Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - Sarepta Therapeutics, Inc. | srpt-ex322_11.htm |
| EX-32.1 - EX-32.1 - Sarepta Therapeutics, Inc. | srpt-ex321_8.htm |
| EX-31.2 - EX-31.2 - Sarepta Therapeutics, Inc. | srpt-ex312_12.htm |
| EX-31.1 - EX-31.1 - Sarepta Therapeutics, Inc. | srpt-ex311_10.htm |
| EX-10.4 - EX-10.4 - Sarepta Therapeutics, Inc. | srpt-ex104_284.htm |
| EX-10.3 - EX-10.3 - Sarepta Therapeutics, Inc. | srpt-ex103_148.htm |
| EX-10.1 - EX-10.1 - Sarepta Therapeutics, Inc. | srpt-ex101_88.htm |
| 10-Q - Q1 2018 10-Q - Sarepta Therapeutics, Inc. | srpt-10q_20180331.htm |
Exhibit 10.2
SPONSORED RESEARCH AGREEMENT
THIS SPONSORED RESEARCH AGREEMENT (this "Agreement"), effective the 12th day of October, 2007 (the "Effective Date"), is entered into by and between AVI BIOPHARMA, INC., an Oregon Corporation, with principal offices located at One SW Columbia, Suite 1105, Portland, Oregon 97258 ("Company"), and CHARLEY'S FUND, INC., a 501(c)(3) tax-exempt public non-profit organization with a mailing address of P.O. Box 297, South Egremont, MA, 01258 (the "Sponsor").
W I T N E S S E T H:
WHEREAS, the Sponsor wishes to promote scientific research leading to exon skipping therapeutics related to Duchenne muscular dystrophy; and
WHEREAS, the Company has developed a proprietary antisense chemistry and has certain employees who possess knowledge, know-how and experience in substantive fields relating to such research and commercialization efforts;
WHEREAS, the Company has a cooperative development and license agreement with Ercole Biotech, Inc., and cross licensing rights to intellectual property important for the freedom to operate;
WHEREAS, the Sponsor is willing to fund such research by the Company, with the objective, as set forth herein, leading to an Investigational New Drug (IND) filing with the Food and Drug Administration; and
WHEREAS, it is the intent of the Sponsor and the Company to complete the research to identify a viable candidate and to demonstrate the safety and efficacy of the putative therapeutic used for exon skipping, sufficiently to file an IND and proceed to human clinical trials required for regulatory approval of said therapeutic,
NOW, THEREFORE, in consideration of the premises herein contained, and for other good and valuable consideration, the parties agree as follows:
|
|
1. |
Definitions |
For purposes of this Agreement, the following definitions apply:
1.1"Affiliates" shall mean any corporation or other entity that controls, is controlled by, or is under common control with, a party. A corporation or other entity shall be regarded as in control of another corporation or entity if it owns or directly or indirectly controls more than 50% of the voting securities or other ownership interest of the other corporation or entity, or if it possesses, directly or indirectly, the power to direct or cause the direction of the management and policies of the corporation or other entity.
1.2"Agreement Period" shall mean the period commencing on the Effective Date of this Agreement and ending upon completion of the Research Project.
|
|
1.4 |
"Confidential Information" shall have the meaning provided in Section 6.1 hereof. |
|
|
1.5 |
"FDA" shall mean the United States Food and Drug Administration. |
1.6"Field" shall mean the treatment or prevention of Duchenne Muscular Dystrophy or any other muscular dystrophy.
|
|
1.7 |
"Invention" shall have the meaning provided in Section 8.1 hereof. |
|
|
1.8 |
"Investigators" shall have the meaning provided in section 2.1 hereof. |
|
|
1.9 |
"Joint Inventions" shall have the meaning provided in Section 8.1 hereof. |
|
|
1.10 |
"Major Market" shall mean any one of the following: |
|
|
(a) |
The United States; |
|
|
(b) |
Japan; or |
|
|
(c) |
Any of the following five (5) European countries: Germany, The United Kingdom, France, Italy, or Netherlands. |
1.11"Net Sales" means the gross amount invoiced for sales of Research Products by Sponsor, its affiliates, and sublicensees, to an independent third party in an arms-length transaction, less:
|
|
(a) |
Trade, quantity and cash discounts allowed; |
|
|
(b) |
Discounts, refunds, rebates, chargebacks, retroactive price adjustments, and any other allowances which effectively reduce the net selling price; |
|
|
(c) |
Credits for actual Research Product returns; |
|
|
(d) |
Any tax imposed on the production, sale, delivery or use of the Research Product, including, without limitation, sales, use, excise or value added taxes. |
|
|
1.12 |
"Option" shall have the meaning provided in Section 9.2 hereof. |
|
|
1.13 |
"Option Term" shall have the meaning provided in Section 9.2 hereof. |
|
|
1.14 |
"Project Funds" shall have the meaning provided in Section 4.1 hereof. |
|
|
1.15 |
"Project Team" shall have the meaning provided in Section 2.1 hereof. |
|
|
1.16 |
"Research Product" shall have the meaning provided for in Section 4.3.6 hereof |
|
|
1.18 |
"Results" shall have the meaning provided in Section 3.4 hereof. |
|
|
1.19 |
"Sponsor Inventions" shall have the meaning provided in Section 8.1 hereof. |
1.20"Study Protocol" shall mean the protocol set forth in Appendix A with attached Gantt chart hereto, which is incorporated herein by reference and made a part hereof as if fully set forth herein, as such protocol may be modified from time to time by mutual written agreement of the Company and the Sponsor.
|
|
2. |
Research |
2.1The Principal Investigator for the Research Project shall be Dr. Patrick Iversen. The Principal Investigator shall be responsible for directing and overseeing the conduct of the Research Project using appropriately qualified collaborating investigators, including Dr. Ryszard Kole, CSO, Ercole Biotech, Dr. Stephen Wilton, University of Western Australia, and Dr. Qi Lu, Carolina's Healthcare Foundation, and scientists and research technicians who are under the Principal Investigator's direction and control and are employed by the Company (collectively, the Principal Investigator, the collaborating investigators, and the Company employees working on the Research Project constitute the "Project Team").
2.2Subject to the terms and conditions of this Agreement, the Company and the Principal Investigator shall perform the Research Project in accordance with the Study Protocol. No change to the Study Protocol shall be effective without the prior written consent of the Sponsor. The Company and the Principal Investigator shall use reasonable efforts to distinguish the research performed under this Agreement from all other work the Principal Investigator performs for other purposes and shall keep records pertaining to such other work separately from the records to be maintained pursuant to Section 2.7 to the extent practicable.
2.3The Company shall provide the Sponsor with written evidence of approval of the Research Project by the responsible body within the Company, if such approval is necessary, and with copies of any documents used in the conduct of the Research Project, including, but not limited to, all documentation required by the Study Protocol.
2.4The Company shall provide the support necessary for the Project Team to complete the Research Project, which support shall include, but is not limited to, human resources, space, dedicated research time, and computing, laboratory, and all other equipment, all in accordance with the Study Protocol attached as Appendix A.
2.5The Company shall also accept and administer the Project Funds. The Company's use of the Project Funds shall be strictly for purposes of the Research Project and shall be subject to the terms and conditions set forth in APPENDIX B, which is incorporated herein by reference and made part of this Agreement. NO PART OF THE PROJECT FUNDS SHALL BE USED FOR INDIRECT EXPENSES OF THE PROJECT TEAM OR THE Company. Except as specified in the Study Protocol relating to work to be conducted by collaborating investigators, no part of the Project Funds shall be transferred to another organization, whether or not the Principal Investigator or any other member of the Project Team becomes associated with that other
organization unless the prior written consent of the Sponsor is obtained by the Company. The Company shall be required to repay to the Sponsor any part of the Project Funds used in contravention of the express terms of this Agreement.
2.6The Company shall ensure that the Research Project shall be conducted in strict compliance with any applicable federal, state, or local laws, regulations, or guidelines pertaining to good research practices and/or good laboratory practices.
2.7The Company shall keep accurate and complete financial and scientific records relating to the Research Project and will make such records reasonably available to the Sponsor for review and/or copying during normal business hours.
2.8The Company shall promptly advise the Sponsor of any changes in the senior personnel comprising the Project Team. If, for any reason, the Principal Investigator (i) ceases to be associated with the Company, (ii) becomes debarred or receives notice of an action or threat of an action with respect to debarment under the provisions of the Generic Drug Enforcement Act of 1992, 21 U.S.C. Section 306(a) and (b), or (iii) otherwise becomes unavailable to work on the Research Project, the Company shall promptly so notify the Sponsor in writing and will propose a qualified replacement scientist at the Company whose appointment as Principal Investigator shall be subject to the approval of Sponsor. The Company shall consult with and reasonably consider and take into account the Sponsor's view with respect to the replacement of the Principal Investigator, provided that, in the case of a proposed replacement chosen by the Company and who is on the Advisory Committee, the Sponsor agrees that it will give its approval to such replacement for the Principal Investigator.
|
|
3. |
Reports to the Sponsor |
3.1During the Agreement Period, the Sponsor may meet with the Principal Investigator from time to time to discuss the planning and progress of the Research Project. An Advisory Committee made up of three members or advisors from the Company, three members or advisors from the Sponsor, a member from Ercole Biotech and an external collaborator will meet once per quarter to review progress of the Research Project. The Company will have final decision-making authority on all drug development, strategic, and other decisions.
3.2During the Agreement Period and for three (3) years thereafter, the Company shall make available to the Sponsor copies of all data and other information generated pursuant to this Agreement including, without limitation, all raw data obtained as a result of studies conducted in the course of the Research Project and all experimental procedures developed under the Research Project.
3.3At least every three (3) months during the conduct of the Research Project, the Company, in coordination with the Principal Investigator, shall provide the Sponsor with an interim written progress report concerning the Research Project.
3.4A final written report setting forth the results achieved under and pursuant to the Research Project and recommendations for next actions shall be submitted by the Company to the Sponsor within ninety (90) days of completion or earlier termination of the research that is the subject of
this Agreement. Such final report shall include a complete summary of the research carried out and detailed experimental results of the research protocols performed in the course of the Research Project (collectively, the "Results").
3.5Each written progress report to the Sponsor, including the final report, shall be accompanied by a financial statement from the Company describing in reasonable detail the disposition to date of the Project Funds.
3.6During the Agreement Period and for five (5) years thereafter, authorized employees and agents of the Sponsor or of the FDA shall have access to the Company and its personnel and records relating to the Research Project for the purpose of determining compliance with this Agreement and the Study Protocol and federal, state, and local laws and regulations and any applicable guidelines for the conduct of research. Such access by employees and agents of the Sponsor shall be on reasonable notice and during normal business hours, and individuals conducting such visits shall be bound by appropriate confidentiality agreements with the Company.
|
|
4. |
Payments and Repayment Rights |
4.1Subject to the terms and conditions of this Agreement including the repayment rights provided for in Section 4.3, the Sponsor shall pay the Company a total amount of Two Million Four Hundred and Fifty-two Thousand Dollars ($2,452,000.00) which amount 1s inclusive of all direct costs of Research Project activities (the "Project Funds") as follows:
Four Hundred Thousand Dollars ($400,000.00) shall be paid within ten (10) days of the parties' execution of this Agreement; Nine Hundred Thousand Dollars ($900,000.00) shall be paid three (3) months from the Effective Date of this Agreement subject to Sponsor's receipt of the first progress report demonstrating completion of that research component of the Research Project; Seven Hundred Thousand Dollars ($700,000.00) shall be paid six (6) months from the Effective Date of this Agreement subject to the Sponsor's receipt of the second progress report demonstrating completion of that research component of the Research Project; and Four Hundred and Fifty-Two Thousand Dollars ($452,000.00) shall be paid nine (9) months from the Effective Date of this Agreement subject to the Sponsor's receipt of the third progress report demonstrating completion of that research component of the Research Project. The Sponsor shall not be obligated to make any payments to the Company in addition to those set forth in this Section 4.1 unless the parties otherwise mutually agree in writing.
4.2The Company shall provide to the Sponsor all information necessary to make the payments described above, including, but not limited to, the name of the payee, its tax identification number, and the name and address of the contact person to whom payments should be sent.
4.3The Company and the Sponsor agree to the following commercial terms with regard to the development and commercialization of a Research Product:
4.3.1The Company shall make a lump sum payment to the Sponsor of Eight Hundred and Seventeen Thousand Three Hundred and Thirty Four Dollars ($817,334.00)
(the "First Payment") within thirty (30) days after the end of the fiscal quarter during which the first commercial sale into a Major Market of the Research Product occurs.
4.3.2The Company shall make a lump sum payment to the Sponsor of Eight Hundred and Seventeen Thousand Three Hundred and Thirty Three Dollars ($817,333.00) (the "Second Payment") within thirty days (30) after the end of the fiscal quarter during which the first anniversary of the first commercial sale into a Major Market of the Research Product occurs.
4.3.3The Company shall make a lump sum payment to the Sponsor of Eight Hundred and Seventeen Thousand Three Hundred and Thirty Three Dollars ($817,333.00) (the "Third Payment") within thirty days (30) after the end of the fiscal quarter during which the second anniversary of the first commercial sale into a Major Market of the Research Product occurs.
4.3.4In the event the Company or one of its Affiliates enters into any sort of partnership (a license agreement, research and development agreement, collaboration or similar arrangement) with a corporate partner that includes the right to sell, distribute promote or market the Research Product or any of the underlying intellectual property and
(i)if, prior to the second anniversary of the first commercial sale of a Research Product in a Major Market country, the corporate partner agrees to pay an upfront cash license fee or similar payment which is earned upon signing, the Company or its Affiliates shall pay to the Sponsor, within thirty (30) days of Company's receipt of such payment from the corporate partner, 15% of the cash received as an upfront fee or the total Project Funds, less any amount already repaid to the Sponsor by the Company, whichever is less.
(ii)and if, thereunder, the Company is entitled to development milestone payments, the Company or its Affiliates shall, within thirty (30) days of receipt of any such payments, make repayments to the Sponsor in the amount of 15% of each individual milestone payment specifically related to the progress for the development of a Research Product, limited to 50% of the Project Funds at each of such milestone payment, until the Project Funds amount is repaid in full.
4.3.5Without limiting the foregoing, in the event that the full amount of the Project Funds have not been repaid to the Sponsor at first commercial sale into a Major Market of the Research Product via the payment mechanisms of Section 4.3.4, the Company shall make payments to the Sponsor as provided for in Sections 4.3.1, 4.3.2, and 4.3.3..
4.3.6"Research Product" shall mean any product containing any molecular candidate arising or derived from the research funded hereunder which is developed as a human therapeutic agent for skipping exon 50 in the indication Duchenne Muscular Dystrophy.
4.3.7Repayments to the Sponsor under all mechanisms in this Article 4 shall not exceed the total Project Funds as provided for in Section 4.1.
5.1The Sponsor strongly encourages the Company, through the efforts of the Principal Investigator, to seek additional funding for the laboratories of the Investigators from the Federal government or other sponsors of research and acknowledges that such additional sponsors may retain rights in and to such funded research.
|
|
6. |
Confidentiality |
6.1The Sponsor and the Company acknowledge that each party may receive confidential technical and business information of the other party ("Confidential Information") during the Agreement Period. Each party hereto agrees that, during the Agreement Period and for a five (5) year period thereafter, that it will maintain in strict confidence, and will not disclose to any third party, any Confidential Information of the other party, whether in oral, written, graphic or electronic form. Each party hereto agrees (i) not to use Confidential Information of the other party except for purpose of conducting Research Project activities in accordance with the Study Protocol or for such other purposes consistent with the intent and terms of this Agreement and (ii) not to disclose Confidential Information of the other party to third parties without the express written permission of the other party, except that (a) each party shall not be prevented from disclosing Confidential Information to its employees, officers, independent contractors and Affiliates requiring access thereto for the purposes of this Agreement provided each such employee, officer, independent contractor or Affiliate is bound by an agreement regarding confidentiality and non-use at least as restrictive as the obligations in this Article 6, and (b) such information may be disclosed insofar as such disclosure is necessary to allow either the Company or the Sponsor, as the case may be, (A) to defend itself against litigation, (B) to file and prosecute patent applications on any Invention in accordance with Article 8 hereof, or (C) to comply with judicial decree, government action or applicable law or regulation, provided that the party shall give prior written notice to the other party so that the other party may attempt to obtain a protective order requiring that the Confidential Information be disclosed only to the extent required by such order, law or regulation, and that it be used only for the purposes for which the decree, action, law or regulation requires such disclosure to be made. The parties agree that no advance notice to the Sponsor is required for AVI’s compliance with its reporting requirements to the Securities and Exchange Commission (SEC). The parties will take all steps necessary to ensure that its employees, officers, independent contractors, and Affiliates comply with the terms and conditions of this Agreement. Notwithstanding the foregoing, such obligation of confidentiality shall not apply to information that the receiving party can establish by competent evidence:
(i)at the time of disclosure is in the public domain;
(ii)has come into the public domain through no fault of the receiving party or its employees and agents;
(iii)was known to the receiving party prior to its disclosure by the disclosing party, as evidenced by the receiving party's written records; or
(iv)is disclosed to the receiving party, without restriction on disclosure, by a third party that is not under an obligation of non-disclosure to the disclosing party.
6.2The Company (including, for purposes of this Section 6.2, the Principal Investigator) shall have the right, and is encouraged, to publish or present the Results of the Research Project, provided the Sponsor has the opportunity to review and comment on any proposed manuscripts or the substance of any presentations describing said Research Project or Results at least thirty (30) days prior to their submission to a third party for publication or review. In the event that the rights to the Research Product have been licensed to Sponsor pursuant to Article 9, then the positions of the Parties with respect to the provisions of this Section 6.2 shall be reversed. The reviewing party shall review any draft and give its comments to the publishing party promptly. The publishing party shall comply with the reviewing party's request to delete references to the reviewing party's Confidential Information in any such publication and the publishing party agrees to withhold publication an additional thirty (30) days to permit the reviewing party to obtain patent or other intellectual property protection, if the reviewing party deems it necessary.
|
|
7. |
Use of the Other Party’s Name; Public Statements |
Each party agrees that it will not at any time during or following expiration or termination of this Agreement use the name of the other party or its employees or any other names, insignia, symbol(s), or logotypes associated with the other party or any variant or variants thereof orally or in any literature, advertising, or other materials without the prior written consent of the other party except for right to publish set forth in Section 6.2 and Company's compliance with its reporting requirements to the SEC, which consent may be withheld at the other party's sole discretion. Notwithstanding the foregoing, the Company agrees that the Sponsor may use the names of the Company, the Principal Investigator and his collaborators in connection with generally publicizing on its website, in press releases or in other publications of the Sponsor provided that such usages are limited to identifying the Company and/or the Principal Investigators and briefly describing the nature of the Research Project and the Sponsor agrees that Company may use Sponsor's name in connection with any board or investor presentation, or press release related thereto, or as may be requested by any funding entity, governmental entity, or academic publisher, or as required by law. Prior to publicizing, both parties agree to give the other party an opportunity to review press releases using the other's name and discussing the work involved in the project.
|
|
8. |
Ownership and Patents |
8.1Company shall own all data obtained in the Research Project, research protocols related to the Research Project, and Results, and shall have the right to submit all such information to support regulatory filings related to the Research Product and other products that may be developed by the Company.
8.2Ownership of any discovery, invention, method, process or other know-how made, conceived or first reduced to practice in the performance of the Research Project during the project period by the Company and its affiliates, the Principal Investigator and/or the Sponsor and all intellectual property arising therefrom (collectively, "Inventions") shall be determined as follows: All Inventions conceived or reduced to practice during the project period solely by employees, agents or consultants of the Company, including, without limitation, the Principal Investigator ("Company Inventions") shall be owned solely by the Company. All Inventions conceived or reduced to practice during the project period solely by employees, agents or consultants of the
Sponsor ("Sponsor Inventions") shall be owned solely by the Sponsor. All Inventions conceived or reduced to practice jointly during the project period by employees, agents or consultants of the Company, on the one hand, and employees, agents or consultants of the Sponsor, on the other hand ("Joint Inventions"), shall vest according to U.S. patent law. The Company represents and warrants that all Company employees and other individuals or entities performing any part of the Research Project are obligated to assign to the Company all inventions and intellectual property rights that are necessary to enable the Company to grant the Sponsor all rights the Company purports to grant under this Agreement.
8.3As soon as the Company reasonably believes a Company Invention or Joint Invention has been conceived or reduced to practice hereunder (and in any event within a reasonable time after its disclosure to the Company Technology Office), the Company shall disclose such invention in writing to the Sponsor in sufficient detail to allow the Sponsor to evaluate its significance.
8.4The Company shall have the first right to prosecute any patent application(s) covering any Company Inventions. Within a reasonable time after disclosure of any such Company Invention to the Sponsor, the Company shall notify the Sponsor in writing if it intends to pursue patent protection for such Company Invention. If the Company elects to pursue patent protection, it shall promptly prepare, file and prosecute any U.S. or foreign application(s) to protect such Company Invention. The Company shall bear all expenses in connection with such preparation, filing, prosecution and maintenance of U.S. and foreign patent applications. For such Company elected patent applications, the Company shall be responsible for making decisions regarding the scope and content of such patent application(s) and the prosecution thereof. The Sponsor shall be responsible for the costs of patent filing and prosecution for Sponsor Inventions or if the Sponsor requests that the Company files a patent application. The Company and Sponsor shall each make reasonable efforts to keep the other advised as to all developments with respect to such application(s).
8.5For Joint Inventions, the Company and Sponsor will negotiate in good faith, at the time of disclosure, the management and prosecution of the invention including any cost related thereto.
8.6Sponsor grants Company an exclusive, worldwide, fully paid-up, royalty-free license under Sponsor's interest in any Joint Inventions and to Sponsor Inventions to make, use and sell Research Products.
|
|
9. |
Granting of Exclusive License |
9.1The parties acknowledge and agree that they intend to use their reasonable best efforts to complete the Research Project as described in the Study Protocol. In the event the Company and its Affiliates and partners elect to discontinue to pursue the development and/or commercialization of a Research Product for reasons other than safety and efficacy, the Company and its Affiliates, at the request of the Sponsor, hereby grants the Sponsor the exclusive royalty-bearing, fully paid up, worldwide license or sublicense as the case may be, with the right to sublicense, to the Research Product, on terms consistent with the requirements of the Ercole Biotech-Isis Pharmaceuticals collaboration agreement and Ercole Biotech-AVI BioPharma collaboration agreement, under patents owned or licensed by the Company or its Affiliates to research, to develop, to use, to sell, to offer for sale, to distribute Research Products, to import, to export and to employ methods covered by any such patents or by Company Inventions or
Company's interest in Joint Inventions relating to the Research Product. In consideration for the exclusive license to use and sell the Research Product
9.1.1if the Sponsor obtains a license to the Research Product while the Research Product is in the research or preclinical phase of the Research Project, a total royalty of 8% of Net Sales of Research Product shall be paid to the Company and its Affiliates by the Sponsor and its licensees.
9.1.2If the Sponsor obtains a license to Research Product during or at the conclusion of Phase I clinical testing, a total royalty of 12% of Net Sales of Research Product shall be paid to the Company and its Affiliates by the Sponsor and its licensees.
9.1.3If the Sponsor obtains a license to Research Product after the initiation of a Phase II clinical trial, a total royalty of 15% of Net Sales of Research Product shall be paid to the Company and its Affiliates by the Sponsor and its licensees.
9.2The Company shall have the right of first refusal to manufacture research, clinical and commercial quantities of Research Product for the Sponsor or the Sponsor's commercial partner. Should the Company not exercise its first right to manufacture, the exclusive license granted to the Sponsor shall be expanded to include the right to develop to make, and to have made, Research Product and to employ methods covered by or incorporating Company Inventions or Company's interest in Joint Inventions which permit the commercialization of the Research Product to the worldwide market. The Company shall transfer the Research Product production process to a mutually agreeable third party contract manufacturer under an agreement that contains appropriate provisions for recovery of technology transfer costs and for limiting disclosure or other use of the Company's technology.
9.3Except as expressly provided herein and including Article 9, nothing in this Agreement shall restrict either party's use, license or exploitation in any way of its interest in its own or any Joint Inventions.
|
|
10. |
Termination |
10.1This Agreement shall remain in effect for the Agreement Period unless extended by written agreement of the parties, or earlier terminated in accordance with this Article 10.
10.2Either party may terminate this Agreement for any material breach of this Agreement by the other party if such breach is not cured within thirty (30) days after the breaching party receives written notice of such breach by the non-breaching party. Such termination shall be effective upon expiration of such thirty (30) day period.
10.3Termination of this Agreement shall not affect the rights and obligations of the parties that shall have accrued prior to termination, including, without limitation, the confidentiality obligations set forth in this Agreement. In the event of any termination of this Agreement prior to expiration of the Agreement Period (other than termination by the Sponsor pursuant to Section 10.2), the Sponsor shall pay the reasonable costs incurred by the Company in winding down and terminating the Research Project, including the costs of the Research Project during the wind-down
period and all costs and non-cancelable commitments made prior to termination. After termination, the Company will submit a final report of all costs incurred and all funds received under this Agreement as set forth in Section 3.4. The report shall be accompanied with a check for any funds remaining which were paid to the Company under Section 4.1, if any. The provisions of Sections 2.7, 3.2, 3.4, 3.5, 3.6, 4.3 and 10.3 and Articles 6, 7, 8, 9, 12, 13, 16, 17, 18, 19, 20 and 21 shall survive termination or expiration of this Agreement.
|
|
11. |
Force Majeure |
Neither party shall be responsible to the other for any failure or delay in performing any of its obligations under this Agreement if such delay or nonperformance is caused by strike, stoppage of labor, lockout or other labor trouble, fire, flood or other weather event, earthquake, accident, explosion, war, act of terrorism, act of God or act of the government of any country or of any local government or any other cause beyond the reasonable control of the defaulting party.
|
|
12. |
Liability and Warranty |
Each party to this agreement agrees to indemnify the other party from damage to persons or property resulting from the negligence on the part of itself, its employees, its agents, or its officers. Neither party assumes any responsibility to the other party for the consequences of any act or omission of any person, firm or corporation not a party to this agreement.
The research results are preliminary in nature. Company makes no representations and extends no warranties of any kind, either expressed or implied, with regards to research results.
|
|
13. |
Independent Contractors |
The Sponsor and the Company shall at all times act as independent parties, and nothing contained in this Agreement shall be construed or implied to create an agency or partnership. Neither party shall have the authority to contract or incur expenses on behalf of the other except as may be expressly authorized by separate written agreement between the parties. The Principal Investigator and members of the Project Team shall not be deemed to be employees of the Sponsor.
|
|
14. |
Other Employment |
The Company warrants that the Principal Investigator is permitted to enter into this Agreement (but not to bind the Company) and that the terms and conditions hereof are consistent with the Principal Investigators' obligations to the Company.
|
|
15. |
Tax Status |
The Company represents and certifies to the Sponsor that it is a publicly traded company.
|
|
16. |
Choice of Law |
This Agreement shall be governed by and shall be construed in accordance with the laws of the Commonwealth of Massachusetts without regard to the conflicts of laws provisions thereof.
If any one or more of the provisions of this Agreement shall be held to be invalid, illegal or unenforceable, the validity, legality or enforceability of the remaining provisions of this Agreement shall not in any way be affected or impaired thereby.
|
|
18. |
Waiver |
The failure of any party hereto to insist upon strict performance of any provisions of this Agreement or to exercise any right hereunder will not constitute a waiver of that provision or right.
|
|
19. |
Notices |
Any notice or communication required or permitted to be given or made under this Agreement by one of the parties to the other shall be in writing and shall be deemed to have been sufficiently given or made for all purposes if such notice or communication is either emailed and its receipt is acknowledged by the recipient, or mailed by certified mail, postage prepaid, addressed to such other party at its respective address as follows:
|
If to the Sponsor: |
Attn: Benjamin D. Seckler, MD, President Charley's Fund, Inc. |
|
|
P.O. Box 297 |
|
|
South Egremont, MA 01258 Email: |
|
|
|
|
If to the Company: |
Attn: Chief Executive Officer |
|
|
AVI BioPharma, Inc. |
|
|
One SW Columbia, Suite 1105, Portland, Oregon 97258 |
|
|
20. |
Assignment |
This Agreement may not be assigned by the Company without prior notice to Sponsor, except in connection with a merger, recapitalization, reorganization, consolidation, sale of securities, sale of assets or any transaction to an affiliate of the Company; provided, however, no such transaction shall relieve Company of its obligations or adversely affect Sponsor's rights hereunder. The Sponsor may assign this Agreement without the Company's consent (i) in connection with a merger, consolidation or sale of all or substantially all of Sponsor's assets or stock, or (ii) to an affiliate of the Sponsor. In addition, the Sponsor may assign all or any part of its rights under Articles 8 and 9 to any third party upon written notice to the Company.
|
|
21. |
Entirety |
This Agreement represents the entire agreement of the parties, and it expressly supersedes all previous written and oral communications between the parties. Except as otherwise expressly provided in this Agreement, no amendment, alteration, or modification of this Agreement or any
Appendix attached hereto shall be valid unless executed in writing by authorized signatories of both parties.
IN WITNESS WHEREOF, the parties hereto have caused this Agreement to be executed by their duly authorized representatives to be effective as of the Effective Date.
|
CHARLEY'S FUND, INC. |
By: /s/ Benjamin D. Seckler |
|
|
Name: Benjamin D. Seckler, MD |
|
|
Title: President |
|
|
|
|
|
|
|
AVI BIOPHARMA, INC. |
By: /s/ K. Michael Forrest |
|
|
Name: K. Michael Forrest |
|
|
Title: Chief Executive Officer |
Preclinical Proposal for Skipping Exon 50 for Duchenne Muscular Dystrophy
This proposal to Charley's Fund describes a research and preclinical development program to select and advance a compound to treat patients with Duchenne muscular dystrophy who could benefit by skipping exon 50 of the dystrophin gene. The research proposed includes a thorough, but rapid evaluation of enhancements to AVI’s standard PMO chemistry that could improve drug potency and the targeting of the drug to critical tissues, including skeletal muscle and the heart. AVI believes that this evaluation will yield a clinical drug candidate with the highest likelihood of having meaningful therapeutic benefit.
|
|
I. |
Background: |
The favorable characteristics of phosphorodiamidate morpholino oligomers (PMOs) include long term stability, simple dose regimens, and an outstanding safety profile, as follows:
1)long term stability- real time stability testing on PMOs stored for 2 years as lyophilized powder or as sterile saline solutions indicate less than 2% loss of active pharmaceutical ingredient (API).
2)PMOs have shown favorable pharmacokinetic properties leading to simple dose regimens. A phase I study with AVI-4065, targeting hepatitis C virus, was recently completed in healthy volunteers with subcutaneous dosing of 50, 100 and 300mg daily for 14 days. The fractional bioavailability based on AUC from this extravascular route appears to be greater than 1.0. The maximal plasma concentrations were predicted with precision from studies in rat and non-human primate. Finally, the apparent elimination half-life observed in plasma was approximately 10 hours but residence time in patients based on urinary clearance was nearly 11 days. We have conducted pharmacokinetic studies with four different PMOs in humans and have never detected a PMO metabolite.
3)The PMO technology has an outstanding safety profile. AVI has completed 17 clinical trials with four different PMO candidates in over 400 subjects and there have been no definitely drug related serious adverse events. It has been ethical and logical with these drugs to restrict volunteer populations to adults (viz., age of majority to 70 years) and not to children or infants. Subjects enrolled in these studies have been racially diverse and approximately half males and half females. Also, it should be noted that no female volunteers in AVI-sponsored clinical trials were pregnant or breast-feeding, and all women of child-bearing potential practiced acceptable birth control for the duration of active study surveillance.
AVI BioPharma is developing a PMO to skip exon 51 for treatment of Duchenne muscular dystrophy. Pharmacokinetic modeling based on animal data suggests that a dose of 100 mg administered subcutaneously once weekly should be effective. However, this will need to be
determined empirically in a dose-ranging clinical study. While we are optimistic that the exon 51 compound, AVI-4658, will be effective in this dose range, the possibility that significantly higher doses will be required to achieve efficacy leads us to search for more potent agents. We believe we can do this for exon 50 through the use of cell penetration enhancers, enhanced potency and improved tissue delivery. These measures have the potential to increase potency several fold. In addition, more potent agents result in less body burden of drug and hence better compliance and less likelihood of toxicity.
|
|
II. |
Applicable Work in Progress |
|
|
A. |
Objective: Identify the Exon 50 sequence. |
Analysis of PMOs in cell culture is well suited to identification of agents capable of inducing molecular events such as exon skipping. These studies provide valuable mechanistic information but are not effective predictors of potency or dose for in vivo activity. Hence, cell culture studies will be used to identify the sequence of the PMO for skipping exon 50. The current plan is to compare the sequence identified by Dr. Steve Wilton (Molecular Therapy, 2007) for skipping exon 50, H50A(+02+30), 5'-CCA CTC AGA GCT CAG ATC TTC TAA CTT CC-3' with that identified by Dr. Qi Lu (who will disclose his sequence to AVI soon). The strength of this approach is that independent evaluation utilizing different endpoints has been employed.
AVI BioPharma research and development group has been building a database of favorable PMO motifs for the past 10 years. This database has provided insights into novel rules for PMO self-interaction which will tend to exclude some selected sequences. We are currently updating this database so we can include evaluation of the selected sequences provided by Drs. Lu and Wilton.
|
|
B. |
Objective: Reduce dose through improved cell delivery and cell penetrance of PMOs. |
AVI BioPharma has been developing peptide conjugates of PMOs for over 10 years in what has been referred to as the CytoPorter™ program. This strategy employs small peptides capable of enhanced cell penetration which are conjugated to the PMOs, referred to as PPMO. The program has utilized a "splice-correction" assay developed by Dr. Ryszard Kole which utilizes a cell line with a beta thalassemia cryptic intron splice acceptor and luciferase as a marker gene expression. This assay led to identification of the peptide (RXR)4XB- in which R is arginine, X is aminohexanoic acid and B is beta alanine. This peptide conjugated to an antisense PMO directed to c-myc is currently the subject of a clinical study of coronary artery bypass grafts (CAGB). In that study, human saphenous veins are exposed to the study drug ex vivo just prior to engraftment into the coronary artery. This peptide has been evaluated for potential toxicity at AVI (Amantana, manuscript in press) and in GLP safety pharmacology studies. Further, Dr. Wilton has evaluated this peptide and found enhanced in vivo potency compared to unconjugated PMO. The limitations have been toxicity reported from animal studies at doses above 15mg/kg dose with repeated administration. However, it is likely that effective human doses will be significantly below this threshold. An evaluation of the published literature for the mdx mouse treated with PMOs suggests that a dose of 2 mg/kg may be therapeutic (see Section E). Since the PPMO compound has been shown to have greater potency than the corresponding PMO, it is reasonable to believe than an efficacious dose will be significantly lower than the maximum tolerated dose.
A comprehensive peptide structure activity study should involve in vivo efficacy and simple endpoint analysis. Collaboration with Dr. Kole over the past 2 years has utilized transgenic mice containing a gene with an artificial intron disruption of GFP expression. These studies have identified peptides which appear to be superior to (RXR)4XB-. The most notable to date has been (RXRRBR)2XB- which demonstrated greater splice correction activity in the heart yes it significantly less toxic than (RXR)4XB-.
This peptide has been provided to Drs. Wilton, Kole and Lu for evaluation in the mdx mouse. So far, Dr. Lu has reported a single dose of 30mg/kg of the PPMO is effective in the mdx mouse and no signs of toxicity have been observed. Dr. Lu indicates this is the smallest effective dose he has observed to date, which tends to confirm our estimations of the utility of PPMO. These studies will continue to evaluate a range of doses and repeated injections.
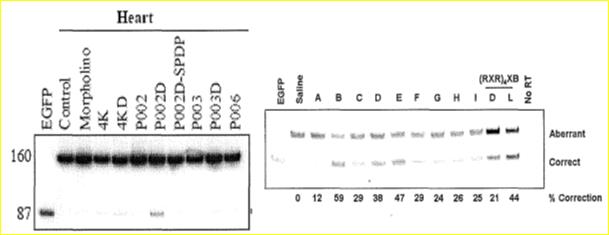
These figures come from Dr. Kole and show splice correction (lower band in each lane) in the heart of the EGFP mouse. On the left, only the P002 peptide synthesized with d-amino acids provided any splice correction in the heart. On the right, peptides B, D, E and (RXR)4XB all show significant splice correction in the heart. The peptide with the least toxicity and greatest efficacy from these effective peptides is peptide B which is (RXRRBR)2XB.
|
|
C. |
Objective: Identify modifications of PMOs that will enhance their inherent potency. |
The highlights of the PMO platform technology are based on neutral charge and no metabolic degradation. Studies are in progress investigating the addition of a positively charged linkage in the PMO at selected sites, referred to as PMO+. This strategy has been used in treating non-human primates in Ebola lethal challenge studies, which have resulted in 70% dose reduction and improved long term survival over neutral PMO treatment. The observed affinity for a PMO binding to RNA is approximately 10-12 M and the PMO+ binding to RNA is approximately 10•13 M, which suggests the enhanced potency is due to greater binding affinity. We intend to evaluate the relative merit of this PMO+ modification for exon 50. The exon 23 sequences for the mdx mouse have been synthesized as PMO+ and are currently in purification at AVI. They will be sent shortly to Drs. Kole, Lu and Wilton for in vivo evaluation.
Finally, studies to date have shown synergy between the PPMO and PMO+ in what we refer to as PPMO+. These two modifications have the potential to reduce the systemic dose to 1% of the dose required for PMO compounds. The current optimal PPMO+ has been synthesized and is currently in purification. These mdx reagents will be sent to Drs. Kole, Lu and Wilton within the next few weeks for their evaluation in the mdx mouse.
|
|
D. |
Objective: Reduce dose through enhanced tissue-specific delivery. |
We have initiated a collaboration with Dr. Hai Fang Yin in the laboratory of Dr. Matthew
J.A. Wood at the University of Oxford. They recently reported use of a homing peptide conjugated to peptide nucleic acid agents. They have identified a muscle-specific homing peptide composed of NH2-ASSLNIA-COOH and more recently a 12-amino acid peptide with heart muscle specific tropism. These peptides may be useful in getting higher concentrations of PMOs into the most relevant tissues for treatment of DMD.
|
|
E. |
Objective: Estimate the dose and dose regimen based on pharmacokinetic studies. |
A study conducted in rat involved IV and IM administration of a single 1.0, 5.0, 25.0, and
125.0mg/kg of an exon 51 30-mer. The apparent plasma elimination half-life is non-linear but increases with increasing dose. The area under the plasma concentration versus time curve is also non-linear but would appear to reach a theoretical maximum of 43 µg-hr/ml. The volume of distribution shows a linear increase with dose exceeding 1 liter/kg at near a 10mg/kg dose, indicating more robust tissue accumulation at larger doses. The plasma pharmacokinetic data are shown in Table 1 below:
TABLE 1. Plasma Pharmacokinetics
|
Dose (mg/kg) |
Route |
Apparent Half-life (hr) |
AUC (µg-hr/ml) |
Vd (I/kg) |
Clpl (ml/min) |
|
1.0 |
IV |
9.31 |
1.3 |
0.23 |
0.07 |
|
5.0 |
IV |
22.78 |
14.38 |
0.73 |
0.17 |
|
25.0 |
IV |
31.51 |
23.71 |
5.22 |
0.40 |
|
125.0 |
IV |
39.84 |
39.31 |
27.88 |
2.20 |
|
1.0 |
IM |
9.41 |
1.45 |
0.32 |
0.07 |
|
5.0 |
IM |
7.31 |
11.79 |
3.33 |
1.01 |
|
25.0 |
IM |
11.94 |
22.38 |
13.64 |
2.61 |
|
125.0 |
IM |
15.85 |
38.49 |
75.79 |
10.89 |
We observed between 10 and 16% of the dose in the urine over the 24 hour post dose interval for IV administration. There is less than 0.2% of the dose excreted in feces so we estimate 84 to 90% of the administered dose in body tissues (Table 2).
TABLE 2. Renal Excretion Data
|
Dose |
Route |
Urine Vol |
Urine |
% Dose |
Tissue |
% Dose |
|
|
(mg/kg) |
|
(ml-24hr) |
(mg) |
Urine |
(mg) |
Tissue |
|
|
1.0 |
IV |
6.67 |
0.05 |
16.03 |
0.15 |
83.97 |
|
|
5.0 |
IV |
22.46 |
0.44 |
12.30 |
0.56 |
87.70 |
|
|
25.0 |
IV |
19.80 |
0.76 |
16.34 |
4.24 |
83.66 |
|
|
125.0 |
IV |
6.80 |
2.65 |
10.00 |
22.35 |
90.00 |
|
|
1.0 |
IM |
8.50 |
0.13 |
33.68 |
0.07 |
66.32 |
|
|
5.0 |
IM |
15.33 |
0.14 |
10.21 |
0.86 |
89.79 |
|
|
25.0 |
IM |
19.01 |
0.92 |
18.81 |
4.08 |
81.19 |
|
|
125.0 |
IM |
16.40 |
8.38 |
3.81 |
16.62 |
96.19 |
|
The muscle accumulation was observed to be linear with dose for either IV or IM administration (r2= 0.999). A rat composed of 47.5% skeletal muscle (Even et al., 2001). Thus, a 250-gram rat has 119 grams (250g * 0.475 = 118.8g) of skeletal muscle. The pharmacokinetic studies utilized a 200mg (0.2g) sample of skeletal muscle recovered 24 hours after a single injection of H51A from either IV or IM routes of administration, homogenized in 1ml of PELB buffer and concentration determined by HPLC FDNA assay. Several of the muscle samples were below the detection limit of the HPLC assay so a liver to muscle ratio was calculated and applied to the lower doses to estimate muscle concentrations. The resulting muscle concentration in ng/ml is calculated [ng ecovery*0.2g/ml]/[0.475*250g]. The molar concentration is estimated from the molecular weight of the exon 51 compound studied (10,391.8 g/mol). We observed the compound in skeletal muscle as indicated in TABLE 3.
TABLE 3. Skeletal Muscle Recovery 24 hours Post Single Dose HSIA
|
Dose (mg/kg) |
Route |
Muscle (mg) |
Muscle (ug/g) |
Muscle µM |
Muscle nM |
|
1.0 |
IV |
0.07 |
0.60 |
0.06 |
5.77 |
|
5.0 |
IV |
0.27 |
2.24 |
0.22 |
21.55 |
|
25.0 |
IV |
2.01 |
16.94 |
1.63 |
163.02 |
|
125.0 |
IV |
10.61 |
89.39 |
8.60 |
860.16 |
|
1.0 |
IM |
0.03 |
0.28 |
0.03 |
2.72 |
|
5.0 |
IM |
0.41 |
3.46 |
0.33 |
33.29 |
|
25.0 |
IM |
1.94 |
16.33 |
1.57 |
157.18 |
|
125.0 |
IM |
7.89 |
66.48 |
6.40 |
639.70 |
There are no simple in vitro assay measures at present that can provide the EC50 for H51A, but based on our observations from cell culture (Wilton, et al, 2006) and other exon skipping inducing oligomers we believe this concentration will be near 100nM. The function of muscle may not require 100 percent exon skipping, so the 100nM value represents a feasible target tissue concentration. Based on the pharmacokinetic data, this would be observed following a dose of 15mg/kg. If we use the equivalent mg/m2 dose in a human (Freireich EJ., et al, 1966) we would administer 2.1mg/kg. In a 50kg boy this dose would be 105mg. This estimate is limited in that the pharmacokinetic studies were conducted in a healthy rat with normal muscle physiology. The muscle of the mdx mouse appears to be more permeable to the oligomer, which would suggest a smaller required dose to produce the 50 percent effect.
Studies reported in the peer reviewed literature utilized both intramuscular injections and systemic routes of administration to induce skipping of exon 23 in the mdx mouse. A summary of these studies is provided in Table 4 below:
TABLE 4. In Vivo Efficacy
|
Ref |
Sequence |
Chemistry |
Dose |
Route |
Endpoints |
Dose Eq. |
|
Mann 2001 |
[+2-18] |
2'0Me-LPF |
1µg |
IM |
Western (WB) |
143nM |
|
|
|
|
|
|
|
|
|
Gebski 2003 |
[+7-18] |
PMO-Leash |
1µg |
IM |
WB- |
116nM |
|
Lu 2003 Nat |
[+2-18] |
2'0Me-BC |
10µg |
IM |
WB, IHC, PCR |
1.4µM |
|
Wells 2003 |
[+2-18] |
2'0Me-elect |
8 µg |
IM |
WB, IHC |
1.14 µM |
|
Lu 2005 |
[+2-18] |
2'0Me |
100mg/kg |
IV |
IHC+ |
8.3mg/kg |
|
Fletcher 2005 |
[+7-18] |
PMO |
25mg/kq |
IP |
IHC, WB, PCR |
2.1mg/kg |
|
Alter 2006 |
[+7-18] |
PMO |
100mg/kg |
IV |
IHC, WB, CK Force |
8.3mg/kg |
|
Fall 2006 |
[+2-18] |
PMO |
10µg |
IM |
IHC, WB, PCR |
1.4µM |
The estimate of the human dose equivalent in the right hand column is based on a 1-gram muscle diffusion area following the intramuscular (IM) injections and the molecular weight of the oligomer to arrive at a molar concentration. These observations indicate an efficacy range from 116 to 1400nM. The interpretation of these data is more complicated due to the use of lipofectins (LPF), block copolymers (BC), leashes and electroporation as delivery enhancers. However, the calculated concentration is based on 100 percent efficient delivery so these observations tend to
indicate efficacy with a PMO can be observed at concentrations as low as 116nM, which is in good agreement with efficacy in cell culture.
There are three studies which employed systemic delivery. The two intravenous (IV) studies employed 100mg/kg doses and the extravascular study used an intrperitoneal (IP) route with 25mg/kg. Robust efficacy was observed in each case and the anticipated human dose based on mg/m2 dose scaling is 2.l mg/kg from an extravascular route of administration. This is in particularly good agreement with independent estimates based on pharmacokinetic observations.
The expression of functional ∆-dystrophin resulting from exon skipping will require active transcription of dystrophin and sufficient concentrations of exon-skipping oligomer in the nucleus of muscle cells. Once an oligomer has induced exon-skipping, the ∆-mRNA can be transported from the nucleus to the cytoplasm for translation. The steady-state of the functional ∆-dystrophin protein will be equal to the rate of translation of ∆-mRNA minus the rate of degradation of the ∆-dystrophin protein. The rate of transcription is relatively fast even for dystrophin, the largest transcript in the human genome, and is equal to 10 - 24 hours/copy. The rate of nuclear transport and the rate of translation are likely to be faster than the rate of transcription, on the order of 1 - 4 hours. Thus the critical features for accumulation of ∆-dystrophin protein will be the half-life of mRNA and the ∆-dystrophin protein.
The ∆-dystrophin protein in the mdx mouse following a single injection of exon skipping oligomer is about 12% of initial concentration at 12 weeks post dose. This would translate to approximately 3 half-lives and indicates a half-life of 4 weeks. (Lu et al., 2003). A single dose of PMO in the mdx mouse will create truncated mRNA that can be observed for over 4 weeks resulting in immunohistochemical evidence of ∆-dystrophin 8 weeks post dose (Fall et al., 2006). The time to steady-state expression of ∆-dystrophin protein will be equivalent to 3 to 5 half-lives of the protein, between 3 and 5 months. If dose is administered at protein half-life intervals, then the interval can be once per 4 weeks. This interval is further supported by the observation of ∆-mRNA persists for 4 weeks.
The apparent plasma half-life of the 30-mer targeting H51A shown in TABLE 1 is approximately 30 hours. The term "apparent" half-life indicates this is not likely to be the actual elimination half-life. Other pharmacokinetic studies with a variety of PMO sequences have plasma half-lives shorter than 30 hours but tissue residence times of 7 to 14 days. The anticipated muscle residence time of a PMO is expected to be greater than 7 days. This would mean a dose interval of 7 days would maintain muscle tissue levels and maximal muscle concentrations should be achieved in approximately one month.
Summary of the rational for the dose regimen:
|
|
1. |
The dose should be extravascular; eg. subcutaneous. |
|
|
2. |
The dose should be 2.1 mg/kg for a 50 kg person or 105mg/dose. |
|
|
3. |
The dose interval should be between 1 and 4 weeks. |
Aim 1: Identify the optimal exon 50 sequence.
The ongoing efforts will provide direction for these studies. If Drs. Wilton and Lu select overlapping sequences, then AVI will prepare the selected sequence for confirmatory studies by both investigators with documented pure, high-quality controlled PMO.
If Wilton and Lu identify non-overlapping PMO sequences then a hierarchy of evaluation methods will be established at AVI to establish methods for comparison of the sequences that will utilize our database of favorable PMO traits to select optima from the unrelated sequences. Further, the potential for enhanced PMOs to alter the target optima can be addressed in cell culture, and Dr. Lu has offered to conduct these studies for exon 50.
Wilton: H50A(+02+30); 5'-CCACTCAGAGCTCAGATCTTCTAACTTCC-3'
Interpretation of data: If the sequences identified by the two independent investigators are identical, then no further interpretation is necessary. This singular sequence will be considered the lead exon 50 agent. If the sequences are not identical, then we will establish a small group of AVI investigators to evaluate the sequences with respect to unique experience and databases created at AVI. These will include:
a)Self-complementarity- this will have been screened by the investigators, but we have identified additional interactions unique to PMO chemistry. Sequences containing greater self- complementarity will receive a lower priority score.
b)Search for SNP- the sequences will be compared to snp databases to ensure there are no known polymorphisms in the targeted region.
c)In vitro translation database search for unfavorable sequence motifs- AVI BioPharma has developed a database of over 100 different sequences. These sequences differ by approximately 1000x in potency allowing us to evaluate unfavorable sequence motifs. Analysis of proposed sequences containing unfavorable motifs will receive lower priority scores.
d)Pharmacokinetic database- AVI BioPharma has developed a database of over 40 sequences that have been evaluated in pharmacokinetic studies. These sequences differ with respect to elimination half-life, renal clearance and volume of distribution, allowing the identification of both favorable and unfavorable motifs. Analysis of proposed sequences containing unfavorable motifs will receive lower priority scores.
e)BLAST search of GenBank for potential interactions with other human genes. Any 14 contiguous base match will receive a lower priority score.
In addition to technical criteria, intellectual property issues will be considered in selecting which sequence to prioritize.
Aim 2: Identify best PMO modality for exon skipping in the mdx mouse model.
The comparison studies for exon 23 in the mdx mouse model are in progress in three laboratories. The results should provide an estimate of the relative potency for PMO, PPMO, PMO+ and PPMO+. The emphasis of the three laboratories tend to differ, and we intend to leave experimental detail to each individual laboratory as a means of providing a greater range of experience. We anticipate the PPMO+ will be the most effective modality. If this is not clearly established, then the lead agent for exon 50 will be the modality with the lowest toxicity and for which we have the greatest experience. The methodology will be reviewed for the experiments to either identify areas of conflict or provide greater confidence due to confirmation.
Use mdx mouse as simple screening tool to compare potency of multiple versions of PMOs:
|
PMO |
anticipate efficacy at 150mg/kg |
|
PMO+ |
anticipate efficacy at 150mg/kg |
|
PPMO |
anticipate efficacy at 15mg/kg |
|
PPMO+ |
anticipate efficacy at 3mg/kg |
This set of studies is currently in progress as materials have been prepared and initial shipments made to Drs. Wilton, Lu, and Kole. Dr. Lu has conducted a pilot study with a single 30mg/kg dose of PPMO and reported by phone that he is seeing robust exon skipping 4 weeks after a single dose. He indicated this is more potent in the mdx mouse than any agents he has evaluated to date and is currently evaluating the uniformity of muscle response. Dr. Wilton has provided initial observations with the PMO+ which clearly show greater potency than the PMO in the mdx mouse. Dr. Kole will meet at AVI next week and discuss his observations with the molecular endpoint of splice altering.
Based on the results from work under Aims 1 and 2, AVI will select a lead compound to take forward into initial pharmacokinetic and toxicity testing described in Aim 4.
Aim 3: Evaluate the influence of homing peptides for PMOs
In parallel with evaluation of the lead compound, additional work will be conducted with another technology that could increase the potency and effectiveness of exon-skipping PMOs.
The muscle homing peptide (MHP), NH2-ASSLNIA-COOH, was identified using a phage display method (Samoylova and Smith 1999). These studies involved intravenous administration of >1012 phage virions (a phage display library) to mice and recovery of phage associated with skeletal muscle. The associated phage were amplified and a second round of selection conducted to identify the 7-amino acid MHP peptide. It is not clear that the small MHP will promote cell penetration, so we propose to combine the MHP with our cell penetrating peptides (CPP) in the following matrix of studies:
MHP-PM0-3'
5'-PMO-MHP
CPP-PMO-MHP
MHP-CPP-PM0-3'
CPP-MHP-PM0-3' 5'-
PMO-CPP-MHP
5'-PMO-MHP-CPP
A peptide that was identified using the same phage display technique with homing to the pancreatic vasculature, NH2-CRV ASVLPC-COOH (Kolonin et al., 2006). This peptide binds to the prolactin receptor (PRLR). We intend to use this peptide as a control for tissue specificity for the MHP. Since the matrix of 8 possibilities in combination with CPP is possible, we will conduct an initial set of studies to evaluate cell uptake into cultured myotomes first. The PMO sequence will be designed to induce exon skipping of dystrophin and the success of the studies will be based on observation of PCR amplified, truncated dystrophin transcripts. Once favorable uptake and efficacy are observed, then the control will be prepared with the same orientations of PMO, CPP and homing peptide.
The optimal MHP/PRLR, CPP and PMO combinations will be evaluated in the mdx mouse targeting skipping of exon 23. If the dose required to induce functional dystrophin production is reduced by a factor of 3, then this approach will be considered sufficiently useful for further evaluation.
Collaborative studies with Dr. HaiFang Yin in the laboratory of Matthew Wood at the University of Oxford will utilize a 12-amino acid peptide they have identified with homing properties to the heart muscle. These studies will follow those proposed to evaluate the MHP. The purpose of these studies will be to investigate the potential therapeutic delivery of exon skipping PMOs to the heart. If successful, this strategy may be used to treat or prevent specific areas of deficit in exon skipping therapy for DMD.
Aim 4. Evaluate the pharmacokinetics and toxicity of selected lead compound
Initial animal studies with the lead compound selected in Aim 2 will utilize doses and dose regimens that are aggressive and based on successful historical animal model studies, e.g. in the range from 30 to 150mg/kg.
AVI BioPharma has substantial experience with studies in rats for initial evaluation of pharmacokinetics. Specifically we will:
1)Develop and validate analytical methodology that is sufficiently sensitive, specific and reproducible to allow measurement and quantitation of candidates agents in body fluids and tissues of animals. A platform method referred to as the FDNA assay has been developed for virtually any PMO agent and utilizes a fluorescently labeled DNA with sequence complementary to the candidate PMO. This method has also been utilized in the case of Ebola virus to simultaneously identify three PMO sequences administered to the animal at the same time. The method is generally
sensitive enough to detect 10ng/ml, is linear with less than 5% coefficient of variance and is reproducible.
2)Conduct plasma protein binding studies and stability studies in various biological fluids and tissues.
3)Determine solubility of these candidates at 30mg/ml or greater. Any candidate that is not soluble at 30mg/ml will be re-synthesized and re-evaluated. If the candidate is confirmed to be poorly soluble (the only cases of this have involved high guanine content), then synthesis of the candidate with inosine substitution of guanine one position at a ti will be conducted to improve solubility.
4)Determine plasma and urinary concentrations of candidates as various times after administration to rats by various routes including intravenous, subcutaneous and intraperitoneal. The data will be fit to pharmacokinetic models and relevant pharmacokinetic parameters (including half-life, renal clearance, volume of distribution, mean residence time and area under the curve) for each route of administration.
5)Tissue distribution studies will also be conducted at various times after administration of the candidate with particular attention to skeletal muscle and the heart muscle, and
6)No PMO metabolite has been detected to date, but this can only be meaningful if studies are sufficient for a mass balance study (e.g., account for greater than 75% of the injected candidate). The studies conducted in rats will also provide data for body and organ weights, blood chemistry, and histology, which will guide future understanding of the potential toxicology of each candidate. These studies will be particularly useful when new PMO modalities (PPMO, PMO+ and PPMO+) are evaluated in an effort to optimize the therapeutic candidate’s efficacy.
Both Drs. Lu and Wilton are also interested in studying gene expression array analysis of PMOs used to induce skipping of exon 50 in a mouse model. This methodology relies on comparison of differences in gene expression between untreated and mice treated chronically with PMOs.
Aim 5: Combination sequences leading to skipping exons 50 - 53.
As is the case with Aim 3, the proposed work under this aim is to continue to research options for the best long-term therapeutic options for exon 50 DMD patients. It is not on the critical path to clinical testing of the initial exon 50 drug candidate, but is proposed to be conducted in parallel.
Dr. Wilton has identified a strategy to skip exons 50-53 and in the near term exons 50-51.
The goal of these studies will be to search for combinations of PMOs that work in synergy. Thus, the total dose required to skip multiple exons should be less than the sum of the doses required to skip them individually. Discussions with Dr. Wilton indicate he is currently supported to evaluate the "cocktail" approach, so our intention is to support his efforts with the most optimal PMO
modalities, experimental design support when needed, pharmacokinetic studies, and analytical support.
Aim 6: GMP Manufacture and GLP Safety Pharmacology
The goal here is to synthesize sufficient quantities of the lead compound for further characterization, including efficacy and formal GLP toxicity testing in in vivo models. While process development is generally advanced at AVI, we will need to evaluate QA/QC methods and methods for recovery, characterization, purification, stability assessment specifically for the selected compound.
The second goal is to perform required benchmarks for IND filing and moving the drug candidate into Phase I/II clinical trials. These studies involve conventional safety pharmacology studies, including a cardiovascular study (in monkeys), pulmonary function study (in monkeys), renal function study (in monkeys), central nervous system (in rats), pharmacokinetic studies in monkeys, conventional toxicology (two species will be required), and genotoxicity studies. Traditional genotoxicity, Ames assay and SCE and micronucleus assays in T-cells are not adequate to evaluate the genotoxicity of the relatively high molecular weight DMD agents. We know these agents do not enter bacterial cells of the Ames assay or T-cells utilized in the other assays. Hence, we propose use of zebrafish embryos for genotoxicity evaluation. This can be done at Oregon State University, a regional center of excellence in zebrafish studies.
Our experience with PMOs suggests a very safe profile and minimal requirement for safety pharmacology studies. However, those agents with newer modalities are likely to require a greater degree of safety pharmacology and toxicology investigation. The specific design of the safety pharmacology and GLP tox studies will be based on the modality chosen and the results obtained in the preliminary pharmacokinetic and toxicity studies conducted in Aim 4.
Aim 7: Prepare and File IND
Based on preclinical safety and efficacy results, AVI’s clinical and regulatory departments will prepare an IND to support initial human clinical testing of the selected exon 50 drug candidate.

Project Cost Estimates
Aim 1: Identify the optimal exon 50 sequence
|
Materials: |
$5,000 |
|
Labor: |
$5,000 |
|
Total: |
$10,000 |
Aim 2: Identify best PMO modality for exon skipping in the mdx mouse model.
|
Materials: |
(already synthesized) |
|
Labor: |
$2,500 |
|
Total: |
$2,500 |
Aim 3: Evaluate the influence of homing peptides for PMOs
|
Materials: |
$15,000 |
|
Animal studies: |
$5,000 |
|
Labor: |
$2,500 |
|
|
$22,500 |
Aim 4: Evaluate the pharmacokinetics and toxicity of optimal PMO modalities (PPMO, PMO+ and PPMO+)
|
Materials: |
$28,000 |
|
Animals: |
$1,000 |
|
Labor: |
$30,000 |
|
Total: |
$59,000 |
Aim 5: Combination sequences leading to skipping exons 50-53
|
Materials: |
$20,000 |
|
Labor: |
$5,000 |
|
Total |
$25,000 |
Aim 6: GMP Manufacture and GLP Safety Pharmacology
|
Materials: |
$1,100,000 |
|
GLP tox studies |
$1,150,000 |
|
Genotox study: |
$30,000 |
|
Labor: |
$20,000 |
|
Total: |
$2,300,000 |
|
Labor: |
$33,000 |
Total Estimated Cost, All Aims:$2,452,000