Attached files
| file | filename |
|---|---|
| EX-32.1 - Qualigen Therapeutics, Inc. | ex32-1.htm |
| EX-31.2 - Qualigen Therapeutics, Inc. | ex31-2.htm |
| EX-31.1 - Qualigen Therapeutics, Inc. | ex31-1.htm |
| EX-23.1 - Qualigen Therapeutics, Inc. | ex23-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| [X] | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
| [ ] | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 001-37428
RITTER PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 26-3474527 | |
(State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
1880 Century Park East, Suite 1000 Los Angeles, California |
90067 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (310) 203-1000
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.001 per share | NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes [ ]
No [X]
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes [ ]
No [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | [ ] | Accelerated filer | [ ] |
| Non-accelerated filer | [ ] (Do not check if a smaller reporting company) | Smaller reporting company | [X] |
| Emerging growth company | [X] |
If an emerging growth company, indicate by check mark if the Registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
As of June 30, 2017 (the last business day of the registrant’s most recently completed second fiscal quarter), the aggregate market value of the registrant’s common stock held by non-affiliates was approximately $8.1 million based upon the closing price for shares of the registrant’s common stock of $0.55 as reported by the NASDAQ Capital Market on that date. For purposes of this calculation, the registrant has assumed that its directors, executive officers and holders of 5% or more of the outstanding common stock are affiliates.
As of March 19, 2018, there were 49,406,521 shares outstanding of the registrant’s common stock, par value $0.001 per share.
DOCUMENTS INCORPORATED BY REFERENCE
None.
RITTER PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2017
Table of Contents
| 2 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
This Annual Report on Form 10-K (“Annual Report”) contains forward-looking statements that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this Annual Report, including statements regarding our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans, objectives of management and expected market growth are forward-looking statements. The words “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These statements are subject to known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
Some of the factors that we believe could cause actual results to differ from those anticipated or predicted include:
| ● | our ability to obtain additional financing on acceptable terms; | |
| ● | the accuracy of our estimates regarding expenses, future revenues and capital requirements; | |
| ● | the success and timing of our preclinical studies and clinical trials; | |
| ● | our ability to obtain and maintain regulatory approval of RP-G28 and any other product candidates we may develop, and the labeling under any approval we may obtain; | |
| ● | regulatory developments in the United States and other countries; | |
| ● | the performance of third-party manufacturers; | |
| ● | our ability to develop and commercialize RP-G28 and any other product candidates we may develop; | |
| ● | our ability to obtain and maintain intellectual property protection for RP-G28 and any other product candidates we may develop; | |
| ● | the successful development of our sales and marketing capabilities; | |
| ● | the potential markets for RP-G28 and any other product candidates we may develop and our ability to serve those markets; | |
| ● | the rate and degree of market acceptance of RP-G28 and any other product candidates we may develop; | |
| ● | the success of competing drugs that are or become available; and | |
| ● | the loss of key scientific or management personnel. |
Although we believe that we have a reasonable basis for each forward-looking statement contained in this Annual Report, we caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained in this Annual Report.
Any forward-looking statement that we make in this Annual Report speaks only as of the date of this Annual Report, and we undertake no obligation to update such statements to reflect events or circumstances after the date of this Annual Report. You should also read carefully the factors described in the “Risk Factors” section of this Annual Report to better understand the risks and uncertainties inherent in our business and underlying any forward-looking statements.
This Annual Report includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third-parties. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data.
| 3 |
Overview
Ritter Pharmaceuticals, Inc. develops novel therapeutic products that modulate the gut microbiome to treat gastrointestinal diseases. We are advancing gut health research by exploring the gut microbiota and translating the functionality of prebiotic-based therapeutics into applications intended to have a meaningful impact on a patient’s health.
Our first novel microbiome modulator, RP-G28, an orally administered, high purity galacto-oligosaccharide, is currently under development for the treatment of lactose intolerance. RP-G28 is designed to selectively stimulate the growth of lactose-metabolizing bacteria in the colon, thereby effectively adapting the gut microbiome to assist in digesting lactose (the sugar found in milk) that reaches the large intestine. RP-G28 has the potential to become the first drug approved by the Food and Drug Administration (“FDA”) for the treatment of lactose intolerance. RP-G28 has been studied in Phase 2a and Phase 2b clinical trials and is a first-in-class compound.
On March 28, 2017, we announced top-line results from our Phase 2b clinical trial of RP-G28 for the treatment of lactose intolerance. The Phase 2b trial was a double-blind, placebo-controlled, three-arm, multi-center study evaluating safety, efficacy and tolerability of two dosing regimens of RP-G28 in patients with lactose intolerance. Enrollment was initiated in March 2016 and the last patient completed dosing in October 2016. The study aimed to evaluate a patient’s ability to consume dairy foods post-treatment with improved tolerance and reduced digestive symptoms. A total of 368 subjects were randomized in the trial with 18 clinical sites participating throughout the United States. Patients underwent a screening period and a 30-day treatment period, followed by a 30-day post-treatment “real world” observation of milk and dairy product consumption period.
A subset of subjects enrolled into a 12-month extension study to evaluate long-term durability of treatment. The extension study also evaluated each participant’s microbiome, expanding knowledge of the effects that RP-G28 may have on adapting the gut microbiota in a beneficial manner. We completed this study in the fourth quarter of 2017.
We held a Type C meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products in March 2017, prior to the unblinding of our Phase 2b data, to discuss our development plans and Phase 2b clinical trial. The focus of the meeting was to obtain the FDA’s feedback on our Phase 2b clinical trial, including our statistical analysis plan (“SAP”), prior to unblinding any data.
We held an End-of-Phase 2 meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products in August 2017. The purpose of the meeting was to obtain the FDA’s feedback on our Phase 3 program. We reached general consensus with the FDA on certain elements of our current Phase 3 program and have received clear guidance and recommendations on many necessary components of our Phase 3 program; including the clinical, non-clinical, and chemistry, manufacturing and controls (CMC) requirements needed to support an NDA submission.
We have incorporated much of this guidance into our Phase 3 program. Our current Phase 3 clinical program will consist of two confirmatory clinical trials of similar trial design as our Phase 2b clinical trial and will include additional components that may allow for claims for durability of effect. These additional trials may be run in parallel. We anticipate that the first Phase 3 clinical trial will begin in the second quarter of 2018.
The Gut Microbiome
The human gut is a relatively under-explored ecosystem but provides a great opportunity for using dietary intervention strategies to reduce the impact of gastrointestinal disease. The human body carries about 100 trillion microorganisms in the intestines, which is 10 times greater than the number of cells in the human body. This microbial population is responsible for a number of beneficial activities such as fermentation, strengthening the immune system, preventing growth of pathogenic bacteria, providing nutrients, and providing hormones. The increasing knowledge of how these microbial populations impact human health provides opportunities for novel therapies to treat an assortment of diseases such as neurological disease, cardiovascular disease, obesity, irritable bowel syndrome, inflammatory bowel disease, colon cancer, allergies, autism and depression.
Lactose Intolerance
Lactose intolerance is a common condition attributed to the absence or insufficient levels of the enzyme lactase, which is needed to properly digest lactose, a complex sugar found in milk and milk-containing foods.
| 4 |
Studies have suggested that lactose intolerance is a widespread condition affecting over one billion people worldwide and over 40 million people in the United States (or 15% of the U.S. population), with an estimated nine million of those individuals demonstrating moderate to severe symptoms.
Current annual spending on over-the-counter lactose intolerance aids in the United States has been estimated at approximately $2.45 billion. However, these options are limited and there is no long-term treatment available.
Unlike many common gastrointestinal conditions, such as irritable bowel syndrome, inflammatory bowel diseases, gastroesophageal reflux disease, or dyspepsia (among many others), lactose intolerance symptoms can be completely abated by avoiding dietary lactose. In this regard, lactose intolerance is an avoidance condition, similar to celiac sprue, food intolerances, or various environmental allergies. However, dairy avoidance may lead to inadequate calcium and vitamin D intake, which can predispose individuals to decreased bone accrual, osteoporosis, hypertension, rickets, osteomalacia, and possibly certain cancers. Although supplements and calcium-rich foods are available, several studies have shown that lactose intolerance patients had an average calcium intake of only 300-388 mg/day, significantly less than the 1000-1200 mg/day adult dietary recommended levels. The 2010 National Institutes of Health conference on lactose intolerance highlighted the long-term consequences of dairy avoidance demonstrating both the importance of treating the condition and the need to find improved solutions for patients.
Diagnosis
Lactose intolerance is often diagnosed by evaluating an individual’s clinical history, which reveals a relationship between lactose ingestion and onset of symptoms. Hydrogen breath tests may also be utilized to diagnose lactose malabsorption and a milk challenge may be used to differentiate between lactose malabsorption and lactose intolerance. Further tests can be conducted to rule out other digestive diseases or conditions, including: stool examination to document the presence of a parasite, blood tests to determine the presence of celiac disease, and intestinal biopsies to determine mucosal problems leading to malabsorption, such as inflammatory bowel disease or ulcerative colitis.
Health Consequences
Substantial evidence indicates that lactose intolerance is a major factor in limiting calcium intake in the diet of individuals who are lactose intolerant. Studies suggest that these individuals avoid milk and dairy products, resulting in an inadequate intake of calcium and significant nutritional and health risks.
At the 2010 National Institute of Health (“NIH”) Consensus Development Conference: Lactose Intolerance and Health, the NIH highlighted numerous health risks tied to lactose intolerance such as: osteoporosis; hypertension; and low bone density. There is substantial evidence indicating that lactose intolerance is a major factor in limiting calcium and nutrient intake in the diet of people who are lactose intolerant. Adequate calcium intake is essential to reducing the risks of osteoporosis and hypertension. In addition, chronic calcium depletion has been linked to increased arterial blood pressure, thereby establishing a relationship between hypertension and low calcium intake. Moreover, there is evidence of a correlation between calcium intake and both colon and breast cancer.
Decreased Calcium Intake Increases the Risk for Hypertension
Numerous published reports show that chronic calcium depletion may lead to increased arterial blood pressure. Many additional papers have corroborated this relationship between hypertension and a low calcium intake.
A growing body of evidence indicates that a nutritionally sound diet rich in fruits, vegetables and a generous component of low-fat dairy foods (sometimes referred to as the DASH diet) is optimal for reducing the risk of hypertension. Several reports have confirmed this finding in middle-aged and elderly women. Further, it appears that the DASH diet with generous low-fat dairy is associated with low prevalence of metabolic syndrome. Studies have suggested that the levels of dairy foods (three to four servings per day) required to achieve these effects are well above current U.S. averages and even further above those of lactose intolerant individuals who are avoiding dairy due to symptoms.
Our History
We were formed as a Nevada limited liability company on March 29, 2004 under the name Ritter Natural Sciences, LLC. Our first prototype, Lactagen™, was an alternative lactose intolerance treatment method. In 2004, clinical testing was conducted, which included a 61-subject double-blind placebo controlled clinical trial. The results were published in the Federation of American Societies for Experimental Biology in May 2005 and demonstrated Lactagen™ to be an effective and safe product for reducing symptoms for nearly 80% of the clinical participants who were on Lactagen™.
| 5 |
In 2008, we expanded our focus by developing a prescription drug development program. We initiated the program by developing RP-G28, a second generation edition of Lactagen™. We believe that RP-G28 enables us to state stronger claims, garner more medical community support and reach a wider market in the effort to treat lactose intolerance.
To help fund the development of RP-G28, we were awarded a grant from the United States government’s Health Care Bill program, the Qualifying Therapeutic Discovery Project, in 2008. The grant program provides support for innovative projects that are determined by the U.S. Department of Health and Human Services to have reasonable potential to result in new therapies that treat areas of unmet medical need and/or prevent, detect or treat chronic or acute diseases and conditions.
On September 16, 2008, we converted into a Delaware corporation under the name Ritter Pharmaceuticals, Inc.
We completed our initial public offering in June 2015. Our common stock is traded on the Nasdaq Capital Market under the trading symbol “RTTR”.
Our Leading Product Candidate — RP-G28
Overview
RP-G28 is a novel highly purified GOS, which is synthesized enzymatically. The product is being developed for the treatment of lactose intolerance. The therapeutic is taken orally (a powder solution mixed in water) for 30 consecutive days. The proposed mechanism of action of RP-G28 is to increase the intestinal growth and colonization of bacteria that can metabolize lactose to compensate for a patient’s intrinsic inability to digest lactose. Once colonization of bacteria has occurred, it is hypothesized that patients will continue to tolerate lactose as long as they maintain their microflora balance. RP-G28 has the potential to become the first FDA-approved drug for the reduction of symptoms associated with lactose intolerance.
Galacto-oligosaccharides (GOS)
RP-G28 is a >95% purified GOS product derived from a commercially available GOS food ingredient, which is designated as generally recognized as safe (GRAS) by the FDA. GOS refers to a group of compounds containing β-linkages of 1 to 6 galactose units with a single glucose on the terminal end and are found at low levels in human milk. GOS is purified to a pharmaceutical grade by minimizing residual glucose, lactose, galactose and other impurities. Further processing includes ultra-filtration, nano-filtration, decolorization, deionization, and concentration to yield GOS 95 syrup, which is the starting material for RP-G28.
GOS products resist hydrolysis by salivary and intestinal enzymes because of the configuration of their glycosidic bonds and reach the colon virtually intact. The undigested GOS enhances the growth of beneficial, lactose metabolizing, colonic bacteria that already exist in the subject’s digestive track, including multiple species and strains of bifidobacteria and lactobacilli. Once colonies of these bacteria have increased, continued lactose exposure should maintain tolerability of lactose without further exposure to RP-G28.
While formal nonclinical studies evaluating the safety of RP-G28 have not been performed, other commercially available GOS products have been evaluated in acute and repeat-dose general toxicology studies, reproductive toxicology studies, juvenile toxicology studies, genetic toxicology studies, and in long-term safety studies.
Clinical studies of GOS products were reviewed as part of the safety evaluation to support the Investigational New Drug Application (“IND”) for RP-G28. These include studies in adults (including pregnant women and geriatrics), children, infants and newborns (preterm and full term). The safety of GOS products in humans has been evaluated in 1316 adults at doses of 2.5 to 20 g/day for up to 12 months, and in 1125 children > 1 year of age at doses of 2.0 to 12 g/day for up to 1 year. Overall, no safety concerns attributable to the consumption of GOS were reported. Where side effects were observed, they were typically mild and limited to increased flatulence, abdominal discomfort, and changes in stool consistency and frequency; however, effects were not consistently observed in all studies. Similar observations of increased flatulence have been reported following the consumption fructo-oligosaccharies (FOS) (15 g/day) over a 7-day period (Alles, 1996), and this symptom represents a localized effect that is expected in association with the consumption of indigestible fiber in large quantities. There were no reports of events in other System Organ Class (SOC) suggestive of systemic toxicity.
The significance of a higher purity GOS, namely RP-G28, was highlighted in a 2010 study by Klaenhammer. The in vitro study concluded that RP-G28 promoted growth of lactobacilli and bifidobacteria, but did not promote multiple strains of E. coli. In contrast, lower purity GOS stimulated both bifidobacteria as well as the strains of E. coli evaluated. (As seen below in Figure 1, NCK 430 (e. coli) grew in the presence of low purity GOS (GOS 2). Alternatively, the higher purity GOS (RP-G28/GOS 1) did not promote the growth of E. coli.).
| 6 |
Figure 1
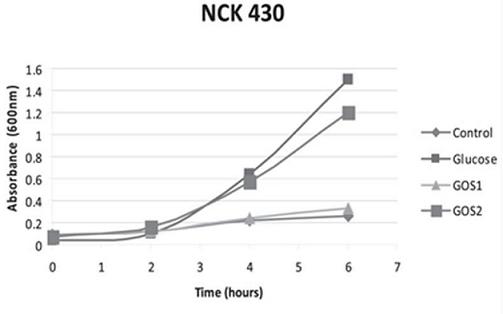
Mechanism of Action
RP-G28 is understood to resist hydrolysis by salivary and intestinal enzymes due to the configuration of their glycosidic bonds and consequently reach the colon virtually intact. The product is then broken down intracellularly by galactosidases, and eventually β-galactosidase hydrolyzes the terminal lactose. This leads to selective alterations in the composition and activity of the microbiome in which RP-G28 enhances the growth of lactose-metabolizing bacteria, including species of Bifidobacteria and Lactobacilli (30). In our Phase 2a Clinical Trial (G28-001), shifts in the fecal microbiome in 82% of participants on treatment and increases in relative abundance of both Bifidobacteria and Lactobacilli were reported. RP-G28 had a bifidogenic effect in 90% of responders, which included species Bifidobacterium longum, Bifidobacterium adolenscentis, Bifidobacterium catenulatum, Bifidobacterium breve, and Bifidobacterium dentium (30). The understood mechanism of action is that by increasing lactose-metabolizing bacteria, less undigested lactose is fermented, and thus reduces gas production and related LI symptoms. Data correlating bacterial taxa and symptom metadata support this proposed hypothesis. In the G28-001 study, microbiome changes correlated with clinical outcomes of improved lactose tolerance in which an increase in Bifidobacterium was associated with decreased pain and cramping outcomes.
Our Market Opportunity
Unmet Medical Needs
Lactose intolerance is a challenging condition to manage. According to a market research study conducted by Objective Insights in April 2012, approximately 60% of lactose intolerant sufferers reported experiencing symptoms daily, or bi-weekly. Not only can symptoms be painful and embarrassing, they can also dramatically affect one’s quality of life, social activities, and health. Currently there are few reliable, or effective, treatments available that provide consistent or satisfactory relief.
Currently, there is no approved prescription treatment for lactose intolerance. Most persons with lactose intolerance avoid ingestion of milk and dairy products while others substitute non-lactose-containing foods in their diet. However, complete avoidance of lactose-containing foods is difficult to achieve (especially for those with moderate to severe symptoms) and can lead to significant long-term morbidity (i.e., dietary deficiencies of calcium and vitamin D).
| 7 |
Treatment Options
Doctors generally recommend the following treatments for the management of lactose intolerance: (1) dairy avoidance; (2) lactase supplements; (3) probiotics/dietary supplements; and (4) dairy substitutes/lactose free products. Despite educating their patients on all viable treatment options, physicians tend to advise their patients to refrain from consuming any dairy products whatsoever. However, in a 2008 survey conducted by Engage Health, 47% of lactose intolerance sufferers reported that this method was not effective (largely due to hidden dairy products in ingredients), and only 30% of lactose intolerance sufferers reported lactase supplements as being effective in managing their lactose intolerance. Further, while probiotics/dietary supplements have been demonstrated to aid and support one’s digestive system, helping break down general foods consumed, they don’t directly help with lactose intolerance. The 2008 survey by Engage Health suggests that the majority of lactose intolerance patients are dissatisfied with current treatment options.
Patients Unsatisfied with Current Management Options

Growing Awareness
Lactose intolerance is a condition that continues to expand as society advances and evolves. It has been estimated that gastroenterologists see approximately 15 new patients with lactose intolerance each month. Education and awareness have increased, and the American diet has greatly changed over the past decade to include more dairy-based goods. As the populace is growing older, the prevalence of lactose intolerance is increasing because more people tend to develop lactose intolerance later in life. Increased education and diagnosis is making more people aware of their allergies and digestive conditions. Physicians may compound the growth of lactose intolerance prevalence and its associated disorders by recommending individuals to avoid dairy products, a practice which in and of itself may increase severity of the intolerance.
| 8 |
Our Competitive Strengths
Market Opportunity
RP-G28 has the potential to become the first approved drug in the United States and Europe for the treatment of lactose intolerance.
Renowned Scientific Team and Management Team
Our leadership team has extensive biotechnology/pharmaceutical expertise in discovering, developing, licensing and commercializing therapeutic products. We have attracted a scientific team comprised of innovative researchers who are renowned in their knowledge and understanding of the host-microbiome in the field of lactose intolerance and gastroenterology.
Patent Portfolio
We have issued patents in the United States, in select countries in Europe (Germany, the United Kingdom, France, Spain, the Netherlands, Spain), and in other jurisdictions, directed to pharmaceutical compositions, methods of making such compositions, and methods of using such compositions for the treatment of lactose intolerance and certain of its symptoms. Additional worldwide patent applications are pending. The patent applications include claims covering pharmaceutical compositions, methods of making, methods of use, formulations and packaging.
In addition, in July 2015 we acquired the rights, title and interest to certain patents and related patent applications with claims covering a process for producing ultra-high purity galacto-oligosaccharide active pharmaceutical ingredients, including RP-G28 from our supplier. See “Manufacturing” for additional details regarding the second amendment to the exclusive supply agreement and our exercise of the exclusive option.
See “Intellectual Property” for additional information regarding our patent portfolio.
Our Growth Strategy
In order to achieve our objective of developing safe and effective applications to treat conditions associated with microbiome disfunctions, our near-term and long-term strategies include the following:
| ● | proceed into Phase 3 clinical trials of RP-G28 for the treatment of lactose intolerance; | |
| ● | complete remaining Phase 3 activities needed for an NDA; | |
| ● | develop and commercialize RP-G28 either by ourselves or in collaboration with others throughout the world; | |
| ● | explore the use of RP-G28 for additional potential therapeutic indications and orphan indications; | |
| ● | establish ourselves as a leader in developing therapeutics that modulates the human gut microbiome; | |
| ● | continue to develop a robust and defensible patent portfolio, including those we own and those we plan to in-license in the future; and | |
| ● | continue to optimize our product development and manufacturing capabilities both internally and externally through outside manufacturers. |
Clinical and Regulatory
IND Application/Phase 1
The IND for RP-G28 was activated initially to support a Phase 2a safety, tolerability and efficacy study in lactose intolerant patients. Standard Phase 1 single and repeat dose safety and tolerability studies in healthy volunteers were not needed because other GOS products that contain similar GOS constituents are generally regarded as safe (GRAS) and therefore supported the safety of RP-G28 in humans.
| 9 |
In 2018, a Phase 1 study will be conducted to understand the potential for systemic absorption of RP-G28 and any impact the presence of food may have on the pharmacokinetic profile of RP-G28. A Phase 1 QT/QTc study may be needed if there is measurable systemic exposure of RP-G28. Phase 1 systemic drug-drug-interaction (DDI) studies may also be needed if there is measurable systemic exposure of RP-G28, or if DDI potential within the gut is suggested by the results of in vitro DDI studies.
Phase 2a Study
We completed a double-blinded, randomized, multi-center, placebo-controlled Phase 2a clinical trial to validate the efficacy, safety and tolerance of RP-G28 compared to a placebo. We evaluated RP-G28 in 62 patients with lactose intolerance over a treatment period of 35 consecutive days. Post-treatment, subjects reintroduced dairy into their diets and were followed for an additional 30 days to evaluate lactose digestion, as measured by hydrogen production and symptom improvements. In order to confirm lactose intolerance and study participation, subjects underwent a 25-gram lactose challenge in the clinic. Lactose intolerance symptoms and hydrogen production via hydrogen breath test were assessed for six hours post-lactose dose. Eligible subjects were required to demonstrate a minimum symptom score and a “positive” hydrogen breath test in order to be eligible for randomization. A “positive” breath test was defined as a hydrogen gas elevation of 20 parts per million (ppm) at two time-points within the six hours following a lactose-loading dose. The primary endpoints included tracking patients’ gastrointestinal symptoms via a patient-reported symptom assessment instrument (a Likert Scale, measuring individual symptoms of flatulence, bloating, cramping, abdominal pain and diarrhea, on a scale of 0 (none) to 10 (worst)) at baseline, day 36 and day 66; as well as the measurement of hydrogen gas levels in their breath following a 25-gram lactose challenge.
Positive trends were seen when the entire per protocol study population was analyzed, including some statistically significant subgroup analysis, suggesting a therapeutically positive effect. Although there were few primary and secondary efficacy endpoints with statistically significant results, the combined data suggest that RP-G28 is exerting a positive therapeutic effect
Key findings of the Phase 2a clinical trial included:
| ● | RP-G28 was well tolerated with no significant study-drug related adverse effects. The benign adverse event safety profile of RP-G28 with dose levels up to 15 gm/day observed in this study is consistent with the known safety of GOS products administered up to 20 gm/day reported in literature. | |
| ● | Subjects in the RP-G28 group reported a reduction in total symptoms after treatment. Reported symptom improvement continued 30 days post-treatment. Improvement in symptoms was assessed in the study using several different measures, including a pain Likert scale and a patient global assessment. Subjects receiving RP-G28 had greater improvement in most of their symptoms (cramps, bloating and gas) following lactose challenge compared to placebo, but the differences were not statistically significant given the small cohort size. However, a clinically meaningful reduction in abdominal pain was seen in subjects receiving RP-G28 compared to placebo. | |
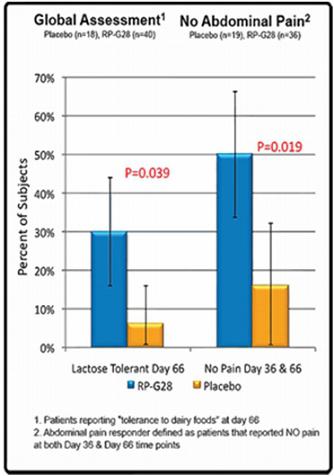
| ● | An analysis of “responders” for abdominal pain (defined as subjects who reported a score of zero in abdominal pain severity following a lactose challenge at Day 36/Hour 6 and Day 66/Hour 6) was performed. In the 55 subjects who noted abdominal pain following the baseline (Day 0) lactose challenge, 50% of RP-G28-treated subjects reported no abdominal pain compared to 17% of the placebo-treated subjects. This difference was statistically significant (p = 0.0190). See Figure 2 below. | |
| ● | An analysis of “responders” for abdominal pain (defined as subjects who reported a greater than 50% decrease in abdominal pain severity following lactose challenge between Day 0/Hour 6 and Day 36/Hour 6) was performed. In the 55 subjects who noted abdominal pain following the baseline (Day 0) lactose challenge, 72.2% of RP-G28-treated subjects reported a >50% reduction in abdominal pain severity compared to 42.1% of the placebo-treated subjects. This difference was also statistically significant (p=0.0288). | |
| ● | Six times as many patients in the treatment group versus the placebo group described themselves as lactose tolerant in the global assessment and did not report symptoms associated with lactose intolerance on Day 66. After completion of study treatment at Day 36, subjects were encouraged to re-introduce dairy foods into their diet. Thirty days later (Day 66), subjects were asked to provide an assessment of their symptom status, i.e., whether they considered themselves still lactose intolerant compared with subjects receiving placebo (Yes/No). As seen below in Figure 2 below, in the 58 subjects providing responses, a significantly larger percentage of subjects receiving RP-G28 (30%) considered themselves no longer lactose intolerant compared with subjects receiving placebo (5.6%); this result was statistically significant (p=0.0389). | |
| ● | Shifts in the fecal microbiome in 82% of participants on treatment (31) and increases in relative abundance of both Bifidobacteria and Lactobacilli were reported. Pre-treatment, three distinct clusters were identified, while post-treatment (Day 66) two distinct clusters were identified, demonstrating a clear shift in certain species represented before and after treatment. RP-G28 had a bifidogenic effect in 90% of responders, which included species Bifidobacterium longum, Bifidobacterium adolenscentis, Bifidobacterium catenulatum, Bifidobacterium breve, and Bifidobacterium dentium (30). |
| 10 |
Figure 2
| 11 |
The clinical results of our Phase 2a study were published in Nutrition Journal in a manuscript entitled “Improving lactose digestion and symptoms of lactose intolerance with a novel galacto-oligosaccharide (RP-G28): a randomized, double-blind clinical trial.” The microbiome results were published in the Proceedings of the National Academy of Science in a manuscript entitled “Impact of short-chain galacto-oligosaccharides on the gut microbiome of lactose-intolerant individuals.”
Type C Meeting with the FDA
We held a Type C meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products on February 2013. The purpose of the meeting was to obtain the FDA’s feedback on the planned Phase 2 program and Phase 3 programs, inform the FDA of our ongoing development plans, gain feedback on relevant clinical trial design and end points related to patient meaningful benefits, and to inform the FDA of the status of our product characterization.
Phase 2b Clinical Trial
Enrollment in our Phase 2b clinical trial of RP-G28 was initiated in March 2016 and completed in August 2016. The final patient completed dosing and all monitoring visits in October 2016.
The Phase 2b trial was a multi-center, randomized, double-blind, placebo-controlled, parallel-group trial of 368 subjects designed to determine the efficacy, safety, and tolerability of two dosing regimens of RP-G28 in subjects with moderate to severe lactose intolerance. Two hundred and forty-seven (247) subjects received RP-G28 while 121 subjects received placebo. Twenty-four (24) subjects were discontinued prematurely from the study and 344 (91.2%) completed the study.
The trial assessed patients with lactose intolerance symptoms as measured on a Likert scale after a lactose challenge. Entry criteria in the Phase 2b trial included a hydrogen breath test to validate lactase deficiency. The Phase 2b trial design included a screening period, a 30-day course treatment period, and a 30-day post-treatment “real world” observation period during which subjects were followed while lactose containing food products were re-introduced into their diets. The study was designed to escalate the dose beyond the 15 gm/day dose level evaluated in the Phase 2a study. Study subjects abstained from lactose containing food products and were then randomized evenly (1:1:1) to receive one of two doses of RP-G28 or placebo for 30 days.
The primary endpoint for the Phase 2b clinical trial was a LI symptom composite score response at day 31. A response was based on change from baseline (Day -7, visit 1) to end of treatment period at day 31 (visit 5), combined average of four maximum symptom scores taken over 0.5, 1, 2, 3, 4, and 5 hours for each symptom (abdominal pain, cramping, bloating, and gas movement) after a lactose challenge test. A response was defined as a 4-point or greater decrease from baseline or a composite score of zero at day 31. The Phase 2b trial further required the collection of fecal samples from patients enrolled to evaluate the baseline and changes to the patient’s microbiome that correlate to symptom reduction and lactose tolerance.
| 12 |
We held a Type C meeting with the FDA in March 2017, to discuss our development plans and Phase 2b clinical trial. The focus of the meeting was to obtain the FDA’s feedback on our Phase 2b clinical trial, including our SAP, prior to unblinding any data.
In order to gather long-term data on subjects exposed to RP-G28, we also offered enrollment in an observational 12-month extension study, G28-003XA, to subjects who completed the Phase 2b protocol. As RP-G28 is expected to provide extended relief from lactose intolerance symptoms beyond the initial 30-day treatment phase, this extension study for the Phase 2b program will assess the long-term treatment effect. The study is also evaluating each participant’s microbiome, expanding our knowledge of the effects that RP-G28 may have on adapting the gut microbiota in a beneficial manner. We completed this study in the fourth quarter of 2017. We intend for the results from this study to support durability of treatment and guide the need to evaluate an additional 30-day course of treatment in subjects who experience the return of lactose intolerance symptoms after an initial course of RP-G28.
Topline results of the Phase 2b clinical trial were announced in March 2017. Due to inconsistent data results from one study site, the data from this site was excluded from the primary analysis population (Efficacy Subset mITT). After excluding the data from the one anomalous study site, results showed a clinically meaningful benefit to subjects in the reduction of lactose intolerance symptoms across a variety of outcome measures. The majority of analyses showed positive outcome measures and the robustness of the data point to a clear drug effect. Treatment patients not only reported meaningful reduced symptoms, but also 30-days after taking the treatment, patients reported adequate relief from lactose intolerance symptoms and satisfaction with the results of the treatment, with RP-G28 preventing or treating their lactose intolerance symptoms. Greater milk and dairy product consumption was also reported by patients.
Because the efficacy data from one study site was found to be significantly different from that of the other study sites, the data from this site was excluded from the primary analysis population (Efficacy Subset mITT. n=296). It was decided that, in addition to the efficacy analysis for the mITT Population, the Efficacy Subset mITT population would be used to perform all efficacy analyses.
In the Efficacy Subset mITT Analysis group, the primary endpoint met statistical significance, (39.7% of the pooled dosing group compared to 25.8% of the placebo group responded (p=0.0159)). Because the primary analysis was statistically significant, the primary endpoint comparison between the high dose group and the placebo group was then tested and also met statistical significance (38.1% of the high dose group, compared to 25.8% of the placebo group responded (p=0.0294)). The comparison between the low dose group and the placebo group further met statistical significance (p=0.0434).
In the entire study population (mITT population), including patients from the excluded study site, taking at least one dose of drug (n=368), the comparison between the pooled treatment groups and the placebo group narrowly missed statistical significance (p=0.0618), (40.1% of the pooled treatment group responded compared to 31.4% of the placebo group). Both low dose and high dose group arms demonstrated a higher proportion of responders than the placebo group.
In the Efficacy Subset Per-protocol population (Efficacy Subset PP), significant and meaningful symptom improvement was consistently seen across key individual lactose intolerance symptoms by patients reporting a ≥4-point improvement from baseline (proportion of subjects on treatment that reported improvement in severity of each symptom). Of the treatment patients, 56.1% reported significant improvement in abdominal pain compared to 45.7% in the placebo group (p=0.1046). Of the treatment patients, 54.5% reported statistically significant improvement in cramping compared to 40.2% in the placebo group (p=0.0257). Of the treatment patients, 55% reported statistically significant improvement in bloating compared to 41.3% in the placebo group (p=0.0282). Finally, 44.4% of treatment patients reported significant improvement in gas movement compared to 32.6% in the placebo group (p=0.0599). See Figure 4 below.
| 13 |
Figure 4

In a more stringent assessment, many patients reported that they experienced complete elimination of lactose intolerance symptoms, scoring a 0 out of 10 on a Likert pain scale post-treatment (Efficacy Subset PP). Of the treatment patients, 37.0% reported complete elimination of abdominal pain compared to 21.7% in the placebo group (p=0.0144). Of the treatment patients, 34.9% reported complete elimination of cramping compared to 16.3% in the placebo group (p=0.0020). Of the treatment patients, 29.6% reported complete elimination of bloating compared to 16.3% in the placebo group (p=0.015). Of the treatment patients, 16.4% reported complete elimination of gas movement compared to 2.2% in the placebo group (p=0.0005). Symptoms of abdominal pain, cramping, bloating and gas movement were then combined into a composite endpoint representing the key symptoms of lactose intolerance. Of the treatment patients, 13% experienced complete elimination of lactose intolerance symptoms compared to 2% in the placebo group (p=0.004). See Figure 5 below.
Figure 5

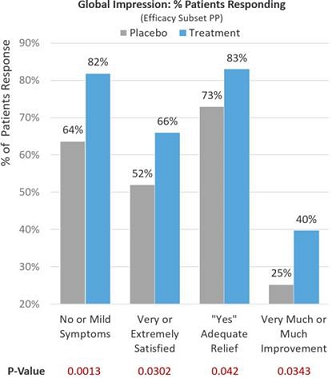
Observing global patient-reported assessments (Efficacy Subset PP) on multiple aspects of their symptom severity and treatment benefit experience 30 days after treatment and adding dairy and milk products back into their diets, 81.9% of treatment patients reported no or mild lactose intolerance symptoms compared to 63.7% in the placebo group (p=0.0013). Of the treatment patients, 66.3% reported being very or extremely satisfied with RP-G28 preventing or treating their lactose intolerance symptoms compared to 51.6% in the placebo group (p=0.0302). Of the treatment patients, 83.2% reported adequate relief from lactose intolerance symptoms compared to 72.5% in the placebo group (p=0.042). Of the treatment patients, 39.7% reported much or very much improvement in their lactose intolerance symptoms compared to 25.3% in the placebo group (p=0.0343). See Figure 6 below.
| 14 |
Figure 6
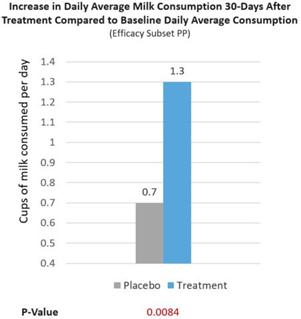
Further, a real-world milk intake assessment was conducted on treatment and placebo group patients (Efficacy Subset PP). At baseline, lactose intolerance patients reported consuming 0.2 cups/d of milk. After RP-G28, treatment patients increased their milk consumption to 1.5 cups/d of milk, consuming 1.3 cups/d more of milk (p=0.0084), 39% more milk consumed per day than placebo patients reported consuming (See Figure 7 below). We believe this is significant because the USDA recommends healthy individuals to consume 1.5 cups/d of milk. Overall, 62% of treatment patients consumed ≥1 cups/d of milk after being treated (p=0.0095). The increase in milk consumption is meaningful for dairy avoiders because it reflects increased lactose tolerance and may lead to more dietary calcium intake post-treatment as milk contains a higher percentage of one’s daily intake of calcium.
Figure 7
No serious adverse events related to treatment were reported and the number of adverse events reported was similar between treatment and placebo groups.
| 15 |
End-of-Phase 2 Meeting with the FDA
We held an End-of-Phase 2 meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products in August 2017. The purpose of the meeting was to obtain the FDA’s feedback on our planned Phase 3 program. We reached general consensus with the FDA on certain elements of our Phase 3 program and have received clear guidance and recommendations on many necessary components of our Phase 3 program; including the clinical, non-clinical, and chemistry, manufacturing and controls (CMC) requirements needed to support an NDA. We have incorporated much of this guidance into our Phase 3 program.
Elements of our Phase 3 program are expected to include the following:
| ● | Trial Design: Will consist of two confirmatory clinical trials of similar trial design and size as our Phase 2b clinical trial and will include additional components that may allow for claims for durability of effect. The trials may be run in parallel. | |
| ● | Protocol design: Will consist of multi-center, randomized, doubled-blind, placebo-controlled, parallel-group trials designed to determine the efficacy, safety and durability of RP-G28 compared to a placebo in subjects with lactose intolerance. The protocol designs include screening to determine lactose intolerance, 30-day course of treatment, and 6-months of post-treatment observation. | |
| ● | Primary endpoint: Will evaluate a patient’s LI symptom composite score (including abdominal pain, cramping, bloating and gas) after a lactose challenge, comparing the mean difference between baseline symptom score to 30-days post-treatment symptom score. | |
| ● | Secondary endpoints: Will evaluate LI signs and symptoms and global assessment outcomes to evaluate and assess a patient’s continued meaningful treatment benefit. |
In preparation for Phase 3, we have had regular communications with the FDA and have received feedback from the FDA on the Phase 3 protocols, the statistical analysis plan (SAP), non-clinical matters, chemistry and controls, as well as other items.
The FDA has provided the following recommendations with respect to our revised Phase 3 protocols, SAP and other items (all of which we intend to implement):
| ● | The FDA has confirmed that two confirmatory pivotal studies are required for a NDA filing. | |
| ● | A primary endpoint was agreed upon. | |
| ● | The FDA has indicated that retreatment must be evaluated in order to provide proper labeling information for clinicians regarding when they should prescribe retreatment, and that we must continue the evaluation of efficacy and safety. | |
| ● | The FDA has indicated that a pharmacokinetic (PK) study, in a fed and fasted state, must be conducted to confirm no to low systemic exposure in order to permit a broad definition of renal classes allowed for inclusion criteria. |
Nonclinical Safety Plans
Given the established safety profile of GOS in humans and the lack of significant safety concerns with RP-G28 administered to subjects in the Phase 2a and Phase 2b clinical trials, it was agreed with the FDA (August 2017 End-of-Phase 2 meeting) that no additional non-clinical safety studies are required to support continued evaluation of RP-G28 in the Phase 3 program. The FDA also agreed that no rat fertility, rat peri-post natal reproductive toxicity, genotoxicity or, importantly, rodent carcinogenicity studies are needed for the NDA submission.
As recommended by the FDA, we will continue to evaluate females of child-bearing potential who are willing to use appropriate contraception throughout the duration of any study. ICH-compliant embryo-fetal development toxicology studies of RP-G28 in the rat and rabbit will be conducted to support the NDA submission. Additional general toxicity studies may also need to be conducted for the NDA submission.
Manufacturing
We do not own or operate manufacturing facilities, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We have an exclusive worldwide agreement (the “Supply Agreement”) to manufacture a higher purity form of GOS (referred to as “Improved GOS”) with Ricerche Sperimentali Montaleor (“RSM”) in connection with our clinical and nonclinical studies we will need to conduct prior to receiving regulatory approval for RPG-28. RSM has also agreed that it will not, except as necessary for RSM to perform its obligations under the Supply Agreement, market or sell Improved GOS, or any galacto-oligosaccharides that are of greater purity to any third party.
| 16 |
Pursuant to the terms of the Supply Agreement, as amended on July 24, 2015, we purchased the exclusive worldwide assignment of all right, title and interest to the Improved GOS (the “Improved GOS IP”) on July 30, 2015 for $800,000. We also issued 100,000 shares of our common stock to RSM pursuant to a stock purchase agreement.
Under the terms of the Supply Agreement, as amended, if we fail to make any future option payment required under the terms of the Supply Agreement, we may be required to return the Improved GOS IP to RSM. The terms of the Supply Agreement, as amended, require us to pay RSM $400,000 within 10 days following FDA approval of a new drug application for the first product owned or controlled by us using Improved GOS as its active pharmaceutical ingredient.
Commercialization
Given our stage of development, we have not yet established a commercial organization or distribution capabilities. RP-G28, if approved, is intended to be prescribed to patients suffering from lactose intolerance. These patients are normally under the care of a gastroenterologist and/or a primary care physician. Our current plan is to evaluate a possible partnership to commercialize RP-G28 for the treatment of lactose intolerance in patients in the United States and Europe if it is approved. We may also build our own commercial infrastructure or utilize contract reimbursement specialists, sales people and medical education specialists, and take other steps to establish the necessary commercial infrastructure at such time as we believe that RP-G28 is approaching marketing approval. Outside of the United States and Europe, subject to obtaining necessary marketing approvals, we will likely seek to commercialize RP-G28 through distribution or other collaboration arrangements for patients suffering from lactose intolerance.
Competition
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Although we know of no drug candidate, other than RP-G28, in advanced clinical trials for treating lactose intolerance, other biopharmaceutical companies may be able to develop compounds or drugs that are able to achieve similar or better results. Our potential competitors include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies and universities and other research institutions. Some of the pharmaceutical and biotechnology companies we expect to compete with include microbiome-based development companies such as Second Genome, Inc., Seres Health, Inc., Enterome SA, Vedanta Biosciences, Inc., and Rebiotix, Inc. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. We will also compete with providers of a wide variety of lactase supplements (the most widely used supplement in the United States being Lactaid®), probiotic/dietary supplements, and lactose-free and dairy-free products. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement.
Intellectual Property
The proprietary nature of, and protection for, our product candidates and our discovery programs, processes and know-how are important to our business. We have sought patent protection in the United States and internationally for uses of RP-G28 and our discovery programs, and any other inventions to which we have rights, where available and when appropriate. Our policy is to pursue, maintain and defend patent rights, whether developed internally or licensed from third parties, and to protect the technology, inventions and improvements that are commercially important to the development of our business. We also rely on trade secrets that may be important to the development of our business. We do not have composition of matter patent protection in the United States for RP-G28, which may result in competitors being able to offer and sell products so long as these competitors do not infringe any other patents that we hold, including patents directed to methods of manufacturing and purified RP-G28 or directed to methods of using RP-G28.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of RP-G28 and any future product candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our product candidates, discovery programs and processes from commercial competition. Furthermore, we cannot be sure that issued patents will not be challenged in court as invalid or in the Patent Office as unpatentable. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors — Risks Relating to Our Intellectual Property.”
| 17 |
Patents and Proprietary Rights Covering Our Drug Candidates
We
strive to protect our product candidates and exclusivity rights, as well as both maintain and fortify our position in the field
of reduction of symptoms associated with lactose intolerance. We believe our intellectual property portfolio consists of early
and broad filings in the area. We have focused on patents and patent applications directed to use of our products in disease treatment.
We have sought and continue to seek the strongest possible intellectual property protection available to us in order to prevent
others from directly competing with us, as well as to exclude competition around our products, their manufacture, and methods
for use of the products in disease treatment. Our intellectual property portfolio directed to RP-G28 contains four issued patents
relating to RP-G28 and its uses. That portfolio also includes at least 15 other related, pending patent applications in the United
States and worldwide. We also own a patent family, including claims generally directed to processes for producing
an improved form of galacto-oligosaccharides (GOS) mixtures (higher purity); this family includes issued patents in United
States (not expiring until 2030), Italy (not expiring until 2029), and China, Germany, and the Netherlands (not expiring
until 2030), as well as applications pending in the United States, Japan, India, and other jurisdictions that, if issued, will
not expire until 2030.
This portfolio includes patents and proprietary rights related to:
| ● | U.S. Patent No. 8,486,668, which has a current expiry date of February 17, 2030, includes claims generally directed to methods for treating lactose intolerance comprising administering, for a predetermined number of days, a high purity galacto-oligosaccharides (GOS) pharmaceutical composition, and wherein the administration leads to a persistent decrease in at least one symptom of lactose intolerance; | |
| ● | U.S. Patent No. 8,492,124, which has a current expiry date of February 17, 2030, includes claims generally directed to methods for treating lactose intolerance comprising administering, for a predetermined number of days, a controlled release pharmaceutical composition that contains galacto-oligosaccharides (GOS), but does not contain a probiotic; | |
| ● | U.S. Patent No. 8,785,160, which has a current expiry date of February 17, 2030, includes claims generally directed to methods for treating lactose intolerance comprising administering a hydrogen breath test, diagnosing lactose intolerance based upon the hydrogen breath test, and administering a high purity galacto-oligosaccharides (GOS) pharmaceutical composition; | |
| ● | U.S. Patent No. 9,200,303, which has a current expiry date of August 6, 2030 (subject to the payment of maintenance fee), includes claims generally directed to the processes for producing ultra-pure galacto-oligosaccharides (GOS) pharmaceutical compositions by utilizing sequential microbiological purifications; | |
| ● | U.S. Patent No. 9,370,532, which has a current expiry date of February 17, 2030, includes claims generally directed to methods for preventing or reducing diarrhea associated with lactose intolerance, and methods for the reduction of severity of diarrhea associated with lactose intolerance, comprising administering a high purity galacto-oligosaccharides (GOS) having 1-10% by weight pentasaccharides and at least a 45% by weight trisaccharides; | |
| ● | U.S. Patent No. 9,579,340, which has a current expiry date of February 17, 2030, includes claims generally directed to an oral dosage form comprising a GOS composition having 95% or more galacto-oligosaccharides (GOS) by weight and less than 5% digestible saccharides by weight, and having 45% by weight trisaccharides; | |
| ● | U.S. Patent No. 9,775,860, which has a current expiry date of February 17, 2030, includes claims generally directed to methods of improving gastrointestinal health, including heartburn, stomach upset, bloating, diarrhea, constipation, or gas by administering a composition having 95% or more GOS by weight and less than 5% digestible saccharides by weight, and having at least 45% by weight trisaccharides; | |
| ● | U.S. Patent No. 9,592,248, which has a current expiry date of February 17, 2030, includes claims generally directed to an oral dosage form having one or more dosing units, each having 0.1 to 10 g of a liquid GOS composition in a gelatin capsule, where the GOS composition has at least about 95% GOS by weight, less than about 5% digestible saccharides by weight, and at least 45% by weight trisaccharides; | |
| ● | U.S. Patent No. 9,808,481, which has a current expiry date of February 17, 2030, includes claims generally directed to a GOS composition having at least 95% by weight GOS and 5% or less by weight digestible saccharides, and having about 5-25% pentasaccharides; |
| 18 |
| ● | United Kingdom Patent No. GB2480042, which has a current expiry date of February 16, 2030, includes claims generally directed to a solid oral unit-dosage form of a high purity galacto-oligosaccharides (GOS); | |
| ● | Canadian Patent No. CA2752800, which has a current expiry date of February 16, 2030 (subject to payment of annuities), includes claims generally directed to the daily use of GOS compositions to increase lactose tolerance or to treat lactose intolerance; | |
| ● | Japanese Patent No. JP6105680, which has a current expiry date of August 6, 2030 (subject to payment of annuities), includes claims generally directed to the production of ultra-pure galacto-oligosaccharides (GOS) pharmaceutical compositions by utilizing sequential microbiological purifications; | |
| ● | European Patent No. EP 2,462,234, validated in six European countries, including Germany, Great Britain, and France, which has a current expiry date of August 6, 2030 (subject to payment of annuities), includes claims generally directed to the processes for producing preparing ultra-pure galacto-oligosaccharides (GOS) pharmaceutical compositions by utilizing sequential microbiological purifications; | |
| ● | Italian Patent No. IT 1,395,068, which has a current expiry date of August 7, 2029 (subject to the payment of annuities), includes claims generally directed to the production of ultra-pure galacto-oligosaccharides (GOS) pharmaceutical compositions by utilizing sequential microbiological purifications; and |
| ● | Chinese Patent No. ZL 201080035013.2, which has a current expiry date of August 6, 2030 (subject to payment of annuities), includes claims generally directed to the production of ultra-pure galacto-oligosaccharides (GOS) pharmaceutical compositions by utilizing sequential microbiological purifications. |
We also are pursuing patent applications. These applications are pending in the United States, Europe, Japan and other jurisdictions, and, if they issue as patents, will not expire until at least 2030, and include claims generally directed to (i) oral dosage forms of a higher purity galacto-ologosaccharides (GOS), (ii) use of galacto-ologosaccharides (GOS) for treating lactose intolerance, and (iii) methods of preventing or reducing certain symptoms of lactose intolerance using galacto-ologosaccharides (GOS) dosage forms. For example, we have paid issue fees (and expect patents to issue in due course) in two United States patent applications: one includes claims directed to an oral dosage forms comprising a prebiotic composition comprising 95% or more galacto-oligosaccharides (GOS) by weight and less than 5% digestible saccharides by weight, in which the GOS comprises at least 45% by weight trisaccharides, and the other includes claims directed to oral dosage forms of GOS, comprising 0.1 to 10 g of a liquid GOS composition encapsulated in a gelatin capsule, in which the GOS composition comprises at least about 95% galacto-oligosaccharides (GOS) by weight, less than about 5% digestible saccharides by weight, and at least 45% by weight trisaccharides.
Trade Secrets
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information. We also seek to preserve the integrity and confidentiality of our data, trade secrets and know-how by maintaining physical security of our premises and physical and electronic security of our information technology systems.
Government Regulation and Product Approval
Governmental authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Our product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the United States and by the European Medicines Agency (the “EMA”) through the Marketing Authorization Application (“MAA”) process before they may be legally marketed in Europe. Our product candidates will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
| 19 |
United States Government Regulation
NDA Approval Processes
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (the “FDCA”) and implemented regulations. Failure to comply with the applicable FDA requirements at any time during the product development process or approval process, or after approval, may subject an applicant to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include:
| ● | refusal to approve pending applications; | |
| ● | withdrawal of an approval; | |
| ● | imposition of a clinical hold; | |
| ● | warning letters; | |
| ● | product seizures and/or condemnation and destruction; | |
| ● | total or partial suspension of production or distribution; or | |
| ● | injunctions, fines, disgorgement, or civil or criminal penalties. |
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of nonclinical laboratory tests, animal studies and formulation studies conducted according to Good Laboratory Practices (“GLPs”) or other applicable regulations; | |
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin; | |
| ● | performance of adequate and well-controlled human clinical trials according to Good Clinical Practices (“GCPs”), to establish the safety and efficacy of the proposed drug for its intended use; | |
| ● | submission to the FDA of a marketing application such as a NDA; | |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Manufacturing Practices (“cGMPs”) to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and | |
| ● | FDA review and approval of the marketing application. |
Once a pharmaceutical candidate is identified for development, it enters the preclinical or nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. A sponsor of an IND must submit the results of the nonclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some nonclinical testing may continue even after the IND is submitted. In addition to including the results of the nonclinical studies, the IND will also include a clinical protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the first phase lends itself to an efficacy determination. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, notifies the sponsor of a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND, and may affect one or more specific studies or all studies conducted under the IND.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCPs. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject selection and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as an amendment to the IND, and progress reports detailing the status of the clinical trials must be submitted to the FDA annually in the IND Annual Report. Sponsors must also report to the FDA, within required timelines, serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigation brochure, or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the drug. An institutional review board (“IRB”) at each institution participating in the clinical trial must review and approve the protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each research subject or the subject’s legal representative, monitor the study until completed and otherwise comply with IRB regulations.
| 20 |
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some products for severe or life-threatening diseases, such as cancer, especially when the product may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. | |
| ● | Phase 2. Clinical trials are performed on a limited patient population intended to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. Although there are no statutory or regulatory definitions for Phase 2a and Phase 2b, Phase 2a is commonly used to describe a Phase 2 clinical trial designed to evaluate efficacy, adverse effects and safety risks and Phase 2b is commonly used to describe a subsequent Phase 2 clinical trial that also evaluates dosage tolerance and optimal dosage. | |
| ● | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product and provide an adequate basis for product labeling. |
Human clinical trials are inherently uncertain and Phase 1, Phase 2 and Phase 3 testing may not be successfully completed. The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to the submission of an IND, at the end of Phase 2 and before an NDA is submitted. Meetings with the FDA may be granted at other times during the development program when requested. For instance, we held a Type C meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products on February 20, 2013. The purpose of the meeting was to obtain the FDA’s feedback on the planned clinical development program and future necessary clinical studies, inform the FDA of our ongoing development plans, gain feedback on relevant clinical trial design and end points related to patient meaningful benefits, and to inform the FDA of the status of our product characterization. Following analysis of the Phase 2a clinical trial, discussions with the FDA during the Type C Meeting in early 2013 about our clinical development plan, and further discussions with our regulatory consultants, we initiated the Phase 2b clinical trial of RPG-28 in March 2016 and completed final enrollment and dosing in October 2016.
FDA meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support the approval of the new drug. Sponsors may request a Special Protocol Assessment (“SPA”), only if the sponsor has already had an end-of-phase 2/pre-phase 3 meeting or end-of-phase 1 meeting as appropriate. The purpose of the SPA is to reach agreement with the FDA on clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
According to published guidance on the SPA process, a sponsor which meets the prerequisites may make a specific request for a SPA and ask specific questions regarding the design and size of the proposed clinical trial. The FDA has a goal to evaluate the protocol within 45 days of the request to assess whether the proposed trial is adequate, and that evaluation may result in discussions and a request for additional information. A SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the testing began.
Concurrent with clinical trials, sponsors usually complete additional animal safety studies and also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing commercial quantities of the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug and the manufacturer must develop methods for testing the identity, strength, quality, purity and potency of the drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its proposed shelf-life.
The results of product development, nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of user fees, but a waiver of such fees may be obtained under specified circumstances. The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination of whether it is sufficiently complete to permit substantive review. It may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
| 21 |
Once the submission is accepted for filing, the FDA begins an in-depth review. NDAs receive either standard or priority review. A drug representing a significant improvement in treatment, prevention or diagnosis of disease may receive priority review. The FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The FDA may refer the NDA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured and tested.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA marketing approval of RP-G28, one of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND, and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for extension must be made prior to expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (“ANDA”), or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric Exclusivity and Pediatric Use
Under the Best Pharmaceuticals for Children Act (“BPCA”), certain drugs may obtain an additional six months of exclusivity, if the sponsor submits information requested in writing by the FDA (a Written Request) relating to the use of the active moiety of the drug in children. The FDA may not issue a Written Request for studies on unapproved or approved indications or where it determines that information relating to the use of a drug in a pediatric population, or part of the pediatric population, may not produce health benefits in that population.
We have not received a Written Request for such pediatric studies, although we may ask the FDA to issue a Written Request for such studies in the future. To receive the six-month pediatric market exclusivity, we would have to receive a Written Request from the FDA, conduct the requested studies in accordance with a written agreement with the FDA or, if there is no written agreement, in accordance with commonly accepted scientific principles, and submit reports of the studies. A Written Request may include studies for indications that are not currently in the labeling if the FDA determines that such information will benefit the public health. The FDA will accept the reports upon its determination that the studies were conducted in accordance with and are responsive to the original Written Request or commonly accepted scientific principles, as appropriate, and that the reports comply with the FDA’s filing requirements.
In addition, the Pediatric Research Equity Act (“PREA”) requires all applications (or supplements to an application) submitted under section 505 of the FDCA (21 U.S.C. §355) for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration to contain a pediatric assessment unless the applicant has obtained a waiver or deferral. It also authorizes the FDA to require holders of approved NDAs for marketed drugs to conduct pediatric studies under certain circumstances. In general, PREA applies only to those drugs developed for diseases and/or conditions that occur in both the adult and pediatric populations. Products intended for pediatric-specific indications will be subject to the requirements of PREA only if they are initially developed for a subset of the relevant pediatric population.
| 22 |
As part of the Food and Drug Administration Safety and Innovation Act, Congress reauthorized both BPCA and PREA, which were slated to expire on September 30, 2012, and made both laws permanent.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States and when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic.
If a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. Orphan drug exclusive marketing rights in the United States also may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
We intend to explore orphan drug designation for RP-G28 for any orphan indication in which there is a medically plausible basis for treatment of the indication through colonic adaptation of gut bacteria.
Post-approval Requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Any drug products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things:
| ● | compliance with cGMPs; | |
| ● | record-keeping requirements; | |
| ● | reporting of adverse experiences with the drug; | |
| ● | providing the FDA with updated safety and efficacy information; | |
| ● | drug sampling and distribution requirements; | |
| ● | notifying the FDA and gaining its approval of specified manufacturing or labeling changes; and | |
| ● | complying with FDA promotion and advertising requirements. |
| 23 |
Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMP and other laws.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be.
Regulation Outside of the United States
In addition to regulations in the United States, we will be subject to regulations of other countries governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of countries outside of the United States before we can commence clinical trials in such countries and approval of the regulators of such countries or economic areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European Union regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology or those medicines intended to treat AIDS, cancer, neurodegenerative disorders or diabetes and optional for those medicines which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessments report, each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
Reimbursement
Sales of our products will depend, in part, on the extent to which the costs of our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the “MMA”) imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
| 24 |
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the “ACA”), enacted in March 2010, is expected to have a significant impact on the health care industry. The ACA is expected to expand coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, the ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the impact of the ACA on pharmaceutical companies, as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet occurred. In addition, some members of the U.S. Congress have been seeking to repeal at least portions of the legislation and we expect they will continue to review and assess this legislation and alternative health care reform proposals. Any legal challenges to the ACA, as well as Congressional efforts to repeal the ACA, add to the uncertainty of the legislative changes enacted as part of the ACA.
In addition, in some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
Employees
As of the date of this Annual Report, we had nine employees, all of whom were full time employees. None of our employees are represented by a labor union, and we consider our relationship with our employees to be good.
Available Information
We file with the Securities and Exchange Commission (“SEC”) annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy and information statements and amendments to reports filed or furnished pursuant to Sections 13(a), 14 and 15(d) of the Securities Exchange Act of 1934, as amended. The public may obtain these filings at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549 or by calling the SEC at 1-800-SEC-0330. The SEC also maintains a website at http://www.sec.gov that contains reports, proxy and information statements and other information regarding Ritter Pharmaceuticals, Inc. and other companies that file materials with the SEC electronically. As soon as practicable after filing with the SEC, we make copies of our reports on Form 10-K, Forms 10-Q and Forms 8-K available to the public, free of charge, through the investor relations tab on our web site, http://www.ritterpharmaceuticals.com/investors.
| 25 |
We operate in a dynamic and rapidly changing business environment that involves multiple risks and substantial uncertainty. The following discussion addresses risks and uncertainties that could cause, or contribute to causing, actual results to differ from expectations in material ways. In evaluating our business, investors should pay particular attention to the risks and uncertainties described below and in other sections of this Annual Report and in our subsequent filings with the SEC. These risks and uncertainties, or other events that we do not currently anticipate or that we currently deem immaterial also may affect our results of operations, cash flows and financial condition. The trading price of our common stock could also decline due to any of these risks, and you could lose all or part of your investment. The following information should be read in conjunction with Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and related notes included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report.
Risks Relating to Our Financial Position and Need for Additional Capital
We have incurred net losses in each year since our inception. Currently, we have no products approved for commercial sale. As a result, our ability to reduce our losses and reach profitability is unknown, and we may never achieve or sustain profitability.
We have incurred net losses in each year since our inception. The accompanying financial statements have been prepared assuming that we will continue as a going concern, which contemplates, among other things, the realization of assets and satisfaction of liabilities in the normal course of business.
To date, we have devoted most of our financial resources to our corporate overhead and research and development, including our drug discovery research, preclinical development activities and clinical trials. We currently have no products that are approved for commercial sale. We expect to continue to incur net losses and negative operating cash flow for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for, RP-G28, prepare for and begin the commercialization of RP-G28, and add infrastructure and personnel to support our product development efforts and operations. We anticipate that any such losses could be significant for the next several years as we begin our Phase 3 clinical trials for RP-G28 and related activities required for regulatory approval of RP-G28. If RP-G28 does not gain regulatory approval, or does not achieve market acceptance, we may never become profitable, unless we are able to develop and market some other product. Our net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. In addition, our expenses could increase if we are required by the FDA or the EMA, to perform studies or trials in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of RP-G28, or any other product candidates we may develop in the future. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues.
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. To complete the work necessary to file an NDA in the United States and a MAA in the European Union for RP-G28, which is currently anticipated to occur in 2019, we will require substantial additional funding. If the FDA or EMA requires that we perform additional nonclinical studies or clinical trials, our expenses would further increase beyond what we currently expect and the anticipated timing of any potential NDA or MAA would likely be delayed.
We will need to secure additional financing in order to complete clinical development of and commercialize RP-G28, if approved, and generally fund our operations. We can provide no assurances that any additional sources of financing will be available to us on favorable terms, if at all. If we are unable to obtain funding on a timely basis, we may be required to significantly curtail our research and development program. We also could be required to seek funds through arrangements with collaborative partners or otherwise that may require us to relinquish rights to RP-G28 or otherwise agree to terms unfavorable to us.
| 26 |
We may sell additional equity or debt securities to fund our operations, which would result in dilution to our stockholders and imposed restrictions on our business.
We may seek additional funding through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. Additional funding may not be available to us on acceptable terms or at all. To the extent that we raise additional funds by issuing equity securities (including any common stock issued to Aspire Capital Fund, LLC (“Aspire Capital”) pursuant to our financing arrangement with Aspire Capital), our stockholders may experience significant dilution. See “Management’s Discussion and Analysis of Financial Condition and Results of Operations — Aspire Capital Financing Arrangement.” Any debt financing, if available, may involve restrictive covenants that impact our ability to conduct business. If we are not able to raise additional capital when required or on acceptable terms, we may have to (i) significantly delay, scale back or discontinue the development and/or commercialization of RP-G28; (ii) seek collaborators for RP-G28 and possibly on terms that are less favorable than might otherwise be available; or (iii) relinquish or otherwise dispose of rights related to RP-G28. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional shares by us, or the possibility of such issuance, may cause the market price of our shares to decline.
Risks Relating to Regulatory Review and Approval of RP-G28
We cannot be certain that RP-G28 will receive regulatory approval, and without regulatory approval we will not be able to market RP-G28.
The development of a product candidate and issues relating to its approval and marketing are subject to extensive regulation by the FDA in the United States, the EMA in Europe, and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market our product candidates in the United States or Europe until we receive approval of a NDA from the FDA or a MAA from the EMA, respectively. We have not submitted any marketing applications for RP-G28.
NDAs and MAAs must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. NDAs and MAAs must also include significant information regarding the chemistry, manufacturing and controls for the product. Obtaining approval of a NDA or a MAA is a lengthy, expensive and uncertain process, and we may not be successful in obtaining approval. The FDA and the EMA review processes can take years to complete and approval is never guaranteed. If we submit a NDA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA. Regulators of other jurisdictions, such as the EMA, have their own procedures for approval of product candidates. Even if a product is approved, the FDA or the EMA, as the case may be, may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time-consuming clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States and Europe also have requirements for approval of drug candidates with which we must comply prior to marketing in those countries. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure that we will be able to obtain regulatory approval in any other country. In addition, delays in approvals or rejections of marketing applications in the United States, Europe or other countries may be based upon many factors, including regulatory requests for additional analyses, reports, data, preclinical studies and clinical trials, regulatory questions regarding different interpretations of data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding our product candidates or other products. Also, regulatory approval for any of our product candidates may be withdrawn.
We have completed a Phase 2a clinical trial and a Phase 2b clinical trial for RP-G28. We held an End-of-Phase 2 meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products in August 2017, regarding the path forward for RP-G28. We reached general consensus with the FDA on many elements of our Phase 3 program and received clear guidance and recommendations on many other necessary components of our Phase 3 program; including the clinical, non-clinical, and chemistry, manufacturing and controls (CMC) requirements needed to support an NDA submission. However, not all clinical, non-clinical, and CMC items have been agreed to with the FDA, and remaining items will need to be reviewed by the agency and agreed to by us.
We will also need to conduct rat and embryo-fetal development toxicity studies.
Additional non-clinical development may be required to be conducted based on future FDA feedback and guidance. We cannot predict whether our future trials and studies will be successful or whether regulators will agree with our conclusions regarding the preclinical studies and clinical trials we have conducted to date.
If we are unable to obtain approval from the FDA, the EMA or other regulatory agencies for RP-G28, we will not be able to market RP-G28. If we are unable to market RP-G28, we may not be able to ever become profitable.
| 27 |
The FDA and other regulatory agencies outside the United States, such as the EMA, may not agree to our proposed endpoint for approval of RP-G28 for the treatment of lactose intolerance in patients, in which case we would need to complete additional clinical trials before seeking market approval.
We do not know if the FDA, the EMA or regulatory authorities in other countries will agree with our final primary endpoint for approval of RP-G28. The FDA, the EMA and regulatory authorities in other countries in which we may seek approval for and market RP-G28, may require additional nonclinical studies and/or clinical trials prior to granting approval, if at all. It may be expensive and time consuming to conduct and complete additional nonclinical studies and clinical trials that the EMA and other regulatory authorities may require us to perform. As such, any requirement by the EMA or other regulatory authorities that we conduct additional nonclinical studies or clinical trials could materially and adversely affect our business, financial condition and results of operations. Furthermore, even if we receive regulatory approval of RP-G28 for the treatment of lactose intolerance in patients, the labeling for RP-G28 in the United States, Europe or other countries in which we seek approval may include limitations that could impact the commercial success of RP-G28.
Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for RP-G28.
Delays in the commencement, enrollment and completion of clinical trials could increase our product development costs or limit the regulatory approval of RP-G28. The commencement, enrollment and completion of clinical trials may be delayed or suspended for a variety of reasons, including:
| ● | inability to obtain sufficient funds required to conduct a clinical trial; | |
| ● | inability to reach agreements on acceptable terms with prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | clinical holds, other regulatory objections to commencing or continuing a clinical trial or the inability to obtain regulatory approval to commence a clinical trial in countries that require such approvals; | |
| ● | discussions with the FDA or non-U.S. regulators regarding the scope or design of our clinical trials; | |
| ● | inability to identify and maintain a sufficient number of trial sites, many of which may already be engaged in other clinical trial programs, including some that may be for the same indications targeted by RP-G28; | |
| ● | inability to obtain approval from institutional review boards (“IRBs”), to conduct a clinical trial at their respective sites; | |
| ● | severe or unexpected drug-related adverse effects experienced by patients; | |
| ● | inability to timely manufacture sufficient quantities of RP-G28 required for a clinical trial; | |
| ● | difficulty recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including meeting the enrollment criteria for our study and competition from other clinical trial programs for the same indications as RP-G28; | |
| ● | the FDA’s rejection of our end points as indicators of efficacy; and | |
| ● | inability to retain enrolled patients after a clinical trial is underway. |
Changes in regulatory requirements and guidance may also occur and we may need to amend clinical trial protocols to reflect these changes with appropriate regulatory authorities. Amendments may require us to resubmit clinical trial protocols to IRBs for re-examination, which may impact the costs, timing or successful completion of a clinical trial. In addition, a clinical trial may be suspended or terminated at any time by us, our future collaborators, the FDA or other regulatory authorities due to a number of factors, including:
| ● | our failure or the failure of our potential future collaborators to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; | |
| ● | unforeseen safety issues or any determination that a clinical trial presents unacceptable health risks; | |
| ● | lack of adequate funding to continue the clinical trial due to unforeseen costs or other business decisions; and | |
| ● | a breach of the terms of any agreement with, or for any other reason by, future collaborators who have responsibility for the clinical development of RP-G28. |
In addition, if we or any of our potential future collaborators are required to conduct additional clinical trials or other nonclinical studies of RP-G28 beyond those contemplated, our ability to obtain regulatory approval of RP-G28 and to generate revenue from its sales would be similarly harmed.
| 28 |
Clinical failure can occur at any stage of clinical development and may prevent us from receiving regulatory approval. The results of earlier clinical trials related to RP-G28 are not necessarily predictive of future results.
Clinical failure can occur at any stage of our clinical development. Clinical trials may produce negative or inconclusive results, and we or our collaborators may decide, or regulators may require us, to conduct additional clinical trials or nonclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval. Successful results from preclinical studies and early clinical trials do not ensure that subsequent clinical trials will generate the same or similar results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. A number of companies in the pharmaceutical industry, including those with greater resources and experience than us, have suffered significant setbacks in later clinical trials, even after seeing promising results in earlier clinical trials.
In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well-advanced. We may be unable to design and execute a clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in composition of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We do not know whether the clinical trials that we or any of our potential future collaborators may conduct will demonstrate the consistent or adequate efficacy and safety that would be required to obtain regulatory approval and market RP-G28. Our Phase 3 clinical trials for RP-G28 may not provide sufficient support for NDA approval.
The FDA may require us to conduct one or more additional clinical trials, possibly involving a larger sample size or a different clinical trial design, or may require longer follow-up periods. If we are unable to bring RP-G28 to market, our ability to create long-term stockholder value will be limited.
RP-G28 may have undesirable side effects which may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
There were no notable differences observed between placebo-treated subjects and RP-G28-treated subjects in the Phase 2b trial. However, unforeseen side effects from RP-G28, could arise at any time during clinical development or, if approved, after the approved product has been marketed. Any undesirable or unacceptable side effects associated with RP-G28 could interrupt, delay or halt clinical trials, and results in delay of, or failure to obtain, marketing approval from the FDA and other regulatory authorities.
In addition:
| ● | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies; | |
| ● | we may be required to change instructions regarding the way the product is administered, conduct additional clinical trials or change the labeling of the product; | |
| ● | we may be subject to limitations on how we may promote the product; | |
| ● | sales of the product may decrease significantly; | |
| ● | regulatory authorities may require us to take our approved product off the market; | |
| ● | we may be subject to litigation or product liability claims; and | |
| ● | our reputation may suffer. |
Any of these events could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of our products.
| 29 |
Reimbursement decisions by third-party payors may have an adverse effect on pricing and market acceptance. If there is not sufficient reimbursement for our products, it is less likely that they will be widely used.
Market acceptance and sales of RP-G28, or any other product candidates we develop in the future, will depend on reimbursement policies and may be affected by, among other things, future healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. We cannot be certain that reimbursement will be available for RP-G28 or any other product candidates that we may develop in the future. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, our products. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize RP-G28, or other product candidates that we develop in the future.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the “MMA”), changed the way Medicare covers and pays for pharmaceutical products. The legislation established Medicare Part D, which expanded Medicare coverage for outpatient prescription drug purchases by the elderly but provided authority for limiting the number of drugs that will be covered in any therapeutic class. The MMA also introduced a new reimbursement methodology based on average sales prices for physician-administered drugs. Any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain in the United States. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
The United States and several other jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. We expect to experience pricing pressures in connection with the sale of RP-G28, and any other product candidates that we develop in the future, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative proposals.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the “ACA”), enacted in March 2010, is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on health industry and impose additional health policy reforms. With regard to pharmaceutical products, among other things, the ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under Medicare Part D program. Although it is too early to determine the full effect of the ACA, the law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA. Congress and President Trump have expressed their intentions to repeal and replace the ACA. President Trump issued an Executive Order and both chambers of Congress passed bills, all with the goal of fulfilling their intensions. However, to date, the Executive Order has had limited effect and the Congressional activities have not resulted in the passage of a law. If a law is enacted, many if not all of the provisions of the ACA may no longer apply to prescription drugs.
If we do not obtain protection under the Hatch-Waxman Act and similar legislation outside of the United States by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of RP-G28, one of our U.S. patents may be eligible for a limited Patent Term Extension under the Drug Price Competition and Patent Term Restoration Act of 1984, which is sometimes referred to as the Hatch-Waxman Act, provided our U.S. patent claims a method of treating lactose intolerance that is approved by the FDA. The Hatch-Waxman Act, 35 U.S.C. §156, permits a patent extension of up to five years as compensation for patent term lost during the FDA regulatory review process. The scope of protection afforded by the patent during the extended term is not commensurate with the scope of the unextended portion of the patent; for example, the “rights derived” from a method of use patent during the extended period are “limited to any use claimed by the patent and approved for the product.” 35 U.S.C. §156(b)(2). We may not be granted an extension because of, for example, failing to apply for the extension within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable statutory and/or regulatory requirements including, for example, the requirement that the patent to be extended “claim” the approved product or a method of using the approved product. Moreover, the applicable period of extension could be less than we request. If we are unable to obtain patent term extension or if the term of any such extension is shorter than we request, the period during which we will be able to exclude others from marketing their versions of our product will be shortened and our competitors may obtain approval of generic products following our patent expiration, and our revenue could be reduced, possibly materially. Similar concerns are associated with obtaining Supplemental Protection Certificates of certain patents issued in Europe and owned by Inalco SpA, to which we have an exclusive options of assignment, based upon patent terms lost during European regulatory review processes. In the event that we are unable to obtain any patent term extension, the issued patents for RP-G28 are expected to expire in 2030, assuming they withstand any challenge toothier validity and/or patentability.
| 30 |
If we market products in a manner that violates healthcare fraud and abuse laws, or if we violate government price reporting laws, we may be subject to civil or criminal penalties.
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal healthcare laws, commonly referred to as “fraud and abuse” laws, have been applied in recent years to restrict certain marketing practices in the pharmaceutical industry. Other jurisdictions such as Europe have similar laws. These laws include false claims and anti-kickback statutes. If we market our products and our products are paid for by governmental programs, it is possible that some of our business activities could be subject to challenge under one or more of these laws.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service covered by Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers or formulary managers on the other. Although there are several statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Most states also have statutes or regulations similar to the federal anti-kickback law and federal false claims laws, which apply to items and services covered by Medicaid and other state programs, or, in several states, apply regardless of the payor. Administrative, civil and criminal sanctions may be imposed under these federal and state laws.
Over the past few years, a number of pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of promotional and marketing activities, such as: providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates.
Any delay or disruption in the manufacture and supply of RP-G28 may negatively impact our operations.
We do not intend to manufacture RP-G28. We have an agreement with RSM, our contract manufacturer, for the production of Improved GOS, the active pharmaceutical ingredient in RP-G28, and the formulation of sufficient quantities of Improved GOS for the clinical and nonclinical studies that we believe we will need to conduct prior to seeking regulatory approval for RP-G28. However, we do not have agreements for commercial supplies of RP-G28 and we may not be able to reach agreement with RSM or any other contract manufacturer for sufficient supplies to commercialize RP-G28 if it is approved.
Reliance on third-party manufacturers entails risks, to which we would not be subject if we manufactured the products ourselves, including:
| ● | the possibility that we are unable to enter into a manufacturing agreement with third parties to manufacture RP-G28; | |
| ● | the possible breach of the manufacturing agreements by third parties because of factors beyond our control; and | |
| ● | the possible termination or nonrenewal of manufacturing agreements by the third parties before we are able to arrange for a qualified replacement third-party manufacturers. |
Any of these factors could cause the delay of approval or commercialization of RP-G28, or cause us to incur higher costs. Furthermore, if RP-G28 is approved and contract manufacturers fail to deliver the required commercial quantities of finished product on a timely basis and at commercially reasonable prices and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our product and could lose potential revenue. It may take several years to establish an alternative source of supply for RP-G28 and to have any such new source approved by the government agencies that regulate our products. In the event we do need to identify alternative manufacturing partners, we may have to secure licenses to manufacturing and/or purification technologies, including third-party patent licenses, to allow us to manufacture RP-G28 that is suitable for the late-stage regulatory review process and/or adequate to manufacture commercial quantities of RP-G28.
| 31 |
If the FDA and EMA and other regulatory agencies do not approve the manufacturing facilities of our future contract manufacturers for commercial production, we may not be able to commercialize RP-G28.
The facilities used by any contract manufacturer to manufacture RP-G28 must be the subject of a satisfactory inspection before the FDA or the regulators in other jurisdictions approve the product candidate manufactured at that facility. We are completely dependent on these third-party manufacturers for compliance with the requirements of U.S. and non-U.S. regulators for the manufacture of our finished products. If our manufacturers cannot successfully manufacture material that conform to our specifications and cGMP requirements of any governmental agency whose jurisdiction to which we are subject, our product candidates will not be approved or, if already approved, may be subject to recalls.
Even if RP-G28 receives regulatory approval, we may still face future development and regulatory difficulties.
RP-G28, and any other product candidates we develop in the future, if approved, will be subject to ongoing regulatory requirements for labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information. In addition, approved products, manufacturers and manufacturers’ facilities are required to comply with extensive FDA and EMA requirements and requirements of other similar agencies, including ensuring that quality control and manufacturing procedures conform to cGMPs. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control. We will also be required to report certain adverse reactions and production problems, if any, to the FDA and EMA and other similar agencies and to comply with certain requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. Accordingly, we may not promote our approved products, if any, for indications or uses for which they are not approved.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of a product, it may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If RP-G28 fails to comply with applicable regulatory requirements, a regulatory agency may:
| ● | issue warning letters; |
| ● | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; | |
| ● | require us or our potential future collaborators to enter into a consent decree or permanent injunction, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; | |
| ● | impose other administrative or judicial civil or criminal penalties; | |
| ● | withdraw regulatory approval; | |
| ● | refuse to approve pending applications or supplements to approved applications filed by us or our potential future collaborators; | |
| ● | impose restrictions on operations, including costly new manufacturing requirements; or | |
| ● | detain, seize and/or condemn and destroy products. |
Risks Relating to the Potential Commercialization of RP-G28
Even if approved, RP-G28 may not achieve broad market acceptance among physicians, patients and healthcare payors, and as a result our revenues generated from its sales may be limited.
The commercial success of RP-G28, if approved, will depend upon its acceptance among the medical community, including physicians, health care payors and patients. The degree of market acceptance of RP-G28 will depend on a number of factors, including:
| ● | limitations or warnings contained in our product candidates’ FDA-approved labeling; | |
| ● | changes in the standard of care or availability of alternative therapies at similar or lower costs for the targeted indications for such product candidates; |
| 32 |
| ● | limitations in the approved clinical indications for RP-G28; | |
| ● | demonstrated clinical safety and efficacy compared to other products; | |
| ● | lack of significant adverse side effects; | |
| ● | sales, marketing and distribution support; | |
| ● | availability of reimbursement from managed care plans and other third-party payors; | |
| ● | timing of market introduction and perceived effectiveness of competitive products; | |
| ● | the degree of cost-effectiveness; | |
| ● | availability of alternative therapies at similar or lower cost, including generics and over-the-counter products; | |
| ● | enforcement by the FDA and EMA of laws and rulings that prohibit the illegal sale of RP-G28 as a dietary supplement; | |
| ● | the extent to which RP-G28 is approved for inclusion on formularies of hospitals and managed care organizations; | |
| ● | whether RP-G28 is designated under physician treatment guidelines for the treatment of or reduction of symptoms associated with the indications for which we have received regulatory approval; | |
| ● | adverse publicity about RP-G28 or favorable publicity about competitive products; | |
| ● | convenience and ease of administration of RP-G28; and | |
| ● | potential product liability claims. |
If RP-G28 is approved, but does not achieve an adequate level of acceptance by physicians, patients, the medical community and healthcare payors, sufficient revenue may not be generated from its sales and we may not become or remain profitable. In addition, efforts to educate the medical community and third-party payors on the benefits of RP-G28 may require significant resources and may never be successful.
We have no internal sales, distribution and/or marketing capabilities at this time and we will have to invest significant resources to develop those capabilities or enter into acceptable third-party sales and marketing arrangements.
We have no internal sales, distribution and/or marketing capabilities at this time. To develop these capabilities, we will have to invest significant amounts of financial and management resources, some of which will be committed prior to any confirmation that RP-G28 will be approved. We could also face a number of additional risks, including:
| ● | we or our third-party sales collaborators may not be able to attract and build an effective marketing or sales force; | |
| ● | the cost of securing or establishing a marketing or sales force may exceed the revenues generated by RP-G28; and | |
| ● | our direct sales and marketing efforts may not be successful. |
We may have limited or no control over the sales, marketing and distribution activities of third parties. Our future revenues could depend heavily on the success of the efforts of third parties.
We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop RP-G28.
Because developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products are expensive, we may seek to enter into collaborations with companies that have more experience. Additionally, if RP-G28 receives marketing approval, we may enter into sales and marketing arrangements with third parties with respect to our unlicensed territories. If we are unable to enter into arrangements on acceptable terms, we may be unable to effectively market and sell RP-G28 in our target markets. We expect to face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement and they may require substantial resources to maintain. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements for the development of RP-G28.
| 33 |
If we collaborate with a third party for the development and commercialization of RP-G28, we can expect to relinquish some or all of the control over the future success of RP-G28. For example, we may relinquish rights to RP-G28 in jurisdictions outside of the United States. Our collaboration partner may not devote sufficient resources to the commercialization of RP-G28 or may otherwise fail in its commercialization. The terms of any collaboration or other arrangement that we establish may not be favorable to us. In addition, any collaboration that we enter into may be unsuccessful in the development and commercialization of RP-G28. In some cases, we may be responsible for continuing preclinical and initial clinical development of RP-G28 or research programs under a collaboration arrangement, and the payment we receive from our collaboration partner may be insufficient to cover the cost of this development. If we are unable to reach agreements with suitable collaborators, we would face increased costs, we may be forced to limit the territories in which we commercialize RP-G28. If we fail to achieve successful collaborations, our operating and financial condition will be materially and adversely affected.
Risks Relating to Our Business and Strategy
We may face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
Although we know of no other drug candidates in advanced clinical trials for treating lactose intolerance, the biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have potential competitors in the United States, Europe and other jurisdictions, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical and generic drug companies and universities and other research institutions. Many of these potential competitors have greater financial and other resources, such as larger research and development staff and more experienced marketing and manufacturing organizations. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, obtaining regulatory approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research, sales and marketing capabilities and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. As a result of all of these factors, these potential competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing drugs for the diseases that we are targeting before we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Some of the pharmaceutical and biotechnology companies we expect to compete with include microbiome based development companies: Second Genome, Inc., Seres Health, Inc., Enterome SA, Vedanta Biosciences, Inc., and Rebiotix Inc. In addition, many universities and private and public research institutes may become active in our target disease areas. These potential competitors may succeed in developing, acquiring or licensing on an exclusive basis, technologies and drug products that are more effective or less costly than RP-G28, which could render RP-G28 obsolete and noncompetitive.
We believe that our ability to successfully compete will depend on, among other things:
| ● | our ability to commercialize and market RP-G28; | |
| ● | the efficacy, safety and reliability of RP-G28; | |
| ● | the price of RP-G28; | |
| ● | adequate levels of reimbursement under private and governmental health insurance plans, including Medicare; | |
| ● | our ability to protect intellectual property rights related to RP-G28; | |
| ● | our ability to manufacture and sell commercial quantities of RP-G28 to the market; and | |
| ● | acceptance of RP-G28 by physicians and other health care providers. |
If our competitors market products that are more effective, safer or less expensive than RP-G28, or that reach the market sooner than RP-G28, we may not achieve commercial success. In addition, the biopharmaceutical industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies or product candidates obsolete, less competitive or not economical.
| 34 |
We depend on third-party contractors for a substantial portion of our operations and may not be able to control their work as effectively as if we performed these functions ourselves.
We outsource substantial portions of our operations to third-party service providers, including the conduct of preclinical studies and clinical trials, collection and analysis of data, and manufacturing. Our agreements with third-party service providers and CROs are on a study-by-study and project-by-project basis. Typically, we may terminate the agreements with notice and are responsible for the supplier’s previously incurred costs. In addition, any CRO that we retain will be subject to the FDA’s and EMA’s regulatory requirements and similar standards outside of the United States and Europe and we do not have control over compliance with these regulations by these providers. Consequently, if these providers do not adhere to applicable governing practices and standards, the development and commercialization of our product candidates could be delayed or stopped, which could severely harm our business and financial condition.
Because we have relied on third parties, our internal capacity to perform these functions is limited to management oversight. Outsourcing these functions involves the risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. Although we have not experienced any significant difficulties with our third-party contractors, it is possible that we could experience difficulties in the future. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. There are a limited number of third-party service providers that specialize or have the expertise required to achieve our business objectives. Identifying, qualifying and managing performance of third-party service providers can be difficult, time consuming and cause delays in our development programs. We currently have a small number of employees, which limits the internal resources we have available to identify and monitor third-party service providers. To the extent we are unable to identify, retain and successfully manage the performance of third-party service providers in the future, our business may be adversely affected, and we may be subject to the imposition of civil or criminal penalties if their conduct of clinical trials violates applicable law.
A variety of risks associated with our possible international business relationships could materially adversely affect our business.
We may enter into agreements with other third parties for the development and commercialization of RP-G28, or other product candidates we develop in the future, in international markets. International business relationships subject us to additional risks that may materially adversely affect our ability to attain or sustain profitable operations, including:
| ● | differing regulatory requirements for drug approvals internationally; | |
| ● | potentially reduced protection for intellectual property rights; | |
| ● | potential third-party patent rights in the United States and/or in countries outside of the United States; | |
| ● | the potential for so-called “parallel importing,” which is what occurs when a local seller, faced with relatively high local prices, opts to import goods from another jurisdiction with relatively low prices, rather than buying them locally; | |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements; | |
| ● | economic weakness, including inflation, or political instability, particularly in non-U.S. economies and markets, including several countries in Europe; | |
| ● | compliance with tax, employment, immigration and labor laws for employees traveling abroad; | |
| ● | taxes in other countries; | |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; | |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and | |
| ● | business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters, including earthquakes, volcanoes, typhoons, floods, hurricanes and fires. |
| 35 |
We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As we advance RP-G28 through clinical trials to commercialization and increase the number of ongoing product development programs, we will need to increase our product development, scientific and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we will need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
| ● | successfully attract and recruit new employees or consultants with the expertise and experience we will require; | |
| ● | manage our clinical programs effectively, which we anticipate being conducted at numerous clinical sites; | |
| ● | develop a marketing and sales infrastructure; and | |
| ● | continue to improve our operational, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel and consultants.
We may not be able to attract or retain qualified management, finance, scientific and clinical personnel and consultants due to the intense competition for qualified personnel and consultants among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain necessary personnel and consultants to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
We are highly dependent on the development, regulatory, commercialization and business development expertise of Michael D. Step, our Chief Executive Officer, Andrew J. Ritter, our Founder and President, and Ira E. Ritter, our Executive Chairman and Chief Strategic Officer. If we were to lose one or more of these key employees, our ability to implement our business strategy successfully could be seriously harmed. Any of our executive officers may terminate their employment at any time. Replacing any of these persons would be difficult and could take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully.
There is also a risk that other obligations could distract our officers and employees from our business, which could have negative impact on our ability to effectuate our business plans.
In addition, we have scientific and clinical advisors and consultants who assist us in formulating our research, development and clinical strategies. Competition to hire and retain consultants from a limited pool is intense. Further, because these advisors are not our employees, they may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us, and typically they will not enter into non-compete agreements with us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. We are required, under Section 404 of the Sarbanes-Oxley Act, to furnish with our annual report on Form 10-K a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment must include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company, as defined in the JOBS Act, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the independent registered public accounting firm’s requirement to attest to the effectiveness of our internal controls over financial reporting.
| 36 |
Our compliance with Section 404 requires that we incur substantial accounting expense and expend significant management efforts. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting once that firm begin its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ stock market, the SEC, or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosure due to error or fraud may occur and not be detected.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading, which could significantly harm our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with the regulations of the FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with health care fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted an employee handbook, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of our product candidates in clinical trials and the sale of any products for which we may obtain marketing approval expose us to the risk of product liability claims. Product liability claims may be brought against us or our potential future collaborators by participants enrolled in our clinical trials, patients, health care providers or others using, administering or selling our products. If we cannot successfully defend ourselves against any such claims, we would incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| ● | withdrawal of clinical trial participants; | |
| ● | termination of clinical trial sites or entire trial programs; |
| 37 |
| ● | costs of related litigation; | |
| ● | substantial monetary awards to patients or other claimants; | |
| ● | decreased demand for our product candidates and loss of revenues; | |
| ● | impairment of our business reputation; | |
| ● | diversion of management and scientific resources from our business operations; and | |
| ● | the inability to commercialize our product candidates. |
We have product liability insurance coverage for our clinical trials in the United States and in selected other jurisdictions where we intend to conduct clinical trials at levels we believe are sufficient and consistent with industry standards for companies at our stage of development. However, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. We intend to expand our insurance coverage for products to include the sale of any commercial products for which we obtain marketing approval, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash resources and adversely affect our business.
Our insurance policies are expensive and only protect us from some business risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability ($2.0 million coverage), employment practices liability, workers’ compensation, and directors’ and officers’ insurance at levels we believe are typical for a company in our industry and at our stage of development. We currently carry clinical trial liability insurance for our clinical trials at levels we believe are sufficient and consistent with industry standards for companies at our stage of development. We do not know, however, if we will be able to maintain insurance with adequate levels of coverage. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our financial position and results of operations.
If we engage in an acquisition, reorganization or business combination, we will incur a variety of risks that could adversely affect our business operations or our stockholders.
From time to time we have considered, and we will continue to consider in the future, strategic business initiatives intended to further the expansion and development of our business. These initiatives may include acquiring businesses, technologies or products or entering into a business combination with another company. If we pursue such a strategy, we could, among other things:
| ● | issue equity securities that would dilute our current stockholders’ percentage ownership; | |
| ● | incur substantial debt that may place strains on our operations; | |
| ● | spend substantial operational, financial and management resources to integrate new businesses, technologies and products; | |
| ● | assume substantial actual or contingent liabilities; | |
| ● | reprioritize our development programs and even cease development and commercialization of our product candidates; or | |
| ● | merge with, or otherwise enter into a business combination with, another company in which our stockholders would receive cash and/or shares of the other company on terms that certain of our stockholders may not deem desirable. |
Although we intend to evaluate and consider acquisitions, reorganizations and business combinations in the future, we have no agreements or understandings with respect to any acquisition, reorganization or business combination at this time.
| 38 |
Risks Relating to Our Intellectual Property
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection. If our patent position does not adequately protect our product candidates, others could compete against us more directly, which would harm our business, possibly materially.
Our commercial success will depend in part on obtaining, maintaining and enforcing patent protection and on developing, preserving and enforcing current trade secret protection. In particular, it will depend in part on our ability to obtain, maintain and enforce patents, especially those directed to methods of using our current product, RP-G28, and other future drug candidates, and those directed to the methods used to develop and manufacture our products, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents (and/or trade secrets) that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will withstand subsequent challenges to their validity, enforceability, and/or patentability, or if they will be commercially useful in protecting our product candidates, discovery programs and processes. Furthermore, we cannot be sure that our existing patents and patent applications will embrace (or “claim”) the particular uses for RP-G28 that will be approved by the FDA.
The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved.
No consistent policy regarding the patentability and/or validity of patent claims related to pharmaceutical patents has emerged, to date, in the United States or in most jurisdictions outside of the United States. Changes in either the patent laws (be they substantive or procedural) or in the interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of any claims that will issue or will be enforceable in the patents that have or may be issued from the patents and applications we currently own or may in the future own or license from third parties. Further, if any patents we obtain, or to which we obtain licenses, are deemed invalid, unpatentable and unenforceable, our ability to commercialize or license our technology could be adversely affected.
In the future others may file patent applications directed to products, uses for products, and manufacturing techniques and related technologies that are similar, identical or competitive to ours or important to our business. We cannot be certain that any patent or patent application owned by a third party will not have priority over patent applications filed or in-licensed by us in the future, or that we or our licensors will not be involved in interference, opposition, inter partes review or invalidity proceedings before U.S. or non-U.S. patent offices or courts.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| ● | others may be able to develop a platform similar to, or better than, ours in a way that does not infringe our patents; | |
| ● | others may be able to make compounds that are similar to our product candidates but that do not infringe our patents; | |
| ● | others may be able to manufacture compounds that are similar or identical to our product candidates using processes that do not infringe our method of making patents; | |
| ● | others may obtain regulatory approval for uses of compounds, similar or identical to our product, that do not infringe our pharmaceutical composition patents or our method of use patents; | |
| ● | we may not be able to obtain licenses for patents that are essential to the process of making the product; |
| ● | we might not have been the first to make the inventions claimed in our issued patents and pending patent applications; | |
| ● | we might not have been the first to file patent applications for these inventions; | |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies; | |
| ● | any patents that we obtain may not provide us with any competitive advantages; | |
| ● | we may not develop additional proprietary technologies that are patentable; or | |
| ● | the patents of others may have an adverse effect on our business. |
| 39 |
Patents directed to pharmaceutical compositions containing RP-G28 or methods of using RP-G28 expire in 2030 if the appropriate maintenance fee renewal, annuity, or other government fees are paid, unless a patent term extension based on regulatory delay is obtained. We expect that expiration in 2030 of some of our pharmaceutical composition and method-of-use patents directed to RP-G28 and its use in treating lactose intolerance will have a limited impact on our ability to protect our intellectual property in the United States, where we have additional issued patents directed to such compositions and uses that extend until 2030. In other countries, our issued patents and pending patent applications directed to compositions containing or methods of using RP-G28 for treating other indications, if issued, would expire in 2030. We will attempt to mitigate the effect of patent expiration by seeking data exclusivity, or the foreign equivalent thereof, in conjunction with product approval, as well as by filing additional patent applications covering improvements in our intellectual property.
We expect that the other patent applications for the RP-G28 portfolio, if issued, and if the appropriate maintenance, renewal, annuity or other governmental fees are paid, would expire in 2030. We own pending applications in the United States, Europe, and certain other countries directed to uses of RP-G28 to treat a variety of disorders, including lactose intolerance. Patent protection, to the extent these patents issue, would be expected to extend to 2030, unless a patent term extension based on regulatory delay is obtained.
Due to the patent laws of a country, or the decisions of a patent examiner in a country, or our own filing strategies, we may not obtain patents directed to all of our product candidates or methods involving these candidates in the parent patent application. We plan to pursue divisional patent applications or continuation patent applications in the United States and other countries to obtain claims directed to inventions that were disclosed but not claimed in the parent patent application.
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or feasible. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
Our patents are not directed to RP-G28 as a composition of matter.
Although we own certain patents and patent applications with claims directed to specific pharmaceutical compositions and methods of using RP-G28 to treat lactose intolerance, we do not have patents directed to RP-G28 as a composition of matter in the United States or elsewhere. As a result, we may be limited in our ability to list our patents in the FDA’s Orange Book if our product or the use of our product, consistent with its FDA-approved label, would not fall within the scope of our patent claims. Also, our competitors may be able to offer and sell products so long as these competitors do not infringe any other patents that we (or third parties) hold, including patents with claims directed to the manufacture of RP-G28, pharmaceutical compositions containing RP-G28, and/or method of using RP-G28. In general, pharmaceutical composition patents and method of use patents are more difficult to enforce than composition of matter patents because, for example, of the risks that FDA may approve alternative uses of the subject compounds not covered by the method of use patents, and others may engage in off-label sale or use of the subject compounds. Physicians are permitted to prescribe an approved product for uses that are not described in the product’s labeling. Although off-label prescriptions may infringe our method of use patents, the practice is common across medical specialties and such infringement is difficult to prevent or prosecute. FDA approval of uses that are not covered by our patents would limit our ability to generate revenue from the sale of RP-G28, if approved for commercial sale. Off-label sales would limit our ability to generate revenue from the sale of RP-G28, if approved for commercial sale.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
If we choose to go to court to stop another party from using the inventions claimed in any patents we obtain, that individual or company may seek a post grant review (including inter partes review) of our patents, and has the right to ask the court to rule that such patents are invalid or should not be enforced against that third party. These lawsuits and administrative proceedings are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents. In addition, there is a risk that the court or administrative body will decide that such patents are not valid or unpatentable and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity/patentability of such patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our patents. In addition, the U.S. Supreme Court and the Court of Appeals for the Federal Circuit have articulated and/or modified certain tests used by the U.S. Patent and Trademark Office (the “USPTO”), in assessing patentability and by the courts in assessing validity and claim scope, which may decrease the likelihood that we will be able to obtain patents and increase the likelihood that others may succeed in challenging any patents we obtain or license.
| 40 |
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our product candidates.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. We cannot guarantee that our products, our methods of manufacture, or our uses of RP-G28 (or our other product candidates), will not infringe third-party patents. Furthermore, a third party may claim that we or our manufacturing or commercialization collaborators are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. There is a risk that a court would decide that we or our commercialization collaborators are infringing the third party’s patents and would order us or our collaborators to stop the activities covered by the patents. In that event, we or our commercialization collaborators may not have a viable way around the patent and may need to halt commercialization of the relevant product. In addition, there is a risk that a court will order us or our collaborators to pay the other party damages for having violated the other party’s patents. In the future, we may agree to indemnify our commercial collaborators against certain intellectual property infringement claims brought by third parties. The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The scope of coverage of a patent is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, the patentee would need to demonstrate, by a preponderance of the evidence that our products or methods infringe the patent claims of the relevant patent, and we would need to demonstrate either that we do not infringe or, by clear and convincing evidence, that the patent claims are invalid; we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and divert management’s time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, which may not be available, defend an infringement action or challenge the validity or enforceability of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, otherwise fail to defend an infringement action successfully or have a court hold that any patent we infringe is invalid or unenforceable, we may incur substantial monetary damages, encounter significant delays in bringing our product candidates to market and we may be precluded from manufacturing or selling our product candidates.
We cannot be certain that others have not filed patent applications for technology claimed in our pending applications, or that we were the first to invent the technology, because:
| ● | some patent applications in the United States may be maintained in secrecy until the patents are issued; | |
| ● | patent applications in the United States are typically not published until at least 18 months after the earliest asserted priority date; and | |
| ● | publications in the scientific literature often lag behind actual discoveries. |
Our competitors may have filed, and may in the future file, patent applications directed to technology similar to ours. Any such patent application may have priority over our patent applications, which could further require us to obtain rights to issued patents directed to such technologies. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if, unbeknownst to us, the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such inventions. Other countries have similar laws that permit secrecy of patent applications, and other parties may be entitled to priority over our applications in such jurisdictions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
| 41 |
Obtaining and maintaining our patent portfolio depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patents could be deemed abandoned or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We employ an outside firm to pay fees due to non-U.S. patent agencies and this outside firm has systems in place to ensure compliance on payment of fees. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers. If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Failure to secure trademark registrations could adversely affect our business.
We have not developed a trademark for our RP-G28 product. Hence, we do not currently own any actual or potential trademark rights associated with our RP-G28 product. If we seek to register additional trademarks, including trademarks associated with our RP-G28 product, our trademark applications may not be allowed for registration or our registered trademarks may not be maintained or enforced. During trademark registration proceedings, we may receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many other jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
Risks Relating to Our Capital Stock
An active trading market for our common stock may not develop or be sustained.
Prior to our initial public offering, there was no public market for our common stock. Since our initial public offering in June 2015, there has been, and we expect that there will continue to be, only a limited volume of trading in our common stock. An active trading market in our common stock may not develop or, if developed, may not be sustained. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market may also reduce the fair market value of your shares. An inactive market may also impair our ability to raise capital to continue to fund operations by selling shares and may impair our ability to acquire other companies or technologies by using our shares as consideration.
| 42 |
Our share price may be volatile, which could subject us to securities class action litigation and prevent you from being able to sell your shares at or above your purchase price.
The market price of shares of our common stock could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including:
| ● | results of our clinical trials; | |
| ● | results of clinical trials of our competitors’ products; | |
| ● | regulatory actions with respect to our products or our competitors’ products; | |
| ● | actual or anticipated fluctuations in our financial condition and operating results; | |
| ● | actual or anticipated changes in our growth rate relative to our competitors; | |
| ● | actual or anticipated fluctuations in our competitors’ operating results or changes in their growth rate; | |
| ● | competition from existing products or new products that may emerge; | |
| ● | announcements by us, our potential future collaborators or our competitors of significant acquisitions, strategic collaborations, joint ventures, or capital commitments; | |
| ● | issuance of new or updated research or reports by securities analysts; | |
| ● | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
| ● | inconsistent trading volume levels of our shares; | |
| ● | additions or departures of key management or scientific personnel; | |
| ● | disputes or other developments related to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; | |
| ● | announcement or expectation of additional financing efforts; | |
| ● | sales of our common stock by us, our insiders or our other stockholders; | |
| ● | market conditions for biopharmaceutical stocks in general; and | |
| ● | general economic and market conditions. |
Furthermore, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations often have been unrelated or disproportionate to the operating performance of those companies. These broad market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes or international currency fluctuations, may negatively impact the market price of shares of our common stock. In addition, such fluctuations could subject us to securities class action litigation, which could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our common stock will depend on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be no assurance that analysts will cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our stock or change their opinion of our stock, our share price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our share price or trading volume to decline.
There is no active, public market for our Series A Preferred stock.
There is no established public trading market for our Series A Preferred stock. We do not intend to apply to list the Series A Preferred stock on a securities exchange. Without an active trading market, the liquidity of the Series A Preferred stock will be limited.
| 43 |
Holders of Series A Preferred have limited voting rights.
Except with respect to certain material changes in the terms of the Series A Preferred stock and certain other matters and except as may be required by Delaware law, holders of Series A Preferred stock have no voting rights.
Future sales of our common stock, or the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is doing well.
Sales by our stockholders of a substantial number of shares of our common stock in the public market could occur in the future. These sales, or the perception in the market that the holders of a large number of shares of common stock intend to sell shares, could reduce the market price of our common stock.
We may sell up to $6.5 million of our shares of common stock to Aspire Capital pursuant to financing arrangement with Aspire Capital. The sale of a substantial number of shares of our common stock by Aspire Capital, or anticipation of such sales, could cause the trading price of our common stock to decline or make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise desire. However, we have the right to control the timing and amount of sales of our shares to Aspire Capital, and we may terminate the financing arrangement at any time at our discretion without any penalty or cost to us.
Exercise of options or warrants or conversion of convertible securities may have a dilutive effect on your percentage ownership and may result in a dilution of your voting power and an increase in the number of shares of common stock eligible for future resale in the public market, which may negatively impact the trading price of our shares of common stock.
The exercise or conversion of some or all of our outstanding options, warrants, or convertible securities could result in significant dilution in the percentage ownership interest of investors in this offering and in the percentage ownership interest of our existing common stockholders and in a significant dilution of voting rights and earnings per share.
Additionally, the issuance of shares of our common stock upon exercise of stock options outstanding under our stock incentive plans will further dilute our stockholders’ voting interests. To the extent options and/or warrants and/or conversion rights are exercised, additional shares of common stock will be issued, and such issuance will dilute stockholders.
Our executive officers, directors and principal stockholders maintain the ability to exert substantial influence over all matters submitted to stockholders for approval.
Our executive officers, directors and principal stockholders beneficially own shares representing approximately 28.5% of our outstanding capital stock. As a result, if these stockholders were to choose to act together, they would be able to exert substantial influence over all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these persons, if they choose to act together, would exert substantial influence over the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of voting power could delay or prevent an acquisition of our Company on terms that other stockholders may desire.
We are an “emerging growth company” and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.” We will remain an emerging growth company until the earlier of (i) the last day of the fiscal year (a) following the fifth anniversary of the date we completed our initial public offering, which was June 29, 2015, (b) in which we have total annual gross revenue of at least $1.07 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, or (ii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
| 44 |
Our failure to meet the continued listing requirements of NASDAQ could result in a de-listing of our common stock.
If we fail to satisfy the continued listing requirements of NASDAQ, such as the corporate governance requirements or the minimum closing bid price requirement, NASDAQ may take steps to de-list our common stock. Such a de-listing would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a de-listing, we would take actions to restore our compliance with NASDAQ’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the NASDAQ minimum bid price requirement or prevent future non-compliance with NASDAQ’s listing requirements.
On June 7, 2017, we received a notice from NASDAQ that, because the closing bid price of our common stock has been below $1.00 per share for 30 consecutive business days, it no longer complies with the minimum bid price requirement for continued listing on The NASDAQ Capital Market. NASDAQ Listing Rule 550(a)(2) requires listed securities to maintain a minimum bid price of $1.00 per share, and Listing Rule 5810(c)(3)(A) provides that a failure to meet the minimum bid price requirement exists if the deficiency continues for a period of 30 consecutive business days.
On November 2, 2017, our board of directors approved a reverse stock split of our outstanding shares of common stock at a ratio within a range of 1-for-8 to 1-for-15, to be determined by the board of directors at a later date, and subject to stockholder approval. At a special meeting of stockholders held on December 20, 2017, our stockholders approved the reverse stock split within a range of 1-for-8 to 1-for-15. On March 1, 2018, our board of directors approved a 1-for-10 reverse stock split, with an anticipated effective date of on or before March 23, 2018.
We have until June 4, 2018 to regain compliance with the minimum bid price requirement. During the compliance period, our shares of common stock will continue to be listed and traded on The Nasdaq Capital Market. To regain compliance, the closing bid price of our common stock must meet or exceed $1.00 per share for a minimum of 10 consecutive business days during the 180 calendar day grace period.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control of our company that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions provide that:
| ● | the authorized number of directors can be changed only by resolution of our board of directors; | |
| ● | our bylaws may be amended or repealed by our board of directors or our stockholders; | |
| ● | stockholders may not call special meetings of the stockholders or fill vacancies on the board of directors; | |
| ● | our board of directors is authorized to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; | |
| ● | our stockholders do not have cumulative voting rights, and therefore our stockholders holding a majority of the shares of common stock outstanding will be able to elect all of our directors; and | |
| ● | our stockholders must comply with advance notice provisions to bring business before or nominate directors for election at a stockholder meeting. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law (the “DGCL”), which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
| 45 |
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful stockholder claims against us and may reduce the amount of money available to us.
As permitted by Section 102(b)(7) of the DGCL, our amended and restated certificate of incorporation limits the liability of our directors to the fullest extent permitted by law. In addition, as permitted by Section 145 of the DGCL, our amended and restated certificate of incorporation and our amended and restated bylaws provide that we shall indemnify, to the fullest extent authorized by the DGCL, each person who is involved in any litigation or other proceeding because such person is or was a director or officer of our company or is or was serving as an officer or director of another entity at our request, against all expense, loss or liability reasonably incurred or suffered in connection therewith. Our amended and restated certificate of incorporation provides that the right to indemnification includes the right to be paid expenses incurred in defending any proceeding in advance of its final disposition, provided, however, that such advance payment will only be made upon delivery to us of an undertaking, by or on behalf of the director or officer, to repay all amounts so advanced if it is ultimately determined that such director is not entitled to indemnification. If we do not pay a proper claim for indemnification in full within 60 days after we receive a written claim for such indemnification, except in the case of a claim for an advancement of expenses, in which case such period is 20 days, our restated certificate of incorporation and our restated bylaws authorize the claimant to bring an action against us and prescribe what constitutes a defense to such action.
Section 145 of the DGCL permits a corporation to indemnify any director or officer of the corporation against expenses (including attorney’s fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person acted in good faith and in a manner that he reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, if he or she had no reason to believe his or her conduct was unlawful. In a derivative action, (i.e., one brought by or on behalf of the corporation), indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or suit if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the court in which the action or suit was brought shall determine that the defendant is fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
The rights conferred in the restated certificate of incorporation and the restated bylaws are not exclusive, and we are authorized to enter into indemnification agreements with our directors, officers, employees and agents and to obtain insurance to indemnify such persons. We have entered into indemnification agreements with each of our officers and directors.
The above limitations on liability and our indemnification obligations limit the personal liability of our directors and officers for monetary damages for breach of their fiduciary duty as directors by shifting the burden of such losses and expenses to us. Although we plan to increase the coverage under our directors’ and officers’ liability insurance, certain liabilities or expenses covered by our indemnification obligations may not be covered by such insurance or the coverage limitation amounts may be exceeded. As a result, we may need to use a significant amount of our funds to satisfy our indemnification obligations, which could severely harm our business and financial condition and limit the funds available to stockholders who may choose to bring a claim against our company.
We have never paid dividends on our common stock and do not anticipate paying dividends for the foreseeable future, and accordingly, stockholders must rely on stock appreciation for any return on their investment.
We have never paid dividends on our common stock and we do not anticipate paying dividends on our common stock for the foreseeable future. Accordingly, any return on an investment in our common stock will be realized, if at all, only when stockholders sell their shares. In addition, our failure to pay dividends may make our stock less attractive to investors, adversely impacting trading volume and price.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As of December 31, 2017, we had federal net operating loss carryforwards (“NOLs”) of approximately $39.0 million which begin to expire in 2028. Our ability to utilize our NOLs may be limited under Section 382 and 383 of the Internal Revenue Code. The limitations apply if an ownership change, as defined by Section 382, occurs. Generally, an ownership change occurs when certain shareholders increase their aggregate ownership by more than 50 percentage points over their lowest ownership percentage in a testing period (typically three years). Although we have not undergone a Section 382 analysis, it is possible that the utilization of the NOLs, could be substantially limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be utilized against future taxes. As a result, we may not be able to take full advantage of these carryforwards for federal and state tax purposes. Future changes in stock ownership may also trigger an ownership change and, consequently, a Section 382 limitation.
| 46 |
Item 1B. Unresolved Staff Comments
None.
On July 9, 2015, we entered into a new lease with Century Park, pursuant to which we are leasing approximately 2,780 square feet of office space in Los Angeles, California for our headquarters. The lease provides for a term of sixty-one (61) months, which commenced October 1, 2015. We pay $9,733 per month in base rent with normal escalations until November 2018, after which our monthly payment will increase ratably per year pursuant to the terms of the lease agreement. We have the option to extend the term of the lease for one five-year term, provided that the rent would be subject to market adjustment at the beginning of the renewal term. We believe that our facility is suitable and adequate for our current needs.
From time to time, we have been and may again become involved in legal proceedings arising in the ordinary course of our business. We are not presently a party to any material litigation.
Item 4. Mine Safety Disclosures
Not Applicable.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our shares of common stock have been listed and traded on The NASDAQ Capital Market under the symbol “RTTR” since June 24, 2015. Prior to that date, there was no public market for our common stock.
The following table sets forth, for the periods indicated, the high and low intra-day sale prices in dollars on The NASDAQ Capital Market for our common stock.
| 2017 | ||||||||
| High | Low | |||||||
| First Quarter | $ | 3.75 | $ | 1.29 | ||||
| Second Quarter | $ | 1.46 | $ | 0.52 | ||||
| Third Quarter | $ | 0.68 | $ | 0.34 | ||||
| Fourth Quarter | $ | 0.42 | $ | 0.28 | ||||
| 2016 | ||||||||
| High | Low | |||||||
| First Quarter | $ | 1.79 | $ | 0.98 | ||||
| Second Quarter | $ | 1.89 | $ | 1.10 | ||||
| Third Quarter | $ | 2.47 | $ | 1.20 | ||||
| Fourth Quarter | $ | 3.26 | $ | 1.62 | ||||
Holders
As of March 16, 2018 there were approximately 36 registered holders of record of our common stock. These figures do not reflect the beneficial ownership or shares held in nominee name.
Dividend Policy
We have never paid or declared any cash dividends on our common stock, and we do not anticipate paying any cash dividends on our common stock in the foreseeable future. We intend to retain all available funds and any future earnings to fund the development and expansion of our business. Any future determination to pay dividends will be at the discretion of our board of directors and will depend upon a number of factors, including our results of operations, financial condition, future prospects, contractual restrictions, restrictions imposed by applicable law and other factors our board of directors deems relevant. See “Risk Factors — Risks Relating to Our Common Stock— We have never paid dividends on our common stock and do not anticipate paying dividends for the foreseeable future, and accordingly, stockholders must rely on stock appreciation for any return on their investment.”
| 47 |
Securities Authorized for Issuance Under Equity Compensation Plans
See Part III, Item 12. “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” for information relating to our equity compensation plans.
Recent Sales of Unregistered Securities
On May 4, 2017, we entered into a common stock purchase agreement with Aspire Capital (the “2017 Aspire Purchase Agreement”), which provides that upon the terms and conditions set forth therein, Aspire Capital is committed to purchase up to an aggregate of $6.5 million of shares of our common stock over the 30-month term of the 2017 Aspire Purchase Agreement. On any trading day on which the closing sale price of our common stock exceeds $0.25, we have the right, in our sole discretion, to present Aspire Capital with a purchase notice, directing Aspire Capital (as principal) to purchase up to 100,000 shares of our common stock per trading day, for up to $6.5 million of our common stock in the aggregate at a per share price, calculated by reference to the prevailing market price of our common stock (as provided in the 2017 Aspire Purchase Agreement). As of the date of this Annual Report, no shares of common stock have been sold to Aspire Capital under the 2017 Aspire Purchase Agreement.
As a condition to the 2017 Aspire Purchase Agreement, we issued 137,324 shares of our common stock to Aspire Capital as a commitment fee (the “Commitment Shares”). The issuance of the Commitment Shares were exempt from registration under the Securities Act of 1933, as amended (the “Securities Act”), pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(a)(2) of the Securities Act.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
Item 6. Selected Financial Data
Not applicable.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis should be read together with our financial statements and the related notes appearing elsewhere in this Annual Report. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. See “Special Note Regarding Forward-Looking Statements and Industry Data” for a discussion of the uncertainties, risks and assumptions associated with these statements. Actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under “Risk Factors” and elsewhere in this Annual Report.
Overview
Ritter Pharmaceuticals, Inc. develops novel therapeutic products that modulate the gut microbiome to treat gastrointestinal diseases. We are advancing gut health research by exploring the metabolic capacity of the gut microbiota and translating the functionality of prebiotic-based therapeutics into applications intended to have a meaningful impact on a patient’s health.
We completed a Phase 2a clinical trial of our leading product candidate, RP-G28, an orally administered, high purity oligosaccharide in November 2011.
We completed a Phase 2b multi-center, randomized, double-blind, placebo-controlled, parallel group trial of RP-G28 in October 2016. Topline results of the trial were announced in March 2017. Due to inconsistent data results from one study site, the data from this site was excluded from the primary analysis population (Efficacy Subset mITT). After excluding the data from the one anomalous study site, results showed a clinically meaningful benefit to subjects in the reduction of lactose intolerance symptoms across a variety of outcome measures. The majority of analyses showed positive outcome measures and the robustness of the data point to a clear drug effect. Treatment patients not only reported meaningful reduced symptoms, but also 30 days after taking the treatment, patients reported adequate relief from lactose intolerance symptoms and satisfaction with the results of the treatment, with RP-G28 preventing or treating their lactose intolerance symptoms. Greater milk and dairy product consumption was also reported by patients.
| 48 |
A subset of subjects from our Phase 2b clinical trial has been rolled into a 12-month extension study to evaluate long-term durability of treatment. The study is also evaluating each participant’s microbiome, expanding our knowledge of the effects that RP-G28 may have on adapting the gut microbiota in a beneficial manner. We completed this study in the fourth quarter of 2017.
We held an End-of-Phase 2 meeting with the FDA’s Division of Gastroenterology and Inborn Errors Products in August 2017. The purpose of the meeting was to obtain the FDA’s feedback on our Phase 3 program. We reached general consensus with the FDA on certain elements of our current Phase 3 program and have received clear guidance and recommendations on many necessary components of our Phase 3 program; including the clinical, non-clinical, and chemistry, manufacturing and controls (CMC) requirements needed to support an NDA submission.
We have incorporated much of this guidance into our Phase 3 program. Our current Phase 3 clinical program will consist of two confirmatory clinical trials of similar trial design and size as our Phase 2b clinical trial and will include additional components that may allow for claims for durability of effect. These additional trials may be run in parallel.
We have devoted substantially all of our resources to development efforts relating to RP-G28, including conducting clinical trials of RP-G28, providing general and administrative support for these operations and protecting our intellectual property. We currently do not have any products approved for sale and we have not generated any revenue from product sales since our inception.
We have incurred net losses in each year since our inception, including net losses of approximately $7.9 million for the year ended December 31, 2017. We had an accumulated deficit of approximately $53.3 million as of December 31, 2017. Substantially all our net losses resulted from costs incurred in connection with our research and development programs, stock-based compensation, and from general and administrative costs associated with our operations.
We expect to continue to incur significant expenses and increasing operating losses for at least the next several years. We anticipate that our expenses will increase substantially as we:
| ● | complete the development of our lead product candidate, RP-G28, for the reduction of symptoms associated with lactose intolerance in patients; | |
| ● | seek to obtain regulatory approvals for RP-G28; | |
| ● | outsource the commercial manufacturing of RP-G28 for any indications for which we receive regulatory approval; | |
| ● | contract with third parties for the sales, marketing and distribution of RP-G28 for any indications for which we receive regulatory approval; | |
| ● | maintain, expand and protect our intellectual property portfolio; | |
| ● | continue our research and development efforts; and | |
| ● | add operational, financial and management information systems and personnel, including personnel to support our product development and commercialization efforts. |
We do not expect to generate revenue from product sales unless and until we successfully complete development and obtain marketing approval for one or more of our product candidates, which we expect will take a number of years and is subject to significant uncertainty. Accordingly, we anticipate that we will need to raise additional capital prior to the commercialization of RP-G28. Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our operating activities through a combination of equity offerings (including shares sold to Aspire Capital pursuant to the 2017 Aspire Purchase Agreement), debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. However, we may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms or at all. Our failure to raise capital or enter into such other arrangements as and when needed would have a negative impact on our financial condition and our ability to develop our product candidates.
Financial Overview
Revenue
We have not generated any revenue since our inception. Our ability to generate revenue in the future will depend almost entirely on our ability to successfully develop, obtain regulatory approval for and then successfully commercialize RP-G28 in the United States. In the event we choose to pursue a partnering arrangement to commercialize RP-G28 or other products outside the United States, we would expect to initiate additional research and development and clinical trial activities in the future.
| 49 |
Research and Development Expenses
Since our inception, we have focused our resources on our research and development activities, including conducting nonclinical studies and clinical trials, manufacturing development efforts and activities related to regulatory filings for RP-G28. Our research and development expenses consist primarily of:
| ● | fees paid to consultants and CROs, including in connection with our nonclinical and clinical trials, and other related clinical trial fees, such as for investigator grants, patient screening, laboratory work, clinical trial database management, clinical trial material management and statistical compilation and analysis; | |
| ● | costs related to acquiring and manufacturing clinical trial materials; | |
| ● | depreciation of equipment, computers and furniture and fixtures; | |
| ● | costs related to compliance with regulatory requirements; and | |
| ● | overhead expenses for personnel in research and development functions. |
From inception through December 31, 2017, we have incurred approximately $22.1 million in research and development expenses. We plan to increase our research and development expenses for the foreseeable future as we continue the development of RP-G28 for the reduction of symptoms associated with lactose intolerance in patients and other indications, subject to the availability of additional funding.
The successful development of RP-G28 is highly uncertain. At this time, we cannot reasonably estimate the nature, timing or costs of the efforts that will be necessary to complete the development of RP-G28 or when, if ever, net cash inflows from RP-G28 may commence. This is due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
| ● | the scope, rate of progress and expense of our ongoing, as well as any additional, clinical trials and other research and development activities; | |
| ● | future clinical trial results; and | |
| ● | the timing and receipt of any regulatory approvals. |
For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of the clinical development of RP-G28 or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
Patent Costs
Patent costs consist primarily of professional fees for legal services to prosecute patents and maintain patent rights.
General and Administrative Expenses
General and administrative expenses include allocation of facilities costs, salaries, benefits, and stock-based compensation for employees, professional fees for directors, fees for independent contractors and accounting and legal services.
We expect that our general and administrative expenses will increase if RP-G28 is approved for commercialization. We believe that these increases will likely include increased costs for director and officer liability insurance, and increased fees for outside consultants, lawyers and accountants, among other expenses.
Interest Income and Interest Expense
Interest income consists of interest earned on our cash.
| 50 |
Critical Accounting Policies and Estimates
This discussion and analysis is based on our financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”). The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to fair value of financial instruments, research and development costs, accrued expenses and stock-based compensation. We base our estimates on historical experience, known trends and events and various other factors we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 3 to our financial statements appearing in “Item 8. Financial Statements and Supplementary Data,” we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our financial condition and results of operations.
Fair Value of Financial Instruments
Fair value measurement guidelines are prescribed by GAAP to value financial instruments. The guidance includes a definition of fair value, prescribes methods for measuring fair value, establishes a fair value hierarchy based on the inputs used to measure fair value and expands disclosures about the use of fair value measurements.
The valuation techniques utilized are based upon observable and unobservable inputs. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect internal market assumptions. Assets are classified in their entirety based on the lowest level of input that is significant to the fair value measurement.
These two types of inputs create the following fair value hierarchy:
Level 1 — Quoted prices in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date. Level 1 primarily consists of financial instruments whose value is based on quoted market prices such as exchange-traded instruments and listed equities.
Level 2 — Inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly. Level 2 includes financial instruments that are valued using models or other valuation methodologies. These models consider various assumptions, including volatility factors, current market prices and contractual prices for the underlying financial instruments. Substantially all of these assumptions are observable in the marketplace, can be derived from observable data or are supported by observable levels at which transactions are executed in the marketplace.
Level 3 — Unobservable inputs for the asset or liability. Financial instruments are considered Level 3 when their fair values are determined using pricing models, discounted cash flows or similar techniques and at least one significant model assumption or input is unobservable
The carrying amounts reported in the balance sheet for cash and cash equivalents, prepaid expenses, accounts payable, accrued expenses, and the notes payable approximate the fair values due to the short-term nature of the instruments.
Research and Development Costs
We expense the cost of research and development as incurred. Research and development expenses comprise costs incurred in performing research and development activities, including clinical study costs, contracted services, and other external costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity is performed or when the goods have been received, rather than when payment is made, in accordance with ASC 730, Research and Development.
Accrued Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued expenses. This process involves reviewing quotations and contracts, identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. The significant estimates in our accrued research and development expenses include fees due to service providers.
| 51 |
We base our expenses on our estimates of the services received and efforts expended pursuant to quotes and contracts with our service providers that conduct research and development on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the research and development expense. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and could result in us reporting amounts that are too high or too low in any particular period.
Stock-Based Compensation
Stock-based compensation cost for equity awards granted to employees and nonemployees is measured at the grant date based on the calculated fair value of the award using the Black-Scholes option-pricing model, and is recognized as an expense, under the straight-line method, over the requisite service period (generally the vesting period of the equity grant). If we determine that other methods are more reasonable, or other methods for calculating these assumptions are prescribed by regulators, the fair value calculated for our stock options could change significantly. Higher volatility and longer expected lives would result in an increase to stock-based compensation expense to non-employees determined at the date of grant.
In addition to the assumptions used in the Black-Scholes option-pricing model, we also estimate a forfeiture rate to calculate the stock-based compensation for our equity awards. We will continue to use judgment in evaluating the expected volatility, expected terms and forfeiture rates utilized for our stock-based compensation calculations on a prospective basis.
Emerging Growth Company Status
On April 5, 2012, the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”) was enacted. Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended (the Securities Act), for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates.
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company,” we intend to rely on certain of these exemptions, including without limitation, (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (ii) complying with any requirement that may be adopted by the PCAOB regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an emerging growth company until the earlier of (i) the last day of the fiscal year (a) following the fifth anniversary of the date we completed our initial public offering, which was June 29, 2015, (b) in which we have total annual gross revenue of at least $1.07 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, and (ii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
| 52 |
Results of Operations
Comparison of the years ended December 31, 2017 and 2016
The following table summarizes the results of our operations for the years ended December 31, 2017 and 2016, together with the changes in those items in dollars and as a percentage:
| For
the Years Ended December 31, | Dollar | Percentage | ||||||||||||||
| 2017 | 2016 | Change | Change | |||||||||||||
| Statement of Operations Data: | ||||||||||||||||
| Operating costs and expenses | ||||||||||||||||
| Research and development | $ | 2,874,184 | $ | 13,292,488 | $ | (10,418,304 | ) | (78 | )% | |||||||
| Patent costs | 250,372 | 272,514 | (22,142 | ) | (8 | )% | ||||||||||
| General and administrative | 4,777,902 | 4,881,725 | (103,823 | ) | (2 | )% | ||||||||||
| Total operating costs and expenses | 7,902,458 | 18,446,727 | (10,544,269 | ) | (57 | )% | ||||||||||
| Loss from operations | (7,902,458 | ) | (18,446,727 | ) | (10,544,269 | ) | (57 | )% | ||||||||
| Other income | ||||||||||||||||
| Interest income | 40,227 | 60,879 | (20,652 | ) | (34 | )% | ||||||||||
| Other (expense) income | (1,627 | ) | 1,214 | (2,841 | ) | (134 | )% | |||||||||
| Total other income | 38,600 | 62,093 | (23,493 | ) | (38 | )% | ||||||||||
| Net loss applicable to common stockholders | $ | (7,863,858 | ) | $ | (18,384,634 | ) | $ | (10,520,776 | ) | (57 | )% | |||||
Research and Development Expenses
Research and development expenses decreased by approximately $10.4 million, or (78%), for the year ended December 31, 2017 as compared to the year ended December 31, 2016. The primary reason for the decrease is that our Phase 2b clinical trial, which was initiated in March 2016, was completed during the fourth quarter of 2016. A decrease of approximately ($11.5) million attributable to fees paid to our third-party CRO for the year ended December 31, 2016 as part of our Phase 2b clinical trial was offset by approximately $0.7 million spent on regulatory consulting during the year ended December 31, 2017 in preparation for our Phase 3 clinical study as well as $0.4 million in fees associated with our extension study to evaluate the long-term durability of treatment.
Patent Costs
Patent costs were approximately in $250,000 and $273,000 for the years ended December 31, 2017 and 2016, respectively, representing a decrease of approximately $22,000, or (8%). The primary reason for the decrease is that our costs and expenses related to the maintenance of patent rights, the prosecution of patents, the application for the issuance of patents, as well as the preparation to file national phase applications in certain foreign countries was higher in 2016 than 2017.
General and Administrative Expenses
General and administrative expenses decreased by approximately $104,000, or (2%), for the year ended December 31, 2017 as compared to the year ended December 31, 2016. The decrease in general and administrative expenses was mainly due to a decrease in stock-based compensation expense of approximately $0.5 million during the year ended December 31, 2017 partially offset by an increase in professional fees of approximately $0.3 million.
Other Income
Interest income decreased by $21,000, or (34%) during the year ended December 31, 2017 as compared to the year ended December 31, 2016, primarily due to use of cash to complete the Phase 2b study in 2016. This was partially offset by the interest earned in the fourth quarter of 2017 from the proceeds of our follow-on offering (described below).
Liquidity and Capital Resources
Sources of Liquidity
Since our inception, we have incurred net losses and negative cash flows from operations, and, as of December 31, 2017, we had an accumulated deficit of approximately $53.3 million. Substantially all our net losses resulted from costs incurred in connection with our research and development programs, stock-based compensation, and from general and administrative costs associated with our operations.
At December 31, 2017, we had working capital of $20.1 million, and cash of $22.6 million. We have not generated any product revenues and have not achieved profitable operations.
| 53 |
Aspire Capital Financing Arrangement
On May 4, 2017, we entered into a common stock purchase agreement with Aspire Capital. The 2017 Aspire Purchase Agreement provides access to us of up to an aggregate of $6.5 million through the sale of shares of our common stock, over a 30-month period. In consideration for entering into the 2017 Aspire Purchase Agreement, we issued to Aspire Capital 137,324 shares of our common stock with an aggregate dollar value equal to $97,500.
Under the 2017 Aspire Purchase Agreement, on any trading day we select, we have the right, in our sole discretion, to present Aspire Capital with a purchase notice (each, a “Purchase Notice”), directing Aspire Capital (as principal) to purchase up to 100,000 shares of our common stock per trading day (which may be increased by as much as an additional 2,000,000 shares per trading day by mutual agreement), up to an aggregate of $6.5 million of our common stock, at a per share price (the “Purchase Price”) equal to the lesser of:
| ● | the lowest sale price of our common stock on the sale date; or | |
| ● | the arithmetic average of the three lowest closing sale prices for our common stock during the ten (10) consecutive trading days ending on the trading day immediately preceding the sale date. |
The aggregate purchase price payable by Aspire Capital on any one purchase date may not exceed $500,000, unless otherwise mutually agreed.
In addition, on any date on which we submit a Purchase Notice to Aspire Capital in an amount equal to 100,000 shares and our stock price is not less than $0.25 per share, we may also, in our sole discretion, present Aspire Capital with a volume-weighted average price purchase notice (each, a “VWAP Purchase Notice”) directing Aspire Capital to purchase an amount of our common stock equal to up to 30% of the aggregate shares of our common stock traded on its principal market on the next trading day (the “VWAP Purchase Date”), as determined by us. The purchase price per share pursuant to such VWAP Purchase Notice is generally 97% of the volume-weighted average price for our common stock traded on its principal market on the VWAP Purchase Date.
We may deliver multiple Purchase Notices and VWAP Purchase Notices to Aspire Capital from time to time during the term of the 2017 Aspire Purchase Agreement, so long as the most recent purchase has been completed.
The Purchase Agreement provides that we may not effect any sales under the 2017 Aspire Purchase Agreement on any date where the closing sale price of our common stock is less than $0.25. There are no trading volume requirements or restrictions under the 2017 Aspire Purchase Agreement, and we control the timing and amount of sales of our common stock to Aspire Capital.
The 2017 Aspire Purchase Agreement provides that the number of shares that may be sold pursuant to Aspire Capital will be limited to 2,842,417 (the “Exchange Cap”), which represents 19.99% of our outstanding shares of common stock as of May 2, 2017, unless stockholder approval or an exception pursuant to the rules of the NASDAQ Capital Market is obtained to issue more than 19.99%. This limitation will not apply if, at any time the Exchange Cap is reached and at all times thereafter, the average price paid for all shares issued under the 2017 Aspire Purchase Agreement is equal to or greater than $0.68, which was the consolidated closing bid price of our common stock on May 4, 2017. We are not required or permitted to issue any shares of common stock under the 2017 Aspire Purchase Agreement if such issuance would breach our obligations under the rules or regulations of the NASDAQ Capital Market.
We expect to use the Aspire facility to complement, rather than replace, other financing that may be required during the next twelve months to continue our operations and support our capital needs.
October 2016 Public Offering
On October 31, 2016, we closed a public offering of 2,127,660 shares of our common stock at a price to the public of $2.35 per share, for net proceeds of approximately $4.4 million, after deducting underwriting discounts and commissions and offering expenses payable by us. The offering was made pursuant to a shelf registration statement on Form S-3 (Registration Number 333-213087).
October 2017 Public Offering
On October 3, 2017, we closed a public offering of (i) 34,550,000 Class A Units consisting of 34,550,000 shares of our common stock and warrants to purchase 34,550,000 shares of our common stock, at a public offering price of $0.40 per unit, and (ii) 9,180 Class B Units consisting of 9,180 shares of our Series A Convertible Preferred stock, with a stated value of $1,000, and convertible into an aggregate of 22,950,000 shares of our common stock, and warrants to purchase 22,950,000 shares of our common stock, at a public offering price of $1,000 per unit. We received approximately $21.0 million in net proceeds from this offering, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. The offering was made pursuant to a shelf registration statement on Form S-3 (Registration Number 333-219147).
| 54 |
Cash Flows
The following table sets forth the significant sources and uses of cash for the periods set forth below:
| For
the Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Net cash (used in) provided by: | ||||||||
| Operating activities | $ | (7,397,912 | ) | $ | (15,208,718 | ) | ||
| Investing activities | (7,678 | ) | (8,063 | ) | ||||
| Financing activities | 22,991,279 | 6,443,497 | ||||||
| Net (decrease) increase in cash | $ | 15,585,689 | $ | (8,773,284 | ) | |||
Operating Activities
The use of cash in both periods resulted primarily from our net losses adjusted for non-cash charges and changes in components of working capital. Net cash used in operating activities was approximately $7.4 million during the year ended December 31, 2017 compared to $15.2 million during the year ended December 31, 2016. The decrease in cash used in operating activities was driven primarily by the completion of the Phase 2b clinical trial in October 2016 with 368 patients at 18 clinical sites.
Investing Activities
Net cash used in investing activities of was approximately $8,000 during the years ended December 31, 2017 and 2016. These expenditures were for purchases of furniture and equipment used in the business.
Financing Activities
Net cash provided by financing activities was approximately $23.0 million during the year ended December 31, 2017 compared to $6.4 million during the year ended December 31, 2016. During the 2017 year, we received proceeds from the October 2017 public offering. The cash provided by financing activities in 2016 was from the October 2016 public offering and the issuance of shares to Aspire Capital.
Future Funding Requirements
To date, we have not generated any revenue. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize RP-G28 or any of our other product candidates. At the same time, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval for, RP-G28. In addition, subject to obtaining regulatory approval of RP-G28, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We anticipate that we will need substantial additional funding in connection with our continuing operations.
Based upon our current operating plan, we believe that our existing cash and cash equivalents, together with interest and any proceeds received from our sale of shares of common stock to Aspire Capital in the future pursuant to the 2017 Aspire Purchase Agreement, will enable us to fund our operating expenses and capital expenditure requirements through 2018.
Our future capital requirements will depend on many factors, including:
| ● | the ability of RP-G28 and any other product candidate that we may develop in the future to progress through clinical development successfully; | |
| ● | the outcome, costs and timing of seeking and obtaining FDA approval; | |
| ● | the willingness of the EMA or other regulatory agencies outside the United States to accept our Phase 3 trials of RP-G28, as well as our other completed and planned clinical and nonclinical studies and other work, as the basis for review and approval of RP-G28 in the European Union for the reduction of symptoms associated with lactose intolerance in patients; | |
| ● | our need to expand our research and development activities; |
| 55 |
| ● | the costs associated with securing and establishing commercialization and manufacturing capabilities; | |
| ● | market acceptance of RP-G28 and any other product candidate that we may develop in the future; | |
| ● | the costs of acquiring, licensing or investing in businesses, products, product candidates and technologies; | |
| ● | our ability to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; | |
| ● | our need and ability to hire additional management and scientific and medical personnel; | |
| ● | the effect of competing technological and market developments; | |
| ● | our need to implement additional internal systems and infrastructure, including financial and reporting systems; | |
| ● | the economic and other terms, timing of and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future; and | |
| ● | the costs of operating as a public company. |
Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our cash needs through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our common stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, commercialization, marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
Contractual Obligations and Commitments
Master Services Agreement
On December 30, 2015, we entered into a Master Service Agreement with Covance, with an effective date of December 29, 2015. Pursuant to the terms of the Master Service Agreement, Covance (or one or more of its affiliates) will provide Phase 1, 2, 3, and 4 clinical services for a clinical study or studies to us, and, at our request, assist us with the design of such studies, in accordance with the terms of separate individual project agreements to be entered into by the parties. The term of the agreement is for three years and will renew automatically for successive one year periods unless Covance is no longer providing services under the agreement or either party has terminated the agreement upon written notice. We may terminate the Master Service Agreement or any individual project agreement entered into under the Master Service Agreement prior to the applicable study’s completion at any time for any reason upon 30 days written notice to Covance, except when the reason for termination is the safety of subjects, in which case it may be terminated immediately. Covance may not terminate any individual project agreement without cause, except when the reason for the termination is the safety of subjects, in which case it may be terminated immediately. In the event of a termination of the Master Service Agreement, Covance will be entitled to full payment for (i) work performed on the applicable project upon through the date work on such project is concluded and (ii) reimbursement for all non-cancellable and non-refundable expenses and financial obligations which Covance (or an affiliate) has incurred or undertaken on our behalf.
Clinical Supply and Cooperation Agreement with Ricerche Sperimentali Montale (“Ricerche”) and Inalco SpA (“Inalco”)
Under the terms of the Supply Agreement with RSM, we are required to pay RSM $400,000 within 10 days following FDA approval of a NDA for the first product owned or controlled by us using Improved GOS as its active pharmaceutical ingredient.
Lease Agreement
We lease office space for our headquarters in California. The lease provides for a term of sixty-one (61) months, commencing on October 1, 2015. We pay $9,733 per month in base rent until November 2018, after which our monthly payment will increase ratably per year pursuant to the terms of the lease agreement. The base rent payments do not include our proportionate share of any operating expenses, including real estate taxes. We have the option to extend the term of the lease for one five-year term, provided that the rent would be subject to market adjustment at the beginning of the renewal term. We will recognize rent expense on a straight-line basis over the lease term.
| 56 |
Rent expense, recognized on a straight-line basis, was approximately $115,000 for the years ended December 31, 2017 and 2016, respectively, and is recorded in general and administrative expenses in the accompanying statement of operations.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements as defined under SEC rules.
Recent Accounting Pronouncements
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update 2016-02, Leases (Topic 842) (“ASU 2016-02”). The provisions of ASU 2016-02 set out the principles for the recognition, measurement, presentation and disclosure of leases for both parties to a contract (i.e., lessees and lessors). The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether the lease expense is recognized based on an effective interest method or on a straight line basis over the term of the lease. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than 12 months regardless of their classification. Leases with a term of 12 months or less will be accounted for under the existing guidance for operating leases today. Topic 842 supersedes the previous lease standard, Topic 840 Leases. The guidance is effective for annual periods and interim periods within those annual periods beginning after December 15, 2018, and is effective for the Company for the year ending December 31, 2019. The Company is currently evaluating the impact that the implementation of this standard will have on the Company’s financial statements.
On March 30, 2016, the FASB issued Accounting Standards Update No. 2016-09, Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting (“ASU 2016-09”). Among other things, ASU 2016-09 requires that entities recognize excess tax benefits and deficiencies related to employee share-based payment transactions as income tax expense or benefit. ASU 2016-09 also eliminates the requirement to reclassify excess tax benefits and deficiencies from operating activities to financing activities in the statement of cash flows. The guidance is effective for the annual periods and interim periods within those annual periods beginning after December 15, 2016. The adoption of this standard did not have a material impact on the Company’s financial statements.
On August 26, 2016, the FASB issued Accounting Standards Update No. 2016-15, Statement of Cash Flows (Topic 230), a consensus of the FASB’s Emerging Issues Task Force (“ASU 2016-15”). The new guidance amends Accounting Standards Codification No. 230 (“ASC 230”) to add or clarify guidance on the classification of certain cash receipts and payments in the statement of cash flows. ASC 230 lacks consistent principles for evaluating the classification of cash payments and receipts in the statement of cash flows. This has led to diversity in practice and, in certain circumstances, financial statement restatements. Therefore, the FASB issued the ASU 2016-15 with the intent of reducing diversity in practice with respect to eight types of cash flows. ASU 2016-15 is effective for annual and interim periods in fiscal years beginning after December 15, 2017, and is effective for the Company for the year ending December 31, 2018. The Company is currently evaluating the impact that the implementation of this standard will have on the Company’s financial statements.
In May 2017, the FASB issued Accounting Standards Update No. 2017-09, Compensation - Stock Compensation (Topic 718): Scope of Modification Accounting (“ASU 2017-09”). ASU 2017-09 provides guidance on the types of changes to the terms or conditions of share-based payment awards to which an entity would be required to apply modification accounting. An entity would not apply modification accounting if the fair value, vesting conditions, and classification of the awards are the same immediately before and after the modification. The amendments are effective for the Company’s interim and annual reporting periods beginning January 1, 2018. The Company does not expect the adoption of ASU 2017-09 to have a material impact on its financial statements.
In July 2017, the FASB issued ASU 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception. The ASU allows companies to exclude a down round feature when determining whether a financial instrument (or embedded conversion feature) is considered indexed to the entity’s own stock. As a result, financial instruments (or embedded conversion features) with down round features may no longer be required to be accounted classified as liabilities. A company will recognize the value of a down round feature only when it is triggered and the strike price has been adjusted downward. For equity-classified freestanding financial instruments, such as warrants, an entity will treat the value of the effect of the down round, when triggered, as a dividend and a reduction of income available to common shareholders in computing basic earnings per share. For convertible instruments with embedded conversion features containing down round provisions, entities will recognize the value of the down round as a beneficial conversion discount to be amortized to earnings. The guidance in ASU 2017-11 is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted, and the guidance is to be applied using a full or modified retrospective approach. The Company adopted this guidance in the current quarter, effective October 1, 2017. As a result, the warrants issued on October 3, in connection with the October 2017 Public Offering, were equity-classified.
| 57 |
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Not applicable.
Item 8. Financial Statements and Supplementary Data.
The financial statements and the reports of our independent registered accounting firm required pursuant to this item are included in Item 15 of this report and are presented beginning on page F-1.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
There has been no change of accountants nor any disagreements with accountants on any matter of accounting principles or practices or financial disclosure required to be reported under this Item.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer (our principal executive officer) and vice president finance (our principal financial officer), evaluated the effectiveness of our disclosures controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act of 1934, as amended (the “Exchange Act”), as of December 31, 2017. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate, to allow timely decisions regarding required disclosure.
Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2017, our principal executive officer and principal financial officer concluded that, as of such date, our disclosure controls and procedures were effective at a reasonable assurance level.
Management’s Annual Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our chief executive officer and vice president finance, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes those policies and procedures that: (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide a reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitation. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgement and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
| 58 |
Management is responsible for establishing and maintaining adequate internal control over our financial reporting, as such term is defined in Rule 13a-15(f) under the Exchange Act. Under the supervision and with the participation of our management, including our chief executive officer and vice president finance, we conducted an evaluation of the effectiveness of our internal control over financial reporting. Management has used the framework set forth in the report entitled “Internal Control – Integrated Framework” published by the Committee of Sponsoring Organizations of the Treadway Commission to evaluate the effectiveness of our internal control over financial reporting. Based on its evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2017, the end of our most recent fiscal year.
Changes in Internal Control over Financial Reporting
We regularly review our system of internal control over financial reporting and make changes to our processes and systems to improve controls and increase efficiency, while ensuring that we maintain an effective internal control environment. Changes may include such activities as implementing new, more efficient systems, consolidating activities, and migrating processes. There were no changes in our internal control over financial reporting that occurred during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
| 59 |
Item 10. Directors, Executive Officers, and Corporate Governance
The Board of Directors
Our board of directors currently consists of eight members. Biographical information with respect to our directors is provided below.
Our directors hold office for one year or until their successors have been duly elected and qualified or until the earlier of their death, resignation or removal. Our amended and restated bylaws provide that the authorized number of directors comprising our board of directors will be fixed, from time to time, by a majority of the total number of directors.
There are no family relationships among any of our directors or executive officers, other than Ira and Andrew Ritter, who are father and son, respectively.
| Name | Position with the Company | Age
as of the Annual Meeting |
Director Since | ||||
| Michael D. Step | Chief Executive Officer and Director | 58 | 2012 | ||||
| Andrew J. Ritter | President and Director | 35 | 2008 | ||||
| Ira E. Ritter | Executive Chairman, Chief Strategic Officer and Director | 69 | 2008 | ||||
| Noah J. Doyle | Director | 50 | 2008 | ||||
| Matthew W. Foehr | Director | 45 | 2015 | ||||
| Paul V. Maier | Director | 70 | 2015 | ||||
| Dr. William M. Merino | Director | 75 | 2017 | ||||
| Gerald T. Proehl | Director | 59 | 2015 |
Michael D. Step became our Chief Executive Officer on October 1, 2014. He has served as a director of the Company since 2012. Mr. Step has over 20 years of business development and corporate development experience in the pharmaceutical industry. Prior to joining the Company as its Chief Executive Officer, Mr. Step served as Senior Vice President of Corporate Development at Santarus, Inc. (“Santarus”), and a member of its executive committee, from 2005 to January 2014, when Santarus was sold to Salix Pharmaceuticals, Ltd. At Santarus, Mr. Step was responsible for corporate development activities. Prior to joining Santarus, he served as Vice President, Corporate Development for Amylin Pharmaceuticals, Inc. (“Amylin”) from 2000 to 2005. In this capacity, he was responsible for leading corporate development activities, including product licensing, strategic planning, and mergers and acquisitions evaluations. Before joining Amylin, Mr. Step served as Senior Director, Business Development at Dura Pharmaceuticals, Inc. (“Dura Pharmaceuticals”) from 1997 to 2000. In this position, his duties included licensing of marketed pharmaceutical products. Prior to joining Dura Pharmaceuticals, he served in corporate development and strategic planning at Hoffmann-La Roche, from 1996 to 1997, and held various sales and management roles at Roche Labs, from 1994 to 1996, and Syntex Labs, from 1992 to 1994. Mr. Step holds a B.A. in political science from Vanderbilt University and a M.B.A. from the University of Southern California.
Qualifications: We believe that Mr. Step is well qualified to serve on our board of directors and as Chief Executive Officer of the Company due to his over 20 years’ experience in the pharmaceutical industry, serving in senior leadership roles within public pharmaceutical companies including in the gastrointestinal disease segment. Mr. Step has served in various executive management positions in sales and sales management, and has had experience with many aspects of pharmaceutical commercialization, strategic planning, business development and licensing providing both strategic and operational vision and guidance. His extensive experience gives him valuable insight into our industry as well as seasoned business judgment.
Andrew J. Ritter served as Co-Founder, President and Chief Executive Officer of the Company’s predecessor in interest from its inception in 2004 until relinquishing the role of Chief Executive Officer to Mr. Step in October 2014. Mr. Ritter was a member of the board of directors of the Company’s predecessor since its inception in 2004 and has been a member of our board of directors since 2008 when the Company was formed. Mr. Ritter has been actively studying the field of lactose intolerance for over 15 years and currently holds over a dozen patents and over twenty pending international patent applications. In addition, he has co-published articles, given presentations at major healthcare and medical conferences, and has been a guest lecturer of entrepreneurship at various graduate and undergraduate schools throughout Los Angeles including: University of Southern California Marshall School of Business, University of California at Los Angeles Anderson School of Business and Pepperdine University Graziadio School of Business and Management. Mr. Ritter served as a Los Angeles City Commissioner on the Commission for Children, Youth and Their Families from 2000 to 2002. He holds a B.A. in Political Science and a minor in Business from the University of Southern California. Mr. Ritter received a Master of Business Administration from the Wharton School of Business.
| 60 |
Qualifications: We believe that Mr. Ritter is well qualified to serve on our board of directors due to his over 15 years of research experience working in lactose intolerance and digestive diseases. Having founded the Company and invented Lactagen™, Mr. Ritter has an in depth knowledge of the Company, and provides senior leadership on the clinical and product development matters facing the Company. Mr. Ritter also brings to the board of directors an extensive scientific and operational background gained previously at Ritter Natural Sciences and over the years at Ritter.
Ira E. Ritter served as Co-Founder, Chief Strategic Officer and Executive Chairman of the Company’s predecessor in interest from its inception in 2004 through the formation of the Company in 2008 and has served in those positions with the Company since 2008. Mr. Ritter has extensive experience creating and building diverse business enterprises and has provided corporate management, strategic planning and financial consulting for a wide range of market segments. Since 2010, Mr. Ritter has also acted as a managing partner of Stonehenge Partners. Mr. Ritter served as President and Vice Chairman of Quality King, Inc., a national wholesale distributor of healthcare products, from 1992 to 2000. From 1998 to 2001, he served as President and Chairman of Rockwood Investments Inc., a business he developed which produced private label health and beauty products for major national retailers, including GNC and K-Mart. He also served as Chairman of ON-TV, a division of Oak Industries, Inc., from 1982 to 1985, where he managed the television division initiating exclusive broadcasts of Los Angeles, Chicago, and New York professional baseball, basketball, and hockey games. During this tenure, he produced the first televised home shopping program and directed development of the largest “pay-per-view” channel system for its time. Mr. Ritter served on the board of directors for Martin Lawrence Art Galleries from 1980 to 1985 helping take it public on The New York Stock Exchange. During his 20 years as a publisher, he produced monthly national consumer magazines focused on health & fitness, women’s issues and the environment. Mr. Ritter also has a long history of public service that includes appointments by three Governors to several State of California Commissions including eight years served as Commissioner on the California Prison Industry Authority. He has guest lectured at University of Southern California Marshall School of Business and Pepperdine University Graziadio School of Business where he also serves as an advisory board member to Pepperdine’s Graduate School of Education and Psychology, Social Entrepreneurship and Change Program. Presently he serves on the board of directors for Vitavis Laboratories. In 1981, Mr. Ritter was honored with the City of Hope’s Man of the Year award.
Qualifications: We believe that Mr. Ritter is well suited to serve on our board of directors due to his over 40 years’ experience overseeing daily operations of diverse business enterprises, and his managing public as well as private companies. Mr. Ritter provides our board of directors with extensive background in operational and strategic planning, as well as general executive and leadership expertise. Mr. Ritter has served on the boards of several companies during his career.
Noah J. Doyle has served as a director of the Company since September 2008. He has been an entrepreneur and investor for over 20 years. Mr. Doyle is the managing director of Javelin GP, LLC, the general partner of Javelin GP, LP, which is the general partner of Javelin and the manager of Javelin SPV. Prior to forming the first Javelin entities in 2008, Mr. Doyle supported over a dozen start-ups as an angel investor, including Keyhole, Inc. (“Keyhole”) (acquired by Google Inc. in 2004), Cantametrix, Inc. (acquired by Gracenote, Inc. in 2002), Amae Software (acquired by Verint Systems, Inc. in 2006), Nuvon, Inc., Aquea Scientific Corporation, Emdigo Inc., Magnacash Inc. (acquired by Yaga, Inc. in 2001), and i-mint India. Mr. Doyle most recently directed the enterprise product line for Google’s geospatial products, Google Earth and Google Maps, from 2004 to 2007. From 2002 to 2004 he managed the Sales and Corporate Development functions at Keyhole, which created the first Web hosted digital earth model. Prior to Keyhole, Mr. Doyle helped establish the Internet loyalty rewards marketplace as a co-founder of MyPoints.com (“MyPoints”), the largest Internet loyalty program with over 6 million active members, where he led product management and business development functions from the company’s inception in 1996 through its initial public offering and subsequent acquisition by United Airlines in 2002. Prior to joining MyPoints, Mr. Doyle was based in Tokyo where he managed overseas sales and marketing for the OEM channel of Matsushita’s (Panasonic) communications equipment subsidiary in Japan, from 1990 to 1994. Mr. Doyle served on the board of directors of MOL Global, Inc. from July 2014 to February 2016. He was also chairman of the management board of the University of California, Berkeley’s campus bookstore, a $17 million retail operation, and also held product management and operations management roles at IBM/Rational (Pure Atria) and Oracle, from 1989 to 1990. Mr. Doyle holds M.B.A. and B.A. Economics degrees, as well as certificates in Management of Technology and Global Management from University of California — Berkeley.
Qualifications: We believe that Mr. Doyle is well suited to serve on our board of directors due to his over 20 years of experience as an entrepreneur and investor. Mr. Doyle has experience as a venture capitalist building and serving on the boards of many public and private emerging companies in leadership roles providing guidance on finance, development and operational growth.
| 61 |
Matthew W. Foehr has served as a director of the Company since February 2015. He currently serves as President and Chief Operating Officer at Ligand Pharmaceuticals Incorporated (“Ligand”), a commercial stage biopharmaceutical company. Prior to joining Ligand in 2011, Mr. Foehr was Vice President and Head of Consumer Dermatology R&D, as well as Acting Chief Scientific Officer of Dermatology, in the Stiefel division of GlaxoSmithKline (“GSK”). Following GSK’s acquisition of Stiefel Laboratories, Inc. (“Stiefel”) in 2009, Mr. Foehr led the R&D integration of Stiefel into GSK. At Stiefel Laboratories, Inc., Mr. Foehr served as Senior Vice President of Global R&D Operations, Senior Vice President of Product Development& Support, and Vice President of Global Supply Chain Technical Services. Prior to joining Stiefel, Mr. Foehr held various executive roles at Connetics Corporation including Senior Vice President of Technical Operations and Vice President of Manufacturing. Currently, he is a member of the board of directors of Viking Therapeutics Inc. Mr. Foehr is the author of multiple scientific publications and is a named inventor on numerous U.S. patents. He received his Bachelor of Science degree in Biology from Santa Clara University.
Qualifications: We believe that Mr. Foehr is well suited to serve on our board of directors due to his more than 20 years of experience in the pharmaceutical industry and his experience managing global operations and research and development programs.
Paul V. Maier has served as a director of the Company since April 2015. From November 2009 through June 2014, Mr. Maier served as the Chief Financial Officer of Sequenom Inc., a publicly held company serving the discovery, clinical research, and diagnostics market. From February 2007 until November 2009, he served as an independent financial consultant. Previously, Mr. Maier was Senior Vice President and Chief Financial Officer of Ligand from 1992 through 2007. From 1990 to 1992, Mr. Maier served as Vice President, Finance of DFS West, a division of DFS Group LP, a private multinational retailer. From 1984 to 1990, Mr. Maier was employed by ICN Pharmaceuticals, a pharmaceutical and biotechnology research products company, where he held various executive positions in finance and general management in ICN as well as SPI Pharmaceuticals, a publicly held subsidiary. Mr. Maier currently serves on the board of directors of International Stem Cell Corporation, Apricus Biosciences, MabVax Therapeutics, and Biological Dynamics. Mr. Maier received an MBA from Harvard Business School and a BS from Pennsylvania State University.
Qualifications: We believe that Mr. Maier is well suited to serve on our board of directors due to his over 25 years of experience as a senior executive in biotechnology and pharmaceutical companies and his extensive experience in finance.
Dr. William M. Merino has served as a director of the Company since January 17, 2017. Dr. Merino served as the Senior Vice President of Worldwide Regulatory Affairs for Warner Lambert Pharmaceuticals from 1987 to 2000, where he was a member of the Office of the Chairman and responsible for the registration and approval of pharmaceuticals products with regulatory agencies around the world. He was also responsible for quality assurance, quality control and drug safety for the company, and led efforts to gain expedited registration of Lipitor in the United States and abroad in 20 other countries. He also has previous experience leading international regulatory affairs at Alcon Pharmaceuticals, G.D. Searle & Co., and Riker Laboratories. Dr. Merino has served as a senior clinical and regulatory advisory to the Company. Dr. Merino received his PhD in Pharmacology from Purdue University.
Qualifications: We believe that Dr. Merino’s deep global experience in drug and device registration and his extensive work with senior members of the FDA as well as several international regulatory authorities will bring important insight and acumen to our board of directors, as the Company continues its interactions with the FDA in an effort to bring RP-G28 to market.
Gerald T. Proehl has served as a director of the Company since April 2015. Currently, Mr. Proehl is President, CEO, Founder and Director of Dermata Therapeutics, LLC, a private biopharmaceutical company. From January 2002 to January 2014, he was the President, Chief Executive Officer and a Director of Santarus, a company that he helped to found in 1999 and sold to Salix Pharmaceuticals in January 2014 for $2.6 billion. From March 2000 through December 2001, Mr. Proehl was President and Chief Operating Officer of Santarus, and from April 1999 to March 2000, Mr. Proehl was Vice President, Marketing and Business Development of Santarus. Prior to joining Santarus, Mr. Proehl was with Hoechst Marion Roussel, Inc. (“Hoechst”), a global pharmaceutical company, for 14 years, where he served in various capacities, including Vice President of Global Marketing. During his career at Hoechst he worked across numerous therapeutic areas, including CNS, cardiovascular, and gastrointestinal. Mr. Proehl currently serves on the board of directors of two other public company boards, Sophiris Bio Inc. and Tenax Therapeutics, Inc. Mr. Proehl also serves on a number of private company boards including Kinetek Sports, Patara Pharma LLC, MDRejuvena, Inc. and Dermata Therapeutics, LLC. He also served on the board of directors of Auspex Pharmaceuticals, Inc. from January 2014 to May 2015. Mr. Proehl holds a B.S. in education from the State University of New York at Cortland, an M.A. in exercise physiology from Wake Forest University and an M.B.A. from Rockhurst College.
Qualifications: We believe that Mr. Proehl is well suited to serve on our board of directors due to his general business and commercial experience in the pharmaceutical industry, as well as his strong background in business operations developed through his leadership at other companies.
| 62 |
Executive Officers
Our Executive Officers as of the date of this Annual Report are as follows:
| Name | Age | Position with the Company | ||
| Michael D. Step | 58 | Chief Executive Officer | ||
| Andrew J. Ritter | 35 | President | ||
| Ira E. Ritter | 69 | Executive Chairman and Chief Strategic Officer | ||
| Jeffrey Benjamin | 53 | Vice President Finance |
Officers serve at the discretion of the board of directors. There are no family relationships among any of our directors or executive officers, other than Ira and Andrew Ritter, who are father and son, respectively. There is no arrangement or understanding between any executive officer and any other person pursuant to which the executive officer was selected.
Michael D. Step. For Mr. Step’s biography, please see above under “Board of Directors.”
Andrew J. Ritter. For Mr. Ritter’s biography, please see above under “Board of Directors.”
Ira E. Ritter. For Mr. Ritter’s biography, please see above under “Board of Directors.”
Jeffrey Benjamin has served as our Vice President, Finance since October 2017. Mr. Benjamin previously served as Interim Corporate Controller for Unified Grocers (acquired by Supervalu Inc. in June 2017) since February 2017. Prior to that he served as a principal consultant for Tatum Consulting from January 2015 to January 2017, Chief Financial Officer of Communications Infrastructure Corporation from March 2013 to January 2015, and Corporate Controller of Liaison Technologies, Inc. from April 2012 to March 2013.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act, and the rules issued thereunder, requires our directors and executive officers and beneficial owners of more than 10% of the outstanding shares of our equity securities to file reports of ownership and changes in beneficial ownership of our equity securities with the SEC. Copies of these reports are furnished to us. We are required to identify any of those individuals who failed to file such reports on a timely basis. Based solely on our review of the copies of such reports furnished to us, and representations from the persons subject to Section 16(a) with respect to the Company, we believe that during 2017 all of our executive officers, directors and 10% stockholders complied with the Section 16(a) requirements.
Code of Business Conduct and Ethics
We adopted a code of business conduct and ethics that applies to all of our employees, officers and directors, including those officers responsible for financial reporting prior to the closing of our initial public offering. The code of business conduct and ethics is available on our website at www.ritterpharmaceuticals.com. Any amendments to the code of business conduct and ethics, or any waivers of its requirements that apply to our principal executive officer, principal financial officer or principal accounting officer, will be disclosed on our website.
Audit Committee
The current members of our audit committee are Matthew W. Foehr, Paul V. Maier and Gerald T. Proehl, with Mr. Maier serving as chairman. Our board of directors has determined that each member of our Audit Committee is independent under Rule 10A-3 of the Exchange Act and the continued listing requirements of NASDAQ, and that each member of our Audit Committee satisfies the other continued listing requirements of NASDAQ for audit committee membership. Our board of directors has also determined that Mr. Maier qualifies as an “audit committee financial expert,” as such term is defined by the SEC, and that he has the requisite level of financial sophistication required by the continued listing requirements of NASDAQ.
| 63 |
Item 11. Executive Compensation
Summary Compensation Table (2017 and 2016)
The following table sets forth the compensation paid or earned for the fiscal years ended December 31, 2017 and 2016 to our named executive officers for each of those years, who are comprised of (1) our principal executive officer for such year, and (2) our next two highest compensated executive officers other than the principal executive officer (whose compensation exceeded $100,000).
| Name and Principal Position | Year | Salary ($) | Bonus ($)(2) | Option Awards(1) ($) | Nonequity Incentive Compensation ($) | All
Other Compensation ($) | Total
($) | |||||||||||||||||||||
| Michael D. Step | 2017 | $ | 411,875 | $ | — | $ | 88,425 | — | $ | — | $ | 500,300 | ||||||||||||||||
| Chief Executive Officer | 2016 | $ | 376,269 | $ | — | $ | 126,280 | — | $ | — | $ | 502,549 | ||||||||||||||||
| Andrew J. Ritter | 2017 | $ | 331,569 | $ | 121,320 | $ | — | — | $ | — | $ | 452,889 | ||||||||||||||||
| President | 2016 | $ | 324,010 | $ | 117,180 | $ | 490,394 | — | $ | — | $ | 931,584 | ||||||||||||||||
| Ira E. Ritter | 2017 | $ | 330,367 | $ | 101,115 | $ | — | — | $ | — | $ | 431,482 | ||||||||||||||||
| Executive Chairman and | 2016 | $ | 308,332 | $ | 97,571 | $ | 490,394 | — | $ | — | $ | 896,297 | ||||||||||||||||
| Chief Strategic Officer | ||||||||||||||||||||||||||||
| (1) | Represent the grant date fair value of the option awards granted during the years presented, determined in accordance with FASB ASC Topic 718. We utilize the Black-Scholes option-pricing model to value awards. Key valuation assumptions include: |
| ● | Expected dividend yield. The expected dividend is assumed to be zero as we have never paid dividends and have no current plans to pay any dividends on our common stock. | |
| ● | Expected stock-price volatility. As our common stock only recently became publicly traded, the expected volatility is derived from the average historical volatilities of publicly traded companies within our industry that we consider to be comparable to our business over a period approximately equal to the expected term. | |
| ● | Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant for zero coupon U.S. Treasury notes with maturities approximately equal to the expected term. | |
| ● | Expected term. The expected term represents the period that the stock-based awards are expected to be outstanding. Our historical share option exercise experience does not provide a reasonable basis upon which to estimate an expected term because of a lack of sufficient data. Therefore, we estimate the expected term by using the simplified method provided by the SEC. The simplified method calculates the expected term as the average of the time-to-vesting and the contractual life of the options. |
In addition to the assumptions used in the Black-Scholes option-pricing model, we also estimate a forfeiture rate to calculate the stock-based compensation for our equity awards. We will continue to use judgment in evaluating the expected volatility, expected terms and forfeiture rates utilized for our stock-based compensation calculations on a prospective basis.
| (2) | Represents annual bonuses earned for 2017 and 2016 based upon the achievement of specific performance goals, pursuant to the terms of their respective offer letters. For 2017, the annual bonuses earned were equal to 90% of the target bonus opportunities for each of Andrew Ritter (target bonus equal to 40% of his base salary) and Ira Ritter (target bonus equal to 35% of his base salary). For 2016, the annual bonuses earned were equal to 90% of the target bonus opportunities for each of Andrew Ritter (target bonus equal to 40% of his base salary) and Ira Ritter (target bonus equal to 35% of his base salary). |
Narrative to Summary Compensation Table
Letter Agreement with Michael D. Step
On December 2, 2014, we entered into a letter agreement (the “Step Letter Agreement”), with Mr. Step, our current Chief Executive Officer, setting forth the terms of his employment. The Step Letter Agreement provides that Mr. Step will be entitled to an annual base salary of $360,000. Pursuant to the Step Letter Agreement, Mr. Step was also entitled to receive three stock options.
| 64 |
The first two options entitle Mr. Step to purchase 646,537 and 73,377 shares of Common Stock of the Company, respectively, for an exercise price of $5.86 per share. Each of these options was immediately exercisable in full as of the date of the grant, with 44/48ths of the total number of shares covered by each option subject to a right of repurchase by the Company upon termination of Mr. Step’s employment with us for any reason. This right of repurchase will lapse over a period of 44 months, with 1/44th of the total number of shares subject to the right of repurchase lapsing on January 1, 2015 and on the first day of each month thereafter. In addition, the right of repurchase will lapse in its entirety upon a termination of the employment of Mr. Step by us without Cause or by Mr. Step with Good Reason and upon a Termination upon a Change in Control.
The third option became exercisable upon the closing of our initial public offering on June 29, 2015. Pursuant to the terms of the agreement, the option is exercisable for a total of 163,799 shares of our Common Stock, which, together with the shares subject to the first option, represent 7.5% of the shares of Common Stock deemed to be outstanding at June 29, 2015 on a fully-diluted basis, after giving effect to the number of shares subject to the third option. Seventy-five percent of the shares subject to the third option are subject to a right of repurchase by us upon termination of Mr. Step’s employment for any reason. This right of repurchase will lapse with respect to 1/36th of the total number of shares subject to the right of repurchase on the first day of each month following the date on which the third option first becomes exercisable. In addition, the right of repurchase will lapse in its entirety upon Mr. Step’s termination of employment under certain circumstances.
For purposes of the Step Letter Agreement, the terms “Cause,” “Good Reason,” and “Termination upon a Change in Control” each have the meanings ascribed to such terms in the Executive Severance & Change in Control Agreement described below.
Offer Letters with Andrew Ritter and Ira Ritter
The compensation terms outlined in the offer letters, which became effective June 29, 2015, superseded and replaced those provided in the Executive Compensation Plan, which is described above, other than certain provisions related to the bonus opportunities. The offer letters provide that Andrew Ritter is entitled to an annual base salary of $310,000 and Ira Ritter is entitled to an annual base salary of $295,000. In accordance with his offer letter, Andrew Ritter also became entitled to receive up to $180,000 payable over a three-year period for tuition reimbursement.
Pursuant to their respective offer letters, Andrew Ritter and Ira Ritter each have the opportunity to earn an annual bonus based upon a percentage of their base salary and the achievement of specific performance as determined by the Company. The initial target bonus opportunities are 40% and 35% of the base salary for Andrew Ritter and Ira Ritter, respectively.
2015 Equity Incentive Plan
On June 15, 2015, our board of directors approved the 2015 Equity Incentive Plan, and on June 17, 2015, the 2015 Equity Incentive Plan was approved by our stockholders. The 2015 Equity Incentive Plan was subsequently amended by the stockholders of the Company on June 3, 2016 and June 2, 2017.
The purposes of the 2015 Equity Incentive Plan are to optimize the profitability and growth of the Company through long-term incentives that are consistent with the Company’s objectives and that link the interests of award recipients (“Grantees”), to those of the Company’s stockholders; to give award recipients an incentive for excellence in individual performance; to promote teamwork among Grantees; and to give the Company flexibility in attracting and retaining key employees, directors and consultants.
Selected employees, officers and directors of the Company or any subsidiary, and consultants, advisors and independent service providers to the Company and any subsidiary who qualify as a “consultant” under the applicable rules of the SEC for registration of shares on a Form S-8 registration statement, are eligible to receive awards under the 2015 Equity Incentive Plan. The plan administrator may also grant awards to individuals in connection with hiring, retention or otherwise before the date the individual first performs services for the Company or any subsidiary; provided, however, that those awards will not become vested or exercisable before the date the individual first performs services for the Company or any subsidiary.
The number of shares of Common Stock that we may issue pursuant to awards under the 2015 Plan is (i) 27,500,000 plus (ii) any shares which were available for grant under the 2008 Stock Plan or the 2009 Stock Plan (collectively, the “Prior Plans”), on the effective date of the 2015 Equity Incentive Plan or are subject to awards under the Prior Plans which, after the effective date of the 2015 Equity Incentive Plan, are forfeited or lapse unexercised or are settled in cash and are not issued under the Prior Plans. No more than 27,500,000 shares of Common Stock may be issued pursuant to incentive stock options intended to qualify under Section 422 of the Internal Revenue Code (the “Code”). No awards may be granted under any Prior Plan; however, any awards granted under any Prior Plan that were outstanding as of the effective date of the 2015 Plan continue to be subject to the terms and conditions of such Prior Plan. The maximum number of shares of Common Stock subject to awards of any combination that may be granted under the 2015 Equity Incentive Plan during any calendar year to any one individual is limited to 3,000,000 shares.
| 65 |
These limits will be appropriately adjusted to reflect any stock dividend, stock split, combination or exchange of shares, merger, consolidation or other distribution and/or similar transactions. To the extent that (i) an award terminates, expires, lapses or is forfeited for any reason, (ii) any award is settled in cash (in whole or in part) without the delivery of shares to the Grantee, or (iii) any shares subject to an award under any Prior Plan terminate, expire, lapse or are forfeited for any reason or an award under any Prior Plan is settled for cash (in whole or in part), then any shares subject to the award, to the extent of such termination, expiration, lapse, forfeiture or cash settlement, will again be available for the grant of an award pursuant to the 2015 Equity Incentive Plan. Any shares tendered or withheld to satisfy the grant or exercise prior or tax withholding obligation pursuant to any award will again be available for the grant of an award pursuant to the 2015 Equity Incentive Plan.
The 2015 Equity Incentive Plan may be administered by a committee or subcommittee of the board of directors as the board of directors may appoint from time to time, or by our full board of directors if no committee is designated or for other specific purposes. At present, the 2015 Equity Incentive Plan is administered by our compensation committee. The plan administrator has the full authority and discretion to administer the 2015 Equity Incentive Plan and to take any action that is necessary or advisable in connection with the administration of the 2015 Equity Incentive Plan, including without limitation the authority and discretion to interpret and administer the 2015 Equity Incentive Plan and any award agreement relating to the 2015 Equity Incentive Plan or any award made thereunder, the authority to designate Grantees to receive awards under the 2015 Equity Incentive Plan and to determine the type or types of awards to be granted to such Grantees, the authority to determine the terms and conditions of awards granted under the 2015 Equity Incentive Plan, and the authority to determine whether, to what extent, and pursuant to what circumstances and award may be settled in, or the exercise price of an award may be paid in, cash, shares, other awards, or other property, or an award may be canceled, forfeited or surrendered. The plan administrator’s determinations will be final and conclusive. The plan administrator may delegate certain of its authority to others as specified in the 2015 Equity Incentive Plan.
The 2015 Equity Incentive Plan provides for grants of stock options (including incentive stock options qualifying under Section 422 of the Code and nonstatutory stock options), restricted stock awards, stock appreciation rights, restricted stock units, performance awards, other stock-based awards or any combination of the foregoing.
Stock options. The 2015 Equity Incentive Plan allows the plan administrator to grant incentive stock options, as that term is defined in Section 422 of the Code, or nonqualified stock options. No incentive stock option award may be granted to any person who is not an employee of the Company or any subsidiary. Options must have an exercise price at least equal to the fair market value of the underlying shares on the date of grant. In addition, in the case of incentive stock options granted to a greater than 10% stockholder of the Company, such exercise price may not be less than 110% of the fair market value of the underlying shares on the date of grant. The option holder may pay the exercise price in cash or by check, by tendering shares of Common Stock (including shares issuable in settlement of the award), payment through a broker or by any other means that the plan administrator approves. Options granted under the 2015 Equity Incentive Plan will have a term of no more than 10 years, or five years in the case of incentive stock options granted to a greater than 10% stockholder of the Company; however, the options will expire earlier if the option holder’s service relationship with us terminates or as otherwise provided in an award agreement.
Restricted stock awards. The 2015 Equity Incentive Plan allows the plan administrator to grant restricted stock awards, which issue to the holder a certain number of shares of Common Stock that are subject to restrictions or conditions as the plan administrator deems appropriate, such as time-based or performance-based criteria, and which become vested upon the lapse or satisfaction of such conditions. The plan administrator may apply limitations to any restricted stock award and establish the purchase price (or provide for no purchase price), provided that if a purchase price is established, it may not be less than par value of the shares to be purchased.
Stock appreciation rights. The 2015 Equity Incentive Plan allows the plan administrator to grant awards of stock appreciation rights, which entitle the holder to receive a payment in cash, in shares of Common Stock, or in a combination of both, having an aggregate value equal to the spread on the date of exercise between the fair market value of the underlying shares on that date and the base price of the shares specified in the grant agreement, multiplied by the number of shares specified in the award being exercised and as otherwise provided in an award agreement. Stock appreciation rights may not have a base price of less than 100% of the fair market value of the underlying shares on the date of grant.
Restricted stock units. The 2015 Equity Incentive Plan allows the plan administrator to grant awards of restricted stock units (“RSUs”), which entitle the holder to a number of shares of Common Stock, a cash payment or some combination thereof, upon satisfaction of vesting and other criteria for issuance or upon such later date as specified in the award agreement, as established by the plan administrator in the award agreement.
Other stock-based awards. The 2015 Equity Incentive Plan allows the plan administrator to grant other stock-based stock awards to eligible participants, including dividend equivalent rights, stock payments and/or deferred stock. A dividend equivalent may be granted alone or in conjunction with another type of award, and generally provides for payment, in cash, Common Stock or some combination thereof, of an amount equal to the dividends that would have been payable with respect to a specified number of underlying shares. A stock payment is an award to a Grantee, only upon satisfaction of performance-based criteria or other criteria specified by the plan administrator, of a specified number of shares of Common Stock, or an option to purchase Common Stock, which may be (but is not required to be) in lieu of base salary, bonus, fees or other cash consideration to the Grantee. A deferred stock award is a grant to a Grantee, only upon satisfaction of performance-based criteria or other criteria specified by the plan administrator, of a specified number of shares of Common Stock.
| 66 |
Performance awards. The 2015 Equity Incentive Plan allows the plan administrator to grant performance awards which become payable in Common Stock, in cash or in a combination of Common Stock and cash, on account of attainment of one or more performance goals established by the plan administrator on one or more specified dates or over a specified period or periods. The plan administrator may establish performance goals relating to any of the following: (i) gross or net earnings (either before or after one or more of the following: interest, taxes, depreciation and amortization); (ii) gross or net sales or revenue; (iii) gross or net income or adjusted income (either before or after taxes); (iv) operating earnings or profit; (v) cash flow (including, but not limited to, operating cash flow and free cash flow); (vi) return on assets; (vii) return on capital; (viii) return on stockholders’ equity; (ix) return on sales; (x) gross or net profit or operating margin; (xi) costs; (xii) funds from operations; (xiii) expenses; (xiv) working capital; (xv) earnings per share or adjusted earnings per share; (xvi) price per share of Common Stock; (xvii) regulatory body approval for commercialization of a product; (xviii) implementation or completion of critical projects; (xix) market share; or (xx) total stockholder return; any of which may be measured either in absolute terms or as compared to any incremental increase or decrease or as compared to results of a peer group or to market performance indicators or indices.
The plan administrator may, in its sole discretion, provide that one or more objectively determinable adjustments will be made to one or more of the performance goals described above, such as adjustments to account for changes in the Company’s or segment’s business (e.g., restructuring, acquisition or disposal or discontinuance of a business segment), accounting or financial reporting (e.g., change in accounting principles, significant income or expense or amortization of assets) or for other unusual or non-recurring events, all as further detailed in the 2015 Equity Incentive Plan. For all awards intended to qualify as performance-based compensation, such determinations shall be made within the time periods prescribed by, and otherwise in compliance with, Section 162(m) of the Code.
Amendment and termination. No award will be granted under the 2015 Equity Incentive Plan after the tenth anniversary of the effective date of the 2015 Equity Incentive Plan. Subject to applicable laws and exchange limitations, our board of directors or the plan administrator may terminate, amend or modify the 2015 Equity Incentive Plan, or any portion thereof, at any time. Stockholder approval will be required to (i) increase the limits imposed on the maximum number of shares which may be issued under the 2015 Equity Incentive Plan or as incentive stock options (other than an appropriate adjustment due to stock dividend, stock split, combination or exchange of shares, merger, consolidation or similar circumstance), (ii) reduce the price per share of any outstanding option or stock appreciation right or cancel any such award in exchange for cash when the exercise price per share exceeds the fair market value of the underlying shares, or (iii) materially change the class of persons who are eligible to participate in the 2015 Equity Incentive Plan; provided, however, that no amendment, suspension or termination of the 2015 Equity Incentive Plan may, without the consent of the Grantee, materially impair any rights or obligations under any award granted or awarded thereunder, unless the award itself otherwise expressly so provides.
Outstanding Equity Awards at 2017 Fiscal Year-End
The following table presents the outstanding equity awards held by each of the named executive officers as of December 31, 2017.
| Name | Number
of Securities Underlying Unexercised Options Exercisable | Number
of Securities Underlying Unexercised Options Unexercisable | Option Exercise Price ($) | Option Expiration Date | ||||||||||
| Michael D. Step | 26,163 | — | $ | 1.14 | 8/16/2022 | |||||||||
| 646,537 | (1) | — | $ | 5.86 | 12/2/2024 | |||||||||
| 73,377 | (2) | — | $ | 5.86 | 12/2/2024 | |||||||||
| 163,799 | (3) | — | $ | 5.86 | 12/2/2024 | |||||||||
| 29,042 | (4) | 52,958 | (4) | $ | 1.54 | 7/5/2026 | ||||||||
| 13,292 | (5) | 44,708 | (5) | $ | 2.89 | 1/25/2027 | ||||||||
| Andrew J. Ritter | 24,878 | (6) | 3,094 | (6) | $ | 1.27 | 9/25/2023 | |||||||
| 20,979 | — | $ | 5.86 | 12/2/2024 | ||||||||||
| 351,354 | (7) | 81,081 | (7) | (7) | 12/2/2024 | |||||||||
| 29,042 | (8) | 52,958 | (8) | $ | 1.54 | 7/5/2026 | ||||||||
| 23,341 | (9) | 116,703 | (9) | $ | 2.60 | 10/25/2026 | ||||||||
| Ira E. Ritter | 24,878 | (10) | 3,094 | (10) | $ | 1.27 | 9/25/2023 | |||||||
| 20,979 | — | $ | 5.86 | 12/2/2024 | ||||||||||
| 351,354 | (11) | 81,081 | (11) | (11) | 12/2/2024 | |||||||||
| 29,042 | (11) | 52,958 | (11) | $ | 1.54 | 7/5/2026 | ||||||||
| 23,341 | (12) | 116,703 | (12) | $ | 2.60 | 10/25/2026 | ||||||||
| 67 |
| (1) | This option was granted to Mr. Step on December 2, 2014 and was immediately exercisable in full as of the date of grant. Of the shares subject to this option, 592,659 shares are subject to a right of repurchase in favor of us at a price of $5.86 per share, which right expires ratably over 44 months commencing January 1, 2015 and in full upon a change of control or upon Mr. Step’s employment termination by us without Cause, subject to his continued employment with us (as described in the stock option award agreement). |
| (2) | This option was granted to Mr. Step on December 2, 2014 and was immediately exercisable in full as of the date of grant. Of the shares subject to this option, 67,262 shares are subject to a right of repurchase in favor of us at a price of $5.86 per share, which rights expires ratably over 44 months commencing January 1, 2015 and in full upon a change of control or upon Mr. Step’s employment termination by us without Cause, subject to his continued employment with us (as described in the stock option award agreement). |
| (3) | This option was granted to Mr. Step on December 2, 2014. The total number of shares issued under this option equaled the number of shares of Common Stock, together with the 646,537 shares subject to the option granted to Mr. Step on December 2, 2014, representing in the aggregate 7.5% of the shares of Common Stock deemed to be outstanding on a fully-diluted basis as of the date that we raised in the aggregate a minimum of $15,000,000 in one or more private and/or public offerings (a “Qualified Financing”), after giving effect to (i) the issuance of the shares issued in the Qualified Financing, (ii) the issuance of this option and (iii) any adjustments. 75% of the shares subject to the third option are subject to a right of repurchase upon termination of Mr. Step’s employment for any reason, which right expires ratably over 36 months commencing with July 1, 2015 and in full upon a change of control or upon Mr. Step’s employment termination by us without Cause, subject to his continued employment with us (as described in the stock option award agreement). |
| (4) | This option was granted to Mr. Step on July 5, 2016 for an aggregate of 82,000 shares. The option vests in 48 equal monthly installments, the first of which vested on July 20, 2016 with the balance vesting on the 20th day of each calendar month thereafter until vested in full. |
| (5) | This option was granted to Mr. Step on January 25, 2017 for an aggregate of 58,000 shares. The option vests in forty-eight (48) equal monthly installments beginning on February 25, 2017 with the balance vesting on the 25th day of each calendar month thereafter until vested in full. |
| (6) | This option was granted to Andrew Ritter on September 25, 2013 for an aggregate of up to 48,951 shares, subject to the achievement of certain milestones. The option included 2,360 shares that vested and became exercisable as of the date of grant (with a balance of 1,137 shares vesting ratably on a monthly basis from September 30, 2013 over 36 months) attributable to the FDA Meeting Bonus milestone. An additional 3,671 shares vested and became exercisable as of June 29, 2015 (with a balance of 6,818 shares vesting ratably on a monthly basis from July 31, 2015 over 36 months) attributable to the Clinical Trial Funding Commitment Bonus Opportunity milestone. An additional 4,895 shares vested and became exercisable as of June 29, 2015 (with a balance of 9,091 shares vesting ratably on a monthly basis beginning July 31, 2015 over 36 months) attributable to the Fundraising Bonus Opportunities milestone. The option for the remaining balance of the 20,979 shares expired unvested as of September 30, 2015. |
| (7) | This option was granted to Andrew Ritter on December 2, 2014 and vests as follows: 25% of the shares vest on September 1, 2015 and the remaining 75% of the shares will vest in 36 equal monthly installments beginning on the last day of the first full month thereafter, subject to his continued employment with us. The exercise price for this option is as follows: (i) $5.86 for the first 152,347 shares; (ii) $9.30 for the next 140,044 shares; and (iii) $13.23 for the remaining 140,043 shares. |
| (8) | This option was granted to Andrew Ritter on July 5, 2016 for an aggregate of 82,000 shares. The option vests in 48 equal monthly installments, the first of which vested on July 20, 2016 with the balance vesting on the 20th day of each calendar month thereafter until vested in full. |
| (9) | This option was granted to Andrew Ritter on October 25, 2016 for an aggregate of 140,044 shares. The option vests ratably in 48 equal monthly installments following the public disclosure of top-line data results from the Company’s Phase 2b clinical trial. |
| 68 |
| (10) | This option was granted to Ira Ritter on September 25, 2013 and is subject to the same vesting schedule as the option granted to Andrew Ritter on this date as reflected in footnote (5) above. |
| (11) | This option was granted to Ira Ritter on December 2, 2014 and is subject to the same vesting schedule as the option granted to Andrew Ritter on this date as reflected in footnote (8) above. |
| (12) | This option was granted to Ira Ritter on July 5, 2016 for an aggregate of 82,000 shares. The option vests in 48 equal monthly installments, the first of which vested on July 20, 2016 with the balance vesting on the 20th day of each calendar month thereafter until vested in full. |
| (13) | This option was granted to Ira Ritter on October 25, 2016 for an aggregate of 140,044 shares. The option vests ratably in 48 equal monthly installments following the public disclosure of top-line data results from the Company’s Phase 2b clinical trial. |
Payments Due Upon Termination of Employment or a Change in Control
Executive Severance & Change in Control Agreements
We have entered into Executive Severance & Change in Control Agreements (the “Severance Agreements”), with each of our named executive officers. The Severance Agreements provide that if we terminate the executive’s employment without Cause, or the executive terminates his employment for Good Reason, the executive will be entitled to: (i) the Accrued Obligations; (ii) an amount equal to twelve (12) months of base salary, as in effect immediately prior to the termination date; (iii) medical, dental benefits provided by the Company to the executive and his spouse and dependents at least equal to the levels of benefits provided to other similarly situated active employees of the Company and its subsidiaries until the earlier of (a) the twelve (12) month anniversary of the date of termination or (b) the date that the executive becomes covered under a subsequent employer’s medical and dental plans; and (iv) acceleration of vesting of all equity and equity-based awards.
Pursuant to the terms of the Severance Agreements, in the event that within one (1) month prior to or the twelve (12) months following a Change in Control, the Company terminates the executive’s employment without Cause, or the executive terminates his employment for Good Reason, then, in lieu of the payments and benefits otherwise due to the executive in the preceding paragraph, the executive will be entitled to: (i) the Accrued Obligations; (ii) an amount equal to the sum of twelve (12) months of base salary, as in effect on the date of termination or the date of the Change in Control, whichever is greater; (iii) medical, dental benefits provided by the Company to the executive and his spouse and dependents at least equal to the level of benefits provided to other similarly situated active employees of the Company and its subsidiaries until the earlier of (a) the twelve (12) month anniversary of the date of termination or (b) the date that the executive becomes covered under a subsequent employer’s medical and dental plans; and (iv) acceleration of vesting of all equity and equity-based awards.
In the event the executive’s employment is terminated by him without Good Reason, by the Company for Cause or due to the executive’s death or disability, the executive and/or his estate or beneficiaries will be solely entitled to the Accrued Obligations.
The executive’s entitlement to the payments (other than the Accrued Obligations) and benefits described above is expressly contingent upon him providing the Company with a signed release satisfactory to the Company.
For purposes of the Severance Agreements:
“Accrued Obligations” means (i) earned but unpaid base salary through the date of termination; (ii) payment of any annual, long-term, or other incentive award which relates to a completed fiscal year or performance period, as applicable, and is payable (but not yet paid) on or before the date of termination; (iii) a lump-sum payment in respect of accrued but unused vacation days at the executive’s per-business-day base salary rate in effect as of the date of termination; and (iv) any unpaid expense or reimbursements due pursuant to Company expense reimbursement policy.
“Cause” means a finding by the Company that the executive has (i) been convicted of a felony or crime involving moral turpitude; (ii) disclosed trade secrets or confidential information of the Company (or any parent or subsidiary) to persons not entitled to receive such information; (iii) engaged in conduct in connection with the executive’s employment or service to the Company (or any parent or subsidiary), that has, or could reasonably be expected to result in, material injury to the business or reputation of the Company (or any parent or subsidiary), including, without limitation, act(s) of fraud, embezzlement, misappropriation and breach of fiduciary duty; (iv) violated the operating and ethics policies of the Company (or any parent or subsidiary) in any material way, including, but not limited to those relating to sexual harassment and the disclosure or misuse of confidential information; (v) engaged in willful and continued negligence in the performance of the duties assigned to the executive by the Company, after the executive has received notice of and failed to cure such negligence; or (vi) breached any material provision of any agreement between the executive and the Company (or any parent or subsidiary), including, without limitation, any confidentiality agreement.
| 69 |
“Change in Control” means the occurrence of any of the following events:
| (i) | Any “person” (as such term is used in Sections 13(d) and 14(d) of the Exchange Act) becomes a “beneficial owner” (as defined in Rule 13d-3 under the Exchange Act), directly or indirectly, of securities of the Company representing more than 50% of the voting power of the then outstanding securities of the Company; provided that a Change of Control will not be deemed to occur as a result of a change of ownership resulting from the death of a shareholder, and a Change of Control will not be deemed to occur as a result of a transaction in which the Company becomes a subsidiary of another corporation and in which the shareholders of the Company, immediately prior to the transaction, will beneficially own, immediately after the transaction, shares entitling such shareholders to more than 50% of all votes to which all shareholders of the parent corporation would be entitled in the election of directors (without consideration of the rights of any class of stock to elect directors by a separate class vote); | |
| (ii) | A change in the effective control of the Company which occurs on the date that a majority of members of the board of directors is replaced during any twelve (12) month period by Directors whose appointment or election is not endorsed by a majority of the members of the board of directors prior to the date of the appointment or election; or | |
| (iii) | The consummation of (A) a merger or consolidation of the Company with another corporation where the shareholders of the Company, immediately prior to the merger or consolidation, will not beneficially own, immediately after the merger or consolidation, shares entitling such shareholders to more than 50% of all votes to which all shareholders of the surviving corporation would be entitled in the election of directors (without consideration of the rights of any class of stock to elect directors by a separate class vote); (B) a sale or other disposition of all or substantially all of the assets of the Company; or (C) a liquidation or dissolution of the Company. |
“Good Reason” means, without the executive’s express written consent, the occurrence of any one or more of the following: (i) a substantial and material diminution in the executive’s duties or responsibilities; (ii) a material reduction in the executive’s Base Salary; or (iii) the relocation of the executive’s principal place of employment to a location more than 50 miles from the executive’ principal work location to a location that is more than 50 miles from the prior location. Notwithstanding the foregoing, a relocation of Mr. Step’s principal place of employment to a location closer to Mr. Step’s principal residence in San Diego, California shall not constitute “Good Reason.” A termination of employment by the executive for Good Reason will be effectuated by giving the Company written notice, or Notice of Termination for Good Reason, not later than 90 days following the occurrence of the circumstance that constitutes Good Reason, setting forth in reasonable detail the specific conduct of the Company that constitutes Good Reason and the specific provision(s) of this Agreement on which the executive relied. The Company will be entitled, during the 30-day period following receipt of a Notice of Termination for Good Reason, to cure the circumstances that gave rise to Good Reason, provided that the Company shall be entitled to waive its right to cure or reduce the cure period by delivery of written notice to that effect to the executive (such 30-day or shorter period, the “Cure Period”). If, during the Cure Period, such circumstance is remedied, the executive will not be permitted to terminate his employment for Good Reason as a result of such circumstance. If, at the end of the Cure Period, the circumstance that constitutes Good Reason has not been remedied, the executive will terminate employment for Good Reason on the date of expiration of the Cure Period.
2009 Stock Plan
The 2009 Stock Plan provides that in the event we merge with or into another corporation, or a Change in Control (as defined below) occurs, each outstanding option and stock purchase right will be assumed or an equivalent option substituted by the successor corporation or a parent or subsidiary of the successor corporation. In the event that the successor corporation in a merger or Change in Control refuses to assume or substitute for the option or stock purchase right, then the optionee will fully vest in and have the right to exercise the option or stock purchase right as to all of the optioned stock, including shares as to which it would not otherwise be vested or exercisable; provided, however, that such exercise will only be permitted as and to the extent it complies with Code Section 409A or does not cause the option or stock purchase right to cease to be exempt from that statute.
For purposes of the 2009 Stock Plan, “Change in Control” means the occurrence of any of the following events:
| (i) | Any “person” (as such term is used in Sections 13(d) and 14(d) of the Exchange Act) becomes the “beneficial owner” (as defined in Rule 13d-3 of the Exchange Act), directly or indirectly, of securities representing fifty percent (50%) or more of the total voting power represented by our then outstanding voting securities; or | |
| (ii) | The consummation of the sale or disposition by us of all or substantially all of our assets; or | |
| (iii) | The consummation of a merger or our consolidation with any other corporation, other than a merger or consolidation which would result in our voting securities outstanding immediately prior thereto continuing to represent (either by remaining outstanding or by being converted into voting securities of the surviving entity or its parent) at least 50% of the total voting power represented by the our voting securities or such surviving entity or its parent outstanding immediately after such merger or consolidation. Notwithstanding the foregoing, only a Change in Control event that also qualifies as a “change in the ownership” or a “change in the effective control” of the Company or a “change in the ownership of a substantial portion” of our assets within the meaning of Treasury Regulation Section 1.409A-3(i)(5) shall be recognized as a Change of Control for purposes of triggering exercise, distribution or settlement rights under any option or stock purchase right granted under the Stock Plan that is subject to Code Section 409A. |
| 70 |
2015 Equity Incentive Plan
The 2015 Equity Incentive Plan provides that notwithstanding any other provision of the 2015 Equity Incentive Plan, in the event of a Change in Control (as defined below), unless otherwise determined by the plan administrator, each outstanding award under the plan will be assumed or an equivalent award substituted by the successor corporation or a parent or subsidiary of the successor corporation. In the event that, or to the extent that, the successor corporation in a Change in Control refuses to assume or substitute for the award, or if the plan administrator determines that such assumption or substitution is not desirable or is only desirable for a portion of any outstanding award, then the plan administrator may take any or all of the following actions: (i) determine that an outstanding award will accelerate and become exercisable, or determine that the restrictions and conditions on an outstanding award will lapse, in whole or in part, as applicable, upon the Change of Control or upon such other event as the plan administrator determines; (ii) require that a Grantee surrender his or her outstanding award, or any portion of such outstanding award, in exchange for a payment by the Company, in cash or stock, as determined by the plan administrator, in an amount equal to the fair market value of the vested portion of the award (with respect to options or stock appreciation rights, or other similar appreciation value awards, such value shall be determined by the amount by which the then fair market value of the shares subject to the Grantee’s unexercised award exceeds the any applicable exercise price or other grant price or base value or the award); or (iii) after giving the Grantee an opportunity to exercise the vested portion of his or her outstanding award, terminate any or all unexercised portion of the award at such time as the plan administrator deems appropriate. Such surrender or termination will take place as of the date of the Change of Control or such other date as the plan administrator may specify.
For purposes of the 2015 Equity Incentive Plan, “Change in Control” means the occurrence of any of the following events:
| (i) | A change in our ownership which occurs on the date that any one person, or more than one person acting as a group, or Person, acquires ownership of our stock that, together with the stock held by such Person, constitutes more than 50% of the total voting power of our stock, except that any change in the ownership of our stock as a result of a private financing that is approved by our board of directors will not be considered a Change in Control; or | |
| (ii) | If we have a class of securities registered pursuant to Section 12 of the Exchange Act, a change in our effective control which occurs on the date that a majority of members of our board of directors is replaced during any twelve (12) month period by directors whose appointment or election is not endorsed by a majority of the members of our board of directors prior to the date of the appointment or election. For purposes of this paragraph (ii), if any Person is considered to be in effective control of our company, the acquisition of additional control of our company by the same Person will not be considered a Change in Control; or | |
| (iii) | A change in the ownership of a substantial portion of our assets which occurs on the date that any Person acquires (or has acquired during the twelve (12) month period ending on the date of the most recent acquisition by such person or persons) assets from us that have a total gross fair market value equal to or more than 50% of the total gross fair market value of all of our assets immediately prior to such acquisition or acquisitions. For purposes of this paragraph (iii), gross fair market value means the value of our assets, or the value of the assets being disposed of, determined without regard to any liabilities associated with such assets. |
Persons will be considered to be acting as a group if they are owners of a corporation that enters into a merger, consolidation, purchase or acquisition of stock, or similar business transaction with us.
Compensation of Directors
Non-Employee Director Compensation Program
Our non-employee directors received the following compensation for their services:
| ● | Annual Cash Retainer — $20,000 | |
| ● | Chairman of the Board Cash Retainer — $15,000 | |
| ● | Audit Committee Chair Retainer — $7,500 | |
| ● | Compensation Committee Chair Retainer — $5,000 |
| 71 |
| ● | Nominating and Corporate Governance Committee Chair Retainer — $3,500 | |
| ● | Initial Equity Grant — 10,000 shares | |
| ● | Annual Equity Grant — 7,000 shares |
Beginning January 23, 2018, our non-employee directors are entitled to receive the following compensation for their services:
| ● | Annual Cash Retainer — $35,000 | |
| ● | Chairman of the Board Cash Retainer — $25,000 | |
| ● | Audit Committee Chair Retainer — $15,000 | |
| ● | Compensation Committee Chair Retainer — $10,000 | |
| ● | Nominating and Corporate Governance Committee Chair Retainer — $7,500 | |
| ● | Initial Equity Grant — 40,000 shares | |
| ● | Annual Equity Grant — 30,000 shares |
2017 Director Compensation
The following table sets forth the compensation paid or earned for the fiscal year ended December 31, 2017 to our non-employee directors. Compensation paid to Michael D. Step, Andrew Ritter, and Ira Ritter is presented as part of the “Summary Compensation Table (2017 and 2016)” above. Our employee directors do not receive compensation for their service as directors. Dr. Merino is not included in the table below, as he was not appointed to our board of directors until January 17, 2017.
| Name of Director | Fees
Earned and Paid in Cash ($) | Option Awards(2) ($) | All
other compensation ($) | Total ($) | ||||||||||||
| Noah Doyle(1) | $ | — | $ | — | — | $ | — | |||||||||
| Matthew W. Foehr | $ | 25,000 | $ | — | — | $ | 25,000 | |||||||||
| Paul V. Maier | $ | 27,500 | $ | — | — | $ | 27,500 | |||||||||
| William M. Merino | $ | 20,000 | 13,068 | 33,068 | ||||||||||||
| Gerald T. Proehl | $ | 23,500 | $ | — | — | $ | 23,500 | |||||||||
| (1) | Mr. Doyle has declined to receive any compensation for his service as director. |
| (2) | Represents the aggregate grant date fair value of the options granted to Dr. Merino on January 25, 2017, determined in accordance with FASB ASC 718. |
We utilize the Black-Scholes option-pricing model to value awards. Key valuation assumptions include:
| ● | Expected dividend yield. The expected dividend is assumed to be zero as we have never paid dividends and have no current plans to pay any dividends on our common stock. | |
| ● | Expected stock-price volatility. As our common stock only recently became publicly traded, the expected volatility is derived from the average historical volatilities of publicly traded companies within our industry that we consider to be comparable to our business over a period approximately equal to the expected term. | |
| ● | Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant for zero coupon U.S. Treasury notes with maturities approximately equal to the expected term. | |
| ● | Expected term. The expected term represents the period that the stock-based awards are expected to be outstanding. Our historical share option exercise experience does not provide a reasonable basis upon which to estimate an expected term because of a lack of sufficient data. Therefore, we estimate the expected term by using the simplified method provided by the SEC. The simplified method calculates the expected term as the average of the time-to-vesting and the contractual life of the options. |
In addition to the assumptions used in the Black-Scholes option-pricing model, we also estimate a forfeiture rate to calculate the stock-based compensation for our equity awards. We will continue to use judgment in evaluating the expected volatility, expected terms and forfeiture rates utilized for our stock-based compensation calculations on a prospective basis.
As of December 31, 2017, each of our non-employee directors (other than Mr. Doyle and Dr. Merino) held option awards to purchase an aggregate of 12,000 shares of our Common Stock and no stock awards. Mr. Doyle held no stock awards or option awards as of December 31, 2017. Dr. Merino held option awards to purchase an aggregate of 12,000 shares of our common stock and no stock awards.
| 72 |
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table sets forth certain information regarding the beneficial ownership of our common stock as of March 1, 2018 by:
| ● | our named executive officers; | |
| ● | each of our directors; | |
| ● | all of our current directors and executive officers as a group; and | |
| ● | each stockholder known by us to own beneficially more than five percent of our common stock. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Shares of common stock that may be acquired by an individual or group within 60 days of March 1, 2018, pursuant to the exercise of options or warrants are deemed to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table. The percentage of beneficial ownership of our common stock is calculated based on an aggregate of 49,406,521 shares outstanding as of March 1, 2018.
Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them, based on information provided to us by such stockholders. Unless otherwise indicated, the address for each director and executive officer listed is: c/o Ritter Pharmaceuticals, Inc., 1880 Century Park East, #1000, Los Angeles, California 90067.
| Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Common Stock Beneficially Owned | ||||||
| Five Percent Stockholders | ||||||||
| Javelin Entities(1) | 7,776,534 | 15.6 | % | |||||
| Aleyska Investment Group L.P.(2) | 4,902,285 | 9.9 | % | |||||
| Aspire Capital Fund LLC (3) | 4,900,000 | 9.9 | % | |||||
| Baker Bros. Funds (4) | 2,800,000 | 5.7 | % | |||||
| Executive Officers, Directors and Director Nominees | ||||||||
| Michael D. Step(5) | 1,044,818 | 2.1 | % | |||||
| Andrew J. Ritter(6) | 1,735,364 | 3.5 | % | |||||
| Ira E. Ritter(7) | 1,735,364 | 3.5 | % | |||||
| Noah J. Doyle(1)(8) | 7,806,761 | 15.7 | % | |||||
| Matthew W. Foehr (9) | 53,833 | * | ||||||
| Paul V. Maier (10) | 18,208 | * | ||||||
| Gerald T. Proehl (11) | 68,208 | * | ||||||
| Dr. William M. Merino(12) | 13,398 | * | ||||||
| All
current executive officers and directors as a group (9 persons)(13) | 16,322,434 | 28.5 | % | |||||
* Represents beneficial ownership of less than 1% of the shares of common stock.
(1) According to a Schedule 13D/A filed with the SEC on November 28, 2017 by Javelin Venture Partners, L.P. (“Javelin”), Javelin Venture Partners I SPV I, LLC (“Javelin SPV”), Javelin Venture Partners GP, L.P. (“Javelin GP, LP”), Javelin Venture Partners GP, LLC (“Javelin GP, LLC”), Noah J. Doyle and Jed Katz, in which the reporting person reported shared voting and dispositive power with respect to 7,776,534 shares. According to the Schedule 13D/A, this number consists of (i) 7,047,804 shares of common stock held directly by Javelin and 83,224 shares of common stock that Javelin has the right to acquire upon exercise of warrants to purchase common stock that are currently exercisable and (ii) 322,753 shares of common stock held directly by Javelin SPV and 322,753 shares of common stock that Javelin SPV has the right to acquire upon exercise of a warrant to purchase common stock that is currently exercisable. Javelin GP, LP serves as the general partner for Javelin and Javelin SPV, Javelin GP, LLC serves as the general partner of Javelin GP, LP, and Noah Doyle and Jed Katz serve as the managers of Javelin GP, LLC. As a result of the application of the beneficial ownership limitation described in this footnote, this number does not include 5,000,000 shares of common stock issuable upon exercise of warrants to purchase common stock owned by Javelin. Under the terms of these warrants, Javelin is not permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Javelin (and its affiliates) beneficially owning more than 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. This limitation is referred to in this footnote as the “beneficial ownership limitation”. Javelin has the right to increase the beneficial ownership limitation in its discretion on 61 days’ prior written notice to us, provided that in no event is Javelin permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Javelin (and its affiliates) beneficially owning in the aggregate more than 19.99% of the number of shares of our common stock outstanding or the combined voting power of our securities outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. The address of the Javelin Entities is One Rincon Center, 101 Spear Street, Suite 255, San Francisco, California 94105. As a Manager of Javelin GP, LLC, Noah Doyle may be deemed the beneficial owner of these shares. Mr. Doyle expressly disclaims beneficial ownership over these shares except to the extent of his pecuniary interest therein.
| 73 |
(2) According to a Schedule 13G/A filed with the SEC on February 14, 2018 by Aleyska Investment Group, L.P., Alyeska Fund GP, LLC and Anand Parekh, in which the reporting persons reported shared voting and dispositive power with respect to 4,999,371 shares. As a result of the application of the beneficial ownership limitation described in this footnote, this number does not include 4,250,000 shares of common stock issuable upon the exercise of warrants to purchase common stock owned by Aleyska Investment Group L.P. (“Aleyska”). Under the terms of these warrants, Aleyska is not permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Aleyska (and its affiliates) beneficially owning more than 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. This limitation is referred to in this footnote as the “beneficial ownership limitation”. Aleyska has the right to increase the beneficial ownership limitation in its discretion on 61 days’ prior written notice to us, provided that in no event is Aleyska permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Aleyska (and its affiliates) beneficially owning in the aggregate more than 19.99% of the number of shares of our common stock outstanding or the combined voting power of our securities outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. The address of Aleyska is 77 West Wacker Drive, 7th Floor, Chicago, IL 60601.
(3) As a result of the application of the beneficial ownership limitation described in this footnote, this number does not include (i) 7,500,000 shares of common stock issuable upon the exercise of warrants to purchase common stock owned by Aspire Capital Fund, LLC (“Aspire”) or (ii) 2,600,000 shares of common stock issuable upon the conversion of 1,040 shares of Series A Convertible preferred stock owned by Aspire. Under the terms of the warrants and Series A Convertible Preferred Stock issued to Aspire, Aspire is not permitted to exercise such warrants to purchase common stock or convert such Series A Convertible Preferred Stock into common stock to the extent that such exercise or conversion would result in Aspire (and its affiliates) beneficially owning more than 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock or conversion of Series A Convertible Preferred Stock. This limitation on the exercise of the warrants and conversion of the Series A Convertible Preferred Stock to purchase common stock issued to Aspire is referred to in this footnote as the “beneficial ownership limitation.” Aspire has the right to increase the beneficial ownership limitation in its discretion on 61 days’ prior written notice to us, provided that in no event is Aspire permitted to exercise such warrants to purchase common stock or convert Series A Convertible Preferred Stock to the extent that such exercise or conversion would result in Aspire (and its affiliates) beneficially owning in the aggregate more than 19.99% of the number of shares of our common stock outstanding or the combined voting power of our securities outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock or conversion of Series A Preferred Stock. The address for Aspire is 155 N. Wacker Drive, Suite 1600, Chicago, IL 60606.
(4) According to a Schedule 13G filed with the SEC on February 13, 2018 by Baker Bros. Advisors LP, Baker Bros. Advisors (GP) LLC, Felix J. Baker, and Julian C. Baker, in which the reporting persons reported sole voting and dispositive power with respect to 2,800,000 shares. According to the Schedule 13G, this number consists of (i) 264,902 shares of our common stock owned by 667, L.P. and (ii) 2,535,098 shares of our common stock owned by Baker Brothers Life Sciences, L.P. (collectively, the “Baker Bros. Funds”). As a result of the application of the beneficial ownership limitation described in this footnote, this number does not include the following: (a) 1,238,287 shares of common stock issuable upon exercise of warrants to purchase common stock owned by 667, L.P. and 11,261,713 shares of common stock issuable upon exercise of warrants to purchase common stock owned by Baker Brothers Life Sciences, L.P. and (b) 1,000,000 shares of common stock issuable upon conversion of 400 shares of Series A Convertible Preferred Stock owned by 667, L.P. and 9,100,000 shares of common stock issuable upon conversion of 3,640 shares of Series A Convertible Preferred Stock owned by Baker Brothers Life Sciences, L.P. The information in this footnote is provided to us by Baker Bros. Advisors LP. Baker Bros. Advisors LP serves as the investment advisor to the Baker Bros. Funds. Baker Bros. Advisors (GP) LLC is the sole general partner of Baker Bros. Advisors LP. Julian C. Baker and Felix J. Baker are principals of Baker Bros. Advisors (GP) LLC. Baker Bros. Advisors LP has complete and unlimited discretion and authority with respect to the investment and voting power of the securities held by the Baker Bros. Funds. Julian C. Baker, Felix J. Baker, Baker Bros. Advisors LP and Baker Bros. Advisors (GP) LLC disclaim beneficial ownership of all shares held by the Baker Bros. Funds, except to the extent of their indirect pecuniary interest therein. Under the terms of the warrants and Series A Convertible Preferred Stock issued to the Baker Bros. Funds, the funds are not permitted to exercise such warrants to purchase common stock or convert such Series A Convertible Preferred Stock into common stock to the extent that such exercise or conversion would result in the Baker Bros. Funds (and their affiliates) beneficially owning more than 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock or conversion of Series A Convertible Preferred Stock. This limitation on the exercise of the warrants and conversion of the Series A Convertible Preferred Stock to purchase common stock issued to the Baker Bros. Funds is referred to in this footnote as the “Beneficial Ownership Limitation.” The Baker Bros. Funds have the right to increase the Beneficial Ownership Limitation in their discretion on 61 days’ prior written notice to us, provided that in no event are the Baker Bros. Funds permitted to exercise such warrants to purchase common stock or convert Series A Convertible Preferred Stock to the extent that such exercise or conversion would result in the Funds (and their affiliates) beneficially owning in the aggregate more than 19.99% of the number of shares of our common stock outstanding or the combined voting power of our securities outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock or conversion of Series A Preferred Stock. The address for the Baker Bros. Funds is 860 Washington Street, 3rd Floor, New York, NY 10014.
| 74 |
(5) Includes 994,818 shares underlying stock option awards held by Mr. Step that are currently exercisable or exercisable within 60 days of March 1, 2018. The number of shares issuable upon exercise of options includes 70,230 shares subject to options that are currently exercisable, but are not subject to vesting within 60 days of March 1, 2018 and accordingly, if exercised, are subject to a repurchase right until vested.
(6) Includes 6,250 shares owned directly, 536,843 shares underlying stock option awards that are currently exercisable or exercisable within 60 days of March 1, 2018 and 1,192,271 shares beneficially owned by Stonehenge Partners LLC (“Stonehenge”), including 187,500 shares that are issuable upon the exercise of warrants that are currently exercisable. Under the terms of the warrants, Stonehenge is not permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Stonehenge (and its affiliates) beneficially owning more than 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. This limitation is referred to in this footnote as the “beneficial ownership limitation”. Stonehenge has the right to increase the beneficial ownership limitation in its discretion on 61 days’ prior written notice to us, provided that in no event is Stonehenge permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Stonehenge (and its affiliates) beneficially owning in the aggregate more than 19.99% of the number of shares of our common stock outstanding or the combined voting power of our securities outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. As a managing partner of Stonehenge, Andrew Ritter may be deemed the beneficial owner of these shares. Andrew Ritter expressly disclaims beneficial ownership of the shares held by Stonehenge.
(7) Includes 536,843 shares underlying stock option awards that are currently exercisable or exercisable within 60 days of March 1, 2018, 6,250 shares held in a retirement plan trust of which the reporting person and his spouse are trustees, and 1,192,271 shares beneficially owned by Stonehenge, including 187,500 shares that are issuable upon the exercise of warrants that are currently exercisable. Under the terms of the warrants, Stonehenge is not permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Stonehenge (and its affiliates) beneficially owning more than 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. This limitation is referred to in this footnote as the “beneficial ownership limitation”. Stonehenge has the right to increase the beneficial ownership limitation in its discretion on 61 days’ prior written notice to us, provided that in no event is Stonehenge permitted to exercise such warrants to purchase common stock to the extent that such exercise would result in Stonehenge (and its affiliates) beneficially owning in the aggregate more than 19.99% of the number of shares of our common stock outstanding or the combined voting power of our securities outstanding immediately after giving effect to the issuance of shares of common stock issuable upon exercise of such warrants to purchase common stock. As a managing partner of Stonehenge, Ira Ritter may be deemed the beneficial owner of these shares. Ira Ritter expressly disclaims beneficial ownership of the shares held by Stonehenge.
(8) Includes 22,727 shares owned directly by Mr. Doyle, 7,500 shares underlying a stock option award held by Mr. Doyle that are currently exercisable or exercisable within 60 days of March 1, 2018 and the shares beneficially owned by the Javelin Entities reflected in footnote (1) above. Mr. Doyle express disclaims beneficial ownership of the share held by the Javelin Entities.
(9) Includes 18,833 shares underlying a stock option award held by Mr. Foehr that are currently exercisable or exercisable within 60 days of March 1, 2018.
| 75 |
(10) Represents shares underlying a stock option award held by Mr. Maier that are currently exercisable or exercisable within 60 days of March 1, 2018.
(11) Includes 18,208 shares underlying a stock option award held by Mr. Proehl that are currently exercisable or exercisable within 60 days of March 1, 2018.
(12) Includes 12,000 shares underlying a stock option award held by Dr. Merino that are currently exercisable or exercisable within 60 days of March 1, 2018.
(13) Includes 7,775,481 shares underlying stock options and warrants that are currently exercisable or exercisable within 60 days of March 1, 2018.
Equity Compensation Plan Information
The following table sets forth aggregate information for the fiscal year ended December 31, 2016, regarding the Company’s compensation plans, including individual compensation agreements, under which equity securities of the Company are authorized for issuance:
| Plan Category | Number
of securities to be issued upon exercise of outstanding options, warrants and rights (#) | Weighted
average exercise price of outstanding options, warrants and rights ($) | Number
of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (#) | |||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders | 2,541,800 | (1) | 5.93 | 26,720,193 | (2) | |||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | 2,541,800 | (1) | 5.93 | 26,720,193 | (2) | |||||||
| (1) | Represents the number of underlying shares of common stock associated with outstanding options that were granted under the 2008 Stock Plan, the 2009 Stock Plan and the 2015 Equity Incentive Plan. |
| (2) | Represents the number of shares of common stock available for future issuance under the 2015 Equity Incentive Plan. As of June 29, 2015, no further awards were permitted to be issued under the 2008 Stock Plan or the 2009 Stock Plan. |
Item 13. Certain Relationships and Related Transactions and Director Independence
Certain Relationships and Related Party Transactions
Our Audit Committee is responsible for reviewing, approving and overseeing any transaction between the Company and its directors, director nominees, executive officers, greater than 5% beneficial owners, and each of their respective immediate family members, where the amount involved exceeds the lesser of (i) $120,000 and (ii) one percent (1%) of the average of our total assets at year-end for the prior two fiscal years. Since January 1, 2016, there have been no such transactions, except as described below.
October 2016 Public Offering
On October 31, 2016, we closed a public offering of 2,127,660 shares of our common stock at a price to the public of $2.35 per share. One of our existing stockholders holding in excess of 5% of our outstanding shares prior to the October 2016 Public Offering, Aspire, purchased shares in the offering for $1,199,999.
October 2017 Public Offering
On October 3, 2017, we closed a public offering of (i) 34,550,000 Class A Units consisting of 34,550,000 shares of our common stock and warrants to purchase 34,550,000 shares of our common stock, at a public offering price of $0.40 per unit, and (ii) 9,180 Class B Units consisting of 9,180 shares of our Series A Convertible Preferred stock, with a stated value of $1,000, and convertible into an aggregate of 22,950,000 shares of our common stock, and warrants to purchase 22,950,000 shares of our common stock, at a public offering price of $1,000 per unit. Two of our existing stockholders holding in excess of 5% of our outstanding shares prior to the October 2017 Public Offering, Javelin and Aleyska, purchased Class A Units in the public offering for $2.0 million and $1.7 million, respectively.
| 76 |
Director Independence
Under NASDAQ’s continued listing requirements, a majority of a listed company’s board of directors must be comprised of independent directors, subject to certain exceptions and phase-in rules. In addition, NASDAQ’s continued listing requirements require that, subject to certain exceptions and phase-in rules, each member of a listed company’s audit, compensation and governance and nominating committees must be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. Under NASDAQ’s continued listing requirements, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Based upon information requested from and provided by each director concerning their background, employment and affiliations, including family relationships, our board of directors has determined that each of Messrs. Doyle, Foehr, Maier, Proehl and Dr. Merino are independent under the applicable rules and regulations of NASDAQ. In making such determinations, the board of directors considered the relationships that each such non-employee director has with our company and all other facts and circumstances the board of directors deemed relevant in determining their independence.
Item 14. Principal Accountant Fees and Services
Fees and Services of Mayer Hoffman McCann P.C.
The following table sets forth the aggregate fees billed to the Company by MHM for the fiscal years ended December 31, 2017 and 2016:
| 2017 | 2016 | |||||||
| Audit Fees(1) | $ | 106,419 | $ | 167,049 | ||||
| Audit-Related Fees | ─ | ─ | ||||||
| Tax Fees | ─ | ─ | ||||||
| All Other Fees(2) | 77,900 | ─ | ||||||
| Total | $ | 184,319 | $ | 167,049 | ||||
| (1) | Audit fees consisted of fees for audit work performed in the audit of financial statements, as well as fees for quarterly reviews and registration statements. |
| (2) | All Other Fees consists of fees paid in connection with our October 2017 public offering. |
The Audit Committee has adopted a formal policy on auditor independence requiring the advance approval by the Audit Committee of all audit and non-audit services provided by our independent registered public accounting firm. In determining whether to approve any services by our independent registered public accounting firm, the Audit Committee reviews the services and the estimated fees, and considers whether approval of the proposed services will have a detrimental impact on the auditor’s independence. On an annual basis, our management reports to the Audit Committee all audit services performed during the previous 12 months and all fees billed by our independent registered public accounting firm for such services.
In fiscal 2017 and 2016, all audit services and the corresponding fees were approved by our board of directors.
| 77 |
Item 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| (a)(1) | Financial Statements. |
The following financial statements of Ritter Pharmaceuticals, Inc., together with the report thereon of Mayer Hoffman McCann P.C., an independent registered public accounting firm, are included in this Annual Report on Form 10-K:
| (a)(2) | Financial Statement Schedules. |
All schedules have been omitted because they are not required or because the required information is given in the Financial Statements or Notes thereto set forth under Item 8 above.
| (a)(3) | Exhibits. |
| 78 |
| 79 |
| 80 |
| 81 |
| 101.INS# | XBRL Instance Document. | |||||||||
| 101.SCH# | XBRL Taxonomy Extension Schema Document. | |||||||||
| 101.CAL# | XBRL Taxonomy Extension Calculation Linkbase Document. | |||||||||
| 101.DEF# | XBRL Taxonomy Extension Definition Linkbase Document. | |||||||||
| 101.LAB# | XBRL Taxonomy Extension Label Linkbase Document. | |||||||||
| 101.PRE# | XBRL Taxonomy Extension Presentation Linkbase Document. |
* Filed herewith.
+ Indicates management contract or compensatory plan or arrangement.
# XBRL (Extensible Business Reporting Language) information is furnished and not filed herewith, is not a part of a registration statement or Prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, is deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934, and otherwise is not subject to liability under these sections.
None.
| 82 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| RITTER PHARMACEUTICALS, INC. | ||
| By: | /s/ Michael D. Step | |
| Name: | Michael D. Step | |
| Title: | Chief Executive Officer | |
Date: March 19, 2018
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Michael D. Step, Andrew J. Ritter and Ira E. Ritter, jointly and severally, his attorneys-in-fact, each with the power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant on March 19, 2018 in the capacities indicated.
| Signature | Title | Date | ||
| /s/ Michael D. Step | Chief Executive Officer and Director | March 19, 2018 | ||
| Michael D. Step | (Principal Executive Officer) | |||
| /s/ Jeffrey S. Benjamin | Vice President Finance | March 19, 2018 | ||
| Jeffrey S. Benjamin | (Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Ira E. Ritter | Executive Chairman, Chief Strategic Officer | March 19, 2018 | ||
| Ira E. Ritter | and Director | |||
| /s/ Andrew J. Ritter | President and Director | March 19, 2018 | ||
| Andrew J. Ritter | ||||
| /s/ Noah Doyle | Director | March 19, 2018 | ||
| Noah Doyle | ||||
| /s/ Matthew W. Foehr | Director | March 19, 2018 | ||
| Matthew W. Foehr | ||||
| /s/ Paul V. Maier | Director | March 19, 2018 | ||
| Paul V. Maier | ||||
| /s/ William M. Merino | Director | March 19, 2018 | ||
| William M. Merino | ||||
| /s/ Gerald T. Proehl | Director | March 19, 2018 | ||
| Gerald T. Proehl |
| 83 |
INDEX TO FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
RITTER PHARMACEUTICALS, INC.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Ritter Pharmaceuticals (“Company”) as of December 31, 2017 and 2016, and the related statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has incurred recurring operating losses and is dependent on additional financing to fund operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are described in Note 1 to the financial statements. The financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ Mayer Hoffman McCann P.C. |
We have served as the Company’s auditor since 2014.
Orange County, CA
March 19, 2018
| F-1 |
RITTER PHARMACEUTICALS, INC.
| December 31, 2017 | December 31, 2016 | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | $ | 22,631,971 | $ | 7,046,282 | ||||
| Prepaid expenses | 167,400 | 156,752 | ||||||
| Total current assets | 22,799,371 | 7,203,034 | ||||||
| Other assets | 10,326 | 10,326 | ||||||
| Property and equipment, net | 23,873 | 23,542 | ||||||
| Total Assets | $ | 22,833,570 | $ | 7,236,902 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 2,237,579 | $ | 1,896,368 | ||||
| Accrued expenses | 454,252 | 1,222,735 | ||||||
| Other liablities | 15,757 | 14,736 | ||||||
| Total current liabilities | 2,707,588 | 3,133,839 | ||||||
| Stockholders’ equity | ||||||||
| Series A preferred stock, $0.001 par value; 15,000,000 shares authorized; 9,140 and 0 shares issued and outstanding as of December 31, 2017 and 2016, respectively | 5,128,536 | - | ||||||
| Common stock, $0.001 par value; 225,000,000 and 25,000,000 shares authorized; 49,406,521 and 11,619,197 shares issued and outstanding as of December 31, 2017 and December 31, 2016, respectively | 49,407 | 11,619 | ||||||
| Additional paid-in capital | 68,279,473 | 49,559,020 | ||||||
| Accumulated deficit | (53,331,434 | ) | (45,467,576 | ) | ||||
| Total stockholders’ equity | 20,125,982 | 4,103,063 | ||||||
| Total Liabilities and Stockholders’ Equity | $ | 22,833,570 | $ | 7,236,902 | ||||
The accompanying notes are an integral part of these financial statements.
| F-2 |
RITTER PHARMACEUTICALS, INC.
| For the Year Ended | ||||||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Operating costs and expenses: | ||||||||
| Research and development | $ | 2,874,184 | $ | 13,292,488 | ||||
| Patent costs | 250,372 | 272,514 | ||||||
| General and administrative | 4,777,902 | 4,881,725 | ||||||
| Total operating costs and expenses | 7,902,458 | 18,446,727 | ||||||
| Operating loss | $ | (7,902,458 | ) | $ | (18,446,727 | ) | ||
| Other income: | ||||||||
| Interest income | 40,227 | 60,879 | ||||||
| Other (expense) income | (1,627 | ) | 1,214 | |||||
| Total other income | 38,600 | 62,093 | ||||||
| Net loss | $ | (7,863,858 | ) | $ | (18,384,634 | ) | ||
| Deemed dividend of Series A preferred stock | (3,111,020 | ) | ― | |||||
| Net loss applicable to common stockholders | $ | (10,974,878 | ) | $ | (18,384,634 | ) | ||
| Net loss per common share – basic and diluted | $ | (0.50 | ) | $ | (2.04 | ) | ||
| Weighted average common shares outstanding – basic and diluted | 22,149,510 | 8,993,317 | ||||||
The accompanying notes are an integral part of these financial statements.
| F-3 |
RITTER PHARMACEUTICALS, INC.
STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
| Stockholders’ Equity | Total | |||||||||||||||||||||||||||
| Series A Preferred Stock | Common Stock | Additional | Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Paid-in Capital | Deficit | Equity | ||||||||||||||||||||||
| Balance at December 31, 2015 | - | - | 8,582,004 | 8,582 | 41,759,355 | (27,082,942 | ) | 14,684,995 | ||||||||||||||||||||
| Issuance of shares upon follow-on offering | 2,127,660 | 2,127 | 4,997,873 | 5,000,000 | ||||||||||||||||||||||||
| Commissions and offering costs of follow-on offering | (598,118 | ) | (598,118 | ) | ||||||||||||||||||||||||
| Issuance of shares under common stock purchase agreement | 888,835 | 889 | 2,025,173 | 2,026,062 | ||||||||||||||||||||||||
| Stock based compensation | 1,359,205 | 1,359,205 | ||||||||||||||||||||||||||
| Exercise of options on common stock | 20,698 | 21 | 15,532 | 15,553 | ||||||||||||||||||||||||
| Net loss | (18,384,634 | ) | (18,384,634 | ) | ||||||||||||||||||||||||
| - | $ | - | 11,619,197 | $ | 11,619 | $ | 49,559,020 | $ | (45,467,576 | ) | $ | 4,103,063 | ||||||||||||||||
| Issuance of shares upon follow-on offering | 9,180 | 5,150,980 | 34,550,000 | 34,550 | 17,844,220 | 23,029,750 | ||||||||||||||||||||||
| Commissions and offering costs of follow-on offering | (2,038,471 | ) | (2,038,471 | ) | ||||||||||||||||||||||||
| Issuance of shares under common stock purchase agreement | 3,137,324 | 3,138 | 1,996,862 | 2,000,000 | ||||||||||||||||||||||||
| Stock based compensation | 895,498 | 895,498 | ||||||||||||||||||||||||||
| Conversion of series A preferred shares | (40 | ) | (22,444 | ) | 100,000 | 100 | 22,344 | - | ||||||||||||||||||||
| Net loss | (7,863,858 | ) | (7,863,858 | ) | ||||||||||||||||||||||||
| Balance at December 31, 2017 | 9,140 | $ | 5,128,536 | 49,406,521 | $ | 49,407 | $ | 68,279,473 | $ | (53,331,434 | ) | $ | 20,125,982 | |||||||||||||||
The accompanying notes are an integral part of these financial statements.
| F-4 |
RITTER PHARMACEUTICALS, INC.
| For the Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (7,863,858 | ) | $ | (18,384,634 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 5,720 | 5,209 | ||||||
| Stock based compensation | 895,498 | 1,359,205 | ||||||
| Loss on disposal of equipment | 1,627 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other assets | (10,648 | ) | 32,384 | |||||
| Accounts payable | 341,211 | 1,157,011 | ||||||
| Accrued expenses | (768,483 | ) | 608,594 | |||||
| Other liabilities | 1,021 | 13,513 | ||||||
| Net cash used in operating activities | (7,397,912 | ) | (15,208,718 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of property and equipment | (7,678 | ) | (8,063 | ) | ||||
| Net cash used in investing activities | (7,678 | ) | (8,063 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from the issuance of shares upon closing of follow-on offering | 23,029,750 | 5,000,000 | ||||||
| Commission and issuance costs of follow-on offering | (2,038,471 | ) | (598,118 | ) | ||||
| Proceeds from the issuance of shares from common stock purchase agreement | 2,000,000 | 2,026,062 | ||||||
| Proceeds from exercising of options on common stock | - | 15,553 | ||||||
| Net cash provided by financing activities | 22,991,279 | 6,443,497 | ||||||
| Net (decrease) increase in cash and cash equivalents | 15,585,689 | (8,773,284 | ) | |||||
| Cash and cash equivalents at beginning of period | 7,046,282 | 15,819,566 | ||||||
| $ | 22,631,971 | $ | 7,046,282 | |||||
| Non-cash financing activities: | ||||||||
| Deemed dividend on Series A Preferred Stock | $ | 3,111,020 | $ | - | ||||
| Cash paid for taxes | $ | 800 | $ | 80,702 | ||||
The accompanying notes are an integral part of these financial statements.
| F-5 |
RITTER PHARMACEUTICALS, INC.
NOTE 1 — ORGANIZATION AND PRINCIPAL ACTIVITIES
Ritter Pharmaceuticals, Inc. (“Ritter” or the “Company”) is a Delaware corporation headquartered in Los Angeles, California. The Company was formed as a Nevada limited liability company on March 29, 2004 under the name Ritter Natural Sciences, LLC, and converted into a Delaware corporation on September 16, 2008.
Ritter Pharmaceuticals, Inc. develops therapeutic products that modulate the human gut microbiome to treat gastrointestinal diseases. The Company conducts human gut health research by exploring metabolic capacity of the gut microbiota and translating the functionality of prebiotic-based therapeutics. The Company’s lead compound, RP-G28 is currently under development for the treatment of lactose intolerance. There currently is no drug approved by the Food and Drug Administration (“FDA”) for the treatment of lactose intolerance, a debilitating disease that affects over 1 billion people worldwide.
The Company currently operates in one business segment focusing on the development and commercialization of RP-G28. The Company is not organized by market and is managed and operated as one business. A single management team reports to the chief operating decision maker, the Chief Executive Officer. The Company does not currently operate any separate lines of business or separate business entities.
NOTE 2 — BASIS OF PRESENTATION
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”) and include all adjustments necessary for the fair presentation of the Company’s financial position for the periods presented.
Going Concern and Liquidity
The accompanying financial statements have been prepared assuming the Company will continue as a going concern, which contemplates, among other things, the realization of assets and satisfaction of liabilities in the normal course of business. The Company has not generated any product revenue and has not achieved profitable operations. The Company had net losses of approximately $7.9 million and $18.4 million for the years ended December 31, 2017 and 2016, respectively, and had net cash used in operating activities of approximately $7.4 million and $15.2 million, for the years ended December 31, 2017 and 2016, respectively. There is no assurance that profitable operations will ever be achieved, and, if achieved, could be sustained on a continuing basis. In addition, development activities, clinical and pre-clinical testing, and commercialization of the Company’s products will require significant financing. These matters, among others, raise substantial doubt about the Company’s ability to continue as a going concern.
Since inception, the operations of the Company have been funded through the sale of common shares, preferred shares and convertible debt. Management cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that the Company raises additional funds by issuing equity securities, the Company’s stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants that impact the Company’s ability to conduct business. If the Company is not able to raise additional capital when required or on acceptable terms, the Company may have to (i) significantly delay, scale back or discontinue the development and/or commercialization of one or more product candidates; (ii) seeks collaborators for product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; (iii) relinquish or otherwise dispose of rights to technologies, product candidates or products that the Company would otherwise seek to develop or commercialize.
The financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
NOTE 3 — SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of financial statements in accordance with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates and such differences may be material to the financial statements. The more significant estimates and assumptions by management include among others; the valuation allowance of deferred tax assets resulting from net operating losses and the valuation of options on the Company’s common stock.
| F-6 |
Cash and Cash Equivalents
Cash consists of amounts held in a financial institution and consists of immediately available fund balances. The funds were maintained at a stable financial institution, generally at amounts in excess of federally insured limits, which represents a concentration of credit risk. The Company has not experienced any losses on deposits of cash and cash equivalents to date.
Property and Equipment
Property and equipment are recorded at cost and depreciated over their estimated useful lives using the straight-line method (see Note 4). Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to income. Maintenance and repairs are charged to expense as incurred while expenditures for refurbishments and improvements that significantly add to the productive capacity or extend the useful life of an asset are capitalized.
Impairment of Long-Lived Assets
The Company periodically assesses the impairment of long lived assets in accordance with Accounting Standards Codification (“ASC”) Topic 360, Property Plant and Equipment. When indicators of impairment are present, the Company evaluates the carrying value of these assets in relation to the operating performance of the business and future undiscounted cash flows expected to result from the use of these assets. No such impairments have been recognized during the years ended December 31, 2017 or 2016.
Clinical Trial and Pre-Clinical Study Accruals
The Company makes estimates of accrued expenses as of each balance sheet date in its financial statements based on the facts and circumstances known to it at that time. Accrued expenses for pre-clinical studies and clinical trials are based on estimates of costs incurred and fees that may be associated with services provided by contract research organizations, clinical trial investigational sites, and other related vendors. Payments under certain contracts with such parties depend on factors such as successful enrollment of patients, site initiation and the completion of milestones. In accruing service fees, management estimates the time period over which services will be performed and the level of effort to be expended in each period. If possible, the Company obtains information regarding unbilled services directly from these service providers. However, the Company may be required to estimate these services based on other information available to it. If the Company underestimates or overestimates the activity or fees associated with a study or service at a given point in time, adjustments to research and development expenses may be necessary in future periods. Historically, estimated accrued liabilities have approximated actual expense incurred. Subsequent changes in estimates may result in a material change in the Company’s accruals.
Research and Development
The Company expenses the cost of research and development as incurred. Research and development expenses comprise costs incurred in performing research and development activities, including clinical trial costs, manufacturing costs for both clinical and pre-clinical materials as well as other contracted services, license fees, and other external costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity is performed or when the goods have been received, rather than when payment is made, in accordance with ASC Topic 730, Research and Development.
Patent Costs
The Company has no historical data to support a probable future economic benefit for the arising patent applications, filing and prosecution costs. Therefore, patent costs are expensed as incurred. Should the Company experience a legal cost to defend a patent in the future, that cost would be capitalized only when it is part of the cost of retaining and obtaining the future economic benefit of the patent. Costs related to an unsuccessful outcome would be expensed.
Stock-based Compensation
Stock-based compensation cost for stock awards issued to employees, members of the Company’s board of directors and non-employees, is measured at the grant date based on the fair value of the award and is recognized as expense over the required service period, which is generally equal to the vesting period. Stock-based compensation is recognized only for those awards that are ultimately expected to vest. Common stock, stock options or warrants issued to non-employees, including consultants and members of the Company’s Scientific Advisory Board as consideration for goods or services received by the Company, are accounted for based on the fair value of the equity instruments issued unless the fair value consideration received can be more reliably measured. The fair value of stock options is determined using the Black-Scholes option-pricing model. The fair value of any options issued to non-employees is recorded as expense over the vesting period. See Note 8 for further information.
| F-7 |
Fair Value Measurements
The Company follows authoritative accounting guidance, which among other things, defines fair value, establishes a consistent framework for measuring fair value and expands disclosure for each major asset and liability category measured at fair value on either a recurring or nonrecurring basis. Fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or liability.
As a basis for considering such assumptions, a three-tier fair value hierarchy has been established, which prioritizes the inputs used in measuring fair value as follows:
| Level 1: | Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities; | |
| Level 2: | Inputs other than quoted prices that are observable for the asset or liability, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. | |
| Level 3: | Unobservable inputs that reflect the reporting entity’s own assumptions. |
As of December 31, 2017 and 2016, the carrying amounts of cash and cash equivalents, prepaid expenses, accounts payable and accrued expenses are generally considered to be representative of their fair value because of the short term nature of these instruments. No transfers between levels have occurred during the periods presented.
Convertible Preferred Stock
The Company follows authoritative accounting guidance to distinguish liabilities from equity when assessing the classification and measurement of preferred stock. Preferred shares subject to mandatory redemptions are considered liabilities and measured at fair value. Conditionally redeemable preferred shares are considered temporary equity. All other preferred shares are considered as stockholders’ equity.
Derivative Financial Instruments
The Company does not use derivative instruments to hedge exposures to cash flow, market, or foreign currency risks. Management evaluates all of the Company’s financial instruments, including warrants, to determine if such instruments are derivatives or contain features that qualify as embedded derivatives. The Company generally uses either the Black-Scholes option-pricing model or a Monte Carlo simulation, as applicable, to value the derivative instruments at inception and subsequent valuation dates when needed. The classification of derivative instruments, including whether such instruments should be recorded as liabilities or as equity, is re-assessed at the end of each reporting period.
Accounting for Income Taxes
Deferred tax assets and liabilities are recognized for the expected future consequences of events that have been reflected in the financial statements. Deferred tax assets and liabilities are determined based on the differences between the book and tax basis of assets and liabilities and operating loss carryforwards, using tax rates expected to be in effect for the years in which the differences are expected to reverse. Such differences arise primarily from stock-based compensation and net operating loss carryforwards. The Company records a valuation allowance to reduce deferred income tax assets when it is more likely than not that some portion or all of the deferred tax asset will not be realized. Prior to September 15, 2008, the Company was a limited liability company and the Company’s tax losses and credits generally flowed directly to the members.
Net Loss Per Share
Basic net loss per share is calculated by dividing net loss by the weighted-average common shares outstanding during the period. Diluted net loss per share is calculated by dividing net loss by the weighted-average common shares outstanding during the period using the treasury stock method or the two-class method, whichever is more dilutive. The potentially dilutive stock options issued under the 2015 Equity Incentive Plan (described in Note 8), Series A Convertible Preferred Stock (described in Note 6) and warrants on the Company’s common stock (described in Notes 6 and 7) are not considered in the computation of diluted net loss per share because they would be anti-dilutive.
| F-8 |
Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. The Company is required to record all components of comprehensive loss in the financial statements in the period in which they are recognized. Net income (loss) and other comprehensive loss, including foreign currency translation adjustments and unrealized gains and losses on investments are reported, net of their related tax effect, to arrive at a comprehensive loss. For the years ended December 31, 2017 and 2016, comprehensive loss was equal to the net loss.
Recent Accounting Pronouncements
In February 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update 2016-02, Leases (Topic 842) (“ASU 2016-02”). The provisions of ASU 2016-02 set out the principles for the recognition, measurement, presentation and disclosure of leases for both parties to a contract (i.e., lessees and lessors). The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether the lease expense is recognized based on an effective interest method or on a straight line basis over the term of the lease. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than 12 months regardless of their classification. Leases with a term of 12 months or less will be accounted for under the existing guidance for operating leases today. Topic 842 supersedes the previous lease standard, Topic 840 Leases. The guidance is effective for annual periods and interim periods within those annual periods beginning after December 15, 2018, and is effective for the Company for the year ending December 31, 2019. The Company is currently evaluating the impact that the implementation of this standard will have on the Company’s financial statements.
On March 30, 2016, the FASB issued Accounting Standards Update No. 2016-09, Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting (“ASU 2016-09”). Among other things, ASU 2016-09 requires that entities recognize excess tax benefits and deficiencies related to employee share-based payment transactions as income tax expense or benefit. ASU 2016-09 also eliminates the requirement to reclassify excess tax benefits and deficiencies from operating activities to financing activities in the statement of cash flows. The guidance is effective for the annual periods and interim periods within those annual periods beginning after December 15, 2016. The adoption of this standard did not have a material impact on the Company’s financial statements.
On August 26, 2016, the FASB issued Accounting Standards Update No. 2016-15, Statement of Cash Flows (Topic 230), a consensus of the FASB’s Emerging Issues Task Force (“ASU 2016-15”). The new guidance amends Accounting Standards Codification No. 230 (“ASC 230”) to add or clarify guidance on the classification of certain cash receipts and payments in the statement of cash flows. ASC 230 lacks consistent principles for evaluating the classification of cash payments and receipts in the statement of cash flows. This has led to diversity in practice and, in certain circumstances, financial statement restatements. Therefore, the FASB issued ASU 2016-15 with the intent of reducing diversity in practice with respect to eight types of cash flows. ASU 2016-15 is effective for annual and interim periods in fiscal years beginning after December 15, 2017, and is effective for the Company for the year ending December 31, 2018. The Company is currently evaluating the impact that the implementation of this standard will have on the Company’s financial statements.
In May 2017, the FASB issued Accounting Standards Update No. 2017-09, Compensation - Stock Compensation (Topic 718): Scope of Modification Accounting (“ASU 2017-09”). ASU 2017-09 provides guidance on the types of changes to the terms or conditions of share-based payment awards to which an entity would be required to apply modification accounting. An entity would not apply modification accounting if the fair value, vesting conditions, and classification of the awards are the same immediately before and after the modification. The amendments are effective for the Company’s interim and annual reporting periods beginning January 1, 2018. The Company does not expect the adoption of ASU 2017-09 to have a material impact on its financial statements.
In July 2017, the FASB issued ASU 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception. The ASU allows companies to exclude a down round feature when determining whether a financial instrument (or embedded conversion feature) is considered indexed to the entity’s own stock. As a result, financial instruments (or embedded conversion features) with down round features may no longer be required to be classified as liabilities. A company will recognize the value of a down round feature only when it is triggered and the strike price has been adjusted downward. For equity-classified freestanding financial instruments, such as warrants, an entity will treat the value of the effect of the down round, when triggered, as a dividend and a reduction of income available to common shareholders in computing basic earnings per share. For convertible instruments with embedded conversion features containing down round provisions, entities will recognize the value of the down round as a beneficial conversion discount to be amortized to earnings. The guidance in ASU 2017-11 is effective for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted, and the guidance is to be applied using a full or modified retrospective approach. The Company early adopted this guidance in the current quarter, effective October 1, 2017. As a result, the warrants issued in 2017, in connection with the October 2017 Public Offering, were equity-classified.
| F-9 |
NOTE 4 — PROPERTY AND EQUIPMENT
Property and equipment consists of the following:
| Estimated Life | December 31, 2017 | December 31, 2016 | ||||||||
| Computer equipment | 5 years | $ | 13,582 | $ | 10,274 | |||||
| Furniture and fixtures | 7 years | 19,158 | 23,325 | |||||||
| Total property and equipment | 32,740 | 33,599 | ||||||||
| Accumulated depreciation | (8,867 | ) | (10,057 | ) | ||||||
| Property and equipment, net | $ | 23,873 | $ | 23,542 | ||||||
Depreciation expense of approximately $5,700 and $5,200 was recognized for the years ended December 31, 2017 and 2016, respectively, and is classified in general and administrative expense in the accompanying Statements of Operations.
NOTE 5 — COMMITMENTS AND CONTINGENCIES
Master Services Agreement
On December 30, 2015, we entered into a Master Service Agreement with Covance, Inc. (“Covance”), with an effective date of December 29, 2015. Pursuant to the terms of the Master Service Agreement, Covance (or one or more of its affiliates) will provide Phase 1, 2, 3, and 4 clinical services for a clinical study or studies to us, and, at our request, assist us with the design of such studies, in accordance with the terms of separate individual project agreements to be entered into by the parties. The term of the agreement is for three years and will renew automatically for successive one year periods unless Covance is no longer providing services under the agreement or either party has terminated the agreement upon written notice. We may terminate the Master Service Agreement or any individual project agreement entered into under the Master Service Agreement prior to the applicable study’s completion at any time for any reason upon 30 days written notice to Covance, except when the reason for termination is the safety of subjects, in which case it may be terminated immediately. Covance may not terminate any individual project agreement without cause, except when the reason for the termination is the safety of subjects, in which case it may be terminated immediately. In the event of a termination of the Master Service Agreement, Covance will be entitled to full payment for (i) work performed on the applicable project upon through the date work on such project is concluded and (ii) reimbursement for all non-cancellable and non-refundable expenses and financial obligations which Covance (or an affiliate) has incurred or undertaken on our behalf.
Clinical Supply and Cooperation Agreement with Ricerche Sperimentali Montale (“Ricerche”) and Inalco SpA (“Inalco”)
Effective July 24, 2015, we entered into an amended Clinical Supply and Cooperation Agreement (the “Amended Supply Agreement”) with Ricerche and Inalco (collectively, “RSM”). The Amended Supply Agreement amends certain terms of the Clinical Supply and Cooperation Agreement, dated December 16, 2009, amended on September 25, 2010 (the “Existing Supply Agreement”).
Under the Existing Supply Agreement, RSM granted us an exclusive worldwide option in a specified field and territory to assignment of all right, title and interest to a purified galacto-oligosaccharides product (“Improved GOS”), the composition of matter of the Improved GOS and any information relating to the Improved GOS, including certain specified technical information and other intellectual property rights (the “Improved GOS IP”). Pursuant to the amended terms, we could exercise the option by paying RSM $800,000 within ten days after the effective date of the Amended Supply Agreement. We exercised this option on July 30, 2015 and RSM transferred the Improved GOS IP to us. Under the terms of the Existing Supply Agreement, if a further option payment of $1 million due in the future is not made, we may be required to return the Improved GOS IP to RSM.
The Amended Supply Agreement also provides that we must pay RSM $400,000 within 10 days following FDA approval of a new drug application for the first product owned or controlled by us using Improved GOS as its active pharmaceutical ingredient.
In consideration for RSM entering into the Amended Supply Agreement, we issued 100,000 shares of our common stock, par value $0.001 per share (the “RSM Shares”), to RSM on November 30, 2015. Fair value of these shares totaling $416,000 was recognized in stockholders’ equity in the balance sheet as of December 31, 2015.
| F-10 |
Lease Agreement
On July 9, 2015, we entered into a lease with Century Park, a California limited partnership, pursuant to which we are leasing approximately 2,780 square feet of office space in Los Angeles, California for our headquarters. The lease provides for a term of sixty-one (61) months, commencing on October 1, 2015. We paid no rent for the first month of the term and will pay base rent of $9,174 per month for months 2 through 13 of the term, with increasing base rent for each twelve month period thereafter under the term of the lease to a maximum of $10,325 per month for months 50 through 61. The base rent payments do not include our proportionate share of any operating expenses, including real estate taxes. We have the option to extend the term of the lease for one five-year term, provided that the rent would be subject to market adjustment at the beginning of the renewal term. We will recognize rent expense on a straight-line basis over the lease term.
Rent expense, recognized on a straight-line basis, was approximately $115,000 and $115,000 for the years ended December 31, 2017 and 2016, respectively, and is recorded in general and administrative expenses in the accompanying statements of operations.
The following table summarizes our lease obligations at December 31, 2017:
| LEASE COMMITMENTS | ||||
| Years ended December 31, | Operating Lease | |||
| 2018 | $ | 117,376 | ||
| 2019 | 120,898 | |||
| 2020 | 103,254 | |||
| Total minimum lease payments | $ | 341,528 | ||
Legal
The Company is not currently involved in any legal matters arising in the normal course of business. From time to time, the Company could become involved in disputes and various litigation matters that arise in the normal course of business. These may include disputes and lawsuits related to intellectual property, licensing, contract law and employee relations matters. Periodically, the Company reviews the status of significant matters, if any exist, and assesses its potential financial exposure. If the potential loss from any claim or legal claim is considered probable and the amount can be estimated, the Company accrues a liability for the estimated loss. Legal proceedings are subject to uncertainties, and the outcomes are difficult to predict. Because of such uncertainties, accruals are based on the best information available at the time. As additional information becomes available, the Company reassesses the potential liability related to pending claims and litigation.
NOTE 6— STOCKHOLDERS’ EQUITY
Authorized Shares
On September 15, 2017, the Company amended its Amended and Restated Certificate of Incorporation to authorize the issuance of up to 225,000,000 shares of common stock, $0.001 par value per share, and 15,000,000 shares of preferred stock, $0.001 par value per share.
As of December 31, 2017, the Company had 49,406,521 shares of common stock and 9,140 shares of Series A convertible preferred stock issued and outstanding. Each share of the Company’s common stock is entitled to one vote, and all shares rank equally as to voting and other matters. Each share of Series A preferred stock is convertible by the holder at $0.40 per share; subject to adjustment for stock splits, stock dividends, subsequent rights offerings, pro rata distributions, and fundamental transactions. Holders are entitled to receive, and the Company shall pay, dividends on outstanding shares of Series A preferred stock, on an as-if-converted-to-common-stock basis, equal to and in the same form as dividends actually paid on outstanding common shares when, as and if such dividends are paid on outstanding common shares. Upon any liquidation, dissolution or winding-up of the Company, whether voluntary or involuntary, the holders of Series A preferred stock shall be entitled to receive out of the assets, whether capital or surplus, of the Company the same amount that a holder of common stock would receive if the Series A preferred stock were fully converted to common stock, which amounts shall be paid pari passu with all common shareholders. Holders of Series A preferred stock have no voting rights. However, as long as any shares of Series A preferred stock are outstanding, the Company shall not, without the affirmative vote of the holders of a majority of the then outstanding shares of Series A preferred stock, (a) alter or change adversely the powers, preferences or rights given to the Series A preferred stock or alter or amend the applicable Certificate of Designation, (b) amend the Company’s certificate of incorporation or other charter documents in any manner that adversely affects any rights of the holders of Series A preferred stock, (c) increase the number of authorized shares of Series A preferred stock, or (d) enter into any agreement with respect to any of the foregoing.
| F-11 |
Aspire Capital Financing Arrangement
On December 18, 2015, the Company entered into a common stock purchase agreement (the “2015 Aspire Purchase Agreement”) with Aspire Capital Fund, LLC (“Aspire Capital”), pursuant to which Aspire Capital was committed to purchase up to an aggregate of $10.0 million of the Company’s shares of common stock over the approximate 30-month term of the 2015 Aspire Purchase Agreement. As of December 31, 2017, the Company had issued an aggregate of 4,577,699 shares of its common stock to Aspire Capital under the 2015 Aspire Purchase Agreement for approximate proceeds of $5.0 million.
On May 4, 2017, the Company terminated the 2015 Aspire Purchase Agreement and entered into a new common stock purchase agreement with Aspire Capital (the “2017 Aspire Purchase Agreement”), which provides that upon the terms and conditions set forth therein, Aspire Capital is committed to purchase up to an aggregate of $6.5 million of shares of the Company’s common stock over the 30-month term of the 2017 Aspire Purchase Agreement. On any trading day on which the closing sale price of the Company’s common stock exceeds $0.25, the Company has the right, in its sole discretion, to present Aspire Capital with a purchase notice, directing Aspire Capital (as principal) to purchase up to 100,000 shares of the Company’s common stock per trading day, for up to $6.5 million of the Company’s common stock in the aggregate at a per share price, calculated by reference to the prevailing market price of the Company’s common stock (as provided in the 2017 Aspire Purchase Agreement).
As a condition to the 2017 Aspire Purchase Agreement, the Company issued 137,324 shares of its common stock to Aspire Capital as a commitment fee. As of the date of this Annual Report, no shares of common stock have been sold to Aspire Capital under the 2017 Aspire Purchase Agreement.
October 2016 Public Offering
On October 31, 2016, the Company closed a public offering, selling 2,127,660 shares of the Company’s common stock at a price to the public of $2.35 per share, for aggregate gross proceeds to the Company of approximately $5.0 million. The Company paid to the underwriters underwriting discounts and commissions of approximately $0.4 million in connection with the offering, and approximately $0.2 million of other expenses in connection with the offering.
This offering was made pursuant to a shelf registration statement on Form S-3, which was declared effective by the SEC on August 23, 2016. The shelf registration statement allows the Company to issue, from time to time at prices and on terms to be determined at or prior to the time of an offering, up to $150,000,000 of any combination of an indeterminate number of shares of common stock, an indeterminate number of shares of preferred stock, an indeterminate principal amount of debt securities, an indeterminate number of warrants, rights and purchase contracts to purchase common stock or debt securities, and an indeterminate number of units. If any debt securities are issued at an original issue discount, then the offering price of such debt securities shall be in such greater principal amount as shall result in an aggregate offering price not to exceed $150,000,000, less the aggregate dollar amount of all securities previously issued hereunder. The securities registered also include such indeterminate number of shares of common stock and preferred stock that may be issued upon conversion or exchange of convertible or exchangeable securities being registered or pursuant to the anti-dilution provisions of any such securities.
October 2017 Public Offering
On October 3, 2017, the Company closed a public offering, selling an aggregate of (i) 34,550,000 Class A Units consisting of 34,550,000 shares of the Company’s common stock and warrants to purchase 34,550,000 shares of the Company’s common stock at a public offering price of $0.40 per unit, and (ii) 9,180 Class B Units consisting of 9,180 shares of Series A Convertible Preferred Stock, with a stated value of $1,000 per unit, and convertible into an aggregate of 22,950,000 shares of the Company’s common stock, and warrants to purchase an aggregate of 22,950,000 shares of the Company’s common stock. The securities were offered by the Company pursuant to a registration statement filed with the SEC that was declared effective on September 28, 2017. The final prospectus relating to the offering was filed with the SEC on October 2, 2017.
The warrants have an exercise price of $0.44, are exercisable upon issuance and expire five years from the date of issuance. The warrant agreements provide for an adjustment to the number of common shares issuable under the warrants and/or adjustment to the exercise price, including but not limited to, if: (a) the Company issues shares of common stock as a dividend or distribution to holders of its common stock; (b) the Company subdivides or combines its common stock; or (c) adjustment of the exercise price upon subsequent equity sales or issuance of new securities by the Company at less than the exercise price.
The Company granted the underwriters a 45-day option to purchase an additional 8,625,000 shares of the Company’s common stock and/or warrants to purchase an additional 8,625,000 shares of the Company’s common stock. As of the closing of the offering, the underwriters have exercised their over-allotment option for warrants to purchase 2,975,000 shares of the Company’s common stock.
| F-12 |
Aggregate gross proceeds to the Company from the public offering were approximately $23.0 million. The Company paid underwriting discounts and commissions of approximately $1.6 million in connection with the offering, and approximately $0.4 million of other expenses in connection with the offering.
The Company early adopted the provisions of ASU 2017-11 in recognizing the warrants. As a result, the exercise price reset provisions were excluded from the assessment of whether the warrants are considered indexed to the Company’s own stock. The warrants otherwise meet the requirements for equity classification, as such were initially classified in Stockholders’ Equity. The Company will recognize the value of the exercise price reset provision if and when it becomes triggered, by recognizing the value of the effect of the exercise price reset as a deemed dividend and a reduction of income available to common shareholders in computing basic earnings per share.
The proceeds received in the October 2017 Public Offering were allocated to each instrument on a relative fair value basis. Total proceeds of $23.0 million were allocated to warrants issued, $10.1 million, Common Stock, $7.8 million and Series A Preferred Stock, $5.1 million. The allocation resulted in an effective conversion price for the Series A preferred stock that was below the quoted market price of the Company’s common stock on the closing date. As such, the Company recognized a beneficial conversion feature equal to the intrinsic value of the conversion feature on the closing date, resulting in a deemed dividend for the Series A preferred stock of approximately $3.1 million recognized on the closing date.
NOTE 7 — WARRANTS
Warrants to purchase an aggregate of 61,053,323 shares of the Company’s common stock were outstanding at December 31, 2017. These warrants are all vested and exercisable, have exercise prices ranging from $0.44 to $9.30 per share, with a weighted average exercise price of $0.52, and expire at various dates through October 2022.
NOTE 8 — STOCK-BASED COMPENSATION
Equity Incentive Plans
The Company has issued equity awards pursuant to its 2015 Equity Incentive Plan (the “2015 Plan”), 2009 Stock Plan and 2008 Stock Plan (collectively the “Plans”). The Plans permit the Company to grant non-statutory stock options, incentive stock options and other equity awards to the Company’s employees, outside directors and consultants; however, incentive stock options may only be granted to the Company’s employees. Beginning June 29, 2015, no further awards may be granted under the 2009 Stock Plan or 2008 Stock Plan. However, to the extent awards under the 2008 Plan or 2009 Plan are forfeited or lapse unexercised or are settled in cash, the common stock subject to such awards will be available for future issuance under the 2015 Equity Incentive Plan.
On June 2, 2017, the stockholders of the Company approved an amendment to the 2015 Plan at the 2017 annual meeting of stockholders, which among other things, increased the number of shares that may be issued pursuant to awards under the 2015 Plan by 838,000 shares of common stock.
On September 15, 2017, the stockholders of the Company approved an amendment to the 2015 Plan at a special meeting of stockholders, which among other things, increased the number of shares that may be issued pursuant to awards under the 2015 Plan by 25,858,711 shares of common stock. As of December 31, 2017, the aggregate number of shares of common stock authorized for issuance under the 2015 Plan, as amended, was 27,500,000, and 26,720,193 shares were available for issuance as of December 31, 2017.
The following represents a summary of the options granted to employees and non-employees that are outstanding at December 31, 2017 and changes during the period then ended:
| Number of Shares | Weighted- Average Exercise Price | Aggregate
Intrinsic Value | Weighted- Average Remaining Contractual Life (in years) | |||||||||||||
| Outstanding at December 31, 2016 | 2,476,924 | $ | 6.01 | $ | 497,351 | 8.3 | ||||||||||
| Options granted | 88,000 | 2.89 | ― | 7.3 | ||||||||||||
| Options forfeited | (23,124 | ) | 2.52 | ― | ― | |||||||||||
| Outstanding at December 31, 2017 | 2,541,800 | 5.93 | ― | 7.3 | ||||||||||||
| Exercisable at December 31, 2017 | 1,718,370 | $ | 6.02 | $ | ― | 7.0 | ||||||||||
| F-13 |
The exercise price for an option issued under the Plans is determined by the Board of Directors, but will be (i) in the case of an incentive stock option (A) granted to an employee who, at the time of grant of such option, is a 10% stockholder, no less than 110% of the fair market value per share on the date of grant; or (B) granted to any other employee, no less than 100% of the fair market value per share on the date of grant; and (ii) in the case of a non-statutory stock option, no less than 100% of the fair market value per share on the date of grant. The options awarded under the Plans will vest as determined by the Board of Directors but will not exceed a ten-year period.
Fair Value of Equity Awards
The Company utilizes the Black-Scholes option pricing model to value awards under its Plans. Key valuation assumptions include:
| ● | Expected dividend yield. The expected dividend is assumed to be zero as the Company has never paid dividends and has no current plans to pay any dividends on the Company’s common stock. |
| ● | Expected stock-price volatility. As the Company’s common stock only recently became publicly traded, the expected volatility is derived from the average historical volatilities of publicly traded companies within the Company’s industry that the Company considers to be comparable to the Company’s business over a period approximately equal to the expected term. |
| ● | Risk-free interest rate. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of grant for zero coupon U.S. Treasury notes with maturities approximately equal to the expected term. |
| ● | Expected term. The expected term represents the period that the stock-based awards are expected to be outstanding. The Company’s historical share option exercise experience does not provide a reasonable basis upon which to estimate an expected term because of a lack of sufficient data. Therefore, the Company estimates the expected term by using the simplified method provided by the SEC. The simplified method calculates the expected term as the average of the time-to-vesting and the contractual life of the options. |
The Company elected to adopt the amendments of ASU 2016-09 (described in Note 3) related to the presentation of excess tax benefits on the statement of cash flows using a prospective transition method and there was no impact to these financial statements.
The material factors incorporated in the Black-Scholes model in estimating the fair value of the options granted for the periods presented were as follows:
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Expected dividend yield | 0.00 | % | 0.00 | % | ||||
| Expected stock-price volatility | 53.08% - 54.08 | % | 53.60% - 59.03 | % | ||||
| Risk-free interest rate | 1.89% - 2.58 | % | 1.29% - 1.78 | % | ||||
| Term of options | 10 | 10 | ||||||
| Stock price | $0.30 - $3.48 | $1.13 - $1.68 | ||||||
Stock-Based Compensation
The Company recognized stock-based compensation expense for services within general and administrative expense in the accompanying statements of operations of approximately $895,000 and $1,359,000 for the years ended December 31, 2017 and 2016, respectively. As of December 31, 2017, there was approximately $414,000 of total unrecognized compensation cost related to unvested stock-based compensation arrangements. This cost is expected to be recognized over a weighted average period of 1.0 years.
The aggregate intrinsic value of stock options exercised during the years ended December 31, 2017 and 2016 was approximately $0 and $56,000, respectively. The cash received from exercised options was approximately $16,000 for the year ended December 31, 2017.
| F-14 |
NOTE 10 — RELATED PARTY TRANSACTIONS
A director of the Company is a managing director of Javelin Venture Partners GP, LLC, the general partner of Javelin Venture Partners GP, L.P., which holds a significant investment in the Company’s common stock and warrants. Two directors of the Company have acted as a managing director of Stonehenge Partners, LLC, which holds significant investment in the Company’s common stock.
Other than disclosed, the Company has not entered into or been a participant in any transaction in which a related party had or will have a direct or indirect material interest.
NOTE 11 — INCOME TAXES
As of December 31, 2017, the Company has net operating loss carryforwards of approximately $39 million available to reduce future taxable income, if any, for Federal and state income tax purposes. The U.S. federal and state net operating loss carryforwards will begin to expire in 2028.
As of December 31, 2017, the Company has Federal and state research and development credit carryforwards of approximately $1.4 million and $800,000, respectively, available to reduce future taxable income, if any, for Federal and state income tax purposes. The Federal credit carryforwards begin to expire in 2029. California credits have no expiration date.
Under the Internal Revenue Code (“IRC”) Sections 382 and 383, annual use of the Company’s net operating loss and research tax credit carryforwards to offset taxable income may be limited based on cumulative changes in ownership. The Company has not completed an analysis to determine whether any such limitations have been triggered as of December 31, 2017. The Company has no income tax affect due to the recognition of a full valuation allowance on the expected tax benefits of future loss carry forwards based on uncertainty surrounding realization of such assets.
A reconciliation of the statutory income tax rates and the Company’s effective tax rate is as follows:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Statutory U.S. federal rate | 34.0 | % | 34.0 | % | ||||
| State income tax, net of federal benefit | 5.8 | % | 5.8 | % | ||||
| Meals & entertainment | (0.2 | )% | (0.1 | )% | ||||
| Valuation allowance | (39.6 | )% | (39.7 | )% | ||||
| Provision for income taxes | 0.0 | % | 0.0 | % | ||||
The tax effects of the temporary differences and carry forwards that give rise to deferred tax assets consist of the following:
| As of December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carry forwards | $ | 10,906,689 | $ | 12,862,811 | ||||
| Patent costs | 325,615 | 363,775 | ||||||
| Accrued Vacation | 21,019 | 31,583 | ||||||
| Research and development credit | 1,932,804 | 1,951,539 | ||||||
| Stock-based compensation | 1,722,380 | 2,095,076 | ||||||
| Other | 8,766 | 7,535 | ||||||
| Gross deferred tax assets | 14,917,273 | 17,312,319 | ||||||
| Valuation allowance | (14,917,273 | ) | (17,312,319 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
The Company did not record any accruals for income tax accounting uncertainties for the years ended December 31, 2017 and 2016.
Authoritative guidance requires companies to accrue interest and related penalties, if applicable, on all tax positions for which reserves have been established consistent with jurisdictional tax laws. The Company’s policy is to recognize interest and penalties that would be assessed in relation to the settlement value of unrecognized tax benefits as a component of income tax expense. The Company did not accrue either interest or penalties from inception through December 31, 2017.
The Company does not have any unrecognized tax benefits that will significantly decrease or increase within 12 months of December 31, 2017.
The Company’s major tax jurisdictions are the United States and California. All of the Company’s tax years will remain open three and four years for examination by the Federal and state tax authorities, respectively, from the date of utilization of the net operating loss. The Company does not have any tax audits pending.
Regarding 2018 federal tax reform, the Company re-measured certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21%. For certain deferred tax assets, the Company has recorded a decrease of approximately $5.5 million, with a corresponding adjustment to valuation allowance for the year ended December 31, 2017. There is no impact on the tax expense.
| F-15 |