COPY OF THE PREVIOUSLY ISSUED REPORT. GHP HORWATH, P.C. HAS CEASED OPERATIONS AND THEREFORE, HAS NOT REISSUED THE REPORT.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors
Nemaura Medical Inc.
We have audited the accompanying consolidated balance sheet of Nemaura Medical Inc. and its subsidiaries (the “Company”) as of March 31, 2016, and the related consolidated statements of comprehensive income/(loss), changes in stockholders’ equity (deficit), and cash flows for each of the years ended March 31, 2016 and 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Nemaura Medical Inc. and its subsidiaries as of March 31, 2016, and the results of their operations and their cash flows for each of the years ended March 31, 2016 and 2015 in conformity with accounting principles generally accepted in the United States of America.
The Company has significant transactions and relationships with related parties that are described in Note 8 to the consolidated financial statements. It is possible that the terms of these transactions may not be the same as those that would result from transactions among unrelated parties.
/s/ GHP Horwath, P.C.
Denver, Colorado
June 13, 2016
F-3
NEMAURA MEDICAL INC.
CONSOLIDATED BALANCE SHEETS
|
|
As of March 31,
|
As of March 31,
|
||||||
|
|
2018
($)
|
2017
($)
|
||||||
|
|
||||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash
|
822,335
|
911,359
|
||||||
|
Fixed rate cash account
|
4,911,551
|
1,867,950
|
||||||
|
Prepaid expenses and other receivables
|
187,139
|
51,086
|
||||||
|
Accrued interest receivable
|
77,508
|
-
|
||||||
|
Total current assets
|
5,998,533
|
2,830,395
|
||||||
|
|
||||||||
|
Other assets:
|
||||||||
|
Property and equipment, net
|
5,770
|
9,161
|
||||||
|
Intangible assets, net of accumulated amortization
|
251,099
|
203,800
|
||||||
|
|
256,869
|
212,961
|
||||||
|
|
||||||||
|
Long term assets:
|
||||||||
|
Fixed rate cash account
|
-
|
4,358,550
|
||||||
|
Total assets
|
6,255,402
|
7,401,906
|
||||||
|
|
||||||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current liabilities
|
||||||||
|
Accounts payable
|
49,912
|
77,530
|
||||||
|
Liability due to related party
|
613,818
|
687,609
|
||||||
|
Other liabilities and accrued expenses
|
147,579
|
87,232
|
||||||
|
|
||||||||
|
Total current liabilities
|
811,309
|
852,371
|
||||||
|
|
||||||||
|
Deferred revenue
|
1,333,128
|
1,183,035
|
||||||
|
1,333,128
|
1,183,035
|
|||||||
|
|
||||||||
|
Total liabilities
|
2,144,437
|
2,035,406
|
||||||
|
|
||||||||
|
Commitments and contingencies
|
||||||||
|
|
||||||||
|
Stockholders’ equity:
|
||||||||
|
Convertible Series A preferred stock, $0.001 par value, 200,000 shares authorized and 137,324 outstanding at March 31, 2018
|
137
|
-
|
||||||
|
Common stock, $0.001 par value, 420,000,000 shares authorized and 67,676,000 shares issued and outstanding at March 31, 2018 (205,000,000 issued and outstanding at March 31, 2017)
|
67,676
|
205,000
|
||||||
|
|
||||||||
|
Additional paid in capital
|
13,056,859
|
12,919,672
|
||||||
|
Accumulated deficit
|
(8,973,082
|
)
|
(7,152,633
|
)
|
||||
|
Accumulated other comprehensive loss
|
(40,625
|
)
|
(605,539
|
)
|
||||
|
Total stockholders’ equity
|
4,110,965
|
5,366,500
|
||||||
|
Total liabilities and stockholders’ equity
|
6,255,402
|
7,401,906
|
||||||
See notes to the consolidated financial statements
F-4
NEMAURA MEDICAL INC
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
|
|
Year Ended March 31,
|
|||||||||||
|
|
2018
($)
|
2017
($)
|
2016
($)
|
|||||||||
|
Revenues
|
||||||||||||
|
Total revenues
|
-
|
-
|
-
|
|||||||||
|
|
||||||||||||
|
|
||||||||||||
|
Operating expenses:
|
||||||||||||
|
Research and development
|
993,833
|
1,034,605
|
1,028,224
|
|||||||||
|
General and administrative
|
915,132
|
516,661
|
511,413
|
|||||||||
|
Total operating expenses
|
1,908,965
|
1,551,266
|
1,539,637
|
|||||||||
|
|
||||||||||||
|
Loss from operations
|
(1,908,965
|
)
|
(1,551,266
|
)
|
(1,539,637
|
)
|
||||||
|
|
||||||||||||
|
Interest income
|
88,516
|
-
|
-
|
|||||||||
|
Net loss
|
(1,820,449
|
)
|
(1,551,266
|
)
|
(1,539,637
|
)
|
||||||
|
|
||||||||||||
|
|
||||||||||||
|
Other comprehensive income/ (loss)
|
||||||||||||
|
Foreign currency translation adjustment
|
564,914
|
(760,999
|
)
|
135,813
|
||||||||
|
Comprehensive loss
|
(1,255,535
|
)
|
(2,312,265
|
)
|
(1,403,824
|
)
|
||||||
|
|
||||||||||||
|
Loss per share
|
||||||||||||
|
Basic and diluted
|
(0.01
|
)
|
*
|
*
|
||||||||
|
|
||||||||||||
|
Weighted average number of shares outstanding
|
150,070,400
|
205,000,000
|
201,726,027
|
|||||||||
* less than $0.01
See notes to the consolidated financial statements
F-5
NEMAURA MEDICAL INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY/(DEFICIT)
YEARS ENDED MARCH 31, 2018 AND 2017
|
|
Common Stock
($)
|
Convertible preferred stock
($)
|
Additional Paid in Capital
($)
|
Accumulated Deficit
($)
|
Accumulated
Other Comprehensive Income
($)
|
Total Stockholders’ Equity
($)
|
||||||||||||||||||
|
|
||||||||||||||||||||||||
|
Balance at April 1, 2015
|
200,000
|
-
|
2,924,672
|
(4,061,730
|
)
|
19,647
|
(917,411
|
)
|
||||||||||||||||
|
|
||||||||||||||||||||||||
|
Common stock issued for cash
|
5,000
|
-
|
9,995,000
|
-
|
-
|
10,000,000
|
||||||||||||||||||
|
Net loss
|
-
|
-
|
(1,539,637
|
)
|
-
|
(1,539,637
|
)
|
|||||||||||||||||
|
Other comprehensive income - foreign currency translation gain
|
-
|
-
|
-
|
-
|
135,813
|
135,813
|
||||||||||||||||||
|
Balance at March 31, 2016
|
205,000
|
12,919,672
|
(5,601,367
|
)
|
155,460
|
7,678,765
|
||||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(1,551,266
|
)
|
-
|
(1,551,266
|
)
|
||||||||||||||||
|
Other comprehensive income - foreign currency translation loss
|
-
|
-
|
-
|
-
|
(760,999
|
)
|
(760,999
|
)
|
||||||||||||||||
|
Balance at March 31, 2017
|
205,000
|
-
|
12,919,672
|
(7,152,633
|
)
|
(605,539
|
)
|
5,366,500
|
||||||||||||||||
|
Cancellation of ordinary stock and issue of convertible preferred stock
|
(137,324
|
)
|
137
|
137,187
|
-
|
-
|
-
|
|||||||||||||||||
|
Net loss
|
-
|
-
|
-
|
(1,820,449
|
)
|
-
|
(1,820,449
|
)
|
||||||||||||||||
|
Other comprehensive income - foreign currency translation gain
|
-
|
-
|
-
|
-
|
564,914
|
564,914
|
||||||||||||||||||
|
Balance at March 31, 2018
|
67,676
|
137
|
13,056,859
|
(8,973,082
|
)
|
(40,625
|
)
|
4,110,965
|
||||||||||||||||
See notes to the consolidated financial statements
F-6
NEMAURA MEDICAL INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
Year Ended March 31
|
|||||||||||
|
|
2018
($)
|
2017
($)
|
2016
($)
|
|||||||||
|
Cash Flows from Operating Activities:
|
||||||||||||
|
Net loss
|
(1,820,449
|
)
|
(1,551,266
|
)
|
(1,539,637
|
)
|
||||||
|
|
||||||||||||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Depreciation and amortization
|
29,256
|
20,433
|
17,404
|
|||||||||
|
Changes in assets and liabilities:
|
||||||||||||
|
Prepaid expenses and other receivables
|
(138,859
|
)
|
85,367
|
224,392
|
||||||||
|
Prepayment to related party for clinical trials
|
-
|
-
|
249,459
|
|||||||||
|
Accounts payable
|
(31,247
|
)
|
2,522
|
(31,279
|
)
|
|||||||
|
Liability due to related party
|
(162,644
|
)
|
270,975
|
-
|
||||||||
|
Accrued expenses
|
60,407
|
(20,859
|
)
|
(129,704
|
)
|
|||||||
|
Accrued interest receivable
|
(73,441
|
)
|
-
|
-
|
||||||||
|
Net cash used in operating activities
|
(2,136,977
|
)
|
(1,192,828
|
)
|
(1,209,365
|
)
|
||||||
|
|
||||||||||||
|
Cash Flows from Investing Activities:
|
||||||||||||
|
Decrease in restricted cash
|
-
|
-
|
-
|
|||||||||
|
Purchase of intangible assets
|
(45,260
|
)
|
(73,070
|
)
|
(78,197
|
)
|
||||||
|
Purchase of property and equipment
|
-
|
(6,519
|
)
|
(9,367
|
)
|
|||||||
|
Fixed rate savings account
|
1,994,475
|
(6,226,500
|
)
|
-
|
||||||||
|
Net cash provided by/ (used in) investing activities
|
1,949,215
|
(6,306,089
|
)
|
(87,564
|
)
|
|||||||
|
|
||||||||||||
|
Cash Flows from Financing Activities:
|
||||||||||||
|
Net proceeds from issuance of common stock
|
-
|
-
|
10,000,000
|
|||||||||
|
Net advances from related party
|
-
|
-
|
299,434
|
|||||||||
|
Net cash provided by financing activities
|
-
|
-
|
10,299,434
|
|||||||||
|
Net (decrease)/increase in cash
|
(187,762
|
)
|
(7,498,917
|
)
|
9,002,505
|
|||||||
|
Effect of exchange rate changes on cash
|
98,738
|
(993,689
|
)
|
46,711
|
||||||||
|
Cash at beginning of year
|
911,359
|
9,403,965
|
354,749
|
|||||||||
|
Cash at end of year
|
822,335
|
911,359
|
9,403,965
|
|||||||||
|
Supplemental disclosure of cash flow information:
|
||||||||||||
|
|
||||||||||||
|
Schedule of non-cash investing and financing transactions:
|
||||||||||||
|
Transfer of property and equipment and intangible assets to related party
|
-
|
-
|
23,428
|
|||||||||
See notes to the consolidated financial statements
F-7
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
NOTE 1 – ORGANIZATION, PRINCIPAL ACTIVITIES AND MANAGEMENT’S PLANS
Nemaura Medical Inc. (“Nemaura” or the “Company”), through its operating subsidiaries, performs medical device research and manufacturing of a continuous glucose monitoring system (“CGM”), named sugarBEAT. The sugarBEAT device is a non-invasive, wireless device for use by persons with Type I and Type II diabetes, and may also be used to screen pre-diabetic patients. The sugarBEAT device extracts analytes, such as glucose, to the surface of the skin in a non-invasive manner where it is measured using unique sensors and interpreted using a unique algorithm.
Nemaura is a Nevada holding company organized in 2013. Nemaura owns one hundred percent (100%) of Region Green Limited, a British Virgin Islands corporation formed (“RGL”) on December 12, 2013. Region Green Limited owns one hundred percent (100%) of the stock in Dermal Diagnostic (Holdings) Limited, an England and Wales corporation (“DDHL”) formed on December 11, 2013, which in turn owns one hundred percent (100%) of Dermal Diagnostics Limited, an England and Wales corporation formed on January 20, 2009 (“DDL”), and one hundred percent (100%) of Trial Clinic Limited, an England and Wales corporation formed on January 12, 2011 (“TCL”).
DDL is a diagnostic medical device company headquartered in Loughborough, Leicestershire, England, and is engaged in the discovery, development and commercialization of diagnostic medical devices. The Company’s initial focus has been on the development of the sugarBEAT device, which consists of a disposable patch containing a sensor, and a non-disposable miniature electronic watch with a re-chargeable power source, which is designed to enable trending or tracking of blood glucose levels. All the Company’s operations and assets are located in England.
The following diagram illustrates Nemaura’s corporate and shareholder structure as of March 31, 2018:
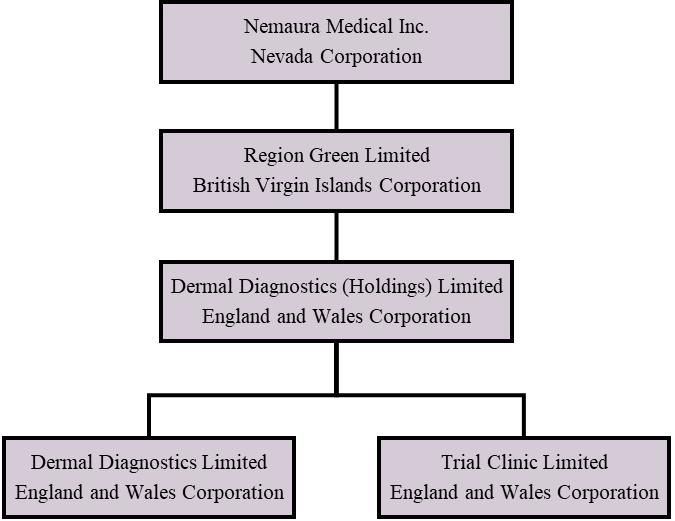
F-8
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
The Company has a limited operating history, recurring losses from operations and an accumulated deficit as of March 31, 2018. The Company expects to continue to incur losses from operations at least until clinical trials are completed later this year, when management expects that the product will become available to be marketed. Management has evaluated the expected expenses to be incurred along with its available cash, and has determined that there is not substantial doubt as to its ability to continue as a going concern for at least one year subsequent to the date of issuance of these financial statements. The Company has approximately $822,000 of readily available cash on hand at March 31, 2018 and approximately $4.9 million that will become available in December 2018 (Note 3b). Early withdrawal may generally be made for liquidity needs.
Management’s strategic plans include the following:
- continuing to advance commercialization of the Company’s principal product, in both UK, European and other international markets;
- pursuing additional capital raising opportunities; and
- continuing to explore and execute prospective partnering or distribution opportunities;
NOTE 2 – BASIS OF PRESENTATION
(a) Basis of presentation
The accompanying consolidated financial statements include the accounts of the Company and the Company’s subsidiaries, DDL, TCL, DDHL and RGL. The consolidated financial statements are prepared in accordance with accounting principles generally accepted in the United States of America, and all significant intercompany balances and transactions have been eliminated on consolidation.
The functional currency for the majority of the Company’s operations is the Great Britain Pound Sterling (“GBP”), and the reporting currency is the US Dollar.
NOTE 3 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
(a) Cash and cash equivalents
The Company considers all highly liquid investments purchased with original maturities of three months or less to be cash equivalents. Cash and cash equivalents consist primarily of cash deposits maintained in the United Kingdom. From time to time, the Company’s cash account balances exceed amounts covered by the Financial Services Compensation Scheme. The Company has never suffered a loss due to such excess balances.
(b) Fixed rate cash accounts:
From time to time the Company invests funds in fixed rate cash savings accounts. These accounts, at the time of the initial investment, provide a higher interest rate than other bank accounts, and also require the Company to maintain the funds in the accounts for a period of time, currently $4.9 million through December 2018. Early withdrawal may generally be made for liquidity needs.
(c) Fair value of financial instruments
The Company’s financial instruments primarily consist of cash, accounts receivable, fixed rate cash accounts, and accounts payable. As of the year-end dates, the estimated fair values of non-related party financial instruments were not materially different from their carrying values as presented, due to their short maturities. The fair value of amounts payable to related parties are not practicable to estimate due to the related party nature of the underlying transactions.
(d) Property and equipment
Property and equipment is stated at cost and depreciated using the straight-line method
over the estimated useful lives of the assets, generally ten years for fixtures and fittings.
(e) Intangible assets
Intangible assets consist of licenses and patents associated with the sugarBEAT device and are amortized on a straight-line basis, generally over their legal lives of up to 20 years and are reviewed for impairment. The Company evaluates its intangible assets (all have finite lives) and other long-lived assets for impairment whenever events or circumstances indicate that they may not be recoverable, or at least annually. Recoverability of finite and other long-lived assets is measured by comparing the carrying amount of an asset group to the future undiscounted net cash flows expected to be generated by that asset group. The Company groups assets for purposes of such review at the lowest level for which identifiable cash flows of the asset group are largely independent of the cash flows of the other groups of assets and liabilities. The amount of impairment to be recognized for finite and other long-lived assets is calculated as the difference between the carrying value and the fair value of the asset group, generally measured by discounting estimated future cash flows. There were no impairment indicators present during the years ended March 31, 2018, 2017 or 2016.
F-9
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
(f) Revenue Recognition
Revenue is recognized when the four basic criteria of revenue recognition are met: (1) a contractual agreement exists; (2) transfer of rights has been completed; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured.
The Company may enter into product development and other agreements and with collaborative partners. The terms of the agreements may include non-refundable signing and licensing fees, milestone payments and royalties on any product sales derived from collaborations.
The Company recognizes up front license payments as revenue upon delivery of the license only if the license has stand alone value to the customer. However, where further performance criteria must be met, revenue is deferred and recognized on a straight line basis over the period the Company is expected to complete its performance obligations (note 4).
Royalty revenue will be recognized upon the sale of the related products provided the Company has no remaining performance obligations under the agreement.
(g) Research and development expenses
The Company charges research and development expenses to operations as incurred. Research and development expenses primarily consist of salaries and related expenses for personnel and outside contractor and consulting services. Other research and development expenses include the costs of materials and supplies used in research and development, prototype manufacturing, clinical studies, related information technology and an allocation of facilities costs.
(h) Income taxes
Income taxes are accounted for under the asset and liability method. Deferred income tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, and operating loss carry forwards. Deferred income tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred income tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is provided to reduce the carrying amount of deferred income tax assets if it is considered more likely than not that some portion, or all, of the deferred income tax assets will not be realized.
The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. The Company has elected to classify interest and penalties related to unrecognized tax benefits as part of income tax expense in the consolidated statements of comprehensive loss. The Company does not have any accrued interest or penalties associated with any unrecognized tax benefits, nor was any interest expense related to unrecognized tax benefits recognized for the years ended March 31, 2018, 2017 and 2016.
In December 2017, the US Tax Cuts and Jobs Act (the”Act”) was signed into law. Generally, this Act reduces corporate rates from a top rate of 35% to a top rate of 21%, effective January 1, 2018. As the Company’s US operations are minimal, and all deferred tax assets are fully allowed for, there is no significant impact to the Company as of and for the three and twelve month periods ended March 31, 2018.
(i) Earnings per share
Basic earnings per share is computed by dividing income available to common stockholders by the weighted-average number of common shares outstanding during the period. There were no potentially dilutive securities as
of March 31, 2018, 2017 and 2016. For the years ended March 31, 2018, 2017 and 2016, warrants to purchase 10 million shares of common stock and preferred stock convertible to 137,324,000 shares of common stock were anti-dilutive and were excluded from the calculation of diluted loss per share.
(j) Use of estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the year. Actual results may differ from those estimates.
(k) Foreign currency translation
The functional currency of the Company is the Great Britain Pound Sterling (“GBP”). The reporting currency is the United States dollar (US$). Stockholders’ equity is translated into United States dollars from GBP at historical exchange rates. Assets and liabilities are translated at the exchange rates as of balance sheet date. Income and expenditures are translated at the average exchange rates prevailing during the reporting period. The translation rates are as follows for each year end to March 31:
|
|
2018
|
2017
|
2016
|
|
Year end GBP : US$ exchange rate
|
1:1.4033
|
1:1.2453
|
1:1.4318
|
|
Average period/yearly GBP : US$ exchange rate
|
1:1.3305
|
1:1.3146
|
1:1.5224
|
Adjustments resulting from translating the financial statements into the United States dollar are recorded as a separate component of accumulated other comprehensive loss in stockholders’ equity.
F-10
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
(l) Recent accounting pronouncements
The Company continually assesses any new accounting pronouncements to determine their applicability. When it is determined that a new accounting pronouncement affects the Company's financial reporting, the Company undertakes a study to determine the consequences of the change to its consolidated financial statements and assures that there are proper controls in place to ascertain that the Company's consolidated financial statements properly reflect the change.
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Updates ("ASU") No. 2014-09, Revenue from Contracts with Customers. ASU 2014-09 has been modified multiple times since its initial release. This ASU outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective. ASU 2014-09, as amended, becomes effective for annual reporting periods beginning after December 15, 2017. Early adoption is permitted. As an Emerging Growth Company, the Company is allowed to adopt new, or updated, accounting standards using the same time frame that applies to private companies. The Company will adopt this standard on April 1, 2019. Management is currently evaluating the impact of adoption of this ASU on the Company’s consolidated financial statements.
In March 2016, the FASB issued ASU No. 2016-02, Leases. The main difference between the provisions of ASU No. 2016-02 and previous U.S. GAAP is the recognition of right-of-use assets and lease liabilities by lessees for those leases classified as operating leases under previous U.S. GAAP. ASU No. 2016- 02 retains a distinction between finance leases and operating leases, and the recognition, measurement, and presentation of expenses and cash flows arising from a lease by a lessee have not significantly changed from previous U.S. GAAP. For leases with a term of 12 months or less, a lessee is permitted to make an accounting policy election by class of underlying asset not to recognize right-of-use assets and lease liabilities. The accounting applied by a lessor is largely unchanged from that applied under previous U.S. GAAP. In transition, lessees and lessors are required to recognize and measure leases at the beginning of the earliest period presented using a modified retrospective approach. This ASU is effective for public business entities in fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018. Early adoption is permitted as of the beginning of any interim or annual reporting period. As an Emerging Growth Company, the Company is allowed to adopt new, or updated, accounting standards using the same time frame that applies to private companies. The Company will adopt this standard on April 1, 2020. Management is currently evaluating the impact of adoption of this ASU on the Company’s consolidated financial statements.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows - Classification of Certain Cash Receipts and Cash Payments. ASU No. 2016-15 is intended to reduce diversity in how certain cash receipts and cash payments are presented in the statement of cash flows. The new guidance clarifies the classification of cash activity related to debt prepayment or debt extinguishment costs, settlement of zero-coupon debt instruments, contingent consideration payments made after a business combination, proceeds from the settlement of insurance claims, proceeds from the settlement of corporate and bank-owned life insurance policies, distributions received from equity-method investments, and beneficial interests in securitization transactions. The guidance also describes a predominance principle pursuant to which cash flows with aspects of more than one class that cannot be separated should be classified based on the activity that is likely to be the predominant source or use of cash flow. This ASU is effective for public entities for annual and interim periods beginning after December 15, 2017. Early adoption is permitted as of the beginning of any interim or annual reporting period. The Company is currently evaluating the impact this standard will have on its financial statements and related disclosures, but does not expect it to have a material effect on the Company's consolidated financial statements and related disclosures.
(m) Risks and Uncertainties:
The Company is in the development stage of one primary product that it expects to introduce to the UK market after completion of clinical trials and CE mark approval (European Union approval of the product). The Company has entered into sales and marketing agreements for the product but has not yet entered into manufacturing agreements. These matters raise uncertainties as regulatory acceptance of the Company’s primary product development efforts and if acceptance is attained, the cost structure to produce the product.
(n) Preferred shares
On October 5, 2017, the Company entered into common stock exchange agreements with each of its three largest shareholders, to exchange, in the aggregate, 137,324,000 shares of the Company’s common stock for 137,324 shares of Series A Convertible Preferred Stock. Each share of Series A Convertible Preferred Stock is convertible into 1,000 shares of the Company’s common stock, automatically upon the occurrence of all of certain triggering events, as set forth in the Certificate of Designation, namely (a) the sugarBEAT® device to be commercialized has CE regulatory approval; (b) retail sales having commenced; and (c) retail sales exceeding USD$5 million, inclusive of advanced sales or voluntarily by the holder after February 7, 2018, if these triggering events have not occurred. Each holder of issued and outstanding Series A Convertible Preferred Stock is entitled to a number of votes equal to the number of shares of common stock into which the Series A Convertible Preferred Stock is convertible. Holders of Series A Convertible Preferred Stock are entitled to vote on any and all matters presented to stockholders of the Company, except as provided by law. The Series A Convertible Preferred Stock has no preference to the common stock as to dividends or distributions of assets upon liquidation or winding up of the Company (which has been agreed to by the holders of the Series A Convertible Preferred Stock). The Company determined that the fair value of the preferred shares issued for the common shares was equivalent to the fair value of the common shares exchanged.
On November 6, 2017, the transaction was consummated and 137,324,000 shares of common stock were cancelled. As a result, the Company has 67,676,000 shares of common stock issued and outstanding.
(o) Subsequent events
We have signed a full commercial agreement with Dallas Burston Ethitronix Limited in May 2018 for all other European territories as part of an equal joint venture agreement. The joint venture intends to seek sub-license rights opportunities to one or more leading companies in the diabetes monitoring space, to leverage their network, infrastructure and resources.
On June 5, 2018, the three holders of our Series A Convertible Preferred Stock (the “Series A Preferred”) each delivered notices of conversion to voluntarily convert their Series A Preferred, in the aggregate amount of 137,324 shares, into 137,324,000 shares of our common stock. The holders had the right to voluntarily convert each share of Series A Preferred into 1,000 shares of common stock of the Company. As a result of the conversion, we currently have 205,000,000 shares of common stock outstanding.
F-11
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
NOTE 4 – LICENSING AGREEMENT
In March 2014, the Company entered into an Exclusive Marketing Rights Agreement with an unrelated third party, that granted to the third party the exclusive right to market and promote the sugarBEAT device and related patches under its own brand in the United Kingdom and the Republic of Ireland, the Channel Islands and the Isle of Man. The Company received a non-refundable, up front cash payment of GBP 1,000,000 (approximately $1.403 million and $1.245 million as of March 31, 2018 and March 31, 2017 respectively) which is wholly non-refundable, upon signing the agreement.
As the Company has continuing performance obligations under the agreement, the up-front fees received from this agreement have been deferred and will be recorded as income over the term of the commercial licensing agreement beginning from the date of clinical evaluation approval. As the Company expects commercialization of the sugarBEAT device to occur in the year ending March 31, 2019, approximately $70,000 of the deferred revenue has been classified as a current liability.
In April 2014, a Letter of Intent was signed with the third party which specified a 10 year term and in November 2015, a Licence, Supply and Distribution agreement with an initial 5 year term was executed. The Company grants the exclusive right to market and promote its product in the United Kingdom, and purchase the product at specified prices.
NOTE 5– PROPERTY AND EQUIPMENT
As of March 31, 2018, and March 31, 2017 property and equipment is summarized as follows.
|
|
March 31, 2018
($)
|
March 31, 2017
($)
|
||||||
|
Fixtures and fittings
|
18,213
|
16,163
|
||||||
|
Less accumulated depreciation
|
(12,443
|
)
|
(7,002
|
)
|
||||
|
|
5,770
|
9,161
|
||||||
NOTE 6 - INTANGIBLE ASSETS
As of March 31, 2018, and March 31, 2017 intangible assets are summarized as follows:
|
|
March 31, 2018
($)
|
March 31, 2017
($)
|
||||||
|
Patents and licenses
|
323,987
|
244,457
|
||||||
|
Less accumulated amortization
|
(72,888
|
)
|
(40,657
|
)
|
||||
|
|
251,099
|
203,800
|
||||||
Estimated amortization expense is approximately $22,000 for each of the next five years.
F-12
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
NOTE 7 – RELATED PARTY TRANSACTIONS
Nemaura Pharma Limited (Pharma) and NDM Technologies Limited (NDM) are entities controlled by the Company’s majority shareholder, DFH Chowdhury.
In accordance with the United States Securities and Exchange Commission (SEC) Staff Accounting Bulletin 55, these financial statements are intended to reflect all costs associated with the operations of DDL and TCL. Pharma has invoiced DDL and TCL for research and development services. In addition, certain operating expenses of DDL and TCL were incurred and paid by Pharma and NDM which have been invoiced to the Company. Certain costs incurred by Pharma and NDM are directly attributable to DDL and TCL and such costs were billed to the Company. Management believes the allocation methodologies used are reasonable. DDL and TCL advanced Pharma certain amounts to cover a portion of the costs.
Following is a summary of activity between the Company and Pharma and NDM for the years ended March 31, 2018, 2017 and 2016. These amounts are unsecured, interest free, and payable on demand.
|
|
Year Ended March 31,
2018
($)
|
Year Ended March 31,
2017
($)
|
Year Ended March 31,
2016
($)
|
|||||||||
|
Balance due from Pharma and NDM at beginning of period
|
(687,609
|
)
|
(494,145
|
)
|
192,484
|
|||||||
|
Amounts advanced to Pharma
|
-
|
-
|
58,197
|
|||||||||
|
Amounts received from Pharma
|
(145,214
|
)
|
(2,480
|
)
|
(228,361
|
)
|
||||||
|
Reduction in prepayments to Pharma for clinical trials
|
-
|
-
|
(247,596
|
)
|
||||||||
|
Amounts invoiced by Pharma to DDL, NM and TCL (1)
|
(842,739
|
)
|
(577,481
|
)
|
(331,714
|
)
|
||||||
|
Amounts invoiced by DDL to Pharma
|
-
|
15,305
|
16,307
|
|||||||||
|
Amounts repaid by DDL to Pharma
|
1,096,767
|
249,060
|
-
|
|||||||||
|
Amounts paid by DDL on behalf of Pharma
|
19,889
|
42,403
|
-
|
|||||||||
|
Sale of fixed and intangible assets to Pharma and NDM
|
-
|
-
|
17,775
|
|||||||||
|
Foreign exchange differences
|
(54,912
|
)
|
79,729
|
28,763
|
||||||||
|
Net balance due to Pharma and NDM at end of the period
|
(613,818
|
)
|
(687,609
|
)
|
(494,145
|
)
|
||||||
| (1) |
These amounts are included primarily in research and development expenses.
|
The Company routinely reviews its statement of cashflows presentation of related party transactions for financing or operating classification based on the underlying nature of the item and intended repayment.
Total costs charged to the Company by Pharma and NDM were $842,739, $577,481 and $331,714 for the years 2018, 2017 and 2016 respectively.
Subsequent to March 31, 2018, the Company made payments to Pharma on the outstanding balances at March 31, 2018 of £279,792.
NOTE 8 – INCOME TAXES
The Company and its subsidiaries file separate income tax returns.
The United States of America
The Company is incorporated in the US, and as a result of the new US Tax Cuts and Jobs Act, is subject to a US federal corporate income tax blended rate of 30.79% for the year ended March 31, 2018. The federal corporate income tax rate for future years is scheduled to be 21%.
British Virgin Islands
RGL is incorporated in the British Virgin Islands (“BVI”). Under the current laws of the BVI, RGL is not subject to tax on income or capital gains. In addition, upon payments of dividends by RGL, no BVI withholding tax is imposed. During the years ended March 31, 2018, 2017 and 2016, there was no income or expenses in the BVI.
F-13
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
UK
DDL, TCL and DDHL are all incorporated in the United Kingdom (UK) and the applicable UK statutory income tax rate for these companies is 19%.
For the years ended March 31, 2018, 2017 and 2016 loss before income tax expense (benefit) arose in the UK and U.S.
|
|
Year ended March 31,
|
|||||||||||
|
|
2018
|
2017
|
2016
|
|||||||||
| $ | $ | $ | ||||||||||
|
Loss before income taxes arising in UK
|
(1,353,243
|
)
|
(1,251,870
|
)
|
(1,300,468
|
)
|
||||||
|
Loss before income taxes arising in United States
|
(467,206
|
)
|
(299,396
|
)
|
(239,169
|
)
|
||||||
|
Total loss before income tax
|
(1,820,449
|
)
|
(1,551,266
|
)
|
(1,539,637
|
)
|
||||||
Reconciliation of our effective tax rate to loss to the statutory U.S federal tax rate is as follows:
|
|
Year ended March 31,
|
|||||||||||||||||||||||
| 2018 | 2017 | 2016 | ||||||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||||||
|
Loss before income taxes
|
(1,820,449
|
)
|
(1,551,266
|
)
|
(1,539,637
|
)
|
||||||||||||||||||
|
Expected tax benefit
|
(561,000
|
)
|
(31
|
%)
|
(527,000
|
)
|
(34
|
%)
|
(523,000
|
)
|
(34
|
%)
|
||||||||||||
|
Foreign tax differential
|
36,000
|
2
|
%
|
217,000
|
14
|
%
|
216,000
|
14
|
%
|
|||||||||||||||
|
Enhanced research and development
|
(215,000
|
)
|
(12
|
%)
|
(198,000
|
)
|
(13
|
%)
|
(177,000
|
)
|
(11
|
%)
|
||||||||||||
|
Other
|
35,000
|
2
|
%
|
-
|
- |
-
|
- | |||||||||||||||||
|
Change in valuation allowance
|
705,000
|
39
|
%
|
455,000
|
29
|
%
|
484,000
|
31
|
%
|
|||||||||||||||
|
Actual income tax benefit
|
- | - | - | - | - | - | ||||||||||||||||||
The tax effects of the temporary differences that give rise to significant portions of deferred income tax assets are presented below:
|
|
As of March 31,
|
|||||||
|
|
2018
|
2017
|
||||||
| $ | $ | |||||||
|
Net operating tax loss carried forwards
|
2,229,000
|
1,818,000 | ||||||
|
Valuation allowance
|
(2,229,000
|
)
|
(1,818,000 | ) | ||||
|
Net deferred tax assets
|
-
|
- | ||||||
For each of the years ended March 31, 2018, 2017 and 2016, the Company did not have unrecognized tax benefits, and therefore no interest or penalties related to unrecognized tax benefits were accrued. Management does not expect that the amount of unrecognized tax benefits will change significantly within the next twelve months.
The Company mainly files income tax returns in the United States and the UK. The Company is subject to U.S. federal income tax examination by tax authorities for tax years beginning in 2014. The UK tax returns for the Company’s UK subsidiaries are open to examination by the UK tax authorities for the tax years beginning in April 1, 2012.
As of March 31, 2018, the Company has net operating losses (NOLs) of approximately $1.4 million in the US and $7.1 million in the UK. These US and UK NOLs may be carried forward indefinitely.
NOTE 9 – STOCKHOLDERS’ EQUITY
In November 2015, the Company issued 5 million shares of common stock and warrants to purchase 10 million shares of common stock for total proceeds of $10 million. The warrants are exercisable at $0.50 per share through to the fifth anniversary of the listing of the Company on a national exchange. The Company listed to the Nasdaq exchange on January 25, 2018.
F-14
NEMAURA MEDICAL INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
March 31, 2018
NOTE 10 - QUARTERLY FINANCIAL INFORMATION (UNAUDITED)
The following is a summary of consolidated quarterly financial information:
|
|
Quarter Ended
|
|||||||||||||||
|
2018
|
June 30
|
Sept. 30
|
Dec. 31
|
March 31
|
||||||||||||
|
Total revenue
|
|
$
|
-
|
|
$
|
-
|
|
$
|
-
|
|
$
|
-
|
||||
|
Loss from operations
|
|
$
|
(417,320
|
)
|
|
$
|
(447,516
|
)
|
|
$
|
(476,353
|
)
|
|
$
|
(567,776
|
)
|
|
Net loss
|
|
$
|
(407,787
|
)
|
|
$
|
(393,031
|
)
|
|
$
|
(466,365
|
)
|
|
$
|
(553,266
|
)
|
|
Basic and diluted loss per share
|
|
$
|
*
|
|
$
|
*
|
|
$
|
*
|
|
$
|
*
|
||||
|
Weighted average number of shares outstanding
|
205,000,000
|
205,000,000
|
121,411,478
|
150,070,400
|
||||||||||||
|
|
Quarter Ended
|
|||||||||||||||
|
2017
|
June 30
|
Sept. 30
|
Dec. 31
|
March 31
|
||||||||||||
|
Total revenue
|
$
|
-
|
$
|
-
|
$
|
-
|
$
|
-
|
||||||||
|
Loss from operations
|
$
|
(494,183
|
)
|
$
|
(322,482
|
)
|
$
|
(375,366
|
)
|
$
|
(359,235
|
)
|
||||
|
Net loss
|
$
|
(494,183
|
)
|
$
|
(322,482
|
)
|
$
|
(375,366
|
)
|
$
|
(359,235
|
)
|
||||
|
Basic and diluted loss per share
|
$
|
*
|
$
|
*
|
$
|
*
|
$
|
*
|
||||||||
|
Weighted average number of shares outstanding
|
205,000,000
|
205,000,000
|
205,000,000
|
205,000,000
|
||||||||||||
* less than $0.01
F-15
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures
Mr. Dewan F.H. Chowdhury, who is our Chief Executive Officer and Mr Iain S. Anderson, who is our Principal Financial and Accounting Officer, have evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company's management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost benefit relationship of possible controls and procedures. Based on this evaluation, management concluded that our disclosure controls and procedures were not effective at the reasonable assurance level due to a material weakness in our internal control over financial reporting, which is described below.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Exchange Act Rule 13a-15(f). Our internal control system is a process designed by, or under the supervision of, our principal executive and principal financial officer, or persons performing similar functions, and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”).
Our internal control over financial reporting includes policies and procedures that pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect transactions and dispositions of assets; provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and that receipts and expenditures are being made only in accordance with the authorization of our management and directors; and provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our consolidated financial statements.
Because of our inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of our internal control over financial reporting as of March 31, 2018. In making this assessment we used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control – Integrated Framework (2013). As a result of its assessment, management identified material weaknesses in our internal control over financial reporting. Based on the material weaknesses as described below, management concluded that our internal control over financial reporting was not effective as of March 31, 2018.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that, there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. As a result of our assessment, management identified the following material weaknesses in internal control over financial reporting as of March 31, 2018:
| · |
Our size has prevented us from being able to employ sufficient resources to enable us to have an adequate level of supervision and segregation of duties within our internal control system. This has resulted in a number of internal control deficiencies. Specifically,
|
| · |
there is a lack of segregation of duties in the processing of financial transactions which could result in inappropriate initiation, processing and review of transactions and the financial reporting of such transactions whether due to errors or fraud;
|
| · |
there is a lack of review and approval of journal entries which could result in the improper initiation and reporting of transactions; and
|
| · |
there is a lack of access controls over the Company’s IT applications which could result in the improper initiation and reporting of significant transactions.
|
| · |
Management has identified that there is a lack of adequate financial expertise related to the assessment of complex transactions and a lack of adequate resources to review out of the ordinary transactions and arrangements of the Company. This could result in the improper reporting of significant transactions or arrangements.
|
| · |
Related party transactions. Specifically, there are limited policies and procedures to ensure that financial statement disclosures reconcile fully to the underlying accounting records and that Board approval of these transactions is not documented.
|
Notwithstanding the identified material weaknesses, management believes the consolidated financial statements included in this Annual Report on Form 10-K fairly represent in all material respects our financial condition, results of operations and cash flows at and for the periods presented in accordance with U.S. GAAP.
30
Remediation of Material Weaknesses
We are in the process of implementing improvements and remedial measures in response to these assessments and recommendations, including:
| · |
Assembling a team from our finance department to be responsible for the preparation of financial statements under U.S. Securities laws, including hiring additional qualified personnel such as a CFO with US listed company experience. In assembling this team, the Company will put in place controls to segregate duties in the processing of key transactions, controls to ensure the review and approval of journal entries and controls to ensure that access to IT systems is limited to authorized users based on the applications and their functions within the organization.
|
| · |
Engaging a third party consulting firm to assist in assessing, designing, implementing, and monitoring controls related to financial statement preparation, IT general controls, journal entries, and significant operating processes.
|
| · |
Organizing regular training sessions on US GAAP for our finance department in the form of workshops, seminars and newsletters as well as requiring our finance personnel to participate in annual in-house or public US GAAP training courses; and
|
| · |
Implementing stronger internal controls and processes over related party transactions including segregating reviews and approvals, as well as continuing efforts to reduce the amount and volume of related party transactions; and
|
| · |
Establishing an audit committee with an “audit committee financial expert” within the definition of the applicable Securities and Exchange Commission. The committee will be helped by an outsourced internal audit department to review our internal control processes, policies and procedures to ensure compliance with the Sarbanes-Oxley Act.
|
In addition of the immediate remediation plan, we will put our effort, in the coming year, in improving our control environment detailed below:
| · |
Ongoing assessment of our current Internal Control Over Financial Reporting against COSO 2013 and the requirements set forth by Sarbanes-Oxley Act section 404. This task will be conducted by an independent expert.
|
| · |
Continued testing of the operating effectiveness of the controls that have been identified and implemented in order to prevent misstatement of the financial statements. In addition, the company will focus on the design and implementation of Key Performance Indicators (KPIs) in order to measure the quality of the processes in place, and the efficiency of the controls.
|
As described below, certain aspects of this plan were implemented in the year ended March 31, 2018 and other aspects are expected to be implemented on, or around, the time that we are prepared to take our sugarBEAT product to market.
Attestation Report of the Registered Public Accounting Firm
As an Emerging Growth Company, we are not required to provide in this Annual Report on Form 10-K, an attestation report of our registered public accounting firm on our internal control over financial reporting.
Changes in Internal Control over Financial Reporting
During the year ended March 31, 2018, we continued to implement the remediation plan discussed above. We have implemented the following changes:
| · |
We have continued to engage with a third party consulting firm to help us assess our current internal control over financial reporting against COSO 2013, as well as identifying a gap analysis, suggest improvements in controls, and assist us in testing our control systems. Further testing has occurred of certain controls, including purchasing processes, payment processes, and month end closing procedures. In addition, an initial assessment of IT general controls has been conducted, with a view to assessing the current situation and strengthening these controls where deemed necessary. The Company has set a target to design and implement controls that will address the material weaknesses by March 31, 2019. The independent advisers have agreed a table of work to complete all controls reviews, implementation and testing in this timeframe and the Company has committed to meeting this timeframe. However, as this process is ongoing and there will need to be sufficient time to ensure implemented controls are operating effectively, there is no assurance that all material weaknesses will be fully remediated by March 31, 2019.
|
| · |
During the quarter ended September 30, 2017, the Board of Directors (“Board”) appointed three independent directors to serve on our Board, each of whom meet the definition of “independent” as set forth under The Nasdaq Stock Market rules. The Board has established an audit committee with each of the independent directors serving as members of the audit committee. The Board has also designated one of the independent directors to serve as Chair of the audit committee who meets the definition of an “audit committee financial expert.”.
|
| · |
During the year, members of our accounting department have attended training courses relating to US accounting practice and changes in taxation laws.
|
There have been no other changes in our internal control over financial reporting during our last fiscal year that have materially affected or are reasonably likely to materially affect our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION.
None.
31
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The following persons are our executive officers and directors, and hold the positions set forth opposite their respective names.
|
Name
|
Age
|
Position
|
Date of Appointment
|
|||
|
Dewan Fazlul Hoque Chowdhury
|
45
|
Chief Executive Officer, President and Director
|
December 24, 2013
|
|||
|
Bashir Timol
|
43
|
Director
Chief Business Officer
|
December 24, 2013
April 9, 2018 |
|||
|
Iain Anderson
|
58
|
Chief Financial Officer
|
December 12, 2016
|
|||
|
Thomas Moore
|
54
|
Independent Director
|
August 3, 2017
|
|||
|
Dr. Salim Natha
|
51
|
Independent Director
|
July 26, 2017
|
|||
|
Timothy Johnson
|
34
|
Independent Director
|
July 17, 2017
|
|||
Our directors hold office until the earlier of their death, resignation or removal or until their successors have been qualified.
Dewan Fazlul Hoque Chowdhury. Dr. Chowdhury has been our President, Chief Executive Officer and a member of our board of directors since our incorporation on January 20, 2009. He is in charge of research and development of our core technologies, product development, innovation and commercialization. He also coordinates and oversees legal compliance; development of the company mission; policy and planning. Prior to establishing the Company, Dr. Chowdhury was the founder and CEO of Microneedle Technologies and Nemaura Pharma Limited where he played a pivotal role in the development, manufacture and launch of a microneedle device used in skin clinics, which is also currently being evaluated for skin cancer drug delivery. Dr. Chowdhury has been responsible for negotiating licensing deals for a transdermal patch to treat Alzheimer’s disease. Additionally, he was involved in negotiations for out-licensing patches to treat Parkinson’s and Hypertension, and in-licensing complementary technologies.
Dr. Chowdhury originally trained as a pharmaceutical scientist and has an MSc in Microsystems and Nanotechnology from Cranfield University, and a Doctorate from the University of Oxford on nano-drug delivery. His experience in the Pharmaceutical Industry includes product development; manufacturing; and technical and corporate management.
Bashir Timol. Mr. Timol has been a Director since Nemaura Medical Inc. was organized on December 24, 2013. He has been a director of Dermal Diagnostics Limited from October 30, 2013. On April 9, 2018 Mr Timol was appointed to the role of Chief Business Officer. Mr, Timol possesses over 10 years’ experience in food and beverage, franchise, and logistic operations. His experience includes constructing sales contracts and having the responsibility for overseeing the key managers in the operation of a large scale retail food chain. He has experience as an entrepreneur investing in and operating a number of retail food chains in the UK, including DIXY Chicken and Costa Coffee. Prior to joining Nemaura Mr. Timol has been employed as a director at SABT 1 Ltd. since March of 2009 and One-E Group since January of 2007. Mr. Timol holds a bachelor degree in Economics from the University of Central Lancashire, UK.
Iain Anderson. Mr Anderson qualified as a Chartered Certified Accountant in 1993 whilst working for Touche Ross (now Deloitte) and received an MBA from Loughborough University in 1999. Having initially worked in accounting practices in audit and accounting roles, he has since worked in industry for a number of businesses, including more than 20 years with American-owned companies. These have included subsidiaries of publicly-owned corporations such as Hitachi, TriMas Corporation, Precision Castparts Corporation and Hospira Inc., with site and regional responsibility for group reporting and SOX compliance. His current responsibilities include the preparation of accounting information, development of the internal control environment of the company and regulatory compliance.
Timothy Johnson. Mr. Johnson was elected as a director in July 2017. He is currently serving in executive positions in Diagnostax advisory, EQIQ and Protech Professional. Mr. Johnson received his first class Masters of Science in Mathematics and Physics from the University of Manchester, UK.
Salim Natha. Dr. Natha was elected as a director in July 2017. He is currently practicing as an Eye Surgeon in the UK National Health Service (NHS), and is the clinical lead for a retinopathy screening programme for over 20,000 diabetics in the Ashton, Wigan and Leigh region. He has published several articles in the medical literature and is a peer reviewer for the English National Diabetic Retinopathy Screening Programme. Dr. Natha graduated with honours from the University of Liverpool Medical School.
Thomas Moore. Mr. Moore was elected as a director in August 2017. He is currently working as a management consultant, having built up three decades of experience in the accountancy and consultancy fields at leading accountancy firms including Grant Thornton, KPMG and PricewaterhouseCoopers. He is a practicing Chartered Tax Adviser and earned his first class Bachelor of Arts in French and Russian from the University of Northumbria, UK.
Family Relationships
There are no family relationships between any of our directors or executive officers.
Involvement in Certain Legal Proceedings.
None.
32
Board of Directors
All directors hold office until the next Annual Meeting of shareholders and until their successors have been duly elected and qualified. Directors are elected at the annual meetings to serve for one-year terms. Officers are elected by, and serve at the discretion of, the Board of Directors. Our Board of Directors shall hold meetings on at least a quarterly basis.
The board of directors complies with the NASDAQ Listing Rules with respect to corporate governance matters. Under the NASDAQ rules we are required to maintain a board of directors comprised of at least 50% independent directors, and an audit committee of at least two members, comprised solely of independent directors who also meet the requirements of Rule 10A-3 under the Securities Exchange Act of 1934.
Director Independence
The board of directors has reviewed the independence of our directors, applying the NASDAQ independence standards. Based on this review, the board of directors determined that each of Thomas Moore, Dr. Salim Natha and Timothy Johnson are independent within the meaning of the NASDAQ rules. In making this determination, our board of directors considered the relationships that each of these non-employee directors has with us and all other facts and circumstances our board of directors deemed relevant in determining their independence. As required under applicable NASDAQ rules, we anticipate that our independent directors will meet on a regular basis as often as necessary to fulfil their responsibilities, including at least annually in executive session without the presence of non-independent directors and management.
Board Committees
Our board of directors has established standing committees in connection with the discharge of its responsibilities. These committees include an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. Our board of directors has adopted written charters for each of these committees. Copies of the charters are available on our website. Our board of directors may establish other committees as it deems necessary or appropriate from time to time.
Audit Committee
Our Audit Committee was established on July 26, 2017 and is comprised of our independent directors: Thomas Moore, Dr. Salim Natha and Timothy Johnson. Mr. Johnson qualifies as the Audit Committee financial expert as defined in Item 407(d)(5) of Regulation S-K promulgated under the Securities Act.
According to its charter, the Audit Committee consists of at least three members, each of whom shall be a non-employee director who has been determined by the Board to meet the independence requirements of NASDAQ, and also Rule 10A-3(b)(1) of the SEC, subject to the exemptions provided in Rule 10A-3(c). The Audit Committee Charter describes the primary functions of the Audit Committee, including the following:
● Oversee the Company’s accounting and financial reporting processes;
● Oversee audits of the Company’s financial statements;
● Discuss policies with respect to risk assessment and risk management, and discuss the Company’s major financial risk exposures and the steps management has taken to monitor and control such exposures;
● Review and discuss with management the Company’s audited financial statements and review with management and the Company’s independent registered public accounting firm the Company’s financial statements prior to the filing with the SEC of any report containing such financial statements.
● Recommend to the board that the Company’s audited financial statements be included in its annual report on Form 10-K for the last fiscal year;
● Meet separately, periodically, with management, with the Company’s internal auditors (or other personnel responsible for the internal audit function) and with the Company’s independent registered public accounting firm;
● Be directly responsible for the appointment, compensation, retention and oversight of the work of any independent registered public accounting firm engaged to prepare or issue an audit report for the Company;
● Take, or recommend that the board take, appropriate action to oversee and ensure the independence of the Company’s independent registered public accounting firm; and
● Review major changes to the Company’s auditing and accounting principles and practices as suggested by the Company’s independent registered public accounting firm, internal auditors or management.
Compensation Committee
The Compensation Committee is responsible for, among other matters:
● reviewing and approving, or recommending to the board of directors to approve the compensation of our CEO and other executive officers and directors reviewing key employee compensation goals, policies, plans and programs;
● administering incentive and equity-based compensation;
● reviewing and approving employment agreements and other similar arrangements between us and our executive officers; and
● appointing and overseeing any compensation consultants or advisors.
Our Compensation Committee was established on July 26, 2017, and currently consists of Thomas Moore, Dr. Salim Natha and Timothy Johnson. Dr. Salim Natha serves as chair of the Compensation Committee.
33
Corporate Governance and Nominating Committee
The Corporate Governance and Nominating Committee is responsible for, among other matters:
● selecting or recommending for selection candidates for directorships;
● evaluating the independence of directors and director nominees;
● reviewing and making recommendations regarding the structure and composition of our board and the board committees;
● developing and recommending to the board corporate governance principles and practices;
● reviewing and monitoring the Company’s Code of Ethics; and
● overseeing the evaluation of the Company’s management.
Our Corporate Governance and Nominating Committee was established on July 26, 2017, and currently consists of Thomas Moore, Dr. Salim Natha and Timothy Johnson. Mr. Johnson serves as chair of the Corporate Governance and Nominating Committee.
Material Changes to Procedures by which Security Holders May Recommend Board Nominees
None.
Board Leadership Structure and Role in Risk Oversight
Mr. Chowdhury holds the positions of chief executive officer and chairman of the board of the Company. The board believes that Mr. Chowdhury’s services as both chief executive officer and chairman of the board is in the best interest of the Company and its shareholders. Mr. Chowdhury possesses detailed and in-depth knowledge of the issues, opportunities and challenges facing the Company in its business and is thus best positioned to develop agendas that ensure that the Board’s time and attention are focused on the most critical matters relating to the business of the Company. His combined role enables decisive leadership, ensures clear accountability, and enhances the Company’s ability to communicate its message and strategy clearly and consistently to the Company’s shareholders, employees and customers.
The board has not designated a lead director. Given the limited number of directors comprising the Board, the independent directors call and plan their executive sessions collaboratively and, between meetings of the Board, communicate with management and one another directly. Under these circumstances, the directors believe designating a lead director to take on responsibility for functions in which they all currently participate might detract from rather than enhance performance of their responsibilities as directors.
Management is responsible for assessing and managing risk, subject to oversight by the board of directors. The board oversees our risk management policies and risk appetite, including operational risks and risks relating to our business strategy and transactions. Various committees of the board assist the board in this oversight responsibility in their respective areas of expertise.
● The Audit Committee assists the board with the oversight of our financial reporting, independent auditors and internal controls. It is charged with identifying any flaws in business management and recommending remedies, detecting fraud risks and implementing anti-fraud measures. The audit committee further discusses Nemaura’s policies with respect to risk assessment and management with respect to financial reporting.
● The Compensation Committee oversees compensation, retention, succession and other human resources-related issues and risks.
● The Corporate Governance and Nominating Committee overviews risks relating to our governance policies and initiatives.
Section 16(a) Beneficial Ownership Reporting Compliance
Under Section 16(a) of the Securities Exchange Act of 1934, as amended (the Exchange Act), the Company's directors, executive officers and persons who own more than ten percent (10%) of our common stock are required to file with the Securities and Exchange Commission (the SEC), initial reports of ownership and reports of changes in ownership of the common stock and other equity securities of the Company. To the Company's knowledge, based solely on a review of copies of such reports furnished to the Company during and/or with respect to year ended March 31, 2018, the Company is not aware of any late or delinquent filings required under Section 16(a) of the Exchange Act in respect of the Company's equity securities, other than the following:
|
Name
|
Form
|
Transaction
|
|||
|
Dewan F.H. Chowdhury
|
4
|
Cancellation of 87,537,000 shares of common stock; Acquisition of 87,537 shares of preferred stock
|
|||
|
Bashir Timol
|
4
|
Cancellation of 27,082,000 shares of common stock; Acquisition of 27,082 shares of preferred stock
|
|||
|
Sufyan Ismail
|
4
|
Cancellation of 22,705,000 shares of common stock; Acquisition of 22,705 shares of preferred stock
|
|||
|
Timothy Johnson
|
3
|
Appointment as a director owning -0- shares
|
|||
|
Salim Natha
|
3
|
Appointment as a director owning 4,006,389 shares
|
|||
|
Thomas J. Moore
|
3
|
Appointment as a director owning -0- shares
|
|||
Code of Ethics
We have adopted a Code of Ethics that applies to our principal executive officer, principal financial officer and other persons performing similar functions. A copy of our Code of Ethics is available on our website. We intend to post amendments to, or waivers from a provision of, our Code of Ethics that apply to our principal executive officer, principal financial officer or persons performing similar functions on our website.
34
ITEM 11. EXECUTIVE COMPENSATION.
Summary Compensation Table
This table provides disclosure, for fiscal years 2018 and 2017, of the compensation paid to our named executive officers.
|
Named Executive Officer
and Principal Position |
Year
|
Salary
($)
|
Bonus
($)
|
All Other Compensation
($)
|
Total ($)
|
||||||||||||
|
|
|
||||||||||||||||
|
Dewan Fazlul Hoque Chowdhury
|
2018
|
106,440
|
-
|
390
|
106,830
|
||||||||||||
|
Chief Executive Officer (Principal Executive Officer)
|
2017
|
105,168
|
-
|
-
|
105,168
|
||||||||||||
|
Iain Anderson
|
2018
|
57,938
|
-
|
297
|
58,235
|
||||||||||||
| Chief Financial Officer (Principal Financial Officer) |
2017
|
34,761
|
-
|
-
|
34,761
|
||||||||||||
Dr. Chowdhury
We entered into an employment agreement with Dr. Chowdhury on November 2, 2013. Dr. Chowdhury’s contract is for an unspecified period. He may leave the Company with notice or the Company may terminate his contract with notice. Termination may be with or without cause. Dr. Chowdhury receives an annual salary of £90,000 pounds sterling or $118,000 USD. Our contract with Dr. Chowdhury does not include any provision for stock options or equity incentives.
Under the executive employment agreement Dr. Chowdhury’s annual salary was adjusted on a pro rata basis to reflect only work that was performed for Nemaura Medical Inc. The disclosure set forth in the table reflects his pro rata compensation from April 1, 2016 through March 31, 2018.
Mr. Anderson
We do not have a written employment contract with our Chief Financial Officer, Iain Anderson. Mr. Anderson had an annual salary of £80,000 (approximately USD106,000), which was increased to £100,000 (approximately USD140,000) in March 2018. These amounts have been prorated for the 2017 and 2018 fiscal years based on actual time working for the Company. Either party may terminate employment by providing the other party with no less than three months' prior notice. Our contract with Mr. Anderson does not include any provision for stock options or equity incentives.
Outstanding Equity Awards for 2018
None. We do not currently have an equity incentive plan.
Potential payments upon termination or change-in-control.
None. Upon termination by us or Mr. Chowdhury, he shall only be entitled to receive his base salary through the date of termination.
35
Director Compensation
Each of our independent directors receive annual fees of £5,000 pounds sterling or $7,000 USD for their service on our board of directors and committees. We currently have no plan for compensating our executive directors for their services in their capacity as directors. Although we have agreements with each of our independent directors to serve on our board, in which we provide for the grant of options, at this time no such option grants have been made and no equity compensation plan has been approved.
|
Name
|
Fees Earned or paid in Cash
($US) (1)
|
Non-Equity Incentive Plan Compensation
($US)
|
All other Compensation
($US) |
Total
($US)
|
||||||||||||
|
Timothy Johnson
|
4,726
|
-0-
|
-0-
|
4,726
|
||||||||||||
|
Salim Natha
|
4,568
|
-0-
|
-0-
|
4,568
|
||||||||||||
|
Thomas Moore
|
4,387
|
-0-
|
-0-
|
4,387
|
||||||||||||
(1) Reflects pro rata amount of fees paid for fiscal 2018 commencing on such director’s date of appointment.
Compensation Committee Interlocks and Insider Participation
No member of our Compensation Committee has at any time been an officer or employee of ours or our subsidiaries. No interlocking relationship exists between our Board of Directors or Compensation Committee and the Board of Directors or Compensation Committee of any other company, nor has any interlocking relationship existed in the past.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCK HOLDER MATTERS.
The following tables set forth certain information as of June 7, 2018 regarding the beneficial ownership of our Common Stock, by (i) each person or entity who, to our knowledge, owns more than 5% of our Common Stock; (ii) our executive officers; (iii) each director; and (iv) all of our executive officers and directors as a group.
Unless otherwise indicated in the footnotes to the following table, each person named in the table has sole voting and investment power and that person’s address is c/o NEMAURA MEDICAL INC., Advanced Technology Innovation Centre, 5 Oakwood Drive, Loughborough, Leicestershire, United Kingdom LE11 3QF. Shares of Common Stock subject to options, warrants, or other rights currently exercisable or exercisable within 60 days of June 7, 2018, are deemed to be beneficially owned and outstanding for computing the share ownership and percentage of the stockholder holding such options, warrants or other rights, but are not deemed outstanding for computing the percentage of any other stockholder.
On June 5, 2018, Mr. Chowdhury, Mr. Timol and Mr. Ismail, the three holders of our Series A Convertible Preferred Stock (the “Series A Preferred”), each delivered notices of conversion to voluntarily convert their Series A Preferred, in the aggregate amount of 137,324 shares, into 137,324,000 shares of our common stock. The holders had the right to voluntarily convert each share of Series A Preferred into 1,000 shares of common stock of the Company. As a result of the conversion, we currently have 205,000,000 shares of common stock outstanding.
Beneficial Ownership
|
Name of Beneficial Owner
|
Shares Beneficially Owned
|
Percentage Total Voting Power1
|
||||||
|
Chowdhury, Dewan F.H.
|
87,537,000
|
43
|
%
|
|||||
|
Timol, Bashir
|
27,082,100
|
13
|
%
|
|||||
|
Iain Anderson
|
0
|
-
|
||||||
|
Timothy Johnson
|
0
|
-
|
||||||
|
Salim Natha
|
4,006,389
|
2
|
%
|
|||||
|
Thomas Moore
|
0
|
- | ||||||
|
Total Officers and Directors as a Group
|
118,625,489
|
58
|
%
|
|||||
|
Holders of 5% or more of our Common Stock
|
||||||||
|
Ismail, Sufyan
|
22,705,250
|
11
|
%
|
|||||
1 Based upon 205,000,000 shares of our Common Stock outstanding.
36
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Nemaura Pharma Limited (Pharma) and NDM Technologies Limited (NDM) are entities controlled by our Chief Executive Officer, President, Chairman of the Board and majority shareholder, Dewan F.H. Chowdhury.
Pharma has invoiced our subsidiaries, Dermal Diagnostics Limited (DDL) and Trial Clinical Limited (TCL) for research and development services. In addition, certain operating expenses of DDL and TCL were incurred and paid by Pharma and NDM which have been invoiced to us. Certain costs incurred by Pharma and NDM are directly attributable to DDL and TCL and such costs were billed to us. Prior to the year ended March 31, 2016, other costs were shared between the organizations. In situations where the costs were shared, expense has been allocated between Pharma and NDM and DDL and TCL using a fixed percentage allocation and were billed to the Company. DDL and TCL advanced Pharma certain amounts to cover a portion of the costs.
Total costs charged to us by Pharma and NDM were $842,739 for the year 2018.
Following is a summary of activity between the Company and Pharma and NDM for the year ended March 31, 2018. These amounts are unsecured, interest free, and payable on demand.
|
|
Year Ended
March 31,
2018
($)
|
|||
|
Balance due from Pharma and NDM at beginning of period
|
(687,609
|
)
|
||
|
Amounts advanced to Pharma
|
-
|
|||
|
Amounts received from Pharma
|
(145,214
|
)
|
||
|
Reduction in prepayments to Pharma for clinical trials
|
-
|
|||
|
Amounts invoiced by Pharma to DDL, NM and TCL (1)
|
(842,739
|
)
|
||
|
Amounts invoiced by DDL to Pharma
|
-
|
|||
|
Amounts repaid by DDL to Pharma
|
1,096,767
|
|||
|
Amounts paid by DDL on behalf of Pharma
|
19,889
|
|||
|
Sale of fixed and intangible assets to Pharma and NDM
|
-
|
|||
|
Foreign exchange differences
|
(54,912
|
)
|
||
|
Net balance due to Pharma and NDM at end of the period
|
(613,818
|
)
|
||
| (1) |
These amounts are included primarily in research and development expenses.
|
REVIEW, APPROVAL OR RATIFICATION OF TRANSACTIONS WITH RELATED PERSONS
It is Company policy to not enter any transaction (other than compensation arrangements in the ordinary course) with any director, executive officer, employee, or principal stockholder or party related to them, unless authorized by a majority of the directors having no interest in the transaction, upon a favorable recommendation by the Audit Committee (or a majority of its disinterested members).
|
ITEM 14.
|
PRINCIPAL ACCOUNTANT FEES AND SERVICES
|
The following table sets forth the aggregate fees billed to us for the fiscal years ended March 31, 2018 and 2017 by Crowe Horwath LLP.
|
|
2018
|
2017
|
||||||
|
Audit Fees
|
$
|
106,000
|
$
|
84,000
|
||||
|
Audit Related Fees
|
-
|
-
|
||||||
|
Tax Fees
|
$
|
6,765
|
$
|
6,000
|
||||
|
Other Fees
|
-
|
-
|
||||||
|
Totals
|
$
|
112,765
|
$
|
90,000
|
||||
Audit fees represent amounts billed for professional services rendered or expected to be rendered for the audit of our annual financial statements.
Audit-related fees represent professional services rendered or expected to be rendered for assurance and related services by the accounting firm that are reasonably related to the performance of the audit or review of our financial statements that are not reported under audit fees
Tax fees represent professional services rendered by the accounting firm for tax compliance.
The Audit Committee approves all auditing services and the terms thereof and non-audit services (other than non-audit services published under Section 10A(g) of the Exchange Act or the applicable rules of the SEC or the Pubic Company Accounting Oversight Board) to be provided to us by the independent auditor; provided, however, the pre-approval requirement is waived with respect to the provisions of non-audit services for us if the "de minimus" provisions of Section 10A(i)(1)(B) of the Exchange Act are satisfied.
Audit Committee Pre-Approval Policy
Under provisions of the Sarbanes-Oxley Act of 2002, our principal accountant may not be engaged to provide non-audit services that are prohibited by law or regulation to be provided by it, and the Audit Committee must pre-approve the engagement of the our principal accountant to provide audit and permissible non-audit services. The Audit Committee has not established any policies or procedures other than those required by applicable laws and regulations.
37
PART IV
|
ITEM 15.
|
EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
|
||
|
(a)
|
Exhibits:
|
||
|
Exhibit No.
|
Description
|
|
|
3.1
|
||
|
3.1(a)*
|
||
|
3.2*
|
||
|
3.3*
|
||
|
4.1
|
||
|
4.2
|
||
|
10.1
|
||
|
10.2
|
||
|
10.3
|
||
|
10.4
|
||
|
10.5+
|
||
|
10.6
|
||
|
10.7
|
||
|
10.8+
|
||
|
14.1
|
||
|
21.1*
|
||
|
23.1*
|
||
| 31.1 | ||
| 31.2 | ||
| 32.1 | ||
| 32.2 | ||
| 101 | Interactive data files pursuant to Rule 405 of Regulation S-T: (i) the Balance Sheets, (ii) the Statements of Comprehensive Loss, (iii) Statements of Stockholders Equity, (iv) the Statement of Cash Flows and (v) the Notes to the Financial Statements | |
| · |
*Filed herewith
|
| · |
+Portions of this Exhibit have been omitted pursuant to a request for confidential treatment.
|
38
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf on June 12, 2018 by the undersigned thereunto duly authorized.
| NEMAURA MEDICAL INC. | |||
|
|
By:
|
/s/ Dewan F. H. Chowdhury | |
| Dewan F. H. Chowdhury | |||
| President, Chief Executive Officer (Principal Executive Officer) | |||
|
|
By:
|
/s/ Iain S Anderson | |
| Iain S Anderson | |||
| Chief Financial Officer (Principal Financial Officer) | |||
Pursuant to the requirements of the Securities and Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant on June 12, 2018, in the capacities indicated.
|
Name
|
Position
|
Date
|
|
|
/s/ Dewan F.H. Chowdhury
|
President, Chief Executive Officer (Principal Executive Officer) Chief Financial Officer
|
June 12, 2018
|
|
|
Dewan F.H, Chowdhury
|
|||
|
/s/ Bashir Timol
|
Director
|
June 12, 2018
|
|
|
Bashir Timol
|
|||
|
/s/ Timothy Johnson
|
Independent Director
|
June 12, 2018
|
|
|
Timothy Johnson
|
|||
|
/s/ Salim Natha
|
Independent Director
|
June 12, 2018
|
|
|
Salim Natha
|
|||
|
/s/ Thomas Moore
|
Independent Director
|
June 12, 2018
|
|
| Thomas Moore |
39