Attached files
| file | filename |
|---|---|
| EX-31.2 - EXHIBIT 31.2 - Cue Biopharma, Inc. | tv489196_ex31-2.htm |
| EX-32.1 - EXHIBIT 32.1 - Cue Biopharma, Inc. | tv489196_ex32-1.htm |
| EX-31.1 - EXHIBIT 31.1 - Cue Biopharma, Inc. | tv489196_ex31-1.htm |
| EX-10.21 - EXHIBIT 10.21 - Cue Biopharma, Inc. | tv489196_ex10-21.htm |
| EX-4.5 - EXHIBIT 4.5 - Cue Biopharma, Inc. | tv489196_ex4-5.htm |
| EX-4.4 - EXHIBIT 4.4 - Cue Biopharma, Inc. | tv489196_ex4-4.htm |
| EX-4.3 - EXHIBIT 4.3 - Cue Biopharma, Inc. | tv489196_ex4-3.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2017
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from __________________ to __________________
Commission file number: 001-38327
| Cue Biopharma, Inc. |
| (Exact Name of Registrant as Specified in Its Charter) |
| Delaware | 47-3324577 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) |
| 675 W. Kendall St., Cambridge, MA | 02142 | |
| (Address of Principal Executive Offices) | (Zip Code) |
(617) 949-2680
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, par value $0.001 per share | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12 (g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | |
| Non-accelerated filer x (Do not check if a smaller reporting company) | Smaller reporting company ¨ | |
| Emerging growth company x |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act): Yes ¨ No x
The registrant completed the initial public offering of its common stock on December 27, 2017. Accordingly, there was no public market for the registrant’s common stock as of June 30, 2017, the last business day of the registrant’s most recently completed second fiscal quarter.
As of March 22, 2018, there were 20,130,766 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The registrant intends to file a definitive proxy statement pursuant to Regulation 14A within 120 days after the end of the fiscal year ended December 31, 2017. Portions of such proxy statement are incorporated by reference into Part III of this Form 10-K.
CUE BIOPHARMA, INC.
TABLE OF CONTENTS
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that are intended to be covered by the “safe harbor” created by those sections. Forward-looking statements, which are based on certain assumptions and describe our future plans, strategies and expectations, can generally be identified by the use of forward-looking terms such as “believe,” “expect,” “may,” “will,” “should,” “would,” “could,” “seek,” “intend,” “plan,” “goal,” “project,” “estimate,” “anticipate,” “strategy”, “future”, “likely” or other comparable terms and references to future periods. All statements other than statements of historical facts included in this Annual Report on Form 10-K regarding our strategies, prospects, financial condition, operations, costs, plans and objectives are forward-looking statements. Examples of forward-looking statements include, among others, statements we make regarding: anticipated results of our drug development efforts, including study results, our expectations regarding regulatory developments and expected future operating results.
Forward-looking statements are neither historical facts nor assurances of future performance. Instead, they are based only on our current beliefs, expectations and assumptions regarding the future of our business, future plans and strategies, projections, anticipated events and trends, the economy and other future conditions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict and many of which are outside of our control. Our actual results and financial condition may differ materially from those indicated in the forward-looking statements. Therefore, you should not rely on any of these forward-looking statements. Important factors that could cause our actual results and financial condition to differ materially from those indicated in the forward-looking statements include, among others, the following:
| · | our limited operating history, limited cash and a history of losses; |
| · | our ability to achieve profitability; |
| · | our ability to secure required Food and Drug Administration (“FDA”) or other governmental approvals for our product candidates and the breadth of any approved indication; |
| · | negative or inconclusive results from our clinical studies or serious and unexpected drug-related side effects or other safety issues experienced by participants in our clinical trials; |
| · | delays and changes in regulatory requirements, policy and guidelines including potential delays in submitting required regulatory applications to the FDA; |
| · | our reliance on licensors, collaborations and strategic alliances; |
| · | our ability to obtain adequate financing to fund our business operations in the future; |
| · | the other risks and uncertainties described in the Risk Factors and in Management’s Discussion; and Analysis of Financial Condition and Results of Operations sections of this Annual Report on Form 10-K. |
Any forward-looking statement made by us in this report is based only on information currently available to us and speaks only as of the date on which it is made. We undertake no obligation to publicly update any forward-looking statement, whether written or oral, that may be made from time to time, whether as a result of new information, future developments or otherwise.
| 1 |
Cue Biopharma, Inc. (“we,” “us,” “our,” “Cue” and the “Company”) was incorporated in the State of Delaware on December 31, 2014 under the name Imagen Biopharma, Inc., and completed its organization, formation and initial capitalization activities effective as of January 1, 2015. In October 2016, the Company changed its name to Cue Biopharma, Inc. The Company’s corporate office and research facilities are located in Cambridge, Massachusetts.
Overview
We are an innovative biopharmaceutical company developing a novel and proprietary class of biologic drugs for the selective modulation of the human immune system to treat a broad range of cancers and autoimmune disorders. We believe our innovative CUE Biologics™ platform approach to selectively modulate disease relevant T cells, provides a transformative solution to the challenges facing prevailing immunotherapeutics. By directly engaging and modulating disease relevant T cells in the patient’s body via an injectable drug, we believe our biologic drug candidates will be able to realize the true potential of immune modulation. Through our proprietary CUE Biologics™ platform, we believe we are uniquely positioned to become a prominent and leading player in immunotherapy, immuno-oncology, and autoimmune disease. While currently in preclinical development, our proprietary platform is intended to allow us to efficiently design and develop drug candidates that specifically and selectively engage disease relevant T cells for therapeutic affect, while minimizing or eliminating unwanted side effects. We have been aggressively seeking patent protection for our pioneering innovations and, combined with a license agreement with the Albert Einstein College of Medicine (“Einstein”), continue to build a robust intellectual property portfolio. This portfolio includes our core technology platform for the engineering of biologics to selectively control T cell activity, which we call CUE Biologics™, a growing portfolio of precision immuno-modulatory drug candidates, and two supporting technologies we call MOD™ and viraTope™ that enable the discovery of costimulatory signaling molecules (ligands) and T cell targeting peptides, respectively.
Background
The human immune system comprises a number of specialized cell types which collectively function to identify and defend the body against foreign threats. Central to the proper functioning of the immune system are the coordinated activities and communications between two specialized cell types, antigen-presenting cells (“APCs”) and T cells. APCs serve to capture and break the proteins from foreign organisms, (e.g., bacteria and viruses), or abnormal proteins (e.g., from genetic mutation in cancer cells) into smaller fragments suitable as signals for scrutiny by the larger immune system, including T cells. In particular, APCs break down proteins into small peptide fragments, which are then paired with a class of host molecules called the major histocompatibility complex (“MHC”) and displayed on the cell surface. Cell surface display of an MHC together with a peptide fragment, also known as a T cell epitope, provides the underlying scaffold surveilled by T cells, allowing for specific recognition. The peptide fragments can be pathogen-derived, tumor-derived, or derived from natural host proteins (self-proteins). Moreover, APCs can recognize certain pathogen associated molecules, such as bacterial toxins, viral proteins, viral nucleic acids, that communicate the pathogenic threat to the APC. The APCs relay this information to T cells through additional costimulatory signals in order to generate a more effective response.
T cells recognize peptide-MHC (“pMHC”) complexes through a specialized cell surface receptor, the T cell receptor (“TCR”). The TCR is unique to each T cell and, as a consequence, each T cell is highly specific for a particular pMHC target. In order to adequately address the universe of potential threats, a very large number (~10,000,000) of distinct T cells with distinct TCRs exist in the human body. Further, any given T cell, specific for a particular T cell peptide, is initially a very small fraction of the total T cell population. Although normally dormant and in limited numbers, T cells bearing specific TCRs can be readily activated and amplified by APCs to generate highly potent T cell responses that involve many millions of T cells. Such activated T cell responses are capable of attacking and clearing viral infections, bacterial infections, and other cellular threats including tumors, as illustrated below. Conversely, the broad, non-specific activation of overly active T cell responses against self or shared antigens can give rise to T cells inappropriately attacking and destroying healthy tissues or cells.
| 2 |
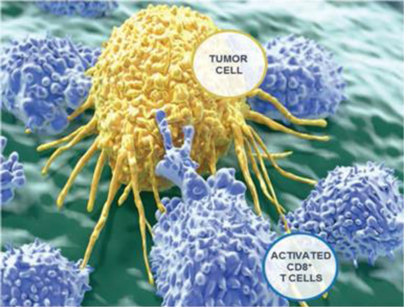
Activated T cells target tumor cells for killing through engagement of the TCR with its cognate pMHC.
TCR engagement by its specific or cognate pMHC delivers an activation signal to the T cell and defines the specificity of the response. However, robust and effective T cell activation requires that the TCR signal be accompanied by additional signals from the APC, collectively referred to as “costimulation.” The sum of these interactions directs the quality and magnitude of the T cell response. Specific costimulatory, as well as coinhibitory signals, may be delivered by the APC to the T cell through specific signaling molecules (ligands) interacting with their cognate receptors at the interface of these two cells, shown below. Based upon the particular nature of the pMHC and costimulatory signal(s), the T cell may differentiate into a variety of cell types, each with a specialized function (e.g., Tc1, Tc2, Th1, Th2, Th17, Treg, Tr1, etc.). Hence communication between APCs and T cells must be capable of precisely identifying threats and generating a response of appropriate quality and magnitude. An insufficient T cell response may result in a persistent pathogenic infection, or in the case of cancer, tumor persistence. Conversely, an excessive or inappropriate T cell response may damage the host acutely (e.g., acute viral hepatitis) or chronically through autoimmune disease (e.g., Type 1 diabetes, celiac disease, rheumatoid arthritis, Graves’ disease, etc.).
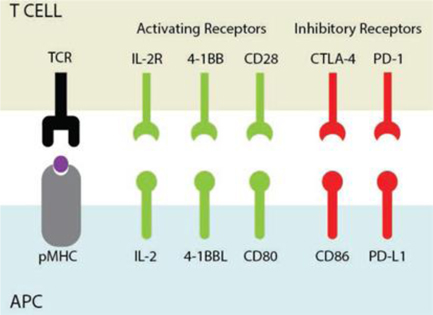
Illustration of the interaction between antigen presenting cells (“APCs”) and T cells. Stimulatory signals are shaded green and inhibitory signals are shaded red.
| 3 |
Cancer is characterized by the uncontrolled proliferation of abnormal cells and is a leading cause of death in developed countries. Cancer cells arise when proteins responsible for regulating cell division, proliferation or death are altered and these changes can occur through several different mechanisms. Cancer cells may express particular proteins (antigens) at much higher levels than normal (collectively referred to as tumor associated antigens, e.g., PSA-1 and Wilms Tumor 1) or express oncogenic (tumor-causing) viral proteins that are responsible for transformation, producing so-called cancer “drivers”, such as HPV E7. Cells can also be rendered oncogenic by the damage of host proteins through mutation (e.g., p53 and KRAS), which result in the expression of cancer neoantigens. Regardless of the nature of oncogenesis, cancer cells can display on their cell surface antigenic peptides, which are often recognized by the immune system.
In addition to being transformed, cancer cells have the ability to evade or modulate immune surveillance. This “immune escape” can be a key factor in their growth, spread, and persistence. Cancer cells employ a variety of approaches to escape immune surveillance or to suppress the effects of an immune response. One example of this is observed when cancer cells evade a normal immune response by expressing cell surface molecules that interact with and inhibit the attacking immune cells. These inhibitory interactions, called “immune checkpoints,” provide the tumor with a shield or buffer from activated and tumor-specific T cells. Checkpoint pathway inhibitors are therapeutic antibodies in immuno-oncology that are designed to block the tumor’s inhibitory shield; they have demonstrated promising clinical results. Another mechanism by which tumors can turn down the immune response is by stimulating the production of CD4+ regulatory T cells (“Tregs”), which in turn inhibit the killer CD8+ T cells that would normally attack the cancer. Thus, through one or more of these different pathways, the cancer cells manipulate and bias the local tumor microenvironment in favor of immune cell suppression. To extend and enhance the observed clinical benefits of therapeutic checkpoint blockade, a concurrent means of specifically activating and expanding a tumor-specific CD8+ T cell population is desired. Of interest, recent results from cancer trials that focus on activating a general (non-specific) immune response have shown that globally stimulated immune cells can attack “self” tissue, thereby triggering a therapeutically-induced autoimmune disease, frequently observed in patients as a consequence of their treatment and can limit further therapy.
In autoimmune and inflammatory diseases, components of the immune system are unable to effectively distinguish between foreign and “self” tissues and so the immune system mounts a response that can result in extensive destruction of normal tissue. Autoimmune and inflammatory diseases often occur in genetically predisposed individuals and can be triggered by select conditions, such as infection or tissue damage. The result is an immune response against healthy tissue, amounting to the second highest cause of chronic illness in the United States and a leading cause of death for women under 65.
In order to eliminate tumor cells and infectious agents, the immune system needs to be amplified, whereas for the effective treatment of autoimmune and inflammatory diseases, the immune response needs to be specifically suppressed. Certain T cell populations are known to be critical to the development and persistence of T cell-mediated autoimmune diseases. In the case of autoimmune diabetes, for example, effector T cells can kill insulin-producing cells of the pancreas through direct and indirect mechanisms. Once most insulin-producing cells have been destroyed by the aberrant T cell response, the patient is no longer able to regulate blood glucose levels and develops diabetes. In addition to disease-causing effector T cells (Teff), there exist populations of regulatory T cells (Tregs), which act to inhibit such dangerous responses in normal individuals. Tregs are able to inhibit the destructive activity of Teff and are part of the immune system’s control apparatus.
We believe that our technology can be used to specifically treat autoimmune disease. We are working on two different approaches: (1) the direct inhibition and/or deletion of the pathogenic autoimmune effector T cells and (2) the control of the effector T cells’ function through the expansion and activation of Tregs.
Immunotherapy
During the last five years, there has been substantial scientific progress in therapeutically modifying the function of immune cells (“immunotherapies”), such as T cells, to either enhance tumor killing in the context of oncology, or protect tissue in the context of autoimmune disease. Immunotherapies are therefore increasingly recognized as an essential aspect of the emerging opportunities in the treatment of cancer and autoimmune disease. Despite the tremendous promise of these therapies, there are a number of continuing challenges. For example, most of the currently used cancer immunotherapies rely on non-specific and general activation of T cells or the inhibition of costimulatory pathways (e.g., checkpoint pathway inhibitors), both of which result in the global, non-specific stimulation of T cells. The global and nonspecific engagement by many current immunotherapeutics results in the activation of a large fraction of disease-irrelevant T cells, rather than selective activation of those T cells which can therapeutically affect the disease by virtue of their antigen-specific TCRs. The net effect of this approach is typically an extremely narrow therapeutic window in which many of the stimulated T cells are not selective towards the tumor, and often recognize “self antigens”. This results in significant toxicity and serious side effects and, in severe cases (e.g., Proleukin™ and Yervoy™), fatalities. Similarly, for the treatment of autoimmune indications, previous immunotherapies have been non-specific and broadly suppress immune function (e.g., Humira™, cyclosporine and methotrexate) and thus potentially predispose patients to deadly infections, and cancer.
| 4 |
Checkpoint pathway inhibitors modulate the costimulatory pathways described and illustrated above by blocking the function of inhibitory receptors which would normally suppress the activation of T cells, so-called “immune checkpoints.” Checkpoint blockade is predominantly achieved through the use of monoclonal antibodies (“mAbs”) directed against the desired checkpoint protein (e.g., mAbs to PD1 or CTLA-4). These mAbs can block the checkpoint proteins such as programmed cell death-1 (e.g., PD-1, Pembrolizumab/ Keytruda (Merck)) and CTLA-4 (e.g., Yervoy/Ipilimumab (Bristol-Myers Squibb)), and hence reactivate the inhibited anti-cancer T cell responses and stimulate T cell proliferation. The checkpoint inhibitors currently on the market have demonstrated significant and durable responses in some patients. However, immunotherapy with checkpoint inhibitors is still limited by modest to low response rates. For example, 34% of advanced melanoma patients respond to anti-PD-1 and 12% of advanced melanoma patients respond to anti-CTLA-4. To enhance the proportion of responding patients, strategies, such as combinations with other existing cancer therapies, are currently being explored. While these strategies have promise, severe side effects including potentially life threatening immune-related adverse events can affect upwards of 24% of patients using anti-CTLA-4 therapy alone, or 54% when used in combination with anti-PD-L1 therapy (e.g., Nivolumab).
An alternative approach to activate an immune response is through the use of bi-specific antibodies that concomitantly engage the TCR on a T cell and an antigen on the tumor cell, examples of which are BiTEs (e.g., Amgen) or Darts (e.g., Macrogenics). These bi-specific antibodies are distinct from the monoclonal antibodies described above in that they are engineered to simultaneously bind to two different types of antigen, rather than one. To activate the T cells, the bi-specific molecules are designed to engage a tumor antigen while also engaging a component of the T cell receptor, called CD3. Since CD3 is expressed on all T cells, dual engagement through bi-specific molecules results in a global, non-specific activation of T cells at the tumor or wherever the antigen is expressed. The non-specific activation of large T cell subsets can give rise to significant toxicity, thereby significantly limiting the acceptable dose and therapeutic range of this approach.
An additional approach to activating an antitumor immune response is cytokine immunotherapy. Cytokines, such as recombinant interleukin-2 (IL-2), can directly stimulate immune effector cells leading to antitumor efficacy, but the clinical benefit with systemic administration is limited by induction of Tregs and by severe toxicities. Recently, concepts for tumor-targeted delivery of IL-2 have shown promise. A Phase 1 trial data with an IL-2 molecule having multiple releasable polyethylene glycol (PEG) chains, NKTR-214, is showing encouraging evidence of anti-tumor activity, and a favorable tolerability profile. The PEGylation leads to a slow release of active IL-2 and loss of IL-2 preferential binding to Tregs. The NKTR-214-induced immune cell changes promote immune cell tumor destruction, with increased numbers of proliferating CD8+ T cells and NK cells in the blood and tumor and accumulation of CD8+ T cells relative to T regulatory immune suppressive cells in tumors. A tumor-antigen-CEA-targeted IL-2 fusion protein also in phase I clinical testing, has been shown to increase natural killer and CD8+ T cells in tumors. These data support the concept of immune cell-specific IL-2 delivery, such as in CUE biologics that include IL-2 costimulatory molecules.
As described above, cancer cells can evade a normal immune response by expressing cell surface inhibitory molecules that interact with and suppress the attacking T cells within the tumor microenvironment. Consequently, more recent bi-specific approaches seek to decorate tumor cells with activating costimulatory signals to potentially overcome tumor resident immune cell suppression. This is achieved by targeting a tumor antigen with one arm and presenting a costimulatory signal for potential tumor resident T cell stimulation through the other. These bispecific molecules have been used in patients with, in some cases, good therapeutic results. However, there are significant limitations. For example, they must first localize to the tumor in order to exert their effects (e.g., those from Altor, Sutro, Covagen, Roche, and Amgen) and have the added limitation of engaging only the subset of T cells which successfully traffic to the tumor within the treatment window. Notably, CUE biologics are designed to engage T cells directly, not requiring tumor targeting, and thus allow for T cell priming and activation in the periphery (e.g., tumor draining lymph node) outside the suppressive tumor microenvironment. Lastly, the use of these tumor directed bi-specifics may be hampered by the drug’s short half life in patients, thus requiring continuous infusion.
| 5 |
A different approach that has shown some promise in tumor therapy is to remove T cells from patients, activate and stimulate them outside the body (ex vivo), which expands them (usually over 7-14 days), and then infuse them back into the patients. These methods are termed cellular therapies, and include adoptive cell therapy (“ACT”) and chimeric antigen receptor T cell (“CAR-T”) therapy (e.g., those from Juno and Kite). At the outset, this is an individualized patient-specific approach. In ACT, native (un-modified) T cells are extracted and purified from a patient, expanded ex vivo and re-introduced to the patient in order to increase the number of T cells available to kill the tumor cell. Similarly, in CAR-T therapy, patient T cells are extracted and purified; however, distinct from ACT, patient T cells are genetically modified to target tumor-specific antigens through the use of chimeric antigen receptors (“CARs”) prior to patient infusion in an attempt to increase specificity. CARs are comprised of an external tumor-specific antibody fragment or engineered TCR (e.g., Adaptimmune) linked to cytoplasmic signaling domains for T cell activation such that upon binding of the tumor antigens, the engineered T cells proliferate and attack cancer cells.
CAR-T-based immunotherapy has demonstrated complete responses of greater than 80% in recent clinical trials of acute lymphoblastic leukemia (“ALL”) patients. However, potent CAR-T cell responses have produced life threatening cytokine release syndromes (“CRS”) in 69% of high burden ALL adults. Severe toxicities and deaths led to a halt in JCAR015, Juno’s lead CAR-T program, and potentially pose a safety challenge to the wider adoption of cellular therapies. Moreover, the technical requirements and expense associated with individualized T cell extraction, ex vivo amplification/modification, and patient re-infusion represent significant scaling and cost challenges that might limit broad usage and eventual commercialization of this therapeutic modality. Despite these safety and scalability considerations, the recent demonstration of impressive clinical survival in patients treated with cellular therapies deserves serious attention.
Our Approach for Next Generation Immunotherapies
We have developed a proprietary platform for the design and development of biologic drugs for in vivo (e.g., directly in the patient’s body) T cell based immunotherapy. In the context of cancer, CUE Biologics are being designed to selectively activate T cells which recognize cancer antigens (e.g., peptides) expressed or amplified in cancer cells (tumor antigens or neoantigens). For the treatment of autoimmune diseases such as Type 1 diabetes, celiac disease, arthritis and others, CUE Biologics are designed to selectively dampen disease-causing T cell responses directed against self-antigens.
CUE Biologics are designed to mimic the signals, or “cues”, of the immune system to generate highly focused T cell responses associated with disease. We accomplish this by the fusion of unique costimulatory signaling molecules (ligands) with a TCR targeting p-MHC complex (“pMHC”). This co-engagement of signals through the TCR and costimulatory receptor mimic and recapitulate the very signals delivered by APCs to T cells during an immune response. In this way CUE Biologics™ allow for the precise targeting of distinct signaling ligands exclusive to the T cell population of interest, resulting in targeted T cell modulation.
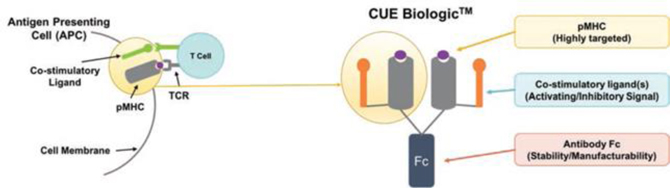
CUE BiologicsTM are designed to mimic Antigen Presenting Cells (“APCs”)
Our therapeutic approach is designed to be administered directly in patients (in vivo) which differs markedly from other T cell therapeutic approaches such as ACT, which requires patients’ T cells to be first harvested, then stimulated and expanded outside the body before being reinfused in an activated state. We believe CUE Biologics represent a breakthrough approach as a disease-specific biologic T cell modulator administered in vivo (in body) rather than the ex vivo (outside the body) approach deployed by current cellular immune therapies. Furthermore, we believe the desired pharmacological effect in patients will be more precisely controlled by directly administering CUE Biologics into the patient for selective modulation of disease relevant T cells.
| 6 |
The therapeutic properties and selective nature of Cue Biopharma’s drug candidates result from the design and optimization of key functional parameters for a given therapeutic framework. Each framework harbors an MHC and one or more costimulatory element(s) optimized to drive a particular type of T cell response, such as stimulation and expansion of a cytolytic T cell response to kill cancer cells, or specific down-regulation and inhibition in the context of autoimmune disease. The targeting of the framework to specific T cell populations is dependent on the specific peptide linked to the MHC. Notably, more than 75 peptides that are expressed by different solid tumors are currently described in the clinical literature. Thus, after finalizing a therapeutic framework (pMHC-ligand-Fc) we believe different tumors can be addressed by changing the targeting peptide, presenting the promise of greatly reducing the time and cost associated with the generation of new CUE molecules to take forward through IND-enabling studies and, potentially, into the clinic.

Illustration of use of different targeting peptides to address different tumor types
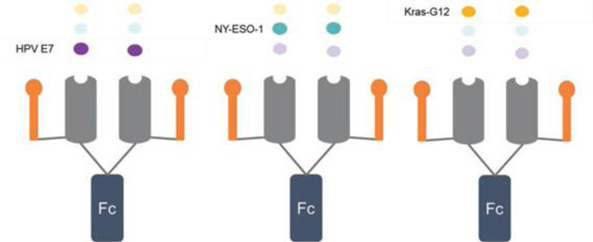
Illustration of use of different targeting peptides to address different indications
Using our CUE Biologics™ platform, we believe we will be able to design biologics that will have certain advantages over existing immunotherapies including increased specificity, reduced toxicity and greater manufacturability. As such, we believe our approach to designing and developing immuno-modulatory biologics represents a breakthrough, next-generation solution to realizing the promise of T cell based immunotherapies.
Competition
Immunotherapy technologies are advancing at a rapid pace and we anticipate competing with companies developing bi-specific antibodies (e.g., Amgen, Inc., Hoffmann-La Roche (Roche), Sutro Biopharma), CAR-T therapies (e.g., Novartis A.G., Juno Therapeutics, Kite Pharma, Inc.), checkpoint inhibitors (e.g., Bristol-Myers Squibb, Merck & Co. and Pfizer, Inc.), antibody drug conjugates (“ADCs”) (e.g., Seattle Genetics, Inc., ImmunoGen, Inc. and Sorrento Therapeutics), and targeted cytokines (Nektar/Bristol Myers Squibb, Roche) many of which have significantly greater financial and other resources than we have.
| 7 |
CUE Biologics™ Drug Candidates
The relative effectiveness of immunotherapies depends on whether a relevant or optimal therapeutic mechanism to engage the immune system has been addressed by the therapy, and it is likely that different immune stimulatory mechanisms will be required to optimally address certain cancers over others. The versatility of the CUE Biologics™ platform allows access to multiple distinct mechanisms with a series of biologic frameworks addressing a variety of conditions and requirements. We have currently designed two promising therapeutic frameworks to support distinct and potent mechanisms of T cell activation: our pMHC/IL-2 based CUE-100 series (to enhance overall numbers of tumor specific T cells) and our pMHC/CD80:4-1BBL based CUE-200 series (to reinvigorate exhausted T cells). We expect to be able to target antigen-specific T cell populations in a variety of indications by a simple peptide exchange into validated CUE Biologics™ frameworks. We continue to evaluate additional constructs from which we will launch further framework series in both oncology and autoimmunity.
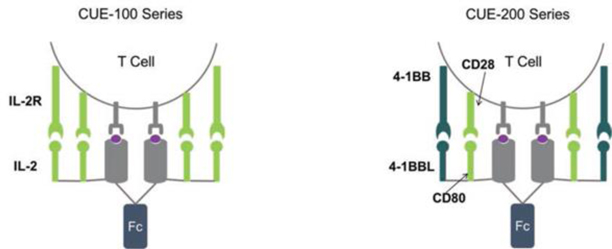
Illustration of CUE-100 and CUE-200 frameworks CUE-101
In furtherance of our efforts to design and test various frameworks addressing a variety of conditions, we are developing and implementing a complementary ex vivo (outside the body) human cancer model assay system for testing and evaluating CUE Biologics™ molecules directly in cancer patient-derived T cells. The ex vivo assays have the potential to provide a powerful tool for demonstrating human translatability by evaluating preclinical proof of mechanism and proof of concept directly in human samples, as well as for defining relevant metrics and critical parameters informing clinical application(s). Through this assay system, we have recently tested various epitopes (CMV and MelanA/MART, shown below) on the CUE-101 framework to support the versatility and robustness of our CUE Biologics™ platform.
| 8 |
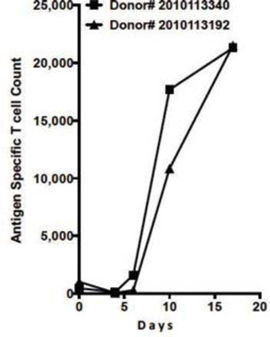
Results from CUE-100 preliminary two-sample human ex vivo study, indicating treatment with CUE:CMV:IL-2 results in activation of antigen-specific T cells.
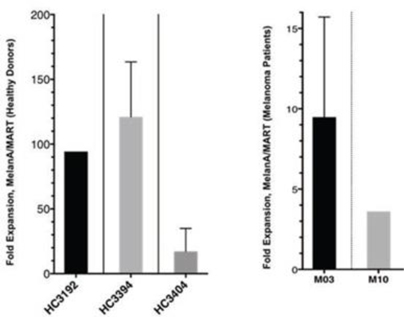
Results from CUE-100 human ex vivo study, indicating treatment with CUE-100 (MelanA/MART) results in activation of antigen-specific T cells in healthy donors and melanoma patients as measured by T cell expansion.
As exemplary of our CUE Biologics™ platform, we have begun development work on our anticipated first clinical candidate, CUE-101 (described below) and are utilizing our ex vivo assay capabilities to generate data for proof of concept with potent, tumor antigen-selective T cell activation. The ex vivo assays, utilizing clinical samples of peripheral blood mononuclear cells (“PBMCs”) for testing and evaluating CUE-101, are expected to provide data to support an IND submission and to inform our phase I study protocol and clinical development strategy. Human PBMCs, including those from cancer patients, are the most relevant model for characterizing the activity and specificity of CUE-101. These studies, investigating the drug levels and duration of drug exposure, for functional and molecular changes (PD effects) in target immune cells and in non-target cells will guide human dosing and PD biomarker monitoring in our phase I trial. In addition to providing potentially insightful data relating to the prospects of antitumor efficacy in patients, the assays are designed to define the drug exposure range for antitumor activity as well as provide evidence for clinical biomarker selection and the biopsy schedule for clinical proof of mechanism.
| 9 |
CUE-101
Our current lead drug candidate, CUE-101, uses the pMHC/IL-2 CUE-100 framework, and is a fusion of a variant form of the cytokine Interleukin-2 (IL-2) and a T cell antigen (pMHC). The targeting peptide contained in the pMHC that is currently used in CUE-101 is derived from the human papilloma virus E7 protein (HPV-E7). CUE-101 is a single, covalently-assembled biologic designed to target and activate T cells specific to HPV-related cancers. Treatments for patients with HPV-related cancers represent are an important unmet clinical need, which accounts for approximately 24,600 cases of cervical, head and neck, and genitoanal cancers in the United States every year leading to approximately 9,000 deaths annually. HPV-driven cancers lead to approximately 225,000 deaths worldwide each year. We believe our drug candidate CUE-101 has the potential to provide patients with a more effective and safer alternative in treating their HPV-driven cancers. It is our current intention to file an IND for CUE-101 in the first quarter of 2019, provided the pre-IND efficacy and safety data generated supports continued development.
Our preclinical data, including a mouse animal model of HPV+ cancer, have generated encouraging results with monotherapy treatment with a murine CUE biologic comprised of IL-2 costimulatory and pMHC-HPV peptide domains. Tumor growth inhibition was observed with treatment in the HPV+ TC-1-Luc tumor model. Studies of this agent in combination with an anti-PD-1 antibody resulted in complete tumor regression in some animals. These results compare favorably to results achieved in the same preclinical study using IL-2 or anti-PD-1 therapy alone, where no regressions were observed. Notably, in the tumor-free mice from the treatment group “cured” by the murine CUE:IL-2 compound combination with anti-PD-1 antibody, immunologic memory was demonstrated whereby no tumor growth observed in animals following tumor cell injection.
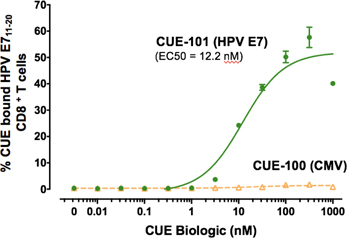
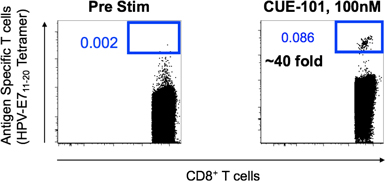
Results from CUE-101 human ex vivo study demonstrating peptide selectivity in target T cell binding and engagement by comparing binding of CUE-101 versus CUE-100 (CMV) to disease specific HPV E711-20 CD8 human T cells.
We have profiled CUE-101 activity in tumor antigen-specific T cells and PBMCs isolated from a healthy HPV+ human donor. These data demonstrate highly targeted engagement of CUE-101 specifically to tumor antigen specific CD8+ T cells (shown above). These data further support that targeted engagement results in T cell receptor and IL-2 receptor activation as measured by increases in receptor signaling molecules. Furthermore, CUE-101 induces antigen specific T cell activation- and cytotoxic T lymphocyte (CTL)- cell surface marker changes and CTL-activity as measured by drug-induced production of IFNg, an established marker of T cell activation and killing potential (shown below). Lastly, CUE-101 has demonstrated expansion of antigen-specific T cells from donor PBMCs as a single agent. These results demonstrate antigen-specific T cell activation and expansion with CUE-101 in human immune cells. The timing and magnitude of changes with drug treatment will guide selection of the most relevant and robust markers and the timing for sampling in phase I patients treated with CUE-101.
| 10 |
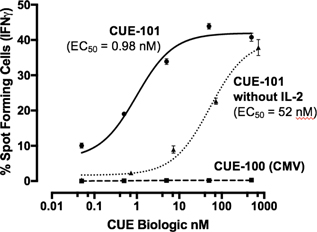
Results from CUE-101 human ex vivo study indicating treatment with CUE-101 (HPV-E7) results in highly specific activation of antigen-specific T cells in a peptide and MOD (IL-2) dependent manner as measured by interferon gamma release,
While this preclinical data from ex vivo analysis of human T cells is highly encouraging, the data represent a single healthy HPV+ subject and may not be representative of results in samples from patients with HPV+ driven malignancies. As described below under “Business — Background,” cancer uses various mechanisms to suppress and alter the immune system. For example, it has been observed from clinical studies that some cancers, including HPV+ cancers, are capable of reducing and altering T cell populations. Therefore, we intend to further study CUE-101 in ex vivo assays with blood derived from multiple HPV+ cancer patients to demonstrate activation and proliferation of HPV-E7 specific T cells. We will also continue to survey other drug constructs targeting various cancer epitopes and plan to select promising candidates for clinical development. It is our current intention to file an IND for CUE-101 by the first quarter of 2019, provided the pre-IND efficacy and safety data generated supports continued development.
Results from CUE-101 human ex vivo study, indicating treatment with CUE-101 results in expansion of antigen-specific T cells as a single agent
While CUE-101 targets the HPV-E7 TCR in cervical/head and neck cancers, we believe our CUE Biologics™ platform may be used to target a large variety of alternative peptides which may allow us to address many tumors with high therapeutic need in the oncology patient population. In support of this, we have recently demonstrated highly potent efficacy in preclinical murine models targeting non-viral epitopes. These data together with the recent human ex vivo experiments using a melanoma specific epitope (MelanA/MART, shown above) in both healthy donor and melanoma patient samples support CUE-100 framework’s ability to activate distinct T cell populations via a simple amino acid peptide antigen exchange on an otherwise validated scaffold, which should reduce the time to clinic (and associated costs) of next-generation biologics. We are currently exploring multiple unique epitopes in the context of the CUE-100 series framework in human ex vivo assays to help guide prioritization of programs.
| 11 |
Extension to Autoimmune Indications
In addition to oncology, we are expanding our technology’s reach to generate highly promising and novel immunotherapeutics for the treatment of debilitating autoimmune disorders. Autoimmune indications may be addressed with our technology through two general strategies: (1) depleting disease causing autoreactive T cells by selectively delivering inhibitory signals or (2) by delivering signals to induce and expand regulatory T cells, which subsequently act to inhibit disease-causing T cells.
CUE Biologics™ frameworks for autoimmune disease will be designed to influence a subset of T cells known as CD4 T cells. CD4 T cells recognize peptides in the context of MHC class II proteins. Therefore, prototypic CUE Biologics™ frameworks in autoimmunity would rely on MHC class II recognition by CD4 T cells. This is distinct from the MHC class I recognition by CD8 T cells that is the basis of our current oncology pipeline. Both pathogenic (i.e., disease causing) and regulatory (disease limiting) CD4 T cell subsets are known to exist in autoimmune disease. Our CUE Biologics™ frameworks in autoimmunity will therefore be intended to treat autoimmune diseases by either depleting pathogenic CD4 T cells or amplifying regulatory CD4 T cell responses to specific disease relevant antigens. Potential autoimmune indications of interest include Type 1 diabetes, arthritis, autoimmune thyroiditis (e.g., Graves’ disease), celiac disease and CNS/neurological autoimmune disorders (e.g., multiple sclerosis, Parkinson’s disease, etc.).
Consistent with our plan to establish strategic partnerships with leading pharmaceutical or biotechnology organizations to further our development efforts, in November 2017 we entered into a Collaboration Agreement with Merck Sharp & Dohme Corp. (“Merck”) for a partnership to research and develop certain of our proprietary biologics that target certain autoimmune diseases. We view this Collaboration Agreement as a component of our development strategy since it will allow us to advance our autoimmune programs in partnership with a world class pharmaceutical company, while also continuing our focus on our more advanced cancer programs. For further information, see below “Business — Our Collaboration Agreement with Merck”.
MOD™ and viraTope™ Technology Platforms
Supporting our CUE Biologics™ platform are two companion discovery platforms: MOD™, a costimulatory optimization and discovery platform, and viraTope™, a T cell epitope discovery platform, both illustrated below.
| 12 |
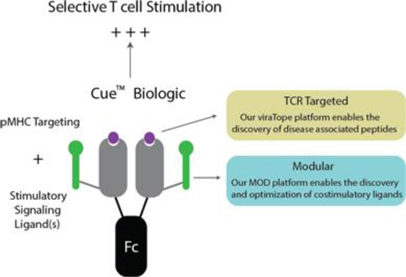
The design of CUE Biologics™ allows for incorporation of antigens and costimulatory molecules discovered through the MODTM platform to develop novel biologics to address new indications in oncology and autoimmune disorders.
We believe that the MOD™ technology platform has a unique ability to optimize existing costimulatory ligands for use in our biologics as well as discover as yet unknown costimulatory signaling molecules. The MOD™ platform represents a high throughput method for determining specific cell surface protein-protein interactions (e.g., signaling receptor:ligand pair(s)). In brief, MOD™ allows for the detection of associations between distinct cell surface query proteins (i.e., ligands) and cell surface expression libraries (i.e., receptors) to first identify molecular engagements and further allows for the mechanistic dissection of complex biochemical function by screening large numbers of mutant molecules. Taken together, we believe that MOD™ can provide powerful tools to first define novel protein-protein interactions associated with T cell activation. Secondly, MOD™ is designed to allow us to modulate these signaling ligands through the rapid screening of mutants in order to dissect biochemical function and alter binding properties (i.e., altered affinities and specificities). A key component of our therapeutic design involves decreasing the binding of the costimulatory element while retaining its biological activity (i.e., affinity attenuation). Affinity attenuation allows the pMHC to drive the engagement with the target T cells and limits off-target engagement and associated collateral toxicity.
The viraTope™ platform addresses the historic difficulty of identifying disease associated T cell signatures through the monitoring of complex T cell repertoires. As discussed previously, at the core of the molecular events comprising a T cell-mediated immune response is the engagement of the T cell receptor (“TCR”) with a small peptide antigen presented by an MHC molecule, referred to as a T cell epitope. This represents the immune system’s targeting mechanism and is a requisite molecular interaction for T cell activation and function, and forms the basis of our targeted immunotherapeutics (i.e., TCR targeting). The viraTope platform is designed to achieve rapid, comprehensive, and quantitative immunomonitoring by interrogating primary T cells with a combinatorial library of pMHC in conjunction with deep sequencing. viraTope’s™ libraries would query T cells with all possible mimotopes, leaving cognate pMHC bound to their respective T cells. Deep sequencing of the bound pMHC would comprehensively enumerate all T cell epitopes recognized by a given T cell sample. In this way, viraTope™ could allow the identification of novel epitopes differentially represented in diseased versus control patients and would further make the frequencies of all known and unknown T cell specificities accessible for prospective, in-study, and retrospective analyses of clinical trials. Thus, the ability to systematically identify the entire ensemble of epitopes for a given disease state represents a unique opportunity for the development of diagnostics and highly targeted therapeutics against infectious diseases, autoimmunity and cancers. We believe that viraTope™ has the ability to comprehensively and quantitatively monitor T cell responses, which could lead to the discovery of novel drug candidates and biomarkers for internal use or to potentially license to strategic partners.
Our Business Strategy
Our primary objective is to become a leading, next-generation immunotherapeutics/biopharmaceutical company developing highly highly selective biologics for precision immune modulation. We plan to do this through coordinated and integrated strategic initiatives. Key elements of our strategy include:
| 13 |
| · | Modular and versatile platform allowing for efficient and rapid drug design, prototyping and optimization. We plan to leverage our CUE Biologics™ platform’s modular capabilities to rapidly and efficiently develop our drug candidates. We believe our platform will provide a highly productive portfolio of promising clinical drug candidates aimed at specifically targeting disease relevant T cells for effective immune modulation. The modular design of our CUE Biologics™ platform provides the flexibility and versatility to construct drug frameworks comprised of various MOD combinations to elicit novel mechanisms of action. As described above under “Our Approach to Next Generation Immunotherapies,” after we establish a successful framework that uses a specific peptide to target a particular disease indication, we expect to be able to use that framework to target additional disease indications by changing the targeting peptide. Therefore, by leveraging the previous work done to establish a framework, we believe we will be able to significantly compress the timeline (by as much as six to twelve months) and capital requirements associated with the development of additional drug candidates. |
| · | Using preclinical data and efficient Phase I clinical study design to accelerate the development process. We recently demonstrated through ex vivo assays using human clinical samples that CUE:IL-2 activates T cells in an antigen specific manner. We plan to continue testing our biologic drug constructs in ex vivo studies with human clinical samples using various cancer relevant epitopes to demonstrate selective activation of T cells specific for various antigens spanning a range of oncology indications. We believe this approach provides meaningful validating data enhancing the quality of our preclinical data package for IND filing. This data also has the potential of increasing the probability for identifying relevant pharmacodynamic (“PD”) biomarkers for patient monitoring and as a potential surrogate marker of anti-tumor activity in the clinical setting. Furthermore, we believe these ex vivo studies will supplement and potentially reduce our reliance on preclinical animal models, providing a more cost and time efficient means of testing our drug candidates’ activities. Given the urgent medical needs we intend to address with our drug candidates, we are planning to design and conduct our Phase I clinical studies to generate safety data and a clinically meaningful data package around efficacy with the aim of approaching the FDA for an accelerated registration study. |
| · | Using our process development and protein biochemistry capabilities as a competitive advantage. We anticipate devoting significant resources to optimizing drug design and process development, including protein engineering and optimization, which are key components to maximizing the value of our current and future drug candidates. Through our core competencies and proprietary CUE Biologics™, MOD™ and viraTope™ technology platforms, we are designing and developing a growing intellectual property portfolio of novel and proprietary immune modulatory biologics. We believe our modular approach to designing biologics, coupled with the protein engineering and optimization capabilities offered by our platform technologies, should enable us to more rapidly and cost-effectively design and optimize potential drug candidates, as compared to more traditional preclinical development processes. Although we have yet to advance any of our drug candidates into the clinic, we believe this efficient preclinical development process positions us well to potentially establish a leading position in the discovery and development of promising next generation immunotherapies. |
| · | Establishing key strategic partnerships with leading pharmaceutical companies. We believe that our CUE Biologics™ platform offers the promise of enabling us to develop multiple drug candidates that address a variety of potential indications within oncology, autoimmune disease and infectious disease. Accordingly, as we continue to evolve and progress our drug candidates through preclinical and early clinical development, we plan to establish strategic partnerships with leading pharmaceutical or biotechnology organizations, such as our partnership with Merck pursuant to the Collaboration Agreement described below. We believe that this will allow us to further enhance our capabilities and capacities to discover and develop multiple, promising drug candidates for unmet medical needs in oncology and autoimmunity in a highly productive and cost-effective manner. |
| · | Leveraging our relationships with Einstein, our scientific founders and other scientific advisors. Our scientific founders and Einstein, as well as our renowned scientific and clinical advisors (“SAB/CABs”), have a history of seminal, pioneering discoveries and possess significant experience in oncology, immunotherapy, immunology, and biophysics, as well as clinical development. We plan to leverage our scientific founders’ and SAB/CABs’ scientific and clinical expertise and guidance as we develop our product pipeline and technologies. |
| 14 |
Our License Agreement with Einstein
Our CUE Biologics™, viraTope™ and MOD™ platforms have all been developed from technology covered by core patent applications licensed to us from Albert Einstein College of Medicine (“Einstein”) pursuant to a license agreement originally entered into on January 14, 2015 and amended and restated on July 31, 2017 (the “Einstein License”). The Einstein License covers certain patent rights relating to: (i) methods for high throughput receptor-ligand identification, which we refer to as our MOD™ platform or technology, (ii) a cellular platform for rapid and comprehensive T cell immunomonitoring, which we refer to as our viraTope™ platform or technology, (iii) CUE Fc fusion constructs and uses thereof, which we refer to as our CUE Biologics™ platform or technology, and (iv) variant PD-L1 polypeptides, T cell modulatory multimeric polypeptides and methods and uses thereof (collectively, the “Patents”). We hold a worldwide exclusive license, with the right to sublicense, import, make, have made, use, provide, offer to sell, and sell all products, processes and services that use the Patents, including certain technology received from Einstein relating thereto (“Licensed Products”).
The Einstein License is a royalty-bearing license obligating us to pay a percentage of proceeds received from sales of categories of Licensed Products at low single digit rates. We have also agreed to share a portion of our proceeds that we derive from other agreements, like sublicense agreements, relating to Licensed Products that we may enter into. The percentage of such proceeds that we are required to pay Einstein ranges from the low to high teens, depending on how far we have developed a Licensed Product before we enter into an agreement relating to the Licensed Product. These percentages are reduced for sales of Licensed Products in countries where a competing product exists and for products or services involving the use or incorporation of technology received from Einstein relating to synapse for targeted T cell activation molecules, receptor ligand identification or platforms for T cell monitoring. In addition to our obligation to pay royalties based upon a percentage of proceeds from sales of Licensed Products, we have also agreed to pay Einstein annual maintenance fees. The maintenance payments are creditable against any royalty payments we pay under the Einstein License. As of December 31, 2017, we had paid to Einstein an aggregate amount of $125,000 in license signing and license maintenance fees under the Einstein License.
Under the Einstein License, we are also obligated to make milestone payments corresponding to: (i) approval of the first IND by the FDA or foreign equivalent for a Licensed Product; (ii) approval of any subsequent IND application or foreign equivalent for a “new indication” for a Licensed Product; (iii) initiation of Phase II clinical trials or foreign equivalent on a Licensed Product; (iv) initiation of Phase II clinical trials or foreign equivalent for a “new indication” for a Licensed Product; (v) initiation of Phase III clinical trials or foreign equivalent on a Licensed Product; (vi) initiation of Phase III clinical trials or foreign equivalent for a “new indication” for a Licensed Product, (vii) the first commercial sale of a Licensed Product; (viii) the first commercial sale of each “new indication” for one of our previously approved Licensed Products; and (ix) cumulative sales of certain Licensed Products reaching certain threshold amounts. The aggregate amount of milestone payments made under the Einstein License may equal up to $1.85 million for each Licensed Product and up to $1.85 million for each new indication of a Licensed Product. Additionally, the aggregate amount of one-time milestone payments based on cumulative sales of all Licensed Products may equal up to $5.75 million.
In addition to our obligations to make the cash payments to Einstein described above, under the Einstein License we were required to issue Einstein 671,572 shares of our Common Stock immediately prior to completion of the intial public offering of our common stock completed on December 27, 2017.
The Einstein License commenced on January 14, 2015 and expires upon the expiration of our last obligation to make royalty payments to Einstein, unless terminated earlier under the provisions thereof. Under the Einstein License, we will be obligated to make royalty payments to Einstein, with respect to certain Licensed Products, for the longer of 15 years from the first sale of such products in each country or for the duration of any market exclusivity period granted by a regulatory agency for such product and, with respect to certain Licensed Products sold by sublicensees, the longer of 10 years from the first sale of such products in each country or for so long as the sublicensee agrees to pay royalties on such products. We have the right to terminate the Einstein License at any time upon sixty (60) days’ written notice to Einstein; provided, however, that we will lose intellectual property rights related to the Patents if we choose to terminate the Einstein License in this manner. Each party has the right to terminate the Einstein License if the other party is in default or breach of any condition of the Einstein License with a right to cure any such breach within sixty (60) days from receipt of notice of such default or breach, unless the other party has disputed the alleged breach in good faith. Either party can also terminate the Einstein License if the other party voluntarily files for bankruptcy or other similar insolvency proceedings, makes a general assignment for the benefit of creditors, or is the subject of an involuntary bankruptcy petition that is not dismissed within ninety (90) days. If we fail to pay any sum that is due and payable to Einstein within thirty (30) days after receiving written notice of our default from Einstein, then Einstein has the option of terminating the Einstein License unless we pay within forty-five (45) days of such notice all delinquent sums with interest. Einstein may also terminate the Einstein License in the event we are convicted of certain felonies relating to the manufacture or use of Licensed Products.
| 15 |
The Einstein License also obligates us to meet certain due diligence requirements (the “Diligence Milestones”) as follows:
| · | update our research and development plan annually; |
| · | submit an IND application to the FDA or similar foreign regulatory agency within a number of years from the Effective Date; |
| · | initiate Phase I clinical trials on a Licensed Product within a number of years from the Effective Date; |
| · | initiate Phase II clinical trials on a Licensed Product within a number of years from the Effective Date; |
| · | initiate Phase III clinical trials on a Licensed Product within a number of years from the Effective Date; |
| · | submit an application for FDA approval to market and sell a Licensed Product within a number of years from the Effective Date; |
| · | have our first commercial sale of an FDA Licensed Product within a number of years from the Effective Date; and |
| · | spend a minimum amount per year on product development until our first commercial sale of a Licensed Product. |
If we fail to meet any of the Diligence Milestones, Einstein will have the right to terminate the Einstein License if such Diligence Milestone is not satisfied within thirty (30) days from receiving a written notice of default from Einstein. Under certain circumstances and upon prior notice to Einstein, we may have the right to an additional extension of our Diligence Milestones if, despite our commercially reasonable efforts we are not able to satisfy the Phase II clinical trial Diligence Milestone or any subsequent Diligence Milestone. As of the date of this prospectus, we have met all required Diligence Milestones.
Our Collaboration Agreement with Merck
On November 14, 2017, we entered into an Exclusive Patent License and Research Collaboration Agreement (the “Collaboration Agreement”) with Merck Sharp & Dohme Corp. (“Merck”) for a partnership to research and develop certain of our proprietary biologics that target certain autoimmune disease indications (the “Initial Indications”). We view this Collaboration Agreement as a component of our development strategy since it will allow us to advance our autoimmune programs in partnership with a world class pharmaceutical company, while also continuing our focus on our more advanced cancer programs. The research program outlined in the Collaboration Agreement entails (1) our research, discovery and development of certain CUE Biologics™ drug candidates up to the point of demonstration of certain biologically relevant effects (“Proof of Mechanism”) and (2) the further development by Merck of the CUE Biologics™ drug candidates that have demonstrated Proof of Mechanism (the “Proposed Product Candidates”) up to the point of demonstration of all or substantially all of the properties outlined in such Proposed Product Candidates’ profiles as described in the Collaboration Agreement.
For the purposes of this collaboration, we have granted to Merck under the Collaboration Agreement an exclusive license under certain of our patent rights, including a sublicense of patent rights licensed from Einstein, to the extent applicable to the specific CUE Biologics™ that are elected to be developed by Merck. From the effective date of the Collaboration Agreement until the earlier of (i) the first achievement of Proof of Mechanism for a Cue Biologic™ drug candidate or (ii) 18 months after we notify the joint steering committee that the first Product Candidate has been synthesized under the research program, we are required to forebear from researching, developing or licensing to a third party rights related to any CUE Biologics™ drug candidate for the treatment of autoimmune diseases other than pursuant to the Collaboration Agreement. In addition, so long as Merck continues product development on a Proposed Product Candidate, we are restricted from conducting any development activities within the Initial Indication covered by such Proposed Product Candidate other than pursuant to the Collaboration Agreement. The Company is not required to forbear at any time, however, from developing other Cue Biologics™ for use in therapeutic areas other than autoimmune diseases, e.g., for use in treating cancer or infectious diseases.
| 16 |
In exchange for the licenses and other rights granted to Merck under the Collaboration Agreement, we received a $2.5 million nonrefundable up-front payment and may be eligible to receive additional funding in developmental milestone payments, as well as tiered royalties if all research, development, regulatory and commercial milestones agreed upon by both parties are successfully achieved. Excluding the upfront payment described above, we are eligible to earn up to $101 million for the achievement of certain research and development milestones, $120 million for the achievement of certain regulatory milestones and $150 million for the achievement of certain commercial milestones, in addition to tiered royalties on sales, if all pre-specified milestones associated with multiple products across the primary disease indication areas are achieved. The Collaboration Agreement requires us to use the first $2.7 million of milestone payments we receive under the agreement to fund contract research. The amount of the royalty payments is a percentage of product sales ranging in the single digits based on the amount of such sales.
The term of the Collaboration Agreement extends until the expiration of all royalty obligations following a product candidate’s receipt of marketing authorization, at which point Merck’s licenses and sublicenses granted under the agreement shall become fully paid-up, perpetual licenses and sublicenses, as applicable. Royalties on each product subject to the Collaboration Agreement shall continue on a country-by-country basis until the expiration of the later of: (1) the last-to-expire patent claiming the compound on which such product is based and (2) a period of ten years after the first commercial sale of such product in such country.
Notwithstanding the foregoing, Merck may terminate the Collaboration Agreement at any time upon 30 days’ notice to the Company. The Collaboration Agreement may also be terminated by either party if the other party is in breach of its obligations thereunder and fails to cure such breach within 90 days after notice or by either party if the other party files for bankruptcy or other similar insolvency proceedings.
Our Intellectual Property
We believe that our current patent applications and any future patents and other proprietary rights that we own, or control through licensing, are and will be essential to our business. We believe that these intellectual property rights will affect our ability to compete effectively with others. We also rely and will rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to develop, maintain and strengthen our competitive position. We seek to protect these, in part, through confidentiality agreements with certain employees, consultants, advisors and other parties. Our success will depend in part on our ability, and the ability of our licensor, to obtain, maintain (including making periodic filings and payments) and enforce patent protection for our/their intellectual property, including those patent applications to which we have secured exclusive rights.
We own or have licensed 14 pending patent applications in the United States (including 11 pending U.S. provisional patent applications), four pending international PCT applications and 34 pending foreign patent applications intended to protect the intellectual property underlying our technology. Our patent applications describe certain features of our technologies, including our CUE Biologics™ platform and drug candidates, viraTope™, MOD™ screening, MOD™ variants and combinations of MODs. We plan to spend considerable resources and focus in the future on obtaining U.S. and foreign patents. We have and will continue to actively protect our intellectual property. No assurances can be given that any of our patent applications will result in the issuance of a patent or that the examination process will not require us to narrow our claims. In addition, any issued patents may be contested, circumvented, found unenforceable or invalid, and we may not be able to successfully enforce our patent rights against third parties. No assurance can be given that others will not independently develop a similar or competing technology or design around any patents that may be issued to us. We intend to expand our international operations in the future and our patent portfolio, copyright, trademark and trade secret protections may not be available or may be limited in foreign countries.
Each of our patents, if and when granted, will generally have a term of 20 years from its respective priority filing date, subject to available extensions. They are thus set to expire no earlier than dates ranging from 2032 to 2037, although there can be no assurance that any of the patent application will be granted.
| 17 |
Target Markets
Our initial focus and objective is to develop drug candidates for cervical/head and neck cancers, hepatocellular carcinomas, and melanoma. We expect that our future developmental roadmap will also focus on new drug candidates that target autoimmune disorders. We currently do not have any products that are developed such that they can be tested for clinical trials or commercial use. According to published reports, in 2016 oncology drugs were an $87.5 billion global market and autoimmune drugs constituted a $47.8 billion global market.
Our Commercialization Strategy
We are a preclinical stage company without a history of revenue or manufacturing, late stage clinical development or marketing experience. Because late stage clinical development, as well as establishing a full manufacturing and distribution structure, is expensive and time consuming, we intend to explore alternative commercialization strategies, including:
| · | developing drug candidates through the earlier stages of clinical development with the objectives of rapid, cost effective risk reduction and value creation and then establishing strategic partnership for late stage clinical development and subsequent commercialization; |
| · | developing a robust pipeline of promising drug candidates at various stages of the development process to establish optionality and regular value inflection opportunities and revenue(s); |
| · | strategically entering into co-development partnership(s) to retain potential for commercialization rights on selected drug candidate(s) and market opportunities; and |
| · | partnering with industry participants to incorporate our technology into new and existing drugs. |
We expect that partnering with pharmaceutical or biotherapeutic companies may accelerate product acceptance into our target market areas and gain the sales and marketing advantages of the partner’s distribution infrastructure. We intend to continue to strengthen our market position and solidify our leadership position in immunotherapy by continuing to improve our technology, broadening our clinical and therapeutic applications, identifying new clinical and therapeutic applications and forming strategic partnerships.
Government Regulation and Product Approval
Therapeutic products are subject to rigorous regulation by the U.S. Food and Drug Administration (the “FDA”) and other governmental agency regulations in the United States and in foreign countries. Noncompliance with applicable requirements can result in import detentions, fines, civil penalties, injunctions, suspensions or losses of regulatory approvals or clearances, recall or seizure of products, operating restrictions, denial of export applications, governmental prohibitions on entering into supply contracts, and criminal prosecution. Failure to obtain regulatory approvals or the restriction, suspension or revocation of regulatory approvals or clearances, as well as any other failure to comply with regulatory requirements, would have a material adverse effect on our business, financial condition and results of operations. In connection with therapeutic approval, we will have to comply with the many requirements associated with preclinical and clinical trials, the FDA application process, the terms of any pre-certification protocols and agreements, FDA manufacturing requirements for prototypes, and testing. Upon approval of a Biologics License Application (“BLA”) and similar approvals in other jurisdictions, there will be additional regulation relating to the packaging, distribution, marking, marketing and claims of our potential products. These later regulations are not only found in federal regulation but many states and, of course, foreign countries.
The FDA Process
The FDA regulates the clinical testing and design of therapeutics to ensure that medical products distributed in the United States are safe and effective for their intended uses. The application process for a new therapeutic is highly regulated.
As a biopharmaceutical company that operates in the United States, we are subject to extensive regulation. Our potential products will be regulated as biologics. With this classification, commercial production of our potential products will need to occur in registered and licensed facilities in compliance with current good manufacturing procedures (“cGMP”) established by the FDA for biologics. The FDA categorizes human cell- or tissue-based products as either minimally manipulated or more than minimally manipulated, and has determined that more than minimally manipulated products require clinical trials to demonstrate product safety and efficacy and the submission of a BLA for marketing authorization.
| 18 |
Government authorities in the United States (at the federal, state and local levels) and in other countries extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of biopharmaceutical products such as those we are developing. Our drug candidates must be approved by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in a foreign country. Generally, our activities in other countries will be subject to regulation that is similar in nature and scope as that imposed in the United States, although there can be important differences. Additionally, some significant aspects of regulation in Europe are addressed in a centralized way, but country-specific regulation remains essential in many respects. The process for obtaining regulatory marketing approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Product Development Process
In the United States, the FDA regulates pharmaceutical and biological products under the Federal Food, Drug and Cosmetic Act (the “FDCA”), the Public Health Services Act (the “PHSA”) and implementing regulations. Products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include, among other actions, refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The FDA has limited experience with commercial development of T cell therapies for cancer. The process required by the FDA before a biological product may be marketed in the United States generally involves the following:
| · | completion of nonclinical laboratory tests and animal studies according to Good Laboratory Practices (“GLPs”) and applicable requirements for the humane use of laboratory animals or other applicable regulations; |
| · | submission to the FDA of an Investigational New Drug Application (an “IND”), which must become effective before human clinical trials may begin; |
| · | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as Good Clinical Practices (“GCPs”), and any additional requirements for the protection of human research patients and their health information, to establish the safety and efficacy of the proposed biological product for its intended use; |
| · | submission to the FDA of a BLA for marketing approval that includes substantive evidence of safety, purity, and potency from results of nonclinical testing and clinical trials; |
| · | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with current Good Marketing Practices (“cGMP”) to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity and, if applicable, the FDA’s current Good Tissue Practices (“cGTPs”) for the use of human cellular and tissue products; |
| · | potential FDA audit of the nonclinical study and clinical trial sites that generated the data in support of the BLA; and |
| · | FDA review and approval, or licensure, of the BLA. |
| 19 |
Before testing any biological drug candidate, including our drug candidates, in humans, the drug candidate enters the preclinical testing stage. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the drug candidate. The conduct of the preclinical tests must comply with federal regulations and requirements, including GLPs. The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a biological drug candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin or that, once begun, issues will not arise that suspend or terminate such trials.
Clinical trials involve the administration of the biological drug candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research patients provide informed consent. Further, each clinical trial must be reviewed and approved by an independent institutional review board (an “IRB”) at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Clinical trials also must be reviewed by an institutional biosafety committee (an “IBC”), a local institutional committee that reviews and oversees basic and clinical research conducted at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| · | Phase 1. The biological product is initially introduced into human subjects to test for safety (adverse effects), determine recommended Phase 2 dosing and evaluate any signals of efficacy for specific targeted diseases. The initial human testing is often conducted in patients, rather than in healthy volunteers, in the case of products for severe or life-threatening diseases, especially when the product is inherently toxic. |
| · | Phase 2. The biological product is evaluated in a limited patient population to identify safety risks, optimize dosing and preliminarily evaluate the efficacy of the product for specific targeted diseases. |
| · | Phase 3. Clinical trials are undertaken in an expanded patient population at geographically dispersed clinical trial sites to further evaluate dosage, clinical efficacy, potency, and safety. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling. |
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA, the National Institutes of Health and the investigators for serious and unexpected adverse events, as well as any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human patients, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research patients are being exposed to an unacceptable health risk, including risks inferred from other unrelated immunotherapy trials. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological product has been associated with unexpected serious harm to patients.
| 20 |
Human immunotherapy products are a new category of therapeutics. Because this is a relatively new and expanding area of novel therapeutic interventions, there can be no assurance as to the length of the trial period, the number of patients the FDA will require to be enrolled in the trials in order to establish the safety, efficacy, purity and potency of immunotherapy products, or that the data generated in these trials will be acceptable to the FDA to support marketing approval.
Concurrently with clinical trials, companies usually complete additional studies and must also develop additional information about the physical characteristics of the biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological drug candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
After the completion of clinical trials of a biological product, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA must include results of product development, laboratory and animal studies, human trials, information on the manufacture and composition of the product, proposed labeling and other relevant information. The FDA may grant deferrals for submission of data or full or partial waivers. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, as amended (the “PDUFA”), each BLA must be accompanied by a significant user fee. The FDA adjusts the PDUFA user fees on an annual basis. PDUFA also imposes an annual product fee for biological products and an annual establishment fee on facilities used to manufacture prescription biological products. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviews a BLA submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, potent, and/or effective for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the biological product approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy (“REMS”) is necessary to assure the safe use of the biological product. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve a BLA without a REMS, if required.
| 21 |
Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. For immunotherapy products, the FDA also will not approve the product if the manufacturer is not in compliance with the cGTPs, to the extent applicable. These are FDA regulations and guidance documents that govern the methods used in, and the facilities and controls used for, the manufacture of human cells, tissues, and cellular and tissue based products (“HCT/Ps”), which are human cells or tissue intended for implantation, transplant, infusion, or transfer into a human recipient. The primary intent of the cGTP requirements is to ensure that cellular tissue based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. FDA regulations also require tissue establishments to register and list their HCT/Ps with the FDA and, when applicable, to evaluate donors through screening and testing. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To assure cGMP, cGTP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA in its present form, the FDA will issue a complete response letter that describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post-marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized.
In addition, under the Pediatric Research Equity Act (the “PREA”), a BLA or supplement to a BLA must contain data to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, the PREA does not apply to any product for an indication for which orphan designation has been granted. However, if only one indication for a product has orphan designation, a pediatric assessment may still be required for any applications to market that same product for the non-orphan indication(s).
Expedited Development and Review Programs
The FDA has various programs, including Fast Track, Breakthrough Therapy Designation, priority review, and accelerated approval, which are intended to expedite or simplify the process for reviewing products, and/or provide for approval on the basis of surrogate endpoints. Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or that the time period for FDA review or approval will not be shortened. Generally, products that may be eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of products to treat serious diseases and fill an unmet medical need. The request may be made at the time of IND submission and generally no later than the pre-BLA or pre-NDA meeting. The FDA will respond within 60 calendar days of receipt of the request. Breakthrough Therapy Designation is available for products that are intended to treat a serious condition where preliminary clinical evidence indicates that the product may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies. The request may be made at the time of IND submission and generally no later than the end-of-Phase 2 meeting. The FDA will respond within 60 calendar days of receipt of the request. Breakthrough Therapy Designation conveys all of the Fast Track program features along with more intensive FDA guidance and interaction and eligibility for rolling review and priority review. Priority review, which is requested at the time of BLA or NDA submission, is designed to give products that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of ten months. Although Fast Track, Breakthrough Therapy Designation and priority review do not affect the standards for approval, the FDA will attempt to expedite review of the application. Accelerated approval provides an earlier approval of products to treat serious diseases, and that fill an unmet medical need based on a surrogate endpoint, which is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome. Discussions with the FDA about the feasibility of an accelerated approval typically begin early in the development of the product in order to identify, among other things, an appropriate endpoint. As a condition of approval, the FDA may require that a sponsor of a product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials.
| 22 |
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug or biologic product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug or biologic for this type of disease or condition will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use will be disclosed publicly by the FDA; the posting will also indicate whether the drug or biologic is no longer designated as an orphan drug. More than one product candidate may receive an orphan drug designation for the same indication. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to seven years of orphan product exclusivity. During the seven-year exclusivity period, the FDA may not approve any other applications to market a product containing the same active moiety for the same disease, except in very limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. A product is clinically superior if it is safer, more effective or makes a major contribution to patient care. Thus, orphan drug exclusivity could block the approval of one of our potential products for seven years if a competitor obtains approval of the same product as defined by the FDA and we are not able to show the clinical superiority of our product candidate or if our product candidate’s indication is determined to be contained within the competitor’s product orphan indication. In addition, the FDA will not recognize orphan drug exclusivity if a sponsor fails to demonstrate upon approval that the product is clinically superior to a previously approved product containing the same active moiety for the same orphan condition, regardless of whether or not the approved product was designated an orphan drug or had orphan drug exclusivity.
Post-Approval Requirements
Any potential products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting products for uses or in patient populations that are not described in the product’s approved uses (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available products for off-label uses, if the physicians deem to be appropriate in their professional medical judgment, it is FDA’s position that manufacturers may not market or promote such off-label uses.
In addition, quality control and manufacturing procedures must continue to conform to applicable manufacturing requirements after approval to ensure the quality and long-term stability of the product. We expect to rely on third parties for the production of clinical and commercial quantities of our potential products in accordance with cGMP regulations. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including, among other things, recall or withdrawal of the product from the market. In addition, changes to the manufacturing process are strictly regulated, and depending on the significance of the change, may require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and claims, are also subject to further FDA review and approval.
| 23 |
The FDA also may require post-marketing testing, known as Phase 4 testing, and surveillance to monitor the effects of an approved product. Discovery of previously unknown problems with a product or the failure to comply with applicable FDA requirements can have negative consequences, including adverse publicity, judicial or administrative enforcement, warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our potential products under development.
U.S. Patent Term Restoration and Marketing Exclusivity
The Biologics Price Competition and Innovation Act (the “BPCIA”) amended the PHSA to authorize the FDA to approve similar versions of innovative biologics, commonly known as biosimilars. A competitor seeking approval of a biosimilar must file an application to establish its molecule as highly similar to an approved innovator biologic, among other requirements. The BPCIA, however, bars the FDA from approving biosimilar applications for 12 years after an innovator biological product receives initial marketing approval. This 12-year period of data exclusivity may be extended by six months, for a total of 12.5 years, if the FDA requests that the innovator company conduct pediatric clinical investigations of the product.
Depending upon the timing, duration and specifics of the FDA approval of the use of our drug candidates, some of our U.S. patents, if granted, may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years, as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application. Only one patent applicable to an approved product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for one of our currently owned or licensed patent applications, if granted, to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant BLA.
Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
Employees
As of December 31, 2017, we had 28 full-time employees and two part-time employees. Substantially all of our employees are in Cambridge, Massachusetts. None of our employees are represented by a labor union or covered by a collective bargaining agreement, and we believe our relationship with our employees is good. Additionally, we utilize independent contractors and other third parties to assist with various aspects of our drug and product development.
| 24 |
We are subject to various risks that may materially harm our business, prospects, financial condition and results of operations. This discussion highlights some of the risks that may affect future operating results. These are the risks and uncertainties we believe are most important for you to consider. We cannot be certain that we will successfully address these risks. If we are unable to address these risks, our business may not grow, our stock price may suffer and we may be unable to stay in business. Additional risks and uncertainties not presently known to us, which we currently deem immaterial or which are similar to those faced by other companies in our industry or business in general, may also impair our business, prospects, results of operations and financial condition. The risks discussed below include forward-looking statements, and our actual results may differ substantially from those discussed in these forward-looking statements.
Risks Related to Our Business
We are a preclinical stage biopharmaceutical company, have no history of generating revenue, have a history of operating losses, and we may never achieve or maintain profitability.
We are a preclinical stage biopharmaceutical company. We have a limited operating history and only a preliminary business plan upon which investors may evaluate our prospects. We have never generated revenues and have a history of losses from operations. As of December 31, 2017, we had an accumulated deficit of approximately $32,820,000. Our ability to achieve revenue-generating operations and, ultimately, achieve profitability will depend on whether we can obtain additional capital when we need it, complete the development of our technology, receive regulatory approval of our planned product candidates and find strategic collaborators that can incorporate our planned products candidates into new or existing drugs which can be successfully commercialized. There can be no assurance that we will ever generate revenues or achieve profitability.
We currently do not have, and may never develop, any FDA-approved or commercialized products.
We currently do not have any products approved by the FDA or any other regulatory agency or any commercialized products and thus have never generated revenue from product sales. We have not yet sought to obtain any regulatory approvals for any planned product candidates in the United States or in any foreign market. Therefore, any estimated timing for our planned product candidates to be commercialized would be highly speculative.
To date, we have invested substantial resources in an exclusive license with Albert Einstein College of Medicine (“Einstein”) that forms the foundation for our planned product candidates and potential applications. For us to develop any products that might ultimately be commercialized, we will have to invest further time and capital in research and product development, regulatory compliance and market development. Therefore, we and our licensor, prospective business partners and other collaborators may never develop any products that can be commercialized. All of our development efforts will require substantial additional funding, none of which may result in any revenue. Our efforts may not lead to commercially successful products for a number of reasons, including:
| · | we and our licensor, prospective business partners and other collaborators may not be able to complete research regarding, and nonclinical and clinical development of, our planned product candidates; |
| · | regulatory approvals and marketing authorizations may not be achieved for our planned product candidates, or the scope of the approved indication may be narrower than sought; |
| · | we and our licensor, prospective business partners and other collaborators may experience delays in our development program, clinical trials and the regulatory approval process; |
| · | our technology may not prove to be safe and effective in clinical or preclinical trials and our planned product candidates may have adverse side effects which outweigh any potential benefit to patients; |
| · | we may not be able to identify suitable collaborators to complete development or commercialization of our potential products; |
| 25 |
| · | we may not be able to maintain, protect or expand our portfolio of intellectual property rights, including patents, trade secrets and know-how; |
| · | any future products that are ultimately approved by the FDA or other regulatory bodies may not be commercially accepted in the marketplace by physicians or patients; |
| · | our future products may not be able to be manufactured in commercial quantities or at an acceptable cost; |
| · | physicians may not receive any reimbursement from third-party payors, or the level of reimbursement may be insufficient to support widespread adoption of any of our future products; and |
| · | rapid technological change may make our technology and future products obsolete. |
Significant additional research and development and clinical testing will be required before we can potentially seek regulatory approval for or commercialize any of our product candidates.
We have product candidates in our oncology preclinical development pipeline, but significant additional research and development activity and clinical testing are required before we and our collaborators will have a chance to achieve a commercially viable product from such candidates. Our research and development efforts remain subject to all of the risks associated with the development of new biopharmaceutical products and treatments based on immune modulation. Development of the underlying technology may be affected by unanticipated technical or other problems, among other research and development issues, and the possible insufficiency of funds needed in order to complete development of these product candidates. Safety, regulatory and efficacy issues, clinical hurdles or other challenges may result in delays and cause us to incur additional expenses that would increase our losses. If we and our collaborators cannot complete, or if we experience significant delays in developing, our potential therapeutics or products for use in potential commercial applications, particularly after incurring significant expenditures, our business may fail and investors may lose the entirety of their investment.
We have no history of conducting clinical trials or commercializing biotechnology products, which may make it difficult to evaluate the prospects for our future viability.
Our operations to date have been limited to financing and staffing our company, conducting research and developing our core technologies, and identifying and optimizing our lead product clinical candidates. Although we have recruited a team that has experience with clinical trials in the United States, as a company, we have no experience conducting clinical trials in any jurisdiction and have not had previous experience commercializing product candidates or submitting an investigational new drug application (“IND”) or a Biologics License Application to the FDA or similar submissions to initiate clinical trials or obtain marketing authorization to foreign regulatory authorities. We cannot be certain that planned clinical trials will begin or be completed on time, if at all, that our planned development programs would be acceptable to the FDA or other regulatory authorities, or that, if regulatory approval is obtained, our product candidates can be successfully commercialized. Clinical trials and commercializing our product candidates will require significant additional financial and management resources, and reliance on third-party clinical investigators, contract research organizations (“CROs”), consultants and collaborators. Relying on third-party clinical investigators, CROs or collaborators may result in delays that are outside of our control.
Furthermore, we may not have the financial resources to continue development of, or to enter into collaborations for, a product candidate if we experience any problems or other unforeseen events that delay or prevent regulatory approval of, or our ability to commercialize, product candidates, including:
| · | negative or inconclusive results from our IND-enabling studies, clinical trials or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program; |
| · | delays in submitting INDs or comparable foreign applications or delays or failure in obtaining the necessary approvals from regulators to commence a clinical trial, or a suspension or termination of a clinical trial once commenced; |
| · | conditions imposed by the FDA or a foreign regulatory authority regarding the number, scope or design of our clinical trials; |
| · | delays in enrolling patients in clinical trials; |
| 26 |
| · | high drop-out rates of patients; |
| · | inadequate supply or quality of clinical trial materials or other supplies necessary to conduct our clinical trials; |
| · | greater than anticipated clinical trial costs; |
| · | poor effectiveness or unacceptable side effects of our product candidates during clinical trials; |
| · | unfavorable FDA or other regulatory agency inspection and review of a clinical trial site; |
| · | failure of our third-party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all; |
| · | serious and unexpected drug-related side effects or other safety issues experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates; |
| · | delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular; or |
| · | varying interpretations of data by the FDA and foreign regulatory authorities. |
We have never dosed any of our product candidates in humans. Our planned clinical trials or those of our collaborators may reveal significant adverse events, toxicities or other side effects not seen in our preclinical studies and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of our product candidates.
In order to obtain marketing approval for any of our product candidates, we must demonstrate the safety and efficacy of the product candidate for the relevant clinical indication or indications through preclinical studies and clinical trials as well as additional supporting data. If our product candidates are associated with undesirable side effects in preclinical studies or clinical trials or have characteristics that are unexpected, we may need to interrupt, delay or abandon their development or limit development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective.
We have not yet initiated any clinical trials or dosed any of our product candidates in humans. We have conducted various preclinical studies of our product candidates, but we do not know the predictive value of these studies for humans, and we cannot guarantee that any positive results in preclinical studies will successfully translate to human patients. It is not uncommon to observe results in human clinical trials that are unexpected based on preclinical testing, and many product candidates fail in clinical trials despite promising preclinical results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for their products. Human patients in clinical trials may suffer significant adverse events or other side effects not observed in our preclinical studies, including, but not limited to, immunogenic responses, organ toxicities such as liver, heart or kidney or other tolerability issues or possibly even death. The observed potency and kinetics of our planned product candidates in preclinical studies may not be observed in human clinical trials. If clinical trials of our planned product candidates fail to demonstrate efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our planned product candidates.
If significant adverse events or other side effects are observed in any of our future clinical trials, we may have difficulty recruiting patients to the clinical trial, patients may drop out of our trial, or we may be required to abandon the trial or our development efforts of that product candidate altogether. We, the FDA or other applicable regulatory authorities, or an Institutional Review Board (“IRB”) may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects in such trials are being exposed to unacceptable health risks or adverse side effects. Some potential therapeutics developed in the biotechnology industry that initially showed therapeutic promise in early-stage studies have later been found to cause side effects that prevented their further development. Even if the side effects do not preclude the drug from obtaining or maintaining marketing approval, undesirable side effects may inhibit market acceptance of the approved product due to its tolerability versus other therapies. Any of these developments could materially harm our business, financial condition and prospects.
| 27 |
Further, if any of our product candidates obtains marketing approval, toxicities associated with our product candidates may also develop after such approval and lead to a requirement to conduct additional clinical safety trials, additional warnings being added to the labeling, significant restrictions on the use of the product or the withdrawal of the product from the market. We cannot predict whether our product candidates will cause toxicities in humans that would preclude or lead to the revocation of regulatory approval based on preclinical studies or early stage clinical testing. However, any such event, were it to occur, would cause substantial harm to our business and financial condition and would result in the diversion of our management’s attention.
We plan to seek collaborations or strategic alliances. However, we may not be able to establish such relationships, and relationships we have established may not provide the expected benefits.
On November 14, 2017, we entered into an Exclusive Patent License and Research Collaboration Agreement (“Collaboration Agreement”) with Merck Sharpe & Dohme Corp. (“Merck”) under which Merck will partner with us in the development of our CUE Biologics™ targeting certain autoimmune diseases. Pursuant to the collaboration agreement, Merck will acquire rights to develop, commercialize and sell CUE Biologics™ relating to autoimmune disease and, in exchange for such rights, has agreed to make payments to us that include a licensing fee, milestone payments and sales royalties. This agreement does not commit Merck to a long-term relationship and it may disengage with us at any time upon 30 days’ notice.
Additionally, we plan to seek strategic alliances or collaborations with other third parties that we believe will complement or augment our development and commercialization efforts with respect to our planned product candidates and any future product candidates that we may develop. In addition, we currently do not have sales, marketing, manufacturing or distribution capabilities or arrangements. In order to commercialize our potential products, we plan to seek development and marketing partners or sublicensees to obtain necessary marketing, manufacturing and distribution capabilities.
Any of these relationships may require us to incur non-recurring and other charges, give up certain rights relating to our intellectual property and research and development activities, increase our near and long-term expenditures, issue securities that dilute our existing stockholders, issue debt which may require liens on our assets and which will increase our monthly expense obligations, or disrupt our management and business. Moreover, we may not be successful in our efforts to establish additional strategic partnerships or other alternative arrangements for our planned product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our planned product candidates as having the requisite potential to demonstrate safety and efficacy. If we are unable to establish additional strategic partnerships or other alternative arrangements to develop our drug candidates, the costs for us to independently develop our drug candidates may be higher than we currently anticipate, which could materially harm our business prospects, financial condition and results of operation.
Further, collaborations involving our planned product candidates are subject to numerous risks, which may include the following:
| · | our collaborators may have significant discretion in determining the efforts and resources that they will apply to our collaboration as compared to their other then-existing collaborations; |
| · | our collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization of our programs based on clinical trial results, changes in their strategic focus due to the acquisition of competitive products, availability of funding or other external factors, such as a business combination that diverts resources or creates competing priorities; |
| · | our collaborators may delay clinical trials, provide insufficient funding for a clinical trial, stop a clinical trial, abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product candidate for clinical testing; |
| · | our collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates; |
| 28 |
| · | a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of each of our potential products; |
| · | our collaborators may not properly maintain or defend our intellectual property rights in accordance with the terms of our contractual arrangements with them or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to other potential liability; |
| · | disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our product candidates, or that result in costly litigation or arbitration that diverts our managements’ attention and our other resources; |
| · | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; and |
| · | our collaborators may own or co-own intellectual property covering our potential products that results from our collaboration with them, and in such case, we would not have the exclusive right to commercialize such intellectual property without our collaborators’ involvement and consent. |
As a result, we may not be able to realize the benefit of collaboration agreements, strategic partnerships or licenses of our technology or potential products, which could delay our product development timelines or otherwise adversely affect our business. We also cannot be certain that, following a strategic transaction or license, we will achieve sufficient revenue or net income to justify such transaction. Any delays in entering into new collaborations or strategic partnership agreements related to our planned product candidates could delay the development and commercialization of our planned product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition, and results of operations.
Our Collaboration Agreement with Merck contains exclusivity and forbearance provisions that restrict our research and development activities.
We have granted to Merck under the Collaboration Agreement an exclusive license under certain of our patent rights, including a sublicense of patent rights licensed from Einstein, to the extent applicable to the specific CUE Biologics™ that are elected to be developed by Merck. From the effective date of the Collaboration Agreement until the earlier of (i) the first achievement of demonstration of certain biologically relevant effects for a Cue Biologic™ drug candidate (“Proof of Mechanism”) or (ii) 18 months after we notify the joint steering committee that the first product candidate has been synthesized under the research program, we are required to forebear from researching, developing or licensing to a third party rights related to any CUE Biologics™ drug candidate for the treatment of autoimmune diseases other than pursuant to the Collaboration Agreement. In addition, so long as Merck continues product development on a CUE Biologics™ drug candidate that has demonstrated Proof of Mechanism (a “Proposed Product Candidate”), we are restricted from conducting any development activities within the initial indication covered by such Proposed Product Candidate (each, an “Initial Indication”) other than pursuant to the Collaboration Agreement. These restrictions on our development activities could impact our ability to successfully develop drug candidates for the Initial Indications, which could harm our future business prospects for commercializing drugs for those Initial Indications.
We may not be successful in our efforts to identify additional product candidates. Due to our limited resources and access to capital, we must prioritize development of certain product candidates; these decisions may prove to be wrong and may adversely affect our business.
Although we intend to explore other therapeutic opportunities, in addition to the product candidates that we are currently developing, we may fail to identify successful product candidates for clinical development for a number of reasons. If we fail to identify additional potential product candidates, our business could be materially harmed.
Research programs to pursue the development of our planned product candidates for additional indications and to identify new product candidates and disease targets require substantial technical, financial and human resources whether or not they are ultimately successful. Our research programs may initially show promise in identifying potential indications and/or product candidates, yet fail to yield results for clinical development for a number of reasons, including:
| 29 |
| · | the research methodology used may not be successful in identifying potential indications and/or product candidates; |
| · | our key platform technologies, CUE Biologics™, MOD™, and viraTope™, may not adequately enable us to design, discover and validate drug candidates; |
| · | potential product candidates may, after further study, be shown to have harmful adverse effects or other characteristics that indicate they are unlikely to be effective drugs; or |
| · | it may take greater human and financial resources than we will possess to identify additional therapeutic opportunities for our product candidates or to develop suitable potential product candidates through internal research programs, thereby limiting our ability to develop, diversify and expand our drug portfolio. |
Because we have limited financial and human resources, we intend to initially focus on research programs and product candidates for a limited set of indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential or a greater likelihood of success. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities.
Accordingly, there can be no assurance that we will ever be able to identify additional therapeutic opportunities for our product candidates or to develop suitable potential product candidates through internal research programs, which could materially adversely affect our future growth and prospects. We may focus our efforts and resources on potential product candidates or other potential programs that ultimately prove to be unsuccessful.
We face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively.
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Our competitors may be able to develop other compounds or drugs that are able to achieve similar or better results than our product candidates. Our competitors may include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, and universities and other research institutions. Many of our competitors have substantially greater financial, technical and other resources than we have, such as a larger research and development staff and experienced marketing and manufacturing organizations, established relationships with CROs and other collaborators, as well as established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors, either alone or with collaborative partners, may succeed in developing, acquiring or licensing on an exclusive basis drug or biologic products that are more effective, safer, more easily commercialized or less costly than our product candidates or may develop proprietary technologies or secure patent protection and, in turn, exclude us from technologies that we may need for the development of our technologies and potential products.
In the field of immunotherapeutics, we will face significant competition from other companies, many of which have greater resources than we have. Immunotherapy technologies are advancing at a rapid pace and we anticipate competing with the largest pharmaceutical companies in the world, such as F. Hoffman-La Roche AG (Roche), Novartis A.G., Johnson & Johnson, Bristol-Myers Squibb and Merck & Co, as well as smaller biopharmaceutical companies like Acceleron Pharma, Inc., Five Prime Therapeutics, Inc., Juno Therapeutics, Inc., Kite Pharma, Inc., Apitope International N.V., Seattle Genetics, Inc., Immatics Biotechnologies GmbH, Sutro Biopharma, Inc., ImmunoGen, Inc., Zyngenia, Inc., Immunocore Limited, and Covagen A.G., which are all currently conducting research in immunotherapeutics and all of which have greater financial and human resources than we currently have.
Even if we obtain regulatory approval of any of our product candidates, we may not be the first to market and that may negatively affect the price or demand for our product candidates. Additionally, we may not be able to implement our business plan if the acceptance of our product candidates is inhibited by price competition or the reluctance of physicians to switch from existing methods of treatment to our product candidates, or if physicians switch to other new drug or biologic products or choose to reserve our product candidates for use in limited circumstances. Furthermore, a competitor could obtain orphan product exclusivity from the FDA with respect to such competitor’s product. If such competitor product is determined to be the same product as one of our product candidates, we may be prevented from obtaining approval from the FDA for such product candidate for the same indication for seven years, except in limited circumstances, and we may be subject to similar restrictions under non-U.S. regulations.
| 30 |
If we lose key management personnel, or if we fail to recruit additional highly skilled personnel, our ability to identify and develop new or next generation product candidates will be impaired, could result in loss of markets or market share and could make us less competitive.
We are highly dependent upon the principal members of our management team, including Daniel Passeri, M.Sc., our President and Chief Executive Officer, Ronald Seidel, Ph.D., our Executive VP of Research and Development, Rodolfo Chaparro, Ph.D., our Executive VP of Immunology, and other members of our scientific and clinical advisory team, including Steven Almo, Ph.D., the Chairman of our Scientific and Clinical Advisory Board. Our team has significant experience and knowledge of oncology drug discovery and development, T cell modulation, protein biochemistry and immunological assays, and the loss of any current or future team member could impair our ability to design, identify, and develop new intellectual property and product candidates and new scientific or product ideas. Additionally, if we lose the services of any of these persons, we would likely be forced to expend significant time and money in the pursuit of replacements, which may result in a delay in the development of our product candidates and the implementation of our business plan and plan of operations and diversion of our management’s attention. We can give no assurance that we could find satisfactory replacements for our current and future key scientific and management employees on terms that would not be unduly expensive or burdensome to us.
To induce valuable personnel to remain at our Company, in addition to salary and cash incentives, we have provided stock options that vest over time. Despite our efforts to retain valuable employees, members of our management, scientific and development teams may terminate their employment with us on short notice. Although we have employment agreements with our key employees, these employment agreements provide for at-will employment, which means that these employees could leave our employment at any time, for or without cause. We do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level and senior managers as well as junior, mid-level and senior scientific and medical and scientific personnel.
Our internal computer systems, or those used by third-party CROs, manufacturers or other contractors or consultants, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems and those of our future CROs, manufacturers and other contractors and consultants are vulnerable to damage from computer viruses and unauthorized access. Although to our knowledge we have not experienced any such material system failure or security breach to date, if such an event were to occur, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information (such as individually identifiable health information), we could incur significant liabilities and the further development and commercialization of our product candidates could be delayed.
Risks Related to Intellectual Property and Other Legal Matters
If we or our licensor are unable to protect our/its intellectual property, then our financial condition, results of operations and the value of our technology and potential products could be adversely affected.
Patents and other proprietary rights are essential to our business, and our ability to compete effectively is dependent upon the proprietary nature of our technologies. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop, maintain and strengthen our competitive position. We seek to protect these, in part, through confidentiality agreements with certain employees, consultants and other parties. Our success will depend in part on the ability of ourselves and our licensor(s) to obtain, to maintain (including making periodic filings and payments) and to enforce patent protection for its intellectual property, particularly those patent applications and other intellectual property to which we have secured exclusive rights. We and our licensor(s) may not successfully prosecute or continue to prosecute the patent applications which we have licensed. Even if patents are issued in respect of pending patent applications, we or our licensor(s) may fail to maintain these patents, may determine not to pursue litigation against entities that are infringing upon these patents, or may pursue such enforcement less aggressively than we ordinarily would. Without adequate protection for the intellectual property that we own or license, others may be able to offer substantially identical products for sale, which could unfavorably affect our competitive business position and harm our business prospects. Even if issued, patents may be challenged, invalidated, or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of the term of patent protection that we may have for our potential products.
| 31 |
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and potential products could be adversely affected.
In addition to our licensed technology, we rely (and will continue to rely) upon, among other things, unpatented proprietary technology, processes, trade secrets, trademarks, and know-how. Any involuntary disclosure to or misappropriation by third parties of our confidential or proprietary information could enable competitors to duplicate or surpass our technological achievements, potentially eroding our competitive position in our market. We seek to protect confidential or proprietary information in part by confidentiality agreements with our employees, consultants and third parties. While we require all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. These agreements may be terminated or breached, and we may not have adequate remedies for any such termination or breach. Furthermore, these agreements may not provide meaningful protection for our trade secrets and know-how in the event of unauthorized use or disclosure. To the extent that any of our staff was previously employed by other pharmaceutical, medical technology or biotechnology companies, those employers may allege violations of trade secrets and other similar claims in relation to their former employee’s therapeutic development activities for us. Any dispute involving such employees may result in liabilities to us.
If we fail to comply with our obligations in the agreements under which we license development or commercialization rights to products or technology from third parties, we could lose license rights that are important to our business.
We hold an exclusive license from Einstein to intellectual property relating to identification of novel immunomodulators, novel epitopes, and novel immunotherapy drugs. This license imposes various developmental milestone obligations on us. If we fail to comply with any obligations under the license agreement and fail to cure such noncompliance, Einstein will have the right to terminate the agreement and our license. The existing patent applications or future patents to which we have rights based on our agreements with Einstein may be too specific and narrowly construed to prevent third parties from developing or designing around the protection provided by these patents. Additionally, we may lose our rights to the anticipated patents and patent applications we license in the event of termination of the license agreement. There is no assurance that we will be successful in meeting all of the milestones in the future on a timely basis or that this important license agreement will not be terminated for other reasons, depriving us of significant rights. The termination of this license agreement would have a material adverse effect on our financial condition, results of operations, and prospects.
If we are unable to patent and protect the intellectual property used in our potential products, others may be able to copy our innovations, which may impair our ability to compete effectively in our markets.
The strength of our anticipated patents will involve complex legal and scientific matters and can be uncertain. As of December 31, 2017, we own or have licensed 14 pending patent applications in the United States (including 11 pending U.S. provisional patent applications), four pending international PCT applications and 34 pending foreign patent applications intended to protect the intellectual property underlying our technology. Our patent applications describe certain features of our technologies, including our CUE Biologics™ platform and specific biologic molecules and drug candidates, viraTope™, MOD™ screening, MOD™ variants and MOD™ combinations. Our anticipated patents may be challenged or fail to result in issued patents and anticipated patents may be too specific and narrowly construed to prevent third parties from developing or designing around the protections provided by our intellectual property and in that event we may lose competitive advantage and our business may suffer. Further, the patent applications that we license or have filed may fail to result in issued patents or the claims may need to be amended. Even after amendment, a patent may not issue. In that event, we may not obtain the exclusive use of the intellectual property that we seek and we may lose competitive advantage, which could result in harm to our business.
| 32 |
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, maintaining and defending patents on product candidates in all countries throughout the world could be prohibitively expensive for us, and our intellectual property rights in some non-U.S. countries can have a different scope and strength than do those in the United States. In addition, the laws of certain non-U.S. countries do not protect intellectual property rights to the same extent as U.S. federal and state laws do. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing drugs made using our inventions in and into the United States or non-U.S. jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own drugs and further, may export otherwise infringing drugs to non-U.S. jurisdictions where we have patent protection, but where enforcement rights are not as strong as those in the United States. These drugs may compete with our product candidates and our patent rights or other intellectual property rights may not be effective or adequate to prevent them from competing.
Many U.S.-based companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of some countries do not favor the enforcement of patents, trade secrets and other intellectual property, particularly those relating to biopharmaceutical products, which could make it difficult in those jurisdictions for us to stop the infringement or misappropriation of our anticipated patents or other intellectual property rights, or the marketing of competing drugs in violation of our proprietary rights. Proceedings to enforce our patent and other intellectual property rights in non-U.S. jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business.
Furthermore, such proceedings could put our anticipated patents at risk of being invalidated, held unenforceable or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke third parties to assert claims of infringement or misappropriation against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop.
Litigation or third-party claims of intellectual property infringement or challenges to the validity of our anticipated patents would require us to use resources to protect our technology and may prevent or delay our development, regulatory approval or commercialization of our product candidates.
If we are the target of claims by third parties asserting that our potential products or intellectual property infringe upon the rights of others we may be forced to incur substantial expenses or divert substantial employee resources from our business. If successful, those claims could result in our having to pay substantial damages or could prevent us from developing one or more product candidates. Further, if a patent infringement suit is brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
If we or our collaborators experience patent infringement claims, or if we elect to avoid potential claims others may be able to assert, we or our collaborators may choose to seek, or be required to seek, a license from the third party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into license agreements on acceptable terms. This could harm our business significantly. The cost to us of any litigation or other proceeding, regardless of its merit, and even if resolved in our favor, could be substantial. Some of our competitors may be able to bear the costs of such litigation or proceedings more effectively than we can because of their having greater financial and human resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Intellectual property litigation and other proceedings may, regardless of their merit, also absorb significant management time and employee resources.
| 33 |
Although we are not currently aware of any litigation or other proceedings or third-party claims of intellectual property infringement, the therapeutic industry is characterized by many suits regarding patents and other intellectual property rights. Other parties may in the future allege that our activities infringe upon their patents or that we are employing their proprietary technology without authorization. We may not have identified all the patents, patent applications or published literature that affect our business either by blocking our ability to commercialize our potential products, by preventing the patentability of one or more aspects of our potential products or those of our licensor or by covering the same or similar technologies that may affect our ability to market our potential products. In addition, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain future licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be unable to further develop and commercialize one or more of our product candidates, which could harm our business significantly.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We will face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any drugs. For example, we may be sued if our product candidates cause or are perceived to cause injury or death or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the drug, negligence, strict liability or a breach of warranties. Claims could also be asserted under state or foreign consumer protection laws. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| · | decreased demand for our potential drugs; |
| · | injury to our reputation and significant negative media attention; |
| · | withdrawal of clinical trial participants and inability to continue clinical trials; |
| · | initiation of investigations by regulators; |
| · | costs to defend the related litigation; |
| · | a diversion of management’s time and our resources; |
| · | substantial monetary awards to trial participants or patients; |
| · | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| · | loss of revenue; |
| · | financial cost; |
| · | exhaustion of any available insurance and our capital resources; and |
| · | the inability to commercialize any product candidate. |
Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of drugs we develop, alone or with collaborators. Our insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our insurance coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Even if our agreements with any future collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
| 34 |
We may be subject to securities litigation, which is expensive and could divert management attention.
The price of our common stock may be volatile, and in the past companies that have experienced volatility in the market price of their common stock have been subject to an increased incidence of securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
Risks Related to Government Regulation
We are subject to regulation in respect of our research and federal funding.
Because our licensor has conducted research under federal grants and we may conduct further research under federal grants, we will be subject to federal regulation in how we conduct our research and the agreement terms relating to those grants. There are also ethical guidelines promulgated by various governments and research institutions that we are required to follow in respect of our research. These guidelines are orientated towards research and experimentation involving humans and animals. Failure to follow the regulations, agreement terms and accepted scientific practices would jeopardize our grants and our results and the use of the results in further research and approval circumstances. Because our licensor has used federal funding, the government retains a “march-in” right in connection with these grants, which is the right to grant additional licenses to practice inventions developed from grant funding. The exercise of these “march-in” rights could result in decreased demand for our future products, which could have a material adverse effect on our results of operations and financial condition. In addition, any failure to comply with applicable laws or regulations could harm our business and divert our management’s attention.
We will be subject to stringent domestic and foreign therapeutic and drug regulation in respect of any potential products. The regulatory approval processes of the FDA and other comparable regulatory authorities outside the United States are lengthy, time-consuming and inherently unpredictable. Any unfavorable regulatory action may materially and adversely affect our future financial condition and business operations.
Our potential products, further development activities and manufacturing and distribution, once developed and determined, will be subject to extensive and rigorous regulation by numerous government agencies, including the FDA and comparable foreign agencies. To varying degrees, each of these agencies monitors and enforces our compliance with laws and regulations governing the development, testing, manufacturing, labeling, marketing, distribution, and the safety and effectiveness of our drugs. The process of obtaining marketing approval or clearance from the FDA and comparable foreign bodies for new products, or for enhancements, expansion of the indications or modifications to existing products, could:
| · | take a significant, indeterminate amount of time; |
| · | require the expenditure of substantial resources; |
| · | involve rigorous preclinical and clinical testing, and possibly post-market surveillance; |
| · | involve modifications, repairs or replacements of our potential products; |
| · | require design changes of our potential products; |
| · | result in limitations on the indicated uses of our potential products; or |
| · | result in our never being granted the regulatory approval we seek. |
| 35 |
Any of these occurrences may cause our operations or potential for success to suffer, harm our competitive standing and result in further losses that adversely affect our financial condition. We will have ongoing responsibilities under FDA and international regulations, both before and after a product is approved and commercially released. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. If the FDA were to conclude that there is non-compliance with applicable laws or regulations, or that any of our potential therapeutics are ineffective or pose an unreasonable health risk, the FDA could ban such drugs, detain or seize such drugs, order a recall, repair, replacement, or refund of purchases of such drugs, or require us to notify health professionals and others that the drugs present unreasonable risks of substantial harm to the public health. Additionally, the FDA may impose other operating restrictions, enjoin and restrain certain violations of applicable law pertaining to therapeutics and assess civil or criminal penalties against us, our officers, our employees, or our collaborative partners. The FDA has increased its scrutiny of the therapeutic industry and U.S. and foreign governments are expected to continue to scrutinize the industry closely with inspections and possibly enforcement actions by the FDA or other agencies. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively commercializing our potential products. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our financial condition and results of operations.
We may seek orphan drug status or breakthrough therapy designation for one or more of our product candidates, but even if either is granted, we may be unable to maintain any benefits associated with orphan drug status or breakthrough therapy designation, including market exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition or for which there is no reasonable expectation that the cost of developing and making available in the United States a drug or biologic for a disease or condition will be recovered from sales in the United States for that drug or biologic. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full Biologics License Application, to market the same drug or biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. In 2012, the FDA established a Breakthrough Therapy Designation which is intended to expedite the development and review of products that treat serious or life-threatening conditions.
We may seek orphan drug status for one or more of our products candidates, but the FDA may not approve any such request. Even if the FDA grants orphan drug status to one or more of our product candidates, exclusive marketing rights in the United States may be limited if we seek FDA marketing approval for an indication broader than the orphan designated indication. Additionally, any product candidate that initially receives orphan drug status designation, may lose such designation if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. In addition, we may seek breakthrough therapy designation for one or more of our product candidates, but there can be no assurance that we will receive such designation. In addition, others may obtain orphan drug status for products addressing the same diseases or conditions as products we are developing, thus limiting our ability to compete in the markets addressing such diseases or conditions for a significant period of time.
We may seek fast-track designation for our drug product candidates. Even if received, fast-track designation may not actually lead to a faster review process.
We aim to benefit from the FDA’s fast track and accelerated approval processes. However, our drug product candidates may not receive an FDA fast-track designation or priority review. Without fast-track designation, submitting a new drug application, or NDA, and getting through the regulatory process to gain marketing approval is a lengthy process. Under fast-track designation, the FDA may initiate review of sections of a fast-track drug’s NDA before the application is complete. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, the fast-track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process. Under the FDA policies, a drug candidate is eligible for priority review, or review within a six-month time frame from the time a complete NDA is accepted for filing, if the drug candidate provides a significant improvement compared to marketed drugs in the treatment, diagnosis or prevention of a disease. A fast-track designated drug candidate would ordinarily meet the FDA’s criteria for priority review.
The fast-track designation for our drug product candidates, if obtained, may not actually lead to a faster review process and a delay in the review process or in the approval of our potential products will delay revenue from their potential sales and will increase the capital necessary to fund these product development programs.
| 36 |
To obtain the necessary approval of our potential products, as a precondition, there will have to be conducted various preclinical and clinical tests, all of which will be costly and time consuming, and may not provide results that will allow us to seek regulatory approval.
The number of preclinical and clinical tests that will be required for regulatory approval varies depending on the disease or condition to be treated, the method of treatment, the nature of the drug, the jurisdiction in which approval is sought and the applicable regulations. Regulatory agencies can delay, limit or deny approval of a product for many reasons. For example, regulatory agencies may:
| · | not deem a therapeutic to be safe or effective; |
| · | interpret data from preclinical and clinical testing differently than we do; |
| · | not approve the manufacturing processes; |
| · | conclude that our drug candidate does not meet quality standards for durability, long-term reliability, biocompatibility, compatibility, or safety; and |
| · | change their approval policies or adopt new regulations. |
The FDA may make requests or suggestions regarding conduct of any clinical trials, resulting in an increased risk of difficulties or delays in obtaining regulatory approval in the United States. Foreign regulatory agencies may similarly have the ability to influence any clinical trials occurring outside the United States. Any of these occurrences could prove materially harmful to our operations and business.
Even if a potential therapeutic is ultimately approved by the various regulatory authorities, it may be approved only for narrow indications which may render it commercially less viable.
Even if a potential therapeutic of ours is approved, it may not be approved for the indications that are necessary or desirable for successful commercialization. Our preference will be to obtain as broad an indication as possible for use in connection with the particular disease and treatment for which it is designed. However, the final classification may be more limited than originally sought. The limitation on use may make the product commercially less viable and more difficult, if not impractical, to market.
Therefore, we may not obtain the revenues that we seek in respect of the proposed product, and we may not be able to become profitable and provide an investment return to our investors.
Even if we receive regulatory approval of our product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our product candidates.
Any regulatory approvals that we receive for our product candidates will require surveillance to monitor the safety and efficacy of the product candidate. The FDA or foreign regulatory agencies may also require a risk evaluation and mitigation strategy in order to approve our product candidates, which could entail requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing processes (“cGMPs”) and current good clinical practices (“cGCPs”) for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| · | restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| 37 |
| · | fines, warning letters or holds on clinical trials; |
| · | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of license approvals; |
| · | product seizure or detention, or refusal to permit the import or export of our product candidates; and |
| · | injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any approval that we may have obtained and we may not achieve or sustain profitability.
Unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives could harm our business in the future.
There is increasing pressure on biotechnology companies to reduce healthcare costs. In the United States, these pressures come from a variety of sources, such as managed care groups and institutional and government purchasers. Increased purchasing power of entities that negotiate on behalf of federal healthcare programs and private sector beneficiaries could increase pricing pressures in the future. Such pressures may also increase the risk of litigation or investigation by the government regarding pricing calculations. The biotechnology industry will likely face greater regulation and political and legal actions in the future.
Adverse pricing limitations may hinder our ability to recoup our investment in one or more future product candidates, even if our future product candidates obtain regulatory approval. Adverse pricing limitations prior to approval will also adversely affect us by reducing our commercial potential. Our ability to commercialize any potential products successfully also will depend in part on the extent to which reimbursement for these products and related treatments becomes available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels.
A significant trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and these third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize in the future and, if reimbursement is available, what the level of reimbursement will be. Reimbursement may impact the demand for, or the price of, any product for which we obtain marketing approval in the future. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize any product candidate that we successfully develop.
There may be significant delays in obtaining reimbursement for approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or regulatory authorities in other countries. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Payment rates may vary according to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower cost products that are already reimbursed and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and profitable payment rates from both government funded and private payors for future products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize potential products and our overall financial condition.
| 38 |
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
Our business operations will subject us to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also may produce hazardous waste products. We expect to generally contract with third parties for the disposal of these materials and wastes. However, we cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties. In addition, we may be required to incur substantial costs to comply with current or future environmental, health and safety laws and regulations.
These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions and we may not have sufficient (or any) insurance to cover any such costs.
Risks Related to Owning Our Common Stock, Our Financial Results and Our Need for Financing
We anticipate future losses and negative cash flow, and it is uncertain if or when we will become profitable.
We do not expect to generate any revenues until we successfully complete development of our first potential products and we are able to successfully commercialize them through sales and licensing. We have not yet demonstrated our ability to generate revenue, and we may never be able to produce revenues or operate on a profitable basis. As a result, we have incurred losses since our inception and expect to experience operating losses and negative cash flow for the foreseeable future. Our planned product candidates may never be approved or become commercially viable. Even if we and our collaborators are able to commercialize our technology, which may include licensing, we may never recover our research and development expenses.
We expect to require additional financing to support our growth and ongoing operations. Additional capital may be difficult to obtain, restrict our operations, require us to relinquish rights to our technologies or product candidates, encumber our assets and result in ongoing debt service cost, or result in additional dilution to our stockholders.
Our business will require additional capital for implementation of our long term business plan and product development and commercialization. As we require additional funds, we may seek to fund our operations through the sale of additional equity securities, debt financing and/or strategic collaboration agreements. We cannot be sure that additional financing from any of these sources will be available when needed or that, if available, the additional financing will be obtained on favorable terms.
Our future funding requirements will depend on many factors, including, but not limited to:
| · | the progress, timing, scope and costs of our clinical trials, including the ability to timely enroll patients in our planned and potential future clinical trials; |
| · | the outcome, timing and cost of regulatory approvals by the FDA and comparable regulatory authorities, including the potential that the FDA or comparable regulatory authorities may require that we perform more studies than those that we currently expect; |
| · | the number and characteristics of product candidates that we may in-license and develop; |
| · | our ability to successfully commercialize our product candidates; |
| · | the amount of sales and other revenues from product candidates that we may commercialize, if any, including the selling prices for such potential products and the availability of adequate third-party reimbursement; |
| 39 |
| · | selling and marketing costs associated with our potential products, including the cost and timing of expanding our marketing and sales capabilities; |
| · | the terms and timing of any potential future collaborations, licensing or other arrangements that we may establish; |
| · | cash requirements of any future acquisitions and/or the development of other product candidates; |
| · | the costs of operating as a public company; |
| · | the cost and timing of completion of commercial-scale, outsourced manufacturing activities; |
| · | the time and cost necessary to respond to technological and market developments; |
| · | any disputes which may occur between us and Einstein, employees, collaborators or other prospective business partners; and |
| · | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights. |
If we raise additional funds by selling shares of our common stock or other equity-linked securities, the ownership interest of our current stockholders will be diluted. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be acceptable to us. If we raise additional funds through debt financing, we may have to grant a security interest on our assets to the future lenders, our debt service costs may be substantial, and the lenders may have a preferential position in connection with any future bankruptcy or liquidation involving the company.
If we are unable to raise additional capital when needed, we may be required to curtail the development of our technology or materially curtail or reduce our operations. We could be forced to sell or dispose of our rights or assets. Any inability to raise adequate funds on commercially reasonable terms could have a material adverse effect on our business, results of operation and financial condition, including the possibility that a lack of funds could cause our business to fail and the Company to dissolve and liquidate with little or no return to investors.
We are an “emerging growth company” under the JOBS Act and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We will remain an “emerging growth company” for up to five years following the initial public offering of our common stock, although we will lose that status sooner if our revenues exceed $1 billion, if we issue more than $1 billion in non-convertible debt in a three year period, or if the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30.
Our status as an “emerging growth company” under the JOBS Act may make it more difficult to raise capital as and when we need it.
Because of the exemptions from various reporting requirements provided to us as an “emerging growth company,” we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our reporting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
| 40 |
Our stock price can be volatile and investors may have difficulty selling their shares.
Our common stock is currently listed on the Nasdaq under the symbol “CUE.” The price of our common stock may fluctuate significantly in response to market and other factors, some of which are beyond our control, including those listed in this “Item 1A. Risk Factors” section and other, unknown factors. Our stock price may also be affected by:
| · | setbacks with respect to our research and development programs; |
| · | announcements of therapeutic innovations or new products by us or our competitors; |
| · | adverse actions taken by regulatory agencies with respect to our clinical trials; |
| · | any adverse changes to our relationship with collaborators; |
| · | results of internal and external studies and clinical trials; |
| · | result of our business development efforts; |
| · | variations in the level of expenses related to our existing product candidates or preclinical and clinical development programs; |
| · | any intellectual property infringement actions in which we may become involved; |
| · | variations in our results of operations; |
| · | press reports, whether or not true, about our business; |
| · | additions to or departures of our management; |
| · | release or expiry of lock-up or other transfer restrictions on our outstanding ordinary shares or our common stock; |
| · | sales or perceived potential sales of additional ordinary shares or our common stock; |
| · | sales of our common stock by us, our executive officers and directors or our stockholders in the future; and |
| · | general economic and market conditions and overall fluctuations in the U.S. equity markets. |
Any of these factors may result in large and sudden changes in the volume and trading price of our common stock. In addition, the stock market, in general, and small pharmaceutical and biotechnology companies have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors beyond our control may negatively affect the market price of our common stock, regardless of our actual operating performance, and cause the price of our common stock to decline rapidly and unexpectedly.
If securities or industry analysts do not publish research reports about our business, or if they issue an adverse opinion about our business, the price of our securities and trading volume could decline.
The trading market for our securities is influenced by the research and reports that industry or securities analysts publish about us or our business. We do not currently have and may never obtain research coverage by securities and industry analysts. If no or few analysts commence research coverage of us, or one or more of the analysts who cover us issues an adverse opinion about our company, the price of our securities would likely decline. If one or more of these analysts ceases research coverage of us or fails to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause the price of our securities or trading volume to decline.
| 41 |
We have not paid dividends in the past and have no immediate plans to pay dividends.
We plan to reinvest all of our earnings, to the extent we have earnings, in order to further develop our technology and potential products and to cover operating costs. We do not plan to pay any cash dividends with respect to our securities in the foreseeable future. We cannot assure you that we would, at any time, generate sufficient surplus cash that would be available for distribution to the holders of our common stock as a dividend.
Concentration of ownership among our executive officers, directors and significant stockholders may prevent new investors from influencing significant corporate decisions.
All decisions with respect to the management of the company will be made by our board of directors and our officers, who beneficially own approximately 16.6% of our common stock at December 31, 2017, as calculated in accordance with Rule 13d-3 promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Such percentage includes the approximately 10.2% of our common stock beneficially owned by MDB Capital Group, LLC (“MDB”), which served as the underwriter in our initial public offering and placement agent in previous private placements of our common stock, and of which three of our seven directors are employees. As a result, management and MDB will be able to exercise a significant level of control over all matters requiring stockholder approval, including the election of directors, amendment of our certificate of incorporation and approval of significant corporate transactions. This control could have the effect of delaying or preventing a change of control of the company or changes in management, in each case, which other stockholders might find favorable, and will make the approval of certain transactions difficult without the support of these significant stockholders.
A significant portion of our total outstanding shares of common stock is restricted from immediate resale but may be sold into the market in the near future. This could cause the market price of our securities to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur in the future. These sales, or the perception in the market that the holders of a large number of securities intend to sell shares, could reduce the market price of our common stock. We currently have outstanding 19,459,194 shares of common stock. Of these outstanding shares, approximately 10,638,484 are restricted under securities laws or as a result of lock-up agreements but will be able to be resold in the near future. We also intend to register all shares of common stock that we may issue under our equity compensation plans. Once we register these shares, they can be freely sold in the public market upon issuance and once vested, subject to 180- and 365-day lock-up periods under certain lock-up agreements.
We expect to continue to incur significant costs as a result of being a public company that reports to the Securities and Exchange Commission and our management will be required to devote substantial time to meet compliance obligations.
As a public company reporting to the Securities and Exchange Commission (“SEC”), we incur significant legal, accounting and other expenses. We are subject to reporting requirements of the Exchange Act and the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), as well as rules subsequently implemented by the SEC that impose significant requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. In addition, there are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Wall Street Reform and Protection Act that increase public companies’ legal and financial compliance costs, make some activities more difficult, time-consuming or costly and may also place undue strain on personnel, systems and resources. Our management and other personnel are expected to devote a substantial amount of time to these compliance initiatives. In addition, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified people to serve on our board of directors, our board committees or as executive officers.
| 42 |
Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable.
Provisions of our amended and restated certificate of incorporation (the “Certificate of Incorporation”) and our amended and restated bylaws (the “Bylaws”) and applicable provisions of Delaware law may delay or discourage transactions involving an actual or potential change in control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. The provisions in our Certificate of Incorporation and Bylaws:
| · | authorize our board of directors to issue preferred stock without stockholder approval and to designate the rights, preferences and privileges of each class; if issued, such preferred stock would increase the number of outstanding shares of our common stock and could include terms that may deter an acquisition of us; |
| · | limit who may call stockholder meetings; |
| · | do not provide for cumulative voting rights; |
| · | provide that all vacancies may be filled only by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| · | provide that stockholders must comply with advance notice procedures with respect to stockholder proposals and the nomination of candidates for director; |
| · | provide that stockholders may only amend our Certificate of Incorporation and Bylaws upon a supermajority vote of stockholders; and |
| · | provide that the Court of Chancery of the State of Delaware will be the exclusive forum for certain legal claims. |
In addition, Section 203 of the Delaware General Corporation Law may limit our ability to engage in any business combination with a person who beneficially owns 15% or more of our outstanding voting stock unless certain conditions are satisfied. This restriction lasts for a period of three years following the share acquisition. These provisions may have the effect of entrenching our management team and may deprive you of the opportunity to sell your shares to potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price of our common stock.
Our Certificate of Incorporation provides, subject to certain exceptions, that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for certain stockholder litigation matters, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or stockholders.
Our Certificate of Incorporation provides, subject to limited exceptions, that the Court of Chancery of the State of Delaware will, to the fullest extent permitted by law, be the sole and exclusive forum for (1) any derivative action or proceeding brought on our behalf; (2) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders; (3) any action asserting a claim against us, any director or our officers and employees arising pursuant to any provision of the Delaware General Corporation Law, our Certificate of Incorporation or our Bylaws, or as to which the Delaware General Corporation Law confers exclusive jurisdiction on the Court of Chancery; or (4) any action asserting a claim against us, any director or our officers or employees that is governed by the internal affairs doctrine. Any person or entity purchasing or otherwise acquiring any interest in shares of our common stock shall be deemed to have notice of and to have consented to the provisions of our Certificate of Incorporation described above. This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or any of our directors, officers, other employees or stockholders which may discourage lawsuits with respect to such claims. Alternatively, if a court were to find the choice of forum provision that will be contained in our Certificate of Incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could materially adversely affect our business, financial condition and results of operations.
| 43 |
If we are unable to implement and maintain effective internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our securities may decrease.
Beginning with our annual report for the year ending December 31, 2018, Section 404 of the Sarbanes-Oxley Act will require that we evaluate and determine the effectiveness of our internal control over financial reporting and provide a management report on our internal control over financial reporting which following our no longer being an “emerging growth company,” must be attested to by our independent registered public accounting firm.
If we are unable to comply with the requirements of Section 404 in a timely manner, if we are unable to assert that our internal control over financial reporting is effective or, once required, provide an attestation report from our independent registered public accounting firm, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could decrease. We could also become subject to stockholder or other third-party litigation as well as investigations by the stock exchange on which our securities are listed, the SEC or other regulatory authorities, which could require additional financial and management resources and could result in fines, trading suspensions or other remedies.
| 44 |
Item 1B. Unresolved Staff Comments
Not applicable.
Our principal office is located in Cambridge, Massachusetts. We currently lease approximately 11,500 square feet of office and laboratory space at a monthly rate of $177,500 under a lease that is due to expire in April 2018. We recently leased additional laboratory space concurrent with this lease for a monthly rate of $50,000.
We have entered into a license agreement that commences May 2018 pursuant to which we will lease approximately 19,800 square feet of office and laboratory space in Cambridge, Massachusetts. We expect to use this space as our new principal executive offices and for general office, research and development, and laboratory uses. The monthly rent payments due under this license agreement will be approximately $297,500 for the first 18 months of the term and increase to approximately $388,400 for the remaining 18 months of the term, which expires on April 30, 2021.
We are not currently a party to any pending legal proceedings that we believe will have a material adverse effect on our business or financial conditions. We may, however, be subject to various claims and legal actions arising in the ordinary course of business from time to time.
Item 4. Mine Safety Disclosures
Not applicable.
| 45 |
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Price Range and Holders of Common Stock
Our shares of common stock have been listed on the Nasdaq Capital Market under the symbol “CUE” since January 2, 2018. Prior to that date, there was no public trading market for our common stock.
On March 22, 2018 the last reported sale price of our common stock was $16.47.
As of March 22, 2018, there were 203 holders of record of our common stock.
Dividend Policy
We have never paid cash dividends on our securities and we do not anticipate paying any cash dividends on our shares of common stock in the foreseeable future. We intend to retain any future earnings for reinvestment in our business. Any future determination to pay cash dividends will be at the discretion of our board of directors, and will be dependent upon our financial condition, results of operations, capital requirements and such other factors as our board of directors deems relevant.
Recent Sales of Unregistered Securities
The following represents a summary of unregistered securities issued by the Company during the time period covered by this report. The Company issued these stock options exercisable for shares of its common stock under its 2016 Omnibus Incentive Plan and 2016 Non-employee Equity Incentive Plan.
| · | Effective March 15, 2017, we granted to certain employees options to purchase an aggregate of 173,000 shares of common stock at an exercise price of $5.00 per share, each with a term of seven years and vesting over four years in eight equal semi-annual installments. |
| · | Effective April 17, 2017, we granted to Ken Pienta, our Chief Medical Officer, options to purchase 150,000 shares of our common stock at an exercise price of $5.00 per shares, with a term of seven years and vesting in annual installments over a four-year period. |
| · | Effective June 14, 2017, we granted to certain employees options to purchase an aggregate of 220,000 shares of our common stock at an exercise price of $5.00 per share, each with a term of seven years and vesting over four years in eight equal semi-annual installments. |
| · | Effective June 14, 2017, we granted to Peter Kiener, our Chairman, an option to purchase 60,000 shares of our common stock at an exercise price of $5.00 per share, with a term of seven years and vesting over five years in equal annual installments. |
| · | Effective June 14, 2017, we granted to Ulrich Weidle, a senior scientific advisor, an option to purchase 100,000 shares of our common stock at an exercise price of $5.00 per share, with a term of seven years and vesting over four years in equal annual installments. |
| · | Effective December 27, 2017, we granted to certain employees options to purchase an aggregate of 136,000 shares of common stock at an exercise price of $7.50 per share, each with a term of seven years and vesting over four years in eight equal semi-annual installments. |
All of the stock options described above were granted in reliance upon an available exemption from the registration requirements of the Securities Act, including those contained in Rule 701 promulgated under Section 3(b) of the Securities Act. Among other things, we relied on the fact that, under Rule 701, companies that are not subject to the reporting requirements of Section 13 or Section 15(d) of the Exchange Act are exempt from registration under the Securities Act with respect to certain offers and sales of securities pursuant to “compensatory benefit plans” as defined under that rule. We believe that all of the options described above were issued pursuant qualifying “compensatory benefit plans”.
| 46 |
Effective December 27, 2017, the Company granted to Colin Sandercock, its Senior Vice President, General Counsel and Secretary, under its 2016 Omnibus Incentive Plan (i) an option to purchase 150,000 shares of our common stock at an exercise price of $7.50 per share, with a term of seven years and vesting over four years in eight equal semi-annual installments and (ii) an option to purchase 100,000 shares of our common stock at an exercise price of $7.50 per share, with a term of seven years and vesting upon the achievement of certain performance goals. We intend to file a registration statement on Form S-8 to register the issuance of the shares issuable upon exercise of these options prior to any such issuance being made.
Use of Proceeds from Registered Securities
On December 14, 2017, our Registration Statement on Form S-1, as amended (File No. 333-220550), was declared effective by the SEC and our Registration Statement on Form S-1 (File No. 333-222211) became effective upon filing with SEC on December 21, 2017. These registration statements, collectively, registered a maximum aggregate $70,000,000 of our common stock in connection with our initial public offering, pursuant to which we sold 8,820,710 shares of common stock at a price to the public of $7.50 per share. The offering closed on December 27, 2017, generating net proceeds of approximately $61.9 million after deducting underwriting commissions of approximately $3.5 million and other offering expenses of $830,000 payable by us. The offering terminated before all of the securities registered on the registration statements had been sold. MDB Capital Group, LLC, of which Christopher Marlett and Anthony DiGiandomenico, are co-founders and owners, served as the underwriter for the offering. No payments were made by us, directly or indirectly, to directors, officers or persons owning ten percent or more of our common stock or to their associates, or to our affiliates, other than underwriting commission paid to MDB Capital Group and payments in the ordinary course of business to officers for salaries and to non-employee directors as compensation for board or board committee service.
There has been no material change in the planned use of proceeds from our initial public offering as described in our final prospectus filed with the SEC pursuant to Rule 424(b) under the Securities Act of 1933, as amended, on December 21, 2017.
| 47 |
Item 6. Selected Financial Data
The data set forth below should be read in conjunction with Item 7 – “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Company’s Financial Statements and notes thereto.
Statement of Operations Data:
| Year Ended | ||||||||||||
| December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Revenue | $ | - | $ | - | $ | - | ||||||
| Operating expenses: | ||||||||||||
| General and administrative | 4,333,542 | 1,970,488 | 425,081 | |||||||||
| Research and development | 18,900,328 | 5,687,847 | 1,503,649 | |||||||||
| Total operating expenses | 23,233,870 | 7,658,335 | 1,928,730 | |||||||||
| Loss from operations | (23,233,870 | ) | (7,658,335 | ) | (1,928,730 | ) | ||||||
| Interest income | 35 | 52 | - | |||||||||
| Net loss | $ | (23,233,835 | ) | $ | (7,658,283 | ) | $ | (1,928,730 | ) | |||
| Net loss per common share – basic and diluted | $ | (2.16 | ) | $ | (1.03 | ) | $ | (0.34 | ) | |||
| Weighted average common shares outstanding – basic and diluted | 10,732,449 | 7,433,433 | 5,658,282 | |||||||||
Balance Sheet Data:
| 2017 | 2016 | |||||||
| Cash | $ | 63,533,649 | $ | 14,925,820 | ||||
| Certificate of deposit | 50,068 | 50,033 | ||||||
| Working capital | 59,718,357 | 14,070,638 | ||||||
| Total assets | 66,953,834 | 16,278,617 | ||||||
| Total stockholders’ equity | 61,607,178 | 15,174,640 | ||||||
| 48 |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion of our financial condition and results of operations should be read in conjunction with the financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K.
Overview
The Company is an innovative biopharmaceutical company developing a novel and proprietary class of biologic drugs for the selective modulation of the human immune system to treat a broad range of cancers and autoimmune disorders. The Company’s corporate offices and research facilities are located in Cambridge, Massachusetts.
The Company’s product candidates are currently in preclinical development, and the Company’s activities are subject to significant risks and uncertainties. The Company has not yet commenced any revenue-generating operations, does not have any cash flows from operations, and will need to raise additional capital to its growth and ongoing business operations.
Plan of Operation
The Company’s technology is in the development phase. The Company believes that its licensed platforms have the potential for creating a robust pipeline of drug candidates addressing multiple medical indications. The Company intends to maximize the value and probability of commercialization of its CUE Biologics™ immunotherapeutics by focusing on research, testing, optimizing, conducting pilot studies, performing early stage clinical development and partnering for more extensive, later stages of clinical development, as well as seeking extensive patent protection and intellectual property development.
Since the Company is a development stage company, the majority of its business activities to date and its planned future activities will be devoted to further research and development.
A fundamental part of the Company’s corporate development strategy is to establish one or more strategic partnerships with leading pharmaceutical or biotechnology organizations that will allow the Company to more fully exploit the potential of its technology platform, such as the one with Merck described below under the heading “Collaboration Agreement with Merck.”
Critical Accounting Policies
The following discussion and analysis of financial condition and results of operations is based upon the Company’s financial statements, which have been prepared in conformity with accounting principles generally accepted in the United States of America (“GAAP”). Certain accounting policies and estimates are particularly important to the understanding of the Company’s financial position and results of operations and require the application of significant judgment by management or can be materially affected by changes from period to period in economic factors or conditions that are outside of the Company’s control. As a result, these issues are subject to an inherent degree of uncertainty. In applying these policies, management uses its judgment to determine the appropriate assumptions to be used in the determination of certain estimates. Those estimates are based on the Company’s historical operations, the future business plans and the projected financial results, the terms of existing contracts, trends in the industry, and information available from other outside sources. For a more complete description of the Company’s significant accounting policies, see Note 3 to the financial statements included in this report.
Research and Development Costs
Research and development expenses consist primarily of compensation costs, fees paid to consultants, outside service providers and organizations (including research institutes at universities), facility costs, and development and clinical trial costs with respect to the Company’s product candidates.
Research and development expenses incurred under contracts are expensed ratably over the life of the underlying contracts, unless the achievement of milestones, the completion of contracted work, or other information indicates that a different expensing schedule is more appropriate. Other research and development expenses are charged to operations as incurred. Payments made pursuant to research and development contracts are initially recorded as research and development contract advances in the Company’s balance sheet and then charged to research and development expenses in the Company’s statement of operations as those contract services are performed. Expenses incurred under research and development contracts in excess of amounts advanced are recorded as research and development contract liabilities in the Company’s balance sheet, with a corresponding charge to research and development expenses in the Company’s statement of operations.
| 49 |
Nonrefundable advance payments for future research and development activities pursuant to an executory contractual arrangement are recorded as advances as described above. Nonrefundable advance payments are recognized as an expense as the related services are performed. The Company evaluates whether it expects the services to be rendered at each quarter end and year end reporting date. If the Company does not expect the services to be rendered, the advance payment is charged to expense. To the extent that a nonrefundable advance payment is for contracted services to be performed within 12 months from the reporting date, such advance is included in current assets; otherwise, such advance is included in non-current assets.
The Company evaluates the status of its research and development agreements and contracts, and the carrying amount of the related assets and liabilities, at each quarter end and year end reporting date, and adjusts the carrying amounts and their classification on the balance sheet as appropriate.
Research and Development Funding Arrangements
The Company’s proprietary biologics are at an early stage and will require substantial time and funding to continue development. There can be no assurances that any of the Company’s biologics will ultimately become commercially viable product candidates. In order to finance its research and development programs, the Company may periodically enter into collaboration agreements with third parties that provide funding for certain aspects of the Company’s ongoing research and development activities. The Company considers various factors in determining the appropriate accounting treatment for such collaboration agreements, including, among others, the risks of and costs associated with the research and development program being funded, the stage of development of the proprietary biologics subject to the research and development program, the likelihood at initiation that the collaboration arrangement will result in an economically successful outcome to the third party, the continuing involvement of the Company in the research and development program and the expenditure of the funds, the transfer of the financial risk associated with the research and development program to the third party, the intended use of the funds and any restrictions thereon, and the probability of any repayment obligations or other forms of consideration if the proprietary biologics subject to the research and development program are not successfully developed and commercialized.
In accordance with ASC 730-20-25-8, to the extent that a collaboration agreement results in a substantive and genuine transfer of financial risk to a third party funding source because any economic benefit that the third party may receive depends solely on the research and development program successfully developing commercially viable product candidates having future economic benefit (which is uncertain at the initiation of the collaboration agreement), the Company will account for such collaboration agreement as a contract to perform research and development services for a third party. The funds received from the third party under such a collaboration agreement will initially be recorded as a deferred credit in the Company’s balance sheet. As the related contractual research and development costs are incurred, the applicable amount of the deferred credit will be credited to operations and will be classified as an offset to such research and development costs in the Company’s statement of operations.
Patent Expenses
The Company is the exclusive worldwide licensee of, and has patent applications pending for, numerous domestic and foreign patents. Due to the significant uncertainty associated with the successful development of one or more commercially viable product candidates based on the Company’s research efforts and any related patent applications, all patent costs, including patent-related legal fees, filing fees and other costs are charged to operations as incurred. Patent expenses are included in general and administrative expenses in the Company’s statement of operations.
Reclassification
Certain comparative figures for the years ended 2016 and 2015 have been adjusted to correct the classification of patent legal costs and intellectual property management fees from research and development expenses to general and administrative expenses. During the years ended December 31, 2016 and 2015, patent legal costs of $366,131 and $178,697, and intellectual property management fees of $90,000 and $38,182, aggregating $456,131 and $216,879, respectively, were reclassified from research and development costs to general and administrative costs. These changes did not impact loss from operations or net loss.
| 50 |
Licensing Fees and Costs
Licensing fees and costs consist primarily of costs relating to the acquisition of the Company’s license agreement with the Albert Einstein College of Medicine, a division of Yeshiva University (“Einstein”), including related royalties, maintenance fees, milestone payments and product development costs. Licensing fees and costs are charged to operations as incurred.
Stock-Based Compensation
The Company periodically issues stock options to officers, directors, employees, Scientific and Clinical Advisory Board members, non-employees and consultants for services rendered. Such issuances vest and expire according to terms established at the issuance date.
Stock-based payments to officers, directors and employees, including grants of employee stock options, are recognized in the financial statements based on their grant date fair values. Stock option grants, which are generally time-vested, are measured at the grant date fair value and charged to operations on a straight-line basis over the vesting period. The fair value of stock options is determined utilizing the Black-Scholes option-pricing model, which is affected by several variables, including the risk-free interest rate, the expected dividend yield, the life of the equity award, the exercise price of the stock option as compared to the fair value of the common stock on the grant date, and the estimated volatility of the common stock over the term of the equity award.1
Stock options granted to members of the Company’s Scientific and Clinical Advisory Board, non-employees and outside advisors and consultants are revalued each reporting period to determine the amount to be recorded as an expense in the respective period. As the stock options vest, they are valued on each vesting date and an adjustment is recorded for the difference between the value already recorded and the value on the date of vesting.
The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. Until the Company has established a trading market for its common stock, estimated volatility is based on the average historical volatilities of comparable public companies in a similar industry. The expected dividend yield is based on the current yield at the grant date; the Company has never declared or paid dividends and has no plans to do so for the foreseeable future. As permitted by Staff Accounting Bulletin No. 107, due to the Company’s lack of history and option activity, management utilizes the simplified method to estimate the expected term of options at the date of grant. The fair value of common stock is determined by reference to either recent or anticipated cash transactions involving the sale of the Company’s common stock.
The Company recognizes the fair value of stock-based compensation in general and administrative expenses and in research and development expenses in the Company’s statement of operations, depending on the type of services provided by the recipient of the equity award. The Company issues new shares of common stock to satisfy stock option exercises.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company accounts for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by GAAP. The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized.
| 51 |
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
The Company is subject to U.S. Federal and Massachusetts state income taxes. As the Company’s net operating losses have yet to be utilized, all previous tax years remain open to examination by Federal and state taxing authorities in which the Company currently operates.
The Company recognizes interest accrued relative to unrecognized tax benefits in interest expense and penalties in operating expense.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”). ASU 2014-09 will eliminate transaction- and industry-specific revenue recognition guidance under current GAAP and replace it with a principle based approach for determining revenue recognition. ASU 2014-09 will require that companies recognize revenue based on the value of transferred goods or services as they occur in the contract. ASU 2014-09 also will require additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. The FASB has issued ASU 2016-08, ASU 2016-10, ASU 2016-11, ASU 2016-12, and ASU 2016-20, all of which clarify certain implementation guidance within ASU 2014-09. ASU 2014-09 is effective for reporting periods beginning after December 15, 2017, with early adoption permitted. Entities will be able to transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. The Company will adopt the provisions of ASU 2014-09 in the quarter beginning January 1, 2018. As the Company is unlikely to generate any sustainable operating revenues in the next several years, the adoption of ASU 2014-09 is not currently expected to have any impact on the Company’s financial statement presentation or disclosures.
In November 2015, the FASB issued Accounting Standards Update No. 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes (“ASU 2015-17”). ASU 2015-17 requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. ASU 2015-17 was effective for financial statements issued for annual periods beginning after December 15, 2016, and interim periods within those annual periods. Earlier application is permitted as of the beginning of an interim or annual reporting period. The Company adopted the provisions of ASU 2015-17 in the quarter beginning January 1, 2017. The adoption of ASU 2015-17 did not have any impact on Company’s financial statement presentation or disclosures.
In February 2016, the FASB issued Accounting Standards Update No. 2016-02, Leases (Topic 842) (“ASU 2016-02”). ASU 2016-02 requires a lessee to record a right-of-use asset and a corresponding lease liability, initially measured at the present value of the lease payments, on the balance sheet for all leases with terms longer than 12 months, as well as the disclosure of key information about leasing arrangements. ASU 2016-02 requires recognition in the statement of operations of a single lease cost, calculated so that the cost of the lease is allocated over the lease term, generally on a straight-line basis. ASU 2016-02 requires classification of all cash payments within operating activities in the statement of cash flows. Disclosures are required to provide the amount, timing and uncertainty of cash flows arising from leases. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early application is permitted. The Company will adopt the provisions of ASU 2016-02 in the quarter beginning January 1, 2019. The Company generally does not finance purchases of property and equipment, but does lease its operating facilities. While the Company is continuing to assess the potential impact of ASU 2016-02, it currently expects that most of its lease commitments will be subject to ASU 2016-02 and accordingly, upon adoption will be recognized as lease liabilities and right-of-use assets in the Company’s balance sheet.
In March 2016, the FASB issued Accounting Standards Update No. 2016-09, Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting (“ASU 2016-09”). ASU 2016-09 requires, among other things, that all income tax effects of awards be recognized in the statement of operations when the awards vest or are settled. ASU 2016-09 also allows for an employer to repurchase more of an employee’s shares than it can today for tax withholding purposes without triggering liability accounting and allows for a policy election to account for forfeitures as they occur. ASU 2016-09 was effective for fiscal years beginning after December 15, 2016, including interim periods within those fiscal years. Early adoption is permitted for any entity in any interim or annual period. The Company adopted the provisions of ASU 2016-09 in the quarter beginning January 1, 2017. The adoption of ASU 2016-09 did not have any impact on the Company’s financial statement presentation or disclosures.
| 52 |
In July 2017, the FASB issued Accounting Standards Update No. 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features; (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception (“ASU 2017-11”). ASU 2017-11 allows companies to exclude a down round feature when determining whether a financial instrument (or embedded conversion feature) is considered indexed to the entity’s own stock. As a result, financial instruments (or embedded conversion features) with down round features may no longer be required to be accounted for as derivative liabilities. A company will recognize the value of a down round feature only when it is triggered and the strike price has been adjusted downward. For equity-classified freestanding financial instruments, an entity will treat the value of the effect of the down round as a dividend and a reduction of income available to common shareholders in computing basic earnings per share. For convertible instruments with embedded conversion features containing down round provisions, entities will recognize the value of the down round as a beneficial conversion discount to be amortized to earnings. ASU 2017-11 is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted. The guidance in ASU 2017-11 can be applied using a full or modified retrospective approach. The adoption of ASU 2017-11 is not currently expected to have any impact on the Company’s financial statement presentation or disclosures.
Management does not believe that any other recently issued, but not yet effective, authoritative guidance, if currently adopted, would have a material impact on the Company’s financial statement presentation or disclosures.
Acquisition of Trademark
On May 4, 2017, the Company entered into a settlement agreement with Cue BioLogics, LLC, an unrelated party, to acquire all right, title and interest in and to the CUE BIOLOGICS mark, and any derivative mark incorporating CUE, throughout the world, together with all associated goodwill and common law rights appurtenant thereto, including, but not limited to, any right, title and interest in any corporate name, company name, business name, trade name, dba, domain name, or other source identifier incorporating CUE (collectively, the “CUE BIOLOGICS Mark”), in exchange for a cash payment by the Company of $175,000.
Accounting Standards Codification (“ASC”) 350-30-20 defines a defensive intangible asset as an acquired intangible asset in a situation in which an entity does not intend to actively use the asset but intends to hold (lock up) the asset to prevent others from obtaining access to the asset. The Company determined that the acquired intangible asset met the definition of a defensive intangible asset and has therefore accounted for the $175,000 payment to Cue BioLogics, LLC for the CUE BIOLOGICS Mark as an acquired intangible asset.
As the Company can renew the underlying rights to the CUE BIOLOGICS trademark indefinitely at nominal cost, this acquired intangible asset has been classified as a non-amortizable intangible asset in Company’s balance sheet at December 31, 2017. The Company evaluates the status of this intangible asset for amortization and impairment at each quarter end and year end reporting date.
Significant Contracts and Agreements Related to Research and Development Activities
License Agreement
On January 14, 2015, the Company entered into a license agreement, as amended and restated on July 31, 2017 (the “Einstein License”), with Einstein for certain patent rights (the “Patents”) relating to the Company’s core technology platform for the engineering of biologics to control T cell activity, precision, immune-modulatory drug candidates, and two supporting technologies that enable the discovery of costimulatory signaling molecules (ligands) and T cell targeting peptides.
| 53 |
The Company holds an exclusive worldwide license, with the right to sublicense, import, make, have made, use, provide, offer to sell, and sell all products, processes and services that use the patents covered by the Einstein License, including certain technology received from Einstein related thereto (the “Licensed Products”). Under the Einstein License, the Company is required to:
| · | Pay royalties based on certain percentage of proceeds, as defined in the Einstein License, from sales of Licensed Products, including sublicense agreements. |
| · | Pay escalating annual maintenance fees, which are non-refundable, but are creditable against the amount due to Einstein for royalties. |
| · | Make significant payments based upon the achievement of certain milestones, as defined in the Einstein License. At December 31, 2017, none of these milestones had been achieved by the Company. |
| · | Incur minimum product development costs per year until the first commercial sale of the first Licensed Product. |
The Company was in compliance with its obligations under the Einstein License at December 31, 2017 and 2016.
The Einstein License expires upon the expiration of the last obligation to make royalty payments to Einstein which may be due with respect to certain Licensed Products, unless terminated earlier under the provisions thereof. The Einstein License includes certain termination provisions if the Company fails to meet its obligations thereunder.
The Company accounts for the costs incurred in connection with the Einstein License in accordance with ASC 730, Research and Development. For the years ended December 31, 2017, 2016, and 2015, costs incurred with respect to the Einstein License aggregated $5,084,708, $31,250, and $127,336 respectively. Such costs are included in research and development costs in the statements of operations.
Pursuant to the Einstein License, the Company issued to Einstein 671,572 shares of common stock of the Company in connection with the consummation of the initial public offering of its common stock on December 27, 2017. Under the Einstein License, the Company must also use its best efforts to file a registration statement covering the resale of the 671,572 shares issued to Einstein no later than 180 days after the consummation of such offering.
The Company accounted for the issuance of these shares in accordance with ASC 730, Research and Development, as a charge to research and development expenses in the statements of operations at their aggregate fair value of $5,036,789, as the Patents acquired from Einstein are for use in the Company’s research and development activities exclusively with respect to its core technology platform, have no alternative future use by the Company, and therefore no separate economic value.
Service Agreement
On October 1, 2015, the Company entered into a service agreement (the “Service Agreement”) with Einstein to support the Company’s ongoing research and development activities. The initial term of the Service Agreement was for three months, which was amended in February 2016 to extend it for the period of time deemed necessary to complete the services pursuant to the terms of the Service Agreement. For the years ended December 31, 2017, 2016, and 2015, costs incurred with respect to the Service Agreement aggregated $0, $80,000, and $200,000, respectively. Such costs are included in research and development expenses in the statements of operations.
Agreements with Catalent Pharma Solutions, LLC
Catalent Pharma Solutions, LLC (“Catalent”) is a global provider of drug delivery technology and development solutions for drugs, biologics and consumer health products.
On March 7, 2017, the Company entered into an agreement with Catalent for Catalent to provide services on a sequential milestone basis with respect to the development and manufacture of the Company’s lead drug candidate, CUE-101. The services under the agreement are designed to support the preparation and filing of an Investigational New Drug Application with the United States Food and Drug Administration to allow for the commencement of a Phase 1 clinical trial of CUE-101 in the United States. The Company incurred total direct costs under this agreement aggregating $2.4 million during the year ended December 31, 2017 and currently estimates that it will incur an additional $2.4 of such costs during the year ended December 31, 2018. The Company expects that certain of these payments will consist of nonrefundable advance payments for which the Company anticipates receiving the contracted services within 12 months from the date of payment. Management periodically reviews and updates the project’s estimated budget and timeline.
| 54 |
On July 5, 2017, the Company entered into a separate Master Services Agreement with Catalent that outlines the terms and conditions under which Catalent will provide contract services with respect to the Company’s research and development activities for a period of five years. The Company may terminate this agreement without cause upon 90 days’ prior written notice. Unless and until terminated, this agreement will automatically be extended for successive one-year periods.
With respect to the $2.4 million of total direct costs the Company had incurred as of December 31, 2017, $1.6 million was charged to research and development expenses in the statement of operations for the year ended December 31, 2017, representing 8.4% of research and development expenses for such period. The remaining $0.8 million of such costs estimated to be incurred is reflected as research and development contract advances in the condensed balance sheet at December 31, 2017. The Company expects to receive the services related to such advance payments by May 31, 2018. Accordingly, advance payments at December 31, 2017 are classified as a current asset and are expected to be charged to research and development expenses in the statement of operations through June 30, 2018.
Collaboration Agreement with Merck
On November 14, 2017, we entered into an Exclusive Patent License and Research Collaboration Agreement (the “Collaboration Agreement”) with Merck Sharp & Dohme Corp. (“Merck”) for a partnership to research and develop certain of our proprietary biologics that target certain autoimmune disease indications (the “Initial Indications”). We view this Collaboration Agreement as a component of our development strategy since it will allow us to advance our autoimmune programs in partnership with a world class pharmaceutical company, while also continuing our focus on our more advanced cancer programs. The research program outlined in the Collaboration Agreement entails (1) our research, discovery and development of certain CUE Biologics™ drug candidates up to the point of demonstration of certain biologically relevant effects (“Proof of Mechanism”) and (2) the further development by Merck of the CUE Biologics™ drug candidates that have demonstrated Proof of Mechanism (the “Proposed Product Candidates”) up to the point of demonstration of all or substantially all of the properties outlined in such Proposed Product Candidates’ profiles as described in the Collaboration Agreement.
For the purposes of this collaboration, we have granted to Merck under the Collaboration Agreement an exclusive license under certain of our patent rights, including a sublicense of patent rights licensed from Einstein, to the extent applicable to the specific CUE Biologics™ that are elected to be developed by Merck. From the effective date of the Collaboration Agreement until the earlier of (i) the first achievement of Proof of Mechanism for a CUE Biologics™ drug candidate or (ii) 18 months after we notify the joint steering committee that the first Product Candidate has been synthesized under the research program, we are required to forebear from researching, developing or licensing to a third party rights related to any CUE Biologics™ drug candidate for the treatment of autoimmune diseases other than pursuant to the Collaboration Agreement. In addition, so long as Merck continues product development on a Proposed Product Candidate, we are restricted from conducting any development activities within the Initial Indication covered by such Proposed Product Candidate other than pursuant to the Collaboration Agreement. The Company is not required to forbear at any time, however, from developing other CUE Biologics™ for use in therapeutic areas other than autoimmune diseases, e.g., for use in treating cancer or infectious diseases.
In exchange for the licenses and other rights granted to Merck under the Collaboration Agreement, Merck paid to us a $2.5 million nonrefundable up-front payment. Additionally, we may be eligible to receive funding in developmental milestone payments, as well as tiered royalties, if all research, development, regulatory and commercial milestones agreed upon by both parties are successfully achieved. Excluding the up-front payment described above, we are eligible to earn up to $101 million for the achievement of certain research and development milestones, $120 million for the achievement of certain regulatory milestones and $150 million for the achievement of certain commercial milestones, in addition to tiered royalties on sales, if all pre-specified milestones associated with multiple products across the primary disease indication areas are achieved. The Collaboration Agreement requires us to use the first $2.7 million of milestone payments we receive under the agreement to fund contract research. The amount of the royalty payments is a percentage of product sales ranging in the single digits based on the amount of such sales.
| 55 |
The term of the Collaboration Agreement extends until the expiration of all royalty obligations following a product candidate’s receipt of marketing authorization, at which point Merck’s licenses and sublicenses granted under the agreement shall become fully paid-up, perpetual licenses and sublicenses, as applicable. Royalties on each product subject to the Collaboration Agreement shall continue on a country-by-country basis until the expiration of the later of: (1) the last-to-expire patent claiming the compound on which such product is based and (2) a period of ten years after the first commercial sale of such product in such country.
Notwithstanding the foregoing, Merck may terminate the Collaboration Agreement at any time upon 30 days’ notice to the Company. The Collaboration Agreement may also be terminated by either party if the other party is in breach of its obligations thereunder and fails to cure such breach within 90 days after notice or by either party if the other party files for bankruptcy or other similar insolvency proceedings.
Results of Operations
Operating Expenses
The Company generally recognizes operating expenses as they are incurred in two general categories, general and administrative expenses and research and development expenses. The Company’s operating expenses also include non-cash components related to depreciation and amortization of property and equipment and stock-based compensation, which are allocated, as appropriate, to general and administrative expenses and research and development expenses.
General and administrative expenses consist of salaries and related expenses for executive, legal, finance, human resources, information technology and administrative personnel, as well as professional fees, insurance costs, and other general corporate expenses. Management expects general and administrative expenses to increase in future periods as the Company adds personnel and incurs additional expenses related to an expansion of its research and development activities and its operation as a public company, including higher legal, accounting, insurance, compliance, compensation and other expenses.
Research and development expenses consist primarily of compensation expenses, fees paid to consultants, outside service providers and organizations (including research institutes at universities), facility expenses, and development and clinical trial expenses with respect to the Company’s product candidates. The Company charges research and development expenses to operations as they are incurred. Management expects research and development expenses to increase in the future as the Company increases its efforts to develop technology for potential future products based on its technology and research.
Years Ended December 31, 2017, 2016 and 2015
The Company’s statements of operations for the years ended December 31, 2017, 2016, and 2015 as discussed herein are presented below.
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Revenue | $ | — | $ | — | $ | — | ||||||
| Operating expenses: | ||||||||||||
| General and administrative | 4,333,542 | 1,970,488 | 425,081 | |||||||||
| Research and development | 18,900,328 | 5,687,847 | 1,503,649 | |||||||||
| Total operating expenses | 23,233,870 | 7,658,335 | 1,928,730 | |||||||||
| Loss from operations | (23,233,870 | ) | (7,658,335 | ) | (1,928,730 | ) | ||||||
| Interest income | 35 | 52 | — | |||||||||
| Net loss | $ | (23,233,835 | ) | $ | (7,658,283 | ) | $ | (1,928,730 | ) | |||
| 56 |
General and Administrative
General and administrative expenses totaled $4,333,542 and $1,970,488 for the years ended December 31, 2017 and 2016, respectively. This increase of $2,363,054 was due primarily to the growth of the Company and its activities. We expect our general and administrative expenses to continue to increase as we expand our operations. General and administrative expenses for the year ended December 31, 2017 consisted of expenses related to employee and board compensation of $1,075,099, stock based compensation of $1,022,962, professional and consulting fees of $1,194,552, rent of $244,678, depreciation and amortization of $15,163, travel of $176,585, and other expenses of $604,503. General and administrative expenses for the year ended December 31, 2016 included expenses related to employee and board compensation of $368,711, professional and consulting fees of $768,586, rent of $117,691, depreciation and amortization of $4,630, stock-based compensation of $439,366, investor relations of $88,629, travel of $63,746, and other expenses of $119,129.
General and administrative expenses totaled $1,970,488 and $425,081 for the years ended December 31, 2016 and 2015, respectively. This increase of $1,545,407 was due primarily to the growth of the Company and its activities. General and administrative expenses for the year ended December 31, 2016 included expenses related to employee and board compensation of $368,711, professional and consulting fees of $768,586, rent of $117,691, depreciation and amortization of $4,630, stock-based compensation of $439,366, investor relations of $88,629, travel of $63,746, and other expenses of $119,129. General and administrative expenses for the year ended December 31, 2015 consisted of expenses related to employee and board compensation of $42,000, professional and consulting fees of $288,156, rent of $42,455, depreciation and amortization of $1,238, travel of $9,948, and other expenses of $41,284.
Research and Development
Research and development expenses totaled $18,900,328 and $5,687,847 for the years ended December 31, 2017 and 2016, respectively. This increase of $13,212,481 was due primarily to the growth of the Company and its activities. We expect our research and development expenses to continue to increase as we expand our development activities. Research and development expenses for the year ended December 31, 2017 included expenses related to employee and Scientific and Clinical Advisory Board compensation of $3,014,833, stock-based compensation of $1,752,401 depreciation and amortization of $404,405, research and laboratory expenses of $6,213,721, rent of $1,730,911, licensing fees of $5,136,118, and other expenses of $647,939. Research and development expenses for the year ended December 31, 2016 included expenses related to employee and Scientific and Clinical Advisory Board compensation of $1,330,625, depreciation and amortization of $197,455, stock-based compensation of $450,190, research and laboratory expenses of $2,651,771, rent of $846,818, licensing fees of $69,465, other professional fees of $103,407, and other expenses of $38,116.
Research and development expenses totaled $5,687,847 and $1,503,649 for the years ended December 31, 2016 and 2015, respectively. This increase of $4,184,198 was due primarily to the growth of the Company and its activities. Research and development expenses for the year ended December 31, 2016 included expenses related to employee and Scientific and Clinical Advisory Board compensation of $1,330,625, depreciation and amortization of $197,455, stock-based compensation of $450,190, research and laboratory expenses of $2,651,771, rent of $846,818, licensing fees of $69,465, other professional fees of $103,407, and other expenses of $38,116. Research and development expenses for the year ended December 31, 2015 included expenses related to employee and Scientific and Clinical Advisory Board compensation of $415,683, depreciation and amortization of $43,577, research and laboratory expenses of $628,198, rent of $319,091, licensing fees of $94,167, and other expenses of $2,933.
Loss from Operations
The Company’s loss from operations was $23,233,870 for the year ended December 31, 2017, as compared to $7,658,335 for the year ended December 31, 2016.
The Company’s loss from operations was $7,658,335 for the year ended December 31, 2016, as compared to $1,928,730 for the year ended December 31, 2015.
| 57 |
Interest Income
Interest income was $35 for the year ended December 31, 2017, as compared to $52 for the year ended December 31, 2016.
Net Loss
As a result of the foregoing, the Company’s net loss was $23,233,835 for the year ended December 31, 2017, as compared to $7,658,283 for the year ended December 31, 2016.
The Company’s net loss was $7,658,283 for the year ended December 31, 2016, as compared to $1,928,730 for the year ended December 31, 2015.
Liquidity and Capital Resources—December 31, 2017 and December 31, 2016
The Company has financed its working capital requirements primarily through private and public offerings of equity securities and cash received in December 2017 from Merck in connection with the Collaboration Agreement. At December 31, 2017 and December 31, 2016, the Company had cash and a certificate of deposit totaling $63,583,717 and $14,975,853, respectively, available to fund the Company’s ongoing business activities. Additional information concerning the Company’s financial condition and results of operations is provided in the financial statements included in this report.
The amounts that the Company actually spends for any specific purpose may vary significantly and will depend on a number of factors, including, but not limited to, the Company’s research and development activities and programs, clinical testing, regulatory approval, market conditions, and changes in or revisions to the Company’s business strategy and technology development plans. Investors will be relying on the judgment of the Company’s management regarding the application of the proceeds from the sale of the Company’s common stock.
The Company believes that its existing cash resources will be sufficient to fund the Company’s projected operating requirements for at least the next 12 months based on current operating plans. Until the Company is able to generate sustainable revenues that generate operating profitability and positive operating cash flows, the Company expects to finance its future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. However, there can be no assurances that the Company will be able to obtain additional financing on acceptable terms and in the amounts necessary to fully fund its future operating requirements, if at all. If the Company is unable to obtain sufficient cash resources to fund its operations, the Company may be forced to reduce or discontinue its operations entirely.
If the Company issues additional equity securities to raise funds, the ownership percentage of the Company’s existing stockholders would be reduced. New investors may demand rights, preferences or privileges senior to those of existing holders of the Company’s common stock. If the Company issues debt securities, the Company may be required to grant security interests in its assets, could have substantial debt service obligations, and lenders may have a senior position (compared to stockholders) in any potential future bankruptcy or liquidation of the Company. Additionally, corporate collaboration and licensing arrangements may require us to incur non-recurring and other charges, give up certain rights relating to our intellectual property and research and development activities, increase our near and long-term expenditures, issue securities that dilute our existing stockholders, issue debt which may require liens on our assets and which will increase our monthly expense obligations, or disrupt our management and business.
Operating Activities
During the year ended December 31, 2017, the Company used cash of $12,191,514 in operating activities, as compared to $5,968,411 in operating activities during the year ended December 31, 2016. The difference between cash used in operating activities and net loss consisted primarily of depreciation and amortization, stock-based compensation, and changes in operating assets and liabilities, and $5,036,789 attributable to the issuance of common stock to Albert Einstein University pursuant to the Einstein license agreement.
| 58 |
Investing Activities
During the year ended December 31, 2017, the Company used cash of $1,204,977 in investing activities, consisting of $1,029,942 for the purchase of office and laboratory equipment, $175,000 for the purchase of the CUE BIOLOGICS Mark and $35 with respect to interest earned on the certificate of deposit. During the year ended December 31, 2016, the Company used cash of $516,012 in investing activities, consisting of $515,979 for the purchase of office and laboratory equipment and $33 with respect to interest earned on the certificate of deposit.
Financing Activities
During the year ended December 31, 2017, the Company generated cash from financing activities of $62,004,320, consisting of the net proceeds from the Company’s initial public offering and $61,995,312, $8,008 from the exercise of stock options, $1,000 from the sale of underwriters warrants. During the year ended December 31, 2016, the Company generated cash from financing activities of $15,005,036, consisting of the net proceeds from the December 2016 common stock private placement.
Principal Commitments
Leased Facilities
On July 29, 2015, the Company entered into an operating lease agreement for its laboratory space for the period from August 1, 2015 through April 30, 2018. The lease contains escalating payments during the lease period. The Company records monthly rent expense on the straight-line basis, equal to the total of the lease payments over the lease term divided by the number of months of the lease term.
On July 30, 2015, the Company entered into an operating lease agreement, as amended, for dedicated vivarium space for the period from August 1, 2015 through March 31, 2018.
On November 14, 2016, June 28, 2017, and January 16, 2018, the Company entered into amendments to the operating lease agreement that each provided the Company with additional laboratory space. These amendments were effective beginning December 1, 2016 and July 1, 2017, and January 16, 2018, respectively, and continue through the expiration of the lease on April 30, 2018.
On January 16, 2018, the Company entered into an amended lease agreement that provided the Company with additional laboratory space. This amendment was effective beginning on January 16, 2018 and continues through the expiration of the lease on April 30, 2018.
On January 22, 2018, the Company entered into an operating lease agreement for laboratory space to commence following the expiration of is current lease agreement described above with a term continuing until April 30, 2021. The monthly rental rate under the lease agreement is $297,495 for the first 18 months and $388,396 for the remainder of the term. Pursuant to the terms of the lease agreement, the Company prepaid three months’ of rent payments upon entering into the lease agreement.
Einstein License Agreement and Einstein Service Agreement
The Company’s commitments with respect to the Einstein License and the Service Agreement are summarized above at “Significant Contracts and Agreements Related to Research and Development Activities”.
Agreements with Catalent
The Company’s commitments with respect to its agreements with Catalent are summarized above at “Significant Contracts and Agreements Related to Research and Development Activities”.
Contractual Commitments and Other Commitments
The following table sets forth the Company’s estimated fixed obligations and commitments to make future payments under existing contracts at December 31, 2017. This table excludes potential milestone and royalty payments due under our Einstein License.
| 59 |
| Payments Due by Period | ||||||||||||||||||||
| Less Than | More Than | |||||||||||||||||||
| Description | Total | One Year | 1 - 3 Years | 3 - 5 Years | 5 Years | |||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Operating lease obligations | 1,004,000 | 1,004,000 | - | - | - | |||||||||||||||
| Total | $ | 1,004,000 | $ | 1,004,000 | $ | - | $ | - | $ | - | ||||||||||
Off-Balance Sheet Transactions
At December 31, 2017, the Company did not have any transactions, obligations or relationships that could be considered off-balance sheet arrangements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Market risk represents the risk of loss that may result from the change in value of financial instruments due to fluctuations in their market price. Market risk is inherent in all financial instruments. The primary quantifiable market risk associated with our financial instruments is sensitivity to changes in interest rates. Interest rate risk represents the potential loss from adverse changes in market interest rates. The primary objective of our investment activities is to preserve principal while maximizing our income from investments and minimizing our market risk. As of December 31, 2017, our portfolio of financial instruments consisted of cash and certificates of deposit. Due to the short term nature of these financial instruments, we believe there is no material exposure to interest rate risk, and/or credit risk, arising from our portfolio of financial instruments.
Our assets and liabilities are denominated in U.S. dollars. Consequently, we have not considered it necessary to use foreign currency contracts or other derivative instruments to manage changes in currency rates. We do not now, nor do we plan to, use derivative financial instruments for speculative or trading purposes. However, these circumstances might change.
Item 8. Financial Statements and Supplementary Data.
| 60 |
INDEX TO FINANCIAL STATEMENTS
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Cue Biopharma, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Cue Biopharma, Inc. (the “Company”) as of December 31, 2017 and 2016, and the related statements of operations, stockholders’ equity and cash flows, for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Gumbiner Savett Inc.
We have served as the Company’s auditor since 2016.
Santa Monica, California
March 29, 2018
| F-2 |
BALANCE SHEETS
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 63,533,649 | $ | 14,925,820 | ||||
| Certificate of deposit | 50,068 | 50,033 | ||||||
| Research and development contract advances | 852,376 | — | ||||||
| Prepaid expenses and other current assets | 403,420 | 162,398 | ||||||
| Deposits, short-term | 225,500 | — | ||||||
| Total current assets | 65,065,013 | 15,138,251 | ||||||
| Property and equipment, net | 1,690,539 | 1,023,366 | ||||||
| Deposits | — | 117,000 | ||||||
| Trademark | 175,000 | — | ||||||
| Long-term services contract | 23,282 | — | ||||||
| Total assets | $ | 66,953,834 | $ | 16,278,617 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | 1,329,124 | $ | 549,963 | ||||
| Accrued compensation and related expenses | 857,315 | 408,559 | ||||||
| Research and development contract liabilities | 623,853 | — | ||||||
| Research and development collaboration agreement – deferred credit | 2,500,000 | — | ||||||
| Current portion of deferred rent | 36,364 | 109,091 | ||||||
| Total current liabilities | 5,346,656 | 1,067,613 | ||||||
| Deferred rent, net of current portion | — | 36,364 | ||||||
| Total liabilities | 5,346,656 | 1,103,977 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred Stock, $0.001 par value, authorized – 10,000,000 shares; issued and outstanding – none | — | — | ||||||
| Common stock, $0.001 par value; authorized – 50,000,000 shares; issued and outstanding – 19,459,194 shares and 10,635,684 shares at December 31, 2017 and 2016, respectively | 19,460 | 10,636 | ||||||
| Common stock to be issued | 671 | — | ||||||
| Additional paid-in capital | 94,407,895 | 24,751,017 | ||||||
| Accumulated deficit | (32,820,848 | ) | (9,587,013 | ) | ||||
| Total stockholders’ equity | 61,607,178 | 15,174,640 | ||||||
| Total liabilities and stockholders’ equity | $ | 66,953,834 | $ | 16,278,617 | ||||
See accompanying notes to financial statements.
| F-3 |
STATEMENTS OF OPERATIONS
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Revenue | $ | — | $ | — | $ | — | ||||||
| Operating expenses: | ||||||||||||
| General and administrative | 4,333,542 | 1,970,488 | 425,081 | |||||||||
| Research and development | 18,900,328 | 5,687,847 | 1,503,649 | |||||||||
| Total operating expenses | 23,233,870 | 7,658,335 | 1,928,730 | |||||||||
| Loss from operations | (23,233,870 | ) | (7,658,335 | ) | (1,928,730 | ) | ||||||
| Interest income | 35 | 52 | — | |||||||||
| Net loss | $ | (23,233,835 | ) | $ | (7,658,283 | ) | $ | (1,928,730 | ) | |||
| Net loss per common share – basic and diluted | $ | (2.16 | ) | $ | (1.03 | ) | $ | (0.34 | ) | |||
| Weighted average common shares outstanding – basic and diluted | 10,732,449 | 7,433,433 | 5,658,282 | |||||||||
See accompanying notes to financial statements.
| F-4 |
STATEMENT OF STOCKHOLDERS’ EQUITY
Years Ended December 31, 2017, 2016 and 2015
| Common Stock | Common Stock (to be issued) | Additional Paid-in | Accumulated | Total | ||||||||||||||||||||||||
| Shares | Par Value | Shares | Par Value | Capital | Deficit | Equity | ||||||||||||||||||||||
| Common stock issued to founders | 3,649,000 | $ | 3,649 | — | $ | — | $ | — | $ | — | $ | 3,649 | ||||||||||||||||
| Common stock issued in private placement | 3,703,704 | 3,704 | — | — | 9,996,296 | — | 10,000,000 | |||||||||||||||||||||
| Costs incurred in connection with private placement of common stock | — | — | — | — | (1,911,529 | ) | — | (1,911,529 | ) | |||||||||||||||||||
| Proceeds from sale of placement agent warrants | — | — | — | — | 1,000 | — | 1,000 | |||||||||||||||||||||
| Fair value of warrants issued in connection with private placement of common stock | — | — | — | — | 773,941 | — | 773,941 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (1,928,730 | ) | (1,928,730 | ) | |||||||||||||||||||
| Balance, December 31, 2015 | 7,352,704 | 7,353 | — | — | 8,859,708 | (1,928,730 | ) | 6,938,331 | ||||||||||||||||||||
| Common stock issued in private placement | 3,282,980 | 3,283 | — | — | 16,411,617 | — | 16,414,900 | |||||||||||||||||||||
| Costs incurred in connection with private placement of common stock | — | — | — | — | (1,409,864 | ) | — | (1,409,864 | ) | |||||||||||||||||||
| Stock-based compensation | — | — | — | — | 889,556 | — | 889,556 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (7,658,283 | ) | (7,658,283 | ) | |||||||||||||||||||
| Balance, December 31, 2016 | 10,635,684 | 10,636 | — | — | 24,751,017 | (9,587,013 | ) | 15,174,640 | ||||||||||||||||||||
| Common stock issued in initial public offering | 8,820,710 | 8,821 | — | — | 66,146,472 | — | 66,155,293 | |||||||||||||||||||||
| Costs incurred in connection with initial public offering | — | — | — | — | (8,396,661 | ) | — | (8,396,661 | ) | |||||||||||||||||||
| Stock-based compensation | — | — | — | — | 2,775,363 | — | 2,775,363 | |||||||||||||||||||||
| Common stock to be issued pursuant to license agreements | — | — | 671,572 | 671 | 5,036,118 | — | 5,036,789 | |||||||||||||||||||||
| Proceeds from sale of placement agent warrants | — | — | — | — | 1,000 | — | 1,000 | |||||||||||||||||||||
| Fair value of warrants issued in connection with initial public offering of common stock | — | — | — | — | 4,086,581 | — | 4,086,581 | |||||||||||||||||||||
| Exercise of stock options | 2,800 | 3 | — | — | 8,005 | — | 8,008 | |||||||||||||||||||||
| Net loss | — | — | — | — | — | (23,233,835 | ) | (23,233,835 | ) | |||||||||||||||||||
| Balance, December 31, 2017 | 19,459,194 | $ | 19,460 | 671,572 | $ | 671 | $ | 94,407,895 | $ | (32,820,848 | ) | $ | 61,607,178 | |||||||||||||||
See accompanying notes to financial statements.
| F-5 |
STATEMENTS OF CASH FLOWS
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net loss | $ | (23,233,835 | ) | $ | (7,658,283 | ) | $ | (1,928,730 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Common stock to be issued pursuant to license agreements | 5,036,789 | — | — | |||||||||
| Depreciation and amortization | 419,568 | 202,085 | 44,815 | |||||||||
| Deferred rent | (109,091 | ) | 65,910 | 79,545 | ||||||||
| Stock-based compensation | 2,775,363 | 889,556 | — | |||||||||
| Loss on disposal of property plant and equipment | 10,677 | — | — | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| (Increase) decrease in – | ||||||||||||
| Research and development contract advances | (852,376 | ) | — | — | ||||||||
| Prepaid expenses and other current assets | (264,304 | ) | (110,951 | ) | (51,447 | ) | ||||||
| Deposits | (108,500 | ) | (18,500 | ) | (98,500 | ) | ||||||
| Increase (decrease) in – | ||||||||||||
| Accounts payable and accrued expenses | 561,586 | 372,148 | 177,815 | |||||||||
| Accrued compensation and related expenses | 448,756 | 289,624 | 118,935 | |||||||||
| Research and development contract liabilities | 623,853 | — | — | |||||||||
| Research and development collaboration agreement – deferred credit | 2,500,000 | — | — | |||||||||
| Net cash used in operating activities | (12,191,514 | ) | (5,968,411 | ) | (1,657,567 | ) | ||||||
| Cash flows from investing activities: | ||||||||||||
| Increase in certificate of deposit | (35 | ) | (33 | ) | (50,000 | ) | ||||||
| Acquisition of trademark | (175,000 | ) | — | — | ||||||||
| Purchases of property and equipment | (1,029,942 | ) | (515,979 | ) | (754,287 | ) | ||||||
| Net cash used in investing activities | (1,204,977 | ) | (516,012 | ) | (804,287 | ) | ||||||
| Cash flows from financing activities: | ||||||||||||
| Proceeds from issuance of common stock to founders | — | — | 3,649 | |||||||||
| Proceeds from private placements of common stock | — | 16,414,900 | 10,000,000 | |||||||||
| Proceeds from sale of placement agent and underwriters warrants | 1,000 | — | 1,000 | |||||||||
| Proceeds from the initial public offering of common stock | 66,155,293 | — | — | |||||||||
| Proceeds from the exercise of stock options | 8,008 | — | — | |||||||||
| Cash payments made for costs incurred in connection with sale of common stock | (4,159,981 | ) | (1,409,864 | ) | (1,137,588 | ) | ||||||
| Net cash provided by financing activities | 62,004,320 | 15,005,036 | 8,867,061 | |||||||||
| Cash: | ||||||||||||
| Net increase | 48,607,829 | 8,520,613 | 6,405,207 | |||||||||
| Balance at beginning of year | 14,925,820 | 6,405,207 | — | |||||||||
| Balance at end of year | $ | 63,533,649 | $ | 14,925,820 | $ | 6,405,207 | ||||||
| Supplemental disclosures of cash flow information: | ||||||||||||
| Cash paid for – | ||||||||||||
| Interest | $ | — | $ | — | $ | — | ||||||
| Income taxes | $ | — | $ | — | $ | — | ||||||
| Non-cash investing and financing activities: | ||||||||||||
| Fair value of warrants issued in connection with issuance of common stock | $ | 4,086,581 | $ | — | $ | 773,941 | ||||||
| Public offering costs included in accounts payable or accrued expenses | $ | 150,099 | $ | — | $ | — | ||||||
| Purchases of property and equipment in accounts payable or accrued expenses | $ | 67,477 | $ | — | $ | — | ||||||
See accompanying notes to financial statements.
| F-6 |
NOTES TO FINANCIAL STATEMENTS
Years Ended December 31, 2017, 2016 and 2015
| 1. | Organization and Basis of Presentation |
Cue Biopharma, Inc. (the “Company”) was incorporated in the State of Delaware on December 31, 2014 under the name Imagen Biopharma, Inc., and completed its organization, formation and initial capitalization activities effective as of January 1, 2015. In October 2016, the Company changed its name to Cue Biopharma, Inc. The Company’s corporate office and research facilities are located in Cambridge, Massachusetts.
On January 14, 2015, in order to implement its business plans, the Company entered into a license agreement with the Albert Einstein College of Medicine, a division of Yeshiva University (“Einstein”), for certain patent rights, as described in Note 5.
The Company is a pre-clinical biopharmaceutical company that is developing a novel and proprietary class of biologic drugs for the selective modulation of the human immune system to treat a broad range of cancers and autoimmune disorders.
Reclassifications
Certain comparative figures for the years ended 2016 and 2015 have been adjusted to correct the classification of patent legal costs and intellectual property management fees from research and development expenses to general and administrative expenses. During the years ended December 31, 2016 and 2015, patent legal costs of $366,131 and $178,697 and intellectual property management fees of $90,000 and $38,182, aggregating $456,131 and $216,879, respectively, were reclassified from research and development costs to general and administrative costs. These changes did not impact loss from operations or net loss.
Risks and Uncertainties
The Company’s operations are subject to a number of factors that may affect its operating results and financial condition. Such factors include, but are not limited to: the Company’s ability to determine candidates for clinical testing, the results of clinical testing and trial activities of the Company’s product candidates, the Company’s ability to obtain regulatory approval to market its product candidates, the Company’s intellectual property, competition from products manufactured and sold or being developed by other companies, the price of, and demand for, the Company’s product candidates if approved for sale, the Company’s ability to negotiate favorable licensing or other manufacturing and marketing agreements for its product candidates, and the Company’s ability to raise capital.
The Company currently has no commercially approved product candidates and there can be no assurance that the Company’s research and development programs will be successfully commercialized. Developing and commercializing a product requires significant time and capital and is subject to regulatory review and approval, as well as competition from other biotechnology and pharmaceutical companies. The Company operates in an environment of rapid change and is dependent upon the continued services of its employees and consultants and obtaining and protecting its intellectual property.
| 2. | Initial Public Offering |
On December 27, 2017, the Company completed an initial public offering (“IPO”) of its common stock, which resulted in the sale of 8,820,710 shares of common stock at a price to the public of $7.50 per share. The Company received net proceeds from the IPO of $61.9 million after deducting underwriting discounts, commissions, and offering costs incurred by the Company.
| 3. | Summary of Significant Accounting Policies |
Use of Estimates
The preparation of financial statements in conformity with United States Generally Accepted Accounting Principles (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Significant estimates include the accounting for potential liabilities, the assumptions utilized in valuing stock-based compensation issued for services, the realization of deferred tax assets, and the impairment of long-lived assets and intangibles. Actual results could differ from those estimates.
| F-7 |
Cash Concentrations
The Company maintains its cash balances with a financial institution in Federally-insured accounts and may periodically have cash balances in excess of insurance limits. The Company maintains its accounts with a financial institution with a high credit rating. The Company has not experienced any losses to date and believes that it is not exposed to any significant credit risk on cash.
Restricted Cash
The Company purchased a $50,000 certificate of deposit to collateralize a credit card account with a commercial bank that was classified as short-term certificate of deposit as of December 31, 2017 and 2016.
Property and Equipment
Property and equipment is recorded at cost. Major improvements are capitalized, while maintenance and repairs are charged to expense as incurred. Gains and losses from disposition of property and equipment are included in income and expense when realized. Amortization of leasehold improvements is provided using the straight-line method over the shorter of the lease term or the useful life of the underlying assets. Depreciation of property and equipment is provided using the straight-line method over the following estimated useful lives:
| Laboratory equipment | 5 years | |||
| Computer equipment | 3 years | |||
| Furniture and fixtures | 3-8 years |
The Company recognizes depreciation and amortization expense in general and administrative expenses and in research and development expenses in the Company’s statements of operations, depending on how each category of property and equipment is utilized in the Company’s business activities.
Trademark
Trademark consists of the Company’s right, title and interest in and to the CUE BIOLOGICS Mark, and any derivative mark incorporating CUE, throughout the world, together with all associated goodwill and common law rights appurtenant thereto, including, but not limited to, any right, title and interest in any corporate name, company name, business name, trade name, dba, domain name, or other source identifier incorporating CUE.
As the Company can renew the underlying rights to the CUE BIOLOGICS Mark indefinitely at nominal cost, this acquired intangible asset has been classified as a non-amortizable intangible asset in the Company’s balance sheet at December 31, 2017. The Company evaluates the status of this intangible asset for amortization and impairment at each quarter end and year end reporting date.
Research and Development Funding Arrangements
The Company’s proprietary biologics are at an early stage and will require substantial time and funding to continue development. There can be no assurances that any of the Company’s biologics will ultimately become commercially viable product candidates. In order to finance its research and development programs, the Company may periodically enter into collaboration agreements with third parties that provide funding for certain aspects of the Company’s ongoing research and development activities. The Company considers various factors in determining the appropriate accounting treatment for such collaboration agreements, including, among others, the risks of and costs associated with the research and development program being funded, the stage of development of the proprietary biologics subject to the research and development program, the likelihood at initiation that the collaboration arrangement will result in an economically successful outcome to the third party, the continuing involvement of the Company in the research and development program and the expenditure of the funds, the transfer of the financial risk associated with the research and development program to the third party, the intended use of the funds and any restrictions thereon, and the probability of any repayment obligations or other forms of consideration if the proprietary biologics subject to the research and development program are not successfully developed and commercialized.
| F-8 |
In accordance with ASC 730-20-25-8, to the extent that a collaboration agreement results in a substantive and genuine transfer of financial risk to a third party funding source because any economic benefit that the third party may receive depends solely on the research and development program successfully developing commercially viable product candidates having future economic benefit (which is uncertain at the initiation of the collaboration agreement), the Company will account for such collaboration agreement as a contract to perform research and development services for a third party. The funds received from the third party under such a collaboration agreement will initially be recorded as a deferred credit in the Company’s balance sheet. As the related contractual research and development costs are incurred, the applicable amount of the deferred credit will be credited to operations and will be classified as an offset to such research and development costs in the Company’s statement of operations.
Research and Development Expenses
Research and development expenses consist primarily of compensation costs, fees paid to consultants, outside service providers and organizations (including research institutes at universities), facility costs, and development and clinical trial costs with respect to the Company’s product candidates.
Research and development expenses incurred under contracts are expensed ratably over the life of the underlying contracts, unless the achievement of milestones, the completion of contracted work, or other information indicates that a different expensing schedule is more appropriate. Other research and development expenses are charged to operations as incurred.
| F-9 |
Payments made pursuant to research and development contracts are initially recorded as research and development contract advances in the Company’s balance sheet and then charged to research and development expenses in the Company’s statement of operations as those contract services are performed. Expenses incurred under research and development contracts in excess of amounts advanced are recorded as research and development contract liabilities in the Company’s balance sheet, with a corresponding charge to research and development expenses in the Company’s statement of operations.
Nonrefundable advance payments for future research and development activities pursuant to an executory contractual arrangement are recorded as advances as described above. Nonrefundable advance payments are recognized as an expense as the related services are performed. The Company evaluates whether it expects the services to be rendered at each quarter end and year end reporting date. If the Company does not expect the services to be rendered, the advance payment is charged to expense. To the extent that a nonrefundable advance payment is for contracted services to be performed within 12 months from the reporting date, such advance is included in current assets; otherwise, such advance is included in non-current assets.
The Company evaluates the status of its research and development agreements and contracts, and the carrying amount of the related assets and liabilities, at each quarter end and year end reporting date, and adjusts the carrying amounts and their classification on the balance sheet as appropriate.
Patent Expenses
The Company is the exclusive worldwide licensee of, and has patent applications pending for, numerous domestic and foreign patents. Due to the significant uncertainty associated with the successful development of one or more commercially viable product candidates based on the Company’s research efforts and any related patent applications, all patent costs, including patent-related legal fees, filing fees and other costs are charged to operations as incurred. For the years ended December 31, 2017, 2016, and 2015, patent expenses were $592,262, $366,131, and $178,697, respectively. Patent expenses are included in general and administrative expenses in the Company’s statement of operations.
Licensing Fees and Costs
Licensing fees and costs consist primarily of costs relating to the acquisition of the Company’s license agreement with Einstein, including related royalties, maintenance fees, milestone payments and product development costs. Licensing fees and costs are charged to operations as incurred.
Long-Lived Assets
The Company reviews long-lived assets, consisting of property and equipment, for impairment at each fiscal year end or when events or changes in circumstances indicate the carrying value of these assets may exceed their current fair values. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized for the amount by which the carrying amount of the asset exceeds the fair value of the assets. Assets to be disposed of are separately presented in the balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and are no longer depreciated. The Company has not historically recorded any impairment to its long-lived assets. In the future, if events or market conditions affect the estimated fair value to the extent that a long-lived asset is impaired, the Company will adjust the carrying value of these long-lived assets in the period in which the impairment occurs. During the years ended December 31, 2017, 2016 and 2015 the company disposed of equipment totaling $15,993, $0 and $0, resulting in a loss of $10,677, $0 and $0, respectively.
Rent Expense and Deferred Rent Liability
Operating lease agreements which contain provisions for future rent increases or periods in which rent payments are reduced or abated are recorded in monthly rent expense in the amount of the total payments over the lease term divided by the number of months of the lease term. The difference between rent expense recorded and the amount paid is credited or charged to a deferred rent liability account. The current portion of deferred rent is included in current liabilities, and the remaining amount is shown in the balance sheet as a non-current liability. Accordingly, rent expense is recorded on a straight-line basis.
| F-10 |
Stock-Based Compensation
The Company periodically issues stock options to officers, directors, employees, Scientific and Clinical Advisory Board members, non-employees and consultants for services rendered. Such issuances vest and expire according to terms established at the issuance date.
Stock-based payments to officers, directors and employees, including grants of employee stock options, are recognized in the financial statements based on their grant date fair values. Stock option grants, which are generally time-vested, are measured at the grant date fair value and charged to operations on a straight-line basis over the vesting period. The fair value of stock options is determined utilizing the Black-Scholes option-pricing model, which is affected by several variables, including the risk-free interest rate, the expected dividend yield, the life of the equity award, the exercise price of the stock option as compared to the fair value of the common stock on the grant date, and the estimated volatility of the common stock over the term of the equity award.
Stock options granted to members of the Company’s Scientific and Clinical Advisory Board, non-employees and outside consultants are revalued each reporting period to determine the amount to be recorded as an expense in the respective period. As the stock options vest, they are valued on each vesting date and an adjustment is recorded for the difference between the value already recorded and the value on the date of vesting.
The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. Until the Company has established a trading market for its common stock, estimated volatility is based on the average historical volatilities of comparable public companies in a similar industry. The expected dividend yield is based on the current yield at the grant date; the Company has never declared or paid dividends and has no plans to do so for the foreseeable future. As permitted by Staff Accounting Bulletin No. 107, due to the Company’s lack of trading history and option activity, management utilizes the simplified method to estimate the expected term of options at the date of grant. The fair value of common stock is determined by reference to either recent or anticipated cash transactions involving the sale of the Company’s common stock. The Company accounts for forfeitures as they occur in accordance with ASU 2016-09, Compensation — Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting (“ASU 2016-09”), and reverses previously recognized stock compensation costs in the period that the award is forfeited.
The Company recognizes the fair value of stock-based compensation in general and administrative expenses and in research and development expenses in the Company’s statement of operations, depending on the type of services provided by the recipient of the equity award. The Company issues new shares of common stock to satisfy stock option exercises.
Income Taxes
The Company accounts for income taxes under an asset and liability approach for financial accounting and reporting for income taxes. Accordingly, the Company recognizes deferred tax assets and liabilities for the expected impact of differences between the financial statements and the tax basis of assets and liabilities.
The Company accounts for uncertainties in income tax law under a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns as prescribed by GAAP. The tax effects of a position are recognized only if it is “more-likely-than-not” to be sustained by the taxing authority as of the reporting date. If the tax position is not considered “more-likely-than-not” to be sustained, then no benefits of the position are recognized.
The Company records a valuation allowance to reduce its deferred tax assets to the amount that is more likely than not to be realized. In the event the Company was to determine that it would be able to realize its deferred tax assets in the future in excess of its recorded amount, an adjustment to the deferred tax assets would be credited to operations in the period such determination was made. Likewise, should the Company determine that it would not be able to realize all or part of its deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to operations in the period such determination was made.
The Company is subject to U.S. Federal and Massachusetts state income taxes. As the Company’s net operating losses have yet to be utilized, all previous tax years remain open to examination by Federal and state taxing authorities in which the Company currently operates.
The Company recognizes interest accrued relative to unrecognized tax benefits in interest expense and penalties in operating expense. During the years ended December 31, 2017, 2016 and 2015, the Company did not recognize any income tax related interest and penalties. The Company did not have any accruals for income tax related interest and penalties at December 31, 2017 and 2016.
| F-11 |
Comprehensive Income (Loss)
Components of comprehensive income or loss, including net income or loss, are reported in the financial statements in the period in which they are recognized. Comprehensive income or loss is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. Net income (loss) and other comprehensive income (loss) are reported net of any related tax effect to arrive at comprehensive income (loss). The Company did not have any items of comprehensive income (loss) for the years ended December 31, 2017, 2016 and 2015.
Earnings (Loss) Per Share
The Company’s computation of earnings (loss) per share (“EPS”) for the respective periods includes basic and diluted EPS. Basic EPS is measured as the income (loss) attributable to common stockholders divided by the weighted average number of common shares outstanding for the period. Diluted EPS is similar to basic EPS but presents the dilutive effect on a per share basis of potential common shares that would result from the exercise of outstanding stock options and warrants as if they had been exercised at the beginning of the periods presented, or issuance date, if later. Potential common shares that have an anti-dilutive effect (i.e., those that increase income per share or decrease loss per share) are excluded from the calculation of diluted EPS. Basic and diluted loss per common share is the same for all periods presented because all outstanding stock options and warrants are anti-dilutive.
At December 31, 2017, 2016 and 2015, the Company excluded the outstanding securities summarized below, which entitle the holders thereof to acquire shares of common stock, from its calculation of earnings per share, as their effect would have been anti-dilutive. The common stock to be issued is not included in the calculation of earnings per share.
| December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Common stock warrants | 1,252,441 | 370,370 | 370,370 | |||||||||
| Common stock options | 2,732,221 | 1,663,221 | — | |||||||||
| Total | 3,984,662 | 2,033,591 | 370,370 | |||||||||
Fair Value of Financial Instruments
The authoritative guidance with respect to fair value established a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three levels, and requires that assets and liabilities carried at fair value be classified and disclosed in one of three categories, as presented below.
Level 1. Observable inputs such as quoted prices in active markets for an identical asset or liability that the Company has the ability to access as of the measurement date. Financial assets and liabilities utilizing Level 1 inputs include active-exchange traded securities and exchange-based derivatives.
Level 2. Inputs, other than quoted prices included within Level 1, which are directly observable for the asset or liability or indirectly observable through corroboration with observable market data. Financial assets and liabilities utilizing Level 2 inputs include fixed income securities, non-exchange based derivatives, mutual funds, and fair-value hedges.
Level 3. Unobservable inputs in which there is little or no market data for the asset or liability which requires the reporting entity to develop its own assumptions. Financial assets and liabilities utilizing Level 3 inputs include infrequently-traded non-exchange-based derivatives and commingled investment funds, and are measured using present value pricing models.
The Company determines the level in the fair value hierarchy within which each fair value measurement falls in its entirety, based on the lowest level input that is significant to the fair value measurement in its entirety. In determining the appropriate levels, the Company performs an analysis of the assets and liabilities at each reporting period end.
| F-12 |
There were no financial instruments that were measured and recorded at fair value on the Company’s balance sheet at December 31, 2017 and 2016.
The carrying value of financial instruments (consisting of cash, a certificate of deposit, accounts payable, accrued compensation and accrued expenses) is considered to be representative of their respective fair values due to the short-term nature of those instruments.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”). ASU 2014-09 will eliminate transaction- and industry-specific revenue recognition guidance under current GAAP and replace it with a principle based approach for determining revenue recognition. ASU 2014-09 will require that companies recognize revenue based on the value of transferred goods or services as they occur in the contract. ASU 2014-09 also will require additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. The FASB has issued ASU 2016-08, ASU 2016-10, ASU 2016-11, ASU 2016-12, and ASU 2016-20, all of which clarify certain implementation guidance within ASU 2014-09. ASU 2014-09 is effective for reporting periods beginning after December 15, 2017, with early adoption permitted. Entities will be able to transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. The Company will adopt the provisions of ASU 2014-09 in the quarter beginning January 1, 2018. As the Company is unlikely to generate any sustainable operating revenues in the next several years, the adoption of ASU 2014-09 is not currently expected to have any impact on the Company’s financial statement presentation or disclosures.
In November 2015, the FASB issued Accounting Standards Update No. 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes (“ASU 2015-17”). ASU 2015-17 requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. ASU 2015-17 is effective for financial statements issued for annual periods beginning after December 15, 2016, and interim periods within those annual periods. The Company adopted the provisions of ASU 2015-17 in the quarter beginning January 1, 2017. The adoption of ASU 2015-17 did not have any impact on Company’s financial statement presentation or disclosures.
In February 2016, the FASB issued Accounting Standards Update No. 2016-02, Leases (Topic 842) (“ASU 2016-02”). ASU 2016-02 requires a lessee to record a right-of-use asset and a corresponding lease liability, initially measured at the present value of the lease payments, on the balance sheet for all leases with terms longer than 12 months, as well as the disclosure of key information about leasing arrangements. ASU 2016-02 requires recognition in the statement of operations of a single lease cost, calculated so that the cost of the lease is allocated over the lease term, generally on a straight-line basis. ASU 2016-02 requires classification of all cash payments within operating activities in the statement of cash flows. Disclosures are required to provide the amount, timing and uncertainty of cash flows arising from leases. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early application is permitted. The Company will adopt the provisions of ASU 2016-02 in the quarter beginning January 1, 2019. The Company generally does not finance purchases of property and equipment, but does lease its operating facilities. While the Company is continuing to assess the potential impact of ASU 2016-02, it currently expects that most of its lease commitments will be subject to ASU 2016-02 and accordingly, upon adoption will be recognized as lease liabilities and right-of-use assets in the Company’s balance sheet.
In March 2016, the FASB issued Accounting Standards Update No. 2016-09, Compensation — Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting (“ASU 2016-09”). ASU 2016-09 requires, among other things, that all income tax effects of awards be recognized in the statement of operations when the awards vest or are settled. ASU 2016-09 also allows for an employer to repurchase more of an employee’s shares than it can today for tax withholding purposes without triggering liability accounting and allows for a policy election to account for forfeitures as they occur. ASU 2016-09 is effective for fiscal years beginning after December 15, 2016, including interim periods within those fiscal years. The Company adopted the provisions of ASU 2016-09 in the quarter beginning January 1, 2017. The adoption of ASU 2016-09 did not have any impact on the Company’s financial statement presentation or disclosures.
| F-13 |
In July 2017, the FASB issued Accounting Standards Update No. 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features; (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception (“ASU 2017-11”). ASU 2017-11 allows companies to exclude a down round feature when determining whether a financial instrument (or embedded conversion feature) is considered indexed to the entity’s own stock. As a result, financial instruments (or embedded conversion features) with down round features may no longer be required to be accounted for as derivative liabilities. A company will recognize the value of a down round feature only when it is triggered and the strike price has been adjusted downward. For equity-classified freestanding financial instruments, an entity will treat the value of the effect of the down round as a dividend and a reduction of income available to common shareholders in computing basic earnings per share. For convertible instruments with embedded conversion features containing down round provisions, entities will recognize the value of the down round as a beneficial conversion discount to be amortized to earnings. ASU 2017-11 is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted. The guidance in ASU 2017-11 can be applied using a full or modified retrospective approach. The adoption of ASU 2017-11 is not currently expected to have any impact on the Company’s financial statement presentation or disclosures.
Management does not believe that any other recently issued, but not yet effective, authoritative guidance, if currently adopted, would have a material impact on the Company’s financial statement presentation or disclosures.
| 4. | Property and Equipment |
Property and equipment as of December 31, 2017 and 2016 is summarized as follows:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Laboratory equipment | $ | 2,205,410 | $ | 1,194,473 | ||||
| Furniture and fixtures | 9,606 | 1,832 | ||||||
| Computer equipment | 82,958 | 44,166 | ||||||
| Leasehold improvements | 53,718 | 29,795 | ||||||
| 2,351,692 | 1,270,266 | |||||||
| Less accumulated depreciation and amortization | (661,153 | ) | (246,900 | ) | ||||
| Net property and equipment | $ | 1,690,539 | $ | 1,023,366 | ||||
Depreciation and amortization expense for the years ended December 31, 2017, 2016 and 2015 was included in the statement of operations as follows:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| General and administrative | $ | 15,163 | $ | 4,630 | $ | 1,238 | ||||||
| Research and development | 404,405 | 197,455 | 43,577 | |||||||||
| Total | $ | 419,568 | $ | 202,085 | $ | 44,815 | ||||||
| 5. | Einstein License and Service Agreement |
License Agreement
On January 14, 2015, the Company entered into a license agreement, as amended on June 2, 2015 (the “Einstein License”), with Einstein for certain patent rights (the “Patents”) relating to the Company’s core technology platform for the engineering of biologics to control T-cell activity, precision, immune-modulatory drug candidates, and two supporting technologies that enable the discovery of costimulatory signaling molecules (ligands) and T-cell targeting peptides. On July 31, 2017, the Company entered into an amended and restated license agreement which modified certain obligations of the parties under the Einstein License.
| F-14 |
Under the Einstein License, the Company holds an exclusive worldwide license, with the right to sublicense, import, make, have made, use, provide, offer to sell, and sell all products, processes and services that use the Patents, including certain technology received from Einstein relating thereto (the “Licensed Products”). Under the Einstein License, the Company is required to:
| · | Pay royalties based on certain percentage of proceeds, as defined in the Einstein License, from sales of Licensed Products, including sublicense agreements. |
| · | Pay escalating annual maintenance fees as follows: $25,000 on January 14, 2017; $50,000 on each of January 14, 2018 and 2019; $75,000 on each of January 14, 2020 and 2021; and $100,000 on January 14, 2022 and each year thereafter. Annual maintenance fees are nonrefundable, but are creditable against the amount due to Einstein for royalties during the 12 month period following each of the due dates for annual maintenance fees. |
| · | Make significant payments up to $5,000,000 based upon the achievement of certain milestones, as defined in the Einstein License. Payments made upon achievement of milestones are nonrefundable and are not creditable against any other payment due to Einstein. At December 31, 2017, none of these milestones had been achieved by the Company. |
| · | Incur a minimum of $250,000 per year of product development costs until the first commercial sale of the first licensed product. |
The Company was in compliance with its obligations under the Einstein License at December 31, 2017 and 2016.
The Einstein License expires upon the expiration of the Company’s last obligation to make royalty payments to Einstein which may be due with respect to certain Licensed Products, unless terminated earlier under the provisions thereof. The Einstein License includes certain termination provisions if the Company fails to meet its obligations thereunder.
The Einstein License required the Company to issue to Einstein a specified number of shares of common stock of the Company on a fully diluted, as converted basis, depending on the achievement of (1) a funding threshold and (2) a liquidity event, each as defined in the Einstein License. The funding threshold was achieved through the completion of the June 15, 2015 private placement as described in Note 8. A liquidity event includes, but is not limited to, an initial public offering of shares of the Company’s common stock; a merger with a public reporting company under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or a company whose shares are listed on a non-U.S. exchange or an affiliate thereof; a merger, consolidation, reorganization, or similar transaction whereby the Company’s stockholders immediately prior to the consummation of the transaction will own less than the majority of the voting power of the resulting corporation after the consummation of the transaction; or a sale of substantially all of the Company’s assets. Accordingly, the Company was required to issue 671,572 shares of the Company’s common stock to Einstein immediately prior to the consummation of an initial public offering by the Company on December 27, 2017. The common stock issued to Einstein upon the occurrence of a liquidity event has certain registration rights, as described in Note 11.
The Company accounted for the issuance of these shares as a charge to research and development expenses in the statement of operations at their aggregate fair value of $5,036,789 on the date of issuance, in accordance with ASC 730, Research and Development, as the Patents acquired from Einstein are for use in the Company’s research and development activities exclusively with respect to its core technology platform and have no alternative future use by the Company, and therefore no separate economic value. The shares issuable in connection with Einstein License were recorded as common stock to be issued at December 31, 2017, and were issued on January 9, 2018.
The Company accounts for the costs incurred in connection with the Einstein License in accordance with ASC 730, Research and Development. For the years ended December 31, 2017, 2016, and 2015, costs incurred with respect to the Einstein License aggregated $5,084,708, $31,250, and $127,336, respectively, and are included in research and development expenses in the statements of operations.
Service Agreement
On October 1, 2015, the Company entered into a service agreement (the “Service Agreement”) with Einstein to support the Company’s ongoing research and development activities. The initial term of the Service Agreement was for three months, which was amended in February 2016 to extend it for the period of time deemed necessary to complete the services pursuant to the terms of the Service Agreement. For the years ended December 31, 2017, 2016, and 2015, costs incurred with respect to the Service Agreement aggregated $0, $80,000, and $200,000, respectively, and are included in research and development expenses in the statement of operations.
| F-15 |
| 6. | Acquisition of Trademark |
On May 4, 2017, the Company entered into a settlement agreement with Cue BioLogics, LLC, an unrelated party, to acquire all right, title and interest in and to the CUE BIOLOGICS mark, and any derivative mark incorporating CUE, throughout the world, together with all associated goodwill and common law rights appurtenant thereto, including, but not limited to, any right, title and interest in any corporate name, company name, business name, trade name, dba, domain name, or other source identifier incorporating CUE (collectively, the “CUE BIOLOGICS Mark”), in exchange for a cash payment by the Company of $175,000.
Accounting Standards Codification (“ASC”) 350-30-20 defines a defensive intangible asset as an acquired intangible asset in a situation in which an entity does not intend to actively use the asset but intends to hold (lock up) the asset to prevent others from obtaining access to the asset. The Company determined that the acquired intangible asset met the definition of a defensive intangible asset and has therefore accounted for the $175,000 payment to Cue BioLogics, LLC for the CUE BIOLOGICS Mark as an acquired intangible asset.
| 7. | Stock-Based Compensation |
Effective March 23, 2016, the Company adopted the 2016 Omnibus Incentive Plan (the “Omnibus Plan”) and the 2016 Non-Employee Equity Incentive Plan (the “Non-Employee Plan”), which are intended to allow the Company to compensate and retain the services of key employees, non-employees, Scientific and Clinical Advisory Board members, and outside advisors and consultants. The plans are under the administration of the Company’s Board of Directors. Under the plans, the Company, at its discretion, may grant stock option awards to certain employees and non-employees through March 23, 2026. The Omnibus Plan and the Non-Employee Plan provide for the grant of a total of 2,000,000 shares and 500,000 shares of common stock, respectively.
On August 13, 2017, the Company’s Board of Directors approved an amendment and restatement of the Company’s 2016 Omnibus Incentive Plan to increase the number of shares authorized for issuance under such plan by 800,000 shares, from 2,000,000 shares to 2,800,000 shares, subject to stockholder approval of such amendment within 12 months following board approval thereof. The Company’s stockholders approved the plan in December 2017. The 2016 Omnibus Incentive Plan, as amended and restated, provides that on the first day of each fiscal year of the Company during the period beginning in fiscal year 2018 and ending on the second day of fiscal year 2027, the number of shares of common stock authorized to be issued under such plan shall be increased by an amount equal to the lesser of (i) the number of shares necessary such that the aggregate number of shares available to be issued under the plan equals 20% of the number of fully diluted outstanding shares on such date (assuming the conversion of all outstanding shares of preferred stock and other outstanding convertible securities and exercise of all outstanding options and warrants to purchase shares) and (ii) an amount to be determined by the Company’s Board of Directors.
Pursuant to the plans, during the year ended December 31, 2017, the Company granted stock options to purchase 1,089,000 shares of the Company’s common stock, of which 150,000 were granted to members of the Scientific and Clinical Advisory Board, 60,000 to independent members of the Company’s Board of Directors, 100,000 to independent consultants, 385,000 to members of management, and 394,000 to other employees. The stock options granted to independent members of the Board of Directors, members of the Scientific and Clinical Advisory Board, and independent consultants were non-qualified stock options, whereas all other stock options were incentive stock options. At December 31, 2017, stock options for 2,362,221 shares of common stock had been granted and 437,779 shares of common stock were reserved for future grants under the Omnibus Plan, and stock options for 370,000 shares of common stock had been granted and 130,000 shares of common stock were reserved for future grants under the Non-Employee Plan. In the aggregate, at December 31, 2017, stock options for a total of 2,732,221 shares of common stock had been granted and 567,779 shares of common stock were reserved for future grants. Such grants are accounted for as share-based compensation in accordance with ASC 718, Compensation - Stock Compensation, and ASC 505-50, Equity-Based Payments to Non-Employees.
For stock options requiring an assessment of value during the years ended December 31, 2017 and 2016, the fair value of each stock option award was estimated using the Black-Scholes option-pricing model utilizing the following assumptions:
| F-16 |
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Risk-free interest rate | 1.71 to 2.22 | % | 1.07 to 2.25 | % | ||||
| Expected dividend yield | 0 | % | 0 | % | ||||
| Expected volatility | 80.9-83.0 | % | 104% to 112 | % | ||||
| Expected life | 3.2 to 7 years | 4.25 to 7 years | ||||||
The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected term of the stock option award; as permitted by Staff Accounting Bulletin 107, due to insufficient history of stock option activity, management has utilized the simplified approach to estimate the expected term of the stock options, which represents the period of time that stock options granted are expected to be outstanding; the expected volatility is based upon historical volatilities of comparable companies in a similar industry; and the expected dividend yield based upon the Company’s current dividend rate and future expectations.
A summary of stock option activity for the years ended December 31, 2017 is as follows:
| Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Stock options outstanding at December 31, 2016 | 1,663,221 | $ | 2.86 | 6.22 | ||||||||
| Granted | 1,089,000 | 5.89 | ||||||||||
| Exercised | (2,800 | ) | 2.86 | |||||||||
| Cancelled | (17,200 | ) | 2.86 | |||||||||
| Stock options outstanding at December 31, 2017 | 2,732,221 | $ | 4.07 | 5.76 | ||||||||
| Stock options exercisable at December 31, 2017 | 566,284 | $ | 3.05 | 5.08 | ||||||||
The Company recognized $2,775,363 and $889,556 in stock-based compensation during the years ended December 31, 2017 and 2016, respectively. As of December 31, 2017, total unrecognized stock-based compensation was approximately $8,900,000, which is expected to be recognized as an operating expense in the Company’s statement of operations through June 2022.
A summary of the exercise prices of common stock options outstanding and exercisable at December 31, 2017 is as follows:
| F-17 |
| Exercise Prices | Options Outstanding (Shares) | Options Exercisable (Shares) | ||||||
| $2.86 | 1,643,221 | 517,159 | ||||||
| $5.00 | 703,000 | 49,125 | ||||||
| $7.50 | 386,000 | — | ||||||
| 2,732,221 | 566,284 | |||||||
The intrinsic value of exercisable but unexercised in-the-money stock options at December 31, 2017 was approximately $2,522,000, based on a fair value of $7.50 per share on December 31, 2017.
Stock-based compensation for the years ended December 31, 2017 and 2016 was included in the statement of operations as follows:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| General and administrative | $ | 1,022,962 | $ | 439,366 | ||||
| Research and development | 1,752,401 | 450,190 | ||||||
| Total | $ | 2,775,363 | $ | 889,556 | ||||
There was no stock-based compensation expense for the year ended December 31, 2015.
| 8. | Stockholders’ Equity |
Preferred Stock
The Company has authorized a total of 10,000,000 shares of preferred stock, par value $0.001 per share, none of which were outstanding at December 31, 2017 and 2016. The Company’s Board of Directors has the authority to issue preferred stock and to determine the rights, preferences, privileges, and restrictions, including voting rights.
Common Stock
The Company has authorized a total of 50,000,000 shares of common stock, par value $0.001 per share, of which 19,459,194 shares and 10,635,684 shares were issued and outstanding at December 31, 2017 and 2016, respectively.
June 15, 2015 Private Placement of Common Stock. On June 15, 2015, the Company sold 3,703,704 shares of common stock in a private placement to accredited investors at $2.70 per share, resulting in gross cash proceeds of $10,000,000. Direct costs of the private placement consisted of a 10% placement agent fee to the placement agent, MDB Capital Group, LLC (“MDB”) (see Note 9), of $1,000,000, and related legal fees and reimbursable expenses of $137,588. In conjunction with this private placement, the Company issued warrants to the placement agent to purchase 370,370 shares of common stock, exercisable at issuance for a period of seven years at $2.70 per share, for a cash consideration of $1,000. The placement agent warrants had a fair value of $773,941, calculated pursuant to the Black-Scholes option-pricing model. Issuance costs of the private placement, including the fair value of placement agent warrants of $773,941, aggregated $1,911,529 and were charged directly to additional paid-in capital. The common stock sold and the placement agent warrants issued in this private placement have certain registration rights, as described in Note 11.
December 22, 2016 Private Placement of Common Stock. On December 22, 2016, the Company sold 3,282,980 shares of common stock in a private placement to accredited investors at $5.00 per share, resulting in gross cash proceeds of $16,414,900. Direct costs of the private placement consisted of a placement agent fee to the placement agent, MDB, of $1,320,745 and related legal fees and reimbursable expenses of $89,119. Issuance costs of the private placement aggregated $1,409,864 and were charged directly to additional paid-in capital. The common stock sold in this private placement has certain registration rights, as described in Note 11.
Down Round Protection. The Company provided investors in the December 22, 2016 private placement of common stock with certain limited protections resulting from one or more issuances of shares of common stock at a per share purchase price below that was paid by the investors in the private placement, terminating on December 31, 2019. If the Company issued additional shares of common stock without consideration, or for consideration per share less than the effective per share price deemed to be in effect immediately prior to such issuance, then concurrently with such issuance, the Company was required to issue to each investor, for no additional consideration additional shares of common stock as specified in the private placement memorandum.
Pursuant to an Irrevocable Waiver and Amendment to Securities Purchase Agreements entered into on December 4, 2017, the Majority Investors in the December 2016 private placement agreed that no shares shall be issuable pursuant to these anti-dilution rights in connection with any issuance of common stock occurring after the Company’s initial public offering.
| F-18 |
Common Stock Warrants
In conjunction with the private placement of common stock on June 15, 2015 at $2.70 per share, the Company issued warrants to the placement agent to purchase 370,370 shares of common stock, exercisable at issuance for a period of seven years at $2.70 per share, for a cash consideration of $1,000. MDB assigned approximately one-half of the warrants to its employees, two of which are officers or directors of the Company. The placement agent warrants have standard anti-dilution protections and cashless exercise rights. The placement agent warrants have certain registration rights, as described in Note 11.
The placement agent warrants were valued pursuant to the Black-Scholes option-pricing model at $773,941, based on the following inputs: risk-free interest rate — 2.11%; expected dividend yield — 0%; expected volatility — 88%; expected life — 7 years; fair value of common stock — $2.70 per share. The expected volatility was determined by reference to the volatility factors of several comparable bio pharmaceutical public companies. These warrants were considered a cost of the private placement offering.
In conjunction with the initial public offering of common stock on December 27, 2017 at $7.50 per share, the Company issued warrants to the underwriter, MDB, to purchase 882,071 shares of common stock, exercisable at issuance for a period of five years at $9.38 per share, for a cash consideration of $1,000. MDB assigned 12,500 warrants to the independent underwriter, Feltl and Company, Inc, and 36,259 shares to Paulson Investment Company LLC for underwriting services. MDB also assigned approximately one half of the remaining warrants to its employees, two of which are current officers or directors of the Company. The underwriters warrants have standard anti-dilution protections and cashless exercise rights. The placement agent warrants have certain registration rights, as described in Note 11.
The underwriters warrants were valued pursuant to the Black-Scholes option-pricing model at $4,086,581, based on the following inputs: risk-free interest rate — 2.22%; expected dividend yield — 0%; expected volatility — 81.6%; expected life — 5 years; fair value of common stock — $7.50 per share. The expected volatility was determined by reference to the volatility factors of several comparable bio pharmaceutical public companies. These warrants were considered a cost of the initial public offering.
A summary of warrant activity for the year ended December 31, 2017 is as follows:
| Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (in Years) | ||||||||||
| Warrants outstanding at December 31, 2016 | 370,370 | $ | 2.70 | |||||||||
| Issued | 882,071 | 9.38 | ||||||||||
| Exercised | — | — | ||||||||||
| Expired | — | — | ||||||||||
| Warrants outstanding at December 31, 2017 | 1,252,441 | $ | 7.40 | 4.8 | ||||||||
| Warrants exercisable at December 31, 2017 | 1,252,441 | $ | 7.40 | 4.8 | ||||||||
The intrinsic value of exercisable but unexercised in-the-money common stock warrants at December 31, 2017 was approximately $1,778,000, based on a fair value of $7.50 per share on December 31, 2017.
| 9. | Related Party Transactions |
During the years ended December 31, 2017, 2016 and 2015, MDB provided investment banking services to the Company (see Notes 2 and 8). For those services, the Company paid MDB cash placement agent fees of $3,473,155 and $1,320,745 in 2017 and 2016, respectively. In 2017, the Company also issued to MDB underwriter warrants to purchase 833,312 shares of the Company’s common stock for a cash consideration of $1,000, exercisable for five years at $9.38 per share. As of December 31, 2017, MDB beneficially owned approximately 10.2% of the Company’s common stock, has served as the underwriter of the Company’s initial public offering as well as the placement agent in previous private placements of the Company’s common stock, and three employees of which are directors of Cue.
| F-19 |
MDB assigned one-half of the warrants that it acquired in conjunction with the June 15, 2015 private placement (see Note 8) to eight employees of MDB, two of which are also current officers or directors of the Company, who received a total of 72,222 of such warrants as part of the normal distribution of warrants received by MDB to its employees.
During the years ended December 31, 2017, 2016 and 2015, the Company incurred expenses amounting to $0, $60,000, and $38,182 respectively, for patent-related services provided by MDB, which is included in general and administrative expenses in the statement of operations. At December 31, 2017 and 2016, $0 and $26,152, respectively, of amounts payable to MDB relating to reimbursable expenses and patent-related services were included in accounts payable.
Beginning in June 2015, the interim Chief Financial Officer of the Company, who is also the Chief Financial Officer of MDB, has been compensated at a rate of $6,000 per month, reflecting an aggregate annual charge to operations for the years ended December 31, 2017, 2016 and 2015 of $72,000, $72,000 and $42,000, respectively.
Information with respect to payments under the Einstein License and the Service Agreement is described in Note 5.
| 10. | Income Taxes |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets as of December 31, 2017 and 2016 are as follows:
| 2017 | 2016 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 8,424,000 | $ | 3,607,000 | ||||
| Research and other credits | 867,000 | 364,000 | ||||||
| Reserves and accruals | 595,000 | 315,000 | ||||||
| Other intangibles | 4,000 | 9,000 | ||||||
| Total gross deferred tax assets | 9,890,000 | 4,295,000 | ||||||
| Less valuation allowance | (9,793,000 | ) | (4,190,000 | ) | ||||
| Total deferred tax assets | 97,000 | 105,000 | ||||||
| Deferred tax liability: | ||||||||
| Depreciation | (97,000 | ) | (105,000 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
In assessing the potential realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the Company attaining future taxable income during the periods in which those temporary differences become deductible. As of December 31, 2017 and 2016, management was unable to determine if it is more likely than not that the Company’s deferred tax assets will be realized, and has therefore recorded a 100% valuation allowance against deferred tax assets at such dates.
No Federal tax provision has been provided for the years ended December 31, 2017, 2016, and 2015 due to the losses incurred during such periods. A reconciliation of the difference between the income tax rate computed by applying the U.S. Federal statutory rate and the effective tax rate for the years ended December 31, 2017, 2016 and 2015 is as follows:
| F-20 |
Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2016 | ||||||||||
| U. S. Federal statutory tax rate | (35 | )% | (35 | )% | (35 | )% | ||||||
| Change in valuation allowance | 16 | % | 36 | % | 37 | % | ||||||
| Tax credits | (2 | )% | (3 | )% | (3 | )% | ||||||
| Stock based compensation | 2 | % | — | — | ||||||||
| Tax reform | 18 | % | — | — | ||||||||
| Other | 1 | % | 2 | % | 1 | % | ||||||
| Effective tax rate | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
The Company has applied the provisions of ASC 740, Income Taxes, which clarifies the accounting for uncertainty in tax positions and requires the recognition of the impact of a tax position in the financial statements if that position is more likely than not of being sustained on a tax return, based on the technical merits of the position, upon examination by the relevant taxing authority. At December 31, 2017 and 2016, the Company had unrecognized tax benefits related to Federal and state research tax credits of approximately $510,000 and $157,000, respectively. The Company is subject to Federal and state income tax examinations by tax authorities for all years since its incorporation in 2014. The Company is currently not under examination by any tax authority.
At December 31, 2017, the Company has available net operating loss carryforwards for Federal and state income tax purposes of approximately $28,700,000 and $29,100,000, respectively, which, if not utilized earlier, will begin to expire in 2035. The Company has Federal research credits of approximately $944,000, which, if not utilized earlier, will begin to expire in 2035, and state research credits of approximately $433,000, which, if not utilized earlier, will begin to expire in 2031. At December 31, 2017 the Company recorded $250,000 in research credits as prepaid to be offset against payroll tax liabilities associated with research and development personnel in 2018.
On December 22, 2017, the Tax Cuts and Jobs Acts was enacted into law. The new tax legislation represents a fundamental and dramatic shift in US taxation. The new legislation contains several key tax provisions that will impact the Company including the reduction of the corporate income tax rate to 21% effective January 1, 2018. ASC 740 requires the Company to recognize the effect of the tax law change in the period of enactment. The lower tax rate requires the Company to remeasure its deferred tax assets and liabilities in fourth quarter of 2017. The total amount of the Company’s deferred tax assets before valuation allowance decreased by $4.3 million as a result of the decrease in the federal tax rate.
ASC 740 requires the Company to recognize the effect of the tax law changes in the period of enactment. The lower corporate income tax rate will require the Company to remeasure its US deferred tax assets and liabilities as well as reassess the realizability of its deferred tax assets and liabilities. The SEC staff has issued SAB 118 which will allow the Company to record provisional amounts during a measurement period which is similar to the measurement period used when accounting for business combinations. The Company will continue to assess the impact of the recently enacted tax law on its business and financial statements and will reflect the provisional impact of the tax law change in the fourth quarter of fiscal 2017.
| 11. | Commitments and Contingencies |
Einstein License and Service Agreement
The Company’s commitments with respect to the Einstein License and the Service Agreement are summarized in Note 5.
Leased Facilities
On July 29, 2015, the Company entered into an operating lease agreement for its laboratory space for the period from August 1, 2015 through April 30, 2018. The lease contains escalating payments during the lease period. The Company records monthly rent expense on the straight-line basis, equal to the total of the lease payments over the lease term divided by the number of months of the lease term.
On November 14, 2016, the Company entered into an amended lease agreement that provided the Company with additional laboratory space. This amendment was effective beginning December 1, 2016 and continues through the expiration of the lease on April 30, 2018.
| F-21 |
On July 30, 2015, the Company entered into an operating lease agreement, as amended, for dedicated vivarium space for the period from August 1, 2015 through March 31, 2018. The operating lease agreement contains an option to increase the amount of space leased for an additional cost.
Future minimum lease payments under these leases at December 31, 2017 are as follows:
| Years Ending December 31, | ||||
| 2018 | $ | 1,004,000 | ||
| Total | $ | 1,004,000 | ||
Total lease expense, excluding the dedicated vivarium space, for the years ended December 31, 2017, 2016 and 2015 was included in the statement of operations as follows:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| General and administrative | $ | 244,678 | $ | 117,691 | $ | 42,455 | ||||||
| Research and development | 1,730,911 | 846,818 | 319,091 | |||||||||
| Total | $ | 1,975,589 | 964,509 | $ | 361,546 | |||||||
Legal Contingencies
The Company may be subject to various legal proceedings from time to time as part of its business. As of December 31, 2017, the Company was not a party to any legal proceedings or threatened legal proceedings, the adverse outcome of which, individually or in the aggregate, would have a material adverse effect on its business, financial condition or results of operations.
Registration Statement Filing Obligations
In conjunction with the sale of common stock on June 15, 2015 and on December 22, 2016 (see Note 8), including placement agent warrants issued to MDB, the Company granted the investors and MDB certain piggy-back registration rights commencing 180 days after the date that the Company becomes a reporting company under the Exchange Act, and for a period of five years thereafter, and a one-time demand registration right commencing 180 days after the date that the Company becomes a reporting company under the Exchange Act. These registration rights terminate for a holder upon the earliest of when all underlying securities held by the holder (i) have been sold pursuant to a registration statement, (ii) have been covered by an effective registration statement which has been effective for an aggregate period of 16 months, or (iii) may be sold by the holder pursuant to Rule 144 of the Securities Act of 1933, as amended, without regard to both the volume limitations for sales as provided in Rule 144 and the limitations for such sales provided in Rule 144(i).
In conjunction with the sale of common stock on December 22, 2016 (see Note 8), the Company granted the investors certain piggy-back and demand registration rights with respect to the common stock commencing six months after an initial public offering by the Company, by joinder to the registration rights agreement entered into by the Company and investors in connection with the Company’s June 15, 2015 private placement of common stock.
In conjunction with the Company’s initial public offering of common stock in December, 2017 (see Note 8), the Company issued warrants to the underwriter, MDB, to purchase shares of common stock. These warrants include certain piggy-back and demand registration rights with respect to the common stock issuable upon exercise thereof. These registration rights terminate for a holder upon the earliest of when all underlying securities held by the holder (i) have been sold pursuant to a registration statement, (ii) have been covered by an effective registration statement which may be kept effective as an evergreen registration statement, or (iii) may be sold by the holder within a 90 day period without registration pursuant to Rule 144 of the Securities Act of 1933, as amended, or consistent with applicable SEC interpretive guidance.
Upon the completion of an initial public offering, the Company is required to use its best efforts to file a registration statement covering the resale of the shares of common stock issued to Einstein (see Note 5) as soon as practicable, but no later than 180 calendar days from the date of the initial public offering, and to cause such registration statement to be declared effective by the United States Securities and Exchange Commission within 120 calendar days from the date of issuance of the shares.
| F-22 |
Agreement with MDB
Effective April 13, 2015, the Company entered into an agreement with MDB to engage such firm as its exclusive placement agent in connection with one or more offerings of the Company’s securities. As consideration for the services to be provided under the agreement, MDB was entitled to a cash fee equal to 10% of the gross proceeds raised in a financing, and warrants for up to 10% of the aggregate securities sold in an offering, exercisable for seven years at 100% of the offering price per share in a private offering and at not less than 120% of the offering price per share in a public offering, each for a cash consideration of $1,000. The agreement provided for the reimbursement of legal expenses incurred by MDB in conjunction with a securities offering.
Effective December 22, 2016, the Company amended the agreement with MDB to modify the consideration that MDB is entitled to receive for its placement agent services, to provide for a cash fee equal to 10% of the initial $10,000,000 of gross proceeds raised in a financing and 5% of the amount in excess of $10,000,000 in gross proceeds raised in a financing, and that MDB shall receive no placement agent warrants.
Agreements with Catalent
Catalent is a global provider of drug delivery technology and development solutions for drugs, biologics and consumer health products.
On March 7, 2017, the Company entered into an agreement with Catalent for Catalent to provide services on a sequential milestone basis with respect to the development and manufacture of the Company’s lead drug candidate, CUE-101. The services under the agreement are designed to support the preparation and filing of an Investigational New Drug Application with the United States Food and Drug Administration to allow for the commencement of a Phase 1 clinical trial of CUE-101 in the United States. The Company currently estimates that it will incur total direct costs under this agreement aggregating approximately $5,875,000, most of which the Company estimates to be incurred during the years ending December 31, 2017 and 2018. The company incurred total direct cost under this agreement aggregating $2.4 million during the year ended December 31, 2017, of which approximately $1.6 million was expensed during the year ended December 31, 2017. The remaining $0.8 million are reflected as research and development advances for which the Company anticipates receiving the contracted services within 12 months from the date of payment. Management periodically reviews and updates the project’s estimated budget and timeline.
On July 5, 2017, the Company entered into a separate Master Services Agreement with Catalent that outlines the terms and conditions under which Catalent will provide contract services with respect to the Company’s research and development activities for a period of five years. The Company may terminate this agreement without cause upon 90 days prior written notice. Unless and until terminated, this agreement will automatically be extended for successive one-year periods.
Collaboration Agreement with Merck
On November 14, 2017, the Company entered into an Exclusive Patent License and Research Collaboration Agreement (the “Collaboration Agreement”) with Merck for a partnership to research and develop certain of the Company’s proprietary biologics that target certain autoimmune disease indications (the “Initial Indications”). The Company views this Collaboration Agreement as a component of its development strategy since it will allow the Company to advance its autoimmune programs in partnership with a world class pharmaceutical company. The research program outlined in the Collaboration Agreement entails (1) research, discovery and development of certain CUE Biologics™ drug candidates up to the point of demonstration of certain biologically relevant effects (“Proof of Mechanism”) and (2) the further development by Merck of the CUE Biologics™ drug candidates that have demonstrated Proof of Mechanism (the “Proposed Product Candidates”) up to the point of demonstration of all or substantially all of the properties outlined in such Proposed Product Candidates’ profiles as described in the Collaboration Agreement.
| F-23 |
For the purposes of this collaboration, the Company has granted to Merck under the Collaboration Agreement an exclusive license under certain of its patent rights, including a sublicense of patent rights licensed from Einstein, to the extent applicable to the specific CUE Biologics™ that are elected to be developed by Merck. From the effective date of the Collaboration Agreement until the earlier of (i) the first achievement of Proof of Mechanism for a CUE Biologics™ drug candidate or (ii) 18 months after we notify the joint steering committee that the first Product Candidate has been synthesized under the research program, the Company is required to forebear from researching, developing or licensing to a third party rights related to any CUE Biologics™ drug candidate for the treatment of autoimmune diseases other than pursuant to the Collaboration Agreement. In addition, so long as Merck continues product development on a Proposed Product Candidate, the Company is restricted from conducting any development activities within the Initial Indication covered by such Proposed Product Candidate other than pursuant to the Collaboration Agreement. The Company is not required to forbear at any time, however, from developing other CUE Biologics™ for use in therapeutic areas other than autoimmune diseases, e.g., for use in treating cancer or infectious diseases.
In exchange for the licenses and other rights granted to Merck under the Collaboration Agreement, Merck paid the Company a $2.5 million nonrefundable up-front payment which was recorded as a deferred credit in the accompanying balance sheet at December 31, 2017. As related research and development costs are incurred, the applicable amount of deferred credit will be recorded as an offset to research and development costs in the statement of operations. The Company may be eligible to receive funding in developmental milestone payments, as well as tiered royalties, if all research, development, regulatory and commercial milestones agreed upon by both parties are successfully achieved.
| 12. | Cue Biopharma 401(k) Plan |
Effective as of January 1, 2017, the Company adopted the Cue Biopharma 401(k) Plan (the “Plan”) for all employees of the Company. Employees may participate in the Plan upon complying with the Plan’s eligibility requirements, subject to limitations imposed by the Internal Revenue Service. Under the Plan, the Company may match employee contributions at its discretion. The Company did not make any contributions to the Plan during the year ended December 31, 2017.
| 13. | Subsequent Events |
On January 16, 2018, the Company entered into an amended lease agreement that provided the Company with additional laboratory space. This amendment was effective beginning on January 16, 2018 and continues through the expiration of the lease on April 30, 2018.
On January 18, 2018, the Company entered into an operating lease agreement for its laboratory and office space at 21 Erie St. Cambridge, Massachusetts for the period from May 1, 2018 through April 30, 2021.The lease contains escalating payments during the lease period.
Future minimum lease payments under this lease are as follows:
| Years Ending December 31, | ||||
| 2018 | $ | 2,380,000 | ||
| 2019 | 3,752,000 | |||
| 2020 | 4,661,000 | |||
| 2021 | 1,554,000 | |||
| Total | $ | 12,347,000 | ||
| F-24 |
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
As of the end of the period covered by this report, management performed, with the participation of our principal executive and principal financial officers, an evaluation of the effectiveness of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act. Our disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, to allow timely decisions regarding required disclosures. Based on the evaluation, our principal executive and principal financial officers concluded that, as of December 31, 2017, our disclosure controls and procedures were effective.
Management’s Report on Internal Control Over Financial Reporting
This Annual Report on Form 10-K does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of the Company’s registered public accounting firm due to a transition period established by rules of the SEC for newly public companies.
Changes in Internal Control Over Financial Reporting
As previously reported in the registration statement on Form S-1 for our initial public offering, in connection with the audit of our financial statements for the year ended December 31, 2016 we identified a material weakness in our internal control over financial reporting. The material weakness related to a lack of effective controls to adequately restrict access and segregation of duties, specifically due to the limited number of staff in our accounting function. During the quarter ended December 31, 2017 we hired additional finance resources and in connection with our review of disclosure controls and procedures as of December 31, 2017 we concluded this material weakness had been remediated. Other than as described in the preceding sentence, for the quarter ended December 31, 2017, there were no changes in our internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
| 61 |
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2018 annual meeting of stockholders.
Item 11. Executive Compensation
The information required by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2018 annual meeting of stockholders.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholders Matters.
The information required by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2018 annual meeting of stockholders.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2018 annual meeting of stockholders.
Item 14. Principal Accountant Fees and Services
The information required by this Item is incorporated by reference to our Proxy Statement on Schedule 14A relating to our 2018 annual meeting of stockholders.
| 62 |
Item 15. Exhibits, Financial Statements and Schedules
| (a) | List of documents filed as part of this report: |
| 1. | Financial Statements (see “Financial Statements and Supplementary Data” at Item 8 and incorporated herein by reference). |
| 2. | Financial Statement Schedules (Schedules to the Financial Statements have been omitted because the information required to be set forth therein is not applicable or is shown in the accompanying Financial Statements or notes thereto) |
| 3. | Exhibits |
The following is a list of exhibits filed as part of this Annual Report on Form 10-K:
| 63 |
* Indicates management compensatory plan, contract or arrangement.
Item 16. Form 10-K Summary
Not applicable.
| 64 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Cue Biopharma, Inc. | ||
| Dated: March 29, 2018 | By: | /s/ Daniel R. Passeri |
|
Daniel R. Passeri Chief Executive Officer and Director (Principal Executive Officer) | ||
POWER OF ATTORNEY AND SIGNATURES
We, the undersigned officers and directors of Cue Biopharma, Inc., hereby severally constitute and appoint Daniel R. Passeri our true and lawful attorney, with full power to him to sign for us and in our names in the capacities indicated below, any amendments to this Annual Report on Form 10-K, and generally to do all things in our names and on our behalf in such capacities to enable Cue Biopharma, Inc. to comply with the provisions of the Securities Exchange Act of 1934, as amended, and all the requirements of the Securities Exchange Commission.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signatures | Title | Date | ||
| /s/ Daniel R. Passeri | Chief Executive Officer and Director | March 29, 2018 | ||
| Daniel R. Passeri | (Principal Executive Officer) | |||
| /s/ Gary Schuman | Interim Chief Financial Officer | March 29, 2018 | ||
| Gary Schuman | (Principal Financial and Accounting Officer) | |||
| /s/ Anthony DiGiandomenico | Director | March 29, 2018 | ||
| Anthony DiGiandomenico | ||||
| /s/ Cameron Gray | Director | March 29, 2018 | ||
| Cameron Gray | ||||
| /s/ Peter A. Kiener | Director | March 29, 2018 | ||
| Peter A. Kiener | ||||
| /s/ Steven McKnight | Director | March 29, 2018 | ||
| Steven McKnight | ||||
| /s/ Christopher Marlett | Director | March 29, 2018 | ||
| Christopher Marlett | ||||
| /s/ Barry Simon | Director | March 29, 2018 | ||
| Barry Simon |
| 65 |