Attached files
| file | filename |
|---|---|
| EX-99.1 - EXHIBIT 99.1 - FENNEC PHARMACEUTICALS INC. | tv488216_ex99-1.htm |
| EX-32.1 - EXHIBIT 32.1 - FENNEC PHARMACEUTICALS INC. | tv488216_ex32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - FENNEC PHARMACEUTICALS INC. | tv488216_ex31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - FENNEC PHARMACEUTICALS INC. | tv488216_ex31-1.htm |
| EX-23.2 - EXHIBIT 23.2 - FENNEC PHARMACEUTICALS INC. | tv488216_ex23-2.htm |
| EX-23.1 - EXHIBIT 23.1 - FENNEC PHARMACEUTICALS INC. | tv488216_ex23-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _____ to _____
Commission File Number: 001-32295
FENNEC PHARMACEUTICALS INC.
(formerly ADHEREX TECHNOLOGIES INC.)
(Exact Name of Registrant as Specified in Its Charter)
| British Columbia, Canada (State or Other Jurisdiction of Incorporation or Organization) |
20-0442384 (I.R.S. Employer Identification No.) |
| PO Box 13628, 68 TW Alexander Drive Research Triangle Park, NC (Address of Principal Executive Offices) |
27709 (Zip Code) |
(919) 636-4530
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO þ
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO þ
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES þ NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
YES þ NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer ¨ (Do not check if a smaller reporting company) |
Smaller reporting company þ | Emerging growth company ¨ |
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ¨ NO þ
The aggregate market value of the voting stock held by non-affiliates of the Registrant, computed by reference to the closing sales price of the Common Shares as reported by the OTCQB on June 30, 2017 (the last business day of the Registrant’s most recently completed second fiscal quarter) was $45,267,296 based upon a total of 7,128,708 shares held as of June 30, 2017 by persons believed to be non-affiliates of the Registrant (for purposes of this calculation, all of the Registrant’s officers, directors and 10% owners known to the Company are deemed to be affiliates of the Registrant).
As of March 16, 2018, there were 18,464,706 shares of the Registrant’s common shares outstanding.
FENNEC PHARMACEUTICALS INC.
2017 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements that involve significant risks and uncertainties. Our actual results, performance or achievements may be materially different from any results, performance or achievements expressed or implied by such forward-looking statements. Words such as “may,” “will,” “expect,” “believe,” “anticipate,” “intend,” “could,” “estimate,” “project,” “plan,” and other similar words are one way to identify such forward-looking statements. Forward-looking statements in this Annual Report include, but are not limited to, statements with respect to (1) our anticipated sources and uses of cash and cash equivalents; (2) our anticipated commencement dates, completion dates and results of clinical trials; (3) our efforts to pursue collaborations with the government, industry groups or other companies; (4) our anticipated progress and costs of our clinical and preclinical research and development programs; (5) our corporate and development strategies; (6) our expected results of operations; (7) our anticipated levels of expenditures; (8) our ability to protect our intellectual property; (9) our ability to fully comply with domestic and international governmental regulation; (10) the anticipated applications and efficacy of our drug candidates; (11) the nature and scope of potential markets for our drug candidates; (12) future legal liability; and (13) our ability to attract and retain key employees. All statements, other than statements of historical fact, included in this Annual Report that address activities, events or developments that we expect or anticipate will or may occur in the future are forward-looking statements. We include forward-looking statements because we believe that it is important to communicate our expectations to our investors. However, all forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties, as discussed below in Item 1A., “Risk Factors.” Although we believe the expectations reflected in the forward-looking statements are based upon reasonable assumptions, we can give no assurance that our expectations will be attained, and we caution you not to place undue reliance on such statements.
Our periodic and current reports are available, free of charge, after the material is electronically filed with, or furnished to, the SEC and EDGAR at http://www.sec.gov/edgar and the Canadian securities regulators on SEDAR, at www.sedar.com. The information provided on our website is not part of this Annual Report and is therefore not incorporated herein by reference.
| Item 1. | Business Overview |
Fennec Pharmaceuticals Inc. (“Fennec,” the “Company,” “we,” “us,” or “our”) is a biopharmaceutical company focused on the development of PEDMARKTM (a unique formulation of Sodium Thiosulfate (“STS”)) for the prevention of platinum-induced ototoxicity in pediatric cancer patients. We incorporated under the Canada Business Corporations Act ("CBCA”) in September 1996. Effective on August 25, 2011, the Company continued from the Canada Business Corporations Act to the Business Corporations Act (British Columbia) (the “Continuance”). The Continuance was approved by the shareholders of Fennec at the Company's June 2011 Annual and Special Meeting and by resolution of the Board of Directors on August 10, 2011. We have three wholly-owned subsidiaries: Oxiquant, Inc. and Fennec Pharmaceuticals, Inc., both Delaware corporations, and Cadherin Biomedical Inc., a Canadian company. With the exception of Fennec Pharmaceuticals, Inc., all subsidiaries are inactive.
On December 12, 2017, the Company announced the completion of an underwritten public offering of 2,352,950 common shares at a public offering price of $8.50 per share. In addition, Fennec issued an additional 135,670 common shares in connection with the partial exercise of the underwriters’ over-allotment option. The approximate total gross proceeds from the offering was $21.2 million.
On June 8, 2017, the Company completed the closing of a non-brokered private placement (the “Offering”) of 1,900,000 common shares for gross proceeds of $7.6 million. Each common share was issued at a price of $4.00.
Lead Product Candidate
The following is our only lead product candidate in the clinical stage of development:
| · | PEDMARKTM (a unique formulation of sodium thiosulfate (STS)) – sodium thiosulfate in a novel formulation, recently announced results of two Phase III clinical trials for the prevention of cisplatin induced hearing loss, or ototoxicity in children including the pivotal Phase III study SIOPEL 6 , “A Multicentre Open Label Randomised Phase 3 Trial of the Efficacy of Sodium Thiosulfate in Reducing Ototoxicity in Patients Receiving Cisplatin Chemotherapy for Standard Risk Hepatoblastoma,” and the proof of concept Phase III study “A Randomized Phase 3 Study of Sodium Thiosulfate for the Prevention of Cisplatin-Induced Ototoxicity in Children”. |
We continue to focus the Company’s resources on the development of PEDMARKTM.
PEDMARKTM
We have licensed from Oregon Health & Science University (“OHSU”) intellectual property rights for the use of PEDMARKTM as a chemoprotectant, and are developing PEDMARKTM as a protectant against the hearing loss often caused by platinum-based anti-cancer agents in children. Preclinical and clinical studies conducted by OHSU and others have indicated that PEDMARKTM can effectively reduce the incidence of hearing loss caused by platinum-based anti-cancer agents. We have received Orphan Drug Designation in the United States for the use of PEDMARKTM in the prevention of platinum-induced ototoxicity in pediatric patients.
| 1 |
Hearing loss among children receiving platinum-based chemotherapy is frequent, permanent and often severely disabling. The incidence of hearing loss in these children depends upon the dose and duration of chemotherapy, and many of these children require lifelong hearing aids. There is currently no established preventive agent for this hearing loss and only expensive, technically difficult and sub-optimal cochlear (inner ear) implants have been shown to provide some benefit. In addition, adults undergoing chemotherapy for several common malignancies, including ovarian cancer, testicular cancer, and particularly head and neck cancer and brain cancer, often receive intensive platinum-based therapy and may experience severe, irreversible hearing loss, particularly in the high frequencies.
The Company estimates based on data from Childhood and Adolescent Cancer Statistics (2014) and Automated Childhood Cancer Information System (ACCIS) that the annual incidence of pediatric solid tumor cases eligible for Platinum-Based therapy collectively in American and European markets is greater than 10,000.
Investigators at OHSU have conducted Phase I and Phase II studies which have shown that STS reduces the hearing loss associated with platinum-based chemotherapy. In one study at OHSU, the need for hearing aids to correct high frequency hearing loss was reduced from about 50% of patients being administered platinum-based chemotherapy to less than 5% of patients being administered platinum-based chemotherapy with STS.
STS has been studied by cooperative groups in two Phase III clinical studies of survival and reduction of ototoxicity, the Clinical Oncology Group (“COG”) Protocol ACCL0431 and the International Society of Pediatric Oncology (“SIOPEL 6”). The COG Protocol ACCL0431 enrolled one of five childhood cancers typically treated with intensive cisplatin therapy for localized and disseminated disease, including newly diagnosed hepatoblastoma, germ cell tumor, osteosarcoma, neuroblastoma, and medulloblastoma/PNET. SIOPEL 6 enrolled only hepatoblastoma patients with localized tumors.
In 2018, Fennec plans to pursue regulatory approval for PEDMARKTM based on the data from the pivotal SIOPEL 6 study along with the proof of principle data from COG ACCL0431. STS has received Orphan Drug Designation in the US in this setting and plans to pursue European Market Exclusivity for Pediatric Use ("PUMA") upon approval. Orphan Drug Designation will allow for 7.5 years of market exclusivity and PUMA will allow for 10 years of market exclusivity.
SIOPEL 6
In October 2007, we announced that our collaborative partner, the International Childhood Liver Tumour Strategy Group, known as SIOPEL, a multi-disciplinary group of specialists under the umbrella of the International Society of Pediatric Oncology, had launched a randomized Phase III clinical trial SIOPEL 6 to investigate whether STS reduces hearing loss in standard risk hepatoblastoma (liver) cancer patients receiving cisplatin as a monotherapy.
The study was initiated in October 2007 initially in the United Kingdom and completed enrollment at the end of 2014. 52 sites from 11 countries enrolled 109 evaluable patients. Under the terms of our agreement, SIOPEL conducts and funds all clinical activities and Fennec provides drug, drug distribution and pharmacovigilance, or safety monitoring, for the study. SIOPEL 6 was completed in December 2014 and the results of the trial were released in October 2017 at SIOP 2017.
The primary objectives of SIOPEL 6 are:
| · | To assess the efficacy of STS to reduce the hearing impairment caused by cisplatin. |
| · | To carefully monitor any potential impact of STS on response to cisplatin and survival. |
SIOPEL 6 - Results - October 2017
Background / Objectives:
Background: Bilateral high-frequency hearing loss is a serious permanent side-effect of cisplatin therapy, particularly debilitating when occurring in young children. STS has been shown to reduce cisplatin induced hearing loss. SIOPEL 6 is a phase III randomized trial to assess the efficacy of STS in reducing ototoxicity in young children treated with cisplatin (Cis) for Standard Risk Hepatoblastoma (SR-HB).
Design / Methods:
Methods: Newly diagnosed patients with SR-HB, defined as tumour limited to PRETEXT I, II or III, no portal or hepatic vein involvement, no intra-abdominal extrahepatic disease, AFP >100ng/ml and no metastases, were randomized to Cis or Cis+STS for 4 preoperative and 2 postoperative courses. Cisplatin 80mg/m2 was administered over 6 hours, STS 20g/m2 was administered intravenously over 15 minutes exactly 6 hours after stopping cisplatin. Tumour response was assessed after 2 and 4 preoperative cycles with serum AFP and liver imaging. In case of progressive disease (PD), STS was to be stopped and doxorubicin 60mg/m2 combined with cisplatin. The primary endpoint is centrally reviewed absolute hearing threshold, at the age of ≥3.5 years by pure tone audiometry.
| 2 |
Results:
Results: One hundred and nine randomized patients (52 Cisplatin only ("Cis") and 57 Cis+STS) are evaluable. The combination of Cis+STS was generally well tolerated. With a follow up time of 52 months for the patients the 3 year Event Free Survival ("EFS") for Cis is 78.8% Cisplatin and 82.1% for the Cis + STS. The 3 year Overall Survival ("OS") is 92.3% for Cis and 98.2% for Cis + STS. Treatment failure defined as Progressive Disease ("PD") at 4 cycles was equivalent in both arms. Among the first 99 evaluable patients, hearing loss occurred in 30/45=67.0% under Cis and in 20/54=37.0% under Cis +STS, corresponding to a relative risk of 0.56(P=0.0033).
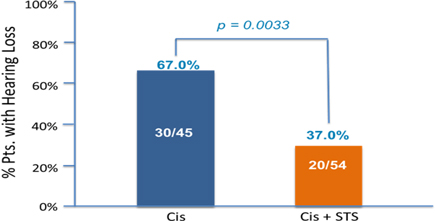
Conclusions:
This randomized phase III trial in SR-HB of cisplatin versus cisplatin plus sodium thiosulfate shows that the addition of sodium thiosulfate significantly reduces the incidence of cisplatin-induced hearing loss without any evidence of tumour protection.
COG ACCL0431
In March 2008, we announced the activation of a Phase III trial with STS to prevent hearing loss in children receiving cisplatin-based chemotherapy in collaboration with the Children’s Oncology Group (“COG ACCL0431”). The goal of this Phase III study was to evaluate in a multi-centered, randomized trial whether STS is an effective and safe means of preventing hearing loss in children receiving cisplatin-based chemotherapy for newly diagnosed germ cell, liver (hepatoblastoma), brain (medulloblastoma), nerve tissue (neuroblastoma) or bone (osteosarcoma) cancers. Eligible children, one to eighteen years of age, who were to receive cisplatin according to their disease-specific regimen and, upon enrollment in this study, were randomized to receive STS or not. Efficacy of STS was determined through comparison of hearing sensitivity at follow-up relative to baseline measurements using standard audiometric techniques. The Children’s Oncology Group is responsible for funding the clinical activities for the study and we are responsible for providing the drug, drug distribution and pharmacovigilance, or safety monitoring, for the study. The trial completed enrollment of 131 pediatric patients in the first quarter of 2012. The final results of COG ACCL0431 were published in Lancet Oncology in December 2016.
COG ACCL0431 - Results
COG Study ACCL0431, “A Randomized Phase III Study of Sodium Thiosulfate for the Prevention of Cisplatin-Induced Ototoxicity in Children,” finished enrollment of 131 patients of which 126 were eligible patients in Q1 2012. The patients had been previously diagnosed with childhood cancers.
The primary endpoint was to evaluate the efficacy of STS for prevention of hearing loss in children receiving cisplatin chemotherapy (hypothesis: 50% relative reduction in hearing loss).
Secondary endpoints included:
| · | Compare change in mean hearing thresholds. |
| · | Compare incidence of other Grade 3/4 toxicities (renal and hematological). |
| · | Monitor Event Free Survival (EFS) and Overall Survival (OS) in two groups. |
125eligible subjects were enrolled with germ cell tumor (32), osteosarcoma (29), neuroblastoma (26), medulloblastoma/pnet (26), hepatoblastoma (7) or other (5). Of these, 104 subjects (64 male and 29 <5 years old) were evaluable for the primary endpoint.
| 3 |
Subjects were randomized either to no treatment (control) or treatment with STS 16 grams/m2 IV over 15 minutes, 6 hours after each cisplatin dose. Hearing was measured using standard audiometry for age and data were reviewed centrally using American Speech-Language-Hearing Association criteria.
The proportion of subjects with hearing loss assessed at 4 weeks post the final cisplatin dose (primary endpoint):
| · | The proportion of hearing loss for STS vs. Control was 28.6% (14/49) vs. 56.4% (31/55), respectively (p=0.004). |
| · | In a predefined subgroup of patients less than 5 years old with 29 eligible subjects: STS vs. Control was 21.4% (3/14) vs. 73.3% (11/15), respectively (p=0.005). |
Conclusions:
| · | STS protects against cisplatin-induced hearing loss in children across a heterogeneous range of tumor types with even stronger efficacy in the protocol predefined subgroup of patients under five years old and is not associated with serious adverse events attributed to its use. |
| · | Further potential clinical use will be informed by the final results of SIOPEL 6 study. |
Intellectual Property
Patents are important to developing and protecting our competitive position. Our general policy is to seek patent protection in the United States, major European countries, Japan, Canada and other jurisdictions as appropriate for our compounds and methods. U.S. patents, as well as most foreign patents, are generally effective for 20 years from the date the earliest (priority) application was filed; however, U.S. patents that issue on applications filed before June 8, 1995 may be effective until 17 years from the issue date, if that is later than the 20 year date. In some cases, the patent term may be extended to recapture a portion of the term lost during the U.S. Food and Drug Administration (“FDA”) regulatory review or because of U.S. Patent and Trademark Office, or USPTO, delays in prosecuting the application. The duration of foreign patents varies similarly, in accordance with local law.
Currently, we have licensed from Oregon Health and Science University 1 U.S. and 9 foreign patents which expire in Europe. All patents expire in 2021, with an additional 2 patents pending.
In addition, periods of marketing exclusivity for STS may also be possible in the United States under orphan drug status and in Europe under European Market Exclusivity for Pediatric Use. We obtained U.S. Orphan Drug Designation for the use of STS in the prevention of platinum-induced ototoxicity in pediatric patients in 2004 which provides 7.5 years of market exclusivity upon approval. We plan to pursue European Market Exclusivity for Pediatric Use upon approval which would allow for 10 years of market exclusivity.
Our success is significantly dependent on our ability to obtain and maintain patent protection for our product candidate, both in the United States and abroad. The patent position of biotechnology and pharmaceutical companies, in general, is highly uncertain and involves complex legal and factual questions, which often results in apparent inconsistencies regarding the breadth of claims allowed and general uncertainty as to their legal interpretation and enforceability. Further, our principal candidate STS, is based on previously known compounds, and the candidates or products that we develop in the future may include or be based on the same or other compounds owned or produced by other parties, some or all of which may not be subject to effective patent protection. In addition, regimens that we may develop for the administration of pharmaceuticals, such as specifications for the frequency, timing and amount of dosages, may not be patentable. Accordingly, our patent applications may not result in patents being issued and issued patents may not afford effective protection. In addition, products or processes that we develop may turn out to be covered by third party patents, in which case we may require a license under such patents if we intend to continue the development of those products or processes.
Our patent position and proprietary rights are subject to certain risks and uncertainties. Please read the “Risk Factors” section of this Annual Report for information about certain risks and uncertainties that may affect our patent position and proprietary rights.
We also rely upon unpatented confidential information to remain competitive. We protect such information principally through confidentiality agreements with our employees, consultants, outside scientific collaborators, and other advisers. In the case of our employees, these agreements also provide, in compliance with relevant law, that inventions and other intellectual property conceived by such employees during their employment shall be our exclusive property.
Corporate Relationships
License Agreement with Oregon Health & Science University
On February 20, 2013, Fennec entered into a new exclusive license agreement with OHSU for exclusive worldwide license rights to intellectual property directed to STS and its use for chemoprotection, including the prevention of ototoxicity induced by platinum chemotherapy, in humans (the "New OHSU Agreement").
The term of the New OHSU Agreement expires on the date of the last to expire claim(s) covered in the patents licensed to us, unless earlier terminated as provided in the agreement. STS is currently protected by methods of use patents that we exclusively licensed from OHSU that expire in Europe, Canada and Australia in 2021 and are currently pending in the United States and Japan. The New OHSU Agreement is terminable by either Fennec or OHSU in the event of a material breach of the agreement by either party after 45 days prior written notice. Fennec has the right to terminate the New OHSU Agreement at any time upon 60 days prior written notice and payment of all fees due to OHSU under the New OHSU Agreement.
| 4 |
On May 18, 2015, Fennec negotiated an amendment ("Amendment 1") to the exclusive license agreement with OHSU. Amendment 1 expands the exclusive license agreement signed with OHSU on February 20, 2013 or New OHSU Agreement to include the use of N-acetylcysteine as a standalone therapy and/or in combination with STS for the prevention of ototoxicity induced by chemotherapeutic agents to treat cancers. Further, Amendment 1 adjusts select milestone payments entered in the OHSU Agreement including but not limited to the royalty rate on net sales for licensed products, royalty rate from sublicensing of the licensed technology and the fee payable upon the regulatory approval of a licensed product. The term of Amendment 1 under the OHSU Agreement expires on the date of the last to expire claim(s) covered in the patents licensed to Fennec or 8 years, whichever is later. In the event a licensed product obtains regulatory approval and is covered by the Orphan Drug Designation, the parties will in good faith amend the term of the agreement.
Competition
Competition in the biotechnology and pharmaceutical industries is intense. We expect that if our product candidate achieves regulatory approval for sale, it will compete on the basis of drug efficacy, safety, patient convenience, reliability, ease of manufacture, price, marketing, distribution, and patent protection, among other variables. Our competitors may develop technologies or drugs that are more effective, safer or more affordable than any we may develop.
There are a number of different approaches to the development of therapeutics for the treatment of cancer that are currently being used and studied. These approaches include: (i) surgery to excise the cancerous tissue; (ii) radiation therapy, which attacks cancerous cells but does not easily distinguish between healthy and diseased cells; (iii) chemotherapy, which works by preventing a cancerous cell from dividing or by killing cells that quickly divide; (iv) immunotherapy, which stimulates the body’s immune system to respond to the disease; and (v) hormone therapy, which may slow the growth of cancer cells or even kill them.
We are aware of a number of companies engaged in the research, development and testing of new cancer therapies or means of increasing the effectiveness of existing therapies, including, among many others, Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Eli Lilly, Eisai, Merck KGaA, Novartis, Johnson & Johnson, Pfizer, Roche, Taiho and Sanofi-Aventis. Some of these companies have products that have already received, or are in the process of receiving, regulatory approval or are in later stages of clinical development than our product. Many of them have much greater financial resources than we do. Many of these companies have marketed drugs or are developing targeted cancer therapeutics which, depending upon the mechanism of action of such agents, could be viewed as competitors.
We are not aware of any commercially available agents that reduce the incidence of hearing loss associated with the use of platinum-based anti-cancer agents, for which purpose we are developing STS. There are several potential competitive agents with activity in preclinical or limited clinical settings. These include: D-methionine, an amino acid that has been shown to protect against hearing loss in experimental settings but was demonstrated to be inferior to STS in comparative studies; SPI-3005, an oral agent primarily being developed by Sound Pharmaceuticals for noise and age-related hearing loss but in early Phase II trials for chemotherapy related hearing loss, which mimics glutathione peroxidase and induces the intracellular induction of glutathione; N-acetylcysteine and amifostine, which have shown effectiveness (but less than STS) in experimental systems; and Vitamin E, salicylate and tiopronin, which have all demonstrated moderate activity in rat models to protect against cisplatin-induced ototoxicity, but no clinical trials have been performed. Cochlear implants, which are small electronic devices that are surgically placed in the inner ear to assist with certain types of deafness, are utilized to offer some relief but are often suboptimal.
Many of our existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. In addition, many of these competitors have extensive experience with preclinical testing and human clinical trials and in obtaining regulatory approvals. In addition, many of the smaller companies that compete with us have formed collaborative relationships with large, established companies to support the research, development, clinical trials and commercialization of any products that they may develop. We may rely on third parties to commercialize the products we develop, and our success will depend in large part on the efforts and competitive merit of these collaborative partners. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for research, clinical development and marketing of products similar to those we seek to develop. These companies and institutions compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our projects. The existence of competitive products, including products or treatments of which we are not aware, or products or treatments that may be developed in the future, may adversely affect the marketability of any products that we may develop.
| 5 |
Government Regulation
The production and manufacture of our product candidate and our research and development activities are subject to significant regulation for safety, efficacy and quality by various governmental authorities around the world. Before new pharmaceutical products may be sold in the U.S. and other countries, clinical trials of the product must be conducted and the results submitted to appropriate regulatory agencies for approval. Clinical trial programs must establish efficacy, determine an appropriate dose and regimen, and define the conditions for safe use. This is a high-risk process that requires stepwise clinical studies in which the candidate product must successfully meet predetermined endpoints. In the U.S., the results of the preclinical and clinical testing of a product are then submitted to the FDA in the form of a Biologics License Application or a New Drug Application. In response to these submissions, the FDA may grant marketing approval, request additional information or deny the application if it determines the application does not provide an adequate basis for approval. Similar submissions are required by authorities in other jurisdictions who independently assess the product and may reach the same or different conclusions.
The receipt of regulatory approval often takes a number of years, involves the expenditure of substantial resources and depends on a number of factors, including the severity of the disease in question, the availability of alternative treatments and the risks and benefits demonstrated in clinical trials. On occasion, regulatory authorities may require larger or additional studies, leading to unanticipated delay or expense. Even after initial approval from the FDA or other regulatory agencies has been obtained, further clinical trials may be required to provide additional data on safety and effectiveness. Additional trials are required to gain clearance for the use of a product as a treatment for indications other than those initially approved. Furthermore, the FDA and other regulatory agencies require companies to disclose clinical trial results. Failure to disclose such results within applicable time periods could result in penalties, including civil monetary penalties.
In Canada, these activities are subject to regulation by Health Canada’s Therapeutic Products Directorate, or TPD, and the rules and regulations promulgated under the Food and Drug Act. In the United States, drugs and biological products are subject to regulation by the FDA. The FDA requires licensing of manufacturing and contract research facilities, carefully controlled research and testing of products and governmental review and approval of results prior to marketing therapeutic products. Additionally, the FDA requires adherence to “Good Laboratory Practices” as well as “Good Clinical Practices” during clinical testing and “Good Manufacturing Practices” and adherence to labeling and supply controls. The systems of new drug approvals in Canada and the United States are substantially similar, and are generally considered to be among the most rigorous in the world.
Generally, the steps required for drug approval in Canada and the United States, specifically in cancer related therapies, include:
| · | Preclinical Studies: Preclinical studies, also known as non-clinical studies, primarily involve evaluations of pharmacology, toxic effects, pharmacokinetics and metabolism of a drug in animals to provide evidence of the relative safety and bioavailability of the drug prior to its administration to humans in clinical studies. A typical program of preclinical studies takes 18 to 24 months to complete. The results of the preclinical studies as well as information related to the chemistry and comprehensive descriptions of proposed human clinical studies are then submitted as part of the Investigational New Drug, application to the FDA, a Clinical Trial Application to the TPD, or similar submission to other foreign regulatory bodies. This is necessary in Canada, the United States and most other countries prior to undertaking clinical studies. Additional preclinical studies are conducted during clinical development to further characterize the toxic effects of a drug prior to submitting a marketing application. |
| · | Phase I Clinical Trials: Most Phase I clinical trials take approximately one year to complete and are usually conducted on a small number of healthy human subjects to evaluate the drug’s safety, tolerability and pharmacokinetics. In some cases, such as cancer indications, Phase I clinical trials are conducted in patients rather than healthy volunteers. |
| · | Phase II Clinical Trials: Phase II clinical trials typically take one to two years to complete and are generally carried out on a relatively small number of patients, generally between 15 and 50, in a specific setting of targeted disease or medical condition, in order to provide an estimate of the drug’s effectiveness in that specific setting. This phase also provides additional safety data and serves to identify possible common short-term side effects and risks in a somewhat larger group of patients. Phase II testing frequently relates to a specific disease, such as breast or lung cancer. Some contemporary methods of developing drugs, particularly molecularly targeted therapies, do not require broad testing in specific diseases, and instead permit testing in subsets of patients expressing the particular marker. In some cases, such as cancer indications, the company sponsoring the new drug may submit a marketing application to seek accelerated approval of the drug based on evidence of the drug’s effect on a “surrogate endpoint” from Phase II clinical trials. A surrogate endpoint is a laboratory finding or physical sign that may not be a direct measurement of how a patient feels, functions or survives, but is still considered likely to predict therapeutic benefit for the patient. If accelerated approval is received, the company sponsoring the new drug must continue testing to demonstrate that the drug indeed provides therapeutic benefit to the patient. |
| · | Phase III Clinical Trials: Phase III clinical trials typically take two to four years to complete and involve tests on a much larger population of patients suffering from the targeted condition or disease. These studies involve conducting controlled testing and/or uncontrolled testing in an expanded patient population, numbering several hundred to several thousand patients, at separate test sites, known as multi-center trials, to establish clinical safety and effectiveness. These trials also generate information from which the overall benefit-risk relationship relating to the drug can be determined and provide a basis for drug labeling. Phase III trials are generally the most time consuming and expensive part of a clinical trial program. In some instances, governmental authorities, such as the FDA, will allow a single Phase III clinical trial to serve as a pivotal efficacy trial to support a Marketing Application. |
| · | Marketing Application: Upon completion of Phase III clinical trials, the pharmaceutical company sponsoring the new drug assembles all the chemistry, preclinical and clinical data and submits it to the TPD or the FDA as part of a New Drug Submission in Canada or a New Drug Application, in the United States. The marketing application is then reviewed by the applicable regulatory body for approval to market the product. The review process generally takes twelve to eighteen months. |
| 6 |
Any clinical trials that we conduct may not be successfully completed, either in a satisfactory time period or at all. The typical time periods described above may vary substantially and may be materially longer. In addition, the FDA and its counterparts in other countries have considerable discretion to discontinue trials if they become aware of any significant safety issues or convincing evidence that a therapy is not effective for the indication being tested. It is possible the FDA and its counterparts in other countries may not (i) allow clinical trials to proceed at any time after receiving an Investigational New Drug, (ii) allow further clinical development phases after authorizing a previous phase, or (iii) approve marketing of a drug after the completion of clinical trials.
While European, U.S. and Canadian regulatory systems require that medical products be safe, effective, and manufactured according to high quality standards, the drug approval process in Europe differs from that in the United States and Canada and may require us to perform additional preclinical or clinical testing regardless of whether FDA or TPD approval has been obtained. The amount of time required to obtain necessary approvals may be longer or shorter than that required for FDA or TPD approval. European Union Regulations and Directives generally classify health care products either as medicinal products, medical devices or in vitro diagnostics. For medicinal products, marketing approval may be sought using either the centralized procedure of the European Agency for the Evaluation of Medicinal Products, or EMEA, or the decentralized, mutual recognition process. The centralized procedure, which is mandatory for some biotechnology derived products, results in an approval recommendation from the EMEA to all member states, while the European Union mutual recognition process involves country by country approval.
Good Clinical Practices
The FDA and other regulatory agencies promulgate regulations and standards, commonly referred to as current Good Clinical Practices for designing, conducting, monitoring, auditing and reporting the results of clinical trials to ensure that the data and results are accurate and that the trial participants are adequately protected. The FDA and other regulatory agencies enforce Good Clinical Practices through periodic inspections of trial sponsors, principal investigators and trial sites. If our study sites fail to comply with applicable Good Clinical Practices, the clinical data generated in our clinical trials may be deemed unreliable and relevant regulatory agencies may require us to perform additional clinical trials before approving our marketing applications.
Good Manufacturing Practices
The FDA and other regulatory agencies regulate and inspect equipment, facilities and processes used in the manufacture of pharmaceutical and biological products prior to approving a product. If, after receiving approval from regulatory agencies, a company makes a material change in manufacturing equipment, location or process, additional regulatory review and approval may be required. All facilities and manufacturing techniques that may be used for the manufacture of our products must comply with applicable regulations governing the production of pharmaceutical products known as "Good Manufacturing Practices."
Orphan Drug Act
Under the U.S. Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which generally is a disease or condition that affects fewer than 200,000 individuals in the U.S. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, i.e., the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years following marketing approval, except in certain very limited circumstances, such as if the later product is shown to be clinically superior to the orphan product. Legislation similar to the U.S. Orphan Drug Act has been enacted in other countries, including within the European Union.
Pediatric Marketing Use Authorization
The PUMA approval is granted by the European Medicines Agency and is intended exclusively for pediatric (patients under 18 years of age) use. PUMA approval is valid in all countries within the European Economic Area. The PUMA process was established to make it more efficient for pharmaceutical companies to market drugs for children. New data for PUMA drugs are protected for 10 years and the applications are, in part, exempt from fees.
Other Laws
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights may be subject to national or supranational antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
Research and Development
Our research and development efforts have been focused on the development of PEDMARKTM since 2013.
| 7 |
We have established relationships with contract research organizations, universities and other institutions, which we utilize to perform many of the day-to-day activities associated with our drug development. Where possible, we have sought to include leading scientific investigators and advisors to enhance our internal capabilities. Research and development issues are reviewed internally by our executive management and supporting scientific team.
Research and development expenses totaled $1.9 million and $0.5 million for the fiscal years ended December 31, 2017 and 2016, respectively. The Company has increased its research and development expenses related to PEDMARKTM as a result of the Company drug manufacturing activities related to the preparation for registration batches.
Our product candidate still requires significant, time-consuming and costly research and development, testing and regulatory clearances. In developing our product candidate, we are subject to risks of failure that are inherent in the development of products based on innovative technologies. For example, it is possible that our product candidate will be ineffective or toxic, or will otherwise fail to receive the necessary regulatory clearances. There is a risk that our product candidate will be uneconomical to manufacture or market or will not achieve market acceptance. There is also a risk that third parties may hold proprietary rights that preclude us from marketing our product candidate or that others will market a superior or equivalent product. As a result of these factors, we are unable to accurately estimate the nature, timing and future costs necessary to complete the development of this product candidate. In addition, we are unable to reasonably estimate the period when material net cash inflows could commence from the sale, licensing or commercialization of such product candidate, if ever.
Employees
At December 31, 2017, we had three employees (our Chief Executive Officer, Chief Financial Officer and Controller). These employees are employed on a full-time basis and there are no part-time employees. The company uses independent contractors to perform certain daily operations of the company.
| Item 1A. | Risk Factors |
An investment in our common shares involves a significant risk of loss. You should carefully read this entire report and should give particular attention to the following risk factors. You should recognize that other significant risks may arise in the future, which we cannot reasonably foresee at this time. Also, the risks that we now foresee might affect us to a greater or different degree than currently expected. There are a number of important factors that could cause our actual results to differ materially from those expressed or implied by any of our forward-looking statements in this Annual Report. These factors include, without limitation, the risk factors listed below and other factors presented throughout this Annual Report and any other documents filed by us with the Securities and Exchange Commission, or the SEC, and the Canadian securities regulators on SEDAR which can be accessed at www.sedar.com.
Risks Related to Our Business
We have a history of significant losses and have had no revenues to date through the sale of our products. If we do not generate significant revenues, we will not achieve profitability.
To date, we have been engaged primarily in research and development activities. We have had no revenues through the sale of our products, and we do not expect to have significant revenues until we are able to either sell our product candidate after obtaining applicable regulatory approvals or we establish collaborations that provide us with up-front payments, licensing fees, milestone payments, royalties or other revenue. We have incurred significant operating losses every year since our inception on September 3, 1996. We reported a loss of approximately $7.0 million (including a non-cash loss on derivative liabilities of $0.13 million) for the year ended December 31, 2017, and reported a net loss of approximately $2.8 million (which included a non-cash gain on derivative liabilities of $0.05 million) for the year ended December 31, 2016. At December 31, 2017, we had an accumulated deficit of approximately $121.4 million. We anticipate incurring substantial additional losses due to the need to spend substantial amounts on activities required for regulatory approval of PEDMARKTM, commercial launch preparation of PEDMARKTM , anticipated research and development activities, and general and administrative expenses, among other factors. We have not commercially introduced any products. Our ability to attain profitability will depend upon our ability to fund and develop products that are safe, effective and commercially viable, to obtain regulatory approval for the manufacture and sale of our product candidate and to license or otherwise market our product candidate successfully. Any revenues generated from such product, assuming it is successfully developed, marketed and sold, may not be realized for a number of years. We may never achieve or sustain profitability on an ongoing basis.
PEDMARKTM is currently our only product candidate and there is no assurance that we will successfully develop PEDMARKTM into a commercially viable product.
Since our formation in September 1996, we have engaged in research and development programs. We have generated no revenue from product sales, do not have any products currently available for sale, and none are expected to be commercially available for sale until we have completed regulatory approval of PEDMARKTM. PEDMARKTM is currently our only product candidate. There can be no assurance that the research we fund and manage will lead PEDMARKTM or any future product candidate to become a commercially viable product. We have completed enrollment of two Phase III studies for PEDMARKTM. We anticipate substantial regulatory review prior to the commercialization of PEDMARKTM.
| 8 |
We anticipate the need for additional capital in the future and if we cannot raise additional capital, we will not be able to fulfill our business plan.
We need to obtain additional funding in the future in order to finance our business strategy, operations and growth. We may not be able to obtain additional financing in sufficient amounts or on acceptable terms when needed. If we fail to arrange for sufficient capital on a timely basis, we may be required to curtail our business activities until we can obtain adequate financing. Debt financing must be repaid regardless of whether or not we generate profits or cash flows from our business activities. Equity financing may result in dilution to existing shareholders and may involve securities that have rights, preferences, or privileges that are senior to our common shares or other securities. If we cannot raise sufficient capital when necessary, we will likely have to curtail operations and you may lose part or all of your investment.
If we do not maintain current or enter into new collaborations with other companies, we might not successfully develop our product candidate or generate sufficient revenues to expand our business.
We currently rely on scientific and research and development collaboration arrangements with academic institutions and other third party collaborators, including an exclusive worldwide license from OHSU for PEDMARKTM. We also rely on collaborators for testing PEDMARKTM, including SIOPEL and the Children’s Oncology Group.
The agreements with OHSU are terminable by either party in the event of an uncured breach by the other party. We may also terminate our agreement with OHSU at any time upon prior written notice of specified durations to the licensor. Termination of any of our collaborative arrangements could materially adversely affect our business. For example, if we are unable to make the necessary payments under these agreements, the licensor might terminate the agreement which might have a material adverse impact. In addition, our collaborators might not perform as agreed in the future.
Since we conduct a significant portion of our research and development through collaborations, our success may depend significantly on the performance of such collaborators, as well as any future collaborators. Collaborators might not commit sufficient resources to the research and development or commercialization of our product candidate. Economic or technological advantages of products being developed by others, among other factors, could lead our collaborators to pursue other product candidates or technologies in preference to those being developed in collaboration with us. The commercial potential of, development stage of and projected resources required to develop our drug candidate will affect our ability to maintain current collaborations or establish new collaborators. There is a risk of dispute with respect to ownership of technology developed under any collaboration. Our management of any collaboration will require significant time and effort as well as an effective allocation of resources. We may not be able to simultaneously manage a large number of collaborations.
Our product candidate is still in development. Due to the long, expensive and unpredictable drug development process, we might not ever successfully develop and commercialize our product candidate.
In order to achieve profitable operations, we, alone or in collaboration with others, must successfully fund, develop, manufacture, introduce and market our product candidate. The time necessary to achieve market success for any individual product is long and uncertain. Our product candidate and research programs are in clinical development and require significant, time-consuming and costly research, testing and regulatory clearances. In developing our product candidate, we are subject to risks of failure that are inherent in the development of therapeutic products based on innovative technologies. The results of preclinical and initial clinical trials are not necessarily predictive of future results. Our product candidate might not be economical to manufacture or market or might not achieve market acceptance. In addition, third parties might hold proprietary rights that preclude us from marketing our product candidates or others might market equivalent or superior products.
We may need to conduct additional human clinical trials to assess our product candidate. If these trials are delayed or are unsuccessful, our development costs will significantly increase and our business prospects may suffer.
Before obtaining regulatory approvals for the commercial sale of our product candidate, we must demonstrate, through preclinical studies with animals and clinical trials with humans, that our product candidate is safe and effective for use in each target indication. To date, we have performed only limited clinical trials. Much of our testing has been conducted on animals or on human cells in the laboratory, and the benefits of treatment seen in animals or on human cells in a laboratory setting may not ultimately be obtained in human clinical trials. As a result, we may need to perform significant additional research and development activities and conduct extensive preclinical and clinical testing prior to any application for commercial use. We may suffer significant setbacks in additional clinical trials, and the trials may demonstrate our product candidate to be unsafe or ineffective. We may also encounter problems in our clinical trials that will cause us to delay, suspend or terminate those clinical trials, which would increase our development costs and harm our financial results and commercial prospects. Identifying and qualifying patients to participate in clinical trials of our potential products is critically important to our success. The timing of our clinical trials depends on, among other things, the speed at which we can recruit patients to participate in testing our product candidate. We have experienced delays in some of our clinical trials and we may experience significant delays in the future. If patients are unwilling to participate in our trials because of competing clinical trials for similar patient populations, perceived risk or any other reason, the timeline for recruiting patients, conducting trials and obtaining regulatory approval of potential products will be delayed. Other factors that may result in significant delays include obtaining regulatory or ethics review board approvals for proposed trials, reaching agreement on acceptable terms with prospective clinical trial sites, and obtaining sufficient quantities of drugs for use in the clinical trials. Such delays could result in the termination of the clinical trials altogether.
| 9 |
Regulatory approval of our product candidate is time-consuming, expensive and uncertain, and could result in unexpectedly high expenses and delay our ability to sell our product.
Development, manufacture and marketing of our product is subject to extensive regulation by governmental authorities in the United States and other countries. This regulation could require us to incur significant unexpected expenses or delay or limit our ability to sell our product candidate. Our clinical studies might be delayed or halted, or additional studies might be required, for various reasons, including:
| · | there is a lack of sufficient funding; |
| · | the drug is not effective; |
| · | patients experience severe side effects during treatment; |
| · | appropriate patients do not enroll in the studies at the rate expected; |
| · | drug supplies are not sufficient to treat the patients in the studies; or |
| · | we decide to modify the drug during testing. |
If regulatory approval of our product is granted, it will be limited to those indications for which the product has been shown to be safe and effective, as demonstrated to the satisfaction of the FDA and foreign regulators through clinical studies. Furthermore, approval might entail ongoing requirements for post-marketing studies. Even if regulatory approval is obtained, labeling and promotional activities are subject to continual scrutiny by the FDA and state and foreign regulatory agencies and, in some circumstances, the Federal Trade Commission. FDA enforcement policy prohibits the marketing of approved products for unapproved, or off-label, uses. These regulations and the FDA’s interpretation of them might impair our ability to effectively market our product.
We and our third-party manufacturers are also required to comply with the applicable current FDA Good Manufacturing Practices regulations, which include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Further, manufacturing facilities must be approved by the FDA before they can be used to manufacture our product, and they are subject to additional FDA inspection. If we fail to comply with any of the FDA’s continuing regulations, we could be subject to reputational harm and sanctions, including:
| · | delays, warning letters and fines; |
| · | product recalls or seizures and injunctions on sales; |
| · | refusal of the FDA to review pending applications; |
| · | total or partial suspension of production; |
| · | withdrawals of previously approved marketing applications; and |
| · | civil penalties and criminal prosecutions. |
In addition, identification of side effects after a drug is on the market or the occurrence of manufacturing problems could cause subsequent withdrawal of approval, reformulation of the drug, additional testing or changes in labeling of the product.
We may be unable to effectively deploy the proceeds from our recent financings for the development of PEDMARKTM.
In December of 2017, the Company announced the completion of an underwritten public offering for gross proceeds of $21.2 million. In June of 2017, the Company announced the closing of a non-brokered private placement for gross proceeds of $7.6 million. Any inability on our part to manage effectively the deployment of this capital could limit our ability to successfully develop PEDMARKTM.
If our licenses to proprietary technology owned by others are terminated or expire, we may suffer increased development costs and delays, and we may not be able to successfully develop our product candidate.
The development of our drug candidate and the manufacture and sale of any products that we develop will involve the use of processes, products and information, some of the rights to which are owned by others. STS is licensed under agreements with OHSU. Although we have obtained licenses or rights with regard to the use of certain processes, products and information, the licenses or rights could be terminated or expire during critical periods and we may not be able to obtain, on favorable terms or at all, licenses or other rights that may be required. Some of these licenses provide for limited periods of exclusivity that may be extended only with the consent of the licensor, which may not be granted.
| 10 |
If we are unable to adequately protect or maintain our patents and licenses related to our product candidate, or if we infringe upon the intellectual property rights of others, we may not be able to successfully develop and commercialize our product candidate.
The value of our technology will depend in part upon our ability, and those of our collaborators, to obtain patent protection or licenses to patents, maintain trade secret protection and operate without infringing on the rights of third parties. Although we have successfully pursued patent applications in the past, it is possible that:
| · | some or all of our pending patent applications, or those we have licensed, may not be allowed; |
| · | proprietary products or processes that we develop in the future may not be patentable; |
| · | any issued patents that we own or license may not provide us with any competitive advantages or may be successfully challenged by third parties; or |
| · | the patents of others may have an adverse effect on our ability to do business. |
It is not possible for us to be certain that we are the original and first creator of inventions encompassed by our pending patent applications or that we were the first to file patent applications for any such inventions. Further, any of our patents, once issued, may be declared by a court to be invalid or unenforceable.
STS is currently protected by methods of use patents that we exclusively licensed from OHSU that expire in Europe in 2021 and are currently pending in the United States. In addition, periods of marketing exclusivity for STS may also be possible in the United States under orphan drug status. We obtained Orphan Drug Designation in the United States for the use of STS in the prevention of platinum-induced ototoxicity in pediatric patients in 2004; if it is subsequently approved, will have seven and a half years of pediatric exclusivity in the United States from the approval date. Refer to the “Description of Business” section of this Annual Report for a further description of the United States Orphan Drug Designation.
We may be required to obtain licenses under patents or other proprietary rights of third parties but the extent to which we may wish or need to do so is unknown. Any such licenses may not be available on terms acceptable to us or at all. If such licenses are obtained, it is likely they would be royalty bearing, which would reduce any future income. If licenses cannot be obtained on an economical basis, we could suffer delays in market introduction of planned products or their introduction could be prevented, in some cases after the expenditure of substantial funds. If we do not obtain such licenses, we would have to design around patents of third parties, potentially causing increased costs and delays in product development and introduction or precluding us from developing, manufacturing or selling our planned products, or our ability to develop, manufacture or sell products requiring such licenses could be foreclosed.
Litigation may also be necessary to enforce or defend patents issued or licensed to us or our collaborators or to determine the scope and validity of a third party’s proprietary rights. We could incur substantial costs if litigation is required to defend ourselves in patent suits brought by third parties, if we participate in patent suits brought against or initiated by our collaborators, or if we initiate such suits. We might not prevail in any such action. An adverse outcome in litigation or an interference to determine priority or other proceeding in a court or patent office could subject us to significant liabilities, require disputed rights to be licensed from other parties or require us or our collaborators to cease using certain technology or products. Any of these events would likely have a material adverse effect on our business, financial condition and results of operations.
Much of our technological know-how that is not patentable may constitute trade secrets. Our confidentiality agreements might not provide for meaningful protection of our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure of information. In addition, others may independently develop or obtain similar technology and may be able to market competing products and obtain regulatory approval through a showing of equivalency to our product that has obtained regulatory approvals, without being required to undertake the same lengthy and expensive clinical studies that we would have already completed.
The vulnerability to off-label use or sale of our product candidate that are covered only by “method of use” patents may cause downward pricing pressure on the product candidate if they are ever commercialized and may make it more difficult for us to enter into collaboration or partnering arrangements for the development of this product candidate.
STS is currently only covered by “method of use” patents, which covers the use of certain compounds to treat specific conditions, and are not covered by “composition of matter” patents, which would cover the chemical composition of the compound. Method of use patents provide less protection than composition of matter patents because of the possibility of off-label competition if other companies develop or market the compound for other uses. If another company markets a drug that we expect to market under the protection of a method of use patent, physicians may prescribe the other company’s drug for use in the indication for which we obtain approval and have a patent, even if the other company’s drug is not approved for such an indication. Off-label use and sales could limit our sales and exert pricing pressure on any product we develop covered only by method of use patents. Also, it may be more difficult to find a collaborator to license or support the development of our product candidate that is only covered by method of use patents.
| 11 |
If our third-party manufacturers breach or terminate their agreements with us, or if we are unable to secure arrangements with third party manufacturers on acceptable terms as needed in the future, we may suffer significant delays and additional costs.
We have no experience manufacturing products and do not currently have the resources to manufacture any products that we may develop. We currently have agreements with contract manufacturers for clinical supplies of PEDMARKTM, including drug substance providers and drug product suppliers, but they might not perform as agreed in the future or may terminate our agreements with them before the end of the required term. Significant additional time and expense would be required to effect a transition to a new contract manufacturer.
We plan to continue to rely on contract manufacturers for the foreseeable future to produce quantities of products and substances necessary for research and development, preclinical trials, human clinical trials and product commercialization, and to perform their obligations in a timely manner and in accordance with applicable government regulations. If we develop any product with commercial potential, we will need to develop the facilities to independently manufacture such product or products or secure arrangements with third parties to manufacture them. We may not be able to independently develop manufacturing capabilities or obtain favorable terms for the manufacture of our product. While we intend to contract for the commercial manufacture of our product candidate, we may not be able to identify and qualify contractors or obtain favorable contracting terms. We or our contract manufacturers may also fail to meet required manufacturing standards, which could result in delays or failures in product delivery, increased costs, injury or death to patients, product recalls or withdrawals and other problems that could significantly hurt our business. We intend to maintain a second source for back-up commercial manufacturing, wherever feasible. However, if a replacement to our future internal or contract manufacturers were required, the ability to establish second-sourcing or find a replacement manufacturer may be difficult due to the lead times generally required to manufacture drugs and the need for FDA compliance inspections and approvals of any replacement manufacturer, all of which factors could result in production delays and additional commercialization costs. Such lead times would vary based on the situation, but might be twelve months or longer.
We may lack the resources necessary to effectively market our product candidate, and we may need to rely on third parties over whom we have little or no control and who may not perform as expected.
We may not have the necessary resources to market our product candidate. If we develop any products with commercial potential, we will either have to develop a marketing capability, including a sales force, which is difficult and expensive to implement successfully, or attempt to enter into a collaboration, merger, joint venture, license or other arrangement with third parties to provide a substantial portion of the financial and other resources needed to market such products. We may not be able to do so on acceptable terms, if at all. If we rely extensively on third parties to market our products, the commercial success of such products may be largely outside of our control.
We conduct our business internationally and are subject to laws and regulations of several countries which may affect our ability to access regulatory agencies and may affect the enforceability and value of our licenses.
We have conducted clinical trials in the United States, Canada, Europe and the Pacific Rim and intend to, or may, conduct future clinical trials in these and other jurisdictions. There can be no assurance that any sovereign government will not establish laws or regulations that will be deleterious to our interests. There is no assurance that we, as a British Columbia corporation, will continue to have access to the regulatory agencies in any jurisdiction where we might want to conduct clinical trials or obtain regulatory approval, and we might not be able to enforce our license or patent rights in foreign jurisdictions. Foreign exchange controls may have a material adverse effect on our business and financial condition, since such controls may limit our ability to flow funds into or out of a particular country to meet obligations under licenses, clinical trial agreements or other collaborations.
Our cash invested in money market funds might be subject to loss.
Even though we believe we take a conservative approach to investing our funds, the nature of financial markets exposes us to investment risk, including the risks that the value and liquidity of our money market investments could deteriorate significantly and the issuers of the investments we hold could be subject to credit rating downgrades. While we have not experienced any loss or write down of our money market investments in the past, we cannot guarantee that such losses will not occur in future periods.
Risks Related to Our Industry
If we are unable to obtain applicable U.S. and/or foreign regulatory approvals, we will be unable to develop and commercialize our drug candidate.
The preclinical studies and clinical trials of our product candidate, as well as the manufacturing, labeling, sale and distribution, export or import, marketing, advertising and promotion of our product candidate, are subject to various regulatory frameworks in the United States, Canada and other countries. Any products that we develop must receive all relevant regulatory approvals and clearances before any marketing, sale or distribution. The regulatory process, which includes extensive preclinical studies and clinical testing to establish product safety and efficacy, can take many years and cost substantial amounts of money. As a result of the length of time, many challenges and costs are associated with the drug development process, and the historical rate of failures for drug candidates is extremely high. Changes in regulatory policy could also cause delays or affect regulatory approval. Any regulatory delays may increase our development costs and negatively impact our competitiveness and prospects. It is possible that we may not be able to obtain regulatory approval of our drug candidate or approvals may take longer and cost more to obtain than expected.
| 12 |
Regulatory approvals, if granted, may entail limitations on the uses for which any product we develop may be marketed, limiting the potential sales for any such products. The granting of product approvals can be withdrawn at any time, and manufacturers of approved products are subject to regular reviews, including for compliance with FDA Good Manufacturing Practices regulations. Failure to comply with any applicable regulatory requirement, which may change from time to time, can result in warning letters, fines, sanctions, penalties, recalling or seizing products, suspension of production, or even criminal prosecution.
Future sales of our product candidate may suffer if they fail to achieve market acceptance.
Even if our product candidate is successfully developed and achieves appropriate regulatory approval, it may not enjoy commercial acceptance or success. Our product candidate may compete with a number of new and traditional drugs and therapies developed by major pharmaceutical and biotechnology companies. Market acceptance is dependent on the product candidate demonstrating clinical efficacy and safety, as well as demonstrating advantages over alternative treatment methods. In addition, market acceptance is influenced by government reimbursement policies and the ability of third parties to pay for such products. Physicians, patients, or the medical community may not accept or utilize any products we may develop.
We face a strong competitive environment. Other companies may develop or commercialize more effective or cheaper products, which may reduce or eliminate the demand for our product candidate.
The biotechnology and pharmaceutical industry, and in particular the field of cancer therapeutics where we are focused, is very competitive. Many companies and research organizations are engaged in the research, development and testing of new cancer therapies or means of increasing the effectiveness of existing therapies, including, among many others, Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Eli Lilly, Eisai, Merck KGaA, Novartis, Johnson & Johnson, Pfizer, Roche, Taiho and Sanofi-Aventis. Many of these companies have marketed drugs or are developing targeted cancer therapeutics, which depending upon the mechanism of action of such agents could be competitors.
Many of our existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. In addition, many of these competitors have extensive experience with preclinical testing and human clinical trials and in obtaining regulatory approvals. Also, some of the smaller companies that compete with us have formed collaborative relationships with large, established companies to support the research, development, clinical trials and commercialization of any products that they may develop. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for research, clinical development and marketing of products similar to those we seek to develop. These companies and institutions compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our projects.
We are likely to face competition in the areas of product efficacy and safety, ease of use and adaptability, as well as pricing, product acceptance, regulatory approvals and intellectual property. Competitors could develop more effective, safer and more affordable products than we do, and they may obtain patent protection or product commercialization before we do or even render our product candidate obsolete. The existence of competitive products, including products or treatments of which we are not aware, or products or treatments that may be developed in the future, may adversely affect the marketability of any product that we develop.
We may face product liability claims that could require us to defend costly lawsuits or incur substantial liabilities that could adversely impact our financial condition, receipt of regulatory approvals for our product candidate and our results of operation.
The use of our product candidate in clinical trials and for commercial applications, if any, may expose us to liability claims in the event that such product candidate causes injury or death or results in other adverse effects. These claims could be made by health care institutions, contract laboratories, and subjects participating in our clinical studies, patients or others using our product candidate. In addition to liability claims, certain serious adverse events could require interruption, delay and/or discontinuation of a clinical trial and potentially prevent further development of our product candidate. We carry clinical trial insurance but the coverage may not be sufficient to protect us from legal expenses and liabilities we might incur. Litigation is very expensive, even if we defend successfully against possible litigation. In addition, our existing coverage may not be adequate if we develop additional products, and future coverage may not be available in sufficient amounts or at reasonable cost. Further, it is possible that we may later reduce or terminate this coverage based on future availability of financial resources. Adverse liability claims may also harm our ability to obtain or maintain regulatory approvals.
| 13 |
We used hazardous materials and chemicals in our research and development, and our failure to comply with laws related to hazardous materials could materially harm us.
Our research and development processes involve the controlled use of hazardous materials, such as flammable organic solvents, corrosive acids and corrosive bases. Accordingly, we are subject to federal, state, local and foreign laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. We could be held liable for any damages that result and any such liability could exceed our resources and may not be covered by our general liability insurance. We currently do not carry insurance specifically for hazardous materials claims. We may be required to incur significant costs to comply with environmental laws and regulations, which may change from time to time. Our current practice is to outsource these activities.
Efforts to reduce product pricing and health care reimbursement and changes to government policies could negatively affect the commercialization of our product candidate.
If our product candidate achieves regulatory approval, we may be materially adversely affected by the continuing efforts of governmental and third-party payers to contain or reduce health care costs. For example, if we succeed in bringing one or more products to market, such products may not be considered cost-effective and the availability of consumer reimbursement may not exist or be sufficient to allow the sale of such products on a competitive basis. The constraints on pricing and availability of competitive products may further limit our pricing and reimbursement policies as well as adversely impact market acceptance and commercialization for the products.
In many markets, the pricing or profitability of healthcare products is subject to government control. In recent years, federal, state, provincial and local officials and legislators have proposed or are proposing a variety of price-based reforms to the healthcare systems in the United States, Canada and elsewhere. Some proposals include measures that would limit or eliminate payments from third-party payors to the consumer for certain medical procedures and treatments or allow government control of pharmaceutical pricing. The adoption of any such proposals or reforms could adversely affect the commercial viability of our product candidates.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the “ACA”, was passed, which substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The expansion of insured among children under the ACA is not expected to be significant to the prospects for our product candidate since Medicaid was more available to children than the general population.
The provisions of the ACA of importance to the pharmaceutical and biotechnology industry are, among others, the following:
| · | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs agents and biologic agents, which is apportioned among these entities according to their market share in certain government healthcare programs; |
| · | an increase in the rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13% of the average manufacturer price for branded and generic drugs, respectively; |
| · | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts to negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| · | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations, unless the drug is subject to discounts under the 340B drug discount program; |
| · | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
| · | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| · | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| · | new requirements under the federal Physician Payments Sunshine Act for drug manufacturers to report information related to payments and other transfers of value made to physicians and teaching hospitals as well as ownership or investment interests held by physicians and their immediate family members; |
| · | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| · | creation of the Independent Payment Advisory Board, which, if and when impaneled, will have authority to recommend certain changes to the Medicare program that could result in reduced payments for prescription drugs; and |
| · | establishment of a Center for Medicare and Medicaid Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. |
| 14 |
Some states are also considering legislation that would control the prices of drugs, and state Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and requiring prior authorization by the state program for use of any drug for which supplemental rebates are not being paid. Managed care organizations continue to seek price discounts and, in some cases, to impose restrictions on the coverage of particular drugs. Government efforts to reduce Medicaid expenses may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products.
Since its enactment, there have been judicial and Congressional challenges to numerous aspects of the ACA, and Congress and the executive branch are seeking to replace the ACA with new federal legislation. There may also be federal and state regulatory changes that impact the ACA or healthcare programs, insurance coverage or reimbursement generally. These efforts have increased uncertainty regarding the availability of healthcare programs, insurance coverage and reimbursement as a general matter as well as for our product candidate, and we cannot predict how these events will impact our business.
In addition, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which have resulted in several recent Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, reduce the price of drugs under Medicare and reform government program reimbursement methodologies for products. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Any significant changes in the healthcare system in the United States, Canada or abroad would likely have a substantial impact on the manner in which we conduct business and could have a material adverse effect on our ability to raise capital and the viability of product commercialization.
Risks Related to Owning Our Common Shares
We may be unable to maintain the listing of our common shares on the Nasdaq Capital Market or the TSX and that would make it more difficult for shareholders to dispose of our common shares.
Our common shares are currently listed on the Nasdaq Capital Market and the Toronto Stock Exchange (the “TSX”). Both the Nasdaq Capital Market and the TSX have rules for continued listing, including minimum market capitalization and other requirements, that we might not meet in the future.
Delisting from the Nasdaq Capital Market or the TSX would make it more difficult for shareholders to dispose of our common shares and more difficult to obtain accurate quotations on our common shares. This could have an adverse effect on the price of our common shares. There can be no assurances that a market maker will make a market in our common shares on the OTCQB or any other stock quotation system after delisting. Furthermore, securities quoted over-the-counter generally have significantly less liquidity than securities traded on a national securities exchange, not only in the number of shares that can be bought and sold, but also through delays in the timing of transactions and lower market prices than might otherwise be obtained. As a result, shareholders might find it difficult to resell shares at prices quoted in the market or at all. Furthermore, because of the limited market and generally low volume of trading in our common shares, our common shares are more likely to be affected by broad market fluctuations, general market conditions, fluctuations in our operating results, changes in the market’s perception of our business, and announcements made by us, our competitors or parties with whom we have business relationships. Our ability to issue additional securities for financing or other purposes, or to otherwise arrange for any financing we may need in the future, may also be materially and adversely affected by the limited market and low trading volume of our common shares.
The market price of our common shares is highly volatile and could cause the value of your investment to significantly decline.
Historically, the market price of our common shares has been highly volatile and the market for our common shares has from time to time experienced significant price and volume fluctuations, some of which are unrelated to our operating performance. From March 15, 2013 to March 16, 2018, the closing trading price of our stock fluctuated from a high of $15.63 Canadian dollars (“CAD”) per share to a low of CAD$0.72 per share on the TSX. From September 13, 2017 to March 16, 2018, the closing trading price of our stock fluctuated from a high of $12.35 per share to a low of $8.26 on the Nasdaq Capital Market. Historically, our common shares have had a low trading volume, and may continue to have a low trading volume in the future. This low volume may contribute to the volatility of the market price of our common shares. It is likely that the market price of our common shares will continue to fluctuate significantly in the future.
The market price of our common shares may be significantly affected by many factors, including without limitation:
| · | the development of our sole product candidate, STS; |
| · | the need to raise additional capital and the terms of any transaction we are able to enter into; |
| · | other external factors generally or stock market trends in the pharmaceutical or biotechnology industries specifically; |
| 15 |
| · | announcements of licensing agreements, joint ventures, collaborations or other strategic alliances that involve our product or those of our competitors; |
| · | innovations related to our or our competitors’ products; |
| · | actual or potential clinical trial results related to our or our competitors’ products; |
| · | our financial results or those of our competitors; |
| · | reports of securities analysts regarding us or our competitors; |
| · | developments or disputes concerning our licensed or owned patents or those of our competitors; |
| · | developments with respect to the efficacy or safety of our product or those of our competitors; and |
| · | health care reforms and reimbursement policy changes nationally and internationally. |
Our existing principal shareholders hold a substantial number of our common shares and may be able to exercise influence in matters requiring approval of our shareholders.
At March 16, 2018, our current shareholders separately representing more than 5% ownership in our Company collectively represented beneficial ownership of approximately 52.49% of our common shares. In particular, Southpoint Capital Advisors LP (“Southpoint Capital”) owns or exercises control over approximately 4.0 million common shares, representing approximately 21.7% of the issued and outstanding common shares. In addition, Essetifin SpA, owns approximately 3.2 million shares, or 17.5% of our common shares. In addition, Manchester Explorer, LP (“Manchester Explorer”), together with its associates, owns approximately 2.5 million shares, or 13.4% of our common shares. Southpoint Capital, Manchester Explorer, our other shareholders representing more than 5% ownership, and other insiders, acting alone or together, might be able to influence the outcomes of matters that require the approval of our shareholders, including but not limited to certain equity transactions (such as a financing), an acquisition or merger with another company, a sale of substantially all of our assets, the election and removal of directors, or amendments to our incorporating documents. These shareholders might make decisions that are adverse to your interests. The concentration of ownership could have the effect of delaying, preventing or deterring a change of control of our Company, which could adversely affect the market price of our common shares or deprive our other shareholders of an opportunity to receive a premium for our common shares as part of a sale of our company.
There are a large number of our common shares underlying outstanding warrants and options, and reserved for issuance under our stock option plan, that may be sold in the market, which could depress the market price of our shares and result in substantial dilution to the holders of our common shares.
The sale or issuance of a substantial amount of our common shares in the future could cause the market price of our common shares to decline. It may also impair our ability to obtain additional financing. At March 16, 2018, we had outstanding warrants to purchase approximately 1.3 million shares ($2.0 million) of our common shares at an average exercise price of $1.50 per common share. In addition, at March 16, 2018, there were approximately 2.3 million common shares issuable upon the exercise of outstanding stock options, of which options to purchase approximately $1.7 million were denominated in Canadian dollars and had a weighted average exercise price of CAD $2.38 per common share and options to purchase approximately $4.3 million were denominated in U.S. dollars and had a weighted average exercise price of $2.70 per common share. We may also issue further warrants as part of any future financings in addition to the additional 2.3 million options to acquire our common shares currently remaining and available for future awards under our stock option plan.
We may need to raise additional funds in the future to continue our operations. Any equity offering could result in significant dilution to the ownership interests of shareholders and may result in dilution of the value of such interests and any debt offering will increase financial risk.
In order to satisfy our anticipated capital requirements to develop our product, we may need to raise additional funds through either the sale of additional equity, the issue of securities convertible into equity, the issuance of debt, the establishment of collaborations that provide us with funding, the out-license or sale of certain aspects of our intellectual property portfolio, or from other sources. The most likely sources of financing that may be available to us in the near term are the sale of common shares and/or securities convertible or exercisable into common shares and the issuance of debt.
We cannot predict the size of future issues of common shares or the future issue of securities convertible or exercisable into common shares or the effect that any such future issues and sales of common shares or other securities will have on the market price of our common shares. Any transaction involving the issue of common shares, or securities convertible or exercisable into common shares, could result in immediate and substantial dilution to present and prospective holders of our common shares. Alternatively, we may rely on debt financing and assume debt obligations that require us to make substantial interest and capital payments and to pledge some or all of our assets as collateral to secure such debt obligations.
| 16 |
We have not paid any dividends since incorporation
and do not anticipate declaring any dividends in the foreseeable future. As a result, you may not be able to recoup your investment
through the payment of dividends on your common shares and the lack of a dividend payable on our common shares might depress the
value of your investment.
For the foreseeable future, we plan to use all available funds to finance the development of our product candidate and operate our business. Our directors will determine if and when dividends should be declared and paid in the future based on our financial position at the relevant time, but since we have no present plans to pay dividends, you should not expect receipt of dividends either for your cash needs or to enhance the value of our common shares held by you.
We may be a passive foreign investment company, or “PFIC,” which could result in adverse United States federal income tax consequences to U.S. investors.
If we are a PFIC for any taxable year (or portion thereof) that is included in the holding period of a U.S. Holder (as such term is defined in the section of this Annual Report “Material U.S. Federal Income Tax Considerations”) of our common shares, the U.S. Holder may be subject to adverse U.S. federal income tax consequences and may be subject to additional reporting requirements. We have not made the analysis necessary to determine whether or not we are currently a PFIC or whether we have ever been a PFIC, and there can be no assurances with respect to our status as a PFIC for our current taxable year or any subsequent taxable year. Moreover, if we are a PFIC for any taxable year, we intend to provide to a U.S. Holder such information as the Internal Revenue Service (“IRS”) may require, including a PFIC annual information statement, in order to enable the U.S. Holder to make and maintain a “qualified electing fund” election. We urge U.S. investors to consult their own tax advisors regarding the possible application of the PFIC rules. For a more detailed explanation of the tax consequences of PFIC classification to U.S. Holders, see the section of this Annual Report entitled “Material U.S. Federal Income Tax Considerations—Tax Consequences if We Are a Passive Foreign Investment Company.” This paragraph is qualified in its entirety by the discussion below under the heading “Material United States Federal Income Tax Considerations.” Each U.S. shareholder should consult its own tax advisors regarding the PFIC rules and the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
The Company has an operating lease in Research Triangle Park, North Carolina utilizing small space within a commercial building. This operating lease is terminable with 30 days’ notice and has no penalties or contingent payments due.
| Item 3. | Legal Proceedings |
None.
| Item 4. | Mine Safety Disclosures |
Not applicable.
| 17 |
| Item 5. | Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer’s Purchases of Equity Securities |
Our common shares currently trade in the U.S. on the Nasdaq Capital Market under the trading symbol “FENC” and in Canada on the TSX under the trading symbol “FRX”. Prior to September 13, 2017, our common shares traded in the U.S. on the OTCQB Market under the trading symbol “FENCF”. The following table sets forth the quarterly high and low market closing prices, and average daily trading volume on the OTCQB, Nasdaq Capital Market (as applicable), and the TSX, for the two most recent full fiscal years:
| Nasdaq
Capital Market/OTCQB (in U.S. dollars) | Toronto Stock Exchange (in Canadian dollars) | |||||||||||||||||||||||
| High $ | Low $ | Volume | High $ | Low $ | Volume | |||||||||||||||||||
| Fiscal 2017: | ||||||||||||||||||||||||
| Quarter ended 12/31/17 | $ | 12.35 | $ | 8.90 | 26,629 | $ | 15.63 | $ | 11.32 | 2,140 | ||||||||||||||
| Quarter ended 09/30/17 | 12.19 | 5.85 | 17,927 | 15.05 | 7.25 | 6,358 | ||||||||||||||||||
| Quarter ended 06/30/17 | 6.35 | 3.00 | 7,295 | 8.02 | 4.03 | 5,256 | ||||||||||||||||||
| Quarter ended 03/31/17 | $ | 3.14 | $ | 1.95 | 1,402 | $ | 4.10 | $ | 2.42 | 2,541 | ||||||||||||||
| Fiscal 2016: | ||||||||||||||||||||||||
| Quarter ended 12/31/16 | $ | 2.18 | $ | 1.64 | 1,821 | $ | 2.85 | $ | 2.21 | 1,576 | ||||||||||||||
| Quarter ended 09/30/16 | 2.30 | 1.85 | 4,469 | 3.15 | 2.41 | 2,060 | ||||||||||||||||||
| Quarter ended 06/30/16 | 3.05 | 1.66 | 1,641 | 3.85 | 2.18 | 6,509 | ||||||||||||||||||
| Quarter ended 03/31/16 | $ | 2.06 | $ | 1.13 | 1,617 | $ | 2.85 | $ | 1.65 | 2,453 | ||||||||||||||
As of March 16, 2018, the last reported sale on the TSX was CAD$12.47 per share and the last reported sale on the Nasdaq Capital Market was $9.57 per share.
Record Holders
As of March 16, 2018, there were approximately 51 shareholders of record of our common shares, one of which was Cede & Co., a nominee for Depository Trust Company, or DTC, and one of which was The Canadian Depository for Securities Limited, or CDS. All of our common shares held by brokerage firms, banks and other financial institutions in the U.S. or Canada as nominees for beneficial owners are considered to be held of record by Cede & Co. in respect of brokerage firms, banks and other financial institutions located in Canada. Cede & Co. and CDS are each considered to be one shareholder of record.
Dividend Policy
We have never declared or paid cash dividends on our common shares. We currently expect to retain future earnings, if any, for use in the operation and expansion of business and do not anticipate paying any cash dividends in the foreseeable future.
Material United States Federal and Canadian Income Tax Consequences
The following discussion sets forth certain material United States and Canadian federal income tax consequences resulting from the acquisition, ownership and disposition of our common shares by a “U.S. Holder”. For purposes of this discussion, a U.S. Holder means any U.S. person who holds common shares. For purposes of our discussion, a U.S. person is:
| · | an individual who is a citizen or resident of the United States; |
| · | a corporation (or other entity taxed as a corporation for U.S. federal income tax purposes) created or organized in the United States or under the laws of the United States or any subdivision thereof; |
| · | an estate, the income of which is includable in gross income for U.S. federal income tax purposes regardless of its source; or |
| · | a trust, if a court within the United States is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have the authority to control all substantial decisions of the trust (or if the trust was in existence on August 20, 1996, and has validly elected to be treated as a U.S. person under applicable Treasury regulations); and |
| · | for purposes of the Income Tax Act (Canada) (the “Tax Act”) is neither resident nor deemed to be resident in Canada and does not use or hold, and is not deemed to use or hold our common shares held by them in connection with carrying on business in Canada (a “Non-Resident Holder”). |
| 18 |
Material U.S. Federal Income Tax Considerations
The following summary describes the material U.S. federal income tax consequences to U.S. Holders (as defined below) of acquiring, owning, and disposing of our common shares acquired pursuant to this Annual Report, subject to the qualifications set forth herein.
General
Tax Consequences Not Addressed
This summary does not address all potential U.S. federal income tax considerations that may be relevant to a particular U.S. Holder. In addition, this summary does not take into account the individual facts and circumstances that may affect the U.S. federal income tax consequences to a particular U.S. Holder, including specific tax consequences under an applicable income tax treaty. Accordingly, this summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. This summary does not address any U.S. federal alternative minimum, U.S. federal estate and gift, U.S. state and local, or non-U.S. tax considerations, and does not discuss tax reporting requirements that may be applicable to any particular U.S. Holder. Each prospective investor should consult a professional tax advisor with respect to the U.S. federal income, U.S. alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences of acquiring, owning, and disposing of our common shares.
Authorities
This summary is based upon the provisions of the Internal Revenue Code of 1986, as amended (the “Code”), the United States Treasury Regulations (whether final, temporary, or proposed) promulgated thereunder, the Convention Between Canada and the United States of America with Respect to Taxes on Income and on Capital, signed September 26, 1980, as amended (the “Canada-U.S. Tax Convention”), and administrative rulings and judicial decisions interpreting the Code and the United States Treasury Regulations, all as currently in effect, and all subject to differing interpretations or change, possibly on a retroactive basis. We have not sought, and will not seek, a ruling from the IRS regarding any matter discussed herein, and no assurance can be given that the IRS would not assert, or that a court would not sustain, a position that is different from, and contrary to, the positions taken in this summary. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation.
U.S. Holders
For purposes of this summary, the term “U.S. Holder” means a beneficial owner of common shares acquired pursuant to this Annual Report that is for U.S. federal income tax purposes:
| · | an individual who is a citizen or resident of the United States (as determined under U.S. federal income tax rules); |
| · | a corporation (or other entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States or of any political subdivision of the United States; |
| · | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| · | a trust that (i) is subject to the primary supervision of a court within the United States and the control of one or more U.S. persons for all substantial decisions or (ii) has a valid election in effect under applicable United States Treasury Regulations to be treated as a U.S. person. |
An individual may be a resident for U.S. federal income tax purposes in any calendar year if the individual was present in the United States for at least 31 days in that calendar year and for an aggregate of at least 183 days during the three-year period ending with the current calendar year. For purposes of this calculation, all of the days present in the current year, one-third of the days present in the immediately preceding year, and one-sixth of the days present in the second preceding year are counted. Residents are taxed for U.S. federal income tax purposes as if they were U.S. citizens.
Non-U.S. Holders Not Addressed
For purposes of this summary, a “non-U.S. Holder” is a beneficial owner of common shares that is not a U.S. Holder and is not a partnership for U.S. federal income tax purposes. This summary does not address the U.S. federal income tax consequences to non-U.S. Holders of acquiring, owning, and disposing of common shares. Each prospective investor should consult a professional tax advisor with respect to the U.S. federal income, U.S. alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences of acquiring, owning, and disposing of our common shares.
| 19 |
Certain U.S. Holders Not Addressed
This summary does not address the U.S. federal income tax considerations applicable U.S. Holders that are subject to special provisions under the Code, including, but not limited to, U.S. Holders that:
| · | are tax-exempt organizations, qualified retirement plans, individual retirement accounts, or other tax-deferred accounts; |
| · | are financial institutions, underwriters, insurance companies, real estate investment trusts, or regulated investment companies; |
| · | are broker-dealers, dealers, or traders in securities or currencies that elect to apply a mark-to-market accounting method; |
| · | have a “functional currency” other than the U.S. dollar; |
| · | own common shares as part of a straddle, hedging transaction, conversion transaction, constructive sale, or other arrangement involving more than one position; |
| · | acquired common shares in connection with the exercise of employee stock options or otherwise as compensation for services; |
| · | hold common shares other than as a capital asset within the meaning of section 1221 of the Code (generally, property held for investment purposes); |
| · | are partnerships or other “pass-through” entities for U.S. federal income tax purposes (or investors in such partnerships or entities); |
| · | own, have owned, or will own (directly, indirectly, or by attribution) 10% or more of the total combined voting power of the outstanding shares of your company; |
| · | are U.S. expatriates or former long-term residents of the United States; |
| · | have been, are, or will be residents or deemed to be residents in Canada for purposes of the Income Tax Act (Canada) (the “Tax Act”); |
| · | use or hold, will use or hold, or that are or will be deemed to use or hold common shares in connection with carrying on a business in Canada; |
| · | are persons whose common shares constitute “taxable Canadian property” under the Tax Act; or |
| · | have a permanent establishment in Canada for the purposes of the Canada-U.S. Tax Convention. |
U.S. Holders that are subject to special provisions under the Code, including, but not limited to, U.S. Holders described immediately above, should consult their own tax advisors regarding the U.S. federal income, U.S. federal alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences of acquiring, owning, and disposing of our common shares.
The following summary is not a substitute for careful tax planning and advice. U.S. Holders of common shares are urged to consult their own tax advisors concerning the U.S. federal income tax consequences of the issues discussed herein, in light of their particular circumstances, as well as any considerations arising under the laws of any foreign, state, local, or other taxing jurisdiction.
General Rules Applicable to the Ownership and Disposition of Common Shares
The following discussion describes the general rules applicable to the ownership and disposition of the common shares but is subject in its entirety to the special rules described below under the headings entitled “Tax Consequences if We Are a Passive Foreign Investment Company” and “Tax Consequences if We are a Controlled Foreign Corporation.”
Distributions on Common Shares
The gross amount of any distribution (including amounts, if any, withheld in respect of Canadian withholding tax) actually or constructively received by a U.S. Holder with respect to our common shares will be taxable to the U.S. Holder as a dividend to the extent of our current or accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions to a U.S. Holder in excess of earnings and profits will be treated first as a return of capital that reduces a U.S. Holder’s tax basis in such common shares (thereby increasing the amount of gain or decreasing the amount of loss that a U.S. Holder would recognize on a subsequent disposition of our common shares), and then as gain from the sale or exchange of such common shares (see “Sale or Other Taxable Disposition of Our Common Shares”). The amount of any distribution of property other than cash will be the fair market value of that property on the date of distribution. In the event we make distributions to holders of common shares, we may or may not calculate our earnings and profits under U.S. federal income tax principles. If we do not do so, any distribution may be required to be regarded as a dividend, even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain. The amount of the dividend will generally be treated as foreign-source dividend income to U.S. Holders.
Non-corporate U.S. Holders, including individuals, will generally be eligible for the preferential U.S. federal rate on “qualified dividend income,” provided that we are a “qualified foreign corporation,” the stock on which the dividend is paid is held for a minimum holding period, and other requirements are satisfied. A “qualified foreign corporation” includes a foreign corporation that is not a PFIC in the year of the distribution or in the prior taxable year and that is eligible for the benefits of an income tax treaty with the United States that contains an exchange of information provision and has been determined by the United States Treasury Department to be satisfactory for purposes of the legislation (such as the Canada-U.S. Tax Convention).
| 20 |
Distributions to U.S. Holders generally will not be eligible for the “dividends received deduction” generally allowed to U.S. corporations in respect of dividends received from other U.S. corporations.
Sale or Other Taxable Disposition of Our Common Shares
Upon the sale, exchange, or other taxable disposition of our common shares, a U.S. Holder generally will recognize gain or loss equal to the difference between the amount realized upon the sale, exchange, or other disposition and such U.S. Holder’s tax basis in such common shares sold or otherwise disposed of. If the U.S. holder receives Canadian dollars in the transaction, the amount realized will be the U.S. dollar value of the Canadian dollars received, which is determined for cash basis taxpayers on the settlement date for the transaction and for accrual basis taxpayers on the trade date (although accrual basis taxpayers can also elect the settlement date). A U.S. Holder’s tax basis in common shares generally will be such holder’s U.S. dollar cost for such common shares. Gain or loss recognized on such sale or other disposition generally will be long-term capital gain or loss if, at the time of the sale or other disposition, the common shares have been held for more than one year.
Preferential tax rates currently apply to long-term capital gain of a U.S. Holder that is an individual, estate, or trust. There are currently no preferential tax rates for long-term capital gain of a corporate U.S. Holder. Deductions for capital losses are subject to significant limitations under the Code. The gain or loss will generally be U.S.-source gain or loss for foreign tax credit purposes.
Additional Medicare Tax on Net Investment Income
Certain U.S. Holders that are individuals, estates, or trusts (other than trusts that are exempt from tax) are subject to a tax of 3.8% on “net investment income” (or undistributed “net investment income,” in the case of estates and trusts) for each taxable year, with such tax applying to the lesser of such income or the excess of such person’s adjusted gross income (with certain adjustments) over a specified amount. Net investment income includes dividends on the common shares and net gains from the disposition of the common shares.
U.S. Holders that are individuals, estates, or trusts should consult their own tax advisors regarding the applicability of this tax to any of their income or gains in respect of the common shares.
Receipt of Foreign Currency
The amount of any distribution paid to a U.S. Holder in foreign currency, or on the sale, exchange, or other taxable disposition of common shares, generally will be equal to the U.S. dollar value of such foreign currency based on the exchange rate applicable on the date of receipt (regardless of whether such foreign currency is converted into U.S. dollars at that time). If the foreign currency received is not converted into U.S. dollars on the date of receipt, a U.S. Holder will have a tax basis in the foreign currency equal to its U.S. dollar value on the date of receipt. Any U.S. Holder who converts or otherwise disposes of the foreign currency after the date of receipt may have a foreign currency exchange gain or loss that would be treated as ordinary income or loss, and generally will be U.S. source income or loss for foreign tax credit purposes. Different rules apply to U.S. Holders who use the accrual method of tax accounting. Each U.S. Holder should consult its own U.S. tax advisors regarding the U.S. federal income tax consequences of receiving, owning, and disposing of foreign currency.
Foreign Tax Credit
Subject to the PFIC rules discussed above, a U.S. Holder that pays (whether directly or through withholding) Canadian income tax with respect to dividends paid on the common shares generally will be entitled, at the election of such U.S. Holder, to receive either a deduction or a credit for such Canadian income tax paid. Generally, a credit will reduce a U.S. Holder’s U.S. federal income tax liability on a dollar-for-dollar basis, whereas a deduction will reduce a U.S. Holder’s income that is subject to U.S. federal income tax. This election is made on a year-by-year basis and applies to all foreign taxes paid (whether directly or through withholding) by a U.S. Holder during a year.
Complex limitations apply to the foreign tax credit, including the general limitation that the credit cannot exceed the proportionate share of a U.S. Holder’s U.S. federal income tax liability that such U.S. Holder’s “foreign source” taxable income bears to such U.S. Holder’s worldwide taxable income. In applying this limitation, a U.S. Holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” Generally, dividends paid by a foreign corporation (including constructive dividends) should be treated as foreign source for this purpose, and gains recognized on the sale of stock of a foreign corporation by a U.S. Holder should be treated as U.S. source for this purpose, except as otherwise provided in an applicable income tax treaty, and if an election is properly made under the Code. However, the amount of a distribution with respect to the common shares that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for Canadian federal income tax purposes, resulting in a reduced foreign tax credit allowance to a U.S. Holder. In addition, this limitation is calculated separately with respect to specific categories of income. The foreign tax credit rules are complex, and each U.S. Holder should consult its own U.S. tax advisors regarding the foreign tax credit rules.
| 21 |
Information Reporting and Backup Withholding
Under U.S. federal income tax law, certain categories of U.S. Holders must file information returns with respect to their investment in, or involvement in, a foreign corporation. For example, certain U.S. Holders who hold certain “specified foreign financial assets” that exceed certain thresholds are required to report information relating to such assets. The definition of “specified foreign financial assets” generally includes not only financial accounts maintained in foreign financial institutions, but also, unless held in accounts maintained by a financial institution, any stock or security issued by a non-U.S. person, any financial instrument or contract held for investment that has an issuer or counterparty other than a U.S. person, and any interest in a foreign entity. U.S. Holders may be subject to these reporting requirements unless their common shares are held in an account at certain financial institutions. Significant penalties may apply for failure to satisfy applicable reporting obligations.
Distributions paid with respect to common shares and proceeds from a sale, exchange, or redemption of common shares made within the United States or through certain U.S.-related financial intermediaries may be subject to information reporting to the IRS and possible U.S. backup withholding (at a rate of 28%). Backup withholding will not apply, however, to a U.S. Holder who furnishes a correct U.S. taxpayer identification number and makes any other required certification on IRS Form W-9 or that is a corporation or other entity that is otherwise exempt from backup withholding. Each U.S. Holder should consult its own tax advisors regarding the application of the U.S. information reporting and backup withholding rules. Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against a holder’s U.S. federal income tax liability, and such holder may obtain a refund of any excess amounts withheld under the backup withholding rules by filing an appropriate claim for refund with the IRS and furnishing any required information in a timely manner.
The discussion of reporting requirements set forth above is not intended to constitute a complete description of all reporting requirements that may apply to a U.S. Holder. A failure to satisfy certain reporting requirements may result in an extension of the time period during which the IRS can assess a tax and, under certain circumstances, such an extension may apply to assessments of amounts unrelated to any unsatisfied reporting requirement. U.S. Holders should consult with their own tax advisors regarding their reporting obligations, if any, as a result of their acquisition, ownership, or disposition of our common shares.
Tax Consequences if We are a Passive Foreign Investment Company
A foreign corporation generally will be treated as a “passive foreign investment company” (“PFIC”) if, after applying certain “look-through” rules, either (i) 75% or more of its gross income is passive income or (ii) 50% or more of the average value of its assets is attributable to assets that produce or are held to produce passive income. Passive income for this purpose generally includes dividends, interest, rents, royalties and gains from securities and commodities transactions. The look-through rules require a foreign corporation that owns at least 25% by value, of the stock of another corporation to treat a proportionate amount of assets and income as held or received directly by the foreign corporation.
We have not made the analysis necessary to determine whether or not we are currently a PFIC or whether we have ever been a PFIC. There can be no assurance that we are not, have never been or will not in the future be a PFIC. If we were to be treated as a PFIC, any gain recognized by a U.S. shareholder upon the sale (or certain other dispositions) of our common shares (or the receipt of certain distributions) generally would be treated as ordinary income, and a U.S. shareholder may be required, in certain circumstances, to pay an interest charge together with tax calculated at maximum rates on certain “excess distributions,” including any gain on the sale or certain dispositions of our common shares. In order to avoid this tax consequence, a U.S. shareholder (i) may be permitted to make a “qualified electing fund” election, in which case, in lieu of such treatment, such shareholder would be required to include in its taxable income certain undistributed amounts of our income or (ii) may elect to mark-to-market our common shares and recognize ordinary income (or possible ordinary loss) each year with respect to such investment and on the sale or other disposition of the common shares. Additionally, if we are deemed to be a PFIC, a U.S. shareholder who acquires our common shares from a decedent will be denied the normally available step-up in tax basis to fair market value for the common shares at the date of the death and instead will have a tax basis equal to the decedent’s tax basis if lower than fair market value. Neither we nor our advisors have the duty to or will undertake to inform U.S. shareholders of changes in circumstances that would cause us to become a PFIC. U.S. shareholders should consult their own tax advisors regarding the application of the PFIC rules including eligibility for and the manner and advisability of making certain elections in the event we are determined to be a PFIC at any point in time after the date of this Annual Report. We intend to take the action necessary for a U.S. shareholder to make a “qualified electing fund” election in the event we are a PFIC.
Further, excess distributions treated as dividends, gains treated as excess distributions and mark-to-market inclusions and deductions, all under the PFIC rules discussed above, are all included in the calculation of net investment income for purposes of the 3.8% tax described above under the subheading entitled “Additional Medicare Tax on Net Investment Income”. United States Treasury Regulations provide, subject to the election described in the following paragraph, that solely for purposes of this additional tax, distributions of previously taxed income will be treated as dividends and included in net investment income subject to the additional 3.8% tax. Additionally, to determine the amount of any capital gain from the sale or other taxable disposition of common shares that will be subject to the additional tax on net investment income, a U.S. Holder who has made a “qualified electing fund” election will be required to recalculate its basis in the common shares excluding basis adjustments resulting from the “qualified electing fund” election. Alternatively, a U.S. Holder may make an election which will be effective with respect to all interests in a PFIC for which a “qualified electing fund” election has been made and which is held in that year or acquired in future years. Under this election, a U.S. Holder pays the additional 3.8% tax on income inclusions resulting from the “qualified electing fund” election and on gains calculated after giving effect to related tax basis adjustments.
| 22 |
Tax Consequences if We are a Controlled Foreign Corporation
A foreign corporation will be treated as a “controlled foreign corporation” (“CFC”) for U.S. federal income tax purposes if, on any day during the taxable year of such foreign corporation, more than 50% of the equity interests in such corporation, measured by reference to the combined voting power or value of the equity of the corporation, is owned directly or by application of the attribution and constructive ownership rules of Sections 958(a) and 958(b) of the Code by United States Shareholders. For this purpose, a “United States Shareholder” is any United States person that possesses directly, or by application of the attribution and constructive ownership rules of Sections 958(a) and 958(b) of the Code, 10% or more of the combined voting power of all classes of equity in such corporation. If a foreign corporation is a CFC for an uninterrupted period of 30 days or more during any taxable year, each United States Shareholder of our Company who owns, directly or indirectly, our common shares on the last day of the taxable year on which we are a CFC will be required to include in its gross income for United States federal income tax purposes its pro rata share of our “Subpart F income,” even if the Subpart F income is not distributed. Subpart F income generally includes passive income but also includes certain related party sales, manufacturing and services income. If we are a CFC, the PFIC rules set forth above, even if we are otherwise considered to be a PFIC, will not be applicable.
United States persons who might, directly, indirectly or constructively, acquire 10% or more of our common shares, and therefore might be a United States Shareholder, should consider the possible application of the CFC rules, and consult a tax advisor with respect to such matter.
Material Canadian Federal Income Tax Considerations
Non-Residents of Canada
The following portion of the summary is generally applicable to a U.S. Holder. Special rules, which are not discussed in this summary, may apply to a U.S. Holder that is an insurer that carries on an insurance business in Canada and elsewhere.
Disposition of Common Shares
Upon the disposition by a U.S. Holder of common shares in our Company, the U.S. Holder will not be subject to tax under the Tax Act in respect of any capital gain realized unless the common shares disposed of constitutes “taxable Canadian property” of the U.S. Holder and the U.S. Holder is not entitled to relief under an applicable tax treaty or convention. Common shares will generally not constitute “taxable Canadian property” of such U.S. Holder unless at any time in the preceding 60 months both of the following statements were true: (a) the U.S. Holder, alone or together with either (i) persons with whom the U.S. Holder does not deal at arm’s length or (ii) partnerships in which the U.S. Holder or a person in (i) holds a membership interest directly or indirectly through one or more partnerships, held shares and/or an option in respect of, or an interest in shares representing 25% or more of the issued shares of any class of our capital stock; and (b) more than 50% of the fair market value of our common stock was derived directly or indirectly from one or any combination of (i) real or immovable property situated in Canada, (ii) Canadian resource properties, (iii) timber resource properties, and (iv) options in respect of, or interests in, or for civil law rights in, property described in any of (i) to (iii).
U.S. Holders whose common shares constitute “taxable Canadian property” should consult their own tax advisors for advice having regard to their particular circumstances.
Dividends Paid on Common Shares
Dividends paid, credited or deemed to have been paid or credited on our common shares held by a U.S. Holder will be subject to a Canadian withholding tax under the Tax Act at a rate of 25% of the gross amount of the dividends, subject to reduction by any applicable tax convention. Under the tax convention between Canada and the United States (the “Tax Treaty”), the rate of withholding tax on dividends generally applicable to U.S. Holders who beneficially own the dividends is reduced to 15%. In the case of U.S. Holders that are corporations that beneficially own at least 10% of our voting shares, the rate of withholding tax on dividends generally is reduced to 5%. So-called “fiscally transparent” entities, such as United States limited liability companies, or LLCs, are not entitled to rely on the terms of the Tax Treaty, however a member of such entity will be considered to have received the dividend directly and to benefit from the reduced rates under the Tax Treaty, where the member is considered under U.S. taxation law to have derived the dividend through that entity and by reason of the entity being a fiscally transparent entity under U.S. taxation law, the treatment of the dividend is the same as its treatment would be if the amount had been derived directly by the member. Members of such entities are regarded as holding their proportionate share of our common shares held by the entity for the purposes of the Tax Treaty.
| 23 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
CAUTIONARY STATEMENT
The discussion below contains forward-looking statements regarding our financial condition and our results of operations that are based upon our annual consolidated financial statements, which have been prepared in accordance with generally accepted accounting principles within the United States, or U.S. GAAP, and applicable U.S. Securities and Exchange Commission, or SEC, regulations for financial information. The preparation of these financial statements requires our management to make estimates and judgments that affect the reported amounts of assets, liabilities, income and expenses, and related disclosure of contingent assets and liabilities. We evaluate our estimates on an ongoing basis. Our estimates are based on historical experience and on various other assumptions that we believe to be reasonable.
Overview
The following is our only lead product candidate in the clinical stage of development:
| · | PEDMARKTM (a unique formulation of sodium thiosulfate (STS)) – sodium thiosulfate in a novel formulation, recently announced results of two Phase III clinical trials for the prevention of cisplatin induced hearing loss, or ototoxicity in children including the pivotal Phase III study SIOPEL 6 , “A Multicentre Open Label Randomised Phase 3 Trial of the Efficacy of Sodium Thiosulfate in Reducing Ototoxicity in Patients Receiving Cisplatin Chemotherapy for Standard Risk Hepatoblastoma,” and the proof of concept Phase III study “A Randomized Phase 3 Study of Sodium Thiosulfate for the Prevention of Cisplatin-Induced Ototoxicity in Children”. |
We continue to focus the Company’s resources on the development of PEDMARKTM.
We have licensed from OHSU intellectual property rights for the use of PEDMARKTM as a chemoprotectant, and are developing PEDMARKTM as a protectant against the hearing loss often caused by platinum-based anti-cancer agents in children. Preclinical and clinical studies conducted by OHSU and others have indicated that PEDMARKTM can effectively reduce the incidence of hearing loss caused by platinum-based anti-cancer agents. We have received Orphan Drug Designation in the United States for the use of PEDMARKTM in the prevention of platinum-induced ototoxicity in pediatric patients.
Hearing loss among children receiving platinum-based chemotherapy is frequent, permanent and often severely disabling. The incidence of hearing loss in these children depends upon the dose and duration of chemotherapy, and many of these children require lifelong hearing aids. There is currently no established preventive agent for this hearing loss and only expensive, technically difficult and sub-optimal cochlear (inner ear) implants have been shown to provide some benefit. In addition, adults undergoing chemotherapy for several common malignancies, including ovarian cancer, testicular cancer, and particularly head and neck cancer and brain cancer, often receive intensive platinum-based therapy and may experience severe, irreversible hearing loss, particularly in the high frequencies.
Investigators at OHSU have conducted Phase I and Phase II studies which have shown that STS reduces the hearing loss associated with platinum-based chemotherapy. In one study at OHSU, the need for hearing aids to correct high frequency hearing loss was reduced from about 50% of patients being administered platinum-based chemotherapy to less than 5% of patients being administered platinum-based chemotherapy with STS. STS has been studied by cooperative groups in two Phase III clinical studies of survival and reduction of ototoxicity, the COG Protocol ACCL0431 and SIOPEL 6. The COG ACCL0431 protocol enrolled one of five childhood cancers typically treated with intensive cisplatin therapy for localized and disseminated disease, including newly diagnosed hepatoblastoma, germ cell tumor, osteosarcoma, neuroblastoma, and medulloblastoma. SIOPEL 6 enrolled only hepatoblastoma patients with localized tumors.
STS has been studied by cooperative groups in two Phase III clinical studies of survival and reduction of ototoxicity, the COG Protocol ACCL0431 and SIOPEL 6. The COG ACCL0431 protocol enrolled one of five childhood cancers typically treated with intensive cisplatin therapy for localized and disseminated disease, including newly diagnosed hepatoblastoma, germ cell tumor, osteosarcoma, neuroblastoma, and medulloblastoma. SIOPEL 6 enrolled only hepatoblastoma patients with localized tumors.
In 2018, Fennec plans to pursue regulatory approval for PEDMARKTM based on the data from SIOPEL 6 study along with the proof of principle data from COG ACCL0431. STS has received Orphan Drug Designation in the US in this setting and plans to pursue European Market Exclusivity for Pediatric Use upon approval.
We have not received and do not expect to have significant revenues from our product candidate until we are either able to sell our product candidate after obtaining applicable regulatory approvals or we establish collaborations that provide us with up-front payments, licensing fees, milestone payments, royalties or other revenue. The Company generated a net loss of $7.0 million for the year ended December 31, 2017 and had a non-cash loss on derivative liabilities of $0.13 million. We generated a net loss of approximately $2.8 million for the year ended December 31, 2016 (there was a non-cash gain on the change in derivative liability of $0.05 million). As of December 31, 2017, our accumulated deficit was approximately $121.4 million.
| 24 |
Our projections of our capital requirements are subject to substantial uncertainty. More capital than we anticipated may be required thereafter. To finance our continuing operations, we may need to raise substantial additional funds through either the sale of additional equity, the issuance of debt, the establishment of collaborations that provide us with funding, the out-license or sale of certain aspects of our intellectual property portfolio or from other sources. Given current economic conditions, we might not be able to raise the necessary capital or such funding may not be available on financially acceptable terms if at all. If we cannot obtain adequate funding in the future, we might be required to further delay, scale back or eliminate certain research and development studies, consider business combinations or even shut down some, or all, of our operations.
Our operating expenses will depend on many factors, including the progress of our drug development efforts and efficiency of our operations and current resources. Our research and development expenses, which include expenses associated with our clinical trials, drug manufacturing to support clinical programs, salaries for research and development personnel, stock-based compensation, consulting fees, sponsored research costs, toxicology studies, license fees, milestone payments, and other fees and costs related to the development of our product candidate, will depend on the availability of financial resources, the results of our clinical trials and any directives from regulatory agencies, which are difficult to predict. Our general and administration expenses include expenses associated with the compensation of employees, stock-based compensation, professional fees, consulting fees, insurance and other administrative matters associated in support of our drug development programs.
On December 12, 2017, the Company announced the completion of a underwritten public offering of 2,352,950 common shares at a public offering price of $8.50 per share. In addition, Fennec issued an additional 135,670 common shares in connection with the partial exercise of the underwriters’ over-allotment option. The approximate total gross proceeds from the offering was $21.2 million.
On June 8, 2017, the Company completed the closing of a non-brokered private placement (the “Offering”) of 1,900,000 common shares for gross proceeds of $7.6 million. Each common share was issued at a price of $4.00.
Results of Operations
Fiscal 2017 versus Fiscal 2016
| Fiscal Year Ended | Fiscal Year Ended | Increase | ||||||||||||||||||
| In thousands of U.S. Dollars | December 31, 2017 | % | December 31, 2016 | % | (Decrease) | |||||||||||||||
| Revenue | $ | - | $ | - | $ | - | ||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 1,936 | 28 | % | 472 | 16 | % | 1,464 | |||||||||||||
| General and administration | 5,015 | 72 | % | 2,399 | 84 | % | 2,616 | |||||||||||||
| Total operating expense | 6,951 | 100 | % | 2,871 | 100 | % | 4,080 | |||||||||||||
| Derivative (loss)/income | (134 | ) | 48 | (182 | ) | |||||||||||||||
| Sale of Eniluracil | - | 40 | (40 | ) | ||||||||||||||||
| Other loss | (8 | ) | (14 | ) | 6 | |||||||||||||||
| Interest income and other, net | 47 | 8 | 39 | |||||||||||||||||
| Net income (loss) | $ | (7,046 | ) | $ | (2,789 | ) | $ | (4,257 | ) | |||||||||||
| · | Research and development expense increased by $1.5 million in fiscal 2017, as compared to fiscal 2016 primarily due to drug manufacturing activities related to the preparation for registration batches as the company prepares to submit new drug application to the FDA and EMA. |
| · | The $2.6 million increase in general and administrative expenses are attributed to a rise in compensation to officers, directors and key contract employees. Most of this increase relates to non-cash equity-based compensation that was granted or vested during the year. Expense associated with equity compensation is directly related to the change in the underlying equity instrument. During fiscal year 2017, the price of the Company’s common shares rose 368%. This had a dramatic impact on the non-cash expense of issuing equity-based compensation. |
| · | Derivative (loss)/income grew by $0.2 million. The company has a very small number of derivative options outstanding. Changes in the valuation associated with these options are not expected to have a significant impact on the Company’s financial statements for the remaining life of these derivatives. The weighted average term of all remaining derivative liabilities is 0.5 years. |
| · | Interest income increased in fiscal 2017, as compared to 2016 due to a higher average cash balance for the comparable periods. |
| 25 |
Quarterly Information
The following table presents selected consolidated financial data for each of the last eight quarters through December 31, 2017, as prepared under U.S. GAAP (dollars in thousands, except per share information).
| Period | Net (Loss)/Income for the Period | Basic Net (Loss)/Income per Common Share | Diluted Net (Loss)/Income per Common Share | |||||||||
| March 31, 2016 | (420 | ) | (0.04 | ) | (0.04 | ) | ||||||
| June 30, 2016 | (724 | ) | (0.06 | ) | (0.06 | ) | ||||||
| September 30, 2016 | (502 | ) | (0.04 | ) | (0.04 | ) | ||||||
| December 31, 2016 | (1,143 | ) | (0.08 | ) | (0.08 | ) | ||||||
| March 31, 2017 | (806 | ) | (0.06 | ) | (0.06 | ) | ||||||
| June 30, 2017 | (1,598 | ) | (0.11 | ) | (0.11 | ) | ||||||
| September 30, 2017 | (2,352 | ) | (0.15 | ) | (0.15 | ) | ||||||
| December 31, 2017 | (2,290 | ) | (0.15 | ) | (0.15 | ) | ||||||
Quarter ended December 31, 2017 versus 2016
| Quarter Ended | Quarter Ended | Increase | ||||||||||||||||||
| In thousands of U.S. Dollars | December 31, 2017 | % | December 31, 2016 | % | (Decrease) | |||||||||||||||
| Revenue | $ | - | $ | - | $ | - | ||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 886 | 35 | % | 174 | 15 | % | 712 | |||||||||||||
| General and administration | 1,629 | 65 | % | 972 | 85 | % | 657 | |||||||||||||
| Total operating expense | 2,515 | 100 | % | 1,146 | 100 | % | 1,369 | |||||||||||||
| Other income | 206 | 1 | 205 | |||||||||||||||||
| Interest income and other, net | 19 | 2 | 17 | |||||||||||||||||
| Net (loss) | $ | (2,290 | ) | $ | (1,143 | ) | $ | (1,147 | ) | |||||||||||
The Company reported a net loss from operations of $2.3 million (which excludes a non-cash gain on derivatives of $0.2 million) for the three months ended December 31, 2017, compared to a net loss from operations of $1.1 million (excluding an immaterial non-cash gain on derivative valuation) in 2016. Research and development expenses totaled $1.0 million for the three months ended December 31, 2017, as compared to a $0.2 million in the same period in 2016 as the Company increased drug manufacturing expense related to the production of registration batches. General and administrative expenses increased by $0.7 million in the three months ended December 31, 2017, as compared to the same period in 2016. The increase relates to compensation in the form of cash, and non-cash equity-based compensation for employees and certain key contract employees.
| Selected Asset and Liability Data (thousands): | As at December 31, 2017 | As at December 31, 2016 | ||||||
| Cash and equivalents | $ | 28,260 | $ | 3,926 | ||||
| Other current assets | 141 | 46 | ||||||
| Current liabilities excluding derivative liability | 1,477 | 369 | ||||||
| Derivative warrant liability | 167 | 33 | ||||||
| Working capital [current assets – current liabilities excluding derivative liability] | 26,924 | 3,603 | ||||||
| Selected Equity: | ||||||||
| Common shares | $ | 103,045 | $ | 74,515 | ||||
| Accumulated deficit | (121,368 | ) | (114,322 | ) | ||||
| Stockholders’ equity | 26,757 | 3,570 | ||||||
Liquidity and Capital Resources
| · | The $24.3 million increase in cash and cash equivalents between December 31, 2017 and December 31, 2016 is due to the $7.6 and $21.2 million (gross proceeds) equity financing completed in June and December, respectively, of 2017, and the $0.6 million cash proceeds from the exercise of 21 warrants and 359 options during 2017. These cash inflows were offset by clinical trial expenses related to our Phase III study of STS, the increase in regulatory and manufacturing activities for STS and our general and administrative expenses. |
| · | The increase in other current assets between December 31, 2017 and December 31, 2016 relates to an increase in pre-paid Director’s and Officer’s Insurance over the prior year and an increase in prepaid expenses to OHSU for research. |
| · | Current liabilities increased primarily due to manufacturing activities associated with production of PEDMARKTM and related regulatory expenses. The company also had payables balances for legal and other professional services related to the December financing. |
| · | Working capital increased between December 31, 2017 and December 31, 2016 by $23.3 million. The increase was a result of the two financings in addition to various warrant and option exercises in 2017. These cash inflows were offset by cash expenditures related to our clinical trials, the commercial development of PEDMARKTM and general and administrative expenses. The Company expects increased cash outflows as it prepares submission batches prior to an FDA filing. |
| 26 |
| Selected Cash Flow Data (dollars and shares in thousands) | Year Ended December 31, 2017 | Year Ended December 31, 2016 | ||||||
| Net cash used in operating activities | $ | (3,641 | ) | $ | (2,124 | ) | ||
| Net cash provided from investing activities | - | - | ||||||
| Net cash provided from financing activities | 27,975 | 5,108 | ||||||
| Net cash flow | $ | 24,334 | $ | 2,984 | ||||
| Number of common shares outstanding | 18,411 | 13,643 | ||||||
The net cash flow used in operating activities for the year ended December 31, 2017 was approximately $3.6 million as compared to $2.1 million in 2016. This increase relates to the commercial development of PEDMARKTM.
We continue to pursue various strategic alternatives including collaborations with other pharmaceutical and biotechnology companies. Our projections of further capital requirements are subject to substantial uncertainty. Our working capital requirements may fluctuate in future periods depending upon numerous factors, including: our ability to obtain additional financial resources; our ability to enter into collaborations that provide us with up-front payments, milestones or other payments; results of our research and development activities; progress or lack of progress in our preclinical studies or clinical trials; unfavorable toxicology in our clinical programs, our drug substance requirements to support clinical programs; change in the focus, direction, or costs of our research and development programs; headcount expense; the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing our patent claims; competitive and technological advances; the potential need to develop, acquire or license new technologies and products; our business development activities; new regulatory requirements implemented by regulatory authorities; the timing and outcome of any regulatory review process; and commercialization activities, if any.
We had cash and cash equivalents of approximately $28.3 million as of December 31, 2017.
Financial Instruments
We invest excess cash and cash equivalents in high credit quality investments held by financial institutions in accordance with our investment policy designed to protect the principal investment. At December 31, 2017, we had approximately $0.28 million in our cash accounts and $27.98 million in our money market accounts. We have not experienced any loss or write down of our money market investments since the inception of the Company.
Our investment policy is to manage investments to achieve, in the order of importance, the financial objectives of preservation of principal, liquidity and return on investment. Investments may be made in U.S. or Canadian obligations and bank securities, commercial paper of U.S. or Canadian industrial companies, utilities, financial institutions and consumer loan companies, and securities of foreign banks provided the obligations are guaranteed or carry ratings appropriate to the policy. Securities must have a minimum Dun & Bradstreet rating of A for bonds or R1 low for commercial paper. The policy also provides for investment limits on concentrations of securities by issuer and maximum-weighted average time to maturity of twelve months. This policy applies to all of our financial resources. The policy risks are primarily the opportunity cost of the conservative nature of the allowable investments. As our main purpose is research and development, we have chosen to avoid investments of a trading or speculative nature.
We classify investments with original maturities at the date of purchase greater than three months which mature at or less than twelve months as current. We carry investments at their fair value with unrealized gains and losses included in other comprehensive income (loss); however we have not held any instruments that were classified as short term investments during the periods presented in this Annual Report.
Off-Balance Sheet Arrangements
Since our inception, we have not had any material off-balance sheet arrangements.
Contractual Obligations and Commitments
None.
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenue and expense during the reporting period. These estimates are based on assumptions and judgments that may be affected by commercial, economic and other factors. Actual results could differ from these estimates.
An accounting policy is considered to be critical if it requires an accounting estimate to be made based on assumptions about matters that are highly uncertain at the time the estimate is made, and if different estimates reasonably could have been used, or changes in the accounting estimates that are reasonably likely to occur periodically, could materially impact the financial statements. The following description of critical accounting policies, judgments and estimates should be read in conjunction with our December 31, 2017 consolidated financial statements.
| 27 |
Stock-based Compensation
The calculation of the fair values of our stock-based compensation plans requires estimates that require management’s judgments. Under ASC 718, the fair value of each stock option is estimated on the grant date using the Black-Scholes option-pricing model. The valuation models require assumptions and estimates to determine expected volatility, expected life, expected dividends and expected risk-free interest rates. The expected volatility was determined using historical volatility of our stock based on the contractual life of the award. The risk-free interest rate assumption was based on the yield on zero-coupon U.S. Treasury strips at the award grant date. We also used historical data to estimate forfeiture experience. In valuing options granted in the year ended December 31, 2017 and fiscal year ended December 31, 2016 we used the following weighted average assumptions:
| Year Ended December 31, 2017 | Year Ended December 31, 2016 | |||
| Expected dividend | 0% | 0% | ||
| Risk-free interest rate | 2.04 – 2.33% | 1.27 – 2.25% | ||
| Expected volatility | 158 – 168% | 134 – 137% | ||
| Expected life | 7 years | 7 years |
Common shares and warrants
Common shares are recorded as the net proceeds received on issuance after deducting all share issuance costs and the relative fair value of investor warrants. Warrants are recorded at relative fair value and are deducted from the proceeds of common shares and recorded on the consolidated statements of stockholders’ equity as additional paid-in capital.
Derivative Instruments
The Company applies ASC Topic 815-40, "Derivatives and Hedging" (ASC 815-40). One of the conclusions reached under ASC 815-40 was that an equity-linked financial instrument would not be considered indexed to the entity's own stock if the strike price is denominated in a currency other than the issuer's functional currency. The conclusion reached under ASC 815-40 clarified the accounting treatment for these and certain other financial instruments. ASC 815-40 specifies that a contract will not be treated as a derivative if it meets the following conditions: (a) indexed to the Company's own stock; and (b) classified in stockholders' equity in the Company's statement of financial position. The Company's outstanding warrants denominated in Canadian dollars are not considered to be indexed to its own stock because the exercise price is denominated in Canadian dollars and the Company's functional currency is United States dollars. Therefore, these warrants have been treated as derivative financial instruments and recorded at their fair value as a liability. All other outstanding convertible instruments are considered to be indexed to the Company's stock, because their exercise price is denominated in the same currency as the Company's functional currency, and are included in stockholders' equity.
The Company's derivative instruments include options to purchase 19 common shares, the exercise prices for which are denominated in a currency other than the Company's functional currency, as follows:
| · | Contractor options to purchase 17 common shares exercisable at CAD$1.62 per whole common share that expire on April 4, 2018; |
| · | Contractor options to purchase 2 common shares exercisable at CAD$2.43 per whole common share that expire on May 18, 2018. |
These options have been recorded at their fair value as a liability at issuance and will continue to be re-measured at fair value as a liability at each subsequent balance sheet date. Any change in value between reporting periods will be recorded as unrealized gain/(loss). These options will continue to be reported as a liability until such time as they are exercised, forfeited or expire. The fair value of these options is estimated using the Black-Scholes option-pricing model.
| Derivative Value at December 31, | (Loss)/Gain on Derivative Instrument December 31, | |||||||||||||||
| Derivative Warrants/Options | 2017 | 2016 | 2017 | 2016 | ||||||||||||
| Warrants expiring March 29, 2016 | - | - | - | 41 | ||||||||||||
| Options (various expiration dates) | 167 | 33 | (134 | ) | 7 | |||||||||||
| Total | 167 | 33 | (134 | ) | 48 | |||||||||||
The value of the derivative liability presented on the balance sheet has typically been influenced by changes in the underlying share price of the Company.
| 28 |
Outstanding Share Information
Our outstanding comparative share data at December 31, 2017 and December 31, 2016 is as follows (in thousands):
| Outstanding Share Type | December 31, 2017 | December 31, 2016 | ||||||
| Common shares | 18,411 | 13,643 | ||||||
| Warrants to purchase common shares | 1,362 | 1,383 | ||||||
| Options to purchase common shares | 2,315 | 2,427 | ||||||
| Total | 22,088 | 17,453 | ||||||
Newly Adopted and Recent Accounting Pronouncements
In February 2017, the FASB issued ASU No. 2017-05, “Other Income - Gains and Losses from the Derecognition of Nonfinancial Assets (Subtopic 610-20): Clarifying the Scope of Asset Derecognition Guidance and Accounting for Partial Sales of Nonfinancial Assets” (“ASU 2017-05”). ASU 2017-05 is meant to clarify the scope of the original guidance within Subtopic 610-20 that was issued in connection with ASU 2014-09, as defined below, which provides guidance for recognizing gains and losses from the transfer of nonfinancial assets in contracts with noncustomers. ASU 2017-05 also added guidance for partial sales of nonfinancial assets. ASU 2017-05 is effective for our fiscal year ending December 31, 2018 and we are required to adopt ASU 2017-05 concurrent with the adoption of ASU 2014-09. Adoption of ASU 2017-05 is required, January 1, 2018. The expected impact of ASU 2017-05 on our consolidated financial statements and disclosures is de minimis.
In May 2017, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update 2017-09, Compensation—Stock Compensation (Topic 718): Scope of Modification Accounting (“ASU 2017-09”). The FASB issued ASU 2017-09 to clarify and reduce both (i) diversity in practice and (ii) cost and complexity when applying the guidance in Topic 718, to a change to the terms and conditions of a share-based payment award. This guidance is effective for the Company as of the fourth quarter of its fiscal year ending December 31, 2018. Early adoption is permitted. The amendments in this ASU should be applied prospectively to an award modified on or after the adoption date. The Company is currently evaluating the impact of this updated standard, but does not believe this update will have a significant impact on its consolidated financial statements.
In May 2014, the FASB issued ASU 2014-9, Revenue from Contracts with Customers (Topic 606), to clarify the principles for recognizing revenue. This update provides a comprehensive new revenue recognition model that requires revenue to be recognized in a manner to depict the transfer of goods or services to a customer at an amount that reflects the consideration expected to be received in exchange for those goods or services. In August 2015, the FASB issued ASU No. 2015-14, Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date, which delayed the effective date of the new standard from January 1, 2017 to January 1, 2018. The FASB also agreed to allow entities to choose to adopt the standard as of the original effective date. In March 2016, the FASB issued ASU No. 2016-08, Revenue from Contracts with Customers (Topic 606): Principal versus Agent Considerations, which clarifies the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued ASU No. 2016-10, Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing, which clarifies certain aspects of identifying performance obligations and licensing implementation guidance. In May 2016, the FASB issued ASU No. 2016-12, Revenue from Contracts with Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients related to disclosures of remaining performance obligations, as well as other amendments to guidance on collectability, non-cash consideration and the presentation of sales and other similar taxes collected from customers. In September 2017, the FASB issued ASU No. 2017-13, Revenue Recognition (Topic 605), Revenue from Contracts with Customers (Topic 606), Leases (Topic 840), and Leases (Topic 842): Amendments to SEC Paragraphs Pursuant to the Staff Announcement at the July 20, 2017 EITF Meeting and Rescission of Prior SEC Staff Announcements and Observer Comments. The amendments in ASU No. 2017-13 amends the early adoption date option for certain companies related to the adoption of ASU No. 2014-09 and ASU No. 2016-02. In November 2017, the FASB issued ASU No. 2017-14, Revenue from Contracts with Customers (Topic 606): Income Statement- Reporting Comprehensive Income (Topic 220), Revenue Recognition (Topic 605), which amends certain SEC paragraphs within the FASB Accounting Standards Codification. These standards have the same effective date and transition date of January 1, 2018. The new revenue standard allows for either full retrospective or modified retrospective application. The Company currently does not have any revenue and therefore this update will not have a significant impact on its consolidated financial statements.
In February 2016, the FASB issued ASU 2016-02, Leases, which amends the accounting guidance related to leases. These changes, which are designed to increase transparency and comparability among organizations for both lessees and lessors, include, among other things, requiring recognition of lease assets and liabilities on the balance sheet and disclosing key information about leasing arrangements. In September 2017, the FASB issued ASU No. 2017-13, Revenue Recognition (Topic 605), Revenue from Contracts with Customers (Topic 606), Leases (Topic 840), and Leases (Topic 842): Amendments to SEC Paragraphs Pursuant to the Staff Announcement at the July 20, 2017 EITF Meeting and Rescission of Prior SEC Staff Announcements and Observer Comments. The amendments in ASU No. 2017-13 amends the early adoption date option for certain companies related to the adoption of ASU No. 2014-09 and ASU No. 2016-02. Adoption and implementation of the guidance is not required by the Company until the beginning of fiscal 2019, although early adoption is permitted. The Company has not yet completed its assessment of the impact that adoption of this guidance will have on its consolidated financial statements.
| 29 |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Money Market Investments
We maintain an investment portfolio consisting of U.S. or Canadian obligations and bank securities and money market investments in compliance with our investment policy. We do not hold any mortgaged-backed investments in our investment portfolio. Securities must have a minimum Dun & Bradstreet rating of A for bonds or R1 low for commercial paper. The policy also provides for investment limits on concentrations of securities by issuer and maximum-weighted average time to maturity of twelve months. This policy applies to all of our financial resources.
At December 31, 2017, we had $27.98 million in money market investments as compared to $3.86 million at December 31, 2016; these investments typically have minimal risk. We have not experienced any loss or write down of our money market investments for the years ended December 31, 2017 and 2016.
Our investment policy is to manage investments to achieve, in the order of importance, the financial objectives of preservation of principal, liquidity and return on investment. Our risk associated with fluctuating interest rates on our investments is minimal and not significant to the results of operations. We currently do not use interest rate derivative instruments to manage exposure to interest rate changes. As the main purpose of the Company is research and development, we have chosen to avoid investments of a trade or speculative nature.
Foreign Currency Exposure
We are subject to foreign currency risks as we purchase goods and services which are denominated in Canadian dollars. To date, we have not employed the use of derivative instruments; however, we do hold Canadian dollars which we use to pay vendors in Canada and other corporate obligations. At December 31, 2017 the company held approximately three hundred twenty-one thousand Canadian dollars.
| Item 8. | Financial Statements and Supplementary Data |
The financial statements required to be filed pursuant to this Item 8 are appended to this Annual Report on Form 10-K. A list of the financial statements filed herewith is found at “Index to Financial Statements” on Page F-1.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
During 2017, the Audit Committee (the “Committee”) of the Board of Directors of the Company conducted a competitive selection process to determine the Company’s independent registered public accounting firm for the fiscal year ending December 31, 2017. This search began after Deloitte LLP (“Deloitte”) advised the Company it would resign as of May 15, 2017. The Committee invited several independent public accounting firms to participate in this process.
The reports of Deloitte on the Company’s consolidated financial statements for the fiscal years ended December 31, 2016 and 2015 did not contain an adverse opinion or disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles. In connection with the audits of the Company's consolidated financial statements for the fiscal years ended December 31, 2016 and 2015, and in the subsequent interim period through May 15, 2017, there were no disagreements with Deloitte on any matters of accounting principles or practices, financial statement disclosure or auditing scope and procedures which, if not resolved to the satisfaction of Deloitte, would have caused Deloitte to make reference to the matter in their report. There were no reportable events (as that term is described in Item 304(a)(1)(v) of Regulation S-K of the Securities Act of 1933, as amended) during the two fiscal years ended December 31, 2016 and 2015, or in the subsequent period through May 15, 2017.
The Company has provided a copy of the foregoing disclosures to Deloitte and requested that Deloitte furnish it with a letter addressed to the Securities and Exchange Commission stating whether Deloitte agrees with the above statements. A copy of Deloitte’s letter, dated May 17, 2017, is filed as Exhibit 16.1 to this Form 10-K.
The Committee approved the appointment of Haskell & White LLP as the Company’s independent registered public accounting firm on May 15, 2017, for the fiscal year ending December 31, 2017. During the two most recent fiscal years and in the subsequent interim period through May 15, 2017, the Company has not consulted with Haskell & White LLP with respect to the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that would have been rendered on the Company’s consolidated financial statements, or any other matters set forth in Item 304(a)(2)(i) or (ii) of Regulation S-K.
| 30 |
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation and supervision of our Chief Executive Officer and Chief Financial Officer, is responsible for our disclosure controls and procedures pursuant to Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified under SEC rules and forms. Disclosure controls and procedures include controls and procedures designed to ensure that information required to be disclosed in our reports filed under the Exchange Act is accumulated and communicated to our principal executive officer and our principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management, including the Chief Executive Officer and the Chief Financial Officer, carried out an evaluation of the effectiveness of our disclosure controls and procedures as of December 31, 2017. Based on this evaluation, our management concluded that as of December 31, 2017 these disclosure controls and procedures were not effective at the reasonable assurance level as a result of the material weaknesses in our internal control over financial reporting, which are described below. As discussed below, our internal control over financial reporting is an integral part of our disclosure controls and procedures.
Management's Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. Internal control over financial reporting is a process designed by, or under the supervision of, our principal executive and principal financial officers, or persons performing similar functions, and effected by our board of directors, management, and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and includes those policies and procedures that:
| 1. | Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; |
| 2. | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
| 3. | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of inherent limitations, no matter how well designed and operated, internal control over financial reporting may not prevent or detect misstatements and can only provide reasonable assurance of achieving the desired control objectives. In addition, the design of internal control over financial reporting must reflect the fact that there are resource constraints and that management is required to apply its judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Our Chief Executive Officer and Chief Financial Officer have performed an evaluation of our internal control over financial reporting under the framework in Internal Control-Integrated Framework (2013), issued by the Committee of Sponsoring Organizations of the Treadway Commission. The objective of this assessment was to determine whether our internal control over financial reporting was effective at December 31, 2017. Based on the results of this evaluation, we have concluded that our internal control over financial reporting was not effective at December 31, 2017 as a result of having identified two material weaknesses in our internal control over financial reporting, as described in further detail below.
Our management has identified a control deficiency due to not maintaining an effective control environment, which is the foundation for the discipline and structure necessary for effective internal control over financial reporting, as evidenced by: (i) a lack of segregation of duties over individuals responsible for certain key control activities; (ii) an insufficient number of personnel appropriately qualified to perform control monitoring activities, including the recognition of the risks and complexities of transactions; and (iii) control activities that are not designed to respond to the risks identified. This control deficiency could result in a misstatement of balance sheet, income and cash flow statement accounts in our interim or annual financial statements that would not be detected. Accordingly, management has determined that this control deficiency constitutes a material weakness.
Our management has also identified another control deficiency that it believes constitutes a material weakness in our control over financial reporting. We did not maintain sufficient personnel with an appropriate level of technical accounting knowledge, experience, and training in the application of U.S. GAAP with regards to unusual transactions commensurate with our complexity and our financial accounting and reporting requirements. This control deficiency could result in a misstatement of the financial statements including disclosure that would not be prevented or detected on a timely basis.
We believe the control deficiencies described herein, individually and when aggregated, represent material weaknesses in our internal control over financial reporting at December 31, 2017 since such deficiencies result in a reasonable possibility that a material misstatement in our annual or interim consolidated financial statements may not be prevented or detected on a timely basis by our internal controls.
| 31 |
These material weaknesses did not result in any material misstatements to the financial statements. However, these material weaknesses could result in misstatement of the aforementioned account balances or disclosures that would result in material misstatements to the annual or interim consolidated financial statements that would not be prevented or detected.
Management’s Remediation Activities
Since the identification of the material weaknesses in 2017, management has begun the evaluation process associated with the remediation of these weaknesses and will continue to take measures, including engaging service providers that may be necessary and advisable to address these weaknesses. In addition, under the direction of the Audit Committee of the Board of Directors, management will continue to review and make necessary changes to the overall design of the Company’s internal control environment, as well as to policies and procedures to improve the overall effectiveness of internal control over financial reporting of the Company.
Changes in Internal Control over Financial Reporting
There were no changes to the Company’s internal control over financial reporting during the fourth quarter of 2017 that materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Inherent Limitation on the Effectiveness of Internal Controls
The effectiveness of any system of internal control over financial reporting is subject to inherent limitations, including the exercise of judgment in designing, implementing, operating, and evaluating the controls and procedures, and the inability to eliminate misconduct completely. Accordingly, any system of internal control over financial reporting can only provide reasonable, not absolute, assurances. In addition, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. We intend to continue to monitor and upgrade our internal controls as necessary or appropriate for our business but cannot assure that such improvements will be sufficient to provide us with effective internal control over financial reporting.
| Item 9B. | Other Information |
None.
| 32 |
| Item 10. | Directors, Executive Officers and Corporate Governance |
The following table sets forth the name of each of our executive officers and directors, such person’s principal occupation or employment, all other positions with us held by such person, if any, the year in which such person became a director of Fennec and such person’s age.
The Corporation has an Audit Committee, a Compensation Committee, and a Governance Committee. The current members of such committees are noted below:
| Name and Province/State and Country of Residence, Position |
Current Principal Occupation and Principal Occupation For Previous Five Years |
Director Since | Age | |||
| Rostislav Raykov, New Jersey, USA Chief Executive Officer, Director |
CEO of Fennec Pharmaceuticals Inc.; Co-Founder and Manager, DCML LLC; previously Portfolio Manager at Alchem Partners; previously Portfolio Manager at John Levin & Company | July 2009 | 42 | |||
| Robert Andrade, Texas, USA Chief Financial Officer |
CFO of Fennec Pharmaceuticals; previously senior analyst at Magnetar Capital; previously Portfolio Manager at Millennium Partners | September 2009- August 2013; November 2015 | 43 | |||
| Chris A. Rallis, North Carolina, USA Director(1)(2) |
Executive in-residence at Pappas Ventures; previously CEO of ImmunoBiosciences | August 2011 | 64 | |||
| Marco Brughera, Milano, Italy Director(2)(3) |
CEO of Leadiant Biosciences SpA; previously Global Head Rare Disease and R&D at Sigma-tau; VP Preclinical Development at Nerviano Medical Sciences. | August, 2016 | 62 | |||
| Adrian J. Haigh, Dublin, Ireland Director(1)(3) |
Senior Vice President and General Manager of EMEA Region at PTC Therapeutics; previously Chief Operating Officer at Gentium GmbH; previously Regional VP Commercial Operations at Biogen Idec | April 2014 | 58 | |||
| Khalid Islam, Zug, Switzerland Chairman of Board, Director(1)(2)(3) |
Founder/co-founder of Sirius Healthcare Partners GMbH; previously Chairman and CEO of Gentium S.p.A.; previously CEO of Arpida AG | April 2014 | 62 |
| (1) | Member of the Audit Committee |
| (2) | Member of the Compensation Committee |
| (3) | Member of the Governance Committee |
Rostislav Raykov
Mr. Raykov has served as a director of Fennec since July 2009 and as Chief Executive Officer since July 2009. From January 2006 to December 2007, Mr. Raykov was a portfolio manager for Alchem Investment Partners and John Levin & Co. Prior to founding Alchem, Mr. Raykov was a portfolio manager and securities analyst for John A. Levin & Co. Event Driven Fund (2002-2005). Prior to joining John A. Levin & Co., Mr. Raykov was a securities analyst for the Merger Fund at Tiedemann Investment Group (1999-2002) and an investment banking analyst at Bear Stearns (1998-1999). Mr. Raykov earned a B.S. in Business Administration from the University of North Carolina at Chapel Hill. As a result of these and other professional experiences, Mr. Raykov has financial expertise and experience with the Company as it has developed within the drug development industry and, as such, is able to provide the Company with unique insight and guidance.
Robert Andrade
Mr. Andrade has served as Chief Financial Officer since November 2015. Mr. Andrade was previously Chief Financial Officer and Director of Fennec from September 2009 until August 2013. In addition to his role with Fennec, Mr. Andrade was a senior analyst at Magnetar Capital, a portfolio manager for Millennium Partners and a senior analyst at Caxton Associates. Mr. Andrade graduated from University of Southern California, where he earned a Masters of Arts degree and Bachelor of Arts degree in economics.
Chris A. Rallis
Mr. Rallis has served as a director of Fennec since August 2011. Mr. Rallis has been an executive-in-residence at Pappas Ventures, a life science venture capital firm since January 2008. Previously, Mr. Rallis was the President and Chief Executive Officer of ImmunoBiosciences, Inc. (“IBI”), a vaccine technology company formerly located in Raleigh, North Carolina from April 2006 through June 2007. Prior to joining IBI, Mr. Rallis served as an executive in residence (part-time) for Pappas Ventures, and as a consultant for Duke University and Panacos Pharmaceuticals, Inc. Mr. Rallis is the former President and Chief Operating Officer (“COO”) and director of Triangle Pharmaceuticals, Inc., which was acquired by Gilead Sciences in January 2003 for approximately $465 million. Prior to assuming the role of President and COO in March 2000, he was Executive Vice President, Business Development and General Counsel. While at Triangle, Mr. Rallis participated in 11 equity financings generating gross proceeds of approximately $500 million. He was also primarily responsible for all business development activities which included a worldwide alliance with Abbott Laboratories and the in-licensing of ten compounds. Before joining Triangle in 1995, Mr. Rallis served in various business development and legal management roles with Burroughs Wellcome Co. over a 13-year period, including Vice President of Strategic Planning and Business Development. Mr. Rallis also serves on the boards of Aeolus Pharmaceuticals, a biopharmaceutical company located in Mission Viejo, California and Tenax Therapeutics, Inc., a biopharmaceutical company located in Morrisville, North Carolina. Mr. Rallis received his A.B. degree in economics from Harvard College and a J.D. from Duke University. As a result of these and other professional experiences, Mr. Rallis possesses particular healthcare industry knowledge and experience which strengthens the Board’s collective qualifications, skills, and experience.
| 33 |
Marco Brughera
Since January 2011, Dr. Brughera has been CEO of Lediant Biosciences SpA and has held several positions for the Sigma-Tau Group, including CEO and Global Head of Sigma Tau Rare Disease, President of Sigma-Tau Research and President of Sigma-Tau Pharmaceuticals. He drove the commercial revival of a lead oncology product line resulting in its successful sale for a total of around $900M. He also successfully out-licensed the Defibrotide US rights to Jazz Pharmaceuticals. From 2004 to 2010, Dr. Brughera served as the Vice President of Preclinical Development at Nerviano Medical Sciences (NMS), a pharmaceutical oncology-focused integrated discovery and development company. He also served as the Managing Director at Accelera, an independent contract research organization with the NMS Group. From 1999 to 2004, Dr. Brughera held several senior level positions in the areas of research and development with Pharmacia and Pfizer. Prior to 1999, he held various positions at Pharmacia & Upjohn and Farmitalia Carlo Erba SpA, an Italian pharmaceutical company. He currently serves on the Board of Solgenix and Lee’s Pharmaceutical and until early 2014 was a member of the Board of Gentium SpA. Dr. Brughera earned his degree in veterinary medicine from the University of Milan and is a European Registered Toxicologist.
Adrian J. Haigh
Mr. Adrian Haigh has been Senior Vice President and General Manager of EMEA Region and Asia Pacific at PTC Therapeutics, Inc. since September 2014. Previously Mr. Haigh served as Senior Vice President, Commercial Operations and Chief Operating Officer of Gentium GmbH since March 2011. Prior to joining Gentium, Mr. Haigh served as Regional Vice President, Commercial Operations at Biogen Idec where he managed several affiliates and also the global distributor business and prior to that was the General Manager of Amgen Nordis and Portugal. He served as the Executive Vice President of Global Marketing and Corporate Planning at EUSA Pharma and joined EUSA from Amgen where he led the international oncology franchise. Mr. Haigh previously has held senior commercial and marketing positions at SmithKline Beecham, Schering Plough, Organon and Novo Nordisk. He has been a Director of Fennec Pharmaceuticals Inc. since April 28, 2014 and a Director at Arch Biopartners Inc. since August 21, 2014. He received a Bachelor of Arts with Honors in Economic History from Huddersfield Polytechnic, West Yorkshire, England and a Diploma in Marketing from the Institute of Marketing. As a result of these and other professional experiences, Mr. Haigh has extensive international oncology development expertise which strengthens the Board’s collective qualifications, skills and experience.
Dr. Khalid Islam
Dr. Khalid Islam was the Chairman and CEO of Gentium S.p.A. (a Nasdaq-listed company; 2009-2014) where he led the transition from a loss-making to a cash-flow positive and profitable company. Under his leadership, the company value increased from US$25 million leading to a successful all cash US$1 billion merger with Jazz Pharmaceuticals, plc. Subsequent to the sale of Gentium, Dr. Islam has been involved from both an advisory and board level in several public and private healthcare related companies. From 1999-2008, Dr. Islam was President and CEO of Arpida AG where he transitioned the early-stage start-up to a SWX-listed company and raised US$300 million in the IPO and follow-ons. From 1987-1999, he held various positions in HMR & MMD (now Sanofi-Aventis). From 1977-1987, Dr. Islam worked in academia at Imperial College (Univ. of London) and in Milan University, where he was a contract professor. Dr. Islam is a graduate of Chelsea College and received his Ph.D. from Imperial College, University of London. He holds several patents and has published over 80 articles in leading journals. He is an advisor to the venture group Kurma Biofund (Paris). He is a founder/co-founder of Sirius Healthcare Partners GmbH (Zurich), PrevAbr LLC (D.C.), BioAim LLC (L.A.) & Life Sciences Management GmbH (Zug). Dr. Islam is Board Chair at Minoryx Therapeutics (Spain). He serves on the board of Karolinska Development (Sweden), MolMed S.p.A. (Italy) and Immunomedics Inc. (IMMU) all of which are traded publicly, and the private company OxThera (Sweden). In the past, he has served as Chairman of the Board of Directors of Pcovery Aps (Copenhagen), Adenium Aps (Copenhagen) and C10 Pharma AS (Oslo).
Audit Committee
On behalf of the Board, the Audit Committee of the Board retains, oversees and evaluates Fennec’s independent auditors, reviews the financial reports and other financial information provided by Fennec, including audited financial statements, and discusses the adequacy of disclosure with management and the auditors. The Audit Committee also reviews the performance of the independent auditors in the annual audit and in assignments unrelated to the audit, assesses the independence of the auditors, and reviews their fees. The Audit Committee is also responsible for reviewing Fennec’s internal controls over financial reporting and disclosure. The Audit Committee operates under a written charter adopted by the Board.
| 34 |
The directors have appointed an Audit Committee consisting of three directors: Chris A. Rallis, Khalid Islam and Adrian Haigh, each of whom is independent and financially literate within the meaning of National Instrument 52-110 – Audit Committees. In addition, the Board has determined that Mr. Rallis qualifies as an “audit committee financial expert,” as defined in Item 407(d)(5) of Regulation S-K promulgated by the SEC based on his business and financial experience described above.
Code of Ethics
In February 2004, Fennec’s Board adopted a Mandate of the Board of Directors, Corporate Governance Guidelines and a Code of Business Conduct and Ethics (the “Conduct and Ethics Code”) applicable to all officers, directors and employees of Fennec. Fennec is committed to adhering to applicable legal requirements and maintaining the highest standards of conduct and integrity. The Conduct and Ethics Code sets out the legal and ethical standards of conduct for personnel of Fennec and addresses topics such as: reporting obligations and procedures; honest and ethical conduct and conflicts of interest; compliance with applicable laws and Company policies and procedures; confidentiality of corporate information; use of corporate assets and opportunities; public disclosure and books and records; and non-retaliation. Fennec undertakes to provide to any person without charge, upon request, a copy of such Conduct and Ethics Code by writing to Attn: Code of Ethics Request, Fennec Pharmaceuticals Inc., 68 TW Alexander Drive, PO Box 13628, Research Triangle Park, North Carolina 27709.
| Item 11. | Executive Compensation |
Summary Compensation Table
The following table sets out certain information respecting the compensation paid to our Executive Officers, for the fiscal years ended December 31, 2017 and December 31, 2016 to our Chief Executive Officer and our Chief Financial Officer.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Option
Awards ($)(1) | Total ($) | |||||||||||||||
| Rostislav Raykov, CEO | 2017 | 262,500 | – | 187,579 | 450,079 | |||||||||||||||
| 2016 | 215,000 | – | 156,885 | 371,885 | ||||||||||||||||
| Robert Andrade, CFO | 2017 | 195,000 | – | 93,788 | 288,788 | |||||||||||||||
| 2016 | 177,500 | – | 268,933 | 446,433 | ||||||||||||||||
| (1) | Represents the aggregate grant date fair value computed in accordance with FASB ASC Topic 718. Dollar value amounts are based on individual grants to each of, Mr. Raykov and Mr. Andrade of 150,000, 100,000 and 75,000, 50,000 options, respectively, on July 5, 2016 and June 27, 2017, at an exercise price of $2.45 and $5.10 per common share, respectively, and will expire on July 5, 2023 and June 27, 2024, respectively. One-third of these options shall vest as of grant the grant date and be exercisable one year after the grant date (the “Vesting Commencement Date”). The remaining two-thirds of the options shall vest monthly at a rate of 1/36th of the remaining grant and shall be exercisable as of the last day of each following month after the Vesting Commencement Date. As of the third anniversary of the grant date, all of the options shall be vested. |
Rostislav Raykov
Mr. Raykov has been employed by Fennec since July 2009. Pursuant to an employment agreement dated May 3, 2010 between Mr. Raykov and Fennec, Mr. Raykov is employed as Fennec’s Chief Executive Officer and: (a) received an initial annual salary in the amount of $140,000, subject to annual adjustment by our Board of Directors, (b) upon approval by shareholders of our amended stock option plan was granted options to purchase up to 5.0% of our common shares estimated by us to be outstanding upon completion of the 2010 Rights Offering, and (c) may receive annual bonuses at the sole discretion of the Board. If Mr. Raykov’s employment terminates due to a change of control of Fennec, Mr. Raykov’s remaining unvested options shall immediately vest and be fully exercisable. If Mr. Raykov is dismissed from employment by us for any reason other than “for cause,” we are obligated to pay Mr. Raykov severance compensation equal to twelve months of salary ($275,000 per year at December 31, 2017) . The initial term of the agreement was for one year and the agreement automatically extends for additional one-year periods unless terminated by either party in accordance with the agreement.
Robert Andrade
Mr. Andrade has been employed by Fennec since November 2015. Mr. Andrade is employed as Fennec’s Chief Financial Officer. Pursuant to an employment agreement dated November 13, 2015, Mr. Andrade (a) receives an initial annual salary in the amount of $165,000, and (b) may receive annual bonuses at the sole discretion of the Board. In addition, conditioned upon the approval of Fennec's shareholders, Fennec will extend Mr. Andrade's existing options to their original expiry date of seven years from issuance. If Mr. Andrade’s employment terminates due to a change of control of the Fennec, Mr. Andrade’s remaining unvested options shall immediately vest and be fully exercisable. If Mr. Andrade is dismissed from employment by us for any reason other than “for cause,” we are obligated to pay Mr. Andrade severance compensation equal to six months of salary ($200,000 per year at December 31, 2017).
| 35 |
In addition to their employment agreements, Mr. Raykov and Mr. Andrade, are a party to a confidentiality and intellectual property agreement with the Company.
In the employment agreements for each of Mr. Andrade and Mr. Raykov “for cause” is generally defined as (1) material breach of the terms of the employment or intellectual property agreements; (2) failure to perform the duties inherent in the Employee’s position in good faith and in a reasonable and appropriate manner; or (3) acts of fraud or embezzlement or other intentional misconduct which adversely affects the Company's business.
Equity Grants, Exercises and Holdings
The following table sets forth information concerning the number and value of unexercised options held by each Named Executive Officer as of December 31, 2017. All executive awards, with the exception of those expiring 6/27/2024 and 07/05/2023, vest and are exercisable immediately. The current Stock Option Plan provides for grants denominated in US and CAD dollars.
| Number of Options | ||||||||||||||||
| Name | Granted | Exercisable | Option Exercise Price | Expiration Date | ||||||||||||
| Rostislav Raykov | 100,000 | - | USD$ | 5.10 | 06/27/2024 | |||||||||||
| 150,000 | 70,833 | USD$ | 2.45 | 07/05/2023 | ||||||||||||
| 25,000 | 25,000 | USD$ | 2.69 | 12/31/2021 | ||||||||||||
| 83,333 | 83,333 | USD$ | 1.59 | 01/24/2021 | ||||||||||||
| 16,666 | 16,666 | USD$ | 0.72 | 08/23/2020 | ||||||||||||
| 50,000 | 50,000 | USD$ | 1.05 | 11/20/2019 | ||||||||||||
| 17,050 | 17,050 | CAD$ | 1.89 | 08/18/2018 | ||||||||||||
| 323,961 | 323,961 | CAD$ | 2.43 | 08/18/2018 | ||||||||||||
| Robert Andrade | 50,000 | - | USD$ | 5.10 | 06/27/2024 | |||||||||||
| 75,000 | 35,416 | USD$ | 2.45 | 07/05/2023 | ||||||||||||
| 17,050 | 17,050 | CAD$ | 1.89 | 08/18/2018 | ||||||||||||
| 323,961 | 323,961 | CAD$ | 2.43 | 08/18/2018 | ||||||||||||
Termination Benefits
In the event of his termination with us other than for cause, we will be obligated to pay Mr. Raykov a one-time severance payment of $275,000. In the event of his termination with us other than for cause, we will be obligated to pay Mr. Andrade a one-time severance payment of $100,000.
Compensation of Directors
Director Compensation Table
The following table summarizes the compensation earned by the Company's non-executive directors for the year ended December 31, 2017.
| Name | Fees paid in Cash | Stock Awards | Option Awards(1)(2) | Total | ||||||||||||
| Dr. Islam | 89,000 | – | 522,710 | 611,710 | ||||||||||||
| Mr. Brughera | 41,500 | – | 98,452 | 139,952 | ||||||||||||
| Mr. Haigh | 44,000 | – | 498,097 | 542,097 | ||||||||||||
| Mr. Rallis | 49,000 | – | 98,452 | 147,452 | ||||||||||||
| Total | $ | 223,500 | $ | – | $ | 1,217,711 | $ | 1,441,211 | ||||||||
| (1) | Represents the aggregate grant date fair value computed in accordance with FASB ASC Topic 718. |
| (2) | Detail of grants are presented in the following table: |
| Name | Date of Grant | Number of Options Granted | Option Exercise Price $USD | |||||||
| Mr. Rallis | June 27, 2017 | 20,000 | 5.10 | |||||||
| Mr. Brughera | June 27, 2017 | 20,000 | 5.10 | |||||||
| Mr. Haigh | June 27, 2017 | 20,000 | 5.10 | |||||||
| Dr. Islam | June 27, 2017 | 25,000 | 5.10 | |||||||
| Mr. Haigh | April 25, 2014(1) | 66,666 | 2.31 | |||||||
| Dr. Islam | April 25, 2014(1) | 66,666 | 2.31 | |||||||
| Total | 218,332 | |||||||||
| 36 |
| (1) | Original grant was for 133,333 options to each of Mr. Haigh and Dr. Islam. The grant’s conditional vesting clause could be satisfied by the Company obtaining either orphan drug designation in the EU or Pediatric Use Marketing Authorization status for PEDMARKTM. On March 7, 2017, the Company received notice that PUMA status had been achieved by Medicine & Healthcare products Regulatory Agency (MHRA). The ruling of the MHRA was reviewed by the Compensation Committee of the Board of Directors on August 10, 2017 and the board voted to vest the remaining portion of the original grants. |
The annual compensation considerations for non-executive directors also include the awarding of stock options. We believe that granting of options to the non-executive directors serves three primary purposes: (1) to recognize the significant time and effort commitments during the past year; (2) to provide long-term incentives for future efforts since the value of the options is directly dependent on the market valuation of the Company; and (3) to retain quality individuals. When determining whether and how many new option grants will be made, the Compensation Committee takes into account the amount and terms of any outstanding options. Fennec does not require its non-executive directors to own a specific amount of common shares.
Each of Adrian J. Haigh, Khalid Islam, Marco Brughera and Chris A. Rallis has entered into an Independent Director Agreement with the Company, which provides for (i) cash compensation as set forth by the Compensation Committee commensurate with that member’s responsibilities. The Compensation Committee may also remunerate members in the form of a grant of options to purchase shares of the Company’s common shares. The options immediately vest when granted and are otherwise subject to the terms and conditions of the Company’s Stock Option Plan, as amended. The Independent Director Agreements also provide for the reimbursement of such director’s reasonable travel and related expenses incurred in the course of attending board meetings.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The following table sets forth information regarding our common shares beneficially owned as of March 16, 2018 by: (i) each of our officers and directors; (ii) all officers and directors as a group; and (iii) each person known by us to beneficially own five percent or more of the outstanding common shares. Except as indicated below, the security holders listed possess sole voting and investment power with respect to the shares beneficially owned by that person. Except as otherwise indicated below, the address for each listed shareholder is c/o Fennec Pharmaceuticals Inc., 68 TW Alexander Drive, PO Box 13628, Research Triangle Park, North Carolina 27709.
| Name | Common shares | Common shares Options Exercisable Within 60 Days | Common shares Purchase Warrants Exercisable Within 60 Days | Total Stock and Stock Based Holdings(1) | % Ownership(1) | |||||||||||||||
| Adrian J. Haigh | – | 173,579 | – | 173,579 | 0.93 | % | ||||||||||||||
| Dr. Khalid Islam | – | 238,825 | – | 238,825 | 1.28 | % | ||||||||||||||
| Robert Andrade | – | 516,011 | – | 516,011 | 2.72 | % | ||||||||||||||
| Marco Brughera | – | 55,545 | – | 55,545 | 0.30 | % | ||||||||||||||
| Chris A. Rallis | – | 131,850 | – | 131,850 | 0.71 | % | ||||||||||||||
| Rostislav Raykov | 40,740 | 866,010 | – | 906,750 | 4.69 | % | ||||||||||||||
| All Officers and Directors as a Group | 40,740 | 1,981,820 | – | 2,022,560 | 9.89 | % | ||||||||||||||
| Southpoint Capital Advisors, LP.(2) | 3,997,214 | – | – | 3,997,214 | 21.65 | % | ||||||||||||||
| Essetifin SpA(3) | 3,225,694 | – | – | 3,225,694 | 17.47 | % | ||||||||||||||
| Manchester Management Company, LLC.(4) | 1,530,588 | – | 999,999 | 2,530,587 | 13.37 | % | ||||||||||||||
| (1) | For purposes of this table “beneficial ownership” is determined in accordance with Rule 13d-3 under the Securities Exchange Act of 1934, pursuant to which a person or group of persons is deemed to have “beneficial ownership” of any common shares that such person or group has the right to acquire within 60 days after March 16, 2018. For purposes of computing the percentage of outstanding common shares held by each person or group of persons named above, any shares that such person or group has the right to acquire within 60 days after March 16, 2018 are deemed outstanding but are not deemed to be outstanding for purposes of computing the percentage ownership of any other person or group. As of March 16, 2018, there were 18,464,706 common shares issued and outstanding. |
| (2) | Southpoint Capital Advisors, LP, 623 Fifth Avenue, Suite 2503, New York, New York 10022. John S. Clark, II holds dispositive power over the shares owned by Southpoint Capital Advisors, LP. |
| (3) | Essetifin SpA, Via Sudafrica 20, Rome, Italy 00144. Mario Artali holds dispositive power over the shares owned by Essetifin SpA. |
| (4) | Manchester Management Company, LLC, 131 Charles Street, 1st Floor, Boston Massachusetts 02114. Includes 1,645,372 shares owned by Manchester Explorer, L.P. and 525,883 shares owned by JEB Partners, L.P. Manchester Management holds dispositive power over the shares held by Manchester Explorer, L.P. and JEB Partners, L.P. Jeb Besser and Morgan Frank hold shared dispositive power over the shares held by Manchester Management Company, LLC. Additionally, Jeb Besser owns 192,666 shares for which he has sole dispositive power and Morgan Frank owns 166,666 shares for which he has sole dispositive power. |
| 37 |
Equity Compensation Plan Information
The following table provides certain information with respect to securities authorized for issuance under equity incentive plans as of December 31, 2017 (share amounts are in thousands):
| Plan Category | (a) Number of securities to be issued upon exercise of outstanding options warrants and rights (*) | (b) Weighted-average exercise price of outstanding options, warrants and rights | (c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in Column (a)) | |||||||||
| Equity compensation plans approved by security holders | 2,315 | USD$ | 2.45 | * | 2,288 | |||||||
| Equity compensation plans not approved by security holders | – | – | – | |||||||||
| Total | 2,315 | – | 2,288 | |||||||||
* The Company’s current stock option plans allow for the issuance of stock options denominated in both U.S. dollars and Canadian dollars. This table presents the number and weighted-average exercise price of outstanding options by the currency associated with the original grants. At December 31, 2017 we had 1,603 stock options denominated in U.S. dollars with a weighted-average exercise price of $2.70 and 712 stock options denominated in CAD dollars with a weighted-average exercise price of CAD$2.38. There were a total of 2,315 stock options outstanding with a combined weighted-average exercise price of USD$2.45 (Canadian denominated exercise prices were converted using the December 31, 2017 exchange rate of 0.7966 CAD/USD. At December 31, 2017, we had 2,288 stock options available for future issuance.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
Related Party Transactions
There were no related party transactions during the years ended December 31, 2017 and 2016.
Director Independence
The Board of Directors is composed of a majority of independent directors. The Board applies the definition of independence found in the rules of the SEC and in Canadian National Instrument 58-101 and National Policy 58-201. The Board has determined that Mr. Brughera, Haigh, Islam, Rallis and Skolsky are “independent.” Mr. Raykov, Chief Executive Officer of the Company is considered to have a material relationship with the Company by virtue of his executive officer position and is therefore not independent. Fennec is of the view that the composition of its Board reflects a diversity of background and experience that are important for effective corporate governance. Other directorships held by Board members are described in this Annual Report under the heading “Directors and Executive Officers.”
| Item 14. | Principal Accounting Fees and Services |
The following presents the aggregate fees for professional services and other services rendered by our independent auditors, Haskell & White LLP and Deloitte LLP in fiscal year 2017 and 2016:
| Fiscal Year 2017 |
Fiscal Year 2016 |
|||||||
| Audit Fees(1) | 138,023 | 65,902 | ||||||
| Audit-Related Fees(2) | – | – | ||||||
| Tax Fees(3) | 14,043 | 10,026 | ||||||
| All Other Fees(4) | – | 1,370 | ||||||
| Total | $ | 152,066 | $ | 77,298 | ||||
| (1) | Audit Fees include fees for the standard audit work that needs to be performed each year in order to issue an opinion on the consolidated financial statements of the Company. It also includes fees for services that can only be provided by the Company’s auditor such as auditing of non-recurring transactions. |
| (2) | Audit-Related Fees include fees assurance and related services that are reasonably related to the performance of the audit or review and are traditionally performed by the independent accountant. |
| (3) | Tax Fees include fees for periodic tax consultations and compliance services in various local, regional and national tax jurisdictions. |
| (4) | All Other Fees include fees for products and services other than Audit Fees, Audit Related Fees and Tax Fees. |
The Audit Committee does not have formal pre-approval policies and procedures; however, prior to the engagement by the registrant, the Audit Committee approved all of the services performed by Haskell & White and Deloitte LLP as required by SEC regulation.
| 38 |
| Item 15. | Exhibits, Financial Statement Schedules |
(a) The following documents are included as part of this Annual Report filed on Form 10-K:
1. Financial Statements – See Index to Financial Statements on page F-1.
2. All schedules are omitted as the information required is inapplicable or the information is presented in the financial statements.
3. Exhibits:
| 39 |
| * | Indicates a management contract or compensatory plan. |
| ** | The Company has received confidential treatment with respect to certain portions of this exhibit. Those portions have been omitted from this exhibit and are filed separately with the U.S. Securities and Exchange Commission. |
| 40 |
Pursuant to the requirements of Section 13 of 15(d) the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Fennec Pharmaceuticals Inc. | |
| By: | /s/ Rostislav Raykov |
| Rostislav Raykov | |
| Chief Executive Officer and Director | |
| Date: March 28, 2018 | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signatures | Title | Date | ||
| /s/ Rostislav Raykov | Chief Executive Officer | March 28, 2018 | ||
| Rostislav Raykov | (principal executive officer) and Director | |||
| /s/ Robert Andrade | Chief Financial Officer | March 28, 2018 | ||
| Robert Andrade | (principal financial officer and principal accounting officer) |
|||
| /s/ Adrian J. Haigh | Director | March 28, 2018 | ||
| Adrian J. Haigh | ||||
| /s/ Dr. Khalid Islam | Director | March 28, 2018 | ||
| Dr. Khalid Islam | ||||
| /s/ Chris A. Rallis | Director | March 28, 2018 | ||
| Chris A. Rallis | ||||
| /s/ Marco Brughera | Director | March 28, 2018 | ||
| Marco Brughera |
Supplemental Information to be Furnished
With Reports Filed Pursuant to Section 15(d) of the Act by Registrants
Which Have Not Registered Securities Pursuant to Section 12 of the Act
The registrant intends to furnish proxy materials to its security holders subsequent to the filing of this annual report on Form 10-K and shall furnish copies of such proxy materials to the Commission when such materials are sent to security holders.
| 41 |
FENNEC PHARMACEUTICALS INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
Fennec Pharmaceuticals Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheet of Fennec Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2017, the related consolidated statements of operations, shareholders’ equity, and cash flows for the year then ended, and the related notes (collectively, the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2017, and the consolidated results of its operations and its cash flows for the year then ended, in conformity with accepted accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission, the PCAOB, and the CPAB.
We conducted our audit in accordance with the standards of the PCAOB and Canadian generally accepted auditing standards. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
| /s/ Haskell & White LLP | |
| HASKELL & WHITE LLP |
We have served as the Company’s auditor since 2017.
Irvine, California
March 28, 2018
| F-2 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of Fennec Pharmaceuticals Inc.
We have audited the accompanying consolidated balance sheet of Fennec Pharmaceuticals Inc. and subsidiaries (the "Company") as of December 31, 2016, and the related consolidated statements of operations, shareholders' equity, and cash flows for the year then ended. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States) and Canadian generally accepted auditing standards. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of Fennec Pharmaceuticals Inc. and subsidiaries as of December 31, 2016, and the results of their operations and their cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company incurred a loss from operations of $2,871,000 during the year ended December 31, 2016 and still has not earned any revenue in its history. At December 31, 2016, the Company had an accumulated deficit of $114,322,000 and had experienced negative cash flows from operating activities in the amount of $2,124,000. These circumstances raise substantial doubt about the Company’s ability to continue as a going concern. Managements’ plans concerning these matters are discussed in Note 1 to the consolidated financial statements. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| /s/ Deloitte LLP | |
| Chartered Professional Accountants | |
| Licensed Public Accountants | |
| Ottawa, Canada | |
| March 28, 2017 |
| F-3 |
Consolidated Balance Sheets
(U.S. dollars and shares in thousands)
| December 31, | December 31, | |||||||
| 2017 | 2016 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 28,260 | $ | 3,926 | ||||
| Prepaid expenses | 128 | 43 | ||||||
| Other current assets | 13 | 3 | ||||||
| Total assets | $ | 28,401 | $ | 3,972 | ||||
| Liabilities and Shareholders' Equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 855 | $ | 244 | ||||
| Accrued liabilities | 622 | 125 | ||||||
| Derivative instruments (Note 5) | 167 | 33 | ||||||
| Total current liabilities | 1,644 | 402 | ||||||
| Total liabilities | 1,644 | 402 | ||||||
| Commitments and Contingencies (Note 9) | ||||||||
| Shareholders' equity: | ||||||||
| Common stock, no par value; unlimited shares authorized; 18,411 shares issued and outstanding (2016-13,643) | 103,045 | 74,515 | ||||||
| Additional paid-in capital | 43,837 | 42,134 | ||||||
| Accumulated deficit | (121,368 | ) | (114,322 | ) | ||||
| Accumulated other comprehensive income | 1,243 | 1,243 | ||||||
| Total shareholders’ equity | 26,757 | 3,570 | ||||||
| Total liabilities and shareholders’ equity | $ | 28,401 | $ | 3,972 | ||||
(The accompanying notes are an integral part of these consolidated financial statements)
| F-4 |
Consolidated Statements of Operations
(U.S. dollars and shares in thousands, except per share information)
| Year Ended | ||||||||
| December 31, | December 31, | |||||||
| 2017 | 2016 | |||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 1,936 | 472 | ||||||
| General and administrative | 5,015 | 2,399 | ||||||
| Loss from operations | (6,951 | ) | (2,871 | ) | ||||
| Other income/(expense): | ||||||||
| Unrealized (loss)/gain on derivatives (Note 5) | (134 | ) | 48 | |||||
| Sale of Eniluracil (Note 8) | - | 40 | ||||||
| Other loss | (8 | ) | (14 | ) | ||||
| Net interest income | 47 | 8 | ||||||
| Total other (loss)/income, net | (95 | ) | 82 | |||||
| Net loss | $ | (7,046 | ) | $ | (2,789 | ) | ||
| Loss per common share, basic and diluted | $ | (0.47 | ) | $ | (0.22 | ) | ||
| Weighted-average number of common shares outstanding, basic (Note 3) | 15,014 | 12,765 | ||||||
(The accompanying notes are an integral part of these consolidated financial statements)
| F-5 |
Consolidated Statements of Cash Flows
(U.S. dollars in thousands)
| Year Ended | ||||||||
| December 31, | December 31, | |||||||
| 2017 | 2016 | |||||||
| Cash flows (used in) provided by: | ||||||||
| Operating activities: | ||||||||
| Net loss | $ | (7,046 | ) | $ | (2,789 | ) | ||
| Adjustments to reconcile net (loss) to net cash used in operating activities: | ||||||||
| Unrealized loss/(gain) on derivatives | 134 | (48 | ) | |||||
| Stock-based compensation - consultants | 741 | 88 | ||||||
| Stock-based compensation - employees | 1,517 | 615 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses | (85 | ) | 33 | |||||
| Other assets | (10 | ) | (2 | ) | ||||
| Accounts payable | 611 | (54 | ) | |||||
| Accrued liabilities | 497 | 33 | ||||||
| Net cash used in operating activities | (3,641 | ) | (2,124 | ) | ||||
| Investing activity: | ||||||||
| Net cash used in investing activity | - | - | ||||||
| Financing activities: | ||||||||
| Issuance of shares, net of issuance costs | 27,381 | 5,000 | ||||||
| Issuance of shares, options exercise | 563 | 6 | ||||||
| Issuance of shares, warrants exercise | 31 | 102 | ||||||
| Net cash provided by financing activities | 27,975 | 5,108 | ||||||
| Increase in cash and cash equivalents | 24,334 | 2,984 | ||||||
| Cash and cash equivalents - Beginning of year | 3,926 | 942 | ||||||
| Cash and cash equivalents - End of year | $ | 28,260 | $ | 3,926 | ||||
(The accompanying notes are an integral part of these consolidated financial statements)
| F-6 |
Consolidated Statements of Shareholders' Equity
(U.S. dollars and shares in thousands)
| Accumulated | ||||||||||||||||||||||||
| Additional | Other | Total | ||||||||||||||||||||||
| Common Stock | Paid-in | Accumulated | Comprehensive | Stockholders' | ||||||||||||||||||||
| Number (Note 7) | Amount | Capital | Deficit | Income | Equity | |||||||||||||||||||
| Balance at December 31, 2015 | 10,940 | $ | 69,401 | $ | 41,437 | $ | (111,533 | ) | $ | 1,243 | $ | 548 | ||||||||||||
| Stock options issued to consultants | - | - | 16 | - | - | 16 | ||||||||||||||||||
| Stock options issued to employees | - | - | 615 | - | - | 615 | ||||||||||||||||||
| Warrants issued to consultants | - | - | 72 | - | - | 72 | ||||||||||||||||||
| Exercise of stock options | 4 | 6 | - | - | - | 6 | ||||||||||||||||||
| Exercise of warrants | 67 | 108 | (6 | ) | - | - | 102 | |||||||||||||||||
| Issuance of securities | 2,632 | 5,000 | - | - | - | 5,000 | ||||||||||||||||||
| Net loss | - | - | - | (2,789 | ) | - | (2,789 | ) | ||||||||||||||||
| Balance at December 31, 2016 | 13,643 | 74,515 | 42,134 | (114,322 | ) | 1,243 | 3,570 | |||||||||||||||||
| Stock options issued to consultants | - | - | 741 | - | - | 741 | ||||||||||||||||||
| Stock options issued to employees | - | - | 1,517 | - | - | 1,517 | ||||||||||||||||||
| Warrants issued to consultants | - | - | - | - | - | - | ||||||||||||||||||
| Exercise of stock options | 359 | 1,107 | (544 | ) | - | - | 563 | |||||||||||||||||
| Exercise of warrants | 21 | 42 | (11 | ) | - | - | 31 | |||||||||||||||||
| Issuance of securities | 4,388 | 27,381 | - | - | - | 27,381 | ||||||||||||||||||
| Net loss | - | - | - | (7,046 | ) | - | (7,046 | ) | ||||||||||||||||
| Balance at December 31, 2017 | 18,411 | $ | 103,045 | $ | 43,837 | $ | (121,368 | ) | $ | 1,243 | $ | 26,757 | ||||||||||||
(The accompanying notes are an integral part of these consolidated financial statements)
| F-7 |
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
| 1. | Nature of Business and Liquidity |
Fennec Pharmaceuticals Inc. (“Fennec”) was originally formed as a British Columbia corporation under the name Adherex Technologies Inc. and subsequently changed its name on September 3, 2014. Fennec, together with its wholly owned subsidiaries Oxiquant, Inc. (“Oxiquant”) and Fennec Pharmaceuticals, Inc., both Delaware corporations, and Cadherin Biomedical Inc. (“CBI”), a Canadian corporation, collectively referred to herein as the “Company,” is a biopharmaceutical company with a product candidate under development for use in the treatment of cancer. With the exception of Fennec Pharmaceuticals, Inc., all subsidiaries are inactive.
These consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (“US GAAP”) that are applicable to a going concern which contemplates that the Company will continue in operation for the foreseeable future and will be able to realize its assets and discharge its liabilities in the normal course of business.
During the year ended December 31, 2016, the Company incurred a net loss from operations of $2,871 and still has not earned any revenue in its history. At December 31, 2016, it had an accumulated deficit of $114,322 and had experienced negative cash flows from operating activities in the amount of $2,124.
Those circumstances raised substantial doubt about the Company’s ability to continue as a going concern within one year after the issue date of the December 31, 2016 consolidated financial statements, The Company was actively seeking to obtain additional funding in the future in order to finance the Company’s business strategy, operations and growth through the issuance of equity, debt or collaboration.
During the year ended December 31, 2017 the Company incurred a net loss from operations of $6,951 and still has not earned any revenue. At December 31, 2017, it had an accumulated deficit of $121,368 and had experienced negative cash flows from operating activities in the amount of $3,641.
On June 8, 2017, the Company completed the closing of a non-brokered private placement (the “Offering”) of 1,900,000 common shares for gross proceeds of $7.6 million ($7,571 net of commissions, fees and issue costs). Each common share was issued at a price of $4.00.
On December 12, 2017, the Company announced the completion of an underwritten public offering of 2,352,950 common shares at a public offering price of $8.50 per share. In addition, Fennec issued an additional 135,670 common shares in connection with the partial exercise of the underwriters’ over-allotment option. The approximate total gross proceeds from the offering was $21.2 million ($19,810 net of commissions, fees and issue costs).
The Company believes the aforementioned raises provide sufficient funding for the Company to carry-out its planned activities for the next twelve to eighteen months as it continues its strategic development of PEDMARKTM.
These financial statements do not reflect the potentially material adjustments in the carrying values of assets and liabilities, the reported expenses, and the balance sheet classifications used, that would be necessary if the going concern assumption were not appropriate.
| 2. | Significant Accounting Policies |
Basis of presentation
The consolidated financial statements include the accounts of Fennec and of all its wholly-owned subsidiaries. All inter-company transactions and balances have been eliminated upon consolidation.
Use of estimates
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that impact the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the consolidated financial statements and the reported amounts of revenue and expense during the reporting period. Significant estimates include the valuation of derivative warrant liability and the valuation of stock-based compensation. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash equivalents consist of highly liquid investments with original maturities at the date of purchase of three months or less.
The Company places its cash and cash equivalents in investments held by highly rated financial institutions in accordance with its investment policy designed to protect the principal investment. At December 31, 2017, the Company had $28,260 in cash and money market accounts (2016- $3,926). Money market investments typically have minimal risks. The Company has not experienced any loss or write-down of its money market investments.
| F-8 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Financial instruments
Financial instruments recognized on the balance sheets at December 31, 2017 and December 31, 2016 consist of cash and cash equivalents, accounts payable, accrued liabilities and derivative instruments, the carrying values of which, with the exception of the derivative instruments, approximate fair value due to their relatively short time to maturity. The Company does not hold or issue financial instruments for trading. The derivative liabilities are carried at fair value.
The Company’s investment policy is to manage investments to achieve, in the order of importance, the financial objectives of preservation of principal, liquidity and return on investment. Investments, when made, are made in U.S. or Canadian bank securities, commercial paper of U.S. or Canadian industrial companies, utilities, financial institutions and consumer loan companies, and securities of foreign banks provided the obligations are guaranteed or carry ratings appropriate to the policy. Securities must have a minimum Dun & Bradstreet rating of A for bonds or R1 low for commercial paper.
The policy risks are primarily the opportunity cost of the conservative nature of the allowable investments. As the main purpose of the Company is research and development, the Company has chosen to avoid investments of a trading or speculative nature.
Common shares and warrants
The Company has warrants outstanding to purchase common shares that were denominated in both United States dollars (“USD”) and Canadian dollars (“CAD”), which resulted in the Company having warrants outstanding that were denominated outside of the Company’s U.S. dollar functional currency.
The Company’s outstanding warrants denominated in Canadian dollars were not considered to be indexed to the Company’s own stock and should therefore be treated as derivative financial instruments and recorded at their fair value as a liability. At December 31, 2017, the derivative liabilities were valued at $167 (2016-$33). There was an unrealized loss on derivative liabilities of $134 for the year ended December 31, 2017(2016 gain-$48).
Revenue recognition
At this time, the Company does not have any revenue.
Research and development costs and investment tax credits
Research costs, including employee compensation, laboratory fees, lab supplies, and research and testing performed under contract by third parties, are expensed as incurred. Development costs, including drug substance costs, clinical study expenses and regulatory expenses are expensed as incurred.
Investment tax credits, which are earned as a result of qualifying research and development expenditures, are recognized when the expenditures are made and their realization is reasonably assured. They are applied to reduce related capital costs and research and development expenses in the year recognized.
Income taxes
The Company accounts for income taxes using the asset and liability method to compute the differences between the tax basis of assets and liabilities and the related financial amounts, using currently enacted tax rates. The Company has deferred tax assets, which are subject to periodic recoverability assessments. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amount that more likely than not will be realized. As of December 31, 2017, we maintained a full valuation allowance against our deferred tax assets.
The provisions of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 740-10, Uncertainty in Income Taxes, address the determination of whether tax benefits claimed or expected to be claimed on a tax return should be recorded in the financial statements. Under ASC 740-10, we may recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by taxing authorities, based on the technical merits of the position.
| F-9 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Foreign currency translation
The U.S. dollar is the functional currency for the Company’s consolidated operations. All gains and losses from currency translations are included in results of operations.
Loss per share
Basic net loss per share is computed by dividing net loss by the weighted average number of common shares outstanding during the year. Diluted net earnings per share is computed using the same method, except the weighted average number of common shares outstanding includes convertible debentures, stock options and warrants, if dilutive, as determined using the if-converted method and treasury methods. Accordingly, options to purchase 2,315 and warrants to purchase 1,362 shares at December 31, 2017, were not included in earnings per share. Such options and warrants would have an antidilutive effect. In 2016, options to purchase 2,427 and warrants to purchase 1,383 shares were excluded from the computation of earnings per share as their inclusion would have been antidilutive.
Recent accounting pronouncements
In February 2017, the FASB issued ASU No. 2017-05, “Other Income - Gains and Losses from the Derecognition of Nonfinancial Assets (Subtopic 610-20): Clarifying the Scope of Asset Derecognition Guidance and Accounting for Partial Sales of Nonfinancial Assets” (“ASU 2017-05”). ASU 2017-05 is meant to clarify the scope of the original guidance within Subtopic 610-20 that was issued in connection with ASU 2014-09, as defined below, which provides guidance for recognizing gains and losses from the transfer of nonfinancial assets in contracts with noncustomers. ASU 2017-05 also added guidance for partial sales of nonfinancial assets. ASU 2017-05 is effective for our fiscal year ending December 31, 2018 and we are required to adopt ASU 2017-05 concurrent with the adoption of ASU 2014-09. Adoption of ASU 2017-05 is required, January 1, 2018. The expected impact of ASU 2017-05 on our consolidated financial statements and disclosures is de minimis.
In May 2017, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update 2017-09, Compensation—Stock Compensation (Topic 718): Scope of Modification Accounting (“ASU 2017-09”). The FASB issued ASU 2017-09 to clarify and reduce both (i) diversity in practice and (ii) cost and complexity when applying the guidance in Topic 718, to a change to the terms and conditions of a share-based payment award. This guidance is effective for the Company as of the fourth quarter of its fiscal year ending December 31, 2018. Early adoption is permitted. The amendments in this ASU should be applied prospectively to an award modified on or after the adoption date. The Company is currently evaluating the impact of this updated standard, but does not believe this update will have a significant impact on its consolidated financial statements.
In May 2014, the FASB issued ASU 2014-9, Revenue from Contracts with Customers (Topic 606), to clarify the principles for recognizing revenue. This update provides a comprehensive new revenue recognition model that requires revenue to be recognized in a manner to depict the transfer of goods or services to a customer at an amount that reflects the consideration expected to be received in exchange for those goods or services. In August 2015, the FASB issued ASU No. 2015-14, Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date, which delayed the effective date of the new standard from January 1, 2017 to January 1, 2018. The FASB also agreed to allow entities to choose to adopt the standard as of the original effective date. In March 2016, the FASB issued ASU No. 2016-08, Revenue from Contracts with Customers (Topic 606): Principal versus Agent Considerations, which clarifies the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued ASU No. 2016-10, Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing, which clarifies certain aspects of identifying performance obligations and licensing implementation guidance. In May 2016, the FASB issued ASU No. 2016-12, Revenue from Contracts with Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients related to disclosures of remaining performance obligations, as well as other amendments to guidance on collectability, non-cash consideration and the presentation of sales and other similar taxes collected from customers. In September 2017, the FASB issued ASU No. 2017-13, Revenue Recognition (Topic 605), Revenue from Contracts with Customers (Topic 606), Leases (Topic 840), and Leases (Topic 842): Amendments to SEC Paragraphs Pursuant to the Staff Announcement at the July 20, 2017 EITF Meeting and Rescission of Prior SEC Staff Announcements and Observer Comments. The amendments in ASU No. 2017-13 amends the early adoption date option for certain companies related to the adoption of ASU No. 2014-09 and ASU No. 2016-02. In November 2017, the FASB issued ASU No. 2017-14, Revenue from Contracts with Customers (Topic 606): Income Statement- Reporting Comprehensive Income (Topic 220), Revenue Recognition (Topic 605), which amends certain SEC paragraphs within the FASB Accounting Standards Codification. These standards have the same effective date and transition date of January 1, 2018. The new revenue standard allows for either full retrospective or modified retrospective application. The Company currently does not have any revenue and therefore this update will not have a significant impact on its consolidated financial statements.
| F-10 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
In February 2016, the FASB issued ASU 2016-02, Leases, which amends the accounting guidance related to leases. These changes, which are designed to increase transparency and comparability among organizations for both lessees and lessors, include, among other things, requiring recognition of lease assets and liabilities on the balance sheet and disclosing key information about leasing arrangements. In September 2017, the FASB issued ASU No. 2017-13, Revenue Recognition (Topic 605), Revenue from Contracts with Customers (Topic 606), Leases (Topic 840), and Leases (Topic 842): Amendments to SEC Paragraphs Pursuant to the Staff Announcement at the July 20, 2017 EITF Meeting and Rescission of Prior SEC Staff Announcements and Observer Comments. The amendments in ASU No. 2017-13 amends the early adoption date option for certain companies related to the adoption of ASU No. 2014-09 and ASU No. 2016-02. Adoption and implementation of the guidance is not required by the Company until the beginning of fiscal 2019, although early adoption is permitted. The Company has not yet completed its assessment of the impact that adoption of this guidance will have on its consolidated financial statements.
| 3. | Loss per Share |
Loss per common share is presented under two formats: basic loss per common share and diluted loss per common share. Basic loss per common share is computed by dividing net loss attributable to common shareholders by the weighted average number of common shares outstanding during the period. Diluted loss per common share is computed by dividing net loss by the weighted average number of common shares outstanding during the period, plus the potentially dilutive impact of common shares equivalents (e.g. stock options and warrants). Dilutive common share equivalents consist of the incremental common shares issuable upon exercise of stock options and warrants. The following table sets forth the computation of basic and diluted net loss per share (in thousands except per share data):
| Year Ended | ||||||||
| December 31, 2017 | December 31, 2016 | |||||||
| Numerator: | ||||||||
| Net loss | $ | (7,046 | ) | $ | (2,789 | ) | ||
| Denominator: | ||||||||
| Weighted-average common shares, basic | 15,014 | 12,765 | ||||||
| Dilutive effect of stock options | – | – | ||||||
| Dilutive effect of warrants | – | – | ||||||
| Incremental dilutive shares | – | – | ||||||
| Weighted-average common shares, dilutive | 15,014 | 12,765 | ||||||
| Net loss per share, basic and diluted | $ | (0.47 | ) | $ | (0.22 | ) | ||
The following outstanding options and warrants were excluded from the computation of basic and diluted net loss per share for the periods presented because including them would have had an anti-dilutive effect:
| Year Ended | ||||||||
| December 31, 2017 | December 31, 2016 | |||||||
| Options to purchase common shares | 2,315 | 2,427 | ||||||
| Warrants to purchase common shares | 1,362 | 1,383 | ||||||
| 4. | Stock options |
The Compensation Committee of the Board of Directors administers the Company’s stock option plan. The Compensation Committee designates eligible participants to be included under the plan and approves the number of options to be granted from time to time under the plan. On June 24, 2010, at the Company’s annual meeting, shareholders approved an amendment to the Company’s Stock Option Plan (the “Plan Maximum Amendment”). The Plan Maximum Amendment relates to changing the maximum number of common shares issuable under the stock option plan from a fixed number of 6,666 to the number of shares that represents twenty-five percent (25%) of the total number of all issued and outstanding common shares. Based upon the current shares outstanding, a maximum of 4,603 options are authorized for issuance under the plan. The option exercise price for all options issued under the plan is based on the fair value of the underlying shares on the date of grant. All options vest within three years or less and are exercisable for a period of seven years from the date of grant. The stock option plan, as amended, allows the issuance of Canadian and U.S. dollar grants. A summary of the stock option transactions, for both the Canadian and U.S. dollar grants, through the year ended December 31, 2017 is below.
Summary of $CAD Option Activity
| Share Prices Reported in $CAD | Number of Options | Range | Weighted Average | |||||||
| Outstanding and exercisable at December 31, 2015 | 1,323 | $ 1.62 – 2.43 | $ | 2.39 | ||||||
| Exercised | – | – | – | |||||||
| Forfeited or expired | (324 | ) | 2.43 | 2.43 | ||||||
| Outstanding and exercisable at December 31, 2016 | 999 | $ 1.62 – 2.43 | $ | 2.38 | ||||||
| Exercised | (196 | ) | 1.89 – 2.43 | 2.36 | ||||||
| Forfeited or expired | (91 | ) | 1.89 – 2.43 | 2.40 | ||||||
| Outstanding and exercisable at December 31, 2017 | 712 | $ 1.89 – 2.43 | $ | 2.38 | ||||||
| F-11 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Summary of $CAD Option Remaining Life
| Price $CAD | Outstanding and Exercisable at December 31, 2017 | Weighted Average Remaining Life (years) | ||||||||
| $ | 1.62 | 17 | 0.26 | |||||||
| $ | 1.89 | 45 | 0.64 | |||||||
| $ | 2.43 | 650 | 0.64 | |||||||
| Total | 712 | 0.63 | ||||||||
Summary of $USD Option Activity
| Number of Options | Range | Weighted Average | ||||||||
| Outstanding and exercisable at December 31, 2015 | 1,094 | $ 0.45 – 3.60 | $ | 1.77 | ||||||
| Granted | 370 | 2.11 – 2.45 | 2.42 | |||||||
| Exercised | (4 | ) | 1.50 | 1.50 | ||||||
| Forfeited or expired | (32 | ) | 1.89 - 2.69 | 2.65 | ||||||
| Outstanding and exercisable at December 31, 2016 | 1,428 | $ 0.45 – 3.60 | $ | 1.93 | ||||||
| Granted | 341 | 3.10 – 10.10 | 5.27 | |||||||
| Exercised | (163 | ) | 0.60 – 2.79 | 1.22 | ||||||
| Forfeited or expired | (3 | ) | 2.79 | 2.79 | ||||||
| Outstanding and exercisable at December 31, 2017 | 1,603 | $ 0.45 – 10.10 | $ | 2.70 | ||||||
Summary of $USD Option Remaining Life
| Price in US Dollars | Number Outstanding and Exercisable at December 31, 2017 | Remaining Life (years) | ||||||||
| $ | 0.45 | 11 | 1.63 | |||||||
| $ | 0.54 | 19 | 2.38 | |||||||
| $ | 0.60 | 17 | 2.26 | |||||||
| $ | 0.72 | 50 | 2.65 | |||||||
| $ | 0.96 | 10 | 2.60 | |||||||
| $ | 1.05 | 110 | 1.97 | |||||||
| $ | 1.13 | 50 | 4.95 | |||||||
| $ | 1.23 | 8 | 4.86 | |||||||
| $ | 1.50 | 15 | 1.34 | |||||||
| $ | 1.59 | 133 | 3.07 | |||||||
| $ | 1.89 | 8 | 0.63 | |||||||
| $ | 2.11 | 36 | 6.00 | |||||||
| $ | 2.30 | 4 | 4.36 | |||||||
| $ | 2.31 | 275 | 3.32 | |||||||
| $ | 2.35 | 4 | 4.59 | |||||||
| $ | 2.40 | 8 | 2.26 | |||||||
| $ | 2.44 | 49 | 5.44 | |||||||
| $ | 2.45 | 285 | 5.51 | |||||||
| $ | 2.51 | 4 | 4.21 | |||||||
| $ | 2.55 | 4 | 3.85 | |||||||
| $ | 2.69 | 118 | 4.00 | |||||||
| $ | 2.79 | 38 | 3.59 | |||||||
| $ | 2.94 | 3 | 2.38 | |||||||
| $ | 3.10 | 10 | 6.26 | |||||||
| $ | 3.60 | 3 | 3.37 | |||||||
| $ | 3.67 | 40 | 6.38 | |||||||
| $ | 5.10 | 250 | 6.49 | |||||||
| $ | 6.72 | 21 | 6.63 | |||||||
| $ | 10.10 | 20 | 6.88 | |||||||
| Total | 1,603 | 4.43 | ||||||||
| F-12 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Stock compensation expense for the fiscal years ended December 31, 2017 and 2016 was $2,258 and $615 respectively. These amounts have been included in the general and administrative expenses for the respective periods. The weighted average fair value per share of options granted and or vested during the fiscal years ended December 31, 2017 and 2016 was $5.27 and $2.42, respectively. The intrinsic value (being the difference between the share price at December 31, 2017 and exercise price) of stock options exercisable at December 31, 2017 was $15,850. The intrinsic value of options exercised during the fiscal year ended December 31, 2017 was $1,827. The fair value of all options vested during the fiscal year ended December 31, 2017 was $2,195.
The fair values of options granted in fiscal years ended December 31, 2017 and 2016 were estimated on the date the options were granted based on the Black-Scholes option-pricing model, using the following weighted average assumptions for all options with a seven-year expiration:
| Year Ended December 31, 2017 | Year Ended December 31, 2016 | |||
| Expected dividend | 0% | 0% | ||
| Risk-free interest rate | 2.04– 2.33% | 1.27 – 2.25% | ||
| Expected volatility | 158 – 168% | 134 – 137% | ||
| Expected life | 7 years | 7 years |
The Company uses the historical volatility and adjusts for available relevant market information pertaining to the Company’s share price.
Modification of Existing US Dollar Denominated Options
In 2016, the Company modified the terms of certain options granted to executives and directors by extending the expiration date by 1 year. The Company recorded option modification expense of approximately $4 included in general and administrative expense. The expense was calculated using the Black-Scholes valuation method. The following table summarizes the effect of the June 8, 2016 transaction:
| Number of Options | Expiration Date | Risk Free Rate | Exercise Price $USD | Share Price $USD | Expected Life (Years) | Volatility | Expense Recognized $USD | |||||||||||||||||||||
| 6 | 11/18/2018 | 0.93 | % | 1.50 | 2.43 | 3.44 | 152 | % | 1 | |||||||||||||||||||
| 17 | 04/04/2020 | 1.08 | % | 0.60 | 2.43 | 3.82 | 155 | % | 1 | |||||||||||||||||||
| 19 | 05/17/2020 | 1.08 | % | 0.54 | 2.43 | 3.94 | 154 | % | 1 | |||||||||||||||||||
| 9 | 11/20/2020 | 1.08 | % | 1.05 | 2.43 | 4.45 | 147 | % | 1 | |||||||||||||||||||
| 51 | 4 | |||||||||||||||||||||||||||
Modification of Existing Canadian Dollar Denominated Options
In 2016, the Company modified the terms of certain options granted to executives and directors by extending the expiration date by a weighted average amount of 1.45 years. The Company recorded option modification expense of approximately $347 included in general and administrative expense. The expense was calculated using the Black-Scholes valuation method with a June 8, 2016 exchange rate of $CAD/$USD 0.7881. The following table summarizes the effect of the June 8, 2016 transaction:
| Number of Options | Expiration Date | Risk Free Rate | Exercise Price $CAD | Share Price $CAD | Expected Life (Years) | Volatility | Expense Recognized $USD | |||||||||||||||||||||
| 17 | 08/18/2018 | 0.52 | % | 1.89 | 3.10 | 2.19 | 94 | % | 9 | |||||||||||||||||||
| 648 | 08/18/2018 | 0.52 | % | 2.43 | 3.10 | 2.19 | 94 | % | 338 | |||||||||||||||||||
| 665 | 347 | |||||||||||||||||||||||||||
Shareholder rights plan
On June 27, 2017, the Company’s shareholders approved a Shareholder Rights Plan Agreement (the "Rights Plan") for the Company. The Rights Plan is to ensure, to the extent possible, that all shareholders of the Corporation are treated fairly and equally in connection with any take-over bid or other acquisition of control of the Corporation. The Rights Plan is designed to require any potential transaction that will result in a person owning, in the aggregate, 20% or more of the outstanding Common Shares to be structured as a formal take-over bid that satisfies certain minimum requirements relating primarily to the manner in which the bid must be made, the minimum number of days the bid must remain open, and the minimum number of shares that must be acquired under the bid.
| F-13 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Registration of Certain Common Shares, Options and Warrants (S-1, S-3 & S-8)
S-1
On August 11, 2017, the Company filed a Form S-1 with the Securities and Exchange Commission to register the 11,943 common shares, which includes 1,383 common shares issuable upon exercise of warrants. The S-1 filing covers shareholders of common shares and warrants from the following transactions:
| · | The Company’s April 2010 private placement of common shares and warrants to purchase common shares; |
| · | The Company’s November 2013 private placement of common shares and warrants to purchase common shares; |
| · | The Company’s February 2016 private placements of warrants to purchase common shares in lieu of payment for services rendered; |
| · | The Company’s May 2016 private placement of common shares; and |
| · | The Company’s June 2017 private placement of common shares. |
S-3
On October 24, 2017, the Company filed an S-3 registration of common shares, pursuant to which the Company may offer from time to time, common shares having an aggregate offering price of up to $90.0 million. Under the Sales Agreement, the Company may sell shares by any method permitted by law and deemed to be an “at-the-market” offering as defined in Rule 415 promulgated under the Securities Act, as amended, including sales made directly on the Nasdaq Capital Market, on any other existing trading market for our common shares or to or through a market maker. The S-3 registration became effective on November 3, 2016. The Company used the S-3 registration to sell shares during a public offering which closed on December 12, 2017. The Company raised $21.2 million total gross proceeds. The Company, may in the future offer shares for up to an aggregate of the remaining limit of the S-3 (approximately $68.8 million).
S-8
On October 24, 2017, the Company filed an S-8 registering its 3,964 outstanding options. The S-8 registration became effective upon filing and remains in effect.
| 5. | Derivative Liabilities |
The Company's derivative instruments include options to purchase 19 common shares, the exercise prices for which are denominated in a currency other than the Company's functional currency, as follows:
| · | Contractor options to purchase 17 common shares exercisable at CAD$1.62 per whole common share that expire on April 4, 2018; |
| · | Contractor options to purchase 2 common shares exercisable at CAD$2.43 per whole common share that expire on May 18, 2018. |
These options have been recorded at their fair value as a liability at issuance and will continue to be re-measured at fair value as a liability at each subsequent balance sheet date. Any change in value between reporting periods will be recorded as unrealized gain/(loss). These options will continue to be reported as a liability until such time as they are exercised, forfeited or expire. The fair value of these options is estimated using the Black-Scholes option-pricing model.
Options issued to contractors in a foreign currency
During the fiscal years ended December 31, 2011 and 2010, the Company issued 36 and 29 (respectively) options to contractors with a Canadian dollar denominated strike price. Consequently, the Company now has derivatives relating to these options since the strike price is denominated in a currency other than the US dollar functional currency of the Company. While there is an exception to this rule for employees in ASU 2010-13 "Compensation-Stock Compensation (Topic 718): Effect of Denominating the exercise price of a share-based payment award in the currency of the market in which the underlying equity security trades", no such exception exists for contractors. These options will be marked to market until the earlier of their expiry or exercise. All Canadian denominated options issued to contractors fully vest at issuance and expire seven years from date of issuance. The fair value of these options at December 31, 2017 and December 31, 2016 was $167 and $33, respectively. The loss for these options for the year ended December, 2017 was $134. There was a gain on these options for the year ended December, 2016 of $7.
| F-14 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
The following is a summary of Canadian denominated contractor option activity for the year ended December 31, 2017 and 2016.
| Share Prices Reported in $CAD | Number of Options Outstanding and Exercisable | Weighted Average Exercise Price | ||||||
| Outstanding and exercisable at December 31, 2015 | 40 | $ | 1.81 | |||||
| Exercised | – | – | ||||||
| Forfeited or expired | – | – | ||||||
| Outstanding and exercisable at December 31, 2016 | 40 | $ | 1.81 | |||||
| Exercised | (21 | ) | 1.90 | |||||
| Forfeited or expired | – | – | ||||||
| Outstanding and exercisable at December 31, 2017 | 19 | $ | 1.71 | |||||
The following table presents the overall change in derivative liability for the year ended December 31, 2017 and December 31, 2016:
| Derivative Value at December 31, | (Loss)/Gain on Derivative Instrument December 31, | |||||||||||||||
| Derivative Warrants/Options | 2017 | 2016 | 2017 | 2016 | ||||||||||||
| Warrants expiring March 29, 2016 | – | – | – | 41 | ||||||||||||
| Options (various expiration dates) | 167 | 33 | (134 | ) | 7 | |||||||||||
| Total | 167 | 33 | (134 | ) | 48 | |||||||||||
| 6. | Fair Value Measurements |
The Company has adopted ASC 820 Fair Value Measurements and Disclosure Topic of the FASB. This Topic applies to certain assets and liabilities that are being measured and reported on a fair value basis. The Fair Value Measurements Topic defines fair value, establishes a framework for measuring fair value in accordance with US GAAP, and expands disclosure about fair value measurements. This Topic enables the reader of the financial statements to assess the inputs used to develop those measurements by establishing a hierarchy for ranking the quality and reliability of the information used to determine fair values. The Topic requires that financial assets and liabilities carried at fair value be classified and disclosed in one of the following three categories:
Level 1: Quoted market prices in active markets for identical assets or liabilities.
Level 2: Observable market based inputs or unobservable inputs that are corroborated by market data.
Level 3: Unobservable inputs that are not corroborated by market data.
Assets/Liabilities Measured at Fair Value on a Recurring Basis
| Fair Value Measurement at December 31, 2017 | ||||||||||||||||
| Quoted Price in Active Market for Identical Instruments | Significant Other Observable Inputs | Significant Unobservable Inputs | ||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Assets | ||||||||||||||||
| Cash and cash equivalents | $ | 275 | (1) | $ | 27,985 | $ | – | $ | 28,260 | |||||||
| Liabilities | ||||||||||||||||
| Derivative liabilities | – | 167 | 167 | |||||||||||||
The Company's financial instruments include cash and cash equivalents and derivative liabilities. Only cash and cash equivalents and derivative liabilities are carried at their fair value. The derivative liabilities are options issued to contractors in a currency other than the functional currency of the Company. The options use the Black Scholes model with the following assumptions: expected dividend 0%; risk-free interest rate of 0.83%; expected volatility of 79%; and a 0.5 year remaining life. The risk free rate was based on Bank of Canada Bond issues of similar term. Expected volatility was estimated by using historical volatility of weekly close share prices for a period equal to the remaining life of the instrument or for a minimum of six months if the remaining life is less than six months.
| (1) | The Company held $275 in cash, of which $255 was in Canadian funds (translated into U.S. dollars). |
| 7. | Stockholders’ Equity |
Authorized capital stock
The Company’s authorized capital stock consists of an unlimited number of shares of no par common shares.
| F-15 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Equity financings
On June 8, 2017, the Company completed the closing of a non-brokered private placement (the “Offering”) of 1,900,000 common shares for gross proceeds of $7.6 million. Each common share was issued at a price of $4.00.
On December 12, 2017, the Company announced the completion of a underwritten public offering of 2,352,950 common shares at a public offering price of $8.50 per share. In addition, Fennec issued an additional 135,670 common shares in connection with the partial exercise of the underwriters’ over-allotment option. The approximate total gross proceeds from the offering was $21.2 million.
Warrants to Purchase Common Shares
At December 31, 2017, the Company had the following warrants outstanding to purchase common shares priced in U.S. dollars with a weighted average price of $1.56 and a weighted average remaining life of 0.9 years:
| Warrant Description | Common Shares Issuable Upon Exercise of Outstanding Warrants at December 31, 2017 | Exercise Price CAD/USD | Expiration Date | |||||||
| Investor warrants(1) | 1,312 | $ | 1.50 USD | November 22, 2018 | ||||||
| Investor warrants(2) | 50 | $ | 3.00 USD | February 2, 2019 | ||||||
| 1,362 | ||||||||||
| (1) | On November 22, 2013, the Company announced it had completed the closing of a non-brokered private placement of 4,000 units, at a price of $0.40 per unit for net proceeds of $1,600. Each unit consisted of one common share of the Company and one common share purchase warrant. Each warrant entitles the holder thereof to acquire one common share of the Company at a price of $0.50 per share for a period of five years from the date of issuance. As a result of the September 3, 2014 share consolidation, each three (3) warrants now entitle the holder thereof to purchase one common share of the Company at a purchase price of $1.50 per whole share for a period of five years from the issue date. |
| (2) | On February 2, 2016 the company issued 50 warrants to Aranea Partners in lieu of cash for investor services. These warrants are fully vested at December 31, 2016 and are redeemable for $3.00 per common share. The fair value of these warrants is estimated using the Black-Scholes pricing model. |
| 8. | Sale of Asset |
On August 29, 2016, Fennec completed the sale of certain intellectual property, data and other assets related to Eniluracil and Adh-1 technologies and development programs to Elion Oncology, LLC for gross proceeds of $40. The Company retained the rights to revenue share payments of 5% of the gross revenues derived from the sold assets until the last to expire patents forming part of such assets.
| 9. | Commitments and Contingencies |
Oregon Health & Science University Agreement
On February 20, 2013, Fennec entered into a new exclusive license agreement with OHSU for exclusive worldwide license rights to intellectual property directed to thiol-based compounds, including STS and their use in oncology (the "New OHSU Agreement"). OHSU will receive certain milestone payments, royalty on net sales for licensed products and a royalty on any consideration received from sublicensing of the licensed technology.
The term of the New OHSU Agreement expires on the date of the last to expire claim(s) covered in the patents licensed to Fennec, unless earlier terminated as provided in the agreement. STS is currently protected by methods of use patents that the Company exclusively licensed from OHSU that expire in Europe in 2021 and are currently pending in the United States. The New OHSU Agreement is terminable by either Fennec or OHSU in the event of a material breach of the agreement by either party after 45 days prior written notice. Fennec also has the right to terminate the New OHSU Agreement at any time upon 60 days prior written notice and payment of all fees due to OHSU under the New OHSU Agreement.
On May 18, 2015, Fennec negotiated an amendment ("Amendment 1") to the exclusive license agreement with OHSU. Amendment 1 expands the exclusive license agreement signed with OHSU on February 20, 2013 ("OHSU Agreement") to include the use of N-acetylcysteine as a standalone therapy and/or in combination with STS for the prevention of ototoxicity induced by chemotherapeutic agents to treat cancers. Further, Amendment 1 adjusts select milestone payments entered in the OHSU Agreement including but not limited to the royalty rate on net sales for licensed products, royalty rate from sublicensing of the licensed technology and the fee payable upon the regulatory approval of a licensed product.
| F-16 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
The term of Amendment 1 under the OHSU Agreement expires on the date of the last to expire claim(s) covered in the patents licensed to Fennec or 8 years, whichever is later. In the event a licensed product obtains regulatory approval and is covered by the Orphan Drug Designation, the parties will in good faith amend the term of the agreement. STS is currently protected by methods of use patents that the Company exclusively licensed from OHSU that expire in Europe in 2021 and are currently pending in the United States. The New OHSU Agreement is terminable by either Fennec or OHSU in the event of a material breach of the agreement by either party after 45 days prior written notice. Fennec also has the right to terminate the New OHSU Agreement at any time upon 60 days prior written notice and payment of all fees due to OHSU under the New OHSU Agreement.
Executive Severance
In the event of his termination with us other than for cause, we will be obligated to pay Mr. Raykov a one-time severance payment of $275,000. In the event of his termination with us other than for cause, we will be obligated to pay Mr. Andrade a one-time severance payment of $100,000.
| 10. | De-recognition of Statute Barred Payables |
The Company had various payables from obligations which existed before current management took over the Company in 2009. These payables, although previously recorded, could not be substantiated as legitimate payables by management. Approximately $79 worth of these payables became statute barred and were therefore written off in 2016 by reversing the original expense entry. This caused the reversal of approximately $23 of general and administrative expense and approximately $56 of research and development expense in the current year. These amounts are presented net in the general and administrative and research and development figures in financial statements.
| 11. | Income Taxes |
The Company operates in both U.S. and Canadian tax jurisdictions. Its income is subject to varying rates of tax and losses incurred in one jurisdiction cannot be used to offset income taxes payable in another. A reconciliation of the combined Canadian federal and provincial income tax rate with the Company’s effective tax rate is as follows:
| Year Ended December 31, | Year Ended December 31, | |||||||
| 2017 | 2016 | |||||||
| Domestic (loss)/gain | $ | (5,277 | ) | $ | (1,771 | ) | ||
| Foreign loss | (1,769 | ) | (1,018 | ) | ||||
| Loss before income taxes | (7,046 | ) | (2,789 | ) | ||||
| Expected statutory rate (recovery) | 26.50 | % | 26.50 | % | ||||
| Expected provision for (recovery of) income tax | (1,867 | ) | (739 | ) | ||||
| Permanent differences | 636 | 156 | ||||||
| Change in valuation allowance | 328 | 583 | ||||||
| Effect of foreign exchange rate differences | – | – | ||||||
| Effect of change in future enacted tax rates | 843 | – | ||||||
| Tax credits and other adjustments | (3 | ) | 1 | |||||
| Effect of tax rate changes and other | 63 | (1 | ) | |||||
| Provision for income taxes | $ | – | $ | – | ||||
The Canadian statutory come tax rate of 26.0 percent is comprised of federal income tax at approximately 15.0 percent and provincial income tax at approximately 11.0 percent.
The primary temporary differences which gave rise to future income taxes (recovery) at December 31, 2017 and December 31, 2016:
| December 31, 2017 | December 31, 2016 | |||||||
| Future tax assets: | ||||||||
| SR&ED expenditures | 2,195 | 2,195 | ||||||
| Income tax loss carryforwards | 19,431 | 19,098 | ||||||
| Non-refundable investment tax credits | 1,263 | 1,263 | ||||||
| Share issue costs | – | 4 | ||||||
| Accrued expenses | – | – | ||||||
| Fixed and intangible assets | 1,031 | 1,032 | ||||||
| Harmonization credit | – | – | ||||||
| 23,920 | 23,592 | |||||||
| Less: valuation allowance | (23,920 | ) | (23,592 | ) | ||||
| Net future tax assets | $ | – | $ | – | ||||
| F-17 |
Fennec Pharmaceuticals Inc.
Notes to the Consolidated Financial Statements
(U.S. dollars and shares in thousands, except per share information)
Tax Cuts and Jobs Act
On December 22, 2017, the United States government enacted comprehensive tax legislation commonly referred to as the Tax Cuts and jobs Act (the “Tax Act”). The Tax Act reduces the corporate tax rate to 21%, effective January 1, 2018. The Securities and Exchange Commission issued Staff Accounting Bulletin No. 118 (“SAB 118”) on December 23, 2017. SAB 118 provides a one-year measurement period from a registrant’s reporting period that includes the United States Tax Act’s enactment date to allow the registrant sufficient time to obtain, prepare and analyze information to complete the accounting required under ASC 740. The ultimate impact of the Tax Act on our reported results may differ from the estimates provided herein, possibly material, due to, among other things, changes in interpretations and assumptions we have made, guidance that may be issued, and other actions we may take as a result of the Tax Act different from presently contemplated.
There are no current income taxes owed, nor are any income taxes expected to be owed in the near term. At December 31, 2017 the Company has unclaimed Scientific Research and Experimental Development ("SR&ED") expenditures, income tax loss carry-forwards and non-refundable investment tax credits. The unclaimed amounts and their expiry dates are as listed below:
| Federal | Province/ State | |||||||
| SR&ED expenditures (no expiry) | $ | 8,283 | $ | - | ||||
| Income tax loss carryforwards (expiry date): | ||||||||
| 2021 | 26 | - | ||||||
| 2022 | 233 | - | ||||||
| 2023 | 133 | - | ||||||
| 2024 | 1,536 | 1,455 | ||||||
| 2025 | 4,795 | 4,768 | ||||||
| 2026 | 20,562 | 20,550 | ||||||
| 2027 | 8,340 | 8,332 | ||||||
| 2028 | 10,840 | 10,823 | ||||||
| 2029 | 8,502 | 8,502 | ||||||
| 2030 | 2,608 | 2,499 | ||||||
| 2031 | 3,378 | 3,224 | ||||||
| 2032 | 3,491 | 3,383 | ||||||
| 2033 | 1,789 | 1,652 | ||||||
| 2034 | 1,812 | 1,606 | ||||||
| 2035 | 1,804 | 1,803 | ||||||
| 2036 | 2,208 | 2,202 | ||||||
| 2037 | 4,631 | 4,511 | ||||||
| Investment tax credits (expiry date): | ||||||||
| 2018 | 10 | |||||||
| 2019 | 8 | |||||||
| 2020 | 96 | |||||||
| 2021 | 55 | |||||||
| 2022 | 548 | |||||||
| 2023 | 399 | |||||||
| 2024 | 178 | |||||||
| 2025 | 199 | |||||||
| 2026 | 86 | |||||||
| 2027 | 90 | |||||||
| 2028 | 50 | |||||||
| F-18 |